
バイオマーカー抽出装置および方法
【課題】本発明は、生物情報学(Bioinformatics)技術に関するものであり、より詳細には配列変異が遺伝子機能に及ぼす毒性分析に基づきにより高信頼性のバイオマーカーを抽出する装置および方法に関するものである。
【解決手段】配列変異が遺伝子機能にいかなる変化を与えるかを分解し、特定疾患の原因になるバイオマーカーを抽出するバイオマーカー抽出装置は、遺伝子サンプルの配列を分解して遺伝子にマッピングされた変異データを抽出する前処理部と、前記変異データに基づき前記遺伝子の機能に発生する障害を定量化した毒性スコアを求める毒性予測器、および前記毒性スコアが所定の臨界値以上の遺伝子が、遺伝子ネットワークに密集している遺伝子ネットワークのサブモジュールを探索するモジュール化部を含み構成することができる。
【解決手段】配列変異が遺伝子機能にいかなる変化を与えるかを分解し、特定疾患の原因になるバイオマーカーを抽出するバイオマーカー抽出装置は、遺伝子サンプルの配列を分解して遺伝子にマッピングされた変異データを抽出する前処理部と、前記変異データに基づき前記遺伝子の機能に発生する障害を定量化した毒性スコアを求める毒性予測器、および前記毒性スコアが所定の臨界値以上の遺伝子が、遺伝子ネットワークに密集している遺伝子ネットワークのサブモジュールを探索するモジュール化部を含み構成することができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、生物情報学(Bioinformatics)技術に関するものであって、より詳細には配列変異が遺伝子機能に及ぼす毒性分解に基づきより高信頼性のバイオマーカーを抽出する装置および方法に関するものである。
【背景技術】
【0002】
ヒトゲノムプロジェクトが完了した後、ヒトのDNA塩基配列が解読され、またそこからヒトの遺伝子の多様な機能が明らかになっている。特に、多様な遺伝子変異が発見されており、これらが人間の形質の差異を起こすだけではなく、特定疾患の原因として作用されることが明らかになるにつれ、ヒトのゲノム分解に関する研究はさらに加速化されつつある。しかし、ヒトのゲノムから発生し得る膨大な遺伝的変異のうち、どれが実質の病因になる変異であるかを調べるのは難しい。
【0003】
最近ではこのような難点を解消するための1つの代案として次世代シーケンシング技術(NGS、Next Generation Sequencing)が研究されている。この技術により個人の全ゲノムの塩基配列化が可能になり、疾患群と正常群の塩基配列および変異比較ツールにより、疾患特異的遺伝子変異を抽出することも可能になった。
【0004】
一方、これとは異なって、DNAシーケンシングの代わりにSNP(single nucleotide polymorphism)チップにより個体群(population)の統計分解に基づくGWAS(Genome Wide Association Study)解析技術も研究されている。この技術によれば、数千〜数万人から得られるSNPデータ解析により、特定疾患群から頻繁に発生する有意の遺伝的変異を抽出することができる。
【0005】
しかし、多様な解析技術によって遺伝的変異を抽出したとしても、このような遺伝的変異を含む遺伝子の実際発現および機能障害があるかどうかを確認するためには追加の実験が要求され、これは時間および費用の面から相当な損失につながる。さらには、個別遺伝子変異情報のみでは特定疾患群を起こす多様な病因に対して十分な説明を提供することができない。
【0006】
これを解決するため、生物学的要素の相互作用を分解し、これに基づき生物システムを解読するいわゆるシステム生物学と呼ばれる技術も研究されている。すなわち、遺伝子から発現する生物学的機能は、生命を維持するために他の遺伝子から発現する機能と結合して行い、頻繁に起こる外部環境の変化にも自らの恒常性を維持することができる。このような技術は、生物学的要素のネットワーク分解により、それぞれの変異を有する遺伝子の機能的位置および相互作用分解を行い、遺伝的変異が周辺要素にいかなる影響を与え、またこのような影響がどのように広がるかを把握することができる。また、遺伝的変異が既知の遺伝子相互作用、遺伝子調節回路、蛋白質相互作用、代謝、信号伝達回路といかなる関連性を有するかを説明できる基礎となる。
【0007】
正常の細胞活動のために必要な細胞内の多様なプロセスは、機能モジュールというより小さくてかつ特殊な蛋白質あるいは遺伝子の集団として作動している。個別遺伝子において個別nsSNP(non-synonymous Single Nucleotide Polymorphism)によって発生する蛋白質機能の毒性予測(toxicity prediction)をする一連の方法としては、SIFT(Sorting Tolerant From Intolerant)、PolyPhen、MAPP(Map Annotator and Pathway Profiler)などが提示されているが、このような方法だけでは高複雑度の疾患に対する病因や疾患マーカー(marker)を見つけることに限界がある。
【0008】
また一般的にSNPのうち蛋白質機能に有害な影響を与えるSNPの割合は非常に低い。したがって、GSEA(Gene set enrichment analysis)とSNPを利用した分解方法の場合、SNPが蛋白質機能に毒性(toxicity)を与えるかどうかに関係なく、SNPにより予測されたすべてのデータを利用するため、実際に特定疾患と密接な関係がない生物学的経路(pathway)や遺伝子セットが統計的に有意であると誤った判断をする可能性が高い。したがって、疾患特異的遺伝子変異を生体分子ネットワークに基づいて分解し、発見した生体分子ネットワークに属する遺伝子の発現パターンを分解し、特定疾患に関係があるバイオマーカーをより正確に特定できる技法の開発が必要である。
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、上記の必要性に鑑みてなされたものであって、従来のゲノム変異あるいは遺伝子発現パターンのうち、それぞれの一部分のみを利用して行っていた相互作用モジュール化および分解の限界を超え、配列変異が遺伝子機能に及ぼす毒性分解に基づき、より高信頼性のバイオマーカーを検出することを目的とする。
【0010】
特に、本発明は、前記バイオマーカーの検出において重要な影響を及ぼす毒性について多角的な方法により定量化した毒性予測技法を開発することを他の目的とする。
【0011】
本発明の技術的課題は、以上で言及した技術的課題に制限されず、言及されていないまた他の技術的課題は次の記載から当業者に明確に理解できるであろう。
【課題を解決するための手段】
【0012】
前記技術的課題を達成するための本発明の一実施形態による、配列変異が遺伝子機能にいかなる変化を与えるかを分解し、特定疾患の原因になるバイオマーカーを抽出するバイオマーカー抽出装置であって、遺伝子サンプルの配列を分解して遺伝子にマッピングされた変異データを抽出する前処理部と、前記変異データに基づき前記遺伝子の機能に発生する障害を定量化した毒性スコアを求める毒性予測器、および前記毒性スコアが所定の臨界値以上の遺伝子が、遺伝子ネットワークに密集している遺伝子ネットワークのサブモジュールを探索するモジュール化部を含む。
【0013】
前記技術的課題を達成するための本発明の一実施形態による、配列変異データが遺伝子の機能に影響を及ぼす障害を定量化するための毒性予測装置であって、前記変異データを複数の毒性予測モデルに適用し、それぞれの毒性を得た後、前記それぞれの毒性に加重値を適用することによって加重毒性を計算する毒性計算部と、前記変異データの頻度に基づき、該当遺伝子変異の有意性(significance)を計算する有意性計算部、および前記加重毒性および前記有意性を組み合わせて毒性スコアを計算するスコア計算部を含む。
【0014】
前記技術的課題を達成するための本発明の一実施形態による、配列変異が遺伝子機能にいかなる変化を与えるかを分解し、特定疾患の原因になるバイオマーカーを抽出する方法であって、遺伝子に含まれた変異データに基づき前記遺伝子の機能に発生する障害を定量化した毒性スコアを求めるステップと、前記毒性スコアが所定の臨界値以上の遺伝子が集められたサブモジュールを遺伝子ネットワークから探索するステップ、および前記探索した複数のサブモジュール間の優先順位を決定するステップを含む。
【0015】
前記技術的課題を達成するための本発明の一実施形態による、配列変異データが遺伝子の機能に影響を及ぼす障害を定量化するための毒性予測方法であって、前記変異データから多様な要素を含む特徴ベクターを生成するステップと、前記生成された特徴ベクターのうちそれぞれの予測モデルに必要な要素を選別するステップと、前記選別した要素の入力を受けて蛋白質配列内で個別のスコアを提供するステップ、および前記予測モデルの出力に加重値を適用して合算し、加重毒性を計算するステップを含む。
【発明の効果】
【0016】
前記のような本発明によれば、疾患群と正常群の比較により得られた疾患特異的配列の変異が該当遺伝子の機能にいかなる変化や障害を起こすかを予測することができる。またこのような発現パターン分解とネットワーク分解を併行することによって個別の遺伝子障害が全体生物システムでの相互作用にいかなる影響を及ぼすかについて情報を提供して疾患メカニズムに基づいたバイオマーカーを発掘できる効果がある。
【0017】
このようなバイオマーカーは特定疾患の診断、特定疾患を治療する薬品の開発、副作用の予防に広く利用される。
【図面の簡単な説明】
【0018】
【図1】本発明の一実施形態によるバイオマーカー抽出装置を図示するブロック図である。
【図2】図1に示す前処理部の細部構成を図示するブロック図である。
【図3】図1に示す毒性予測器の細部構成を図示するブロック図である。
【図4】図3に示す毒性計算部の細部構成を図示するブロック図である。
【図5】毒性計算部で使用されるマッピング関数の一例を示す図である。
【図6】モジュール化部でサブモジュールの探索を行う過程を詳細に図示するフローチャートである。
【図7】遺伝子サブモジュールと特定の遺伝子集合内に共通して存在する遺伝子個数から有意性を検証する概念を示す図である。
【発明を実施するための形態】
【0019】
以下、本発明のバイオマーカー抽出装置および方法を実施するための形態の具体例を、図面を参照しながら説明する。本発明の本発明の利点、特徴、及びそれらを達成する方法は、添付される図面と共に詳細に後述する実施形態を参照すれば明確になるであろう。しかし、本発明は、以下で開示する実施形態に限定されるものではなく、異なる多様な形態で具現することが可能である。本実施形態は、単に本発明の開示が完全になるように、本発明が属する技術分野で通常の知識を有する者に対して発明の範疇を完全に知らしめるために提供するものであり、本発明は、請求項の範疇によってのみ定義される。明細書全体において同一参照符号は同一構成要素を指す。
【0020】
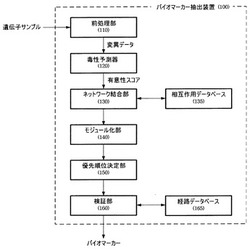
