
ノイラミン酸誘導体、シアリダーゼ活性阻害剤及び抗インフルエンザ薬

【課題】ウイルスのシアリダーゼ活性を選択的に阻害し、ヒトのシアリダーゼ活性を阻害し難い、ノイラミン酸誘導体、シアリダーゼ活性阻害剤及び抗インフルエンザ薬を提供する。
【解決手段】本発明のノイラミン酸誘導体は、下記化学式(1)(ただし、R1及びR2は、水素又は炭素数1〜8の分岐してもよいアルキル基若しくは炭素数1〜8の分岐してもよいシクロアルキル基を示し、R3は水素又はフッ素を示す)又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とする。

【解決手段】本発明のノイラミン酸誘導体は、下記化学式(1)(ただし、R1及びR2は、水素又は炭素数1〜8の分岐してもよいアルキル基若しくは炭素数1〜8の分岐してもよいシクロアルキル基を示し、R3は水素又はフッ素を示す)又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ノイラミン酸誘導体、並びにそれを有効成分として含有するシアリダーゼ活性阻害剤及び抗インフルエンザ薬に関する。
【背景技術】
【0002】
インフルエンザウイルスは鳥類等が感染し、伝播の過程できわめて変異しやすく、タミフルのような既存の薬剤に対する耐性が発現する。こうして新型ウイルスが発生すると、世界同時流行(パンデミック)となる場合もあり、社会的、経済的に甚大な被害をもたらす。ウイルス感染にはワクチンが有効であるが、インフルエンザウイルスやHIVのような、きわめて変異しやすいウイルスにおいては、ワクチンのみでは対応できない場合もあり、それを補うものとして、抗ウイルス薬の開発が社会的急務となっている。
【0003】
ウイルスの表面には、ヘマグルチニン(HA)及びノイラミニダーゼ(NA)(シアリダーゼとも呼ばれるが、ウイルス学の分野ではノイラミニダーゼと呼ばれる場合が多い。)という2つの重要な糖タンパク質スパイクが存在しており、ウイルスの侵入や複製に重要な役割をしている。すなわち、ヘマグルチニンは糖複合体の残基に結合するシアル酸の末端を認識し、結合する。これにより、宿主細胞に付着したり、侵入したりするのを媒介する。
一方、ノイラミニダーゼは、ウイルス分子表面のヘマグルチニンに結合する糖タンパク質端末のシアル酸(I)(下記構造式化1参照)を加水分解する反応を触媒し(これをシアリダーゼ活性という)、これにより宿主細胞からのウイルスの放出過程を促進する。さらに、ノイラミニダーゼは呼吸器系の粘液を通じてウイルス分子の移動を促し、ウイルス分子の凝集を防ぎ、ウイルスの出芽(発芽)・遊離を促進する。このため、シアリダーゼ活性を阻害する薬剤は、インフルエンザの治療予防に効果があると考えられている。
【0004】
【化1】
【0005】
現在まで、シアリダーゼ活性を阻害してインフルエンザを予防する薬剤として、下記構造式(化1)に示すオセルタミビル(2)(登録商標名:タミフル)や、ザナミビル(3)(登録商標名:リレンザ)が開発され、さらには、それらの様々な誘導体が開発されている(特許文献1及び2参照)。
【0006】
【化2】
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開2001−354635
【特許文献2】特開2008−297320
【発明の概要】
【発明が解決しようとする課題】
【0008】
しかしながら、従来のインフルエンザを予防薬剤では、精神神経疾患、高血糖症、出血性疾患、皮膚疾患、肝臓と腎臓疾患、突然死等の症例と関連する、重篤な副作用の症例が報告されており、問題となっていた。そして、それらの副作用の原因として、上記従来のシアリダーゼ活性阻害剤が、ウイルスのノイラミニダーゼのみならず、ヒトのシアリダーゼに対しても活性阻害を有することに起因するのではないかと考えられている。なぜならば、ヒトのシアリダーゼとウイルスのノイラミニダーゼとは、三次元構造及び活性部位の構造において顕著な類似性を示すからである。このため、ウイルスのシアリダーゼ活性を選択的に阻害し、ヒトのシアリダーゼ活性を阻害しない薬剤の開発が望まれていた。
【0009】
本発明は、上記従来の実情に鑑みてなされたものであり、ウイルスのノイラミニダーゼの活性を選択的に阻害し、ヒトのシアリダーゼ活性を阻害し難い、ノイラミン酸誘導体、シアリダーゼ活性阻害剤及び抗インフルエンザ薬を提供することを解決すべき課題としている。
【課題を解決するための手段】
【0010】
本発明者は、上記課題を解決すべく、ザナミビル(3)のグリセロール部分の末端をアミドに替えた化合物を合成し、そのシアリダーゼ活性について調べた。その結果、ウイルスのシアリダーゼ活性を選択的に阻害し、ヒトのシアリダーゼ活性はほとんど阻害しないことを見出し、本発明を完成するに至った。
【0011】
すなわち、本発明のノイラミン酸誘導体は、下記化学式(1)(ただし、R1及びR2は、水素又は炭素数1〜8の分岐してもよいアルキル基若しくは炭素数1〜8の分岐してもよいシクロアルキル基を示し、R3は水素又はフッ素を示す)又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とする。
【0012】
【化3】
【0013】
本発明者らは、上記化学式(1)におけるR1=n−ブチル基、R2=メチル基、R3=水素であるノイラミン酸誘導体や、R1=シクロペンチル基、R2=メチル基、R3=水素であるノイラミン酸誘導体が、ウイルスのシアリダーゼ活性を確実に阻害し、ヒトのシアリダーゼ活性をほとんど阻害しないことを確認している。
【0014】
以上の結果から、本発明のノイラミン酸誘導体をシアリダーゼ活性阻害剤に利用することができる。すなわち、本発明のシアリダーゼ活性阻害剤は、本発明のノイラミン酸誘導体を有効成分として含有することを特徴とする。
【0015】
さらには、本発明のノイラミン酸誘導体を有効成分とする抗インフルエンザ薬は、シアリダーゼ活性阻害効果が高く、ウイルスのシアリダーゼ活性を選択的に阻害することから、副作用が少ないという利点を有する。
【発明を実施するための形態】
【実施例】
【0016】
(実施例1)
実施例1では、下記化4の合成経路に従ってノイラミン酸誘導体(11)を合成した。
【0017】
【化4】
【0018】
(ステップ1)
5-Acetamido-4-azido-7,8,9-tri-O-acetyl-2,3,4,5-tetradeoxy-D-glycero-D-galacto-
non-2-enopyranosonic acid methyl ester (4)の合成
出発物質となる化合物(4)はMalcolm Chandlerらの方法(J. Chem. Soc. Perkin Trans. 1, 1173-1180 (1995)参照)に従い、下記の合成経路で合成を行った。
【0019】
【化5】
【0020】
(ステップ2)
5-Acetamido-4-(N-tert-butoxycarbonyl)amino-7,8,9-tri-O-acetyl-2,3,4,5-tetradeoxy-D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (5)の合成
ステップ1で合成した化合物(4)(7.95g, 15mmol)を酢酸エチル(250ml)に溶解させ、リンドラ―触媒(4g)、二炭酸ジ-tert-ブチル(20ml)を順次加え、水素雰囲気化、1気圧、50℃で2日間撹拌した。反応が完了した後、反応混合物をセライトろ過し、メタノールで洗浄した。続いてエバポレータで溶媒留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(酢酸エチル100%)で精製し、化合物(5)(9.1 g (98%))を白色泡状物として得た。得られた化合物(5)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CDCl3)δ 1.41 (9H, s, NHCOC(CH3)3), 1.92, 2.05, 2.07, 2.10 (12H, 4 × s, 3 × OCOCH3, 1 ×NHCOCH3), 3.78 (3H, s, COOCH3 ), 4.13 to 4.23 (3H, m, H-5, H-6, H-9a), 4.46 (1H, td, J 4,NH 9.6 , J 4,3 2.1, J 4,5 9.6, H-4), 4.66 to 4.70 (2H, m, NH4, H-9b), 5.30 (1H, ddd, J 8,7 7.5, J 8,9a 5.5, J 8,9b 2.8, H-8), 5.46 (1H, dd, J 7,8 4.8, J 7,6 1.3, H-7), 5.54 (1H, d, J NH,5 8.9, NH5), 6.90 (1H, d, J 3,4 2.1, H-3)
HR-MS (Positive): Calcd. for C23H34N2O12 (M+Na)+, 553.2009, Found 553.2012.
