
ヒトにおける早漏を治療するための方法
本発明は薬理学、医用薬および医用薬化学の分野に属し、性機能不全を治療するための方法および組成物を提供する;より具体的には、本発明はヒトにおける早漏の治療に関する。
【発明の詳細な説明】
【背景技術】
【0001】
発明の背景
原始的な早漏は男性の最も一般的な性障害と見なされる。これは、ヒトの本能的要求の満足のために必要な性的調節を達成する能力の喪失を惹起し得る。最近、このような性的調節の喪失によって引き起こされる様々な症状を発現する症例数がかなり多いことが明らかとなっている。男性における早漏を原因とする性的な問題は、家庭内のひずみに加えて、自信の喪失に起因する無気力など社会的な難題に至る。早漏は、挿入前、挿入時または挿入直後の持続的または反復性の射精を含む。
【0002】
本来、女性は、少なくとも性的活動の開始時には男性よりも著しく弱く性交を経験するように進化している。従って、性交中に最大限まで緊張した全神経系の自然な弛緩を与えるオルガスムに達するためには女性はより長い時間をかけなければならない。今日まで、ヒトの性生活においては接触の感覚が重要な役割を担っている;性感帯は接触に特に敏感であり、それらの中でも先ず第一に、例えば、口腔、直腸、女性性器および乳頭の近くなど皮膚が粘膜と接する部位である。女性の性感帯は女性の身体表面全体であり得る。このような場合は、身体の任意の部分に接触することによって女性に扇情的な感情を惹起することが可能である。しかし、性感帯は陰核、小陰唇および膣などの厳密に限定された場所に局在する場合が一般的である。さらに、生殖器とは別に、多くのこのような過敏な部位がある。これらは唇、耳、瞼、首、乳頭などである。いくつかの場合には、これらの部位はそれらへの単なる接触によって女性にオルガスムを誘発し得るほど敏感である。
【0003】
しかし、男性の場合は、性感帯は専ら生殖器および隣接部分に限定される。従って、性体験のある男性パートナーが時にこれらの部位の探索において全くの探求の旅を試みざるを得ないということは驚くべきことではなく、これがなければ、誰も女性の性的反射の複雑な装置を活性化させることはできない。これが男性がしばしばオルガスムに達するために比較にならない短い時間を必要とする一つの理由であり、通常はだ男性自身のためばかりでなく彼のパートナーのためにも性交を終了する。性交の開始時に、男性は自身が既にあるレベルの興奮にあることを見出し、勃起にはこのことが不可欠であり、これがなければこの性交は全く不可能となる。オルガスム後直ちに射精に伴う萎縮が起こるため男性は彼のパートナーのことを考えて行為を継続することはできず、膣内でのさらなる摩擦は全て不可能である。
【0004】
理想的な性交は、膣内への陰茎の挿入後、双方の当事者が同時にオルガスムの境界に達して、それを交差させて、共に性交を終了することであろう(図1)。時にこれは、性交経験のある女性が行為開始時に逸した興奮を補ってその代わりに彼女のパートナーと共に最後のラインに達することができる場合に起こる。青年および中年男性における正常な射精の典型は陰茎の腟内への挿入後2〜6分で推移する。
【0005】
早漏は、現代人の性的行為において非常に頻繁に発生する。陰茎の腟内への挿入後すぐに、時には2〜3回の動作後に生じて(図2)、射精およびオルガスムが起こり、勃起が萎縮して性的行為が終了するという事実に関する。このような状況では明らかに女性は刺激されただけであり、発散の可能性は全くないはずである。明らかに、不十分な勃起によるかまたは早漏によるかに関わらず、任意の種類の男性のインポテンスの存在下において女性パートナーの性的満足および正常なリラクゼーションの可能性は全くないはずである。
【0006】
陰茎の勃起は3段階の永続可能なプロセスであり得る:1)血管拡張;2)内因性平滑筋弛緩物質の放出;および3)初期開始部位から遠位へのこれらの影響の連続。これは「カスケード効果」(Andersson et al 1995)と呼ばれている。パパベリンはアヘンアルカロイドであり、恐らくサイクリックGMPホスホジエステラーゼ阻害によって平滑筋弛緩剤として作用する。これは陰茎の血管系の筋系を弛緩させて、血流を増加させる(Papaverine Topical Gel Treatment For Erectile Dysfunction, Urology, Vol. 133(2)(1995), pp. 361-365)。インポテンスの治療において有用であることが見出されたもう一つの化合物は、陰茎への動脈流入量を増加させるように作用して、また静脈からの流出量を制限し得る天然の化合物であるプロスタグランジンE1である。プロスタグランジンE1は、陰茎で局所的に代謝されること、および低血圧などの全身症状を惹起する可能性が低いことから、インポテンスの治療のために注射として使用されるその他の化合物よりも好ましい。修飾血管組織として陰茎海綿体(ccp)は、通常の血管組織と同一の範囲のオートクリンおよびパラクリン調節物質を産生および分泌する。
【0007】
しかし、ccpの平滑筋緊張度は、血管壁と同じ方法で調節されるとは思われない。現在、ccpの緊張度または収縮性はアドレナリン作動性の制御によって調節されて、局所性にNOおよびエンドテリンを産生すると仮定されている。ccpにおいて、大半の試験はNO(Rajfer et al 1992; Burnett 1995)、血管作用性小腸ペプチド(VIP)、カルシトニン遺伝子関連ペプチド(CGRP)、ならびに通常およびccpの血管平滑筋に対しても同様の効果を持つ副交感神経支配の弛緩効果を観察することを目的としている。
【0008】
ccpへの血液の流入が類洞腔を結合させる正常な陰茎勃起中、小柱状組織は勃起を維持するために海綿体を取り巻く厚い線維組織に対して小さな海綿体静脈を圧迫する。血流量におけるこれらの変化を介在するために、シナプス後性副交感ニューロンおよびより軽度ではあるが、内皮細胞から一酸化窒素が放出されて、動脈および小柱状平滑筋においてαアドレナリン作動性ニューロンが阻害される。容易に拡散可能な一酸化窒素は、平滑筋細胞を弛緩させるためにグアニル酸シクラーゼによって海綿体内の増加するサイクリックグアノシン一リン酸(GMP)の形成を刺激する。
【0009】
最近、男性勃起不全の治療のためにシルデナフィルのクエン酸塩の経口使用が米国食品医薬品局(FDA)によって承認されている。シルデナフィルの組成物は最初に欧州特許EP 0463756において開示されて、米国または欧州の一国以外のその他の国ではシルデナフィルをカバーする物質特許(composition of matter patent)はない。シルデナフィルは、海綿体で形成されるサイクリックGMPを代謝する主なイソ酵素であるサイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)の選択的阻害剤であることが報告されている(Boolell et al 1996)。シルデナフィルは海綿体におけるPDE5の強力な阻害物質であるため、陰茎において、特に性的刺激と共に、海綿体の血流量を増加させることによって一酸化窒素の影響を増大させると考えられる。現在の推奨用量である25〜100mgのシルデナフィルが性的刺激の不在化では殆ど効果がない限り、シルデナフィルは性的刺激に対する自然の勃起反応を回復するのであってこのような刺激の不在下で勃起を惹起するものではないと考えられている(Goldstein 1998)。サイクリックGMPが平滑筋の弛緩を刺激する局所的なメカニズムは解明されていない。
【0010】
ヒトの男性における正常な射精機能は精液の噴出性の順行輸送を促進するための平滑筋および横紋筋の収縮の協調シーケンスを意味する。この過程は、陰茎幹から上位中枢への内陰部神経を介しての求心性神経刺激の伝達で始まる。射精反射を完了するためには遠心性の刺激が脊髄の脊柱から伝達されるが、下腹神経叢または交感神経叢を含むために腰部レベルから発する。下腸間膜動脈神経節からは、短いアドレナリン作動性節後線維が精嚢、精管膨大部および膀胱頚部で終結する。精嚢の交感神経支配は後部尿道への精液の放出に至る。適時の膀胱頚部閉鎖はこの精液ボーラスの逆流を防いで、この精液ボーラスは骨盤底の球海綿体筋および坐骨海綿体筋の間代性収縮によって順行性の方向に促される。 射精は中枢的な統合された末梢誘発反射であり、α1-アドレナリン作動性レセプター活性化の結果として生じる。早漏の治療のための有効な薬理学的薬剤は存在するが、それらは、例えばクロミプラミンおよびフェノキシベンザミンなど重度の副作用を被る。その他の治療は効果が限られている(メトクロプラミドなど)。
【0011】
デキストロメトルファン(よくDMとして略記される)は(+)-3-メトキシ-N-メチルモルフィナンの一般名である(図3)。これは咳嗽シロップとして広く使用されていて、Rodd 1960(論文の完全な引用は以下に示す)およびGoodman and Gilman's Pharmacological Basis of Therapeuticsなどの参照に記載されている。簡単に言うと、DMは、大半のオピエートの分子コアを形成するモルフィナン環構造の右旋性鏡像異性体(鏡像)を含む非常用性のオピオイドである。DMはシグマレセプターとして公知のニューロンレセプターの一種にて作用する。これらはしばしばシグマオピエートレセプターと記載されるが、それらがオピエートレセプターであるかどうかに関しては若干の疑問があり、非常に多くの研究者がそれらを単にシグマレセプターまたは高親和性デキストロメトルファンレセプターと呼ぶ。それらは抑制性レセプターであり、DMまたはその他のシグマアゴニストによるそれらの活性化が特定の種類の神経シグナルの抑制を引き起こすことを意味する。デキストロメトルファンは、興奮性アミノ酸(EAA)レセプターの一種であるN-メチル-D-アスパラギン酸(NMDA)レセプターとして公知のもう一つの種類のレセプターでも作用する。シグマレセプターでのそのアゴニストの活性とは異なり、DMはNMDAレセプターにおいてアンタゴニストとして作用して、これはDMがNMDAレセプターを介して調節される神経インパルスの伝達を抑制することを意味する。NMDAレセプターは興奮性レセプターであるため、DMのNMDAアンタゴニストとしての活性も特定の種類の神経シグナルの抑制を引き起こし、これがある種の咳嗽に関与する可能性がある。NMDAアンタゴニストとしての活性故に、DMおよびその代謝物の1つであるデキストロルファンは、卒中発作、心停止、および仮死などの事象によって引き起こされる虚血(低血流)および低酸素症(不十分な酸素供給)に誘発される特定の種類の興奮毒性脳損傷の可能性のある治療として積極的に評価されている。デキストロメトルファンおよびデキストロルファンの抗興奮毒性活性、ならびにこれらの薬剤によるNMDAレセプターの遮断については、Choi 1987、Wong et al 1988、Steinberg et al 1988、および米国特許第4,806,543号(Choi 1989)などの論文等で考察されている。デキストロメトルファンは、神経カルシウムチャネルにおいて活性を抑制することも報告されている(Carpenter et al 1988)。デキストロメトルファンおよびそれが相互作用するレセプターいついては、Tortella et al 1989、Leander 1989、Koyuncuoglu & Saydam 1990、Ferkany et al 1988、George et al 1988、Prince & Feeser 1988、Feeser et al 1988、Craviso and Musacchio 1983、およびMusacchio et al 1988においてより詳細に考察されている。
【0012】
DMは血流からかなり速やかに消失する(例えば、Vetticaden et al 1989およびRamachander et al 1977を参照されたい)。DMは肝臓においてO-脱メチル化と呼ばれる酵素過程によってデキストロルファンおよび3-メモキシモルフィナンと呼ばれる2つの代謝物に転換される;この過程では2つのペンダントメチル基の一つが水素によって置き換えられる。2つ目のメチル基が除去されると、形成される代謝物は5-ヒドロキシモルフィナンと呼ばれる。デキストロルファンおよび5-ヒドロキシモルフィナンは、尿血流を介して体内からかなり速やかに排出されるグルクロニドまたは硫酸抱合体を形成するために肝臓においてその他の化合物に共有結合性に結合する(主にグルクロン酸またはグルタチオンのようなイオウ含有化合物)。この酵素は、何年も前にデブリソキンのヒドロキシル化反応を実行することが発見されたことから、通常はデブリソキンヒドロキシラーゼと呼ばれる。これは様々な論文においてP450DBまたはP450-2D6とも呼ばれている。これは、数年前にスパルテインを代謝することが示されたスパルテインモノオキシゲナーゼと呼ばれる酵素と明らかに同一である;最近まで、科学者は単一のイソ酵素がデブリソキンおよびスパルテインの双方、ならびにデキストロメトルファンおよびその他の様々な基質の酸化を主に司ると思われることを認識しなかった。デブリソキンヒドロキシラーゼは「チトクロームP450」酵素、または「チトクロームオキシダーゼ」酵素として公知の酵素ファミリーに属する。化学物質の一酸素添加はチトクロームP450(P450)に帰するとされている。450nm付近に還元一酸化炭素吸収スペクトルの最大を示すモノオキシゲナーゼ酵素を含むこれらの血液タンパク質は、内因性および外因性化合物のヒドロキシル化を含む様々な酸化反応を触媒することが示されている(Jachau, 1990)。P450が酸素移動反応を触媒することができるメカニズムに関して膨大な量の研究が実施されている(Testa and Jenner, 1981;Guengerich, 1992;Brosen et al, 1990;Murray et al, 1990;およびPorter et al, 1991)。
【0013】
P450反応サイクルは簡単に言うと次のように進行する:三価鉄型のチトクロームへの基質分子(RH)の最初の結合によって二元複合体が形成されて、三価鉄酵素のスピン平衡が低スピンから高スピン状態へ推移する。この立体配置は、二価鉄のP450−基質複合体を形成するためにフラボタンパク質レダクターゼから電子をより容易に受け取ることを示唆するいくつかの証拠が示されている。しかし、必ずしも全てのP450が高スピン含有量と還元速度の相関性を示す訳ではない。実際、P450基質の性状、酵素/基質複合体のトポグラフィー、および酸化可能原子の電位を含む複数の要因がそれぞれ還元速度の調節において役割を果たすことが提唱されている。分子の酸素は、二価鉄酸素錯体を形成するために、後にP450レダクターゼからの(または、多分、いくつかのケースでは、チトクロームb5およびそのレダクターゼを介して還元されたニコチンアミドアデニンジヌクレオチドからの)第二の電子によって還元される二価鉄P450−基質複合体に結合する。還元型二価鉄酸素錯体の二酸素結合が開裂すると、酸素の一方の原子は基質に挿入されて、もう一方の酸素原子は水に還元されて、二価鉄血液タンパク質は元に戻る。
【0014】
P450酵素ファミリーの個々のメンバーおよび関連する混合機能オキシダーゼ活性は、脳、副腎、腎、精巣、卵巣、肺および皮膚を含む肝外組織において記載されている。個々のP450は、同様に選択された科学的分類による誘導能に関して特徴付けられている。P450 1A1および1A2サブファミリーのような特異的P450酵素の誘導は、mRNA転写および酵素活性の発現の増加の調節過程に関して幅広く研究されている。ベータ-ナフタフラボン(beta-NF)、3-メチルコラントレン(3-MC)、アロクロール1254(ACLR)、および2,3,7,8-テトラクロロジベンゾ-p-ダイオキシン(TCDD)のような材料は指定されたP450 1Aサブファミリーを持つP450酵素の誘導物質として分類されている材料である(Murray et al, 1990;およびGuengerich, 1989)。
【0015】
多くの化合物がデブリソキンヒドロキシラーゼ(スパルテインモノオキシゲナーゼ)イソ酵素の活性を阻害する(Inaba et al 1985)。これらの阻害物質の最も強力なものはキニンの右旋性立体異性体であるキニジンであり(図3)、これは通常は不整脈を治療するために用いられる。Inaba et al(1986)およびNielsen et al(1990)はキニジンがインビボにおける動物試験においてスパルテインの酸化を阻害する能力について考察していて、またBrinn et al(1986)、Brosen et al(1987)およびBroly et al(1989)はキニジンが肝細胞調製物中でDM代謝を阻害する能力について考察している。例外的に強力であり、かつ、容易に実証されるデブリソキンヒドロキシラーゼの阻害に加えて、その他のチトクロームP450イソ酵素も、様々なレベルの結合親和性で、キニジンに抑制される可能性が高い。従って、キニジンはデブリソキンヒドロキシラーゼに対してその最も顕著な効果を発揮するものの、同様に多くのその他のチトクロームP450酵素を抑制し、それによって患者は正常かつ望ましい肝臓の活性をより全般的に喪失する可能性が高い。デキストロメトルファンの主たる酸化代謝産物はデキストロルファンであり、これは神経学者の間ではデキストロメトルファンと全く同じ方法で活性であると広く信じられている;報告によると、双方の薬剤はシグマアゴニスト、NMDAアンタゴニストおよびカルシウムチャネルアンタゴニストである。デブリソキンヒドロキシラーゼを阻害する化合物のDMと組み合わせた投与は、DMのみを投与される患者に比して患者の血液中のDMの濃度および安定性の大幅な増大が惹起される;またDMと組み合わせたデブリソキンヒドロキシラーゼ阻害剤の投与はヒトにおけるDMの検出可能な作用に対して明確で実質的な影響を有する。
【0016】
トラマドールは、(+/-)-トランス(RR,SS)-2-[(ジメチルアミノ)メチル]-1-(3-メトキシフェニル)シクロヘキサノールの化学名を持ち、文献中ではしばしばシス(RS,SR)ジアステレオマーとして誤記される。トラマドールは、オピエート由来でなく、NSAIDでもない中枢作用性の二成分鎮痛薬である。これは、骨関節炎および術後鎮痛のような慢性疼痛の状況、ならびに歯痛などの急性疼痛において中等度の疼痛を管理するために用いられる。
【0017】
トラマドールはラセミ化合物であり、等量の(+)-および(-)-鏡像異性体からなる。トラマドールの純粋な鏡像異性体はラセミ体に比して異なる薬学的プロフィールおよび効果を持つことが公知である。(+)-鏡像異性体はμ-オピエートレセプターとの結合によるオピエート様鎮痛作用によって識別されて、双方の鏡像異性体は5-ヒドロキシトリプタミン(セロトニン)およびノルアドレナリン(ノルエピネフリン)の再取り込みを阻害し、これはトラマドールラセミ混合物のそれよりも強力であり、(-)-鏡像異性体ではノルアドレナリン再取り込みの明確な阻害が観察される。(+)-および(-)-トラマドールについては、モデルに応じて、2つの鏡像異性体がそれらの個々の作用を相互に強化および促進することが示されている(Raffa, R. et al., 1993;Grond S et al, 1995、およびWiebalck A et al., 1998)。トラマドールの強力な鎮痛作用が鏡像異性体の作用のこの相互に依存性の強化に基づくと結論づけられることは明らかである。トラマドールの主な活性代謝物であるO-デスメチルトラマドール(M1)はμ-オピエートレセプターに対して高い親和性を示して親薬剤の少なくとも2倍の鎮痛強度を有する。O-デスメチル-N-モノ-デスメチルトラマドール(以下の本文のいくつかの場所および文献中ではM5と記載される)はトラマドールのインビボにおける代謝物である(1RS,2RS)-2[ジメチルアミノ)メチル]-1-(3-メトキシフェニル)シクロヘキサノールの一つとして公知である(Lintz et al., 1981)。中枢神経系に対する影響、例えば鎮痛作用は脳室内投与の場合よりも静脈内投与において明らかに低いことから、M5はごく限られた程度で血液-脳関門を通過する。
【0018】
トラマドールは化学的にはオピエートと無関係であるという事実にも関わらず、トラマドールの投与に付随する有害な副作用は、高用量で用いられた場合には、オピエートのそれとほぼ同じである。
【0019】
カフェインはコーヒーアラビカ(Coffea arabica)またはコーヒーノキ(coffee plant)の葉および種子、ならびにテアシネンシス(Thea sinensis)またはチャ(tea)の葉から得られるアルカロイドである。カフェインはメチル化されたキサンチンであり、化学的には3,7-ジヒドロ-1,3,7-トリメチル-1H-プリン-2,6-ジオンと示される(図3)。カフェインは天然で生じるが、市販用薬品の用途のために合成的に調製される。カフェインは世界中で最も広い活性物質である。成人による平均カフェイン摂取量は異なる文化および国の間で一日一人あたり80から400mgまで様々である(Daly 1998)。カフェインは、覚醒状態の亢進、精神運動反応時間の短縮、ならびに睡眠潜時および起きている時間の増大を含む多くの薬理学的反応を引き起こして、知的活動に影響する可能性もある(Nehlig 1992)。さらに、カフェインは平滑筋の弛緩を惹起して、胃酸の分泌およびカテコールアミンの放出を促進し、代謝活性を高める(Fredholm 1999)。
【0020】
カフェインは本質的には無毒性である。FDAは、この化合物の過量投与の結果として致命的なカフェイン中毒がこれまで報告されていないことを指摘している。成人におけるカフェインの短期致死量は5〜10gである。中等度の用量ではカフェインはヒトの胎児においてほとんどまたは全く発生毒性のリスクを持たない。カフェインの摂取が癌の発生または冠動脈心疾患の発生率の増大に必然的に関連することを示す証拠はない。カフェインは経口、経直腸または非経口投与後に容易に吸収される。1時間以内に最大血漿中濃度に到達する。カフェインの血漿中半減期は3〜7時間である。
【0021】
カフェインはFDAの刺激物質の基準を満たす、医師の処方なしで購入できる唯一の刺激物質である。FDAは、カフェインは安全かつ有効であるということで意見が一致している。推奨用量は100〜200mgであり、3または4時間毎よりもより頻繁に投与されるべきではない。FDAは、多くのコーヒー抽出物の刺激性とは対照的に、カフェイン自体は通常の用量において胃腸管の刺激を惹起しないと述べている。これは、薬剤が刺激特性のために使用される場合に有利である。FDAは、その刊行物内で、ヒトが眠気を催しているまたは疲労している場合にカフェインが覚醒を回復することは証拠によって証明されていると述べている。
【0022】
ホスホジエステラーゼの阻害はカフェインの作用に寄与し得るが(Daly 1998)、このキサンチンの大半の薬理学的作用はA1、A2A、A2BおよびA3サブタイプと命名されたアデノシンレセプターの拮抗作用に起因すること(Fredholm 1999)を示す証拠が集まりつつある。カフェインはA2Aレセプターにおいて最も強力に作用して、A1レセプターがこれに次いで近く、続いてA2Bレセプター(Klotz 1998; Ongini 1996)であり、ヒトA3レセプターでは弱いアンタゴニストとして作用する。カフェインによるアデノシンレセプター、つまり、A1およびA2Aレセプタータイプの遮断は、様々な生理学的過程に対する内因性アデノシンの作用を阻害する(Fredholm 1995)。通常の条件下において、アデノシンの血中量は血小板におけるA2Aレセプターを緊張性に活性化するために十分であると思われる。近年、A2Aレセプターノックアウトマウスでは血小板凝集が増加することが報告されて、血小板機能におけるこのレセプターサブタイプの重要性を示している(Ledent 1997)。従って、カフェインは血小板におけるこれらの緊張性に活性化されるA2Aレセプターを遮断して、アデノシンによって介在されるそれらの機能を変化させることができると考えられる。
【0023】
長年、コーヒーを飲むことと心臓血管疾患、特に冠動脈心疾患との関連性が疑われているが、最近ではコーヒーまたはカフェインの摂取は冠動脈心疾患または卒中発作のリスクを増大させないことが実証されている(Grobbee 1990;Jee 1999)。
【0024】
カフェインは複数の鎮痛調製物に含まれる。これが全く合理的である限り、それは痛覚過敏を引き起こす知覚神経終末または終末近くのアデノシンA2Aレセプターの存在に関連し得る(Ledent et al., 1997)。実際、カフェインは特定の種類のC線維介在性疼痛において痛覚鈍麻効果を持つ(Myers et al., 1997)。鎮痛効果は小さい(Battig and Welzl, 1993)。しかし、疼痛の条件下において、カフェインは気分および頭の冴えを高めることによって間接的な有利な効果を持つことができる(Lieberman et al., 1987)。この試験では、気分および覚醒状態の双方が、アスピリンの単独投与またはプラセボよりもカフェインと併用したアスピリンによってさらに改善されることが見出された。様々な量のカフェインと併用して、鎮痛薬であるアスピリン、アセトアミノフェンおよびフェナセチンの1つまたは複数を含む組成物がこれまでに販売されている。複数のケースでは、非麻薬性鎮痛薬/カフェインの併用製剤が麻薬性鎮痛薬であるコデイン、プロキシフェンまたはオキシコドンの1つをさらに含んでいる。これらの組み合わせの例には、Excedrin(商標)、SK-65(商標)、Darvon(商標)、Anacin(商標)として、またコデインであるTabloid(商標)のブランドで市販されている公知の製剤が含まれる。
【0025】
カフェインが特定の種類の疼痛において鎮痛特性を持つ可能性があるということは排除できず、これは頭痛の症例であり得て(Ward et al., 1991)、二重盲検条件下においてカフェインによって著しく、かつ、用量依存性に頭痛が軽減する。効果はアセトアミノフェンのそれとほぼ等しく、それはしばしばカフェインと組み合わせられて、気分に対する影響または自己報告されたコーヒー摂取との関連性は示されなかった。総評(Migliardi et al., 1994)の通り、患者は頭痛の治療に関してカフェインフリーの調製物よりもカフェインを含有する鎮痛薬を優れていると判定する。さらに、カフェインは、i.c.v.注射後にマウスにおいて疼痛関連性の行動を拮抗することができることから(Ghelardini et al., 1997)、脳において抗侵害受容効果を発揮する可能性がある。さらに、この効果はコリン作動性伝達を抑制するアデノシンA1レセプターの緊張性阻害活性の拮抗作用に関連する可能性がある(Rainnie et al., 1994; Carter et al., 1995を参照)。
【0026】
上記の通り、睡眠はヒトにおけるカフェインの影響に最も敏感な生理学的機能の1つであると想われる。就寝時に摂取するカフェインは睡眠に対してネガティブに作用することは周知である(Snel, 1993を参照されたい)。一般に、睡眠に対して顕著な影響を及ぼすためには200mgを超えるカフェインが必要である。最も顕著な影響は、総睡眠時間の短縮、睡眠潜時の延長、最初の軽睡眠EEG段階の増加、およびその後の熟睡EEG段階の減少、さらに睡眠段階間の推移回数の増加である。
【0027】
現在、早漏の優れた治療は、Master & Johnsonプロトコルに基づく行動二チームセックス療法、または個々の心理療法としての心理療法である(Rifelli and Moro. Sessuologia Clinica. Bologna, 1989)。これまでの早漏治療の方法には、心理療法、局所麻酔、および装置の使用が含まれる(米国特許第5,535,758号、第5,063,915号、第5,327,910号、および第5,468,212号)。これらの方法はすべて、重大な欠点を持ち得る。心理療法は一部の患者においてのみ有利であり、特に遠隔地域ではすべての患者に対応できないかもしれない専門のセラピストが必要である。さらに、心理療法は非心理学的原因に起因する早漏は緩和することができない。麻酔性物質は組織の感受性を低下させて、それによって性的満足を減じる。また、局所麻酔薬は性的パートナーに伝達されて、それによってパートナーの感受性および満足をも減少させる可能性がある。装置に関しては、これらは使いにくくて不便であり使用にまごつく可能性がある。装置は非常に目に付きやすく、苦しんでいるパートナーが隠したがり得るその状態を暴露する。さらに、装置は一方または双方のパートナーに刺激を引き起こす可能性がある。
【0028】
