
ハリコンドリンB類似体
本発明は、薬剤活性を持つハリコンドリンB類似体、あるいはそれらの結晶形、ならびに合成中間体としてさらなる有用性を持つハリコンドリンB類似体を含む。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願
本出願は、2008年4月4日に出願された米国特許仮出願番号第61/042,643号および2008年4月11日に出願された米国特許仮出願番号61/071,111号の優先権を主張し、これらは、その全てが参照によりここに組み込まれる。
【0002】
発明の分野
本発明は薬剤活性を持つハリコンドリンB類似体に関する。ある実施態様において、この類似体は結晶体を持ち、そして/あるいは、強力な抗癌活性を持つ化合物であるハリコンドリンB類似体E7389の合成における合成中間体としてのさらなる有用性を持つ。
【化1】

【背景技術】
【0003】
背景技術
強力な抗癌剤であるハリコンドリンBは、カイメン、Halichondria okadaiより最初に単離された。それはまた、Axinella sp.、Phakellia carteriおよびLissondendryx sp.においても発見された。ハリコンドリンBは、インビトロ(in vitro)において、チューブリンの重合化、微小管重合、ベータ−チューブリン架橋、GTPおよびビンブラスチンのチューブリンへの結合、ならびにチューブリン依存性GTP加水分解を阻害する事を示した。それはまた、インビトロ(in vitro)およびインビボ(in vivo)において抗癌活性を示した。
【発明の概要】
【発明が解決しようとする課題】
【0004】
Aicherらは、1992年にハリコンドリンBの全合成を発表した(Aicher,T.らのJ.Am.Chem.Soc.114:3162−3164およびZheng,W.J.らのJ.Bioorg.Med.Chem.Lett.、2004、14、5551を参照)。ハリコンドリンBおよびさまざまな類似体の合成は、米国特許第6,214,865号、第6,365,759号、第6,469,182号および第6,653,341号;米国特許出願公開2004/0198806、2006/0104984:ならびに特許協力条約に基づく国際出願公開2005/118565に記載され、それらすべては、参照によりその全体がここに組み入れられる。
【課題を解決するための手段】
【0005】
本発明は薬剤活性を持つハリコンドリンB類似体に関する。類似体は結晶体を持ち、そして/あるいは強力な抗癌活性を持つ化合物であるハリコンドリンB類似体、E7389の合成における合成中間体としてのさらなる有用性を持つ。従って、本発明は、ER−076349一水和物の結晶体;ER−809681の結晶体;化合物ER−820057もしくはER−820057の結晶体;化合物ER−818906もしくはER−818906の結晶体;化合物ER−819531もしくはER−819531の結晶体;ならびに化合物ER−111197もしくはER−111197の結晶体に関する。本発明はまた、それらの化合物を含有する医薬組成物、その化合物を投与する事による癌の治療方法に関する。
【図面の簡単な説明】
【0006】
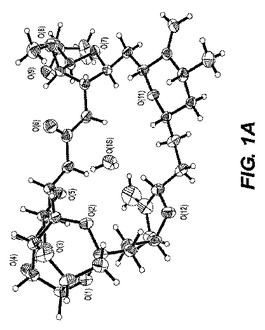
【図1−A】図1−Aは、ER−076349−00一水和物の単結晶構造のORTEP図である。
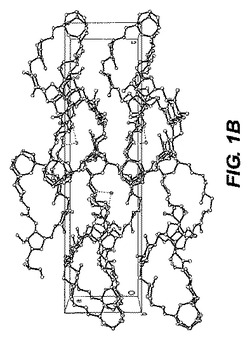
【図1−B】図1−Bは、ER−076349一水和物のb軸に沿った結晶パッキング図である。
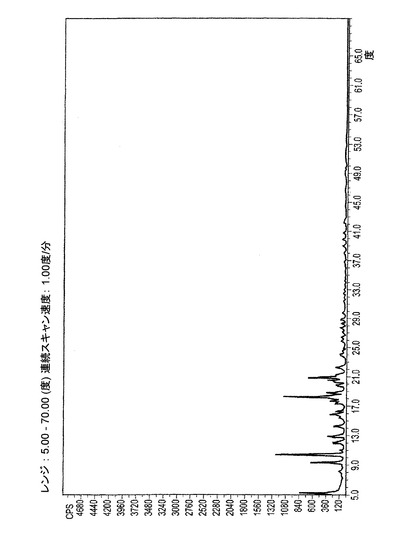
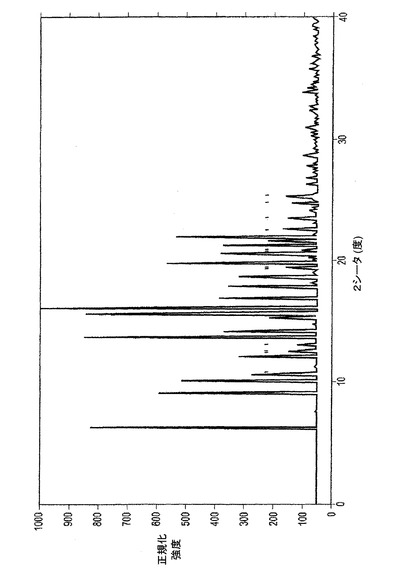
【図1−C】図1−Cは、イソプロピルアルコールより結晶化されたER−076349一水和物の粉末X線回折(XRPD)パターンである。
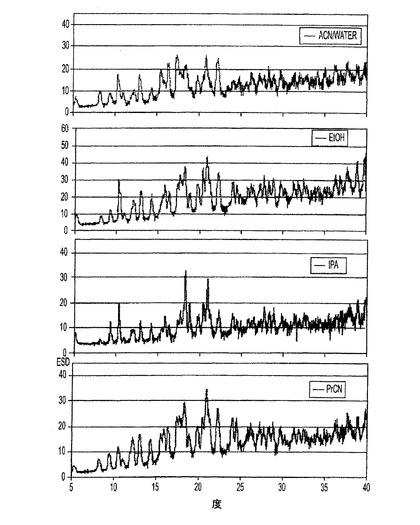
【図1−D】図1−Dは、さまざまな溶媒からのER−076349一水和物のXRPDパターンの描画である。
【図1−E】図1−Eは、エタノールより結晶化されたER−076349一水和物のDSCの描画である。
【図1−F】図1−Fは、イソプロパノールより結晶化されたER−076349一水和物のDSCの描画である。
【図1−G】図1−Gは、プロピオニトリルより結晶化されたER−076349一水和物のDSCの描画である。
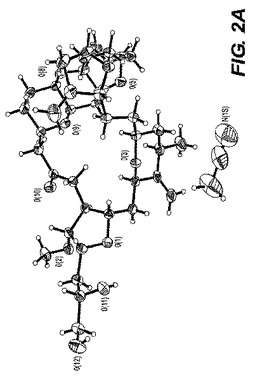
【図2−A】図2−Aは、ER−818906モノ−アセトニトリル溶媒和物の単結晶構造のORTEP図である。
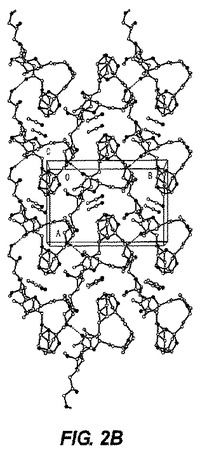
【図2−B】図2−Bは、ER−818906モノ−アセトニトリル溶媒和物のc軸に沿った結晶パッキング図である。
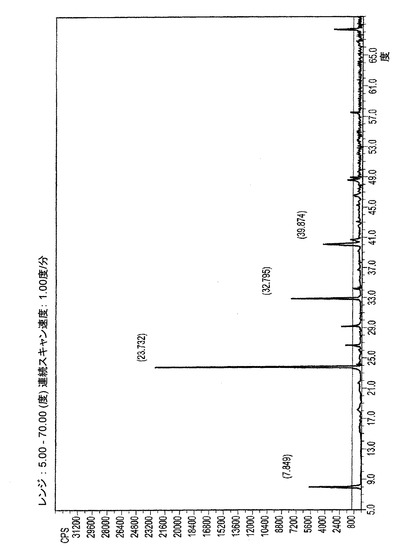
【図2−C】図2−Cは、ER−818906(epi−C20ジオール)モノ−アセトニトリル溶媒和物のXRPDパターンである。
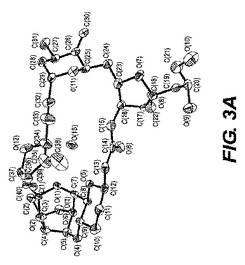
【図3−A】図3−Aは、ER−819531(epi−C23ジオール)一水和物の単結晶構造のORTEP図である。
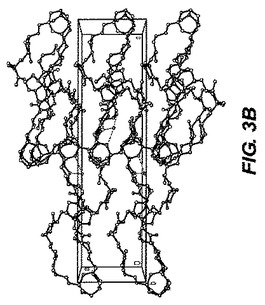
【図3−B】図3−Bは、ER−819531(epi−C23ジオール)一水和物のb軸に沿った結晶パッキング図である。
【図3−C】図3−Cは、ER−819531(epi−C23ジオール)のXRPDパターンである。
【図4−A】図4−Aは、ER−820057(epi−C23アミン)のXRPDパターンである。
【図5−A】図5−Aは、ER−111107(epi−C34ジオール)一水和物の単結晶構造のORTEP図である。
【図5−B】図5−Bは、ER−111107(epi−C34ジオール)一水和物のa軸に沿った結晶パッキング図である。
【図6−A】図6−Aは、ER−809681(エポキシド)の単結晶構造のORTEP図である。
【図6−B】図6−Bは、ER−809681(エポキシド)のb軸に沿った結晶パッキング図である。
【図6−C】図6−Cは、ER−809681(エポキシド)のXRPDパターンである。
【図6−D】図6−Dは、ER−809681(エポキシド)の理論上のXRPDパターンである。
【発明を実施するための形態】
【0007】
米国特許第6,214,865号に記載されるように、ハリコンドリンB類似体、ER−086526(化合物B−1939として認識される)は強力な抗癌剤である。ER−086526は、中間体であるジオール化合物、ER−076349(化合物B−1793として認識される)より調製される。米国特許第6,214,865号は、ER−076349(B−1793)の調製ならびに、いくつかの他のハリコンドリンB類似体の調製における使用について記載する。
【化2】
【化3】
【0008】
ER−076349を介する、ER−086526への代替的な合成経路は、特許協力条約に基づく国際出願公開2005/118565に記載される。国際出願公開2005/118565によると、ER−076349は、ER−086526へと、以下のように転換される:
【化4】
【0009】
国際出願公開2005/118565の方法において、以下の図に示されるように、ER−086526を生成するまでに、ER−076349はその場でトシラート、ER−082892を生成するために反応し、それはそののち、その場でエポキシド、ER−809681へと転換される。
【化5】
【0010】
ER−076349はさまざまな溶媒により一水和物として結晶化する。エポキシド、ER−809681はまた、結晶化合物として単離し得る。結晶のER−076349一水和物(実施例1)および結晶のER−809681(実施例6)は、本発明の別の実施態様を表わす。
【0011】
米国特許第6,214,865号および国際特許出願公開2005/118565に記載されるもののような多段階の合成において、中間体が調製され、望まない副産物もしくは不純物はより早いステップより持ち越される。しばしば、ろ過、分離および/もしくは精製ステップは望まない副産物もしくは不純物を除去するのに導入される。そのようなステップを組み入れることはコストの増大させるだけでなく、全体の合成の収率を下げることにもなる。多段階合成において結晶中間体を得ることはこれらの問題に対処し得る。結晶中間体は、高純度の中間体が他の精製ステップの必要性を減少させ得るし、そして合成プロセスのコストを減少させ得るといういくつかの利点をもたらす。ER−086526の調製において、高純度中間体として結晶体ER−076349一水和物を得る事は、そのC25エピマーからの分離ができるようにする。バルクの結晶体ER−076349において、C25エピマーの量は一般的に0.1重量%である。
【0012】
本発明は、いくつかER−076349のジアステレオマーの結晶体に関する。一つもしくはそれ以上のジアステレオマーは、同一のキラル炭素原子を持つ化合物の多段階の合成を占める共通の不純物である。さまざまなジアステレオマーを分析する事は、所望の化合物の調製において発生する、もしくは発生し得るエピマーの不純物を同定するために、有用である。エピマーの不純物を同定する事で、不純物を減少させ、避け、定量し、もしくは除去するための方法の改善を導くことができる。さまざまなエピマーの結晶体は、特定のエピマーの詳細な分析およびその合成における同定をもたらす。さらに、特定のエピマーの結晶化は、その除去の手段であり得、そしてそれにより所望の化合物を精製し得る。ER−076349のエピマーの結晶体は、ER−086526の合成におけるそれらの必要性に対応する。ER−076249に関係する以下の化合物:ER−809681;ER−820057;ER−818906;ER−819531;およびER−111187の結晶体は、本発明のさらなる実施態様を表わし、これらのすべては抗癌活性を示す。本開示のこの目的のために、化合物と化合物の結晶体とのこれらの群は集合的にハリコンドリンBと呼ばれ、そして個別にはハリコンドリンB類似体と呼ばれる。
【0013】
本発明によるハリコンドリンB類似体の治療と組み合わせて使用できる数多くの種類の抗癌のアプローチが存在する。それらは、例えば、化学療法剤(下記参照)、生物剤(例えば、ホルモン剤、サイトカイン(例えば、インターロイキン、インターフェロン、顆粒球コロニー刺激因子(G−CSF)、マクロファージコロニー刺激因子(GM−CSF)、ケモカイン、ワクチン抗原および抗体)、抗血管新生剤(例えば、アンジオスタチンおよびエンドスタチン)、放射線療法ならびに外科手術を含む。
【0014】
当業者によって判定し得るように、当分野においてこれらのアプローチが使用されると知られているのと同じ種類の癌と同様に他のものを治療するために、本発明の方法を使用できる。さらにこれらのアプローチは、その使用において当分野において知られるものと同様のパラメータ(例えば投薬計画および投与量)に従って実施し得る。しかしながら、当分野に知られるように、これらのアプローチと併用する追加的なハリコンドリンB類似体の使用のために、これらのパラメータを補正する事が望ましいであろう。当業者が判定し得るように、例えば、薬剤が通常で単独の治療剤として投与されるとしたら、本発明によるハリコンドリンB類似体と組み合わせられるとき、薬剤の投与量を減らすことが望ましいであろう。本発明の方法ならびにそれらの方法において使用できる組成物の例は、以下に示される。
