
ヒトインシュリン様成長因子−1受容体に対する完全ヒト抗体
【課題】ヒトインシュリン-様成長因子-I受容体(IGF-IR)に結合するヒト抗体、これらの抗体の誘導体(Fab、単一鎖抗体、二重特異性抗体又は融合タンパク質)、及び治療法及び診断法の提供。
【解決手段】インシュリン様成長因子−I受容体(IGF−IR)に特異的に結合し、かつ以下、(i)IGF−I又はIGF−IIからIGF−IRへの結合を阻害する。(ii)IGF−I又はIGF−IIによってIGF−IRの活性化を中和する。(iii)IGF−IR表面受容体を少なくとも約80%減少させる。及び、(iv)1×10−10M−1以下のKdでIGF−IRに結合する、からなる群より選ばれる少なくとも1つの性質を有する、単離ヒト抗体又はその断片。
【解決手段】インシュリン様成長因子−I受容体(IGF−IR)に特異的に結合し、かつ以下、(i)IGF−I又はIGF−IIからIGF−IRへの結合を阻害する。(ii)IGF−I又はIGF−IIによってIGF−IRの活性化を中和する。(iii)IGF−IR表面受容体を少なくとも約80%減少させる。及び、(iv)1×10−10M−1以下のKdでIGF−IRに結合する、からなる群より選ばれる少なくとも1つの性質を有する、単離ヒト抗体又はその断片。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、本発明は、インシュリン様成長因子-I受容体(IGF-IR)に特異的な抗体又はその断片、及び抗体をコードする単離又は精製ポリヌクレオチド配列に関する。
【背景技術】
【0002】
本出願は、2003年5月1日に出願された米国仮出願第60/467,177号の利益を請求する。
【0003】
背景
インシュリン様成長因子受容体(IGF-IR)は、標準的な胎児及び出生後の成長及び発達に不可欠な、至る所に存在する膜貫通型チロシンキナーゼ受容体である。IGF-IRは、細胞増殖、細胞分化、細胞サイズの変化及を刺激し、細胞をアポトーシスから保護することができる。それは、細胞形質転換の準義務的であるとも考えられてきた(Adamsら, Cell. Mol. Life Sci. 57: 1050-93 (2000); Baserga, Oncogene 19: 5574-81 (2000)で調べられる)。IGF-IRは、ほとんどの細胞種に存在し、成長因子IGF-I及びIGF-IIのシグナル分子(以下、IGF類と総称される)として役立つ。IGF-IRはまた、それがIGF類に結合する親和性が3オーダー低いにもかかわらず、インシュリンに結合する。IGF-IRは、ジスルフィド結合によって共有結合的に結合された2個のアルファ鎖及び2個のベータ鎖を含む、前形成ヘテロ-四量体である。受容体サブユニットは、180kdの単一ポリペプチド鎖の一部として合成され、次いで、アルファ(130kd)及びベータ(95kd)サブニットにタンパク質分解される。完全なアルファ鎖は細胞外にあり、リガンド結合部位を含む。ベータ鎖は、膜貫通型ドメイン、チロシンキナーゼドメイン及び細胞の分化及び形質転換に必要なC-末端伸張を有するが、マイトジェンシグナル伝達及びアポトーシスからの保護は不可欠ではない。
【0004】
IGF-IRは、インシュリン受容体(IR)に高い類似性を有し、特にベータ鎖配列(70%相同性)内にある。この相同性のため、これらの受容体が1個のIR二量体及び1個のIGF-IR二量体を含むハイブリッドを形成することができることが、近年の研究で証明されている(Pandini ら, Clin. Canc. Res. 5: 1935-19 (1999))。ハイブリッドの形成は、両方の標準的な形質転換細胞で起こり、ハイブリッド含有量は、細胞内の2個のホモダイマー受容体(IR及びIGF-IR)の濃度に依拠する。39個の乳癌検体の一研究では、IR及びIGF-IRは全ての腫瘍試料で過剰発現したが、ハイブリッド受容体含有量は、常に、両ホモ-受容体のレベルを約3倍越えた(Pandiniら, Clin. Canc. Res. 5: 1935-44 (1999))。ハイブリッド受容体は、IR及びIGF-IRの対から成るが、ハイブリッドは、IGF-IRの親和性に類似した親和性でIGF類に選択的に結合し、インシュリンには弱く結合するにすぎない(Siddle及びSoos, The IGF System. Humana Press. pp. 199-225. 1999)。そのため、これらのハイブリッドはIGF類に結合し、両方の標準的な形質転換細胞にシグナルを形質導入することができる。
【0005】
第二のIGF受容体、IGF-IIR又はマンノース-6-リン酸(M6P)受容体はまた、高親和性でIGF-IIリガンドに結合するが、チロシンキナーゼ活性を有しない(Oatesら, Breast Cancer Res. Treat. 47: 269-81 (1998))。それはIGF-IIRの分解を招くため、このリガンドの成長促進効果を拮抗するIGF-IIの沈降物であると考えられる。腫瘍細胞のIGF-IIRの喪失は、IGF-IIのIGF-IRとの結合に与える拮抗作用の緩和によって、成長性を亢進することができる(Byrdら, J.Biol. Chem. 274: 24408-16 (1999))。
【0006】
IGF-Iの内分泌発現は、主に成長ホルモンによって調節され、肝臓で起こるが、近年の証拠は、多くの他の組織種もIGF-Iを発現できることを示唆している。従って、多くの腫瘍細胞種の場合には、このリガンドは内分泌調節及び傍分泌調節並びに自己分泌調節を受ける(Yu, H.及びRohan, J., J. Natl. Cancer Inst. 92: 1472-89 (2000))。
【0007】
IGFとの特異的結合親和性を有する6種のIGF結合タンパク質(IGFBP)は、血清中で特定されてきた(Yu, H及びRohan, J., J. Natl. Cancer Inst. 92: 1472-89 (2000))。翻訳後修飾の結果として、結合タンパク質の分子構造によって決定されるように、IGFBPは、IGFの作用を亢進又は阻害することができる。それらの主な役割は、IGFの輸送、IGFのタンパク質分解からの保護及びIGFとIGF-IRとの相互作用の調節である。血清IGF-Iのたったの約1%が遊離リガンドとして存在し、残余はIGFBPと会合している(Yu, H.及びRohan, J., J. Natl. Cancer Inst. 92: 1472-89 (2000))。
【0008】
リガンド(IGF)の結合において、IGF-IRは、ベータ鎖の触媒ドメイン内の保存的チロシン残基で自己ホスホリル化される。ベータ鎖内の別のチロシン残基の次のホスホリル化は、シグナルカスケードに匹敵する下流分子の採用のためのドッキング部位を提供する。IGFシグナルの形質導入の主な経路は、マイトジェン活性化プロテインキナーゼ(MAPK)及びホスファチジルイノシトール 3-キナーゼ(PI3K)である(BlakesleyらのThe IGF System. Humana Press. 143-163 (1999)を参照されたい)。MARK経路は、IGF類刺激の後に誘発されるマイトジェンシグナルに主に起因し、PI3Kは、抗-アポトーシス又は生存過程のIGF-依存誘起に起因する。
【0009】
IGF-IRシグナル伝達の重要な役割は、その抗-アポトーシス又は生存機能である。活性化IGF-IRシグナルは、PI3K、Aktの下流ホスホリル化又はタンパク質キナーゼBをシグナル伝達する。Aktは、ホスホリル化によって、プログラムされた細胞死の開始に必須であり、アポトーシスの開始を阻害するBAD等の分子を効果的に阻害できる(Dattaら, Cell 91: 231-41 (1997))。アポトーシスは、標準的な発達過程に匹敵する重要な細胞機構である(Oppenheim, Annu. Rev. Neurosci. 14: 453-501 (1991))。それは、重度の障害性細胞の除去に影響を与え、腫瘍形成を促進する変異原生障害の潜在的固執を軽減するための重要な機構である。このために、IGFのシグナル伝達の活性化は、マウストランスジェニックモデルでの自然発生癌形成を促進することができる、ことが証明されている(DiGiovanniら, Cancer Res. 60: 1561-70 (2000))。更に、IGF過剰発現は、化学療法誘導細胞死由来の細胞を救援することができ、腫瘍細胞薬物耐性において重要な因子となる(Goochら, Breast Cancer Res. Treat. 56: 1-10 (1999))。従って、IGFシグナル伝達経路の調節は、化学療法剤に対する腫瘍細胞の感受性を増加させることが判っている(Beniniら, Clinical Cancer Res. 7: 1790-97 (2001))。
【0010】
多数の調査及び臨床的研究は、癌の発症、維持及び進行におけるIGF-IR及びそのリガンド(IGF)に関与してきた。腫瘍細胞では、受容体の過剰発現は、しばしばIGFリガンドの過剰発現に呼応しており、これらのシグナルの相乗作用をもたらし、結果として細胞増殖及び生存を亢進する。IGF-I及びIGF-IIは、幅広い癌細胞株、例えば前立腺癌(Nickersonら, Cancer Res. 61: 6276-80 (2001) ; Hellawellら, Cancer Res. 62: 2942-50 (2002) )、乳癌(Goochら, Breast Cancer Res. Treat. 56: 1-10 (1999))、肺癌、結腸癌(Hassan及びMacaulay, Ann. Oncol. 13: 349-56 (2002) )、胃癌、白血病、膵癌、脳腫瘍、骨髄腫(Ge及びRudikoff, Blood 96: 2856-61 (2000)、黒色腫(All-Ericssonら, Invest. Ophthalmol. Vis. Sci. 43: 1-8 (2002))及び卵巣癌(Macaulay, Br. J. Cancer 65: 311-20 (1990)を参照されたい)の強力なマイトジェンであることが判っており、この効果はIGF-IRによって仲介される。血清中のIGF-Iの高循環レベルは、乳癌、前立腺癌及び結腸癌の高い危険性に関係している(Pollak, Eur. J. Cancer 36: 1224-28 (2000))。結腸癌のマウスモデルにおいて、in vivoでの循環IGF-Iレベルの増加は、腫瘍成長及び転移の発生率の顕著な増加をもたらす(Wuら, Cancer Res. 62: 1030-35 (2002))。トランスジェニックマウスの表皮基底細胞でのIGF-Iの構成的発現は、自然発生的腫瘍形成を促進することが判っている(DiGiovanniら, Cancer Res. 60: 1561-1570, 2000; Bolら,Oncogene 14: 1725-1734 (1997))。細胞株及び腫瘍でのIGF-IIの過剰発現は高頻度で生じ、IGF-II遺伝子のゲノムインプリンティングの喪失から起こる(Yaginumaら, Oncology 54: 502-7 (1997))。受容体過剰発現は、多種多様のヒト腫瘍種、例えば肺癌(Quinnら, J.Biol. Chem. 271: 11477-83 (1996))、乳癌(Cullenら, Cancer Res. 50: 48-53 (1990); Peyrat及びBonneterre, Cancer Res. 22: 59-67 (1992); Lee and Yee, Biomed Pharmacother. 49: 415-21 (1995))、肉腫(van Valenら, J. Cancer Res. Clin. Oncol. 118: 269-75 (1992); Scotlandiら, Cancer Res. 56: 4570-74 (1996))、前立腺癌(Nickersonら ,Cancer Res. 61: 6276-80 (2001))及び結腸癌(Hassan及びMacaulay, Ann. OticoL 13: 349-56 (2002))において証明されてきた。加えて、高変異原生癌細胞は、転移する傾向が少ない腫瘍細胞に比べて、IGF-II及びIGF-IRを高発現することが判っている(Guerraら, Int. J. Cancer 65: 812-20 (1996))。細胞増殖及び形質転換でのIGF-IRの重要な役割は、マウス胚繊維芽細胞由来のIGF-IRノックアウトの実験で証明された。これらの主な細胞は、10%血清を含む培地で著しく低い速度で成長し、SV40 Large T抗原を含む様々な癌遺伝子によって形質転換されない(Sellら, Mol. Cell. Biol. 3604-12 (1994))。近年、いくつかの形の乳癌における薬物ハーセプチンへの耐性は、当該癌ではIGF-IRシグナル伝達の活性化に起因する、ことが証明された(Luら, J. Natl. Cancer Inst. 93: 1852-57 (2001))。そのため、IGF-IRの過剰発現又は活性化は、腫瘍形成での主な決定基であるだけでなく、腫瘍細胞において薬物耐性でもある。
【0011】
IGF系の活性化はまた、癌、例えば末端肥大症(Drange及びMelmed. In : The IGF System. Humana Press. 699-720 (1999))、網膜新生血管(Smithら, Nature Med. 12: 1390-95 (1999))、及び乾癬(Wraightら, Nature Biotech. 18: 521-26 (2000))に加えて、いくつかの病理学的条件に関与している。最近の研究では、IGF-IRを標的とするアンチセンスオリゴヌクレオチド調製は、マウスモデルでの乾癬皮膚移植における表皮細胞の高増殖を顕著に阻害する点で効果的であった。これは、抗-IGF-IR療法がこの慢性的疾患の治療に効果的であることを示唆している。
【発明の概要】
【発明が解決しようとする課題】
【0012】
細胞でのIGF-IRシグナル伝達経路を阻害するために様々な戦略が開発されてきた。アンチセンスオリゴヌクレオチドは、乾癬について上に示したように、in vitro及び実験的マウスで効果的であった。加えて、in vitro及びin vivoで抗-増殖活性を有する、IGF-IRを標的とする阻害ペプチドが産生されている(Pietrzkowskiら, Cancer Res. 52: 6447-51 (1992); Haylorら, J. Am. Soc. Nephrol. 11: 2027-35 (2000))。IGF-IRのC-末端由来の合成ペプチド配列は、アポトーシスを誘導し、腫瘍成長を顕著に阻害することが判っている(Reissら, J. Cell. Phys. 181: 124-35 (1999))。腫瘍細胞株で過剰発現させた場合に、リガンドを野生型IGF-IRと競合し、in vitro及びin vivoでの腫瘍細胞成長を効果的に阻害する、様々なIGF-IRの優性遺伝子-陰性突然変異体も作製されている(Scotlandiら, Int. J. Cancer 101: 11-6 (2002); Seelyら, BMC Cancer 2: 15 (2002))。加えて、溶解型IGF-IRはまた、in vivoで腫瘍成長を阻害することが証明されている(D'Ambrosioら, Cancer Res. 56: 4013-20 (1996))。ヒトIGF-IRに対する抗体はまた、乳癌(Artega andOsborne, Cancer Res. 49: 6237-41 (1989))、ユーイング骨肉腫(Scotlandiら, Cancer Res. 58: 4127-31 (1998))及び黒色腫(Furlanettoら, Cancer Res. 53: 2522-26 (1993))由来の細胞株を含む、in vitroでの腫瘍細胞増殖及びin vivoでの腫瘍形成を阻害することが判っている。抗体は、主に、それらが、1)特定のタンパク質抗原に対して高い選択性を有することができる、2)抗原に高親和性結合を示すことができる、3)in vivoで長半減期を示す、ために魅力的な治療法であり、またそれらは天然の免疫産物であるため、4)in vivoでの毒性が低い(Park及びSmolen. In : Advances in Protein Chemistry. Academic Press. pp: 360-421 (2001))。しかしながら、非-ヒト起源、例えばマウス由来の抗体は、反復適用に従って治療抗体に対して直接的免疫応答をもたらし、抗体の効力を中和する。ヒトではネズミ又はキメラ抗体よりも免疫原生が低い可能性があり、天然免疫-応答抗体に類似しているので、完全ヒト抗体は、ヒト治療薬の成功の最も高い可能性を提供する。このために、治療用途のための、高親和性ヒト抗-IGF-IRモノクローナル抗体を開発する必要がある。
【課題を解決するための手段】
【0013】
発明の要約
本発明は、ヒトIGF-I受容体に特異的に結合するヒトモノクローナル抗体及びその断片を提供する。抗体は、(i)IGF-I又はIGF-IIのIGF-IRへの結合を阻害する;(ii)IGF-I又はIGF-IIによってIGF-IRの活性化を中和する;(iii)IGF-IR表面受容体を少なくとも約80%減少させる;及び(iv)約3×10-10M-1以下のKdでIGF-IRに結合する、からなる群より選ばれる少なくとも1つの性質を有する。より好ましい実施態様では、本発明の抗体は、IGF-IR表面受容体を少なくとも約85%、より好ましくは少なくとも約90%減少させる。更に、抗体は、リガンド-介在受容体自己ホスホリル化、及びMAPK及びAkt経路を介する下流細胞シグナル伝達を阻害する。本発明の抗体は、単独又は抗新形成剤と組み合わせて使用され、特に腫瘍性疾患及び高増殖疾患に有用である。
【0014】
本発明は、抗体又はその断片をコードする単離ポリヌクレオチド、当該ポリヌクレオチド配列を含む発現ベクター及び発現のための宿主細胞を提供する。
【0015】
更に、本発明は、医薬組成物及び腫瘍及び高増殖疾患の治療のために診断方法及び治療方法を提供する。当該方法は、抗新形成剤の投与又は治療を更に含むことができる。
【図面の簡単な説明】
【0016】
【図1】図1は、2F8重鎖可変ドメインのヌクレオチド配列を示す。
【図2】図2は、2F8重鎖可変ドメインのアミノ酸配列を示す。CDRは太字で下線を引いてある。
【図3】図3は、完全2F8重鎖のヌクレオチド配列を示す(下線: 分泌シグナル配列; 斜体: IgG1不変領域)。
【図4】図4は、完全2F8重鎖のアミノ酸配列を示す(下線: 分泌シグナル配列; 太字: CDR; 斜体:IgG1不変領域)。
【図5】図5は、2F8軽鎖可変ドメインのヌクレオチド配列を示す。
【図6】図6は、2F8軽鎖可変ドメインのアミノ酸配列を示す。CDRは太字で下線を引いてある。
【図7】図7は、完全2F8軽鎖のヌクレオチド配列を示す(下線: 分泌シグナル配列; 斜体: IgG1不変領域)。
【図8】図8は、完全2F8軽鎖のアミノ酸配列を示す(下線: 分泌シグナル配列; 太字: CDR; 斜体:IgG1不変領域)。
【図9】図9は、A12軽鎖可変ドメインのヌクレオチド配列を示す。
【図10】図10は、A12軽鎖可変ドメインのアミノ酸配列を示す。CDRは太字で下線を引いてある。
【図11】図11は、完全A12軽鎖のヌクレオチド配列を示す(下線: 分泌シグナル配列; 斜体: IgG1不変領域)。
【図12】図12は、完全A12軽鎖のアミノ酸配列を示す(下線: 分泌シグナル配列; 太字: CDR; 斜体:IgG1不変領域)。
【図13】図13は、抗体2F8及びA12のVH及びVL CDR配列を示す。VL CDRの相違に下線を引いてある。
【図14】図14は、2F8及びA12の軽鎖可変領域アミノ酸配列間の相同性を示す。配列の相違を四角で囲み、CDRを強調してある。
【図15】図15は、固定化溶解性IGF-IRへのIGF-Iの結合を阻害する抗体2F8及びA12の能力を測定するアッセイの結果を示す。カッパ及びラムダ軽鎖不変領域を有するA12抗体を試験した。
【図16】図16は、IGF-IのMCF7細胞への結合を阻害する抗体2F8及びA12(ラムダ軽鎖)の能力を測定するアッセイの結果を示す。
【図17】図17は、インシュリンのZR75-I細胞への結合を阻害する抗体2F8及びA12の能力を測定するアッセイの結果を示す。
【図18A】図18Aは、MCF7乳癌細胞依存性有糸分裂生起アッセイにおける3H-チミジン取り込みに与えるA12抗体の影響を示す。
【図18B】図18Bは、BxPC-3膵臓癌細胞依存性有糸分裂生起アッセイにおける3H-チミジン取り込みに与えるA12抗体の影響を示す。
【図18C】図18Cは、HT-29結腸癌細胞依存性有糸分裂生起アッセイにおける3H-チミジン取り込みに与えるA12抗体の影響を示す。
【図19A】図19Aは、IGF-I介在受容体ホスホリル化の阻害を示す。抗体A12及び2F8によるMCF7乳癌細胞での阻害。
【図19B】図19Bは、IGF-I介在受容体ホスホリル化の阻害を示す。抗体A12によるHT-29結腸癌細胞及びBxPC-3膵臓癌細胞での阻害。
【図20A】図20Aは、抗体A12及び2F8による下流エフェクター分子のIGF-I介在MAPKホスホリル化の阻害を示す。
【図20B】図20Bは、抗体A12及び2F8による下流エフェクター分子のIGF-I介在Aktホスホリル化の阻害を示す。
【図21】図21は、ヒト及びネズミIGF-IR陽性及び陰性細胞株への抗体A12の結合を示す。MCF7: ヒト乳癌細胞株; R-: マウス胚繊維芽細胞; HEL: ヒト白血病細胞; ルイス肺: マウス肺癌細胞。
【図22A】図22Aは、MCF7細胞上のIGF-IRへの結合後の標識抗体A12の内在化を示す。
【図22B】図22Bは、細胞表面会合IGF-IRの減少を示す。
【図22C】図22Cは、A12で長期処理後の総細胞IGF-IRの分解を示す。
【図23】図23は、抗体SF8及びCPT-11(イリノテカン)の単独又は併用によるヌードマウスでのHT-29ヒト結腸腫瘍成長の阻害を示す。
【図24】図24は、ヌードマウスでのHT-29ヒト結腸腫瘍成長に与える抗体A12の効果を示す。
【図25】図25は、ヌードマウスでのMCF7ヒト乳癌成長に与える抗体A12の効果を示す。
【図26】図26は、抗体A12、ゲムシタビン又はCPT-11(イリノテカン)を単独又は併用するヌードマウスでのBxPC-3膵臓癌異種移植の阻害を示す。
【図27】図27は、抗体A12、パクリタキセル又はCPT-11(イリノテカン)を単独又は併用するヌードマウスでのHT-29結腸癌異種移植の阻害を示す。
【発明を実施するための形態】
【0017】
発明の詳細な説明
本発明のある実施態様では、ヒト抗体が提供される。抗体はまた、その他の化学剤及び生物剤、例えば特に限定されないが、抗新形成剤、及び/又は細胞成長を介在するその他の受容体もしくは受容体基質の阻害剤である薬剤、と組み合わせて使用することができる。本発明は、トポイソメラーゼ機能の阻害剤である抗新形成剤に更に関する。かかる薬剤の選択は、IGF-IRに特異的な抗体と組み合わせた治療方法での使用に有利である。
【0018】
天然抗体は典型的には、2個の同一重鎖及び2個の同一軽鎖を有し、各軽鎖は共有結合的に鎖間ジスルフィド結合によって重鎖に結合し、及び複数のジスルフィド結合は2個の重鎖に互いに更に結合する。個々の鎖は、類似のサイズ(110〜125アミノ酸)及び構造を有し、機能は異なるドメインに折畳むことができる。軽鎖は、1可変ドメイン(VL)及び/又は1不変ドメイン(Css)を含むことができる。この重鎖はまた、1可変ドメイン(VH)及び/又は抗体のクラス又はアイソタイプによって3又は4の不変ドメイン(CH1、CH2、CH3及びCH4)を含むことができる。ヒトでは、アイソタイプはIgA、IgD、IgE、IgG及びIgMであり、IgGは更にサブクラス又はサブタイプ(IgA1-2及びIgG1-4)に細分される。