【0021】
(ステップ3)
5-Acetamido-4-(N-tert-butoxycarbonyl)amino-2,3,4,5-tetradeoxy-D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (6)の合成
ステップ2で合成した化合物(5)(5.3g,10mmol)をメタノール(50ml)に溶解させ、触媒量のナトリウムメトキシド加え、室温で一晩撹拌した。反応が完了した後、Dowex-50×8(H+)を用いて、PH7になるよう調製した。反応混合物をろ過した後エバポレータで溶媒留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム:メタノール=1:20)で精製し、化合物(6)(3.03g,75%)を白色固体として得た。得られた化合物(6)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CD3OD)δ 1.44 (9H, s, NHCOC(CH3)3), 1.98 (3H, s, NHCOCH3), 3.57 to 3.67 (2H, m, H-7, H-9a), 3.76 (3H, s, COOCH3 ), 3.81 (1H, dd, J 9b,9a 11.2, J 9b,8 3.1, H-9b), 3.87 (1H, ddd, J 8,7 11.2, J 8,9a 4.5, J 8,9b 2.2, H-8), 4.05 (1H, dd, J 5,4 8.7, J 5,6 9.8, H-5), 4.22 (1H, dd, J 6,5 9.8, J 6,7 <1, H-6), 4.46 (1H, dd, J 4,5 8.7, J 4,3 2.2, H-4), 5.82 (1H, d, J 3,4 2.2, H-3);
HR-MS (Positive): Calcd. for C17H28N2O9 (M+Na)+, 427.1693, Found 427.1673.
【0022】
(ステップ4)
5-Acetamido-4-(N-tert-butoxycarbonyl)amino-2,3,4,5-tetradeoxy-9-O-(p-toluene sulphonyl)-D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (7)の合成
ステップ3で合成した化合物(6)(2.23g,4mmol)と、p-トルエンスルホニルクロリド(1.15g, 18 mmol)を二股フラスコに入れ、シリカゲルが入っているデシゲータ中で減圧下、一晩乾燥させた。化合物(6)を脱水ピリジン(15 mL)に溶解させ、p-トルエンスルホニルクロリドを、−10℃で12時間以上かけて加え、0℃にて一晩撹拌した。反応混合物に脱水メタノールを加えて反応を停止させ、30分撹拌した後、エバポレータで溶媒留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(メタノール/クロロホルム=0、1/20の順)で精製し、化合物(7)(2.49g ,81%)を白色固体として得た。得られた化合物(6)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CD3OD)δ 1.43 (9H, s, NHCOC(CH3)3), 1.98 (3H, s, NHCOCH3), 2.45 (3H, s, Ar-CH3 ), 3.51 (1H, dd, J 7,6 <1, J 7,8 9.1, H-7), 3.76 (3H, s, COOCH3 ), 3.96 to 4.16 (4H, m, H-5, H-6, H-8, H-9a), 4.29 (1H, dd, J 9b,9a 9.6, J 9b,8 1.3, H-9b), 4.44 (1H, dd, J 4,3 2.1, J 4,5 9.0, H-4), 5.81 (1H, d, J 3,4 2.1, H-3), 7.43 (2H, d, H-2', H-6'), 7.80 (2H, d, H-3', H-5')
HR-MS (Positive): Calcd. for C24H34N2O11S (M+Na)+, 581.1781, Found 581.1791.
【0023】
(ステップ5)
5-Acetamido-9-azido-4-(N-tert-butoxycarbonyl)amino-2,3,4,5,9-pentadeoxy-)-D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (8)の合成
ステップ4で合成した化合物(7)(1.11g,2mmol)をDMF(10ml)に溶解させ、アジ化ナトリウム(650mg, 10mmol)及び18-クラウン-6-エーテル(105mg, 0.4 mmol)を加えて50℃で一晩撹拌した。その後、室温まで冷却し、エバポレータで溶媒留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(メタノール/クロロホルム=0、1/20の順)で精製し、化合物(8)(770mg,92%)を白色泡状物として得た。得られた化合物(8)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CD3OD)δ 1.44 (9H, s, NHCOC(CH3)3), 1.99 (3H, s, NHCOCH3), 3.38 (1H, dd, J 9a,9b 12.8, J 9a,8 6, H-9a), 3.52 to 3.56 (2H, m, H-7, H-9b), 3.77 (3H, s, COOCH3 ), 4.01 to 4.06 (2H, m, H-5, H-8), 4.22 (1H, dd, J 6,7 <1, J 6,5 11.0, H-6), 4.46 (1H, dd, J 4,3 2.3, J 4,5 9.6, H-4), 5.81 (1H, d, J 3,4 2.3, H-3)
HR-MS (Positive): Calcd. for C17H27N5O8 (M+Na)+, 452.1757, Found 452.1756.
【0024】
(ステップ6)
5-Acetamido-4-(N-tert-butoxycarbonyl)amino-2,3,4,5,9-pentadeoxy-9-pentanamido-D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (9)の合成
ステップ5で合成したアジド化合物(8)(243mg, 0.5 mmol)とN-ヒドロキシスクシンイミジルバレリルエステル(Valeryl NHS ester )(200mg, 1mmol)とを脱水テトラヒドロフラン(3ml)に溶解させ、トリメチルホスフィン(1M in THF, 1mL, 1mmol)を、窒素が発生するまで室温で滴下した後、一晩撹拌した。そして、エバポレータで溶媒留去し、残渣に水(20ml)を加えて、酢酸エチル(20 ml)で抽出した。水層を酢酸エチル(10 ml)で再度抽出し、先に抽出した酢酸エチル層と合わせ、飽和炭酸水素ナトリウム溶液、水(20mL)で順次洗浄し、硫酸ナトリウムを加えて乾燥させた。ろ過した後、エバポレータで溶媒留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(メタノール/クロロホルム=0、1/20の順)で精製し、化合物(9)(204mg,74%)を白色固体として得た。得られた化合物(9)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CD3OD)δ 0.93 (3H, t, H-4'), 1.36 (2H, sex, , H-3'), 1.43 (9H, s, NHCOC(CH3)3), 1.59 (2H, p, H-2'), 1.97 (3H, s, NHCOCH3), 2.23 (2H, t, H-1'), 3.32 (1H, H-9a, mixed with CD3OD peak), 3.43 (1H, dd, J 7,6 <1, J 7,8 8.9, H-7), 3.56 (1H, dd, J 9b,9a 13.7, J 9b,8 2.8, H-9b), 3.76 (3H, s, COOCH3 ), 3.92 (1H, ddd, J 8,7 9.6, J 8,9a 6.2, J 8,9b 2.8, H-8), 4.04 (1H, dd, J 5,4 = J 5,6 10.3, H-5), 4.25 (1H, dd, J 6,5 11.0, J 6,7 <1, H-6), 4.47 (1H, dd, J 4,5 8.9 , J 4,3 2.0, H-4), 5.82 (1H, d, J 3,4 2.0, H-3)
HR-MS (Positive): Calcd. for C22H37N3O9 (M+Na)+, 510.2427, Found 510.2482.
【0025】
(ステップ7)
5-Acetamido-4-(N,N'-bis-tert-butoxycarbonyl)guanidino-2,3,4,5,9-pentadeoxy-9-pentanamido-D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (10)の合成
ステップ6で合成した化合物(9)(195mg,0.4mmol)をジクロロメタン(2ml)に溶解させ、トリフルオロ酢酸(2mL)を室温にて加えた。出発原料の消失を確認した後、エバポレータで溶媒留去した。残渣をN,N'−ジメチルホルムアミド(2ml)に溶解させ、トリエチルアミン(165μl, 1.2mmol)を加えた。N,N’-bis-(tert-butoxycarbonyl)-1H-pyrazole-1-carboxamidine (Bis-BocPCH)(185mg, 0.6mmol)をジメチルホルムアミド(2ml)に溶解させた溶液を、反応溶液に滴下し、50℃にて16時間攪拌した。エバポレータで溶媒留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(メタノール/クロロホルム=4/10、9/10の順)で精製し、化合物(10)(141mg,56%) を白色固体として得た。得られた化合物(10)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CD3OD)δ 0.93 (3H, t, H-4'), 1.36 (2H, sex, , H-3'), 1.47, 1.52 (18H, 2 × s, 2 ×, NHCOC(CH3)3), 1.60 (2H, p, H-2'), 1.95 (3H, s, NHCOCH3), 2.23 (2H, t, H-1'), 3.34 (1H, dd, J 9a,9b 13.7, J 9a,8 6.9, H-9a), 3.45 (1H, dd, J 7,6 <1, J 7,8 8.4, H-7), 3.59 (1H, dd, J 9b,9a 13.7, J 9b,8 2.8, H-9b), 3.78 (3H, s, COOCH3 ), 3.93 (1H, ddd, J 8,7 8.4, J 8,9a 6.9, J 8,9b 2.8, H-8), 4.21 (1H, dd, J 5,4 = J 5,6 10.3, H-5), 4.30 (1H, dd, J 6,5 10.3, J 6,7 <1, H-6), 5.06 (1H, dd, J 4,5 9.6 , J 4,3 2.0, H-4), 5.89 (1H, d, J 3,4 2.0, H-3); HR-MS (Positive): Calcd. for C28H47N5O11 (M+Na)+, 652.3170, Found 652.3232.
