
ネラメキサンの製造方法
【課題】1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩を製造する方法であって、次のステップ(i)から(iv)から選択される二つ以上のステップを具備する方法;ステップ(i)においては、塩化メチルマグネシウム存在下でイソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換する;ステップ(ii)においては、塩化メチルマグネシウム存在下で3,3,5,5−テトラメチルシクロヘキサノンを1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する;ステップ(iii)においては、酸性溶液中でクロロアセトニトリル存在下で1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する;ステップ(iv)においては、水中でチオ尿素存在下で1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン(ネラメキサン)又はその薬学的に許容される塩を製造する方法に関する。
【背景技術】
【0002】
1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン(ネラメキサン)及びその薬理学的に許容される塩は、耳鳴症や眼振症のような疾患及び症状を患う患者の持続的療法にとって重要な薬剤である。
【0003】
これらの薬剤を製造する複数の方法が知られている。
【0004】
ある方法においては、次の反応スキームによる5つのステップを含む反応順序で、市販のイソフォロンをネラメキサンに変換する(非特許文献1)。
【化1】

【0005】
前記反応順序の第一ステップにおいては、塩化銅触媒を用いたヨウ化メチルマグネシウムの共役付加によって、イソフォロン(1)を3,3,5,5−テトラメチルシクロヘキサノン(2)に変換する。
【0006】
また、塩化第一銅存在下でイソフォロンに臭化メチルマグネシウムを添加することにより、3,3,5,5−テトラメチルシクロヘキサノンが収率82.5重量%で生成し得ることが知られている。副産物として、1,3,5,5−テトラメチルシクロヘキサジエンが収率6.9重量%で生成する(非特許文献2)。
【0007】
同文献の実験の部(2313頁)の開示では、塩化第二鉄存在下でイソフォロンに塩化メチルマグネシウムを添加している。しかしながら、3,3,5,5−テトラメチルシクロヘキサノンは形成されず、それとは全く異なる生成物が単離されてしまう。
【0008】
第二ステップにおいては、ヨウ化メチルマグネシウムを用いたグリニャール反応によって、3,3,5,5−テトラメチルシクロヘキサノン(2)を1,3,3,5,5−ペンタメチルシクロヘキサノール(3)に変換する。
【0009】
非特許文献3による開示では、3,3,5,5−テトラメチルシクロヘキサノンとハロゲン化メチルマグネシウムとの反応により1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを製造している。前記生成物はシリカゲルカラムクロマトグラフィーによって精製された。
【0010】
第三ステップにおいては、リッター反応によって、クロロアセトニトリルにより前記シクロヘキサノール(3)を1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサン(6)に変換する。
【0011】
続いて第四ステップにおいては、チオ尿素を用いてアミド(6)中のクロロアセトアミド基を開裂し、生じたアミンを前記反応順序の最終第五ステップにおいて塩酸により酸性化することで、塩酸塩型のネラメキサン(1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン)(7)が生ずる。
【0012】
アミド(6)中のクロロアセトアミド基の開裂は、イルゲンソンス(Jirgensons)らによっても詳しく研究されている(非特許文献4)。それによると、エタノールと酢酸との5:1混合物中で1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを還流する。10時間の反応後、反応混合物を水で希釈し、生成する沈殿物を単離する。ろ液をアルカリ性化し、ヘキサンで抽出する。塩酸の添加後、塩酸塩型の1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンが収率89重量%で単離される。
【0013】
前記文献によれば、前記反応順序の5つのステップによる総収量は約50重量%である。
【先行技術文献】
【非特許文献】
【0014】
【非特許文献1】ダニス(Danysz)ら、「カレント・ファーマシューティカル・デザイン(Current Pharmaceutical Design)」、2002年、第8巻、p.835−843
【非特許文献2】カラシュ(Kharasch)ら、「ジャーナル・オブ・ザ・アメリカン・ケミカル・ソサエティー(Journal of the American Chemical Society)」、1941年、第63巻、p.2308
【非特許文献3】イルゲンソンス(Jirgensons)ら、「ヨーロピアン・ジャーナル・オブ・メディシナル・ケミストリー(European Journal of Medicinal Chemistry)」、2000年、第35巻、p.555−565
【非特許文献4】イルゲンソンス(Jirgensons)ら、「シンセシス(Synthesis)」、2000年、第12号、p.1709−1712
【発明の概要】
【発明が解決しようとする課題】
【0015】
本発明の目的の一つは、経済的な工業規模での有利な実施を可能とする1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩の製造方法を提供するために、上記で参照した各反応ステップのうち一つ又は複数のステップを改良することである。また別の目的は、ネラメキサン又はその薬理学的に許容される塩の製造過程で生ずる廃棄物及び/又は未反応化学物質の量を極少化することである。さらなる目的は、ネラメキサン又はその薬理学的に許容される塩について収率及び/又は選択性、及び/又は生成物品質を最適化又は改善することである。そのような改良された方法は、経済的な工業規模でネラメキサン又はその薬理学的に許容される塩を有利に製造するための必要条件であると考えてよい。
【課題を解決するための手段】
【0016】
本発明は、次のステップ(i)から(iv)から選択される二つ以上のステップを具備する、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩を製造する方法に関する。ステップ(i)においては、塩化メチルマグネシウム存在下でイソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換する。ステップ(ii)においては、塩化メチルマグネシウム存在下で3,3,5,5−テトラメチルシクロヘキサノンを1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iii)においては、酸性溶液中でクロロアセトニトリル存在下で1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iv)においては、水中でチオ尿素存在下で1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。
【0017】
一つの実施形態においては、ステップ(i)及び(ii)を選択する。
【0018】
一つの実施形態においては、ステップ(i)及び(iii)を選択する。
【0019】
一つの実施形態においては、ステップ(i)及び(iv)を選択する。
【0020】
一つの実施形態においては、ステップ(ii)及び(iii)を選択する。
【0021】
一つの実施形態においては、ステップ(ii)及び(iv)を選択する。
【0022】
一つの実施形態においては、ステップ(iii)及び(iv)を選択する。
【0023】
一つの実施形態においては、ステップ(i)、(ii)、及び(iii)を選択する。
【0024】
一つの実施形態においては、ステップ(i)、(ii)、及び(iv)を選択する。
【0025】
一つの実施形態においては、ステップ(i)、(iii)、及び(iv)を選択する。
【0026】
一つの実施形態においては、ステップ(ii)、(iii)、及び(iv)を選択する。
【0027】
一つの実施形態においては、ステップ(i)、(ii)、(iii)、及び(iv)を選択する。
【0028】
一つの実施形態においては、3,3,5,5−テトラチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち少なくとも一つは精製ステップに供さない。
【0029】
一つの実施形態においては、本方法はさらにステップ(v)を具備する。ステップ(v)においては、酸存在下で1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンをその薬理学的に許容される塩に変換する。
【0030】
一つの実施形態においては、前記酸はメタンスルホン酸である。
【0031】
一つの実施形態においては、前記塩化メチルマグネシウムには塩化エチルマグネシウムが含まれない。
【0032】
本発明は一つの態様においては、1−アミノ−1−エチル−3,3,5,5−テトラメチルシクロヘキサン及び1−アミノ−3−エチル−1,3,5,5−テトラメチルシクロヘキサン、又はそれらの薬理学的に許容される塩を実質的に含まない、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩に関する。
【0033】
本発明のステップ(i)から(iv)を含む反応順序においては、蒸留、再結晶、又はクロマトグラフィーのような古典的な精製法によって3,3,5,5−テトラメチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち一つ又は複数を精製しなくてもよいことを開示する。従って、前記化合物のうち一つ又は複数を精製ステップに供さず、未精製形態で使用する。
【発明の効果】
【0034】
一つ又は複数の未精製の反応中間体を使用することによって医療用途に十分な純度の前記目標化合物(すなわちネラメキサン又は薬理学的に許容される塩形態のネラメキサン)が得られるということは、過去には期待できなかった。蒸留、再結晶、又はクロマトグラフィーのような反応中間体の複雑な洗浄ステップは一般的に生成物の損失の原因となる。しかし本発明の方法によればこのステップが省略可能であるために、ネラメキサン又はその薬理学的に許容される塩の収率を60重量%以上とすることが可能である。よって、ネラメキサンを生産する本発明の新規簡易法は有利で経済的な工業規模で実施し得る。
【図面の簡単な説明】
【0035】
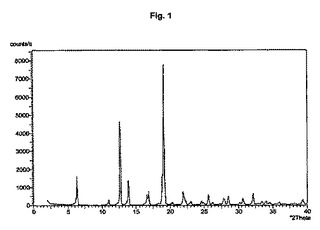
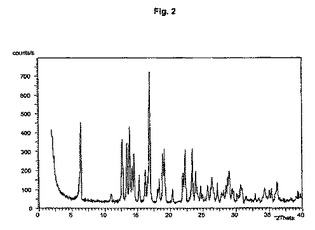
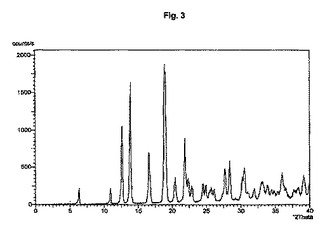
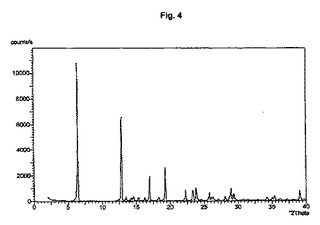
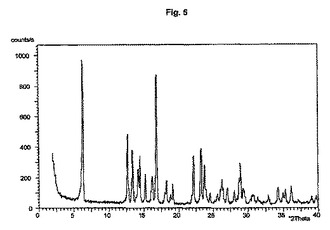
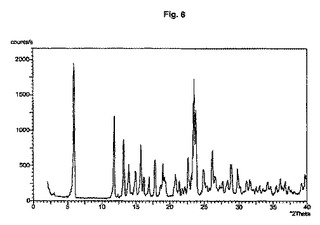
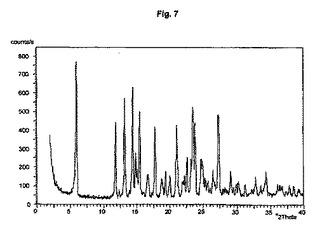
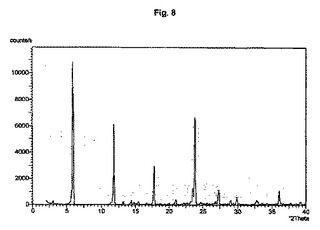
図1から10は、結晶形A、A’、B、C、D、及びEの粉末X線回折のダイアグラムを示す。x軸は2Θ(°)/d[Å]、y軸は回折強度(相対単位)である。
【図1】結晶形Aのダイアグラムを示す。
【図2】結晶形A(粉砕試料)のダイアグラムを示す。
【図3】結晶形A’ のダイアグラムを示す。
【図4】結晶形Bのダイアグラムを示す。
【図5】結晶形B(粉砕試料)のダイアグラムを示す。
【図6】結晶形Cのダイアグラムを示す。
【図7】結晶形C(粉末試料)のダイアグラムを示す。
【図8】結晶形Dのダイアグラムを示す。
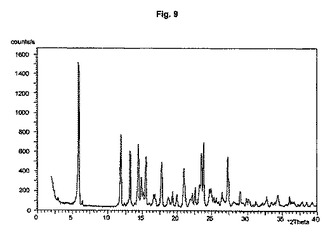
【図9】結晶形D(粉砕試料)のダイアグラムを示す。
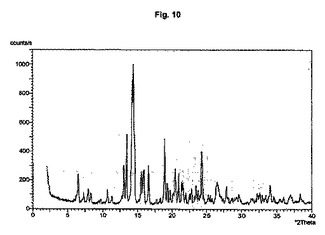
【図10】結晶形Eのダイアグラムを示す。
【発明を実施するための形態】
【0036】
本発明は、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン(ネラメキサン)又はその薬理学的に許容される塩を製造する方法に関する。
【0037】
本発明は具体的には、次のステップ(i)から(iv)から選択される二つ以上のステップを具備する、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩を製造する方法に関する。ステップ(i)においては、塩化メチルマグネシウム存在下でイソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換する。ステップ(ii)においては、塩化メチルマグネシウム存在下で3,3,5,5−テトラメチルシクロヘキサノンを1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iii)においては、酸性溶液中でクロロアセトニトリル存在下で1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iv)においては、水中でチオ尿素存在下で1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。
【0038】
ステップ(i):塩化メチルマグネシウム存在下でのイソフォロンから3,3,5,5−テトラメチルシクロヘキサノンへの変換
ステップ(i)における変換は、イソフォロンと塩化メチルマグネシウムとの反応によって実施する。
【0039】
そのようなグリニャール試薬は、マグネシウムと塩化メチルとから作製してもよい。
【0040】
一つの実施形態においては、ステップ(i)における変換は、銅化合物の存在下で実施する。前記銅化合物は、イソフォロンに対するグリニャール試薬の1,4−共役付加を1,2−付加よりも優先的に進めるための触媒として働き得る。
【0041】
一つの実施形態においては、前記銅化合物はハロゲン化第一銅である。一つの実施形態においては、前記ハロゲン化第一銅はヨウ化第一銅、臭化第一銅、又は塩化第一銅からなる群から選択する。
【0042】
一つの実施形態においては、前記第一銅化合物(例えば塩化第一銅又はヨウ化第一銅のようなハロゲン化第一銅)はリチウム化合物存在下で提供される。
【0043】
一つの実施形態においては、前記リチウム化合物は塩化リチウムのようなハロゲン化リチウムである。
【0044】
一つの実施形態においては、塩化第一銅又はヨウ化第一銅が塩化リチウム存在下で提供される。
【0045】
一つの実施形態においては、塩化第一銅又はヨウ化第一銅存在下で塩化メチルマグネシウムを反応させる。
【0046】
一つの実施形態においては、ヨウ化第一銅又は塩化第一銅と塩化リチウムとの存在下におけるイソフォロンと塩化メチルマグネシウムとの反応によって、ステップ(i)の変換反応を実施する。
【0047】
一つの実施形態においては、ハロゲン化第一銅とハロゲン化リチウムとのモル比は1:1.5から1:2.5の範囲である。
【0048】
一つの実施形態においては、塩化第一銅と塩化リチウムとのモル比又はヨウ化第一銅と塩化リチウムとのモル比は、それぞれ約1:1.5から1:2.5、又は1:2である。
【0049】
通常、ステップ(i)の反応は溶媒中で実施する。
【0050】
一つの実施形態においては、ステップ(i)の反応に使用する溶媒はエーテルであるか又はエーテルを含む溶媒である。
【0051】
適当なエーテルは、ジエチルエーテル、1,4−ジオキサン、及びテトラヒドロフランからなる群から選択してもよい。
【0052】
一つの実施形態においては、前記エーテルはテトラヒドロフランである。
【0053】
一つの実施形態においては、ステップ(i)で使用する前記溶媒はテトラヒドロフランであるか、又はテトラヒドロフランを含む。
【0054】
一つの実施形態においては、塩化メチルマグネシウムと、塩化第一銅又はヨウ化第一銅と、塩化リチウムとの存在下、テトラヒドロフラン中でイソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換する。
【0055】
一つの実施形態においては、塩化メチルマグネシウムとヨウ化第一銅と塩化リチウムとの存在下、テトラヒドロフラン中でイソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換する。
【0056】
一つの実施形態においては、イソフォロンと、ハロゲン化第一銅のような第一銅化合物(例えばヨウ化第一銅又は塩化第一銅)と、及び任意にハロゲン化リチウムのようなリチウム化合物(例えば塩化リチウム)とを溶媒中に用意し、前記混合物にグリニャール試薬を任意で溶媒に溶解したものを添加する。
【0057】
また別の実施形態においては、塩化メチルマグネシウムと、ハロゲン化第一銅のような銅化合物(例えばヨウ化第一銅又は塩化第一銅)とを、任意にハロゲン化リチウムのようなリチウム化合物(例えば塩化リチウム)の存在下で反応させる。一つの実施形態においては、前記混合物をイソフォロンに添加する。また別の実施形態においては、イソフォロンを前記混合物に添加する。
【0058】
また別の実施形態においては、塩化メチルマグネシウムと、ヨウ化第一銅又は塩化第一銅のような銅化合物とを反応させる。
【0059】
一つの実施形態においては、イソフォロンとヨウ化第一銅と塩化リチウムとの混合物を、テトラヒドロフラン中に用意する。テトラヒドロフランに溶解した塩化メチルマグネシウムをこの混合物に添加する。
【0060】
一つの実施形態においては、テトラヒドロフラン中の塩化メチルマグネシウム濃度は、塩化メチルマグネシウム及びテトラヒドロフランの総量を基準として15から30重量%又は20から25重量%である。
【0061】
一つの実施形態においては、テトラヒドロフラン中の塩化メチルマグネシウム濃度は、塩化メチルマグネシウム及びテトラヒドロフランの総量を基準として23重量%である。
【0062】
一つの実施形態においては、1モル当量のイソフォロン当たり、1モル当量よりも多い塩化メチルマグネシウムを使用する。
【0063】
一つの実施形態においては、1モル当量のイソフォロン当たり、1.0から1.75モル当量の塩化メチルマグネシウム又は1.2から1.5モル当量の塩化メチルマグネシウムを使用する。
【0064】
一つの実施形態においては、テトラヒドロフラン中の塩化メチルマグネシウム濃度は、塩化メチルマグネシウム及びテトラヒドロフランの総量を基準として23重量%であり、塩化メチルマグネシウム及びテトラヒドロフランの量を基準として10重量%の触媒(1モル当量のヨウ化第一銅及び2モル当量の塩化リチウム)を使用する。
