
ヒトのプログラムされたデスレセプターPD−1に対する抗体
【課題】ヒトおよびカニクイザルのPD−1に結合する単離された抗体および抗体断片の提供。
【解決手段】ヒトおよびカニクイザルのPD−1に結合する単離された抗体および抗体断片せある、いくつかの実施形態において抗体または抗体断片は、ヒトPD−L1およびヒトPD−L2のヒトPD−1への結合を遮断する。いくつかの実施形態において本発明のPD−1抗体または抗体断片は、特定な配列からなるアミノ酸配列のCDR(抗体相補性決定領域)を含む。
【解決手段】ヒトおよびカニクイザルのPD−1に結合する単離された抗体および抗体断片せある、いくつかの実施形態において抗体または抗体断片は、ヒトPD−L1およびヒトPD−L2のヒトPD−1への結合を遮断する。いくつかの実施形態において本発明のPD−1抗体または抗体断片は、特定な配列からなるアミノ酸配列のCDR(抗体相補性決定領域)を含む。
【発明の詳細な説明】
【背景技術】
【0001】
(発明の背景)
プログラムされたデスレセプター1(PD−1)は、活性化されたT細胞およびB細胞において主に発現される免疫抑制受容体である。そのリガンドとの相互作用はin vitroおよびin vivoの両方でT細胞応答を減弱させることが示されている。PD−1とそのリガンドの1つPD−L1との相互作用の遮断は、腫瘍特異的CD8+T細胞免疫を増強することが示されており、したがって免疫系による腫瘍細胞の排除において有益でありうる。
【0002】
PD−1(遺伝子Pdcd1によってコードされる)は、CD28およびCTLA−4に関連する免疫グロブリンスーパーファミリーの一員である。PD−1は、そのリガンド(PD−L1および/またはPD−L2)との結合で抗原受容体シグナル伝達を負に制御することが示されている。マウスPD−1の構造は解析されており、ヒトPD−L1を伴うマウスPD−1の共結晶構造も同様である(非特許文献1;非特許文献2)。PD−1などのファミリーの構成員は、リガンド結合に関与するIg可変型(V型)ドメインおよびシグナル伝達分子の結合に関与する細胞質側尾部を含有するI型膜貫通型糖タンパク質である。PD−1の細胞質側尾部は、2個のチロシンに基づくシグナル伝達モチーフ、ITIM(免疫受容体チロシンベース抑制モチーフ)およびITSM(免疫受容体チロシンベーススイッチモチーフ)を含有する。
【0003】
T細胞の刺激に続いて、PD−1は、その細胞質側尾部内のITSMモチーフにチロシンホスファターゼSHP−2を補充し、CD3T細胞シグナル伝達カスケードに関与するCD3ζ、PKCθおよびZAP70などのエフェクター分子の脱リン酸化を導く。PD−1がT細胞応答を下方調節する機構は、CTLA−4の機構と、両分子が重複した一連のシグナル伝達タンパク質を制御することから似てはいるが異なっている(非特許文献3)。Bennettおよび協力者らは、T細胞シグナル伝達のPD−1介在抑制が活性化シグナルおよび抑制シグナルの両方が同じ表面にある場合にだけ有効であることを示し、PD−1シグナル伝達機構が時空間的に決定されることを示唆している(非特許文献4)。
【0004】
PD−1は、活性化されたリンパ球(末梢CD4+およびCD8+T細胞、B細胞ならびに単球)で発現されることが示され、胸腺の発達の際にCD4−CD8−(ダブルネガティブ)T細胞およびNK−T細胞で発現されることも示されている。
【0005】
PD−1に対するリガンド(PD−L1およびPD−L2)は、恒常的に発現されているか、または、非造血性組織および種々の腫瘍型を含むさまざまな細胞型において誘導されうる。PD−L1は、B細胞、T細胞、骨髄系細胞および樹状細胞(DC)において発現されるが、微小血管内皮細胞および、心臓、肺などの非リンパ器官などの末梢細胞においても発現される。対照的にPD−L2は、マクロファージおよびDCにおいてのみ見いだされる。PD−1リガンドの発現様式は、末梢性寛容の維持におけるPD−1の役割について示唆的であり、末梢における自己反応性T細胞およびB細胞の応答を制御するために役立ちうる。両リガンドは、IgVおよびIgC様ドメインの両方を細胞外領域に含むI型膜貫通型受容体である。両リガンドは、未知のシグナル伝達モチーフを有する短い細胞質側領域を含む。
【0006】
今日までに多数の研究が、PD−1とそのリガンドとの相互作用がin vitroおよびin vivoにおいてリンパ球増殖の抑制を導くことを示している。PD−1/PD−L1相互作用の破壊は、T細胞増殖およびサイトカイン産生を増大させ、かつ細胞周期の進行を遮断することが示されている。Pdcd1−/−マウスでの最初の研究は、劇的な免疫表現型を同定しなかった。しかし高齢マウスは、Pdcdl欠損を戻し交配した株によって異なる自然発症の自己免疫疾患を発症した。これらは、ループス様増殖性関節炎(C57BL/6)(非特許文献5)、胎児性心筋炎(BALB/c)(非特許文献6)、およびI型糖尿病(NOD)(非特許文献7)を含む。総合的に、ノックアウト動物の分析は、PD−1が主に末梢性寛容の誘導および制御において機能するという理解を導いている。したがってPD−1経路の治療用遮断は、免疫寛容の克服において有益でありうる。そのような選択的遮断は、癌または感染症の治療およびワクチン接種(予防用または治療用のいずれでも)の際の免疫を高めることにおいて有用でありうる。
【0007】
癌におけるPD−1の役割は、文献において立証されている。腫瘍微小環境が有効な免疫性の破壊から腫瘍細胞を保護できることが知られている。近年PD−L1は、多数のマウスおよびヒトの腫瘍において発現される(かつPD−L1陰性腫瘍細胞系の大部分においてIFNγによって誘導可能である)ことが示されており、免疫回避を介在すると推定される(非特許文献8;非特許文献9)。
【0008】
ヒトにおいて(腫瘍浸潤リンパ球における)PD−1および/または(腫瘍細胞における)PD−L1の発現は、免疫組織化学によって評価された多数の原発性腫瘍の生検において見いだされている。そのような組織として、肺、肝臓、卵巣、子宮頸部、皮膚、大腸、神経膠腫、膀胱、乳房、腎臓、食道、胃、口腔扁平上皮細胞、尿路上皮細胞および膵臓の癌ならびに頭頸部の腫瘍が挙げられる(非特許文献10;非特許文献11;非特許文献12;非特許文献9;非特許文献13;非特許文献14;非特許文献15)。より著しくは、腫瘍細胞におけるPD−リガンド発現は、複数の腫瘍型にわたって癌患者の予後不良と関連している(非特許文献16で概説されている)。
【0009】
PD−1/PD−L1相互作用の遮断は、腫瘍特異的T細胞免疫の増強を導くことができ、したがって免疫系による腫瘍細胞の排除において有益でありえた。この課題に取り組むために、多数の研究が実施された。侵襲性膵臓癌のマウスモデルにおいて、T.Nomiら(非特許文献17)は、PD−1/PD−L1遮断の治療効果を示した。PD−1またはPD−L1のいずれかに対する抗体の投与は、腫瘍増殖を顕著に抑制した。抗体遮断は、腫瘍反応性CD8+T細胞の腫瘍への浸潤を効果的に促進し、IFNγ、グランザイムBおよびパーフォリンを含む抗腫瘍エフェクターの上方制御を生じた。さらに該著者らは、PD−1遮断は相乗効果をもたらすために化学療法と効果的に組み合わせうることを示した。マウスでの扁平上皮癌のモデルを使用する他の研究において、PD−1またはPD−L1の抗体遮断は、腫瘍増殖を顕著に抑制した(非特許文献18)。
【0010】
他の研究においてPD−L1でのマウス肥満細胞腫系への形質移入は、腫瘍特異的CTLクローンと同時培養した場合に腫瘍細胞の溶解の減少を生じた。抗PD−L1 mAbを加えた場合に溶解は回復した(非特許文献8)。in vivoにおいてPD−1/PD−L1相互作用の遮断は、マウス腫瘍モデルにおいて養子T細胞移入療法の効果を増大させることが示された(非特許文献9)。癌治療におけるPD−1の役割についてのさらなる証拠は、PD−1ノックアウトマウスで実施された実験によってもたらされる。PD−L1発現骨髄腫細胞は、野生型動物においてのみ増殖し(腫瘍の増殖およびそれに伴う動物の死を生じる)、PD−1欠損マウスにおいては増殖しない(非特許文献8)。
【0011】
ヒトでの研究において、非特許文献19)は、完全なヒト抗PD−1抗体を使用するPD−1遮断は、ワクチン抗原およびワクチン接種された個体由来の細胞を使用するex vivo刺激アッセイにおいて腫瘍特異的CD8+T細胞(CTL)の絶対数を増加させることを示した。同様の研究においてPD−L1の抗体遮断は、腫瘍関連抗原特異的細胞傷害性T細胞の細胞溶解活性の増強および腫瘍特異的TH細胞によるサイトカイン産生の増大を生じた(非特許文献20)。同著者らは、抗CTLA−4遮断と組み合わせて使用する場合にPD−L1遮断がin vitroでの腫瘍特異的T細胞応答を増大させることを示した。
【0012】
総合的に、PD−1/PD−L1経路は、癌治療用の抗体療法の開発のための十分に検証された標的である。抗PD−1抗体は、慢性ウイルス感染においても有用でありうる。急性ウイルス感染後に生成されたメモリーCD8+T細胞は、高度に機能的でありかつ防御免疫の重要な成分を構成する。対照的に慢性の感染は、ウイルス特異的T細胞応答の機能障害(消耗)の程度の変化によってしばしば特徴付けられ、この欠陥が宿主が持続性の病原体を排除できない主な理由である。感染の初期において機能的なエフェクターT細胞が最初に生成されるが、慢性的な感染の過程においてそれらは徐々に機能を失う。Barberら(非特許文献21)は、LCMVの研究室株で感染させたマウスが血液中および他の組織における高レベルのウイルスを生じる慢性感染を発症することを示した。これらのマウスは、最初強いT細胞応答を発現したが、T細胞の消耗により最終的には感染に屈服した。著者らは、慢性的に感染したマウスにおけるエフェクターT細胞の数および機能の低下は、PD−1とPD−L1との間の相互作用を遮断する抗体の注射によって回復できることを見いだした。
【0013】
近年、PD−1がHIVに感染した個体由来のT細胞で高度に発現されていることならびに受容体発現がT細胞機能障害および疾患の進行と関連することが示されている(非特許文献22;非特許文献23)。両方の研究においてリガンドPD−L1の遮断は、in vitroでHIV特異的IFNγ産生細胞の増殖を顕著に増加させた。
【0014】
他の研究もウイルス感染の管理におけるPD−1経路の重要性に関連している。PD−1ノックアウトマウスは、野生型マウスよりも優れたアデノウイルス感染の管理を示す(非特許文献24)。同様にHBV特異的T細胞のHBVトランスジェニック動物への養子移入は、肝炎を惹起する(非特許文献25)。これらの動物の疾患の状態は、肝臓での抗原認識および肝臓細胞によるPD−1の上方制御の結果として変動する。
【先行技術文献】
【非特許文献】
【0015】
【非特許文献1】Zhang, X.ら、Immunity20巻:337〜347頁(2004年)
【非特許文献2】Linら、Proc.Natl.Acad.Sci.USA105巻:3011〜6頁(2008年)
【非特許文献3】Parryら、Mol.Cell Biol.25巻:9543〜9553頁
【非特許文献4】Bennett F.ら、J Immunol.170巻:711〜8頁(2003年)
【非特許文献5】Nishimura H.ら、Int.Immunol.10巻:1563〜1572頁(1998年)
【非特許文献6】Nishimura H.ら、Science291巻:319〜322頁(2001年)
【非特許文献7】Wang J.ら、Proc.Natl.Acad.Sci.U.S.A 102巻:11823〜11828頁(2005年)
【非特許文献8】Iwai Y.ら、Proc.Natl.Acad.Sci.U.S.A.99巻:12293〜12297頁(2002年)
【非特許文献9】Strome S.E.ら、Cancer Res.、63巻:6501〜6505頁(2003年)
【非特許文献10】Brown J.A.ら、J.Immunol.170巻:1257〜1266頁(2003年)
【非特許文献11】Dong H.ら、Nat.Med.8巻:793〜800頁(2002年)
【非特許文献12】Wintterleら、Cancer Res.63巻:7462〜7467頁(2003年)
【非特許文献13】Thompson R.H.ら、Cancer Res.66巻:3381〜5頁(2006年)
【非特許文献14】Thompsonら、Clin.Cancer Res.13巻:1757〜61頁(2007年)
【非特許文献15】Nomi T.ら、Clin.Cancer Res.13巻:2151〜7頁(2007年)
【非特許文献16】Okazaki and Honjo、Int.Immunol.19巻:813〜824頁(2007年)
【非特許文献17】Clin.Cancer Res.13巻:2151〜2157頁(2007年)
【非特許文献18】Tsushima F.ら、Oral Oncol.42巻:268〜274頁(2006年)
【非特許文献19】R.M.Wongら(Int.Immunol.19巻:1223〜1234頁(2007年)
【非特許文献20】Blank C.ら、Int.J.Cancer119巻:317〜327頁(2006年)
【非特許文献21】Barberら、Nature439巻:682〜687頁(2006年)
【非特許文献22】Dayら、Nature443巻:350〜4頁(2006年)
【非特許文献23】Trautmann L.ら、Nat.Med.12巻:1198〜202頁(2006年)
【非特許文献24】Iwaiら、J.Exp.Med.198巻:39〜50頁(2003年)
【非特許文献25】Isogawa M.ら、Immunity23巻:53〜63頁(2005年)
【発明の概要】
【課題を解決するための手段】
【0016】
(発明の簡単な要旨)
本発明は、ヒトおよびカニクイザルのPD−1に結合する単離された抗体および抗体断片を提供する。いくつかの実施形態において抗体または抗体断片は、ヒトPD−L1およびヒトPD−L2のヒトPD−1への結合を遮断する。いくつかの実施形態において本発明のPD−1抗体または抗体断片は、配列番号9、10、11、12、13、14、15、16、17、18、19および20から選択される1つまたは複数のCDR(抗体相補性決定領域)を含み、さらなる実施形態においては、配列番号12、13、14、18、19および20の1つまたは複数の重鎖CDRならびに/または配列番号9、10、11、15、16および17の軽鎖CDRを含む。いくつかの実施形態において抗体または抗体断片は、キメラ抗体、ヒト抗体、ヒト化抗体またはその断片である。
本発明は、例えば以下の項目を提供する。
(項目1)
a.配列番号9、10、11、15、16および17からなる群から選択される少なくとも1つのCDR、もしくは任意の該配列の変異体、ならびに/または
b.配列番号12、13、14、18、19および20からなる群から選択される少なくとも1つのCDR、もしくは任意の該配列の変異体
を含む、ヒトPD−1に結合する単離された抗体または抗体断片。
(項目2)
a.配列番号9、10および11の軽鎖CDR、もしくは任意の該配列の変異体、ならびに配列番号12、13および14の重鎖CDR、もしくは任意の該配列の変異体、または
b.配列番号15、16および17の軽鎖CDR、もしくは任意の該配列の変異体、ならびに配列番号18、19および20の重鎖CDR、もしくは任意の該配列の変異体
を含む、項目1に記載の抗体または抗体断片。
(項目3)
a.i.配列番号5もしくはその変異体、
ii.配列番号7もしくはその変異体、
iii.配列番号30のアミノ酸残基20から139もしくはその変異体、および
iv.配列番号30のアミノ酸残基20から139に少なくとも90%の相同性を有するアミノ酸配列
からなる群から選択されるアミノ酸配列を含む重鎖可変領域
を含み、
b.i.配列番号6もしくはその変異体、
ii.配列番号8もしくはその変異体、
iii.配列番号32のアミノ酸残基20から130もしくはその変異体、
iv.配列番号33のアミノ酸残基20から130もしくはその変異体、
v.配列番号34のアミノ酸残基20から130もしくはその変異体、および
vi.配列番号32、33もしくは34のアミノ酸残基20から130に少なくとも90%の相同性を有するアミノ酸配列
からなる群から選択されるアミノ酸配列を含む軽鎖可変領域
をさらに含む、項目2に記載の抗体または抗体断片。
(項目4)
a.i.配列番号31のアミノ酸残基20から466もしくはその変異体、および
ii.配列番号35のアミノ酸残基20から469もしくはその変異体
からなる群から選択されるアミノ酸配列を含む重鎖、
ならびに
b.i.配列番号36のアミノ酸残基20から237もしくはその変異体、
ii.配列番号37のアミノ酸残基20から237もしくはその変異体、および
iii.配列番号38のアミノ酸残基20から237もしくはその変異体
からなる群から選択されるアミノ酸配列を含む軽鎖
を含む、項目1に記載の抗体。
(項目5)
(1つまたは複数の)任意の前記変異体が3個までの保存的に改変されたアミノ酸置換を含みうる、項目1から4のいずれかに記載の抗体または抗体断片。
(項目6)
a.ヒト重鎖定常領域、もしくは20個までの保存的に改変されたアミノ酸置換を含むその変異体、および/または
b.ヒト軽鎖定常領域、もしくは20個までの保存的に改変されたアミノ酸置換を含むその変異体
をさらに含む、項目1から3のいずれかに記載の抗体。
(項目7)
前記ヒト重鎖定常領域がγ4もしくはγ1ヒト重鎖定常領域、または20個までの保存的に改変されたアミノ酸置換を含むその変異体を含む、項目6に記載の抗体。
(項目8)
a.約100pMのKDまたはそれより低いKDでヒトPD−1に結合する、
b.約30pMのKDまたはそれより低いKDでヒトPD−1に結合する、
c.配列番号31のアミノ酸配列を含む重鎖および配列番号32のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合する、
d.配列番号31のアミノ酸配列を含む重鎖および配列番号33のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合する、
e.約7.5×105 1/M・sまたはそれより速いkassocでヒトPD−1に結合する、
f.約1×106 1/M・sまたはそれより速いkassocでヒトPD−1に結合する、
g.約2×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合する、
h.約2.7×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合する、
i.約3×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合する、
j.約1nMのIC50またはそれより低いIC50でヒトPD−L1またはヒトPD−L2のヒトPD−1への結合を遮断する、
項目1から7のいずれかに記載の抗体または抗体断片。
(項目9)
項目1から4に記載のいずれかの抗体とPD−1上の結合エピトープについて競合し、以下の特徴:
a.約100pMのKDまたはそれより低いKDでヒトPD−1に結合する、
b.約30pMのKDまたはそれより低いKDでヒトPD−1に結合する、
c.配列番号31のアミノ酸配列を含む重鎖および配列番号32のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合する、
d.配列番号31のアミノ酸配列を含む重鎖および配列番号33のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合する、
e.約7.5×105 1/M・sまたはそれより速いkassocでヒトPD−1に結合する、
f.約1×106 1/M・sまたはそれより速いkassocでヒトPD−1に結合する、
g.約2×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合する、
h.約2.7×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合する、
i.約3×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合する、または
j.約1nMのIC50またはそれより低いIC50でヒトPD−L1またはヒトPD−L2のヒトPD−1への結合を遮断する、
のうち1つを有する
単離された抗体または抗体断片。
(項目10)
a.キメラ抗体もしくはその断片、
b.ヒト抗体もしくはその断片、
c.ヒト化抗体もしくはその断片、または
d.Fab、Fab’、Fab’−SH、Fv、scFv、F(ab’)2およびダイアボディーからなる群から選択される抗体断片
である、項目1から9のいずれかに記載の抗体または抗体断片。
(項目11)
T細胞の活性化を増大させる、項目1から10のいずれかに記載の抗体または抗体断片。
(項目12)
項目1から7のいずれか一項に記載の抗体または抗体断片をコードする単離されたポリヌクレオチド。
(項目13)
項目12に記載の単離されたポリヌクレオチドを含む発現ベクター。
(項目14)
項目13に記載の発現ベクターを含む宿主細胞。
(項目15)
a.前記核酸配列が発現される条件下で、培地中で項目14に記載の宿主細胞を培養し、それにより前記軽鎖および前記重鎖可変領域を含むポリペプチドを産生するステップと、
b.該宿主細胞または該培地から該ポリペプチドを回収するステップと
を含む、項目1から11のいずれか一項に記載の抗体または抗体断片を産生する方法。
(項目16)
薬学的に許容される担体または希釈剤と組み合わせて、項目1から11のいずれか一項に記載の抗体または抗体断片を含む組成物。
(項目17)
免疫細胞を項目1から11に記載の抗体のいずれか1つと接触させるステップを含む、該免疫細胞の活性を増大させる方法。
(項目18)
治療有効量の項目1から11のいずれか一項に記載の抗体または抗体断片を、それを必要とする対象に投与するステップを含む、免疫細胞の活性を増大させる方法。
(項目19)
a.癌の治療のため、
b.感染症もしくは感染性疾患の治療のため、または
c.ワクチンアジュバントとして
使用される、項目18に記載の方法。
(項目20)
前記対象由来の試料においてex vivoでT細胞活性化を測定するステップをさらに含み、T細胞活性における増大が前記治療を継続すべきであることを示すか、または前記治療が成功する可能性を予測する、項目18に記載の方法。
(項目21)
T細胞活性における前記増大が
a.IL−2、TNFα、IL−17、IFNγ、GM−CSF、RANTES、IL−6、IL−8、IL−5およびIL−13からなる群から選択される1つまたは複数のサイトカインのSEB誘発産生を測定するステップ、または
b.IL−2、TNFα、IL−17、IFNγ、GM−CSF、RANTES、IL−6、IL−8、IL−5およびIL−13からなる群から選択されるサイトカインのTT誘発産生を測定するステップ
によって決定される、項目20に記載の方法。
(項目22)
a.免疫細胞活性化を増大させるための、
b.癌を治療するための、または
c.感染症もしくは感染性疾患を治療するための
薬剤の調製のための、項目1から11のいずれか一項に記載の抗体または抗体断片の使用。
(項目23)
診断的使用のための、項目1から11のいずれか一項に記載の抗体または抗体断片の使用。
【0017】
一実施形態において本発明は、配列番号9、10および11のCDR、もしくは任意の該配列の変異体を含む軽鎖、ならびに/または配列番号12、13および14のCDR、もしくは任意の該配列の変異体を含む重鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0018】
他の実施形態において本発明は、配列番号15、16および17のCDR、もしくは任意の該配列の変異体を含む軽鎖、ならびに/または配列番号18、19および20のCDR、もしくは任意の該配列の変異体を含む重鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0019】
一実施形態において本発明は、配列番号5の重鎖可変領域もしくはその変異体、および/または配列番号6もしくはその変異体を含む軽鎖可変領域を含む抗体または抗原結合断片を含む。
【0020】
一実施形態において本発明は、配列番号7の重鎖可変領域もしくはその変異体、および/または配列番号8もしくはその変異体を含む軽鎖可変領域を含む抗体または抗原結合断片を提供する。
【0021】
一実施形態において本発明は、配列番号30のアミノ酸残基20から139もしくはその変異体を含む重鎖可変領域、および/または配列番号32のアミノ酸残基20から130もしくはその変異体を含む軽鎖可変領域を含む抗体または抗原結合断片を含む。
【0022】
一実施形態において本発明は、配列番号30のアミノ酸残基20から139もしくはその変異体を含む重鎖可変領域、および/または配列番号33のアミノ酸残基20から130もしくはその変異体を含む軽鎖可変領域を含む抗体または抗原結合断片を含む。
【0023】
一実施形態において本発明は、配列番号30のアミノ酸残基20から139もしくはその変異体を含む重鎖可変領域、および/または配列番号34のアミノ酸残基20から130もしくはその変異体を含む軽鎖可変領域を含む抗体または抗原結合断片を含む。
【0024】
一実施形態において本発明は、配列番号30のアミノ酸残基20から139に少なくとも90%の相同性を有するアミノ酸配列を含む重鎖可変領域、および/または配列番号32、33または34のアミノ酸残基20から130に少なくとも90%の相同性を有するアミノ酸配列を含む軽鎖可変領域を含む抗体または抗原結合断片を含む。
【0025】
一実施形態において本発明は、配列番号31のアミノ酸残基20から466もしくはその変異体を含む重鎖、および/または配列番号36のアミノ酸残基20から237もしくはその変異体を含む軽鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0026】
一実施形態において本発明は、配列番号31のアミノ酸残基20から466もしくはその変異体を含む重鎖、および/または配列番号37のアミノ酸残基20から237もしくはその変異体を含む軽鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0027】
一実施形態において本発明は、配列番号31のアミノ酸残基20から466もしくはその変異体を含む重鎖、および/または配列番号38のアミノ酸残基20から237もしくはその変異体を含む軽鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0028】
一実施形態において本発明は、配列番号35のアミノ酸残基20から469もしくはその変異体を含む重鎖、および/または配列番号36のアミノ酸残基20から237もしくはその変異体を含む軽鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0029】
一実施形態において本発明は、配列番号35のアミノ酸残基20から469もしくはその変異体を含む重鎖、および/または配列番号37のアミノ酸残基20から237もしくはその変異体を含む軽鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0030】
一実施形態において本発明は、配列番号35のアミノ酸残基20から469もしくはその変異体を含む重鎖、および/または配列番号38のアミノ酸残基20から237もしくはその変異体を含む軽鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0031】
任意の上記実施形態において本発明の抗体または抗体断片の変異体は、1個、2個または3個の保存的に改変されたアミノ酸置換を含みうる。
【0032】
任意の上記実施形態において本発明の抗体または抗体断片は、ヒト重鎖定常領域もしくは20個までの保存的に改変されたアミノ酸置換を含むその変異体、および/またはヒト軽鎖定常領域もしくは20個までの保存的に改変されたアミノ酸置換を含むその変異体、を含みうる。いくつかの実施形態において変異体は、10個までの保存的に改変されたアミノ酸置換を含みうる。いくつかの実施形態において変異体は、5個までの保存的に改変されたアミノ酸置換を含みうる。いくつかの実施形態において変異体は、3個までの保存的に改変されたアミノ酸置換を含みうる。任意の上記実施形態においてヒト重鎖定常領域またはその変異体は、IgG1またはIgG4アイソタイプのものでありうる。
【0033】
任意の上に記載の実施形態において本発明の抗体または抗体断片は、約100pM以下のKDでヒトPD−1に結合できる。他の実施形態において抗体または抗体断片は、約30pM以下のKDでヒトPD−1に結合できる。他の実施形態において抗体または抗体断片は、配列番号31のアミノ酸配列を含む重鎖および配列番号32のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合できる。他の実施形態において抗体または抗体断片は、配列番号31のアミノ酸配列を含む重鎖および配列番号33のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合できる。
【0034】
任意の上に記載の実施形態において本発明の抗体または抗体断片は、約7.5×105 1/M・sまたはそれより速いkassocでヒトPD−1に結合できる。一実施形態において抗体または抗体断片は、約1×106 1/M・sまたはそれより速いkassocでヒトPD−1に結合できる。
【0035】
任意の上に記載の実施形態において抗体または抗体断片は、約2×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合できる。一実施形態において抗体または抗体断片は、約2.7×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合できる。一実施形態において抗体または抗体断片は、約3×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合できる。
【0036】
KD、kassocおよびkdissoc値は、任意の利用可能な方法を使用して測定されうる。好ましい実施形態において解離(dissaociation)定数は、生物光学干渉法(bio−light interferometry)(例えば実施例2で記載されるForteBio Octet法)を使用して測定される。他の好ましい実施形態において解離(disassociation)定数は、表面プラズモン共鳴(例えばBiacore)またはKinexaを使用して測定されうる。
【0037】
さらに任意の上に記載の実施形態において本発明の抗体または抗体断片は、ヒトPD−L1またはヒトPD−L2のヒトPD−1への結合を約1nM以下のIC50で遮断できる。リガンド結合の遮断は、例えば本明細書の(hereub)実施例に記載のFACS法またはFMAT法などの当技術分野において公知の任意の方法を使用して測定することができ、かつIC50を算出されうる。
【0038】
本発明は、ヒトPD−1上の結合エピトープに対して上に記載の任意の抗体と競合し、かつヒトPD−L1またはPD−L2のヒトPD−1への結合を約1nM以下のIC50で遮断する抗体または抗体断片も含む。
【0039】
本発明は、ヒトPD−1上の結合エピトープについて上に記載の任意の抗体と競合し、かつ約100pM以下のKDでヒトPD−1に結合する抗体または抗体断片も含む。一実施形態において抗体または抗体断片は、約30pM以下のKDでヒトPD−1に結合する。
【0040】
本発明は、ヒトPD−1上の結合エピトープについて上に記載の任意の抗体と競合し、かつ配列番号31のアミノ酸配列を含む重鎖および配列番号32のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合する抗体または抗体断片も含む。
【0041】
本発明は、ヒトPD−1上の結合エピトープについて上に記載の任意の抗体と競合し、かつ配列番号31のアミノ酸配列を含む重鎖および配列番号33のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合する抗体または抗体断片も含む。
【0042】
本発明は、ヒトPD−1上の結合エピトープについて上に記載の任意の抗体と競合し、かつ約7.5×105 1/M・sまたはそれより速いkassocでヒトPD−1に結合する抗体または抗体断片も含む。一実施形態において抗体または抗体断片は、約1×106 1/M・sまたはそれより速いkassocでヒトPD−1に結合できる。
【0043】
本発明は、ヒトPD−1上の結合エピトープについて上に記載の任意の抗体と競合し、かつ約2×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合する抗体または抗体断片も含む。一実施形態において抗体または抗体断片は、約2.7×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合できる。一実施形態において抗体または抗体断片は、約3×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合できる。
【0044】
いくつかの実施形態において本発明の抗体または抗体断片は、キメラ抗体またはキメラ抗体の断片である。
【0045】
いくつかの実施形態において本発明の抗体または抗体断片は、ヒト抗体またはヒト抗体の断片である。
【0046】
いくつかの実施形態において本発明の抗体または抗体断片は、ヒト化抗体またはヒト化抗体の断片である。