図1は、本発明の一実施形態によるバイオマーカー抽出装置100を図示するブロック図である。バイオマーカー抽出装置100は前処理部110、毒性予測器120、ネットワーク結合部130、モジュール化部140、優先順位決定部150、検証部160を含み構成される。ここで、ネットワーク結合部130、検証部160は実施形態によっては省略できる構成要素である。また、相互作用データベース135は、ネットワーク結合部130と連動し、経路(pathway)データベース165は検証部160と連動する。このように図1(後述する図2〜図4も同様である)に図示する機能ブロックはハードウェアシステムで動作することができ、前記ハードウェアシステムはパーソナルコンピュータ(携帯用または固定された装置を問わない)または通信ネットワークに接続されるサーバ-クライアント装置である。例えば、前記ブリックはソフトウェアモジュールとして実現し、プロセッサとメモリを備えるハードウェアシステムで動作することができる。前記メモリは、前記機能ブロックに関するモジュールをロードして前記プロセッサに提供するが、前記プロセッサが前記ロードしたモジュールを処理する方式によりバイオマーカー抽出装置100が実現することができる。
【0021】
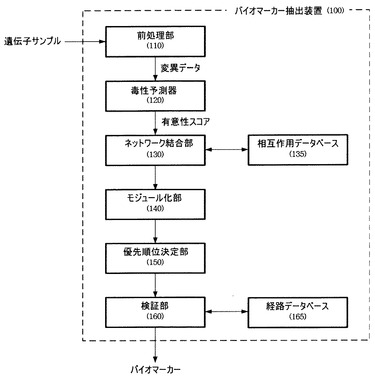
前処理部110は、遺伝子サンプルから遺伝子にマッピングされた変異データ(variation)を抽出する。具体的には、前処理部110は図2に図示するように、疾患群比較部112、変異抽出部114、変異データベース115および変異マッピング部116を含み、構成される。
【0022】
具体的には、疾患群比較部112は疾患群の変異と正常群の変異を抽出して両者間の比較により疾患群に存在する変異を獲得する。変異抽出部114は、前記獲得した疾患群変異と既知の変異データベース115を比較し、新たな変異のみを抽出する。また、変異マッピング部116は、前記抽出した新たな変異のうち蛋白質発現時にアミノ酸が変化する種類、すなわち非同義置換(non-synonymous)のみを抽出してこれを機能遺伝子にマッピングする。
【0023】
シーケンシングデータから遺伝子型が分解された遺伝子変異データは、GFF3またはGVFなどのファイル形式で保存され、現在はGFF3 (Genetic Feature Format)が最も広く使用されている。表1は、遺伝子変異データをGFF3ファイル形式に整理した例である。
【0024】
【表1】

【0025】
遺伝子変異データは、遺伝子変異の染色体番号(Chr)、該当染色体での変異開始位置(Start)、該当染色体での変異終了位置(End)情報と属性を含む。属性(attributes)は、使用した標準ゲノムでの該当位置の遺伝子型(reference)およびシーケンシング対象の遺伝子型(genotype)および他の付加情報を含む。前記遺伝子型情報が異型接合性(heterozygocity)を有する場合、シンボルYを活用して2個の塩基配列を同時に表示する。
【0026】
特定疾患群から得られた遺伝子変異データから、正常群から得られた遺伝子変異データおよび既知の遺伝子変異に関する情報(例:dbSNP、1000ゲノムプロジェクト(100 genome project)など)を除去すると、該当疾患に特異的遺伝子変異情報のみが残る。疾患群比較部112は、このような方式により疾患群に存在する変異を獲得し、変異抽出部114は、前記獲得した疾患群変異と既知の変異データベース115を比較して新たな変異のみを抽出することができる。
【0027】
変位マッピング部116は、前記新たな変異を該当疾患に特異的遺伝子変異情報に対して知らされている遺伝子にマッピングし、各遺伝子変異が該当遺伝子のイントロンに位置するのか、該当遺伝子が発現させる蛋白質のアミノ酸を変化させるのか、STOPコドン(codon)を生成するのかなどの情報を抽出する。
【0028】
次の表2は、遺伝子に変異をマッピングしたマッピング情報を例示的に示す。
【0029】
【表2】
【0030】
表2では、例えば、K105Kは塩基配列の変化があったにもかかわらず、蛋白質でのアミノ酸の変異がない変異を、V203Mは蛋白質配列の203番目のVがMに置換したことを意味する。また、変異領域がイントロン(intron)である場合にはコーディングを行わないため、蛋白質IDおよびアミノ酸の変化情報が記録されていない。また、「NP_xxxxxx」は、蛋白質配列に関するIDの一種であり、米国の国立生物工学情報センター(NCBIの)参照配列ID(refseq ID)である。
【0031】
再び、図1を参照すると、前処理部110によって抽出した変異データは毒性予測器120に提供する。毒性予測器120は、前記変異データに基づき該当遺伝子の機能に発生する障害を定量化する。前記変異データは、遺伝子にマッピングされた遺伝子変異情報、特に、蛋白質コーディング領域でアミノ酸の置換を起こす変異を含む。
【0032】
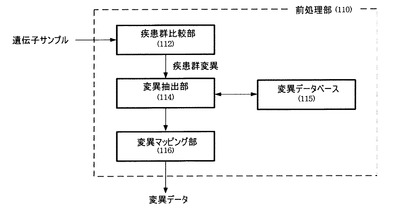
これのため、毒性予測器120は図3に図示するように、毒性計算部170、有意性計算部180およびスコア計算部190を含んで構成される。
【0033】
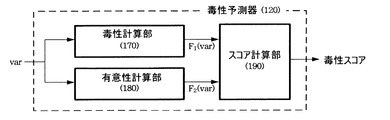
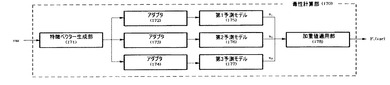
毒性計算部170は、入力された変異データ(var)を複数の毒性予測モデルに適用し、それぞれの毒性を得た後、それぞれの毒性に加重値を適用し、合算することによって変異データの毒性(加重毒性)を計算する。図4は、毒性計算部170の具体的な構成を示す。
【0034】
特徴ベクター生成部171は、入力された変異データから多様な要素を含む特徴ベクターを生成する。このような特徴ベクターを構成する要素としては、例えば遺伝子変異とマッピングされる遺伝子および蛋白質で該当位置でのアミノ酸の様々な生物種間の保存スコア(conservation score)、アミノ酸置換が起こす生化学性質の変化(hydrophobicity)、蛋白質の構造的特徴の変化(蛋白質相互作用インターフェースの変化、アミノ酸サイズなど)、イントロンのスプライス部位(intron splice junction)(情報)の有無、5-UTR(five prime untranslated region)の変異位置などがある。
【0035】
アダプタ(172、173、174)は、前記生成した特徴ベクターのうちそれぞれの予測モデルに必要な要素を選別し、該当予測モデル(175、176、177)に提供する。前記予測モデルは蛋白質配列内で個別のnsSNP(non-synonymous Single Nucleotide Polymorphism)を見つけるように研究された従来の技法である。nsSNPは、遺伝的変異のうち蛋白質配列を構成するアミノ酸の置換を誘発する遺伝的変異を意味する。またnsSNPは、蛋白質本来の機能に悪影響を及ぼす可能性があるため、慎重に扱われる。このような予測モデルとしてはSIFT(Sorting Tolerant From Intolerant)、PolyPhen、MAPP((Map Annotator and Pathway Profiler)などを使用することができ、これ以外のモデルを使用する場合もある。また、図4では予測モデルが3個であるものを例示しているが、任意の複数の予測モデルを使用できるのはもちろんである。
【0036】
以下、現在知られている代表的な予測モデルであるSIFT、PolyPhenおよびMAPPについて簡略に説明する。
【0037】
SIFTツールの基本前提は、重要なアミノ酸はタンパク質ファミリー(protein family)内に保存される傾向があり、保存性が高い位置のアミノ酸の置換は蛋白質本来機能に影響を与える。SIFTは、蛋白質配列データベースから入力配列と配列類似性を有する蛋白質配列を獲得した後、これらの配列を利用してPSSM(position specific scoring matrices)を生成し、入力配列の各アミノ酸の配列保存点数、アミノ酸の疎水性、配列位置でのアミノ酸の存在確率を計算し、各アミノ酸置換が有する毒性を見つける。
【0038】
ただし、SIFTは入力された蛋白質配列が配列類似性検索によって集めた他の配列蛋白質と高度に類似する場合にPSSMが非常に高く保存された性向に生成されるため、有害ではないアミノ酸置換も毒性が高いものと誤って予測する誤謬が発生し得る。SIFTは約69%の敏感性(sensitivity)と共に13%の特殊性(specificity)を表す。
【0039】
配列類似性と蛋白質の特性情報、蛋白質構造データを結合してアミノ酸置換による毒性を予測する進化した方法としてPolyPhenがある。PolyPhenはSIFTで利用する配列保存性データ以外にスイスプロット(Swiss-Prot)の特徴テーブルと蛋白質構造を追加に利用する。PolyPhenはPSIC(Position Specific Independent Counts)スコアの差異値、アミノ酸置換部位および置換形態を総合してアミノ酸置換の毒性を予測する。PolyPhenは約68%の敏感性と16%の特殊性を表す。
【0040】
一方、蛋白質配列の類似性とアミノ酸の物理化学的な特性を結合してアミノ酸置換の類似性を予測するモデルとしてMAPPが開発された。MAPPは配列類似性を示すタンパク質ファミリーを利用して配列の整列を行い、各位置に存在するアミノ酸の配列差異とアミノ酸が有する物理化学的特性(疎水性、極性、容積)とを共に考慮し、蛋白質機能に影響を及ぼす可能があるすべてのアミノ酸置換を予測する。
【0041】
以上のような多様な予測モデル(175、176、177)を利用すると、蛋白質配列内で個別のnsSNPを検索することによって該当遺伝子変異が有するスコアを得ることができる。それぞれの予測モデルから得たスコア(s1, s2, s3)は加重値適用部178に提供する。
【0042】
加重値適用部178は、前記それぞれのスコア(s1, s2, s3)を0〜1間の値に正規化した後、それぞれの正規化したスコアに加重値を乗じ、これを合算して毒性(F1(var))を計算する。前記加重値は、既知の疾患遺伝子変異を学習データとして活用して経験から得られる値である。したがって、毒性(F1(var))は次の式1のように計算する。
【0043】
【数1】
【0044】
加重値適用部178は、このように計算した毒性を再び0〜1間の値に正規化することもできる。
【0045】
一方、疾患群に含まれた多数のサンプルのうち反復された位置から発見した遺伝子変異は重要な変異と判断することができるため、各遺伝子変異の頻度により有意性を判断することができる。再び図3を参照すると、有意性計算部180は前記遺伝子変異の頻度、すなわち確率分布に基づき、該当遺伝子変異の有意性を計算する。前記遺伝子変異の発生確率(p(var))は、疾患群サンプルから該当遺伝子変異が発見された確率を意味し、例えば、最尤推定(MLE、Maximum Likelihood Estimation)またはベイズ確率(Bayesian probability)推論により得られる。
【0046】
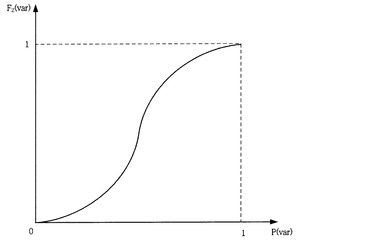
