
バキュロウイルスベクター発現系におけるタンパク質の発現
【課題】バキュロベクターウイルス発現系においてタンパク質抗原の発現を増加するためのワクチン製造方法の提供。
【解決手段】下記工程を含む古典的ブタ熱ウイルスE2バキュロウイルスタンパク質を含むワクチンの製造方法。当該タンパク質をエンコードする組換えバキュロウイルを選択する工程、少なくとも2リットルの十分な容量を備える培養容器において増殖培地中で昆虫細胞を増殖する工程、1×105〜1×106個/mlの密度の当該細胞を0.01未満のMOIにて当該バキュロウイルスの接種物で感染する工程、前記細胞の約半分以上が細胞変性効果を示すまで前記細胞を培養し、増殖培地中のE2たんぱく質の最終濃度が100〜300μg/mlのときに、細胞性物質を除去する工程、免疫アフィニティ精製することなく、ワクチンを調製する工程。当該製造法はさらに、組換え卵胞刺激ホルモンの製造に適用可能である。
【解決手段】下記工程を含む古典的ブタ熱ウイルスE2バキュロウイルスタンパク質を含むワクチンの製造方法。当該タンパク質をエンコードする組換えバキュロウイルを選択する工程、少なくとも2リットルの十分な容量を備える培養容器において増殖培地中で昆虫細胞を増殖する工程、1×105〜1×106個/mlの密度の当該細胞を0.01未満のMOIにて当該バキュロウイルスの接種物で感染する工程、前記細胞の約半分以上が細胞変性効果を示すまで前記細胞を培養し、増殖培地中のE2たんぱく質の最終濃度が100〜300μg/mlのときに、細胞性物質を除去する工程、免疫アフィニティ精製することなく、ワクチンを調製する工程。当該製造法はさらに、組換え卵胞刺激ホルモンの製造に適用可能である。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
本発明は、バキュロウイルスベクター発現系においてタンパク質またはポリぺプチドの発現を増加するための方法に関する。組換えDNA技術が開発されたとき、遺伝子改変された細菌を使用する大規模のタンパク質生成に関して期待が高かった。しかし、大多数の商業的に魅力的なタンパク質は、それらが生物学的に活性なタンパク質になり得る前に翻訳後修飾を必ず受ける。したがって、動物細胞が、組換えタンパク質を生成するために現在より頻繁に使用される。
【0002】
動物細胞の中で、昆虫細胞は組換えタンパク質の生成のために益々重要である。昆虫細胞を使用する簡便なおよび用途の広いバキュロウイルスベクター系が開発された。昆虫細胞の生理学に対する情報はかなり少ないが、バキュロウイルス組換え技術を介して生成されるワクチンは一般に十分に受容される。1つの例は、臨床試行において可能なAIDSワクチンとしてのヒト免疫不全ウイルスI型の、バキュロウイルスにより発現されるgp160エンベロープタンパク質の使用である。
【0003】
現在まで、昆虫細胞におけるバキュロウイルスにより発現されるタンパク質の大規模の生成は、約10リットルまでのバイオリアクターに制限される。規模を大きくするために、懸濁培養は最も良好な可能性を提供する。大規模生成において(Tramperら、Rec. Adv. Biotech.、1992、263〜284;PowerおよびNielsen、Cytotechnology 20:209〜219、1996を参照のこと)、特別な重要視が細胞増殖およびウイルス生成を影響する因子に与えられるべきである。このような因子の変化は、組換えタンパク質の生成の最終的なレベルを大きく影響する。
【0004】
バキュロウイルスは、閉塞体中に一般に閉塞される桿状ウイルス粒子により特徴付けられる。
【0005】
バキュロウイルスは、閉塞体中に一般に閉塞される桿状ウイルス粒子により特徴づけられる(多角体としてまた呼ばれる)。バキュロウイルス科は、閉塞ウイルス:核多角体病ウイルス(NPV)および顆粒症ウイルス(GV)の2つの属を含有する真正バキュロウイルス、ならびに非閉塞ウイルスを含有するヌードバキュロウイルス亜科の2つの亜科に分けられる。Autographa californica(Ac)NPVの細胞および分子生物学がより詳細に研究された。
【0006】
多くのタンパク質が、そのタンパク質をコードする組換えバキュロウイルスで感染された昆虫細胞において発現されてきた。コードすることは、このようなウイルスが非相同タンパク質をコードする核酸配列を提供され、そしてしばしば、プロモーターのような調節核酸配列をさらに提供されることを意味する。最も頻繁には、ポリヘドリンプロモーターが外来遺伝子を発現するために使用されるが、p10プロモーターは等しく十分に適切であり、そして同様に使用される。
【0007】
いくつかの細胞株が、組換えバキュロウイルスでの感染に利用可能である。細胞株SF−21は、アワトウヨウの幼虫(Spodoptera frugiperda)の卵巣組識に由来した。アメリカンタイプカルチャーコレクション(CRL 1711)から利用可能であるクローン単離物のSF−9は、組換えウイルスのインビトロ生成のためのおおむね標準的な細胞株であり、そして組換えウイルスを生成することにおいて優れていると言われる。他の細胞株は、たとえば、Hi−Five細胞株ならびにイラクサキンウワバ(Trichoplusia ni)から得られるTn−368およびTn−368A細胞株である。昆虫細胞が増殖する最も広範に使用される培地としては、TNM−FH、BML−TC/10、およびIPL-41が挙げられる。これらの培地は、通常、哺乳動物血清、特に胎児ウシ血清のようなおおむね規定された成分で補充される。血清置換はまた、昆虫細胞培養に適用されており、そしてEx−Cell 400(商標)およびSf900のような血清非含有培地が市販される。
【0008】
昆虫細胞は一般に固体支持体上でおよび懸濁液中で増殖するが、固体支持体上で増殖される場合にウイルスのより高い収量を与えることが報告される。感染は、細胞が指数関数増殖期において感染される場合に最も効果的であるが、細胞当たりの生成される多角体およびウイルスの量は、細胞周期の異なる段階で感染された細胞間で有意に変化しない。細胞密度は、ウイルス生成に対して大きな影響を有する。昆虫細胞は接触阻害の形態を示し得、より高い細胞密度で減少されたウイルス生成を生じる。
【0009】
初回感染多重度(m.o.i.)は、細胞当たりの感染性ウイルスの数であり、一般に感染の終了時での感染された細胞の画分および細胞当たりの多角体の数の両方に影響する。ウイルス生成についての至適なm.o.i.は一般に、約20〜30であると考えられる。β-ガラクトシダーゼを発現する組換えバキュロウイルスでの研究(LicariおよびBailey、Biotech.Bioeng.、37:238〜246、1991)において、Sf−9細胞は、0と100との間のm.o.i.値で感染された。β-ガラクトシダーゼの収量は増加され、そして細胞密度は、m.o.i.の増加とともに減少した。m.o.i.を増加することおよび減少することは、感染された細胞当たりの組換えタンパク質の最大に達成可能な収量に対して制限された効果のみを有すると一般に考えられる。しかし、低いm.o.i.を選択することは、感染に必要なウイルスストックの減少を許容し、そしてバキュロウイルスの欠陥妨害粒子の生成の危険性を最小にする。昆虫細胞のバッチ培養が高いm.o.i.(>5)で感染される場合、次の感染プロセスは本質的に同時進行される(すなわち、全ての細胞は、同時に感染周期を経る)。細胞がバッチ培養においてm.o.i.1〜5で感染される場合、培養物はもはや同時進行されない。全ての細胞が感染されそして所望のタンパク質の生成が終わるまで、培養物は、最初は非感染細胞と個々の感染周期における異なる時点の細胞とからなる。一般的にはこのような培養物において生成レベルははるかに低い。培養物の挙動は、それぞれ生成の異なる相にある個々の細胞の組み合わせの挙動であり;至適な状態に及ばない生成レベルを説明する。連続的な培養において、非感染細胞が連続的に添加され、そして培養物は明らかに非同時進行的に感染される。
【0010】
数学的モデルの設計を介して、低いm.o.i.で細胞を感染する場合または連続的な培養系中でウイルスを増殖する場合に観察される挙動のような複雑な挙動を予測することが可能であると考えられる。同じ現象についての純粋に経験的な分析は、不可能でないにしても非常に困難であると考えられる。現在、3つのモデル、Licari & Baileyモデル、de Gooijerモデル、およびPower & Nielsenモデルが知られる。これらは、それらの複雑性およびそれらを詳しく開発した努力にもかかわらず、全て第1世代のモデルであり、ポリヘドリンプロモーターの制御下で発現されるモデル組換えタンパク質(β-ガラクトシダーゼ)を発現するバキュロウィルスの挙動について推論する。これらは、大部分、DNAおよびRNA集積ならびに感染周期を無視して、ブラックボックス系としてみる場合、バキュロウイルスと昆虫細胞との間の相互作用についての本発明者らの定量的な理解をまとめる。結合および最初の感染プロセスはなお、定量的に十分には理解されておらず、この分野におけるさらなる研究が非常に必要とされる。
【0011】
バキュロウイルス発現系は、組換えタンパク質の生成のための強力な道具を提供する。いくつかの細胞培養配置が、大規模生成のために使用され得る。しかし、これらの系は、それらの可能性の十分な利点を採用するためにさらなる至適化を必要とする。商業的な適用について、大規模および低コストの生成が極めて重要である。大規模細胞培養において報告される多角体生成系は、価格競合製品についての商業的要求を満たすために劇的に改善されるべきである。
【0012】
本発明は、所望のタンパク質の増加されたまたは改善された収量を許容する、バキュロウイルス発現系を使用する大規模で組換えタンパク質を生成するための方法を提供する。本発明は、昆虫細胞培養物において、組換えタンパク質を生成するおよび組換えタンパク質の収量を改善するための方法を提供し、当該タンパク質をコードする組換えバキュロウイルスを選択する工程、培養容器において増殖培地中で昆虫細胞を増殖する工程、および0.01未満の感染多重度で細胞を感染する工程を包含する。
【0013】
好ましい実施態様において、本発明は昆虫細胞培養物において生成される組換えタンパク質の収量を増加するための方法を提供し、当該タンパク質をコードする組換えバキュロウイルスを選択する工程、培養容器において増殖培地中で昆虫細胞を増殖する工程、および0.01未満の感染多重度を用いて少なくとも1つのバキュロウイルスの接種物で細胞を感染する工程を包含する。漸増する収量は、いくつかの研究グループの主題である。たとえば、Chenら(Drug metabolism and Disposition:24 399〜405頁、1997)は、同時感染アプローチのためにMOIを至適化する可能性を研究し、これによって2つの異なるバキュロウイルス発現タンパク質が昆虫細胞培養物において生成された。本明細書中に記載される結果と反対に、彼らは、彼らの2つのタンパク質について約0.015〜0.03の最も良好なMOIを見出した。MOIを0.01未満に減少することにより、Chenらの同時感染系の収量は減少した。Radfordら(Cytotechnology 24、73〜81、1997)は、同時感染系を研究することによって妨げられず、1よりも大きいMOIが常に最終的なプロセス収量を最大にするために使用されるべきであることを明らかに示し、本発明の知見に反対することを再度教示する。彼らは、低いMOIを使用して細胞当たりより大きな量のタンパク質およびウイルスを生成することは不可能であることを述べ、そしてその代わりに感染時間(TOI)を調節することを示唆する。
【0014】
Nguyenら(J.Biotech.31、205〜217、1993)のように他は、MOIの変化における解決を見出さないが、流加培養法を適用することによって収量を増加することにねらいを向けるか、またはウイルスが増殖する温度を変化し(Wuら、Cytotechnology、9、141〜147、1992;Kingら、Biotechnol. Bioeng、38、1091〜1099、1991)、そして0.01未満のMOI下でのウイルスの増殖を回避する。
【0015】
これらの初期の結果は、本明細書中の記載において提供される結果とは明らかに異なり(たとえば図1を参照のこと)、本明細書中の記載において培養物は0.01未満(たとえば、0.003、0.001、またはさらに0.0001)のMOIで感染され、0.01または0.1のMOIで感染される培養物よりも高い収量を達成する。
【0016】
本発明の好ましい実施態様は、単層培養において増殖されない昆虫細胞培養物において組換えタンパク質を生成するおよび組換えタンパク質の収量を改善するための方法を提供する。小量でバキュロウイルス発現ベクター系においてタンパク質を生成するための従来の実験室の方法は、培養フラスコ中の昆虫細胞の単層培養物を使用する。これらの静置培養物は、同時進行性の感染を確実にするために、通常、高いMOIで感染される。単層がコンフルエントになる前に全ての細胞が感染されることが極めて重要である。なぜなら、接触阻害が細胞中の代謝変化に導き、これが最終産物の収量を影響し得るからである。単層培養物中の細胞密度を正確に確立することは困難であるので、MOI実験を行うことは不可能である。培養物を感染するために低いMOI(0.01未満)を使用することはさらにより役に立たない。感染時の細胞密度の過剰評価および過小評価の両方は、有意に至適な状態に及ばないタンパク質の収量を導く。日常的に、高いMOIを使用し、これは全細胞集団の同時進行性の感染を保証する。このことは、至適な収量を達成するために、大量のウイルスストックが単層培養物を感染するために必要とされることを意味する。
【0017】
単層培養物を使用するタンパク質生成の規模拡大は、より多くの組織培養フラスコを使用することを単に意味する。単層培養物を使用する大量のタンパク質の生成は、非常に労働集中的である。さらに、溶解された酸素濃度およびpHのような重要な培養パラメーターを調節および/またはモニタすることは可能でない。
【0018】
本発明は今や、全ての昆虫細胞培養物に適切な方法を提供する。本発明は、低いMOIが単層培養物以外の昆虫培養物における収量を至適化するために有益であるという洞察を提供する。
【0019】
異なるタイプの培養容器が、単層培養以外において昆虫細胞を培養するために使用され得る。全ての発酵器設計の目的は、十分な通気および高い細胞密度を達成するが、可能な限り低い剪断力を維持することである(Tramperら、Recent advantages in biotechnology(Vardar-SukanおよびSukan編 1992)。文献において記載される大多数の場合において、気体噴霧器を備えた攪拌タンクバイオリアクターが使用される。このタイプの容器において、均一性が回転翼を使用することによって達成される。このタイプの発酵器は、異なる方法において操作され得る。先ず第1に、バッチ法である。これは、最も簡単なおよび単純な方法である。細胞が培養され、ウイルスが添加され、そして産物が感染の終了時に採集される。より複雑な方法は、流加培養法である。栄養素の濃縮混合物が培養容器に添加され、より高い細胞密度およびより高い容量の産物収量が達成される(Nguyenら、1993 Journal of Biotechnology 第31巻、205〜217頁)。これは制限される栄養素がなんであるのか常には明らかでないので、より複雑化される。1つの栄養素制限をこの基質を添加することによって取り消すことにより、別の基質制限が直接的に導かれ得、したがって必ずしもより高い産物収量が導かれないかもしれない。
【0020】
混合する異なる方法が、空中補給バイオリアクターにおいて使用される(Wuら、1992 Cytotechnology 第9巻 141〜147頁)。このタイプの容器は2つのシリンダからなる。最も小さな直径を備えるシリンダ(ドラフト管)は、より大きな直径を備えるシリンダ(降下管)の内側に配置される。中央のシリンダにおいて空気および別の気体が散布され、上昇流を作製する。発酵器の上部で、気体は液体を残し、そして液体は中央のシリンダの外側を下がる。この方法において、通気および均一性の両方が、ドラフト管および降下管における密度の差異に起因して気体のみの噴霧を使用することによって達成される。この方法は、ずり応力を減少し得る。しかし、連続的な通気速度が、正確な混合を達成するために必要とされる。
【0021】
発酵器中の生存細胞密度を増加するための別の方法は、スピンフィルタまたは別の細胞保持デバイスを含むことによる。これは灌流と呼ばれる。これは、発酵器中に細胞を保持しながら、発酵器から不用な培地を除去し新鮮な培地を添加することを許容する。この方法は、多くの特別の装置およびより困難な発酵器操作を生じる(Caronら、1994 Biotechnology and Bioengineering 第43巻、881〜891頁)。さらに、多孔質床およびゲルマトリクス中の固定化のような方法が報告される。このタイプの方法は、マトリクス上のまたは内側の細胞の固定化に依存し、細胞培養物を希釈することなく不用な培地の除去および新鮮な培地の添加を可能にさせる。細胞密度、接触阻害、および単層培養の他の関連の問題が処理可能である場合、本発明の方法は、上述の種々の培養系において適用され得るのみでなく、単層培養においてもまた適用され得る。
【0022】
たとえば、本発明の好ましい実施態様は、昆虫細胞培養物において組換えタンパク質を生成するおよび組換えタンパク質の収量を改善するための方法が提供され、当該タンパク質をコードする組換えバキュロウイルスを選択する工程、少なくとも2リットルを含むのに十分な容量を備える培養容器において増殖培地中で昆虫細胞を増殖する工程、および当該バキュロウイルス/細胞の0.01未満のPFUのm.o.i.で少なくとも1つのバキュロウイルスの接種物で昆虫細胞を感染する工程を包含する。本発明は、たとえば1〜5のm.o.i.よりもかなり低い感染多重度が使用され、非同時進行的に感染される培養物を導く方法を提供する。本発明により提供されるようなm.o.i.を使用してバキュロウイルスで昆虫培養物を感染することによって、至適な平衡が、細胞の複製の速度とウイルスの複製の速度との間で達成され、それによって所望のタンパク質の至適な発現を許容する。本発明によって提供される方法は、同様にm.o.i.を調整することによってより高いおよびより低い細胞密度に容易に調整され得、ここでは本発明によって提供される効率および密度にしたがって、複製の種々の相における細胞を感染するために利用可能であるウイルス粒子の相対比が残存する。本発明の方法の好ましい実施態様は、少なくとも10、より好ましくは少なくとも20、より好ましくは少なくとも50または250リットルの増殖培地を含むのに十分な容量を備える培養容器において細胞を増殖し、それによって非相同タンパク質を発現するバキュロウイルス培養物の規模拡大を許容する工程を包含する。たとえば、存在する増殖培地の容量について必要とされるよりも大きな容量を備える培養培地を使用し得る。たとえば、20〜70リットルの細胞培養物を培養するために100Lの培養容器を使用し得る。本発明の方法の好ましい実施態様は、1×105〜5×106細胞/ml、より好ましくは5×105〜1.5×106細胞/mlの細胞密度で細胞を感染し、それによって容易に操作可能な範囲内にウイルス接種の実際の容量を維持する工程を包含する。本発明の方法のなお別の実施態様は、0.005未満(たとえば、0.003、0.001、0.0005、または0.00025)のm.o.i.で細胞を感染し、それによって接種物をできるだけ小さく維持する工程を包含する。本発明の方法の好ましい実施態様はp10プロモーターの制御下で所望のタンパク質を発現する組換えバキュロウイルスを選択する工程を包含する。P10プロモーターは、細胞溶解において役割を果たし、p10タンパク質の不在は、感染された細胞をインタクトなままにさせ、そして感染された細胞からの多角体の放出を妨ぎ、それによって再感染速度を減少するが、感染力自身は減少しない。ウイルス感染の全プロセスは、ポリヘドリン遺伝子、したがって多角体がなお存在するという事実に起因して、現在視覚的にチェックされ得る。ウイルス感染は、細胞核中に集積する高密度のタンパク質粒子として観察され得る。本発明の方法の別の実施態様は、バッチ培養系において昆虫細胞を増殖し、それによって未だ未感染の細胞の感染および複製を損ない得る欠陥妨害バキュロウイルスウイルス粒子の集積を最小化する工程を包含する。本発明により提供される別の方法は、懸濁液中で、好ましくは攪拌され得る(中程度に)発酵器のような培養容器中で昆虫細胞を増殖する工程を包含する。(攪拌された)懸濁培養物の使用は、特にウイルス増殖を視覚的にチェックするために所望のタンパク質がp10プロモーターの制御下にある組換えバキュロウイルスの使用と組み合わされる場合(しかし、CPEを観察するようなチェックする他の方法(たとえば、ポリヘドリンプロモーターを使用する場合において)がまた利用可能である)、培養のより良好な制御を許容する。
【0023】
懸濁液の中程度の攪拌は、基質勾配が作られないおよび細胞が高すぎる剪断力を受けない均一な培養を保証する。さらに、攪拌は、感染された細胞から非感染細胞へのウイルスの効率的な移動を生じ、細胞のウイルス感染のより高い効率を与える。最初は約0.1〜0.3%の細胞のみが感染されるので、残りの99.7〜99.9%の細胞は成長および増殖を許容される。
【0024】
本発明は、種々の起源の組換えタンパク質を発現および生成するための方法を提供する。本発明によって提供される例は、ペスチウイルス由来のタンパク質を、増殖培地中に、少なくとも100、120、または150μg/ml、より好ましくは少なくとも200、またはさらに300μg/mlの収穫の当該タンパク質の濃度に生成することである。所望のタンパク質はまた、獣医学的および医学的使用のために、たとえば、ワクチン中におよび診断試験において組込まれた抗原性物質を調製するために使用され得る。本発明の方法によって生成される所望のタンパク質は、たとえば、ワクチンを調製するために使用され得る。
【0025】
本発明によって提供される例は、ペスチウイルスE2タンパク質またはそのフラグメントであり、これはたとえば、ペスチウイルス感染(たとえば、ブタにおける伝統的なブタ熱)に対するワクチンを調製するために使用され得る。好ましい実施態様において、本発明は、組換えペスチウイルスのE2タンパク質もしくはErnsタンパク質、またはそのフラグメントを含有し、イムノアフィニティ精製されておらずおよび好ましくは1用量での単回のワクチン接種後にPD95レベルでペスチウイルスに対して防御を付与するという点で特徴付けられるワクチンを提供する。これは、CSFVワクチン接種に特に関連する。適用される場合、CSFVワクチン接種は一般に、CSFVの発生が生じた領域において大規模なキャンペーンの間に行われる。これは、比較的短い期間における多数の動物の迅速なワクチン接種を必要とする。このような大規模なキャンペーンにおいて、十分な防御レベル(野生型ウイルス感染に対して防御されるブタの数)が迅速に達成されることが差し迫って重要である。防御を達成するために第2のワクチン接種について、第1のワクチン接種後に数週間待つことは、疾患の制御を大きく妨げおよび遅延する。組換え的に発現されるE2タンパク質を生成するための種々の方法の間には、バキュロウイルスにおいて発現されるE2(フラグメント)と比較する場合であっても、差異が存在する。初期に報告されたE2タンパク質生成培養において、E2タンパク質(フラグメント)の収量は、20〜90μg/mlの間で変化し(Hulstら、J.Virol. 5435〜5442、1993;HulstおよびMoormann、Cytotechnology 20:271〜279、1996)、単回注射のワクチン接種のために必要なおよび関連のE2抗原質量を得るためにモノクローナル抗体でのイムノアフィニティ精製をさらに必要とする。別の方法(EP 0389034において記載されるE2のフラグメントを使用する)は、さらなるイムノアフィニティ精製を伴わずに昆虫細胞の上清から採集されたE2を使用し、満足する(防御的な)免疫応答が得られる前に2回注射されるE2ベースのワクチンを得る。E2抗原を含有するワクチン(ProcilisRPesti)が現在登録されているが、所望の免疫応答を提供するために4週間の間隔で少なくとも2回のワクチン接種を行うので、このワクチンは2回適用されなくてはならず、それによって、所望の免疫応答を提供するために4週間の間隔で少なくとも2回のワクチン接種を行うので、このワクチンでの伝統的なブタ熱感染に対するワクチン接種の有用性は、妨げられる。