【0019】
一般的に、可変ドメインは、1抗体から次の抗体まで、特に抗原結合部位の位置ではかなりのアミノ酸配列変化を示す。超可変又は相補性決定部位(CDR)と呼ばれる3領域は、VL及びVHの各ドメインに見られ、当該領域は、フレームワークと呼ばれる変化の少ない領域によって支持されている。
【0020】
VL及びVHドメインからなる抗体の部分は、Fv(可変断片)を示し、抗原結合部位を構成する。単一鎖Fv(scFv)は、VLドメイン及びVHドメインを1ポリペプチド鎖上に含む抗体断片であり、1ドメインのN末端及びその他のドメインのC末端は、適応性のあるリンカーで結合されている(米国特許第4,946,778号明細書(Ladnerら); 国際公開第 WO 88/09344号パンフレット(Hustonら)を参照されたい)。国際公開第 WO92/01047号パンフレット(McCaffertyら)は、溶解性組換え遺伝子ディスプレイパッケージ、例えばバクテリオファージの表面上のscFv断片のディスプレイを記載する。
【0021】
単一鎖抗体を産生するために使用されるペプチドは、正確な3次元折畳み及びVL及びVHドメインの会合が起こることを確実にするために選択された適合性のあるペプチドでよい。リンカーは一般的に、10〜50アミノ酸残基である。好ましくは、リンカーは10〜30アミノ酸残基である。より好ましくは、リンカーは12〜30残基である。最も好ましくは、リンカーは15〜25アミノ酸残基である。かかるリンカーペプチドの非限定的例は、(Gly-Gly-Gly-Gly-Ser)3(配列番号33)である。
【0022】
Fab(抗原結合断片)は、VL-CL及びVH-CH1ドメインからなる抗体の断片を言う。パパインによる完全抗体の消化によって得られたかかる断片は、それによって2個の重鎖が通常結合される、抗体のヒンジ部位を保持しない。当該断片は、一価であり、単にFabと言われる。あるいは、ペプシンによる消化は、ヒンジ部位を保持する断片を生じる。2個の重鎖を結合する完全な鎖間ジスルフィド結合を有するこのような断片は、二価であり、F(ab')2と言われる。F(ab')2のジスルフィド結合が緩和され、重鎖が分離される場合に、一価のFab'が生成する。それらは二価であるため、完全抗体及びF(ab')2断片は、一価のFab又はFab'断片よりも高い抗原親和力を有する。国際公開第 WO 92/01047号パンフレット(McCaffertyら)は、溶解性組換え遺伝子ディスプレイパッケージ、例えばバクテリオファージの表面上のFab断片のディスプレイを記載する。
【0023】
Fc(結晶化可能断片)は、一対の重鎖不変ドメインからなる抗体の部分又は断片を言う。IgG抗体例えばFcは、重鎖CH2及びCH3ドメインからなる。IgA又はIgM抗体のFcは、CH4ドメインを更に含む。Fcは、Fc受容体結合、補体依存性細胞障害活性の活性化及び抗体依存性細胞障害活性(ADCC)の活性化に関連する。複数のIgG様タンパク質の複合体であるIgA及びIgMのような抗体に関して、複合体形成はFc不変ドメインを必要とする。
【0024】
最後に、互いにかつFcに対してFabが可動性である場合には、2個の重鎖の共有結合のための複数のジスルフィド結合を含めて、ヒンジ部位は、抗体のFab及びFc部分を分離する。
【0025】
結合特異性を保持する抗体型が開発されているが、望ましいその他の特徴、例えば生物特異性、多価性(2個より多い結合部位)、コンパクトサイズ(例えば結合ドメインのみ)を有する。
【0026】
単一鎖抗体は、それら由来する完全抗体の不変ドメインのいくつか又は全てが欠落している。そのため、それらは、完全抗体の使用に関連する問題のいくつかを解消することができる。例えば、単一鎖抗体は、重鎖不変領域及び他の生物分子間の特定の望ましくない相互作用がない傾向にある。加えて、単一鎖抗体は、完全抗体よりもかなり小さく、完全抗体よりも透過性が高い。そのため、より効果的に単一鎖抗体を抗原結合部位に配置し及び当該部位を標的することができる。更に、かなり小さいサイズの単一鎖抗体は、完全抗体に比べて、レシピエントにおける好ましくない免疫応答を誘起しないようである。
【0027】
複数の単一鎖抗体であって、各単一鎖が第一のペプチドリンカーによって共有結合的に結合された1VH及び1VLドメインを有する抗体は、少なくとも1以上のペプチドリンカーによって共有結合的に結合されて、単一特異的でも又は多重特異的でもよい多価単一鎖抗体を形成する。多価単一鎖抗体の各鎖は、可変軽鎖断片及び可変重鎖断片を含み、ペプチドリンカーによって少なくとも1つの他の鎖に結合される。ペプチドリンカーは、少なくとも15アミノ酸残基からなる。アミノ酸残基の最大数は約100である。
【0028】
2個の単一鎖抗体は混合されて、二価二量体としても知られている二重特異性抗体を形成することができる。二重特異性抗体は、2個の鎖及び2個の結合部位を有し、単一特異的でも又は多特異的でもよい。二重特異性抗体の各鎖は、VLドメインに結合したVHドメインを含む。ドメインは、十分に短いために同一鎖上のドメイン間でペアリングが起こらないリンカーと結合される。それによって、異なった鎖上の相補性ドメイン間でのペアリングを推進して2個の抗原結合部位を再現する。
【0029】
3個の単一鎖抗体は混合されて三価三量体としても知られている三重特異性抗体(triabodies)を形成することができる。三重特異性抗体は、VL又はVHドメインのカルボキシル末端に直接連結したVL又はVHドメインのアミノ酸末端で、すなわちリンカー配列を用いることなく、構築される。三重特異性抗体は、環状の頭-尾形式に配置されたポリペプチドを有する3個のFv頭部を有する。三重特異性抗体の可能な立体配置は、3個の結合部位と平面関係にあり、当該部位は1つの平面内にその他と120度の角度にある。三重特異性抗体は、単一特異的でも二重特異的でも又は三重特異的でもよい。
【0030】
従って、本発明の抗体及びその断片は、特に限定されないが、抗原に特異的に結合する天然抗体、二価断片例えば(Fab')2、一価断片例えばFab、単一鎖抗体、単一鎖Fv(scFv)、単一ドメイン抗体、多価単一鎖抗体、二重特異性抗体、三重特異性抗体などを含む。
【0031】
本発明の抗体及び特にその可変ドメインは、当該分野で公知の方法によって得られる。これらの方法は、例えば、Kohler及びMilstein, Nature, 256: 495-497 (1975)及びCampbell, Monoclonal Antibody Technology, The Production and Characterization of Rodent and Human Hybridomas, Burdonら著, Laboratory Techniques in Biochemistry and Molecular Biology, 第13巻, Elsevier Science Publishers, Amsterdam (1985)によって記載されている免疫法;例えばHuseら, Science, 246,1275-81 (1989)によって記載されている組換えDNA法を含む。抗体は、scFv又はFabの形態でのVH及びVLドメインの組み合わせを生じるファージディスプレイ・ライブラリーからも得られる。VH及びVLドメインは、合成、半合成又は天然由来のヌクレオチドによってコードされる。ある実施態様では、ヒト抗体断片を生じるファージディスプレイ・ライブラリーが好ましい。ヒト抗体の他の起源は、ヒト免疫グロブリン遺伝子を発現するように設計されたトランスジェニックマウスである。
【0032】
抗体断片は、完全抗体を開裂することによって、又は当該断片をコードするDNAを発現することによって作製することができる。抗体断片は、Lamoyiら, J. Inimutiol. Metliods, 56: 235-243 (1983)及びParham, J. Immunol. 131: 2895-2902 (1983)によって記載の方法によって調製することができる。かかる断片は、1又は両Fab断片もしくはF(ab')2断片を含んでもよい。かかる断片は、単一鎖断片可変領域抗体、すなわちscFv、二重特異性抗体又は他の抗体断片を含んでもよい。かかる機能性等価体を製造する方法は、PCT出願の国際公開 WO 93/21319号パンフレット、欧州特許出願第239,400号明細書; PCT出願の国際公開第 WO 89/09622号パンフレット; 欧州特許出願第338,745号;及び欧州特許出願 EP 332,424号に開示されている。
【0033】
本発明の抗体又はその断片はIGF-IRに特異的である。抗体特異性とは、特定の抗原エピトープに対する抗体の選択的認識を言う。本発明の抗体又はその断片は、例えば単一特異性でも二重特異性でもよい。二重特異性抗体(BsAb)は、2種の異なった抗原結合特異性又は部位を有する抗体である。抗体が1より多い特異性を有する場合には、認識エピトープは、単一抗原又は1抗原より多い抗原と関連する。従って、本発明は二重特異性抗体又はその断片を提供する。これらは2種の異なった抗原に結合し、IGF-IRに対して少なくとも1つの特異性を有する。
【0034】
本抗体又はその断片のIGF-IRに対する特異性は、親和性及び/又は親和力に基づいて決定される。抗原と抗体との平衡解離定数によって表される親和性(Kd)は、抗原決定部位及び抗体結合部位間の結合強度を測定する。親和力は、抗体と抗原との間の結合強度を測定する。親和力は、抗体上のその抗原結合部位を有するエピトープ間の親和性、及び抗体価に関連する。この抗体価とは、特定のエピトープに特異的な抗原結合部位の数を言う。抗体は典型的には、10-5〜10-11 リットル/molの解離定数(Kd)で結合する。10-4 リットル/molより大きいKdは、一般的に非特異的結合を示すと考えられる。Kd値が小さくなればなるほど、抗原決定基及び抗体結合部位間の結合強度は強くなる。
【0035】
本発明の抗体又はその断片はまた、結合特性が直接的突然変異、親和性成熟、ファージディスプレイ又は鎖シャフリングによって改善されたものを含む。親和性及び特異性は、改変、又はCDR及び/又はFW残基を突然変異及び所望の特性を有する抗原結合部位をスクリーニングすることによって改良することができる(例えばYangら, J. Mol. Biol., (1995) 254: 392-403を参照されたい)。1つの方法は、2〜20アミノ酸の小集団、そうでなければ同一の抗原結合部位において、当該アミノ酸のサブセットが特定の位置で見出されるように、個々の残基又は残基の組み合わせを無作為化することである。あるいは、突然変異はエラープローンPCR法によって一定の範囲の残基を誘発することができる(例えばHawkinsら, J. Mol. Biol., (1992) 226: 889-96を参照されたい)。別の実施例では、重及び軽鎖可変領域遺伝子を含むファージディスプレイ・ベクターは、E. coliの突然変異誘発株で増殖することができる(例えば、Lowら, J. Mol. Biol., (1996) 250: 359-68を参照されたい)。これらの突然変異誘発方法は、当業者に公知の多くの方法の例示に過ぎない。
【0036】
本発明の抗体又はその断片の等価物は、完全長抗-IGF-IR抗体の可変又は超可変領域のアミノ酸配列と実質的に同一のアミノ酸を有するポリペプチドを含む。Pearson及びLipman (Proc. Natl. Acad. Sci. USA (1988) 85: 2444-8)に従うFASTA調査法によって決定されているように、実質的に同一のアミノ酸配列は、本明細書では、もう1つのアミノ酸配列に対して、少なくとも70%、好ましくは少なくとも約80%及びより好ましくは少なくとも約90%の相同性を有する配列として定義されている。
【0037】
保存的アミノ酸置換は、ペプチド、ポリペプチドもしくはタンパク質、又はその断片の1又は2アミノ酸を置換する方法によるアミノ酸組成物における変更と定義される。置換は、実質的にペプチド、ポリペプチドもしくはタンパク質の特性(例えば、電荷、等電点、親和性、親和力、立体配置、溶解性)又は活性を実質的に変更しないように、一般的に類似の性質(例えば酸性、塩基性、芳香族性、サイズ、正電荷又は負電荷、極性、非極性)を有するアミノ酸についてである。かかる保存的アミノ酸置換について行われる典型的置換は、以下のアミノ酸の群間である:グリシン(G)、アラニン(A)、バリン(V)、ロイシン(L)及びイソロイシン(I);アスパラギン酸(D)及びグルタミン酸(E); アラニン(A)、セリン(S)及びスレオニン(T); ヒスチジン(H)、リジン(K)及びアルギニン(R): アスパラギン(N)及びグルタミン(Q); フェニルアラニン(F)、チロシン(Y)及びトリプトファン(W)。
【0038】
保存的アミノ酸置換は、例えば、分子の選択的及び/又は特異的結合特性に主に起因する超可変領域のフランキング領域、及び分子のタンパク質の部分例えば可変重鎖カセットでなされる。
【0039】
本発明の抗体の各ドメインは、重又は軽鎖可変ドメインを有する完全抗体でもよく、又は天然ドメイン又は例えばin vitroで国際公開第WO 93/11236 (Griffithsら)号パンフレットに記載の方法を用いて構築された合成ドメインの機能性等価体、突然変異体又は誘導体でもよい。例えば、少なくとも1つのアミノ酸が欠落している抗体可変ドメインに対応するドメインと組み合わせることができる。重要な特徴は、相補性ドメインを会合して抗原結合部位を形成する各ドメインの能力である。従って、可変重及び軽鎖断片の用語は、特異性に重大な効果を与えない変異体を排除するものと解釈されるべきではない。
【0040】
好ましい実施態様では、本発明の抗-IGF-IR抗体は1以上の以下の性質を示すヒト抗体である。
【0041】
1) 抗体はIGF-IRの外部ドメインに結合し、IGF-I又はIGF-IIのIGF-IRへの結合を阻害する。例えば、阻害は、精製又は膜結合受容体を用いる直接的結合アッセイを用いて決定することができる。この実施態様において、本発明の抗体又はその断片は、好ましくは、少なくともIGF-IR(IGF-I及びIGF-II)の天然リガンドのような強さでIGF-IRに結合する。
【0042】
2) 抗体はIGF-IRを中和する。リガンド例えばIGF-I又はIGF-IIの、IGF-IRの外部、細胞外ドメインへの結合は、ベータサブユニットの自己ホスホリル化及びIFG-IRサブユニット例えばMAPK、Akt及びIRS-1のホスホリル化を刺激する。
【0043】
IGF-IRの中和は、阻害、減少、不活性化及び/又はシグナル形質導入に通常関連するこれらの活性の1以上の破壊を含む。更に、これは、IGF-IR/IRへテロダイマー及びIGF-IRホモダイマーの阻害を含む。従って、IGF-IRの中和は、様々な効果、例えば阻害、減少、不活性化及び/又は成長破壊(増殖及び分化)、拮抗作用(血管補充、侵入及び転移)、及び細胞運動及び転移(細胞接着及び侵襲性)を有する。
【0044】
IGF-IR中和の1測定は、受容体のチロシンキナーゼ活性の阻害である。チロシンキナーゼ阻害は、周知の方法を用いて測定することができる;例えば組換えキナーゼ受容体の自己ホスホリル化レベル及び/又は天然もしくは合成基質のホスホリル化を測定することである。従って、ホスホリル化アッセイは、本発明の分脈において中和抗体の測定に有用である。例えば、ホスホリル化は、ELISAアッセイ又はウェスタンブロットにおいてホスホチロシンに特異的な抗体を用いて検出することができる。チロシンキナーゼ活性のためのいくつかのアッセイは、Panekら, J. Pharmacol. Exp. Thera. 283: 1433-44 (1997)及びBatleyら, Life Sci. 62: 143-50 (1998)に記載されている。本発明の抗体は、リガンドに応答する細胞において、IGF-IRのチロシンホスホリル化を少なくとも約75%、好ましくは少なくとも約85%及びより好ましくは少なくとも約90%減少させる。
【0045】
IGF-IR中和の別の測定は、IGF-IRの下流基質のホスホリル化の阻害である。従って、MAPK、Akt又はIRS-1のホスホリル化レベルを測定することができる。基質ホスホリル化の減少は、少なくとも約50%、好ましくは少なくとも約65%、より好ましくは少なくとも約80%である。
【0046】
加えて、タンパク質発現の検出方法は、IGF-IR中和を側定するために利用することができる。この測定されるタンパク質はIGF-IRキナーゼ活性によって調節される。これらの方法は、タンパク質発現検出のための免疫組織化学(IHC)、遺伝子増幅の検出のための蛍光in situハイブリダイジェーション(FISH)、競争的放射性リガンド結合アッセイ、固体マトリックスブロッティング法、例えばノーザン及びサザンブロット、逆転写酵素ポリメラーゼ鎖反応((RT-PCR)及びELISAを含む。例えば、Grandis ら, Cancer, 78: 1284-92 (1996); Shimizuら, Japan J. Cancer Res., 85: 567-71 (1994); Sauterら, Am. J. Path., 148: 1047-53 (1996); Collins, Glia 15: 289-96 (1995); Radinskyら, Clin. Cancer Res. 1: 19-31 (1995); Petridesら, Cancer Res. 50: 3934-39 (1990); Hoffmannら, Anticancer Res. 17: 4419-26 (1997); Wikstrandraら, Cancer Res. 55: 3140-48 (1995)を参照されたい。
【0047】
In vivoアッセイは、IGF-IR中和を測定するために利用することもできる。例えば、受容体チロシンキナーゼ阻害は、阻害剤の存在及び非存在下、受容体リガンドで刺激された細胞株を用いるマイトジェンアッセイによって観察できる。例えば、IGF-I又はIGF-11で刺激されたMCF7(American Type Culture Collection (ATCC), Rockville, MD)は、IGF-IR阻害をアッセイするために使用することができる。別の方法は、IGF-IR発現腫瘍細胞又はトランスフェクトされてIGF-IRを発現する細胞の成長阻害試験を含む。阻害は、腫瘍細胞、例えばマウスに注射されたヒト腫瘍細胞を用いて観察することもできる。
【0048】
本発明は、IGF-IR中和の任意の特定のメカニズムによって制限されない。本発明の抗-IGF-IR抗体は、IGF-IR細胞表面受容体、リガンドの切断部位(例えばIGF-I又はIGF-II)及び受容体関連チロシンキナーゼによって仲介される次のシグナル形質導入に外部から結合することができ、及びシグナル形質導入カスケードにおけるIGF-IR及び他の下流タンパク質のホスホリル化を防ぐことができる。
【0049】
3) 抗体はIGF-IRの調節を下げる。細胞表面に存在するIGF-IR量は、受容体タンパク質産生、内在化及び分解に依拠する。細胞表面に存在するIGF-IR量は、受容体又は受容体に結合する分子の内在化を検出することによって、直接的に測定することができる。例えば、受容体内在化は、IGF-IRを発現する細胞を標識抗体と接触することにより測定することができる。次いで、膜結合抗体は剥ぎ取られ、回収され及びカウントされる。内在化抗体は、細胞を溶解し、溶解物中の標識を検出することによって測定される。
【0050】
別の方法は、抗-IGF-IR抗体又は他の基質で処理した後に、細胞表面に存在する受容体量を、例えば、IGF-IRの表面発現用に染色された細胞の蛍光活性化細胞選別分析によって、直接測定することである。染色細胞を37℃でインキュベートし、終始、蛍光強度を測定する。対照として、染色集団の一部は4℃(受容体内在化が停止する条件)でインキュベートすることができる。
【0051】
実施例に記載したように、細胞表面IGF-IRは、IGF-IRに特異的でありかつ試験抗体の結合を妨害しない又は当該結合と競争しない異なった抗体を用いて検出及び測定することができる(Burtrumら Cancer Res. 63: 8912-21 (2003))。IGF-IR発現細胞の本発明の抗体による処理は。細胞表面IGF-IRの減少をもたらす。好ましい実施態様では、この減少は、本発明の抗体での処理に対する応答において、少なくとも約70%、より好ましくは少なくとも約80%及びより好ましくは少なくとも約90%である。顕著な減少は、たった4時間内に観察することができる。
【0052】
低調節の別の測定は、細胞に存在する総受容体タンパク質の減少であり、内部受容体の分解を反映する。従って、細胞(特に癌細胞)の本発明の抗体による処理は、総細胞IGF-IRの減少を招く。好ましい実施態様では、当該減少は、少なくとも約70%、より好ましくは少なくとも約80%及び更により好ましくは少なくとも約90%である。
【0053】
本発明の抗体は、約3 x 10-l0 M-1以下、好ましくは約1 x 10-10 M-1以下及びより好ましくは約3 x 10-11 M-1以下のKdでIGF-IRに結合する。
【0054】
本発明の実施態様では、抗体は腫瘍成長を阻害する。例えば、皮下異種移植腫瘍は、癌細胞株の細胞の免疫不全マウスに注射することによって樹立することができる。次いで、マウスを、抗体の例えば3週間ごとの腹腔内注射によって処理し、腫瘍サイズを一定間隔で測定する。対照注射に比べて、本発明の抗体は腫瘍成長を阻害する。好ましい実施態様では、本発明の抗体は、抗新形成剤と併用する場合に腫瘍退縮を促進する。更に、以下に示すように、より好ましい実施態様では、本発明の抗体は、単一療法で用いられる場合に腫瘍退縮を促進する。腫瘍退縮の促進は、抗体の有効量又は抗体及び抗新形成剤の併用の有効量の投与が腫瘍のサイズ又は壊死の減少をもたらす、ことを意味する。本発明の好ましい実施態様では、腫瘍退縮は、少なくとも約20日間、より好ましくは少なくとも約40日間、より好ましくは少なくとも約60日間の期間、観察及び持続することができる。腫瘍退縮は、特定の治療計画を経験する患者群の平均として測定することができ、腫瘍退縮治療群の患者数によって測定することができる。
【0055】
本発明の好ましい抗体又はその断片は、配列番号14、配列番号16、配列番号18、配列番号20、配列番号22、配列番号24、配列番号26、配列番号28及び配列番号30からなる群より選ばれる、1、2、3、4、5及び/6の相補性決定部位((CDR)を有するヒト抗体である。好ましくは、本発明の抗体(又はその断片)は、配列番号14、配列番号16及び配列番号18のCDRを有する。あるいは及び好ましくは、本抗体又はその断片は、配列番号20、配列番号22及び配列番号24のCDRを有する。あるいは及び好ましくは、本抗体又はその断片は、配列番号26、配列番号28及び配列番号30のCDRを有する。CDRのアミノ酸配列は以下の表1に記載されている。
【0056】
【表1】

【0057】
