
ヒストンデアセチラーゼの阻害剤
本発明は、細胞増殖性疾患を治療するための化合物および方法を提供する。本発明は、ヒストンデアセチラーゼ酵素活性の新規な阻害剤、これらの阻害剤および薬学上許容可能なキャリヤー、賦形剤または希釈剤を含んでなる化合物の組成物、およびイン・ビトロおよび治療上で細胞増殖を阻害するためのこれらの化合物の使用方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
発明の背景
発明の分野
本発明は、ヒストンデアセチラーゼの阻害に関する。更に詳細には、本発明は、ヒストンデアセチラーゼ酵素活性を阻害する化合物および方法に関する。
【背景技術】
【0002】
関連技術の概要
真核生物細胞では、核DNAはヒストンと会合して、クロマチンと呼ばれるコンパクトな複合体を形成している。ヒストンは、真核生物種中で一般に高度に保存される塩基性タンパク質のファミリーを構成する。H2A、H2B、H3、およびH4と呼ばれるコアヒストン同士が会合して、タンパク質コアを形成する。DNAはこのタンパク質コアの周りを取り巻き、ヒストンの塩基性アミノ酸がDNAの負に帯電したリン酸基と相互作用している。DNAの約146の塩基対がヒストンコアに巻き付いて、クロマチンの反復構造モティーフであるヌクレオソーム粒子を形成している。
【0003】
Csordas, Biochem. J., 286:23-38 (1990)では、ヒストンをN-末端リシン残基のα,ε-アミノ基の翻訳後アセチル化、すなわちヒストンアセチルトランスフェラーゼ(HAT1)によって触媒される反応に付すことが教示されている。アセチル化によってリジン側鎖の正電荷が中和され、クロマチン構造に衝撃を与えることが考えられる。実際に、Taunton et al., Science, 272:408-411 (1996)には、転写因子のクロマチンへの接近がヒストン超アセチル化によって増すことが教示されている。Taunton et al.は、更にアセチル化ヒストンH4でのエンリッチメントはゲノムの転写的にサイレントな領域で見られたことも教示している。
【0004】
ヒストンアセチル化は可逆的修飾であり、脱アセチル化はヒストンデアセチラーゼ (HDAC)と呼ばれる酵素のファミリーによって触媒される。Grozinger et al., Proc. Natl. Acad. Sci. USA, 96: 4868-4873 (1999)では、HDACが酵母Rpd3様タンパク質によって表される第一のクラスと、酵母Hdal様タンパク質によって表される第二のクラスの2つのクラスに分類されることが教示されている。Grozinger et al.は、ヒトHDAC1、HDAC2およびHDAC3タンパク質が第一のクラスのHDACに属し、HDAC4、HDAC5およびHDAC6と命名された第二のクラスのHDACに属する新規なタンパク質も開示している。Kao et al., Genes & Dev., 14: 55-66 (2000),には、第二のクラスのHDACの新たな一員であるHDAC7が開示されている。Van den Wyngaert, FEBS, 478: 77-83 (2000)には、第一のクラスのHDACの新たな一員であるHDAC8が開示されている。
【0005】
Richon et al., Proc. Natl. Acad. Sci. USA, 95: 3003-3007 (1998)には、HDAC活性がStreptomyces hygroscopicusから単離される天然産物であるトリコスタチンA (TSA)および合成化合物であるスベロイルアニリドヒドロキサム酸(SAHA)によって阻害されることが開示されている。Yoshida and Beppu, Exper. Cell Res., 177: 122-131 (1988)には、TSAが細胞サイクルのG1およびG2期においてラット線維芽細胞の増殖を停止し、細胞サイクル調節においてHDACが関係していることが教示されている。実際に、Finnin et al., Nature, 401: 188-193 (1999)では、TSAおよびSAHAが細胞成長を阻害し、末端分化を誘発し、マウスにおける腫瘍の形成を予防することが教示されている。Suzuki et al., 米国特許第6,174,905号明細書、欧州特許第0847992号明細書、日本国特許第258863/96号明細書、および日本国特許出願第10138957号明細書には、細胞分化を誘発し且つHDACを阻害するベンズアミド誘導体が開示されている。Delorme et al., WO 01/38322号明細書およびPCT IB01/00683号明細書には、HDAC阻害剤として働く他の化合物が開示されている。
【0006】
これらの知見は、HDAC活性の阻害が細胞サイクル調節への介入の新規な方法であり、HDAC阻害剤が細胞増殖性疾患または疾病の治療に大きな治療能力を有することを示唆している。これまでのところ、ヒストンデアセチラーゼの阻害剤は当該技術分野ではほとんど知られていない。従って、他のHDAC阻害剤を同定し、強力なHDAC阻害活性に必要な構造的特徴を同定する必要がある。
【発明の開示】
【発明が解決しようとする課題】
【0007】
発明の概要
本発明は、細胞増殖性疾患の治療のための化合物および方法を提供する。本発明は、ヒストンデアセチラーゼ酵素活性の新規な阻害剤を提供する。
【0008】
第一の様相では、本発明は、ヒストンデアセチラーゼの阻害剤として有用な化合物を提供する。
【0009】
第二の様相では、本発明は、本発明によるヒストンデアセチラーゼの阻害剤、および薬学上許容可能なキャリヤー、賦形剤、または希釈剤を含んでなる医薬組成物を提供する。
【0010】
第三の様相では、本発明は、細胞におけるヒストンデアセチラーゼを阻害する方法であって、ヒストンデアセチラーゼの阻害が所望な細胞を本発明のヒストンデアセチラーゼの阻害剤と接触させることを含んでなる、方法を提供する。
【0011】
上記の説明は本発明のある種の様相を単にまとめたものであり、制限しようとするものではない。これらの様相および他の様相および態様を、下記において更に詳細に説明する。
【課題を解決するための手段】
【0012】
好ましい態様の詳細な説明
本発明は、ヒストンデアセチラーゼ酵素活性を阻害するための化合物および方法を提供する。本発明は、細胞増殖性疾患および疾病を治療するための組成物および方法も提供する。本明細書で参照される特許および科学文献によって、当業者に利用可能な知識が確立される。本明細書で引用される発行された特許明細書、特許出願明細書、および参考文献は、それぞれが参考として引用されることが具体的且つ個別的に示される場合と同程度まで参考として本明細書に引用される。不一致の場合には、本明細書の開示内容が優先する。
【0013】
第一の様相の一態様では、本発明は、下式
【化1】

(式中、Arはアリールまたはヘテロアリールであり、それらのそれぞれは場合によっては1-3個の置換基によって置換されている)
を有する化合物、または薬学上許容可能なその塩を含んでなる。
【0014】
好ましくは、Arは、[0013]節の化合物において、アリールまたはピリジニルである。
【0015】
Arの好ましい置換基としては、ハロ、場合によってはハロで置換されたC1-C6-ヒドロカルビル、場合によってはハロで置換されたC1-C6-ヒドロカルビルオキシが挙げられる。特に好ましい置換基としては、フルオロ、クロロ、メトキシ、シクロプロピルオキシ、およびシクロペンチルオキシが挙げられる。
【0016】
[0013]節に記載の化合物の好ましい態様では、Arは下記の
【化2】
から選択される。
【0017】
[0013]節の好ましい化合物としては、下表6の化合物が挙げられる。
【0018】
第一の様相のもう一つの態様では、本発明は、下式
【化3】
(式中、Xは-N(R1)-、-O-、または-S-であるか、またはXは窒素を含むヘテロシクリルであり、但し、窒素は構造Vにおける隣接カルボニルに共有結合し且つ場合によっては1-3個の置換基で置換されており、
RおよびR1は独立して-H、または 場合によっては置換されたa) C1-C6-ヒドロカルビルまたはb) R2-L-であり、但し、R2はアリールまたはヘテロアリールであり、LはCO-C6-ヒドロカルビル-L1-CO-C6-ヒドロカルビルであり、L1は共有結合、-O-、-S-、または-NH-である)
を有する化合物、および薬学上許容可能なその塩を含んでなる。
【0019】
好ましくは、 [0018]節に記載の化合物では、Xは-NH-、-O-、モルホリン-4-イル、ピペリジン-1-イル、ピペリジン-1-イル、またはピロリジン-1-イルである。
【0020】
[0018]節に記載の化合物のもう一つの好ましい態様では、Xは-N(R1)-であり、但し、R1は場合によっては置換メチルまたはエチルである。好ましくは、R1はシアノエチルまたはピリジニルメチルである。
【0021】
好ましくは、 [0018]節に記載の化合物では、RはR2-L-であって、但し、R2はフェニル、ピリジニル、インジル、またはインドリルであり、Lは共有結合、メチル、エチル、またはオキシエチルである。
【0022】
Rの好ましい置換基としては、メトキシおよびヒドロキシが挙げられる。
【0023】
[0018]節に記載の化合物の好ましい態様では、R-X-の組合せは、下記の
【化4】
から選択される。
【0024】
[0018]節に記載の好ましい化合物としては、表7に挙げられているものが挙げられる。
【0025】
第一の様相のもう一つの態様では、本発明は、下式
【化5】
(式中、Yは-N(R4)-、-O-、-S-、-N(R4)SO2-、-SO2-N(R4)-、-SO2-、-N(R4)-C(O)-、-C(O)-N(R4)-、-NHC(O)NH-、-N(R4)C(O)O-、-OC(O)N(R4)-、または共有結合であり、
R1、R2、およびR3は独立して-Hであるか、またはRa-C0-C6-ヒドロカルビルであって、但し、Raは-HであるまたはRaはアリールまたはヘテロアリールであり、それらのそれぞれは、場合によっては1-3個の置換基で置換されており、
R4は-H、-C(O)-Rb、-C(O)O-Rb、-C(O)NH-Rb、またはRc-CO-C6-ヒドロカルビルであり、
但し、
Rbは-Hまたは-C1-C6-ヒドロカルビルであり、
Rcは-H、またはアリールまたはヘテロアリールであり、それらのそれぞれは場合によっては1-3個の置換基で置換されている)
の化合物、および薬学上許容可能なその塩を含んでなる。
【0026】
[0025]節の化合物では、R2およびR3は好ましくは両方とも-Hである。
【0027】
[0025]節の化合物では、Yは好ましくは-NH-、-SO2-NH-、または-N(R4)-であり、但し、R4は-C(O)O-C1-C6-ヒドロカルビルである。
【0028】
[0025]節の化合物では、R1は好ましくはアリール、ベンゾチアゾリル、ピリミジニル、トリアゾリル、ベンゾジオキソレニル、またはピリジニルである。
【0029】
Rlの好ましい置換基としては、C1-C6-ヒドロカルビル、C1-C6-ヒドロカルビルオキシ(例えば、メトキシおよびシクロプロピルオキシ)ハロ、メチルチオ、およびアセチルが挙げられる。
【0030】
[0025]節に記載の化合物の好ましい態様では、R1-Y-は、下記の
【化6】
から選択される。
【0031】
[0025]節に記載の好ましい化合物としては、表8に記載の化合物が挙げられる。
【0032】
第一の様相のもう一つの態様では、本発明は、式
【化7】
(式中、Ar1が場合によっては-NO2、CH3O-、およびモルホリニル(例えば、モルホリン-4-イル)から独立して選択される1-3個の置換基で置換されているアリールまたはヘテロアリールである)
の化合物、および薬学上許容可能なその塩を含んでなる。
【0033】
[0032]節に記載の化合物の好ましい態様では、Ar1はアリール(例えば、フェニル)である。
【0034】
[0032]節に記載の化合物の好ましい態様では、Ar1は、
【化8】
から選択される。
【0035】
[0032]節に記載の好ましい化合物としては、表9に記載の化合物が挙げられる。
【0036】
第二の様相では、本発明は、[0013]-[0035]節の一つに記載の化合物と薬学上許容可能なキャリヤー、賦形剤または希釈剤を含んでなる組成物を含んでなる。
【0037】
第三の様相では、本発明は、細胞中のヒストンデアセチラーゼを阻害する方法であって、ヒストンデアセチラーゼの阻害が所望な細胞を[0013]-[0036]節の一つに記載のヒストンデアセチラーゼの阻害剤と接触させることを含んでなる方法を提供する。
【0038】
もう一つの様相では、本発明は、細胞増殖性疾患または疾病に罹っている哺乳類(好ましくは、ヒト)に[0036]節に記載の組成物の治療上有効量を投与することを含んでなる。
【0039】
本発明の目的のために、(特に断らない限り)下記の定義を用いる。
【0040】
本明細書で用いられる「ヒストンデアセチラーゼ」および「HDAC」という用語は、ヒストンのN-末端におけるリジン残基の -アミノ基からアセチル基を除去する酵素のファミリーの任意の一つを指すものと解釈される。文脈によって指示されない限り、「ヒストン」という用語は任意の種由来のH1、H2A、H2B、H3、H4、およびH5などの任意のヒストンタンパク質を表すことを意味する。好ましいヒストンデアセチラーゼとしては、クラスIおよびクラスIIの酵素が挙げられる。好ましくは、ヒストンデアセチラーゼは、HDAC-1、HDAC-2、HDAC-3、HDAC-4、HDAC-5、HDAC-6、HDAC-7、およびHDAC-8などのヒトHDACであるが、これらに限定されない。幾つかの他の好ましい態様では、ヒストンデアセチラーゼは原生動物または真菌供給源に由来する。
【0041】
「ヒストンデアセチラーゼ阻害剤」および「ヒストンデアセチラーゼの阻害剤」という用語は、本明細書で定義される構造を有し、ヒストンデアセチラーゼと相互作用してその酵素活性を阻害することができる化合物を同定するのに用いられる。「ヒストンデアセチラーゼ酵素活性を阻害する」とは、ヒストンからアセチル基を除去するヒストンデアセチラーゼの能力を減少させることを意味する。幾つかの好ましい態様では、このようなヒストンデアセチラーゼ活性の減少は少なくとも約50%であり、更に好ましくは、少なくとも約75%であり、更に一層好ましくは、少なくとも約90%である。他の好ましい態様では、ヒストンデアセチラーゼ活性は、少なくとも95%、更に好ましくは、少なくとも99%減少する。
【0042】
好ましくは、このような阻害は特異的であり、すなわち、ヒストンデアセチラーゼ阻害剤は、別の関係のない生物学的効果を生成するのに要する阻害剤の濃度より低い濃度でヒストンからアセチル基を除去するヒストンデアセチラーゼの能力を減少させる。好ましくは、ヒストンデアセチラーゼ阻害活性に要する阻害剤の濃度は、関係のない生物学的効果を生成するのに要する濃度の少なくとも2分の1以下であり、更に好ましくは、少なくとも5分の1以下であり、更に一層好ましくは、少なくとも1O分の1以下であり、最も好ましくは、少なくとも2O分の1以下である。
【0043】
簡単にするため、化学残基は、主として一価の化学残基(例えば、アルキル、アリールなど)として本明細書中で定義され、表される。しかしながら、このような用語は、当業者には明らかな適当な構造的環境下では相当する多価残基を送達するのにも用いられる。例えば、「アルキル」残基は一般に一価の基(例えば、CH3-CH2-)を表すが、ある環境では二価の結合残基が「アルキル」であることができ、この場合には、当業者であれば、アルキルが二価の基(例えば、-CH2-CH2-)であり、これは「アルキレン」という用語と同等であることを理解されるであろう。(同様に、二価残基が必要であり且つ「アリール」として表される環境では、当業者であれば、「アリール」という用語が相当する二価の残基であるアリーレンを表すことを理解されるであろう。)総ての原子は、結合形成のための標準的な結合価数(すなわち、炭素については4、Nについては3、Oについては2、およびSについてはSの酸化状態によって2、4または6)を有することが理解される。ときには、残基は、例えば(A)a-B- (式中、aは0または1である)として定義することができる。このような場合には、aが0であるときには、残基はB-であり、aが1であるときには、残基はA-B-である。また、本明細書に開示される残基の数は多数の互変異性形態で存在し、それらの総ては任意の所定の互変異性構造によって包含されると解釈される。
【0044】
「ヒドロカルビル」という用語は、直線状、分岐状または環状アルキル、アルケニル、またはアルキニルであって、それぞれ本明細書で定義されているものを表す。「C0」ヒドロカルビルとは、共有結合を表すのに用いられる。従って、「C0-C3-ヒドロカルビル」としては、共有結合、メチル、エチル、エテニル、エチニル、プロピル、プロペニル、プロピニル、およびシクロプロピルが挙げられる。
【0045】
本明細書で用いられる「アルキル」という用語は、1-12個の炭素原子、好ましくは、1-8個の炭素原子、更に好ましくは、1-6個の炭素原子を有し、場合によっては1、2または3個の置換基で置換されている直鎖状および分岐鎖状脂肪族基を表す。好ましいアルキル基としては、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、第二ブチル、第三ブチル、ペンチル、およびヘキシルが挙げられるが、これらに限定されない。(「C0-C3-アルキル」におけるような)「C0」アルキルは、(「C0」ヒドロカルビルのような)共有結合である。
【0046】
本明細書で用いられる「アルケニル」という用語は、1個以上の炭素-炭素二重結合を有し、2-12個の炭素原子、好ましくは、2-8個の炭素原子、更に好ましくは、2-6個の炭素原子を有し、場合によっては1、2または3個の置換基で置換されている不飽和の直鎖状または分岐鎖状脂肪族基を意味する。好ましいアルケニル基としては、エテニル、プロペニル、ブテニル、ペンテニル、およびヘキセニルが挙げられるが、これらに限定されない。
【0047】
本明細書で用いられる「アルキニル」という用語は、1個以上の炭素-炭素三重結合を有し、2-12個の炭素原子、好ましくは、2-8個の炭素原子、更に好ましくは、2-6個の炭素原子を有し、場合によっては1、2または3個の置換基で置換されている不飽和の直鎖状または分岐鎖状脂肪族基を意味する。好ましいアルキニル基としては、エチニル、プロピニル、ブチニル、ペンチニル、およびヘキシニルが挙げられるが、これらに限定されない。
【0048】
「アルキレン」、「アルケニレン」または「アルキニレン」基とは、2個の他の化学基の間にあってこれらを結合する役割をする上記で定義したアルキル、アルケニルまたはアルキニル基である。好ましいアルキレン基としては、メチレン、エチレン、プロピレン、およびブチレンが挙げられるが、これらに限定されない。好ましいアルケニレン基としては、エテニレン、プロペニレン、およびブテニレンが挙げられるが、これらに限定されない。好ましいアルキニレン基としては、エチニレン、プロピニレン、およびブチニレンが挙げられるが、これらに限定されない。
【0049】
本明細書で用いられる「シクロアルキル」という用語は、3-12個の炭素、好ましくは、3-8個の炭素、更に好ましくは、3-6個の炭素を有する飽和および部分不飽和の環状炭化水素基を包含し、このシクロアルキル基は更に場合によっては置換されている。好ましいシクロアルキル基としては、シクロプロピル、シクロブチル、シクロペンチル、シクロペンテニル、シクロヘキシル、シクロヘキセニル、シクロヘプチル、およびシクロオクチルが挙げられるが、これらに限定されない。
【0050】
「ヘテロアルキル」という用語は、上記で定義した通りのアルキル基であって、鎖の1個以上の炭素原子がO、SおよびNからなる群から選択されるヘテロ原子によって置換されているものを表す。
【0051】
「アリール」基は、1-3個の芳香族環を含んでなり、場合によっては置換されているC6-C14芳香族残基である。好ましくは、アリール基はC6-C10アリール基である。好ましいアリール基としては、フェニル、ナフチル、アントラセニル、およびフルオレニルが挙げられるが、これらに限定されない。「アラールキル」または「アリールアルキル」基は、アルキル基に共有結合したアリール基であって、それらのいずれが独立して場合によっては置換されているかまたは未置換であることができるものを含んでなる。好ましくは、アラールキル基は (C1-C6)アルク(C6-C10)アリールであり、ベンジル、フェネチル、およびナフチルメチルが挙げられるが、これらに限定されない。
【0052】
「ヘテロシクリル」または「複素環状」基は、約3-約12個の原子であって、1個以上の原子がN、OおよびSからなる群から選択される環構造である。複素環状基は、場合によっては1個以上の位置の炭素上で置換されている。複素環状基も、独立して場合によっては窒素上でアルキル、アリール、アラールキル、アルキルカルボニル、アルキルスルホニル、アリールカルボニル、アリールスルホニル、アルコキシカルボニル、アラールコキシカルボニルで置換されており、または硫黄上でオキソまたは低級アルキルで置換されている。好ましい複素環状基としては、エポキシ、アジリジニル、テトラヒドロフラニル、ピロリジニル、ピペリジニル、ピペラジニル、チアゾリジニル、オキサゾリジニル、オキサゾリジノニル、およびモルホリノが挙げられるが、これらに限定されない。ある種の好ましい態様では、複素環状基はアリール、ヘテロアリールまたはシクロアルキル基に融合している。このような融合複素環の例としては、テトラヒドロキノリンおよびジヒドロベンゾフランが挙げられるが、これらに限定されない。この用語の範囲から具体的に除外されるものは、別の環OまたはS原子に隣接している環Oおよび/またはS原子を有する化合物である。
【0053】
本明細書で用いられる「ヘテロアリール」という用語は、5-14個の環原子、好ましくは、5、6、9または10個の環原子を有し、環状配列において共有された6、10または14個のπ電子を有し、且つ炭素原子の他にN、OおよびSからなる群から選択される環当たり1-3個のヘテロ原子を有する基を表す。「ヘテロアリール」という用語は、単環性および二環性基を包含することも意味する。例えば、ヘテロアリール基は、ピリミジニル、ピリジニル、ベンズイミダゾリル、チエニル、ベンゾチアゾリル、ベンゾフラニル、およびインドリニルであってもよい。「ヘテロアラールキル」または「ヘテロアリールアルキル」基は、アルキル基に共有結合したヘテロアリール基であって、それらのいずれかが独立して場合によっては置換されておりまたは未置換であるものを含んでなる。好ましいヘテロアルキル基は、C1-C6アルキル基および5、6、9または10個の環原子を有するヘテロアリール基を含んでなる。この用語の範囲から具体的に除外されるものは、隣接した環Oおよび/またはS原子を有する化合物である。好ましいヘテロアラールキル基としては、ピリジルメチル、ピリジルエチル、ピロリルメチル、ピロリルエチル、イミダゾリルメチル、イミダゾリルエチル、チアゾリルメチル、およびチアゾリルエチルが挙げられる。この用語の範囲から具体的に除外されるものは、隣接した環Oおよび/またはS原子を有する化合物である。
【0054】
「アリーレン」、「ヘテロアリーレン」または「ヘテロシクリレン」基は、2個の他の化学基の間にあってこれらを結合する役割をする上記で定義した通りのアリール、ヘテロアリールまたはヘテロシクリル基である。
【0055】
好ましいヘテロシクリルおよびヘテロアリールとしては、アクリジニル、アゾシニル、ベンズイミダゾリル、ベンゾフラニル、ベンゾチオフラニル、ベンゾチオフェニル、ベンズオキサゾリル、ベンズチアゾリル、ベンズトリアゾリル、ベンズテトラゾリル、ベンズイソキサゾリル、ベンズイソチアゾリル、ベンズイミダゾリニル、カルバゾリル、4aH-カルバゾリル、カルボリニル、クロマニル、クロメニル、シンノリニル、デカヒドロキノリニル、2H,6H-1,5,2-ジチアジニル、ジヒドロフロ[2,3-b]テトラヒドロフラン、フラニル、フラザニル、イミダゾリジニル、イミダゾリニル、イミダゾリル、1H-インダゾリル、インドレニル、インドリニル、インドリジニル、インドリル、3H-インドリル、イソベンゾフラニル、イソクロマニル、イソインダゾリル、イソインドリニル、イソインドリル、イソキノリニル、イソチアゾリル、イソキサゾリル、メチレンジオキシフェニル、モルホリニル、ナフチリジニル、オクタヒドロイソキノリニル、オキサジアゾリル、1,2,3-オキサジアゾリル、1,2,4-オキサジアゾリル、1,2,5-オキサジアゾリル、1,3,4-オキサジアゾリル、オキサゾリジニル、オキサゾリル、オキサゾリジニル、ピリミジニル、フェナントリジニル、フェナントロリニル、フェナジニル、フェノチアジニル、フェノキサチイニル、フェノキサジニル、フタラジニル、ピペラジニル、ピペリジニル、ピペリドニル、4-ピペリドニル、ピペロニル、プテリジニル、プリニル、ピラニル、ピラジニル、ピラゾリジニル、ピラゾリニル、ピラゾリル、ピリダジニル、ピリドキサゾール、ピリドイミダゾール、ピリドチアゾール、ピリジニル、ピリジル、ピリミジニル、ピロリジニル、ピロリニル、2H-ピロリル、ピロリル、キナゾリニル、キノリニル、4H-キノリジニル、キノキサリニル、キヌクリジニル、テトラヒドロフラニル、テトラヒドロイソキノリニル、テトラヒドロキノリニル、テトラゾリル、6H-1,2,5-チアジアジニル、1,2,3-チアジアゾリル、1,2,4-チアジアゾリル、1,2,5-チアジアゾリル、1,3,4-チアジアゾリル、チアントレニル、チアゾリル、チエニル、チエノチアゾリル、チエノオキサゾリル、チエノイミダゾリル、チオフェニル、トリアジニル、1,2,3-トリアゾリル、1,2,4-トリアゾリル、1,2,5-トリアゾリル、1,3,4-トリアゾリル、およびキサンテニルが挙げられるが、これらに限定されない。
【0056】
本明細書で用いられる様に、残基 (例えば、シクロアルキル、ヒドロカルビル、アリール、ヘテロアリール、複素環状、尿素など)が「場合によっては置換されている」と記載されるときには、その基は、場合によっては1-4個、好ましくは1-3個、更に好ましくは1または2個の非水素置換基を有することを意味する。適当な置換基としては、ハロ、ヒドロキシ、オキソ(例えば、オキソで置換された環-CH-は-C(O)-)ニトロ、ハロ-ヒドロカルビル、ヒドロカルビル、アリール、アラールキル、アルコキシ、アリールオキシ、アミノ、アシルアミノ、アルキルカルバモイル、アリールカルバモイル、アミノアルキル、アシル、カルボキシ、ヒドロキシアルキル、アルカンスルホニル、アレンスルホニル、アルカンスルホンアミド、アレンスルホンアミド、アラールキルスルホンアミド、アルキルカルボニル、アシルオキシ、シアノ、およびウレイド基が挙げられるが、これらに限定されない。好ましい置換基であって、(特に断らない限り)それら自身が更に置換基を持たないものは、
(a) ハロ、シアノ、オキソ、カルボキシ、ホルミル、ニトロ、アミノ、アミジノ、グアニジノ、
(b) C1-C5アルキルまたはアルケニルまたはアリールアルキルイミノ、カルバモイル、アジド、カルボキサミド、メルカプト、ヒドロキシ、ヒドロキシアルキル、アルキルアリール、アリールアルキル、C1-C8アルキル、C1-C8アルケニル、C1-C8アルコキシ、C1-C8アルコキシカルボニル、アリールオキシカルボニル、C2-C8 アシル, C2-C8アシルアミノ、C1-C8アルキルチオ、アリールアルキルチオ、アリールチオ、C1-C8アルキルスルフィニル、アリールアルキルスルフィニル、アリールスルフィニル、C1-C8アルキルスルホニル、アリールアルキルスルホニル、アリールスルホニル、CO-C6N-アルキルカルバモイル、C2-C15 N,N-ジアルキルカルバモイル、C3-C7シクロアルキル、アロイル、アリールオキシ、アリールアルキルエーテル、アリール、シクロアルキルまたは複素環または別のアリール環に融合したアリール、C3-C7複素環、またはシクロアルキル、ヘテロシクリルまたはアリールに融合またはスピロ融合したこれらの環のいずれかであって、上記のそれぞれが更に場合によっては上記(a)に挙げた1個以上の残基で置換されているもの、および
(c) -(CH2)s-NR30R31(式中、sは0(この場合には、窒素は置換される残基に直接結合している)- 6であり、R30およびR31はそれぞれ独立して水素、シアノ、オキソ、カルボキサミド、アミジノ、C1-C8ヒドロキシアルキル、C1-C3アルキルアリール、アリール-C1-C3アルキル、C1-C8アルキル、C1-C8アルケニル、C1-C8アルコキシ、C1-C8アルコキシカルボニル、アリールオキシカルボニル、アリール-C1-C3アルコキシカルボニル、C2-C8アシル、C1-C8アルキルスルホニル、アリールアルキルスルホニル、アリールスルホニル、アロイル、アリール、シクロアルキル、ヘテロシクリル、またはヘテロアリールであって、上記のそれぞれが更に場合によっては上記(a)に挙げた1個以上の残基で置換されているもの、または
R30およびR31が、それらが結合しているNと一緒になってヘテロシクリルまたはヘテロアリールを形成し、それらのそれぞれは場合によっては上記(a)からの1-3個の置換基で置換されているもの
である。
【0057】
更に、環状残基(すなわち、シクロアルキル, ヘテロシクリル, アリール, ヘテロアリール)上の置換基としては、親環状残基に融合した5-6員の単環状および9-14員の二環状残基であって、二または三環性の融合環系を形成するものが挙げられる。例えば、場合によっては置換されるフェニルとしては、下記のものが挙げられる。
【化9】
【0058】
「ハロヒドロカルビル」とは、ヒドロカルビル残基であって、1個-全部の水素が1個以上のハロゲンで置き換わっているものである。
【0059】
本明細書で用いられる「ハロゲン」または「ハロ」という用語は、塩素、臭素、フッ素、またはヨウ素を表す。本明細書で用いられる「アシル」という用語は、アルキルカルボニルまたはアリールカルボニル置換基を表す。「アシルアミノ」という用語は、窒素原子に結合したアミド基(すなわち、R-CO-NH-)を表す。「カルバモイル」という用語は、カルボニル炭素原子に結合したアミド基(すなわち、NH2-CO-)を表す。アシルアミノまたはカルバモイル置換基の窒素原子は、更に置換されている。「スルホンアミド」という用語は、硫黄または窒素原子によって結合されているスルホンアミド置換基を表す。「アミノ」という用語は、NH2、アルキルアミノ、アリールアミノおよび環状アミノ基を包含することを意味する。本明細書で用いられる「ウレイド」という用語は、置換または未置換尿素残基を表す。
【0060】
本明細書で用いられる「ラジカル」という用語は、1個以上の不対電子を含んでなる化学残基を意味する。
【0061】
置換される残基は、1個以上の水素が独立して別の化学置換基に置換されているものである。非制限的例としては、置換フェニルとしては、2-フルオロフェニル、3,4-ジクロロフェニル、3-クロロ-4-フルオロ-フェニル、2-フルオロ-3-プロピルフェニルが挙げられる。別の非制限的例としては、置換n-オクチルとしては、2,4-ジメチル-5-エチル-オクチルおよび3-シクロペンチル-オクチルが挙げられる。この定義に包含されるものは、カルボニル(-CO-)を形成するため酸素で置換されたメチレン(-CH2-)である。
【0062】