【0026】
(ステップ8)
5-Acetamido-4-guanidino-2,3,4,5,9-pentadeoxy-9-pentanamido-D-glycero-D-galacto-non-2-enopyranosonic acid Trifluoroacetic Acid Salt (11)の合成
ステップ7で合成した化合物(10) (100mg, 0.16mmol)を、メタノール/THF混合溶媒(5/1)2mlに溶かし、LiOH(25mg)を水1mlに溶かしたアルカリ溶液を、0℃に冷却した反応液に加え、12時間撹拌を行う。反応終了後、反応混合物をイオン交換樹脂(Dowex-50×8 (H+))に通してpH7とした後、反応混合物をろ過し、エバポレートして、黄色固体として得た。これをジクロロメタン(2mL)とトリフルオロ酢酸(2mL)に室温下で溶かした。出発物質が消滅していることを確認した後、エバポレータで乾燥させ、こうして得られた粗成物は逆相カラムクロマトグラフィーにより、0〜20% メタノール水溶液(0.1% のトリフルオロ酢酸を含む)によって精製した。精製したフラクションを乾燥させ、凍結乾燥し、白色固体としてノイラミン酸誘導体(11)を(64mg,76%) で得た。得られたノイラミン酸誘導体(11)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (D2O)δ 0.90 (3H, t, H-4'), 1.32 (2H, sex, , H-3'), 1.57 (2H, p, H-2'), 2.03 (3H, s, NHCOCH3), 2.29 (2H, t, H-1'), 3.36 (1H, dd, J 9a,9b 14.4, J 9a,8 6.8, H-9a), 3.56 to 3.58 (2H, m, H-7, H-9b), 3.98 (1H, ddd, J 8,7 12.3, J 8,9a 10.3, J 8,9b 3.4, H-8), 4.22 (1H, dd, J 5,4 = J 5,6 10.3, H-5), 4.38 (1H, dd, J 6,5 10.3, J 6,7 <1, H-6), 4.44 (1H, dd, J 4,5 9.6 , J 4,3 2.7, H-4), 5.64 (1H, d, J 3,4 2.1, H-3); HR-MS (Negative): Calcd. For C19H29F3N5O9 (M-H)-, 528.1923, Found 528.1783.
【0027】
(実施例2)
実施例2では、下記化4の合成経路に従ってノイラミン酸誘導体(14)を合成した。
【0028】
【化6】
【0029】
(ステップ1)
5-Acetamido-4-(N-tert-butoxycarbonyl)amino-2,3,4,5,9-pentadeoxy-9-cyclopropane-carboxamido-D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (12) の合成
上記実施例1における中間化合物であるアジド化合物(8) (243mg, 0.5 mmol)とシクロプロパンカルボンのN-ヒドロキシコハク酸イミジルエステル(シクロプロピルNHS エステル) (185mg, 1mmol)とを乾燥THF(3ml)に溶かし、撹拌しながらトリメチルホスフィン (1M in THF, 1mL, 1mmol) を室温下で窒素の発生が完了するまで滴下した。そして、反応混合物を1昼夜撹拌し他後、揮発成分を減圧下で留去させ、残渣を酢酸エチル(20 ml)と水 (20 ml)とで抽出した。水相を分離し、さらに酢酸エチル(10ml)で抽出した。そして、酢酸エチル抽出物は飽和NaHCO3及び水(20mL)で洗浄し、無水Na2SO4を加えて乾燥し、濾過し、減圧下で濃縮した。こうして得られた粗成物をシリカゲルカラムクロマトグラフィー(0〜5% メタノール/酢酸エチル)で精製し、化合物(12)190mg (収率81%)を白色粉体として得た。得られた化合物(12)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CD3OD)δ 0.76 to 0.90 (4H, 2 × m, H-2', H-3'), 1.45 (9H, s, NHCOC(CH3)3), 1.57 to 1.61 (1H, m, H-1'), 2.01 (3H, s, NHCOCH3), 3.39 to 3.44 (2H, m, H-7, H-9b), 3.61 (1H, dd, J 9b,9a 13.7, J 9b,8 3.4, H-9b), 3.78 (3H, s, COOCH3 ), 3.94 (1H, ddd, J 8,7 9.6, J 8,9a 6.2, J 8,9b 3.4, H-8), 4.05 (1H, dd, J 5,4 = J 5,6 10.3, H-5), 4.23 (1H, dd, J 6,5 10.3, J 6,7 <1, H-6), 4.46 (1H, dd, J 4,5 10.3, J 4,3 2.7, H-4), 5.82 (1H, d, J 3,4 2.0, H-3); HR-MS (Positive): Calcd. for C21H33N3O9 (M+Na)+, 494.2114, Found 494.2149.
【0030】
(ステップ2)
5-Acetamido-4-(N,N'-bis-tert-butoxycarbonyl)guanidino-2,3,4,5,9-pentadeoxy-9-cyclopropanecarboxamido D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (13)の合成
上記のようにして得られた化合物(12)(188mg,0.4mmol)をジクロルメタン(2mL)とトリフルオロ酢酸(2mL)の混合物に室温下で加え、出発物質の消失を確認した後、反応液をエバポレートして、乾燥した。残渣をジクロルメタン (2ml)とトリエチルアミン (165μl,1.2mmol)の混合液に加えた。N,N'-bis-(tert-butoxycarbonyl)-1H-pyrazole-1-carboxamidine (Bis-BocPCH)(185mg, 0.6mmol) のジクロルメタン (2ml)溶液を反応液に滴下し、50℃で16時間撹拌を行なった後、真空下で溶媒を留去し、シリカゲルカラムクロマトグラフィー(40〜90% 酢酸エチル/ヘキサン)で精製し、化合物(13)146mg (収率61%)を白色粉体として得た。得られた化合物(13)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CD3OD)δ 0.74 to 0.86 (4H, 2 × m, H-2', H-3'), 1.47, 1.52 (18H, 2 × s, 2 ×, NHCOC(CH3)3), 1.61 to 1.65 (2H, m, H-1'), 1.95 (3H, s, NHCOCH3), 3.37 (1H, dd, J 9a,9b 13.6, J 9a,8 6.2, H-9a), 3.45 (1H, dd, J 7,6 <1, J 7,8 8.9, H-7), 3.58 to 3.61 (1H, m, H-9b), 3.77 (3H, s, COOCH3 ), 3.93 (1H, ddd, J 8,7 9.6, J 8,9a 6.8, J 8,9b 3.4, H-8), 4.21 (1H, dd, J 5,4 = J 5,6 10.3, H-5), 4.31 (1H, dd, J 6,5 11.0, J 6,7 <1, H-6), 5.04 (1H, dd, J 4,5 10.3 , J 4,3 2.7, H-4), 5.89 (1H, d, J 3,4 2.1, H-3); HR-MS (Positive): Calcd. for C27H43N5O11 (M+Na)+, 636.2857, Found 636.2884.
【0031】
(ステップ3)
5-Acetamido-4-guanidino-2,3,4,5,9-pentadeoxy-9-cyclopropanecarboxamido-D-glycero-D-galacto-non-2-enopyranosonic acid Trifluoroacetic Acid Salt (14)の合成
上記のようにして得られた化合物(13)(100mg, 0.16mmol)をメタノール/THF(5/1)混合溶媒2mlに溶かし、0℃に冷却下、LiOH (25mg)/H2O(1ml)のアルカリ液を滴下し、12時間撹拌する。反応終了後、反応混合物をイオン交換樹脂(Dowex-50×8 (H+))に通してpH7とした後、反応混合物をろ過し、エバポレートして、黄白色固体として得た。これをジクロロメタン(2mL)とトリフルオロ酢酸(2mL)に室温下で溶かした。出発物質が消滅していることを確認した後、エバポレータで乾燥させ、こうして得られた粗成物は逆相カラムクロマトグラフィーにより、0〜20% メタノール水溶液(0.1% のトリフルオロ酢酸を含む)によって精製した。精製したフラクションを乾燥させ、凍結乾燥し、白色固体としてノイラミン酸誘導体(14)を67mg (収率83%)で得た。得られたノイラミン酸誘導体(14)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (D2O)δ 0.83 to 0.84 (4H, 2 × m, H-2', H-3'), 1.63 to 1.66 (2H, m, H-1'), 2.02 (3H, s, NHCOCH3), 3.34 (1H, dd, J 9a,9b 13.7, J 9a,8 7.6, H-9a), 3.55 (1H, dd, J 7,8 8.9 , J 7,6 1.4, H-7), 3.59 (1H, dd, J 9b,9a 13.7, J 9b,8 2.8, H-9b), 3.97 (1H, ddd, J 8,7 12.3, J 8,9a 6.9, J 8,9b 3.4, H-8), 4.20 (1H, dd, J 5,4 = J 5,6 10.3, H-5), 4.36 (1H, dd, J 6,5 10.3, J 6,7 1.4, H-6), 4.42 (1H, dd, J 4,5 9.6 , J 4,3 2.7, H-4), 5.61 (1H, d, J 3,4 2.0, H-3); HR-MS (Negative): Calcd. For C18H25F3N5O9 (M-H)-, 512.1610, Found 512.1729.
