
ナフトキノン誘導体化合物
【課題】チロシンホスファターゼの阻害剤として有用な新規化合物、並びに、それを有効成分として含有する、チロシンホスファターゼ阻害剤、医薬組成物、細胞増殖阻害剤、細胞周期進行阻害剤、抗菌剤、イミペネム活性増強剤、及び細菌増殖阻害剤、並びに、上述の化合物を用いたイミペネム活性増強方法、及び細菌の増殖阻害方法、並びに、上述の化合物の活性増強剤及び活性増強方法を提供すること。
【解決手段】本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩は、チロシンホスファターゼに対する阻害活性、細胞増殖阻害作用、細胞周期進行に対する阻害作用、抗菌作用、イミペネムの活性を増強する作用、細菌増殖を阻害する作用を有する。また、イミペネムはナフトキノン誘導体化合物又はその薬理学的に許容される塩の活性を増強する作用を有する。
【解決手段】本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩は、チロシンホスファターゼに対する阻害活性、細胞増殖阻害作用、細胞周期進行に対する阻害作用、抗菌作用、イミペネムの活性を増強する作用、細菌増殖を阻害する作用を有する。また、イミペネムはナフトキノン誘導体化合物又はその薬理学的に許容される塩の活性を増強する作用を有する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ナフトキノン誘導体化合物又はその薬理学的に許容される塩、並びに、それを有効成分として含有する、チロシンホスファターゼ阻害剤、医薬組成物、細胞増殖阻害剤、細胞周期進行阻害剤、抗菌剤、イミペネム活性増強剤、及び細菌増殖阻害剤、並びに、上述の化合物を用いたイミペネム活性増強方法、及び細菌の増殖阻害方法、並びに、上述の化合物の活性増強剤及び活性増強方法に関する。
【背景技術】
【0002】
チロシンホスファターゼに対する阻害剤は、2型糖尿病、肥満症、腫瘍、神経疾患等のチロシンホスファターゼの過剰発現又は活性化に起因する疾患に有用であるとされており、各種チロシンホスファターゼに対する阻害剤の開発が求められている。従来、チロシンホスファターゼであるCdc25に対する阻害剤として、いくつかのナフトキノンアナログが知られている(例えば、非特許文献1〜4参照)。また、チロシンホスファターゼであるPTP1Bに対する阻害剤として、いくつかの化合物やクロレラの細胞壁破砕物等が知られている(例えば、特許文献1〜5参照)。
【非特許文献1】Bioorg. Med. Chem. Lett. 1998, 8(18), 2507-10
【非特許文献2】J. Antibiot. 1999, 52, 256-262
【非特許文献3】Bull Chem Soc Jpn. 2004, 77, 1925-30
【非特許文献4】Bioorg. Med. Chem. Lett. 2005, 15(1), 61-5
【特許文献1】特開平11−222474号公報
【特許文献2】特開2000−256330号公報
【特許文献3】特開2002−121186号公報
【特許文献4】特開2005−89322号公報
【特許文献5】特表平11−508919号公報
【発明の開示】
【発明が解決しようとする課題】
【0003】
本発明は、チロシンホスファターゼの阻害剤として有用な新規化合物、並びにそれを有効成分として含有する、チロシンホスファターゼ阻害剤、医薬組成物、細胞増殖阻害剤、細胞周期進行阻害剤、抗菌剤、イミペネム活性増強剤、及び細菌増殖阻害剤、並びに、上述の化合物を用いたイミペネム活性増強方法、及び細菌の増殖阻害方法、並びに、上述の化合物の活性増強剤及び活性増強方法に関する。
【課題を解決するための手段】
【0004】
本発明者らは、上記課題を解決すべく、下記の一般式(II)で表されるナフトキノン誘導体化合物(式中、R5は水素原子又はアルキル基であり、R6はヒドロキシエチル基、ヒドロキシプロピル基、ヒドロキシヘキシル基、アリールアルコール基、又はアルキルカルボニル基であり、R7は水素原子、アシル基、又はブチルジメチルシリル基であり、前記R6と前記R7とで環状を構成しない。)として、下式(f)〜(j)で表される化合物(以下、それぞれを「化合物(f)」、「化合物(g)」、「化合物(h)」、「化合物(i)」、及び「化合物(j)」と称する。)を用いてチロシンホスファターゼの酵素活性に与える影響を調べた。その結果、化合物(f)〜(j)で表される化合物はCdc25Aホスファターゼやプロテインチロシンホスファターゼ1B等のチロシンホスファターゼの酵素活性を阻害することを見出した。
【化1】

【化2】
【0005】
また、上記と同様に一般式(III)で表されるナフトキノン誘導体化合物(式中、R8は水素原子又はアシル基である。)として、下式(k)及び(l)で表される化合物(以下、それぞれを「化合物(k)」及び「化合物(l)」と称する。)を用いてチロシンホスファターゼの酵素活性に与える影響を調べた。その結果、化合物(k)及び(l)で表される化合物はCdc25Aホスファターゼやプロテインチロシンホスファターゼ1B等のチロシンホスファターゼの酵素活性を阻害することが明らかになった。
【化3】
【化4】
【0006】
また、同様に一般式(IV)で表されるナフトキノン誘導体化合物(式中、R9はプロピル基、ブチル基、へキシル基、又はアルキニルオキシアルキル基である。)として、下式(m)〜(p)で表される化合物(以下、それぞれを「化合物(m)」、「化合物(n)」、「化合物(o)」、及び「化合物(p)」と称する。)を用いてチロシンホスファターゼの酵素活性に与える影響を調べた。その結果、化合物(m)〜(p)で表される化合物はCdc25Aホスファターゼやプロテインチロシンホスファターゼ1B等のチロシンホスファターゼの酵素活性を阻害することが明らかになった。
【化5】
【化6】
【0007】
ところで、Cdc25AはCdk2/CyclinEやCdk2/CyclinAに作用し、細胞周期の調節においてG1/Sの移行に機能することが知られている。そこで、本発明者らは、Cdc25Aホスファターゼを阻害する上述の化合物が細胞周期の進行を阻害するかどうかを調べるため、化合物(f)〜(p)と、下式(q)で表される化合物(以下、「化合物(q)」と称する。)とを用いて細胞周期の進行に与える影響を調べた。その結果、化合物(j)及び(q)で表される化合物が細胞周期のG1期で進行を停止することが明らかになった。
【化7】
【0008】
また、本発明者らは、上述の化合物が細胞増殖を抑制するかどうかを調べるため、化合物(j)〜(q)を用いて細胞増殖抑制活性を調べた。その結果、化合物(j)及び化合物(m)〜(p)が細胞増殖抑制活性を有することが明らかになった。
【0009】
さらに、本発明者らは、下式(r)、(s)、(v)、及び(w)で表される化合物(以下、それぞれを「化合物(r)」、「化合物(s)」、「化合物(v)」、及び「化合物(w)」と称する。)が単独で抗MRSA(メチシリン耐性黄色ブドウ球菌)作用を有すること、化合物(r)、(s)、(v)、及び(w)、並びに下式(u)で表される化合物(以下、「化合物(u)」と称する。)を、MRSAには単独で効果がないイミペネムと協同してMRSAに作用させると抗MRSA活性が増強することを見出した。
【化8】
【0010】
このようにして、本発明者らは本発明を完成するに至った。
【0011】
すなわち、本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩は、下記の一般式(I)で表されることを特徴とする。
【化9】
【0012】
式(I)中、R3がアリールアルコール基又はアルキルカルボニル基である場合には、R1は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R2はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R4はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、又は2−ジメタン−1,3−ジオキサン−4−アルキル基であり、式中、R4がアルキニルオキシアルキル基又はブチルジメチルシリルオキシアルケニル基である場合には、R1は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R2はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R3は水素原子、ヒドロキシアルキル基、ハロゲン原子、又はアリールアルコール基であり、式中、R1がアシルオキシ基である場合には、R2はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R3は水素原子、ヒドロキシアルキル基、又はハロゲン原子であり、R4はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、又は2−ジメタン−1,3−ジオキサン−4−アルキル基であり、式中、R1及びR2がメトキシ基であり、R4がヒドロキシブタ−2−エニル基である場合には、R3はヒドロキシエチル基、ヒドロキシプロピル基、又はヒドロキシヘキシル基であり、式中、R2がメトキシ基であり、R3が水素原子であり、R4がメトキシメチル基である場合には、R1はヒドロキシル基であり、式中、R1及びR2がメトキシ基であり、R3が水素原子である場合には、R4はへキシルオキシメチル基などである。なお、本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩は、R3とR4とで環状を構成しないことが好ましい。
【0013】
また、本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩は、上述の一般式(II)〜(IV)で表されることを特徴とする。なお、式(II)中、R5は水素原子又はアルキル基であり、R6はヒドロキシエチル基、ヒドロキシプロピル基、ヒドロキシヘキシル基、アリールアルコール基、又はアルキルカルボニル基であり、R7は水素原子、アシル基、又はブチルジメチルシリル基であり、前記R6と前記R7とで環状を構成しない。式(III)中、R8は水素原子又はアシル基である。式(IV)中、R9はプロピル基、ブチル基、へキシル基、又はアルキニルオキシアルキル基である。前記ナフトキノン誘導体化合物としては、上述の化合物(a)〜(q)であることが好ましい。
【0014】
本発明に係るチロシンホスファターゼ阻害剤は、上述のナフトキノン誘導体化合物又はその薬理学的に許容される塩を有効成分として含有する。前記チロシンホスファターゼとしては、例えば、Cdc25Aホスファターゼ、プロテインチロシンホスファターゼ1Bなどである。
【0015】
また、本発明に係る医薬組成物は、上述のナフトキノン誘導体化合物又はその薬理学的に許容される塩を有効成分として含有する。前記医薬組成物は、例えば、プロテインチロシンホスファターゼ1Bの過剰発現又は活性化に起因する疾患、Cdc25Aホスファターゼの過剰発現又は活性化に起因する疾患などの予防又は改善に有用である。前記プロテインチロシンホスファターゼ1Bの過剰発現又は活性化に起因する疾患としては、例えば、2型糖尿病、肥満症、神経疾患(例えば、アルツハイマー病、パーキンソン病など)、腫瘍(例えば、卵巣癌、乳癌など)、骨髄性白血病等が挙げられる。また、前記Cdc25Aホスファターゼの過剰発現又は活性化に起因する疾患としては、例えば、腫瘍などが挙げられる。
【0016】
さらに、本発明に係る細胞増殖阻害剤は、下記の一般式(V)で表されるナフトキノン誘導体化合物又はその薬理学的に許容される塩を有効成分として含有する。
【化10】
【0017】
式(V)中、R12がアリールアルコール基又はアルキルカルボニル基である場合には、R10は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R11はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R13はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、又は2−ジメタン−1,3−ジオキサン−4−アルキル基であり、式(V)中、R13がアルキニルオキシアルキル基又はブチルジメチルシリルオキシアルケニル基である場合には、R10は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R11はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R12は水素原子、ヒドロキシアルキル基、ハロゲン原子、又はアリールアルコール基であり、式(V)中、R10及びR11がメトキシ基であり、R12がヒドロキシブタ−2−エニル基である場合には、R13はヒドロキシエチル基、ヒドロキシプロピル基、又はヒドロキシヘキシル基であり、式(V)中、R10及びR11がメトキシ基であり、R12が水素原子である場合には、R13はへキシルオキシメチル基などである。なお、本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩は、R12とR13とで環状を構成しないことが好ましい。前記ナフトキノン誘導体化合物としては、上述の化合物(j)又は化合物(m)〜(p)のいずれかであることが好ましい。
【0018】
本発明に係る細胞周期進行阻害剤は、細胞周期のG1期の進行を阻害するものであって、下記の一般式(VI)で表される化合物又はその薬理学的に許容される塩を有効成分として含有する。
【化11】
【0019】
式(VI)中、R14はヒドロキシアルキル基、アルキルカルボニル基、又はアリールアルコール基であり、R15はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、2−ジメタン−1,3−ジオキサン−4−アルキル基、アルキニルオキシアルキル基、ブチルジメチルシリルオキシアルケニル基などである。なお、本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩は、R14とR15とで環状を構成しないことが好ましい。前記化合物としては、上述の化合物(j)や化合物(q)を用いることが好ましい。
【0020】
本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩は、下記の一般式(VII)で表されるもの又は化合物(r)であることを特徴とする。
【化12】
【0021】
式(VII)中、R16はヒドロキシル基又はアルコキシル基であり、R17は水素原子又はハロゲン原子であり、R18はヒドロキシル基又はアルコキシル基である。前記一般式(VII)で表されるナフトキノン誘導体化合物としては、例えば、上述の化合物(S)、化合物(t)、化合物(u)などである。
【0022】
本発明に係る抗菌剤は、下記の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩を有効成分として含有する。
【化13】
【0023】
式(VIII)中、R19及びR20はアルコキシル基である。前記一般式(VIII)で表されるナフトキノン誘導体化合物としては、例えば、化合物(s)などである。本発明に係る抗菌剤は、さらにイミペネムを有効成分として含有してもよい。また、本発明に係る抗菌剤は、化合物(u)又はその薬理学的に許容される塩、及びイミペネムを有効成分として含有する。なお、本発明に係る抗菌剤は、例えば、MRSA(メチシリン耐性黄色ブドウ球菌)などのグラム陽性菌に対しても、グラム陰性菌に対しても抗菌作用を有する。
【0024】
本発明に係るイミペネムの活性を増強するイミペネム活性増強剤は、上述の一般式(VII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩を有効成分として含有する。なお、前記一般式(VII)で表されるナフトキノン誘導体化合物としては、例えば、化合物(s)、化合物(u)等である。前記イミペネムの活性は、例えば、細菌に対する抗菌活性などである。前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0025】
また、本発明に係る細菌増殖阻害剤は、上述の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩を有効成分として含有する。前記一般式(VIII)で表されるナフトキノン誘導体化合物は例えば、化合物(s)などである。本発明に係る細菌増殖阻害剤は、さらに、イミペネムを有効成分として含有してもよい。本発明に係る細菌増殖阻害剤は、化合物(u)又はその薬理学的に許容される塩、及びイミペネムを有効成分として含有してもよい。なお、前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0026】
さらに、本発明に係る医薬組成物は、細菌の感染又は増殖に起因する疾患を改善する医薬組成物であって、上述の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩を有効成分として含有する。前記一般式(VIII)で表されるナフトキノン誘導体化合物は例えば、化合物(s)などである。本発明に係る医薬組成物は、さらに、イミペネムを有効成分として含有してもよい。本発明に係る医薬組成物は、化合物(u)又はその薬理学的に許容される塩、及びイミペネムを有効成分として含有してもよい。前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0027】
なお、前記MRSAの感染または増殖に起因する疾患(以下、「MRSA感染症」と称する場合がある。)には、例えば、脳炎、肺炎、敗血症、腹膜炎、腸炎、骨髄炎、胆管炎、皮膚化膿症、褥瘡感染、MRSAが産生する毒素(例えば、エンテロトキシン、TSST-1など)による食中毒、トキシックショック症候群等が含まれる。また、前記グラム陽性菌またはグラム陰性菌の感染または増殖に起因する疾患(以下、「グラム陽性菌またはグラム陰性菌の感染症」と称する場合がある。)には、例えば、中毒症、歯周病、炎症性疾患、血管炎、IV型アレルギ-性疾患、ブドウ球菌性熱傷様皮膚症候群、リッタ-病、新生児における水疱性インペチゴ、腫瘍、肺炎、関節炎、髄膜炎、各種化膿性疾患、腸炎、髄膜炎、菌血症、眼感染、食中毒、呼吸器感染症、中耳炎、副鼻腔炎、咽頭炎、猩紅熱、急性糸状体腎炎、リウマチ熱、膿痂疹、激症型感染、齲歯、尿路感染症、創感染、胆道感染症、細菌感染によるアトピ-性疾患(アトピ-性皮膚炎など)の重症化などが含まれる。
【0028】
本発明に係るナフトキノン誘導体化合物活性増強剤は、上述の一般式(VII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩の活性を増強する薬剤であって、イミペネムを有効成分として含有する。前記一般式(VII)で表されるナフトキノン誘導体化合物は例えば、化合物(s)、化合物(u)等である。前記活性は、例えば、細菌に対する抗菌活性などである。前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0029】
また、本発明に係るイミペネムの活性を増強するイミペネム活性増強方法は、 前記イミペネムと協同させて、上述の一般式(VII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩を作用させる工程を含む。前記一般式(VII)で表されるナフトキノン誘導体化合物は例えば、化合物(s)、化合物(u)等である。前記イミペネムの活性は、例えば、細菌に対する抗菌活性などである。前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0030】
さらに、本発明に係るナフトキノン誘導体化合物活性増強方法は、上述の一般式(VII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩の活性を増強する方法であって、前記ナフトキノン誘導体化合物と協同させてイミペネムを作用させる工程を含む。前記一般式(VII)で表されるナフトキノン誘導体化合物は例えば、化合物(s)、化合物(u)等である。前記活性は、例えば、細菌に対する抗菌活性などである。前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0031】
本発明に係る細菌の増殖阻害方法は、上述の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩を作用させる工程を含む。前記一般式(VIII)で表されるナフトキノン誘導体化合物は例えば、化合物(s)などである。本発明に係る細菌の増殖阻害方法は、さらに、イミペネムを協同して作用させることとしてもよい。本発明に係る細菌の増殖阻害方法は、化合物(u)又はその薬理学的に許容される塩、及びイミペネムを協同して作用させることとしてもよい。なお、前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0032】
本発明に係る細菌の感染または増殖に起因する疾患を改善する方法は、上述の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩を作用させるように、ヒト又はヒト以外の脊椎動物(例えば、マウス、ラットなど)に投与する工程を含む。前記一般式(VIII)で表されるナフトキノン誘導体化合物は例えば、化合物(s)などである。本発明に係る細菌の感染または増殖に起因する疾患を改善する方法は、さらに、イミペネムを協同して作用させることとしてもよい。本発明に係る細菌の感染または増殖に起因する疾患を改善する方法は、化合物(u)又はその薬理学的に許容される塩、及びイミペネムを協同して作用させることとしてもよい。前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0033】
なお、前記MRSAの感染または増殖に起因する疾患には、例えば、脳炎、肺炎、敗血症、腹膜炎、腸炎、骨髄炎、胆管炎、皮膚化膿症、褥瘡感染、MRSAが産生する毒素(例えば、エンテロトキシン、TSST-1など)による食中毒、トキシックショック症候群等が含まれる。また、前記グラム陽性菌またはグラム陰性菌の感染または増殖に起因する疾患には、例えば、中毒症、歯周病、炎症性疾患、血管炎、IV型アレルギ-性疾患、ブドウ球菌性熱傷様皮膚症候群、リッタ-病、新生児における水疱性インペチゴ、腫瘍、肺炎、関節炎、髄膜炎、各種化膿性疾患、腸炎、髄膜炎、菌血症、眼感染、食中毒、呼吸器感染症、中耳炎、副鼻腔炎、咽頭炎、猩紅熱、急性糸状体腎炎、リウマチ熱、膿痂疹、激症型感染、齲歯、尿路感染症、創感染、胆道感染症、細菌感染によるアトピ-性疾患(アトピ-性皮膚炎など)の重症化などが含まれる。
【発明の効果】
【0034】
本発明によれば、チロシンホスファターゼの阻害剤として有用な新規化合物、並びに、それを有効成分として含有する、チロシンホスファターゼ阻害剤、医薬組成物、細胞増殖阻害剤、細胞周期進行阻害剤、抗菌剤、イミペネム活性増強剤、及び細菌増殖阻害剤、並びに、上述の化合物を用いたイミペネム活性増強方法、及び細菌の増殖阻害方法、並びに、上述の化合物の活性増強剤及び活性増強方法を提供することができる。
【発明を実施するための最良の形態】
【0035】
以下、上記知見に基づき完成した本発明の実施の形態を、実施例を挙げながら詳細に説明する。実施の形態及び実施例に特に説明がない場合には、J. Sambrook, E. F. Fritsch & T. Maniatis (Ed.), Molecular cloning, a laboratory manual (3rd edition), Cold Spring Harbor Press, Cold Spring Harbor, New York (2001); F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J.G. Seidman, J. A. Smith, K. Struhl (Ed.), Current Protocols in Molecular Biology, John Wiley & Sons Ltd.などの標準的なプロトコール集に記載の方法、あるいはそれを修飾したり、改変した方法を用いる。また、市販の試薬キットや測定装置を用いている場合には、特に説明が無い場合、それらに添付のプロトコールを用いる。
【0036】
なお、本発明の目的、特徴、利点、及びそのアイデアは、本明細書の記載により、当業者には明らかであり、本明細書の記載から、当業者であれば、容易に本発明を再現できる。以下に記載された発明の実施の形態及び具体的な実施例などは、本発明の好ましい実施態様を示すものであり、例示又は説明のために示されているのであって、本発明をそれらに限定するものではない。本明細書で開示されている本発明の意図並びに範囲内で、本明細書の記載に基づき、様々な改変並びに修飾ができることは、当業者にとって明らかである。
【0037】
===本発明に係るナフトキノン誘導体化合物の薬理作用===
上述のように、チロシンホスファターゼに対する阻害剤は、2型糖尿病、肥満症、腫瘍、神経疾患((アルツハイマー病、パーキンソン病)、骨髄性白血病等のチロシンホスファターゼの過剰発現又は活性化に起因する疾患に有用であるとされている。そこで、本発明者らは、上述の化合物(f)〜(p)がチロシンホスファターゼの酵素活性を阻害するかどうかを調べた。その結果、化合物(f)〜(p)は、Cdc25Aホスファターゼやプロテインチロシンホスファターゼ1B等のチロシンホスファターゼに対して優れた阻害活性(IC50=10μg/ml以下)を有することが明らかになった。
【0038】
このことから、上述の化合物(f)〜(p)と類似の構造を有する上述の一般式(I)で表されるナフトキノン誘導体化合物、特に上述の一般式(II)〜(IV)で表されるナフトキノン誘導体化合物は、Cdc25Aホスファターゼ、プロテインチロシンホスファターゼ1B等のチロシンホスファターゼ阻害活性を有していると考えられる。また、上述の一般式(I)で表されるナフトキノン誘導体化合物の薬理学的に許容される塩も同様にCdc25Aホスファターゼ、プロテインチロシンホスファターゼ1B等のチロシンホスファターゼ阻害活性を有していると考えられる。従って、上述の一般式(I)で表されるナフトキノン誘導体化合物又はその薬理学的に許容される塩は、Cdc25Aホスファターゼ、プロテインチロシンホスファターゼ1B等のチロシンホスファターゼの阻害剤として有用であり、チロシンホスファターゼの過剰発現又は活性化に起因する疾患の予防又は改善に有用であると考えられる。
【0039】
また、前記チロシンホスファターゼの過剰発現又は活性化に起因する疾患としては、例えば、2型糖尿病、肥満症、腫瘍、神経疾患(例えば、アルツハイマー病、パーキンソン病)、骨髄性白血病等のプロテインチロシンホスファターゼ(PTP)1Bの過剰発現又は活性化に起因する疾患、腫瘍等のCdc25Aホスファターゼの過剰発現又は活性化に起因する疾患などを挙げることができる。
【0040】
ところで、Cdc25AはCdk2/CyclinEやCdk2/CyclinAに作用し、細胞周期の調節においてG1/Sの移行に機能することが知られている。そこで、本発明者らは、上述の化合物(f)〜(q)で表される化合物を用いて細胞周期の進行に与える影響を調べた。その結果、上述の化合物(j)及び(q)が、細胞周期のG1期の進行に対して優れた阻害作用を有することが明らかになった。このことから、細胞周期のG1期の進行を阻害するためには、上述の一般式(I)で表されるナフトキノン誘導体化合物において、少なくともR1がメトキシ基であり、R2がヒドロキシル基であり、R3がヒドロキシル又はオキソを有する基であることが必要であると考えられる。従って、上述の化合物(j)及び(q)と類似の構造を有する上述の一般式(VI)で表されるナフトキノン誘導体化合物や、その薬理学的に許容される塩は、細胞周期のG1期の進行を阻害する細胞周期進行阻害剤として有用であると考えられる。
【0041】
また、本発明者らは、上述の化合物が細胞増殖を抑制するかどうかを調べるため、化合物(j)〜(q)を用いて細胞増殖抑制活性を調べた。その結果、化合物(j)及び化合物(m)〜(p)が細胞増殖抑制活性を有することが明らかになった。従って、上述の化合物(j)及び化合物(m)〜(p)と類似の構造を有する上述の一般式(V)で表されるナフトキノン誘導体化合物や、その薬理学的に許容される塩は、細胞増殖抑制剤として有用であると考えられる。
【0042】
さらに、本発明者らの鋭意努力の結果、化合物(r)、(s)、(v)、及び(w)が単独で抗MRSA(メチシリン耐性黄色ブドウ球菌)作用を有すること、化合物(r)、(s)、(u)、(v)、及び(w)をイミペネムと協同してMRSAに作用させることにより、抗MRSA活性が増強することが明らかになった。従って、上述の一般式(VIII)で表されるナフトキノン誘導体化合物、化合物(r)、(v)、及び(w)、並びにそれらの薬理学的に許容される塩、並びに、化合物(u)とイミペネムとの混合物は、MRSAなどのグラム陽性菌、グラム陰性菌等の細菌感染症の改善(予防、抑制、及び治療を含む。)に有用であり、特に、MRSA感染症、例えば、脳炎、肺炎、敗血症、腹膜炎、腸炎、骨髄炎、胆管炎、皮膚化膿症、褥瘡感染、MRSAが産生する毒素(例えば、エンテロトキシン、TSST-1など)による食中毒、トキシックショック症候群等のMRSA感染症の改善(予防、抑制、及び治療を含む。)に有用である。また、上述の一般式(VIII)で表されるナフトキノン誘導体化合物、化合物(r)、(v)、及び(w)、並びにそれらの薬理学的に許容される塩、並びに、化合物(u)とイミペネムとの混合物は、同様に、グラム陽性菌またはグラム陰性菌の感染症、例えば、中毒症、歯周病、炎症性疾患、血管炎、IV型アレルギ-性疾患、ブドウ球菌性熱傷様皮膚症候群、リッタ-病、新生児における水疱性インペチゴ、腫瘍、肺炎、関節炎、髄膜炎、各種化膿性疾患、腸炎、髄膜炎、菌血症、眼感染、食中毒、呼吸器感染症、中耳炎、副鼻腔炎、咽頭炎、猩紅熱、急性糸状体腎炎、リウマチ熱、膿痂疹、激症型感染、齲歯、尿路感染症、創感染、胆道感染症、細菌感染によるアトピー性疾患(アトピー性皮膚炎など)の重症化等にも有用であると考えられる。
【0043】
さらに、化合物(r)、(s)、(v)、及び(w)をイミペネムとともに用いることにより抗MRSA作用が増強するので、これらの化合物とイミペネムとの使用はMRSA感染症の改善に最も効果的であるといえる。従って、上述の一般式(VII)で表されるナフトキノン誘導体化合物、化合物(r)、(v)、及び(w)、並びにそれらの薬理学的に許容される塩は、イミペネム活性増強のための薬剤または医薬組成物として使用できる。また、両者を含有する薬剤または医薬組成物は、抗菌剤または抗生物質として利用でき、特にMRSAに対して有効である。この際、協同して作用させるように、添加または投与されるのであれば、薬剤又は医薬組成物の剤形は、両者が別々に剤形化されていても、両者が単剤になっていてもよい。ここで、「協同して作用させる」とは、両者を添加又は投与することにより両者の相乗効果(例えば、MRSA増殖阻害活性)を生じさせることである。従って、時間的には両者は同時に添加又は投与されても、前後して添加又は投与されてもよい。
【0044】
===本発明に係るナフトキノン誘導体化合物の製造方法===
本発明に係るナフトキノン誘導体化合物は、以下の反応工程式に準じて反応を行ったり、以下の反応工程式の各工程や常法を適宜組み合わせたり、使用する物質を適宜変更したりすることにより、製造することができる。なお、本発明に係るナフトキノン誘導体化合物の薬理学的に許容される塩、すなわち、アルカリ金属塩(ナトリウム塩、カリウム塩など)、アルカリ土類金属塩(マグネシウム塩、カルシウム塩など)、その他の金属塩(アルミニウム塩など)、無機塩(塩酸塩、アンモニウム塩、アミン類など)、有機塩(グルコサミン塩など)等の公知の塩を含むナフトキノン誘導体化合物は、常法に従って製造することができる。
【0045】
まず、上述の一般式(III)で表されるナフトキノン誘導体化合物(式中、R8は水素原子又はアシル基である。)の製造方法の一例を反応工程式1に基づいて説明する。なお、反応工程式1中のR21はアシル基である。
【化14】
【0046】
<工程1a:化合物(2)の製造>
化合物(1)をジメチルホルムアミドに溶解し、水素化ナトリウムおよびヨウ化メチルを加える。撹拌後、氷冷下飽和塩化アンモニウム水溶液を加え、反応液を酢酸エチルで希釈した。これを飽和食塩水にて洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより化合物(2)を得ることができる。
【0047】
<工程2a:化合物(3)の製造>
化合物(2)をジクロロメタンに溶解し、水、t-ブチルアルコール、DDQ(2,3-ジクロロ-5,6-ジシアノ-1,4-ベンゾキノン)を順次加える。撹拌後、反応液をクロロホルムで希釈し、飽和炭酸水素ナトリウム水溶液を加える。有機層を飽和食塩水にて洗浄し、さらに水層をクロロホルムで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより化合物(3)を得ることができる。
【0048】
<工程3a:化合物(k)の製造>
化合物(3)をメタノールに溶解し、水酸化ナトリウム水溶液を加える。撹拌後、反応液を酢酸エチルで希釈し、塩酸で洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより化合物(k)を得ることができる。
【0049】
<工程4a:一般式(IX)で表される化合物の製造>
化合物(k)にカルボン酸無水物(alkanoic anhydride)、ピリジンを加えて撹拌する。反応液を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(IX)で表される化合物を得ることができる。
【0050】
次に、上述の一般式(IV)で表されるナフトキノン誘導体化合物の製造方法の一例を反応工程式2に基づいて説明する。なお、反応工程式2中のR9はプロピル基、ブチル基、へキシル基、又はアルキニルオキシアルキル基である。
【化15】
【0051】
<工程5a:化合物(4)の製造>
化合物(1)をジメチルホルムアミドに溶解し、炭酸カリウムおよびヨウ化メチルを加える。撹拌後、反応液を酢酸エチルで希釈する。有機層を飽和食塩水で洗浄し、さらに水層を酢酸エチルで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧濃縮して得られる粗生成物をクロマトグラフィーにて精製することにより化合物(4)を得ることができる。
【0052】
<工程6a:一般式(X)で表される化合物の製造>
化合物(4)をジメチルホルムアミドに溶解し、テトラn-ブチルアンモニウムヨージド(必要に応じて添加する。)、水素化ナトリウム、R9X(Xはハロゲン原子である。)を順次加える。撹拌後、反応液を酢酸エチルで希釈し、飽和塩化アンモニウム水溶液で洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(X)で表される化合物を得ることができる。
【0053】
<工程7a:一般式(IV)で表される化合物の製造>
一般式(X)で表される化合物を溶媒(例えば、ジクロロメタン、1,4-ジオキサンなど)に溶解し、水、t-ブチルアルコール、DDQを順次加える。撹拌後、反応液をクロロホルムで希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加える。有機層を飽和食塩水にて洗浄し、さらに水層をクロロホルムで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(IV)で表される化合物を得ることができる。
【0054】
また、上述の一般式(II)で表されるナフトキノン誘導体化合物の例として一般式(XVIII)で表される化合物を挙げ、その製造方法を反応工程式3に基づいて説明する。なお、反応工程式3中のR22はアルキル基であり、R24はヒドロキシエチル基、ヒドロキシプロピル基、ヒドロキシヘキシル基、又はアリールアルコール基である。
【化16】
【0055】
<工程8a:化合物(5)の製造>
化合物(1)を無水テトラヒドロフランに溶解し、臭化水素酸ペルブロミドピリジン錯体(Pyr・HBr3)を加える。撹拌後、飽和チオ硫酸ナトリウム水溶液を加え、反応液を酢酸エチルで希釈する。これを飽和食塩水にて洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより化合物(5)を得ることができる。
【0056】
<工程9a:化合物(6)の製造>
化合物(5)をジクロロメタンに溶解し、ジメチルスルホキシド、トリエチルアミン、三酸化硫黄ピリジン錯体(SO3・Pyr)を順次加える。そして、反応液をクロロホルムで希釈し、飽和塩化アンモニウム水溶液を加える。有機層を飽和食塩水にて洗浄し、さらに水層をクロロホルムで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧濃縮して得られる粗生成物をクロマトグラフィーにて精製することにより化合物(6)を得ることができる。
【0057】
<工程10a:一般式(XI)で表される化合物の製造>
化合物(6)をジメチルホルムアミドに溶解し、炭酸カリウムおよびヨウ化アルキルを加える。撹拌した後、反応液を酢酸エチルで希釈する。有機層を飽和食塩水で洗浄し、さらに水層を酢酸エチルで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧濃縮して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XI)で表される化合物を得ることができる。
【0058】
<工程11a:一般式(XII)で表される化合物の製造>
水素化ナトリウムにジメチルスルホキシドを加えて撹拌した後、反応液を無水テトラヒドロフランで希釈する。その後、ジメチルスルホキシドに溶解させたヨウ化トリメチルスルホニウムを加え、その後さらに、ジメチルスルホキシドに溶解させた一般式(XI)で表される化合物を加えて撹拌する。反応液を酢酸エチルで希釈した後、飽和塩化アンモニウム水溶液にて洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XII)で表される化合物を得ることができる。
【0059】
<工程12a:一般式(XIII)で表される化合物の製造>
