
バリオールアミンの立体選択的合成を行うための物質及び方法
【課題】高純度のバリオールアミンを大量調製する実用的な方法を提供する。
【解決手段】本発明は、バリオールアミンを調製するための中間化合物及びその調製方法に関する。この中間化合物は、下記の式(21)に示される化学構造式を有する。異性体を含む式(14)の化合物をC−アシル化することにより式(21)の二環性カルバミン酸塩誘導体を生成させ、その後、式(21)の化合物からのR基除去及び/又は加水分解により、99%以上の純度を有するバリオールアミンを得る。本発明で得られた高純度のバリオールアミンは、高純度の糖尿病治療薬ボグリボースの調製に更に用いることができる。得られたボグリボースは、HPLCによる測定で99.5%以上の純度を有する。

【解決手段】本発明は、バリオールアミンを調製するための中間化合物及びその調製方法に関する。この中間化合物は、下記の式(21)に示される化学構造式を有する。異性体を含む式(14)の化合物をC−アシル化することにより式(21)の二環性カルバミン酸塩誘導体を生成させ、その後、式(21)の化合物からのR基除去及び/又は加水分解により、99%以上の純度を有するバリオールアミンを得る。本発明で得られた高純度のバリオールアミンは、高純度の糖尿病治療薬ボグリボースの調製に更に用いることができる。得られたボグリボースは、HPLCによる測定で99.5%以上の純度を有する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、製薬中間化合物及びその調製方法に関し、特にバリオールアミン(valiolamine)の中間化合物及びその調製方法に関する。
【背景技術】
【0002】
バリオールアミン(1)は、(1S)−(1(ヒドロキシ),2,4,5/1,3)−5−アミノ−1−C−ヒドロキシメチル−1,2,3,4−シクロヘキサンテトロールとして化学的に知られ、α−D−グルコシダーゼに対して強い阻害活性を有する(J.Antibiot.,1984,37,1301−1307)。
【化1】
【0003】
その誘導体もα−D−グルコシダーゼに対して強い阻害活性を持ち、このうちのいくつかの化合物は既に薬剤として開発され、ボグリボース(2)のように臨床治療における糖尿病治療の重要な薬物となっている(J.Med.Chem.,1986,29,1038−1046)。バリオールアミンは、ボグリボース調製における重要な中間体である(反応経路1)(EP56194,1982)。
【化2】
これを反応経路1とする。
【0004】
現在、バリオールアミンの調製方法は大きく2つに分けられ、1つは生合成に基づいた方法、そしてもう1つは全化学合成による方法である。
【0005】
生合成に基づいた方法は、最初はstreptomyces hygroscopicus subsp. Limoneus IFO 12703を用いた微生物発酵を利用して、直接バリオールアミンを生み出すものであった(J.Antibiot.,1984,37,1301−1307)。しかし、その後、微生物によるバリダマイシンAの分解によりバリエナミン及びバリダミンが生み出され得ることが見いだされ(特開昭57−054593号公報、特開昭58−152496号公報、特開昭62−181793号公報、WO2005098014)、そしてアカルボース及びその誘導体からのバリエナミン調製法が開示された(WO2006107134、WO2004000782、WO2005151967)。バリエナミン及びバリダミンは、更にバリオールアミンへと変換され得る(特開昭57−179174号公報、CN1683320、特開昭58−046044号公報)。
【0006】
反応経路2は、特開昭57−179174号公報で開示されたバリエナミン(3)からバリオールアミン(1)を調製する際の反応経路図である。
【化3】
これを反応経路2とする。
【0007】
反応経路3に示されるように、CN1683320は、バリエナミン(3)からバリオールアミン(1)を調製する別の方法を開示している。
【化4】
これを反応経路3とする。
【0008】
この方法は、まずバリエナミン(3)をアセチル化することにより化合物(7)を生成させ、次いで選択的エポキシ化により化合物(8)を生成させ、最後に開環及び脱アセチル化によりバリオールアミン(1)を生成させるものである。反応経路2及び反応経路3は共に、原料として発酵により生産されたバリエナミン(3)を利用するが、バリエナミンの発酵的生産には特定の細菌しか利用できないため、大規模に応用することができない。また、発酵液の面倒な後処理、低収率、多量の廃液に対する環境処分要求により、工業化する利点に欠ける。
【0009】
反応経路4は、多段階反応によるバリダミン(9)からのバリオールアミン(1)の調製経路である。
【化5】
これを反応経路4とする。
【0010】
この合成経路において、バリオールアミン(1)は、9つの化学反応ステップを経て調製される。このような長い反応経路は、低収率、高コスト、大量生産における製品の乏しい競争力、及び低工業的価値を導く。
【0011】
これに対し、全化学合成に基づく方法は、短時間で大量に生産することができ、条件が制御しやすく、高収率、低コスト、そして明らかな工業的利点を備える。初期の合成方法は、文献J.Org.Chem.1992,57,3651−3658に開示されており、化合物(12)とヒドロキシルアミンとを縮合させることによりオキシム(13)を生成するステップと、その後それを水素化させることにより5’アノマーを含む粗テトラベンジルバリオールアミンを形成するステップと、ベンジル基を脱離させることによりバリオールアミン(1)及びその5’アミノアノマー(15)を生成するステップとを含む。ここで、アノマー(15)の含有量は、6%程度である。純粋なバリオールアミン(1)を得るためには、綿密なカラムクロマトグラフィ単離が必要とされ、これが大量工業生産を行う際の障壁となる。
【化6】
これを反応経路5とする。
【0012】
特開2003−146957号公報は、反応経路6に示されるように、グルコース(16)から出発して、10ステップ以上を経てバリオールアミン(1)を調製する方法を開示している。
【化7】
これを反応経路6とする。
【0013】
この反応経路は、低収率及び高コストであるため、その工業製品は低競争力なものとなる。
【0014】
WO2005/049547及びWO2005/092834は、バリオールアミン(1)の合成方法を開示している。この方法は、反応経路7に示されるように、化合物(12)と酢酸アンモニウムとを還元アミノ化反応させることにより粗テトラベンジルバリオールアミン(14’)を生成させるステップと,その後ベンジル基を除去することによりバリオールアミン(1)を生成させるステップとを含む。
【化8】
これを反応経路7とする。
【0015】
この方法により得られたテトラベンジル−バリオールアミン(14’)は、僅か88%の純度しか持たないため、更なるベンジル基の除去の後、より純度の高いバリオールアミン(1)を得るためにカラムクロマトグラフィ単離を行う必要がある。これにより、純度を98.7%まで向上することができる。
【0016】
バリオールアミンは、1つの反応ステップによりボグリボースの調製に利用することができるため、バリオールアミンの品質は、最終産物であるボグリボースの品質に直接影響を与える。そのため、高純度のボグリボースの調製は、高度に精製されたバリオールアミンを必要とする。バリオールアミンの現在の調製方法の調査より、生合成に基づく方法は、原材料が簡単に入手できないという不利な点をもつため、この方法の普及及び利用は制限される。また、原材料の調製は発酵工程を必要とするため、大規模な発酵、低収率、不安定な菌種、面倒で複雑な分離処理、きつい労働、大量の廃液、環境汚染といった不利な点を有する。これに対し、全化学合成に基づく方法は、短いサイクル過程、大量生産の容易さ、低コスト、比較的清浄で安全、及び制御しやすいといった利点を有する。
【発明の開示】
【発明が解決しようとする課題】
【0017】
しかしながら、既存の全化学合成方法には、長い反応過程や低い産物純度(特に、低純度の問題)、5’アノマー除去の難しさ、分離及び精製の困難さといった欠点がある。従って、高純度のバリオールアミンを大量に調製する実用的な方法が、依然として早急に求められている。
【課題を解決するための手段】
【0018】
本発明は、高純度なバリオールアミン(1)を調製するための中間化合物を提供する。この化合物は、下記の構造式を有する。
【化9】
【0019】
式中、Rはヒドロキシル保護基であり、例えば、ベンゾイル基、ベンジル基、t−ブチルジメチルシリル基であり、好ましくはベンジル基である。
【0020】
特に、Rがベンジル基である場合、前記化合物は、下記式(21’)のテトラベンジル−バリオールアミン−1,5−カルバミン酸塩となり、その化学名称は、(1S,5S,6S,7R,8S)6,7,8−トリ−O−ベンジル−1−ベンジルオキシメチル−3−オキソ−2−オキサ−4−アザビシクロ[3.3.1]ノナンとなる。
【化10】
【0021】
更に、本発明は、式(21)の化合物を調製する方法を提供する。すなわち、異性体を含有する式(14)の化合物は、C−アシル化を受けることにより、式(21)の二環性化合物バリオールアミン−1,5−カルバミン酸塩誘導体を生成する。
【化11】
【0022】
この調製法は、更に再結晶化により式(21)の化合物を精製することにより、その結晶産物を得るステップを含む。
【0023】
例えば、異性体を含有する式(14’)の化合物テトラベンジル−バリオールアミンは、C−アシル化を受けることにより、式(21’)の二環性化合物テトラベンジル−バリオールアミン−1,5−カルバミン酸塩を生成する。
【化12】
【0024】
式(21’)の化合物は、更に再結晶化により精製され、式(21’)の化合物の結晶を与える。
【0025】
従って、本発明のもう一つの目的は、式(21’)の化合物の結晶性形態を提供することである。この結晶は、下記の物理的特性を備える。
粉末X線回折(Cu)において、2θ=4.70±0.2°、6.52±0.2°、8.38±0.2°、9.40±0.2°、13.06±0.2°、15.64±0.2°、16.14±0.2°、16.82±0.2°、18.40±0.2°、20.60±0.2°、22.64±0.2°、23.24±0.2°、24.02±0.2°、25.36±0.2°、及び27.12±0.2°に特徴的なピークを有し、
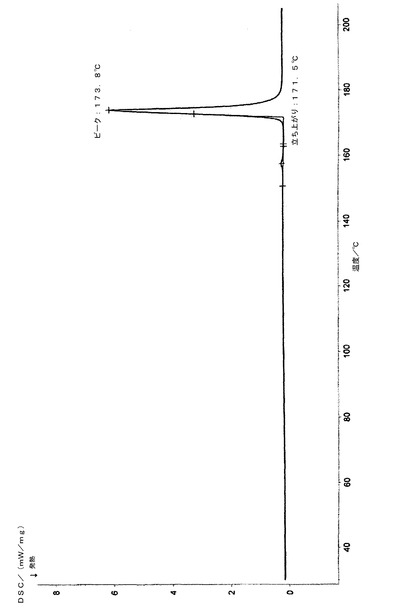
示差走査熱量測定(DSC)分析において、約173.8℃に特徴的な吸収ピークを持ち、
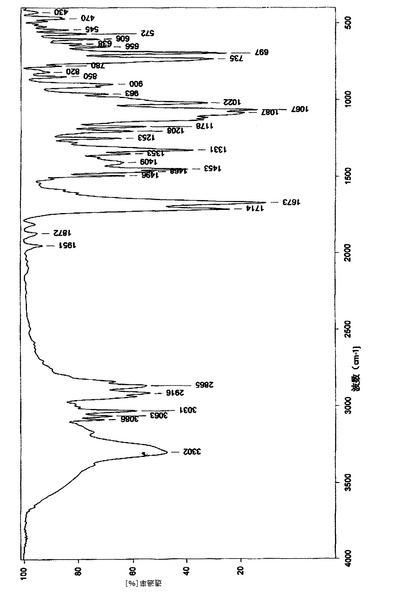
