
ハイドロフルオロカーボン溶媒中で化合物の立体選択的製造のための酵素法
二次化合物を立体選択的に製造するための方法が記載されている。本方法は、一次化合物を二次化合物へ変換するために、生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で、少くとも1種の一次化合物を含む基質を試薬と反応させることからなる。

【発明の詳細な説明】
【発明の分野】
【0001】
本発明は、一次化合物の触媒変換による二次化合物の製造方法に関する。更に詳しくは、本発明は、生物学的触媒の存在下で、一次化合物を含めた基質を試薬と反応させることにより、二次化合物を立体選択的に製造するための方法に関する。
【背景技術】
【0002】
触媒とは、反応で自らが消費されることなく、反応速度を増すように働く物質である。酵素は、多くの場合に、反応が拡散限定されるような程度まで、十分有効に反応活性化エネルギーを低下させられる、天然触媒である。
【0003】
酵素触媒作用の顕著な特徴は、生物学的機能を決定することが知られた基質特異性である。一部の酵素は1種のみの生物学的基質を利用し、絶対基質特異性を示すと言われている。例えば、グルコキナーゼはATPからグルコースへのリン酸の転移を触媒し、他の糖へは転移させない。他の酵素はかなり広い基質特異性を示し、それらの天然基質と多くの場合に非類似である構造関連分子も利用しうる。これらの酵素は、関連基特異性を示すと言われている。この種の酵素の例はCandida cylindracea(C.cylindracea)リパーゼであり、これは様々なアシルドナーとアシルアクセプターとのエステル交換反応を触媒する。化学的特異性に加えて、酵素は立体化学的特異性も示す。
【0004】
国際生化学会は、触媒する反応のタイプに応じて、酵素を6カテゴリーに分類した。
【0005】
オキシドレダクターゼは、酸化および還元反応を触媒する。更に詳しくは、それらはC‐H、C‐CおよびC=C結合の酸素添加と、H原子相当物の除去または付加を触媒する。
【0006】
トランスフェラーゼは、アルデヒド、ケトン、アシル、糖、ホスホリルまたはメチル基のような様々な基の転移を触媒する。
【0007】
ヒドロラーゼは、特に、加水分解によるエステル、アミド、ラクトン、ラクタム、エポキシド、ニトリル、無水物およびグリコシドの形成を触媒する。
【0008】
リアーゼは、C=C、C=NおよびC=O結合での小分子の付加‐脱離を触媒する。
【0009】
イソメラーゼは、ラセミ化およびエピマー化のような異性化反応を触媒する。
【0010】
リガーゼは、C‐O、C‐S、C‐NおよびC‐C結合の形成および開裂と同時に、三リン酸開裂を触媒する。
【0011】
性質上、一部の酵素は細胞膜中の脂質層またはその内部で機能する。リパーゼは、例えば、水‐脂質界面で活性である。脂質層は、作業酵素向けの非水性および非極性環境をもたらす。
【0012】
酵素触媒は、立体選択性を利用するために、相当数のプロセスで商業的にも用いられている。例えば、ヒドロラーゼ種(プロテアーゼおよびリパーゼ)の酵素は、プロキラルおよび中心対称化合物からキラル化合物への変換と、メソ化合物の脱対称化において、二級アルコールおよびカルボン酸のラセミ混合物の分割のために、商業的に用いられている。酵素は、ヘキサンのような非極性有機溶媒中で、最も効果的に働く。溶媒の極性を高めると、酵素の急速な不活化および/または反応速度の著しい低下を招きやすい。
【0013】
生成物へ至る反応速度、選択性および/または変換性を改善することで、商業的酵素触媒プロセスに改良を加えることが望ましいであろう。広範囲の反応基質を溶解でき、反応に際して酵素の不活化を軽減し、広範囲の基質で効果的に所定の酵素を利用しうるような、溶媒を用いることも望ましいであろう。
【0014】
特に、現在商業用の公知プロセスよりも効率的に一次化合物を二次化合物へ立体選択的に変換しうる酵素触媒プロセスについて、必要性がある。
【発明の具体的説明】
【0015】
本発明によると、生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で、少くとも1種の一次化合物を含む出発物質または基質を試薬と反応させることからなる、二次化合物を立体選択的に製造するための方法が提供される。
【0016】
本発明の方法は、例えば、非キラル化合物、化合物のラセミ混合物、鏡像的純粋物質、メソ化合物、プロキラル化合物または中心対称化合物のうち、少くとも1種の一次化合物を、特定のキラル二次化合物へ立体選択的に変換する。このことは、一次化合物が、原則的に反応すると立体異性体の混合物を形成しうるが、生物学的触媒の影響下で優先的または選択的に反応して、主に、好ましくは排他的に、1種のエナンチオマーを生じることを、我々は意味している。特に、我々は、1種の特定エナンチオマーを主に、好ましくは排他的に得る方法に言及している。更に詳しくは、出発物質または基質の変換とは、望ましいエナンチオマーが50%以上、更に好ましくは70%以上、特に90%以上のエナンチオマー過剰率で形成されることをいう。
【0017】
本発明の方法は、高い立体選択性で、一次化合物から二次化合物へ良好な変換を行える。変換性および立体選択性は、ヘキサンのような従来の炭化水素溶媒を用いる公知の商業法で得られる場合よりも、優れている。更に、該方法は従来の炭化水素溶媒で行われる方法よりも速く進行する。
【0018】
本方法で用いられている(ハイドロ)フルオロカーボン溶媒は、同反応がヘキサンのような従来の炭化水素溶媒を用いて行われる場合よりも、生物学的触媒の分解を減らせる、とも考えられている。ひいては、このことから、触媒が変化するまで長時間にわたり連続プロセスを行わせられ、またはバッチプロセスにおいては触媒をより多くの回数にわたり再使用しうるのである。
【0019】
本発明の方法は、少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で行われる。“(ハイドロ)フルオロカーボン”という用語について、我々はハイドロフルオロカーボンおよびペルフルオロカーボンからなる群より選択される化合物を意味している。“ハイドロフルオロカーボン”という用語について、我々は炭素、水素およびフッ素原子のみを有する化合物を意味している。ハイドロフルオロカーボン溶媒が好ましい。
【0020】
溶媒は通常液体状態であるが、我々は超臨界液の使用を除外しているわけではない。室温で気体である1種以上の低沸点化合物を溶媒が含んでいる場合は、溶媒を適度な低温に冷却することにより、および/またはプロセスのある時点でそれを大気圧以上に付すことにより、望ましい液体状態が得られる。反応させる基質と(ハイドロ)フルオロカーボン溶媒が混合される前または後に、必要であればプロセスに際して連続的に、これら手段の一方または双方が適用される。
【0021】
適切な(ハイドロ)フルオロカーボンは、C1‐10、特にC1‐5、とりわけC1‐4(ハイドロ)フルオロカーボンから選択される。
【0022】
好ましいペルフルオロカーボンには、ヘキサフルオロエタン(R‐116)およびオクタフルオロプロパン(R‐218)がある。
【0023】
好ましいハイドロフルオロカーボンは、C1‐10、特にC1‐5、とりわけC1‐4ハイドロフルオロアルカンから選択される。適切なC1‐4ハイドロフルオロアルカンには、ハイドロフルオロメタン、例えばトリフルオロメタン(R‐23)、フルオロメタン(R‐41)およびジフルオロメタン(R‐32);ハイドロフルオロエタン、例えばペンタフルオロエタン(R‐125)、1,1,1‐トリフルオロエタン(R‐143a)、1,1,2,2‐テトラフルオロエタン(R‐134)、1,1,1,2‐テトラフルオロエタン(R‐134a)および1,1‐ジフルオロエタン(R‐152a);ハイドロフルオロプロパン、例えば1,1,1,3,3‐ペンタフルオロプロパン(R‐245fa)、1,1,2,2,3‐ペンタフルオロプロパン(R‐245ca)、1,1,1,2,3‐ペンタフルオロプロパン(R‐245eb)、1,1,2,3,3‐ペンタフルオロプロパン(R‐245ea)、1,1,1,2,3,3‐ヘキサフルオロプロパン(R‐236ea)、1,1,1,2,2,3‐ヘキサフルオロプロパン(R‐236cb)、1,1,1,3,3,3‐ヘキサフルオロプロパン(R‐236fa)、1,1,1,2,3,3,3‐ヘプタフルオロプロパン(R‐227ea)および1,1,1,2,2,3,3‐ヘプタフルオロプロパン(R‐227ca);およびハイドロフルオロブタン、例えば1,1,1,3,3‐ペンタフルオロブタン(R‐356mfc)がある。好ましいハイドロフルオロカーボンはR‐32、R‐134a、R‐134、R‐152a、R‐143a、R‐125、R‐245fa、R‐236eaおよびR‐227eaであり、これらはすべて低沸点であって、プロセスの最後に比較的容易に反応混合物から除去される。これらのうちR‐32およびR‐134aが特に好ましく、R‐134aが最も好ましい。
【0024】
2種以上の(ハイドロ)フルオロカーボンの混合物からなる溶媒も、所望であれば用いてよい。
【0025】
本発明の方法で用いられる溶媒は、(ハイドロ)フルオロカーボンに加えて、有機補助溶媒も含んでよい。
【0026】
適切な補助溶媒には、特に無フッ素、一般的には無ハロゲンの化合物がある。適切な無ハロゲン補助溶媒は、典型的には200℃以下、例えば−85〜200℃の範囲の沸点を有している。好ましい補助溶媒は、120℃以下、例えば−85〜120℃の範囲、更に好ましくは100℃以下、例えば−70〜100℃の範囲、特に10℃以下、例えば−60〜10℃の範囲の沸点を有している。2種以上の補助溶媒の混合物も、所望であれば用いてよい。
【0027】
適切な補助溶媒はC2‐6、特にC2‐4炭化水素化合物から選択され、それについて我々は炭素および水素原子のみを有した化合物を意味している。適切な炭化水素にはアルカンおよびシクロアルカンがあり、エタン、n‐プロパン、i‐プロパン、n‐ブタン、i‐ブタンおよびn‐ペンタンのようなアルカンが好ましい。
【0028】
他の適切な補助溶媒には炭化水素エーテルがあり、それについて我々は式R1‐O‐R2を有する化合物を意味し、ここでR1およびR2は、独立して、炭素および水素原子のみを有したヒドロカルビル基、例えばC1‐6、特にC1‐3アルキル基である。適切なジアルキルエーテルには、ジメチルエーテル、メチルエチルエーテルおよびジエチルエーテルがある。
【0029】
更に別の適切な補助溶媒は、アミド、スルホキシド、アルコール、ケトン、カルボン酸、カルボン酸誘導体、無機酸およびニトロ化合物から選択される。
【0030】
適切なアミド補助溶媒には、N,N′‐ジアルキルアミドおよびアルキルアミド、例えばジメチルホルムアミドおよびホルムアミドがある。
【0031】
適切なスルホキシド補助溶媒には、ジアルキルスルホキシド、例えばジメチルスルホキシドがある。
【0032】
適切なアルコール補助溶媒には、脂肪族アルコール、特にアルカノールがある。適切なアルカノールは、C1‐6、特にC1‐3アルカノール、例えばメタノール、エタノール、1‐プロパノールおよび2‐プロパノールから選択される。
【0033】
適切なケトン補助溶媒には、脂肪族ケトン、特にジアルキルケトン、例えばアセトンがある。
【0034】
適切なカルボン酸補助溶媒には、ギ酸および酢酸がある。
【0035】
補助溶媒として使用上適切なカルボン酸誘導体には、無水物、例えば無水酢酸、およびC1‐6、特にC1‐3アルカン酸のC1‐6、特にC1‐3アルキルエステル、酢酸エチルがある。
【0036】
補助溶媒として使用上適切なニトロ化合物には、ニトロアルカンおよびニトロアリール化合物、例えばニトロメタンおよびニトロベンゼンがある。
【0037】
好ましいというわけではないが、有機補助溶媒が用いられる場合、溶媒混合物は典型的には80.0〜99.0重量%の(ハイドロ)フルオロカーボンおよび1〜20重量%の補助溶媒からなる。好ましくは、溶媒混合物は90.0〜99.0重量%の(ハイドロ)フルオロカーボンおよび1〜10.0重量%の補助溶媒からなる。補助溶媒の極性が増してくると、酵素の不活化と共にあらゆる問題を避ける上で、補助溶媒を少なく用いることが通常望ましい。
【0038】
ほとんどの酵素の適正な機能のためには水が必要であるため、本発明の方法は典型的には少くとも少量の水の存在下で行われる。しかしながら、用いられる水の量は、通常、水が反応系で分離相を形成しないようなものとされる。これは、本方法の目的が、主に(ハイドロ)フルオロカーボン溶媒からなる環境下で、酵素機能を発揮させることにあるからである。好ましくは、水の量は、用いられる溶媒の飽和レベル以下に保たれる。更に好ましくは、反応は溶媒の総重量に対して1重量%以下の水の存在下で行われる。
【0039】
本発明の方法は、生物学的触媒の存在下で行われる。“生物学的触媒”とは、我々は生体組織または系でみられる触媒を意味している。本発明の方法で使用向けの具体的な生物学的触媒は、酵素およびアブザイムである。生物学的触媒は、もちろん、二次化合物への基質の立体選択的変換を触媒できねばならない。
【0040】
典型的には、本発明の方法は単一触媒の存在下で行われるが、我々は触媒の混合物が用いられる可能性を除外していない。
【0041】
本方法で使用上好ましい酵素は、前記6種の酵素のいずれから選択してもよい。
【0042】
各酵素は、それらが通常ホスト生物に存在しているか、またはそこで過剰発現により生産される生体組織から、それらが単離されている、という意味で分離している。これらの分離酵素はそのまま用いても、あるいは、例えばFitzpatrick,P.A.,Klibanov,A.M.,J.Am.Chem.Soc.,1991,113,3166で記載されているような標準文献プロセスを用いてそれらは凍結乾燥してもよい。しかしながら、少くとも一部の酵素が事前に凍結乾燥することなく(ハイドロ)フルオロカーボン溶媒中で有効に機能でき、そのため重要な処理ステップを省ける可能性をもたらすことを、我々は発見したのである。
【0043】
酵素は、凍結乾燥させてもまたはそうでなくても、標準文献プロセスを用いて通常固定される。例えば、酵素は、例えば物理的吸着または結合により、固体の不溶性マトリックスへ固定される。適切なマトリックスとしては、特にガラス、珪藻土、シリカおよび有機ポリマー、例えばポリスチレンおよびポリアクリレートホモポリマーおよびコポリマーがある。
【0044】
一方、酵素は、生細胞培養物、例えばLactobacillus acidophilusのような全細胞培養物、休止細胞培養物、例えば温水で活性化されうる乾燥パン酵母、または酵素および必要補因子を含有した非生細胞培養物、例えば死酵母の一部でもよい。酵素を含有した全細胞培養物は、標準文献プロセスを用いて、例えば物理的吸着または結合により、固体の不溶性マトリックスへ通常固定される。上記のマトリックスがこの目的のために用いうる。
【0045】
本発明の方法で使用上好ましい酵素には、ヒドロラーゼカテゴリーに属するものがある。具体的な酵素は、Subtilisin carlsbergおよびSubtilisin BPNのようなプロテアーゼ、Porcine pancreaticリパーゼ、Candida antarctica BリパーゼおよびPseudomonas cepaciaリパーゼのようなリパーゼ、Aspergillus orgzeaのα‐およびβ‐ガラクトシダーゼのようなグリコシダーゼである。
【0046】
アブザイムとは、触媒抗体、即ち特定の化学反応を触媒しうる抗体である。適切なアブザイムはアルドラーゼ抗体38C2である。
【0047】
アブザイムは、酵素に関して前記されたように、凍結乾燥および/または固定される。
【0048】
本発明の方法は、許容しうる反応速度および成分溶解性をもたらして、生物学的触媒、一次化合物および二次化合物の著しい分解を避けられる温度で通常行われる。典型的には、本方法は−60〜120℃の範囲、好ましくは−30〜80℃の範囲、特に0〜60℃の範囲の温度、例えば約20℃で行われる。
【0049】
本方法は、大気圧、大気圧以下または大気圧以上で行われる。正確な作業圧力は、とりわけ、用いられる溶媒、特にその沸点に依存する。好ましい作業圧力は、0.1〜200バールの範囲、更に好ましくは0.5〜30バールの範囲、特に1〜15バールの範囲である。
【0050】
(ハイドロ)フルオロカーボン溶媒対反応させる基質の重量比は、好ましくは1:1〜1000:1の範囲、更に好ましくは1:1〜500:1の範囲、特に1:1〜10:1の範囲である。生物学的触媒は、典型的には非常に少量で、例えば基質に対して触媒10−3〜10−4モル%程度で用いられる。正確な量は、酵素の活性に依存する。
【0051】
本発明の方法は、様々な立体選択的変換に通常適用しうる。医薬化合物の製造で中間体として用いうる化合物を製造する際に、それは特に有用である。
【0052】
一態様において、本発明の方法は、混合物を形成するエナンチオマーの一方を優先的または選択的に反応させて、新たなエナンチオマー化合物を形成させ、一方で他のエナンチオマーを大部分または完全に未反応のままにするように、生物学的触媒および(ハイドロ)フルオロカーボン溶媒の存在下で混合物を試薬と反応させることにより、ラセミ混合物またはラセミ改変物を分割するために用いられる。
【0053】
したがって、本発明の一態様では、ラセミ混合物を形成するエナンチオマーの一方を優先的または選択的に新たなエナンチオマー化合物へ変換するように、生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で該混合物を試薬と反応させることからなる、ラセミ混合物の分割方法が提供される。
【0054】