【0065】
一つの実施形態においては、1モル当量のイソフォロン当たり、0.1から0.25モル当量の塩化リチウムと0.05から0.125モル当量のヨウ化第一銅とを使用する。
【0066】
一つの実施形態においては、温度管理下で前記添加を実施する。
【0067】
一つの実施形態においては、温度が比較的狭い温度範囲に維持されるように前記添加を実施する。
【0068】
一つの実施形態においては、−5℃から20℃、又は0℃から20℃、又は−5℃から15℃、又は1℃から10℃の温度でステップ(i)の変換を実施する。
【0069】
一つの実施形態においては、グリニャール試薬とイソフォロンとの反応は通常比較的速く進行する。前記反応は通常、使用する反応温度に依存して3時間、2時間、又は1時間の短時間で終了する。
【0070】
グリニャール試薬とイソフォロンとの反応後、グリニャール試薬が過剰量使用されている場合にそれを分解するため、及び塩基性マグネシウム化合物を分解するために、前記混合物を水で処理してもよい。
【0071】
一つの実施形態においては、3,3,5,5−テトラメチルシクロヘキサノンの形成を補助するために、塩酸のような酸又はアンモニウム塩を添加する。
【0072】
一つの実施形態においては、形成された有機層を水層から分離する。次に揮発性有機化合物を減圧下で除去することにより、前記有機層を濃縮してもよい。残留物が粗3,3,5,5−テトラメチルシクロヘキサノンである。
【0073】
一つの実施形態においては、ステップ(i)で形成される生成物は、塩化メチレン、トルエン又は石油エーテルのような適当な有機溶媒で前記水性混合物を抽出することによって生成及び単離される。抽出後、前記溶媒を蒸留除去する。生成し単離された状態の粗3,3,5,5−テトラメチルシクロヘキサノンを含む残留液を、精製ステップにかけることなく、反応順序のステップ(ii)に使用する。
【0074】
また別の実施形態においては、抽出の次に前記抽出物を従来の方法によって乾燥してもよい。例えば硫酸ナトリウムによって抽出物を乾燥してよい。前記硫酸塩をろ過分離した後、溶媒を蒸留除去してもよい。生成し単離された状態の粗3,3,5,5−テトラメチルシクロヘキサノンを含む残留物を、精製ステップにかけることなく、反応順序のステップ(ii)に使用してよい。
【0075】
一つの実施形態においては、ステップ(i)で生成し単離された状態の粗3,3,5,5−テトラメチルシクロヘキサノンの収率は88から96重量%の範囲である。
【0076】
一つの実施形態においては、前記粗生成物には93重量%以上の量の目標化合物が含まれることがガス液体クロマトグラフィーにより測定できる。
【0077】
一つの実施形態においては、3,3,5,5−テトラメチルシクロヘキサノンを精製してもよい。一つの実施形態においては、3,3,5,5−テトラメチルシクロヘキサノンを蒸留してもよい。
【0078】
また別の実施形態においては、ステップ(i)で得られた状態の粗3,3,5,5−テトラメチルシクロヘキサノンをステップ(ii)で使用する。
【0079】
ステップ(ii):塩化メチルマグネシウム存在下での3,3,5,5−テトラメチルシクロヘキサノンから1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンへの変換
ステップ(ii)における3,3,5,5−テトラメチルシクロヘキサノンから1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンへの変換は、塩化メチルマグネシウムを用いて実施する。
【0080】
通常、ステップ(ii)の反応は溶媒中で実施する。
【0081】
一つの実施形態においては、前記溶媒はエーテルを含むか又はエーテルである。
【0082】
エーテルは、ジエチルエーテル、1,4−ジオキサン、又はテトラヒドロフランから選択してもよい。
【0083】
一つの実施形態においては、前記エーテルはテトラヒドロフランである。
【0084】
本発明の方法の一つの実施形態においては、3,3,5,5−テトラメチルシクロヘキサノンに塩化メチルマグネシウムを添加する。
【0085】
また別の実施形態においては、塩化メチルマグネシウムにテトラメチルシクロヘキサノンを添加する。
【0086】
一つの実施形態においては、3,3,5,5−テトラメチルシクロヘキサノンのテトラヒドロフラン溶液に塩化メチルマグネシウムのテトラヒドロフラン溶液を添加する。
【0087】
また別の実施形態においては、塩化メチルマグネシウムのテトラヒドロフラン溶液に3,3,5,5−テトラメチルシクロヘキサノンのテトラヒドロフラン溶液を添加する。
【0088】
一つの実施形態においては、テトラヒドロフラン中の塩化メチルマグネシウム濃度は、塩化メチルマグネシウム及びテトラヒドロフランの総量を基準として15から30重量%又は20から25重量%である。
【0089】
一つの実施形態においては、テトラヒドロフラン中の塩化メチルマグネシウム濃度は、塩化メチルマグネシウム及びテトラヒドロフランの総量を基準として23重量%である。
【0090】
従って一つの実施形態においては、塩化メチルマグネシウムとテトラヒドロフランとを含む混合物を、3,3,5,5−テトラメチルシクロヘキサノンとテトラヒドロフランとを含む混合物と反応させる。
【0091】
一つの実施形態においては、塩化メチルマグネシウムのテトラヒドロフラン溶液に3,3,5,5−テトラメチルシクロヘキサノンのテトラヒドロフラン溶液を添加する。1モル当量の3,3,5,5−テトラメチルシクロヘキサノン当たり、1.2から1.75モル当量の塩化メチルマグネシウム量が含まれる。
【0092】
一つの実施形態においては、ステップ(i)で得られた3,3,5,5−テトラメチルシクロヘキサノンのテトラヒドロフラン溶液を、塩化メチルマグネシウムのテトラヒドロフラン溶液に添加する。
【0093】
一つの実施形態においては、ステップ(i)で得られた3,3,5,5−テトラメチルシクロヘキサノンの1モル当量当たり、1.2から1.75モル当量の塩化メチルマグネシウムを使用する。
【0094】
また別の実施形態においては、ステップ(i)で得られた3,3,5,5−テトラメチルシクロヘキサノンを含むテトラヒドロフラン溶液に、塩化メチルマグネシウムを含むテトラヒドロフラン溶液を添加する。
【0095】
一つの実施形態においては、温度管理下で前記変換を実施する。
【0096】
一つの実施形態においては、温度が比較的狭い温度範囲に維持されるように前記変換を実施する。
【0097】
一つの実施形態においては、−5℃から30℃、又は0℃から30℃、又は0℃から20℃、又は0℃から25℃、又は0℃から20℃、又は5℃から20℃、又は10℃から25℃、又は15℃から25℃の温度でステップ(ii)の変換を実施する。
【0098】
ステップ(ii)で形成される1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを単離するためには、ステップ(i)の3,3,5,5−テトラメチルシクロヘキサノンの単離に関連する上記の方法と基本的に同じ方法を使用してよい。
【0099】
一つの実施形態においては、粗1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンの収率は90重量%から100重量%の範囲である。
【0100】
一つの実施形態においては、前記粗生成物には94重量%以上の量の目標化合物1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンが含まれることがガス液体クロマトグラフィーにより測定できる。
【0101】
一つの実施形態においては、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを精製してもよい。一つの実施形態においては、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを蒸留するか、又はクロマトグラフィーに供してもよい。
【0102】
また別の実施形態においては、ステップ(iii)で使用する1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンは粗生成物の状態である。
【0103】
ステップ(iii):酸性溶液中、クロロアセトニトリル存在下での1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンから1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンへの変換
ステップ(iii)における変換は、酸性溶液中における1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンとクロロアセトニトリルとの反応によって実施する。
【0104】
ステップ(iii)のリッター反応は、従来技術において参照された方法に従って実施してもよい。
【0105】
一つの実施形態においては、前記酸は硫酸、硝酸、リン酸、酢酸、又はそれらの混合物からなる群から選択する。
【0106】
一つの実施形態においては、前記酸は濃酸の状態で使用する。
【0107】
一つの実施形態においては、硫酸と酢酸とを使用する。一つの実施形態においては、硫酸は濃硫酸であり、酢酸は氷酢酸である。
【0108】
一つの実施形態においては、ステップ(ii)で生成し単離された状態の1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンとクロロアセトニトリルとを酢酸中に用意し、前記混合物に硫酸を添加する。
【0109】
また別の実施形態においては、ステップ(ii)で生成し単離された状態の1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを酢酸中に用意し、前記混合物にクロロアセトニトリルと硫酸との混合物を添加する。
【0110】
一つの実施形態においては、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンと酢酸とを重量比1:1.5から1:2.5で用意する。
【0111】
一つの実施形態においては、前記シクロヘキサノールと酢酸とを重量比約1:2で用意する。
【0112】
また別の実施形態においては、1モル当量の1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン当たり1.5から2.5モル当量のクロロアセトニトリルと2.5から3.5モル当量の硫酸とを使用する。
【0113】
また別の実施形態においては、1モル当量の1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン当たり約2モル当量のクロロアセトニトリルと3モル当量の硫酸とを使用する。
【0114】
一つの実施形態においては、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンと酢酸とを重量比1:1.5から1:2.5で用意し、1モル当量の1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン当たり1.5から2.5モル当量のクロロアセトニトリルと2.5から3.5モル当量の硫酸とを使用する。
【0115】
一つの実施形態においては、前記シクロヘキサノールと酢酸とを重量比約1:2で用意し、2モル当量のクロロアセトニトリルと3モル当量の硫酸とを使用する。
【0116】
一つの実施形態においては、0℃から30℃、又は0℃から20℃、又は0℃から15℃、又は5℃から10℃の範囲に反応温度が維持されるように、硫酸の添加又はクロロアセトニトリルと硫酸との混合物の添加を実施する。
【0117】
通常、前記反応は目標化合物の方向に比較的速く進行する。一つの実施形態においては、前記反応は2時間又は1時間という短時間で終了し得る。
【0118】
反応混合物を仕上げるために、前記反応終了後には前記混合物を水、氷又は氷水に注いでもよい。沈殿する1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンをろ過によって単離してもよい。
【0119】
付着する酸を除去するために、前記沈殿物を水で洗浄してもよい。
【0120】
一つの実施形態においては、粗生成物の収率は98から100重量%の範囲である。
【0121】
一つの実施形態においては、1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを精製してもよい。一つの実施形態においては、1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを再結晶してもよい。
【0122】
一つの実施形態においては、ステップ(iii)で得られた1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを、粗製物すなわち未精製生成物の状態でステップ(iv)に使用してもよい。前記化合物は乾燥状態又は未乾燥状態で使用してよい。
【0123】
ステップ(iv):水中、チオ尿素存在下での1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンから1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンへの変換
ステップ(iv)における変換は、水中における1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンとチオ尿素との反応によって実施する。
【0124】
一つの実施形態においては、ステップ(iv)で使用する混合物にはさらに有機溶媒が含まれる。
【0125】
一つの実施形態においては、前記有機溶媒はステップ(iv)の反応条件下で水混和性である、アルコールのような溶媒である。
【0126】
一つの実施形態においては、前記有機溶媒はメタノール、エタノール、プロパノール、ブタノール、及びエチレングリコールからなる群から選択されるアルコールである。
【0127】
一つの実施形態においては、前記有機溶媒の量は水の量を基準として0から200重量%である。また別の実施形態においては、前記有機溶媒の量は水の量を基準として0から150重量%、又は0から100重量%、又は0から50重量%、又は0から10重量%、又は0から5重量%である。
【0128】
また別の実施形態においては、ステップ(iv)で用いる混合物は実質的に有機溶媒を含まない。
【0129】
「実質的に有機溶媒を含まない」という語は、前記混合物の有機溶媒含有量が水の量を基準として0から5重量%、又は0から3重量%、又は0から1重量%であることを想定する。
【0130】
一つの実施形態においては、チオ尿素と水との重量比は1:0.5から1:50、又は1:1から1:20、又は1:2から1:10の範囲である。
【0131】
ステップ(iv)の反応は酸の添加なしに実施し得るが、そのような化合物の添加によって1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンから1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンへの変換は促進され得る。
【0132】
従って一つの実施形態においては、ステップ(iv)の混合物にはさらに酸が含まれる。
【0133】
使用し得る酸は、塩酸、硫酸、リン酸、p−トルエンスルホン酸、メタンスルホン酸、酢酸、及び安息香酸であるが、これらに限定されない。つまり無機酸及び有機酸を使用し得る。
【0134】
何らかの酸を使用する場合、その量の範囲は比較的広範である。
【0135】
一つの実施形態においては、前記混合物は水の量を基準として0.1から20重量%の量の酸を含む。
【0136】
一つの実施形態においては、使用する酸は塩酸である。
【0137】
1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンの変換をさらに促進するために、ステップ(iv)で使用する混合物を加熱する。
【0138】
「加熱」という語とは、ステップ(iv)で使用する混合物を環境温度よりも高温に調節することを想定する。
【0139】
一つの実施形態においては、ステップ(iv)で使用する混合物を50℃から前記混合物の還流温度までの範囲の温度に加熱する。
【0140】
また別の実施形態においては、前記混合物を80℃から前記混合物の還流温度までの範囲の温度に加熱する。
【0141】
さらにまた別の実施形態においては、前記混合物をその還流温度まで加熱する。
【0142】
ステップ(iv)において実質的に有機溶媒を含まない混合物を使用する場合、還流温度は通常100℃前後、すなわち95から105℃の範囲である。ステップ(iv)において有機溶媒を含む混合物を使用する場合の還流温度は、使用する有機溶媒の量及び沸点によって、水は含むが有機溶媒は実質的に含まない混合物の還流温度よりも高温又は低温であり得る。
【0143】
ステップ(iv)の1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンから1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンへの変換は、例えばガス液体クロマトグラフィーのような一般的なクロマトグラフィー法によって管理し得る。
【0144】
一つの実施形態においては、ステップ(iv)において、1モルの1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサン当たり1.0から2モルのチオ尿素と、1から3モルの酸と、チオ尿素及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンの量を基準として500から1,500重量%の水とを還流温度で使用する。
【0145】
一つの実施形態においては、前記1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンと、約1.2モル当量のチオ尿素及び2モル当量の塩酸とを、8倍量(チオ尿素及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを基準とした重量比)の水中で還流温度で反応させる。
【0146】
通常、ステップ(iv)の水を含む混合物中での変換は比較的速く進行する。
【0147】
一つの実施形態においては、実質的に有機溶媒を含まない水中でステップ(iv)を実施し、還流温度、すなわち100℃前後の温度で加熱を行い、酸を添加する場合、変換は2時間でも終了し得、又は1時間でさえ終了し得る。
【0148】
一つの実施形態においては、前記変換は6時間、5時間、4時間、3時間、又は3時間よりもさらに短時間でも終了する。
【0149】
前記変換が酸によって触媒される場合、生成するアミンの少なくとも一部はアミノ基のプロトン化によって水に溶解し、塩を形成する。
【0150】
一つの実施形態においては、生成するアミンを単離するために、本発明の方法はさらに前記混合物へのアルカリ添加を具備する。これはpH値を7以上に調節して前記混合物から1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンを分離するためである。