複数の異なる抗うつ剤化合物の全身投与による早漏を治療するための方法が述べられている(米国特許第4,507,323号、第4.940,731号、第5,151,448号、および第5,276,042号;PCT公報W095/13072)。しかし、これらの薬剤はすべての患者には有効でない可能性があり、これらの薬剤の副作用によって治療が中止となったり患者のコンプライアンスが損なわれたりすることがある。疾患の状態またはその他の薬剤との有害な相互作用はこれらの化合物の使用に禁忌を示す場合があり、または射精の開始を遅延させるためには有効でないかもしれない低用量を要求する場合がある。さらに、抗うつ剤療法に関連する精神的不健康の徴候が患者にこのような治療の開始または継続に対して二の足を踏ませる可能性がある。抗うつ剤であるフルオキセチンの投与は早漏を治療するために特許請求されている(米国特許第5,151,448号)。しかし、フルオキセチンの投与は多くの望ましくない局面を持ち得る。肝または腎機能障害を持つ患者は、肝での代謝および腎を介しての排出のために、フルオキセチンを使用できない可能性がある。フルオキセチン投与中に肺、腎または肝に関わる全身性の事象が発生していて、過量投与から死亡が発生している。さらに、経口フルオキセチン投与の副作用には、脱毛、悪心、嘔吐、消化不良、下痢、食欲不振、不安、神経過敏、不眠症、傾眠状態、疲労、頭痛、振戦、浮動性めまい、痙攣、発汗、そう痒症、および皮疹が含まれる。フルオキセチンは、しばしば、肝によるそれらの代謝を障害することによって、一連の薬剤と相互作用する。
【0029】
米国特許第4,940,731号は、早漏の治療のためのセルトラリンの経口または非経口投与について記載する。セルトラリンはフルオキセチンと同一の問題の多くを共有することが認識されている(Martindale, The Extra Pharmacopoeia, 31st edition, at p. 333 (London: The Royal Pharmaceutical Society, 1996)を参照されたい)。セルトラリンは肝で代謝されて、尿および糞便中に排出される。従って、肝硬変の患者は低用量を使用しなければならないし、腎機能不全の患者にセルトラリンを投与する際には注意しなければならない。モノアミンオキシダーゼ阻害剤を使用中の個体は、記憶の変化、錯乱、易刺激性、悪寒、発熱および筋固縮に至る毒性のリスクがあるため、セルトラリンを使用できない。セルトラリン経口投与に起因する副作用には、悪心、下痢、消化不良、不眠症、傾眠、発汗、口内乾燥、振戦および躁病が含まれる。昏睡、痙攣、便失禁および女性化乳房の稀な事例が、セルトラリン療法を受けている患者で発生している。米国特許第5,276,042号は、早漏の治療のためのパラキセチンの投与について記載している。パラキセチンは主に尿中に排出されて、肝および腎機能障害を持つ患者では低用量が推奨される。セルトラリンと同様にパラキセチンも、モノアミンオキシダーゼ阻害剤の投与を受けている患者には与えられない。パラキセチンの経口投与による副作用には、低ナトリウム血症、無力症、発汗、悪心、食欲減退、中咽頭の疾患、傾眠、浮動性めまい、不眠症、振戦、不安、ツチ骨の可動性障害、脱力および錯感覚が含まれる。従って、専門の心理療法を必要とせず、便利に、かつ、戸惑うことなく使用することができて、しかも事前の治療方法に関連する問題に関与しない早漏治療の方法が必要である。
【0030】
米国特許第6,037,360号は、様々なセロトニンアゴニストおよびアンタゴニストの投与が早漏の治療に有効であることを開示している。セロトニン阻害剤を用いた治療中に最も頻繁に発現する有害作用は、例えば、悪心、下痢/軟便、便秘などの胃腸障害である。(Drugs 43 (Suppl. 2), 1992)。悪心は、発生率の点で主たる有害作用である。さらに、セロトニン阻害剤の投与後に患者が消化不良に苦しむことは頻繁に観察されている。
【0031】
米国特許第5,707,999号は、2つの特異的α1-遮断薬であるアルフゾシンおよびテトラゾシンが心因性早漏の治療に有効であり、該薬剤は心理療法からの利点がないことが証明された患者において有効であることが明らかとなったことを示す。しかし、テラゾシンおよびその類似体は頭痛、悪心、体重増加、浮動性めまい、傾眠、呼吸困難および霧視を含む複数の副作用を持つ。
【0032】
米国特許第6,037,346号は勃起不全の治療のためのホスホジエステラーゼ阻害剤の局所投与を開示して、好ましい投与モードは経尿道として特許請求される。薬学的製剤およびキットも示されている。米国特許出願第2002/0037828 Al号は、早漏治療のためのホスホジエステラーゼ阻害剤の使用を開示する。
【0033】
米国特許第4,656,177号および第4,777,174号は、非麻薬性鎮痛剤/非ステロイド系抗炎症剤および/または麻薬鎮痛剤ならびにカフェインの組み合わせを開示する。組成物は、疼痛緩和物質が単独で投与される場合よりもより強力かつより速やかな鎮痛反応を示す。
【0034】
米国特許第4,777,174号は、非麻薬性鎮痛剤/非ステロイド系抗炎症剤および/または麻薬鎮痛剤ならびにカフェインの組み合わせを開示する。組成物は、疼痛緩和物質が単独で投与される場合よりもより強力かつより速やかな鎮痛反応を示す。
【0035】
米国特許第5,248,678号は、カフェインなどのアデノシンレセプターアンタゴニストおよびギャバペンチンなどのGABAアゴニストの有効量を患者に投与する工程を含む、昏睡状態の患者または近昏睡状態の患者の覚醒(arousal)覚醒状態(alertness)を増大させる方法を示す。
【0036】
これまで、トラマドールなどのμ-オピエート鎮痛薬およびデキストロメトルファンの鎮痛促進量の組み合わせ、またはもっと詳しく言うならば、その他の任意のNMDAレセプターアンタゴニストがヒトにおいて早漏を治療するために効果的に用いることができるという認識または評価はなかった。さらに、これまで、トラマドールなどのμ-オピエート鎮痛薬、サイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)阻害剤、およびデキストロメトルファンの鎮痛促進量の組み合わせ、またはもっと詳しく言うならば、その他の任意のNMDAレセプターアンタゴニストがヒトにおいて早漏を治療するために効果的に用いることができるという認識または評価はなかった。さらに、これまで、トラマドールなどのμ-オピエート鎮痛薬、サイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)阻害剤、カフェイン、およびデキストロメトルファンの鎮痛促進量の組み合わせ、またはもっと詳しく言うならば、その他の任意のNMDAレセプターアンタゴニストがヒトにおいて早漏を治療するために効果的に用いることができるという認識または評価はなかった。
【0037】
従って、本発明の目的は、現在有効な治療法に付随する有害な副作用を伴わずに提供する早漏の治療のための方法および組成物を提供することである。驚くべきことに、現在、デキストロメトルファンなどの無毒性NMDAレセプターアンタゴニストとトラマドールなどのμ-オピエート鎮痛薬の組み合わせが早漏に対して顕著な対症効果を示すことが見出されている。驚くべきことに、現在、デキストロメトルファンなどの無毒性NMDAレセプターアンタゴニストとトラマドールなどのμ-オピエート鎮痛薬およびシルデナフィルのようなサイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)の組み合わせも早漏に対して顕著な対症効果を示すことが見出されている。本発明のこれらおよびその他の目的および特徴は後続の説明から明らかとなるであろう。
【発明の開示】
【0038】
発明の概要
一つの局面において、本発明はヒトまたはその他の哺乳動物における性機能不全を効果的に治療する方法を提供する。方法は、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、およびb)μ-オピエートレセプターのアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニストであるμ-オピエート鎮痛薬または薬学的に許容されるその塩を含むある量の作用物質を、このような治療を必要とする患者に投与する工程を含む。組み合わせた量の作用物質は性機能不全を治療するために有効である。
【0039】
本発明に従って、性機能不全は早漏または早漏を状態の要素として含む性機能不全であり得る。
【0040】
作用物質は別々に、または組み合わせて投与することができる。3つまたはそれよりも多い作用物質が関与する場合、作用物質は様々な組み合わせで投与することができる。例えば、3つの作用物質は一緒に投与して良く、または2つの作用物質を一緒に投与して第三の作用物質は別に投与されても良い。
【0041】
作用物質は好ましくは性的活動の前に投与される。投与は、経口的に、インプラントによって、非経口的に、皮下で、舌下で、経直腸的に、局所的にまたは吸入を介して行われ得る。好ましい態様において、作用物質は経口的に投与される。
【0042】
NMDAレセプターアンタゴニストは、デキストロメトルファン、デキストロルファン、ケタミン、アマンタジン、メマンチン、エリプロジル、イフェンプロジル、フェンシクリジン、MK-801、ジゾシルピン、CCPエン、フルピルチン、またはそれらの誘導体もしくは塩であり得る。好ましくは、アンタゴニストはデキストロメトルファンである。
【0043】
μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストは、(1R,2Rまたは1S,2S)-(ジメチルアミノメチル)-1-(3-メトキシフェニル)-シクロヘキサノール(トラマドール)、そのN-酸化物誘導体(「トラマドールN-酸化物」)、およびそのO-デスメチル誘導体(「O-デスメチルトラマドール」)、またはそれらの混合物、立体異性体もしくはラセミ体の任意の1つであってよい。好ましい態様において、μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストはトラマドールである。
【0044】
作用物質は、タブレット、経口投与のための多粒子(multiparticulate)製剤;経口投与のための液剤、徐放性製剤、懸濁剤もしくはエリキシル剤、注射可能製剤、移植可能な装置、局所用調製物、固体状態および/もしくはデポー型の経皮送達装置、坐剤、口腔内タブレット、または副鼻腔に吸入もしくは注入されるように意図された放出制御粒子製剤、もしくはスプレー、ミストもしくはその他の局所ビヒクルなどの吸入製剤の剤形として投与することができる。剤形は、タブレットまたはカプセルとして製剤化される固形経口剤形としてさらに限定されてもよい。
【0045】
本発明に従って、μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合は、約15:1〜1:15、約10:1〜1:10、約5:1〜1:5、または約1:2であり得る。
【0046】
本発明の特定の態様において、ホスホジエステラーゼ(PDE)阻害剤または薬学的に許容されるその塩が作用物質の1つとして含まれる。好ましくはPDE阻害剤は5型ホスホジエステラーゼ阻害剤である。PDE阻害剤は、シルデナフィル、アミノフィリン、テオフィリン、アムリノン、ミルリノン、ベスナリノン、ビンポセチン、ペモベンダン、シロスタミド、エノキシモン、ペロキシモン、ロリプラム、R020-1724、ザニプラスト、ジピリダモール、MY5445もしくはIC-351、または薬学的に許容されるその塩であり得る。NMDAレセプターアンタゴニスト対ホスホジエステラーゼ阻害剤対μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストの割合は、約90:1:1〜1:90:1〜1:1:90であって良い。
【0047】
特定の態様において、チトクロームP450阻害剤または薬学的に許容されるその塩は作用物質の1つとして含まれる。好ましくはチトクロームP450阻害剤はデブリソキンヒドロキシラーゼ阻害剤である。阻害剤は、キニジン、キニン、ナフチリジン、キサンチン、フェノキシアミノアルカン、カルバモイルイミダゾール、グアニジンイミダゾール、シメチジン(N-シアノ-N'-メチル-N"-[2[[(5-メチル-1H-イミダゾール-4イル)メチル]チオ]エチル]グアニジン)、キノリン、クロロキン(7-クロロ-4-(4-ジエチルアミノ-1-メチルブチルアミノ)キノリン)、プリマキン(8-(4-アミノ-1-メチルブチルアミノ)-6-メトキシキノリン)、トリフルオロメチルオキシムエーテル、5-メトキシ-1-[4-(トリフルオロメチル)-フェニル]-1ペンタノンO-(2-アミノエチル)オキシムとしても公知であるフルボキサミン、または薬学的に許容されるその塩であることができる。NMDAレセプターアンタゴニスト対チトクロームP450阻害剤対μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストの割合は、約90:1:1〜1:90:1〜1:1:90であって良い。
【0048】
特定の態様において、カフェインは作用物質の1つとして含まれる。
【0049】
本発明のさらなる態様において、ホスホジエステラーゼ阻害剤およびチトクロームP450阻害剤の双方が作用物質として含まれる。本発明のさらなる態様において、ホスホジエステラーゼ阻害剤およびカフェインの双方が作用物質として含まれる。本発明のさらなる態様において、カフェインおよびチトクロームP450阻害剤の双方が作用物質として含まれる。本発明のさらなる態様において、ホスホジエステラーゼ阻害剤、カフェイン、およびチトクロームP450阻害剤の双方が作用物質として含まれる。
【0050】
もう一つの局面において、本発明は作用物質の組み合わせの治療上有効量を含む薬学的組成物を提供する。組み合わせは、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)V型ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩を含む。特定の態様において、薬学的組成物はカフェインを作用物質としてさらに含む。
【0051】
本発明は、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)チトクロームP450阻害剤または薬学的に許容されるその塩を含む、作用物質の組み合わせの治療上有効量を含む薬学的組成物も提供する。特定の態様において、薬学的組成物はカフェインを作用物質としてさらに含む。
【0052】
本発明は、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、c)V型ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩、およびd)チトクロームP450阻害剤または薬学的に許容されるその塩を含む、作用物質の組み合わせの治療上有効量を含む薬学的組成物をさらに提供する。特定の態様において、薬学的組成物はカフェインを作用物質としてさらに含む。
【0053】
もう一つの局面において、本発明は作用物質の組み合わせの治療上有効量を含む薬学的組成物を提供する。組み合わせは、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)カフェインを含む。
【0054】
本発明に従って、本出願人らは、これまでに、早漏の治療においてヒトに投与する組成物(組み合わせおよび製剤)を開発した。これらの組成物(組み合わせおよび製剤)は複数の無毒性有効投与量を用いるか、組み合わせるか、または組み入れる(場合によって)。各投与量は、例えば、例えばトラマドール(またはその塩)であるμ-オピエート鎮痛薬の薬剤の無毒性有効投与量、デキストロメトルファン(好ましくはデキストロメトルファン水和物またはその塩)のようなNMDAレセプターアンタゴニストの無毒性有効投与量、、PDE5阻害剤(例えば、シルデナフィル)、および任意で例えばキニジンのようなチトクロームP450阻害剤(好ましくはキニジン水和物またはその塩)の無毒性有効投与量を含む。
【0055】
本出願人らは、早漏および勃起の治療においてヒトに投与される組成物(組み合わせおよび製剤)も開発した。これらの組成物(組み合わせおよび製剤)は、多くの無毒性有効投与量を用いるか、組み合わせるか、または組み入れる(場合によっては)。各投与量は、例えばシルデナフィル(またはその塩)であるサイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)を阻害する薬剤の無毒性有効投与量、デキストロメトルファン(好ましくはデキストロメトルファン水和物またはその塩)のようなNMDAレセプターアンタゴニストの無毒性有効投与量、カフェイン、および例えばトラマドール(またはその塩)のようなμ-オピエート鎮痛薬の無毒性有効投与量を含む。
【0056】
従って、本発明のさらなる局面は、トラマドールのようなμ-オピエート鎮痛薬、ヒトにおける抗興奮毒性活性に関与するデキストロメトルファンのようなNMDAレセプターアンタゴニスト、および任意でキニジンのようなチトクロームP450阻害剤の組み合わせはヒト男性において射精の開始の遅延に極めて有効であることの開示である。
【0057】
本発明のさらにもう一つの局面に従って、本出願人らは、それぞれが経口または非経口投与に適した薬学的賦形剤と共に作用物質の治療上有効量を含む多くの投与量を含む薬学的組成物(組み合わせおよび製剤)を提供している。量は、患者に対して無毒性である様式においてヒト男性における早漏の疾患および状態を治療するおよび緩解を助けるために有効である。作用物質の治療上有効投与量には、例えばトラマドールであるμ-オピエート鎮痛薬、ならびにデキストロメトルファンならびに/またはその塩(例えば、臭化水素酸塩)、および/もしくはその同族体、類似体、誘導体、複合体、プロドラッグ、エステル、および/もしくは断片などのNMDAレセプターアンタゴニストの無毒性有効投与量、ならびに任意で例えばキニジン(好ましくは、キニジン水和物またはその塩)のようなチトクロームP450阻害剤の無毒性有効投与量が含まれる。
【0058】
本発明のもう一つの局面は、各々の薬理学的活性物質が経口的に投与される方法を提供することである。本発明のさらなる局面は、各々の薬理学的活性物質が非経口的に投与される方法を提供することである。
【0059】
本発明は、早漏を治療するための方法であって、このような治療を必要とする個体にトラマドールのようなμ-オピエート鎮痛薬、デキストロメトルファンのようなNMDAレセプターアンタゴニスト、および任意でナフチリジン、キサンチン、フェノキシアミノアルカン、カルバモイルイミダゾール、シメチジン(N-シアノ-N'-メチル-N"-[2[[(5-メチル-1H-イミダゾール-4イル)メチル]チオ]エチル]グアニジン)のようなグアニジンイミダゾール、クロロキン(7-クロロ-4-(4-ジエチルアミノ-1-メチルブチルアミノ)キノリン)およびプリマキン(8-(4-アミノ-1-メチルブチルアミノ)-6-メトキシキノリン)のようなキノリン、5-メトキシ-1-[4-(トリフルオロメチル)-フェニル]-1ペンタノンO-(2-アミノエチル)オキシムとしても公知であるフルボキサミンのようなトリフルオロメチルオキシムエーテルなどのチトクロームP450の酸化的活性を阻害する作用物質を含む薬学的製剤を投与する工程を含む方法を提供する。薬学的製剤の投与は、作用物質が早漏の治療において有効であるように予め定められた投与計画の状況において実施される。薬剤送達は、経口、非経口、口腔、経直腸、局所、経皮、経尿道および海綿体内注射を含む、早漏からの緩和を示すために有効な任意の経路を介して実施してよい。
【0060】
本発明に従って、本発明の方法を実施するための薬学的製剤が提供される。薬学的製剤は、有効量のトラマドールのような特定のμ-オピエート鎮痛薬、デキストロメトルファンのようなNMDAレセプターアンタゴニスト、任意でチトクロームP450阻害剤、薬学的に許容される担体またはビヒクル、および任意で(即ち、局所、経皮または経尿道製剤の場合)エンハンサーを含む。例えば、賦形剤、界面活性剤、保存料(例えば、抗酸化剤)、安定剤、酵素阻害剤、キレート化物質など、その他の種類の成分も製剤に組み入れることができて、これは薬学的製剤の調製および薬剤送達の当業者によって理解されるであろう。
【0061】
本発明のさらにもう一つの局面は、性的刺激下でヒトにおいて陰茎の勃起を促進するシルデナフィルのようなサイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)阻害剤、ヒトにおいて抗興奮毒性活性に関与するデキストロメトルファンのようなNMDAレセプターアンタゴニスト、トラマドールのようなμ-オピエート鎮痛薬、および任意でキニジンのようなチトクロームP450阻害剤の組み合わせが勃起および射精の問題を持つ男性において射精の開始の遅延に非常に有効であることを示す開示である。
【0062】
従って、本発明のさらにもう一つの局面に従って、本発明者らは、各々が、経口または非経口投与に適した薬学的賦形剤と共に、例えばシルデナフィルなどのサイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)を阻害する薬剤の治療上有効な(患者に対して無毒性でありヒト男性における早漏の疾患および状態を治療するおよび緩解を助けるための)投与量、ならびにデキストロメトルファンおよび/またはその塩(例えば、臭化水素酸塩)および/もしくはその同族体、類似体、誘導体、複合体、プロドラッグ、エステル、および/もしくは断片などのNMDAレセプターアンタゴニストの無毒性有効投与量、ならびにトラマドールおよび/またはその塩(例えば、臭化水素酸塩)および/もしくはその同族体、類似体、誘導体、複合体、プロドラッグ、エステル、および/もしくは断片などのμ-オピエート鎮痛薬の無毒性有効投与量を含む複数の投与量を含む薬学的組成物(組み合わせおよび製剤)を提供している。薬剤送達は、経口、非経口、口腔、経直腸、局所、経皮、経尿道および海綿体内注射を含む、早漏からの緩和を示すために有効な任意の経路を介して実施してよい。
【0063】
チトクロームP450阻害剤を含む組成物と同様に、PDE5阻害剤を含む組成物は薬学的に許容される担体またはビヒクル、および任意で(即ち、局所、経皮または経尿道製剤において)エンハンサーも含むことができる。例えば、賦形剤、界面活性剤、保存料(例えば、抗酸化剤)、安定剤、酵素阻害剤、キレート化物質など、その他の種類の成分も製剤に組み入れることができて、これは薬学的製剤の調製および薬剤送達の当業者によって理解されるであろう。
【0064】
本発明の実践において、NMDAアンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩は、NMDAアンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩の投与間隔が、トラマドールまたはその他のμ-オピエートアゴニストもしくはアゴニスト/アンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩の投与間隔と一部重複するように、トラマドールまたはその他のμ-オピエートアゴニストもしくはアゴニスト/アンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩の投与の前に、同時に、または投与後に投与することができる。
【0065】
同様に、サイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)阻害剤、チトクロームP450阻害剤、および/または少なくとも1つの薬学的に許容されるその塩は、サイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)阻害剤、チトクロームP450阻害剤、および/または少なくとも1つの薬学的に許容されるその塩の投与間隔が、トラマドールまたはその他のμ-オピエートアゴニストもしくはアゴニスト/アンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩の投与間隔、ならびにNMDAアンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩の投与間隔と一部重複するように、トラマドールまたはその他のμ-オピエートアゴニストもしくはアゴニスト/アンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩、ならびにNMDAアンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩の投与前、同時に、または投与後に投与することができる。カフェインも、早漏を治療するために、PDE5阻害剤、μ-オピエートアンタゴニストまたはチトクロームP450阻害剤の投与前、投与中または投与後に投与され得る。本発明の組成物および方法においてカフェインを使用することのさらなる利点は、オピエート鎮痛薬の使用者が経験する可能性がある傾眠状態または鎮静状態を相殺するために用いることができることである。
【0066】
本発明は、以下の番号を付けた文における態様を参照することによってさらに理解され得る:
1.ヒトまたはその他の哺乳動物における性機能不全を効果的に治療する方法であって、このような治療を必要とする患者にa)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、およびb)μ-オピエートレセプターのアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩を含む、ある量の作用物質を投与して、それによって該作用物質の組み合わせた量が性機能不全を治療するために有効である工程を含む方法。
2.性機能不全が早漏である、文1の方法。
3.作用物質が別々に投与される、文1の方法。
4.作用物質が組み合わせて投与される、文1の方法。
5.作用物質が性的活動の前に投与される、文1の方法。
6.作用物質が経口的に、インプラントによって、非経口的に、皮下で、舌下で、経直腸的に、局所的に、または吸入を介して投与される、文1の方法。
7.作用物質が経口的に投与される、文6の方法。
8. NMDAレセプターアンタゴニストがデキストロメトルファン、デキストロルファン、ケタミン、アマンタジン、メマンチン、エリプロジル、イフェンプロジル、フェンシクリジン、MK-801、ジゾシルピン、CCPエン、フルピルチン、またはその誘導体もしくは塩である、文1の方法。
9.NMDAレセプターアンタゴニストがデキストロメトルファンである、文8の方法。
10.μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストが、(1R,2Rまたは1S,2S)-(ジメチルアミノメチル)-1-(3-メトキシフェニル)-シクロヘキサノール(トラマドール)、そのN-酸化物誘導体(「トラマドールN-酸化物」)、およびそのO-デスメチル誘導体(「O-デスメチルトラマドール」)、またはその混合物、立体異性体もしくはラセミ体の任意の1つである、文1の方法。
11.μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストがトラマドールである、文10の方法。
12.作用物質が、タブレット、経口投与のための多粒子製剤;経口投与のための液剤、徐放性製剤、懸濁剤もしくはエリキシル剤、注射可能製剤、移植可能な装置、局所用調製物、固体状態および/もしくはデポー型の経皮送達装置、坐剤、口腔内タブレット、または副鼻腔に吸入もしくは注入されるように意図された放出制御粒子製剤、もしくはスプレー、ミストもしくはその他の局所ビヒクルなどの吸入製剤からなる群より選択される剤形で投与される、文1の方法。
13.剤形がタブレットまたはカプセルとして製剤化される固形経口剤形としてさらに限定される、文12の方法。
14.μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約15:1〜1:15である、文1の方法。
15.μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約10:1〜1:10である、文14の方法。
16.μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約5:1〜1:5である、文15の方法。
17.μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約1:2である、文16の方法。
18.ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩が作用物質として含まれる、文1の方法。
19.ホスホジエステラーゼ阻害剤がV型ホスホジエステラーゼ阻害剤である、文18の方法。
20.ホスホジエステラーゼ阻害剤が、シルデナフィル、アミノフィリン、テオフィリン、アムリノン、ミルリノン、ベスナリノン、ビンポセチン、ペモベンダン、シロスタミド、エノキシモン、ペロキシモン、ロリプラム、R020-1724、ザニプラスト、ジピリダモール、MY5445もしくはIC-351、または薬学的に許容されるその塩である、文18の方法。