ベンゼン中において、臭化亜鉛を加熱還流し、この混合物にベンゼンに溶解させた一般式(XII)で表される化合物を加える。反応液を酢酸エチルで希釈した後、水、飽和食塩水にて洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去してアルデヒドの粗生成物を得る。
【0060】
ジエチルホスホノ酢酸エチルを無水テトラヒドロフランに溶解し、水素化ナトリウムを加えて撹拌する。その後、無水テトラヒドロフランに溶解させたアルデヒドを加えて撹拌する。反応液に飽和塩化アンモニウム水溶液を加え、酢酸エチルで希釈する。有機層を飽和食塩水で洗浄し、さらに水層を酢酸エチルで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧濃縮して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XIII)で表される化合物を得ることができる。
【0061】
<工程13a:一般式(XIV)で表される化合物の製造>
一般式(XIII)で表される化合物を無水テトラヒドロフランに溶解し、水素化ジイソブチルアルミニウムを滴下する。滴下終了後撹拌し、塩酸を加える。反応液を酢酸エチルで希釈した後、飽和炭酸水素ナトリウム水溶液、飽和食塩水にて洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XIV)で表される化合物を得ることができる。
【0062】
<工程14a:一般式(XV)で表される化合物の製造>
一般式(XIV)で表される化合物をジメチルホルムアミドに溶解し、イミダゾール、塩化t-ブチルジメチルシリルを順次加え、撹拌する。反応液を酢酸エチルで希釈した後、飽和食塩水で洗浄し、有機層を無水硫酸ナトリウムで脱水する。溶媒を減圧濃縮して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XV)で表される化合物を得ることができる。
【0063】
<工程15a:一般式(XVI)で表される化合物の製造>
一般式(XV)で表される化合物を無水テトラヒドロフランに溶解し、n-ブチルリチウム(ヘキサン溶液)を滴下する。さらにR23CHO(R23はメチル基、エチル基、ペンチル基、又はアリール基である。)を加えて撹拌する。反応液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで希釈した後、飽和食塩水で洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮することにより一般式(XVI)で表される化合物の粗生成物を得ることができる。
【0064】
<工程16a:一般式(XVII)で表される化合物の製造>
一般式(XVI)で表される化合物の粗生成物をジクロロメタンに溶解し、水、t-ブチルアルコール、DDQを順次加える。撹拌後、反応液をクロロホルムで希釈し、飽和炭酸水素ナトリウム水溶液を加える。有機層を飽和食塩水にて洗浄し、さらに水層をクロロホルムで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XVII)で表される化合物を得ることができる。
【0065】
<工程17a:一般式(XVIII)で表される化合物の製造>
一般式(XVII)で表される化合物を無水テトラヒドロフランに溶解し、酢酸に溶解させたフッ化テトラn-ブチルアンモニウム(テトラヒドロフラン溶液)を加える。撹拌後、反応液を酢酸エチルで希釈する。次に、水、飽和食塩水で洗浄し、有機層を無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XVIII)で表される化合物を得ることができる。なお、上述の反応工程式1〜3において用いる化合物(1)は、例えば、文献(Bull. Chem. Soc. Jpn. 2004, 77, 1925-1930)に記載の方法に準じて製造することができる。
【0066】
次に、上述の一般式(II)で表されるナフトキノン誘導体化合物の例として一般式(XXIV)で表される化合物を挙げ、その製造方法を反応工程式4に基づいて説明する。なお、反応工程式4中のR22はアルキル基であり、R25はヒドロキシアルキル基又はアリールアルコール基であり、R26はアルキルカルボニル基である。
【化17】
【0067】
<工程18a:一般式(XIX)で表される化合物の製造>
上述の一般式(XV)で表される化合物を無水テトラヒドロフランに溶解し、n−ブチルリチウム(ヘキサン溶液)を滴下する。さらにアルキルアルデヒド又はアリールアルデヒドを加えて撹拌する。反応液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで希釈した後飽和食塩水で洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコールの粗生成物を得る。
【0068】
二級アルコールを無水テトラヒドロフランに溶解し、フッ化テトラn−ブチルアンモニウムを加える。撹拌後、反応液を酢酸エチルで希釈する。水、飽和食塩水で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XIX)で表される化合物を得ることができる。
【0069】
<工程19a:一般式(XX)で表される化合物の製造>
一般式(XIX)で表される化合物をジクロロメタンに溶解し、ピリジンおよび無水酢酸を加える。撹拌後、溶媒をトルエン共沸により減圧留去して得られる残留物をクロマトグラフィーにて精製することにより一般式(XX)で表される化合物を得ることができる。
【0070】
<工程20a:一般式(XXI)で表される化合物の製造>
一般式(XX)で表される化合物をジクロロメタンに溶解し、水、t-ブチルアルコール、DDQを順次加える。撹拌後、反応液をクロロホルムで希釈し、飽和炭酸水素ナトリウム水溶液を加える。有機層を飽和食塩水にて洗浄し、さらに水層をクロロホルムで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXII)で表される化合物を得ることができる。
【0071】
<工程21a:一般式(XXI)で表される化合物の製造>
三臭化ホウ素をジクロロメタンに溶解し、ジクロロメタンに溶解させた一般式(XXI)で表される化合物を加えて撹拌する。反応液をクロロホルムで希釈した後、水、飽和食塩水で洗浄し、有機層を無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXII)で表される化合物を得ることができる。
【0072】
<工程22a:一般式(XXIII)で表される化合物の製造>
一般式(XXII)で表される化合物を無水テトラヒドロフランに溶解し、ジメチルスルホキシド、IBX(o-Iodoxylbenzoic Acid)を加える。撹拌後、反応液をヘキサンで希釈してセライト濾過し、ヘキサン:酢酸エチル=3:1の混合溶媒で洗浄して減圧濃縮する。得られた粗生成物をクロマトグラフィーにて精製することにより一般式(XXIII)で表される化合物を得ることができる。
【0073】
<工程23a:一般式(XXIV)で表される化合物の製造>
一般式(XXIII)で表される化合物をメタノールに溶解し、炭酸カリウムを加える。撹拌後、反応液を酢酸エチルで希釈する。飽和塩化アンモニウム水溶液、飽和食塩水で順次洗浄した後、有機層を無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXIV)で表される化合物を得ることができる。
【0074】
次に、上述の一般式(VII)で表されるナフトキノン誘導体化合物の例として化合物(u)で表される化合物を挙げ、その製造方法を反応工程式5に基づいて説明する。なお、反応工程式5中のR22はアルキル基である。
【化18】
【0075】
<工程24a:一般式(XXV)で表される化合物の製造>
ベンゼン中、臭化亜鉛を加熱還流し、この混合物に、ベンゼンに溶解させた一般式(XII)で表される化合物を加える。この反応液を酢酸エチルで希釈した後、水、飽和食塩水で洗浄し、有機層を無水硫酸ナトリウムで脱水する。溶媒を減圧留去してアルデヒドの粗生成物を得た。
【0076】
得られたアルデヒドを無水テトラヒドロフランに溶解し、臭化メチルマグネシウム(テトラヒドロフラン溶液に溶解)を滴下する。撹拌後、反応液を酢酸エチルで希釈し、飽和炭酸水素ナトリウム水溶液を加える。有機層を飽和食塩水にて洗浄し、さらに水層を酢酸エチルで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水後、溶媒を減圧留去して二級アルコールの粗生成物を得る。
【0077】
得られた二級アルコールを無水テトラヒドロフランに溶解し、ジメチルスルホキシド、IBXを順次加えて撹拌し、反応液をヘキサンで希釈する。沈殿物をセライト濾過し、ヘキサン:酢酸エチル=3:1の混合溶媒で洗浄したろ液を減圧濃縮して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXV)で表される化合物を得ることができる。
【0078】
<工程25a:一般式(XXVI)で表される化合物の製造>
ベンゼン中、一般式(XXV)で表される化合物、エチレングリコール、及び4-トルエンスルホン酸一水和物を加熱還流する。反応液を酢酸エチルで希釈した後、飽和炭酸水素ナトリウム水溶液、飽和食塩水等で洗浄し、有機層を無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXVI)で表される化合物を得ることができる。
【0079】
<工程26a:一般式(XXVII)で表される化合物の製造>
一般式(XXVI)で表される化合物を無水テトラヒドロフランに溶解し、n-ブチルリチウムを滴下する。さらにn-ブチルアルデヒドを加えて撹拌する。反応液に飽和炭酸水素ナトリウム水溶液を加えた後、酢酸エチルで希釈し、飽和食塩水で洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコールの粗生成物を得る。
【0080】
得られた二級アルコールをベンゼンに溶解し、4-トルエンスルホン酸一水和物を加える。撹拌後、酢酸エチルで希釈し、飽和炭酸水素ナトリウム水溶液、飽和食塩水等で洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮してピラノナフタレンの粗生成物を得る。
【0081】
得られたピラノナフタレンを1,4-ジオキサンに溶解し、水、DDQを順次加える。撹拌後、反応液をクロロホルムで希釈し、飽和炭酸水素ナトリウム水溶液を加える。有機層を飽和食塩水にて洗浄し、さらに水層をクロロホルムで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXVII)で表される化合物を得ることができる。
【0082】
<工程27a:一般式(XXVIII)で表される化合物の製造>
一般式(XXVII)で表される化合物をメタノールに溶解し、Pd/C(パラジウムカーボン触媒)を加える。水素気流下にて撹拌後、沈殿物をセライト濾過し、メタノールで洗浄する。洗浄したろ液を減圧濃縮して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXVIII)で表される化合物を得ることができる。
【0083】
<工程28a:一般式(XXIX)で表される化合物の製造>
一般式(XXVIII)で表される化合物をジクロロメタンに溶解し、ピリジン、臭化水素酸ペルブロミドピリジン錯体(Pyr・HBr3)を加える。撹拌後、反応液をクロロホルムで希釈し、塩酸を加える。有機層を飽和食塩水にて洗浄し、さらに水層をクロロホルムで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXIX)で表される化合物を得ることができる。
【0084】
<工程29a:化合物(u)の製造>
エタノール中、一般式(XXIX)で表される化合物、及び濃塩酸を加熱還流する。反応液をクロロホルムで希釈した後、水で洗浄し、有機層を無水硫酸ナトリウムで脱水する。溶媒を減圧留去することにより化合物(u)を得ることができる。
【0085】
==本発明に係るナフトキノン誘導体化合物等を用いた薬剤・医薬品==
本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩及び/又はイミペネムを有効成分として含有する組成物は、医薬品として、ヒト、ヒト以外の脊椎動物に投与してもよいし、試薬として実験用に用いてもよい。本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩を有効成分として含有する医薬品は、錠剤、カプセル剤、顆粒剤、散剤、シロップ剤などの製剤にして、経口投与してもよいし、注射剤、坐剤などの製剤にして、腹腔内や静脈内への注射により非経口投与してもよい。本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩を有効成分として含有する医薬品の製剤化は、従来使用されている製剤添加物(例えば、賦形剤、結合剤、滑沢剤、崩壊剤、矯味矯臭剤、溶剤、安定剤など)を用いて、常法で行うことができる。
【実施例】
【0086】
以下、実施例を用いてより詳細に説明する。なお、実施例において、核磁気共鳴スペクトル(1H-NMRおよび13C-NMR)はJNM GX-400とEX-270(日本電子製)を用いて測定した。また、赤外吸収スペクトルはModel A-202(日本分光製)を用いて測定した。
【0087】
シリカゲル薄層クロマトグラフィーはkieselgel 60F254シリカゲル(Merck製)を使用し、リンモリブデン硫酸で呈色した。シリカゲルカラムクロマトグラフィーはシリカゲル60N(関東化学製)を用いた。なお、各反応は特に記載のない限り、アルゴン中で反応を行った。
【0088】
[実施例1]
<4-ベンジルオキシ-1,2,5-トリメトキシ-7-メトキシメチルナフタレンの製造>
8-ベンジルオキシ-3-ヒドロキシメチル-5,6-ジメトキシナフトール(図1中の化合物(1);159 mg)をジメチルホルムアミド(3 mL)に溶解し、0℃において水素化ナトリウム(121 mg)およびヨウ化メチル(0.18 mL)を加えた。1.5時間撹拌した後、氷冷下飽和塩化アンモニウム水溶液を加え、反応液を酢酸エチル(20 mL)で希釈した。これを飽和食塩水(5 mL)にて洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1〜1:1)にて精製し、無色針状晶として4-ベンジルオキシ-1,2,5-トリメトキシ-7-メトキシメチルナフタレン(図1中の化合物(2);111 mg,65%)を得た。
1HNMR (270 MHz, CDCl3) δ 3.45 (s, 3H), 3.94 (s, 6H), 3.97 (s, 3H), 4.60 (s, 2H), 5.17 (s, 2H), 6.75 (s, 1H), 6.78 (s, 1H), 7.35 (d, 1H, J= 7.6 Hz), 7.40 (t, 2H, J= 7.6 Hz), 7.59 (d, 2H, J= 7.6 Hz), 7.65 (s, 1H).
【0089】
[実施例2]
<2,5-ジメトキシ-7-メトキシメチルナフタレン-1,4-ジオンの製造>
実施例1により得られた化合物(2)(78.4 mg)をジクロロメタン(2 mL)に溶解し、0℃にて水(1 mL)、t-ブチルアルコール(1 mL)、DDQ(135 mg)を順次加えた。2時間撹拌した後、反応液をクロロホルム(20 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(10 mL)にて洗浄し、さらに水層をクロロホルム(10 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム:酢酸エチル=1:1)にて精製し、2,5-ジメトキシ-7-メトキシメチルナフタレン-1,4-ジオン(図1中の化合物(3);47.1 mg,84%)を得た。
1HNMR (270 MHz, CDCl3) δ 3.46 (s, 3H), 3.86 (s, 3H), 4.01 (s, 3H), 4.54 (s, 2H), 6.07 (s, 1H), 7.34 (s, 1H), 7.69 (s, 1H).
【0090】
[実施例3]
<2-ヒドロキシ-5-メトキシ-7-メトキシメチルナフタレン-1,4-ジオンの製造>
実施例2により得られた化合物(3)(10.8 mg)をメタノール(4 mL)に溶解し、0 ℃にて水酸化ナトリウム水溶液(1 M、0.25 mL)を加えた。室温で4時間撹拌した後、反応液を酢酸エチル(30 mL)で希釈した。1M塩酸(10 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール=5:1)にて精製し、2-ヒドロキシ-5-メトキシ-7-メトキシメチルナフタレン-1,4-ジオン(図1中の化合物(k);6.9 mg,68%)を得た。
1HNMR (270 MHz, CDCl3) δ 3.47 (s, 3H), 4.01 (s, 3H), 4.55 (s, 2H), 6.25 (s, 1H), 7.38 (s, 1H), 7.70 (s. 1H).
【0091】
[実施例4]
<5-メトキシ-7-メトキシメチル-1,4-ジオキソ-2-ナフチルアセテートの製造>
実施例3により得られた化合物(k)(5.4 mg)に室温にて無水酢酸(0.5 mL)、ピリジン(0.1 mL)を加えた。7時間撹拌した後、反応液を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール=10:1)にて精製し、5-メトキシ-7-メトキシメチル-1,4-ジオキソ-2-ナフチルアセテート(図1中の化合物(l);3.0 mg,49%)を得た。
1HNMR (270 MHz, CDCl3) δ 2.37 (s, 3H), 3.46 (s, 3H), 4.02 (s, 3H), 4.56 (s, 2H), 6.64 (s, 1H), 7.37 (s, 1H), 7.66 (s. 1H).
【0092】
[実施例5]
<(5-ベンジルオキシ-4,7,8-トリメトキシ-2-ナフチル)-メタン-1-オ−ルの製造>
8-ベンジルオキシ-3-ヒドロキシメチル-5,6-ジメトキシナフトール(図2中の化合物(1);120 mg)をジメチルホルムアミド(3 mL)に溶解し、0℃において炭酸カリウム(383 mg)およびヨウ化メチル(0.2 mL)を加えた。10時間撹拌した後、反応液を酢酸エチル(25 mL)で希釈した。有機層を飽和食塩水(10 mL)で洗浄し、さらに水層を酢酸エチル(10 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧濃縮して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、(5-ベンジルオキシ-4,7,8-トリメトキシ-2-ナフチル)-メタン-1-オ−ル(図2中の化合物(4);53.4 mg,43%)を得た。
1HNMR (270 MHz, CDCl3) δ 3.90 (s, 3H), 3.91 (s, 3H), 3.93 (s, 3H), 4.77 (s, 2H), 5.13 (s, 2H), 6.70 (s, 1H), 6.72 (s, 1H), 7.34 (d, 1H, J= 7.0 Hz), 7.41 (t, 2H, J= 7.0 Hz), 7.56 (s, 1H), 7.60 (complex, 2H).
【0093】
[実施例6]
<4-ベンジルオキシ-7-ヘキシルオキシメチル-1,2,5-トリメトキシナフタレンの製造>
実施例5により得られた化合物(4)(8.6 mg)をジメチルホルムアミド(1 mL)に溶解し、0℃にてテトラn-ブチルアンモニウムヨージド(10 mg)、水素化ナトリウム(15 mg)、1-ブロモヘキサン(40μL)を順次加えた。室温で13時間撹拌した後、反応液を酢酸エチル(25 mL)で希釈した。飽和塩化アンモニウム水溶液(15 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)にて精製し、4-ベンジルオキシ-7-ヘキシルオキシメチル-1,2,5-トリメトキシナフタレン(図2中の化合物(7);6.6 mg,62%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.88 (t, 3H, J= 6.8 Hz), 1.27-1.42 (complex, 6H), 1.56-1.67 (complex, 2H), 3.50 (t, 2H, J= 6.5 Hz), 3.90 (s, 3H), 3.93 (s, 3H), 3.95 (s, 3H), 4.62 (s, 2H), 5.15 (s, 2H), 6.72 (s, 1H), 6.77 (s, 1H), 7.32-7.44 (complex, 3H), 7.55 (s. 1H), 7.58 (brs, 2H).
【0094】
[実施例7]
<7-ヘキシルオキシメチル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例6により得られた化合物(7)(6.6 mg)をジクロロメタン(1 mL)に溶解し、0℃にて水(0.3 mL)、t-ブチルアルコール(0.3 mL)、DDQ (18.0 mg)を順次加えた。室温で1時間撹拌した後、反応液をクロロホルム(25 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(5 mL)にて洗浄し、さらに水層をクロロホルム(15 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム:酢酸エチル=1:1)にて精製し、7-ヘキシルオキシメチル-2,5-ジメトキシナフタレン-1,4-ジオン(図2中の化合物(m);1.3 mg,33%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.94 (t, 3H, J= 7.0 Hz), 1.26-1.53 (complex, 6H), 1.55-1.68 (complex, 2H), 3.53 (t, 2H, J= 6.5 Hz), 3.86 (s, 3H), 4.01 (s, 3H), 4.58 (s, 2H), 6.07 (s, 1H), 7.71 (s, 1H).
【0095】
[実施例8]
<4-ベンジルオキシ-1,2,5-トリメトキシ-7-(プロパ-2-イニルオキシメチル)ナフタレンの製造>
実施例5により得られた化合物(4)(8.4 mg)をジメチルホルムアミド(1 mL)に溶解し、0℃にてテトラn-ブチルアンモニウムヨージド(10 mg)、水素化ナトリウム(10 mg)、臭化プロパルギル (70μL)を順次加えた。室温で30分間撹拌した後、反応液を酢酸エチル(30 mL)で希釈した。飽和塩化アンモニウム水溶液(15 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)にて精製し、4-ベンジルオキシ-1,2,5-トリメトキシ-7-(プロパ-2-イニルオキシメチル)ナフタレン(図2中の化合物(8);7.6 mg,82%)を得た。
1HNMR (270 MHz, CDCl3) δ 2.49 (t, 1H, J= 2.4 Hz), 3.90 (s, 3H), 3.94 (s, 3H), 3.95 (s, 3H), 4.21 (d, 2H, J= 2.4 Hz), 4.73 (s, 2H), 5.15 (s, 2H), 6.73 (s, 1H), 6.76 (s, 1H), 7.32-7.44 (complex, 3H), 7.56 (d, 2H, J= 7.0 Hz), 7.63 (s, 1H).
【0096】
[実施例9]
<2,5-ジメトキシ-7-(プロパ-2-イニルオキシメチル)ナフタレン-1,4-ジオンの製造>
実施例8により得られた化合物(8)(7.6 mg)をジクロロメタン(1 mL)に溶解し、0℃にて水(0.3 mL)、t-ブチルアルコール(0.3 mL)、DDQ (19.7 mg)を順次加えた。室温で15分間撹拌した後、反応液をクロロホルム(30 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(20 mL)を加えた。有機層を飽和食塩水(10 mL)にて洗浄し、さらに水層をクロロホルム(15 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム:酢酸エチル=1:1)にて精製し、2,5-ジメトキシ-7-(プロパ-2-イニルオキシメチル)ナフタレン-1,4-ジオン(図2中の化合物(n);3.2 mg,58%)を得た。
1HNMR (270 MHz, CDCl3) δ 2.51 (t, 1H, J= 2.4 Hz), 3.86 (s, 3H), 4.02 (s, 3H), 4.27 (d, 2H, J= 2.4 Hz), 4.70 (s, 2H), 6.08 (s, 1H), 7.36 (s, 1H), 7.73 (s, 1H).
【0097】
[実施例10]
<4-ベンジルオキシ-1,2,5-トリメトキシ-7-[(2-メチルプロポキシ)メチル]ナフタレンの製造>
実施例5により得られた化合物(4)(11.1 mg)をジメチルホルムアミド(1 mL)に溶解し、0℃にてテトラn-ブチルアンモニウムヨージド(10 mg)、水素化ナトリウム(10 mg)、臭化イソブチル (70μL)を順次加えた。室温で18時間撹拌した後、反応液を酢酸エチル(25 mL)で希釈した。飽和塩化アンモニウム水溶液(10 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)にて精製し、4-ベンジルオキシ-1,2,5-トリメトキシ-7-[(2-メチルプロポキシ)メチル]ナフタレン(図2中の化合物(9);9.2 mg,71%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.94 (d, 6H, J= 6.5 Hz), 1.94 (m, 1H), 3.26 (d, 2H, J= 6.5 Hz), 3.90 (s, 3H), 3.93 (s, 3H), 3.94 (s, 3H), 4.63 (s, 2H), 5.15 (s, 2H), 6.72 (s, 1H), 6.78 (s, 1H), 7.32-7.43 (complex, 3H), 7.55 (s, 1H), 7.59 (s, 2H).
【0098】
[実施例11]
<2,5-ジメトキシ-7-[(2-メチルプロポキシ)メチル]ナフタレン-1,4-ジオンの製造>
実施例10により得られた化合物(9)(9.2 mg)をジクロロメタン(1 mL)に溶解し、0℃にて水(0.3 mL)、t-ブチルアルコール(0.3 mL)、DDQ (23.0 mg)を順次加えた。室温で35分間撹拌した後、反応液をクロロホルム(20 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(20 mL)を加えた。有機層を飽和食塩水(10 mL)にて洗浄し、さらに水層をクロロホルム(15 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム:酢酸エチル=1:1)にて精製し、2,5-ジメトキシ-7-[(2-メチルプロポキシ)メチル]ナフタレン-1,4-ジオン(図2中の化合物(o);2.4 mg,35%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.96 (d, 6H, J= 4.3 Hz), 1.96 (m, 1H), 3.29 (d, 2H, J= 4.3 Hz), 3.86 (s, 3H), 3.99 (s, 3H), 4.59 (s, 2H), 6.08 (s, 1H), 7.37 (s, 1H), 7.71 (s, 1H).
【0099】
[実施例12]
<4-ベンジルオキシ-1,2,5-トリメトキシ-7-プロポキシメチルナフタレンの製造>
実施例5により得られた化合物(4)(11.3 mg)をジメチルホルムアミド(1 mL)に溶解し、0℃にて水素化ナトリウム(10 mg)、ヨウ化n-プロピル (100μL)を順次加えた。室温で12時間撹拌した後、反応液を酢酸エチル(30 mL)で希釈した。飽和塩化アンモニウム水溶液(15 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)にて精製し、4-ベンジルオキシ-1,2,5-トリメトキシ-7-プロポキシメチルナフタレン(図2中の化合物(10);8.6 mg,68%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.96 (t, 3H, J= 7.6 Hz), 1.58-1.73 (complex, 2H), 3.47 (t, 2H, J= 6.8 Hz), 3.90 (s, 3H), 3.93 (s, 3H), 3.95 (s, 3H), 4.63 (s, 2H), 5.15 (s, 2H), 6.72 (s, 1H), 6.78 (s, 1H), 7.32-7.43 (complex, 3H), 7.55 (s, 1H), 7.59 (d, 2H, J= 4.9 Hz).
【0100】
[実施例13]
<2,5-ジメトキシ-7プロポキシメチルナフタレン-1,4-ジオンの製造>
実施例12により得られた化合物(10)(7.0 mg)を1,4-ジオキサン(1 mL)に溶解し、0℃にて水(0.5 mL)、DDQ (15 mg)を順次加えた。室温で20分間撹拌した後、反応液をクロロホルム(20 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(10 mL)にて洗浄し、さらに水層をクロロホルム(15 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、2,5-ジメトキシ-7プロポキシメチルナフタレン-1,4-ジオン(図2中の化合物(p);4.8 mg,76%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.98 (t, 3H, J= 7.6 Hz), 1.59-1.75 (complex, 2H), 3.50 (t, 2H, J= 6.8 Hz), 3.86 (s, 3H), 4.02 (s, 3H), 4.59 (s, 2H), 6.07 (s, 1H), 7.37 (s, 1H), 7.71 (s, 1H).
【0101】
[実施例14]
<8-ベンジルオキシ-2-ブロモ-3-ヒドロキシメチル-5,6-ジメトキシナフトールの製造>
8-ベンジルオキシ-3-ヒドロキシメチル-5,6-ジメトキシナフトール(図3中の化合物(1);2.42 g)を無水テトラヒドロフラン(50 mL)に溶解し、0℃において臭化水素酸ペルブロミドピリジン錯体(Pyr・HBr3)(2.52 g)を加えた。1時間撹拌した後、飽和チオ硫酸ナトリウム水溶液(30 mL)を加え、反応液を酢酸エチル(100 mL)で希釈した。これを飽和食塩水(20 mL)にて洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1)にて精製し、黄褐色針状晶として8-ベンジルオキシ-2-ブロモ-3-ヒドロキシメチル-5,6-ジメトキシナフトール(図3中の化合物(5);2.60 g,87%)を得た。
1HNMR (270 MHz, CDCl3) δ 2.25 (br, 1H), 3.90 (s, 3H), 3.97 (s, 3H), 4.85 (s, 2H), 5.26 (s, 2H), 6.74 (s, 1H), 7.44-7.47 (complex, 5H), 7.65 (s, 1H), 9.99 (s, 1H).
13CNMR (67.80 MHz, DMSO-d6) δ 56.7, 56.9, 60.4, 63.1, 63.8, 71.4, 97.7, 99.6, 101.4, 109.5, 128.3, 128.5, 128.7, 128.9, 135.5, 136.0, 140.1, 148.2, 149.7, 150.1.
IR (disk) 3504, 3354, 3056, 2972, 2940, 2881, 2360, 1612 cm-1.
m.p. 165℃ dec. (hexane-EtOAc)
【0102】
[実施例15]
<5-ベンジルオキシ-3-ブロモ-4-ヒドロキシ-7,8-ジメトキシナフタレン-2-カルバルデヒドの製造>
実施例14により得られた化合物(5)(1.35 g)をジクロロメタン(10 mL)に溶解し、室温にてジメチルスルホキシド(9 mL)、トリエチルアミン(2.7 mL)、三酸化硫黄ピリジン錯体(SO3・Pyr)(1.51 g)を順次加えた。20分後、反応液をクロロホルム(200 mL)で希釈し、氷冷下、飽和塩化アンモニウム水溶液(50 mL)を加えた。有機層を飽和食塩水(30 mL)にて洗浄し、さらに水層をクロロホルム(100 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧濃縮して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム)にて精製し、黄色針状晶として5-ベンジルオキシ-3-ブロモ-4-ヒドロキシ-7,8-ジメトキシナフタレン-2-カルバルデヒド(図3中の化合物(6);1.24 g,93%)を得た。
1HNMR (270 MHz, CDCl3) δ 3.92 (s, 3H), 3.97 (s, 3H), 5.27 (s, 2H), 6.85 (s, 1H), 7.44-7.47 (complex, 5H), 8.17 (s, 1H), 10.09 (s, 1H), 10.48 (s, 1H).
13CNMR (67.80 MHz, CDCl3) δ 57.1, 61.5, 72.6, 99.6, 99.8, 103.1, 116.2, 116.3, 128.2, 129.1, 129.2, 131.9, 134.0, 134.3, 148.7, 150.6, 151.6, 151.7, 192.3.
IR (disk) 3303, 2950, 1703, 1606 cm-1.
m.p. 174℃ dec. (hexane-EtOAc)
【0103】
[実施例16]
<5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシナフタレン-2-カルバルデヒドの製造>
実施例15により得られた化合物(6)(449 mg)をジメチルホルムアミド(10 mL)に溶解し、0℃にて炭酸カリウム(463 mg)およびヨウ化メチル(0.3 mL)を加えた。室温にて1時間撹拌した後、反応液を酢酸エチル(50 mL)で希釈した。有機層を飽和食塩水(10 mL)で洗浄し、さらに水層を酢酸エチル(30 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧濃縮して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、黄色板状晶として5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシナフタレン-2-カルバルデヒド(図3中の化合物(11);437 mg,94%)を得た。
1HNMR (400 MHz, CDCl3) δ 3.80 (s, 3H), 3.95 (s, 3H), 3.96 (s, 3H), 5.20 (s, 2H), 6.91 (s, 1H), 7.38 (d, 1H, J= 7.2 Hz), 7.43 (t, 2H, J= 7.2 Hz), 7.56 (d, 2H, J= 7.2 Hz), 8.50 (s, 1H), 10.50 (s, 1H).
13CNMR (100.40 MHz, CDCl3) δ 56.9, 61.5, 61.9, 62.0, 62.1, 72.8, 100.5, 103.3, 113.8, 119.6, 121.4, 127.7, 128.1, 128.6, 129.9, 131.5, 136.3, 138.9, 149.0, 151.0, 154.1, 192.2.
IR (disk) 2943, 2848, 1687, 1612, 1577 cm-1.
m.p. 129-130℃ (hexane-EtOAc)
【0104】
[実施例17]
<8-ベンジルオキシ-2-ブロモ-1,5,6-トリメトキシ-3-オキシラン-2-イルナフタレンの製造>
水素化ナトリウム(517 mg)にジメチルスルホキシド(5 mL)を加え、室温で2時間撹拌した後、反応液を無水テトラヒドロフラン(50 mL)で希釈した。その後、0℃にてジメチルスルホキシド(10 mL)に溶解させたヨウ化トリメチルスルホニウム(2.75 g)を加え、さらに40分後、ジメチルスルホキシド(25 mL)に溶解させた化合物(11)(1.89 g)を加えて45分間撹拌した。反応液を酢酸エチル(50 mL)で希釈した後、飽和塩化アンモニウム水溶液(30 mL)にて洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)にて精製し、淡黄色針状晶として8-ベンジルオキシ-2-ブロモ-1,5,6-トリメトキシ-3-オキシラン-2-イルナフタレン(図3中の化合物(12);1.77 g,92%)を得た。
【0105】
1HNMR (400 MHz, CDCl3) δ 2.72 (dd, 1H, J= 2.8, 6.0 Hz), 3.25 (dd, 1H, J= 4.0, 6.0 Hz), 3.78 (s, 3H), 3.89 (s, 3H), 3.95 (s, 3H), 4.26 (dd, 1H, J= 2.8, 4.0 Hz), 5.18 (s, 2H), 6.78 (s, 1H), 7.37 (d, 1H, J= 7.2 Hz), 7.43 (t, 2H, J= 7.2 Hz), 7.56 (d, 2H, J= 7.2 Hz), 7.79 (s, 1H).
13CNMR (100.40 MHz, CDCl3) δ 51.1, 52.9, 56.9, 61.2, 61.9, 72.6, 100.4, 100.5, 113.5, 114.4, 116.7, 127.7, 128.0, 128.5, 130.8, 136.1, 136.6, 137.3, 143.9, 148.6, 151.1, 153.2.