別の実施態様では、本発明の抗体又はその断片は、配列番号1の重鎖可変領域及び/又は配列番号5又は配列番号6から選ばれる軽鎖可変領域を有する。IMC-A12は、本発明の特に好ましい抗体である。この抗体は、ヒトVH及びVLフレームワーク領域(FW)、及びCDRを有する。IMC-A12 (配列番号:1)のVH可変ドメインは、配列番号14、16及び18に対応する3CDRを有し、及びVLドメイン(配列番号:5)は、配列番号20、22及び24に対応する3CDRを有する。IMC-2F8は本発明の別の好ましい抗体である。この抗体はまた、ヒトVH及びVLフレームワーク領域(FW)及びCDRを有する。IMC-2F8のVH可変ドメインは、IMC-A12のVH可変ドメインと同一である。IMC-2F8 (配列番号9)のVLドメインは、配列番号26、28及び30に対応する3CDRを有する。
【0058】
別の実施態様では、本発明の抗体は、IGF-IRへの結合をIMC-A12及び/又はIMC-2F8と競う。すなわち、抗体は、同一又は類似の重複エピトープに結合する。
【0059】
本発明はまた、前述の抗体又はその断片をコードする単離ポリペプチドを提供する。本発明は、表2に記載の、1、2、3、4、5及び/又は全ての6CDRをコードする配列を有する核酸を含む。
【0060】
【表2】
【0061】
ヒト抗体をコードするDNAは、対応するヒト抗体領域から実質的に又はもっぱら得られる、CDR以外の、ヒト不変領域及び可変領域をコードする組換えDNA、及びヒト由来のCDRをコードするDNAによって調製することができる(例えば、重鎖可変ドメインCDRについては、配列番号13、15及び17 、及び軽鎖可変ドメインCDRについては、配列番号19、21及び23、又は配列番号25、27及び29)。
【0062】
抗体断片をコードするその他の好適なDNA起源は、完全長抗体を発現する任意の細胞、例えばハイブリドーマ及び脾臓細胞を含む。当該断片は抗体等価物としてそれ自体使用することができ、上記のように等価物に組み換えることができる。DNA組換え及びこの項で記載の他の方法は、公知の方法によって実行することができる。DNAの他の起源は、当該分野で公知のファージディスプレイ・ライブラリーから作製された単一鎖抗体又はFabである。
【0063】
加えて、本発明は発現配列、プロモーター及びエンハンサー配列に動作可能に連結された、先に記載のポリヌクレオチド配列を含む発現ベクターを提供する。原核生物、例えば細菌及び真核系、例えば特に限定されないが酵母及び哺乳動物細胞培養系における、抗体ポリペプチドの効率的合成のための様々な発現ベクターが開発されている。本発明のベクターは、染色体、非染色体及び合成DNA配列の部分を含むことができる。
【0064】
任意の好適な発現ベクターを使用することができる。例えば、真核性クローニングベクターは、E. coli例えばcolEl、pCRl、pBR322、pMB9、pUC、pKSM及びRP4由来のプラスミドを含む。原核性ベクターはまた、ファージDNA例えばM13及び他の線状単一-鎖DNA配列の誘導体を含む。酵母において有用なベクターの例は、211プラスミドである。哺乳動物細胞における発現に好適なベクターは、SV-40、アデノウイルス、レトロウイルス-由来DNA配列及び機能性哺乳動物性ベクター例えば上記のベクターの組み合わせから得られるシャトルベクターの周知の誘導体、及び機能性プラスミド及びファージDNAを含む。
【0065】
追加の真核性発現ベクターは当該分野で公知である(例えば、P. J. Southern及びP. Berg, J. Mol. Appl. Genet. 1: 327-41 (1982); Subramaniら, Mol. Cell. Biol. 1: 854-64 (1981); Kaufmann及びSharp, "Amplification And Expression of Sequences Cotransfected with a Modular Dihydrofolate Reductase Complementary DNAGene," J. Mol. Biol. 159: 601-21 (1982); Kaufmann及びSharp, Mol. Cell. Biol. 159: 601-64 (1982); Scahillら, "Expression And Characterization Of The Product Of A Human Immune Interferon DNA Gene In Chinese Hamster Ovary Cells, "Proc. Nat'l Acad. Sci. USA80, 4654-59 (1983); Urlaub and Chasin, Proc. Nat'l Acad. Sci. USA 77: 4216-20 (1980))。
【0066】
本発明に有用な発現ベクターは、発現されるDNA配列又は断片に作動的に連結される、少なくとも1つの発現コントロール配列を含む。コントロール配列は、クローン化DNA配列の発現を制御し及び調節するために、ベクターに挿入される。有用な発現コントロール配列の例は、lac系、trp系、tac系、trc系、ラムダファージの主なオペレーター及びプロモーター、fdコートタンパク質のコントロール部位、酵母の解糖プロモーター例えば3-ホスホグリセレートキナーゼのプロモーター、酵母酸ホスファターゼのプロモーター例えばPho5、酵母アルファ-交配因子のプロモーター、及びポリオーマ、アデノウイルス、レトロウイルス及びシミアンウイルス由来のプロモーター例えば初期及び後期プロモーター又はSV40、並びに原核細胞もしくは真核細胞及びそれらのウイルス又はそれらの組み合わせの遺伝子発現を制御することが知られている他の配列である。
【0067】
酵母での遺伝子コンストラクトの発現が望まれる場合には、酵母で使用するための好適な選択遺伝子は、酵母プラスミドYRp7に存在するtrp1遺伝子である。Stinchcombら, Nature, 282: 39 (1979); Kingsmanら, Gene, 7: 141 (1979)。当該trp1遺伝子は、トリプトファンでの成長能力欠損酵素の突然変異株についての選択マーカー、例えばATCC番号44076又はPEP4-1を提供する。Jones, Genetics, 85: 12 (1977)。次いで、酵母宿主細胞ゲノム中のtrp1損傷の存在は、トリプトファンの非存在下での成長による形質転換を検出するために効果的な環境を提供する。同様に、Leu2-欠損酵母株(ATCC 20,622又は38,626)は、Leu2遺伝子を有する公知のプラスミドにより考慮される。
【0068】
本発明はまた、先述の発現ベクターを含む組換え宿主細胞を提供する。本発明の抗体は、ハイブリドーマ以外の細胞株で発現することができる。本発明に従うポリペプチドをコードする配列を含む核酸は、好適な哺乳動物宿主細胞の形質転換に用いることができる。
【0069】
具体的な選択細胞株は、高発現レベル、対象のタンパク質の構成性発現及び宿主タンパク質由来の最小限の汚染に基づいて選択される。発現のために宿主として入手可能な哺乳動物細胞株は、当該分野で公知であり、多くの不死化細胞株、例えば特に制限されないがCOS-7細胞、チャイニーズハムスター卵巣(CHO)細胞、ベビーハムスター腎臓(BHK)及びリンパ起源の細胞株例えばリンパ腫、骨髄腫又はハイブリドーマ細胞を含む多くのその他の細胞株を含む。好適な追加の真核細胞は酵母及び他の菌類を含む。有用な原核生物宿主は、例えばE. coli例えばE. coli SG-936、E. coli HB 101、E. coli W3110、E. coli X1776、E. coli X2282、E. coli DHI及びE. coli MRC1、シュードモナス、バシラス属例えば枯草菌及びストレプトミセスを含む。
【0070】
これらの本組換え宿主細胞は、抗体又は抗体断片を発現させる条件下で細胞を培養し、宿主細胞又は宿主細胞周囲の培地から抗体又はその断片を精製することによって、抗体又は抗体断片を産生するために使用することができる。組換え宿主細胞における分泌のための発現抗体又はその断片のターゲッティングは、対象の抗体-コーディング遺伝子の5'末端にある、シグナル性又は分泌性リーダーペプチド-コーディング配列(Shokriら, ApplMicrobiol Biotechnol. 60: 654-64 (2003) Nielsenら, Prot. Eng. 10: 1-6 (1997)及びvonHeinjeら, Nucl. Acids Res. 14: 4683-90 (1986)を参照されたい)を挿入することによって促進することができる。これらの分泌性リーダーペプチド要素は、原核生物又は真核生物配列のいずれかから得られる。従って、好適には、宿主細胞質ゾルからのポリペプチドの移動及び分泌を培地へ方向付けるために、アミノ酸がポリペプチドのN-末端に結合している、分泌性リーダーペプチドが使用される。
【0071】
形質転換宿主細胞は、吸収可能な炭素源(糖類例えばグルコース又はラクトース)、窒素源(アミノ酸、ぺプチド、タンパク質又はそれらの分解産物例えばペプトン、アンモニウム塩等)及び無機塩(硫酸塩、リン酸塩及び/又はナトリウム、カリウム、マグネシウム及びカルシウムの炭酸塩)を含む液体培地中で、当該分野で公知の方法によって培養される。培地は、例えば成長-促進物質、例えば痕跡元素、例えば鉄、亜鉛、マンガン等を更に含む。
【0072】
本発明における抗体の調製のための別の実施態様は、挿入ヒト抗体産生ゲノムの実質的部分を有し及び内因性抗体の産生に欠けるトランスジェニック動物における、本発明に従う抗体をコードする核酸の発現である。トランスジェニック動物は、例えばマウス、ヤギ及びウサギを含むが特に限定されない。本発明の更なる1態様は、抗体-コーディング遺伝子、例えば泌乳中にポリペプチドを分泌するための動物の乳腺、の発現を含む。
【0073】
以下の実施例に記載のように、本発明に従う高親和性抗-IGF-IR抗体は、ヒト重鎖及び軽鎖可変領域遺伝子から構築されるファージディスプレイ・ライブラリーより単離することができる。例えば、本発明の可変ドメインは、転位可変領域遺伝子を含む末梢血リンパ球から得られる。あるいは、可変ドメイン部分例えばCDR及びFW領域は、異なったヒト配列から得られる。選択の3順後の90%を越える回収クローンは、IGF-IRに特異的である。選別FabのIGF-IRに対する結合親和性は、nMのレンジである。これは、ハイブリドーマ技術を用いて産生された多くの二価抗-IFG-IRモノクローナル抗体と同程度に高い。
【0074】
本発明の抗体及びその断片は、例えば天然抗体又はFab又はscFvファージディスプレイ・ライブラリーから得られる。単一ドメイン抗体は、天然抗体又はハイブリドーマ由来のVH又はVLドメインから選ばれることによって得られ、又はVHドメインのライブラリー又はVLドメインのライブラリーから選択される。単一ドメイン抗体の一次結合決定基であるアミノ酸残基は、CDRと定義されるKabat内にあるが、他の残基及び例えばVH-VLヘテロダイマーのVH-VL表面に埋もれている残基を含んでもよいことが、理解できるだろう。
【0075】
本発明の抗体はまた、結合特性が直接的突然変異、親和性成熟法、ファージディスプレイ又は鎖シャフリングによって改良されてきた抗体を含む。親和性及び特異性は、CDRを突然変異させ及び所望の性質を有する抗原結合部位をスクリーニングすることによって修飾又は改良することができる(例えばYangら, J. Mol. Biol., 254: 392-403 (1995)を参照されたい)。CDRは様々な方法で突然変異する。1方法は、個々の残基又は残基の組み合わせを無作為化し、別の抗原結合部位の集団において、20アミノ酸全てが特定の位置に見出されることである。あるいは、突然変異は、エラープローンPCR法によってCDR残基の範囲に渡って誘発される(例えばHawkinsら, J. Mol. Biol., 226: 889-896(1992)を参照されたい)。例えば、重鎖及び軽鎖可変領域遺伝子を含むファージディスプレイ・ベクターは、E. coliの突然変異株中で増殖することができる(例えばLowら, J. Mol. Biol., 250: 359-368 (1996)を参照されたい)。これらの突然変異誘発方法は、当業者に公知の多くの方法の例示である。
【0076】
本発明のIGF-IR結合抗体を特定するために使用されるタンパク質は、好ましくはIGF-RIであり、より好ましくはIGF-RIの細胞外ドメインである。IGF-RIの細胞外ドメインは、遊離でも又は別の分子に結合してもよい。
【0077】
本発明の抗体は、追加のアミノ酸残基に融合することができる。かかるアミノ酸残基は、おそらく単離を促進するためにペプチドタグでもよい。特定の器官又は組織に対する抗体の自動誘導のための他のアミノ酸残基も考慮される。
【0078】
本発明の別の局面では、抗-IGF-IR抗体又は抗体の断片は、抗新形成剤又は検出可能なシグナル-形成剤に化学的又は生合成的に結合することができる。以下に例示するように、本発明の抗体は、IGF-IRを産生する細胞に結合する際に効率よく吸収される。抗体に結合された抗新形成剤は、抗体が結合された腫瘍を破壊又は損傷する任意の薬剤、又は抗体が結合された細胞環境を含む。例えば、抗腫溶剤は、毒物例えば化学療法剤又は放射性同位体である。好適な化学療法剤は当業者に公知であり、アンスラサイクリン(例えば、ダウノマイシン及びドキソルビシン)、メトトレキサート、ビンクリスチン、ネオカルチノスタチン、シスプラチン、クロラムブシル、シトシン アラビノシド、5-フルオロウリジン、メルファラン、リシン及びカリチェア ミシン)を含む。化学療法剤は、一般的な方法を用いて抗体に結合される(例えばHermentin及びSeiler, Behrihglnst. Mitt. 82: 197-215 (1988)参照されたい)。
【0079】
検出可能なシグナル-形成剤は、in vivo及びin vitroでの診断目的に有用である。シグナル-形成剤は、外部手段、通常、電磁放射線の測定によって検出できる測定可能なシグナルを作製する。ほとんどの場合には、シグナル形成剤は酵素又はクロモフォアであり、蛍光、リン光又は化学ルミネセンスによって光を放つ。クロモフォアは、紫外又は可視領域で光を吸収する色素を含み、酵素触媒反応の基質又は分解産物でもよい。
【0080】
本発明は、標的又はレポーター部分が結合された本発明の抗-IGF-IR抗体又は抗体断片を更に考慮する。標的部分は結合ペアの第一のメンバーである。例えば、抗新形成剤はかかるペアの第二メンバーに結合し、それによって抗原結合タンパク質が結合する部位に導かれる。かかる結合ペアの一般的例は、アビジン及びビオチンである。好ましい実施態様では、ビオチンは本発明の抗原結合タンパク質に結合し、それにより、アビジン又はストレプトアビジンに結合される抗新形成剤又は他の部分のために標的を提供する。あるいは、ビオチン又は別のかかる部分は、本発明の抗原結合タンパク質に結合し、レポーターとして、例えば検出可能なシグナル-形成剤がアビジン又はストレプトアビジンに結合される診断系において使用される。
【0081】
抗新形成剤を使用するための好適な放射性同位体もまた当業者に公知である。例えば、131I又は211Atが用いられる。これらの同位体は、一般的な方法を用いて抗体に結合される(例えばPedleyら, Br. J. Cancer 68, 69-73 (1993)を参照されたい)。あるいは、抗体に結合される抗新形成剤は、プロドラッグを活性化する酵素である。このようにして、プロドラッグは投与されるが、抗体複合体が投与されると直ちに細胞障害性形態に変換される腫瘍部位に到達するまでその活性化形態を維持する。事実、抗体-酵素複合体は患者に投与され、治療される部位に局在化される。次いで、細胞障害性への変換が治療組織部位で生じるように、プロドラッグは患者に投与される。あるいは、抗体に結合された抗新形成剤は、サイトカイン例えばインターロイキン-2 (IL-2)、インターロイキン-4(IL-4)又は腫瘍壊死因子アルファ(TNF-α)である。抗体は、腫瘍にサイトカインをターゲットするので、サイトカインは他の組織に影響を与えることなく腫瘍の損傷又は破壊を介在する。サイトカインは、一般的な組換えDNA法を用いるDNAレベルで抗体に融合する。
【0082】
前述の抗体の有効量を哺乳動物に投与することによって哺乳動物における腫瘍成長を治療する方法もまた、本発明によって提供される。IGF-IRシグナル伝達経路は、多くの種類の癌の発症における病原体であることが広く証明されている。IGF-I及びIGF-IIは、幅広い癌細胞株例えば前立腺癌、乳癌、結腸癌、骨髄腫、卵巣癌、膵臓癌及び肺癌のための強力なマイトジェンであることが判っている。更に、高転移性癌細胞は、転移する傾向の低い腫瘍細胞よりも高レベルのIGF-IR及びIGF-IIを発現することが判明している。
【0083】
本発明に従って治療される好適な腫瘍は、好ましくはIGF-IRを発現する。任意の特定のメカニズムに拘束される意図ではないが、本発明によって治療され又は抑制される疾患及び症状は、例えば病原性血管新生又は腫瘍成長がIGF-IR傍分泌及び/又は自己分泌ループを介して刺激される疾患等を含む。例えば、高転移性腫瘍は、IGF-II及びIGF-IRを発現する傾向にある。
【0084】
本発明の実施態様では、抗-IGF-IR抗体は1以上の他の抗新形成剤と組み合わせて投与される。併用療法の例としては、例えば米国特許第6,217,866号明細書(Schlessingerら) (抗新形成剤と組み合わせた抗-EGFR抗体); 国際公開第 WO 99/60023号パンフレット(Waksalら) (照射と組み合わせた抗-EGFR抗体)を参照されたい。任意の好適な抗新形成剤、例えば化学療法剤、照射又はそれらの組み合わせが用いられる。抗新形成剤は、アルキル化剤又は抗代謝剤でもよい。アルキル化剤の例は特に限定されないがシスプラチン、シクロホスファミド、メルファラン及びダカルバジンである。抗代謝剤の例は、特に限定されないが、ドキソルビシン、ダウノルビシン及びパクリタキセル、ゲムシタビン、及びトポイソメラーゼ阻害剤イリノテカン(CPT-11)、アミノカンプトテシン、カンプトテシン、DX-8951f、及びトポテカン(トポイソメラーゼI)及びエトポシド(VP-16)及びテニポリシド(VM-26)( トポイソメラーゼII)を含む。抗新形成剤が照射である場合には、照射起源は、治療患者に対して外部放射線(外部放射線療法-EBRT)でも又は内部放射線(短療法-BT)でもよい。投与される抗新形成剤の用量は多数の因子、例えば薬剤の種類、治療される腫瘍の種類及び重度、及び薬剤の投与経路に依拠する。しかしながら、強調しておくが、本発明はいずれかの特定の用量に限定されない。
【0085】
当該分野で現在公知又は評価されている抗新形成剤は、様々なクラス、例えば分裂阻害剤、アルキル化剤、抗代謝剤、挿入抗生物質、成長因子阻害剤、細胞サイクル阻害剤、酵素、トポイソメラーゼ阻害剤、抗生存剤(anti survival agents)、生物応答改変剤、抗ホルモン剤及び抗管形成剤に群分けすることができる。
【0086】
これらのクラスでは、本明細書に報告されているデータは、IGF-IRに結合する抗体と併用される場合に、トポイソメラーゼ阻害剤が特に有効な抗新形成剤である、ことを示唆している。従って、本発明の実施態様は、トポイソメラーゼ阻害剤がIGF-IRに結合する抗体と組み合わせて投与される方法を含む。阻害剤は、トポイソメラーゼI又はトポイソメラーゼIIの阻害剤でもよい。トポイソメラーゼI阻害剤は、イリノテカン(CPT-11)、アミノカンプトテシン、カンプトテシン、DX-8951f、トポテカンを含む。トポイソメラーゼII阻害剤は、エトポシド(VP-16)及びテニポシド(VM-26)を含む。他の物質はトポイソメラーゼ活性及び抗新形成剤としての効力について現在評価中である。好ましい実施態様では、トポイソメラーゼ阻害剤は、イリノテカン(CPT-11)である。組み合わせて使用される抗体は、IGF-IRに結合し、かつ以下の性質を少なくとも1つ有する本発明の抗体である:(i)IGF-I又はIGF-IIのIGF-IRへの結合を阻害する;(ii)IGF-I又はIGF-IIによってIGF-IRの活性化を中和する;(iii)IGF-IR表面受容体を減少させる;及び約1×10-10M-1以下のKdでIGF-IRに結合する。より好ましい実施態様では、トポイソメラーゼ阻害剤を組み合わせて使用される抗体は、上記のヒト抗体の特徴を有する。
【0087】
本発明の抗-IGF-IR抗体は、腫瘍成長又は脈管形成に関連する他の受容体を中和する抗体と共に投与することができる。本発明の実施態様では、抗-IGF-IR抗体は、EGFRに特異的に結合する受容体アンタゴニストを組み合わせて使用される。特に好ましいのは、EGFRの細胞外ドメインに結合し、1以上のそのリガンドの結合を妨害し及び/又はEGFRのリガンド-誘起活性化を中和する抗原結合タンパク質である。EGFRアンタゴニストは、EGFR又はEGFRのリガンドに結合し、EGFRのそのリガンドへの結合を阻害する抗体でよい。EGFRに対するリガンドは、例えばEGF、TGF-α、アンヘレグリン(amphiregulin)、ヘパリン-結合EGF(HB-EGF)及びベータセルリンを含む。TGF-αは脈管形成を促進する点でより強力であることが明らかになっているが、EGF及びTGF-αは、EGFR介在刺激をもたらす主な内因性リガンドであると考えられる。EGFRアンタゴニストは、リガンドへの結合を阻害することもできるし又は阻害しないこともできるEGFRの細胞外部分に外的に結合することができ、又はチロシンキナーゼドメインに内的に結合することができる、ことは理解できよう。
EGFRに結合するEGFRアンタゴニストの例は、制限なく、生物分子、例えばEGFRに特異的な抗体(及びその機能性等価物)、及び小分子、例えばEGFRの細胞ドメインに直接作用する合成キナーゼ阻害剤を含む。
【0088】
かかる受容体の別の例は、VEGFRである。本発明の実施態様では、抗-IGF-IR抗体は、VEGFRアンタゴニストと組み合わせて使用される。本発明の1実施態様では、抗-IGF-IR抗体は、VEGFR-1/Flt-1受容体に特異的に結合する受容体アンタゴニストと組み合わせて使用される。別の実施態様では、抗-IGF-IR抗体は、VEGFR-2/KDR受容体に特異的に結合する受容体アンタゴニストと組み合わせて使用される。特に好ましくは、VEGFR-1又はVEGFR-2の細胞外ドメインと結合し、そのリガンドによって結合を妨害し(VEGFR-2は、VEGFによって最も強く刺激される;VEGFR-1は、VEGFではなく、P1GFによって最も強く刺激される)及び/又はリガンド誘起活性を中和する抗原結合タンパク質である。例えば、IMC-1121は、VEGFR-2に結合し、これを中和するヒト抗体である(国際公開第 WO 03/075840号パンフレット; Zhu)。別の例は、MAb 6.12である。これは、溶解性でかつ細胞表面発現VEGFR-1に結合するscFvである。ScFv 6.12は、マウスモノクローナル抗体MAb 6.12のVL及びVHドメインを含む。MAb 6.12を産生するハイブリドーマ細胞株は、「特許手続上の微生物の寄託の国際的承認に関するブタペスト条約」及びそれに基づく規則(ブタペスト条約)の規定のもと、ATCC番号PTA-3344として寄託されている。
【0089】
腫瘍形成に関連する成長因子受容体の他の例は、血小板-由来成長因子(PDGFR)、神経成長因子(NGFR)及び繊維芽細胞成長因子(FGFR)である。
【0090】