【0151】
前記実施形態においては、好ましくは前記混合物を冷却した後、アルカリ添加後にアミンが水相から分離して、相分離され得る。
【0152】
また別の実施形態においては、アルカリ添加後に有機相と水相とを形成する前記混合物から、水非混和性の有機溶媒によって前記アミンを抽出し得る。適当な溶媒は、塩化メチレン、トルエン、又は石油エーテルのような溶媒である。抽出後、硫酸ナトリウム等を用いて前記抽出物を乾燥してもよい。蒸発法によって溶媒を除去した後、粗アミンが得られる。
【0153】
一つの実施形態においては、粗生成物の収率は大体において理論値の95重量%よりも高く、又はほとんど定量的である。前記粗生成物は通常、ガス液体クロマトグラフィー試験によれば、95重量%以上、97重量%以上、又は99重量%以上という非常に多量の目標化合物を含む。
【0154】
一つの実施形態においては、必要に応じて蒸留により前記粗アミンをさらに精製する。
【0155】
ステップ(iv)で生成し単離された生成物は、それ以上精製することなく本発明の方法のステップ(v)に使用してもよい。
【0156】
しかしながら一つの実施形態においては、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンよりも揮発性が高い化合物を粗生成物から蒸留除去し、残留物をステップ(v)に使用することも可能である。
【0157】
一つの実施形態においては、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンを蒸留精製する。
【0158】
ステップ(iv)で用いる方法によれば、従来技術の方法において開示された反応時間と比較して反応時間がかなり短縮されるということが、期せずして明らかとなった。さらに背景技術のセクションで参照した水の添加と沈殿物のろ過とが不要となるため、本発明の方法によれば生成するアミンの仕上げがかなり単純化される。アミンの収率は高く、ほとんど定量的である。すなわち、本新規方法は経済的な工業規模で有利に実施し得る。
【0159】
ステップ(i)から(iv)のうち二つ以上のステップの選択
一つの実施形態においては、ステップ(i)から(iv)のうち二つのステップを選択する。
【0160】
従って一つの実施形態においては、ステップ(i)及び(ii)を選択する。
【0161】
また別の実施形態においては、ステップ(i)及び(iii)を選択する。
【0162】
また別の実施形態においては、ステップ(i)及び(iv)を選択する。
【0163】
また別の実施形態においては、ステップ(ii)及び(iii)を選択する。
【0164】
また別の実施形態においては、ステップ(ii)及び(iv)を選択する。
【0165】
また別の実施形態においては、ステップ(iii)及び(iv)を選択する。
【0166】
一つの実施形態においては、ステップ(i)から(iv)のうち三つのステップを選択する。
【0167】
従って一つの実施形態においては、ステップ(i)、(ii)、及び(iii)を選択する。
【0168】
一つの実施形態においては、ステップ(i)、(ii)、及び(iv)を選択する。
【0169】
また別の実施形態においては、ステップ(i)、(iii)、及び(iv)を選択する。
【0170】
また別の実施形態においては、ステップ(ii)、(iii)、及び(iv)を選択する。
【0171】
一つの実施形態においては、ステップ(i)から(iv)のうち四つのステップを選択する。
【0172】
従って一つの実施形態においては、ステップ(i)、(ii)、(iii)、及び(iv)を選択する。
【0173】
一つの実施形態においては、本方法は次のステップ(i)から(iv)から選択される二つ以上のステップを具備する。ステップ(i)においては、塩化メチルマグネシウム存在下かつヨウ化銅(I)と塩化リチウムとテトラヒドロフランとの存在下で、イソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換する。ステップ(ii)においては、塩化メチルマグネシウム存在下かつテトラヒドロフラン存在下で、3,3,5,5−テトラメチルシクロヘキサノンを1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iii)においては、クロロアセトニトリル存在下かつ酢酸と硫酸との存在下で1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iv)においては、チオ尿素存在下かつ水と塩酸との存在下で1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。
【0174】
未精製の3,3,5,5−テトラメチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンの使用
一つの実施形態においては、ステップ(i)から(iii)のそれぞれで得られた状態の3,3,5,5−テトラチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち少なくとも一つは精製ステップに供さない。
【0175】
従って本発明の方法では、反応中間体である3,3,5,5−テトラチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち少なくとも一つは、反応順序の先行ステップで得られたままの形態で該当の反応ステップにおいて使用することを想定する。すなわちステップ(i)から(iii)の反応順序で製造された反応中間体の少なくとも一つは、精製ステップに供さない。
【0176】
「精製ステップ」という語は、各反応ステップで生成した化合物の再結晶、蒸留、クロマトグラフィー、又はそれらの組み合わせを包含する。
【0177】
「精製ステップを経ていない」という語では、標準的な仕上げのステップは許容される。そのようなステップは、前記化合物(本明細書中では3,3,5,5−テトラチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサン)と溶媒とを含む混合物から前記溶媒を蒸留除去すること、溶媒によって前記化合物を水相から抽出すること、前記化合物と溶媒とを含む混合物を無水硫酸ナトリウムなどを用いて乾燥すること、前記化合物を減圧下で乾燥すること、及び固体化合物を液剤で洗浄することなどである。
【0178】
「再結晶」、「蒸留」、又は「クロマトグラフィー」による精製とは、有機化合物のような化学物質を精製するために実験室規模及び工業規模の両方で用いる古典的な方法である。
【0179】
再結晶とは、混合物中に含まれる複数化合物の単一溶媒又は混合溶媒に対する溶解度の差異に基づいて、混合物を分離する方法である。ある化合物を再結晶によって精製する場合、前記化合物を適当な溶媒に溶解した後に冷却する。その結果、所望の精製化合物が溶液から沈降(再結晶)する。しかし、所望の化合物が不溶性である別の溶媒を、所望の化合物の沈殿が始まるまで前記溶液に添加することも可能である。従って本発明の趣旨における「再結晶」という語は、精製化合物を形成させるために、化合物を溶解状態にし、前記溶解状態から自発的沈殿を生ぜしめるか又は人為的に沈殿させることを意味する。
【0180】
蒸留とは、沸騰液体混合物中における、混合物中に含まれる複数化合物の揮発度の差異に基づいて、混合物を分離する方法である。従って本発明の趣旨においては、「精製」という語の定義について使用する「蒸留」という語は、先ず化合物を液相から気相に相転移させる必要があり、次に凝縮させて精製物を形成させることを意味する。
【0181】
化学におけるクロマトグラフィーとは、混合物中に含まれる複数化合物の固定相−移動相間での分配の差異に基づいて混合物を分離し、精製物を形成させる方法である。一般的な方法はカラムクロマトグラフィーであり、種々の製造法に使用し得る。
【0182】
従って、ステップ(i)から(iii)の反応順序において製造した3,3,5,5−テトラチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち少なくとも一つは上記の精製ステップの何れにも供さず、前記の精製ステップを用いることなしに以降の各ステップ(ii)から(iv)で使用する。
【0183】
つまり本発明の方法の一つの実施形態においては、3,3,5,5−テトラチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち一つは、再結晶、蒸留、又はクロマトグラフィーの何れにも供さない。
【0184】
一つの実施形態においては、ステップ(i)で得られた3,3,5,5−テトラチルシクロヘキサノールは精製ステップに供さない。
【0185】
ステップ(i)で得られてステップ(ii)に使用する3,3,5,5−テトラチルシクロヘキサノンは、環境温度(25℃)においては液体である。一つの実施形態においては、前記化合物は蒸留に供さない。つまり、精製化合物を形成するために3,3,5,5−テトラチルシクロヘキサノンを液相から気相に相転移して凝縮させることはしない。
【0186】
前記化合物はクロマトグラフィーにも供さない。つまり、精製物を形成するために3,3,5,5−テトラチルシクロヘキサノンを固定相−移動相間に分配することはしない。
【0187】
ステップ(i)で得られた状態の粗化合物は高収率及び高純度であるので、反応順序の次のステップ(ii)で使用する3,3,5,5−テトラチルシクロヘキサノンは粗生成物のままでもよい。次のステップ(ii)で前記粗生成物の使用が可能であるのは、イソフォロンと塩化メチルマグネシウムとの反応が、ヨウ化メチルマグネシウム又は臭化メチルマグネシウムとの反応とは逆に上記の副産物の形成を限界まで抑制するためである。
【0188】
従ってイソフォロンから3,3,5,5−テトラチルシクロヘキサノンへの変換に塩化メチルマグネシウムを使用することは、臭化メチルマグネシウム及びヨウ化メチルマグネシウムの各々を使用することよりも有利である。これは具体的には、副産物が抑制されること、及び/又は高収率が達成可能であること、及び/又は得られた化合物を背景技術のセクションに記載の反応順序のステップ(ii)において粗生成状態で利用できることに関係する。蒸留、再結晶、又はクロマトグラフィーのような化合物の複雑な洗浄ステップは一般的に生成物の損失の原因となるが、本発明の方法によればこのステップを省略することが可能である。また、背景技術のセクションで参照したネラメキサン又はその薬理学的に許容される塩を製造する反応順序における次のステップ(ii)において、前記目標化合物は未精製形態で使用し得る。これらの理由から、本新規簡易方法は有利かつ経済的な工業規模で実施し得る。
【0189】
また別の実施形態においては、ステップ(ii)で得られた1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンは精製ステップに供さない。
【0190】
ステップ(ii)で得られてステップ(iii)に使用する1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンは、環境温度においては液体である。一つの実施形態においては、前記化合物は蒸留に供さない。つまり、精製物を形成するために1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを液相から気相に相転移して凝縮させることはしない。
【0191】
前記化合物はクロマトグラフィーにも供さない。つまり、精製物を形成するために1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを固定相−移動相間に分配することはしない。
【0192】
ステップ(ii)で得られる粗化合物は高収率及び高純度であるので、反応順序の次のステップ(iii)で使用する1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンは粗生成物のままでもよい。
【0193】
この実施形態においては、3,3,5,5−テトラチルシクロヘキサノンから1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンへの変換に塩化メチルマグネシウムを使用することは、臭化メチルマグネシウム及びヨウ化メチルマグネシウムの各々を使用することよりも有利である。これは具体的には、得られた化合物を背景技術のセクションに記載の反応順序において粗生成状態で利用できることと高収率が達成可能であることとに関係する。蒸留、再結晶、又はクロマトグラフィーのような反応中間体の複雑な洗浄ステップは一般的に生成物の損失の原因となるが、本発明の方法によればこのステップを省略することが可能である。また、背景技術のセクションで参照したネラメキサン又はその薬理学的に許容される塩を製造する反応順序における次の反応ステップ(iii)において、前記目標化合物は未精製形態で使用し得る。これらの理由から、本新規簡易方法は有利かつ経済的な工業規模で実施し得る。
【0194】
一つの実施形態においては、ステップ(iii)で得られた1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンは精製ステップに供さない。
【0195】
ステップ(iii)で得られてステップ(iv)に使用する1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンは、環境温度においては固体である。一つの実施形態においては、前記化合物は再結晶に供さない。つまり、精製物を形成するために1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを溶解状態にした上で前記溶解状態から自発的沈殿を生ぜしめるか又は人為的に沈殿させることはしない。
【0196】
前記化合物はクロマトグラフィーにも供さない。つまり、精製物を形成するために1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを固定相−移動相間に分配することはしない。
【0197】
ハロゲン化メチルマグネシウムと3,3,5,5−テトラメチルシクロヘキサノンとの反応で得られたままの1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを、蒸留又はクロマトグラフィーのような精製ステップにかけることなくステップ(iii)に使用することによって、収率90から100重量%という高収率の粗1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンが生成することが見い出されている。蒸留、再結晶、又はクロマトグラフィーのような反応中間体の複雑な洗浄ステップは一般的に生成物の損失の原因となるが、本発明の方法によればこのステップを省略することが可能である。また、背景技術のセクションで参照したネラメキサン又はその薬理学的に許容される塩を製造する反応順序における次の反応ステップ(iv)において、前記目標化合物は未精製形態で使用し得る。これらの理由から、本新規簡易方法は有利かつ経済的な工業規模で実施し得る。
【0198】
一つの実施形態においては、ステップ(i)で得られた3,3,5,5−テトラメチルシクロヘキサノン及びステップ(ii)で得られた1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンは精製ステップに供さない。
【0199】
また別の実施形態においては、ステップ(i)で得られた3,3,5,5−テトラメチルシクロヘキサノン、ステップ(ii)で得られた1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及びステップ(iii)で得られた1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンは精製ステップに供さない。
【0200】
一つの実施形態においては、ステップ(ii)で得られた1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン及びステップ(iii)で得られた1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンは精製ステップに供さない。
【0201】
また別の実施形態においては、ステップ(i)で得られた3,3,5,5−テトラメチルシクロヘキサノン及びステップ(iii)で得られた1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンは精製ステップに供さない。
【0202】
一つの実施形態においては、本方法は次のステップ(i)から(iv)から選択される二つ以上のステップを具備する。ステップ(i)においては、塩化メチルマグネシウム存在下かつヨウ化銅(I)と塩化リチウムとテトラヒドロフランとの存在下で、イソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換する。ステップ(ii)においては、塩化メチルマグネシウム存在下かつテトラヒドロフラン存在下で、ステップ(i)で得られた3,3,5,5−テトラメチルシクロヘキサノンを1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iii)においては、クロロアセトニトリル存在下かつ酢酸と硫酸との存在下で、ステップ(ii)で得られた1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iv)においては、チオ尿素存在下かつ水と塩酸との存在下で、ステップ(iii)で得られた1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。前記の3,3,5,5−テトラメチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち少なくとも一つは、精製ステップを経ない。
【0203】
ステップ(v):酸存在下での1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンからその塩への変換
一つの実施形態においては、適当な酸の添加によって1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンをその薬理学的に許容される塩に変換する。
【0204】
従って、本方法は追加のステップ(v)を具備する。ステップ(v)においては、酸存在下で1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンをその薬理学的に許容される塩に変換する。
【0205】
本開示の趣旨のため、「薬理学的に許容される塩」という語は、哺乳動物(例えばヒト)への投与時に生理学的に許容され、有害な反応を通常もたらさないネラメキサン塩を指す。「薬理学的に許容される塩」という語は、通常は哺乳動物、特にヒトに対する使用について、連邦政府又は州政府の規制当局により承認されるもの、又は米国薬局方もしくは他の一般に承認された薬局方のリストに記載されるものを意味する。
【0206】