IR (disk) 2931, 2858, 1612, 1593, 1574 cm-1.
m.p. 133-134℃ (hexane-EtOAc)
【0106】
[実施例18]
<エチル(2E)-4-[5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ(2-ナフチル)]ブタ-2-エノアートの製造>
ベンゼン(17 mL)中、臭化亜鉛(708 mg)を40分間加熱還流し、この混合物にベンゼン(10 mL)に溶解させた化合物(12)(468 mg)を加えた。20分後反応液を室温に戻し、酢酸エチル(30 mL)で希釈した後、水、飽和食塩水(各々20 mL)にて洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去してアルデヒドの粗生成物を得た。
【0107】
ジエチルホスホノ酢酸エチル(1.1 mL)を無水テトラヒドロフラン(10 mL)に溶解し、氷冷下、水素化ナトリウム(216 mg)を加え、室温にて1.5時間撹拌した。その後、無水テトラヒドロフラン(10 mL)に溶解させたアルデヒドを−78℃にて加え、25分間撹拌した。反応液に飽和塩化アンモニウム水溶液(20 mL)を加え、酢酸エチル(30 mL)で希釈した。有機層を飽和食塩水(10 mL)で洗浄し、さらに水層を酢酸エチル(20 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧濃縮して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1)にて精製し、エチル(2E)-4-[5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ(2-ナフチル)]ブタ-2-エノアート(図3中の化合物(13);462 mg,85%)を得た。
【0108】
1HNMR (270 MHz, CDCl3) δ 1.27 (t, 3H, J= 7.0 Hz), 3.77 (s, 3H), 3.82 (d, 2H, J= 6.2 Hz), 3.90 (s, 3H), 3.95 (s, 3H), 4.18 (q, 2H, J= 7.0 Hz), 5.18 (s, 2H), 5.81 (d, 1H, J= 16 Hz), 7.18 (dt, 1H, J= 6.2, 16 Hz), 7.36 (d, 1H, J= 7.2 Hz), 7.42 (t, 2H, J= 7.2 Hz), 7.56 (d, 2H, J= 7.2 Hz), 7.74 (s, 1H).
13CNMR (67.80 MHz, CDCl3) δ 14.3, 39.5, 56.8, 60.2, 61.1, 61.8, 72.5, 100.1, 116.1, 116.3, 118.4, 122.7, 127.6, 127.9, 128.4, 130.3, 130.5, 136.4, 136.5, 136.7, 140.1, 145.7, 148.4, 151.0, 153.7, 166.2.
IR (film) 2937, 2846, 1716, 1614, 1591, 1573 cm-1.
【0109】
[実施例19]
<(2E)-4-[5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ(2-ナフチル)]ブタ-2-エン-1-オールの製造>
実施例18により得られた化合物(13)(147 mg)を無水テトラヒドロフラン(3 mL)に溶解し、−78℃にて水素化ジイソブチルアルミニウム(1.01 M、トルエン溶液、1.5 mL)を滴下した。滴下終了後35分間撹拌し、4M 塩酸(10 mL)を加えて0℃に戻した。反応液を酢酸エチル(20 mL)で希釈した後、飽和炭酸水素ナトリウム水溶液(10 mL)、飽和食塩水(5 mL)にて洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、淡黄色板状晶として(2E)-4-[5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ(2-ナフチル)]ブタ-2-エン-1-オール(図3中の化合物(14);125 mg,93%)を得た。
【0110】
1HNMR (270 MHz, CDCl3) δ 1.82 (br, 1H), 3.66 (d, 2H, J= 6.2 Hz), 3.77 (s, 3H), 3.90 (s, 3H), 3.94 (s, 3H), 4.13 (q, 2H, J= 5.4 Hz), 5.17 (s, 2H), 5.75 (dt, 1H, J= 5.4, 16 Hz), 5.95 (dt, 1H, J= 6.2, 16 Hz), 6.74 (s, 1H), 7.37 (d, 1H, J= 7.2 Hz), 7.42 (t, 2H, J= 7.2 Hz), 7.56 (d, 2H, J= 7.2 Hz), 7.73 (s, 1H).
13CNMR (67.80 MHz, CDCl3) δ 39.6, 56.8, 61.1, 61.7, 63.4, 63.5, 72.5, 99.8, 115.8, 116.6, 117.7, 127.6, 127.8, 128.3, 129.5, 129.6, 130.5, 131.0, 131.1, 136.7, 138.5, 148.2, 151.0, 153.3.
IR (disk) 3521, 2902, 2860, 1614, 1589, 1574 cm-1.
m.p. 104-105℃ (hexane-EtOAc)
【0111】
[実施例20]
<1-{(2E)-4-[5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ(2-ナフチル) ]ブタ-2-エニルオキシ}-1,1,2,2-テトラメチル-1-シラプロパンの製造>
実施例19により得られた化合物(14)(125 mg)をジメチルホルムアミド(3 mL)に溶解し、0℃にてイミダゾール(180 mg)、塩化t-ブチルジメチルシリル(215 mg)を順次加え、15分間撹拌した。反応液を酢酸エチル(15 mL)で希釈した後、飽和食塩水(10 mL)で洗浄し、有機層を無水硫酸ナトリウムで脱水した。溶媒を減圧濃縮して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1)にて精製し、1-{(2E)-4-[5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ(2-ナフチル) ]ブタ-2-エニルオキシ}-1,1,2,2-テトラメチル-1-シラプロパン(図3中の化合物(15);149 mg,96%)を得た。
【0112】
1HNMR (400 MHz, CDCl3) δ 0.07 (s, 6H), 0.90 (s, 9H), 3.65 (d, 2H, J= 6.8 Hz), 3.76 (s, 3H), 3.89 (s, 3H), 3.94 (s, 3H), 4.19 (q, 2H, J= 5.2 Hz), 5.18 (s, 2H), 5.67 (dt, 1H, J= 5.2, 15 Hz), 5.93 (dt, 1H, J= 6.8, 15 Hz), 6.74 (s, 1H), 7.36 (d, 1H, J= 7.2 Hz), 7.42 (t, 2H, J= 7.2 Hz), 7.56 (d, 2H, J= 7.2 Hz), 7.73 (s, 1H).
13CNMR (100.40 MHz, CDCl3) δ -5.0, 18.5, 25.7, 26.0, 39.6, 56.9, 61.3, 61.7, 63.7, 72.6, 99.9, 115.9, 116.8, 117.7, 127.6, 127.7, 127.9, 128.5, 130.6, 131.6, 135.3, 136.8, 137.0, 139.0, 148.2, 151.1.
IR (film) 2931, 2894, 2854, 1614, 1591, 1576 cm-1.
【0113】
[実施例21]
<7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシエチル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例20により得られた化合物(15)(65.3 mg)を無水テトラヒドロフラン(2 mL)に溶解し、−78℃にてn-ブチルリチウム(1.59 M、ヘキサン溶液、0.4 mL)を滴下した。さらにアセトアルデヒド(1.0 mL)を加え、15分間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液 (10 mL)を加えて0 ℃に戻し、酢酸エチル(30 mL)で希釈した後、飽和食塩水(5 mL)で洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコール(図4中の化合物(16))の粗生成物を得た。
【0114】
二級アルコールの粗生成物をジクロロメタン(2 mL)に溶解し、0℃にて水(0.2 mL)、t-ブチルアルコール(0.2 mL)、DDQ (40.1 mg)を順次加えた。15分間撹拌した後、反応液をクロロホルム(20 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(5 mL)にて洗浄し、さらに水層をクロロホルム(20 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシエチル-2,5-ジメトキシナフタレン-1,4-ジオン(図4中の化合物(a);10.5 mg,29%)を得た。
【0115】
1HNMR (400 MHz, CDCl3) δ 0.05 (s, 6H), 0.88 (s, 9H), 1.58 (d, 3H, J= 5.2 Hz), 3.54 (complex, 2H), 3.85 (s, 3H), 3.98 (s, 3H), 4.15 (d, 2H, J= 4.0 Hz), 5.18 (m, 1H), 5.55 (m, 1H), 5.77 (m, 1H), 6.08 (s, 1H), 7.78 (s, 1H).
【0116】
[実施例22]
<7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシエチル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例21により得られた化合物(a)(10.5 mg)を無水テトラヒドロフラン(1.5 mL)に溶解し、0℃にて酢酸(13μL)に溶解させたフッ化テトラn-ブチルアンモニウム(1.0 M、テトラヒドロフラン溶液、0.2 mL)を加えた。室温で12時間撹拌した後、反応液を酢酸エチル(25 mL)で希釈した。水(10 mL)、飽和食塩水(5 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシエチル-2,5-ジメトキシナフタレン-1,4-ジオン(図4中の化合物(f);7.4 mg,95%)を得た。
1HNMR (400 MHz, CDCl3) δ 1.59 (d, 3H, J= 7.2 Hz), 3.55 (complex, 2H), 3.88 (s, 3H), 3.97 (s, 3H), 4.12 (d, 2H, J= 4.8 Hz), 5.18 (t, 1H, J= 7.2 Hz), 5.64 (dt, 1H, J= 6.0, 15 Hz), 5.84 (dt, 1H, J= 6.0, 15 Hz), 6.08 (s, 1H), 7.76 (s, 1H).
【0117】
[実施例23]
<7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシプロピル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例20により得られた化合物(15)(93.9 mg)を無水テトラヒドロフラン(5 mL)に溶解し、−78℃にてn-ブチルリチウム(1.59 M、ヘキサン溶液、0.5 mL)を滴下した。さらにプロピオンアルデヒド(0.23 mL)を加え、15分間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液 (15 mL)を加えて0℃に戻し、酢酸エチル(40 mL)で希釈した後、飽和食塩水(5 mL)で洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコールの粗生成物(図5中の化合物(17))を得た。
【0118】
二級アルコールの粗生成物をジクロロメタン(2 mL)に溶解し、0℃にて水(0.8 mL)、t-ブチルアルコール(0.3 mL)、DDQ (120 mg)を順次加えた。40分間撹拌した後、反応液をクロロホルム(25 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(5 mL)にて洗浄し、さらに水層をクロロホルム(20 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシプロピル-2,5-ジメトキシナフタレン-1,4-ジオン(図5中の化合物(b);24.3 mg,33%)を得た。
【0119】
1HNMR (400 MHz, CDCl3) δ 0.05 (s, 6H), 0.88 (s, 9H), 1.04 (t, 3H, J= 8.0 Hz), 1.74 (complex, 2H), 3.53 (d, 2H, J= 6.0 Hz), 3.86 (s, 3H), 3.93 (s, 3H), 4.15 (d, 2H, J= 4.0 Hz), 4.85 (m, 1H), 5.57 (dt, 1H, J= 5.2, 15 Hz), 5.78 (dt, 1H, J= 5.2, 15 Hz), 6.06 (s, 1H), 7.77 (s, 1H).
【0120】
[実施例24]
<7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシプロピル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例23により得られた化合物(b)(24.3 mg)を無水テトラヒドロフラン(3 mL)に溶解し、0℃にて酢酸(33μL)に溶解させたフッ化テトラn-ブチルアンモニウム(1.0 M、テトラヒドロフラン溶液、0.53 mL)を加えた。室温で11時間撹拌した後、反応液を酢酸エチル(40 mL)で希釈した。水(15 mL)、飽和食塩水(5 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシプロピル-2,5-ジメトキシナフタレン-1,4-ジオン(図5中の化合物(g);13.1 mg,72%)を得た。
1HNMR (270 MHz, CDCl3) δ 1.05 (t, 3H, J= 7.2 Hz), 1.75 (complex, 2H), 3.54 (d, 2H, J= 5.4 Hz), 3.87 (s, 3H), 3.94 (s, 3H), 4.11 (d, 2H, J= 4.8 Hz), 4.86 (m, 1H), 5.65 (dt, 1H, J= 5.4, 15 Hz), 5.83 (dt, 1H, J= 5.4, 15 Hz), 6.07 (s, 1H), 7.76 (s, 1H).
【0121】
[実施例25]
<7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシヘキシル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例20により得られた化合物(15)(93.5 mg)を無水テトラヒドロフラン(3 mL)に溶解し、−78℃にてn-ブチルリチウム(1.59 M、ヘキサン溶液、0.5 mL)を滴下した。さらにn-ヘキシルアルデヒド(0.60 mL)を加え、35分間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液 (15 mL)を加えて0℃に戻し、酢酸エチル(40 mL)で希釈した後、飽和食塩水(5 mL)で洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコールの粗生成物(図6中の化合物(18))を得た。
【0122】
二級アルコールの粗生成物をジクロロメタン(3 mL)に溶解し、0℃にて水(0.5 mL)、t-ブチルアルコール(0.5 mL)、DDQ (112 mg)を順次加えた。30分間撹拌した後、反応液をクロロホルム(20 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(5 mL)にて洗浄し、さらに水層をクロロホルム(15 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシヘキシル-2,5-ジメトキシナフタレン-1,4-ジオン(図6中の化合物(c);49.3 mg,62%)を得た。
【0123】
1HNMR (400 MHz, CDCl3) δ 0.05 (s, 6H), 0.88 (s, 9H), 0.91 (complex, 3H), 1.31 (complex, 6H), 1.63 (m, 1H), 1.92 (m, 1H), 3.52 (d, 2H, J= 5.6 Hz), 3.86 (s, 3H), 3.93 (s, 3H), 4.13 (d, 2H, J= 4.4 Hz), 4.94 (m, 1H), 5.56 (dt, 1H, J= 4.8, 15 Hz), 5.78 (dt, 1H, J= 4.8, 15 Hz), 6.06 (s, 1H), 7.76 (s, 1H).
【0124】
[実施例26]
<7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシヘキシル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例25により得られた化合物(c)(49.3 mg)を無水テトラヒドロフラン(3 mL)に溶解し、0℃にて酢酸(50μL)に溶解させたフッ化テトラn-ブチルアンモニウム(1.0 M、テトラヒドロフラン溶液、0.50 mL)を加えた。室温で15時間撹拌した後、反応液を酢酸エチル(45 mL)で希釈した。水(15 mL)、飽和食塩水(10 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシヘキシル-2,5-ジメトキシナフタレン-1,4-ジオン(図6中の化合物(h);29.0 mg,76%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.88 (complex, 3H), 1.31 (complex, 5H), 1.68 (complex, 3H), 1.94 (m, 1H), 3.53 (d, 2H, J= 5.9 Hz), 3.87 (s, 3H), 3.93 (s, 3H), 4.11 (d, 2H, J= 4.3 Hz), 4.94 (m, 1H), 5.63 (dt, 1H, J= 5.4, 15 Hz), 5.82 (dt, 1H, J= 5.4, 15 Hz), 6.07 (s, 1H), 7.75 (s, 1H).
【0125】
[実施例27]
<7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシフェニルメチル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例20により得られた化合物(15)(92.4 mg)を無水テトラヒドロフラン(2 mL)に溶解し、−78℃にてn-ブチルリチウム(1.59 M、ヘキサン溶液、0.6 mL)を滴下した。さらにベンズアルデヒド(0.60 mL)を加え、10分間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液 (10 mL)を加えて0℃に戻し、酢酸エチル(45 mL)で希釈した後、飽和食塩水(5 mL)で洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコールの粗生成物(図7中の化合物(19))を得た。
【0126】
二級アルコールの粗生成物をジクロロメタン(4 mL)に溶解し、0℃にて水(1.0 mL)、t-ブチルアルコール(0.5 mL)、DDQ (94.9 mg)を順次加えた。35分間撹拌した後、反応液をクロロホルム(25 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(10 mL)にて洗浄し、さらに水層をクロロホルム(15 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシフェニルメチル-2,5-ジメトキシナフタレン-1,4-ジオン(図7中の化合物(d);30.4 mg,25%)を得た。
【0127】
1HNMR (400 MHz, CDCl3) δ 0.05 (s, 6H), 0.93 (s, 9H), 3.29 (s, 3H), 3.65 (d, 2H, J= 6.4 Hz), 3.93 (s, 3H), 3.93 (s, 3H), 4.05 (d, 1H, J= 11 Hz), 4.17 (d, 2H, J= 3.6 Hz), 5.66 (dt, 1H, J= 4.8, 15 Hz), 5.86 (dt, 1H, J= 4.8, 15 Hz), 6.12 (s, 1H), 6.20 (d, 1H, J= 11 Hz), 7.28-7.42 (complex, 5H), 7.95 (s, 1H).
【0128】
[実施例28]
<7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシフェニルメチル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例27により得られた化合物(d)(30.4 mg)を無水テトラヒドロフラン(2 mL)に溶解し、0℃にて酢酸(50μL)に溶解させたフッ化テトラn-ブチルアンモニウム(1.0 M、テトラヒドロフラン溶液、0.50 mL)を加えた。室温で4時間撹拌した後、反応液を酢酸エチル(40 mL)で希釈した。水(15 mL)、飽和食塩水(10 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:3)にて精製し、7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシフェニルメチル-2,5-ジメトキシナフタレン-1,4-ジオン(図7中の化合物(i);14.9 mg,63%)を得た。
1HNMR (400 MHz, CDCl3) δ 3.22 (s, 3H), 3.59 (d, 2H, J= 6.0 Hz), 3.87 (s, 3H), 4.05 (d, 2H, J= 5.6 Hz), 4.09 (d, 1H, J= 10 Hz), 4.17 (d, 2H, J= 3.6 Hz), 5.63 (dt, 1H, J= 5.6, 15 Hz), 5.80 (dt, 1H, J= 5.6, 15 Hz), 6.06 (s, 1H), 6.13 (d, 1H, J= 10 Hz), 7.22-7.34 (complex, 5H), 7.86 (s, 1H).
【0129】
[実施例29]
<(2E)-4-[5-ベンジルオキシ-3-ヒドロキシペンチル-4,7,8-トリメトキシ (2-ナフチル)]ブタ-2-エン-1-オールの製造>
実施例20により得られた化合物(15)(1.06 g)を無水テトラヒドロフラン(30 mL)に溶解し、−78℃にてn-ブチルリチウム(1.57 M、ヘキサン溶液、3.4 mL)を滴下した。さらにn-バレルアルデヒド(1.0 mL)を加え、10分間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液(15 mL)を加え0℃に戻し、酢酸エチル(30 mL)で希釈した後飽和食塩水(10 mL)で洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコールの粗生成物を得た。
【0130】
二級アルコールを無水テトラヒドロフラン(20 mL)に溶解し、0℃にてフッ化テトラn-ブチルアンモニウム(1.0 M、テトラヒドロフラン溶液、9.0 mL)を加えた。室温で1.5時間撹拌した後、反応液を酢酸エチル(30 mL)で希釈した。水(10 mL)、飽和食塩水(5 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1)にて精製し、(2E)-4-[5-ベンジルオキシ-3-ヒドロキシペンチル-4,7,8-トリメトキシ (2-ナフチル)]ブタ-2-エン-1-オール(図8中の化合物(20);693 mg,80%)を得た。
【0131】
1HNMR (270 MHz, CDCl3) δ 0.92 (t, 3H, J= 6.2 Hz), 1.37-1.39 (complex, 2H), 1.63-1.80 (complex, 2H), 1.92-2.04 (complex, 2H), 3.57 (d, 2H, J= 5.4 Hz), 3.80 (s, 3H), 3.90 (s, 3H), 3.93 (s, 3H), 4.09 (d, 2H, J= 5.6 Hz), 4.99 (m, 1H), 5.15 (d, 2H, J= 12 Hz), 5.64 (dt, 1H, J= 5.6, 15 Hz), 5.91 (dt, 1H, J= 5.4, 15 Hz), 6.73 (s, 1H), 7.36 (d, 1H, J= 7.2 Hz), 7.41 (t, 2H, J= 7.2 Hz), 7.53 (d, 2H, J= 7.2 Hz), 7.65 (s, 1H).
13CNMR (67.80 MHz, CDCl3) δ 14.2, 22.8, 29.2,36.6, 39.0, 50.3, 56.9, 61.1, 63.4, 64.0, 71.1, 72.9, 100.3, 113.7, 115.1, 118.2, 127.4, 127.8, 128.4, 130.7, 131.0, 131.2, 136.8, 136.9, 137.0, 147.9, 151.1, 154.8, 163.6.
IR (film) 3408, 2933, 2858, 1620, 1574 cm-1.
【0132】
[実施例30]
<(2E)-4-[5-ベンジルオキシ-3-ヒドロキシペンチル-4,7,8-トリメトキシ(2-ナフチル) ]ブタ-2-エニルアセテートの製造>
実施例29により得られた化合物(20)(111 mg)をジクロロメタン(5 mL)に溶解し、氷浴中、ピリジン(36μL)および無水酢酸(15μL)を加えた。室温で2日間撹拌した後、溶媒をトルエン共沸により減圧留去して得られる残留物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、(2E)-4-[5-ベンジルオキシ-3-ヒドロキシペンチル-4,7,8-トリメトキシ(2-ナフチル) ]ブタ-2-エニルアセテート(図8中の化合物(21);70.5 mg,58%)を得た。
【0133】
1HNMR (400 MHz, CDCl3) δ 0.93 (t, 3H, J= 6.8 Hz), 1.34-1.44, (complex, 4H), 1.63-1.83 (complex, 2H), 2.04 (s, 3H), 3.62 (d, 2H, J= 6.4 Hz), 3.80 (s, 3H), 3.90 (s, 3H), 3.94 (s, 3H), 4.55 (d, 2H, J= 6.4 Hz), 5.01 (m, 1H), 5.12 (d, 1H, J= 11 Hz), 5.19 (d, 1H, J= 11 Hz), 5.61 (dt, 1H, J= 6.4, 15 Hz), 6.01 (dt, 1H, J= 6.0, 15 Hz), 6.74 (s, 1H), 7.36 (d, 1H, J= 7.2 Hz), 7.42 (t, 2H, J= 7.2 Hz), 7.53 (d, 2H, J= 7.2 Hz), 7.66 (s, 1H)
13CNMR (100.40 MHz, CDCl3) δ 14.2, 21.0, 22.7, 29.2, 36.4, 38.9, 56.9, 61.0, 63.9, 64.8, 70.9, 72.8, 100.2, 115.0, 118.3, 125.4, 127.4, 127.8, 128.2, 128.4, 130.9, 131.2, 134.2, 136.6, 136.8, 137.0, 147.9, 151.1, 154.8, 170.6.
IR (film) 3480, 2933, 2857, 1739, 1620, 1573 cm-1
【0134】
[実施例31]
<(2E)-4-[3-ヒドロキシペンチル-4,7-ジメトキシ-5,8-ジオキソ(2-ナフチル)]ブタ-2-エニルアセテートの製造>
実施例30により得られた化合物(21)(60.4 mg)をジクロロメタン(4 mL)に溶解し、0℃にて水(2 mL)、t-ブチルアルコール(2 mL)、DDQ(83.4 mg)を順次加えた。20分間撹拌した後、反応液をクロロホルム(20 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(10 mL)にて洗浄し、さらに水層をクロロホルム(10 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、(2E)-4-[3-ヒドロキシペンチル-4,7-ジメトキシ-5,8-ジオキソ(2-ナフチル)]ブタ-2-エニルアセテート(図8中の化合物(v);37.4 mg,78%)を得た。
【0135】
1HNMR (400 MHz, CDCl3) δ 0.90 (t, 3H, J= 7.2 Hz), 1.28-1.37, (complex, 4H), 1.57-1.68 (complex, 2H), 2.03 (s, 3H), 3.56 (d, 2H, J= 6.4 Hz), 3.85 (s, 3H), 3.91 (s, 3H), 4.51 (d, 2H, J= 6.0 Hz), 4.94 (m, 1H), 5.58 (dt, 1H, J= 6.4, 15 Hz), 5.88 (dt, 1H, J= 6.0, 15 Hz), 6.06 (s, 1H), 7.74 (s, 1H)
13CNMR (100.40 MHz, CDCl3) δ 14.1, 20.9, 22.6, 28.8, 36.3, 37.7, 56.3, 63.4, 64.4, 70.5, 111.7, 125.1, 126.6, 126.8, 127.4, 128.4, 131.7, 132.1, 144.3, 158.7, 170.6. 179.6, 183.7.
IR (film) 3502, 2935, 1737, 1683, 1648, 1619 cm-1
【0136】
[実施例32]
<(2E)-4-[4-ヒドロキシ-3-ヒドロキシペンチル-7-メトキシ-5,8-ジオキソ(2-ナフチル)]ブタ-2-エニルアセテートの製造>
三臭化ホウ素(1M、ジクロロメタン溶液、0.22 mL)をジクロロメタン(5 mL)に溶解し、−78℃にてジクロロメタン(3.5 mL)に溶解させた化合物(v)(83.2 mg)を加えて25分間撹拌した。反応液をクロロホルム(15 mL)で希釈した後、水、飽和食塩水(各々10 mL)で洗浄し、有機層を無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲル薄層クロマトグラフィー(ヘキサン:酢酸エチル=1:2)にて精製し、(2E)-4-[4-ヒドロキシ-3-ヒドロキシペンチル-7-メトキシ-5,8-ジオキソ(2-ナフチル)]ブタ-2-エニルアセテート(図8中の化合物(w);23.7 mg,29%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.90 (t, 3H, J= 6.8 Hz), 1.33-1.42 (complex, 4H), 1.50-1.76 (complex, 2H), 2.05 (s, 3H), 3.50 (d, 2H, J= 5.8 Hz), 3.92 (s, 3H), 4.52 (d, 2H, J= 6.2 Hz), 4.86 (m, 1H), 5.59 (dt, 1H, J= 5.8, 15 Hz), 5.87 (dt, 1H, J= 6.2, 15 Hz), 6.07 (s, 1H), 7.48 (s, 1H), 13.11 (s, 1H)
13CNMR (67.80 MHz, CDCl3) δ 14.1, 21.0, 22.7, 28.6, 36.3, 36.4, 49.2, 49.3, 56.7, 64.4, 70.6, 109.3, 121.7, 126.8, 129.0, 131.6, 138.4, 144.4, 159.5, 161.0, 190.7, 206.5.
IR (film) 3534, 2956, 1739, 1683, 1625 cm-1
【0137】
[実施例33]
<(2E)-4-(4-ヒドロキシ-7-メトキシ-5,8-ジオキソ-3-ペンタノイル(2-ナフチル))ブタ-2-エニルアセテートの製造>
実施例32により得られた化合物(w)(2.6 mg)を無水テトラヒドロフラン(0.9 mL)に溶解し、0℃にてジメチルスルホキシド(0.1 mL)、IBX (20.6 mg)を加えた。室温で7時間撹拌した後、反応液をヘキサン(20 mL)で希釈した。沈殿物をセライト濾過し、ヘキサン:酢酸エチル=3:1の混合溶媒で洗浄したろ液を減圧濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、(2E)-4-(4-ヒドロキシ-7-メトキシ-5,8-ジオキソ-3-ペンタノイル(2-ナフチル))ブタ-2-エニルアセテート(図8中の化合物(e);2.7 mg)を得た。
1HNMR (270 MHz, CDCl3) δ 0.94 (t, 3H, J= 7.3 Hz), 1.37 (complex, 2H), 1.68 (complex, 2H), 2.11 (s, 3H), 2.86 (t, 2H, J= 7.6 Hz), 3.36 (d, 2H, J= 6.2 Hz), 3.93 (s, 3H), 4.52 (d, 2H, J= 6.2 Hz), 5.62 (dt, 1H, J= 5.9, 15 Hz), 5.81 (dt, 1H, J= 5.9, 15 Hz), 6.11 (s, 1H), 7.52 (s, 1H), 12.5 (s. 1H).
【0138】
[実施例34]
<7-((2E)-4-ヒドロキシブタ-2-エニル)-5-ヒドロキシ-2-メトキシ-6-ペンタノイルナフタレン-1,4-ジオンの製造>
実施例33により得られた化合物(e)(2.7 mg)をメタノール(1 mL)に溶解し、0℃にて炭酸カリウム(11.3 mg)を加えた。室温で30分間撹拌した後、反応液を酢酸エチル(30 mL)で希釈した。飽和塩化アンモニウム水溶液(10 mL)、飽和食塩水(5 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、7-((2E)-4-ヒドロキシブタ-2-エニル)-5-ヒドロキシ-2-メトキシ-6-ペンタノイルナフタレン-1,4-ジオン(図8中の化合物(j);2.3 mg、96%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.94 (t, 3H, J= 7.3 Hz), 1.34 (complex, 2H), 1.67 (complex, 2H), 2.87 (t, 2H, J= 7.3 Hz), 3.35 (d, 2H, J= 5.4 Hz), 3.93 (s, 3H), 4.20 (complex, 2H), 5.64-5.83 (complex, 2H), 6.11 (s, 1H), 7.53 (s, 1H), 12.5 (s. 1H).
