
ネコ由来β2ミクログロブリンの測定方法および測定キット、ならびに、そのための抗体および抗体産生細胞株
【課題】より高感度でネコ由来のβ2ミクログロブリンに特異的な抗体の組み合わせによる測定方法およびキットを提供する。
【解決手段】細胞株Mouse-Mouse hybridoma β2-m mAb3により産生された第1の抗体と、細胞株Mouse-Mouse hybridoma β2-m mAb4により産生された第2の抗体とを用いて、酵素免疫測定法、蛍光免疫測定法、放射性同位体免疫測定法、免疫比濁法およびラテックス免疫比濁法からなる群より選択されるいずれかの方法により、検体中のネコ由来β2ミクログロブリン濃度を測定する方法、ならびに、細胞株Mouse-Mouse hybridoma β2-m mAb3により産生された第1の抗体と、細胞株Mouse-Mouse hybridoma β2-m mAb4により産生された第2の抗体とを含む、ネコ由来β2ミクログロブリン濃度測定用キット。
【解決手段】細胞株Mouse-Mouse hybridoma β2-m mAb3により産生された第1の抗体と、細胞株Mouse-Mouse hybridoma β2-m mAb4により産生された第2の抗体とを用いて、酵素免疫測定法、蛍光免疫測定法、放射性同位体免疫測定法、免疫比濁法およびラテックス免疫比濁法からなる群より選択されるいずれかの方法により、検体中のネコ由来β2ミクログロブリン濃度を測定する方法、ならびに、細胞株Mouse-Mouse hybridoma β2-m mAb3により産生された第1の抗体と、細胞株Mouse-Mouse hybridoma β2-m mAb4により産生された第2の抗体とを含む、ネコ由来β2ミクログロブリン濃度測定用キット。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ネコ由来のβ2ミクログロブリンの測定方法および測定キット、ならびに、そのための抗体および抗体産生細胞株に関する。
【背景技術】
【0002】
近年、少子化に伴い、ペットを飼う世帯は増加の一途をたどっている。しかしながら、ペットの性質に即した飼い方がなされていないケースも少なくはない。特に、偏食の結果、ペットが糖尿病などの成人病的症状を引き起こしてしまい、ペットを獣医に通院させるケースまで見られる。
【0003】
このような現状から、近年はペットの診断に関する事業が拡大しつつある。仮に、ペットの腎症を早期に発見することができれば、獣医師は、飼い主によるペットの飼い方、特に食事の与え方について改善を指導できるようになる。一般に、腎症のマーカーの1つとして、β2ミクログロブリン(β2-m)が挙げられる。
【0004】
β2ミクログロブリンは、たとえばヒト由来の場合には、ヒトの全身の細胞で産生されており、細胞内外の環境変化にはほとんど影響を受けないで一定の産生量で細胞外に分泌され、近年では、糖尿病性腎症などの早期診断の指標として有用であるとする報告例がみられる。
【0005】
しかしながら、ネコ由来のβ2ミクログロブリンに関しては、当該タンパクに特異的な抗体が存在しないどころ、当該タンパクのアミノ酸配列すら解明されていないのが現状である。
【0006】
本発明者は、ネコ由来のβ2ミクログロブリンのアミノ酸配列を解明すると共に、当該タンパクに特異的に結合するモノクローナル抗体を産生することに成功し、特許出願を行っている(特願2009−286712、本明細書中において「先願」と呼称する)。
【0007】
しかしながら、これらの抗体は、高感度でネコ由来のβ2ミクログロブリンの濃度を測定するには十分ではなかった。
【先行技術文献】
【非特許文献】
【0008】
【非特許文献1】Journal of Veterinary Internal Medicine. 22(5):1111-1117, 2008
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、上記課題を解決するためになされたものであって、その目的とするところは、より高感度でネコ由来のβ2ミクログロブリンに特異的な抗体の組み合わせによる測定方法およびキット、ならびに、そのための抗体および抗体産生細胞株を提供することである。
【課題を解決するための手段】
【0010】
本発明者は、鋭意研究の結果、この抗体を産生する細胞を取得における、マウスの取得箇所等により、ネコ由来のβ2ミクログロブリンの抗体への結合力が異なる点を発見し、本発明を完成するに至った。すなわち、本発明は以下のとおりである。
【0011】
本発明は、細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)により産生された第1の抗体と、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生された第2の抗体を用いてサンドイッチ酵素免疫測定法により、検体中のネコ由来β2ミクログロブリン濃度を測定する方法を提供する。
【0012】
本発明は、さらに、細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)により産生された第1の抗体と、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生された第2の抗体とを含む、ネコ由来β2ミクログロブリン濃度を測定するキットについても提供する。
【0013】
また本発明は、新規な抗体産生細胞株である細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)についても提供する。
【0014】
本発明はさらに、配列番号1で表わされるアミノ酸配列を有するタンパク質を抗原とし、細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)または細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生されたものである、ネコ由来β2ミクログロブリンに特異的に結合する抗体についても提供する。
【発明の効果】
【0015】
本発明によれば、より確実に検体中のネコ由来β2ミクログロブリン濃度を高感度で測定することができるようになる。
【図面の簡単な説明】
【0016】
【図1】参考例1において、アニーリング温度を変えた条件でPCRを行い、電気泳動を行った結果を示す写真である。
【図2】参考例1において、形質転換された大腸菌から抽出したpcDNA-Fβ2mをアガロースゲル電気泳動した結果を示す写真である。
【図3】参考例1において、pcDNA-Fβ2mのシークエンス解析結果を模式的に示す図である。
【図4】参考例1におけるSDS-PAGEの結果を示す写真である。
【図5】参考例1における、GST融合タンパク溶液の尿素濃度ごとのクロマトグラムを模式的に示す図である。
【図6】参考例1において、各尿素濃度のリガンド結合分画について、SDS−PAGEで比較した結果を示す写真である。
【図7】参考例1おいて、リガンド結合分画に各濃度でDTTを添加し、透析した後、PreScission Proteaseを反応させた結果を示す写真である。
【図8】参考例1におけるHPLCのクロマトグラムを模式的に示す図である。
【図9】HPLC後の各分画についてのSDS−PAGEの結果を示す写真である。
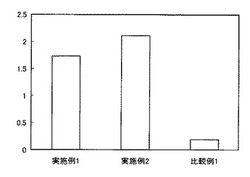
【図10】実施例1〜4および比較例1の酵素免疫測定法(サンドイッチ法)による測定結果を示すグラフであり、左側の縦軸は吸光度を示している。
【発明を実施するための形態】
【0017】
本発明によれば、細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)により産生された第1の抗体と、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生された第2の抗体とを用いてサンドイッチ酵素免疫測定法により、検体中のネコ由来β2ミクログロブリン濃度を測定する方法が提供される。
【0018】
ここで、検体とは、ネコ由来の血液、尿、骨髄液およびネコ由来細胞の懸濁液の他、ネコの血液透析で用いた透析液も含まれる。
【0019】
また、ネコ由来β2ミクログロブリンとは、配列番号1で示されるアミノ酸配列を有するタンパク質をいう。本発明者は、ネコの遺伝子の中でβ2ミクログロブリンをコードする構造遺伝子(本明細書中において「β2-m遺伝子」と呼称する。)を初めて特定し、当該β2-m遺伝子によりネコ由来のβ2ミクログロブリン(本明細書中において「β2-m」と呼称する。)のアミノ酸配列も初めて解析し、特許出願をしている(特願2009−286712)。配列番号1に示されるアミノ酸配列を有する本発明のタンパク質が、本発明者によって初めてアミノ酸配列が特定されたネコ由来のβ2-mである。
【0020】
また、本発明者は、このネコ由来β2ミクログロブリンを抗原とする抗体を産生する細胞の作製に成功し、独立行政法人 産業技術総合研究所 特許生物寄託センターに寄託を行っている(受託番号:FERM P-21879、FERM P-21880)。本発明は、上述の抗体よりもより好感度にβ2-mを測定することに成功し、改めて、平成23年1月12日付けで独立行政法人 産業技術総合研究所 特許生物寄託センターに寄託を行ったものである(受領番号:FERM AP-22051、受領番号:FERM AP-22052)。本発明は、この新規な抗体産生細胞株である細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)についても提供する。
【0021】
また本発明は、配列番号1で表わされるアミノ酸配列を有するタンパク質(ネコ由来のβ2-m)を抗原とし、細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)により産生されたものである、ネコ由来β2ミクログロブリンに特異的に結合する抗体(第1の抗体)、ならびに、配列番号1で表わされるアミノ酸配列を有するタンパク質(ネコ由来のβ2-m)を抗原とし、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生されたものである、ネコ由来β2ミクログロブリンに特異的に結合する抗体(第2の抗体)についても提供する。
【0022】
本発明の第1の抗体および第2の抗体は、酵素免疫測定法(サンドイッチ法)、蛍光免疫測定法(サンドイッチ法)、放射性同位体免疫測定法(サンドイッチ法)、免疫比濁法およびラテックス免疫比濁法の2つの抗体として用いられる。
【0023】
酵素免疫測定法(サンドイッチ法)は、タンパク定量における代表的な測定方法であり、当業者であれば容易に実践できる測定方法である。酵素免疫測定法(サンドイッチ法)は、基本的には、固相に固定する抗体(捕獲抗体)と、当該捕獲抗体に結合した抗原に対してさらに結合する抗体(一次抗体)の2種類を用いる。この上で、発色または発光反応を行うための酵素標識が一次抗体になされている。または、一次抗体に酵素標識がなされていない場合は、一次抗体にさらに結合可能な抗体であって、酵素標識がなされている抗体(二次抗体)を用いてもよい。二次抗体としては、IgGおよびIgMなどが利用できる可能性がある。
【0024】
なお、ここでいう酵素標識とは、発色または発光反応に利用可能な酵素を直接一次抗体に標識する態様の他、ある特定の結合様式を介して反応時に間接的に酵素を修飾する態様も含むことは、当業者であれば常識である。特定の結合様式としては、例えば、アビジン−ビオチン結合が代表的である。より詳しく説明すると、ビオチンを修飾の一次抗体と、アビジン修飾ペルオキシダーゼの組み合わせがこの態様に該当し、アビジン−ビオチン結合という結合様式を介してペルオキシダーゼは間接的に一次抗体に修飾している。
【0025】
一次抗体等に標識される酵素は、当業者が適宜選択することができ、ペルオキシダーゼの他、アルカリホスファターゼ、ルシフェラーゼおよびβ−ガラクトシダーゼ等を用いることができる。酵素に用いる基質についても、用いる酵素の種類に応じて当業者が適宜選択することができ、例えば、酵素がペルオキシダーゼの場合、テトラメチルベンジリン、2,2’−アジノビス(3−エチルベゾチアゾリン−6−スルホン酸)アンモニウム塩、4−アミノアンチピリン、4−アミノアンチピリン塩酸塩、5−アミノサリチル酸、2,4−ジクロロフェノール、N,N−ジメチル−m−トルイジンおよびN,N−ジメチルアニリン等が挙げられ、酵素がアルカリフォスファターゼの場合、ブルーテトラゾリウム、5−ブロモ−4−クロロ−3−インドリルリン酸2ナトリウム塩、5−ブロモ−4−クロロ−3−インドリルリン酸p−トルイジン塩およびニトロブルーテトラゾリウム等が挙げられる、酵素がルシフェラーゼの場合、D−(−)−ルシフェリンが挙げられ、酵素がβ−ガラクトシダーゼの場合、5−ブロモ−4−クロロ−3−インドリルβ−D−ガラクトシダーゼ、5−ブロモ−6−クロロ−3−インドリルβ−D−ガラクトシダーゼ、5−ブロモ−3−インドリルβ−D−ガラクトシダーゼおよび6−クロロ−3−インドリルβ−D−ガラクトシダーゼ等が挙げられる。