【0047】
いくつかの実施形態において本発明の抗体断片は、Fab、Fab’、Fab’−SH、Fv、scFvまたはF(ab’)2抗体断片である。
【0048】
いくつかの実施形態において本発明の抗体断片は、ダイアボディーである。
【0049】
本発明は、ヒトPD−1に結合する上に記載の抗体または抗体断片のいずれか1つを含む二重特異性抗体も含む。
【0050】
いくつかの実施形態において本発明の単離された抗PD−1抗体および抗体断片は、当業者に公知の典型的な手段によって測定されるT細胞活性化(免疫細胞増殖の増大、サイトカイン分泌の増大またはCD25および/もしくはCD69などの活性化マーカーの発現を非限定的に含む)を増大させる。
【0051】
上に記載の任意の実施形態において本発明の抗体または抗体断片は、ex vivoまたはin vivoでのブドウ球菌エンテロトキシンBまたは破傷風トキソイドでの刺激後に免疫応答を増強できる。増大した免疫活性化は、例えば免疫細胞(T細胞など)の増殖または免疫細胞によるサイトカイン産生(例えばT細胞によるINFγもしくはIL−2の産生)を定量することである当業者に公知の方法を使用して決定できる。
【0052】
本発明は、本発明の抗PD−1抗体および抗体断片をコードする核酸も含む。配列番号5から20および30〜38において開示のアミノ酸配列(リーダー配列を含むまたは含まない)のいずれか1つをコードする核酸は、本発明に含まれる。配列番号1から4および21から29を含む核酸(リーダー配列をコードする核酸を含むまたは含まない)も、本発明に含まれる。
【0053】
本発明は、本発明の抗体または抗体断片をコードする核酸を含む細胞および発現ベクターも含む。さらに本発明は、(a)核酸配列が発現される条件下で、培地中で本発明の抗体または抗体断片をコードする核酸を含む宿主細胞を培養し、それにより軽鎖および重鎖可変領域を含むポリペプチドを産生するステップと、(b)宿主細胞または培地からポリペプチドを回収するステップとを含む、本発明の抗体または抗体断片を産生する方法を含む。
【0054】
本発明は、薬学的に許容される担体または希釈剤と組み合わせて、本発明の抗体または抗体断片を含む組成物も含む。
【0055】
本発明は、本発明の抗体または抗体断片の治療有効量をそれを必要とする対象に投与するステップを含む、免疫細胞の活性を増大させる方法も含む。一実施形態においてこの方法は、癌を治療するために使用されうる。他の実施形態においてこの方法は、感染症または感染性疾患を治療するために使用されうる。さらに他の実施形態においてこの方法は、ワクチンアジュバントとして使用されうる。いくつかの実施形態においてこの方法は、第2の治療用薬物または治療様式をさらに投与するステップを含む。
【0056】
いくつかの実施形態において本発明は、免疫細胞の活性を増大させる方法であって、治療有効量の本発明の抗体または抗体断片を、それを必要とする対象に投与するステップを含み、対象由来の試料におけるT細胞活性化をex vivoで測定するステップをさらに含み、T細胞活性における増大が治療を継続すべきであることを示す方法を含む。他の実施形態において本発明は、免疫細胞の活性を増大させる方法であって、治療有効量の本発明の抗体または抗体断片を、それを必要とする対象に投与するステップを含み、対象由来の試料におけるT細胞活性化をex vivoで測定するステップをさらに含み、T細胞活性化における増大が治療が成功する可能性を予測する方法を含む。一実施形態においてT細胞活性における増大は、(i)IL−2、TNFα、IL−17、IFNγ、GM−CSF、RANTES、IL−6、IL−8、IL−5およびIL−13からなる群から選択される1つまたは複数のサイトカインのSEB誘発産生を測定するステップ、または(ii)IL−2、TNFα、IL−17、IFNγ、GM−CSF、RANTES、IL−6、IL−8、IL−5およびIL−13からなる群から選択されるサイトカインのTT誘発産生を測定するステップによって決定される。
【0057】
本発明は、免疫応答を増大させる薬物の調製のための本発明の抗PD−1抗体または抗体断片の使用も含む。
【0058】
本発明は、癌を治療するための薬物の調製のための本発明の抗PD−1抗体または抗体断片の使用も含む。
【0059】
本発明は、ワクチンアジュバントとしての本発明の抗PD−1抗体または抗体断片の使用も含む。
【0060】
本発明は、細菌性毒素または放射性毒素などの治療剤に連結した本発明の抗PD−1抗体または抗体断片を含む免疫複合体も含む。細胞毒作用物質の非限定的な例として、タキソール、サイトカラシンB、マイトマイシン、エトポシドおよびビンクリスチンまたは他の代謝拮抗剤、アルキル化剤、抗生物質および抗有糸分裂剤が挙げられる。
【0061】
本発明は、免疫細胞と本発明の抗体または抗体断片のいずれか1つとを接触させるステップを含む、免疫細胞の活性を増大させるまたは免疫細胞の下方調節を低減する方法も含む。この方法は、癌もしくは感染性疾患(慢性ウイルス性感染症など)を治療するために使用されることができ、または予防用もしくは治療用ワクチンのためのアジュバントとして使用されうる。
【0062】
本発明は、免疫細胞を抗原および抗PD−1抗体または抗体断片と、抗原への免疫応答が増大されるかまたは増強されるように接触させるステップを含む抗原への免疫応答を増大させる方法も含む。この方法は、in vivo(対象中で)またはex vivoで実施されうる。
【0063】
いくつかの実施形態において抗PD−1抗体または抗体断片は、第2の治療薬または治療様式と組み合わされうる。一実施形態において抗PD−1抗体または抗体断片は、組換えサイトカインまたは分泌された免疫性因子の適用を伴う癌治療と組み合わされうる。組合せの非限定的な例として、抗PD−1抗体と組換えIL−2または組換えIFNα2とを黒色腫または腎細胞癌の治療のために組み合わせることが挙げられる。組換えIL−2は、癌患者においてT細胞の増殖を増強する。組換えIFNα2は、癌細胞の増殖を抑制するが、治療を受ける患者において癌細胞、抗原提示細胞および他の体細胞上のPD−1に対する抑制型リガンドの発現も増大させる。抗PD−1は、癌または感染性疾患の治療のために有用であると考えられうる他のサイトカインと組み合わされうる。
【0064】
いくつかの実施形態において抗PD−1抗体または抗体断片は、癌または感染性疾患を予防または治療するためにワクチンと組み合わされうる。非限定的な例として抗PD−1は、治療される癌または感染症に関連する1つもしくは複数の抗原を含有するタンパク質、ペプチドもしくはDNAのワクチンと、またはそのような抗原でパルスした樹状細胞を含むワクチンと組み合わされうる。他の実施形態は、(弱毒化)癌細胞ワクチンまたはまるごとのウイルスワクチンを伴う抗PD−1の使用を含む。一実施形態は、GM−CSFを分泌するように工学的に操作されたまるごとの細胞の癌ワクチンと抗PD−1療法との組合せを含む。
【0065】
いくつかの実施形態において抗PD−1抗体または抗体断片は、癌または感染性疾患のケアの標準法であると考えられる治療と組み合わされうる。そのような組合せについての論理的根拠は、抗PD−1によって同時に増大する免疫活性化がケア治療の標準法への初期の臨床反応を誘発または促進し、持続的臨床反応および疾患の長期免疫管理を誘導することである。
【0066】
一実施形態において抗PD−1抗体または抗体断片での治療は、化学療法と組み合わされうる。細胞傷害性薬物を使用する化学療法は、癌細胞死を生じ、それにより腫瘍抗原の放出が増大する。腫瘍抗原の可用性のそのような増大は、抗PD−1治療との相乗作用を生じうる。非限定的な例は、黒色腫の治療のためのデカルバジンもしくはテモゾロミドまたは膵臓癌のためのゲムシタビンの使用によって提供される。
【0067】
一実施形態において抗PD−1抗体または抗体断片での治療は、放射線療法と組み合わされうる。放射線療法は、癌細胞死を生じ、免疫細胞の提示および活性化のための腫瘍抗原の可用性が増大する。
【0068】
別の実施形態において抗PD−1抗体または抗体断片での治療は、対象から癌細胞を除く手術と組み合わされうる。
【0069】
他の実施形態において抗PD−L1抗体または抗体断片は、ホルモン剥奪もしくは血管新生の抑制のために使用される標的薬剤、または腫瘍細胞において活性な標的タンパク質を含み、PD−1遮断と相乗効果を生じうる療法と組み合わされることができ、全ては、腫瘍細胞死および免疫刺激腫瘍抗原の可用性の増強を生じる。抗PD−1抗体または抗体断片との組合せにおいて、増大したT細胞活性化は、癌の持続的な免疫管理を生じうる。
【0070】
いくつかの実施形態において抗PD−1抗体または抗体断片は、癌または感染性疾患の治療のために有用である他の治療用抗体と組み合わされうる。非限定的な例は、抗PD−1抗体とHer2/neuを標的とするまたはEGF受容体を標的とする抗体との組合せによって提供される。他の非限定的な例において抗PD−1抗体または抗体断片は、VEGFまたはその受容体を標的とする治療と組み合わされる。他の実施形態において抗PD−1抗体または抗体断片は、抗CTLA−4と組み合わされる。さらに他の非限定的な例において抗PD−1は、RSVを標的とする抗体と組み合わされる。
【図面の簡単な説明】
【0071】
【図1】ハイブリドーマ上清由来の固定化抗体は、固定化した抗CD3および可溶性抗CD28で刺激したジャーカットE6.2.11細胞によるIL−2分泌を低減できることを示す実験結果を示すグラフである。
【図2】ヒトPD−1に対する抗体がPD−1に結合することを示す実験結果を示すグラフである。図2Aは、タンパク質ELISAにおける、精製PD−1/Fcへの抗PD−1抗体の用量依存的結合を示すグラフである。図2Bは、CELISAにおける、hPD−1を形質移入したCHO細胞の表面で発現されるPD−1への抗PD−1抗体の用量依存的結合を示すグラフである。
【図3】PD−1に対する抗体がヒトPD−1を形質移入したCHO細胞へのPD−L1およびPD−L2の結合について競合することを示すFMAT実験の結果を示すグラフである。図3Aは、hPD−1.08AおよびhPD−1.09ならびに少ない程度でのJ116によるPD−L1の結合の用量依存的な抑制を示すグラフである。図3Bは、PD−L2の用量依存的な抑制を示すグラフである。
【図4】健常ドナー血液細胞によるSEB刺激IL−2産生は、抗PD−1、抗PD−L1または抗CTLA−4抗体の存在下で増強されることを示す実験結果を示す棒グラフである。棒は、ドナー間のIL−2における平均倍数増加(±SEM)を示す。各棒内の数値は、示したドナー数を示す。マウス(m)IgG1は、抗PD−1.08A(08A)、抗PD−1.09A(09A)および抗PD−L1についてのアイソタイプ対照である。マウス(m)IgG2は、抗CTLA−4についてのアイソタイプ対照である。各IL−2値を倍数変化を決定するためにそれ自体の対照と比較する(IL−2の倍数変化4は、SEB単独の場合と比較した場合のIL−2産生における400%増大を意味する)。なし=SEB単独
【図5】リコール抗原破傷風トキソイドで刺激された場合に抗PD−1抗体がT細胞増殖およびサイトカイン分泌(IL−2およびIFNγ)を、促進することを示す実験結果を示すグラフである。図5は、濃度依存的IFNγ分泌を示す。
【図6】生物光学干渉法によって測定した抗PD−1抗体についてのkassocおよびkdissoc速度を示すグラフである。斜線は、理論的に算出されるKD値を示す。抗体は、右側にKDによる昇順で列挙される。
【図7】健常ドナー血液細胞によるSEB刺激IL−2産生は、25μg/mlマウス(09A)、またはヒト化抗PD−1抗体(h409A11、h409A16およびh409A17)の存在下で増大されることを示す実験結果を示す棒グラフである。棒は、ドナー3名の間のIL−2における平均倍数増加(±SEM)を示す。マウス(m)IgG1は、抗PD−1.09A(09A)についてのアイソタイプ対照である。ヒト(h)IgG4は、h409A11、h409A16およびh409A17抗体についてのアイソタイプ対照である。各IL−2値を倍数変化を決定するためにそれ自体の対照と比較する。なし=SEB単独
【発明を実施するための形態】
【0072】
(発明の詳細な説明)
略語および定義
本発明の詳細な記載および実施例を通じて以下の略語が使用される。
【0073】
hPD−1.08A マウスモノクローナル抗hPD−1抗体
hPD−1.09A マウスモノクローナル抗hPD−1抗体
08A−VH hPD−1.08Aハイブリドーマから単離されたVH
08A−VK hPD−1.08Aハイブリドーマから単離されたVK
09A−VH hPD−1.09Aハイブリドーマから単離されたVH
09A−VK hPD−1.09Aハイブリドーマから単離されたVK
c109A hPD−1.09A抗体のキメラIgG1バージョン
c109A−VH hIgG1定常領域に融合しているマウス09A−VHからなるキメラ重鎖
c109A−VK ヒトκ定常領域に融合しているマウス09A−VKからなるキメラ軽鎖
109A−H 復帰突然変異を含まないヒト化IgG1 09A重鎖配列
409A−H FWR復帰突然変異を含まないヒト化IgG4−09A重鎖配列
K09A−L−11 長さ11AAのCDR1を独自に有するフレームワークを含むヒト化09A−κ配列
K09A−L−16 長さ16AAのCDR1を独自に有するフレームワークを含むヒト化09A−κ配列
K09A−L−17 長さ17AAのCDR1を独自に有するフレームワークを含むヒト化09A−κ配列
h409A11 409A−Hの配列を含む重鎖およびK09A−L−11の配列を含む軽鎖を含む09A抗体のヒト化IgG4バージョン
h409A16 409A−Hの配列を含む重鎖およびK09A−L−16の配列を含む軽鎖を含む09A抗体のヒト化IgG4バージョン
h409A17 409A−Hの配列を含む重鎖およびK09A−L−17の配列を含む軽鎖を含む09A抗体のヒト化IgG4バージョン
hPD−1 ヒトPD−1タンパク質
CDR 免疫グロブリン可変領域中の相補性決定領域、Kabat番号付け方式を使用して定義される
EC50 50%有効性または結合を生じる濃度
ELISA 酵素結合免疫吸着検定法
FW 抗体フレームワーク領域、CDR領域を除く免疫グロブリン可変領域
HRP 西洋わさびペルオキシダーゼ
IL−2 インターロイキン2
IFN インターフェロン
IC50 50%抑制を生じる濃度
IgG 免疫グロブリンG
Kabat Elvin A Kabatによって開発された免疫グロブリン配列比較(alignment)および番号付け方式
mAb モノクローナル抗体
MES 2−(N−モルホリノ)エタンスルホン酸
NHS 正常ヒト血清
PCR ポリメラーゼ連鎖反応
SAM ヒツジ抗マウス(IgG)ポリクローナル抗体
V領域 異なる抗体間の配列において可変であるIgG鎖のセグメント。軽鎖におけるKabat残基109および重鎖における113に及ぶ。
【0074】
VH 免疫グロブリン重鎖可変領域
VK 免疫グロブリンκ軽鎖可変領域。
【0075】
「抗体」は、リガンドのその受容体への結合の抑制または受容体のリガンド誘発シグナル伝達の抑制によるなどの所望の生物学的活性を示す任意の形態の抗体を指す。したがって「抗体」は、最も広義で使用され、具体的には非限定的にモノクローナル抗体(全長モノクローナル抗体含む)、ポリクローナル抗体および多重特異性抗体(例えば二重特異性抗体)に及ぶ。
【0076】
「抗体断片」および「抗体結合断片」は、典型的には親抗体の抗原結合領域または可変領域(例えば1つまたは複数のCDR)の少なくとも一部を含む抗原結合断片および抗体の類似物を意味する。抗体断片は、親抗体の結合特異性の少なくとも一部を保持する。典型的には抗体断片は、活性をモルベースで表す場合に親の結合活性の少なくとも10%の結合活性を保持する。好ましくは抗体断片は、標的に対して親抗体の結合親和性の少なくとも20%、50%、70%、80%、90%、95%もしくは100%またはそれを超える結合親和性を保持する。抗体断片の例として、それだけに限らないがFab、Fab’、F(ab’)2およびFv断片、ダイアボディー、直鎖抗体、例えばsc−Fv、ユニボディー(Genmab由来の技術)である単鎖抗体分子、ナノボディー(Domantis由来の技術)、ドメイン抗体(Ablynx由来の技術)、ならびに抗体断片から形成された多重特異性抗体が挙げられる。工学的に操作された抗体変異体は、Holliger and Hudson(2005年)Nat.Biotechnol.23巻:1126〜1136頁において概説されている。
【0077】
「Fab断片」は、軽鎖1つならびに1つの重鎖のCH1および可変領域で構成されている。Fab分子の重鎖は、他の重鎖分子とジスルフィド結合を形成できない。
【0078】
「Fc領域」は、抗体のCH1およびCH2ドメインを含む2つの重鎖断片を含有する。2つの重鎖断片は、2つ以上のジスルフィド結合およびCH3ドメインの疎水性相互作用によって維持している。
【0079】
「Fab’断片」は、軽鎖1つさらに、VHドメインおよびCH1ドメインならびにCH1ドメインとCH2ドメインとの間の領域も、F(ab’)2分子を形成するために2つのFab’断片の2つの重鎖間に鎖間ジスルフィド結合が形成されうるように含有する1つの重鎖の一部を含有する。
【0080】
「F(ab’)2断片」は、軽鎖2つ、およびCH1ドメインとCH2ドメインとの間の定常領域の一部を含有する重鎖2つを、鎖間ジスルフィド結合が2つの重鎖の間に形成されるように含有する。したがってF(ab’)2断片は、2つの重鎖間のジスルフィド結合によって維持される2つのFab’断片から構成される。
【0081】
「Fv領域」は、重鎖および軽鎖の両方由来の可変領域を含むが、定常領域を欠損している。
【0082】
「単鎖Fv抗体」(または「scFv抗体」)は、抗体のVHおよびVLドメインを含む抗体断片を指し、これらのドメインは、1本のポリペプチド鎖に存在する。一般にFvポリペプチドは、scFvが抗原結合のための所望の構造を形成できるようにするVHドメインとVLドメインの間のポリペプチドリンカーをさらに含む。scFvについての概説として、Pluckthun (1994年)THE PHARMACOLOGY OF MONOCLONAL ANTIBODIES、113巻、Rosenburg and Moore eds.Springer−Verlag、New York、269〜315頁を参照されたい。国際特許出願公開WO88/01649ならびに米国特許第4,946,778号および第5,260,203号も参照されたい。
【0083】
「ダイアボディー」は、2個の抗原結合部位を有する小さな抗体断片である。断片は、同じポリペプチド鎖中で軽鎖可変ドメイン(VL)に連結した重鎖可変ドメイン(VH)を含む(VH−VLまたはVL−VH)。同じ鎖上の2つのドメイン間で対合させるには短すぎるリンカーを使用することによって、ドメインは他の鎖の相補性ドメインと対合させられ、2つの抗原結合部位を作り出す。ダイアボディーは、例えばEP404,097、WO93/11161およびHolligerら、(1993年)Proc.Natl.Acad.Sci.USA 90巻:6444〜6448頁においてさらに十分に記載されている。
【0084】
「ドメイン抗体断片」は、重鎖の可変領域または軽鎖の可変領域だけを含有する免疫学的に機能性の免疫グロブリン断片である。場合により2つ以上のVH領域が二価ドメイン抗体断片を作り出すようにペプチドリンカーに共有結合的に結合する。二価ドメイン抗体断片の2つのVH領域は、同じまたは異なる抗原を標的にできる。
【0085】
本発明の抗体断片は、例えば通常重鎖間ジスルフィド結合に関与するヒンジシステインの少なくとも1つが本明細書に記載のとおりに変更されている場合に、ジスルフィド結合能力が低下している重鎖の二量体化(または多量体化)を可能にする定常領域の十分な部分を含みうる。他の実施形態において抗体断片、例えばFc領域を含むものは、FcRn結合、抗体半減期調節、ADCC機能および/または補体結合など(例えば抗体がADCC機能または補体結合に必要なグリコシル化特性を有する場合)の、無傷の抗体中に存在する場合にFc領域に通常付随する少なくとも1つの生物学的機能を保持する。
【0086】
用語「キメラ」抗体は、重鎖および/または軽鎖の一部が特定の種に由来するかまたは特定の抗体のクラスもしくはサブクラスに属する抗体の対応する配列と同一であるかまたは相同である一方で、(1つまたは複数の)鎖の残部が他の種に由来するかまたは他の抗体のクラスもしくはサブクラスに属する抗体の対応する配列と同一であるかまたは相同である、ならびに所望の生物学的活性を示す範囲でそのような抗体の断片である抗体を指す(例えば米国特許第4,816,567号およびMorrisonら、1984年、Proc.Natl.Acad.Sci.USA 81巻:6851〜6855頁を参照されたい)。
【0087】
非ヒト(例えばマウス)抗体の「ヒト化」形態は、非ヒト免疫グロブリン由来の最少配列を含有するキメラ抗体である。大部分についてヒト化抗体は、レシピエントの超可変領域由来の残基が、所望の特異性、親和性および適応力を有するマウス、ラットまたはヒトではない霊長類などのヒトではない種(ドナー抗体)の超可変領域由来の残基によって置き換えられたヒト免疫グロブリン(レシピエント抗体)である。場合により、ヒト免疫グロブリンのFvフレームワーク領域(FR)残基は、対応する非ヒト残基によって置き換えられる。さらにヒト化抗体は、レシピエント抗体またはドナー抗体において見いだされない残基を含みうる。これらの改変は、抗体の性能をさらに改良するために行われる。一般にヒト化抗体は、超可変ループの全てまたは実質的に全てが非ヒト免疫グロブリンの超可変ループに対応し、およびFR領域の全てまたは実質的に全てがヒト免疫グロブリン配列のものである、少なくとも1つ、および典型的には2つの可変ドメインの実質的に全てを含む。ヒト化抗体は、任意選択で免疫グロブリン定常領域(Fc)の少なくとも一部、典型的にはヒト免疫グロブリンのそれも含む。さらなる詳細については、Jonesら、Nature 321巻:522〜525頁(1986年);Riechmannら、Nature332巻:323〜329頁(1988年)およびPresta、Curr.Op.Struct.Biol.2巻:593〜596頁(1992年)を参照されたい。
【0088】
本明細書において使用される用語「超可変領域」は、抗原結合に関与する抗体のアミノ酸残基を指す。超可変領域は、配列比較によって定義される「相補性決定領域」または「CDR」由来のアミノ酸残基、例えば軽鎖可変ドメイン中の24〜34(L1)、50〜56(L2)および89〜97(L3)残基、ならびに重鎖可変ドメイン中の31〜35(H1)、50〜65(H2)および95〜102(H3)残基(Kabatら、1991年、Sequences of proteins of Immunological Interest、5th Ed.Public Health Service、National Institutes of Health、Bethesda、Md.を参照されたい)、さらに/または構造的に定義される「超可変ループ」(HVL)例えば軽鎖可変ドメイン中の26〜32(L1)、50〜52(L2)および91〜96(L3)残基、ならびに重鎖可変ドメイン中の26〜32(H1)、53〜55(H2)および96〜101(H3)残基由来の残基(Chothia and Leskl、1987年、J.Mol.Biol.196巻:901〜917頁を参照されたい)を含む。「フレームワーク」または「FR」残基は、本明細書において定義する超可変領域残基以外の可変ドメイン残基である。
【0089】
「ヒト抗体」は、ヒトによって産生される、かつ/または本明細書において開示するヒト抗体を作製するための技術のいずれかを使用して作られている抗体のアミノ酸配列に対応するアミノ酸配列を有する抗体である。この定義は、具体的には非ヒト抗原結合残基を含むヒト化抗体を排除する。
【0090】
「単離された」抗体は、その天然の環境の成分から同定され、分離され、かつ/または回収されたものである。その天然の環境の混入成分は、抗体の診断または治療用使用を妨げる物質であり、酵素、ホルモンおよび他のタンパク質性または非タンパク質性の溶質を含みうる。いくつかの実施形態において抗体は、(1)ローリー法によって決定されるように抗体で95重量%を超えて、および最も好ましくは99重量%を超えるまで、(2)スピニングカップ配列決定装置の使用により、N末端の少なくとも15残基もしくは内部アミノ酸配列を得るために十分な程度まで、または(3)クーマシーブルー、もしくは好ましくは銀染色を使用する還元もしくは非還元条件下でのSDS−PAGEにより均一になるまで、精製される。単離された抗体は、抗体の天然の環境の少なくとも1つの成分が存在しないことになることから、組換え細胞内でインサイチュの抗体を含む。しかし通常は、単離された抗体は、少なくとも1つの精製ステップによって調製される。
【0091】
「単離された」核酸分子は、抗体核酸の天然の供給源においてそれが通常関連している少なくとも1つの混入核酸分子から同定および分離される核酸分子である。単離された核酸分子は、それが天然で見いだされる形態もしくは状態にある以外のものである。したがって単離された核酸分子は、天然の細胞内に存在する場合の核酸分子とは区別される。しかし単離された核酸分子は、例えば核酸分子が天然の細胞においてとは異なる染色体中の位置にある場合に、通常抗体を発現する細胞内に含まれる核酸分子を含む。
【0092】
本明細書において使用する用語「モノクローナル抗体」は、実質的に同質の抗体の集団から得た抗体を指す、すなわち集団を構成する個々の抗体は、少量で存在しうる天然に生じる可能性のある変異を除いて同一である。モノクローナル抗体は、高度に特異的であり、単一の抗原部位に対して誘導されている。さらに、典型的には異なる決定基(エピトープ)に対して誘導された異なる抗体を含む従来の(ポリクローナル)抗体調製物とは対照的に、各モノクローナル抗体は抗原上の単一の決定基に対して誘導されている。修飾語「モノクローナル」は、実質的に同質な抗体の集団から得られる抗体の特性を示し、特定の方法による抗体産生を必要とするとは解釈されない。例えば本発明により使用されるモノクローナル抗体は、Kohlerら、1975年、Nature 256巻:495頁によって最初に記載されたハイブリドーマ法によって作製されてもよく、または組換えDNA法(例えば米国特許第4,816,567号を参照されたい)によって作製されてもよい。「モノクローナル抗体」は、例えばClacksonら、1991年、Nature352巻:624〜628頁およびMarksら、1991年、J.Mol Biol.222巻:581〜597頁に記載された技術を使用するファージ抗体ライブラリーから単離されてもよい。本明細書におけるモノクローナル抗体は、詳細には「キメラ」抗体を含む。
【0093】
本明細書において使用する用語「免疫細胞」は、造血系由来であり、かつ免疫応答において役割を演じる細胞を含む。免疫細胞として、B細胞およびT細胞などのリンパ球;ナチュラルキラー細胞;単球、マクロファージ、好酸球、マスト細胞、好塩基球および顆粒球などの骨髄系細胞が挙げられる。
【0094】
本明細書において使用する「免疫複合体」は、細菌性毒素、細胞毒性薬物または放射性毒素などの治療用成分に結合した抗PD−1抗体またはその断片を指す。毒性成分は、当技術分野において利用可能な方法を使用して本発明の抗体に結合されうる。
【0095】
以下の核酸多義性記号は、本明細書において使用される、R=AまたはG、Y=CまたはT、M=AまたはC、K=GまたはT、S=GまたはC、およびW=AまたはT。
【0096】
本明細書において使用する配列「変異体」は、本開示の配列とは1つまたは複数のアミノ酸残基で異なるが、得られる分子の生物学的活性を保持している配列を指す。
【0097】
「保存的に改変された変異体」または「保存的アミノ酸置換」は、当業者に公知のアミノ酸の置換を指し、得られる分子の生物学的活性を変えることなく一般に作製されうる。当業者は、一般にポリペプチドの非必須領域における単一のアミノ酸の置換が生物学的活性を実質的に変えないことを認識する(例えばWatsonら、Molecular Biology of the Gene、The Benjamin/Cummings Pub.Co.、224頁(4th Edition 1987年)を参照されたい)。そのような例示的な置換は、好ましくは以下に列挙するものに従って行われる:
【0098】
【数1】

本明細書において使用する、2つの配列間で「%同一性」は、配列が共有する同一の位置の数の関数を指し(すなわち、%相同=同一の位置の数/位置の総数×100)、2つの配列の最適な配列比較ために導入される必要があるギャップの数および各ギャップの長さを考慮する。配列の比較および2つの配列間の同一性百分率の決定は、数学的アルゴリズムを使用して達成されうる。例えば2つのアミノ酸配列間の同一性百分率は、ALIGNプログラム(バージョン2.0)に組み入れられているE.MeyersおよびW.Miller(Comput.Appl.Biosci.、4巻:11〜17頁(1988年))のアルゴリズムを使用して、PAM120重み残基表、ギャップ長ペナルティー12、およびギャップペナルティー4を使用して決定されうる。加えて2つのアミノ酸配列間の同一性百分率は、GCGソフトウェアパッケージ(www.gcg.comで入手可能)中のGAPプログラムに組み入れられているNeedlemanおよびWunsch(J.Mol Biol.48巻:444〜453頁(1970年))アルゴリズムを使用して、Blossum62マトリックスまたはPAM250マトリックスのいずれか、ならびにギャップ重み16、14、12、10、8、6または4および長さ重み1、2、3、4、5または6を使用して決定されうる。
【0099】
本明細書において使用する用語「約」は、当業者によって決定される特定の値に対する許容可能な誤差範囲内の値を指し、その値がどのように測定または決定されたかに、すなわち測定系の限界にある程度依存する。例えば「約」は、当技術分野において一実施あたり1以内の標準偏差または1を超える標準偏差を意味できる。代替えとして「約」または「を本質的に含む」は、20%までの範囲を意味できる。さらに具体的に生物学的システムまたはプロセスに関して、用語は値の一桁まで、または5倍までを意味できる。本出願書類および本特許請求の範囲において具体的な値が提供される場合、他に明記しない限り、「約」または「を本質的に含む」の意味は、具体的な値について許容可能な誤差範囲内であると推定されるべきである。
【0100】
リガンド/受容体、抗体/抗原、または他の結合対に関する場合「特異的に」結合するは、タンパク質および/または他の生物体の不均質な集団におけるタンパク質、例えばPD−1、の存在の決定要因である結合反応を示す。したがって、指定の条件下では、特定のリガンド/抗原は特定の受容体/抗体に結合し、試料中に存在する他のタンパク質には顕著な量では結合しない。
【0101】
動物、ヒト、実験対象、細胞、組織、器官または生体液に適用される場合に「投与」および「処置」は、外因性の薬学的、治療用、診断用の薬剤または組成物の動物、ヒト、対象、細胞、組織、器官または生体液への接触を指す。「投与」および「処置」は、例えば治療用、薬物動態学的、診断用、研究用および実験用の方法を指すことができる。細胞の処理は、試薬の細胞への接触、および液が細胞に接触している場合に試薬の液への接触を包含する。「投与」および「処置」は、例えば細胞の、試薬による、診断用、結合組成物による、または他の細胞によるin vitroおよびex vivoの処置、も意味する。
【0102】
「有効量」は、医学的状態の症状または兆候を改善または予防するために十分な量を包含する。有効量は、診断を可能にするまたは促進するために十分な量も意味する。具体的な対象についての有効量は、治療される状態、患者の健康全般、投与の方法経路および用量、ならびに副作用(side affect)の重症度などの要因に依存して変動できる。有効量は、顕著な副作用または毒性作用を避ける最大用量または投薬手順でありうる。効果は、診断的指標またはパラメーターの改善を少なくとも5%、通常は少なくとも10%、より通常には少なくとも20%、最も通常は少なくとも30%、好ましくは少なくとも40%、より好ましくは少なくとも50%、最も好ましくは少なくとも60%、理想的には少なくとも70%、より理想的には少なくとも80%、および最も理想的には少なくとも90%生じ、100%は正常な対象によって示される診断パラメーターと定義される(例えば、Maynardら(1996年)A Handbook of SOPs for Good Clinical Practice、Interpharm Press、Boca Raton、FL;Dent(2001年)Good Laboratory and Good Clinical Practice、Urch Publ.、London、UKを参照されたい)。
【0103】
モノクローナル抗体
PD−1に対するモノクローナル抗体は、被験体にPD−1抗原を注射し、次いで所望の配列または機能特性を有する抗体を発現しているハイブリドーマを単離する当技術分野における知識および技術によって作製できる。
【0104】
モノクローナル抗体をコードするDNAは、従来の手段を使用して(例えばモノクローナル抗体の重鎖および軽鎖をコードする遺伝子に特異的に結合できるオリゴヌクレオチドプローブを使用することによって)容易に単離され、かつ配列決定される。ハイブリドーマは、そのようなDNAの好ましい供給源として役立つ。単離されるとDNAは、発現ベクターに入ることができ、次いで組換え宿主細胞におけるモノクローナル抗体の合成を得るために、他に免疫グロブリンタンパク質を産生しないE.coli細胞、サルCOS細胞、チャイニーズハムスター卵巣(CHO)細胞または骨髄腫細胞などの宿主細胞に形質移入される。抗体の組換え産生は、より詳細に以下に記載される。