【0026】
これらの問題(これらは、本発明により解決される)は、特に、ワクチンが調製される開始材料(たとえば、細胞培養上清)中の関連の抗原性物質(この場合においてE2タンパク質(フラグメント))の低い濃度に関連する。理論的に、抗原質量をさらに集積し得え、精製および濃縮法は当該分野において知られるが、これは商業的に魅力的なワクチンの生成を導かず、用量当たり高いコストを引き起こす。別の例は、ペスチウイルスErnsタンパク質であり、これはまたワクチン中でおよび診断試験において、または他の(治療学的)物質中で使用され得る。
【0027】
たとえば、Ruggliら(Virus Genes 10:115〜126、1995)は、単層昆虫細胞培養物中でE2を発現するバキュロウイルスを、5〜10μg/106細胞以下の最大収量に増殖した。さらに、Moserら(Vet.Microbiol. 51:41〜53)は、5のMOIを使用して単層昆虫細胞培養物中でRuglliらのE2を増殖し、そしてELISAの目的のために未濃縮の形態において十分な抗原を生成し得ない。彼らの経験において、ニッケルキレートアフィニティクロマトグラフィによるタンパク質のさらなる精製が、ELISAの操作を単純にし、そしてELISAの質を改善するために前もって必要である。したがって調製されたE2タンパク質を用いるワクチン調製は意図されていない。
【0028】
ワクチンが研究の目的であった場合、ワクチン接種は、防御のある基準を達成するために免疫精製されたE2タンパク質を使用して、2回行われることが必要であったことが見出された。たとえば、Hulstら(J.Virol. 67:5435〜5442、1993)、WO 95/35380、およびvan Rijnら(J. Gen. Virol. 77:2737〜2745、1996)において、E2は単層中で生成され、そしてイムノアフィニティ精製されて、防御ワクチンを達成した。
【0029】
本発明によって提供される例は、組換えCSFV E2タンパク質(フラグメント)を含有するワクチンであり、タンパク質は今や十分に多くの量が生成され得、動物が1用量で単回のワクチン接種を受けた2〜3週間以内に伝統的なブタ熱ウイルス感染に対する防御を付与する(95%の防御用量レベル(PD95)で)ワクチン中に取り込まれる前にイムノアフィニティ精製される必要がもはやない。本発明によって提供される方法は、組換えペスチウイルスE2タンパク質もしくはErnsタンパク質またはそのフラグメントを含有するワクチンを提供し、ワクチンは、1用量での単回のワクチン接種後にペスチウイルス感染に対して防御を付与し、一方タンパク質(フラグメント)は、イムノアフィニティによって精製されていない。本発明はまた、本発明によって提供されるタンパク質を含有し、さらにアジュバントを含有するワクチンを提供する。適切なアジュバント(たとえば、フロインドアジュバント、または水酸化アルミニウム、または乳濁液(たとえば、油中水乳濁液、二重油中水乳濁液、または水中油乳濁液))は当業者に公知である。所望のタンパク質はまた、他の物質(たとえば、獣医学的または医学的使用)を調製するために使用され得る。本発明によって提供されるなお別の例は、卵胞刺激ホルモン(FSH、αユニットおよび/またはβユニット、ならびにその複合体およびフラグメント)のようなホルモン様物質であり、これはたとえば、培養物中でαユニットを発現する1つのバキュロウイルスでおよび/またはβユニットを発現する別のバキュロウイルスで、昆虫細胞培養物を感染することによって生成され得る。本発明の方法はまた、生物殺虫剤としての(組換え)バキュロウイルスの大規模および低コストの生成のために使用され得る。好ましい実施態様は、生物殺虫剤の生成において外来遺伝子の発現のためにp10プロモーターを利用する組換えウイルスの使用である。なぜなら、昆虫は一般に多角体遺伝子を欠陥するバキュロウイルスによりあまり十分には感染されないからである。本発明の方法を用いて、他の組換えタンパク質を発現する他の組換えバキュロウイルスを培養すること、ならびに殺虫剤の、医学的、治療学的、および/または抗原性の物質または産物中に組込むためのこのようなウイルスタンパク質の生成および/または使用は、当業者の範囲内である。本発明はさらに、実施例において説明されるが、それらに制限されない。
【0030】
実施例
フラビウイルス科のペスチウイルス属は、従来、伝統的なブタ熱ウイルス(CSFV)、ボーダ病ウイルス(BDV)、およびウシウイルス性下痢ウイルス(BVDV)からなる。いくつかのBVDVおよびCSFV株のゲノムは配列決定されている(Renardら、1987 EP特許出願第0208672号;Collettら、1988、Virology 165、191〜199;DengおよびBrock、1992、Virology 1991、865〜679;Meyersら、1989、Virology 171、555〜567;Moormannら、1990、Virology 177、184〜188)。BDVについて、未だ不完全なゲノムヌクレオチド配列のみが利用可能である。ペスチウイルスゲノムは、1つの大きなオープンリーディングフレームを含有する約12.5キロベースのポジティブ鎖RNA分子である。オープンリーディングフレームは、約4,000アミノ酸の推定のポリタンパク質に翻訳され、これはウイルスおよび細胞にコードされるプロテアーゼによりプロセスされる。オープンリーディングフレームは、2つの高度に保存された小さな非翻訳領域により隣接され、これらはおそらくゲノムの複製に関与する。5'非コード領域はまた、翻訳の開始において役割を果たす。
【0031】
細胞およびウイルスのプロテアーゼによって、翻訳と同時におよび翻訳後にプロセスされるポリタンパク質は、ウイルスの構造タンパク質および非構造タンパク質の全てを含む(C.M. Rice:Fields Virology、第3版、1996 フラビウイルス科:ウイルスおよびそれらの複製:第30章:931〜959頁を参照のこと)。ウイルス構造タンパク質、特にエンベロープタンパク質のErns、E1、およびE2はポリタンパク質のN末端部分に位置される。非構造タンパク質、特にセリンプロテアーゼNS3ならびにRNAレプリカーゼ複合体NS5AおよびNA5Bは、ポリタンパク質のC末端部分に位置される。
【0032】
ペスチウイルスで感染される動物は、Erns、E2、およびNS3に対する抗体を発生する。しかし、E2に対して指向される抗体のみが強力にウイルスを中和化し、一方ErnsおよびNS3に対して指向される抗体は、ごく低いウイルス中和化能力を有するか、または全く有しない。この知見は、CSFサブユニットマーカーワクチンとしてのE2の適切性を評価することを開始するように本発明者らを促した。この設定において、Ernsおよび/またはNS3は、E2マーカーワクチンを付随する診断試験の開発のために使用され得た。
【0033】
今日までに、BDVおよびBVDVは、異なる種から単離されているが、CSFVはブタに制限されるようである。ペスチウイルスは構造的におよび抗原的に密接に関連する。エンベロープ糖タンパク質のE2は最も免疫原性であり、およびペスチウイルスの最も可変性のタンパク質である。本発明者らは、多くの異なるペスチウイルス株のE2遺伝子をクローン化し、シカおよびキリンからの株を含んだ。E2遺伝子を一過的に発現し、モノクローナル抗体で特徴づけし、配列決定し、そしてP.A. van Rijnら、1997、Virology、237:337〜348に比較した。これらのデータに基づいて、本発明者らは、ペスチウイルス属内に6つの主要な群を示し得た。4つの群は規定された遺伝子型に対応したが、2つの他の群は、ペスチウイルス属内の新規な遺伝子型であった。1つの群は、ブタから単離されたCSFV株を含む。第2の群は、ヒツジから単離されたBDV株Moredun、L83、およびX818、ならびにブタ由来のF株からなる。第3の群は、ヒツジ由来のBD78株、ブタ由来の5250株、およびウシ由来の178003株を含む。E2に基づいて、これらのウイルスは、ウシウイルス性下痢の急性の重篤な発生と関連するBVDV株(いわゆる、2型BVDV)に非常に類似する。第4の群は、ウシに優先的に由来するBVDV株からなる。このBVDV群は、2つの亜型または亜群のBVDV−1aおよび1bに分けられ得:BVDV−1aは、NADLおよびOregonのような米国に由来するウイルス、ならびに欧州に由来する150022および1138のような他のウイルスを含む。亜群BVDV−1bは、Osloss株およびいくつかのオランダ単離物を含む。第5および第6の「群」は、2つの新規な遺伝子型として提唱され得、それぞれ、Deer株およびGiraffe株を含む。
【0034】
獣医学的使用のためのマーカーワクチンの開発は、家畜のウイルス疾患の経済的に重要な制御および根絶のための新しい概念を導いた。マーカーワクチンの使用は、ワクチン接種された動物と野外ウイルスに感染された動物との間の血清学的な区別を許容し、それによってウイルスを制御して排除をできる。大部分のメンバにおいて、たとえば、アウエスキー病(AD)に対するEUワクチン接種の状況は、gEネガティブなワクチンのみで許容される。AD野外ウイルスで感染された動物は、gEに対する抗体を発生する。これらの抗体を検出するELISA試験が、感染された動物の検出のために(続いて感染された動物は集団から除去され得る)、および(長期間の)ワクチン接種キャンペーンの間、集団における野外ウイルス感染の状況をモニタするために使用される。最終的に、目的は、集団の野外ウイルスがない状態を達成することである。次いで、ワクチン接種は中止され、そしてこの状況を保護する血清学的な監視プログラムが行われる。したがって、マーカーワクチンでのワクチン接種は、感染された個々の動物のみのではなく感染された集団の撲滅にかかる根絶運動の費用を有意に減少し、および十分に大きな規模で、および十分に長期間、結果的に行われる場合であっても、ブタ集団の野外ウイルスがない状態を達成するためには、撲滅よりも早い方法であり得る。
【0035】
現在および今後、ADのような現在の風土病が根絶された場合、たとえば、集団から根絶された疾患および日常的なワクチン接種がない疾患の発生を制御するために、マーカーワクチンはなお使用される。このような場合において、動物集団が血清学的にネイティブである場合、非常に伝染性の疾患が爆発的に広がり得、次いで莫大な経済的な損失を引き起こし得る。
【0036】
バキュロウイルス系が非相同タンパク質の高レベルの発現を支持するという多くの例が示された。高レベルの発現は非常に有利である。なぜなら、デッドサブユニットワクチンは、一般に、大量の抗原が適用される場合に防御的な免疫応答を誘発し得るのみであるからである。本発明者らの最初のアプローチにおいて、2つの組換えバキュロウイルス(一方はTMRを伴ってE2を発現する(BacE2[+])および他方はTMRを伴わないでE2を発現する(BacE2[−]))が構築された(Hulstら、1993、J. Virol. 67:5435〜5442)。この刊行物および他の比較可能な刊行物において、E2は未だその古い名称のE1で表されることに注意のこと。TMRを伴わないE2は、細胞培養培地に分泌されるが、TMRを伴うE2は分泌されなかった。さらに、両方のE2は、E2上の4つの抗原性ドメインのそれぞれを提示するモノクローナル抗体と同一に反応する。このことは、細胞会合性E2および分泌性E2の抗原特性が同一であることを示唆し、そして分泌性E2が細胞会合性E2よりも非常に高いレベル(10倍以上の高さ)に生成されたので、ブタにおけるワクチン接種試行において分泌性の免疫アフィニティ精製されたE2を試験することが決定された。二重水-油アジュバント中に20μg E2/mlおよび100μg E2/mlを含有する2つのワクチン処方物を調製した。2つのSPFブタの4つの群のうち、2つの群を20μg E2で、および2つの群を100μg E2で筋肉内にワクチン接種した。28日後、20μg E2でワクチン接種された1つの群および100μg E2でワクチン接種された1つの群を、同じ用量のE2で再ワクチン接種した。42日目に、全ての動物を病原性CSFV株のBresciaで鼻腔内に攻撃した。適用されたワクチン用量にかかわらず、全てのワクチン接種されたブタは、異なるレベルにであったが28日目で中和化抗体をマウントした。攻撃の日にちの42日まで、抗体力価は、全ての動物において、それらが追加免疫されようがなかろうが高レベルに上昇したままであった。それゆえ、20μgの免疫アフィニティ精製されたE2の用量で一回のみワクチン接種された動物であっても、CSFに対して既に完全に防御されたということは驚くことではない(Hulstら、1993 J. Virol. 67:5435〜3442)。
【0037】
ペスチウイルスで感染された動物は、第2のウイルスエンベロープ糖タンパク質のErnsに対する抗体を必ず生成するので(Kwangら、1992、Vet.Microbiol. 32:281〜292)、これはE2と組み合わせて診断試験を開発するために最も適切なウイルスタンパク質の1つである。しかし、ErnsがCSFに対するブタの防御のために重要な抗原であり得たことを示唆する1つの報告がまたある(Konigら、1995、J. Virol. 69:6479〜6486)。これらの研究において、Ernsは生存ウイルスワクシニアベクターで発現された。注射部位あたり5×107PFUのワクシニア−Erns組換えウイルスで3つの異なる経路(皮内、静脈内、および腹腔内)により同時にワクチン接種された動物は、ワクチン接種の5週間後での致死用量のCSFV株Alfortでの攻撃でも生存し、CSFの任意の徴候を示さなかった。
【0038】
デッドサブユニットワクチンとしてのErnsの適切性を評価するために、バキュロウイルスにより発現されたBrescia株のErns(Hulstら、1994、Virology、200:558〜565)を、ブタにおいて試験した。6匹のSPFブタの群を、それぞれ、5.0および20μg Ernsで筋肉内に1回ワクチン接種した(Moormannら、1996、Proc. 14th Intern. Pig. Vet. Society Congress、25〜29頁、Bologna、Italy)。4匹のワクチン接種されなかったSPFブタの第3の群は、コントロールとして作用した。ワクチン接種の3週間後、全ての動物を、105 TCID50のBehring株で筋肉内に攻撃した。攻撃の5日後以内に、最も低い用量のErnsでワクチン接種された全ての動物は、CSFの重篤な徴候を発症した。攻撃の14日後以内に、2匹の動物が死亡し、3匹は瀕死の状態になったときに殺傷され、1匹は見かけ上、回復した。回復した1匹を含むこれらの動物のうちの5匹は、IFTにおいてポジティブであった。攻撃後、最も高い容量のErnsでワクチン接種された全ての動物は、数日間多かれ少なかれCSFの重篤な徴候および高熱を示したが、14日以内に、6匹のうちの4匹の動物が回復し、1匹の動物は死亡し、そして残りの1匹は瀕死の状態になったときに殺傷された。4匹の生存する動物のうちの1匹のみが、IFTにおいてポジティブであるようであった。対照的に、全てのコントロール動物が、攻撃後5日以内にCSFの重篤な徴候を示し、攻撃後14日以内に死亡し、IFTにおいてポジティブであった(Moormannら、1996、Proc. 14th Intern. Pig. Vet. Society Congress、25〜29頁、Bologna、Italy)。
【0039】
本発明者らは、バキュロウイルスにより発現されたErnsは、バキュロウイルスにより発現されたE2投与よりも非常に低い効率であるが、非相同CSFV攻撃に対してブタを防御し得ることを結論した。
【0040】
卵胞刺激ホルモン(FSH)は、下垂体(黄体形成ホルモン、LH;甲状腺刺激ホルモン、TSH)においてまたは胎盤(ヒト絨毛性ゴナドトロピン、hCG;妊馬血清ゴナドトロピン、PMSG)においてのいずれかで生成される糖タンパク質ホルモンのファミリに属する。種内で、これらのホルモンのそれぞれは共通のαサブユニットからなり、これはホルモン特異的なβサブユニットに非共有結合される。単独でまたはLHと組み合わせて投与された精製されたFSHは、ウシを含む多くの種において過剰排卵応答を誘導するために広範に使用される。
【0041】
ウシにおける問題は、ウシFSHがウシ下垂体から実質的な量において精製することが困難であることである。この理由のために、ヒツジのFSH(oFSH)、ブタのFSH(pFSH)、またはウマのFSH(eFSH、PMSG)起源のFSHがウシの過剰排卵処理のために一般に使用される。しかし、ウシにおいてウシ海綿状脳症(BSE)、およびおそらくヒトにおいてクロイツフェルト−ヤコブ病(CJD)の変形を引き起こし得るプリオン様タンパク質の潜在的な存在に起因して、脳組識に由来する材料の適用は危険がなくはない。さらに、胎盤由来の材料の使用は、生物学的半減期が非常に長いという不利益を有し、これは特異的な抗体の注射によるその中和化を必要とする。最後に、全ての現在使用されるFSH調製物は、過剰排卵の結果において観察される大きな変分を少なくとも一部担うと考えられるいくつかのLH活性を含む。
【0042】
これらの理由のために、ウシの過剰排卵処理は、昆虫細胞(バキュロウイルス発現系)のような非哺乳動物細胞において生成される組換えウシFSH(
rbFSH)を適用することにより、有益であり得るようである。
【0043】
材料と方法
1.SF21細胞の産生細胞培養物(PCS)の調製例
SF21作業用細胞種子(WCS)1.5mlを容れた凍結バイアル(細胞総数、4〜10×106細胞/ml)を20〜30℃の温度で融解する。融解後、バイアルの内容物を、無血清培地SF900II8.5mlを含む15ml容量ファルコンチューブに移し、懸濁させる。懸濁後、ファルコンチューブの内容物を100〜200×gで10分間遠沈し、細胞を沈殿させる。培地を廃棄し、ペレットを4〜6mlのSF900IIに懸濁する。懸濁した細胞を、10mlのSF900IIを容れた100ml容量の振盪フラスコに移す。フラスコを軌道旋回式シェーカの台上にのせ、40〜80rpm、26〜30℃で3〜7日間、細胞を培養する。細胞の増殖と細胞生存度は、工程途上サンプルを取出しモニタする。細胞密度が1.0〜6.5×106細胞/mlに達したとき、それぞれ100mlのSF900IIを容れた2個の500ml容量振盪フラスコに移す。これは10倍希釈の細胞に相当する。再度、フラスコを軌道旋回式シェーカの台上にのせ、40〜80rpm、26〜30℃で3〜7日間、細胞を培養する。細胞の増殖と細胞生存度は定常的に工程内コントロールによりする。細胞密度が1.0〜6.5×106細胞/mlに達したとき、二度目として、今回はそれぞれ100mlのSF900IIを容れた6〜11個の500ml容量振盪フラスコに移す。過剰の細胞材料は廃棄する。また、この場合、10倍の希釈を達成する。フラスコを軌道旋回式シェーカの台上にのせ、40〜80rpm、26〜30℃で3〜7日間、細胞を培養する。細胞の増殖と細胞生存度は定常的に工程内コントロールによりモニタする。細胞密度が1.0〜6.5×106細胞/mlに達したとき、該細胞を含む懸濁液500mlまたは1000mlを、それぞれ約4.5リットルまたは9リットルのSF900IIを容れた5リットルまたは10リットル醗酵槽に移す。これは10倍希釈の細胞に相当する。この細胞を26〜30℃で3〜7日間培養する。この懸濁液を定常的に攪拌する(50〜100rpm)。細胞の増殖と細胞生存度は定常的に工程内コントロールによりモニタする。細胞密度が1.0〜6.5×106細胞/mlに達したとき、5リットル容量醗酵槽の内容物を、約40リットルのSF900IIを容れた50L醗酵槽に移す。この細胞を、密度が±5〜15×105細胞/mlに達するまで26〜30℃で培養する。この懸濁液を定常的に攪拌する(50〜100rpm)。サンプルを工程内コントロール用に採取する。
【0044】
2.E2抗原の産生例
上記の細胞懸濁液に、±107TCID50/mlを含む作業種子ウイルス(WVS)(BacE2[−])1〜2mlを添加する。細胞の70〜100%が細胞変性効果を示すまで、28℃にて3〜8日間培養する。培養の間に工程内コントロール用にサンプルを採取する。次に、この懸濁液を精密濾過による細胞除去により清澄とする。得られた濾液(すなわち、抗原溶液)を集め、不活化工程(3.3)を始めるまで≦−20℃にて保存する。工程内コントロール用にサンプルを採取する。抗原含量を定量するために、サンプルを、たとえば、抗原質量を定量するために、酵素結合免疫アッセイで、または水中タンパク質容量あたりの実質重量を定量するために、タンパク質アッセイで、あるいはかかる方法の併用により試験する。
【0045】
3.ウイルス不活化例
ウイルスに臭化2−ブロモエチルアンモニウム(BEA)を10ミリモル/Lの濃度に加えることにより不活化する。pHを8.2〜8.7に、温度を34〜37℃に調節することにより、BEAを臭化2−ブロモエチル−インミン(BEI)に変換する。BEIはウイルスを不活化する活性成分である。ウイルス殺滅は工程内コントロール用サンプルを採取することによりチェックする。不活化には24〜72時間を要する。不活化後、10000リットルあたり1個未満の感染性粒子が存在する。ウイルスの不活化後、チオ硫酸ナトリウムを5〜25ミリモル/L添加し、pHを5.7〜6.3に調整することによりBEIを中和する。工程内コントロール用にサンプルを採取する。不活化し、中和した抗原溶液を1リットルおよび/または5リットル容量のバッグに移し、≦−20℃にて保存する。
【0046】
4.製剤化例
凍結した抗原溶液を22〜28℃で融解し、SF900IIまたはPBSにより希釈し、50μg/mlの抗原溶液とする。抗微生物剤としてチオメルサールを100μg/mlまで加える。工程内コントロール用にサンプルを採取する。この溶液(すなわち、第1水相)を2〜8℃にて<3日間保存する。この間、マルコール(Marcol)52とモンタニド(Montanide)80とを混合する(9:1)ことにより油相を調製する。また、この溶液も2〜8℃にて3日を超えない範囲で保存する。油相は0.22μm−無菌フィルタにより無菌濾過する。工程内コントロール用にサンプルを採取する。最後に、第2水相をリン酸バッファー塩溶液(PBS)またはSF900IIをモンタノックス(Montanox)80と混合することにより調整し(98:2)、チオメルサールを最終濃度100μg/mlまで添加する。この溶液を使用時まで2〜8℃にて保存する(<3日間)。使用前、第1水相を無菌濾過する。工程内コントロール用にサンプルを採取する。第1水相と油相とを混合(1:1.1)することにより第1エマルジョンを調製する。このエマルジョンを2〜8℃にて3日以内保存する。工程内コントロール用にサンプルを採取する。二重油中水エマルジョンは、第1エマルジョンを第2水相で乳化することにより(2.1:1)調製する。二重エマルジョンはバイアルに詰めるまで隔離保存室内に2〜8℃にて保存する。工程内コントロール用にサンプルを採取する。