【0015】
とりわけ、例えば、代謝拮抗物質、抗生剤、アルキル化剤、植物性アルカロイド、ホルモン剤、抗凝固剤、抗血栓剤、および他の天然性剤を含むいくつかの異なる種類の化学療法剤を、本発明によるハリコンドリンB治療と組み合わせて使用できる。これらの類の薬剤およびそれらの使用によって治療できる癌の具体的な非限定的例は、以下に示される。
【0016】
ハリコンドリンB類似体と併用できる代謝拮抗剤は、例えば、メトレキサート、プリン代謝拮抗薬(例えば、メルカプトプリン、チオグアニン、リン酸フルダラビン、クラドリビン、およびペントスタチン)、ならびにピリミジン拮抗薬(例えば、ゲムシタビン、カペシタビン、フルオロウラシル(例えば5−FU)、シタラビンおよびアザシチジン)を含む。特定の種類の癌の治療でのこれらの薬剤の使用は、当分野で周知であり、これらの薬剤はハリコンドリンB類似体と組み合わせて、これらの種類の癌および他の種類の癌の治療に使用できる。具体的な非限定的例として、ハリコンドリンB類似体は、非小細胞肺癌、膵臓癌もしくは転移性乳癌の治療においてゲムシタビンと併用できる。さらなる例において、ハリコンドリンB類似体は、乳癌もしくは大腸癌においてカペシタビンと併用できる。
【0017】
上述のように、ハリコンドリンB類似体と併用できる他の類の化学療法剤は、抗癌抗生剤を含む。これらは、例えば、アントラサイクリン(例えばドキソルビシン、エピルビシン、ダウノルビシンおよびイダルビシン)、アドリアマイシン、ダクチノマイシン、イダルビンシン(idarubincin)、プリカマイシン、マイトマイシンならびにブレオマイシンを含む。上述の薬剤と同様、特定の種類の癌の治療におけるこれらの薬剤の使用は当分野に周知であり、これらは、これらの種類の癌および他の種類の癌の治療のためにハリコンドリンB類似体と併用できる。具体的な非限定例として、ドキソルビシンのようなアントラサイクリンは、乳癌もしくは膵臓癌の治療のためにハリコンドリンB療法と併用できる。代替的に、第三の薬剤、シクロホスファミドもこの方法において使用できる。
【0018】
アルキル化剤は、本発明によるハリコンドリンB類似体と組み合わせて投与できる他の類の化学療法剤からなる。そのような薬剤の例は、プロカルバジン、ダカルバジン、アルトレタミン、シスプラチン、カルボプラチンおよびニトロソウレアを含む。ハリコンドリンB類似体は、これらの薬剤を治療するのに使用すると当分野で知られている癌の治療、ならびに他の癌の治療においてこれらの薬剤を併用できる。例えば、ハリコンドリンB類似体は、非小細胞肺癌もしくは卵巣癌の治療においてカルボプラチンと併用できる。
【0019】
本発明によるハリコンドリンB類似体と併用できるさらなる種類の化学療法剤は、ビンブラスチン、ビンクリスチン、エトポシド、テニポシド、トポテカン、イリノテカン、パクリタキセルおよびドセタキセルのような植物アルカロイドである。具体的な非限定例として、ハリコンドリンB類似体は結腸直腸癌の治療において、イリノテカンと併用でき、あるいは卵巣癌もしくは非小細胞肺癌の治療においてトポテカンと併用できる。
【0020】
本発明によるハリコンドリンB類似体療法と併用できるさらなる種類の抗癌剤は、抗凝固剤および抗血栓剤である。例えば、ヘパリン(例えば低分子量ヘパリンもしくはヘパラン硫酸)もしくはワルファリンを使用できる。治療中の患者における、例えば注射もしくは経口によるこれらの薬剤の使用は当分野に周知であり、そして当業者によって本発明において使用するよう容易に適応できる。
【0021】
抗癌剤を投与する数多くのアプローチは当分野に公知であり、本発明において使用するように容易に適応できる。ハリコンドリンB類似体と併用して投与できる一つもしくはそれ以上の薬剤の場合、例えば、総合的な治療の投薬計画の一部として、その薬剤を一つの組成物として一緒に、もしくは別々に投与できる。全身投与のために、その薬剤を例えば、静脈内注射(持続的もしくはボーラス)によって投与できる。そのような投与の適切なスケジュールや投与量は、たとえば動物における前臨床試験およびヒトにおける臨床試験(例えばフェーズI試験)に基づいて、当業者によって容易に決定できる。さらに、当分野に周知のように、同様の薬剤を用いる治療の分析、ならびに、患者における血球数(例えば好中球および血小板)および生体信号のような要素を観察することも利用できる。
【0022】
化学療法剤を投与するのに使われる多くの投薬計画は、例えば、薬剤の静脈内注射、ならびに患者が治療の任意の副作用から復帰する期間を置いて(例えば1〜4週間)後に続くこの治療の繰り返しを含む。それぞれの投与において療法の薬剤を用いること、もしくは代替的に一つの薬剤のみ(もしくは薬剤の一部分)を含む地医療を施すことが望ましいであろう。
【0023】
本発明に含まれる治療の投薬計画の具体的な非限定的例として、ハリコンドリンB類似体(例えば、0.01〜5mg/m2)を患者へと0.5〜3時間で静脈内注射によって投与でき、そして他の薬剤(例えばゲムシタビンを例えば500〜900mg/m2)の0.5〜3時間かけての静脈内注射が後に続く。当業者によって許容できるか効果的であるか決定されるに従って、この治療のコースを2〜3週ごとに繰り返す事ができる。この方法の変法として、上述のように最初の日に両方の薬剤によって治療を実施し、しかしながらそのあと、次の週に第二の薬剤(例えばゲムシタビン)のみを用いての治療が後に続くようにできる。
【0024】
さらに、当分野によく知られているように、本発明の方法を用いる治療は、癌化学療法剤の共通の副作用である吐き気および嘔吐を軽減するのに用いられる抗嘔吐剤の投与と組み合わせて実施できることができる。そのような薬剤の例は強トランキライザ(例えば、クロルプロマジンおよびプロクロルペラジンのようなフェノチアジン)、ドーパミン拮抗剤(例えば、メトクロプラミド)、セレトニン拮抗剤(例えばオンダンセトロンおよびグラニセトロン)、カンナビノイド(例えばドロナビノール)、ならびにベンゾジアゼピン系鎮静剤を含む。
【0025】
上述の癌に加えて、本発明の方法及び組成物を、以下の種類の癌ならびに他のものの治療に用いる事ができる:皮膚(例えば、扁平上皮癌、基底細胞癌もしくはメラノーマ)、前立腺、脳および神経、頭頸部、精巣、肺、肝臓(例えば肝癌)、腎臓、膀胱、胃腸、骨、内分泌系(例えば、甲状腺および下垂体腫瘍)、ならびにリンパ系(例えばホジキンリンパ腫および非ホジキンリンパ腫)の癌。本発明の方法を用いて治療できる他の種類の癌は繊維肉種、神経外胚葉性腫瘍、中皮腫、類表皮癌、およびカポジ肉腫を含む。
【0026】
本発明はまた、ハリコンドリンB類似体を、上述の任意の薬剤のようなさらなる治療薬剤と組み合わせて含む組成物をまた含む。それらの組成物における薬剤は、好ましくは患者へと投与できるように調合されている(例えば生理食塩水中に)か、あるいは代替的に投与の前にさらなる加工を必要とする形状であっても良い。例えば、組成物は凍結乾燥状態において、もしくは、希釈を必要とする濃縮状態において、薬剤を含む事ができる。化学療法において使用するための薬剤の調合は当分野における標準的な方法(例えば、Remington’s Pharmaceutical Science(第18版)、A.Gennaro編、1990、Mack Publishing Co., Eaton,Paを参照)を用いて実施できる。
【実施例】
【0027】
単結晶X線回析データ集積および構造解析
データはOxford Cryostream低温装置を備えたBruker SMART APEX CCD(電荷結合素子)系の回析計を用いて絶対温度193度で集積された。データは半球が集積されるように30秒間、毎フレーム0.3°のオメガスキャンを用いて測定された。0.76Åの最大解像度で、全部で1271フレームが集積された。最初の50フレームは、減衰を監視するためにデータ集積の最後に再集積された。セル(cell)パラメータはSMARTソフトウェア(SMART V5.625 (NT)CCD検出系のためのソフトウェア;Bruker Analytical X−ray System、Madison、WI(2001))を用いて抽出された。データ整理は、Lpおよび減衰を補正するSAINTソフトウェア(SAINT V6.22(NT)CCD検出系のためのソフトウェア;Bruker Analytical X−ray System、Madison、WI(2001))を用いて行われた。構造は、SHELXTL−PC V6.10(SHELXTL 6.1(PC版)、構造解析および分子グラフィックスのためのプログラムライブラリ;Bruker Analytical X−ray System、Madison、WI(2000))に含まれるSHELXS−97プログラム(Sheldric,G.M.SHELXS−90、結晶構造解析のためのプログラム、ゲッチンゲン大学、ドイツ、1990)を用いる直接法によって解析された。
【0028】
粉末X線解析(XRPD)データ集積手順
少量の結晶化物質が下記のXRPD回析試験のために用いられた。物質は粉砕されておらず、選択配向効果を有するかもしれない。Scintag Diffractometer上の水晶板を用いる。データは、銅放射線を用いて、5〜70度の2シータレンジで通常の粉末回析条件によって取得された。バックグランド補正は実施されなかった。表1はデータ取得パラメータを列挙する。
【0029】
【表1】
【0030】
実施例1:結晶ER−076349一水和物の解析
エタノール、イソプロパノール(IPA)、プロピオニトリルのXRPDパターンはアセトニトリル/水サンプルと同一である。一水和物としての解析はこの比較に基づく。アセトニトリル/水のX線ユニットセルの結晶は単一の水分子の存在を示した。
【0031】
A.ER−076349一水和物の結晶化の手順
エタノール、イソプロパノールおよびプロピオニトリルからのER−076349の結晶化(XRPDサンプル):ER−076349は溶媒(5倍容量)に溶解された。溶液は室温において蒸発され、XRPD試験に適した結晶は16から24時間かけて形成された。
【0032】
アセトニトリル/水溶液からのER−076349の結晶化(XRPDサンプル):ER−076349はアセトニトリル(1倍容量)に溶解された。この溶液に水/アセトニトリル溶液(10倍容量、2部の水、1部のアセトニトリル)がゆっくりと添加された;もし溶液が濁るときは、最小量のアセトニトリルを添加してもよい。溶液はER−076349(0.004重量)を種子として入れられた。混合物は室温で攪拌され、溶液は不活性ガス流下において、アセトニトリルが5%以下になるまで蒸発された。固形物は水(5倍容量)の中でスラリー化され、混合物はろ過された。ろ過された固形物は高真空および不活性ガス流下で、12から24時間乾燥された。
【0033】
アセトニトリル/水溶液からのER−076349の結晶化(単結晶):ER−076349はアセトニトリル(5倍容量)に溶解された。水(0.5倍容量)がそののち添加された。コンテナは室温に保管され、4週間のゆっくりとした溶液の蒸発が発散され、X線構造解析に適した単結晶がゆっくりと形成された。
【0034】
B.ER−076349一水和物の単結晶X線解析