【0026】
本発明の第1の抗体は、捕獲抗体、一次抗体のいずれとして用いられてもよく、限定されるものではない。換言すれば、第2の抗体は、第1の抗体が捕獲抗体である場合は一次抗体であり、第1の抗体が一次抗体である場合は捕獲抗体となる。この点は後述する実施例においても実証されている。
【0027】
また、本発明の第1の抗体および第2の抗体が酵素免疫測定法(サンドイッチ法)で測定できるということは、当業者であれば、蛍光免疫測定法(サンドイッチ法)、放射性同位体免疫測定法(サンドイッチ法)、免疫比濁法およびラテックス免疫比濁法にも応用することができる。例えば、測定法として、蛍光免疫測定法(サンドイッチ法)を用いる場合は、上述の酵素免疫測定法(サンドイッチ法)における酵素標識の代わりに蛍光標識を、また、放射性同位体免疫測定法(サンドイッチ法)を用いる場合は、上述の酵素免疫測定法(サンドイッチ法)における酵素標識の代わりに放射性同位体標識を適用すればよい。また、免疫比濁法およびラテックス免疫比濁法に至っては、反応系においてこれら2つの抗体を用いればよいだけのことである。ラテックス免疫比濁法におけるラテックスの材料も、当業者であれば適宜選択できるものであるが、一般的に用いられているポリスチレンを用いればよい。
【0028】
本発明は、さらに上述した本発明の第1の抗体、第2の抗体を利用した測定診断キットについても提供するものである。本発明の診断キットは、上述の測定法に応じて、当該測定が利用できる内容で組み合わされるものであり、例えば、測定法として酵素免疫測定法(サンドイッチ法)を選択する場合、本発明の抗体以外に、ウェル、色原性基質溶液、反応停止液、洗浄液、標準溶液などを含むよう設計される。
【実施例】
【0029】
以下、参考例、実施例および実験例を挙げて本発明をより詳細に説明するが、本発明はこれらに限定されるものではない。
【0030】
<参考例>
以下、参考例としてネコ由来β2-mの合成と、当該β2-mを用いた抗体産生ハイブリドーマの作製、ならびに本発明の抗体の作製を示す。これら参考例は、実質的に先願の手法と同一である。
【0031】
<参考例1:ネコ由来β2-mの合成>
(1)プライマーの設計
以下の上流側プライマーおよび下流側プライマーを設計した。ベクターとしてpGEX-6P-1(GEヘルスケアバイオサイエンス)を用いるため、上流側プライマーの5’末端にBam HI、下流側プライマーの5’末端にSal Iの制限酵素認識配列を加え、それぞれ以下の塩基配列となるように作製した。なお、NはT、A、CまたはGを示している。
【0032】
・上流側プライマー:5'-NNNGGATCCGTCCAGCATTCCAAAGGTTCAGGT-3'(配列番号2)
・下流側プライマー:5'-NNNGTCGACTTACATGTCTCGATCCCACTTAACGACCTT-3'(配列番号3)
(2)PCR法
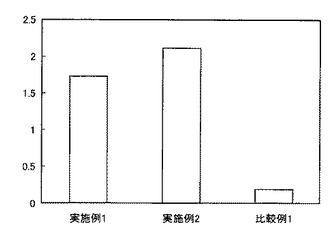
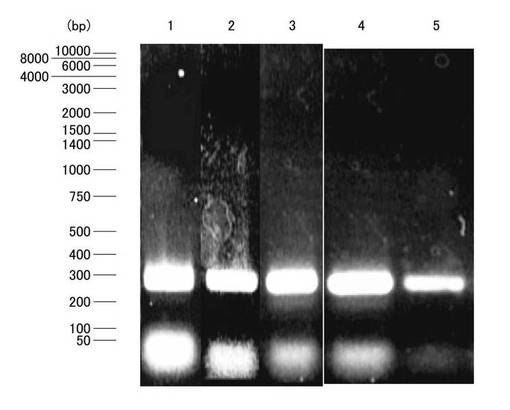
上述のfirst strand cDNA 4μl、GoTaqR Green Master Mix(Promega)12.5μl、上流側プライマー1μl、下流側プライマー1μlおよびRNase free H2O 6.5μlをPCRチューブ内で混和し、サーマルクライマーを用いてPCRを行った。PCRの条件は95℃2分間加温後、95℃で45秒間、primer pairのアニーリング温度で45秒間、72℃で1分間のサイクルを35サイクル、その後72℃で7分間のプログラムとした。また、PCRの最適な条件を見つけるため、アニーリング温度の検討を行った。アニーリング温度が、77.5℃で行ったRT-PCRでは、目的とするβ2-m cDNAと思われる約300bpのバンドの他に約50bpのバンドが出現した。ここで、図1は、アニーリング温度を変えた条件でPCRを行い、2% アガロースゲルで電気泳動を行った結果を示す写真であり、レーン1〜3はそれぞれアニーリング温度を77.5℃、80℃、85℃とした場合、レーン4、5はプライマーの添加量を変えた場合である。この約50bpのバンドは、アニーリング温度が77.5℃、80℃および85℃と高くなるにつれて減少したが、消失しなかった。したがって、アニーリング温度を85℃に設定しプライマーの添加量を変え、レーン5ではプライマーの添加量を半分に落としたところ、約50bpのバンドが少なくなり、β2-m cDNAのバンドと思われる約300bpのほぼ単一なバンドのPCR産物が得られ、ベクタープラスミドへの挿入cDNAとした。
【0033】
(3)ゲルからのDNA抽出
QIAquick Gel Extraction Kit(QIAGEN)を用いて、添付のプロトコールに従ってゲルからのDNA抽出を行った。アガロースゲルの目的のDNAバンドを切り出し、ゲルの重量を測定した。切り出したゲルを、3倍量のQGバッファーを添加され50℃の恒温槽(TR-2A、ASONE)内で10分間加温し、ゲルを完全に溶解させた後、ゲルと同量のイソプロパノールを加え、よく混和させた。DNA溶液を、キットに付属のカラムのセットされた2mlのコレクションチューブに加え、室温で13400×g、1分間遠心分離した。その後、コレクションチューブ内の濾液を捨てた後、再びカラムに0.75mlのPEバッファーを添加し、室温で15700×g、1分間遠心分離にて洗浄後、濾液を除去し、さらに1分間遠心分離した。その後、カラムを新しい1.5mlのマイクロチューブにセットし、EBバッファー 50μlを加えた後、室温で1分間放置し、15700×gで1分間の遠心分離により抽出液を回収し、得られた溶液をDNA抽出溶液とした。
【0034】
(4)PCR産物の濃縮
DNA抽出溶液をフェノールと等量混合後、15700×gで5分間遠心分離後、核酸を含む水層を分離した。その水層に、クロロホルムを等量混合し、15700×gで5分間遠心分離させた後、上清を分離した。次に分離後の溶液は、2.5倍量の100%エタノールが加えられ−80℃で30分静置し、その後15700×gで5分間遠心分離、上清を除去し、沈渣を得た。沈渣に70%エタノールを添加し、15700×gで5分間遠心分離後、上清を除去し、PCR産物の濃縮試料とした。
【0035】
(5)β2-m cDNA組み込みベクターの作成および大腸菌の形質転換法
PCR産物の濃縮試料を、BamHI(タカラバイオ株式会社)5μl、SalI(タカラバイオ株式会社)5μl、H.Buffer(500mM Tris-HCl、pH7.5、100mM MgCl2、10mM Dithiothreitol、1000mM NaCl)5μlおよびRNase free H2O 35μlに混和させた。また、pGEX6P-1 5μl(2.5μg)を、BamHI 5μl、SalI 5μl、 H.Buffer 5μlおよびRNase free H2O 30μlと混和させた。各溶液を、37℃で1晩インキュベートすることで制限酵素処理を施した後、アガロースゲル電気泳動を行い、QIAquick Gel Extraction Kitを使用して、各DNAバンドの抽出を行った。ライゲーションはDNA Ligation Kit(タカラバイオ株式会社)を用いて行った。すなわち、5μlのLigation Mixと1μlの制限酵素処理されたβ2-m cDNA溶液および4μlのpGEX6P-1が混和され、16℃で1晩静置された。そして、この反応溶液を用いて、β2-m cDNAのトランスフェクションを行った。反応溶液2.5μlを、E.coli JM109 Competent Cells(タカラバイオ株式会社)25μlに添加し氷上で30分間静置させ、次に42℃の恒温槽で45秒間Heat-Shockを与え、直ちに2分間氷冷後SOC培地(2% tryptone、0.5% Yeast extract、10mM NaCl、2.5mM KCl、10mM MgSO4、10mM MgCl2、20mM glucose)250μlを緩やかに加え、37℃1時間保温された。β2-m cDNAトランスフェクト溶液100μlをアンピシリン加LB培地にそれぞれ塗布し、37℃で1晩静置後、コロニーを釣菌し、アンピシリン加LB液体培地1.2mlに混和後、37℃で1晩培養した。培養後の液体培地を13400×gで1分間遠心分離後、上清を完全に除去し、得られた沈渣からプラスミド抽出を行った。
【0036】
(6)プラスミド抽出
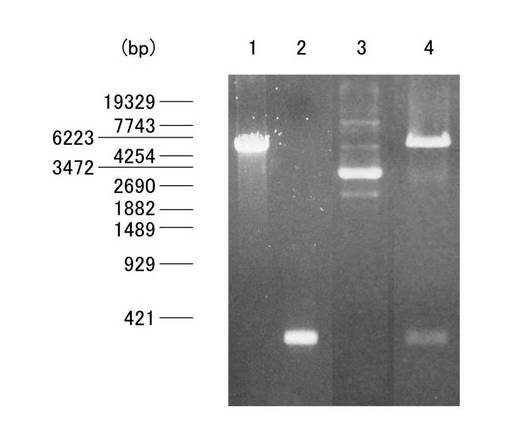
QIAPrep Spin Mini Kit50(QIAGEN)を用い、添付のプロトコールに従ってプラスミド抽出を行った。具体的には、まず、上述の得られた沈渣を、buffer P1 250μlで溶解させた後、buffer P2 250μl を添加し、その後緩やかに転倒混和させることで溶菌させた。中性化N3 buffer 350μlの添加により溶菌反応を停止させた後、15700×gで10分間遠心分離し、上清を付属のカラム付きコレクションチューブに添加した。カラムを、5900×gで1分間遠心分離した後、濾液を除去し、Binding Buffer 500μlを加えた。続いて、9300×gで1分間遠心洗浄後、濾液を除去し、エタノール含有脱塩buffer 750μlを加え、9300×gで1分間遠心洗浄後、新しいチューブに移した。カラムのメンブレンに、Elution Buffer 50μlを添加し、9300×gで1分間遠心分離後、プラスミド抽出溶液を得、このプラスミドをpcDNA-Fβ2mとし、アガロースゲル電気泳動法により確認した。図2は、形質転換された大腸菌から抽出したpcDNA-Fβ2mをアガロースゲル電気泳動した結果を示す写真である。図2において、レーン1、2は、それぞれ制限酵素(BamHIおよびSal1)で処理されたプラスミド(pGEX6p-1)およびβ2-m cDNAであり、レーン3はpcDNA-Fβ2m、レーン4はpcDNA-Fβ2mを制限酵素(BamHIおよびSal1)で処理した試料についての結果を示している。図2に示されるように、約3000bpに太いバンドが、約8000bp、約5000bpおよび約2000bpに薄いバンドが確認された。また、抽出後のpcDNA-Fβ2mをBamH1およびSal1で制限酵素処理を行った試料を泳動したレーン4では、約5000bpと約300bpのバンドが確認された。この約5000bpと約300bpの二つのバンドは、形質導入前のβ2-m cDNAおよびpGEX6P-1の泳動結果と比較するとほぼ同じ分子量のバンドであった。また、pcDNA-Fβ2mのサブクローニングの成否は、T7プライマーを用いてDye Deoxy Terminator Cycle Sequencing kit(Applied Biosystems)およびApplied Biosystems 3130xl Genetic Analyzer(Applied Biosystems)を使用したシークエンス解析により行った。図3は、pcDNA-Fβ2mのシークエンス解析結果を模式的に示す図である。図3に示す結果から、組み込んだβ2-m cDNAの塩基配列が正しくベクターに組み込まれていたことが確認された。
【0037】
(7)GST融合タンパク発現の確認
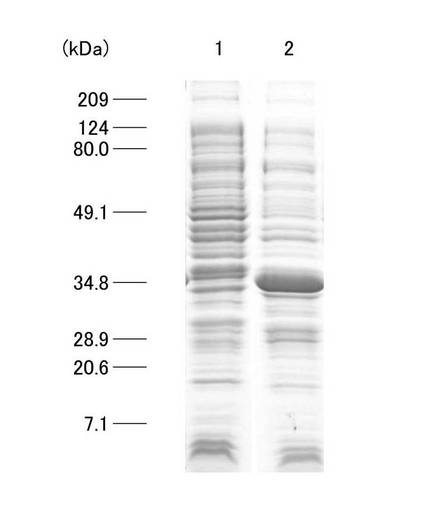