【0047】
5.充填および施栓例
清浄室内において、二重エマルジョン溶液をクラスAゾーンにて無菌的に充填する。充填量は50、100または250mlバイアルに、それぞれ51、102または255mlである。充填容量は濾過容積の重量をチェックすることにより定常的にモニタする。充填直後、バイアルには栓をし、キャップを被せる。最後に、バイアルは隔離保存室内に2〜8℃で保存し、その後の品質管理を開始する。
【0048】
【表1】

【0049】
【表2】
【0050】
【表3】
【0051】
【表4】
【0052】
【表5】
【0053】
6.E2安定性実験例
目的
異なる正常条件下、長期間培養後の感染細胞培養物(MOI*0.0001)におけるE2タンパク質の安定性を検討するため。細胞培養におけるバキュロウイルス感染過程の溶菌性のために、感染サイクルの後期培地にプロテアーゼが放出されていると思われる。このことがE2タンパク質の分解と容量タンパク質レベルの低下に至らしめるのか否かが、本実験の命題である。
【0054】
材料と方法
培地またはSF900IIと記載されている場合、0.2%プルロニックF68含有SF900II培地を意味する。
【0055】
SF21細胞を容れたバイアルを融解し、100ml容量振盪フラスコ中、10mlのSF900IIにより、インキュベータ内軌道旋回式シェーカ台座上、培養する。細胞培養が必要とする細胞密度に達したところで、細胞を500ml振盪フラスコ中、10倍に希釈して100mlとする。この細胞培養が必要とする細胞密度に達したところで、再度、10倍に希釈し100mlとし、過剰の細胞性物質を廃棄した。この細胞培養が必要とする細胞密度に達したところで、これを10倍に希釈し、3個の500ml振盪フラスコに各フラスコ100ml容量ずつ入れた。過剰の物質を廃棄した。振盪フラスコをIKS軌道旋回式シェーカ台座に載せ、28℃のインキュベータ中70rpmで攪拌する。細胞密度1ml当たり0.612×105細胞で、100倍希釈のウイルス懸濁液63μlをフラスコ番号2および3に加える。振盪フラスコ番号1は未感染ブランクとして維持する。培養物を感染させたMOIは(0.063×0.01×0.8×107)/(100×0.612×105)=0.00008である。
【0056】
使用した100倍希釈ウイルス懸濁液は以下に概説したように調製した。MSVバイアル253をQCにより融解し、TC100培地に100倍希釈し、10%FBS(ロット番号97QC0139)を添加し、0.5mlずつに分割して−70℃に保存した。
【0057】
細胞培養条件を決定するために、定期的にサンプルを感染振盪フラスコ培養物と非感染振盪フラスコの両方から採取する。サンプル採取は以下に概括したように実施した。
【0058】
細胞物質約1.1mlを15ml容量のファルコンチューブ中、IECセントラGP8R遠心分離機により1000rpmで6分間遠心分離する。次いで、上清をゲルマン・アクロディスク・シリンジフィルタにより0.45μmで濾過し、なお残存する細胞を廃棄する。サンプル0.4mlを後の分析用にナルゲン低温バイアル中に−70℃で保存する。
【0059】
結果と結論
図1のデータおよび図2のグラフに見られるように、フラスコ2および3の細胞増殖曲線はおおよそ類似している。191hpiの後、それ以上の細胞計数はなされなかったが、その理由は事実上すべての細胞が死滅し、感染したからである。フラスコ2の139hpiサンプルはグラフから除外する。このサンプルの細胞計数は、前記のサンプルおよび以下のサンプル両方よりもはるかに高い。このことは、恐らくある種の沈殿がサンプル採取の前後に起こり、その結果、トリパンブルー溶液と混合したサンプルが不正確となったことを示している。この仮定は生存率と感染率両方が予測した結果と一致するという事実により確認される。このことは、感染培養物それ自体にはなんら異常なことが起こっていないことを意味する。この不正確なサンプル採取は、サンプル中のE2濃度になんら影響を与えることはないだろう。というのは、E2は培地には存在するが、細胞内には存在しないからである。
【0060】
フラスコ2のE2濃度は非常に急速に上昇して、139hpi近辺で>190μg/mlの最大値となるが、そこからゆっくり低下して215hpiで170μg/mlとなる。次いで、E2−含量がさらに急速に降下して、306.5hpiで<100μg/mlの最終レベルとなる。
【0061】
フラスコ3のプロフィールはおおよそ同じである。最大E2タンパク質含量もまた139hpi付近で到達するが、フラスコ2よりもわずかに高いだけである。E2濃度は139hpiでの233μg/mlから191hpiでの212μg/mlにゆっくりと低下するが、それ以後はより急速に低下し、215hpiで141μg/mlに至る。フラスコ2および3は良好な相関性を示す。
【0062】
t=44.25hpiで、振盪フラスコ番号1(ブランク)についても計測した。生存細胞密度は2.435×106細胞/ml、死滅細胞密度は0.190×106細胞/mlであり、総細胞密度は2.625×106細胞/mlであって、生存度は92.8%となった。生存細胞密度はフラスコ2および3両方よりも有意に高く、感染が細胞増殖をすでに減速しつつあることを示している。このブランクを次いで濾過した。
【0063】
以下の表はフラスコ2のサンプル抜粋物について実施した免疫ブロッティングアッセイの結果を示す。
【0064】
【表6】
【0065】
この表に見られるように、エピトープV3およびV8は、0および44hpiサンプルでなお検出不可である。この結果はE2−ELISAの結果とよく相関する。V3およびV8はともにE2バンドに検出されるので、115hpi以降のサンプルにはインタクトのE2が検出される。
【0066】
191hpi以降には、より小さいタンパク質からの第2バンドがゲル上に目視できる。2つの個々のブロットにおけるV3とV8での免疫ブロッティングは、分解産物がV3エピトープを含み、V8エピトープを含まないことを示す。V8はブロット上のいずれの場所にも検出されないが、未処理E2バンドには検出される。このアッセイは、E2タンパク質の分解がプロテアーゼの存在により起こることを明瞭に示している。事実、これがE2タンパク質含量降下の原因であり、この降下はバルクE2抗原溶液の精密濾過工程の際に観察される。この下流の処理加工段階で、細胞はウイルスの不活化が始まる前に除かれる。
【0067】
したがって、恐らくプロテアーゼの存在がE2タンパク質分解の原因である。
7.MOI実験例
目的
MOI(感染多重度、細胞当たりのウイルス数)、最大細胞密度、および容量計測E2含量間の関係をさらに検討すること。一方で、完全な細胞母集団の感染は培地が消費し尽くされるまでに完了しなければならないが、他方で、低細胞濃度の総母集団の感染は最適以下のタンパク質収量に至る。
【0068】
材料と方法
培地というときは、SF900II培地を意味する。
【0069】
細胞懸濁液76mlを、1000ml容量ボトル中、444mlのSF900IIに希釈し、ゆるやかに混合する。培養物希釈直前の生存細胞密度は、3.41×106細胞/ml、死滅細胞密度は0.190×106細胞/mlであり、生存率は94.8%となる。この希釈により、細胞密度は±5×105細胞/mlとなる。
【0070】
さらに、ゲンタマイシンを加え、最終濃度10μg/mlとした。細胞懸濁液を50mlずつ250ml振盪フラスコ中に9分割し、過剰の細胞は廃棄した。最初の50mlを振盪フラスコ1に移し、最後の50mlを振盪フラスコ5に移した。フラスコ番号1および6はMOI=0(ブランク)に使用し、番号2および7はMOI=0.000001用とし、番号3および8はMOI=0.00001用とし、番号4および9はMOI=0.0001用とし、番号5はMOI=0.001とする。
【0071】
ウイルス原液の調製は以下のようにする。希釈範囲は、100倍希釈したウイルス懸濁液を入れたバイアルから調製する(該バイアルは1ml当たり0.8×105プラーク形成単位(pfu)を含む)。この100倍希釈したウイルス原液は以下のように調製した。マスター種子ウイルスを融解し、TC100培地に100倍希釈し、10%FBSを添加して、0.5mlに分割、−70℃にて保存する。
【0072】
この100倍希釈したウイルス溶液0.4mlをSF900II培地3.6mlで希釈して希釈ファクタ10-1とし、この溶液1mlを培地9mlに加えて希釈ファクタ10-2とし、この溶液1mlを培地9mlに加えて希釈ファクタ10-3とし、そしてこの溶液1mlを10倍に希釈して希釈ファクタ10-4とする。細胞培養物を含む振盪フラスコ1および6(ブランクフラスコ)に培地3.1mlを加える。ウイルス希釈液10-2、10-3および10-4をそれぞれ振盪フラスコ4と9、3と8、および2と7に加える。これらの振盪フラスコにはすでに細胞培養物を入れてある。
【0073】
3.1mlのウイルス希釈液10-2には、3.1×10-2×0.8×105=2.5×103pfuが存在する。50mlの細胞懸濁液を容れた振盪フラスコには、0.5×106×50=2.5×107の細胞が存在する。そこで、50mlの細胞懸濁液に3.1mlのウイルス希釈液10-2を加えると、MOIが2.5×103/2.5×107=0.0001となる。他のウイルス希釈液について同等の計算をすると、細胞培養物50mlに添加したウイルス希釈液10-3と10-4については、MOI計数0.00001と0.000001が得られる。最終的に、2.85mlの希釈液10-1を振盪フラスコ番号5に加えると、MOIが0.001ではなく0.00092となる。
【0074】
サンプル3.1mlを、ウイルス添加直後、各振盪フラスコに取る。サンプル中の細胞は15mlファルコンチューブ中、IECセントラGP8R遠心分離機により遠沈する(10分、1000rpm)。サンプル採取は低MOIから始めて高MOIについて実施し、高MOIの感染細胞懸濁液から低MOI感染細胞懸濁液にウイルスが伝播するのを防止する。遠心分離後、サンプルを0.45μmで濾過し(上清になお存在する可能性のある細胞を除去するため)、3個のナルゲン低温バイアルに分割し、後の試験用に−70℃にて保存する。
【0075】
振盪フラスコ1および5の細胞密度はウイルス添加直後に定量した。これらは細胞接種すべき最初と最後のフラスコであった。細胞密度が殆ど一致したので、すべてのフラスコは両方の細胞計数平均値に等しい細胞密度を有すると見なした。当初の生存細胞密度は0.423×106細胞/mlであり、一方、0.012×106細胞が死滅していたので、生存率は97.3%となった。
【0076】
すべてのフラスコをラボテック300軌道旋回式シェーカ台座に載せ、28℃の室内にて約75rpmで攪拌した。
【0077】
23、95、125、144、173および194hpi(感染後の時間)に、すべてのフラスコからサンプル採取した。細胞密度を定量したサンプルは容量が3.3mlあり、その0.2mlを用いて細胞密度を定量した。残り3.1mlはt=0サンプルのために上記のように取扱う。計測しなかった培養物サンプルは容量が3.1mlあった。194hpiでの細胞密度を定量し、この実験を終えた。残りの細胞懸濁液(約30ml)は、その1ml、3部をナルゲン低温バイアルに、約6mlの4部を15mlファルコンチューブに入れ、保存した。
【0078】
結果と結論
この結果は、MOIと最適細胞密度との間に、さらに重要なことは、MOIとE2タンパク質収率との間に良好な相関があることを示す。このことは、MOI 0.001で感染した培養物の最大容量E2タンパク質収率を100%とした場合のグラフに見て取れる。それは容量E2タンパク質収率がMOIの減少とともに増大することを示す。0.0001のMOIまでは、細胞密度は、最大達成可能細胞密度に達する前に感染される。0.00001またはそれ以下のMOIを用いると、培養物の感染が終了する前に培地が消耗されてしまうという感染結果となり、最適以下のタンパク質収率となる。したがって、引き出される結論は、感染過程とE2の産生が完了するまでに培地が消尽しない限り、使用されるMOIが低い程、E2タンパク質収率が高くなるということである。
bFSHαおよび/またはbHSFβを発現する組換えバキュロウイルス
伝達ベクターpDW−アルファ−9.1およびpDW−ベータ−3.1を構築した。Sf21細胞はpDW−アルファ−9.1またはpDW−ベータ−3.1、および細胞外ウイルス粒子から単離した野生型(wt)AcNPV/M021DNA(PCT出願:WO96/25696)により同時移入した。β−ガラクトシダーゼ発現ポリヘドリン陽性プラークを単離し、bFSHαまたはbFSHβを発現するかについて、培地のELISAにより分析した。一つのプラーク精製したbFSHαウイルス(AcNPVα3.4)および一つのプラーク精製したbFSHβ(AcNPVβ1.4)を用い、それぞれ約107と108のTCID50をもつウイルス原液を調製した。FSHの産生は上記方法により生じ、産生レベルは17〜33μg/mlとなった。
【0079】
PD50試験
病毒力の強いCSFV株ブレスシア(Brescia)100LD50の攻撃に対し、予防接種したブタの95%(PD50)を予防する、1回の予防接種後に必要とするE2用量(μg)の評価。
【0080】
動物
26頭の特異病原体陰性(SPF)ブタ(6〜7週令)を到着順に無作為に、8頭ずつの3群(A〜C)と2頭の1群(D)に分けた。これらの動物をID−DLOの高度封じ込め設備内にある家畜小屋B18、B19、B20およびB21に収容した。動物は3日間順化させる。動物は1日に1度、飼い葉桶で完全食物ペレット(ホープ・ファームス)として給餌し、水は給水器(niple ad libitum)から飲むことができた。
【0081】
予防接種および攻撃
二重油中水型アジュバント添加ワクチンの3種の製剤を上記のように調製し、それぞれ異なるE2抗原濃度、32.0、8.0および2.0μg/投与量とした。ブタには筋肉内に一度接種したが、各ブタは左耳の後方2cmのところに1回量(A:2μgのE2、B:8μgのE2、C:32μgのE2、D:0μgのE2)を受けた。対照群DはDOEアジュバントのみを接種し、前哨用のブタにはまったく接種しなかった。予防接種の3週間後に前哨用を除く各動物に、CSFV株ブレスシア456610の100 50%致死量(=100Va LD50)を鼻腔内攻撃した。攻撃直前に前哨用をその群から隔離し、24時間後に戻した。接種物のウイルス含量は家畜小屋から戻した後に採取したサンプルの滴定により定量した。
【0082】
臨床的観察
ブタは動物飼育係が毎日チェックし、異常が見つかり次第記録し、要すれば、監督者の獣医を呼んだ。各群は給餌時および小屋の清掃前およびその間に1日少なくとも15分間観察した。
【0083】
食餌摂取の減少した群または個々の動物に注目した。
体温(直腸)は予防接種後9日間、また、攻撃後20日間記録した。
【0084】
攻撃後の血液分析
EDTA−血液サンプルを攻撃後、0、2、4、7、10および14日目に採取し、白血球および血小板数の変化をモニタした。血中の白血球(血球減少症)および血小板(血小板減少症)数減少はCSFの典型的な徴候の一つである。通常のブタでは、白血球細胞と血小板に対する正常細胞計数は、それぞれ11〜23×109/Lおよび320〜720×109/Lの範囲にある。SPFのブタでは、これらの値がやや小さい;6〜12×109/Lおよび300〜700×109/L。双方言及の範囲は各ブタで変動する。血液細胞の分析はメドニックRCA570クールタ計数器により実施した。血球減少症および血小板減少は、細胞/血小板計数が、好ましくは1日以上、上記の最小数よりも顕著に低い場合と定義した。。
【0085】
ウイルス拡散/排出およびウイルス検出
体温、白血球細胞と血小板計数、および前哨のセロコンバーションをパラメーターとして使用し、接種した動物からこれらの動物へとウイルスが伝播したことを検出した。死後の組織サンプルは以下の器官から採取した:扁桃腺、脾臓、腎臓、および回腸について、ウイルス抗原の存在につき直接免疫蛍光法により試験した。これら組織サンプルからの低温維持切片(厚さ4μm、各器官2切片)を固定化し、ポリクローナルブタ抗ペスチウイルスFITC−接合血清と培養した。洗浄後、切片を蛍光顕微鏡下で読取った。結果を陽性(=蛍光性)または陰性(=非蛍光性)として表した。
【0086】
血清学的応答
すべてのブタの血清を予防接種後、0、2および3週目、および死亡時に採取した。サンプルを−20℃にて保存し、ウイルス中和試験(VNT)およびセディテスト(CeditestR)(CSF特異抗体を検出するためのELISA)によりアッセイした。血清中のCSFV中和抗体タイタをミクロタイタシステムにより定量した。血清の段階2倍希釈液を、30〜300TCID50含有のCSFV(ブレスシア株)懸濁液等容量と混合した。CO2インキュベータ内37℃で1時間培養した後、ウエル当たり約25.000PK15細胞を添加した。4日後に、マイクロタイタプレートを上記のように処理し、顕微鏡下に読取った。CSFV中和タイタは、すべてのウイルスを中和する最高希釈の逆数として表した。
【0087】
統計的評価
95%防御的用量の決定は、≧50のCSFV中和Abタイタが完全な防御を表わすという仮定にもとづいた。
【0088】
結果
本セクション全体を通して動物数については表7または表8に示す。
【0089】
予防接種後の臨床的観察
予防接種はブタに何ら悪影響を及ぼさず、食餌摂取も体温も正常のままであった。A群およびC群には、軽い下痢、食欲不振、およびうつ症状が、予防接種後3日目に見られた。A群からの1頭(no.448)は全期間にわたり定常的に嘔吐し、成長が遅れた。B群からのブタno.438は一度嘔吐した。ブタno.434(C群、前哨)は体温がわずかに上昇したが、この動物は右後肢の跛行をわずらっていた。食餌摂取量は、予防接種から攻撃までの期間、1日1群当たり3kgから6kgに増加した。
【0090】
攻撃後の臨床的観察
非予防接種対照動物は、攻撃後3日目(no.59)および6日目(no.60)にCSFの徴候を示し始めた。発熱、群がり行動、震え、食欲喪失およびうつ症状が攻撃から10日後の死亡時まで見られた。さらに、この動物は、チアノーゼ、後肢不全麻痺、下痢および重症の嘔吐を示した。両動物は瀕死の状態のときに殺した。
【0091】
2μgのE2で予防接種したブタ(A群)はすべて攻撃の2〜3日後に病気の徴候を示し始め、主に、発熱、群がり、うつ症状および食欲不振を示した。発熱は2〜10日に及び、最高体温(Tmax)は40.7〜42.2℃で変動した。7日目以降、ブタの内、443〜446は発熱が収まり、食餌摂取量も攻撃前の量まで増大した。しかし、448のブタは9日目に死亡しているのが見つかり、447のブタは急性のCSF(痙攣、後肢不全麻痺)に懸り、同日瀕死の状態であるときに殺した。両前哨用は攻撃7〜9日後まで正常であったが、その後、急性のCSFとなり、20日目、瀕死の状態であるときに殺した。
【0092】
B群およびC群の動物は、疾患の臨床徴候および持続期間が、E2抗原の有効量増大とともに軽くなった。
【0093】
B群では、発熱(435〜439)が27日目まで続き、Tmaxは40.2℃と41.7℃の間で変動した。ブタno.436は非常に軽度の臨床徴候で攻撃に抵抗した。1頭のブタ(no.440)は18日後に急性のCSFで死亡したが、瀕死の状態であるときに殺した。残り、B群からの5頭の動物は回復した。両前哨用は攻撃後11〜12日目まで正常であったが、その後、急性のCSFとなり、20日目に死亡させた。
【0094】
C群では、4〜6日目から6頭の内2頭(430、431)に軽い発熱が見られ、Tmaxは40.5℃〜41.2℃で変動した。臨床的徴候は、攻撃後6日目にわずかなうつ症状が見られた以外、注目すべきものはなかった。従前どおり、攻撃ブタno.430は嘔吐した。
【0095】
両前哨用は実験終了時まで正常であった。
攻撃後の血液分析
A群からは、ブタ(450)1頭のみ(前哨用)が明瞭な白血球減少および血小板減少を示した。ブタno.445および446は攻撃後、7日目および10日目に、また、ブタ449は10日目および14日目に血小板減少となった。B群からのブタ1頭(440)は白血球減少および血小板減少を示したが、他は正常であった。両前哨用は14日目に血小板減少となり始める徴候を示した。C群では、前哨用も含めて、白血球減少も血小板減少も検出しなかった。
【0096】
両対照動物(no.59および60)は7日目以降から血小板減少となった。
攻撃後のウイルス拡散およびウイルス抗原検出
家畜小屋から戻って後、接種量を逆滴定して、ウイルス力価を2.55TCID50/mlとした。CSFVウイルス抗原(表7)は、A群およびB群から選択した対照と前哨用の組織サンプルすべてに検出された。A群およびB群において、6頭の接種した動物の内、1頭が陽性であった。C群では前哨用も含めて陽性がなかった。
【0097】
血清学的応答
CSFVでのセロコンバーションはVNTにおいて力価≧25と定義した。VNTに用いたブレスシアウイルスの量を定量するための逆滴定の結果は、41TCID50/ウエルであった。
【0098】
対照(D)または前哨用動物は本実験の間、セロコンバーションを受けなかった(表8)。A群の動物6頭の内2頭が3週間後にセロコンバーションを受けた。しかし、6頭の内4頭は攻撃後、追加刺激を受けた(表8)。B群およびC群は予防接種後21日目にセロコンバーションを受けたが、この動物はすべて攻撃後に追加刺激を受けた。後者のみが、当該動物において攻撃ウイルスの複製が成功していることを物語っている。
【0099】
結論
動物1頭当たりのPD95投与量を一回につきDOEアジュバント2ml中32μgのE2と決定した。この投与量により、毒性の非常に高いCSFV株を攻撃したことによる疾患の臨床徴候が最小に止まり、接触した動物へのウイルス拡散も発生しない。要約すると、6頭のブタ4群(A〜D)につき、筋肉内投与により、アジュバント(DOE)2ml当たりそれぞれ32(A)、8(B)、2(C)および0μg(D)で予防接種した。2頭の非予防接種ブタを各群(A〜C)内に置き、接触感染の原因となるウイルスの排出を検出した(前哨用)。3週間後、鼻腔内投与により毒性の非常に高いCSFV株ブレスシア100LD50で動物を攻撃した。臨床学的、ウイルス学的および血清学的パラメーターを測定し、E2サブユニットワクチンの効能を評価した。予測どおりに、C群の動物は他の群の動物よりもより良好に攻撃に抵抗した。ウイルス抗原は、E2の最大投与量(32μg)を接種した動物の組織サンプルに検出されず、この群には疾病も存在しなかった。PD95用量は、≧50のNPLA力価(予防接種後21日目)が完全な予防効果を与える(ウイルスの拡散がない)という仮定にもとづき計算した。この結果がPD95用量32μg/頭である。
【0100】
