
ヒトアンチトロンビンIII変異体
【発明の詳細な説明】
【0001】
【産業上の利用分野】ヒトアンチトロンビンIII (AT III)のアミノ酸配列中、1個または複数個のアミノ酸を他のアミノ酸に変換させることにより、ヘパリン非存在下でも高い抗プロテアーゼ活性を有するAT III変異体に関するもので、血栓性疾病の治療薬として利用される。
【0002】
【従来の技術】ヘパリンをはじめとするグリコサミノグリカンの有する血液凝固阻害活性は、血中のアンチトロンビンIII (AT III)およびヘパリンコファクターII(HCII)により媒介される。AT IIIおよびHCIIは、いわゆるセルピンと総称されるセリンプロテアーゼインヒビターである。これらのうちAT IIIは先天的あるいは後天的な理由による血中レベルの低下により血栓症が発症するという報告が多く、一連のセリンプロテアーゼにより構成される血液凝固系の調節因子として生理的に重要な役割を担っている。
【0003】ヒトAT IIIは主に肝臓で合成され正常血漿中に約 150μg /mlの濃度で存在する、分子量約 60kd の糖蛋白であり、トロンビンおよびXa因子をはじめとする凝固、線溶系に携わるセリンプロテアーゼを阻害することが知られている。ヒトAT IIIの一次構造は、アミノ酸配列の直接決定(Petersen, T. E. et al., In: The Physiological Inhibitors of Blood Coagulation and Fibrinolysis, Elsevier Science Publishers, Amsterdam, 43, 1979)ならびにcDNAのクローニング(Bock, S. C. et al., Nucl. Acids Res., 10, 8113, 1982 ; Prochownik, E.V. et al., J. Biol. Chem., 258, 8389, 1982 ; Chandra, T. et al., Proc. Natl. Acad. Sci. USA, 80, 1845, 1983)により明らかにされている。これらの報告によると、ヒトAT IIIは前駆体蛋白質より32残基のシグナルペプチドが切断除去されて分泌生成される、 432アミノ酸からなる一本鎖糖蛋白である。分子内にN結合性の糖鎖付加を受ける部位を4ケ所含んでおり、分子量の約15%は糖である。
【0004】AT IIIはトロンビンなどのセリンプロテアーゼと1:1で反応し、安定な複合体を形成することにより、これらのプロテアーゼの活性を阻害する。この際AT IIIは分子中の 393番目のArg 残基と 394番目の Ser残基の間のペプチド結合がプロテアーゼにより切断され、この結果新たに生じた末端のArg 残基とプロテアーゼの活性中心の Ser残基との間にアシル結合が生じると考えられている。一般的に、この Arg (393)−Ser(394)を反応部位と呼ぶ。
【0005】AT IIIによるプロテアーゼの阻害反応は比較的緩やかに進行するが、反応系にへパリンが存在すると、その反応速度は劇的に加速され、AT IIIによるトロンビンの阻害速度はヘパリンの添加により1000倍以上となる。この作用機構はヘパリンがAT III中の特定部位(ヘパリン結合部位)に結合することにより、AT IIIの高次構造に変化をもたらしプロテアーゼと相互作用しやすい構造をとらせるとともに、プロテアーゼもそのヘパリン分子に結合することにより3者複合体を形成しやすくなるためと考えられている。また生理的には血管内皮細胞表面に存在するヘパリン様物質が、同様の作用を現すことによりAT IIIによる血液凝固系の制御機構に重要な役割を担っていると考えられている。
【0006】種々の原因により引き起こされる血栓症の予防、治療にはいわゆる抗凝固薬が使用されており、ヘパリンは現在でも極めて重要な抗凝固薬の一つである。しかしヘパリンの使用により時に重篤な副作用が生じることが報告されている(Amerena, J. et al., Adverse Drag React. Acute. Poisoning Rev., 9, 1, 1990 ;Levine, M. N., et al. Semi. in Thrombos. Hemostas., 12, 39, 1986 ; Kelton, J. G. et al., ibid, 12, 59, 1986 ; Levine, M. N., ibid, 12, 63, 1986)。代表的なものとして出血、血小板減少症、副腎機能障害、過敏症、投与部位の壊死、骨粗鬆症等があげられる。このため産婦人科領域あるいは外科手術後など出血の危険性が高い場合、あるいは長期にわたる投与においてはその使用は慎重に行われるべきである。さらにヘパリンは試験管内で好中球のエラスターゼによるAT IIIの不活性化を促進するという報告もあり(Jordan, R. E. et al, Science, 237, 777, 1987 ; Jordan, R. E. et al., J. Biol. Chem., 264, 10493,1989)、重篤な感染症あるい敗血症などの好中球のエラスターゼが病態に関与していると考えられる場合の投与においては考慮されるべきである。またヘパリンの抗凝固作用はあくまでもAT IIIを介したものであり、血中のAT III濃度が低下しているような病態ではその効果は期待されない。
【0007】一方、ヒトAT IIIは血漿由来の濃縮製剤という形で、先天的なAT III欠乏に基づく血栓形成傾向、およびAT III低下を伴う汎発性血管内凝固症候群(DIC)において臨床的に用いられている。しかし先に述べたように、ヘパリン非存在下におけるAT IIIの抗凝固活性は漸進的なもので、単独の使用は補充療法的な意味合いが強く、抗凝固薬としての有用性は限定される。そこでAT IIIの抗凝固薬としての有用性を高めるために、AT IIIとヘパリンの併用あるいはAT IIIとヘパリンの複合体を調製し、これを用いるといった方法が検討されている。しかしこれらの方法においても前述のヘパリンの有する欠点を除くことができないことは明らかである。
【0008】上記の如く、AT IIIには反応部位およびヘパリン結合部位という2つの機能部位が存在する。反応部位近傍のアミノ酸配列はプロテアーゼインヒビターとしての機能発現に重要な役割を担っているとともに、各種プロテアーゼに対する阻害特異性を決定する上でも重要であることが多くの報告により明らかとなっている。例えば先天性AT III異常症のなかで 382位のAla が Thrに置換したAT III Hamilton (Devraj-Kizuk, R. et al., Blood, 72, 1518, 1988) 、 384位のAla が Proに置換したAT III Cambridge I (Perry, P. J. et al., FEBS Lett, 254, 174, 1989) 、 393位のArg がHis に置換したAT III Glasgow (Erdjument, H. et al., J. Biol. Chem., 263, 5589, 1988)、同じくPro に置換したAT III Pescara (Lane, D. A. et al., J. Biol. Chem., 264, 10200, 1989)、 394位のSer がLeu に置換したAT III Denver(Stephens, A. W. et al.,J. Biol. Chem., 262, 1044, 1987)などにおいては異常AT III分子はいずれも抗プロテアーゼ活性を失っており、これらの患者は血栓症を発症している。
【0009】一方、ヘパリン結合部位すなわちヘパリンとの結合に直接関わっているアミノ酸は、やはりAT IIIの先天性分子異常症に関する研究およびアミノ酸残基の化学修飾の結果より明らかとなってきた。分子異常症では7位のIle が Asnに変換したAT III Rouen III (Brennan, S. O. et al., FEBS Lett., 237, 118, 1988) 、24位のArg が Cysに置換されたAT III Rouen IV (Borg, J. Y. et al., FEBS Lett., 266, 163, 1990) 、41位のPro が Leuに置換されたAT III Basel (Chang, J. Y. and Tran, T. H., J. Biol. Chem., 261, 1174, 1986)、47位のArg が Cysに置換されたAT III Toyama (Koide, T. et al., Proc. Natl. Acad. Sci. USA, 81, 289, 1984) 、 129位のArg が Glnに置換されたAT III Geneva (Gandrille, S. et al., J. Biol. Chem., 265, 18997, 1990)等が報告されている。これらの異常AT IIIではいずれもヘパリン親和性が低下しており、生理的に正常な機能を持ち得ないために血栓症を発症している。さらにアミノ酸の化学修飾の実験より49位のTrp 、114 位の Lys, 125位の Lys、 129位の Arg、 136位の Lys、 145位の Arg等のアミノ酸がヘパリンとの結合に直接関与していると考えられている(Blackburn, M. N. et al., J. Biol. Chem., 259, 939,1984 ; Peterson, C. et al., J. Biol. Chem., 262, 8061, 1987 ; Sun, X. J.and Chang, J. Y., Biochemistry, 29, 8957, 1990 )。
【0010】これらの知見をもとにこれまでAT IIIのアミノ酸を変換することによりATIIIの改良が試みられている。たとえば Zettlemeissl らはAT III中の糖鎖付加部位のアミノ酸を変換することによって、ヘパリン結合/ヘパリン活性化の性質を向上させるAT III変異体、および反応部位のアミノ酸を変換することにより酵素特異性を変化させたAT III変異体の製造法を開示している(特開平2−262598)。また Dijkemaらは反応部位のアミノ酸を変換することによって抗トロンビン/抗Xa活性の変化したAT III変異体の製造法を示した(EP90/01026)。しかしながら、臨床的に満足しうるAT III変異体は見いだされておらず、ヘパリン非存在下でのトロンビンあるいはXa因子阻害活性を上昇させたAT III変異体の作製が強く望まれている。
【0011】
【発明が解決しようする課題】本発明はヘパリン非存在下でも高い抗トロンビン活性を有する、新規なAT III変異体を提供することにある。さらに遺伝子組換え法による大量製造法をも提供することにある。
【0012】
【課題を解決するための手段】ヘパリンによるAT IIIの抗プロテア−ゼ活性増強のメカニズムは現在次のように考えられている。まずヘパリンがAT IIIのヘパリン結合部位に結合することにより、AT IIIの立体構造をプロテア−ゼとより反応しやすい構造へと変換する。同時にプロテア−ゼもそのヘパリン結合部位において同一のヘパリン分子と結合することにより、結果的にAT IIIとプロテア−ゼの複合体形成速度が上昇する(Pletcher,C.H. and Nelsestuen,G.L.,J.Biol.Chem., 258,1086,1988)。この仮説に従うと、AT IIIのヘパリン結合部位にヘパリンが結合することにより引き起こされる、反応部位の立体構造の変化が、抗プロテア−ゼ活性の増強において重要であると考えられる。このことは反応部位近傍のアミノ酸配列を人為的に変換することで反応部位の立体構造を変化させることにより、ヘパリン非存在下でのプロテア−ゼ活性が上昇したAT III変異体を製造することが可能であることを示している。
【0013】一方このような考えに基づきヘパリン非存在下の抗トロンビン活性が上昇したAT III変異体が得られたならば、そのAT III変異体にとってヘパリンと結合するということは重要な性質とはなり得ないと考えられる。したがってヘパリン結合部位に変異を来したために機能異常となった前述のAT III TOYAMA、 GENEVA などと異なり、ヘパリン結合部位にアミノ酸置換を導入しヘパリン親和性を低下させても機能的に影響は少ないと考えられる。むしろ血管内皮細胞表面のヘパリン様物質との相互作用が低下することにより、血中半減期の延長、好中球エラスタ−ゼによる失活の回避などがもたらされ、臨床的な有用性が増すことも考えられる。このような考えに基づき、本発明者らはAT IIIの改良を鋭意研究の結果、目的とする新規なAT III変異体作製に成功し本発明を完成した。
【0014】本発明は、変異させたヒトアンチトロンビンIII (AT III)であって、(1)11-14 位、41-47 位、125-133 位および 384-398位の四つの領域のアミノ酸が、それぞれの領域、単独で又は組み合わせで少なくとも一個、他のアミノ酸に変換されていることを特徴とするAT III変異体。
(2)384-398 位の領域のアミノ酸が少なくとも一個、他のアミノ酸に変換され、さらに 11-14位、41-49 位および 125-133位の三つの領域のアミノ酸が、各領域単独又は組み合わせで少なくとも一個、他のアミノ酸に変換された組み合わせであることを特徴とする(1)記載のAT III変異体。
(3)384-398 位の領域のアミノ酸が少なくとも一個、他のアミノ酸に変換され、さらに 11-14位および 41-47位の二つの領域のアミノ酸が、各領域単独又は組み合わせで少なくとも一個、他のアミノ酸に変換された組み合わせであることを特徴とする(1)記載のAT III変異体。
(4)384-398 位の領域のアミノ酸が少なくとも一個、他のアミノ酸に変換され、さらに 11-14位および 125-133位の二つの領域のアミノ酸が、各領域単独又は組み合わせで少なくとも一個、他のアミノ酸に変換された組み合わせであることを特徴とする(1)記載のAT III変異体。
(5)384-398 位の領域のアミノ酸が少なくとも一個、他のアミノ酸に変換され、さらに 41-47位および 125-133位の二つの領域のアミノ酸が、各領域単独又は組み合わせで少なくとも一個、他のアミノ酸に変換された組み合わせであることを特徴とする(1)記載のAT III変異体。
【0015】(6)11-14 位および 384-398位の二領域のアミノ酸がそれぞれの領域において少なくとも一個、他のアミノ酸に変換された(1)記載のAT III変異体。
(7)41-47 位および 384-398位の二領域のアミノ酸がそれぞれの領域において少なくとも一個、他のアミノ酸に変換された(1)記載のAT III変異体。
(8)125-133 位および 384-398位の二領域のアミノ酸がそれぞれの領域において少なくとも一個、他のアミノ酸に変換された(1)記載のAT III変異体。
(9)384-398 位の領域のアミノ酸が少なくとも一個、他のアミノ酸に変換された(1)記載のAT III変異体。
(10)四つの領域のアミノ酸がAla, Gly,Trp, Pro, Leu, Val, Phe, Tyr, Ile,Glu, Ser, Gln, Asn および Argから選ばれるアミノ酸に変換されている(1)記載のAT III変異体。
【0016】(11)384-398 位の領域のアミノ酸がAla, Pro, Leu, Val, Gly, Arg, Glu および Pheから選ばれるアミノ酸に変換され、さらに他の領域も変換されていてもよい(1)記載のAT III変異体。
(12)390-392 位の領域のアミノ酸がAla, Pro, Leu, Val, および Pheから選ばれるアミノ酸に変換され、さらに他の領域も変換されていてもよい(1)記載のAT III変異体。
(13)392 位Gly が Proに変換され、さらに他の領域も変換されていてもよい(1)記載のATIII 変異体。
(14)390 位 Ileが Alaに、 391位 Alaが Phe, Val または Leuに、および 392位Gly がPro に、単独または組み合わせで変換され、さらに他の領域も変換されていてもよい(1)記載のAT III変異体。
(15)384 位 Alaが Glyに、387 位Ala が Pheに、389 位 Valが Proに、397 位Proが Argに、または 398位 AsnがGlu または Argに、単独または組み合わせで変換され、さらに他の領域も変換されていてもよい(1)記載のAT III変異体。
【0017】(16)11位 Lysが Ileに、14位 Aspが Serに、単独または組み合わせで変換され、さらに他の領域も変換されていてもよい(1)または(14)記載のAT III変異体。
(17)125 位 Lysが Glnに、129 位 Argが Glnに、 132位 Argが Glnに、または133位 Lysが Asnまたは Glnに、単独または組み合わせで変換され、さらに他の領域も変換されていてもよい(1)または(14)記載のAT III変異体。
【0018】(18)下記アミノ酸に変換された変異体から選択される(1)記載のAT III変異体。
(a)392 位 Glyが Proに変換されたAT III変異体。
(b)391-392 位 Ala-Glyが Phe-Proに変換されたAT III変異体。
(c)390-391 位 Ile-Alaが Ala-Leuに変換されたAT III変異体。
(d)125 位 Lysが Glnに、 391-392位 Ala-Glyが Phe-Proに変換されたAT III変異体。
(e)132-133 位 Arg-Lysが Gln-Asnに、390-391 位 Ile-Alaが Ala-Leuに変換されたAT III変異体。
(f)132-133 位 Arg-Lysが Gln-Asnに、391-392 位 Ala-Glyが Phe-Proに変換されたAT III変異体。
(g)133 位Lys がAsn に、391-392 位 Ala-Glyが Phe-Proに変換されたAT III変異体。
【0019】(19)上記のATIII 変異体をコードするDNA、それらDNAを組み込んだ発現可能なベクター、これらベクターにより形質転換された宿主細胞、宿主細胞を用いるAT III変異体の製造方法およびこれらAT IIIを用いる医薬組成物に関する。
【0020】これらのAT III変異体はいずれも動物細胞を宿主として発現され製造され、得られた変異体は以下に示すようにヘパリン非存在下の抗トロンビン活性がヒト血漿由来のAT IIIあるいは天然型の組換えAT IIIにくらべ上昇していた。また動物実験においてもヒト血漿由来のAT IIIにくらべ強い薬効を示し、臨床上の有用性が高いものと期待される。
【0021】以下に本発明を詳細に説明する。
1)ヒトAT III cDNAの単離ヒトAT IIIは主に肝臓において合成されるので、市販のヒト肝臓cDNAライブラリー(λgt 11 、クローンテック社)を用いればよい。クローニングする方法は公知の方法、例えばヒトAT IIIアミノ酸配列に対応する合成オリゴヌクレオチドをプローブとして用いるプラークハイブリダイゼイション法(Sambrook, J. et al., Molecular Cloning, Cold Spring Harbor Laboratory, 1989 )などが挙げられる。得られたクローンは必要に応じて、例えば pUC 18 などのプラスミドにサブクローニングする。このようにして得られたcDNAの塩基配列はマキサムギルバート法(Maxam, A. M. and Gilbert, W., Proc. Natl. Acad. Sci. USA, 74, 560, 1977)またはジデオキシ法(Sanger, F., Science, 214, 1205, 1981)によって決定、確認することができる。得られたAT IIIcDNAのコ−ディング領域の塩基配列とそれに基づいて演繹されたアミノ酸配列は配列番号1に示した。
【0022】2)部位特異的変異導入法変異導入方法は、例えばZollerらの方法(Zoller,M. and Smith,M., Methodsin Enzymology, 100, 468, 1983)、Kramerらの方法(Kramer,W. and Fritz,H-JMethods in Enzymology, 154, 350, 1987)およびVandeyarらの方法(Vandeyar,M.A. et al., Gene., 65, 129, 1988) などが挙げられる。Kramerらの方法はgapped duplex 法と呼ばれるもので、 M13ファージのアンバー変異体M13tv18 、M13tv19 などをベクターとして用いることができる。これらのベクターにAT IIIをコードするDNAをクローニングし、この一本鎖DNAとアンバー変異の入っていない M13の二本鎖DNAの断片( M13mpP をPvu IIにて切断して得たベクター断片)とを変性後アニーリングさせgapped duplex DNAを得る。次にこのDNAに導入したい変異を含む合成オリゴヌクレオチドをハイブリダイズさせ、DNAポリメラーゼとDNAリガーゼを作用させることによってギャップを埋めた後、大腸菌のmutS株(BMH71-18mutS)にトランスフェクションし、supOの大腸菌でのみ増殖できるノンアンバーファージを選択することにより目的とする変異が導入されたファージを効率よく得ることができる。実際の操作には市販のキットを用いてもよい(宝酒造;Mutan-G)。一方、Vandeyarらの方法はAT IIIをコードするDNAをクローニングした M13の一本鎖DNAに、導入したい変異を含むオリゴヌクレオチドをハイブリダイズさせる。これを鋳型としdATP、 dGTP、 dTTP、 および5-methyl-dCTP を基質として T7 DNAポリメラーゼを作用させ二本鎖DNAを合成した後、 T4 DNAリガーゼを用いて閉環状の二本鎖DNAとする。つぎにこの二本鎖DNAを制限酵素 Mspl で処理したのち、エキソヌクレアーゼIII にて処理することにより変異が導入された鎖のみからなる環状一本鎖DNAを得る。これをメチル化DNAに特異的な制限システムを持たない大腸菌(SDM株)に導入することにより目的とするクローンを効率よく得る。こちらの方法でも実際の操作には市販のキットを用いてもよい(United States Biochemical Corporation ; T7-GEN In Vitro Mutagenesis Kit)。導入したい変異を含む合成オリゴヌクレオチドはDNA合成機(ABI社 380A)を用いてフォスフォアミダイト法にて合成することができる。
