
ネビボロールの製造方法
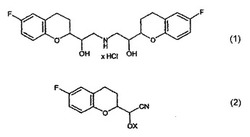
【課題】 本発明の主題はラセミ型の有効成分であるネビボロールを製造する方法である。
【解決手段】 本方法では、ジアステレオマーシアノヒドリンを生じさせ、分離しそしてその分離したジアステレオマーに変換、好適にはシアノ基に還元をある程度または完全に受けさせるか或はピンナー鹸化を受けさせた後、それらを互いにカップリングさせる。
【解決手段】 本方法では、ジアステレオマーシアノヒドリンを生じさせ、分離しそしてその分離したジアステレオマーに変換、好適にはシアノ基に還元をある程度または完全に受けさせるか或はピンナー鹸化を受けさせた後、それらを互いにカップリングさせる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ラセミ型の製薬学的物質であるネビボロール(nebivolol)を遊離塩基または製薬学的に適合し得る塩、好適には塩酸塩として製造する方法に関する。
【背景技術】
【0002】
塩酸塩として市場に出ている式I,
【0003】
【化1】

【0004】
で表される製薬学的物質であるネビボロールは、ベータ受容体遮断薬の製薬学的物質群に属する。この分子はキラル中心を4個有しかつ同時に中心対称的(centrosymmetrically)構造を有する。認可された製薬学的物質であるネビボロールはラセミ体として用いられる。それは対称的構造を有することから、考慮すべきジアステレオマーは、8種類のジアステレオマー(理論的には16種類の鏡像異性体による)ではなく、正式には、原則として10種類の異性体を伴う6種類(R*R*R*R*,R*R*S*S*,R*R*R*S*,R*R*S*R*,R*S*R*S*およびR*S*S*R*)のみである。後者の中で、R*R*R*S*と表示する高純度のジアステレオマー(これは絶対配置がRRRSおよびSSSRの鏡像異性体の1:1の混合物である)が実際に認可された有効成分である。前記ネビボロールの両方の鏡像異性体が異なるプロファイルを示すことがそれが示す薬理学的作用に対して相乗的に貢献していることから極めて特殊な状況がもたらされる。従って、可能な最高水準として、高純度の鏡像異性体を直接合成するのではなく、必要な相対的キラル中心配置を有するラセミ型ジアステレオマーを合成することを考慮することができる。
【0005】
ラセミ型ネビボロール自身を合成する目的で直接利用可能な合成は文献に見られない。確かに、そのような成分の合成方法はいくつか記述されてはいるが、個々の立体異性体に分離する目的で各場合ともクロマトグラフィー段階が用いられており(例えば特許文献1)、それの説明は不明瞭なままであることから、技術的に使用可能であることをそれから引き出すのは原則として不可能である。注目すべきは、ネビボロールの高純度鏡像異性体を多少とも有利に合成する方法はかなり数多く見られる(例えば非特許文献1、2)。そのような合成法を前記ラセミ型有効成分の製造に移行させるには鏡像異性体を個別に製造する必要があるが、しかしながら、その後にそれらを1:1で混合しなければならない。従って、合成計画の目的にとって、ラセミ型の有効成分を製造する方が高純度の鏡像異性体を製造するよりも価値がある。
【0006】
ネビボロールは、中心元素以外に、正式には、相対的立体化学は別として同じ2個の分子部分で構成されている。それらの2個のキラル中心の相対的立体化学はR*R*およびR*S*として示される。実際に相対的な形態を有するが、ジアステレオマー中間体生成物をこれらの異なる形態に関して以下にAおよびBと識別する。全く選択不能なネビボロール合成法(このような方法では可能なあらゆる立体異性体が生じそしてそれらを分離す
る)ではなく、キラル中心の相対的立体化学AおよびBを有する2種類の中間体生成物を連結させることで合成を実施する方策を提供する。
【0007】
【特許文献1】EP 145067
【非特許文献1】S.Chandrasekhar,V.Reddy,Tetrahedron,56(2000),6339−6344
【非特許文献2】C.W.Johannes,M.S.Visser,G.S.Weatherhead,A.H.Hoveyda,J.Am.Chem.Soc.120(1998),8340−8347
【発明の開示】
【0008】
本発明の主題は、ネビボロールの製造方法であり、ここでは、一般式2
【0009】
【化2】
【0010】
[式中、Xは、X=Hの意味を有するか、或はXは、シアノヒドリンの典型的な保護基、例えばベンジル型(例えばベンジルまたは4−メトキシベンジル)またはアセタール型保護基(例えばテトラヒドロピラニルまたはMEM)、好適にはシリル保護基、極めて特に好適にはt−ブチルジメチルシリル保護基を意味する]
で表されるラセミ型のジアステレオマーシアノヒドリン(即ち以下に示すジアステレオマーAおよびB)を中間体生成物として用いる。
【0011】
本発明に従う方法の他の有利な態様を従属請求項に従って開示する。
【0012】
本発明は、更に、結晶性のラセミ型ジアステレオマーAとしてのα−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリルにも関する。
【0013】
本発明は、また、油状物状のラセミ型ジアステレオマーBとしてのα−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリルにも関する。
【0014】
前記シアノヒドリンの製造は、アルデヒド3
【0015】
【化3】
【0016】
にシアノヒドリン反応を受けさせることで実施可能である。
【0017】
また、前記アルデヒド3も、例えばエステル4
【0018】
【化4】
【0019】
のピラン環に還元を受けさせた後にエステル基に還元を直接または間接的(即ちアルコールを用いて)に受けさせることでアルデヒドを生じさせることなどで得ることができる。
【0020】
前記シアノヒドリン反応は立体選択的または非立体選択的に実施可能であるが、好適な変法では、非立体選択的合成をO−誘導体化を用いて実施する。シリル保護基を持つ式2で表される化合物のジアステレオマーもまた前記アルデヒド3から相当するシアン化シリル(シアン化t−ブチルジメチルシリルが好適である)を用いて1段階で生じさせることができる。カップリングに先立って前記成分の追加的合成を導入し、その結果として生じた配置AおよびBを有するジアステレオマーシアノヒドリン2(両方とも追加的合成に必要である)の分離を実施する。本発明の観点で特に好適なジアステレオマーは、結晶化特性が明瞭に異なる誘導体化ジアステレオマーAおよびB、好適には一方のジアステレオマーが結晶形態で存在しそしてもう一方が油状物であるジアステレオマーである。このようにすると、有機溶媒中でダイジェッションまたは熟成(digestion)により、技術的に極めて特に簡潔でありかつ効率の良いジアステレオマー分離を行うことが可能になる。本発明の観点でt−ブチルジメチルシリル保護基が極めて特に好適である。前記2種類のジアステレオマーの中の一方(A)は特に良好に結晶化する一方、もう一方のジアステレオマー(B)は油状物として得られ、非極性溶媒、好適には低級アルカン、特にヘキサンを用いてそれを分離すると、Aの比率が低くても、結晶化で冷却することでそれを容易に分離することができる。非極性溶媒、好適には低級アルカン、特にヘキサンを用いた再結晶化によって、結晶性のジアステレオマーをより高い純度にすることができる。
【0021】
連結部を生じさせる目的で、配置AまたはBを有する2個の分子部分をいろいろな様式で更に反応させる。その目的でシアノヒドリンを中間体生成物として用いるのが特に有利であることが分かる、と言うのは、シアノヒドリン基をいろいろな様式で更に反応させることで、前記2個の分子部分(AおよびB)の連結に最適な条件を得ることができるからである。さらなる反応に好適な条件は下記である:Oが保護されている一般式5[式中、Xは、式2で示した意味を有する]で表されるアルデヒドを生じさせる還元ばかりでなく、Oが保護されているか或はOが保護されていない一般式6[式中、Xは、式2で示した意味を有する]で表されるアミノアルコールを生じさせる還元、およびOが保護されていない一般式7[式中、Rは、分枝もしくは非分枝低級アルキル、または置換もしくは非置換ベンジルを意味する]で表されるヒドロキシルエステルを生じさせるピンナー(Pinne)鹸化。