上記で定義した「未置換」残基(例えば、未置換シクロアルキル、未置換ヘテロアリールなど)は、(上記の)残基の定義が別に提供する任意の置換基のいずれも持たない上記で定義した残基を意味する。従って、例えば、「アリール」はフェニル、およびハロゲンで置換されたフェニルを包含するが、「未置換アリール」はハロゲンで置換されたフェニルを包含しない。
【0063】
本発明の好ましい態様は、本明細書に特に記載された好ましい態様の組合せも包含する。
【0064】
合成
一般式Iの化合物を、工程図1および2に示した合成経路に従って調製した。幾つかの態様では、4-アセチル安息香酸を、水酸化ナトリウムの水溶液(1N)の存在下でメタノール(MeOH)のような溶媒中で芳香族および/またはヘテロ芳香族アルデヒドと反応させて、濾過、またはpH=5-6になるまで酸性化および濾過の後にカルコンIIを得た。化合物IIを、最初にトリエチルアミン(Et3N)の存在下にてN,N-ジメチルホルムアミド(DMF)のような溶媒中で、カップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)で処理した。イン・シテューで形成した生成する活性化エステル中間体を、最後に1,2-フェニレンジアミンと反応させて、化合物Iをえた(工程図1)。
【化10】
【0065】
あるいは、幾つかの他の態様では、4-アセチル安息香酸を、最初にトリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェートで処理した。次に、イン・シテューで形成した生成する活性化エステル中間体を、t-ブチル(2-アミノ-フェニル)-カルバメートと反応させ、一般のアセトフェノン誘導体IIIを得た。カルコンIVは、化合物IIIと適当な芳香族および/またはヘテロ芳香族アルデヒドを水酸化ナトリウムの水溶液(1N)の存在下でメタノールのような溶媒中でClaisen-Schmidt縮合によって調製した。アニリンIVのN-Boc保護基を、最終的にジクロロメタン(CH2Cl2)のような溶媒中でトリフルオロ酢酸の含水溶液(TFA, 95%、水中)によって開裂して、化合物Iを得た(工程図2)。
【化11】
【0066】
一般式Vの化合物は、工程図3および4に示した合成経路に従って調製した。ある種の好ましい態様では、メチル4-ホルミルベンゾエートを、テトラヒドロフラン(THF)とヘキサンのような溶媒混合物中でt-ブチルアセテートのアニオンと反応させた後、新たな脱水剤としての2-クロロ-4,6-ジメトキシ-1,3,5-トリアジンで処理することによって、純粋なトランス-α,β-不飽和エステルVIに転換した。t-ブチルエステルVIの酸加水分解は、ジクロロメタンのような溶媒中でトリフルオロ酢酸の含水溶液(95%、水中)によって行い、化合物VIIを得た。化合物IXの形成は、RXHの親核性により2種類の相補的方法によって行った。方法Aでは、カルボン酸VIIを、最初にトリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)を用いて、安定な活性化エステルVIIIに転換した。次に、この安定な活性化エステルVIIIを、トリエチルアミンの存在下にてジクロロメタンのような溶媒中で弱い親核試薬(例えば、RXH=アニリンまたはアミノヘテロアリール)と反応させて、化合物IXを得た。方法Bでは、カルボン酸VIIからイン・シテューで形成された同じ活性化エステルVIII中間体を、次にトリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中で強い親核試薬(例えば、RXH=アミン、アルコール、チオール、ヒドロキシルアミンおよび誘導体、またはヒドラジンおよび誘導体)と反応させて、化合物IXを得た。
【化12】
【0067】
メチルエステルIXの塩基加水分解は、テトラヒドロフランのような溶媒中で水酸化リチウムの水溶液によって行い、化合物Xを得た。カルボン酸Xを、最後にトリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)で処理した。次に、イン・シテューで形成された生成する活性化エステル中間体を1,2-フェニレンジアミンと反応させて、化合物Vを得た(工程図3)。
【0068】
あるいは、幾つかの他の態様では、触媒量のN,N-ジメチルホルムアミドの存在下にてジクロロメタンのような溶媒中で塩化チオニル(SOCl2)を用いて酸塩化物中間体に転換した。次に、生成する酸塩化物中間体をt-ブチル(2-アミノ-フェニル)-カルバメートと反応させて、一般的なベンズアルデヒド誘導体XIを得た。
【化13】
【0069】
アルデヒドXIのWittigオレフィン化は、トルエンのような溶媒中でメチル(トリフェニル-ホスホラニリデン)アセテートを用いて行い、トランス-α,β-不飽和エステルXIIを得た。メチルエステルXIIの塩基加水分解は、テトラヒドロフランのような溶媒中で水酸化リチウムの水溶液を用いて行い、化合物XIIIを得た。カルボン酸 XIIIを、トリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)で処理した。次に、イン・シテューで形成された生成する活性化エステル中間体を親核試薬R1XHと反応させ、化合物XIVを得た。アニリンXIVのN-Boc保護基は、最終的にジクロロメタンのような溶媒中でトリフルオロ酢酸の含水溶液(95%、水中)によって開裂させ、化合物Vを得た(工程図4)。
【0070】
一般式XVの化合物は、工程図5-7に示す合成経路に従って調製した。従って、アルデヒドXIのWittigオレフィン化は、トルエンのような溶媒中で(トリフェニルホスホラニリデン)-アセトアルデヒド試薬を用いて行い、トランス-α,β-不飽和アルデヒドXVIを得た。ある種の好ましい態様では、化合物XVIIの形成は、R1XHの親核性によって記載した方法に従って還元的アミノ化によって行った。アルデヒドXVIを、最初に触媒量のジブチルスズ二塩化物の存在下にてテトラヒドロフランのような溶媒中で弱親核試薬(例えば、R1XH=アニリンまたはアミノヘテロアリール)と混合した。イン・シテューで形成された生成するイミニウム中間体を次に還元試薬フェニルシランと反応させ、化合物XVIIを得た。
【化14】
【0071】
アニリンXVIIのN-Boc保護基を、最終的にジクロロメタンのような溶媒中でトリフルオロ酢酸の含水溶液(95%、水中)によって開裂し、化合物XVを得た(工程図5)。
【0072】
あるいは、幾つかの他の態様では、トランス-α,β-不飽和アルデヒドXVIをエタノールのような溶媒中で還元試薬水素化ホウ素ナトリウムによって第一アリルアルコールXVIIIに還元した。このアルコールXVIIIを、次に、トリフェニルホスフィンおよびジエチルアゾジカルボキシレート(DEAD)の存在下にてテトラヒドロフランのような溶媒中でMitsunobu型反応によって親核試薬R1XHと反応させ、化合物XVIIを得た。アニリンXVIIのN-Boc保護基を、最終的にジクロロメタンのような溶媒中でトリフルオロ酢酸の含水溶液(95%、水中)によって開裂し、化合物XVを得た(工程図6)。
【化15】
【0073】
更に、幾つかの他の態様では、メチル4-ホルミルベンゾエートのWittigオレフィン化を、トルエンのような溶媒中で(トリフェニルホスホラニリデン)-アセトアルデヒド試薬またはジクロロメタン/水のような二相性媒質中TDA-1{トリス[2-(2-メトキシエトキシ)エチル]アミン)および炭酸カリウムの存在下にて(1,3-ジオキソラン-2-イル)メチルトリフェニルホスホニウムブロミド試薬を用いて行った後、酸加水分解によりトランス-α,β-不飽和アルデヒドXIXを得た。アルデヒドXIXを、最初にジクロロメタンまたは1,2-ジクロロエタンのような溶媒中で親核試薬(R1R2NH)と混合した。次に、イン・シテューで形成される生成するイミニウム中間体を還元試薬水素化トリアセトキシホウ素ナトリウム[NaBH(OAc)3]と反応させて、化合物XXを得た。経路Aでは、メチルエステルの塩基加水分解と化合物XX (R1=アルキル、R2=H)の第二アミンの保護は、1,4-ジオキサンのような溶媒中で水酸化ナトリウムの水溶液(1N)と保護試薬ジ-第三ブチルジカーボネート[(Boc)2O]の存在下にて同時に行い、化合物XXIを得た。カルボン酸XXIを、最初にトリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)で処理した。次に、イン・シテューで形成される生成する活性化エステル中間体を1,2-フェニレンジアミンと反応させ、化合物XXIIを得た。アミンXXIIのN-Boc保護基を、最終的にジクロロメタンのような溶媒中でトリフルオロ酢酸の含水溶液(95%、水中)で開裂し、化合物XVを得た(工程図7)。
【化16】
【0074】
経路Bでは、メチルエステルXXを、塩基加水分解および1,2-フェニレンジアミンでカップリングした後に、直接最終化合物XVに転換した(工程図7)。
【0075】
一般式XXIVの化合物は、工程図8および9に示される合成経路に従って調製した。幾つかの態様では、4-ホルミル安息香酸を、水酸化ナトリウムの水溶液(1N)の存在下にてメタノール(MeOH)のような溶媒中でアリールおよび/またはヘテロアリールメチルケトンと反応させ、濾過後にカルコンXXIIIを得た。化合物XXIIIを、最初にトリエチルアミン(Et3N)の存在下にてN,N-ジメチルホルムアミド(DMF)のような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)で処理した。イン・シテューで形成される生成する活性化エステル中間体を、最後に1,2-フェニレンジアミンと反応させて、化合物XXIVを得た(工程図8)。
【化17】
【0076】
あるいは、幾つかの他の態様では、カルコンXXVを、水酸化ナトリウムの水溶液(1N)の存在下にてメタノールのような溶媒中でベンズアルデヒド誘導体XIと適当なアリールおよび/またはヘテロアリールメチルケトンとのClaisen-Schmidt縮合によって調製した。アニリンXXVのN-Boc保護基を、最後にジクロロメタン(CH2Cl2)のような溶媒中でトリフルオロ酢酸の含水溶液(TFA 95%、水中)によって開裂し、化合物XXIVを得た(工程図9)。
【化18】
【0077】
一般式XXVIIおよびXXIXの化合物は、工程図10に示される合成経路に従って調製した。従って、化合物XXIIIの二重結合の選択的還元は、N,N-ジメチルホルムアミドのような溶媒中で還元試薬ベンゼンスルホニルヒドラジドを用いることによって行い、化合物XXVIを得た。カルボン酸XXVIを、最初にトリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)で処理した。イン・シテューで形成される生成する活性化エステル中間体を、最後に1,2-フェニレンジアミンと反応させ、化合物XXVIIを得た。
【化19】
【0078】
更に、α,β-不飽和ケトンXXIIIの飽和化合物 XXVIIIへの完全な還元は、N,N-ジメチルアセタミド(DMA)のような溶媒中で10%パラジウム/炭(Degussa型)によって触媒される水素化によって行った。次に、カルボン酸XXVIIIを、最初にトリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)で処理した。イン・シテューで形成される生成する活性化エステル中間体を、最後に1,2-フェニレンジアミンと反応させて、化合物XXIXを得た(工程図10)。
【0079】
一般式XXXの化合物は、工程図11に示される合成経路に従って調製した。従って、メチル4-ブロモベンゾエートと(トリメチルシリル)アセチレンとの間のSonogashira型反応は、THFのような溶媒中でEt3Nの存在下にて触媒量のパラジウム触媒とヨウ化銅とによって行って、保護されたアルキンXXXIを得た。XXXIのTMS基の塩基性脱保護は、メタノールの存在下にて炭酸カリウムによって行い、アルキンXXXIIを得た。XXXIIの三重結合のハイドロボレーションは、THFのような溶媒中でカテコールボラン試薬によって行った後、ボロネート中間体を酸加水分解によってボロン酸XXXIIIを得た。アリルアミンXXXIVは、1,4-ジオキサンのような溶媒中で、ビニルボロン酸XXXIIIをアミノ化合物(R1R2NH)とアルデヒド(R3CHO)との予備形成混合物と反応させることによるPetasis型反応によって合成した。最後に、メチルエステルXXIVを、塩基加水分解および1,2-フェニレンジアミンとのカップリングの後、最終化合物XXXに転換した。
【化20】
【0080】
ヒストンデアセチラーゼの阻害
第三の様相では、本発明は、細胞においてヒストンデアセチラーゼを阻害する方法であって、ヒストンデアセチラーゼの阻害が所望な細胞を本発明によるヒストンデアセチラーゼの阻害剤と接触させることを含んでなる方法を提供する。
【0081】
ヒストンデアセチラーゼの酵素活性の測定は、既知の方法を用いて行うことができる。例えば、Yoshida et al., J.Biol. Chem., 265: 17174-17179 (1990)には、トリコスタチンAで処理した細胞におけるアセチル化ヒストンの検出によるヒストンデアセチラーゼ酵素活性の評価が記載されている。Taunton et al., Science, 272: 408-411 (1996)にも、同様に、内在性および組換えHDAC-1を用いてヒストンデアセチラーゼ酵素活性を測定する方法が記載されている。
【0082】
幾つかの好ましい態様では、ヒストンデアセチラーゼ阻害剤は、細胞中の総てのヒストンデアセチラーゼと相互作用し且つこの活性を減少させる。本発明のこの様相による幾つかの他の好ましい態様では、ヒストンデアセチラーゼ阻害剤は、細胞中の総てより少数のヒストンデアセチラーゼと相互作用し且つこの活性を減少させる。ある種の好ましい態様では、阻害剤は、1種類のヒストンデアセチラーゼ(例えば、HDAC-1)と相互作用し且つこの活性を減少させるが、他のヒストンデアセチラーゼ(例えば、HDAC-2、HDAC-3、HDAC-4、HDAC-5、HDAC-6、HDAC-7およびHDAC-8)とは相互作用もせずこれらの活性を減少させもしない。下記のように、ある種の特に好ましいヒストンデアセチラーゼ阻害剤は、腫瘍形成に関与するヒストンデアセチラーゼと相互作用し且つこの酵素活性を減少させるものである。ある種の他の好ましいヒストンデアセチラーゼ阻害剤は、真菌ヒストンデアセチラーゼと相互作用し且つこの酵素活性を減少させる。
【0083】
好ましくは、本発明の第三の様相による方法は、接触した細胞の細胞増殖の阻害を生じる。「細胞増殖を阻害する」という用語は、阻害剤と接触しなかった細胞と比較して接触した細胞の成長を遅延させるヒストンデアセチラーゼの阻害剤の能力を表すのに用いられる。細胞増殖は、Coulter Cell Counter (Coulter, Miami, FL)または血球計を用いて接触したおよび接触しなかった細胞を計数することによって行うことができる。細胞が固体成長である場合(例えば、固形腫瘍または器官)には、このような細胞増殖の評価はカリパスで成長を測定し、接触した細胞の成長の程度を接触しなかった細胞と比較することによって行うことができる。
【0084】
好ましくは、阻害剤と接触した細胞の成長は、接触しなかった細胞と比較して少なくとも50%遅延する。更に好ましくは、細胞増殖は100%阻害される(すなわち、接触した細胞の数は増加しない)。最も好ましくは、「細胞増殖を阻害する」という用語は、接触した細胞の数または大きさが接触しなかった細胞と比較して減少することを包含する。従って、接触した細胞での細胞増殖を阻害する本発明によるヒストンデアセチラーゼの阻害剤は、接触した細胞に成長遅延、成長停止、細胞死プログラム(すなわち、アポトーシス)、または壊死による細胞死を誘発することがある。
【0085】
本発明によるヒストンデアセチラーゼ阻害剤の細胞増殖阻害能は、様々な生物学的過程におけるヒストンデアセチラーゼの役割を研究するための有用な研究手段となっている。例えば、本発明によるヒストンデアセチラーゼ阻害剤の細胞増殖阻害能によって、非同時成長細胞の集団を同期させることができる。例えば、本発明のヒストンデアセチラーゼ阻害剤は、イン・ビトロで成長した非腫瘍細胞の集団を細胞サイクルのGlまたはG2期に停止させるのに用いることができる。このような同期により、例えば、細胞サイクルのGlまたはG2期に発現した遺伝子および/または遺伝子産物を同定することができる。培養細胞のこのような同期は、トランスフェクション効率が変化し且つトランスフェクションされる細胞の特定の細胞サイクル期によって変化する新規なトランスフェクションプロトコールの効力の試験にも有用であることがある。本発明のヒストンデアセチラーゼ阻害剤を用いることによって細胞の集団を同期することができ、これにより、増加したトランスフェクション効率の検出が促進される。
【0086】
幾つかの好ましい態様では、接触した細胞は腫瘍細胞である。「腫瘍細胞」という用語は、異常な細胞成長を示す細胞を表すのに用いられる。好ましくは、腫瘍細胞の異常な細胞成長は細胞成長の増加である。腫瘍性細胞は、増殖性細胞、イン・ビトロでの成長の接触阻害を欠いている細胞、イン・ビボで転移が不可能な良性腫瘍細胞、またはイン・ビボで転移することができ且つ除去を試みた後に再発する可能性がある癌細胞であることもある。「腫瘍形成」という用語は、細胞増殖を誘発し、腫瘍性成長が展開されることを表すのに用いられる。幾つかの態様では、ヒストンデアセチラーゼ阻害剤は、接触した細胞での細胞分化を誘発する。従って、腫瘍性細胞をヒストンデアセチラーゼの阻害剤と接触させることにより、分化が誘発され、接触した細胞より系統発生的に進化した非腫瘍性娘細胞が産生されることがある。
【0087】
幾つかの好ましい態様では、接触した細胞は動物である。従って、本発明は、動物における細胞増殖性疾患または疾病の治療方法であって、このような治療を必要とする動物に本発明のヒストンデアセチラーゼ阻害剤の治療上有効量を投与することを含んでなる方法を提供する。好ましくは、動物は哺乳類であり、更に好ましくは、家畜である。最も好ましくは、動物はヒトである。
【0088】
「細胞増殖性疾患または疾病」という用語は、異常な細胞成長、好ましくは細胞増殖の異常増加を特徴とする任意の疾病を表すことを意味する。このような細胞増殖性疾患または疾病としては、癌、再狭窄、および乾癬が挙げられるが、これらに限定されない。特に好ましい態様では、本発明は、動物における腫瘍性細胞増殖を阻害する方法であって、動物体に少なくとも1個の腫瘍性細胞が含まれている動物に本発明のヒストンデアセチラーゼ阻害剤の治療上有効量を投与することを含んでなる、方法を提供する。
【0089】
本発明の幾つかの化合物は、原生動物由来のヒストンデアセチラーゼに対して阻害剤活性を有すると考えられている。従って、本発明は、原生動物による疾患または感染症を治療しまたは予防する方法であって、このような治療を必要とする動物に本発明によるヒストンデアセチラーゼ阻害剤の治療上有効量を投与することを含んでなる方法も提供する。好ましくは、動物は哺乳類であり、更に好ましくはヒトである。好ましくは、本発明のこの態様によって用いられるヒストンデアセチラーゼ阻害剤は、哺乳類のヒストンデアセチラーゼ、特にヒトのヒストンデアセチラーゼを阻害するより大幅に原生動物のヒストンデアセチラーゼを阻害する。
【0090】
本発明は、真菌性疾患または感染症の治療方法であって、このような治療を必要とする動物に本発明のヒストンデアセチラーゼ阻害剤の治療上有効量を投与することを含んでなる方法も提供する。好ましくは、動物は哺乳類であり、更に好ましくは、ヒトである。好ましくは、本発明のこの態様によって用いられるヒストンデアセチラーゼ阻害剤は、真菌性ヒストンデアセチラーゼを、哺乳類のヒストンデアセチラーゼ、特にヒトのヒストンデアセチラーゼを阻害するよりも大幅に阻害する。
【0091】
「治療上有効量」という用語は、患者の細胞でヒストンデアセチラーゼ活性を阻害するのに十分な投薬量、または患者の細胞増殖を阻害しまたは細胞分化を誘発するのに十分な投薬量を表すことを意味する。投与は、非経口、経口、舌下、経皮、局所、経鼻、気管内、または直腸内など任意の経路が挙げられるが、これらに限定されない。ある種の特に好ましい態様では、本発明の化合物は、病院施設で静脈内に投与される。ある種の他の好ましい態様では、投与は、好ましくは、経口によることができる。
【0092】
全身投与の際は、ヒストンデアセチラーゼ阻害剤は、好ましくは、阻害剤の血中濃度が約0.01μM-約100μM、更に好ましくは、約0.05μM-約50μM、更に一層好ましくは、約0.1μM-約25μM、更に一層好ましくは、約0.5μM-約25μMとなるのに十分な投薬量で投与される。局所化投与では、これより遙かに低濃度で有効であることができ、また遙かに高濃度を用いることもできる。当業者であれば、治療効果を生じるのに必要なヒストンデアセチラーゼ阻害剤の投薬量は、治療を行う組織、器官、または特定の動物または患者によってかなり変動することがあることを理解するであろう。
【0093】
本発明の第三の様相のある種の好ましい態様では、この方法は、細胞をヒストンデアセチラーゼの発現を阻害するアンチセンスオリゴヌクレオチドと接触させることをも含んでなる。核酸レベルの阻害剤 (例えば、アンチセンスオリゴヌクレオチド)とタンパク質レベルの阻害剤(すなわち、ヒストンデアセチラーゼ酵素活性阻害剤)を併用すると、阻害効果が向上し、それにより、これらの阻害剤を個々に用いるときに必要な量と比較して、所定の阻害効果を得るのに要する阻害剤の量が減少する。本発明のこの様相によるアンチセンスオリゴヌクレオチドは、HDAC-1、HDAC-2、HDAC-3、HDAC-4、HDAC-5、HDAC-6、HDAC7、および/またはHDAC-8をコードするRNAまたは二本鎖DNAの領域に相補的である(例えば、HDAC-1についてはGenBank Accession Number U50079、HDAC-2についてはGenBank Accession Number U31814およびHDAC-3についてはGenBank Accession Number U75697を参照されたい)。
【0094】
本発明の目的に対して、「オリゴヌクレオチド」という用語は、2種類以上のデオキシリボヌクレオシド、リボヌクレオシド、または2'-置換リボヌクレオシド残基、またはそれらの任意の組合せのポリマーを包含する。好ましくは、このようなオリゴヌクレオチドは約6-約100ヌクレオシド残基を有し、更に好ましくは、約8-約50ヌクレオシド残基、最も好ましくは、約12-約30ヌクレオシド残基を有する。ヌクレオシド残基は、多数の既知のヌクレオシド間結合のいずれかによって互いにカップリングすることができる。このようなヌクレオシド間結合としては、ホスホロチオエート、ホスホロジチオエート、アルキルホスホネート、アルキルホスホノチオエート、ホスホトリエステル、ホスホルアミデート、シロキサン、カーボネート、カルボキシメチルエステル、アセトアミデート、カルバメート、チオエーテル、架橋ホスホルアミデート、架橋メチレンホスホネート、架橋ホスホロチオエート、およびスルホンヌクレオシド間結合が挙げられるが、これらに限定されない。ある種の好ましい態様では、これらのヌクレオシド間結合は、ホスホジエステル、ホスホトリエステル、ホスホロチオエート、またはホスホルアミデート結合、またはそれらの組合せであることができる。オリゴヌクレオチドという用語は、化学的に修飾した塩基または糖を有するおよび/または親油性基、インターカレート剤、ジアミン、およびアダマンタンなどのこれらに限定されない追加の置換基を有するポリマーも包含する。
【0095】
本発明の目的のために、「2'-置換リボヌクレオシド」という用語は、ペントース残基の2'位のヒドロキシル基が置換されて2'-O-置換リボヌクレオシドを産生するリボヌクレオシドを包含する。好ましくは、このような置換は、1-6個の飽和または不飽和炭素原子を有する低級アルキル基によるもの、または2-6個の炭素原子を有するアリールまたはアリル基によるものであって、このようなアルキル、アリールまたはアリル基は未置換であってもよく、または例えば、ハロ、ヒドロキシ、トリフルオロメチル、シアノ、ニトロ、アシル、アシルオキシ、アルコキシ、カルボキシル、カルボアルコキシルまたはアミノ基で置換されていてもよい。「2'-置換リボヌクレオシド」という用語は、2'-ヒドロキシル基がアミノ基またはハロ基、好ましくは、フルオロで置換されているリボヌクレオシドも包含する。
【0096】
本発明のこの様相で用いられる特に好ましいアンチセンスオリゴヌクレオチドとしては、キメラオリゴヌクレオチドおよびハイブリッドオリゴヌクレオチドが挙げられる。
【0097】
本発明の目的のために、「キメラオリゴヌクレオチド」とは、2種類以上のヌクレオシド間結合を有するオリゴヌクレオチドを表す。このようなキメラオリゴヌクレオチドの一つの好ましい例は、ホスホロチオエート、ホスホジエステルまたはホスホロジチオエート領域を含んでなる、好ましくは、約2-約12個のヌクレオチド、およびアルキルホスホネートまたはアルキルホスホノチオエート領域を含んでなるキメラオリゴヌクレオチドである(例えば、Pederson et al. 米国特許第5,635,377号明細書および第5,366,878号明細書を参照)。好ましくは、このようなキメラオリゴヌクレオチドは、ホスホジエステルおよびホスホロチオエート結合、またはそれらの組合せから選択される少なくとも3個の連続したヌクレオシド間結合を含む。
【0098】
本発明の目的のために、「ハイブリッドオリゴヌクレオチド」とは、2種類以上のヌクレオシドを有するオリゴヌクレオチドを表す。このようなハイブリッドオリゴヌクレオチドの好ましい一例は、リボヌクレオチドまたは2'-置換リボヌクレオチド領域を含んでなり、好ましくは、約2-約12個の2'-置換ヌクレオチドと1個のデオキシリボヌクレオチド領域を含んでなる。好ましくは、このようなハイブリッドオリゴヌクレオチドは、少なくとも3個の連続したデオキシリボヌクレオシドを含み、またリボヌクレオシド、2'-置換リボヌクレオシド、好ましくは、2'-O-置換リボヌクレオシド、またはそれらの組合せも含む(例えば、Metelev and Agrawal, 米国特許第5,652,355号明細書参照)。
【0099】
本発明で用いられるアンチセンスオリゴヌクレオチドの正確なヌクレオチド配列および化学構造は、オリゴヌクレオチドが目的とする遺伝子発現を阻害する能力を保持する限り、変化することができる。これは、特定のアンチセンスオリゴヌクレオチドが活性であるかどうかを試験することによって容易に決定される。この目的のための有用な評価法としては、遺伝子の産物をコードするmRNAの定量、遺伝子の産物についてのウェスタンブロット分析、酵素活性遺伝子産物についての活性分析法、またはソフトアーガー成長分析法、またはレポーター遺伝子構築物分析法、またはイン・ビボ腫瘍成長分析法が挙げられ、これらはいずれも本明細書またはRamchandani et al. (1997) Proc. Natl. Acad. Sci. USA 94: 684-689に詳細に記載されている。
【0100】
本発明で用いられるアンチセンスオリゴヌクレオチドは、H-ホスホネート化学、ホスホルアミダイト化学、またはH-ホスホネート化学とホスホルアミダイト化学の組合せ(すなわち、幾つかのサイクルについてH-ホスホネート化学および他のサイクルについてホスホルアミダイト化学)などの周知の化学的方法を用いて適当な固形支持体上で好都合に合成することができる。適当な固形支持体としては、コントロールド-ポアグラス(controlled-pore glass; CPG)のような固相オリゴヌクレオチド合成に用いられる標準的固形支持体のいずれかが挙げられる(例えば、 Pon, R. T. (1993) Methods in Molec. Biol. 20:465-496参照)。
【0101】
特に好ましいオリゴヌクレオチドは、表1に示されるヌクレオチド配列を包含する約13-約35個のヌクレオチドのヌクレオチド配列を有する。更に一層特に好ましいオリゴヌクレオチドは、表1に示されるヌクレオチド配列の約15-約26個のヌクレオチドのヌクレオチド配列を有する。
【0102】
【表1】
【0103】
下記の実施例は、本発明のある種の好ましい態様を更に例示しようとするものであり、発明の範囲を制限しようとするものではない。
実施例
(実施例1)
【0104】
N-(2-アミノ-フェニル)-4-[3-(3,4-ジクロロ-フェニル)-アクリロイル]-ベンズアミド (Ia)
工程1: 4-[3-(3,4-ジクロロ-フェニル)-アクリロイル]-安息香酸 (IIa)
4-アセチル安息香酸 (1.71g, 10.44ミリモル)、3,4-ジクロロベンズアルデヒド(2.05g, 11.49ミリモル)またはアルデヒド(1.1当量)をMeOH (50ml)中で室温にて攪拌懸濁したものに、NaOH (26.1ml, 1N、H2O中)の溶液を加えた。19時間後、反応混合物を濾別し、MeOHで洗浄し、乾燥して、標題化合物 IIa (3.22g, 10.03ミリモル, 収率96%)を黄色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): 8.35 (s, 1H), AB系(δA=8.13, δB=8.01, J=8.4Hz, 4H), 8.12 (d, J=15.8Hz, 1H), 7.93 (d, J=7.9Hz, 1H), 7.76 (d, J=8.3Hz, 1H), 7.73 (d, 15.8Hz, 1H)。
【0105】
工程2: N-(2-アミノ-フェニル)-4-[3-(3,4-ジクロロ-フェニル)-アクリロイル]-ベンズアミド (Ia)
IIa (300mg, 0.93ミリモル)を無水DMF (15ml)に窒素下で室温にて攪拌溶解したものに、Et3N (156μl, 1.12ミリモル)およびBOP試薬(454mg, 1.03ミリモル)をそれぞれ加えた。30分後、1,2-フェニレンジアミン(111mg, 1.03ミリモル)、Et3N (391μl, 2.80ミリモル)を無水DMF (2ml)に溶解したものを滴加した。21時間後、反応混合物をNH4Clの飽和水溶液に空けて、AcOEtで希釈した。分離後、有機層を飽和NH4Cl、H2Oおよび塩水で連続して洗浄して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/CH2Cl2: 10/90→20/90)、標題化合物Ia(237mg, 0.58ミリモル、収率62%)を黄色粉末として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): 9.90 (s, 1H), 8.40-8.30 (m, 3H), 8.25-8.10 (m, 3H), 7.97 (d, J=8.8Hz, 1H), 7.85-7.75 (m, 2H), 7.23 (d, J=7.5Hz, 1H), 7.03 (t, J=7.3Hz, 1H), 6.84 (d, J=7.9Hz, 1H), 6.65 (t, J=7.5Hz, 1H), 4.99 (s, 2H)。
(実施例2および10)
【0106】
実施例2および10 (化合物Ib, Ij)は、実施例1の化合物Iaについて記載したのと同じ手続きを用いて調製した(工程図1)。
(実施例3)
【0107】
N-(2-アミノ-フェニル)-4-[3-(2,6-ジクロロ-フェニル)-アクリロイル]-ベンズアミド(Ic)