【0023】3)AT IIIcDNA変異導入用鋳型の調製上記1)で得たAT IIIcDNAのコ−ディング領域の前後に制限酵素切断部位を導入し、変異導入のための鋳型を調製する。制限酵素としては公知のものから適宜選択すればよく、本発明の場合はATIII コ−ディング領域の直前にHindIII切断部位、直後にBgl II切断部位を導入した。まず上記1)で得たAT IIIcDNAを含むプラスミドをEcoRIにて切断し、AT III全コーディング領域を含む 1.5kbの断片を得る。この断片をファージM13tv18のRF(Replicative Form、二本鎖DNA)をEcoRIにて切断し開環したものに導入する。こうして得られたクローンのうちAT IIIのセンス鎖を含む一本鎖DNAを鋳型としてKramerらの方法に従い、Hind IIIおよびBgl IIの酵素切断部位をそれぞれ含む2種の合成オリゴヌクレオチドをプライマーとして、制限酵素切断部位をAT IIIcDNAのコ−ディング領域の前後に導入する。次いで、このようにして得られたクローンからAT IIIcDNA配列を含む。断片を適切なプラスミドに挿入し、変異導入のための鋳型を調製する。
【0024】本発明の場合は、前記クローンをHind IIIおよびEcoRIにて切断して得られる約 1.5kbのAT III全コーディング配列を含むDNA断片を同酵素にて切断したプラスミドM13tv19RFあるいはM13mp19に挿入することにより、変異導入の鋳型を調製することができる。さらに、AT IIIのcDNAには1ケ所制限酵素 SacI認識部位(配列番号1の 721〜726 位の塩基部分)が存在し、反応部位とヘパリン結合部位を分断しうることから、上記クローンをHind IIIおよび SacIにて切断して得たAT IIIのN端側、すなわちヘパリン結合部位を含むDNA断片を同じ酵素で切断したプラスミドM13tv19またはM13mp19などに導入し、ヘパリン結合部位への変異導入用鋳型を調製することができる。反応部位についてもEcoRIおよび SacIを用い、同様な操作で可能である。
4)目的とする位置への変異導入AT IIIアミノ酸配列において、変換させたい位置のアミノ酸を目的とする他のアミノ酸(以下、目的アミノ酸と称す)に変換させるには、前記の公知の方法に従い目的アミノ酸をコードするDNAを含む合成オリゴヌクレオチドと、3)に記載した適当なプラスミドを鋳型として用いることにより実施することができる。例えばAT III 392位 Glyを Proに変換させる時は、表1記載のAT1Rオリゴヌクレオチドを、 391-392位 Ala-Glyを Phe-Proに変換させる時は、表1記載のAT5Rオリゴヌクレオチドを用いればよい。また各部位での変換させるアミノ酸が複数個でそれぞれの位置が離れている場合は、1個ずつ順に変異導入の操作を行うことにより、いくつでも変異を導入することができる。
【0025】本発明において使用したオリゴヌクレオチドの代表例を表1および表2に、アミノ酸変換部位と目的アミノ酸を表3および表4に記載した。目的アミノ酸をコードする塩基コドンは表1および表2記載のコドンに限定されるものではなく、目的アミノ酸をコードするコドンであれば、いずれも使用できる。
【0026】
【表1】

【0027】
【表2】
【0028】
【表3】
【0029】
【表4】
【0030】5)反応部位近傍の変異とヘパリン結合部位の変異の組み合わせ前記の如く、AT IIIcDNAには1ケ所 SacI切断部位が存在し、これが反応部位とヘパリン結合部位の間に位置する。それゆえに上記4)に記載された方法により得られた変異AT IIIDNAを含むプラスミドをHind IIIおよび SacIまたは SacIおよび BglIIにて切断することにより、ヘパリン結合部位を含む断片と反応部位を含む断片を得ることができる。反応部位またはヘパリン結合部位を変異させたAT III DNAを制限酵素にて処理し、反応部位変異DNA断片とヘパリン結合部位変異DNA断片を調製し、それぞれの変異DNA断片を適切なプラスミドに連結することにより両部位を変異させたAT III変異DNAの調製ができ、この方法により両部位の変異のいかなる組合せも可能である。両変異DNA断片を連結構築するプラスミドは宿主での発現に適しているものならばいずれも使用でき、例えばpSV2、pK4Kなどが挙げられる。表4に記載した記号で、2G35R とは 2G 変異DNA断片と35R 変異断片の組み合せにより得られる変異体を意味する。
【0031】6)AT III変異体組換え発現ベクターとその形質転換体上記記載の方法により得られたAT III変異体をコ−ドするDNAを適切なベクターに組み込み、該ベクターを適切な宿主細胞に移入することにより形質転換体を得ることができる。これを常法により培養し、培養物よりAT III変異体を大量に生産することができる。AT III変異体をコードするDNAをAT III変異体の発現に適したベクターのプロモーター下流に制限酵素とDNAリガーゼを用いる公知の方法により再結合して組換え発現ベクターを作製することができる。ベクターは宿主内で複製、増幅可能であれば特に限定されない。プロモーターおよびターミネーターに関してもAT III変異体をコードする塩基配列の発現に用いられる宿主に対応したものであれば特に限定されず、宿主に応じて適切な組み合わせも可能である。
【0032】このようにして得られた組換え発現ベクターはコンピテントセル法(Hanahan,D., J. Mol. Biol., 166, 557, 1983 )、リン酸カルシウム法( Wigler,M. etal., Cell, 11, 222, 1977) などにより宿主に導入し、形質転換体が作製される。宿主としては大腸菌および動物細胞などが用いられ、得られた形質転換体はその宿主に応じた適切な培地中で培養される。培養は通常20℃〜45℃、pH5〜8の範囲で行われ、必要に応じて通気、攪拌が行われる。培養物からのAT III変異体の分離、精製は公知の分離、精製法を適宜組み合わせて実施すれば良い。これらの公知の方法としては塩析、溶媒沈殿法、透析、ゲル濾過法、電気泳動法、イオン交換クロマトグラフィ、アフィニティクロマトグラフィー、逆相高速液体クロマトグラフィなどが挙げられる。このようにして得られたAT III変異体はヘパリン非存在下で天然型AT IIIより高い抗トロンビン活性を有し、ラットでのin vivo抗血栓作用においても天然型AT IIIより強い作用を示した。
【0033】
【発明の効果】
(1)抗トロンビン活性テストチームAT III2キット(第一化学薬品)を利用して、本発明のAT III変異体の抗トロンビン活性を測定した。すなわち、トロンビンの合成基質(S−2238)を用いて、トロンビンに対する阻害活性をヘパリンの非存在下において測定した。対照としてAT III血漿濃縮製剤(アンスロビンP;ヘキストジャパン社)を用いた。本測定において、緩衝液として 0.1%ウシ血清アルブミンおよび0.15M塩化ナトリウム含有 50mM トリス塩酸緩衝液(pH7.5 )を用い、種々の濃度に調製した検体と一定量のトロンビン(ウシ由来)を37℃で5分間反応させた。反応後、合成基質S−2238を添加し2分間に遊離してくるp−ニトロアニリン量を波長 405nmの吸光度変化で測定することにより、残存するトロンビン活性を求めた。この条件下、トロンビンの活性を50%阻害するAT III変異体の濃度(以下IC50)を算出した。表5に各変異体のIC50 値を示した。ヘパリン非存在下におけるヒトAT III濃縮製剤のIC50 値は13.0×10-8M であり、天然型組換えATIII もほぼ同じ値を示した。これに対し、本発明のAT III変異体のIC50 値は明らかに低い値を示し、ヘパリン非存在下の抗トロンビン活性の上昇が認められた。
【0034】
【表5】
【0035】(2)ヘパリン親和性高速液体クロマトグラフィ法によって、ヘパリン−5PW (7.5mm ×75mm:東ソウ)を用いて、本発明のAT III変異体について、ヘパリンに対する親和性の強さを比較検討した。すなわち、移動相に50mMトリス塩酸緩衝液(pH7.5 )を用い、流速1ml/minで30分間に塩化ナトリウム濃度を0M から2M 濃度まで直線的に上昇させた。検出は、波長 280nmの吸収で行い、検体が溶出されてくるまでの時間で比較した。表6に示したように、ヒトAT III及び天然型組換えAT IIIの主ピークが溶出されるまでの時間は22.3分と23.1分であり、大きな差は認められなかった。また、反応部位近傍の変異体においてもヒトAT IIIおよび天然型組換えAT IIIと比較して顕著な差は認められなかった。一方、反応部位近傍とヘパリン結合部位の両部位変異体については、いずれも主ピークが溶出されるまでの時間が有意に短縮しており、ヘパリン結合部位の変異を導入することによりヘパリンに対する親和性が低下することが確認された。
【0036】
【表6】
【0037】(3)AT III変異体の抗血栓作用血漿由来AT III濃縮製剤(アンスロビンP、ヘキストジャパン社)および天然型組換えAT IIIを対照として、本発明のAT III変異体の抗血栓作用を以下のように測定した。方法は Peters ら(Peters, R. F. et al., Thromb. Haemostas., 65, 268、1991)の報告に改良を加えて行った。すなわち、麻酔したSprague-Dawley系雄ラット(200 〜300g)の頸動静脈に生理食塩液を満たしたアトム静脈カテーテル(4Fr, 3.5cm、アトム社)をカニュレーションし、shunt を作製した。血流を遮断した状態で shuntの動脈側に脈波ピックアップ(MPP-3 、日本光電)を装着し、血流の変化をポリグラフ記録計で実験中モニターした。計算量の検体材料を1mlになるように生理食塩液で希釈して同ラットの大腿静脈より単回急速投与した後、shunt を開いて血流を開通させた。shunt を開いてから、shunt 内に血栓が形成されて閉塞するまでの時間を測定し閉塞時間とした。結果を表7および表8に示す。これにより本発明のAT III変異体は血漿由来AT III濃縮製剤および天然型組換えヒトAT IIIに比較して強い抗血栓作用を有することが判明した。
【0038】
【表7】
【0039】
【表8】
【0040】以上の結果から、本発明のAT III変異体は血液凝固阻止剤として血栓形成を抑制し、血栓性疾患の予防治療薬として期待される。
【0041】(4)AT III変異体の実験的汎発性血管内凝固症(DIC)モデルにおける効果血漿由来AT III濃縮製剤を対照として、本発明のAT III変異体の実験的汎発性血管内凝固症(DIC)モデルにおける効果を以下のように検討した。 方法は、杉島らの報告(杉島忠志ら, 臨床と研究, 62, 274, 1985 )に改良を加えて行った。すなわち、麻酔したSprague-Dawley系雄性ラット(200 〜300g)の頸静脈にアトム静脈カテーテル(3Fr ,アトム社)をカニュレーションし、組織トロンボプラスチン(トロンボレルS,ベーリングベルケ社)を1時間かけて持続投与することによって、モデルを作製した。試験検体は、組織トロンボプラスチンの投与を開始する直前に同ラットの大腿動脈より単回急速投与した。組織トロンボプラスチン投与終了30分後に、下行大動脈より3.8%クエン酸ナトリウムを1/10量加えて採血した。採血後、直ちに血液0.5ml を自動血球数装置用採血容器(東亞医用電子株式会社)に分取し、TECHNICON 社 H・1 System にて血小板数を測定した。残りの血液より遠心分離(3000rpm 10min )にて血漿を得て、血漿中のフィブリノーゲン量を測定した。血漿中フィブリノーゲン量は、トロンビン時間法(フィブリノーゲンB−テストワコー、和光純薬)にて測定した。結果を表9に示す。これより本発明のAT III変異体は、組織トロンボプラスチン誘発の実験的DICモデルにおいて、血小板数の減少および血漿中フィブリノーゲン量の低下に対して、血漿由来AT III濃縮製剤に比較して強い効果を示すことが判明した。以上の結果から、本発明のAT III変異体はDIC治療剤として期待される。
【0042】
【表9】
【0043】このAT III変異体は経口的、局所的、静注的、もしくは筋注的、皮下注的などにより投与することができるが、局所もしくは静注投与が好ましい。投与量は0.1〜 100mg/kg、好ましくは 0.5〜20mg/kgであり、体重に応じて1〜50mlの生理食塩水に溶解して用いる。また製剤の形としては、水和剤、水溶剤、錠剤、カプセル剤、粉剤、座剤などが使用でき、これら製剤の担体としては薬学的に許容される賦形剤、崩壊剤、滑沢剤、分散剤など通常の医薬品に使用されているものが用いられる。
【0044】
【実施例】以下の実施例により本発明を詳細に且つ具体的に説明するが、本発明はこれらの実施例に限定されるものではない。
【0045】実施例1ヒトAT IIIをコードするDNA配列のクローニング市販のヒト肝臓cDNAライブラリー(λgt 11 、クローンテック社)を原料として、常法に従いプローブとして32P標識合成オリゴヌクレオチドを用いてスクリーニングした。合成オリゴヌクレオチドの配列は Chandraらの報告をもとに、AT IIIの 314位から 322位のアミノ酸に対応する塩基配列部分とした。スクリーニングの結果、#2および#6という2種のクローンが得られた。それぞれのクローンより制限酵素EcoRI にてDNA断片を回収し M13mp18にサブクローニングし塩基配列を決定したところ、#2のクローンは33番目のアミノ酸に対応する配列から polyAまでの約 1.3kbを含み、一方#6のクローンは翻訳開始コドンから 348番目のアミノ酸に対応する約 1.1kbを含むことが確認された。次にこれらのクローンよりEcoRI にてインサートを切り出し、それぞれ EcoRIにて切断した pUC18にサブクローニングした。このようにして#2のクローンより pUC-Hを、#6のクローンより pUC-Lを作製した。次いでpUC-L をNcoIおよびHind IIIで切断し生じた約 3.7kbのDNA断片(pUC18 およびアンチトロンビンIII のN末側に対応する約 1.0kbの配列を含む)と、pUC-H を同じくNcoIおよびHind IIIで切断し生じた約 0.5kbのDNA断片(アンチトロンビン IIIのC末側に対応する配列を含む)を結合し、AT IIIの翻訳開始コドンより終始コドンまでの全コーディング配列を含むAT III FLpUCというプラスミドを得た。このプラスミドに含まれるAT IIIcDNAの開始コドンから終始コドンまでの全配列を配列番号1に示した。
【0046】実施例2制限酵素切断部位の導入実施例1で得られたプラスミドAT III FLpUCを材料としてAT IIIコーディング配列の直前に制限酵素 Hind III 切断部位、直後に制限酵素 BglII切断部位を導入したDNAを作製した。まず初めにAT III FLpUCをEcoRI にて切断し、AT III全コーディング領域を含む約 1.5kbの断片を得た。この断片を先に述べたM13tv18 のRFをEcoRI にて切断し開環したものに導入した。こうして得られたクローンのうちAT IIIのセンス鎖を一本鎖DNAとして与えるクローンを tvATRとした。次のこの tvATRの一本鎖DNAを鋳型とし、各酵素の切断部位をそれぞれ含む下記2種の合成オリゴヌクレオチドをプライマーとして、Kramerらの方法に従い制限酵素切断部位を導入した。
【0047】
実際の操作は市販のキット(宝酒造、Mutan G )を用いた。すなわち約 0.5μg の tvATRの一本鎖DNAと 0.2μg のキット添付 dsDNA(M13mp18 のマルチクローニングサイトを含む PvuII断片を欠失させたファージ M13mpP のRFDNA を PvuIIで切断し直鎖化したもの)を 20mM Tris ・ HCl pH8-10mM MgCl2-50mM NaCl-1mM DTT 中 100℃3分間、65℃10分間、37℃10分間静置し gapped duplexを形成させた。この1/10量をとり、T4ポリヌクレオチドキナーゼにて5'端をリン酸化したAT5H、AT3B各 5pmolと混和し(計3μl )65℃15分間、37℃15分間静置した。これにキット添付の緩衝液(50mM Tris ・ HCl pH8-60mM酢酸アンモニウム -5mM MgC12-5mM DTT-1mM NAD-0.5mM each dNTPs(A、C、G、T)25μl 、60U の E.coliDNAリガーゼ、1UのT4DNA ポリメラーゼを加え25℃約2時間静置した。3μlの0.2M EDTA pH8 を加え65℃5分間加熱したのち一部をとり Hanahanの方法(Hanahan, D., J. Mol. Biol., 166, 557, 1983)によって調製した大腸菌 BMH71-18mutS 株のコンピテントセルにトランスフェクションした。大腸菌MV1184株を指示菌として得られたプラークを拾い、常法に従い培養し RFDNAを調製した。このDNAを制限酵素 Hind III および BglIIにて切断し、新たな切断部位が生じているクローンにつきダイデオキシ法にて塩基配列を決定し、目的とする変異が導入されていることを確認した。こうして得られたクローンをAT5H3Bとした。
【0048】このAT5H3Bを Hind III および EcoRIによって切断し得られる約 1.5kbのDNA断片を、やはり Hind III および EcoRIにて切断し、開環した M13tv19RFに挿入したクローンを tv19-5H3Bとした。またプラスミド pSV2-dhfr(Lee, F. et al., Nature, 294, 228, 1981 ; Subramani, S. et al., Mol. Cell. Biol., 1,854, 1981 )をHind IIIおよび Bg1IIにて切断しマウスジヒドロ葉酸還元酵素(dhfr)をコードする領域を除いたDNA断片と、AT5H3Bをやはり Hind III と Bg1IIにて切断して得た、AT IIIの全コーディング配列を含むDNA断片とを結合しプラスミド pSV2-5H3Bを得た。さらに pSV2-5H3BをHind IIIおよび SacI にて切断して得たAT IIIのN端側をコードする約730bp のDNA断片を、やはりHind III および SacI にて切断し開環した M13tv19、および M13mp19に導入してそれぞれtv19-ATN、mp19-ATNとした。
【0049】実施例3a)1R変異体DNAの作製AT IIIの 392番目の Glyを Proで置換したAT III変異体1R(表3)をコードする配列を部位特異的変異導入法によって得た。すなわち、実施例2で得られたAT5H3Bの一本鎖DNAを鋳型としてKramerらの方法に従って、合成オリゴヌクレオチドAT1R(表1)を作用させることによって目的とするクローン 1Rmutを得た。操作は市販のキット(Mutan G )を用いて、実施例2に記載した方法と同様に行った。得られたプラークを12個拾って解析したところ、5個が目的とするクローンであった。得られたクローンの RFDNAを Hind III および BglIIにて切断し生じた約1.4kb のDNA断片を実施例2と同様プラスミド pSV2-dhfr中のマウスDHFR遺伝子と入れ換えプラスミド pSV2-1Rを構築した。
【0050】b)他の反応部位近傍変異体DNAの作製1R以外の反応部位近傍への変異導入は実施例2で得られた tV19-5H3Bを鋳型として上述のKramerらの方法に従って行った。この方法で5R、26R 、28R 、29R 、30R 、39R 、40R 、46R 、48R 、49R 、50R 、27R 、7R、34R 、35R 、38R 、9R、19R 、24R 、2R' 、5R' 、6R' の変異を導入した。それぞれのATIII 変異体の反応部位近傍のアミノ酸配列を表3に、変異導入に使用した合成オリゴヌクレオチドの配列を表1および表2に示す。操作は実施例2と同様にキット添付のマニュアルに従い変異導入反応を行ったのち得られたクローンを数個拾い、塩基配列を確認し目的とする変異が導入されたクローンを得た。それぞれのクローンより Hind III 、Bgl IIにて約1.4kb のDNA断片を得て、5R、26R 、28R 、30R、27R 、7R、19R 、24R 、2R' 、5R' 、6R' については実施例2および3a)と同様 pSV2-dhfrのマウスDHFR遺伝子と入れ換え、それぞれプラスミド pSV2-5R、pSV2-26R、pSV2-28R、pSV2-30R、pSV2-27R、pSV2-7R 、pSV2-19R、pSV2-24R、pSV2-2R'、pSV2-5R'、pSV2-6R'とした。また 39R、 40R、 46R、 48R、 49R、 50R、 34R、 35R、 38RについてはそれぞれのDNA断片を後述のプラスミドpK4K中のNKAF遺伝子の一部と入れ換え、それぞれプラスミド pK4K-39R 、 pK4K-40R 、pK4K-46R 、 pK4K-48R 、 pK4K-49R 、 pK4K-50R 、 pK4K-34R 、 pK4K-35R 、pK4K-38R とした。なお 29R、9Rについてはプラスミド pSV2-29R 、 pSV2-9RよりHind IIIおよび BglIIにて約 1.4kbのDNA断片を再び単離し、同様にプラスミド pK4K-29R 、pK4K-9R を構築した。