赤外線(IR)スペクトル分析において、図4に示されるような赤外線スペクトルを与え、
168〜172℃の融点を有する。
【0026】
本発明の更にもう一つの目的は、式(21)の化合物を用いて、高純度のバリオールアミン(1)を調製する方法を提供することである。この方法は、式(21)の化合物及び/又はその結晶を加水分解することにより式(22)の化合物を生成させるステップと、R基を除去することにより高純度のバリオールアミン(1)を得るステップとを備えた、又は、式(21)の化合物及び/又はその結晶のR基を除去することにより式(6)の化合物を生成させるステップと、それを加水分解してバリオールアミン(1)を得るステップとを備えたものである。
【0027】
その反応経路は下記の通りである。
【化13】
これを反応経路8とする。
【0028】
式中、R基はヒドロキシル保護基であり、例えば、ベンゾイル基、ベンジル、t−ブチルジメチルシリル基であり、好ましくはベンジル基である。
【0029】
本発明によれば、バリオールアミン(1)の前駆体化合物(21)を立体選択的に合成した後、R基を除去してから加水分解、又は加水分解をしてからR基の除去を行い、高純度のバリオールアミン(1)を得ることができる。本発明により得られた高純度のバリオールアミンは、高純度のボグリボース(2)の調製に利用することができる。従って、本発明は、ボグリボース(2)の調製方法も提供することになる。その反応経路は、下記の通りである。
【化14】
これを反応経路9とする。
【0030】
例えば、異性体を含有するテトラベンジル−バリオールアミン(14’)は、C−アシル化を受けることにより、二環性化合物(21’)、すなわち、テトラベンジル−バリオールアミン−1,5−カルバミン酸塩(化学名称:(1S,5S,6S,7R,8S)6,7,8−トリ−O−ベンジル−1−ベンジルオキシメチル−3−オキソ−2−オキサ−4−アザビシクロ[3.3.1]ノナン)を与える。この環状化合物(21’)は、まず加水分解によって開環されることによりテトラベンジル−バリオールアミン(22’)を生成させた後、ベンジル基の除去により高純度のバリオールアミン(1)を与える、又は、テトラベンジル−バリオールアミン−1,5−カルバミン酸塩(21’)は、まず脱ベンジル化されることにより化合物(6)を生成させた後、加水分解されてバリオールアミン(1)を与える。この反応経路は、下記の通りである。
【化15】
これを反応経路10とする。
【0031】
本発明の重要な特色は、バリオールアミン(1)が鍵となる中間体(21)を介して調製されることである。式(21)の二環性化合物の形成により、1’(1−C)のS配置ヒドロキシル基は、カルボニル基を介して5’(5−C)のS配置アミノ基と連結される。これにより、生成された二環性化合物(21)は、非常に安定な結晶となり、極めて容易に単離及び精製される。しかしながら、5’のR配置アミノ基は、1’のS配置ヒドロキシル基と連結され得ないため、環状化合物を形成することができない。その結果、式(21)の化合物の結晶化精製過程において、5’にR配置アミノ基を有するアノマー誘導体及び他の不純物は徹底的に除かれ、高純度の環状化合物(21)を得るという精製目的を達成することができる。
【0032】
本発明では、式(14)の化合物と対応するC−アシル化試薬とを反応させることにより二環性化合物(21)の高純度結晶を得て、これによりバリオールアミン前駆体の立体選択的合成という目的を達し、その後、R基及びカルボニル保護基を除去することにより、高純度のバリオールアミン(1)を得る。この反応により生成されたバリオールアミンは、不純物が少なく、特に、5’アミノアノマー(15)を含有していない。
【0033】
特に、異性体を含有し得る式(14)の化合物は、ホスゲン、トリクロロメチルクロロギ酸エステル、ビス(トリクロロメチル)炭酸塩のようなC−アシル化試薬と反応される。これにより、式(14)の化合物は、1’(1−C)のS配置ヒドロキシル基と、5’(5−C)のS配置アミノ基とがカルボニル基により分子内縮合で結ばれ、二環性化合物(21)を与える。一方、5’にR配置のアミノ基を持つアノマーは、そのような分子内縮合反応を行うことができない。この反応は立体選択的であり、得られた二環性化合物(21)は、容易に結晶化及び精製され、他の不純物、アノマー、及びその誘導体から分離され得る。これにより、高い純度を有するバリオールアミン前駆体を調製することが可能となる。
【0034】
不純物を含む又は異性体を含有し得る式(14)の化合物は、例えば、対応するアジ化水素酸塩(20)の還元(特開2003−146957号公報)、又は化合物(12)の還元アミノ化(WO2005/049547、WO2005/092834)、又は化合物(12)のオキシム化及びその後の還元(J.Org.Chem.1992,57,3651−3658)といった、先行文献に開示された方法に従って調製され得る。これらの方法を用いて得られた式(14)の化合物は、カラムクロマトグラフィのような他の手段により精製する必要はなく、本発明に直接用いることができる。
【0035】
得られた不純物を含む化合物(14)(例えば、化合物(14’))は、非プロトン性極性溶媒又は非極性溶媒に溶解される。ここで、非プロトン性極性溶媒は、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、テトラヒドロフラン、1,4−ジオキサン、エチルエーテル、ジイソプロピルエーテル、ジエチレングリコールジメチルエーテル、酢酸エチル、アセトン、2−ブタノン、シクロヘキサノン、ジメチルスルホキシド、又はこれらの混合物より構成される群から選択される1つ又は複数の種類の溶媒である。非極性溶媒は、四塩化炭素、シクロヘキサン、ヘキサンより構成される群から選択される。その後、有機塩基でも無機塩基でもよい塩基が加えられる。有機塩基は、トリエチルアミン、N−メチルモルホリン、ピリジン、及び4−ジメチルアミノピリジンより構成される群から選択され、無機塩基は、炭酸カリウム、炭酸ナトリウム、重炭酸ナトリウム、水酸化カリウム、水酸化ナトリウム、水酸化リチウム、及び炭酸リチウムより構成される群から選択される。−20〜50℃の温度範囲において、非プロトン性極性溶媒又は非極性溶媒中のC−アシル化試薬は、一滴ずつ添加される。対応するC−アシル化試薬は、ホスゲン、トリクロロメチルクロロギ酸エステル、及びビス(トリクロロメチル)炭酸塩より構成される群から選択され、好ましくは固体であり、計量が容易で、かつ危険性の低いビス(トリクロロメチル)炭酸塩である。非プロトン性極性溶媒は、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、テトラヒドロフラン、1,4−ジオキサン、エチルエーテル、ジイソプロピルエーテル、ジエチレングリコールジメチルエーテル、酢酸エチル、アセトン、2−ブタノン、シクロヘキサノン、ジメチルスルホキシド、又はこれらの混合物より構成される群から選択される1つ又は複数の種類の溶媒であり、非極性溶媒は、四塩化炭素、シクロヘキサン、又はヘキサンである。非プロトン性極性溶媒が好ましく、ジクロロメタン、ジイソプロピルエーテル、テトラヒドロフランがより好ましい。C−アシル化試薬の添加後、反応は同じ条件で1〜24時間、好ましくは8〜10時間行われ、反応終了後、反応液は通常の方法により処理される。例えば、反応液は水に注入され、有機溶媒で抽出される。対応する有機溶媒は、クロロホルム、ジクロロメタン、酢酸エチル、エチルエーテル、イソプロピルエーテル、ブタノン、又はシクロヘキサノン等の非プロトン性極性溶媒である。その後、抽出液は、水、2N塩酸溶液、5%炭酸ナトリウム水性溶液、飽和塩水で連続的に洗浄され、無水硫酸ナトリウムを用いて乾燥される。抽出溶媒は回収され、得られた固体が粗環状産物(21’)である。
【0036】
粗環状産物(21’)は、再結晶化により効率的に精製され、純度99%以上の白色結晶を与える。再結晶化の溶媒は、メタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノール、イソブタノールのようなC1〜C5の低級脂肪族アルコール、又はエーテル、イソプロピルエーテル、テトラヒドロフラン、1,4−ジオキサンのようなエーテル類、又は酢酸エチル、酢酸メチルのようなエステル類、又はアセトン、ブタノン、シクロヘキサノンのようなケトン類であり、これらの中から1つ又は複数選択される。
【0037】
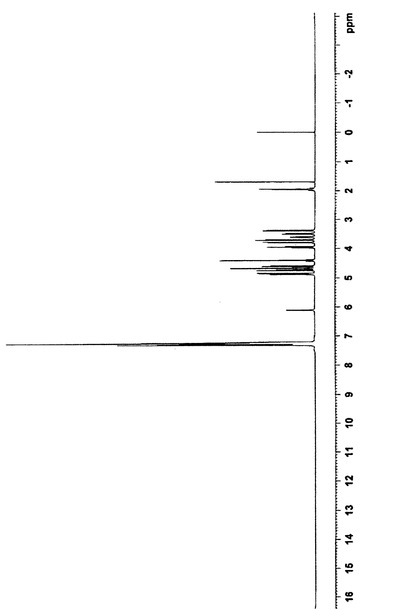
得られた産物(21’)は、白色針状の結晶形態を持ち、168〜172℃の融点を有し、その構造は、1H−NMRスペクトル、13C−NMRスペクトル、赤外線(IR)スペクトル、マススペクトル、元素分析、比旋光度、及び示差走査熱量測定分析により確認された。
【0038】
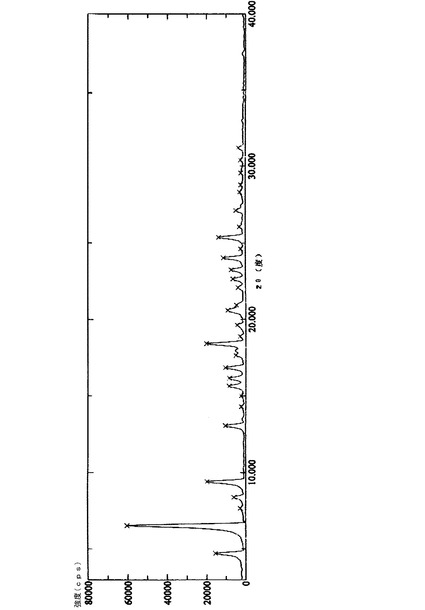
更に、この結晶の特性は、その粉末のX線回折パターンにより調べることができる。具体的には、この結晶は、X線回折パターンにおいて2θ=4.70±0.2°、6.52±0.2°、9.40±0.2°、18.40±0.2°、24.02±0.2°、及び25.36±0.2°にX線回折ピークを与え、更に2θ=8.38±0.2°、13.06±0.2°、15.64±0.2°、16.14±0.2°、16.82±0.2°、20.60±0.2°、22.64±0.2°、23.24±0.2°、及び27.12±0.2°にもX線回折ピークを与える。
【0039】