工程1:t-ブチル[2-(4-アセチル-ベンゾイルアミノ)-フェニル]-カルバメート (III)
4-アセチル安息香酸 (395mg, 2.41ミリモル)を無水DMF (15ml)中で窒素下にて室温で攪拌溶解したものに、Et3N (369μl, 2.65ミリモル)とBOP試薬 (1.171g, 2.65ミリモル)をそれぞれ加えた。30分後、t-ブチル(2-アミノ-フェニル)-カルバメート (551mg, 2.65ミリモル)、Et3N (1.01ml, 7.22ミリモル)を無水DMF (5ml)に溶解したものを滴加した。19時間後、反応混合物をNH4Clの飽和水溶液に空けて、AcOEtで希釈した。分離後、有機層を飽和NH4Cl、H2Oおよび塩水で連続して洗浄し、無水MgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 40/6O→50/50)、標題化合物 III (500mg, 1.41ミリモル, 59%収率)を黄色固形物として得た。1H NMR (300 MHz,CDCl3) δ(ppm): 9.47 (bs,1H), 8.1O-8.00 (m, 4H), 7.91 (d, J=7.9Hz, 1H), 7.33-7.15 (m, 3H), 6.67 (s,1H), 2.67 (s, 3H), 1.53 (s,9H)。
【0108】
工程2: t-ブチル(2-{4-[3-(2,6-ジクロロ-フェニル)-アクリロイル]-ベンゾイルアミノ}-フェニル)-カルバメート (IVc)
III (150mg, 0.42ミリモル)、2,6-ジクロロベンズアルデヒド(148mg, 0.85ミリモル)またはアルデヒド(1.5-2.0当量)をMeOH (10ml)に室温にて攪拌溶解したものに、NaOH (1.7ml, 1N、H2O中)の溶液を加えた。淡黄色沈澱が生じた。3日後、反応混合物を濾別し、H2Oで洗浄した。次に、固形残渣をAcOEtに溶解し、無水MgSO4上で乾燥し、濾過して、濃縮した。粗製残渣を最後にシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 20/80→40/60)、標題化合物IVc (185mg, 0.36ミリモル, 85%収率)を淡黄色フォームとして得た。1H NMR (300 MHz, CDCl3) δ(ppm): 9.48 (bs,1H), 8.11 (s, 4H), 7.96 (d, J=17.1Hz, 1H), 7.89 (d, J=8.8Hz, 1H), 7.68 (d, J=16.3Hz, 1H), 7.42 (d, J=7.9Hz, 2H), 7.35-7.15 (m, 4H), 6.68 (s,1H), 1.54 (s, 9H)。
【0109】
工程3: N-(2-アミノ-フェニル)-4-[3-(2,6-ジクロロ-フェニル)-アクリロイル]-ベンズアミド (Ic)
IVc (135mg, 0.26ミリモル)をCH2Cl2 (10ml)に室温にて攪拌溶解したものに、トリフルオロ酢酸 (2ml, 95%水溶液)を加えた。16時間後、反応混合物を濃縮し、シリカゲル上でフラッシュクロマトグラフィーによって直接精製し(AcOEt/ CH2Cl2: 15/85)、標題化合物 Ic (90mg, 0.22ミリモル, 83%収率)を橙色固形物として得た。1H NMR (300 MHz, DMSO-d6) δ(ppm): 9.89 (s,1H), AB系(δA=8.23, δB=8.19, J=8.5Hz, 4H), 7.90 (d, J=16.3Hz, 1H), 7.78 (d, J=16.3Hz, 1H), 7.67 (d, J=7.9Hz, 2H), 7.55-7.45 (m,1H), 7.23 (d, J=7.5Hz, 1H), 7.03 (t, J=7.5Hz, 1H), 6.83 (d, J=7.9Hz, 1H), 6.64 (t, J=7.3Hz, 1H), 5.00 (s, 2H)。
(実施例4−9)
【0110】
実施例4-9 (化合物Id-Ii)は、実施例3の化合物Icについて記載したのと同じ手続きを用いて調製した(工程図2)。
【0111】
【表2−1】
【表2−2】
(実施例11)
【0112】
N-(2-アミノ-フェニル)-4-[2-(3,4,5-トリメトキシ-フェニルカルバモイル)-ビニル]-ベンズアミド (Va)
工程1: メチル 4-(2-t-ブトキシカルボニル-ビニル)-ベンゾエート (VI)
無水i-Pr2NH (1.76ml, 12.49ミリモル)を0℃で窒素下にて攪拌した無水THF (30ml)に溶解したものに、n-BuLi (5.36ml, 13.40ミリモル, 2.5M、ヘキサン中)の溶液を徐々に加えた。30分後、LDAを-78℃に冷却し、t-ブチルアセテート(1.64ml, 12.18ミリモル)を滴加した。30分後、メチル 4-ホルミルベンゾエート (1.00g, 6.09ミリモル)を無水THF (10ml)に溶解したものを徐々に加えた。2時間後、2-クロロ-4,6-ジメトキシ-1,3,5-トリアジン(1.604g, 9.14ミリモル)を無水THF (10ml)に溶解したものを加えた。次いで、温度を、一晩で室温まで上昇させた。懸濁液を生じた。反応混合物をNH4Clの飽和水溶液に空けて、AcOEtで希釈した。分離後、有機層をH2Oおよび塩水で連続して洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。粗生成物をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 10/90→15/85)、標題生成物VI (785mg, 3.00ミリモル, 49%収率)を白色固形物として得た。1H NMR(300 MHz, CDCl3) δ(ppm): AB系(δA=8.04, δB=7.57, J=8.4Hz, 4H), 7.60 (d, J=15.4Hz, 1H), 6.46 (d, J=15.8Hz, 1H), 3.93 (s, 3H), 1.54 (s, 9H)。13C NMR (75 MHz, CDCl3)δ(ppm): 166.72, 166.01, 142.31, 139.18, 131.33, 130.26, 127.99, 122.87, 81.11, 52.46, 28.40。
【0113】
工程2: メチル 4-(2-カルボキシ-ビニル)-ベンゾエート (VII)
VI (745mg, 2.84ミリモル)をCH2Cl2 (10ml)に室温にて攪拌溶解したものに、トリフルオロ酢酸 (6ml, 95%、水中)を加えた。27時間後、反応混合物を濃縮し、水中で粉砕した。1時間後、懸濁液を濾別し、H2Oで洗浄し、乾燥して、標題化合物 VII (556mg, 2.70ミリモル, 95%収率)を灰白色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): AB系(δA=8.01, δB=7.88, J=8.1Hz, 4H), 7.68 (d, J=15.8 Hz, 1H), 6.70 (d, J=16.3Hz, 1H), 3.90 (s, 3H)。
【0114】
方法A, 工程3: メチル 4-[2-(ベンゾトリアゾール-1-イルオキシカルボニル)-ビニル]-ベンゾエート (VIII)
VII (264mg, 1.28ミリモル)を無水DMF (10ml)に窒素下で室温にて攪拌溶解したものに、Et3N (196μl, 1.41ミリモル)とBOP試薬 (680mg, 1.1.54ミリモル)をそれぞれ加えた。数分後、沈澱を生じた。3時間後、反応混合物をNH4Clの飽和水溶液に空けて、AcOEtで希釈した。分離後、有機層を飽和NH4Cl、H2Oおよび塩水で連続して洗浄し、少し濃縮し、ヘキサンを加えた。懸濁液を濾別し、ヘキサンで洗浄した。固形物を水中で粉砕し、濾別し、水で洗浄し、乾燥して、標題化合物 VIII (346mg, 1.07ミリモル, 84%収率)を淡黄色固形物として得た(シリカゲル上では安定でない!)。1H NMR (300 MHz, CDCl3) δ(ppm): 8.56 (d, J=8.3Hz, 1H), 8.21-8.02 (m, 3H), 7.9O-7.72 (m, 4H), 7.62 (t, J=7.4Hz, 1H), 3.97 (s, 3H)。
【0115】
工程4: メチル4-[2- (3,4,5-トリメトキシ-フェニルカルバモイル)-ビニル]-ベンゾエート (IXa)
VIII (150mg, 0.46ミリモル)を無水CH2Cl2 (10ml)に窒素下で室温にて攪拌懸濁したものに、Et3N (194μl, 1.39ミリモル)および3,4,5-トリメトキシアニリン(94mg, 0.51ミリモル)またはArNH2(1.1-1.2当量)をそれぞれ加えた。反応混合物を、60℃に加熱した。20時間後、反応混合物を濃縮し、AcOEtで希釈し、NH4Clの飽和水溶液、H2Oおよび塩水で連続して洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。粗生成物をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ CH2Cl2: 15/85→20/80)、標題生成物IXa (130mg, 0.35ミリモル, 75%収率)を黄色固形物として得た。1H NMR (300 MHz, アセトン-d6)δ(ppm): 9.42 (bs, 1H), AB系 (δA=8.09, δB=7.78, J=8.1Hz, 4H), 7.75 (d, J=15.6Hz, 1H), 7.21 (s, 2H), 7.00 (d, J=15.8Hz, 1H), 3.94 (s, 3H), 3.85 (s, 6H), 3.73 (s, 3H)。
【0116】
工程5: 4-[2-(3,4,5-トリメトキシ-フェニルカルバモイル)-ビニル]-ベンゾエート (Xa)
IXa (125mg, 0.34ミリモル)をTHF (5ml)に室温にて攪拌溶解したものに、LiOH.H2O (35mg, 0.84ミリモル)を水(5ml)に溶解したものを加えた。1.5日後、反応混合物を濃縮し、水で希釈し、1N HClでpH 4-5まで酸性にして、沈澱を得た。10分間攪拌した後、懸濁液を濾別し、水で洗浄し、乾燥して、標題化合物 Xa (110mg, 0.31ミリモル, 91%収率)を淡黄色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm) : 10.29 (s, 1H), AB系 (δA=8.04, δB=7.76, J=8.4Hz, 4H), 7.65 (d, J=15.8Hz, 1H), 7.13 (s, 2H), 6.94 (d, J=15.8Hz, 1H), 3.81 (s, 6H), 3.67 (s, 3H)。
【0117】
工程6: N-(2-アミノ-フェニル)-4-[2-(3,4,5-トリメトキシ-フェニルカルバモイル)-ビニル]-ベンズアミド (Va)
Xa(110mg, 0.31ミリモル)を無水DMF (3ml)に窒素下で室温にて攪拌溶解したものに、Et3N (47μl, 0.34ミリモル)およびBOP試薬 (163mg, 0.37ミリモル)をそれぞれ加えた。30分後、1,2-フェニレンジアミン (37mg, 0.34ミリモル), Et3N(129μl, 0.92ミリモル)を無水DMF (1ml)に溶解したものを滴加した。3時間後、反応混合物をNH4Clの飽和水溶液に空けて、AcOEtで希釈した。分離後、有機層を飽和NH4Cl、H2Oおよび塩水で連続して洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/CH2Cl2: 50/50→80/20)、標題化合物 Va (98mg, 0.22ミリモル,71%収率)を黄色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): 10.27 (s, 1H), 9.76 (s, 1H), AB系(δA=8.09, δB= 7.78, J=7.9Hz, 4H), 7.71 (d, J=15.8Hz, 1H), 7.22 (d, J=7.5Hz, 1H), 7.14 (s, 2H), 7.02 (t, J=7.0Hz, 1H), 6.95 (d, J=15.8Hz, 1H), 6.83 (d, J=7.9Hz, 1H), 6.65 (t, J=7.5Hz, 1H), 4.97 (bs, 2H), 3.81 (s, 6H), 3.68 (s, 3H)。
(実施例12)
【0118】
N-(2-アミノ-フェニル)-4-{2-[(ピリジン-3-イルメチル)-カルバモイル]-ビニル}-ベンズアミド (Vb)
方法B, 工程3: メチル4-[2-(ピリジン-3-イルメチル)-カルバモイル)-ビニル]-ベンゾエート (Vb)
VIII (140mg, 0.68ミリモル)を無水DMF (5ml)に窒素下で室温にて攪拌溶解したものに、Et3N(104μl, 0.75ミリモル)とBOP試薬 (331mg, 0.75ミリモル)をそれぞれ加えた。30分後、3-(アミノメチル)ピリジン (90μl, 0.88ミリモル)またはR1R2NH (1.2-1.3当量)、Et3N (284μl, 2.04ミリモル)を無水DMF (2ml)に溶解したものを滴加した。4時間後、反応混合物をNH4Clの飽和水溶液に空けて、AcOEtで希釈した。分離後、有機層を飽和NH4Cl、H2Oおよび塩水で連続して洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(MeOH/ CH2Cl2: 5/95→7/93)、標題化合物 IXb (185mg, 0.62ミリモル, 92%収率)を白色固形物として得た。1H NMR (300 MHz, CDCl3)δ(ppm): 8.67-8.44 (m, 2H), AB系(δA=8.03,δB=7.55, J=8.4Hz, 4H), 7.78-7.64 (m, 2H), 7.33-7.26 (m, 1H), 6.54 (d, J=15.8Hz, 1H), 6.38 (bs, 1H), 4.61 (d, J=6.2Hz, 2H), 3.92 (s, 3H)。
【0119】
工程4: N-(2-アミノ-フェニル)-4-{2-[(ピリジン-3-イルメチル)-カルバモイル]-ビニル}-ベンズアミド (Vb)
標題化合物Vbは、IXbから実施例10の工程5および6(工程図3)と同じ手続きによって2段階で得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): 9.74 (s, 1H), 8.79 (t, J=5.7Hz, 1H), 8.58 (s, 1H), 8.52 (d, J=4.0Hz, 1H), 8.06 (d, J=7.9Hz, 2H), 7.83-7.68 (m, 3H), 7.59 (d, J=15.8Hz, 1H), 7.41 (dd, J=7.9, 4.7Hz, 1H), 7.21 (d,J=7.9Hz, 1H), 7.02 (t, J=7.0Hz, 1H), 6.83 (d, J=15.8Hz, 1H), 6.82 (d, J=7.5Hz, 1H), 6.64 (t,J=7.3Hz, 1H), 4.96 (bs, 2H), 4.48 (d, J=5.7Hz, 2H)。
(実施例13−15)
【0120】
実施例13-15 (化合物Vc-Ve)は、実施例12の化合物Vbについて記載したのと同じ手続きを用いて調製した(工程図3)。
(実施例16)
【0121】
N-(2-アミノ-フェニル)-4-[2-(2-ピリジン-3-イル-エチルカルバモイル)-ビニル]-ベンズアミド (Vf)
工程1: t-ブチル[2-(4-ホルミル-ベンゾイルアミノ)-フェニル]-カルバメート (XI)
4-カルボキシベンズアルデヒド(3.00g, 19.98ミリモル)を無水CH2Cl2(10ml)に窒素下で室温にて攪拌懸濁したものに、塩化チオニル (2.19ml, 29.97ミリモル)と無水DMF(387μl, 5.00ミリモル)をそれぞれ加えた。反応混合物を5時間還流した。次に、反応混合物を室温まで放冷し、濃縮し、無水CH2Cl2(20ml)で窒素下にて希釈した。この溶液を、t-ブチル(2-アミノ-フェニル)-カルバミン酸エステル(4.575g, 21.98ミリモル)、Et3N (8.36ml, 59.95ミリモル)を無水CH2Cl2(50ml)に窒素下で-20℃で冷却混合したものにカニューレ導入した。1時間後、反応混合物を室温まで温度を上昇させた。1時間後、これをNH4Clの飽和水溶液に空けて、CH2Cl2で抽出した。一緒に合わせた有機層を連続してMgSO4上で乾燥し、濾過して、濃縮した。粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 30/70→40/60)、標題化合物 XI (4.80g, 14.11ミリモル, 71%収率)を淡黄色固形物として得た。1H NMR (300 MHz,CDCl3)δ(ppm): 10.11 (s, 1H), 9.58 (bs, 1H), AB系(δA=8.14, δB=7.99, J=8.1Hz, 4H), 7.89 (d, J=7.9Hz, 1H), 7.35-7.10 (m, 3H), 6.75 (s, 1H), 1.53 (s, 9H)。
【0122】
工程2: メチル3-[4-(2-t-ブトキシカルボニルアミノ-フェニルカルバモイル)-フェニル]-アクリレート (XII)
化合物XI (500mg, 1.47ミリモル)、メチル(トリフェニル-ホスホラニリデン)アセテート (590mg, 1.76ミリモル)を無水トルエン(20ml)に攪拌懸濁したものを、窒素下で90℃で加熱した。2日後、反応混合物を濃縮し、直ちにシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 30/70→40/60)、標題化合物Xll (568mg, 1.43ミリモル, 97%収率)を淡黄色フォームとして得た。1H NMR (300 MHz, CDCl3) δ(ppm): 9.32 (bs,1H), AB系(δA=7.99, δB=7.62, J=8.4Hz, 4H), 7.87 (d, J=7.9Hz, 1H), 7.73 (d, J=15.8Hz, 1H), 7.32-7.13 (m, 3H), 6.69 (bs, 1H), 6.53 (d, J=16.3Hz, 1H), 3.83 (s, 3H), 1.53 (s, 9H)。
【0123】
工程3: 3-[4-(2-t-ブトキシカルボニルアミノ-フェニルカルバモイル)-フェニル]-アクリル酸(XIII)
化合物XII (560mg, 1.41ミリモル)をTHF (20ml)に室温にて攪拌溶解したものに、LiOH.H2O (148mg, 3.53ミリモル)を水 (20ml)に溶解したものを加えた。23時間後、反応混合物を濃縮し、水で希釈し、1N HClでpH 4-5になるまで酸性にして、白色沈澱を得た。15分間攪拌した後、懸濁液を濾別し、水で洗浄し、乾燥して、標題化合物XIII (495mg, 1.29ミリモル, 92%収率)を白色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm):9.92 (s, 1H), 8.72 (bs, 1H), AB系(δA=8.02,δB=7.90, J=7.9Hz, 4H), 7.69 (d, J=16.3Hz, 1H), 7.62-7.53 (m, 2H), 7.3O-7.13 (m, 2H), 6.72 (d,J=16.3Hz, 1H), 1.48 (s, 9H)。
【0124】
工程4: t-ブチル(2-{4-[2-(2-ピリジン-3-イル-エチルカルバモイル)-ビニル]-ベンゾイルアミノ}-フェニル)-カルバメート (XIVf)
化合物XIII (80mg, 0.21ミリモル)を無水DMF (3ml)に窒素下で室温にて攪拌溶解したものに、Et3N (35μl, 0.25ミリモル)およびBOP試薬 (102mg, 0.23ミリモル)をそれぞれ加えた。30分後、3-(2-アミノエチル)ピリジン (51mg, 0.42ミリモル)またはRXH (1.5-2.0当量)、Et3N (87μl, 0.63ミリモル)を無水DMF(1ml)に溶解したものを滴加した。3-5時間後、反応混合物をNH4Clの飽和水溶液に空けて、AcOEtで希釈した。分離後、有機層を連続して飽和NH4Cl、H2Oおよび塩水で洗浄し、MgSO4上で乾燥し、濾過して、濃縮し、標題化合物 XIVfを得た。これは、更に精製することなく次工程に用いた。
【0125】
工程5: N-(2-アミノ-フェニル)-4-[2-(2-ピリジン-3-イル-エチルカルバモイル)-ビニルl-ベンズアミド (Vf)
XIVfをCH2Cl2 (15ml)に室温にて攪拌溶解したものに、トリフルオロ酢酸 (2ml, 95%、水中)を加えた。18時間後、反応混合物を濃縮し、水に溶解し、NaHCO3の飽和水溶液でpH=7になるまで中和した。淡黄色沈澱を生じた。数分後、懸濁液を濾別し、H2Oで洗浄し、乾燥して、標題化合物 Vf (69mg, 0.18ミリモル, 2工程についての収率85%)を淡黄色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): 9.72 (s, 1H), 8.53-8.41 (m, 2H), 8.29 (t, J=5.5Hz, 1H), 8.05 (d, J=8.4Hz, 2H), 7.8O-7.63 (m, 3H), 7.51 (d, J=15.8Hz, 1H), 7.37 (dd, J=7.5, 4.8Hz, 1H), 7.21 (d, J=7.5Hz, 1H), 7.02 (t, J=7.5Hz, 1H), 6.82 (d, J=7.5Hz, 1H), 6.76 (d, J=15.8Hz, 1H), 6.64 (t, J=7.3Hz, 1H), 4.95 (bs, 2H), 3.51 (dd, J=6.8Hz, 2H), 2.86 (t, J=6.8Hz, 2H)。
(実施例17−26)
【0126】
実施例17-26 (化合物Vg-Vp)は、実施例16の化合物Vfについて記載したのと同じ手続きを用いて調製した(工程図4)。
【0127】
【表3−1】
【表3−2】
【表3−3】
(実施例27)
【0128】
N-(2-アミノ-フェニル)-4-[3-(3-シクロペンチルオキシ-4-メトキシ-フェニルアミノ)-プロペニル]-ベンズアミド (XVa)
工程1: t-ブチル {2-[4-(3-オキソ-プロペニル)-ベンゾイルアミノ]-フェニル}-カルバメート (XVI)
化合物XI (4.00g, 11.75ミリモル)、(トリフェニルホスホラニリデン)-アセトアルデヒド(3.60g, 11.83ミリモル)を無水トルエン (100ml)中で攪拌懸濁したものを、窒素下にて80℃で加熱した。2日後、反応混合物を濃縮し、シリカゲル上でフラッシュクロマトグラフィーによって直接精製し(AcOEt/ヘキサン: 30/70)、標題化合物 XVI (3.70g, 10.10ミリモル, 86%収率)を黄色の粘稠な固形物として得た(ジエンを僅かに混入)。1H NMR (300 MHz, CDCl3)δ(ppm): 9.75 (d, J=7.8Hz, 1H), 9.49 (bs, 1H), AB系(δA=8.03, δB=7.65, J=8.4Hz, 4H), 7.85-7.72 (m, 1H), 7.52 (d, J=15.6Hz, 1H), 7.33-7.05 (m, 3H), 7.05-6.90 (m,1H), 6.78(dd, J=15.6, 7.8Hz, 1H), 1.53 (s, 9H)。
【0129】
工程2: t-ブチル (2-{4-[3-(3-シクロペンチルオキシ-4-メトキシ-フェニルアミノ)-プロペニル]-ベンゾイルアミノ}-フェニル)-カルバメート (XVIIa)
化合物XVI (210mg, 0.57ミリモル)、3-シクロペンチルオキシ-4-メトキシ-アニリン (125mg, 0.60ミリモル)またはArNH2 (1.05-1.2当量)を無水THF (7ml)に窒素下で室温にて攪拌溶解したものに、二塩化ジブチルスズ(3.5mg, 0.01ミリモル)を加えた。10分後、フェニルシラン (78μl, 0.63ミリモル)を滴加した。3日後、反応混合物を濃縮し、シリカゲル上でフラッシュクロマトグラフィーによって直接精製し(AcOEt/ヘキサン: 30/70→50/50)、標題化合物XVIIaを黄色の粘稠な油状生成物として得た。
【0130】
工程3: N-(2-アミノ-フェニル)-4-[3-(3-シクロペンチルオキシ-4-メトキシ-フェニルアミノ)-プロペニル]-ベンズアミド (XVa)
XVIIaをCH2Cl2(30ml)に室温にて攪拌溶解したものに、トリフルオロ酢酸 (5ml, 95%、水中)を加えた。16時間後、反応混合物を濃縮し、水に溶解し、NaOH(1N)の水溶液で塩基性にし、pH=8とした。ベージュ色沈澱が生じた。15分後、懸濁液を濾別し、H2Oで洗浄し、風乾した。粗生成物をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ CH2Cl2: 15/85→20/80+εNH4OH)、標題化合物 XVa (145mg, 0.32ミリモル, 55%、二段階についての収率)を黄色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): ロータマーの混合物、9.67および9.63 (2s, 1H), 7.98 (d, J=7.9Hz, 2H), 7.57および7.51 (2d, J=7.9Hz, 2H), 7.20 (d, J=7.9Hz, 1H), 7.01 (t, J=7.7Hz, 1H), 6.82 (d, J=7.9Hz, 1H), 6.77-6.67 (m, 2H), 6.63 (t, J=7.5Hz, 1H), 6.54 (dt, J=16.3, 5.2Hz, 1H), 6.35および6.30 (2d, J=2.0Hz, 1H), 6.15および6.06 (2dd, J=8.6, 2.0Hz, 1H), 5.98および5.57 (2t, J=5.5Hz, 1H), 4.92 (bs, 2H), 4.78-4.63 (m, 1H), 4.32および3.87 (2d, J=5.7Hz, 2H), 3.65および3.62 (2s, 3H), 1.95-1.45 (m, 8H)。
(実施例28−32)
【0131】
実施例28-32 (化合物XVb-XVf)は、実施例27の化合物XVaについて記載したのと同じ手続きを用いて調製した(工程図5)。
(実施例33)
【0132】
N-(2-アミノ-フェニル)-4-[3-(4-トリル-スルホニルアミノ)-プロペニル]-ベンズアミド (XVg)
工程1: t-ブチル {2-[4-(3-ヒドロキシ-プロペニル)-ベンゾイルアミノ]-フェニル}-カルバメート (XVIII)
化合物XVI (1.00g, 2.79ミリモル)をエタノール(15ml)に窒素下で攪拌溶解したものに、水素化ホウ素ナトリウム (110mg, 2.73ミリモル)を加えた。5分後、反応混合物を水で反応停止させ、AcOEtで希釈した。分離後、有機層を塩水で連続して洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 40/60)、標題化合物 XVIII (910mg, 2.29ミリモル, 82%収率)を淡黄色固形物として得た(ジエンが僅かに混入)。1H NMR (300 MHz, CDCl3)δ(ppm): 9.20 (s, 1H), 7.90 (d, J=7.8Hz, 2H), 7.75 (d, J=7.5Hz, 1H), 7.43 (d, J=8.4Hz, 2H), 7.32-7.08 (m, 3H), 6.94 (s, 1H), 6.65 (d, J=15.9Hz, 1H), 6.45 (td, J=15.9, 5.4Hz, 1H), 4.35 (d, J=5.4Hz, 2H), 1.92 (s, 1H), 1.51 (s, 9H)。
【0133】
工程2: t-ブチル (2-{4-[3-(4-トリル-スルホニルアミノ)-プロペニル]-ベンゾイルアミノ}-フェニル)-カルバメート(XVIIg)
N-Boc-4-トリルスルホンアミド (221mg, 0.81ミリモル)とPPh3 (427mg, 1.63ミリモル)を無水THF(4ml)に窒素下で攪拌溶解したものに、化合物XVIII (200mg, 0.54ミリモル)を無水THF(1ml)に溶解したものおよびジエチルアゾジカルボキシレート(DEAD) (214μl, 1.36ミリモル)を加えた。16時間後、反応混合物を水で反応停止し、AcOEtで希釈した。分離後、有機層を水および塩水で連続して洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 40/60)、標題化合物XVIIg (337 mg)を得た。
【0134】
工程3: N-(2-アミノ-フェニル)-4-[3-(4-トリル-スルホニルアミノ)-プロペニル]-ベンズアミド (XVg)
XVIIgをCH2Cl2 (20ml)に室温にて攪拌溶解したものに、トリフルオロ酢酸 (2ml, 95%、水中)を加えた。16時間後、反応混合物を濃縮し、水に溶解し、NaHCO3の飽和水溶液で塩基性にした。水性層をAcOEtで抽出した。合わせた有機層を塩水で連続洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。粗製残渣をできるだけ少量のAcOEt/MeOH(95/5)の混合物で可溶化させ、ヘキサンで共沈させた。灰白色沈澱生成物を生じた。数分後、懸濁液を濾別し、ヘキサンで洗浄し、乾燥して、標題化合物XVg (173mg, 0.41ミリモル, 76%、二段階についての収率)を灰白色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): 9.64 (s, 1H), AB系(δA=7.93, δB=7.72, J=8.4Hz, 4H), 7.84 (s, 1H), 7.41 (t, J=8.4Hz, 4H), 7.16 (d, J=7.8Hz, 1H), 6.97 (t, J=7.5Hz, 1H), 6.77 (d, J=7.5Hz, 1H), 6.59 (t, J=7.8Hz, 1H), 6.52 (d, J=15.6Hz, 1H), 6.21 (dt, J=15.6, 5.7Hz, 1H), 4.89 (s, 2H), 3.60 (bs, 2H), 2.08 (s, 3H)。
(実施例34)
【0135】
N-(2-アミノ-フェニル)-4-{3-[(ピリジン-3-イルメチル)-アミノ]-プロペニル}-ベンズアミド (XVh)
工程1: メチル4-(3-オキソ-プロペニル)-ベンゾエート (XIX)
方法A: 化合物メチル4-ホルミルベンゾエート (4.00g, 24.37ミリモル)、(トリフェニルホスホラニリデン)-アセトアルデヒド(7.56g, 24.85ミリモル)を無水トルエン (100ml)に攪拌懸濁したものを、窒素下にて8O-90℃で加熱した。1日後、反応混合物を濃縮し、シリカゲル上でフラッシュクロマトグラフィーによって直接精製し(AcOEt/ヘキサン: 20/80→30/70)、標題化合物 XIX (2.52g, 13.25ミリモル, 54%収率)を淡黄色固形物(ジエンが僅かに混入)として得た。1H NMR (500 MHz, CDCl3)δ(ppm): 9.76 (d, J=7.3Hz, 1H), AB系(δA=8.11, δB=7.64, J=8.1Hz, 4H), 7.51 (d, J=15.6Hz, 1H), 6.79 (dd, J=15.8, 7.6Hz, 1H), 3.95 (s, 3H)。