このように得られた確率(p(var))をそのまま有意性として利用することもできるが、実際有意性として利用するためにはマッピング関数により修正を行う必要がある。マッピング関数は図5に図示するように、0〜1間の確率(p(var))を0〜1の間の有意性(F2(var))に変換する関数である。マッピング関数の形態は多様に設定できるが、好ましくは図5のように、0と1の近くでは相対的に小さい勾配を有し、0.5の近くでは相対的に大きい勾配を有する。すなわち、有意性は0と1の近くの確率に比べ、0.5の近くではより高い敏感度を有する。このようなマッピング関数は例えば式2のように表示される。ここで、αは常数である。
【0047】
【数2】
【0048】
毒性計算部170から得られた毒性(0〜1間の値)と、有意性計算部180から得られた有意性(0〜1間の値)は最終的にスコア計算部190に提供する。スコア計算部190は前記毒性および有意性を組合わせて最終的な毒性スコアを計算する。例えば、毒性スコア(f(var))は次の式3のように毒性および有意性の合算により求めることができるが、これに限らず、毒性および有意性のうち少なくとも1つ以上を反映できる多様な計算式により求めることができる。すなわち、毒性と有意性は共に使用するときに好ましい効果があるが、これに限らず、独立的に使用することもできる。
【0049】
【数3】
【0050】
このように各々遺伝子変異は特定遺伝子にマッピングされ、それぞれの遺伝子の毒性がどの程度であるかを推定することに使用される。しかし、大きな影響を与える1つの遺伝子変異も重要であるが、比較的に少ない影響を与える多数の遺伝子変異が含まれる遺伝子も有意であるといえる。したがって、スコア計算部190は1つの遺伝子内に含まれた遺伝子変異の毒性スコア(f(var))を遺伝子の長さで割った値により最終毒性スコアを求めることもできる。この場合には、最終毒性スコア(s(Gene))は次の式4のように求めることができる。
【0051】
【数4】
【0052】
1つの遺伝子内に存在する遺伝子変異が有する毒性スコアの合計(Σf(var))は、遺伝子長さで割ることによって最終毒性(s(Gene))が求められる。これは前述したように、遺伝子内に大きな影響を与える1つの遺伝子変異だけではなく、多数の遺伝子変異を総合的に考慮するためであり、また毒性スコアの合計(Σf(var))が同一であれば、遺伝子の長さが短いほど最終毒性スコアが大きくなることを意味する。すなわち、単位遺伝子の長さ当たりにより高い毒性スコアは遺伝子変異が該当遺伝子に対してより有意な毒性を示すと推測することができる。
【0053】
再び図1を参照すると、ネットワーク結合部130は毒性予測器120から得られた遺伝子から発現する蛋白質を既知の蛋白質相互作用データベース135と結合して相互作用ネットワークを生成する。一般的には、遺伝子から得られた遺伝子変異の実際の発現は、生体機能を示す蛋白質単位で行われるといえる。すなわち、前記遺伝子変異が有害であるとしても実際の蛋白質単位では潜在的な毒性が発現しない場合もあり、多様な遺伝子変異の組合せによって多様な発現形態が現れる。このような相互作用ネットワークは遺伝子、蛋白質および酵素の順で結合が行われることにより遺伝子ノードの数が増加されることがある。相互作用ネットワークの結合過程のより詳細は、例えば、Automated Network Analysis Identifies Core Pathways in Clioblastoma (www.plosone.org、February 2010,volume 5,issue 2,e8918)を参照して分かる。本発明において、相互作用ネットワークの結合過程は遺伝子単位での毒性のみを求める場合、省略することもできる。
【0054】
モジュール化部140は、毒性スコアが所定の臨界値以上の遺伝子が、遺伝子ネットワーク上に密集している遺伝子ネットワークのサブモジュールを探索する。より具体的には、モジュール化部140は遺伝子毒性が臨界値以上の遺伝子がネットワーク上に密集しているサブモジュールを見つけるために密集分布を統計的検定を行う。前記統計的検定手法の例として、超幾何分布(hypergeometic distribution)を利用し得る。
【0055】
遺伝子ネットワーク上の全体遺伝子個数がNであり、遺伝子ネットワークの全体遺伝子のうち毒性スコアが所定の臨界値を上回る遺伝子の個数がnであり、遺伝子ネットワークのサブモジュール内に存在する遺伝子の個数がmであると仮定すると、遺伝子ネットワークのサブモジュールで毒性スコアが前記臨界値を上回る遺伝子の個数がk個になる確率(P(X=k))は次の式5のとおりである。
【0056】
【数5】
【0057】
ここで、
はN個のうちn個を組み合わせた個数、すなわちNCnを意味する。
【0058】
したがって、遺伝子ネットワークのサブモジュールで毒性スコアが前記臨界値を上回る遺伝子の個数がk個を超過する確率(p)は次の式6によりに計算することができる。
【0059】
【数6】
【0060】
前記確率(p)値は、特定サブモジュール内に毒性スコアが所定の臨界値を超過する遺伝子個数が特定個数(k)を超過して密集している確率を意味する。前記臨界値は多様な方法により決定できるが、一例としては、全体遺伝子の毒性スコア分布で所定の百分位数(1 percentile、5 percentile、10 percentileなど)を基準に決定することができる。このように、特定サブモジュールに対して前記確率(p)が高いほどより有意のサブモジュールとして評価することができる。
【0061】
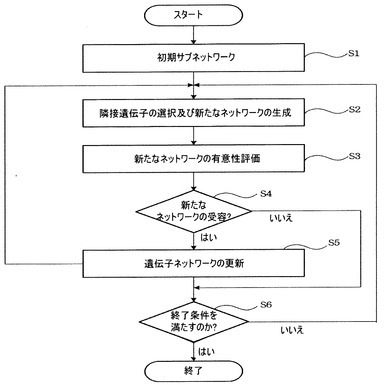
モジュール化部140は、通常のgreedy探索アルゴリズム、確率的探索アルゴリズム(例、simulated annealing)などを活用して実際にサブモジュールを探索することができ、その具体的な過程は図6に示すフローチャートを参照して説明する。
【0062】
先に、モジュール化部140は初期サブネットワークを設定する(S1)。初期ネットワークは、毒性スコアが有意であるすべての遺伝子(例、毒性スコアが上位5%以内の遺伝子)を単一のノードとして有するネットワークを意味する。このような初期ネットワークを構成するノードを始めとして探索アルゴリズムを適用して最適の有意性を有する遺伝子ネットワークサブモジュールを探索する。
【0063】
モジュール化部140は隣接遺伝子(現在遺伝子と直接連結された遺伝子)を選択して結合して新たなネットワークを生成し(S2)、新たなネットワークの有意性を評価する(S3)。すなわち、前記初期ノードの隣接遺伝子を新たなノードに結合し、新たなネットワークを生成した後、結合したノードで構成された単位(サブモジュールの予備ステップ)に対して有意性を評価する。このような有意性は例えば、前述した超幾何分布での確率(p)を利用して評価することができる。
【0064】
モジュール化部140は、このように構成した新たなネットワークが有意である場合(S4のはい)には現在のネットワークを前記有意のネットワークに更新し(S5)、再びステップS2に戻る。仮に、有意ではない場合(S4のいいえ)にはネットワークの更新を行わず、終了条件を満たしているかを確認し、終了条件を満たしていれば(S6のはい)サブモジュールの探索を終了する。前記終了条件が満たされていなければ(S6のいいえ)ステップS2に戻る。
【0065】
このようなネットワーク更新過程が終了すると、最終に更新された遺伝子ネットワークに含まれるサブモジュールを確定(探索完了)することができる。
【0066】
再び図1を参照すると、優先順位決定部150はモジュール化部140によって探索した複数のサブモジュール間の優先順位を決定する。すなわち、遺伝子変異に基づいて発見した遺伝子ネットワークのサブモジュールに対して遺伝子発現データの変化とどれほど相関関係を有するかを評価し、該当サブモジュールの優先順位を決定する。
【0067】
探索したサブモジュールに対する遺伝子発現パターンの分解は、該当サブモジュールだけではなく、該当サブモジュールに直接連結された遺伝子を含んで行うことが好ましい。これは転写調節因子のような遺伝子に変異が発生した際には、自身の発現より転写調節因子がターゲットにするターゲット遺伝子の発現パターンに変化が生じやすいからである。
【0068】
正常および疾患の二つの条件から調査された遺伝子発現データは、前処理により二つの条件の間での発現差異をZ-スコア(z-score)の形態で計算することができる。例えば、それぞれのサブモジュールに対して、前記サブモジュールに直接連結された遺伝子集合をGとすると、サブモジュールの優先度を評価する指数(es)は次の式7により求める。
【0069】
【数7】
【0070】
ここで、ziは該当サブモジュールに直接連結された遺伝子の集合において、それぞれの遺伝子が有する毒性スコアのZ-スコア値を意味し、|G|は該当サブモジュールに直接連結された遺伝子集合のサイズ(遺伝子の個数)を意味する。Z-スコアは統計学分野で知られているように、現在変数(x)から平均(μ)を減じた後、標準偏差(σ)で割った値であり、現在変数(毒性スコア)が平均(μ)から何σだけ離隔されているかを示す指標である。このような過程により得られた優先順位で並べ替えたサブモジュールは結局、特定遺伝子の発現との相関関係を示すバイオマーカーとしての機能を有する。したがって、疾患群と正常群の比較により得られた疾患特異的配列の変異は該当遺伝子の機能にいかなる変化を与え、また障害を起こすかを予測することができ、さらに個別遺伝子障害が全体生物システムでの相互作用にいかなる影響を及ぼすかについての情報を提供することができる。
【0071】
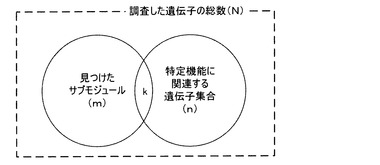
検証部160は、前記得られた優先順位で並べ替えたサブモジュールを既知の経路データベース165と比較して機能別の関連性を評価する。最も広く使用される方法は、超幾何分布を使用する方法であり、それぞれのサブモジュールに対して経路データベース165から特定生物学的機能単位で抽出した遺伝子集合に対して重畳する遺伝子数字の有意性を計算する。すなわち、図7に図示するように、調査した遺伝子の総数(N)、特定生物学的機能と関連する遺伝子集合の遺伝子個数(n)、遺伝子ネットワークから見つけたサブモジュール内の遺伝子個数(m)および遺伝子サブモジュールと特定遺伝子集合内に共通に存在する遺伝子個数(k)から有意性を示す確率を計算する。前記確率が高いほど最終に得たサブモジュールはより高い有意性を有する。
【0072】
図1〜図4の各構成要素は、ソフトウェア(software)またはFPGA(field-programmable gate array)やASIC(application-specific integrated circuit)のようなハードウェア(hardware)を意味する。しかし、前記構成要素は、ソフトウェアまたはハードウェアに限定されず、アドレッシング(addressing)できる保存媒体にあるように構成することもでき、1つまたはそれ以上のプロセッサを実行させるように構成することもできる。前記構成要素から提供される機能は、さらに細分化した構成要素によって実現することができ、複数の構成要素を組み合わせて特定の機能を行う1つの構成要素として実現することもできる。