予防接種2週間および3週間後の100LD50毒性CSFVによる攻撃に対し、その防御効果のレベルをさらに評価するために、6頭のブタ2群(A〜B)を32μgのE2により筋肉内経路で予防接種した。2頭の非予防接種ブタを各群内(A〜B)に置き、接触感染の原因となる予防接種ブタからのウイルスの排出を検出した。2週(A)および3週(B)後、予防接種した動物および対照の動物を毒性の非常に高いCSFV株ブレスシア100LD50で鼻腔内経路により攻撃した。予防接種の2週間および3週間後、臨床学的、ウイルス学的および血清学的パラメーターを測定し、E2サブユニットワクチンの効能を評価した。予測どおりに、B群はA群よりも臨床徴候が少なく、攻撃に抵抗した。前哨用へのウイルスの伝播はA群にのみ起こり、両群からの動物すべての白血球フラクションにウイルスが検出された。B群の動物1頭のみが予防接種の14日目にセロコンバーションを受けていた(no.414)。同一群からの動物1頭は21日後にもセロコンバーションを受けなかったが、防御されており、発熱は2日間のみであり、攻撃後11日以内にセロコンバーションを受けた。対照と前哨用の1頭(A群)のみが白血球減少症と血小板減少症に罹患した。A群およびB群動物からの組織サンプルいずれにもウイルス抗原は検出されなかった。結論として、動物はすべて単回投与での予防接種後2週間および3週間目の攻撃に対し防御されていた。
【0101】
予防接種3ヵ月および6ヵ月後の100LD50毒性CSFVによる攻撃に対し、その予防効果のレベルをさらに評価するために、6頭のブタ2群(A〜B)を32μgのE2により筋肉内経路で予防接種した。2頭の非予防接種ブタを各群内(A〜B)に置き、接触感染の原因となる予防接種ブタからのウイルスの排出を検出した。3ヵ月(A)および6ヵ月(B)後、予防接種した動物および対照の動物を毒性の非常に高いCSFV株ブレスシア100LD50で鼻腔内経路により攻撃した。この期間の後、臨床学的、ウイルス学的および血清学的パラメーターを測定し、E2サブユニットワクチンの効能を評価した。A群およびB群からの動物はすべて攻撃に抵抗し、臨床徴候はわずかであった。そして対照のみが白血球減少症と血小板減少症に罹患した。前哨用へのウイルスの伝播は起こらなかった。また、両群から選択された白血球フラクションのいずれにもウイルスは検出されなかった。B群の動物1頭のみが予防接種後の28日目にセロコンバーションを受けていた(no.1983)。A群、B群および前哨用動物のいずれの組織サンプルにもウイルス抗原は検出されなかった。結論として、すべての動物が単回投与での予防接種後3ヵ月および6ヵ月目の攻撃に対し防御されており、ウイルス伝播は起こらなかった。
0 BVDV株4800、150022および178003を用い、実験的E2サブユニットワクチンを生成させた。これらの株のE2遺伝子はバキュロウイルス発現系において発現させた(ハルスト(Hulst)ら、1993、J. Viol. 67:5435〜5442)(リジュン(P.A.van Rijn)ら、1996、I)、Gen. Virol.、77:2737〜2745)。スポドプテラ・フルギペルダ(Spodoptera frugiperda)細胞系SF21を、組換えバキュロウイルスの増殖とE2タンパク質の生産に用いた。SF21細胞は無血清SF900培地(ギブコBRL)中28℃にて増殖した。SF21細胞の集密単層を組換えバキュロウイルスにより、細胞当たり5TCID50(50%組織培養感染用量)の感染多重度にて感染させた。4〜5日後、培養は80〜90%の細胞変性効果を示した。細胞を1500×gで10分間遠心分離し、バキュロウイルスとE2を含むその上清を収集し、−20℃にて保存する。バキュロウイルスを不活化するために、臭化2−ブロモエチル−インミン(BEI)での処理を標準手法に従い実施した。手短に説明すると、BEI 600μlと1M−NaOH600μlとを混合した。20℃、30分後、上清150mlを加え、20℃で24時間攪拌した。次いで、25%チオ硫酸20mlを加えた。バキュロウイルスの不活化はSF21細胞上、上清の滴定により確認した。
【0102】
1 実験的BVDVワクチンは二重油中水エマルジョン中、E2含有上清からなっていた。該油は90%マルコール52(エッソ)および10%モンタニド80(セピック)を含んでいた。水性フラクション(PBS+)は98%リン酸バッファ塩溶液、2%モンタノックス80(セピック)および100ppmチオメルサールを含んでいた。上清、油およびPBS+は、10:11:10の比で用いた。まず、上清と油を乳化し、次いでPBS+を加え、乳化した。対照のワクチンは、野生型バキュロウイルスで感染させたSF21細胞の上清から同様に調製した。調製後、ワクチンは無菌であると判定された。
【0103】
2 3種ワクチンの抗原量は、発現されたタンパク質の同等性の故にほぼ同じであろうとする我々の当初の仮定は、間違っていた。抗原量の差は恐らく昆虫細胞での発現の差に原因がある。ワクチン150022は用量当たり55μgのE2を含んでおり、2回の予防接種(0日および21日目)後、胎児に対し防御的であった(ブラッシェ(Brusche)ら、1997、Vaccine、印刷中)。ワクチン4800およびワクチン178003は用量当たり12μgと17μgのE2をそれぞれ含んでおり、胎児に対し防御的ではなかった。この結果はE2タンパク質量と胎児の防御との間に相関性のあるこを示唆している。しかし、E2糖タンパク質の免疫原性の差と、攻撃株が胎盤を通過する能力のないことが、攻撃が異なる結果をもたらすことに対する説明となるかも知れない。非相同BVDVの攻撃に対し、該ワクチンは防御しなかった。
【0104】
3 この研究の結果はBVDVサブユニットワクチンのさらにる開発のために将来有望である。その理由は我々が、エンベロープ糖タンパク質E2での予防接種により胎児の防御を達成し得ることを示したからである。さらにそれはマーカーワクチンであり、予防接種した動物と野外ウイルス株との間に識別を可能とする。これは将来のBVDV撲滅プログラムに照らして有利である。
【0105】
検討
4 この開発された生産工程は、昆虫細胞内バキュロウイルスに挿入された遺伝子がエンコードする非相同タンパク質の最適生産を達成することを目的とする。バキュロウイルス発現ベクター系(BEVS)は異なる組換えタンパク質の産生に非常によく適合する。その理由は、正しいタンパク質の折りたたみと正確な翻訳後プロセシングが、動物とヒトへの応用のために生物学的に活性なタンパク質に至らしめるからである。バキュロウイルスは2つの非本質的遺伝子を含み、それは非常に強力なプロモーター、p10およびポリヘドリンプロモーターから転写される。p10遺伝子の欠失突然変異誘発(バン・オアーズ(van Oers)ら、J. Gen Virol. 74:563〜574;1993)は、それが細胞培養におけるウイルスの複製にとって必須ではないが、p10が細胞溶解において一つの役割を演じており、p10タンパク質が存在しないと、感染した細胞が損なわれないままに存在することの原因となり、感染細胞からの多角体の放出を妨ぎ、それによって再感染率を降下させるが、それ自体感染性ではないということを示した。遺伝子産物(p10(またはフィブリリン)およびポリヘドリン)は感染段階の後期に発現される。これらの遺伝子を、必要とするタンパク質の遺伝子に置換えても(ポリヘドリンプロモーターの場合)、理論的に、昆虫細胞培養の総タンパク質生産収率は50%までとなるが、発現レベルは培養1リットル当たりポリヘドリンタンパク質1グラムを超えるものとなる(マイオレラ(Maiorella)ら、(1988) 組換えタンパク質生産のための大規模昆虫細胞培養。Bio/technology、6、1406〜1410)。感染多重度(MOI、1ml当たりのウイルス密度と1ml当たりの細胞密度の比)は、タンパク質生産の最適化にとって重要なパラメーターである。感染多重度の低い感染であることの明白な理由は、ウイルス植菌量をできるだけ少なくしていることにある。もし感染が、50L醗酵槽内でMOIを0.1として1ml当たり2百万の細胞密度で起こるならば、その場合は、1×1010のウイルスが必要となる。このことは約1リットルの植菌量を必要とすることである。さらに、かかる植菌量を造るための余分な生産工程が必要となる。付着培養の2つの主な欠点は、効率の悪い培地の使用と酸素量が制限されることである。水性液での酸素溶解度が比較的乏しいという事実のために、単層培養での細胞にとって、酸素は制限のある基質である。静置培養での最大細胞密度の決定因子は利用可能な表面である。その表面が細胞の単層で一旦被われると、細胞増殖は停止してしまうことになる。その結果、細胞密度は培地1ml当たり約1×106に留まる。懸濁培養では酸素の制限が存在せず、他の基質の利用可能性が細胞密度にとっての制限因子となる。この制限は酸素の制限よりもより高い細胞密度で起こり、したがって、細胞密度が静置培養におけるよりもより高くなる。酸素の制限は、溶解酸素濃度をモニタする酸素電極を使用することにより防止される。酸素がある値以下に落ちると、酸素は自動的に、培養槽の上部空間を経て散布によりまたは添加により、懸濁液に添加される。懸濁液を適度に攪拌することは均一な培養を保証し、そこでは基質の勾配が形成されず、また、細胞も過度に高い剪断力にさらされることがない。さらに、攪拌は感染細胞のウイルスを非感染細胞に効率的に移動させることになり、細胞のウイルス感染効率を高めることとなる。最初、細胞はわずかに約0.1〜0.3%が感染しているだけなので、残り99.7〜99.9%の細胞は増殖し、複製することが可能となる。感染細胞内で産生されるウイルス粒子は、感染後12〜20時間で放出される(ディーおよびシュラ(Dee,K.U. and Shuler, M.L.)1997、Biotechnology Progress, 13、14〜24)。細胞当たりに放出されるウイルス粒子数は100〜200であり、それが新たなサイクルにおいてそれまで非感染であった細胞を感染させる。ウイルス粒子が十分に産生され、培養物内に存在するすべての細胞を感染させるまでには、数回のサイクルを要する。目指すところは、培地が使い尽くされ、すべての代謝工程が停止するようになるまでに、最終の感染経路のタンパク質産生を完遂することである。また、最適以下の細胞密度で完全な感染を達成させることも望ましくない。というのは、培地が効率的に使用されなければ非相同タンパク質の容量収率も低くなるからである。ウイルス感染の全工程は、ポリヘドリン遺伝子がなお存在するという事実により、目視によりチェックすることができる。ウイルスの感染は細胞核に蓄積する密集したタンパク質粒子として観察することができる。細胞に対する剪断損傷を防止するためには、プルロニックF−68を最終濃度が0.2%となるように培地に添加する。さらに、要すれば、消泡剤Aを加えて、過度の泡立ちを防止するが、これもまた細胞死を招く。50Lの培養に添加される消泡剤Aの総量は平均で20mlである。細胞を感染させる最適ウイルス密度は計算することができる。この密度は、細胞の増殖率、新たなウイルス粒子が放出されるまでの感染後の時間、および感染細胞当たりに産生される新たなウイルス粒子の数に依存する。本発明により提供される方法の例証となるのはペスチウイルスE2タンパク質の産生である。このタンパク質は活性の高い抗原として知られていたが、ワクチン用または診断試験用のE2(フラグメント)の生産は常に不成功である。PD50は2μgと低い(特に、免疫原E2フラグメントを用いる場合)にもかかわらず、問題は、多数動物群の防御的予防接種を1頭当たり単回の予防接種で可能とするために、商業的に魅力のある方法で、十分な抗原性部分をもつワクチンの投与に十分な量を如何に集めるかである。この問題はとりわけ、CSFVの予防接種に関連性がある。投与に際しては、CSFVの予防接種は、一般に、CSFVが突然発生した地域での集団活動で実施される。この場合には、膨大な数の動物を、比較的短期間で、迅速に予防接種することを求められる。かかる集団活動においては、適切な防御レベル(野生型ウイルスの感染に対し防御されるブタの数)を迅速に達成することが差し迫って重要である。防御を達成するために、最初の予防接種後二回目の予防接種まて数週間も待つことは、この疾患の制御を著しく妨害し、遅らせる。組換えにより発現したE2タンパク質を産生させる様々な方法の間には、バキュロウイルスで発現したE2(フラグメント)を比較するだけでも、違いが存在する。初期に報告されたE2タンパク質産生培養において、E2タンパク質(フラグメント)の収量は20〜90μg/mlの間で変動する(ハルスト(Hulst)ら、J. Vir. 5435〜5442、1993;ハルストおよびムーアマン(Hulst and Moormann)、Cytotechnology、20:271〜279、1996)が、単回投与予防接種に必要な関連するE2抗原性部分を得るためには、モノクローナル抗体による免疫アフィニティ精製をさらに必要とする。他の方法(EP0389034に記載されたE2のフラグメントを使用する)は、この方法では昆虫細胞の上清から取得したE2をさらに免疫アフィニティ精製することなしに使用するが、それがE2ベースのワクチンであって、満足すべき(防御)免疫応答が得られるまでに、2回注射する。これらの問題は、とりわけ、該ワクチンを調製する出発原料、たとえば、細胞培養上清における関連する抗原性物質、この場合にはE2タンパク質(フラグメント)、の低濃度に関係する。理論的には、技術上既知の精製・濃縮法により抗原性部分をさらに蓄積することができるが、しかし、この方法では商業的に魅力のあるワクチン生産法に導き得ず、しかも1用量当たり高コストの原因ともなる。本発明方法により提供される方法を用いる生産工程では、我々の50L容量醗酵槽において、定常的に約200〜300μg/mlのCSFV E2タンパク質フラグメントを生成するに至り、理論的に、1回の培養当たり100,000頭の動物を予防接種するのに十分な量である。細胞はおおよそ107TCID50/mlを含むウイルス菌量1〜10mlにより、0.5〜1.5×106細胞/mlの密度で感染される。この培養では、好ましくは、細胞の >50〜80%がCPEを示したときに収得する。これは感染後約100〜150時間である。初期E2タンパク質生産培養においては、細胞はMOI> 1で(同時に)免疫されるが、タンパク質収量は本発明により提供されるものより3倍低くなる。本発明により提供される方法では、50L容量の醗酵槽は、5Lの醗酵槽内で増殖した細胞懸濁液5Lにより、または10Lの醗酵槽内で増殖した懸濁液のすべて10Lにより接種する。初期細胞密度は約3×105細胞/mlである。細胞は、この懸濁液にウイルスを加える前に、計算した細胞密度まで増殖させる。下流のプロセシングは精密濾過による細胞と多面体の除去とともに始まる。中空糸精密濾過装置を醗酵槽に結合し、原料を0.22μmの孔径をもつ濾過モジュールにポンプ輸送する。保持流動体は醗酵槽を再循環させ、浸透する流動体は100Lの容器に収集する。濾過が完了したとき、現段階で無細胞ではあるが、感染性のバキュロウイルスをなお含む抗原溶液を100L容器中で不活化する。一般に、臭化2−ブロモエチル−アンモニウム(BEA)を、最終濃度が8〜12mMとなるように該懸濁液に加える。2M−NaOHを添加して、pHを約5.8から8.2〜8.7に上昇させる。このpH移動はBEAをBEI(臭化2−ブロモエチル−インミン)に変換する。これはDNA不活化剤である。pHを注意深くモニタして、6〜10に調整し、温度は34〜39℃に維持する。不活化の6時間後に、抗原溶液を第2不活化容器に移す。これにより、容器中のすべての物質がBEIと接触し、その結果、不活化されるであろうことが保証される。ウイルスを含むが、BEIを含まない液体のしずくが第1容器には存在し、第2容器には存在しないはずである。104〜107pfu/mlの濃度で存在するバキュロウイルスは、<10-7pfu/mlの値(10m3当たり<1個のウイルス粒子)まで分解される。これは、宿主動物を感染させ得るウイルスにもとづくウイルスに対して用いたのと同じ水準である(FMD同様)。ブタはバキュロウイルスに対して宿主ではない。不活化した抗原の大部分は、水−油−水エマルジョンに剤形化するまで≦−20℃に保存する。32μgのE2を含む単回投与物(投与容量2ml)は、古典的なブタコレラに対し、予防接種後2週間から少なくとも6ヵ月まで防御効果( >PD95)を与えるに十分である。生産工程は、該工程のスケールアップが簡単で、250Lまたはそれより大きい醗酵槽の使用が可能となるような方法で設計する。静置培養生産のスケールアップもまた簡単である(さらに多くの組織培養フラスコを使用するだけである)。しかし、50L醗酵槽にて定常的に達成される総タンパク質生産には、約7000個のT175組織培養フラスコを必要とする。細胞の増殖と感染は、酸素およびpH−電極が存在するので、醗酵槽中でより良好にモニタし、調整することができる。組織培養フラスコでは、フラスコ同士の変動が多分存在するが、それを量的に決めることはできない。不活化は静置培養生産にて実施したように、容器中でさらにより正確にモニタし、調整することができる。BEVSで発現される他のタンパク質の容量産生レベルは(我々の研究所にて)、もし細胞が本発明により提供される方法で増殖するならば、著しく改善される。たとえば、ウシFSHの収率は、10Lの醗酵槽を用いたとき、3〜4のファクタで増加する。細胞は2種の組換えバキュロウイルスそれぞれについて0.003のMOIで、1.1×106細胞/mlの細胞密度で同時感染させた。BVDVの異なるE2の、およびErnsタンパク質またはBVDVおよび/またはCSFVの収率は、振盪フラスコ中の懸濁培養において3倍に増加した。この方法は他の組換えタンパク質にも適用可能である。
【0106】
【表7】
【0107】
【表8】
【図面の簡単な説明】
【0108】
【図1】長期培養実験におけるE2の安定性のデータを示す図である。
【図2】培養実験におけるE2の安定性のグラフである。
【図2−1】培養実験におけるE2の安定性のグラフである。
【図2−2】培養実験におけるE2の安定性のグラフである。
【図3】MO1実験のデータを示す図である。
【図3−1】MO1実験のデータを示す図である。
【図3−2】MO1実験のデータを示す図である。
【図4】MO1実験のグラフである。
【図4−1】MO1実験のグラフである。
【図4−2】MO1実験のグラフである。
【図4−3】MO1実験のグラフである。
【図4−4】MO1実験のグラフである。
【図4−5】MO1実験のグラフである。
【図4−6】MO1実験のグラフである。
【発明の詳細な説明】
【0001】
本発明は、バキュロウイルスベクター発現系においてタンパク質またはポリぺプチドの発現を増加するための方法に関する。組換えDNA技術が開発されたとき、遺伝子改変された細菌を使用する大規模のタンパク質生成に関して期待が高かった。しかし、大多数の商業的に魅力的なタンパク質は、それらが生物学的に活性なタンパク質になり得る前に翻訳後修飾を必ず受ける。したがって、動物細胞が、組換えタンパク質を生成するために現在より頻繁に使用される。
【0002】
動物細胞の中で、昆虫細胞は組換えタンパク質の生成のために益々重要である。昆虫細胞を使用する簡便なおよび用途の広いバキュロウイルスベクター系が開発された。昆虫細胞の生理学に対する情報はかなり少ないが、バキュロウイルス組換え技術を介して生成されるワクチンは一般に十分に受容される。1つの例は、臨床試行において可能なAIDSワクチンとしてのヒト免疫不全ウイルスI型の、バキュロウイルスにより発現されるgp160エンベロープタンパク質の使用である。
【0003】
現在まで、昆虫細胞におけるバキュロウイルスにより発現されるタンパク質の大規模の生成は、約10リットルまでのバイオリアクターに制限される。規模を大きくするために、懸濁培養は最も良好な可能性を提供する。大規模生成において(Tramperら、Rec. Adv. Biotech.、1992、263〜284;PowerおよびNielsen、Cytotechnology 20:209〜219、1996を参照のこと)、特別な重要視が細胞増殖およびウイルス生成を影響する因子に与えられるべきである。このような因子の変化は、組換えタンパク質の生成の最終的なレベルを大きく影響する。
【0004】
バキュロウイルスは、閉塞体中に一般に閉塞される桿状ウイルス粒子により特徴付けられる。
【0005】
バキュロウイルスは、閉塞体中に一般に閉塞される桿状ウイルス粒子により特徴づけられる(多角体としてまた呼ばれる)。バキュロウイルス科は、閉塞ウイルス:核多角体病ウイルス(NPV)および顆粒症ウイルス(GV)の2つの属を含有する真正バキュロウイルス、ならびに非閉塞ウイルスを含有するヌードバキュロウイルス亜科の2つの亜科に分けられる。Autographa californica(Ac)NPVの細胞および分子生物学がより詳細に研究された。
【0006】
多くのタンパク質が、そのタンパク質をコードする組換えバキュロウイルスで感染された昆虫細胞において発現されてきた。コードすることは、このようなウイルスが非相同タンパク質をコードする核酸配列を提供され、そしてしばしば、プロモーターのような調節核酸配列をさらに提供されることを意味する。最も頻繁には、ポリヘドリンプロモーターが外来遺伝子を発現するために使用されるが、p10プロモーターは等しく十分に適切であり、そして同様に使用される。
【0007】
いくつかの細胞株が、組換えバキュロウイルスでの感染に利用可能である。細胞株SF−21は、アワトウヨウの幼虫(Spodoptera frugiperda)の卵巣組識に由来した。アメリカンタイプカルチャーコレクション(CRL 1711)から利用可能であるクローン単離物のSF−9は、組換えウイルスのインビトロ生成のためのおおむね標準的な細胞株であり、そして組換えウイルスを生成することにおいて優れていると言われる。他の細胞株は、たとえば、Hi−Five細胞株ならびにイラクサキンウワバ(Trichoplusia ni)から得られるTn−368およびTn−368A細胞株である。昆虫細胞が増殖する最も広範に使用される培地としては、TNM−FH、BML−TC/10、およびIPL-41が挙げられる。これらの培地は、通常、哺乳動物血清、特に胎児ウシ血清のようなおおむね規定された成分で補充される。血清置換はまた、昆虫細胞培養に適用されており、そしてEx−Cell 400(商標)およびSf900のような血清非含有培地が市販される。
【0008】
昆虫細胞は一般に固体支持体上でおよび懸濁液中で増殖するが、固体支持体上で増殖される場合にウイルスのより高い収量を与えることが報告される。感染は、細胞が指数関数増殖期において感染される場合に最も効果的であるが、細胞当たりの生成される多角体およびウイルスの量は、細胞周期の異なる段階で感染された細胞間で有意に変化しない。細胞密度は、ウイルス生成に対して大きな影響を有する。昆虫細胞は接触阻害の形態を示し得、より高い細胞密度で減少されたウイルス生成を生じる。
【0009】