【0136】
方法B: TDA-1 (6.278g, 19.41ミリモル)および10%炭酸カリウム水溶液 (100ml)をCH2Cl2 (100ml)に室温で激しく攪拌したエマルションに、(1,3-ジオキソラン-2-イル)メチルトリフェニルホスホニウムブロミド(10g, 23.29ミリモル)およびメチル4-ホルミルベンゾエート (3.187g, 19.41ミリモル)をそれぞれ加えた。18時間攪拌後、反応混合物をCH2Cl2で抽出し、合わせた有機層を濃縮した。次に、10% HCl水溶液(100ml)を加えて、混合物を室温で一晩攪拌した。反応混合物を、水で希釈し、CH2Cl2で抽出した。合わせた有機層を連続的にMgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン:20/80→30/70)、AcOEt/ヘキサン中で粉砕し、標題化合物 XIX (2.50g, 13.14ミリモル, 68%収率)を結晶性固形物として得た(純粋なトランス型であり、ジエンを含まない)。
【0137】
工程2: メチル4-{3-[(ピリジン-3-イルメチル)-アミノ]-プロペニル}-ベンゾエート (XXh)
化合物XIX (300mg, 1.58ミリモル)および3-(アミノメチル)ピリジン(193μl, 0.60ミリモル)またはRNH2(1.1-1.2当量)を無水ジクロロメタン(15ml)に窒素下で溶解したものを1時間攪拌し、水素化トリアセトキシホウ素ナトリウム(401mg, 1.89ミリモル)を加えた。64時間後、反応混合物をK2CO3の水溶液(10%)で反応停止し、ジクロロメタンで抽出した。合わせた有機層を、MgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(MeOH/CH2Cl2: 5/95 + εNH4OH)、標題化合物 XXh (188mg, 0.66ミリモル, 42%収率)を暗黄色油状生成物として得た。
【0138】
工程3: 4-[3-(第三ブトキシカルボニル-ピリジン-3-イルメチル-アミノ)-プロペニル]-安息香酸 (XXIh)
XXh (187mg, 0.66ミリモル)を1,4-ジオキサン(7ml)に室温にて攪拌溶解したものに、(Boc)2O (173mg, 0.80ミリモル)およびNaOHの水溶液(3.3ml, 1N)をそれぞれ加えた。24時間後、反応混合物を濃縮し、水で希釈し、HClの水溶液(1N)で中和した(pH=6-7)。生成する淡黄色懸濁液を、ジクロロメタンで抽出した。合わせた有機層をMgSO4上で乾燥し、濾過し、濃縮して、標題化合物 XXIh (160mg, 0.43ミリモル, 66%収率)を黄色固形物として得た。
【0139】
工程4: t-ブチル{3-[4-(2-アミノ-フェニルカルバモイル)-フェニル]-アリル}-ピリジン-3-イルメチル-カルバメート(XXIIh)
標題化合物XXIIh (実施例34)は、実施例11、工程6と同じ手続きによって一段階でXXIhから淡黄色フォームとして得た。
【0140】
工程5: N-(2-アミノ-フェニル)-4-{3-[(ピリジン-3-イルメチル)-アミノ]-プロペニル}-ベンズアミド (XVh)
XXIIh (77mg, 0.17ミリモル)をジクロロメタン(10ml)中で室温にて攪拌溶解したものに、TFA (2ml, 95%、水中)を加えた。4.5時間後、反応混合物を濃縮し、水で希釈し、NaOHの水溶液(1N)で塩基性にし(pH=9)、ジクロロメタンで抽出した。合わせた有機層を、MgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(MeOH/CH2Cl2: 10/90 + εNH4OH)、標題化合物 XVh (35mg, 0.10ミリモル, 58%収率)を黄色粉末として得た。1H NMR (400MHz, DMSO-d6)δ(ppm): 9.64 (s, 1H), 8.55 (s, 1H), 8.44 (d, J=3.9Hz, 1H), AB系(δA=7.94, δB=7.55, J=8.0Hz, 4H), 7.78 (d, J=7.4Hz, 1H), 7.36 (dd, J=7.0, 5.1Hz, 1H), 7.16 (d, J=7.4Hz, 1H), 6.98 (t, J=7.4Hz, 1H), 6.79 (d, J=7.8Hz, 1H), 6.65-6.55(m, 2H), 6.51 (dt, J=16.0, 5.9Hz, 1H), 4.93 (bs, 2H), 3.77 (s, 2H)。
(実施例35−36)
【0141】
実施例35-36 (化合物 XVi-XVj)は、実施例34の化合物XVhについて記載したのと同じ手続きを用いて調製した(工程図7,経路B)。
【0142】
【表4−1】
【表4−2】
(実施例37)
【0143】
N-(2-アミノ-フェニル)-4-(3-オキソ-3-フェニル-プロペニル)-ベンズアミド (XXIVa)
工程1: 4-(3-オキソ-3-フェニル-プロペニル)-安息香酸(XIIIa)
4-ホルミル安息香酸 (2.58g, 17ミリモル)およびアセトフェノン (2.0ml, 17ミリモル)またはアセトフェノン誘導体 (1.O-1.1当量)をMeOH (100ml)中で室温にて攪拌懸濁したものに、NaOHの溶液(34ml, 1N、H2O中)を加えた。16時間後、反応混合物を濃HClで酸性にし(pH=1-2)、濾別し、H2Oで洗浄し、乾燥して、標題化合物XXIIIa (3.73g, 14.6ミリモル, 86%収率)を黄色固形物として得た。
【0144】
工程2: N-(2-アミノ-フェニル)-4-(3-オキソ-3-フェニル-プロペニル)-ベンズアミド (XXIVa)
標題化合物XXIVaは、実施例1、工程2と同じ手続きによって一段階でXXIIIaから得た(工程図1)。1H NMR (300 MHz, DMSO-d6) δ(ppm): 9.77 (s, 1H), 8.21 (d, J=7.0Hz, 2H), 8.06 (m, 5H), 7.82 (d, J=15.4Hz, 1H), 7.71 (t, J=7.3Hz, 1H), 7.60 (t, J=7.3Hz, 2H), 7.18 (d, J=7.9Hz, 1H), 6.99 (t, J=7.0Hz, 1H), 6.80 (d, J=7.5Hz, 1H), 6.61 (t, J=7.3Hz, 1H), 4.95 (bs, 2H)。
(実施例38−41)
【0145】
実施例38-41 (化合物 XXIVb-XXIVe)は、実施例37の化合物XXIVaについて記載したのと同じ手続きを用いて調製した(工程図8)。
(実施例42)
【0146】
N-(2-アミノ-フェニル)-4-[3-(4-モルホリン-4-イル-フェニル)-3-オキソ-プロペニル]-ベンズアミド (XXIVf)
工程1: t-ブチル(2-{4-[3-(4-モルホリン-4-イル-フェニル)-3-オキソ-プロペニル]-ベンゾイルアミノ}-フェニル)-カルバメート (XXVf)
XI (210mg, 0.62ミリモル)、4'-モルホリノアセトフェノン (227mg, 1.11ミリモル)またはアセトフェノン誘導体 (1.5-2.0当量)をMeOH (10ml)に室温にて攪拌溶解したものに、NaOHの溶液(1.9ml, 1N、H2O中)を加えた。沈澱を生じた。3日後、反応混合物を濾別し、MeOHで洗浄し、風乾し、真空乾燥して、標題化合物 XXVf (295mg, 0.56ミリモル, 90%収率)を黄色固形物として得た。
【0147】
工程2: N-(2-アミノ-フェニル)-4-[3-(4-モルホリン-4-イル-フェニル)-3-オキソ-プロペニル]-ベンズアミド(XXIVf)
XXVf (285mg, 0.54ミリモル)をCH2Cl2 (10ml)に室温にて攪拌溶解したものに、トリフルオロ酢酸 (2ml, 95%、水中)を加えた。17時間後、反応混合物を濃縮し、AcOEtで希釈し、飽和NaHCO3、H2O、飽和NH4Cl、H2Oおよび塩水で連続的に洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。粗生成物をAcOEt/ヘキサンの混合物中で共沈させ、粉砕した。数時間後、懸濁液を濾別し、ヘキサンで洗浄し、乾燥して、標題化合物XXIVf (210mg, 0.49ミリモル,91%収率)を黄色-橙色固形物として得た。1H NMR (300 MHz, DMSO-d6) δ(ppm): 9.78 (s, 1H), 8.25-7.94 (m, 7H), 7.76 (d, J=15.4Hz, 1H), 7.22 (d, J=7.5Hz, 1H), 7.09 (d, J=8.8Hz, 2H), 7.03 (t, J=7.7Hz, 1H), 6.83 (d, J=7.5Hz, 1H), 6.65 (t, J=7.5Hz, 1H), 4.97 (bs, 2H), 3.88-3.70(m, 4H), 3.48-3.30 (m, 4H)。
【0148】
【表5】
(実施例43)
【0149】
N-(2-アミノ-フェニル)-4-(3-オキソ-3-フェニル-プロピル)-ベンズアミド (XXVIIa)
工程1: 4-(3-オキソ-3-フェニル-プロピル)-安息香酸(XXVIa)
カルコンXXIIIa (1.29g, 5.13ミリモル)をDMF (20ml)に室温にて攪拌溶解したものに、フェニルスルホニルヒドラジン(1.76g, 10.26ミリモル)を加えた。反応混合物を110℃で15時間攪拌し、冷却して、濃縮した。残りの油状残渣を、NH4Clの飽和水溶液とAcOEtとの間で分配した。分離後、有機層を乾燥し、部分蒸発させ、濾過した。濾液をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 50/50→75/25)、物質を得て、これを第二のカラム精製の後(MeOH/CH2Cl2: 5/95)、標題化合物XXVIa (400mg, 1.59ミリモル, 31%収率)を得た。
【0150】
工程2: N-(2-アミノ-フェニル)-4-(3-オキソ-3-フェニル-プロピル)-ベンズアミド (XXVIIa)
標題化合物XXVIIaは、実施例1、工程2と同じ手続きによって一段階でXXVIaから得た(工程図1)。1H NMR (300 MHz, DMSO-d6)δ(ppm): 9.59 (s, 1H), 8.00 (d, J=7.5Hz, 2H), 7.90 (d, J= 7.9Hz, 2H), 7.64 (t, J=7.5Hz, 2H), 7.43 (d, J= 7.9Hz, 2H), 7.16 (d, J=7.5Hz, 1H), 6.97 (t, J= 7.0Hz, 1H), 6.78 (d, J=7.0Hz, 1H), 6.59 (t, J= 7.5Hz, 1H), 4.88 (bs, 2H), 3.44 (t, J=7.3Hz, 2H), 3.03 (t, J=7.3Hz, 2H)。
(実施例44)
【0151】
N-(2-アミノ-フェニル)-4-(3-フェニル-プロピル)-ベンズアミド (XXIXa)
工程1: 4-(3-フェニル-プロピル)-安息香酸(XXVIIIa)
XXIIIa (1.34g, 5.31ミリモル)を25ml DMAに室温にて攪拌溶解したものを、10% Pd/C (600mg, Degussa型)上で1気圧で3時間水素化した。触媒をセライトパッドを介して濾過によって除去した後、溶液を濃縮し、残渣を水で処理した。沈澱後、懸濁液を濾別し、H2Oで洗浄し、乾燥して、標題化合物XXVIIIa (1.13g, 4.72ミリモル, 89%収率)を得た。
【0152】
工程2: N-(2-アミノ-フェニル)-4-(3-フェニル-プロピル)-ベンズアミド(XXIXa)
標題化合物XXIXaは、実施例1、工程2と同じ手続きによって一段階でXXVIIIaから得た(工程図1)。1H NMR (300 MHz, DMSO-d6)δ(ppm): 9.60 (s, 1H), 7.91 (d, J=7.9Hz, 2H), 7.34 (d, J=8.4Hz, 2H), 7.28 (d, J=7.5Hz, 2H), 7.23-7.15 (m, 4H), 6.97 (t, J=7.0Hz, 1H), 6.78 (d, J=7.5Hz, 1H), 6.59 (t, J=7.3Hz, 1H), 4.88 (bs, 2H), 2.71-2.59 (m, 4H), 1.92 (m, 2H)。
(実施例45)
【0153】
N-(2-アミノ-フェニル)-4-[3-(1,3-ジヒドロ-イソインドール-2-イル)-プロペニル]-ベンズアミド (XXXa)
工程1: メチル4-トリメチルシラニルエチニル-ベンゾエート (XXXI)
メチル4-ブロモベンゾエート (8.84g, 41.11ミリモル)、Pd(PPh3)2Cl2(840mg, 1.20ミリモル)およびCuI (455mg, 2.39ミリモル)を無水THF (200ml)に室温にて攪拌溶解したものに、窒素を15分間飽和した。次に、溶液を窒素下で0℃に冷却し、トリメチルシリルアセチレン (7.2ml, 50.91ミリモル)およびトリエチルアミン (22ml, 157.8ミリモル)を連続して加えた。反応混合物を、室温まで温度上昇させた。2時間後、Pd(PPh3)2Cl2(100 mg)およびCuI (80 mg)およびトリメチルシリルアセチレン (0.5ml)を再度加え、反応混合物を一晩攪拌した。次に、反応混合物をAcOEtで希釈し、NH4Clの飽和水溶液および塩水で連続的に洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 5/95→10/90)、標題化合物 XXXI (9.05g, 38.95ミリモル, 94%収率)を黄色の粘稠な固形物として得た。1H NMR: (400 MHz, CDCl3)δ(ppm): AB系(δA=7.67, δB=7.22, JAB=8.5Hz, 4H), 3.63 (s, 3H), 0.00 (s, 9H)。
【0154】
工程2: メチル4-エチニル-ベンゾエート(XXXII)
XXXI (9.05g, 38.95ミリモル)を0℃で窒素下にてMeOH (280ml)に攪拌溶解したものに、炭酸カリウム(1.62g, 11.72ミリモル)を加えた。3時間後、反応混合物を濃縮し、シリカゲル上でフラッシュクロマトグラフィーによって直接精製し(CH2Cl2: 100)、標題化合物XXXII (6.16g, 38.46ミリモル, 98%収率)を淡黄色固形物として得た。1H NMR: (400 MHz,CDCl3)δ(ppm): AB系(δA=7.98, δB=7.54, JAB=8.6Hz, 4H), 3.93 (s, 3H), 3.24 (s, 1H)。
【0155】
工程3: β-(4-メトキシカルボニル)-スチリルボロン酸(XXXIII)
XXXII (6.16g, 38.46ミリモル)を無水THF (15ml)に窒素下で室温にて攪拌溶解したものに、カテコールボラン(4.52ml, 42.80ミリモル)を加えた。反応混合物を70℃に4時間加熱し、カテコールボラン (2ml)を再度加えた。1.5時間後、反応混合物を室温まで冷却し、2N HClの水溶液 (50ml)を加え、一晩攪拌した。次に、これをRotavap上で濃縮し、濾別し、濾過ケーキをトルエン中で粉砕した。濾過後、中間固形物をTHF (50ml)に溶解し、2N HClの水溶液 (150ml)を加えた。生成懸濁液を一晩かけて40℃まで加熱し、濾別し、水で洗浄し、風乾および真空乾燥して、標題化合物XXXIII (3.10g, 15.05ミリモル, 39%収率)を灰白色の綿毛状固形物として得た。1H NMR: (400 MHz, DMSO-d6)δ(ppm): AB系(δA=7.96, δB=7.63, JAB=8.4Hz, 4H), 7.94 (s, 2H), 7.32 (d, J=18.2Hz, 1H), 6.30 (d, J=18.2Hz, 1H), 3.88 (s, 3H)。
【0156】
工程4: メチル4-[3-(1,3-ジヒドロ-イソインドール-2-イル)-プロペニル]-ベンゾエート (XXXIVa)
イソインドリン(116mg, 0.97ミリモル)およびパラホルムアルデヒド(32mg, 1.07ミリモル)を無水1,4-ジオキサン(10ml)中で窒素下にて90℃で15分間予熱して攪拌溶解したものに、XXXIII (245mg, 1.17ミリモル)を加えた。90℃で一晩攪拌した後、反応混合物を室温まで放冷し、2N HCl水溶液(30ml)を加え、30分間振盪した。次に、水性混合物をEt2Oで抽出し、2N NaOH(50ml)で塩基性にし、CH2Cl2で抽出した。合わせたジクロロメタン層をMgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(MeOH/ CH2Cl2: 5/95)、標題化合物XXXIVa (135mg, 0.46ミリモル, 48%収率)を灰白色固形物として得た。1H NMR (400 MHz, DMSO-d6)δ(ppm): AB系(δA=7.93, δB=7.64, JAB=8.4Hz, 4H), 7.29-7.17 (m, 4H), 6.75 (d, J=15.8Hz, 1H), 6.62 (dt, J=16.0, 6.3Hz, 1H), 3.94 (s, 4H), 3.88 (s, 3H), 3.55 (dd, J=6.1, 1.0Hz, 2H)。
【0157】
工程5: N-(2-アミノ-フェニル)-4-[3-(1,3-ジヒドロ-イソインドール-2-イル)-プロペニル]-ベンズアミド (XXXa)
標題化合物XXXaを、実施例11、工程5および6と同じ手続きによって二段階でXXXIVaから得た(工程図3)。1H NMR (400 MHz, DMSO-d6)δ(ppm): 9.67 (s, 1H), AB系 (δA=7.98, δB=7.63, JAB=8.3Hz, 4H), 7.3O-7.15 (m,5H), 7.00 (td, J=7.6, 1.5Hz, 1H), 6.81 (dd, J=8.0, 1.4Hz, 1H), 6.75 (d, J=15.8Hz, 1H), 6.66-6.56 (m, 2H), 4.93 (s, 2H), 3.95 (s, 4H), 3.56 (dd, J=6.2, 0.9Hz, 2H)。
(実施例46−49)
【0158】
実施例46-49 (化合物 XXXb-XXXe)を、実施例45の化合物XXXaについて記載したのと同じ手続き用いて調製した(工程図11)。
【0159】
【表6】
(実施例50)
【0160】
ヒストンデアセチラーゼ酵素活性の阻害
1. ヒトHDAC-1: 評価1
HDAC阻害剤を、Baculovirus昆虫細胞発現系から発現し精製したクローニングした組換えヒトHDAC-1酵素に対してスクリーニングした。デアセチラーゼ評価のため、20,000cpmの[3H]-代謝標識したアセチル化ヒストン基質(M. Yoshida et al., J. Biol. Chem. 265 (28): 17174-17179 (1990))を、30μgのクローニングした組換えhHDAC-1と共に37℃で10分間インキュベーションした。反応は、酢酸(0.04 M, 最終濃度)およびHCl (250 mM, 最終濃度)を加えて停止した。混合物を酢酸エチルで抽出し、放出された[3H]-酢酸をシンチレーション計数法によって定量した。阻害研究のため、酵素を化合物と共に4℃で30分間予備インキュベーションした後、酵素評価を開始した。HDAC酵素阻害剤についてのIC50値を、個々の化合物を用いる用量応答曲線を行い且つ最大阻害の50%を生じる阻害剤の濃度を測定することによって決定した。この手続きを用いて評価した典型的化合物についてのIC50を、表7-10の第三欄に示す(括弧内のデーターを除く)。
【0161】
2. ヒトHDAC-1: 評価2
代替法では、下記のプロトコールを用いて、本発明の化合物を評価した。この評価では、用いた緩衝剤は25mM HEPES, pH 8.0、137mM NaCl、2.7mM KCl、1mM MgCl2であり、基質はDMSO中での50mM原液中のBoc-Lys(Ac)-AMCである。酵素原液は、緩衝剤1ml当たり4.08μgであった。
【0162】
化合物を、酵素(20μl, 4.08μg/ml)と共に室温にて10分間予備インキュベーションした(評価プレートに移すため、2μl/DMSOを13μl/緩衝剤に希釈)(35μl予備インキュベーション容積)。混合物を、室温で5分間予備インキュベーションした。反応は、温度を37℃にし、16μlの基質を加えることによって開始した。総反応容積は50μlであった。反応を、20分後にBiomolによって指示された方法で調製した50μlの顕色剤(Fluor-de-Lys顕色剤, カタログ番号KI-105)を加えることによって停止した。プレートを暗所で室温にて10分間インキュベーションした後、読み取りを行った(λEx=360nm, λEm=470nm, カットオフフィルター435nm)。
【0163】
この手続きを用いて評価した典型的な化合物のIC50値を、表9の第三欄(括弧内[]データー)に示す。
【0164】
3. MTT評価
HCT116 細胞(2000/ウェル)を、化合物処理の1日前に96-ウェル組織培養プレートにまいた。様々な濃度の化合物を、細胞に加えた。細胞を、5% C02インキュベーターで37℃で72時間インキュベーションした。MTT(3-[4,5-ジメチルチアゾール-2-イル]-2,5 ジフェニルテトラゾリウムブロミド, Sigma)を0.5mg/mlの最終濃度で加え、細胞と共に4時間インキュベーションした後、可溶化緩衝剤(50% N,N-ジメチルホルムアミド, 20% SDS, pH 4.7) 1容を培養細胞に加えた。一晩インキュベーションした後、可溶化した色素をMR 700プレートリーダー(Dynatech Laboratories Inc.)を用いて、630nMでのリファレンスを用いて570nMでの比色計の読みによって定量した。OD値を、関連細胞系の標準増殖曲線によって細胞数に転換した。細胞数を溶媒処理した細胞数の50%まで減少させる濃度を、MTT IC50として決定する。典型的な化合物についてのIC50値を、表7-10の第四欄に示す。
【0165】
4. 免疫ブロットによる総細胞におけるヒストンH4アセチル化
培養で増殖するT24ヒト膀胱癌細胞を、HDAC阻害剤と共に16時間インキュベーションした。M. Yoshida et al.によって記載された通りの培養時間の後、ヒストンを細胞から抽出した(J. Biol. Chem. 265 (28): 17174-17179 (1990))。全ヒストンタンパク質20gをSDS/PAGEに装填し、ニトロセルロース膜に移した。膜を、アセチル化ヒストンH-4 に特異的なポリクローナル抗体(Upstate Biotech Inc.)でプローブした後、西洋ワサビペルオキシダーゼに接合した二次抗体(Sigma)でプローブした。エンハンスト・ケミルミネッセンス(Enhanced Chemiluminescence; ECL)(Amersham)検出を、Kodakフィルム(Eastman Kodak)を用いて行った。アセチル化H-4シグナルを、デンシトメトリーによって定量した。典型的データーを、表7-10の第五欄に示す。データーは、アセチル化H-4シグナルを50%だけ減少させるのに有効な濃度(EC50)として示される。
【0166】
【表7−1】
【表7−2】
【0167】
【表8−1】
【表8−2】
【表8−3】
【0168】
【表9−1】
【表9−2】
【表9−3】
【0169】
【表10】
【0170】
(実施例51)
イン・ビボでのヒト腫瘍異種移植片に対するヒストンデアセチラーゼ阻害剤の抗腫瘍効果
8-10週齢の雌CD1ヌードマウス(Taconic Labs, Great Barrington, NY)の側腹部に2x106個のプレコンディショニングしたHCT116ヒト結腸直腸癌細胞を皮下投与した。これらの細胞のプレコンディショニングは、同一系統のヌードマウスで少なくとも3回連続的に腫瘍を移植することによって行った。その後、約30mgの腫瘍断片を摘出し、Forene麻酔(Abbott Labs, Geneva, Switzerland)によりマウスの左側腹部に皮下移植した。腫瘍が100mm3の平均容積に達したときに、ヒストンデアセチラーゼ阻害剤をPBS、DMSO/水、またはTween 80/水のような適当なビヒクルに溶解したものを10mg/kgの出発用量でマウスに静脈内、皮下、または腹腔内に毎日投与した。HDAC阻害剤の最適用量を、標準的プロトコールによって用量応答実験によって確立した。腫瘍容積を、標準的方法によって輸液後2日毎に計算した(例えば、Meyer et al., Int. J. Cancer 43: 851-856 (1989))。本発明によるHDAC阻害剤を投与したところ、ビヒクルだけを投与したコントロール(すなわち、HDAC阻害剤なし)と比較して腫瘍重量および容積が有意に減少し、これらの化合物のサブセットは毒性を示した。一例として、化合物XVjについての結果を、図1に示す。
【図面の簡単な説明】
【0171】
【図1】上記の実施例51に記載のイン・ビボでのヒト腫瘍異種移植片に対する本発明によるヒストンデアセチラーゼ阻害剤の抗腫瘍効果。
【技術分野】
【0001】
発明の背景
発明の分野
本発明は、ヒストンデアセチラーゼの阻害に関する。更に詳細には、本発明は、ヒストンデアセチラーゼ酵素活性を阻害する化合物および方法に関する。
【背景技術】
【0002】
関連技術の概要
真核生物細胞では、核DNAはヒストンと会合して、クロマチンと呼ばれるコンパクトな複合体を形成している。ヒストンは、真核生物種中で一般に高度に保存される塩基性タンパク質のファミリーを構成する。H2A、H2B、H3、およびH4と呼ばれるコアヒストン同士が会合して、タンパク質コアを形成する。DNAはこのタンパク質コアの周りを取り巻き、ヒストンの塩基性アミノ酸がDNAの負に帯電したリン酸基と相互作用している。DNAの約146の塩基対がヒストンコアに巻き付いて、クロマチンの反復構造モティーフであるヌクレオソーム粒子を形成している。
【0003】
Csordas, Biochem. J., 286:23-38 (1990)では、ヒストンをN-末端リシン残基のα,ε-アミノ基の翻訳後アセチル化、すなわちヒストンアセチルトランスフェラーゼ(HAT1)によって触媒される反応に付すことが教示されている。アセチル化によってリジン側鎖の正電荷が中和され、クロマチン構造に衝撃を与えることが考えられる。実際に、Taunton et al., Science, 272:408-411 (1996)には、転写因子のクロマチンへの接近がヒストン超アセチル化によって増すことが教示されている。Taunton et al.は、更にアセチル化ヒストンH4でのエンリッチメントはゲノムの転写的にサイレントな領域で見られたことも教示している。
【0004】
ヒストンアセチル化は可逆的修飾であり、脱アセチル化はヒストンデアセチラーゼ (HDAC)と呼ばれる酵素のファミリーによって触媒される。Grozinger et al., Proc. Natl. Acad. Sci. USA, 96: 4868-4873 (1999)では、HDACが酵母Rpd3様タンパク質によって表される第一のクラスと、酵母Hdal様タンパク質によって表される第二のクラスの2つのクラスに分類されることが教示されている。Grozinger et al.は、ヒトHDAC1、HDAC2およびHDAC3タンパク質が第一のクラスのHDACに属し、HDAC4、HDAC5およびHDAC6と命名された第二のクラスのHDACに属する新規なタンパク質も開示している。Kao et al., Genes & Dev., 14: 55-66 (2000),には、第二のクラスのHDACの新たな一員であるHDAC7が開示されている。Van den Wyngaert, FEBS, 478: 77-83 (2000)には、第一のクラスのHDACの新たな一員であるHDAC8が開示されている。
【0005】
Richon et al., Proc. Natl. Acad. Sci. USA, 95: 3003-3007 (1998)には、HDAC活性がStreptomyces hygroscopicusから単離される天然産物であるトリコスタチンA (TSA)および合成化合物であるスベロイルアニリドヒドロキサム酸(SAHA)によって阻害されることが開示されている。Yoshida and Beppu, Exper. Cell Res., 177: 122-131 (1988)には、TSAが細胞サイクルのG1およびG2期においてラット線維芽細胞の増殖を停止し、細胞サイクル調節においてHDACが関係していることが教示されている。実際に、Finnin et al., Nature, 401: 188-193 (1999)では、TSAおよびSAHAが細胞成長を阻害し、末端分化を誘発し、マウスにおける腫瘍の形成を予防することが教示されている。Suzuki et al., 米国特許第6,174,905号明細書、欧州特許第0847992号明細書、日本国特許第258863/96号明細書、および日本国特許出願第10138957号明細書には、細胞分化を誘発し且つHDACを阻害するベンズアミド誘導体が開示されている。Delorme et al., WO 01/38322号明細書およびPCT IB01/00683号明細書には、HDAC阻害剤として働く他の化合物が開示されている。
【0006】
これらの知見は、HDAC活性の阻害が細胞サイクル調節への介入の新規な方法であり、HDAC阻害剤が細胞増殖性疾患または疾病の治療に大きな治療能力を有することを示唆している。これまでのところ、ヒストンデアセチラーゼの阻害剤は当該技術分野ではほとんど知られていない。従って、他のHDAC阻害剤を同定し、強力なHDAC阻害活性に必要な構造的特徴を同定する必要がある。
【発明の開示】
【発明が解決しようとする課題】
【0007】
発明の概要
本発明は、細胞増殖性疾患の治療のための化合物および方法を提供する。本発明は、ヒストンデアセチラーゼ酵素活性の新規な阻害剤を提供する。
【0008】
第一の様相では、本発明は、ヒストンデアセチラーゼの阻害剤として有用な化合物を提供する。
【0009】
第二の様相では、本発明は、本発明によるヒストンデアセチラーゼの阻害剤、および薬学上許容可能なキャリヤー、賦形剤、または希釈剤を含んでなる医薬組成物を提供する。
【0010】
第三の様相では、本発明は、細胞におけるヒストンデアセチラーゼを阻害する方法であって、ヒストンデアセチラーゼの阻害が所望な細胞を本発明のヒストンデアセチラーゼの阻害剤と接触させることを含んでなる、方法を提供する。
【0011】
上記の説明は本発明のある種の様相を単にまとめたものであり、制限しようとするものではない。これらの様相および他の様相および態様を、下記において更に詳細に説明する。
【課題を解決するための手段】
【0012】
好ましい態様の詳細な説明