【0073】
以上添付する図面を参照して本発明の実施形態について説明したが、本発明が属する技術分野で通常の知識を有する者は、本発明がその技術的思想や必須の特徴を変更しない範囲で、他の具体的な形態で実施され得ることを理解することができる。したがって、上記実施形態はすべての面において例示的なものであり、限定するものではないと理解しなければならない。
【符号の説明】
【0074】
100 バイオマーカー抽出装置
110 前処理部
120 毒性予測器
130 ネットワーク結合部
135 相互作用データベース
140 モジュール化部
150 優先順位決定部
160 検証部
165 経路データベース
170 毒性計算部
180 有意性計算部
190 スコア計算部
【技術分野】
【0001】
本発明は、生物情報学(Bioinformatics)技術に関するものであって、より詳細には配列変異が遺伝子機能に及ぼす毒性分解に基づきより高信頼性のバイオマーカーを抽出する装置および方法に関するものである。
【背景技術】
【0002】
ヒトゲノムプロジェクトが完了した後、ヒトのDNA塩基配列が解読され、またそこからヒトの遺伝子の多様な機能が明らかになっている。特に、多様な遺伝子変異が発見されており、これらが人間の形質の差異を起こすだけではなく、特定疾患の原因として作用されることが明らかになるにつれ、ヒトのゲノム分解に関する研究はさらに加速化されつつある。しかし、ヒトのゲノムから発生し得る膨大な遺伝的変異のうち、どれが実質の病因になる変異であるかを調べるのは難しい。
【0003】
最近ではこのような難点を解消するための1つの代案として次世代シーケンシング技術(NGS、Next Generation Sequencing)が研究されている。この技術により個人の全ゲノムの塩基配列化が可能になり、疾患群と正常群の塩基配列および変異比較ツールにより、疾患特異的遺伝子変異を抽出することも可能になった。
【0004】
一方、これとは異なって、DNAシーケンシングの代わりにSNP(single nucleotide polymorphism)チップにより個体群(population)の統計分解に基づくGWAS(Genome Wide Association Study)解析技術も研究されている。この技術によれば、数千〜数万人から得られるSNPデータ解析により、特定疾患群から頻繁に発生する有意の遺伝的変異を抽出することができる。
【0005】
しかし、多様な解析技術によって遺伝的変異を抽出したとしても、このような遺伝的変異を含む遺伝子の実際発現および機能障害があるかどうかを確認するためには追加の実験が要求され、これは時間および費用の面から相当な損失につながる。さらには、個別遺伝子変異情報のみでは特定疾患群を起こす多様な病因に対して十分な説明を提供することができない。
【0006】
これを解決するため、生物学的要素の相互作用を分解し、これに基づき生物システムを解読するいわゆるシステム生物学と呼ばれる技術も研究されている。すなわち、遺伝子から発現する生物学的機能は、生命を維持するために他の遺伝子から発現する機能と結合して行い、頻繁に起こる外部環境の変化にも自らの恒常性を維持することができる。このような技術は、生物学的要素のネットワーク分解により、それぞれの変異を有する遺伝子の機能的位置および相互作用分解を行い、遺伝的変異が周辺要素にいかなる影響を与え、またこのような影響がどのように広がるかを把握することができる。また、遺伝的変異が既知の遺伝子相互作用、遺伝子調節回路、蛋白質相互作用、代謝、信号伝達回路といかなる関連性を有するかを説明できる基礎となる。
【0007】
正常の細胞活動のために必要な細胞内の多様なプロセスは、機能モジュールというより小さくてかつ特殊な蛋白質あるいは遺伝子の集団として作動している。個別遺伝子において個別nsSNP(non-synonymous Single Nucleotide Polymorphism)によって発生する蛋白質機能の毒性予測(toxicity prediction)をする一連の方法としては、SIFT(Sorting Tolerant From Intolerant)、PolyPhen、MAPP(Map Annotator and Pathway Profiler)などが提示されているが、このような方法だけでは高複雑度の疾患に対する病因や疾患マーカー(marker)を見つけることに限界がある。
【0008】
また一般的にSNPのうち蛋白質機能に有害な影響を与えるSNPの割合は非常に低い。したがって、GSEA(Gene set enrichment analysis)とSNPを利用した分解方法の場合、SNPが蛋白質機能に毒性(toxicity)を与えるかどうかに関係なく、SNPにより予測されたすべてのデータを利用するため、実際に特定疾患と密接な関係がない生物学的経路(pathway)や遺伝子セットが統計的に有意であると誤った判断をする可能性が高い。したがって、疾患特異的遺伝子変異を生体分子ネットワークに基づいて分解し、発見した生体分子ネットワークに属する遺伝子の発現パターンを分解し、特定疾患に関係があるバイオマーカーをより正確に特定できる技法の開発が必要である。
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、上記の必要性に鑑みてなされたものであって、従来のゲノム変異あるいは遺伝子発現パターンのうち、それぞれの一部分のみを利用して行っていた相互作用モジュール化および分解の限界を超え、配列変異が遺伝子機能に及ぼす毒性分解に基づき、より高信頼性のバイオマーカーを検出することを目的とする。
【0010】
特に、本発明は、前記バイオマーカーの検出において重要な影響を及ぼす毒性について多角的な方法により定量化した毒性予測技法を開発することを他の目的とする。
【0011】
本発明の技術的課題は、以上で言及した技術的課題に制限されず、言及されていないまた他の技術的課題は次の記載から当業者に明確に理解できるであろう。
【課題を解決するための手段】
【0012】
前記技術的課題を達成するための本発明の一実施形態による、配列変異が遺伝子機能にいかなる変化を与えるかを分解し、特定疾患の原因になるバイオマーカーを抽出するバイオマーカー抽出装置であって、遺伝子サンプルの配列を分解して遺伝子にマッピングされた変異データを抽出する前処理部と、前記変異データに基づき前記遺伝子の機能に発生する障害を定量化した毒性スコアを求める毒性予測器、および前記毒性スコアが所定の臨界値以上の遺伝子が、遺伝子ネットワークに密集している遺伝子ネットワークのサブモジュールを探索するモジュール化部を含む。
【0013】
前記技術的課題を達成するための本発明の一実施形態による、配列変異データが遺伝子の機能に影響を及ぼす障害を定量化するための毒性予測装置であって、前記変異データを複数の毒性予測モデルに適用し、それぞれの毒性を得た後、前記それぞれの毒性に加重値を適用することによって加重毒性を計算する毒性計算部と、前記変異データの頻度に基づき、該当遺伝子変異の有意性(significance)を計算する有意性計算部、および前記加重毒性および前記有意性を組み合わせて毒性スコアを計算するスコア計算部を含む。
【0014】
前記技術的課題を達成するための本発明の一実施形態による、配列変異が遺伝子機能にいかなる変化を与えるかを分解し、特定疾患の原因になるバイオマーカーを抽出する方法であって、遺伝子に含まれた変異データに基づき前記遺伝子の機能に発生する障害を定量化した毒性スコアを求めるステップと、前記毒性スコアが所定の臨界値以上の遺伝子が集められたサブモジュールを遺伝子ネットワークから探索するステップ、および前記探索した複数のサブモジュール間の優先順位を決定するステップを含む。
【0015】
前記技術的課題を達成するための本発明の一実施形態による、配列変異データが遺伝子の機能に影響を及ぼす障害を定量化するための毒性予測方法であって、前記変異データから多様な要素を含む特徴ベクターを生成するステップと、前記生成された特徴ベクターのうちそれぞれの予測モデルに必要な要素を選別するステップと、前記選別した要素の入力を受けて蛋白質配列内で個別のスコアを提供するステップ、および前記予測モデルの出力に加重値を適用して合算し、加重毒性を計算するステップを含む。
【発明の効果】
【0016】
前記のような本発明によれば、疾患群と正常群の比較により得られた疾患特異的配列の変異が該当遺伝子の機能にいかなる変化や障害を起こすかを予測することができる。またこのような発現パターン分解とネットワーク分解を併行することによって個別の遺伝子障害が全体生物システムでの相互作用にいかなる影響を及ぼすかについて情報を提供して疾患メカニズムに基づいたバイオマーカーを発掘できる効果がある。
【0017】
このようなバイオマーカーは特定疾患の診断、特定疾患を治療する薬品の開発、副作用の予防に広く利用される。
【図面の簡単な説明】
【0018】
【図1】本発明の一実施形態によるバイオマーカー抽出装置を図示するブロック図である。
【図2】図1に示す前処理部の細部構成を図示するブロック図である。
【図3】図1に示す毒性予測器の細部構成を図示するブロック図である。
【図4】図3に示す毒性計算部の細部構成を図示するブロック図である。
【図5】毒性計算部で使用されるマッピング関数の一例を示す図である。
【図6】モジュール化部でサブモジュールの探索を行う過程を詳細に図示するフローチャートである。
【図7】遺伝子サブモジュールと特定の遺伝子集合内に共通して存在する遺伝子個数から有意性を検証する概念を示す図である。
【発明を実施するための形態】
【0019】
以下、本発明のバイオマーカー抽出装置および方法を実施するための形態の具体例を、図面を参照しながら説明する。本発明の本発明の利点、特徴、及びそれらを達成する方法は、添付される図面と共に詳細に後述する実施形態を参照すれば明確になるであろう。しかし、本発明は、以下で開示する実施形態に限定されるものではなく、異なる多様な形態で具現することが可能である。本実施形態は、単に本発明の開示が完全になるように、本発明が属する技術分野で通常の知識を有する者に対して発明の範疇を完全に知らしめるために提供するものであり、本発明は、請求項の範疇によってのみ定義される。明細書全体において同一参照符号は同一構成要素を指す。
【0020】
図1は、本発明の一実施形態によるバイオマーカー抽出装置100を図示するブロック図である。バイオマーカー抽出装置100は前処理部110、毒性予測器120、ネットワーク結合部130、モジュール化部140、優先順位決定部150、検証部160を含み構成される。ここで、ネットワーク結合部130、検証部160は実施形態によっては省略できる構成要素である。また、相互作用データベース135は、ネットワーク結合部130と連動し、経路(pathway)データベース165は検証部160と連動する。このように図1(後述する図2〜図4も同様である)に図示する機能ブロックはハードウェアシステムで動作することができ、前記ハードウェアシステムはパーソナルコンピュータ(携帯用または固定された装置を問わない)または通信ネットワークに接続されるサーバ-クライアント装置である。例えば、前記ブリックはソフトウェアモジュールとして実現し、プロセッサとメモリを備えるハードウェアシステムで動作することができる。前記メモリは、前記機能ブロックに関するモジュールをロードして前記プロセッサに提供するが、前記プロセッサが前記ロードしたモジュールを処理する方式によりバイオマーカー抽出装置100が実現することができる。