0.10×0.10×0.04mmの寸法の無色のプレート結晶が非常に少量のパラトン油(paratone oil)を用いて0.2nmのナイロンループ上に取り付けられた。図1−Aに示される単結晶構造は上述の手順を用いて測定された。表2は、結晶データおよびER−076349一水和物の単結晶構造を測定するのに用いられた構造精密化パラメータを列挙する。構造は消滅則の分析によってスペースグループP212121(#19)中で解析された。全ての非水素原子は異方的に精密化された。水素は幾何学的方法によって計算され、ライディングモデル(riding model)として精密化された。回析試験に用いられる化粧はデータ集積の間、分解を示さなかった。全ての描写は50%楕円において実施された。図1−BはER−076349一水和物のb軸に沿った結晶パッキング図であり、結晶中の水素結合、点線の最適の図を示す。
【0035】
【表2】
【0036】
C.ER−076349一水和物の粉末X線回析による解析
図1−Cは、上述のようにイソプロピルアルコールから結晶化されたER−076349一水和物のXRPD図である。表3AはXRPD図中の相対強度のピークのリストを示す。
【0037】
【表3A】
【0038】
図1−dは、アセトニトリルおよび水、エタノール、イソプロパノール、ならびにプロピオニトリルを含むさまざまな再結晶化溶媒より得られたER−076349一水和物の個別のXRPDのプロットをまとめたものである。上述のものと同じパターン取得の方法が用いられた。
【0039】
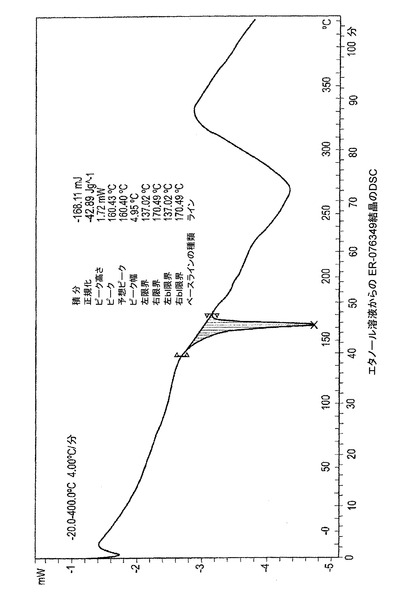
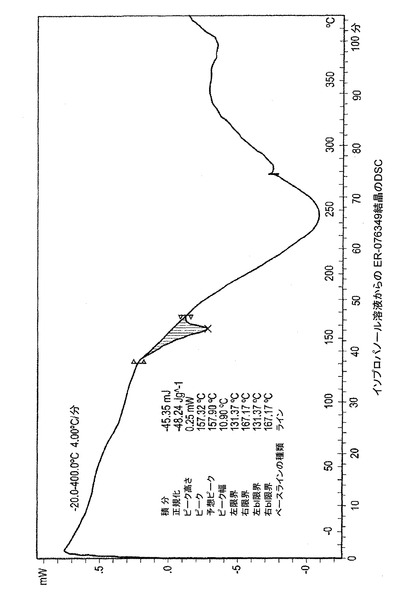
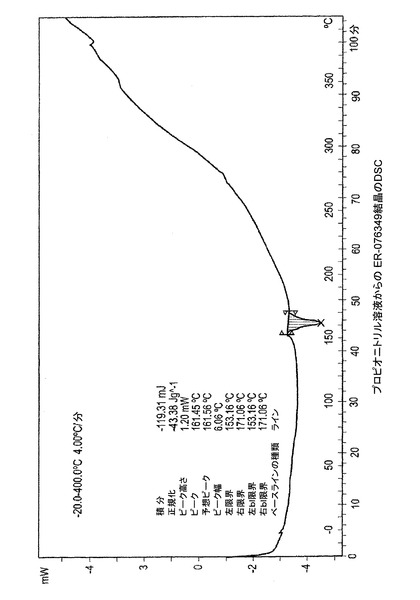
図1−E、1−Fおよび1−Gは、ER−076349一水和物の示差走査熱量計(DSC)のサーモグラムを描写する。図1−Eはエタノール溶液より結晶化されたER−076349のDSCのプロットを描写する。DSCは160℃において吸熱ピークを示す。図1−Fはイソプロパノール溶液から結晶化されたER−076349のDSCのプロットを描写する。DSCは157℃において吸熱ピークを示す。図1−Gはプロピオニトリル溶液より結晶化されたER−076349のDSCのプロットを描写する。DSCは161℃において吸熱ピークを示す。
【0040】
実施例2:ER−818906(epi−C20ジオール)、モノ−アセトニトリル溶媒和物
A.X線およびXRPD結晶サンプルの生成のための結晶化手順
E0818906(1重量)はアセトニトリル(10倍容量)に懸濁され、完全に溶解するのを助けるためゆるやかに加熱された。溶液は室温に置かれ、30分後に結晶成長が始まった。X線構造解析にふさわしい単結晶サンプルは16から24時間後に生成した。ER−818906(epi−C20ジオール)の化学構造は以下に示される。
【化6】
【0041】
B.ER−818906(epi−C20ジオール)モノ−アセトニトリル溶媒和物の単結晶X線解析
0.12×0.12×0.10mmの寸法の無色のブロック結晶は非常に少量のパラトン油を用いて0.2nmのナイロンループ上に取り付けられた。図2−Aに示される単結晶構造は上述の手順を用いて測定された。表3Bは結晶データおよびER−818906モノ−アセトニトリル溶媒和物の単結晶構造を測定するのに用いられた構造精密化パラメータを列挙する。構造はスペースグループP21(#4)において解析された。全ての非水素原子は異方的に精密化された。水素は幾何学的方法によって計算され、ライディングモデルとして精密化された。フラック(Flack)パラメータは結晶を分析するのに用いられた。もしフラックパラメータが0に近い時、構造要素精密化によって与えられる全体の構造がほぼ正しく;その値が1.0に近いとき、倒置構造(他の光学異生体)がほぼ正しく;もしその値が0.5に近い時、結晶はほぼラセミ体である。Flack、H.D.Acta Cryst. A39.1983、876−881を参照。フラックパラメータは0.3(4)へと精密化された。この化合物のキラリティはその合成の源により確かめられた。回析試験において用いられる結晶はデータ集積の間、分解を示さなかった。全ての描写は50%楕円において実施された。図2−BはER−818906モノ−アセトニトリル溶媒和物のc軸に沿った結晶パッキング図である。
【0042】
【表3B】
【0043】
C.ER−818906(epi−C20ジオール)モノ−アセトニトリル溶媒和物の粉末X線回析(XRPD)による解析
実施例2Aにおける単結晶試験のサンプルを取り出したのち、少量の物質が残った。XRPDパターンは上述のように取得された。XRPDパターンは図2−Cに示される。表4はXRPD中の相対強度のピークのリストを示す。
【0044】
【表4】
【0045】
実施例3:ER−819531(epi−C23ジオール)一水和物の解析
A.XRPD品質サンプルのための結晶化手順
ER−819531(1重量)はアセトニトリル(10倍容量)に室温で溶解され、温度は−20℃まで低下し、この温度は16時間維持された。粉末XRPD試験に適した結晶のクラスターは16時間後に生成した。ER−819531(epi−C23ジオール)の化学構造は下に示される。
【化7】
【0046】
B.ER−819531(epi−C23ジオール)一水和物の単結晶X線解析
ER−819531サンプルの一部はアセトニトリル、数滴のトルエン、および1滴の水の混合物中に溶解された。これはのちに少量のバイアル中でゆっくりと蒸発された。これは単結晶を生じ、その一つはこの試験に用いられるよう選択された。0.12×0.12×0.04mmの寸法の無色のブロック結晶は非常に少量のパラトン油を用いてナイロンループ上に組み込まれた。図3−Aに示される、この単結晶構造は上述の手順を用いて解析された。表5は結晶データおよびER−819531一水和物の単結晶構造を測定するのに用いられた構造精密化パラメータを列挙する。構造は消滅則の分析によってスペースグループP212121(#19)中で解析された。全ての非水素原子は異方的に精密化された。水素は幾何学的方法によって計算され、ライディングモデルとして精密化された。フラックパラメータは結晶を分析するのに用いられた。もしフラックパラメータが0に近い時、構造要素精密化によって与えられる全体の構造がほぼ正しく;その値が1.0に近いとき、倒置構造(他の光学異生体)がほぼ正しく;もしその値が0.5に近い時、結晶はほぼラセミ体である。Flack、H.D.Acta Cryst. A39.1983、876−881を参照。フラックパラメータは0.50(16)へと精密化された。この化合物のキラリティはその合成の源により確かめられた。回析試験において用いられる結晶はデータ集積の間、分解を示さなかった。全ての描写は50%楕円において実施された。図3−Bはb軸に沿った結晶パッキング図であり、それは結晶中の水素結合を点線で示す。
【0047】
【表5】
【0048】
C.ER−819531(epi−C23ジオール)一水和物の粉末X線回析(XRPD)による解析
ER−819531一水和物は上述のように得られた。XRPDパターンは図3−Cに示される。表6はXRPDパターン中の相対強度のピークのリストを示す。
【0049】
【表6】
【0050】
実施例4:ER−820057(epi−C23アミン)の解析
A.ER−820057結晶サンプルの調製
ER820057はアセトニトリル(10倍容量)中に室温で溶解され、溶液は室温に置かれた。結晶成長は2時間後に観察された。粉末XRPD試験にふさわしい結晶サンプルは16から24時間後に単離された。ER−820057(epi−C23アミン)の化学構造は以下に示される。
【化8】
【0051】
B.ER−820057(epi−C23アミン)の粉末X線回析(XRPD)による解析
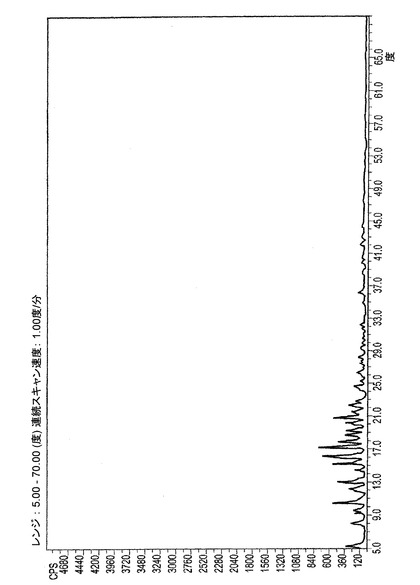
ER−820057のXRPDは上述のように得られた。XRPDパターンは図4−Aに示される。表7はXRPDパターンの相対強度のピークのリストを示す。
【0052】
【表7】
【0053】
実施例5:ER−111197(epi−C34ジオール)一水和物の解析
A.X線品質の結晶の生成のための結晶化手順
ER−111197(1重量)はアセトニトリル(8倍容量)に溶解された。水(0.8倍容量)が添加された。溶液は室温で2〜3日間蒸発するよう放置された。結晶が生成しない場合、溶液は乾燥するまで蒸発され、この手順が結晶が出来るまで繰り返された。ER−111197(epi−C34ジオール)の化学構造は以下に示される。
【化9】
【0054】
B.R−111197(epi−C34ジオール)一水和物の単結晶X線解析
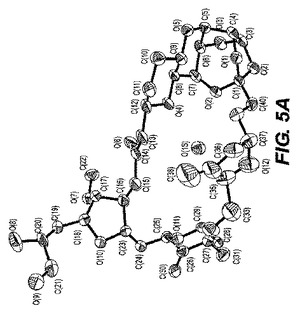
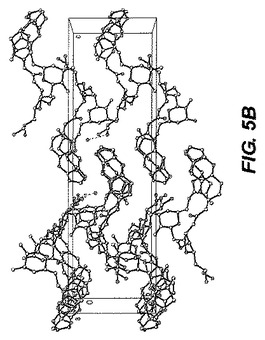
0.22×0.14×0.04mmの寸法の無色の針状結晶が、非常に少量のパラトン油を用いてナイロンループに組み込まれた。図5−Aに示される単結晶構造は、上述の手順を用いて解析された。表7Bは結晶データおよびER−111197一水和物の単結晶構造を測定するのに用いられた構造精密化パラメータを列挙する。構造は消滅則の分析によってスペースグループP212121(#19)中で解析された。全ての非水素原子は異方的に精密化された。水素は幾何学的方法によって計算され、ライディングモデルとして精密化された。フラックパラメータは結晶を分析するのに用いられた。もしフラックパラメータが0に近い時、構造要素精密化によって与えられる全体の構造がほぼ正しく;その値が1.0に近いとき、倒置構造(他の光学異生体)がほぼ正しく;もしその値が0.5に近い時、結晶はほぼラセミ体である。Flack、H.D.Acta Cryst. A39.1983、876−881を参照。フラックパラメータは0.3(13)へと精密化された。この化合物のキラリティはその合成の源により確かめられた。回析試験において用いられる結晶はデータ集積の間、分解を示さなかった。全ての描写は50%楕円において実施された。図5−Bはa軸に沿った結晶パッキング図であり、それは結晶中の水素結合を点線で示す。
【0055】
【表7B】
【0056】
C.ER−111197(epi−C34ジオール)一水和物のシミュレートされた粉末X線解析スペクトル
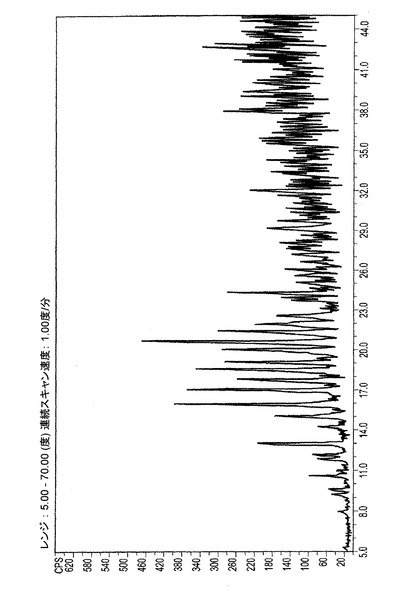
単結晶のデータから生成されたER−111197のシミュレートされたXRPDスペクトルは図5−Cに示される。
【0057】
実施例6:ER−809681(エポキシド)
A.X線品質の結晶を生成するための結晶化手順
ER−809681−00(50mg)はジクロロメタン(0.25ml)に溶解された。ペンタン(0.5ml)が添加された。溶液は視覚的に均一になるまで攪拌された。溶液は室温で18時間、ゆっくりと蒸発された。X線結晶学的試験にふさわしい結晶は上清の液体をデカントすることによって集められた。ER−809681(エポキシド)の化学構造は下に示される。
【化10】
【0058】
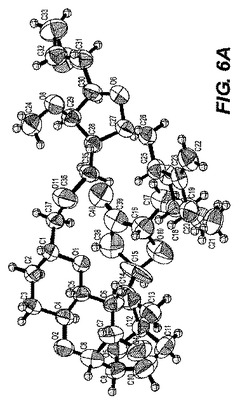
B.ER−809681(エポキシド)の単結晶X線解析
X線解析方法は上述のものから変更され、以下の節に詳細に説明される。
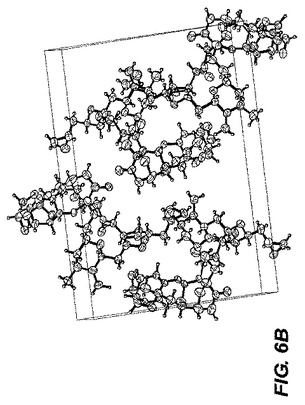