本発明のこの態様に従い分割されるラセミ混合物は、RおよびSアルコール、RおよびSカルボン酸またはエステル、RおよびSアミノ酸エステル、RおよびSアミン、RおよびSチオール、またはRおよびSアミドのラセミ混合物である。好ましくは、それはRおよびSアミノ酸エステルの混合物である。この特殊な分割は、RまたはSエナンチオマーのキラル炭素に付いた官能基を優先的または選択的に変換することにより行われる。生物学的触媒は、好ましくは酵素である。
【0055】
特別な態様において、本方法は、異なるアルコキシ基をもたらすアルカノールとの反応で、RまたはSエナンチオマーのアルコキシ基が優先的に、好ましくは選択的に交換されるエステル交換反応により、ラセミN‐P‐dl‐フェニルアラニンアルキルエステル(Pは保護基を表わす)を分割するために用いられる。通常、エステル交換反応をうけるのはSエナンチオマーである。好ましい保護基はアセチルおよびトリフルオロアセチルであり、好ましいアルキルエステルはプロピルエステルであり、そのため好ましいラセミ混合物はN‐アセチル‐dl‐フェニルアラニンプロピルエステルおよびN‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルである。好ましいアルカノールはメタノールである。生物学的触媒は、好ましくは酵素、更に好ましくはプロテアーゼ、更に一層好ましくはSubtilisin carlsbergである。
【0056】
N‐P‐dl‐フェニルアラニンアルキルエステル対アルカノールのモル比は、好ましくは1:0.1〜1:100の範囲、更に好ましくは1:1〜1:50の範囲、特に1:1〜1:10の範囲である。
【0057】
反応時間は、典型的には0.1〜48時間の範囲、好ましくは1〜36時間の範囲、特に1〜24時間の範囲である。
【0058】
ラセミN‐P‐dl‐フェニルアラニンアルキルエステルのRまたはSエナンチオマー(通常Sエナンチオマー)の優先的/選択的エステル交換とは、望ましいエナンチオマーが典型的には50%以上、好ましくは70%以上、特に90%以上、例えば100%のエナンチオマー過剰率で形成されるようなものをいう。
【0059】
Sエナンチオマーの100%エナンチオマー過剰率と仮定した、メタノールを用いるラセミN‐アセチル‐dl‐フェニルアラニンプロピルエステルの分割が、式(1)で示されている。
【化1】
【0060】
(Sエナンチオマーが100%エナンチオマー過剰率で形成される、と再び仮定した)メタノールを用いるN‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルの分割も、同様に進行するであろう。
【0061】
他の態様において、本発明の方法は、RまたはSエナンチオマーのOH基が試薬との反応で優先的に、好ましくは選択的に交換されるエステル交換反応により、ラセミ1‐フェニルエタノールを分割するために用いられる。用いられる試薬は、好ましくはアシルドナー、例えばビニルまたはイソプロペニルアルカノエートのようなエノールエステル、またはアルコキシエノールエステルである。好ましい試薬は酢酸ビニルである。通常、エステル交換反応をうけるのはRエナンチオマーである。生物学的触媒は、好ましくはリパーゼ、例えばCandida antarctica Bリパーゼである。
【0062】
1‐フェニルエタノール対アシルドナーのモル比は、好ましくは1:0.1〜1:100の範囲、更に好ましくは1:1〜1:50の範囲、特に1:20である。
【0063】
反応時間は、典型的には0.1〜48時間の範囲、好ましくは1〜36時間の範囲、特に1〜24時間の範囲である。
【0064】
ラセミ1‐フェニルエタノールのRまたはSエナンチオマー(通常Rエナンチオマー)の優先的/選択的エステル交換とは、望ましいエナンチオマーが典型的には50%以上、好ましくは70%以上、特に90%以上、例えば100%のエナンチオマー過剰率で形成されるようなものをいう。
【0065】
Rエナンチオマーの100%エナンチオマー過剰率と仮定した、酢酸ビニルを用いるラセミ1‐フェニルエタノールの分割が、式(2)で示されている。
【化2】
【0066】
他の態様において、本発明の方法は、生物学的触媒および(ハイドロ)フルオロカーボン溶媒の存在下でメソ化合物を試薬と反応させることにより、メソ化合物から優先的に、好ましくは選択的に特定のエナンチオマーを製造するために用いられる。鏡像と重ね合わせられるため対称的であるメソ化合物は、エナンチオマー生成物へ変換されることから、メソ化合物の反応も脱対称化と称される。もちろん、エナンチオマーはその鏡像と重ね合わせられない。
【0067】
したがって、本発明の別な態様では、生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下でメソ化合物を試薬と反応させることからなる、メソ化合物から優先的または選択的に特定のエナンチオマーを製造するための方法が提供される。
【0068】
本方法は、キラル炭素の1つに付いた官能基を優先的または選択的に置換または変換することにより行われる。
【0069】
メソ化合物は好ましくはシス‐4‐シクロペンテン‐1,3‐ジオールであり、用いられる試薬は好ましくはアシルドナー、例えばビニルまたはイソプロペニルアルカノエートのようなエノールエステル、またはアルコキシエノールエステルである。好ましい試薬は酢酸ビニルである。しかしながら、他のメソ化合物および他の試薬も用いてよい。
【0070】
生物学的触媒は好ましくは酵素であり、メソ化合物がシス‐4‐シクロペンテン‐1,3‐ジオールであるとき、酵素は好ましくはリパーゼ、更に好ましくはブタ膵臓リパーゼ、Candida antarctica BリパーゼまたはPseudomonas cepaciaリパーゼである。
【0071】
反応は、ヒンダードアミン、特に三級アミン、例えばトリエチルアミンの存在下で行われる。アミンの存在は、速い反応速度および大きな変換率に寄与しうる。しかしながら、酵素を減少させると、粗反応混合物の下流精製を簡単にできる。
【0072】
メソ シス‐4‐シクロペンテン‐1,3‐ジオールと酢酸ビニルとの反応は、式(3)で示されているように進行する。
【化3】
【0073】
本プロセスは2段階で生じると考えられている。第一段階はエナンチオマー モノアセテート生成物、即ち(1R,3S)‐(+)‐4‐シクロペンテン‐1,3‐ジオール‐1‐アセテート(a)、(1S,3R)‐(−)‐4‐シクロペンテン‐1,3‐ジオール‐1‐アセテート(b)またはエナンチオマー(a)および(b)の混合物の立体選択的形成であり、一方のエナンチオマーが過剰である。ブタ膵臓リパーゼ、Candida antarctica BリパーゼまたはPseudomonas cepaciaリパーゼが酵素として用いられる場合、エナンチオマー(b)が優先的に、多くの場合は排他的に形成される傾向がある。
【0074】
第二段階では、モノアセテート(a)および/または(b)が更に反応を続け、別な酢酸ビニル分子との反応で、ジアセテート シス‐4‐シクロペンテン‐1,3‐ジアセテートを形成する。もちろん、ジアセテートは他のメソ化合物である。
【0075】
双方のモノアセテート生成物とも、プロスタグランジン、プロスタサイクリンおよびトロンボキサンの合成において重要な出発物質である。
【0076】
シス‐4‐シクロペンテン‐1,3‐ジオール対酢酸ビニルのモル比は、好ましくは1:0.1〜1:100の範囲、更に好ましくは1:1〜1:50の範囲、特に1:1〜1:20の範囲である。
【0077】
反応時間は、典型的には0.1〜48時間の範囲、好ましくは1〜36時間の範囲、特に1〜24時間の範囲である。
【0078】
シス‐4‐シクロペンテン‐1,3‐ジオールと酢酸ビニルとの反応は、優先的/選択的に形成されるエナンチオマー(通常(1S,3R)‐(−)‐4‐シクロペンテン‐1,3‐ジオール‐1‐アセテート)が50%以上、更に好ましくは70%以上、特に90%以上、例えば100%のエナンチオマー過剰率で形成されるように、通常進行する。
【0079】
更に他の態様において、本発明の方法は、生物学的触媒および(ハイドロ)フルオロカーボン溶媒の存在下でプロキラル化合物を試薬と反応させることにより、プロキラル化合物から優先的に、好ましくは選択的に特定のエナンチオマーを製造するために用いられる。光学的に不活性な前駆体が非対称の光学的に活性な生成物へ変換されることから、プロキラル化合物の反応も脱対称化と称される。
【0080】
したがって、本発明の別な態様では、生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下でプロキラル化合物を試薬と反応させることからなる、プロキラル化合物から優先的または選択的に特定のエナンチオマーを製造するための方法が提供される。
【0081】
本方法は、少くとも1つの非キラル炭素原子を、キラル中心に異なる4つの官能基を有するキラル炭素原子へ、優先的または選択的に変換することにより行われる。
【0082】
プロキラル化合物は好ましくは2‐エチルプロパン‐1,3‐ジオールであり、用いられる試薬は好ましくはアシルドナー、例えばビニルまたはイソプロペニルアルカノエートのようなエノールエステル、またはアルコキシエノールエステルである。好ましい試薬は酢酸ビニルである。しかしながら、他のプロキラル化合物および他の試薬も用いてよい。
【0083】
生物学的触媒は好ましくは酵素であり、プロキラル化合物が2‐エチルプロパン‐1,3‐ジオールであるとき、酵素は好ましくはリパーゼ、更に好ましくはPseudomonas cepaciaリパーゼである。
【0084】
2‐エチルプロパン‐1,3‐ジオールと酢酸ビニルとの反応は、式(4)で示されているように進行する。
【化4】
【0085】
式(4)で示されているように、プロキラル 2‐エチルプロパン‐1,3‐ジオールは、最初にモノアセテート化合物1‐ヒドロキシ‐3‐アセトキシ‐2‐エチルプロパンへ変換される。この変換は排他的なRまたはSエナンチオマーの形成をもたらすか、または2種のうち一方が優先的な2種エナンチオマーの混合物の形成をもたらす。Pseudomonas cepaciaリパーゼが酵素として用いられる場合、Rエナンチオマーが優先的に、多くの場合は排他的に形成される傾向がある。
【0086】
その後も、モノアセテートは更に反応を続け、別の酢酸ビニル分子との反応で、ジアセテート 2‐エチルプロパン‐1,3‐ジアセテートを形成する。もちろん、ジアセテートもプロキラルである。
【0087】
双方のモノアセテート生成物とも、(Faber,K.,Biotransformations in Organic Chemistry,Springer-Verlag,1997で記載されているように)血小板活性化因子の合成において重要な構成単位である。
【0088】
2‐エチルプロパン‐1,3‐ジオール対酢酸ビニルのモル比は、好ましくは1:0.1〜1:100の範囲、更に好ましくは1:1〜1:50の範囲、特に1:1〜1:10の範囲である。
【0089】
反応時間は、典型的には0.1〜48時間の範囲、好ましくは1〜36時間の範囲、特に1〜24時間の範囲である。
【0090】
2‐エチルプロパン‐1,3‐ジオールと酢酸ビニルとの反応は、優先的/選択的に形成されるエナンチオマー(通常Rエナンチオマー)が50%以上、更に好ましくは70%以上、特に90%以上、例えば100%のエナンチオマー過剰率で形成されるように、通常進行する。
【0091】
本発明の方法は、バッチ式でまたは連続的に操作される。環境以下の沸点を有する(ハイドロ)フルオロカーボン溶媒が用いられる場合、反応容器は典型的には高圧に耐えうる圧力容器とする。
【0092】
バッチプロセスでは、例えば、(ハイドロ)フルオロカーボンが環境温度で気体であればフラッシュ蒸発により、または粗反応混合物を得た後で精製するために、必要であれば、望ましい二次化合物を単離するために蒸留により、(ハイドロ)フルオロカーボン溶媒がプロセスの最後に除去される。
【0093】
連続プロセスでは、(ハイドロ)フルオロカーボン溶媒および反応剤を含有した反応剤ストリームが、触媒を含有した反応容器へ連続的に運ばれる。典型的には、反応剤ストリームは固定触媒の層へ送られる。次いで、反応容器を出た粗反応混合物は、(ハイドロ)フルオロカーボン溶媒を除去して、該プロセスで形成された1種以上の望ましい二次化合物を回収するために、例えば溶媒エバポレーターで処理される。除去された(ハイドロ)フルオロカーボン溶媒は、所望であれば溶媒インファントリー(solvent infantry)を最少に抑えるため、濃縮してリサイクルしてもよい。未反応出発物質も、所望であればリサイクルしてよい。
【0094】
溶媒がリサイクルされる場合、低沸点溶媒用に適した回収系は、これについて我々は25℃以下、例えば0℃以下の沸点を有する溶媒を意味するのであるが、本プロセスから生じた粗反応混合物が送られるエバポレーター、エバポレーターで生じた蒸気を圧縮するためのコンプレッサー、およびコンプレッサーから生じた圧縮蒸気を冷却するためのコンデンサーからなる。溶媒は、コンプレッサーから吸引で誘導されるフラッシュ蒸発により、エバポレーターで粗反応混合物から除去され、次いでこうして生成した溶媒蒸気はコンプレッサー、例えばダイヤフラムコンプレッサーへ送られ、そこでそれが圧縮される。コンプレッサーから、溶媒蒸気はコンデンサーへ送られて、そこでそれが冷却され、本プロセスへ再供給するための液体形へ、または可能であれば本プロセスへ溶媒を供給する溶媒リザーバーへ戻される。コンデンサーは、コイル巻管の形をとりうるが、溶媒を蒸発させるために必要なエネルギーの少くとも一部を凝縮の潜熱が供給するように、エバポレーターの内部に配置しうる。
【0095】
低沸点溶媒用に適した別の回収系は、本プロセスから生じた反応混合物が送られて、溶媒が蒸発されるエバポレーターと、エバポレーターで生じた蒸気が冷却され、本プロセスへ再供給するための液体形へ、または可能であれば本プロセスへ溶媒を供給する溶媒リザーバーへ戻されるコンデンサーを含んでなる、溶媒リサイクル回路からなる。エバポレーターの加熱およびコンデンサーの冷却は独立して行えるが、好ましい態様ではエバポレーターを加熱してコンデンサーを冷却するために外部熱ポンプ系が用いられる。外部熱ポンプ系はエバポレーター、コンプレッサー、コンデンサーおよび伸縮バルブからなり、これらは熱媒液が流動させられる回路に順次配置されている。外部熱ポンプ系のエバポレーターは、コイル巻管の形をとりうるが、エバポレーターで熱媒液の蒸発がコンデンサーを冷却して、溶媒リサイクル回路へ送られる溶媒蒸気を凝縮するように、溶媒リサイクル回路のコンデンサーの内部または外部付近に配置される。次いで、外部熱ポンプ系のエバポレーターで生じた蒸気は圧縮され、コンデンサーへ送られ、そこでそれが凝縮して、発熱する。外部熱ポンプ系のコンデンサーは、これもコイル巻管の形をとりうるが、溶媒を蒸発させて溶媒リサイクル回路へ送るために必要な熱を熱媒液の凝縮に伴う凝縮潜熱が供給するように、溶媒リサイクル回路のエバポレーターの内部または外部付近に配置される。次いで、凝縮された熱媒液は伸縮バルブからエバポレーターへ戻され、こうして外部熱ポンプ系でサイクルを終える。
【0096】
外部熱ポンプ系の代わりとして、溶媒凝縮の熱をエバポレーター容器へ移して溶媒蒸発用の熱を供給するために、外部循環熱媒液が用いうる。
【0097】
本発明のプロセスが完了したとき、望ましい生成物を単離するため、粗反応混合物は精製ステップへ付される。次いで、例えば医薬化合物を得るために、純粋な生成物が1回以上の別な合成ステップへ付される。一方、粗反応混合物は別な合成に直接用いてもよい。適切な精製技術には、クロマトグラフィー、結晶化および蒸留のような、化学合成で常用されるものがある。
【0098】
本発明がここでは実例で示されているが、但し下記例で制限されることはない。
【0099】
一般操作
N‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルの製造
下記のように、Curphey,T.J.,J.Org.Chem.,1979,44,2805-2807で開示された方法を用いて、ラセミ N‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルを製造した:
オーブン乾燥フラスコにフェニルアラニンを加えた。次いで、フラスコをN2ガスでパージし、DMF(溶媒)、ジイソプロピルエチルアミン(1当量)およびトリフルオロ酢酸エチル(1.25当量)を加えた。溶液を50℃で17時間攪拌し、次いでヨウ化プロピル(1.25当量)を加えた。溶液を更に72時間攪拌した。粗生成物を再抽出し、勾配溶離を用いてカラムクロマトグラフィーにより単離した。ヘキサン400mlから出発し、9:1へキサン:酢酸エチル300ml、次いで4:1へキサン:酢酸エチル300ml、次いで3.5:1へキサン:酢酸エチル200ml、最後に2:1へキサン:酢酸エチル200mlを加えることにより、極性を徐々に増した。単離収量は4.17g、28%であった。次いで単離生成物をKugelrohr蒸留、次いで石油スピリットおよび酢酸エチル(各9:1)の混合液からの再結晶化で更に精製した。精製されたN‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルは白色結晶固体物であった。生成物の同定はNMRおよびGC‐質量スペクトル測定により行った。
【0100】
2‐エチルプロパン‐1,3‐ジオールの製造
2‐エチルプロパン‐1,3‐ジオールを次のように製造した:
ジエチルエチルマロネート(2.0g、10.7mmol)の溶液に、0℃で乾燥エタノール中LiAlH4(2.5当量)の懸濁液を加えた。反応混合液を攪拌しながら室温まで加温し、1時間後、更に1時間還流した。氷浴で冷却後、蒸留水1mlを攪拌しながら反応混合液へ加え、次いで2M NaOH溶液1mlを加えた。次いで、混合液を濾過し、濾液を酢酸エチルで洗浄した。合わせた洗液を減圧下で蒸発させて、黄色油状物を得、溶媒として5:1酢酸エチル:ヘキサンを用いてシリカフラッシュクロマトグラフィーにより精製した。生成物は75%収率で油状物として得た。生成物の同定はNMRおよびGC‐質量スペクトル測定により行った。