本発明は、ヒストンデアセチラーゼ酵素活性を阻害するための化合物および方法を提供する。本発明は、細胞増殖性疾患および疾病を治療するための組成物および方法も提供する。本明細書で参照される特許および科学文献によって、当業者に利用可能な知識が確立される。本明細書で引用される発行された特許明細書、特許出願明細書、および参考文献は、それぞれが参考として引用されることが具体的且つ個別的に示される場合と同程度まで参考として本明細書に引用される。不一致の場合には、本明細書の開示内容が優先する。
【0013】
第一の様相の一態様では、本発明は、下式
【化1】
(式中、Arはアリールまたはヘテロアリールであり、それらのそれぞれは場合によっては1-3個の置換基によって置換されている)
を有する化合物、または薬学上許容可能なその塩を含んでなる。
【0014】
好ましくは、Arは、[0013]節の化合物において、アリールまたはピリジニルである。
【0015】
Arの好ましい置換基としては、ハロ、場合によってはハロで置換されたC1-C6-ヒドロカルビル、場合によってはハロで置換されたC1-C6-ヒドロカルビルオキシが挙げられる。特に好ましい置換基としては、フルオロ、クロロ、メトキシ、シクロプロピルオキシ、およびシクロペンチルオキシが挙げられる。
【0016】
[0013]節に記載の化合物の好ましい態様では、Arは下記の
【化2】
から選択される。
【0017】
[0013]節の好ましい化合物としては、下表6の化合物が挙げられる。
【0018】
第一の様相のもう一つの態様では、本発明は、下式
【化3】
(式中、Xは-N(R1)-、-O-、または-S-であるか、またはXは窒素を含むヘテロシクリルであり、但し、窒素は構造Vにおける隣接カルボニルに共有結合し且つ場合によっては1-3個の置換基で置換されており、
RおよびR1は独立して-H、または 場合によっては置換されたa) C1-C6-ヒドロカルビルまたはb) R2-L-であり、但し、R2はアリールまたはヘテロアリールであり、LはCO-C6-ヒドロカルビル-L1-CO-C6-ヒドロカルビルであり、L1は共有結合、-O-、-S-、または-NH-である)
を有する化合物、および薬学上許容可能なその塩を含んでなる。
【0019】
好ましくは、 [0018]節に記載の化合物では、Xは-NH-、-O-、モルホリン-4-イル、ピペリジン-1-イル、ピペリジン-1-イル、またはピロリジン-1-イルである。
【0020】
[0018]節に記載の化合物のもう一つの好ましい態様では、Xは-N(R1)-であり、但し、R1は場合によっては置換メチルまたはエチルである。好ましくは、R1はシアノエチルまたはピリジニルメチルである。
【0021】
好ましくは、 [0018]節に記載の化合物では、RはR2-L-であって、但し、R2はフェニル、ピリジニル、インジル、またはインドリルであり、Lは共有結合、メチル、エチル、またはオキシエチルである。
【0022】
Rの好ましい置換基としては、メトキシおよびヒドロキシが挙げられる。
【0023】
[0018]節に記載の化合物の好ましい態様では、R-X-の組合せは、下記の
【化4】
から選択される。
【0024】
[0018]節に記載の好ましい化合物としては、表7に挙げられているものが挙げられる。
【0025】
第一の様相のもう一つの態様では、本発明は、下式
【化5】
(式中、Yは-N(R4)-、-O-、-S-、-N(R4)SO2-、-SO2-N(R4)-、-SO2-、-N(R4)-C(O)-、-C(O)-N(R4)-、-NHC(O)NH-、-N(R4)C(O)O-、-OC(O)N(R4)-、または共有結合であり、
R1、R2、およびR3は独立して-Hであるか、またはRa-C0-C6-ヒドロカルビルであって、但し、Raは-HであるまたはRaはアリールまたはヘテロアリールであり、それらのそれぞれは、場合によっては1-3個の置換基で置換されており、
R4は-H、-C(O)-Rb、-C(O)O-Rb、-C(O)NH-Rb、またはRc-CO-C6-ヒドロカルビルであり、
但し、
Rbは-Hまたは-C1-C6-ヒドロカルビルであり、
Rcは-H、またはアリールまたはヘテロアリールであり、それらのそれぞれは場合によっては1-3個の置換基で置換されている)
の化合物、および薬学上許容可能なその塩を含んでなる。
【0026】
[0025]節の化合物では、R2およびR3は好ましくは両方とも-Hである。
【0027】
[0025]節の化合物では、Yは好ましくは-NH-、-SO2-NH-、または-N(R4)-であり、但し、R4は-C(O)O-C1-C6-ヒドロカルビルである。
【0028】
[0025]節の化合物では、R1は好ましくはアリール、ベンゾチアゾリル、ピリミジニル、トリアゾリル、ベンゾジオキソレニル、またはピリジニルである。
【0029】
Rlの好ましい置換基としては、C1-C6-ヒドロカルビル、C1-C6-ヒドロカルビルオキシ(例えば、メトキシおよびシクロプロピルオキシ)ハロ、メチルチオ、およびアセチルが挙げられる。
【0030】
[0025]節に記載の化合物の好ましい態様では、R1-Y-は、下記の
【化6】
から選択される。
【0031】
[0025]節に記載の好ましい化合物としては、表8に記載の化合物が挙げられる。
【0032】
第一の様相のもう一つの態様では、本発明は、式
【化7】
(式中、Ar1が場合によっては-NO2、CH3O-、およびモルホリニル(例えば、モルホリン-4-イル)から独立して選択される1-3個の置換基で置換されているアリールまたはヘテロアリールである)
の化合物、および薬学上許容可能なその塩を含んでなる。
【0033】
[0032]節に記載の化合物の好ましい態様では、Ar1はアリール(例えば、フェニル)である。
【0034】
[0032]節に記載の化合物の好ましい態様では、Ar1は、
【化8】
から選択される。
【0035】
[0032]節に記載の好ましい化合物としては、表9に記載の化合物が挙げられる。
【0036】
第二の様相では、本発明は、[0013]-[0035]節の一つに記載の化合物と薬学上許容可能なキャリヤー、賦形剤または希釈剤を含んでなる組成物を含んでなる。
【0037】
第三の様相では、本発明は、細胞中のヒストンデアセチラーゼを阻害する方法であって、ヒストンデアセチラーゼの阻害が所望な細胞を[0013]-[0036]節の一つに記載のヒストンデアセチラーゼの阻害剤と接触させることを含んでなる方法を提供する。
【0038】
もう一つの様相では、本発明は、細胞増殖性疾患または疾病に罹っている哺乳類(好ましくは、ヒト)に[0036]節に記載の組成物の治療上有効量を投与することを含んでなる。
【0039】
本発明の目的のために、(特に断らない限り)下記の定義を用いる。
【0040】
本明細書で用いられる「ヒストンデアセチラーゼ」および「HDAC」という用語は、ヒストンのN-末端におけるリジン残基の -アミノ基からアセチル基を除去する酵素のファミリーの任意の一つを指すものと解釈される。文脈によって指示されない限り、「ヒストン」という用語は任意の種由来のH1、H2A、H2B、H3、H4、およびH5などの任意のヒストンタンパク質を表すことを意味する。好ましいヒストンデアセチラーゼとしては、クラスIおよびクラスIIの酵素が挙げられる。好ましくは、ヒストンデアセチラーゼは、HDAC-1、HDAC-2、HDAC-3、HDAC-4、HDAC-5、HDAC-6、HDAC-7、およびHDAC-8などのヒトHDACであるが、これらに限定されない。幾つかの他の好ましい態様では、ヒストンデアセチラーゼは原生動物または真菌供給源に由来する。
【0041】
「ヒストンデアセチラーゼ阻害剤」および「ヒストンデアセチラーゼの阻害剤」という用語は、本明細書で定義される構造を有し、ヒストンデアセチラーゼと相互作用してその酵素活性を阻害することができる化合物を同定するのに用いられる。「ヒストンデアセチラーゼ酵素活性を阻害する」とは、ヒストンからアセチル基を除去するヒストンデアセチラーゼの能力を減少させることを意味する。幾つかの好ましい態様では、このようなヒストンデアセチラーゼ活性の減少は少なくとも約50%であり、更に好ましくは、少なくとも約75%であり、更に一層好ましくは、少なくとも約90%である。他の好ましい態様では、ヒストンデアセチラーゼ活性は、少なくとも95%、更に好ましくは、少なくとも99%減少する。
【0042】
好ましくは、このような阻害は特異的であり、すなわち、ヒストンデアセチラーゼ阻害剤は、別の関係のない生物学的効果を生成するのに要する阻害剤の濃度より低い濃度でヒストンからアセチル基を除去するヒストンデアセチラーゼの能力を減少させる。好ましくは、ヒストンデアセチラーゼ阻害活性に要する阻害剤の濃度は、関係のない生物学的効果を生成するのに要する濃度の少なくとも2分の1以下であり、更に好ましくは、少なくとも5分の1以下であり、更に一層好ましくは、少なくとも1O分の1以下であり、最も好ましくは、少なくとも2O分の1以下である。
【0043】
簡単にするため、化学残基は、主として一価の化学残基(例えば、アルキル、アリールなど)として本明細書中で定義され、表される。しかしながら、このような用語は、当業者には明らかな適当な構造的環境下では相当する多価残基を送達するのにも用いられる。例えば、「アルキル」残基は一般に一価の基(例えば、CH3-CH2-)を表すが、ある環境では二価の結合残基が「アルキル」であることができ、この場合には、当業者であれば、アルキルが二価の基(例えば、-CH2-CH2-)であり、これは「アルキレン」という用語と同等であることを理解されるであろう。(同様に、二価残基が必要であり且つ「アリール」として表される環境では、当業者であれば、「アリール」という用語が相当する二価の残基であるアリーレンを表すことを理解されるであろう。)総ての原子は、結合形成のための標準的な結合価数(すなわち、炭素については4、Nについては3、Oについては2、およびSについてはSの酸化状態によって2、4または6)を有することが理解される。ときには、残基は、例えば(A)a-B- (式中、aは0または1である)として定義することができる。このような場合には、aが0であるときには、残基はB-であり、aが1であるときには、残基はA-B-である。また、本明細書に開示される残基の数は多数の互変異性形態で存在し、それらの総ては任意の所定の互変異性構造によって包含されると解釈される。
【0044】
「ヒドロカルビル」という用語は、直線状、分岐状または環状アルキル、アルケニル、またはアルキニルであって、それぞれ本明細書で定義されているものを表す。「C0」ヒドロカルビルとは、共有結合を表すのに用いられる。従って、「C0-C3-ヒドロカルビル」としては、共有結合、メチル、エチル、エテニル、エチニル、プロピル、プロペニル、プロピニル、およびシクロプロピルが挙げられる。
【0045】
本明細書で用いられる「アルキル」という用語は、1-12個の炭素原子、好ましくは、1-8個の炭素原子、更に好ましくは、1-6個の炭素原子を有し、場合によっては1、2または3個の置換基で置換されている直鎖状および分岐鎖状脂肪族基を表す。好ましいアルキル基としては、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、第二ブチル、第三ブチル、ペンチル、およびヘキシルが挙げられるが、これらに限定されない。(「C0-C3-アルキル」におけるような)「C0」アルキルは、(「C0」ヒドロカルビルのような)共有結合である。
【0046】
本明細書で用いられる「アルケニル」という用語は、1個以上の炭素-炭素二重結合を有し、2-12個の炭素原子、好ましくは、2-8個の炭素原子、更に好ましくは、2-6個の炭素原子を有し、場合によっては1、2または3個の置換基で置換されている不飽和の直鎖状または分岐鎖状脂肪族基を意味する。好ましいアルケニル基としては、エテニル、プロペニル、ブテニル、ペンテニル、およびヘキセニルが挙げられるが、これらに限定されない。
【0047】
本明細書で用いられる「アルキニル」という用語は、1個以上の炭素-炭素三重結合を有し、2-12個の炭素原子、好ましくは、2-8個の炭素原子、更に好ましくは、2-6個の炭素原子を有し、場合によっては1、2または3個の置換基で置換されている不飽和の直鎖状または分岐鎖状脂肪族基を意味する。好ましいアルキニル基としては、エチニル、プロピニル、ブチニル、ペンチニル、およびヘキシニルが挙げられるが、これらに限定されない。
【0048】
「アルキレン」、「アルケニレン」または「アルキニレン」基とは、2個の他の化学基の間にあってこれらを結合する役割をする上記で定義したアルキル、アルケニルまたはアルキニル基である。好ましいアルキレン基としては、メチレン、エチレン、プロピレン、およびブチレンが挙げられるが、これらに限定されない。好ましいアルケニレン基としては、エテニレン、プロペニレン、およびブテニレンが挙げられるが、これらに限定されない。好ましいアルキニレン基としては、エチニレン、プロピニレン、およびブチニレンが挙げられるが、これらに限定されない。
【0049】
本明細書で用いられる「シクロアルキル」という用語は、3-12個の炭素、好ましくは、3-8個の炭素、更に好ましくは、3-6個の炭素を有する飽和および部分不飽和の環状炭化水素基を包含し、このシクロアルキル基は更に場合によっては置換されている。好ましいシクロアルキル基としては、シクロプロピル、シクロブチル、シクロペンチル、シクロペンテニル、シクロヘキシル、シクロヘキセニル、シクロヘプチル、およびシクロオクチルが挙げられるが、これらに限定されない。
【0050】
「ヘテロアルキル」という用語は、上記で定義した通りのアルキル基であって、鎖の1個以上の炭素原子がO、SおよびNからなる群から選択されるヘテロ原子によって置換されているものを表す。
【0051】
「アリール」基は、1-3個の芳香族環を含んでなり、場合によっては置換されているC6-C14芳香族残基である。好ましくは、アリール基はC6-C10アリール基である。好ましいアリール基としては、フェニル、ナフチル、アントラセニル、およびフルオレニルが挙げられるが、これらに限定されない。「アラールキル」または「アリールアルキル」基は、アルキル基に共有結合したアリール基であって、それらのいずれが独立して場合によっては置換されているかまたは未置換であることができるものを含んでなる。好ましくは、アラールキル基は (C1-C6)アルク(C6-C10)アリールであり、ベンジル、フェネチル、およびナフチルメチルが挙げられるが、これらに限定されない。
【0052】
「ヘテロシクリル」または「複素環状」基は、約3-約12個の原子であって、1個以上の原子がN、OおよびSからなる群から選択される環構造である。複素環状基は、場合によっては1個以上の位置の炭素上で置換されている。複素環状基も、独立して場合によっては窒素上でアルキル、アリール、アラールキル、アルキルカルボニル、アルキルスルホニル、アリールカルボニル、アリールスルホニル、アルコキシカルボニル、アラールコキシカルボニルで置換されており、または硫黄上でオキソまたは低級アルキルで置換されている。好ましい複素環状基としては、エポキシ、アジリジニル、テトラヒドロフラニル、ピロリジニル、ピペリジニル、ピペラジニル、チアゾリジニル、オキサゾリジニル、オキサゾリジノニル、およびモルホリノが挙げられるが、これらに限定されない。ある種の好ましい態様では、複素環状基はアリール、ヘテロアリールまたはシクロアルキル基に融合している。このような融合複素環の例としては、テトラヒドロキノリンおよびジヒドロベンゾフランが挙げられるが、これらに限定されない。この用語の範囲から具体的に除外されるものは、別の環OまたはS原子に隣接している環Oおよび/またはS原子を有する化合物である。
【0053】
本明細書で用いられる「ヘテロアリール」という用語は、5-14個の環原子、好ましくは、5、6、9または10個の環原子を有し、環状配列において共有された6、10または14個のπ電子を有し、且つ炭素原子の他にN、OおよびSからなる群から選択される環当たり1-3個のヘテロ原子を有する基を表す。「ヘテロアリール」という用語は、単環性および二環性基を包含することも意味する。例えば、ヘテロアリール基は、ピリミジニル、ピリジニル、ベンズイミダゾリル、チエニル、ベンゾチアゾリル、ベンゾフラニル、およびインドリニルであってもよい。「ヘテロアラールキル」または「ヘテロアリールアルキル」基は、アルキル基に共有結合したヘテロアリール基であって、それらのいずれかが独立して場合によっては置換されておりまたは未置換であるものを含んでなる。好ましいヘテロアルキル基は、C1-C6アルキル基および5、6、9または10個の環原子を有するヘテロアリール基を含んでなる。この用語の範囲から具体的に除外されるものは、隣接した環Oおよび/またはS原子を有する化合物である。好ましいヘテロアラールキル基としては、ピリジルメチル、ピリジルエチル、ピロリルメチル、ピロリルエチル、イミダゾリルメチル、イミダゾリルエチル、チアゾリルメチル、およびチアゾリルエチルが挙げられる。この用語の範囲から具体的に除外されるものは、隣接した環Oおよび/またはS原子を有する化合物である。
【0054】
「アリーレン」、「ヘテロアリーレン」または「ヘテロシクリレン」基は、2個の他の化学基の間にあってこれらを結合する役割をする上記で定義した通りのアリール、ヘテロアリールまたはヘテロシクリル基である。
【0055】
好ましいヘテロシクリルおよびヘテロアリールとしては、アクリジニル、アゾシニル、ベンズイミダゾリル、ベンゾフラニル、ベンゾチオフラニル、ベンゾチオフェニル、ベンズオキサゾリル、ベンズチアゾリル、ベンズトリアゾリル、ベンズテトラゾリル、ベンズイソキサゾリル、ベンズイソチアゾリル、ベンズイミダゾリニル、カルバゾリル、4aH-カルバゾリル、カルボリニル、クロマニル、クロメニル、シンノリニル、デカヒドロキノリニル、2H,6H-1,5,2-ジチアジニル、ジヒドロフロ[2,3-b]テトラヒドロフラン、フラニル、フラザニル、イミダゾリジニル、イミダゾリニル、イミダゾリル、1H-インダゾリル、インドレニル、インドリニル、インドリジニル、インドリル、3H-インドリル、イソベンゾフラニル、イソクロマニル、イソインダゾリル、イソインドリニル、イソインドリル、イソキノリニル、イソチアゾリル、イソキサゾリル、メチレンジオキシフェニル、モルホリニル、ナフチリジニル、オクタヒドロイソキノリニル、オキサジアゾリル、1,2,3-オキサジアゾリル、1,2,4-オキサジアゾリル、1,2,5-オキサジアゾリル、1,3,4-オキサジアゾリル、オキサゾリジニル、オキサゾリル、オキサゾリジニル、ピリミジニル、フェナントリジニル、フェナントロリニル、フェナジニル、フェノチアジニル、フェノキサチイニル、フェノキサジニル、フタラジニル、ピペラジニル、ピペリジニル、ピペリドニル、4-ピペリドニル、ピペロニル、プテリジニル、プリニル、ピラニル、ピラジニル、ピラゾリジニル、ピラゾリニル、ピラゾリル、ピリダジニル、ピリドキサゾール、ピリドイミダゾール、ピリドチアゾール、ピリジニル、ピリジル、ピリミジニル、ピロリジニル、ピロリニル、2H-ピロリル、ピロリル、キナゾリニル、キノリニル、4H-キノリジニル、キノキサリニル、キヌクリジニル、テトラヒドロフラニル、テトラヒドロイソキノリニル、テトラヒドロキノリニル、テトラゾリル、6H-1,2,5-チアジアジニル、1,2,3-チアジアゾリル、1,2,4-チアジアゾリル、1,2,5-チアジアゾリル、1,3,4-チアジアゾリル、チアントレニル、チアゾリル、チエニル、チエノチアゾリル、チエノオキサゾリル、チエノイミダゾリル、チオフェニル、トリアジニル、1,2,3-トリアゾリル、1,2,4-トリアゾリル、1,2,5-トリアゾリル、1,3,4-トリアゾリル、およびキサンテニルが挙げられるが、これらに限定されない。
【0056】
本明細書で用いられる様に、残基 (例えば、シクロアルキル、ヒドロカルビル、アリール、ヘテロアリール、複素環状、尿素など)が「場合によっては置換されている」と記載されるときには、その基は、場合によっては1-4個、好ましくは1-3個、更に好ましくは1または2個の非水素置換基を有することを意味する。適当な置換基としては、ハロ、ヒドロキシ、オキソ(例えば、オキソで置換された環-CH-は-C(O)-)ニトロ、ハロ-ヒドロカルビル、ヒドロカルビル、アリール、アラールキル、アルコキシ、アリールオキシ、アミノ、アシルアミノ、アルキルカルバモイル、アリールカルバモイル、アミノアルキル、アシル、カルボキシ、ヒドロキシアルキル、アルカンスルホニル、アレンスルホニル、アルカンスルホンアミド、アレンスルホンアミド、アラールキルスルホンアミド、アルキルカルボニル、アシルオキシ、シアノ、およびウレイド基が挙げられるが、これらに限定されない。好ましい置換基であって、(特に断らない限り)それら自身が更に置換基を持たないものは、
(a) ハロ、シアノ、オキソ、カルボキシ、ホルミル、ニトロ、アミノ、アミジノ、グアニジノ、
(b) C1-C5アルキルまたはアルケニルまたはアリールアルキルイミノ、カルバモイル、アジド、カルボキサミド、メルカプト、ヒドロキシ、ヒドロキシアルキル、アルキルアリール、アリールアルキル、C1-C8アルキル、C1-C8アルケニル、C1-C8アルコキシ、C1-C8アルコキシカルボニル、アリールオキシカルボニル、C2-C8 アシル, C2-C8アシルアミノ、C1-C8アルキルチオ、アリールアルキルチオ、アリールチオ、C1-C8アルキルスルフィニル、アリールアルキルスルフィニル、アリールスルフィニル、C1-C8アルキルスルホニル、アリールアルキルスルホニル、アリールスルホニル、CO-C6N-アルキルカルバモイル、C2-C15 N,N-ジアルキルカルバモイル、C3-C7シクロアルキル、アロイル、アリールオキシ、アリールアルキルエーテル、アリール、シクロアルキルまたは複素環または別のアリール環に融合したアリール、C3-C7複素環、またはシクロアルキル、ヘテロシクリルまたはアリールに融合またはスピロ融合したこれらの環のいずれかであって、上記のそれぞれが更に場合によっては上記(a)に挙げた1個以上の残基で置換されているもの、および
(c) -(CH2)s-NR30R31(式中、sは0(この場合には、窒素は置換される残基に直接結合している)- 6であり、R30およびR31はそれぞれ独立して水素、シアノ、オキソ、カルボキサミド、アミジノ、C1-C8ヒドロキシアルキル、C1-C3アルキルアリール、アリール-C1-C3アルキル、C1-C8アルキル、C1-C8アルケニル、C1-C8アルコキシ、C1-C8アルコキシカルボニル、アリールオキシカルボニル、アリール-C1-C3アルコキシカルボニル、C2-C8アシル、C1-C8アルキルスルホニル、アリールアルキルスルホニル、アリールスルホニル、アロイル、アリール、シクロアルキル、ヘテロシクリル、またはヘテロアリールであって、上記のそれぞれが更に場合によっては上記(a)に挙げた1個以上の残基で置換されているもの、または
R30およびR31が、それらが結合しているNと一緒になってヘテロシクリルまたはヘテロアリールを形成し、それらのそれぞれは場合によっては上記(a)からの1-3個の置換基で置換されているもの
である。
【0057】
更に、環状残基(すなわち、シクロアルキル, ヘテロシクリル, アリール, ヘテロアリール)上の置換基としては、親環状残基に融合した5-6員の単環状および9-14員の二環状残基であって、二または三環性の融合環系を形成するものが挙げられる。例えば、場合によっては置換されるフェニルとしては、下記のものが挙げられる。
【化9】
【0058】
「ハロヒドロカルビル」とは、ヒドロカルビル残基であって、1個-全部の水素が1個以上のハロゲンで置き換わっているものである。
【0059】
本明細書で用いられる「ハロゲン」または「ハロ」という用語は、塩素、臭素、フッ素、またはヨウ素を表す。本明細書で用いられる「アシル」という用語は、アルキルカルボニルまたはアリールカルボニル置換基を表す。「アシルアミノ」という用語は、窒素原子に結合したアミド基(すなわち、R-CO-NH-)を表す。「カルバモイル」という用語は、カルボニル炭素原子に結合したアミド基(すなわち、NH2-CO-)を表す。アシルアミノまたはカルバモイル置換基の窒素原子は、更に置換されている。「スルホンアミド」という用語は、硫黄または窒素原子によって結合されているスルホンアミド置換基を表す。「アミノ」という用語は、NH2、アルキルアミノ、アリールアミノおよび環状アミノ基を包含することを意味する。本明細書で用いられる「ウレイド」という用語は、置換または未置換尿素残基を表す。
【0060】
本明細書で用いられる「ラジカル」という用語は、1個以上の不対電子を含んでなる化学残基を意味する。
【0061】
置換される残基は、1個以上の水素が独立して別の化学置換基に置換されているものである。非制限的例としては、置換フェニルとしては、2-フルオロフェニル、3,4-ジクロロフェニル、3-クロロ-4-フルオロ-フェニル、2-フルオロ-3-プロピルフェニルが挙げられる。別の非制限的例としては、置換n-オクチルとしては、2,4-ジメチル-5-エチル-オクチルおよび3-シクロペンチル-オクチルが挙げられる。この定義に包含されるものは、カルボニル(-CO-)を形成するため酸素で置換されたメチレン(-CH2-)である。
【0062】
上記で定義した「未置換」残基(例えば、未置換シクロアルキル、未置換ヘテロアリールなど)は、(上記の)残基の定義が別に提供する任意の置換基のいずれも持たない上記で定義した残基を意味する。従って、例えば、「アリール」はフェニル、およびハロゲンで置換されたフェニルを包含するが、「未置換アリール」はハロゲンで置換されたフェニルを包含しない。
【0063】
本発明の好ましい態様は、本明細書に特に記載された好ましい態様の組合せも包含する。
【0064】
合成
一般式Iの化合物を、工程図1および2に示した合成経路に従って調製した。幾つかの態様では、4-アセチル安息香酸を、水酸化ナトリウムの水溶液(1N)の存在下でメタノール(MeOH)のような溶媒中で芳香族および/またはヘテロ芳香族アルデヒドと反応させて、濾過、またはpH=5-6になるまで酸性化および濾過の後にカルコンIIを得た。化合物IIを、最初にトリエチルアミン(Et3N)の存在下にてN,N-ジメチルホルムアミド(DMF)のような溶媒中で、カップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)で処理した。イン・シテューで形成した生成する活性化エステル中間体を、最後に1,2-フェニレンジアミンと反応させて、化合物Iをえた(工程図1)。
【化10】
【0065】
あるいは、幾つかの他の態様では、4-アセチル安息香酸を、最初にトリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェートで処理した。次に、イン・シテューで形成した生成する活性化エステル中間体を、t-ブチル(2-アミノ-フェニル)-カルバメートと反応させ、一般のアセトフェノン誘導体IIIを得た。カルコンIVは、化合物IIIと適当な芳香族および/またはヘテロ芳香族アルデヒドを水酸化ナトリウムの水溶液(1N)の存在下でメタノールのような溶媒中でClaisen-Schmidt縮合によって調製した。アニリンIVのN-Boc保護基を、最終的にジクロロメタン(CH2Cl2)のような溶媒中でトリフルオロ酢酸の含水溶液(TFA, 95%、水中)によって開裂して、化合物Iを得た(工程図2)。
【化11】
【0066】
一般式Vの化合物は、工程図3および4に示した合成経路に従って調製した。ある種の好ましい態様では、メチル4-ホルミルベンゾエートを、テトラヒドロフラン(THF)とヘキサンのような溶媒混合物中でt-ブチルアセテートのアニオンと反応させた後、新たな脱水剤としての2-クロロ-4,6-ジメトキシ-1,3,5-トリアジンで処理することによって、純粋なトランス-α,β-不飽和エステルVIに転換した。t-ブチルエステルVIの酸加水分解は、ジクロロメタンのような溶媒中でトリフルオロ酢酸の含水溶液(95%、水中)によって行い、化合物VIIを得た。化合物IXの形成は、RXHの親核性により2種類の相補的方法によって行った。方法Aでは、カルボン酸VIIを、最初にトリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)を用いて、安定な活性化エステルVIIIに転換した。次に、この安定な活性化エステルVIIIを、トリエチルアミンの存在下にてジクロロメタンのような溶媒中で弱い親核試薬(例えば、RXH=アニリンまたはアミノヘテロアリール)と反応させて、化合物IXを得た。方法Bでは、カルボン酸VIIからイン・シテューで形成された同じ活性化エステルVIII中間体を、次にトリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中で強い親核試薬(例えば、RXH=アミン、アルコール、チオール、ヒドロキシルアミンおよび誘導体、またはヒドラジンおよび誘導体)と反応させて、化合物IXを得た。
【化12】
【0067】
メチルエステルIXの塩基加水分解は、テトラヒドロフランのような溶媒中で水酸化リチウムの水溶液によって行い、化合物Xを得た。カルボン酸Xを、最後にトリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)で処理した。次に、イン・シテューで形成された生成する活性化エステル中間体を1,2-フェニレンジアミンと反応させて、化合物Vを得た(工程図3)。
【0068】
あるいは、幾つかの他の態様では、触媒量のN,N-ジメチルホルムアミドの存在下にてジクロロメタンのような溶媒中で塩化チオニル(SOCl2)を用いて酸塩化物中間体に転換した。次に、生成する酸塩化物中間体をt-ブチル(2-アミノ-フェニル)-カルバメートと反応させて、一般的なベンズアルデヒド誘導体XIを得た。
【化13】
【0069】
アルデヒドXIのWittigオレフィン化は、トルエンのような溶媒中でメチル(トリフェニル-ホスホラニリデン)アセテートを用いて行い、トランス-α,β-不飽和エステルXIIを得た。メチルエステルXIIの塩基加水分解は、テトラヒドロフランのような溶媒中で水酸化リチウムの水溶液を用いて行い、化合物XIIIを得た。カルボン酸 XIIIを、トリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)で処理した。次に、イン・シテューで形成された生成する活性化エステル中間体を親核試薬R1XHと反応させ、化合物XIVを得た。アニリンXIVのN-Boc保護基は、最終的にジクロロメタンのような溶媒中でトリフルオロ酢酸の含水溶液(95%、水中)によって開裂させ、化合物Vを得た(工程図4)。
【0070】
一般式XVの化合物は、工程図5-7に示す合成経路に従って調製した。従って、アルデヒドXIのWittigオレフィン化は、トルエンのような溶媒中で(トリフェニルホスホラニリデン)-アセトアルデヒド試薬を用いて行い、トランス-α,β-不飽和アルデヒドXVIを得た。ある種の好ましい態様では、化合物XVIIの形成は、R1XHの親核性によって記載した方法に従って還元的アミノ化によって行った。アルデヒドXVIを、最初に触媒量のジブチルスズ二塩化物の存在下にてテトラヒドロフランのような溶媒中で弱親核試薬(例えば、R1XH=アニリンまたはアミノヘテロアリール)と混合した。イン・シテューで形成された生成するイミニウム中間体を次に還元試薬フェニルシランと反応させ、化合物XVIIを得た。
【化14】
【0071】
アニリンXVIIのN-Boc保護基を、最終的にジクロロメタンのような溶媒中でトリフルオロ酢酸の含水溶液(95%、水中)によって開裂し、化合物XVを得た(工程図5)。
【0072】