【0032】
<評 価>
(ウイルスシアリダーゼに対する活性阻害の測定)
上記のようにして合成したノイラミン酸誘導体(11)及びノイラミン酸誘導体(14)について、インフルエンザA型ウイルスのシダリダーゼに対する活性阻害を、4-methylumbelliferyl N-acetylneuraminic酸(MUNA)を用いた蛍光測定法によって評価した。
すなわち、20mMの酢酸ナトリウム緩衝液(pH6.0)15μlと、5段階の濃度に調整した所定量のノイラミン酸誘導体と、2ユニットのシアリダーゼを含むウイルス5μlとを混合し、37℃で15分インキュベーションを行った。ここで、ウイルスシアリダーゼの1ユニットとは、pH6.0で37℃で1分につき1nmolのMUの生成をするために必要な酵素量と定義される。
次に、5μlのMUNAを加え、さらに37℃で15分インキュベーションを行った。
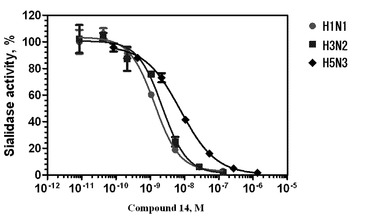
最後に、200mM炭酸ナトリウム−重炭酸塩緩衝液(pH10.6)200μlを加えて反応を終了させた。リリースされるノイラミン酸と等しい量のMUは、プレートリーダー(ミトラLB940、バートルドTechnologies、Pforzheim、ドイツ)によって、355nmの励起と460nmの蛍光によって検出された。IC50値は、GraphPad Prismソフトウェア(サンディエゴ、CA、USA)で非線形回帰分析法を使用して計算した。また、比較のため、オセルタミビル(2)(登録商標名:タミフル)及びザナミビル(3)(登録商標名:リレンザ)の活性阻害についても、同様に測定した。結果を表1並びに図3及び図4に示す。
【0033】
【表1】
【0034】
図1及び表1に示すように、ノイラミン酸誘導体(11)は、H1N1、H3N2及びH5N3のウイルスのシアリダーゼに対して、高い活性阻害作用を奏することが分かった。また、ノイラミン酸誘導体(14)は、図2及び表1に示すように、活性阻害の効果は、ノイラミン酸誘導体(11)よりも顕著であり、ザナミビル(3)やオセルタミビル(2)と同程度であった。
以上の結果は、ノイラミン酸誘導体(11)及びノイラミン酸誘導体(14)がシアリダーゼ活性阻害剤及び抗ウイルス薬として充分な効果を有することを明らかにしている。
【0035】
(ヒト−シアリダーゼ(NEU1、NEU2及びNEU4)に対する活性阻害の測定)
上記のようにして合成したノイラミン酸誘導体(11)及びノイラミン酸誘導体(14)について、3種類(NEU1、NEU2、及びNEU4)のヒト−シアリダーゼに対する活性阻害を、4-methylumbelliferyl N-acetylneuraminic酸(MUNA)を用いた蛍光測定法によって評価した。
すなわち、50μLの反応液において、500mMの酢酸ナトリウム緩衝液(NEU1及びNEU4に対してはpH4.6、NEU2に対してはpH5.5)10μL、5段階の濃度に調整した所定量のノイラミン酸誘導体、50μgの子牛由来のアルブミン、20nmolのMUNA、所定量の組み換え型ヒト−シアリダーゼを混合し、37℃で30分間インキュベーションを行った。そしてリリースされるノイラミン酸と等しい量のMUを、ウイルスシアリダーゼに対する活性阻害の測定の場合と同様の方法で測定した。
なお、IC50値は、マイクロソフトエクセルによる非線形回帰分析法を用いて計算した。
また、比較のため、ザナミビル(3)(登録商標名:リレンザ)の活性阻害についても同様に測定した。
【0036】
(ヒト−シアリダーゼ(NEU3)に対する活性阻害の測定)
上記のようにして合成したノイラミン酸誘導体(11)及びノイラミン酸誘導体(14)について、ヒト−シアリダーゼNEU3に対する活性阻害を、malononitrileを用いた高速液体クロマトグラフィーによる蛍光測定法によって評価した。
すなわち、5μLの反応液において、500mMの酢酸ナトリウム緩衝液(pH4.5)10μL、5段階の濃度に調整した所定量のノイラミン酸誘導体、50μgの子牛由来のアルブミン、5nmolのガングリオシドGM3、50μgのトリトンX−100、所定量の組み換え型ヒト−シアリダーゼを混合し、37℃で30分間インキュベーションを行った。
最後に、200mM炭酸ナトリウム−重炭酸塩緩衝液(pH10.6)200μlを加えて反応を終了させた。そしてGM3からリリースされたシアル酸をmalononitrileを用いた高速液体クロマトグラフィーによる蛍光測定法によって測定した。
なお、IC50値は、マイクロソフトエクセルによる非線形回帰分析法を用いて計算した。
また、比較のため、ザナミビル(3)(登録商標名:リレンザ)の活性阻害についても同様に測定した。
【0037】
【表2】
【0038】
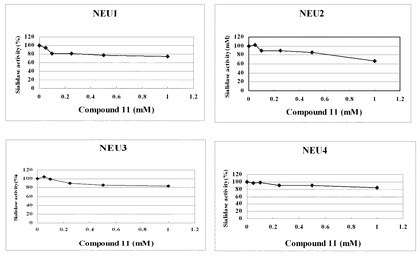
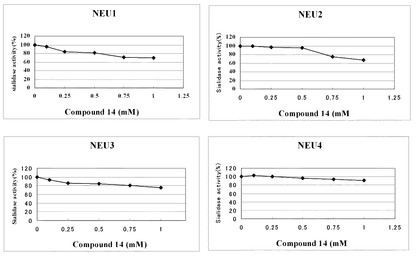
結果を表2並びに図3及び図4に示す。
表2及び図3に示すように、ノイラミン酸誘導体(11)は、ヒト由来の4種類のシアリダーゼ(NEU1、NEU2、NEU3及びNEU4)のいずれに対しても、ヒト−シアリダーゼ活性阻害(IC50)はほとんど認められない程度であった。
また、表2及び図4に示すように、ノイラミン酸誘導体(14)についても、ヒト由来の4種類のシアリダーゼ(NEU1、NEU2、NEU3及びNEU4)のいずれに対しても、ヒト−シアリダーゼ活性阻害(IC50)はほとんど認められない程度であった。
以上の結果は、ノイラミン酸誘導体(11)及びノイラミン酸誘導体(14)を抗ウイルス薬として用いた場合においても、副作用の可能性が極めて少ないことを示している。
【0039】
この発明は上記発明の実施の態様及び実施例の説明に何ら限定されるものではない。特許請求の範囲を逸脱せず、当業者が容易に想到できる範囲で種々の変形態様もこの発明に含まれる。
【図面の簡単な説明】
【0040】
【図1】ノイラミン酸誘導体(11)のウイルス(H1N1、H3N2及びH5N3)シアリダーゼに対する活性阻害の測定結果を示すグラフである。
【図2】ノイラミン酸誘導体(14)のウイルス(H1N1、H3N2及びH5N3)シアリダーゼに対する活性阻害の測定結果を示すグラフである。
【図3】ノイラミン酸誘導体(11)のヒト−シアリダーゼ(NEU1、NEU2、NEU3及びNEU4)に対する活性阻害の測定結果を示すグラフである。
【図4】ノイラミン酸誘導体(14)のヒト−シアリダーゼ(NEU1、NEU2、NEU3及びNEU4)に対する活性阻害の測定結果を示すグラフである。
【産業上の利用可能性】
【0041】
本発明のノイラミン酸誘導体及びシアリダーゼ活性阻害剤は、シアリダーゼの機能を調べる生物学的ツールとして利用することができる。また、本発明のノイラミン酸誘導体は、ウイルスのシアリダーゼ活性を選択的に阻害し、ヒトのシアリダーゼ活性をほとんど阻害しないことから、副作用の少ない抗インフルエンザ薬等に利用できる。
【技術分野】
【0001】
本発明は、ノイラミン酸誘導体、並びにそれを有効成分として含有するシアリダーゼ活性阻害剤及び抗インフルエンザ薬に関する。
【背景技術】
【0002】
インフルエンザウイルスは鳥類等が感染し、伝播の過程できわめて変異しやすく、タミフルのような既存の薬剤に対する耐性が発現する。こうして新型ウイルスが発生すると、世界同時流行(パンデミック)となる場合もあり、社会的、経済的に甚大な被害をもたらす。ウイルス感染にはワクチンが有効であるが、インフルエンザウイルスやHIVのような、きわめて変異しやすいウイルスにおいては、ワクチンのみでは対応できない場合もあり、それを補うものとして、抗ウイルス薬の開発が社会的急務となっている。
【0003】
ウイルスの表面には、ヘマグルチニン(HA)及びノイラミニダーゼ(NA)(シアリダーゼとも呼ばれるが、ウイルス学の分野ではノイラミニダーゼと呼ばれる場合が多い。)という2つの重要な糖タンパク質スパイクが存在しており、ウイルスの侵入や複製に重要な役割をしている。すなわち、ヘマグルチニンは糖複合体の残基に結合するシアル酸の末端を認識し、結合する。これにより、宿主細胞に付着したり、侵入したりするのを媒介する。
一方、ノイラミニダーゼは、ウイルス分子表面のヘマグルチニンに結合する糖タンパク質端末のシアル酸(I)(下記構造式化1参照)を加水分解する反応を触媒し(これをシアリダーゼ活性という)、これにより宿主細胞からのウイルスの放出過程を促進する。さらに、ノイラミニダーゼは呼吸器系の粘液を通じてウイルス分子の移動を促し、ウイルス分子の凝集を防ぎ、ウイルスの出芽(発芽)・遊離を促進する。このため、シアリダーゼ活性を阻害する薬剤は、インフルエンザの治療予防に効果があると考えられている。