21.NMDAレセプターアンタゴニスト対ホスホジエステラーゼ阻害剤対μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストの割合が約90:1:1〜1:90:1〜1:1:90である、文18の方法。
22.チトクロームP450阻害剤または薬学的に許容されるその塩が作用物質として含まれる、文1または18の方法。
23.チトクロームP450阻害剤がデブリソキンヒドロキシラーゼ阻害剤である、文22の方法。
24.チトクロームP450阻害剤が、キニジン、キニン、ナフチリジン、キサンチン、フェノキシアミノアルカン、カルバモイルイミダゾール、グアニジンイミダゾール、シメチジン(N-シアノ-N'-メチル-N"-[2[[(5-メチル-1H-イミダゾール-4イル)メチル]チオ]エチル]グアニジン)、キノリン、クロロキン(7-クロロ-4-(4-ジエチルアミノ-1-メチルブチルアミノ)キノリン)、プリマキン(8-(4-アミノ-1-メチルブチルアミノ)-6-メトキシキノリン)、トリフルオロメチルオキシムエーテル、5-メトキシ-1-[4-(トリフルオロメチル)-フェニル]-1ペンタノンO-(2-アミノエチル)オキシムとしても公知であるフルボキサミン、または薬学的に許容されるその塩である、分22の方法。
25.NMDAレセプターアンタゴニスト対チトクロームP450阻害剤対μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストの割合が約90:1:1〜1:90:1〜1:1:90である、文22の方法。
26.カフェインが作用物質として含まれる、文1、18または22の方法。
27.作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)V型ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩を含む、薬学的組成物。
28.作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)チトクロームP450阻害剤または薬学的に許容されるその塩を含む、薬学的組成物。
29.作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、c)V型ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩、およびd)チトクロームP450阻害剤または薬学的に許容されるその塩を含む、薬学的組成物。
30.カフェインをさらに含む、文27、28または29の薬学的組成物。
31.作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)カフェインを含む薬学的組成物。
【0067】
本発明のさらなる目的、利点および新規の特徴は、一部は以下の説明に記載されて、また一部は以下の検討により当業者に明らかとなるであると考えられ、または本発明の実践によって学ばれ得る。
【0068】
例証的態様の説明
本発明を詳細に説明する前に、薬剤または薬剤送達系は変化し得るものであり、本発明は特定の薬剤または薬剤送達系に限定されないということが理解されるべきである。本明細書で用いられる用語は特定の態様のみを説明することを目的とするものであり、限定することを意図するものではないことも理解されるべきである。本明細書で用いられるように、「一つの(a)」、「一つの(an)」、および「その(the)」という単数形は、文脈からそうでないことが明示される場合を除いて、複数の指示物を含む。従って、「一つの(a)薬理学的活性物質」への言及は2つまたはそれよりも多くの薬理学的活性物質の組み合わせなどを含む。本発明について説明する場合、次の用語は以下に示す定義に従って用いられる。
【0069】
「活性物質」、「薬剤」および「薬理学的活性物質」という用語は、本明細書では、生体(ヒトまたは動物)に投与された際に所望の薬理学的効果を誘発する化学的材料または化合物を指すために互換的に用いられる。同じく所望の薬理学的効果を誘発することが具体的に記載される、それらの化合物の誘導体および類似体または化合物のクラスも含まれる。
【0070】
「局所投与」という用語は、皮膚または粘膜への局所的な薬剤または薬理学的活性物質の送達を示すために慣例的な意味で用いられる。
【0071】
本明細書で用いられるように「担体」または「ビヒクル」は薬剤の投与に適した担体材料を示す。本明細書において有用な担体およびビヒクルには、例えば、任意の液体、ゲル、溶媒、液体希釈剤、可溶化剤など、無毒性で、かつ、組成物のその他の成分と有害な様式で相互作用しない、当技術分野において公知の任意のこのような材料が含まれる。
【0072】
薬剤または薬理学的活性物質の「有効」量は、無毒性であるが、所望の効果を示すために十分な薬剤または作用物質の量を意味する。
【0073】
本明細書で用いられるように「早漏」という用語は、男性がパートナーを満足させるために十分な程度で射精過程を制御することができない性機能不全を意味する。一般に、「早漏」は、性交前または性交中に最小限の刺激で射精を持続または反復することを指す。この用語は、例えば、米国特許第5,151,448号およびMale Infertility and Sexual Dysfunction at p. 356(New York: Springer- Verlag, 1997)に示されるように、「先天性」または「生涯」早漏、および「原発性」または「後天性」早漏の双方を含む。Diagnostic and Statistical Manual of Mental Disorders(Washington, D.C.: American Psychiatric Association, 1994)も参照されたい。
【0074】
「NSAID」という用語は、シクロオキシゲナーゼ酵素と結合することによってプロスタグランジンの産生を阻害する非ステロイド系の物質を指す。アセトアミノフェンは抗炎症特性は持たないが末梢および視床下部の温度調節中枢においてシクロオキシゲナーゼ酵素と結合するので、化合物のアセトアミノフェンはこのカテゴリーに含まれる。
【0075】
本明細書で用いられるように「シルデナフィル」という用語は、この化合物の遊離塩基型、および有機カルボン酸、有機スルホン酸または無機の酸と共に形成される薬学的に許容されるその酸付加塩を含む。本発明のために、有機カルボン酸塩である3.5mg/mlの水溶解度を持つクエン酸シルデナフィルが特に好ましい。「シルデナフィル」に対する言及にはクエン酸シルデナフィルが含まれる。
【0076】
本明細書で用いられるように「カフェイン」という用語は、無水粉末としてのカフェインばかりでなく、無毒性で薬学的に許容され、ならびに本明細書に記載されるように用いられた場合に鎮痛または抗炎症反応を促進および増強することができるカフェインの任意の塩もしくは誘導体、またはそれらの配合された任意の混合物も包含することを意図する(本発明の組成物において有用であることが証明され得るカフェインの塩、誘導体および混合物の説明については、例えば、The Merck Index, ninth edition, Merck & Co., Inc. Rahway, N.J. (1976), pp. 207-208を参照されたい)。それにも関わらず、現在のことろ無水粉末塩基としてのカフェインが好ましく、カフェインの具体的な量が以下に記載される場合、このような量は無水塩基のmgにて示される。
【0077】
本発明者らは早漏の有効な治療を探索していた。前記の先行技術に基づいて、本発明者らは、勃起に何の問題もない男性において射精の過程を制御して満足な性交を持つためには(以下、クラスIと記載する)、性的パートナーが最大の性的満足に達するために十分な性交の時間を持つように射精の過程を遅延させなければならないと推論した。
【0078】
勃起の問題を持ち、一旦勃起が達成されると射精を制御できない男性の場合は、次の2つの段階が好ましいはずである;(1)男性が性的パートナーによる刺激によって十分に勃起するようにシルデナフィルのような特定の医薬物質を介して勃起が達成されなければならない;(2)性的パートナーが最大の性的満足に達するために十分な性交の時間を持つように射精の過程が遅延されなければならない(以下、クラスIIと記載する)。
【0079】
DMの投与はヒトにおいて抗興奮毒性作用を持ち、ヒト男性へのDMの投与は射精の過程に対して影響を及ぼすであろうということが理解される。トラマドールは、その神経シグナルに対する作用のために、鎮痛効果を持つことも理解される。さらに、デブリソキンヒドロキシラーゼ阻害剤またはチトクロームP450阻害剤をDMと同時に投与するとヒト臨床試験において観察可能なDMの治療効果が実質的に増大し、次いで、早漏治療のための作用物質としてのDMの有効性はチトクロームオキシダーゼ阻害剤の同時投与によっても増強されることができることが理解される。これらの観察に基づいて、本発明者らはこれらの作用物質の組み合わせの投与がクラスIの男性の早漏に対して治療効果を持つであろうと推論した。驚いたことに、本発明者らは、これまでに、これらの作用物質の摂取が実際に早漏に対して著しい効果を持ち、それらは最大オルガスムに達するために性交を延長させることを発見した。さらに、本発明者らは、これらの作用物質を用いて性交中に複数のオルガスムを持つことができることを観察した。
【0080】
加えて、本発明者らは、シルデナフィル、トラマドールおよびDMの摂取がクラスIIの男性の早漏に対して顕著な効果を持ち、それらが最大オルガスムに達するために性交を延長させることを見出している。さらに、本発明者らは、これらの作用物質を用いて性交中に複数のオルガスムを持つことができることを観察した。さらに、本発明者らはトラマドールをシルデナフィルおよびDMと共に摂取してもクラスIIの患者において早漏治療におけるシルデナフィルおよびDMの組み合わせの治療的効果に影響はないことを見出している。
【0081】
加えて、本発明の特定の態様において、上記の組成物へのカフェインの添加は、オピエート鎮痛薬の一部の使用者が経験する傾眠状態または鎮静状態を相殺する利点がある。
【0082】
クラスIの男性における早漏を治療するための活性物質
クラスIの男性における早漏を治療するために本発明の方法を実践するために、選択された薬理学的活性物質を個体に投与する。活性物質は、経口的、非経口的、口腔内、経直腸的、または海綿体内注射によってもしくは尿道への送達によって局所的に投与され得る。適切な薬理学的活性物質は、μ-オピエート鎮痛薬であるトラマドール、その代謝物、その塩を含む。
【0083】
(+/-)-トラマドールはコデインの合成4-フェニル-ピペリジン類似体である(Shipton EA 2000)。これは、オピエートレセプターに低親和性を持つ中枢性鎮痛薬である。そのμレセプターに関する選択性は最近実証されたものであり、肝臓のO-脱メチル化によって産生されるトラマドールのM1代謝物はオピエートレセプターに対して親薬剤よりも高い親和性を示す。このM1誘導体(O-デメチルトラマドール)の産生速度は、デブリソキン型の多型性イソ酵素であるチトクロームP450 2D6(CYP2D6)に影響される。1つのメカニズムがμ-オピエートレセプターに対するその弱い親和性に関連する(モルフィンの1/6000、d-プロポキシフェンの1/100、コデインの1/10、およびデキストロメトルファンと同等)。さらに、その他のオピエートとは対照的に、トラマドールの鎮痛作用はオピエートアンタゴニストであるナロキソンによってごく一部が阻害されるのみであり、このことはもう一つの作用メカニズムの存在を示唆する。このことは、ノルアドレナリン(ノルエピネフリン)およびセロトニン(5-ヒドロキシトリプタミン;5-HT)の再取り込みを阻害して、脊髄レベルでの侵害受容インパルスを遮断することによって鎮痛作用に大きく寄与するするモノアミン作動性の活性の発見によって実証された(Dayer et al. 1994 & 1997)。
【0084】
(+/-)-トラマドールは2つの鏡像異性体のラセミ混合物であり、それぞれの鏡像異性体は様々なレセプターに対して異なる親和性を示す。(+/-)-トラマドールはμレセプターの選択的アゴニストであり、セロトニンの再取り込みを優先的に阻害する一方、(-)-トラマドールは主としてノルアドレナリンの再取り込みを阻害する。これらの2つの鏡像異性体の作用は双方とも相補的かつ相乗的であり、その結果、(+/-)-トラマドールの鎮痛効果が得られる。経口投与後、トラマドールは68%の生物学的利用能を示して、2時間以内に最高血清中濃度に到達した。排出動態は2コンパートメントとして説明され得、100mg単回経口投与後の半減期はトラマドールが5.1時間、M1誘導体が9時間である。このことが、トラマドールによる反復投与治療中に観察される親薬剤およびそのM1誘導体の約2倍の蓄積の原因である。トラマドールの推奨一日投与量は4〜6時間毎に50〜100mgであり、最大用量は400mg/日である。トラマドール100mgの単回経口投与後の鎮痛効果の期間は約6時間である。有害影響、特に悪心は、用量依存性であり、従って、負荷用量が高ければ発現する可能性がかなり高い。治療の最初の数日の期間中にこの用量を減量することは忍容性向上の重要な要因である。その他の有害影響は概ねオピエートのそれとほぼ等しいが、それらは通常はより軽度であり、呼吸抑制、不快気分および便秘が含まれ得る。トラマドールはその他の鎮痛薬、特に末梢作用を持つ鎮痛薬と同時に投与することができるが、CNS機能を抑制する薬剤はトラマドールの鎮静効果を増強する可能性がある。トラマドールは、依存に至る可能性は極めて低い薬力学および薬物動態特性を持つ。このことは様々な対照試験および市販後調査試験によって確認されて、試験では耐性を示す患者またはトラマドール乱用の事例の数が極端に少ないことが報告された(Raffa et al. 1993; Lee et al. 1993)。
【0085】
本発明の特定の態様において、早漏は、呼吸抑制、睡眠パターンの障害、食欲減退、発作、ならびに心理的および/または身体的依存などの、従来の鎮痛薬に付随する有害な副作用なしに治療することができる。
【0086】
それは疼痛の制御に関しては安全かつ有効な作用物質であることが証明されているものの、その使用に伴って有害作用が生じ得る。4mg/kgのトラマドールの小児への偶発的投与後に発作的活動の発生が報告されている(Tobias 1997)。
【0087】
デキストロメトルファンはシグマレセプターとして公知のニューロンレセプターのクラスに作用する。それらは抑制性レセプターであり、これはDMまたはその他のシグマアゴニストによるそれらの活性化が特定の種類の神経シグナルの抑制を引き起こすことを意味する。デキストロメトルファンはN-メチル-D-アスパラギン酸(NMDA)レセプターとして公知の、興奮性アミノ酸(EAA)レセプターの一種であるもう一つのクラスのレセプターにも作用する。シグマレセプターへのそのアゴニスト活性とは異なり、DMはNMDAレセプターにおいてアンタゴニストとして作用し、これはDMがNMDAレセプターを介して調節される神経インパルスの伝達を抑制することを意味する。NMDAレセプターは興奮性レセプターであるため、DMのNMDAアンタゴニストとしての活性も特定の種類の神経シグナルの抑制を誘発する。デキストロメトルファンおよびデキストロルファンの抗興奮毒性活性、ならびにこれらの薬剤によるNMDAレセプターの遮断については、Choi 1987、Wong et al 1988、Steinberg et al 1988、および米国特許第4,806,543号(Choi 1989)などの文献等で考察されている。デキストロメトルファンは、神経カルシウムチャネルにおいて活性を抑制することも報告されている(Carpenter et al 1988)。デキストロメトルファンおよびそれが相互作用するレセプターについては、Tortella et al 1989、Leander 1989、Koyuncuoglu & Saydam 1990、Ferkany et al 1988、George et al 1988、Prince & Feeser 1988、Feeser et al 1988、Craviso and Musacchio 1983、およびMusacchio et al 1988においてより詳細に考察されている。
【0088】
DMは血流からかなり速やかに消失する(例えば、Vetticaden et al 1989およびRamachander et al 1977を参照されたい)。DMは、肝臓においてO-脱メチル化と呼ばれる酵素過程によってデキストロルファンおよび3-メトキシモルフィナンと呼ばれる2つの代謝物に転換される。この酵素は、何年も前にデブリソキンのヒドロキシル化反応を実行するために発見されたことから、通常はデブリソキンヒドロキシラーゼと呼ばれる。これは様々な論文においてP450DBまたはP450-2D6とも呼ばれている。多くの化合物がデブリソキンヒドロキシラーゼ(スパルテインモノオキシゲナーゼ)イソ酵素の活性を阻害する;Inaba et al 1985を参照されたい。これらの阻害物質の最も強力なものはキニンの右旋性立体異性体であるキニジンであり、これは通常は心不整脈を治療するために用いられる。Inaba et al(1986)およびNielsen et al(1990)はキニジンがインビボ動物試験においてスパルテインの酸化を阻害する能力について考察し、Brinn et al(1986)、Brosen et al(1987)、およびBroly et al(1989)はキニジンが肝細胞調製物中でのDM代謝を阻害する能力について考察している。例外的に強力であり、かつ、容易に実証されるデブリソキンヒドロキシラーゼの阻害に加えて、その他のチトクロームP450イソ酵素も、様々なレベルの結合親和性で、キニジンによって抑制される可能性が高い。さらに、キニジンはデブリソキンヒドロキシラーゼに対して最も顕著な効果を発揮するものの、同様に多くのその他のチトクロームP450酵素を抑制して、それによって患者は正常かつ望ましい肝臓の活性をより全般的に喪失する可能性が高い。デキストロメトルファンの主たる酸化代謝産物はデキストロルファンであり、これは神経学者の間ではデキストロメトルファンと全く同じ様式で活性であると広く信じられている;報告によると、双方の薬剤はシグマアゴニスト、NMDAアンタゴニストおよびカルシウムチャネルアンタゴニストである。DMと組み合わせたデブリソキンヒドロキシラーゼを阻害する化合物の投与は、DMのみを投与される患者に比して患者の血液中のDMの濃度および安定性の大幅な増大が惹起される;かつDMと組み合わせたデブリソキンヒドロキシラーゼ阻害剤の投与はヒトにおけるDMの検出可能な作用に対して明確で実質的な影響を有する。デキストロメトルファンの活性の増強にはデブリソキンヒドロキシラーゼ阻害剤が好ましいが、ナフチリジン、キサンチン、フェノキシアミノアルカン、カルバモイルイミダゾール、シメチジン(N-シアノ-N'-メチル-N"-[2[[(5-メチル-1H-イミダゾール-4イル)メチル]チオ]エチル]グアニジン)などのグアニジンイミダゾール、クロロキン(7-クロロ-4-(4-ジエチルアミノ-1-メチルブチルアミノ)キノリン)およびプリマキン(8-(4-アミノ-1-メチルブチルアミノ)-6-メトキシキノリン)などのキノリン、5-メトキシ-1-[4-(トリフルオロメチル)-フェニル]-1ペンタノンO-(2-アミノエチル)オキシムとしても公知であるフルボキサミンなどのトリフルオロメチルオキシムエーテルなど、チトクロームP450の酸化的活性を阻害するその他の作用物質もデキストロメトルファンの活性の増強に有用であり得る。
【0089】
カフェインはいくつかの鎮痛剤に含まれている。これが全く合理的である限り、それは痛覚過敏を引き起こす知覚神経終末のまたは終末近くのアデノシンA2Aレセプターの存在に関連し得る(Ledent et al., 1997)。実際、カフェインは特定の種類のC線維介在性疼痛において痛覚鈍麻作用を持つ(Myers et al., 1997)。鎮痛効果は小さい(Battig and Welzl, 1993)。カフェインは気分および頭の冴えを高めることによって間接的に有益な効果を持ち得る(Lieberman et al., 1987)。この試験では、気分および覚醒状態の双方が、アスピリンの単独投与またはプラセボよりもカフェインと併用したアスピリンによってさらに改善することが見出された。
【0090】
総説(Migliardi et al., 1994)の通り、患者は頭痛の治療に関してカフェインフリーの調剤品よりもカフェインを含有する鎮痛薬を優れていると判定する。さらに、カフェインは、i.c.v.注射後にマウスにおいて疼痛関連性の行動に拮抗することができることから(Ghelardini et al., 1997)、脳において抗侵害受容効果を発揮する可能性がある。さらに、この効果はコリン作動性伝達を抑制するアデノシンA1レセプターの持続性阻害活性の拮抗作用に関連する可能性がある(Rainnie et al., 1994; Carter et al., 1995)。
【0091】
上記の通り、睡眠はヒトにおけるカフェインの影響に対する最も敏感な生理学的機能の1つであると考えられる。就寝時に摂取するカフェインは睡眠に対してネガティブに作用することは周知である(Snel, 1993を参照されたい)。一般に、睡眠に対して顕著な影響を及ぼすためには200mgを超えるカフェインが必要である。最も顕著な影響は、総睡眠時間の短縮、睡眠潜時の延長、最初の軽睡眠EEG段階の増加、およびその後の熟睡EEG段階の減少、さらに睡眠段階間の推移回数の増加である。
【0092】
本発明を実践するために、本発明において用いられ得るμ-オピエート鎮痛薬剤の非限定的なリストにはトラマドール、その代謝物、その塩、その複合体が含まれる。
【0093】
本発明において用いられ得るNMDAアンタゴニストには、デキストロメトルファン、ケタミンおよびアマンチジン、ならびにその代謝物、塩および複合体が含まれる。
【0094】
本発明において用いられ得るカフェイン類似体の非限定的なリストには、キサンチン、ヒポキサンチン(6-ヒドロキシプリン)、1-メチルキサンチン、3-メチルキサンチン、7-メチルキサンチン、アザキサンチン(8-アザ-2,6-ジヒドロキシプリン)、テオフィリンおよびテオブロミンが含まれる。
【0095】
経口組み合わせ投与単位は、患者に投与される組み合わせ用量に伴う副作用が最小限または実質的に皆無である限り、好ましくは、約30から200ミリグラム(mg)以下の範囲、好ましくは約60〜約120mgの範囲のデキストロメトルファン、ならびに約30〜約500mgの範囲、好ましくは約30〜約200mgの範囲のトラマドールを含む。カフェインは、約30〜200mg以下、好ましくは約60mg〜約100mgの範囲の用量で組成物に含まれ得る。
【0096】
特に好ましい経口組み合わせ投与単位は、約120mgのデキストロメトルファンおよび100mg以下のトラマドール、より好ましくは、約90mgのデキストロメトルファンおよび約100mg以下のトラマドールを含む。もう一つの好ましい経口組み合わせ投与単位は、約120mgのデキストロメトルファン、約100mgのカフェイン、および100mg以下のトラマドール、より好ましくは約90mgのデキストロメトルファン、約60mgのカフェイン、および約100mg以下のトラマドールを含む。
【0097】
または、デキストロメトルファンおよびトラマドールは、それぞれの各薬剤の順次的な投与を実践するための単独の活性成分として、前記の組成物において別々に製剤化されてもよい。
【0098】
または、デキストロメトルファン、カフェインおよびトラマドールは、それぞれの各薬剤の順次的な投与を実践するための単独の活性成分として、前記の組成物において別々に製剤化されてもよい。
【0099】
順次的投与療法において、トラマドール、カフェインおよびデキストロメトルファンはそれぞれ別々の投与量にて投与される。患者が投与される総組み合わせ用量に伴う好ましくない副作用が最小限または実質的に皆無である限り、トラマドールの順次的投与では、投与単位は、好ましくは約10〜約500mgの範囲、より好ましくは約20mg〜約300mgの範囲のトラマドールを含み、カフェインの投与における投与単位は好ましくは約10〜約400mgの範囲、より好ましくは約30mg〜約200mgの範囲のカフェインを含み、デキストロメトルファンの投与の投与単位は好ましくは約30から120mg以下の範囲、より好ましくは約60〜約90mgの範囲のデキストロメトルファンを含む。
【0100】
特に好ましい順次的投与の投与単位は、約30〜約100mgの範囲のトラマドールを含み、約30〜約135mgの範囲のデキストロメトルファンを含む。好ましくは、各薬剤は経口的に投与される。または、各薬剤は異なる経口的経路によって投与されてもよい;即ち、一つは摂取されて、他方は舌下または口内パッチにより投与されてもよい。
【0101】
トラマドール、カフェインおよびデキストロメトルファンの有効な順次的投与のため、各薬剤の放出には、デキストロメトルファンによる有利な射精遅延を最大限とするように時間差が設けられることが好ましい。
【0102】
特に好ましい順次的投与の投与単位は、約30〜約100mgの範囲のトラマドール、30〜100mgの範囲のカフェイン、および約30〜約135mgの範囲のデキストロメトルファンを含む。好ましくは、各薬剤は経口的に投与される。または、各薬剤は異なる経口的経路によって投与されてもよい;即ち、一つは摂取されて、他方は舌下または口内パッチにより投与されてもよい。
【0103】
トラマドール、カフェインおよびデキストロメトルファンの有効な順次的投与のため、各薬剤の放出には、デキストロメトルファンによる有利な射精遅延を最大限とするように時間差が設けられることが好ましい。
【0104】
デキストロメトルファンの効果を増強するために、任意でキニジンのようなチトクロームP450酵素阻害剤の有効量を組み合わせ投与単位として、または順次的投与の投与単位として患者に投与してもよい。チトクロームP450阻害剤がデキストロメトルファンの効果を増強するために投与される場合、デキストロメトルファンの投与量は有効性を最大限として副作用を最小限とするように適切に調節することができる。経口組み合わせ投与単位は、好ましくはキニジンを約50から200ミリグラム(mg)以下の範囲で、好ましくは約90〜約120mgの範囲で含み得る。
【0105】
クラスIIの男性の早漏を治療するための活性物質
射精と共に勃起の問題を持つクラスIIの男性における早漏を治療するために本発明の方法を実践するために、選択された薬理学的活性物質を個体に投与する。活性物質は、経口的、非経口的、口腔内、経直腸的、または海綿体内注射によってもしくは尿道への送達によって局所的に投与され得る。適切な薬理学的活性物質には、シルデナフィルのようなサイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)阻害剤、カフェイン、デキストロメトルファンのような抗興奮毒性物質、トラマドールのようなμ-オピエート鎮痛薬、ならびに任意でキニン、キニジン、ナフチリジン、キサンチン、フェノキシアミノアルカン、カルバモイルイミダゾール、シメチジン(N-シアノ-N'-メチル-N"-[2[[(5-メチル-1H-イミダゾール-4イル)メチル]チオ]エチル]グアニジン)のようなグアニジンイミダゾール、クロロキン(7-クロロ-4-(4-ジエチルアミノ-1-メチルブチルアミノ)キノリン)およびプリマキン(8-(4-アミノ-1-メチルブチルアミノ)-6-メトキシキノリン)のようなキノリン、5-メトキシ-1-[4-(トリフルオロメチル)-フェニル]-1ペンタノンO-(2-アミノエチル)オキシムとしても公知であるフルボキサミンのようなトリフルオロメチルオキシムエーテルなどのチトクロームP450阻害剤が含まれる。
【0106】
シルデナフィルは、化学的には1-[[3-(6,7-ジヒドロ-1-メチル-7-オキソ-3-プロピル-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)-4-エトキシフェニル]スルホニル]-4-メチルピペラジンと呼ばれ、以下の構造式を持つ:図3。
【0107】
クエン酸シルデナフィルは、現在、経口投与用の25mg、50mgおよび100mgシルデナフィルに相当するタブレットとして製剤化されたViagra(商標)(Pfizer Labs, N.Y.)の名称で販売されるインポテンスのための市販薬の活性成分である。製造業者によると、活性成分であるクエン酸シルデナフィルに加えて、各タブレットは次の活性成分を含む:微晶性セルロース、無水第二リン酸カルシウム、クロスカルメロースナトリウム、ステアリン酸マグネシウム、ヒドロキシプロピルメチルセルロース、二酸化チタン、ラクトース、トリアセチン、およびFD&C青色2号アルミニウムレーキ。
【0108】