0.30×0.10×0.10mmのおおよその寸法を持つC40H56O11の無色のブロック結晶はガラス線維上に組み込まれた。単結晶は図6−Aに示される。すべての解析は、グラファイトで単色化されたCu−Kα放射線を持つRigaku RAXIS RAPID imaging plate area detector上で行われた。指数化は15秒の曝露による3つの振動より実施された。結晶と検出器の距離は127.40mmであった。寸法を持つ一次斜方晶系のセルに対応するデータ集積のためのセル定数および配向マトリックス:a=9.4700(3)Å;b=17.6327(5)Å;c=23.1790(6)Å;V=3879.47(19)Å3。Z=4でかつF.W.=712.88の時、計算上の密度は1.223g/cm3である。h00:h±2n;0k0:k±2n;001:1±2nの消滅則は特異的にスペースグループをp212121(#19)と定める。結晶パッケージ図は図6−Bに示される。
【0059】
データは23±1℃において136.5°の最大2θ値で集積された。全体で30振幅の画像が集積された。χ=54.0°かつφ=15.0°において80.0から260.0°の30.0ステップのωスキャンを用いてデータのスイープ(sweep)が実施された。χ=54.0°かつφ=105.0°において80.0から260.0°の30.0ステップのωスキャンを用いて2度目のスイープが実施された。照射線量率は15.0[秒/°]であった。χ=54.0°かつφ=270.0°において80.0から260.0°の30.0ステップのωスキャンを用いて他のスイープが実施された。照射線量率は15.0[秒/°]であった。χ=0.0°かつφ=0.0°において80.0から260.0°の30.0ステップのωスキャンを用いて他のスイープが実施された。照射線量率は15.0[秒/°]であった。結晶と検出器の距離は127.40mmであった。読み出しは0.100mmピクセルモードで実施された。
【0060】
データ解析:集積された16412の反射のうち、934がユニークであった(Rint=0.043);同等の反射は融合された。Cu−Kα放射の線吸収係数、μは7.229cm−1であった。0.748から0.930の範囲の透過率が生じる実験的な吸収の補正が適応された。データはローレンツ偏光効果のために補正された。
【0061】
構造解析と精密化:構造は直接方を用いて解析され(SHELX97:Sheldric,G.M.(1997)、フーリエ法を用いて拡張された。DIRDIF99:Beurskens,P.T.,Admiraal,G.,Beurskens,G.,Bosman,W.P.,de Gelder,R.,Israel,R. and Smits,J.M.M.(1999)、”The DIRDIF−99プログラムシステム”、Technical Report of the Crystallography Laboratory,”University of Nijmegen, The Netherland.を参照。非水素原子は異方的に精密化された。水素原子はライディングモデルを用いて精密化された。Σw(Fo2−Fc2)2(式中、w=最小二乗重量)で表わされるF2のフルマトリックス最小二乗精密化の最後のサイクルは、7045の観察された反射と461の変数パラメータに基づく。それは、R1=Σ||Fo|−|Fc||/Σ|Fo|=0.0630かつwR2=[Σ(w(Fo2−Fc2)2)/Σw(Fo2)2]1/2=0.2168という計量されてないアグリーメント・ファクタ(agreement factor)および計量されたアグリーメント・ファクタに収斂する。
【0062】
単位重量の観測の標準偏差は1.10であり、[Σw(Fo2−Fc2)2/(No−Nv)]1/2(式中、No=観察の数およびNv=変数の数)として計算される理論上の単位重量の標準偏差はである。最終の差フーリエ図上の最大ピークおよび最小ピークは、それぞれ0.20および−0.28e−/Å3に対応する。
【0063】
中性子散乱因子は、CromerとWaber,”International Tables for X−ray Crystallography”Vol.IV,The Kynoch Press, Birmingham,England,表2.2A(1974)より得た。異常分散効果はFcalc中に含まれ(Ibers,J.A.およびHamilton,W.C.;Acta Crystallogr.,17,781(1964)を参照)、ΔfおよびΔf’’の値はCreagh,D.C.およびMcAuley,W.J.” International Tables for Crystallography”Vol.C,(A.J.C.Wilson編)、Kluwer Academic Publishers, Boston,表4.2.6.8、219〜222ページ(1992)より得られた。質量減衰係数の値はCreagh,D.C.およびHubbell,J.H.;” International Tables for Crystallography”Vol.C,(A.J.C.Wilson編)、Kluwer Academic Publishers, Boston,表4.2.4.3、200〜206ページ(1992)より得られた。すべての計算はCrystalStructure結晶学ソフトウェアパッケージを用いて実施された。CrystalStructure3.7.0:Crystal Structure Analysis Package,Rigaku and Rigaku/MSC(2000−2005)、9009 New Trails Dr. The Woodlands TX77381米国;ならびにCRYSTALS Issure 10:Watkin, D.J.,Prout,C.K. Carruthers, J.R.およびBetteridge, P.W.、Chemical Crystallography Laboratory,Oxford,英国(1996)を参照。これの例外は精密化の計算であり、SHELX97:Sheldrick,G.M.(1997)を用いて実施された。
【0064】
【表7c】
【表7d】
【表7e】
【0065】
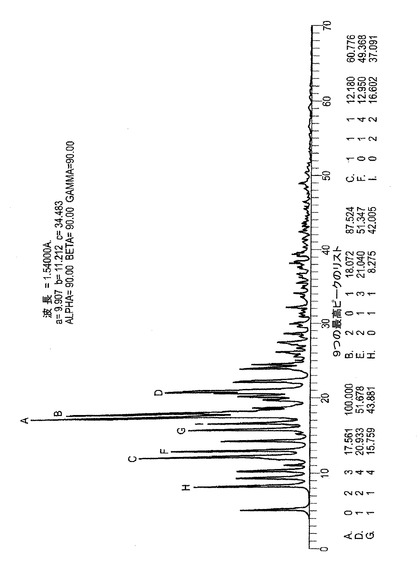
C.ER−809681(エポキシド)の粉末X線解析(XRPD)による解析
この化合b通のXRPD集積方法は上述のものとデータスキャンレンジが2−70°である点でのみ異なる。XRPDパターンは図6−Cに示される。
【0066】
【表8】
【0067】
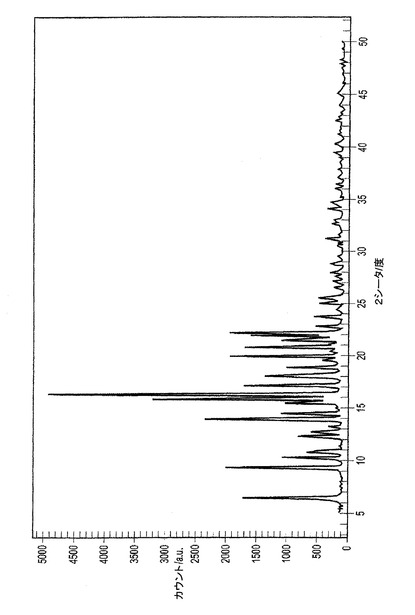
D.シミュレートされたXRPDスペクトル
単結晶データより生成されるシミュレートされたXRPDパターンは図6Dに示される。
【0068】
上述の化合物の、上述の結晶形における抗癌活性は、LiuJ.らのAnticancer Research、27:1509−1511(2007)に記載されるような細胞増殖阻害試験を用いて測定された。IC50nMの単位での活性データは下の表9に記載される。MES−SAおよびMES−SA/Dx5−Rx1子宮肉腫はAmerican Type Culture Collectionより入手し、推奨される条件で増殖された。
【0069】
細胞増殖阻害試験
細胞は96穴プレートに10%FBSならびにペニシリン、ストレプトマイシンおよびL−グルタミンを補った推奨される培地中に7.5×103細胞/ウェルで播種された。4時間の培養の後、試験化合物はそれぞれのウェルに0から10μMの範囲の一連の濃度になるように添加された。細胞は4日間37℃で培養された。細胞増殖はメチレンブルーに基づいたマイクロ培養試験(Towle M.J.らの”In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B.”Cancer Res 61:1013−1021、2001を参照)の修正したもの(FinlayG.J.らの”A semiautomated microculture method for investigating growth inhibitory effects of cytotoxic compounds on exponentially growing carcinoma cells”,Anal Biochem 139:272−277、1984を参照)を用いて測定された。培地は除去され細胞は100μlのメチレンブルー(500μg/ml)で45分間染色された。水で染色したあと、線食された細胞は100μlのサルコシン(1mg/ml)に溶解され、90分間緩やかに攪拌された。このプレートはA600−A405で観測された。
【0070】
P−グリコプロテイン介在多薬剤耐性(MDR)に対する化合物の感受性
一対の子宮肉種の細胞株が用いられた:MES−SA(MDRのない親細胞株)、DX5−Rx1(長期間ドキソルビシンへと曝露されたMES−SA由来の細胞株)。亜株はP−グリコプロテインを高発現している。療法の細胞株は10%FBSならびにペニシリン、ストレプトマイシンおよびL−グルタミンを補ったMcCoy’s5A中に7.5×103細胞/ウェルで播種され、細胞増殖が上述のように測定された。親株および耐性細胞株において取得されたIC50値の比は耐性の倍数を意味し、それぞれ比較される二つの細胞株に対するP−グリコプロテイン介在MDRへの化合物の感受性の指標をもたらす。
【0071】
【表9】
【技術分野】
【0001】
関連出願
本出願は、2008年4月4日に出願された米国特許仮出願番号第61/042,643号および2008年4月11日に出願された米国特許仮出願番号61/071,111号の優先権を主張し、これらは、その全てが参照によりここに組み込まれる。
【0002】
発明の分野
本発明は薬剤活性を持つハリコンドリンB類似体に関する。ある実施態様において、この類似体は結晶体を持ち、そして/あるいは、強力な抗癌活性を持つ化合物であるハリコンドリンB類似体E7389の合成における合成中間体としてのさらなる有用性を持つ。
【化1】
【背景技術】
【0003】
背景技術
強力な抗癌剤であるハリコンドリンBは、カイメン、Halichondria okadaiより最初に単離された。それはまた、Axinella sp.、Phakellia carteriおよびLissondendryx sp.においても発見された。ハリコンドリンBは、インビトロ(in vitro)において、チューブリンの重合化、微小管重合、ベータ−チューブリン架橋、GTPおよびビンブラスチンのチューブリンへの結合、ならびにチューブリン依存性GTP加水分解を阻害する事を示した。それはまた、インビトロ(in vitro)およびインビボ(in vivo)において抗癌活性を示した。
【発明の概要】
【発明が解決しようとする課題】
【0004】
Aicherらは、1992年にハリコンドリンBの全合成を発表した(Aicher,T.らのJ.Am.Chem.Soc.114:3162−3164およびZheng,W.J.らのJ.Bioorg.Med.Chem.Lett.、2004、14、5551を参照)。ハリコンドリンBおよびさまざまな類似体の合成は、米国特許第6,214,865号、第6,365,759号、第6,469,182号および第6,653,341号;米国特許出願公開2004/0198806、2006/0104984:ならびに特許協力条約に基づく国際出願公開2005/118565に記載され、それらすべては、参照によりその全体がここに組み入れられる。
【課題を解決するための手段】
【0005】
本発明は薬剤活性を持つハリコンドリンB類似体に関する。類似体は結晶体を持ち、そして/あるいは強力な抗癌活性を持つ化合物であるハリコンドリンB類似体、E7389の合成における合成中間体としてのさらなる有用性を持つ。従って、本発明は、ER−076349一水和物の結晶体;ER−809681の結晶体;化合物ER−820057もしくはER−820057の結晶体;化合物ER−818906もしくはER−818906の結晶体;化合物ER−819531もしくはER−819531の結晶体;ならびに化合物ER−111197もしくはER−111197の結晶体に関する。本発明はまた、それらの化合物を含有する医薬組成物、その化合物を投与する事による癌の治療方法に関する。