【0021】
前処理部110は、遺伝子サンプルから遺伝子にマッピングされた変異データ(variation)を抽出する。具体的には、前処理部110は図2に図示するように、疾患群比較部112、変異抽出部114、変異データベース115および変異マッピング部116を含み、構成される。
【0022】
具体的には、疾患群比較部112は疾患群の変異と正常群の変異を抽出して両者間の比較により疾患群に存在する変異を獲得する。変異抽出部114は、前記獲得した疾患群変異と既知の変異データベース115を比較し、新たな変異のみを抽出する。また、変異マッピング部116は、前記抽出した新たな変異のうち蛋白質発現時にアミノ酸が変化する種類、すなわち非同義置換(non-synonymous)のみを抽出してこれを機能遺伝子にマッピングする。
【0023】
シーケンシングデータから遺伝子型が分解された遺伝子変異データは、GFF3またはGVFなどのファイル形式で保存され、現在はGFF3 (Genetic Feature Format)が最も広く使用されている。表1は、遺伝子変異データをGFF3ファイル形式に整理した例である。
【0024】
【表1】
【0025】
遺伝子変異データは、遺伝子変異の染色体番号(Chr)、該当染色体での変異開始位置(Start)、該当染色体での変異終了位置(End)情報と属性を含む。属性(attributes)は、使用した標準ゲノムでの該当位置の遺伝子型(reference)およびシーケンシング対象の遺伝子型(genotype)および他の付加情報を含む。前記遺伝子型情報が異型接合性(heterozygocity)を有する場合、シンボルYを活用して2個の塩基配列を同時に表示する。
【0026】
特定疾患群から得られた遺伝子変異データから、正常群から得られた遺伝子変異データおよび既知の遺伝子変異に関する情報(例:dbSNP、1000ゲノムプロジェクト(100 genome project)など)を除去すると、該当疾患に特異的遺伝子変異情報のみが残る。疾患群比較部112は、このような方式により疾患群に存在する変異を獲得し、変異抽出部114は、前記獲得した疾患群変異と既知の変異データベース115を比較して新たな変異のみを抽出することができる。
【0027】
変位マッピング部116は、前記新たな変異を該当疾患に特異的遺伝子変異情報に対して知らされている遺伝子にマッピングし、各遺伝子変異が該当遺伝子のイントロンに位置するのか、該当遺伝子が発現させる蛋白質のアミノ酸を変化させるのか、STOPコドン(codon)を生成するのかなどの情報を抽出する。
【0028】
次の表2は、遺伝子に変異をマッピングしたマッピング情報を例示的に示す。
【0029】
【表2】
【0030】
表2では、例えば、K105Kは塩基配列の変化があったにもかかわらず、蛋白質でのアミノ酸の変異がない変異を、V203Mは蛋白質配列の203番目のVがMに置換したことを意味する。また、変異領域がイントロン(intron)である場合にはコーディングを行わないため、蛋白質IDおよびアミノ酸の変化情報が記録されていない。また、「NP_xxxxxx」は、蛋白質配列に関するIDの一種であり、米国の国立生物工学情報センター(NCBIの)参照配列ID(refseq ID)である。
【0031】
再び、図1を参照すると、前処理部110によって抽出した変異データは毒性予測器120に提供する。毒性予測器120は、前記変異データに基づき該当遺伝子の機能に発生する障害を定量化する。前記変異データは、遺伝子にマッピングされた遺伝子変異情報、特に、蛋白質コーディング領域でアミノ酸の置換を起こす変異を含む。
【0032】
これのため、毒性予測器120は図3に図示するように、毒性計算部170、有意性計算部180およびスコア計算部190を含んで構成される。
【0033】
毒性計算部170は、入力された変異データ(var)を複数の毒性予測モデルに適用し、それぞれの毒性を得た後、それぞれの毒性に加重値を適用し、合算することによって変異データの毒性(加重毒性)を計算する。図4は、毒性計算部170の具体的な構成を示す。
【0034】
特徴ベクター生成部171は、入力された変異データから多様な要素を含む特徴ベクターを生成する。このような特徴ベクターを構成する要素としては、例えば遺伝子変異とマッピングされる遺伝子および蛋白質で該当位置でのアミノ酸の様々な生物種間の保存スコア(conservation score)、アミノ酸置換が起こす生化学性質の変化(hydrophobicity)、蛋白質の構造的特徴の変化(蛋白質相互作用インターフェースの変化、アミノ酸サイズなど)、イントロンのスプライス部位(intron splice junction)(情報)の有無、5-UTR(five prime untranslated region)の変異位置などがある。
【0035】
アダプタ(172、173、174)は、前記生成した特徴ベクターのうちそれぞれの予測モデルに必要な要素を選別し、該当予測モデル(175、176、177)に提供する。前記予測モデルは蛋白質配列内で個別のnsSNP(non-synonymous Single Nucleotide Polymorphism)を見つけるように研究された従来の技法である。nsSNPは、遺伝的変異のうち蛋白質配列を構成するアミノ酸の置換を誘発する遺伝的変異を意味する。またnsSNPは、蛋白質本来の機能に悪影響を及ぼす可能性があるため、慎重に扱われる。このような予測モデルとしてはSIFT(Sorting Tolerant From Intolerant)、PolyPhen、MAPP((Map Annotator and Pathway Profiler)などを使用することができ、これ以外のモデルを使用する場合もある。また、図4では予測モデルが3個であるものを例示しているが、任意の複数の予測モデルを使用できるのはもちろんである。
【0036】
以下、現在知られている代表的な予測モデルであるSIFT、PolyPhenおよびMAPPについて簡略に説明する。
【0037】
SIFTツールの基本前提は、重要なアミノ酸はタンパク質ファミリー(protein family)内に保存される傾向があり、保存性が高い位置のアミノ酸の置換は蛋白質本来機能に影響を与える。SIFTは、蛋白質配列データベースから入力配列と配列類似性を有する蛋白質配列を獲得した後、これらの配列を利用してPSSM(position specific scoring matrices)を生成し、入力配列の各アミノ酸の配列保存点数、アミノ酸の疎水性、配列位置でのアミノ酸の存在確率を計算し、各アミノ酸置換が有する毒性を見つける。
【0038】
ただし、SIFTは入力された蛋白質配列が配列類似性検索によって集めた他の配列蛋白質と高度に類似する場合にPSSMが非常に高く保存された性向に生成されるため、有害ではないアミノ酸置換も毒性が高いものと誤って予測する誤謬が発生し得る。SIFTは約69%の敏感性(sensitivity)と共に13%の特殊性(specificity)を表す。
【0039】
配列類似性と蛋白質の特性情報、蛋白質構造データを結合してアミノ酸置換による毒性を予測する進化した方法としてPolyPhenがある。PolyPhenはSIFTで利用する配列保存性データ以外にスイスプロット(Swiss-Prot)の特徴テーブルと蛋白質構造を追加に利用する。PolyPhenはPSIC(Position Specific Independent Counts)スコアの差異値、アミノ酸置換部位および置換形態を総合してアミノ酸置換の毒性を予測する。PolyPhenは約68%の敏感性と16%の特殊性を表す。
【0040】
一方、蛋白質配列の類似性とアミノ酸の物理化学的な特性を結合してアミノ酸置換の類似性を予測するモデルとしてMAPPが開発された。MAPPは配列類似性を示すタンパク質ファミリーを利用して配列の整列を行い、各位置に存在するアミノ酸の配列差異とアミノ酸が有する物理化学的特性(疎水性、極性、容積)とを共に考慮し、蛋白質機能に影響を及ぼす可能があるすべてのアミノ酸置換を予測する。
【0041】
以上のような多様な予測モデル(175、176、177)を利用すると、蛋白質配列内で個別のnsSNPを検索することによって該当遺伝子変異が有するスコアを得ることができる。それぞれの予測モデルから得たスコア(s1, s2, s3)は加重値適用部178に提供する。
【0042】
加重値適用部178は、前記それぞれのスコア(s1, s2, s3)を0〜1間の値に正規化した後、それぞれの正規化したスコアに加重値を乗じ、これを合算して毒性(F1(var))を計算する。前記加重値は、既知の疾患遺伝子変異を学習データとして活用して経験から得られる値である。したがって、毒性(F1(var))は次の式1のように計算する。
【0043】
【数1】
【0044】
加重値適用部178は、このように計算した毒性を再び0〜1間の値に正規化することもできる。
【0045】
一方、疾患群に含まれた多数のサンプルのうち反復された位置から発見した遺伝子変異は重要な変異と判断することができるため、各遺伝子変異の頻度により有意性を判断することができる。再び図3を参照すると、有意性計算部180は前記遺伝子変異の頻度、すなわち確率分布に基づき、該当遺伝子変異の有意性を計算する。前記遺伝子変異の発生確率(p(var))は、疾患群サンプルから該当遺伝子変異が発見された確率を意味し、例えば、最尤推定(MLE、Maximum Likelihood Estimation)またはベイズ確率(Bayesian probability)推論により得られる。
【0046】
このように得られた確率(p(var))をそのまま有意性として利用することもできるが、実際有意性として利用するためにはマッピング関数により修正を行う必要がある。マッピング関数は図5に図示するように、0〜1間の確率(p(var))を0〜1の間の有意性(F2(var))に変換する関数である。マッピング関数の形態は多様に設定できるが、好ましくは図5のように、0と1の近くでは相対的に小さい勾配を有し、0.5の近くでは相対的に大きい勾配を有する。すなわち、有意性は0と1の近くの確率に比べ、0.5の近くではより高い敏感度を有する。このようなマッピング関数は例えば式2のように表示される。ここで、αは常数である。
【0047】
【数2】
【0048】
毒性計算部170から得られた毒性(0〜1間の値)と、有意性計算部180から得られた有意性(0〜1間の値)は最終的にスコア計算部190に提供する。スコア計算部190は前記毒性および有意性を組合わせて最終的な毒性スコアを計算する。例えば、毒性スコア(f(var))は次の式3のように毒性および有意性の合算により求めることができるが、これに限らず、毒性および有意性のうち少なくとも1つ以上を反映できる多様な計算式により求めることができる。すなわち、毒性と有意性は共に使用するときに好ましい効果があるが、これに限らず、独立的に使用することもできる。