【0101】
R134aおよびR‐32はIneos Fluor Ltd.から提供され、更に精製することなく用いた。双方の溶媒とも、標準プラスチック被覆10mlガラスエーロゾルボトル中で反応を行わせることにより、自己圧力下で液体状態に維持した。
【0102】
酵素は、Aldrich Chemical Company、Sigma Chemical CompanyまたはFluka Chemical Companyから入手し、更に処理することなく、またはFitzpatrick,P.A.,Klibanov,A.M.,J.Am.Chem.Soc.,1991,113,3166で記載された操作を用いる凍結乾燥後に用いた。
【0103】
ハイドロフルオロカーボン(R‐134a、R‐32およびR‐227ea)はIneos Fluor Limitedから提供された。他の全化学物質および溶媒はAldrich Chemical CompanyまたはSigma Chemical Companyから購入し、更に精製することなく用いた。
【0104】
エーロゾルはIneos Fluor Limitedから提供された。
【0105】
ガスクロマトグラムは、HP SE‐54キャピラリーカラム(25m×0.21mm内径)装備のShimadzu GC‐17a機器を用いて記録した。キラルガスクロマトグラムは、Chiraldex GTAキャピラリーカラム(30m×0.25mm内径)装備のChrompack CP9001機器で得た。フレームイオン化検出器を双方の場合で用い、標準溶液を用いて個別物質毎に応答ファクターを較正した。反応混合液から採取されたサンプルをジクロロメタン溶媒に溶解させ、必要であればナフタレンを内部標準として用いた。
【実施例】
【0106】
例1 ラセミ N‐アセチル‐dl‐フェニルアラニンプロピルエステルのSubtilisin carlsberg触媒分割
この例では、Subtilisin carlsbergを用いてラセミ混合物のSエナンチオマーを対応メチルエステルへ変換することによる、ラセミ N‐アセチル‐dl‐フェニルアラニンプロピルエステルの分割を研究した。反応は前記で更に詳細に説明されてきた。
【0107】
10mM N‐アセチル‐dl‐フェニルアラニンプロピルエステルおよび200mMメタノールの溶液をへキサン、テトラヒドロフランおよびアセトニトリルの各々で調製した。各溶液4mlに凍結乾燥Subtilisin carlsberg4.0mgを加えた。得られた懸濁液を室温で攪拌し、収率およびエナンチオマー過剰率の双方に関するガスクロマトグラフィー分析用にサンプルを定期的に採取した。
【0108】
同反応は、溶媒としてR‐134aおよびR‐32を用いても研究した。10mM N‐アセチル‐dl‐フェニルアラニンプロピルエステル、200mMメタノールおよび凍結乾燥Subtilisin carlsberg4.0mgの2種混合液をガラスエーロゾルボトル中で調製した。次いでエーロゾルボトルにキャップを取り付け、キャップを適所でかしめ、秤量済みの液体ハイドロフルオロカーボン溶媒を大型圧力容器からエーロゾルバルブを介して導入した。次いで、得られた懸濁液を室温で磁気攪拌し、収率およびエナンチオマー過剰率の双方に関するガスクロマトグラフィー分析用に反応混合液のサンプルを定期的に採取した。反応混合液の一部をバルブからサンプルバイアル中へ出すことにより、サンプルを取り出した。ハイドロフルオロカーボン溶媒は本プロセスで蒸発し、反応混合液の低揮発性残渣をサンプルバイアル中に残した。次いで、必要であればGC分析用に、内部標準を含有した既知量の溶媒中へこの残渣を溶解させた。
【0109】
反応剤および生成物は、試験溶媒の各々で良好な溶解性を示した。結果は表1に掲載されている。
【表1】
【0110】
R‐134aが、ヘキサンのような従来の溶媒よりも、速い反応および大きな最終変換率を示すことは、表1から明らかである。ヘキサンは、例1のプロセスで従来から最良の溶媒として通常みなされている。R‐32はアセトニトリルおよびテトラヒドロフランの各々と比較して良い性能を示し、約1時間以内の早い反応段階ではヘキサンに近い。R‐134aおよびR‐32は双方とも優れたエナンチオ選択性を示している。
【0111】
例2 ラセミ N‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルのSubtilisin carlsberg触媒分割
N‐アセチル‐dl‐フェニルアラニンプロピルエステルの代わりに10mM N‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルを用いて、例1を繰返した。この例は、基質特異性に対する酵素‐溶媒ペアの感受性を試験するために行った。結果は表2に掲載されている。
【0112】
【表2】
【0113】
フッ素化N保護基の使用のおかげで、R‐134aが、ヘキサンで得られる場合と比較して変換に明瞭な改善を呈することは、明らかである。加えて、溶媒がR‐134aである場合、本プロセスは72時間を過ぎても適度な速度で続くようであり、一方へキサンで観察される速度は著しく低い。これは、R‐134aがヘキサンよりも低度で酵素を分解していることを示唆しているのであろう。この特性は、従来の有機溶媒よりも大きな程度で、ハイドロフルオロカーボン溶媒中における酵素の再使用を可能にしており、経済的利益につながる。
【0114】
このプロセスでのテトラヒドロフラン中の反応速度は、例1と比較して有意に低い。これは、ハイドロフルオロカーボン溶媒が、テトラヒドロフランのような類似極性の従来溶媒とは対照的に、広範囲の物質と大きな効力で酵素を機能させうることを示している。
【0115】
例3 メソ シス‐4‐シクロペンテン‐1,3‐ジオールのリパーゼ触媒反応
この例では、ブタ膵臓リパーゼを用いたR‐134aおよびテトラヒドロフラン中におけるシス‐4‐シクロペンテン‐1,3‐ジオールと酢酸ビニルとの酵素触媒反応を比較した。この反応は前記で更に詳細に説明されており、一方の特定エナンチオマーの優先的形成をもたらす。行う方法は、Theil et al.,Tetrahedron,1991,47,7569で記載されたものであった。
【0116】
ジオール(1.0012g、1.0mmol)およびトリエチルアミン(0.070g、0.7mmol)をブタ膵臓リパーゼ(PPL)0.5gおよび酢酸ビニル(0.600g、7mmol)へ加えた。溶媒2mlを直ちに加え、反応混合液を規定時間にわたり室温で磁気攪拌した。R‐134aを用いた反応は、例1で記載されたものと全く同一の技術を用いて、ガラスエーロゾルボトル中で行った。収率およびエナンチオマー過剰率の双方に関するGC分析用に物質を反応混合液から採取し、結果を表3に掲載している。
【0117】
【表3】
【0118】
表3は、トリエチルアミンの存在下で、ブタ膵臓リパーゼを用いた脱対称化反応において、R‐134aがテトラヒドロフランと同程度に効率的かつ選択的な溶媒であることを示している。テトラヒドロフランは、従来の非水性溶媒の中で最も有効であることが、Theilらにより発見されていた。
【0119】
例4 メソ シス‐4‐シクロペンテン‐1,3‐ジオールのリパーゼ触媒反応
Pseudomonas cepaciaリパーゼを用いて、例3を繰返した。R‐32も研究し、この反応も、R‐134a反応のように、ガラスエーロゾルボトル中で行った。ハイドロフルオロカーボン溶媒の場合は、トリエチルアミンを加えなかった。得られた結果は表4で示されている。
【0120】
【表4】
【0121】
表4の結果から、R‐134aおよびR‐32は双方とも、反応の早期段階で、キラルモノエステル(b)の生成について、テトラヒドロフランよりも高度の選択性を示すようである。これは、2時間の段階まで、生成物の著しく高いエナンチオマー過剰率で示されている。この選択性改善に加えて、R‐134aおよびR‐32は添加トリエチルアミンの不在下で高度で速い変換を示し、おそらくより簡単な下流生成物単離および精製操作をもたらすであろう。
【0122】
例5 2‐エチルプロパン‐1,3‐ジオールのリパーゼ触媒反応
この例では、Pseudomonas cepaciaリパーゼを用いたR‐134a、R‐32およびクロロホルム中における2‐エチルプロパン‐1,3‐ジオールと酢酸ビニルとの酵素触媒反応を研究した。この反応は前記で更に詳細に説明されており、一方の特定エナンチオマーの優先的形成をもたらす。
【0123】
得られた結果は、(Gill et al.,Tetrahedron,1991,47,7569で開示されたような)クロロホルム中で得られた文献データと比較した。
【0124】
ジオール1.0mmol、酢酸ビニル3.9mmolおよびPseudomonas cepaciaリパーゼ0.01112gを溶媒2mlと混合し、室温で19時間にわたり磁気攪拌した。R‐134aおよびR‐32を用いた反応を、例1で記載されたものと全く同一の技術を用いて、ガラスエーロゾルボトル中で行った。混合液をサンプリングし、収率およびエナンチオマー過剰率の双方に関してGCにより分析し、結果を表5に掲載している。
【0125】
【表5】
【0126】
例3および4の反応の場合のように、R‐134aおよびR‐32中での変換は、従来の溶媒、クロロホルムで観察される場合よりも有意に高度のエナンチオ選択性を明らかに示している。
【0127】
例6 ラセミ 1‐フェニルエタノールのリパーゼ触媒分割
この例では、Candida antarctica Bリパーゼを用いてラセミ体のRエナンチオマーを対応アセテートへ変換することによるラセミ 1‐フェニルエタノールの分割を研究した。本プロセスは、様々なハイドロフルオロカーボン溶媒とヘキサンを用いて行った。
【0128】
溶媒としてハイドロフルオロカーボンを用いる反応は、次のように行った:
Novozym 435(0.0095g;95単位‐10,000単位/g(固定Candida antarctica Bリパーゼ))をエーロゾル中1‐フェニルエタノール(0.0620g;0.5mmol)および酢酸ビニル(0.8609g;10mmol)へ加えた。エーロゾルを密封し、R‐134a(6.0500g;5.00ml)、R‐32(4.8000g;5.00ml)またはR‐227ea(6.93000g;5.00ml)で充填した。反応液を室温(約20℃)で磁気攪拌した。エーロゾルの反転とバルブの弛緩によりサンプルを定期的に抜き取り、少量(約50μL)の反応溶液をガラスバイアル中へ取り出した。次いで、サンプルをジクロロメタン(0.1ml)に溶解し、ガスクロマトグラフィーにより分析した。
【0129】
溶媒としてヘキサン用いる反応は、次のように行った:
Novozym 435(0.0095g;95単位‐10,000単位/g(固定Candida antarctica Bリパーゼ))をSuppelcoTMバイアル中1‐フェニルエタノール(0.0620g;0.5mmol)および酢酸ビニル(0.8609g;10mmol)へ加えた。次いで、ヘキサン(5.00ml)を加えた。反応液を室温(約20℃)で磁気攪拌した。Hamiltonシリンジ(1μL)を用いてサンプル1μLを定期的に抜き取り、ガスクロマトグラフィーにより分析した。
【0130】
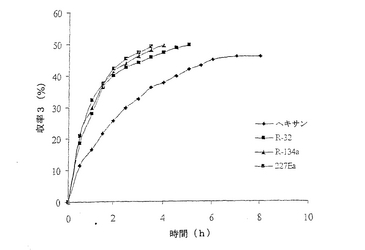
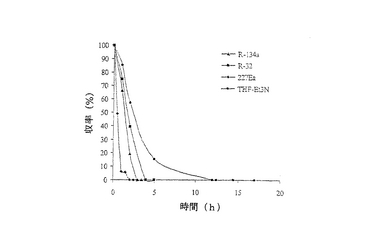
結果は表6に掲載されている。各溶媒中での反応の時間経過が図1に掲載されている。
【0131】
【表6】
【0132】
これまでは、リパーゼで触媒されるエステル交換反応はヘキサンのような非極性疎水性溶媒中で最も効率的である(それより極性の高い溶媒は、酵素からそれに必須の水を奪うことがある)、と広く考えられてきた(G.Kirchner,M.P.Scollar,A.M.Klibanov,J.Am.Chem.Soc.1985,107,7072-7076およびA.Zaks,A.M.Klibanov,Proc.Natl.Acad.Sci.USA,1985,82,3192-3196)。様々なハイドロフルオロカーボン中における1‐フェニルエタノールの分割を、ヘキサン中同一条件下で1‐フェニルエタノールの分割と比較した。収率およびエナンチオマー過剰率(e.e.)双方に関して、反応が試験されたすべてのハイドロフルオロカーボンで優れていることは、表6の結果から明らかである。
【0133】
図1は、この例で研究された溶媒での経時的プロットである。図1は、試験されたハイドロフルオロカーボンで、Novozym 435の優れた活性を明らかに示している;ハイドロフルオロカーボン溶媒中での反応速度はヘキサンの場合より大きい。R‐32を用いた場合は、各々(S‐1およびR‐3)>99%のエナンチオマー過剰率で、各エナンチオマー50%の分割収率が得られた。反応がR‐134aまたはR‐227ea中で行われた場合にも、同様の結果が得られた。しかしながら、反応がヘキサン中で行われた場合には、得られた収率および光学的純度が低かった。
【0134】
例7 Candida antarctica BリパーゼまたはPseudomonas cepaciaリパーゼを用いたメソ シス‐4‐シクロペンテン‐1,3‐ジオールのリパーゼ触媒反応
この例では、Candida antarctica BリパーゼまたはPseudomonas cepaciaリパーゼを用いたシス‐4‐シクロペンテン‐1,3‐ジオールと酢酸ビニルとの酵素触媒反応を研究した。反応は、R‐134a、R‐32、R‐227eaおよびTHF‐Et3Nの各々で行った。
【0135】
溶媒としてハイドロフルオロカーボンを用いる反応は、次のように行った:
Novozym 435(0.0010g;10単位‐10,000単位/g(固定Candida antarctica Bリパーゼ))またはPseudomonas cepaciaリパーゼ(0.0050g;0.463単位‐92.6単位/g(粉末凍結乾燥酵素))をエーロゾル中シス‐4‐シクロペンテン‐1,3‐ジオール(0.0050g;0.05mmol)および酢酸ビニル(0.0869g;1mmol)へ加えた。エーロゾルを密封し、R‐134a(6.0500g;5.00ml)、R‐32(4.8000g;5.00ml)またはR‐227ea(6.93000g;5.00ml)で充填した。反応液を室温(約20℃)で磁気攪拌した。エーロゾルの反転とバルブの弛緩によりサンプルを定期的に抜き取り、少量(約50μL)の反応溶液をガラスバイアル中へ取り出した。次いで、サンプルをジクロロメタン(0.1ml)に溶解し、ガスクロマトグラフィーにより分析した。
【0136】
無水THF‐Et3N中の反応は、次のように行った:
Novozym 435(0.0010g;10単位‐10,000単位/g(固定Candida antarctica B))またはPseudomonas cepaciaリパーゼ(0.0050g;0.463単位‐92.6単位/g(粉末凍結乾燥酵素))をSuppelcoTMバイアルで無水THF(5.00ml)中シス‐4‐シクロペンテン‐1,3‐ジオール(0.0050g;0.05mmol)、酢酸ビニル(0.0869g;1mmol)およびトリエチルアミン(0.0101g;0.1mmol(Et3N))の溶液(5.00ml)へ加えた。反応液を室温(約20℃)で磁気攪拌した。Hamiltonシリンジ(1μL)を用いてサンプル1μLを定期的に抜き取り、ガスクロマトグラフィーにより分析した。
【0137】
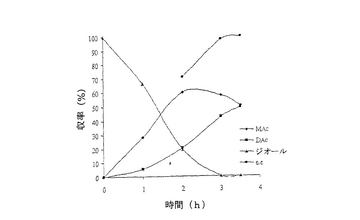
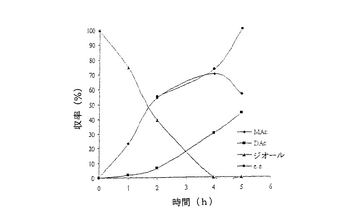
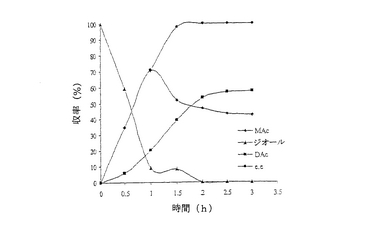
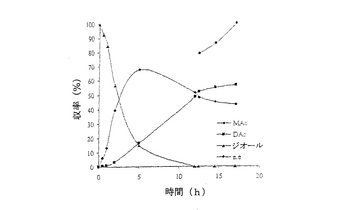
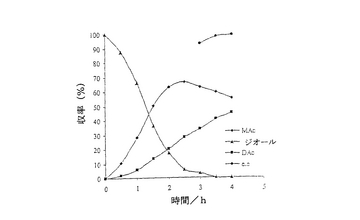
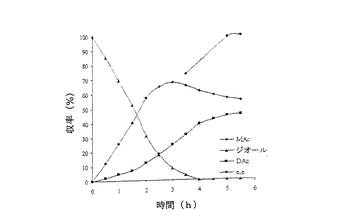
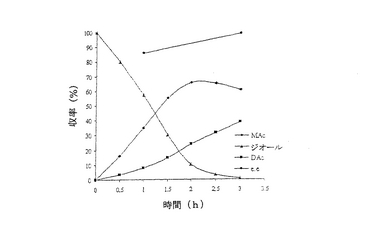
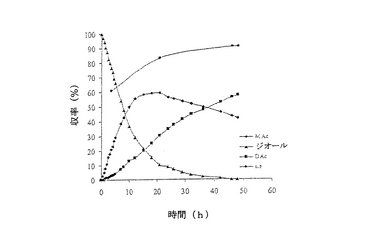
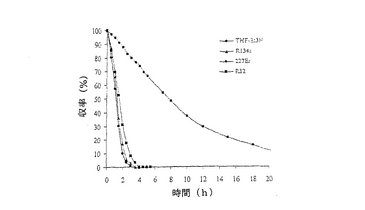
結果は表7に掲載されている。各酵素に関する各溶媒中での反応の時間経過が図2〜11に掲載されている。図2〜9で、MAcはモノアセテートを表わし、DAcはジアセテートを表わし、ジオールはシス‐4‐シクロペンテン‐1,3‐ジオールを表わし、e.e.はエナンチオマー過剰率を表わす。
【0138】
【表7】
【0139】
これまでは、リパーゼで触媒されるシス‐4‐シクロペンテン‐1,3‐ジオールと酢酸ビニルとの反応で最良の溶媒系はTHF‐Et3N系である、と広く考えられてきた(F.Theil,H.Schick,G.Winter,G.Reck,Tetrahedron,1991,47,7569-7582;S.R.Ghorpade,R.K.Kharul,R.R.Joshi,U.R.Kalkote,T.Ravindranathan,Tetrahedron Asymmetry,1999,10,891-899;C.R.Johnson,S.J.Bis,Tetrahedron Lett.1992,33,7287-7290)。したがって、様々なハイドロフルオロカーボン中におけるシス‐4‐シクロペンテン‐1,3‐ジオールの脱対称化を、THF‐Et3N中同一条件下で行われた同様の反応と比較した。Pseudomonas cepaciaリパーゼを用いた反応は、R‐32またはR‐134a中で行われたとき、THF‐Et3N中で行われた反応よりも優れていることは、(モノアセテート生成物の大きな収率で立証されているように)表7に掲載された結果から明らかである。反応がR‐227ea中で行われたとき、モノアセテート生成物の収率は、反応がTHF‐Et3N中で行われたときに得られる場合とおおよそ匹敵するが、その収率へはかなり短い時間で到達する。Novozym 435リパーゼが用いられたとき、モノアセテート生成物の優れた収率は、用いられたすべてのハイドロフルオロカーボン溶媒で得られ、短い反応時間でありながらも、THF‐Et3N中で反応を行う場合と比較して大きなエナンチオマー過剰率を呈した。