トランスフェクトされた大腸菌を、37℃、1晩LB培地で培養後、100μlをIsopropl-β-D-thiogalactopyranoside(IPTG:0.1mM)20μlと混和させ、37℃で約2時間振盪培養(BR40-LF、TAITEC)した。振盪培養後の大腸菌溶液を、15700×gで1分間遠心分離後、上清を除去し、沈渣に可溶化液(50mM Tris-Hcl 50μl、1×RIPA Lysis Buffer(Up State)100μl、Protease Inhibitor 140μl、H2O 710μl)30μlを加え可溶化後、15700×gで5分遠心分離し、上清と沈渣に分けた。上清30μlに、2×SB溶液(2% SDS、40% Glycerol、0.6% BPB、25mM Tris-HCl Buffer(pH6.8、20℃))30μl、2ME 1μlを加え、95℃で3分間加温した。沈渣に、SB溶液20μlを加え、超音波破砕機(UR-20P、TOMY SEIKO CO,LTD)で5秒間破砕後、95℃で3分間加温した。その後、上清および沈渣について、SDS−PAGEにてGST融合タンパク質発現の確認およびGST融合タンパクの大腸菌での溶解性を確認した。GST融合タンパク誘導発現後に得られた大腸菌をソニケーションし、遠心分離して得られた上清および沈渣について、SDS−PAGEにて泳動させた。図4は、SDS−PAGEの結果を示す写真であり、レーン1は上清、レーン2は沈渣についての結果を示している。GST融合タンパクの分子量は、約37kDaであり、上清には顕著なバンドの確認はできなかったが、沈渣では、明瞭な太いバンドが確認された。そのため、GST融合タンパクは、不溶性画分に発現したことが確認された。
【0038】
(8)SDS−PAGE法
コンパクトPAGE(AE-7300、ATTO)を用いてLaemmliの方法に準拠し、これに以下に示す修正を加えてSDS−PAGEを実施した。具体的には、分離ゲルの組成は、15% acrylamide、0.2% N,N-methylene-bis-acrylamide、0.1% SDS、375mM Tris-HCl buffer(pH8.8, 20℃)とした。ゲルは2・4連ゲル作製器(AE-7344、ATTO)を用いて作製した。電極緩衝液の組成は、0.1% SDS、129mM glycine、25mM Tris(pH8.3、20℃)とした。泳動用試料(SB)の組成は、1% SDS、20% glycerol、0.3% BPB、12.5mM Tris-HCl Buffer(pH6.8、20℃)とした。また、マーカーとして、プレステインドSDS−PAGEスタンダード(Broad)マーカー(BIO-RAD)もしくはSDS−PAGEスタンダード(Broad)マーカー(BIO-RAD)が用いられた。泳動は、Tris-Gly/PAGE Highモードで30分泳動させた後、Tris-Gly/PAGE Lowモードにして、下部イオン界面をゲル下端から1〜2mm上方の位置に移動したときに終了した。SDS−PAGE終了後のゲルには、Oakley法に準拠した銀染色法を施した。すなわち、ゲルを30% ethanol、10% acetic acid溶液にて固定後、洗浄し、20 % ethanolに5分間2回浸漬させた。20% ethanol除去後、5% glutaraldehyde溶液にて4分間反応させ、純水で洗浄後、20% ethanolに4分間2回浸漬させた。その後、純水で洗浄し、アンモニア性硝酸銀溶液にて5分間反応させ、純水で洗浄後、0.005% citric acid、0.019% formaldehyde溶液で発色させた。発色確認後のゲルは、20% ethanol、10% acetic acid溶液にて5分間固定させ、20% ethanolに5分間2回浸漬後、写真を撮影した。なお、銀染色法はすべて遮光条件下にて実施した。
【0039】
(9)GST融合タンパク質の発現誘導と単離
GST融合タンパク質の発現が確認された大腸菌をアンピシリン加LB寒天培地に塗布し、コロニーを釣菌後、3mlのアンピシリン加LB液体培地に加え1晩37℃で振盪培養した。続いて、その培養液3mlをアンピシリン加LB液体培地250mlに加え、37℃で約150分振盪培養後、0.1mM IPTG 2.5mlを添加し、約2時間、37℃で振盪培養したGST融合タンパク質のタンパク発現後のタンパク組成を分析した。GST融合タンパク発現誘導後の培養液を6000×gで15分間遠心分離した沈渣に、20mlの0.5mM EDTA、0.4M NaCl、5mM MgCl2、5%グリセロール、0.5mM phenylmethylsulfonyl fluoride(PMSF)、1mM dithiothreitol(DTT)および1mg/mlリゾチーム加50mM Tris-HCl(pH8.0)を懸濁させ、4℃で1時間静置後、凍結融解を2回行った。続いて、Nonidet P-40を0.5%添加し、超音波破砕機で20秒間×5回破砕後、9300×gで20分間遠心分離し、上清を除去し、沈渣を得た。得られた沈渣を、10mlの8M Urea、0.5mM DTT加Phosphate buffer saline(PBS:140mM NaCl、2.7mM KCl、10mM Na2PO4、1.8mM KH2PO4、pH 7.3)に再懸濁後4℃で1時間静置し、9000×gで20分間遠心分離し、上清を得た。この上清をGST融合タンパク溶液とした。
【0040】
(10)アフイニティークロマトグラフィー法
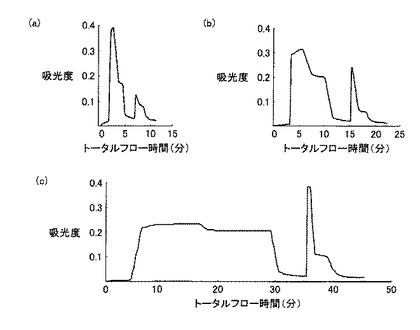
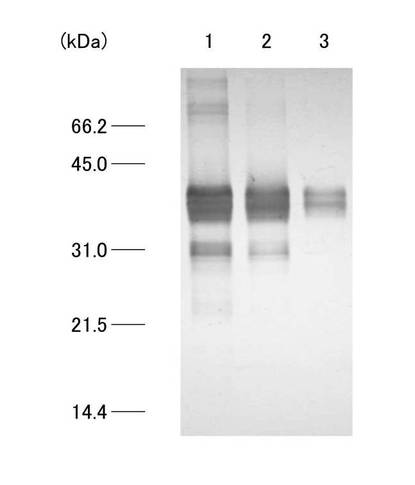
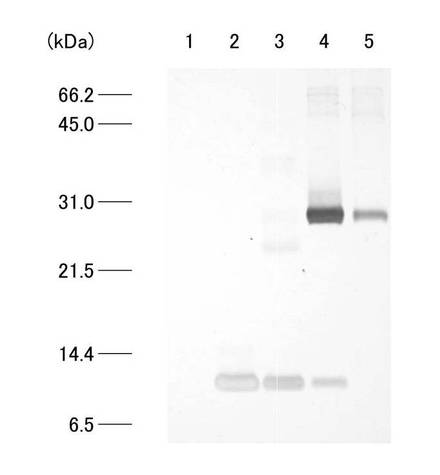
20mlのGST融合タンパク溶液を0.5M Urea 加PBSで平衡化させたGSTrap HP カラム(GEヘルスケアバイオサイエンス)にペリスタポンプ(SJ-1211L、ATTO)を用い、流速0.3ml/minで添加した。なお、カラムに添加するGST融合タンパク溶液について、尿素濃度の違いによるカラムへのGST融合タンパクの吸着量の比較を行った。カラムを、0.5M Urea加PBSで洗浄後、10mM reduced glutathione、1M Urea 加50mM Tris-HCl(pH8.0)で溶出させた。GST融合タンパク溶出液の吸光度を紫外部吸光度モニター(AC-5100L、ATTO)を用いて吸光波長220nmでモニターし、記録計(R-01A、RIKADENKI)で記録した。得られたGST融合タンパク溶出液2mlを、濃度が1mMになるようにDTTを添加し混和後、分子量13kDaカットの透析膜(UC30-32-100、三光純薬株式会社)に入れ、150mM NaCl、1mM EDTA加50mM Tris-HCl(pH7.5)2Lを用いて約6時間透析した。尿素濃度の違い(2M、1Mおよび0.5M)によるGST融合タンパク質のHiTrap affinity カラムへの結合量の比較を行った。各尿素濃度のGST融合タンパク溶液のクロマトグラムを図5(a)、(b)、(c)にそれぞれ示す。図5(a)、(b)、(c)は、それぞれ、尿素濃度2M、1Mおよび0.5Mのタンパク試料を用いたクロマトグラムを示している。各クロマトグラムは、前半部分に試料添加による吸光度変化が見られ、洗浄バッファーの添加により吸光度が減少した。その後、溶出バッファー添加によりシャープなピークが確認されたが、尿素濃度の減少に伴い吸光度の増加がみられ、尿素濃度0.5Mで最もシャープな分画が観察された。各尿素濃度のリガンド結合分画について、SDS−PAGEで比較した結果を図6に示す。図6において、レーン1は図5(c)で得られたリガンド結合分画、レーン2は図5(b)で得られたリガンド結合分画、レーン3は図5(a)で得られたリガンド結合分画についての結果が示されている。図6に示されるように、尿素濃度が高くなるに従いGST融合タンパク質の結合量の減少が確認され、尿素濃度0.5Mで行ったアフィニティークロマトグラフィーのリガンド結合分画(図5(c)で得られたもの)(以下、「C4分画」と呼称する)を以下の実験で用いた。
【0041】
(11)プロテアーゼ処理
透析後のGST融合タンパク溶出液について、DC Protein Assay(Bio-Rad)を用いてタンパク定量を行い、タンパク量200μgに対し、PreScission Protease(GE ヘルスケアバイオサイエンス)を1μl添加して混和させた後、4℃で6時間以上反応させ、高速液体クロマトグラフィー(HPLC)用の試料とした。PreScission Protease反応に及ぼす透析およびDTTの効果について検討を行った。また、C4分画を用いてPreScission Protease活性に与えるDTT濃度の影響を検討した。C4分画に最終濃度1mM、2.5mMおよび5mMになるようにDTTを添加し、透析後、PreScission Proteaseを反応させた結果を図7に示す。図7において、レーン1は1mM DTTを添加した場合、レーン2は2.5mM DTTを添加した場合、レーン3は5mM DTTを添加した場合をそれぞれ示している。いずれの濃度のDDT添加においても約11kDaのβ2-mのバンドは認められたが、1mMのDTT添加では約15kDaのバンドの消失、2.5mMおよび5mMのDTT添加では、β2-mのバンド以外に約13kDaのバンドが確認された。上記の結果より、次の精製ステップである高速液体クロマトグラフィー法(HPLC)では、1mMのDDTを添加したC4分画を透析後にPreScission Protease処理した試料を用いた。
【0042】
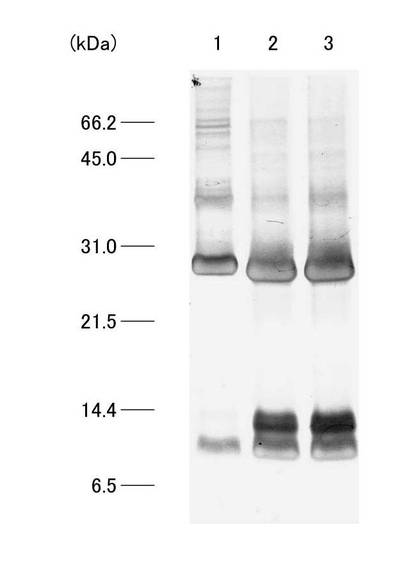
(12)HPLC法
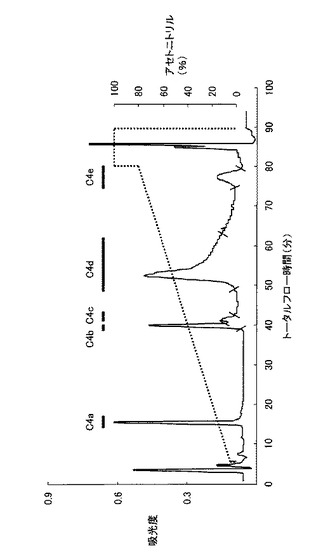
HPLCシステムは、システムコントローラー(SCL-10A VP、Shimadzu)、送液ユニット(LC-10AD VP、Shimadzu)、紫外部分光高度計(SPD-10A VP、Shimadzu)、カラムオーブン(CTO-10A VP、Shimadzu)および脱気ユニット(DGU-14A、Shimadzu)から構成され、カラムはMightysilRP-18 GP250-4.6(Cat.No.25415-96、関東化学)を使用した。HPLCの分離条件は、移動相の流速を1ml/min、試料添加量は400μlとし0.1%Trifluoroacetic Acid(TFA)溶液で平衡化させカラムを0.1%TFA加acetonitrile溶液を用いて、acetonitrile濃度0〜80%のライナーグラジエントで行った。なお、溶出液は、吸光波長220nmで吸光度をモニターし、検出されたピークを分取し、濃縮遠心機(CC-181、TOMY)にて1時間遠心分離後、凍結乾燥機(FDU-540、EYELA)にて乾燥させた後−20℃で保存した。また、溶出された各分画のタンパクを、SDS−PAGE法により分析した。図8は、HPLCのクロマトグラムを模式的に示す図である。プロテアーゼ処理後のC4分画は、主に5つの分画として溶出された。これらの5つの分画は溶出された順にC4a、C4b、C4c、C4dおよびC4eとし、SDS−PAGEにより分析した。図9は、HPLC後の各分画についてのSDS−PAGEの結果を示す写真である。図9に示すように、C4a分画ではバンドが確認されず、C4b分画では約11kDaのバンドが、C4c分画では約11kDa、約25kDa、約27kDaのバンドが、C4d分画では約11kDa、約27kDaのバンドが、C4e分画では約27kDaのバンドが確認された。目的とするβ2-mと思われるタンパク質は、C4b分画において単一のバンドとして検出された。また、C4b分画の溶出時のacetonitrile濃度は39.5%であった。この溶出液を組み換えネコβ2-mとして、濃縮遠心し、凍結乾燥後、−80℃で保存した。
【0043】
<参考例2:抗体産生ハイブリドーマ、抗rFeβ2-m抗体の作製>
参考例1で合成したタンパクを組み替え型ネコβ2-m(rFeβ2-m)の抗原としてモノクローナル抗体を作製するにあたり、まずは抗体産生ハイブリドーマを作製した。
【0044】
(1)抗体産生ハイブリドーマの作製
(1−1)免疫法
免疫法は、精製rFeβ2-mを抗原としてBalb/cマウスの後肢肉球(footpad)の皮下に注射することにより行った。免疫は5日間隔で4回行い、初回から第3回目までの免疫は抗原溶液100μl(1mg/ml)とアジュバントを等量混合させてエマルジョン化させた抗原液200μl(50μg/foot)を、また、最終免疫では抗原溶液20μl(10μg/foot)のみを用いて行った。また、アジュバントは初回免疫ではAdjuvant Complete Freund(和光純薬工業株式会社)を、第2回目から第3回目の免疫ではAdjuvant Incomplete Freund(和光純薬工業株式会社)を用いた。
【0045】
(1−2)細胞融合