【0105】
さらなる実施形態において抗体または抗体断片は、McCaffertyら、1990年、Nature、348巻:552〜554頁に記載されている技術を使用して生成される抗体ファージライブラリーから単離されうる。Clacksonら、1991年、Nature、352巻:624〜628頁およびMarksら、1991年、J.Mol.Biol.222巻:581〜597頁は、ファージライブラリーを使用するマウスおよびヒトの抗体の単離をそれぞれ記載している。次の刊行物は、鎖シャッフリングによる高親和性(nM範囲)ヒト抗体の産生(Marksら、1992年、Bio/Technology、10巻:779〜783頁)、ならび非常に大きなファージライブラリーを構築するための戦略としてのコンビナトリアル感染およびin vivo組換えを記載している(Waterhouseら、1993年、Nuc.Acids.Res.21巻:2265〜2266頁)。したがってこれらの技術は、モノクローナル抗体の単離のための伝統的なモノクローナル抗体ハイブリドーマ技術の実行可能な代替法である。
【0106】
キメラ抗体
抗体DNAは、例えばヒト重鎖および軽鎖の定常ドメインのコード配列を相同なマウス配列の代わりに置換することによって(米国特許第4,816,567号;Morrisonら、1984年、Proc.Natl Acad.Sci.USA、81巻:6851頁)、または免疫グロブリンではない物質(例えばタンパク質ドメイン)のコード配列の全てまたは一部を免疫グロブリンコード配列に共有結合させることによっても改変されうる。典型的には、そのような免疫グロブリンではない物質は、抗体の定常ドメインの代わりをするか、または、抗原に対する特異性を有する1つの抗原結合部位および異なる抗原に対する特異性を有する他の抗原結合部位とを含むキメラ二価抗体を作るために抗体の1つの抗原結合部位の可変ドメインの代わりをする。
【0107】
ヒト化抗体およびヒト抗体
ヒト化抗体は、ヒトではない供給源由来の1つまたは複数のアミノ酸残基を有する。非ヒトアミノ酸残基は、しばしば「移入」残基と称され、典型的には「移入」可変ドメインから取り込まれる。ヒト化は、一般にWinterおよび協力者らの方法(Jonesら、1986年、Nature321巻:522〜525頁;Riechmannら、1988年、Nature、332巻:323〜327頁;Verhoeyenら、1988年、Science 239巻:1534〜1536頁)に従って、げっ歯類CDRまたはCDR配列をヒト抗体の対応する配列の代わりに置換することによって実施されうる。したがってそのような「ヒト化」抗体は、無傷のより実質的に少ないヒト可変ドメインがヒトではない種由来の対応する配列によって置換されているキメラ抗体(米国特許第4,816,567号)である。実際にヒト化抗体は、典型的には、いくつかのCDR残基および場合によりいくつかのFR残基がヒトではない、例えばげっ歯類の抗体中の類似部位由来の残基によって置換されているヒト抗体である。
【0108】
ヒト化抗体の作製において使用される軽鎖および重鎖の両方のヒト可変ドメインの選択は、抗原性を低減するために非常に重要である。いわゆる「最適な」方法により、げっ歯類抗体の可変ドメインの配列は、既知のヒト可変ドメイン配列の完全なライブラリーに対して選別される。その結果げっ歯類の配列に最も近いヒト配列が、ヒト化抗体用のヒトフレームワーク(FR)として受けいれられる(Simsら、1987年、J.Immunol.151巻:2296頁;Chothiaら、1987年、J.Mol.Biol.196巻:901頁)。他の方法は、軽鎖および重鎖の特定のサブグループの全てのヒト抗体の共通配列由来の特定のフレームワークを使用する。同じフレームワークがいくつかの異なるヒト化抗体用に使用されうる(Carterら、1992年、Proc.Natl.Acad.Sci.USA 89巻:4285頁;Prestaら、1993、 J.Immnol.151巻:2623頁)。
【0109】
抗体が抗原に対する高い親和性および他の有益な生物学的特性を保持してヒト化されることは、さらに重要である。この目標を達成するために、好ましい方法によりヒト化抗体は、親配列およびヒト化配列の3次元モデルを使用する親配列および種々の概念的なヒト化産物の分析の工程によって調製される。3次元免疫グロブリンモデルは、一般的に入手可能であり、当業者によく知られている。選択された候補免疫グロブリン配列の有望な3次元立体配置構造を例示および表示するコンピュータープログラムは、利用可能である。これらの表示の検査は、候補免疫グロブリン配列の機能における残基の可能性のある役割の分析、すなわち候補免疫グロブリンがその抗原に結合する能力に影響する残基の分析、を可能にする。このようにFR残基は、(1つまたは複数の)標的抗原に対する親和性の増大などの所望の抗体の特性が達成されるように、レシピエント配列および移入配列から選択および組み合わせられることができる。一般にCDR残基は、直接かつ最も実質的に抗原結合に影響を及ぼすことに関与する。
【0110】
抗体のヒト化は、直接的なタンパク質工学的作業である。ほとんど全てのマウス抗体は、CDR移植によってヒト化されることができ、抗原結合は保持される。Lo, Benny, K.C.、editor、in Antibody Engineering:Methods and Protocols、248巻、Humana Press、New Jersey、2004年を参照されたい。
【0111】
代替えとして、免疫化において、内在性の免疫グロブリンを産生することなくヒト抗体の完全なレパートリーを産生できるトランスジェニック動物(例えばマウス)を産生することがいまや可能である。例えば、キメラおよび生殖系列変異体マウスでの抗体重鎖連結領域(JH)遺伝子のホモ欠損は、内在性の抗体産生の完全な抑制を生じることが記載されている。ヒト生殖系列免疫グロブリン遺伝子アレイの、そのような生殖系列変異マウスへの移入は、抗原曝露でのヒト抗体の産生を生じる。例えばJakobovitsら、1993年、Proc.Natl.Acad.Sci.USA 90巻:2551頁;Jakobovitsら、1993年、Nature 362巻:255〜258頁;Bruggermannら、1993年、Year in Immunology 7巻:33頁およびDuchosalら、1992年、Nature 355巻:258頁を参照されたい。ヒト抗体は、ファージディスプレイライブラリーにも由来できる(Hoogenboomら、1991年J.Mol.Biol.227巻:381頁;Marksら、J.Mol.Biol.1991年、222巻:581〜597頁;Vaughanら、1996年、Nature Biotech 14巻:309頁)。
【0112】
抗体精製
組換え技術を使用する場合、抗体は細胞内、周辺質間隙に産生されるか、または培地に直接分泌されうる。抗体が細胞内に産生される場合、最初の段階として宿主細胞または溶解断片のいずれかの粒子細片は、例えば遠心分離または限外濾過によって除去される。Carterら、1992年、Bio/Technology 10巻:163〜167頁は、E.coliの周辺質間隙に分泌される抗体の単離のための手順を記載している。簡潔には、細胞ペーストを酢酸ナトリウム(pH3.5)、EDTAおよびフッ化フェニルメチルスルホニル(PMSF)の存在下で約30分間かけて融解する。細胞細片は、遠心分離によって除去されうる。抗体が培地に分泌される場合は、そのような発現系由来の上清を一般的には最初に、市販で入手可能なタンパク質濃縮用フィルター、例えばAmiconまたはMillipore,Pellicon限外ろ過ユニットを使用して濃縮する。PMSFなどのプロテアーゼ阻害剤は、任意の前述のステップにタンパク質分解を阻害するために含まれてよく、抗生物質は外来性の混入物の増殖を防ぐために含まれてよい。
【0113】
細胞から調製された抗体組成物は、例えば、ヒドロキシアパタイトクロマトグラフィー、ゲル電気泳動、透析およびアフィニティークロマトグラフィーを使用して、好ましい精製技術であるアフィニティークロマトグラフィーを用いて精製されうる。プロテインAの親和性リガンドとしての適合性は、種および抗体中に存在する任意の免疫グロブリンFc領域のアイソタイプに依存する。プロテインAは、ヒトγ1、γ2またはγ4重鎖に基づく抗体を精製するために使用されうる(Lindmarkら、1983年、J.Immunol.Meth.62巻:1〜13頁)。プロテインGは、全てのマウスアイソタイプおよびヒトγ3に対して推奨される(Gussら、1986年、EMBO J 5巻:15671575頁)。親和性リガンドが結合するマトリックスは、最も頻繁にはアガロースであるが、他のマトリックスも利用可能である。細孔制御されたガラスまたはポリ(スチレンジビニル)ベンゼンなどの機械的に安定なマトリックスは、アガロースで達成されうるよりもより早い流速およびより短い処理時間を可能にする。抗体がCH3ドメインを含む場合は、Bakerbond ABX(商標)レジン(J.T.Baker、Phillipsburg、N.J.)が、精製のために有用である。イオン交換カラムでの分画、エタノール沈殿、逆相HPLC、シリカ上でのクロマトグラフィー、ヘパリンSEPHAROSETM上でのクロマトグラフィー、陰イオンまたは陽イオン交換樹脂(ポリアスパラギン酸カラムなど)上でのクロマトグラフィー、等電点電気泳動、SDS−PAGEおよび硫酸アンモニウム沈殿などのタンパク質精製のための他の技術も回収される抗体に依存して利用可能である。
【0114】
一実施形態において糖タンパク質は、調製物からフコース含有糖タンパク質を除き、したがってフコースを含まない糖タンパク質を富化するためにレクチン基質での吸着(例えばレクチン親和性カラム)を使用して精製されうる。
【0115】
医薬製剤
本発明は、本発明のPD−1抗体または抗体断片の医薬製剤を含む。医薬組成物または滅菌組成物を調製するために、抗体およびその断片は、薬学的に許容される担体または賦形剤と混合される、例えばRemington’s Pharmaceutical Sciences and U.S.Pharmacopeia: National Formulary、Mack Publishing Company、Easton、PA(1984年)を参照されたい。治療剤および診断剤の製剤は、生理学的に許容される単体、賦形剤または安定化剤と、例えば凍結乾燥粉末、スラリー、水性溶液または懸濁液の形態で混合することによって調製されうる(例えばHardmanら、(2001年)Goodman and Gilman’s The Pharmacological Basis of Therapeutics、McGraw−Hill、New York、NY;Gennaro(2000年)Remington:The Science and Practice of Pharmacy、Lippincott、Williams、and Wilkins、New York、NY;Avisら(eds.)(1993年)Pharmaceutical Dosage Forms:Parenteral Medications、Marcel Dekker、NY;Liebermanら(eds.)(1990年))Pharmaceutical Dosage Forms:Tablets、Marcel Dekker、NY;Liebermanら(eds.)(1990年)Pharmaceutical Dosage Forms:Disperse Systems、Marcel Dekker、NY;Weiner and Kotkoskie (2000年)Excipient Toxicity and Safety、Marcel Dekker、Inc.、New York、NYを参照されたい)。
【0116】
単独または免疫抑制剤との組合せで投与される抗体組成物の毒性および治療効果は、例えばLD50(集団の50%致死量)およびED50(集団の50%において治療的に有効な用量)を決定するための、細胞培養物または実験動物での標準的薬学的手順によって決定されうる。毒性量と治療有効量との間の用量比が、治療指数であり、LD50とED50との比として表されうる。これらの細胞培養アッセイおよび動物の研究から得られるデータは、ヒトでの使用のための投与量範囲を製剤化することにおいて使用されうる。そのような化合物の投与量は、好ましくは、毒性が殆どないか、または無しでのED50を含む循環濃度の範囲内にある。投与量は、使用する投与形態および使用する投与経路に依存してこの範囲内で変えることができる。
【0117】
投与の適切な経路は、筋肉内、静脈内または皮下投与などの非経口投与および経口投与を含む。医薬組成物中で使用されるまたは本発明の方法を実行するための抗体の投与は、経口摂取、吸入、局所投与または皮膚、皮下、腹腔内、非経口、動脈内または静脈内注射などのさまざまな従来の方法で実施されうる。一実施形態において本発明の結合化合物は、静脈内に投与される。他の実施形態において本発明の結合化合物は、皮下に投与される。
【0118】
代替えとして、例えば、しばしばデポー製剤または徐放性製剤での作用部位に直接での抗体の注射を介して、抗体を全身的様式よりは局所に投与できる。さらに標的化した薬物送達系で抗体を投与できる。
【0119】
抗体、サイトカインおよび小分子の適切な用量の選択における指針は、入手可能である(例えばWawrzynczak(1996年)Antibody Therapy、Bios Scientific Pub.Ltd、Oxfordshire、UK;Kresina(ed.)(1991年)Monoclonal Antibodies、Cytokines and Arthritis、Marcel Dekker、New York、NY;Bach(ed.)(1993年)Monoclonal Antibodies and Peptide Therapy in Autoimmune Diseases、Marcel Dekker、New York、NY;Baertら、(2003年)New Engl.J.Med.348巻:601〜608頁;Milgromら、(1999年)New Engl.J.Med.341巻:1966〜1973頁;Slamonら、(2001年)New Engl.J.Med.344巻:783〜792頁;Beniaminovitzら、(2000年)New Engl.J.Med.342巻:613〜619頁;Ghoshら、(2003年)New Engl.J.Med.348巻:24〜32頁;Lipskyら、(2000年)New Engl.J.Med.343巻:1594〜1602頁を参照されたい)。
【0120】
適切な用量の決定は、臨床医によって、例えば治療に影響するまたは治療に影響すると予測される当技術分野において公知または疑われるパラメーターまたは因子を使用して行われる。一般に、用量は、最適用量より幾分か少ない量から始め、その後任意の負の副作用と比較して所望のまたは最適な効果が達成されるまで少量ずつ増加させる。重要な診断指標は、症状、例えば炎症または産生される炎症性サイトカインのレベルの測定を含む。
【0121】
抗体、抗体断片およびサイトカインは、持続注入によって、または例えば1日、1週間もしくは1週間に1〜7回の間隔での投薬によって提供されうる。用量は静脈内、皮下、腹腔内、皮膚、局所、経口、経鼻、直腸、筋肉内、脳内、脊髄内、または吸引によって提供されうる。好ましい投与プロトコールは、重大な望ましくない副作用を避ける最高の用量または投与頻度を含むものである。1週間の合計用量は、一般的には体重1kgあたり少なくとも0.05μg、より一般的には体重1kgあたり少なくとも0.2μg、最も一般的には体重1kgあたり少なくとも0.5μg、典型的には体重1kgあたり少なくとも1μg、より典型的には体重1kgあたり少なくとも10μg、最も典型的には体重1kgあたり少なくとも100μg、好ましくは体重1kgあたり少なくとも0.2mg、より好ましくは体重1kgあたり少なくとも1.0mg、最も好ましくは体重1kgあたり少なくとも2.0mg、最適には体重1kgあたり少なくとも10mg、より最適には体重1kgあたり少なくとも25mg、最も最適には体重1kgあたり少なくとも50mgである(例えばYangら、(2003年)New Engl.J.Med.349巻:427〜434頁;Heroldら、(2002年)New Engl.J.Med.346巻:1692〜1698頁;Liuら、(1999年)J.Neural.Neurosurg.Psych.67巻:451〜456頁;Portieljiら、(20003)Cancer Immunol.Immunother.52巻:133〜144頁を参照されたい)。小分子治療、例えばペプチド模倣物、天然物または有機化学物質の望ましい用量は、1kgあたりのモルベースでの抗体またはポリペプチドについてとほぼ同じである。
【0122】
本明細書において使用する「抑制」、または「治療する」または「治療」は、疾患に関連する症状の発症の遅延および/または該疾患で発症するもしくは発症が予測されるそのような症状の重症度の低減を含む。用語は、さらに発症している症状の改善、追加的症状の予防、およびそのような症状の元となる原因の改善または予防を含む。したがって用語は、有益な結果が疾患を有する脊椎動物対象に与えられていることを意味する。
【0123】
本明細書において使用する用語「治療有効量」または「有効量」は、単独でまたは追加的治療剤との組合せで細胞、組織、または対象に投与された場合に治療される疾患または状態を予防または改善するために有効である、抗PD−1抗体またはその断片の量を指す。治療有効量は、さらに、症状の改善、例えば関連する医学的状態の治療、治癒、予防もしくは改善またはそのような状態の治療、治癒、予防もしくは改善の速度の増加を生じるために十分な化合物の量を指す。単独で投与される個々の活性成分に適用される場合、治療有効量は、成分単独でのそれを意味する。組合せに適用される場合治療有効量は、組合せで連続的にまたは同時に投与されるかにかかわらず、治療効果を生じる活性成分の組み合わされた量を指す。治療剤の有効量は、典型的には少なくとも10%、通常は少なくとも20%、好ましくは少なくとも約30%、より好ましくは少なくとも40%および最も好ましくは少なくとも50%症状を低減する。
【0124】
第2の治療剤を伴う同時投与または治療のための方法は、当技術分野において十分に周知であり、例えばHardmanら、(eds.)(2001年)Goodman and Gilman’s The Pharmacological Basis of Therapeutics、10th ed.、McGraw−Hill、New York、NY;Poole and Peterson(eds.)(2001年)Pharmacotherapeutics for Advanced Practice:A Practical Approach、Lippincott、Williams & Wilkins、Phila.、PA;Chabner and Longo(eds.)(2001年)Cancer Chemotherapy and Biotherapy、Lippincott、Williams & Wilkins、Phila.、PAを参照されたい。
【0125】
本発明の医薬組成物は、細胞毒性剤、細胞増殖抑制剤、抗血管新生剤または代謝拮抗剤、腫瘍標的剤、免疫刺激剤または免疫調節剤または、細胞毒性剤、細胞増殖抑制剤もしくは他の毒性作用物質と結合した抗体を非限定的に含む他の薬物も含有できる。医薬組成物は、手術、化学療法および放射線などの他の治療様式と共にも使用されうる。
【0126】
典型的な獣医学的、実験的または研究用対象として、サル、イヌ、ネコ、ラット、マウス、ウサギ、モルモット、ウマおよびヒトが挙げられる。
【0127】
本発明の抗体および抗体断片についての治療的使用
ヒトPD−1に特異的に結合する本発明の抗体または抗原結合断片は、免疫応答を増大、増強、刺激または上方制御するために使用されうる。本発明の抗体および抗体断片は、T細胞媒介の免疫応答の増大によって治療されうる障害を有する対象を治療するために特に適切である。好ましい対象は、免疫応答を増強する必要のあるヒト患者を含む。
【0128】
癌
本発明の抗体または抗原結合断片は、癌を治療する(すなわち腫瘍細胞の増殖または生存を抑制する)ために使用されうる。本発明の抗体を使用して増殖が抑制されうる好ましい癌は、典型的には免疫療法に応答する癌を含むが、免疫療法にこれまで関連がなかった癌も含む。治療について好ましい癌の非限定的例として、黒色腫(例えば転移性悪性黒色腫)、腎臓癌(例えば明細胞癌)、前立腺癌(例えばホルモン難治性前立腺腺癌)、膵臓腺癌、乳癌、結腸癌、肺癌(例えば非小細胞肺癌)、食道癌、頭頸部の扁平上皮癌、肝臓癌、卵巣癌、子宮頸癌、甲状腺癌、神経膠芽腫、神経膠腫、白血病、リンパ腫および他の腫瘍性悪性疾患が挙げられる。追加的に、本発明は、本発明の抗体を使用して増殖が抑制されうる難治性または再発性悪性疾患を含む。
【0129】
本発明の抗体または抗体断片は、単独であるいは、他の抗腫瘍性剤もしくは免疫原性薬剤(例えば、弱毒化された癌性細胞、腫瘍抗原(組換えタンパク質、ペプチドおよび炭水化物分子を含む))、腫瘍由来抗原もしくは核酸でパルスされた樹状細胞などの抗原提示細胞、免疫刺激サイトカイン(例えばIL−2、IFNa2、GM−CSF_)および非限定的にGM−CSFなどの免疫刺激サイトカインをコードする遺伝子で形質移入された細胞、標準的癌治療法(例えば化学療法、放射線療法もしくは手術)、または他の抗体(非限定的にVEGF、EGFR、Her2/neu、VEGF受容体、他の増殖因子受容体、CD20、CD40、CTLA−4、OX−40、4−IBBおよびICOSに対する抗体を含む)との組合せで使用されうる。
【0130】
感染性疾患
本発明の抗体または抗体断片は、感染症および感染性疾患を予防または治療するためにも使用されうる。抗体または抗体断片は、病原体、毒素および自己抗原への免疫応答を刺激するために、単独でまたはワクチンとの組合せで使用されうる。抗体またはその抗原結合断片は、非限定的にヒト免疫不全ウイルス、肝炎ウイルスクラスA、BおよびC、エプスタインバーウイルス(Eppstein Barr virus)、ヒト(hyman)サイトメガロウイルス、ヒトパピローマウイルス、ヘルペスウイルスなどの、ヒトに感染性のウイルスへの免疫応答を刺激するために使用されうる。抗体またはその抗原結合断片は、細菌性または真菌性寄生生物および他の病原体での感染への免疫応答を刺激するために使用されうる。
【0131】
ワクチン接種アジュバント
本発明の抗体または抗体断片は、他の組換えタンパク質および/またはペプチド(腫瘍抗原または癌細胞など)と結合してこれらのタンパク質への免疫応答を増大させるために(すなわちワクチン接種手順において)使用されうる。
【0132】
例えば抗PD−1抗体およびその抗体断片は、抗PD−1抗体と目的の抗原(例えばワクチン)との同時投与によって抗原特異的免疫応答を刺激するために使用されうる。したがって他の態様において本発明は、(i)抗原、および(ii)本発明の抗PD−1抗体またはその抗原結合成分を、対象における抗原への免疫応答が増強されるように対象に投与するステップを含む、対象において抗原への免疫応答を増強する方法を提供する。抗原は、例えば腫瘍抗原、ウイルス抗原、細菌抗原または病原体由来抗原でありうる。そのような抗原の非限定的例として、腫瘍抗原、またはウイルス、細菌もしくは他の病原体由来の抗原が限定なく挙げられる。
【0133】
Th2介在性疾患
本発明の抗PD−1抗体および抗体断片は、喘息およびアレルギーなどのTh2介在性疾患を治療するためにも使用されうる。これは、本発明の抗体がTh1応答の誘発を支援できるという発見に基づく。したがって本発明の抗体は、より(mofre)バランスのとれた免疫応答を作り出すためにTh2介在性疾患において使用されうる。
【0134】
T細胞のex−vivo活性化
本発明の抗体および抗原断片は、抗原特異的T細胞のex vivo活性化および増殖、ならびに腫瘍に対する抗原特異的T細胞を増加させるためのこれらの細胞のレシピエントへの養子移入のためにも使用されうる。これらの方法は、CMVなどの感染性物質へのT細胞応答を活性化させるためにも使用されうる。抗PD−1抗体の存在下でのex vivo活性化は、養子移入されたT細胞の頻度および活性を増大させることが期待されうる。
【0135】
他の組合せ療法
既に記載されたとおり本発明の抗PD−1抗体は、1つまたは複数の他の治療剤、例えば細胞傷害性剤、放射毒性剤または免疫抑制剤と同時投与されうる。抗体は、該薬剤(免疫複合体として)と結合されうるか、または該薬剤とは別に投与されうる。後者の場合(別々の投与)、抗体は該薬剤の前、後もしくは同時に投与されることができ、または他の既知の療法と同時投与されうる。
【0136】
本発明の抗体および抗原結合断片は、ドナーに移植された腫瘍特異的T細胞の効果を増大させるためにも使用されうる。
【0137】
本発明の抗体および抗体断片の非治療用使用
非治療用使用についての抗PD−1抗体の市場は、San Diego、Clifornia、USAのeBioscienceによって販売されている、フローサイトメトリー分析、免疫組織化学およびin vitro機能アッセイにおける使用のためのJ116およびJ105モノクローナル抗hPD−1抗体、ならびにMinneapolis、MN、USAのR&D Systemsによって販売されている、フローサイトメトリー、ウェスタンブロットおよびELISAにおける使用のためのmab1086、モノクローナル抗hPD−1抗体、の商業的販売によって示されるとおり既に存在する。本発明の抗体は、J116、J105および/またはMab1086によって現在果たされている任意の非治療用目的のために使用されうる。
【0138】
本発明の抗体は、親和性精製剤として使用されうる。
【0139】
抗体は、例えば特定の細胞、組織または血清中のPD−1の発現を検出するための、診断用アッセイにおいても有用である。診断用の適用において、抗体は典型的には(直接または間接のいずれかで)検出可能成分で標識される。以下の分類:ビオチン、蛍光色素、放射性ヌクレオチド、酵素、ヨウ素および生合成標識に一般に分けられ得る多数の標識が使用可能である。
【0140】
本発明の抗体は、競合的結合アッセイ、直接および間接サンドイッチアッセイならびに免疫沈降アッセイなどの任意の既知のアッセイ法において使用されうる。Zola, Monoclonal Antibodies.A Manual of Techniques、147〜158頁(CRC Press, Inc.1987年)
抗体は、in vivo診断アッセイのためにも使用されうる。一般に抗体は、放射性核種(111In、99Tc、4C、3H、125I、3H、32P35Sまたは18Fなど)で標識して、免疫シンチグラフィー(immunoscintiography)、または陽電子放出断層撮影を使用して、抗原またはそれを発現する細胞を限局化し得る。
【0141】
材料の寄託
ヒト化抗体h409A11、h409A16およびh409A17の重鎖および軽鎖の可変領域をコードするDNA構築物は、American Type Culture Collection Patent Depository(10801 University Blvd.、Manassas、VA)に寄託されている。h409A−11、h409A−16およびh409A−17の重鎖をコードするDNAを含有するプラスミドは、2008年6月9日に寄託され、081469_SPD−Hと認定された。h409A11の軽鎖をコードするDNAを含有するプラスミドは、2008年6月9日に寄託され、0801470_SPD−L−11と認定された。h409A16の軽鎖をコードするDNAを含有するプラスミドは、2008年6月9日に寄託され、0801471_SPD−L−16と認定された。h409A17の軽鎖をコードするDNAを含有するプラスミドは、2008年6月9日に寄託され、0801472_SPD−L−17と指定された。寄託は、特許手続きのための微生物寄託の国際認識に関するブダペスト条約の規定およびその規則(ブダペスト条約)に基づいて行われた。
【0142】
上に記載された明細書は、当業者に本発明を実施可能にするために十分であると考えられる。寄託された実施形態は、本発明の一態様の単一の例示の目的であり、機能的に同等である任意の培養物は本発明の範囲内であることから、本発明は、寄託された培養物によって範囲を限定されない。本明細書の材料の寄託は、本明細書に含まれる書面による説明が、その最良の様式を含む本発明の任意の態様の実施を可能にするために不適切であるとの承認を構成するものでなく、本特許請求の範囲をそれが提示する具体的な例示に限定すると解釈されるべきでもない。実際に、本明細書に示されかつ記載されるものに加えての本発明の種々の変更は、上の記載から当業者に明らかになり、かつ添付の特許請求の範囲内である。
【0143】
本発明は、以下の実施例を参照することによってより完全に理解される。しかしそれらは、本発明の範囲を限定するとして解釈されるべきではない。本明細書において述べる全ての文献および特許の引用は、明確に参照として本明細書に組み込まれる。
【実施例】
【0144】
(実施例1)
抗PD−1抗体の免疫化および選択
hPD−1cDNAでのマウスの免疫化
ヒトPD−1(「hPD−1」)受容体に対する抗体を生成するために、hPD−1受容体のオープンリーディングフレームをコードするcDNAをPCRによって得て、ベクターpcDNA3.1(Invitrogen、Carlsbad、CA)にサブクローン化した。次にCHO−K1細胞をhPD−1で安定に形質移入し、発現をフローサイトメトリーを使用してモニターした。ヒトPD−1を膜上に発現するCHO−K1クローンを単離し、CHO−hPD1と名付けた。
【0145】
Helios gene gun(BioRad)およびDNA coated gold bullets(BioRad)を製造者の説明書に従って使用して遺伝子銃免疫化によってマウスを免疫化した。簡潔には1μmの金粒子をhPD−1 cDNA(pcDNA3.1にクローン化した)と、指示のある場合はマウスFlt3LおよびマウスGM−CSF用の市販の発現ベクター(どちらもAldevron、Fargo NDから)とで2:1:1の割合でコートした。合計1μgのプラスミドDNAを500μgのgold bulletをコートするために使用した。
【0146】
具体的には、7〜8週齢の雌性BALB/Cマウスを、遺伝子銃によって、2、3または4サイクルのショットを両耳に受けさせて耳で免疫化した(表Iを参照されたい)。マウス1匹は、5×106CHO−hPD1細胞を用いた最終追加免疫を腹腔に受けた。およそ1:1000の抗hPD−1力価がマウス血清において2回のDNA免疫化後に、CHO−hPD1対CHO−K1親細胞を使用する細胞ELISAによって検出可能であった。最終免疫化の4日後、マウスを屠殺し、赤血球除去脾臓細胞集団を以前記載されたとおり(Steenbakkersら、1992年、J.Immunol.Meth.152巻:69〜77頁;Steenbakkersら、1994年、Mol.Biol.Rep.19巻:125〜134頁)調製し、−140℃で凍結した。
【0147】
【表1】
抗PD−1抗体産生B細胞の選択
抗ヒトPD−1抗体産生B細胞クローンを選択するために、hPD−1 DNA免疫化マウス、すなわちマウス730、731および738(表1を参照されたい)由来の赤血球除去脾臓細胞2×107個をB細胞培養のためにプールした。脾臓細胞をDMEM/HAM’s F12/10%子ウシ血清(Hyclone、Logan、UT、USA)で37℃、1時間、プラスチック培養フラスコ中で単球を除くためにインキュベートした。非接着細胞を1回のCHO−K1細胞でのネガティブパニング、続いてCHO−hPD1細胞でのポジティブパニングにかけた。両方の選択手順は、21cm2ペトリ皿またはT25培養フラスコでコンフルエントに増殖した培養物において37℃、1時間実施した(細胞培養物は使用前に合計線量2000RADまで照射した)。ポジティブパニング後、未結合細胞を、0.132%CaCl2.2H2Oおよび0.1%MgCl2.6H2Oを補充したPBSでの10回洗浄によって除去した。最後に結合性B細胞をトリプシン処理によって回収した。
【0148】
選択したB細胞を培養し、Steenbakkersら、1994年、Mol.Biol.Rep.19巻:125〜134頁に記載のとおり不死化した。簡潔には、選択したB細胞を、平底96ウェル組織培養プレート中の7.5%(v/v)T細胞上清および50000照射(2500RAD)EL−4 B5栄養細胞と最終容量200μLのDMEM/HAM’s F12/10%子ウシ血清中で混合した。8日目に、上清をそれらの抗PD−1反応性についてCHO−hPD−1細胞ELISAによって以下の手順を使用して選別した。CHO−K1およびCHO−hPD−1細胞をコンフルエントまで平底96ウェルプレートで、50μL DMEM/HAM’s F12、10%FBS中で培養した。次に50μLの免疫グロブリン含有上清を37℃、1時間で加えた。PBS−Tweenで3回洗浄後、DMEM/HAM’s F12/10%FBS中の100μL(1:1000希釈)ヤギ抗マウス西洋わさびペルオキシダーゼ(HRP、Southern、Birmingham、AL、USA)を37℃、1時間で加えた。PBS−Tweenで3回洗浄後、固定化免疫グロブリンをUPO/TMB(Biomerieux、Boxtel、Netherlands)で可視化した。