【0140】
これは図2〜11でも示されている。例えば、シス‐4‐シクロペンテン‐1,3‐ジオールの消費を表わす曲線の勾配で示されるように、図10はハイドロフルオロカーボン溶媒中で優れた反応速度を示している。同様の結論は図11の検分からも得られる。ハイドロフルオロカーボン溶媒中の反応速度が、THF‐Et3N中の場合よりもかなり大きくみえたことは、明らかである。シス‐4‐シクロペンテン‐1,3‐ジオールのPseudomonas cepaciaリパーゼ触媒脱対称化では、R‐32中での反応が最も効率的であり、99%エナンチオマー過剰率で、60%のモノアセテート生成物収率を示している。
【0141】
シス‐4‐シクロペンテン‐1,3‐ジオールのNovozym 435リパーゼ触媒脱対称化では、227ea中での反応が最も効率的であると判明し、99%エナンチオマー過剰率で、61%のモノアセテート生成物収率を示している。
【図面の簡単な説明】
【0142】
【図1】例6で研究された反応の経時的プロットである。
【図2】例7で研究されたような、R‐134a中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのPseudomonas cepacia触媒脱対称化の経時的プロットである。
【図3】例7で研究されたような、R‐32中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのPseudomonas cepacia触媒脱対称化の経時的プロットである。
【図4】例7で研究されたような、R‐227ea中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのPseudomonas cepacia触媒脱対称化の経時的プロットである。
【図5】例7で研究されたような、THF‐Et3N中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのPseudomonas cepacia触媒脱対称化の経時的プロットである。
【図6】例7で研究されたような、R‐134a中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのNovozym 435触媒脱対称化の経時的プロットである。
【図7】例7で研究されたような、R‐32中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのNovozym 435触媒脱対称化の経時的プロットである。
【図8】例7で研究されたような、R‐227ea中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのNovozym 435触媒脱対称化の経時的プロットである。
【図9】例7で研究されたような、THF‐Et3N中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのNovozym 435触媒脱対称化の経時的プロットである。
【図10】ジオールの消費を示す、例7で用いられた全4種の溶媒中における、シス‐4‐シクロペンテン‐1,3‐ジオールのPseudomonas cepacia触媒脱対称化の経時的プロットである。
【図11】ジオールの消費を示す、例7で用いられた全4種の溶媒中における、シス‐4‐シクロペンテン‐1,3‐ジオールのNovozym 435触媒脱対称化の経時的プロットである。
【発明の分野】
【0001】
本発明は、一次化合物の触媒変換による二次化合物の製造方法に関する。更に詳しくは、本発明は、生物学的触媒の存在下で、一次化合物を含めた基質を試薬と反応させることにより、二次化合物を立体選択的に製造するための方法に関する。
【背景技術】
【0002】
触媒とは、反応で自らが消費されることなく、反応速度を増すように働く物質である。酵素は、多くの場合に、反応が拡散限定されるような程度まで、十分有効に反応活性化エネルギーを低下させられる、天然触媒である。
【0003】
酵素触媒作用の顕著な特徴は、生物学的機能を決定することが知られた基質特異性である。一部の酵素は1種のみの生物学的基質を利用し、絶対基質特異性を示すと言われている。例えば、グルコキナーゼはATPからグルコースへのリン酸の転移を触媒し、他の糖へは転移させない。他の酵素はかなり広い基質特異性を示し、それらの天然基質と多くの場合に非類似である構造関連分子も利用しうる。これらの酵素は、関連基特異性を示すと言われている。この種の酵素の例はCandida cylindracea(C.cylindracea)リパーゼであり、これは様々なアシルドナーとアシルアクセプターとのエステル交換反応を触媒する。化学的特異性に加えて、酵素は立体化学的特異性も示す。
【0004】
国際生化学会は、触媒する反応のタイプに応じて、酵素を6カテゴリーに分類した。
【0005】
オキシドレダクターゼは、酸化および還元反応を触媒する。更に詳しくは、それらはC‐H、C‐CおよびC=C結合の酸素添加と、H原子相当物の除去または付加を触媒する。
【0006】
トランスフェラーゼは、アルデヒド、ケトン、アシル、糖、ホスホリルまたはメチル基のような様々な基の転移を触媒する。
【0007】
ヒドロラーゼは、特に、加水分解によるエステル、アミド、ラクトン、ラクタム、エポキシド、ニトリル、無水物およびグリコシドの形成を触媒する。
【0008】
リアーゼは、C=C、C=NおよびC=O結合での小分子の付加‐脱離を触媒する。
【0009】
イソメラーゼは、ラセミ化およびエピマー化のような異性化反応を触媒する。
【0010】
リガーゼは、C‐O、C‐S、C‐NおよびC‐C結合の形成および開裂と同時に、三リン酸開裂を触媒する。
【0011】
性質上、一部の酵素は細胞膜中の脂質層またはその内部で機能する。リパーゼは、例えば、水‐脂質界面で活性である。脂質層は、作業酵素向けの非水性および非極性環境をもたらす。
【0012】
酵素触媒は、立体選択性を利用するために、相当数のプロセスで商業的にも用いられている。例えば、ヒドロラーゼ種(プロテアーゼおよびリパーゼ)の酵素は、プロキラルおよび中心対称化合物からキラル化合物への変換と、メソ化合物の脱対称化において、二級アルコールおよびカルボン酸のラセミ混合物の分割のために、商業的に用いられている。酵素は、ヘキサンのような非極性有機溶媒中で、最も効果的に働く。溶媒の極性を高めると、酵素の急速な不活化および/または反応速度の著しい低下を招きやすい。
【0013】
生成物へ至る反応速度、選択性および/または変換性を改善することで、商業的酵素触媒プロセスに改良を加えることが望ましいであろう。広範囲の反応基質を溶解でき、反応に際して酵素の不活化を軽減し、広範囲の基質で効果的に所定の酵素を利用しうるような、溶媒を用いることも望ましいであろう。
【0014】
特に、現在商業用の公知プロセスよりも効率的に一次化合物を二次化合物へ立体選択的に変換しうる酵素触媒プロセスについて、必要性がある。
【発明の具体的説明】
【0015】
本発明によると、生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で、少くとも1種の一次化合物を含む出発物質または基質を試薬と反応させることからなる、二次化合物を立体選択的に製造するための方法が提供される。
【0016】
本発明の方法は、例えば、非キラル化合物、化合物のラセミ混合物、鏡像的純粋物質、メソ化合物、プロキラル化合物または中心対称化合物のうち、少くとも1種の一次化合物を、特定のキラル二次化合物へ立体選択的に変換する。このことは、一次化合物が、原則的に反応すると立体異性体の混合物を形成しうるが、生物学的触媒の影響下で優先的または選択的に反応して、主に、好ましくは排他的に、1種のエナンチオマーを生じることを、我々は意味している。特に、我々は、1種の特定エナンチオマーを主に、好ましくは排他的に得る方法に言及している。更に詳しくは、出発物質または基質の変換とは、望ましいエナンチオマーが50%以上、更に好ましくは70%以上、特に90%以上のエナンチオマー過剰率で形成されることをいう。
【0017】
本発明の方法は、高い立体選択性で、一次化合物から二次化合物へ良好な変換を行える。変換性および立体選択性は、ヘキサンのような従来の炭化水素溶媒を用いる公知の商業法で得られる場合よりも、優れている。更に、該方法は従来の炭化水素溶媒で行われる方法よりも速く進行する。
【0018】
本方法で用いられている(ハイドロ)フルオロカーボン溶媒は、同反応がヘキサンのような従来の炭化水素溶媒を用いて行われる場合よりも、生物学的触媒の分解を減らせる、とも考えられている。ひいては、このことから、触媒が変化するまで長時間にわたり連続プロセスを行わせられ、またはバッチプロセスにおいては触媒をより多くの回数にわたり再使用しうるのである。
【0019】
本発明の方法は、少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で行われる。“(ハイドロ)フルオロカーボン”という用語について、我々はハイドロフルオロカーボンおよびペルフルオロカーボンからなる群より選択される化合物を意味している。“ハイドロフルオロカーボン”という用語について、我々は炭素、水素およびフッ素原子のみを有する化合物を意味している。ハイドロフルオロカーボン溶媒が好ましい。
【0020】
溶媒は通常液体状態であるが、我々は超臨界液の使用を除外しているわけではない。室温で気体である1種以上の低沸点化合物を溶媒が含んでいる場合は、溶媒を適度な低温に冷却することにより、および/またはプロセスのある時点でそれを大気圧以上に付すことにより、望ましい液体状態が得られる。反応させる基質と(ハイドロ)フルオロカーボン溶媒が混合される前または後に、必要であればプロセスに際して連続的に、これら手段の一方または双方が適用される。
【0021】
適切な(ハイドロ)フルオロカーボンは、C1‐10、特にC1‐5、とりわけC1‐4(ハイドロ)フルオロカーボンから選択される。
【0022】
好ましいペルフルオロカーボンには、ヘキサフルオロエタン(R‐116)およびオクタフルオロプロパン(R‐218)がある。
【0023】
好ましいハイドロフルオロカーボンは、C1‐10、特にC1‐5、とりわけC1‐4ハイドロフルオロアルカンから選択される。適切なC1‐4ハイドロフルオロアルカンには、ハイドロフルオロメタン、例えばトリフルオロメタン(R‐23)、フルオロメタン(R‐41)およびジフルオロメタン(R‐32);ハイドロフルオロエタン、例えばペンタフルオロエタン(R‐125)、1,1,1‐トリフルオロエタン(R‐143a)、1,1,2,2‐テトラフルオロエタン(R‐134)、1,1,1,2‐テトラフルオロエタン(R‐134a)および1,1‐ジフルオロエタン(R‐152a);ハイドロフルオロプロパン、例えば1,1,1,3,3‐ペンタフルオロプロパン(R‐245fa)、1,1,2,2,3‐ペンタフルオロプロパン(R‐245ca)、1,1,1,2,3‐ペンタフルオロプロパン(R‐245eb)、1,1,2,3,3‐ペンタフルオロプロパン(R‐245ea)、1,1,1,2,3,3‐ヘキサフルオロプロパン(R‐236ea)、1,1,1,2,2,3‐ヘキサフルオロプロパン(R‐236cb)、1,1,1,3,3,3‐ヘキサフルオロプロパン(R‐236fa)、1,1,1,2,3,3,3‐ヘプタフルオロプロパン(R‐227ea)および1,1,1,2,2,3,3‐ヘプタフルオロプロパン(R‐227ca);およびハイドロフルオロブタン、例えば1,1,1,3,3‐ペンタフルオロブタン(R‐356mfc)がある。好ましいハイドロフルオロカーボンはR‐32、R‐134a、R‐134、R‐152a、R‐143a、R‐125、R‐245fa、R‐236eaおよびR‐227eaであり、これらはすべて低沸点であって、プロセスの最後に比較的容易に反応混合物から除去される。これらのうちR‐32およびR‐134aが特に好ましく、R‐134aが最も好ましい。
【0024】
2種以上の(ハイドロ)フルオロカーボンの混合物からなる溶媒も、所望であれば用いてよい。
【0025】
本発明の方法で用いられる溶媒は、(ハイドロ)フルオロカーボンに加えて、有機補助溶媒も含んでよい。
【0026】
適切な補助溶媒には、特に無フッ素、一般的には無ハロゲンの化合物がある。適切な無ハロゲン補助溶媒は、典型的には200℃以下、例えば−85〜200℃の範囲の沸点を有している。好ましい補助溶媒は、120℃以下、例えば−85〜120℃の範囲、更に好ましくは100℃以下、例えば−70〜100℃の範囲、特に10℃以下、例えば−60〜10℃の範囲の沸点を有している。2種以上の補助溶媒の混合物も、所望であれば用いてよい。
【0027】
適切な補助溶媒はC2‐6、特にC2‐4炭化水素化合物から選択され、それについて我々は炭素および水素原子のみを有した化合物を意味している。適切な炭化水素にはアルカンおよびシクロアルカンがあり、エタン、n‐プロパン、i‐プロパン、n‐ブタン、i‐ブタンおよびn‐ペンタンのようなアルカンが好ましい。
【0028】
他の適切な補助溶媒には炭化水素エーテルがあり、それについて我々は式R1‐O‐R2を有する化合物を意味し、ここでR1およびR2は、独立して、炭素および水素原子のみを有したヒドロカルビル基、例えばC1‐6、特にC1‐3アルキル基である。適切なジアルキルエーテルには、ジメチルエーテル、メチルエチルエーテルおよびジエチルエーテルがある。
【0029】
更に別の適切な補助溶媒は、アミド、スルホキシド、アルコール、ケトン、カルボン酸、カルボン酸誘導体、無機酸およびニトロ化合物から選択される。
【0030】
適切なアミド補助溶媒には、N,N′‐ジアルキルアミドおよびアルキルアミド、例えばジメチルホルムアミドおよびホルムアミドがある。
【0031】
適切なスルホキシド補助溶媒には、ジアルキルスルホキシド、例えばジメチルスルホキシドがある。
【0032】
適切なアルコール補助溶媒には、脂肪族アルコール、特にアルカノールがある。適切なアルカノールは、C1‐6、特にC1‐3アルカノール、例えばメタノール、エタノール、1‐プロパノールおよび2‐プロパノールから選択される。
【0033】
適切なケトン補助溶媒には、脂肪族ケトン、特にジアルキルケトン、例えばアセトンがある。
【0034】
適切なカルボン酸補助溶媒には、ギ酸および酢酸がある。
【0035】
補助溶媒として使用上適切なカルボン酸誘導体には、無水物、例えば無水酢酸、およびC1‐6、特にC1‐3アルカン酸のC1‐6、特にC1‐3アルキルエステル、酢酸エチルがある。
【0036】
補助溶媒として使用上適切なニトロ化合物には、ニトロアルカンおよびニトロアリール化合物、例えばニトロメタンおよびニトロベンゼンがある。
【0037】
好ましいというわけではないが、有機補助溶媒が用いられる場合、溶媒混合物は典型的には80.0〜99.0重量%の(ハイドロ)フルオロカーボンおよび1〜20重量%の補助溶媒からなる。好ましくは、溶媒混合物は90.0〜99.0重量%の(ハイドロ)フルオロカーボンおよび1〜10.0重量%の補助溶媒からなる。補助溶媒の極性が増してくると、酵素の不活化と共にあらゆる問題を避ける上で、補助溶媒を少なく用いることが通常望ましい。
【0038】
ほとんどの酵素の適正な機能のためには水が必要であるため、本発明の方法は典型的には少くとも少量の水の存在下で行われる。しかしながら、用いられる水の量は、通常、水が反応系で分離相を形成しないようなものとされる。これは、本方法の目的が、主に(ハイドロ)フルオロカーボン溶媒からなる環境下で、酵素機能を発揮させることにあるからである。好ましくは、水の量は、用いられる溶媒の飽和レベル以下に保たれる。更に好ましくは、反応は溶媒の総重量に対して1重量%以下の水の存在下で行われる。
【0039】
本発明の方法は、生物学的触媒の存在下で行われる。“生物学的触媒”とは、我々は生体組織または系でみられる触媒を意味している。本発明の方法で使用向けの具体的な生物学的触媒は、酵素およびアブザイムである。生物学的触媒は、もちろん、二次化合物への基質の立体選択的変換を触媒できねばならない。
【0040】
典型的には、本発明の方法は単一触媒の存在下で行われるが、我々は触媒の混合物が用いられる可能性を除外していない。
【0041】
本方法で使用上好ましい酵素は、前記6種の酵素のいずれから選択してもよい。
【0042】
各酵素は、それらが通常ホスト生物に存在しているか、またはそこで過剰発現により生産される生体組織から、それらが単離されている、という意味で分離している。これらの分離酵素はそのまま用いても、あるいは、例えばFitzpatrick,P.A.,Klibanov,A.M.,J.Am.Chem.Soc.,1991,113,3166で記載されているような標準文献プロセスを用いてそれらは凍結乾燥してもよい。しかしながら、少くとも一部の酵素が事前に凍結乾燥することなく(ハイドロ)フルオロカーボン溶媒中で有効に機能でき、そのため重要な処理ステップを省ける可能性をもたらすことを、我々は発見したのである。
【0043】
酵素は、凍結乾燥させてもまたはそうでなくても、標準文献プロセスを用いて通常固定される。例えば、酵素は、例えば物理的吸着または結合により、固体の不溶性マトリックスへ固定される。適切なマトリックスとしては、特にガラス、珪藻土、シリカおよび有機ポリマー、例えばポリスチレンおよびポリアクリレートホモポリマーおよびコポリマーがある。