【0022】
【化5】
【0023】
ある時点で、式2で表される結晶性のシアノヒドリンAおよび式2で表される油状物状のシアノヒドリンBの両方に還元を受けさせることでアルデヒド5またはヒドロキシエステル7を生じさせ、そして別の時点で、アミン6を生じさせる。この場合、製造および次に行う反応シーケンスを実施する時に収率に差が生じる。
【0024】
ネビボロールを生じさせる連結および反応を、好適には、アルデヒドとアミンの反応をシッフ塩基生成による還元アミノ化で起こさせた後に好適には1槽反応で還元を限られた還元力を有する複合水素化物、例えばシアノホウ水素化ナトリウムまたはトリアセトキシホウ水素化ナトリウムなどを用いて起こさせることで一般式8,
【0025】
【化6】
【0026】
[式中、Xは、式2で示した意味を有する]
で表される化合物を生じさせることを通して実施するか、或は一般式7で表されるエステルと一般式6で表されるアミンを反応させて一般式9
【0027】
【化7】
【0028】
[式中、Xは、化合物2で示した意味を有する]
で表されるアミドを生じさせた後にアミド9に還元を受けさせて一般式10
【0029】
【化8】
【0030】
[式中、Xは、化合物2で示した意味を有する]
で表されるアミンを生じさせることを通して実施する。
【0031】
式8および10で表される化合物を場合により前以て精製しておいた後、保護基を開裂させ、そして場合により、その塩酸塩および/または遊離塩基を再結晶化で精製してネビボロールの塩基または製薬学的に受け入れられる塩を製薬学的品質で生じさせることを通してネビボロールを得る。
【0032】
このカップリングで場合によりOが保護されているか或はOが保護されていない誘導体を使用、特にOが保護されているアルデヒドとOが保護されていないアミンのカップリングを実施すると、式10で表されるモノ保護ネビボロールを精製することで、場合によりカップリング後に少量存在する可能性のある副生成物であるジアステレオマーを特に効率良く分離することが可能になる。X=t−ブチルジメチルシリルの場合のそのような合成変法を下記の式図で示す。
【0033】
【化9】
【0034】
配置Aを有する分子と配置Bを有する分子をカップリングさせると均一なジアステレオマーではなく、むしろ2種類のジアステレオマーの混合物が生じることを注目すべきである。R*R*配置を有する化合物とR*S*配置を有する化合物をカップリングさせると、即ちジアステレオマーR*R*R*S*(鏡像異性体RRRSとSSSRで構成されるネビボロール)とR*R*S*R*(鏡像異性体RRSRとSSRSで構成される)が生
じる。実際、ラセミ型ネビボロールは、遊離塩基としてかつ塩酸塩として非常に容易に結晶化する化合物である。前記カップリングを1:1の比率で実施した時に生じるR*R*S*R*配置を有するジアステレオマーを遊離塩基の再結晶化および塩酸塩の再結晶化で特に容易に取り出すことができることが特に有利である。このように、必要でないジアステレオマーが示す溶解性が有意に高いことを基にして、前記ジアステレオマー混合物を再結晶化させることで高純度のネビボロールを得ることができる。
【0035】
本発明の説明を本発明の実施に可能な態様を基にして以下により詳細に行う。
【0036】
実施例1
(R*,R*/R*,S*)6−フルオロ−3,4−ジヒドロ−α−ヒドロキシ−2H−2−[1]ベンゾピランアセトニトリル−ラセミ型ジアステレオマー混合物
33gの(R*S*)6−フルオロ−3,4−ジヒドロ−2H−[1]ベンゾピラン−2−カルボアルデヒドを150mlのMTBEと150mlの80%酢酸に入れることで生じさせた溶液を23.8gのシアン化カリウムと混合した後、アルゴン雰囲気下室温で1時間撹拌する。次に、この反応混合物を600mlの冷却しておいた飽和炭酸ナトリウム水溶液に穏やかに撹拌しながら滴下した後、固体状の炭酸ナトリウムを反応溶液が中性様式で反応するまで加える。次に、それにMTBEを用いた抽出を数回受けさせ、その有機相を水で洗浄し、Na2SO4で乾燥させ、1滴の燐酸と混合した後、溶媒を真空下で除去する。
【0037】
収量:36.1g(95%),黄色がかった油状物。
1H−NMR:(CDCl3):δ(ppm)=6.82−6.74(m,3H),4.20−4.14(dd,1H),4.20−4.14(m,1H),2.84−2.79(m,2H),2.21−2.02(m,1H),2.00−1.88(m,1H).
【0038】
実施例2
(R*,R*/R*,S*)− −α−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2−2H−[1]ベンゾピランアセトニトリル−ラセミ型のジアステレオマー混合物
変法1:
25gのイミダゾールを150mlの無水ジメチルホルムアミドに入れることで生じさせた0℃の溶液に31.5gの塩化t−ブチルジメチルシリルを加えた後、その混合物を0℃で15分間撹拌する。次に、36gのシアノヒドリン(実施例1)を150mlの無水ジメチルホルムアミドに入れることで生じさせた溶液を滴下する。この滴下が終了した後、それを0℃で更に15分間撹拌した後、室温にして、2時間撹拌する。次に、その反応混合物を飽和NaHCO3溶液とMTBEの間で分離させ、その水相にMTBEを用いた抽出を受けさせ、その有機相を一緒にして水で洗浄し、Na2SO4で乾燥させた後、真空下で蒸発させることで濃縮する。トルエンを用いた洗浄を数回実施することでDMFを除去した後、生成物を高真空下で乾燥させる。
【0039】
収量:53.1g(95%),黄色の油状物;1H−NMR:(CDCl3):δ(ppm)=6.84−6.78(m,3H),4.75/4.62(dd,1H),4.25−4.07(m,1H),2.89−2.85(m,2H),2.37−2.17(m,1H),2.03−1.85(m,1H),0.97(d,9H),0.23(t,6H).
【0040】
変法2:シアン化t−ブチルジメチルシリルを使用
(R*S*)6−フルオロ−3,4−ジヒドロ−2H−[1]ベンゾピラン−2−カルボアルデヒド(2g)を20mlの無水DCMに入れることで生じさせた溶液に1.77
gのヨウ化亜鉛を加える。−10℃で1.88gのシアン化t−ブチルジメチルシリルを加えた後、その混合物を前記温度で2時間撹拌する。次に、その反応混合物を5%のNaHCO3溶液とMTBEの間で分離させ、その水相にMTBEを用いた抽出を2回受けさせ、その有機相を一緒にして水で洗浄し、Na-2SO4で乾燥させた後、真空下で蒸発させることで濃縮する。生成物を高真空下で乾燥させる。
【0041】
収量:3.1g(87%),黄色の油状物。
1H−NMR:(CDCl3):δ(ppm)=6.83−6.79(m,3H),4.73/4.61(dd,1H),4.23−4.07(m,1H),2.90−2.85(m,2H),2.37−2.15(m,1H),2.01−1.83(m,1H),0.96(d,9H),0.21(t,6H).
【0042】
実施例3
α−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリル−結晶性のラセミ型ジアステレオマーA
実施例2で得た16gのラセミ型ジアステレオマー混合物を20x量の石油状物エーテルに溶解させ、−45℃にした後、結晶性ジアステレオマー成分の種晶を加える。4時間後、母液を傾斜法で除去し、少量の深凍結石油状物エーテルを用いて結晶を熟成させ、前記エーテルを傾斜法で除去し、その母液を一緒にして蒸発による濃縮を元々の溶液の体積の1/3になるまで実施し、種晶を再び加えた後、前記手順を繰り返すことで生成物を2回目の収穫で得る。さらなる精製を行う目的で、R*,S*−ジアステレオマー(残存するR*,R*−ジアステレオマー含有量が8%)を再び石油状物エーテルに加熱しながら溶解させ、低温で析出させることで結晶性画分を得る。
【0043】
収量:結晶画分:6.5g=40%,白色結晶;引火点=66℃.