【0004】
【化1】
【0005】
現在まで、シアリダーゼ活性を阻害してインフルエンザを予防する薬剤として、下記構造式(化1)に示すオセルタミビル(2)(登録商標名:タミフル)や、ザナミビル(3)(登録商標名:リレンザ)が開発され、さらには、それらの様々な誘導体が開発されている(特許文献1及び2参照)。
【0006】
【化2】
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開2001−354635
【特許文献2】特開2008−297320
【発明の概要】
【発明が解決しようとする課題】
【0008】
しかしながら、従来のインフルエンザを予防薬剤では、精神神経疾患、高血糖症、出血性疾患、皮膚疾患、肝臓と腎臓疾患、突然死等の症例と関連する、重篤な副作用の症例が報告されており、問題となっていた。そして、それらの副作用の原因として、上記従来のシアリダーゼ活性阻害剤が、ウイルスのノイラミニダーゼのみならず、ヒトのシアリダーゼに対しても活性阻害を有することに起因するのではないかと考えられている。なぜならば、ヒトのシアリダーゼとウイルスのノイラミニダーゼとは、三次元構造及び活性部位の構造において顕著な類似性を示すからである。このため、ウイルスのシアリダーゼ活性を選択的に阻害し、ヒトのシアリダーゼ活性を阻害しない薬剤の開発が望まれていた。
【0009】
本発明は、上記従来の実情に鑑みてなされたものであり、ウイルスのノイラミニダーゼの活性を選択的に阻害し、ヒトのシアリダーゼ活性を阻害し難い、ノイラミン酸誘導体、シアリダーゼ活性阻害剤及び抗インフルエンザ薬を提供することを解決すべき課題としている。
【課題を解決するための手段】
【0010】
本発明者は、上記課題を解決すべく、ザナミビル(3)のグリセロール部分の末端をアミドに替えた化合物を合成し、そのシアリダーゼ活性について調べた。その結果、ウイルスのシアリダーゼ活性を選択的に阻害し、ヒトのシアリダーゼ活性はほとんど阻害しないことを見出し、本発明を完成するに至った。
【0011】
すなわち、本発明のノイラミン酸誘導体は、下記化学式(1)(ただし、R1及びR2は、水素又は炭素数1〜8の分岐してもよいアルキル基若しくは炭素数1〜8の分岐してもよいシクロアルキル基を示し、R3は水素又はフッ素を示す)又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とする。
【0012】
【化3】
【0013】
本発明者らは、上記化学式(1)におけるR1=n−ブチル基、R2=メチル基、R3=水素であるノイラミン酸誘導体や、R1=シクロペンチル基、R2=メチル基、R3=水素であるノイラミン酸誘導体が、ウイルスのシアリダーゼ活性を確実に阻害し、ヒトのシアリダーゼ活性をほとんど阻害しないことを確認している。
【0014】
以上の結果から、本発明のノイラミン酸誘導体をシアリダーゼ活性阻害剤に利用することができる。すなわち、本発明のシアリダーゼ活性阻害剤は、本発明のノイラミン酸誘導体を有効成分として含有することを特徴とする。
【0015】
さらには、本発明のノイラミン酸誘導体を有効成分とする抗インフルエンザ薬は、シアリダーゼ活性阻害効果が高く、ウイルスのシアリダーゼ活性を選択的に阻害することから、副作用が少ないという利点を有する。
【発明を実施するための形態】
【実施例】
【0016】
(実施例1)
実施例1では、下記化4の合成経路に従ってノイラミン酸誘導体(11)を合成した。
【0017】
【化4】
【0018】
(ステップ1)
5-Acetamido-4-azido-7,8,9-tri-O-acetyl-2,3,4,5-tetradeoxy-D-glycero-D-galacto-
non-2-enopyranosonic acid methyl ester (4)の合成
出発物質となる化合物(4)はMalcolm Chandlerらの方法(J. Chem. Soc. Perkin Trans. 1, 1173-1180 (1995)参照)に従い、下記の合成経路で合成を行った。
【0019】
【化5】
【0020】
(ステップ2)
5-Acetamido-4-(N-tert-butoxycarbonyl)amino-7,8,9-tri-O-acetyl-2,3,4,5-tetradeoxy-D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (5)の合成
ステップ1で合成した化合物(4)(7.95g, 15mmol)を酢酸エチル(250ml)に溶解させ、リンドラ―触媒(4g)、二炭酸ジ-tert-ブチル(20ml)を順次加え、水素雰囲気化、1気圧、50℃で2日間撹拌した。反応が完了した後、反応混合物をセライトろ過し、メタノールで洗浄した。続いてエバポレータで溶媒留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(酢酸エチル100%)で精製し、化合物(5)(9.1 g (98%))を白色泡状物として得た。得られた化合物(5)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CDCl3)δ 1.41 (9H, s, NHCOC(CH3)3), 1.92, 2.05, 2.07, 2.10 (12H, 4 × s, 3 × OCOCH3, 1 ×NHCOCH3), 3.78 (3H, s, COOCH3 ), 4.13 to 4.23 (3H, m, H-5, H-6, H-9a), 4.46 (1H, td, J 4,NH 9.6 , J 4,3 2.1, J 4,5 9.6, H-4), 4.66 to 4.70 (2H, m, NH4, H-9b), 5.30 (1H, ddd, J 8,7 7.5, J 8,9a 5.5, J 8,9b 2.8, H-8), 5.46 (1H, dd, J 7,8 4.8, J 7,6 1.3, H-7), 5.54 (1H, d, J NH,5 8.9, NH5), 6.90 (1H, d, J 3,4 2.1, H-3)
HR-MS (Positive): Calcd. for C23H34N2O12 (M+Na)+, 553.2009, Found 553.2012.
【0021】
(ステップ3)
5-Acetamido-4-(N-tert-butoxycarbonyl)amino-2,3,4,5-tetradeoxy-D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (6)の合成
ステップ2で合成した化合物(5)(5.3g,10mmol)をメタノール(50ml)に溶解させ、触媒量のナトリウムメトキシド加え、室温で一晩撹拌した。反応が完了した後、Dowex-50×8(H+)を用いて、PH7になるよう調製した。反応混合物をろ過した後エバポレータで溶媒留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム:メタノール=1:20)で精製し、化合物(6)(3.03g,75%)を白色固体として得た。得られた化合物(6)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CD3OD)δ 1.44 (9H, s, NHCOC(CH3)3), 1.98 (3H, s, NHCOCH3), 3.57 to 3.67 (2H, m, H-7, H-9a), 3.76 (3H, s, COOCH3 ), 3.81 (1H, dd, J 9b,9a 11.2, J 9b,8 3.1, H-9b), 3.87 (1H, ddd, J 8,7 11.2, J 8,9a 4.5, J 8,9b 2.2, H-8), 4.05 (1H, dd, J 5,4 8.7, J 5,6 9.8, H-5), 4.22 (1H, dd, J 6,5 9.8, J 6,7 <1, H-6), 4.46 (1H, dd, J 4,5 8.7, J 4,3 2.2, H-4), 5.82 (1H, d, J 3,4 2.2, H-3);
HR-MS (Positive): Calcd. for C17H28N2O9 (M+Na)+, 427.1693, Found 427.1673.