【0044】
一方、酵素は、生細胞培養物、例えばLactobacillus acidophilusのような全細胞培養物、休止細胞培養物、例えば温水で活性化されうる乾燥パン酵母、または酵素および必要補因子を含有した非生細胞培養物、例えば死酵母の一部でもよい。酵素を含有した全細胞培養物は、標準文献プロセスを用いて、例えば物理的吸着または結合により、固体の不溶性マトリックスへ通常固定される。上記のマトリックスがこの目的のために用いうる。
【0045】
本発明の方法で使用上好ましい酵素には、ヒドロラーゼカテゴリーに属するものがある。具体的な酵素は、Subtilisin carlsbergおよびSubtilisin BPNのようなプロテアーゼ、Porcine pancreaticリパーゼ、Candida antarctica BリパーゼおよびPseudomonas cepaciaリパーゼのようなリパーゼ、Aspergillus orgzeaのα‐およびβ‐ガラクトシダーゼのようなグリコシダーゼである。
【0046】
アブザイムとは、触媒抗体、即ち特定の化学反応を触媒しうる抗体である。適切なアブザイムはアルドラーゼ抗体38C2である。
【0047】
アブザイムは、酵素に関して前記されたように、凍結乾燥および/または固定される。
【0048】
本発明の方法は、許容しうる反応速度および成分溶解性をもたらして、生物学的触媒、一次化合物および二次化合物の著しい分解を避けられる温度で通常行われる。典型的には、本方法は−60〜120℃の範囲、好ましくは−30〜80℃の範囲、特に0〜60℃の範囲の温度、例えば約20℃で行われる。
【0049】
本方法は、大気圧、大気圧以下または大気圧以上で行われる。正確な作業圧力は、とりわけ、用いられる溶媒、特にその沸点に依存する。好ましい作業圧力は、0.1〜200バールの範囲、更に好ましくは0.5〜30バールの範囲、特に1〜15バールの範囲である。
【0050】
(ハイドロ)フルオロカーボン溶媒対反応させる基質の重量比は、好ましくは1:1〜1000:1の範囲、更に好ましくは1:1〜500:1の範囲、特に1:1〜10:1の範囲である。生物学的触媒は、典型的には非常に少量で、例えば基質に対して触媒10−3〜10−4モル%程度で用いられる。正確な量は、酵素の活性に依存する。
【0051】
本発明の方法は、様々な立体選択的変換に通常適用しうる。医薬化合物の製造で中間体として用いうる化合物を製造する際に、それは特に有用である。
【0052】
一態様において、本発明の方法は、混合物を形成するエナンチオマーの一方を優先的または選択的に反応させて、新たなエナンチオマー化合物を形成させ、一方で他のエナンチオマーを大部分または完全に未反応のままにするように、生物学的触媒および(ハイドロ)フルオロカーボン溶媒の存在下で混合物を試薬と反応させることにより、ラセミ混合物またはラセミ改変物を分割するために用いられる。
【0053】
したがって、本発明の一態様では、ラセミ混合物を形成するエナンチオマーの一方を優先的または選択的に新たなエナンチオマー化合物へ変換するように、生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で該混合物を試薬と反応させることからなる、ラセミ混合物の分割方法が提供される。
【0054】
本発明のこの態様に従い分割されるラセミ混合物は、RおよびSアルコール、RおよびSカルボン酸またはエステル、RおよびSアミノ酸エステル、RおよびSアミン、RおよびSチオール、またはRおよびSアミドのラセミ混合物である。好ましくは、それはRおよびSアミノ酸エステルの混合物である。この特殊な分割は、RまたはSエナンチオマーのキラル炭素に付いた官能基を優先的または選択的に変換することにより行われる。生物学的触媒は、好ましくは酵素である。
【0055】
特別な態様において、本方法は、異なるアルコキシ基をもたらすアルカノールとの反応で、RまたはSエナンチオマーのアルコキシ基が優先的に、好ましくは選択的に交換されるエステル交換反応により、ラセミN‐P‐dl‐フェニルアラニンアルキルエステル(Pは保護基を表わす)を分割するために用いられる。通常、エステル交換反応をうけるのはSエナンチオマーである。好ましい保護基はアセチルおよびトリフルオロアセチルであり、好ましいアルキルエステルはプロピルエステルであり、そのため好ましいラセミ混合物はN‐アセチル‐dl‐フェニルアラニンプロピルエステルおよびN‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルである。好ましいアルカノールはメタノールである。生物学的触媒は、好ましくは酵素、更に好ましくはプロテアーゼ、更に一層好ましくはSubtilisin carlsbergである。
【0056】
N‐P‐dl‐フェニルアラニンアルキルエステル対アルカノールのモル比は、好ましくは1:0.1〜1:100の範囲、更に好ましくは1:1〜1:50の範囲、特に1:1〜1:10の範囲である。
【0057】
反応時間は、典型的には0.1〜48時間の範囲、好ましくは1〜36時間の範囲、特に1〜24時間の範囲である。
【0058】
ラセミN‐P‐dl‐フェニルアラニンアルキルエステルのRまたはSエナンチオマー(通常Sエナンチオマー)の優先的/選択的エステル交換とは、望ましいエナンチオマーが典型的には50%以上、好ましくは70%以上、特に90%以上、例えば100%のエナンチオマー過剰率で形成されるようなものをいう。
【0059】
Sエナンチオマーの100%エナンチオマー過剰率と仮定した、メタノールを用いるラセミN‐アセチル‐dl‐フェニルアラニンプロピルエステルの分割が、式(1)で示されている。
【化1】
【0060】
(Sエナンチオマーが100%エナンチオマー過剰率で形成される、と再び仮定した)メタノールを用いるN‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルの分割も、同様に進行するであろう。
【0061】
他の態様において、本発明の方法は、RまたはSエナンチオマーのOH基が試薬との反応で優先的に、好ましくは選択的に交換されるエステル交換反応により、ラセミ1‐フェニルエタノールを分割するために用いられる。用いられる試薬は、好ましくはアシルドナー、例えばビニルまたはイソプロペニルアルカノエートのようなエノールエステル、またはアルコキシエノールエステルである。好ましい試薬は酢酸ビニルである。通常、エステル交換反応をうけるのはRエナンチオマーである。生物学的触媒は、好ましくはリパーゼ、例えばCandida antarctica Bリパーゼである。
【0062】
1‐フェニルエタノール対アシルドナーのモル比は、好ましくは1:0.1〜1:100の範囲、更に好ましくは1:1〜1:50の範囲、特に1:20である。
【0063】
反応時間は、典型的には0.1〜48時間の範囲、好ましくは1〜36時間の範囲、特に1〜24時間の範囲である。
【0064】
ラセミ1‐フェニルエタノールのRまたはSエナンチオマー(通常Rエナンチオマー)の優先的/選択的エステル交換とは、望ましいエナンチオマーが典型的には50%以上、好ましくは70%以上、特に90%以上、例えば100%のエナンチオマー過剰率で形成されるようなものをいう。
【0065】
Rエナンチオマーの100%エナンチオマー過剰率と仮定した、酢酸ビニルを用いるラセミ1‐フェニルエタノールの分割が、式(2)で示されている。
【化2】
【0066】
他の態様において、本発明の方法は、生物学的触媒および(ハイドロ)フルオロカーボン溶媒の存在下でメソ化合物を試薬と反応させることにより、メソ化合物から優先的に、好ましくは選択的に特定のエナンチオマーを製造するために用いられる。鏡像と重ね合わせられるため対称的であるメソ化合物は、エナンチオマー生成物へ変換されることから、メソ化合物の反応も脱対称化と称される。もちろん、エナンチオマーはその鏡像と重ね合わせられない。
【0067】
したがって、本発明の別な態様では、生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下でメソ化合物を試薬と反応させることからなる、メソ化合物から優先的または選択的に特定のエナンチオマーを製造するための方法が提供される。
【0068】
本方法は、キラル炭素の1つに付いた官能基を優先的または選択的に置換または変換することにより行われる。
【0069】
メソ化合物は好ましくはシス‐4‐シクロペンテン‐1,3‐ジオールであり、用いられる試薬は好ましくはアシルドナー、例えばビニルまたはイソプロペニルアルカノエートのようなエノールエステル、またはアルコキシエノールエステルである。好ましい試薬は酢酸ビニルである。しかしながら、他のメソ化合物および他の試薬も用いてよい。
【0070】
生物学的触媒は好ましくは酵素であり、メソ化合物がシス‐4‐シクロペンテン‐1,3‐ジオールであるとき、酵素は好ましくはリパーゼ、更に好ましくはブタ膵臓リパーゼ、Candida antarctica BリパーゼまたはPseudomonas cepaciaリパーゼである。
【0071】
反応は、ヒンダードアミン、特に三級アミン、例えばトリエチルアミンの存在下で行われる。アミンの存在は、速い反応速度および大きな変換率に寄与しうる。しかしながら、酵素を減少させると、粗反応混合物の下流精製を簡単にできる。
【0072】
メソ シス‐4‐シクロペンテン‐1,3‐ジオールと酢酸ビニルとの反応は、式(3)で示されているように進行する。
【化3】
【0073】
本プロセスは2段階で生じると考えられている。第一段階はエナンチオマー モノアセテート生成物、即ち(1R,3S)‐(+)‐4‐シクロペンテン‐1,3‐ジオール‐1‐アセテート(a)、(1S,3R)‐(−)‐4‐シクロペンテン‐1,3‐ジオール‐1‐アセテート(b)またはエナンチオマー(a)および(b)の混合物の立体選択的形成であり、一方のエナンチオマーが過剰である。ブタ膵臓リパーゼ、Candida antarctica BリパーゼまたはPseudomonas cepaciaリパーゼが酵素として用いられる場合、エナンチオマー(b)が優先的に、多くの場合は排他的に形成される傾向がある。
【0074】
第二段階では、モノアセテート(a)および/または(b)が更に反応を続け、別な酢酸ビニル分子との反応で、ジアセテート シス‐4‐シクロペンテン‐1,3‐ジアセテートを形成する。もちろん、ジアセテートは他のメソ化合物である。
【0075】
双方のモノアセテート生成物とも、プロスタグランジン、プロスタサイクリンおよびトロンボキサンの合成において重要な出発物質である。
【0076】
シス‐4‐シクロペンテン‐1,3‐ジオール対酢酸ビニルのモル比は、好ましくは1:0.1〜1:100の範囲、更に好ましくは1:1〜1:50の範囲、特に1:1〜1:20の範囲である。
【0077】
反応時間は、典型的には0.1〜48時間の範囲、好ましくは1〜36時間の範囲、特に1〜24時間の範囲である。
【0078】
シス‐4‐シクロペンテン‐1,3‐ジオールと酢酸ビニルとの反応は、優先的/選択的に形成されるエナンチオマー(通常(1S,3R)‐(−)‐4‐シクロペンテン‐1,3‐ジオール‐1‐アセテート)が50%以上、更に好ましくは70%以上、特に90%以上、例えば100%のエナンチオマー過剰率で形成されるように、通常進行する。
【0079】
更に他の態様において、本発明の方法は、生物学的触媒および(ハイドロ)フルオロカーボン溶媒の存在下でプロキラル化合物を試薬と反応させることにより、プロキラル化合物から優先的に、好ましくは選択的に特定のエナンチオマーを製造するために用いられる。光学的に不活性な前駆体が非対称の光学的に活性な生成物へ変換されることから、プロキラル化合物の反応も脱対称化と称される。
【0080】
したがって、本発明の別な態様では、生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下でプロキラル化合物を試薬と反応させることからなる、プロキラル化合物から優先的または選択的に特定のエナンチオマーを製造するための方法が提供される。
【0081】
本方法は、少くとも1つの非キラル炭素原子を、キラル中心に異なる4つの官能基を有するキラル炭素原子へ、優先的または選択的に変換することにより行われる。
【0082】
プロキラル化合物は好ましくは2‐エチルプロパン‐1,3‐ジオールであり、用いられる試薬は好ましくはアシルドナー、例えばビニルまたはイソプロペニルアルカノエートのようなエノールエステル、またはアルコキシエノールエステルである。好ましい試薬は酢酸ビニルである。しかしながら、他のプロキラル化合物および他の試薬も用いてよい。
【0083】
生物学的触媒は好ましくは酵素であり、プロキラル化合物が2‐エチルプロパン‐1,3‐ジオールであるとき、酵素は好ましくはリパーゼ、更に好ましくはPseudomonas cepaciaリパーゼである。
【0084】
2‐エチルプロパン‐1,3‐ジオールと酢酸ビニルとの反応は、式(4)で示されているように進行する。
【化4】
【0085】
式(4)で示されているように、プロキラル 2‐エチルプロパン‐1,3‐ジオールは、最初にモノアセテート化合物1‐ヒドロキシ‐3‐アセトキシ‐2‐エチルプロパンへ変換される。この変換は排他的なRまたはSエナンチオマーの形成をもたらすか、または2種のうち一方が優先的な2種エナンチオマーの混合物の形成をもたらす。Pseudomonas cepaciaリパーゼが酵素として用いられる場合、Rエナンチオマーが優先的に、多くの場合は排他的に形成される傾向がある。
【0086】
その後も、モノアセテートは更に反応を続け、別の酢酸ビニル分子との反応で、ジアセテート 2‐エチルプロパン‐1,3‐ジアセテートを形成する。もちろん、ジアセテートもプロキラルである。
【0087】
双方のモノアセテート生成物とも、(Faber,K.,Biotransformations in Organic Chemistry,Springer-Verlag,1997で記載されているように)血小板活性化因子の合成において重要な構成単位である。
【0088】
2‐エチルプロパン‐1,3‐ジオール対酢酸ビニルのモル比は、好ましくは1:0.1〜1:100の範囲、更に好ましくは1:1〜1:50の範囲、特に1:1〜1:10の範囲である。
【0089】
反応時間は、典型的には0.1〜48時間の範囲、好ましくは1〜36時間の範囲、特に1〜24時間の範囲である。
【0090】
2‐エチルプロパン‐1,3‐ジオールと酢酸ビニルとの反応は、優先的/選択的に形成されるエナンチオマー(通常Rエナンチオマー)が50%以上、更に好ましくは70%以上、特に90%以上、例えば100%のエナンチオマー過剰率で形成されるように、通常進行する。
【0091】
本発明の方法は、バッチ式でまたは連続的に操作される。環境以下の沸点を有する(ハイドロ)フルオロカーボン溶媒が用いられる場合、反応容器は典型的には高圧に耐えうる圧力容器とする。
【0092】
バッチプロセスでは、例えば、(ハイドロ)フルオロカーボンが環境温度で気体であればフラッシュ蒸発により、または粗反応混合物を得た後で精製するために、必要であれば、望ましい二次化合物を単離するために蒸留により、(ハイドロ)フルオロカーボン溶媒がプロセスの最後に除去される。
【0093】
連続プロセスでは、(ハイドロ)フルオロカーボン溶媒および反応剤を含有した反応剤ストリームが、触媒を含有した反応容器へ連続的に運ばれる。典型的には、反応剤ストリームは固定触媒の層へ送られる。次いで、反応容器を出た粗反応混合物は、(ハイドロ)フルオロカーボン溶媒を除去して、該プロセスで形成された1種以上の望ましい二次化合物を回収するために、例えば溶媒エバポレーターで処理される。除去された(ハイドロ)フルオロカーボン溶媒は、所望であれば溶媒インファントリー(solvent infantry)を最少に抑えるため、濃縮してリサイクルしてもよい。未反応出発物質も、所望であればリサイクルしてよい。
【0094】
溶媒がリサイクルされる場合、低沸点溶媒用に適した回収系は、これについて我々は25℃以下、例えば0℃以下の沸点を有する溶媒を意味するのであるが、本プロセスから生じた粗反応混合物が送られるエバポレーター、エバポレーターで生じた蒸気を圧縮するためのコンプレッサー、およびコンプレッサーから生じた圧縮蒸気を冷却するためのコンデンサーからなる。溶媒は、コンプレッサーから吸引で誘導されるフラッシュ蒸発により、エバポレーターで粗反応混合物から除去され、次いでこうして生成した溶媒蒸気はコンプレッサー、例えばダイヤフラムコンプレッサーへ送られ、そこでそれが圧縮される。コンプレッサーから、溶媒蒸気はコンデンサーへ送られて、そこでそれが冷却され、本プロセスへ再供給するための液体形へ、または可能であれば本プロセスへ溶媒を供給する溶媒リザーバーへ戻される。コンデンサーは、コイル巻管の形をとりうるが、溶媒を蒸発させるために必要なエネルギーの少くとも一部を凝縮の潜熱が供給するように、エバポレーターの内部に配置しうる。
【0095】
低沸点溶媒用に適した別の回収系は、本プロセスから生じた反応混合物が送られて、溶媒が蒸発されるエバポレーターと、エバポレーターで生じた蒸気が冷却され、本プロセスへ再供給するための液体形へ、または可能であれば本プロセスへ溶媒を供給する溶媒リザーバーへ戻されるコンデンサーを含んでなる、溶媒リサイクル回路からなる。エバポレーターの加熱およびコンデンサーの冷却は独立して行えるが、好ましい態様ではエバポレーターを加熱してコンデンサーを冷却するために外部熱ポンプ系が用いられる。外部熱ポンプ系はエバポレーター、コンプレッサー、コンデンサーおよび伸縮バルブからなり、これらは熱媒液が流動させられる回路に順次配置されている。外部熱ポンプ系のエバポレーターは、コイル巻管の形をとりうるが、エバポレーターで熱媒液の蒸発がコンデンサーを冷却して、溶媒リサイクル回路へ送られる溶媒蒸気を凝縮するように、溶媒リサイクル回路のコンデンサーの内部または外部付近に配置される。次いで、外部熱ポンプ系のエバポレーターで生じた蒸気は圧縮され、コンデンサーへ送られ、そこでそれが凝縮して、発熱する。外部熱ポンプ系のコンデンサーは、これもコイル巻管の形をとりうるが、溶媒を蒸発させて溶媒リサイクル回路へ送るために必要な熱を熱媒液の凝縮に伴う凝縮潜熱が供給するように、溶媒リサイクル回路のエバポレーターの内部または外部付近に配置される。次いで、凝縮された熱媒液は伸縮バルブからエバポレーターへ戻され、こうして外部熱ポンプ系でサイクルを終える。