【0139】
[実施例35]
<GST-Cdc25Aの調製>
GST融合タンパク質発現ベクターであるpGEX-2T(Amersham Biosciences社)にヒトcdc25A(hcdc25A) のcDNAを組み込みpGEX-2T-hcdc25Aを作製した。これらのプラスミドで形質転換した各大腸菌BL21株を37℃にてOD600=0.6〜0.8になるまで培養し、0.1mMのイソプロピル-1-チオ-β-D-チオガラクトピラノシド(IPTG) (Stratagene社)を添加することによりGST-Cdc25Aタンパク質の発現をそれぞれ誘導した。さらに37℃にて4時間培養後、菌体を回収し可溶化溶液(MT-PBS(150mM NaCl, 20mM Na2HPO4, 4mM NaH2PO4)、0.1% トリトンX-100、10μg/mL ロイペプチン、1mM フェニルメチルスルホニルフルオライド(PMSF))に懸濁した。懸濁した大腸菌を超音波破砕器により破砕後、遠心操作により可溶画分を得た。この可溶画分に、MT-PBSにより平衡化済みのグルタチオンアガロース (SIGMA社)を加え、4℃にて1時間GST-Cdc25Aをそれぞれ吸着させた。遠心操作によりグルタチオンアガロースを回収し、MT-PBSで4回洗浄後、溶出液(50mM Tris-HCl (pH8.0)、20mM 還元型グルタチオン)で4℃にて1時間攪拌することによりGST-Cdc25Aを溶出した。その後、遠心操作により上清を回収し、これを酵素(GST-Cdc25A)として以下の実施例に用いた。
また、PP1B(BIOMOL Research Laboratories社)およびPP2A(Promega社)を酵素として以下の実施例に用いた。
【0140】
[実施例36]
<ホスファターゼアッセイ>
上述の実施例により得られた化合物(f)〜(p)、並びに、上述の化合物(q)及び下式(x)で表される化合物(以下、「化合物(x)」と称する。)のホスファターゼ活性に対する阻害効果を調べた。なお、本実施例ではCdc25Aの基質として、3-O-メチルフルオレセインホスフェート(3-O-methylfluorescein phosphate;OMFP(SIGMA製))と4-ニトロフェニルホスフェート(4-nitrophenyl phosphate;pNPP(SIGMA製))とを用い、PTP1B及びPP2Aの基質としてpNPPを用いた。
【化19】
【0141】
<pNPPを用いた場合>
酵素(Cdc25A、PTP1B、及びPP2A) 30μl (30μg)と各濃度の化合物(化合物(f)〜(q)、化合物(x)及びNa3VO4(ホスファターゼ阻害剤のポジティブコントロールとして使用))1μlをassay buffer(50mM Tris-HCl (pH7.5)) 60μl中に懸濁し、室温にて15分間プレインキュベートした。そこに基質(5mM pNPP) 10μlを加え37℃にて60分間反応させた。その後、MICRO PLATE READER(TOSOH製)を用いて波長405nmでの吸光度を測定し、化合物の各酵素(Cdc25A、PTP1B、及びGST-PP2A)に対する阻害活性を評価した。なお、阻害活性の評価は阻害率(%)を算出することにより行った。
【0142】
<OMFPを用いた場合>
酵素(Cdc25A) 30μl (15μg)と各濃度の化合物(化合物(f)〜(q)、化合物(x)及びNa3VO4)1μlをassay buffer(50mM Tris-HCl (pH7.5)) 60μl中に懸濁し、室温にて15分間プレインキュベートした。そこに基質(最終濃度で25μM OMFP) 10μlを加え、室温にて30分間反応させた。その後、Fluoroskan Ascent(Labsystem製)を用いて3-O-メチルフルオレセインの生成量(励起波長355nm、吸収波長538nm)を測定し、化合物の酵素(Cdc25A)に対する阻害活性を評価した。なお、阻害活性の評価は阻害率(%)を算出することにより行った。
【0143】
以上の結果を表1に示す。なお、表1に示す各数値はIC50(μg/ml)の値をそれぞれ示す(但し、Na3VO4はμMで示す。)。
【表1】
【0144】
表1に示すように、化合物(q)は特定の基質(pNPP)を用いた時にのみCdc25Aに対する阻害活性を示し、化合物(x)はCdc25Aのみに対して阻害活性を示し、それ以外には阻害活性を示さないことがわかった。これに対して、化合物(f)〜(p)はチロシンホスファターゼ(Cdc25A及びPTP1B)のみに対して阻害活性を示し、プロテインホスファターゼ(PP2A)には阻害活性を示さないことがわかった。これらのことから、化合物(f)〜(p)は、チロシンホスファターゼを特異的に阻害する阻害剤として有用であることが明らかになった。
【0145】
[実施例37]
<細胞周期解析>
Cdc25AはCdk2/CyclinEやCdk2/CyclinAに作用するホスファターゼであり、細胞周期の調節においてG1/Sの移行に必要であることが知られている。そこで、Cdc25Aホスファターゼを阻害する化合物(f)〜(q)及び化合物(x)が細胞周期の進行を阻害するかどうかを調べた。
【0146】
5x105 個のマウス繊維芽細胞であるNIH3T3細胞を、0.2%牛胎児血清(MOREGATE社製)を含むダルベッコ改変イーグル培地(DMEM;10 ml)に加えて2日間培養し、静止期に同調した。その後、メタノールに溶解した各化合物(化合物(f)〜(q)、化合物(x)及びNa3VO4)を最終濃度で3μg/mL(Na3VO4は10又は100μM)となるように添加して15分間培養し、最終濃度が10%になるように牛胎児血清を添加した。血清刺激後、20時間培養した細胞を回収し、1mlのリン酸緩衝溶液に懸濁した。なお、コントロールとして、各化合物を含有していない等量のメタノールを添加して培養し、牛胎児血清で刺激した後、0時間(G0)又は20時間(20h cont.)培養した細胞をそれぞれ回収し、1mlのリン酸緩衝溶液に懸濁したものをそれぞれ準備した。懸濁後、50μgのヨウ化プロピジウム(WAKO社製)を添加して染色されたDNAの含有量をフローサイトメーター(EPICS ALTRA:BECKMAN COULTER社製)により測定した。その結果を図9に示す。
【0147】
図9に示すように、化合物(j)及び(q)は細胞周期のG1期で進行を停止させる作用を有することが明らかになった。そこで、上記と同様の方法により、化合物(j)及び(q)を用いて各濃度(1,3,10μg/mL)における細胞周期の進行阻害の程度を評価した。図10に示すように、化合物(j)及び(q)は少なくとも3μg/mLで細胞周期のG1期の進行を阻害できることが明らかになった。
【0148】
[実施例38]
<細胞増殖に対する抑制効果>
次に、化合物(f)〜(q)の細胞増殖に対する抑制効果を調べるため、化合物(j)〜(q)を用いてMTTアッセイを行った。48ウェルプレートに、NIH3T3細胞の懸濁液(1×105個/ml;10%牛胎児血清を含むDMEM)を500μlずつ添加して37℃で24時間培養し、メタノールに溶解した各化合物(化合物(j)〜(q))の溶液(0.03、0.1、0.3、1、3 mg/mL)を5μL添加して37℃で48時間培養した(なお、コントロールとして、メタノールのみを5μL添加して37℃で48時間培養したものを準備した。)。その後、MTT(3-(4,5-dimethlthiazol-2-yl)-2,5-diphenyltetrazolium bromide;Sigma)溶液(PBS-で5mg/mlに調整したもの)を25μlずつ添加して37℃で4時間培養した。培養後、培地を除去し、フォルマザン沈殿(細胞のミトコンドリアに存在するコハク酸脱水素酵素により、黄色水溶性のMTTが還元された暗青色不溶性の物質)を500μlのジメチルスルホキシドで溶解し、マイクロプレートリーダー(東ソー株式会社)を用いて吸光度(測定波長570 nm,リファレンス側620 nm)を測定し、増殖阻害作用を評価した。
【0149】
その結果を表2に示す。また、各化合物で刺激した細胞に細胞死が引き起こされていないかどうかを確認するため、顕微鏡により細胞の形態を観察した。
【表2】
【0150】
表2に示すように、化合物(j)及び化合物(m)〜(p)は、細胞増殖を抑制する作用を有しており、特に化合物(j)は優れた細胞増殖抑制作用を有していることが明らかになった。また、顕微鏡の形態観察により、多量の化合物(j)又は化合物(n)〜(q)のいずれかを用いると細胞死を引き起こすことが明らかになった。
【0151】
[実施例39]
<1-(5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ-2-ナフチル)アセトンの製造>
ベンゼン(6 mL)中、臭化亜鉛(257 mg)を30分間加熱還流し、この混合物にベンゼン(5 mL)に溶解させた化合物(12)(153 mg)を加えた。1.5時間後反応液を室温に戻し、酢酸エチル(35 mL)で希釈した後、水、飽和食塩水(各々15 mL)にて洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去してアルデヒドの粗生成物を得た。
【0152】
アルデヒドの粗生成物を無水テトラヒドロフラン(5 mL)に溶解し、-20℃にて臭化メチルマグネシウム(0.93 M、テトラヒドロフラン溶液、2 mL)を滴下した。徐々に昇温しながら30分間撹拌した後、反応液を酢酸エチル(20 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(5 mL)にて洗浄し、さらに水層を酢酸エチル(20 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水後、溶媒を減圧留去して二級アルコールの粗生成物を得た。
【0153】
二級アルコールの粗生成物を無水テトラヒドロフラン(2.7 mL)に溶解し、20 ℃にてジメチルスルホキシド(0.3 mL)、IBX (353 mg)を順次加えた。室温にて23時間撹拌した後、反応液をヘキサン(20 mL)で希釈した。沈殿物をセライト濾過し、ヘキサン:酢酸エチル=3:1の混合溶媒で洗浄したろ液を減圧濃縮して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1)にて精製し、1-(5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ-2-ナフチル)アセトン(図11中の化合物(22);99.2 mg、63%)を得た。
1HNMR (270 MHz, CDCl3) δ 2.25 (s, 3H), 3.77 (s, 3H), 3.90 (s, 3H), 3.95 (s, 3H), 4.03 (s, 2H), 5.18 (s, 2H), 6.77 (s, 1H), 7.35-7.45 (complex, 3H), 7.56 (d, 2H, J= 7.0 Hz), 7.76 (s. 1H).
【0154】
[実施例40]
<8-ベンジルオキシ-2-ブロモ-1,5,6-トリメトキシ-3-(2-メチル(1,3-ジオキソラン-2-イル)メチル)ナフタレンの製造>
ベンゼン(30 mL)中、化合物(22)(761 mg)、エチレングリコール(1.0 mL)、4-トルエンスルホン酸一水和物(663 mg)を4時間加熱還流した。反応液を室温に戻し、酢酸エチル(35 mL)で希釈した後、飽和炭酸水素ナトリウム水溶液(20 mL)、飽和食塩水(10 mL)で洗浄し、有機層を無水硫酸ナトリウムで脱水後、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1)にて精製し、8-ベンジルオキシ-2-ブロモ-1,5,6-トリメトキシ-3-(2-メチル(1,3-ジオキソラン-2-イル)メチル)ナフタレン(図11中の化合物(23);765 mg、92%)を得た。
1HNMR (400 MHz, CDCl3) δ 1.43 (s, 3H), 3.37 (s, 2H), 3.78 (s, 4H), 3.90 (s, 3H), 3.95 (s, 3H), 5.18 (s, 2H), 6.75 (s, 1H), 7.36 (d, 1H, J= 7.6 Hz), 7.42 (t, 2H, J= 7.2 Hz), 7.57 (d, 2H, J= 7.6 Hz) 7.91 (s, 1H).
【0155】
[実施例41]
<7,10-ジメトキシ-3-メチル-1-プロピルベンゾ[2,1-g]イソクロメン-6,9-ジオンの製造>
化合物(23)(432 mg)を無水テトラヒドロフラン(10 mL)に溶解し、-78℃にてn-ブチルリチウム(1.56 M、ヘキサン溶液、2.8 mL)を滴下した。さらにn-ブチルアルデヒド(1.2 mL)を加え、10分間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液 (15 mL)を加えて0℃に戻し、酢酸エチル(35 mL)で希釈した後、飽和食塩水(10 mL)で洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコールの粗生成物を得た。
【0156】
二級アルコールの粗生成物をベンゼン(15 mL)に溶解し、0℃にて4-トルエンスルホン酸一水和物(396 mg)を加えた。室温で10分間撹拌した後、酢酸エチル(25 mL)で希釈し飽和炭酸水素ナトリウム水溶液(15 mL)、飽和食塩水(5 mL)で洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮してピラノナフタレンの粗生成物を得た。
【0157】
ピラノナフタレンの粗生成物を1,4-ジオキサン(4 mL)に溶解し、0℃にて水(1 mL)、DDQ (362 mg)を順次加えた。40分間撹拌した後、反応液をクロロホルム(25 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(10 mL)にて洗浄し、さらに水層をクロロホルム(20 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)にて精製し、7,10-ジメトキシ-3-メチル-1-プロピルベンゾ[2,1-g]イソクロメン-6,9-ジオン(図1中の化合物(r);75.5 mg、27%)を得た。
【0158】
1HNMR (270 MHz, CDCl3) δ 0.95 (t, 3H, J= 7.0 Hz), 1.38-1.58 (complex, 3H), 1.96 (s, 3H), 2.04 (m, 1H), 3.86 (s, 3H), 3.88 (s, 3H), 5.76 (dd, 1H, J= 3.0, 9.5 Hz), 5.66 (s, 1H), 6.03 (s, 1H), 7.47 (s. 1H).
【0159】
[実施例42]
<7,10-ジメトキシ-3-メチル-1-プロピルベンゾ[2,1-g]イソクロマン-6,9-ジオンの製造>
化合物(r)(36.2 mg)をメタノール(3 mL)に溶解し、Pd/C (10 mg)を加えた。水素気流下室温にて1時間撹拌した後、沈殿物をセライト濾過し、メタノールで洗浄したろ液を減圧濃縮して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、7,10-ジメトキシ-3-メチル-1-プロピルベンゾ[2,1-g]イソクロマン-6,9-ジオン(図11中の化合物(s);31.3 mg、86%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.88 (t, 3H, J= 7.3 Hz), 1.00 (t, 3H, J= 7.3 Hz), 1.34 (d, 2H, J= 5.9 Hz), 1.35 (d, 2H, J= 6.2 Hz), 1.20-2.05 (complex, 8H), 2.66-2.87 (complex, 4H), 3.64 (m, 1H), 3.84 (s, 3H), 3.87 (s, 3H), 3.88 (s, 6H), 4.05 (m, 1H), 5.00 (dd, 1H, J= 3.2, 9.7 Hz), 5.06 (d, 1H, J= 3.5 Hz), 6.07 (s, 2H), 7.69 (s, 1H), 7.70 (s. 1H).
【0160】
[実施例43]
<8-ブロモ-7,10-ジメトキシ-3-メチル-1-プロピルベンゾ[1,2-g]イソクロマン-6,9-ジオンの製造>
化合物(s)(31.3 mg)をジクロロメタン(2 mL)に溶解し、0℃にてピリジン(30 μL)、臭化水素酸ペルブロミドピリジン錯体(Pyr・HBr3)(42.4 mg)を加えた。室温で4時間撹拌した後、反応液をクロロホルムで希釈し、1M塩酸(15 mL)を加えた。有機層を飽和食塩水(5 mL)にて洗浄し、さらに水層をクロロホルム(15 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム:酢酸エチル=1:1)にて精製し、8-ブロモ-7,10-ジメトキシ-3-メチル-1-プロピルベンゾ[1,2-g]イソクロマン-6,9-ジオン(図11中の化合物(t);16.8 mg、36%)及び化合物(s)(12.3 mg、39%)を得た。
1HNMR (400 MHz, CDCl3) δ 0.88 (t, 3H, J= 7.3 Hz), 1.00 (t, 3H, J= 7.3 Hz), 1.34 (d, 2H, J= 5.9 Hz), 1.35 (d, 2H, J= 6.2 Hz), 1.31-2.04 (complex, 8H), 2.67-2.86 (complex, 4H), 3.76 (m, 1H), 3.84 (s, 3H), 3.87 (s, 3H), 4.05 (m, 1H), 4.25 (s, 6H), 5.00 (dd, 1H, J= 3.6, 11 Hz), 5.06 (m, 1H), 7.63 (s, 1H), 7.64 (s. 1H).
【0161】
[実施例44]
<8-クロロ-7,10-ジヒドロキシ-3-メチル-1-プロピルベンゾ[1,2-g]イソクロマン-6,9-ジオンの製造>
エタノール(2 mL)中、化合物(t)(3.8 mg)、及び濃塩酸(4 mL)を15時間加熱還流した。反応液を室温に戻し、クロロホルム(40 mL)で希釈した後、水(30 mL)で洗浄し、有機層を無水硫酸ナトリウムで脱水した。溶媒を減圧留去してクロロナフトキノンの粗生成物(図1中の化合物(u))を得た。
1HNMR (270 MHz, CDCl3) δ 0.88 (t, 3H, J= 7.3 Hz), 1.00 (t, 3H, J= 7.2 Hz), 1.34 (d, 2H, J= 6.0 Hz), 1.35 (d, 2H, J= 6.1 Hz), 1.25-2.05 (complex, 8H), 2.61-2.81 (complex, 4H), 3.65 (m, 1H), 4.03 (m, 1H), 5.02 (complex, 2H), 7.43 (s, 2H), 12.4 (s. 1H), 12.6 (s, 1H).
【0162】
[実施例45]
<MRSAに対する抗菌作用及びイミペネム活性増強作用>
本実施例では、検定菌として、臨床分離されたMRSA K24株を用いた。検定プレートは、Mueller-Hinton Broth (DIFCO社)に1.5%のAgar (清水食品株式会社)を添加して作製した対照用プレート、及び上記組成にイミペネム(萬有製薬株式会社、商品名:チエナム筋注用力価0.5)を最終濃度10μg/mLとなるように添加して作製した検定用プレートを用いた。接種菌液はMueller-Hinton Brothで一晩培養した検定菌培養液を、同培地で、0.5Mc Farand (約108cfu/mL)になるように調製したものを用いた。この接種菌液をNCCLS法に従い、滅菌綿棒で各プレートに塗抹した。検定菌に対する各プレートでの抗菌活性は、ペーパーディスク法により、10μgの検定薬剤(化合物(r)、(s)、(v)、及び(w))のメタノール溶液を染み込ませた6mmのペーパーディスクを各プレートにのせて、37℃で20時間培養した。その後、検定菌に対する抗菌活性の指標として、プレートにおける阻止円径 (ペーパーディスクの中心を通る直径(mm))を測定した。
【0163】
その結果を表3に示す。
【表3】
【0164】
表3に示すように、化合物(r)、(s)、(v)、及び(w)は単独で抗MRSA作用を有することが明らかになった。また、10μg/mLのイミペネム単独では抗MRSA作用を有しないが、化合物(r)、(s)、(u)、(v)、及び(w)をイミペネムと協同して検定菌に作用させると、抗MRSA作用が増強することが明らかになった。このことから、化合物(r)、(s)、(u)、(v)、及び(w)がイミペネムの活性を増強する作用を有すること、あるいは、イミペネムがこれらの化合物の活性を増強する作用を有することが示唆された。
【図面の簡単な説明】
【0165】
【図1】本発明の一実施例において、化合物(k)及び(l)の製造過程を示す図である。
【図2】本発明の一実施例において、化合物(m)〜(p)の製造過程を示す図である。
【図3】本発明の一実施例において、化合物(15)の製造過程を示す図である。
【図4】本発明の一実施例において、化合物(f)の製造過程を示す図である。
【図5】本発明の一実施例において、化合物(g)の製造過程を示す図である。
【図6】本発明の一実施例において、化合物(h)の製造過程を示す図である。
【図7】本発明の一実施例において、化合物(i)の製造過程を示す図である。
【図8】本発明の一実施例において、化合物(j)の製造過程を示す図である。
【図9】本発明の一実施例において、化合物(f)〜(q)、化合物(x)及びNa3VO4が細胞周期の進行に与える影響を調べた結果を示す図である。
【図10】本発明の一実施例において、各濃度の化合物(化合物(j)又は(q))が細胞周期の進行に与える影響を調べた結果を示す図である。
【図11】本発明の一実施例において、化合物(r)、化合物(s)、及び化合物(u)の製造過程を示す図である。
【技術分野】
【0001】
本発明は、ナフトキノン誘導体化合物又はその薬理学的に許容される塩、並びに、それを有効成分として含有する、チロシンホスファターゼ阻害剤、医薬組成物、細胞増殖阻害剤、細胞周期進行阻害剤、抗菌剤、イミペネム活性増強剤、及び細菌増殖阻害剤、並びに、上述の化合物を用いたイミペネム活性増強方法、及び細菌の増殖阻害方法、並びに、上述の化合物の活性増強剤及び活性増強方法に関する。
【背景技術】
【0002】
チロシンホスファターゼに対する阻害剤は、2型糖尿病、肥満症、腫瘍、神経疾患等のチロシンホスファターゼの過剰発現又は活性化に起因する疾患に有用であるとされており、各種チロシンホスファターゼに対する阻害剤の開発が求められている。従来、チロシンホスファターゼであるCdc25に対する阻害剤として、いくつかのナフトキノンアナログが知られている(例えば、非特許文献1〜4参照)。また、チロシンホスファターゼであるPTP1Bに対する阻害剤として、いくつかの化合物やクロレラの細胞壁破砕物等が知られている(例えば、特許文献1〜5参照)。
【非特許文献1】Bioorg. Med. Chem. Lett. 1998, 8(18), 2507-10
【非特許文献2】J. Antibiot. 1999, 52, 256-262
【非特許文献3】Bull Chem Soc Jpn. 2004, 77, 1925-30
【非特許文献4】Bioorg. Med. Chem. Lett. 2005, 15(1), 61-5
【特許文献1】特開平11−222474号公報
【特許文献2】特開2000−256330号公報
【特許文献3】特開2002−121186号公報
【特許文献4】特開2005−89322号公報
【特許文献5】特表平11−508919号公報
【発明の開示】
【発明が解決しようとする課題】
【0003】
本発明は、チロシンホスファターゼの阻害剤として有用な新規化合物、並びにそれを有効成分として含有する、チロシンホスファターゼ阻害剤、医薬組成物、細胞増殖阻害剤、細胞周期進行阻害剤、抗菌剤、イミペネム活性増強剤、及び細菌増殖阻害剤、並びに、上述の化合物を用いたイミペネム活性増強方法、及び細菌の増殖阻害方法、並びに、上述の化合物の活性増強剤及び活性増強方法に関する。
【課題を解決するための手段】
【0004】
本発明者らは、上記課題を解決すべく、下記の一般式(II)で表されるナフトキノン誘導体化合物(式中、R5は水素原子又はアルキル基であり、R6はヒドロキシエチル基、ヒドロキシプロピル基、ヒドロキシヘキシル基、アリールアルコール基、又はアルキルカルボニル基であり、R7は水素原子、アシル基、又はブチルジメチルシリル基であり、前記R6と前記R7とで環状を構成しない。)として、下式(f)〜(j)で表される化合物(以下、それぞれを「化合物(f)」、「化合物(g)」、「化合物(h)」、「化合物(i)」、及び「化合物(j)」と称する。)を用いてチロシンホスファターゼの酵素活性に与える影響を調べた。その結果、化合物(f)〜(j)で表される化合物はCdc25Aホスファターゼやプロテインチロシンホスファターゼ1B等のチロシンホスファターゼの酵素活性を阻害することを見出した。
【化1】
【化2】
【0005】
また、上記と同様に一般式(III)で表されるナフトキノン誘導体化合物(式中、R8は水素原子又はアシル基である。)として、下式(k)及び(l)で表される化合物(以下、それぞれを「化合物(k)」及び「化合物(l)」と称する。)を用いてチロシンホスファターゼの酵素活性に与える影響を調べた。その結果、化合物(k)及び(l)で表される化合物はCdc25Aホスファターゼやプロテインチロシンホスファターゼ1B等のチロシンホスファターゼの酵素活性を阻害することが明らかになった。
【化3】
【化4】
【0006】
また、同様に一般式(IV)で表されるナフトキノン誘導体化合物(式中、R9はプロピル基、ブチル基、へキシル基、又はアルキニルオキシアルキル基である。)として、下式(m)〜(p)で表される化合物(以下、それぞれを「化合物(m)」、「化合物(n)」、「化合物(o)」、及び「化合物(p)」と称する。)を用いてチロシンホスファターゼの酵素活性に与える影響を調べた。その結果、化合物(m)〜(p)で表される化合物はCdc25Aホスファターゼやプロテインチロシンホスファターゼ1B等のチロシンホスファターゼの酵素活性を阻害することが明らかになった。
【化5】
【化6】
【0007】
ところで、Cdc25AはCdk2/CyclinEやCdk2/CyclinAに作用し、細胞周期の調節においてG1/Sの移行に機能することが知られている。そこで、本発明者らは、Cdc25Aホスファターゼを阻害する上述の化合物が細胞周期の進行を阻害するかどうかを調べるため、化合物(f)〜(p)と、下式(q)で表される化合物(以下、「化合物(q)」と称する。)とを用いて細胞周期の進行に与える影響を調べた。その結果、化合物(j)及び(q)で表される化合物が細胞周期のG1期で進行を停止することが明らかになった。
【化7】
【0008】
また、本発明者らは、上述の化合物が細胞増殖を抑制するかどうかを調べるため、化合物(j)〜(q)を用いて細胞増殖抑制活性を調べた。その結果、化合物(j)及び化合物(m)〜(p)が細胞増殖抑制活性を有することが明らかになった。
【0009】
さらに、本発明者らは、下式(r)、(s)、(v)、及び(w)で表される化合物(以下、それぞれを「化合物(r)」、「化合物(s)」、「化合物(v)」、及び「化合物(w)」と称する。)が単独で抗MRSA(メチシリン耐性黄色ブドウ球菌)作用を有すること、化合物(r)、(s)、(v)、及び(w)、並びに下式(u)で表される化合物(以下、「化合物(u)」と称する。)を、MRSAには単独で効果がないイミペネムと協同してMRSAに作用させると抗MRSA活性が増強することを見出した。
【化8】
【0010】
このようにして、本発明者らは本発明を完成するに至った。
【0011】
すなわち、本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩は、下記の一般式(I)で表されることを特徴とする。
【化9】
【0012】
式(I)中、R3がアリールアルコール基又はアルキルカルボニル基である場合には、R1は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R2はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R4はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、又は2−ジメタン−1,3−ジオキサン−4−アルキル基であり、式中、R4がアルキニルオキシアルキル基又はブチルジメチルシリルオキシアルケニル基である場合には、R1は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R2はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R3は水素原子、ヒドロキシアルキル基、ハロゲン原子、又はアリールアルコール基であり、式中、R1がアシルオキシ基である場合には、R2はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R3は水素原子、ヒドロキシアルキル基、又はハロゲン原子であり、R4はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、又は2−ジメタン−1,3−ジオキサン−4−アルキル基であり、式中、R1及びR2がメトキシ基であり、R4がヒドロキシブタ−2−エニル基である場合には、R3はヒドロキシエチル基、ヒドロキシプロピル基、又はヒドロキシヘキシル基であり、式中、R2がメトキシ基であり、R3が水素原子であり、R4がメトキシメチル基である場合には、R1はヒドロキシル基であり、式中、R1及びR2がメトキシ基であり、R3が水素原子である場合には、R4はへキシルオキシメチル基などである。なお、本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩は、R3とR4とで環状を構成しないことが好ましい。
【0013】
また、本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩は、上述の一般式(II)〜(IV)で表されることを特徴とする。なお、式(II)中、R5は水素原子又はアルキル基であり、R6はヒドロキシエチル基、ヒドロキシプロピル基、ヒドロキシヘキシル基、アリールアルコール基、又はアルキルカルボニル基であり、R7は水素原子、アシル基、又はブチルジメチルシリル基であり、前記R6と前記R7とで環状を構成しない。式(III)中、R8は水素原子又はアシル基である。式(IV)中、R9はプロピル基、ブチル基、へキシル基、又はアルキニルオキシアルキル基である。前記ナフトキノン誘導体化合物としては、上述の化合物(a)〜(q)であることが好ましい。
【0014】
本発明に係るチロシンホスファターゼ阻害剤は、上述のナフトキノン誘導体化合物又はその薬理学的に許容される塩を有効成分として含有する。前記チロシンホスファターゼとしては、例えば、Cdc25Aホスファターゼ、プロテインチロシンホスファターゼ1Bなどである。
【0015】
また、本発明に係る医薬組成物は、上述のナフトキノン誘導体化合物又はその薬理学的に許容される塩を有効成分として含有する。前記医薬組成物は、例えば、プロテインチロシンホスファターゼ1Bの過剰発現又は活性化に起因する疾患、Cdc25Aホスファターゼの過剰発現又は活性化に起因する疾患などの予防又は改善に有用である。前記プロテインチロシンホスファターゼ1Bの過剰発現又は活性化に起因する疾患としては、例えば、2型糖尿病、肥満症、神経疾患(例えば、アルツハイマー病、パーキンソン病など)、腫瘍(例えば、卵巣癌、乳癌など)、骨髄性白血病等が挙げられる。また、前記Cdc25Aホスファターゼの過剰発現又は活性化に起因する疾患としては、例えば、腫瘍などが挙げられる。
【0016】
さらに、本発明に係る細胞増殖阻害剤は、下記の一般式(V)で表されるナフトキノン誘導体化合物又はその薬理学的に許容される塩を有効成分として含有する。
【化10】
【0017】
式(V)中、R12がアリールアルコール基又はアルキルカルボニル基である場合には、R10は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R11はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R13はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、又は2−ジメタン−1,3−ジオキサン−4−アルキル基であり、式(V)中、R13がアルキニルオキシアルキル基又はブチルジメチルシリルオキシアルケニル基である場合には、R10は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R11はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R12は水素原子、ヒドロキシアルキル基、ハロゲン原子、又はアリールアルコール基であり、式(V)中、R10及びR11がメトキシ基であり、R12がヒドロキシブタ−2−エニル基である場合には、R13はヒドロキシエチル基、ヒドロキシプロピル基、又はヒドロキシヘキシル基であり、式(V)中、R10及びR11がメトキシ基であり、R12が水素原子である場合には、R13はへキシルオキシメチル基などである。なお、本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩は、R12とR13とで環状を構成しないことが好ましい。前記ナフトキノン誘導体化合物としては、上述の化合物(j)又は化合物(m)〜(p)のいずれかであることが好ましい。
【0018】
本発明に係る細胞周期進行阻害剤は、細胞周期のG1期の進行を阻害するものであって、下記の一般式(VI)で表される化合物又はその薬理学的に許容される塩を有効成分として含有する。
【化11】
【0019】
式(VI)中、R14はヒドロキシアルキル基、アルキルカルボニル基、又はアリールアルコール基であり、R15はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、2−ジメタン−1,3−ジオキサン−4−アルキル基、アルキニルオキシアルキル基、ブチルジメチルシリルオキシアルケニル基などである。なお、本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩は、R14とR15とで環状を構成しないことが好ましい。前記化合物としては、上述の化合物(j)や化合物(q)を用いることが好ましい。
【0020】
本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩は、下記の一般式(VII)で表されるもの又は化合物(r)であることを特徴とする。
【化12】
【0021】
式(VII)中、R16はヒドロキシル基又はアルコキシル基であり、R17は水素原子又はハロゲン原子であり、R18はヒドロキシル基又はアルコキシル基である。前記一般式(VII)で表されるナフトキノン誘導体化合物としては、例えば、上述の化合物(S)、化合物(t)、化合物(u)などである。
【0022】
本発明に係る抗菌剤は、下記の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩を有効成分として含有する。
【化13】
【0023】
式(VIII)中、R19及びR20はアルコキシル基である。前記一般式(VIII)で表されるナフトキノン誘導体化合物としては、例えば、化合物(s)などである。本発明に係る抗菌剤は、さらにイミペネムを有効成分として含有してもよい。また、本発明に係る抗菌剤は、化合物(u)又はその薬理学的に許容される塩、及びイミペネムを有効成分として含有する。なお、本発明に係る抗菌剤は、例えば、MRSA(メチシリン耐性黄色ブドウ球菌)などのグラム陽性菌に対しても、グラム陰性菌に対しても抗菌作用を有する。
【0024】
本発明に係るイミペネムの活性を増強するイミペネム活性増強剤は、上述の一般式(VII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩を有効成分として含有する。なお、前記一般式(VII)で表されるナフトキノン誘導体化合物としては、例えば、化合物(s)、化合物(u)等である。前記イミペネムの活性は、例えば、細菌に対する抗菌活性などである。前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0025】
また、本発明に係る細菌増殖阻害剤は、上述の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩を有効成分として含有する。前記一般式(VIII)で表されるナフトキノン誘導体化合物は例えば、化合物(s)などである。本発明に係る細菌増殖阻害剤は、さらに、イミペネムを有効成分として含有してもよい。本発明に係る細菌増殖阻害剤は、化合物(u)又はその薬理学的に許容される塩、及びイミペネムを有効成分として含有してもよい。なお、前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0026】
さらに、本発明に係る医薬組成物は、細菌の感染又は増殖に起因する疾患を改善する医薬組成物であって、上述の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩を有効成分として含有する。前記一般式(VIII)で表されるナフトキノン誘導体化合物は例えば、化合物(s)などである。本発明に係る医薬組成物は、さらに、イミペネムを有効成分として含有してもよい。本発明に係る医薬組成物は、化合物(u)又はその薬理学的に許容される塩、及びイミペネムを有効成分として含有してもよい。前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0027】
なお、前記MRSAの感染または増殖に起因する疾患(以下、「MRSA感染症」と称する場合がある。)には、例えば、脳炎、肺炎、敗血症、腹膜炎、腸炎、骨髄炎、胆管炎、皮膚化膿症、褥瘡感染、MRSAが産生する毒素(例えば、エンテロトキシン、TSST-1など)による食中毒、トキシックショック症候群等が含まれる。また、前記グラム陽性菌またはグラム陰性菌の感染または増殖に起因する疾患(以下、「グラム陽性菌またはグラム陰性菌の感染症」と称する場合がある。)には、例えば、中毒症、歯周病、炎症性疾患、血管炎、IV型アレルギ-性疾患、ブドウ球菌性熱傷様皮膚症候群、リッタ-病、新生児における水疱性インペチゴ、腫瘍、肺炎、関節炎、髄膜炎、各種化膿性疾患、腸炎、髄膜炎、菌血症、眼感染、食中毒、呼吸器感染症、中耳炎、副鼻腔炎、咽頭炎、猩紅熱、急性糸状体腎炎、リウマチ熱、膿痂疹、激症型感染、齲歯、尿路感染症、創感染、胆道感染症、細菌感染によるアトピ-性疾患(アトピ-性皮膚炎など)の重症化などが含まれる。
【0028】
本発明に係るナフトキノン誘導体化合物活性増強剤は、上述の一般式(VII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩の活性を増強する薬剤であって、イミペネムを有効成分として含有する。前記一般式(VII)で表されるナフトキノン誘導体化合物は例えば、化合物(s)、化合物(u)等である。前記活性は、例えば、細菌に対する抗菌活性などである。前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0029】
また、本発明に係るイミペネムの活性を増強するイミペネム活性増強方法は、 前記イミペネムと協同させて、上述の一般式(VII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩を作用させる工程を含む。前記一般式(VII)で表されるナフトキノン誘導体化合物は例えば、化合物(s)、化合物(u)等である。前記イミペネムの活性は、例えば、細菌に対する抗菌活性などである。前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0030】