追加の他の実施態様では、IGF-IR抗体は、1以上の好適なアジュバント、例えばサイトカイン(例えばIL-10及びIL-13)又は他の免疫刺激因子、例えば特に限定されないがケモカイン、腫瘍関連抗原及びペプチドを組み合わせて投与することができる。例えば上記のLarriveeらを参照されたい。しかしながら、抗-IGF-IR抗体のみの投与は、治療上有効な方法で腫瘍進行を抑制、阻害又は減じるには十分である、ことが理解されよう。
【0091】
併用療法では、抗-IGF-IR抗体は、別の薬剤での治療開始前、間又は後に投与される。また、その任意の組み合わせ、すなわち抗新形成剤治療開始前及び間、前後、間及び後、又は前、間及び後に投与される。例えば、抗-IGF-IR抗体は、照射療法の開始前の1〜30日、好ましくは3〜20日、より好ましくは5〜12日に投与することができる。本発明の好ましい実施態様では、化学療法は、抗体療法と併用して、又はより好ましくは抗体療法の次に投与することができる。
【0092】
本発明では、任意の好適な方法又は経路は、本発明の抗-IGF-IR抗体を投与するため、及び場合により、抗新形成剤及び/又は他の受容体のアンタゴニストを同時投与するために用いることができる。本発明に従って利用される抗新形成剤投薬計画は、患者の腫瘍症状の治療のために最適であると考えられるいずれの投薬計画も含む。異なった悪性腫瘍は、特異的抗腫瘍抗体及び特異的抗新形成剤の使用を必要としてもよい。それは、患者を基準として決定されるだろう。投与経路は、例えば、経口、静脈内、腹腔内、皮下又は筋肉内投与を含む。投与されるアンタゴニストの用量は、多数の要素、例えばアンタゴニストの種類、治療される腫瘍の種類及び重度、及びアンタゴニストの投与経路に依拠する。しかしながら、強調しておくが、本発明は、任意の特定の方法又は投与経路に限定されない。
【0093】
受容体に特異的に結合し、リガンド-毒内在化の後に毒性で致命的ペイロードを送達する、本発明の抗体-IGF-IR抗体は、複合体して投与することができることに留意されたい。抗体-薬物/小分子複合体は、互いに又はペプチド又は非ペプチド性のリンカーを介して直接結合される。
【0094】
本発明の別の局面では、本発明の抗-IGF-IR抗体は、1以上の抗新形成剤又は抗血管由来薬物に化学的又は生合成的に結合することができる。
【0095】
本発明は、標的又はレポーター部分が結合された抗-IGF-IR抗体を更に考慮する。標的部分は結合ペアの第一のメンバーである。例えば抗新形成剤は、かかるペアの第二のメンバーに結合し、それによって抗-IGF-IR抗体が結合される部位に指向される。かかる結合ペアの一般的例は、アビジン及びビオチンである。好ましい実施態様では、ビオチンは、抗-IGF-IR抗体に結合し、それによってアビジン又はストレプトアビジンに結合される抗新形成剤又は他の部分に対する標的を提供する。あるいは、ビオチン又は別のかかる部分は、本発明の抗-IGF-IR抗体に結合され、レポーターとして、例えば検出可能なシグナル-形成剤がアビジン又はストレプトアビジンに結合する診断系において、用いられる。
【0096】
本発明の抗-IGF-IR抗体は、予防又治療目的で哺乳動物に用いられる場合には、薬学的に許容される担体を更に含む組成物の形態で投与されるだろう。好適な薬学的に許容される担体は、例えば1以上の水、生理食塩水、リン酸緩衝液、デキストロース、グリセロール、エタノール等、及びそれらの組み合わせを含む。薬学的に許容される担体は、貯蔵安定性又は結合タンパク質の効果を亢進させる、補助物質、例えば湿潤剤又は乳化剤、保存剤又は緩衝液の少量を更に含むことができる。注射組成物は、当該分野で公知であり、哺乳動物に投与した後に活性剤の迅速、徐放又は遅延放出を提供することができるように調合することができる。
【0097】
本発明はまた、ヒト抗-IGF-IR抗体の治療上有効量を含む腫瘍成長及び/又は脈菅形成を阻害するためのキットを含む。キットは、任意の好適なアンタゴニスト、例えば腫瘍形成又は脈菅形成に関連する別の成長因子受容体(例えば、上記のEGFR、VEGFR-l/Flt-1、VEGFR-2、PDGFR、NGFR、FGFR等)を更に含む。あるいは又は加えて、本発明のキットは抗新形成剤を更に含む。本発明の分脈における好適な抗新形成剤の例は、本明細書に記載されている。本発明のキットはアジュバントを更に含むことができ、その例は上に記載されてもいる。
【0098】
更に、本発明の範囲内には、当該分野で周知の研究法又は診断法のためのin vivo及びin vitroでの本発明の抗体の使用を含む。当該診断法は、本発明の抗体を含むキットを含む。
【0099】
従って、本発明の受容体抗体は、当業者に周知の研究、診断、予防又は治療方法にin vivo及びin vitroで使用することができる。当然のことながら、本明細書に開示の発明の本質における変更が当業者によってなされることは理解及び予想され、またかかる修飾が本発明の範囲内に含まれることになるのは意図されている。
【実施例】
【0100】
以下の実施例は本発明を更に説明するが、本発明の範囲を限定するものと解釈されるべきではない。一般的な方法、例えばベクター及びプラスミドの構築、ポリペプチドをコードする遺伝子のかかるベクター及びプラスミドへの挿入、プラスミドの宿主細胞への導入、及び遺伝子及び遺伝子産物の発現及びその決定に用いられる方法の詳細な記載は、多数の刊行物、例えばSambrook, Jら, (1989) Molecular Cloning: A Laboratory Manual,第二版, Cold Spring Harbor Laboratory Press; 及びColigan, J.ら (1994) Current Protocols in Immunology, Wiley & Sonsから得られる。本明細書に記載の全ての文献はその全体が援用されている。
【0101】
抗-ヒトIGF-IRモノクローナル抗体の選択及び設計
ヒトIGF-I受容体に対する高親和性抗体を単離するために、ヒトIGF-IRの組換え細胞外部分を用い、3.7 x 1010固有クローンを含むヒト野生型(非-免疫)バクテリオファージFabライブラリーをスクリーニングした(de Haardら, J. Biol. Chem. 274: 18218-30 (1999))。溶解性IGF-IR(50μg/ml)を試験菅にコートし、37℃で1時間、3%ミルク/PBSでブロックした。ファージを、ファージ培養を記録するためのライブラリーストックを培養し、M13K07ヘルパーファージで救援し、及びアンピシリン及びカナマイシン選択を含む2YTAK培地で30℃で終夜増幅することによって調製した。得られたファージ調製物を4% PEG/0.5M NaCl中に沈殿させ、3%ミルク/PBSで再懸濁させた。次いで、固定受容体を室温で1時間、ファージ調製物でインキュベートした。その後、試験管をPBST(0.1% Tween-20を含むPS)で10回洗浄した後、PBSで10回洗浄した。結合ファージを100 mMトリエチルアミンの新鮮調製溶液1 mlで、室温で10分間溶出した。溶出ファージを37℃で30分間静置させて、10 mlの中間-記録ファージTG1細胞でインキュベートし、30分間攪拌した。感染TG1細胞をペレットにし、いくつかの大2YAGプレートに置き、30℃で終夜インキュベートした。プレート上の全てのコロニーを3〜5 mlの2YTA培地にすくい取り、グリセロール(最終濃度:10%)で混合し、等分し、-70℃で保存した。第二順選択では、100μlのファージストックを25 mlの2YTAG培地に加え、中間-記録期まで培養した。上記処理に続く選択のために、培養物をM13K07ヘルパーファージで救援し、増幅し、沈殿させ、使用した。但し、試験管に固定されたIGF-IRは低濃度(5μg/ml)であり、結合処理に続く洗浄回数を増やした。合計で2順の選択を行った。
【0102】
個々のTG1クローンを取り、96ウェルプレートで37℃で培養し、上記のM13K07ヘルパーファージで救援した。増幅ファージ調製物は、室温で1時間、18%ミルク/PBSの1/6体積でブロックし、IGF-IR(1μg/ml x 100μl)でコートしたMaxi-sorb 96-ウェル マイクロタイタープレート(Nunc)に添加した。室温で1時間インキュベーション後、プレートをPBSTで3回洗浄し、マウス抗-M13ファージ-HRP複合体(Amersham Pharmacia Biotech, Piscataway, NJ)でインキュベートした。プレートを5回洗浄し、TMBペルオキシダーゼ基質(KPL, Gaithersburg, MD)を加え、マイクロプレートリーダー(Molecular Device, Sunnyvale, CA)を使って450nmでの吸収を測定した。選択の2順では、80%の独立クローンはIGF-IR結合に陽性であった。
【0103】
第二順選択後の抗-IGF-IR Fabクローンの多様性を制限酵素消化パターン(すなわちDNAフィンガープリント)によって分析した。個々のクローンのFab遺伝子挿入は、以下のプライマーを用いるPCR増幅であった:ファージベクター内の独自のFab遺伝子をフランキングする配列に特異的な、リバースPUC19(5'-AGCGGATAACAATTTCACACAGG-3';配列番号31)及びfdtet配列(5'-GTCGTCTTTCCAGACGTTAGT-3';配列番号32)。各増幅産物を通常の切断酵素、BstNIで消化し、3%アガロースゲルで分析した。25個の個々のパターンを全て同定した。各消化パターン由来の代表的クローンのDNA配列をジデオキシヌクレオチド配列決定法によって決定した。
【0104】
IGF-IR及び独自のDNAプロファイルへの正結合を示す個々のクローン由来のプラスミドを使用し、非サプレッサーE. coli宿主HB2151を形質転換した。細胞を1 mMイソプピル-1-β-D-ガラクトピラノシド(IPTG, Sigma)を含む2YTA培地で30℃で培養することによって、HB2151におけるFab断片の発現を誘起した。20% (w/v)ショ糖、200 mM NaCl、1 mM EDTA及び0.1 mM PMSF含む25 mM Tris (pH 7.5)中で細胞ペレットを再懸濁し、次いで4℃で1時間緩やかに攪拌しながらインキュベーションすることによって、細胞のペリプラスム抽出物を調製した。15,000 rpmで15分間遠心分離した後、溶解性Fabタンパク質を、プロティンGカラムを用いるアフィニティークロマトグラフィーによって上清から精製し、製造者のプロトコール(Amersham Pharmacia Biotech)に従った。
【0105】
Fabクローン結合候補化合物を、96ストリップ(strip)-ウェルプレートにコートされた固定IGF-IR(100μg/ml)に対する放射性同位体標識ヒトIGF-Iリガンドの競争的ブロッキングでスクリーニングした。Fab調製物を希釈し、PBS/0.1% BSA中で、室温で0.5〜1時間、IGR-IRプレートでインキュベートした。次いで、40 pMの125I-IGF-Iを加え、プレートを更に90分間インキュベートした。次いで、ウェルを氷冷PBS/0.1% BSAで3回洗浄し、乾燥し、ガンマシンチレーションカウンタで測定した。単一ポイントアッセイにおいて、対照放射性同位体リガンド結合の阻害が30%より高かった候補化合物を選択し、in vitroでのブロッキング力価を決定した。4クローンを同定した。当然のことながら、Fabクローン2G8のみが50%を超えるリガンド結合を阻害し及び約200 nMのIC50を有することが分かった、また完全長IgG1型への変換のために選択した。2F8の重鎖可変領域配列及び翻訳アミノ酸配列をそれぞれ図1及び2に示す。完全長IgG1として設計された2F8重鎖のDNA配列及び翻訳ポリペプチド配列をそれぞれ図3及び4に示す。
【0106】
Fab 2F8塩基配列決定法は、このFabがラムダ軽鎖不変領域にあると決定した。2F8軽鎖のDNA配列及び翻訳アミノ酸配列をそれぞれ図5及び6に示した。完全長ラムダ軽鎖型の配列を図7及び8に示す。結合動的解析をBIAcoreユニットを用いて2F8 IgGで行った。この抗体は0.5〜1 nM (0.5-1 x 10-9M)の親和性でIGF-IRに結合すると決定した。
【0107】
この抗体の親和性を向上させるために、第二世代Fabファージライブラリーを、そこで2F8重鎖が保存され、軽鎖が108種の独自の種類を越える多様性に変化するように作製した。この方法は、軽鎖シャフリングと呼ばれ、所与の標的抗原のための親和性成熟選択抗体に対して首尾良く利用されている(Chamesら, J. Immunol. 169: 1110-18 (2002))。次いで、このライブラリーを上記処理後のヒトIGF-IR(10μg/ml)への結合のためにスクリーニングし、パニング(panning)処理は、高親和性結合Fabを向上させるために低IGF-IR濃度((2μg/ml)を用いて追加の3順を反復した。7クローンを4順後に分析した。7種全ては同一のDNA及び制限消化プロファイルを含んだ。単一単離FabをA12と表わし、ラムダ軽鎖不変領域を有することが明らかになった。軽鎖DNA配列を図9に、アミノ酸配列を図10に示す。完全ラムダ軽鎖配列及び翻訳ポリペプチド配列をそれぞれ図11及び12に示す。2G8及びA12軽鎖のアミノ酸配列比較により、2種の可変領域は全部で11アミノ酸が異なることを決定した(図13及び14に言及)。9種の変化はCDR領域内に存在し、ほとんど(6アミノ酸残基)はCDR3内に生じた。
【0108】
ヒトIGF-IR及びそれらのリガンドのブロッキング活性に対する2抗体(完全IgG)親和性の比較を表3に示した。結合結果は、ヒトIGF-IR ELISAによって決定し、飽和に対して50%結合活性を達成するために必要な抗体力価の濃度を表わす。ブロッキング結果は、固定ヒトIGF-IRに対する125I-IGF-Iリガンドの50%結合を阻害するために必要な抗体レベルを示す。親和性を製造者の使用書(Pharmacia BIACORE 3000)に従うBIAcore分析によって測定した。溶解性IGF-IRをセンサーチップに固定し、抗体結合反応速度を決定した。
【0109】
【表3】
【0110】
抗体A12を作製するためにSF8軽鎖中で生じる抗体変化は、2G8よりもIGF-IRに対するA12の顕著に高い親和性に影響をもたらした。これに伴い、この増加は、受容体に対するより高いA12結合能をもたらした。これは、ELISAによって測定され、固定受容体のためのリガンドのブロッキング活性が少なくとも3倍増加した。図15は、受容体ブロッキングアッセイにおける2種の抗-IGF-IR抗体の代表的力価を示す。軽鎖がヒトラムダ類又はカッパ類の不変領域について設計されたか否かに関らず、A12の活性は同一であった。ラムダ類の軽鎖で設計した抗体A12を全ての以下の過程で利用した。このアッセイでは、A12は、寒冷リガンドとの競争よりもIGF-IRへの放射性同位体標識IGF-Iの結合を大幅に阻害した。2F8の活性を寒冷リガントとの競争と比較した。これは、2種の抗体(表3を参照されたい)及びIGF-Iの相対的親和性(0.5〜1 nM)と一致した。
【0111】
Fabクローン由来の完全ヒトIgG1抗-IGF-IR抗体の設計及び発現
Fab 2F8及びFab A12の重及び軽鎖遺伝子をコードするDNA配列を、製造者の教示に従うBoerhinger Mannheim Expandキットを用いるポリメラーゼチェーン反応(PCR)によって増幅した。フォワード及びリバースプライマーは、哺乳動物発現ベクターへのクローニング用の制限エンドヌクレアーゼ部位の配列を含んだ。重鎖用の受容ベクターは、強力な真核生物性プロモーター及び3’ポリアデニル化配列が隣接する、完全ヒトガンマ1不変領域cDNA配列を含んだ。2F8又はA12の完全長ラムダ軽鎖配列をそれぞれ、哺乳動物細胞中の発現の真核性調節要素のみを有する第二ベクターにクローン化した。選択マーカーは、プラスミドの哺乳動物細胞へのトランスフェクション後の安定DNA構成要素の選択用ベクター上にも存在する。フォワードプライマーはまた、発現抗体の適正な分泌のための強力な哺乳動物シグナルペプチド配列をコードする配列を用いて設計した。適正にクローン化された免疫グロブリン遺伝子配列の同定後に、DNAを塩基配列決定し、一時的トランスフェクションにおける発現を試験した。製造者の仕様書に従い、リポフェクチンを用いて一時的トランスフェクションをCOS7霊長類細胞株に行った。24時間又は48時間トランスフェクション後に、完全IgG抗体の発現を抗-ヒト-Fc結合ELISAによって調整培養上清中で検出した。ELISAプレート(96ウェル)を100 ng/ウェルのヤギ抗ヒトFc-特異的ポリクローナル抗体(Sigma)で被覆することによって調製し、4℃で終夜、5%ミルク/PBSでブロックした。次いで、プレートをPBSで5回洗浄した。調整上清をウェルに加え、室温で1.5時間インキュベーションした。結合抗体をヤギ抗ヒトラムダ軽鎖-HRP抗体(Sigma)で検出し、TMB試薬及び上記のマイクロプレートリーダーで視覚化した。抗-IGF-IR抗体の大規模調製を、COS細胞への大規模な一時的トランスフェクションによって、リポフェクション法のスケールアップによって、又は好適な宿主細胞例えばマウス骨髄腫細胞株(NSO, Sp2/0)又はチャイニーズハムスター卵巣細胞株(CHO)への安定トランスフェクションによって達成した。抗-IGF-IR抗体をコードするプラスミドを、エレクトロポレーション法によって宿主細胞へトランスフェクトし、約2週間、好適な薬物選択培地中で選択した。抗-Fc ELISAによる抗体発現及び血清なしの細胞培養培地に拡張された陽性クローンについて、安定選択コロニーをスクリーニングした。最大2週間の期間、スピナーフラスコ又はバイオリアクターの懸濁培養中で、安定選択細胞由来の抗体産生を実行した。一時的又は安定トランスフェクションによって産生した抗体をProAアフィニィークロマトグラフィー(Harlow and Lane. Antibodies. A Laboratory Manual. Cold Spring Harbor Press.1988)で精製し、中性緩衝生理食塩水中へ溶出し、定量した。
【0112】
ヒト腫瘍細胞における抗-IGF-IRモノクローナル抗体のリガンドブロッキング活性の測定
【0113】
次いで、抗-IGF-IR抗体について、ヒト腫瘍細胞を用いて、野生型IGF-IRに対する放射性同位体標識リガンドのブロッキングを試験した。アッセイ条件は、Arteaga and Osborne(Cancer Res. 49: 6237-41 (1989))に若干の変更を加えて行った。MCF7ヒト乳癌細胞を24ウェル皿に播種し、終夜培養した。サブコンフルエント単層を2〜3回、結合緩衝液(Iscove's培地/0.1% BSA)中で洗浄し、結合緩衝液に抗体を添加した。室温で当該抗体で短時間インキュベーションを行った後、40 pM 125I-IGF-I(約40,000 cpm/ウェル)を各ウェルに加え、穏やかに攪拌しながら更に1時間インキュベートした。次いで、ウェルを氷冷PBS/0.1 % BSAで3回洗浄した。次いで、単層を200μl 0.5N NaOHで溶解し、ガンマカウンタでカウントした。結果を図16に図式的に示す。ヒト腫瘍細胞上で、抗体A12は、IC50 3 nM (0.45llg/ml)でIGF-IRへのリガンド結合を阻害した。これは、寒冷IGF-I リガンドの阻害活性(IC50 = 1 nM)よりも少し低かったが、寒冷IGF-IIの阻害活性(IC50 = 9 nM)よりも高かった。2種のIGFリガンドで観察された相違は、リガンドIGF-IよりもIGF-IRについてのIGF-IIの低い結合反応速度に起因するらしい(Janssonら, J Biol Chem. 272: 8189-97 (1997))。抗体2F8のIC50は30 nM (4.5 g/ml)であると決定した。我々は、続いて、いくつかの異なったヒト腫瘍型におけるA12抗体のIGF-Iリガンドブロッキング活性を測定した。結果を表4に示す。抗体A12は内生的細胞IGF-IRに対する結合に効果的であり、及び乳癌、膵臓癌及び結腸癌組織を含むヒト腫瘍型の範囲に結合するリガンドの阻害に効果的であった。
【0114】
【表4】
【0115】
IGF-IRは、インシュリン受容体(IR)に対してかなりの相同性を共有する。抗-IGF-IR抗体がこのIGF-IRに特異的であり、インシュリン結合をブロックしないか否かを決定するために、細胞型ブロッキングアッセイをヒトZR-75I乳癌細胞で行った。インシュリンは、僅かであるがIRよりも3オーダー低くIGF-IRに結合することができるため、我々は、MCF7細胞と比べてIR対IGF-IR比が高いヒト乳癌細胞株ZR-75Iを利用した。この細胞株の利用によって、我々は、細胞に対するインシュリン結合が特異的IR結合のよりよい指標となると結論した。アッセイはMCF7細胞について上記の方法で行い、結果を表17に示す。寒冷インシュリンは放射性同位体標識インシュリンの細胞への結合を滴定することができるが、2F8も高親和性抗体A12抗体も、200 nM抗体濃度でさえもインシュリン結合をブロックせず、IGF-IRへのこれらの抗体の選択的結合と一致し、IRへのこれらの抗体の選択的結合とは一致しなかった。
【0116】
リガンド-依存的細胞有糸分裂生起の抗体-介在阻害
IGF-IRへのIGF-I結合のブロッキングが阻害細胞増殖を阻害するか否かを決定するために、マイトジェンアッセイをMCF7乳癌細胞、BxPC-3膵臓癌細胞及びHT-29結腸癌細胞で行った。アッセイは、若干の変更加えて、Pragerら(Proc. Natl. Acad. Sci. U. S. A. 91: 2181-85 (1994)に従って行った。細胞を96-ウェル組織培養プレートに5000〜10000細胞/ウェルで播き、終夜吸着させた。次いで、培地を血清なしの培地で交換し、37℃で終夜インキュベートした。次いで、細胞を抗体A12の存在又は非存在下でIGF-Iでインキュベートし、更に37℃で終夜インキュベートした。次いで、0.25μCi [3H]チミジンを各ウェル加え、37℃で5時間インキュベートした。上清を吸引し、細胞をトリプシン処理で5分間懸濁した。次いで、細胞をフィルターで回収し、セル・ハーベスターを用いて水で3回洗浄した。乾燥後、フィルターをシンチレーションカウンタで読むために処理した。結果を図18A、B及びCに示す(それぞれ、MCF7、BxPC-3及びHT-29)。IGF-Iデータポイントは最大のマイトジェン反応を達成するために必要な量を決定するリガンドの滴定を示す。様々な癌細胞型に対する抗体A12の活性の測定では、IGF-Iを5nMの濃度で添加し、抗体は200 nM〜0.05 nMの力価を示した。抗体A12は、用量依存的にIGF-Iリガンドに反応して、IC50 6nMで、MCF7有糸分裂生起を阻害した。
【0117】
次いで、いくつかの追加のヒト腫瘍細胞株に対するマイトジェン阻害を抗体A12について試験した。結果を表5に示す。抗体A12は、乳癌、結腸癌及び多発性骨髄腫を含む多数のヒト腫瘍細胞株のIGF-Iリガンド-介在有糸分裂生起の阻害に効果的であった。
【0118】
【表5】
【0119】
IGF-I指向受容体ホスホリル化及び下流シグナル伝達の抗体-介在阻害