最終免疫から3日後、膝窩リンパ節を摘出し、リンパ球を回収後、GenomONE-CF(石原産業株式会社)を用いて、細胞融合を行った。また、ミエローマ細胞としてはP3X63-Ag8.653(大日本住友製薬株式会社)を用いた。融合方法は添付のプロトコールに従って行った。具体的には、まず、リンパ球とミエローマ細胞とを細胞数が5:1の比率になるように混合し、1000rpm、4℃で5分間遠心した後、上清を除去した。そこに、氷冷した融合用緩衝液をリンパ球108cellsあたり1ml添加し、均一に懸濁した後、氷冷したHVJ-Envelope懸濁液を細胞混合液1mlあたり25μl添加した。細胞懸濁液を氷上で5分間静置した後、1000rpm、4℃5分間遠心し、上清を除去せずに細胞がペレット化した状態のまま37℃で15分間インキュベートした。
【0046】
インキュベート終了後、37℃に加温した増殖用培地をリンパ球108cells当たり50ml加え、懸濁後、96穴プレート(96 Well Cell Culture Plate:Greiner bio-one)に100μl/wellで播種した。なお、増殖用培地としてRPMI1640(Invitrogen)にペニシリンG(PG;明治製薬株式会社)10万IU/ml、ストレプトマイシン(SM;明治製薬株式会社)100mg/ml、7.5% Bri Clone(IL-6、ヒト、ブライクローン;Cat. No. BR-001、大日本住友製薬株式会社)、10% 非働化ウシ胎仔血清(FBS;株式会社ニチレイ)を加えたものを用い、添加、懸濁の際は穏やかに操作した。24時間培養後、培養培地を上記の増殖用培地に2% HAT(Invitrogen)を添加したHAT培地に交換した。
【0047】
(2)抗体産生ハイブリドーマのスクリーニング
得られたハイブリドーマについて、細胞融合から1週間後にELISA法を用いた一次スクリーニングを行い、この結果、反応陽性となったwellのハイブリドーマのみをWestern blotting法を用いた二次スクリーニングで確認した。
【0048】
(2−1)一次スクリーニング
rFeβ2-mを抗原としたELISA法を用いて、抗体産生ハイブリドーマの一次スクリーニングを行った。ELISAプレートとしては、96 Well ELISA Microplate(Greiner bio-one)を使用した。また、プレートの洗浄には自動洗浄機(Auto Mini Washer AMW-8、バイオテック株式会社)を用い、洗浄液としてはPBS(1.37M NaCl、27mM KCl、100mM Na2HPO4、18mM KH2PO4、pH7.4、25℃)を使用した。固相として、PBSで3μg/mlに調整したrFeβ2-mを50μl/wellでプレートに添加し、4℃で一晩反応させた。固相反応終了後、プレートの抗原液を捨て、ブロッキング液として0.5% Bovine Serum Albumin(BSA;和光純薬工業株式会社)を添加したPBSを150μl/wellで加え、37℃で60分間反応させた。ブロッキング反応終了後、プレートを1回洗浄し、一次抗体として各ハイブリドーマ培養の培養上精を50μl/wellで加え、37℃で60分間反応させた。一次抗体反応終了後、プレートを1回洗浄し、二次抗体として0.1% BSAを添加したPBSで1000倍に希釈したペルオキシダーゼ標識抗マウスIgG抗体(SIGMA-ALDRICH)を50μl/wellで加え、37℃で60分間反応させた。二次抗体反応終了後、プレートを3回洗浄し、基質液として0.04% o-フェニレンジアミン、0.04% H2O2を添加したPBSを150μl/wellで加え、室温、遮光下で30〜60分間反応された。基質反応終了後、3M H2SO4を反応停止液として50μl/wellで加え、1分間振盪後、Microplate Reader(Model 550、BIO-RAD)で波長490nmにおける吸光度を測定した。吸光度の高かった陽性wellの細胞を、24穴プレート(24 Well Cell Culture Plate;Greiner bio-one)に移して培養した。
【0049】
(2−2)二次スクリーニング
rFeβ2-mを抗原としたWestern blotting法で確認し、抗体産生ハイブリドーマの二次スクリーニングを行った。Lowryの方法に基づき、DC Protein Assay Kit(BIO-RAD)を用いて、Microplate Readerで波長655nmにおける吸光度を測定し、タンパク質を定量した。検量線はBSAを用いて作製した。Western blotting法はTowbinらの方法に準拠し、以下のように実施した。転写膜はポリビニリデンジフルオリド(PVDF)膜(BIO-RAD)を使用した。PVDF膜は100% methanolに10秒間、さらに転写用電極buffer(25mM Tris-HCl(pH8.3、20℃)、192mM glycine、5% methanol)に30分間浸潤し、泳動に供した。転写装置の組み立ては、陽極電極板上に下から順に濾紙(BIO-RAD)、PVDF膜、SDS−PAGE終了後のゲル、濾紙を重層し、その上に陰極電極板を固定した。なお、濾紙は予め電極bufferに2〜3分浸しておいた。転写条件は1.9mA/cm2の定電流で60分間とした。転写終了後のPVDF膜は10mM Tris-HCl(pH7.5、20℃)、140mM NaCl、0.01% Tween20(TBST)に0.5% BSAを加え、室温で60分間振盪し、ブロッキング操作を行った。ブロッキング終了後、TBSTで5分間、2回振盪洗浄し、一次抗体として細胞の培養上精を用い、室温で90分振盪反応させた。一次抗体反応終了後、TBSTで5分間、2回振盪洗浄した後、TBSTで1000倍希釈したペルオキシダーゼ標識抗マウスIgG抗体を、室温で60分間振盪反応させた。二次抗体反応終了後、TBSTで5分間、2回振盪洗浄し、0.06% 3,3-diaminobenzidine tetra-hydrochloride、0.03% H2O2、50mM Tris-HCl(pH7.6、20℃)を基質反応液として使用し、1〜5分間反応させた。基質反応終了後、水洗し反応を停止させた後、乾燥して保存した。反応陽性を示したハイブリドーマについては後述する限界希釈法によりクローニングを行った。
【0050】
(3)クローニング
ハイブリドーマのクローニングには限界希釈法を用いた。具体的には、スクリーニング後のハイブリドーマを2cells/100μlとなるようにHAT培地で希釈し、100μl/wellで96穴プレートに播種した。ハイブリドーマはセミコンフルエントになったところで24穴プレートに拡大培養し、再びセミコンフルエントになるまで培養した後、二次スクリーニングと同様にrFeβ2-mを抗原としたWestern blotting法で確認した。このクローニング操作を2回行った。また、ハイブリドーマを長期間継代培養することにより抗体産生能が減少するのを防ぐため、クローニング毎に細胞凍結保存液(セルバンカー(BLC-1)、十慈フィールド株式会社)を用いて保存した。
【0051】
(4)抗体産生ハイブリドーマの大量培養および抗rFeβ2-m・mAbの採取と精製
クローニングが終了したハイブリドーマを、浮遊細胞培養フラスコ(フィルタートップSCフラスコ250ml 75cm2;Greiner bio-one)を用いて大量培養した。なお、培養は37℃、5% CO2、5日間、CO2インキュベーター(十慈フィールド株式会社)で行い、培地としてはHAT培地を用いた。大量培養されたハイブリドーマを無血清RPMIで懸濁し、ヌードマウス(Balb/c-nu)の腹腔内に2×107cells/headで投与した。投与してから10〜20日後、腹水を採取した。ヌードマウスから採取した腹水を室温で1時間、あるいは4℃で一晩静置した後、3000rpm、4℃で5分間遠心し、腹水中のフィブリン、ハイブリドーマ、赤血球などを除去した。分離した上清を50%の硫安で塩析させた。具体的には、氷上で撹拌しながら上清と等量の飽和硫酸アンモニウム溶液を徐々に滴下し、滴下後さらに1時間撹拌した。これを10000rpm、4℃で10分間遠心し、沈殿物を20mM リン酸ナトリウムbuffer(pH7.0)に溶解した。塩析後のグロブリン溶液を、20mM リン酸ナトリウムbuffer(pH7.0)で平衡化したSephadex G-25 Fine(GEヘルスケアバイオサイエンス)カラム(内径1.5cm、長さ30cm)を用いて脱塩した。クロマトグラフィーの流速をペリスタポンプ(SJ-1211L、ATTO)で0.5ml/minに調節した。脱塩後のグロブリン溶液を、エコカラム(内径2.5cm、長さ10.0cm:BIO-RAD)に充填したProtein G Sepharose 4 Fast Flow(GEヘルスケアバイオサイエンス)を用いたアフィニティークロマトグラフィー法により精製した。具体的には、脱塩後のグロブリン溶液を20mM リン酸ナトリウムbuffer(pH7.0)で平衡化されたカラムに流速0.5ml/minで添加し、その後カラムを100mM glycine(pH3.0)で溶出させた。溶出液は直ちに10分の1量の1M Tris-HCl(pH9.0)で中和した。精製後の溶出液を50mM 酢酸アンモニウム(pH7.0)で平衡化させたSephadex G-25 Fineカラム(内径2cm、長さ30cm)で脱塩させた後、Freeze Dryer(FDU540、EYELA東京理化器械株式会社)を用いて凍結乾燥し、−20℃で保存した。
【0052】
<実施例1〜4、比較例1>
後述する実験例に用いる実施例1〜4および比較例1の抗体の組み合わせを表1に示す。
【0053】
【表1】

【0054】
表1中、β2-m mAb3産生抗体は、細胞株Mouse-Mouse hybridoma β2-m mAb3(受託番号:FERM AP-22051)により産生された抗体を指し、β2-m mAb4産生抗体は、細胞株Mouse-Mouse hybridoma β2-m mAb4(受託番号:FERM AP-22052)により産生された抗体を指し、β2-m mAb1産生抗体は、細胞株Mouse-Mouse hybridoma β2-m mAb1(受託番号:FERM P-21879)により産生された抗体を指し、β2-m mAb2 産生抗体は、細胞株Mouse-Mouse hybridoma β2-m mAb2(受託番号:FERM P-21880)により産生された抗体を指す。
【0055】
<実験例>
サンドイッチ酵素免疫測定法により、実施例1〜4および比較例1の評価を行った。具体的には、1匹の健常なネコの尿中のβ2-mの測定を行った。具体的には、捕捉抗体を0.5mg/wellで固相し、ブロッキングした後、1匹の健常なネコの尿を2時間反応させた。洗浄後、ビオチン標識した一次抗体を2時間反応させた後、アビジンペルオキシダーゼを1時間反応させ、tetramethylbenzidineを用いて基質反応を行い、450nmにおける吸光度の測定を行った。図10はその結果を示すグラフであり、縦軸は吸光度である。図10から明らかなように、実施例1〜4の抗体の組み合わせによる吸光度は、比較例と比較して大幅に高かった。この結果から、本発明の抗体の組み合わせは、ネコβ2-mに対してより高い結合力を示すことが明らかであり、高感度のネコβ2-mの測定が可能であることを示した。
【0056】
今回開示された実施の形態および実験例はすべての点で例示であって制限的なものではないと考えられるべきである。本発明の範囲は上記した説明ではなくて特許請求の範囲によって示され、特許請求の範囲と均等の意味および範囲内でのすべての変更が含まれることが意図される。
【技術分野】
【0001】
本発明は、ネコ由来のβ2ミクログロブリンの測定方法および測定キット、ならびに、そのための抗体および抗体産生細胞株に関する。
【背景技術】
【0002】
近年、少子化に伴い、ペットを飼う世帯は増加の一途をたどっている。しかしながら、ペットの性質に即した飼い方がなされていないケースも少なくはない。特に、偏食の結果、ペットが糖尿病などの成人病的症状を引き起こしてしまい、ペットを獣医に通院させるケースまで見られる。
【0003】
このような現状から、近年はペットの診断に関する事業が拡大しつつある。仮に、ペットの腎症を早期に発見することができれば、獣医師は、飼い主によるペットの飼い方、特に食事の与え方について改善を指導できるようになる。一般に、腎症のマーカーの1つとして、β2ミクログロブリン(β2-m)が挙げられる。
【0004】
β2ミクログロブリンは、たとえばヒト由来の場合には、ヒトの全身の細胞で産生されており、細胞内外の環境変化にはほとんど影響を受けないで一定の産生量で細胞外に分泌され、近年では、糖尿病性腎症などの早期診断の指標として有用であるとする報告例がみられる。