【図面の簡単な説明】
【0006】
【図1−A】図1−Aは、ER−076349−00一水和物の単結晶構造のORTEP図である。
【図1−B】図1−Bは、ER−076349一水和物のb軸に沿った結晶パッキング図である。
【図1−C】図1−Cは、イソプロピルアルコールより結晶化されたER−076349一水和物の粉末X線回折(XRPD)パターンである。
【図1−D】図1−Dは、さまざまな溶媒からのER−076349一水和物のXRPDパターンの描画である。
【図1−E】図1−Eは、エタノールより結晶化されたER−076349一水和物のDSCの描画である。
【図1−F】図1−Fは、イソプロパノールより結晶化されたER−076349一水和物のDSCの描画である。
【図1−G】図1−Gは、プロピオニトリルより結晶化されたER−076349一水和物のDSCの描画である。
【図2−A】図2−Aは、ER−818906モノ−アセトニトリル溶媒和物の単結晶構造のORTEP図である。
【図2−B】図2−Bは、ER−818906モノ−アセトニトリル溶媒和物のc軸に沿った結晶パッキング図である。
【図2−C】図2−Cは、ER−818906(epi−C20ジオール)モノ−アセトニトリル溶媒和物のXRPDパターンである。
【図3−A】図3−Aは、ER−819531(epi−C23ジオール)一水和物の単結晶構造のORTEP図である。
【図3−B】図3−Bは、ER−819531(epi−C23ジオール)一水和物のb軸に沿った結晶パッキング図である。
【図3−C】図3−Cは、ER−819531(epi−C23ジオール)のXRPDパターンである。
【図4−A】図4−Aは、ER−820057(epi−C23アミン)のXRPDパターンである。
【図5−A】図5−Aは、ER−111107(epi−C34ジオール)一水和物の単結晶構造のORTEP図である。
【図5−B】図5−Bは、ER−111107(epi−C34ジオール)一水和物のa軸に沿った結晶パッキング図である。
【図6−A】図6−Aは、ER−809681(エポキシド)の単結晶構造のORTEP図である。
【図6−B】図6−Bは、ER−809681(エポキシド)のb軸に沿った結晶パッキング図である。
【図6−C】図6−Cは、ER−809681(エポキシド)のXRPDパターンである。
【図6−D】図6−Dは、ER−809681(エポキシド)の理論上のXRPDパターンである。
【発明を実施するための形態】
【0007】
米国特許第6,214,865号に記載されるように、ハリコンドリンB類似体、ER−086526(化合物B−1939として認識される)は強力な抗癌剤である。ER−086526は、中間体であるジオール化合物、ER−076349(化合物B−1793として認識される)より調製される。米国特許第6,214,865号は、ER−076349(B−1793)の調製ならびに、いくつかの他のハリコンドリンB類似体の調製における使用について記載する。
【化2】
【化3】
【0008】
ER−076349を介する、ER−086526への代替的な合成経路は、特許協力条約に基づく国際出願公開2005/118565に記載される。国際出願公開2005/118565によると、ER−076349は、ER−086526へと、以下のように転換される:
【化4】
【0009】
国際出願公開2005/118565の方法において、以下の図に示されるように、ER−086526を生成するまでに、ER−076349はその場でトシラート、ER−082892を生成するために反応し、それはそののち、その場でエポキシド、ER−809681へと転換される。
【化5】
【0010】
ER−076349はさまざまな溶媒により一水和物として結晶化する。エポキシド、ER−809681はまた、結晶化合物として単離し得る。結晶のER−076349一水和物(実施例1)および結晶のER−809681(実施例6)は、本発明の別の実施態様を表わす。
【0011】
米国特許第6,214,865号および国際特許出願公開2005/118565に記載されるもののような多段階の合成において、中間体が調製され、望まない副産物もしくは不純物はより早いステップより持ち越される。しばしば、ろ過、分離および/もしくは精製ステップは望まない副産物もしくは不純物を除去するのに導入される。そのようなステップを組み入れることはコストの増大させるだけでなく、全体の合成の収率を下げることにもなる。多段階合成において結晶中間体を得ることはこれらの問題に対処し得る。結晶中間体は、高純度の中間体が他の精製ステップの必要性を減少させ得るし、そして合成プロセスのコストを減少させ得るといういくつかの利点をもたらす。ER−086526の調製において、高純度中間体として結晶体ER−076349一水和物を得る事は、そのC25エピマーからの分離ができるようにする。バルクの結晶体ER−076349において、C25エピマーの量は一般的に0.1重量%である。
【0012】
本発明は、いくつかER−076349のジアステレオマーの結晶体に関する。一つもしくはそれ以上のジアステレオマーは、同一のキラル炭素原子を持つ化合物の多段階の合成を占める共通の不純物である。さまざまなジアステレオマーを分析する事は、所望の化合物の調製において発生する、もしくは発生し得るエピマーの不純物を同定するために、有用である。エピマーの不純物を同定する事で、不純物を減少させ、避け、定量し、もしくは除去するための方法の改善を導くことができる。さまざまなエピマーの結晶体は、特定のエピマーの詳細な分析およびその合成における同定をもたらす。さらに、特定のエピマーの結晶化は、その除去の手段であり得、そしてそれにより所望の化合物を精製し得る。ER−076349のエピマーの結晶体は、ER−086526の合成におけるそれらの必要性に対応する。ER−076249に関係する以下の化合物:ER−809681;ER−820057;ER−818906;ER−819531;およびER−111187の結晶体は、本発明のさらなる実施態様を表わし、これらのすべては抗癌活性を示す。本開示のこの目的のために、化合物と化合物の結晶体とのこれらの群は集合的にハリコンドリンBと呼ばれ、そして個別にはハリコンドリンB類似体と呼ばれる。
【0013】
本発明によるハリコンドリンB類似体の治療と組み合わせて使用できる数多くの種類の抗癌のアプローチが存在する。それらは、例えば、化学療法剤(下記参照)、生物剤(例えば、ホルモン剤、サイトカイン(例えば、インターロイキン、インターフェロン、顆粒球コロニー刺激因子(G−CSF)、マクロファージコロニー刺激因子(GM−CSF)、ケモカイン、ワクチン抗原および抗体)、抗血管新生剤(例えば、アンジオスタチンおよびエンドスタチン)、放射線療法ならびに外科手術を含む。
【0014】
当業者によって判定し得るように、当分野においてこれらのアプローチが使用されると知られているのと同じ種類の癌と同様に他のものを治療するために、本発明の方法を使用できる。さらにこれらのアプローチは、その使用において当分野において知られるものと同様のパラメータ(例えば投薬計画および投与量)に従って実施し得る。しかしながら、当分野に知られるように、これらのアプローチと併用する追加的なハリコンドリンB類似体の使用のために、これらのパラメータを補正する事が望ましいであろう。当業者が判定し得るように、例えば、薬剤が通常で単独の治療剤として投与されるとしたら、本発明によるハリコンドリンB類似体と組み合わせられるとき、薬剤の投与量を減らすことが望ましいであろう。本発明の方法ならびにそれらの方法において使用できる組成物の例は、以下に示される。
【0015】
とりわけ、例えば、代謝拮抗物質、抗生剤、アルキル化剤、植物性アルカロイド、ホルモン剤、抗凝固剤、抗血栓剤、および他の天然性剤を含むいくつかの異なる種類の化学療法剤を、本発明によるハリコンドリンB治療と組み合わせて使用できる。これらの類の薬剤およびそれらの使用によって治療できる癌の具体的な非限定的例は、以下に示される。
【0016】
ハリコンドリンB類似体と併用できる代謝拮抗剤は、例えば、メトレキサート、プリン代謝拮抗薬(例えば、メルカプトプリン、チオグアニン、リン酸フルダラビン、クラドリビン、およびペントスタチン)、ならびにピリミジン拮抗薬(例えば、ゲムシタビン、カペシタビン、フルオロウラシル(例えば5−FU)、シタラビンおよびアザシチジン)を含む。特定の種類の癌の治療でのこれらの薬剤の使用は、当分野で周知であり、これらの薬剤はハリコンドリンB類似体と組み合わせて、これらの種類の癌および他の種類の癌の治療に使用できる。具体的な非限定的例として、ハリコンドリンB類似体は、非小細胞肺癌、膵臓癌もしくは転移性乳癌の治療においてゲムシタビンと併用できる。さらなる例において、ハリコンドリンB類似体は、乳癌もしくは大腸癌においてカペシタビンと併用できる。
【0017】
上述のように、ハリコンドリンB類似体と併用できる他の類の化学療法剤は、抗癌抗生剤を含む。これらは、例えば、アントラサイクリン(例えばドキソルビシン、エピルビシン、ダウノルビシンおよびイダルビシン)、アドリアマイシン、ダクチノマイシン、イダルビンシン(idarubincin)、プリカマイシン、マイトマイシンならびにブレオマイシンを含む。上述の薬剤と同様、特定の種類の癌の治療におけるこれらの薬剤の使用は当分野に周知であり、これらは、これらの種類の癌および他の種類の癌の治療のためにハリコンドリンB類似体と併用できる。具体的な非限定例として、ドキソルビシンのようなアントラサイクリンは、乳癌もしくは膵臓癌の治療のためにハリコンドリンB療法と併用できる。代替的に、第三の薬剤、シクロホスファミドもこの方法において使用できる。
【0018】
アルキル化剤は、本発明によるハリコンドリンB類似体と組み合わせて投与できる他の類の化学療法剤からなる。そのような薬剤の例は、プロカルバジン、ダカルバジン、アルトレタミン、シスプラチン、カルボプラチンおよびニトロソウレアを含む。ハリコンドリンB類似体は、これらの薬剤を治療するのに使用すると当分野で知られている癌の治療、ならびに他の癌の治療においてこれらの薬剤を併用できる。例えば、ハリコンドリンB類似体は、非小細胞肺癌もしくは卵巣癌の治療においてカルボプラチンと併用できる。
【0019】
本発明によるハリコンドリンB類似体と併用できるさらなる種類の化学療法剤は、ビンブラスチン、ビンクリスチン、エトポシド、テニポシド、トポテカン、イリノテカン、パクリタキセルおよびドセタキセルのような植物アルカロイドである。具体的な非限定例として、ハリコンドリンB類似体は結腸直腸癌の治療において、イリノテカンと併用でき、あるいは卵巣癌もしくは非小細胞肺癌の治療においてトポテカンと併用できる。
【0020】
本発明によるハリコンドリンB類似体療法と併用できるさらなる種類の抗癌剤は、抗凝固剤および抗血栓剤である。例えば、ヘパリン(例えば低分子量ヘパリンもしくはヘパラン硫酸)もしくはワルファリンを使用できる。治療中の患者における、例えば注射もしくは経口によるこれらの薬剤の使用は当分野に周知であり、そして当業者によって本発明において使用するよう容易に適応できる。
【0021】