IGF-Iシグナル伝達に対する抗−IGF-IR抗体の阻害効果を視覚化するために、抗体A12又は2F8の存在又は非存在下で、受容体自己ホスホリル化及び下流エフェクター分子ホスホリル化分析を行った。MCF7ヒト乳癌細胞株をその高IGF-IR多様性に起因する使用のために選択した。細胞を10 cm又は6ウェル培養皿に播き、70〜80%コンフルエンスになるまで培養した。次いで、単層をPBSで2回洗浄し、血清なしの培地で終夜培養した。抗-IGF-IR抗体を新鮮な血清なし培地(100 nM〜10 nM)で加え、リガンド(10 nM)の添加前に30分間インキュベートした。細胞を10分間リガンドと共にインキュベートし、その後氷上に置き、氷冷PBSで洗浄した。細胞を溶解液(50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% TritonX-100, 1 mM EDTA, 1 mM PMSF, 0.5 mM Na3V04, 1 /ml ロイペプチン, 1μg/ml ペプスタチン, 及び1μg/ml アプロチニン)を添加して溶解し、氷上の遠心管に15分間ですくい取った。次いで溶解物を4℃で遠心して清浄した。次いで、その溶解物から溶解IGF-IRを免疫沈降させた(IP)。1μg/mlの抗体3B7(Santa Cruz)又はA12を、4℃で400μlの溶解物と共にインキュベートした。次いで、4℃で2時間、免疫複合体をProA-セファロースビーズの添加によって沈殿させ、ペレット化し、溶解緩衝液で3回洗浄した。ProAビーズに結合したIPをすくい取り、変性ゲルランニング緩衝液に加えた。溶解物又はIPを変性ゲル電気泳動のために処理し、4〜12%アクリルアミドゲルで泳動し、Towbinら (Biotechnology 24: 145-9 (1992))に従うウェスタンブロットによってナイロン又はニトロセルロース膜にブロットした。チロシンホスホリル化タンパク質を抗-p-チロシン抗体(Cell Signaling #9411)及び抗-マウス-HRP第二抗体を用いてブロットで検出した。IGF-IRをモノクローナル抗体C-20(Santa Cruz)で検出した。Aktホスホリル化のために、リン酸-Aktを抗体#559029で検出し、総Aktを#559028(BDPhanningen)で検出した。MARKホスホリル化のために、リン酸-p44/42を#9101で、総p44/42を#9102(Cell Signaling Tech.)で検出した。バンドは、X-線フィルムを用いECL試薬で視覚化した。
【0120】
図19Aから明らかなように、MCF7細胞におけるIGF-IRの自己-ホスホリル化は血清除去後に停止され、2F8又はA12のいずれかの添加は、受容体ホスホリル化を誘起しなかった。これは、検出可能なアゴニスト活性の欠落を証明した。10nM IGF-Iを添加した場合に、IGF-IRホスホリル化は強く誘起された。抗体2F8は、IGF-IRホスホリル化を約50%低減させた、一方、高親和性抗体A12はほとんど完全にホスホリル化を阻害した。同様に、抗体A12は、HT-29結腸癌細胞及びBxPC-3膵臓癌細胞においてIGF-IRの自己ホスホリル化を阻害した(図19B)。
【0121】
IGF-Iに応答する下流エフェクターシグナル伝達もまた抗-IGF-IR抗体によって阻害された(図20)。MAPKホスホリル化は、2F8及びA12によってかなりの程度阻害された。抗-アポトーシス分子Aktのホスホリル化は、2F8を有するIGF-IR抗体遮断に対して感受性が低かった。それは、Aktホスホリル化をいくらか減少させたにすぎなかた。A12は、濃度lO nMでさえも顕著にAktホスホリル化を阻害した。抗体A12は同等に、市販の抗体3B7として可溶化IGF-IRの免疫沈降に優れたが、A12は、ウェスタンブロット転移後のナイロン膜上に固定された変性IGF-IRを検出することができなかった。
【0122】
モノクローナル抗体A12の腫瘍細胞株に対するFACS結合解析
A12は内生IGF-IRを免疫沈降することができるので、我々は、A12を蛍光活性細胞選別法(FACS)に検出抗体として用いることができるか否かを決定する点に興味を持った。ヒト腫瘍細胞株を培地で培養し、氷冷PBSですくい取り、測定した。一次抗体A12(0.5μg)を250μl PBS/5% FBS中の約5百万細胞に加え、氷上で1時間インキュベートした。次いで、細胞をPBS/5% FBSで3 mlに希釈し、ペレット化し、上清を吸引した。次いで、二次フィコエリトリン(PE)-標識ヤギ抗-ヒトIgG F(ab)2断片を250μl PBS/5% FBSに1:200で加え、氷上で60分間インキュベートした。その後、既に述べたように、細胞を再度希釈し、ペレット化した後、500μl PBS/5% FBSに再懸濁した。次いで、Epics XLユニット(Coulter)でFACS解析を行った。図21から明らかなように、抗体A12は完全にヒト乳癌細胞株MCF7及びヒト白血病細胞数HELをシフトした。IGF-IR陰性マウス胚繊維芽細胞(R-細胞) (R. Basera, Thomas Jefferson University, Philadelphia, PAから入手)は負の対照として働いた。A12はこれらの細胞に結合できず、IGF-IRに対する抗体結合特異性の指標である。しかしながら、A12はマウス腫瘍細胞ルイス肺腫瘍に結合し、当該腫瘍を部分的にシフトした。これは、この抗-ヒトIGF-IR抗体がマウスIGF-IRに対していくらか交差反応性を有することを示唆するものである。
【0123】
抗体A12の結合後のIGF-I受容体内在化
抗体A12は、高親和性を有するヒト腫瘍細胞に野生型IGF-IRを結合することが明らかとなった。抗体A12は、製造者の教示に従うIODO-ビーズ(Pierce)を用いて125ヨウ素で放射標識した。MCF7ヒト乳癌細胞を6-ウェルプレートに播き、50%コンフルエンスになるまで終夜培養した。125I-A12の1μgを各ウェルに加え、37℃でインキュベートするか又は氷上で4℃に維持した。プレートを30分間、90分間又は180分間インキュベートし、各時点について三点で行った。4℃での培養を180分間維持した。各時点で、ウェルを1 x w/PBSで洗浄した後、100 mMグリシン-HCl、2M尿素で5分間すくい取った。すくい取った材料は膜結合抗体を示し、氷上においてカウントした。次いで、ウェルをPBSで3回洗浄し、細胞を1N NaOH/1% TritonX100で溶解した。溶解画分は内在化抗体を示した。次いで、すくい取られた溶解画分をガンマカウンタで読み、標準偏差を用いてプロットした。図22に示すように、内在的放射活性のレベルは、37℃で培養された細胞中で時間と共に増加したが、膜輸送をおそらく遅らせる4℃に維持された細胞中では、取り込みはほとんど観察されなかった。これは、IGF-IRへの結合の際に、抗体A12は迅速に内在化され、強力に表面結合受容体の枯渇をもたらす、ことを証明した。
【0124】
IGF-IR表面受容体多様性をFACSで決定した。接着性MCF7細胞を37℃で4時間、50 nMのIGF-Iの抗体A12で処理した。細胞を氷冷PBS/5% BSAで2回洗浄し、1 x 106 細胞を染色チューブに等分し、氷上に置いた。次いで、非ブロッキングマウスモノクローナル抗体(Ab-1, NeoMarkers, Fremont, CA)に特異的なIGF-Iアルファ-ポリペプチドを4℃で2時間、細胞でインキュベートした。PBS/BSA洗浄後、細胞を氷上で1時間、抗-マウスIgGフィコエリトリン-複合化第二抗体(PharMingen, BD Biosciences)でインキュベートした。PBS/BSA洗浄後、FACSvantage SEフローサイトメトリー(BD Bioscience)を用いる蛍光活性化細胞選別アッセイによって、細胞を分析した。図22bから明らかなように、A12への暴露は、MCF7細胞の蛍光強度に顕著な減少をもたらした。これは、内在化に起因する表面受容体低調節の指標である。計算した平均蛍光強度比は、A12によるインキュベーション後に90%のIGF-IR表面染色の減少を示した。4℃でA12で細胞をインキュベートした場合には、このシフトは見られず、エネルギー依存性抗体-介在受容体内在化処理と一致した。IGI-Iへの細胞の暴露は、表面IGF-IR蛍光強度における顕著な変化を起こさず、IGF-IR分解にリガンドの効果をほとんど及ぼさないことを示すウェスタンブロッティング解析と一致した。
【0125】
全細胞IGF-IRは、IGF-I又はA12による処理に応答するHT-29細胞及びBxPC-3細胞について決定した。図22cに示すように、培地へのIGF-Iの添加はIGF-IR発現に何の影響を及ぼさなかった。反対に、抗体A12の添加は、3〜6時間後の細胞のIGF-IRレベルの重大な枯渇をもたらした。
【0126】
単独又はイリノテカン(CPT-11)と併用したヒト結腸癌の成長阻害
抗-IGF-IR抗体がin vivoでヌードマウス異種移植モデルにおいてヒト腫瘍成長を阻害することができるか否かを決定することに興味を持った。細胞培養中の2〜3百万個の生存HT-29ヒト結腸癌細胞の皮下投与によって、腫瘍を、3〜4週齢の胸腺欠損マウス(nu/nu)に誘発させた。腫瘍を樹立し、腫瘍体積が200 mm3に達した時に抗体処置を開始した。10動物に1処理群当たりの腫瘍細胞を注射した。抗体を0.5 ml TBS中の1 mg又は0.5 mgで3日間ごとに腹腔内投与(IP)した。薬物イリノテカン(CPT-11) (LKT Laboratories)を抗体処置の開始から4週に1回IP (100 mg/kg)投与した。対照動物には、クラスが一致した無関係なヒトIgG抗体を与えた。高さ、幅及び長さを測定するVernierキャリパーを用いて一定の間隔で腫瘍測定を行い、計算し、総腫瘍体積を決定した。対照腫瘍が3000mm3に達した時に試験を終了した。図23で示されるように、毎3日間の0.5 mg又は1 mgの抗体2F8用量は、このモデルにおいて顕著な阻害効果を示した(P<0.05)。0.5又は1 mgの2F8での処置群の腫瘍サイズ間には全く統計上の誤差はなく、その反応はCPT-11単独で処置しものと同様であった。2F8及びCPT-11を一緒に与えた場合には、より高い腫瘍成長阻害(72%減少)を示した。これは、抗-IGF-IR抗体療法が腫瘍成長に与える化学療法剤CPT-11の抗腫瘍活性を亢進することができたことを証明するものである。
【0127】
ヒト結腸癌に対するin vivoでの抗体A12の抗腫瘍活性
抗体A12は、抗体2F8よりもIGF-IRに対して10倍より高い親和性を有する。顕著な腫瘍阻害が抗体2F8を用いてin vivoで観察されたので、我々は、マウス異種移植モデルにおけるヒト結腸癌株H-29の腫瘍成長に与えるA12の活性を試験した。腫瘍を先述のようにして誘発させ、腫瘍樹立後(大きさ200 mm3)直ちに抗体処置を開始した。次いで、抗体処置を実験期間中毎3日間、1 mg、100μg又は10μgの濃度で行った。1処置群につき10動物を使用し、対照動物にはクラス一致IgG対照抗体を与えた。図24に示すように、抗体A12は、対照に比べて腫瘍成長が74%減少した(P<0.05)。これは、A12がこの異種移植モデルにおいて結腸癌成長の阻害に単一治療剤として有効である、ことを証明した。この実験では、明確な用量-応答効果が認められた。100μg A12の用量で抗腫瘍活性も観察された。
【0128】
異種腫瘍モデルにおけるin vivoでのヒト乳癌に対する抗体A12活性
抗体A12は、in vitroで、MCF7細胞のIGF-依存的細胞分裂誘起刺激及び増幅を示した。In vivoでのMCF7腫瘍成長に対するその活性を利用するために、マウス異種移植腫瘍モデルを利用した。MCF7細胞をエストロゲン依存的ヒト腫瘍から最初に単離し、in vivoでの維持及び成長のためにエストロゲンを外部から添加した。ヌードマウスに生分解性エストロゲンペレットを移植した(0.72 mg 17-β-エストラジオール/ペレット, 60日間放出)。加えて、皮下腫瘍細胞注射時には、マウスにはまた、ゴマ種油の50μl懸濁液中の0.5 mgエストラジオールで皮下的に右脇腹に注射した。抗体処置開始前に腫瘍を約150 mm3のサイズに樹立した。抗体を毎3日間、1 mg、100μg又は10μgで投与し、実験期間中、継続した。29日目に、1 mgのA12による動物処置は腫瘍成長が89%減少した(図25)。この処置群では、明らかに樹立腫瘍が最小限であった。当該モデルでは、用量依存的反応がA12処置について認められた。この試験は、A12がin vivoでのヒト乳癌細胞の成長を顕著に減少させる点で有効である、ことを証明した。更に、1 mg/用量で抗体のみで処置すると、腫瘍退縮が観察され、試験終了(50日間)まで続いた。
【0129】
BxPC-3膵臓癌異種移植における、A12のCPT-11又はゲムシタビンとの併用効果
胸腺nu/nuマウスに、Matrigelと1:1混合した2x106 BxPC-3ヒト膵臓癌細胞皮下に投与した。20日後、腫瘍が約200〜300 mm3に達した時に、マウスを無作為化し、処置群に分類した:1) TBS対照; 2) mAb A12 1mg/用量、3回/週; 3) 2 mg イリノテカン、毎7日に1回; 4) mAb A12及びイリノテカン; 5) 2.5 mg ゲムシタビン、毎7日に1回; 6) mAb A12及びゲンシタビン。全ての処置を腹腔内注射によって投与した。式:体積 =(π/σ)lxw2を使って、腫瘍測定を週に2回記録した。
【0130】
18日目に、1群3動物を屠殺し、腫瘍を中間組織的評価のために切除した。TBS群をのぞいて44日目まで残りの動物について処置及び腫瘍測定を継続した。潰瘍形成及び壊死のため、当該対照群を37日目に屠殺した。試験終了時に、最終腫瘍測定を記録し、動物を屠殺し、1群4動物の腫瘍を最終組織的評価のために切除した。
【0131】
BxPC-3腫瘍はmAb A12に対し非常に反応性が高く、8動物中の2動物は処置の5週間後に部分的に腫瘍退縮した。BxPC-3腫瘍のイリノテカン又はゲムシタビンのみへの反応は、抗体と比較できるものであったが、腫瘍退縮は見られなかった。抗体A12をイリノテカン又はゲムシタビンと併用した場合には、薬剤を単独で使用した場合に比べてより効果的であった(図26)。イリノテカン及びMab A12はより効果的な組み合わせである。A12及びイリノテカンの組み合わせについては、9動物中の3動物が部分的に腫瘍退縮を示した。しかしながら、イリノテカンを与えた動物では、腫瘍退縮はなかった。
【0132】
【表6】
【0133】
HT-29結腸癌異種移植におけるCPT-11又はパクリタキセルと併用したA12の効果
雌性胸腺nu/nuマウスに、Matrigelとの0.4 ml混合1:1中に5 x 106 細胞を含むHT-29ヒト結腸癌細胞懸濁液を皮下に投与した。腫瘍が約200 mm3に達した時に、マウスを無作為化し、処置群に分類した:1) TBS対照; 2) mAb A12 1mg/用量、3回/週; 3) 2 mg イリノテカン、毎7日に1回; 4) mAb A12及びイリノテカン; 5) 121μg パクリタキセルの0.2 ml、毎7日に1回; 6) mAb A12及びパクリタキセル。全ての処置を腹腔内注射によって投与した。
【0134】
処置を6週間継続し、式:体積 =(π/σ)lxw2を使って、腫瘍測定を週に2回記録した。
21日目に、1群4動物を屠殺し、腫瘍を組織的評価のために切除した。40日まで残りの動物について処置及び腫瘍測定を継続した。その際に最終測定を記録し、動物を屠殺し、1群4匹の腫瘍を最終組織的評価のために切除した。
【0135】
単剤mAb A12、CPT-11又はパクリタキセルは顕著に(P<0.02)、TBS対照群に比べてHT-29異種移植片の成長を阻害した(図27)。mAb A12とCPT-11又はパクリタキセルのいずれかとの併用療法は、いずれかの単独処置に比べて腫瘍成長において顕著な阻害を示した(P<0.003)。
【0136】
単一療法群では腫瘍退縮は観察されなかった。対照腫瘍はそのままで、40日目での単一療法群と同様に22日目に高度に血管新生を示した。しかしながら、CPT-11及びmAbA12を用いる併用療法は、8動物中6動物において部分的に腫瘍退縮が起こり、パクリタキセル及びmAbA12を用いる併用療法は、8動物中3動物において腫瘍退縮が起こった。
【0137】
【表7】
【0138】
本明細書に開示される本発明の原理におけるバリエーションが当業者によってなされることは理解および予想され、かかる修飾が本発明の範囲に含まれるものであることが意図される。
【技術分野】
【0001】
本発明は、本発明は、インシュリン様成長因子-I受容体(IGF-IR)に特異的な抗体又はその断片、及び抗体をコードする単離又は精製ポリヌクレオチド配列に関する。
【背景技術】
【0002】
本出願は、2003年5月1日に出願された米国仮出願第60/467,177号の利益を請求する。
【0003】
背景
インシュリン様成長因子受容体(IGF-IR)は、標準的な胎児及び出生後の成長及び発達に不可欠な、至る所に存在する膜貫通型チロシンキナーゼ受容体である。IGF-IRは、細胞増殖、細胞分化、細胞サイズの変化及を刺激し、細胞をアポトーシスから保護することができる。それは、細胞形質転換の準義務的であるとも考えられてきた(Adamsら, Cell. Mol. Life Sci. 57: 1050-93 (2000); Baserga, Oncogene 19: 5574-81 (2000)で調べられる)。IGF-IRは、ほとんどの細胞種に存在し、成長因子IGF-I及びIGF-IIのシグナル分子(以下、IGF類と総称される)として役立つ。IGF-IRはまた、それがIGF類に結合する親和性が3オーダー低いにもかかわらず、インシュリンに結合する。IGF-IRは、ジスルフィド結合によって共有結合的に結合された2個のアルファ鎖及び2個のベータ鎖を含む、前形成ヘテロ-四量体である。受容体サブユニットは、180kdの単一ポリペプチド鎖の一部として合成され、次いで、アルファ(130kd)及びベータ(95kd)サブニットにタンパク質分解される。完全なアルファ鎖は細胞外にあり、リガンド結合部位を含む。ベータ鎖は、膜貫通型ドメイン、チロシンキナーゼドメイン及び細胞の分化及び形質転換に必要なC-末端伸張を有するが、マイトジェンシグナル伝達及びアポトーシスからの保護は不可欠ではない。
【0004】
IGF-IRは、インシュリン受容体(IR)に高い類似性を有し、特にベータ鎖配列(70%相同性)内にある。この相同性のため、これらの受容体が1個のIR二量体及び1個のIGF-IR二量体を含むハイブリッドを形成することができることが、近年の研究で証明されている(Pandini ら, Clin. Canc. Res. 5: 1935-19 (1999))。ハイブリッドの形成は、両方の標準的な形質転換細胞で起こり、ハイブリッド含有量は、細胞内の2個のホモダイマー受容体(IR及びIGF-IR)の濃度に依拠する。39個の乳癌検体の一研究では、IR及びIGF-IRは全ての腫瘍試料で過剰発現したが、ハイブリッド受容体含有量は、常に、両ホモ-受容体のレベルを約3倍越えた(Pandiniら, Clin. Canc. Res. 5: 1935-44 (1999))。ハイブリッド受容体は、IR及びIGF-IRの対から成るが、ハイブリッドは、IGF-IRの親和性に類似した親和性でIGF類に選択的に結合し、インシュリンには弱く結合するにすぎない(Siddle及びSoos, The IGF System. Humana Press. pp. 199-225. 1999)。そのため、これらのハイブリッドはIGF類に結合し、両方の標準的な形質転換細胞にシグナルを形質導入することができる。
【0005】
第二のIGF受容体、IGF-IIR又はマンノース-6-リン酸(M6P)受容体はまた、高親和性でIGF-IIリガンドに結合するが、チロシンキナーゼ活性を有しない(Oatesら, Breast Cancer Res. Treat. 47: 269-81 (1998))。それはIGF-IIRの分解を招くため、このリガンドの成長促進効果を拮抗するIGF-IIの沈降物であると考えられる。腫瘍細胞のIGF-IIRの喪失は、IGF-IIのIGF-IRとの結合に与える拮抗作用の緩和によって、成長性を亢進することができる(Byrdら, J.Biol. Chem. 274: 24408-16 (1999))。
【0006】
IGF-Iの内分泌発現は、主に成長ホルモンによって調節され、肝臓で起こるが、近年の証拠は、多くの他の組織種もIGF-Iを発現できることを示唆している。従って、多くの腫瘍細胞種の場合には、このリガンドは内分泌調節及び傍分泌調節並びに自己分泌調節を受ける(Yu, H.及びRohan, J., J. Natl. Cancer Inst. 92: 1472-89 (2000))。
【0007】
IGFとの特異的結合親和性を有する6種のIGF結合タンパク質(IGFBP)は、血清中で特定されてきた(Yu, H及びRohan, J., J. Natl. Cancer Inst. 92: 1472-89 (2000))。翻訳後修飾の結果として、結合タンパク質の分子構造によって決定されるように、IGFBPは、IGFの作用を亢進又は阻害することができる。それらの主な役割は、IGFの輸送、IGFのタンパク質分解からの保護及びIGFとIGF-IRとの相互作用の調節である。血清IGF-Iのたったの約1%が遊離リガンドとして存在し、残余はIGFBPと会合している(Yu, H.及びRohan, J., J. Natl. Cancer Inst. 92: 1472-89 (2000))。
【0008】
リガンド(IGF)の結合において、IGF-IRは、ベータ鎖の触媒ドメイン内の保存的チロシン残基で自己ホスホリル化される。ベータ鎖内の別のチロシン残基の次のホスホリル化は、シグナルカスケードに匹敵する下流分子の採用のためのドッキング部位を提供する。IGFシグナルの形質導入の主な経路は、マイトジェン活性化プロテインキナーゼ(MAPK)及びホスファチジルイノシトール 3-キナーゼ(PI3K)である(BlakesleyらのThe IGF System. Humana Press. 143-163 (1999)を参照されたい)。MARK経路は、IGF類刺激の後に誘発されるマイトジェンシグナルに主に起因し、PI3Kは、抗-アポトーシス又は生存過程のIGF-依存誘起に起因する。
【0009】
IGF-IRシグナル伝達の重要な役割は、その抗-アポトーシス又は生存機能である。活性化IGF-IRシグナルは、PI3K、Aktの下流ホスホリル化又はタンパク質キナーゼBをシグナル伝達する。Aktは、ホスホリル化によって、プログラムされた細胞死の開始に必須であり、アポトーシスの開始を阻害するBAD等の分子を効果的に阻害できる(Dattaら, Cell 91: 231-41 (1997))。アポトーシスは、標準的な発達過程に匹敵する重要な細胞機構である(Oppenheim, Annu. Rev. Neurosci. 14: 453-501 (1991))。それは、重度の障害性細胞の除去に影響を与え、腫瘍形成を促進する変異原生障害の潜在的固執を軽減するための重要な機構である。このために、IGFのシグナル伝達の活性化は、マウストランスジェニックモデルでの自然発生癌形成を促進することができる、ことが証明されている(DiGiovanniら, Cancer Res. 60: 1561-70 (2000))。更に、IGF過剰発現は、化学療法誘導細胞死由来の細胞を救援することができ、腫瘍細胞薬物耐性において重要な因子となる(Goochら, Breast Cancer Res. Treat. 56: 1-10 (1999))。従って、IGFシグナル伝達経路の調節は、化学療法剤に対する腫瘍細胞の感受性を増加させることが判っている(Beniniら, Clinical Cancer Res. 7: 1790-97 (2001))。