式(22)の化合物は、化合物(21’)結晶のアルカリ媒体中での加水分解のように、精製された結晶性産物の塩基性加水分解により得られる。アルカリ媒体中の塩基は、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化カルシウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウムのような無機塩基であってもよい。溶媒は、10%〜90%(v/v)濃度のメタノール、エタノール、イソプロパノールのようなC1〜C5の短鎖脂肪族アルコールを含む水性溶液であってもよい。使用される媒体は、10%〜90%(v/v)の濃度のアセトン、テトラヒドロフラン、1,4−ジオキサン、DMF(N,N−ジメチルホルムアミド)、DME(N,N−ジメチルアセトアミド)の水性溶液であってもよく、好ましくはC1〜C5の短鎖脂肪族アルコールの水性溶液であり、より好ましくは75〜80%(水:エタノール)(v/v)濃度のエタノール、イソプロパノールである。反応温度は室温から還流温度、反応時間は2〜48時間であり、好ましくは24時間である。反応終了点は、薄層クロマトグラフィ(TLC)により調べられ、原材料がなくなる点が反応終了点とされる。反応終了後、反応液は、一般的な方法で処理される。例えば、反応液を氷水に注入し、有機溶媒で抽出する。このとき、対応する有機溶媒は、クロロホルム、ジクロロメタン、酢酸エチル、エチルエーテル、イソプロピルエーテル、ブタノン、シクロヘキサノンのような非プロトン性極性溶媒であり、抽出液の洗浄は、水、2N塩酸溶液、5%炭酸ナトリウム水性溶液、飽和塩水で連続的に行われ、無水硫酸ナトリウムにより乾燥された後、抽出溶媒が回収され、得られた無色の油状物質がテトラベンジル−バリオールアミン(22’)である。別の方法としては、まず反応液を濃縮した後、得られたものを有機溶媒及び水で層抽出し、抽出液を水、2N塩酸溶液、5%炭酸ナトリウム水性溶液、飽和塩水で連続的に洗浄し、無水硫酸ナトリウムを用いて乾燥した後、抽出溶媒を回収する。このとき、対応する有機溶媒は、クロロホルム、塩化メチレン、トルエン、エチルエーテル、イソプロピルエーテル等であり、好ましくはトルエン、塩化メチレンである。得られた無色の粘性の高い油が式(22’)の化合物であり、この物質は次の反応に直接用いることができる。
【0040】
バリオールアミン(1)は、脱ベンジル化反応により式(22’)の化合物から調製され得る。特に、式(22’)の化合物は、液体アンモニア/金属リチウムによる脱ベンジル化(Synthesis,1999,(4),571−573参照)、又は三臭化ホウ素による脱ベンジル化(J.Am.Chem.Soc.,1999,121,6973−6983参照)、又はパラジウム−炭素/水素による脱ベンジル化(WO2005/049547;J.Am.Chem.Soc.,1999,121,6973−6983参照)、又はパラジウムブラック/ギ酸による脱ベンジル化(J.Org.Chem.1992,57,3651−3658参照)により脱ベンジル化される。これらの方法により簡単にベンジル基は除去され、バリオールアミンを得ることができる。
【0041】
本発明の方法により得られたバリオールアミン(1)は、白色結晶としてメタノールから結晶化される。その構造は、1H−NMRスペクトル、13C−NMRスペクトル、赤外線(IR)スペクトル、マススペクトル、及び比旋光度により確認される。その純度は、高圧液体クロマトグラフィ(HPLC)による測定で>99%であり、とりわけ、99.5%以上に達し得る。
【0042】
また、バリオールアミン(1)は、本発明により与えられた化合物(21)のR基(例えば、ベンジル基)の除去及びその後の加水分解を含む反応経路により調製され得る(反応経路8参照)。例えば、脱ベンジル化の方法は、液体アンモニア/金属リチウム(Synthesis,1999,(4),571−573参照)、又は三臭化ホウ素(J.Am.Chem.Soc.,1999,121,6973−6983参照)、又はパラジウム−炭素/水素(WO2005/049547;J.Am.Chem.Soc.,1999,121,6973−6983参照)、又はパラジウムブラック/ギ酸(J.Org.Chem.1992,57,3651−3658参照)を利用して行われ得る。本発明は、パラジウム−炭素/水素を用いた脱ベンジル化法を提供する。すなわち、環状化合物(21’)をエタノール/テトラヒドロフラン(1:1,v/v)に溶解し、10%パラジウム−炭素を加え、10気圧水素の存在下で10時間反応させ、脱ベンジル化産物(6)を得る。化合物(6)は、更に加水分解されて、バリオールアミン(1)を生成させる。このときの特別な加水分解条件に関しては、文献J.Med.Chem.,1986,29,1038−1046、又は米国特許US4803303を参照することができる。本発明においては、加水分解は水酸化バリウムの水性溶液中で行われ、8時間還流される。この反応の終了後、pHがほぼ中性となるまで二酸化炭素ガスが添加される。過剰な苛性バリタを除去し、炭酸バリウム沈殿をろ過した後、水性溶液は濃縮され、メタノールを加えることにより白色結晶状のバリオールアミンを分離させる。
【0043】
上記の脱ベンジル化及びその後の加水分解を含む方法により調製されたバリオールアミンは、加水分解及びその後の脱ベンジル化により得られたバリオールアミンと同一である。その純度は、HPLCによる測定で99%以上であり、とりわけ、99.5%以上に達する。
【発明の効果】
【0044】
本発明で調製された高純度のバリオールアミンは、更に高純度の糖尿病治療薬ボグリボースの調製に用いることができる。得られたボグリボースは、HPLCによる測定で99.5%以上の純度を有し、とりわけ、99.8%以上に達する。
【図面の簡単な説明】
【0045】
【図1】式(21’)の化合物の結晶の粉末X線回折パターンを示す図。
【図2】式(21’)の化合物の結晶の粉末X線回折パターンを示す図。
【図3】式(21’)の化合物の結晶の粉末X線回折パターンを示す図。
【図4】式(21’)の化合物の結晶の赤外線(IR)スペクトルを示す図。
【図5】式(21’)の化合物の結晶の1H−NMRスペクトルを示す図。
【図6】式(21’)の化合物の結晶の示差走査熱量曲線を示す図。
【発明を実施するための形態】
【0046】
検出器:
核磁気共鳴装置
核磁気共鳴装置は、Bruker社製AVANCE AV500超電導核磁気共鳴装置である。
元素分析装置
元素分析装置は、ELEMENTAR社製vario EL元素分析装置である。
旋光計
POLARTRONIC HHW5旋光計
粉末X線回折
粉末X線回折装置は、Rigaku社(日本)のD/max−rC型である。
X線は、銅ターゲット(銅ターゲットアノードを備えたX線管)より発せられる。
発散スリット1°、受光スリット0.15mm、散乱スリット1°、管電圧40KV、管電流50mA
示差走査熱量測定
示差走査熱量測定器は、NETZSCH DSC204型である。
試料重量:3.11mg
温度範囲:30〜200℃
加熱速度:10℃/min
赤外線(IR)スペクトル
赤外線(IR)分光計は、Bruker EQUINOX55型であり、臭化カリウム錠剤法による測定を採用している。
【0047】
(実施例1)粗テトラベンジル−バリオールアミン(14’)の調製
J.Org.Chem.1992,57,3651−3658の方法を参照し、50gの化合物(12)、100gの塩酸ヒドロキシルアミン、及び50gの無水酢酸ナトリウムを1000mlのメタノール中で混合し、一晩撹拌しながら反応させた。次いで、反応液を濃縮し、2000mlの酢酸エチル及び500mlの水により層分離させた。酢酸エチル層を2N塩酸溶液、飽和重炭酸ナトリウム溶液、及び飽和塩水で連続的に洗浄し、無水硫酸ナトリウムを用いて乾燥させた。酢酸エチルを回収し、得られた無色の油を500mlのメタノールに溶解し、15gのラネーニッケルを加え、10気圧水素下で5時間水素化させた。反応液をろ過し、そのろ液を濃縮して50gの赤褐色の油、すなわち、産物(14’)を得た。産物(14’)は、次の反応に直接用いることができる。
【0048】
(実施例2)テトラベンジルバリオールアミン−1,5−カルバミン酸塩(21’)の調製
上で得られた50gの化合物(14’)を500mlの二塩化メチレンに溶解し、100mlのトリエチルアミンを加え、12gのビス(トリクロロメチル)炭酸塩をゆっくり滴下した後、2時間撹拌しながら反応させた。反応液を氷水に注入し、二塩化メチレン層を分離し、2N塩酸溶液、5%炭酸ナトリウム溶液、及び飽和塩水で連続的に洗浄し、無水硫酸ナトリウムを用いて乾燥させた。二塩化メチレンを回収し、得られた結晶を化合物(21’)の粗産物とした。この粗産物を1000mlの無水エタノールで再結晶化させ、40gの白色針状結晶を得た。HPLC純度:99.8%、融点:170−172℃、1H−NMR(CDCl3,500MHz):δ1.92(2H,m,9−CH2),δ3.46(1H,dd,J=2.7,8.7Hz,6−CH),δ3.38(1H,d,J=8.9,1−CH2OBn),δ3.64(1H,m,5−CH),δ3.68(1H,d,J=9.4,8−CH),δ3.79(1H,d,J=8.9,1−CH2OBn),δ3.93(1H,d,J=9.1,7−CH),δ4.39(1H,d,J=11.7,1−OCH2Ph),δ4.42(1H,d,J=11.7,1−OCH2Ph),δ4.58(1H,d,J=11.5,8−OCH2),δ4.61(1H,d,J=12.0,6−OCH2),δ4.65(1H,d,J=10.8,7−OCH2),δ4.69(1H,d,J=11.4,6−OCH2),δ4.80(1H,d,J=10.8,7−OCH2),δ4.86(1H,d,J=11.4,8−OCH2),δ6.81(1H,d,J=4.5,−NH),δ7.15−7.35(20H,m,Ar−H)、13C−NMR(CDCl3,125MHz):δ27.5,47.5,71.2,72.1,73.3,75.2,75.9,81.8,81.9,82.4,83.2,137.5,137.9,138.4,138.5,153.2、[α]20D+34.0°(c1,CHCl3);元素分析:C36H37NO6,計算値(%)C,74.59;H,6.43;N,2.42;実験値(%)C,74.20;H,6.39;N,2.33;赤外線(IR)(KBr)ν3302,3086,3063,3031,2916,2865,1714,1673,1453,1331,1087,1067,735,697;MS(EI)m/e:580(M++1),579(M+.),488,91(100);粉末X線回折グラフを図1に示し、関連データを表1に示す。化合物の赤外線(IR)スペクトルを図4に示し、1H−NMRスペクトルを図5に示し、示差走査熱量測定パターンを図6に示す。
【表1】
【0049】
(実施例3)テトラベンジル−バリオールアミン−1,5−カルバミン酸塩(21’)の調製
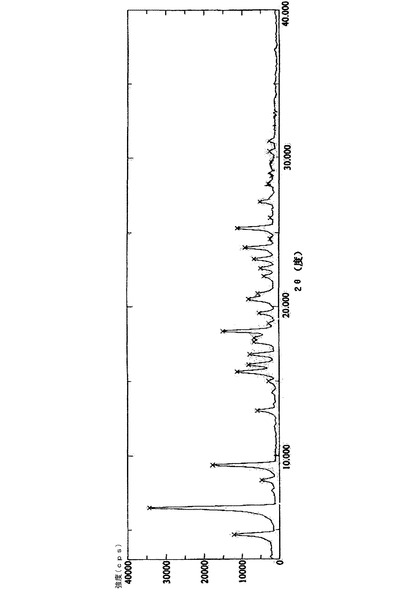
実施例1で得られた25gの化合物(14’)を250mlのクロロホルムに溶解し、60gの炭酸カリウムを加えた後、0℃で撹拌しながら5gのトリクロロメチルクロロギ酸エステルを一滴ずつゆっくり添加し、2時間撹拌しながら反応させた。反応液を氷水に注入し、クロロホルム層を分離し、それを2N塩酸溶液、5%炭酸ナトリウム溶液、及び飽和塩水で連続的に洗浄した後、無水硫酸ナトリウムを用いて乾燥させた。クロロホルムを回収し、得られた結晶を化合物(21’)の粗産物とした。この粗産物を800mlのメタノールを用いて再結晶化させ、約18gの白色針状結晶を得た。融点:169〜171℃、HPLC純度:99.7% その構造確認に関するデータは、実施例2のものと同一である。その粉末X線回折パターンを図2に示し、関連するデータを表2に示す。