【0096】
外部熱ポンプ系の代わりとして、溶媒凝縮の熱をエバポレーター容器へ移して溶媒蒸発用の熱を供給するために、外部循環熱媒液が用いうる。
【0097】
本発明のプロセスが完了したとき、望ましい生成物を単離するため、粗反応混合物は精製ステップへ付される。次いで、例えば医薬化合物を得るために、純粋な生成物が1回以上の別な合成ステップへ付される。一方、粗反応混合物は別な合成に直接用いてもよい。適切な精製技術には、クロマトグラフィー、結晶化および蒸留のような、化学合成で常用されるものがある。
【0098】
本発明がここでは実例で示されているが、但し下記例で制限されることはない。
【0099】
一般操作
N‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルの製造
下記のように、Curphey,T.J.,J.Org.Chem.,1979,44,2805-2807で開示された方法を用いて、ラセミ N‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルを製造した:
オーブン乾燥フラスコにフェニルアラニンを加えた。次いで、フラスコをN2ガスでパージし、DMF(溶媒)、ジイソプロピルエチルアミン(1当量)およびトリフルオロ酢酸エチル(1.25当量)を加えた。溶液を50℃で17時間攪拌し、次いでヨウ化プロピル(1.25当量)を加えた。溶液を更に72時間攪拌した。粗生成物を再抽出し、勾配溶離を用いてカラムクロマトグラフィーにより単離した。ヘキサン400mlから出発し、9:1へキサン:酢酸エチル300ml、次いで4:1へキサン:酢酸エチル300ml、次いで3.5:1へキサン:酢酸エチル200ml、最後に2:1へキサン:酢酸エチル200mlを加えることにより、極性を徐々に増した。単離収量は4.17g、28%であった。次いで単離生成物をKugelrohr蒸留、次いで石油スピリットおよび酢酸エチル(各9:1)の混合液からの再結晶化で更に精製した。精製されたN‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルは白色結晶固体物であった。生成物の同定はNMRおよびGC‐質量スペクトル測定により行った。
【0100】
2‐エチルプロパン‐1,3‐ジオールの製造
2‐エチルプロパン‐1,3‐ジオールを次のように製造した:
ジエチルエチルマロネート(2.0g、10.7mmol)の溶液に、0℃で乾燥エタノール中LiAlH4(2.5当量)の懸濁液を加えた。反応混合液を攪拌しながら室温まで加温し、1時間後、更に1時間還流した。氷浴で冷却後、蒸留水1mlを攪拌しながら反応混合液へ加え、次いで2M NaOH溶液1mlを加えた。次いで、混合液を濾過し、濾液を酢酸エチルで洗浄した。合わせた洗液を減圧下で蒸発させて、黄色油状物を得、溶媒として5:1酢酸エチル:ヘキサンを用いてシリカフラッシュクロマトグラフィーにより精製した。生成物は75%収率で油状物として得た。生成物の同定はNMRおよびGC‐質量スペクトル測定により行った。
【0101】
R134aおよびR‐32はIneos Fluor Ltd.から提供され、更に精製することなく用いた。双方の溶媒とも、標準プラスチック被覆10mlガラスエーロゾルボトル中で反応を行わせることにより、自己圧力下で液体状態に維持した。
【0102】
酵素は、Aldrich Chemical Company、Sigma Chemical CompanyまたはFluka Chemical Companyから入手し、更に処理することなく、またはFitzpatrick,P.A.,Klibanov,A.M.,J.Am.Chem.Soc.,1991,113,3166で記載された操作を用いる凍結乾燥後に用いた。
【0103】
ハイドロフルオロカーボン(R‐134a、R‐32およびR‐227ea)はIneos Fluor Limitedから提供された。他の全化学物質および溶媒はAldrich Chemical CompanyまたはSigma Chemical Companyから購入し、更に精製することなく用いた。
【0104】
エーロゾルはIneos Fluor Limitedから提供された。
【0105】
ガスクロマトグラムは、HP SE‐54キャピラリーカラム(25m×0.21mm内径)装備のShimadzu GC‐17a機器を用いて記録した。キラルガスクロマトグラムは、Chiraldex GTAキャピラリーカラム(30m×0.25mm内径)装備のChrompack CP9001機器で得た。フレームイオン化検出器を双方の場合で用い、標準溶液を用いて個別物質毎に応答ファクターを較正した。反応混合液から採取されたサンプルをジクロロメタン溶媒に溶解させ、必要であればナフタレンを内部標準として用いた。
【実施例】
【0106】
例1 ラセミ N‐アセチル‐dl‐フェニルアラニンプロピルエステルのSubtilisin carlsberg触媒分割
この例では、Subtilisin carlsbergを用いてラセミ混合物のSエナンチオマーを対応メチルエステルへ変換することによる、ラセミ N‐アセチル‐dl‐フェニルアラニンプロピルエステルの分割を研究した。反応は前記で更に詳細に説明されてきた。
【0107】
10mM N‐アセチル‐dl‐フェニルアラニンプロピルエステルおよび200mMメタノールの溶液をへキサン、テトラヒドロフランおよびアセトニトリルの各々で調製した。各溶液4mlに凍結乾燥Subtilisin carlsberg4.0mgを加えた。得られた懸濁液を室温で攪拌し、収率およびエナンチオマー過剰率の双方に関するガスクロマトグラフィー分析用にサンプルを定期的に採取した。
【0108】
同反応は、溶媒としてR‐134aおよびR‐32を用いても研究した。10mM N‐アセチル‐dl‐フェニルアラニンプロピルエステル、200mMメタノールおよび凍結乾燥Subtilisin carlsberg4.0mgの2種混合液をガラスエーロゾルボトル中で調製した。次いでエーロゾルボトルにキャップを取り付け、キャップを適所でかしめ、秤量済みの液体ハイドロフルオロカーボン溶媒を大型圧力容器からエーロゾルバルブを介して導入した。次いで、得られた懸濁液を室温で磁気攪拌し、収率およびエナンチオマー過剰率の双方に関するガスクロマトグラフィー分析用に反応混合液のサンプルを定期的に採取した。反応混合液の一部をバルブからサンプルバイアル中へ出すことにより、サンプルを取り出した。ハイドロフルオロカーボン溶媒は本プロセスで蒸発し、反応混合液の低揮発性残渣をサンプルバイアル中に残した。次いで、必要であればGC分析用に、内部標準を含有した既知量の溶媒中へこの残渣を溶解させた。
【0109】
反応剤および生成物は、試験溶媒の各々で良好な溶解性を示した。結果は表1に掲載されている。
【表1】
【0110】
R‐134aが、ヘキサンのような従来の溶媒よりも、速い反応および大きな最終変換率を示すことは、表1から明らかである。ヘキサンは、例1のプロセスで従来から最良の溶媒として通常みなされている。R‐32はアセトニトリルおよびテトラヒドロフランの各々と比較して良い性能を示し、約1時間以内の早い反応段階ではヘキサンに近い。R‐134aおよびR‐32は双方とも優れたエナンチオ選択性を示している。
【0111】
例2 ラセミ N‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルのSubtilisin carlsberg触媒分割
N‐アセチル‐dl‐フェニルアラニンプロピルエステルの代わりに10mM N‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルを用いて、例1を繰返した。この例は、基質特異性に対する酵素‐溶媒ペアの感受性を試験するために行った。結果は表2に掲載されている。
【0112】
【表2】
【0113】
フッ素化N保護基の使用のおかげで、R‐134aが、ヘキサンで得られる場合と比較して変換に明瞭な改善を呈することは、明らかである。加えて、溶媒がR‐134aである場合、本プロセスは72時間を過ぎても適度な速度で続くようであり、一方へキサンで観察される速度は著しく低い。これは、R‐134aがヘキサンよりも低度で酵素を分解していることを示唆しているのであろう。この特性は、従来の有機溶媒よりも大きな程度で、ハイドロフルオロカーボン溶媒中における酵素の再使用を可能にしており、経済的利益につながる。
【0114】
このプロセスでのテトラヒドロフラン中の反応速度は、例1と比較して有意に低い。これは、ハイドロフルオロカーボン溶媒が、テトラヒドロフランのような類似極性の従来溶媒とは対照的に、広範囲の物質と大きな効力で酵素を機能させうることを示している。
【0115】
例3 メソ シス‐4‐シクロペンテン‐1,3‐ジオールのリパーゼ触媒反応
この例では、ブタ膵臓リパーゼを用いたR‐134aおよびテトラヒドロフラン中におけるシス‐4‐シクロペンテン‐1,3‐ジオールと酢酸ビニルとの酵素触媒反応を比較した。この反応は前記で更に詳細に説明されており、一方の特定エナンチオマーの優先的形成をもたらす。行う方法は、Theil et al.,Tetrahedron,1991,47,7569で記載されたものであった。
【0116】
ジオール(1.0012g、1.0mmol)およびトリエチルアミン(0.070g、0.7mmol)をブタ膵臓リパーゼ(PPL)0.5gおよび酢酸ビニル(0.600g、7mmol)へ加えた。溶媒2mlを直ちに加え、反応混合液を規定時間にわたり室温で磁気攪拌した。R‐134aを用いた反応は、例1で記載されたものと全く同一の技術を用いて、ガラスエーロゾルボトル中で行った。収率およびエナンチオマー過剰率の双方に関するGC分析用に物質を反応混合液から採取し、結果を表3に掲載している。
【0117】
【表3】
【0118】
表3は、トリエチルアミンの存在下で、ブタ膵臓リパーゼを用いた脱対称化反応において、R‐134aがテトラヒドロフランと同程度に効率的かつ選択的な溶媒であることを示している。テトラヒドロフランは、従来の非水性溶媒の中で最も有効であることが、Theilらにより発見されていた。
【0119】
例4 メソ シス‐4‐シクロペンテン‐1,3‐ジオールのリパーゼ触媒反応
Pseudomonas cepaciaリパーゼを用いて、例3を繰返した。R‐32も研究し、この反応も、R‐134a反応のように、ガラスエーロゾルボトル中で行った。ハイドロフルオロカーボン溶媒の場合は、トリエチルアミンを加えなかった。得られた結果は表4で示されている。
【0120】
【表4】
【0121】
表4の結果から、R‐134aおよびR‐32は双方とも、反応の早期段階で、キラルモノエステル(b)の生成について、テトラヒドロフランよりも高度の選択性を示すようである。これは、2時間の段階まで、生成物の著しく高いエナンチオマー過剰率で示されている。この選択性改善に加えて、R‐134aおよびR‐32は添加トリエチルアミンの不在下で高度で速い変換を示し、おそらくより簡単な下流生成物単離および精製操作をもたらすであろう。
【0122】
例5 2‐エチルプロパン‐1,3‐ジオールのリパーゼ触媒反応
この例では、Pseudomonas cepaciaリパーゼを用いたR‐134a、R‐32およびクロロホルム中における2‐エチルプロパン‐1,3‐ジオールと酢酸ビニルとの酵素触媒反応を研究した。この反応は前記で更に詳細に説明されており、一方の特定エナンチオマーの優先的形成をもたらす。
【0123】
得られた結果は、(Gill et al.,Tetrahedron,1991,47,7569で開示されたような)クロロホルム中で得られた文献データと比較した。
【0124】
ジオール1.0mmol、酢酸ビニル3.9mmolおよびPseudomonas cepaciaリパーゼ0.01112gを溶媒2mlと混合し、室温で19時間にわたり磁気攪拌した。R‐134aおよびR‐32を用いた反応を、例1で記載されたものと全く同一の技術を用いて、ガラスエーロゾルボトル中で行った。混合液をサンプリングし、収率およびエナンチオマー過剰率の双方に関してGCにより分析し、結果を表5に掲載している。
【0125】
【表5】
【0126】
例3および4の反応の場合のように、R‐134aおよびR‐32中での変換は、従来の溶媒、クロロホルムで観察される場合よりも有意に高度のエナンチオ選択性を明らかに示している。
【0127】
例6 ラセミ 1‐フェニルエタノールのリパーゼ触媒分割
この例では、Candida antarctica Bリパーゼを用いてラセミ体のRエナンチオマーを対応アセテートへ変換することによるラセミ 1‐フェニルエタノールの分割を研究した。本プロセスは、様々なハイドロフルオロカーボン溶媒とヘキサンを用いて行った。
【0128】
溶媒としてハイドロフルオロカーボンを用いる反応は、次のように行った:
Novozym 435(0.0095g;95単位‐10,000単位/g(固定Candida antarctica Bリパーゼ))をエーロゾル中1‐フェニルエタノール(0.0620g;0.5mmol)および酢酸ビニル(0.8609g;10mmol)へ加えた。エーロゾルを密封し、R‐134a(6.0500g;5.00ml)、R‐32(4.8000g;5.00ml)またはR‐227ea(6.93000g;5.00ml)で充填した。反応液を室温(約20℃)で磁気攪拌した。エーロゾルの反転とバルブの弛緩によりサンプルを定期的に抜き取り、少量(約50μL)の反応溶液をガラスバイアル中へ取り出した。次いで、サンプルをジクロロメタン(0.1ml)に溶解し、ガスクロマトグラフィーにより分析した。
【0129】
溶媒としてヘキサン用いる反応は、次のように行った:
Novozym 435(0.0095g;95単位‐10,000単位/g(固定Candida antarctica Bリパーゼ))をSuppelcoTMバイアル中1‐フェニルエタノール(0.0620g;0.5mmol)および酢酸ビニル(0.8609g;10mmol)へ加えた。次いで、ヘキサン(5.00ml)を加えた。反応液を室温(約20℃)で磁気攪拌した。Hamiltonシリンジ(1μL)を用いてサンプル1μLを定期的に抜き取り、ガスクロマトグラフィーにより分析した。
【0130】
結果は表6に掲載されている。各溶媒中での反応の時間経過が図1に掲載されている。
【0131】
【表6】
【0132】
これまでは、リパーゼで触媒されるエステル交換反応はヘキサンのような非極性疎水性溶媒中で最も効率的である(それより極性の高い溶媒は、酵素からそれに必須の水を奪うことがある)、と広く考えられてきた(G.Kirchner,M.P.Scollar,A.M.Klibanov,J.Am.Chem.Soc.1985,107,7072-7076およびA.Zaks,A.M.Klibanov,Proc.Natl.Acad.Sci.USA,1985,82,3192-3196)。様々なハイドロフルオロカーボン中における1‐フェニルエタノールの分割を、ヘキサン中同一条件下で1‐フェニルエタノールの分割と比較した。収率およびエナンチオマー過剰率(e.e.)双方に関して、反応が試験されたすべてのハイドロフルオロカーボンで優れていることは、表6の結果から明らかである。
【0133】
図1は、この例で研究された溶媒での経時的プロットである。図1は、試験されたハイドロフルオロカーボンで、Novozym 435の優れた活性を明らかに示している;ハイドロフルオロカーボン溶媒中での反応速度はヘキサンの場合より大きい。R‐32を用いた場合は、各々(S‐1およびR‐3)>99%のエナンチオマー過剰率で、各エナンチオマー50%の分割収率が得られた。反応がR‐134aまたはR‐227ea中で行われた場合にも、同様の結果が得られた。しかしながら、反応がヘキサン中で行われた場合には、得られた収率および光学的純度が低かった。
【0134】
例7 Candida antarctica BリパーゼまたはPseudomonas cepaciaリパーゼを用いたメソ シス‐4‐シクロペンテン‐1,3‐ジオールのリパーゼ触媒反応
この例では、Candida antarctica BリパーゼまたはPseudomonas cepaciaリパーゼを用いたシス‐4‐シクロペンテン‐1,3‐ジオールと酢酸ビニルとの酵素触媒反応を研究した。反応は、R‐134a、R‐32、R‐227eaおよびTHF‐Et3Nの各々で行った。
【0135】
溶媒としてハイドロフルオロカーボンを用いる反応は、次のように行った:
Novozym 435(0.0010g;10単位‐10,000単位/g(固定Candida antarctica Bリパーゼ))またはPseudomonas cepaciaリパーゼ(0.0050g;0.463単位‐92.6単位/g(粉末凍結乾燥酵素))をエーロゾル中シス‐4‐シクロペンテン‐1,3‐ジオール(0.0050g;0.05mmol)および酢酸ビニル(0.0869g;1mmol)へ加えた。エーロゾルを密封し、R‐134a(6.0500g;5.00ml)、R‐32(4.8000g;5.00ml)またはR‐227ea(6.93000g;5.00ml)で充填した。反応液を室温(約20℃)で磁気攪拌した。エーロゾルの反転とバルブの弛緩によりサンプルを定期的に抜き取り、少量(約50μL)の反応溶液をガラスバイアル中へ取り出した。次いで、サンプルをジクロロメタン(0.1ml)に溶解し、ガスクロマトグラフィーにより分析した。