インビトロ試験から、シルデナフィルは、心収縮性の制御に関与するPDE3のようなその他の公知のホスホジエステラーゼに対してよりも5型ホスホジエステラーゼ(PDE5)の阻害に関して約4,000倍選択的であることが知られている。報告によると、シルデナフィルは網膜において見出された酵素であるPDE6に比べてPDE5に対する力価として僅か約10倍であり、この低い選択性が高い用量または血漿中濃度で観察される色覚に関連する異常の理由であると考えられる。
【0109】
市販のViagra(商標)製剤として投与されるシルデナフィルは経口投与後に速やかに吸収されることが報告されて、絶対生物学的利用能は約40%である。その薬物動態は推奨用量範囲を通して用量に比例する。Viagra(商標)の製造業者の製品資料に基づいて、空腹時には経口投与の30〜120分(中央値 60分)以内に最大観察血漿中濃度に到達する。Viagra(商標)製剤を高脂肪食と共に服用すると、吸収速度が低下して、Tmaxは平均60分遅延して、Cmaxは平均29%低下する。シルデナフィルの平均定常状態分布容積(Vss)は報告によると105Lであり、組織内に分布することを示している。投与後90分における健常志願者の精液中シルデナフィル報告測定値に基づくと、投与量の0.001%未満が患者の精液に出現した。
【0110】
驚くべきことに、本発明の組成物と共に用いられるデキストロメトルファン、トラマドールおよびシルデナフィルの組み合わせの治療上有効量は早漏の遅延によりシルデナフィルの有利な勃起誘発効果を最大限とする。
【0111】
経口組み合わせ投与単位は、患者に投与される組み合わせ用量に伴う望ましくない副作用が最小限または実質的に皆無である限り、好ましくは、約10〜300ミリグラム(mg)以下の範囲、好ましくは約30および約200mgの範囲のデキストロメトルファン、約10〜約200ミリグラム(mg)の範囲、好ましくは約30〜約150mgの範囲のトラマドール、ならびに約10〜約150mgの範囲、好ましくは約15〜約100mgの範囲のシルデナフィルを含む。特に好ましい経口組み合わせ投与単位は、約150mgのデキストロメトルファン、200mg以下のトラマドール、より好ましくは約100mgのトラマドール、および150mg以下のシルデナフィル、より好ましくは約135mgのデキストロメトルファン、約100mgのトラマドールおよび約100mg以下のシルデナフィルを含む。
【0112】
または、デキストロメトルファン、トラマドールおよびシルデナフィルは、それぞれの各薬剤の順次的な投与を実践するための単独の活性成分として、前記の組成物において別々に製剤化してもよい。
【0113】
順次的投与療法において、シルデナフィル、トラマドールおよびデキストロメトルファンはそれぞれ別々の投与量にて投与される。患者が投与される総組み合わせ用量に伴う好ましくない副作用が最小限または実質的に皆無である限り、シルデナフィルの順次的投与では、投与単位は、好ましくは約10〜約300mgの範囲、より好ましくは約25〜約200mgの範囲のシルデナフィルを含み、トラマドールの投与における投与単位は好ましくは約20〜約400mg以下の範囲、より好ましくは約30〜約200mgの範囲のトラマドールを含み、デキストロメトルファンの投与では、投与単位は好ましくは約30〜500mg以下の範囲、より好ましくは約60〜約300mgの範囲のデキストロメトルファンを含む。
【0114】
シルデナフィルの特に好ましい順次的投与の投与単位は、約50〜約150mgの範囲のシルデナフィルを含み、トラマドールについては約50〜約200mgの範囲のトラマドールを含み、およびデキストロメトルファンについては約45〜約200mgの範囲のデキストロメトルファンを含む。好ましくは、各薬剤は経口的に投与される。または、各薬剤は異なる経口的経路によって投与することができる;即ち、一つは摂取されて、他方は舌下または口内パッチにより投与することができる。
【0115】
望ましい場合、吸収、従って生物学的利用能を促進するために、シクロデキストリン、特にβ-シクロデキストリン、またはヒドロキシプロピル-β-シクロデキストリン(HPBCD)のようなその誘導体などの吸収促進物質を加えてもよい。シクロデキストリンは、6個、7個または8個のグルコピラノース環から構成されて、それぞれ、α、βおよびγシクロデキストリンとして公知である環式非還元性オリゴ糖のグループである。シクロデキストリンは、分子包接化合物を形成する特性を有する空洞内包環式化合物のクラスであり、共有結合を形成することなくもう一つの化学的化合物を係留または捕捉する。HPBCDは、内部空洞を含むドーナツ状の分子構造を有する環式ポリマーである。
【0116】
ヒドロキシプロピル-β-シクロデキストリンは、最大約15またはそれ以上までの置換度(D.S.)を有する対応するヒドロキシプロピル誘導体を与えるために、酸化プロピレンと縮合することによってβ-シクロデキストリンから誘導される市販の化合物である。本発明のために、約5〜7のD.S.値が好ましい。
【0117】
このような適切なヒドロキシプロピル-β-シクロデキストリンの調製は、とりわけ、International Journal of Pharmaceutics, 29, 73-82 (1986)およびJournal of Pharmaceutical Sciences, 75 (6), 571-572 (1986)に記載されている。シクロデキストリンのポリエーテルであるヒドロキシプロピル-β-シクロデキストリンも公知であり本発明のために適切であり、Grameraらに対する米国特許第3,459,731号に記載されるように過剰な酸化ヒドロキシプロピレンのβ-シクロデキストリンを用いた縮合によって得られる。ヒドロキシプロピレン-β-シクロデキストリン(HPBCD)は特に好ましいシクロデキストリン要素であるが、それらに限定されるものではない。HPBCDの組成物中の重量パーセントは、好ましくは組成物全体の約1〜約10重量パーセントの範囲内である。
【0118】
特にシルデナフィルの場合は、HPBCDが生物学的利用能を促進することが見出されている。従って、所望の治療効果を比較的低い用量のシルデナフィルで達成することができ、それによって有害な影響の可能性が最小限に抑えられる。
【0119】
シルデナフィル、トラマドールおよびデキストロメトルファンの効果的な順次的投与のために、各薬剤の放出には、性的刺激時のデキストロメトルファンおよびトラマドールによる有利な勃起の延長ならびにシルデナフィルによる勃起の維持を最大限とするように時間差が設けられることが好ましい。
【0120】
デキストロメトルファン、トラマドールおよびシルデナフィル療法の有利な効果を増強するために、より少ない量の勃起誘発物質を含めることができる。本明細書で用いられるように「勃起誘発物質」という用語は、テストステロン、デヒドロエピアンドロステロン(DHEA)などのような副腎ステロイドを指す。好ましくは、勃起誘発物質は投与されるシルデナフィルの重量の約5〜約10重量パーセントの範囲で、より好ましくは約6〜約8重量パーセントの範囲の量で加えられる。
【0121】
オピエート鎮痛薬の一部の使用者が経験する傾眠状態または鎮静状態を相殺するために、組成物にカフェインを加えてもよい。
【0122】
デキストロメトルファンの効果を増強するために、任意でキニジンのようなチトクロームP450酵素阻害剤の有効量を組み合わせ投与単位、または順次的投与の投与単位のいずれかとして患者に投与してもよい。チトクロームP450阻害剤がデキストロメトルファンの効果を増強するために投与される場合、デキストロメトルファンの投与量は有効性を最大限として副作用を最小限とするように適切に調節することができる。経口組み合わせ投与単位は、好ましくは、キニジンを約50から200ミリグラム(mg)以下の範囲で、好ましくは約90〜約120mgの範囲で含み得る。経口組み合わせ投与単位は、好ましくは、キニジンを約50から200ミリグラム(mg)以下の範囲で、好ましくは約90〜約120mgの範囲で含み得る。
【0123】
活性物質は薬学的に許容される塩、エステル、アミドもしくはプロドラッグ、またはその組み合わせの形で投与され得る。しかし、不活性エステル、アミドまたはプロドラッグ型の活性型への転換は標的組織または細胞への到達前または到達後直ちに起こらなければならない。活性物質の塩、エステル、アミドおよびプロドラッグは、合成有機化学の技術分野の業者に公知であり、例えば、J. March, Advanced Organic Chemistry: Reactions, Mechanisms and Structure, 4th Ed.(New York: Wiley-Interscience, 1992)に記載されるような標準的手順を用いて調製してよい。例えば、酸付加塩は遊離塩基(典型的には中性の型の薬剤が中性の--NH2基を持つ)から適切な酸との反応を伴う従来の方法を用いて調製される。一般に、薬剤の塩基型はメタノールまたはエタノールのような極性有機溶媒に溶解して、そこに酸が加えられる。形成される塩は沈殿させるか、または極性の低い溶媒を加えることによって溶液から取り出すことができる。酸付加塩の調製に適切な酸には、例えば、酢酸、プロピオン酸、グリコール酸、ピルビン酸、シュウ酸、リンゴ酸、マロン酸、コハク酸、マレイン酸、フマル酸、酒石酸、クエン酸、安息香酸、桂皮酸、マンデル酸、メタンスルホン酸、エタンスルホン酸、p-トルエンスルホン酸、サリチル酸などの有機酸、および塩酸、臭化水素酸、硫酸、硝酸、リン酸などの無機酸が含まれる。酸付加塩は適切な塩基での処理によって遊離塩基に戻すことができる。逆に、薬剤に含まれ得る酸部分の塩基性塩の調製は、水酸化ナトリウム、水酸化カリウム、水酸化アンモニウム、水酸化カルシウム、トリメチルアミンなどのような薬学的に許容される塩基を用いて同様の方法で調製される。エステルの調製は、薬剤の分子構造内に含まれ得るヒドロキシル基および/またはカルボキシル基の官能化を伴う。エステルは、典型的には遊離アルコール基のアシル置換された誘導体、つまり、式RCOOHのカルボン酸から誘導される部分であって、Rはアルキルであり、好ましくは低級アルキルである。エステルは、望ましい場合、通常の水素化分解または加水分解の手順を用いることによって遊離酸に戻すことができる。アミドおよびプロドラッグの調製は類似の方法で実施することができる。活性物質のその他の誘導体および類似体は合成有機化学の技術分野の業者に公知の標準的方法を用いて調製され得るか、または関連する文献の参照より推論され得る。さらに、キラル活性物質は鏡像異性的に純粋な型であり得るか、または鏡像異性混合物として投与されてもよい。
【0124】
意図する投与方法に応じて、薬学的組成物は例えばタブレット、坐剤、丸剤、カプセル、粉剤、液体、懸濁剤、クリーム、軟膏、ローション剤などのような固体、半固体または液体の剤形であり得、好ましくは正確な用量の単回投与に適した単位剤形であり得る。この組成物は薬学的に許容される担体と組み合わせて、選択された薬剤の有効量を含み、さらに、その他の医薬物質、アジュバント、希釈剤、緩衝剤などを含み得る。従って、化合物は、通常の無毒性の薬学的に許容される担体、アジュバントおよびビヒクルを含む投与製剤として経口的に、非経口的に、経皮的に、経直腸的に、経鼻的に、口腔内に、局所的に、または埋め込み式リザーバーを介して投与することができる。本明細書で用いられるように「非経口的」という用語は、皮下、静脈内および筋肉内注射を含むことを意図する。勿論、投与される活性化合物の量は治療しようとする被験体、被験体の体重、投与様式、および処方する医師の判断に依存する。
【0125】
固形組成物に関して、無毒性の一般的固形担体には、例えば医薬品グレードのマンニトール、ラクトース、澱粉、ステアリン酸マグネシウム、サッカリンナトリウム、タルク、セルロース、ブドウ糖、ショ糖、炭酸マグネシウムなどが含まれる。薬学的に投与可能な液体組成物は、例えば、本明細書に記載される活性化合物および任意で薬学的アジュバントを、例えば水、生理食塩液、デキストロース水溶液、グリセロール、エタノールなどのような賦形剤に溶解、分散させるなどして、それによって溶液または懸濁液を生成することによって調製することができる。望ましい場合、投与される薬学的組成物は、例えば酢酸ナトリウム、モノラウリル酸ソルビタン、トリエタノールアミン酢酸ナトリウム、オレイン酸トリエタノールアミンなどの湿潤剤または乳化剤、pH緩衝剤などの無毒性の補助物質も含むことができる。このような剤形を調製する実際の方法は当業者に公知であるか、または明らかであると考えられる;例えば、上記において引用したRemington's Pharmaceutical Sciencesを参照されたい。経口投与のためには、組成物は一般にタブレットもしくはカプセルの形をとり、または水性もしくは非水性の液剤、懸濁剤もしくはシロップ剤としてもよい。タブレットおよびカプセルは好ましい経口投与形である。経口使用のためのタブレットおよびカプセルは、一般に、ラクトースおよびコーンスターチのような一般的に用いられる1種類または数種類の担体を含む。ステアリン酸マグネシウムのような潤滑剤も一般的に加えられる。液体懸濁剤を用いる場合、活性物質は乳化剤および懸濁剤と組み合わせることができる。着香料、着色料および/または甘味料も加えてよい。本明細書において経口製剤に組み入れるためのその他の選択的成分には防腐剤、懸濁剤、増粘剤などが含まれるが、これらに限定されるものではない。
【0126】
非経口的投与が使用される場合は一般に注射を特徴とする。注射可能な製剤は、流動性の液剤または懸濁剤のいずれかとしての一般的な形、注射前に液体への溶解もしくは懸濁に適した固形剤として、または乳剤として調製することができる。好ましくは、適切な担体、分散剤または湿潤剤、および懸濁剤を用いて当技術分野において公知の手法に従って、無菌の注射可能な懸濁剤が製剤化される。無菌の注射可能な製剤は、無毒性の非経口的に許容される希釈剤または溶媒中の無菌の注射可能な液剤または懸濁剤であり得る。使用することのできる許容されるビヒクルおよび溶媒には、水、リンガー液および等張塩化ナトリウム液がある。さらに、無菌の固定油、脂肪酸エステルまたはポリオルも溶媒または懸濁媒として一般的に用いられる。ごく最近に修正された非経口投与のためのアプローチは、一定レベルの用量が維持されるような徐放性または持続放出性の系の使用を含む。例えば、米国特許第3,710,795号を参照されたい。
【0127】
活性物質は経尿道の薬剤送達に適した薬学的製剤として投与することができる。製剤は、水、シリコン、ワックス、ワセリン、ポリエチレングリコール(「PEG」)、プロピレングリコール(「PG」)、リポソーム、マンニトールおよびラクトースのような糖、ならびに/またはその他の様々な材料のような1つまたは複数の選択された担体または賦形剤を含むが、ポリエチレングリコールおよびその誘導体が特に好ましい。投与される薬剤に応じて、尿道投与剤形に経尿道浸透促進剤を組み入れることが望ましい場合もある。適切な経尿道浸透促進剤の例には、ジメチルスルホキシド(「DMSO」)、ジメチルホルムアミド(「DMF」)、N,N-ジメチルアセトアミド(「DMA」)、デシルメチルスルホキシド(「C10 MSO」)、モノラウリン酸ポリエチレングリコール(「PEGML」)、モノラウリン酸グリセロール、レシチン、1-置換アザシクロヘプタン-2-オン類、特に1-n-ドデシルシクラザシクロヘプタン-2-オン(Nelson Research & Development Co. Irvine, Calif.からのAzone(登録商標)の商標で入手可能)、SEPA(登録商標)(Macrochem Co., Lexington, Mass.から販売)、アルコール(例えば、エタノール)、界面活性剤(Tergitol(登録商標)、Nonoxynol-9(登録商標)、およびTWEEN-80(登録商標))などが含まれる。
【0128】
本発明はその好ましい具体的な態様と関連して記載されていて、前記の説明および以下の例は本発明の範囲を例示することを意図するものであって、限定することを意図するものではない。本発明の範囲内のその他の局面、利点および改変は、本発明が属する技術分野における業者には明らかである。
【0129】
実施例1
カプセル製剤
カプセル製剤の各1カプセル中の次の成分を正確に秤量して、乳棒および乳鉢を用いて細かい均質な粉末まで粉砕した。これらの粉末を100メッシュの篩に通して、硬質ゼラチンカプセルに充填した。各カプセル製剤の組成は以下に示す。

【0130】
実施例2
シルデナフィル含有カプセル製剤
カプセル製剤の各1カプセル中の次の成分を正確に秤量して、乳棒および乳鉢を用いて細かい均質な粉末まで粉砕した。これらの粉末を100メッシュの篩に通して、硬質ゼラチンカプセルに充填した。各カプセル製剤の組成は以下に示す。
【0131】
実施例3
被験体は健康状態良好な40歳、白人男性であった。被験体は自身の性的活動に満足を示していたが、さらなる「持久力」を望んでいた。被験体は、性的活動を営む約1時間前に被験物である実施例2のカプセル製剤1の2カプセルを服用した。被験体は、自身の意志を働かせるだけでクライマックスを延期させることができたと報告した。被験体は、身体的に必要な動きを行うことができる限り、性交を続けることができたことを述べた。
【0132】
実施例4
被験体は健康状態の優れた31歳、白人男性であった。被験体は、性的活動の開始約3時間前に被験物である実施例1のカプセル製剤2の2カプセルを服用した。被験体は、自身の精力が著しく増強されて、中間の弛緩期間なく2回のクライマックスに到達したと報告した。
【0133】
実施例5
49歳の白人男性は会社を設立して複数の製品を製造し、会社のための資本を確保して製品を市場に出すための戦略を立てているところであった。彼は一日に長時間働くことが常であった。この患者は、ガールフレンドとの性的活動中に自分が射精を制御できず、その結果、女性パートナーから失望されたことに気が付いた。早漏の問題のために、この患者は可能な場合は必ず女性パートナーとの性的接触を避けるようにして、彼のパートナーは性的経験を満たされないと感じて立腹することもあった。この患者は実施例2の製剤1のカプセルを提供されて、性交の約3時間前に2カプセルおよび性交の約1時間前に1カプセルを服用するように勧められた。患者は最初の晩に2カプセルを服用して、彼の証言によると、彼は陰茎に若干のしびれを感じて、性交前の性行為を行うことによって女性パートナーの性的感情を起こさせることができて、しかも、彼のパートナーはほぼ30分間にわたって何ら射精を伴うことなく彼の陰茎で性交前の性行為を行うことができた。彼は20分を超えて性交することができて、彼のパートナーは疲労困憊を感じた。彼の女性パートナーは非常に恍惚状態であり、彼は同晩に2回の性交を行うことができた。患者は、そうしない場合にはストレス性の身体のために、良好で健全な性交を持ちたい場合には必ず定期的にカプセルを服用している。
【0134】
実施例6
ガールフレンドと同棲している白人男性、40歳は、彼の性的活動に対する本発明の組成物の効果に強い関心を持っていた。この被験体は18:00頃に実施例2の製剤1として記載されるカプセル2個を服用した。18:45に被験体は顔面の若干の潮紅感と共に生殖器の重感を報告した。性的活動は19:15に開始した。被験体は、通常経験するよりも勃起の硬直度が高く膨満感が増した感覚があったと報告した。被験体は、彼の通常の経験よりも70%ほど持久力が増したと報告した。性交は約90分間継続して、通常のオルガスムより力強く終了した。
【0135】
実施例7
クラスIIの男性に対するシルデナフィル、デキストロメトルファンおよびトラマドールの効果
クラスIIの男性の早漏を治療するためのシルデナフィル、トラマドールおよびデキストロメトルファン組成物の有効性を実証するために、早漏および勃起の問題を持つ21〜57歳の年齢群から30名の志願者を選択した。志願者に実施例2の製剤1のカプセルを与えた。志願者には性的行為の2〜3時間前に2カプセルを服用するように依頼して、性的行為の前後に表に示す書式に記入するよう依頼した。試験は8週間にわたって実施して、結果をまとめて性的満足について分析した。結果は、80%を超える志願者が早漏の問題に関して本発明の組成物に非常に満足したことを示している。
【0136】
次の刊行物は参照により本明細書に関連する部分に組み入れられる。
【0137】
参考文献
【図面の簡単な説明】
【0138】
【図1】通常の性交中のオルガスムレベルを示すグラフである。通常の性交中の男性および女性のオルガスムレベルが示される。オルガスムレベルは、性交中の身体的および感情的な興奮を示す恣意的な量である。
【図2】早漏の症例におけるオルガスムレベルを示すグラフである。早漏の症例における男性および女性のオルガスムレベルが示される。オルガスムレベルは、性交中の身体的および感情的な興奮を示す恣意的な量である。
【図3】トラマドール、キニジン、デキストロメトルファン、カフェインおよびシルデナフィルの化学構造を示す。
【背景技術】
【0001】
発明の背景
原始的な早漏は男性の最も一般的な性障害と見なされる。これは、ヒトの本能的要求の満足のために必要な性的調節を達成する能力の喪失を惹起し得る。最近、このような性的調節の喪失によって引き起こされる様々な症状を発現する症例数がかなり多いことが明らかとなっている。男性における早漏を原因とする性的な問題は、家庭内のひずみに加えて、自信の喪失に起因する無気力など社会的な難題に至る。早漏は、挿入前、挿入時または挿入直後の持続的または反復性の射精を含む。
【0002】
本来、女性は、少なくとも性的活動の開始時には男性よりも著しく弱く性交を経験するように進化している。従って、性交中に最大限まで緊張した全神経系の自然な弛緩を与えるオルガスムに達するためには女性はより長い時間をかけなければならない。今日まで、ヒトの性生活においては接触の感覚が重要な役割を担っている;性感帯は接触に特に敏感であり、それらの中でも先ず第一に、例えば、口腔、直腸、女性性器および乳頭の近くなど皮膚が粘膜と接する部位である。女性の性感帯は女性の身体表面全体であり得る。このような場合は、身体の任意の部分に接触することによって女性に扇情的な感情を惹起することが可能である。しかし、性感帯は陰核、小陰唇および膣などの厳密に限定された場所に局在する場合が一般的である。さらに、生殖器とは別に、多くのこのような過敏な部位がある。これらは唇、耳、瞼、首、乳頭などである。いくつかの場合には、これらの部位はそれらへの単なる接触によって女性にオルガスムを誘発し得るほど敏感である。
【0003】
しかし、男性の場合は、性感帯は専ら生殖器および隣接部分に限定される。従って、性体験のある男性パートナーが時にこれらの部位の探索において全くの探求の旅を試みざるを得ないということは驚くべきことではなく、これがなければ、誰も女性の性的反射の複雑な装置を活性化させることはできない。これが男性がしばしばオルガスムに達するために比較にならない短い時間を必要とする一つの理由であり、通常はだ男性自身のためばかりでなく彼のパートナーのためにも性交を終了する。性交の開始時に、男性は自身が既にあるレベルの興奮にあることを見出し、勃起にはこのことが不可欠であり、これがなければこの性交は全く不可能となる。オルガスム後直ちに射精に伴う萎縮が起こるため男性は彼のパートナーのことを考えて行為を継続することはできず、膣内でのさらなる摩擦は全て不可能である。
【0004】
理想的な性交は、膣内への陰茎の挿入後、双方の当事者が同時にオルガスムの境界に達して、それを交差させて、共に性交を終了することであろう(図1)。時にこれは、性交経験のある女性が行為開始時に逸した興奮を補ってその代わりに彼女のパートナーと共に最後のラインに達することができる場合に起こる。青年および中年男性における正常な射精の典型は陰茎の腟内への挿入後2〜6分で推移する。
【0005】
早漏は、現代人の性的行為において非常に頻繁に発生する。陰茎の腟内への挿入後すぐに、時には2〜3回の動作後に生じて(図2)、射精およびオルガスムが起こり、勃起が萎縮して性的行為が終了するという事実に関する。このような状況では明らかに女性は刺激されただけであり、発散の可能性は全くないはずである。明らかに、不十分な勃起によるかまたは早漏によるかに関わらず、任意の種類の男性のインポテンスの存在下において女性パートナーの性的満足および正常なリラクゼーションの可能性は全くないはずである。
【0006】
陰茎の勃起は3段階の永続可能なプロセスであり得る:1)血管拡張;2)内因性平滑筋弛緩物質の放出;および3)初期開始部位から遠位へのこれらの影響の連続。これは「カスケード効果」(Andersson et al 1995)と呼ばれている。パパベリンはアヘンアルカロイドであり、恐らくサイクリックGMPホスホジエステラーゼ阻害によって平滑筋弛緩剤として作用する。これは陰茎の血管系の筋系を弛緩させて、血流を増加させる(Papaverine Topical Gel Treatment For Erectile Dysfunction, Urology, Vol. 133(2)(1995), pp. 361-365)。インポテンスの治療において有用であることが見出されたもう一つの化合物は、陰茎への動脈流入量を増加させるように作用して、また静脈からの流出量を制限し得る天然の化合物であるプロスタグランジンE1である。プロスタグランジンE1は、陰茎で局所的に代謝されること、および低血圧などの全身症状を惹起する可能性が低いことから、インポテンスの治療のために注射として使用されるその他の化合物よりも好ましい。修飾血管組織として陰茎海綿体(ccp)は、通常の血管組織と同一の範囲のオートクリンおよびパラクリン調節物質を産生および分泌する。
【0007】
しかし、ccpの平滑筋緊張度は、血管壁と同じ方法で調節されるとは思われない。現在、ccpの緊張度または収縮性はアドレナリン作動性の制御によって調節されて、局所性にNOおよびエンドテリンを産生すると仮定されている。ccpにおいて、大半の試験はNO(Rajfer et al 1992; Burnett 1995)、血管作用性小腸ペプチド(VIP)、カルシトニン遺伝子関連ペプチド(CGRP)、ならびに通常およびccpの血管平滑筋に対しても同様の効果を持つ副交感神経支配の弛緩効果を観察することを目的としている。
【0008】
ccpへの血液の流入が類洞腔を結合させる正常な陰茎勃起中、小柱状組織は勃起を維持するために海綿体を取り巻く厚い線維組織に対して小さな海綿体静脈を圧迫する。血流量におけるこれらの変化を介在するために、シナプス後性副交感ニューロンおよびより軽度ではあるが、内皮細胞から一酸化窒素が放出されて、動脈および小柱状平滑筋においてαアドレナリン作動性ニューロンが阻害される。容易に拡散可能な一酸化窒素は、平滑筋細胞を弛緩させるためにグアニル酸シクラーゼによって海綿体内の増加するサイクリックグアノシン一リン酸(GMP)の形成を刺激する。
【0009】
最近、男性勃起不全の治療のためにシルデナフィルのクエン酸塩の経口使用が米国食品医薬品局(FDA)によって承認されている。シルデナフィルの組成物は最初に欧州特許EP 0463756において開示されて、米国または欧州の一国以外のその他の国ではシルデナフィルをカバーする物質特許(composition of matter patent)はない。シルデナフィルは、海綿体で形成されるサイクリックGMPを代謝する主なイソ酵素であるサイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)の選択的阻害剤であることが報告されている(Boolell et al 1996)。シルデナフィルは海綿体におけるPDE5の強力な阻害物質であるため、陰茎において、特に性的刺激と共に、海綿体の血流量を増加させることによって一酸化窒素の影響を増大させると考えられる。現在の推奨用量である25〜100mgのシルデナフィルが性的刺激の不在化では殆ど効果がない限り、シルデナフィルは性的刺激に対する自然の勃起反応を回復するのであってこのような刺激の不在下で勃起を惹起するものではないと考えられている(Goldstein 1998)。サイクリックGMPが平滑筋の弛緩を刺激する局所的なメカニズムは解明されていない。
【0010】
ヒトの男性における正常な射精機能は精液の噴出性の順行輸送を促進するための平滑筋および横紋筋の収縮の協調シーケンスを意味する。この過程は、陰茎幹から上位中枢への内陰部神経を介しての求心性神経刺激の伝達で始まる。射精反射を完了するためには遠心性の刺激が脊髄の脊柱から伝達されるが、下腹神経叢または交感神経叢を含むために腰部レベルから発する。下腸間膜動脈神経節からは、短いアドレナリン作動性節後線維が精嚢、精管膨大部および膀胱頚部で終結する。精嚢の交感神経支配は後部尿道への精液の放出に至る。適時の膀胱頚部閉鎖はこの精液ボーラスの逆流を防いで、この精液ボーラスは骨盤底の球海綿体筋および坐骨海綿体筋の間代性収縮によって順行性の方向に促される。 射精は中枢的な統合された末梢誘発反射であり、α1-アドレナリン作動性レセプター活性化の結果として生じる。早漏の治療のための有効な薬理学的薬剤は存在するが、それらは、例えばクロミプラミンおよびフェノキシベンザミンなど重度の副作用を被る。その他の治療は効果が限られている(メトクロプラミドなど)。