【0022】
(ステップ4)
5-Acetamido-4-(N-tert-butoxycarbonyl)amino-2,3,4,5-tetradeoxy-9-O-(p-toluene sulphonyl)-D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (7)の合成
ステップ3で合成した化合物(6)(2.23g,4mmol)と、p-トルエンスルホニルクロリド(1.15g, 18 mmol)を二股フラスコに入れ、シリカゲルが入っているデシゲータ中で減圧下、一晩乾燥させた。化合物(6)を脱水ピリジン(15 mL)に溶解させ、p-トルエンスルホニルクロリドを、−10℃で12時間以上かけて加え、0℃にて一晩撹拌した。反応混合物に脱水メタノールを加えて反応を停止させ、30分撹拌した後、エバポレータで溶媒留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(メタノール/クロロホルム=0、1/20の順)で精製し、化合物(7)(2.49g ,81%)を白色固体として得た。得られた化合物(6)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CD3OD)δ 1.43 (9H, s, NHCOC(CH3)3), 1.98 (3H, s, NHCOCH3), 2.45 (3H, s, Ar-CH3 ), 3.51 (1H, dd, J 7,6 <1, J 7,8 9.1, H-7), 3.76 (3H, s, COOCH3 ), 3.96 to 4.16 (4H, m, H-5, H-6, H-8, H-9a), 4.29 (1H, dd, J 9b,9a 9.6, J 9b,8 1.3, H-9b), 4.44 (1H, dd, J 4,3 2.1, J 4,5 9.0, H-4), 5.81 (1H, d, J 3,4 2.1, H-3), 7.43 (2H, d, H-2', H-6'), 7.80 (2H, d, H-3', H-5')
HR-MS (Positive): Calcd. for C24H34N2O11S (M+Na)+, 581.1781, Found 581.1791.
【0023】
(ステップ5)
5-Acetamido-9-azido-4-(N-tert-butoxycarbonyl)amino-2,3,4,5,9-pentadeoxy-)-D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (8)の合成
ステップ4で合成した化合物(7)(1.11g,2mmol)をDMF(10ml)に溶解させ、アジ化ナトリウム(650mg, 10mmol)及び18-クラウン-6-エーテル(105mg, 0.4 mmol)を加えて50℃で一晩撹拌した。その後、室温まで冷却し、エバポレータで溶媒留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(メタノール/クロロホルム=0、1/20の順)で精製し、化合物(8)(770mg,92%)を白色泡状物として得た。得られた化合物(8)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CD3OD)δ 1.44 (9H, s, NHCOC(CH3)3), 1.99 (3H, s, NHCOCH3), 3.38 (1H, dd, J 9a,9b 12.8, J 9a,8 6, H-9a), 3.52 to 3.56 (2H, m, H-7, H-9b), 3.77 (3H, s, COOCH3 ), 4.01 to 4.06 (2H, m, H-5, H-8), 4.22 (1H, dd, J 6,7 <1, J 6,5 11.0, H-6), 4.46 (1H, dd, J 4,3 2.3, J 4,5 9.6, H-4), 5.81 (1H, d, J 3,4 2.3, H-3)
HR-MS (Positive): Calcd. for C17H27N5O8 (M+Na)+, 452.1757, Found 452.1756.
【0024】
(ステップ6)
5-Acetamido-4-(N-tert-butoxycarbonyl)amino-2,3,4,5,9-pentadeoxy-9-pentanamido-D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (9)の合成
ステップ5で合成したアジド化合物(8)(243mg, 0.5 mmol)とN-ヒドロキシスクシンイミジルバレリルエステル(Valeryl NHS ester )(200mg, 1mmol)とを脱水テトラヒドロフラン(3ml)に溶解させ、トリメチルホスフィン(1M in THF, 1mL, 1mmol)を、窒素が発生するまで室温で滴下した後、一晩撹拌した。そして、エバポレータで溶媒留去し、残渣に水(20ml)を加えて、酢酸エチル(20 ml)で抽出した。水層を酢酸エチル(10 ml)で再度抽出し、先に抽出した酢酸エチル層と合わせ、飽和炭酸水素ナトリウム溶液、水(20mL)で順次洗浄し、硫酸ナトリウムを加えて乾燥させた。ろ過した後、エバポレータで溶媒留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(メタノール/クロロホルム=0、1/20の順)で精製し、化合物(9)(204mg,74%)を白色固体として得た。得られた化合物(9)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CD3OD)δ 0.93 (3H, t, H-4'), 1.36 (2H, sex, , H-3'), 1.43 (9H, s, NHCOC(CH3)3), 1.59 (2H, p, H-2'), 1.97 (3H, s, NHCOCH3), 2.23 (2H, t, H-1'), 3.32 (1H, H-9a, mixed with CD3OD peak), 3.43 (1H, dd, J 7,6 <1, J 7,8 8.9, H-7), 3.56 (1H, dd, J 9b,9a 13.7, J 9b,8 2.8, H-9b), 3.76 (3H, s, COOCH3 ), 3.92 (1H, ddd, J 8,7 9.6, J 8,9a 6.2, J 8,9b 2.8, H-8), 4.04 (1H, dd, J 5,4 = J 5,6 10.3, H-5), 4.25 (1H, dd, J 6,5 11.0, J 6,7 <1, H-6), 4.47 (1H, dd, J 4,5 8.9 , J 4,3 2.0, H-4), 5.82 (1H, d, J 3,4 2.0, H-3)
HR-MS (Positive): Calcd. for C22H37N3O9 (M+Na)+, 510.2427, Found 510.2482.
【0025】
(ステップ7)
5-Acetamido-4-(N,N'-bis-tert-butoxycarbonyl)guanidino-2,3,4,5,9-pentadeoxy-9-pentanamido-D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (10)の合成
ステップ6で合成した化合物(9)(195mg,0.4mmol)をジクロロメタン(2ml)に溶解させ、トリフルオロ酢酸(2mL)を室温にて加えた。出発原料の消失を確認した後、エバポレータで溶媒留去した。残渣をN,N'−ジメチルホルムアミド(2ml)に溶解させ、トリエチルアミン(165μl, 1.2mmol)を加えた。N,N’-bis-(tert-butoxycarbonyl)-1H-pyrazole-1-carboxamidine (Bis-BocPCH)(185mg, 0.6mmol)をジメチルホルムアミド(2ml)に溶解させた溶液を、反応溶液に滴下し、50℃にて16時間攪拌した。エバポレータで溶媒留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(メタノール/クロロホルム=4/10、9/10の順)で精製し、化合物(10)(141mg,56%) を白色固体として得た。得られた化合物(10)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CD3OD)δ 0.93 (3H, t, H-4'), 1.36 (2H, sex, , H-3'), 1.47, 1.52 (18H, 2 × s, 2 ×, NHCOC(CH3)3), 1.60 (2H, p, H-2'), 1.95 (3H, s, NHCOCH3), 2.23 (2H, t, H-1'), 3.34 (1H, dd, J 9a,9b 13.7, J 9a,8 6.9, H-9a), 3.45 (1H, dd, J 7,6 <1, J 7,8 8.4, H-7), 3.59 (1H, dd, J 9b,9a 13.7, J 9b,8 2.8, H-9b), 3.78 (3H, s, COOCH3 ), 3.93 (1H, ddd, J 8,7 8.4, J 8,9a 6.9, J 8,9b 2.8, H-8), 4.21 (1H, dd, J 5,4 = J 5,6 10.3, H-5), 4.30 (1H, dd, J 6,5 10.3, J 6,7 <1, H-6), 5.06 (1H, dd, J 4,5 9.6 , J 4,3 2.0, H-4), 5.89 (1H, d, J 3,4 2.0, H-3); HR-MS (Positive): Calcd. for C28H47N5O11 (M+Na)+, 652.3170, Found 652.3232.
【0026】
(ステップ8)
5-Acetamido-4-guanidino-2,3,4,5,9-pentadeoxy-9-pentanamido-D-glycero-D-galacto-non-2-enopyranosonic acid Trifluoroacetic Acid Salt (11)の合成
ステップ7で合成した化合物(10) (100mg, 0.16mmol)を、メタノール/THF混合溶媒(5/1)2mlに溶かし、LiOH(25mg)を水1mlに溶かしたアルカリ溶液を、0℃に冷却した反応液に加え、12時間撹拌を行う。反応終了後、反応混合物をイオン交換樹脂(Dowex-50×8 (H+))に通してpH7とした後、反応混合物をろ過し、エバポレートして、黄色固体として得た。これをジクロロメタン(2mL)とトリフルオロ酢酸(2mL)に室温下で溶かした。出発物質が消滅していることを確認した後、エバポレータで乾燥させ、こうして得られた粗成物は逆相カラムクロマトグラフィーにより、0〜20% メタノール水溶液(0.1% のトリフルオロ酢酸を含む)によって精製した。精製したフラクションを乾燥させ、凍結乾燥し、白色固体としてノイラミン酸誘導体(11)を(64mg,76%) で得た。得られたノイラミン酸誘導体(11)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (D2O)δ 0.90 (3H, t, H-4'), 1.32 (2H, sex, , H-3'), 1.57 (2H, p, H-2'), 2.03 (3H, s, NHCOCH3), 2.29 (2H, t, H-1'), 3.36 (1H, dd, J 9a,9b 14.4, J 9a,8 6.8, H-9a), 3.56 to 3.58 (2H, m, H-7, H-9b), 3.98 (1H, ddd, J 8,7 12.3, J 8,9a 10.3, J 8,9b 3.4, H-8), 4.22 (1H, dd, J 5,4 = J 5,6 10.3, H-5), 4.38 (1H, dd, J 6,5 10.3, J 6,7 <1, H-6), 4.44 (1H, dd, J 4,5 9.6 , J 4,3 2.7, H-4), 5.64 (1H, d, J 3,4 2.1, H-3); HR-MS (Negative): Calcd. For C19H29F3N5O9 (M-H)-, 528.1923, Found 528.1783.