【0010】
多数の調査及び臨床的研究は、癌の発症、維持及び進行におけるIGF-IR及びそのリガンド(IGF)に関与してきた。腫瘍細胞では、受容体の過剰発現は、しばしばIGFリガンドの過剰発現に呼応しており、これらのシグナルの相乗作用をもたらし、結果として細胞増殖及び生存を亢進する。IGF-I及びIGF-IIは、幅広い癌細胞株、例えば前立腺癌(Nickersonら, Cancer Res. 61: 6276-80 (2001) ; Hellawellら, Cancer Res. 62: 2942-50 (2002) )、乳癌(Goochら, Breast Cancer Res. Treat. 56: 1-10 (1999))、肺癌、結腸癌(Hassan及びMacaulay, Ann. Oncol. 13: 349-56 (2002) )、胃癌、白血病、膵癌、脳腫瘍、骨髄腫(Ge及びRudikoff, Blood 96: 2856-61 (2000)、黒色腫(All-Ericssonら, Invest. Ophthalmol. Vis. Sci. 43: 1-8 (2002))及び卵巣癌(Macaulay, Br. J. Cancer 65: 311-20 (1990)を参照されたい)の強力なマイトジェンであることが判っており、この効果はIGF-IRによって仲介される。血清中のIGF-Iの高循環レベルは、乳癌、前立腺癌及び結腸癌の高い危険性に関係している(Pollak, Eur. J. Cancer 36: 1224-28 (2000))。結腸癌のマウスモデルにおいて、in vivoでの循環IGF-Iレベルの増加は、腫瘍成長及び転移の発生率の顕著な増加をもたらす(Wuら, Cancer Res. 62: 1030-35 (2002))。トランスジェニックマウスの表皮基底細胞でのIGF-Iの構成的発現は、自然発生的腫瘍形成を促進することが判っている(DiGiovanniら, Cancer Res. 60: 1561-1570, 2000; Bolら,Oncogene 14: 1725-1734 (1997))。細胞株及び腫瘍でのIGF-IIの過剰発現は高頻度で生じ、IGF-II遺伝子のゲノムインプリンティングの喪失から起こる(Yaginumaら, Oncology 54: 502-7 (1997))。受容体過剰発現は、多種多様のヒト腫瘍種、例えば肺癌(Quinnら, J.Biol. Chem. 271: 11477-83 (1996))、乳癌(Cullenら, Cancer Res. 50: 48-53 (1990); Peyrat及びBonneterre, Cancer Res. 22: 59-67 (1992); Lee and Yee, Biomed Pharmacother. 49: 415-21 (1995))、肉腫(van Valenら, J. Cancer Res. Clin. Oncol. 118: 269-75 (1992); Scotlandiら, Cancer Res. 56: 4570-74 (1996))、前立腺癌(Nickersonら ,Cancer Res. 61: 6276-80 (2001))及び結腸癌(Hassan及びMacaulay, Ann. OticoL 13: 349-56 (2002))において証明されてきた。加えて、高変異原生癌細胞は、転移する傾向が少ない腫瘍細胞に比べて、IGF-II及びIGF-IRを高発現することが判っている(Guerraら, Int. J. Cancer 65: 812-20 (1996))。細胞増殖及び形質転換でのIGF-IRの重要な役割は、マウス胚繊維芽細胞由来のIGF-IRノックアウトの実験で証明された。これらの主な細胞は、10%血清を含む培地で著しく低い速度で成長し、SV40 Large T抗原を含む様々な癌遺伝子によって形質転換されない(Sellら, Mol. Cell. Biol. 3604-12 (1994))。近年、いくつかの形の乳癌における薬物ハーセプチンへの耐性は、当該癌ではIGF-IRシグナル伝達の活性化に起因する、ことが証明された(Luら, J. Natl. Cancer Inst. 93: 1852-57 (2001))。そのため、IGF-IRの過剰発現又は活性化は、腫瘍形成での主な決定基であるだけでなく、腫瘍細胞において薬物耐性でもある。
【0011】
IGF系の活性化はまた、癌、例えば末端肥大症(Drange及びMelmed. In : The IGF System. Humana Press. 699-720 (1999))、網膜新生血管(Smithら, Nature Med. 12: 1390-95 (1999))、及び乾癬(Wraightら, Nature Biotech. 18: 521-26 (2000))に加えて、いくつかの病理学的条件に関与している。最近の研究では、IGF-IRを標的とするアンチセンスオリゴヌクレオチド調製は、マウスモデルでの乾癬皮膚移植における表皮細胞の高増殖を顕著に阻害する点で効果的であった。これは、抗-IGF-IR療法がこの慢性的疾患の治療に効果的であることを示唆している。
【発明の概要】
【発明が解決しようとする課題】
【0012】
細胞でのIGF-IRシグナル伝達経路を阻害するために様々な戦略が開発されてきた。アンチセンスオリゴヌクレオチドは、乾癬について上に示したように、in vitro及び実験的マウスで効果的であった。加えて、in vitro及びin vivoで抗-増殖活性を有する、IGF-IRを標的とする阻害ペプチドが産生されている(Pietrzkowskiら, Cancer Res. 52: 6447-51 (1992); Haylorら, J. Am. Soc. Nephrol. 11: 2027-35 (2000))。IGF-IRのC-末端由来の合成ペプチド配列は、アポトーシスを誘導し、腫瘍成長を顕著に阻害することが判っている(Reissら, J. Cell. Phys. 181: 124-35 (1999))。腫瘍細胞株で過剰発現させた場合に、リガンドを野生型IGF-IRと競合し、in vitro及びin vivoでの腫瘍細胞成長を効果的に阻害する、様々なIGF-IRの優性遺伝子-陰性突然変異体も作製されている(Scotlandiら, Int. J. Cancer 101: 11-6 (2002); Seelyら, BMC Cancer 2: 15 (2002))。加えて、溶解型IGF-IRはまた、in vivoで腫瘍成長を阻害することが証明されている(D'Ambrosioら, Cancer Res. 56: 4013-20 (1996))。ヒトIGF-IRに対する抗体はまた、乳癌(Artega andOsborne, Cancer Res. 49: 6237-41 (1989))、ユーイング骨肉腫(Scotlandiら, Cancer Res. 58: 4127-31 (1998))及び黒色腫(Furlanettoら, Cancer Res. 53: 2522-26 (1993))由来の細胞株を含む、in vitroでの腫瘍細胞増殖及びin vivoでの腫瘍形成を阻害することが判っている。抗体は、主に、それらが、1)特定のタンパク質抗原に対して高い選択性を有することができる、2)抗原に高親和性結合を示すことができる、3)in vivoで長半減期を示す、ために魅力的な治療法であり、またそれらは天然の免疫産物であるため、4)in vivoでの毒性が低い(Park及びSmolen. In : Advances in Protein Chemistry. Academic Press. pp: 360-421 (2001))。しかしながら、非-ヒト起源、例えばマウス由来の抗体は、反復適用に従って治療抗体に対して直接的免疫応答をもたらし、抗体の効力を中和する。ヒトではネズミ又はキメラ抗体よりも免疫原生が低い可能性があり、天然免疫-応答抗体に類似しているので、完全ヒト抗体は、ヒト治療薬の成功の最も高い可能性を提供する。このために、治療用途のための、高親和性ヒト抗-IGF-IRモノクローナル抗体を開発する必要がある。
【課題を解決するための手段】
【0013】
発明の要約
本発明は、ヒトIGF-I受容体に特異的に結合するヒトモノクローナル抗体及びその断片を提供する。抗体は、(i)IGF-I又はIGF-IIのIGF-IRへの結合を阻害する;(ii)IGF-I又はIGF-IIによってIGF-IRの活性化を中和する;(iii)IGF-IR表面受容体を少なくとも約80%減少させる;及び(iv)約3×10-10M-1以下のKdでIGF-IRに結合する、からなる群より選ばれる少なくとも1つの性質を有する。より好ましい実施態様では、本発明の抗体は、IGF-IR表面受容体を少なくとも約85%、より好ましくは少なくとも約90%減少させる。更に、抗体は、リガンド-介在受容体自己ホスホリル化、及びMAPK及びAkt経路を介する下流細胞シグナル伝達を阻害する。本発明の抗体は、単独又は抗新形成剤と組み合わせて使用され、特に腫瘍性疾患及び高増殖疾患に有用である。
【0014】
本発明は、抗体又はその断片をコードする単離ポリヌクレオチド、当該ポリヌクレオチド配列を含む発現ベクター及び発現のための宿主細胞を提供する。
【0015】
更に、本発明は、医薬組成物及び腫瘍及び高増殖疾患の治療のために診断方法及び治療方法を提供する。当該方法は、抗新形成剤の投与又は治療を更に含むことができる。
【図面の簡単な説明】
【0016】
【図1】図1は、2F8重鎖可変ドメインのヌクレオチド配列を示す。
【図2】図2は、2F8重鎖可変ドメインのアミノ酸配列を示す。CDRは太字で下線を引いてある。
【図3】図3は、完全2F8重鎖のヌクレオチド配列を示す(下線: 分泌シグナル配列; 斜体: IgG1不変領域)。
【図4】図4は、完全2F8重鎖のアミノ酸配列を示す(下線: 分泌シグナル配列; 太字: CDR; 斜体:IgG1不変領域)。
【図5】図5は、2F8軽鎖可変ドメインのヌクレオチド配列を示す。
【図6】図6は、2F8軽鎖可変ドメインのアミノ酸配列を示す。CDRは太字で下線を引いてある。
【図7】図7は、完全2F8軽鎖のヌクレオチド配列を示す(下線: 分泌シグナル配列; 斜体: IgG1不変領域)。
【図8】図8は、完全2F8軽鎖のアミノ酸配列を示す(下線: 分泌シグナル配列; 太字: CDR; 斜体:IgG1不変領域)。
【図9】図9は、A12軽鎖可変ドメインのヌクレオチド配列を示す。
【図10】図10は、A12軽鎖可変ドメインのアミノ酸配列を示す。CDRは太字で下線を引いてある。
【図11】図11は、完全A12軽鎖のヌクレオチド配列を示す(下線: 分泌シグナル配列; 斜体: IgG1不変領域)。
【図12】図12は、完全A12軽鎖のアミノ酸配列を示す(下線: 分泌シグナル配列; 太字: CDR; 斜体:IgG1不変領域)。
【図13】図13は、抗体2F8及びA12のVH及びVL CDR配列を示す。VL CDRの相違に下線を引いてある。
【図14】図14は、2F8及びA12の軽鎖可変領域アミノ酸配列間の相同性を示す。配列の相違を四角で囲み、CDRを強調してある。
【図15】図15は、固定化溶解性IGF-IRへのIGF-Iの結合を阻害する抗体2F8及びA12の能力を測定するアッセイの結果を示す。カッパ及びラムダ軽鎖不変領域を有するA12抗体を試験した。
【図16】図16は、IGF-IのMCF7細胞への結合を阻害する抗体2F8及びA12(ラムダ軽鎖)の能力を測定するアッセイの結果を示す。
【図17】図17は、インシュリンのZR75-I細胞への結合を阻害する抗体2F8及びA12の能力を測定するアッセイの結果を示す。
【図18A】図18Aは、MCF7乳癌細胞依存性有糸分裂生起アッセイにおける3H-チミジン取り込みに与えるA12抗体の影響を示す。
【図18B】図18Bは、BxPC-3膵臓癌細胞依存性有糸分裂生起アッセイにおける3H-チミジン取り込みに与えるA12抗体の影響を示す。
【図18C】図18Cは、HT-29結腸癌細胞依存性有糸分裂生起アッセイにおける3H-チミジン取り込みに与えるA12抗体の影響を示す。
【図19A】図19Aは、IGF-I介在受容体ホスホリル化の阻害を示す。抗体A12及び2F8によるMCF7乳癌細胞での阻害。
【図19B】図19Bは、IGF-I介在受容体ホスホリル化の阻害を示す。抗体A12によるHT-29結腸癌細胞及びBxPC-3膵臓癌細胞での阻害。
【図20A】図20Aは、抗体A12及び2F8による下流エフェクター分子のIGF-I介在MAPKホスホリル化の阻害を示す。
【図20B】図20Bは、抗体A12及び2F8による下流エフェクター分子のIGF-I介在Aktホスホリル化の阻害を示す。
【図21】図21は、ヒト及びネズミIGF-IR陽性及び陰性細胞株への抗体A12の結合を示す。MCF7: ヒト乳癌細胞株; R-: マウス胚繊維芽細胞; HEL: ヒト白血病細胞; ルイス肺: マウス肺癌細胞。
【図22A】図22Aは、MCF7細胞上のIGF-IRへの結合後の標識抗体A12の内在化を示す。
【図22B】図22Bは、細胞表面会合IGF-IRの減少を示す。
【図22C】図22Cは、A12で長期処理後の総細胞IGF-IRの分解を示す。
【図23】図23は、抗体SF8及びCPT-11(イリノテカン)の単独又は併用によるヌードマウスでのHT-29ヒト結腸腫瘍成長の阻害を示す。
【図24】図24は、ヌードマウスでのHT-29ヒト結腸腫瘍成長に与える抗体A12の効果を示す。
【図25】図25は、ヌードマウスでのMCF7ヒト乳癌成長に与える抗体A12の効果を示す。
【図26】図26は、抗体A12、ゲムシタビン又はCPT-11(イリノテカン)を単独又は併用するヌードマウスでのBxPC-3膵臓癌異種移植の阻害を示す。
【図27】図27は、抗体A12、パクリタキセル又はCPT-11(イリノテカン)を単独又は併用するヌードマウスでのHT-29結腸癌異種移植の阻害を示す。
【発明を実施するための形態】
【0017】
発明の詳細な説明
本発明のある実施態様では、ヒト抗体が提供される。抗体はまた、その他の化学剤及び生物剤、例えば特に限定されないが、抗新形成剤、及び/又は細胞成長を介在するその他の受容体もしくは受容体基質の阻害剤である薬剤、と組み合わせて使用することができる。本発明は、トポイソメラーゼ機能の阻害剤である抗新形成剤に更に関する。かかる薬剤の選択は、IGF-IRに特異的な抗体と組み合わせた治療方法での使用に有利である。
【0018】
天然抗体は典型的には、2個の同一重鎖及び2個の同一軽鎖を有し、各軽鎖は共有結合的に鎖間ジスルフィド結合によって重鎖に結合し、及び複数のジスルフィド結合は2個の重鎖に互いに更に結合する。個々の鎖は、類似のサイズ(110〜125アミノ酸)及び構造を有し、機能は異なるドメインに折畳むことができる。軽鎖は、1可変ドメイン(VL)及び/又は1不変ドメイン(Css)を含むことができる。この重鎖はまた、1可変ドメイン(VH)及び/又は抗体のクラス又はアイソタイプによって3又は4の不変ドメイン(CH1、CH2、CH3及びCH4)を含むことができる。ヒトでは、アイソタイプはIgA、IgD、IgE、IgG及びIgMであり、IgGは更にサブクラス又はサブタイプ(IgA1-2及びIgG1-4)に細分される。
【0019】
一般的に、可変ドメインは、1抗体から次の抗体まで、特に抗原結合部位の位置ではかなりのアミノ酸配列変化を示す。超可変又は相補性決定部位(CDR)と呼ばれる3領域は、VL及びVHの各ドメインに見られ、当該領域は、フレームワークと呼ばれる変化の少ない領域によって支持されている。
【0020】
VL及びVHドメインからなる抗体の部分は、Fv(可変断片)を示し、抗原結合部位を構成する。単一鎖Fv(scFv)は、VLドメイン及びVHドメインを1ポリペプチド鎖上に含む抗体断片であり、1ドメインのN末端及びその他のドメインのC末端は、適応性のあるリンカーで結合されている(米国特許第4,946,778号明細書(Ladnerら); 国際公開第 WO 88/09344号パンフレット(Hustonら)を参照されたい)。国際公開第 WO92/01047号パンフレット(McCaffertyら)は、溶解性組換え遺伝子ディスプレイパッケージ、例えばバクテリオファージの表面上のscFv断片のディスプレイを記載する。
【0021】
単一鎖抗体を産生するために使用されるペプチドは、正確な3次元折畳み及びVL及びVHドメインの会合が起こることを確実にするために選択された適合性のあるペプチドでよい。リンカーは一般的に、10〜50アミノ酸残基である。好ましくは、リンカーは10〜30アミノ酸残基である。より好ましくは、リンカーは12〜30残基である。最も好ましくは、リンカーは15〜25アミノ酸残基である。かかるリンカーペプチドの非限定的例は、(Gly-Gly-Gly-Gly-Ser)3(配列番号33)である。
【0022】
Fab(抗原結合断片)は、VL-CL及びVH-CH1ドメインからなる抗体の断片を言う。パパインによる完全抗体の消化によって得られたかかる断片は、それによって2個の重鎖が通常結合される、抗体のヒンジ部位を保持しない。当該断片は、一価であり、単にFabと言われる。あるいは、ペプシンによる消化は、ヒンジ部位を保持する断片を生じる。2個の重鎖を結合する完全な鎖間ジスルフィド結合を有するこのような断片は、二価であり、F(ab')2と言われる。F(ab')2のジスルフィド結合が緩和され、重鎖が分離される場合に、一価のFab'が生成する。それらは二価であるため、完全抗体及びF(ab')2断片は、一価のFab又はFab'断片よりも高い抗原親和力を有する。国際公開第 WO 92/01047号パンフレット(McCaffertyら)は、溶解性組換え遺伝子ディスプレイパッケージ、例えばバクテリオファージの表面上のFab断片のディスプレイを記載する。
【0023】
Fc(結晶化可能断片)は、一対の重鎖不変ドメインからなる抗体の部分又は断片を言う。IgG抗体例えばFcは、重鎖CH2及びCH3ドメインからなる。IgA又はIgM抗体のFcは、CH4ドメインを更に含む。Fcは、Fc受容体結合、補体依存性細胞障害活性の活性化及び抗体依存性細胞障害活性(ADCC)の活性化に関連する。複数のIgG様タンパク質の複合体であるIgA及びIgMのような抗体に関して、複合体形成はFc不変ドメインを必要とする。
【0024】
最後に、互いにかつFcに対してFabが可動性である場合には、2個の重鎖の共有結合のための複数のジスルフィド結合を含めて、ヒンジ部位は、抗体のFab及びFc部分を分離する。
【0025】
結合特異性を保持する抗体型が開発されているが、望ましいその他の特徴、例えば生物特異性、多価性(2個より多い結合部位)、コンパクトサイズ(例えば結合ドメインのみ)を有する。
【0026】
単一鎖抗体は、それら由来する完全抗体の不変ドメインのいくつか又は全てが欠落している。そのため、それらは、完全抗体の使用に関連する問題のいくつかを解消することができる。例えば、単一鎖抗体は、重鎖不変領域及び他の生物分子間の特定の望ましくない相互作用がない傾向にある。加えて、単一鎖抗体は、完全抗体よりもかなり小さく、完全抗体よりも透過性が高い。そのため、より効果的に単一鎖抗体を抗原結合部位に配置し及び当該部位を標的することができる。更に、かなり小さいサイズの単一鎖抗体は、完全抗体に比べて、レシピエントにおける好ましくない免疫応答を誘起しないようである。
【0027】
複数の単一鎖抗体であって、各単一鎖が第一のペプチドリンカーによって共有結合的に結合された1VH及び1VLドメインを有する抗体は、少なくとも1以上のペプチドリンカーによって共有結合的に結合されて、単一特異的でも又は多重特異的でもよい多価単一鎖抗体を形成する。多価単一鎖抗体の各鎖は、可変軽鎖断片及び可変重鎖断片を含み、ペプチドリンカーによって少なくとも1つの他の鎖に結合される。ペプチドリンカーは、少なくとも15アミノ酸残基からなる。アミノ酸残基の最大数は約100である。
【0028】
2個の単一鎖抗体は混合されて、二価二量体としても知られている二重特異性抗体を形成することができる。二重特異性抗体は、2個の鎖及び2個の結合部位を有し、単一特異的でも又は多特異的でもよい。二重特異性抗体の各鎖は、VLドメインに結合したVHドメインを含む。ドメインは、十分に短いために同一鎖上のドメイン間でペアリングが起こらないリンカーと結合される。それによって、異なった鎖上の相補性ドメイン間でのペアリングを推進して2個の抗原結合部位を再現する。
【0029】
3個の単一鎖抗体は混合されて三価三量体としても知られている三重特異性抗体(triabodies)を形成することができる。三重特異性抗体は、VL又はVHドメインのカルボキシル末端に直接連結したVL又はVHドメインのアミノ酸末端で、すなわちリンカー配列を用いることなく、構築される。三重特異性抗体は、環状の頭-尾形式に配置されたポリペプチドを有する3個のFv頭部を有する。三重特異性抗体の可能な立体配置は、3個の結合部位と平面関係にあり、当該部位は1つの平面内にその他と120度の角度にある。三重特異性抗体は、単一特異的でも二重特異的でも又は三重特異的でもよい。
【0030】
従って、本発明の抗体及びその断片は、特に限定されないが、抗原に特異的に結合する天然抗体、二価断片例えば(Fab')2、一価断片例えばFab、単一鎖抗体、単一鎖Fv(scFv)、単一ドメイン抗体、多価単一鎖抗体、二重特異性抗体、三重特異性抗体などを含む。
【0031】
本発明の抗体及び特にその可変ドメインは、当該分野で公知の方法によって得られる。これらの方法は、例えば、Kohler及びMilstein, Nature, 256: 495-497 (1975)及びCampbell, Monoclonal Antibody Technology, The Production and Characterization of Rodent and Human Hybridomas, Burdonら著, Laboratory Techniques in Biochemistry and Molecular Biology, 第13巻, Elsevier Science Publishers, Amsterdam (1985)によって記載されている免疫法;例えばHuseら, Science, 246,1275-81 (1989)によって記載されている組換えDNA法を含む。抗体は、scFv又はFabの形態でのVH及びVLドメインの組み合わせを生じるファージディスプレイ・ライブラリーからも得られる。VH及びVLドメインは、合成、半合成又は天然由来のヌクレオチドによってコードされる。ある実施態様では、ヒト抗体断片を生じるファージディスプレイ・ライブラリーが好ましい。ヒト抗体の他の起源は、ヒト免疫グロブリン遺伝子を発現するように設計されたトランスジェニックマウスである。
【0032】
抗体断片は、完全抗体を開裂することによって、又は当該断片をコードするDNAを発現することによって作製することができる。抗体断片は、Lamoyiら, J. Inimutiol. Metliods, 56: 235-243 (1983)及びParham, J. Immunol. 131: 2895-2902 (1983)によって記載の方法によって調製することができる。かかる断片は、1又は両Fab断片もしくはF(ab')2断片を含んでもよい。かかる断片は、単一鎖断片可変領域抗体、すなわちscFv、二重特異性抗体又は他の抗体断片を含んでもよい。かかる機能性等価体を製造する方法は、PCT出願の国際公開 WO 93/21319号パンフレット、欧州特許出願第239,400号明細書; PCT出願の国際公開第 WO 89/09622号パンフレット; 欧州特許出願第338,745号;及び欧州特許出願 EP 332,424号に開示されている。