【0051】実施例4ヘパリン結合部位変異体DNAの作製ヘパリン結合部位の変異のうち1G、2G、8Gの変異導入は実施例2で得られた tv19-5H3Bを鋳型としてKramerらの方法に従って行った。用いた合成オリゴヌクレオチドの配列は表2に示す。目的とする変異が導入されたクローンをそれぞれ 1Gmut、2Gmut 、8Gmut とした。これらのクローンより Hind III 、Bgl IIにより約 1.4kbのDNA断片を切り出し、反応部位の変異の場合と同様にプラスミド pSV2-1G、pSV2-2G 、pSV2-8G を得た。また pSV2-1Gを Hind III および SacIにより切断し得られる約 730bpのDNA断片を、同じ酵素で切断した M13tv19に導入し tv19-1GN とした。1F, 2F, 3F, 7Gの変異導入は実施例2で得られたtv19-ATNを鋳型として同様に行った。目的とする変異が導入された M13クローンをそれぞれ 1Fmut, 2Fmut, 3Fmut, 7Gmut とした。
【0052】9Gの変異導入は mp19-ATN を鋳型として Vandeyar らの方法にて行った。実際の操作はキット(USB社;T7-GEN In vitro mutagenesis system)添付のマニュアルに従った。初めに1μg の mp 19-ATN一本鎖DNAと、T4ポリヌクレオチドキナーゼにて5'端をリン酸化した合成オリゴヌクレオチド AT9G 2pmol を40mMTris-HCl pH7.5-20mM MgC12-50mM NaCl中65℃5分間加熱後、室温となるまで徐冷した。次にこの反応液(10μl )に 10X Synthesis mix(100mM Tirs- HCl pH7.5-20mM DTT-5mM dATP-5mM dGTP-5mM dTTP-5mM 5-Methyl-dCTP-10mM ATP)を2μl 、2.5UのT7DNA ポリメラーゼ、5UのT4DNA リガーゼを加え最終液量20μl として37℃1時間静置した。この操作により変異の導入された鎖のみメチル化されたRFDNA が合成される。反応液を70℃10分間加熱して酵素を失活させたのち制限酵素MspIおよびHhaIを各5U加え37℃45分間反応させた。この操作でMspIにより二本鎖DNAのうち鋳型としてメチル化されていないDNA鎖にのみ切れ込みが入るとともに二本鎖DNAに変換されなかった鋳型の一本鎖DNAがHhaIにより切断される。次にこの反応液に50U のエキソヌクレアーゼIII を加え37℃45分間反応させることによって、切れ込みの入った鋳型鎖のみが分解され、結果として変異が導入されたDNA鎖が濃縮される。70℃10分間加熱し反応を止めた後、メチル化されたDNAに特異的な制限システムを持たない(mcrAB )大腸菌SDM株に常法でトランスフェクションした。得られたプラークを数個拾いDNAを得て塩基配列を決定することによって、目的とする変異の導入されたクローンを選択した。こうして得られたクローンよりHind IIIおよびSacIにて約 730bpのDNA断片を単離し、同じ酵素にて切断して同サイズの断片を除いた pSV2-5H3Bに導入して pSV2-9Gとした。
【0053】12G の変異は1Gの変異が導入されているDNAを鋳型としてAT2Gの合成オリゴヌクレオチド(表2)によってさらに変異を導入することによって得た。すなわちtv19-1GNを鋳型としてKramerらの方法によって行った。目的とする変異が導入されていることを確認したクローンを12Gmutとした。127Gの変異は12Gmutの一本鎖DNAを鋳型として、前記のVandeyarらの方法によって合成オリゴヌクレオチドAT7Gを作用させることによって得た。はやり目的とする変異が導入されていることを確認したクローンを127Gmut とした。
【0054】実施例5反応部位近傍とヘパリン結合部位の両部位変異体DNAの作製a)1G5R変異体DNAの作製ヘパリン結合部位の変異1Gに反応部位近傍の変異5Rを組み合わせた1G5R変異体のDNAは以下のように作製した。実施例4で得られた1Gmut のRFDNA をHind IIIおよびSacIにて切断し、約 730bpのヘパリン結合部位に変異を含むDNA断片を調製した。また実施例3で得られた pSV2-5RをSacIおよび BglIIにて切断し、約 670bpの反応部位近傍に変異を含むDNA断片を調製した。これらDNA断片を組み合わせて、Hind IIIおよびBgl II にてマウスDHFR遺伝子を除去した pSV2-dhfrに導入して pSV2-1G5Rを作製した。さらに、この pSV2-1G5RをHind IIIおよび Bgl II にて切断し生じた約1.4kbのDNA断片を、後述のプラスミド pK4K をHind IIIおよび BamHIにて切断しNKAF遺伝子の一部を除去したものに導入し、pK4K-1G5R を作製した。これら変異を含むDNA断片の調製および両変異DNA断片の組み合わせによる pSV2-1G5RおよびpK4K-1G5R の構築は公知の方法に準じて行った。プラスミド pK4K-1G5Rを含有する大腸菌(Escherichia coli)HB101-pK4K-1G5R は、通産省工業技術院微生物工業技術研究所に寄託された(受託番号 FERM BP-3806)。
【0055】b)2G5R変異体DNAの作製ヘパリン結合部位の変異2Gに反応部位近傍の変異5Rを組み合わせた2G5R変異体のDNAは以下のように作製した。実施例4で得られた2Gmut のRFDNA をHind IIIおよびSacIにて切断し、約 730bpのヘパリン結合部位に変異を含むDNA断片を調製した。また実施例3で得られた pSV2-5RをSacIおよび Bgl II にて切断し、約 670bpの反応部位近傍に変異を含むDNA断片を調製した。これらDNA断片を組み合わせて、Hind IIIおよび BglIIにてマウスDHFR遺伝子を除去した pSV2-dhfrに導入して pSV2-2G5Rを作製した。さらに、この pSV2-2G5RをHind IIIおよび BglIIにて切断し生じた約 1.4kbのDNA断片を、後述のプラスミド pK4K をHind IIIおよび BamHIにて切断しNKAF遺伝子の一部を除去したものに導入し、pK4K-2G5R を作製した。プラスミド pK4K-2G5Rを含有する大腸菌(Escherichia coli)HB101-pK4K-2G5R は、通産省工業技術院微生物工業技術研究所に寄託された(受託番号 FERM BP-3807)。
【0056】c)他の両部位変異体DNAの作製実施例3および4で得られた各部位変異体DNAを用いた。ヘパリン結合部位の変異を含むDNA断片として、 pSV2-1G、pSV2-2G 、pSV2-9G 1Fmut, 2Fmut,3Fmut, 7Gmut、12Gmut、127Gmut をそれぞれHind IIIおよびSacIにて切断して生じた約 730bpのDNA断片を用意した。また反応部位の変異を含むDNA断片として pSV2-1R、pSV2-5R (あるいはpSV2-1G5R )および pSV2-30R をSacIおよびBgl II にて切断して約 670bpのDNA断片を得た。さらに pK4K-35R をSacIおよび XhoIIにて切断し同じく約 670bpのDNA断片を得た(pK4KをHind IIIおよび BamHIにて切断しNKAF遺伝子の一部を除去したプラスミドに、AT III変異体遺伝子をHind III-BglII断片として導入したDNAでは、 BglII切断端と BamHI切断端とが連結される結果、再度 BglIIにて切断することができない。しかしこの部位は XhoIIにて切断可能である)。以上示したDNA断片を組み合わせて、pK4KをHind IIIおよび BamHIにて切断しNKAF遺伝子の一部を除去したプラスミドに導入することによってpK4K-1G30R、pK4K-1G35R、pK4K-2G30R、pK4K-2G35R、pK4K-1F5R, pK4K-2F5R, pK4K-3F5R, pK4K-7G5R、pK4K-7G30R、pK4K-7G35R、pK4K-9G5R 、pK4K-9G30R、pK4K-9G35R、pK4K-12G5R、pK4K-12G30R 、pK4K-12G35R 、pK4K-127G5R 、pK4K-127G30R、pK4K-127G35Rを構築した。またHind IIIおよび BglII にてマウスDHFR遺伝子を除去した pSV2-dhfrを用いて同様に pSV2-1G1R、pSV2-2G1R を構築した。
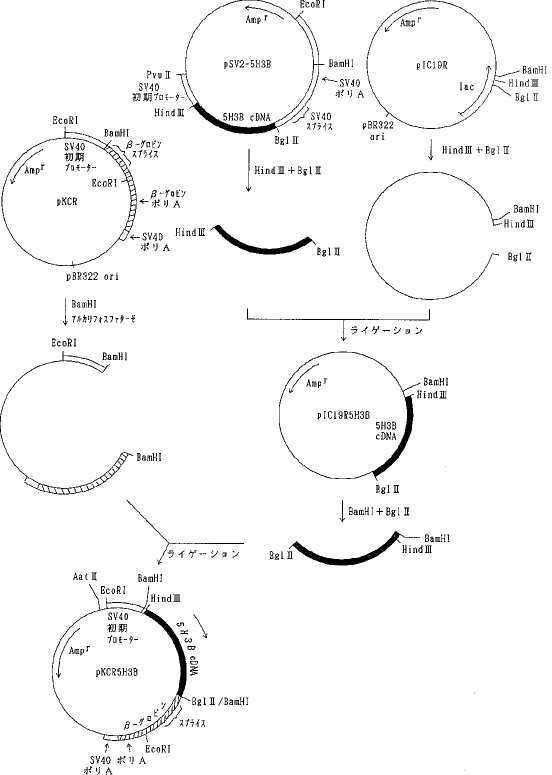
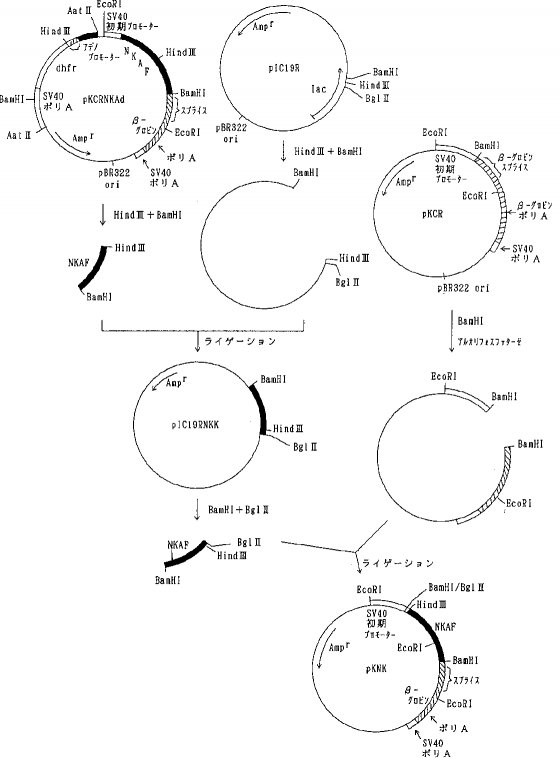
【0057】実施例6動物細胞用発現ベクターの構築a)天然型組換えAT IIIおよび1Rの発現ベクターの構築組換えナチュラルキラー細胞活性化因子(NKAF)をコードするcDNAを含むプラスミド pNK8308(特開平2-231083明細書に開示)を Bgl II と BamHIで消化しアガロース電気泳動を行って約 0.75kb のNKAF cDNA断片を単離した。プラスミド pKCR (O Hare, K. et. al., Proc. Natl. Acad. Sci. USA, 78, 1527, 1981)を BamHIで消化した後、アルカリフォスファターゼで脱リン酸して得られたベクターDNAをNKAF cDNA断片とT4DNA リガーゼを加えて結合させ(ライゲーションし)pKCRNKを得た(図1)。
【0058】プラスミド pUC19を PstI で消化した後、常法に従ってT4DNA ポリメラーゼで処理し、3'および5'の両末端を平滑末端とし(ブラントエンド化し)、ライゲーションして pUC19Pst-を得た。次いで、 pUC19Pst-を BamHIで消化した後アルカリフォスファターゼで脱リン酸して得られたベクターDNAとプラスミド pAdD26SV(A)(no.3) (Kaufmann, R. and Sharp, P., Mol. Cell. Biol., 2,1304, 1982) を BamHIで消化し、アガロース電気泳動を行って単離した約 2.4kbのDNA断片(アデノウイルスプロモーター、マウスジヒドロ葉酸還元酵素(DHFR)遺伝子、SV40ポリAシグナルを含む)をライゲーションして pUC19Pst-Adを得た(図2)。さらに pUC19Pst-AdをPstIで消化し、T4DNA ポリメラーゼでブラントエンド化しライゲーションして pUC19Pst-AdPst-を得た。そして、pAdD26SV(A) (no.3)を BamHIで消化し、脱リン酸し、アガロース電気泳動を行って単離したテトラサイクリン耐性遺伝子を含む約 2.9kbのDNA断片と pUC19Pst-AdPst-を BamHIで消化しアガロース電気泳動を行って単離したアデノウイルスプロモーター、マウスDHFR遺伝子、SV40ポリAシグナルを含む約 2.4kbのDNA断片をライゲーションして pAdPst-を得た(図3)。pAdPst- をEcoRI で消化した後、DNAポリメラーゼIクレノウフラグメントで処理し、ブラントエンド化した。ついで、PstIで消化した後、T4DNA ポリメラーゼでブラントエンド化した。これにAat IIリンカーを加えてライゲーションさせ、反応生成物をAat IIで消化し、アガロース電気泳動を行い、アデノウイルスプロモーター、マウスDHFR遺伝子、SV40ポリAシグナルを含む約 2.7kbのDNA断片を得た。このDNA断片をpKCRNKをAat IIで消化し、脱リン酸したDNAとライゲーションして pKCRNKAd を得た(図4)。実施例2で得られたプラスミド pSV2-5H3BをHind IIIと BglIIで消化し、ヒトAT IIIcDNAを含む約 1.4kbのDNA断片を単離し、プラスミド pIC19R (Marsh, J. L. et al., Gene, 32, 481, 1984)をHind IIIと Bgl II で消化して得られたベクターDNAとライゲーションして pIC19R5H3B を得た。次いでpIC19R5H3BをBamHI と Bgl II で消化し、5H3BcDNAを含む約 1.4kbのDNA断片を単離し、pKCRをBamHI 消化、脱リン酸したベクターDNAとライゲーションして pKCR5H3B を得た(図5)。
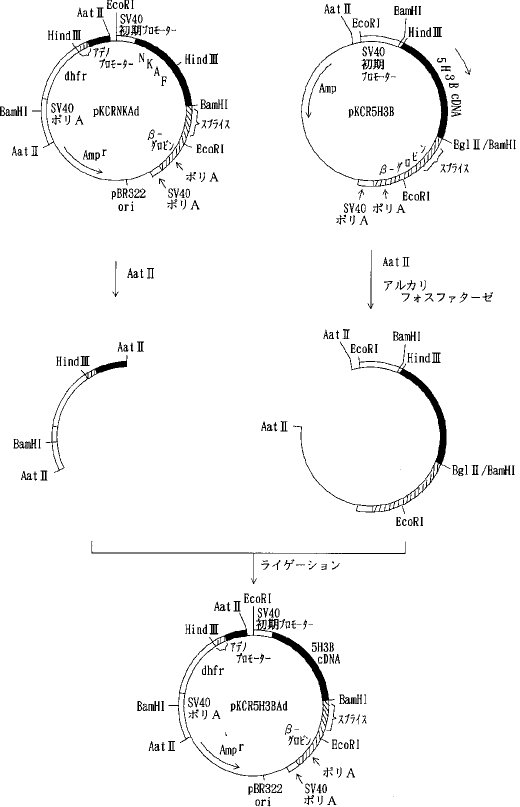
【0059】pKCRNKAdをAat IIで消化し、アデノウイルスプロモーター、マウスDHFR遺伝子、SV40ポリAシグナルを含む約 2.7kbのDNA断片を単離し、pKCR5H3BをAat II消化、脱リン酸したベクターDNAとライゲーションしてpKCR5H3BAdを得た(図6)。 pKCR5H3BAd は実施例7に示すように、動物細胞において天然型組換えAT IIIを発現させるために用いた。同様に、実施例3 a)で得られた pSV2-1Rを出発材料としてpKCR1RAdを得た。pKCR1RAdは実施例7に示すように、動物細胞において変異体1Rを発現させるために用いた。
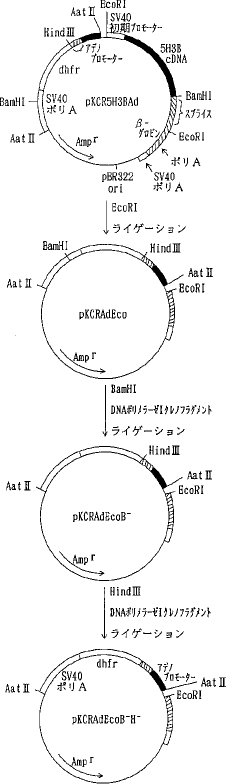
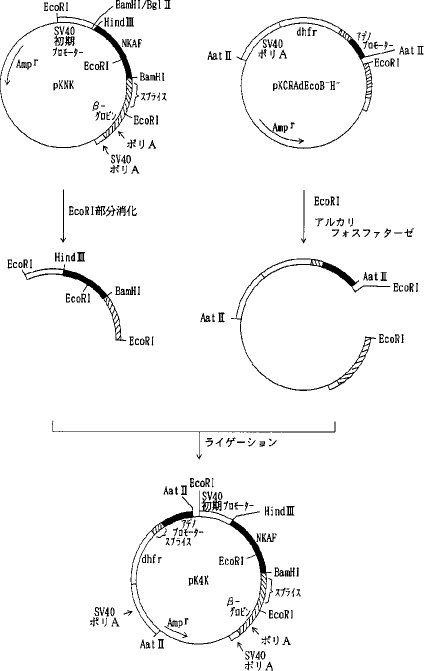
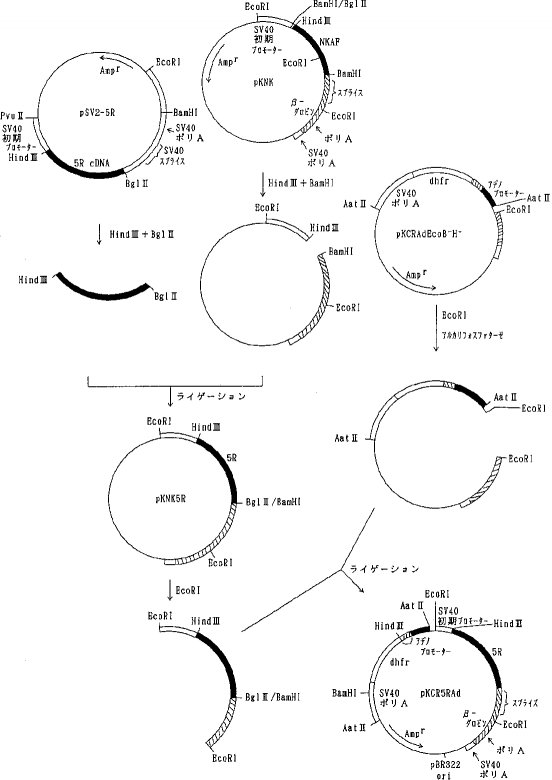
【0060】b)各種変異体の動物細胞での発現用ベクターの構築pKCR5H3BAdをEcoRI 消化した後、セルフライゲーションさせSV40プロモーター、NKAF遺伝子の一部、ウサギβ−グロビン遺伝子の一部を除いた pKCRAdEcoを選択した。pKCRAdEco を BamHI消化、DNAポリメラーゼIクレノウフラグメントでブラントエンド化し、ライゲーションして、pKCRAdEcoB- を得た。次いでpKCRAdEcoB- をHind III消化、DNAポリメラーゼ Iクレノウフラグメントでブラントエンド化してライゲーションして、pKCRAdEcoB-H- を得た(図7)。pKCRNKAdをHind IIIと BamHIで消化し、NKAF遺伝子の一部を含む約 0.4kbのDNA断片を単離し、pIC19RをHind IIIと BamHIで消化したベクターDNAとライゲーションして pIC19RNKKを得た。 pIC19RNKKを BglIIと BamHIで消化し、NKAF遺伝子の一部を含む 0.4kbのDNA断片を単離し、pKCRを BamHI消化、脱リン酸したベクターDNAとライゲーションしてpKNKを得た(図8)。pKNKを EcoRIで部分消化し、SV40プロモーター、NKAF遺伝子の一部、ウサギβ−グロビン遺伝子の一部を含む約 1.5kbのDNA断片を単離し、pKCRAdEcoB-H-を EcoRI消化、脱リン酸したベクターDNAとライゲーションしてpK4Kを得た(図9)。
【0061】pK4Kは図9に示すごとく、SV40初期遺伝子のプロモーター、SV40の複製開始領域、NKAF遺伝子の一部、ウサギβ−グロビン遺伝子の一部(スプライシングおよびポリAシグナル)、SV40初期遺伝子のポリAシグナル、タイプIIアデノウイルスの主要後期遺伝子プロモーターと5'スプライスシグナル、ウサギイムノグロブリン3'スプライスシグナル、マウスDHFR遺伝子、SV40初期遺伝子のポリAシグナル、pBR322の複製開始領域およびpBR322由来のβラクタマーゼ遺伝子(Amp γ)を含み、アデノウイルスの主要後期遺伝子プロモーターの下流にdhfrが接続され、SV40初期遺伝子のプロモーター下流にNKAF遺伝子の一部が接続されている。AT III変異体の遺伝子を pK4K のNKAF遺伝子の一部をHind IIIと BamHIで切り出したあとに挿入することによって動物細胞における発現用ベクターを構築することができる。実際に、変異体1G5Rおよび2G5Rの発現ベクターは実施例5a)、b)に示したように、それぞれ pSV2-1G5Rおよび pSV2-2G5Rと pK4K を用いて上記の方法により作製し、pK4K-1G5R 、pK4K-2G5R とした。実施例3b)および実施例5c)に示したように、他の変異体についても同様にpK4Kを用いて発現ベクターを構築した。
【0062】c)変異体5Rおよび7Rの発現ベクターの構築実施例3 b)で得られたプラスミド pSV2-5RをHind IIIと BglIIで消化して5R遺伝子を含む約 1.4kbのDNA断片を単離し、pKNKをHind IIIと BamHIで消化、NKAF遺伝子の一部を除いたベクターDNAとライゲーションして pKNK5R を得た。pKNK5Rを EcoRI消化し、SV40初期遺伝子のプロモーター、5R遺伝子、ウサギβ−グロビン遺伝子の一部を含む約 1.5kbのDNA断片を単離し、pKCRAdEcoB-H-を EcoRI 消化、脱リン酸したベクターDNAとライゲーションして pKCR5RAdを得た(図10)。同様にして実施例3 b)で得られた pSV2-7Rを用いて pKCR7RAd を得た。
【0063】実施例7AT III変異体の動物細胞による発現a)CHO 細胞による発現CHO 細胞(dhfr欠損株、Urlaub, G. and Chasin, L. A., Proc. Natl. Acad.Sci. USA. 77, 4216, 1980)を 7×105 cells /5ml /25cm2 培養フラスコで植え込み、翌日、実施例6 a)で得られたプラスミド pKCRlRAd 3μg を CellPhect (Pharmacia 社製キット)を使ったリン酸カルシウム法でトランスフェクションした。培地はハム F12培地とダルベッコ変法イーグル培地の1:1混合物(DF培地)に牛胎児血清を10%になるように加えて用いた。3日後、細胞をトリプシン処理し、選択培地(ヒポキサンチンおよびチミジン不含DF培地+10%透析牛胎児血清)で希釈して25cm2 培養フラスコ1本分の細胞を4枚の培養用24ウェルプレートの各ウェルに1mlずつ分注した。次いで3〜4日毎に選択培地にて培地交換を行いながら培養を続行した。これらの条件下で生存する細胞のみがマウスDHFR遺伝子により形質転換を受けた細胞である。約2週間後、生じたコロニーをウェル内でトリプシン処理して分散し、培地を加えてさらに3−4日培養後、培養液を交換し、翌日、培養上清中に含まれる1Rの量を EIA法で測定した。数十ng/ml/day 程度以上を発現している個々のクロ−ンを50nMのメソトレキセート(MTX) を含む選択培地に移して2−3週間培養した。さらに MTXの濃度を順次、100nM 、400nM および1000nMへと上げて同様に培養を続けた。1000nM MTX中で増殖させたクローンのうち発現量の高いものについて96ウェルプレートを用いた限界希釈法によってクローニングを行った。こうして得られた代表的なクローン 110-6はコンフルエントに増殖した状態で 0.3ml培地/cm2 の条件で1日当たり約10μg /mlの1Rを培養上清中に分泌した。同様に実施例6 a)で得た pKCR5H3BAd を用いて、天然型組換えATIII を発現する CHO細胞を得た。
【0064】b)BHK 細胞による各種変異体の発現i)pSV2ベクターを使用した場合プラスミド pSV2-dhfr中のマウスDHFR遺伝子とAT III変異体DNAとを入れ換えることによって構築された、実施例3および実施例5に示したプラスミドは、pSV2-dhfr とともに動物細胞に導入することにより(コトランスフェクション)各変異体の発現のために用いることができる。BHK 細胞(tk-ts13 株、Waechter, D. E. and Baserga, R., Proc. Natl. Acad. Sci. USA, 79, 1106, 1982 )を 5×105 cells /5ml /25cm2 培養フラスコで植え込み、翌日、実施例3 b)に示した変異体 28Rの遺伝子を導入したプラスミド pSV2-28R 7μg を pSV2-dhfr 3.5μg とともに CellPhectを使ったリン酸カルシウム法でトランスフェクションした。培地はダルベッコ変法イーグル培地に牛胎児血清を5%になるように加えて用いた。3日後、細胞をトリプシン処理し、200nM MTX を含む培地で75cm2 培養フラスコに継代した。2−3日毎に培養液を交換し10日間培養を続けた後、100nM MTX を含む培地で 175cm2 培養フラスコに継代した。さらに2−3日毎に培養液を交換し10日間培養した後、96ウェルプレートを用いた限界希釈法によって発現量の高い細胞株をクローニングした。こうして得られたクローン#4はコンフルエントに増殖した状態で 0.3ml培地/cm2 の条件で1日当たり約 0.7μg /mlの 28Rを培養上清中に分泌した。実施例3および実施例5に示した、pSV2を用いて作製した他の変異体DNAを含むプラスミドについても同様に発現細胞を得た。
【0065】ii)その他のベクターを使用した場合BHK 細胞(tk-ts13 株)を 3×105cells/5ml/25cm2 培養フラスコで植え込み、翌日、実施例5 b)で得た2G5R変異体の遺伝子を導入したプラスミド pK4K -2G5R 3 μg を CellPhectを使ったリン酸カルシウム法でトランスフェクションした。培地はダルベッコ変法イーグル培地に牛胎児血清を5%になるように加えて用いた。2日後、細胞をトリプシン処理し、250nM MTX を含む培地で希釈して25cm2 培養フラスコ一本分の細胞を12枚の培養用24ウェルプレートの各ウェルに分注した。次いで3−4日毎に培地交換を行いながら培養を続行した。12日後、生じたコロニーをウェル内でトリプシン処理して分散し、培地を加えてさらに6日間培養後、培養液を交換し、翌日、培養上清中に含まれる各変異体の量を EIA法で測定し、発現量の高い細胞株をクローニングした。こうして得られたクローン6−5はコンフルエントに増殖した状態で 0.3ml培地/cm2 の条件で1日当たり約16μg /mlの2G5Rを培養上清中に分泌した。