あるいは、幾つかの他の態様では、トランス-α,β-不飽和アルデヒドXVIをエタノールのような溶媒中で還元試薬水素化ホウ素ナトリウムによって第一アリルアルコールXVIIIに還元した。このアルコールXVIIIを、次に、トリフェニルホスフィンおよびジエチルアゾジカルボキシレート(DEAD)の存在下にてテトラヒドロフランのような溶媒中でMitsunobu型反応によって親核試薬R1XHと反応させ、化合物XVIIを得た。アニリンXVIIのN-Boc保護基を、最終的にジクロロメタンのような溶媒中でトリフルオロ酢酸の含水溶液(95%、水中)によって開裂し、化合物XVを得た(工程図6)。
【化15】
【0073】
更に、幾つかの他の態様では、メチル4-ホルミルベンゾエートのWittigオレフィン化を、トルエンのような溶媒中で(トリフェニルホスホラニリデン)-アセトアルデヒド試薬またはジクロロメタン/水のような二相性媒質中TDA-1{トリス[2-(2-メトキシエトキシ)エチル]アミン)および炭酸カリウムの存在下にて(1,3-ジオキソラン-2-イル)メチルトリフェニルホスホニウムブロミド試薬を用いて行った後、酸加水分解によりトランス-α,β-不飽和アルデヒドXIXを得た。アルデヒドXIXを、最初にジクロロメタンまたは1,2-ジクロロエタンのような溶媒中で親核試薬(R1R2NH)と混合した。次に、イン・シテューで形成される生成するイミニウム中間体を還元試薬水素化トリアセトキシホウ素ナトリウム[NaBH(OAc)3]と反応させて、化合物XXを得た。経路Aでは、メチルエステルの塩基加水分解と化合物XX (R1=アルキル、R2=H)の第二アミンの保護は、1,4-ジオキサンのような溶媒中で水酸化ナトリウムの水溶液(1N)と保護試薬ジ-第三ブチルジカーボネート[(Boc)2O]の存在下にて同時に行い、化合物XXIを得た。カルボン酸XXIを、最初にトリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)で処理した。次に、イン・シテューで形成される生成する活性化エステル中間体を1,2-フェニレンジアミンと反応させ、化合物XXIIを得た。アミンXXIIのN-Boc保護基を、最終的にジクロロメタンのような溶媒中でトリフルオロ酢酸の含水溶液(95%、水中)で開裂し、化合物XVを得た(工程図7)。
【化16】
【0074】
経路Bでは、メチルエステルXXを、塩基加水分解および1,2-フェニレンジアミンでカップリングした後に、直接最終化合物XVに転換した(工程図7)。
【0075】
一般式XXIVの化合物は、工程図8および9に示される合成経路に従って調製した。幾つかの態様では、4-ホルミル安息香酸を、水酸化ナトリウムの水溶液(1N)の存在下にてメタノール(MeOH)のような溶媒中でアリールおよび/またはヘテロアリールメチルケトンと反応させ、濾過後にカルコンXXIIIを得た。化合物XXIIIを、最初にトリエチルアミン(Et3N)の存在下にてN,N-ジメチルホルムアミド(DMF)のような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)で処理した。イン・シテューで形成される生成する活性化エステル中間体を、最後に1,2-フェニレンジアミンと反応させて、化合物XXIVを得た(工程図8)。
【化17】
【0076】
あるいは、幾つかの他の態様では、カルコンXXVを、水酸化ナトリウムの水溶液(1N)の存在下にてメタノールのような溶媒中でベンズアルデヒド誘導体XIと適当なアリールおよび/またはヘテロアリールメチルケトンとのClaisen-Schmidt縮合によって調製した。アニリンXXVのN-Boc保護基を、最後にジクロロメタン(CH2Cl2)のような溶媒中でトリフルオロ酢酸の含水溶液(TFA 95%、水中)によって開裂し、化合物XXIVを得た(工程図9)。
【化18】
【0077】
一般式XXVIIおよびXXIXの化合物は、工程図10に示される合成経路に従って調製した。従って、化合物XXIIIの二重結合の選択的還元は、N,N-ジメチルホルムアミドのような溶媒中で還元試薬ベンゼンスルホニルヒドラジドを用いることによって行い、化合物XXVIを得た。カルボン酸XXVIを、最初にトリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)で処理した。イン・シテューで形成される生成する活性化エステル中間体を、最後に1,2-フェニレンジアミンと反応させ、化合物XXVIIを得た。
【化19】
【0078】
更に、α,β-不飽和ケトンXXIIIの飽和化合物 XXVIIIへの完全な還元は、N,N-ジメチルアセタミド(DMA)のような溶媒中で10%パラジウム/炭(Degussa型)によって触媒される水素化によって行った。次に、カルボン酸XXVIIIを、最初にトリエチルアミンの存在下にてN,N-ジメチルホルムアミドのような溶媒中でカップリング試薬ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP試薬)で処理した。イン・シテューで形成される生成する活性化エステル中間体を、最後に1,2-フェニレンジアミンと反応させて、化合物XXIXを得た(工程図10)。
【0079】
一般式XXXの化合物は、工程図11に示される合成経路に従って調製した。従って、メチル4-ブロモベンゾエートと(トリメチルシリル)アセチレンとの間のSonogashira型反応は、THFのような溶媒中でEt3Nの存在下にて触媒量のパラジウム触媒とヨウ化銅とによって行って、保護されたアルキンXXXIを得た。XXXIのTMS基の塩基性脱保護は、メタノールの存在下にて炭酸カリウムによって行い、アルキンXXXIIを得た。XXXIIの三重結合のハイドロボレーションは、THFのような溶媒中でカテコールボラン試薬によって行った後、ボロネート中間体を酸加水分解によってボロン酸XXXIIIを得た。アリルアミンXXXIVは、1,4-ジオキサンのような溶媒中で、ビニルボロン酸XXXIIIをアミノ化合物(R1R2NH)とアルデヒド(R3CHO)との予備形成混合物と反応させることによるPetasis型反応によって合成した。最後に、メチルエステルXXIVを、塩基加水分解および1,2-フェニレンジアミンとのカップリングの後、最終化合物XXXに転換した。
【化20】
【0080】
ヒストンデアセチラーゼの阻害
第三の様相では、本発明は、細胞においてヒストンデアセチラーゼを阻害する方法であって、ヒストンデアセチラーゼの阻害が所望な細胞を本発明によるヒストンデアセチラーゼの阻害剤と接触させることを含んでなる方法を提供する。
【0081】
ヒストンデアセチラーゼの酵素活性の測定は、既知の方法を用いて行うことができる。例えば、Yoshida et al., J.Biol. Chem., 265: 17174-17179 (1990)には、トリコスタチンAで処理した細胞におけるアセチル化ヒストンの検出によるヒストンデアセチラーゼ酵素活性の評価が記載されている。Taunton et al., Science, 272: 408-411 (1996)にも、同様に、内在性および組換えHDAC-1を用いてヒストンデアセチラーゼ酵素活性を測定する方法が記載されている。
【0082】
幾つかの好ましい態様では、ヒストンデアセチラーゼ阻害剤は、細胞中の総てのヒストンデアセチラーゼと相互作用し且つこの活性を減少させる。本発明のこの様相による幾つかの他の好ましい態様では、ヒストンデアセチラーゼ阻害剤は、細胞中の総てより少数のヒストンデアセチラーゼと相互作用し且つこの活性を減少させる。ある種の好ましい態様では、阻害剤は、1種類のヒストンデアセチラーゼ(例えば、HDAC-1)と相互作用し且つこの活性を減少させるが、他のヒストンデアセチラーゼ(例えば、HDAC-2、HDAC-3、HDAC-4、HDAC-5、HDAC-6、HDAC-7およびHDAC-8)とは相互作用もせずこれらの活性を減少させもしない。下記のように、ある種の特に好ましいヒストンデアセチラーゼ阻害剤は、腫瘍形成に関与するヒストンデアセチラーゼと相互作用し且つこの酵素活性を減少させるものである。ある種の他の好ましいヒストンデアセチラーゼ阻害剤は、真菌ヒストンデアセチラーゼと相互作用し且つこの酵素活性を減少させる。
【0083】
好ましくは、本発明の第三の様相による方法は、接触した細胞の細胞増殖の阻害を生じる。「細胞増殖を阻害する」という用語は、阻害剤と接触しなかった細胞と比較して接触した細胞の成長を遅延させるヒストンデアセチラーゼの阻害剤の能力を表すのに用いられる。細胞増殖は、Coulter Cell Counter (Coulter, Miami, FL)または血球計を用いて接触したおよび接触しなかった細胞を計数することによって行うことができる。細胞が固体成長である場合(例えば、固形腫瘍または器官)には、このような細胞増殖の評価はカリパスで成長を測定し、接触した細胞の成長の程度を接触しなかった細胞と比較することによって行うことができる。
【0084】
好ましくは、阻害剤と接触した細胞の成長は、接触しなかった細胞と比較して少なくとも50%遅延する。更に好ましくは、細胞増殖は100%阻害される(すなわち、接触した細胞の数は増加しない)。最も好ましくは、「細胞増殖を阻害する」という用語は、接触した細胞の数または大きさが接触しなかった細胞と比較して減少することを包含する。従って、接触した細胞での細胞増殖を阻害する本発明によるヒストンデアセチラーゼの阻害剤は、接触した細胞に成長遅延、成長停止、細胞死プログラム(すなわち、アポトーシス)、または壊死による細胞死を誘発することがある。
【0085】
本発明によるヒストンデアセチラーゼ阻害剤の細胞増殖阻害能は、様々な生物学的過程におけるヒストンデアセチラーゼの役割を研究するための有用な研究手段となっている。例えば、本発明によるヒストンデアセチラーゼ阻害剤の細胞増殖阻害能によって、非同時成長細胞の集団を同期させることができる。例えば、本発明のヒストンデアセチラーゼ阻害剤は、イン・ビトロで成長した非腫瘍細胞の集団を細胞サイクルのGlまたはG2期に停止させるのに用いることができる。このような同期により、例えば、細胞サイクルのGlまたはG2期に発現した遺伝子および/または遺伝子産物を同定することができる。培養細胞のこのような同期は、トランスフェクション効率が変化し且つトランスフェクションされる細胞の特定の細胞サイクル期によって変化する新規なトランスフェクションプロトコールの効力の試験にも有用であることがある。本発明のヒストンデアセチラーゼ阻害剤を用いることによって細胞の集団を同期することができ、これにより、増加したトランスフェクション効率の検出が促進される。
【0086】
幾つかの好ましい態様では、接触した細胞は腫瘍細胞である。「腫瘍細胞」という用語は、異常な細胞成長を示す細胞を表すのに用いられる。好ましくは、腫瘍細胞の異常な細胞成長は細胞成長の増加である。腫瘍性細胞は、増殖性細胞、イン・ビトロでの成長の接触阻害を欠いている細胞、イン・ビボで転移が不可能な良性腫瘍細胞、またはイン・ビボで転移することができ且つ除去を試みた後に再発する可能性がある癌細胞であることもある。「腫瘍形成」という用語は、細胞増殖を誘発し、腫瘍性成長が展開されることを表すのに用いられる。幾つかの態様では、ヒストンデアセチラーゼ阻害剤は、接触した細胞での細胞分化を誘発する。従って、腫瘍性細胞をヒストンデアセチラーゼの阻害剤と接触させることにより、分化が誘発され、接触した細胞より系統発生的に進化した非腫瘍性娘細胞が産生されることがある。
【0087】
幾つかの好ましい態様では、接触した細胞は動物である。従って、本発明は、動物における細胞増殖性疾患または疾病の治療方法であって、このような治療を必要とする動物に本発明のヒストンデアセチラーゼ阻害剤の治療上有効量を投与することを含んでなる方法を提供する。好ましくは、動物は哺乳類であり、更に好ましくは、家畜である。最も好ましくは、動物はヒトである。
【0088】
「細胞増殖性疾患または疾病」という用語は、異常な細胞成長、好ましくは細胞増殖の異常増加を特徴とする任意の疾病を表すことを意味する。このような細胞増殖性疾患または疾病としては、癌、再狭窄、および乾癬が挙げられるが、これらに限定されない。特に好ましい態様では、本発明は、動物における腫瘍性細胞増殖を阻害する方法であって、動物体に少なくとも1個の腫瘍性細胞が含まれている動物に本発明のヒストンデアセチラーゼ阻害剤の治療上有効量を投与することを含んでなる、方法を提供する。
【0089】
本発明の幾つかの化合物は、原生動物由来のヒストンデアセチラーゼに対して阻害剤活性を有すると考えられている。従って、本発明は、原生動物による疾患または感染症を治療しまたは予防する方法であって、このような治療を必要とする動物に本発明によるヒストンデアセチラーゼ阻害剤の治療上有効量を投与することを含んでなる方法も提供する。好ましくは、動物は哺乳類であり、更に好ましくはヒトである。好ましくは、本発明のこの態様によって用いられるヒストンデアセチラーゼ阻害剤は、哺乳類のヒストンデアセチラーゼ、特にヒトのヒストンデアセチラーゼを阻害するより大幅に原生動物のヒストンデアセチラーゼを阻害する。
【0090】
本発明は、真菌性疾患または感染症の治療方法であって、このような治療を必要とする動物に本発明のヒストンデアセチラーゼ阻害剤の治療上有効量を投与することを含んでなる方法も提供する。好ましくは、動物は哺乳類であり、更に好ましくは、ヒトである。好ましくは、本発明のこの態様によって用いられるヒストンデアセチラーゼ阻害剤は、真菌性ヒストンデアセチラーゼを、哺乳類のヒストンデアセチラーゼ、特にヒトのヒストンデアセチラーゼを阻害するよりも大幅に阻害する。
【0091】
「治療上有効量」という用語は、患者の細胞でヒストンデアセチラーゼ活性を阻害するのに十分な投薬量、または患者の細胞増殖を阻害しまたは細胞分化を誘発するのに十分な投薬量を表すことを意味する。投与は、非経口、経口、舌下、経皮、局所、経鼻、気管内、または直腸内など任意の経路が挙げられるが、これらに限定されない。ある種の特に好ましい態様では、本発明の化合物は、病院施設で静脈内に投与される。ある種の他の好ましい態様では、投与は、好ましくは、経口によることができる。
【0092】
全身投与の際は、ヒストンデアセチラーゼ阻害剤は、好ましくは、阻害剤の血中濃度が約0.01μM-約100μM、更に好ましくは、約0.05μM-約50μM、更に一層好ましくは、約0.1μM-約25μM、更に一層好ましくは、約0.5μM-約25μMとなるのに十分な投薬量で投与される。局所化投与では、これより遙かに低濃度で有効であることができ、また遙かに高濃度を用いることもできる。当業者であれば、治療効果を生じるのに必要なヒストンデアセチラーゼ阻害剤の投薬量は、治療を行う組織、器官、または特定の動物または患者によってかなり変動することがあることを理解するであろう。
【0093】
本発明の第三の様相のある種の好ましい態様では、この方法は、細胞をヒストンデアセチラーゼの発現を阻害するアンチセンスオリゴヌクレオチドと接触させることをも含んでなる。核酸レベルの阻害剤 (例えば、アンチセンスオリゴヌクレオチド)とタンパク質レベルの阻害剤(すなわち、ヒストンデアセチラーゼ酵素活性阻害剤)を併用すると、阻害効果が向上し、それにより、これらの阻害剤を個々に用いるときに必要な量と比較して、所定の阻害効果を得るのに要する阻害剤の量が減少する。本発明のこの様相によるアンチセンスオリゴヌクレオチドは、HDAC-1、HDAC-2、HDAC-3、HDAC-4、HDAC-5、HDAC-6、HDAC7、および/またはHDAC-8をコードするRNAまたは二本鎖DNAの領域に相補的である(例えば、HDAC-1についてはGenBank Accession Number U50079、HDAC-2についてはGenBank Accession Number U31814およびHDAC-3についてはGenBank Accession Number U75697を参照されたい)。
【0094】
本発明の目的に対して、「オリゴヌクレオチド」という用語は、2種類以上のデオキシリボヌクレオシド、リボヌクレオシド、または2'-置換リボヌクレオシド残基、またはそれらの任意の組合せのポリマーを包含する。好ましくは、このようなオリゴヌクレオチドは約6-約100ヌクレオシド残基を有し、更に好ましくは、約8-約50ヌクレオシド残基、最も好ましくは、約12-約30ヌクレオシド残基を有する。ヌクレオシド残基は、多数の既知のヌクレオシド間結合のいずれかによって互いにカップリングすることができる。このようなヌクレオシド間結合としては、ホスホロチオエート、ホスホロジチオエート、アルキルホスホネート、アルキルホスホノチオエート、ホスホトリエステル、ホスホルアミデート、シロキサン、カーボネート、カルボキシメチルエステル、アセトアミデート、カルバメート、チオエーテル、架橋ホスホルアミデート、架橋メチレンホスホネート、架橋ホスホロチオエート、およびスルホンヌクレオシド間結合が挙げられるが、これらに限定されない。ある種の好ましい態様では、これらのヌクレオシド間結合は、ホスホジエステル、ホスホトリエステル、ホスホロチオエート、またはホスホルアミデート結合、またはそれらの組合せであることができる。オリゴヌクレオチドという用語は、化学的に修飾した塩基または糖を有するおよび/または親油性基、インターカレート剤、ジアミン、およびアダマンタンなどのこれらに限定されない追加の置換基を有するポリマーも包含する。
【0095】
本発明の目的のために、「2'-置換リボヌクレオシド」という用語は、ペントース残基の2'位のヒドロキシル基が置換されて2'-O-置換リボヌクレオシドを産生するリボヌクレオシドを包含する。好ましくは、このような置換は、1-6個の飽和または不飽和炭素原子を有する低級アルキル基によるもの、または2-6個の炭素原子を有するアリールまたはアリル基によるものであって、このようなアルキル、アリールまたはアリル基は未置換であってもよく、または例えば、ハロ、ヒドロキシ、トリフルオロメチル、シアノ、ニトロ、アシル、アシルオキシ、アルコキシ、カルボキシル、カルボアルコキシルまたはアミノ基で置換されていてもよい。「2'-置換リボヌクレオシド」という用語は、2'-ヒドロキシル基がアミノ基またはハロ基、好ましくは、フルオロで置換されているリボヌクレオシドも包含する。
【0096】
本発明のこの様相で用いられる特に好ましいアンチセンスオリゴヌクレオチドとしては、キメラオリゴヌクレオチドおよびハイブリッドオリゴヌクレオチドが挙げられる。
【0097】
本発明の目的のために、「キメラオリゴヌクレオチド」とは、2種類以上のヌクレオシド間結合を有するオリゴヌクレオチドを表す。このようなキメラオリゴヌクレオチドの一つの好ましい例は、ホスホロチオエート、ホスホジエステルまたはホスホロジチオエート領域を含んでなる、好ましくは、約2-約12個のヌクレオチド、およびアルキルホスホネートまたはアルキルホスホノチオエート領域を含んでなるキメラオリゴヌクレオチドである(例えば、Pederson et al. 米国特許第5,635,377号明細書および第5,366,878号明細書を参照)。好ましくは、このようなキメラオリゴヌクレオチドは、ホスホジエステルおよびホスホロチオエート結合、またはそれらの組合せから選択される少なくとも3個の連続したヌクレオシド間結合を含む。
【0098】
本発明の目的のために、「ハイブリッドオリゴヌクレオチド」とは、2種類以上のヌクレオシドを有するオリゴヌクレオチドを表す。このようなハイブリッドオリゴヌクレオチドの好ましい一例は、リボヌクレオチドまたは2'-置換リボヌクレオチド領域を含んでなり、好ましくは、約2-約12個の2'-置換ヌクレオチドと1個のデオキシリボヌクレオチド領域を含んでなる。好ましくは、このようなハイブリッドオリゴヌクレオチドは、少なくとも3個の連続したデオキシリボヌクレオシドを含み、またリボヌクレオシド、2'-置換リボヌクレオシド、好ましくは、2'-O-置換リボヌクレオシド、またはそれらの組合せも含む(例えば、Metelev and Agrawal, 米国特許第5,652,355号明細書参照)。
【0099】
本発明で用いられるアンチセンスオリゴヌクレオチドの正確なヌクレオチド配列および化学構造は、オリゴヌクレオチドが目的とする遺伝子発現を阻害する能力を保持する限り、変化することができる。これは、特定のアンチセンスオリゴヌクレオチドが活性であるかどうかを試験することによって容易に決定される。この目的のための有用な評価法としては、遺伝子の産物をコードするmRNAの定量、遺伝子の産物についてのウェスタンブロット分析、酵素活性遺伝子産物についての活性分析法、またはソフトアーガー成長分析法、またはレポーター遺伝子構築物分析法、またはイン・ビボ腫瘍成長分析法が挙げられ、これらはいずれも本明細書またはRamchandani et al. (1997) Proc. Natl. Acad. Sci. USA 94: 684-689に詳細に記載されている。
【0100】
本発明で用いられるアンチセンスオリゴヌクレオチドは、H-ホスホネート化学、ホスホルアミダイト化学、またはH-ホスホネート化学とホスホルアミダイト化学の組合せ(すなわち、幾つかのサイクルについてH-ホスホネート化学および他のサイクルについてホスホルアミダイト化学)などの周知の化学的方法を用いて適当な固形支持体上で好都合に合成することができる。適当な固形支持体としては、コントロールド-ポアグラス(controlled-pore glass; CPG)のような固相オリゴヌクレオチド合成に用いられる標準的固形支持体のいずれかが挙げられる(例えば、 Pon, R. T. (1993) Methods in Molec. Biol. 20:465-496参照)。
【0101】
特に好ましいオリゴヌクレオチドは、表1に示されるヌクレオチド配列を包含する約13-約35個のヌクレオチドのヌクレオチド配列を有する。更に一層特に好ましいオリゴヌクレオチドは、表1に示されるヌクレオチド配列の約15-約26個のヌクレオチドのヌクレオチド配列を有する。
【0102】
【表1】
【0103】
下記の実施例は、本発明のある種の好ましい態様を更に例示しようとするものであり、発明の範囲を制限しようとするものではない。
実施例
(実施例1)
【0104】
N-(2-アミノ-フェニル)-4-[3-(3,4-ジクロロ-フェニル)-アクリロイル]-ベンズアミド (Ia)
工程1: 4-[3-(3,4-ジクロロ-フェニル)-アクリロイル]-安息香酸 (IIa)
4-アセチル安息香酸 (1.71g, 10.44ミリモル)、3,4-ジクロロベンズアルデヒド(2.05g, 11.49ミリモル)またはアルデヒド(1.1当量)をMeOH (50ml)中で室温にて攪拌懸濁したものに、NaOH (26.1ml, 1N、H2O中)の溶液を加えた。19時間後、反応混合物を濾別し、MeOHで洗浄し、乾燥して、標題化合物 IIa (3.22g, 10.03ミリモル, 収率96%)を黄色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): 8.35 (s, 1H), AB系(δA=8.13, δB=8.01, J=8.4Hz, 4H), 8.12 (d, J=15.8Hz, 1H), 7.93 (d, J=7.9Hz, 1H), 7.76 (d, J=8.3Hz, 1H), 7.73 (d, 15.8Hz, 1H)。
【0105】
工程2: N-(2-アミノ-フェニル)-4-[3-(3,4-ジクロロ-フェニル)-アクリロイル]-ベンズアミド (Ia)
IIa (300mg, 0.93ミリモル)を無水DMF (15ml)に窒素下で室温にて攪拌溶解したものに、Et3N (156μl, 1.12ミリモル)およびBOP試薬(454mg, 1.03ミリモル)をそれぞれ加えた。30分後、1,2-フェニレンジアミン(111mg, 1.03ミリモル)、Et3N (391μl, 2.80ミリモル)を無水DMF (2ml)に溶解したものを滴加した。21時間後、反応混合物をNH4Clの飽和水溶液に空けて、AcOEtで希釈した。分離後、有機層を飽和NH4Cl、H2Oおよび塩水で連続して洗浄して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/CH2Cl2: 10/90→20/90)、標題化合物Ia(237mg, 0.58ミリモル、収率62%)を黄色粉末として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): 9.90 (s, 1H), 8.40-8.30 (m, 3H), 8.25-8.10 (m, 3H), 7.97 (d, J=8.8Hz, 1H), 7.85-7.75 (m, 2H), 7.23 (d, J=7.5Hz, 1H), 7.03 (t, J=7.3Hz, 1H), 6.84 (d, J=7.9Hz, 1H), 6.65 (t, J=7.5Hz, 1H), 4.99 (s, 2H)。
(実施例2および10)
【0106】
実施例2および10 (化合物Ib, Ij)は、実施例1の化合物Iaについて記載したのと同じ手続きを用いて調製した(工程図1)。
(実施例3)
【0107】
N-(2-アミノ-フェニル)-4-[3-(2,6-ジクロロ-フェニル)-アクリロイル]-ベンズアミド(Ic)
工程1:t-ブチル[2-(4-アセチル-ベンゾイルアミノ)-フェニル]-カルバメート (III)
4-アセチル安息香酸 (395mg, 2.41ミリモル)を無水DMF (15ml)中で窒素下にて室温で攪拌溶解したものに、Et3N (369μl, 2.65ミリモル)とBOP試薬 (1.171g, 2.65ミリモル)をそれぞれ加えた。30分後、t-ブチル(2-アミノ-フェニル)-カルバメート (551mg, 2.65ミリモル)、Et3N (1.01ml, 7.22ミリモル)を無水DMF (5ml)に溶解したものを滴加した。19時間後、反応混合物をNH4Clの飽和水溶液に空けて、AcOEtで希釈した。分離後、有機層を飽和NH4Cl、H2Oおよび塩水で連続して洗浄し、無水MgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 40/6O→50/50)、標題化合物 III (500mg, 1.41ミリモル, 59%収率)を黄色固形物として得た。1H NMR (300 MHz,CDCl3) δ(ppm): 9.47 (bs,1H), 8.1O-8.00 (m, 4H), 7.91 (d, J=7.9Hz, 1H), 7.33-7.15 (m, 3H), 6.67 (s,1H), 2.67 (s, 3H), 1.53 (s,9H)。
【0108】
工程2: t-ブチル(2-{4-[3-(2,6-ジクロロ-フェニル)-アクリロイル]-ベンゾイルアミノ}-フェニル)-カルバメート (IVc)
III (150mg, 0.42ミリモル)、2,6-ジクロロベンズアルデヒド(148mg, 0.85ミリモル)またはアルデヒド(1.5-2.0当量)をMeOH (10ml)に室温にて攪拌溶解したものに、NaOH (1.7ml, 1N、H2O中)の溶液を加えた。淡黄色沈澱が生じた。3日後、反応混合物を濾別し、H2Oで洗浄した。次に、固形残渣をAcOEtに溶解し、無水MgSO4上で乾燥し、濾過して、濃縮した。粗製残渣を最後にシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 20/80→40/60)、標題化合物IVc (185mg, 0.36ミリモル, 85%収率)を淡黄色フォームとして得た。1H NMR (300 MHz, CDCl3) δ(ppm): 9.48 (bs,1H), 8.11 (s, 4H), 7.96 (d, J=17.1Hz, 1H), 7.89 (d, J=8.8Hz, 1H), 7.68 (d, J=16.3Hz, 1H), 7.42 (d, J=7.9Hz, 2H), 7.35-7.15 (m, 4H), 6.68 (s,1H), 1.54 (s, 9H)。
【0109】
工程3: N-(2-アミノ-フェニル)-4-[3-(2,6-ジクロロ-フェニル)-アクリロイル]-ベンズアミド (Ic)
IVc (135mg, 0.26ミリモル)をCH2Cl2 (10ml)に室温にて攪拌溶解したものに、トリフルオロ酢酸 (2ml, 95%水溶液)を加えた。16時間後、反応混合物を濃縮し、シリカゲル上でフラッシュクロマトグラフィーによって直接精製し(AcOEt/ CH2Cl2: 15/85)、標題化合物 Ic (90mg, 0.22ミリモル, 83%収率)を橙色固形物として得た。1H NMR (300 MHz, DMSO-d6) δ(ppm): 9.89 (s,1H), AB系(δA=8.23, δB=8.19, J=8.5Hz, 4H), 7.90 (d, J=16.3Hz, 1H), 7.78 (d, J=16.3Hz, 1H), 7.67 (d, J=7.9Hz, 2H), 7.55-7.45 (m,1H), 7.23 (d, J=7.5Hz, 1H), 7.03 (t, J=7.5Hz, 1H), 6.83 (d, J=7.9Hz, 1H), 6.64 (t, J=7.3Hz, 1H), 5.00 (s, 2H)。
(実施例4−9)
【0110】
実施例4-9 (化合物Id-Ii)は、実施例3の化合物Icについて記載したのと同じ手続きを用いて調製した(工程図2)。
【0111】
【表2−1】
【表2−2】
(実施例11)
【0112】
N-(2-アミノ-フェニル)-4-[2-(3,4,5-トリメトキシ-フェニルカルバモイル)-ビニル]-ベンズアミド (Va)
工程1: メチル 4-(2-t-ブトキシカルボニル-ビニル)-ベンゾエート (VI)
無水i-Pr2NH (1.76ml, 12.49ミリモル)を0℃で窒素下にて攪拌した無水THF (30ml)に溶解したものに、n-BuLi (5.36ml, 13.40ミリモル, 2.5M、ヘキサン中)の溶液を徐々に加えた。30分後、LDAを-78℃に冷却し、t-ブチルアセテート(1.64ml, 12.18ミリモル)を滴加した。30分後、メチル 4-ホルミルベンゾエート (1.00g, 6.09ミリモル)を無水THF (10ml)に溶解したものを徐々に加えた。2時間後、2-クロロ-4,6-ジメトキシ-1,3,5-トリアジン(1.604g, 9.14ミリモル)を無水THF (10ml)に溶解したものを加えた。次いで、温度を、一晩で室温まで上昇させた。懸濁液を生じた。反応混合物をNH4Clの飽和水溶液に空けて、AcOEtで希釈した。分離後、有機層をH2Oおよび塩水で連続して洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。粗生成物をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 10/90→15/85)、標題生成物VI (785mg, 3.00ミリモル, 49%収率)を白色固形物として得た。1H NMR(300 MHz, CDCl3) δ(ppm): AB系(δA=8.04, δB=7.57, J=8.4Hz, 4H), 7.60 (d, J=15.4Hz, 1H), 6.46 (d, J=15.8Hz, 1H), 3.93 (s, 3H), 1.54 (s, 9H)。13C NMR (75 MHz, CDCl3)δ(ppm): 166.72, 166.01, 142.31, 139.18, 131.33, 130.26, 127.99, 122.87, 81.11, 52.46, 28.40。