【0033】
本発明の抗体又はその断片はIGF-IRに特異的である。抗体特異性とは、特定の抗原エピトープに対する抗体の選択的認識を言う。本発明の抗体又はその断片は、例えば単一特異性でも二重特異性でもよい。二重特異性抗体(BsAb)は、2種の異なった抗原結合特異性又は部位を有する抗体である。抗体が1より多い特異性を有する場合には、認識エピトープは、単一抗原又は1抗原より多い抗原と関連する。従って、本発明は二重特異性抗体又はその断片を提供する。これらは2種の異なった抗原に結合し、IGF-IRに対して少なくとも1つの特異性を有する。
【0034】
本抗体又はその断片のIGF-IRに対する特異性は、親和性及び/又は親和力に基づいて決定される。抗原と抗体との平衡解離定数によって表される親和性(Kd)は、抗原決定部位及び抗体結合部位間の結合強度を測定する。親和力は、抗体と抗原との間の結合強度を測定する。親和力は、抗体上のその抗原結合部位を有するエピトープ間の親和性、及び抗体価に関連する。この抗体価とは、特定のエピトープに特異的な抗原結合部位の数を言う。抗体は典型的には、10-5〜10-11 リットル/molの解離定数(Kd)で結合する。10-4 リットル/molより大きいKdは、一般的に非特異的結合を示すと考えられる。Kd値が小さくなればなるほど、抗原決定基及び抗体結合部位間の結合強度は強くなる。
【0035】
本発明の抗体又はその断片はまた、結合特性が直接的突然変異、親和性成熟、ファージディスプレイ又は鎖シャフリングによって改善されたものを含む。親和性及び特異性は、改変、又はCDR及び/又はFW残基を突然変異及び所望の特性を有する抗原結合部位をスクリーニングすることによって改良することができる(例えばYangら, J. Mol. Biol., (1995) 254: 392-403を参照されたい)。1つの方法は、2〜20アミノ酸の小集団、そうでなければ同一の抗原結合部位において、当該アミノ酸のサブセットが特定の位置で見出されるように、個々の残基又は残基の組み合わせを無作為化することである。あるいは、突然変異はエラープローンPCR法によって一定の範囲の残基を誘発することができる(例えばHawkinsら, J. Mol. Biol., (1992) 226: 889-96を参照されたい)。別の実施例では、重及び軽鎖可変領域遺伝子を含むファージディスプレイ・ベクターは、E. coliの突然変異誘発株で増殖することができる(例えば、Lowら, J. Mol. Biol., (1996) 250: 359-68を参照されたい)。これらの突然変異誘発方法は、当業者に公知の多くの方法の例示に過ぎない。
【0036】
本発明の抗体又はその断片の等価物は、完全長抗-IGF-IR抗体の可変又は超可変領域のアミノ酸配列と実質的に同一のアミノ酸を有するポリペプチドを含む。Pearson及びLipman (Proc. Natl. Acad. Sci. USA (1988) 85: 2444-8)に従うFASTA調査法によって決定されているように、実質的に同一のアミノ酸配列は、本明細書では、もう1つのアミノ酸配列に対して、少なくとも70%、好ましくは少なくとも約80%及びより好ましくは少なくとも約90%の相同性を有する配列として定義されている。
【0037】
保存的アミノ酸置換は、ペプチド、ポリペプチドもしくはタンパク質、又はその断片の1又は2アミノ酸を置換する方法によるアミノ酸組成物における変更と定義される。置換は、実質的にペプチド、ポリペプチドもしくはタンパク質の特性(例えば、電荷、等電点、親和性、親和力、立体配置、溶解性)又は活性を実質的に変更しないように、一般的に類似の性質(例えば酸性、塩基性、芳香族性、サイズ、正電荷又は負電荷、極性、非極性)を有するアミノ酸についてである。かかる保存的アミノ酸置換について行われる典型的置換は、以下のアミノ酸の群間である:グリシン(G)、アラニン(A)、バリン(V)、ロイシン(L)及びイソロイシン(I);アスパラギン酸(D)及びグルタミン酸(E); アラニン(A)、セリン(S)及びスレオニン(T); ヒスチジン(H)、リジン(K)及びアルギニン(R): アスパラギン(N)及びグルタミン(Q); フェニルアラニン(F)、チロシン(Y)及びトリプトファン(W)。
【0038】
保存的アミノ酸置換は、例えば、分子の選択的及び/又は特異的結合特性に主に起因する超可変領域のフランキング領域、及び分子のタンパク質の部分例えば可変重鎖カセットでなされる。
【0039】
本発明の抗体の各ドメインは、重又は軽鎖可変ドメインを有する完全抗体でもよく、又は天然ドメイン又は例えばin vitroで国際公開第WO 93/11236 (Griffithsら)号パンフレットに記載の方法を用いて構築された合成ドメインの機能性等価体、突然変異体又は誘導体でもよい。例えば、少なくとも1つのアミノ酸が欠落している抗体可変ドメインに対応するドメインと組み合わせることができる。重要な特徴は、相補性ドメインを会合して抗原結合部位を形成する各ドメインの能力である。従って、可変重及び軽鎖断片の用語は、特異性に重大な効果を与えない変異体を排除するものと解釈されるべきではない。
【0040】
好ましい実施態様では、本発明の抗-IGF-IR抗体は1以上の以下の性質を示すヒト抗体である。
【0041】
1) 抗体はIGF-IRの外部ドメインに結合し、IGF-I又はIGF-IIのIGF-IRへの結合を阻害する。例えば、阻害は、精製又は膜結合受容体を用いる直接的結合アッセイを用いて決定することができる。この実施態様において、本発明の抗体又はその断片は、好ましくは、少なくともIGF-IR(IGF-I及びIGF-II)の天然リガンドのような強さでIGF-IRに結合する。
【0042】
2) 抗体はIGF-IRを中和する。リガンド例えばIGF-I又はIGF-IIの、IGF-IRの外部、細胞外ドメインへの結合は、ベータサブユニットの自己ホスホリル化及びIFG-IRサブユニット例えばMAPK、Akt及びIRS-1のホスホリル化を刺激する。
【0043】
IGF-IRの中和は、阻害、減少、不活性化及び/又はシグナル形質導入に通常関連するこれらの活性の1以上の破壊を含む。更に、これは、IGF-IR/IRへテロダイマー及びIGF-IRホモダイマーの阻害を含む。従って、IGF-IRの中和は、様々な効果、例えば阻害、減少、不活性化及び/又は成長破壊(増殖及び分化)、拮抗作用(血管補充、侵入及び転移)、及び細胞運動及び転移(細胞接着及び侵襲性)を有する。
【0044】
IGF-IR中和の1測定は、受容体のチロシンキナーゼ活性の阻害である。チロシンキナーゼ阻害は、周知の方法を用いて測定することができる;例えば組換えキナーゼ受容体の自己ホスホリル化レベル及び/又は天然もしくは合成基質のホスホリル化を測定することである。従って、ホスホリル化アッセイは、本発明の分脈において中和抗体の測定に有用である。例えば、ホスホリル化は、ELISAアッセイ又はウェスタンブロットにおいてホスホチロシンに特異的な抗体を用いて検出することができる。チロシンキナーゼ活性のためのいくつかのアッセイは、Panekら, J. Pharmacol. Exp. Thera. 283: 1433-44 (1997)及びBatleyら, Life Sci. 62: 143-50 (1998)に記載されている。本発明の抗体は、リガンドに応答する細胞において、IGF-IRのチロシンホスホリル化を少なくとも約75%、好ましくは少なくとも約85%及びより好ましくは少なくとも約90%減少させる。
【0045】
IGF-IR中和の別の測定は、IGF-IRの下流基質のホスホリル化の阻害である。従って、MAPK、Akt又はIRS-1のホスホリル化レベルを測定することができる。基質ホスホリル化の減少は、少なくとも約50%、好ましくは少なくとも約65%、より好ましくは少なくとも約80%である。
【0046】
加えて、タンパク質発現の検出方法は、IGF-IR中和を側定するために利用することができる。この測定されるタンパク質はIGF-IRキナーゼ活性によって調節される。これらの方法は、タンパク質発現検出のための免疫組織化学(IHC)、遺伝子増幅の検出のための蛍光in situハイブリダイジェーション(FISH)、競争的放射性リガンド結合アッセイ、固体マトリックスブロッティング法、例えばノーザン及びサザンブロット、逆転写酵素ポリメラーゼ鎖反応((RT-PCR)及びELISAを含む。例えば、Grandis ら, Cancer, 78: 1284-92 (1996); Shimizuら, Japan J. Cancer Res., 85: 567-71 (1994); Sauterら, Am. J. Path., 148: 1047-53 (1996); Collins, Glia 15: 289-96 (1995); Radinskyら, Clin. Cancer Res. 1: 19-31 (1995); Petridesら, Cancer Res. 50: 3934-39 (1990); Hoffmannら, Anticancer Res. 17: 4419-26 (1997); Wikstrandraら, Cancer Res. 55: 3140-48 (1995)を参照されたい。
【0047】
In vivoアッセイは、IGF-IR中和を測定するために利用することもできる。例えば、受容体チロシンキナーゼ阻害は、阻害剤の存在及び非存在下、受容体リガンドで刺激された細胞株を用いるマイトジェンアッセイによって観察できる。例えば、IGF-I又はIGF-11で刺激されたMCF7(American Type Culture Collection (ATCC), Rockville, MD)は、IGF-IR阻害をアッセイするために使用することができる。別の方法は、IGF-IR発現腫瘍細胞又はトランスフェクトされてIGF-IRを発現する細胞の成長阻害試験を含む。阻害は、腫瘍細胞、例えばマウスに注射されたヒト腫瘍細胞を用いて観察することもできる。
【0048】
本発明は、IGF-IR中和の任意の特定のメカニズムによって制限されない。本発明の抗-IGF-IR抗体は、IGF-IR細胞表面受容体、リガンドの切断部位(例えばIGF-I又はIGF-II)及び受容体関連チロシンキナーゼによって仲介される次のシグナル形質導入に外部から結合することができ、及びシグナル形質導入カスケードにおけるIGF-IR及び他の下流タンパク質のホスホリル化を防ぐことができる。
【0049】
3) 抗体はIGF-IRの調節を下げる。細胞表面に存在するIGF-IR量は、受容体タンパク質産生、内在化及び分解に依拠する。細胞表面に存在するIGF-IR量は、受容体又は受容体に結合する分子の内在化を検出することによって、直接的に測定することができる。例えば、受容体内在化は、IGF-IRを発現する細胞を標識抗体と接触することにより測定することができる。次いで、膜結合抗体は剥ぎ取られ、回収され及びカウントされる。内在化抗体は、細胞を溶解し、溶解物中の標識を検出することによって測定される。
【0050】
別の方法は、抗-IGF-IR抗体又は他の基質で処理した後に、細胞表面に存在する受容体量を、例えば、IGF-IRの表面発現用に染色された細胞の蛍光活性化細胞選別分析によって、直接測定することである。染色細胞を37℃でインキュベートし、終始、蛍光強度を測定する。対照として、染色集団の一部は4℃(受容体内在化が停止する条件)でインキュベートすることができる。
【0051】
実施例に記載したように、細胞表面IGF-IRは、IGF-IRに特異的でありかつ試験抗体の結合を妨害しない又は当該結合と競争しない異なった抗体を用いて検出及び測定することができる(Burtrumら Cancer Res. 63: 8912-21 (2003))。IGF-IR発現細胞の本発明の抗体による処理は。細胞表面IGF-IRの減少をもたらす。好ましい実施態様では、この減少は、本発明の抗体での処理に対する応答において、少なくとも約70%、より好ましくは少なくとも約80%及びより好ましくは少なくとも約90%である。顕著な減少は、たった4時間内に観察することができる。
【0052】
低調節の別の測定は、細胞に存在する総受容体タンパク質の減少であり、内部受容体の分解を反映する。従って、細胞(特に癌細胞)の本発明の抗体による処理は、総細胞IGF-IRの減少を招く。好ましい実施態様では、当該減少は、少なくとも約70%、より好ましくは少なくとも約80%及び更により好ましくは少なくとも約90%である。
【0053】
本発明の抗体は、約3 x 10-l0 M-1以下、好ましくは約1 x 10-10 M-1以下及びより好ましくは約3 x 10-11 M-1以下のKdでIGF-IRに結合する。
【0054】
本発明の実施態様では、抗体は腫瘍成長を阻害する。例えば、皮下異種移植腫瘍は、癌細胞株の細胞の免疫不全マウスに注射することによって樹立することができる。次いで、マウスを、抗体の例えば3週間ごとの腹腔内注射によって処理し、腫瘍サイズを一定間隔で測定する。対照注射に比べて、本発明の抗体は腫瘍成長を阻害する。好ましい実施態様では、本発明の抗体は、抗新形成剤と併用する場合に腫瘍退縮を促進する。更に、以下に示すように、より好ましい実施態様では、本発明の抗体は、単一療法で用いられる場合に腫瘍退縮を促進する。腫瘍退縮の促進は、抗体の有効量又は抗体及び抗新形成剤の併用の有効量の投与が腫瘍のサイズ又は壊死の減少をもたらす、ことを意味する。本発明の好ましい実施態様では、腫瘍退縮は、少なくとも約20日間、より好ましくは少なくとも約40日間、より好ましくは少なくとも約60日間の期間、観察及び持続することができる。腫瘍退縮は、特定の治療計画を経験する患者群の平均として測定することができ、腫瘍退縮治療群の患者数によって測定することができる。
【0055】
本発明の好ましい抗体又はその断片は、配列番号14、配列番号16、配列番号18、配列番号20、配列番号22、配列番号24、配列番号26、配列番号28及び配列番号30からなる群より選ばれる、1、2、3、4、5及び/6の相補性決定部位((CDR)を有するヒト抗体である。好ましくは、本発明の抗体(又はその断片)は、配列番号14、配列番号16及び配列番号18のCDRを有する。あるいは及び好ましくは、本抗体又はその断片は、配列番号20、配列番号22及び配列番号24のCDRを有する。あるいは及び好ましくは、本抗体又はその断片は、配列番号26、配列番号28及び配列番号30のCDRを有する。CDRのアミノ酸配列は以下の表1に記載されている。
【0056】
【表1】
【0057】
別の実施態様では、本発明の抗体又はその断片は、配列番号1の重鎖可変領域及び/又は配列番号5又は配列番号6から選ばれる軽鎖可変領域を有する。IMC-A12は、本発明の特に好ましい抗体である。この抗体は、ヒトVH及びVLフレームワーク領域(FW)、及びCDRを有する。IMC-A12 (配列番号:1)のVH可変ドメインは、配列番号14、16及び18に対応する3CDRを有し、及びVLドメイン(配列番号:5)は、配列番号20、22及び24に対応する3CDRを有する。IMC-2F8は本発明の別の好ましい抗体である。この抗体はまた、ヒトVH及びVLフレームワーク領域(FW)及びCDRを有する。IMC-2F8のVH可変ドメインは、IMC-A12のVH可変ドメインと同一である。IMC-2F8 (配列番号9)のVLドメインは、配列番号26、28及び30に対応する3CDRを有する。
【0058】
別の実施態様では、本発明の抗体は、IGF-IRへの結合をIMC-A12及び/又はIMC-2F8と競う。すなわち、抗体は、同一又は類似の重複エピトープに結合する。
【0059】
本発明はまた、前述の抗体又はその断片をコードする単離ポリペプチドを提供する。本発明は、表2に記載の、1、2、3、4、5及び/又は全ての6CDRをコードする配列を有する核酸を含む。
【0060】
【表2】
【0061】
ヒト抗体をコードするDNAは、対応するヒト抗体領域から実質的に又はもっぱら得られる、CDR以外の、ヒト不変領域及び可変領域をコードする組換えDNA、及びヒト由来のCDRをコードするDNAによって調製することができる(例えば、重鎖可変ドメインCDRについては、配列番号13、15及び17 、及び軽鎖可変ドメインCDRについては、配列番号19、21及び23、又は配列番号25、27及び29)。
【0062】
抗体断片をコードするその他の好適なDNA起源は、完全長抗体を発現する任意の細胞、例えばハイブリドーマ及び脾臓細胞を含む。当該断片は抗体等価物としてそれ自体使用することができ、上記のように等価物に組み換えることができる。DNA組換え及びこの項で記載の他の方法は、公知の方法によって実行することができる。DNAの他の起源は、当該分野で公知のファージディスプレイ・ライブラリーから作製された単一鎖抗体又はFabである。
【0063】
加えて、本発明は発現配列、プロモーター及びエンハンサー配列に動作可能に連結された、先に記載のポリヌクレオチド配列を含む発現ベクターを提供する。原核生物、例えば細菌及び真核系、例えば特に限定されないが酵母及び哺乳動物細胞培養系における、抗体ポリペプチドの効率的合成のための様々な発現ベクターが開発されている。本発明のベクターは、染色体、非染色体及び合成DNA配列の部分を含むことができる。
【0064】
任意の好適な発現ベクターを使用することができる。例えば、真核性クローニングベクターは、E. coli例えばcolEl、pCRl、pBR322、pMB9、pUC、pKSM及びRP4由来のプラスミドを含む。原核性ベクターはまた、ファージDNA例えばM13及び他の線状単一-鎖DNA配列の誘導体を含む。酵母において有用なベクターの例は、211プラスミドである。哺乳動物細胞における発現に好適なベクターは、SV-40、アデノウイルス、レトロウイルス-由来DNA配列及び機能性哺乳動物性ベクター例えば上記のベクターの組み合わせから得られるシャトルベクターの周知の誘導体、及び機能性プラスミド及びファージDNAを含む。
【0065】
追加の真核性発現ベクターは当該分野で公知である(例えば、P. J. Southern及びP. Berg, J. Mol. Appl. Genet. 1: 327-41 (1982); Subramaniら, Mol. Cell. Biol. 1: 854-64 (1981); Kaufmann及びSharp, "Amplification And Expression of Sequences Cotransfected with a Modular Dihydrofolate Reductase Complementary DNAGene," J. Mol. Biol. 159: 601-21 (1982); Kaufmann及びSharp, Mol. Cell. Biol. 159: 601-64 (1982); Scahillら, "Expression And Characterization Of The Product Of A Human Immune Interferon DNA Gene In Chinese Hamster Ovary Cells, "Proc. Nat'l Acad. Sci. USA80, 4654-59 (1983); Urlaub and Chasin, Proc. Nat'l Acad. Sci. USA 77: 4216-20 (1980))。
【0066】
本発明に有用な発現ベクターは、発現されるDNA配列又は断片に作動的に連結される、少なくとも1つの発現コントロール配列を含む。コントロール配列は、クローン化DNA配列の発現を制御し及び調節するために、ベクターに挿入される。有用な発現コントロール配列の例は、lac系、trp系、tac系、trc系、ラムダファージの主なオペレーター及びプロモーター、fdコートタンパク質のコントロール部位、酵母の解糖プロモーター例えば3-ホスホグリセレートキナーゼのプロモーター、酵母酸ホスファターゼのプロモーター例えばPho5、酵母アルファ-交配因子のプロモーター、及びポリオーマ、アデノウイルス、レトロウイルス及びシミアンウイルス由来のプロモーター例えば初期及び後期プロモーター又はSV40、並びに原核細胞もしくは真核細胞及びそれらのウイルス又はそれらの組み合わせの遺伝子発現を制御することが知られている他の配列である。
【0067】
酵母での遺伝子コンストラクトの発現が望まれる場合には、酵母で使用するための好適な選択遺伝子は、酵母プラスミドYRp7に存在するtrp1遺伝子である。Stinchcombら, Nature, 282: 39 (1979); Kingsmanら, Gene, 7: 141 (1979)。当該trp1遺伝子は、トリプトファンでの成長能力欠損酵素の突然変異株についての選択マーカー、例えばATCC番号44076又はPEP4-1を提供する。Jones, Genetics, 85: 12 (1977)。次いで、酵母宿主細胞ゲノム中のtrp1損傷の存在は、トリプトファンの非存在下での成長による形質転換を検出するために効果的な環境を提供する。同様に、Leu2-欠損酵母株(ATCC 20,622又は38,626)は、Leu2遺伝子を有する公知のプラスミドにより考慮される。
【0068】
本発明はまた、先述の発現ベクターを含む組換え宿主細胞を提供する。本発明の抗体は、ハイブリドーマ以外の細胞株で発現することができる。本発明に従うポリペプチドをコードする配列を含む核酸は、好適な哺乳動物宿主細胞の形質転換に用いることができる。
【0069】
具体的な選択細胞株は、高発現レベル、対象のタンパク質の構成性発現及び宿主タンパク質由来の最小限の汚染に基づいて選択される。発現のために宿主として入手可能な哺乳動物細胞株は、当該分野で公知であり、多くの不死化細胞株、例えば特に制限されないがCOS-7細胞、チャイニーズハムスター卵巣(CHO)細胞、ベビーハムスター腎臓(BHK)及びリンパ起源の細胞株例えばリンパ腫、骨髄腫又はハイブリドーマ細胞を含む多くのその他の細胞株を含む。好適な追加の真核細胞は酵母及び他の菌類を含む。有用な原核生物宿主は、例えばE. coli例えばE. coli SG-936、E. coli HB 101、E. coli W3110、E. coli X1776、E. coli X2282、E. coli DHI及びE. coli MRC1、シュードモナス、バシラス属例えば枯草菌及びストレプトミセスを含む。
【0070】
これらの本組換え宿主細胞は、抗体又は抗体断片を発現させる条件下で細胞を培養し、宿主細胞又は宿主細胞周囲の培地から抗体又はその断片を精製することによって、抗体又は抗体断片を産生するために使用することができる。組換え宿主細胞における分泌のための発現抗体又はその断片のターゲッティングは、対象の抗体-コーディング遺伝子の5'末端にある、シグナル性又は分泌性リーダーペプチド-コーディング配列(Shokriら, ApplMicrobiol Biotechnol. 60: 654-64 (2003) Nielsenら, Prot. Eng. 10: 1-6 (1997)及びvonHeinjeら, Nucl. Acids Res. 14: 4683-90 (1986)を参照されたい)を挿入することによって促進することができる。これらの分泌性リーダーペプチド要素は、原核生物又は真核生物配列のいずれかから得られる。従って、好適には、宿主細胞質ゾルからのポリペプチドの移動及び分泌を培地へ方向付けるために、アミノ酸がポリペプチドのN-末端に結合している、分泌性リーダーペプチドが使用される。