1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンからその薬理学的に許容される塩への変換は、不活性な有機溶媒中で前記塩基と1モル当量以上の選択された酸とを混合することによって、従来の方法で達成する。前記塩の単離は、前記塩の溶解度が低い非極性溶媒(例えばエーテル)を用いた沈殿誘導のような、当分野の公知技術によって実施する。非毒性であって所望の薬理活性に実質的に干渉しない限り、前記塩の種類は重要ではない。
【0207】
薬理学的に許容される塩の例は、塩酸、臭化水素酸、メタンスルホン酸、酢酸、コハク酸、マレイン酸、クエン酸、及び関連する酸で形成されるものである。
【0208】
さらなる薬理学的に許容される塩には、ヨウ化水素酸、過塩素酸、硫酸、硝酸、リン酸、プロピオン酸、グリコール酸、乳酸、ピルビン酸、マロン酸、フマル酸、酒石酸、安息香酸、炭酸、桂皮酸、マンデル酸、エタンスルホン酸、ヒドロキシエタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸、シクロヘキサンスルファミン酸、サリシクリック酸、p−アミノサリチル酸、2−フェノキシ安息香酸、及び2−アセトキシ安息香酸で形成されるような酸付加塩が含まれるが、これらに限定されない。
【0209】
一つの実施形態においては、ステップ(iv)で生成し単離した1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンを、酸の添加前に単一溶媒又は混合溶媒に溶解又は分散もしくは懸濁する。
【0210】
適当な溶媒は、アセトン、アニソール、酢酸ブチル、t−ブチルメチルエーテル、クメン、ジメチルスルホキシド、酢酸エチル、エチルエーテル、ギ酸エチル、ヘプタン、酢酸イソブチル、酢酸イソプロピル、酢酸メチル、メチルエチルケトン、メチルイソブチルケトン、ペンタン、酢酸プロピル、テトラヒドロフラン、1,1−ジエトキシプロパン、1,1−ジメトキシメタン、2,2−ジメトキシプロパン、イソオクタン、イソプロピルエーテル、メチル−i−プロピルケトン、及びメチルテトラヒドロフランのような溶媒である。
【0211】
一つの実施形態においては、メチルエチルケトンと水とのような、溶媒と水との混合物を使用してもよい。
【0212】
溶解又は分散もしくは懸濁後に、塩を形成させるために適当な酸を添加する。前記酸も、上記の溶媒の一つ又は複数に溶解又は分散もしくは懸濁してもよい。
【0213】
前記沈殿及び/又は結晶の塩は、ろ過によって反応混合物から分離してよい。
【0214】
前記沈殿物に付着する溶媒は、乾燥及び/又は減圧によって除去し得る。
【0215】
一つの実施形態においては、使用する酸は塩酸であって、ステップ(v)で生成する塩は塩化物である。
【0216】
一つの実施形態においては、前記酸はメタンスルホン酸である。ステップ(v)で生成する塩はメシレートである。前記メシレートの融点は、示差走査熱量計により昇温速度10K/分で測定したところ173.1℃である。
【0217】
一つの実施形態においては、塩の収率は95重量%以上であって、98.5重量%以上の純度である。
【0218】
また別の実施形態においては、前記純度は99.9重量%以上である。
【0219】
一つの実施形態においては、ステップ(i)から(v)を具備する一連の反応の総収率は65重量%以上である。
【0220】
また別の実施形態においては、使用する酸は、臭化水素酸、酢酸、クエン酸、マレイン酸、又はコハク酸であって、生成する塩は、臭化物、酢酸塩(融点142.2℃)、モノクエン酸塩(融点151.5℃)、モノマレイン酸塩(融点160.1℃)、又はモノコハク酸塩(融点177.2℃)である。
【0221】
1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン塩は、多形又は擬多形で存在してもよい。
【0222】
「多形」という語は、固体物質が複数の形態又は結晶構造で存在し得ることを指す。
【0223】
「擬多形」という語は、水和又は溶媒和の結果として固体物質が異なるタイプの結晶を形成し得ることを指す。
【0224】
ネラメキサン塩酸塩は二つの多形及び三つの擬多形水和物の形で存在し得る。
【0225】
本開示の趣旨においては、前記の二つの多形を結晶形A及び結晶形Eと呼ぶ。
【0226】
本開示の趣旨においては、前記の三つの擬多形は、結晶形Bと呼ぶ一水和物、結晶形Cと呼ぶセスキ水和物、及び結晶形Dと呼ぶ三水和物である。
【0227】
一つの実施形態においては、約50℃、100mbarでネラメキサン塩酸塩を乾燥することによって結晶形Aを製造してよい。結晶形Aには約0.7重量%以下の量の水が含まれていてよい。しかし前記結晶形は完全に乾燥していてもよく、そのような結晶形は本明細書では結晶形A’と呼ぶ。
【0228】
結晶形A及び結晶形Eは互変二形関係にある。すなわち、これらは温度変化によって可逆的に相互変換し得る。低温型A(融点221℃)は少なくとも70℃までは熱力学的に安定である。70℃よりも高温では、低温型Aは高温型E(融点241℃)に転移する。
【0229】
25℃で約50%よりも高い相対湿度(r.h.)では、結晶形Aは水和物に変換し得る。結晶形Cは擬多形の中では最も安定な結晶形である。25℃で約25%よりも低い相対湿度、及び40℃で約33%よりも低い相対湿度では、結晶形Cは結晶形Aに変換し得る。次に安定な水和物は結晶形Bである。結晶形Dは水中に懸濁した状態でのみ安定である。
【0230】
一つの態様においては、本発明は1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン塩酸塩の結晶形A、結晶形A’、又は結晶形Eに関する。
【0231】
また別の態様においては、本発明は1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン塩酸塩の結晶形B、結晶形C、又は結晶形Dに関する。
【0232】
結晶多形及び擬多形は、粉末X線回折によって分析できる。結晶形A、B、及びDの試料は通常、3つから4つの強いピークを示す。粉砕試料のピーク強度は、非粉砕試料と比較して顕著な変化を示す。
【0233】
副産物としての1−アミノ−1−エチル−3,3,5,5−テトラメチルシクロヘキサン及び1−アミノ−3−エチル−1,3,5,5−テトラメチルシクロヘキサン
塩化メチルマグネシウムのようなメチルマグネシウム・グリニャール試薬を用いてステップ(i)の変換を実施する場合、ステップ(i)から(iv)又はステップ(i)から(v)の反応順序の一つの実施形態においては、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン及びその塩に加えて、目標化合物である1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキシルアミン又はその塩とは異なるアミノ化合物がさらに生成し得る。
【0234】
一つの実施形態においては、三つの副産物が生成し得る。それらは例えばガスクロマトグラフィー分析によって検出できる。
【0235】
一つの実施形態においては、副産物として1−アミノ−3−エチル−1,3,5,5−テトラメチルシクロヘキサンが生成し得る。この化合物は二つのキラル中心を持つため、二つのジアステレオマーが検出され得る。
【0236】
一つの実施形態においては、さらに1−アミノ−1−エチル−3,3,5,5−テトラメチルシクロヘキサンが生成する。
【0237】
一つの実施形態においては、1−アミノ−3−エチル−1,3,5,5−テトラメチルシクロヘキサンの生成原因を、ステップ(i)においてメチル基ではなくエチル基がイソフォロンに付加されて各シクロへキサノンが生じたことに帰属することができる。前記付加後にステップ(ii)から(iv)又はステップ(i)から(v)に類似の反応順序を実施する場合、それぞれ前記アミン又はその塩が形成される。
【0238】
一つの実施形態においては、1−アミノ−1−エチル−3,3,5,5−テトラメチルシクロヘキサンの生成原因を、ステップ(ii)においてメチル基ではなくエチル基が各シクロへキサノンのカルボニル基に付加されたことに帰属することができる。前記付加後にステップ(iii)から(iv)又はステップ(iii)から(v)に類似の反応順序を実施する場合、それぞれ前記アミン又はその塩が形成される。
【0239】
一つの実施形態においては、前記副産物の生成原因を、使用したメチルマグネシウム・グリニャール試薬にエチルマグネシウム・グリニャール試薬がコンタミネーションしたことに帰属することができる。
【0240】
一つの実施形態においては、塩化エチルマグネシウムのようなエチルマグネシウム・グリニャール試薬を含まない精製したメチルマグネシウム・グリニャール試薬を使用することによって、前記副産物の生成を抑制し得る。
【0241】
一つの実施形態においては、塩化メチルマグネシウムに含まれる塩化エチルマグネシウムは、塩化メチルマグネシウムと塩化エチルマグネシウムとの総量を基準として1重量%未満、0.5重量%未満、又は0.1重量%未満である。
【0242】
一つの実施形態においては、ステップ(iv)で得られたアミンを精製することにより、目標化合物から不必要な副産物を除去してもよい。一つの実施形態においては、前記アミンを蒸留精製して副産物を除去してもよい。
【0243】
また別の実施形態においては、ステップ(v)によって得られた塩を精製する。一つの実施形態においては、再結晶のステップによって前記塩を精製してもよい。適当な溶媒は、例えばステップ(v)で使用する溶媒から選択する。一つの実施形態においては前記溶媒はアニソールである。一つの実施形態においては前記塩はメシレートである。
【0244】
本発明は一つの態様においては、1−アミノ−1−エチル−3,3,5,5−テトラメチルシクロヘキサン、1−アミノ−3−エチル−1,3,5,5−テトラメチルシクロヘキサン、又はそれらの薬理学的に許容される塩を実質的に含まない、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩に関する。
【0245】
「実質的に含まない」という語は、前記副産物と1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩との総量を基準として、前記副産物の量が0.5重量%未満であることを指す。
【0246】
背景技術のセクションで参照した反応順序によると、目標化合物であるネラメキサンは約51%の収率で得られる。それに対応する本願のステップ(i)から(iv)を具備する反応順序の総収率は、65重量%以上である。実施例1から5によれば、約88%もの総収率で前記目標化合物が得られる。すなわち本発明の反応順序は、従来の反応順序と比較してネラメキサンの収率を改善する。一つ又は複数の未精製の反応中間体を使用することによって、医療用途に十分な純度の前記目標化合物、すなわちネラメキサン又は薬理学的に許容される塩形態のネラメキサンが得られるということは、過去には期待できなかった。蒸留、再結晶、又はクロマトグラフィーのような反応中間体の複雑な洗浄ステップは一般的に生成物の損失の原因となるが、本発明の方法によればこのステップを省略可能である。それゆえに、ネラメキサンを生産する本発明の新規簡易法は有利で経済的な工業規模で実施し得る。
【0247】
[実施例1]
139gのイソフォロンと19gのヨウ化銅(I)と8.4gの塩化リチウムと1,550gのテトラヒドロフランとの撹拌混合物に、93gの塩化メチルマグネシウムと372gのテトラヒドロフランとの混合物を滴加する。無機化合物は滴下前に溶解する。前記混合物の温度が5〜15℃に維持されるように滴下速度を選択する。滴加が終了した後、混合物を60分間撹拌する。次に、過剰の塩化メチルマグネシウムを分解するため、及び塩基性マグネシウム化合物を分解するために、希釈した塩酸を添加する。前記混合物を石油エーテルで二回抽出する。前記複数の抽出物を混合し、アンモニアで洗浄する。次に溶媒を蒸留除去する。粗目標化合物の収量は定量的である(153g)。前記粗生成物中の3,3,5,5−テトラメチルシクロヘキサノンの含有量は、ガス液体クロマトグラフィー試験によれば約91重量%である。前記粗生成物中には、約2重量%の未反応イソフォロンと、イソフォロンに対するグリニャール試薬の1,2−付加により生成する1重量%未満の1,3,5,5−テトラメチルシクロヘキサノール、又は前記化合物から生成するオレフィン、及び1重量%の1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンが含まれる。
【0248】
[実施例2]
実施例1で得られた153gの3,3,5,5−テトラメチルシクロヘキサノンと153gのテトラヒドロフランとの混合物を、93gの塩化メチルマグネシウムと372gのテトラヒドロフランとの撹拌混合物に滴加する。混合物の温度が5〜15℃に維持されるように滴下速度を選択する。滴加が終了した後、前記混合物を60分間撹拌する。次に、過剰の塩化メチルマグネシウムを分解するため、及び塩基性マグネシウム化合物を分解するために、希釈した塩酸を添加する。前記混合物を石油エーテルで二回抽出する。前記複数の抽出物を混合し、溶媒を蒸留除去する。1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンの粗収量は定量的である(170g)。前記粗生成物中の目標化合物含有量は、ガス液体クロマトグラフィー試験によれば約95重量%である。
【0249】
[実施例3]
320gの氷酢酸と、実施例2で得られた170gの粗1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンと、150gのクロロアセトニトリルとの撹拌混合物に、294gの濃硫酸を滴加する。反応混合物の温度が5から10℃に維持されるように滴下速度を選択する。滴加が終了した後、混合物をさらに60分間撹拌する。次に前記混合物を氷水に注ぐ。沈殿する目標化合物1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンをろ過分離する。乾燥後、230gの目標化合物が得られる。収率はほぼ定量的である(94重量%)。
【0250】
[実施例4]
実施例3に従って製造した245gの1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンと、91gのチオ尿素と、2,700gの水と、220gの塩酸(33%酸)との混合物を、還流下で加熱する。6時間の反応後に混合物を環境温度まで冷却し、水酸化ナトリウムを添加して混合物のpH値を7以上に調節する。次に、前記混合物を石油エーテルで二回抽出する。前記複数の抽出物を混合する。石油エーテルを蒸留除去した後、収率97重量%(159g)の粗1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンが得られる。前記粗生成物中の目標化合物含有量はガス液体クロマトグラフィー試験によれば97重量%であった。前記生成物を蒸留精製する。
【0251】
[実施例5]
1,860gの酢酸エチル中の、実施例4で得られた1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン169gの混合物に、101gのメタンスルホン酸を滴加する。滴加は、温度が0から5℃に維持されるように行う。前記混合物を60分間撹拌した後、沈殿物をろ過して酢酸エチルで洗浄し、減圧下で乾燥する。生成物の収率は241g(91重量%)である。
【技術分野】
【0001】
本発明は、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン(ネラメキサン)又はその薬学的に許容される塩を製造する方法に関する。
【背景技術】
【0002】
1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン(ネラメキサン)及びその薬理学的に許容される塩は、耳鳴症や眼振症のような疾患及び症状を患う患者の持続的療法にとって重要な薬剤である。
【0003】
これらの薬剤を製造する複数の方法が知られている。
【0004】
ある方法においては、次の反応スキームによる5つのステップを含む反応順序で、市販のイソフォロンをネラメキサンに変換する(非特許文献1)。
【化1】
【0005】
前記反応順序の第一ステップにおいては、塩化銅触媒を用いたヨウ化メチルマグネシウムの共役付加によって、イソフォロン(1)を3,3,5,5−テトラメチルシクロヘキサノン(2)に変換する。
【0006】
また、塩化第一銅存在下でイソフォロンに臭化メチルマグネシウムを添加することにより、3,3,5,5−テトラメチルシクロヘキサノンが収率82.5重量%で生成し得ることが知られている。副産物として、1,3,5,5−テトラメチルシクロヘキサジエンが収率6.9重量%で生成する(非特許文献2)。
【0007】
同文献の実験の部(2313頁)の開示では、塩化第二鉄存在下でイソフォロンに塩化メチルマグネシウムを添加している。しかしながら、3,3,5,5−テトラメチルシクロヘキサノンは形成されず、それとは全く異なる生成物が単離されてしまう。
【0008】
第二ステップにおいては、ヨウ化メチルマグネシウムを用いたグリニャール反応によって、3,3,5,5−テトラメチルシクロヘキサノン(2)を1,3,3,5,5−ペンタメチルシクロヘキサノール(3)に変換する。
【0009】
非特許文献3による開示では、3,3,5,5−テトラメチルシクロヘキサノンとハロゲン化メチルマグネシウムとの反応により1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを製造している。前記生成物はシリカゲルカラムクロマトグラフィーによって精製された。
【0010】
第三ステップにおいては、リッター反応によって、クロロアセトニトリルにより前記シクロヘキサノール(3)を1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサン(6)に変換する。
【0011】
続いて第四ステップにおいては、チオ尿素を用いてアミド(6)中のクロロアセトアミド基を開裂し、生じたアミンを前記反応順序の最終第五ステップにおいて塩酸により酸性化することで、塩酸塩型のネラメキサン(1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン)(7)が生ずる。
【0012】
アミド(6)中のクロロアセトアミド基の開裂は、イルゲンソンス(Jirgensons)らによっても詳しく研究されている(非特許文献4)。それによると、エタノールと酢酸との5:1混合物中で1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを還流する。10時間の反応後、反応混合物を水で希釈し、生成する沈殿物を単離する。ろ液をアルカリ性化し、ヘキサンで抽出する。塩酸の添加後、塩酸塩型の1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンが収率89重量%で単離される。
【0013】
前記文献によれば、前記反応順序の5つのステップによる総収量は約50重量%である。
【先行技術文献】
【非特許文献】
【0014】