抗癌剤を投与する数多くのアプローチは当分野に公知であり、本発明において使用するように容易に適応できる。ハリコンドリンB類似体と併用して投与できる一つもしくはそれ以上の薬剤の場合、例えば、総合的な治療の投薬計画の一部として、その薬剤を一つの組成物として一緒に、もしくは別々に投与できる。全身投与のために、その薬剤を例えば、静脈内注射(持続的もしくはボーラス)によって投与できる。そのような投与の適切なスケジュールや投与量は、たとえば動物における前臨床試験およびヒトにおける臨床試験(例えばフェーズI試験)に基づいて、当業者によって容易に決定できる。さらに、当分野に周知のように、同様の薬剤を用いる治療の分析、ならびに、患者における血球数(例えば好中球および血小板)および生体信号のような要素を観察することも利用できる。
【0022】
化学療法剤を投与するのに使われる多くの投薬計画は、例えば、薬剤の静脈内注射、ならびに患者が治療の任意の副作用から復帰する期間を置いて(例えば1〜4週間)後に続くこの治療の繰り返しを含む。それぞれの投与において療法の薬剤を用いること、もしくは代替的に一つの薬剤のみ(もしくは薬剤の一部分)を含む地医療を施すことが望ましいであろう。
【0023】
本発明に含まれる治療の投薬計画の具体的な非限定的例として、ハリコンドリンB類似体(例えば、0.01〜5mg/m2)を患者へと0.5〜3時間で静脈内注射によって投与でき、そして他の薬剤(例えばゲムシタビンを例えば500〜900mg/m2)の0.5〜3時間かけての静脈内注射が後に続く。当業者によって許容できるか効果的であるか決定されるに従って、この治療のコースを2〜3週ごとに繰り返す事ができる。この方法の変法として、上述のように最初の日に両方の薬剤によって治療を実施し、しかしながらそのあと、次の週に第二の薬剤(例えばゲムシタビン)のみを用いての治療が後に続くようにできる。
【0024】
さらに、当分野によく知られているように、本発明の方法を用いる治療は、癌化学療法剤の共通の副作用である吐き気および嘔吐を軽減するのに用いられる抗嘔吐剤の投与と組み合わせて実施できることができる。そのような薬剤の例は強トランキライザ(例えば、クロルプロマジンおよびプロクロルペラジンのようなフェノチアジン)、ドーパミン拮抗剤(例えば、メトクロプラミド)、セレトニン拮抗剤(例えばオンダンセトロンおよびグラニセトロン)、カンナビノイド(例えばドロナビノール)、ならびにベンゾジアゼピン系鎮静剤を含む。
【0025】
上述の癌に加えて、本発明の方法及び組成物を、以下の種類の癌ならびに他のものの治療に用いる事ができる:皮膚(例えば、扁平上皮癌、基底細胞癌もしくはメラノーマ)、前立腺、脳および神経、頭頸部、精巣、肺、肝臓(例えば肝癌)、腎臓、膀胱、胃腸、骨、内分泌系(例えば、甲状腺および下垂体腫瘍)、ならびにリンパ系(例えばホジキンリンパ腫および非ホジキンリンパ腫)の癌。本発明の方法を用いて治療できる他の種類の癌は繊維肉種、神経外胚葉性腫瘍、中皮腫、類表皮癌、およびカポジ肉腫を含む。
【0026】
本発明はまた、ハリコンドリンB類似体を、上述の任意の薬剤のようなさらなる治療薬剤と組み合わせて含む組成物をまた含む。それらの組成物における薬剤は、好ましくは患者へと投与できるように調合されている(例えば生理食塩水中に)か、あるいは代替的に投与の前にさらなる加工を必要とする形状であっても良い。例えば、組成物は凍結乾燥状態において、もしくは、希釈を必要とする濃縮状態において、薬剤を含む事ができる。化学療法において使用するための薬剤の調合は当分野における標準的な方法(例えば、Remington’s Pharmaceutical Science(第18版)、A.Gennaro編、1990、Mack Publishing Co., Eaton,Paを参照)を用いて実施できる。
【実施例】
【0027】
単結晶X線回析データ集積および構造解析
データはOxford Cryostream低温装置を備えたBruker SMART APEX CCD(電荷結合素子)系の回析計を用いて絶対温度193度で集積された。データは半球が集積されるように30秒間、毎フレーム0.3°のオメガスキャンを用いて測定された。0.76Åの最大解像度で、全部で1271フレームが集積された。最初の50フレームは、減衰を監視するためにデータ集積の最後に再集積された。セル(cell)パラメータはSMARTソフトウェア(SMART V5.625 (NT)CCD検出系のためのソフトウェア;Bruker Analytical X−ray System、Madison、WI(2001))を用いて抽出された。データ整理は、Lpおよび減衰を補正するSAINTソフトウェア(SAINT V6.22(NT)CCD検出系のためのソフトウェア;Bruker Analytical X−ray System、Madison、WI(2001))を用いて行われた。構造は、SHELXTL−PC V6.10(SHELXTL 6.1(PC版)、構造解析および分子グラフィックスのためのプログラムライブラリ;Bruker Analytical X−ray System、Madison、WI(2000))に含まれるSHELXS−97プログラム(Sheldric,G.M.SHELXS−90、結晶構造解析のためのプログラム、ゲッチンゲン大学、ドイツ、1990)を用いる直接法によって解析された。
【0028】
粉末X線解析(XRPD)データ集積手順
少量の結晶化物質が下記のXRPD回析試験のために用いられた。物質は粉砕されておらず、選択配向効果を有するかもしれない。Scintag Diffractometer上の水晶板を用いる。データは、銅放射線を用いて、5〜70度の2シータレンジで通常の粉末回析条件によって取得された。バックグランド補正は実施されなかった。表1はデータ取得パラメータを列挙する。
【0029】
【表1】
【0030】
実施例1:結晶ER−076349一水和物の解析
エタノール、イソプロパノール(IPA)、プロピオニトリルのXRPDパターンはアセトニトリル/水サンプルと同一である。一水和物としての解析はこの比較に基づく。アセトニトリル/水のX線ユニットセルの結晶は単一の水分子の存在を示した。
【0031】
A.ER−076349一水和物の結晶化の手順
エタノール、イソプロパノールおよびプロピオニトリルからのER−076349の結晶化(XRPDサンプル):ER−076349は溶媒(5倍容量)に溶解された。溶液は室温において蒸発され、XRPD試験に適した結晶は16から24時間かけて形成された。
【0032】
アセトニトリル/水溶液からのER−076349の結晶化(XRPDサンプル):ER−076349はアセトニトリル(1倍容量)に溶解された。この溶液に水/アセトニトリル溶液(10倍容量、2部の水、1部のアセトニトリル)がゆっくりと添加された;もし溶液が濁るときは、最小量のアセトニトリルを添加してもよい。溶液はER−076349(0.004重量)を種子として入れられた。混合物は室温で攪拌され、溶液は不活性ガス流下において、アセトニトリルが5%以下になるまで蒸発された。固形物は水(5倍容量)の中でスラリー化され、混合物はろ過された。ろ過された固形物は高真空および不活性ガス流下で、12から24時間乾燥された。
【0033】
アセトニトリル/水溶液からのER−076349の結晶化(単結晶):ER−076349はアセトニトリル(5倍容量)に溶解された。水(0.5倍容量)がそののち添加された。コンテナは室温に保管され、4週間のゆっくりとした溶液の蒸発が発散され、X線構造解析に適した単結晶がゆっくりと形成された。
【0034】
B.ER−076349一水和物の単結晶X線解析
0.10×0.10×0.04mmの寸法の無色のプレート結晶が非常に少量のパラトン油(paratone oil)を用いて0.2nmのナイロンループ上に取り付けられた。図1−Aに示される単結晶構造は上述の手順を用いて測定された。表2は、結晶データおよびER−076349一水和物の単結晶構造を測定するのに用いられた構造精密化パラメータを列挙する。構造は消滅則の分析によってスペースグループP212121(#19)中で解析された。全ての非水素原子は異方的に精密化された。水素は幾何学的方法によって計算され、ライディングモデル(riding model)として精密化された。回析試験に用いられる化粧はデータ集積の間、分解を示さなかった。全ての描写は50%楕円において実施された。図1−BはER−076349一水和物のb軸に沿った結晶パッキング図であり、結晶中の水素結合、点線の最適の図を示す。
【0035】
【表2】
【0036】
C.ER−076349一水和物の粉末X線回析による解析
図1−Cは、上述のようにイソプロピルアルコールから結晶化されたER−076349一水和物のXRPD図である。表3AはXRPD図中の相対強度のピークのリストを示す。
【0037】
【表3A】
【0038】
図1−dは、アセトニトリルおよび水、エタノール、イソプロパノール、ならびにプロピオニトリルを含むさまざまな再結晶化溶媒より得られたER−076349一水和物の個別のXRPDのプロットをまとめたものである。上述のものと同じパターン取得の方法が用いられた。
【0039】
図1−E、1−Fおよび1−Gは、ER−076349一水和物の示差走査熱量計(DSC)のサーモグラムを描写する。図1−Eはエタノール溶液より結晶化されたER−076349のDSCのプロットを描写する。DSCは160℃において吸熱ピークを示す。図1−Fはイソプロパノール溶液から結晶化されたER−076349のDSCのプロットを描写する。DSCは157℃において吸熱ピークを示す。図1−Gはプロピオニトリル溶液より結晶化されたER−076349のDSCのプロットを描写する。DSCは161℃において吸熱ピークを示す。
【0040】
実施例2:ER−818906(epi−C20ジオール)、モノ−アセトニトリル溶媒和物
A.X線およびXRPD結晶サンプルの生成のための結晶化手順
E0818906(1重量)はアセトニトリル(10倍容量)に懸濁され、完全に溶解するのを助けるためゆるやかに加熱された。溶液は室温に置かれ、30分後に結晶成長が始まった。X線構造解析にふさわしい単結晶サンプルは16から24時間後に生成した。ER−818906(epi−C20ジオール)の化学構造は以下に示される。
【化6】
【0041】
B.ER−818906(epi−C20ジオール)モノ−アセトニトリル溶媒和物の単結晶X線解析
0.12×0.12×0.10mmの寸法の無色のブロック結晶は非常に少量のパラトン油を用いて0.2nmのナイロンループ上に取り付けられた。図2−Aに示される単結晶構造は上述の手順を用いて測定された。表3Bは結晶データおよびER−818906モノ−アセトニトリル溶媒和物の単結晶構造を測定するのに用いられた構造精密化パラメータを列挙する。構造はスペースグループP21(#4)において解析された。全ての非水素原子は異方的に精密化された。水素は幾何学的方法によって計算され、ライディングモデルとして精密化された。フラック(Flack)パラメータは結晶を分析するのに用いられた。もしフラックパラメータが0に近い時、構造要素精密化によって与えられる全体の構造がほぼ正しく;その値が1.0に近いとき、倒置構造(他の光学異生体)がほぼ正しく;もしその値が0.5に近い時、結晶はほぼラセミ体である。Flack、H.D.Acta Cryst. A39.1983、876−881を参照。フラックパラメータは0.3(4)へと精密化された。この化合物のキラリティはその合成の源により確かめられた。回析試験において用いられる結晶はデータ集積の間、分解を示さなかった。全ての描写は50%楕円において実施された。図2−BはER−818906モノ−アセトニトリル溶媒和物のc軸に沿った結晶パッキング図である。
【0042】