【0136】
無水THF‐Et3N中の反応は、次のように行った:
Novozym 435(0.0010g;10単位‐10,000単位/g(固定Candida antarctica B))またはPseudomonas cepaciaリパーゼ(0.0050g;0.463単位‐92.6単位/g(粉末凍結乾燥酵素))をSuppelcoTMバイアルで無水THF(5.00ml)中シス‐4‐シクロペンテン‐1,3‐ジオール(0.0050g;0.05mmol)、酢酸ビニル(0.0869g;1mmol)およびトリエチルアミン(0.0101g;0.1mmol(Et3N))の溶液(5.00ml)へ加えた。反応液を室温(約20℃)で磁気攪拌した。Hamiltonシリンジ(1μL)を用いてサンプル1μLを定期的に抜き取り、ガスクロマトグラフィーにより分析した。
【0137】
結果は表7に掲載されている。各酵素に関する各溶媒中での反応の時間経過が図2〜11に掲載されている。図2〜9で、MAcはモノアセテートを表わし、DAcはジアセテートを表わし、ジオールはシス‐4‐シクロペンテン‐1,3‐ジオールを表わし、e.e.はエナンチオマー過剰率を表わす。
【0138】
【表7】
【0139】
これまでは、リパーゼで触媒されるシス‐4‐シクロペンテン‐1,3‐ジオールと酢酸ビニルとの反応で最良の溶媒系はTHF‐Et3N系である、と広く考えられてきた(F.Theil,H.Schick,G.Winter,G.Reck,Tetrahedron,1991,47,7569-7582;S.R.Ghorpade,R.K.Kharul,R.R.Joshi,U.R.Kalkote,T.Ravindranathan,Tetrahedron Asymmetry,1999,10,891-899;C.R.Johnson,S.J.Bis,Tetrahedron Lett.1992,33,7287-7290)。したがって、様々なハイドロフルオロカーボン中におけるシス‐4‐シクロペンテン‐1,3‐ジオールの脱対称化を、THF‐Et3N中同一条件下で行われた同様の反応と比較した。Pseudomonas cepaciaリパーゼを用いた反応は、R‐32またはR‐134a中で行われたとき、THF‐Et3N中で行われた反応よりも優れていることは、(モノアセテート生成物の大きな収率で立証されているように)表7に掲載された結果から明らかである。反応がR‐227ea中で行われたとき、モノアセテート生成物の収率は、反応がTHF‐Et3N中で行われたときに得られる場合とおおよそ匹敵するが、その収率へはかなり短い時間で到達する。Novozym 435リパーゼが用いられたとき、モノアセテート生成物の優れた収率は、用いられたすべてのハイドロフルオロカーボン溶媒で得られ、短い反応時間でありながらも、THF‐Et3N中で反応を行う場合と比較して大きなエナンチオマー過剰率を呈した。
【0140】
これは図2〜11でも示されている。例えば、シス‐4‐シクロペンテン‐1,3‐ジオールの消費を表わす曲線の勾配で示されるように、図10はハイドロフルオロカーボン溶媒中で優れた反応速度を示している。同様の結論は図11の検分からも得られる。ハイドロフルオロカーボン溶媒中の反応速度が、THF‐Et3N中の場合よりもかなり大きくみえたことは、明らかである。シス‐4‐シクロペンテン‐1,3‐ジオールのPseudomonas cepaciaリパーゼ触媒脱対称化では、R‐32中での反応が最も効率的であり、99%エナンチオマー過剰率で、60%のモノアセテート生成物収率を示している。
【0141】
シス‐4‐シクロペンテン‐1,3‐ジオールのNovozym 435リパーゼ触媒脱対称化では、227ea中での反応が最も効率的であると判明し、99%エナンチオマー過剰率で、61%のモノアセテート生成物収率を示している。
【図面の簡単な説明】
【0142】
【図1】例6で研究された反応の経時的プロットである。
【図2】例7で研究されたような、R‐134a中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのPseudomonas cepacia触媒脱対称化の経時的プロットである。
【図3】例7で研究されたような、R‐32中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのPseudomonas cepacia触媒脱対称化の経時的プロットである。
【図4】例7で研究されたような、R‐227ea中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのPseudomonas cepacia触媒脱対称化の経時的プロットである。
【図5】例7で研究されたような、THF‐Et3N中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのPseudomonas cepacia触媒脱対称化の経時的プロットである。
【図6】例7で研究されたような、R‐134a中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのNovozym 435触媒脱対称化の経時的プロットである。
【図7】例7で研究されたような、R‐32中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのNovozym 435触媒脱対称化の経時的プロットである。
【図8】例7で研究されたような、R‐227ea中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのNovozym 435触媒脱対称化の経時的プロットである。
【図9】例7で研究されたような、THF‐Et3N中におけるシス‐4‐シクロペンテン‐1,3‐ジオールのNovozym 435触媒脱対称化の経時的プロットである。
【図10】ジオールの消費を示す、例7で用いられた全4種の溶媒中における、シス‐4‐シクロペンテン‐1,3‐ジオールのPseudomonas cepacia触媒脱対称化の経時的プロットである。
【図11】ジオールの消費を示す、例7で用いられた全4種の溶媒中における、シス‐4‐シクロペンテン‐1,3‐ジオールのNovozym 435触媒脱対称化の経時的プロットである。
【特許請求の範囲】
【請求項1】
生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で、少くとも1種の一次化合物を含む基質を試薬と反応させることからなる、二次化合物の立体選択的な製造方法。
【請求項2】
生物学的触媒が酵素である、請求項1に記載の方法。
【請求項3】
酵素がヒドロラーゼである、請求項2に記載の方法。
【請求項4】
酵素が、プロテアーゼおよびリパーゼから選択される、請求項3に記載の方法。
【請求項5】
酵素が全細胞培養物の一部である、請求項2〜4のいずれか一項に記載の方法。
【請求項6】
生物学的触媒がアブザイムである、請求項1に記載の方法。
【請求項7】
基質が、50%以上のエナンチオマー過剰率で、エナンチオマーを形成するように反応する、請求項1〜6のいずれか一項に記載の方法。
【請求項8】
ラセミ混合物を形成するエナンチオマーの一方を優先的または選択的に新たなエナンチオマー化合物に変換するように、生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で該混合物を試薬と反応させることからなる、ラセミ混合物の分割方法。
【請求項9】
ラセミ混合物が、RおよびSアルコール、RおよびSカルボン酸、RおよびSカルボン酸エステル、RおよびSアミノ酸エステル、RおよびSアミン、RおよびSチオール、またはRおよびSアミドの混合物である、請求項8に記載の方法。
【請求項10】
ラセミ混合物がRおよびSアミノ酸エステルの混合物、またはRおよびSアルコールの混合物である、請求項9に記載の方法。
【請求項11】
ラセミ混合物がN‐P‐dl‐フェニルアラニンアルキルエステルの混合物であり、ここでPは保護基を表わし、試薬がアルカノールである、請求項10に記載の方法。
【請求項12】
ラセミ混合物がN‐アセチル‐dl‐フェニルアラニンプロピルエステルの混合物、またはN‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルの混合物であり、アルカノールがメタノールである、請求項11に記載の方法。
【請求項13】
ラセミ混合物が1‐フェニルエタノールの混合物であり、試薬が酢酸ビニルである、請求項10に記載の方法。
【請求項14】
新たなエナンチオマー化合物が、50%以上のエナンチオマー過剰率で形成される、請求項8〜13のいずれか一項に記載の方法。
【請求項15】
生物学的触媒が酵素である、請求項8〜14のいずれか一項に記載の方法。
【請求項16】
酵素がヒドロラーゼである、請求項14に記載の方法。
【請求項17】
酵素がプロテアーゼである、請求項16に記載の方法。
【請求項18】
酵素がSubtilisin carlsbergである、請求項17に記載の方法。
【請求項19】
生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下でメソ化合物を試薬と反応させることからなる、メソ化合物からの優先的または選択的な特定エナンチオマーの製造方法。
【請求項20】
メソ化合物がシス‐4‐シクロペンテン‐1,3‐ジオールであり、試薬がアシルドナーである、請求項19に記載の方法。
【請求項21】
アシルドナーがエノールエステルである、請求項20に記載の方法。
【請求項22】
アシルドナーが酢酸ビニルである、請求項20に記載の方法。
【請求項23】
反応がヒンダードアミンの存在下で行われる、請求項20〜22のいずれか一項に記載の方法。
【請求項24】
ヒンダードアミンが三級アミンである、請求項23に記載の方法。
【請求項25】
特定エナンチオマーが、50%以上のエナンチオマー過剰率で形成される、請求項19〜24のいずれか一項に記載の方法。
【請求項26】
生物学的触媒が酵素である、請求項19〜25のいずれか一項に記載の方法。
【請求項27】
酵素がヒドロラーゼである、請求項26に記載の方法。
【請求項28】
酵素がリパーゼである、請求項27に記載の方法。
【請求項29】
酵素がブタ膵臓リパーゼ、Candida antarctica BリパーゼまたはPseudomonas cepaciaリパーゼである、請求項28に記載の方法。
【請求項30】
生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下でプロキラル化合物を試薬と反応させることからなる、プロキラル化合物からの優先的または選択的な特定エナンチオマーの製造方法。
【請求項31】
プロキラル化合物が2‐エチルプロパン‐1,3‐ジオールであり、試薬がアシルドナーである、請求項30に記載の方法。
【請求項32】
アシルドナーがエノールエステルである、請求項31に記載の方法。
【請求項33】
アシルドナーが酢酸ビニルである、請求項31に記載の方法。
【請求項34】
特定エナンチオマーが、50%以上のエナンチオマー過剰率で形成される、請求項30〜33のいずれか一項に記載の方法。
【請求項35】
生物学的触媒が酵素である、請求項30〜34のいずれか一項に記載の方法。
【請求項36】
酵素がヒドロラーゼである、請求項35に記載の方法。
【請求項37】
酵素がリパーゼである、請求項36に記載の方法。
【請求項38】
酵素がPseudomonas cepaciaリパーゼである、請求項37に記載の方法。
【請求項39】
溶媒が少くとも1種のC1‐10(ハイドロ)フルオロアルカンを含んでなる、請求項1〜38のいずれか一項に記載の方法。
【請求項40】
少くとも1種のC1‐10(ハイドロ)フルオロアルカンが、ジフルオロメタン(R‐32)、ペンタフルオロエタン(R‐125)、1,1,1‐トリフルオロエタン(R‐143a)、1,1,2,2‐テトラフルオロエタン(R‐134)、1,1,1,2‐テトラフルオロエタン(R‐134a)、1,1‐ジフルオロエタン(R‐152a)、1,1,1,3,3‐ペンタフルオロプロパン(R‐245fa)、1,1,1,2,3,3‐ヘキサフルオロプロパン(R‐236ea)および1,1,1,2,3,3,3‐ヘプタフルオロプロパン(R‐227ea)からなる群より選択される、請求項39に記載の方法。
【請求項41】
溶媒が、ジフルオロメタン(R‐32)および1,1,1,2‐テトラフルオロエタン(R‐134a)のうち少くとも1種を含んでなる、請求項40に記載の方法。
【請求項42】
少くとも1種の(ハイドロ)フルオロカーボンが補助溶媒と併用される、請求項1〜41のいずれか一項に記載の方法。
【請求項43】
補助溶媒が無ハロゲンである、請求項42に記載の方法。
【請求項44】
溶媒が液体状態にある、請求項1〜43のいずれか一項に記載の方法。
【請求項45】
反応系で水が単独の水相を形成するために必要とされるよりも低いレベルにおいて、水の存在下で行われる、請求項1〜44のいずれか一項に記載の方法。
【請求項46】
用いられる水の量が、溶媒の飽和レベル未満である、請求項45に記載の方法。
【請求項47】
用いられる水の量が、溶媒の総重量に対して水1重量%未満である、請求項45に記載の方法。
【特許請求の範囲】
【請求項1】
生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で、少くとも1種の一次化合物を含む基質を試薬と反応させることからなり、反応系で水が単独の水相を形成するために必要とされるよりも低いレベルにおいて、水の存在下で行われる、二次化合物の立体選択的な製造方法。
【請求項2】
生物学的触媒が酵素である、請求項1に記載の方法。
【請求項3】
酵素がヒドロラーゼである、請求項2に記載の方法。
【請求項4】
酵素が、プロテアーゼおよびリパーゼから選択される、請求項3に記載の方法。
【請求項5】
酵素が全細胞培養物の一部である、請求項2〜4のいずれか一項に記載の方法。
【請求項6】
生物学的触媒がアブザイムである、請求項1に記載の方法。
【請求項7】
基質が、50%以上のエナンチオマー過剰率で、エナンチオマーを形成するように反応する、請求項1〜6のいずれか一項に記載の方法。
【請求項8】
ラセミ混合物を形成するエナンチオマーの一方を優先的または選択的に新たなエナンチオマー化合物に変換するように、生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で該混合物を試薬と反応させることからなる、ラセミ混合物の分割方法。
【請求項9】
ラセミ混合物が、RおよびSアルコール、RおよびSカルボン酸、RおよびSカルボン酸エステル、RおよびSアミノ酸エステル、RおよびSアミン、RおよびSチオール、またはRおよびSアミドの混合物である、請求項8に記載の方法。
【請求項10】
ラセミ混合物がRおよびSアミノ酸エステルの混合物、またはRおよびSアルコールの混合物である、請求項9に記載の方法。
【請求項11】
ラセミ混合物がN‐P‐dl‐フェニルアラニンアルキルエステルの混合物であり、ここでPは保護基を表わし、試薬がアルカノールである、請求項10に記載の方法。
【請求項12】
ラセミ混合物がN‐アセチル‐dl‐フェニルアラニンプロピルエステルの混合物、またはN‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルの混合物であり、アルカノールがメタノールである、請求項11に記載の方法。
【請求項13】
ラセミ混合物が1‐フェニルエタノールの混合物であり、試薬が酢酸ビニルである、請求項10に記載の方法。
【請求項14】
新たなエナンチオマー化合物が、50%以上のエナンチオマー過剰率で形成される、請求項8〜13のいずれか一項に記載の方法。
【請求項15】
生物学的触媒が酵素である、請求項8〜14のいずれか一項に記載の方法。
【請求項16】
酵素がヒドロラーゼである、請求項14に記載の方法。
【請求項17】
酵素がプロテアーゼである、請求項16に記載の方法。
【請求項18】
酵素がSubtilisin carlsbergである、請求項17に記載の方法。
【請求項19】
生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下でメソ化合物を試薬と反応させることからなる、メソ化合物からの優先的または選択的な特定エナンチオマーの製造方法。
【請求項20】
メソ化合物がシス‐4‐シクロペンテン‐1,3‐ジオールであり、試薬がアシルドナーである、請求項19に記載の方法。
【請求項21】
アシルドナーがエノールエステルである、請求項20に記載の方法。
【請求項22】
アシルドナーが酢酸ビニルである、請求項20に記載の方法。
【請求項23】
反応がヒンダードアミンの存在下で行われる、請求項20〜22のいずれか一項に記載の方法。
【請求項24】
ヒンダードアミンが三級アミンである、請求項23に記載の方法。
【請求項25】
特定エナンチオマーが、50%以上のエナンチオマー過剰率で形成される、請求項19〜24のいずれか一項に記載の方法。
【請求項26】
生物学的触媒が酵素である、請求項19〜25のいずれか一項に記載の方法。
【請求項27】
酵素がヒドロラーゼである、請求項26に記載の方法。
【請求項28】
酵素がリパーゼである、請求項27に記載の方法。
【請求項29】
酵素がブタ膵臓リパーゼ、Candida antarctica BリパーゼまたはPseudomonas cepaciaリパーゼである、請求項28に記載の方法。
【請求項30】