【0011】
デキストロメトルファン(よくDMとして略記される)は(+)-3-メトキシ-N-メチルモルフィナンの一般名である(図3)。これは咳嗽シロップとして広く使用されていて、Rodd 1960(論文の完全な引用は以下に示す)およびGoodman and Gilman's Pharmacological Basis of Therapeuticsなどの参照に記載されている。簡単に言うと、DMは、大半のオピエートの分子コアを形成するモルフィナン環構造の右旋性鏡像異性体(鏡像)を含む非常用性のオピオイドである。DMはシグマレセプターとして公知のニューロンレセプターの一種にて作用する。これらはしばしばシグマオピエートレセプターと記載されるが、それらがオピエートレセプターであるかどうかに関しては若干の疑問があり、非常に多くの研究者がそれらを単にシグマレセプターまたは高親和性デキストロメトルファンレセプターと呼ぶ。それらは抑制性レセプターであり、DMまたはその他のシグマアゴニストによるそれらの活性化が特定の種類の神経シグナルの抑制を引き起こすことを意味する。デキストロメトルファンは、興奮性アミノ酸(EAA)レセプターの一種であるN-メチル-D-アスパラギン酸(NMDA)レセプターとして公知のもう一つの種類のレセプターでも作用する。シグマレセプターでのそのアゴニストの活性とは異なり、DMはNMDAレセプターにおいてアンタゴニストとして作用して、これはDMがNMDAレセプターを介して調節される神経インパルスの伝達を抑制することを意味する。NMDAレセプターは興奮性レセプターであるため、DMのNMDAアンタゴニストとしての活性も特定の種類の神経シグナルの抑制を引き起こし、これがある種の咳嗽に関与する可能性がある。NMDAアンタゴニストとしての活性故に、DMおよびその代謝物の1つであるデキストロルファンは、卒中発作、心停止、および仮死などの事象によって引き起こされる虚血(低血流)および低酸素症(不十分な酸素供給)に誘発される特定の種類の興奮毒性脳損傷の可能性のある治療として積極的に評価されている。デキストロメトルファンおよびデキストロルファンの抗興奮毒性活性、ならびにこれらの薬剤によるNMDAレセプターの遮断については、Choi 1987、Wong et al 1988、Steinberg et al 1988、および米国特許第4,806,543号(Choi 1989)などの論文等で考察されている。デキストロメトルファンは、神経カルシウムチャネルにおいて活性を抑制することも報告されている(Carpenter et al 1988)。デキストロメトルファンおよびそれが相互作用するレセプターいついては、Tortella et al 1989、Leander 1989、Koyuncuoglu & Saydam 1990、Ferkany et al 1988、George et al 1988、Prince & Feeser 1988、Feeser et al 1988、Craviso and Musacchio 1983、およびMusacchio et al 1988においてより詳細に考察されている。
【0012】
DMは血流からかなり速やかに消失する(例えば、Vetticaden et al 1989およびRamachander et al 1977を参照されたい)。DMは肝臓においてO-脱メチル化と呼ばれる酵素過程によってデキストロルファンおよび3-メモキシモルフィナンと呼ばれる2つの代謝物に転換される;この過程では2つのペンダントメチル基の一つが水素によって置き換えられる。2つ目のメチル基が除去されると、形成される代謝物は5-ヒドロキシモルフィナンと呼ばれる。デキストロルファンおよび5-ヒドロキシモルフィナンは、尿血流を介して体内からかなり速やかに排出されるグルクロニドまたは硫酸抱合体を形成するために肝臓においてその他の化合物に共有結合性に結合する(主にグルクロン酸またはグルタチオンのようなイオウ含有化合物)。この酵素は、何年も前にデブリソキンのヒドロキシル化反応を実行することが発見されたことから、通常はデブリソキンヒドロキシラーゼと呼ばれる。これは様々な論文においてP450DBまたはP450-2D6とも呼ばれている。これは、数年前にスパルテインを代謝することが示されたスパルテインモノオキシゲナーゼと呼ばれる酵素と明らかに同一である;最近まで、科学者は単一のイソ酵素がデブリソキンおよびスパルテインの双方、ならびにデキストロメトルファンおよびその他の様々な基質の酸化を主に司ると思われることを認識しなかった。デブリソキンヒドロキシラーゼは「チトクロームP450」酵素、または「チトクロームオキシダーゼ」酵素として公知の酵素ファミリーに属する。化学物質の一酸素添加はチトクロームP450(P450)に帰するとされている。450nm付近に還元一酸化炭素吸収スペクトルの最大を示すモノオキシゲナーゼ酵素を含むこれらの血液タンパク質は、内因性および外因性化合物のヒドロキシル化を含む様々な酸化反応を触媒することが示されている(Jachau, 1990)。P450が酸素移動反応を触媒することができるメカニズムに関して膨大な量の研究が実施されている(Testa and Jenner, 1981;Guengerich, 1992;Brosen et al, 1990;Murray et al, 1990;およびPorter et al, 1991)。
【0013】
P450反応サイクルは簡単に言うと次のように進行する:三価鉄型のチトクロームへの基質分子(RH)の最初の結合によって二元複合体が形成されて、三価鉄酵素のスピン平衡が低スピンから高スピン状態へ推移する。この立体配置は、二価鉄のP450−基質複合体を形成するためにフラボタンパク質レダクターゼから電子をより容易に受け取ることを示唆するいくつかの証拠が示されている。しかし、必ずしも全てのP450が高スピン含有量と還元速度の相関性を示す訳ではない。実際、P450基質の性状、酵素/基質複合体のトポグラフィー、および酸化可能原子の電位を含む複数の要因がそれぞれ還元速度の調節において役割を果たすことが提唱されている。分子の酸素は、二価鉄酸素錯体を形成するために、後にP450レダクターゼからの(または、多分、いくつかのケースでは、チトクロームb5およびそのレダクターゼを介して還元されたニコチンアミドアデニンジヌクレオチドからの)第二の電子によって還元される二価鉄P450−基質複合体に結合する。還元型二価鉄酸素錯体の二酸素結合が開裂すると、酸素の一方の原子は基質に挿入されて、もう一方の酸素原子は水に還元されて、二価鉄血液タンパク質は元に戻る。
【0014】
P450酵素ファミリーの個々のメンバーおよび関連する混合機能オキシダーゼ活性は、脳、副腎、腎、精巣、卵巣、肺および皮膚を含む肝外組織において記載されている。個々のP450は、同様に選択された科学的分類による誘導能に関して特徴付けられている。P450 1A1および1A2サブファミリーのような特異的P450酵素の誘導は、mRNA転写および酵素活性の発現の増加の調節過程に関して幅広く研究されている。ベータ-ナフタフラボン(beta-NF)、3-メチルコラントレン(3-MC)、アロクロール1254(ACLR)、および2,3,7,8-テトラクロロジベンゾ-p-ダイオキシン(TCDD)のような材料は指定されたP450 1Aサブファミリーを持つP450酵素の誘導物質として分類されている材料である(Murray et al, 1990;およびGuengerich, 1989)。
【0015】
多くの化合物がデブリソキンヒドロキシラーゼ(スパルテインモノオキシゲナーゼ)イソ酵素の活性を阻害する(Inaba et al 1985)。これらの阻害物質の最も強力なものはキニンの右旋性立体異性体であるキニジンであり(図3)、これは通常は不整脈を治療するために用いられる。Inaba et al(1986)およびNielsen et al(1990)はキニジンがインビボにおける動物試験においてスパルテインの酸化を阻害する能力について考察していて、またBrinn et al(1986)、Brosen et al(1987)およびBroly et al(1989)はキニジンが肝細胞調製物中でDM代謝を阻害する能力について考察している。例外的に強力であり、かつ、容易に実証されるデブリソキンヒドロキシラーゼの阻害に加えて、その他のチトクロームP450イソ酵素も、様々なレベルの結合親和性で、キニジンに抑制される可能性が高い。従って、キニジンはデブリソキンヒドロキシラーゼに対してその最も顕著な効果を発揮するものの、同様に多くのその他のチトクロームP450酵素を抑制し、それによって患者は正常かつ望ましい肝臓の活性をより全般的に喪失する可能性が高い。デキストロメトルファンの主たる酸化代謝産物はデキストロルファンであり、これは神経学者の間ではデキストロメトルファンと全く同じ方法で活性であると広く信じられている;報告によると、双方の薬剤はシグマアゴニスト、NMDAアンタゴニストおよびカルシウムチャネルアンタゴニストである。デブリソキンヒドロキシラーゼを阻害する化合物のDMと組み合わせた投与は、DMのみを投与される患者に比して患者の血液中のDMの濃度および安定性の大幅な増大が惹起される;またDMと組み合わせたデブリソキンヒドロキシラーゼ阻害剤の投与はヒトにおけるDMの検出可能な作用に対して明確で実質的な影響を有する。
【0016】
トラマドールは、(+/-)-トランス(RR,SS)-2-[(ジメチルアミノ)メチル]-1-(3-メトキシフェニル)シクロヘキサノールの化学名を持ち、文献中ではしばしばシス(RS,SR)ジアステレオマーとして誤記される。トラマドールは、オピエート由来でなく、NSAIDでもない中枢作用性の二成分鎮痛薬である。これは、骨関節炎および術後鎮痛のような慢性疼痛の状況、ならびに歯痛などの急性疼痛において中等度の疼痛を管理するために用いられる。
【0017】
トラマドールはラセミ化合物であり、等量の(+)-および(-)-鏡像異性体からなる。トラマドールの純粋な鏡像異性体はラセミ体に比して異なる薬学的プロフィールおよび効果を持つことが公知である。(+)-鏡像異性体はμ-オピエートレセプターとの結合によるオピエート様鎮痛作用によって識別されて、双方の鏡像異性体は5-ヒドロキシトリプタミン(セロトニン)およびノルアドレナリン(ノルエピネフリン)の再取り込みを阻害し、これはトラマドールラセミ混合物のそれよりも強力であり、(-)-鏡像異性体ではノルアドレナリン再取り込みの明確な阻害が観察される。(+)-および(-)-トラマドールについては、モデルに応じて、2つの鏡像異性体がそれらの個々の作用を相互に強化および促進することが示されている(Raffa, R. et al., 1993;Grond S et al, 1995、およびWiebalck A et al., 1998)。トラマドールの強力な鎮痛作用が鏡像異性体の作用のこの相互に依存性の強化に基づくと結論づけられることは明らかである。トラマドールの主な活性代謝物であるO-デスメチルトラマドール(M1)はμ-オピエートレセプターに対して高い親和性を示して親薬剤の少なくとも2倍の鎮痛強度を有する。O-デスメチル-N-モノ-デスメチルトラマドール(以下の本文のいくつかの場所および文献中ではM5と記載される)はトラマドールのインビボにおける代謝物である(1RS,2RS)-2[ジメチルアミノ)メチル]-1-(3-メトキシフェニル)シクロヘキサノールの一つとして公知である(Lintz et al., 1981)。中枢神経系に対する影響、例えば鎮痛作用は脳室内投与の場合よりも静脈内投与において明らかに低いことから、M5はごく限られた程度で血液-脳関門を通過する。
【0018】
トラマドールは化学的にはオピエートと無関係であるという事実にも関わらず、トラマドールの投与に付随する有害な副作用は、高用量で用いられた場合には、オピエートのそれとほぼ同じである。
【0019】
カフェインはコーヒーアラビカ(Coffea arabica)またはコーヒーノキ(coffee plant)の葉および種子、ならびにテアシネンシス(Thea sinensis)またはチャ(tea)の葉から得られるアルカロイドである。カフェインはメチル化されたキサンチンであり、化学的には3,7-ジヒドロ-1,3,7-トリメチル-1H-プリン-2,6-ジオンと示される(図3)。カフェインは天然で生じるが、市販用薬品の用途のために合成的に調製される。カフェインは世界中で最も広い活性物質である。成人による平均カフェイン摂取量は異なる文化および国の間で一日一人あたり80から400mgまで様々である(Daly 1998)。カフェインは、覚醒状態の亢進、精神運動反応時間の短縮、ならびに睡眠潜時および起きている時間の増大を含む多くの薬理学的反応を引き起こして、知的活動に影響する可能性もある(Nehlig 1992)。さらに、カフェインは平滑筋の弛緩を惹起して、胃酸の分泌およびカテコールアミンの放出を促進し、代謝活性を高める(Fredholm 1999)。
【0020】
カフェインは本質的には無毒性である。FDAは、この化合物の過量投与の結果として致命的なカフェイン中毒がこれまで報告されていないことを指摘している。成人におけるカフェインの短期致死量は5〜10gである。中等度の用量ではカフェインはヒトの胎児においてほとんどまたは全く発生毒性のリスクを持たない。カフェインの摂取が癌の発生または冠動脈心疾患の発生率の増大に必然的に関連することを示す証拠はない。カフェインは経口、経直腸または非経口投与後に容易に吸収される。1時間以内に最大血漿中濃度に到達する。カフェインの血漿中半減期は3〜7時間である。
【0021】
カフェインはFDAの刺激物質の基準を満たす、医師の処方なしで購入できる唯一の刺激物質である。FDAは、カフェインは安全かつ有効であるということで意見が一致している。推奨用量は100〜200mgであり、3または4時間毎よりもより頻繁に投与されるべきではない。FDAは、多くのコーヒー抽出物の刺激性とは対照的に、カフェイン自体は通常の用量において胃腸管の刺激を惹起しないと述べている。これは、薬剤が刺激特性のために使用される場合に有利である。FDAは、その刊行物内で、ヒトが眠気を催しているまたは疲労している場合にカフェインが覚醒を回復することは証拠によって証明されていると述べている。
【0022】
ホスホジエステラーゼの阻害はカフェインの作用に寄与し得るが(Daly 1998)、このキサンチンの大半の薬理学的作用はA1、A2A、A2BおよびA3サブタイプと命名されたアデノシンレセプターの拮抗作用に起因すること(Fredholm 1999)を示す証拠が集まりつつある。カフェインはA2Aレセプターにおいて最も強力に作用して、A1レセプターがこれに次いで近く、続いてA2Bレセプター(Klotz 1998; Ongini 1996)であり、ヒトA3レセプターでは弱いアンタゴニストとして作用する。カフェインによるアデノシンレセプター、つまり、A1およびA2Aレセプタータイプの遮断は、様々な生理学的過程に対する内因性アデノシンの作用を阻害する(Fredholm 1995)。通常の条件下において、アデノシンの血中量は血小板におけるA2Aレセプターを緊張性に活性化するために十分であると思われる。近年、A2Aレセプターノックアウトマウスでは血小板凝集が増加することが報告されて、血小板機能におけるこのレセプターサブタイプの重要性を示している(Ledent 1997)。従って、カフェインは血小板におけるこれらの緊張性に活性化されるA2Aレセプターを遮断して、アデノシンによって介在されるそれらの機能を変化させることができると考えられる。
【0023】
長年、コーヒーを飲むことと心臓血管疾患、特に冠動脈心疾患との関連性が疑われているが、最近ではコーヒーまたはカフェインの摂取は冠動脈心疾患または卒中発作のリスクを増大させないことが実証されている(Grobbee 1990;Jee 1999)。
【0024】
カフェインは複数の鎮痛調製物に含まれる。これが全く合理的である限り、それは痛覚過敏を引き起こす知覚神経終末または終末近くのアデノシンA2Aレセプターの存在に関連し得る(Ledent et al., 1997)。実際、カフェインは特定の種類のC線維介在性疼痛において痛覚鈍麻効果を持つ(Myers et al., 1997)。鎮痛効果は小さい(Battig and Welzl, 1993)。しかし、疼痛の条件下において、カフェインは気分および頭の冴えを高めることによって間接的な有利な効果を持つことができる(Lieberman et al., 1987)。この試験では、気分および覚醒状態の双方が、アスピリンの単独投与またはプラセボよりもカフェインと併用したアスピリンによってさらに改善されることが見出された。様々な量のカフェインと併用して、鎮痛薬であるアスピリン、アセトアミノフェンおよびフェナセチンの1つまたは複数を含む組成物がこれまでに販売されている。複数のケースでは、非麻薬性鎮痛薬/カフェインの併用製剤が麻薬性鎮痛薬であるコデイン、プロキシフェンまたはオキシコドンの1つをさらに含んでいる。これらの組み合わせの例には、Excedrin(商標)、SK-65(商標)、Darvon(商標)、Anacin(商標)として、またコデインであるTabloid(商標)のブランドで市販されている公知の製剤が含まれる。
【0025】
カフェインが特定の種類の疼痛において鎮痛特性を持つ可能性があるということは排除できず、これは頭痛の症例であり得て(Ward et al., 1991)、二重盲検条件下においてカフェインによって著しく、かつ、用量依存性に頭痛が軽減する。効果はアセトアミノフェンのそれとほぼ等しく、それはしばしばカフェインと組み合わせられて、気分に対する影響または自己報告されたコーヒー摂取との関連性は示されなかった。総評(Migliardi et al., 1994)の通り、患者は頭痛の治療に関してカフェインフリーの調製物よりもカフェインを含有する鎮痛薬を優れていると判定する。さらに、カフェインは、i.c.v.注射後にマウスにおいて疼痛関連性の行動を拮抗することができることから(Ghelardini et al., 1997)、脳において抗侵害受容効果を発揮する可能性がある。さらに、この効果はコリン作動性伝達を抑制するアデノシンA1レセプターの緊張性阻害活性の拮抗作用に関連する可能性がある(Rainnie et al., 1994; Carter et al., 1995を参照)。
【0026】
上記の通り、睡眠はヒトにおけるカフェインの影響に最も敏感な生理学的機能の1つであると想われる。就寝時に摂取するカフェインは睡眠に対してネガティブに作用することは周知である(Snel, 1993を参照されたい)。一般に、睡眠に対して顕著な影響を及ぼすためには200mgを超えるカフェインが必要である。最も顕著な影響は、総睡眠時間の短縮、睡眠潜時の延長、最初の軽睡眠EEG段階の増加、およびその後の熟睡EEG段階の減少、さらに睡眠段階間の推移回数の増加である。
【0027】
現在、早漏の優れた治療は、Master & Johnsonプロトコルに基づく行動二チームセックス療法、または個々の心理療法としての心理療法である(Rifelli and Moro. Sessuologia Clinica. Bologna, 1989)。これまでの早漏治療の方法には、心理療法、局所麻酔、および装置の使用が含まれる(米国特許第5,535,758号、第5,063,915号、第5,327,910号、および第5,468,212号)。これらの方法はすべて、重大な欠点を持ち得る。心理療法は一部の患者においてのみ有利であり、特に遠隔地域ではすべての患者に対応できないかもしれない専門のセラピストが必要である。さらに、心理療法は非心理学的原因に起因する早漏は緩和することができない。麻酔性物質は組織の感受性を低下させて、それによって性的満足を減じる。また、局所麻酔薬は性的パートナーに伝達されて、それによってパートナーの感受性および満足をも減少させる可能性がある。装置に関しては、これらは使いにくくて不便であり使用にまごつく可能性がある。装置は非常に目に付きやすく、苦しんでいるパートナーが隠したがり得るその状態を暴露する。さらに、装置は一方または双方のパートナーに刺激を引き起こす可能性がある。
【0028】
複数の異なる抗うつ剤化合物の全身投与による早漏を治療するための方法が述べられている(米国特許第4,507,323号、第4.940,731号、第5,151,448号、および第5,276,042号;PCT公報W095/13072)。しかし、これらの薬剤はすべての患者には有効でない可能性があり、これらの薬剤の副作用によって治療が中止となったり患者のコンプライアンスが損なわれたりすることがある。疾患の状態またはその他の薬剤との有害な相互作用はこれらの化合物の使用に禁忌を示す場合があり、または射精の開始を遅延させるためには有効でないかもしれない低用量を要求する場合がある。さらに、抗うつ剤療法に関連する精神的不健康の徴候が患者にこのような治療の開始または継続に対して二の足を踏ませる可能性がある。抗うつ剤であるフルオキセチンの投与は早漏を治療するために特許請求されている(米国特許第5,151,448号)。しかし、フルオキセチンの投与は多くの望ましくない局面を持ち得る。肝または腎機能障害を持つ患者は、肝での代謝および腎を介しての排出のために、フルオキセチンを使用できない可能性がある。フルオキセチン投与中に肺、腎または肝に関わる全身性の事象が発生していて、過量投与から死亡が発生している。さらに、経口フルオキセチン投与の副作用には、脱毛、悪心、嘔吐、消化不良、下痢、食欲不振、不安、神経過敏、不眠症、傾眠状態、疲労、頭痛、振戦、浮動性めまい、痙攣、発汗、そう痒症、および皮疹が含まれる。フルオキセチンは、しばしば、肝によるそれらの代謝を障害することによって、一連の薬剤と相互作用する。
【0029】
米国特許第4,940,731号は、早漏の治療のためのセルトラリンの経口または非経口投与について記載する。セルトラリンはフルオキセチンと同一の問題の多くを共有することが認識されている(Martindale, The Extra Pharmacopoeia, 31st edition, at p. 333 (London: The Royal Pharmaceutical Society, 1996)を参照されたい)。セルトラリンは肝で代謝されて、尿および糞便中に排出される。従って、肝硬変の患者は低用量を使用しなければならないし、腎機能不全の患者にセルトラリンを投与する際には注意しなければならない。モノアミンオキシダーゼ阻害剤を使用中の個体は、記憶の変化、錯乱、易刺激性、悪寒、発熱および筋固縮に至る毒性のリスクがあるため、セルトラリンを使用できない。セルトラリン経口投与に起因する副作用には、悪心、下痢、消化不良、不眠症、傾眠、発汗、口内乾燥、振戦および躁病が含まれる。昏睡、痙攣、便失禁および女性化乳房の稀な事例が、セルトラリン療法を受けている患者で発生している。米国特許第5,276,042号は、早漏の治療のためのパラキセチンの投与について記載している。パラキセチンは主に尿中に排出されて、肝および腎機能障害を持つ患者では低用量が推奨される。セルトラリンと同様にパラキセチンも、モノアミンオキシダーゼ阻害剤の投与を受けている患者には与えられない。パラキセチンの経口投与による副作用には、低ナトリウム血症、無力症、発汗、悪心、食欲減退、中咽頭の疾患、傾眠、浮動性めまい、不眠症、振戦、不安、ツチ骨の可動性障害、脱力および錯感覚が含まれる。従って、専門の心理療法を必要とせず、便利に、かつ、戸惑うことなく使用することができて、しかも事前の治療方法に関連する問題に関与しない早漏治療の方法が必要である。
【0030】
米国特許第6,037,360号は、様々なセロトニンアゴニストおよびアンタゴニストの投与が早漏の治療に有効であることを開示している。セロトニン阻害剤を用いた治療中に最も頻繁に発現する有害作用は、例えば、悪心、下痢/軟便、便秘などの胃腸障害である。(Drugs 43 (Suppl. 2), 1992)。悪心は、発生率の点で主たる有害作用である。さらに、セロトニン阻害剤の投与後に患者が消化不良に苦しむことは頻繁に観察されている。
【0031】
米国特許第5,707,999号は、2つの特異的α1-遮断薬であるアルフゾシンおよびテトラゾシンが心因性早漏の治療に有効であり、該薬剤は心理療法からの利点がないことが証明された患者において有効であることが明らかとなったことを示す。しかし、テラゾシンおよびその類似体は頭痛、悪心、体重増加、浮動性めまい、傾眠、呼吸困難および霧視を含む複数の副作用を持つ。
【0032】
米国特許第6,037,346号は勃起不全の治療のためのホスホジエステラーゼ阻害剤の局所投与を開示して、好ましい投与モードは経尿道として特許請求される。薬学的製剤およびキットも示されている。米国特許出願第2002/0037828 Al号は、早漏治療のためのホスホジエステラーゼ阻害剤の使用を開示する。
【0033】
米国特許第4,656,177号および第4,777,174号は、非麻薬性鎮痛剤/非ステロイド系抗炎症剤および/または麻薬鎮痛剤ならびにカフェインの組み合わせを開示する。組成物は、疼痛緩和物質が単独で投与される場合よりもより強力かつより速やかな鎮痛反応を示す。
【0034】
米国特許第4,777,174号は、非麻薬性鎮痛剤/非ステロイド系抗炎症剤および/または麻薬鎮痛剤ならびにカフェインの組み合わせを開示する。組成物は、疼痛緩和物質が単独で投与される場合よりもより強力かつより速やかな鎮痛反応を示す。
【0035】
米国特許第5,248,678号は、カフェインなどのアデノシンレセプターアンタゴニストおよびギャバペンチンなどのGABAアゴニストの有効量を患者に投与する工程を含む、昏睡状態の患者または近昏睡状態の患者の覚醒(arousal)覚醒状態(alertness)を増大させる方法を示す。
【0036】
これまで、トラマドールなどのμ-オピエート鎮痛薬およびデキストロメトルファンの鎮痛促進量の組み合わせ、またはもっと詳しく言うならば、その他の任意のNMDAレセプターアンタゴニストがヒトにおいて早漏を治療するために効果的に用いることができるという認識または評価はなかった。さらに、これまで、トラマドールなどのμ-オピエート鎮痛薬、サイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)阻害剤、およびデキストロメトルファンの鎮痛促進量の組み合わせ、またはもっと詳しく言うならば、その他の任意のNMDAレセプターアンタゴニストがヒトにおいて早漏を治療するために効果的に用いることができるという認識または評価はなかった。さらに、これまで、トラマドールなどのμ-オピエート鎮痛薬、サイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)阻害剤、カフェイン、およびデキストロメトルファンの鎮痛促進量の組み合わせ、またはもっと詳しく言うならば、その他の任意のNMDAレセプターアンタゴニストがヒトにおいて早漏を治療するために効果的に用いることができるという認識または評価はなかった。
【0037】
従って、本発明の目的は、現在有効な治療法に付随する有害な副作用を伴わずに提供する早漏の治療のための方法および組成物を提供することである。驚くべきことに、現在、デキストロメトルファンなどの無毒性NMDAレセプターアンタゴニストとトラマドールなどのμ-オピエート鎮痛薬の組み合わせが早漏に対して顕著な対症効果を示すことが見出されている。驚くべきことに、現在、デキストロメトルファンなどの無毒性NMDAレセプターアンタゴニストとトラマドールなどのμ-オピエート鎮痛薬およびシルデナフィルのようなサイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)の組み合わせも早漏に対して顕著な対症効果を示すことが見出されている。本発明のこれらおよびその他の目的および特徴は後続の説明から明らかとなるであろう。
【発明の開示】
【0038】
発明の概要
一つの局面において、本発明はヒトまたはその他の哺乳動物における性機能不全を効果的に治療する方法を提供する。方法は、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、およびb)μ-オピエートレセプターのアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニストであるμ-オピエート鎮痛薬または薬学的に許容されるその塩を含むある量の作用物質を、このような治療を必要とする患者に投与する工程を含む。組み合わせた量の作用物質は性機能不全を治療するために有効である。
【0039】
本発明に従って、性機能不全は早漏または早漏を状態の要素として含む性機能不全であり得る。
【0040】
作用物質は別々に、または組み合わせて投与することができる。3つまたはそれよりも多い作用物質が関与する場合、作用物質は様々な組み合わせで投与することができる。例えば、3つの作用物質は一緒に投与して良く、または2つの作用物質を一緒に投与して第三の作用物質は別に投与されても良い。