【0149】
このB細胞培養物から、13個のhPD−1反応性の上清が同定され、プラスチック上に固定化された場合にジャーカットT細胞活性化を抑制することを示し、陽性ウェルからのB細胞クローンを、刊行された手順(Steenbakkersら、1992年、J.Immunol.Meth.152巻:69〜77頁;Steenbakkersら、1994年、Mol.Biol.Rep.19巻:125〜134頁)に従ってミニ電気融合によって不死化した。具体的には、B細胞を106個のNS−1骨髄腫細胞と混合し、血清をDMEM/HAM’s F12での洗浄によって除去した。次に細胞をプロナーゼ溶液で3分間処理し、続いて融合培地で洗浄した。電気融合を50μL融合容器で、30s、2MHz、400V/cmの交流電場に続いて、10μs、3kV/cmのスクウェア、高電場パルスおよび再度の30s、2MHz、400V/cmの交流電場によって実施した。最後に融合容器の内容物をハイブリドーマ選択培地に移し、限界希釈条件下で96ウェルプレートに蒔いた。融合の14日後、培養物をハイブリドーマの増殖について検査し、hPD−1への抗体反応性の存在について選別した。この手順は、hPD−1.05A、hPD−1.06B、hPD−1.08A、hPD−1.09AおよびhPD−1.13Aと名付ける5種の異なる抗hPD−1ハイブリドーマをもたらし、それらの完全性を保護するために限界希釈によってサブクローン化し、抗体を産生するためにさらに培養した。これらのハイブリドーマから得た上清は、抗CD3/抗CD28刺激におけるジャーカットE6.2.11細胞からのIL−2産生を強く抑制した(図1および以下の本文を参照されたい)。
【0150】
ジャーカットE6.1細胞(American Type Culture Collection)を限界希釈によって標準的方法を使用してサブクローン化し、サブクローンをCD3およびCD28の交差結合(cross−linking)でIL−2を産生する増大した能力について検査した。高IL−2産生サブクローンを得て、次にジャーカットE6.2.11と名付け、さらなるアッセイにおいて使用した。Costar3370 96ウェルアッセイプレートを5μg/mLヒツジ抗マウスIg(SAM)で一晩、4℃でコートした。過剰量のSAMを除去し、プレートを1ウェルあたり200μLのPBS/10%ウシ胎仔血清で1時間、室温でブロックした。PBSで3回洗浄後、ウェルを1ウェルあたり100μLの抗CD3(OKT3、10または60ng/mL)で1時間、37℃でコートした。PBSで3回洗浄後、1ウェルあたり50μLのPBS/10%ウシ胎仔血清および1ウェルあたり50μLのB細胞またはハイブリドーマ上清を30分間、37℃で加えた。PBSで3回洗浄後、1ウェルあたり120μLの細胞懸濁液、ジャーカットE6.2.11細胞(DMEM/F12/10%ウシ胎仔血清中の、1ウェルあたり細胞2×105個+0.5μg/mL抗CD28(Sanquin #M1650、Central Laboratory for Bloodtransfusion、Amsterdam、NL))を加えた。6時間培養後、上清をIL−2産生について標準的サンドイッチELISAをPharmingenからの抗hIL−2捕捉抗体とビオチン化検出抗体との対および検出試薬としてストレプトアビジン−西洋わさびペルオキシダーゼ(Southern Biotech)で使用して検査した。これらの抗体の効力をPD−L1と比較して決定するために、mAbの小さな群を大規模に産生させた。mAbをプロテインGアフィニティークロマトグラフィーを使用して精製した(実施例2を参照されたい)。精製した抗体、hPD−L1/Fc(組換えヒトB7−H1/Fcキメラ、R&D systems)またはネガティブコントロールとしてのマウスIgG1κ(Sigmaから)を同じ濃度でプレートに抗CD3と共に上に記載のとおりコートした。ジャーカットE6.2.11細胞および抗CD28を6時間加え、T細胞活性化を上清に産生されたIL−2によって測定した。抗体のうち2つ(hPD1.08AおよびhPD1.09A)は、固定化PD−L1/Fcと比較して8〜10倍強い抑制を示した。
【0151】
(実施例2)
マウス抗PD−1抗体の精製および特徴付け
抗PD−1産生ハイブリドーマの安定化および抗PD−1抗体の精製
クローン細胞集団を各ハイブリドーマから、それらを複数回(>4)の限界希釈にさらすことによって得た。次いで安定なハイブリドーマを無血清条件でCELLineバイオリアクター(Integra−biosciences)を使用して6から8日間培養した。細胞を内側の容器の無血清培地中に1mLあたり細胞3×106個の密度で15mLに蒔き、およそ1mLあたり細胞4×107個に8日間かけて増殖させた。外側の容器を10%までのBCS(仔ウシ血清)で補充した培地で満たした。6日目から8日目に、内側の容器の培養物を回収し、15mL SF培地で洗浄し、ハイブリドーマと再播種した。バイオリアクター上清および洗浄液を合わせ、遠心分離で澄ませた。得られた上清を0.22μM濾過膜で濾過した。抗体精製のために、上清を1:1で高塩濃度の結合緩衝液(1M グリシン/2M NaCl、pH9.0)に希釈し、mAbをプロテインG HiTrap 5mLカラム(GEhealthcare)を使用して精製した。PBSで洗浄後、結合抗体を0.1Mグリシン pH=2.7を使用して溶出し、3M Trisを使用するpH中和が続いた。最後に緩衝液をPD−10ゲル濾過カラム(GEhealthcare)を使用してPBSに交換し、抗体をUltra−15遠心濃縮器(Amicon)を使用して濃縮し、分光光度法を使用して定量した。
【0152】
市販の抗体
以下の市販の抗体を本明細書に記載の種々の研究において使用した:抗PD−1抗体クローンJ116(#14−9989)はeBioscienceから購入した。抗CTLA−4クローン14D3(mAb 16−1529)はeBioscienceから購入した。抗PD−1クローン192106(mAb1086)はR&D systemsから購入した(#mAb1086)。アイソタイプ対照抗体mIgG1、κ、クローンMOPC21はSigmaから購入した(#M9269)。アイソタイプ対照mIgG1κ(mAb 16−4714)およびIgG2aκ(mAb 16−4724)はeBioscienceから購入した。
【0153】
結合活性
タンパク質に基づく、および細胞に基づくELISA(「CELISA」)実験を見かけの結合親和性(EC50値として報告される)を決定するために使用した。場合により、抗PD−1抗体の結合を市販の抗PD−1抗体J116(eBioscience)およびMab1086(R&D systems)のそれと比較した。
【0154】
タンパク質ELISAを、抗体のヒトPD−1/Fcへの相対結合の決定のために使用した。hPD−1/Fc(R&D Systems)をMaxisorp 96ウェルプレート(Nunc)に4時間、室温(または4℃、一晩)でのインキュベーションによって固定化した。非特異的結合部位をPBST中の3%BSAとの1時間、室温でのインキュベーションによってブロックした。コーティング後、プレートをPBSTで3回洗浄した。抗PD−1抗体の希釈物を結合緩衝液(0.1%Tween20および0.3%BSAを含有するPBS)中に調製し、固定化融合タンパク質と1時間、25℃でインキュベートした。結合後、プレートをPBSTで3回洗浄し、結合緩衝液中に1/4000で希釈したペルオキシダーゼ標識ヤギ抗マウスIgG(Southern Biotech)と1時間、25℃でインキュベートし、再び洗浄し、TMBを使用して発色させた。ELISA結果を図2に示す。最大半量の結合での濃度を相対結合親和性の測定値として報告する(表II)。
【0155】
CHO−hPD−1細胞への結合もCELISAによって評価した。CELISAのためにCHO−hPD−1細胞を80から100パーセントコンフルエントまで50μL培地(DMEM/HAM’S 12F、10%FBS)中で培養した。次に種々の濃度の精製mAbを含有する50μL培地を1時間、37℃で加えた。PBS−Tweenでの3回洗浄後、100μLヤギ−抗マウス−HRP(Southern Biotech cat #1030−05)(培地中に1:1000希釈)を1時間、37℃で加えた。PBS−Tweenでさらに3回洗浄後、固定化免疫グロブリンを比色定量のペルオキシダーゼ基質TMB(BD Biosciences)で可視化した。ペルオキシダーゼ活性による吸光度(450nm)の増加をマイクロタイタープレートリーダーで測定した。図2は、抗体hPD−1.08AおよびhPD−1.09Aにおける濃度と結合との間の用量応答関係を示す。タンパク質および細胞の結合研究の結果を表IIにまとめる。
【0156】
生物光学干渉法(ForteBio)による動態解析
抗体の結合特性をさらに特徴付けるために、それぞれをOctet system(ForteBio、Menlo Park、CA)での生物光学干渉法を使用して概要を示し、結合動態を明らかにし、および平衡結合定数を算出した。このアッセイは、PD−1Fc融合タンパク質(R&D systems)の、標準的アミン化学を使用するアミン反応性バイオセンサー(Fortebio)へのカップリングによって実施された。次いで抗PD−1mAbのバイオセンサーへの結合およびバイオセンサーからの解離を種々の抗体濃度で観察した。具体的には、アミン反応性バイオセンサーを、0.1M MES pH=5.5を含むウェルに5分間浸漬することによって予め湿らせた。次いでバイオセンサーを0.1M NHS/0.4M EDC混合物を5分間使用して活性化した。PD−1/Fc融合タンパク質(R&D systems)を、バイオセンサーを0.1M MES中の12μg/mL PD−1/Fcの溶液に7.5分間浸漬することによってカップリングさせた。バイオセンサーの表面を1Mエタノールアミン溶液を5分間使用してクエンチさせた。バイオセンサーをPBS中に5分間、平衡化させた。抗PD−1mAbの会合を、種々の抗体濃度(PBS中、10〜80nM精製抗体>99% SDS−PAGEによる)を含むウェル中にバイオセンサーを置き、干渉(interferometry)を30分間モニターすることによって観察した。解離は、バイオセンサーをPBS中に移し、干渉シグナルを60分間モニターした後に測定した。観察された会合速度および解離速度(kobsおよびkd)を、検査した全ての濃度を含む1:1結合の包括的フィットモデルを使用して適合させ、平衡結合定数KDを算出した。動態研究からの結果を表IIおよび下の図6に示す。
【0157】
【表2】
モノクローナル抗体のうちの2つ、hPD−1.08AおよびhPD−1.09Aは、このアッセイを使用して検査した任意の他のmAbよりも相当に強く結合し、hPD−1.08AおよびhPD−1.09Aについて決定されたKDがそれぞれ24pMおよび22pMであった。検査した他の抗PD−1抗体と比較して、親和性の増大は、hPD−1.08AおよびhPD−1.09Aについて測定されたより遅い解離速度および顕著に早い会合速度による。
【0158】
リガンド遮断
フローサイトメトリーを使用して研究したリガンド結合の遮断。ヒトPD−1を発現するCHO細胞を接着培養フラスコから解離させ、種々の濃度の抗PD−1抗体、および一定濃度(600ng/mL)の未標識hPD−L1/Fcまたは組換えヒトPD−L2/Fc融合タンパク質(どちらもR&D Systemsから)と96ウェルプレートで混合した。混合物を30分間氷上で平衡化させ、FACS緩衝液(1%BCSおよび0.1%アジ化ナトリウムを含有するPBS)で3回洗浄し、FITC標識ヤギ抗ヒトFcとさらに15分間、氷上でインキュベートした。細胞を再びFACS緩衝液で洗浄し、フローサイトメトリーによって分析した。データを非線形回帰を使用するPrism(GraphPad Software、San Diego、CA)で分析し、IC50値を算出した。
【0159】
算出したIC50値を表IIにまとめる。抗体05A、06Bおよび13Aは、hPD−1の結合について600pMから3nMの間のKDを示すと確定された。強力な結合にもかかわらず、これらの抗体それぞれは、hPD−1へのhPD−L1の結合の遮断についてIC50>10nMを示した。市販で入手できる抗PD−1抗体J116(eBiosciences)は、結合について、この実験の範囲外に算出されたIC50(>100,nM)を有してPD−L1と弱く競合した。対照マウスIgG1は、PD−1結合についてPD−L1と競合しない。対照的に高親和性抗体hPD−1.08AおよびhPD−1.09Aは、1nMより低いIC50値でPD−L1結合を抑制したが、PD−L2結合はIC50値1〜2nM程度で遮断した(表II)。PD−L2は、PD−L1よりも2から6倍高い親和性でPD−1に結合するとより早く報告された。(Youngnak P.ら、2003年、Biochem.Biophys.Res.Commun.307巻、672〜677頁)
リガンド遮断は、蛍光比色微量容積アッセイ(fluorometric microvolume assay)技術(FMAT)を使用する同質的競合アッセイおよび検出を使用して確認された。簡潔には、CHO.hPD−1を接着培養フラスコから解離させ、種々の濃度の抗PD−1抗体および一定濃度(600ng/mL)のhPD−L1/FcまたはhPD−L2/Fc融合タンパク質(どちらもR&D Systemsから)と混合し、96ウェルプレート中で蛍光色素(AlexaFluor647、Invitrogen)で標識した。混合物を90分間、37℃で平衡化させ、AB8200Cellular Detection Analyzer(Applied Biosystems、Foster City、CA)を使用して読み取った。データは非線形回帰を使用するPrism(GraphPad Software、San Diego、CA)で分析し、IC50値を算出した。図3は、リガンド遮断の程度は抗体濃度によって決定されることを示す用量応答実験の結果を示す。hPD−L1/FcおよびhPD−L2/Fcの両方のCHO−hPD−1細胞への結合は、hPD−1.08A、hPD−1.09Aおよび(より少ない程度で)J116によって用量依存的な様式で完全に抑制されうる。算出されたIC50データを表IIにまとめる。フローサイトメトリーを使用して得られた結果を確認して、高親和性抗体hPD−1.08AおよびhPD−1.09Aは、PD−L1結合を1nMより低いIC50値で抑制した。
【0160】
種の交差反応
抗体の種の交差反応性を評価するために、マウスおよびcynomolgus macaque PD−1受容体をPCRによってクローン化し、安定に形質移入したCHO−K1細胞を作製した。抗体をカニクイザル受容体への結合についてCELISAを使用して検査した。市販の抗体J116、hPD−1.08AおよびhPD−1.09Aは、ヒトおよびカニクイザルPD−1に同等な親和性で結合し、hPD−L1/FcおよびhPD−L2/FcのカニクイザルPD−1への結合をヒトPD−1と比較して同様の有効性で遮断することが見いだされた。カニクイザルPD−1の細胞外部分のアミノ酸配列がヒトPF−1のそれと97%同一であることが見いだされたことから、これは驚くべきことではない。cynomolgous macaque由来のPD−1に加えて、hPD−1.08AおよびhPD−1.09Aは、rhesus macaques由来のPD−1も実施例3に記載のSEM刺激血液細胞培養物中で機能的に遮断した。検査した抗体で、使用したいずれのアッセイにおいても検出可能な親和性でマウスPD−1に結合したものはなかった。
【0161】
要約として、ジャーカット機能を調整する能力に基づいて単離した5個の抗PD−1モノクローナル抗体を精製し、かつ特徴付けた。これらの抗体は、PD−1に強く結合し(解離定数20pMから3nMの範囲で)、さまざまなIC50値でPD−L1およびPD−L2の両方と相互作用の遮断をできた。これらの抗hPD−1mAbの4個は、市販で入手可能な最も良い抗PD−1mAbより相当に良かった。受容体アンタゴニストとして作用する溶液中に加えた場合に各抗体は、最終的にはT細胞応答を増強した(実施例3を参照されたい)。
【0162】
(実施例3)
抗PD−1抗体の機能的プロファイリング
SEBへのヒトT細胞応答は、hPD−1.08AおよびhPD−1.09Aによって増強される。
【0163】
抗PD−1抗体をT細胞活性を増強するそれらの能力についてin vitroで、健常ボランティア由来の血液細胞を使用して検査した。ヒトPD−1受容体を遮断することの機能的結果を特徴付けるために使用したアッセイは、Vβ3およびVβ8 T細胞受容体鎖を発現する全てのT細胞に結合し、かつ活性化するためにブドウ球菌エンテロトキシンB(SEB)を利用した。健常人ドナー血液を得て、培地中に1:10で希釈した。希釈した全血を96ウェル丸底プレートに蒔き(1ウェルあたり150μl)、30〜60分間、mAbとさまざまな濃度でプレインキュベートした。次いでSEBを10ng/mLから10μg/mLの範囲の種々の濃度で加えた。培養2から4日後に上清を回収し、産生されたIL−2の量をELISA(実施例1に記載)を使用して、または標準的多重技術(Luminex platform−Biosourceサイトカイン検出キット)を使用して定量した。100ng/mLから最大10μg/mLまでのSEBの滴定は、全血細胞によるIL−2産生を顕著に刺激した。通常、1μg/mLのSEBでの刺激2〜4日後にELISAによって、ドナーに依存して、100から1000pg/mLのIL−2が検出可能であった。hPD−1.08AおよびhPD−1.09Aの添加は、対照マウスIgG1を超えて、検査した最高抗体濃度(25μg/mL)で平均2から4倍にIL−2産生を増強した。刺激指数は、一連の独立の健常ボランティアで実施した実験について平均した(図4)。これらの実験は、hPD−1.08AおよびhPD−1.09Aの両方が希釈全血細胞のSEB刺激でIL−2産生を増強したことを示した。PD−1およびPD−L1の両方(しかしPD−L2ではない)の発現レベルは、全血細胞のSEB刺激後に徐々に上方制御された(フローサイトメトリーによる定量)。抗PD−L1モノクローナル抗体(クローンMIH5、Ebioscience #16−5982)および抗CTLA−4(クローン14D3、eBioscience #16−1529)も、同様の条件下でIL−2産生の増加を誘発し、同時刺激経路の操作後にT細胞活性を定量するためのSEB刺激アッセイの使用をさらに検証した発見を導いた(図4)。抗PD−1抗体によって増強されたIL−2産生は、用量依存的であることが見いだされた。IL−2に加えてLuminex technologyによって、TNTα、IL−17、IL−7、IL−6およびIFNγのレベルもhPD−1.08AおよびhPD−1.09Aによって顕著に調節されることが見いだされた。これらの実験の結果は、hPD−1.08AおよびhPD−1.09AをヒトT細胞応答を刺激するために使用できることを示す。
【0164】
抗PD−1抗体hPD−1.09AをT細胞活性を増強するその能力についてin vitroで癌患者由来の血液細胞を使用してさらに検査した。進行黒色腫(1名)または前立腺癌(3名)を有する患者由来の血液を上の手順に従って検査した。サイトカイン定量の結果を表IIIに、細胞を25μg/mL hPD−1.09Aの存在下で刺激した場合を抗体非存在下でのSEB刺激と比較して、産生されたサイトカインの増加倍数で表す。要約すると、hPD−1.09Aは、4名の各患者についてSEB誘発IL−2産生を2から3.5倍増加させたことが見いだされた。同様にTNTα、IL−17およびIFNγの産生は、増強され、IL−5およびIL−13の産生は減少した。これらの実験は、hPD−1.09Aが癌患者においてT細胞応答を刺激する能力を有することを示す。さらにこれらの実験は、Th1応答に対する選好性を示唆する。
【0165】
【表3】
TTチャレンジに対するヒトリコールT細胞応答は、hPD−1.08AおよびhPD−1.09Aによって増強される。
【0166】
抗ヒトPD−1抗体遮断受容体のその天然のリガンドとの相互作用の機能的効果を要約するために使用された他のアッセイは、破傷風トキソイド(TT)抗原を健常人ドナー血液における既存のメモリーT細胞を刺激するために使用した。この目的を達成するために、新鮮調製PBMC(細胞2×105個)を96ウェル丸底プレートの完全RPMI1640培地(5%熱不活性化ヒト血清を含む)に蒔き、さまざまな濃度の検査抗体とプレインキュベートし、100ng/mLの濃度のTT(Astarte Biologics)で刺激した。細胞を3〜7日間、37℃、5%CO2でインキュベートし、その後上清を回収した。サイトカイン濃度をELISA(eBioscienceからのIL−2およびIFNγ ELISA検出抗体対セット)および多重分析(Luminex platform−Biosourceサイトカイン検出キット)によって決定した。PD−1の遮断は、抗原単独と比較して、増殖を増強し、かつIFNγおよびIL−2を含むサイトカイン産生を顕著に増強した(図5)。Luminex分析は、サイトカインGM−CSF、RANTESおよびIL−6の産生がPD−1遮断で増加することを明らかにした。
【0167】
ホルマリン固定パラフィン包埋ヒト細胞でのヒトPD−1の染色
SEB刺激血液細胞がPD−1の増強された発現をフローサイトメトリーで示したことから、これらの細胞を、hPD−1.09Aが組織学的使用のためのホルマリン固定パラフィン包埋組織においてPD−1を検出できるかどうかを決定するために使用した。ヒトドナー末梢血単核球を、0.1μg/mL SEBで3日間刺激し、その後非接着細胞(主にリンパ球)を回収し、PBSで2回洗浄し、遠心分離(1100rpm、5分間)した。細胞を10分間、4%ホルムアルデヒドで固定し、細胞沈渣をアガロースに包埋し、エタノール(70%、80%、96%および100%と続いて)およびキシレン中で脱水し、その後パラフィンに包埋した。切片(4μm)をガラススライドにマウントし、水和させ(キシレン、エタノール100%、96%、80%、70%、PBS緩衝液)、その後加熱したクエン酸緩衝液への抗原回収を標準的方法を使用して実施した。ペルオキシダーゼ活性を0.3%H2O2を含む100%メタノールを使用して遮断し、スライドを水およびPBS、0.1%Tweenでリンスした。切片をhPD−1.09Aと1.5時間、室温でインキュベートし、PBS−Tweenでリンスし、標準的検出法が続いた。スライドをヘマトキシリンで30秒間、室温で対比染色し、キシレンで脱水し、マウントして顕微鏡検査した。これらの実験は、SEB刺激PBCM培養物由来のリンパ球は、未刺激のPBMC培養物とは対照的に、hPD−1.09Aで(アイソタイプ対照と比較した場合)強く染まり、hPD−1.09Aが診断試薬として有用であることを示した。
【0168】
(実施例4)
抗PD−1抗体配列および続くヒト化
免疫グロブリンcDNAのクローニング
縮重プライマーPCRに基づく方法を使用して、ハイブリドーマhPD−1.08AおよびhPD−1.09Aによって発現されるマウス抗体の可変領域をコードするDNA配列を決定した。簡潔には、重鎖および軽鎖についての遺伝子特異的cDNAをiScript Select cDNA合成キット(Biorad #1708896)を製造者の説明書に従って使用して生成した。使用したPCRプライマーは、Ig−プライマーセット(Novagen #69831−3)に基づいた。縮重PCR反応は、TaqポリメラーゼをNovagenプライマーセット手順書に従って使用して実施した。PCR産物をアガロースゲル電気泳動によって分析した。重鎖および軽鎖の可変領域の両方について予測される増幅産物サイズは、約500塩基対である。適切なバンドをもたらした反応からのTaq−増幅PCR産物2μlをpCR4 TOPOベクター(Invitrogen #K4595−40)にクローン化し、DH5−αE.coliに製造者による指示のとおり形質転換した。クローンをユニバーサルM13順方向および逆方向プライマーを使用してコロニーPCRによって選別し、各反応から2から3クローンをDNA配列分析用に選択した。
【0169】
クローンをユニバーサルプライマーM13順方向、M13逆方向、T3およびT7を使用して両方向に配列決定した。各クローンについての各配列決定反応の結果をSeqmanを使用して分析した。共通配列を、NCBI Ig−Blast(http://www.ncbi.nlm.nih.gov/projects/igblast/)を使用して、生殖系列および再配列Ig可変領域配列のデータベースに対して探索した。hPD−1.08AについてのBlastの結果は、ストップコドンが導入されていない生産性(インフレーム)に再配列された重鎖を同定した。2種の異なる配列、1つはストップコドンが導入されていない生産性(インフレーム)に再配列された軽鎖、他方はFR4領域中にストップコドンに至るフレームシフトを含有する非生産的に再配列された配列、をコードする軽鎖クローンが同定された。観察された非生産的増殖不能転写物は、おそらく骨髄腫融合相手由来であり(Carroll W.L.ら、Mol.Immunol.25巻:991〜995頁(1988年)排除した。
【0170】
hPD−1.09AについてのBlastの結果は、ストップコドンが導入されていない生産性(インフレーム)に再配列された重鎖および軽鎖を同定した。発現されたタンパク質のアミノ酸配列は、質量分析によって確認した。配列を添付の配列表で開示し、表IVに列挙する。
【0171】
【表4−1】
【0172】
【表4−2】
CDRおよびフレームワーク領域は、Kabat E.A.ら、1991年、Sequences of proteins of Immunological interest、In: NIH Publication No.91−3242、US Department of Health and Human Services、Bethesda、MDに従って注釈を付けた。
【0173】
キメラc109A抗体の構築および発現
キメラ軽鎖および重鎖を、マウスhPD−1.09A VLおよびVH領域のPCRでクローン化したcDNAをヒトκおよびIgG1定常領域にそれぞれ連結することによって構築した。マウスcDNA配列の5’および3’末端を、各鎖に適切なリーダー配列および既存の組換え抗体発現ベクターへのクローニングを可能にする制限部位を付加するために設計したPCRプライマーを使用して改変した。
【0174】
COS−7細胞(1mLあたり107個で0.7mL)に各キメラ重鎖および軽鎖発現プラスミド10μgを電気穿孔した。次いでこれらの細胞を8mL 増殖培地中で3日間培養した。サンドイッチELISAを、COS−7形質移入体由来の上清中の抗体濃度を測定するために使用した。これは、形質移入されたCOS−7細胞は、3種の別々の形質移入体で約295ng/mLのキメラIgG1−κ抗体を分泌したことを示した。
【0175】
形質移入されたCOS−7細胞によって産生されたキメラ抗体の結合は、PD−1結合ELISAおよびCELISA(実施例2を参照されたい)を使用して測定し、マウス抗体に匹敵する親和性でPD−1に結合することを示した。
【0176】
ヒト化抗体設計
hPD−1.09A抗体をMRCT(Cambridge UK)によりCDR移植技術を使用してヒト化した(例えば、米国特許第5,225,539号を参照されたい)。簡潔には、マウス抗体hPD−1.09Aの可変鎖配列をResearch Collaboratory for Structural Bioinformatics(RCSB)タンパク質データバンクにおいて入手可能な配列と比較した。hPD−1.09Aの相同性モデルを最も近いVHおよびVK構造に基づいて生成した。hPD−1.09Aに最も高い同一性を有するヒト配列を同定し、分析した(Foote and Winter、J.Mol.Biol.224巻:487〜499頁(1992年);Morea V.ら、Methods 20巻:267〜270頁(2000年);Chothia C.ら、J.Mol.Biol.186巻:651〜663頁(1985年))。CDR移植重鎖および軽鎖を構築するために最も適切なヒトフレームワークを同定した。
【0177】
重鎖について、genbankアクセッション番号AB063829でコードされるフレームワークを最も適切であると決定した。hPD−1.09A VK配列の分析は、そのCDR1の長さ(15残基)が任意のヒトVKにおいて見いだされないことを示す。このため3種の異なるCDR1の長さ(11、16および17残基)のフレームワークを、どのCDR1の長さがhPD−1.09A VKの挙動を再生するのかを検査するために分析した。その構造中において重要である選択された残基においてhPD−1.09A VKに最も高い同一性を有し、CDR1の長さ11、16および17を有するヒトVK配列を同定した。genbankアクセッション番号M29469のフレームワークをK109A−L−11に基づいて選択した。genbankアクセッション番号AB064135由来のフレームワークをK09A−L−16に基づいて選択し、genbankアクセッション番号X72431由来のフレームワークをK09A−L−17に基づいて選択した。
【0178】
ストレートグラフト(straight graft)を各鎖に対する発現構築物を生成するために実施した。109A−H、K09A−L−11、K09A−L−16およびK09A−L−17のDNAおよびタンパク質配列を添付の配列表(表IV)において開示する。
【0179】
セリン241(Kabat番号)がプロリンに転換された安定化Adair変異を有する(Angal S.ら、Mol.Immuol.30巻:105〜108頁(1993年))ヒト化h109A抗体のIgG4バージョンを生成した。この配列は、配列番号23および31で開示される。
【0180】
(実施例5)
ヒト化抗PD−1抗体の結合特性および機能特性
産生および精製
ヒト化抗体h409A11、h409A16およびh409A17をCHO−S細胞の一過的形質移入によって産生させた。細胞をCD−CHO(Gibco)およびC5467培地(Sigma)で8日間、振盪フラスコ中で増殖させた。抗体をプロテインAクロマトグラフィーによって細胞上清から精製し、洗浄し、1M酢酸を使用して溶出し、3M Trisを使用して中和した。最後に緩衝液を、1M Tris塩基でpH5.5に調整した100mM酢酸に交換した。
【0181】
結合および動態解析
見かけの結合親和性を決定するために(EC50値として報告される)タンパク質に基づく、および細胞に基づくELISAを実施例2に記載のとおり実施した。ヒト化抗PD−1抗体は、PD−1/Fcおよび細胞で発現されるPD−1にマウス親抗体に匹敵するEC50値でそれぞれ結合した(表V)。
【0182】
抗体の動態学的な結合特性も実施例2に記載のとおり生物光学干渉法を使用して実施した(図6)。ヒト化抗体の2つh409A11およびh409A16は、h409A11およびh409A16について決定されたKDがそれぞれ29pMおよび27pMであって(表V)、このアッセイを使用して検査した他のいずれのmAbよりも相当強く結合した。検査した他の抗PD−1抗体と比較して、増大した親和性は、主により遅い解離速度による。マウス親抗体と同様に、ヒト化抗PD−1抗体h409A11、h409A16は、120pMより低く決定されたKDでカニクイザル(cynomolgous)PD−1への結合を示した。
【0183】
リガンド遮断
PD−L1およびPD−L2のPD−1への結合を遮断するヒト化抗体の能力を、実施例2に記載のとおりFMAT競合アッセイを使用する同質的競合アッセイおよび検出を使用して測定した。
【0184】
hPD−L1/FcおよびhPD−L2/Fcの両方のCHO−hPD1細胞への結合は、検査した任意のヒト化抗体によって用量依存的に完全に抑制し得る。算出されたIC50データを表Vにまとめる。親マウス抗体hPD−1.09Aと同様に、ヒト化mAb、h409A11、h409A16およびh409A17のそれぞれは、PD−L1およびPD−L2結合を1nMより低いIC50値で抑制した。マウス親抗体と同様に、ヒト化抗PD−1抗体h409A11、h409A16およびh409A17は、約1nMより低く算出されたIC50でカニクイザルPD−1へのリガンド結合の抑制を示した。
【0185】
【表5】
SEBへのヒトT細胞の応答はヒト化mAbによって増強される。
【0186】
ヒト化抗PD−1抗体をT細胞活性を増強させるそれらの能力についてin vitroで、実施例3に記載のとおり健常人ボランティア由来の血液細胞を使用して検査した。上清を培養4日後に回収し、産生されたIL−2の量をELISAを使用して定量した。ヒト化PD−1抗体は、SEBによって刺激されたIL−2産生を増大させる能力を示した(図7)。追加的にヒト化PD−1抗体は、実施例3において記載されたのと同様に、癌患者の血液においてSEB誘発IL−2産生を増大させた。
【0187】
要約として、ヒト化mAb h409A11、h409A16およびh409A17は、ヒト化過程において全ての機能的活性を保持した。h409A11およびh409A16mAbは、ヒト化においてマウスの親抗体hPD109Aの親和性を全て保持した。
【背景技術】
【0001】
(発明の背景)
プログラムされたデスレセプター1(PD−1)は、活性化されたT細胞およびB細胞において主に発現される免疫抑制受容体である。そのリガンドとの相互作用はin vitroおよびin vivoの両方でT細胞応答を減弱させることが示されている。PD−1とそのリガンドの1つPD−L1との相互作用の遮断は、腫瘍特異的CD8+T細胞免疫を増強することが示されており、したがって免疫系による腫瘍細胞の排除において有益でありうる。
【0002】