さらに、本発明に係るナフトキノン誘導体化合物活性増強方法は、上述の一般式(VII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩の活性を増強する方法であって、前記ナフトキノン誘導体化合物と協同させてイミペネムを作用させる工程を含む。前記一般式(VII)で表されるナフトキノン誘導体化合物は例えば、化合物(s)、化合物(u)等である。前記活性は、例えば、細菌に対する抗菌活性などである。前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0031】
本発明に係る細菌の増殖阻害方法は、上述の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩を作用させる工程を含む。前記一般式(VIII)で表されるナフトキノン誘導体化合物は例えば、化合物(s)などである。本発明に係る細菌の増殖阻害方法は、さらに、イミペネムを協同して作用させることとしてもよい。本発明に係る細菌の増殖阻害方法は、化合物(u)又はその薬理学的に許容される塩、及びイミペネムを協同して作用させることとしてもよい。なお、前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0032】
本発明に係る細菌の感染または増殖に起因する疾患を改善する方法は、上述の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは化合物(r)、化合物(v)、若しくは化合物(w)、又はその薬理学的に許容される塩を作用させるように、ヒト又はヒト以外の脊椎動物(例えば、マウス、ラットなど)に投与する工程を含む。前記一般式(VIII)で表されるナフトキノン誘導体化合物は例えば、化合物(s)などである。本発明に係る細菌の感染または増殖に起因する疾患を改善する方法は、さらに、イミペネムを協同して作用させることとしてもよい。本発明に係る細菌の感染または増殖に起因する疾患を改善する方法は、化合物(u)又はその薬理学的に許容される塩、及びイミペネムを協同して作用させることとしてもよい。前記細菌としては、MRSAなどのグラム陽性菌でも、グラム陰性菌でもよい。
【0033】
なお、前記MRSAの感染または増殖に起因する疾患には、例えば、脳炎、肺炎、敗血症、腹膜炎、腸炎、骨髄炎、胆管炎、皮膚化膿症、褥瘡感染、MRSAが産生する毒素(例えば、エンテロトキシン、TSST-1など)による食中毒、トキシックショック症候群等が含まれる。また、前記グラム陽性菌またはグラム陰性菌の感染または増殖に起因する疾患には、例えば、中毒症、歯周病、炎症性疾患、血管炎、IV型アレルギ-性疾患、ブドウ球菌性熱傷様皮膚症候群、リッタ-病、新生児における水疱性インペチゴ、腫瘍、肺炎、関節炎、髄膜炎、各種化膿性疾患、腸炎、髄膜炎、菌血症、眼感染、食中毒、呼吸器感染症、中耳炎、副鼻腔炎、咽頭炎、猩紅熱、急性糸状体腎炎、リウマチ熱、膿痂疹、激症型感染、齲歯、尿路感染症、創感染、胆道感染症、細菌感染によるアトピ-性疾患(アトピ-性皮膚炎など)の重症化などが含まれる。
【発明の効果】
【0034】
本発明によれば、チロシンホスファターゼの阻害剤として有用な新規化合物、並びに、それを有効成分として含有する、チロシンホスファターゼ阻害剤、医薬組成物、細胞増殖阻害剤、細胞周期進行阻害剤、抗菌剤、イミペネム活性増強剤、及び細菌増殖阻害剤、並びに、上述の化合物を用いたイミペネム活性増強方法、及び細菌の増殖阻害方法、並びに、上述の化合物の活性増強剤及び活性増強方法を提供することができる。
【発明を実施するための最良の形態】
【0035】
以下、上記知見に基づき完成した本発明の実施の形態を、実施例を挙げながら詳細に説明する。実施の形態及び実施例に特に説明がない場合には、J. Sambrook, E. F. Fritsch & T. Maniatis (Ed.), Molecular cloning, a laboratory manual (3rd edition), Cold Spring Harbor Press, Cold Spring Harbor, New York (2001); F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J.G. Seidman, J. A. Smith, K. Struhl (Ed.), Current Protocols in Molecular Biology, John Wiley & Sons Ltd.などの標準的なプロトコール集に記載の方法、あるいはそれを修飾したり、改変した方法を用いる。また、市販の試薬キットや測定装置を用いている場合には、特に説明が無い場合、それらに添付のプロトコールを用いる。
【0036】
なお、本発明の目的、特徴、利点、及びそのアイデアは、本明細書の記載により、当業者には明らかであり、本明細書の記載から、当業者であれば、容易に本発明を再現できる。以下に記載された発明の実施の形態及び具体的な実施例などは、本発明の好ましい実施態様を示すものであり、例示又は説明のために示されているのであって、本発明をそれらに限定するものではない。本明細書で開示されている本発明の意図並びに範囲内で、本明細書の記載に基づき、様々な改変並びに修飾ができることは、当業者にとって明らかである。
【0037】
===本発明に係るナフトキノン誘導体化合物の薬理作用===
上述のように、チロシンホスファターゼに対する阻害剤は、2型糖尿病、肥満症、腫瘍、神経疾患((アルツハイマー病、パーキンソン病)、骨髄性白血病等のチロシンホスファターゼの過剰発現又は活性化に起因する疾患に有用であるとされている。そこで、本発明者らは、上述の化合物(f)〜(p)がチロシンホスファターゼの酵素活性を阻害するかどうかを調べた。その結果、化合物(f)〜(p)は、Cdc25Aホスファターゼやプロテインチロシンホスファターゼ1B等のチロシンホスファターゼに対して優れた阻害活性(IC50=10μg/ml以下)を有することが明らかになった。
【0038】
このことから、上述の化合物(f)〜(p)と類似の構造を有する上述の一般式(I)で表されるナフトキノン誘導体化合物、特に上述の一般式(II)〜(IV)で表されるナフトキノン誘導体化合物は、Cdc25Aホスファターゼ、プロテインチロシンホスファターゼ1B等のチロシンホスファターゼ阻害活性を有していると考えられる。また、上述の一般式(I)で表されるナフトキノン誘導体化合物の薬理学的に許容される塩も同様にCdc25Aホスファターゼ、プロテインチロシンホスファターゼ1B等のチロシンホスファターゼ阻害活性を有していると考えられる。従って、上述の一般式(I)で表されるナフトキノン誘導体化合物又はその薬理学的に許容される塩は、Cdc25Aホスファターゼ、プロテインチロシンホスファターゼ1B等のチロシンホスファターゼの阻害剤として有用であり、チロシンホスファターゼの過剰発現又は活性化に起因する疾患の予防又は改善に有用であると考えられる。
【0039】
また、前記チロシンホスファターゼの過剰発現又は活性化に起因する疾患としては、例えば、2型糖尿病、肥満症、腫瘍、神経疾患(例えば、アルツハイマー病、パーキンソン病)、骨髄性白血病等のプロテインチロシンホスファターゼ(PTP)1Bの過剰発現又は活性化に起因する疾患、腫瘍等のCdc25Aホスファターゼの過剰発現又は活性化に起因する疾患などを挙げることができる。
【0040】
ところで、Cdc25AはCdk2/CyclinEやCdk2/CyclinAに作用し、細胞周期の調節においてG1/Sの移行に機能することが知られている。そこで、本発明者らは、上述の化合物(f)〜(q)で表される化合物を用いて細胞周期の進行に与える影響を調べた。その結果、上述の化合物(j)及び(q)が、細胞周期のG1期の進行に対して優れた阻害作用を有することが明らかになった。このことから、細胞周期のG1期の進行を阻害するためには、上述の一般式(I)で表されるナフトキノン誘導体化合物において、少なくともR1がメトキシ基であり、R2がヒドロキシル基であり、R3がヒドロキシル又はオキソを有する基であることが必要であると考えられる。従って、上述の化合物(j)及び(q)と類似の構造を有する上述の一般式(VI)で表されるナフトキノン誘導体化合物や、その薬理学的に許容される塩は、細胞周期のG1期の進行を阻害する細胞周期進行阻害剤として有用であると考えられる。
【0041】
また、本発明者らは、上述の化合物が細胞増殖を抑制するかどうかを調べるため、化合物(j)〜(q)を用いて細胞増殖抑制活性を調べた。その結果、化合物(j)及び化合物(m)〜(p)が細胞増殖抑制活性を有することが明らかになった。従って、上述の化合物(j)及び化合物(m)〜(p)と類似の構造を有する上述の一般式(V)で表されるナフトキノン誘導体化合物や、その薬理学的に許容される塩は、細胞増殖抑制剤として有用であると考えられる。
【0042】
さらに、本発明者らの鋭意努力の結果、化合物(r)、(s)、(v)、及び(w)が単独で抗MRSA(メチシリン耐性黄色ブドウ球菌)作用を有すること、化合物(r)、(s)、(u)、(v)、及び(w)をイミペネムと協同してMRSAに作用させることにより、抗MRSA活性が増強することが明らかになった。従って、上述の一般式(VIII)で表されるナフトキノン誘導体化合物、化合物(r)、(v)、及び(w)、並びにそれらの薬理学的に許容される塩、並びに、化合物(u)とイミペネムとの混合物は、MRSAなどのグラム陽性菌、グラム陰性菌等の細菌感染症の改善(予防、抑制、及び治療を含む。)に有用であり、特に、MRSA感染症、例えば、脳炎、肺炎、敗血症、腹膜炎、腸炎、骨髄炎、胆管炎、皮膚化膿症、褥瘡感染、MRSAが産生する毒素(例えば、エンテロトキシン、TSST-1など)による食中毒、トキシックショック症候群等のMRSA感染症の改善(予防、抑制、及び治療を含む。)に有用である。また、上述の一般式(VIII)で表されるナフトキノン誘導体化合物、化合物(r)、(v)、及び(w)、並びにそれらの薬理学的に許容される塩、並びに、化合物(u)とイミペネムとの混合物は、同様に、グラム陽性菌またはグラム陰性菌の感染症、例えば、中毒症、歯周病、炎症性疾患、血管炎、IV型アレルギ-性疾患、ブドウ球菌性熱傷様皮膚症候群、リッタ-病、新生児における水疱性インペチゴ、腫瘍、肺炎、関節炎、髄膜炎、各種化膿性疾患、腸炎、髄膜炎、菌血症、眼感染、食中毒、呼吸器感染症、中耳炎、副鼻腔炎、咽頭炎、猩紅熱、急性糸状体腎炎、リウマチ熱、膿痂疹、激症型感染、齲歯、尿路感染症、創感染、胆道感染症、細菌感染によるアトピー性疾患(アトピー性皮膚炎など)の重症化等にも有用であると考えられる。
【0043】
さらに、化合物(r)、(s)、(v)、及び(w)をイミペネムとともに用いることにより抗MRSA作用が増強するので、これらの化合物とイミペネムとの使用はMRSA感染症の改善に最も効果的であるといえる。従って、上述の一般式(VII)で表されるナフトキノン誘導体化合物、化合物(r)、(v)、及び(w)、並びにそれらの薬理学的に許容される塩は、イミペネム活性増強のための薬剤または医薬組成物として使用できる。また、両者を含有する薬剤または医薬組成物は、抗菌剤または抗生物質として利用でき、特にMRSAに対して有効である。この際、協同して作用させるように、添加または投与されるのであれば、薬剤又は医薬組成物の剤形は、両者が別々に剤形化されていても、両者が単剤になっていてもよい。ここで、「協同して作用させる」とは、両者を添加又は投与することにより両者の相乗効果(例えば、MRSA増殖阻害活性)を生じさせることである。従って、時間的には両者は同時に添加又は投与されても、前後して添加又は投与されてもよい。
【0044】
===本発明に係るナフトキノン誘導体化合物の製造方法===
本発明に係るナフトキノン誘導体化合物は、以下の反応工程式に準じて反応を行ったり、以下の反応工程式の各工程や常法を適宜組み合わせたり、使用する物質を適宜変更したりすることにより、製造することができる。なお、本発明に係るナフトキノン誘導体化合物の薬理学的に許容される塩、すなわち、アルカリ金属塩(ナトリウム塩、カリウム塩など)、アルカリ土類金属塩(マグネシウム塩、カルシウム塩など)、その他の金属塩(アルミニウム塩など)、無機塩(塩酸塩、アンモニウム塩、アミン類など)、有機塩(グルコサミン塩など)等の公知の塩を含むナフトキノン誘導体化合物は、常法に従って製造することができる。
【0045】
まず、上述の一般式(III)で表されるナフトキノン誘導体化合物(式中、R8は水素原子又はアシル基である。)の製造方法の一例を反応工程式1に基づいて説明する。なお、反応工程式1中のR21はアシル基である。
【化14】
【0046】
<工程1a:化合物(2)の製造>
化合物(1)をジメチルホルムアミドに溶解し、水素化ナトリウムおよびヨウ化メチルを加える。撹拌後、氷冷下飽和塩化アンモニウム水溶液を加え、反応液を酢酸エチルで希釈した。これを飽和食塩水にて洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより化合物(2)を得ることができる。
【0047】
<工程2a:化合物(3)の製造>
化合物(2)をジクロロメタンに溶解し、水、t-ブチルアルコール、DDQ(2,3-ジクロロ-5,6-ジシアノ-1,4-ベンゾキノン)を順次加える。撹拌後、反応液をクロロホルムで希釈し、飽和炭酸水素ナトリウム水溶液を加える。有機層を飽和食塩水にて洗浄し、さらに水層をクロロホルムで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより化合物(3)を得ることができる。
【0048】
<工程3a:化合物(k)の製造>
化合物(3)をメタノールに溶解し、水酸化ナトリウム水溶液を加える。撹拌後、反応液を酢酸エチルで希釈し、塩酸で洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより化合物(k)を得ることができる。
【0049】
<工程4a:一般式(IX)で表される化合物の製造>
化合物(k)にカルボン酸無水物(alkanoic anhydride)、ピリジンを加えて撹拌する。反応液を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(IX)で表される化合物を得ることができる。
【0050】
次に、上述の一般式(IV)で表されるナフトキノン誘導体化合物の製造方法の一例を反応工程式2に基づいて説明する。なお、反応工程式2中のR9はプロピル基、ブチル基、へキシル基、又はアルキニルオキシアルキル基である。
【化15】
【0051】
<工程5a:化合物(4)の製造>
化合物(1)をジメチルホルムアミドに溶解し、炭酸カリウムおよびヨウ化メチルを加える。撹拌後、反応液を酢酸エチルで希釈する。有機層を飽和食塩水で洗浄し、さらに水層を酢酸エチルで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧濃縮して得られる粗生成物をクロマトグラフィーにて精製することにより化合物(4)を得ることができる。
【0052】
<工程6a:一般式(X)で表される化合物の製造>
化合物(4)をジメチルホルムアミドに溶解し、テトラn-ブチルアンモニウムヨージド(必要に応じて添加する。)、水素化ナトリウム、R9X(Xはハロゲン原子である。)を順次加える。撹拌後、反応液を酢酸エチルで希釈し、飽和塩化アンモニウム水溶液で洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(X)で表される化合物を得ることができる。
【0053】
<工程7a:一般式(IV)で表される化合物の製造>
一般式(X)で表される化合物を溶媒(例えば、ジクロロメタン、1,4-ジオキサンなど)に溶解し、水、t-ブチルアルコール、DDQを順次加える。撹拌後、反応液をクロロホルムで希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加える。有機層を飽和食塩水にて洗浄し、さらに水層をクロロホルムで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(IV)で表される化合物を得ることができる。
【0054】
また、上述の一般式(II)で表されるナフトキノン誘導体化合物の例として一般式(XVIII)で表される化合物を挙げ、その製造方法を反応工程式3に基づいて説明する。なお、反応工程式3中のR22はアルキル基であり、R24はヒドロキシエチル基、ヒドロキシプロピル基、ヒドロキシヘキシル基、又はアリールアルコール基である。
【化16】
【0055】
<工程8a:化合物(5)の製造>
化合物(1)を無水テトラヒドロフランに溶解し、臭化水素酸ペルブロミドピリジン錯体(Pyr・HBr3)を加える。撹拌後、飽和チオ硫酸ナトリウム水溶液を加え、反応液を酢酸エチルで希釈する。これを飽和食塩水にて洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより化合物(5)を得ることができる。
【0056】
<工程9a:化合物(6)の製造>
化合物(5)をジクロロメタンに溶解し、ジメチルスルホキシド、トリエチルアミン、三酸化硫黄ピリジン錯体(SO3・Pyr)を順次加える。そして、反応液をクロロホルムで希釈し、飽和塩化アンモニウム水溶液を加える。有機層を飽和食塩水にて洗浄し、さらに水層をクロロホルムで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧濃縮して得られる粗生成物をクロマトグラフィーにて精製することにより化合物(6)を得ることができる。
【0057】
<工程10a:一般式(XI)で表される化合物の製造>
化合物(6)をジメチルホルムアミドに溶解し、炭酸カリウムおよびヨウ化アルキルを加える。撹拌した後、反応液を酢酸エチルで希釈する。有機層を飽和食塩水で洗浄し、さらに水層を酢酸エチルで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧濃縮して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XI)で表される化合物を得ることができる。
【0058】
<工程11a:一般式(XII)で表される化合物の製造>
水素化ナトリウムにジメチルスルホキシドを加えて撹拌した後、反応液を無水テトラヒドロフランで希釈する。その後、ジメチルスルホキシドに溶解させたヨウ化トリメチルスルホニウムを加え、その後さらに、ジメチルスルホキシドに溶解させた一般式(XI)で表される化合物を加えて撹拌する。反応液を酢酸エチルで希釈した後、飽和塩化アンモニウム水溶液にて洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XII)で表される化合物を得ることができる。
【0059】
<工程12a:一般式(XIII)で表される化合物の製造>
ベンゼン中において、臭化亜鉛を加熱還流し、この混合物にベンゼンに溶解させた一般式(XII)で表される化合物を加える。反応液を酢酸エチルで希釈した後、水、飽和食塩水にて洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去してアルデヒドの粗生成物を得る。
【0060】
ジエチルホスホノ酢酸エチルを無水テトラヒドロフランに溶解し、水素化ナトリウムを加えて撹拌する。その後、無水テトラヒドロフランに溶解させたアルデヒドを加えて撹拌する。反応液に飽和塩化アンモニウム水溶液を加え、酢酸エチルで希釈する。有機層を飽和食塩水で洗浄し、さらに水層を酢酸エチルで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧濃縮して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XIII)で表される化合物を得ることができる。
【0061】
<工程13a:一般式(XIV)で表される化合物の製造>
一般式(XIII)で表される化合物を無水テトラヒドロフランに溶解し、水素化ジイソブチルアルミニウムを滴下する。滴下終了後撹拌し、塩酸を加える。反応液を酢酸エチルで希釈した後、飽和炭酸水素ナトリウム水溶液、飽和食塩水にて洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XIV)で表される化合物を得ることができる。
【0062】
<工程14a:一般式(XV)で表される化合物の製造>
一般式(XIV)で表される化合物をジメチルホルムアミドに溶解し、イミダゾール、塩化t-ブチルジメチルシリルを順次加え、撹拌する。反応液を酢酸エチルで希釈した後、飽和食塩水で洗浄し、有機層を無水硫酸ナトリウムで脱水する。溶媒を減圧濃縮して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XV)で表される化合物を得ることができる。
【0063】
<工程15a:一般式(XVI)で表される化合物の製造>
一般式(XV)で表される化合物を無水テトラヒドロフランに溶解し、n-ブチルリチウム(ヘキサン溶液)を滴下する。さらにR23CHO(R23はメチル基、エチル基、ペンチル基、又はアリール基である。)を加えて撹拌する。反応液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで希釈した後、飽和食塩水で洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮することにより一般式(XVI)で表される化合物の粗生成物を得ることができる。
【0064】
<工程16a:一般式(XVII)で表される化合物の製造>
一般式(XVI)で表される化合物の粗生成物をジクロロメタンに溶解し、水、t-ブチルアルコール、DDQを順次加える。撹拌後、反応液をクロロホルムで希釈し、飽和炭酸水素ナトリウム水溶液を加える。有機層を飽和食塩水にて洗浄し、さらに水層をクロロホルムで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XVII)で表される化合物を得ることができる。
【0065】
<工程17a:一般式(XVIII)で表される化合物の製造>
一般式(XVII)で表される化合物を無水テトラヒドロフランに溶解し、酢酸に溶解させたフッ化テトラn-ブチルアンモニウム(テトラヒドロフラン溶液)を加える。撹拌後、反応液を酢酸エチルで希釈する。次に、水、飽和食塩水で洗浄し、有機層を無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XVIII)で表される化合物を得ることができる。なお、上述の反応工程式1〜3において用いる化合物(1)は、例えば、文献(Bull. Chem. Soc. Jpn. 2004, 77, 1925-1930)に記載の方法に準じて製造することができる。
【0066】
次に、上述の一般式(II)で表されるナフトキノン誘導体化合物の例として一般式(XXIV)で表される化合物を挙げ、その製造方法を反応工程式4に基づいて説明する。なお、反応工程式4中のR22はアルキル基であり、R25はヒドロキシアルキル基又はアリールアルコール基であり、R26はアルキルカルボニル基である。
【化17】
【0067】
<工程18a:一般式(XIX)で表される化合物の製造>
上述の一般式(XV)で表される化合物を無水テトラヒドロフランに溶解し、n−ブチルリチウム(ヘキサン溶液)を滴下する。さらにアルキルアルデヒド又はアリールアルデヒドを加えて撹拌する。反応液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで希釈した後飽和食塩水で洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコールの粗生成物を得る。
【0068】
二級アルコールを無水テトラヒドロフランに溶解し、フッ化テトラn−ブチルアンモニウムを加える。撹拌後、反応液を酢酸エチルで希釈する。水、飽和食塩水で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XIX)で表される化合物を得ることができる。
【0069】
<工程19a:一般式(XX)で表される化合物の製造>
一般式(XIX)で表される化合物をジクロロメタンに溶解し、ピリジンおよび無水酢酸を加える。撹拌後、溶媒をトルエン共沸により減圧留去して得られる残留物をクロマトグラフィーにて精製することにより一般式(XX)で表される化合物を得ることができる。
【0070】
<工程20a:一般式(XXI)で表される化合物の製造>
一般式(XX)で表される化合物をジクロロメタンに溶解し、水、t-ブチルアルコール、DDQを順次加える。撹拌後、反応液をクロロホルムで希釈し、飽和炭酸水素ナトリウム水溶液を加える。有機層を飽和食塩水にて洗浄し、さらに水層をクロロホルムで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXII)で表される化合物を得ることができる。
【0071】
<工程21a:一般式(XXI)で表される化合物の製造>
三臭化ホウ素をジクロロメタンに溶解し、ジクロロメタンに溶解させた一般式(XXI)で表される化合物を加えて撹拌する。反応液をクロロホルムで希釈した後、水、飽和食塩水で洗浄し、有機層を無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXII)で表される化合物を得ることができる。
【0072】
<工程22a:一般式(XXIII)で表される化合物の製造>
一般式(XXII)で表される化合物を無水テトラヒドロフランに溶解し、ジメチルスルホキシド、IBX(o-Iodoxylbenzoic Acid)を加える。撹拌後、反応液をヘキサンで希釈してセライト濾過し、ヘキサン:酢酸エチル=3:1の混合溶媒で洗浄して減圧濃縮する。得られた粗生成物をクロマトグラフィーにて精製することにより一般式(XXIII)で表される化合物を得ることができる。
【0073】
<工程23a:一般式(XXIV)で表される化合物の製造>
一般式(XXIII)で表される化合物をメタノールに溶解し、炭酸カリウムを加える。撹拌後、反応液を酢酸エチルで希釈する。飽和塩化アンモニウム水溶液、飽和食塩水で順次洗浄した後、有機層を無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXIV)で表される化合物を得ることができる。
【0074】
次に、上述の一般式(VII)で表されるナフトキノン誘導体化合物の例として化合物(u)で表される化合物を挙げ、その製造方法を反応工程式5に基づいて説明する。なお、反応工程式5中のR22はアルキル基である。
【化18】
【0075】
<工程24a:一般式(XXV)で表される化合物の製造>
ベンゼン中、臭化亜鉛を加熱還流し、この混合物に、ベンゼンに溶解させた一般式(XII)で表される化合物を加える。この反応液を酢酸エチルで希釈した後、水、飽和食塩水で洗浄し、有機層を無水硫酸ナトリウムで脱水する。溶媒を減圧留去してアルデヒドの粗生成物を得た。
【0076】
得られたアルデヒドを無水テトラヒドロフランに溶解し、臭化メチルマグネシウム(テトラヒドロフラン溶液に溶解)を滴下する。撹拌後、反応液を酢酸エチルで希釈し、飽和炭酸水素ナトリウム水溶液を加える。有機層を飽和食塩水にて洗浄し、さらに水層を酢酸エチルで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水後、溶媒を減圧留去して二級アルコールの粗生成物を得る。
【0077】
得られた二級アルコールを無水テトラヒドロフランに溶解し、ジメチルスルホキシド、IBXを順次加えて撹拌し、反応液をヘキサンで希釈する。沈殿物をセライト濾過し、ヘキサン:酢酸エチル=3:1の混合溶媒で洗浄したろ液を減圧濃縮して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXV)で表される化合物を得ることができる。
【0078】
<工程25a:一般式(XXVI)で表される化合物の製造>
ベンゼン中、一般式(XXV)で表される化合物、エチレングリコール、及び4-トルエンスルホン酸一水和物を加熱還流する。反応液を酢酸エチルで希釈した後、飽和炭酸水素ナトリウム水溶液、飽和食塩水等で洗浄し、有機層を無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXVI)で表される化合物を得ることができる。
【0079】
<工程26a:一般式(XXVII)で表される化合物の製造>
一般式(XXVI)で表される化合物を無水テトラヒドロフランに溶解し、n-ブチルリチウムを滴下する。さらにn-ブチルアルデヒドを加えて撹拌する。反応液に飽和炭酸水素ナトリウム水溶液を加えた後、酢酸エチルで希釈し、飽和食塩水で洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコールの粗生成物を得る。
【0080】
得られた二級アルコールをベンゼンに溶解し、4-トルエンスルホン酸一水和物を加える。撹拌後、酢酸エチルで希釈し、飽和炭酸水素ナトリウム水溶液、飽和食塩水等で洗浄する。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮してピラノナフタレンの粗生成物を得る。
【0081】
得られたピラノナフタレンを1,4-ジオキサンに溶解し、水、DDQを順次加える。撹拌後、反応液をクロロホルムで希釈し、飽和炭酸水素ナトリウム水溶液を加える。有機層を飽和食塩水にて洗浄し、さらに水層をクロロホルムで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXVII)で表される化合物を得ることができる。
【0082】
<工程27a:一般式(XXVIII)で表される化合物の製造>
一般式(XXVII)で表される化合物をメタノールに溶解し、Pd/C(パラジウムカーボン触媒)を加える。水素気流下にて撹拌後、沈殿物をセライト濾過し、メタノールで洗浄する。洗浄したろ液を減圧濃縮して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXVIII)で表される化合物を得ることができる。
【0083】
<工程28a:一般式(XXIX)で表される化合物の製造>
一般式(XXVIII)で表される化合物をジクロロメタンに溶解し、ピリジン、臭化水素酸ペルブロミドピリジン錯体(Pyr・HBr3)を加える。撹拌後、反応液をクロロホルムで希釈し、塩酸を加える。有機層を飽和食塩水にて洗浄し、さらに水層をクロロホルムで洗浄した後、有機層を合し無水硫酸ナトリウムで脱水する。溶媒を減圧留去して得られる粗生成物をクロマトグラフィーにて精製することにより一般式(XXIX)で表される化合物を得ることができる。
【0084】
<工程29a:化合物(u)の製造>
エタノール中、一般式(XXIX)で表される化合物、及び濃塩酸を加熱還流する。反応液をクロロホルムで希釈した後、水で洗浄し、有機層を無水硫酸ナトリウムで脱水する。溶媒を減圧留去することにより化合物(u)を得ることができる。
【0085】
==本発明に係るナフトキノン誘導体化合物等を用いた薬剤・医薬品==
本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩及び/又はイミペネムを有効成分として含有する組成物は、医薬品として、ヒト、ヒト以外の脊椎動物に投与してもよいし、試薬として実験用に用いてもよい。本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩を有効成分として含有する医薬品は、錠剤、カプセル剤、顆粒剤、散剤、シロップ剤などの製剤にして、経口投与してもよいし、注射剤、坐剤などの製剤にして、腹腔内や静脈内への注射により非経口投与してもよい。本発明に係るナフトキノン誘導体化合物又はその薬理学的に許容される塩を有効成分として含有する医薬品の製剤化は、従来使用されている製剤添加物(例えば、賦形剤、結合剤、滑沢剤、崩壊剤、矯味矯臭剤、溶剤、安定剤など)を用いて、常法で行うことができる。
【実施例】
【0086】
以下、実施例を用いてより詳細に説明する。なお、実施例において、核磁気共鳴スペクトル(1H-NMRおよび13C-NMR)はJNM GX-400とEX-270(日本電子製)を用いて測定した。また、赤外吸収スペクトルはModel A-202(日本分光製)を用いて測定した。
【0087】
シリカゲル薄層クロマトグラフィーはkieselgel 60F254シリカゲル(Merck製)を使用し、リンモリブデン硫酸で呈色した。シリカゲルカラムクロマトグラフィーはシリカゲル60N(関東化学製)を用いた。なお、各反応は特に記載のない限り、アルゴン中で反応を行った。
【0088】
[実施例1]
<4-ベンジルオキシ-1,2,5-トリメトキシ-7-メトキシメチルナフタレンの製造>
8-ベンジルオキシ-3-ヒドロキシメチル-5,6-ジメトキシナフトール(図1中の化合物(1);159 mg)をジメチルホルムアミド(3 mL)に溶解し、0℃において水素化ナトリウム(121 mg)およびヨウ化メチル(0.18 mL)を加えた。1.5時間撹拌した後、氷冷下飽和塩化アンモニウム水溶液を加え、反応液を酢酸エチル(20 mL)で希釈した。これを飽和食塩水(5 mL)にて洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1〜1:1)にて精製し、無色針状晶として4-ベンジルオキシ-1,2,5-トリメトキシ-7-メトキシメチルナフタレン(図1中の化合物(2);111 mg,65%)を得た。
1HNMR (270 MHz, CDCl3) δ 3.45 (s, 3H), 3.94 (s, 6H), 3.97 (s, 3H), 4.60 (s, 2H), 5.17 (s, 2H), 6.75 (s, 1H), 6.78 (s, 1H), 7.35 (d, 1H, J= 7.6 Hz), 7.40 (t, 2H, J= 7.6 Hz), 7.59 (d, 2H, J= 7.6 Hz), 7.65 (s, 1H).
【0089】
[実施例2]
<2,5-ジメトキシ-7-メトキシメチルナフタレン-1,4-ジオンの製造>
実施例1により得られた化合物(2)(78.4 mg)をジクロロメタン(2 mL)に溶解し、0℃にて水(1 mL)、t-ブチルアルコール(1 mL)、DDQ(135 mg)を順次加えた。2時間撹拌した後、反応液をクロロホルム(20 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(10 mL)にて洗浄し、さらに水層をクロロホルム(10 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム:酢酸エチル=1:1)にて精製し、2,5-ジメトキシ-7-メトキシメチルナフタレン-1,4-ジオン(図1中の化合物(3);47.1 mg,84%)を得た。
1HNMR (270 MHz, CDCl3) δ 3.46 (s, 3H), 3.86 (s, 3H), 4.01 (s, 3H), 4.54 (s, 2H), 6.07 (s, 1H), 7.34 (s, 1H), 7.69 (s, 1H).
【0090】
[実施例3]
<2-ヒドロキシ-5-メトキシ-7-メトキシメチルナフタレン-1,4-ジオンの製造>
実施例2により得られた化合物(3)(10.8 mg)をメタノール(4 mL)に溶解し、0 ℃にて水酸化ナトリウム水溶液(1 M、0.25 mL)を加えた。室温で4時間撹拌した後、反応液を酢酸エチル(30 mL)で希釈した。1M塩酸(10 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール=5:1)にて精製し、2-ヒドロキシ-5-メトキシ-7-メトキシメチルナフタレン-1,4-ジオン(図1中の化合物(k);6.9 mg,68%)を得た。
1HNMR (270 MHz, CDCl3) δ 3.47 (s, 3H), 4.01 (s, 3H), 4.55 (s, 2H), 6.25 (s, 1H), 7.38 (s, 1H), 7.70 (s. 1H).
【0091】
[実施例4]
<5-メトキシ-7-メトキシメチル-1,4-ジオキソ-2-ナフチルアセテートの製造>
実施例3により得られた化合物(k)(5.4 mg)に室温にて無水酢酸(0.5 mL)、ピリジン(0.1 mL)を加えた。7時間撹拌した後、反応液を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール=10:1)にて精製し、5-メトキシ-7-メトキシメチル-1,4-ジオキソ-2-ナフチルアセテート(図1中の化合物(l);3.0 mg,49%)を得た。
1HNMR (270 MHz, CDCl3) δ 2.37 (s, 3H), 3.46 (s, 3H), 4.02 (s, 3H), 4.56 (s, 2H), 6.64 (s, 1H), 7.37 (s, 1H), 7.66 (s. 1H).
【0092】
[実施例5]
<(5-ベンジルオキシ-4,7,8-トリメトキシ-2-ナフチル)-メタン-1-オ−ルの製造>
8-ベンジルオキシ-3-ヒドロキシメチル-5,6-ジメトキシナフトール(図2中の化合物(1);120 mg)をジメチルホルムアミド(3 mL)に溶解し、0℃において炭酸カリウム(383 mg)およびヨウ化メチル(0.2 mL)を加えた。10時間撹拌した後、反応液を酢酸エチル(25 mL)で希釈した。有機層を飽和食塩水(10 mL)で洗浄し、さらに水層を酢酸エチル(10 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧濃縮して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、(5-ベンジルオキシ-4,7,8-トリメトキシ-2-ナフチル)-メタン-1-オ−ル(図2中の化合物(4);53.4 mg,43%)を得た。
1HNMR (270 MHz, CDCl3) δ 3.90 (s, 3H), 3.91 (s, 3H), 3.93 (s, 3H), 4.77 (s, 2H), 5.13 (s, 2H), 6.70 (s, 1H), 6.72 (s, 1H), 7.34 (d, 1H, J= 7.0 Hz), 7.41 (t, 2H, J= 7.0 Hz), 7.56 (s, 1H), 7.60 (complex, 2H).
【0093】
[実施例6]
<4-ベンジルオキシ-7-ヘキシルオキシメチル-1,2,5-トリメトキシナフタレンの製造>
実施例5により得られた化合物(4)(8.6 mg)をジメチルホルムアミド(1 mL)に溶解し、0℃にてテトラn-ブチルアンモニウムヨージド(10 mg)、水素化ナトリウム(15 mg)、1-ブロモヘキサン(40μL)を順次加えた。室温で13時間撹拌した後、反応液を酢酸エチル(25 mL)で希釈した。飽和塩化アンモニウム水溶液(15 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)にて精製し、4-ベンジルオキシ-7-ヘキシルオキシメチル-1,2,5-トリメトキシナフタレン(図2中の化合物(7);6.6 mg,62%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.88 (t, 3H, J= 6.8 Hz), 1.27-1.42 (complex, 6H), 1.56-1.67 (complex, 2H), 3.50 (t, 2H, J= 6.5 Hz), 3.90 (s, 3H), 3.93 (s, 3H), 3.95 (s, 3H), 4.62 (s, 2H), 5.15 (s, 2H), 6.72 (s, 1H), 6.77 (s, 1H), 7.32-7.44 (complex, 3H), 7.55 (s. 1H), 7.58 (brs, 2H).