【0005】
しかしながら、ネコ由来のβ2ミクログロブリンに関しては、当該タンパクに特異的な抗体が存在しないどころ、当該タンパクのアミノ酸配列すら解明されていないのが現状である。
【0006】
本発明者は、ネコ由来のβ2ミクログロブリンのアミノ酸配列を解明すると共に、当該タンパクに特異的に結合するモノクローナル抗体を産生することに成功し、特許出願を行っている(特願2009−286712、本明細書中において「先願」と呼称する)。
【0007】
しかしながら、これらの抗体は、高感度でネコ由来のβ2ミクログロブリンの濃度を測定するには十分ではなかった。
【先行技術文献】
【非特許文献】
【0008】
【非特許文献1】Journal of Veterinary Internal Medicine. 22(5):1111-1117, 2008
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、上記課題を解決するためになされたものであって、その目的とするところは、より高感度でネコ由来のβ2ミクログロブリンに特異的な抗体の組み合わせによる測定方法およびキット、ならびに、そのための抗体および抗体産生細胞株を提供することである。
【課題を解決するための手段】
【0010】
本発明者は、鋭意研究の結果、この抗体を産生する細胞を取得における、マウスの取得箇所等により、ネコ由来のβ2ミクログロブリンの抗体への結合力が異なる点を発見し、本発明を完成するに至った。すなわち、本発明は以下のとおりである。
【0011】
本発明は、細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)により産生された第1の抗体と、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生された第2の抗体を用いてサンドイッチ酵素免疫測定法により、検体中のネコ由来β2ミクログロブリン濃度を測定する方法を提供する。
【0012】
本発明は、さらに、細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)により産生された第1の抗体と、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生された第2の抗体とを含む、ネコ由来β2ミクログロブリン濃度を測定するキットについても提供する。
【0013】
また本発明は、新規な抗体産生細胞株である細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)についても提供する。
【0014】
本発明はさらに、配列番号1で表わされるアミノ酸配列を有するタンパク質を抗原とし、細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)または細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生されたものである、ネコ由来β2ミクログロブリンに特異的に結合する抗体についても提供する。
【発明の効果】
【0015】
本発明によれば、より確実に検体中のネコ由来β2ミクログロブリン濃度を高感度で測定することができるようになる。
【図面の簡単な説明】
【0016】
【図1】参考例1において、アニーリング温度を変えた条件でPCRを行い、電気泳動を行った結果を示す写真である。
【図2】参考例1において、形質転換された大腸菌から抽出したpcDNA-Fβ2mをアガロースゲル電気泳動した結果を示す写真である。
【図3】参考例1において、pcDNA-Fβ2mのシークエンス解析結果を模式的に示す図である。
【図4】参考例1におけるSDS-PAGEの結果を示す写真である。
【図5】参考例1における、GST融合タンパク溶液の尿素濃度ごとのクロマトグラムを模式的に示す図である。
【図6】参考例1において、各尿素濃度のリガンド結合分画について、SDS−PAGEで比較した結果を示す写真である。
【図7】参考例1おいて、リガンド結合分画に各濃度でDTTを添加し、透析した後、PreScission Proteaseを反応させた結果を示す写真である。
【図8】参考例1におけるHPLCのクロマトグラムを模式的に示す図である。
【図9】HPLC後の各分画についてのSDS−PAGEの結果を示す写真である。
【図10】実施例1〜4および比較例1の酵素免疫測定法(サンドイッチ法)による測定結果を示すグラフであり、左側の縦軸は吸光度を示している。
【発明を実施するための形態】
【0017】
本発明によれば、細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)により産生された第1の抗体と、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生された第2の抗体とを用いてサンドイッチ酵素免疫測定法により、検体中のネコ由来β2ミクログロブリン濃度を測定する方法が提供される。
【0018】
ここで、検体とは、ネコ由来の血液、尿、骨髄液およびネコ由来細胞の懸濁液の他、ネコの血液透析で用いた透析液も含まれる。
【0019】
また、ネコ由来β2ミクログロブリンとは、配列番号1で示されるアミノ酸配列を有するタンパク質をいう。本発明者は、ネコの遺伝子の中でβ2ミクログロブリンをコードする構造遺伝子(本明細書中において「β2-m遺伝子」と呼称する。)を初めて特定し、当該β2-m遺伝子によりネコ由来のβ2ミクログロブリン(本明細書中において「β2-m」と呼称する。)のアミノ酸配列も初めて解析し、特許出願をしている(特願2009−286712)。配列番号1に示されるアミノ酸配列を有する本発明のタンパク質が、本発明者によって初めてアミノ酸配列が特定されたネコ由来のβ2-mである。
【0020】
また、本発明者は、このネコ由来β2ミクログロブリンを抗原とする抗体を産生する細胞の作製に成功し、独立行政法人 産業技術総合研究所 特許生物寄託センターに寄託を行っている(受託番号:FERM P-21879、FERM P-21880)。本発明は、上述の抗体よりもより好感度にβ2-mを測定することに成功し、改めて、平成23年1月12日付けで独立行政法人 産業技術総合研究所 特許生物寄託センターに寄託を行ったものである(受領番号:FERM AP-22051、受領番号:FERM AP-22052)。本発明は、この新規な抗体産生細胞株である細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)についても提供する。
【0021】
また本発明は、配列番号1で表わされるアミノ酸配列を有するタンパク質(ネコ由来のβ2-m)を抗原とし、細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)により産生されたものである、ネコ由来β2ミクログロブリンに特異的に結合する抗体(第1の抗体)、ならびに、配列番号1で表わされるアミノ酸配列を有するタンパク質(ネコ由来のβ2-m)を抗原とし、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生されたものである、ネコ由来β2ミクログロブリンに特異的に結合する抗体(第2の抗体)についても提供する。
【0022】
本発明の第1の抗体および第2の抗体は、酵素免疫測定法(サンドイッチ法)、蛍光免疫測定法(サンドイッチ法)、放射性同位体免疫測定法(サンドイッチ法)、免疫比濁法およびラテックス免疫比濁法の2つの抗体として用いられる。
【0023】
酵素免疫測定法(サンドイッチ法)は、タンパク定量における代表的な測定方法であり、当業者であれば容易に実践できる測定方法である。酵素免疫測定法(サンドイッチ法)は、基本的には、固相に固定する抗体(捕獲抗体)と、当該捕獲抗体に結合した抗原に対してさらに結合する抗体(一次抗体)の2種類を用いる。この上で、発色または発光反応を行うための酵素標識が一次抗体になされている。または、一次抗体に酵素標識がなされていない場合は、一次抗体にさらに結合可能な抗体であって、酵素標識がなされている抗体(二次抗体)を用いてもよい。二次抗体としては、IgGおよびIgMなどが利用できる可能性がある。
【0024】
なお、ここでいう酵素標識とは、発色または発光反応に利用可能な酵素を直接一次抗体に標識する態様の他、ある特定の結合様式を介して反応時に間接的に酵素を修飾する態様も含むことは、当業者であれば常識である。特定の結合様式としては、例えば、アビジン−ビオチン結合が代表的である。より詳しく説明すると、ビオチンを修飾の一次抗体と、アビジン修飾ペルオキシダーゼの組み合わせがこの態様に該当し、アビジン−ビオチン結合という結合様式を介してペルオキシダーゼは間接的に一次抗体に修飾している。
【0025】
一次抗体等に標識される酵素は、当業者が適宜選択することができ、ペルオキシダーゼの他、アルカリホスファターゼ、ルシフェラーゼおよびβ−ガラクトシダーゼ等を用いることができる。酵素に用いる基質についても、用いる酵素の種類に応じて当業者が適宜選択することができ、例えば、酵素がペルオキシダーゼの場合、テトラメチルベンジリン、2,2’−アジノビス(3−エチルベゾチアゾリン−6−スルホン酸)アンモニウム塩、4−アミノアンチピリン、4−アミノアンチピリン塩酸塩、5−アミノサリチル酸、2,4−ジクロロフェノール、N,N−ジメチル−m−トルイジンおよびN,N−ジメチルアニリン等が挙げられ、酵素がアルカリフォスファターゼの場合、ブルーテトラゾリウム、5−ブロモ−4−クロロ−3−インドリルリン酸2ナトリウム塩、5−ブロモ−4−クロロ−3−インドリルリン酸p−トルイジン塩およびニトロブルーテトラゾリウム等が挙げられる、酵素がルシフェラーゼの場合、D−(−)−ルシフェリンが挙げられ、酵素がβ−ガラクトシダーゼの場合、5−ブロモ−4−クロロ−3−インドリルβ−D−ガラクトシダーゼ、5−ブロモ−6−クロロ−3−インドリルβ−D−ガラクトシダーゼ、5−ブロモ−3−インドリルβ−D−ガラクトシダーゼおよび6−クロロ−3−インドリルβ−D−ガラクトシダーゼ等が挙げられる。
【0026】
本発明の第1の抗体は、捕獲抗体、一次抗体のいずれとして用いられてもよく、限定されるものではない。換言すれば、第2の抗体は、第1の抗体が捕獲抗体である場合は一次抗体であり、第1の抗体が一次抗体である場合は捕獲抗体となる。この点は後述する実施例においても実証されている。
【0027】
また、本発明の第1の抗体および第2の抗体が酵素免疫測定法(サンドイッチ法)で測定できるということは、当業者であれば、蛍光免疫測定法(サンドイッチ法)、放射性同位体免疫測定法(サンドイッチ法)、免疫比濁法およびラテックス免疫比濁法にも応用することができる。例えば、測定法として、蛍光免疫測定法(サンドイッチ法)を用いる場合は、上述の酵素免疫測定法(サンドイッチ法)における酵素標識の代わりに蛍光標識を、また、放射性同位体免疫測定法(サンドイッチ法)を用いる場合は、上述の酵素免疫測定法(サンドイッチ法)における酵素標識の代わりに放射性同位体標識を適用すればよい。また、免疫比濁法およびラテックス免疫比濁法に至っては、反応系においてこれら2つの抗体を用いればよいだけのことである。ラテックス免疫比濁法におけるラテックスの材料も、当業者であれば適宜選択できるものであるが、一般的に用いられているポリスチレンを用いればよい。
【0028】
本発明は、さらに上述した本発明の第1の抗体、第2の抗体を利用した測定診断キットについても提供するものである。本発明の診断キットは、上述の測定法に応じて、当該測定が利用できる内容で組み合わされるものであり、例えば、測定法として酵素免疫測定法(サンドイッチ法)を選択する場合、本発明の抗体以外に、ウェル、色原性基質溶液、反応停止液、洗浄液、標準溶液などを含むよう設計される。
【実施例】
【0029】
以下、参考例、実施例および実験例を挙げて本発明をより詳細に説明するが、本発明はこれらに限定されるものではない。
【0030】
<参考例>
以下、参考例としてネコ由来β2-mの合成と、当該β2-mを用いた抗体産生ハイブリドーマの作製、ならびに本発明の抗体の作製を示す。これら参考例は、実質的に先願の手法と同一である。
【0031】
<参考例1:ネコ由来β2-mの合成>
(1)プライマーの設計