【非特許文献1】ダニス(Danysz)ら、「カレント・ファーマシューティカル・デザイン(Current Pharmaceutical Design)」、2002年、第8巻、p.835−843
【非特許文献2】カラシュ(Kharasch)ら、「ジャーナル・オブ・ザ・アメリカン・ケミカル・ソサエティー(Journal of the American Chemical Society)」、1941年、第63巻、p.2308
【非特許文献3】イルゲンソンス(Jirgensons)ら、「ヨーロピアン・ジャーナル・オブ・メディシナル・ケミストリー(European Journal of Medicinal Chemistry)」、2000年、第35巻、p.555−565
【非特許文献4】イルゲンソンス(Jirgensons)ら、「シンセシス(Synthesis)」、2000年、第12号、p.1709−1712
【発明の概要】
【発明が解決しようとする課題】
【0015】
本発明の目的の一つは、経済的な工業規模での有利な実施を可能とする1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩の製造方法を提供するために、上記で参照した各反応ステップのうち一つ又は複数のステップを改良することである。また別の目的は、ネラメキサン又はその薬理学的に許容される塩の製造過程で生ずる廃棄物及び/又は未反応化学物質の量を極少化することである。さらなる目的は、ネラメキサン又はその薬理学的に許容される塩について収率及び/又は選択性、及び/又は生成物品質を最適化又は改善することである。そのような改良された方法は、経済的な工業規模でネラメキサン又はその薬理学的に許容される塩を有利に製造するための必要条件であると考えてよい。
【課題を解決するための手段】
【0016】
本発明は、次のステップ(i)から(iv)から選択される二つ以上のステップを具備する、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩を製造する方法に関する。ステップ(i)においては、塩化メチルマグネシウム存在下でイソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換する。ステップ(ii)においては、塩化メチルマグネシウム存在下で3,3,5,5−テトラメチルシクロヘキサノンを1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iii)においては、酸性溶液中でクロロアセトニトリル存在下で1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iv)においては、水中でチオ尿素存在下で1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。
【0017】
一つの実施形態においては、ステップ(i)及び(ii)を選択する。
【0018】
一つの実施形態においては、ステップ(i)及び(iii)を選択する。
【0019】
一つの実施形態においては、ステップ(i)及び(iv)を選択する。
【0020】
一つの実施形態においては、ステップ(ii)及び(iii)を選択する。
【0021】
一つの実施形態においては、ステップ(ii)及び(iv)を選択する。
【0022】
一つの実施形態においては、ステップ(iii)及び(iv)を選択する。
【0023】
一つの実施形態においては、ステップ(i)、(ii)、及び(iii)を選択する。
【0024】
一つの実施形態においては、ステップ(i)、(ii)、及び(iv)を選択する。
【0025】
一つの実施形態においては、ステップ(i)、(iii)、及び(iv)を選択する。
【0026】
一つの実施形態においては、ステップ(ii)、(iii)、及び(iv)を選択する。
【0027】
一つの実施形態においては、ステップ(i)、(ii)、(iii)、及び(iv)を選択する。
【0028】
一つの実施形態においては、3,3,5,5−テトラチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち少なくとも一つは精製ステップに供さない。
【0029】
一つの実施形態においては、本方法はさらにステップ(v)を具備する。ステップ(v)においては、酸存在下で1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンをその薬理学的に許容される塩に変換する。
【0030】
一つの実施形態においては、前記酸はメタンスルホン酸である。
【0031】
一つの実施形態においては、前記塩化メチルマグネシウムには塩化エチルマグネシウムが含まれない。
【0032】
本発明は一つの態様においては、1−アミノ−1−エチル−3,3,5,5−テトラメチルシクロヘキサン及び1−アミノ−3−エチル−1,3,5,5−テトラメチルシクロヘキサン、又はそれらの薬理学的に許容される塩を実質的に含まない、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩に関する。
【0033】
本発明のステップ(i)から(iv)を含む反応順序においては、蒸留、再結晶、又はクロマトグラフィーのような古典的な精製法によって3,3,5,5−テトラメチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち一つ又は複数を精製しなくてもよいことを開示する。従って、前記化合物のうち一つ又は複数を精製ステップに供さず、未精製形態で使用する。
【発明の効果】
【0034】
一つ又は複数の未精製の反応中間体を使用することによって医療用途に十分な純度の前記目標化合物(すなわちネラメキサン又は薬理学的に許容される塩形態のネラメキサン)が得られるということは、過去には期待できなかった。蒸留、再結晶、又はクロマトグラフィーのような反応中間体の複雑な洗浄ステップは一般的に生成物の損失の原因となる。しかし本発明の方法によればこのステップが省略可能であるために、ネラメキサン又はその薬理学的に許容される塩の収率を60重量%以上とすることが可能である。よって、ネラメキサンを生産する本発明の新規簡易法は有利で経済的な工業規模で実施し得る。
【図面の簡単な説明】
【0035】
図1から10は、結晶形A、A’、B、C、D、及びEの粉末X線回折のダイアグラムを示す。x軸は2Θ(°)/d[Å]、y軸は回折強度(相対単位)である。
【図1】結晶形Aのダイアグラムを示す。
【図2】結晶形A(粉砕試料)のダイアグラムを示す。
【図3】結晶形A’ のダイアグラムを示す。
【図4】結晶形Bのダイアグラムを示す。
【図5】結晶形B(粉砕試料)のダイアグラムを示す。
【図6】結晶形Cのダイアグラムを示す。
【図7】結晶形C(粉末試料)のダイアグラムを示す。
【図8】結晶形Dのダイアグラムを示す。
【図9】結晶形D(粉砕試料)のダイアグラムを示す。
【図10】結晶形Eのダイアグラムを示す。
【発明を実施するための形態】
【0036】
本発明は、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン(ネラメキサン)又はその薬理学的に許容される塩を製造する方法に関する。
【0037】
本発明は具体的には、次のステップ(i)から(iv)から選択される二つ以上のステップを具備する、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩を製造する方法に関する。ステップ(i)においては、塩化メチルマグネシウム存在下でイソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換する。ステップ(ii)においては、塩化メチルマグネシウム存在下で3,3,5,5−テトラメチルシクロヘキサノンを1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iii)においては、酸性溶液中でクロロアセトニトリル存在下で1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iv)においては、水中でチオ尿素存在下で1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。
【0038】
ステップ(i):塩化メチルマグネシウム存在下でのイソフォロンから3,3,5,5−テトラメチルシクロヘキサノンへの変換
ステップ(i)における変換は、イソフォロンと塩化メチルマグネシウムとの反応によって実施する。
【0039】
そのようなグリニャール試薬は、マグネシウムと塩化メチルとから作製してもよい。
【0040】
一つの実施形態においては、ステップ(i)における変換は、銅化合物の存在下で実施する。前記銅化合物は、イソフォロンに対するグリニャール試薬の1,4−共役付加を1,2−付加よりも優先的に進めるための触媒として働き得る。
【0041】
一つの実施形態においては、前記銅化合物はハロゲン化第一銅である。一つの実施形態においては、前記ハロゲン化第一銅はヨウ化第一銅、臭化第一銅、又は塩化第一銅からなる群から選択する。
【0042】
一つの実施形態においては、前記第一銅化合物(例えば塩化第一銅又はヨウ化第一銅のようなハロゲン化第一銅)はリチウム化合物存在下で提供される。
【0043】
一つの実施形態においては、前記リチウム化合物は塩化リチウムのようなハロゲン化リチウムである。
【0044】
一つの実施形態においては、塩化第一銅又はヨウ化第一銅が塩化リチウム存在下で提供される。
【0045】
一つの実施形態においては、塩化第一銅又はヨウ化第一銅存在下で塩化メチルマグネシウムを反応させる。
【0046】
一つの実施形態においては、ヨウ化第一銅又は塩化第一銅と塩化リチウムとの存在下におけるイソフォロンと塩化メチルマグネシウムとの反応によって、ステップ(i)の変換反応を実施する。
【0047】
一つの実施形態においては、ハロゲン化第一銅とハロゲン化リチウムとのモル比は1:1.5から1:2.5の範囲である。
【0048】
一つの実施形態においては、塩化第一銅と塩化リチウムとのモル比又はヨウ化第一銅と塩化リチウムとのモル比は、それぞれ約1:1.5から1:2.5、又は1:2である。
【0049】
通常、ステップ(i)の反応は溶媒中で実施する。
【0050】
一つの実施形態においては、ステップ(i)の反応に使用する溶媒はエーテルであるか又はエーテルを含む溶媒である。
【0051】
適当なエーテルは、ジエチルエーテル、1,4−ジオキサン、及びテトラヒドロフランからなる群から選択してもよい。
【0052】
一つの実施形態においては、前記エーテルはテトラヒドロフランである。
【0053】
一つの実施形態においては、ステップ(i)で使用する前記溶媒はテトラヒドロフランであるか、又はテトラヒドロフランを含む。
【0054】
一つの実施形態においては、塩化メチルマグネシウムと、塩化第一銅又はヨウ化第一銅と、塩化リチウムとの存在下、テトラヒドロフラン中でイソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換する。
【0055】
一つの実施形態においては、塩化メチルマグネシウムとヨウ化第一銅と塩化リチウムとの存在下、テトラヒドロフラン中でイソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換する。
【0056】
一つの実施形態においては、イソフォロンと、ハロゲン化第一銅のような第一銅化合物(例えばヨウ化第一銅又は塩化第一銅)と、及び任意にハロゲン化リチウムのようなリチウム化合物(例えば塩化リチウム)とを溶媒中に用意し、前記混合物にグリニャール試薬を任意で溶媒に溶解したものを添加する。
【0057】
また別の実施形態においては、塩化メチルマグネシウムと、ハロゲン化第一銅のような銅化合物(例えばヨウ化第一銅又は塩化第一銅)とを、任意にハロゲン化リチウムのようなリチウム化合物(例えば塩化リチウム)の存在下で反応させる。一つの実施形態においては、前記混合物をイソフォロンに添加する。また別の実施形態においては、イソフォロンを前記混合物に添加する。
【0058】
また別の実施形態においては、塩化メチルマグネシウムと、ヨウ化第一銅又は塩化第一銅のような銅化合物とを反応させる。
【0059】
一つの実施形態においては、イソフォロンとヨウ化第一銅と塩化リチウムとの混合物を、テトラヒドロフラン中に用意する。テトラヒドロフランに溶解した塩化メチルマグネシウムをこの混合物に添加する。
【0060】
一つの実施形態においては、テトラヒドロフラン中の塩化メチルマグネシウム濃度は、塩化メチルマグネシウム及びテトラヒドロフランの総量を基準として15から30重量%又は20から25重量%である。
【0061】
一つの実施形態においては、テトラヒドロフラン中の塩化メチルマグネシウム濃度は、塩化メチルマグネシウム及びテトラヒドロフランの総量を基準として23重量%である。
【0062】
一つの実施形態においては、1モル当量のイソフォロン当たり、1モル当量よりも多い塩化メチルマグネシウムを使用する。
【0063】
一つの実施形態においては、1モル当量のイソフォロン当たり、1.0から1.75モル当量の塩化メチルマグネシウム又は1.2から1.5モル当量の塩化メチルマグネシウムを使用する。
【0064】
一つの実施形態においては、テトラヒドロフラン中の塩化メチルマグネシウム濃度は、塩化メチルマグネシウム及びテトラヒドロフランの総量を基準として23重量%であり、塩化メチルマグネシウム及びテトラヒドロフランの量を基準として10重量%の触媒(1モル当量のヨウ化第一銅及び2モル当量の塩化リチウム)を使用する。
【0065】
一つの実施形態においては、1モル当量のイソフォロン当たり、0.1から0.25モル当量の塩化リチウムと0.05から0.125モル当量のヨウ化第一銅とを使用する。
【0066】
一つの実施形態においては、温度管理下で前記添加を実施する。
【0067】
一つの実施形態においては、温度が比較的狭い温度範囲に維持されるように前記添加を実施する。
【0068】
一つの実施形態においては、−5℃から20℃、又は0℃から20℃、又は−5℃から15℃、又は1℃から10℃の温度でステップ(i)の変換を実施する。
【0069】
一つの実施形態においては、グリニャール試薬とイソフォロンとの反応は通常比較的速く進行する。前記反応は通常、使用する反応温度に依存して3時間、2時間、又は1時間の短時間で終了する。
【0070】
グリニャール試薬とイソフォロンとの反応後、グリニャール試薬が過剰量使用されている場合にそれを分解するため、及び塩基性マグネシウム化合物を分解するために、前記混合物を水で処理してもよい。
【0071】
一つの実施形態においては、3,3,5,5−テトラメチルシクロヘキサノンの形成を補助するために、塩酸のような酸又はアンモニウム塩を添加する。
【0072】
一つの実施形態においては、形成された有機層を水層から分離する。次に揮発性有機化合物を減圧下で除去することにより、前記有機層を濃縮してもよい。残留物が粗3,3,5,5−テトラメチルシクロヘキサノンである。
【0073】
一つの実施形態においては、ステップ(i)で形成される生成物は、塩化メチレン、トルエン又は石油エーテルのような適当な有機溶媒で前記水性混合物を抽出することによって生成及び単離される。抽出後、前記溶媒を蒸留除去する。生成し単離された状態の粗3,3,5,5−テトラメチルシクロヘキサノンを含む残留液を、精製ステップにかけることなく、反応順序のステップ(ii)に使用する。
【0074】
また別の実施形態においては、抽出の次に前記抽出物を従来の方法によって乾燥してもよい。例えば硫酸ナトリウムによって抽出物を乾燥してよい。前記硫酸塩をろ過分離した後、溶媒を蒸留除去してもよい。生成し単離された状態の粗3,3,5,5−テトラメチルシクロヘキサノンを含む残留物を、精製ステップにかけることなく、反応順序のステップ(ii)に使用してよい。
【0075】
一つの実施形態においては、ステップ(i)で生成し単離された状態の粗3,3,5,5−テトラメチルシクロヘキサノンの収率は88から96重量%の範囲である。
【0076】
一つの実施形態においては、前記粗生成物には93重量%以上の量の目標化合物が含まれることがガス液体クロマトグラフィーにより測定できる。
【0077】
一つの実施形態においては、3,3,5,5−テトラメチルシクロヘキサノンを精製してもよい。一つの実施形態においては、3,3,5,5−テトラメチルシクロヘキサノンを蒸留してもよい。
【0078】
また別の実施形態においては、ステップ(i)で得られた状態の粗3,3,5,5−テトラメチルシクロヘキサノンをステップ(ii)で使用する。
【0079】
ステップ(ii):塩化メチルマグネシウム存在下での3,3,5,5−テトラメチルシクロヘキサノンから1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンへの変換
ステップ(ii)における3,3,5,5−テトラメチルシクロヘキサノンから1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンへの変換は、塩化メチルマグネシウムを用いて実施する。
【0080】
通常、ステップ(ii)の反応は溶媒中で実施する。
【0081】
一つの実施形態においては、前記溶媒はエーテルを含むか又はエーテルである。
【0082】
エーテルは、ジエチルエーテル、1,4−ジオキサン、又はテトラヒドロフランから選択してもよい。
【0083】
一つの実施形態においては、前記エーテルはテトラヒドロフランである。
【0084】
本発明の方法の一つの実施形態においては、3,3,5,5−テトラメチルシクロヘキサノンに塩化メチルマグネシウムを添加する。
【0085】
また別の実施形態においては、塩化メチルマグネシウムにテトラメチルシクロヘキサノンを添加する。
【0086】
一つの実施形態においては、3,3,5,5−テトラメチルシクロヘキサノンのテトラヒドロフラン溶液に塩化メチルマグネシウムのテトラヒドロフラン溶液を添加する。
【0087】
また別の実施形態においては、塩化メチルマグネシウムのテトラヒドロフラン溶液に3,3,5,5−テトラメチルシクロヘキサノンのテトラヒドロフラン溶液を添加する。
【0088】
一つの実施形態においては、テトラヒドロフラン中の塩化メチルマグネシウム濃度は、塩化メチルマグネシウム及びテトラヒドロフランの総量を基準として15から30重量%又は20から25重量%である。
【0089】
一つの実施形態においては、テトラヒドロフラン中の塩化メチルマグネシウム濃度は、塩化メチルマグネシウム及びテトラヒドロフランの総量を基準として23重量%である。
【0090】
従って一つの実施形態においては、塩化メチルマグネシウムとテトラヒドロフランとを含む混合物を、3,3,5,5−テトラメチルシクロヘキサノンとテトラヒドロフランとを含む混合物と反応させる。
【0091】
一つの実施形態においては、塩化メチルマグネシウムのテトラヒドロフラン溶液に3,3,5,5−テトラメチルシクロヘキサノンのテトラヒドロフラン溶液を添加する。1モル当量の3,3,5,5−テトラメチルシクロヘキサノン当たり、1.2から1.75モル当量の塩化メチルマグネシウム量が含まれる。
【0092】