【0049】
【数3】
【0050】
このように各々遺伝子変異は特定遺伝子にマッピングされ、それぞれの遺伝子の毒性がどの程度であるかを推定することに使用される。しかし、大きな影響を与える1つの遺伝子変異も重要であるが、比較的に少ない影響を与える多数の遺伝子変異が含まれる遺伝子も有意であるといえる。したがって、スコア計算部190は1つの遺伝子内に含まれた遺伝子変異の毒性スコア(f(var))を遺伝子の長さで割った値により最終毒性スコアを求めることもできる。この場合には、最終毒性スコア(s(Gene))は次の式4のように求めることができる。
【0051】
【数4】
【0052】
1つの遺伝子内に存在する遺伝子変異が有する毒性スコアの合計(Σf(var))は、遺伝子長さで割ることによって最終毒性(s(Gene))が求められる。これは前述したように、遺伝子内に大きな影響を与える1つの遺伝子変異だけではなく、多数の遺伝子変異を総合的に考慮するためであり、また毒性スコアの合計(Σf(var))が同一であれば、遺伝子の長さが短いほど最終毒性スコアが大きくなることを意味する。すなわち、単位遺伝子の長さ当たりにより高い毒性スコアは遺伝子変異が該当遺伝子に対してより有意な毒性を示すと推測することができる。
【0053】
再び図1を参照すると、ネットワーク結合部130は毒性予測器120から得られた遺伝子から発現する蛋白質を既知の蛋白質相互作用データベース135と結合して相互作用ネットワークを生成する。一般的には、遺伝子から得られた遺伝子変異の実際の発現は、生体機能を示す蛋白質単位で行われるといえる。すなわち、前記遺伝子変異が有害であるとしても実際の蛋白質単位では潜在的な毒性が発現しない場合もあり、多様な遺伝子変異の組合せによって多様な発現形態が現れる。このような相互作用ネットワークは遺伝子、蛋白質および酵素の順で結合が行われることにより遺伝子ノードの数が増加されることがある。相互作用ネットワークの結合過程のより詳細は、例えば、Automated Network Analysis Identifies Core Pathways in Clioblastoma (www.plosone.org、February 2010,volume 5,issue 2,e8918)を参照して分かる。本発明において、相互作用ネットワークの結合過程は遺伝子単位での毒性のみを求める場合、省略することもできる。
【0054】
モジュール化部140は、毒性スコアが所定の臨界値以上の遺伝子が、遺伝子ネットワーク上に密集している遺伝子ネットワークのサブモジュールを探索する。より具体的には、モジュール化部140は遺伝子毒性が臨界値以上の遺伝子がネットワーク上に密集しているサブモジュールを見つけるために密集分布を統計的検定を行う。前記統計的検定手法の例として、超幾何分布(hypergeometic distribution)を利用し得る。
【0055】
遺伝子ネットワーク上の全体遺伝子個数がNであり、遺伝子ネットワークの全体遺伝子のうち毒性スコアが所定の臨界値を上回る遺伝子の個数がnであり、遺伝子ネットワークのサブモジュール内に存在する遺伝子の個数がmであると仮定すると、遺伝子ネットワークのサブモジュールで毒性スコアが前記臨界値を上回る遺伝子の個数がk個になる確率(P(X=k))は次の式5のとおりである。
【0056】
【数5】
【0057】
ここで、
はN個のうちn個を組み合わせた個数、すなわちNCnを意味する。
【0058】
したがって、遺伝子ネットワークのサブモジュールで毒性スコアが前記臨界値を上回る遺伝子の個数がk個を超過する確率(p)は次の式6によりに計算することができる。
【0059】
【数6】
【0060】
前記確率(p)値は、特定サブモジュール内に毒性スコアが所定の臨界値を超過する遺伝子個数が特定個数(k)を超過して密集している確率を意味する。前記臨界値は多様な方法により決定できるが、一例としては、全体遺伝子の毒性スコア分布で所定の百分位数(1 percentile、5 percentile、10 percentileなど)を基準に決定することができる。このように、特定サブモジュールに対して前記確率(p)が高いほどより有意のサブモジュールとして評価することができる。
【0061】
モジュール化部140は、通常のgreedy探索アルゴリズム、確率的探索アルゴリズム(例、simulated annealing)などを活用して実際にサブモジュールを探索することができ、その具体的な過程は図6に示すフローチャートを参照して説明する。
【0062】
先に、モジュール化部140は初期サブネットワークを設定する(S1)。初期ネットワークは、毒性スコアが有意であるすべての遺伝子(例、毒性スコアが上位5%以内の遺伝子)を単一のノードとして有するネットワークを意味する。このような初期ネットワークを構成するノードを始めとして探索アルゴリズムを適用して最適の有意性を有する遺伝子ネットワークサブモジュールを探索する。
【0063】
モジュール化部140は隣接遺伝子(現在遺伝子と直接連結された遺伝子)を選択して結合して新たなネットワークを生成し(S2)、新たなネットワークの有意性を評価する(S3)。すなわち、前記初期ノードの隣接遺伝子を新たなノードに結合し、新たなネットワークを生成した後、結合したノードで構成された単位(サブモジュールの予備ステップ)に対して有意性を評価する。このような有意性は例えば、前述した超幾何分布での確率(p)を利用して評価することができる。
【0064】
モジュール化部140は、このように構成した新たなネットワークが有意である場合(S4のはい)には現在のネットワークを前記有意のネットワークに更新し(S5)、再びステップS2に戻る。仮に、有意ではない場合(S4のいいえ)にはネットワークの更新を行わず、終了条件を満たしているかを確認し、終了条件を満たしていれば(S6のはい)サブモジュールの探索を終了する。前記終了条件が満たされていなければ(S6のいいえ)ステップS2に戻る。
【0065】
このようなネットワーク更新過程が終了すると、最終に更新された遺伝子ネットワークに含まれるサブモジュールを確定(探索完了)することができる。
【0066】
再び図1を参照すると、優先順位決定部150はモジュール化部140によって探索した複数のサブモジュール間の優先順位を決定する。すなわち、遺伝子変異に基づいて発見した遺伝子ネットワークのサブモジュールに対して遺伝子発現データの変化とどれほど相関関係を有するかを評価し、該当サブモジュールの優先順位を決定する。
【0067】
探索したサブモジュールに対する遺伝子発現パターンの分解は、該当サブモジュールだけではなく、該当サブモジュールに直接連結された遺伝子を含んで行うことが好ましい。これは転写調節因子のような遺伝子に変異が発生した際には、自身の発現より転写調節因子がターゲットにするターゲット遺伝子の発現パターンに変化が生じやすいからである。
【0068】
正常および疾患の二つの条件から調査された遺伝子発現データは、前処理により二つの条件の間での発現差異をZ-スコア(z-score)の形態で計算することができる。例えば、それぞれのサブモジュールに対して、前記サブモジュールに直接連結された遺伝子集合をGとすると、サブモジュールの優先度を評価する指数(es)は次の式7により求める。
【0069】
【数7】
【0070】
ここで、ziは該当サブモジュールに直接連結された遺伝子の集合において、それぞれの遺伝子が有する毒性スコアのZ-スコア値を意味し、|G|は該当サブモジュールに直接連結された遺伝子集合のサイズ(遺伝子の個数)を意味する。Z-スコアは統計学分野で知られているように、現在変数(x)から平均(μ)を減じた後、標準偏差(σ)で割った値であり、現在変数(毒性スコア)が平均(μ)から何σだけ離隔されているかを示す指標である。このような過程により得られた優先順位で並べ替えたサブモジュールは結局、特定遺伝子の発現との相関関係を示すバイオマーカーとしての機能を有する。したがって、疾患群と正常群の比較により得られた疾患特異的配列の変異は該当遺伝子の機能にいかなる変化を与え、また障害を起こすかを予測することができ、さらに個別遺伝子障害が全体生物システムでの相互作用にいかなる影響を及ぼすかについての情報を提供することができる。
【0071】
検証部160は、前記得られた優先順位で並べ替えたサブモジュールを既知の経路データベース165と比較して機能別の関連性を評価する。最も広く使用される方法は、超幾何分布を使用する方法であり、それぞれのサブモジュールに対して経路データベース165から特定生物学的機能単位で抽出した遺伝子集合に対して重畳する遺伝子数字の有意性を計算する。すなわち、図7に図示するように、調査した遺伝子の総数(N)、特定生物学的機能と関連する遺伝子集合の遺伝子個数(n)、遺伝子ネットワークから見つけたサブモジュール内の遺伝子個数(m)および遺伝子サブモジュールと特定遺伝子集合内に共通に存在する遺伝子個数(k)から有意性を示す確率を計算する。前記確率が高いほど最終に得たサブモジュールはより高い有意性を有する。
【0072】
図1〜図4の各構成要素は、ソフトウェア(software)またはFPGA(field-programmable gate array)やASIC(application-specific integrated circuit)のようなハードウェア(hardware)を意味する。しかし、前記構成要素は、ソフトウェアまたはハードウェアに限定されず、アドレッシング(addressing)できる保存媒体にあるように構成することもでき、1つまたはそれ以上のプロセッサを実行させるように構成することもできる。前記構成要素から提供される機能は、さらに細分化した構成要素によって実現することができ、複数の構成要素を組み合わせて特定の機能を行う1つの構成要素として実現することもできる。
【0073】
以上添付する図面を参照して本発明の実施形態について説明したが、本発明が属する技術分野で通常の知識を有する者は、本発明がその技術的思想や必須の特徴を変更しない範囲で、他の具体的な形態で実施され得ることを理解することができる。したがって、上記実施形態はすべての面において例示的なものであり、限定するものではないと理解しなければならない。
【符号の説明】
【0074】
100 バイオマーカー抽出装置
110 前処理部
120 毒性予測器
130 ネットワーク結合部
135 相互作用データベース
140 モジュール化部
150 優先順位決定部
160 検証部
165 経路データベース
170 毒性計算部
180 有意性計算部
190 スコア計算部
【特許請求の範囲】
【請求項1】
配列変異が遺伝子機能にいかなる変化を与えるかを分解し、特定疾患の原因になるバイオマーカーを抽出するバイオマーカー抽出装置であって、
遺伝子サンプルの配列を分解して遺伝子にマッピングされた変異データを抽出する前処理部と、
前記変異データに基づき前記遺伝子の機能に発生する障害を定量化した毒性スコアを求める毒性予測器、および