【0041】
作用物質は好ましくは性的活動の前に投与される。投与は、経口的に、インプラントによって、非経口的に、皮下で、舌下で、経直腸的に、局所的にまたは吸入を介して行われ得る。好ましい態様において、作用物質は経口的に投与される。
【0042】
NMDAレセプターアンタゴニストは、デキストロメトルファン、デキストロルファン、ケタミン、アマンタジン、メマンチン、エリプロジル、イフェンプロジル、フェンシクリジン、MK-801、ジゾシルピン、CCPエン、フルピルチン、またはそれらの誘導体もしくは塩であり得る。好ましくは、アンタゴニストはデキストロメトルファンである。
【0043】
μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストは、(1R,2Rまたは1S,2S)-(ジメチルアミノメチル)-1-(3-メトキシフェニル)-シクロヘキサノール(トラマドール)、そのN-酸化物誘導体(「トラマドールN-酸化物」)、およびそのO-デスメチル誘導体(「O-デスメチルトラマドール」)、またはそれらの混合物、立体異性体もしくはラセミ体の任意の1つであってよい。好ましい態様において、μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストはトラマドールである。
【0044】
作用物質は、タブレット、経口投与のための多粒子(multiparticulate)製剤;経口投与のための液剤、徐放性製剤、懸濁剤もしくはエリキシル剤、注射可能製剤、移植可能な装置、局所用調製物、固体状態および/もしくはデポー型の経皮送達装置、坐剤、口腔内タブレット、または副鼻腔に吸入もしくは注入されるように意図された放出制御粒子製剤、もしくはスプレー、ミストもしくはその他の局所ビヒクルなどの吸入製剤の剤形として投与することができる。剤形は、タブレットまたはカプセルとして製剤化される固形経口剤形としてさらに限定されてもよい。
【0045】
本発明に従って、μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合は、約15:1〜1:15、約10:1〜1:10、約5:1〜1:5、または約1:2であり得る。
【0046】
本発明の特定の態様において、ホスホジエステラーゼ(PDE)阻害剤または薬学的に許容されるその塩が作用物質の1つとして含まれる。好ましくはPDE阻害剤は5型ホスホジエステラーゼ阻害剤である。PDE阻害剤は、シルデナフィル、アミノフィリン、テオフィリン、アムリノン、ミルリノン、ベスナリノン、ビンポセチン、ペモベンダン、シロスタミド、エノキシモン、ペロキシモン、ロリプラム、R020-1724、ザニプラスト、ジピリダモール、MY5445もしくはIC-351、または薬学的に許容されるその塩であり得る。NMDAレセプターアンタゴニスト対ホスホジエステラーゼ阻害剤対μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストの割合は、約90:1:1〜1:90:1〜1:1:90であって良い。
【0047】
特定の態様において、チトクロームP450阻害剤または薬学的に許容されるその塩は作用物質の1つとして含まれる。好ましくはチトクロームP450阻害剤はデブリソキンヒドロキシラーゼ阻害剤である。阻害剤は、キニジン、キニン、ナフチリジン、キサンチン、フェノキシアミノアルカン、カルバモイルイミダゾール、グアニジンイミダゾール、シメチジン(N-シアノ-N'-メチル-N"-[2[[(5-メチル-1H-イミダゾール-4イル)メチル]チオ]エチル]グアニジン)、キノリン、クロロキン(7-クロロ-4-(4-ジエチルアミノ-1-メチルブチルアミノ)キノリン)、プリマキン(8-(4-アミノ-1-メチルブチルアミノ)-6-メトキシキノリン)、トリフルオロメチルオキシムエーテル、5-メトキシ-1-[4-(トリフルオロメチル)-フェニル]-1ペンタノンO-(2-アミノエチル)オキシムとしても公知であるフルボキサミン、または薬学的に許容されるその塩であることができる。NMDAレセプターアンタゴニスト対チトクロームP450阻害剤対μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストの割合は、約90:1:1〜1:90:1〜1:1:90であって良い。
【0048】
特定の態様において、カフェインは作用物質の1つとして含まれる。
【0049】
本発明のさらなる態様において、ホスホジエステラーゼ阻害剤およびチトクロームP450阻害剤の双方が作用物質として含まれる。本発明のさらなる態様において、ホスホジエステラーゼ阻害剤およびカフェインの双方が作用物質として含まれる。本発明のさらなる態様において、カフェインおよびチトクロームP450阻害剤の双方が作用物質として含まれる。本発明のさらなる態様において、ホスホジエステラーゼ阻害剤、カフェイン、およびチトクロームP450阻害剤の双方が作用物質として含まれる。
【0050】
もう一つの局面において、本発明は作用物質の組み合わせの治療上有効量を含む薬学的組成物を提供する。組み合わせは、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)V型ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩を含む。特定の態様において、薬学的組成物はカフェインを作用物質としてさらに含む。
【0051】
本発明は、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)チトクロームP450阻害剤または薬学的に許容されるその塩を含む、作用物質の組み合わせの治療上有効量を含む薬学的組成物も提供する。特定の態様において、薬学的組成物はカフェインを作用物質としてさらに含む。
【0052】
本発明は、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、c)V型ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩、およびd)チトクロームP450阻害剤または薬学的に許容されるその塩を含む、作用物質の組み合わせの治療上有効量を含む薬学的組成物をさらに提供する。特定の態様において、薬学的組成物はカフェインを作用物質としてさらに含む。
【0053】
もう一つの局面において、本発明は作用物質の組み合わせの治療上有効量を含む薬学的組成物を提供する。組み合わせは、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)カフェインを含む。
【0054】
本発明に従って、本出願人らは、これまでに、早漏の治療においてヒトに投与する組成物(組み合わせおよび製剤)を開発した。これらの組成物(組み合わせおよび製剤)は複数の無毒性有効投与量を用いるか、組み合わせるか、または組み入れる(場合によって)。各投与量は、例えば、例えばトラマドール(またはその塩)であるμ-オピエート鎮痛薬の薬剤の無毒性有効投与量、デキストロメトルファン(好ましくはデキストロメトルファン水和物またはその塩)のようなNMDAレセプターアンタゴニストの無毒性有効投与量、、PDE5阻害剤(例えば、シルデナフィル)、および任意で例えばキニジンのようなチトクロームP450阻害剤(好ましくはキニジン水和物またはその塩)の無毒性有効投与量を含む。
【0055】
本出願人らは、早漏および勃起の治療においてヒトに投与される組成物(組み合わせおよび製剤)も開発した。これらの組成物(組み合わせおよび製剤)は、多くの無毒性有効投与量を用いるか、組み合わせるか、または組み入れる(場合によっては)。各投与量は、例えばシルデナフィル(またはその塩)であるサイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)を阻害する薬剤の無毒性有効投与量、デキストロメトルファン(好ましくはデキストロメトルファン水和物またはその塩)のようなNMDAレセプターアンタゴニストの無毒性有効投与量、カフェイン、および例えばトラマドール(またはその塩)のようなμ-オピエート鎮痛薬の無毒性有効投与量を含む。
【0056】
従って、本発明のさらなる局面は、トラマドールのようなμ-オピエート鎮痛薬、ヒトにおける抗興奮毒性活性に関与するデキストロメトルファンのようなNMDAレセプターアンタゴニスト、および任意でキニジンのようなチトクロームP450阻害剤の組み合わせはヒト男性において射精の開始の遅延に極めて有効であることの開示である。
【0057】
本発明のさらにもう一つの局面に従って、本出願人らは、それぞれが経口または非経口投与に適した薬学的賦形剤と共に作用物質の治療上有効量を含む多くの投与量を含む薬学的組成物(組み合わせおよび製剤)を提供している。量は、患者に対して無毒性である様式においてヒト男性における早漏の疾患および状態を治療するおよび緩解を助けるために有効である。作用物質の治療上有効投与量には、例えばトラマドールであるμ-オピエート鎮痛薬、ならびにデキストロメトルファンならびに/またはその塩(例えば、臭化水素酸塩)、および/もしくはその同族体、類似体、誘導体、複合体、プロドラッグ、エステル、および/もしくは断片などのNMDAレセプターアンタゴニストの無毒性有効投与量、ならびに任意で例えばキニジン(好ましくは、キニジン水和物またはその塩)のようなチトクロームP450阻害剤の無毒性有効投与量が含まれる。
【0058】
本発明のもう一つの局面は、各々の薬理学的活性物質が経口的に投与される方法を提供することである。本発明のさらなる局面は、各々の薬理学的活性物質が非経口的に投与される方法を提供することである。
【0059】
本発明は、早漏を治療するための方法であって、このような治療を必要とする個体にトラマドールのようなμ-オピエート鎮痛薬、デキストロメトルファンのようなNMDAレセプターアンタゴニスト、および任意でナフチリジン、キサンチン、フェノキシアミノアルカン、カルバモイルイミダゾール、シメチジン(N-シアノ-N'-メチル-N"-[2[[(5-メチル-1H-イミダゾール-4イル)メチル]チオ]エチル]グアニジン)のようなグアニジンイミダゾール、クロロキン(7-クロロ-4-(4-ジエチルアミノ-1-メチルブチルアミノ)キノリン)およびプリマキン(8-(4-アミノ-1-メチルブチルアミノ)-6-メトキシキノリン)のようなキノリン、5-メトキシ-1-[4-(トリフルオロメチル)-フェニル]-1ペンタノンO-(2-アミノエチル)オキシムとしても公知であるフルボキサミンのようなトリフルオロメチルオキシムエーテルなどのチトクロームP450の酸化的活性を阻害する作用物質を含む薬学的製剤を投与する工程を含む方法を提供する。薬学的製剤の投与は、作用物質が早漏の治療において有効であるように予め定められた投与計画の状況において実施される。薬剤送達は、経口、非経口、口腔、経直腸、局所、経皮、経尿道および海綿体内注射を含む、早漏からの緩和を示すために有効な任意の経路を介して実施してよい。
【0060】
本発明に従って、本発明の方法を実施するための薬学的製剤が提供される。薬学的製剤は、有効量のトラマドールのような特定のμ-オピエート鎮痛薬、デキストロメトルファンのようなNMDAレセプターアンタゴニスト、任意でチトクロームP450阻害剤、薬学的に許容される担体またはビヒクル、および任意で(即ち、局所、経皮または経尿道製剤の場合)エンハンサーを含む。例えば、賦形剤、界面活性剤、保存料(例えば、抗酸化剤)、安定剤、酵素阻害剤、キレート化物質など、その他の種類の成分も製剤に組み入れることができて、これは薬学的製剤の調製および薬剤送達の当業者によって理解されるであろう。
【0061】
本発明のさらにもう一つの局面は、性的刺激下でヒトにおいて陰茎の勃起を促進するシルデナフィルのようなサイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)阻害剤、ヒトにおいて抗興奮毒性活性に関与するデキストロメトルファンのようなNMDAレセプターアンタゴニスト、トラマドールのようなμ-オピエート鎮痛薬、および任意でキニジンのようなチトクロームP450阻害剤の組み合わせが勃起および射精の問題を持つ男性において射精の開始の遅延に非常に有効であることを示す開示である。
【0062】
従って、本発明のさらにもう一つの局面に従って、本発明者らは、各々が、経口または非経口投与に適した薬学的賦形剤と共に、例えばシルデナフィルなどのサイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)を阻害する薬剤の治療上有効な(患者に対して無毒性でありヒト男性における早漏の疾患および状態を治療するおよび緩解を助けるための)投与量、ならびにデキストロメトルファンおよび/またはその塩(例えば、臭化水素酸塩)および/もしくはその同族体、類似体、誘導体、複合体、プロドラッグ、エステル、および/もしくは断片などのNMDAレセプターアンタゴニストの無毒性有効投与量、ならびにトラマドールおよび/またはその塩(例えば、臭化水素酸塩)および/もしくはその同族体、類似体、誘導体、複合体、プロドラッグ、エステル、および/もしくは断片などのμ-オピエート鎮痛薬の無毒性有効投与量を含む複数の投与量を含む薬学的組成物(組み合わせおよび製剤)を提供している。薬剤送達は、経口、非経口、口腔、経直腸、局所、経皮、経尿道および海綿体内注射を含む、早漏からの緩和を示すために有効な任意の経路を介して実施してよい。
【0063】
チトクロームP450阻害剤を含む組成物と同様に、PDE5阻害剤を含む組成物は薬学的に許容される担体またはビヒクル、および任意で(即ち、局所、経皮または経尿道製剤において)エンハンサーも含むことができる。例えば、賦形剤、界面活性剤、保存料(例えば、抗酸化剤)、安定剤、酵素阻害剤、キレート化物質など、その他の種類の成分も製剤に組み入れることができて、これは薬学的製剤の調製および薬剤送達の当業者によって理解されるであろう。
【0064】
本発明の実践において、NMDAアンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩は、NMDAアンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩の投与間隔が、トラマドールまたはその他のμ-オピエートアゴニストもしくはアゴニスト/アンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩の投与間隔と一部重複するように、トラマドールまたはその他のμ-オピエートアゴニストもしくはアゴニスト/アンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩の投与の前に、同時に、または投与後に投与することができる。
【0065】
同様に、サイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)阻害剤、チトクロームP450阻害剤、および/または少なくとも1つの薬学的に許容されるその塩は、サイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)阻害剤、チトクロームP450阻害剤、および/または少なくとも1つの薬学的に許容されるその塩の投与間隔が、トラマドールまたはその他のμ-オピエートアゴニストもしくはアゴニスト/アンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩の投与間隔、ならびにNMDAアンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩の投与間隔と一部重複するように、トラマドールまたはその他のμ-オピエートアゴニストもしくはアゴニスト/アンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩、ならびにNMDAアンタゴニストおよび/または少なくとも1つの薬学的に許容されるその塩の投与前、同時に、または投与後に投与することができる。カフェインも、早漏を治療するために、PDE5阻害剤、μ-オピエートアンタゴニストまたはチトクロームP450阻害剤の投与前、投与中または投与後に投与され得る。本発明の組成物および方法においてカフェインを使用することのさらなる利点は、オピエート鎮痛薬の使用者が経験する可能性がある傾眠状態または鎮静状態を相殺するために用いることができることである。
【0066】
本発明は、以下の番号を付けた文における態様を参照することによってさらに理解され得る:
1.ヒトまたはその他の哺乳動物における性機能不全を効果的に治療する方法であって、このような治療を必要とする患者にa)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、およびb)μ-オピエートレセプターのアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩を含む、ある量の作用物質を投与して、それによって該作用物質の組み合わせた量が性機能不全を治療するために有効である工程を含む方法。
2.性機能不全が早漏である、文1の方法。
3.作用物質が別々に投与される、文1の方法。
4.作用物質が組み合わせて投与される、文1の方法。
5.作用物質が性的活動の前に投与される、文1の方法。
6.作用物質が経口的に、インプラントによって、非経口的に、皮下で、舌下で、経直腸的に、局所的に、または吸入を介して投与される、文1の方法。
7.作用物質が経口的に投与される、文6の方法。
8. NMDAレセプターアンタゴニストがデキストロメトルファン、デキストロルファン、ケタミン、アマンタジン、メマンチン、エリプロジル、イフェンプロジル、フェンシクリジン、MK-801、ジゾシルピン、CCPエン、フルピルチン、またはその誘導体もしくは塩である、文1の方法。
9.NMDAレセプターアンタゴニストがデキストロメトルファンである、文8の方法。
10.μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストが、(1R,2Rまたは1S,2S)-(ジメチルアミノメチル)-1-(3-メトキシフェニル)-シクロヘキサノール(トラマドール)、そのN-酸化物誘導体(「トラマドールN-酸化物」)、およびそのO-デスメチル誘導体(「O-デスメチルトラマドール」)、またはその混合物、立体異性体もしくはラセミ体の任意の1つである、文1の方法。
11.μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストがトラマドールである、文10の方法。
12.作用物質が、タブレット、経口投与のための多粒子製剤;経口投与のための液剤、徐放性製剤、懸濁剤もしくはエリキシル剤、注射可能製剤、移植可能な装置、局所用調製物、固体状態および/もしくはデポー型の経皮送達装置、坐剤、口腔内タブレット、または副鼻腔に吸入もしくは注入されるように意図された放出制御粒子製剤、もしくはスプレー、ミストもしくはその他の局所ビヒクルなどの吸入製剤からなる群より選択される剤形で投与される、文1の方法。
13.剤形がタブレットまたはカプセルとして製剤化される固形経口剤形としてさらに限定される、文12の方法。
14.μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約15:1〜1:15である、文1の方法。
15.μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約10:1〜1:10である、文14の方法。
16.μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約5:1〜1:5である、文15の方法。
17.μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約1:2である、文16の方法。
18.ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩が作用物質として含まれる、文1の方法。
19.ホスホジエステラーゼ阻害剤がV型ホスホジエステラーゼ阻害剤である、文18の方法。
20.ホスホジエステラーゼ阻害剤が、シルデナフィル、アミノフィリン、テオフィリン、アムリノン、ミルリノン、ベスナリノン、ビンポセチン、ペモベンダン、シロスタミド、エノキシモン、ペロキシモン、ロリプラム、R020-1724、ザニプラスト、ジピリダモール、MY5445もしくはIC-351、または薬学的に許容されるその塩である、文18の方法。
21.NMDAレセプターアンタゴニスト対ホスホジエステラーゼ阻害剤対μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストの割合が約90:1:1〜1:90:1〜1:1:90である、文18の方法。
22.チトクロームP450阻害剤または薬学的に許容されるその塩が作用物質として含まれる、文1または18の方法。
23.チトクロームP450阻害剤がデブリソキンヒドロキシラーゼ阻害剤である、文22の方法。
24.チトクロームP450阻害剤が、キニジン、キニン、ナフチリジン、キサンチン、フェノキシアミノアルカン、カルバモイルイミダゾール、グアニジンイミダゾール、シメチジン(N-シアノ-N'-メチル-N"-[2[[(5-メチル-1H-イミダゾール-4イル)メチル]チオ]エチル]グアニジン)、キノリン、クロロキン(7-クロロ-4-(4-ジエチルアミノ-1-メチルブチルアミノ)キノリン)、プリマキン(8-(4-アミノ-1-メチルブチルアミノ)-6-メトキシキノリン)、トリフルオロメチルオキシムエーテル、5-メトキシ-1-[4-(トリフルオロメチル)-フェニル]-1ペンタノンO-(2-アミノエチル)オキシムとしても公知であるフルボキサミン、または薬学的に許容されるその塩である、分22の方法。
25.NMDAレセプターアンタゴニスト対チトクロームP450阻害剤対μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストの割合が約90:1:1〜1:90:1〜1:1:90である、文22の方法。
26.カフェインが作用物質として含まれる、文1、18または22の方法。
27.作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)V型ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩を含む、薬学的組成物。
28.作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)チトクロームP450阻害剤または薬学的に許容されるその塩を含む、薬学的組成物。
29.作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、c)V型ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩、およびd)チトクロームP450阻害剤または薬学的に許容されるその塩を含む、薬学的組成物。
30.カフェインをさらに含む、文27、28または29の薬学的組成物。
31.作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)カフェインを含む薬学的組成物。
【0067】
本発明のさらなる目的、利点および新規の特徴は、一部は以下の説明に記載されて、また一部は以下の検討により当業者に明らかとなるであると考えられ、または本発明の実践によって学ばれ得る。
【0068】
例証的態様の説明
本発明を詳細に説明する前に、薬剤または薬剤送達系は変化し得るものであり、本発明は特定の薬剤または薬剤送達系に限定されないということが理解されるべきである。本明細書で用いられる用語は特定の態様のみを説明することを目的とするものであり、限定することを意図するものではないことも理解されるべきである。本明細書で用いられるように、「一つの(a)」、「一つの(an)」、および「その(the)」という単数形は、文脈からそうでないことが明示される場合を除いて、複数の指示物を含む。従って、「一つの(a)薬理学的活性物質」への言及は2つまたはそれよりも多くの薬理学的活性物質の組み合わせなどを含む。本発明について説明する場合、次の用語は以下に示す定義に従って用いられる。
【0069】
「活性物質」、「薬剤」および「薬理学的活性物質」という用語は、本明細書では、生体(ヒトまたは動物)に投与された際に所望の薬理学的効果を誘発する化学的材料または化合物を指すために互換的に用いられる。同じく所望の薬理学的効果を誘発することが具体的に記載される、それらの化合物の誘導体および類似体または化合物のクラスも含まれる。
【0070】
「局所投与」という用語は、皮膚または粘膜への局所的な薬剤または薬理学的活性物質の送達を示すために慣例的な意味で用いられる。
【0071】
本明細書で用いられるように「担体」または「ビヒクル」は薬剤の投与に適した担体材料を示す。本明細書において有用な担体およびビヒクルには、例えば、任意の液体、ゲル、溶媒、液体希釈剤、可溶化剤など、無毒性で、かつ、組成物のその他の成分と有害な様式で相互作用しない、当技術分野において公知の任意のこのような材料が含まれる。
【0072】
薬剤または薬理学的活性物質の「有効」量は、無毒性であるが、所望の効果を示すために十分な薬剤または作用物質の量を意味する。
【0073】
本明細書で用いられるように「早漏」という用語は、男性がパートナーを満足させるために十分な程度で射精過程を制御することができない性機能不全を意味する。一般に、「早漏」は、性交前または性交中に最小限の刺激で射精を持続または反復することを指す。この用語は、例えば、米国特許第5,151,448号およびMale Infertility and Sexual Dysfunction at p. 356(New York: Springer- Verlag, 1997)に示されるように、「先天性」または「生涯」早漏、および「原発性」または「後天性」早漏の双方を含む。Diagnostic and Statistical Manual of Mental Disorders(Washington, D.C.: American Psychiatric Association, 1994)も参照されたい。
【0074】
「NSAID」という用語は、シクロオキシゲナーゼ酵素と結合することによってプロスタグランジンの産生を阻害する非ステロイド系の物質を指す。アセトアミノフェンは抗炎症特性は持たないが末梢および視床下部の温度調節中枢においてシクロオキシゲナーゼ酵素と結合するので、化合物のアセトアミノフェンはこのカテゴリーに含まれる。
【0075】
本明細書で用いられるように「シルデナフィル」という用語は、この化合物の遊離塩基型、および有機カルボン酸、有機スルホン酸または無機の酸と共に形成される薬学的に許容されるその酸付加塩を含む。本発明のために、有機カルボン酸塩である3.5mg/mlの水溶解度を持つクエン酸シルデナフィルが特に好ましい。「シルデナフィル」に対する言及にはクエン酸シルデナフィルが含まれる。
【0076】
本明細書で用いられるように「カフェイン」という用語は、無水粉末としてのカフェインばかりでなく、無毒性で薬学的に許容され、ならびに本明細書に記載されるように用いられた場合に鎮痛または抗炎症反応を促進および増強することができるカフェインの任意の塩もしくは誘導体、またはそれらの配合された任意の混合物も包含することを意図する(本発明の組成物において有用であることが証明され得るカフェインの塩、誘導体および混合物の説明については、例えば、The Merck Index, ninth edition, Merck & Co., Inc. Rahway, N.J. (1976), pp. 207-208を参照されたい)。それにも関わらず、現在のことろ無水粉末塩基としてのカフェインが好ましく、カフェインの具体的な量が以下に記載される場合、このような量は無水塩基のmgにて示される。
【0077】
本発明者らは早漏の有効な治療を探索していた。前記の先行技術に基づいて、本発明者らは、勃起に何の問題もない男性において射精の過程を制御して満足な性交を持つためには(以下、クラスIと記載する)、性的パートナーが最大の性的満足に達するために十分な性交の時間を持つように射精の過程を遅延させなければならないと推論した。
【0078】
勃起の問題を持ち、一旦勃起が達成されると射精を制御できない男性の場合は、次の2つの段階が好ましいはずである;(1)男性が性的パートナーによる刺激によって十分に勃起するようにシルデナフィルのような特定の医薬物質を介して勃起が達成されなければならない;(2)性的パートナーが最大の性的満足に達するために十分な性交の時間を持つように射精の過程が遅延されなければならない(以下、クラスIIと記載する)。
【0079】