1H−NMR:(CDCl3):δ(ppm)6.83−6.73(m,3H),4.71(d,1H),4.09−4.05(m,1H),2.91−2.79(m,2H),2.30−2.25(m,1H),1.97−1.88(m,1H),0.94(s,9H),0.23(s,3H),0.19(s,3H).
【0044】
実施例4
α−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリル−油状物状のラセミ型ジアステレオマーB
ジアステレオマーAの結晶化で2回取り出した母液に蒸発による濃縮を受けさせることでジアステレオマーB(残存するジアステレオマーの含有量=10%)を得る;収量:9.0g=55%,黄色の油状物;1H−NMR:(CDCl3):δ(ppm),6.83−6.72(m,3H),4.58(d,1H),4.18−4.14(m,1H),2.90−2.77(m,2H),2.22−2.17(m,1H),1.93−1.85(m,1H),0.92(d,9H),0.24(s,3H),0.16(s,3H).
【0045】
実施例5
α−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトアルデヒド−ラセミ型のジアステレオマーB
実施例4で得た8gの油状物状ラセミ型ジアステレオマーシアノヒドリンBを80mlのトルエンに入れることで生じさせた溶液を5℃にした後、トルエン中1.5Mの水素化ジイソブチルアルミニウム溶液(18.25ml)と混合する。その反応混合物を室温で1時間撹拌する。次に、その反応混合物を600mlの1N塩酸に加えた後、100mlのMTBEで希釈する。相分離を起こさせた後、その水相に200mlずつのMTBEを用いた抽出を更に3回受けさせる。その有機相を一緒にして飽和塩化ナトリウム溶液で洗
浄し、Na2SO4で乾燥させた後、溶媒を真空下で除去する。
【0046】
収量:7.1g(88%),黄色の油状物.
1H−NMR:(CDCl3):δ(ppm)=9.79(s,1H),6.84−6.77(m,3H),4.32−4.25(m,1H),3.73−3.65(m,1H),2.84−2.79(m,2H),2.00−1.91(m,2H),0.98(d,9H),0.25(t,6H).
【0047】
実施例6a
6−フルオロ−3,4−ジヒドロ−α−ヒドロキシ−2H−2−[1]ベンゾピランエタンアミン−ラセミ型ジアステレオマーA
α−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリルの結晶性ラセミ型ジアステレオマーA(5g)を50mlのトルエンに入れることで生じさせた−20℃の溶液にトルエン中1.5MのDIBAL溶液を20.7ml滴下する。それを室温に加熱して15分間撹拌する。次に、その反応混合物を0℃に冷却した後、1.18gの水素化リチウムアルミニウムと混合する。次に、その反応混合物を室温で14時間撹拌する。次に、トルエン中1.5MのDIBAL溶液を更に20.7ml加え、室温で1時間そして35℃で1時間撹拌する。その反応混合物を酢酸エチルが400mlでMeOHが20mlの冷溶液に加える。この目的で、300mlの酒石酸カリウム溶液および30mlの2N NaOHを加えて1時間撹拌した。その反応混合物をセライトの上に置いて濾過し、その残留物とセライトを150mlずつの酢酸エチルで3回再洗浄する。相分離を起こさせ、その有機相を水で洗浄し、乾燥させた後、溶媒を真空下で除去する。結晶性の粗生成物を3.49g得る。この後者の再結晶化をMTBEを用いて実施する。
【0048】
収量:2.15g(65.5%),白色の固体;引火点=124℃.
1H−NMR:(CDCl3):δ(ppm)=6.81−6.77(m,3H),4.02−3.95(m,1H),3.71−3.65(m,1H),3.00−2.81(m,4H),2.04−1.85(m,5H).
【0049】
実施例6b
6−フルオロ−3,4−ジヒドロ−α−ヒドロキシ−2H−2−[1]ベンゾピランエタンアミン ラセミ型ジアステレオマーB
α−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリルのラセミ型ジアステレオマーB(4g)を40mlのトルエンに入れることで生じさせた−40℃の溶液にトルエン中1.5MのDIBAL溶液を16.6ml滴下する。それを室温に加熱して15分間撹拌する。次に、その反応混合物を0℃に冷却した後、1.18gの水素化リチウムアルミニウムと混合する。次に、その反応混合物を室温で14時間撹拌する。次に、トルエン中の1.5MのDIBAL溶液を更に16.6ml加え、室温で1時間そして35℃で1時間撹拌する。その反応混合物を酢酸エチルが300mlでMeOHが20mlの冷溶液に加える。これに250mlの酒石酸カリウム溶液および25mlの2N NaOHを加えた後、1時間撹拌する。その反応混合物をセライトの上に置いて濾過し、その残留物とセライトを120mlずつの酢酸エチルで3回再洗浄する。相分離を起こさせた後、その有機相を水で洗浄し、乾燥させた後、溶媒を真空下で除去する。結晶性の粗生成物を2.4g得る。後者の再結晶化をMTBEを用いて実施する。
【0050】
収量:1.7g(64.6%),白色の固体:1H−NMR:(CDCl3):δ(ppm)=6.78−6.65(m,3H),3.87−3.84(m,1H),3.67−3.63(m,1H),3.00−2.71(m,4H),2.51(bs,1H),
2.14−2.10(m,1H),1.84−1.76(m,1H).
【0051】
実施例7
N−{2−[(t−ブチルジメチルシリル)オキシ]−2−(6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピラニル)エチル]−6−フルオロ−3,4−ジヒドロ−α−ヒドロキシ−2H−2[1]ベンゾピランエタンアミン−ラセミ型A/Bジアステレオマー
6−フルオロ−3,4−ジヒドロ−α−ヒドロキシ−2H−2−[1]ベンゾピランエタンアミンのラセミ型ジアステレオマーA(2.28g)とα−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトアルデヒドのラセミ型ジアステレオマーB(7g)を250mlのTHFと50mlのメタノールに入れることで生じさせた溶液に0.81gのシアノホウ水素化ナトリウムを加える。1mlの氷酢酸を加えた後、室温で14時間撹拌する。次に、シアノホウ水素化ナトリウムを更に0.81g加える。それを室温で更に18時間撹拌する。その反応混合物を400mlの5%NaHCO3溶液と混合し、5分間撹拌した後、酢酸エチルを用いた抽出を数回実施する。その有機相を乾燥させた後、溶媒を真空下で除去する。そのようにして得た粗生成物(10.1g)をフラッシュクロマトグラフィー(可動溶媒DCM/MeOH 50+1)で精製する。黄色の油状物を4.2g得る。
【0052】
1H−NMR:(CDCl3):δ(ppm)=6.81−6.75(m,6H),4.09−3.95(m,4H),2.92−2.65(m,8H),2.10−1.98(m,2H),1.87−1.72(m,2H),0.94(s,9H),0.18(s,3H),0.13(s,3H).
【0053】
実施例8
(R*,R*,R*,S*)−α,α’−[イミノビス(メチレン)ビス[6−フルオロ−3,4−ジヒドロ−2H−1−ベンゾピラン−2−メタノール]]の塩酸塩−ネビボロール
4.2gのラセミ型A/BジアステレオマーであるN−{2−[(t−ブチルジメチルシリル)オキシ]−2−(6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピラニル)エチル}−6−フルオロ−3,4−ジヒドロ−α−ヒドロキシ−2H−2−[1]ベンゾピランエタンアミンを75mlのTHFと75mlのMeOHに入れることで生じさせた溶液を25mlの6N塩酸を混合した後、70℃で1時間撹拌する。次に、それに蒸発による濃縮を元々の体積の半分になるまで受けさせ、350mlの水を用いた希釈そして石油状物エーテルを用いた抽出を受けさせる。その水相を2NのNaOHで塩基性にした後、酢酸エチルを用いた抽出を数回実施する。その有機相を一緒にして水で洗浄し、乾燥させた後、溶媒を真空下で除去する。
【0054】
収量:3.2g(97%),黄色の油状物。
【0055】
その油状物を30mlのメタノールで取り上げ、ジエチルエーテル中2NのHCl(6ml)と混合した後、−20℃で放置することで結晶化させる。メタノールを用いた再結晶化で痕跡量の混入物であるジアステレオマーを分離することができる。
【0056】
収量:1.15g(32.1%);1H−NMR:(MeOD):δ(ppm)=6.84−6.75(m,6H),4.14−4.11(m,1H),4.04−4.01(m,2H),3.95−3.91(m,1H),3.53(dd,1H),3.44−3.34(m,2H),3.26(dd,1H),2.96−2.78(m,4H),2.29−2.22(m,1H),2.05−1.89(m,2H),1.84−1.74(m,1H).