前記毒性スコアが所定の臨界値以上の遺伝子が、遺伝子ネットワークに密集している遺伝子ネットワークのサブモジュールを探索するモジュール化部を含むバイオマーカー抽出装置。
【請求項2】
前記前処理部は、
疾患群の変異と正常群の変異を比較し、前記分解した遺伝子サンプルから前記疾患群に存在する変異を獲得する疾患群比較部と、
前記獲得した疾患群変異と既知の変異データベースを比較して新たな変異を抽出する変異抽出部、および
前記抽出した新たな変異を機能遺伝子にマッピングする変異マッピング部を含む請求項1に記載のバイオマーカー抽出装置。
【請求項3】
前記変異マッピング部は、
前記抽出した新たな変異のうち蛋白質発現時のアミノ酸が変化する種類のみを抽出して前記機能遺伝子にマッピングする請求項2に記載のバイオマーカー抽出装置。
【請求項4】
前記毒性予測器は、
前記変異データを複数の毒性予測モデルに適用し、それぞれの毒性を得た後、前記それぞれの毒性に加重値を適用することによって加重毒性を計算する毒性計算部を含む請求項1に記載のバイオマーカー抽出装置。
【請求項5】
前記毒性計算部は、
前記変異データから多様な要素を含む特徴ベクターを生成する特徴ベクター生成部と、
前記生成した特徴ベクターのうちそれぞれの予測モデルに必要な要素を選別するアダプタと、
前記選別した要素の入力を受けて蛋白質配列内で個別のnsSNP(non-synonymous Single Nucleotide Polymorphism)を発見できるようにする2以上の予測モデル、および
前記予測モデルの出力に加重値を適用して合算する加重値適用部を含む請求項4に記載のバイオマーカー抽出装置。
【請求項6】
前記加重値適用部は、
前記予測モデルの出力を0〜1間の値に正規化した後、前記加重値を適用して合算し、前記合算した結果を0〜1間の値に正規化する請求項5に記載のバイオマーカー抽出装置。
【請求項7】
前記特徴ベクターは、
遺伝子変異とマッピングする遺伝子および蛋白質で該当位置でのアミノ酸の様々な生物種間の保存スコア(conservation score)、アミノ酸置換が起こす生化学性質の変化(hydrophobicity)、蛋白質の構造的特徴の変化、イントロンのスプライス部位(intron splice junction)(情報)の有無の有無、5-UTR(five prime untranslated region)の変異位置のうち2つ以上を含む請求項5に記載のバイオマーカー抽出装置。
【請求項8】
前記予測モデルは、
SIFT(Sorting Tolerant From Intolerant)、PolyPhen、MAPP((Map Annotator and Pathway Profiler)のうち少なくとも1つ以上を含む請求項5に記載のバイオマーカー抽出装置。
【請求項9】
前記毒性予測器は、
前記変異データの頻度に基づき該当遺伝子変異の有意性を計算する有意性計算部、および
前記加重毒性および前記有意性を組み合わせて毒性スコアを計算するスコア計算部をさらに含む請求項4に記載のバイオマーカー抽出装置。
【請求項10】
前記有意性計算部は、
前記該当遺伝子変異が疾患群サンプルから発見された確率により有意性を計算し、
前記確率は、最尤推定またはベイズ確率である請求項9に記載のバイオマーカー抽出装置。
【請求項11】
前記スコア計算部は、
1つの遺伝子内で遺伝子変異が有する毒性スコアの合計を前記遺伝子の長さで割って最終毒性スコアを求める請求項9に記載のバイオマーカー抽出装置。
【請求項12】
前記モジュール化部は、
現在の遺伝子ノードの集合に対する隣接遺伝子の結合が有意であるかに基づき、遺伝子ネットワークを更新する過程を繰り返すことによって前記サブモジュールを探索する請求項1に記載のバイオマーカー抽出装置。
【請求項13】
前記モジュール化部は、
前記毒性スコアが所定の臨界値を上回る遺伝子個数に対する超幾何分布から得られた確率を利用して前記有意性を判断する請求項12に記載のバイオマーカー抽出装置。
【請求項14】
前記所定の臨界値は、
全体遺伝子の毒性スコア分布で所定の百分位数を基準に決定される請求項13に記載のバイオマーカー抽出装置。
【請求項15】
前記毒性スコアを求めた遺伝子から発現する蛋白質を既知の蛋白質相互作用データベースに結合し、相互作用ネットワークを生成するネットワーク結合部をさらに含む請求項1に記載のバイオマーカー抽出装置。
【請求項16】
前記モジュール化部によって探索した複数のサブモジュール間の優先順位を、Z-スコア(z-score)を基準にして決定する優先順位決定部をさらに含む請求項1に記載のバイオマーカー抽出装置。
【請求項17】
前記優先順位で並べ替えたサブモジュールを既知の経路データベースと比較し、機能別関連性を評価する検証部をさらに含む請求項16に記載のバイオマーカー抽出装置。
【請求項18】
配列変異データが遺伝子の機能に影響を及ぼす障害を定量化するための毒性予測装置であって、
前記変異データを複数の毒性予測モデルに適用し、それぞれの毒性を得た後、前記それぞれの毒性に加重値を適用することによって加重毒性を計算する毒性計算部と、
前記変異データの頻度に基づき、該当遺伝子変異の有意性を計算する有意性計算部、および
前記加重毒性および前記有意性を組み合わせて毒性スコアを計算するスコア計算部を含む毒性予測装置。
【請求項19】
前記毒性計算部は、
前記変異データから多様な要素を含む特徴ベクターを生成する特徴ベクター生成部と、
前記生成された特徴ベクターのうちそれぞれの予測モデルに必要な要素を選別するアダプタと、
前記選別した要素の入力を受けて蛋白質配列内で個別のnsSNP(non-synonymous Single Nucleotide Polymorphism)を発見できるようにする2つ以上の予測モデル、および
前記予測モデルの出力に加重値を適用して合算する加重値適用部を含む請求項18に記載の毒性予測装置。
【請求項20】
前記加重値適用部は、
前記予測モデルの出力を0〜1間の値に正規化する後、前記加重値を適用して合算し、前記合算された結果を0〜1間の値に正規化する請求項19に記載の毒性予測装置。
【請求項21】
前記特徴ベクターは、
遺伝子変異とマッピングする遺伝子および蛋白質で該当位置でのアミノ酸の様々な生物種間の保存スコア(conservation score)、アミノ酸置換が起こす生化学性質の変化(hydrophobicity)、蛋白質の構造的特徴変化、イントロンスプライス接合点(intron splice junction)存在の有無、5-UTR(five prime untranslated region)変異位置のうち2つ以上を含む請求項19に記載の毒性予測装置。
【請求項22】
前記予測モデルは、
SIFT(Sorting Tolerant From Intolerant)、PolyPhen、MAPP((Map Annotator and Pathway Profiler)のうち少なくとも1つ以上を含む請求項19に記載の毒性予測装置。
【請求項23】
前記有意性計算部は、
前記該当遺伝子変異が疾患群サンプルから発見された確率により有意性を計算し、前記確率は、最尤推定またはベイズ確率である請求項18に記載の毒性予測装置。
【請求項24】
前記スコア計算部は、
1つの遺伝子内で遺伝子変異が有する毒性スコアの合計を前記遺伝子の長さで割って最終毒性スコアを求める請求項18に記載の毒性予測装置。
【請求項25】
配列変異が遺伝子機能にいかなる変化を与えるかを分解し、特定疾患の原因になるバイオマーカーを抽出する方法であって、
バイオマーカー抽出装置が遺伝子に含まれた変異データに基づき前記遺伝子の機能に発生する障害を定量化した毒性スコアを求めるステップと、
前記バイオマーカー抽出装置が、前記毒性スコアが所定の臨界値以上の遺伝子が集められたサブモジュールを遺伝子ネットワークから探索するステップ、および
前記バイオマーカー抽出装置が前記探索した複数のサブモジュール間の優先順位を決定するステップを含むバイオマーカー抽出方法。
【請求項26】
前記優先順位を決定するステップは、
前記バイオマーカー抽出装置が前記サブモジュールが有する各々のZ-スコア(z-score)を基準に高いZ-スコアを有するサブモジュールに高い優先順位を決定するステップを含む請求項25に記載のバイオマーカー抽出方法。
【請求項27】
前記バイオマーカー抽出装置が前記毒性スコアを求めた遺伝子から発現する蛋白質を既知の蛋白質相互作用データベースに結合して相互作用ネットワークを生成するステップをさらに含む請求項25に記載のバイオマーカー抽出方法。
【請求項28】
前記バイオマーカー抽出装置が前記優先順位で並べ替えたサブモジュールを既知の経路データベースと比較し、機能別の関連性を評価するステップをさらに含む請求項25に記載のバイオマーカー抽出方法。
【請求項29】
配列変異データが遺伝子の機能に影響を及ぼす障害を定量化するための毒性予測方法であって、
バイオマーカー抽出装置が前記変異データから多様な要素を含む特徴ベクターを生成するステップと、
前記バイオマーカー抽出装置が前記生成された特徴ベクターのうちそれぞれの予測モデルに必要な要素を選別するステップと、
前記バイオマーカー抽出装置が前記選別した要素の入力を受けて蛋白質配列内で個別のスコアを提供するステップ、および
前記バイオマーカー抽出装置が前記予測モデルの出力に加重値を適用して合算し、加重毒性を計算するステップを含む毒性予測方法。
【請求項30】
前記加重値は、
既知の疾患遺伝子変異を学習データとして活用して経験から得られる値である請求項29に記載の毒性予測方法。
【請求項31】
前記加重毒性を得るステップは、
前記バイオマーカー抽出装置が前記予測モデルの出力を0〜1間の値に正規化する後、前記加重値を適用して合算し、前記合算した結果を0〜1間の値に正規化するステップを含む請求項19に記載の毒性予測方法。
【請求項32】
前記バイオマーカー抽出装置が前記変異データの頻度に基づき、該当遺伝子変異の有意性を計算するステップ、および
前記バイオマーカー抽出装置が前記加重毒性および前記有意性を組み合わせて毒性スコアを計算するステップをさらに含む請求項29に記載の毒性予測方法。
【請求項33】
前記有意性を計算するステップは、最尤推定またはベイズ確率に基づき、該当遺伝子変異が疾患群サンプルから発見された確率によって、前記有意性を計算するステップを含む請求項32に記載の毒性予測方法。
【請求項34】
前記バイオマーカー抽出装置が1つの遺伝子内で遺伝子変異が有する毒性スコアの合計を前記遺伝子の長さで割って最終毒性スコアを求めるステップをさらに含む請求項32に記載の毒性予測方法。
【請求項1】
配列変異が遺伝子機能にいかなる変化を与えるかを分解し、特定疾患の原因になるバイオマーカーを抽出するバイオマーカー抽出装置であって、
遺伝子サンプルの配列を分解して遺伝子にマッピングされた変異データを抽出する前処理部と、
前記変異データに基づき前記遺伝子の機能に発生する障害を定量化した毒性スコアを求める毒性予測器、および
前記毒性スコアが所定の臨界値以上の遺伝子が、遺伝子ネットワークに密集している遺伝子ネットワークのサブモジュールを探索するモジュール化部を含むバイオマーカー抽出装置。
【請求項2】
前記前処理部は、