【表2】
【0050】
(実施例4)テトラベンジルバリオールアミン−1,5−カルバミン酸塩(21’)の調製
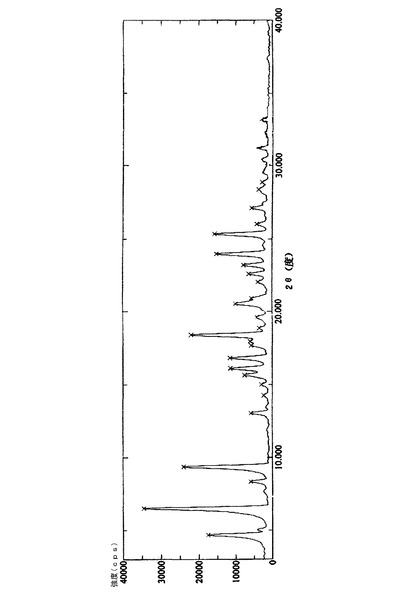
実施例1で得られた25gの化合物(14’)を250mlのクロロホルムに溶解し、50mlのピリジンを加え、−10℃で撹拌しながらホスゲンガスをゆっくりと加えた。薄層クロマトグラフィ(TLC)で開始物質(14’)の消失を確認した後、反応液を氷水に注入し、クロロホルム層を分離し、これを2N塩酸溶液、5%炭酸ナトリウム溶液、及び飽和塩水で連続的に洗浄した後、無水硫酸ナトリウムを用いて乾燥させた。クロロホルムを回収し、得られた結晶を化合物(21’)の粗産物とした。この粗産物を300mlのn−プロパノールを用いて再結晶化させ、約19gの白色針状結晶を得た。融点:168〜170℃、HPLC純度:99.6% その構造確認に関するデータは、実施例2のものと同一である。その粉末X線回折パターンを図3に示し、関連するデータを表3に示す。
【表3】
【0051】
(実施例5)バリオールアミンの調製
テトラベンジル−バリオールアミン(22’)の調製
126gの化合物(21’)、80gの水酸化ナトリウム、800mlの水、及び3400mlのエタノールを混合し、24時間還流させた。反応終了後、エタノールを回収し、それを1000mlトルエンで抽出した。そのトルエン層を、水、2N塩酸溶液、5%炭酸ナトリウム溶液、及び飽和塩水で連続的に洗浄し、無水硫酸ナトリウムを用いて乾燥させた。トルエンを回収し、得られた無色の粘性の高い油(約120g)を産物(22’)とした。この産物(22’)は、次の反応に直接用いることができる。
【0052】
バリオールアミンの調製
得られた約50gの油(22’)を1000mlのメタノールに溶解し、50mlの94%ギ酸、5gのパラジウムブラックを加え、撹拌しながら室温で18時間反応させ、反応液をろ過した。そのろ液を濃縮し、強酸性イオン交換樹脂カラムにロードし、水、次いで0.5Nアンモニア水で溶出した。溶出された液体を乾燥するまで濃縮し、メタノールを加えて白色結晶、すなわち、バリオールアミンを分離させた。このバリオールアミンは、更にメタノールを用いて再結晶化させることにより、HPLCによる測定で99.6%の純度を有する白色結晶とすることができる。
【0053】
(実施例6)バリオールアミンの調製
バリオールアミン−1,5−カルバミン酸塩(6)(化学名称:(1S,5S,6S,7R,8S)6,7,8−トリヒドロキシ−1−ヒドロキシメチル−3−オキソ−2−オキサ−4−アザビシクロ[3.3.1]ノナン)の調製
20gのテトラベンジルバリオールアミン−1,5−カルバミン酸塩(21’)を500mlのテトラヒドロフラン/エタノール(1:1)(v/v)に溶解し、5gの10%パラジウム/炭素を加え、室温、10気圧水素下で水素化脱ベンジル化反応を10時間行った。この反応終了後、反応液をろ過し、そのろ液を減圧下で濃縮し、反応溶媒を回収した。これをメタノールで再結晶化させ、約8gの白色結晶、すなわち、バリオールアミン−1,5−カルバミン酸塩(6)を得た。
【0054】
バリオールアミンの調製
5gのバリオールアミン−1,5−カルバミン酸塩を150mlの水に溶解し、20gの苛性バリタを加え、70〜80℃の温度に保持したまま撹拌しながら3時間反応させた。その反応液を20℃まで冷却し、30分間二酸化炭素を添加した。生成した沈殿を除去し、水で洗浄した。そのろ液及び洗液を合わせ、減圧下で蒸発させて水分を除いた。その産物をメタノールを用いて再結晶化させ、4gの白色粉末結晶状のバリオールアミンを得た。HPLC純度:99.8%
【0055】
(実施例7)ボグリボースの調製
上記実施例5で得られた2.0gのバリオールアミンを50mlのN,N−ジメチルホルムアミドに溶解し、3.4gの1,3−ジヒドロキシアセトン、1.5mlの2mol/L HCl、及び2.6gのシアノ水素化ホウ素ナトリウムを加え、撹拌しながら室温で16時間反応させた。その反応液を濃縮し、その濃縮物を100mlの水に溶解して、2mol/L HClで酸性化、30〜40分間撹拌して冷却した後、250mlのDowex 50W×8(H+)クロマトグラフィカラムへとロードした。水で洗浄した後、その産物を0.5mol/Lのアンモニア水で完全に溶出させた。その溶出液を減圧下で濃縮し、それを400mlの無水エタノール中で30分間還流し、少し冷却させてから0.2gの活性炭を加え、15分間還流した後ろ過した。そのろ液を室温まで冷却して、得られた白色粉末をろ過し、40℃で12時間真空乾燥させ、結晶性粉末として1.9gのボグリボースを得た。融点:164.1〜165.7℃、HPLC純度:99.8%
【0056】
(実施例8)ボグリボースの調製
上記実施例6で得られた3.0gのバリオールアミンを75mlのN,N−ジメチルホルムアミドに溶解し、5.1gの1,3−ジヒドロキシアセトン、2.3mlの2mol/L HCl、及び4gのシアノ水素化ホウ素ナトリウムを加え、撹拌しながら室温で16時間反応させた。その反応液を濃縮し、その濃縮物を150mlの水に溶解、2mol/L HClで酸性化させ、30〜40分間撹拌して冷却した後、375mlのDowex 50W×8(H+)クロマトグラフィカラムへとロードした。水で洗浄した後、その産物を0.5mol/Lのアンモニアで完全に溶出させた。その溶出液を減圧下で濃縮し、それを600mlの無水エタノール中で30分間還流し、少し冷却させてから0.3gの活性炭を加え、15分間還流した後ろ過した。そのろ液を室温まで冷却して、得られた白色粉末をろ過し、40℃で12時間真空乾燥させ、結晶性粉末として2.8gのボグリボースを得た。融点:164.3〜165.8℃、HPLC純度:99.9%
【技術分野】
【0001】
本発明は、製薬中間化合物及びその調製方法に関し、特にバリオールアミン(valiolamine)の中間化合物及びその調製方法に関する。
【背景技術】
【0002】
バリオールアミン(1)は、(1S)−(1(ヒドロキシ),2,4,5/1,3)−5−アミノ−1−C−ヒドロキシメチル−1,2,3,4−シクロヘキサンテトロールとして化学的に知られ、α−D−グルコシダーゼに対して強い阻害活性を有する(J.Antibiot.,1984,37,1301−1307)。
【化1】
【0003】
その誘導体もα−D−グルコシダーゼに対して強い阻害活性を持ち、このうちのいくつかの化合物は既に薬剤として開発され、ボグリボース(2)のように臨床治療における糖尿病治療の重要な薬物となっている(J.Med.Chem.,1986,29,1038−1046)。バリオールアミンは、ボグリボース調製における重要な中間体である(反応経路1)(EP56194,1982)。
【化2】
これを反応経路1とする。
【0004】
現在、バリオールアミンの調製方法は大きく2つに分けられ、1つは生合成に基づいた方法、そしてもう1つは全化学合成による方法である。
【0005】
生合成に基づいた方法は、最初はstreptomyces hygroscopicus subsp. Limoneus IFO 12703を用いた微生物発酵を利用して、直接バリオールアミンを生み出すものであった(J.Antibiot.,1984,37,1301−1307)。しかし、その後、微生物によるバリダマイシンAの分解によりバリエナミン及びバリダミンが生み出され得ることが見いだされ(特開昭57−054593号公報、特開昭58−152496号公報、特開昭62−181793号公報、WO2005098014)、そしてアカルボース及びその誘導体からのバリエナミン調製法が開示された(WO2006107134、WO2004000782、WO2005151967)。バリエナミン及びバリダミンは、更にバリオールアミンへと変換され得る(特開昭57−179174号公報、CN1683320、特開昭58−046044号公報)。
【0006】
反応経路2は、特開昭57−179174号公報で開示されたバリエナミン(3)からバリオールアミン(1)を調製する際の反応経路図である。
【化3】
これを反応経路2とする。
【0007】
反応経路3に示されるように、CN1683320は、バリエナミン(3)からバリオールアミン(1)を調製する別の方法を開示している。
【化4】
これを反応経路3とする。
【0008】
この方法は、まずバリエナミン(3)をアセチル化することにより化合物(7)を生成させ、次いで選択的エポキシ化により化合物(8)を生成させ、最後に開環及び脱アセチル化によりバリオールアミン(1)を生成させるものである。反応経路2及び反応経路3は共に、原料として発酵により生産されたバリエナミン(3)を利用するが、バリエナミンの発酵的生産には特定の細菌しか利用できないため、大規模に応用することができない。また、発酵液の面倒な後処理、低収率、多量の廃液に対する環境処分要求により、工業化する利点に欠ける。
【0009】
反応経路4は、多段階反応によるバリダミン(9)からのバリオールアミン(1)の調製経路である。
【化5】
これを反応経路4とする。
【0010】
この合成経路において、バリオールアミン(1)は、9つの化学反応ステップを経て調製される。このような長い反応経路は、低収率、高コスト、大量生産における製品の乏しい競争力、及び低工業的価値を導く。
【0011】
これに対し、全化学合成に基づく方法は、短時間で大量に生産することができ、条件が制御しやすく、高収率、低コスト、そして明らかな工業的利点を備える。初期の合成方法は、文献J.Org.Chem.1992,57,3651−3658に開示されており、化合物(12)とヒドロキシルアミンとを縮合させることによりオキシム(13)を生成するステップと、その後それを水素化させることにより5’アノマーを含む粗テトラベンジルバリオールアミンを形成するステップと、ベンジル基を脱離させることによりバリオールアミン(1)及びその5’アミノアノマー(15)を生成するステップとを含む。ここで、アノマー(15)の含有量は、6%程度である。純粋なバリオールアミン(1)を得るためには、綿密なカラムクロマトグラフィ単離が必要とされ、これが大量工業生産を行う際の障壁となる。
【化6】
これを反応経路5とする。
【0012】
特開2003−146957号公報は、反応経路6に示されるように、グルコース(16)から出発して、10ステップ以上を経てバリオールアミン(1)を調製する方法を開示している。
【化7】
これを反応経路6とする。
【0013】
この反応経路は、低収率及び高コストであるため、その工業製品は低競争力なものとなる。
【0014】