【0057】
要約として、本発明に従う方法を用いると所望の相対的キラル中心配置を有するネビボロールのラセミ型ジアステレオマーが高い収率および満足される純度で得られると述べることができる。これは、本発明に従い、相当するジアステレオマーシアノヒドリンを生じさせ、分離しそしてその分離したジアステレオマーに変換、好適にはシアノ基に還元をある程度または完全に受けさせるか或はピンナー鹸化を受けさせた後、それらを互いにカップリングさせることで実施可能である。
【技術分野】
【0001】
本発明は、ラセミ型の製薬学的物質であるネビボロール(nebivolol)を遊離塩基または製薬学的に適合し得る塩、好適には塩酸塩として製造する方法に関する。
【背景技術】
【0002】
塩酸塩として市場に出ている式I,
【0003】
【化1】
【0004】
で表される製薬学的物質であるネビボロールは、ベータ受容体遮断薬の製薬学的物質群に属する。この分子はキラル中心を4個有しかつ同時に中心対称的(centrosymmetrically)構造を有する。認可された製薬学的物質であるネビボロールはラセミ体として用いられる。それは対称的構造を有することから、考慮すべきジアステレオマーは、8種類のジアステレオマー(理論的には16種類の鏡像異性体による)ではなく、正式には、原則として10種類の異性体を伴う6種類(R*R*R*R*,R*R*S*S*,R*R*R*S*,R*R*S*R*,R*S*R*S*およびR*S*S*R*)のみである。後者の中で、R*R*R*S*と表示する高純度のジアステレオマー(これは絶対配置がRRRSおよびSSSRの鏡像異性体の1:1の混合物である)が実際に認可された有効成分である。前記ネビボロールの両方の鏡像異性体が異なるプロファイルを示すことがそれが示す薬理学的作用に対して相乗的に貢献していることから極めて特殊な状況がもたらされる。従って、可能な最高水準として、高純度の鏡像異性体を直接合成するのではなく、必要な相対的キラル中心配置を有するラセミ型ジアステレオマーを合成することを考慮することができる。
【0005】
ラセミ型ネビボロール自身を合成する目的で直接利用可能な合成は文献に見られない。確かに、そのような成分の合成方法はいくつか記述されてはいるが、個々の立体異性体に分離する目的で各場合ともクロマトグラフィー段階が用いられており(例えば特許文献1)、それの説明は不明瞭なままであることから、技術的に使用可能であることをそれから引き出すのは原則として不可能である。注目すべきは、ネビボロールの高純度鏡像異性体を多少とも有利に合成する方法はかなり数多く見られる(例えば非特許文献1、2)。そのような合成法を前記ラセミ型有効成分の製造に移行させるには鏡像異性体を個別に製造する必要があるが、しかしながら、その後にそれらを1:1で混合しなければならない。従って、合成計画の目的にとって、ラセミ型の有効成分を製造する方が高純度の鏡像異性体を製造するよりも価値がある。
【0006】
ネビボロールは、中心元素以外に、正式には、相対的立体化学は別として同じ2個の分子部分で構成されている。それらの2個のキラル中心の相対的立体化学はR*R*およびR*S*として示される。実際に相対的な形態を有するが、ジアステレオマー中間体生成物をこれらの異なる形態に関して以下にAおよびBと識別する。全く選択不能なネビボロール合成法(このような方法では可能なあらゆる立体異性体が生じそしてそれらを分離す
る)ではなく、キラル中心の相対的立体化学AおよびBを有する2種類の中間体生成物を連結させることで合成を実施する方策を提供する。
【0007】
【特許文献1】EP 145067
【非特許文献1】S.Chandrasekhar,V.Reddy,Tetrahedron,56(2000),6339−6344
【非特許文献2】C.W.Johannes,M.S.Visser,G.S.Weatherhead,A.H.Hoveyda,J.Am.Chem.Soc.120(1998),8340−8347
【発明の開示】
【0008】
本発明の主題は、ネビボロールの製造方法であり、ここでは、一般式2
【0009】
【化2】
【0010】
[式中、Xは、X=Hの意味を有するか、或はXは、シアノヒドリンの典型的な保護基、例えばベンジル型(例えばベンジルまたは4−メトキシベンジル)またはアセタール型保護基(例えばテトラヒドロピラニルまたはMEM)、好適にはシリル保護基、極めて特に好適にはt−ブチルジメチルシリル保護基を意味する]
で表されるラセミ型のジアステレオマーシアノヒドリン(即ち以下に示すジアステレオマーAおよびB)を中間体生成物として用いる。
【0011】
本発明に従う方法の他の有利な態様を従属請求項に従って開示する。
【0012】
本発明は、更に、結晶性のラセミ型ジアステレオマーAとしてのα−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリルにも関する。
【0013】
本発明は、また、油状物状のラセミ型ジアステレオマーBとしてのα−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリルにも関する。
【0014】
前記シアノヒドリンの製造は、アルデヒド3
【0015】
【化3】
【0016】
にシアノヒドリン反応を受けさせることで実施可能である。
【0017】
また、前記アルデヒド3も、例えばエステル4
【0018】
【化4】
【0019】
のピラン環に還元を受けさせた後にエステル基に還元を直接または間接的(即ちアルコールを用いて)に受けさせることでアルデヒドを生じさせることなどで得ることができる。
【0020】
前記シアノヒドリン反応は立体選択的または非立体選択的に実施可能であるが、好適な変法では、非立体選択的合成をO−誘導体化を用いて実施する。シリル保護基を持つ式2で表される化合物のジアステレオマーもまた前記アルデヒド3から相当するシアン化シリル(シアン化t−ブチルジメチルシリルが好適である)を用いて1段階で生じさせることができる。カップリングに先立って前記成分の追加的合成を導入し、その結果として生じた配置AおよびBを有するジアステレオマーシアノヒドリン2(両方とも追加的合成に必要である)の分離を実施する。本発明の観点で特に好適なジアステレオマーは、結晶化特性が明瞭に異なる誘導体化ジアステレオマーAおよびB、好適には一方のジアステレオマーが結晶形態で存在しそしてもう一方が油状物であるジアステレオマーである。このようにすると、有機溶媒中でダイジェッションまたは熟成(digestion)により、技術的に極めて特に簡潔でありかつ効率の良いジアステレオマー分離を行うことが可能になる。本発明の観点でt−ブチルジメチルシリル保護基が極めて特に好適である。前記2種類のジアステレオマーの中の一方(A)は特に良好に結晶化する一方、もう一方のジアステレオマー(B)は油状物として得られ、非極性溶媒、好適には低級アルカン、特にヘキサンを用いてそれを分離すると、Aの比率が低くても、結晶化で冷却することでそれを容易に分離することができる。非極性溶媒、好適には低級アルカン、特にヘキサンを用いた再結晶化によって、結晶性のジアステレオマーをより高い純度にすることができる。
【0021】
連結部を生じさせる目的で、配置AまたはBを有する2個の分子部分をいろいろな様式で更に反応させる。その目的でシアノヒドリンを中間体生成物として用いるのが特に有利であることが分かる、と言うのは、シアノヒドリン基をいろいろな様式で更に反応させることで、前記2個の分子部分(AおよびB)の連結に最適な条件を得ることができるからである。さらなる反応に好適な条件は下記である:Oが保護されている一般式5[式中、Xは、式2で示した意味を有する]で表されるアルデヒドを生じさせる還元ばかりでなく、Oが保護されているか或はOが保護されていない一般式6[式中、Xは、式2で示した意味を有する]で表されるアミノアルコールを生じさせる還元、およびOが保護されていない一般式7[式中、Rは、分枝もしくは非分枝低級アルキル、または置換もしくは非置換ベンジルを意味する]で表されるヒドロキシルエステルを生じさせるピンナー(Pinne)鹸化。