DMの投与はヒトにおいて抗興奮毒性作用を持ち、ヒト男性へのDMの投与は射精の過程に対して影響を及ぼすであろうということが理解される。トラマドールは、その神経シグナルに対する作用のために、鎮痛効果を持つことも理解される。さらに、デブリソキンヒドロキシラーゼ阻害剤またはチトクロームP450阻害剤をDMと同時に投与するとヒト臨床試験において観察可能なDMの治療効果が実質的に増大し、次いで、早漏治療のための作用物質としてのDMの有効性はチトクロームオキシダーゼ阻害剤の同時投与によっても増強されることができることが理解される。これらの観察に基づいて、本発明者らはこれらの作用物質の組み合わせの投与がクラスIの男性の早漏に対して治療効果を持つであろうと推論した。驚いたことに、本発明者らは、これまでに、これらの作用物質の摂取が実際に早漏に対して著しい効果を持ち、それらは最大オルガスムに達するために性交を延長させることを発見した。さらに、本発明者らは、これらの作用物質を用いて性交中に複数のオルガスムを持つことができることを観察した。
【0080】
加えて、本発明者らは、シルデナフィル、トラマドールおよびDMの摂取がクラスIIの男性の早漏に対して顕著な効果を持ち、それらが最大オルガスムに達するために性交を延長させることを見出している。さらに、本発明者らは、これらの作用物質を用いて性交中に複数のオルガスムを持つことができることを観察した。さらに、本発明者らはトラマドールをシルデナフィルおよびDMと共に摂取してもクラスIIの患者において早漏治療におけるシルデナフィルおよびDMの組み合わせの治療的効果に影響はないことを見出している。
【0081】
加えて、本発明の特定の態様において、上記の組成物へのカフェインの添加は、オピエート鎮痛薬の一部の使用者が経験する傾眠状態または鎮静状態を相殺する利点がある。
【0082】
クラスIの男性における早漏を治療するための活性物質
クラスIの男性における早漏を治療するために本発明の方法を実践するために、選択された薬理学的活性物質を個体に投与する。活性物質は、経口的、非経口的、口腔内、経直腸的、または海綿体内注射によってもしくは尿道への送達によって局所的に投与され得る。適切な薬理学的活性物質は、μ-オピエート鎮痛薬であるトラマドール、その代謝物、その塩を含む。
【0083】
(+/-)-トラマドールはコデインの合成4-フェニル-ピペリジン類似体である(Shipton EA 2000)。これは、オピエートレセプターに低親和性を持つ中枢性鎮痛薬である。そのμレセプターに関する選択性は最近実証されたものであり、肝臓のO-脱メチル化によって産生されるトラマドールのM1代謝物はオピエートレセプターに対して親薬剤よりも高い親和性を示す。このM1誘導体(O-デメチルトラマドール)の産生速度は、デブリソキン型の多型性イソ酵素であるチトクロームP450 2D6(CYP2D6)に影響される。1つのメカニズムがμ-オピエートレセプターに対するその弱い親和性に関連する(モルフィンの1/6000、d-プロポキシフェンの1/100、コデインの1/10、およびデキストロメトルファンと同等)。さらに、その他のオピエートとは対照的に、トラマドールの鎮痛作用はオピエートアンタゴニストであるナロキソンによってごく一部が阻害されるのみであり、このことはもう一つの作用メカニズムの存在を示唆する。このことは、ノルアドレナリン(ノルエピネフリン)およびセロトニン(5-ヒドロキシトリプタミン;5-HT)の再取り込みを阻害して、脊髄レベルでの侵害受容インパルスを遮断することによって鎮痛作用に大きく寄与するするモノアミン作動性の活性の発見によって実証された(Dayer et al. 1994 & 1997)。
【0084】
(+/-)-トラマドールは2つの鏡像異性体のラセミ混合物であり、それぞれの鏡像異性体は様々なレセプターに対して異なる親和性を示す。(+/-)-トラマドールはμレセプターの選択的アゴニストであり、セロトニンの再取り込みを優先的に阻害する一方、(-)-トラマドールは主としてノルアドレナリンの再取り込みを阻害する。これらの2つの鏡像異性体の作用は双方とも相補的かつ相乗的であり、その結果、(+/-)-トラマドールの鎮痛効果が得られる。経口投与後、トラマドールは68%の生物学的利用能を示して、2時間以内に最高血清中濃度に到達した。排出動態は2コンパートメントとして説明され得、100mg単回経口投与後の半減期はトラマドールが5.1時間、M1誘導体が9時間である。このことが、トラマドールによる反復投与治療中に観察される親薬剤およびそのM1誘導体の約2倍の蓄積の原因である。トラマドールの推奨一日投与量は4〜6時間毎に50〜100mgであり、最大用量は400mg/日である。トラマドール100mgの単回経口投与後の鎮痛効果の期間は約6時間である。有害影響、特に悪心は、用量依存性であり、従って、負荷用量が高ければ発現する可能性がかなり高い。治療の最初の数日の期間中にこの用量を減量することは忍容性向上の重要な要因である。その他の有害影響は概ねオピエートのそれとほぼ等しいが、それらは通常はより軽度であり、呼吸抑制、不快気分および便秘が含まれ得る。トラマドールはその他の鎮痛薬、特に末梢作用を持つ鎮痛薬と同時に投与することができるが、CNS機能を抑制する薬剤はトラマドールの鎮静効果を増強する可能性がある。トラマドールは、依存に至る可能性は極めて低い薬力学および薬物動態特性を持つ。このことは様々な対照試験および市販後調査試験によって確認されて、試験では耐性を示す患者またはトラマドール乱用の事例の数が極端に少ないことが報告された(Raffa et al. 1993; Lee et al. 1993)。
【0085】
本発明の特定の態様において、早漏は、呼吸抑制、睡眠パターンの障害、食欲減退、発作、ならびに心理的および/または身体的依存などの、従来の鎮痛薬に付随する有害な副作用なしに治療することができる。
【0086】
それは疼痛の制御に関しては安全かつ有効な作用物質であることが証明されているものの、その使用に伴って有害作用が生じ得る。4mg/kgのトラマドールの小児への偶発的投与後に発作的活動の発生が報告されている(Tobias 1997)。
【0087】
デキストロメトルファンはシグマレセプターとして公知のニューロンレセプターのクラスに作用する。それらは抑制性レセプターであり、これはDMまたはその他のシグマアゴニストによるそれらの活性化が特定の種類の神経シグナルの抑制を引き起こすことを意味する。デキストロメトルファンはN-メチル-D-アスパラギン酸(NMDA)レセプターとして公知の、興奮性アミノ酸(EAA)レセプターの一種であるもう一つのクラスのレセプターにも作用する。シグマレセプターへのそのアゴニスト活性とは異なり、DMはNMDAレセプターにおいてアンタゴニストとして作用し、これはDMがNMDAレセプターを介して調節される神経インパルスの伝達を抑制することを意味する。NMDAレセプターは興奮性レセプターであるため、DMのNMDAアンタゴニストとしての活性も特定の種類の神経シグナルの抑制を誘発する。デキストロメトルファンおよびデキストロルファンの抗興奮毒性活性、ならびにこれらの薬剤によるNMDAレセプターの遮断については、Choi 1987、Wong et al 1988、Steinberg et al 1988、および米国特許第4,806,543号(Choi 1989)などの文献等で考察されている。デキストロメトルファンは、神経カルシウムチャネルにおいて活性を抑制することも報告されている(Carpenter et al 1988)。デキストロメトルファンおよびそれが相互作用するレセプターについては、Tortella et al 1989、Leander 1989、Koyuncuoglu & Saydam 1990、Ferkany et al 1988、George et al 1988、Prince & Feeser 1988、Feeser et al 1988、Craviso and Musacchio 1983、およびMusacchio et al 1988においてより詳細に考察されている。
【0088】
DMは血流からかなり速やかに消失する(例えば、Vetticaden et al 1989およびRamachander et al 1977を参照されたい)。DMは、肝臓においてO-脱メチル化と呼ばれる酵素過程によってデキストロルファンおよび3-メトキシモルフィナンと呼ばれる2つの代謝物に転換される。この酵素は、何年も前にデブリソキンのヒドロキシル化反応を実行するために発見されたことから、通常はデブリソキンヒドロキシラーゼと呼ばれる。これは様々な論文においてP450DBまたはP450-2D6とも呼ばれている。多くの化合物がデブリソキンヒドロキシラーゼ(スパルテインモノオキシゲナーゼ)イソ酵素の活性を阻害する;Inaba et al 1985を参照されたい。これらの阻害物質の最も強力なものはキニンの右旋性立体異性体であるキニジンであり、これは通常は心不整脈を治療するために用いられる。Inaba et al(1986)およびNielsen et al(1990)はキニジンがインビボ動物試験においてスパルテインの酸化を阻害する能力について考察し、Brinn et al(1986)、Brosen et al(1987)、およびBroly et al(1989)はキニジンが肝細胞調製物中でのDM代謝を阻害する能力について考察している。例外的に強力であり、かつ、容易に実証されるデブリソキンヒドロキシラーゼの阻害に加えて、その他のチトクロームP450イソ酵素も、様々なレベルの結合親和性で、キニジンによって抑制される可能性が高い。さらに、キニジンはデブリソキンヒドロキシラーゼに対して最も顕著な効果を発揮するものの、同様に多くのその他のチトクロームP450酵素を抑制して、それによって患者は正常かつ望ましい肝臓の活性をより全般的に喪失する可能性が高い。デキストロメトルファンの主たる酸化代謝産物はデキストロルファンであり、これは神経学者の間ではデキストロメトルファンと全く同じ様式で活性であると広く信じられている;報告によると、双方の薬剤はシグマアゴニスト、NMDAアンタゴニストおよびカルシウムチャネルアンタゴニストである。DMと組み合わせたデブリソキンヒドロキシラーゼを阻害する化合物の投与は、DMのみを投与される患者に比して患者の血液中のDMの濃度および安定性の大幅な増大が惹起される;かつDMと組み合わせたデブリソキンヒドロキシラーゼ阻害剤の投与はヒトにおけるDMの検出可能な作用に対して明確で実質的な影響を有する。デキストロメトルファンの活性の増強にはデブリソキンヒドロキシラーゼ阻害剤が好ましいが、ナフチリジン、キサンチン、フェノキシアミノアルカン、カルバモイルイミダゾール、シメチジン(N-シアノ-N'-メチル-N"-[2[[(5-メチル-1H-イミダゾール-4イル)メチル]チオ]エチル]グアニジン)などのグアニジンイミダゾール、クロロキン(7-クロロ-4-(4-ジエチルアミノ-1-メチルブチルアミノ)キノリン)およびプリマキン(8-(4-アミノ-1-メチルブチルアミノ)-6-メトキシキノリン)などのキノリン、5-メトキシ-1-[4-(トリフルオロメチル)-フェニル]-1ペンタノンO-(2-アミノエチル)オキシムとしても公知であるフルボキサミンなどのトリフルオロメチルオキシムエーテルなど、チトクロームP450の酸化的活性を阻害するその他の作用物質もデキストロメトルファンの活性の増強に有用であり得る。
【0089】
カフェインはいくつかの鎮痛剤に含まれている。これが全く合理的である限り、それは痛覚過敏を引き起こす知覚神経終末のまたは終末近くのアデノシンA2Aレセプターの存在に関連し得る(Ledent et al., 1997)。実際、カフェインは特定の種類のC線維介在性疼痛において痛覚鈍麻作用を持つ(Myers et al., 1997)。鎮痛効果は小さい(Battig and Welzl, 1993)。カフェインは気分および頭の冴えを高めることによって間接的に有益な効果を持ち得る(Lieberman et al., 1987)。この試験では、気分および覚醒状態の双方が、アスピリンの単独投与またはプラセボよりもカフェインと併用したアスピリンによってさらに改善することが見出された。
【0090】
総説(Migliardi et al., 1994)の通り、患者は頭痛の治療に関してカフェインフリーの調剤品よりもカフェインを含有する鎮痛薬を優れていると判定する。さらに、カフェインは、i.c.v.注射後にマウスにおいて疼痛関連性の行動に拮抗することができることから(Ghelardini et al., 1997)、脳において抗侵害受容効果を発揮する可能性がある。さらに、この効果はコリン作動性伝達を抑制するアデノシンA1レセプターの持続性阻害活性の拮抗作用に関連する可能性がある(Rainnie et al., 1994; Carter et al., 1995)。
【0091】
上記の通り、睡眠はヒトにおけるカフェインの影響に対する最も敏感な生理学的機能の1つであると考えられる。就寝時に摂取するカフェインは睡眠に対してネガティブに作用することは周知である(Snel, 1993を参照されたい)。一般に、睡眠に対して顕著な影響を及ぼすためには200mgを超えるカフェインが必要である。最も顕著な影響は、総睡眠時間の短縮、睡眠潜時の延長、最初の軽睡眠EEG段階の増加、およびその後の熟睡EEG段階の減少、さらに睡眠段階間の推移回数の増加である。
【0092】
本発明を実践するために、本発明において用いられ得るμ-オピエート鎮痛薬剤の非限定的なリストにはトラマドール、その代謝物、その塩、その複合体が含まれる。
【0093】
本発明において用いられ得るNMDAアンタゴニストには、デキストロメトルファン、ケタミンおよびアマンチジン、ならびにその代謝物、塩および複合体が含まれる。
【0094】
本発明において用いられ得るカフェイン類似体の非限定的なリストには、キサンチン、ヒポキサンチン(6-ヒドロキシプリン)、1-メチルキサンチン、3-メチルキサンチン、7-メチルキサンチン、アザキサンチン(8-アザ-2,6-ジヒドロキシプリン)、テオフィリンおよびテオブロミンが含まれる。
【0095】
経口組み合わせ投与単位は、患者に投与される組み合わせ用量に伴う副作用が最小限または実質的に皆無である限り、好ましくは、約30から200ミリグラム(mg)以下の範囲、好ましくは約60〜約120mgの範囲のデキストロメトルファン、ならびに約30〜約500mgの範囲、好ましくは約30〜約200mgの範囲のトラマドールを含む。カフェインは、約30〜200mg以下、好ましくは約60mg〜約100mgの範囲の用量で組成物に含まれ得る。
【0096】
特に好ましい経口組み合わせ投与単位は、約120mgのデキストロメトルファンおよび100mg以下のトラマドール、より好ましくは、約90mgのデキストロメトルファンおよび約100mg以下のトラマドールを含む。もう一つの好ましい経口組み合わせ投与単位は、約120mgのデキストロメトルファン、約100mgのカフェイン、および100mg以下のトラマドール、より好ましくは約90mgのデキストロメトルファン、約60mgのカフェイン、および約100mg以下のトラマドールを含む。
【0097】
または、デキストロメトルファンおよびトラマドールは、それぞれの各薬剤の順次的な投与を実践するための単独の活性成分として、前記の組成物において別々に製剤化されてもよい。
【0098】
または、デキストロメトルファン、カフェインおよびトラマドールは、それぞれの各薬剤の順次的な投与を実践するための単独の活性成分として、前記の組成物において別々に製剤化されてもよい。
【0099】
順次的投与療法において、トラマドール、カフェインおよびデキストロメトルファンはそれぞれ別々の投与量にて投与される。患者が投与される総組み合わせ用量に伴う好ましくない副作用が最小限または実質的に皆無である限り、トラマドールの順次的投与では、投与単位は、好ましくは約10〜約500mgの範囲、より好ましくは約20mg〜約300mgの範囲のトラマドールを含み、カフェインの投与における投与単位は好ましくは約10〜約400mgの範囲、より好ましくは約30mg〜約200mgの範囲のカフェインを含み、デキストロメトルファンの投与の投与単位は好ましくは約30から120mg以下の範囲、より好ましくは約60〜約90mgの範囲のデキストロメトルファンを含む。
【0100】
特に好ましい順次的投与の投与単位は、約30〜約100mgの範囲のトラマドールを含み、約30〜約135mgの範囲のデキストロメトルファンを含む。好ましくは、各薬剤は経口的に投与される。または、各薬剤は異なる経口的経路によって投与されてもよい;即ち、一つは摂取されて、他方は舌下または口内パッチにより投与されてもよい。
【0101】
トラマドール、カフェインおよびデキストロメトルファンの有効な順次的投与のため、各薬剤の放出には、デキストロメトルファンによる有利な射精遅延を最大限とするように時間差が設けられることが好ましい。
【0102】
特に好ましい順次的投与の投与単位は、約30〜約100mgの範囲のトラマドール、30〜100mgの範囲のカフェイン、および約30〜約135mgの範囲のデキストロメトルファンを含む。好ましくは、各薬剤は経口的に投与される。または、各薬剤は異なる経口的経路によって投与されてもよい;即ち、一つは摂取されて、他方は舌下または口内パッチにより投与されてもよい。
【0103】
トラマドール、カフェインおよびデキストロメトルファンの有効な順次的投与のため、各薬剤の放出には、デキストロメトルファンによる有利な射精遅延を最大限とするように時間差が設けられることが好ましい。
【0104】
デキストロメトルファンの効果を増強するために、任意でキニジンのようなチトクロームP450酵素阻害剤の有効量を組み合わせ投与単位として、または順次的投与の投与単位として患者に投与してもよい。チトクロームP450阻害剤がデキストロメトルファンの効果を増強するために投与される場合、デキストロメトルファンの投与量は有効性を最大限として副作用を最小限とするように適切に調節することができる。経口組み合わせ投与単位は、好ましくはキニジンを約50から200ミリグラム(mg)以下の範囲で、好ましくは約90〜約120mgの範囲で含み得る。
【0105】
クラスIIの男性の早漏を治療するための活性物質
射精と共に勃起の問題を持つクラスIIの男性における早漏を治療するために本発明の方法を実践するために、選択された薬理学的活性物質を個体に投与する。活性物質は、経口的、非経口的、口腔内、経直腸的、または海綿体内注射によってもしくは尿道への送達によって局所的に投与され得る。適切な薬理学的活性物質には、シルデナフィルのようなサイクリックGMP特異的5型ホスホジエステラーゼ(PDE5)阻害剤、カフェイン、デキストロメトルファンのような抗興奮毒性物質、トラマドールのようなμ-オピエート鎮痛薬、ならびに任意でキニン、キニジン、ナフチリジン、キサンチン、フェノキシアミノアルカン、カルバモイルイミダゾール、シメチジン(N-シアノ-N'-メチル-N"-[2[[(5-メチル-1H-イミダゾール-4イル)メチル]チオ]エチル]グアニジン)のようなグアニジンイミダゾール、クロロキン(7-クロロ-4-(4-ジエチルアミノ-1-メチルブチルアミノ)キノリン)およびプリマキン(8-(4-アミノ-1-メチルブチルアミノ)-6-メトキシキノリン)のようなキノリン、5-メトキシ-1-[4-(トリフルオロメチル)-フェニル]-1ペンタノンO-(2-アミノエチル)オキシムとしても公知であるフルボキサミンのようなトリフルオロメチルオキシムエーテルなどのチトクロームP450阻害剤が含まれる。
【0106】
シルデナフィルは、化学的には1-[[3-(6,7-ジヒドロ-1-メチル-7-オキソ-3-プロピル-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)-4-エトキシフェニル]スルホニル]-4-メチルピペラジンと呼ばれ、以下の構造式を持つ:図3。
【0107】
クエン酸シルデナフィルは、現在、経口投与用の25mg、50mgおよび100mgシルデナフィルに相当するタブレットとして製剤化されたViagra(商標)(Pfizer Labs, N.Y.)の名称で販売されるインポテンスのための市販薬の活性成分である。製造業者によると、活性成分であるクエン酸シルデナフィルに加えて、各タブレットは次の活性成分を含む:微晶性セルロース、無水第二リン酸カルシウム、クロスカルメロースナトリウム、ステアリン酸マグネシウム、ヒドロキシプロピルメチルセルロース、二酸化チタン、ラクトース、トリアセチン、およびFD&C青色2号アルミニウムレーキ。
【0108】
インビトロ試験から、シルデナフィルは、心収縮性の制御に関与するPDE3のようなその他の公知のホスホジエステラーゼに対してよりも5型ホスホジエステラーゼ(PDE5)の阻害に関して約4,000倍選択的であることが知られている。報告によると、シルデナフィルは網膜において見出された酵素であるPDE6に比べてPDE5に対する力価として僅か約10倍であり、この低い選択性が高い用量または血漿中濃度で観察される色覚に関連する異常の理由であると考えられる。
【0109】
市販のViagra(商標)製剤として投与されるシルデナフィルは経口投与後に速やかに吸収されることが報告されて、絶対生物学的利用能は約40%である。その薬物動態は推奨用量範囲を通して用量に比例する。Viagra(商標)の製造業者の製品資料に基づいて、空腹時には経口投与の30〜120分(中央値 60分)以内に最大観察血漿中濃度に到達する。Viagra(商標)製剤を高脂肪食と共に服用すると、吸収速度が低下して、Tmaxは平均60分遅延して、Cmaxは平均29%低下する。シルデナフィルの平均定常状態分布容積(Vss)は報告によると105Lであり、組織内に分布することを示している。投与後90分における健常志願者の精液中シルデナフィル報告測定値に基づくと、投与量の0.001%未満が患者の精液に出現した。
【0110】
驚くべきことに、本発明の組成物と共に用いられるデキストロメトルファン、トラマドールおよびシルデナフィルの組み合わせの治療上有効量は早漏の遅延によりシルデナフィルの有利な勃起誘発効果を最大限とする。
【0111】
経口組み合わせ投与単位は、患者に投与される組み合わせ用量に伴う望ましくない副作用が最小限または実質的に皆無である限り、好ましくは、約10〜300ミリグラム(mg)以下の範囲、好ましくは約30および約200mgの範囲のデキストロメトルファン、約10〜約200ミリグラム(mg)の範囲、好ましくは約30〜約150mgの範囲のトラマドール、ならびに約10〜約150mgの範囲、好ましくは約15〜約100mgの範囲のシルデナフィルを含む。特に好ましい経口組み合わせ投与単位は、約150mgのデキストロメトルファン、200mg以下のトラマドール、より好ましくは約100mgのトラマドール、および150mg以下のシルデナフィル、より好ましくは約135mgのデキストロメトルファン、約100mgのトラマドールおよび約100mg以下のシルデナフィルを含む。
【0112】
または、デキストロメトルファン、トラマドールおよびシルデナフィルは、それぞれの各薬剤の順次的な投与を実践するための単独の活性成分として、前記の組成物において別々に製剤化してもよい。
【0113】
順次的投与療法において、シルデナフィル、トラマドールおよびデキストロメトルファンはそれぞれ別々の投与量にて投与される。患者が投与される総組み合わせ用量に伴う好ましくない副作用が最小限または実質的に皆無である限り、シルデナフィルの順次的投与では、投与単位は、好ましくは約10〜約300mgの範囲、より好ましくは約25〜約200mgの範囲のシルデナフィルを含み、トラマドールの投与における投与単位は好ましくは約20〜約400mg以下の範囲、より好ましくは約30〜約200mgの範囲のトラマドールを含み、デキストロメトルファンの投与では、投与単位は好ましくは約30〜500mg以下の範囲、より好ましくは約60〜約300mgの範囲のデキストロメトルファンを含む。
【0114】
シルデナフィルの特に好ましい順次的投与の投与単位は、約50〜約150mgの範囲のシルデナフィルを含み、トラマドールについては約50〜約200mgの範囲のトラマドールを含み、およびデキストロメトルファンについては約45〜約200mgの範囲のデキストロメトルファンを含む。好ましくは、各薬剤は経口的に投与される。または、各薬剤は異なる経口的経路によって投与することができる;即ち、一つは摂取されて、他方は舌下または口内パッチにより投与することができる。
【0115】
望ましい場合、吸収、従って生物学的利用能を促進するために、シクロデキストリン、特にβ-シクロデキストリン、またはヒドロキシプロピル-β-シクロデキストリン(HPBCD)のようなその誘導体などの吸収促進物質を加えてもよい。シクロデキストリンは、6個、7個または8個のグルコピラノース環から構成されて、それぞれ、α、βおよびγシクロデキストリンとして公知である環式非還元性オリゴ糖のグループである。シクロデキストリンは、分子包接化合物を形成する特性を有する空洞内包環式化合物のクラスであり、共有結合を形成することなくもう一つの化学的化合物を係留または捕捉する。HPBCDは、内部空洞を含むドーナツ状の分子構造を有する環式ポリマーである。
【0116】
ヒドロキシプロピル-β-シクロデキストリンは、最大約15またはそれ以上までの置換度(D.S.)を有する対応するヒドロキシプロピル誘導体を与えるために、酸化プロピレンと縮合することによってβ-シクロデキストリンから誘導される市販の化合物である。本発明のために、約5〜7のD.S.値が好ましい。
【0117】
このような適切なヒドロキシプロピル-β-シクロデキストリンの調製は、とりわけ、International Journal of Pharmaceutics, 29, 73-82 (1986)およびJournal of Pharmaceutical Sciences, 75 (6), 571-572 (1986)に記載されている。シクロデキストリンのポリエーテルであるヒドロキシプロピル-β-シクロデキストリンも公知であり本発明のために適切であり、Grameraらに対する米国特許第3,459,731号に記載されるように過剰な酸化ヒドロキシプロピレンのβ-シクロデキストリンを用いた縮合によって得られる。ヒドロキシプロピレン-β-シクロデキストリン(HPBCD)は特に好ましいシクロデキストリン要素であるが、それらに限定されるものではない。HPBCDの組成物中の重量パーセントは、好ましくは組成物全体の約1〜約10重量パーセントの範囲内である。
【0118】
特にシルデナフィルの場合は、HPBCDが生物学的利用能を促進することが見出されている。従って、所望の治療効果を比較的低い用量のシルデナフィルで達成することができ、それによって有害な影響の可能性が最小限に抑えられる。
【0119】
シルデナフィル、トラマドールおよびデキストロメトルファンの効果的な順次的投与のために、各薬剤の放出には、性的刺激時のデキストロメトルファンおよびトラマドールによる有利な勃起の延長ならびにシルデナフィルによる勃起の維持を最大限とするように時間差が設けられることが好ましい。
【0120】
デキストロメトルファン、トラマドールおよびシルデナフィル療法の有利な効果を増強するために、より少ない量の勃起誘発物質を含めることができる。本明細書で用いられるように「勃起誘発物質」という用語は、テストステロン、デヒドロエピアンドロステロン(DHEA)などのような副腎ステロイドを指す。好ましくは、勃起誘発物質は投与されるシルデナフィルの重量の約5〜約10重量パーセントの範囲で、より好ましくは約6〜約8重量パーセントの範囲の量で加えられる。
【0121】
オピエート鎮痛薬の一部の使用者が経験する傾眠状態または鎮静状態を相殺するために、組成物にカフェインを加えてもよい。
【0122】
デキストロメトルファンの効果を増強するために、任意でキニジンのようなチトクロームP450酵素阻害剤の有効量を組み合わせ投与単位、または順次的投与の投与単位のいずれかとして患者に投与してもよい。チトクロームP450阻害剤がデキストロメトルファンの効果を増強するために投与される場合、デキストロメトルファンの投与量は有効性を最大限として副作用を最小限とするように適切に調節することができる。経口組み合わせ投与単位は、好ましくは、キニジンを約50から200ミリグラム(mg)以下の範囲で、好ましくは約90〜約120mgの範囲で含み得る。経口組み合わせ投与単位は、好ましくは、キニジンを約50から200ミリグラム(mg)以下の範囲で、好ましくは約90〜約120mgの範囲で含み得る。
【0123】