以下の上流側プライマーおよび下流側プライマーを設計した。ベクターとしてpGEX-6P-1(GEヘルスケアバイオサイエンス)を用いるため、上流側プライマーの5’末端にBam HI、下流側プライマーの5’末端にSal Iの制限酵素認識配列を加え、それぞれ以下の塩基配列となるように作製した。なお、NはT、A、CまたはGを示している。
【0032】
・上流側プライマー:5'-NNNGGATCCGTCCAGCATTCCAAAGGTTCAGGT-3'(配列番号2)
・下流側プライマー:5'-NNNGTCGACTTACATGTCTCGATCCCACTTAACGACCTT-3'(配列番号3)
(2)PCR法
上述のfirst strand cDNA 4μl、GoTaqR Green Master Mix(Promega)12.5μl、上流側プライマー1μl、下流側プライマー1μlおよびRNase free H2O 6.5μlをPCRチューブ内で混和し、サーマルクライマーを用いてPCRを行った。PCRの条件は95℃2分間加温後、95℃で45秒間、primer pairのアニーリング温度で45秒間、72℃で1分間のサイクルを35サイクル、その後72℃で7分間のプログラムとした。また、PCRの最適な条件を見つけるため、アニーリング温度の検討を行った。アニーリング温度が、77.5℃で行ったRT-PCRでは、目的とするβ2-m cDNAと思われる約300bpのバンドの他に約50bpのバンドが出現した。ここで、図1は、アニーリング温度を変えた条件でPCRを行い、2% アガロースゲルで電気泳動を行った結果を示す写真であり、レーン1〜3はそれぞれアニーリング温度を77.5℃、80℃、85℃とした場合、レーン4、5はプライマーの添加量を変えた場合である。この約50bpのバンドは、アニーリング温度が77.5℃、80℃および85℃と高くなるにつれて減少したが、消失しなかった。したがって、アニーリング温度を85℃に設定しプライマーの添加量を変え、レーン5ではプライマーの添加量を半分に落としたところ、約50bpのバンドが少なくなり、β2-m cDNAのバンドと思われる約300bpのほぼ単一なバンドのPCR産物が得られ、ベクタープラスミドへの挿入cDNAとした。
【0033】
(3)ゲルからのDNA抽出
QIAquick Gel Extraction Kit(QIAGEN)を用いて、添付のプロトコールに従ってゲルからのDNA抽出を行った。アガロースゲルの目的のDNAバンドを切り出し、ゲルの重量を測定した。切り出したゲルを、3倍量のQGバッファーを添加され50℃の恒温槽(TR-2A、ASONE)内で10分間加温し、ゲルを完全に溶解させた後、ゲルと同量のイソプロパノールを加え、よく混和させた。DNA溶液を、キットに付属のカラムのセットされた2mlのコレクションチューブに加え、室温で13400×g、1分間遠心分離した。その後、コレクションチューブ内の濾液を捨てた後、再びカラムに0.75mlのPEバッファーを添加し、室温で15700×g、1分間遠心分離にて洗浄後、濾液を除去し、さらに1分間遠心分離した。その後、カラムを新しい1.5mlのマイクロチューブにセットし、EBバッファー 50μlを加えた後、室温で1分間放置し、15700×gで1分間の遠心分離により抽出液を回収し、得られた溶液をDNA抽出溶液とした。
【0034】
(4)PCR産物の濃縮
DNA抽出溶液をフェノールと等量混合後、15700×gで5分間遠心分離後、核酸を含む水層を分離した。その水層に、クロロホルムを等量混合し、15700×gで5分間遠心分離させた後、上清を分離した。次に分離後の溶液は、2.5倍量の100%エタノールが加えられ−80℃で30分静置し、その後15700×gで5分間遠心分離、上清を除去し、沈渣を得た。沈渣に70%エタノールを添加し、15700×gで5分間遠心分離後、上清を除去し、PCR産物の濃縮試料とした。
【0035】
(5)β2-m cDNA組み込みベクターの作成および大腸菌の形質転換法
PCR産物の濃縮試料を、BamHI(タカラバイオ株式会社)5μl、SalI(タカラバイオ株式会社)5μl、H.Buffer(500mM Tris-HCl、pH7.5、100mM MgCl2、10mM Dithiothreitol、1000mM NaCl)5μlおよびRNase free H2O 35μlに混和させた。また、pGEX6P-1 5μl(2.5μg)を、BamHI 5μl、SalI 5μl、 H.Buffer 5μlおよびRNase free H2O 30μlと混和させた。各溶液を、37℃で1晩インキュベートすることで制限酵素処理を施した後、アガロースゲル電気泳動を行い、QIAquick Gel Extraction Kitを使用して、各DNAバンドの抽出を行った。ライゲーションはDNA Ligation Kit(タカラバイオ株式会社)を用いて行った。すなわち、5μlのLigation Mixと1μlの制限酵素処理されたβ2-m cDNA溶液および4μlのpGEX6P-1が混和され、16℃で1晩静置された。そして、この反応溶液を用いて、β2-m cDNAのトランスフェクションを行った。反応溶液2.5μlを、E.coli JM109 Competent Cells(タカラバイオ株式会社)25μlに添加し氷上で30分間静置させ、次に42℃の恒温槽で45秒間Heat-Shockを与え、直ちに2分間氷冷後SOC培地(2% tryptone、0.5% Yeast extract、10mM NaCl、2.5mM KCl、10mM MgSO4、10mM MgCl2、20mM glucose)250μlを緩やかに加え、37℃1時間保温された。β2-m cDNAトランスフェクト溶液100μlをアンピシリン加LB培地にそれぞれ塗布し、37℃で1晩静置後、コロニーを釣菌し、アンピシリン加LB液体培地1.2mlに混和後、37℃で1晩培養した。培養後の液体培地を13400×gで1分間遠心分離後、上清を完全に除去し、得られた沈渣からプラスミド抽出を行った。
【0036】
(6)プラスミド抽出
QIAPrep Spin Mini Kit50(QIAGEN)を用い、添付のプロトコールに従ってプラスミド抽出を行った。具体的には、まず、上述の得られた沈渣を、buffer P1 250μlで溶解させた後、buffer P2 250μl を添加し、その後緩やかに転倒混和させることで溶菌させた。中性化N3 buffer 350μlの添加により溶菌反応を停止させた後、15700×gで10分間遠心分離し、上清を付属のカラム付きコレクションチューブに添加した。カラムを、5900×gで1分間遠心分離した後、濾液を除去し、Binding Buffer 500μlを加えた。続いて、9300×gで1分間遠心洗浄後、濾液を除去し、エタノール含有脱塩buffer 750μlを加え、9300×gで1分間遠心洗浄後、新しいチューブに移した。カラムのメンブレンに、Elution Buffer 50μlを添加し、9300×gで1分間遠心分離後、プラスミド抽出溶液を得、このプラスミドをpcDNA-Fβ2mとし、アガロースゲル電気泳動法により確認した。図2は、形質転換された大腸菌から抽出したpcDNA-Fβ2mをアガロースゲル電気泳動した結果を示す写真である。図2において、レーン1、2は、それぞれ制限酵素(BamHIおよびSal1)で処理されたプラスミド(pGEX6p-1)およびβ2-m cDNAであり、レーン3はpcDNA-Fβ2m、レーン4はpcDNA-Fβ2mを制限酵素(BamHIおよびSal1)で処理した試料についての結果を示している。図2に示されるように、約3000bpに太いバンドが、約8000bp、約5000bpおよび約2000bpに薄いバンドが確認された。また、抽出後のpcDNA-Fβ2mをBamH1およびSal1で制限酵素処理を行った試料を泳動したレーン4では、約5000bpと約300bpのバンドが確認された。この約5000bpと約300bpの二つのバンドは、形質導入前のβ2-m cDNAおよびpGEX6P-1の泳動結果と比較するとほぼ同じ分子量のバンドであった。また、pcDNA-Fβ2mのサブクローニングの成否は、T7プライマーを用いてDye Deoxy Terminator Cycle Sequencing kit(Applied Biosystems)およびApplied Biosystems 3130xl Genetic Analyzer(Applied Biosystems)を使用したシークエンス解析により行った。図3は、pcDNA-Fβ2mのシークエンス解析結果を模式的に示す図である。図3に示す結果から、組み込んだβ2-m cDNAの塩基配列が正しくベクターに組み込まれていたことが確認された。
【0037】
(7)GST融合タンパク発現の確認
トランスフェクトされた大腸菌を、37℃、1晩LB培地で培養後、100μlをIsopropl-β-D-thiogalactopyranoside(IPTG:0.1mM)20μlと混和させ、37℃で約2時間振盪培養(BR40-LF、TAITEC)した。振盪培養後の大腸菌溶液を、15700×gで1分間遠心分離後、上清を除去し、沈渣に可溶化液(50mM Tris-Hcl 50μl、1×RIPA Lysis Buffer(Up State)100μl、Protease Inhibitor 140μl、H2O 710μl)30μlを加え可溶化後、15700×gで5分遠心分離し、上清と沈渣に分けた。上清30μlに、2×SB溶液(2% SDS、40% Glycerol、0.6% BPB、25mM Tris-HCl Buffer(pH6.8、20℃))30μl、2ME 1μlを加え、95℃で3分間加温した。沈渣に、SB溶液20μlを加え、超音波破砕機(UR-20P、TOMY SEIKO CO,LTD)で5秒間破砕後、95℃で3分間加温した。その後、上清および沈渣について、SDS−PAGEにてGST融合タンパク質発現の確認およびGST融合タンパクの大腸菌での溶解性を確認した。GST融合タンパク誘導発現後に得られた大腸菌をソニケーションし、遠心分離して得られた上清および沈渣について、SDS−PAGEにて泳動させた。図4は、SDS−PAGEの結果を示す写真であり、レーン1は上清、レーン2は沈渣についての結果を示している。GST融合タンパクの分子量は、約37kDaであり、上清には顕著なバンドの確認はできなかったが、沈渣では、明瞭な太いバンドが確認された。そのため、GST融合タンパクは、不溶性画分に発現したことが確認された。
【0038】
(8)SDS−PAGE法
コンパクトPAGE(AE-7300、ATTO)を用いてLaemmliの方法に準拠し、これに以下に示す修正を加えてSDS−PAGEを実施した。具体的には、分離ゲルの組成は、15% acrylamide、0.2% N,N-methylene-bis-acrylamide、0.1% SDS、375mM Tris-HCl buffer(pH8.8, 20℃)とした。ゲルは2・4連ゲル作製器(AE-7344、ATTO)を用いて作製した。電極緩衝液の組成は、0.1% SDS、129mM glycine、25mM Tris(pH8.3、20℃)とした。泳動用試料(SB)の組成は、1% SDS、20% glycerol、0.3% BPB、12.5mM Tris-HCl Buffer(pH6.8、20℃)とした。また、マーカーとして、プレステインドSDS−PAGEスタンダード(Broad)マーカー(BIO-RAD)もしくはSDS−PAGEスタンダード(Broad)マーカー(BIO-RAD)が用いられた。泳動は、Tris-Gly/PAGE Highモードで30分泳動させた後、Tris-Gly/PAGE Lowモードにして、下部イオン界面をゲル下端から1〜2mm上方の位置に移動したときに終了した。SDS−PAGE終了後のゲルには、Oakley法に準拠した銀染色法を施した。すなわち、ゲルを30% ethanol、10% acetic acid溶液にて固定後、洗浄し、20 % ethanolに5分間2回浸漬させた。20% ethanol除去後、5% glutaraldehyde溶液にて4分間反応させ、純水で洗浄後、20% ethanolに4分間2回浸漬させた。その後、純水で洗浄し、アンモニア性硝酸銀溶液にて5分間反応させ、純水で洗浄後、0.005% citric acid、0.019% formaldehyde溶液で発色させた。発色確認後のゲルは、20% ethanol、10% acetic acid溶液にて5分間固定させ、20% ethanolに5分間2回浸漬後、写真を撮影した。なお、銀染色法はすべて遮光条件下にて実施した。
【0039】
(9)GST融合タンパク質の発現誘導と単離