初回感染多重度(m.o.i.)は、細胞当たりの感染性ウイルスの数であり、一般に感染の終了時での感染された細胞の画分および細胞当たりの多角体の数の両方に影響する。ウイルス生成についての至適なm.o.i.は一般に、約20〜30であると考えられる。β-ガラクトシダーゼを発現する組換えバキュロウイルスでの研究(LicariおよびBailey、Biotech.Bioeng.、37:238〜246、1991)において、Sf−9細胞は、0と100との間のm.o.i.値で感染された。β-ガラクトシダーゼの収量は増加され、そして細胞密度は、m.o.i.の増加とともに減少した。m.o.i.を増加することおよび減少することは、感染された細胞当たりの組換えタンパク質の最大に達成可能な収量に対して制限された効果のみを有すると一般に考えられる。しかし、低いm.o.i.を選択することは、感染に必要なウイルスストックの減少を許容し、そしてバキュロウイルスの欠陥妨害粒子の生成の危険性を最小にする。昆虫細胞のバッチ培養が高いm.o.i.(>5)で感染される場合、次の感染プロセスは本質的に同時進行される(すなわち、全ての細胞は、同時に感染周期を経る)。細胞がバッチ培養においてm.o.i.1〜5で感染される場合、培養物はもはや同時進行されない。全ての細胞が感染されそして所望のタンパク質の生成が終わるまで、培養物は、最初は非感染細胞と個々の感染周期における異なる時点の細胞とからなる。一般的にはこのような培養物において生成レベルははるかに低い。培養物の挙動は、それぞれ生成の異なる相にある個々の細胞の組み合わせの挙動であり;至適な状態に及ばない生成レベルを説明する。連続的な培養において、非感染細胞が連続的に添加され、そして培養物は明らかに非同時進行的に感染される。
【0010】
数学的モデルの設計を介して、低いm.o.i.で細胞を感染する場合または連続的な培養系中でウイルスを増殖する場合に観察される挙動のような複雑な挙動を予測することが可能であると考えられる。同じ現象についての純粋に経験的な分析は、不可能でないにしても非常に困難であると考えられる。現在、3つのモデル、Licari & Baileyモデル、de Gooijerモデル、およびPower & Nielsenモデルが知られる。これらは、それらの複雑性およびそれらを詳しく開発した努力にもかかわらず、全て第1世代のモデルであり、ポリヘドリンプロモーターの制御下で発現されるモデル組換えタンパク質(β-ガラクトシダーゼ)を発現するバキュロウィルスの挙動について推論する。これらは、大部分、DNAおよびRNA集積ならびに感染周期を無視して、ブラックボックス系としてみる場合、バキュロウイルスと昆虫細胞との間の相互作用についての本発明者らの定量的な理解をまとめる。結合および最初の感染プロセスはなお、定量的に十分には理解されておらず、この分野におけるさらなる研究が非常に必要とされる。
【0011】
バキュロウイルス発現系は、組換えタンパク質の生成のための強力な道具を提供する。いくつかの細胞培養配置が、大規模生成のために使用され得る。しかし、これらの系は、それらの可能性の十分な利点を採用するためにさらなる至適化を必要とする。商業的な適用について、大規模および低コストの生成が極めて重要である。大規模細胞培養において報告される多角体生成系は、価格競合製品についての商業的要求を満たすために劇的に改善されるべきである。
【0012】
本発明は、所望のタンパク質の増加されたまたは改善された収量を許容する、バキュロウイルス発現系を使用する大規模で組換えタンパク質を生成するための方法を提供する。本発明は、昆虫細胞培養物において、組換えタンパク質を生成するおよび組換えタンパク質の収量を改善するための方法を提供し、当該タンパク質をコードする組換えバキュロウイルスを選択する工程、培養容器において増殖培地中で昆虫細胞を増殖する工程、および0.01未満の感染多重度で細胞を感染する工程を包含する。
【0013】
好ましい実施態様において、本発明は昆虫細胞培養物において生成される組換えタンパク質の収量を増加するための方法を提供し、当該タンパク質をコードする組換えバキュロウイルスを選択する工程、培養容器において増殖培地中で昆虫細胞を増殖する工程、および0.01未満の感染多重度を用いて少なくとも1つのバキュロウイルスの接種物で細胞を感染する工程を包含する。漸増する収量は、いくつかの研究グループの主題である。たとえば、Chenら(Drug metabolism and Disposition:24 399〜405頁、1997)は、同時感染アプローチのためにMOIを至適化する可能性を研究し、これによって2つの異なるバキュロウイルス発現タンパク質が昆虫細胞培養物において生成された。本明細書中に記載される結果と反対に、彼らは、彼らの2つのタンパク質について約0.015〜0.03の最も良好なMOIを見出した。MOIを0.01未満に減少することにより、Chenらの同時感染系の収量は減少した。Radfordら(Cytotechnology 24、73〜81、1997)は、同時感染系を研究することによって妨げられず、1よりも大きいMOIが常に最終的なプロセス収量を最大にするために使用されるべきであることを明らかに示し、本発明の知見に反対することを再度教示する。彼らは、低いMOIを使用して細胞当たりより大きな量のタンパク質およびウイルスを生成することは不可能であることを述べ、そしてその代わりに感染時間(TOI)を調節することを示唆する。
【0014】
Nguyenら(J.Biotech.31、205〜217、1993)のように他は、MOIの変化における解決を見出さないが、流加培養法を適用することによって収量を増加することにねらいを向けるか、またはウイルスが増殖する温度を変化し(Wuら、Cytotechnology、9、141〜147、1992;Kingら、Biotechnol. Bioeng、38、1091〜1099、1991)、そして0.01未満のMOI下でのウイルスの増殖を回避する。
【0015】
これらの初期の結果は、本明細書中の記載において提供される結果とは明らかに異なり(たとえば図1を参照のこと)、本明細書中の記載において培養物は0.01未満(たとえば、0.003、0.001、またはさらに0.0001)のMOIで感染され、0.01または0.1のMOIで感染される培養物よりも高い収量を達成する。
【0016】
本発明の好ましい実施態様は、単層培養において増殖されない昆虫細胞培養物において組換えタンパク質を生成するおよび組換えタンパク質の収量を改善するための方法を提供する。小量でバキュロウイルス発現ベクター系においてタンパク質を生成するための従来の実験室の方法は、培養フラスコ中の昆虫細胞の単層培養物を使用する。これらの静置培養物は、同時進行性の感染を確実にするために、通常、高いMOIで感染される。単層がコンフルエントになる前に全ての細胞が感染されることが極めて重要である。なぜなら、接触阻害が細胞中の代謝変化に導き、これが最終産物の収量を影響し得るからである。単層培養物中の細胞密度を正確に確立することは困難であるので、MOI実験を行うことは不可能である。培養物を感染するために低いMOI(0.01未満)を使用することはさらにより役に立たない。感染時の細胞密度の過剰評価および過小評価の両方は、有意に至適な状態に及ばないタンパク質の収量を導く。日常的に、高いMOIを使用し、これは全細胞集団の同時進行性の感染を保証する。このことは、至適な収量を達成するために、大量のウイルスストックが単層培養物を感染するために必要とされることを意味する。
【0017】
単層培養物を使用するタンパク質生成の規模拡大は、より多くの組織培養フラスコを使用することを単に意味する。単層培養物を使用する大量のタンパク質の生成は、非常に労働集中的である。さらに、溶解された酸素濃度およびpHのような重要な培養パラメーターを調節および/またはモニタすることは可能でない。
【0018】
本発明は今や、全ての昆虫細胞培養物に適切な方法を提供する。本発明は、低いMOIが単層培養物以外の昆虫培養物における収量を至適化するために有益であるという洞察を提供する。
【0019】
異なるタイプの培養容器が、単層培養以外において昆虫細胞を培養するために使用され得る。全ての発酵器設計の目的は、十分な通気および高い細胞密度を達成するが、可能な限り低い剪断力を維持することである(Tramperら、Recent advantages in biotechnology(Vardar-SukanおよびSukan編 1992)。文献において記載される大多数の場合において、気体噴霧器を備えた攪拌タンクバイオリアクターが使用される。このタイプの容器において、均一性が回転翼を使用することによって達成される。このタイプの発酵器は、異なる方法において操作され得る。先ず第1に、バッチ法である。これは、最も簡単なおよび単純な方法である。細胞が培養され、ウイルスが添加され、そして産物が感染の終了時に採集される。より複雑な方法は、流加培養法である。栄養素の濃縮混合物が培養容器に添加され、より高い細胞密度およびより高い容量の産物収量が達成される(Nguyenら、1993 Journal of Biotechnology 第31巻、205〜217頁)。これは制限される栄養素がなんであるのか常には明らかでないので、より複雑化される。1つの栄養素制限をこの基質を添加することによって取り消すことにより、別の基質制限が直接的に導かれ得、したがって必ずしもより高い産物収量が導かれないかもしれない。
【0020】
混合する異なる方法が、空中補給バイオリアクターにおいて使用される(Wuら、1992 Cytotechnology 第9巻 141〜147頁)。このタイプの容器は2つのシリンダからなる。最も小さな直径を備えるシリンダ(ドラフト管)は、より大きな直径を備えるシリンダ(降下管)の内側に配置される。中央のシリンダにおいて空気および別の気体が散布され、上昇流を作製する。発酵器の上部で、気体は液体を残し、そして液体は中央のシリンダの外側を下がる。この方法において、通気および均一性の両方が、ドラフト管および降下管における密度の差異に起因して気体のみの噴霧を使用することによって達成される。この方法は、ずり応力を減少し得る。しかし、連続的な通気速度が、正確な混合を達成するために必要とされる。
【0021】
発酵器中の生存細胞密度を増加するための別の方法は、スピンフィルタまたは別の細胞保持デバイスを含むことによる。これは灌流と呼ばれる。これは、発酵器中に細胞を保持しながら、発酵器から不用な培地を除去し新鮮な培地を添加することを許容する。この方法は、多くの特別の装置およびより困難な発酵器操作を生じる(Caronら、1994 Biotechnology and Bioengineering 第43巻、881〜891頁)。さらに、多孔質床およびゲルマトリクス中の固定化のような方法が報告される。このタイプの方法は、マトリクス上のまたは内側の細胞の固定化に依存し、細胞培養物を希釈することなく不用な培地の除去および新鮮な培地の添加を可能にさせる。細胞密度、接触阻害、および単層培養の他の関連の問題が処理可能である場合、本発明の方法は、上述の種々の培養系において適用され得るのみでなく、単層培養においてもまた適用され得る。
【0022】
たとえば、本発明の好ましい実施態様は、昆虫細胞培養物において組換えタンパク質を生成するおよび組換えタンパク質の収量を改善するための方法が提供され、当該タンパク質をコードする組換えバキュロウイルスを選択する工程、少なくとも2リットルを含むのに十分な容量を備える培養容器において増殖培地中で昆虫細胞を増殖する工程、および当該バキュロウイルス/細胞の0.01未満のPFUのm.o.i.で少なくとも1つのバキュロウイルスの接種物で昆虫細胞を感染する工程を包含する。本発明は、たとえば1〜5のm.o.i.よりもかなり低い感染多重度が使用され、非同時進行的に感染される培養物を導く方法を提供する。本発明により提供されるようなm.o.i.を使用してバキュロウイルスで昆虫培養物を感染することによって、至適な平衡が、細胞の複製の速度とウイルスの複製の速度との間で達成され、それによって所望のタンパク質の至適な発現を許容する。本発明によって提供される方法は、同様にm.o.i.を調整することによってより高いおよびより低い細胞密度に容易に調整され得、ここでは本発明によって提供される効率および密度にしたがって、複製の種々の相における細胞を感染するために利用可能であるウイルス粒子の相対比が残存する。本発明の方法の好ましい実施態様は、少なくとも10、より好ましくは少なくとも20、より好ましくは少なくとも50または250リットルの増殖培地を含むのに十分な容量を備える培養容器において細胞を増殖し、それによって非相同タンパク質を発現するバキュロウイルス培養物の規模拡大を許容する工程を包含する。たとえば、存在する増殖培地の容量について必要とされるよりも大きな容量を備える培養培地を使用し得る。たとえば、20〜70リットルの細胞培養物を培養するために100Lの培養容器を使用し得る。本発明の方法の好ましい実施態様は、1×105〜5×106細胞/ml、より好ましくは5×105〜1.5×106細胞/mlの細胞密度で細胞を感染し、それによって容易に操作可能な範囲内にウイルス接種の実際の容量を維持する工程を包含する。本発明の方法のなお別の実施態様は、0.005未満(たとえば、0.003、0.001、0.0005、または0.00025)のm.o.i.で細胞を感染し、それによって接種物をできるだけ小さく維持する工程を包含する。本発明の方法の好ましい実施態様はp10プロモーターの制御下で所望のタンパク質を発現する組換えバキュロウイルスを選択する工程を包含する。P10プロモーターは、細胞溶解において役割を果たし、p10タンパク質の不在は、感染された細胞をインタクトなままにさせ、そして感染された細胞からの多角体の放出を妨ぎ、それによって再感染速度を減少するが、感染力自身は減少しない。ウイルス感染の全プロセスは、ポリヘドリン遺伝子、したがって多角体がなお存在するという事実に起因して、現在視覚的にチェックされ得る。ウイルス感染は、細胞核中に集積する高密度のタンパク質粒子として観察され得る。本発明の方法の別の実施態様は、バッチ培養系において昆虫細胞を増殖し、それによって未だ未感染の細胞の感染および複製を損ない得る欠陥妨害バキュロウイルスウイルス粒子の集積を最小化する工程を包含する。本発明により提供される別の方法は、懸濁液中で、好ましくは攪拌され得る(中程度に)発酵器のような培養容器中で昆虫細胞を増殖する工程を包含する。(攪拌された)懸濁培養物の使用は、特にウイルス増殖を視覚的にチェックするために所望のタンパク質がp10プロモーターの制御下にある組換えバキュロウイルスの使用と組み合わされる場合(しかし、CPEを観察するようなチェックする他の方法(たとえば、ポリヘドリンプロモーターを使用する場合において)がまた利用可能である)、培養のより良好な制御を許容する。
【0023】
懸濁液の中程度の攪拌は、基質勾配が作られないおよび細胞が高すぎる剪断力を受けない均一な培養を保証する。さらに、攪拌は、感染された細胞から非感染細胞へのウイルスの効率的な移動を生じ、細胞のウイルス感染のより高い効率を与える。最初は約0.1〜0.3%の細胞のみが感染されるので、残りの99.7〜99.9%の細胞は成長および増殖を許容される。
【0024】
本発明は、種々の起源の組換えタンパク質を発現および生成するための方法を提供する。本発明によって提供される例は、ペスチウイルス由来のタンパク質を、増殖培地中に、少なくとも100、120、または150μg/ml、より好ましくは少なくとも200、またはさらに300μg/mlの収穫の当該タンパク質の濃度に生成することである。所望のタンパク質はまた、獣医学的および医学的使用のために、たとえば、ワクチン中におよび診断試験において組込まれた抗原性物質を調製するために使用され得る。本発明の方法によって生成される所望のタンパク質は、たとえば、ワクチンを調製するために使用され得る。
【0025】
本発明によって提供される例は、ペスチウイルスE2タンパク質またはそのフラグメントであり、これはたとえば、ペスチウイルス感染(たとえば、ブタにおける伝統的なブタ熱)に対するワクチンを調製するために使用され得る。好ましい実施態様において、本発明は、組換えペスチウイルスのE2タンパク質もしくはErnsタンパク質、またはそのフラグメントを含有し、イムノアフィニティ精製されておらずおよび好ましくは1用量での単回のワクチン接種後にPD95レベルでペスチウイルスに対して防御を付与するという点で特徴付けられるワクチンを提供する。これは、CSFVワクチン接種に特に関連する。適用される場合、CSFVワクチン接種は一般に、CSFVの発生が生じた領域において大規模なキャンペーンの間に行われる。これは、比較的短い期間における多数の動物の迅速なワクチン接種を必要とする。このような大規模なキャンペーンにおいて、十分な防御レベル(野生型ウイルス感染に対して防御されるブタの数)が迅速に達成されることが差し迫って重要である。防御を達成するために第2のワクチン接種について、第1のワクチン接種後に数週間待つことは、疾患の制御を大きく妨げおよび遅延する。組換え的に発現されるE2タンパク質を生成するための種々の方法の間には、バキュロウイルスにおいて発現されるE2(フラグメント)と比較する場合であっても、差異が存在する。初期に報告されたE2タンパク質生成培養において、E2タンパク質(フラグメント)の収量は、20〜90μg/mlの間で変化し(Hulstら、J.Virol. 5435〜5442、1993;HulstおよびMoormann、Cytotechnology 20:271〜279、1996)、単回注射のワクチン接種のために必要なおよび関連のE2抗原質量を得るためにモノクローナル抗体でのイムノアフィニティ精製をさらに必要とする。別の方法(EP 0389034において記載されるE2のフラグメントを使用する)は、さらなるイムノアフィニティ精製を伴わずに昆虫細胞の上清から採集されたE2を使用し、満足する(防御的な)免疫応答が得られる前に2回注射されるE2ベースのワクチンを得る。E2抗原を含有するワクチン(ProcilisRPesti)が現在登録されているが、所望の免疫応答を提供するために4週間の間隔で少なくとも2回のワクチン接種を行うので、このワクチンは2回適用されなくてはならず、それによって、所望の免疫応答を提供するために4週間の間隔で少なくとも2回のワクチン接種を行うので、このワクチンでの伝統的なブタ熱感染に対するワクチン接種の有用性は、妨げられる。
【0026】
これらの問題(これらは、本発明により解決される)は、特に、ワクチンが調製される開始材料(たとえば、細胞培養上清)中の関連の抗原性物質(この場合においてE2タンパク質(フラグメント))の低い濃度に関連する。理論的に、抗原質量をさらに集積し得え、精製および濃縮法は当該分野において知られるが、これは商業的に魅力的なワクチンの生成を導かず、用量当たり高いコストを引き起こす。別の例は、ペスチウイルスErnsタンパク質であり、これはまたワクチン中でおよび診断試験において、または他の(治療学的)物質中で使用され得る。
【0027】
たとえば、Ruggliら(Virus Genes 10:115〜126、1995)は、単層昆虫細胞培養物中でE2を発現するバキュロウイルスを、5〜10μg/106細胞以下の最大収量に増殖した。さらに、Moserら(Vet.Microbiol. 51:41〜53)は、5のMOIを使用して単層昆虫細胞培養物中でRuglliらのE2を増殖し、そしてELISAの目的のために未濃縮の形態において十分な抗原を生成し得ない。彼らの経験において、ニッケルキレートアフィニティクロマトグラフィによるタンパク質のさらなる精製が、ELISAの操作を単純にし、そしてELISAの質を改善するために前もって必要である。したがって調製されたE2タンパク質を用いるワクチン調製は意図されていない。
【0028】
ワクチンが研究の目的であった場合、ワクチン接種は、防御のある基準を達成するために免疫精製されたE2タンパク質を使用して、2回行われることが必要であったことが見出された。たとえば、Hulstら(J.Virol. 67:5435〜5442、1993)、WO 95/35380、およびvan Rijnら(J. Gen. Virol. 77:2737〜2745、1996)において、E2は単層中で生成され、そしてイムノアフィニティ精製されて、防御ワクチンを達成した。
【0029】
本発明によって提供される例は、組換えCSFV E2タンパク質(フラグメント)を含有するワクチンであり、タンパク質は今や十分に多くの量が生成され得、動物が1用量で単回のワクチン接種を受けた2〜3週間以内に伝統的なブタ熱ウイルス感染に対する防御を付与する(95%の防御用量レベル(PD95)で)ワクチン中に取り込まれる前にイムノアフィニティ精製される必要がもはやない。本発明によって提供される方法は、組換えペスチウイルスE2タンパク質もしくはErnsタンパク質またはそのフラグメントを含有するワクチンを提供し、ワクチンは、1用量での単回のワクチン接種後にペスチウイルス感染に対して防御を付与し、一方タンパク質(フラグメント)は、イムノアフィニティによって精製されていない。本発明はまた、本発明によって提供されるタンパク質を含有し、さらにアジュバントを含有するワクチンを提供する。適切なアジュバント(たとえば、フロインドアジュバント、または水酸化アルミニウム、または乳濁液(たとえば、油中水乳濁液、二重油中水乳濁液、または水中油乳濁液))は当業者に公知である。所望のタンパク質はまた、他の物質(たとえば、獣医学的または医学的使用)を調製するために使用され得る。本発明によって提供されるなお別の例は、卵胞刺激ホルモン(FSH、αユニットおよび/またはβユニット、ならびにその複合体およびフラグメント)のようなホルモン様物質であり、これはたとえば、培養物中でαユニットを発現する1つのバキュロウイルスでおよび/またはβユニットを発現する別のバキュロウイルスで、昆虫細胞培養物を感染することによって生成され得る。本発明の方法はまた、生物殺虫剤としての(組換え)バキュロウイルスの大規模および低コストの生成のために使用され得る。好ましい実施態様は、生物殺虫剤の生成において外来遺伝子の発現のためにp10プロモーターを利用する組換えウイルスの使用である。なぜなら、昆虫は一般に多角体遺伝子を欠陥するバキュロウイルスによりあまり十分には感染されないからである。本発明の方法を用いて、他の組換えタンパク質を発現する他の組換えバキュロウイルスを培養すること、ならびに殺虫剤の、医学的、治療学的、および/または抗原性の物質または産物中に組込むためのこのようなウイルスタンパク質の生成および/または使用は、当業者の範囲内である。本発明はさらに、実施例において説明されるが、それらに制限されない。
【0030】
実施例
フラビウイルス科のペスチウイルス属は、従来、伝統的なブタ熱ウイルス(CSFV)、ボーダ病ウイルス(BDV)、およびウシウイルス性下痢ウイルス(BVDV)からなる。いくつかのBVDVおよびCSFV株のゲノムは配列決定されている(Renardら、1987 EP特許出願第0208672号;Collettら、1988、Virology 165、191〜199;DengおよびBrock、1992、Virology 1991、865〜679;Meyersら、1989、Virology 171、555〜567;Moormannら、1990、Virology 177、184〜188)。BDVについて、未だ不完全なゲノムヌクレオチド配列のみが利用可能である。ペスチウイルスゲノムは、1つの大きなオープンリーディングフレームを含有する約12.5キロベースのポジティブ鎖RNA分子である。オープンリーディングフレームは、約4,000アミノ酸の推定のポリタンパク質に翻訳され、これはウイルスおよび細胞にコードされるプロテアーゼによりプロセスされる。オープンリーディングフレームは、2つの高度に保存された小さな非翻訳領域により隣接され、これらはおそらくゲノムの複製に関与する。5'非コード領域はまた、翻訳の開始において役割を果たす。
【0031】