活性物質は薬学的に許容される塩、エステル、アミドもしくはプロドラッグ、またはその組み合わせの形で投与され得る。しかし、不活性エステル、アミドまたはプロドラッグ型の活性型への転換は標的組織または細胞への到達前または到達後直ちに起こらなければならない。活性物質の塩、エステル、アミドおよびプロドラッグは、合成有機化学の技術分野の業者に公知であり、例えば、J. March, Advanced Organic Chemistry: Reactions, Mechanisms and Structure, 4th Ed.(New York: Wiley-Interscience, 1992)に記載されるような標準的手順を用いて調製してよい。例えば、酸付加塩は遊離塩基(典型的には中性の型の薬剤が中性の--NH2基を持つ)から適切な酸との反応を伴う従来の方法を用いて調製される。一般に、薬剤の塩基型はメタノールまたはエタノールのような極性有機溶媒に溶解して、そこに酸が加えられる。形成される塩は沈殿させるか、または極性の低い溶媒を加えることによって溶液から取り出すことができる。酸付加塩の調製に適切な酸には、例えば、酢酸、プロピオン酸、グリコール酸、ピルビン酸、シュウ酸、リンゴ酸、マロン酸、コハク酸、マレイン酸、フマル酸、酒石酸、クエン酸、安息香酸、桂皮酸、マンデル酸、メタンスルホン酸、エタンスルホン酸、p-トルエンスルホン酸、サリチル酸などの有機酸、および塩酸、臭化水素酸、硫酸、硝酸、リン酸などの無機酸が含まれる。酸付加塩は適切な塩基での処理によって遊離塩基に戻すことができる。逆に、薬剤に含まれ得る酸部分の塩基性塩の調製は、水酸化ナトリウム、水酸化カリウム、水酸化アンモニウム、水酸化カルシウム、トリメチルアミンなどのような薬学的に許容される塩基を用いて同様の方法で調製される。エステルの調製は、薬剤の分子構造内に含まれ得るヒドロキシル基および/またはカルボキシル基の官能化を伴う。エステルは、典型的には遊離アルコール基のアシル置換された誘導体、つまり、式RCOOHのカルボン酸から誘導される部分であって、Rはアルキルであり、好ましくは低級アルキルである。エステルは、望ましい場合、通常の水素化分解または加水分解の手順を用いることによって遊離酸に戻すことができる。アミドおよびプロドラッグの調製は類似の方法で実施することができる。活性物質のその他の誘導体および類似体は合成有機化学の技術分野の業者に公知の標準的方法を用いて調製され得るか、または関連する文献の参照より推論され得る。さらに、キラル活性物質は鏡像異性的に純粋な型であり得るか、または鏡像異性混合物として投与されてもよい。
【0124】
意図する投与方法に応じて、薬学的組成物は例えばタブレット、坐剤、丸剤、カプセル、粉剤、液体、懸濁剤、クリーム、軟膏、ローション剤などのような固体、半固体または液体の剤形であり得、好ましくは正確な用量の単回投与に適した単位剤形であり得る。この組成物は薬学的に許容される担体と組み合わせて、選択された薬剤の有効量を含み、さらに、その他の医薬物質、アジュバント、希釈剤、緩衝剤などを含み得る。従って、化合物は、通常の無毒性の薬学的に許容される担体、アジュバントおよびビヒクルを含む投与製剤として経口的に、非経口的に、経皮的に、経直腸的に、経鼻的に、口腔内に、局所的に、または埋め込み式リザーバーを介して投与することができる。本明細書で用いられるように「非経口的」という用語は、皮下、静脈内および筋肉内注射を含むことを意図する。勿論、投与される活性化合物の量は治療しようとする被験体、被験体の体重、投与様式、および処方する医師の判断に依存する。
【0125】
固形組成物に関して、無毒性の一般的固形担体には、例えば医薬品グレードのマンニトール、ラクトース、澱粉、ステアリン酸マグネシウム、サッカリンナトリウム、タルク、セルロース、ブドウ糖、ショ糖、炭酸マグネシウムなどが含まれる。薬学的に投与可能な液体組成物は、例えば、本明細書に記載される活性化合物および任意で薬学的アジュバントを、例えば水、生理食塩液、デキストロース水溶液、グリセロール、エタノールなどのような賦形剤に溶解、分散させるなどして、それによって溶液または懸濁液を生成することによって調製することができる。望ましい場合、投与される薬学的組成物は、例えば酢酸ナトリウム、モノラウリル酸ソルビタン、トリエタノールアミン酢酸ナトリウム、オレイン酸トリエタノールアミンなどの湿潤剤または乳化剤、pH緩衝剤などの無毒性の補助物質も含むことができる。このような剤形を調製する実際の方法は当業者に公知であるか、または明らかであると考えられる;例えば、上記において引用したRemington's Pharmaceutical Sciencesを参照されたい。経口投与のためには、組成物は一般にタブレットもしくはカプセルの形をとり、または水性もしくは非水性の液剤、懸濁剤もしくはシロップ剤としてもよい。タブレットおよびカプセルは好ましい経口投与形である。経口使用のためのタブレットおよびカプセルは、一般に、ラクトースおよびコーンスターチのような一般的に用いられる1種類または数種類の担体を含む。ステアリン酸マグネシウムのような潤滑剤も一般的に加えられる。液体懸濁剤を用いる場合、活性物質は乳化剤および懸濁剤と組み合わせることができる。着香料、着色料および/または甘味料も加えてよい。本明細書において経口製剤に組み入れるためのその他の選択的成分には防腐剤、懸濁剤、増粘剤などが含まれるが、これらに限定されるものではない。
【0126】
非経口的投与が使用される場合は一般に注射を特徴とする。注射可能な製剤は、流動性の液剤または懸濁剤のいずれかとしての一般的な形、注射前に液体への溶解もしくは懸濁に適した固形剤として、または乳剤として調製することができる。好ましくは、適切な担体、分散剤または湿潤剤、および懸濁剤を用いて当技術分野において公知の手法に従って、無菌の注射可能な懸濁剤が製剤化される。無菌の注射可能な製剤は、無毒性の非経口的に許容される希釈剤または溶媒中の無菌の注射可能な液剤または懸濁剤であり得る。使用することのできる許容されるビヒクルおよび溶媒には、水、リンガー液および等張塩化ナトリウム液がある。さらに、無菌の固定油、脂肪酸エステルまたはポリオルも溶媒または懸濁媒として一般的に用いられる。ごく最近に修正された非経口投与のためのアプローチは、一定レベルの用量が維持されるような徐放性または持続放出性の系の使用を含む。例えば、米国特許第3,710,795号を参照されたい。
【0127】
活性物質は経尿道の薬剤送達に適した薬学的製剤として投与することができる。製剤は、水、シリコン、ワックス、ワセリン、ポリエチレングリコール(「PEG」)、プロピレングリコール(「PG」)、リポソーム、マンニトールおよびラクトースのような糖、ならびに/またはその他の様々な材料のような1つまたは複数の選択された担体または賦形剤を含むが、ポリエチレングリコールおよびその誘導体が特に好ましい。投与される薬剤に応じて、尿道投与剤形に経尿道浸透促進剤を組み入れることが望ましい場合もある。適切な経尿道浸透促進剤の例には、ジメチルスルホキシド(「DMSO」)、ジメチルホルムアミド(「DMF」)、N,N-ジメチルアセトアミド(「DMA」)、デシルメチルスルホキシド(「C10 MSO」)、モノラウリン酸ポリエチレングリコール(「PEGML」)、モノラウリン酸グリセロール、レシチン、1-置換アザシクロヘプタン-2-オン類、特に1-n-ドデシルシクラザシクロヘプタン-2-オン(Nelson Research & Development Co. Irvine, Calif.からのAzone(登録商標)の商標で入手可能)、SEPA(登録商標)(Macrochem Co., Lexington, Mass.から販売)、アルコール(例えば、エタノール)、界面活性剤(Tergitol(登録商標)、Nonoxynol-9(登録商標)、およびTWEEN-80(登録商標))などが含まれる。
【0128】
本発明はその好ましい具体的な態様と関連して記載されていて、前記の説明および以下の例は本発明の範囲を例示することを意図するものであって、限定することを意図するものではない。本発明の範囲内のその他の局面、利点および改変は、本発明が属する技術分野における業者には明らかである。
【0129】
実施例1
カプセル製剤
カプセル製剤の各1カプセル中の次の成分を正確に秤量して、乳棒および乳鉢を用いて細かい均質な粉末まで粉砕した。これらの粉末を100メッシュの篩に通して、硬質ゼラチンカプセルに充填した。各カプセル製剤の組成は以下に示す。
【0130】
実施例2
シルデナフィル含有カプセル製剤
カプセル製剤の各1カプセル中の次の成分を正確に秤量して、乳棒および乳鉢を用いて細かい均質な粉末まで粉砕した。これらの粉末を100メッシュの篩に通して、硬質ゼラチンカプセルに充填した。各カプセル製剤の組成は以下に示す。
【0131】
実施例3
被験体は健康状態良好な40歳、白人男性であった。被験体は自身の性的活動に満足を示していたが、さらなる「持久力」を望んでいた。被験体は、性的活動を営む約1時間前に被験物である実施例2のカプセル製剤1の2カプセルを服用した。被験体は、自身の意志を働かせるだけでクライマックスを延期させることができたと報告した。被験体は、身体的に必要な動きを行うことができる限り、性交を続けることができたことを述べた。
【0132】
実施例4
被験体は健康状態の優れた31歳、白人男性であった。被験体は、性的活動の開始約3時間前に被験物である実施例1のカプセル製剤2の2カプセルを服用した。被験体は、自身の精力が著しく増強されて、中間の弛緩期間なく2回のクライマックスに到達したと報告した。
【0133】
実施例5
49歳の白人男性は会社を設立して複数の製品を製造し、会社のための資本を確保して製品を市場に出すための戦略を立てているところであった。彼は一日に長時間働くことが常であった。この患者は、ガールフレンドとの性的活動中に自分が射精を制御できず、その結果、女性パートナーから失望されたことに気が付いた。早漏の問題のために、この患者は可能な場合は必ず女性パートナーとの性的接触を避けるようにして、彼のパートナーは性的経験を満たされないと感じて立腹することもあった。この患者は実施例2の製剤1のカプセルを提供されて、性交の約3時間前に2カプセルおよび性交の約1時間前に1カプセルを服用するように勧められた。患者は最初の晩に2カプセルを服用して、彼の証言によると、彼は陰茎に若干のしびれを感じて、性交前の性行為を行うことによって女性パートナーの性的感情を起こさせることができて、しかも、彼のパートナーはほぼ30分間にわたって何ら射精を伴うことなく彼の陰茎で性交前の性行為を行うことができた。彼は20分を超えて性交することができて、彼のパートナーは疲労困憊を感じた。彼の女性パートナーは非常に恍惚状態であり、彼は同晩に2回の性交を行うことができた。患者は、そうしない場合にはストレス性の身体のために、良好で健全な性交を持ちたい場合には必ず定期的にカプセルを服用している。
【0134】
実施例6
ガールフレンドと同棲している白人男性、40歳は、彼の性的活動に対する本発明の組成物の効果に強い関心を持っていた。この被験体は18:00頃に実施例2の製剤1として記載されるカプセル2個を服用した。18:45に被験体は顔面の若干の潮紅感と共に生殖器の重感を報告した。性的活動は19:15に開始した。被験体は、通常経験するよりも勃起の硬直度が高く膨満感が増した感覚があったと報告した。被験体は、彼の通常の経験よりも70%ほど持久力が増したと報告した。性交は約90分間継続して、通常のオルガスムより力強く終了した。
【0135】
実施例7
クラスIIの男性に対するシルデナフィル、デキストロメトルファンおよびトラマドールの効果
クラスIIの男性の早漏を治療するためのシルデナフィル、トラマドールおよびデキストロメトルファン組成物の有効性を実証するために、早漏および勃起の問題を持つ21〜57歳の年齢群から30名の志願者を選択した。志願者に実施例2の製剤1のカプセルを与えた。志願者には性的行為の2〜3時間前に2カプセルを服用するように依頼して、性的行為の前後に表に示す書式に記入するよう依頼した。試験は8週間にわたって実施して、結果をまとめて性的満足について分析した。結果は、80%を超える志願者が早漏の問題に関して本発明の組成物に非常に満足したことを示している。
【0136】
次の刊行物は参照により本明細書に関連する部分に組み入れられる。
【0137】
参考文献
【図面の簡単な説明】
【0138】
【図1】通常の性交中のオルガスムレベルを示すグラフである。通常の性交中の男性および女性のオルガスムレベルが示される。オルガスムレベルは、性交中の身体的および感情的な興奮を示す恣意的な量である。
【図2】早漏の症例におけるオルガスムレベルを示すグラフである。早漏の症例における男性および女性のオルガスムレベルが示される。オルガスムレベルは、性交中の身体的および感情的な興奮を示す恣意的な量である。
【図3】トラマドール、キニジン、デキストロメトルファン、カフェインおよびシルデナフィルの化学構造を示す。
【特許請求の範囲】
【請求項1】
以下の工程を含む、ヒトまたはその他の哺乳動物における性機能不全を効果的に治療する方法:a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、およびb)μ-オピエートレセプターのアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩を含む、ある量の作用物質を、治療を必要とする患者に投与する工程であって、それによって組み合わせた量の該作用物質が性機能不全を治療するために有効である、工程。
【請求項2】
性機能不全が早漏である、請求項1記載の方法。
【請求項3】
作用物質が別々に投与される、請求項1記載の方法。
【請求項4】
作用物質が組み合わせて投与される、請求項1記載の方法。
【請求項5】
作用物質が性的活動の前に投与される、請求項1記載の方法。
【請求項6】
作用物質が経口的に、インプラントによって、非経口的に、皮下で、舌下で、経直腸的に、局所的に、または吸入を介して投与される、請求項1記載の方法。
【請求項7】
作用物質が経口的に投与される、請求項6記載の方法。
【請求項8】
NMDAレセプターアンタゴニストがデキストロメトルファン、デキストロルファン、ケタミン、アマンタジン、メマンチン、エリプロジル、イフェンプロジル、フェンシクリジン、MK-801、ジゾシルピン、CCPエン、フルピルチン、またはその誘導体もしくは塩である、請求項1記載の方法。
【請求項9】
NMDAレセプターアンタゴニストがデキストロメトルファンである、請求項8記載の方法。
【請求項10】
μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストが、(1R,2Rまたは1S,2S)-(ジメチルアミノメチル)-1-(3-メトキシフェニル)-シクロヘキサノール(トラマドール)、そのN-酸化物誘導体(「トラマドールN-酸化物」)、およびそのO-デスメチル誘導体(「O-デスメチルトラマドール」)、またはその混合物、立体異性体もしくはラセミ体の任意の1つである、請求項1記載の方法。
【請求項11】
μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストがトラマドールである、請求項10記載の方法。
【請求項12】
作用物質が、タブレット、経口投与のための多粒子(multiparticulate)製剤;経口投与のための液剤、徐放性製剤、懸濁剤もしくはエリキシル剤、注射可能製剤、移植可能な装置、局所用調製物、固体状態および/もしくはデポー型の経皮送達装置、坐剤、口腔内タブレット、または副鼻腔に吸入もしくは注入されるように意図された放出制御粒子製剤、もしくはスプレー、ミストもしくはその他の局所ビヒクルなどの吸入製剤からなる群より選択される剤形で投与される、請求項1記載の方法。
【請求項13】
剤形がタブレットまたはカプセルとして製剤化される固形経口剤形としてさらに限定される、請求項12記載の方法。
【請求項14】
μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約15:1〜1:15である、請求項1記載の方法。
【請求項15】
μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約10:1〜1:10である、請求項14記載の方法。
【請求項16】
μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約5:1〜1:5である、請求項15記載の方法。
【請求項17】
μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約1:2である、請求項16記載の方法。
【請求項18】
ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩が作用物質として含まれる、請求項1記載の方法。
【請求項19】
ホスホジエステラーゼ阻害剤がV型ホスホジエステラーゼ阻害剤である、請求項18記載の方法。
【請求項20】
ホスホジエステラーゼ阻害剤が、シルデナフィル、アミノフィリン、テオフィリン、アムリノン、ミルリノン、ベスナリノン、ビンポセチン、ペモベンダン、シロスタミド、エノキシモン、ペロキシモン、ロリプラム、R020-1724、ザニプラスト、ジピリダモール、MY5445もしくはIC-351、または薬学的に許容されるその塩である、請求項18記載の方法。
【請求項21】
NMDAレセプターアンタゴニスト対ホスホジエステラーゼ阻害剤対μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストの割合が約90:1:1〜1:90:1〜1:1:90である、請求項18記載の方法。
【請求項22】
チトクロームP450阻害剤または薬学的に許容されるその塩が作用物質として含まれる、請求項1または18記載の方法。
【請求項23】
チトクロームP450阻害剤がデブリソキンヒドロキシラーゼ阻害剤である、請求項22記載の方法。
【請求項24】
チトクロームP450阻害剤が、キニジン、キニン、ナフチリジン、キサンチン、フェノキシアミノアルカン、カルバモイルイミダゾール、グアニジンイミダゾール、シメチジン(N-シアノ-N'-メチル-N"-[2[[(5-メチル-1H-イミダゾール-4イル)メチル]チオ]エチル]グアニジン)、キノリン、クロロキン(7-クロロ-4-(4-ジエチルアミノ-1-メチルブチルアミノ)キノリン)、プリマキン(8-(4-アミノ-1-メチルブチルアミノ)-6-メトキシキノリン)、トリフルオロメチルオキシムエーテル、5-メトキシ-1-[4-(トリフルオロメチル)-フェニル]-1ペンタノンO-(2-アミノエチル)オキシムとしても公知であるフルボキサミン、または薬学的に許容されるその塩である、請求項22記載の方法。
【請求項25】
NMDAレセプターアンタゴニスト対チトクロームP450阻害剤対μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストの割合が約90:1:1〜1:90:1〜1:1:90である、請求項22記載の方法。
【請求項26】
カフェインが作用物質として含まれる、請求項1、18または22のいずれか一項記載の方法。
【請求項27】
作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)V型ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩を含む、薬学的組成物。
【請求項28】
作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)チトクロームP450阻害剤または薬学的に許容されるその塩を含む、薬学的組成物。
【請求項29】
作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、c)V型ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩、およびd)チトクロームP450阻害剤または薬学的に許容されるその塩を含む、薬学的組成物。
【請求項30】
カフェインをさらに含む、請求項27、28または29のいずれか一項記載の薬学的組成物。
【請求項31】
作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)カフェインを含む、薬学的組成物。
【請求項1】
以下の工程を含む、ヒトまたはその他の哺乳動物における性機能不全を効果的に治療する方法:a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、およびb)μ-オピエートレセプターのアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩を含む、ある量の作用物質を、治療を必要とする患者に投与する工程であって、それによって組み合わせた量の該作用物質が性機能不全を治療するために有効である、工程。
【請求項2】
性機能不全が早漏である、請求項1記載の方法。
【請求項3】
作用物質が別々に投与される、請求項1記載の方法。
【請求項4】
作用物質が組み合わせて投与される、請求項1記載の方法。
【請求項5】
作用物質が性的活動の前に投与される、請求項1記載の方法。
【請求項6】
作用物質が経口的に、インプラントによって、非経口的に、皮下で、舌下で、経直腸的に、局所的に、または吸入を介して投与される、請求項1記載の方法。
【請求項7】
作用物質が経口的に投与される、請求項6記載の方法。
【請求項8】
NMDAレセプターアンタゴニストがデキストロメトルファン、デキストロルファン、ケタミン、アマンタジン、メマンチン、エリプロジル、イフェンプロジル、フェンシクリジン、MK-801、ジゾシルピン、CCPエン、フルピルチン、またはその誘導体もしくは塩である、請求項1記載の方法。
【請求項9】
NMDAレセプターアンタゴニストがデキストロメトルファンである、請求項8記載の方法。
【請求項10】
μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストが、(1R,2Rまたは1S,2S)-(ジメチルアミノメチル)-1-(3-メトキシフェニル)-シクロヘキサノール(トラマドール)、そのN-酸化物誘導体(「トラマドールN-酸化物」)、およびそのO-デスメチル誘導体(「O-デスメチルトラマドール」)、またはその混合物、立体異性体もしくはラセミ体の任意の1つである、請求項1記載の方法。
【請求項11】
μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストがトラマドールである、請求項10記載の方法。
【請求項12】
作用物質が、タブレット、経口投与のための多粒子(multiparticulate)製剤;経口投与のための液剤、徐放性製剤、懸濁剤もしくはエリキシル剤、注射可能製剤、移植可能な装置、局所用調製物、固体状態および/もしくはデポー型の経皮送達装置、坐剤、口腔内タブレット、または副鼻腔に吸入もしくは注入されるように意図された放出制御粒子製剤、もしくはスプレー、ミストもしくはその他の局所ビヒクルなどの吸入製剤からなる群より選択される剤形で投与される、請求項1記載の方法。
【請求項13】
剤形がタブレットまたはカプセルとして製剤化される固形経口剤形としてさらに限定される、請求項12記載の方法。
【請求項14】
μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約15:1〜1:15である、請求項1記載の方法。
【請求項15】
μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約10:1〜1:10である、請求項14記載の方法。
【請求項16】
μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約5:1〜1:5である、請求項15記載の方法。
【請求項17】
μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストに対するNMDAレセプターアンタゴニストの割合が約1:2である、請求項16記載の方法。
【請求項18】
ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩が作用物質として含まれる、請求項1記載の方法。
【請求項19】
ホスホジエステラーゼ阻害剤がV型ホスホジエステラーゼ阻害剤である、請求項18記載の方法。
【請求項20】
ホスホジエステラーゼ阻害剤が、シルデナフィル、アミノフィリン、テオフィリン、アムリノン、ミルリノン、ベスナリノン、ビンポセチン、ペモベンダン、シロスタミド、エノキシモン、ペロキシモン、ロリプラム、R020-1724、ザニプラスト、ジピリダモール、MY5445もしくはIC-351、または薬学的に許容されるその塩である、請求項18記載の方法。
【請求項21】
NMDAレセプターアンタゴニスト対ホスホジエステラーゼ阻害剤対μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストの割合が約90:1:1〜1:90:1〜1:1:90である、請求項18記載の方法。
【請求項22】
チトクロームP450阻害剤または薬学的に許容されるその塩が作用物質として含まれる、請求項1または18記載の方法。
【請求項23】
チトクロームP450阻害剤がデブリソキンヒドロキシラーゼ阻害剤である、請求項22記載の方法。
【請求項24】
チトクロームP450阻害剤が、キニジン、キニン、ナフチリジン、キサンチン、フェノキシアミノアルカン、カルバモイルイミダゾール、グアニジンイミダゾール、シメチジン(N-シアノ-N'-メチル-N"-[2[[(5-メチル-1H-イミダゾール-4イル)メチル]チオ]エチル]グアニジン)、キノリン、クロロキン(7-クロロ-4-(4-ジエチルアミノ-1-メチルブチルアミノ)キノリン)、プリマキン(8-(4-アミノ-1-メチルブチルアミノ)-6-メトキシキノリン)、トリフルオロメチルオキシムエーテル、5-メトキシ-1-[4-(トリフルオロメチル)-フェニル]-1ペンタノンO-(2-アミノエチル)オキシムとしても公知であるフルボキサミン、または薬学的に許容されるその塩である、請求項22記載の方法。
【請求項25】
NMDAレセプターアンタゴニスト対チトクロームP450阻害剤対μ-オピエートレセプターアゴニスト、部分アゴニストまたはアゴニスト/アンタゴニストの割合が約90:1:1〜1:90:1〜1:1:90である、請求項22記載の方法。
【請求項26】
カフェインが作用物質として含まれる、請求項1、18または22のいずれか一項記載の方法。
【請求項27】
作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)V型ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩を含む、薬学的組成物。
【請求項28】
作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)チトクロームP450阻害剤または薬学的に許容されるその塩を含む、薬学的組成物。
【請求項29】
作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、c)V型ホスホジエステラーゼ阻害剤または薬学的に許容されるその塩、およびd)チトクロームP450阻害剤または薬学的に許容されるその塩を含む、薬学的組成物。
【請求項30】
カフェインをさらに含む、請求項27、28または29のいずれか一項記載の薬学的組成物。
【請求項31】
作用物質の組み合わせの治療上有効量を含む薬学的組成物であって、組み合わせが、a)NMDAレセプターアンタゴニストまたは薬学的に許容されるその塩、b)μ-オピエートレセプターアゴニスト、部分アゴニストもしくはアゴニスト/アンタゴニスト、または薬学的に許容されるその塩、およびc)カフェインを含む、薬学的組成物。
【図1】

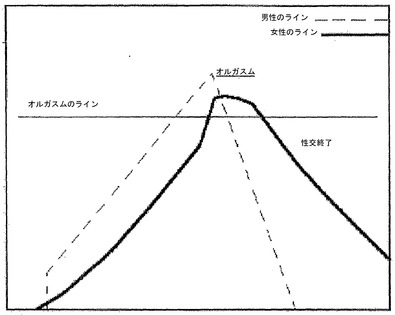
【図2】
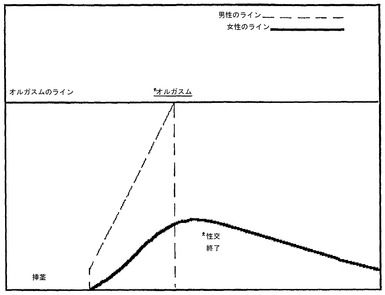
【図3】
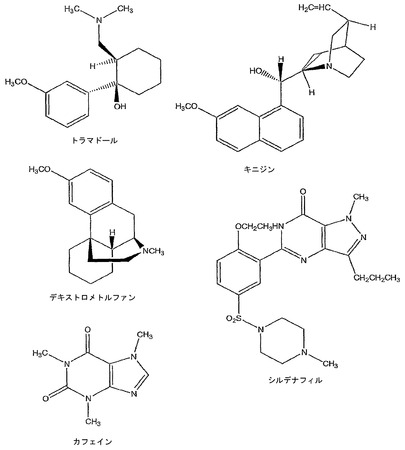
【図2】
【図3】
【公表番号】特表2009−519350(P2009−519350A)
【公表日】平成21年5月14日(2009.5.14)
【国際特許分類】
【出願番号】特願2008−545930(P2008−545930)
【出願日】平成18年12月11日(2006.12.11)
【国際出願番号】PCT/US2006/061873
【国際公開番号】WO2007/070779
【国際公開日】平成19年6月21日(2007.6.21)
【出願人】(308036169)トリニティ ラボラトリーズ インコーポレイテッド (2)
【Fターム(参考)】
【公表日】平成21年5月14日(2009.5.14)
【国際特許分類】
【出願日】平成18年12月11日(2006.12.11)
【国際出願番号】PCT/US2006/061873
【国際公開番号】WO2007/070779
【国際公開日】平成19年6月21日(2007.6.21)
【出願人】(308036169)トリニティ ラボラトリーズ インコーポレイテッド (2)
【Fターム(参考)】
[ Back to top ]