GST融合タンパク質の発現が確認された大腸菌をアンピシリン加LB寒天培地に塗布し、コロニーを釣菌後、3mlのアンピシリン加LB液体培地に加え1晩37℃で振盪培養した。続いて、その培養液3mlをアンピシリン加LB液体培地250mlに加え、37℃で約150分振盪培養後、0.1mM IPTG 2.5mlを添加し、約2時間、37℃で振盪培養したGST融合タンパク質のタンパク発現後のタンパク組成を分析した。GST融合タンパク発現誘導後の培養液を6000×gで15分間遠心分離した沈渣に、20mlの0.5mM EDTA、0.4M NaCl、5mM MgCl2、5%グリセロール、0.5mM phenylmethylsulfonyl fluoride(PMSF)、1mM dithiothreitol(DTT)および1mg/mlリゾチーム加50mM Tris-HCl(pH8.0)を懸濁させ、4℃で1時間静置後、凍結融解を2回行った。続いて、Nonidet P-40を0.5%添加し、超音波破砕機で20秒間×5回破砕後、9300×gで20分間遠心分離し、上清を除去し、沈渣を得た。得られた沈渣を、10mlの8M Urea、0.5mM DTT加Phosphate buffer saline(PBS:140mM NaCl、2.7mM KCl、10mM Na2PO4、1.8mM KH2PO4、pH 7.3)に再懸濁後4℃で1時間静置し、9000×gで20分間遠心分離し、上清を得た。この上清をGST融合タンパク溶液とした。
【0040】
(10)アフイニティークロマトグラフィー法
20mlのGST融合タンパク溶液を0.5M Urea 加PBSで平衡化させたGSTrap HP カラム(GEヘルスケアバイオサイエンス)にペリスタポンプ(SJ-1211L、ATTO)を用い、流速0.3ml/minで添加した。なお、カラムに添加するGST融合タンパク溶液について、尿素濃度の違いによるカラムへのGST融合タンパクの吸着量の比較を行った。カラムを、0.5M Urea加PBSで洗浄後、10mM reduced glutathione、1M Urea 加50mM Tris-HCl(pH8.0)で溶出させた。GST融合タンパク溶出液の吸光度を紫外部吸光度モニター(AC-5100L、ATTO)を用いて吸光波長220nmでモニターし、記録計(R-01A、RIKADENKI)で記録した。得られたGST融合タンパク溶出液2mlを、濃度が1mMになるようにDTTを添加し混和後、分子量13kDaカットの透析膜(UC30-32-100、三光純薬株式会社)に入れ、150mM NaCl、1mM EDTA加50mM Tris-HCl(pH7.5)2Lを用いて約6時間透析した。尿素濃度の違い(2M、1Mおよび0.5M)によるGST融合タンパク質のHiTrap affinity カラムへの結合量の比較を行った。各尿素濃度のGST融合タンパク溶液のクロマトグラムを図5(a)、(b)、(c)にそれぞれ示す。図5(a)、(b)、(c)は、それぞれ、尿素濃度2M、1Mおよび0.5Mのタンパク試料を用いたクロマトグラムを示している。各クロマトグラムは、前半部分に試料添加による吸光度変化が見られ、洗浄バッファーの添加により吸光度が減少した。その後、溶出バッファー添加によりシャープなピークが確認されたが、尿素濃度の減少に伴い吸光度の増加がみられ、尿素濃度0.5Mで最もシャープな分画が観察された。各尿素濃度のリガンド結合分画について、SDS−PAGEで比較した結果を図6に示す。図6において、レーン1は図5(c)で得られたリガンド結合分画、レーン2は図5(b)で得られたリガンド結合分画、レーン3は図5(a)で得られたリガンド結合分画についての結果が示されている。図6に示されるように、尿素濃度が高くなるに従いGST融合タンパク質の結合量の減少が確認され、尿素濃度0.5Mで行ったアフィニティークロマトグラフィーのリガンド結合分画(図5(c)で得られたもの)(以下、「C4分画」と呼称する)を以下の実験で用いた。
【0041】
(11)プロテアーゼ処理
透析後のGST融合タンパク溶出液について、DC Protein Assay(Bio-Rad)を用いてタンパク定量を行い、タンパク量200μgに対し、PreScission Protease(GE ヘルスケアバイオサイエンス)を1μl添加して混和させた後、4℃で6時間以上反応させ、高速液体クロマトグラフィー(HPLC)用の試料とした。PreScission Protease反応に及ぼす透析およびDTTの効果について検討を行った。また、C4分画を用いてPreScission Protease活性に与えるDTT濃度の影響を検討した。C4分画に最終濃度1mM、2.5mMおよび5mMになるようにDTTを添加し、透析後、PreScission Proteaseを反応させた結果を図7に示す。図7において、レーン1は1mM DTTを添加した場合、レーン2は2.5mM DTTを添加した場合、レーン3は5mM DTTを添加した場合をそれぞれ示している。いずれの濃度のDDT添加においても約11kDaのβ2-mのバンドは認められたが、1mMのDTT添加では約15kDaのバンドの消失、2.5mMおよび5mMのDTT添加では、β2-mのバンド以外に約13kDaのバンドが確認された。上記の結果より、次の精製ステップである高速液体クロマトグラフィー法(HPLC)では、1mMのDDTを添加したC4分画を透析後にPreScission Protease処理した試料を用いた。
【0042】
(12)HPLC法
HPLCシステムは、システムコントローラー(SCL-10A VP、Shimadzu)、送液ユニット(LC-10AD VP、Shimadzu)、紫外部分光高度計(SPD-10A VP、Shimadzu)、カラムオーブン(CTO-10A VP、Shimadzu)および脱気ユニット(DGU-14A、Shimadzu)から構成され、カラムはMightysilRP-18 GP250-4.6(Cat.No.25415-96、関東化学)を使用した。HPLCの分離条件は、移動相の流速を1ml/min、試料添加量は400μlとし0.1%Trifluoroacetic Acid(TFA)溶液で平衡化させカラムを0.1%TFA加acetonitrile溶液を用いて、acetonitrile濃度0〜80%のライナーグラジエントで行った。なお、溶出液は、吸光波長220nmで吸光度をモニターし、検出されたピークを分取し、濃縮遠心機(CC-181、TOMY)にて1時間遠心分離後、凍結乾燥機(FDU-540、EYELA)にて乾燥させた後−20℃で保存した。また、溶出された各分画のタンパクを、SDS−PAGE法により分析した。図8は、HPLCのクロマトグラムを模式的に示す図である。プロテアーゼ処理後のC4分画は、主に5つの分画として溶出された。これらの5つの分画は溶出された順にC4a、C4b、C4c、C4dおよびC4eとし、SDS−PAGEにより分析した。図9は、HPLC後の各分画についてのSDS−PAGEの結果を示す写真である。図9に示すように、C4a分画ではバンドが確認されず、C4b分画では約11kDaのバンドが、C4c分画では約11kDa、約25kDa、約27kDaのバンドが、C4d分画では約11kDa、約27kDaのバンドが、C4e分画では約27kDaのバンドが確認された。目的とするβ2-mと思われるタンパク質は、C4b分画において単一のバンドとして検出された。また、C4b分画の溶出時のacetonitrile濃度は39.5%であった。この溶出液を組み換えネコβ2-mとして、濃縮遠心し、凍結乾燥後、−80℃で保存した。
【0043】
<参考例2:抗体産生ハイブリドーマ、抗rFeβ2-m抗体の作製>
参考例1で合成したタンパクを組み替え型ネコβ2-m(rFeβ2-m)の抗原としてモノクローナル抗体を作製するにあたり、まずは抗体産生ハイブリドーマを作製した。
【0044】
(1)抗体産生ハイブリドーマの作製
(1−1)免疫法
免疫法は、精製rFeβ2-mを抗原としてBalb/cマウスの後肢肉球(footpad)の皮下に注射することにより行った。免疫は5日間隔で4回行い、初回から第3回目までの免疫は抗原溶液100μl(1mg/ml)とアジュバントを等量混合させてエマルジョン化させた抗原液200μl(50μg/foot)を、また、最終免疫では抗原溶液20μl(10μg/foot)のみを用いて行った。また、アジュバントは初回免疫ではAdjuvant Complete Freund(和光純薬工業株式会社)を、第2回目から第3回目の免疫ではAdjuvant Incomplete Freund(和光純薬工業株式会社)を用いた。
【0045】
(1−2)細胞融合
最終免疫から3日後、膝窩リンパ節を摘出し、リンパ球を回収後、GenomONE-CF(石原産業株式会社)を用いて、細胞融合を行った。また、ミエローマ細胞としてはP3X63-Ag8.653(大日本住友製薬株式会社)を用いた。融合方法は添付のプロトコールに従って行った。具体的には、まず、リンパ球とミエローマ細胞とを細胞数が5:1の比率になるように混合し、1000rpm、4℃で5分間遠心した後、上清を除去した。そこに、氷冷した融合用緩衝液をリンパ球108cellsあたり1ml添加し、均一に懸濁した後、氷冷したHVJ-Envelope懸濁液を細胞混合液1mlあたり25μl添加した。細胞懸濁液を氷上で5分間静置した後、1000rpm、4℃5分間遠心し、上清を除去せずに細胞がペレット化した状態のまま37℃で15分間インキュベートした。
【0046】
インキュベート終了後、37℃に加温した増殖用培地をリンパ球108cells当たり50ml加え、懸濁後、96穴プレート(96 Well Cell Culture Plate:Greiner bio-one)に100μl/wellで播種した。なお、増殖用培地としてRPMI1640(Invitrogen)にペニシリンG(PG;明治製薬株式会社)10万IU/ml、ストレプトマイシン(SM;明治製薬株式会社)100mg/ml、7.5% Bri Clone(IL-6、ヒト、ブライクローン;Cat. No. BR-001、大日本住友製薬株式会社)、10% 非働化ウシ胎仔血清(FBS;株式会社ニチレイ)を加えたものを用い、添加、懸濁の際は穏やかに操作した。24時間培養後、培養培地を上記の増殖用培地に2% HAT(Invitrogen)を添加したHAT培地に交換した。
【0047】
(2)抗体産生ハイブリドーマのスクリーニング
得られたハイブリドーマについて、細胞融合から1週間後にELISA法を用いた一次スクリーニングを行い、この結果、反応陽性となったwellのハイブリドーマのみをWestern blotting法を用いた二次スクリーニングで確認した。
【0048】
(2−1)一次スクリーニング
rFeβ2-mを抗原としたELISA法を用いて、抗体産生ハイブリドーマの一次スクリーニングを行った。ELISAプレートとしては、96 Well ELISA Microplate(Greiner bio-one)を使用した。また、プレートの洗浄には自動洗浄機(Auto Mini Washer AMW-8、バイオテック株式会社)を用い、洗浄液としてはPBS(1.37M NaCl、27mM KCl、100mM Na2HPO4、18mM KH2PO4、pH7.4、25℃)を使用した。固相として、PBSで3μg/mlに調整したrFeβ2-mを50μl/wellでプレートに添加し、4℃で一晩反応させた。固相反応終了後、プレートの抗原液を捨て、ブロッキング液として0.5% Bovine Serum Albumin(BSA;和光純薬工業株式会社)を添加したPBSを150μl/wellで加え、37℃で60分間反応させた。ブロッキング反応終了後、プレートを1回洗浄し、一次抗体として各ハイブリドーマ培養の培養上精を50μl/wellで加え、37℃で60分間反応させた。一次抗体反応終了後、プレートを1回洗浄し、二次抗体として0.1% BSAを添加したPBSで1000倍に希釈したペルオキシダーゼ標識抗マウスIgG抗体(SIGMA-ALDRICH)を50μl/wellで加え、37℃で60分間反応させた。二次抗体反応終了後、プレートを3回洗浄し、基質液として0.04% o-フェニレンジアミン、0.04% H2O2を添加したPBSを150μl/wellで加え、室温、遮光下で30〜60分間反応された。基質反応終了後、3M H2SO4を反応停止液として50μl/wellで加え、1分間振盪後、Microplate Reader(Model 550、BIO-RAD)で波長490nmにおける吸光度を測定した。吸光度の高かった陽性wellの細胞を、24穴プレート(24 Well Cell Culture Plate;Greiner bio-one)に移して培養した。