【0066】実施例6に示した pKCR1RAd 、pKCR5RAd、pKCR7RAdおよび実施例3、実施例5に示した、pK4Kを用いて作製した他の変異体DNAを含むプラスミドについても同様に発現細胞を得た。また、実施例6に示したpKCR5H3BAdを用いて同様に天然型組換えAT IIIを発現する細胞を得た。これらの内一部の発現細胞についてはさらに 1000nM MTX を含む培地に移して培養した。1000nM MTXを含む培地中で培養させたクローンのうち一部の発現量の高いものについては96ウェルプレートを用いた限界希釈法によってクローニングを行った。こうして得られた代表的なクローンの発現量を表10に示した
【0067】
【表10】
【0068】実施例8変異体発現細胞の培養および変異体の精製実施例7で得られたAT III変異体発現細胞をローラーボトル(1750cm2 )にて培養した。培地として5%牛胎児血清を加えたダルベッコ変法イーグル培地にMTXを加えたもの(終濃度 250nMまたは1000nM)を用いた。 300mlの培地に細胞を接種し、37℃で培養し、培養開始より3〜4日後より毎日同量の培地で培地交換し、培養上清を集めた。AT III変異体の精製は、抗ヒトAT IIIモノクローナル抗体を担体に結合した抗体カラムによるアフィニティークロマトグラフィーにて行った。すなわちあらかじめ50mM Tris ・ HCl pH7.5-0.5M NaCl にて平衡化した抗体カラムに上記培養上清をチャージし、同上バッファーにて洗浄後、0.2M Glycine ・ HCl pH2.5にて溶出した。溶出画分は直ちに1/2容量の1M Tris ・ HCl pH8.0 にて中和した。得られた画分をダルベッコ PBS(−)に対して透析後、限外濃縮してその後の実験に供した。一部の変異体については、抗体カラムの溶出画分を限外濃縮後、セファクリルS-200 にチャージし、ダルベッコ PBS(−)にてゲルろ過した。得られた活性画分を濃縮後、その後の実験に供した。なお、培養、精製過程でのAT III変異体の定量は抗AT III抗体を用いた EIA法にて行った。対照として用いた天然型組換えAT IIIについても同様に培養、精製を行った。
【0069】
【配列表】
配列番号:1配列の長さ:1395配列の型:核酸鎖の数:二本鎖トポロジー:直鎖状配列の種類:cDNA起源生物名:ホモサピエンス(Homo sapiens)配列の特徴特徴を表す記号:CDS存在位置:1..1395特徴を決定した方法:E特徴を表す記号:sig peptide存在位置:1..96特徴を決定した方法:S特徴を表す記号:mat peptide存在位置:97..1395特徴を決定した方法:S配列ATG TAT TCC AAT GTG ATA GGA ACT GTA ACC TCT GGA AAA AGG AAG GTT 48Met Tyr Ser Asn Val Ile Gly Thr Val Thr Ser Gly Lys Arg Lys Val -30 -25 -20 TAT CTT TTG TCC TTG CTG CTC ATT GGC TTC TGG GAC TGC GTG ACC TGT 96Tyr Leu Leu Ser Leu Leu Leu Ile Gly Phe Trp Asp Cys Val Thr Cys -15 -10 -5 CAC GGG AGC CCT GTG GAC ATC TGC ACA GCC AAG CCG CGG GAC ATT CCC 144His Gly Ser Pro Val Asp Ile Cys Thr Ala Lys Pro Arg Asp Ile Pro 1 5 10 15 ATG AAT CCC ATG TGC ATT TAC CGC TCC CCG GAG AAG AAG GCA ACT GAG 192Met Asn Pro Met Cys Ile Tyr Arg Ser Pro Glu Lys Lys Ala Thr Glu 20 25 30 GAT GAG GGC TCA GAA CAA AAG ATC CCG GAG GCC ACC AAC CGG CGT GTC 240Asp Glu Gly Ser Glu Gln Lys Ile Pro Glu Ala Thr Asn Arg Arg Val 35 40 45 TGG GAA CTG TCC AAG GCC AAT TCC CGC TTT GCT ACC ACT TTC TAT CAG 288Trp Glu Leu Ser Lys Ala Asn Ser Arg Phe Ala Thr Thr Phe Tyr Gln 50 55 60 CAC CTG GCA GAT TCC AAG AAT GAC AAT GAT AAC ATT TTC CTG TCA CCC 336His Leu Ala Asp Ser Lys Asn Asp Asn Asp Asn Ile Phe Leu Ser Pro 65 70 75 80 CTG AGT ATC TCC ACG GCT TTT GCT ATG ACC AAG CTG GGT GCC TGT AAT 384Leu Ser Ile Ser Thr Ala Phe Ala Met Thr Lys Leu Gly Ala Cys Asn 85 90 95 GAC ACC CTC CAG CAA CTG ATG GAG GTA TTT AAG TTT GAC ACC ATA TCT 432Asp Thr Leu Gln Gln Leu Met Glu Val Phe Lys Phe Asp Thr Ile Ser 100 105 110 GAG AAA ACA TCT GAT CAG ATC CAC TTC TTC TTT GCC AAA CTG AAC TGC 480Glu Lys Thr Ser Asp Gln Ile His Phe Phe Phe Ala Lys Leu Asn Cys 115 120 125 CGA CTC TAT CGA AAA GCC AAC AAA TCC TCC AAG TTA GTA TCA GCC AAT 528Arg Leu Tyr Arg Lys Ala Asn Lys Ser Ser Lys Leu Val Ser Ala Asn 130 135 140 CGC CTT TTT GGA GAC AAA TCC CTT ACC TTC AAT GAG ACC TAC CAG GAC 576Arg Leu Phe Gly Asp Lys Ser Leu Thr Phe Asn Glu Thr Tyr Gln Asp 145 150 155 160 ATC AGT GAG TTG GTA TAT GGA GCC AAG CTC CAG CCC CTG GAC TTC AAG 624Ile Ser Glu Leu Val Tyr Gly Ala Lys Leu Gln Pro Leu Asp Phe Lys 165 170 175 GAA AAT GCA GAG CAA TCC AGA GCG GCC ATC AAC AAA TGG GTG TCC AAT 672Glu Asn Ala Glu Gln Ser Arg Ala Ala Ile Asn Lys Trp Val Ser Asn 180 185 190 AAG ACC GAA GGC CGA ATC ACC GAT GTC ATT CCC TCG GAA GCC ATC AAT 720Lys Thr Glu Gly Arg Ile Thr Asp Val Ile Pro Ser Glu Ala Ile Asn 195 200 205 GAG CTC ACT GTT CTG GTG CTG GTT AAC ACC ATT TAC TTC AAG GGC CTG 768Glu Leu Thr Val Leu Val Leu Val Asn Thr Ile Tyr Phe Lys Gly Leu 210 215 220 TGG AAG TCA AAG TTC AGC CCT GAG AAC ACA AGG AAG GAA CTG TTC TAC 816Trp Lys Ser Lys Phe Ser Pro Glu Asn Thr Arg Lys Glu Leu Phe Tyr 225 230 235 240 AAG GCT GAT GGA GAG TCG TGT TCA GCA TCT ATG ATG TAC CAG GAA GGC 864Lys Ala Asp Gly Glu Ser Cys Ser Ala Ser Met Met Tyr Gln Glu Gly 245 250 255 AAG TTC CGT TAT CGG CGC GTG GCT GAA GGC ACC CAG GTG CTT GAG TTG 912Lys Phe Arg Tyr Arg Arg Val Ala Glu Gly Thr Gln Val Leu Glu Leu 260 265 270 CCC TTC AAA GGT GAT GAC ATC ACC ATG GTC CTC ATC TTG CCC AAG CCT 960Pro Phe Lys Gly Asp Asp Ile Thr Met Val Leu Ile Leu Pro Lys Pro 275 280 285 GAG AAG AGC CTG GCC AAG GTT GAG AAG GAA CTC ACC CCA GAA GTG CTG 1008Glu Lys Ser Leu Ala Lys Val Glu Lys Glu Leu Thr Pro Glu Val Leu 290 295 300 CAG GAG TGG CTG GAT GAA TTG GAG GAG ATG ATG CTG GTG GTC CAC ATG 1056Gln Glu Trp Leu Asp Glu Leu Glu Glu Met Met Leu Val Val His Met 305 310 315 320 CCC CGC TTC CGC ATT GAG GAC GGC TTC AGT TTG AAG GAG CAG CTG CAA 1104Pro Arg Phe Arg Ile Glu Asp Gly Phe Ser Leu Lys Glu Gln Leu Gln 325 330 335 GAC ATG GGC CTT GTC GAT CTG TTC AGC CCT GAA AAG TCC AAA CTC CCA 1152Asp Met Gly Leu Val Asp Leu Phe Ser Pro Glu Lys Ser Lys Leu Pro 340 345 350 GGT ATT GTT GCA GAA GGC CGA GAT GAC CTC TAT GTC TCA GAT GCA TTC 1200Gly Ile Val Ala Glu Gly Arg Asp Asp Leu Tyr Val Ser Asp Ala Phe 355 360 365 CAT AAG GCA TTT CTT GAG GTA AAC GAA GAA GGC AGT GAA GCA GCT GCA 1248His Lys Ala Phe Leu Glu Val Asn Glu Glu Gly Ser Glu Ala Ala Ala 370 375 380 AGT ACC GCT GTT GTG ATT GCT GGC CGT TCG CTA AAC CCC AAC AGG GTG 1296Ser Thr Ala Val Val Ile Ala Gly Arg Ser Leu Asn Pro Asn Arg Val 385 390 395 400 ACT TTC AAG GCC AAC AGG CCT TTC CTG GTT TTT ATA AGA GAA GTT CCT 1344Thr Phe Lys Ala Asn Arg Pro Phe Leu Val Phe Ile Arg Glu Val Pro 405 410 415 CTG AAC ACT ATT ATC TTC ATG GGC AGA GTA GCC AAC CCT TGT GTT AAG 1392Leu Asn Thr Ile Ile Phe Met Gly Arg Val Ala Asn Pro Cys Val Lys 420 425 430 TAA 1395
【図面の簡単な説明】
【図1】 pKCRNKの構築図
【図2】 pUC19st-Adの構築図
【図3】 pAdPst- の構築図
【図4】 pKCRNKAdの構築図
【図5】 pKCR5H3Bの構築図
【図6】 pKCR5H3BAdの構築図
【図7】 pKCRAdEcoB-H- の構築図
【図8】 pKNKの構築図
【図9】 pK4Kの構築図
【図10】 pKCR5RAdの構築図
【0001】
【産業上の利用分野】ヒトアンチトロンビンIII (AT III)のアミノ酸配列中、1個または複数個のアミノ酸を他のアミノ酸に変換させることにより、ヘパリン非存在下でも高い抗プロテアーゼ活性を有するAT III変異体に関するもので、血栓性疾病の治療薬として利用される。
【0002】
【従来の技術】ヘパリンをはじめとするグリコサミノグリカンの有する血液凝固阻害活性は、血中のアンチトロンビンIII (AT III)およびヘパリンコファクターII(HCII)により媒介される。AT IIIおよびHCIIは、いわゆるセルピンと総称されるセリンプロテアーゼインヒビターである。これらのうちAT IIIは先天的あるいは後天的な理由による血中レベルの低下により血栓症が発症するという報告が多く、一連のセリンプロテアーゼにより構成される血液凝固系の調節因子として生理的に重要な役割を担っている。
【0003】ヒトAT IIIは主に肝臓で合成され正常血漿中に約 150μg /mlの濃度で存在する、分子量約 60kd の糖蛋白であり、トロンビンおよびXa因子をはじめとする凝固、線溶系に携わるセリンプロテアーゼを阻害することが知られている。ヒトAT IIIの一次構造は、アミノ酸配列の直接決定(Petersen, T. E. et al., In: The Physiological Inhibitors of Blood Coagulation and Fibrinolysis, Elsevier Science Publishers, Amsterdam, 43, 1979)ならびにcDNAのクローニング(Bock, S. C. et al., Nucl. Acids Res., 10, 8113, 1982 ; Prochownik, E.V. et al., J. Biol. Chem., 258, 8389, 1982 ; Chandra, T. et al., Proc. Natl. Acad. Sci. USA, 80, 1845, 1983)により明らかにされている。これらの報告によると、ヒトAT IIIは前駆体蛋白質より32残基のシグナルペプチドが切断除去されて分泌生成される、 432アミノ酸からなる一本鎖糖蛋白である。分子内にN結合性の糖鎖付加を受ける部位を4ケ所含んでおり、分子量の約15%は糖である。
【0004】AT IIIはトロンビンなどのセリンプロテアーゼと1:1で反応し、安定な複合体を形成することにより、これらのプロテアーゼの活性を阻害する。この際AT IIIは分子中の 393番目のArg 残基と 394番目の Ser残基の間のペプチド結合がプロテアーゼにより切断され、この結果新たに生じた末端のArg 残基とプロテアーゼの活性中心の Ser残基との間にアシル結合が生じると考えられている。一般的に、この Arg (393)−Ser(394)を反応部位と呼ぶ。
【0005】AT IIIによるプロテアーゼの阻害反応は比較的緩やかに進行するが、反応系にへパリンが存在すると、その反応速度は劇的に加速され、AT IIIによるトロンビンの阻害速度はヘパリンの添加により1000倍以上となる。この作用機構はヘパリンがAT III中の特定部位(ヘパリン結合部位)に結合することにより、AT IIIの高次構造に変化をもたらしプロテアーゼと相互作用しやすい構造をとらせるとともに、プロテアーゼもそのヘパリン分子に結合することにより3者複合体を形成しやすくなるためと考えられている。また生理的には血管内皮細胞表面に存在するヘパリン様物質が、同様の作用を現すことによりAT IIIによる血液凝固系の制御機構に重要な役割を担っていると考えられている。
【0006】種々の原因により引き起こされる血栓症の予防、治療にはいわゆる抗凝固薬が使用されており、ヘパリンは現在でも極めて重要な抗凝固薬の一つである。しかしヘパリンの使用により時に重篤な副作用が生じることが報告されている(Amerena, J. et al., Adverse Drag React. Acute. Poisoning Rev., 9, 1, 1990 ;Levine, M. N., et al. Semi. in Thrombos. Hemostas., 12, 39, 1986 ; Kelton, J. G. et al., ibid, 12, 59, 1986 ; Levine, M. N., ibid, 12, 63, 1986)。代表的なものとして出血、血小板減少症、副腎機能障害、過敏症、投与部位の壊死、骨粗鬆症等があげられる。このため産婦人科領域あるいは外科手術後など出血の危険性が高い場合、あるいは長期にわたる投与においてはその使用は慎重に行われるべきである。さらにヘパリンは試験管内で好中球のエラスターゼによるAT IIIの不活性化を促進するという報告もあり(Jordan, R. E. et al, Science, 237, 777, 1987 ; Jordan, R. E. et al., J. Biol. Chem., 264, 10493,1989)、重篤な感染症あるい敗血症などの好中球のエラスターゼが病態に関与していると考えられる場合の投与においては考慮されるべきである。またヘパリンの抗凝固作用はあくまでもAT IIIを介したものであり、血中のAT III濃度が低下しているような病態ではその効果は期待されない。
【0007】一方、ヒトAT IIIは血漿由来の濃縮製剤という形で、先天的なAT III欠乏に基づく血栓形成傾向、およびAT III低下を伴う汎発性血管内凝固症候群(DIC)において臨床的に用いられている。しかし先に述べたように、ヘパリン非存在下におけるAT IIIの抗凝固活性は漸進的なもので、単独の使用は補充療法的な意味合いが強く、抗凝固薬としての有用性は限定される。そこでAT IIIの抗凝固薬としての有用性を高めるために、AT IIIとヘパリンの併用あるいはAT IIIとヘパリンの複合体を調製し、これを用いるといった方法が検討されている。しかしこれらの方法においても前述のヘパリンの有する欠点を除くことができないことは明らかである。
【0008】上記の如く、AT IIIには反応部位およびヘパリン結合部位という2つの機能部位が存在する。反応部位近傍のアミノ酸配列はプロテアーゼインヒビターとしての機能発現に重要な役割を担っているとともに、各種プロテアーゼに対する阻害特異性を決定する上でも重要であることが多くの報告により明らかとなっている。例えば先天性AT III異常症のなかで 382位のAla が Thrに置換したAT III Hamilton (Devraj-Kizuk, R. et al., Blood, 72, 1518, 1988) 、 384位のAla が Proに置換したAT III Cambridge I (Perry, P. J. et al., FEBS Lett, 254, 174, 1989) 、 393位のArg がHis に置換したAT III Glasgow (Erdjument, H. et al., J. Biol. Chem., 263, 5589, 1988)、同じくPro に置換したAT III Pescara (Lane, D. A. et al., J. Biol. Chem., 264, 10200, 1989)、 394位のSer がLeu に置換したAT III Denver(Stephens, A. W. et al.,J. Biol. Chem., 262, 1044, 1987)などにおいては異常AT III分子はいずれも抗プロテアーゼ活性を失っており、これらの患者は血栓症を発症している。
【0009】一方、ヘパリン結合部位すなわちヘパリンとの結合に直接関わっているアミノ酸は、やはりAT IIIの先天性分子異常症に関する研究およびアミノ酸残基の化学修飾の結果より明らかとなってきた。分子異常症では7位のIle が Asnに変換したAT III Rouen III (Brennan, S. O. et al., FEBS Lett., 237, 118, 1988) 、24位のArg が Cysに置換されたAT III Rouen IV (Borg, J. Y. et al., FEBS Lett., 266, 163, 1990) 、41位のPro が Leuに置換されたAT III Basel (Chang, J. Y. and Tran, T. H., J. Biol. Chem., 261, 1174, 1986)、47位のArg が Cysに置換されたAT III Toyama (Koide, T. et al., Proc. Natl. Acad. Sci. USA, 81, 289, 1984) 、 129位のArg が Glnに置換されたAT III Geneva (Gandrille, S. et al., J. Biol. Chem., 265, 18997, 1990)等が報告されている。これらの異常AT IIIではいずれもヘパリン親和性が低下しており、生理的に正常な機能を持ち得ないために血栓症を発症している。さらにアミノ酸の化学修飾の実験より49位のTrp 、114 位の Lys, 125位の Lys、 129位の Arg、 136位の Lys、 145位の Arg等のアミノ酸がヘパリンとの結合に直接関与していると考えられている(Blackburn, M. N. et al., J. Biol. Chem., 259, 939,1984 ; Peterson, C. et al., J. Biol. Chem., 262, 8061, 1987 ; Sun, X. J.and Chang, J. Y., Biochemistry, 29, 8957, 1990 )。