【0022】
【化5】
【0023】
ある時点で、式2で表される結晶性のシアノヒドリンAおよび式2で表される油状物状のシアノヒドリンBの両方に還元を受けさせることでアルデヒド5またはヒドロキシエステル7を生じさせ、そして別の時点で、アミン6を生じさせる。この場合、製造および次に行う反応シーケンスを実施する時に収率に差が生じる。
【0024】
ネビボロールを生じさせる連結および反応を、好適には、アルデヒドとアミンの反応をシッフ塩基生成による還元アミノ化で起こさせた後に好適には1槽反応で還元を限られた還元力を有する複合水素化物、例えばシアノホウ水素化ナトリウムまたはトリアセトキシホウ水素化ナトリウムなどを用いて起こさせることで一般式8,
【0025】
【化6】
【0026】
[式中、Xは、式2で示した意味を有する]
で表される化合物を生じさせることを通して実施するか、或は一般式7で表されるエステルと一般式6で表されるアミンを反応させて一般式9
【0027】
【化7】
【0028】
[式中、Xは、化合物2で示した意味を有する]
で表されるアミドを生じさせた後にアミド9に還元を受けさせて一般式10
【0029】
【化8】
【0030】
[式中、Xは、化合物2で示した意味を有する]
で表されるアミンを生じさせることを通して実施する。
【0031】
式8および10で表される化合物を場合により前以て精製しておいた後、保護基を開裂させ、そして場合により、その塩酸塩および/または遊離塩基を再結晶化で精製してネビボロールの塩基または製薬学的に受け入れられる塩を製薬学的品質で生じさせることを通してネビボロールを得る。
【0032】
このカップリングで場合によりOが保護されているか或はOが保護されていない誘導体を使用、特にOが保護されているアルデヒドとOが保護されていないアミンのカップリングを実施すると、式10で表されるモノ保護ネビボロールを精製することで、場合によりカップリング後に少量存在する可能性のある副生成物であるジアステレオマーを特に効率良く分離することが可能になる。X=t−ブチルジメチルシリルの場合のそのような合成変法を下記の式図で示す。
【0033】
【化9】
【0034】
配置Aを有する分子と配置Bを有する分子をカップリングさせると均一なジアステレオマーではなく、むしろ2種類のジアステレオマーの混合物が生じることを注目すべきである。R*R*配置を有する化合物とR*S*配置を有する化合物をカップリングさせると、即ちジアステレオマーR*R*R*S*(鏡像異性体RRRSとSSSRで構成されるネビボロール)とR*R*S*R*(鏡像異性体RRSRとSSRSで構成される)が生
じる。実際、ラセミ型ネビボロールは、遊離塩基としてかつ塩酸塩として非常に容易に結晶化する化合物である。前記カップリングを1:1の比率で実施した時に生じるR*R*S*R*配置を有するジアステレオマーを遊離塩基の再結晶化および塩酸塩の再結晶化で特に容易に取り出すことができることが特に有利である。このように、必要でないジアステレオマーが示す溶解性が有意に高いことを基にして、前記ジアステレオマー混合物を再結晶化させることで高純度のネビボロールを得ることができる。
【0035】
本発明の説明を本発明の実施に可能な態様を基にして以下により詳細に行う。
【0036】
実施例1
(R*,R*/R*,S*)6−フルオロ−3,4−ジヒドロ−α−ヒドロキシ−2H−2−[1]ベンゾピランアセトニトリル−ラセミ型ジアステレオマー混合物
33gの(R*S*)6−フルオロ−3,4−ジヒドロ−2H−[1]ベンゾピラン−2−カルボアルデヒドを150mlのMTBEと150mlの80%酢酸に入れることで生じさせた溶液を23.8gのシアン化カリウムと混合した後、アルゴン雰囲気下室温で1時間撹拌する。次に、この反応混合物を600mlの冷却しておいた飽和炭酸ナトリウム水溶液に穏やかに撹拌しながら滴下した後、固体状の炭酸ナトリウムを反応溶液が中性様式で反応するまで加える。次に、それにMTBEを用いた抽出を数回受けさせ、その有機相を水で洗浄し、Na2SO4で乾燥させ、1滴の燐酸と混合した後、溶媒を真空下で除去する。
【0037】
収量:36.1g(95%),黄色がかった油状物。
1H−NMR:(CDCl3):δ(ppm)=6.82−6.74(m,3H),4.20−4.14(dd,1H),4.20−4.14(m,1H),2.84−2.79(m,2H),2.21−2.02(m,1H),2.00−1.88(m,1H).
【0038】
実施例2
(R*,R*/R*,S*)− −α−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2−2H−[1]ベンゾピランアセトニトリル−ラセミ型のジアステレオマー混合物
変法1:
25gのイミダゾールを150mlの無水ジメチルホルムアミドに入れることで生じさせた0℃の溶液に31.5gの塩化t−ブチルジメチルシリルを加えた後、その混合物を0℃で15分間撹拌する。次に、36gのシアノヒドリン(実施例1)を150mlの無水ジメチルホルムアミドに入れることで生じさせた溶液を滴下する。この滴下が終了した後、それを0℃で更に15分間撹拌した後、室温にして、2時間撹拌する。次に、その反応混合物を飽和NaHCO3溶液とMTBEの間で分離させ、その水相にMTBEを用いた抽出を受けさせ、その有機相を一緒にして水で洗浄し、Na2SO4で乾燥させた後、真空下で蒸発させることで濃縮する。トルエンを用いた洗浄を数回実施することでDMFを除去した後、生成物を高真空下で乾燥させる。
【0039】
収量:53.1g(95%),黄色の油状物;1H−NMR:(CDCl3):δ(ppm)=6.84−6.78(m,3H),4.75/4.62(dd,1H),4.25−4.07(m,1H),2.89−2.85(m,2H),2.37−2.17(m,1H),2.03−1.85(m,1H),0.97(d,9H),0.23(t,6H).
【0040】
変法2:シアン化t−ブチルジメチルシリルを使用
(R*S*)6−フルオロ−3,4−ジヒドロ−2H−[1]ベンゾピラン−2−カルボアルデヒド(2g)を20mlの無水DCMに入れることで生じさせた溶液に1.77
gのヨウ化亜鉛を加える。−10℃で1.88gのシアン化t−ブチルジメチルシリルを加えた後、その混合物を前記温度で2時間撹拌する。次に、その反応混合物を5%のNaHCO3溶液とMTBEの間で分離させ、その水相にMTBEを用いた抽出を2回受けさせ、その有機相を一緒にして水で洗浄し、Na-2SO4で乾燥させた後、真空下で蒸発させることで濃縮する。生成物を高真空下で乾燥させる。
【0041】
収量:3.1g(87%),黄色の油状物。
1H−NMR:(CDCl3):δ(ppm)=6.83−6.79(m,3H),4.73/4.61(dd,1H),4.23−4.07(m,1H),2.90−2.85(m,2H),2.37−2.15(m,1H),2.01−1.83(m,1H),0.96(d,9H),0.21(t,6H).
【0042】
実施例3
α−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリル−結晶性のラセミ型ジアステレオマーA
実施例2で得た16gのラセミ型ジアステレオマー混合物を20x量の石油状物エーテルに溶解させ、−45℃にした後、結晶性ジアステレオマー成分の種晶を加える。4時間後、母液を傾斜法で除去し、少量の深凍結石油状物エーテルを用いて結晶を熟成させ、前記エーテルを傾斜法で除去し、その母液を一緒にして蒸発による濃縮を元々の溶液の体積の1/3になるまで実施し、種晶を再び加えた後、前記手順を繰り返すことで生成物を2回目の収穫で得る。さらなる精製を行う目的で、R*,S*−ジアステレオマー(残存するR*,R*−ジアステレオマー含有量が8%)を再び石油状物エーテルに加熱しながら溶解させ、低温で析出させることで結晶性画分を得る。
【0043】
収量:結晶画分:6.5g=40%,白色結晶;引火点=66℃.