【0027】
(実施例2)
実施例2では、下記化4の合成経路に従ってノイラミン酸誘導体(14)を合成した。
【0028】
【化6】
【0029】
(ステップ1)
5-Acetamido-4-(N-tert-butoxycarbonyl)amino-2,3,4,5,9-pentadeoxy-9-cyclopropane-carboxamido-D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (12) の合成
上記実施例1における中間化合物であるアジド化合物(8) (243mg, 0.5 mmol)とシクロプロパンカルボンのN-ヒドロキシコハク酸イミジルエステル(シクロプロピルNHS エステル) (185mg, 1mmol)とを乾燥THF(3ml)に溶かし、撹拌しながらトリメチルホスフィン (1M in THF, 1mL, 1mmol) を室温下で窒素の発生が完了するまで滴下した。そして、反応混合物を1昼夜撹拌し他後、揮発成分を減圧下で留去させ、残渣を酢酸エチル(20 ml)と水 (20 ml)とで抽出した。水相を分離し、さらに酢酸エチル(10ml)で抽出した。そして、酢酸エチル抽出物は飽和NaHCO3及び水(20mL)で洗浄し、無水Na2SO4を加えて乾燥し、濾過し、減圧下で濃縮した。こうして得られた粗成物をシリカゲルカラムクロマトグラフィー(0〜5% メタノール/酢酸エチル)で精製し、化合物(12)190mg (収率81%)を白色粉体として得た。得られた化合物(12)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CD3OD)δ 0.76 to 0.90 (4H, 2 × m, H-2', H-3'), 1.45 (9H, s, NHCOC(CH3)3), 1.57 to 1.61 (1H, m, H-1'), 2.01 (3H, s, NHCOCH3), 3.39 to 3.44 (2H, m, H-7, H-9b), 3.61 (1H, dd, J 9b,9a 13.7, J 9b,8 3.4, H-9b), 3.78 (3H, s, COOCH3 ), 3.94 (1H, ddd, J 8,7 9.6, J 8,9a 6.2, J 8,9b 3.4, H-8), 4.05 (1H, dd, J 5,4 = J 5,6 10.3, H-5), 4.23 (1H, dd, J 6,5 10.3, J 6,7 <1, H-6), 4.46 (1H, dd, J 4,5 10.3, J 4,3 2.7, H-4), 5.82 (1H, d, J 3,4 2.0, H-3); HR-MS (Positive): Calcd. for C21H33N3O9 (M+Na)+, 494.2114, Found 494.2149.
【0030】
(ステップ2)
5-Acetamido-4-(N,N'-bis-tert-butoxycarbonyl)guanidino-2,3,4,5,9-pentadeoxy-9-cyclopropanecarboxamido D-glycero-D-galacto-non-2-enopyranosonic acid methyl ester (13)の合成
上記のようにして得られた化合物(12)(188mg,0.4mmol)をジクロルメタン(2mL)とトリフルオロ酢酸(2mL)の混合物に室温下で加え、出発物質の消失を確認した後、反応液をエバポレートして、乾燥した。残渣をジクロルメタン (2ml)とトリエチルアミン (165μl,1.2mmol)の混合液に加えた。N,N'-bis-(tert-butoxycarbonyl)-1H-pyrazole-1-carboxamidine (Bis-BocPCH)(185mg, 0.6mmol) のジクロルメタン (2ml)溶液を反応液に滴下し、50℃で16時間撹拌を行なった後、真空下で溶媒を留去し、シリカゲルカラムクロマトグラフィー(40〜90% 酢酸エチル/ヘキサン)で精製し、化合物(13)146mg (収率61%)を白色粉体として得た。得られた化合物(13)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (CD3OD)δ 0.74 to 0.86 (4H, 2 × m, H-2', H-3'), 1.47, 1.52 (18H, 2 × s, 2 ×, NHCOC(CH3)3), 1.61 to 1.65 (2H, m, H-1'), 1.95 (3H, s, NHCOCH3), 3.37 (1H, dd, J 9a,9b 13.6, J 9a,8 6.2, H-9a), 3.45 (1H, dd, J 7,6 <1, J 7,8 8.9, H-7), 3.58 to 3.61 (1H, m, H-9b), 3.77 (3H, s, COOCH3 ), 3.93 (1H, ddd, J 8,7 9.6, J 8,9a 6.8, J 8,9b 3.4, H-8), 4.21 (1H, dd, J 5,4 = J 5,6 10.3, H-5), 4.31 (1H, dd, J 6,5 11.0, J 6,7 <1, H-6), 5.04 (1H, dd, J 4,5 10.3 , J 4,3 2.7, H-4), 5.89 (1H, d, J 3,4 2.1, H-3); HR-MS (Positive): Calcd. for C27H43N5O11 (M+Na)+, 636.2857, Found 636.2884.
【0031】
(ステップ3)
5-Acetamido-4-guanidino-2,3,4,5,9-pentadeoxy-9-cyclopropanecarboxamido-D-glycero-D-galacto-non-2-enopyranosonic acid Trifluoroacetic Acid Salt (14)の合成
上記のようにして得られた化合物(13)(100mg, 0.16mmol)をメタノール/THF(5/1)混合溶媒2mlに溶かし、0℃に冷却下、LiOH (25mg)/H2O(1ml)のアルカリ液を滴下し、12時間撹拌する。反応終了後、反応混合物をイオン交換樹脂(Dowex-50×8 (H+))に通してpH7とした後、反応混合物をろ過し、エバポレートして、黄白色固体として得た。これをジクロロメタン(2mL)とトリフルオロ酢酸(2mL)に室温下で溶かした。出発物質が消滅していることを確認した後、エバポレータで乾燥させ、こうして得られた粗成物は逆相カラムクロマトグラフィーにより、0〜20% メタノール水溶液(0.1% のトリフルオロ酢酸を含む)によって精製した。精製したフラクションを乾燥させ、凍結乾燥し、白色固体としてノイラミン酸誘導体(14)を67mg (収率83%)で得た。得られたノイラミン酸誘導体(14)の1H-NMRデータ及び、HR-MSデータを以下に示す。
1H NMR (D2O)δ 0.83 to 0.84 (4H, 2 × m, H-2', H-3'), 1.63 to 1.66 (2H, m, H-1'), 2.02 (3H, s, NHCOCH3), 3.34 (1H, dd, J 9a,9b 13.7, J 9a,8 7.6, H-9a), 3.55 (1H, dd, J 7,8 8.9 , J 7,6 1.4, H-7), 3.59 (1H, dd, J 9b,9a 13.7, J 9b,8 2.8, H-9b), 3.97 (1H, ddd, J 8,7 12.3, J 8,9a 6.9, J 8,9b 3.4, H-8), 4.20 (1H, dd, J 5,4 = J 5,6 10.3, H-5), 4.36 (1H, dd, J 6,5 10.3, J 6,7 1.4, H-6), 4.42 (1H, dd, J 4,5 9.6 , J 4,3 2.7, H-4), 5.61 (1H, d, J 3,4 2.0, H-3); HR-MS (Negative): Calcd. For C18H25F3N5O9 (M-H)-, 512.1610, Found 512.1729.