PD−1(遺伝子Pdcd1によってコードされる)は、CD28およびCTLA−4に関連する免疫グロブリンスーパーファミリーの一員である。PD−1は、そのリガンド(PD−L1および/またはPD−L2)との結合で抗原受容体シグナル伝達を負に制御することが示されている。マウスPD−1の構造は解析されており、ヒトPD−L1を伴うマウスPD−1の共結晶構造も同様である(非特許文献1;非特許文献2)。PD−1などのファミリーの構成員は、リガンド結合に関与するIg可変型(V型)ドメインおよびシグナル伝達分子の結合に関与する細胞質側尾部を含有するI型膜貫通型糖タンパク質である。PD−1の細胞質側尾部は、2個のチロシンに基づくシグナル伝達モチーフ、ITIM(免疫受容体チロシンベース抑制モチーフ)およびITSM(免疫受容体チロシンベーススイッチモチーフ)を含有する。
【0003】
T細胞の刺激に続いて、PD−1は、その細胞質側尾部内のITSMモチーフにチロシンホスファターゼSHP−2を補充し、CD3T細胞シグナル伝達カスケードに関与するCD3ζ、PKCθおよびZAP70などのエフェクター分子の脱リン酸化を導く。PD−1がT細胞応答を下方調節する機構は、CTLA−4の機構と、両分子が重複した一連のシグナル伝達タンパク質を制御することから似てはいるが異なっている(非特許文献3)。Bennettおよび協力者らは、T細胞シグナル伝達のPD−1介在抑制が活性化シグナルおよび抑制シグナルの両方が同じ表面にある場合にだけ有効であることを示し、PD−1シグナル伝達機構が時空間的に決定されることを示唆している(非特許文献4)。
【0004】
PD−1は、活性化されたリンパ球(末梢CD4+およびCD8+T細胞、B細胞ならびに単球)で発現されることが示され、胸腺の発達の際にCD4−CD8−(ダブルネガティブ)T細胞およびNK−T細胞で発現されることも示されている。
【0005】
PD−1に対するリガンド(PD−L1およびPD−L2)は、恒常的に発現されているか、または、非造血性組織および種々の腫瘍型を含むさまざまな細胞型において誘導されうる。PD−L1は、B細胞、T細胞、骨髄系細胞および樹状細胞(DC)において発現されるが、微小血管内皮細胞および、心臓、肺などの非リンパ器官などの末梢細胞においても発現される。対照的にPD−L2は、マクロファージおよびDCにおいてのみ見いだされる。PD−1リガンドの発現様式は、末梢性寛容の維持におけるPD−1の役割について示唆的であり、末梢における自己反応性T細胞およびB細胞の応答を制御するために役立ちうる。両リガンドは、IgVおよびIgC様ドメインの両方を細胞外領域に含むI型膜貫通型受容体である。両リガンドは、未知のシグナル伝達モチーフを有する短い細胞質側領域を含む。
【0006】
今日までに多数の研究が、PD−1とそのリガンドとの相互作用がin vitroおよびin vivoにおいてリンパ球増殖の抑制を導くことを示している。PD−1/PD−L1相互作用の破壊は、T細胞増殖およびサイトカイン産生を増大させ、かつ細胞周期の進行を遮断することが示されている。Pdcd1−/−マウスでの最初の研究は、劇的な免疫表現型を同定しなかった。しかし高齢マウスは、Pdcdl欠損を戻し交配した株によって異なる自然発症の自己免疫疾患を発症した。これらは、ループス様増殖性関節炎(C57BL/6)(非特許文献5)、胎児性心筋炎(BALB/c)(非特許文献6)、およびI型糖尿病(NOD)(非特許文献7)を含む。総合的に、ノックアウト動物の分析は、PD−1が主に末梢性寛容の誘導および制御において機能するという理解を導いている。したがってPD−1経路の治療用遮断は、免疫寛容の克服において有益でありうる。そのような選択的遮断は、癌または感染症の治療およびワクチン接種(予防用または治療用のいずれでも)の際の免疫を高めることにおいて有用でありうる。
【0007】
癌におけるPD−1の役割は、文献において立証されている。腫瘍微小環境が有効な免疫性の破壊から腫瘍細胞を保護できることが知られている。近年PD−L1は、多数のマウスおよびヒトの腫瘍において発現される(かつPD−L1陰性腫瘍細胞系の大部分においてIFNγによって誘導可能である)ことが示されており、免疫回避を介在すると推定される(非特許文献8;非特許文献9)。
【0008】
ヒトにおいて(腫瘍浸潤リンパ球における)PD−1および/または(腫瘍細胞における)PD−L1の発現は、免疫組織化学によって評価された多数の原発性腫瘍の生検において見いだされている。そのような組織として、肺、肝臓、卵巣、子宮頸部、皮膚、大腸、神経膠腫、膀胱、乳房、腎臓、食道、胃、口腔扁平上皮細胞、尿路上皮細胞および膵臓の癌ならびに頭頸部の腫瘍が挙げられる(非特許文献10;非特許文献11;非特許文献12;非特許文献9;非特許文献13;非特許文献14;非特許文献15)。より著しくは、腫瘍細胞におけるPD−リガンド発現は、複数の腫瘍型にわたって癌患者の予後不良と関連している(非特許文献16で概説されている)。
【0009】
PD−1/PD−L1相互作用の遮断は、腫瘍特異的T細胞免疫の増強を導くことができ、したがって免疫系による腫瘍細胞の排除において有益でありえた。この課題に取り組むために、多数の研究が実施された。侵襲性膵臓癌のマウスモデルにおいて、T.Nomiら(非特許文献17)は、PD−1/PD−L1遮断の治療効果を示した。PD−1またはPD−L1のいずれかに対する抗体の投与は、腫瘍増殖を顕著に抑制した。抗体遮断は、腫瘍反応性CD8+T細胞の腫瘍への浸潤を効果的に促進し、IFNγ、グランザイムBおよびパーフォリンを含む抗腫瘍エフェクターの上方制御を生じた。さらに該著者らは、PD−1遮断は相乗効果をもたらすために化学療法と効果的に組み合わせうることを示した。マウスでの扁平上皮癌のモデルを使用する他の研究において、PD−1またはPD−L1の抗体遮断は、腫瘍増殖を顕著に抑制した(非特許文献18)。
【0010】
他の研究においてPD−L1でのマウス肥満細胞腫系への形質移入は、腫瘍特異的CTLクローンと同時培養した場合に腫瘍細胞の溶解の減少を生じた。抗PD−L1 mAbを加えた場合に溶解は回復した(非特許文献8)。in vivoにおいてPD−1/PD−L1相互作用の遮断は、マウス腫瘍モデルにおいて養子T細胞移入療法の効果を増大させることが示された(非特許文献9)。癌治療におけるPD−1の役割についてのさらなる証拠は、PD−1ノックアウトマウスで実施された実験によってもたらされる。PD−L1発現骨髄腫細胞は、野生型動物においてのみ増殖し(腫瘍の増殖およびそれに伴う動物の死を生じる)、PD−1欠損マウスにおいては増殖しない(非特許文献8)。
【0011】
ヒトでの研究において、非特許文献19)は、完全なヒト抗PD−1抗体を使用するPD−1遮断は、ワクチン抗原およびワクチン接種された個体由来の細胞を使用するex vivo刺激アッセイにおいて腫瘍特異的CD8+T細胞(CTL)の絶対数を増加させることを示した。同様の研究においてPD−L1の抗体遮断は、腫瘍関連抗原特異的細胞傷害性T細胞の細胞溶解活性の増強および腫瘍特異的TH細胞によるサイトカイン産生の増大を生じた(非特許文献20)。同著者らは、抗CTLA−4遮断と組み合わせて使用する場合にPD−L1遮断がin vitroでの腫瘍特異的T細胞応答を増大させることを示した。
【0012】
総合的に、PD−1/PD−L1経路は、癌治療用の抗体療法の開発のための十分に検証された標的である。抗PD−1抗体は、慢性ウイルス感染においても有用でありうる。急性ウイルス感染後に生成されたメモリーCD8+T細胞は、高度に機能的でありかつ防御免疫の重要な成分を構成する。対照的に慢性の感染は、ウイルス特異的T細胞応答の機能障害(消耗)の程度の変化によってしばしば特徴付けられ、この欠陥が宿主が持続性の病原体を排除できない主な理由である。感染の初期において機能的なエフェクターT細胞が最初に生成されるが、慢性的な感染の過程においてそれらは徐々に機能を失う。Barberら(非特許文献21)は、LCMVの研究室株で感染させたマウスが血液中および他の組織における高レベルのウイルスを生じる慢性感染を発症することを示した。これらのマウスは、最初強いT細胞応答を発現したが、T細胞の消耗により最終的には感染に屈服した。著者らは、慢性的に感染したマウスにおけるエフェクターT細胞の数および機能の低下は、PD−1とPD−L1との間の相互作用を遮断する抗体の注射によって回復できることを見いだした。
【0013】
近年、PD−1がHIVに感染した個体由来のT細胞で高度に発現されていることならびに受容体発現がT細胞機能障害および疾患の進行と関連することが示されている(非特許文献22;非特許文献23)。両方の研究においてリガンドPD−L1の遮断は、in vitroでHIV特異的IFNγ産生細胞の増殖を顕著に増加させた。
【0014】
他の研究もウイルス感染の管理におけるPD−1経路の重要性に関連している。PD−1ノックアウトマウスは、野生型マウスよりも優れたアデノウイルス感染の管理を示す(非特許文献24)。同様にHBV特異的T細胞のHBVトランスジェニック動物への養子移入は、肝炎を惹起する(非特許文献25)。これらの動物の疾患の状態は、肝臓での抗原認識および肝臓細胞によるPD−1の上方制御の結果として変動する。
【先行技術文献】
【非特許文献】
【0015】
【非特許文献1】Zhang, X.ら、Immunity20巻:337〜347頁(2004年)
【非特許文献2】Linら、Proc.Natl.Acad.Sci.USA105巻:3011〜6頁(2008年)
【非特許文献3】Parryら、Mol.Cell Biol.25巻:9543〜9553頁
【非特許文献4】Bennett F.ら、J Immunol.170巻:711〜8頁(2003年)
【非特許文献5】Nishimura H.ら、Int.Immunol.10巻:1563〜1572頁(1998年)
【非特許文献6】Nishimura H.ら、Science291巻:319〜322頁(2001年)
【非特許文献7】Wang J.ら、Proc.Natl.Acad.Sci.U.S.A 102巻:11823〜11828頁(2005年)
【非特許文献8】Iwai Y.ら、Proc.Natl.Acad.Sci.U.S.A.99巻:12293〜12297頁(2002年)
【非特許文献9】Strome S.E.ら、Cancer Res.、63巻:6501〜6505頁(2003年)
【非特許文献10】Brown J.A.ら、J.Immunol.170巻:1257〜1266頁(2003年)
【非特許文献11】Dong H.ら、Nat.Med.8巻:793〜800頁(2002年)
【非特許文献12】Wintterleら、Cancer Res.63巻:7462〜7467頁(2003年)
【非特許文献13】Thompson R.H.ら、Cancer Res.66巻:3381〜5頁(2006年)
【非特許文献14】Thompsonら、Clin.Cancer Res.13巻:1757〜61頁(2007年)
【非特許文献15】Nomi T.ら、Clin.Cancer Res.13巻:2151〜7頁(2007年)
【非特許文献16】Okazaki and Honjo、Int.Immunol.19巻:813〜824頁(2007年)
【非特許文献17】Clin.Cancer Res.13巻:2151〜2157頁(2007年)
【非特許文献18】Tsushima F.ら、Oral Oncol.42巻:268〜274頁(2006年)
【非特許文献19】R.M.Wongら(Int.Immunol.19巻:1223〜1234頁(2007年)
【非特許文献20】Blank C.ら、Int.J.Cancer119巻:317〜327頁(2006年)
【非特許文献21】Barberら、Nature439巻:682〜687頁(2006年)
【非特許文献22】Dayら、Nature443巻:350〜4頁(2006年)
【非特許文献23】Trautmann L.ら、Nat.Med.12巻:1198〜202頁(2006年)
【非特許文献24】Iwaiら、J.Exp.Med.198巻:39〜50頁(2003年)
【非特許文献25】Isogawa M.ら、Immunity23巻:53〜63頁(2005年)
【発明の概要】
【課題を解決するための手段】
【0016】
(発明の簡単な要旨)
本発明は、ヒトおよびカニクイザルのPD−1に結合する単離された抗体および抗体断片を提供する。いくつかの実施形態において抗体または抗体断片は、ヒトPD−L1およびヒトPD−L2のヒトPD−1への結合を遮断する。いくつかの実施形態において本発明のPD−1抗体または抗体断片は、配列番号9、10、11、12、13、14、15、16、17、18、19および20から選択される1つまたは複数のCDR(抗体相補性決定領域)を含み、さらなる実施形態においては、配列番号12、13、14、18、19および20の1つまたは複数の重鎖CDRならびに/または配列番号9、10、11、15、16および17の軽鎖CDRを含む。いくつかの実施形態において抗体または抗体断片は、キメラ抗体、ヒト抗体、ヒト化抗体またはその断片である。
本発明は、例えば以下の項目を提供する。
(項目1)
a.配列番号9、10、11、15、16および17からなる群から選択される少なくとも1つのCDR、もしくは任意の該配列の変異体、ならびに/または
b.配列番号12、13、14、18、19および20からなる群から選択される少なくとも1つのCDR、もしくは任意の該配列の変異体
を含む、ヒトPD−1に結合する単離された抗体または抗体断片。
(項目2)
a.配列番号9、10および11の軽鎖CDR、もしくは任意の該配列の変異体、ならびに配列番号12、13および14の重鎖CDR、もしくは任意の該配列の変異体、または
b.配列番号15、16および17の軽鎖CDR、もしくは任意の該配列の変異体、ならびに配列番号18、19および20の重鎖CDR、もしくは任意の該配列の変異体
を含む、項目1に記載の抗体または抗体断片。
(項目3)
a.i.配列番号5もしくはその変異体、
ii.配列番号7もしくはその変異体、
iii.配列番号30のアミノ酸残基20から139もしくはその変異体、および
iv.配列番号30のアミノ酸残基20から139に少なくとも90%の相同性を有するアミノ酸配列
からなる群から選択されるアミノ酸配列を含む重鎖可変領域
を含み、
b.i.配列番号6もしくはその変異体、
ii.配列番号8もしくはその変異体、
iii.配列番号32のアミノ酸残基20から130もしくはその変異体、
iv.配列番号33のアミノ酸残基20から130もしくはその変異体、
v.配列番号34のアミノ酸残基20から130もしくはその変異体、および
vi.配列番号32、33もしくは34のアミノ酸残基20から130に少なくとも90%の相同性を有するアミノ酸配列
からなる群から選択されるアミノ酸配列を含む軽鎖可変領域
をさらに含む、項目2に記載の抗体または抗体断片。
(項目4)
a.i.配列番号31のアミノ酸残基20から466もしくはその変異体、および
ii.配列番号35のアミノ酸残基20から469もしくはその変異体
からなる群から選択されるアミノ酸配列を含む重鎖、
ならびに
b.i.配列番号36のアミノ酸残基20から237もしくはその変異体、
ii.配列番号37のアミノ酸残基20から237もしくはその変異体、および
iii.配列番号38のアミノ酸残基20から237もしくはその変異体
からなる群から選択されるアミノ酸配列を含む軽鎖
を含む、項目1に記載の抗体。
(項目5)
(1つまたは複数の)任意の前記変異体が3個までの保存的に改変されたアミノ酸置換を含みうる、項目1から4のいずれかに記載の抗体または抗体断片。
(項目6)
a.ヒト重鎖定常領域、もしくは20個までの保存的に改変されたアミノ酸置換を含むその変異体、および/または
b.ヒト軽鎖定常領域、もしくは20個までの保存的に改変されたアミノ酸置換を含むその変異体
をさらに含む、項目1から3のいずれかに記載の抗体。
(項目7)
前記ヒト重鎖定常領域がγ4もしくはγ1ヒト重鎖定常領域、または20個までの保存的に改変されたアミノ酸置換を含むその変異体を含む、項目6に記載の抗体。
(項目8)
a.約100pMのKDまたはそれより低いKDでヒトPD−1に結合する、
b.約30pMのKDまたはそれより低いKDでヒトPD−1に結合する、
c.配列番号31のアミノ酸配列を含む重鎖および配列番号32のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合する、
d.配列番号31のアミノ酸配列を含む重鎖および配列番号33のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合する、
e.約7.5×105 1/M・sまたはそれより速いkassocでヒトPD−1に結合する、
f.約1×106 1/M・sまたはそれより速いkassocでヒトPD−1に結合する、
g.約2×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合する、
h.約2.7×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合する、
i.約3×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合する、
j.約1nMのIC50またはそれより低いIC50でヒトPD−L1またはヒトPD−L2のヒトPD−1への結合を遮断する、
項目1から7のいずれかに記載の抗体または抗体断片。
(項目9)
項目1から4に記載のいずれかの抗体とPD−1上の結合エピトープについて競合し、以下の特徴:
a.約100pMのKDまたはそれより低いKDでヒトPD−1に結合する、
b.約30pMのKDまたはそれより低いKDでヒトPD−1に結合する、
c.配列番号31のアミノ酸配列を含む重鎖および配列番号32のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合する、
d.配列番号31のアミノ酸配列を含む重鎖および配列番号33のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合する、
e.約7.5×105 1/M・sまたはそれより速いkassocでヒトPD−1に結合する、
f.約1×106 1/M・sまたはそれより速いkassocでヒトPD−1に結合する、
g.約2×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合する、
h.約2.7×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合する、
i.約3×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合する、または
j.約1nMのIC50またはそれより低いIC50でヒトPD−L1またはヒトPD−L2のヒトPD−1への結合を遮断する、
のうち1つを有する
単離された抗体または抗体断片。
(項目10)
a.キメラ抗体もしくはその断片、
b.ヒト抗体もしくはその断片、
c.ヒト化抗体もしくはその断片、または
d.Fab、Fab’、Fab’−SH、Fv、scFv、F(ab’)2およびダイアボディーからなる群から選択される抗体断片
である、項目1から9のいずれかに記載の抗体または抗体断片。
(項目11)
T細胞の活性化を増大させる、項目1から10のいずれかに記載の抗体または抗体断片。
(項目12)
項目1から7のいずれか一項に記載の抗体または抗体断片をコードする単離されたポリヌクレオチド。
(項目13)
項目12に記載の単離されたポリヌクレオチドを含む発現ベクター。
(項目14)
項目13に記載の発現ベクターを含む宿主細胞。
(項目15)
a.前記核酸配列が発現される条件下で、培地中で項目14に記載の宿主細胞を培養し、それにより前記軽鎖および前記重鎖可変領域を含むポリペプチドを産生するステップと、
b.該宿主細胞または該培地から該ポリペプチドを回収するステップと
を含む、項目1から11のいずれか一項に記載の抗体または抗体断片を産生する方法。
(項目16)
薬学的に許容される担体または希釈剤と組み合わせて、項目1から11のいずれか一項に記載の抗体または抗体断片を含む組成物。
(項目17)
免疫細胞を項目1から11に記載の抗体のいずれか1つと接触させるステップを含む、該免疫細胞の活性を増大させる方法。
(項目18)
治療有効量の項目1から11のいずれか一項に記載の抗体または抗体断片を、それを必要とする対象に投与するステップを含む、免疫細胞の活性を増大させる方法。
(項目19)
a.癌の治療のため、
b.感染症もしくは感染性疾患の治療のため、または
c.ワクチンアジュバントとして
使用される、項目18に記載の方法。
(項目20)
前記対象由来の試料においてex vivoでT細胞活性化を測定するステップをさらに含み、T細胞活性における増大が前記治療を継続すべきであることを示すか、または前記治療が成功する可能性を予測する、項目18に記載の方法。
(項目21)
T細胞活性における前記増大が
a.IL−2、TNFα、IL−17、IFNγ、GM−CSF、RANTES、IL−6、IL−8、IL−5およびIL−13からなる群から選択される1つまたは複数のサイトカインのSEB誘発産生を測定するステップ、または
b.IL−2、TNFα、IL−17、IFNγ、GM−CSF、RANTES、IL−6、IL−8、IL−5およびIL−13からなる群から選択されるサイトカインのTT誘発産生を測定するステップ
によって決定される、項目20に記載の方法。
(項目22)
a.免疫細胞活性化を増大させるための、
b.癌を治療するための、または
c.感染症もしくは感染性疾患を治療するための
薬剤の調製のための、項目1から11のいずれか一項に記載の抗体または抗体断片の使用。
(項目23)
診断的使用のための、項目1から11のいずれか一項に記載の抗体または抗体断片の使用。
【0017】
一実施形態において本発明は、配列番号9、10および11のCDR、もしくは任意の該配列の変異体を含む軽鎖、ならびに/または配列番号12、13および14のCDR、もしくは任意の該配列の変異体を含む重鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0018】
他の実施形態において本発明は、配列番号15、16および17のCDR、もしくは任意の該配列の変異体を含む軽鎖、ならびに/または配列番号18、19および20のCDR、もしくは任意の該配列の変異体を含む重鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0019】
一実施形態において本発明は、配列番号5の重鎖可変領域もしくはその変異体、および/または配列番号6もしくはその変異体を含む軽鎖可変領域を含む抗体または抗原結合断片を含む。
【0020】
一実施形態において本発明は、配列番号7の重鎖可変領域もしくはその変異体、および/または配列番号8もしくはその変異体を含む軽鎖可変領域を含む抗体または抗原結合断片を提供する。
【0021】
一実施形態において本発明は、配列番号30のアミノ酸残基20から139もしくはその変異体を含む重鎖可変領域、および/または配列番号32のアミノ酸残基20から130もしくはその変異体を含む軽鎖可変領域を含む抗体または抗原結合断片を含む。
【0022】
一実施形態において本発明は、配列番号30のアミノ酸残基20から139もしくはその変異体を含む重鎖可変領域、および/または配列番号33のアミノ酸残基20から130もしくはその変異体を含む軽鎖可変領域を含む抗体または抗原結合断片を含む。
【0023】
一実施形態において本発明は、配列番号30のアミノ酸残基20から139もしくはその変異体を含む重鎖可変領域、および/または配列番号34のアミノ酸残基20から130もしくはその変異体を含む軽鎖可変領域を含む抗体または抗原結合断片を含む。
【0024】
一実施形態において本発明は、配列番号30のアミノ酸残基20から139に少なくとも90%の相同性を有するアミノ酸配列を含む重鎖可変領域、および/または配列番号32、33または34のアミノ酸残基20から130に少なくとも90%の相同性を有するアミノ酸配列を含む軽鎖可変領域を含む抗体または抗原結合断片を含む。
【0025】
一実施形態において本発明は、配列番号31のアミノ酸残基20から466もしくはその変異体を含む重鎖、および/または配列番号36のアミノ酸残基20から237もしくはその変異体を含む軽鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0026】
一実施形態において本発明は、配列番号31のアミノ酸残基20から466もしくはその変異体を含む重鎖、および/または配列番号37のアミノ酸残基20から237もしくはその変異体を含む軽鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0027】
一実施形態において本発明は、配列番号31のアミノ酸残基20から466もしくはその変異体を含む重鎖、および/または配列番号38のアミノ酸残基20から237もしくはその変異体を含む軽鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0028】
一実施形態において本発明は、配列番号35のアミノ酸残基20から469もしくはその変異体を含む重鎖、および/または配列番号36のアミノ酸残基20から237もしくはその変異体を含む軽鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0029】
一実施形態において本発明は、配列番号35のアミノ酸残基20から469もしくはその変異体を含む重鎖、および/または配列番号37のアミノ酸残基20から237もしくはその変異体を含む軽鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0030】
一実施形態において本発明は、配列番号35のアミノ酸残基20から469もしくはその変異体を含む重鎖、および/または配列番号38のアミノ酸残基20から237もしくはその変異体を含む軽鎖を含み、ヒトPD−1に結合する単離された抗体または抗体断片を提供する。
【0031】
任意の上記実施形態において本発明の抗体または抗体断片の変異体は、1個、2個または3個の保存的に改変されたアミノ酸置換を含みうる。
【0032】
任意の上記実施形態において本発明の抗体または抗体断片は、ヒト重鎖定常領域もしくは20個までの保存的に改変されたアミノ酸置換を含むその変異体、および/またはヒト軽鎖定常領域もしくは20個までの保存的に改変されたアミノ酸置換を含むその変異体、を含みうる。いくつかの実施形態において変異体は、10個までの保存的に改変されたアミノ酸置換を含みうる。いくつかの実施形態において変異体は、5個までの保存的に改変されたアミノ酸置換を含みうる。いくつかの実施形態において変異体は、3個までの保存的に改変されたアミノ酸置換を含みうる。任意の上記実施形態においてヒト重鎖定常領域またはその変異体は、IgG1またはIgG4アイソタイプのものでありうる。
【0033】
任意の上に記載の実施形態において本発明の抗体または抗体断片は、約100pM以下のKDでヒトPD−1に結合できる。他の実施形態において抗体または抗体断片は、約30pM以下のKDでヒトPD−1に結合できる。他の実施形態において抗体または抗体断片は、配列番号31のアミノ酸配列を含む重鎖および配列番号32のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合できる。他の実施形態において抗体または抗体断片は、配列番号31のアミノ酸配列を含む重鎖および配列番号33のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合できる。
【0034】
任意の上に記載の実施形態において本発明の抗体または抗体断片は、約7.5×105 1/M・sまたはそれより速いkassocでヒトPD−1に結合できる。一実施形態において抗体または抗体断片は、約1×106 1/M・sまたはそれより速いkassocでヒトPD−1に結合できる。
【0035】
任意の上に記載の実施形態において抗体または抗体断片は、約2×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合できる。一実施形態において抗体または抗体断片は、約2.7×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合できる。一実施形態において抗体または抗体断片は、約3×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合できる。
【0036】
KD、kassocおよびkdissoc値は、任意の利用可能な方法を使用して測定されうる。好ましい実施形態において解離(dissaociation)定数は、生物光学干渉法(bio−light interferometry)(例えば実施例2で記載されるForteBio Octet法)を使用して測定される。他の好ましい実施形態において解離(disassociation)定数は、表面プラズモン共鳴(例えばBiacore)またはKinexaを使用して測定されうる。
【0037】
さらに任意の上に記載の実施形態において本発明の抗体または抗体断片は、ヒトPD−L1またはヒトPD−L2のヒトPD−1への結合を約1nM以下のIC50で遮断できる。リガンド結合の遮断は、例えば本明細書の(hereub)実施例に記載のFACS法またはFMAT法などの当技術分野において公知の任意の方法を使用して測定することができ、かつIC50を算出されうる。
【0038】
本発明は、ヒトPD−1上の結合エピトープに対して上に記載の任意の抗体と競合し、かつヒトPD−L1またはPD−L2のヒトPD−1への結合を約1nM以下のIC50で遮断する抗体または抗体断片も含む。
【0039】
本発明は、ヒトPD−1上の結合エピトープについて上に記載の任意の抗体と競合し、かつ約100pM以下のKDでヒトPD−1に結合する抗体または抗体断片も含む。一実施形態において抗体または抗体断片は、約30pM以下のKDでヒトPD−1に結合する。
【0040】
本発明は、ヒトPD−1上の結合エピトープについて上に記載の任意の抗体と競合し、かつ配列番号31のアミノ酸配列を含む重鎖および配列番号32のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合する抗体または抗体断片も含む。
【0041】
本発明は、ヒトPD−1上の結合エピトープについて上に記載の任意の抗体と競合し、かつ配列番号31のアミノ酸配列を含む重鎖および配列番号33のアミノ酸配列を含む軽鎖を有する抗体と同程度のKDでヒトPD−1に結合する抗体または抗体断片も含む。
【0042】
本発明は、ヒトPD−1上の結合エピトープについて上に記載の任意の抗体と競合し、かつ約7.5×105 1/M・sまたはそれより速いkassocでヒトPD−1に結合する抗体または抗体断片も含む。一実施形態において抗体または抗体断片は、約1×106 1/M・sまたはそれより速いkassocでヒトPD−1に結合できる。
【0043】
本発明は、ヒトPD−1上の結合エピトープについて上に記載の任意の抗体と競合し、かつ約2×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合する抗体または抗体断片も含む。一実施形態において抗体または抗体断片は、約2.7×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合できる。一実施形態において抗体または抗体断片は、約3×10−5 1/sまたはそれより遅いkdissocでヒトPD−1に結合できる。
【0044】
いくつかの実施形態において本発明の抗体または抗体断片は、キメラ抗体またはキメラ抗体の断片である。
【0045】
いくつかの実施形態において本発明の抗体または抗体断片は、ヒト抗体またはヒト抗体の断片である。
【0046】
いくつかの実施形態において本発明の抗体または抗体断片は、ヒト化抗体またはヒト化抗体の断片である。
【0047】
いくつかの実施形態において本発明の抗体断片は、Fab、Fab’、Fab’−SH、Fv、scFvまたはF(ab’)2抗体断片である。
【0048】
いくつかの実施形態において本発明の抗体断片は、ダイアボディーである。
【0049】
本発明は、ヒトPD−1に結合する上に記載の抗体または抗体断片のいずれか1つを含む二重特異性抗体も含む。
【0050】
いくつかの実施形態において本発明の単離された抗PD−1抗体および抗体断片は、当業者に公知の典型的な手段によって測定されるT細胞活性化(免疫細胞増殖の増大、サイトカイン分泌の増大またはCD25および/もしくはCD69などの活性化マーカーの発現を非限定的に含む)を増大させる。
【0051】
上に記載の任意の実施形態において本発明の抗体または抗体断片は、ex vivoまたはin vivoでのブドウ球菌エンテロトキシンBまたは破傷風トキソイドでの刺激後に免疫応答を増強できる。増大した免疫活性化は、例えば免疫細胞(T細胞など)の増殖または免疫細胞によるサイトカイン産生(例えばT細胞によるINFγもしくはIL−2の産生)を定量することである当業者に公知の方法を使用して決定できる。
【0052】
本発明は、本発明の抗PD−1抗体および抗体断片をコードする核酸も含む。配列番号5から20および30〜38において開示のアミノ酸配列(リーダー配列を含むまたは含まない)のいずれか1つをコードする核酸は、本発明に含まれる。配列番号1から4および21から29を含む核酸(リーダー配列をコードする核酸を含むまたは含まない)も、本発明に含まれる。