生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下でプロキラル化合物を試薬と反応させることからなる、プロキラル化合物からの優先的または選択的な特定エナンチオマーの製造方法。
【請求項31】
プロキラル化合物が2‐エチルプロパン‐1,3‐ジオールであり、試薬がアシルドナーである、請求項30に記載の方法。
【請求項32】
アシルドナーがエノールエステルである、請求項31に記載の方法。
【請求項33】
アシルドナーが酢酸ビニルである、請求項31に記載の方法。
【請求項34】
特定エナンチオマーが、50%以上のエナンチオマー過剰率で形成される、請求項30〜33のいずれか一項に記載の方法。
【請求項35】
生物学的触媒が酵素である、請求項30〜34のいずれか一項に記載の方法。
【請求項36】
酵素がヒドロラーゼである、請求項35に記載の方法。
【請求項37】
酵素がリパーゼである、請求項36に記載の方法。
【請求項38】
酵素がPseudomonas cepaciaリパーゼである、請求項37に記載の方法。
【請求項39】
溶媒が少くとも1種のC1‐10(ハイドロ)フルオロアルカンを含んでなる、請求項1〜38のいずれか一項に記載の方法。
【請求項40】
少くとも1種のC1‐10(ハイドロ)フルオロアルカンが、ジフルオロメタン(R‐32)、ペンタフルオロエタン(R‐125)、1,1,1‐トリフルオロエタン(R‐143a)、1,1,2,2‐テトラフルオロエタン(R‐134)、1,1,1,2‐テトラフルオロエタン(R‐134a)、1,1‐ジフルオロエタン(R‐152a)、1,1,1,3,3‐ペンタフルオロプロパン(R‐245fa)、1,1,1,2,3,3‐ヘキサフルオロプロパン(R‐236ea)および1,1,1,2,3,3,3‐ヘプタフルオロプロパン(R‐227ea)からなる群より選択される、請求項39に記載の方法。
【請求項41】
溶媒が、ジフルオロメタン(R‐32)および1,1,1,2‐テトラフルオロエタン(R‐134a)のうち少くとも1種を含んでなる、請求項40に記載の方法。
【請求項42】
少くとも1種の(ハイドロ)フルオロカーボンが補助溶媒と併用される、請求項1〜41のいずれか一項に記載の方法。
【請求項43】
補助溶媒が無ハロゲンである、請求項42に記載の方法。
【請求項44】
溶媒が液体状態にある、請求項1〜43のいずれか一項に記載の方法。
【請求項45】
用いられる水の量が、溶媒の飽和レベル未満である、請求項1〜44のいずれか一項に記載の方法。
【請求項46】
用いられる水の量が、溶媒の総重量に対して水1重量%未満である、請求項1〜45のいずれか一項に記載の方法。
【請求項1】
生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で、少くとも1種の一次化合物を含む基質を試薬と反応させることからなる、二次化合物の立体選択的な製造方法。
【請求項2】
生物学的触媒が酵素である、請求項1に記載の方法。
【請求項3】
酵素がヒドロラーゼである、請求項2に記載の方法。
【請求項4】
酵素が、プロテアーゼおよびリパーゼから選択される、請求項3に記載の方法。
【請求項5】
酵素が全細胞培養物の一部である、請求項2〜4のいずれか一項に記載の方法。
【請求項6】
生物学的触媒がアブザイムである、請求項1に記載の方法。
【請求項7】
基質が、50%以上のエナンチオマー過剰率で、エナンチオマーを形成するように反応する、請求項1〜6のいずれか一項に記載の方法。
【請求項8】
ラセミ混合物を形成するエナンチオマーの一方を優先的または選択的に新たなエナンチオマー化合物に変換するように、生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で該混合物を試薬と反応させることからなる、ラセミ混合物の分割方法。
【請求項9】
ラセミ混合物が、RおよびSアルコール、RおよびSカルボン酸、RおよびSカルボン酸エステル、RおよびSアミノ酸エステル、RおよびSアミン、RおよびSチオール、またはRおよびSアミドの混合物である、請求項8に記載の方法。
【請求項10】
ラセミ混合物がRおよびSアミノ酸エステルの混合物、またはRおよびSアルコールの混合物である、請求項9に記載の方法。
【請求項11】
ラセミ混合物がN‐P‐dl‐フェニルアラニンアルキルエステルの混合物であり、ここでPは保護基を表わし、試薬がアルカノールである、請求項10に記載の方法。
【請求項12】
ラセミ混合物がN‐アセチル‐dl‐フェニルアラニンプロピルエステルの混合物、またはN‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルの混合物であり、アルカノールがメタノールである、請求項11に記載の方法。
【請求項13】
ラセミ混合物が1‐フェニルエタノールの混合物であり、試薬が酢酸ビニルである、請求項10に記載の方法。
【請求項14】
新たなエナンチオマー化合物が、50%以上のエナンチオマー過剰率で形成される、請求項8〜13のいずれか一項に記載の方法。
【請求項15】
生物学的触媒が酵素である、請求項8〜14のいずれか一項に記載の方法。
【請求項16】
酵素がヒドロラーゼである、請求項14に記載の方法。
【請求項17】
酵素がプロテアーゼである、請求項16に記載の方法。
【請求項18】
酵素がSubtilisin carlsbergである、請求項17に記載の方法。
【請求項19】
生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下でメソ化合物を試薬と反応させることからなる、メソ化合物からの優先的または選択的な特定エナンチオマーの製造方法。
【請求項20】
メソ化合物がシス‐4‐シクロペンテン‐1,3‐ジオールであり、試薬がアシルドナーである、請求項19に記載の方法。
【請求項21】
アシルドナーがエノールエステルである、請求項20に記載の方法。
【請求項22】
アシルドナーが酢酸ビニルである、請求項20に記載の方法。
【請求項23】
反応がヒンダードアミンの存在下で行われる、請求項20〜22のいずれか一項に記載の方法。
【請求項24】
ヒンダードアミンが三級アミンである、請求項23に記載の方法。
【請求項25】
特定エナンチオマーが、50%以上のエナンチオマー過剰率で形成される、請求項19〜24のいずれか一項に記載の方法。
【請求項26】
生物学的触媒が酵素である、請求項19〜25のいずれか一項に記載の方法。
【請求項27】
酵素がヒドロラーゼである、請求項26に記載の方法。
【請求項28】
酵素がリパーゼである、請求項27に記載の方法。
【請求項29】
酵素がブタ膵臓リパーゼ、Candida antarctica BリパーゼまたはPseudomonas cepaciaリパーゼである、請求項28に記載の方法。
【請求項30】
生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下でプロキラル化合物を試薬と反応させることからなる、プロキラル化合物からの優先的または選択的な特定エナンチオマーの製造方法。
【請求項31】
プロキラル化合物が2‐エチルプロパン‐1,3‐ジオールであり、試薬がアシルドナーである、請求項30に記載の方法。
【請求項32】
アシルドナーがエノールエステルである、請求項31に記載の方法。
【請求項33】
アシルドナーが酢酸ビニルである、請求項31に記載の方法。
【請求項34】
特定エナンチオマーが、50%以上のエナンチオマー過剰率で形成される、請求項30〜33のいずれか一項に記載の方法。
【請求項35】
生物学的触媒が酵素である、請求項30〜34のいずれか一項に記載の方法。
【請求項36】
酵素がヒドロラーゼである、請求項35に記載の方法。
【請求項37】
酵素がリパーゼである、請求項36に記載の方法。
【請求項38】
酵素がPseudomonas cepaciaリパーゼである、請求項37に記載の方法。
【請求項39】
溶媒が少くとも1種のC1‐10(ハイドロ)フルオロアルカンを含んでなる、請求項1〜38のいずれか一項に記載の方法。
【請求項40】
少くとも1種のC1‐10(ハイドロ)フルオロアルカンが、ジフルオロメタン(R‐32)、ペンタフルオロエタン(R‐125)、1,1,1‐トリフルオロエタン(R‐143a)、1,1,2,2‐テトラフルオロエタン(R‐134)、1,1,1,2‐テトラフルオロエタン(R‐134a)、1,1‐ジフルオロエタン(R‐152a)、1,1,1,3,3‐ペンタフルオロプロパン(R‐245fa)、1,1,1,2,3,3‐ヘキサフルオロプロパン(R‐236ea)および1,1,1,2,3,3,3‐ヘプタフルオロプロパン(R‐227ea)からなる群より選択される、請求項39に記載の方法。
【請求項41】
溶媒が、ジフルオロメタン(R‐32)および1,1,1,2‐テトラフルオロエタン(R‐134a)のうち少くとも1種を含んでなる、請求項40に記載の方法。
【請求項42】
少くとも1種の(ハイドロ)フルオロカーボンが補助溶媒と併用される、請求項1〜41のいずれか一項に記載の方法。
【請求項43】
補助溶媒が無ハロゲンである、請求項42に記載の方法。
【請求項44】
溶媒が液体状態にある、請求項1〜43のいずれか一項に記載の方法。
【請求項45】
反応系で水が単独の水相を形成するために必要とされるよりも低いレベルにおいて、水の存在下で行われる、請求項1〜44のいずれか一項に記載の方法。
【請求項46】
用いられる水の量が、溶媒の飽和レベル未満である、請求項45に記載の方法。
【請求項47】
用いられる水の量が、溶媒の総重量に対して水1重量%未満である、請求項45に記載の方法。
【特許請求の範囲】
【請求項1】
生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で、少くとも1種の一次化合物を含む基質を試薬と反応させることからなり、反応系で水が単独の水相を形成するために必要とされるよりも低いレベルにおいて、水の存在下で行われる、二次化合物の立体選択的な製造方法。
【請求項2】
生物学的触媒が酵素である、請求項1に記載の方法。
【請求項3】
酵素がヒドロラーゼである、請求項2に記載の方法。
【請求項4】
酵素が、プロテアーゼおよびリパーゼから選択される、請求項3に記載の方法。
【請求項5】
酵素が全細胞培養物の一部である、請求項2〜4のいずれか一項に記載の方法。
【請求項6】
生物学的触媒がアブザイムである、請求項1に記載の方法。
【請求項7】
基質が、50%以上のエナンチオマー過剰率で、エナンチオマーを形成するように反応する、請求項1〜6のいずれか一項に記載の方法。
【請求項8】
ラセミ混合物を形成するエナンチオマーの一方を優先的または選択的に新たなエナンチオマー化合物に変換するように、生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下で該混合物を試薬と反応させることからなる、ラセミ混合物の分割方法。
【請求項9】
ラセミ混合物が、RおよびSアルコール、RおよびSカルボン酸、RおよびSカルボン酸エステル、RおよびSアミノ酸エステル、RおよびSアミン、RおよびSチオール、またはRおよびSアミドの混合物である、請求項8に記載の方法。
【請求項10】
ラセミ混合物がRおよびSアミノ酸エステルの混合物、またはRおよびSアルコールの混合物である、請求項9に記載の方法。
【請求項11】
ラセミ混合物がN‐P‐dl‐フェニルアラニンアルキルエステルの混合物であり、ここでPは保護基を表わし、試薬がアルカノールである、請求項10に記載の方法。
【請求項12】
ラセミ混合物がN‐アセチル‐dl‐フェニルアラニンプロピルエステルの混合物、またはN‐トリフルオロアセチル‐dl‐フェニルアラニンプロピルエステルの混合物であり、アルカノールがメタノールである、請求項11に記載の方法。
【請求項13】
ラセミ混合物が1‐フェニルエタノールの混合物であり、試薬が酢酸ビニルである、請求項10に記載の方法。
【請求項14】
新たなエナンチオマー化合物が、50%以上のエナンチオマー過剰率で形成される、請求項8〜13のいずれか一項に記載の方法。
【請求項15】
生物学的触媒が酵素である、請求項8〜14のいずれか一項に記載の方法。
【請求項16】
酵素がヒドロラーゼである、請求項14に記載の方法。
【請求項17】
酵素がプロテアーゼである、請求項16に記載の方法。
【請求項18】
酵素がSubtilisin carlsbergである、請求項17に記載の方法。
【請求項19】
生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下でメソ化合物を試薬と反応させることからなる、メソ化合物からの優先的または選択的な特定エナンチオマーの製造方法。
【請求項20】
メソ化合物がシス‐4‐シクロペンテン‐1,3‐ジオールであり、試薬がアシルドナーである、請求項19に記載の方法。
【請求項21】
アシルドナーがエノールエステルである、請求項20に記載の方法。
【請求項22】
アシルドナーが酢酸ビニルである、請求項20に記載の方法。
【請求項23】
反応がヒンダードアミンの存在下で行われる、請求項20〜22のいずれか一項に記載の方法。
【請求項24】
ヒンダードアミンが三級アミンである、請求項23に記載の方法。
【請求項25】
特定エナンチオマーが、50%以上のエナンチオマー過剰率で形成される、請求項19〜24のいずれか一項に記載の方法。
【請求項26】
生物学的触媒が酵素である、請求項19〜25のいずれか一項に記載の方法。
【請求項27】
酵素がヒドロラーゼである、請求項26に記載の方法。
【請求項28】
酵素がリパーゼである、請求項27に記載の方法。
【請求項29】
酵素がブタ膵臓リパーゼ、Candida antarctica BリパーゼまたはPseudomonas cepaciaリパーゼである、請求項28に記載の方法。
【請求項30】
生物学的触媒および少くとも1種の(ハイドロ)フルオロカーボンを含む溶媒の存在下でプロキラル化合物を試薬と反応させることからなる、プロキラル化合物からの優先的または選択的な特定エナンチオマーの製造方法。
【請求項31】
プロキラル化合物が2‐エチルプロパン‐1,3‐ジオールであり、試薬がアシルドナーである、請求項30に記載の方法。
【請求項32】
アシルドナーがエノールエステルである、請求項31に記載の方法。
【請求項33】
アシルドナーが酢酸ビニルである、請求項31に記載の方法。
【請求項34】
特定エナンチオマーが、50%以上のエナンチオマー過剰率で形成される、請求項30〜33のいずれか一項に記載の方法。
【請求項35】
生物学的触媒が酵素である、請求項30〜34のいずれか一項に記載の方法。
【請求項36】
酵素がヒドロラーゼである、請求項35に記載の方法。
【請求項37】
酵素がリパーゼである、請求項36に記載の方法。
【請求項38】
酵素がPseudomonas cepaciaリパーゼである、請求項37に記載の方法。
【請求項39】
溶媒が少くとも1種のC1‐10(ハイドロ)フルオロアルカンを含んでなる、請求項1〜38のいずれか一項に記載の方法。
【請求項40】
少くとも1種のC1‐10(ハイドロ)フルオロアルカンが、ジフルオロメタン(R‐32)、ペンタフルオロエタン(R‐125)、1,1,1‐トリフルオロエタン(R‐143a)、1,1,2,2‐テトラフルオロエタン(R‐134)、1,1,1,2‐テトラフルオロエタン(R‐134a)、1,1‐ジフルオロエタン(R‐152a)、1,1,1,3,3‐ペンタフルオロプロパン(R‐245fa)、1,1,1,2,3,3‐ヘキサフルオロプロパン(R‐236ea)および1,1,1,2,3,3,3‐ヘプタフルオロプロパン(R‐227ea)からなる群より選択される、請求項39に記載の方法。
【請求項41】
溶媒が、ジフルオロメタン(R‐32)および1,1,1,2‐テトラフルオロエタン(R‐134a)のうち少くとも1種を含んでなる、請求項40に記載の方法。
【請求項42】
少くとも1種の(ハイドロ)フルオロカーボンが補助溶媒と併用される、請求項1〜41のいずれか一項に記載の方法。
【請求項43】
補助溶媒が無ハロゲンである、請求項42に記載の方法。
【請求項44】
溶媒が液体状態にある、請求項1〜43のいずれか一項に記載の方法。
【請求項45】
用いられる水の量が、溶媒の飽和レベル未満である、請求項1〜44のいずれか一項に記載の方法。
【請求項46】
用いられる水の量が、溶媒の総重量に対して水1重量%未満である、請求項1〜45のいずれか一項に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2006−514833(P2006−514833A)
【公表日】平成18年5月18日(2006.5.18)
【国際特許分類】
【出願番号】特願2006−500260(P2006−500260)
【出願日】平成16年3月18日(2004.3.18)
【国際出願番号】PCT/GB2004/001180
【国際公開番号】WO2004/083444
【国際公開日】平成16年9月30日(2004.9.30)
【出願人】(505353319)イネオス、フラウアー、ホールディングス、リミテッド (13)
【氏名又は名称原語表記】INEOS FLUOR HOLDINGS LIMITED
【Fターム(参考)】
【公表日】平成18年5月18日(2006.5.18)
【国際特許分類】
【出願日】平成16年3月18日(2004.3.18)
【国際出願番号】PCT/GB2004/001180
【国際公開番号】WO2004/083444
【国際公開日】平成16年9月30日(2004.9.30)
【出願人】(505353319)イネオス、フラウアー、ホールディングス、リミテッド (13)
【氏名又は名称原語表記】INEOS FLUOR HOLDINGS LIMITED
【Fターム(参考)】
[ Back to top ]