【表3B】
【0043】
C.ER−818906(epi−C20ジオール)モノ−アセトニトリル溶媒和物の粉末X線回析(XRPD)による解析
実施例2Aにおける単結晶試験のサンプルを取り出したのち、少量の物質が残った。XRPDパターンは上述のように取得された。XRPDパターンは図2−Cに示される。表4はXRPD中の相対強度のピークのリストを示す。
【0044】
【表4】
【0045】
実施例3:ER−819531(epi−C23ジオール)一水和物の解析
A.XRPD品質サンプルのための結晶化手順
ER−819531(1重量)はアセトニトリル(10倍容量)に室温で溶解され、温度は−20℃まで低下し、この温度は16時間維持された。粉末XRPD試験に適した結晶のクラスターは16時間後に生成した。ER−819531(epi−C23ジオール)の化学構造は下に示される。
【化7】
【0046】
B.ER−819531(epi−C23ジオール)一水和物の単結晶X線解析
ER−819531サンプルの一部はアセトニトリル、数滴のトルエン、および1滴の水の混合物中に溶解された。これはのちに少量のバイアル中でゆっくりと蒸発された。これは単結晶を生じ、その一つはこの試験に用いられるよう選択された。0.12×0.12×0.04mmの寸法の無色のブロック結晶は非常に少量のパラトン油を用いてナイロンループ上に組み込まれた。図3−Aに示される、この単結晶構造は上述の手順を用いて解析された。表5は結晶データおよびER−819531一水和物の単結晶構造を測定するのに用いられた構造精密化パラメータを列挙する。構造は消滅則の分析によってスペースグループP212121(#19)中で解析された。全ての非水素原子は異方的に精密化された。水素は幾何学的方法によって計算され、ライディングモデルとして精密化された。フラックパラメータは結晶を分析するのに用いられた。もしフラックパラメータが0に近い時、構造要素精密化によって与えられる全体の構造がほぼ正しく;その値が1.0に近いとき、倒置構造(他の光学異生体)がほぼ正しく;もしその値が0.5に近い時、結晶はほぼラセミ体である。Flack、H.D.Acta Cryst. A39.1983、876−881を参照。フラックパラメータは0.50(16)へと精密化された。この化合物のキラリティはその合成の源により確かめられた。回析試験において用いられる結晶はデータ集積の間、分解を示さなかった。全ての描写は50%楕円において実施された。図3−Bはb軸に沿った結晶パッキング図であり、それは結晶中の水素結合を点線で示す。
【0047】
【表5】
【0048】
C.ER−819531(epi−C23ジオール)一水和物の粉末X線回析(XRPD)による解析
ER−819531一水和物は上述のように得られた。XRPDパターンは図3−Cに示される。表6はXRPDパターン中の相対強度のピークのリストを示す。
【0049】
【表6】
【0050】
実施例4:ER−820057(epi−C23アミン)の解析
A.ER−820057結晶サンプルの調製
ER820057はアセトニトリル(10倍容量)中に室温で溶解され、溶液は室温に置かれた。結晶成長は2時間後に観察された。粉末XRPD試験にふさわしい結晶サンプルは16から24時間後に単離された。ER−820057(epi−C23アミン)の化学構造は以下に示される。
【化8】
【0051】
B.ER−820057(epi−C23アミン)の粉末X線回析(XRPD)による解析
ER−820057のXRPDは上述のように得られた。XRPDパターンは図4−Aに示される。表7はXRPDパターンの相対強度のピークのリストを示す。
【0052】
【表7】
【0053】
実施例5:ER−111197(epi−C34ジオール)一水和物の解析
A.X線品質の結晶の生成のための結晶化手順
ER−111197(1重量)はアセトニトリル(8倍容量)に溶解された。水(0.8倍容量)が添加された。溶液は室温で2〜3日間蒸発するよう放置された。結晶が生成しない場合、溶液は乾燥するまで蒸発され、この手順が結晶が出来るまで繰り返された。ER−111197(epi−C34ジオール)の化学構造は以下に示される。
【化9】
【0054】
B.R−111197(epi−C34ジオール)一水和物の単結晶X線解析
0.22×0.14×0.04mmの寸法の無色の針状結晶が、非常に少量のパラトン油を用いてナイロンループに組み込まれた。図5−Aに示される単結晶構造は、上述の手順を用いて解析された。表7Bは結晶データおよびER−111197一水和物の単結晶構造を測定するのに用いられた構造精密化パラメータを列挙する。構造は消滅則の分析によってスペースグループP212121(#19)中で解析された。全ての非水素原子は異方的に精密化された。水素は幾何学的方法によって計算され、ライディングモデルとして精密化された。フラックパラメータは結晶を分析するのに用いられた。もしフラックパラメータが0に近い時、構造要素精密化によって与えられる全体の構造がほぼ正しく;その値が1.0に近いとき、倒置構造(他の光学異生体)がほぼ正しく;もしその値が0.5に近い時、結晶はほぼラセミ体である。Flack、H.D.Acta Cryst. A39.1983、876−881を参照。フラックパラメータは0.3(13)へと精密化された。この化合物のキラリティはその合成の源により確かめられた。回析試験において用いられる結晶はデータ集積の間、分解を示さなかった。全ての描写は50%楕円において実施された。図5−Bはa軸に沿った結晶パッキング図であり、それは結晶中の水素結合を点線で示す。
【0055】
【表7B】
【0056】
C.ER−111197(epi−C34ジオール)一水和物のシミュレートされた粉末X線解析スペクトル
単結晶のデータから生成されたER−111197のシミュレートされたXRPDスペクトルは図5−Cに示される。
【0057】
実施例6:ER−809681(エポキシド)
A.X線品質の結晶を生成するための結晶化手順
ER−809681−00(50mg)はジクロロメタン(0.25ml)に溶解された。ペンタン(0.5ml)が添加された。溶液は視覚的に均一になるまで攪拌された。溶液は室温で18時間、ゆっくりと蒸発された。X線結晶学的試験にふさわしい結晶は上清の液体をデカントすることによって集められた。ER−809681(エポキシド)の化学構造は下に示される。
【化10】
【0058】
B.ER−809681(エポキシド)の単結晶X線解析
X線解析方法は上述のものから変更され、以下の節に詳細に説明される。
0.30×0.10×0.10mmのおおよその寸法を持つC40H56O11の無色のブロック結晶はガラス線維上に組み込まれた。単結晶は図6−Aに示される。すべての解析は、グラファイトで単色化されたCu−Kα放射線を持つRigaku RAXIS RAPID imaging plate area detector上で行われた。指数化は15秒の曝露による3つの振動より実施された。結晶と検出器の距離は127.40mmであった。寸法を持つ一次斜方晶系のセルに対応するデータ集積のためのセル定数および配向マトリックス:a=9.4700(3)Å;b=17.6327(5)Å;c=23.1790(6)Å;V=3879.47(19)Å3。Z=4でかつF.W.=712.88の時、計算上の密度は1.223g/cm3である。h00:h±2n;0k0:k±2n;001:1±2nの消滅則は特異的にスペースグループをp212121(#19)と定める。結晶パッケージ図は図6−Bに示される。
【0059】
データは23±1℃において136.5°の最大2θ値で集積された。全体で30振幅の画像が集積された。χ=54.0°かつφ=15.0°において80.0から260.0°の30.0ステップのωスキャンを用いてデータのスイープ(sweep)が実施された。χ=54.0°かつφ=105.0°において80.0から260.0°の30.0ステップのωスキャンを用いて2度目のスイープが実施された。照射線量率は15.0[秒/°]であった。χ=54.0°かつφ=270.0°において80.0から260.0°の30.0ステップのωスキャンを用いて他のスイープが実施された。照射線量率は15.0[秒/°]であった。χ=0.0°かつφ=0.0°において80.0から260.0°の30.0ステップのωスキャンを用いて他のスイープが実施された。照射線量率は15.0[秒/°]であった。結晶と検出器の距離は127.40mmであった。読み出しは0.100mmピクセルモードで実施された。
【0060】
データ解析:集積された16412の反射のうち、934がユニークであった(Rint=0.043);同等の反射は融合された。Cu−Kα放射の線吸収係数、μは7.229cm−1であった。0.748から0.930の範囲の透過率が生じる実験的な吸収の補正が適応された。データはローレンツ偏光効果のために補正された。
【0061】
構造解析と精密化:構造は直接方を用いて解析され(SHELX97:Sheldric,G.M.(1997)、フーリエ法を用いて拡張された。DIRDIF99:Beurskens,P.T.,Admiraal,G.,Beurskens,G.,Bosman,W.P.,de Gelder,R.,Israel,R. and Smits,J.M.M.(1999)、”The DIRDIF−99プログラムシステム”、Technical Report of the Crystallography Laboratory,”University of Nijmegen, The Netherland.を参照。非水素原子は異方的に精密化された。水素原子はライディングモデルを用いて精密化された。Σw(Fo2−Fc2)2(式中、w=最小二乗重量)で表わされるF2のフルマトリックス最小二乗精密化の最後のサイクルは、7045の観察された反射と461の変数パラメータに基づく。それは、R1=Σ||Fo|−|Fc||/Σ|Fo|=0.0630かつwR2=[Σ(w(Fo2−Fc2)2)/Σw(Fo2)2]1/2=0.2168という計量されてないアグリーメント・ファクタ(agreement factor)および計量されたアグリーメント・ファクタに収斂する。
【0062】
単位重量の観測の標準偏差は1.10であり、[Σw(Fo2−Fc2)2/(No−Nv)]1/2(式中、No=観察の数およびNv=変数の数)として計算される理論上の単位重量の標準偏差はである。最終の差フーリエ図上の最大ピークおよび最小ピークは、それぞれ0.20および−0.28e−/Å3に対応する。
【0063】
中性子散乱因子は、CromerとWaber,”International Tables for X−ray Crystallography”Vol.IV,The Kynoch Press, Birmingham,England,表2.2A(1974)より得た。異常分散効果はFcalc中に含まれ(Ibers,J.A.およびHamilton,W.C.;Acta Crystallogr.,17,781(1964)を参照)、ΔfおよびΔf’’の値はCreagh,D.C.およびMcAuley,W.J.” International Tables for Crystallography”Vol.C,(A.J.C.Wilson編)、Kluwer Academic Publishers, Boston,表4.2.6.8、219〜222ページ(1992)より得られた。質量減衰係数の値はCreagh,D.C.およびHubbell,J.H.;” International Tables for Crystallography”Vol.C,(A.J.C.Wilson編)、Kluwer Academic Publishers, Boston,表4.2.4.3、200〜206ページ(1992)より得られた。すべての計算はCrystalStructure結晶学ソフトウェアパッケージを用いて実施された。CrystalStructure3.7.0:Crystal Structure Analysis Package,Rigaku and Rigaku/MSC(2000−2005)、9009 New Trails Dr. The Woodlands TX77381米国;ならびにCRYSTALS Issure 10:Watkin, D.J.,Prout,C.K. Carruthers, J.R.およびBetteridge, P.W.、Chemical Crystallography Laboratory,Oxford,英国(1996)を参照。これの例外は精密化の計算であり、SHELX97:Sheldrick,G.M.(1997)を用いて実施された。