【0010】これらの知見をもとにこれまでAT IIIのアミノ酸を変換することによりATIIIの改良が試みられている。たとえば Zettlemeissl らはAT III中の糖鎖付加部位のアミノ酸を変換することによって、ヘパリン結合/ヘパリン活性化の性質を向上させるAT III変異体、および反応部位のアミノ酸を変換することにより酵素特異性を変化させたAT III変異体の製造法を開示している(特開平2−262598)。また Dijkemaらは反応部位のアミノ酸を変換することによって抗トロンビン/抗Xa活性の変化したAT III変異体の製造法を示した(EP90/01026)。しかしながら、臨床的に満足しうるAT III変異体は見いだされておらず、ヘパリン非存在下でのトロンビンあるいはXa因子阻害活性を上昇させたAT III変異体の作製が強く望まれている。
【0011】
【発明が解決しようする課題】本発明はヘパリン非存在下でも高い抗トロンビン活性を有する、新規なAT III変異体を提供することにある。さらに遺伝子組換え法による大量製造法をも提供することにある。
【0012】
【課題を解決するための手段】ヘパリンによるAT IIIの抗プロテア−ゼ活性増強のメカニズムは現在次のように考えられている。まずヘパリンがAT IIIのヘパリン結合部位に結合することにより、AT IIIの立体構造をプロテア−ゼとより反応しやすい構造へと変換する。同時にプロテア−ゼもそのヘパリン結合部位において同一のヘパリン分子と結合することにより、結果的にAT IIIとプロテア−ゼの複合体形成速度が上昇する(Pletcher,C.H. and Nelsestuen,G.L.,J.Biol.Chem., 258,1086,1988)。この仮説に従うと、AT IIIのヘパリン結合部位にヘパリンが結合することにより引き起こされる、反応部位の立体構造の変化が、抗プロテア−ゼ活性の増強において重要であると考えられる。このことは反応部位近傍のアミノ酸配列を人為的に変換することで反応部位の立体構造を変化させることにより、ヘパリン非存在下でのプロテア−ゼ活性が上昇したAT III変異体を製造することが可能であることを示している。
【0013】一方このような考えに基づきヘパリン非存在下の抗トロンビン活性が上昇したAT III変異体が得られたならば、そのAT III変異体にとってヘパリンと結合するということは重要な性質とはなり得ないと考えられる。したがってヘパリン結合部位に変異を来したために機能異常となった前述のAT III TOYAMA、 GENEVA などと異なり、ヘパリン結合部位にアミノ酸置換を導入しヘパリン親和性を低下させても機能的に影響は少ないと考えられる。むしろ血管内皮細胞表面のヘパリン様物質との相互作用が低下することにより、血中半減期の延長、好中球エラスタ−ゼによる失活の回避などがもたらされ、臨床的な有用性が増すことも考えられる。このような考えに基づき、本発明者らはAT IIIの改良を鋭意研究の結果、目的とする新規なAT III変異体作製に成功し本発明を完成した。
【0014】本発明は、変異させたヒトアンチトロンビンIII (AT III)であって、(1)11-14 位、41-47 位、125-133 位および 384-398位の四つの領域のアミノ酸が、それぞれの領域、単独で又は組み合わせで少なくとも一個、他のアミノ酸に変換されていることを特徴とするAT III変異体。
(2)384-398 位の領域のアミノ酸が少なくとも一個、他のアミノ酸に変換され、さらに 11-14位、41-49 位および 125-133位の三つの領域のアミノ酸が、各領域単独又は組み合わせで少なくとも一個、他のアミノ酸に変換された組み合わせであることを特徴とする(1)記載のAT III変異体。
(3)384-398 位の領域のアミノ酸が少なくとも一個、他のアミノ酸に変換され、さらに 11-14位および 41-47位の二つの領域のアミノ酸が、各領域単独又は組み合わせで少なくとも一個、他のアミノ酸に変換された組み合わせであることを特徴とする(1)記載のAT III変異体。
(4)384-398 位の領域のアミノ酸が少なくとも一個、他のアミノ酸に変換され、さらに 11-14位および 125-133位の二つの領域のアミノ酸が、各領域単独又は組み合わせで少なくとも一個、他のアミノ酸に変換された組み合わせであることを特徴とする(1)記載のAT III変異体。
(5)384-398 位の領域のアミノ酸が少なくとも一個、他のアミノ酸に変換され、さらに 41-47位および 125-133位の二つの領域のアミノ酸が、各領域単独又は組み合わせで少なくとも一個、他のアミノ酸に変換された組み合わせであることを特徴とする(1)記載のAT III変異体。
【0015】(6)11-14 位および 384-398位の二領域のアミノ酸がそれぞれの領域において少なくとも一個、他のアミノ酸に変換された(1)記載のAT III変異体。
(7)41-47 位および 384-398位の二領域のアミノ酸がそれぞれの領域において少なくとも一個、他のアミノ酸に変換された(1)記載のAT III変異体。
(8)125-133 位および 384-398位の二領域のアミノ酸がそれぞれの領域において少なくとも一個、他のアミノ酸に変換された(1)記載のAT III変異体。
(9)384-398 位の領域のアミノ酸が少なくとも一個、他のアミノ酸に変換された(1)記載のAT III変異体。
(10)四つの領域のアミノ酸がAla, Gly,Trp, Pro, Leu, Val, Phe, Tyr, Ile,Glu, Ser, Gln, Asn および Argから選ばれるアミノ酸に変換されている(1)記載のAT III変異体。
【0016】(11)384-398 位の領域のアミノ酸がAla, Pro, Leu, Val, Gly, Arg, Glu および Pheから選ばれるアミノ酸に変換され、さらに他の領域も変換されていてもよい(1)記載のAT III変異体。
(12)390-392 位の領域のアミノ酸がAla, Pro, Leu, Val, および Pheから選ばれるアミノ酸に変換され、さらに他の領域も変換されていてもよい(1)記載のAT III変異体。
(13)392 位Gly が Proに変換され、さらに他の領域も変換されていてもよい(1)記載のATIII 変異体。
(14)390 位 Ileが Alaに、 391位 Alaが Phe, Val または Leuに、および 392位Gly がPro に、単独または組み合わせで変換され、さらに他の領域も変換されていてもよい(1)記載のAT III変異体。
(15)384 位 Alaが Glyに、387 位Ala が Pheに、389 位 Valが Proに、397 位Proが Argに、または 398位 AsnがGlu または Argに、単独または組み合わせで変換され、さらに他の領域も変換されていてもよい(1)記載のAT III変異体。
【0017】(16)11位 Lysが Ileに、14位 Aspが Serに、単独または組み合わせで変換され、さらに他の領域も変換されていてもよい(1)または(14)記載のAT III変異体。
(17)125 位 Lysが Glnに、129 位 Argが Glnに、 132位 Argが Glnに、または133位 Lysが Asnまたは Glnに、単独または組み合わせで変換され、さらに他の領域も変換されていてもよい(1)または(14)記載のAT III変異体。
【0018】(18)下記アミノ酸に変換された変異体から選択される(1)記載のAT III変異体。
(a)392 位 Glyが Proに変換されたAT III変異体。
(b)391-392 位 Ala-Glyが Phe-Proに変換されたAT III変異体。
(c)390-391 位 Ile-Alaが Ala-Leuに変換されたAT III変異体。
(d)125 位 Lysが Glnに、 391-392位 Ala-Glyが Phe-Proに変換されたAT III変異体。
(e)132-133 位 Arg-Lysが Gln-Asnに、390-391 位 Ile-Alaが Ala-Leuに変換されたAT III変異体。
(f)132-133 位 Arg-Lysが Gln-Asnに、391-392 位 Ala-Glyが Phe-Proに変換されたAT III変異体。
(g)133 位Lys がAsn に、391-392 位 Ala-Glyが Phe-Proに変換されたAT III変異体。
【0019】(19)上記のATIII 変異体をコードするDNA、それらDNAを組み込んだ発現可能なベクター、これらベクターにより形質転換された宿主細胞、宿主細胞を用いるAT III変異体の製造方法およびこれらAT IIIを用いる医薬組成物に関する。
【0020】これらのAT III変異体はいずれも動物細胞を宿主として発現され製造され、得られた変異体は以下に示すようにヘパリン非存在下の抗トロンビン活性がヒト血漿由来のAT IIIあるいは天然型の組換えAT IIIにくらべ上昇していた。また動物実験においてもヒト血漿由来のAT IIIにくらべ強い薬効を示し、臨床上の有用性が高いものと期待される。
【0021】以下に本発明を詳細に説明する。
1)ヒトAT III cDNAの単離ヒトAT IIIは主に肝臓において合成されるので、市販のヒト肝臓cDNAライブラリー(λgt 11 、クローンテック社)を用いればよい。クローニングする方法は公知の方法、例えばヒトAT IIIアミノ酸配列に対応する合成オリゴヌクレオチドをプローブとして用いるプラークハイブリダイゼイション法(Sambrook, J. et al., Molecular Cloning, Cold Spring Harbor Laboratory, 1989 )などが挙げられる。得られたクローンは必要に応じて、例えば pUC 18 などのプラスミドにサブクローニングする。このようにして得られたcDNAの塩基配列はマキサムギルバート法(Maxam, A. M. and Gilbert, W., Proc. Natl. Acad. Sci. USA, 74, 560, 1977)またはジデオキシ法(Sanger, F., Science, 214, 1205, 1981)によって決定、確認することができる。得られたAT IIIcDNAのコ−ディング領域の塩基配列とそれに基づいて演繹されたアミノ酸配列は配列番号1に示した。
【0022】2)部位特異的変異導入法変異導入方法は、例えばZollerらの方法(Zoller,M. and Smith,M., Methodsin Enzymology, 100, 468, 1983)、Kramerらの方法(Kramer,W. and Fritz,H-JMethods in Enzymology, 154, 350, 1987)およびVandeyarらの方法(Vandeyar,M.A. et al., Gene., 65, 129, 1988) などが挙げられる。Kramerらの方法はgapped duplex 法と呼ばれるもので、 M13ファージのアンバー変異体M13tv18 、M13tv19 などをベクターとして用いることができる。これらのベクターにAT IIIをコードするDNAをクローニングし、この一本鎖DNAとアンバー変異の入っていない M13の二本鎖DNAの断片( M13mpP をPvu IIにて切断して得たベクター断片)とを変性後アニーリングさせgapped duplex DNAを得る。次にこのDNAに導入したい変異を含む合成オリゴヌクレオチドをハイブリダイズさせ、DNAポリメラーゼとDNAリガーゼを作用させることによってギャップを埋めた後、大腸菌のmutS株(BMH71-18mutS)にトランスフェクションし、supOの大腸菌でのみ増殖できるノンアンバーファージを選択することにより目的とする変異が導入されたファージを効率よく得ることができる。実際の操作には市販のキットを用いてもよい(宝酒造;Mutan-G)。一方、Vandeyarらの方法はAT IIIをコードするDNAをクローニングした M13の一本鎖DNAに、導入したい変異を含むオリゴヌクレオチドをハイブリダイズさせる。これを鋳型としdATP、 dGTP、 dTTP、 および5-methyl-dCTP を基質として T7 DNAポリメラーゼを作用させ二本鎖DNAを合成した後、 T4 DNAリガーゼを用いて閉環状の二本鎖DNAとする。つぎにこの二本鎖DNAを制限酵素 Mspl で処理したのち、エキソヌクレアーゼIII にて処理することにより変異が導入された鎖のみからなる環状一本鎖DNAを得る。これをメチル化DNAに特異的な制限システムを持たない大腸菌(SDM株)に導入することにより目的とするクローンを効率よく得る。こちらの方法でも実際の操作には市販のキットを用いてもよい(United States Biochemical Corporation ; T7-GEN In Vitro Mutagenesis Kit)。導入したい変異を含む合成オリゴヌクレオチドはDNA合成機(ABI社 380A)を用いてフォスフォアミダイト法にて合成することができる。
【0023】3)AT IIIcDNA変異導入用鋳型の調製上記1)で得たAT IIIcDNAのコ−ディング領域の前後に制限酵素切断部位を導入し、変異導入のための鋳型を調製する。制限酵素としては公知のものから適宜選択すればよく、本発明の場合はATIII コ−ディング領域の直前にHindIII切断部位、直後にBgl II切断部位を導入した。まず上記1)で得たAT IIIcDNAを含むプラスミドをEcoRIにて切断し、AT III全コーディング領域を含む 1.5kbの断片を得る。この断片をファージM13tv18のRF(Replicative Form、二本鎖DNA)をEcoRIにて切断し開環したものに導入する。こうして得られたクローンのうちAT IIIのセンス鎖を含む一本鎖DNAを鋳型としてKramerらの方法に従い、Hind IIIおよびBgl IIの酵素切断部位をそれぞれ含む2種の合成オリゴヌクレオチドをプライマーとして、制限酵素切断部位をAT IIIcDNAのコ−ディング領域の前後に導入する。次いで、このようにして得られたクローンからAT IIIcDNA配列を含む。断片を適切なプラスミドに挿入し、変異導入のための鋳型を調製する。
【0024】本発明の場合は、前記クローンをHind IIIおよびEcoRIにて切断して得られる約 1.5kbのAT III全コーディング配列を含むDNA断片を同酵素にて切断したプラスミドM13tv19RFあるいはM13mp19に挿入することにより、変異導入の鋳型を調製することができる。さらに、AT IIIのcDNAには1ケ所制限酵素 SacI認識部位(配列番号1の 721〜726 位の塩基部分)が存在し、反応部位とヘパリン結合部位を分断しうることから、上記クローンをHind IIIおよび SacIにて切断して得たAT IIIのN端側、すなわちヘパリン結合部位を含むDNA断片を同じ酵素で切断したプラスミドM13tv19またはM13mp19などに導入し、ヘパリン結合部位への変異導入用鋳型を調製することができる。反応部位についてもEcoRIおよび SacIを用い、同様な操作で可能である。
4)目的とする位置への変異導入AT IIIアミノ酸配列において、変換させたい位置のアミノ酸を目的とする他のアミノ酸(以下、目的アミノ酸と称す)に変換させるには、前記の公知の方法に従い目的アミノ酸をコードするDNAを含む合成オリゴヌクレオチドと、3)に記載した適当なプラスミドを鋳型として用いることにより実施することができる。例えばAT III 392位 Glyを Proに変換させる時は、表1記載のAT1Rオリゴヌクレオチドを、 391-392位 Ala-Glyを Phe-Proに変換させる時は、表1記載のAT5Rオリゴヌクレオチドを用いればよい。また各部位での変換させるアミノ酸が複数個でそれぞれの位置が離れている場合は、1個ずつ順に変異導入の操作を行うことにより、いくつでも変異を導入することができる。
【0025】本発明において使用したオリゴヌクレオチドの代表例を表1および表2に、アミノ酸変換部位と目的アミノ酸を表3および表4に記載した。目的アミノ酸をコードする塩基コドンは表1および表2記載のコドンに限定されるものではなく、目的アミノ酸をコードするコドンであれば、いずれも使用できる。
【0026】
【表1】
【0027】
【表2】
【0028】
【表3】
【0029】
【表4】
【0030】5)反応部位近傍の変異とヘパリン結合部位の変異の組み合わせ前記の如く、AT IIIcDNAには1ケ所 SacI切断部位が存在し、これが反応部位とヘパリン結合部位の間に位置する。それゆえに上記4)に記載された方法により得られた変異AT IIIDNAを含むプラスミドをHind IIIおよび SacIまたは SacIおよび BglIIにて切断することにより、ヘパリン結合部位を含む断片と反応部位を含む断片を得ることができる。反応部位またはヘパリン結合部位を変異させたAT III DNAを制限酵素にて処理し、反応部位変異DNA断片とヘパリン結合部位変異DNA断片を調製し、それぞれの変異DNA断片を適切なプラスミドに連結することにより両部位を変異させたAT III変異DNAの調製ができ、この方法により両部位の変異のいかなる組合せも可能である。両変異DNA断片を連結構築するプラスミドは宿主での発現に適しているものならばいずれも使用でき、例えばpSV2、pK4Kなどが挙げられる。表4に記載した記号で、2G35R とは 2G 変異DNA断片と35R 変異断片の組み合せにより得られる変異体を意味する。
【0031】6)AT III変異体組換え発現ベクターとその形質転換体上記記載の方法により得られたAT III変異体をコ−ドするDNAを適切なベクターに組み込み、該ベクターを適切な宿主細胞に移入することにより形質転換体を得ることができる。これを常法により培養し、培養物よりAT III変異体を大量に生産することができる。AT III変異体をコードするDNAをAT III変異体の発現に適したベクターのプロモーター下流に制限酵素とDNAリガーゼを用いる公知の方法により再結合して組換え発現ベクターを作製することができる。ベクターは宿主内で複製、増幅可能であれば特に限定されない。プロモーターおよびターミネーターに関してもAT III変異体をコードする塩基配列の発現に用いられる宿主に対応したものであれば特に限定されず、宿主に応じて適切な組み合わせも可能である。
【0032】このようにして得られた組換え発現ベクターはコンピテントセル法(Hanahan,D., J. Mol. Biol., 166, 557, 1983 )、リン酸カルシウム法( Wigler,M. etal., Cell, 11, 222, 1977) などにより宿主に導入し、形質転換体が作製される。宿主としては大腸菌および動物細胞などが用いられ、得られた形質転換体はその宿主に応じた適切な培地中で培養される。培養は通常20℃〜45℃、pH5〜8の範囲で行われ、必要に応じて通気、攪拌が行われる。培養物からのAT III変異体の分離、精製は公知の分離、精製法を適宜組み合わせて実施すれば良い。これらの公知の方法としては塩析、溶媒沈殿法、透析、ゲル濾過法、電気泳動法、イオン交換クロマトグラフィ、アフィニティクロマトグラフィー、逆相高速液体クロマトグラフィなどが挙げられる。このようにして得られたAT III変異体はヘパリン非存在下で天然型AT IIIより高い抗トロンビン活性を有し、ラットでのin vivo抗血栓作用においても天然型AT IIIより強い作用を示した。
【0033】
【発明の効果】
(1)抗トロンビン活性テストチームAT III2キット(第一化学薬品)を利用して、本発明のAT III変異体の抗トロンビン活性を測定した。すなわち、トロンビンの合成基質(S−2238)を用いて、トロンビンに対する阻害活性をヘパリンの非存在下において測定した。対照としてAT III血漿濃縮製剤(アンスロビンP;ヘキストジャパン社)を用いた。本測定において、緩衝液として 0.1%ウシ血清アルブミンおよび0.15M塩化ナトリウム含有 50mM トリス塩酸緩衝液(pH7.5 )を用い、種々の濃度に調製した検体と一定量のトロンビン(ウシ由来)を37℃で5分間反応させた。反応後、合成基質S−2238を添加し2分間に遊離してくるp−ニトロアニリン量を波長 405nmの吸光度変化で測定することにより、残存するトロンビン活性を求めた。この条件下、トロンビンの活性を50%阻害するAT III変異体の濃度(以下IC50)を算出した。表5に各変異体のIC50 値を示した。ヘパリン非存在下におけるヒトAT III濃縮製剤のIC50 値は13.0×10-8M であり、天然型組換えATIII もほぼ同じ値を示した。これに対し、本発明のAT III変異体のIC50 値は明らかに低い値を示し、ヘパリン非存在下の抗トロンビン活性の上昇が認められた。
【0034】
【表5】
【0035】(2)ヘパリン親和性高速液体クロマトグラフィ法によって、ヘパリン−5PW (7.5mm ×75mm:東ソウ)を用いて、本発明のAT III変異体について、ヘパリンに対する親和性の強さを比較検討した。すなわち、移動相に50mMトリス塩酸緩衝液(pH7.5 )を用い、流速1ml/minで30分間に塩化ナトリウム濃度を0M から2M 濃度まで直線的に上昇させた。検出は、波長 280nmの吸収で行い、検体が溶出されてくるまでの時間で比較した。表6に示したように、ヒトAT III及び天然型組換えAT IIIの主ピークが溶出されるまでの時間は22.3分と23.1分であり、大きな差は認められなかった。また、反応部位近傍の変異体においてもヒトAT IIIおよび天然型組換えAT IIIと比較して顕著な差は認められなかった。一方、反応部位近傍とヘパリン結合部位の両部位変異体については、いずれも主ピークが溶出されるまでの時間が有意に短縮しており、ヘパリン結合部位の変異を導入することによりヘパリンに対する親和性が低下することが確認された。