一つの実施形態においては、ステップ(i)で得られた3,3,5,5−テトラメチルシクロヘキサノンのテトラヒドロフラン溶液を、塩化メチルマグネシウムのテトラヒドロフラン溶液に添加する。
【0093】
一つの実施形態においては、ステップ(i)で得られた3,3,5,5−テトラメチルシクロヘキサノンの1モル当量当たり、1.2から1.75モル当量の塩化メチルマグネシウムを使用する。
【0094】
また別の実施形態においては、ステップ(i)で得られた3,3,5,5−テトラメチルシクロヘキサノンを含むテトラヒドロフラン溶液に、塩化メチルマグネシウムを含むテトラヒドロフラン溶液を添加する。
【0095】
一つの実施形態においては、温度管理下で前記変換を実施する。
【0096】
一つの実施形態においては、温度が比較的狭い温度範囲に維持されるように前記変換を実施する。
【0097】
一つの実施形態においては、−5℃から30℃、又は0℃から30℃、又は0℃から20℃、又は0℃から25℃、又は0℃から20℃、又は5℃から20℃、又は10℃から25℃、又は15℃から25℃の温度でステップ(ii)の変換を実施する。
【0098】
ステップ(ii)で形成される1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを単離するためには、ステップ(i)の3,3,5,5−テトラメチルシクロヘキサノンの単離に関連する上記の方法と基本的に同じ方法を使用してよい。
【0099】
一つの実施形態においては、粗1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンの収率は90重量%から100重量%の範囲である。
【0100】
一つの実施形態においては、前記粗生成物には94重量%以上の量の目標化合物1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンが含まれることがガス液体クロマトグラフィーにより測定できる。
【0101】
一つの実施形態においては、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを精製してもよい。一つの実施形態においては、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを蒸留するか、又はクロマトグラフィーに供してもよい。
【0102】
また別の実施形態においては、ステップ(iii)で使用する1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンは粗生成物の状態である。
【0103】
ステップ(iii):酸性溶液中、クロロアセトニトリル存在下での1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンから1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンへの変換
ステップ(iii)における変換は、酸性溶液中における1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンとクロロアセトニトリルとの反応によって実施する。
【0104】
ステップ(iii)のリッター反応は、従来技術において参照された方法に従って実施してもよい。
【0105】
一つの実施形態においては、前記酸は硫酸、硝酸、リン酸、酢酸、又はそれらの混合物からなる群から選択する。
【0106】
一つの実施形態においては、前記酸は濃酸の状態で使用する。
【0107】
一つの実施形態においては、硫酸と酢酸とを使用する。一つの実施形態においては、硫酸は濃硫酸であり、酢酸は氷酢酸である。
【0108】
一つの実施形態においては、ステップ(ii)で生成し単離された状態の1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンとクロロアセトニトリルとを酢酸中に用意し、前記混合物に硫酸を添加する。
【0109】
また別の実施形態においては、ステップ(ii)で生成し単離された状態の1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを酢酸中に用意し、前記混合物にクロロアセトニトリルと硫酸との混合物を添加する。
【0110】
一つの実施形態においては、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンと酢酸とを重量比1:1.5から1:2.5で用意する。
【0111】
一つの実施形態においては、前記シクロヘキサノールと酢酸とを重量比約1:2で用意する。
【0112】
また別の実施形態においては、1モル当量の1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン当たり1.5から2.5モル当量のクロロアセトニトリルと2.5から3.5モル当量の硫酸とを使用する。
【0113】
また別の実施形態においては、1モル当量の1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン当たり約2モル当量のクロロアセトニトリルと3モル当量の硫酸とを使用する。
【0114】
一つの実施形態においては、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンと酢酸とを重量比1:1.5から1:2.5で用意し、1モル当量の1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン当たり1.5から2.5モル当量のクロロアセトニトリルと2.5から3.5モル当量の硫酸とを使用する。
【0115】
一つの実施形態においては、前記シクロヘキサノールと酢酸とを重量比約1:2で用意し、2モル当量のクロロアセトニトリルと3モル当量の硫酸とを使用する。
【0116】
一つの実施形態においては、0℃から30℃、又は0℃から20℃、又は0℃から15℃、又は5℃から10℃の範囲に反応温度が維持されるように、硫酸の添加又はクロロアセトニトリルと硫酸との混合物の添加を実施する。
【0117】
通常、前記反応は目標化合物の方向に比較的速く進行する。一つの実施形態においては、前記反応は2時間又は1時間という短時間で終了し得る。
【0118】
反応混合物を仕上げるために、前記反応終了後には前記混合物を水、氷又は氷水に注いでもよい。沈殿する1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンをろ過によって単離してもよい。
【0119】
付着する酸を除去するために、前記沈殿物を水で洗浄してもよい。
【0120】
一つの実施形態においては、粗生成物の収率は98から100重量%の範囲である。
【0121】
一つの実施形態においては、1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを精製してもよい。一つの実施形態においては、1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを再結晶してもよい。
【0122】
一つの実施形態においては、ステップ(iii)で得られた1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを、粗製物すなわち未精製生成物の状態でステップ(iv)に使用してもよい。前記化合物は乾燥状態又は未乾燥状態で使用してよい。
【0123】
ステップ(iv):水中、チオ尿素存在下での1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンから1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンへの変換
ステップ(iv)における変換は、水中における1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンとチオ尿素との反応によって実施する。
【0124】
一つの実施形態においては、ステップ(iv)で使用する混合物にはさらに有機溶媒が含まれる。
【0125】
一つの実施形態においては、前記有機溶媒はステップ(iv)の反応条件下で水混和性である、アルコールのような溶媒である。
【0126】
一つの実施形態においては、前記有機溶媒はメタノール、エタノール、プロパノール、ブタノール、及びエチレングリコールからなる群から選択されるアルコールである。
【0127】
一つの実施形態においては、前記有機溶媒の量は水の量を基準として0から200重量%である。また別の実施形態においては、前記有機溶媒の量は水の量を基準として0から150重量%、又は0から100重量%、又は0から50重量%、又は0から10重量%、又は0から5重量%である。
【0128】
また別の実施形態においては、ステップ(iv)で用いる混合物は実質的に有機溶媒を含まない。
【0129】
「実質的に有機溶媒を含まない」という語は、前記混合物の有機溶媒含有量が水の量を基準として0から5重量%、又は0から3重量%、又は0から1重量%であることを想定する。
【0130】
一つの実施形態においては、チオ尿素と水との重量比は1:0.5から1:50、又は1:1から1:20、又は1:2から1:10の範囲である。
【0131】
ステップ(iv)の反応は酸の添加なしに実施し得るが、そのような化合物の添加によって1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンから1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンへの変換は促進され得る。
【0132】
従って一つの実施形態においては、ステップ(iv)の混合物にはさらに酸が含まれる。
【0133】
使用し得る酸は、塩酸、硫酸、リン酸、p−トルエンスルホン酸、メタンスルホン酸、酢酸、及び安息香酸であるが、これらに限定されない。つまり無機酸及び有機酸を使用し得る。
【0134】
何らかの酸を使用する場合、その量の範囲は比較的広範である。
【0135】
一つの実施形態においては、前記混合物は水の量を基準として0.1から20重量%の量の酸を含む。
【0136】
一つの実施形態においては、使用する酸は塩酸である。
【0137】
1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンの変換をさらに促進するために、ステップ(iv)で使用する混合物を加熱する。
【0138】
「加熱」という語とは、ステップ(iv)で使用する混合物を環境温度よりも高温に調節することを想定する。
【0139】
一つの実施形態においては、ステップ(iv)で使用する混合物を50℃から前記混合物の還流温度までの範囲の温度に加熱する。
【0140】
また別の実施形態においては、前記混合物を80℃から前記混合物の還流温度までの範囲の温度に加熱する。
【0141】
さらにまた別の実施形態においては、前記混合物をその還流温度まで加熱する。
【0142】
ステップ(iv)において実質的に有機溶媒を含まない混合物を使用する場合、還流温度は通常100℃前後、すなわち95から105℃の範囲である。ステップ(iv)において有機溶媒を含む混合物を使用する場合の還流温度は、使用する有機溶媒の量及び沸点によって、水は含むが有機溶媒は実質的に含まない混合物の還流温度よりも高温又は低温であり得る。
【0143】
ステップ(iv)の1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンから1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンへの変換は、例えばガス液体クロマトグラフィーのような一般的なクロマトグラフィー法によって管理し得る。
【0144】
一つの実施形態においては、ステップ(iv)において、1モルの1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサン当たり1.0から2モルのチオ尿素と、1から3モルの酸と、チオ尿素及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンの量を基準として500から1,500重量%の水とを還流温度で使用する。
【0145】
一つの実施形態においては、前記1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンと、約1.2モル当量のチオ尿素及び2モル当量の塩酸とを、8倍量(チオ尿素及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを基準とした重量比)の水中で還流温度で反応させる。
【0146】
通常、ステップ(iv)の水を含む混合物中での変換は比較的速く進行する。
【0147】
一つの実施形態においては、実質的に有機溶媒を含まない水中でステップ(iv)を実施し、還流温度、すなわち100℃前後の温度で加熱を行い、酸を添加する場合、変換は2時間でも終了し得、又は1時間でさえ終了し得る。
【0148】
一つの実施形態においては、前記変換は6時間、5時間、4時間、3時間、又は3時間よりもさらに短時間でも終了する。
【0149】
前記変換が酸によって触媒される場合、生成するアミンの少なくとも一部はアミノ基のプロトン化によって水に溶解し、塩を形成する。
【0150】
一つの実施形態においては、生成するアミンを単離するために、本発明の方法はさらに前記混合物へのアルカリ添加を具備する。これはpH値を7以上に調節して前記混合物から1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンを分離するためである。
【0151】
前記実施形態においては、好ましくは前記混合物を冷却した後、アルカリ添加後にアミンが水相から分離して、相分離され得る。
【0152】
また別の実施形態においては、アルカリ添加後に有機相と水相とを形成する前記混合物から、水非混和性の有機溶媒によって前記アミンを抽出し得る。適当な溶媒は、塩化メチレン、トルエン、又は石油エーテルのような溶媒である。抽出後、硫酸ナトリウム等を用いて前記抽出物を乾燥してもよい。蒸発法によって溶媒を除去した後、粗アミンが得られる。
【0153】
一つの実施形態においては、粗生成物の収率は大体において理論値の95重量%よりも高く、又はほとんど定量的である。前記粗生成物は通常、ガス液体クロマトグラフィー試験によれば、95重量%以上、97重量%以上、又は99重量%以上という非常に多量の目標化合物を含む。
【0154】
一つの実施形態においては、必要に応じて蒸留により前記粗アミンをさらに精製する。
【0155】
ステップ(iv)で生成し単離された生成物は、それ以上精製することなく本発明の方法のステップ(v)に使用してもよい。
【0156】
しかしながら一つの実施形態においては、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンよりも揮発性が高い化合物を粗生成物から蒸留除去し、残留物をステップ(v)に使用することも可能である。
【0157】
一つの実施形態においては、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンを蒸留精製する。
【0158】
ステップ(iv)で用いる方法によれば、従来技術の方法において開示された反応時間と比較して反応時間がかなり短縮されるということが、期せずして明らかとなった。さらに背景技術のセクションで参照した水の添加と沈殿物のろ過とが不要となるため、本発明の方法によれば生成するアミンの仕上げがかなり単純化される。アミンの収率は高く、ほとんど定量的である。すなわち、本新規方法は経済的な工業規模で有利に実施し得る。
【0159】
ステップ(i)から(iv)のうち二つ以上のステップの選択
一つの実施形態においては、ステップ(i)から(iv)のうち二つのステップを選択する。
【0160】
従って一つの実施形態においては、ステップ(i)及び(ii)を選択する。
【0161】
また別の実施形態においては、ステップ(i)及び(iii)を選択する。
【0162】
また別の実施形態においては、ステップ(i)及び(iv)を選択する。
【0163】
また別の実施形態においては、ステップ(ii)及び(iii)を選択する。
【0164】
また別の実施形態においては、ステップ(ii)及び(iv)を選択する。
【0165】
また別の実施形態においては、ステップ(iii)及び(iv)を選択する。
【0166】
一つの実施形態においては、ステップ(i)から(iv)のうち三つのステップを選択する。
【0167】
従って一つの実施形態においては、ステップ(i)、(ii)、及び(iii)を選択する。
【0168】
一つの実施形態においては、ステップ(i)、(ii)、及び(iv)を選択する。
【0169】
また別の実施形態においては、ステップ(i)、(iii)、及び(iv)を選択する。
【0170】
また別の実施形態においては、ステップ(ii)、(iii)、及び(iv)を選択する。
【0171】
一つの実施形態においては、ステップ(i)から(iv)のうち四つのステップを選択する。
【0172】
従って一つの実施形態においては、ステップ(i)、(ii)、(iii)、及び(iv)を選択する。
【0173】
一つの実施形態においては、本方法は次のステップ(i)から(iv)から選択される二つ以上のステップを具備する。ステップ(i)においては、塩化メチルマグネシウム存在下かつヨウ化銅(I)と塩化リチウムとテトラヒドロフランとの存在下で、イソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換する。ステップ(ii)においては、塩化メチルマグネシウム存在下かつテトラヒドロフラン存在下で、3,3,5,5−テトラメチルシクロヘキサノンを1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iii)においては、クロロアセトニトリル存在下かつ酢酸と硫酸との存在下で1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iv)においては、チオ尿素存在下かつ水と塩酸との存在下で1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。
【0174】
未精製の3,3,5,5−テトラメチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンの使用
一つの実施形態においては、ステップ(i)から(iii)のそれぞれで得られた状態の3,3,5,5−テトラチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち少なくとも一つは精製ステップに供さない。
【0175】
従って本発明の方法では、反応中間体である3,3,5,5−テトラチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち少なくとも一つは、反応順序の先行ステップで得られたままの形態で該当の反応ステップにおいて使用することを想定する。すなわちステップ(i)から(iii)の反応順序で製造された反応中間体の少なくとも一つは、精製ステップに供さない。
【0176】