【0094】
[実施例7]
<7-ヘキシルオキシメチル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例6により得られた化合物(7)(6.6 mg)をジクロロメタン(1 mL)に溶解し、0℃にて水(0.3 mL)、t-ブチルアルコール(0.3 mL)、DDQ (18.0 mg)を順次加えた。室温で1時間撹拌した後、反応液をクロロホルム(25 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(5 mL)にて洗浄し、さらに水層をクロロホルム(15 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム:酢酸エチル=1:1)にて精製し、7-ヘキシルオキシメチル-2,5-ジメトキシナフタレン-1,4-ジオン(図2中の化合物(m);1.3 mg,33%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.94 (t, 3H, J= 7.0 Hz), 1.26-1.53 (complex, 6H), 1.55-1.68 (complex, 2H), 3.53 (t, 2H, J= 6.5 Hz), 3.86 (s, 3H), 4.01 (s, 3H), 4.58 (s, 2H), 6.07 (s, 1H), 7.71 (s, 1H).
【0095】
[実施例8]
<4-ベンジルオキシ-1,2,5-トリメトキシ-7-(プロパ-2-イニルオキシメチル)ナフタレンの製造>
実施例5により得られた化合物(4)(8.4 mg)をジメチルホルムアミド(1 mL)に溶解し、0℃にてテトラn-ブチルアンモニウムヨージド(10 mg)、水素化ナトリウム(10 mg)、臭化プロパルギル (70μL)を順次加えた。室温で30分間撹拌した後、反応液を酢酸エチル(30 mL)で希釈した。飽和塩化アンモニウム水溶液(15 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)にて精製し、4-ベンジルオキシ-1,2,5-トリメトキシ-7-(プロパ-2-イニルオキシメチル)ナフタレン(図2中の化合物(8);7.6 mg,82%)を得た。
1HNMR (270 MHz, CDCl3) δ 2.49 (t, 1H, J= 2.4 Hz), 3.90 (s, 3H), 3.94 (s, 3H), 3.95 (s, 3H), 4.21 (d, 2H, J= 2.4 Hz), 4.73 (s, 2H), 5.15 (s, 2H), 6.73 (s, 1H), 6.76 (s, 1H), 7.32-7.44 (complex, 3H), 7.56 (d, 2H, J= 7.0 Hz), 7.63 (s, 1H).
【0096】
[実施例9]
<2,5-ジメトキシ-7-(プロパ-2-イニルオキシメチル)ナフタレン-1,4-ジオンの製造>
実施例8により得られた化合物(8)(7.6 mg)をジクロロメタン(1 mL)に溶解し、0℃にて水(0.3 mL)、t-ブチルアルコール(0.3 mL)、DDQ (19.7 mg)を順次加えた。室温で15分間撹拌した後、反応液をクロロホルム(30 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(20 mL)を加えた。有機層を飽和食塩水(10 mL)にて洗浄し、さらに水層をクロロホルム(15 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム:酢酸エチル=1:1)にて精製し、2,5-ジメトキシ-7-(プロパ-2-イニルオキシメチル)ナフタレン-1,4-ジオン(図2中の化合物(n);3.2 mg,58%)を得た。
1HNMR (270 MHz, CDCl3) δ 2.51 (t, 1H, J= 2.4 Hz), 3.86 (s, 3H), 4.02 (s, 3H), 4.27 (d, 2H, J= 2.4 Hz), 4.70 (s, 2H), 6.08 (s, 1H), 7.36 (s, 1H), 7.73 (s, 1H).
【0097】
[実施例10]
<4-ベンジルオキシ-1,2,5-トリメトキシ-7-[(2-メチルプロポキシ)メチル]ナフタレンの製造>
実施例5により得られた化合物(4)(11.1 mg)をジメチルホルムアミド(1 mL)に溶解し、0℃にてテトラn-ブチルアンモニウムヨージド(10 mg)、水素化ナトリウム(10 mg)、臭化イソブチル (70μL)を順次加えた。室温で18時間撹拌した後、反応液を酢酸エチル(25 mL)で希釈した。飽和塩化アンモニウム水溶液(10 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)にて精製し、4-ベンジルオキシ-1,2,5-トリメトキシ-7-[(2-メチルプロポキシ)メチル]ナフタレン(図2中の化合物(9);9.2 mg,71%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.94 (d, 6H, J= 6.5 Hz), 1.94 (m, 1H), 3.26 (d, 2H, J= 6.5 Hz), 3.90 (s, 3H), 3.93 (s, 3H), 3.94 (s, 3H), 4.63 (s, 2H), 5.15 (s, 2H), 6.72 (s, 1H), 6.78 (s, 1H), 7.32-7.43 (complex, 3H), 7.55 (s, 1H), 7.59 (s, 2H).
【0098】
[実施例11]
<2,5-ジメトキシ-7-[(2-メチルプロポキシ)メチル]ナフタレン-1,4-ジオンの製造>
実施例10により得られた化合物(9)(9.2 mg)をジクロロメタン(1 mL)に溶解し、0℃にて水(0.3 mL)、t-ブチルアルコール(0.3 mL)、DDQ (23.0 mg)を順次加えた。室温で35分間撹拌した後、反応液をクロロホルム(20 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(20 mL)を加えた。有機層を飽和食塩水(10 mL)にて洗浄し、さらに水層をクロロホルム(15 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム:酢酸エチル=1:1)にて精製し、2,5-ジメトキシ-7-[(2-メチルプロポキシ)メチル]ナフタレン-1,4-ジオン(図2中の化合物(o);2.4 mg,35%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.96 (d, 6H, J= 4.3 Hz), 1.96 (m, 1H), 3.29 (d, 2H, J= 4.3 Hz), 3.86 (s, 3H), 3.99 (s, 3H), 4.59 (s, 2H), 6.08 (s, 1H), 7.37 (s, 1H), 7.71 (s, 1H).
【0099】
[実施例12]
<4-ベンジルオキシ-1,2,5-トリメトキシ-7-プロポキシメチルナフタレンの製造>
実施例5により得られた化合物(4)(11.3 mg)をジメチルホルムアミド(1 mL)に溶解し、0℃にて水素化ナトリウム(10 mg)、ヨウ化n-プロピル (100μL)を順次加えた。室温で12時間撹拌した後、反応液を酢酸エチル(30 mL)で希釈した。飽和塩化アンモニウム水溶液(15 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)にて精製し、4-ベンジルオキシ-1,2,5-トリメトキシ-7-プロポキシメチルナフタレン(図2中の化合物(10);8.6 mg,68%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.96 (t, 3H, J= 7.6 Hz), 1.58-1.73 (complex, 2H), 3.47 (t, 2H, J= 6.8 Hz), 3.90 (s, 3H), 3.93 (s, 3H), 3.95 (s, 3H), 4.63 (s, 2H), 5.15 (s, 2H), 6.72 (s, 1H), 6.78 (s, 1H), 7.32-7.43 (complex, 3H), 7.55 (s, 1H), 7.59 (d, 2H, J= 4.9 Hz).
【0100】
[実施例13]
<2,5-ジメトキシ-7プロポキシメチルナフタレン-1,4-ジオンの製造>
実施例12により得られた化合物(10)(7.0 mg)を1,4-ジオキサン(1 mL)に溶解し、0℃にて水(0.5 mL)、DDQ (15 mg)を順次加えた。室温で20分間撹拌した後、反応液をクロロホルム(20 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(10 mL)にて洗浄し、さらに水層をクロロホルム(15 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、2,5-ジメトキシ-7プロポキシメチルナフタレン-1,4-ジオン(図2中の化合物(p);4.8 mg,76%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.98 (t, 3H, J= 7.6 Hz), 1.59-1.75 (complex, 2H), 3.50 (t, 2H, J= 6.8 Hz), 3.86 (s, 3H), 4.02 (s, 3H), 4.59 (s, 2H), 6.07 (s, 1H), 7.37 (s, 1H), 7.71 (s, 1H).
【0101】
[実施例14]
<8-ベンジルオキシ-2-ブロモ-3-ヒドロキシメチル-5,6-ジメトキシナフトールの製造>
8-ベンジルオキシ-3-ヒドロキシメチル-5,6-ジメトキシナフトール(図3中の化合物(1);2.42 g)を無水テトラヒドロフラン(50 mL)に溶解し、0℃において臭化水素酸ペルブロミドピリジン錯体(Pyr・HBr3)(2.52 g)を加えた。1時間撹拌した後、飽和チオ硫酸ナトリウム水溶液(30 mL)を加え、反応液を酢酸エチル(100 mL)で希釈した。これを飽和食塩水(20 mL)にて洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1)にて精製し、黄褐色針状晶として8-ベンジルオキシ-2-ブロモ-3-ヒドロキシメチル-5,6-ジメトキシナフトール(図3中の化合物(5);2.60 g,87%)を得た。
1HNMR (270 MHz, CDCl3) δ 2.25 (br, 1H), 3.90 (s, 3H), 3.97 (s, 3H), 4.85 (s, 2H), 5.26 (s, 2H), 6.74 (s, 1H), 7.44-7.47 (complex, 5H), 7.65 (s, 1H), 9.99 (s, 1H).
13CNMR (67.80 MHz, DMSO-d6) δ 56.7, 56.9, 60.4, 63.1, 63.8, 71.4, 97.7, 99.6, 101.4, 109.5, 128.3, 128.5, 128.7, 128.9, 135.5, 136.0, 140.1, 148.2, 149.7, 150.1.
IR (disk) 3504, 3354, 3056, 2972, 2940, 2881, 2360, 1612 cm-1.
m.p. 165℃ dec. (hexane-EtOAc)
【0102】
[実施例15]
<5-ベンジルオキシ-3-ブロモ-4-ヒドロキシ-7,8-ジメトキシナフタレン-2-カルバルデヒドの製造>
実施例14により得られた化合物(5)(1.35 g)をジクロロメタン(10 mL)に溶解し、室温にてジメチルスルホキシド(9 mL)、トリエチルアミン(2.7 mL)、三酸化硫黄ピリジン錯体(SO3・Pyr)(1.51 g)を順次加えた。20分後、反応液をクロロホルム(200 mL)で希釈し、氷冷下、飽和塩化アンモニウム水溶液(50 mL)を加えた。有機層を飽和食塩水(30 mL)にて洗浄し、さらに水層をクロロホルム(100 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧濃縮して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム)にて精製し、黄色針状晶として5-ベンジルオキシ-3-ブロモ-4-ヒドロキシ-7,8-ジメトキシナフタレン-2-カルバルデヒド(図3中の化合物(6);1.24 g,93%)を得た。
1HNMR (270 MHz, CDCl3) δ 3.92 (s, 3H), 3.97 (s, 3H), 5.27 (s, 2H), 6.85 (s, 1H), 7.44-7.47 (complex, 5H), 8.17 (s, 1H), 10.09 (s, 1H), 10.48 (s, 1H).
13CNMR (67.80 MHz, CDCl3) δ 57.1, 61.5, 72.6, 99.6, 99.8, 103.1, 116.2, 116.3, 128.2, 129.1, 129.2, 131.9, 134.0, 134.3, 148.7, 150.6, 151.6, 151.7, 192.3.
IR (disk) 3303, 2950, 1703, 1606 cm-1.
m.p. 174℃ dec. (hexane-EtOAc)
【0103】
[実施例16]
<5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシナフタレン-2-カルバルデヒドの製造>
実施例15により得られた化合物(6)(449 mg)をジメチルホルムアミド(10 mL)に溶解し、0℃にて炭酸カリウム(463 mg)およびヨウ化メチル(0.3 mL)を加えた。室温にて1時間撹拌した後、反応液を酢酸エチル(50 mL)で希釈した。有機層を飽和食塩水(10 mL)で洗浄し、さらに水層を酢酸エチル(30 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧濃縮して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、黄色板状晶として5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシナフタレン-2-カルバルデヒド(図3中の化合物(11);437 mg,94%)を得た。
1HNMR (400 MHz, CDCl3) δ 3.80 (s, 3H), 3.95 (s, 3H), 3.96 (s, 3H), 5.20 (s, 2H), 6.91 (s, 1H), 7.38 (d, 1H, J= 7.2 Hz), 7.43 (t, 2H, J= 7.2 Hz), 7.56 (d, 2H, J= 7.2 Hz), 8.50 (s, 1H), 10.50 (s, 1H).
13CNMR (100.40 MHz, CDCl3) δ 56.9, 61.5, 61.9, 62.0, 62.1, 72.8, 100.5, 103.3, 113.8, 119.6, 121.4, 127.7, 128.1, 128.6, 129.9, 131.5, 136.3, 138.9, 149.0, 151.0, 154.1, 192.2.
IR (disk) 2943, 2848, 1687, 1612, 1577 cm-1.
m.p. 129-130℃ (hexane-EtOAc)
【0104】
[実施例17]
<8-ベンジルオキシ-2-ブロモ-1,5,6-トリメトキシ-3-オキシラン-2-イルナフタレンの製造>
水素化ナトリウム(517 mg)にジメチルスルホキシド(5 mL)を加え、室温で2時間撹拌した後、反応液を無水テトラヒドロフラン(50 mL)で希釈した。その後、0℃にてジメチルスルホキシド(10 mL)に溶解させたヨウ化トリメチルスルホニウム(2.75 g)を加え、さらに40分後、ジメチルスルホキシド(25 mL)に溶解させた化合物(11)(1.89 g)を加えて45分間撹拌した。反応液を酢酸エチル(50 mL)で希釈した後、飽和塩化アンモニウム水溶液(30 mL)にて洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)にて精製し、淡黄色針状晶として8-ベンジルオキシ-2-ブロモ-1,5,6-トリメトキシ-3-オキシラン-2-イルナフタレン(図3中の化合物(12);1.77 g,92%)を得た。
【0105】
1HNMR (400 MHz, CDCl3) δ 2.72 (dd, 1H, J= 2.8, 6.0 Hz), 3.25 (dd, 1H, J= 4.0, 6.0 Hz), 3.78 (s, 3H), 3.89 (s, 3H), 3.95 (s, 3H), 4.26 (dd, 1H, J= 2.8, 4.0 Hz), 5.18 (s, 2H), 6.78 (s, 1H), 7.37 (d, 1H, J= 7.2 Hz), 7.43 (t, 2H, J= 7.2 Hz), 7.56 (d, 2H, J= 7.2 Hz), 7.79 (s, 1H).
13CNMR (100.40 MHz, CDCl3) δ 51.1, 52.9, 56.9, 61.2, 61.9, 72.6, 100.4, 100.5, 113.5, 114.4, 116.7, 127.7, 128.0, 128.5, 130.8, 136.1, 136.6, 137.3, 143.9, 148.6, 151.1, 153.2.
IR (disk) 2931, 2858, 1612, 1593, 1574 cm-1.
m.p. 133-134℃ (hexane-EtOAc)
【0106】
[実施例18]
<エチル(2E)-4-[5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ(2-ナフチル)]ブタ-2-エノアートの製造>
ベンゼン(17 mL)中、臭化亜鉛(708 mg)を40分間加熱還流し、この混合物にベンゼン(10 mL)に溶解させた化合物(12)(468 mg)を加えた。20分後反応液を室温に戻し、酢酸エチル(30 mL)で希釈した後、水、飽和食塩水(各々20 mL)にて洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去してアルデヒドの粗生成物を得た。
【0107】
ジエチルホスホノ酢酸エチル(1.1 mL)を無水テトラヒドロフラン(10 mL)に溶解し、氷冷下、水素化ナトリウム(216 mg)を加え、室温にて1.5時間撹拌した。その後、無水テトラヒドロフラン(10 mL)に溶解させたアルデヒドを−78℃にて加え、25分間撹拌した。反応液に飽和塩化アンモニウム水溶液(20 mL)を加え、酢酸エチル(30 mL)で希釈した。有機層を飽和食塩水(10 mL)で洗浄し、さらに水層を酢酸エチル(20 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧濃縮して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1)にて精製し、エチル(2E)-4-[5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ(2-ナフチル)]ブタ-2-エノアート(図3中の化合物(13);462 mg,85%)を得た。
【0108】
1HNMR (270 MHz, CDCl3) δ 1.27 (t, 3H, J= 7.0 Hz), 3.77 (s, 3H), 3.82 (d, 2H, J= 6.2 Hz), 3.90 (s, 3H), 3.95 (s, 3H), 4.18 (q, 2H, J= 7.0 Hz), 5.18 (s, 2H), 5.81 (d, 1H, J= 16 Hz), 7.18 (dt, 1H, J= 6.2, 16 Hz), 7.36 (d, 1H, J= 7.2 Hz), 7.42 (t, 2H, J= 7.2 Hz), 7.56 (d, 2H, J= 7.2 Hz), 7.74 (s, 1H).
13CNMR (67.80 MHz, CDCl3) δ 14.3, 39.5, 56.8, 60.2, 61.1, 61.8, 72.5, 100.1, 116.1, 116.3, 118.4, 122.7, 127.6, 127.9, 128.4, 130.3, 130.5, 136.4, 136.5, 136.7, 140.1, 145.7, 148.4, 151.0, 153.7, 166.2.
IR (film) 2937, 2846, 1716, 1614, 1591, 1573 cm-1.
【0109】
[実施例19]
<(2E)-4-[5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ(2-ナフチル)]ブタ-2-エン-1-オールの製造>
実施例18により得られた化合物(13)(147 mg)を無水テトラヒドロフラン(3 mL)に溶解し、−78℃にて水素化ジイソブチルアルミニウム(1.01 M、トルエン溶液、1.5 mL)を滴下した。滴下終了後35分間撹拌し、4M 塩酸(10 mL)を加えて0℃に戻した。反応液を酢酸エチル(20 mL)で希釈した後、飽和炭酸水素ナトリウム水溶液(10 mL)、飽和食塩水(5 mL)にて洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、淡黄色板状晶として(2E)-4-[5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ(2-ナフチル)]ブタ-2-エン-1-オール(図3中の化合物(14);125 mg,93%)を得た。
【0110】
1HNMR (270 MHz, CDCl3) δ 1.82 (br, 1H), 3.66 (d, 2H, J= 6.2 Hz), 3.77 (s, 3H), 3.90 (s, 3H), 3.94 (s, 3H), 4.13 (q, 2H, J= 5.4 Hz), 5.17 (s, 2H), 5.75 (dt, 1H, J= 5.4, 16 Hz), 5.95 (dt, 1H, J= 6.2, 16 Hz), 6.74 (s, 1H), 7.37 (d, 1H, J= 7.2 Hz), 7.42 (t, 2H, J= 7.2 Hz), 7.56 (d, 2H, J= 7.2 Hz), 7.73 (s, 1H).
13CNMR (67.80 MHz, CDCl3) δ 39.6, 56.8, 61.1, 61.7, 63.4, 63.5, 72.5, 99.8, 115.8, 116.6, 117.7, 127.6, 127.8, 128.3, 129.5, 129.6, 130.5, 131.0, 131.1, 136.7, 138.5, 148.2, 151.0, 153.3.
IR (disk) 3521, 2902, 2860, 1614, 1589, 1574 cm-1.
m.p. 104-105℃ (hexane-EtOAc)
【0111】
[実施例20]
<1-{(2E)-4-[5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ(2-ナフチル) ]ブタ-2-エニルオキシ}-1,1,2,2-テトラメチル-1-シラプロパンの製造>
実施例19により得られた化合物(14)(125 mg)をジメチルホルムアミド(3 mL)に溶解し、0℃にてイミダゾール(180 mg)、塩化t-ブチルジメチルシリル(215 mg)を順次加え、15分間撹拌した。反応液を酢酸エチル(15 mL)で希釈した後、飽和食塩水(10 mL)で洗浄し、有機層を無水硫酸ナトリウムで脱水した。溶媒を減圧濃縮して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1)にて精製し、1-{(2E)-4-[5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ(2-ナフチル) ]ブタ-2-エニルオキシ}-1,1,2,2-テトラメチル-1-シラプロパン(図3中の化合物(15);149 mg,96%)を得た。
【0112】
1HNMR (400 MHz, CDCl3) δ 0.07 (s, 6H), 0.90 (s, 9H), 3.65 (d, 2H, J= 6.8 Hz), 3.76 (s, 3H), 3.89 (s, 3H), 3.94 (s, 3H), 4.19 (q, 2H, J= 5.2 Hz), 5.18 (s, 2H), 5.67 (dt, 1H, J= 5.2, 15 Hz), 5.93 (dt, 1H, J= 6.8, 15 Hz), 6.74 (s, 1H), 7.36 (d, 1H, J= 7.2 Hz), 7.42 (t, 2H, J= 7.2 Hz), 7.56 (d, 2H, J= 7.2 Hz), 7.73 (s, 1H).
13CNMR (100.40 MHz, CDCl3) δ -5.0, 18.5, 25.7, 26.0, 39.6, 56.9, 61.3, 61.7, 63.7, 72.6, 99.9, 115.9, 116.8, 117.7, 127.6, 127.7, 127.9, 128.5, 130.6, 131.6, 135.3, 136.8, 137.0, 139.0, 148.2, 151.1.
IR (film) 2931, 2894, 2854, 1614, 1591, 1576 cm-1.
【0113】
[実施例21]
<7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシエチル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例20により得られた化合物(15)(65.3 mg)を無水テトラヒドロフラン(2 mL)に溶解し、−78℃にてn-ブチルリチウム(1.59 M、ヘキサン溶液、0.4 mL)を滴下した。さらにアセトアルデヒド(1.0 mL)を加え、15分間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液 (10 mL)を加えて0 ℃に戻し、酢酸エチル(30 mL)で希釈した後、飽和食塩水(5 mL)で洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコール(図4中の化合物(16))の粗生成物を得た。
【0114】
二級アルコールの粗生成物をジクロロメタン(2 mL)に溶解し、0℃にて水(0.2 mL)、t-ブチルアルコール(0.2 mL)、DDQ (40.1 mg)を順次加えた。15分間撹拌した後、反応液をクロロホルム(20 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(5 mL)にて洗浄し、さらに水層をクロロホルム(20 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシエチル-2,5-ジメトキシナフタレン-1,4-ジオン(図4中の化合物(a);10.5 mg,29%)を得た。
【0115】
1HNMR (400 MHz, CDCl3) δ 0.05 (s, 6H), 0.88 (s, 9H), 1.58 (d, 3H, J= 5.2 Hz), 3.54 (complex, 2H), 3.85 (s, 3H), 3.98 (s, 3H), 4.15 (d, 2H, J= 4.0 Hz), 5.18 (m, 1H), 5.55 (m, 1H), 5.77 (m, 1H), 6.08 (s, 1H), 7.78 (s, 1H).
【0116】
[実施例22]
<7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシエチル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例21により得られた化合物(a)(10.5 mg)を無水テトラヒドロフラン(1.5 mL)に溶解し、0℃にて酢酸(13μL)に溶解させたフッ化テトラn-ブチルアンモニウム(1.0 M、テトラヒドロフラン溶液、0.2 mL)を加えた。室温で12時間撹拌した後、反応液を酢酸エチル(25 mL)で希釈した。水(10 mL)、飽和食塩水(5 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシエチル-2,5-ジメトキシナフタレン-1,4-ジオン(図4中の化合物(f);7.4 mg,95%)を得た。
1HNMR (400 MHz, CDCl3) δ 1.59 (d, 3H, J= 7.2 Hz), 3.55 (complex, 2H), 3.88 (s, 3H), 3.97 (s, 3H), 4.12 (d, 2H, J= 4.8 Hz), 5.18 (t, 1H, J= 7.2 Hz), 5.64 (dt, 1H, J= 6.0, 15 Hz), 5.84 (dt, 1H, J= 6.0, 15 Hz), 6.08 (s, 1H), 7.76 (s, 1H).
【0117】
[実施例23]
<7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシプロピル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例20により得られた化合物(15)(93.9 mg)を無水テトラヒドロフラン(5 mL)に溶解し、−78℃にてn-ブチルリチウム(1.59 M、ヘキサン溶液、0.5 mL)を滴下した。さらにプロピオンアルデヒド(0.23 mL)を加え、15分間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液 (15 mL)を加えて0℃に戻し、酢酸エチル(40 mL)で希釈した後、飽和食塩水(5 mL)で洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコールの粗生成物(図5中の化合物(17))を得た。
【0118】
二級アルコールの粗生成物をジクロロメタン(2 mL)に溶解し、0℃にて水(0.8 mL)、t-ブチルアルコール(0.3 mL)、DDQ (120 mg)を順次加えた。40分間撹拌した後、反応液をクロロホルム(25 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(5 mL)にて洗浄し、さらに水層をクロロホルム(20 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシプロピル-2,5-ジメトキシナフタレン-1,4-ジオン(図5中の化合物(b);24.3 mg,33%)を得た。
【0119】
1HNMR (400 MHz, CDCl3) δ 0.05 (s, 6H), 0.88 (s, 9H), 1.04 (t, 3H, J= 8.0 Hz), 1.74 (complex, 2H), 3.53 (d, 2H, J= 6.0 Hz), 3.86 (s, 3H), 3.93 (s, 3H), 4.15 (d, 2H, J= 4.0 Hz), 4.85 (m, 1H), 5.57 (dt, 1H, J= 5.2, 15 Hz), 5.78 (dt, 1H, J= 5.2, 15 Hz), 6.06 (s, 1H), 7.77 (s, 1H).
【0120】
[実施例24]
<7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシプロピル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例23により得られた化合物(b)(24.3 mg)を無水テトラヒドロフラン(3 mL)に溶解し、0℃にて酢酸(33μL)に溶解させたフッ化テトラn-ブチルアンモニウム(1.0 M、テトラヒドロフラン溶液、0.53 mL)を加えた。室温で11時間撹拌した後、反応液を酢酸エチル(40 mL)で希釈した。水(15 mL)、飽和食塩水(5 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシプロピル-2,5-ジメトキシナフタレン-1,4-ジオン(図5中の化合物(g);13.1 mg,72%)を得た。
1HNMR (270 MHz, CDCl3) δ 1.05 (t, 3H, J= 7.2 Hz), 1.75 (complex, 2H), 3.54 (d, 2H, J= 5.4 Hz), 3.87 (s, 3H), 3.94 (s, 3H), 4.11 (d, 2H, J= 4.8 Hz), 4.86 (m, 1H), 5.65 (dt, 1H, J= 5.4, 15 Hz), 5.83 (dt, 1H, J= 5.4, 15 Hz), 6.07 (s, 1H), 7.76 (s, 1H).
【0121】
[実施例25]
<7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシヘキシル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例20により得られた化合物(15)(93.5 mg)を無水テトラヒドロフラン(3 mL)に溶解し、−78℃にてn-ブチルリチウム(1.59 M、ヘキサン溶液、0.5 mL)を滴下した。さらにn-ヘキシルアルデヒド(0.60 mL)を加え、35分間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液 (15 mL)を加えて0℃に戻し、酢酸エチル(40 mL)で希釈した後、飽和食塩水(5 mL)で洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコールの粗生成物(図6中の化合物(18))を得た。
【0122】
二級アルコールの粗生成物をジクロロメタン(3 mL)に溶解し、0℃にて水(0.5 mL)、t-ブチルアルコール(0.5 mL)、DDQ (112 mg)を順次加えた。30分間撹拌した後、反応液をクロロホルム(20 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(5 mL)にて洗浄し、さらに水層をクロロホルム(15 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシヘキシル-2,5-ジメトキシナフタレン-1,4-ジオン(図6中の化合物(c);49.3 mg,62%)を得た。
【0123】
1HNMR (400 MHz, CDCl3) δ 0.05 (s, 6H), 0.88 (s, 9H), 0.91 (complex, 3H), 1.31 (complex, 6H), 1.63 (m, 1H), 1.92 (m, 1H), 3.52 (d, 2H, J= 5.6 Hz), 3.86 (s, 3H), 3.93 (s, 3H), 4.13 (d, 2H, J= 4.4 Hz), 4.94 (m, 1H), 5.56 (dt, 1H, J= 4.8, 15 Hz), 5.78 (dt, 1H, J= 4.8, 15 Hz), 6.06 (s, 1H), 7.76 (s, 1H).
【0124】
[実施例26]
<7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシヘキシル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例25により得られた化合物(c)(49.3 mg)を無水テトラヒドロフラン(3 mL)に溶解し、0℃にて酢酸(50μL)に溶解させたフッ化テトラn-ブチルアンモニウム(1.0 M、テトラヒドロフラン溶液、0.50 mL)を加えた。室温で15時間撹拌した後、反応液を酢酸エチル(45 mL)で希釈した。水(15 mL)、飽和食塩水(10 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシヘキシル-2,5-ジメトキシナフタレン-1,4-ジオン(図6中の化合物(h);29.0 mg,76%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.88 (complex, 3H), 1.31 (complex, 5H), 1.68 (complex, 3H), 1.94 (m, 1H), 3.53 (d, 2H, J= 5.9 Hz), 3.87 (s, 3H), 3.93 (s, 3H), 4.11 (d, 2H, J= 4.3 Hz), 4.94 (m, 1H), 5.63 (dt, 1H, J= 5.4, 15 Hz), 5.82 (dt, 1H, J= 5.4, 15 Hz), 6.07 (s, 1H), 7.75 (s, 1H).
【0125】
[実施例27]
<7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシフェニルメチル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例20により得られた化合物(15)(92.4 mg)を無水テトラヒドロフラン(2 mL)に溶解し、−78℃にてn-ブチルリチウム(1.59 M、ヘキサン溶液、0.6 mL)を滴下した。さらにベンズアルデヒド(0.60 mL)を加え、10分間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液 (10 mL)を加えて0℃に戻し、酢酸エチル(45 mL)で希釈した後、飽和食塩水(5 mL)で洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコールの粗生成物(図7中の化合物(19))を得た。
【0126】
二級アルコールの粗生成物をジクロロメタン(4 mL)に溶解し、0℃にて水(1.0 mL)、t-ブチルアルコール(0.5 mL)、DDQ (94.9 mg)を順次加えた。35分間撹拌した後、反応液をクロロホルム(25 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(10 mL)にて洗浄し、さらに水層をクロロホルム(15 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、7-[(2E)-4-(1,1,2,2-テトラメチル-1-シラプロポキシ)ブタ-2-エニル]-6-ヒドロキシフェニルメチル-2,5-ジメトキシナフタレン-1,4-ジオン(図7中の化合物(d);30.4 mg,25%)を得た。
【0127】
1HNMR (400 MHz, CDCl3) δ 0.05 (s, 6H), 0.93 (s, 9H), 3.29 (s, 3H), 3.65 (d, 2H, J= 6.4 Hz), 3.93 (s, 3H), 3.93 (s, 3H), 4.05 (d, 1H, J= 11 Hz), 4.17 (d, 2H, J= 3.6 Hz), 5.66 (dt, 1H, J= 4.8, 15 Hz), 5.86 (dt, 1H, J= 4.8, 15 Hz), 6.12 (s, 1H), 6.20 (d, 1H, J= 11 Hz), 7.28-7.42 (complex, 5H), 7.95 (s, 1H).
【0128】
[実施例28]
<7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシフェニルメチル-2,5-ジメトキシナフタレン-1,4-ジオンの製造>
実施例27により得られた化合物(d)(30.4 mg)を無水テトラヒドロフラン(2 mL)に溶解し、0℃にて酢酸(50μL)に溶解させたフッ化テトラn-ブチルアンモニウム(1.0 M、テトラヒドロフラン溶液、0.50 mL)を加えた。室温で4時間撹拌した後、反応液を酢酸エチル(40 mL)で希釈した。水(15 mL)、飽和食塩水(10 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:3)にて精製し、7-((2E)-4-ヒドロキシブタ-2-エニル)-6-ヒドロキシフェニルメチル-2,5-ジメトキシナフタレン-1,4-ジオン(図7中の化合物(i);14.9 mg,63%)を得た。
1HNMR (400 MHz, CDCl3) δ 3.22 (s, 3H), 3.59 (d, 2H, J= 6.0 Hz), 3.87 (s, 3H), 4.05 (d, 2H, J= 5.6 Hz), 4.09 (d, 1H, J= 10 Hz), 4.17 (d, 2H, J= 3.6 Hz), 5.63 (dt, 1H, J= 5.6, 15 Hz), 5.80 (dt, 1H, J= 5.6, 15 Hz), 6.06 (s, 1H), 6.13 (d, 1H, J= 10 Hz), 7.22-7.34 (complex, 5H), 7.86 (s, 1H).