細胞およびウイルスのプロテアーゼによって、翻訳と同時におよび翻訳後にプロセスされるポリタンパク質は、ウイルスの構造タンパク質および非構造タンパク質の全てを含む(C.M. Rice:Fields Virology、第3版、1996 フラビウイルス科:ウイルスおよびそれらの複製:第30章:931〜959頁を参照のこと)。ウイルス構造タンパク質、特にエンベロープタンパク質のErns、E1、およびE2はポリタンパク質のN末端部分に位置される。非構造タンパク質、特にセリンプロテアーゼNS3ならびにRNAレプリカーゼ複合体NS5AおよびNA5Bは、ポリタンパク質のC末端部分に位置される。
【0032】
ペスチウイルスで感染される動物は、Erns、E2、およびNS3に対する抗体を発生する。しかし、E2に対して指向される抗体のみが強力にウイルスを中和化し、一方ErnsおよびNS3に対して指向される抗体は、ごく低いウイルス中和化能力を有するか、または全く有しない。この知見は、CSFサブユニットマーカーワクチンとしてのE2の適切性を評価することを開始するように本発明者らを促した。この設定において、Ernsおよび/またはNS3は、E2マーカーワクチンを付随する診断試験の開発のために使用され得た。
【0033】
今日までに、BDVおよびBVDVは、異なる種から単離されているが、CSFVはブタに制限されるようである。ペスチウイルスは構造的におよび抗原的に密接に関連する。エンベロープ糖タンパク質のE2は最も免疫原性であり、およびペスチウイルスの最も可変性のタンパク質である。本発明者らは、多くの異なるペスチウイルス株のE2遺伝子をクローン化し、シカおよびキリンからの株を含んだ。E2遺伝子を一過的に発現し、モノクローナル抗体で特徴づけし、配列決定し、そしてP.A. van Rijnら、1997、Virology、237:337〜348に比較した。これらのデータに基づいて、本発明者らは、ペスチウイルス属内に6つの主要な群を示し得た。4つの群は規定された遺伝子型に対応したが、2つの他の群は、ペスチウイルス属内の新規な遺伝子型であった。1つの群は、ブタから単離されたCSFV株を含む。第2の群は、ヒツジから単離されたBDV株Moredun、L83、およびX818、ならびにブタ由来のF株からなる。第3の群は、ヒツジ由来のBD78株、ブタ由来の5250株、およびウシ由来の178003株を含む。E2に基づいて、これらのウイルスは、ウシウイルス性下痢の急性の重篤な発生と関連するBVDV株(いわゆる、2型BVDV)に非常に類似する。第4の群は、ウシに優先的に由来するBVDV株からなる。このBVDV群は、2つの亜型または亜群のBVDV−1aおよび1bに分けられ得:BVDV−1aは、NADLおよびOregonのような米国に由来するウイルス、ならびに欧州に由来する150022および1138のような他のウイルスを含む。亜群BVDV−1bは、Osloss株およびいくつかのオランダ単離物を含む。第5および第6の「群」は、2つの新規な遺伝子型として提唱され得、それぞれ、Deer株およびGiraffe株を含む。
【0034】
獣医学的使用のためのマーカーワクチンの開発は、家畜のウイルス疾患の経済的に重要な制御および根絶のための新しい概念を導いた。マーカーワクチンの使用は、ワクチン接種された動物と野外ウイルスに感染された動物との間の血清学的な区別を許容し、それによってウイルスを制御して排除をできる。大部分のメンバにおいて、たとえば、アウエスキー病(AD)に対するEUワクチン接種の状況は、gEネガティブなワクチンのみで許容される。AD野外ウイルスで感染された動物は、gEに対する抗体を発生する。これらの抗体を検出するELISA試験が、感染された動物の検出のために(続いて感染された動物は集団から除去され得る)、および(長期間の)ワクチン接種キャンペーンの間、集団における野外ウイルス感染の状況をモニタするために使用される。最終的に、目的は、集団の野外ウイルスがない状態を達成することである。次いで、ワクチン接種は中止され、そしてこの状況を保護する血清学的な監視プログラムが行われる。したがって、マーカーワクチンでのワクチン接種は、感染された個々の動物のみのではなく感染された集団の撲滅にかかる根絶運動の費用を有意に減少し、および十分に大きな規模で、および十分に長期間、結果的に行われる場合であっても、ブタ集団の野外ウイルスがない状態を達成するためには、撲滅よりも早い方法であり得る。
【0035】
現在および今後、ADのような現在の風土病が根絶された場合、たとえば、集団から根絶された疾患および日常的なワクチン接種がない疾患の発生を制御するために、マーカーワクチンはなお使用される。このような場合において、動物集団が血清学的にネイティブである場合、非常に伝染性の疾患が爆発的に広がり得、次いで莫大な経済的な損失を引き起こし得る。
【0036】
バキュロウイルス系が非相同タンパク質の高レベルの発現を支持するという多くの例が示された。高レベルの発現は非常に有利である。なぜなら、デッドサブユニットワクチンは、一般に、大量の抗原が適用される場合に防御的な免疫応答を誘発し得るのみであるからである。本発明者らの最初のアプローチにおいて、2つの組換えバキュロウイルス(一方はTMRを伴ってE2を発現する(BacE2[+])および他方はTMRを伴わないでE2を発現する(BacE2[−]))が構築された(Hulstら、1993、J. Virol. 67:5435〜5442)。この刊行物および他の比較可能な刊行物において、E2は未だその古い名称のE1で表されることに注意のこと。TMRを伴わないE2は、細胞培養培地に分泌されるが、TMRを伴うE2は分泌されなかった。さらに、両方のE2は、E2上の4つの抗原性ドメインのそれぞれを提示するモノクローナル抗体と同一に反応する。このことは、細胞会合性E2および分泌性E2の抗原特性が同一であることを示唆し、そして分泌性E2が細胞会合性E2よりも非常に高いレベル(10倍以上の高さ)に生成されたので、ブタにおけるワクチン接種試行において分泌性の免疫アフィニティ精製されたE2を試験することが決定された。二重水-油アジュバント中に20μg E2/mlおよび100μg E2/mlを含有する2つのワクチン処方物を調製した。2つのSPFブタの4つの群のうち、2つの群を20μg E2で、および2つの群を100μg E2で筋肉内にワクチン接種した。28日後、20μg E2でワクチン接種された1つの群および100μg E2でワクチン接種された1つの群を、同じ用量のE2で再ワクチン接種した。42日目に、全ての動物を病原性CSFV株のBresciaで鼻腔内に攻撃した。適用されたワクチン用量にかかわらず、全てのワクチン接種されたブタは、異なるレベルにであったが28日目で中和化抗体をマウントした。攻撃の日にちの42日まで、抗体力価は、全ての動物において、それらが追加免疫されようがなかろうが高レベルに上昇したままであった。それゆえ、20μgの免疫アフィニティ精製されたE2の用量で一回のみワクチン接種された動物であっても、CSFに対して既に完全に防御されたということは驚くことではない(Hulstら、1993 J. Virol. 67:5435〜3442)。
【0037】
ペスチウイルスで感染された動物は、第2のウイルスエンベロープ糖タンパク質のErnsに対する抗体を必ず生成するので(Kwangら、1992、Vet.Microbiol. 32:281〜292)、これはE2と組み合わせて診断試験を開発するために最も適切なウイルスタンパク質の1つである。しかし、ErnsがCSFに対するブタの防御のために重要な抗原であり得たことを示唆する1つの報告がまたある(Konigら、1995、J. Virol. 69:6479〜6486)。これらの研究において、Ernsは生存ウイルスワクシニアベクターで発現された。注射部位あたり5×107PFUのワクシニア−Erns組換えウイルスで3つの異なる経路(皮内、静脈内、および腹腔内)により同時にワクチン接種された動物は、ワクチン接種の5週間後での致死用量のCSFV株Alfortでの攻撃でも生存し、CSFの任意の徴候を示さなかった。
【0038】
デッドサブユニットワクチンとしてのErnsの適切性を評価するために、バキュロウイルスにより発現されたBrescia株のErns(Hulstら、1994、Virology、200:558〜565)を、ブタにおいて試験した。6匹のSPFブタの群を、それぞれ、5.0および20μg Ernsで筋肉内に1回ワクチン接種した(Moormannら、1996、Proc. 14th Intern. Pig. Vet. Society Congress、25〜29頁、Bologna、Italy)。4匹のワクチン接種されなかったSPFブタの第3の群は、コントロールとして作用した。ワクチン接種の3週間後、全ての動物を、105 TCID50のBehring株で筋肉内に攻撃した。攻撃の5日後以内に、最も低い用量のErnsでワクチン接種された全ての動物は、CSFの重篤な徴候を発症した。攻撃の14日後以内に、2匹の動物が死亡し、3匹は瀕死の状態になったときに殺傷され、1匹は見かけ上、回復した。回復した1匹を含むこれらの動物のうちの5匹は、IFTにおいてポジティブであった。攻撃後、最も高い容量のErnsでワクチン接種された全ての動物は、数日間多かれ少なかれCSFの重篤な徴候および高熱を示したが、14日以内に、6匹のうちの4匹の動物が回復し、1匹の動物は死亡し、そして残りの1匹は瀕死の状態になったときに殺傷された。4匹の生存する動物のうちの1匹のみが、IFTにおいてポジティブであるようであった。対照的に、全てのコントロール動物が、攻撃後5日以内にCSFの重篤な徴候を示し、攻撃後14日以内に死亡し、IFTにおいてポジティブであった(Moormannら、1996、Proc. 14th Intern. Pig. Vet. Society Congress、25〜29頁、Bologna、Italy)。
【0039】
本発明者らは、バキュロウイルスにより発現されたErnsは、バキュロウイルスにより発現されたE2投与よりも非常に低い効率であるが、非相同CSFV攻撃に対してブタを防御し得ることを結論した。
【0040】
卵胞刺激ホルモン(FSH)は、下垂体(黄体形成ホルモン、LH;甲状腺刺激ホルモン、TSH)においてまたは胎盤(ヒト絨毛性ゴナドトロピン、hCG;妊馬血清ゴナドトロピン、PMSG)においてのいずれかで生成される糖タンパク質ホルモンのファミリに属する。種内で、これらのホルモンのそれぞれは共通のαサブユニットからなり、これはホルモン特異的なβサブユニットに非共有結合される。単独でまたはLHと組み合わせて投与された精製されたFSHは、ウシを含む多くの種において過剰排卵応答を誘導するために広範に使用される。
【0041】
ウシにおける問題は、ウシFSHがウシ下垂体から実質的な量において精製することが困難であることである。この理由のために、ヒツジのFSH(oFSH)、ブタのFSH(pFSH)、またはウマのFSH(eFSH、PMSG)起源のFSHがウシの過剰排卵処理のために一般に使用される。しかし、ウシにおいてウシ海綿状脳症(BSE)、およびおそらくヒトにおいてクロイツフェルト−ヤコブ病(CJD)の変形を引き起こし得るプリオン様タンパク質の潜在的な存在に起因して、脳組識に由来する材料の適用は危険がなくはない。さらに、胎盤由来の材料の使用は、生物学的半減期が非常に長いという不利益を有し、これは特異的な抗体の注射によるその中和化を必要とする。最後に、全ての現在使用されるFSH調製物は、過剰排卵の結果において観察される大きな変分を少なくとも一部担うと考えられるいくつかのLH活性を含む。
【0042】
これらの理由のために、ウシの過剰排卵処理は、昆虫細胞(バキュロウイルス発現系)のような非哺乳動物細胞において生成される組換えウシFSH(
rbFSH)を適用することにより、有益であり得るようである。
【0043】
材料と方法
1.SF21細胞の産生細胞培養物(PCS)の調製例
SF21作業用細胞種子(WCS)1.5mlを容れた凍結バイアル(細胞総数、4〜10×106細胞/ml)を20〜30℃の温度で融解する。融解後、バイアルの内容物を、無血清培地SF900II8.5mlを含む15ml容量ファルコンチューブに移し、懸濁させる。懸濁後、ファルコンチューブの内容物を100〜200×gで10分間遠沈し、細胞を沈殿させる。培地を廃棄し、ペレットを4〜6mlのSF900IIに懸濁する。懸濁した細胞を、10mlのSF900IIを容れた100ml容量の振盪フラスコに移す。フラスコを軌道旋回式シェーカの台上にのせ、40〜80rpm、26〜30℃で3〜7日間、細胞を培養する。細胞の増殖と細胞生存度は、工程途上サンプルを取出しモニタする。細胞密度が1.0〜6.5×106細胞/mlに達したとき、それぞれ100mlのSF900IIを容れた2個の500ml容量振盪フラスコに移す。これは10倍希釈の細胞に相当する。再度、フラスコを軌道旋回式シェーカの台上にのせ、40〜80rpm、26〜30℃で3〜7日間、細胞を培養する。細胞の増殖と細胞生存度は定常的に工程内コントロールによりする。細胞密度が1.0〜6.5×106細胞/mlに達したとき、二度目として、今回はそれぞれ100mlのSF900IIを容れた6〜11個の500ml容量振盪フラスコに移す。過剰の細胞材料は廃棄する。また、この場合、10倍の希釈を達成する。フラスコを軌道旋回式シェーカの台上にのせ、40〜80rpm、26〜30℃で3〜7日間、細胞を培養する。細胞の増殖と細胞生存度は定常的に工程内コントロールによりモニタする。細胞密度が1.0〜6.5×106細胞/mlに達したとき、該細胞を含む懸濁液500mlまたは1000mlを、それぞれ約4.5リットルまたは9リットルのSF900IIを容れた5リットルまたは10リットル醗酵槽に移す。これは10倍希釈の細胞に相当する。この細胞を26〜30℃で3〜7日間培養する。この懸濁液を定常的に攪拌する(50〜100rpm)。細胞の増殖と細胞生存度は定常的に工程内コントロールによりモニタする。細胞密度が1.0〜6.5×106細胞/mlに達したとき、5リットル容量醗酵槽の内容物を、約40リットルのSF900IIを容れた50L醗酵槽に移す。この細胞を、密度が±5〜15×105細胞/mlに達するまで26〜30℃で培養する。この懸濁液を定常的に攪拌する(50〜100rpm)。サンプルを工程内コントロール用に採取する。
【0044】
2.E2抗原の産生例
上記の細胞懸濁液に、±107TCID50/mlを含む作業種子ウイルス(WVS)(BacE2[−])1〜2mlを添加する。細胞の70〜100%が細胞変性効果を示すまで、28℃にて3〜8日間培養する。培養の間に工程内コントロール用にサンプルを採取する。次に、この懸濁液を精密濾過による細胞除去により清澄とする。得られた濾液(すなわち、抗原溶液)を集め、不活化工程(3.3)を始めるまで≦−20℃にて保存する。工程内コントロール用にサンプルを採取する。抗原含量を定量するために、サンプルを、たとえば、抗原質量を定量するために、酵素結合免疫アッセイで、または水中タンパク質容量あたりの実質重量を定量するために、タンパク質アッセイで、あるいはかかる方法の併用により試験する。
【0045】
3.ウイルス不活化例
ウイルスに臭化2−ブロモエチルアンモニウム(BEA)を10ミリモル/Lの濃度に加えることにより不活化する。pHを8.2〜8.7に、温度を34〜37℃に調節することにより、BEAを臭化2−ブロモエチル−インミン(BEI)に変換する。BEIはウイルスを不活化する活性成分である。ウイルス殺滅は工程内コントロール用サンプルを採取することによりチェックする。不活化には24〜72時間を要する。不活化後、10000リットルあたり1個未満の感染性粒子が存在する。ウイルスの不活化後、チオ硫酸ナトリウムを5〜25ミリモル/L添加し、pHを5.7〜6.3に調整することによりBEIを中和する。工程内コントロール用にサンプルを採取する。不活化し、中和した抗原溶液を1リットルおよび/または5リットル容量のバッグに移し、≦−20℃にて保存する。
【0046】
4.製剤化例
凍結した抗原溶液を22〜28℃で融解し、SF900IIまたはPBSにより希釈し、50μg/mlの抗原溶液とする。抗微生物剤としてチオメルサールを100μg/mlまで加える。工程内コントロール用にサンプルを採取する。この溶液(すなわち、第1水相)を2〜8℃にて<3日間保存する。この間、マルコール(Marcol)52とモンタニド(Montanide)80とを混合する(9:1)ことにより油相を調製する。また、この溶液も2〜8℃にて3日を超えない範囲で保存する。油相は0.22μm−無菌フィルタにより無菌濾過する。工程内コントロール用にサンプルを採取する。最後に、第2水相をリン酸バッファー塩溶液(PBS)またはSF900IIをモンタノックス(Montanox)80と混合することにより調整し(98:2)、チオメルサールを最終濃度100μg/mlまで添加する。この溶液を使用時まで2〜8℃にて保存する(<3日間)。使用前、第1水相を無菌濾過する。工程内コントロール用にサンプルを採取する。第1水相と油相とを混合(1:1.1)することにより第1エマルジョンを調製する。このエマルジョンを2〜8℃にて3日以内保存する。工程内コントロール用にサンプルを採取する。二重油中水エマルジョンは、第1エマルジョンを第2水相で乳化することにより(2.1:1)調製する。二重エマルジョンはバイアルに詰めるまで隔離保存室内に2〜8℃にて保存する。工程内コントロール用にサンプルを採取する。
【0047】
5.充填および施栓例
清浄室内において、二重エマルジョン溶液をクラスAゾーンにて無菌的に充填する。充填量は50、100または250mlバイアルに、それぞれ51、102または255mlである。充填容量は濾過容積の重量をチェックすることにより定常的にモニタする。充填直後、バイアルには栓をし、キャップを被せる。最後に、バイアルは隔離保存室内に2〜8℃で保存し、その後の品質管理を開始する。
【0048】
【表1】
【0049】
【表2】
【0050】
【表3】
【0051】
【表4】
【0052】
【表5】
【0053】
6.E2安定性実験例
目的
異なる正常条件下、長期間培養後の感染細胞培養物(MOI*0.0001)におけるE2タンパク質の安定性を検討するため。細胞培養におけるバキュロウイルス感染過程の溶菌性のために、感染サイクルの後期培地にプロテアーゼが放出されていると思われる。このことがE2タンパク質の分解と容量タンパク質レベルの低下に至らしめるのか否かが、本実験の命題である。
【0054】
材料と方法
培地またはSF900IIと記載されている場合、0.2%プルロニックF68含有SF900II培地を意味する。
【0055】
SF21細胞を容れたバイアルを融解し、100ml容量振盪フラスコ中、10mlのSF900IIにより、インキュベータ内軌道旋回式シェーカ台座上、培養する。細胞培養が必要とする細胞密度に達したところで、細胞を500ml振盪フラスコ中、10倍に希釈して100mlとする。この細胞培養が必要とする細胞密度に達したところで、再度、10倍に希釈し100mlとし、過剰の細胞性物質を廃棄した。この細胞培養が必要とする細胞密度に達したところで、これを10倍に希釈し、3個の500ml振盪フラスコに各フラスコ100ml容量ずつ入れた。過剰の物質を廃棄した。振盪フラスコをIKS軌道旋回式シェーカ台座に載せ、28℃のインキュベータ中70rpmで攪拌する。細胞密度1ml当たり0.612×105細胞で、100倍希釈のウイルス懸濁液63μlをフラスコ番号2および3に加える。振盪フラスコ番号1は未感染ブランクとして維持する。培養物を感染させたMOIは(0.063×0.01×0.8×107)/(100×0.612×105)=0.00008である。
【0056】
使用した100倍希釈ウイルス懸濁液は以下に概説したように調製した。MSVバイアル253をQCにより融解し、TC100培地に100倍希釈し、10%FBS(ロット番号97QC0139)を添加し、0.5mlずつに分割して−70℃に保存した。
【0057】
細胞培養条件を決定するために、定期的にサンプルを感染振盪フラスコ培養物と非感染振盪フラスコの両方から採取する。サンプル採取は以下に概括したように実施した。
【0058】
細胞物質約1.1mlを15ml容量のファルコンチューブ中、IECセントラGP8R遠心分離機により1000rpmで6分間遠心分離する。次いで、上清をゲルマン・アクロディスク・シリンジフィルタにより0.45μmで濾過し、なお残存する細胞を廃棄する。サンプル0.4mlを後の分析用にナルゲン低温バイアル中に−70℃で保存する。
【0059】
結果と結論
図1のデータおよび図2のグラフに見られるように、フラスコ2および3の細胞増殖曲線はおおよそ類似している。191hpiの後、それ以上の細胞計数はなされなかったが、その理由は事実上すべての細胞が死滅し、感染したからである。フラスコ2の139hpiサンプルはグラフから除外する。このサンプルの細胞計数は、前記のサンプルおよび以下のサンプル両方よりもはるかに高い。このことは、恐らくある種の沈殿がサンプル採取の前後に起こり、その結果、トリパンブルー溶液と混合したサンプルが不正確となったことを示している。この仮定は生存率と感染率両方が予測した結果と一致するという事実により確認される。このことは、感染培養物それ自体にはなんら異常なことが起こっていないことを意味する。この不正確なサンプル採取は、サンプル中のE2濃度になんら影響を与えることはないだろう。というのは、E2は培地には存在するが、細胞内には存在しないからである。
【0060】
フラスコ2のE2濃度は非常に急速に上昇して、139hpi近辺で>190μg/mlの最大値となるが、そこからゆっくり低下して215hpiで170μg/mlとなる。次いで、E2−含量がさらに急速に降下して、306.5hpiで<100μg/mlの最終レベルとなる。
【0061】
フラスコ3のプロフィールはおおよそ同じである。最大E2タンパク質含量もまた139hpi付近で到達するが、フラスコ2よりもわずかに高いだけである。E2濃度は139hpiでの233μg/mlから191hpiでの212μg/mlにゆっくりと低下するが、それ以後はより急速に低下し、215hpiで141μg/mlに至る。フラスコ2および3は良好な相関性を示す。
【0062】
t=44.25hpiで、振盪フラスコ番号1(ブランク)についても計測した。生存細胞密度は2.435×106細胞/ml、死滅細胞密度は0.190×106細胞/mlであり、総細胞密度は2.625×106細胞/mlであって、生存度は92.8%となった。生存細胞密度はフラスコ2および3両方よりも有意に高く、感染が細胞増殖をすでに減速しつつあることを示している。このブランクを次いで濾過した。
【0063】
以下の表はフラスコ2のサンプル抜粋物について実施した免疫ブロッティングアッセイの結果を示す。
【0064】
【表6】
【0065】
この表に見られるように、エピトープV3およびV8は、0および44hpiサンプルでなお検出不可である。この結果はE2−ELISAの結果とよく相関する。V3およびV8はともにE2バンドに検出されるので、115hpi以降のサンプルにはインタクトのE2が検出される。
【0066】
191hpi以降には、より小さいタンパク質からの第2バンドがゲル上に目視できる。2つの個々のブロットにおけるV3とV8での免疫ブロッティングは、分解産物がV3エピトープを含み、V8エピトープを含まないことを示す。V8はブロット上のいずれの場所にも検出されないが、未処理E2バンドには検出される。このアッセイは、E2タンパク質の分解がプロテアーゼの存在により起こることを明瞭に示している。事実、これがE2タンパク質含量降下の原因であり、この降下はバルクE2抗原溶液の精密濾過工程の際に観察される。この下流の処理加工段階で、細胞はウイルスの不活化が始まる前に除かれる。