WO2005/049547及びWO2005/092834は、バリオールアミン(1)の合成方法を開示している。この方法は、反応経路7に示されるように、化合物(12)と酢酸アンモニウムとを還元アミノ化反応させることにより粗テトラベンジルバリオールアミン(14’)を生成させるステップと,その後ベンジル基を除去することによりバリオールアミン(1)を生成させるステップとを含む。
【化8】
これを反応経路7とする。
【0015】
この方法により得られたテトラベンジル−バリオールアミン(14’)は、僅か88%の純度しか持たないため、更なるベンジル基の除去の後、より純度の高いバリオールアミン(1)を得るためにカラムクロマトグラフィ単離を行う必要がある。これにより、純度を98.7%まで向上することができる。
【0016】
バリオールアミンは、1つの反応ステップによりボグリボースの調製に利用することができるため、バリオールアミンの品質は、最終産物であるボグリボースの品質に直接影響を与える。そのため、高純度のボグリボースの調製は、高度に精製されたバリオールアミンを必要とする。バリオールアミンの現在の調製方法の調査より、生合成に基づく方法は、原材料が簡単に入手できないという不利な点をもつため、この方法の普及及び利用は制限される。また、原材料の調製は発酵工程を必要とするため、大規模な発酵、低収率、不安定な菌種、面倒で複雑な分離処理、きつい労働、大量の廃液、環境汚染といった不利な点を有する。これに対し、全化学合成に基づく方法は、短いサイクル過程、大量生産の容易さ、低コスト、比較的清浄で安全、及び制御しやすいといった利点を有する。
【発明の開示】
【発明が解決しようとする課題】
【0017】
しかしながら、既存の全化学合成方法には、長い反応過程や低い産物純度(特に、低純度の問題)、5’アノマー除去の難しさ、分離及び精製の困難さといった欠点がある。従って、高純度のバリオールアミンを大量に調製する実用的な方法が、依然として早急に求められている。
【課題を解決するための手段】
【0018】
本発明は、高純度なバリオールアミン(1)を調製するための中間化合物を提供する。この化合物は、下記の構造式を有する。
【化9】
【0019】
式中、Rはヒドロキシル保護基であり、例えば、ベンゾイル基、ベンジル基、t−ブチルジメチルシリル基であり、好ましくはベンジル基である。
【0020】
特に、Rがベンジル基である場合、前記化合物は、下記式(21’)のテトラベンジル−バリオールアミン−1,5−カルバミン酸塩となり、その化学名称は、(1S,5S,6S,7R,8S)6,7,8−トリ−O−ベンジル−1−ベンジルオキシメチル−3−オキソ−2−オキサ−4−アザビシクロ[3.3.1]ノナンとなる。
【化10】
【0021】
更に、本発明は、式(21)の化合物を調製する方法を提供する。すなわち、異性体を含有する式(14)の化合物は、C−アシル化を受けることにより、式(21)の二環性化合物バリオールアミン−1,5−カルバミン酸塩誘導体を生成する。
【化11】
【0022】
この調製法は、更に再結晶化により式(21)の化合物を精製することにより、その結晶産物を得るステップを含む。
【0023】
例えば、異性体を含有する式(14’)の化合物テトラベンジル−バリオールアミンは、C−アシル化を受けることにより、式(21’)の二環性化合物テトラベンジル−バリオールアミン−1,5−カルバミン酸塩を生成する。
【化12】
【0024】
式(21’)の化合物は、更に再結晶化により精製され、式(21’)の化合物の結晶を与える。
【0025】
従って、本発明のもう一つの目的は、式(21’)の化合物の結晶性形態を提供することである。この結晶は、下記の物理的特性を備える。
粉末X線回折(Cu)において、2θ=4.70±0.2°、6.52±0.2°、8.38±0.2°、9.40±0.2°、13.06±0.2°、15.64±0.2°、16.14±0.2°、16.82±0.2°、18.40±0.2°、20.60±0.2°、22.64±0.2°、23.24±0.2°、24.02±0.2°、25.36±0.2°、及び27.12±0.2°に特徴的なピークを有し、
示差走査熱量測定(DSC)分析において、約173.8℃に特徴的な吸収ピークを持ち、
赤外線(IR)スペクトル分析において、図4に示されるような赤外線スペクトルを与え、
168〜172℃の融点を有する。
【0026】
本発明の更にもう一つの目的は、式(21)の化合物を用いて、高純度のバリオールアミン(1)を調製する方法を提供することである。この方法は、式(21)の化合物及び/又はその結晶を加水分解することにより式(22)の化合物を生成させるステップと、R基を除去することにより高純度のバリオールアミン(1)を得るステップとを備えた、又は、式(21)の化合物及び/又はその結晶のR基を除去することにより式(6)の化合物を生成させるステップと、それを加水分解してバリオールアミン(1)を得るステップとを備えたものである。
【0027】
その反応経路は下記の通りである。
【化13】
これを反応経路8とする。
【0028】
式中、R基はヒドロキシル保護基であり、例えば、ベンゾイル基、ベンジル、t−ブチルジメチルシリル基であり、好ましくはベンジル基である。
【0029】
本発明によれば、バリオールアミン(1)の前駆体化合物(21)を立体選択的に合成した後、R基を除去してから加水分解、又は加水分解をしてからR基の除去を行い、高純度のバリオールアミン(1)を得ることができる。本発明により得られた高純度のバリオールアミンは、高純度のボグリボース(2)の調製に利用することができる。従って、本発明は、ボグリボース(2)の調製方法も提供することになる。その反応経路は、下記の通りである。
【化14】
これを反応経路9とする。
【0030】
例えば、異性体を含有するテトラベンジル−バリオールアミン(14’)は、C−アシル化を受けることにより、二環性化合物(21’)、すなわち、テトラベンジル−バリオールアミン−1,5−カルバミン酸塩(化学名称:(1S,5S,6S,7R,8S)6,7,8−トリ−O−ベンジル−1−ベンジルオキシメチル−3−オキソ−2−オキサ−4−アザビシクロ[3.3.1]ノナン)を与える。この環状化合物(21’)は、まず加水分解によって開環されることによりテトラベンジル−バリオールアミン(22’)を生成させた後、ベンジル基の除去により高純度のバリオールアミン(1)を与える、又は、テトラベンジル−バリオールアミン−1,5−カルバミン酸塩(21’)は、まず脱ベンジル化されることにより化合物(6)を生成させた後、加水分解されてバリオールアミン(1)を与える。この反応経路は、下記の通りである。
【化15】
これを反応経路10とする。
【0031】
本発明の重要な特色は、バリオールアミン(1)が鍵となる中間体(21)を介して調製されることである。式(21)の二環性化合物の形成により、1’(1−C)のS配置ヒドロキシル基は、カルボニル基を介して5’(5−C)のS配置アミノ基と連結される。これにより、生成された二環性化合物(21)は、非常に安定な結晶となり、極めて容易に単離及び精製される。しかしながら、5’のR配置アミノ基は、1’のS配置ヒドロキシル基と連結され得ないため、環状化合物を形成することができない。その結果、式(21)の化合物の結晶化精製過程において、5’にR配置アミノ基を有するアノマー誘導体及び他の不純物は徹底的に除かれ、高純度の環状化合物(21)を得るという精製目的を達成することができる。
【0032】
本発明では、式(14)の化合物と対応するC−アシル化試薬とを反応させることにより二環性化合物(21)の高純度結晶を得て、これによりバリオールアミン前駆体の立体選択的合成という目的を達し、その後、R基及びカルボニル保護基を除去することにより、高純度のバリオールアミン(1)を得る。この反応により生成されたバリオールアミンは、不純物が少なく、特に、5’アミノアノマー(15)を含有していない。
【0033】
特に、異性体を含有し得る式(14)の化合物は、ホスゲン、トリクロロメチルクロロギ酸エステル、ビス(トリクロロメチル)炭酸塩のようなC−アシル化試薬と反応される。これにより、式(14)の化合物は、1’(1−C)のS配置ヒドロキシル基と、5’(5−C)のS配置アミノ基とがカルボニル基により分子内縮合で結ばれ、二環性化合物(21)を与える。一方、5’にR配置のアミノ基を持つアノマーは、そのような分子内縮合反応を行うことができない。この反応は立体選択的であり、得られた二環性化合物(21)は、容易に結晶化及び精製され、他の不純物、アノマー、及びその誘導体から分離され得る。これにより、高い純度を有するバリオールアミン前駆体を調製することが可能となる。
【0034】
不純物を含む又は異性体を含有し得る式(14)の化合物は、例えば、対応するアジ化水素酸塩(20)の還元(特開2003−146957号公報)、又は化合物(12)の還元アミノ化(WO2005/049547、WO2005/092834)、又は化合物(12)のオキシム化及びその後の還元(J.Org.Chem.1992,57,3651−3658)といった、先行文献に開示された方法に従って調製され得る。これらの方法を用いて得られた式(14)の化合物は、カラムクロマトグラフィのような他の手段により精製する必要はなく、本発明に直接用いることができる。
【0035】
得られた不純物を含む化合物(14)(例えば、化合物(14’))は、非プロトン性極性溶媒又は非極性溶媒に溶解される。ここで、非プロトン性極性溶媒は、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、テトラヒドロフラン、1,4−ジオキサン、エチルエーテル、ジイソプロピルエーテル、ジエチレングリコールジメチルエーテル、酢酸エチル、アセトン、2−ブタノン、シクロヘキサノン、ジメチルスルホキシド、又はこれらの混合物より構成される群から選択される1つ又は複数の種類の溶媒である。非極性溶媒は、四塩化炭素、シクロヘキサン、ヘキサンより構成される群から選択される。その後、有機塩基でも無機塩基でもよい塩基が加えられる。有機塩基は、トリエチルアミン、N−メチルモルホリン、ピリジン、及び4−ジメチルアミノピリジンより構成される群から選択され、無機塩基は、炭酸カリウム、炭酸ナトリウム、重炭酸ナトリウム、水酸化カリウム、水酸化ナトリウム、水酸化リチウム、及び炭酸リチウムより構成される群から選択される。−20〜50℃の温度範囲において、非プロトン性極性溶媒又は非極性溶媒中のC−アシル化試薬は、一滴ずつ添加される。対応するC−アシル化試薬は、ホスゲン、トリクロロメチルクロロギ酸エステル、及びビス(トリクロロメチル)炭酸塩より構成される群から選択され、好ましくは固体であり、計量が容易で、かつ危険性の低いビス(トリクロロメチル)炭酸塩である。非プロトン性極性溶媒は、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、テトラヒドロフラン、1,4−ジオキサン、エチルエーテル、ジイソプロピルエーテル、ジエチレングリコールジメチルエーテル、酢酸エチル、アセトン、2−ブタノン、シクロヘキサノン、ジメチルスルホキシド、又はこれらの混合物より構成される群から選択される1つ又は複数の種類の溶媒であり、非極性溶媒は、四塩化炭素、シクロヘキサン、又はヘキサンである。非プロトン性極性溶媒が好ましく、ジクロロメタン、ジイソプロピルエーテル、テトラヒドロフランがより好ましい。C−アシル化試薬の添加後、反応は同じ条件で1〜24時間、好ましくは8〜10時間行われ、反応終了後、反応液は通常の方法により処理される。例えば、反応液は水に注入され、有機溶媒で抽出される。対応する有機溶媒は、クロロホルム、ジクロロメタン、酢酸エチル、エチルエーテル、イソプロピルエーテル、ブタノン、又はシクロヘキサノン等の非プロトン性極性溶媒である。その後、抽出液は、水、2N塩酸溶液、5%炭酸ナトリウム水性溶液、飽和塩水で連続的に洗浄され、無水硫酸ナトリウムを用いて乾燥される。抽出溶媒は回収され、得られた固体が粗環状産物(21’)である。