「精製ステップ」という語は、各反応ステップで生成した化合物の再結晶、蒸留、クロマトグラフィー、又はそれらの組み合わせを包含する。
【0177】
「精製ステップを経ていない」という語では、標準的な仕上げのステップは許容される。そのようなステップは、前記化合物(本明細書中では3,3,5,5−テトラチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサン)と溶媒とを含む混合物から前記溶媒を蒸留除去すること、溶媒によって前記化合物を水相から抽出すること、前記化合物と溶媒とを含む混合物を無水硫酸ナトリウムなどを用いて乾燥すること、前記化合物を減圧下で乾燥すること、及び固体化合物を液剤で洗浄することなどである。
【0178】
「再結晶」、「蒸留」、又は「クロマトグラフィー」による精製とは、有機化合物のような化学物質を精製するために実験室規模及び工業規模の両方で用いる古典的な方法である。
【0179】
再結晶とは、混合物中に含まれる複数化合物の単一溶媒又は混合溶媒に対する溶解度の差異に基づいて、混合物を分離する方法である。ある化合物を再結晶によって精製する場合、前記化合物を適当な溶媒に溶解した後に冷却する。その結果、所望の精製化合物が溶液から沈降(再結晶)する。しかし、所望の化合物が不溶性である別の溶媒を、所望の化合物の沈殿が始まるまで前記溶液に添加することも可能である。従って本発明の趣旨における「再結晶」という語は、精製化合物を形成させるために、化合物を溶解状態にし、前記溶解状態から自発的沈殿を生ぜしめるか又は人為的に沈殿させることを意味する。
【0180】
蒸留とは、沸騰液体混合物中における、混合物中に含まれる複数化合物の揮発度の差異に基づいて、混合物を分離する方法である。従って本発明の趣旨においては、「精製」という語の定義について使用する「蒸留」という語は、先ず化合物を液相から気相に相転移させる必要があり、次に凝縮させて精製物を形成させることを意味する。
【0181】
化学におけるクロマトグラフィーとは、混合物中に含まれる複数化合物の固定相−移動相間での分配の差異に基づいて混合物を分離し、精製物を形成させる方法である。一般的な方法はカラムクロマトグラフィーであり、種々の製造法に使用し得る。
【0182】
従って、ステップ(i)から(iii)の反応順序において製造した3,3,5,5−テトラチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち少なくとも一つは上記の精製ステップの何れにも供さず、前記の精製ステップを用いることなしに以降の各ステップ(ii)から(iv)で使用する。
【0183】
つまり本発明の方法の一つの実施形態においては、3,3,5,5−テトラチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち一つは、再結晶、蒸留、又はクロマトグラフィーの何れにも供さない。
【0184】
一つの実施形態においては、ステップ(i)で得られた3,3,5,5−テトラチルシクロヘキサノールは精製ステップに供さない。
【0185】
ステップ(i)で得られてステップ(ii)に使用する3,3,5,5−テトラチルシクロヘキサノンは、環境温度(25℃)においては液体である。一つの実施形態においては、前記化合物は蒸留に供さない。つまり、精製化合物を形成するために3,3,5,5−テトラチルシクロヘキサノンを液相から気相に相転移して凝縮させることはしない。
【0186】
前記化合物はクロマトグラフィーにも供さない。つまり、精製物を形成するために3,3,5,5−テトラチルシクロヘキサノンを固定相−移動相間に分配することはしない。
【0187】
ステップ(i)で得られた状態の粗化合物は高収率及び高純度であるので、反応順序の次のステップ(ii)で使用する3,3,5,5−テトラチルシクロヘキサノンは粗生成物のままでもよい。次のステップ(ii)で前記粗生成物の使用が可能であるのは、イソフォロンと塩化メチルマグネシウムとの反応が、ヨウ化メチルマグネシウム又は臭化メチルマグネシウムとの反応とは逆に上記の副産物の形成を限界まで抑制するためである。
【0188】
従ってイソフォロンから3,3,5,5−テトラチルシクロヘキサノンへの変換に塩化メチルマグネシウムを使用することは、臭化メチルマグネシウム及びヨウ化メチルマグネシウムの各々を使用することよりも有利である。これは具体的には、副産物が抑制されること、及び/又は高収率が達成可能であること、及び/又は得られた化合物を背景技術のセクションに記載の反応順序のステップ(ii)において粗生成状態で利用できることに関係する。蒸留、再結晶、又はクロマトグラフィーのような化合物の複雑な洗浄ステップは一般的に生成物の損失の原因となるが、本発明の方法によればこのステップを省略することが可能である。また、背景技術のセクションで参照したネラメキサン又はその薬理学的に許容される塩を製造する反応順序における次のステップ(ii)において、前記目標化合物は未精製形態で使用し得る。これらの理由から、本新規簡易方法は有利かつ経済的な工業規模で実施し得る。
【0189】
また別の実施形態においては、ステップ(ii)で得られた1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンは精製ステップに供さない。
【0190】
ステップ(ii)で得られてステップ(iii)に使用する1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンは、環境温度においては液体である。一つの実施形態においては、前記化合物は蒸留に供さない。つまり、精製物を形成するために1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを液相から気相に相転移して凝縮させることはしない。
【0191】
前記化合物はクロマトグラフィーにも供さない。つまり、精製物を形成するために1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを固定相−移動相間に分配することはしない。
【0192】
ステップ(ii)で得られる粗化合物は高収率及び高純度であるので、反応順序の次のステップ(iii)で使用する1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンは粗生成物のままでもよい。
【0193】
この実施形態においては、3,3,5,5−テトラチルシクロヘキサノンから1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンへの変換に塩化メチルマグネシウムを使用することは、臭化メチルマグネシウム及びヨウ化メチルマグネシウムの各々を使用することよりも有利である。これは具体的には、得られた化合物を背景技術のセクションに記載の反応順序において粗生成状態で利用できることと高収率が達成可能であることとに関係する。蒸留、再結晶、又はクロマトグラフィーのような反応中間体の複雑な洗浄ステップは一般的に生成物の損失の原因となるが、本発明の方法によればこのステップを省略することが可能である。また、背景技術のセクションで参照したネラメキサン又はその薬理学的に許容される塩を製造する反応順序における次の反応ステップ(iii)において、前記目標化合物は未精製形態で使用し得る。これらの理由から、本新規簡易方法は有利かつ経済的な工業規模で実施し得る。
【0194】
一つの実施形態においては、ステップ(iii)で得られた1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンは精製ステップに供さない。
【0195】
ステップ(iii)で得られてステップ(iv)に使用する1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンは、環境温度においては固体である。一つの実施形態においては、前記化合物は再結晶に供さない。つまり、精製物を形成するために1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを溶解状態にした上で前記溶解状態から自発的沈殿を生ぜしめるか又は人為的に沈殿させることはしない。
【0196】
前記化合物はクロマトグラフィーにも供さない。つまり、精製物を形成するために1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを固定相−移動相間に分配することはしない。
【0197】
ハロゲン化メチルマグネシウムと3,3,5,5−テトラメチルシクロヘキサノンとの反応で得られたままの1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを、蒸留又はクロマトグラフィーのような精製ステップにかけることなくステップ(iii)に使用することによって、収率90から100重量%という高収率の粗1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンが生成することが見い出されている。蒸留、再結晶、又はクロマトグラフィーのような反応中間体の複雑な洗浄ステップは一般的に生成物の損失の原因となるが、本発明の方法によればこのステップを省略することが可能である。また、背景技術のセクションで参照したネラメキサン又はその薬理学的に許容される塩を製造する反応順序における次の反応ステップ(iv)において、前記目標化合物は未精製形態で使用し得る。これらの理由から、本新規簡易方法は有利かつ経済的な工業規模で実施し得る。
【0198】
一つの実施形態においては、ステップ(i)で得られた3,3,5,5−テトラメチルシクロヘキサノン及びステップ(ii)で得られた1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンは精製ステップに供さない。
【0199】
また別の実施形態においては、ステップ(i)で得られた3,3,5,5−テトラメチルシクロヘキサノン、ステップ(ii)で得られた1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及びステップ(iii)で得られた1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンは精製ステップに供さない。
【0200】
一つの実施形態においては、ステップ(ii)で得られた1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン及びステップ(iii)で得られた1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンは精製ステップに供さない。
【0201】
また別の実施形態においては、ステップ(i)で得られた3,3,5,5−テトラメチルシクロヘキサノン及びステップ(iii)で得られた1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンは精製ステップに供さない。
【0202】
一つの実施形態においては、本方法は次のステップ(i)から(iv)から選択される二つ以上のステップを具備する。ステップ(i)においては、塩化メチルマグネシウム存在下かつヨウ化銅(I)と塩化リチウムとテトラヒドロフランとの存在下で、イソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換する。ステップ(ii)においては、塩化メチルマグネシウム存在下かつテトラヒドロフラン存在下で、ステップ(i)で得られた3,3,5,5−テトラメチルシクロヘキサノンを1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iii)においては、クロロアセトニトリル存在下かつ酢酸と硫酸との存在下で、ステップ(ii)で得られた1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。ステップ(iv)においては、チオ尿素存在下かつ水と塩酸との存在下で、ステップ(iii)で得られた1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する。前記の3,3,5,5−テトラメチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち少なくとも一つは、精製ステップを経ない。
【0203】
ステップ(v):酸存在下での1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンからその塩への変換
一つの実施形態においては、適当な酸の添加によって1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンをその薬理学的に許容される塩に変換する。
【0204】
従って、本方法は追加のステップ(v)を具備する。ステップ(v)においては、酸存在下で1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンをその薬理学的に許容される塩に変換する。
【0205】
本開示の趣旨のため、「薬理学的に許容される塩」という語は、哺乳動物(例えばヒト)への投与時に生理学的に許容され、有害な反応を通常もたらさないネラメキサン塩を指す。「薬理学的に許容される塩」という語は、通常は哺乳動物、特にヒトに対する使用について、連邦政府又は州政府の規制当局により承認されるもの、又は米国薬局方もしくは他の一般に承認された薬局方のリストに記載されるものを意味する。
【0206】
1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンからその薬理学的に許容される塩への変換は、不活性な有機溶媒中で前記塩基と1モル当量以上の選択された酸とを混合することによって、従来の方法で達成する。前記塩の単離は、前記塩の溶解度が低い非極性溶媒(例えばエーテル)を用いた沈殿誘導のような、当分野の公知技術によって実施する。非毒性であって所望の薬理活性に実質的に干渉しない限り、前記塩の種類は重要ではない。
【0207】
薬理学的に許容される塩の例は、塩酸、臭化水素酸、メタンスルホン酸、酢酸、コハク酸、マレイン酸、クエン酸、及び関連する酸で形成されるものである。
【0208】
さらなる薬理学的に許容される塩には、ヨウ化水素酸、過塩素酸、硫酸、硝酸、リン酸、プロピオン酸、グリコール酸、乳酸、ピルビン酸、マロン酸、フマル酸、酒石酸、安息香酸、炭酸、桂皮酸、マンデル酸、エタンスルホン酸、ヒドロキシエタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸、シクロヘキサンスルファミン酸、サリシクリック酸、p−アミノサリチル酸、2−フェノキシ安息香酸、及び2−アセトキシ安息香酸で形成されるような酸付加塩が含まれるが、これらに限定されない。
【0209】
一つの実施形態においては、ステップ(iv)で生成し単離した1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンを、酸の添加前に単一溶媒又は混合溶媒に溶解又は分散もしくは懸濁する。
【0210】
適当な溶媒は、アセトン、アニソール、酢酸ブチル、t−ブチルメチルエーテル、クメン、ジメチルスルホキシド、酢酸エチル、エチルエーテル、ギ酸エチル、ヘプタン、酢酸イソブチル、酢酸イソプロピル、酢酸メチル、メチルエチルケトン、メチルイソブチルケトン、ペンタン、酢酸プロピル、テトラヒドロフラン、1,1−ジエトキシプロパン、1,1−ジメトキシメタン、2,2−ジメトキシプロパン、イソオクタン、イソプロピルエーテル、メチル−i−プロピルケトン、及びメチルテトラヒドロフランのような溶媒である。
【0211】
一つの実施形態においては、メチルエチルケトンと水とのような、溶媒と水との混合物を使用してもよい。
【0212】
溶解又は分散もしくは懸濁後に、塩を形成させるために適当な酸を添加する。前記酸も、上記の溶媒の一つ又は複数に溶解又は分散もしくは懸濁してもよい。
【0213】
前記沈殿及び/又は結晶の塩は、ろ過によって反応混合物から分離してよい。
【0214】
前記沈殿物に付着する溶媒は、乾燥及び/又は減圧によって除去し得る。
【0215】
一つの実施形態においては、使用する酸は塩酸であって、ステップ(v)で生成する塩は塩化物である。
【0216】
一つの実施形態においては、前記酸はメタンスルホン酸である。ステップ(v)で生成する塩はメシレートである。前記メシレートの融点は、示差走査熱量計により昇温速度10K/分で測定したところ173.1℃である。
【0217】
一つの実施形態においては、塩の収率は95重量%以上であって、98.5重量%以上の純度である。
【0218】
また別の実施形態においては、前記純度は99.9重量%以上である。
【0219】
一つの実施形態においては、ステップ(i)から(v)を具備する一連の反応の総収率は65重量%以上である。
【0220】
また別の実施形態においては、使用する酸は、臭化水素酸、酢酸、クエン酸、マレイン酸、又はコハク酸であって、生成する塩は、臭化物、酢酸塩(融点142.2℃)、モノクエン酸塩(融点151.5℃)、モノマレイン酸塩(融点160.1℃)、又はモノコハク酸塩(融点177.2℃)である。
【0221】
1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン塩は、多形又は擬多形で存在してもよい。
【0222】
「多形」という語は、固体物質が複数の形態又は結晶構造で存在し得ることを指す。
【0223】
「擬多形」という語は、水和又は溶媒和の結果として固体物質が異なるタイプの結晶を形成し得ることを指す。
【0224】
ネラメキサン塩酸塩は二つの多形及び三つの擬多形水和物の形で存在し得る。
【0225】
本開示の趣旨においては、前記の二つの多形を結晶形A及び結晶形Eと呼ぶ。
【0226】
本開示の趣旨においては、前記の三つの擬多形は、結晶形Bと呼ぶ一水和物、結晶形Cと呼ぶセスキ水和物、及び結晶形Dと呼ぶ三水和物である。
【0227】
一つの実施形態においては、約50℃、100mbarでネラメキサン塩酸塩を乾燥することによって結晶形Aを製造してよい。結晶形Aには約0.7重量%以下の量の水が含まれていてよい。しかし前記結晶形は完全に乾燥していてもよく、そのような結晶形は本明細書では結晶形A’と呼ぶ。
【0228】
結晶形A及び結晶形Eは互変二形関係にある。すなわち、これらは温度変化によって可逆的に相互変換し得る。低温型A(融点221℃)は少なくとも70℃までは熱力学的に安定である。70℃よりも高温では、低温型Aは高温型E(融点241℃)に転移する。
【0229】
25℃で約50%よりも高い相対湿度(r.h.)では、結晶形Aは水和物に変換し得る。結晶形Cは擬多形の中では最も安定な結晶形である。25℃で約25%よりも低い相対湿度、及び40℃で約33%よりも低い相対湿度では、結晶形Cは結晶形Aに変換し得る。次に安定な水和物は結晶形Bである。結晶形Dは水中に懸濁した状態でのみ安定である。
【0230】
一つの態様においては、本発明は1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン塩酸塩の結晶形A、結晶形A’、又は結晶形Eに関する。