【0067】
したがって、恐らくプロテアーゼの存在がE2タンパク質分解の原因である。
7.MOI実験例
目的
MOI(感染多重度、細胞当たりのウイルス数)、最大細胞密度、および容量計測E2含量間の関係をさらに検討すること。一方で、完全な細胞母集団の感染は培地が消費し尽くされるまでに完了しなければならないが、他方で、低細胞濃度の総母集団の感染は最適以下のタンパク質収量に至る。
【0068】
材料と方法
培地というときは、SF900II培地を意味する。
【0069】
細胞懸濁液76mlを、1000ml容量ボトル中、444mlのSF900IIに希釈し、ゆるやかに混合する。培養物希釈直前の生存細胞密度は、3.41×106細胞/ml、死滅細胞密度は0.190×106細胞/mlであり、生存率は94.8%となる。この希釈により、細胞密度は±5×105細胞/mlとなる。
【0070】
さらに、ゲンタマイシンを加え、最終濃度10μg/mlとした。細胞懸濁液を50mlずつ250ml振盪フラスコ中に9分割し、過剰の細胞は廃棄した。最初の50mlを振盪フラスコ1に移し、最後の50mlを振盪フラスコ5に移した。フラスコ番号1および6はMOI=0(ブランク)に使用し、番号2および7はMOI=0.000001用とし、番号3および8はMOI=0.00001用とし、番号4および9はMOI=0.0001用とし、番号5はMOI=0.001とする。
【0071】
ウイルス原液の調製は以下のようにする。希釈範囲は、100倍希釈したウイルス懸濁液を入れたバイアルから調製する(該バイアルは1ml当たり0.8×105プラーク形成単位(pfu)を含む)。この100倍希釈したウイルス原液は以下のように調製した。マスター種子ウイルスを融解し、TC100培地に100倍希釈し、10%FBSを添加して、0.5mlに分割、−70℃にて保存する。
【0072】
この100倍希釈したウイルス溶液0.4mlをSF900II培地3.6mlで希釈して希釈ファクタ10-1とし、この溶液1mlを培地9mlに加えて希釈ファクタ10-2とし、この溶液1mlを培地9mlに加えて希釈ファクタ10-3とし、そしてこの溶液1mlを10倍に希釈して希釈ファクタ10-4とする。細胞培養物を含む振盪フラスコ1および6(ブランクフラスコ)に培地3.1mlを加える。ウイルス希釈液10-2、10-3および10-4をそれぞれ振盪フラスコ4と9、3と8、および2と7に加える。これらの振盪フラスコにはすでに細胞培養物を入れてある。
【0073】
3.1mlのウイルス希釈液10-2には、3.1×10-2×0.8×105=2.5×103pfuが存在する。50mlの細胞懸濁液を容れた振盪フラスコには、0.5×106×50=2.5×107の細胞が存在する。そこで、50mlの細胞懸濁液に3.1mlのウイルス希釈液10-2を加えると、MOIが2.5×103/2.5×107=0.0001となる。他のウイルス希釈液について同等の計算をすると、細胞培養物50mlに添加したウイルス希釈液10-3と10-4については、MOI計数0.00001と0.000001が得られる。最終的に、2.85mlの希釈液10-1を振盪フラスコ番号5に加えると、MOIが0.001ではなく0.00092となる。
【0074】
サンプル3.1mlを、ウイルス添加直後、各振盪フラスコに取る。サンプル中の細胞は15mlファルコンチューブ中、IECセントラGP8R遠心分離機により遠沈する(10分、1000rpm)。サンプル採取は低MOIから始めて高MOIについて実施し、高MOIの感染細胞懸濁液から低MOI感染細胞懸濁液にウイルスが伝播するのを防止する。遠心分離後、サンプルを0.45μmで濾過し(上清になお存在する可能性のある細胞を除去するため)、3個のナルゲン低温バイアルに分割し、後の試験用に−70℃にて保存する。
【0075】
振盪フラスコ1および5の細胞密度はウイルス添加直後に定量した。これらは細胞接種すべき最初と最後のフラスコであった。細胞密度が殆ど一致したので、すべてのフラスコは両方の細胞計数平均値に等しい細胞密度を有すると見なした。当初の生存細胞密度は0.423×106細胞/mlであり、一方、0.012×106細胞が死滅していたので、生存率は97.3%となった。
【0076】
すべてのフラスコをラボテック300軌道旋回式シェーカ台座に載せ、28℃の室内にて約75rpmで攪拌した。
【0077】
23、95、125、144、173および194hpi(感染後の時間)に、すべてのフラスコからサンプル採取した。細胞密度を定量したサンプルは容量が3.3mlあり、その0.2mlを用いて細胞密度を定量した。残り3.1mlはt=0サンプルのために上記のように取扱う。計測しなかった培養物サンプルは容量が3.1mlあった。194hpiでの細胞密度を定量し、この実験を終えた。残りの細胞懸濁液(約30ml)は、その1ml、3部をナルゲン低温バイアルに、約6mlの4部を15mlファルコンチューブに入れ、保存した。
【0078】
結果と結論
この結果は、MOIと最適細胞密度との間に、さらに重要なことは、MOIとE2タンパク質収率との間に良好な相関があることを示す。このことは、MOI 0.001で感染した培養物の最大容量E2タンパク質収率を100%とした場合のグラフに見て取れる。それは容量E2タンパク質収率がMOIの減少とともに増大することを示す。0.0001のMOIまでは、細胞密度は、最大達成可能細胞密度に達する前に感染される。0.00001またはそれ以下のMOIを用いると、培養物の感染が終了する前に培地が消耗されてしまうという感染結果となり、最適以下のタンパク質収率となる。したがって、引き出される結論は、感染過程とE2の産生が完了するまでに培地が消尽しない限り、使用されるMOIが低い程、E2タンパク質収率が高くなるということである。
bFSHαおよび/またはbHSFβを発現する組換えバキュロウイルス
伝達ベクターpDW−アルファ−9.1およびpDW−ベータ−3.1を構築した。Sf21細胞はpDW−アルファ−9.1またはpDW−ベータ−3.1、および細胞外ウイルス粒子から単離した野生型(wt)AcNPV/M021DNA(PCT出願:WO96/25696)により同時移入した。β−ガラクトシダーゼ発現ポリヘドリン陽性プラークを単離し、bFSHαまたはbFSHβを発現するかについて、培地のELISAにより分析した。一つのプラーク精製したbFSHαウイルス(AcNPVα3.4)および一つのプラーク精製したbFSHβ(AcNPVβ1.4)を用い、それぞれ約107と108のTCID50をもつウイルス原液を調製した。FSHの産生は上記方法により生じ、産生レベルは17〜33μg/mlとなった。
【0079】
PD50試験
病毒力の強いCSFV株ブレスシア(Brescia)100LD50の攻撃に対し、予防接種したブタの95%(PD50)を予防する、1回の予防接種後に必要とするE2用量(μg)の評価。
【0080】
動物
26頭の特異病原体陰性(SPF)ブタ(6〜7週令)を到着順に無作為に、8頭ずつの3群(A〜C)と2頭の1群(D)に分けた。これらの動物をID−DLOの高度封じ込め設備内にある家畜小屋B18、B19、B20およびB21に収容した。動物は3日間順化させる。動物は1日に1度、飼い葉桶で完全食物ペレット(ホープ・ファームス)として給餌し、水は給水器(niple ad libitum)から飲むことができた。
【0081】
予防接種および攻撃
二重油中水型アジュバント添加ワクチンの3種の製剤を上記のように調製し、それぞれ異なるE2抗原濃度、32.0、8.0および2.0μg/投与量とした。ブタには筋肉内に一度接種したが、各ブタは左耳の後方2cmのところに1回量(A:2μgのE2、B:8μgのE2、C:32μgのE2、D:0μgのE2)を受けた。対照群DはDOEアジュバントのみを接種し、前哨用のブタにはまったく接種しなかった。予防接種の3週間後に前哨用を除く各動物に、CSFV株ブレスシア456610の100 50%致死量(=100Va LD50)を鼻腔内攻撃した。攻撃直前に前哨用をその群から隔離し、24時間後に戻した。接種物のウイルス含量は家畜小屋から戻した後に採取したサンプルの滴定により定量した。
【0082】
臨床的観察
ブタは動物飼育係が毎日チェックし、異常が見つかり次第記録し、要すれば、監督者の獣医を呼んだ。各群は給餌時および小屋の清掃前およびその間に1日少なくとも15分間観察した。
【0083】
食餌摂取の減少した群または個々の動物に注目した。
体温(直腸)は予防接種後9日間、また、攻撃後20日間記録した。
【0084】
攻撃後の血液分析
EDTA−血液サンプルを攻撃後、0、2、4、7、10および14日目に採取し、白血球および血小板数の変化をモニタした。血中の白血球(血球減少症)および血小板(血小板減少症)数減少はCSFの典型的な徴候の一つである。通常のブタでは、白血球細胞と血小板に対する正常細胞計数は、それぞれ11〜23×109/Lおよび320〜720×109/Lの範囲にある。SPFのブタでは、これらの値がやや小さい;6〜12×109/Lおよび300〜700×109/L。双方言及の範囲は各ブタで変動する。血液細胞の分析はメドニックRCA570クールタ計数器により実施した。血球減少症および血小板減少は、細胞/血小板計数が、好ましくは1日以上、上記の最小数よりも顕著に低い場合と定義した。。
【0085】
ウイルス拡散/排出およびウイルス検出
体温、白血球細胞と血小板計数、および前哨のセロコンバーションをパラメーターとして使用し、接種した動物からこれらの動物へとウイルスが伝播したことを検出した。死後の組織サンプルは以下の器官から採取した:扁桃腺、脾臓、腎臓、および回腸について、ウイルス抗原の存在につき直接免疫蛍光法により試験した。これら組織サンプルからの低温維持切片(厚さ4μm、各器官2切片)を固定化し、ポリクローナルブタ抗ペスチウイルスFITC−接合血清と培養した。洗浄後、切片を蛍光顕微鏡下で読取った。結果を陽性(=蛍光性)または陰性(=非蛍光性)として表した。
【0086】
血清学的応答
すべてのブタの血清を予防接種後、0、2および3週目、および死亡時に採取した。サンプルを−20℃にて保存し、ウイルス中和試験(VNT)およびセディテスト(CeditestR)(CSF特異抗体を検出するためのELISA)によりアッセイした。血清中のCSFV中和抗体タイタをミクロタイタシステムにより定量した。血清の段階2倍希釈液を、30〜300TCID50含有のCSFV(ブレスシア株)懸濁液等容量と混合した。CO2インキュベータ内37℃で1時間培養した後、ウエル当たり約25.000PK15細胞を添加した。4日後に、マイクロタイタプレートを上記のように処理し、顕微鏡下に読取った。CSFV中和タイタは、すべてのウイルスを中和する最高希釈の逆数として表した。
【0087】
統計的評価
95%防御的用量の決定は、≧50のCSFV中和Abタイタが完全な防御を表わすという仮定にもとづいた。
【0088】
結果
本セクション全体を通して動物数については表7または表8に示す。
【0089】
予防接種後の臨床的観察
予防接種はブタに何ら悪影響を及ぼさず、食餌摂取も体温も正常のままであった。A群およびC群には、軽い下痢、食欲不振、およびうつ症状が、予防接種後3日目に見られた。A群からの1頭(no.448)は全期間にわたり定常的に嘔吐し、成長が遅れた。B群からのブタno.438は一度嘔吐した。ブタno.434(C群、前哨)は体温がわずかに上昇したが、この動物は右後肢の跛行をわずらっていた。食餌摂取量は、予防接種から攻撃までの期間、1日1群当たり3kgから6kgに増加した。
【0090】
攻撃後の臨床的観察
非予防接種対照動物は、攻撃後3日目(no.59)および6日目(no.60)にCSFの徴候を示し始めた。発熱、群がり行動、震え、食欲喪失およびうつ症状が攻撃から10日後の死亡時まで見られた。さらに、この動物は、チアノーゼ、後肢不全麻痺、下痢および重症の嘔吐を示した。両動物は瀕死の状態のときに殺した。
【0091】
2μgのE2で予防接種したブタ(A群)はすべて攻撃の2〜3日後に病気の徴候を示し始め、主に、発熱、群がり、うつ症状および食欲不振を示した。発熱は2〜10日に及び、最高体温(Tmax)は40.7〜42.2℃で変動した。7日目以降、ブタの内、443〜446は発熱が収まり、食餌摂取量も攻撃前の量まで増大した。しかし、448のブタは9日目に死亡しているのが見つかり、447のブタは急性のCSF(痙攣、後肢不全麻痺)に懸り、同日瀕死の状態であるときに殺した。両前哨用は攻撃7〜9日後まで正常であったが、その後、急性のCSFとなり、20日目、瀕死の状態であるときに殺した。
【0092】
B群およびC群の動物は、疾患の臨床徴候および持続期間が、E2抗原の有効量増大とともに軽くなった。
【0093】
B群では、発熱(435〜439)が27日目まで続き、Tmaxは40.2℃と41.7℃の間で変動した。ブタno.436は非常に軽度の臨床徴候で攻撃に抵抗した。1頭のブタ(no.440)は18日後に急性のCSFで死亡したが、瀕死の状態であるときに殺した。残り、B群からの5頭の動物は回復した。両前哨用は攻撃後11〜12日目まで正常であったが、その後、急性のCSFとなり、20日目に死亡させた。
【0094】
C群では、4〜6日目から6頭の内2頭(430、431)に軽い発熱が見られ、Tmaxは40.5℃〜41.2℃で変動した。臨床的徴候は、攻撃後6日目にわずかなうつ症状が見られた以外、注目すべきものはなかった。従前どおり、攻撃ブタno.430は嘔吐した。
【0095】
両前哨用は実験終了時まで正常であった。
攻撃後の血液分析
A群からは、ブタ(450)1頭のみ(前哨用)が明瞭な白血球減少および血小板減少を示した。ブタno.445および446は攻撃後、7日目および10日目に、また、ブタ449は10日目および14日目に血小板減少となった。B群からのブタ1頭(440)は白血球減少および血小板減少を示したが、他は正常であった。両前哨用は14日目に血小板減少となり始める徴候を示した。C群では、前哨用も含めて、白血球減少も血小板減少も検出しなかった。
【0096】
両対照動物(no.59および60)は7日目以降から血小板減少となった。
攻撃後のウイルス拡散およびウイルス抗原検出
家畜小屋から戻って後、接種量を逆滴定して、ウイルス力価を2.55TCID50/mlとした。CSFVウイルス抗原(表7)は、A群およびB群から選択した対照と前哨用の組織サンプルすべてに検出された。A群およびB群において、6頭の接種した動物の内、1頭が陽性であった。C群では前哨用も含めて陽性がなかった。
【0097】
血清学的応答
CSFVでのセロコンバーションはVNTにおいて力価≧25と定義した。VNTに用いたブレスシアウイルスの量を定量するための逆滴定の結果は、41TCID50/ウエルであった。
【0098】
対照(D)または前哨用動物は本実験の間、セロコンバーションを受けなかった(表8)。A群の動物6頭の内2頭が3週間後にセロコンバーションを受けた。しかし、6頭の内4頭は攻撃後、追加刺激を受けた(表8)。B群およびC群は予防接種後21日目にセロコンバーションを受けたが、この動物はすべて攻撃後に追加刺激を受けた。後者のみが、当該動物において攻撃ウイルスの複製が成功していることを物語っている。
【0099】
結論
動物1頭当たりのPD95投与量を一回につきDOEアジュバント2ml中32μgのE2と決定した。この投与量により、毒性の非常に高いCSFV株を攻撃したことによる疾患の臨床徴候が最小に止まり、接触した動物へのウイルス拡散も発生しない。要約すると、6頭のブタ4群(A〜D)につき、筋肉内投与により、アジュバント(DOE)2ml当たりそれぞれ32(A)、8(B)、2(C)および0μg(D)で予防接種した。2頭の非予防接種ブタを各群(A〜C)内に置き、接触感染の原因となるウイルスの排出を検出した(前哨用)。3週間後、鼻腔内投与により毒性の非常に高いCSFV株ブレスシア100LD50で動物を攻撃した。臨床学的、ウイルス学的および血清学的パラメーターを測定し、E2サブユニットワクチンの効能を評価した。予測どおりに、C群の動物は他の群の動物よりもより良好に攻撃に抵抗した。ウイルス抗原は、E2の最大投与量(32μg)を接種した動物の組織サンプルに検出されず、この群には疾病も存在しなかった。PD95用量は、≧50のNPLA力価(予防接種後21日目)が完全な予防効果を与える(ウイルスの拡散がない)という仮定にもとづき計算した。この結果がPD95用量32μg/頭である。
【0100】
予防接種2週間および3週間後の100LD50毒性CSFVによる攻撃に対し、その防御効果のレベルをさらに評価するために、6頭のブタ2群(A〜B)を32μgのE2により筋肉内経路で予防接種した。2頭の非予防接種ブタを各群内(A〜B)に置き、接触感染の原因となる予防接種ブタからのウイルスの排出を検出した。2週(A)および3週(B)後、予防接種した動物および対照の動物を毒性の非常に高いCSFV株ブレスシア100LD50で鼻腔内経路により攻撃した。予防接種の2週間および3週間後、臨床学的、ウイルス学的および血清学的パラメーターを測定し、E2サブユニットワクチンの効能を評価した。予測どおりに、B群はA群よりも臨床徴候が少なく、攻撃に抵抗した。前哨用へのウイルスの伝播はA群にのみ起こり、両群からの動物すべての白血球フラクションにウイルスが検出された。B群の動物1頭のみが予防接種の14日目にセロコンバーションを受けていた(no.414)。同一群からの動物1頭は21日後にもセロコンバーションを受けなかったが、防御されており、発熱は2日間のみであり、攻撃後11日以内にセロコンバーションを受けた。対照と前哨用の1頭(A群)のみが白血球減少症と血小板減少症に罹患した。A群およびB群動物からの組織サンプルいずれにもウイルス抗原は検出されなかった。結論として、動物はすべて単回投与での予防接種後2週間および3週間目の攻撃に対し防御されていた。
【0101】
予防接種3ヵ月および6ヵ月後の100LD50毒性CSFVによる攻撃に対し、その予防効果のレベルをさらに評価するために、6頭のブタ2群(A〜B)を32μgのE2により筋肉内経路で予防接種した。2頭の非予防接種ブタを各群内(A〜B)に置き、接触感染の原因となる予防接種ブタからのウイルスの排出を検出した。3ヵ月(A)および6ヵ月(B)後、予防接種した動物および対照の動物を毒性の非常に高いCSFV株ブレスシア100LD50で鼻腔内経路により攻撃した。この期間の後、臨床学的、ウイルス学的および血清学的パラメーターを測定し、E2サブユニットワクチンの効能を評価した。A群およびB群からの動物はすべて攻撃に抵抗し、臨床徴候はわずかであった。そして対照のみが白血球減少症と血小板減少症に罹患した。前哨用へのウイルスの伝播は起こらなかった。また、両群から選択された白血球フラクションのいずれにもウイルスは検出されなかった。B群の動物1頭のみが予防接種後の28日目にセロコンバーションを受けていた(no.1983)。A群、B群および前哨用動物のいずれの組織サンプルにもウイルス抗原は検出されなかった。結論として、すべての動物が単回投与での予防接種後3ヵ月および6ヵ月目の攻撃に対し防御されており、ウイルス伝播は起こらなかった。
0 BVDV株4800、150022および178003を用い、実験的E2サブユニットワクチンを生成させた。これらの株のE2遺伝子はバキュロウイルス発現系において発現させた(ハルスト(Hulst)ら、1993、J. Viol. 67:5435〜5442)(リジュン(P.A.van Rijn)ら、1996、I)、Gen. Virol.、77:2737〜2745)。スポドプテラ・フルギペルダ(Spodoptera frugiperda)細胞系SF21を、組換えバキュロウイルスの増殖とE2タンパク質の生産に用いた。SF21細胞は無血清SF900培地(ギブコBRL)中28℃にて増殖した。SF21細胞の集密単層を組換えバキュロウイルスにより、細胞当たり5TCID50(50%組織培養感染用量)の感染多重度にて感染させた。4〜5日後、培養は80〜90%の細胞変性効果を示した。細胞を1500×gで10分間遠心分離し、バキュロウイルスとE2を含むその上清を収集し、−20℃にて保存する。バキュロウイルスを不活化するために、臭化2−ブロモエチル−インミン(BEI)での処理を標準手法に従い実施した。手短に説明すると、BEI 600μlと1M−NaOH600μlとを混合した。20℃、30分後、上清150mlを加え、20℃で24時間攪拌した。次いで、25%チオ硫酸20mlを加えた。バキュロウイルスの不活化はSF21細胞上、上清の滴定により確認した。
【0102】
1 実験的BVDVワクチンは二重油中水エマルジョン中、E2含有上清からなっていた。該油は90%マルコール52(エッソ)および10%モンタニド80(セピック)を含んでいた。水性フラクション(PBS+)は98%リン酸バッファ塩溶液、2%モンタノックス80(セピック)および100ppmチオメルサールを含んでいた。上清、油およびPBS+は、10:11:10の比で用いた。まず、上清と油を乳化し、次いでPBS+を加え、乳化した。対照のワクチンは、野生型バキュロウイルスで感染させたSF21細胞の上清から同様に調製した。調製後、ワクチンは無菌であると判定された。
【0103】
2 3種ワクチンの抗原量は、発現されたタンパク質の同等性の故にほぼ同じであろうとする我々の当初の仮定は、間違っていた。抗原量の差は恐らく昆虫細胞での発現の差に原因がある。ワクチン150022は用量当たり55μgのE2を含んでおり、2回の予防接種(0日および21日目)後、胎児に対し防御的であった(ブラッシェ(Brusche)ら、1997、Vaccine、印刷中)。ワクチン4800およびワクチン178003は用量当たり12μgと17μgのE2をそれぞれ含んでおり、胎児に対し防御的ではなかった。この結果はE2タンパク質量と胎児の防御との間に相関性のあるこを示唆している。しかし、E2糖タンパク質の免疫原性の差と、攻撃株が胎盤を通過する能力のないことが、攻撃が異なる結果をもたらすことに対する説明となるかも知れない。非相同BVDVの攻撃に対し、該ワクチンは防御しなかった。
【0104】
3 この研究の結果はBVDVサブユニットワクチンのさらにる開発のために将来有望である。その理由は我々が、エンベロープ糖タンパク質E2での予防接種により胎児の防御を達成し得ることを示したからである。さらにそれはマーカーワクチンであり、予防接種した動物と野外ウイルス株との間に識別を可能とする。これは将来のBVDV撲滅プログラムに照らして有利である。
【0105】
検討