【0049】
(2−2)二次スクリーニング
rFeβ2-mを抗原としたWestern blotting法で確認し、抗体産生ハイブリドーマの二次スクリーニングを行った。Lowryの方法に基づき、DC Protein Assay Kit(BIO-RAD)を用いて、Microplate Readerで波長655nmにおける吸光度を測定し、タンパク質を定量した。検量線はBSAを用いて作製した。Western blotting法はTowbinらの方法に準拠し、以下のように実施した。転写膜はポリビニリデンジフルオリド(PVDF)膜(BIO-RAD)を使用した。PVDF膜は100% methanolに10秒間、さらに転写用電極buffer(25mM Tris-HCl(pH8.3、20℃)、192mM glycine、5% methanol)に30分間浸潤し、泳動に供した。転写装置の組み立ては、陽極電極板上に下から順に濾紙(BIO-RAD)、PVDF膜、SDS−PAGE終了後のゲル、濾紙を重層し、その上に陰極電極板を固定した。なお、濾紙は予め電極bufferに2〜3分浸しておいた。転写条件は1.9mA/cm2の定電流で60分間とした。転写終了後のPVDF膜は10mM Tris-HCl(pH7.5、20℃)、140mM NaCl、0.01% Tween20(TBST)に0.5% BSAを加え、室温で60分間振盪し、ブロッキング操作を行った。ブロッキング終了後、TBSTで5分間、2回振盪洗浄し、一次抗体として細胞の培養上精を用い、室温で90分振盪反応させた。一次抗体反応終了後、TBSTで5分間、2回振盪洗浄した後、TBSTで1000倍希釈したペルオキシダーゼ標識抗マウスIgG抗体を、室温で60分間振盪反応させた。二次抗体反応終了後、TBSTで5分間、2回振盪洗浄し、0.06% 3,3-diaminobenzidine tetra-hydrochloride、0.03% H2O2、50mM Tris-HCl(pH7.6、20℃)を基質反応液として使用し、1〜5分間反応させた。基質反応終了後、水洗し反応を停止させた後、乾燥して保存した。反応陽性を示したハイブリドーマについては後述する限界希釈法によりクローニングを行った。
【0050】
(3)クローニング
ハイブリドーマのクローニングには限界希釈法を用いた。具体的には、スクリーニング後のハイブリドーマを2cells/100μlとなるようにHAT培地で希釈し、100μl/wellで96穴プレートに播種した。ハイブリドーマはセミコンフルエントになったところで24穴プレートに拡大培養し、再びセミコンフルエントになるまで培養した後、二次スクリーニングと同様にrFeβ2-mを抗原としたWestern blotting法で確認した。このクローニング操作を2回行った。また、ハイブリドーマを長期間継代培養することにより抗体産生能が減少するのを防ぐため、クローニング毎に細胞凍結保存液(セルバンカー(BLC-1)、十慈フィールド株式会社)を用いて保存した。
【0051】
(4)抗体産生ハイブリドーマの大量培養および抗rFeβ2-m・mAbの採取と精製
クローニングが終了したハイブリドーマを、浮遊細胞培養フラスコ(フィルタートップSCフラスコ250ml 75cm2;Greiner bio-one)を用いて大量培養した。なお、培養は37℃、5% CO2、5日間、CO2インキュベーター(十慈フィールド株式会社)で行い、培地としてはHAT培地を用いた。大量培養されたハイブリドーマを無血清RPMIで懸濁し、ヌードマウス(Balb/c-nu)の腹腔内に2×107cells/headで投与した。投与してから10〜20日後、腹水を採取した。ヌードマウスから採取した腹水を室温で1時間、あるいは4℃で一晩静置した後、3000rpm、4℃で5分間遠心し、腹水中のフィブリン、ハイブリドーマ、赤血球などを除去した。分離した上清を50%の硫安で塩析させた。具体的には、氷上で撹拌しながら上清と等量の飽和硫酸アンモニウム溶液を徐々に滴下し、滴下後さらに1時間撹拌した。これを10000rpm、4℃で10分間遠心し、沈殿物を20mM リン酸ナトリウムbuffer(pH7.0)に溶解した。塩析後のグロブリン溶液を、20mM リン酸ナトリウムbuffer(pH7.0)で平衡化したSephadex G-25 Fine(GEヘルスケアバイオサイエンス)カラム(内径1.5cm、長さ30cm)を用いて脱塩した。クロマトグラフィーの流速をペリスタポンプ(SJ-1211L、ATTO)で0.5ml/minに調節した。脱塩後のグロブリン溶液を、エコカラム(内径2.5cm、長さ10.0cm:BIO-RAD)に充填したProtein G Sepharose 4 Fast Flow(GEヘルスケアバイオサイエンス)を用いたアフィニティークロマトグラフィー法により精製した。具体的には、脱塩後のグロブリン溶液を20mM リン酸ナトリウムbuffer(pH7.0)で平衡化されたカラムに流速0.5ml/minで添加し、その後カラムを100mM glycine(pH3.0)で溶出させた。溶出液は直ちに10分の1量の1M Tris-HCl(pH9.0)で中和した。精製後の溶出液を50mM 酢酸アンモニウム(pH7.0)で平衡化させたSephadex G-25 Fineカラム(内径2cm、長さ30cm)で脱塩させた後、Freeze Dryer(FDU540、EYELA東京理化器械株式会社)を用いて凍結乾燥し、−20℃で保存した。
【0052】
<実施例1〜4、比較例1>
後述する実験例に用いる実施例1〜4および比較例1の抗体の組み合わせを表1に示す。
【0053】
【表1】
【0054】
表1中、β2-m mAb3産生抗体は、細胞株Mouse-Mouse hybridoma β2-m mAb3(受託番号:FERM AP-22051)により産生された抗体を指し、β2-m mAb4産生抗体は、細胞株Mouse-Mouse hybridoma β2-m mAb4(受託番号:FERM AP-22052)により産生された抗体を指し、β2-m mAb1産生抗体は、細胞株Mouse-Mouse hybridoma β2-m mAb1(受託番号:FERM P-21879)により産生された抗体を指し、β2-m mAb2 産生抗体は、細胞株Mouse-Mouse hybridoma β2-m mAb2(受託番号:FERM P-21880)により産生された抗体を指す。
【0055】
<実験例>
サンドイッチ酵素免疫測定法により、実施例1〜4および比較例1の評価を行った。具体的には、1匹の健常なネコの尿中のβ2-mの測定を行った。具体的には、捕捉抗体を0.5mg/wellで固相し、ブロッキングした後、1匹の健常なネコの尿を2時間反応させた。洗浄後、ビオチン標識した一次抗体を2時間反応させた後、アビジンペルオキシダーゼを1時間反応させ、tetramethylbenzidineを用いて基質反応を行い、450nmにおける吸光度の測定を行った。図10はその結果を示すグラフであり、縦軸は吸光度である。図10から明らかなように、実施例1〜4の抗体の組み合わせによる吸光度は、比較例と比較して大幅に高かった。この結果から、本発明の抗体の組み合わせは、ネコβ2-mに対してより高い結合力を示すことが明らかであり、高感度のネコβ2-mの測定が可能であることを示した。
【0056】
今回開示された実施の形態および実験例はすべての点で例示であって制限的なものではないと考えられるべきである。本発明の範囲は上記した説明ではなくて特許請求の範囲によって示され、特許請求の範囲と均等の意味および範囲内でのすべての変更が含まれることが意図される。
【特許請求の範囲】
【請求項1】
細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)により産生された第1の抗体と、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生された第2の抗体とを用いて、酵素免疫測定法(サンドイッチ法)、蛍光免疫測定法(サンドイッチ法)、放射性同位体免疫測定法(サンドイッチ法)、免疫比濁法およびラテックス免疫比濁法からなる群より選択されるいずれかの方法により、検体中のネコ由来β2ミクログロブリン濃度を測定する方法。
【請求項2】
細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)により産生された第1の抗体と、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生された第2の抗体とを含む、ネコ由来β2ミクログロブリン濃度測定用キット。
【請求項3】
細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)。
【請求項4】
細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)。
【請求項5】
配列番号1で表わされるアミノ酸配列を有するタンパク質を抗原とし、細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)により産生されたものである、ネコ由来β2ミクログロブリンに特異的に結合する抗体。
【請求項6】
配列番号1で表わされるアミノ酸配列を有するタンパク質を抗原とし、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生されたものである、ネコ由来β2ミクログロブリンに特異的に結合する抗体。
【請求項1】
細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)により産生された第1の抗体と、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生された第2の抗体とを用いて、酵素免疫測定法(サンドイッチ法)、蛍光免疫測定法(サンドイッチ法)、放射性同位体免疫測定法(サンドイッチ法)、免疫比濁法およびラテックス免疫比濁法からなる群より選択されるいずれかの方法により、検体中のネコ由来β2ミクログロブリン濃度を測定する方法。
【請求項2】
細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)により産生された第1の抗体と、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生された第2の抗体とを含む、ネコ由来β2ミクログロブリン濃度測定用キット。
【請求項3】
細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)。
【請求項4】
細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)。
【請求項5】
配列番号1で表わされるアミノ酸配列を有するタンパク質を抗原とし、細胞株Mouse-Mouse hybridoma β2-m mAb3(受領番号:FERM AP-22051)により産生されたものである、ネコ由来β2ミクログロブリンに特異的に結合する抗体。
【請求項6】
配列番号1で表わされるアミノ酸配列を有するタンパク質を抗原とし、細胞株Mouse-Mouse hybridoma β2-m mAb4(受領番号:FERM AP-22052)により産生されたものである、ネコ由来β2ミクログロブリンに特異的に結合する抗体。
【図3】


【図5】
【図8】
【図10】
【図1】
【図2】
【図4】
【図6】
【図7】
【図9】
【図5】
【図8】
【図10】
【図1】
【図2】
【図4】
【図6】
【図7】
【図9】
【公開番号】特開2012−189504(P2012−189504A)
【公開日】平成24年10月4日(2012.10.4)
【国際特許分類】
【出願番号】特願2011−54496(P2011−54496)
【出願日】平成23年3月11日(2011.3.11)
【出願人】(598041566)学校法人北里研究所 (180)
【出願人】(000135036)ニプロ株式会社 (583)
【Fターム(参考)】
【公開日】平成24年10月4日(2012.10.4)
【国際特許分類】
【出願日】平成23年3月11日(2011.3.11)
【出願人】(598041566)学校法人北里研究所 (180)
【出願人】(000135036)ニプロ株式会社 (583)
【Fターム(参考)】
[ Back to top ]