【0036】
粗環状産物(21’)は、再結晶化により効率的に精製され、純度99%以上の白色結晶を与える。再結晶化の溶媒は、メタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノール、イソブタノールのようなC1〜C5の低級脂肪族アルコール、又はエーテル、イソプロピルエーテル、テトラヒドロフラン、1,4−ジオキサンのようなエーテル類、又は酢酸エチル、酢酸メチルのようなエステル類、又はアセトン、ブタノン、シクロヘキサノンのようなケトン類であり、これらの中から1つ又は複数選択される。
【0037】
得られた産物(21’)は、白色針状の結晶形態を持ち、168〜172℃の融点を有し、その構造は、1H−NMRスペクトル、13C−NMRスペクトル、赤外線(IR)スペクトル、マススペクトル、元素分析、比旋光度、及び示差走査熱量測定分析により確認された。
【0038】
更に、この結晶の特性は、その粉末のX線回折パターンにより調べることができる。具体的には、この結晶は、X線回折パターンにおいて2θ=4.70±0.2°、6.52±0.2°、9.40±0.2°、18.40±0.2°、24.02±0.2°、及び25.36±0.2°にX線回折ピークを与え、更に2θ=8.38±0.2°、13.06±0.2°、15.64±0.2°、16.14±0.2°、16.82±0.2°、20.60±0.2°、22.64±0.2°、23.24±0.2°、及び27.12±0.2°にもX線回折ピークを与える。
【0039】
式(22)の化合物は、化合物(21’)結晶のアルカリ媒体中での加水分解のように、精製された結晶性産物の塩基性加水分解により得られる。アルカリ媒体中の塩基は、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化カルシウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウムのような無機塩基であってもよい。溶媒は、10%〜90%(v/v)濃度のメタノール、エタノール、イソプロパノールのようなC1〜C5の短鎖脂肪族アルコールを含む水性溶液であってもよい。使用される媒体は、10%〜90%(v/v)の濃度のアセトン、テトラヒドロフラン、1,4−ジオキサン、DMF(N,N−ジメチルホルムアミド)、DME(N,N−ジメチルアセトアミド)の水性溶液であってもよく、好ましくはC1〜C5の短鎖脂肪族アルコールの水性溶液であり、より好ましくは75〜80%(水:エタノール)(v/v)濃度のエタノール、イソプロパノールである。反応温度は室温から還流温度、反応時間は2〜48時間であり、好ましくは24時間である。反応終了点は、薄層クロマトグラフィ(TLC)により調べられ、原材料がなくなる点が反応終了点とされる。反応終了後、反応液は、一般的な方法で処理される。例えば、反応液を氷水に注入し、有機溶媒で抽出する。このとき、対応する有機溶媒は、クロロホルム、ジクロロメタン、酢酸エチル、エチルエーテル、イソプロピルエーテル、ブタノン、シクロヘキサノンのような非プロトン性極性溶媒であり、抽出液の洗浄は、水、2N塩酸溶液、5%炭酸ナトリウム水性溶液、飽和塩水で連続的に行われ、無水硫酸ナトリウムにより乾燥された後、抽出溶媒が回収され、得られた無色の油状物質がテトラベンジル−バリオールアミン(22’)である。別の方法としては、まず反応液を濃縮した後、得られたものを有機溶媒及び水で層抽出し、抽出液を水、2N塩酸溶液、5%炭酸ナトリウム水性溶液、飽和塩水で連続的に洗浄し、無水硫酸ナトリウムを用いて乾燥した後、抽出溶媒を回収する。このとき、対応する有機溶媒は、クロロホルム、塩化メチレン、トルエン、エチルエーテル、イソプロピルエーテル等であり、好ましくはトルエン、塩化メチレンである。得られた無色の粘性の高い油が式(22’)の化合物であり、この物質は次の反応に直接用いることができる。
【0040】
バリオールアミン(1)は、脱ベンジル化反応により式(22’)の化合物から調製され得る。特に、式(22’)の化合物は、液体アンモニア/金属リチウムによる脱ベンジル化(Synthesis,1999,(4),571−573参照)、又は三臭化ホウ素による脱ベンジル化(J.Am.Chem.Soc.,1999,121,6973−6983参照)、又はパラジウム−炭素/水素による脱ベンジル化(WO2005/049547;J.Am.Chem.Soc.,1999,121,6973−6983参照)、又はパラジウムブラック/ギ酸による脱ベンジル化(J.Org.Chem.1992,57,3651−3658参照)により脱ベンジル化される。これらの方法により簡単にベンジル基は除去され、バリオールアミンを得ることができる。
【0041】
本発明の方法により得られたバリオールアミン(1)は、白色結晶としてメタノールから結晶化される。その構造は、1H−NMRスペクトル、13C−NMRスペクトル、赤外線(IR)スペクトル、マススペクトル、及び比旋光度により確認される。その純度は、高圧液体クロマトグラフィ(HPLC)による測定で>99%であり、とりわけ、99.5%以上に達し得る。
【0042】
また、バリオールアミン(1)は、本発明により与えられた化合物(21)のR基(例えば、ベンジル基)の除去及びその後の加水分解を含む反応経路により調製され得る(反応経路8参照)。例えば、脱ベンジル化の方法は、液体アンモニア/金属リチウム(Synthesis,1999,(4),571−573参照)、又は三臭化ホウ素(J.Am.Chem.Soc.,1999,121,6973−6983参照)、又はパラジウム−炭素/水素(WO2005/049547;J.Am.Chem.Soc.,1999,121,6973−6983参照)、又はパラジウムブラック/ギ酸(J.Org.Chem.1992,57,3651−3658参照)を利用して行われ得る。本発明は、パラジウム−炭素/水素を用いた脱ベンジル化法を提供する。すなわち、環状化合物(21’)をエタノール/テトラヒドロフラン(1:1,v/v)に溶解し、10%パラジウム−炭素を加え、10気圧水素の存在下で10時間反応させ、脱ベンジル化産物(6)を得る。化合物(6)は、更に加水分解されて、バリオールアミン(1)を生成させる。このときの特別な加水分解条件に関しては、文献J.Med.Chem.,1986,29,1038−1046、又は米国特許US4803303を参照することができる。本発明においては、加水分解は水酸化バリウムの水性溶液中で行われ、8時間還流される。この反応の終了後、pHがほぼ中性となるまで二酸化炭素ガスが添加される。過剰な苛性バリタを除去し、炭酸バリウム沈殿をろ過した後、水性溶液は濃縮され、メタノールを加えることにより白色結晶状のバリオールアミンを分離させる。
【0043】
上記の脱ベンジル化及びその後の加水分解を含む方法により調製されたバリオールアミンは、加水分解及びその後の脱ベンジル化により得られたバリオールアミンと同一である。その純度は、HPLCによる測定で99%以上であり、とりわけ、99.5%以上に達する。
【発明の効果】
【0044】
本発明で調製された高純度のバリオールアミンは、更に高純度の糖尿病治療薬ボグリボースの調製に用いることができる。得られたボグリボースは、HPLCによる測定で99.5%以上の純度を有し、とりわけ、99.8%以上に達する。
【図面の簡単な説明】
【0045】
【図1】式(21’)の化合物の結晶の粉末X線回折パターンを示す図。
【図2】式(21’)の化合物の結晶の粉末X線回折パターンを示す図。
【図3】式(21’)の化合物の結晶の粉末X線回折パターンを示す図。
【図4】式(21’)の化合物の結晶の赤外線(IR)スペクトルを示す図。
【図5】式(21’)の化合物の結晶の1H−NMRスペクトルを示す図。
【図6】式(21’)の化合物の結晶の示差走査熱量曲線を示す図。
【発明を実施するための形態】
【0046】
検出器:
核磁気共鳴装置
核磁気共鳴装置は、Bruker社製AVANCE AV500超電導核磁気共鳴装置である。
元素分析装置
元素分析装置は、ELEMENTAR社製vario EL元素分析装置である。
旋光計
POLARTRONIC HHW5旋光計
粉末X線回折
粉末X線回折装置は、Rigaku社(日本)のD/max−rC型である。
X線は、銅ターゲット(銅ターゲットアノードを備えたX線管)より発せられる。
発散スリット1°、受光スリット0.15mm、散乱スリット1°、管電圧40KV、管電流50mA
示差走査熱量測定
示差走査熱量測定器は、NETZSCH DSC204型である。
試料重量:3.11mg
温度範囲:30〜200℃
加熱速度:10℃/min
赤外線(IR)スペクトル
赤外線(IR)分光計は、Bruker EQUINOX55型であり、臭化カリウム錠剤法による測定を採用している。
【0047】
(実施例1)粗テトラベンジル−バリオールアミン(14’)の調製
J.Org.Chem.1992,57,3651−3658の方法を参照し、50gの化合物(12)、100gの塩酸ヒドロキシルアミン、及び50gの無水酢酸ナトリウムを1000mlのメタノール中で混合し、一晩撹拌しながら反応させた。次いで、反応液を濃縮し、2000mlの酢酸エチル及び500mlの水により層分離させた。酢酸エチル層を2N塩酸溶液、飽和重炭酸ナトリウム溶液、及び飽和塩水で連続的に洗浄し、無水硫酸ナトリウムを用いて乾燥させた。酢酸エチルを回収し、得られた無色の油を500mlのメタノールに溶解し、15gのラネーニッケルを加え、10気圧水素下で5時間水素化させた。反応液をろ過し、そのろ液を濃縮して50gの赤褐色の油、すなわち、産物(14’)を得た。産物(14’)は、次の反応に直接用いることができる。