1H−NMR:(CDCl3):δ(ppm)6.83−6.73(m,3H),4.71(d,1H),4.09−4.05(m,1H),2.91−2.79(m,2H),2.30−2.25(m,1H),1.97−1.88(m,1H),0.94(s,9H),0.23(s,3H),0.19(s,3H).
【0044】
実施例4
α−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリル−油状物状のラセミ型ジアステレオマーB
ジアステレオマーAの結晶化で2回取り出した母液に蒸発による濃縮を受けさせることでジアステレオマーB(残存するジアステレオマーの含有量=10%)を得る;収量:9.0g=55%,黄色の油状物;1H−NMR:(CDCl3):δ(ppm),6.83−6.72(m,3H),4.58(d,1H),4.18−4.14(m,1H),2.90−2.77(m,2H),2.22−2.17(m,1H),1.93−1.85(m,1H),0.92(d,9H),0.24(s,3H),0.16(s,3H).
【0045】
実施例5
α−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトアルデヒド−ラセミ型のジアステレオマーB
実施例4で得た8gの油状物状ラセミ型ジアステレオマーシアノヒドリンBを80mlのトルエンに入れることで生じさせた溶液を5℃にした後、トルエン中1.5Mの水素化ジイソブチルアルミニウム溶液(18.25ml)と混合する。その反応混合物を室温で1時間撹拌する。次に、その反応混合物を600mlの1N塩酸に加えた後、100mlのMTBEで希釈する。相分離を起こさせた後、その水相に200mlずつのMTBEを用いた抽出を更に3回受けさせる。その有機相を一緒にして飽和塩化ナトリウム溶液で洗
浄し、Na2SO4で乾燥させた後、溶媒を真空下で除去する。
【0046】
収量:7.1g(88%),黄色の油状物.
1H−NMR:(CDCl3):δ(ppm)=9.79(s,1H),6.84−6.77(m,3H),4.32−4.25(m,1H),3.73−3.65(m,1H),2.84−2.79(m,2H),2.00−1.91(m,2H),0.98(d,9H),0.25(t,6H).
【0047】
実施例6a
6−フルオロ−3,4−ジヒドロ−α−ヒドロキシ−2H−2−[1]ベンゾピランエタンアミン−ラセミ型ジアステレオマーA
α−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリルの結晶性ラセミ型ジアステレオマーA(5g)を50mlのトルエンに入れることで生じさせた−20℃の溶液にトルエン中1.5MのDIBAL溶液を20.7ml滴下する。それを室温に加熱して15分間撹拌する。次に、その反応混合物を0℃に冷却した後、1.18gの水素化リチウムアルミニウムと混合する。次に、その反応混合物を室温で14時間撹拌する。次に、トルエン中1.5MのDIBAL溶液を更に20.7ml加え、室温で1時間そして35℃で1時間撹拌する。その反応混合物を酢酸エチルが400mlでMeOHが20mlの冷溶液に加える。この目的で、300mlの酒石酸カリウム溶液および30mlの2N NaOHを加えて1時間撹拌した。その反応混合物をセライトの上に置いて濾過し、その残留物とセライトを150mlずつの酢酸エチルで3回再洗浄する。相分離を起こさせ、その有機相を水で洗浄し、乾燥させた後、溶媒を真空下で除去する。結晶性の粗生成物を3.49g得る。この後者の再結晶化をMTBEを用いて実施する。
【0048】
収量:2.15g(65.5%),白色の固体;引火点=124℃.
1H−NMR:(CDCl3):δ(ppm)=6.81−6.77(m,3H),4.02−3.95(m,1H),3.71−3.65(m,1H),3.00−2.81(m,4H),2.04−1.85(m,5H).
【0049】
実施例6b
6−フルオロ−3,4−ジヒドロ−α−ヒドロキシ−2H−2−[1]ベンゾピランエタンアミン ラセミ型ジアステレオマーB
α−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリルのラセミ型ジアステレオマーB(4g)を40mlのトルエンに入れることで生じさせた−40℃の溶液にトルエン中1.5MのDIBAL溶液を16.6ml滴下する。それを室温に加熱して15分間撹拌する。次に、その反応混合物を0℃に冷却した後、1.18gの水素化リチウムアルミニウムと混合する。次に、その反応混合物を室温で14時間撹拌する。次に、トルエン中の1.5MのDIBAL溶液を更に16.6ml加え、室温で1時間そして35℃で1時間撹拌する。その反応混合物を酢酸エチルが300mlでMeOHが20mlの冷溶液に加える。これに250mlの酒石酸カリウム溶液および25mlの2N NaOHを加えた後、1時間撹拌する。その反応混合物をセライトの上に置いて濾過し、その残留物とセライトを120mlずつの酢酸エチルで3回再洗浄する。相分離を起こさせた後、その有機相を水で洗浄し、乾燥させた後、溶媒を真空下で除去する。結晶性の粗生成物を2.4g得る。後者の再結晶化をMTBEを用いて実施する。
【0050】
収量:1.7g(64.6%),白色の固体:1H−NMR:(CDCl3):δ(ppm)=6.78−6.65(m,3H),3.87−3.84(m,1H),3.67−3.63(m,1H),3.00−2.71(m,4H),2.51(bs,1H),
2.14−2.10(m,1H),1.84−1.76(m,1H).
【0051】
実施例7
N−{2−[(t−ブチルジメチルシリル)オキシ]−2−(6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピラニル)エチル]−6−フルオロ−3,4−ジヒドロ−α−ヒドロキシ−2H−2[1]ベンゾピランエタンアミン−ラセミ型A/Bジアステレオマー
6−フルオロ−3,4−ジヒドロ−α−ヒドロキシ−2H−2−[1]ベンゾピランエタンアミンのラセミ型ジアステレオマーA(2.28g)とα−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトアルデヒドのラセミ型ジアステレオマーB(7g)を250mlのTHFと50mlのメタノールに入れることで生じさせた溶液に0.81gのシアノホウ水素化ナトリウムを加える。1mlの氷酢酸を加えた後、室温で14時間撹拌する。次に、シアノホウ水素化ナトリウムを更に0.81g加える。それを室温で更に18時間撹拌する。その反応混合物を400mlの5%NaHCO3溶液と混合し、5分間撹拌した後、酢酸エチルを用いた抽出を数回実施する。その有機相を乾燥させた後、溶媒を真空下で除去する。そのようにして得た粗生成物(10.1g)をフラッシュクロマトグラフィー(可動溶媒DCM/MeOH 50+1)で精製する。黄色の油状物を4.2g得る。
【0052】
1H−NMR:(CDCl3):δ(ppm)=6.81−6.75(m,6H),4.09−3.95(m,4H),2.92−2.65(m,8H),2.10−1.98(m,2H),1.87−1.72(m,2H),0.94(s,9H),0.18(s,3H),0.13(s,3H).