【0129】
[実施例29]
<(2E)-4-[5-ベンジルオキシ-3-ヒドロキシペンチル-4,7,8-トリメトキシ (2-ナフチル)]ブタ-2-エン-1-オールの製造>
実施例20により得られた化合物(15)(1.06 g)を無水テトラヒドロフラン(30 mL)に溶解し、−78℃にてn-ブチルリチウム(1.57 M、ヘキサン溶液、3.4 mL)を滴下した。さらにn-バレルアルデヒド(1.0 mL)を加え、10分間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液(15 mL)を加え0℃に戻し、酢酸エチル(30 mL)で希釈した後飽和食塩水(10 mL)で洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコールの粗生成物を得た。
【0130】
二級アルコールを無水テトラヒドロフラン(20 mL)に溶解し、0℃にてフッ化テトラn-ブチルアンモニウム(1.0 M、テトラヒドロフラン溶液、9.0 mL)を加えた。室温で1.5時間撹拌した後、反応液を酢酸エチル(30 mL)で希釈した。水(10 mL)、飽和食塩水(5 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1)にて精製し、(2E)-4-[5-ベンジルオキシ-3-ヒドロキシペンチル-4,7,8-トリメトキシ (2-ナフチル)]ブタ-2-エン-1-オール(図8中の化合物(20);693 mg,80%)を得た。
【0131】
1HNMR (270 MHz, CDCl3) δ 0.92 (t, 3H, J= 6.2 Hz), 1.37-1.39 (complex, 2H), 1.63-1.80 (complex, 2H), 1.92-2.04 (complex, 2H), 3.57 (d, 2H, J= 5.4 Hz), 3.80 (s, 3H), 3.90 (s, 3H), 3.93 (s, 3H), 4.09 (d, 2H, J= 5.6 Hz), 4.99 (m, 1H), 5.15 (d, 2H, J= 12 Hz), 5.64 (dt, 1H, J= 5.6, 15 Hz), 5.91 (dt, 1H, J= 5.4, 15 Hz), 6.73 (s, 1H), 7.36 (d, 1H, J= 7.2 Hz), 7.41 (t, 2H, J= 7.2 Hz), 7.53 (d, 2H, J= 7.2 Hz), 7.65 (s, 1H).
13CNMR (67.80 MHz, CDCl3) δ 14.2, 22.8, 29.2,36.6, 39.0, 50.3, 56.9, 61.1, 63.4, 64.0, 71.1, 72.9, 100.3, 113.7, 115.1, 118.2, 127.4, 127.8, 128.4, 130.7, 131.0, 131.2, 136.8, 136.9, 137.0, 147.9, 151.1, 154.8, 163.6.
IR (film) 3408, 2933, 2858, 1620, 1574 cm-1.
【0132】
[実施例30]
<(2E)-4-[5-ベンジルオキシ-3-ヒドロキシペンチル-4,7,8-トリメトキシ(2-ナフチル) ]ブタ-2-エニルアセテートの製造>
実施例29により得られた化合物(20)(111 mg)をジクロロメタン(5 mL)に溶解し、氷浴中、ピリジン(36μL)および無水酢酸(15μL)を加えた。室温で2日間撹拌した後、溶媒をトルエン共沸により減圧留去して得られる残留物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、(2E)-4-[5-ベンジルオキシ-3-ヒドロキシペンチル-4,7,8-トリメトキシ(2-ナフチル) ]ブタ-2-エニルアセテート(図8中の化合物(21);70.5 mg,58%)を得た。
【0133】
1HNMR (400 MHz, CDCl3) δ 0.93 (t, 3H, J= 6.8 Hz), 1.34-1.44, (complex, 4H), 1.63-1.83 (complex, 2H), 2.04 (s, 3H), 3.62 (d, 2H, J= 6.4 Hz), 3.80 (s, 3H), 3.90 (s, 3H), 3.94 (s, 3H), 4.55 (d, 2H, J= 6.4 Hz), 5.01 (m, 1H), 5.12 (d, 1H, J= 11 Hz), 5.19 (d, 1H, J= 11 Hz), 5.61 (dt, 1H, J= 6.4, 15 Hz), 6.01 (dt, 1H, J= 6.0, 15 Hz), 6.74 (s, 1H), 7.36 (d, 1H, J= 7.2 Hz), 7.42 (t, 2H, J= 7.2 Hz), 7.53 (d, 2H, J= 7.2 Hz), 7.66 (s, 1H)
13CNMR (100.40 MHz, CDCl3) δ 14.2, 21.0, 22.7, 29.2, 36.4, 38.9, 56.9, 61.0, 63.9, 64.8, 70.9, 72.8, 100.2, 115.0, 118.3, 125.4, 127.4, 127.8, 128.2, 128.4, 130.9, 131.2, 134.2, 136.6, 136.8, 137.0, 147.9, 151.1, 154.8, 170.6.
IR (film) 3480, 2933, 2857, 1739, 1620, 1573 cm-1
【0134】
[実施例31]
<(2E)-4-[3-ヒドロキシペンチル-4,7-ジメトキシ-5,8-ジオキソ(2-ナフチル)]ブタ-2-エニルアセテートの製造>
実施例30により得られた化合物(21)(60.4 mg)をジクロロメタン(4 mL)に溶解し、0℃にて水(2 mL)、t-ブチルアルコール(2 mL)、DDQ(83.4 mg)を順次加えた。20分間撹拌した後、反応液をクロロホルム(20 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(10 mL)にて洗浄し、さらに水層をクロロホルム(10 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(酢酸エチル)にて精製し、(2E)-4-[3-ヒドロキシペンチル-4,7-ジメトキシ-5,8-ジオキソ(2-ナフチル)]ブタ-2-エニルアセテート(図8中の化合物(v);37.4 mg,78%)を得た。
【0135】
1HNMR (400 MHz, CDCl3) δ 0.90 (t, 3H, J= 7.2 Hz), 1.28-1.37, (complex, 4H), 1.57-1.68 (complex, 2H), 2.03 (s, 3H), 3.56 (d, 2H, J= 6.4 Hz), 3.85 (s, 3H), 3.91 (s, 3H), 4.51 (d, 2H, J= 6.0 Hz), 4.94 (m, 1H), 5.58 (dt, 1H, J= 6.4, 15 Hz), 5.88 (dt, 1H, J= 6.0, 15 Hz), 6.06 (s, 1H), 7.74 (s, 1H)
13CNMR (100.40 MHz, CDCl3) δ 14.1, 20.9, 22.6, 28.8, 36.3, 37.7, 56.3, 63.4, 64.4, 70.5, 111.7, 125.1, 126.6, 126.8, 127.4, 128.4, 131.7, 132.1, 144.3, 158.7, 170.6. 179.6, 183.7.
IR (film) 3502, 2935, 1737, 1683, 1648, 1619 cm-1
【0136】
[実施例32]
<(2E)-4-[4-ヒドロキシ-3-ヒドロキシペンチル-7-メトキシ-5,8-ジオキソ(2-ナフチル)]ブタ-2-エニルアセテートの製造>
三臭化ホウ素(1M、ジクロロメタン溶液、0.22 mL)をジクロロメタン(5 mL)に溶解し、−78℃にてジクロロメタン(3.5 mL)に溶解させた化合物(v)(83.2 mg)を加えて25分間撹拌した。反応液をクロロホルム(15 mL)で希釈した後、水、飽和食塩水(各々10 mL)で洗浄し、有機層を無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲル薄層クロマトグラフィー(ヘキサン:酢酸エチル=1:2)にて精製し、(2E)-4-[4-ヒドロキシ-3-ヒドロキシペンチル-7-メトキシ-5,8-ジオキソ(2-ナフチル)]ブタ-2-エニルアセテート(図8中の化合物(w);23.7 mg,29%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.90 (t, 3H, J= 6.8 Hz), 1.33-1.42 (complex, 4H), 1.50-1.76 (complex, 2H), 2.05 (s, 3H), 3.50 (d, 2H, J= 5.8 Hz), 3.92 (s, 3H), 4.52 (d, 2H, J= 6.2 Hz), 4.86 (m, 1H), 5.59 (dt, 1H, J= 5.8, 15 Hz), 5.87 (dt, 1H, J= 6.2, 15 Hz), 6.07 (s, 1H), 7.48 (s, 1H), 13.11 (s, 1H)
13CNMR (67.80 MHz, CDCl3) δ 14.1, 21.0, 22.7, 28.6, 36.3, 36.4, 49.2, 49.3, 56.7, 64.4, 70.6, 109.3, 121.7, 126.8, 129.0, 131.6, 138.4, 144.4, 159.5, 161.0, 190.7, 206.5.
IR (film) 3534, 2956, 1739, 1683, 1625 cm-1
【0137】
[実施例33]
<(2E)-4-(4-ヒドロキシ-7-メトキシ-5,8-ジオキソ-3-ペンタノイル(2-ナフチル))ブタ-2-エニルアセテートの製造>
実施例32により得られた化合物(w)(2.6 mg)を無水テトラヒドロフラン(0.9 mL)に溶解し、0℃にてジメチルスルホキシド(0.1 mL)、IBX (20.6 mg)を加えた。室温で7時間撹拌した後、反応液をヘキサン(20 mL)で希釈した。沈殿物をセライト濾過し、ヘキサン:酢酸エチル=3:1の混合溶媒で洗浄したろ液を減圧濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、(2E)-4-(4-ヒドロキシ-7-メトキシ-5,8-ジオキソ-3-ペンタノイル(2-ナフチル))ブタ-2-エニルアセテート(図8中の化合物(e);2.7 mg)を得た。
1HNMR (270 MHz, CDCl3) δ 0.94 (t, 3H, J= 7.3 Hz), 1.37 (complex, 2H), 1.68 (complex, 2H), 2.11 (s, 3H), 2.86 (t, 2H, J= 7.6 Hz), 3.36 (d, 2H, J= 6.2 Hz), 3.93 (s, 3H), 4.52 (d, 2H, J= 6.2 Hz), 5.62 (dt, 1H, J= 5.9, 15 Hz), 5.81 (dt, 1H, J= 5.9, 15 Hz), 6.11 (s, 1H), 7.52 (s, 1H), 12.5 (s. 1H).
【0138】
[実施例34]
<7-((2E)-4-ヒドロキシブタ-2-エニル)-5-ヒドロキシ-2-メトキシ-6-ペンタノイルナフタレン-1,4-ジオンの製造>
実施例33により得られた化合物(e)(2.7 mg)をメタノール(1 mL)に溶解し、0℃にて炭酸カリウム(11.3 mg)を加えた。室温で30分間撹拌した後、反応液を酢酸エチル(30 mL)で希釈した。飽和塩化アンモニウム水溶液(10 mL)、飽和食塩水(5 mL)で洗浄した後、有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、7-((2E)-4-ヒドロキシブタ-2-エニル)-5-ヒドロキシ-2-メトキシ-6-ペンタノイルナフタレン-1,4-ジオン(図8中の化合物(j);2.3 mg、96%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.94 (t, 3H, J= 7.3 Hz), 1.34 (complex, 2H), 1.67 (complex, 2H), 2.87 (t, 2H, J= 7.3 Hz), 3.35 (d, 2H, J= 5.4 Hz), 3.93 (s, 3H), 4.20 (complex, 2H), 5.64-5.83 (complex, 2H), 6.11 (s, 1H), 7.53 (s, 1H), 12.5 (s. 1H).
【0139】
[実施例35]
<GST-Cdc25Aの調製>
GST融合タンパク質発現ベクターであるpGEX-2T(Amersham Biosciences社)にヒトcdc25A(hcdc25A) のcDNAを組み込みpGEX-2T-hcdc25Aを作製した。これらのプラスミドで形質転換した各大腸菌BL21株を37℃にてOD600=0.6〜0.8になるまで培養し、0.1mMのイソプロピル-1-チオ-β-D-チオガラクトピラノシド(IPTG) (Stratagene社)を添加することによりGST-Cdc25Aタンパク質の発現をそれぞれ誘導した。さらに37℃にて4時間培養後、菌体を回収し可溶化溶液(MT-PBS(150mM NaCl, 20mM Na2HPO4, 4mM NaH2PO4)、0.1% トリトンX-100、10μg/mL ロイペプチン、1mM フェニルメチルスルホニルフルオライド(PMSF))に懸濁した。懸濁した大腸菌を超音波破砕器により破砕後、遠心操作により可溶画分を得た。この可溶画分に、MT-PBSにより平衡化済みのグルタチオンアガロース (SIGMA社)を加え、4℃にて1時間GST-Cdc25Aをそれぞれ吸着させた。遠心操作によりグルタチオンアガロースを回収し、MT-PBSで4回洗浄後、溶出液(50mM Tris-HCl (pH8.0)、20mM 還元型グルタチオン)で4℃にて1時間攪拌することによりGST-Cdc25Aを溶出した。その後、遠心操作により上清を回収し、これを酵素(GST-Cdc25A)として以下の実施例に用いた。
また、PP1B(BIOMOL Research Laboratories社)およびPP2A(Promega社)を酵素として以下の実施例に用いた。
【0140】
[実施例36]
<ホスファターゼアッセイ>
上述の実施例により得られた化合物(f)〜(p)、並びに、上述の化合物(q)及び下式(x)で表される化合物(以下、「化合物(x)」と称する。)のホスファターゼ活性に対する阻害効果を調べた。なお、本実施例ではCdc25Aの基質として、3-O-メチルフルオレセインホスフェート(3-O-methylfluorescein phosphate;OMFP(SIGMA製))と4-ニトロフェニルホスフェート(4-nitrophenyl phosphate;pNPP(SIGMA製))とを用い、PTP1B及びPP2Aの基質としてpNPPを用いた。
【化19】
【0141】
<pNPPを用いた場合>
酵素(Cdc25A、PTP1B、及びPP2A) 30μl (30μg)と各濃度の化合物(化合物(f)〜(q)、化合物(x)及びNa3VO4(ホスファターゼ阻害剤のポジティブコントロールとして使用))1μlをassay buffer(50mM Tris-HCl (pH7.5)) 60μl中に懸濁し、室温にて15分間プレインキュベートした。そこに基質(5mM pNPP) 10μlを加え37℃にて60分間反応させた。その後、MICRO PLATE READER(TOSOH製)を用いて波長405nmでの吸光度を測定し、化合物の各酵素(Cdc25A、PTP1B、及びGST-PP2A)に対する阻害活性を評価した。なお、阻害活性の評価は阻害率(%)を算出することにより行った。
【0142】
<OMFPを用いた場合>
酵素(Cdc25A) 30μl (15μg)と各濃度の化合物(化合物(f)〜(q)、化合物(x)及びNa3VO4)1μlをassay buffer(50mM Tris-HCl (pH7.5)) 60μl中に懸濁し、室温にて15分間プレインキュベートした。そこに基質(最終濃度で25μM OMFP) 10μlを加え、室温にて30分間反応させた。その後、Fluoroskan Ascent(Labsystem製)を用いて3-O-メチルフルオレセインの生成量(励起波長355nm、吸収波長538nm)を測定し、化合物の酵素(Cdc25A)に対する阻害活性を評価した。なお、阻害活性の評価は阻害率(%)を算出することにより行った。
【0143】
以上の結果を表1に示す。なお、表1に示す各数値はIC50(μg/ml)の値をそれぞれ示す(但し、Na3VO4はμMで示す。)。
【表1】
【0144】
表1に示すように、化合物(q)は特定の基質(pNPP)を用いた時にのみCdc25Aに対する阻害活性を示し、化合物(x)はCdc25Aのみに対して阻害活性を示し、それ以外には阻害活性を示さないことがわかった。これに対して、化合物(f)〜(p)はチロシンホスファターゼ(Cdc25A及びPTP1B)のみに対して阻害活性を示し、プロテインホスファターゼ(PP2A)には阻害活性を示さないことがわかった。これらのことから、化合物(f)〜(p)は、チロシンホスファターゼを特異的に阻害する阻害剤として有用であることが明らかになった。
【0145】
[実施例37]
<細胞周期解析>
Cdc25AはCdk2/CyclinEやCdk2/CyclinAに作用するホスファターゼであり、細胞周期の調節においてG1/Sの移行に必要であることが知られている。そこで、Cdc25Aホスファターゼを阻害する化合物(f)〜(q)及び化合物(x)が細胞周期の進行を阻害するかどうかを調べた。
【0146】
5x105 個のマウス繊維芽細胞であるNIH3T3細胞を、0.2%牛胎児血清(MOREGATE社製)を含むダルベッコ改変イーグル培地(DMEM;10 ml)に加えて2日間培養し、静止期に同調した。その後、メタノールに溶解した各化合物(化合物(f)〜(q)、化合物(x)及びNa3VO4)を最終濃度で3μg/mL(Na3VO4は10又は100μM)となるように添加して15分間培養し、最終濃度が10%になるように牛胎児血清を添加した。血清刺激後、20時間培養した細胞を回収し、1mlのリン酸緩衝溶液に懸濁した。なお、コントロールとして、各化合物を含有していない等量のメタノールを添加して培養し、牛胎児血清で刺激した後、0時間(G0)又は20時間(20h cont.)培養した細胞をそれぞれ回収し、1mlのリン酸緩衝溶液に懸濁したものをそれぞれ準備した。懸濁後、50μgのヨウ化プロピジウム(WAKO社製)を添加して染色されたDNAの含有量をフローサイトメーター(EPICS ALTRA:BECKMAN COULTER社製)により測定した。その結果を図9に示す。
【0147】
図9に示すように、化合物(j)及び(q)は細胞周期のG1期で進行を停止させる作用を有することが明らかになった。そこで、上記と同様の方法により、化合物(j)及び(q)を用いて各濃度(1,3,10μg/mL)における細胞周期の進行阻害の程度を評価した。図10に示すように、化合物(j)及び(q)は少なくとも3μg/mLで細胞周期のG1期の進行を阻害できることが明らかになった。
【0148】
[実施例38]
<細胞増殖に対する抑制効果>
次に、化合物(f)〜(q)の細胞増殖に対する抑制効果を調べるため、化合物(j)〜(q)を用いてMTTアッセイを行った。48ウェルプレートに、NIH3T3細胞の懸濁液(1×105個/ml;10%牛胎児血清を含むDMEM)を500μlずつ添加して37℃で24時間培養し、メタノールに溶解した各化合物(化合物(j)〜(q))の溶液(0.03、0.1、0.3、1、3 mg/mL)を5μL添加して37℃で48時間培養した(なお、コントロールとして、メタノールのみを5μL添加して37℃で48時間培養したものを準備した。)。その後、MTT(3-(4,5-dimethlthiazol-2-yl)-2,5-diphenyltetrazolium bromide;Sigma)溶液(PBS-で5mg/mlに調整したもの)を25μlずつ添加して37℃で4時間培養した。培養後、培地を除去し、フォルマザン沈殿(細胞のミトコンドリアに存在するコハク酸脱水素酵素により、黄色水溶性のMTTが還元された暗青色不溶性の物質)を500μlのジメチルスルホキシドで溶解し、マイクロプレートリーダー(東ソー株式会社)を用いて吸光度(測定波長570 nm,リファレンス側620 nm)を測定し、増殖阻害作用を評価した。
【0149】
その結果を表2に示す。また、各化合物で刺激した細胞に細胞死が引き起こされていないかどうかを確認するため、顕微鏡により細胞の形態を観察した。
【表2】
【0150】
表2に示すように、化合物(j)及び化合物(m)〜(p)は、細胞増殖を抑制する作用を有しており、特に化合物(j)は優れた細胞増殖抑制作用を有していることが明らかになった。また、顕微鏡の形態観察により、多量の化合物(j)又は化合物(n)〜(q)のいずれかを用いると細胞死を引き起こすことが明らかになった。
【0151】
[実施例39]
<1-(5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ-2-ナフチル)アセトンの製造>
ベンゼン(6 mL)中、臭化亜鉛(257 mg)を30分間加熱還流し、この混合物にベンゼン(5 mL)に溶解させた化合物(12)(153 mg)を加えた。1.5時間後反応液を室温に戻し、酢酸エチル(35 mL)で希釈した後、水、飽和食塩水(各々15 mL)にて洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧留去してアルデヒドの粗生成物を得た。
【0152】
アルデヒドの粗生成物を無水テトラヒドロフラン(5 mL)に溶解し、-20℃にて臭化メチルマグネシウム(0.93 M、テトラヒドロフラン溶液、2 mL)を滴下した。徐々に昇温しながら30分間撹拌した後、反応液を酢酸エチル(20 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(5 mL)にて洗浄し、さらに水層を酢酸エチル(20 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水後、溶媒を減圧留去して二級アルコールの粗生成物を得た。
【0153】
二級アルコールの粗生成物を無水テトラヒドロフラン(2.7 mL)に溶解し、20 ℃にてジメチルスルホキシド(0.3 mL)、IBX (353 mg)を順次加えた。室温にて23時間撹拌した後、反応液をヘキサン(20 mL)で希釈した。沈殿物をセライト濾過し、ヘキサン:酢酸エチル=3:1の混合溶媒で洗浄したろ液を減圧濃縮して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1)にて精製し、1-(5-ベンジルオキシ-3-ブロモ-4,7,8-トリメトキシ-2-ナフチル)アセトン(図11中の化合物(22);99.2 mg、63%)を得た。
1HNMR (270 MHz, CDCl3) δ 2.25 (s, 3H), 3.77 (s, 3H), 3.90 (s, 3H), 3.95 (s, 3H), 4.03 (s, 2H), 5.18 (s, 2H), 6.77 (s, 1H), 7.35-7.45 (complex, 3H), 7.56 (d, 2H, J= 7.0 Hz), 7.76 (s. 1H).
【0154】
[実施例40]
<8-ベンジルオキシ-2-ブロモ-1,5,6-トリメトキシ-3-(2-メチル(1,3-ジオキソラン-2-イル)メチル)ナフタレンの製造>
ベンゼン(30 mL)中、化合物(22)(761 mg)、エチレングリコール(1.0 mL)、4-トルエンスルホン酸一水和物(663 mg)を4時間加熱還流した。反応液を室温に戻し、酢酸エチル(35 mL)で希釈した後、飽和炭酸水素ナトリウム水溶液(20 mL)、飽和食塩水(10 mL)で洗浄し、有機層を無水硫酸ナトリウムで脱水後、溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1)にて精製し、8-ベンジルオキシ-2-ブロモ-1,5,6-トリメトキシ-3-(2-メチル(1,3-ジオキソラン-2-イル)メチル)ナフタレン(図11中の化合物(23);765 mg、92%)を得た。
1HNMR (400 MHz, CDCl3) δ 1.43 (s, 3H), 3.37 (s, 2H), 3.78 (s, 4H), 3.90 (s, 3H), 3.95 (s, 3H), 5.18 (s, 2H), 6.75 (s, 1H), 7.36 (d, 1H, J= 7.6 Hz), 7.42 (t, 2H, J= 7.2 Hz), 7.57 (d, 2H, J= 7.6 Hz) 7.91 (s, 1H).
【0155】
[実施例41]
<7,10-ジメトキシ-3-メチル-1-プロピルベンゾ[2,1-g]イソクロメン-6,9-ジオンの製造>
化合物(23)(432 mg)を無水テトラヒドロフラン(10 mL)に溶解し、-78℃にてn-ブチルリチウム(1.56 M、ヘキサン溶液、2.8 mL)を滴下した。さらにn-ブチルアルデヒド(1.2 mL)を加え、10分間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液 (15 mL)を加えて0℃に戻し、酢酸エチル(35 mL)で希釈した後、飽和食塩水(10 mL)で洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮して二級アルコールの粗生成物を得た。
【0156】
二級アルコールの粗生成物をベンゼン(15 mL)に溶解し、0℃にて4-トルエンスルホン酸一水和物(396 mg)を加えた。室温で10分間撹拌した後、酢酸エチル(25 mL)で希釈し飽和炭酸水素ナトリウム水溶液(15 mL)、飽和食塩水(5 mL)で洗浄した。有機層を無水硫酸ナトリウムで脱水し、溶媒を減圧濃縮してピラノナフタレンの粗生成物を得た。
【0157】
ピラノナフタレンの粗生成物を1,4-ジオキサン(4 mL)に溶解し、0℃にて水(1 mL)、DDQ (362 mg)を順次加えた。40分間撹拌した後、反応液をクロロホルム(25 mL)で希釈し、飽和炭酸水素ナトリウム水溶液(15 mL)を加えた。有機層を飽和食塩水(10 mL)にて洗浄し、さらに水層をクロロホルム(20 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)にて精製し、7,10-ジメトキシ-3-メチル-1-プロピルベンゾ[2,1-g]イソクロメン-6,9-ジオン(図1中の化合物(r);75.5 mg、27%)を得た。
【0158】
1HNMR (270 MHz, CDCl3) δ 0.95 (t, 3H, J= 7.0 Hz), 1.38-1.58 (complex, 3H), 1.96 (s, 3H), 2.04 (m, 1H), 3.86 (s, 3H), 3.88 (s, 3H), 5.76 (dd, 1H, J= 3.0, 9.5 Hz), 5.66 (s, 1H), 6.03 (s, 1H), 7.47 (s. 1H).
【0159】
[実施例42]
<7,10-ジメトキシ-3-メチル-1-プロピルベンゾ[2,1-g]イソクロマン-6,9-ジオンの製造>
化合物(r)(36.2 mg)をメタノール(3 mL)に溶解し、Pd/C (10 mg)を加えた。水素気流下室温にて1時間撹拌した後、沈殿物をセライト濾過し、メタノールで洗浄したろ液を減圧濃縮して得られる粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、7,10-ジメトキシ-3-メチル-1-プロピルベンゾ[2,1-g]イソクロマン-6,9-ジオン(図11中の化合物(s);31.3 mg、86%)を得た。
1HNMR (270 MHz, CDCl3) δ 0.88 (t, 3H, J= 7.3 Hz), 1.00 (t, 3H, J= 7.3 Hz), 1.34 (d, 2H, J= 5.9 Hz), 1.35 (d, 2H, J= 6.2 Hz), 1.20-2.05 (complex, 8H), 2.66-2.87 (complex, 4H), 3.64 (m, 1H), 3.84 (s, 3H), 3.87 (s, 3H), 3.88 (s, 6H), 4.05 (m, 1H), 5.00 (dd, 1H, J= 3.2, 9.7 Hz), 5.06 (d, 1H, J= 3.5 Hz), 6.07 (s, 2H), 7.69 (s, 1H), 7.70 (s. 1H).
【0160】
[実施例43]
<8-ブロモ-7,10-ジメトキシ-3-メチル-1-プロピルベンゾ[1,2-g]イソクロマン-6,9-ジオンの製造>
化合物(s)(31.3 mg)をジクロロメタン(2 mL)に溶解し、0℃にてピリジン(30 μL)、臭化水素酸ペルブロミドピリジン錯体(Pyr・HBr3)(42.4 mg)を加えた。室温で4時間撹拌した後、反応液をクロロホルムで希釈し、1M塩酸(15 mL)を加えた。有機層を飽和食塩水(5 mL)にて洗浄し、さらに水層をクロロホルム(15 mL)で洗浄した後、有機層を合し無水硫酸ナトリウムで脱水した。溶媒を減圧留去して得られる粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム:酢酸エチル=1:1)にて精製し、8-ブロモ-7,10-ジメトキシ-3-メチル-1-プロピルベンゾ[1,2-g]イソクロマン-6,9-ジオン(図11中の化合物(t);16.8 mg、36%)及び化合物(s)(12.3 mg、39%)を得た。
1HNMR (400 MHz, CDCl3) δ 0.88 (t, 3H, J= 7.3 Hz), 1.00 (t, 3H, J= 7.3 Hz), 1.34 (d, 2H, J= 5.9 Hz), 1.35 (d, 2H, J= 6.2 Hz), 1.31-2.04 (complex, 8H), 2.67-2.86 (complex, 4H), 3.76 (m, 1H), 3.84 (s, 3H), 3.87 (s, 3H), 4.05 (m, 1H), 4.25 (s, 6H), 5.00 (dd, 1H, J= 3.6, 11 Hz), 5.06 (m, 1H), 7.63 (s, 1H), 7.64 (s. 1H).
【0161】
[実施例44]
<8-クロロ-7,10-ジヒドロキシ-3-メチル-1-プロピルベンゾ[1,2-g]イソクロマン-6,9-ジオンの製造>
エタノール(2 mL)中、化合物(t)(3.8 mg)、及び濃塩酸(4 mL)を15時間加熱還流した。反応液を室温に戻し、クロロホルム(40 mL)で希釈した後、水(30 mL)で洗浄し、有機層を無水硫酸ナトリウムで脱水した。溶媒を減圧留去してクロロナフトキノンの粗生成物(図1中の化合物(u))を得た。
1HNMR (270 MHz, CDCl3) δ 0.88 (t, 3H, J= 7.3 Hz), 1.00 (t, 3H, J= 7.2 Hz), 1.34 (d, 2H, J= 6.0 Hz), 1.35 (d, 2H, J= 6.1 Hz), 1.25-2.05 (complex, 8H), 2.61-2.81 (complex, 4H), 3.65 (m, 1H), 4.03 (m, 1H), 5.02 (complex, 2H), 7.43 (s, 2H), 12.4 (s. 1H), 12.6 (s, 1H).