【0036】
【表6】
【0037】(3)AT III変異体の抗血栓作用血漿由来AT III濃縮製剤(アンスロビンP、ヘキストジャパン社)および天然型組換えAT IIIを対照として、本発明のAT III変異体の抗血栓作用を以下のように測定した。方法は Peters ら(Peters, R. F. et al., Thromb. Haemostas., 65, 268、1991)の報告に改良を加えて行った。すなわち、麻酔したSprague-Dawley系雄ラット(200 〜300g)の頸動静脈に生理食塩液を満たしたアトム静脈カテーテル(4Fr, 3.5cm、アトム社)をカニュレーションし、shunt を作製した。血流を遮断した状態で shuntの動脈側に脈波ピックアップ(MPP-3 、日本光電)を装着し、血流の変化をポリグラフ記録計で実験中モニターした。計算量の検体材料を1mlになるように生理食塩液で希釈して同ラットの大腿静脈より単回急速投与した後、shunt を開いて血流を開通させた。shunt を開いてから、shunt 内に血栓が形成されて閉塞するまでの時間を測定し閉塞時間とした。結果を表7および表8に示す。これにより本発明のAT III変異体は血漿由来AT III濃縮製剤および天然型組換えヒトAT IIIに比較して強い抗血栓作用を有することが判明した。
【0038】
【表7】
【0039】
【表8】
【0040】以上の結果から、本発明のAT III変異体は血液凝固阻止剤として血栓形成を抑制し、血栓性疾患の予防治療薬として期待される。
【0041】(4)AT III変異体の実験的汎発性血管内凝固症(DIC)モデルにおける効果血漿由来AT III濃縮製剤を対照として、本発明のAT III変異体の実験的汎発性血管内凝固症(DIC)モデルにおける効果を以下のように検討した。 方法は、杉島らの報告(杉島忠志ら, 臨床と研究, 62, 274, 1985 )に改良を加えて行った。すなわち、麻酔したSprague-Dawley系雄性ラット(200 〜300g)の頸静脈にアトム静脈カテーテル(3Fr ,アトム社)をカニュレーションし、組織トロンボプラスチン(トロンボレルS,ベーリングベルケ社)を1時間かけて持続投与することによって、モデルを作製した。試験検体は、組織トロンボプラスチンの投与を開始する直前に同ラットの大腿動脈より単回急速投与した。組織トロンボプラスチン投与終了30分後に、下行大動脈より3.8%クエン酸ナトリウムを1/10量加えて採血した。採血後、直ちに血液0.5ml を自動血球数装置用採血容器(東亞医用電子株式会社)に分取し、TECHNICON 社 H・1 System にて血小板数を測定した。残りの血液より遠心分離(3000rpm 10min )にて血漿を得て、血漿中のフィブリノーゲン量を測定した。血漿中フィブリノーゲン量は、トロンビン時間法(フィブリノーゲンB−テストワコー、和光純薬)にて測定した。結果を表9に示す。これより本発明のAT III変異体は、組織トロンボプラスチン誘発の実験的DICモデルにおいて、血小板数の減少および血漿中フィブリノーゲン量の低下に対して、血漿由来AT III濃縮製剤に比較して強い効果を示すことが判明した。以上の結果から、本発明のAT III変異体はDIC治療剤として期待される。
【0042】
【表9】
【0043】このAT III変異体は経口的、局所的、静注的、もしくは筋注的、皮下注的などにより投与することができるが、局所もしくは静注投与が好ましい。投与量は0.1〜 100mg/kg、好ましくは 0.5〜20mg/kgであり、体重に応じて1〜50mlの生理食塩水に溶解して用いる。また製剤の形としては、水和剤、水溶剤、錠剤、カプセル剤、粉剤、座剤などが使用でき、これら製剤の担体としては薬学的に許容される賦形剤、崩壊剤、滑沢剤、分散剤など通常の医薬品に使用されているものが用いられる。
【0044】
【実施例】以下の実施例により本発明を詳細に且つ具体的に説明するが、本発明はこれらの実施例に限定されるものではない。
【0045】実施例1ヒトAT IIIをコードするDNA配列のクローニング市販のヒト肝臓cDNAライブラリー(λgt 11 、クローンテック社)を原料として、常法に従いプローブとして32P標識合成オリゴヌクレオチドを用いてスクリーニングした。合成オリゴヌクレオチドの配列は Chandraらの報告をもとに、AT IIIの 314位から 322位のアミノ酸に対応する塩基配列部分とした。スクリーニングの結果、#2および#6という2種のクローンが得られた。それぞれのクローンより制限酵素EcoRI にてDNA断片を回収し M13mp18にサブクローニングし塩基配列を決定したところ、#2のクローンは33番目のアミノ酸に対応する配列から polyAまでの約 1.3kbを含み、一方#6のクローンは翻訳開始コドンから 348番目のアミノ酸に対応する約 1.1kbを含むことが確認された。次にこれらのクローンよりEcoRI にてインサートを切り出し、それぞれ EcoRIにて切断した pUC18にサブクローニングした。このようにして#2のクローンより pUC-Hを、#6のクローンより pUC-Lを作製した。次いでpUC-L をNcoIおよびHind IIIで切断し生じた約 3.7kbのDNA断片(pUC18 およびアンチトロンビンIII のN末側に対応する約 1.0kbの配列を含む)と、pUC-H を同じくNcoIおよびHind IIIで切断し生じた約 0.5kbのDNA断片(アンチトロンビン IIIのC末側に対応する配列を含む)を結合し、AT IIIの翻訳開始コドンより終始コドンまでの全コーディング配列を含むAT III FLpUCというプラスミドを得た。このプラスミドに含まれるAT IIIcDNAの開始コドンから終始コドンまでの全配列を配列番号1に示した。
【0046】実施例2制限酵素切断部位の導入実施例1で得られたプラスミドAT III FLpUCを材料としてAT IIIコーディング配列の直前に制限酵素 Hind III 切断部位、直後に制限酵素 BglII切断部位を導入したDNAを作製した。まず初めにAT III FLpUCをEcoRI にて切断し、AT III全コーディング領域を含む約 1.5kbの断片を得た。この断片を先に述べたM13tv18 のRFをEcoRI にて切断し開環したものに導入した。こうして得られたクローンのうちAT IIIのセンス鎖を一本鎖DNAとして与えるクローンを tvATRとした。次のこの tvATRの一本鎖DNAを鋳型とし、各酵素の切断部位をそれぞれ含む下記2種の合成オリゴヌクレオチドをプライマーとして、Kramerらの方法に従い制限酵素切断部位を導入した。
【0047】
実際の操作は市販のキット(宝酒造、Mutan G )を用いた。すなわち約 0.5μg の tvATRの一本鎖DNAと 0.2μg のキット添付 dsDNA(M13mp18 のマルチクローニングサイトを含む PvuII断片を欠失させたファージ M13mpP のRFDNA を PvuIIで切断し直鎖化したもの)を 20mM Tris ・ HCl pH8-10mM MgCl2-50mM NaCl-1mM DTT 中 100℃3分間、65℃10分間、37℃10分間静置し gapped duplexを形成させた。この1/10量をとり、T4ポリヌクレオチドキナーゼにて5'端をリン酸化したAT5H、AT3B各 5pmolと混和し(計3μl )65℃15分間、37℃15分間静置した。これにキット添付の緩衝液(50mM Tris ・ HCl pH8-60mM酢酸アンモニウム -5mM MgC12-5mM DTT-1mM NAD-0.5mM each dNTPs(A、C、G、T)25μl 、60U の E.coliDNAリガーゼ、1UのT4DNA ポリメラーゼを加え25℃約2時間静置した。3μlの0.2M EDTA pH8 を加え65℃5分間加熱したのち一部をとり Hanahanの方法(Hanahan, D., J. Mol. Biol., 166, 557, 1983)によって調製した大腸菌 BMH71-18mutS 株のコンピテントセルにトランスフェクションした。大腸菌MV1184株を指示菌として得られたプラークを拾い、常法に従い培養し RFDNAを調製した。このDNAを制限酵素 Hind III および BglIIにて切断し、新たな切断部位が生じているクローンにつきダイデオキシ法にて塩基配列を決定し、目的とする変異が導入されていることを確認した。こうして得られたクローンをAT5H3Bとした。
【0048】このAT5H3Bを Hind III および EcoRIによって切断し得られる約 1.5kbのDNA断片を、やはり Hind III および EcoRIにて切断し、開環した M13tv19RFに挿入したクローンを tv19-5H3Bとした。またプラスミド pSV2-dhfr(Lee, F. et al., Nature, 294, 228, 1981 ; Subramani, S. et al., Mol. Cell. Biol., 1,854, 1981 )をHind IIIおよび Bg1IIにて切断しマウスジヒドロ葉酸還元酵素(dhfr)をコードする領域を除いたDNA断片と、AT5H3Bをやはり Hind III と Bg1IIにて切断して得た、AT IIIの全コーディング配列を含むDNA断片とを結合しプラスミド pSV2-5H3Bを得た。さらに pSV2-5H3BをHind IIIおよび SacI にて切断して得たAT IIIのN端側をコードする約730bp のDNA断片を、やはりHind III および SacI にて切断し開環した M13tv19、および M13mp19に導入してそれぞれtv19-ATN、mp19-ATNとした。
【0049】実施例3a)1R変異体DNAの作製AT IIIの 392番目の Glyを Proで置換したAT III変異体1R(表3)をコードする配列を部位特異的変異導入法によって得た。すなわち、実施例2で得られたAT5H3Bの一本鎖DNAを鋳型としてKramerらの方法に従って、合成オリゴヌクレオチドAT1R(表1)を作用させることによって目的とするクローン 1Rmutを得た。操作は市販のキット(Mutan G )を用いて、実施例2に記載した方法と同様に行った。得られたプラークを12個拾って解析したところ、5個が目的とするクローンであった。得られたクローンの RFDNAを Hind III および BglIIにて切断し生じた約1.4kb のDNA断片を実施例2と同様プラスミド pSV2-dhfr中のマウスDHFR遺伝子と入れ換えプラスミド pSV2-1Rを構築した。
【0050】b)他の反応部位近傍変異体DNAの作製1R以外の反応部位近傍への変異導入は実施例2で得られた tV19-5H3Bを鋳型として上述のKramerらの方法に従って行った。この方法で5R、26R 、28R 、29R 、30R 、39R 、40R 、46R 、48R 、49R 、50R 、27R 、7R、34R 、35R 、38R 、9R、19R 、24R 、2R' 、5R' 、6R' の変異を導入した。それぞれのATIII 変異体の反応部位近傍のアミノ酸配列を表3に、変異導入に使用した合成オリゴヌクレオチドの配列を表1および表2に示す。操作は実施例2と同様にキット添付のマニュアルに従い変異導入反応を行ったのち得られたクローンを数個拾い、塩基配列を確認し目的とする変異が導入されたクローンを得た。それぞれのクローンより Hind III 、Bgl IIにて約1.4kb のDNA断片を得て、5R、26R 、28R 、30R、27R 、7R、19R 、24R 、2R' 、5R' 、6R' については実施例2および3a)と同様 pSV2-dhfrのマウスDHFR遺伝子と入れ換え、それぞれプラスミド pSV2-5R、pSV2-26R、pSV2-28R、pSV2-30R、pSV2-27R、pSV2-7R 、pSV2-19R、pSV2-24R、pSV2-2R'、pSV2-5R'、pSV2-6R'とした。また 39R、 40R、 46R、 48R、 49R、 50R、 34R、 35R、 38RについてはそれぞれのDNA断片を後述のプラスミドpK4K中のNKAF遺伝子の一部と入れ換え、それぞれプラスミド pK4K-39R 、 pK4K-40R 、pK4K-46R 、 pK4K-48R 、 pK4K-49R 、 pK4K-50R 、 pK4K-34R 、 pK4K-35R 、pK4K-38R とした。なお 29R、9Rについてはプラスミド pSV2-29R 、 pSV2-9RよりHind IIIおよび BglIIにて約 1.4kbのDNA断片を再び単離し、同様にプラスミド pK4K-29R 、pK4K-9R を構築した。
【0051】実施例4ヘパリン結合部位変異体DNAの作製ヘパリン結合部位の変異のうち1G、2G、8Gの変異導入は実施例2で得られた tv19-5H3Bを鋳型としてKramerらの方法に従って行った。用いた合成オリゴヌクレオチドの配列は表2に示す。目的とする変異が導入されたクローンをそれぞれ 1Gmut、2Gmut 、8Gmut とした。これらのクローンより Hind III 、Bgl IIにより約 1.4kbのDNA断片を切り出し、反応部位の変異の場合と同様にプラスミド pSV2-1G、pSV2-2G 、pSV2-8G を得た。また pSV2-1Gを Hind III および SacIにより切断し得られる約 730bpのDNA断片を、同じ酵素で切断した M13tv19に導入し tv19-1GN とした。1F, 2F, 3F, 7Gの変異導入は実施例2で得られたtv19-ATNを鋳型として同様に行った。目的とする変異が導入された M13クローンをそれぞれ 1Fmut, 2Fmut, 3Fmut, 7Gmut とした。
【0052】9Gの変異導入は mp19-ATN を鋳型として Vandeyar らの方法にて行った。実際の操作はキット(USB社;T7-GEN In vitro mutagenesis system)添付のマニュアルに従った。初めに1μg の mp 19-ATN一本鎖DNAと、T4ポリヌクレオチドキナーゼにて5'端をリン酸化した合成オリゴヌクレオチド AT9G 2pmol を40mMTris-HCl pH7.5-20mM MgC12-50mM NaCl中65℃5分間加熱後、室温となるまで徐冷した。次にこの反応液(10μl )に 10X Synthesis mix(100mM Tirs- HCl pH7.5-20mM DTT-5mM dATP-5mM dGTP-5mM dTTP-5mM 5-Methyl-dCTP-10mM ATP)を2μl 、2.5UのT7DNA ポリメラーゼ、5UのT4DNA リガーゼを加え最終液量20μl として37℃1時間静置した。この操作により変異の導入された鎖のみメチル化されたRFDNA が合成される。反応液を70℃10分間加熱して酵素を失活させたのち制限酵素MspIおよびHhaIを各5U加え37℃45分間反応させた。この操作でMspIにより二本鎖DNAのうち鋳型としてメチル化されていないDNA鎖にのみ切れ込みが入るとともに二本鎖DNAに変換されなかった鋳型の一本鎖DNAがHhaIにより切断される。次にこの反応液に50U のエキソヌクレアーゼIII を加え37℃45分間反応させることによって、切れ込みの入った鋳型鎖のみが分解され、結果として変異が導入されたDNA鎖が濃縮される。70℃10分間加熱し反応を止めた後、メチル化されたDNAに特異的な制限システムを持たない(mcrAB )大腸菌SDM株に常法でトランスフェクションした。得られたプラークを数個拾いDNAを得て塩基配列を決定することによって、目的とする変異の導入されたクローンを選択した。こうして得られたクローンよりHind IIIおよびSacIにて約 730bpのDNA断片を単離し、同じ酵素にて切断して同サイズの断片を除いた pSV2-5H3Bに導入して pSV2-9Gとした。
【0053】12G の変異は1Gの変異が導入されているDNAを鋳型としてAT2Gの合成オリゴヌクレオチド(表2)によってさらに変異を導入することによって得た。すなわちtv19-1GNを鋳型としてKramerらの方法によって行った。目的とする変異が導入されていることを確認したクローンを12Gmutとした。127Gの変異は12Gmutの一本鎖DNAを鋳型として、前記のVandeyarらの方法によって合成オリゴヌクレオチドAT7Gを作用させることによって得た。はやり目的とする変異が導入されていることを確認したクローンを127Gmut とした。
【0054】実施例5反応部位近傍とヘパリン結合部位の両部位変異体DNAの作製a)1G5R変異体DNAの作製ヘパリン結合部位の変異1Gに反応部位近傍の変異5Rを組み合わせた1G5R変異体のDNAは以下のように作製した。実施例4で得られた1Gmut のRFDNA をHind IIIおよびSacIにて切断し、約 730bpのヘパリン結合部位に変異を含むDNA断片を調製した。また実施例3で得られた pSV2-5RをSacIおよび BglIIにて切断し、約 670bpの反応部位近傍に変異を含むDNA断片を調製した。これらDNA断片を組み合わせて、Hind IIIおよびBgl II にてマウスDHFR遺伝子を除去した pSV2-dhfrに導入して pSV2-1G5Rを作製した。さらに、この pSV2-1G5RをHind IIIおよび Bgl II にて切断し生じた約1.4kbのDNA断片を、後述のプラスミド pK4K をHind IIIおよび BamHIにて切断しNKAF遺伝子の一部を除去したものに導入し、pK4K-1G5R を作製した。これら変異を含むDNA断片の調製および両変異DNA断片の組み合わせによる pSV2-1G5RおよびpK4K-1G5R の構築は公知の方法に準じて行った。プラスミド pK4K-1G5Rを含有する大腸菌(Escherichia coli)HB101-pK4K-1G5R は、通産省工業技術院微生物工業技術研究所に寄託された(受託番号 FERM BP-3806)。
【0055】b)2G5R変異体DNAの作製ヘパリン結合部位の変異2Gに反応部位近傍の変異5Rを組み合わせた2G5R変異体のDNAは以下のように作製した。実施例4で得られた2Gmut のRFDNA をHind IIIおよびSacIにて切断し、約 730bpのヘパリン結合部位に変異を含むDNA断片を調製した。また実施例3で得られた pSV2-5RをSacIおよび Bgl II にて切断し、約 670bpの反応部位近傍に変異を含むDNA断片を調製した。これらDNA断片を組み合わせて、Hind IIIおよび BglIIにてマウスDHFR遺伝子を除去した pSV2-dhfrに導入して pSV2-2G5Rを作製した。さらに、この pSV2-2G5RをHind IIIおよび BglIIにて切断し生じた約 1.4kbのDNA断片を、後述のプラスミド pK4K をHind IIIおよび BamHIにて切断しNKAF遺伝子の一部を除去したものに導入し、pK4K-2G5R を作製した。プラスミド pK4K-2G5Rを含有する大腸菌(Escherichia coli)HB101-pK4K-2G5R は、通産省工業技術院微生物工業技術研究所に寄託された(受託番号 FERM BP-3807)。
【0056】c)他の両部位変異体DNAの作製実施例3および4で得られた各部位変異体DNAを用いた。ヘパリン結合部位の変異を含むDNA断片として、 pSV2-1G、pSV2-2G 、pSV2-9G 1Fmut, 2Fmut,3Fmut, 7Gmut、12Gmut、127Gmut をそれぞれHind IIIおよびSacIにて切断して生じた約 730bpのDNA断片を用意した。また反応部位の変異を含むDNA断片として pSV2-1R、pSV2-5R (あるいはpSV2-1G5R )および pSV2-30R をSacIおよびBgl II にて切断して約 670bpのDNA断片を得た。さらに pK4K-35R をSacIおよび XhoIIにて切断し同じく約 670bpのDNA断片を得た(pK4KをHind IIIおよび BamHIにて切断しNKAF遺伝子の一部を除去したプラスミドに、AT III変異体遺伝子をHind III-BglII断片として導入したDNAでは、 BglII切断端と BamHI切断端とが連結される結果、再度 BglIIにて切断することができない。しかしこの部位は XhoIIにて切断可能である)。以上示したDNA断片を組み合わせて、pK4KをHind IIIおよび BamHIにて切断しNKAF遺伝子の一部を除去したプラスミドに導入することによってpK4K-1G30R、pK4K-1G35R、pK4K-2G30R、pK4K-2G35R、pK4K-1F5R, pK4K-2F5R, pK4K-3F5R, pK4K-7G5R、pK4K-7G30R、pK4K-7G35R、pK4K-9G5R 、pK4K-9G30R、pK4K-9G35R、pK4K-12G5R、pK4K-12G30R 、pK4K-12G35R 、pK4K-127G5R 、pK4K-127G30R、pK4K-127G35Rを構築した。またHind IIIおよび BglII にてマウスDHFR遺伝子を除去した pSV2-dhfrを用いて同様に pSV2-1G1R、pSV2-2G1R を構築した。
【0057】実施例6動物細胞用発現ベクターの構築a)天然型組換えAT IIIおよび1Rの発現ベクターの構築組換えナチュラルキラー細胞活性化因子(NKAF)をコードするcDNAを含むプラスミド pNK8308(特開平2-231083明細書に開示)を Bgl II と BamHIで消化しアガロース電気泳動を行って約 0.75kb のNKAF cDNA断片を単離した。プラスミド pKCR (O Hare, K. et. al., Proc. Natl. Acad. Sci. USA, 78, 1527, 1981)を BamHIで消化した後、アルカリフォスファターゼで脱リン酸して得られたベクターDNAをNKAF cDNA断片とT4DNA リガーゼを加えて結合させ(ライゲーションし)pKCRNKを得た(図1)。