疾患群の変異と正常群の変異を比較し、前記分解した遺伝子サンプルから前記疾患群に存在する変異を獲得する疾患群比較部と、
前記獲得した疾患群変異と既知の変異データベースを比較して新たな変異を抽出する変異抽出部、および
前記抽出した新たな変異を機能遺伝子にマッピングする変異マッピング部を含む請求項1に記載のバイオマーカー抽出装置。
【請求項3】
前記変異マッピング部は、
前記抽出した新たな変異のうち蛋白質発現時のアミノ酸が変化する種類のみを抽出して前記機能遺伝子にマッピングする請求項2に記載のバイオマーカー抽出装置。
【請求項4】
前記毒性予測器は、
前記変異データを複数の毒性予測モデルに適用し、それぞれの毒性を得た後、前記それぞれの毒性に加重値を適用することによって加重毒性を計算する毒性計算部を含む請求項1に記載のバイオマーカー抽出装置。
【請求項5】
前記毒性計算部は、
前記変異データから多様な要素を含む特徴ベクターを生成する特徴ベクター生成部と、
前記生成した特徴ベクターのうちそれぞれの予測モデルに必要な要素を選別するアダプタと、
前記選別した要素の入力を受けて蛋白質配列内で個別のnsSNP(non-synonymous Single Nucleotide Polymorphism)を発見できるようにする2以上の予測モデル、および
前記予測モデルの出力に加重値を適用して合算する加重値適用部を含む請求項4に記載のバイオマーカー抽出装置。
【請求項6】
前記加重値適用部は、
前記予測モデルの出力を0〜1間の値に正規化した後、前記加重値を適用して合算し、前記合算した結果を0〜1間の値に正規化する請求項5に記載のバイオマーカー抽出装置。
【請求項7】
前記特徴ベクターは、
遺伝子変異とマッピングする遺伝子および蛋白質で該当位置でのアミノ酸の様々な生物種間の保存スコア(conservation score)、アミノ酸置換が起こす生化学性質の変化(hydrophobicity)、蛋白質の構造的特徴の変化、イントロンのスプライス部位(intron splice junction)(情報)の有無の有無、5-UTR(five prime untranslated region)の変異位置のうち2つ以上を含む請求項5に記載のバイオマーカー抽出装置。
【請求項8】
前記予測モデルは、
SIFT(Sorting Tolerant From Intolerant)、PolyPhen、MAPP((Map Annotator and Pathway Profiler)のうち少なくとも1つ以上を含む請求項5に記載のバイオマーカー抽出装置。
【請求項9】
前記毒性予測器は、
前記変異データの頻度に基づき該当遺伝子変異の有意性を計算する有意性計算部、および
前記加重毒性および前記有意性を組み合わせて毒性スコアを計算するスコア計算部をさらに含む請求項4に記載のバイオマーカー抽出装置。
【請求項10】
前記有意性計算部は、
前記該当遺伝子変異が疾患群サンプルから発見された確率により有意性を計算し、
前記確率は、最尤推定またはベイズ確率である請求項9に記載のバイオマーカー抽出装置。
【請求項11】
前記スコア計算部は、
1つの遺伝子内で遺伝子変異が有する毒性スコアの合計を前記遺伝子の長さで割って最終毒性スコアを求める請求項9に記載のバイオマーカー抽出装置。
【請求項12】
前記モジュール化部は、
現在の遺伝子ノードの集合に対する隣接遺伝子の結合が有意であるかに基づき、遺伝子ネットワークを更新する過程を繰り返すことによって前記サブモジュールを探索する請求項1に記載のバイオマーカー抽出装置。
【請求項13】
前記モジュール化部は、
前記毒性スコアが所定の臨界値を上回る遺伝子個数に対する超幾何分布から得られた確率を利用して前記有意性を判断する請求項12に記載のバイオマーカー抽出装置。
【請求項14】
前記所定の臨界値は、
全体遺伝子の毒性スコア分布で所定の百分位数を基準に決定される請求項13に記載のバイオマーカー抽出装置。
【請求項15】
前記毒性スコアを求めた遺伝子から発現する蛋白質を既知の蛋白質相互作用データベースに結合し、相互作用ネットワークを生成するネットワーク結合部をさらに含む請求項1に記載のバイオマーカー抽出装置。
【請求項16】
前記モジュール化部によって探索した複数のサブモジュール間の優先順位を、Z-スコア(z-score)を基準にして決定する優先順位決定部をさらに含む請求項1に記載のバイオマーカー抽出装置。
【請求項17】
前記優先順位で並べ替えたサブモジュールを既知の経路データベースと比較し、機能別関連性を評価する検証部をさらに含む請求項16に記載のバイオマーカー抽出装置。
【請求項18】
配列変異データが遺伝子の機能に影響を及ぼす障害を定量化するための毒性予測装置であって、
前記変異データを複数の毒性予測モデルに適用し、それぞれの毒性を得た後、前記それぞれの毒性に加重値を適用することによって加重毒性を計算する毒性計算部と、
前記変異データの頻度に基づき、該当遺伝子変異の有意性を計算する有意性計算部、および
前記加重毒性および前記有意性を組み合わせて毒性スコアを計算するスコア計算部を含む毒性予測装置。
【請求項19】
前記毒性計算部は、
前記変異データから多様な要素を含む特徴ベクターを生成する特徴ベクター生成部と、
前記生成された特徴ベクターのうちそれぞれの予測モデルに必要な要素を選別するアダプタと、
前記選別した要素の入力を受けて蛋白質配列内で個別のnsSNP(non-synonymous Single Nucleotide Polymorphism)を発見できるようにする2つ以上の予測モデル、および
前記予測モデルの出力に加重値を適用して合算する加重値適用部を含む請求項18に記載の毒性予測装置。
【請求項20】
前記加重値適用部は、
前記予測モデルの出力を0〜1間の値に正規化する後、前記加重値を適用して合算し、前記合算された結果を0〜1間の値に正規化する請求項19に記載の毒性予測装置。
【請求項21】
前記特徴ベクターは、
遺伝子変異とマッピングする遺伝子および蛋白質で該当位置でのアミノ酸の様々な生物種間の保存スコア(conservation score)、アミノ酸置換が起こす生化学性質の変化(hydrophobicity)、蛋白質の構造的特徴変化、イントロンスプライス接合点(intron splice junction)存在の有無、5-UTR(five prime untranslated region)変異位置のうち2つ以上を含む請求項19に記載の毒性予測装置。
【請求項22】
前記予測モデルは、
SIFT(Sorting Tolerant From Intolerant)、PolyPhen、MAPP((Map Annotator and Pathway Profiler)のうち少なくとも1つ以上を含む請求項19に記載の毒性予測装置。
【請求項23】
前記有意性計算部は、
前記該当遺伝子変異が疾患群サンプルから発見された確率により有意性を計算し、前記確率は、最尤推定またはベイズ確率である請求項18に記載の毒性予測装置。
【請求項24】
前記スコア計算部は、
1つの遺伝子内で遺伝子変異が有する毒性スコアの合計を前記遺伝子の長さで割って最終毒性スコアを求める請求項18に記載の毒性予測装置。
【請求項25】
配列変異が遺伝子機能にいかなる変化を与えるかを分解し、特定疾患の原因になるバイオマーカーを抽出する方法であって、
バイオマーカー抽出装置が遺伝子に含まれた変異データに基づき前記遺伝子の機能に発生する障害を定量化した毒性スコアを求めるステップと、
前記バイオマーカー抽出装置が、前記毒性スコアが所定の臨界値以上の遺伝子が集められたサブモジュールを遺伝子ネットワークから探索するステップ、および
前記バイオマーカー抽出装置が前記探索した複数のサブモジュール間の優先順位を決定するステップを含むバイオマーカー抽出方法。
【請求項26】
前記優先順位を決定するステップは、
前記バイオマーカー抽出装置が前記サブモジュールが有する各々のZ-スコア(z-score)を基準に高いZ-スコアを有するサブモジュールに高い優先順位を決定するステップを含む請求項25に記載のバイオマーカー抽出方法。
【請求項27】
前記バイオマーカー抽出装置が前記毒性スコアを求めた遺伝子から発現する蛋白質を既知の蛋白質相互作用データベースに結合して相互作用ネットワークを生成するステップをさらに含む請求項25に記載のバイオマーカー抽出方法。
【請求項28】
前記バイオマーカー抽出装置が前記優先順位で並べ替えたサブモジュールを既知の経路データベースと比較し、機能別の関連性を評価するステップをさらに含む請求項25に記載のバイオマーカー抽出方法。
【請求項29】
配列変異データが遺伝子の機能に影響を及ぼす障害を定量化するための毒性予測方法であって、
バイオマーカー抽出装置が前記変異データから多様な要素を含む特徴ベクターを生成するステップと、
前記バイオマーカー抽出装置が前記生成された特徴ベクターのうちそれぞれの予測モデルに必要な要素を選別するステップと、
前記バイオマーカー抽出装置が前記選別した要素の入力を受けて蛋白質配列内で個別のスコアを提供するステップ、および
前記バイオマーカー抽出装置が前記予測モデルの出力に加重値を適用して合算し、加重毒性を計算するステップを含む毒性予測方法。
【請求項30】
前記加重値は、
既知の疾患遺伝子変異を学習データとして活用して経験から得られる値である請求項29に記載の毒性予測方法。
【請求項31】
前記加重毒性を得るステップは、
前記バイオマーカー抽出装置が前記予測モデルの出力を0〜1間の値に正規化する後、前記加重値を適用して合算し、前記合算した結果を0〜1間の値に正規化するステップを含む請求項19に記載の毒性予測方法。
【請求項32】
前記バイオマーカー抽出装置が前記変異データの頻度に基づき、該当遺伝子変異の有意性を計算するステップ、および
前記バイオマーカー抽出装置が前記加重毒性および前記有意性を組み合わせて毒性スコアを計算するステップをさらに含む請求項29に記載の毒性予測方法。
【請求項33】
前記有意性を計算するステップは、最尤推定またはベイズ確率に基づき、該当遺伝子変異が疾患群サンプルから発見された確率によって、前記有意性を計算するステップを含む請求項32に記載の毒性予測方法。
【請求項34】
前記バイオマーカー抽出装置が1つの遺伝子内で遺伝子変異が有する毒性スコアの合計を前記遺伝子の長さで割って最終毒性スコアを求めるステップをさらに含む請求項32に記載の毒性予測方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2012−94143(P2012−94143A)
【公開日】平成24年5月17日(2012.5.17)
【国際特許分類】
【出願番号】特願2011−232749(P2011−232749)
【出願日】平成23年10月24日(2011.10.24)
【出願人】(510294195)サムソン エスディーエス カンパニー リミテッド (33)
【Fターム(参考)】
【公開日】平成24年5月17日(2012.5.17)
【国際特許分類】
【出願日】平成23年10月24日(2011.10.24)
【出願人】(510294195)サムソン エスディーエス カンパニー リミテッド (33)
【Fターム(参考)】
[ Back to top ]