【0053】
実施例8
(R*,R*,R*,S*)−α,α’−[イミノビス(メチレン)ビス[6−フルオロ−3,4−ジヒドロ−2H−1−ベンゾピラン−2−メタノール]]の塩酸塩−ネビボロール
4.2gのラセミ型A/BジアステレオマーであるN−{2−[(t−ブチルジメチルシリル)オキシ]−2−(6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピラニル)エチル}−6−フルオロ−3,4−ジヒドロ−α−ヒドロキシ−2H−2−[1]ベンゾピランエタンアミンを75mlのTHFと75mlのMeOHに入れることで生じさせた溶液を25mlの6N塩酸を混合した後、70℃で1時間撹拌する。次に、それに蒸発による濃縮を元々の体積の半分になるまで受けさせ、350mlの水を用いた希釈そして石油状物エーテルを用いた抽出を受けさせる。その水相を2NのNaOHで塩基性にした後、酢酸エチルを用いた抽出を数回実施する。その有機相を一緒にして水で洗浄し、乾燥させた後、溶媒を真空下で除去する。
【0054】
収量:3.2g(97%),黄色の油状物。
【0055】
その油状物を30mlのメタノールで取り上げ、ジエチルエーテル中2NのHCl(6ml)と混合した後、−20℃で放置することで結晶化させる。メタノールを用いた再結晶化で痕跡量の混入物であるジアステレオマーを分離することができる。
【0056】
収量:1.15g(32.1%);1H−NMR:(MeOD):δ(ppm)=6.84−6.75(m,6H),4.14−4.11(m,1H),4.04−4.01(m,2H),3.95−3.91(m,1H),3.53(dd,1H),3.44−3.34(m,2H),3.26(dd,1H),2.96−2.78(m,4H),2.29−2.22(m,1H),2.05−1.89(m,2H),1.84−1.74(m,1H).
【0057】
要約として、本発明に従う方法を用いると所望の相対的キラル中心配置を有するネビボロールのラセミ型ジアステレオマーが高い収率および満足される純度で得られると述べることができる。これは、本発明に従い、相当するジアステレオマーシアノヒドリンを生じさせ、分離しそしてその分離したジアステレオマーに変換、好適にはシアノ基に還元をある程度または完全に受けさせるか或はピンナー鹸化を受けさせた後、それらを互いにカップリングさせることで実施可能である。
【特許請求の範囲】
【請求項1】
ラセミ型の製薬学的物質であるネビボロールを遊離塩基としてか或は製薬学的に適合し得る塩、好適には式1
【化1】
に従う塩酸塩として製造する方法であって、合成を一般式2の以下に示すジアステレオマーAおよびB:
【化2】
[式中、Xは、水素に相当するか、或はXは、シアノヒドリンの典型的な保護基を意味する]
で表されるラセミ型のジアステレオマーであるシアノヒドリンを用いて完成することを特徴とする方法。
【請求項2】
前記シアノヒドリンの典型的な保護基Xがベンジル型保護基、例えばベンジルまたは4−メトキシベンジル保護基など、またはアセタール型保護基、例えばテトラヒドロピラニルまたはMEM保護基などである請求項1記載の方法。
【請求項3】
前記シアノヒドリンの典型的な保護基Xがシリル保護基、好適にはt−ブチルジメチルシリル保護基である請求項1記載の方法。
【請求項4】
前記ジアステレオマーAとBを結晶化で互いに分離する請求項1から3の1項記載の方法。
【請求項5】
結晶性ジアステレオマーAを非結晶性ジアステレオマーからダイジェッション(digestion)により分離する請求項4記載の方法。
【請求項6】
ネビボロールをもたらすための連結部を生じさせる目的で、配置AおよびBを有する一般式2で表される2種類のジアステレオマーをいろいろな方法で更に反応させるが、反応として、一般式5
【化3】
[式中、Xは、式2で示した意味を有する]
で表されるアルデヒドを生じさせる還元ばかりでなく、一般式6,
【化4】
[式中、Xは、式2で示した意味を有する]
で表されるアミノアルコールを生じさせる還元、および一般式7,
【化5】
[式中、Rは、分枝もしくは非分枝低級アルキル、または置換もしくは非置換ベンジルを意味する]
で表されるヒドロキシルエステルを生じさせるピンナー(Pinner)鹸化が好適である請求項1から5の1項記載の方法。
【請求項7】
前記一般式5で表されるアルデヒドを生じさせる還元を複合水素化物、例えば水素化ジイソブチルアルミニウムなどを用いて実施する請求項6記載の方法。
【請求項8】
前記式6で表されるアミンを生じさせる還元を複合水素化物、例えば水素化リチウムアルミニウムなどを好適には水素化ジイソブチルアルミニウムと組み合わせて用いて実施する請求項6記載の方法。
【請求項9】
前記式7で表されるヒドロキシエステルを生じさせるピンナー鹸化をアルコールROHを過剰量で存在させて無水エーテル、好適にはジエチルエーテル、THFまたはMTBEの中に冷却を温度が10℃未満になるように行いながら酸としての塩酸を気体として導入するか或はエーテル中の溶液として添加することで実施する請求項6記載の方法。
【請求項10】
前記合成シーケンス中に、一般式2で表されるジアステレオマーBに還元を受けさせることでXが式2で示した意味を有する式5で表されるアルデヒドを生じさせ、そして一般式2で表されるジアステレオマーAに還元を受けさせることでXが式2で示した意味を有する式6で表されるアミノアルコールを生じさせ、そしてこの2種類の成分を互いに還元アミノ化で反応させることで、保護基Xで保護されている一般式8
【化6】
で表されるネビボロールを生じさせ、その後、場合により前以て精製しておいた後、前記保護基Xを開裂させることでネビボロールを得、そして場合により、前記塩酸塩または別の容易に結晶化する塩および/または遊離塩基を再結晶化させることで精製して製薬学的
品質にする請求項1から9の1項記載の方法。
【請求項11】
前記合成シーケンス中に、一般式2で表されるジアステレオマーBに還元を受けさせることで酸素が保護されている式5で表されるアルデヒド(ここで、Xは、水素を除く、式2で示した意味を有する)を生じさせ、そして一般式2で表されるジアステレオマーAに還元を受けさせることでXが水素と同じ意味を有する式6で表されるアミノアルコールを生じさせ、そしてこの2種類の成分を互いに還元アミノ化で反応させることで、保護基Xで保護されている一般式10
【化7】
で表されるネビボロールを生じさせる請求項1から9の1項記載の方法。
【請求項12】
式2に従うジアステレオマーシアノヒドリンAおよびBを更に反応させることでネビボロールを生じさせる時に、一般式2で表されるジアステレオマーAに還元を受けさせることで式5で表されるアルデヒドを生じさせそして一般式2で表されるジアステレオマーBに還元を受けさせることで式6で表されるアミノアルコールを生じさせる請求項10または11記載の方法。
【請求項13】
ネビボロールを生じさせる目的で、式2で表されるジアステレオマーシアノヒドリンAおよびBのさらなる反応を実施し、一般式7で表されるエステルと一般式6で表されるアミンを反応させて一般式9,
【化8】
[式中、Xは、化合物2で示した意味を有する]
で表されるアミドを生じさせた後、それに還元を受けさせることで一般式10,
【化9】
[式中、Xは、化合物2で示した意味を有する]
で表されるアミンを生じさせ、それを場合により前以て精製しておいた後、保護基を開裂
させることでネビボロールを得、そして場合により前記塩酸塩または別の容易に結晶化する塩および/または遊離塩基を再結晶化させることで精製して製薬学的品質にする請求項1から12の1項記載の方法。
【請求項14】
式2で表されるジアステレオマーシアノヒドリンAおよびBを更に反応させてネビボロールを生じさせる目的で、一般式2で表されるジアステレオマーBにピンナー鹸化による還元を受けさせることで式7で表されるヒドロキシエステルを生じさせそして一般式2で表されるジアステレオマーAに還元を受けさせることで酸素が保護されている一般式6で表されるアミノアルコールを生じさせる請求項1から13の1項記載の方法。
【請求項15】
式2で表されるジアステレオマーシアノヒドリンAおよびBを更に反応させてネビボロールを生じさせる目的で、一般式2で表されるジアステレオマーAにピンナー鹸化による還元を受けさせることで式7で表されるヒドロキシエステルを生じさせそして一般式2で表されるジアステレオマーBに還元を受けさせることで酸素が保護されている一般式6で表されるアミノアルコールを生じさせる請求項1から14の1項記載の方法。
【請求項16】
一般式2で表されるシアノヒドリンを製造する方法であって、式3