【0053】
本発明は、本発明の抗体または抗体断片をコードする核酸を含む細胞および発現ベクターも含む。さらに本発明は、(a)核酸配列が発現される条件下で、培地中で本発明の抗体または抗体断片をコードする核酸を含む宿主細胞を培養し、それにより軽鎖および重鎖可変領域を含むポリペプチドを産生するステップと、(b)宿主細胞または培地からポリペプチドを回収するステップとを含む、本発明の抗体または抗体断片を産生する方法を含む。
【0054】
本発明は、薬学的に許容される担体または希釈剤と組み合わせて、本発明の抗体または抗体断片を含む組成物も含む。
【0055】
本発明は、本発明の抗体または抗体断片の治療有効量をそれを必要とする対象に投与するステップを含む、免疫細胞の活性を増大させる方法も含む。一実施形態においてこの方法は、癌を治療するために使用されうる。他の実施形態においてこの方法は、感染症または感染性疾患を治療するために使用されうる。さらに他の実施形態においてこの方法は、ワクチンアジュバントとして使用されうる。いくつかの実施形態においてこの方法は、第2の治療用薬物または治療様式をさらに投与するステップを含む。
【0056】
いくつかの実施形態において本発明は、免疫細胞の活性を増大させる方法であって、治療有効量の本発明の抗体または抗体断片を、それを必要とする対象に投与するステップを含み、対象由来の試料におけるT細胞活性化をex vivoで測定するステップをさらに含み、T細胞活性における増大が治療を継続すべきであることを示す方法を含む。他の実施形態において本発明は、免疫細胞の活性を増大させる方法であって、治療有効量の本発明の抗体または抗体断片を、それを必要とする対象に投与するステップを含み、対象由来の試料におけるT細胞活性化をex vivoで測定するステップをさらに含み、T細胞活性化における増大が治療が成功する可能性を予測する方法を含む。一実施形態においてT細胞活性における増大は、(i)IL−2、TNFα、IL−17、IFNγ、GM−CSF、RANTES、IL−6、IL−8、IL−5およびIL−13からなる群から選択される1つまたは複数のサイトカインのSEB誘発産生を測定するステップ、または(ii)IL−2、TNFα、IL−17、IFNγ、GM−CSF、RANTES、IL−6、IL−8、IL−5およびIL−13からなる群から選択されるサイトカインのTT誘発産生を測定するステップによって決定される。
【0057】
本発明は、免疫応答を増大させる薬物の調製のための本発明の抗PD−1抗体または抗体断片の使用も含む。
【0058】
本発明は、癌を治療するための薬物の調製のための本発明の抗PD−1抗体または抗体断片の使用も含む。
【0059】
本発明は、ワクチンアジュバントとしての本発明の抗PD−1抗体または抗体断片の使用も含む。
【0060】
本発明は、細菌性毒素または放射性毒素などの治療剤に連結した本発明の抗PD−1抗体または抗体断片を含む免疫複合体も含む。細胞毒作用物質の非限定的な例として、タキソール、サイトカラシンB、マイトマイシン、エトポシドおよびビンクリスチンまたは他の代謝拮抗剤、アルキル化剤、抗生物質および抗有糸分裂剤が挙げられる。
【0061】
本発明は、免疫細胞と本発明の抗体または抗体断片のいずれか1つとを接触させるステップを含む、免疫細胞の活性を増大させるまたは免疫細胞の下方調節を低減する方法も含む。この方法は、癌もしくは感染性疾患(慢性ウイルス性感染症など)を治療するために使用されることができ、または予防用もしくは治療用ワクチンのためのアジュバントとして使用されうる。
【0062】
本発明は、免疫細胞を抗原および抗PD−1抗体または抗体断片と、抗原への免疫応答が増大されるかまたは増強されるように接触させるステップを含む抗原への免疫応答を増大させる方法も含む。この方法は、in vivo(対象中で)またはex vivoで実施されうる。
【0063】
いくつかの実施形態において抗PD−1抗体または抗体断片は、第2の治療薬または治療様式と組み合わされうる。一実施形態において抗PD−1抗体または抗体断片は、組換えサイトカインまたは分泌された免疫性因子の適用を伴う癌治療と組み合わされうる。組合せの非限定的な例として、抗PD−1抗体と組換えIL−2または組換えIFNα2とを黒色腫または腎細胞癌の治療のために組み合わせることが挙げられる。組換えIL−2は、癌患者においてT細胞の増殖を増強する。組換えIFNα2は、癌細胞の増殖を抑制するが、治療を受ける患者において癌細胞、抗原提示細胞および他の体細胞上のPD−1に対する抑制型リガンドの発現も増大させる。抗PD−1は、癌または感染性疾患の治療のために有用であると考えられうる他のサイトカインと組み合わされうる。
【0064】
いくつかの実施形態において抗PD−1抗体または抗体断片は、癌または感染性疾患を予防または治療するためにワクチンと組み合わされうる。非限定的な例として抗PD−1は、治療される癌または感染症に関連する1つもしくは複数の抗原を含有するタンパク質、ペプチドもしくはDNAのワクチンと、またはそのような抗原でパルスした樹状細胞を含むワクチンと組み合わされうる。他の実施形態は、(弱毒化)癌細胞ワクチンまたはまるごとのウイルスワクチンを伴う抗PD−1の使用を含む。一実施形態は、GM−CSFを分泌するように工学的に操作されたまるごとの細胞の癌ワクチンと抗PD−1療法との組合せを含む。
【0065】
いくつかの実施形態において抗PD−1抗体または抗体断片は、癌または感染性疾患のケアの標準法であると考えられる治療と組み合わされうる。そのような組合せについての論理的根拠は、抗PD−1によって同時に増大する免疫活性化がケア治療の標準法への初期の臨床反応を誘発または促進し、持続的臨床反応および疾患の長期免疫管理を誘導することである。
【0066】
一実施形態において抗PD−1抗体または抗体断片での治療は、化学療法と組み合わされうる。細胞傷害性薬物を使用する化学療法は、癌細胞死を生じ、それにより腫瘍抗原の放出が増大する。腫瘍抗原の可用性のそのような増大は、抗PD−1治療との相乗作用を生じうる。非限定的な例は、黒色腫の治療のためのデカルバジンもしくはテモゾロミドまたは膵臓癌のためのゲムシタビンの使用によって提供される。
【0067】
一実施形態において抗PD−1抗体または抗体断片での治療は、放射線療法と組み合わされうる。放射線療法は、癌細胞死を生じ、免疫細胞の提示および活性化のための腫瘍抗原の可用性が増大する。
【0068】
別の実施形態において抗PD−1抗体または抗体断片での治療は、対象から癌細胞を除く手術と組み合わされうる。
【0069】
他の実施形態において抗PD−L1抗体または抗体断片は、ホルモン剥奪もしくは血管新生の抑制のために使用される標的薬剤、または腫瘍細胞において活性な標的タンパク質を含み、PD−1遮断と相乗効果を生じうる療法と組み合わされることができ、全ては、腫瘍細胞死および免疫刺激腫瘍抗原の可用性の増強を生じる。抗PD−1抗体または抗体断片との組合せにおいて、増大したT細胞活性化は、癌の持続的な免疫管理を生じうる。
【0070】
いくつかの実施形態において抗PD−1抗体または抗体断片は、癌または感染性疾患の治療のために有用である他の治療用抗体と組み合わされうる。非限定的な例は、抗PD−1抗体とHer2/neuを標的とするまたはEGF受容体を標的とする抗体との組合せによって提供される。他の非限定的な例において抗PD−1抗体または抗体断片は、VEGFまたはその受容体を標的とする治療と組み合わされる。他の実施形態において抗PD−1抗体または抗体断片は、抗CTLA−4と組み合わされる。さらに他の非限定的な例において抗PD−1は、RSVを標的とする抗体と組み合わされる。
【図面の簡単な説明】
【0071】
【図1】ハイブリドーマ上清由来の固定化抗体は、固定化した抗CD3および可溶性抗CD28で刺激したジャーカットE6.2.11細胞によるIL−2分泌を低減できることを示す実験結果を示すグラフである。
【図2】ヒトPD−1に対する抗体がPD−1に結合することを示す実験結果を示すグラフである。図2Aは、タンパク質ELISAにおける、精製PD−1/Fcへの抗PD−1抗体の用量依存的結合を示すグラフである。図2Bは、CELISAにおける、hPD−1を形質移入したCHO細胞の表面で発現されるPD−1への抗PD−1抗体の用量依存的結合を示すグラフである。
【図3】PD−1に対する抗体がヒトPD−1を形質移入したCHO細胞へのPD−L1およびPD−L2の結合について競合することを示すFMAT実験の結果を示すグラフである。図3Aは、hPD−1.08AおよびhPD−1.09ならびに少ない程度でのJ116によるPD−L1の結合の用量依存的な抑制を示すグラフである。図3Bは、PD−L2の用量依存的な抑制を示すグラフである。
【図4】健常ドナー血液細胞によるSEB刺激IL−2産生は、抗PD−1、抗PD−L1または抗CTLA−4抗体の存在下で増強されることを示す実験結果を示す棒グラフである。棒は、ドナー間のIL−2における平均倍数増加(±SEM)を示す。各棒内の数値は、示したドナー数を示す。マウス(m)IgG1は、抗PD−1.08A(08A)、抗PD−1.09A(09A)および抗PD−L1についてのアイソタイプ対照である。マウス(m)IgG2は、抗CTLA−4についてのアイソタイプ対照である。各IL−2値を倍数変化を決定するためにそれ自体の対照と比較する(IL−2の倍数変化4は、SEB単独の場合と比較した場合のIL−2産生における400%増大を意味する)。なし=SEB単独
【図5】リコール抗原破傷風トキソイドで刺激された場合に抗PD−1抗体がT細胞増殖およびサイトカイン分泌(IL−2およびIFNγ)を、促進することを示す実験結果を示すグラフである。図5は、濃度依存的IFNγ分泌を示す。
【図6】生物光学干渉法によって測定した抗PD−1抗体についてのkassocおよびkdissoc速度を示すグラフである。斜線は、理論的に算出されるKD値を示す。抗体は、右側にKDによる昇順で列挙される。
【図7】健常ドナー血液細胞によるSEB刺激IL−2産生は、25μg/mlマウス(09A)、またはヒト化抗PD−1抗体(h409A11、h409A16およびh409A17)の存在下で増大されることを示す実験結果を示す棒グラフである。棒は、ドナー3名の間のIL−2における平均倍数増加(±SEM)を示す。マウス(m)IgG1は、抗PD−1.09A(09A)についてのアイソタイプ対照である。ヒト(h)IgG4は、h409A11、h409A16およびh409A17抗体についてのアイソタイプ対照である。各IL−2値を倍数変化を決定するためにそれ自体の対照と比較する。なし=SEB単独
【発明を実施するための形態】
【0072】
(発明の詳細な説明)
略語および定義
本発明の詳細な記載および実施例を通じて以下の略語が使用される。
【0073】
hPD−1.08A マウスモノクローナル抗hPD−1抗体
hPD−1.09A マウスモノクローナル抗hPD−1抗体
08A−VH hPD−1.08Aハイブリドーマから単離されたVH
08A−VK hPD−1.08Aハイブリドーマから単離されたVK
09A−VH hPD−1.09Aハイブリドーマから単離されたVH
09A−VK hPD−1.09Aハイブリドーマから単離されたVK
c109A hPD−1.09A抗体のキメラIgG1バージョン
c109A−VH hIgG1定常領域に融合しているマウス09A−VHからなるキメラ重鎖
c109A−VK ヒトκ定常領域に融合しているマウス09A−VKからなるキメラ軽鎖
109A−H 復帰突然変異を含まないヒト化IgG1 09A重鎖配列
409A−H FWR復帰突然変異を含まないヒト化IgG4−09A重鎖配列
K09A−L−11 長さ11AAのCDR1を独自に有するフレームワークを含むヒト化09A−κ配列
K09A−L−16 長さ16AAのCDR1を独自に有するフレームワークを含むヒト化09A−κ配列
K09A−L−17 長さ17AAのCDR1を独自に有するフレームワークを含むヒト化09A−κ配列
h409A11 409A−Hの配列を含む重鎖およびK09A−L−11の配列を含む軽鎖を含む09A抗体のヒト化IgG4バージョン
h409A16 409A−Hの配列を含む重鎖およびK09A−L−16の配列を含む軽鎖を含む09A抗体のヒト化IgG4バージョン
h409A17 409A−Hの配列を含む重鎖およびK09A−L−17の配列を含む軽鎖を含む09A抗体のヒト化IgG4バージョン
hPD−1 ヒトPD−1タンパク質
CDR 免疫グロブリン可変領域中の相補性決定領域、Kabat番号付け方式を使用して定義される
EC50 50%有効性または結合を生じる濃度
ELISA 酵素結合免疫吸着検定法
FW 抗体フレームワーク領域、CDR領域を除く免疫グロブリン可変領域
HRP 西洋わさびペルオキシダーゼ
IL−2 インターロイキン2
IFN インターフェロン
IC50 50%抑制を生じる濃度
IgG 免疫グロブリンG
Kabat Elvin A Kabatによって開発された免疫グロブリン配列比較(alignment)および番号付け方式
mAb モノクローナル抗体
MES 2−(N−モルホリノ)エタンスルホン酸
NHS 正常ヒト血清
PCR ポリメラーゼ連鎖反応
SAM ヒツジ抗マウス(IgG)ポリクローナル抗体
V領域 異なる抗体間の配列において可変であるIgG鎖のセグメント。軽鎖におけるKabat残基109および重鎖における113に及ぶ。
【0074】
VH 免疫グロブリン重鎖可変領域
VK 免疫グロブリンκ軽鎖可変領域。
【0075】
「抗体」は、リガンドのその受容体への結合の抑制または受容体のリガンド誘発シグナル伝達の抑制によるなどの所望の生物学的活性を示す任意の形態の抗体を指す。したがって「抗体」は、最も広義で使用され、具体的には非限定的にモノクローナル抗体(全長モノクローナル抗体含む)、ポリクローナル抗体および多重特異性抗体(例えば二重特異性抗体)に及ぶ。
【0076】
「抗体断片」および「抗体結合断片」は、典型的には親抗体の抗原結合領域または可変領域(例えば1つまたは複数のCDR)の少なくとも一部を含む抗原結合断片および抗体の類似物を意味する。抗体断片は、親抗体の結合特異性の少なくとも一部を保持する。典型的には抗体断片は、活性をモルベースで表す場合に親の結合活性の少なくとも10%の結合活性を保持する。好ましくは抗体断片は、標的に対して親抗体の結合親和性の少なくとも20%、50%、70%、80%、90%、95%もしくは100%またはそれを超える結合親和性を保持する。抗体断片の例として、それだけに限らないがFab、Fab’、F(ab’)2およびFv断片、ダイアボディー、直鎖抗体、例えばsc−Fv、ユニボディー(Genmab由来の技術)である単鎖抗体分子、ナノボディー(Domantis由来の技術)、ドメイン抗体(Ablynx由来の技術)、ならびに抗体断片から形成された多重特異性抗体が挙げられる。工学的に操作された抗体変異体は、Holliger and Hudson(2005年)Nat.Biotechnol.23巻:1126〜1136頁において概説されている。
【0077】
「Fab断片」は、軽鎖1つならびに1つの重鎖のCH1および可変領域で構成されている。Fab分子の重鎖は、他の重鎖分子とジスルフィド結合を形成できない。
【0078】
「Fc領域」は、抗体のCH1およびCH2ドメインを含む2つの重鎖断片を含有する。2つの重鎖断片は、2つ以上のジスルフィド結合およびCH3ドメインの疎水性相互作用によって維持している。
【0079】
「Fab’断片」は、軽鎖1つさらに、VHドメインおよびCH1ドメインならびにCH1ドメインとCH2ドメインとの間の領域も、F(ab’)2分子を形成するために2つのFab’断片の2つの重鎖間に鎖間ジスルフィド結合が形成されうるように含有する1つの重鎖の一部を含有する。
【0080】
「F(ab’)2断片」は、軽鎖2つ、およびCH1ドメインとCH2ドメインとの間の定常領域の一部を含有する重鎖2つを、鎖間ジスルフィド結合が2つの重鎖の間に形成されるように含有する。したがってF(ab’)2断片は、2つの重鎖間のジスルフィド結合によって維持される2つのFab’断片から構成される。
【0081】
「Fv領域」は、重鎖および軽鎖の両方由来の可変領域を含むが、定常領域を欠損している。
【0082】
「単鎖Fv抗体」(または「scFv抗体」)は、抗体のVHおよびVLドメインを含む抗体断片を指し、これらのドメインは、1本のポリペプチド鎖に存在する。一般にFvポリペプチドは、scFvが抗原結合のための所望の構造を形成できるようにするVHドメインとVLドメインの間のポリペプチドリンカーをさらに含む。scFvについての概説として、Pluckthun (1994年)THE PHARMACOLOGY OF MONOCLONAL ANTIBODIES、113巻、Rosenburg and Moore eds.Springer−Verlag、New York、269〜315頁を参照されたい。国際特許出願公開WO88/01649ならびに米国特許第4,946,778号および第5,260,203号も参照されたい。
【0083】
「ダイアボディー」は、2個の抗原結合部位を有する小さな抗体断片である。断片は、同じポリペプチド鎖中で軽鎖可変ドメイン(VL)に連結した重鎖可変ドメイン(VH)を含む(VH−VLまたはVL−VH)。同じ鎖上の2つのドメイン間で対合させるには短すぎるリンカーを使用することによって、ドメインは他の鎖の相補性ドメインと対合させられ、2つの抗原結合部位を作り出す。ダイアボディーは、例えばEP404,097、WO93/11161およびHolligerら、(1993年)Proc.Natl.Acad.Sci.USA 90巻:6444〜6448頁においてさらに十分に記載されている。
【0084】
「ドメイン抗体断片」は、重鎖の可変領域または軽鎖の可変領域だけを含有する免疫学的に機能性の免疫グロブリン断片である。場合により2つ以上のVH領域が二価ドメイン抗体断片を作り出すようにペプチドリンカーに共有結合的に結合する。二価ドメイン抗体断片の2つのVH領域は、同じまたは異なる抗原を標的にできる。
【0085】
本発明の抗体断片は、例えば通常重鎖間ジスルフィド結合に関与するヒンジシステインの少なくとも1つが本明細書に記載のとおりに変更されている場合に、ジスルフィド結合能力が低下している重鎖の二量体化(または多量体化)を可能にする定常領域の十分な部分を含みうる。他の実施形態において抗体断片、例えばFc領域を含むものは、FcRn結合、抗体半減期調節、ADCC機能および/または補体結合など(例えば抗体がADCC機能または補体結合に必要なグリコシル化特性を有する場合)の、無傷の抗体中に存在する場合にFc領域に通常付随する少なくとも1つの生物学的機能を保持する。
【0086】
用語「キメラ」抗体は、重鎖および/または軽鎖の一部が特定の種に由来するかまたは特定の抗体のクラスもしくはサブクラスに属する抗体の対応する配列と同一であるかまたは相同である一方で、(1つまたは複数の)鎖の残部が他の種に由来するかまたは他の抗体のクラスもしくはサブクラスに属する抗体の対応する配列と同一であるかまたは相同である、ならびに所望の生物学的活性を示す範囲でそのような抗体の断片である抗体を指す(例えば米国特許第4,816,567号およびMorrisonら、1984年、Proc.Natl.Acad.Sci.USA 81巻:6851〜6855頁を参照されたい)。
【0087】
非ヒト(例えばマウス)抗体の「ヒト化」形態は、非ヒト免疫グロブリン由来の最少配列を含有するキメラ抗体である。大部分についてヒト化抗体は、レシピエントの超可変領域由来の残基が、所望の特異性、親和性および適応力を有するマウス、ラットまたはヒトではない霊長類などのヒトではない種(ドナー抗体)の超可変領域由来の残基によって置き換えられたヒト免疫グロブリン(レシピエント抗体)である。場合により、ヒト免疫グロブリンのFvフレームワーク領域(FR)残基は、対応する非ヒト残基によって置き換えられる。さらにヒト化抗体は、レシピエント抗体またはドナー抗体において見いだされない残基を含みうる。これらの改変は、抗体の性能をさらに改良するために行われる。一般にヒト化抗体は、超可変ループの全てまたは実質的に全てが非ヒト免疫グロブリンの超可変ループに対応し、およびFR領域の全てまたは実質的に全てがヒト免疫グロブリン配列のものである、少なくとも1つ、および典型的には2つの可変ドメインの実質的に全てを含む。ヒト化抗体は、任意選択で免疫グロブリン定常領域(Fc)の少なくとも一部、典型的にはヒト免疫グロブリンのそれも含む。さらなる詳細については、Jonesら、Nature 321巻:522〜525頁(1986年);Riechmannら、Nature332巻:323〜329頁(1988年)およびPresta、Curr.Op.Struct.Biol.2巻:593〜596頁(1992年)を参照されたい。
【0088】
本明細書において使用される用語「超可変領域」は、抗原結合に関与する抗体のアミノ酸残基を指す。超可変領域は、配列比較によって定義される「相補性決定領域」または「CDR」由来のアミノ酸残基、例えば軽鎖可変ドメイン中の24〜34(L1)、50〜56(L2)および89〜97(L3)残基、ならびに重鎖可変ドメイン中の31〜35(H1)、50〜65(H2)および95〜102(H3)残基(Kabatら、1991年、Sequences of proteins of Immunological Interest、5th Ed.Public Health Service、National Institutes of Health、Bethesda、Md.を参照されたい)、さらに/または構造的に定義される「超可変ループ」(HVL)例えば軽鎖可変ドメイン中の26〜32(L1)、50〜52(L2)および91〜96(L3)残基、ならびに重鎖可変ドメイン中の26〜32(H1)、53〜55(H2)および96〜101(H3)残基由来の残基(Chothia and Leskl、1987年、J.Mol.Biol.196巻:901〜917頁を参照されたい)を含む。「フレームワーク」または「FR」残基は、本明細書において定義する超可変領域残基以外の可変ドメイン残基である。
【0089】
「ヒト抗体」は、ヒトによって産生される、かつ/または本明細書において開示するヒト抗体を作製するための技術のいずれかを使用して作られている抗体のアミノ酸配列に対応するアミノ酸配列を有する抗体である。この定義は、具体的には非ヒト抗原結合残基を含むヒト化抗体を排除する。
【0090】
「単離された」抗体は、その天然の環境の成分から同定され、分離され、かつ/または回収されたものである。その天然の環境の混入成分は、抗体の診断または治療用使用を妨げる物質であり、酵素、ホルモンおよび他のタンパク質性または非タンパク質性の溶質を含みうる。いくつかの実施形態において抗体は、(1)ローリー法によって決定されるように抗体で95重量%を超えて、および最も好ましくは99重量%を超えるまで、(2)スピニングカップ配列決定装置の使用により、N末端の少なくとも15残基もしくは内部アミノ酸配列を得るために十分な程度まで、または(3)クーマシーブルー、もしくは好ましくは銀染色を使用する還元もしくは非還元条件下でのSDS−PAGEにより均一になるまで、精製される。単離された抗体は、抗体の天然の環境の少なくとも1つの成分が存在しないことになることから、組換え細胞内でインサイチュの抗体を含む。しかし通常は、単離された抗体は、少なくとも1つの精製ステップによって調製される。
【0091】
「単離された」核酸分子は、抗体核酸の天然の供給源においてそれが通常関連している少なくとも1つの混入核酸分子から同定および分離される核酸分子である。単離された核酸分子は、それが天然で見いだされる形態もしくは状態にある以外のものである。したがって単離された核酸分子は、天然の細胞内に存在する場合の核酸分子とは区別される。しかし単離された核酸分子は、例えば核酸分子が天然の細胞においてとは異なる染色体中の位置にある場合に、通常抗体を発現する細胞内に含まれる核酸分子を含む。
【0092】
本明細書において使用する用語「モノクローナル抗体」は、実質的に同質の抗体の集団から得た抗体を指す、すなわち集団を構成する個々の抗体は、少量で存在しうる天然に生じる可能性のある変異を除いて同一である。モノクローナル抗体は、高度に特異的であり、単一の抗原部位に対して誘導されている。さらに、典型的には異なる決定基(エピトープ)に対して誘導された異なる抗体を含む従来の(ポリクローナル)抗体調製物とは対照的に、各モノクローナル抗体は抗原上の単一の決定基に対して誘導されている。修飾語「モノクローナル」は、実質的に同質な抗体の集団から得られる抗体の特性を示し、特定の方法による抗体産生を必要とするとは解釈されない。例えば本発明により使用されるモノクローナル抗体は、Kohlerら、1975年、Nature 256巻:495頁によって最初に記載されたハイブリドーマ法によって作製されてもよく、または組換えDNA法(例えば米国特許第4,816,567号を参照されたい)によって作製されてもよい。「モノクローナル抗体」は、例えばClacksonら、1991年、Nature352巻:624〜628頁およびMarksら、1991年、J.Mol Biol.222巻:581〜597頁に記載された技術を使用するファージ抗体ライブラリーから単離されてもよい。本明細書におけるモノクローナル抗体は、詳細には「キメラ」抗体を含む。
【0093】
本明細書において使用する用語「免疫細胞」は、造血系由来であり、かつ免疫応答において役割を演じる細胞を含む。免疫細胞として、B細胞およびT細胞などのリンパ球;ナチュラルキラー細胞;単球、マクロファージ、好酸球、マスト細胞、好塩基球および顆粒球などの骨髄系細胞が挙げられる。
【0094】
本明細書において使用する「免疫複合体」は、細菌性毒素、細胞毒性薬物または放射性毒素などの治療用成分に結合した抗PD−1抗体またはその断片を指す。毒性成分は、当技術分野において利用可能な方法を使用して本発明の抗体に結合されうる。
【0095】
以下の核酸多義性記号は、本明細書において使用される、R=AまたはG、Y=CまたはT、M=AまたはC、K=GまたはT、S=GまたはC、およびW=AまたはT。
【0096】
本明細書において使用する配列「変異体」は、本開示の配列とは1つまたは複数のアミノ酸残基で異なるが、得られる分子の生物学的活性を保持している配列を指す。
【0097】
「保存的に改変された変異体」または「保存的アミノ酸置換」は、当業者に公知のアミノ酸の置換を指し、得られる分子の生物学的活性を変えることなく一般に作製されうる。当業者は、一般にポリペプチドの非必須領域における単一のアミノ酸の置換が生物学的活性を実質的に変えないことを認識する(例えばWatsonら、Molecular Biology of the Gene、The Benjamin/Cummings Pub.Co.、224頁(4th Edition 1987年)を参照されたい)。そのような例示的な置換は、好ましくは以下に列挙するものに従って行われる:
【0098】
【数1】
本明細書において使用する、2つの配列間で「%同一性」は、配列が共有する同一の位置の数の関数を指し(すなわち、%相同=同一の位置の数/位置の総数×100)、2つの配列の最適な配列比較ために導入される必要があるギャップの数および各ギャップの長さを考慮する。配列の比較および2つの配列間の同一性百分率の決定は、数学的アルゴリズムを使用して達成されうる。例えば2つのアミノ酸配列間の同一性百分率は、ALIGNプログラム(バージョン2.0)に組み入れられているE.MeyersおよびW.Miller(Comput.Appl.Biosci.、4巻:11〜17頁(1988年))のアルゴリズムを使用して、PAM120重み残基表、ギャップ長ペナルティー12、およびギャップペナルティー4を使用して決定されうる。加えて2つのアミノ酸配列間の同一性百分率は、GCGソフトウェアパッケージ(www.gcg.comで入手可能)中のGAPプログラムに組み入れられているNeedlemanおよびWunsch(J.Mol Biol.48巻:444〜453頁(1970年))アルゴリズムを使用して、Blossum62マトリックスまたはPAM250マトリックスのいずれか、ならびにギャップ重み16、14、12、10、8、6または4および長さ重み1、2、3、4、5または6を使用して決定されうる。
【0099】
本明細書において使用する用語「約」は、当業者によって決定される特定の値に対する許容可能な誤差範囲内の値を指し、その値がどのように測定または決定されたかに、すなわち測定系の限界にある程度依存する。例えば「約」は、当技術分野において一実施あたり1以内の標準偏差または1を超える標準偏差を意味できる。代替えとして「約」または「を本質的に含む」は、20%までの範囲を意味できる。さらに具体的に生物学的システムまたはプロセスに関して、用語は値の一桁まで、または5倍までを意味できる。本出願書類および本特許請求の範囲において具体的な値が提供される場合、他に明記しない限り、「約」または「を本質的に含む」の意味は、具体的な値について許容可能な誤差範囲内であると推定されるべきである。
【0100】
リガンド/受容体、抗体/抗原、または他の結合対に関する場合「特異的に」結合するは、タンパク質および/または他の生物体の不均質な集団におけるタンパク質、例えばPD−1、の存在の決定要因である結合反応を示す。したがって、指定の条件下では、特定のリガンド/抗原は特定の受容体/抗体に結合し、試料中に存在する他のタンパク質には顕著な量では結合しない。
【0101】
動物、ヒト、実験対象、細胞、組織、器官または生体液に適用される場合に「投与」および「処置」は、外因性の薬学的、治療用、診断用の薬剤または組成物の動物、ヒト、対象、細胞、組織、器官または生体液への接触を指す。「投与」および「処置」は、例えば治療用、薬物動態学的、診断用、研究用および実験用の方法を指すことができる。細胞の処理は、試薬の細胞への接触、および液が細胞に接触している場合に試薬の液への接触を包含する。「投与」および「処置」は、例えば細胞の、試薬による、診断用、結合組成物による、または他の細胞によるin vitroおよびex vivoの処置、も意味する。
【0102】
「有効量」は、医学的状態の症状または兆候を改善または予防するために十分な量を包含する。有効量は、診断を可能にするまたは促進するために十分な量も意味する。具体的な対象についての有効量は、治療される状態、患者の健康全般、投与の方法経路および用量、ならびに副作用(side affect)の重症度などの要因に依存して変動できる。有効量は、顕著な副作用または毒性作用を避ける最大用量または投薬手順でありうる。効果は、診断的指標またはパラメーターの改善を少なくとも5%、通常は少なくとも10%、より通常には少なくとも20%、最も通常は少なくとも30%、好ましくは少なくとも40%、より好ましくは少なくとも50%、最も好ましくは少なくとも60%、理想的には少なくとも70%、より理想的には少なくとも80%、および最も理想的には少なくとも90%生じ、100%は正常な対象によって示される診断パラメーターと定義される(例えば、Maynardら(1996年)A Handbook of SOPs for Good Clinical Practice、Interpharm Press、Boca Raton、FL;Dent(2001年)Good Laboratory and Good Clinical Practice、Urch Publ.、London、UKを参照されたい)。
【0103】
モノクローナル抗体
PD−1に対するモノクローナル抗体は、被験体にPD−1抗原を注射し、次いで所望の配列または機能特性を有する抗体を発現しているハイブリドーマを単離する当技術分野における知識および技術によって作製できる。
【0104】
モノクローナル抗体をコードするDNAは、従来の手段を使用して(例えばモノクローナル抗体の重鎖および軽鎖をコードする遺伝子に特異的に結合できるオリゴヌクレオチドプローブを使用することによって)容易に単離され、かつ配列決定される。ハイブリドーマは、そのようなDNAの好ましい供給源として役立つ。単離されるとDNAは、発現ベクターに入ることができ、次いで組換え宿主細胞におけるモノクローナル抗体の合成を得るために、他に免疫グロブリンタンパク質を産生しないE.coli細胞、サルCOS細胞、チャイニーズハムスター卵巣(CHO)細胞または骨髄腫細胞などの宿主細胞に形質移入される。抗体の組換え産生は、より詳細に以下に記載される。
【0105】
さらなる実施形態において抗体または抗体断片は、McCaffertyら、1990年、Nature、348巻:552〜554頁に記載されている技術を使用して生成される抗体ファージライブラリーから単離されうる。Clacksonら、1991年、Nature、352巻:624〜628頁およびMarksら、1991年、J.Mol.Biol.222巻:581〜597頁は、ファージライブラリーを使用するマウスおよびヒトの抗体の単離をそれぞれ記載している。次の刊行物は、鎖シャッフリングによる高親和性(nM範囲)ヒト抗体の産生(Marksら、1992年、Bio/Technology、10巻:779〜783頁)、ならび非常に大きなファージライブラリーを構築するための戦略としてのコンビナトリアル感染およびin vivo組換えを記載している(Waterhouseら、1993年、Nuc.Acids.Res.21巻:2265〜2266頁)。したがってこれらの技術は、モノクローナル抗体の単離のための伝統的なモノクローナル抗体ハイブリドーマ技術の実行可能な代替法である。
【0106】
キメラ抗体