【0113】
工程2: メチル 4-(2-カルボキシ-ビニル)-ベンゾエート (VII)
VI (745mg, 2.84ミリモル)をCH2Cl2 (10ml)に室温にて攪拌溶解したものに、トリフルオロ酢酸 (6ml, 95%、水中)を加えた。27時間後、反応混合物を濃縮し、水中で粉砕した。1時間後、懸濁液を濾別し、H2Oで洗浄し、乾燥して、標題化合物 VII (556mg, 2.70ミリモル, 95%収率)を灰白色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): AB系(δA=8.01, δB=7.88, J=8.1Hz, 4H), 7.68 (d, J=15.8 Hz, 1H), 6.70 (d, J=16.3Hz, 1H), 3.90 (s, 3H)。
【0114】
方法A, 工程3: メチル 4-[2-(ベンゾトリアゾール-1-イルオキシカルボニル)-ビニル]-ベンゾエート (VIII)
VII (264mg, 1.28ミリモル)を無水DMF (10ml)に窒素下で室温にて攪拌溶解したものに、Et3N (196μl, 1.41ミリモル)とBOP試薬 (680mg, 1.1.54ミリモル)をそれぞれ加えた。数分後、沈澱を生じた。3時間後、反応混合物をNH4Clの飽和水溶液に空けて、AcOEtで希釈した。分離後、有機層を飽和NH4Cl、H2Oおよび塩水で連続して洗浄し、少し濃縮し、ヘキサンを加えた。懸濁液を濾別し、ヘキサンで洗浄した。固形物を水中で粉砕し、濾別し、水で洗浄し、乾燥して、標題化合物 VIII (346mg, 1.07ミリモル, 84%収率)を淡黄色固形物として得た(シリカゲル上では安定でない!)。1H NMR (300 MHz, CDCl3) δ(ppm): 8.56 (d, J=8.3Hz, 1H), 8.21-8.02 (m, 3H), 7.9O-7.72 (m, 4H), 7.62 (t, J=7.4Hz, 1H), 3.97 (s, 3H)。
【0115】
工程4: メチル4-[2- (3,4,5-トリメトキシ-フェニルカルバモイル)-ビニル]-ベンゾエート (IXa)
VIII (150mg, 0.46ミリモル)を無水CH2Cl2 (10ml)に窒素下で室温にて攪拌懸濁したものに、Et3N (194μl, 1.39ミリモル)および3,4,5-トリメトキシアニリン(94mg, 0.51ミリモル)またはArNH2(1.1-1.2当量)をそれぞれ加えた。反応混合物を、60℃に加熱した。20時間後、反応混合物を濃縮し、AcOEtで希釈し、NH4Clの飽和水溶液、H2Oおよび塩水で連続して洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。粗生成物をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ CH2Cl2: 15/85→20/80)、標題生成物IXa (130mg, 0.35ミリモル, 75%収率)を黄色固形物として得た。1H NMR (300 MHz, アセトン-d6)δ(ppm): 9.42 (bs, 1H), AB系 (δA=8.09, δB=7.78, J=8.1Hz, 4H), 7.75 (d, J=15.6Hz, 1H), 7.21 (s, 2H), 7.00 (d, J=15.8Hz, 1H), 3.94 (s, 3H), 3.85 (s, 6H), 3.73 (s, 3H)。
【0116】
工程5: 4-[2-(3,4,5-トリメトキシ-フェニルカルバモイル)-ビニル]-ベンゾエート (Xa)
IXa (125mg, 0.34ミリモル)をTHF (5ml)に室温にて攪拌溶解したものに、LiOH.H2O (35mg, 0.84ミリモル)を水(5ml)に溶解したものを加えた。1.5日後、反応混合物を濃縮し、水で希釈し、1N HClでpH 4-5まで酸性にして、沈澱を得た。10分間攪拌した後、懸濁液を濾別し、水で洗浄し、乾燥して、標題化合物 Xa (110mg, 0.31ミリモル, 91%収率)を淡黄色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm) : 10.29 (s, 1H), AB系 (δA=8.04, δB=7.76, J=8.4Hz, 4H), 7.65 (d, J=15.8Hz, 1H), 7.13 (s, 2H), 6.94 (d, J=15.8Hz, 1H), 3.81 (s, 6H), 3.67 (s, 3H)。
【0117】
工程6: N-(2-アミノ-フェニル)-4-[2-(3,4,5-トリメトキシ-フェニルカルバモイル)-ビニル]-ベンズアミド (Va)
Xa(110mg, 0.31ミリモル)を無水DMF (3ml)に窒素下で室温にて攪拌溶解したものに、Et3N (47μl, 0.34ミリモル)およびBOP試薬 (163mg, 0.37ミリモル)をそれぞれ加えた。30分後、1,2-フェニレンジアミン (37mg, 0.34ミリモル), Et3N(129μl, 0.92ミリモル)を無水DMF (1ml)に溶解したものを滴加した。3時間後、反応混合物をNH4Clの飽和水溶液に空けて、AcOEtで希釈した。分離後、有機層を飽和NH4Cl、H2Oおよび塩水で連続して洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/CH2Cl2: 50/50→80/20)、標題化合物 Va (98mg, 0.22ミリモル,71%収率)を黄色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): 10.27 (s, 1H), 9.76 (s, 1H), AB系(δA=8.09, δB= 7.78, J=7.9Hz, 4H), 7.71 (d, J=15.8Hz, 1H), 7.22 (d, J=7.5Hz, 1H), 7.14 (s, 2H), 7.02 (t, J=7.0Hz, 1H), 6.95 (d, J=15.8Hz, 1H), 6.83 (d, J=7.9Hz, 1H), 6.65 (t, J=7.5Hz, 1H), 4.97 (bs, 2H), 3.81 (s, 6H), 3.68 (s, 3H)。
(実施例12)
【0118】
N-(2-アミノ-フェニル)-4-{2-[(ピリジン-3-イルメチル)-カルバモイル]-ビニル}-ベンズアミド (Vb)
方法B, 工程3: メチル4-[2-(ピリジン-3-イルメチル)-カルバモイル)-ビニル]-ベンゾエート (Vb)
VIII (140mg, 0.68ミリモル)を無水DMF (5ml)に窒素下で室温にて攪拌溶解したものに、Et3N(104μl, 0.75ミリモル)とBOP試薬 (331mg, 0.75ミリモル)をそれぞれ加えた。30分後、3-(アミノメチル)ピリジン (90μl, 0.88ミリモル)またはR1R2NH (1.2-1.3当量)、Et3N (284μl, 2.04ミリモル)を無水DMF (2ml)に溶解したものを滴加した。4時間後、反応混合物をNH4Clの飽和水溶液に空けて、AcOEtで希釈した。分離後、有機層を飽和NH4Cl、H2Oおよび塩水で連続して洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(MeOH/ CH2Cl2: 5/95→7/93)、標題化合物 IXb (185mg, 0.62ミリモル, 92%収率)を白色固形物として得た。1H NMR (300 MHz, CDCl3)δ(ppm): 8.67-8.44 (m, 2H), AB系(δA=8.03,δB=7.55, J=8.4Hz, 4H), 7.78-7.64 (m, 2H), 7.33-7.26 (m, 1H), 6.54 (d, J=15.8Hz, 1H), 6.38 (bs, 1H), 4.61 (d, J=6.2Hz, 2H), 3.92 (s, 3H)。
【0119】
工程4: N-(2-アミノ-フェニル)-4-{2-[(ピリジン-3-イルメチル)-カルバモイル]-ビニル}-ベンズアミド (Vb)
標題化合物Vbは、IXbから実施例10の工程5および6(工程図3)と同じ手続きによって2段階で得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): 9.74 (s, 1H), 8.79 (t, J=5.7Hz, 1H), 8.58 (s, 1H), 8.52 (d, J=4.0Hz, 1H), 8.06 (d, J=7.9Hz, 2H), 7.83-7.68 (m, 3H), 7.59 (d, J=15.8Hz, 1H), 7.41 (dd, J=7.9, 4.7Hz, 1H), 7.21 (d,J=7.9Hz, 1H), 7.02 (t, J=7.0Hz, 1H), 6.83 (d, J=15.8Hz, 1H), 6.82 (d, J=7.5Hz, 1H), 6.64 (t,J=7.3Hz, 1H), 4.96 (bs, 2H), 4.48 (d, J=5.7Hz, 2H)。
(実施例13−15)
【0120】
実施例13-15 (化合物Vc-Ve)は、実施例12の化合物Vbについて記載したのと同じ手続きを用いて調製した(工程図3)。
(実施例16)
【0121】
N-(2-アミノ-フェニル)-4-[2-(2-ピリジン-3-イル-エチルカルバモイル)-ビニル]-ベンズアミド (Vf)
工程1: t-ブチル[2-(4-ホルミル-ベンゾイルアミノ)-フェニル]-カルバメート (XI)
4-カルボキシベンズアルデヒド(3.00g, 19.98ミリモル)を無水CH2Cl2(10ml)に窒素下で室温にて攪拌懸濁したものに、塩化チオニル (2.19ml, 29.97ミリモル)と無水DMF(387μl, 5.00ミリモル)をそれぞれ加えた。反応混合物を5時間還流した。次に、反応混合物を室温まで放冷し、濃縮し、無水CH2Cl2(20ml)で窒素下にて希釈した。この溶液を、t-ブチル(2-アミノ-フェニル)-カルバミン酸エステル(4.575g, 21.98ミリモル)、Et3N (8.36ml, 59.95ミリモル)を無水CH2Cl2(50ml)に窒素下で-20℃で冷却混合したものにカニューレ導入した。1時間後、反応混合物を室温まで温度を上昇させた。1時間後、これをNH4Clの飽和水溶液に空けて、CH2Cl2で抽出した。一緒に合わせた有機層を連続してMgSO4上で乾燥し、濾過して、濃縮した。粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 30/70→40/60)、標題化合物 XI (4.80g, 14.11ミリモル, 71%収率)を淡黄色固形物として得た。1H NMR (300 MHz,CDCl3)δ(ppm): 10.11 (s, 1H), 9.58 (bs, 1H), AB系(δA=8.14, δB=7.99, J=8.1Hz, 4H), 7.89 (d, J=7.9Hz, 1H), 7.35-7.10 (m, 3H), 6.75 (s, 1H), 1.53 (s, 9H)。
【0122】
工程2: メチル3-[4-(2-t-ブトキシカルボニルアミノ-フェニルカルバモイル)-フェニル]-アクリレート (XII)
化合物XI (500mg, 1.47ミリモル)、メチル(トリフェニル-ホスホラニリデン)アセテート (590mg, 1.76ミリモル)を無水トルエン(20ml)に攪拌懸濁したものを、窒素下で90℃で加熱した。2日後、反応混合物を濃縮し、直ちにシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 30/70→40/60)、標題化合物Xll (568mg, 1.43ミリモル, 97%収率)を淡黄色フォームとして得た。1H NMR (300 MHz, CDCl3) δ(ppm): 9.32 (bs,1H), AB系(δA=7.99, δB=7.62, J=8.4Hz, 4H), 7.87 (d, J=7.9Hz, 1H), 7.73 (d, J=15.8Hz, 1H), 7.32-7.13 (m, 3H), 6.69 (bs, 1H), 6.53 (d, J=16.3Hz, 1H), 3.83 (s, 3H), 1.53 (s, 9H)。
【0123】
工程3: 3-[4-(2-t-ブトキシカルボニルアミノ-フェニルカルバモイル)-フェニル]-アクリル酸(XIII)
化合物XII (560mg, 1.41ミリモル)をTHF (20ml)に室温にて攪拌溶解したものに、LiOH.H2O (148mg, 3.53ミリモル)を水 (20ml)に溶解したものを加えた。23時間後、反応混合物を濃縮し、水で希釈し、1N HClでpH 4-5になるまで酸性にして、白色沈澱を得た。15分間攪拌した後、懸濁液を濾別し、水で洗浄し、乾燥して、標題化合物XIII (495mg, 1.29ミリモル, 92%収率)を白色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm):9.92 (s, 1H), 8.72 (bs, 1H), AB系(δA=8.02,δB=7.90, J=7.9Hz, 4H), 7.69 (d, J=16.3Hz, 1H), 7.62-7.53 (m, 2H), 7.3O-7.13 (m, 2H), 6.72 (d,J=16.3Hz, 1H), 1.48 (s, 9H)。
【0124】
工程4: t-ブチル(2-{4-[2-(2-ピリジン-3-イル-エチルカルバモイル)-ビニル]-ベンゾイルアミノ}-フェニル)-カルバメート (XIVf)
化合物XIII (80mg, 0.21ミリモル)を無水DMF (3ml)に窒素下で室温にて攪拌溶解したものに、Et3N (35μl, 0.25ミリモル)およびBOP試薬 (102mg, 0.23ミリモル)をそれぞれ加えた。30分後、3-(2-アミノエチル)ピリジン (51mg, 0.42ミリモル)またはRXH (1.5-2.0当量)、Et3N (87μl, 0.63ミリモル)を無水DMF(1ml)に溶解したものを滴加した。3-5時間後、反応混合物をNH4Clの飽和水溶液に空けて、AcOEtで希釈した。分離後、有機層を連続して飽和NH4Cl、H2Oおよび塩水で洗浄し、MgSO4上で乾燥し、濾過して、濃縮し、標題化合物 XIVfを得た。これは、更に精製することなく次工程に用いた。
【0125】
工程5: N-(2-アミノ-フェニル)-4-[2-(2-ピリジン-3-イル-エチルカルバモイル)-ビニルl-ベンズアミド (Vf)
XIVfをCH2Cl2 (15ml)に室温にて攪拌溶解したものに、トリフルオロ酢酸 (2ml, 95%、水中)を加えた。18時間後、反応混合物を濃縮し、水に溶解し、NaHCO3の飽和水溶液でpH=7になるまで中和した。淡黄色沈澱を生じた。数分後、懸濁液を濾別し、H2Oで洗浄し、乾燥して、標題化合物 Vf (69mg, 0.18ミリモル, 2工程についての収率85%)を淡黄色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): 9.72 (s, 1H), 8.53-8.41 (m, 2H), 8.29 (t, J=5.5Hz, 1H), 8.05 (d, J=8.4Hz, 2H), 7.8O-7.63 (m, 3H), 7.51 (d, J=15.8Hz, 1H), 7.37 (dd, J=7.5, 4.8Hz, 1H), 7.21 (d, J=7.5Hz, 1H), 7.02 (t, J=7.5Hz, 1H), 6.82 (d, J=7.5Hz, 1H), 6.76 (d, J=15.8Hz, 1H), 6.64 (t, J=7.3Hz, 1H), 4.95 (bs, 2H), 3.51 (dd, J=6.8Hz, 2H), 2.86 (t, J=6.8Hz, 2H)。
(実施例17−26)
【0126】
実施例17-26 (化合物Vg-Vp)は、実施例16の化合物Vfについて記載したのと同じ手続きを用いて調製した(工程図4)。
【0127】
【表3−1】
【表3−2】
【表3−3】
(実施例27)
【0128】
N-(2-アミノ-フェニル)-4-[3-(3-シクロペンチルオキシ-4-メトキシ-フェニルアミノ)-プロペニル]-ベンズアミド (XVa)
工程1: t-ブチル {2-[4-(3-オキソ-プロペニル)-ベンゾイルアミノ]-フェニル}-カルバメート (XVI)
化合物XI (4.00g, 11.75ミリモル)、(トリフェニルホスホラニリデン)-アセトアルデヒド(3.60g, 11.83ミリモル)を無水トルエン (100ml)中で攪拌懸濁したものを、窒素下にて80℃で加熱した。2日後、反応混合物を濃縮し、シリカゲル上でフラッシュクロマトグラフィーによって直接精製し(AcOEt/ヘキサン: 30/70)、標題化合物 XVI (3.70g, 10.10ミリモル, 86%収率)を黄色の粘稠な固形物として得た(ジエンを僅かに混入)。1H NMR (300 MHz, CDCl3)δ(ppm): 9.75 (d, J=7.8Hz, 1H), 9.49 (bs, 1H), AB系(δA=8.03, δB=7.65, J=8.4Hz, 4H), 7.85-7.72 (m, 1H), 7.52 (d, J=15.6Hz, 1H), 7.33-7.05 (m, 3H), 7.05-6.90 (m,1H), 6.78(dd, J=15.6, 7.8Hz, 1H), 1.53 (s, 9H)。
【0129】
工程2: t-ブチル (2-{4-[3-(3-シクロペンチルオキシ-4-メトキシ-フェニルアミノ)-プロペニル]-ベンゾイルアミノ}-フェニル)-カルバメート (XVIIa)
化合物XVI (210mg, 0.57ミリモル)、3-シクロペンチルオキシ-4-メトキシ-アニリン (125mg, 0.60ミリモル)またはArNH2 (1.05-1.2当量)を無水THF (7ml)に窒素下で室温にて攪拌溶解したものに、二塩化ジブチルスズ(3.5mg, 0.01ミリモル)を加えた。10分後、フェニルシラン (78μl, 0.63ミリモル)を滴加した。3日後、反応混合物を濃縮し、シリカゲル上でフラッシュクロマトグラフィーによって直接精製し(AcOEt/ヘキサン: 30/70→50/50)、標題化合物XVIIaを黄色の粘稠な油状生成物として得た。
【0130】
工程3: N-(2-アミノ-フェニル)-4-[3-(3-シクロペンチルオキシ-4-メトキシ-フェニルアミノ)-プロペニル]-ベンズアミド (XVa)
XVIIaをCH2Cl2(30ml)に室温にて攪拌溶解したものに、トリフルオロ酢酸 (5ml, 95%、水中)を加えた。16時間後、反応混合物を濃縮し、水に溶解し、NaOH(1N)の水溶液で塩基性にし、pH=8とした。ベージュ色沈澱が生じた。15分後、懸濁液を濾別し、H2Oで洗浄し、風乾した。粗生成物をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ CH2Cl2: 15/85→20/80+εNH4OH)、標題化合物 XVa (145mg, 0.32ミリモル, 55%、二段階についての収率)を黄色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): ロータマーの混合物、9.67および9.63 (2s, 1H), 7.98 (d, J=7.9Hz, 2H), 7.57および7.51 (2d, J=7.9Hz, 2H), 7.20 (d, J=7.9Hz, 1H), 7.01 (t, J=7.7Hz, 1H), 6.82 (d, J=7.9Hz, 1H), 6.77-6.67 (m, 2H), 6.63 (t, J=7.5Hz, 1H), 6.54 (dt, J=16.3, 5.2Hz, 1H), 6.35および6.30 (2d, J=2.0Hz, 1H), 6.15および6.06 (2dd, J=8.6, 2.0Hz, 1H), 5.98および5.57 (2t, J=5.5Hz, 1H), 4.92 (bs, 2H), 4.78-4.63 (m, 1H), 4.32および3.87 (2d, J=5.7Hz, 2H), 3.65および3.62 (2s, 3H), 1.95-1.45 (m, 8H)。
(実施例28−32)
【0131】
実施例28-32 (化合物XVb-XVf)は、実施例27の化合物XVaについて記載したのと同じ手続きを用いて調製した(工程図5)。
(実施例33)
【0132】
N-(2-アミノ-フェニル)-4-[3-(4-トリル-スルホニルアミノ)-プロペニル]-ベンズアミド (XVg)
工程1: t-ブチル {2-[4-(3-ヒドロキシ-プロペニル)-ベンゾイルアミノ]-フェニル}-カルバメート (XVIII)
化合物XVI (1.00g, 2.79ミリモル)をエタノール(15ml)に窒素下で攪拌溶解したものに、水素化ホウ素ナトリウム (110mg, 2.73ミリモル)を加えた。5分後、反応混合物を水で反応停止させ、AcOEtで希釈した。分離後、有機層を塩水で連続して洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 40/60)、標題化合物 XVIII (910mg, 2.29ミリモル, 82%収率)を淡黄色固形物として得た(ジエンが僅かに混入)。1H NMR (300 MHz, CDCl3)δ(ppm): 9.20 (s, 1H), 7.90 (d, J=7.8Hz, 2H), 7.75 (d, J=7.5Hz, 1H), 7.43 (d, J=8.4Hz, 2H), 7.32-7.08 (m, 3H), 6.94 (s, 1H), 6.65 (d, J=15.9Hz, 1H), 6.45 (td, J=15.9, 5.4Hz, 1H), 4.35 (d, J=5.4Hz, 2H), 1.92 (s, 1H), 1.51 (s, 9H)。
【0133】
工程2: t-ブチル (2-{4-[3-(4-トリル-スルホニルアミノ)-プロペニル]-ベンゾイルアミノ}-フェニル)-カルバメート(XVIIg)
N-Boc-4-トリルスルホンアミド (221mg, 0.81ミリモル)とPPh3 (427mg, 1.63ミリモル)を無水THF(4ml)に窒素下で攪拌溶解したものに、化合物XVIII (200mg, 0.54ミリモル)を無水THF(1ml)に溶解したものおよびジエチルアゾジカルボキシレート(DEAD) (214μl, 1.36ミリモル)を加えた。16時間後、反応混合物を水で反応停止し、AcOEtで希釈した。分離後、有機層を水および塩水で連続して洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 40/60)、標題化合物XVIIg (337 mg)を得た。
【0134】
工程3: N-(2-アミノ-フェニル)-4-[3-(4-トリル-スルホニルアミノ)-プロペニル]-ベンズアミド (XVg)
XVIIgをCH2Cl2 (20ml)に室温にて攪拌溶解したものに、トリフルオロ酢酸 (2ml, 95%、水中)を加えた。16時間後、反応混合物を濃縮し、水に溶解し、NaHCO3の飽和水溶液で塩基性にした。水性層をAcOEtで抽出した。合わせた有機層を塩水で連続洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。粗製残渣をできるだけ少量のAcOEt/MeOH(95/5)の混合物で可溶化させ、ヘキサンで共沈させた。灰白色沈澱生成物を生じた。数分後、懸濁液を濾別し、ヘキサンで洗浄し、乾燥して、標題化合物XVg (173mg, 0.41ミリモル, 76%、二段階についての収率)を灰白色固形物として得た。1H NMR (300 MHz, DMSO-d6)δ(ppm): 9.64 (s, 1H), AB系(δA=7.93, δB=7.72, J=8.4Hz, 4H), 7.84 (s, 1H), 7.41 (t, J=8.4Hz, 4H), 7.16 (d, J=7.8Hz, 1H), 6.97 (t, J=7.5Hz, 1H), 6.77 (d, J=7.5Hz, 1H), 6.59 (t, J=7.8Hz, 1H), 6.52 (d, J=15.6Hz, 1H), 6.21 (dt, J=15.6, 5.7Hz, 1H), 4.89 (s, 2H), 3.60 (bs, 2H), 2.08 (s, 3H)。
(実施例34)
【0135】
N-(2-アミノ-フェニル)-4-{3-[(ピリジン-3-イルメチル)-アミノ]-プロペニル}-ベンズアミド (XVh)
工程1: メチル4-(3-オキソ-プロペニル)-ベンゾエート (XIX)
方法A: 化合物メチル4-ホルミルベンゾエート (4.00g, 24.37ミリモル)、(トリフェニルホスホラニリデン)-アセトアルデヒド(7.56g, 24.85ミリモル)を無水トルエン (100ml)に攪拌懸濁したものを、窒素下にて8O-90℃で加熱した。1日後、反応混合物を濃縮し、シリカゲル上でフラッシュクロマトグラフィーによって直接精製し(AcOEt/ヘキサン: 20/80→30/70)、標題化合物 XIX (2.52g, 13.25ミリモル, 54%収率)を淡黄色固形物(ジエンが僅かに混入)として得た。1H NMR (500 MHz, CDCl3)δ(ppm): 9.76 (d, J=7.3Hz, 1H), AB系(δA=8.11, δB=7.64, J=8.1Hz, 4H), 7.51 (d, J=15.6Hz, 1H), 6.79 (dd, J=15.8, 7.6Hz, 1H), 3.95 (s, 3H)。
【0136】
方法B: TDA-1 (6.278g, 19.41ミリモル)および10%炭酸カリウム水溶液 (100ml)をCH2Cl2 (100ml)に室温で激しく攪拌したエマルションに、(1,3-ジオキソラン-2-イル)メチルトリフェニルホスホニウムブロミド(10g, 23.29ミリモル)およびメチル4-ホルミルベンゾエート (3.187g, 19.41ミリモル)をそれぞれ加えた。18時間攪拌後、反応混合物をCH2Cl2で抽出し、合わせた有機層を濃縮した。次に、10% HCl水溶液(100ml)を加えて、混合物を室温で一晩攪拌した。反応混合物を、水で希釈し、CH2Cl2で抽出した。合わせた有機層を連続的にMgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン:20/80→30/70)、AcOEt/ヘキサン中で粉砕し、標題化合物 XIX (2.50g, 13.14ミリモル, 68%収率)を結晶性固形物として得た(純粋なトランス型であり、ジエンを含まない)。
【0137】
工程2: メチル4-{3-[(ピリジン-3-イルメチル)-アミノ]-プロペニル}-ベンゾエート (XXh)
化合物XIX (300mg, 1.58ミリモル)および3-(アミノメチル)ピリジン(193μl, 0.60ミリモル)またはRNH2(1.1-1.2当量)を無水ジクロロメタン(15ml)に窒素下で溶解したものを1時間攪拌し、水素化トリアセトキシホウ素ナトリウム(401mg, 1.89ミリモル)を加えた。64時間後、反応混合物をK2CO3の水溶液(10%)で反応停止し、ジクロロメタンで抽出した。合わせた有機層を、MgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(MeOH/CH2Cl2: 5/95 + εNH4OH)、標題化合物 XXh (188mg, 0.66ミリモル, 42%収率)を暗黄色油状生成物として得た。
【0138】
工程3: 4-[3-(第三ブトキシカルボニル-ピリジン-3-イルメチル-アミノ)-プロペニル]-安息香酸 (XXIh)
XXh (187mg, 0.66ミリモル)を1,4-ジオキサン(7ml)に室温にて攪拌溶解したものに、(Boc)2O (173mg, 0.80ミリモル)およびNaOHの水溶液(3.3ml, 1N)をそれぞれ加えた。24時間後、反応混合物を濃縮し、水で希釈し、HClの水溶液(1N)で中和した(pH=6-7)。生成する淡黄色懸濁液を、ジクロロメタンで抽出した。合わせた有機層をMgSO4上で乾燥し、濾過し、濃縮して、標題化合物 XXIh (160mg, 0.43ミリモル, 66%収率)を黄色固形物として得た。
【0139】
工程4: t-ブチル{3-[4-(2-アミノ-フェニルカルバモイル)-フェニル]-アリル}-ピリジン-3-イルメチル-カルバメート(XXIIh)
標題化合物XXIIh (実施例34)は、実施例11、工程6と同じ手続きによって一段階でXXIhから淡黄色フォームとして得た。
【0140】
工程5: N-(2-アミノ-フェニル)-4-{3-[(ピリジン-3-イルメチル)-アミノ]-プロペニル}-ベンズアミド (XVh)
XXIIh (77mg, 0.17ミリモル)をジクロロメタン(10ml)中で室温にて攪拌溶解したものに、TFA (2ml, 95%、水中)を加えた。4.5時間後、反応混合物を濃縮し、水で希釈し、NaOHの水溶液(1N)で塩基性にし(pH=9)、ジクロロメタンで抽出した。合わせた有機層を、MgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(MeOH/CH2Cl2: 10/90 + εNH4OH)、標題化合物 XVh (35mg, 0.10ミリモル, 58%収率)を黄色粉末として得た。1H NMR (400MHz, DMSO-d6)δ(ppm): 9.64 (s, 1H), 8.55 (s, 1H), 8.44 (d, J=3.9Hz, 1H), AB系(δA=7.94, δB=7.55, J=8.0Hz, 4H), 7.78 (d, J=7.4Hz, 1H), 7.36 (dd, J=7.0, 5.1Hz, 1H), 7.16 (d, J=7.4Hz, 1H), 6.98 (t, J=7.4Hz, 1H), 6.79 (d, J=7.8Hz, 1H), 6.65-6.55(m, 2H), 6.51 (dt, J=16.0, 5.9Hz, 1H), 4.93 (bs, 2H), 3.77 (s, 2H)。
(実施例35−36)
【0141】
実施例35-36 (化合物 XVi-XVj)は、実施例34の化合物XVhについて記載したのと同じ手続きを用いて調製した(工程図7,経路B)。
【0142】
【表4−1】
【表4−2】
(実施例37)
【0143】
N-(2-アミノ-フェニル)-4-(3-オキソ-3-フェニル-プロペニル)-ベンズアミド (XXIVa)
工程1: 4-(3-オキソ-3-フェニル-プロペニル)-安息香酸(XIIIa)
4-ホルミル安息香酸 (2.58g, 17ミリモル)およびアセトフェノン (2.0ml, 17ミリモル)またはアセトフェノン誘導体 (1.O-1.1当量)をMeOH (100ml)中で室温にて攪拌懸濁したものに、NaOHの溶液(34ml, 1N、H2O中)を加えた。16時間後、反応混合物を濃HClで酸性にし(pH=1-2)、濾別し、H2Oで洗浄し、乾燥して、標題化合物XXIIIa (3.73g, 14.6ミリモル, 86%収率)を黄色固形物として得た。
【0144】
工程2: N-(2-アミノ-フェニル)-4-(3-オキソ-3-フェニル-プロペニル)-ベンズアミド (XXIVa)