4 この開発された生産工程は、昆虫細胞内バキュロウイルスに挿入された遺伝子がエンコードする非相同タンパク質の最適生産を達成することを目的とする。バキュロウイルス発現ベクター系(BEVS)は異なる組換えタンパク質の産生に非常によく適合する。その理由は、正しいタンパク質の折りたたみと正確な翻訳後プロセシングが、動物とヒトへの応用のために生物学的に活性なタンパク質に至らしめるからである。バキュロウイルスは2つの非本質的遺伝子を含み、それは非常に強力なプロモーター、p10およびポリヘドリンプロモーターから転写される。p10遺伝子の欠失突然変異誘発(バン・オアーズ(van Oers)ら、J. Gen Virol. 74:563〜574;1993)は、それが細胞培養におけるウイルスの複製にとって必須ではないが、p10が細胞溶解において一つの役割を演じており、p10タンパク質が存在しないと、感染した細胞が損なわれないままに存在することの原因となり、感染細胞からの多角体の放出を妨ぎ、それによって再感染率を降下させるが、それ自体感染性ではないということを示した。遺伝子産物(p10(またはフィブリリン)およびポリヘドリン)は感染段階の後期に発現される。これらの遺伝子を、必要とするタンパク質の遺伝子に置換えても(ポリヘドリンプロモーターの場合)、理論的に、昆虫細胞培養の総タンパク質生産収率は50%までとなるが、発現レベルは培養1リットル当たりポリヘドリンタンパク質1グラムを超えるものとなる(マイオレラ(Maiorella)ら、(1988) 組換えタンパク質生産のための大規模昆虫細胞培養。Bio/technology、6、1406〜1410)。感染多重度(MOI、1ml当たりのウイルス密度と1ml当たりの細胞密度の比)は、タンパク質生産の最適化にとって重要なパラメーターである。感染多重度の低い感染であることの明白な理由は、ウイルス植菌量をできるだけ少なくしていることにある。もし感染が、50L醗酵槽内でMOIを0.1として1ml当たり2百万の細胞密度で起こるならば、その場合は、1×1010のウイルスが必要となる。このことは約1リットルの植菌量を必要とすることである。さらに、かかる植菌量を造るための余分な生産工程が必要となる。付着培養の2つの主な欠点は、効率の悪い培地の使用と酸素量が制限されることである。水性液での酸素溶解度が比較的乏しいという事実のために、単層培養での細胞にとって、酸素は制限のある基質である。静置培養での最大細胞密度の決定因子は利用可能な表面である。その表面が細胞の単層で一旦被われると、細胞増殖は停止してしまうことになる。その結果、細胞密度は培地1ml当たり約1×106に留まる。懸濁培養では酸素の制限が存在せず、他の基質の利用可能性が細胞密度にとっての制限因子となる。この制限は酸素の制限よりもより高い細胞密度で起こり、したがって、細胞密度が静置培養におけるよりもより高くなる。酸素の制限は、溶解酸素濃度をモニタする酸素電極を使用することにより防止される。酸素がある値以下に落ちると、酸素は自動的に、培養槽の上部空間を経て散布によりまたは添加により、懸濁液に添加される。懸濁液を適度に攪拌することは均一な培養を保証し、そこでは基質の勾配が形成されず、また、細胞も過度に高い剪断力にさらされることがない。さらに、攪拌は感染細胞のウイルスを非感染細胞に効率的に移動させることになり、細胞のウイルス感染効率を高めることとなる。最初、細胞はわずかに約0.1〜0.3%が感染しているだけなので、残り99.7〜99.9%の細胞は増殖し、複製することが可能となる。感染細胞内で産生されるウイルス粒子は、感染後12〜20時間で放出される(ディーおよびシュラ(Dee,K.U. and Shuler, M.L.)1997、Biotechnology Progress, 13、14〜24)。細胞当たりに放出されるウイルス粒子数は100〜200であり、それが新たなサイクルにおいてそれまで非感染であった細胞を感染させる。ウイルス粒子が十分に産生され、培養物内に存在するすべての細胞を感染させるまでには、数回のサイクルを要する。目指すところは、培地が使い尽くされ、すべての代謝工程が停止するようになるまでに、最終の感染経路のタンパク質産生を完遂することである。また、最適以下の細胞密度で完全な感染を達成させることも望ましくない。というのは、培地が効率的に使用されなければ非相同タンパク質の容量収率も低くなるからである。ウイルス感染の全工程は、ポリヘドリン遺伝子がなお存在するという事実により、目視によりチェックすることができる。ウイルスの感染は細胞核に蓄積する密集したタンパク質粒子として観察することができる。細胞に対する剪断損傷を防止するためには、プルロニックF−68を最終濃度が0.2%となるように培地に添加する。さらに、要すれば、消泡剤Aを加えて、過度の泡立ちを防止するが、これもまた細胞死を招く。50Lの培養に添加される消泡剤Aの総量は平均で20mlである。細胞を感染させる最適ウイルス密度は計算することができる。この密度は、細胞の増殖率、新たなウイルス粒子が放出されるまでの感染後の時間、および感染細胞当たりに産生される新たなウイルス粒子の数に依存する。本発明により提供される方法の例証となるのはペスチウイルスE2タンパク質の産生である。このタンパク質は活性の高い抗原として知られていたが、ワクチン用または診断試験用のE2(フラグメント)の生産は常に不成功である。PD50は2μgと低い(特に、免疫原E2フラグメントを用いる場合)にもかかわらず、問題は、多数動物群の防御的予防接種を1頭当たり単回の予防接種で可能とするために、商業的に魅力のある方法で、十分な抗原性部分をもつワクチンの投与に十分な量を如何に集めるかである。この問題はとりわけ、CSFVの予防接種に関連性がある。投与に際しては、CSFVの予防接種は、一般に、CSFVが突然発生した地域での集団活動で実施される。この場合には、膨大な数の動物を、比較的短期間で、迅速に予防接種することを求められる。かかる集団活動においては、適切な防御レベル(野生型ウイルスの感染に対し防御されるブタの数)を迅速に達成することが差し迫って重要である。防御を達成するために、最初の予防接種後二回目の予防接種まて数週間も待つことは、この疾患の制御を著しく妨害し、遅らせる。組換えにより発現したE2タンパク質を産生させる様々な方法の間には、バキュロウイルスで発現したE2(フラグメント)を比較するだけでも、違いが存在する。初期に報告されたE2タンパク質産生培養において、E2タンパク質(フラグメント)の収量は20〜90μg/mlの間で変動する(ハルスト(Hulst)ら、J. Vir. 5435〜5442、1993;ハルストおよびムーアマン(Hulst and Moormann)、Cytotechnology、20:271〜279、1996)が、単回投与予防接種に必要な関連するE2抗原性部分を得るためには、モノクローナル抗体による免疫アフィニティ精製をさらに必要とする。他の方法(EP0389034に記載されたE2のフラグメントを使用する)は、この方法では昆虫細胞の上清から取得したE2をさらに免疫アフィニティ精製することなしに使用するが、それがE2ベースのワクチンであって、満足すべき(防御)免疫応答が得られるまでに、2回注射する。これらの問題は、とりわけ、該ワクチンを調製する出発原料、たとえば、細胞培養上清における関連する抗原性物質、この場合にはE2タンパク質(フラグメント)、の低濃度に関係する。理論的には、技術上既知の精製・濃縮法により抗原性部分をさらに蓄積することができるが、しかし、この方法では商業的に魅力のあるワクチン生産法に導き得ず、しかも1用量当たり高コストの原因ともなる。本発明方法により提供される方法を用いる生産工程では、我々の50L容量醗酵槽において、定常的に約200〜300μg/mlのCSFV E2タンパク質フラグメントを生成するに至り、理論的に、1回の培養当たり100,000頭の動物を予防接種するのに十分な量である。細胞はおおよそ107TCID50/mlを含むウイルス菌量1〜10mlにより、0.5〜1.5×106細胞/mlの密度で感染される。この培養では、好ましくは、細胞の >50〜80%がCPEを示したときに収得する。これは感染後約100〜150時間である。初期E2タンパク質生産培養においては、細胞はMOI> 1で(同時に)免疫されるが、タンパク質収量は本発明により提供されるものより3倍低くなる。本発明により提供される方法では、50L容量の醗酵槽は、5Lの醗酵槽内で増殖した細胞懸濁液5Lにより、または10Lの醗酵槽内で増殖した懸濁液のすべて10Lにより接種する。初期細胞密度は約3×105細胞/mlである。細胞は、この懸濁液にウイルスを加える前に、計算した細胞密度まで増殖させる。下流のプロセシングは精密濾過による細胞と多面体の除去とともに始まる。中空糸精密濾過装置を醗酵槽に結合し、原料を0.22μmの孔径をもつ濾過モジュールにポンプ輸送する。保持流動体は醗酵槽を再循環させ、浸透する流動体は100Lの容器に収集する。濾過が完了したとき、現段階で無細胞ではあるが、感染性のバキュロウイルスをなお含む抗原溶液を100L容器中で不活化する。一般に、臭化2−ブロモエチル−アンモニウム(BEA)を、最終濃度が8〜12mMとなるように該懸濁液に加える。2M−NaOHを添加して、pHを約5.8から8.2〜8.7に上昇させる。このpH移動はBEAをBEI(臭化2−ブロモエチル−インミン)に変換する。これはDNA不活化剤である。pHを注意深くモニタして、6〜10に調整し、温度は34〜39℃に維持する。不活化の6時間後に、抗原溶液を第2不活化容器に移す。これにより、容器中のすべての物質がBEIと接触し、その結果、不活化されるであろうことが保証される。ウイルスを含むが、BEIを含まない液体のしずくが第1容器には存在し、第2容器には存在しないはずである。104〜107pfu/mlの濃度で存在するバキュロウイルスは、<10-7pfu/mlの値(10m3当たり<1個のウイルス粒子)まで分解される。これは、宿主動物を感染させ得るウイルスにもとづくウイルスに対して用いたのと同じ水準である(FMD同様)。ブタはバキュロウイルスに対して宿主ではない。不活化した抗原の大部分は、水−油−水エマルジョンに剤形化するまで≦−20℃に保存する。32μgのE2を含む単回投与物(投与容量2ml)は、古典的なブタコレラに対し、予防接種後2週間から少なくとも6ヵ月まで防御効果( >PD95)を与えるに十分である。生産工程は、該工程のスケールアップが簡単で、250Lまたはそれより大きい醗酵槽の使用が可能となるような方法で設計する。静置培養生産のスケールアップもまた簡単である(さらに多くの組織培養フラスコを使用するだけである)。しかし、50L醗酵槽にて定常的に達成される総タンパク質生産には、約7000個のT175組織培養フラスコを必要とする。細胞の増殖と感染は、酸素およびpH−電極が存在するので、醗酵槽中でより良好にモニタし、調整することができる。組織培養フラスコでは、フラスコ同士の変動が多分存在するが、それを量的に決めることはできない。不活化は静置培養生産にて実施したように、容器中でさらにより正確にモニタし、調整することができる。BEVSで発現される他のタンパク質の容量産生レベルは(我々の研究所にて)、もし細胞が本発明により提供される方法で増殖するならば、著しく改善される。たとえば、ウシFSHの収率は、10Lの醗酵槽を用いたとき、3〜4のファクタで増加する。細胞は2種の組換えバキュロウイルスそれぞれについて0.003のMOIで、1.1×106細胞/mlの細胞密度で同時感染させた。BVDVの異なるE2の、およびErnsタンパク質またはBVDVおよび/またはCSFVの収率は、振盪フラスコ中の懸濁培養において3倍に増加した。この方法は他の組換えタンパク質にも適用可能である。
【0106】
【表7】
【0107】
【表8】
【図面の簡単な説明】
【0108】
【図1】長期培養実験におけるE2の安定性のデータを示す図である。
【図2】培養実験におけるE2の安定性のグラフである。
【図2−1】培養実験におけるE2の安定性のグラフである。
【図2−2】培養実験におけるE2の安定性のグラフである。
【図3】MO1実験のデータを示す図である。
【図3−1】MO1実験のデータを示す図である。
【図3−2】MO1実験のデータを示す図である。
【図4】MO1実験のグラフである。
【図4−1】MO1実験のグラフである。
【図4−2】MO1実験のグラフである。
【図4−3】MO1実験のグラフである。
【図4−4】MO1実験のグラフである。
【図4−5】MO1実験のグラフである。
【図4−6】MO1実験のグラフである。
【特許請求の範囲】
【請求項1】
培養容器中の増殖培地において昆虫細胞を増殖させ、
膜貫通領域を伴わない古典的ブタ熱ウイルスE2たんぱく質をエンコードする組換えバキュロウイルス接種物で、0.0001〜0.01の前記細胞の感染多重度にて、前記細胞を感染させ、
前記細胞の約半分以上が細胞変性効果を示すまで前記細胞を培養し、
増殖培地から細胞性物質を除去し、
前記細胞性物質から、免疫アフィニティ精製することなく、単回接種で古典的ブタ熱に対してPD95のレベルの防御力を付与し得るE2サブユニットワクチンを調製する
ことを特徴とする膜貫通領域を伴わない古典的ブタ熱ウイルスE2たんぱく質を含むE2サブユニットワクチンの製造方法。
【請求項2】
前記細胞を5×105〜1.5×106個/mlの密度にて感染させることを特徴とする請求項1記載のE2サブユニットワクチンの製造方法。
【請求項3】
前記感染多重度が0.001〜0.005であることを特徴とする請求項1記載のE2サブユニットワクチンの製造方法。
【請求項4】
前記感染多重度が0.001〜0.003であることを特徴とする請求項3記載のE2サブユニットワクチンの製造方法。
【請求項5】
増殖培地中のE2たんぱく質の最終濃度が100〜300μg/mlのときに、細胞性物質を除去することを特徴とする請求項1記載のE2サブユニットワクチンの製造方法。
【請求項6】
アジュバントを含めることによってE2サブユニットワクチンを調製することを特徴とする請求項1記載のE2サブユニットワクチンの製造方法。
【請求項7】
前記アジュバントが二重油中水型エマルジョンから成ることを特徴とする請求項6記載のE2サブユニットワクチンの製造方法。
【請求項8】
培養容器中の増殖培地において昆虫細胞を増殖させ、
膜貫通領域を伴わない古典的ブタ熱ウイルスE2たんぱく質をエンコードする組換えバキュロウイルス接種物で、0.0001〜0.01の前記細胞の感染多重度にて、任意の時点で前記細胞を感染させ、
前記細胞の約半分以上が細胞変性効果を示すまで前記細胞を培養し、
増殖培地から細胞性物質を除去し、
前記細胞性物質から、免疫アフィニティ精製することなく、単回接種で古典的ブタ熱に対してPD95のレベルの防御力を付与し得るE2サブユニットワクチンを調製する
ことを特徴とする膜貫通領域を伴わない古典的ブタ熱ウイルスE2たんぱく質を含むE2サブユニットワクチンの製造方法。
【請求項9】
前記細胞を5×105〜1.5×106個/mlの密度にて感染させることを特徴とする請求項8記載のE2サブユニットワクチンの製造方法。
【請求項10】
前記感染多重度が0.001〜0.005であることを特徴とする請求項8記載のE2サブユニットワクチンの製造方法。
【請求項11】
前記感染多重度が0.001〜0.003であることを特徴とする請求項10記載のE2サブユニットワクチンの製造方法。
【請求項12】
増殖培地中のE2たんぱく質またはそのフラグメントの最終濃度が100〜300μg/mlのときに、細胞性物質を除去することを特徴とする請求項8記載のE2サブユニットワクチンの製造方法。
【請求項13】
アジュバントを加えることによってE2サブユニットワクチンを調製することを特徴とする請求項8記載のE2サブユニットワクチンの製造方法。
【請求項14】
前記アジュバントが二重油中水型エマルジョンから成ることを特徴とする請求項13記載のE2サブユニットワクチンの製造方法。
【請求項15】
バキュロウイルス系にて産生される組換え古典的ブタ熱ウイルス(CSFV)E2糖たんぱく質を含むE2サブユニットワクチンにおいて、
前記E2糖たんぱく質は、免疫アフィニティ精製されず、32μgの単回接種でCSFV感染に対してPD95のレベルの防御力を付与し、かつ
前記E2糖たんぱく質は膜貫通領域(TMR)を欠くことを特徴とするE2サブユニットワクチン。
【請求項16】
さらにアジュバントを含むことを特徴とする請求項15記載のE2サブユニットワクチン。
【請求項17】
前記アジュバントが二重油中水型エマルジョンから成ることを特徴とする請求項16記載のE2サブユニットワクチン。
【請求項1】
培養容器中の増殖培地において昆虫細胞を増殖させ、
膜貫通領域を伴わない古典的ブタ熱ウイルスE2たんぱく質をエンコードする組換えバキュロウイルス接種物で、0.0001〜0.01の前記細胞の感染多重度にて、前記細胞を感染させ、
前記細胞の約半分以上が細胞変性効果を示すまで前記細胞を培養し、
増殖培地から細胞性物質を除去し、
前記細胞性物質から、免疫アフィニティ精製することなく、単回接種で古典的ブタ熱に対してPD95のレベルの防御力を付与し得るE2サブユニットワクチンを調製する
ことを特徴とする膜貫通領域を伴わない古典的ブタ熱ウイルスE2たんぱく質を含むE2サブユニットワクチンの製造方法。
【請求項2】
前記細胞を5×105〜1.5×106個/mlの密度にて感染させることを特徴とする請求項1記載のE2サブユニットワクチンの製造方法。
【請求項3】
前記感染多重度が0.001〜0.005であることを特徴とする請求項1記載のE2サブユニットワクチンの製造方法。
【請求項4】
前記感染多重度が0.001〜0.003であることを特徴とする請求項3記載のE2サブユニットワクチンの製造方法。
【請求項5】
増殖培地中のE2たんぱく質の最終濃度が100〜300μg/mlのときに、細胞性物質を除去することを特徴とする請求項1記載のE2サブユニットワクチンの製造方法。
【請求項6】
アジュバントを含めることによってE2サブユニットワクチンを調製することを特徴とする請求項1記載のE2サブユニットワクチンの製造方法。
【請求項7】
前記アジュバントが二重油中水型エマルジョンから成ることを特徴とする請求項6記載のE2サブユニットワクチンの製造方法。
【請求項8】
培養容器中の増殖培地において昆虫細胞を増殖させ、
膜貫通領域を伴わない古典的ブタ熱ウイルスE2たんぱく質をエンコードする組換えバキュロウイルス接種物で、0.0001〜0.01の前記細胞の感染多重度にて、任意の時点で前記細胞を感染させ、
前記細胞の約半分以上が細胞変性効果を示すまで前記細胞を培養し、
増殖培地から細胞性物質を除去し、
前記細胞性物質から、免疫アフィニティ精製することなく、単回接種で古典的ブタ熱に対してPD95のレベルの防御力を付与し得るE2サブユニットワクチンを調製する
ことを特徴とする膜貫通領域を伴わない古典的ブタ熱ウイルスE2たんぱく質を含むE2サブユニットワクチンの製造方法。
【請求項9】
前記細胞を5×105〜1.5×106個/mlの密度にて感染させることを特徴とする請求項8記載のE2サブユニットワクチンの製造方法。
【請求項10】
前記感染多重度が0.001〜0.005であることを特徴とする請求項8記載のE2サブユニットワクチンの製造方法。
【請求項11】
前記感染多重度が0.001〜0.003であることを特徴とする請求項10記載のE2サブユニットワクチンの製造方法。
【請求項12】
増殖培地中のE2たんぱく質またはそのフラグメントの最終濃度が100〜300μg/mlのときに、細胞性物質を除去することを特徴とする請求項8記載のE2サブユニットワクチンの製造方法。
【請求項13】
アジュバントを加えることによってE2サブユニットワクチンを調製することを特徴とする請求項8記載のE2サブユニットワクチンの製造方法。
【請求項14】
前記アジュバントが二重油中水型エマルジョンから成ることを特徴とする請求項13記載のE2サブユニットワクチンの製造方法。
【請求項15】
バキュロウイルス系にて産生される組換え古典的ブタ熱ウイルス(CSFV)E2糖たんぱく質を含むE2サブユニットワクチンにおいて、
前記E2糖たんぱく質は、免疫アフィニティ精製されず、32μgの単回接種でCSFV感染に対してPD95のレベルの防御力を付与し、かつ
前記E2糖たんぱく質は膜貫通領域(TMR)を欠くことを特徴とするE2サブユニットワクチン。
【請求項16】
さらにアジュバントを含むことを特徴とする請求項15記載のE2サブユニットワクチン。
【請求項17】
前記アジュバントが二重油中水型エマルジョンから成ることを特徴とする請求項16記載のE2サブユニットワクチン。
【図1】

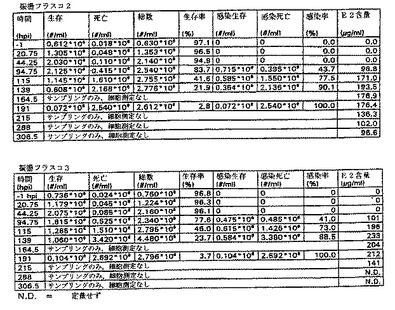
【図2】
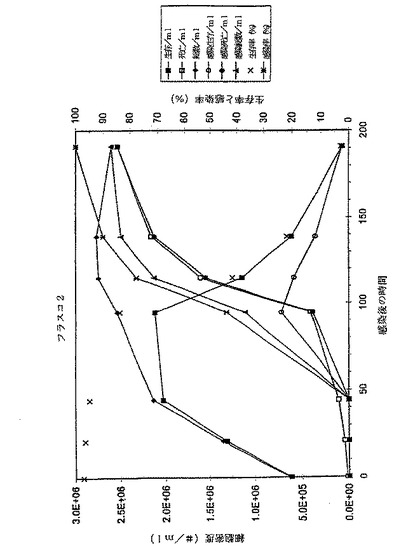
【図2−1】
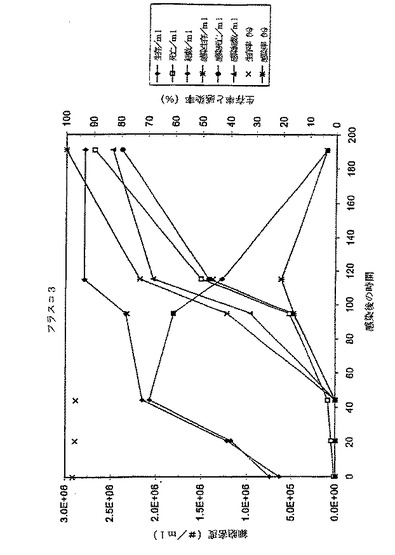
【図2−2】
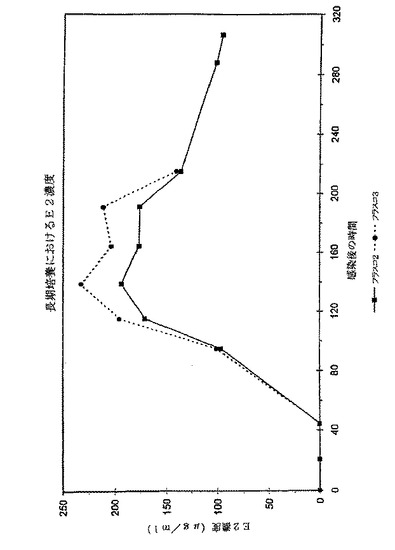
【図3】
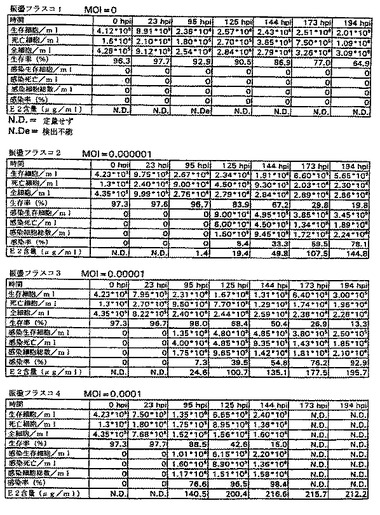
【図3−1】
【図3−2】
【図4】
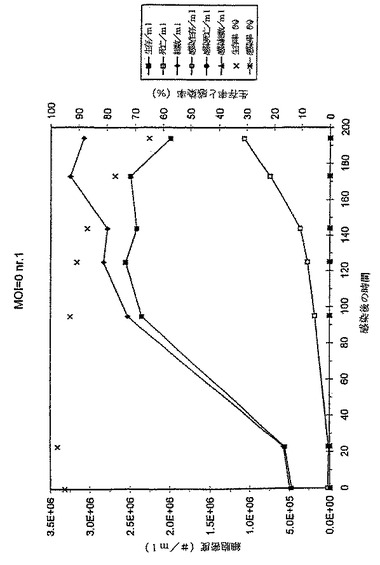
【図4−1】
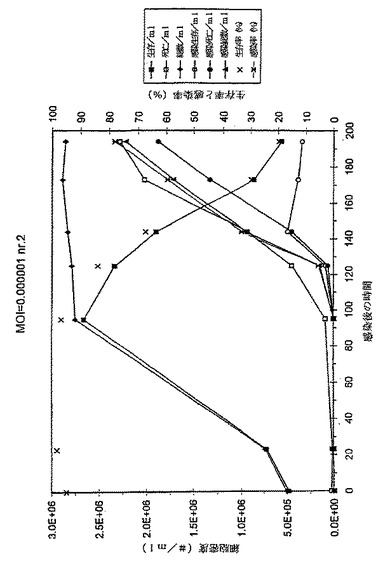
【図4−2】
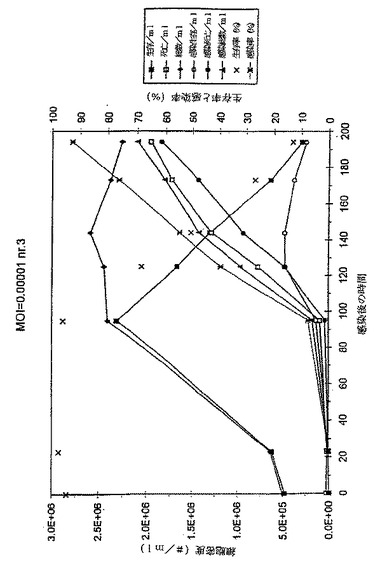
【図4−3】
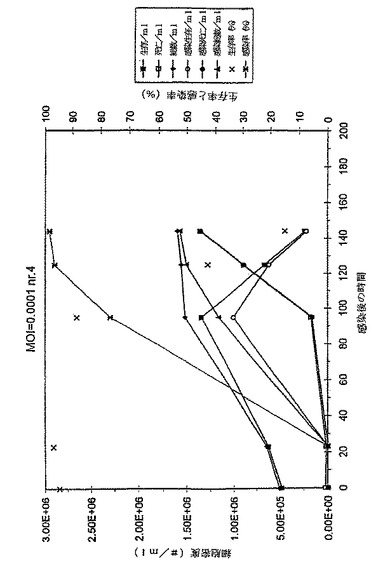
【図4−4】

【図4−5】

【図4−6】
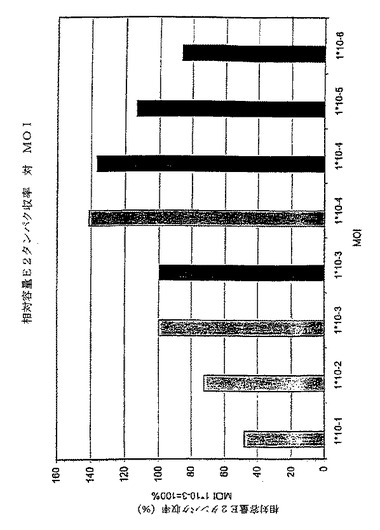
【図2】
【図2−1】
【図2−2】
【図3】
【図3−1】
【図3−2】
【図4】
【図4−1】
【図4−2】
【図4−3】
【図4−4】
【図4−5】
【図4−6】
【公開番号】特開2007−254482(P2007−254482A)
【公開日】平成19年10月4日(2007.10.4)
【国際特許分類】
【出願番号】特願2007−122898(P2007−122898)
【出願日】平成19年5月7日(2007.5.7)
【分割の表示】特願2000−539155(P2000−539155)の分割
【原出願日】平成10年12月17日(1998.12.17)
【出願人】(500343887)スティヒティング インスティチュート フォール ディールハウデレイ エン ディールゲゾントヘイト (1)
【氏名又は名称原語表記】Stichting Instituut voor Dierhouderij en Diergezondheid
【住所又は居所原語表記】Edelhertweg 15,Lelystad NETHERLANDS
【出願人】(500343898)バイエル アクチエンゲゼルシャフト (1)
【氏名又は名称原語表記】Bayer Aktiengesellschaft
【住所又は居所原語表記】Konzernbereich RP,Patente und Lizenzen,Leverkusen GERMANY
【Fターム(参考)】
【公開日】平成19年10月4日(2007.10.4)
【国際特許分類】
【出願日】平成19年5月7日(2007.5.7)
【分割の表示】特願2000−539155(P2000−539155)の分割
【原出願日】平成10年12月17日(1998.12.17)
【出願人】(500343887)スティヒティング インスティチュート フォール ディールハウデレイ エン ディールゲゾントヘイト (1)
【氏名又は名称原語表記】Stichting Instituut voor Dierhouderij en Diergezondheid
【住所又は居所原語表記】Edelhertweg 15,Lelystad NETHERLANDS
【出願人】(500343898)バイエル アクチエンゲゼルシャフト (1)
【氏名又は名称原語表記】Bayer Aktiengesellschaft
【住所又は居所原語表記】Konzernbereich RP,Patente und Lizenzen,Leverkusen GERMANY
【Fターム(参考)】
[ Back to top ]