【0064】
【表7c】
【表7d】
【表7e】
【0065】
C.ER−809681(エポキシド)の粉末X線解析(XRPD)による解析
この化合b通のXRPD集積方法は上述のものとデータスキャンレンジが2−70°である点でのみ異なる。XRPDパターンは図6−Cに示される。
【0066】
【表8】
【0067】
D.シミュレートされたXRPDスペクトル
単結晶データより生成されるシミュレートされたXRPDパターンは図6Dに示される。
【0068】
上述の化合物の、上述の結晶形における抗癌活性は、LiuJ.らのAnticancer Research、27:1509−1511(2007)に記載されるような細胞増殖阻害試験を用いて測定された。IC50nMの単位での活性データは下の表9に記載される。MES−SAおよびMES−SA/Dx5−Rx1子宮肉腫はAmerican Type Culture Collectionより入手し、推奨される条件で増殖された。
【0069】
細胞増殖阻害試験
細胞は96穴プレートに10%FBSならびにペニシリン、ストレプトマイシンおよびL−グルタミンを補った推奨される培地中に7.5×103細胞/ウェルで播種された。4時間の培養の後、試験化合物はそれぞれのウェルに0から10μMの範囲の一連の濃度になるように添加された。細胞は4日間37℃で培養された。細胞増殖はメチレンブルーに基づいたマイクロ培養試験(Towle M.J.らの”In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B.”Cancer Res 61:1013−1021、2001を参照)の修正したもの(FinlayG.J.らの”A semiautomated microculture method for investigating growth inhibitory effects of cytotoxic compounds on exponentially growing carcinoma cells”,Anal Biochem 139:272−277、1984を参照)を用いて測定された。培地は除去され細胞は100μlのメチレンブルー(500μg/ml)で45分間染色された。水で染色したあと、線食された細胞は100μlのサルコシン(1mg/ml)に溶解され、90分間緩やかに攪拌された。このプレートはA600−A405で観測された。
【0070】
P−グリコプロテイン介在多薬剤耐性(MDR)に対する化合物の感受性
一対の子宮肉種の細胞株が用いられた:MES−SA(MDRのない親細胞株)、DX5−Rx1(長期間ドキソルビシンへと曝露されたMES−SA由来の細胞株)。亜株はP−グリコプロテインを高発現している。療法の細胞株は10%FBSならびにペニシリン、ストレプトマイシンおよびL−グルタミンを補ったMcCoy’s5A中に7.5×103細胞/ウェルで播種され、細胞増殖が上述のように測定された。親株および耐性細胞株において取得されたIC50値の比は耐性の倍数を意味し、それぞれ比較される二つの細胞株に対するP−グリコプロテイン介在MDRへの化合物の感受性の指標をもたらす。
【0071】
【表9】
【特許請求の範囲】
【請求項1】
化合物:
【化11】
の一水和物結晶。
【請求項2】
9.3°2θ±0.2°2θ、10.4°2θ±0.2°2θ、および13.0°2θ±0.2°2θにピークを持つ粉末X線回析パターンによって特徴付けられる、請求項1に記載の一水和物結晶。
【請求項3】
約157から約161℃の範囲の開始温度を持つ示差走査熱量計サーモグラムによってさらに特徴付けられる、請求項2に記載の一水和物結晶。
【請求項4】
治療に効果的な量の請求項1に記載の一水和物結晶および医薬として許容できる担体を含有する医薬組成物。
【請求項5】
化学療法剤を更に含有する請求項4に記載の組成物。
【請求項6】
前記化学療法剤が、代謝拮抗物質、抗生剤、アルキル化剤、植物性アルカロイドおよびホルモン剤からなる群より選択される、請求項5に記載の組成物。
【請求項7】
化合物:
【化12】
の結晶形。
【請求項8】
6.5°2θ±0.2°2θ、9.3°2θ±0.2°2θ、および17.1°2θ±0.2°2θにピークを持つ粉末X線回析パターンによって特徴付けられる、請求項7に記載の結晶形。
【請求項9】
化合物:
【化13】
の結晶形。
【請求項10】
13.0°2θ±0.2°2θ、15.0°2θ±0.2°2θ、および15.9°2θ±0.2°2θにピークを持つ粉末X線回析パターンによって特徴付けられる、請求項9に記載の結晶形。
【請求項11】
治療に効果的な量の請求項9に記載の化合物の結晶形および医薬として許容できる担体を含有する医薬組成物。
【請求項12】
化合物:
【化14】
のモノ−アセトニトリル溶媒和物結晶。
【請求項13】
7.9°2θ±0.2°2θ、23.7°2θ±0.2°2θ、32.8°2θ±0.2°2θおよび40.0°2θ±0.2°2θにピークを持つ粉末X線回析パターンによって特徴付けられる、請求項12に記載の溶媒和物結晶。
【請求項14】
化合物:
【化15】
の一水和物結晶。
【請求項15】
前記一水和物結晶がラセミ体である、請求項14に記載の一水和物結晶。
【請求項16】
8.1°2θ±0.2°2θ、13.1°2θ±0.2°2θおよび14.3°2θ±0.2°2θにピークを持つ粉末X線回析パターンによって特徴付けられる、請求項14に記載の一水和物結晶。
【請求項17】
治療に効果的な量の請求項14に記載の化合物の一水和物結晶および医薬として許容できる担体を含有する医薬組成物。
【請求項18】
化合物:
【化16】
の一水和物結晶。
【請求項19】
8.3°2θ±0.2°2θ、15.8°2θ±0.2°2θおよび16.6°2θ±0.2°2θにピークを持つ粉末X線回析パターンによって特徴付けられる、請求項18に記載の一水和物結晶。
【請求項20】
治療に効果的な量の請求項18に記載の化合物の一水和物結晶および医薬として許容できる担体を含有する医薬組成物。
【請求項1】
化合物:
【化11】
の一水和物結晶。
【請求項2】
9.3°2θ±0.2°2θ、10.4°2θ±0.2°2θ、および13.0°2θ±0.2°2θにピークを持つ粉末X線回析パターンによって特徴付けられる、請求項1に記載の一水和物結晶。
【請求項3】
約157から約161℃の範囲の開始温度を持つ示差走査熱量計サーモグラムによってさらに特徴付けられる、請求項2に記載の一水和物結晶。
【請求項4】
治療に効果的な量の請求項1に記載の一水和物結晶および医薬として許容できる担体を含有する医薬組成物。
【請求項5】
化学療法剤を更に含有する請求項4に記載の組成物。
【請求項6】
前記化学療法剤が、代謝拮抗物質、抗生剤、アルキル化剤、植物性アルカロイドおよびホルモン剤からなる群より選択される、請求項5に記載の組成物。
【請求項7】
化合物:
【化12】
の結晶形。
【請求項8】
6.5°2θ±0.2°2θ、9.3°2θ±0.2°2θ、および17.1°2θ±0.2°2θにピークを持つ粉末X線回析パターンによって特徴付けられる、請求項7に記載の結晶形。
【請求項9】
化合物:
【化13】
の結晶形。
【請求項10】
13.0°2θ±0.2°2θ、15.0°2θ±0.2°2θ、および15.9°2θ±0.2°2θにピークを持つ粉末X線回析パターンによって特徴付けられる、請求項9に記載の結晶形。
【請求項11】
治療に効果的な量の請求項9に記載の化合物の結晶形および医薬として許容できる担体を含有する医薬組成物。
【請求項12】
化合物:
【化14】
のモノ−アセトニトリル溶媒和物結晶。
【請求項13】
7.9°2θ±0.2°2θ、23.7°2θ±0.2°2θ、32.8°2θ±0.2°2θおよび40.0°2θ±0.2°2θにピークを持つ粉末X線回析パターンによって特徴付けられる、請求項12に記載の溶媒和物結晶。
【請求項14】
化合物:
【化15】
の一水和物結晶。
【請求項15】
前記一水和物結晶がラセミ体である、請求項14に記載の一水和物結晶。
【請求項16】
8.1°2θ±0.2°2θ、13.1°2θ±0.2°2θおよび14.3°2θ±0.2°2θにピークを持つ粉末X線回析パターンによって特徴付けられる、請求項14に記載の一水和物結晶。
【請求項17】
治療に効果的な量の請求項14に記載の化合物の一水和物結晶および医薬として許容できる担体を含有する医薬組成物。
【請求項18】
化合物:
【化16】
の一水和物結晶。
【請求項19】
8.3°2θ±0.2°2θ、15.8°2θ±0.2°2θおよび16.6°2θ±0.2°2θにピークを持つ粉末X線回析パターンによって特徴付けられる、請求項18に記載の一水和物結晶。
【請求項20】
治療に効果的な量の請求項18に記載の化合物の一水和物結晶および医薬として許容できる担体を含有する医薬組成物。
【図1−A】

【図1−B】
【図1−C】
【図1−D】
【図1−E】
【図1−F】
【図1−G】
【図2−A】
【図2−B】
【図2−C】
【図3−A】
【図3−B】
【図3−C】
【図4−A】
【図5−A】
【図5−B】
【図5−C】
【図6−A】
【図6−B】
【図6−C】
【図6−D】
【図1−B】
【図1−C】
【図1−D】
【図1−E】
【図1−F】
【図1−G】
【図2−A】
【図2−B】
【図2−C】
【図3−A】
【図3−B】
【図3−C】
【図4−A】
【図5−A】
【図5−B】
【図5−C】
【図6−A】
【図6−B】
【図6−C】
【図6−D】
【公表番号】特表2011−516493(P2011−516493A)
【公表日】平成23年5月26日(2011.5.26)
【国際特許分類】
【出願番号】特願2011−503203(P2011−503203)
【出願日】平成21年4月3日(2009.4.3)
【国際出願番号】PCT/US2009/039432
【国際公開番号】WO2009/124237
【国際公開日】平成21年10月8日(2009.10.8)
【出願人】(506137147)エーザイ・アール・アンド・ディー・マネジメント株式会社 (215)
【Fターム(参考)】
【公表日】平成23年5月26日(2011.5.26)
【国際特許分類】
【出願日】平成21年4月3日(2009.4.3)
【国際出願番号】PCT/US2009/039432
【国際公開番号】WO2009/124237
【国際公開日】平成21年10月8日(2009.10.8)
【出願人】(506137147)エーザイ・アール・アンド・ディー・マネジメント株式会社 (215)
【Fターム(参考)】
[ Back to top ]