【化10】
で表されるアルデヒドを反応させて相当するシアノヒドリンを生じさせた後、保護基X、好適にはt−ブチルジメチルシリル保護基を導入する方法。
【請求項17】
結晶性のラセミ型ジアステレオマーAとしてのα−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリル。
【請求項18】
油状物状のラセミ型ジアステレオマーBとしてのα−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリル。
【請求項1】
ラセミ型の製薬学的物質であるネビボロールを遊離塩基としてか或は製薬学的に適合し得る塩、好適には式1
【化1】
に従う塩酸塩として製造する方法であって、合成を一般式2の以下に示すジアステレオマーAおよびB:
【化2】
[式中、Xは、水素に相当するか、或はXは、シアノヒドリンの典型的な保護基を意味する]
で表されるラセミ型のジアステレオマーであるシアノヒドリンを用いて完成することを特徴とする方法。
【請求項2】
前記シアノヒドリンの典型的な保護基Xがベンジル型保護基、例えばベンジルまたは4−メトキシベンジル保護基など、またはアセタール型保護基、例えばテトラヒドロピラニルまたはMEM保護基などである請求項1記載の方法。
【請求項3】
前記シアノヒドリンの典型的な保護基Xがシリル保護基、好適にはt−ブチルジメチルシリル保護基である請求項1記載の方法。
【請求項4】
前記ジアステレオマーAとBを結晶化で互いに分離する請求項1から3の1項記載の方法。
【請求項5】
結晶性ジアステレオマーAを非結晶性ジアステレオマーからダイジェッション(digestion)により分離する請求項4記載の方法。
【請求項6】
ネビボロールをもたらすための連結部を生じさせる目的で、配置AおよびBを有する一般式2で表される2種類のジアステレオマーをいろいろな方法で更に反応させるが、反応として、一般式5
【化3】
[式中、Xは、式2で示した意味を有する]
で表されるアルデヒドを生じさせる還元ばかりでなく、一般式6,
【化4】
[式中、Xは、式2で示した意味を有する]
で表されるアミノアルコールを生じさせる還元、および一般式7,
【化5】
[式中、Rは、分枝もしくは非分枝低級アルキル、または置換もしくは非置換ベンジルを意味する]
で表されるヒドロキシルエステルを生じさせるピンナー(Pinner)鹸化が好適である請求項1から5の1項記載の方法。
【請求項7】
前記一般式5で表されるアルデヒドを生じさせる還元を複合水素化物、例えば水素化ジイソブチルアルミニウムなどを用いて実施する請求項6記載の方法。
【請求項8】
前記式6で表されるアミンを生じさせる還元を複合水素化物、例えば水素化リチウムアルミニウムなどを好適には水素化ジイソブチルアルミニウムと組み合わせて用いて実施する請求項6記載の方法。
【請求項9】
前記式7で表されるヒドロキシエステルを生じさせるピンナー鹸化をアルコールROHを過剰量で存在させて無水エーテル、好適にはジエチルエーテル、THFまたはMTBEの中に冷却を温度が10℃未満になるように行いながら酸としての塩酸を気体として導入するか或はエーテル中の溶液として添加することで実施する請求項6記載の方法。
【請求項10】
前記合成シーケンス中に、一般式2で表されるジアステレオマーBに還元を受けさせることでXが式2で示した意味を有する式5で表されるアルデヒドを生じさせ、そして一般式2で表されるジアステレオマーAに還元を受けさせることでXが式2で示した意味を有する式6で表されるアミノアルコールを生じさせ、そしてこの2種類の成分を互いに還元アミノ化で反応させることで、保護基Xで保護されている一般式8
【化6】
で表されるネビボロールを生じさせ、その後、場合により前以て精製しておいた後、前記保護基Xを開裂させることでネビボロールを得、そして場合により、前記塩酸塩または別の容易に結晶化する塩および/または遊離塩基を再結晶化させることで精製して製薬学的
品質にする請求項1から9の1項記載の方法。
【請求項11】
前記合成シーケンス中に、一般式2で表されるジアステレオマーBに還元を受けさせることで酸素が保護されている式5で表されるアルデヒド(ここで、Xは、水素を除く、式2で示した意味を有する)を生じさせ、そして一般式2で表されるジアステレオマーAに還元を受けさせることでXが水素と同じ意味を有する式6で表されるアミノアルコールを生じさせ、そしてこの2種類の成分を互いに還元アミノ化で反応させることで、保護基Xで保護されている一般式10
【化7】
で表されるネビボロールを生じさせる請求項1から9の1項記載の方法。
【請求項12】
式2に従うジアステレオマーシアノヒドリンAおよびBを更に反応させることでネビボロールを生じさせる時に、一般式2で表されるジアステレオマーAに還元を受けさせることで式5で表されるアルデヒドを生じさせそして一般式2で表されるジアステレオマーBに還元を受けさせることで式6で表されるアミノアルコールを生じさせる請求項10または11記載の方法。
【請求項13】
ネビボロールを生じさせる目的で、式2で表されるジアステレオマーシアノヒドリンAおよびBのさらなる反応を実施し、一般式7で表されるエステルと一般式6で表されるアミンを反応させて一般式9,
【化8】
[式中、Xは、化合物2で示した意味を有する]
で表されるアミドを生じさせた後、それに還元を受けさせることで一般式10,
【化9】
[式中、Xは、化合物2で示した意味を有する]
で表されるアミンを生じさせ、それを場合により前以て精製しておいた後、保護基を開裂
させることでネビボロールを得、そして場合により前記塩酸塩または別の容易に結晶化する塩および/または遊離塩基を再結晶化させることで精製して製薬学的品質にする請求項1から12の1項記載の方法。
【請求項14】
式2で表されるジアステレオマーシアノヒドリンAおよびBを更に反応させてネビボロールを生じさせる目的で、一般式2で表されるジアステレオマーBにピンナー鹸化による還元を受けさせることで式7で表されるヒドロキシエステルを生じさせそして一般式2で表されるジアステレオマーAに還元を受けさせることで酸素が保護されている一般式6で表されるアミノアルコールを生じさせる請求項1から13の1項記載の方法。
【請求項15】
式2で表されるジアステレオマーシアノヒドリンAおよびBを更に反応させてネビボロールを生じさせる目的で、一般式2で表されるジアステレオマーAにピンナー鹸化による還元を受けさせることで式7で表されるヒドロキシエステルを生じさせそして一般式2で表されるジアステレオマーBに還元を受けさせることで酸素が保護されている一般式6で表されるアミノアルコールを生じさせる請求項1から14の1項記載の方法。
【請求項16】
一般式2で表されるシアノヒドリンを製造する方法であって、式3
【化10】
で表されるアルデヒドを反応させて相当するシアノヒドリンを生じさせた後、保護基X、好適にはt−ブチルジメチルシリル保護基を導入する方法。
【請求項17】
結晶性のラセミ型ジアステレオマーAとしてのα−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリル。
【請求項18】
油状物状のラセミ型ジアステレオマーBとしてのα−[(t−ブチルジメチルシリル)オキシ]−6−フルオロ−3,4−ジヒドロ−2H−2−[1]ベンゾピランアセトニトリル。
【公表番号】特表2009−502750(P2009−502750A)
【公表日】平成21年1月29日(2009.1.29)
【国際特許分類】
【出願番号】特願2008−521739(P2008−521739)
【出願日】平成18年7月17日(2006.7.17)
【国際出願番号】PCT/AT2006/000303
【国際公開番号】WO2007/009143
【国際公開日】平成19年1月25日(2007.1.25)
【出願人】(508020340)フアルマコン−フオルシユング・ウント・ベラトウング・ゲゼルシヤフト・ミツト・ベシユレンクテル・ハフツング (1)
【Fターム(参考)】
【公表日】平成21年1月29日(2009.1.29)
【国際特許分類】
【出願日】平成18年7月17日(2006.7.17)
【国際出願番号】PCT/AT2006/000303
【国際公開番号】WO2007/009143
【国際公開日】平成19年1月25日(2007.1.25)
【出願人】(508020340)フアルマコン−フオルシユング・ウント・ベラトウング・ゲゼルシヤフト・ミツト・ベシユレンクテル・ハフツング (1)
【Fターム(参考)】
[ Back to top ]