抗体DNAは、例えばヒト重鎖および軽鎖の定常ドメインのコード配列を相同なマウス配列の代わりに置換することによって(米国特許第4,816,567号;Morrisonら、1984年、Proc.Natl Acad.Sci.USA、81巻:6851頁)、または免疫グロブリンではない物質(例えばタンパク質ドメイン)のコード配列の全てまたは一部を免疫グロブリンコード配列に共有結合させることによっても改変されうる。典型的には、そのような免疫グロブリンではない物質は、抗体の定常ドメインの代わりをするか、または、抗原に対する特異性を有する1つの抗原結合部位および異なる抗原に対する特異性を有する他の抗原結合部位とを含むキメラ二価抗体を作るために抗体の1つの抗原結合部位の可変ドメインの代わりをする。
【0107】
ヒト化抗体およびヒト抗体
ヒト化抗体は、ヒトではない供給源由来の1つまたは複数のアミノ酸残基を有する。非ヒトアミノ酸残基は、しばしば「移入」残基と称され、典型的には「移入」可変ドメインから取り込まれる。ヒト化は、一般にWinterおよび協力者らの方法(Jonesら、1986年、Nature321巻:522〜525頁;Riechmannら、1988年、Nature、332巻:323〜327頁;Verhoeyenら、1988年、Science 239巻:1534〜1536頁)に従って、げっ歯類CDRまたはCDR配列をヒト抗体の対応する配列の代わりに置換することによって実施されうる。したがってそのような「ヒト化」抗体は、無傷のより実質的に少ないヒト可変ドメインがヒトではない種由来の対応する配列によって置換されているキメラ抗体(米国特許第4,816,567号)である。実際にヒト化抗体は、典型的には、いくつかのCDR残基および場合によりいくつかのFR残基がヒトではない、例えばげっ歯類の抗体中の類似部位由来の残基によって置換されているヒト抗体である。
【0108】
ヒト化抗体の作製において使用される軽鎖および重鎖の両方のヒト可変ドメインの選択は、抗原性を低減するために非常に重要である。いわゆる「最適な」方法により、げっ歯類抗体の可変ドメインの配列は、既知のヒト可変ドメイン配列の完全なライブラリーに対して選別される。その結果げっ歯類の配列に最も近いヒト配列が、ヒト化抗体用のヒトフレームワーク(FR)として受けいれられる(Simsら、1987年、J.Immunol.151巻:2296頁;Chothiaら、1987年、J.Mol.Biol.196巻:901頁)。他の方法は、軽鎖および重鎖の特定のサブグループの全てのヒト抗体の共通配列由来の特定のフレームワークを使用する。同じフレームワークがいくつかの異なるヒト化抗体用に使用されうる(Carterら、1992年、Proc.Natl.Acad.Sci.USA 89巻:4285頁;Prestaら、1993、 J.Immnol.151巻:2623頁)。
【0109】
抗体が抗原に対する高い親和性および他の有益な生物学的特性を保持してヒト化されることは、さらに重要である。この目標を達成するために、好ましい方法によりヒト化抗体は、親配列およびヒト化配列の3次元モデルを使用する親配列および種々の概念的なヒト化産物の分析の工程によって調製される。3次元免疫グロブリンモデルは、一般的に入手可能であり、当業者によく知られている。選択された候補免疫グロブリン配列の有望な3次元立体配置構造を例示および表示するコンピュータープログラムは、利用可能である。これらの表示の検査は、候補免疫グロブリン配列の機能における残基の可能性のある役割の分析、すなわち候補免疫グロブリンがその抗原に結合する能力に影響する残基の分析、を可能にする。このようにFR残基は、(1つまたは複数の)標的抗原に対する親和性の増大などの所望の抗体の特性が達成されるように、レシピエント配列および移入配列から選択および組み合わせられることができる。一般にCDR残基は、直接かつ最も実質的に抗原結合に影響を及ぼすことに関与する。
【0110】
抗体のヒト化は、直接的なタンパク質工学的作業である。ほとんど全てのマウス抗体は、CDR移植によってヒト化されることができ、抗原結合は保持される。Lo, Benny, K.C.、editor、in Antibody Engineering:Methods and Protocols、248巻、Humana Press、New Jersey、2004年を参照されたい。
【0111】
代替えとして、免疫化において、内在性の免疫グロブリンを産生することなくヒト抗体の完全なレパートリーを産生できるトランスジェニック動物(例えばマウス)を産生することがいまや可能である。例えば、キメラおよび生殖系列変異体マウスでの抗体重鎖連結領域(JH)遺伝子のホモ欠損は、内在性の抗体産生の完全な抑制を生じることが記載されている。ヒト生殖系列免疫グロブリン遺伝子アレイの、そのような生殖系列変異マウスへの移入は、抗原曝露でのヒト抗体の産生を生じる。例えばJakobovitsら、1993年、Proc.Natl.Acad.Sci.USA 90巻:2551頁;Jakobovitsら、1993年、Nature 362巻:255〜258頁;Bruggermannら、1993年、Year in Immunology 7巻:33頁およびDuchosalら、1992年、Nature 355巻:258頁を参照されたい。ヒト抗体は、ファージディスプレイライブラリーにも由来できる(Hoogenboomら、1991年J.Mol.Biol.227巻:381頁;Marksら、J.Mol.Biol.1991年、222巻:581〜597頁;Vaughanら、1996年、Nature Biotech 14巻:309頁)。
【0112】
抗体精製
組換え技術を使用する場合、抗体は細胞内、周辺質間隙に産生されるか、または培地に直接分泌されうる。抗体が細胞内に産生される場合、最初の段階として宿主細胞または溶解断片のいずれかの粒子細片は、例えば遠心分離または限外濾過によって除去される。Carterら、1992年、Bio/Technology 10巻:163〜167頁は、E.coliの周辺質間隙に分泌される抗体の単離のための手順を記載している。簡潔には、細胞ペーストを酢酸ナトリウム(pH3.5)、EDTAおよびフッ化フェニルメチルスルホニル(PMSF)の存在下で約30分間かけて融解する。細胞細片は、遠心分離によって除去されうる。抗体が培地に分泌される場合は、そのような発現系由来の上清を一般的には最初に、市販で入手可能なタンパク質濃縮用フィルター、例えばAmiconまたはMillipore,Pellicon限外ろ過ユニットを使用して濃縮する。PMSFなどのプロテアーゼ阻害剤は、任意の前述のステップにタンパク質分解を阻害するために含まれてよく、抗生物質は外来性の混入物の増殖を防ぐために含まれてよい。
【0113】
細胞から調製された抗体組成物は、例えば、ヒドロキシアパタイトクロマトグラフィー、ゲル電気泳動、透析およびアフィニティークロマトグラフィーを使用して、好ましい精製技術であるアフィニティークロマトグラフィーを用いて精製されうる。プロテインAの親和性リガンドとしての適合性は、種および抗体中に存在する任意の免疫グロブリンFc領域のアイソタイプに依存する。プロテインAは、ヒトγ1、γ2またはγ4重鎖に基づく抗体を精製するために使用されうる(Lindmarkら、1983年、J.Immunol.Meth.62巻:1〜13頁)。プロテインGは、全てのマウスアイソタイプおよびヒトγ3に対して推奨される(Gussら、1986年、EMBO J 5巻:15671575頁)。親和性リガンドが結合するマトリックスは、最も頻繁にはアガロースであるが、他のマトリックスも利用可能である。細孔制御されたガラスまたはポリ(スチレンジビニル)ベンゼンなどの機械的に安定なマトリックスは、アガロースで達成されうるよりもより早い流速およびより短い処理時間を可能にする。抗体がCH3ドメインを含む場合は、Bakerbond ABX(商標)レジン(J.T.Baker、Phillipsburg、N.J.)が、精製のために有用である。イオン交換カラムでの分画、エタノール沈殿、逆相HPLC、シリカ上でのクロマトグラフィー、ヘパリンSEPHAROSETM上でのクロマトグラフィー、陰イオンまたは陽イオン交換樹脂(ポリアスパラギン酸カラムなど)上でのクロマトグラフィー、等電点電気泳動、SDS−PAGEおよび硫酸アンモニウム沈殿などのタンパク質精製のための他の技術も回収される抗体に依存して利用可能である。
【0114】
一実施形態において糖タンパク質は、調製物からフコース含有糖タンパク質を除き、したがってフコースを含まない糖タンパク質を富化するためにレクチン基質での吸着(例えばレクチン親和性カラム)を使用して精製されうる。
【0115】
医薬製剤
本発明は、本発明のPD−1抗体または抗体断片の医薬製剤を含む。医薬組成物または滅菌組成物を調製するために、抗体およびその断片は、薬学的に許容される担体または賦形剤と混合される、例えばRemington’s Pharmaceutical Sciences and U.S.Pharmacopeia: National Formulary、Mack Publishing Company、Easton、PA(1984年)を参照されたい。治療剤および診断剤の製剤は、生理学的に許容される単体、賦形剤または安定化剤と、例えば凍結乾燥粉末、スラリー、水性溶液または懸濁液の形態で混合することによって調製されうる(例えばHardmanら、(2001年)Goodman and Gilman’s The Pharmacological Basis of Therapeutics、McGraw−Hill、New York、NY;Gennaro(2000年)Remington:The Science and Practice of Pharmacy、Lippincott、Williams、and Wilkins、New York、NY;Avisら(eds.)(1993年)Pharmaceutical Dosage Forms:Parenteral Medications、Marcel Dekker、NY;Liebermanら(eds.)(1990年))Pharmaceutical Dosage Forms:Tablets、Marcel Dekker、NY;Liebermanら(eds.)(1990年)Pharmaceutical Dosage Forms:Disperse Systems、Marcel Dekker、NY;Weiner and Kotkoskie (2000年)Excipient Toxicity and Safety、Marcel Dekker、Inc.、New York、NYを参照されたい)。
【0116】
単独または免疫抑制剤との組合せで投与される抗体組成物の毒性および治療効果は、例えばLD50(集団の50%致死量)およびED50(集団の50%において治療的に有効な用量)を決定するための、細胞培養物または実験動物での標準的薬学的手順によって決定されうる。毒性量と治療有効量との間の用量比が、治療指数であり、LD50とED50との比として表されうる。これらの細胞培養アッセイおよび動物の研究から得られるデータは、ヒトでの使用のための投与量範囲を製剤化することにおいて使用されうる。そのような化合物の投与量は、好ましくは、毒性が殆どないか、または無しでのED50を含む循環濃度の範囲内にある。投与量は、使用する投与形態および使用する投与経路に依存してこの範囲内で変えることができる。
【0117】
投与の適切な経路は、筋肉内、静脈内または皮下投与などの非経口投与および経口投与を含む。医薬組成物中で使用されるまたは本発明の方法を実行するための抗体の投与は、経口摂取、吸入、局所投与または皮膚、皮下、腹腔内、非経口、動脈内または静脈内注射などのさまざまな従来の方法で実施されうる。一実施形態において本発明の結合化合物は、静脈内に投与される。他の実施形態において本発明の結合化合物は、皮下に投与される。
【0118】
代替えとして、例えば、しばしばデポー製剤または徐放性製剤での作用部位に直接での抗体の注射を介して、抗体を全身的様式よりは局所に投与できる。さらに標的化した薬物送達系で抗体を投与できる。
【0119】
抗体、サイトカインおよび小分子の適切な用量の選択における指針は、入手可能である(例えばWawrzynczak(1996年)Antibody Therapy、Bios Scientific Pub.Ltd、Oxfordshire、UK;Kresina(ed.)(1991年)Monoclonal Antibodies、Cytokines and Arthritis、Marcel Dekker、New York、NY;Bach(ed.)(1993年)Monoclonal Antibodies and Peptide Therapy in Autoimmune Diseases、Marcel Dekker、New York、NY;Baertら、(2003年)New Engl.J.Med.348巻:601〜608頁;Milgromら、(1999年)New Engl.J.Med.341巻:1966〜1973頁;Slamonら、(2001年)New Engl.J.Med.344巻:783〜792頁;Beniaminovitzら、(2000年)New Engl.J.Med.342巻:613〜619頁;Ghoshら、(2003年)New Engl.J.Med.348巻:24〜32頁;Lipskyら、(2000年)New Engl.J.Med.343巻:1594〜1602頁を参照されたい)。
【0120】
適切な用量の決定は、臨床医によって、例えば治療に影響するまたは治療に影響すると予測される当技術分野において公知または疑われるパラメーターまたは因子を使用して行われる。一般に、用量は、最適用量より幾分か少ない量から始め、その後任意の負の副作用と比較して所望のまたは最適な効果が達成されるまで少量ずつ増加させる。重要な診断指標は、症状、例えば炎症または産生される炎症性サイトカインのレベルの測定を含む。
【0121】
抗体、抗体断片およびサイトカインは、持続注入によって、または例えば1日、1週間もしくは1週間に1〜7回の間隔での投薬によって提供されうる。用量は静脈内、皮下、腹腔内、皮膚、局所、経口、経鼻、直腸、筋肉内、脳内、脊髄内、または吸引によって提供されうる。好ましい投与プロトコールは、重大な望ましくない副作用を避ける最高の用量または投与頻度を含むものである。1週間の合計用量は、一般的には体重1kgあたり少なくとも0.05μg、より一般的には体重1kgあたり少なくとも0.2μg、最も一般的には体重1kgあたり少なくとも0.5μg、典型的には体重1kgあたり少なくとも1μg、より典型的には体重1kgあたり少なくとも10μg、最も典型的には体重1kgあたり少なくとも100μg、好ましくは体重1kgあたり少なくとも0.2mg、より好ましくは体重1kgあたり少なくとも1.0mg、最も好ましくは体重1kgあたり少なくとも2.0mg、最適には体重1kgあたり少なくとも10mg、より最適には体重1kgあたり少なくとも25mg、最も最適には体重1kgあたり少なくとも50mgである(例えばYangら、(2003年)New Engl.J.Med.349巻:427〜434頁;Heroldら、(2002年)New Engl.J.Med.346巻:1692〜1698頁;Liuら、(1999年)J.Neural.Neurosurg.Psych.67巻:451〜456頁;Portieljiら、(20003)Cancer Immunol.Immunother.52巻:133〜144頁を参照されたい)。小分子治療、例えばペプチド模倣物、天然物または有機化学物質の望ましい用量は、1kgあたりのモルベースでの抗体またはポリペプチドについてとほぼ同じである。
【0122】
本明細書において使用する「抑制」、または「治療する」または「治療」は、疾患に関連する症状の発症の遅延および/または該疾患で発症するもしくは発症が予測されるそのような症状の重症度の低減を含む。用語は、さらに発症している症状の改善、追加的症状の予防、およびそのような症状の元となる原因の改善または予防を含む。したがって用語は、有益な結果が疾患を有する脊椎動物対象に与えられていることを意味する。
【0123】
本明細書において使用する用語「治療有効量」または「有効量」は、単独でまたは追加的治療剤との組合せで細胞、組織、または対象に投与された場合に治療される疾患または状態を予防または改善するために有効である、抗PD−1抗体またはその断片の量を指す。治療有効量は、さらに、症状の改善、例えば関連する医学的状態の治療、治癒、予防もしくは改善またはそのような状態の治療、治癒、予防もしくは改善の速度の増加を生じるために十分な化合物の量を指す。単独で投与される個々の活性成分に適用される場合、治療有効量は、成分単独でのそれを意味する。組合せに適用される場合治療有効量は、組合せで連続的にまたは同時に投与されるかにかかわらず、治療効果を生じる活性成分の組み合わされた量を指す。治療剤の有効量は、典型的には少なくとも10%、通常は少なくとも20%、好ましくは少なくとも約30%、より好ましくは少なくとも40%および最も好ましくは少なくとも50%症状を低減する。
【0124】
第2の治療剤を伴う同時投与または治療のための方法は、当技術分野において十分に周知であり、例えばHardmanら、(eds.)(2001年)Goodman and Gilman’s The Pharmacological Basis of Therapeutics、10th ed.、McGraw−Hill、New York、NY;Poole and Peterson(eds.)(2001年)Pharmacotherapeutics for Advanced Practice:A Practical Approach、Lippincott、Williams & Wilkins、Phila.、PA;Chabner and Longo(eds.)(2001年)Cancer Chemotherapy and Biotherapy、Lippincott、Williams & Wilkins、Phila.、PAを参照されたい。
【0125】
本発明の医薬組成物は、細胞毒性剤、細胞増殖抑制剤、抗血管新生剤または代謝拮抗剤、腫瘍標的剤、免疫刺激剤または免疫調節剤または、細胞毒性剤、細胞増殖抑制剤もしくは他の毒性作用物質と結合した抗体を非限定的に含む他の薬物も含有できる。医薬組成物は、手術、化学療法および放射線などの他の治療様式と共にも使用されうる。
【0126】
典型的な獣医学的、実験的または研究用対象として、サル、イヌ、ネコ、ラット、マウス、ウサギ、モルモット、ウマおよびヒトが挙げられる。
【0127】
本発明の抗体および抗体断片についての治療的使用
ヒトPD−1に特異的に結合する本発明の抗体または抗原結合断片は、免疫応答を増大、増強、刺激または上方制御するために使用されうる。本発明の抗体および抗体断片は、T細胞媒介の免疫応答の増大によって治療されうる障害を有する対象を治療するために特に適切である。好ましい対象は、免疫応答を増強する必要のあるヒト患者を含む。
【0128】
癌
本発明の抗体または抗原結合断片は、癌を治療する(すなわち腫瘍細胞の増殖または生存を抑制する)ために使用されうる。本発明の抗体を使用して増殖が抑制されうる好ましい癌は、典型的には免疫療法に応答する癌を含むが、免疫療法にこれまで関連がなかった癌も含む。治療について好ましい癌の非限定的例として、黒色腫(例えば転移性悪性黒色腫)、腎臓癌(例えば明細胞癌)、前立腺癌(例えばホルモン難治性前立腺腺癌)、膵臓腺癌、乳癌、結腸癌、肺癌(例えば非小細胞肺癌)、食道癌、頭頸部の扁平上皮癌、肝臓癌、卵巣癌、子宮頸癌、甲状腺癌、神経膠芽腫、神経膠腫、白血病、リンパ腫および他の腫瘍性悪性疾患が挙げられる。追加的に、本発明は、本発明の抗体を使用して増殖が抑制されうる難治性または再発性悪性疾患を含む。
【0129】
本発明の抗体または抗体断片は、単独であるいは、他の抗腫瘍性剤もしくは免疫原性薬剤(例えば、弱毒化された癌性細胞、腫瘍抗原(組換えタンパク質、ペプチドおよび炭水化物分子を含む))、腫瘍由来抗原もしくは核酸でパルスされた樹状細胞などの抗原提示細胞、免疫刺激サイトカイン(例えばIL−2、IFNa2、GM−CSF_)および非限定的にGM−CSFなどの免疫刺激サイトカインをコードする遺伝子で形質移入された細胞、標準的癌治療法(例えば化学療法、放射線療法もしくは手術)、または他の抗体(非限定的にVEGF、EGFR、Her2/neu、VEGF受容体、他の増殖因子受容体、CD20、CD40、CTLA−4、OX−40、4−IBBおよびICOSに対する抗体を含む)との組合せで使用されうる。
【0130】
感染性疾患
本発明の抗体または抗体断片は、感染症および感染性疾患を予防または治療するためにも使用されうる。抗体または抗体断片は、病原体、毒素および自己抗原への免疫応答を刺激するために、単独でまたはワクチンとの組合せで使用されうる。抗体またはその抗原結合断片は、非限定的にヒト免疫不全ウイルス、肝炎ウイルスクラスA、BおよびC、エプスタインバーウイルス(Eppstein Barr virus)、ヒト(hyman)サイトメガロウイルス、ヒトパピローマウイルス、ヘルペスウイルスなどの、ヒトに感染性のウイルスへの免疫応答を刺激するために使用されうる。抗体またはその抗原結合断片は、細菌性または真菌性寄生生物および他の病原体での感染への免疫応答を刺激するために使用されうる。
【0131】
ワクチン接種アジュバント
本発明の抗体または抗体断片は、他の組換えタンパク質および/またはペプチド(腫瘍抗原または癌細胞など)と結合してこれらのタンパク質への免疫応答を増大させるために(すなわちワクチン接種手順において)使用されうる。
【0132】
例えば抗PD−1抗体およびその抗体断片は、抗PD−1抗体と目的の抗原(例えばワクチン)との同時投与によって抗原特異的免疫応答を刺激するために使用されうる。したがって他の態様において本発明は、(i)抗原、および(ii)本発明の抗PD−1抗体またはその抗原結合成分を、対象における抗原への免疫応答が増強されるように対象に投与するステップを含む、対象において抗原への免疫応答を増強する方法を提供する。抗原は、例えば腫瘍抗原、ウイルス抗原、細菌抗原または病原体由来抗原でありうる。そのような抗原の非限定的例として、腫瘍抗原、またはウイルス、細菌もしくは他の病原体由来の抗原が限定なく挙げられる。
【0133】
Th2介在性疾患
本発明の抗PD−1抗体および抗体断片は、喘息およびアレルギーなどのTh2介在性疾患を治療するためにも使用されうる。これは、本発明の抗体がTh1応答の誘発を支援できるという発見に基づく。したがって本発明の抗体は、より(mofre)バランスのとれた免疫応答を作り出すためにTh2介在性疾患において使用されうる。
【0134】
T細胞のex−vivo活性化
本発明の抗体および抗原断片は、抗原特異的T細胞のex vivo活性化および増殖、ならびに腫瘍に対する抗原特異的T細胞を増加させるためのこれらの細胞のレシピエントへの養子移入のためにも使用されうる。これらの方法は、CMVなどの感染性物質へのT細胞応答を活性化させるためにも使用されうる。抗PD−1抗体の存在下でのex vivo活性化は、養子移入されたT細胞の頻度および活性を増大させることが期待されうる。
【0135】
他の組合せ療法
既に記載されたとおり本発明の抗PD−1抗体は、1つまたは複数の他の治療剤、例えば細胞傷害性剤、放射毒性剤または免疫抑制剤と同時投与されうる。抗体は、該薬剤(免疫複合体として)と結合されうるか、または該薬剤とは別に投与されうる。後者の場合(別々の投与)、抗体は該薬剤の前、後もしくは同時に投与されることができ、または他の既知の療法と同時投与されうる。
【0136】
本発明の抗体および抗原結合断片は、ドナーに移植された腫瘍特異的T細胞の効果を増大させるためにも使用されうる。
【0137】
本発明の抗体および抗体断片の非治療用使用
非治療用使用についての抗PD−1抗体の市場は、San Diego、Clifornia、USAのeBioscienceによって販売されている、フローサイトメトリー分析、免疫組織化学およびin vitro機能アッセイにおける使用のためのJ116およびJ105モノクローナル抗hPD−1抗体、ならびにMinneapolis、MN、USAのR&D Systemsによって販売されている、フローサイトメトリー、ウェスタンブロットおよびELISAにおける使用のためのmab1086、モノクローナル抗hPD−1抗体、の商業的販売によって示されるとおり既に存在する。本発明の抗体は、J116、J105および/またはMab1086によって現在果たされている任意の非治療用目的のために使用されうる。
【0138】
本発明の抗体は、親和性精製剤として使用されうる。
【0139】
抗体は、例えば特定の細胞、組織または血清中のPD−1の発現を検出するための、診断用アッセイにおいても有用である。診断用の適用において、抗体は典型的には(直接または間接のいずれかで)検出可能成分で標識される。以下の分類:ビオチン、蛍光色素、放射性ヌクレオチド、酵素、ヨウ素および生合成標識に一般に分けられ得る多数の標識が使用可能である。
【0140】
本発明の抗体は、競合的結合アッセイ、直接および間接サンドイッチアッセイならびに免疫沈降アッセイなどの任意の既知のアッセイ法において使用されうる。Zola, Monoclonal Antibodies.A Manual of Techniques、147〜158頁(CRC Press, Inc.1987年)
抗体は、in vivo診断アッセイのためにも使用されうる。一般に抗体は、放射性核種(111In、99Tc、4C、3H、125I、3H、32P35Sまたは18Fなど)で標識して、免疫シンチグラフィー(immunoscintiography)、または陽電子放出断層撮影を使用して、抗原またはそれを発現する細胞を限局化し得る。
【0141】
材料の寄託
ヒト化抗体h409A11、h409A16およびh409A17の重鎖および軽鎖の可変領域をコードするDNA構築物は、American Type Culture Collection Patent Depository(10801 University Blvd.、Manassas、VA)に寄託されている。h409A−11、h409A−16およびh409A−17の重鎖をコードするDNAを含有するプラスミドは、2008年6月9日に寄託され、081469_SPD−Hと認定された。h409A11の軽鎖をコードするDNAを含有するプラスミドは、2008年6月9日に寄託され、0801470_SPD−L−11と認定された。h409A16の軽鎖をコードするDNAを含有するプラスミドは、2008年6月9日に寄託され、0801471_SPD−L−16と認定された。h409A17の軽鎖をコードするDNAを含有するプラスミドは、2008年6月9日に寄託され、0801472_SPD−L−17と指定された。寄託は、特許手続きのための微生物寄託の国際認識に関するブダペスト条約の規定およびその規則(ブダペスト条約)に基づいて行われた。
【0142】
上に記載された明細書は、当業者に本発明を実施可能にするために十分であると考えられる。寄託された実施形態は、本発明の一態様の単一の例示の目的であり、機能的に同等である任意の培養物は本発明の範囲内であることから、本発明は、寄託された培養物によって範囲を限定されない。本明細書の材料の寄託は、本明細書に含まれる書面による説明が、その最良の様式を含む本発明の任意の態様の実施を可能にするために不適切であるとの承認を構成するものでなく、本特許請求の範囲をそれが提示する具体的な例示に限定すると解釈されるべきでもない。実際に、本明細書に示されかつ記載されるものに加えての本発明の種々の変更は、上の記載から当業者に明らかになり、かつ添付の特許請求の範囲内である。
【0143】
本発明は、以下の実施例を参照することによってより完全に理解される。しかしそれらは、本発明の範囲を限定するとして解釈されるべきではない。本明細書において述べる全ての文献および特許の引用は、明確に参照として本明細書に組み込まれる。
【実施例】
【0144】
(実施例1)
抗PD−1抗体の免疫化および選択
hPD−1cDNAでのマウスの免疫化
ヒトPD−1(「hPD−1」)受容体に対する抗体を生成するために、hPD−1受容体のオープンリーディングフレームをコードするcDNAをPCRによって得て、ベクターpcDNA3.1(Invitrogen、Carlsbad、CA)にサブクローン化した。次にCHO−K1細胞をhPD−1で安定に形質移入し、発現をフローサイトメトリーを使用してモニターした。ヒトPD−1を膜上に発現するCHO−K1クローンを単離し、CHO−hPD1と名付けた。
【0145】
Helios gene gun(BioRad)およびDNA coated gold bullets(BioRad)を製造者の説明書に従って使用して遺伝子銃免疫化によってマウスを免疫化した。簡潔には1μmの金粒子をhPD−1 cDNA(pcDNA3.1にクローン化した)と、指示のある場合はマウスFlt3LおよびマウスGM−CSF用の市販の発現ベクター(どちらもAldevron、Fargo NDから)とで2:1:1の割合でコートした。合計1μgのプラスミドDNAを500μgのgold bulletをコートするために使用した。
【0146】
具体的には、7〜8週齢の雌性BALB/Cマウスを、遺伝子銃によって、2、3または4サイクルのショットを両耳に受けさせて耳で免疫化した(表Iを参照されたい)。マウス1匹は、5×106CHO−hPD1細胞を用いた最終追加免疫を腹腔に受けた。およそ1:1000の抗hPD−1力価がマウス血清において2回のDNA免疫化後に、CHO−hPD1対CHO−K1親細胞を使用する細胞ELISAによって検出可能であった。最終免疫化の4日後、マウスを屠殺し、赤血球除去脾臓細胞集団を以前記載されたとおり(Steenbakkersら、1992年、J.Immunol.Meth.152巻:69〜77頁;Steenbakkersら、1994年、Mol.Biol.Rep.19巻:125〜134頁)調製し、−140℃で凍結した。
【0147】
【表1】
抗PD−1抗体産生B細胞の選択
抗ヒトPD−1抗体産生B細胞クローンを選択するために、hPD−1 DNA免疫化マウス、すなわちマウス730、731および738(表1を参照されたい)由来の赤血球除去脾臓細胞2×107個をB細胞培養のためにプールした。脾臓細胞をDMEM/HAM’s F12/10%子ウシ血清(Hyclone、Logan、UT、USA)で37℃、1時間、プラスチック培養フラスコ中で単球を除くためにインキュベートした。非接着細胞を1回のCHO−K1細胞でのネガティブパニング、続いてCHO−hPD1細胞でのポジティブパニングにかけた。両方の選択手順は、21cm2ペトリ皿またはT25培養フラスコでコンフルエントに増殖した培養物において37℃、1時間実施した(細胞培養物は使用前に合計線量2000RADまで照射した)。ポジティブパニング後、未結合細胞を、0.132%CaCl2.2H2Oおよび0.1%MgCl2.6H2Oを補充したPBSでの10回洗浄によって除去した。最後に結合性B細胞をトリプシン処理によって回収した。
【0148】
選択したB細胞を培養し、Steenbakkersら、1994年、Mol.Biol.Rep.19巻:125〜134頁に記載のとおり不死化した。簡潔には、選択したB細胞を、平底96ウェル組織培養プレート中の7.5%(v/v)T細胞上清および50000照射(2500RAD)EL−4 B5栄養細胞と最終容量200μLのDMEM/HAM’s F12/10%子ウシ血清中で混合した。8日目に、上清をそれらの抗PD−1反応性についてCHO−hPD−1細胞ELISAによって以下の手順を使用して選別した。CHO−K1およびCHO−hPD−1細胞をコンフルエントまで平底96ウェルプレートで、50μL DMEM/HAM’s F12、10%FBS中で培養した。次に50μLの免疫グロブリン含有上清を37℃、1時間で加えた。PBS−Tweenで3回洗浄後、DMEM/HAM’s F12/10%FBS中の100μL(1:1000希釈)ヤギ抗マウス西洋わさびペルオキシダーゼ(HRP、Southern、Birmingham、AL、USA)を37℃、1時間で加えた。PBS−Tweenで3回洗浄後、固定化免疫グロブリンをUPO/TMB(Biomerieux、Boxtel、Netherlands)で可視化した。
【0149】
このB細胞培養物から、13個のhPD−1反応性の上清が同定され、プラスチック上に固定化された場合にジャーカットT細胞活性化を抑制することを示し、陽性ウェルからのB細胞クローンを、刊行された手順(Steenbakkersら、1992年、J.Immunol.Meth.152巻:69〜77頁;Steenbakkersら、1994年、Mol.Biol.Rep.19巻:125〜134頁)に従ってミニ電気融合によって不死化した。具体的には、B細胞を106個のNS−1骨髄腫細胞と混合し、血清をDMEM/HAM’s F12での洗浄によって除去した。次に細胞をプロナーゼ溶液で3分間処理し、続いて融合培地で洗浄した。電気融合を50μL融合容器で、30s、2MHz、400V/cmの交流電場に続いて、10μs、3kV/cmのスクウェア、高電場パルスおよび再度の30s、2MHz、400V/cmの交流電場によって実施した。最後に融合容器の内容物をハイブリドーマ選択培地に移し、限界希釈条件下で96ウェルプレートに蒔いた。融合の14日後、培養物をハイブリドーマの増殖について検査し、hPD−1への抗体反応性の存在について選別した。この手順は、hPD−1.05A、hPD−1.06B、hPD−1.08A、hPD−1.09AおよびhPD−1.13Aと名付ける5種の異なる抗hPD−1ハイブリドーマをもたらし、それらの完全性を保護するために限界希釈によってサブクローン化し、抗体を産生するためにさらに培養した。これらのハイブリドーマから得た上清は、抗CD3/抗CD28刺激におけるジャーカットE6.2.11細胞からのIL−2産生を強く抑制した(図1および以下の本文を参照されたい)。