【0162】
[実施例45]
<MRSAに対する抗菌作用及びイミペネム活性増強作用>
本実施例では、検定菌として、臨床分離されたMRSA K24株を用いた。検定プレートは、Mueller-Hinton Broth (DIFCO社)に1.5%のAgar (清水食品株式会社)を添加して作製した対照用プレート、及び上記組成にイミペネム(萬有製薬株式会社、商品名:チエナム筋注用力価0.5)を最終濃度10μg/mLとなるように添加して作製した検定用プレートを用いた。接種菌液はMueller-Hinton Brothで一晩培養した検定菌培養液を、同培地で、0.5Mc Farand (約108cfu/mL)になるように調製したものを用いた。この接種菌液をNCCLS法に従い、滅菌綿棒で各プレートに塗抹した。検定菌に対する各プレートでの抗菌活性は、ペーパーディスク法により、10μgの検定薬剤(化合物(r)、(s)、(v)、及び(w))のメタノール溶液を染み込ませた6mmのペーパーディスクを各プレートにのせて、37℃で20時間培養した。その後、検定菌に対する抗菌活性の指標として、プレートにおける阻止円径 (ペーパーディスクの中心を通る直径(mm))を測定した。
【0163】
その結果を表3に示す。
【表3】
【0164】
表3に示すように、化合物(r)、(s)、(v)、及び(w)は単独で抗MRSA作用を有することが明らかになった。また、10μg/mLのイミペネム単独では抗MRSA作用を有しないが、化合物(r)、(s)、(u)、(v)、及び(w)をイミペネムと協同して検定菌に作用させると、抗MRSA作用が増強することが明らかになった。このことから、化合物(r)、(s)、(u)、(v)、及び(w)がイミペネムの活性を増強する作用を有すること、あるいは、イミペネムがこれらの化合物の活性を増強する作用を有することが示唆された。
【図面の簡単な説明】
【0165】
【図1】本発明の一実施例において、化合物(k)及び(l)の製造過程を示す図である。
【図2】本発明の一実施例において、化合物(m)〜(p)の製造過程を示す図である。
【図3】本発明の一実施例において、化合物(15)の製造過程を示す図である。
【図4】本発明の一実施例において、化合物(f)の製造過程を示す図である。
【図5】本発明の一実施例において、化合物(g)の製造過程を示す図である。
【図6】本発明の一実施例において、化合物(h)の製造過程を示す図である。
【図7】本発明の一実施例において、化合物(i)の製造過程を示す図である。
【図8】本発明の一実施例において、化合物(j)の製造過程を示す図である。
【図9】本発明の一実施例において、化合物(f)〜(q)、化合物(x)及びNa3VO4が細胞周期の進行に与える影響を調べた結果を示す図である。
【図10】本発明の一実施例において、各濃度の化合物(化合物(j)又は(q))が細胞周期の進行に与える影響を調べた結果を示す図である。
【図11】本発明の一実施例において、化合物(r)、化合物(s)、及び化合物(u)の製造過程を示す図である。
【特許請求の範囲】
【請求項1】
下記の一般式(I)で表されるナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化1】
(式中、R3がアリールアルコール基又はアルキルカルボニル基である場合には、R1は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R2はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R4はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、又は2−ジメタン−1,3−ジオキサン−4−アルキル基であり、
式中、R4がアルキニルオキシアルキル基又はブチルジメチルシリルオキシアルケニル基である場合には、R1は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R2はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R3は水素原子、ヒドロキシアルキル基、ハロゲン原子、又はアリールアルコール基であり、
式中、R1がアシルオキシ基である場合には、R2はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R3は水素原子、ヒドロキシアルキル基、又はハロゲン原子であり、R4はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、又は2−ジメタン−1,3−ジオキサン−4−アルキル基であり、
式中、R1及びR2がメトキシ基であり、R4がヒドロキシブタ−2−エニル基である場合には、R3はヒドロキシエチル基、ヒドロキシプロピル基、又はヒドロキシヘキシル基であり、
式中、R2がメトキシ基であり、R3が水素原子であり、R4がメトキシメチル基である場合には、R1はヒドロキシル基であり、
式中、R1及びR2がメトキシ基であり、R3が水素原子である場合には、R4はへキシルオキシメチル基であり、
前記R3と前記R4とで環状を構成しない。)
【請求項2】
下記の一般式(II)で表されるナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化2】
(式中、R5は水素原子又はアルキル基であり、R6はヒドロキシエチル基、ヒドロキシプロピル基、ヒドロキシヘキシル基、アリールアルコール基、又はアルキルカルボニル基であり、R7は水素原子、アシル基、又はブチルジメチルシリル基であり、前記R6と前記R7とで環状を構成しない。)
【請求項3】
下記の一般式(III)で表されるナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化3】
(式中、R8は水素原子又はアシル基である。)
【請求項4】
下記の一般式(IV)で表されるナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化4】
(式中、R9はプロピル基、ブチル基、へキシル基、又はアルキニルオキシアルキル基である。)
【請求項5】
前記化合物が下式(a)〜(j)で表される化合物であることを特徴とする請求項2に記載のナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化5】
【請求項6】
前記化合物が下式(k)又は(l)で表される化合物であることを特徴とする請求項3に記載のナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化6】
【請求項7】
前記化合物が下式(m)〜(p)で表される化合物であることを特徴とする請求項4に記載のナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化7】
【請求項8】
請求項1〜7のいずれかに記載のナフトキノン誘導体化合物又はその薬理学的に許容される塩を有効成分として含有するチロシンホスファターゼ阻害剤。
【請求項9】
前記チロシンホスファターゼが、Cdc25Aホスファターゼ又はプロテインチロシンホスファターゼ1Bであることを特徴とする請求項8に記載の阻害剤。
【請求項10】
請求項1〜7のいずれかに記載のナフトキノン誘導体化合物又はその薬理学的に許容される塩を有効成分として含有する医薬組成物。
【請求項11】
プロテインチロシンホスファターゼ1Bの過剰発現又は活性化に起因する疾患を予防又は改善することを特徴とする請求項10に記載の医薬組成物。
【請求項12】
前記疾患が、2型糖尿病、肥満症、神経疾患、腫瘍、及び骨髄性白血病からなるグループから選ばれるいずれかの疾患であることを特徴とする請求項11に記載の医薬組成物。
【請求項13】
Cdc25Aホスファターゼの過剰発現又は活性化に起因する疾患を予防又は改善することを特徴とする請求項10に記載の医薬組成物。
【請求項14】
前記疾患が、腫瘍であることを特徴とする請求項13に記載の医薬組成物。
【請求項15】
下記の一般式(V)で表される化合物又はその薬理学的に許容される塩を有効成分として含有する細胞増殖阻害剤。
【化8】
(式中、R12がアリールアルコール基又はアルキルカルボニル基である場合には、R10は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R11はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R13はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、又は2−ジメタン−1,3−ジオキサン−4−アルキル基であり、
式中、R13がアルキニルオキシアルキル基又はブチルジメチルシリルオキシアルケニル基である場合には、R10は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R11はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R12は水素原子、ヒドロキシアルキル基、ハロゲン原子、又はアリールアルコール基であり、
式中、R10及びR11がメトキシ基であり、R12がヒドロキシブタ−2−エニル基である場合には、R13はヒドロキシエチル基、ヒドロキシプロピル基、又はヒドロキシヘキシル基であり、
式中、R10及びR11がメトキシ基であり、R12が水素原子である場合には、R13はへキシルオキシメチル基であり、
前記R12と前記R13とで環状を構成しない。)
【請求項16】
前記化合物が、下式(j)で表される化合物又は下式(m)〜(p)のいずれかに表される化合物であることを特徴とする請求項15に記載の細胞増殖阻害剤。
【化9】
【請求項17】
細胞周期のG1期の進行を阻害する細胞周期進行阻害剤であって、
下記の一般式(VI)で表される化合物又はその薬理学的に許容される塩を有効成分として含有する細胞周期進行阻害剤。
【化10】
(式中、R14はヒドロキシアルキル基、アルキルカルボニル基、又はアリールアルコール基であり、R15はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、2−ジメタン−1,3−ジオキサン−4−アルキル基、アルキニルオキシアルキル基、又はブチルジメチルシリルオキシアルケニル基であり、前記R14と前記R15とで環状を構成しない。)
【請求項18】
前記化合物が、下式(j)又は(q)で表される化合物であることを特徴とする請求項17に記載の細胞周期進行阻害剤。
【化11】
【請求項19】
下記の一般式(VII)で表されるナフトキノン誘導体化合物若しくは下式(r)で表される化合物又はその薬理学的に許容される塩。
【化12】
(式中、R16はヒドロキシル基又はアルコキシル基であり、R17は水素原子又はハロゲン原子であり、R18はヒドロキシル基又はアルコキシル基である。)
【請求項20】
前記一般式(VII)で表されるナフトキノン誘導体化合物が、下式(s)〜(u)で表される化合物であることを特徴とする請求項19に記載のナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化13】
【請求項21】
下記の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩を有効成分として含有する抗菌剤。
【化14】
(式中、R19及びR20はアルコキシル基である。)
【請求項22】
前記一般式(VIII)で表されるナフトキノン誘導体化合物が、下式(s)で表される化合物であることを特徴とする請求項21に記載の抗菌剤。
【化15】
【請求項23】
さらに、イミペネムを含むことを特徴とする請求項21又は22に記載の抗菌剤。
【請求項24】
下式(u)で表される化合物又はその薬理学的に許容される塩、及びイミペネムを有効成分として含有する抗菌剤。
【化16】
【請求項25】
MRSA(メチシリン耐性黄色ブドウ球菌)に対して抗菌作用を有することを特徴とする請求項21〜24のいずれかに記載の抗菌剤。
【請求項26】
イミペネムの活性を増強するイミペネム活性増強剤であって、
下記の一般式(VII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩を有効成分として含有するイミペネム活性増強剤。
【化17】
(式中、R16はヒドロキシル基又はアルコキシル基であり、R17は水素原子又はハロゲン原子であり、R18はヒドロキシル基又はアルコキシル基である。)
【請求項27】
前記一般式(VII)で表されるナフトキノン誘導体化合物が、下式(s)又は下式(u)で表される化合物であることを特徴とする請求項26に記載のイミペネム活性増強剤。
【化18】
【請求項28】
前記イミペネムの活性が抗菌活性であることを特徴とする請求項26又は27に記載のイミペネム活性増強剤。
【請求項29】
前記抗菌活性がMRSA(メチシリン耐性黄色ブドウ球菌)に対する抗菌活性であることを特徴とする請求項28に記載のイミペネム活性増強剤。
【請求項30】
細菌の増殖を阻害する薬剤であって、
下記の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩を有効成分として含有することを特徴とする薬剤。
【化19】
(式中、R19及びR20はアルコキシル基である。)
【請求項31】
前記一般式(VIII)で表されるナフトキノン誘導体化合物が、下式(s)で表される化合物であることを特徴とする請求項30に記載の薬剤。
【化20】
【請求項32】
さらに、イミペネムを含むことを特徴とする請求項30又は31に記載の薬剤。
【請求項33】
細菌の増殖を阻害する薬剤であって、
下式(u)で表される化合物又はその薬理学的に許容される塩、及びイミペネムを有効成分として含有する薬剤。
【化21】
【請求項34】
前記細菌がMRSA(メチシリン耐性黄色ブドウ球菌)であることを特徴とする請求項30〜33のいずれかに記載の薬剤。
【請求項35】
細菌の感染又は増殖に起因する疾患を改善する医薬組成物であって、下記の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩を有効成分として含有することを特徴とする医薬組成物。
【化22】
(式中、R19及びR20はアルコキシル基である。)
【請求項36】
前記一般式(VIII)で表されるナフトキノン誘導体化合物が、下式(s)で表される化合物であることを特徴とする請求項35に記載の医薬組成物。
【化23】
【請求項37】
さらに、イミペネムを含むことを特徴とする請求項35又は36に記載の医薬組成物。
【請求項38】
細菌の感染又は増殖に起因する疾患を改善する医薬組成物であって、下式(u)で表される化合物又はその薬理学的に許容される塩、及びイミペネムを有効成分として含有する医薬組成物。
【化24】
【請求項39】
前記細菌がMRSA(メチシリン耐性黄色ブドウ球菌)であることを特徴とする請求項35〜38のいずれかに記載の医薬組成物。
【請求項40】
前記疾患が、脳炎、肺炎、敗血症、腹膜炎、腸炎、骨髄炎、胆管炎、皮膚化膿症、褥瘡感染、並びに、MRSAが産生する毒素による食中毒及びトキシックショック症候群から選ばれる一つまたは複数の疾患であることを特徴とする請求項39に記載の医薬組成物。
【請求項41】
下記の一般式(VII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩の活性を増強するナフトキノン誘導体化合物活性増強剤であって、
イミペネムを有効成分として含有するナフトキノン誘導体化合物活性増強剤。
【化25】
(式中、R16はヒドロキシル基又はアルコキシル基であり、R17は水素原子又はハロゲン原子であり、R18はヒドロキシル基又はアルコキシル基である。)
【請求項42】
前記一般式(VII)で表されるナフトキノン誘導体化合物が、下式(s)又は下式(u)で表される化合物であることを特徴とする請求項41に記載のナフトキノン誘導体化合物活性増強剤。
【化26】
【請求項43】
前記活性が抗菌活性であることを特徴とする請求項41又は42に記載のナフトキノン誘導体化合物活性増強剤。
【請求項44】
前記抗菌活性が、MRSA(メチシリン耐性黄色ブドウ球菌)に対する抗菌活性であることを特徴とする請求項43に記載のナフトキノン誘導体化合物活性増強剤。
【請求項45】
イミペネムの活性を増強するイミペネム活性増強方法であって、
前記イミペネムと協同させて、下記の一般式(VII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩を作用させることを特徴とするイミペネム活性増強方法。
【化27】
(式中、R16はヒドロキシル基又はアルコキシル基であり、R17は水素原子又はハロゲン原子であり、R18はヒドロキシル基又はアルコキシル基である。)
【請求項46】
前記一般式(VII)で表されるナフトキノン誘導体化合物が、下式(s)又は下式(u)で表される化合物であることを特徴とする請求項45に記載のイミペネム活性増強方法。
【化28】
【請求項47】
前記イミペネムの活性が抗菌活性であることを特徴とする請求項45又は46に記載のイミペネム活性増強方法。
【請求項48】
前記抗菌活性がMRSA(メチシリン耐性黄色ブドウ球菌)に対する抗菌活性であることを特徴とする請求項47に記載のイミペネム活性増強方法。
【請求項49】
下記の一般式(VII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩の活性を増強するナフトキノン誘導体化合物活性増強方法であって、
前記ナフトキノン誘導体化合物と協同させてイミペネムを作用させることを特徴とするナフトキノン誘導体化合物活性増強方法。
【化29】
(式中、R16はヒドロキシル基又はアルコキシル基であり、R17は水素原子又はハロゲン原子であり、R18はヒドロキシル基又はアルコキシル基である。)
【請求項50】
前記一般式(VII)で表されるナフトキノン誘導体化合物が、下式(s)又は下式(u)で表される化合物であることを特徴とする請求項49に記載のナフトキノン誘導体化合物活性増強方法。
【化30】
【請求項51】
前記活性が抗菌活性であることを特徴とする請求項49又は50に記載のナフトキノン誘導体化合物活性増強方法。
【請求項52】
前記抗菌活性が、MRSA(メチシリン耐性黄色ブドウ球菌)に対する抗菌活性であることを特徴とする請求項51に記載のナフトキノン誘導体化合物活性増強方法。
【請求項53】
下記の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩を作用させることを特徴とする細菌の増殖阻害方法。
【化31】
(式中、R19及びR20はアルコキシル基である。)
【請求項54】
前記一般式(VIII)で表されるナフトキノン誘導体化合物が、下式(s)で表される化合物であることを特徴とする請求項53に記載の細菌の増殖阻害方法。
【化32】
【請求項55】
さらに、イミペネムを協同して作用させることを特徴とする請求項53又は54に記載の細菌の増殖阻害方法。
【請求項56】
下式(u)で表される化合物又はその薬理学的に許容される塩、及びイミペネムを協同して作用させることを特徴とする細菌の増殖阻害方法。
【化33】
【請求項57】
前記細菌がMRSA(メチシリン耐性黄色ブドウ球菌)であることを特徴とする請求項53〜56のいずれかに記載の細菌の増殖阻害方法。
【請求項1】
下記の一般式(I)で表されるナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化1】
(式中、R3がアリールアルコール基又はアルキルカルボニル基である場合には、R1は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R2はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R4はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、又は2−ジメタン−1,3−ジオキサン−4−アルキル基であり、
式中、R4がアルキニルオキシアルキル基又はブチルジメチルシリルオキシアルケニル基である場合には、R1は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R2はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R3は水素原子、ヒドロキシアルキル基、ハロゲン原子、又はアリールアルコール基であり、
式中、R1がアシルオキシ基である場合には、R2はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R3は水素原子、ヒドロキシアルキル基、又はハロゲン原子であり、R4はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、又は2−ジメタン−1,3−ジオキサン−4−アルキル基であり、
式中、R1及びR2がメトキシ基であり、R4がヒドロキシブタ−2−エニル基である場合には、R3はヒドロキシエチル基、ヒドロキシプロピル基、又はヒドロキシヘキシル基であり、
式中、R2がメトキシ基であり、R3が水素原子であり、R4がメトキシメチル基である場合には、R1はヒドロキシル基であり、
式中、R1及びR2がメトキシ基であり、R3が水素原子である場合には、R4はへキシルオキシメチル基であり、
前記R3と前記R4とで環状を構成しない。)
【請求項2】
下記の一般式(II)で表されるナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化2】
(式中、R5は水素原子又はアルキル基であり、R6はヒドロキシエチル基、ヒドロキシプロピル基、ヒドロキシヘキシル基、アリールアルコール基、又はアルキルカルボニル基であり、R7は水素原子、アシル基、又はブチルジメチルシリル基であり、前記R6と前記R7とで環状を構成しない。)
【請求項3】
下記の一般式(III)で表されるナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化3】
(式中、R8は水素原子又はアシル基である。)
【請求項4】
下記の一般式(IV)で表されるナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化4】
(式中、R9はプロピル基、ブチル基、へキシル基、又はアルキニルオキシアルキル基である。)
【請求項5】
前記化合物が下式(a)〜(j)で表される化合物であることを特徴とする請求項2に記載のナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化5】
【請求項6】
前記化合物が下式(k)又は(l)で表される化合物であることを特徴とする請求項3に記載のナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化6】
【請求項7】
前記化合物が下式(m)〜(p)で表される化合物であることを特徴とする請求項4に記載のナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化7】
【請求項8】
請求項1〜7のいずれかに記載のナフトキノン誘導体化合物又はその薬理学的に許容される塩を有効成分として含有するチロシンホスファターゼ阻害剤。
【請求項9】
前記チロシンホスファターゼが、Cdc25Aホスファターゼ又はプロテインチロシンホスファターゼ1Bであることを特徴とする請求項8に記載の阻害剤。
【請求項10】
請求項1〜7のいずれかに記載のナフトキノン誘導体化合物又はその薬理学的に許容される塩を有効成分として含有する医薬組成物。
【請求項11】
プロテインチロシンホスファターゼ1Bの過剰発現又は活性化に起因する疾患を予防又は改善することを特徴とする請求項10に記載の医薬組成物。
【請求項12】
前記疾患が、2型糖尿病、肥満症、神経疾患、腫瘍、及び骨髄性白血病からなるグループから選ばれるいずれかの疾患であることを特徴とする請求項11に記載の医薬組成物。
【請求項13】
Cdc25Aホスファターゼの過剰発現又は活性化に起因する疾患を予防又は改善することを特徴とする請求項10に記載の医薬組成物。
【請求項14】
前記疾患が、腫瘍であることを特徴とする請求項13に記載の医薬組成物。
【請求項15】
下記の一般式(V)で表される化合物又はその薬理学的に許容される塩を有効成分として含有する細胞増殖阻害剤。
【化8】
(式中、R12がアリールアルコール基又はアルキルカルボニル基である場合には、R10は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R11はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R13はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、又は2−ジメタン−1,3−ジオキサン−4−アルキル基であり、
式中、R13がアルキニルオキシアルキル基又はブチルジメチルシリルオキシアルケニル基である場合には、R10は水素原子、アルキル基、ヒドロキシル基、アルコキシル基、又はヒドロキシアルキル基であり、R11はヒドロキシル基、アシルオキシ基、又はアルコキシル基であり、R12は水素原子、ヒドロキシアルキル基、ハロゲン原子、又はアリールアルコール基であり、
式中、R10及びR11がメトキシ基であり、R12がヒドロキシブタ−2−エニル基である場合には、R13はヒドロキシエチル基、ヒドロキシプロピル基、又はヒドロキシヘキシル基であり、
式中、R10及びR11がメトキシ基であり、R12が水素原子である場合には、R13はへキシルオキシメチル基であり、
前記R12と前記R13とで環状を構成しない。)
【請求項16】
前記化合物が、下式(j)で表される化合物又は下式(m)〜(p)のいずれかに表される化合物であることを特徴とする請求項15に記載の細胞増殖阻害剤。
【化9】
【請求項17】
細胞周期のG1期の進行を阻害する細胞周期進行阻害剤であって、
下記の一般式(VI)で表される化合物又はその薬理学的に許容される塩を有効成分として含有する細胞周期進行阻害剤。
【化10】
(式中、R14はヒドロキシアルキル基、アルキルカルボニル基、又はアリールアルコール基であり、R15はアシル基、ヒドロキシアルキル基、ヒドロキシアルケニル基、カルボキシル基、アルコキシカルボニル基、アルコキシアルキル基、アルコキシアルケニル基、フェノキシアルキル基、フェノキシアルケニル基、カルボキシアルケニル基、アルコキシカルボニルアルケニル基、アシルオキシアルケニル基、ヒドロキシアルキルオキシランアルキル基、アルコキシアルキルオキシランアルキル基、ジヒドロキシアルキル基、ヒドロキシアルコキシアルキル基、オキシラン基、2−ジメタン−1,3−ジオキサン−4−アルキル基、アルキニルオキシアルキル基、又はブチルジメチルシリルオキシアルケニル基であり、前記R14と前記R15とで環状を構成しない。)
【請求項18】
前記化合物が、下式(j)又は(q)で表される化合物であることを特徴とする請求項17に記載の細胞周期進行阻害剤。
【化11】
【請求項19】
下記の一般式(VII)で表されるナフトキノン誘導体化合物若しくは下式(r)で表される化合物又はその薬理学的に許容される塩。
【化12】
(式中、R16はヒドロキシル基又はアルコキシル基であり、R17は水素原子又はハロゲン原子であり、R18はヒドロキシル基又はアルコキシル基である。)
【請求項20】
前記一般式(VII)で表されるナフトキノン誘導体化合物が、下式(s)〜(u)で表される化合物であることを特徴とする請求項19に記載のナフトキノン誘導体化合物又はその薬理学的に許容される塩。
【化13】
【請求項21】
下記の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩を有効成分として含有する抗菌剤。
【化14】
(式中、R19及びR20はアルコキシル基である。)
【請求項22】
前記一般式(VIII)で表されるナフトキノン誘導体化合物が、下式(s)で表される化合物であることを特徴とする請求項21に記載の抗菌剤。
【化15】
【請求項23】
さらに、イミペネムを含むことを特徴とする請求項21又は22に記載の抗菌剤。
【請求項24】
下式(u)で表される化合物又はその薬理学的に許容される塩、及びイミペネムを有効成分として含有する抗菌剤。
【化16】
【請求項25】
MRSA(メチシリン耐性黄色ブドウ球菌)に対して抗菌作用を有することを特徴とする請求項21〜24のいずれかに記載の抗菌剤。
【請求項26】
イミペネムの活性を増強するイミペネム活性増強剤であって、
下記の一般式(VII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩を有効成分として含有するイミペネム活性増強剤。
【化17】
(式中、R16はヒドロキシル基又はアルコキシル基であり、R17は水素原子又はハロゲン原子であり、R18はヒドロキシル基又はアルコキシル基である。)
【請求項27】
前記一般式(VII)で表されるナフトキノン誘導体化合物が、下式(s)又は下式(u)で表される化合物であることを特徴とする請求項26に記載のイミペネム活性増強剤。
【化18】
【請求項28】
前記イミペネムの活性が抗菌活性であることを特徴とする請求項26又は27に記載のイミペネム活性増強剤。
【請求項29】
前記抗菌活性がMRSA(メチシリン耐性黄色ブドウ球菌)に対する抗菌活性であることを特徴とする請求項28に記載のイミペネム活性増強剤。
【請求項30】
細菌の増殖を阻害する薬剤であって、
下記の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩を有効成分として含有することを特徴とする薬剤。
【化19】
(式中、R19及びR20はアルコキシル基である。)
【請求項31】
前記一般式(VIII)で表されるナフトキノン誘導体化合物が、下式(s)で表される化合物であることを特徴とする請求項30に記載の薬剤。
【化20】
【請求項32】
さらに、イミペネムを含むことを特徴とする請求項30又は31に記載の薬剤。
【請求項33】
細菌の増殖を阻害する薬剤であって、
下式(u)で表される化合物又はその薬理学的に許容される塩、及びイミペネムを有効成分として含有する薬剤。
【化21】
【請求項34】
前記細菌がMRSA(メチシリン耐性黄色ブドウ球菌)であることを特徴とする請求項30〜33のいずれかに記載の薬剤。
【請求項35】
細菌の感染又は増殖に起因する疾患を改善する医薬組成物であって、下記の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩を有効成分として含有することを特徴とする医薬組成物。
【化22】
(式中、R19及びR20はアルコキシル基である。)
【請求項36】
前記一般式(VIII)で表されるナフトキノン誘導体化合物が、下式(s)で表される化合物であることを特徴とする請求項35に記載の医薬組成物。
【化23】
【請求項37】
さらに、イミペネムを含むことを特徴とする請求項35又は36に記載の医薬組成物。
【請求項38】
細菌の感染又は増殖に起因する疾患を改善する医薬組成物であって、下式(u)で表される化合物又はその薬理学的に許容される塩、及びイミペネムを有効成分として含有する医薬組成物。
【化24】
【請求項39】
前記細菌がMRSA(メチシリン耐性黄色ブドウ球菌)であることを特徴とする請求項35〜38のいずれかに記載の医薬組成物。
【請求項40】
前記疾患が、脳炎、肺炎、敗血症、腹膜炎、腸炎、骨髄炎、胆管炎、皮膚化膿症、褥瘡感染、並びに、MRSAが産生する毒素による食中毒及びトキシックショック症候群から選ばれる一つまたは複数の疾患であることを特徴とする請求項39に記載の医薬組成物。
【請求項41】
下記の一般式(VII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩の活性を増強するナフトキノン誘導体化合物活性増強剤であって、
イミペネムを有効成分として含有するナフトキノン誘導体化合物活性増強剤。
【化25】
(式中、R16はヒドロキシル基又はアルコキシル基であり、R17は水素原子又はハロゲン原子であり、R18はヒドロキシル基又はアルコキシル基である。)
【請求項42】
前記一般式(VII)で表されるナフトキノン誘導体化合物が、下式(s)又は下式(u)で表される化合物であることを特徴とする請求項41に記載のナフトキノン誘導体化合物活性増強剤。
【化26】
【請求項43】
前記活性が抗菌活性であることを特徴とする請求項41又は42に記載のナフトキノン誘導体化合物活性増強剤。
【請求項44】
前記抗菌活性が、MRSA(メチシリン耐性黄色ブドウ球菌)に対する抗菌活性であることを特徴とする請求項43に記載のナフトキノン誘導体化合物活性増強剤。
【請求項45】
イミペネムの活性を増強するイミペネム活性増強方法であって、
前記イミペネムと協同させて、下記の一般式(VII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩を作用させることを特徴とするイミペネム活性増強方法。
【化27】
(式中、R16はヒドロキシル基又はアルコキシル基であり、R17は水素原子又はハロゲン原子であり、R18はヒドロキシル基又はアルコキシル基である。)
【請求項46】
前記一般式(VII)で表されるナフトキノン誘導体化合物が、下式(s)又は下式(u)で表される化合物であることを特徴とする請求項45に記載のイミペネム活性増強方法。
【化28】
【請求項47】
前記イミペネムの活性が抗菌活性であることを特徴とする請求項45又は46に記載のイミペネム活性増強方法。
【請求項48】
前記抗菌活性がMRSA(メチシリン耐性黄色ブドウ球菌)に対する抗菌活性であることを特徴とする請求項47に記載のイミペネム活性増強方法。
【請求項49】
下記の一般式(VII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩の活性を増強するナフトキノン誘導体化合物活性増強方法であって、
前記ナフトキノン誘導体化合物と協同させてイミペネムを作用させることを特徴とするナフトキノン誘導体化合物活性増強方法。
【化29】
(式中、R16はヒドロキシル基又はアルコキシル基であり、R17は水素原子又はハロゲン原子であり、R18はヒドロキシル基又はアルコキシル基である。)
【請求項50】
前記一般式(VII)で表されるナフトキノン誘導体化合物が、下式(s)又は下式(u)で表される化合物であることを特徴とする請求項49に記載のナフトキノン誘導体化合物活性増強方法。
【化30】
【請求項51】
前記活性が抗菌活性であることを特徴とする請求項49又は50に記載のナフトキノン誘導体化合物活性増強方法。
【請求項52】
前記抗菌活性が、MRSA(メチシリン耐性黄色ブドウ球菌)に対する抗菌活性であることを特徴とする請求項51に記載のナフトキノン誘導体化合物活性増強方法。
【請求項53】
下記の一般式(VIII)で表されるナフトキノン誘導体化合物若しくは下式(r)、下式(v)、若しくは下式(w)で表される化合物、又はその薬理学的に許容される塩を作用させることを特徴とする細菌の増殖阻害方法。
【化31】
(式中、R19及びR20はアルコキシル基である。)
【請求項54】
前記一般式(VIII)で表されるナフトキノン誘導体化合物が、下式(s)で表される化合物であることを特徴とする請求項53に記載の細菌の増殖阻害方法。
【化32】
【請求項55】
さらに、イミペネムを協同して作用させることを特徴とする請求項53又は54に記載の細菌の増殖阻害方法。
【請求項56】
下式(u)で表される化合物又はその薬理学的に許容される塩、及びイミペネムを協同して作用させることを特徴とする細菌の増殖阻害方法。
【化33】
【請求項57】
前記細菌がMRSA(メチシリン耐性黄色ブドウ球菌)であることを特徴とする請求項53〜56のいずれかに記載の細菌の増殖阻害方法。
【図1】


【図2】
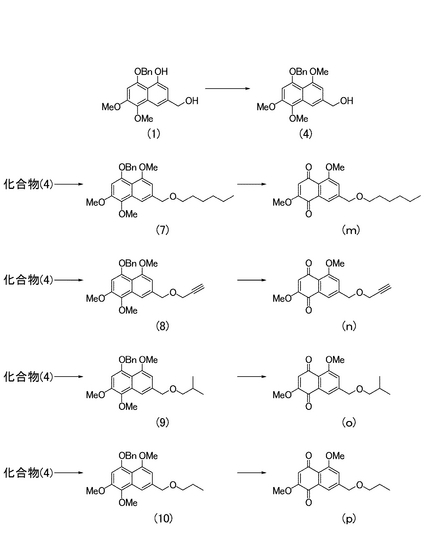
【図3】
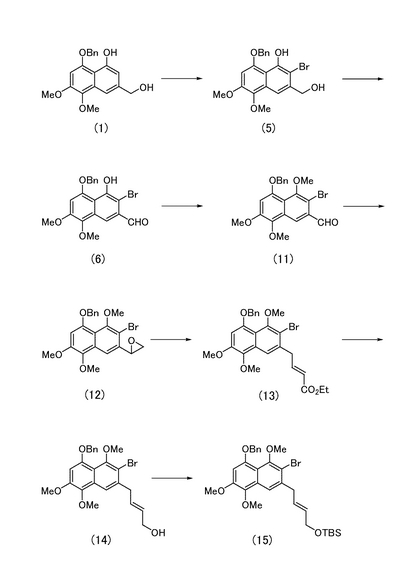
【図4】
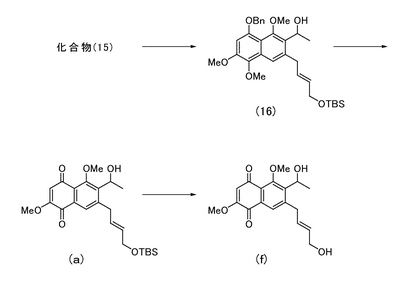
【図5】
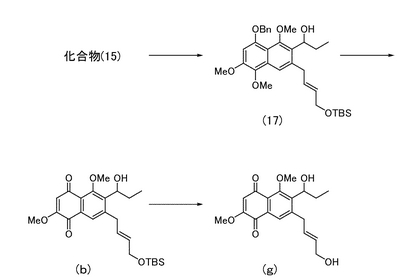
【図6】
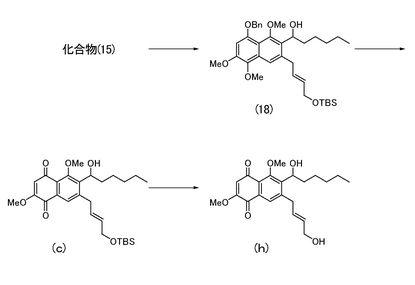
【図7】
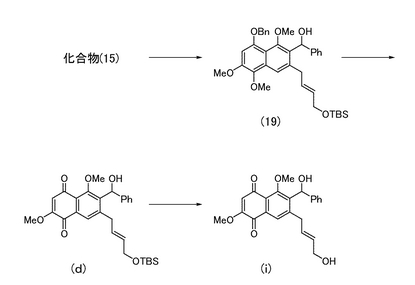
【図8】
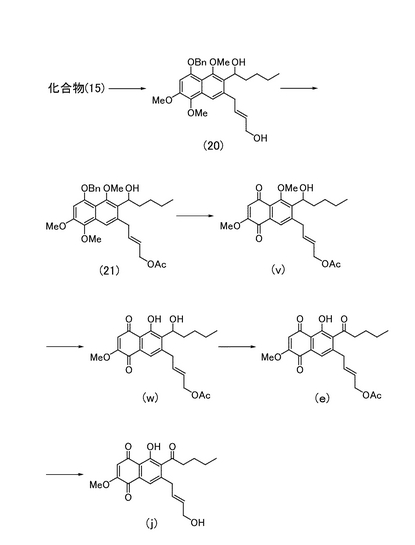
【図11】
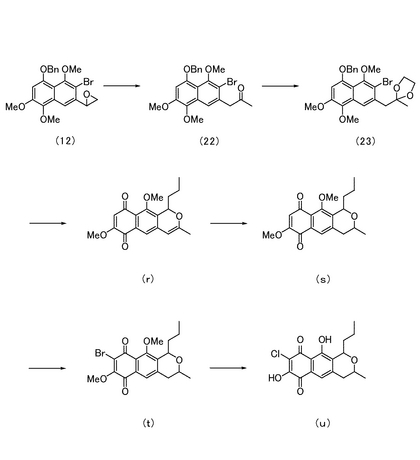
【図9】
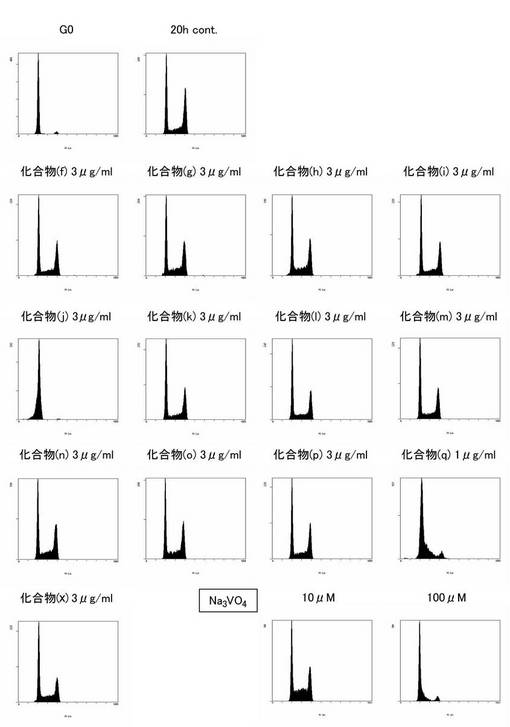
【図10】
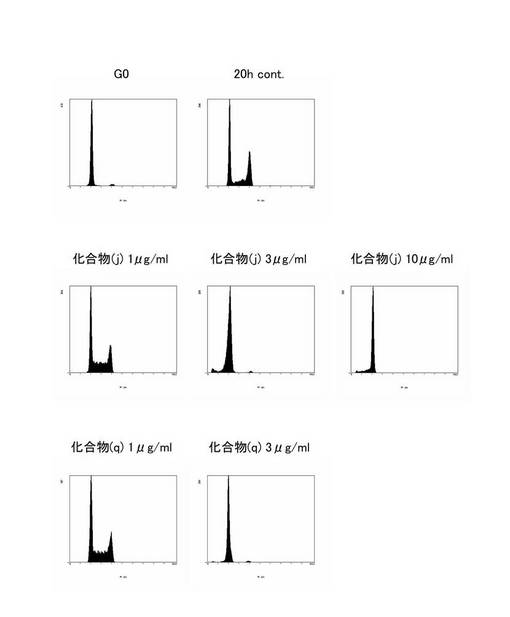
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図11】
【図9】
【図10】
【公開番号】特開2006−316001(P2006−316001A)
【公開日】平成18年11月24日(2006.11.24)
【国際特許分類】
【出願番号】特願2005−140550(P2005−140550)
【出願日】平成17年5月13日(2005.5.13)
【出願人】(899000079)学校法人慶應義塾 (742)
【Fターム(参考)】
【公開日】平成18年11月24日(2006.11.24)
【国際特許分類】
【出願日】平成17年5月13日(2005.5.13)
【出願人】(899000079)学校法人慶應義塾 (742)
【Fターム(参考)】
[ Back to top ]