【0058】プラスミド pUC19を PstI で消化した後、常法に従ってT4DNA ポリメラーゼで処理し、3'および5'の両末端を平滑末端とし(ブラントエンド化し)、ライゲーションして pUC19Pst-を得た。次いで、 pUC19Pst-を BamHIで消化した後アルカリフォスファターゼで脱リン酸して得られたベクターDNAとプラスミド pAdD26SV(A)(no.3) (Kaufmann, R. and Sharp, P., Mol. Cell. Biol., 2,1304, 1982) を BamHIで消化し、アガロース電気泳動を行って単離した約 2.4kbのDNA断片(アデノウイルスプロモーター、マウスジヒドロ葉酸還元酵素(DHFR)遺伝子、SV40ポリAシグナルを含む)をライゲーションして pUC19Pst-Adを得た(図2)。さらに pUC19Pst-AdをPstIで消化し、T4DNA ポリメラーゼでブラントエンド化しライゲーションして pUC19Pst-AdPst-を得た。そして、pAdD26SV(A) (no.3)を BamHIで消化し、脱リン酸し、アガロース電気泳動を行って単離したテトラサイクリン耐性遺伝子を含む約 2.9kbのDNA断片と pUC19Pst-AdPst-を BamHIで消化しアガロース電気泳動を行って単離したアデノウイルスプロモーター、マウスDHFR遺伝子、SV40ポリAシグナルを含む約 2.4kbのDNA断片をライゲーションして pAdPst-を得た(図3)。pAdPst- をEcoRI で消化した後、DNAポリメラーゼIクレノウフラグメントで処理し、ブラントエンド化した。ついで、PstIで消化した後、T4DNA ポリメラーゼでブラントエンド化した。これにAat IIリンカーを加えてライゲーションさせ、反応生成物をAat IIで消化し、アガロース電気泳動を行い、アデノウイルスプロモーター、マウスDHFR遺伝子、SV40ポリAシグナルを含む約 2.7kbのDNA断片を得た。このDNA断片をpKCRNKをAat IIで消化し、脱リン酸したDNAとライゲーションして pKCRNKAd を得た(図4)。実施例2で得られたプラスミド pSV2-5H3BをHind IIIと BglIIで消化し、ヒトAT IIIcDNAを含む約 1.4kbのDNA断片を単離し、プラスミド pIC19R (Marsh, J. L. et al., Gene, 32, 481, 1984)をHind IIIと Bgl II で消化して得られたベクターDNAとライゲーションして pIC19R5H3B を得た。次いでpIC19R5H3BをBamHI と Bgl II で消化し、5H3BcDNAを含む約 1.4kbのDNA断片を単離し、pKCRをBamHI 消化、脱リン酸したベクターDNAとライゲーションして pKCR5H3B を得た(図5)。
【0059】pKCRNKAdをAat IIで消化し、アデノウイルスプロモーター、マウスDHFR遺伝子、SV40ポリAシグナルを含む約 2.7kbのDNA断片を単離し、pKCR5H3BをAat II消化、脱リン酸したベクターDNAとライゲーションしてpKCR5H3BAdを得た(図6)。 pKCR5H3BAd は実施例7に示すように、動物細胞において天然型組換えAT IIIを発現させるために用いた。同様に、実施例3 a)で得られた pSV2-1Rを出発材料としてpKCR1RAdを得た。pKCR1RAdは実施例7に示すように、動物細胞において変異体1Rを発現させるために用いた。
【0060】b)各種変異体の動物細胞での発現用ベクターの構築pKCR5H3BAdをEcoRI 消化した後、セルフライゲーションさせSV40プロモーター、NKAF遺伝子の一部、ウサギβ−グロビン遺伝子の一部を除いた pKCRAdEcoを選択した。pKCRAdEco を BamHI消化、DNAポリメラーゼIクレノウフラグメントでブラントエンド化し、ライゲーションして、pKCRAdEcoB- を得た。次いでpKCRAdEcoB- をHind III消化、DNAポリメラーゼ Iクレノウフラグメントでブラントエンド化してライゲーションして、pKCRAdEcoB-H- を得た(図7)。pKCRNKAdをHind IIIと BamHIで消化し、NKAF遺伝子の一部を含む約 0.4kbのDNA断片を単離し、pIC19RをHind IIIと BamHIで消化したベクターDNAとライゲーションして pIC19RNKKを得た。 pIC19RNKKを BglIIと BamHIで消化し、NKAF遺伝子の一部を含む 0.4kbのDNA断片を単離し、pKCRを BamHI消化、脱リン酸したベクターDNAとライゲーションしてpKNKを得た(図8)。pKNKを EcoRIで部分消化し、SV40プロモーター、NKAF遺伝子の一部、ウサギβ−グロビン遺伝子の一部を含む約 1.5kbのDNA断片を単離し、pKCRAdEcoB-H-を EcoRI消化、脱リン酸したベクターDNAとライゲーションしてpK4Kを得た(図9)。
【0061】pK4Kは図9に示すごとく、SV40初期遺伝子のプロモーター、SV40の複製開始領域、NKAF遺伝子の一部、ウサギβ−グロビン遺伝子の一部(スプライシングおよびポリAシグナル)、SV40初期遺伝子のポリAシグナル、タイプIIアデノウイルスの主要後期遺伝子プロモーターと5'スプライスシグナル、ウサギイムノグロブリン3'スプライスシグナル、マウスDHFR遺伝子、SV40初期遺伝子のポリAシグナル、pBR322の複製開始領域およびpBR322由来のβラクタマーゼ遺伝子(Amp γ)を含み、アデノウイルスの主要後期遺伝子プロモーターの下流にdhfrが接続され、SV40初期遺伝子のプロモーター下流にNKAF遺伝子の一部が接続されている。AT III変異体の遺伝子を pK4K のNKAF遺伝子の一部をHind IIIと BamHIで切り出したあとに挿入することによって動物細胞における発現用ベクターを構築することができる。実際に、変異体1G5Rおよび2G5Rの発現ベクターは実施例5a)、b)に示したように、それぞれ pSV2-1G5Rおよび pSV2-2G5Rと pK4K を用いて上記の方法により作製し、pK4K-1G5R 、pK4K-2G5R とした。実施例3b)および実施例5c)に示したように、他の変異体についても同様にpK4Kを用いて発現ベクターを構築した。
【0062】c)変異体5Rおよび7Rの発現ベクターの構築実施例3 b)で得られたプラスミド pSV2-5RをHind IIIと BglIIで消化して5R遺伝子を含む約 1.4kbのDNA断片を単離し、pKNKをHind IIIと BamHIで消化、NKAF遺伝子の一部を除いたベクターDNAとライゲーションして pKNK5R を得た。pKNK5Rを EcoRI消化し、SV40初期遺伝子のプロモーター、5R遺伝子、ウサギβ−グロビン遺伝子の一部を含む約 1.5kbのDNA断片を単離し、pKCRAdEcoB-H-を EcoRI 消化、脱リン酸したベクターDNAとライゲーションして pKCR5RAdを得た(図10)。同様にして実施例3 b)で得られた pSV2-7Rを用いて pKCR7RAd を得た。
【0063】実施例7AT III変異体の動物細胞による発現a)CHO 細胞による発現CHO 細胞(dhfr欠損株、Urlaub, G. and Chasin, L. A., Proc. Natl. Acad.Sci. USA. 77, 4216, 1980)を 7×105 cells /5ml /25cm2 培養フラスコで植え込み、翌日、実施例6 a)で得られたプラスミド pKCRlRAd 3μg を CellPhect (Pharmacia 社製キット)を使ったリン酸カルシウム法でトランスフェクションした。培地はハム F12培地とダルベッコ変法イーグル培地の1:1混合物(DF培地)に牛胎児血清を10%になるように加えて用いた。3日後、細胞をトリプシン処理し、選択培地(ヒポキサンチンおよびチミジン不含DF培地+10%透析牛胎児血清)で希釈して25cm2 培養フラスコ1本分の細胞を4枚の培養用24ウェルプレートの各ウェルに1mlずつ分注した。次いで3〜4日毎に選択培地にて培地交換を行いながら培養を続行した。これらの条件下で生存する細胞のみがマウスDHFR遺伝子により形質転換を受けた細胞である。約2週間後、生じたコロニーをウェル内でトリプシン処理して分散し、培地を加えてさらに3−4日培養後、培養液を交換し、翌日、培養上清中に含まれる1Rの量を EIA法で測定した。数十ng/ml/day 程度以上を発現している個々のクロ−ンを50nMのメソトレキセート(MTX) を含む選択培地に移して2−3週間培養した。さらに MTXの濃度を順次、100nM 、400nM および1000nMへと上げて同様に培養を続けた。1000nM MTX中で増殖させたクローンのうち発現量の高いものについて96ウェルプレートを用いた限界希釈法によってクローニングを行った。こうして得られた代表的なクローン 110-6はコンフルエントに増殖した状態で 0.3ml培地/cm2 の条件で1日当たり約10μg /mlの1Rを培養上清中に分泌した。同様に実施例6 a)で得た pKCR5H3BAd を用いて、天然型組換えATIII を発現する CHO細胞を得た。
【0064】b)BHK 細胞による各種変異体の発現i)pSV2ベクターを使用した場合プラスミド pSV2-dhfr中のマウスDHFR遺伝子とAT III変異体DNAとを入れ換えることによって構築された、実施例3および実施例5に示したプラスミドは、pSV2-dhfr とともに動物細胞に導入することにより(コトランスフェクション)各変異体の発現のために用いることができる。BHK 細胞(tk-ts13 株、Waechter, D. E. and Baserga, R., Proc. Natl. Acad. Sci. USA, 79, 1106, 1982 )を 5×105 cells /5ml /25cm2 培養フラスコで植え込み、翌日、実施例3 b)に示した変異体 28Rの遺伝子を導入したプラスミド pSV2-28R 7μg を pSV2-dhfr 3.5μg とともに CellPhectを使ったリン酸カルシウム法でトランスフェクションした。培地はダルベッコ変法イーグル培地に牛胎児血清を5%になるように加えて用いた。3日後、細胞をトリプシン処理し、200nM MTX を含む培地で75cm2 培養フラスコに継代した。2−3日毎に培養液を交換し10日間培養を続けた後、100nM MTX を含む培地で 175cm2 培養フラスコに継代した。さらに2−3日毎に培養液を交換し10日間培養した後、96ウェルプレートを用いた限界希釈法によって発現量の高い細胞株をクローニングした。こうして得られたクローン#4はコンフルエントに増殖した状態で 0.3ml培地/cm2 の条件で1日当たり約 0.7μg /mlの 28Rを培養上清中に分泌した。実施例3および実施例5に示した、pSV2を用いて作製した他の変異体DNAを含むプラスミドについても同様に発現細胞を得た。
【0065】ii)その他のベクターを使用した場合BHK 細胞(tk-ts13 株)を 3×105cells/5ml/25cm2 培養フラスコで植え込み、翌日、実施例5 b)で得た2G5R変異体の遺伝子を導入したプラスミド pK4K -2G5R 3 μg を CellPhectを使ったリン酸カルシウム法でトランスフェクションした。培地はダルベッコ変法イーグル培地に牛胎児血清を5%になるように加えて用いた。2日後、細胞をトリプシン処理し、250nM MTX を含む培地で希釈して25cm2 培養フラスコ一本分の細胞を12枚の培養用24ウェルプレートの各ウェルに分注した。次いで3−4日毎に培地交換を行いながら培養を続行した。12日後、生じたコロニーをウェル内でトリプシン処理して分散し、培地を加えてさらに6日間培養後、培養液を交換し、翌日、培養上清中に含まれる各変異体の量を EIA法で測定し、発現量の高い細胞株をクローニングした。こうして得られたクローン6−5はコンフルエントに増殖した状態で 0.3ml培地/cm2 の条件で1日当たり約16μg /mlの2G5Rを培養上清中に分泌した。
【0066】実施例6に示した pKCR1RAd 、pKCR5RAd、pKCR7RAdおよび実施例3、実施例5に示した、pK4Kを用いて作製した他の変異体DNAを含むプラスミドについても同様に発現細胞を得た。また、実施例6に示したpKCR5H3BAdを用いて同様に天然型組換えAT IIIを発現する細胞を得た。これらの内一部の発現細胞についてはさらに 1000nM MTX を含む培地に移して培養した。1000nM MTXを含む培地中で培養させたクローンのうち一部の発現量の高いものについては96ウェルプレートを用いた限界希釈法によってクローニングを行った。こうして得られた代表的なクローンの発現量を表10に示した
【0067】
【表10】
【0068】実施例8変異体発現細胞の培養および変異体の精製実施例7で得られたAT III変異体発現細胞をローラーボトル(1750cm2 )にて培養した。培地として5%牛胎児血清を加えたダルベッコ変法イーグル培地にMTXを加えたもの(終濃度 250nMまたは1000nM)を用いた。 300mlの培地に細胞を接種し、37℃で培養し、培養開始より3〜4日後より毎日同量の培地で培地交換し、培養上清を集めた。AT III変異体の精製は、抗ヒトAT IIIモノクローナル抗体を担体に結合した抗体カラムによるアフィニティークロマトグラフィーにて行った。すなわちあらかじめ50mM Tris ・ HCl pH7.5-0.5M NaCl にて平衡化した抗体カラムに上記培養上清をチャージし、同上バッファーにて洗浄後、0.2M Glycine ・ HCl pH2.5にて溶出した。溶出画分は直ちに1/2容量の1M Tris ・ HCl pH8.0 にて中和した。得られた画分をダルベッコ PBS(−)に対して透析後、限外濃縮してその後の実験に供した。一部の変異体については、抗体カラムの溶出画分を限外濃縮後、セファクリルS-200 にチャージし、ダルベッコ PBS(−)にてゲルろ過した。得られた活性画分を濃縮後、その後の実験に供した。なお、培養、精製過程でのAT III変異体の定量は抗AT III抗体を用いた EIA法にて行った。対照として用いた天然型組換えAT IIIについても同様に培養、精製を行った。
【0069】
【配列表】
配列番号:1配列の長さ:1395配列の型:核酸鎖の数:二本鎖トポロジー:直鎖状配列の種類:cDNA起源生物名:ホモサピエンス(Homo sapiens)配列の特徴特徴を表す記号:CDS存在位置:1..1395特徴を決定した方法:E特徴を表す記号:sig peptide存在位置:1..96特徴を決定した方法:S特徴を表す記号:mat peptide存在位置:97..1395特徴を決定した方法:S配列ATG TAT TCC AAT GTG ATA GGA ACT GTA ACC TCT GGA AAA AGG AAG GTT 48Met Tyr Ser Asn Val Ile Gly Thr Val Thr Ser Gly Lys Arg Lys Val -30 -25 -20 TAT CTT TTG TCC TTG CTG CTC ATT GGC TTC TGG GAC TGC GTG ACC TGT 96Tyr Leu Leu Ser Leu Leu Leu Ile Gly Phe Trp Asp Cys Val Thr Cys -15 -10 -5 CAC GGG AGC CCT GTG GAC ATC TGC ACA GCC AAG CCG CGG GAC ATT CCC 144His Gly Ser Pro Val Asp Ile Cys Thr Ala Lys Pro Arg Asp Ile Pro 1 5 10 15 ATG AAT CCC ATG TGC ATT TAC CGC TCC CCG GAG AAG AAG GCA ACT GAG 192Met Asn Pro Met Cys Ile Tyr Arg Ser Pro Glu Lys Lys Ala Thr Glu 20 25 30 GAT GAG GGC TCA GAA CAA AAG ATC CCG GAG GCC ACC AAC CGG CGT GTC 240Asp Glu Gly Ser Glu Gln Lys Ile Pro Glu Ala Thr Asn Arg Arg Val 35 40 45 TGG GAA CTG TCC AAG GCC AAT TCC CGC TTT GCT ACC ACT TTC TAT CAG 288Trp Glu Leu Ser Lys Ala Asn Ser Arg Phe Ala Thr Thr Phe Tyr Gln 50 55 60 CAC CTG GCA GAT TCC AAG AAT GAC AAT GAT AAC ATT TTC CTG TCA CCC 336His Leu Ala Asp Ser Lys Asn Asp Asn Asp Asn Ile Phe Leu Ser Pro 65 70 75 80 CTG AGT ATC TCC ACG GCT TTT GCT ATG ACC AAG CTG GGT GCC TGT AAT 384Leu Ser Ile Ser Thr Ala Phe Ala Met Thr Lys Leu Gly Ala Cys Asn 85 90 95 GAC ACC CTC CAG CAA CTG ATG GAG GTA TTT AAG TTT GAC ACC ATA TCT 432Asp Thr Leu Gln Gln Leu Met Glu Val Phe Lys Phe Asp Thr Ile Ser 100 105 110 GAG AAA ACA TCT GAT CAG ATC CAC TTC TTC TTT GCC AAA CTG AAC TGC 480Glu Lys Thr Ser Asp Gln Ile His Phe Phe Phe Ala Lys Leu Asn Cys 115 120 125 CGA CTC TAT CGA AAA GCC AAC AAA TCC TCC AAG TTA GTA TCA GCC AAT 528Arg Leu Tyr Arg Lys Ala Asn Lys Ser Ser Lys Leu Val Ser Ala Asn 130 135 140 CGC CTT TTT GGA GAC AAA TCC CTT ACC TTC AAT GAG ACC TAC CAG GAC 576Arg Leu Phe Gly Asp Lys Ser Leu Thr Phe Asn Glu Thr Tyr Gln Asp 145 150 155 160 ATC AGT GAG TTG GTA TAT GGA GCC AAG CTC CAG CCC CTG GAC TTC AAG 624Ile Ser Glu Leu Val Tyr Gly Ala Lys Leu Gln Pro Leu Asp Phe Lys 165 170 175 GAA AAT GCA GAG CAA TCC AGA GCG GCC ATC AAC AAA TGG GTG TCC AAT 672Glu Asn Ala Glu Gln Ser Arg Ala Ala Ile Asn Lys Trp Val Ser Asn 180 185 190 AAG ACC GAA GGC CGA ATC ACC GAT GTC ATT CCC TCG GAA GCC ATC AAT 720Lys Thr Glu Gly Arg Ile Thr Asp Val Ile Pro Ser Glu Ala Ile Asn 195 200 205 GAG CTC ACT GTT CTG GTG CTG GTT AAC ACC ATT TAC TTC AAG GGC CTG 768Glu Leu Thr Val Leu Val Leu Val Asn Thr Ile Tyr Phe Lys Gly Leu 210 215 220 TGG AAG TCA AAG TTC AGC CCT GAG AAC ACA AGG AAG GAA CTG TTC TAC 816Trp Lys Ser Lys Phe Ser Pro Glu Asn Thr Arg Lys Glu Leu Phe Tyr 225 230 235 240 AAG GCT GAT GGA GAG TCG TGT TCA GCA TCT ATG ATG TAC CAG GAA GGC 864Lys Ala Asp Gly Glu Ser Cys Ser Ala Ser Met Met Tyr Gln Glu Gly 245 250 255 AAG TTC CGT TAT CGG CGC GTG GCT GAA GGC ACC CAG GTG CTT GAG TTG 912Lys Phe Arg Tyr Arg Arg Val Ala Glu Gly Thr Gln Val Leu Glu Leu 260 265 270 CCC TTC AAA GGT GAT GAC ATC ACC ATG GTC CTC ATC TTG CCC AAG CCT 960Pro Phe Lys Gly Asp Asp Ile Thr Met Val Leu Ile Leu Pro Lys Pro 275 280 285 GAG AAG AGC CTG GCC AAG GTT GAG AAG GAA CTC ACC CCA GAA GTG CTG 1008Glu Lys Ser Leu Ala Lys Val Glu Lys Glu Leu Thr Pro Glu Val Leu 290 295 300 CAG GAG TGG CTG GAT GAA TTG GAG GAG ATG ATG CTG GTG GTC CAC ATG 1056Gln Glu Trp Leu Asp Glu Leu Glu Glu Met Met Leu Val Val His Met 305 310 315 320 CCC CGC TTC CGC ATT GAG GAC GGC TTC AGT TTG AAG GAG CAG CTG CAA 1104Pro Arg Phe Arg Ile Glu Asp Gly Phe Ser Leu Lys Glu Gln Leu Gln 325 330 335 GAC ATG GGC CTT GTC GAT CTG TTC AGC CCT GAA AAG TCC AAA CTC CCA 1152Asp Met Gly Leu Val Asp Leu Phe Ser Pro Glu Lys Ser Lys Leu Pro 340 345 350 GGT ATT GTT GCA GAA GGC CGA GAT GAC CTC TAT GTC TCA GAT GCA TTC 1200Gly Ile Val Ala Glu Gly Arg Asp Asp Leu Tyr Val Ser Asp Ala Phe 355 360 365 CAT AAG GCA TTT CTT GAG GTA AAC GAA GAA GGC AGT GAA GCA GCT GCA 1248His Lys Ala Phe Leu Glu Val Asn Glu Glu Gly Ser Glu Ala Ala Ala 370 375 380 AGT ACC GCT GTT GTG ATT GCT GGC CGT TCG CTA AAC CCC AAC AGG GTG 1296Ser Thr Ala Val Val Ile Ala Gly Arg Ser Leu Asn Pro Asn Arg Val 385 390 395 400 ACT TTC AAG GCC AAC AGG CCT TTC CTG GTT TTT ATA AGA GAA GTT CCT 1344Thr Phe Lys Ala Asn Arg Pro Phe Leu Val Phe Ile Arg Glu Val Pro 405 410 415 CTG AAC ACT ATT ATC TTC ATG GGC AGA GTA GCC AAC CCT TGT GTT AAG 1392Leu Asn Thr Ile Ile Phe Met Gly Arg Val Ala Asn Pro Cys Val Lys 420 425 430 TAA 1395