【0150】
ジャーカットE6.1細胞(American Type Culture Collection)を限界希釈によって標準的方法を使用してサブクローン化し、サブクローンをCD3およびCD28の交差結合(cross−linking)でIL−2を産生する増大した能力について検査した。高IL−2産生サブクローンを得て、次にジャーカットE6.2.11と名付け、さらなるアッセイにおいて使用した。Costar3370 96ウェルアッセイプレートを5μg/mLヒツジ抗マウスIg(SAM)で一晩、4℃でコートした。過剰量のSAMを除去し、プレートを1ウェルあたり200μLのPBS/10%ウシ胎仔血清で1時間、室温でブロックした。PBSで3回洗浄後、ウェルを1ウェルあたり100μLの抗CD3(OKT3、10または60ng/mL)で1時間、37℃でコートした。PBSで3回洗浄後、1ウェルあたり50μLのPBS/10%ウシ胎仔血清および1ウェルあたり50μLのB細胞またはハイブリドーマ上清を30分間、37℃で加えた。PBSで3回洗浄後、1ウェルあたり120μLの細胞懸濁液、ジャーカットE6.2.11細胞(DMEM/F12/10%ウシ胎仔血清中の、1ウェルあたり細胞2×105個+0.5μg/mL抗CD28(Sanquin #M1650、Central Laboratory for Bloodtransfusion、Amsterdam、NL))を加えた。6時間培養後、上清をIL−2産生について標準的サンドイッチELISAをPharmingenからの抗hIL−2捕捉抗体とビオチン化検出抗体との対および検出試薬としてストレプトアビジン−西洋わさびペルオキシダーゼ(Southern Biotech)で使用して検査した。これらの抗体の効力をPD−L1と比較して決定するために、mAbの小さな群を大規模に産生させた。mAbをプロテインGアフィニティークロマトグラフィーを使用して精製した(実施例2を参照されたい)。精製した抗体、hPD−L1/Fc(組換えヒトB7−H1/Fcキメラ、R&D systems)またはネガティブコントロールとしてのマウスIgG1κ(Sigmaから)を同じ濃度でプレートに抗CD3と共に上に記載のとおりコートした。ジャーカットE6.2.11細胞および抗CD28を6時間加え、T細胞活性化を上清に産生されたIL−2によって測定した。抗体のうち2つ(hPD1.08AおよびhPD1.09A)は、固定化PD−L1/Fcと比較して8〜10倍強い抑制を示した。
【0151】
(実施例2)
マウス抗PD−1抗体の精製および特徴付け
抗PD−1産生ハイブリドーマの安定化および抗PD−1抗体の精製
クローン細胞集団を各ハイブリドーマから、それらを複数回(>4)の限界希釈にさらすことによって得た。次いで安定なハイブリドーマを無血清条件でCELLineバイオリアクター(Integra−biosciences)を使用して6から8日間培養した。細胞を内側の容器の無血清培地中に1mLあたり細胞3×106個の密度で15mLに蒔き、およそ1mLあたり細胞4×107個に8日間かけて増殖させた。外側の容器を10%までのBCS(仔ウシ血清)で補充した培地で満たした。6日目から8日目に、内側の容器の培養物を回収し、15mL SF培地で洗浄し、ハイブリドーマと再播種した。バイオリアクター上清および洗浄液を合わせ、遠心分離で澄ませた。得られた上清を0.22μM濾過膜で濾過した。抗体精製のために、上清を1:1で高塩濃度の結合緩衝液(1M グリシン/2M NaCl、pH9.0)に希釈し、mAbをプロテインG HiTrap 5mLカラム(GEhealthcare)を使用して精製した。PBSで洗浄後、結合抗体を0.1Mグリシン pH=2.7を使用して溶出し、3M Trisを使用するpH中和が続いた。最後に緩衝液をPD−10ゲル濾過カラム(GEhealthcare)を使用してPBSに交換し、抗体をUltra−15遠心濃縮器(Amicon)を使用して濃縮し、分光光度法を使用して定量した。
【0152】
市販の抗体
以下の市販の抗体を本明細書に記載の種々の研究において使用した:抗PD−1抗体クローンJ116(#14−9989)はeBioscienceから購入した。抗CTLA−4クローン14D3(mAb 16−1529)はeBioscienceから購入した。抗PD−1クローン192106(mAb1086)はR&D systemsから購入した(#mAb1086)。アイソタイプ対照抗体mIgG1、κ、クローンMOPC21はSigmaから購入した(#M9269)。アイソタイプ対照mIgG1κ(mAb 16−4714)およびIgG2aκ(mAb 16−4724)はeBioscienceから購入した。
【0153】
結合活性
タンパク質に基づく、および細胞に基づくELISA(「CELISA」)実験を見かけの結合親和性(EC50値として報告される)を決定するために使用した。場合により、抗PD−1抗体の結合を市販の抗PD−1抗体J116(eBioscience)およびMab1086(R&D systems)のそれと比較した。
【0154】
タンパク質ELISAを、抗体のヒトPD−1/Fcへの相対結合の決定のために使用した。hPD−1/Fc(R&D Systems)をMaxisorp 96ウェルプレート(Nunc)に4時間、室温(または4℃、一晩)でのインキュベーションによって固定化した。非特異的結合部位をPBST中の3%BSAとの1時間、室温でのインキュベーションによってブロックした。コーティング後、プレートをPBSTで3回洗浄した。抗PD−1抗体の希釈物を結合緩衝液(0.1%Tween20および0.3%BSAを含有するPBS)中に調製し、固定化融合タンパク質と1時間、25℃でインキュベートした。結合後、プレートをPBSTで3回洗浄し、結合緩衝液中に1/4000で希釈したペルオキシダーゼ標識ヤギ抗マウスIgG(Southern Biotech)と1時間、25℃でインキュベートし、再び洗浄し、TMBを使用して発色させた。ELISA結果を図2に示す。最大半量の結合での濃度を相対結合親和性の測定値として報告する(表II)。
【0155】
CHO−hPD−1細胞への結合もCELISAによって評価した。CELISAのためにCHO−hPD−1細胞を80から100パーセントコンフルエントまで50μL培地(DMEM/HAM’S 12F、10%FBS)中で培養した。次に種々の濃度の精製mAbを含有する50μL培地を1時間、37℃で加えた。PBS−Tweenでの3回洗浄後、100μLヤギ−抗マウス−HRP(Southern Biotech cat #1030−05)(培地中に1:1000希釈)を1時間、37℃で加えた。PBS−Tweenでさらに3回洗浄後、固定化免疫グロブリンを比色定量のペルオキシダーゼ基質TMB(BD Biosciences)で可視化した。ペルオキシダーゼ活性による吸光度(450nm)の増加をマイクロタイタープレートリーダーで測定した。図2は、抗体hPD−1.08AおよびhPD−1.09Aにおける濃度と結合との間の用量応答関係を示す。タンパク質および細胞の結合研究の結果を表IIにまとめる。
【0156】
生物光学干渉法(ForteBio)による動態解析
抗体の結合特性をさらに特徴付けるために、それぞれをOctet system(ForteBio、Menlo Park、CA)での生物光学干渉法を使用して概要を示し、結合動態を明らかにし、および平衡結合定数を算出した。このアッセイは、PD−1Fc融合タンパク質(R&D systems)の、標準的アミン化学を使用するアミン反応性バイオセンサー(Fortebio)へのカップリングによって実施された。次いで抗PD−1mAbのバイオセンサーへの結合およびバイオセンサーからの解離を種々の抗体濃度で観察した。具体的には、アミン反応性バイオセンサーを、0.1M MES pH=5.5を含むウェルに5分間浸漬することによって予め湿らせた。次いでバイオセンサーを0.1M NHS/0.4M EDC混合物を5分間使用して活性化した。PD−1/Fc融合タンパク質(R&D systems)を、バイオセンサーを0.1M MES中の12μg/mL PD−1/Fcの溶液に7.5分間浸漬することによってカップリングさせた。バイオセンサーの表面を1Mエタノールアミン溶液を5分間使用してクエンチさせた。バイオセンサーをPBS中に5分間、平衡化させた。抗PD−1mAbの会合を、種々の抗体濃度(PBS中、10〜80nM精製抗体>99% SDS−PAGEによる)を含むウェル中にバイオセンサーを置き、干渉(interferometry)を30分間モニターすることによって観察した。解離は、バイオセンサーをPBS中に移し、干渉シグナルを60分間モニターした後に測定した。観察された会合速度および解離速度(kobsおよびkd)を、検査した全ての濃度を含む1:1結合の包括的フィットモデルを使用して適合させ、平衡結合定数KDを算出した。動態研究からの結果を表IIおよび下の図6に示す。
【0157】
【表2】
モノクローナル抗体のうちの2つ、hPD−1.08AおよびhPD−1.09Aは、このアッセイを使用して検査した任意の他のmAbよりも相当に強く結合し、hPD−1.08AおよびhPD−1.09Aについて決定されたKDがそれぞれ24pMおよび22pMであった。検査した他の抗PD−1抗体と比較して、親和性の増大は、hPD−1.08AおよびhPD−1.09Aについて測定されたより遅い解離速度および顕著に早い会合速度による。
【0158】
リガンド遮断
フローサイトメトリーを使用して研究したリガンド結合の遮断。ヒトPD−1を発現するCHO細胞を接着培養フラスコから解離させ、種々の濃度の抗PD−1抗体、および一定濃度(600ng/mL)の未標識hPD−L1/Fcまたは組換えヒトPD−L2/Fc融合タンパク質(どちらもR&D Systemsから)と96ウェルプレートで混合した。混合物を30分間氷上で平衡化させ、FACS緩衝液(1%BCSおよび0.1%アジ化ナトリウムを含有するPBS)で3回洗浄し、FITC標識ヤギ抗ヒトFcとさらに15分間、氷上でインキュベートした。細胞を再びFACS緩衝液で洗浄し、フローサイトメトリーによって分析した。データを非線形回帰を使用するPrism(GraphPad Software、San Diego、CA)で分析し、IC50値を算出した。
【0159】
算出したIC50値を表IIにまとめる。抗体05A、06Bおよび13Aは、hPD−1の結合について600pMから3nMの間のKDを示すと確定された。強力な結合にもかかわらず、これらの抗体それぞれは、hPD−1へのhPD−L1の結合の遮断についてIC50>10nMを示した。市販で入手できる抗PD−1抗体J116(eBiosciences)は、結合について、この実験の範囲外に算出されたIC50(>100,nM)を有してPD−L1と弱く競合した。対照マウスIgG1は、PD−1結合についてPD−L1と競合しない。対照的に高親和性抗体hPD−1.08AおよびhPD−1.09Aは、1nMより低いIC50値でPD−L1結合を抑制したが、PD−L2結合はIC50値1〜2nM程度で遮断した(表II)。PD−L2は、PD−L1よりも2から6倍高い親和性でPD−1に結合するとより早く報告された。(Youngnak P.ら、2003年、Biochem.Biophys.Res.Commun.307巻、672〜677頁)
リガンド遮断は、蛍光比色微量容積アッセイ(fluorometric microvolume assay)技術(FMAT)を使用する同質的競合アッセイおよび検出を使用して確認された。簡潔には、CHO.hPD−1を接着培養フラスコから解離させ、種々の濃度の抗PD−1抗体および一定濃度(600ng/mL)のhPD−L1/FcまたはhPD−L2/Fc融合タンパク質(どちらもR&D Systemsから)と混合し、96ウェルプレート中で蛍光色素(AlexaFluor647、Invitrogen)で標識した。混合物を90分間、37℃で平衡化させ、AB8200Cellular Detection Analyzer(Applied Biosystems、Foster City、CA)を使用して読み取った。データは非線形回帰を使用するPrism(GraphPad Software、San Diego、CA)で分析し、IC50値を算出した。図3は、リガンド遮断の程度は抗体濃度によって決定されることを示す用量応答実験の結果を示す。hPD−L1/FcおよびhPD−L2/Fcの両方のCHO−hPD−1細胞への結合は、hPD−1.08A、hPD−1.09Aおよび(より少ない程度で)J116によって用量依存的な様式で完全に抑制されうる。算出されたIC50データを表IIにまとめる。フローサイトメトリーを使用して得られた結果を確認して、高親和性抗体hPD−1.08AおよびhPD−1.09Aは、PD−L1結合を1nMより低いIC50値で抑制した。
【0160】
種の交差反応
抗体の種の交差反応性を評価するために、マウスおよびcynomolgus macaque PD−1受容体をPCRによってクローン化し、安定に形質移入したCHO−K1細胞を作製した。抗体をカニクイザル受容体への結合についてCELISAを使用して検査した。市販の抗体J116、hPD−1.08AおよびhPD−1.09Aは、ヒトおよびカニクイザルPD−1に同等な親和性で結合し、hPD−L1/FcおよびhPD−L2/FcのカニクイザルPD−1への結合をヒトPD−1と比較して同様の有効性で遮断することが見いだされた。カニクイザルPD−1の細胞外部分のアミノ酸配列がヒトPF−1のそれと97%同一であることが見いだされたことから、これは驚くべきことではない。cynomolgous macaque由来のPD−1に加えて、hPD−1.08AおよびhPD−1.09Aは、rhesus macaques由来のPD−1も実施例3に記載のSEM刺激血液細胞培養物中で機能的に遮断した。検査した抗体で、使用したいずれのアッセイにおいても検出可能な親和性でマウスPD−1に結合したものはなかった。
【0161】
要約として、ジャーカット機能を調整する能力に基づいて単離した5個の抗PD−1モノクローナル抗体を精製し、かつ特徴付けた。これらの抗体は、PD−1に強く結合し(解離定数20pMから3nMの範囲で)、さまざまなIC50値でPD−L1およびPD−L2の両方と相互作用の遮断をできた。これらの抗hPD−1mAbの4個は、市販で入手可能な最も良い抗PD−1mAbより相当に良かった。受容体アンタゴニストとして作用する溶液中に加えた場合に各抗体は、最終的にはT細胞応答を増強した(実施例3を参照されたい)。
【0162】
(実施例3)
抗PD−1抗体の機能的プロファイリング
SEBへのヒトT細胞応答は、hPD−1.08AおよびhPD−1.09Aによって増強される。
【0163】
抗PD−1抗体をT細胞活性を増強するそれらの能力についてin vitroで、健常ボランティア由来の血液細胞を使用して検査した。ヒトPD−1受容体を遮断することの機能的結果を特徴付けるために使用したアッセイは、Vβ3およびVβ8 T細胞受容体鎖を発現する全てのT細胞に結合し、かつ活性化するためにブドウ球菌エンテロトキシンB(SEB)を利用した。健常人ドナー血液を得て、培地中に1:10で希釈した。希釈した全血を96ウェル丸底プレートに蒔き(1ウェルあたり150μl)、30〜60分間、mAbとさまざまな濃度でプレインキュベートした。次いでSEBを10ng/mLから10μg/mLの範囲の種々の濃度で加えた。培養2から4日後に上清を回収し、産生されたIL−2の量をELISA(実施例1に記載)を使用して、または標準的多重技術(Luminex platform−Biosourceサイトカイン検出キット)を使用して定量した。100ng/mLから最大10μg/mLまでのSEBの滴定は、全血細胞によるIL−2産生を顕著に刺激した。通常、1μg/mLのSEBでの刺激2〜4日後にELISAによって、ドナーに依存して、100から1000pg/mLのIL−2が検出可能であった。hPD−1.08AおよびhPD−1.09Aの添加は、対照マウスIgG1を超えて、検査した最高抗体濃度(25μg/mL)で平均2から4倍にIL−2産生を増強した。刺激指数は、一連の独立の健常ボランティアで実施した実験について平均した(図4)。これらの実験は、hPD−1.08AおよびhPD−1.09Aの両方が希釈全血細胞のSEB刺激でIL−2産生を増強したことを示した。PD−1およびPD−L1の両方(しかしPD−L2ではない)の発現レベルは、全血細胞のSEB刺激後に徐々に上方制御された(フローサイトメトリーによる定量)。抗PD−L1モノクローナル抗体(クローンMIH5、Ebioscience #16−5982)および抗CTLA−4(クローン14D3、eBioscience #16−1529)も、同様の条件下でIL−2産生の増加を誘発し、同時刺激経路の操作後にT細胞活性を定量するためのSEB刺激アッセイの使用をさらに検証した発見を導いた(図4)。抗PD−1抗体によって増強されたIL−2産生は、用量依存的であることが見いだされた。IL−2に加えてLuminex technologyによって、TNTα、IL−17、IL−7、IL−6およびIFNγのレベルもhPD−1.08AおよびhPD−1.09Aによって顕著に調節されることが見いだされた。これらの実験の結果は、hPD−1.08AおよびhPD−1.09AをヒトT細胞応答を刺激するために使用できることを示す。
【0164】
抗PD−1抗体hPD−1.09AをT細胞活性を増強するその能力についてin vitroで癌患者由来の血液細胞を使用してさらに検査した。進行黒色腫(1名)または前立腺癌(3名)を有する患者由来の血液を上の手順に従って検査した。サイトカイン定量の結果を表IIIに、細胞を25μg/mL hPD−1.09Aの存在下で刺激した場合を抗体非存在下でのSEB刺激と比較して、産生されたサイトカインの増加倍数で表す。要約すると、hPD−1.09Aは、4名の各患者についてSEB誘発IL−2産生を2から3.5倍増加させたことが見いだされた。同様にTNTα、IL−17およびIFNγの産生は、増強され、IL−5およびIL−13の産生は減少した。これらの実験は、hPD−1.09Aが癌患者においてT細胞応答を刺激する能力を有することを示す。さらにこれらの実験は、Th1応答に対する選好性を示唆する。
【0165】
【表3】
TTチャレンジに対するヒトリコールT細胞応答は、hPD−1.08AおよびhPD−1.09Aによって増強される。
【0166】
抗ヒトPD−1抗体遮断受容体のその天然のリガンドとの相互作用の機能的効果を要約するために使用された他のアッセイは、破傷風トキソイド(TT)抗原を健常人ドナー血液における既存のメモリーT細胞を刺激するために使用した。この目的を達成するために、新鮮調製PBMC(細胞2×105個)を96ウェル丸底プレートの完全RPMI1640培地(5%熱不活性化ヒト血清を含む)に蒔き、さまざまな濃度の検査抗体とプレインキュベートし、100ng/mLの濃度のTT(Astarte Biologics)で刺激した。細胞を3〜7日間、37℃、5%CO2でインキュベートし、その後上清を回収した。サイトカイン濃度をELISA(eBioscienceからのIL−2およびIFNγ ELISA検出抗体対セット)および多重分析(Luminex platform−Biosourceサイトカイン検出キット)によって決定した。PD−1の遮断は、抗原単独と比較して、増殖を増強し、かつIFNγおよびIL−2を含むサイトカイン産生を顕著に増強した(図5)。Luminex分析は、サイトカインGM−CSF、RANTESおよびIL−6の産生がPD−1遮断で増加することを明らかにした。
【0167】
ホルマリン固定パラフィン包埋ヒト細胞でのヒトPD−1の染色
SEB刺激血液細胞がPD−1の増強された発現をフローサイトメトリーで示したことから、これらの細胞を、hPD−1.09Aが組織学的使用のためのホルマリン固定パラフィン包埋組織においてPD−1を検出できるかどうかを決定するために使用した。ヒトドナー末梢血単核球を、0.1μg/mL SEBで3日間刺激し、その後非接着細胞(主にリンパ球)を回収し、PBSで2回洗浄し、遠心分離(1100rpm、5分間)した。細胞を10分間、4%ホルムアルデヒドで固定し、細胞沈渣をアガロースに包埋し、エタノール(70%、80%、96%および100%と続いて)およびキシレン中で脱水し、その後パラフィンに包埋した。切片(4μm)をガラススライドにマウントし、水和させ(キシレン、エタノール100%、96%、80%、70%、PBS緩衝液)、その後加熱したクエン酸緩衝液への抗原回収を標準的方法を使用して実施した。ペルオキシダーゼ活性を0.3%H2O2を含む100%メタノールを使用して遮断し、スライドを水およびPBS、0.1%Tweenでリンスした。切片をhPD−1.09Aと1.5時間、室温でインキュベートし、PBS−Tweenでリンスし、標準的検出法が続いた。スライドをヘマトキシリンで30秒間、室温で対比染色し、キシレンで脱水し、マウントして顕微鏡検査した。これらの実験は、SEB刺激PBCM培養物由来のリンパ球は、未刺激のPBMC培養物とは対照的に、hPD−1.09Aで(アイソタイプ対照と比較した場合)強く染まり、hPD−1.09Aが診断試薬として有用であることを示した。
【0168】
(実施例4)
抗PD−1抗体配列および続くヒト化
免疫グロブリンcDNAのクローニング
縮重プライマーPCRに基づく方法を使用して、ハイブリドーマhPD−1.08AおよびhPD−1.09Aによって発現されるマウス抗体の可変領域をコードするDNA配列を決定した。簡潔には、重鎖および軽鎖についての遺伝子特異的cDNAをiScript Select cDNA合成キット(Biorad #1708896)を製造者の説明書に従って使用して生成した。使用したPCRプライマーは、Ig−プライマーセット(Novagen #69831−3)に基づいた。縮重PCR反応は、TaqポリメラーゼをNovagenプライマーセット手順書に従って使用して実施した。PCR産物をアガロースゲル電気泳動によって分析した。重鎖および軽鎖の可変領域の両方について予測される増幅産物サイズは、約500塩基対である。適切なバンドをもたらした反応からのTaq−増幅PCR産物2μlをpCR4 TOPOベクター(Invitrogen #K4595−40)にクローン化し、DH5−αE.coliに製造者による指示のとおり形質転換した。クローンをユニバーサルM13順方向および逆方向プライマーを使用してコロニーPCRによって選別し、各反応から2から3クローンをDNA配列分析用に選択した。
【0169】
クローンをユニバーサルプライマーM13順方向、M13逆方向、T3およびT7を使用して両方向に配列決定した。各クローンについての各配列決定反応の結果をSeqmanを使用して分析した。共通配列を、NCBI Ig−Blast(http://www.ncbi.nlm.nih.gov/projects/igblast/)を使用して、生殖系列および再配列Ig可変領域配列のデータベースに対して探索した。hPD−1.08AについてのBlastの結果は、ストップコドンが導入されていない生産性(インフレーム)に再配列された重鎖を同定した。2種の異なる配列、1つはストップコドンが導入されていない生産性(インフレーム)に再配列された軽鎖、他方はFR4領域中にストップコドンに至るフレームシフトを含有する非生産的に再配列された配列、をコードする軽鎖クローンが同定された。観察された非生産的増殖不能転写物は、おそらく骨髄腫融合相手由来であり(Carroll W.L.ら、Mol.Immunol.25巻:991〜995頁(1988年)排除した。
【0170】
hPD−1.09AについてのBlastの結果は、ストップコドンが導入されていない生産性(インフレーム)に再配列された重鎖および軽鎖を同定した。発現されたタンパク質のアミノ酸配列は、質量分析によって確認した。配列を添付の配列表で開示し、表IVに列挙する。
【0171】
【表4−1】
【0172】
【表4−2】
CDRおよびフレームワーク領域は、Kabat E.A.ら、1991年、Sequences of proteins of Immunological interest、In: NIH Publication No.91−3242、US Department of Health and Human Services、Bethesda、MDに従って注釈を付けた。
【0173】
キメラc109A抗体の構築および発現
キメラ軽鎖および重鎖を、マウスhPD−1.09A VLおよびVH領域のPCRでクローン化したcDNAをヒトκおよびIgG1定常領域にそれぞれ連結することによって構築した。マウスcDNA配列の5’および3’末端を、各鎖に適切なリーダー配列および既存の組換え抗体発現ベクターへのクローニングを可能にする制限部位を付加するために設計したPCRプライマーを使用して改変した。
【0174】
COS−7細胞(1mLあたり107個で0.7mL)に各キメラ重鎖および軽鎖発現プラスミド10μgを電気穿孔した。次いでこれらの細胞を8mL 増殖培地中で3日間培養した。サンドイッチELISAを、COS−7形質移入体由来の上清中の抗体濃度を測定するために使用した。これは、形質移入されたCOS−7細胞は、3種の別々の形質移入体で約295ng/mLのキメラIgG1−κ抗体を分泌したことを示した。
【0175】
形質移入されたCOS−7細胞によって産生されたキメラ抗体の結合は、PD−1結合ELISAおよびCELISA(実施例2を参照されたい)を使用して測定し、マウス抗体に匹敵する親和性でPD−1に結合することを示した。
【0176】
ヒト化抗体設計
hPD−1.09A抗体をMRCT(Cambridge UK)によりCDR移植技術を使用してヒト化した(例えば、米国特許第5,225,539号を参照されたい)。簡潔には、マウス抗体hPD−1.09Aの可変鎖配列をResearch Collaboratory for Structural Bioinformatics(RCSB)タンパク質データバンクにおいて入手可能な配列と比較した。hPD−1.09Aの相同性モデルを最も近いVHおよびVK構造に基づいて生成した。hPD−1.09Aに最も高い同一性を有するヒト配列を同定し、分析した(Foote and Winter、J.Mol.Biol.224巻:487〜499頁(1992年);Morea V.ら、Methods 20巻:267〜270頁(2000年);Chothia C.ら、J.Mol.Biol.186巻:651〜663頁(1985年))。CDR移植重鎖および軽鎖を構築するために最も適切なヒトフレームワークを同定した。
【0177】
重鎖について、genbankアクセッション番号AB063829でコードされるフレームワークを最も適切であると決定した。hPD−1.09A VK配列の分析は、そのCDR1の長さ(15残基)が任意のヒトVKにおいて見いだされないことを示す。このため3種の異なるCDR1の長さ(11、16および17残基)のフレームワークを、どのCDR1の長さがhPD−1.09A VKの挙動を再生するのかを検査するために分析した。その構造中において重要である選択された残基においてhPD−1.09A VKに最も高い同一性を有し、CDR1の長さ11、16および17を有するヒトVK配列を同定した。genbankアクセッション番号M29469のフレームワークをK109A−L−11に基づいて選択した。genbankアクセッション番号AB064135由来のフレームワークをK09A−L−16に基づいて選択し、genbankアクセッション番号X72431由来のフレームワークをK09A−L−17に基づいて選択した。
【0178】
ストレートグラフト(straight graft)を各鎖に対する発現構築物を生成するために実施した。109A−H、K09A−L−11、K09A−L−16およびK09A−L−17のDNAおよびタンパク質配列を添付の配列表(表IV)において開示する。
【0179】
セリン241(Kabat番号)がプロリンに転換された安定化Adair変異を有する(Angal S.ら、Mol.Immuol.30巻:105〜108頁(1993年))ヒト化h109A抗体のIgG4バージョンを生成した。この配列は、配列番号23および31で開示される。
【0180】
(実施例5)
ヒト化抗PD−1抗体の結合特性および機能特性
産生および精製
ヒト化抗体h409A11、h409A16およびh409A17をCHO−S細胞の一過的形質移入によって産生させた。細胞をCD−CHO(Gibco)およびC5467培地(Sigma)で8日間、振盪フラスコ中で増殖させた。抗体をプロテインAクロマトグラフィーによって細胞上清から精製し、洗浄し、1M酢酸を使用して溶出し、3M Trisを使用して中和した。最後に緩衝液を、1M Tris塩基でpH5.5に調整した100mM酢酸に交換した。
【0181】
結合および動態解析
見かけの結合親和性を決定するために(EC50値として報告される)タンパク質に基づく、および細胞に基づくELISAを実施例2に記載のとおり実施した。ヒト化抗PD−1抗体は、PD−1/Fcおよび細胞で発現されるPD−1にマウス親抗体に匹敵するEC50値でそれぞれ結合した(表V)。
【0182】
抗体の動態学的な結合特性も実施例2に記載のとおり生物光学干渉法を使用して実施した(図6)。ヒト化抗体の2つh409A11およびh409A16は、h409A11およびh409A16について決定されたKDがそれぞれ29pMおよび27pMであって(表V)、このアッセイを使用して検査した他のいずれのmAbよりも相当強く結合した。検査した他の抗PD−1抗体と比較して、増大した親和性は、主により遅い解離速度による。マウス親抗体と同様に、ヒト化抗PD−1抗体h409A11、h409A16は、120pMより低く決定されたKDでカニクイザル(cynomolgous)PD−1への結合を示した。
【0183】
リガンド遮断
PD−L1およびPD−L2のPD−1への結合を遮断するヒト化抗体の能力を、実施例2に記載のとおりFMAT競合アッセイを使用する同質的競合アッセイおよび検出を使用して測定した。
【0184】
hPD−L1/FcおよびhPD−L2/Fcの両方のCHO−hPD1細胞への結合は、検査した任意のヒト化抗体によって用量依存的に完全に抑制し得る。算出されたIC50データを表Vにまとめる。親マウス抗体hPD−1.09Aと同様に、ヒト化mAb、h409A11、h409A16およびh409A17のそれぞれは、PD−L1およびPD−L2結合を1nMより低いIC50値で抑制した。マウス親抗体と同様に、ヒト化抗PD−1抗体h409A11、h409A16およびh409A17は、約1nMより低く算出されたIC50でカニクイザルPD−1へのリガンド結合の抑制を示した。
【0185】
【表5】
SEBへのヒトT細胞の応答はヒト化mAbによって増強される。
【0186】
ヒト化抗PD−1抗体をT細胞活性を増強させるそれらの能力についてin vitroで、実施例3に記載のとおり健常人ボランティア由来の血液細胞を使用して検査した。上清を培養4日後に回収し、産生されたIL−2の量をELISAを使用して定量した。ヒト化PD−1抗体は、SEBによって刺激されたIL−2産生を増大させる能力を示した(図7)。追加的にヒト化PD−1抗体は、実施例3において記載されたのと同様に、癌患者の血液においてSEB誘発IL−2産生を増大させた。
【0187】
要約として、ヒト化mAb h409A11、h409A16およびh409A17は、ヒト化過程において全ての機能的活性を保持した。h409A11およびh409A16mAbは、ヒト化においてマウスの親抗体hPD109Aの親和性を全て保持した。
【特許請求の範囲】
【請求項1】
明細書中に記載の発明。
【請求項1】
明細書中に記載の発明。
【図1】

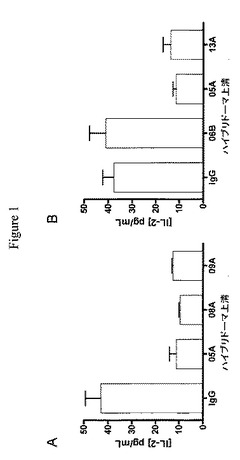
【図2】
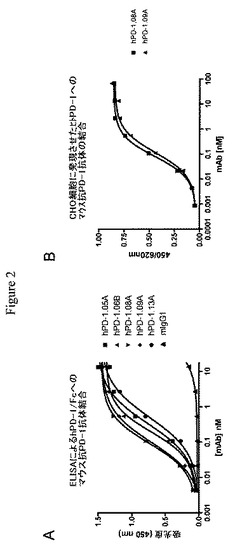
【図3】
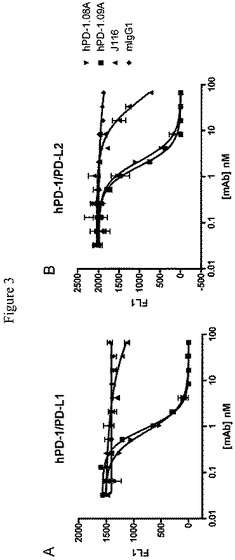
【図4】
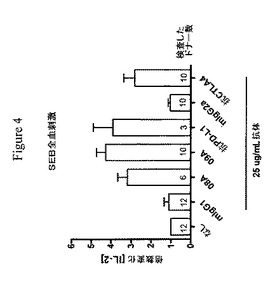
【図5】
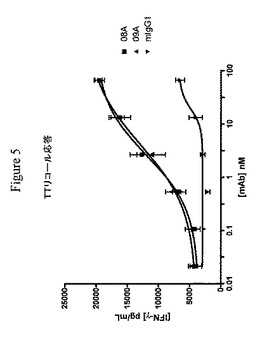
【図6】
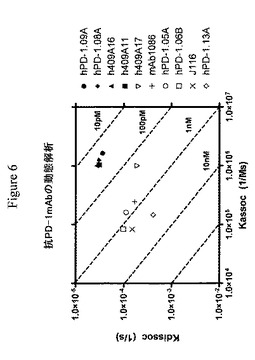
【図7】
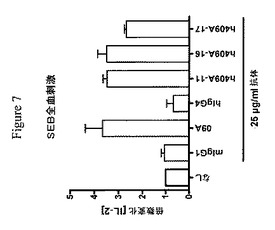
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2012−254092(P2012−254092A)
【公開日】平成24年12月27日(2012.12.27)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−185573(P2012−185573)
【出願日】平成24年8月24日(2012.8.24)
【分割の表示】特願2010−513201(P2010−513201)の分割
【原出願日】平成20年6月13日(2008.6.13)
【出願人】(512059936)エム・エス・ディー・オス・ベー・フェー (24)
【Fターム(参考)】
【公開日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願番号】特願2012−185573(P2012−185573)
【出願日】平成24年8月24日(2012.8.24)
【分割の表示】特願2010−513201(P2010−513201)の分割
【原出願日】平成20年6月13日(2008.6.13)
【出願人】(512059936)エム・エス・ディー・オス・ベー・フェー (24)
【Fターム(参考)】
[ Back to top ]