【0231】
また別の態様においては、本発明は1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン塩酸塩の結晶形B、結晶形C、又は結晶形Dに関する。
【0232】
結晶多形及び擬多形は、粉末X線回折によって分析できる。結晶形A、B、及びDの試料は通常、3つから4つの強いピークを示す。粉砕試料のピーク強度は、非粉砕試料と比較して顕著な変化を示す。
【0233】
副産物としての1−アミノ−1−エチル−3,3,5,5−テトラメチルシクロヘキサン及び1−アミノ−3−エチル−1,3,5,5−テトラメチルシクロヘキサン
塩化メチルマグネシウムのようなメチルマグネシウム・グリニャール試薬を用いてステップ(i)の変換を実施する場合、ステップ(i)から(iv)又はステップ(i)から(v)の反応順序の一つの実施形態においては、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン及びその塩に加えて、目標化合物である1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキシルアミン又はその塩とは異なるアミノ化合物がさらに生成し得る。
【0234】
一つの実施形態においては、三つの副産物が生成し得る。それらは例えばガスクロマトグラフィー分析によって検出できる。
【0235】
一つの実施形態においては、副産物として1−アミノ−3−エチル−1,3,5,5−テトラメチルシクロヘキサンが生成し得る。この化合物は二つのキラル中心を持つため、二つのジアステレオマーが検出され得る。
【0236】
一つの実施形態においては、さらに1−アミノ−1−エチル−3,3,5,5−テトラメチルシクロヘキサンが生成する。
【0237】
一つの実施形態においては、1−アミノ−3−エチル−1,3,5,5−テトラメチルシクロヘキサンの生成原因を、ステップ(i)においてメチル基ではなくエチル基がイソフォロンに付加されて各シクロへキサノンが生じたことに帰属することができる。前記付加後にステップ(ii)から(iv)又はステップ(i)から(v)に類似の反応順序を実施する場合、それぞれ前記アミン又はその塩が形成される。
【0238】
一つの実施形態においては、1−アミノ−1−エチル−3,3,5,5−テトラメチルシクロヘキサンの生成原因を、ステップ(ii)においてメチル基ではなくエチル基が各シクロへキサノンのカルボニル基に付加されたことに帰属することができる。前記付加後にステップ(iii)から(iv)又はステップ(iii)から(v)に類似の反応順序を実施する場合、それぞれ前記アミン又はその塩が形成される。
【0239】
一つの実施形態においては、前記副産物の生成原因を、使用したメチルマグネシウム・グリニャール試薬にエチルマグネシウム・グリニャール試薬がコンタミネーションしたことに帰属することができる。
【0240】
一つの実施形態においては、塩化エチルマグネシウムのようなエチルマグネシウム・グリニャール試薬を含まない精製したメチルマグネシウム・グリニャール試薬を使用することによって、前記副産物の生成を抑制し得る。
【0241】
一つの実施形態においては、塩化メチルマグネシウムに含まれる塩化エチルマグネシウムは、塩化メチルマグネシウムと塩化エチルマグネシウムとの総量を基準として1重量%未満、0.5重量%未満、又は0.1重量%未満である。
【0242】
一つの実施形態においては、ステップ(iv)で得られたアミンを精製することにより、目標化合物から不必要な副産物を除去してもよい。一つの実施形態においては、前記アミンを蒸留精製して副産物を除去してもよい。
【0243】
また別の実施形態においては、ステップ(v)によって得られた塩を精製する。一つの実施形態においては、再結晶のステップによって前記塩を精製してもよい。適当な溶媒は、例えばステップ(v)で使用する溶媒から選択する。一つの実施形態においては前記溶媒はアニソールである。一つの実施形態においては前記塩はメシレートである。
【0244】
本発明は一つの態様においては、1−アミノ−1−エチル−3,3,5,5−テトラメチルシクロヘキサン、1−アミノ−3−エチル−1,3,5,5−テトラメチルシクロヘキサン、又はそれらの薬理学的に許容される塩を実質的に含まない、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩に関する。
【0245】
「実質的に含まない」という語は、前記副産物と1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩との総量を基準として、前記副産物の量が0.5重量%未満であることを指す。
【0246】
背景技術のセクションで参照した反応順序によると、目標化合物であるネラメキサンは約51%の収率で得られる。それに対応する本願のステップ(i)から(iv)を具備する反応順序の総収率は、65重量%以上である。実施例1から5によれば、約88%もの総収率で前記目標化合物が得られる。すなわち本発明の反応順序は、従来の反応順序と比較してネラメキサンの収率を改善する。一つ又は複数の未精製の反応中間体を使用することによって、医療用途に十分な純度の前記目標化合物、すなわちネラメキサン又は薬理学的に許容される塩形態のネラメキサンが得られるということは、過去には期待できなかった。蒸留、再結晶、又はクロマトグラフィーのような反応中間体の複雑な洗浄ステップは一般的に生成物の損失の原因となるが、本発明の方法によればこのステップを省略可能である。それゆえに、ネラメキサンを生産する本発明の新規簡易法は有利で経済的な工業規模で実施し得る。
【0247】
[実施例1]
139gのイソフォロンと19gのヨウ化銅(I)と8.4gの塩化リチウムと1,550gのテトラヒドロフランとの撹拌混合物に、93gの塩化メチルマグネシウムと372gのテトラヒドロフランとの混合物を滴加する。無機化合物は滴下前に溶解する。前記混合物の温度が5〜15℃に維持されるように滴下速度を選択する。滴加が終了した後、混合物を60分間撹拌する。次に、過剰の塩化メチルマグネシウムを分解するため、及び塩基性マグネシウム化合物を分解するために、希釈した塩酸を添加する。前記混合物を石油エーテルで二回抽出する。前記複数の抽出物を混合し、アンモニアで洗浄する。次に溶媒を蒸留除去する。粗目標化合物の収量は定量的である(153g)。前記粗生成物中の3,3,5,5−テトラメチルシクロヘキサノンの含有量は、ガス液体クロマトグラフィー試験によれば約91重量%である。前記粗生成物中には、約2重量%の未反応イソフォロンと、イソフォロンに対するグリニャール試薬の1,2−付加により生成する1重量%未満の1,3,5,5−テトラメチルシクロヘキサノール、又は前記化合物から生成するオレフィン、及び1重量%の1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンが含まれる。
【0248】
[実施例2]
実施例1で得られた153gの3,3,5,5−テトラメチルシクロヘキサノンと153gのテトラヒドロフランとの混合物を、93gの塩化メチルマグネシウムと372gのテトラヒドロフランとの撹拌混合物に滴加する。混合物の温度が5〜15℃に維持されるように滴下速度を選択する。滴加が終了した後、前記混合物を60分間撹拌する。次に、過剰の塩化メチルマグネシウムを分解するため、及び塩基性マグネシウム化合物を分解するために、希釈した塩酸を添加する。前記混合物を石油エーテルで二回抽出する。前記複数の抽出物を混合し、溶媒を蒸留除去する。1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンの粗収量は定量的である(170g)。前記粗生成物中の目標化合物含有量は、ガス液体クロマトグラフィー試験によれば約95重量%である。
【0249】
[実施例3]
320gの氷酢酸と、実施例2で得られた170gの粗1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンと、150gのクロロアセトニトリルとの撹拌混合物に、294gの濃硫酸を滴加する。反応混合物の温度が5から10℃に維持されるように滴下速度を選択する。滴加が終了した後、混合物をさらに60分間撹拌する。次に前記混合物を氷水に注ぐ。沈殿する目標化合物1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンをろ過分離する。乾燥後、230gの目標化合物が得られる。収率はほぼ定量的である(94重量%)。
【0250】
[実施例4]
実施例3に従って製造した245gの1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンと、91gのチオ尿素と、2,700gの水と、220gの塩酸(33%酸)との混合物を、還流下で加熱する。6時間の反応後に混合物を環境温度まで冷却し、水酸化ナトリウムを添加して混合物のpH値を7以上に調節する。次に、前記混合物を石油エーテルで二回抽出する。前記複数の抽出物を混合する。石油エーテルを蒸留除去した後、収率97重量%(159g)の粗1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンが得られる。前記粗生成物中の目標化合物含有量はガス液体クロマトグラフィー試験によれば97重量%であった。前記生成物を蒸留精製する。
【0251】
[実施例5]
1,860gの酢酸エチル中の、実施例4で得られた1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン169gの混合物に、101gのメタンスルホン酸を滴加する。滴加は、温度が0から5℃に維持されるように行う。前記混合物を60分間撹拌した後、沈殿物をろ過して酢酸エチルで洗浄し、減圧下で乾燥する。生成物の収率は241g(91重量%)である。
【特許請求の範囲】
【請求項1】
次のステップ(i)から(iv)から選択される二つ以上のステップを具備することを特徴とする、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩を製造する方法であって:
ステップ(i)においては塩化メチルマグネシウム存在下でイソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換し;
ステップ(ii)においては塩化メチルマグネシウム存在下で3,3,5,5−テトラメチルシクロヘキサノンを1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換し;
ステップ(iii)においては酸性溶液中でクロロアセトニトリル存在下で1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンに変換し;
ステップ(iv)においては水中でチオ尿素存在下で1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する方法。
【請求項2】
請求項1の方法であって、ステップ(i)及び(ii)が選択されることを特徴とする方法。
【請求項3】
請求項1又は2の方法であって、ステップ(i)及び(iii)が選択されることを特徴とする方法。
【請求項4】
請求項1から3のいずれか一つの方法であって、ステップ(i)及び(iv)が選択されることを特徴とする方法。
【請求項5】
請求項1から4のいずれか一つの方法であって、ステップ(ii)及び(iii)が選択されることを特徴とする方法。
【請求項6】
請求項1から5のいずれか一つの方法であって、ステップ(ii)及び(iv)が選択されることを特徴とする方法。
【請求項7】
請求項1から6のいずれか一つの方法であって、ステップ(iii)及び(iv)が選択されることを特徴とする方法。
【請求項8】
請求項1から7のいずれか一つの方法であって、ステップ(i)、(ii)、及び(iii)が選択されることを特徴とする方法。
【請求項9】
請求項1から8のいずれか一つの方法であって、ステップ(i)、(ii)、及び(iv)が選択されることを特徴とする方法。
【請求項10】
請求項1から9のいずれか一つの方法であって、ステップ(i)、(iii)、及び(iv)が選択されることを特徴とする方法。
【請求項11】
請求項1から10のいずれか一つの方法であって、ステップ(ii)、(iii)、及び(iv)が選択されることを特徴とする方法。
【請求項12】
請求項1から11のいずれか一つの方法であって、ステップ(i)、(ii)、(iii)、及び(iv)が選択されることを特徴とする方法。
【請求項13】
請求項1から12のいずれか一つの方法であって、3,3,5,5−テトラメチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち少なくとも一つが精製ステップを経ないことを特徴とする方法。
【請求項14】
さらにステップ(v)を具備することを特徴とする、請求項1から13のいずれか一つの方法であって:
ステップ(v)においては酸存在下で1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンをその薬理学的に許容される塩に変換する方法。
【請求項15】
請求項14の方法であって、前記酸がメタンスルホン酸であることを特徴とする方法。
【請求項16】
請求項1から15のいずれか一つの方法であって、前記塩化メチルマグネシウムが塩化エチルマグネシウムを含まないことを特徴とする方法。
【請求項17】
1−アミノ−1−エチル−3,3,5,5−テトラメチルシクロヘキサン及び1−アミノ−3−エチル−1,3,5,5−テトラメチルシクロヘキサン、又はそれらの薬理学的に許容される塩を実質的に含まないことを特徴とする、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩。
【請求項1】
次のステップ(i)から(iv)から選択される二つ以上のステップを具備することを特徴とする、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩を製造する方法であって:
ステップ(i)においては塩化メチルマグネシウム存在下でイソフォロンを3,3,5,5−テトラメチルシクロヘキサノンに変換し;
ステップ(ii)においては塩化メチルマグネシウム存在下で3,3,5,5−テトラメチルシクロヘキサノンを1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換し;
ステップ(iii)においては酸性溶液中でクロロアセトニトリル存在下で1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサンを1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンに変換し;
ステップ(iv)においては水中でチオ尿素存在下で1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンを1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンに変換する方法。
【請求項2】
請求項1の方法であって、ステップ(i)及び(ii)が選択されることを特徴とする方法。
【請求項3】
請求項1又は2の方法であって、ステップ(i)及び(iii)が選択されることを特徴とする方法。
【請求項4】
請求項1から3のいずれか一つの方法であって、ステップ(i)及び(iv)が選択されることを特徴とする方法。
【請求項5】
請求項1から4のいずれか一つの方法であって、ステップ(ii)及び(iii)が選択されることを特徴とする方法。
【請求項6】
請求項1から5のいずれか一つの方法であって、ステップ(ii)及び(iv)が選択されることを特徴とする方法。
【請求項7】
請求項1から6のいずれか一つの方法であって、ステップ(iii)及び(iv)が選択されることを特徴とする方法。
【請求項8】
請求項1から7のいずれか一つの方法であって、ステップ(i)、(ii)、及び(iii)が選択されることを特徴とする方法。
【請求項9】
請求項1から8のいずれか一つの方法であって、ステップ(i)、(ii)、及び(iv)が選択されることを特徴とする方法。
【請求項10】
請求項1から9のいずれか一つの方法であって、ステップ(i)、(iii)、及び(iv)が選択されることを特徴とする方法。
【請求項11】
請求項1から10のいずれか一つの方法であって、ステップ(ii)、(iii)、及び(iv)が選択されることを特徴とする方法。
【請求項12】
請求項1から11のいずれか一つの方法であって、ステップ(i)、(ii)、(iii)、及び(iv)が選択されることを特徴とする方法。
【請求項13】
請求項1から12のいずれか一つの方法であって、3,3,5,5−テトラメチルシクロヘキサノン、1−ヒドロキシ−1,3,3,5,5−ペンタメチルシクロヘキサン、及び1−クロロアセトアミド−1,3,3,5,5−ペンタメチルシクロヘキサンのうち少なくとも一つが精製ステップを経ないことを特徴とする方法。
【請求項14】
さらにステップ(v)を具備することを特徴とする、請求項1から13のいずれか一つの方法であって:
ステップ(v)においては酸存在下で1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサンをその薬理学的に許容される塩に変換する方法。
【請求項15】
請求項14の方法であって、前記酸がメタンスルホン酸であることを特徴とする方法。
【請求項16】
請求項1から15のいずれか一つの方法であって、前記塩化メチルマグネシウムが塩化エチルマグネシウムを含まないことを特徴とする方法。
【請求項17】
1−アミノ−1−エチル−3,3,5,5−テトラメチルシクロヘキサン及び1−アミノ−3−エチル−1,3,5,5−テトラメチルシクロヘキサン、又はそれらの薬理学的に許容される塩を実質的に含まないことを特徴とする、1−アミノ−1,3,3,5,5−ペンタメチルシクロヘキサン又はその薬理学的に許容される塩。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公表番号】特表2012−531388(P2012−531388A)
【公表日】平成24年12月10日(2012.12.10)
【国際特許分類】
【出願番号】特願2012−516586(P2012−516586)
【出願日】平成22年6月28日(2010.6.28)
【国際出願番号】PCT/EP2010/003922
【国際公開番号】WO2011/000539
【国際公開日】平成23年1月6日(2011.1.6)
【出願人】(509295413)メルツ・ファルマ・ゲーエムベーハー・ウント・コ・カーゲーアーアー (14)
【Fターム(参考)】
【公表日】平成24年12月10日(2012.12.10)
【国際特許分類】
【出願日】平成22年6月28日(2010.6.28)
【国際出願番号】PCT/EP2010/003922
【国際公開番号】WO2011/000539
【国際公開日】平成23年1月6日(2011.1.6)
【出願人】(509295413)メルツ・ファルマ・ゲーエムベーハー・ウント・コ・カーゲーアーアー (14)
【Fターム(参考)】
[ Back to top ]