標題化合物XXIVaは、実施例1、工程2と同じ手続きによって一段階でXXIIIaから得た(工程図1)。1H NMR (300 MHz, DMSO-d6) δ(ppm): 9.77 (s, 1H), 8.21 (d, J=7.0Hz, 2H), 8.06 (m, 5H), 7.82 (d, J=15.4Hz, 1H), 7.71 (t, J=7.3Hz, 1H), 7.60 (t, J=7.3Hz, 2H), 7.18 (d, J=7.9Hz, 1H), 6.99 (t, J=7.0Hz, 1H), 6.80 (d, J=7.5Hz, 1H), 6.61 (t, J=7.3Hz, 1H), 4.95 (bs, 2H)。
(実施例38−41)
【0145】
実施例38-41 (化合物 XXIVb-XXIVe)は、実施例37の化合物XXIVaについて記載したのと同じ手続きを用いて調製した(工程図8)。
(実施例42)
【0146】
N-(2-アミノ-フェニル)-4-[3-(4-モルホリン-4-イル-フェニル)-3-オキソ-プロペニル]-ベンズアミド (XXIVf)
工程1: t-ブチル(2-{4-[3-(4-モルホリン-4-イル-フェニル)-3-オキソ-プロペニル]-ベンゾイルアミノ}-フェニル)-カルバメート (XXVf)
XI (210mg, 0.62ミリモル)、4'-モルホリノアセトフェノン (227mg, 1.11ミリモル)またはアセトフェノン誘導体 (1.5-2.0当量)をMeOH (10ml)に室温にて攪拌溶解したものに、NaOHの溶液(1.9ml, 1N、H2O中)を加えた。沈澱を生じた。3日後、反応混合物を濾別し、MeOHで洗浄し、風乾し、真空乾燥して、標題化合物 XXVf (295mg, 0.56ミリモル, 90%収率)を黄色固形物として得た。
【0147】
工程2: N-(2-アミノ-フェニル)-4-[3-(4-モルホリン-4-イル-フェニル)-3-オキソ-プロペニル]-ベンズアミド(XXIVf)
XXVf (285mg, 0.54ミリモル)をCH2Cl2 (10ml)に室温にて攪拌溶解したものに、トリフルオロ酢酸 (2ml, 95%、水中)を加えた。17時間後、反応混合物を濃縮し、AcOEtで希釈し、飽和NaHCO3、H2O、飽和NH4Cl、H2Oおよび塩水で連続的に洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。粗生成物をAcOEt/ヘキサンの混合物中で共沈させ、粉砕した。数時間後、懸濁液を濾別し、ヘキサンで洗浄し、乾燥して、標題化合物XXIVf (210mg, 0.49ミリモル,91%収率)を黄色-橙色固形物として得た。1H NMR (300 MHz, DMSO-d6) δ(ppm): 9.78 (s, 1H), 8.25-7.94 (m, 7H), 7.76 (d, J=15.4Hz, 1H), 7.22 (d, J=7.5Hz, 1H), 7.09 (d, J=8.8Hz, 2H), 7.03 (t, J=7.7Hz, 1H), 6.83 (d, J=7.5Hz, 1H), 6.65 (t, J=7.5Hz, 1H), 4.97 (bs, 2H), 3.88-3.70(m, 4H), 3.48-3.30 (m, 4H)。
【0148】
【表5】
(実施例43)
【0149】
N-(2-アミノ-フェニル)-4-(3-オキソ-3-フェニル-プロピル)-ベンズアミド (XXVIIa)
工程1: 4-(3-オキソ-3-フェニル-プロピル)-安息香酸(XXVIa)
カルコンXXIIIa (1.29g, 5.13ミリモル)をDMF (20ml)に室温にて攪拌溶解したものに、フェニルスルホニルヒドラジン(1.76g, 10.26ミリモル)を加えた。反応混合物を110℃で15時間攪拌し、冷却して、濃縮した。残りの油状残渣を、NH4Clの飽和水溶液とAcOEtとの間で分配した。分離後、有機層を乾燥し、部分蒸発させ、濾過した。濾液をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 50/50→75/25)、物質を得て、これを第二のカラム精製の後(MeOH/CH2Cl2: 5/95)、標題化合物XXVIa (400mg, 1.59ミリモル, 31%収率)を得た。
【0150】
工程2: N-(2-アミノ-フェニル)-4-(3-オキソ-3-フェニル-プロピル)-ベンズアミド (XXVIIa)
標題化合物XXVIIaは、実施例1、工程2と同じ手続きによって一段階でXXVIaから得た(工程図1)。1H NMR (300 MHz, DMSO-d6)δ(ppm): 9.59 (s, 1H), 8.00 (d, J=7.5Hz, 2H), 7.90 (d, J= 7.9Hz, 2H), 7.64 (t, J=7.5Hz, 2H), 7.43 (d, J= 7.9Hz, 2H), 7.16 (d, J=7.5Hz, 1H), 6.97 (t, J= 7.0Hz, 1H), 6.78 (d, J=7.0Hz, 1H), 6.59 (t, J= 7.5Hz, 1H), 4.88 (bs, 2H), 3.44 (t, J=7.3Hz, 2H), 3.03 (t, J=7.3Hz, 2H)。
(実施例44)
【0151】
N-(2-アミノ-フェニル)-4-(3-フェニル-プロピル)-ベンズアミド (XXIXa)
工程1: 4-(3-フェニル-プロピル)-安息香酸(XXVIIIa)
XXIIIa (1.34g, 5.31ミリモル)を25ml DMAに室温にて攪拌溶解したものを、10% Pd/C (600mg, Degussa型)上で1気圧で3時間水素化した。触媒をセライトパッドを介して濾過によって除去した後、溶液を濃縮し、残渣を水で処理した。沈澱後、懸濁液を濾別し、H2Oで洗浄し、乾燥して、標題化合物XXVIIIa (1.13g, 4.72ミリモル, 89%収率)を得た。
【0152】
工程2: N-(2-アミノ-フェニル)-4-(3-フェニル-プロピル)-ベンズアミド(XXIXa)
標題化合物XXIXaは、実施例1、工程2と同じ手続きによって一段階でXXVIIIaから得た(工程図1)。1H NMR (300 MHz, DMSO-d6)δ(ppm): 9.60 (s, 1H), 7.91 (d, J=7.9Hz, 2H), 7.34 (d, J=8.4Hz, 2H), 7.28 (d, J=7.5Hz, 2H), 7.23-7.15 (m, 4H), 6.97 (t, J=7.0Hz, 1H), 6.78 (d, J=7.5Hz, 1H), 6.59 (t, J=7.3Hz, 1H), 4.88 (bs, 2H), 2.71-2.59 (m, 4H), 1.92 (m, 2H)。
(実施例45)
【0153】
N-(2-アミノ-フェニル)-4-[3-(1,3-ジヒドロ-イソインドール-2-イル)-プロペニル]-ベンズアミド (XXXa)
工程1: メチル4-トリメチルシラニルエチニル-ベンゾエート (XXXI)
メチル4-ブロモベンゾエート (8.84g, 41.11ミリモル)、Pd(PPh3)2Cl2(840mg, 1.20ミリモル)およびCuI (455mg, 2.39ミリモル)を無水THF (200ml)に室温にて攪拌溶解したものに、窒素を15分間飽和した。次に、溶液を窒素下で0℃に冷却し、トリメチルシリルアセチレン (7.2ml, 50.91ミリモル)およびトリエチルアミン (22ml, 157.8ミリモル)を連続して加えた。反応混合物を、室温まで温度上昇させた。2時間後、Pd(PPh3)2Cl2(100 mg)およびCuI (80 mg)およびトリメチルシリルアセチレン (0.5ml)を再度加え、反応混合物を一晩攪拌した。次に、反応混合物をAcOEtで希釈し、NH4Clの飽和水溶液および塩水で連続的に洗浄し、MgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(AcOEt/ヘキサン: 5/95→10/90)、標題化合物 XXXI (9.05g, 38.95ミリモル, 94%収率)を黄色の粘稠な固形物として得た。1H NMR: (400 MHz, CDCl3)δ(ppm): AB系(δA=7.67, δB=7.22, JAB=8.5Hz, 4H), 3.63 (s, 3H), 0.00 (s, 9H)。
【0154】
工程2: メチル4-エチニル-ベンゾエート(XXXII)
XXXI (9.05g, 38.95ミリモル)を0℃で窒素下にてMeOH (280ml)に攪拌溶解したものに、炭酸カリウム(1.62g, 11.72ミリモル)を加えた。3時間後、反応混合物を濃縮し、シリカゲル上でフラッシュクロマトグラフィーによって直接精製し(CH2Cl2: 100)、標題化合物XXXII (6.16g, 38.46ミリモル, 98%収率)を淡黄色固形物として得た。1H NMR: (400 MHz,CDCl3)δ(ppm): AB系(δA=7.98, δB=7.54, JAB=8.6Hz, 4H), 3.93 (s, 3H), 3.24 (s, 1H)。
【0155】
工程3: β-(4-メトキシカルボニル)-スチリルボロン酸(XXXIII)
XXXII (6.16g, 38.46ミリモル)を無水THF (15ml)に窒素下で室温にて攪拌溶解したものに、カテコールボラン(4.52ml, 42.80ミリモル)を加えた。反応混合物を70℃に4時間加熱し、カテコールボラン (2ml)を再度加えた。1.5時間後、反応混合物を室温まで冷却し、2N HClの水溶液 (50ml)を加え、一晩攪拌した。次に、これをRotavap上で濃縮し、濾別し、濾過ケーキをトルエン中で粉砕した。濾過後、中間固形物をTHF (50ml)に溶解し、2N HClの水溶液 (150ml)を加えた。生成懸濁液を一晩かけて40℃まで加熱し、濾別し、水で洗浄し、風乾および真空乾燥して、標題化合物XXXIII (3.10g, 15.05ミリモル, 39%収率)を灰白色の綿毛状固形物として得た。1H NMR: (400 MHz, DMSO-d6)δ(ppm): AB系(δA=7.96, δB=7.63, JAB=8.4Hz, 4H), 7.94 (s, 2H), 7.32 (d, J=18.2Hz, 1H), 6.30 (d, J=18.2Hz, 1H), 3.88 (s, 3H)。
【0156】
工程4: メチル4-[3-(1,3-ジヒドロ-イソインドール-2-イル)-プロペニル]-ベンゾエート (XXXIVa)
イソインドリン(116mg, 0.97ミリモル)およびパラホルムアルデヒド(32mg, 1.07ミリモル)を無水1,4-ジオキサン(10ml)中で窒素下にて90℃で15分間予熱して攪拌溶解したものに、XXXIII (245mg, 1.17ミリモル)を加えた。90℃で一晩攪拌した後、反応混合物を室温まで放冷し、2N HCl水溶液(30ml)を加え、30分間振盪した。次に、水性混合物をEt2Oで抽出し、2N NaOH(50ml)で塩基性にし、CH2Cl2で抽出した。合わせたジクロロメタン層をMgSO4上で乾燥し、濾過して、濃縮した。次に、粗製残渣をシリカゲル上でフラッシュクロマトグラフィーによって精製し(MeOH/ CH2Cl2: 5/95)、標題化合物XXXIVa (135mg, 0.46ミリモル, 48%収率)を灰白色固形物として得た。1H NMR (400 MHz, DMSO-d6)δ(ppm): AB系(δA=7.93, δB=7.64, JAB=8.4Hz, 4H), 7.29-7.17 (m, 4H), 6.75 (d, J=15.8Hz, 1H), 6.62 (dt, J=16.0, 6.3Hz, 1H), 3.94 (s, 4H), 3.88 (s, 3H), 3.55 (dd, J=6.1, 1.0Hz, 2H)。
【0157】
工程5: N-(2-アミノ-フェニル)-4-[3-(1,3-ジヒドロ-イソインドール-2-イル)-プロペニル]-ベンズアミド (XXXa)
標題化合物XXXaを、実施例11、工程5および6と同じ手続きによって二段階でXXXIVaから得た(工程図3)。1H NMR (400 MHz, DMSO-d6)δ(ppm): 9.67 (s, 1H), AB系 (δA=7.98, δB=7.63, JAB=8.3Hz, 4H), 7.3O-7.15 (m,5H), 7.00 (td, J=7.6, 1.5Hz, 1H), 6.81 (dd, J=8.0, 1.4Hz, 1H), 6.75 (d, J=15.8Hz, 1H), 6.66-6.56 (m, 2H), 4.93 (s, 2H), 3.95 (s, 4H), 3.56 (dd, J=6.2, 0.9Hz, 2H)。
(実施例46−49)
【0158】
実施例46-49 (化合物 XXXb-XXXe)を、実施例45の化合物XXXaについて記載したのと同じ手続き用いて調製した(工程図11)。
【0159】
【表6】
(実施例50)
【0160】
ヒストンデアセチラーゼ酵素活性の阻害
1. ヒトHDAC-1: 評価1
HDAC阻害剤を、Baculovirus昆虫細胞発現系から発現し精製したクローニングした組換えヒトHDAC-1酵素に対してスクリーニングした。デアセチラーゼ評価のため、20,000cpmの[3H]-代謝標識したアセチル化ヒストン基質(M. Yoshida et al., J. Biol. Chem. 265 (28): 17174-17179 (1990))を、30μgのクローニングした組換えhHDAC-1と共に37℃で10分間インキュベーションした。反応は、酢酸(0.04 M, 最終濃度)およびHCl (250 mM, 最終濃度)を加えて停止した。混合物を酢酸エチルで抽出し、放出された[3H]-酢酸をシンチレーション計数法によって定量した。阻害研究のため、酵素を化合物と共に4℃で30分間予備インキュベーションした後、酵素評価を開始した。HDAC酵素阻害剤についてのIC50値を、個々の化合物を用いる用量応答曲線を行い且つ最大阻害の50%を生じる阻害剤の濃度を測定することによって決定した。この手続きを用いて評価した典型的化合物についてのIC50を、表7-10の第三欄に示す(括弧内のデーターを除く)。
【0161】
2. ヒトHDAC-1: 評価2
代替法では、下記のプロトコールを用いて、本発明の化合物を評価した。この評価では、用いた緩衝剤は25mM HEPES, pH 8.0、137mM NaCl、2.7mM KCl、1mM MgCl2であり、基質はDMSO中での50mM原液中のBoc-Lys(Ac)-AMCである。酵素原液は、緩衝剤1ml当たり4.08μgであった。
【0162】
化合物を、酵素(20μl, 4.08μg/ml)と共に室温にて10分間予備インキュベーションした(評価プレートに移すため、2μl/DMSOを13μl/緩衝剤に希釈)(35μl予備インキュベーション容積)。混合物を、室温で5分間予備インキュベーションした。反応は、温度を37℃にし、16μlの基質を加えることによって開始した。総反応容積は50μlであった。反応を、20分後にBiomolによって指示された方法で調製した50μlの顕色剤(Fluor-de-Lys顕色剤, カタログ番号KI-105)を加えることによって停止した。プレートを暗所で室温にて10分間インキュベーションした後、読み取りを行った(λEx=360nm, λEm=470nm, カットオフフィルター435nm)。
【0163】
この手続きを用いて評価した典型的な化合物のIC50値を、表9の第三欄(括弧内[]データー)に示す。
【0164】
3. MTT評価
HCT116 細胞(2000/ウェル)を、化合物処理の1日前に96-ウェル組織培養プレートにまいた。様々な濃度の化合物を、細胞に加えた。細胞を、5% C02インキュベーターで37℃で72時間インキュベーションした。MTT(3-[4,5-ジメチルチアゾール-2-イル]-2,5 ジフェニルテトラゾリウムブロミド, Sigma)を0.5mg/mlの最終濃度で加え、細胞と共に4時間インキュベーションした後、可溶化緩衝剤(50% N,N-ジメチルホルムアミド, 20% SDS, pH 4.7) 1容を培養細胞に加えた。一晩インキュベーションした後、可溶化した色素をMR 700プレートリーダー(Dynatech Laboratories Inc.)を用いて、630nMでのリファレンスを用いて570nMでの比色計の読みによって定量した。OD値を、関連細胞系の標準増殖曲線によって細胞数に転換した。細胞数を溶媒処理した細胞数の50%まで減少させる濃度を、MTT IC50として決定する。典型的な化合物についてのIC50値を、表7-10の第四欄に示す。
【0165】
4. 免疫ブロットによる総細胞におけるヒストンH4アセチル化
培養で増殖するT24ヒト膀胱癌細胞を、HDAC阻害剤と共に16時間インキュベーションした。M. Yoshida et al.によって記載された通りの培養時間の後、ヒストンを細胞から抽出した(J. Biol. Chem. 265 (28): 17174-17179 (1990))。全ヒストンタンパク質20gをSDS/PAGEに装填し、ニトロセルロース膜に移した。膜を、アセチル化ヒストンH-4 に特異的なポリクローナル抗体(Upstate Biotech Inc.)でプローブした後、西洋ワサビペルオキシダーゼに接合した二次抗体(Sigma)でプローブした。エンハンスト・ケミルミネッセンス(Enhanced Chemiluminescence; ECL)(Amersham)検出を、Kodakフィルム(Eastman Kodak)を用いて行った。アセチル化H-4シグナルを、デンシトメトリーによって定量した。典型的データーを、表7-10の第五欄に示す。データーは、アセチル化H-4シグナルを50%だけ減少させるのに有効な濃度(EC50)として示される。
【0166】
【表7−1】
【表7−2】
【0167】
【表8−1】
【表8−2】
【表8−3】
【0168】
【表9−1】
【表9−2】
【表9−3】
【0169】
【表10】
【0170】
(実施例51)
イン・ビボでのヒト腫瘍異種移植片に対するヒストンデアセチラーゼ阻害剤の抗腫瘍効果
8-10週齢の雌CD1ヌードマウス(Taconic Labs, Great Barrington, NY)の側腹部に2x106個のプレコンディショニングしたHCT116ヒト結腸直腸癌細胞を皮下投与した。これらの細胞のプレコンディショニングは、同一系統のヌードマウスで少なくとも3回連続的に腫瘍を移植することによって行った。その後、約30mgの腫瘍断片を摘出し、Forene麻酔(Abbott Labs, Geneva, Switzerland)によりマウスの左側腹部に皮下移植した。腫瘍が100mm3の平均容積に達したときに、ヒストンデアセチラーゼ阻害剤をPBS、DMSO/水、またはTween 80/水のような適当なビヒクルに溶解したものを10mg/kgの出発用量でマウスに静脈内、皮下、または腹腔内に毎日投与した。HDAC阻害剤の最適用量を、標準的プロトコールによって用量応答実験によって確立した。腫瘍容積を、標準的方法によって輸液後2日毎に計算した(例えば、Meyer et al., Int. J. Cancer 43: 851-856 (1989))。本発明によるHDAC阻害剤を投与したところ、ビヒクルだけを投与したコントロール(すなわち、HDAC阻害剤なし)と比較して腫瘍重量および容積が有意に減少し、これらの化合物のサブセットは毒性を示した。一例として、化合物XVjについての結果を、図1に示す。
【図面の簡単な説明】
【0171】
【図1】上記の実施例51に記載のイン・ビボでのヒト腫瘍異種移植片に対する本発明によるヒストンデアセチラーゼ阻害剤の抗腫瘍効果。
【特許請求の範囲】
【請求項1】
下記の式
【化1】
(式中、Arはアリールまたはヘテロアリールであり、それらのそれぞれは場合によっては1-3個の置換基によって置換されている)
を有する化合物、または薬学上許容可能なその塩。
【請求項2】
Arがアリールまたはピリジニルである、請求項1に記載の化合物。
【請求項3】
Arがフェニルである、請求項1に記載の化合物。
【請求項4】
Arがハロ、場合によってはハロで置換されたC1-C6-ヒドロカルビル、場合によってはハロで置換されたC1-C6-ヒドロカルビルオキシからなる群から選択される1-3個の置換基で置換されている、請求項1に記載の化合物。
【請求項5】
Arが下記の
【化2】
の1つから選択される、請求項1に記載の化合物。
【請求項6】
下式
【化3】
(式中、Xは-N(R1)-、-O-、または-S-であるか、またはXは窒素を含むヘテロシクリルであり、但し、窒素は構造Vにおける隣接カルボニルに共有結合し且つ場合によっては1-3個の置換基で置換されており、
RおよびR1は独立して-H、または 場合によっては置換されたa) C1-C6-ヒドロカルビルまたはb) R2-L-であり、但し、R2はアリールまたはヘテロアリールであり、LはCO-C6-ヒドロカルビル-L1-CO-C6-ヒドロカルビルであり、L1は共有結合、-O-、-S-、または-NH-である)
を有する化合物、または薬学上許容可能なその塩。
【請求項7】
Xが-NH-、-O-、モルホリン-4-イル、ピペリジン-1-イル、ピペリジン-1-イル、またはピロリジン-1-イルである、請求項6に記載の化合物。
【請求項8】
Xが-N(R1)-であり、但し、R1は場合によっては置換メチルまたはエチルである、請求項6に記載の化合物。
【請求項9】
Xが-N(R1)-であり、但し、R1は シアノエチルまたはピリジニルメチルである、請求項6に記載の化合物。
【請求項10】
Xが-N(R1)-であり、但し、RがR2-L-であり、R2がフェニル、ピリジニル、インジル、またはインドリルであり、Lが共有結合、メチル、エチル、またはオキシエチルである、請求項6に記載の化合物。
【請求項11】
R-X-の組合せが下記の
【化4】
から選択される、請求項6に記載の化合物。
【請求項12】
第三の様相では、本発明は、下式
【化5】
(式中、Yは-N(R4)-、-O-、-S-、-N(R4)SO2-、-SO2-N(R4)-、-SO2-、-N(R4)-C(O)-、-C(O)-N(R4)-、-NHC(O)NH-、-N(R4)C(O)O-、-OC(O)N(R4)-、または共有結合であり、
R1、R2、およびR3は独立して-Hであるか、またはRa-C0-C6-ヒドロカルビルであって、但し、Raは-Hであるか、またはRaはアリールまたはヘテロアリールであり、それらのそれぞれは、場合によっては1-3個の置換基で置換されており、
R4は-H、-C(O)-Rb、-C(O)O-Rb、-C(O)NH-Rb、またはRc-CO-C6-ヒドロカルビルであり、
但し、
Rbは-Hまたは-C1-C6-ヒドロカルビルであり、
Rcは-H、またはアリールまたはヘテロアリールであり、それらのそれぞれは場合によっては1-3個の置換基で置換されている)
の化合物、または薬学上許容可能なその塩を含んでなる。
【請求項13】
R2およびR3が両方とも-Hである、請求項12に記載の化合物。
【請求項14】
Yが-NH-、-SO2-NH-、または-N(R4)-であり、但し、R4は-C(O)O-C1-C6-ヒドロカルビルである、請求項12に記載の化合物。
【請求項15】
R1がアリール、ベンゾチアゾリル、ピリミジニル、トリアゾリル、ベンゾジオキソレニルまたはピリジニルであって、それらのそれぞれが場合によっては 1-3個の置換基で置換されている、請求項12に記載の化合物。
【請求項16】
R1がC1-C6-ヒドロカルビル、C1-C6-ヒドロカルビルオキシ、ハロ、メチルチオ、およびアセチルから独立して選択された1-3個の置換基で置換されている、請求項15に記載の化合物。
【請求項17】
下記の
【化6】
から選択される、請求項12に記載の化合物。
【請求項18】
式
【化7】
(式中、Ar1が場合によっては-NO2、CH3O-、およびモルホリニル(例えば、モルホリン-4-イル)から独立して選択される1-3個の置換基で置換されているアリールまたはヘテロアリールである)
の化合物、または薬学上許容可能なその塩。
【請求項19】
Ar1が、場合によっては-NO2、CH3O-、およびモルホリニル(例えば、モルホリン-4-イル)から独立して選択される1-3個の置換基で置換されているアリールである、請求項18に記載の化合物。
【請求項20】
Ar1が、場合によっては-NO2、CH3O-、およびモルホリニル(例えば、モルホリン-4-イル)から独立して選択される1-3個の置換基で置換されているフェニルである、請求項18に記載の化合物。
【請求項21】
下式
【化8】
から選択される、請求項18に記載の化合物。
【請求項22】
請求項1-21のいずれか一項に記載の化合物、および薬学上許容可能なキャリヤー、賦形剤、または希釈剤を含んでなる、組成物。
【請求項23】
細胞のヒストンデアセチラーゼを阻害する方法であって、ヒストンデアセチラーゼの阻害が所望な細胞を請求項1-21のいずれか一項に記載のヒストンデアセチラーゼの阻害剤と接触させることを含んでなる、方法。
【請求項24】
細胞増殖性疾患または疾病にかかっている哺乳類を請求項22に記載の組成物の治療上有効量で治療する方法。
【請求項25】
哺乳類がヒトである、請求項24に記載の方法。
【請求項1】
下記の式
【化1】
(式中、Arはアリールまたはヘテロアリールであり、それらのそれぞれは場合によっては1-3個の置換基によって置換されている)
を有する化合物、または薬学上許容可能なその塩。
【請求項2】
Arがアリールまたはピリジニルである、請求項1に記載の化合物。
【請求項3】
Arがフェニルである、請求項1に記載の化合物。
【請求項4】
Arがハロ、場合によってはハロで置換されたC1-C6-ヒドロカルビル、場合によってはハロで置換されたC1-C6-ヒドロカルビルオキシからなる群から選択される1-3個の置換基で置換されている、請求項1に記載の化合物。
【請求項5】
Arが下記の
【化2】
の1つから選択される、請求項1に記載の化合物。
【請求項6】
下式
【化3】
(式中、Xは-N(R1)-、-O-、または-S-であるか、またはXは窒素を含むヘテロシクリルであり、但し、窒素は構造Vにおける隣接カルボニルに共有結合し且つ場合によっては1-3個の置換基で置換されており、
RおよびR1は独立して-H、または 場合によっては置換されたa) C1-C6-ヒドロカルビルまたはb) R2-L-であり、但し、R2はアリールまたはヘテロアリールであり、LはCO-C6-ヒドロカルビル-L1-CO-C6-ヒドロカルビルであり、L1は共有結合、-O-、-S-、または-NH-である)
を有する化合物、または薬学上許容可能なその塩。
【請求項7】
Xが-NH-、-O-、モルホリン-4-イル、ピペリジン-1-イル、ピペリジン-1-イル、またはピロリジン-1-イルである、請求項6に記載の化合物。
【請求項8】
Xが-N(R1)-であり、但し、R1は場合によっては置換メチルまたはエチルである、請求項6に記載の化合物。
【請求項9】
Xが-N(R1)-であり、但し、R1は シアノエチルまたはピリジニルメチルである、請求項6に記載の化合物。
【請求項10】
Xが-N(R1)-であり、但し、RがR2-L-であり、R2がフェニル、ピリジニル、インジル、またはインドリルであり、Lが共有結合、メチル、エチル、またはオキシエチルである、請求項6に記載の化合物。
【請求項11】
R-X-の組合せが下記の
【化4】
から選択される、請求項6に記載の化合物。
【請求項12】
第三の様相では、本発明は、下式
【化5】
(式中、Yは-N(R4)-、-O-、-S-、-N(R4)SO2-、-SO2-N(R4)-、-SO2-、-N(R4)-C(O)-、-C(O)-N(R4)-、-NHC(O)NH-、-N(R4)C(O)O-、-OC(O)N(R4)-、または共有結合であり、
R1、R2、およびR3は独立して-Hであるか、またはRa-C0-C6-ヒドロカルビルであって、但し、Raは-Hであるか、またはRaはアリールまたはヘテロアリールであり、それらのそれぞれは、場合によっては1-3個の置換基で置換されており、
R4は-H、-C(O)-Rb、-C(O)O-Rb、-C(O)NH-Rb、またはRc-CO-C6-ヒドロカルビルであり、
但し、
Rbは-Hまたは-C1-C6-ヒドロカルビルであり、
Rcは-H、またはアリールまたはヘテロアリールであり、それらのそれぞれは場合によっては1-3個の置換基で置換されている)
の化合物、または薬学上許容可能なその塩を含んでなる。
【請求項13】
R2およびR3が両方とも-Hである、請求項12に記載の化合物。
【請求項14】
Yが-NH-、-SO2-NH-、または-N(R4)-であり、但し、R4は-C(O)O-C1-C6-ヒドロカルビルである、請求項12に記載の化合物。
【請求項15】
R1がアリール、ベンゾチアゾリル、ピリミジニル、トリアゾリル、ベンゾジオキソレニルまたはピリジニルであって、それらのそれぞれが場合によっては 1-3個の置換基で置換されている、請求項12に記載の化合物。
【請求項16】
R1がC1-C6-ヒドロカルビル、C1-C6-ヒドロカルビルオキシ、ハロ、メチルチオ、およびアセチルから独立して選択された1-3個の置換基で置換されている、請求項15に記載の化合物。
【請求項17】
下記の
【化6】
から選択される、請求項12に記載の化合物。
【請求項18】
式
【化7】
(式中、Ar1が場合によっては-NO2、CH3O-、およびモルホリニル(例えば、モルホリン-4-イル)から独立して選択される1-3個の置換基で置換されているアリールまたはヘテロアリールである)
の化合物、または薬学上許容可能なその塩。
【請求項19】
Ar1が、場合によっては-NO2、CH3O-、およびモルホリニル(例えば、モルホリン-4-イル)から独立して選択される1-3個の置換基で置換されているアリールである、請求項18に記載の化合物。
【請求項20】
Ar1が、場合によっては-NO2、CH3O-、およびモルホリニル(例えば、モルホリン-4-イル)から独立して選択される1-3個の置換基で置換されているフェニルである、請求項18に記載の化合物。
【請求項21】
下式
【化8】
から選択される、請求項18に記載の化合物。
【請求項22】
請求項1-21のいずれか一項に記載の化合物、および薬学上許容可能なキャリヤー、賦形剤、または希釈剤を含んでなる、組成物。
【請求項23】
細胞のヒストンデアセチラーゼを阻害する方法であって、ヒストンデアセチラーゼの阻害が所望な細胞を請求項1-21のいずれか一項に記載のヒストンデアセチラーゼの阻害剤と接触させることを含んでなる、方法。
【請求項24】
細胞増殖性疾患または疾病にかかっている哺乳類を請求項22に記載の組成物の治療上有効量で治療する方法。
【請求項25】
哺乳類がヒトである、請求項24に記載の方法。
【図1】

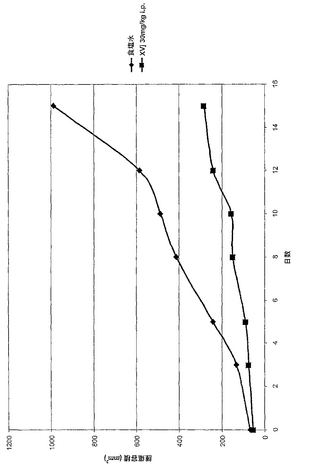
【公表番号】特表2006−503082(P2006−503082A)
【公表日】平成18年1月26日(2006.1.26)
【国際特許分類】
【出願番号】特願2004−543863(P2004−543863)
【出願日】平成15年10月16日(2003.10.16)
【国際出願番号】PCT/CA2003/001557
【国際公開番号】WO2004/035525
【国際公開日】平成16年4月29日(2004.4.29)
【出願人】(504098163)メシルジーン、インコーポレイテッド (13)
【Fターム(参考)】
【公表日】平成18年1月26日(2006.1.26)
【国際特許分類】
【出願日】平成15年10月16日(2003.10.16)
【国際出願番号】PCT/CA2003/001557
【国際公開番号】WO2004/035525
【国際公開日】平成16年4月29日(2004.4.29)
【出願人】(504098163)メシルジーン、インコーポレイテッド (13)
【Fターム(参考)】
[ Back to top ]