【0032】
<評 価>
(ウイルスシアリダーゼに対する活性阻害の測定)
上記のようにして合成したノイラミン酸誘導体(11)及びノイラミン酸誘導体(14)について、インフルエンザA型ウイルスのシダリダーゼに対する活性阻害を、4-methylumbelliferyl N-acetylneuraminic酸(MUNA)を用いた蛍光測定法によって評価した。
すなわち、20mMの酢酸ナトリウム緩衝液(pH6.0)15μlと、5段階の濃度に調整した所定量のノイラミン酸誘導体と、2ユニットのシアリダーゼを含むウイルス5μlとを混合し、37℃で15分インキュベーションを行った。ここで、ウイルスシアリダーゼの1ユニットとは、pH6.0で37℃で1分につき1nmolのMUの生成をするために必要な酵素量と定義される。
次に、5μlのMUNAを加え、さらに37℃で15分インキュベーションを行った。
最後に、200mM炭酸ナトリウム−重炭酸塩緩衝液(pH10.6)200μlを加えて反応を終了させた。リリースされるノイラミン酸と等しい量のMUは、プレートリーダー(ミトラLB940、バートルドTechnologies、Pforzheim、ドイツ)によって、355nmの励起と460nmの蛍光によって検出された。IC50値は、GraphPad Prismソフトウェア(サンディエゴ、CA、USA)で非線形回帰分析法を使用して計算した。また、比較のため、オセルタミビル(2)(登録商標名:タミフル)及びザナミビル(3)(登録商標名:リレンザ)の活性阻害についても、同様に測定した。結果を表1並びに図3及び図4に示す。
【0033】
【表1】
【0034】
図1及び表1に示すように、ノイラミン酸誘導体(11)は、H1N1、H3N2及びH5N3のウイルスのシアリダーゼに対して、高い活性阻害作用を奏することが分かった。また、ノイラミン酸誘導体(14)は、図2及び表1に示すように、活性阻害の効果は、ノイラミン酸誘導体(11)よりも顕著であり、ザナミビル(3)やオセルタミビル(2)と同程度であった。
以上の結果は、ノイラミン酸誘導体(11)及びノイラミン酸誘導体(14)がシアリダーゼ活性阻害剤及び抗ウイルス薬として充分な効果を有することを明らかにしている。
【0035】
(ヒト−シアリダーゼ(NEU1、NEU2及びNEU4)に対する活性阻害の測定)
上記のようにして合成したノイラミン酸誘導体(11)及びノイラミン酸誘導体(14)について、3種類(NEU1、NEU2、及びNEU4)のヒト−シアリダーゼに対する活性阻害を、4-methylumbelliferyl N-acetylneuraminic酸(MUNA)を用いた蛍光測定法によって評価した。
すなわち、50μLの反応液において、500mMの酢酸ナトリウム緩衝液(NEU1及びNEU4に対してはpH4.6、NEU2に対してはpH5.5)10μL、5段階の濃度に調整した所定量のノイラミン酸誘導体、50μgの子牛由来のアルブミン、20nmolのMUNA、所定量の組み換え型ヒト−シアリダーゼを混合し、37℃で30分間インキュベーションを行った。そしてリリースされるノイラミン酸と等しい量のMUを、ウイルスシアリダーゼに対する活性阻害の測定の場合と同様の方法で測定した。
なお、IC50値は、マイクロソフトエクセルによる非線形回帰分析法を用いて計算した。
また、比較のため、ザナミビル(3)(登録商標名:リレンザ)の活性阻害についても同様に測定した。
【0036】
(ヒト−シアリダーゼ(NEU3)に対する活性阻害の測定)
上記のようにして合成したノイラミン酸誘導体(11)及びノイラミン酸誘導体(14)について、ヒト−シアリダーゼNEU3に対する活性阻害を、malononitrileを用いた高速液体クロマトグラフィーによる蛍光測定法によって評価した。
すなわち、5μLの反応液において、500mMの酢酸ナトリウム緩衝液(pH4.5)10μL、5段階の濃度に調整した所定量のノイラミン酸誘導体、50μgの子牛由来のアルブミン、5nmolのガングリオシドGM3、50μgのトリトンX−100、所定量の組み換え型ヒト−シアリダーゼを混合し、37℃で30分間インキュベーションを行った。
最後に、200mM炭酸ナトリウム−重炭酸塩緩衝液(pH10.6)200μlを加えて反応を終了させた。そしてGM3からリリースされたシアル酸をmalononitrileを用いた高速液体クロマトグラフィーによる蛍光測定法によって測定した。
なお、IC50値は、マイクロソフトエクセルによる非線形回帰分析法を用いて計算した。
また、比較のため、ザナミビル(3)(登録商標名:リレンザ)の活性阻害についても同様に測定した。
【0037】
【表2】
【0038】
結果を表2並びに図3及び図4に示す。
表2及び図3に示すように、ノイラミン酸誘導体(11)は、ヒト由来の4種類のシアリダーゼ(NEU1、NEU2、NEU3及びNEU4)のいずれに対しても、ヒト−シアリダーゼ活性阻害(IC50)はほとんど認められない程度であった。
また、表2及び図4に示すように、ノイラミン酸誘導体(14)についても、ヒト由来の4種類のシアリダーゼ(NEU1、NEU2、NEU3及びNEU4)のいずれに対しても、ヒト−シアリダーゼ活性阻害(IC50)はほとんど認められない程度であった。
以上の結果は、ノイラミン酸誘導体(11)及びノイラミン酸誘導体(14)を抗ウイルス薬として用いた場合においても、副作用の可能性が極めて少ないことを示している。
【0039】
この発明は上記発明の実施の態様及び実施例の説明に何ら限定されるものではない。特許請求の範囲を逸脱せず、当業者が容易に想到できる範囲で種々の変形態様もこの発明に含まれる。
【図面の簡単な説明】
【0040】
【図1】ノイラミン酸誘導体(11)のウイルス(H1N1、H3N2及びH5N3)シアリダーゼに対する活性阻害の測定結果を示すグラフである。
【図2】ノイラミン酸誘導体(14)のウイルス(H1N1、H3N2及びH5N3)シアリダーゼに対する活性阻害の測定結果を示すグラフである。
【図3】ノイラミン酸誘導体(11)のヒト−シアリダーゼ(NEU1、NEU2、NEU3及びNEU4)に対する活性阻害の測定結果を示すグラフである。
【図4】ノイラミン酸誘導体(14)のヒト−シアリダーゼ(NEU1、NEU2、NEU3及びNEU4)に対する活性阻害の測定結果を示すグラフである。
【産業上の利用可能性】
【0041】
本発明のノイラミン酸誘導体及びシアリダーゼ活性阻害剤は、シアリダーゼの機能を調べる生物学的ツールとして利用することができる。また、本発明のノイラミン酸誘導体は、ウイルスのシアリダーゼ活性を選択的に阻害し、ヒトのシアリダーゼ活性をほとんど阻害しないことから、副作用の少ない抗インフルエンザ薬等に利用できる。
【特許請求の範囲】
【請求項1】
下記化学式(1)(ただし、R1及びR2は、水素又は炭素数1〜8の分岐してもよいアルキル基若しくは炭素数1〜8の分岐してもよいシクロアルキル基を示し、R3は水素又はフッ素を示す)又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とするノイラミン酸誘導体。
【化1】
【請求項2】
R1はn−ブチル基であり、R2はメチル基であり、R3は水素であることを特徴とする請求項1記載のノイラミン酸誘導体。
【請求項3】
R1はシクロペンチル基であり、R2はメチル基であり、R3は水素であることを特徴とする請求項1記載のノイラミン酸誘導体。
【請求項4】
請求項1乃至3のいずれか1項に記載のノイラミン酸誘導体を有効成分として含有することを特徴とするシアリダーゼ活性阻害剤。
【請求項5】
請求項1乃至3のいずれか1項に記載のノイラミン酸誘導体を有効成分として含有することを特徴とする抗インフルエンザ薬。
【請求項1】
下記化学式(1)(ただし、R1及びR2は、水素又は炭素数1〜8の分岐してもよいアルキル基若しくは炭素数1〜8の分岐してもよいシクロアルキル基を示し、R3は水素又はフッ素を示す)又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなることを特徴とするノイラミン酸誘導体。
【化1】
【請求項2】
R1はn−ブチル基であり、R2はメチル基であり、R3は水素であることを特徴とする請求項1記載のノイラミン酸誘導体。
【請求項3】
R1はシクロペンチル基であり、R2はメチル基であり、R3は水素であることを特徴とする請求項1記載のノイラミン酸誘導体。
【請求項4】
請求項1乃至3のいずれか1項に記載のノイラミン酸誘導体を有効成分として含有することを特徴とするシアリダーゼ活性阻害剤。
【請求項5】
請求項1乃至3のいずれか1項に記載のノイラミン酸誘導体を有効成分として含有することを特徴とする抗インフルエンザ薬。
【図1】


【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2011−136964(P2011−136964A)
【公開日】平成23年7月14日(2011.7.14)
【国際特許分類】
【出願番号】特願2009−298788(P2009−298788)
【出願日】平成21年12月28日(2009.12.28)
【出願人】(304019399)国立大学法人岐阜大学 (289)
【出願人】(500433225)学校法人中部大学 (105)
【出願人】(591074736)宮城県 (60)
【Fターム(参考)】
【公開日】平成23年7月14日(2011.7.14)
【国際特許分類】
【出願日】平成21年12月28日(2009.12.28)
【出願人】(304019399)国立大学法人岐阜大学 (289)
【出願人】(500433225)学校法人中部大学 (105)
【出願人】(591074736)宮城県 (60)
【Fターム(参考)】
[ Back to top ]