【図面の簡単な説明】
【図1】 pKCRNKの構築図
【図2】 pUC19st-Adの構築図
【図3】 pAdPst- の構築図
【図4】 pKCRNKAdの構築図
【図5】 pKCR5H3Bの構築図
【図6】 pKCR5H3BAdの構築図
【図7】 pKCRAdEcoB-H- の構築図
【図8】 pKNKの構築図
【図9】 pK4Kの構築図
【図10】 pKCR5RAdの構築図
【特許請求の範囲】
【請求項1】 変異させたヒトアンチトロンビンIII (AT III)であって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、AT IIIアミノ酸配列 390位 IleをAla,LeuまたはGlyに変換(以下変換(i)という)、391位 AlaをPhe, Ile, Gly, Tyr, Trp, ValまたはLeuに変換(以下変換(ii)という)、392位GlyをProに変換(以下変換(iii)という)から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、AT III変異体。
【請求項2】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、AT IIIアミノ酸配列125位LysをGlnに変換(以下変換(iv)という)させ、更に、上記変換(i)、(ii)、(iii)から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、AT III変異体。
【請求項3】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、AT IIIアミノ酸配列132位Argを Glnに変換(以下変換(v)という)させ、133位 LysをAsnまたはGlnに変換(以下変換(vi)という)させ、更に、上記変換(i)、(ii)、(iii)から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、AT III変異体。
【請求項4】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、上記変換(ii)及び(iii)が行われ、更に上記変換(v)、(vi) から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、AT III変異体。
【請求項5】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、AT IIIアミノ酸配列 129位Argを Glnに変換(以下変換(vii)という)させ、更に、上記変換(i)、(ii)、(iii)から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、AT III変異体。
【請求項6】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、AT IIIアミノ酸配列 11位 LysをIleに変換(以下変換(viii)という)させ、14位 AspをSerに変換(以下変換(ix)という)させ、更に、上記変換(i)、(ii)、(iii)から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、ATIII変異体。
【請求項7】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、上記変換(iv)、(v)及び(vi)が行われ、更に上記変換(i)、(ii)、(iii)から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、AT III変異体。
【請求項8】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、上記変換(iv)、(vii)、(v)及び(vi)が行われ、更に上記変換(i)、(ii)、(iii)から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、AT III変異体。
【請求項9】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、AT IIIアミノ酸配列 384位のAlaをGlyに変換(以下変換(x)という)、387位のAlaをPheに変換(以下変換(xi)という)、389位のValをProに変換(以下変換(xii)という)、398位のAsnをGluまたはArgに変換(以下変換(xiii)という)のいずれかの変換が単独で行われ、それ以外の変換が行われていない、AT III変異体。
【請求項10】 下記(1)〜(35)から選ばれるいずれかの変換が行われ、それ以外の変換が行われていない請求項1〜9のいずれか一項に記載のAT III変異体。
(1) 392位のGlyをProに変換(2) 391, 392位のAla-GlyをPhe-Proに変換(3) 391, 392位のAla-GlyをIle-Proに変換(4) 391, 392位のAla-GlyをGly-Proに変換(5) 391, 392位のAla-GlyをTyr-Proに変換(6) 391, 392位のAla-GlyをTrp-Proに変換(7) 391, 392位のAla-GlyをVal-Proに変換(8) 391, 392位のAla-GlyをLeu-Proに変換(9) 390, 391, 392位のIle-Ala-GlyをAla-Val-Proに変換(10) 390, 391, 392位のIle-Ala-GlyをLeu-Phe-Proに変換(11) 390, 391, 392位のIle-Ala-GlyをAla-Tyr-Proに変換(12) 390, 391, 392位のIle-Ala-GlyをAla-Trp-Proに変換(13) 390, 391, 392位のIle-Ala-GlyをLeu-Trp-Proに変換(14) 390, 391位のIle-AlaをAla-Valに変換(15) 390, 391位のIle-AlaをAla-Ileに変換(16) 390, 391位のIle-AlaをAla-Leuに変換(17) 390, 391位のIle-AlaをGly-Leuに変換(18) 384位のAlaをGlyに変換(19) 389位のValをProに変換(20) 387位のAlaをPheに変換(21) 398位のAsnをGluに変換(22) 398位のAsnをArgに変換(23) 125位のLysをGlnに、392位のGlyをProに変換(24) 125位のLysをGlnに、391, 392位のAla-GlyをPhe-Proに変換(25) 132, 133位のArg-LysをGln-Asnに、392位のGlyをProに変換(26) 132, 133位のArg-LysをGln-Asnに、391, 392位のAla-GlyをPhe-Proに変換(27) 132, 133位のArg-LysをGln-Asnに、391, 392位のAla-GlyをVal-Proに変換(28) 132, 133位のArg-LysをGln-Asnに、390, 391位のIle-AlaをAla-Leuに変換(29) 129位のArgをGlnに、391, 392位のAla-GlyをPhe-Proに変換(30) 11位のLysをIleに、14位のAspをSerに、391, 392位のAla-GlyをPhe-Proに変換(31) 125位のLysをGlnに、129位のArgをGlnに、132, 133位のArg-LysをGln-Asnに、391, 392位のAla-GlyをPhe-Proに変換(32) 132位のArgをGlnに、391, 392位のAla-GlyをPhe-Proに変換(33) 133位のLysをAsnに、391, 392位のAla-GlyをPhe-Proに変換(34) 133位のLysをGlnに、391, 392位のAla-GlyをPhe-Proに変換(35) 125位のLysをGlnに、132, 133位のArg-LysをGln-Asnに、391, 392位のAla-GlyをPhe-Proに変換
【請求項11】 アミノ酸の変換が上記(1)〜(31)から選ばれるいずれかの変換で、それ以外の変換が行われていない、請求項10記載のAT III変異体。
【請求項12】 アミノ酸の変換が上記(2), (7), (16), (24), (26), (27),(28), (29), (30)あるいは(31)のいずれかの変換で、それ以外の変換が行われていない、請求項11記載のAT III変異体。
【請求項13】 請求項1〜12のいずれか一項に記載のAT III変異体をコードするDNA。
【請求項14】 請求項13記載のDNAを含有するAT III変異体を発現可能なベクター。
【請求項15】 請求項14記載のベクターにより形質転換された宿主細胞。
【請求項16】 形質転換された宿主細胞が大腸菌または動物細胞である請求項15記載の宿主細胞。
【請求項17】 請求項15又は16記載の宿主細胞を培養し、産生されたAT III変異体を培養物中より分取することを特徴とするAT III変異体の製造方法。
【請求項18】 請求項1〜12のいずれか一項に記載のAT III変異体と薬学的に許容しうる担体とを含有する血栓性疾病用医薬組成物。
【請求項1】 変異させたヒトアンチトロンビンIII (AT III)であって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、AT IIIアミノ酸配列 390位 IleをAla,LeuまたはGlyに変換(以下変換(i)という)、391位 AlaをPhe, Ile, Gly, Tyr, Trp, ValまたはLeuに変換(以下変換(ii)という)、392位GlyをProに変換(以下変換(iii)という)から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、AT III変異体。
【請求項2】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、AT IIIアミノ酸配列125位LysをGlnに変換(以下変換(iv)という)させ、更に、上記変換(i)、(ii)、(iii)から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、AT III変異体。
【請求項3】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、AT IIIアミノ酸配列132位Argを Glnに変換(以下変換(v)という)させ、133位 LysをAsnまたはGlnに変換(以下変換(vi)という)させ、更に、上記変換(i)、(ii)、(iii)から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、AT III変異体。
【請求項4】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、上記変換(ii)及び(iii)が行われ、更に上記変換(v)、(vi) から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、AT III変異体。
【請求項5】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、AT IIIアミノ酸配列 129位Argを Glnに変換(以下変換(vii)という)させ、更に、上記変換(i)、(ii)、(iii)から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、AT III変異体。
【請求項6】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、AT IIIアミノ酸配列 11位 LysをIleに変換(以下変換(viii)という)させ、14位 AspをSerに変換(以下変換(ix)という)させ、更に、上記変換(i)、(ii)、(iii)から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、ATIII変異体。
【請求項7】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、上記変換(iv)、(v)及び(vi)が行われ、更に上記変換(i)、(ii)、(iii)から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、AT III変異体。
【請求項8】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、上記変換(iv)、(vii)、(v)及び(vi)が行われ、更に上記変換(i)、(ii)、(iii)から選ばれる変換が単独または組み合わせて行われ、それ以外の変換が行われていない、AT III変異体。
【請求項9】 変異させたAT IIIであって、天然型AT IIIと比較してヘパリン非存在下での抗トロンビン活性が高いことを特徴とし、AT IIIアミノ酸配列 384位のAlaをGlyに変換(以下変換(x)という)、387位のAlaをPheに変換(以下変換(xi)という)、389位のValをProに変換(以下変換(xii)という)、398位のAsnをGluまたはArgに変換(以下変換(xiii)という)のいずれかの変換が単独で行われ、それ以外の変換が行われていない、AT III変異体。
【請求項10】 下記(1)〜(35)から選ばれるいずれかの変換が行われ、それ以外の変換が行われていない請求項1〜9のいずれか一項に記載のAT III変異体。
(1) 392位のGlyをProに変換(2) 391, 392位のAla-GlyをPhe-Proに変換(3) 391, 392位のAla-GlyをIle-Proに変換(4) 391, 392位のAla-GlyをGly-Proに変換(5) 391, 392位のAla-GlyをTyr-Proに変換(6) 391, 392位のAla-GlyをTrp-Proに変換(7) 391, 392位のAla-GlyをVal-Proに変換(8) 391, 392位のAla-GlyをLeu-Proに変換(9) 390, 391, 392位のIle-Ala-GlyをAla-Val-Proに変換(10) 390, 391, 392位のIle-Ala-GlyをLeu-Phe-Proに変換(11) 390, 391, 392位のIle-Ala-GlyをAla-Tyr-Proに変換(12) 390, 391, 392位のIle-Ala-GlyをAla-Trp-Proに変換(13) 390, 391, 392位のIle-Ala-GlyをLeu-Trp-Proに変換(14) 390, 391位のIle-AlaをAla-Valに変換(15) 390, 391位のIle-AlaをAla-Ileに変換(16) 390, 391位のIle-AlaをAla-Leuに変換(17) 390, 391位のIle-AlaをGly-Leuに変換(18) 384位のAlaをGlyに変換(19) 389位のValをProに変換(20) 387位のAlaをPheに変換(21) 398位のAsnをGluに変換(22) 398位のAsnをArgに変換(23) 125位のLysをGlnに、392位のGlyをProに変換(24) 125位のLysをGlnに、391, 392位のAla-GlyをPhe-Proに変換(25) 132, 133位のArg-LysをGln-Asnに、392位のGlyをProに変換(26) 132, 133位のArg-LysをGln-Asnに、391, 392位のAla-GlyをPhe-Proに変換(27) 132, 133位のArg-LysをGln-Asnに、391, 392位のAla-GlyをVal-Proに変換(28) 132, 133位のArg-LysをGln-Asnに、390, 391位のIle-AlaをAla-Leuに変換(29) 129位のArgをGlnに、391, 392位のAla-GlyをPhe-Proに変換(30) 11位のLysをIleに、14位のAspをSerに、391, 392位のAla-GlyをPhe-Proに変換(31) 125位のLysをGlnに、129位のArgをGlnに、132, 133位のArg-LysをGln-Asnに、391, 392位のAla-GlyをPhe-Proに変換(32) 132位のArgをGlnに、391, 392位のAla-GlyをPhe-Proに変換(33) 133位のLysをAsnに、391, 392位のAla-GlyをPhe-Proに変換(34) 133位のLysをGlnに、391, 392位のAla-GlyをPhe-Proに変換(35) 125位のLysをGlnに、132, 133位のArg-LysをGln-Asnに、391, 392位のAla-GlyをPhe-Proに変換
【請求項11】 アミノ酸の変換が上記(1)〜(31)から選ばれるいずれかの変換で、それ以外の変換が行われていない、請求項10記載のAT III変異体。
【請求項12】 アミノ酸の変換が上記(2), (7), (16), (24), (26), (27),(28), (29), (30)あるいは(31)のいずれかの変換で、それ以外の変換が行われていない、請求項11記載のAT III変異体。
【請求項13】 請求項1〜12のいずれか一項に記載のAT III変異体をコードするDNA。
【請求項14】 請求項13記載のDNAを含有するAT III変異体を発現可能なベクター。
【請求項15】 請求項14記載のベクターにより形質転換された宿主細胞。
【請求項16】 形質転換された宿主細胞が大腸菌または動物細胞である請求項15記載の宿主細胞。
【請求項17】 請求項15又は16記載の宿主細胞を培養し、産生されたAT III変異体を培養物中より分取することを特徴とするAT III変異体の製造方法。
【請求項18】 請求項1〜12のいずれか一項に記載のAT III変異体と薬学的に許容しうる担体とを含有する血栓性疾病用医薬組成物。
【図1】

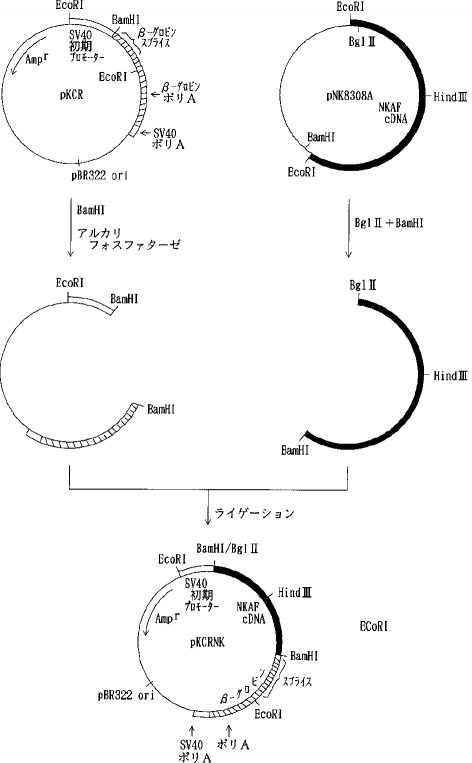
【図2】
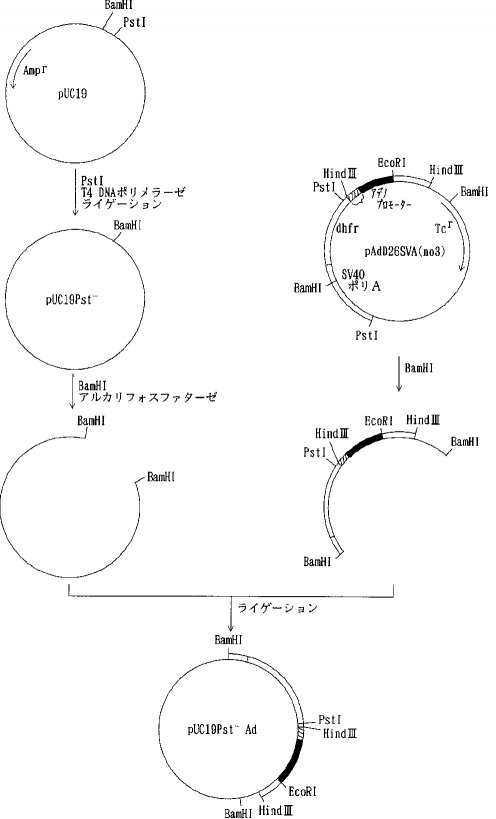
【図3】
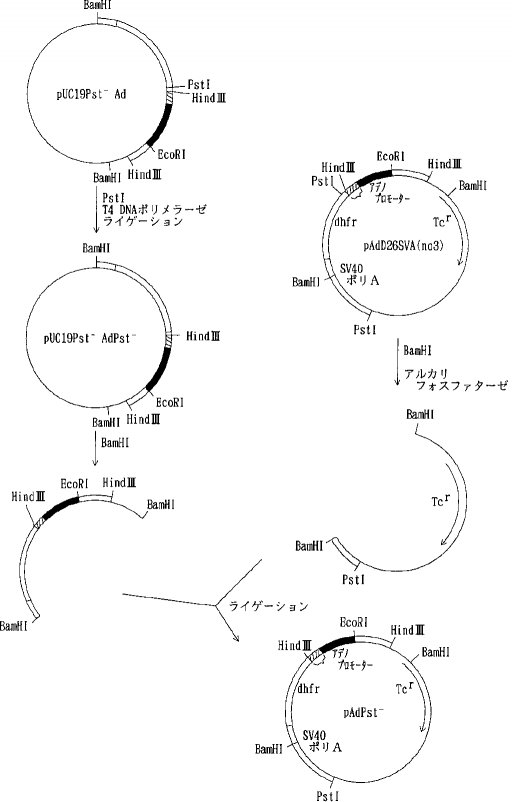
【図4】
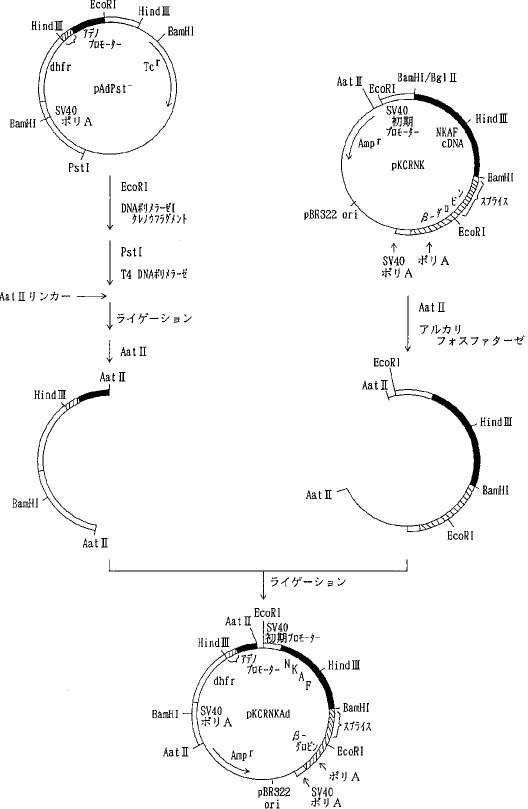
【図5】
【図6】
【図7】
【図9】
【図8】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図9】
【図8】
【図10】
【特許番号】特許第3479539号(P3479539)
【登録日】平成15年10月3日(2003.10.3)
【発行日】平成15年12月15日(2003.12.15)
【国際特許分類】
【出願番号】特願平5−31855
【出願日】平成5年2月22日(1993.2.22)
【公開番号】特開平5−339292
【公開日】平成5年12月21日(1993.12.21)
【審査請求日】平成12年2月7日(2000.2.7)
【出願人】(000000217)エーザイ株式会社 (102)
【参考文献】
【文献】特開 平2−262598(JP,A)
【登録日】平成15年10月3日(2003.10.3)
【発行日】平成15年12月15日(2003.12.15)
【国際特許分類】
【出願日】平成5年2月22日(1993.2.22)
【公開番号】特開平5−339292
【公開日】平成5年12月21日(1993.12.21)
【審査請求日】平成12年2月7日(2000.2.7)
【出願人】(000000217)エーザイ株式会社 (102)
【参考文献】
【文献】特開 平2−262598(JP,A)
[ Back to top ]