【0048】
(実施例2)テトラベンジルバリオールアミン−1,5−カルバミン酸塩(21’)の調製
上で得られた50gの化合物(14’)を500mlの二塩化メチレンに溶解し、100mlのトリエチルアミンを加え、12gのビス(トリクロロメチル)炭酸塩をゆっくり滴下した後、2時間撹拌しながら反応させた。反応液を氷水に注入し、二塩化メチレン層を分離し、2N塩酸溶液、5%炭酸ナトリウム溶液、及び飽和塩水で連続的に洗浄し、無水硫酸ナトリウムを用いて乾燥させた。二塩化メチレンを回収し、得られた結晶を化合物(21’)の粗産物とした。この粗産物を1000mlの無水エタノールで再結晶化させ、40gの白色針状結晶を得た。HPLC純度:99.8%、融点:170−172℃、1H−NMR(CDCl3,500MHz):δ1.92(2H,m,9−CH2),δ3.46(1H,dd,J=2.7,8.7Hz,6−CH),δ3.38(1H,d,J=8.9,1−CH2OBn),δ3.64(1H,m,5−CH),δ3.68(1H,d,J=9.4,8−CH),δ3.79(1H,d,J=8.9,1−CH2OBn),δ3.93(1H,d,J=9.1,7−CH),δ4.39(1H,d,J=11.7,1−OCH2Ph),δ4.42(1H,d,J=11.7,1−OCH2Ph),δ4.58(1H,d,J=11.5,8−OCH2),δ4.61(1H,d,J=12.0,6−OCH2),δ4.65(1H,d,J=10.8,7−OCH2),δ4.69(1H,d,J=11.4,6−OCH2),δ4.80(1H,d,J=10.8,7−OCH2),δ4.86(1H,d,J=11.4,8−OCH2),δ6.81(1H,d,J=4.5,−NH),δ7.15−7.35(20H,m,Ar−H)、13C−NMR(CDCl3,125MHz):δ27.5,47.5,71.2,72.1,73.3,75.2,75.9,81.8,81.9,82.4,83.2,137.5,137.9,138.4,138.5,153.2、[α]20D+34.0°(c1,CHCl3);元素分析:C36H37NO6,計算値(%)C,74.59;H,6.43;N,2.42;実験値(%)C,74.20;H,6.39;N,2.33;赤外線(IR)(KBr)ν3302,3086,3063,3031,2916,2865,1714,1673,1453,1331,1087,1067,735,697;MS(EI)m/e:580(M++1),579(M+.),488,91(100);粉末X線回折グラフを図1に示し、関連データを表1に示す。化合物の赤外線(IR)スペクトルを図4に示し、1H−NMRスペクトルを図5に示し、示差走査熱量測定パターンを図6に示す。
【表1】
【0049】
(実施例3)テトラベンジル−バリオールアミン−1,5−カルバミン酸塩(21’)の調製
実施例1で得られた25gの化合物(14’)を250mlのクロロホルムに溶解し、60gの炭酸カリウムを加えた後、0℃で撹拌しながら5gのトリクロロメチルクロロギ酸エステルを一滴ずつゆっくり添加し、2時間撹拌しながら反応させた。反応液を氷水に注入し、クロロホルム層を分離し、それを2N塩酸溶液、5%炭酸ナトリウム溶液、及び飽和塩水で連続的に洗浄した後、無水硫酸ナトリウムを用いて乾燥させた。クロロホルムを回収し、得られた結晶を化合物(21’)の粗産物とした。この粗産物を800mlのメタノールを用いて再結晶化させ、約18gの白色針状結晶を得た。融点:169〜171℃、HPLC純度:99.7% その構造確認に関するデータは、実施例2のものと同一である。その粉末X線回折パターンを図2に示し、関連するデータを表2に示す。
【表2】
【0050】
(実施例4)テトラベンジルバリオールアミン−1,5−カルバミン酸塩(21’)の調製
実施例1で得られた25gの化合物(14’)を250mlのクロロホルムに溶解し、50mlのピリジンを加え、−10℃で撹拌しながらホスゲンガスをゆっくりと加えた。薄層クロマトグラフィ(TLC)で開始物質(14’)の消失を確認した後、反応液を氷水に注入し、クロロホルム層を分離し、これを2N塩酸溶液、5%炭酸ナトリウム溶液、及び飽和塩水で連続的に洗浄した後、無水硫酸ナトリウムを用いて乾燥させた。クロロホルムを回収し、得られた結晶を化合物(21’)の粗産物とした。この粗産物を300mlのn−プロパノールを用いて再結晶化させ、約19gの白色針状結晶を得た。融点:168〜170℃、HPLC純度:99.6% その構造確認に関するデータは、実施例2のものと同一である。その粉末X線回折パターンを図3に示し、関連するデータを表3に示す。
【表3】
【0051】
(実施例5)バリオールアミンの調製
テトラベンジル−バリオールアミン(22’)の調製
126gの化合物(21’)、80gの水酸化ナトリウム、800mlの水、及び3400mlのエタノールを混合し、24時間還流させた。反応終了後、エタノールを回収し、それを1000mlトルエンで抽出した。そのトルエン層を、水、2N塩酸溶液、5%炭酸ナトリウム溶液、及び飽和塩水で連続的に洗浄し、無水硫酸ナトリウムを用いて乾燥させた。トルエンを回収し、得られた無色の粘性の高い油(約120g)を産物(22’)とした。この産物(22’)は、次の反応に直接用いることができる。
【0052】
バリオールアミンの調製
得られた約50gの油(22’)を1000mlのメタノールに溶解し、50mlの94%ギ酸、5gのパラジウムブラックを加え、撹拌しながら室温で18時間反応させ、反応液をろ過した。そのろ液を濃縮し、強酸性イオン交換樹脂カラムにロードし、水、次いで0.5Nアンモニア水で溶出した。溶出された液体を乾燥するまで濃縮し、メタノールを加えて白色結晶、すなわち、バリオールアミンを分離させた。このバリオールアミンは、更にメタノールを用いて再結晶化させることにより、HPLCによる測定で99.6%の純度を有する白色結晶とすることができる。
【0053】
(実施例6)バリオールアミンの調製
バリオールアミン−1,5−カルバミン酸塩(6)(化学名称:(1S,5S,6S,7R,8S)6,7,8−トリヒドロキシ−1−ヒドロキシメチル−3−オキソ−2−オキサ−4−アザビシクロ[3.3.1]ノナン)の調製
20gのテトラベンジルバリオールアミン−1,5−カルバミン酸塩(21’)を500mlのテトラヒドロフラン/エタノール(1:1)(v/v)に溶解し、5gの10%パラジウム/炭素を加え、室温、10気圧水素下で水素化脱ベンジル化反応を10時間行った。この反応終了後、反応液をろ過し、そのろ液を減圧下で濃縮し、反応溶媒を回収した。これをメタノールで再結晶化させ、約8gの白色結晶、すなわち、バリオールアミン−1,5−カルバミン酸塩(6)を得た。
【0054】
バリオールアミンの調製
5gのバリオールアミン−1,5−カルバミン酸塩を150mlの水に溶解し、20gの苛性バリタを加え、70〜80℃の温度に保持したまま撹拌しながら3時間反応させた。その反応液を20℃まで冷却し、30分間二酸化炭素を添加した。生成した沈殿を除去し、水で洗浄した。そのろ液及び洗液を合わせ、減圧下で蒸発させて水分を除いた。その産物をメタノールを用いて再結晶化させ、4gの白色粉末結晶状のバリオールアミンを得た。HPLC純度:99.8%
【0055】
(実施例7)ボグリボースの調製
上記実施例5で得られた2.0gのバリオールアミンを50mlのN,N−ジメチルホルムアミドに溶解し、3.4gの1,3−ジヒドロキシアセトン、1.5mlの2mol/L HCl、及び2.6gのシアノ水素化ホウ素ナトリウムを加え、撹拌しながら室温で16時間反応させた。その反応液を濃縮し、その濃縮物を100mlの水に溶解して、2mol/L HClで酸性化、30〜40分間撹拌して冷却した後、250mlのDowex 50W×8(H+)クロマトグラフィカラムへとロードした。水で洗浄した後、その産物を0.5mol/Lのアンモニア水で完全に溶出させた。その溶出液を減圧下で濃縮し、それを400mlの無水エタノール中で30分間還流し、少し冷却させてから0.2gの活性炭を加え、15分間還流した後ろ過した。そのろ液を室温まで冷却して、得られた白色粉末をろ過し、40℃で12時間真空乾燥させ、結晶性粉末として1.9gのボグリボースを得た。融点:164.1〜165.7℃、HPLC純度:99.8%
【0056】
(実施例8)ボグリボースの調製
上記実施例6で得られた3.0gのバリオールアミンを75mlのN,N−ジメチルホルムアミドに溶解し、5.1gの1,3−ジヒドロキシアセトン、2.3mlの2mol/L HCl、及び4gのシアノ水素化ホウ素ナトリウムを加え、撹拌しながら室温で16時間反応させた。その反応液を濃縮し、その濃縮物を150mlの水に溶解、2mol/L HClで酸性化させ、30〜40分間撹拌して冷却した後、375mlのDowex 50W×8(H+)クロマトグラフィカラムへとロードした。水で洗浄した後、その産物を0.5mol/Lのアンモニアで完全に溶出させた。その溶出液を減圧下で濃縮し、それを600mlの無水エタノール中で30分間還流し、少し冷却させてから0.3gの活性炭を加え、15分間還流した後ろ過した。そのろ液を室温まで冷却して、得られた白色粉末をろ過し、40℃で12時間真空乾燥させ、結晶性粉末として2.8gのボグリボースを得た。融点:164.3〜165.8℃、HPLC純度:99.9%
【特許請求の範囲】
【請求項1】
バリオールアミンを調製するための中間化合物であって、下記式(21)に示される化学構造を有することを特徴とする中間化合物。
【化1】
ここに、Rはヒドロキシル保護基である。
【請求項2】
前記Rはベンジル基(Bn)であり、前記中間化合物は下記式(21’)の化合物テトラベンジル−バリオールアミン−1,5−カルバミン酸塩(化学名称:(1S,5S,6S,7R,8S)6,7,8−トリ−O−ベンジル−1−ベンジルオキシメチル−3−オキソ−2−オキサ−4−アザビシクロ[3.3.1]ノナン)であることを特徴とする請求項1に記載の中間化合物。
【化2】
【請求項3】
粉末X線回折(Cu)において、2θ=4.70±0.2°、6.52±0.2°、8.38±0.2°、9.40±0.2°、13.06±0.2°、15.64±0.2°、16.14±0.2°、16.82±0.2°、18.40±0.2°、20.60±0.2°、22.64±0.2°、23.24±0.2°、24.02±0.2°、25.36±0.2°、及び27.12±0.2°に特性ピークを与えることを特徴とする請求項2に記載の中間化合物の結晶。
【請求項4】
前記結晶は、示差走査熱量(DSC)分析において、173.8℃に吸収ピークを与えることを特徴とする請求項3に記載の中間化合物の結晶。
【請求項5】
前記結晶は、赤外線(IR)スペクトル分析において、図4に示されるような赤外線(IR)スペクトルパターンを与えることを特徴とする請求項3に記載の中間化合物の結晶。
【請求項6】
前記結晶は、168〜172℃に融点を有することを特徴とする請求項3に記載の中間化合物の結晶。
【請求項7】
請求項1に記載の中間化合物を調製する方法であって、
式(14)の化合物は、C−アシル化され、下記式(21)の二環性化合物バリオールアミン−1,5−カルバミン酸塩誘導体を与えるステップを備えたことを特徴とする方法。
【化3】
ここに、Rはヒドロキシ保護基である。
【請求項8】
前記式(14)の化合物は、非プロトン性極性溶媒又は非極性溶媒に溶解された後、塩基を加えられ、非プロトン性極性溶媒又は非極性溶媒中で−20〜50℃の温度範囲においてC−アシル化試薬を添加され、1〜24時間反応させられることを特徴とする請求項7に記載の方法。
【請求項9】
前記非プロトン性極性溶媒は、二塩化メチレン、クロロホルム、1,2−ジクロロエタン、テトラヒドロフラン、1,4−ジオキサン、エチルエーテル、ジイソプロピルエーテル、ジエチレングリコールジメチルエーテル、酢酸エチル、アセトン、2−ブタノン、シクロヘキサノン、及びジメチルスルホキシドより構成される群から1つ又は複数選択され、
前記非極性溶媒は、四塩化炭素、シクロヘキサン、及びn−ヘキサンより構成される群から選択され、
前記塩基は、有機塩基又は無機塩基であり、
前記有機塩基は、トリエチルアミン、N−メチルモルホリン、ピリジン、及びDMAPより構成される群から選択され、
前記無機塩基は、炭酸カリウム、炭酸ナトリウム、重炭酸ナトリウム、水酸化カリウム、水酸化ナトリウム、水酸化リチウム、及び炭酸リチウムより構成される群から選択され、
前記C−アシル化試薬は、ホスゲン、トリクロロメチルクロロギ酸エステル、及びビス(トリクロロメチル)炭酸塩より構成される群から選択されることを特徴とする請求項8に記載の方法。
【請求項10】
前記C−アシル化試薬は、ビス(トリクロロメチル)炭酸塩であることを特徴とする請求項9に記載の方法。
【請求項11】
前記式(21)の化合物は、更に再結晶化により精製され、化合物(21)の結晶を与えることを特徴とする請求項7に記載の方法。
【請求項12】
前記式(14’)の化合物テトラベンジル−バリオールアミンは、C−アシル化され、下記の二環性化合物(21’)テトラベンジル−バリオールアミン−1,5−カルバミン酸塩を与えることを特徴とする請求項7に記載の方法。
【化4】
【請求項13】
前記化合物(21’)は、更に再結晶化により精製され、化合物(21’)の結晶を与えることを特徴とする請求項12に記載の方法。
【請求項14】
バリオールアミン(1)を調製する方法であって、下記の反応経路のように、
式(21)の化合物及び/又はその結晶を加水分解することにより式(22)の化合物を生成させた後、R基を除去することによりバリオールアミン(1)を得るステップを備えた、又は、
式(21)の化合物及び/又はその結晶のR基を除去することにより式(6)の化合物を生成させた後、加水分解することによりバリオールアミン(1)を得るステップを備えたことを特徴とする方法。
【化5】
ここに、Rはヒドロキシ保護基である。
【請求項15】
前記Rはベンジル基であり、反応経路が下記であることを特徴とする請求項14に記載の方法。
【化6】
【請求項16】
ボグリボースを調製する方法であって、下記の反応経路のように、
1)C−アシル化により式(14)の化合物から式(21)の二環性化合物バリオールアミン−1,5−カルバミン酸塩誘導体を調製するステップと、
2)前記式(21)の化合物からバリオールアミン(1)を調製するステップと、
3)前記バリオールアミン(1)からボグリボース(2)を調製するステップと、を備えたことを特徴とする方法。
【化7】
ここに、Rはヒドロキシ保護基である。
【請求項17】
前記Rは、ベンジル基であることを特徴とする請求項16に記載のボグリボース調製方法。
【請求項1】
バリオールアミンを調製するための中間化合物であって、下記式(21)に示される化学構造を有することを特徴とする中間化合物。
【化1】
ここに、Rはヒドロキシル保護基である。
【請求項2】
前記Rはベンジル基(Bn)であり、前記中間化合物は下記式(21’)の化合物テトラベンジル−バリオールアミン−1,5−カルバミン酸塩(化学名称:(1S,5S,6S,7R,8S)6,7,8−トリ−O−ベンジル−1−ベンジルオキシメチル−3−オキソ−2−オキサ−4−アザビシクロ[3.3.1]ノナン)であることを特徴とする請求項1に記載の中間化合物。
【化2】
【請求項3】
粉末X線回折(Cu)において、2θ=4.70±0.2°、6.52±0.2°、8.38±0.2°、9.40±0.2°、13.06±0.2°、15.64±0.2°、16.14±0.2°、16.82±0.2°、18.40±0.2°、20.60±0.2°、22.64±0.2°、23.24±0.2°、24.02±0.2°、25.36±0.2°、及び27.12±0.2°に特性ピークを与えることを特徴とする請求項2に記載の中間化合物の結晶。
【請求項4】
前記結晶は、示差走査熱量(DSC)分析において、173.8℃に吸収ピークを与えることを特徴とする請求項3に記載の中間化合物の結晶。
【請求項5】
前記結晶は、赤外線(IR)スペクトル分析において、図4に示されるような赤外線(IR)スペクトルパターンを与えることを特徴とする請求項3に記載の中間化合物の結晶。
【請求項6】
前記結晶は、168〜172℃に融点を有することを特徴とする請求項3に記載の中間化合物の結晶。
【請求項7】
請求項1に記載の中間化合物を調製する方法であって、
式(14)の化合物は、C−アシル化され、下記式(21)の二環性化合物バリオールアミン−1,5−カルバミン酸塩誘導体を与えるステップを備えたことを特徴とする方法。
【化3】
ここに、Rはヒドロキシ保護基である。
【請求項8】
前記式(14)の化合物は、非プロトン性極性溶媒又は非極性溶媒に溶解された後、塩基を加えられ、非プロトン性極性溶媒又は非極性溶媒中で−20〜50℃の温度範囲においてC−アシル化試薬を添加され、1〜24時間反応させられることを特徴とする請求項7に記載の方法。
【請求項9】
前記非プロトン性極性溶媒は、二塩化メチレン、クロロホルム、1,2−ジクロロエタン、テトラヒドロフラン、1,4−ジオキサン、エチルエーテル、ジイソプロピルエーテル、ジエチレングリコールジメチルエーテル、酢酸エチル、アセトン、2−ブタノン、シクロヘキサノン、及びジメチルスルホキシドより構成される群から1つ又は複数選択され、
前記非極性溶媒は、四塩化炭素、シクロヘキサン、及びn−ヘキサンより構成される群から選択され、
前記塩基は、有機塩基又は無機塩基であり、
前記有機塩基は、トリエチルアミン、N−メチルモルホリン、ピリジン、及びDMAPより構成される群から選択され、
前記無機塩基は、炭酸カリウム、炭酸ナトリウム、重炭酸ナトリウム、水酸化カリウム、水酸化ナトリウム、水酸化リチウム、及び炭酸リチウムより構成される群から選択され、
前記C−アシル化試薬は、ホスゲン、トリクロロメチルクロロギ酸エステル、及びビス(トリクロロメチル)炭酸塩より構成される群から選択されることを特徴とする請求項8に記載の方法。
【請求項10】
前記C−アシル化試薬は、ビス(トリクロロメチル)炭酸塩であることを特徴とする請求項9に記載の方法。
【請求項11】
前記式(21)の化合物は、更に再結晶化により精製され、化合物(21)の結晶を与えることを特徴とする請求項7に記載の方法。
【請求項12】
前記式(14’)の化合物テトラベンジル−バリオールアミンは、C−アシル化され、下記の二環性化合物(21’)テトラベンジル−バリオールアミン−1,5−カルバミン酸塩を与えることを特徴とする請求項7に記載の方法。
【化4】
【請求項13】
前記化合物(21’)は、更に再結晶化により精製され、化合物(21’)の結晶を与えることを特徴とする請求項12に記載の方法。
【請求項14】
バリオールアミン(1)を調製する方法であって、下記の反応経路のように、
式(21)の化合物及び/又はその結晶を加水分解することにより式(22)の化合物を生成させた後、R基を除去することによりバリオールアミン(1)を得るステップを備えた、又は、
式(21)の化合物及び/又はその結晶のR基を除去することにより式(6)の化合物を生成させた後、加水分解することによりバリオールアミン(1)を得るステップを備えたことを特徴とする方法。
【化5】
ここに、Rはヒドロキシ保護基である。
【請求項15】
前記Rはベンジル基であり、反応経路が下記であることを特徴とする請求項14に記載の方法。
【化6】
【請求項16】
ボグリボースを調製する方法であって、下記の反応経路のように、
1)C−アシル化により式(14)の化合物から式(21)の二環性化合物バリオールアミン−1,5−カルバミン酸塩誘導体を調製するステップと、
2)前記式(21)の化合物からバリオールアミン(1)を調製するステップと、
3)前記バリオールアミン(1)からボグリボース(2)を調製するステップと、を備えたことを特徴とする方法。
【化7】
ここに、Rはヒドロキシ保護基である。
【請求項17】
前記Rは、ベンジル基であることを特徴とする請求項16に記載のボグリボース調製方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2011−530487(P2011−530487A)
【公表日】平成23年12月22日(2011.12.22)
【国際特許分類】
【出願番号】特願2011−521422(P2011−521422)
【出願日】平成20年8月8日(2008.8.8)
【国際出願番号】PCT/CN2008/001436
【国際公開番号】WO2010/015107
【国際公開日】平成22年2月11日(2010.2.11)
【出願人】(511033139)▲無▼▲錫▼▲薬▼▲興▼▲医▼▲薬▼科技有限公司 (1)
【Fターム(参考)】
【公表日】平成23年12月22日(2011.12.22)
【国際特許分類】
【出願日】平成20年8月8日(2008.8.8)
【国際出願番号】PCT/CN2008/001436
【国際公開番号】WO2010/015107
【国際公開日】平成22年2月11日(2010.2.11)
【出願人】(511033139)▲無▼▲錫▼▲薬▼▲興▼▲医▼▲薬▼科技有限公司 (1)
【Fターム(参考)】
[ Back to top ]