【0071】
形質転換宿主細胞は、吸収可能な炭素源(糖類例えばグルコース又はラクトース)、窒素源(アミノ酸、ぺプチド、タンパク質又はそれらの分解産物例えばペプトン、アンモニウム塩等)及び無機塩(硫酸塩、リン酸塩及び/又はナトリウム、カリウム、マグネシウム及びカルシウムの炭酸塩)を含む液体培地中で、当該分野で公知の方法によって培養される。培地は、例えば成長-促進物質、例えば痕跡元素、例えば鉄、亜鉛、マンガン等を更に含む。
【0072】
本発明における抗体の調製のための別の実施態様は、挿入ヒト抗体産生ゲノムの実質的部分を有し及び内因性抗体の産生に欠けるトランスジェニック動物における、本発明に従う抗体をコードする核酸の発現である。トランスジェニック動物は、例えばマウス、ヤギ及びウサギを含むが特に限定されない。本発明の更なる1態様は、抗体-コーディング遺伝子、例えば泌乳中にポリペプチドを分泌するための動物の乳腺、の発現を含む。
【0073】
以下の実施例に記載のように、本発明に従う高親和性抗-IGF-IR抗体は、ヒト重鎖及び軽鎖可変領域遺伝子から構築されるファージディスプレイ・ライブラリーより単離することができる。例えば、本発明の可変ドメインは、転位可変領域遺伝子を含む末梢血リンパ球から得られる。あるいは、可変ドメイン部分例えばCDR及びFW領域は、異なったヒト配列から得られる。選択の3順後の90%を越える回収クローンは、IGF-IRに特異的である。選別FabのIGF-IRに対する結合親和性は、nMのレンジである。これは、ハイブリドーマ技術を用いて産生された多くの二価抗-IFG-IRモノクローナル抗体と同程度に高い。
【0074】
本発明の抗体及びその断片は、例えば天然抗体又はFab又はscFvファージディスプレイ・ライブラリーから得られる。単一ドメイン抗体は、天然抗体又はハイブリドーマ由来のVH又はVLドメインから選ばれることによって得られ、又はVHドメインのライブラリー又はVLドメインのライブラリーから選択される。単一ドメイン抗体の一次結合決定基であるアミノ酸残基は、CDRと定義されるKabat内にあるが、他の残基及び例えばVH-VLヘテロダイマーのVH-VL表面に埋もれている残基を含んでもよいことが、理解できるだろう。
【0075】
本発明の抗体はまた、結合特性が直接的突然変異、親和性成熟法、ファージディスプレイ又は鎖シャフリングによって改良されてきた抗体を含む。親和性及び特異性は、CDRを突然変異させ及び所望の性質を有する抗原結合部位をスクリーニングすることによって修飾又は改良することができる(例えばYangら, J. Mol. Biol., 254: 392-403 (1995)を参照されたい)。CDRは様々な方法で突然変異する。1方法は、個々の残基又は残基の組み合わせを無作為化し、別の抗原結合部位の集団において、20アミノ酸全てが特定の位置に見出されることである。あるいは、突然変異は、エラープローンPCR法によってCDR残基の範囲に渡って誘発される(例えばHawkinsら, J. Mol. Biol., 226: 889-896(1992)を参照されたい)。例えば、重鎖及び軽鎖可変領域遺伝子を含むファージディスプレイ・ベクターは、E. coliの突然変異株中で増殖することができる(例えばLowら, J. Mol. Biol., 250: 359-368 (1996)を参照されたい)。これらの突然変異誘発方法は、当業者に公知の多くの方法の例示である。
【0076】
本発明のIGF-IR結合抗体を特定するために使用されるタンパク質は、好ましくはIGF-RIであり、より好ましくはIGF-RIの細胞外ドメインである。IGF-RIの細胞外ドメインは、遊離でも又は別の分子に結合してもよい。
【0077】
本発明の抗体は、追加のアミノ酸残基に融合することができる。かかるアミノ酸残基は、おそらく単離を促進するためにペプチドタグでもよい。特定の器官又は組織に対する抗体の自動誘導のための他のアミノ酸残基も考慮される。
【0078】
本発明の別の局面では、抗-IGF-IR抗体又は抗体の断片は、抗新形成剤又は検出可能なシグナル-形成剤に化学的又は生合成的に結合することができる。以下に例示するように、本発明の抗体は、IGF-IRを産生する細胞に結合する際に効率よく吸収される。抗体に結合された抗新形成剤は、抗体が結合された腫瘍を破壊又は損傷する任意の薬剤、又は抗体が結合された細胞環境を含む。例えば、抗腫溶剤は、毒物例えば化学療法剤又は放射性同位体である。好適な化学療法剤は当業者に公知であり、アンスラサイクリン(例えば、ダウノマイシン及びドキソルビシン)、メトトレキサート、ビンクリスチン、ネオカルチノスタチン、シスプラチン、クロラムブシル、シトシン アラビノシド、5-フルオロウリジン、メルファラン、リシン及びカリチェア ミシン)を含む。化学療法剤は、一般的な方法を用いて抗体に結合される(例えばHermentin及びSeiler, Behrihglnst. Mitt. 82: 197-215 (1988)参照されたい)。
【0079】
検出可能なシグナル-形成剤は、in vivo及びin vitroでの診断目的に有用である。シグナル-形成剤は、外部手段、通常、電磁放射線の測定によって検出できる測定可能なシグナルを作製する。ほとんどの場合には、シグナル形成剤は酵素又はクロモフォアであり、蛍光、リン光又は化学ルミネセンスによって光を放つ。クロモフォアは、紫外又は可視領域で光を吸収する色素を含み、酵素触媒反応の基質又は分解産物でもよい。
【0080】
本発明は、標的又はレポーター部分が結合された本発明の抗-IGF-IR抗体又は抗体断片を更に考慮する。標的部分は結合ペアの第一のメンバーである。例えば、抗新形成剤はかかるペアの第二メンバーに結合し、それによって抗原結合タンパク質が結合する部位に導かれる。かかる結合ペアの一般的例は、アビジン及びビオチンである。好ましい実施態様では、ビオチンは本発明の抗原結合タンパク質に結合し、それにより、アビジン又はストレプトアビジンに結合される抗新形成剤又は他の部分のために標的を提供する。あるいは、ビオチン又は別のかかる部分は、本発明の抗原結合タンパク質に結合し、レポーターとして、例えば検出可能なシグナル-形成剤がアビジン又はストレプトアビジンに結合される診断系において使用される。
【0081】
抗新形成剤を使用するための好適な放射性同位体もまた当業者に公知である。例えば、131I又は211Atが用いられる。これらの同位体は、一般的な方法を用いて抗体に結合される(例えばPedleyら, Br. J. Cancer 68, 69-73 (1993)を参照されたい)。あるいは、抗体に結合される抗新形成剤は、プロドラッグを活性化する酵素である。このようにして、プロドラッグは投与されるが、抗体複合体が投与されると直ちに細胞障害性形態に変換される腫瘍部位に到達するまでその活性化形態を維持する。事実、抗体-酵素複合体は患者に投与され、治療される部位に局在化される。次いで、細胞障害性への変換が治療組織部位で生じるように、プロドラッグは患者に投与される。あるいは、抗体に結合された抗新形成剤は、サイトカイン例えばインターロイキン-2 (IL-2)、インターロイキン-4(IL-4)又は腫瘍壊死因子アルファ(TNF-α)である。抗体は、腫瘍にサイトカインをターゲットするので、サイトカインは他の組織に影響を与えることなく腫瘍の損傷又は破壊を介在する。サイトカインは、一般的な組換えDNA法を用いるDNAレベルで抗体に融合する。
【0082】
前述の抗体の有効量を哺乳動物に投与することによって哺乳動物における腫瘍成長を治療する方法もまた、本発明によって提供される。IGF-IRシグナル伝達経路は、多くの種類の癌の発症における病原体であることが広く証明されている。IGF-I及びIGF-IIは、幅広い癌細胞株例えば前立腺癌、乳癌、結腸癌、骨髄腫、卵巣癌、膵臓癌及び肺癌のための強力なマイトジェンであることが判っている。更に、高転移性癌細胞は、転移する傾向の低い腫瘍細胞よりも高レベルのIGF-IR及びIGF-IIを発現することが判明している。
【0083】
本発明に従って治療される好適な腫瘍は、好ましくはIGF-IRを発現する。任意の特定のメカニズムに拘束される意図ではないが、本発明によって治療され又は抑制される疾患及び症状は、例えば病原性血管新生又は腫瘍成長がIGF-IR傍分泌及び/又は自己分泌ループを介して刺激される疾患等を含む。例えば、高転移性腫瘍は、IGF-II及びIGF-IRを発現する傾向にある。
【0084】
本発明の実施態様では、抗-IGF-IR抗体は1以上の他の抗新形成剤と組み合わせて投与される。併用療法の例としては、例えば米国特許第6,217,866号明細書(Schlessingerら) (抗新形成剤と組み合わせた抗-EGFR抗体); 国際公開第 WO 99/60023号パンフレット(Waksalら) (照射と組み合わせた抗-EGFR抗体)を参照されたい。任意の好適な抗新形成剤、例えば化学療法剤、照射又はそれらの組み合わせが用いられる。抗新形成剤は、アルキル化剤又は抗代謝剤でもよい。アルキル化剤の例は特に限定されないがシスプラチン、シクロホスファミド、メルファラン及びダカルバジンである。抗代謝剤の例は、特に限定されないが、ドキソルビシン、ダウノルビシン及びパクリタキセル、ゲムシタビン、及びトポイソメラーゼ阻害剤イリノテカン(CPT-11)、アミノカンプトテシン、カンプトテシン、DX-8951f、及びトポテカン(トポイソメラーゼI)及びエトポシド(VP-16)及びテニポリシド(VM-26)( トポイソメラーゼII)を含む。抗新形成剤が照射である場合には、照射起源は、治療患者に対して外部放射線(外部放射線療法-EBRT)でも又は内部放射線(短療法-BT)でもよい。投与される抗新形成剤の用量は多数の因子、例えば薬剤の種類、治療される腫瘍の種類及び重度、及び薬剤の投与経路に依拠する。しかしながら、強調しておくが、本発明はいずれかの特定の用量に限定されない。
【0085】
当該分野で現在公知又は評価されている抗新形成剤は、様々なクラス、例えば分裂阻害剤、アルキル化剤、抗代謝剤、挿入抗生物質、成長因子阻害剤、細胞サイクル阻害剤、酵素、トポイソメラーゼ阻害剤、抗生存剤(anti survival agents)、生物応答改変剤、抗ホルモン剤及び抗管形成剤に群分けすることができる。
【0086】
これらのクラスでは、本明細書に報告されているデータは、IGF-IRに結合する抗体と併用される場合に、トポイソメラーゼ阻害剤が特に有効な抗新形成剤である、ことを示唆している。従って、本発明の実施態様は、トポイソメラーゼ阻害剤がIGF-IRに結合する抗体と組み合わせて投与される方法を含む。阻害剤は、トポイソメラーゼI又はトポイソメラーゼIIの阻害剤でもよい。トポイソメラーゼI阻害剤は、イリノテカン(CPT-11)、アミノカンプトテシン、カンプトテシン、DX-8951f、トポテカンを含む。トポイソメラーゼII阻害剤は、エトポシド(VP-16)及びテニポシド(VM-26)を含む。他の物質はトポイソメラーゼ活性及び抗新形成剤としての効力について現在評価中である。好ましい実施態様では、トポイソメラーゼ阻害剤は、イリノテカン(CPT-11)である。組み合わせて使用される抗体は、IGF-IRに結合し、かつ以下の性質を少なくとも1つ有する本発明の抗体である:(i)IGF-I又はIGF-IIのIGF-IRへの結合を阻害する;(ii)IGF-I又はIGF-IIによってIGF-IRの活性化を中和する;(iii)IGF-IR表面受容体を減少させる;及び約1×10-10M-1以下のKdでIGF-IRに結合する。より好ましい実施態様では、トポイソメラーゼ阻害剤を組み合わせて使用される抗体は、上記のヒト抗体の特徴を有する。
【0087】
本発明の抗-IGF-IR抗体は、腫瘍成長又は脈管形成に関連する他の受容体を中和する抗体と共に投与することができる。本発明の実施態様では、抗-IGF-IR抗体は、EGFRに特異的に結合する受容体アンタゴニストを組み合わせて使用される。特に好ましいのは、EGFRの細胞外ドメインに結合し、1以上のそのリガンドの結合を妨害し及び/又はEGFRのリガンド-誘起活性化を中和する抗原結合タンパク質である。EGFRアンタゴニストは、EGFR又はEGFRのリガンドに結合し、EGFRのそのリガンドへの結合を阻害する抗体でよい。EGFRに対するリガンドは、例えばEGF、TGF-α、アンヘレグリン(amphiregulin)、ヘパリン-結合EGF(HB-EGF)及びベータセルリンを含む。TGF-αは脈管形成を促進する点でより強力であることが明らかになっているが、EGF及びTGF-αは、EGFR介在刺激をもたらす主な内因性リガンドであると考えられる。EGFRアンタゴニストは、リガンドへの結合を阻害することもできるし又は阻害しないこともできるEGFRの細胞外部分に外的に結合することができ、又はチロシンキナーゼドメインに内的に結合することができる、ことは理解できよう。
EGFRに結合するEGFRアンタゴニストの例は、制限なく、生物分子、例えばEGFRに特異的な抗体(及びその機能性等価物)、及び小分子、例えばEGFRの細胞ドメインに直接作用する合成キナーゼ阻害剤を含む。
【0088】
かかる受容体の別の例は、VEGFRである。本発明の実施態様では、抗-IGF-IR抗体は、VEGFRアンタゴニストと組み合わせて使用される。本発明の1実施態様では、抗-IGF-IR抗体は、VEGFR-1/Flt-1受容体に特異的に結合する受容体アンタゴニストと組み合わせて使用される。別の実施態様では、抗-IGF-IR抗体は、VEGFR-2/KDR受容体に特異的に結合する受容体アンタゴニストと組み合わせて使用される。特に好ましくは、VEGFR-1又はVEGFR-2の細胞外ドメインと結合し、そのリガンドによって結合を妨害し(VEGFR-2は、VEGFによって最も強く刺激される;VEGFR-1は、VEGFではなく、P1GFによって最も強く刺激される)及び/又はリガンド誘起活性を中和する抗原結合タンパク質である。例えば、IMC-1121は、VEGFR-2に結合し、これを中和するヒト抗体である(国際公開第 WO 03/075840号パンフレット; Zhu)。別の例は、MAb 6.12である。これは、溶解性でかつ細胞表面発現VEGFR-1に結合するscFvである。ScFv 6.12は、マウスモノクローナル抗体MAb 6.12のVL及びVHドメインを含む。MAb 6.12を産生するハイブリドーマ細胞株は、「特許手続上の微生物の寄託の国際的承認に関するブタペスト条約」及びそれに基づく規則(ブタペスト条約)の規定のもと、ATCC番号PTA-3344として寄託されている。
【0089】
腫瘍形成に関連する成長因子受容体の他の例は、血小板-由来成長因子(PDGFR)、神経成長因子(NGFR)及び繊維芽細胞成長因子(FGFR)である。
【0090】
追加の他の実施態様では、IGF-IR抗体は、1以上の好適なアジュバント、例えばサイトカイン(例えばIL-10及びIL-13)又は他の免疫刺激因子、例えば特に限定されないがケモカイン、腫瘍関連抗原及びペプチドを組み合わせて投与することができる。例えば上記のLarriveeらを参照されたい。しかしながら、抗-IGF-IR抗体のみの投与は、治療上有効な方法で腫瘍進行を抑制、阻害又は減じるには十分である、ことが理解されよう。
【0091】
併用療法では、抗-IGF-IR抗体は、別の薬剤での治療開始前、間又は後に投与される。また、その任意の組み合わせ、すなわち抗新形成剤治療開始前及び間、前後、間及び後、又は前、間及び後に投与される。例えば、抗-IGF-IR抗体は、照射療法の開始前の1〜30日、好ましくは3〜20日、より好ましくは5〜12日に投与することができる。本発明の好ましい実施態様では、化学療法は、抗体療法と併用して、又はより好ましくは抗体療法の次に投与することができる。
【0092】
本発明では、任意の好適な方法又は経路は、本発明の抗-IGF-IR抗体を投与するため、及び場合により、抗新形成剤及び/又は他の受容体のアンタゴニストを同時投与するために用いることができる。本発明に従って利用される抗新形成剤投薬計画は、患者の腫瘍症状の治療のために最適であると考えられるいずれの投薬計画も含む。異なった悪性腫瘍は、特異的抗腫瘍抗体及び特異的抗新形成剤の使用を必要としてもよい。それは、患者を基準として決定されるだろう。投与経路は、例えば、経口、静脈内、腹腔内、皮下又は筋肉内投与を含む。投与されるアンタゴニストの用量は、多数の要素、例えばアンタゴニストの種類、治療される腫瘍の種類及び重度、及びアンタゴニストの投与経路に依拠する。しかしながら、強調しておくが、本発明は、任意の特定の方法又は投与経路に限定されない。
【0093】
受容体に特異的に結合し、リガンド-毒内在化の後に毒性で致命的ペイロードを送達する、本発明の抗体-IGF-IR抗体は、複合体して投与することができることに留意されたい。抗体-薬物/小分子複合体は、互いに又はペプチド又は非ペプチド性のリンカーを介して直接結合される。
【0094】
本発明の別の局面では、本発明の抗-IGF-IR抗体は、1以上の抗新形成剤又は抗血管由来薬物に化学的又は生合成的に結合することができる。
【0095】
本発明は、標的又はレポーター部分が結合された抗-IGF-IR抗体を更に考慮する。標的部分は結合ペアの第一のメンバーである。例えば抗新形成剤は、かかるペアの第二のメンバーに結合し、それによって抗-IGF-IR抗体が結合される部位に指向される。かかる結合ペアの一般的例は、アビジン及びビオチンである。好ましい実施態様では、ビオチンは、抗-IGF-IR抗体に結合し、それによってアビジン又はストレプトアビジンに結合される抗新形成剤又は他の部分に対する標的を提供する。あるいは、ビオチン又は別のかかる部分は、本発明の抗-IGF-IR抗体に結合され、レポーターとして、例えば検出可能なシグナル-形成剤がアビジン又はストレプトアビジンに結合する診断系において、用いられる。
【0096】
本発明の抗-IGF-IR抗体は、予防又治療目的で哺乳動物に用いられる場合には、薬学的に許容される担体を更に含む組成物の形態で投与されるだろう。好適な薬学的に許容される担体は、例えば1以上の水、生理食塩水、リン酸緩衝液、デキストロース、グリセロール、エタノール等、及びそれらの組み合わせを含む。薬学的に許容される担体は、貯蔵安定性又は結合タンパク質の効果を亢進させる、補助物質、例えば湿潤剤又は乳化剤、保存剤又は緩衝液の少量を更に含むことができる。注射組成物は、当該分野で公知であり、哺乳動物に投与した後に活性剤の迅速、徐放又は遅延放出を提供することができるように調合することができる。
【0097】
本発明はまた、ヒト抗-IGF-IR抗体の治療上有効量を含む腫瘍成長及び/又は脈菅形成を阻害するためのキットを含む。キットは、任意の好適なアンタゴニスト、例えば腫瘍形成又は脈菅形成に関連する別の成長因子受容体(例えば、上記のEGFR、VEGFR-l/Flt-1、VEGFR-2、PDGFR、NGFR、FGFR等)を更に含む。あるいは又は加えて、本発明のキットは抗新形成剤を更に含む。本発明の分脈における好適な抗新形成剤の例は、本明細書に記載されている。本発明のキットはアジュバントを更に含むことができ、その例は上に記載されてもいる。
【0098】
更に、本発明の範囲内には、当該分野で周知の研究法又は診断法のためのin vivo及びin vitroでの本発明の抗体の使用を含む。当該診断法は、本発明の抗体を含むキットを含む。
【0099】
従って、本発明の受容体抗体は、当業者に周知の研究、診断、予防又は治療方法にin vivo及びin vitroで使用することができる。当然のことながら、本明細書に開示の発明の本質における変更が当業者によってなされることは理解及び予想され、またかかる修飾が本発明の範囲内に含まれることになるのは意図されている。
【実施例】
【0100】
以下の実施例は本発明を更に説明するが、本発明の範囲を限定するものと解釈されるべきではない。一般的な方法、例えばベクター及びプラスミドの構築、ポリペプチドをコードする遺伝子のかかるベクター及びプラスミドへの挿入、プラスミドの宿主細胞への導入、及び遺伝子及び遺伝子産物の発現及びその決定に用いられる方法の詳細な記載は、多数の刊行物、例えばSambrook, Jら, (1989) Molecular Cloning: A Laboratory Manual,第二版, Cold Spring Harbor Laboratory Press; 及びColigan, J.ら (1994) Current Protocols in Immunology, Wiley & Sonsから得られる。本明細書に記載の全ての文献はその全体が援用されている。
【0101】
抗-ヒトIGF-IRモノクローナル抗体の選択及び設計
ヒトIGF-I受容体に対する高親和性抗体を単離するために、ヒトIGF-IRの組換え細胞外部分を用い、3.7 x 1010固有クローンを含むヒト野生型(非-免疫)バクテリオファージFabライブラリーをスクリーニングした(de Haardら, J. Biol. Chem. 274: 18218-30 (1999))。溶解性IGF-IR(50μg/ml)を試験菅にコートし、37℃で1時間、3%ミルク/PBSでブロックした。ファージを、ファージ培養を記録するためのライブラリーストックを培養し、M13K07ヘルパーファージで救援し、及びアンピシリン及びカナマイシン選択を含む2YTAK培地で30℃で終夜増幅することによって調製した。得られたファージ調製物を4% PEG/0.5M NaCl中に沈殿させ、3%ミルク/PBSで再懸濁させた。次いで、固定受容体を室温で1時間、ファージ調製物でインキュベートした。その後、試験管をPBST(0.1% Tween-20を含むPS)で10回洗浄した後、PBSで10回洗浄した。結合ファージを100 mMトリエチルアミンの新鮮調製溶液1 mlで、室温で10分間溶出した。溶出ファージを37℃で30分間静置させて、10 mlの中間-記録ファージTG1細胞でインキュベートし、30分間攪拌した。感染TG1細胞をペレットにし、いくつかの大2YAGプレートに置き、30℃で終夜インキュベートした。プレート上の全てのコロニーを3〜5 mlの2YTA培地にすくい取り、グリセロール(最終濃度:10%)で混合し、等分し、-70℃で保存した。第二順選択では、100μlのファージストックを25 mlの2YTAG培地に加え、中間-記録期まで培養した。上記処理に続く選択のために、培養物をM13K07ヘルパーファージで救援し、増幅し、沈殿させ、使用した。但し、試験管に固定されたIGF-IRは低濃度(5μg/ml)であり、結合処理に続く洗浄回数を増やした。合計で2順の選択を行った。
【0102】
個々のTG1クローンを取り、96ウェルプレートで37℃で培養し、上記のM13K07ヘルパーファージで救援した。増幅ファージ調製物は、室温で1時間、18%ミルク/PBSの1/6体積でブロックし、IGF-IR(1μg/ml x 100μl)でコートしたMaxi-sorb 96-ウェル マイクロタイタープレート(Nunc)に添加した。室温で1時間インキュベーション後、プレートをPBSTで3回洗浄し、マウス抗-M13ファージ-HRP複合体(Amersham Pharmacia Biotech, Piscataway, NJ)でインキュベートした。プレートを5回洗浄し、TMBペルオキシダーゼ基質(KPL, Gaithersburg, MD)を加え、マイクロプレートリーダー(Molecular Device, Sunnyvale, CA)を使って450nmでの吸収を測定した。選択の2順では、80%の独立クローンはIGF-IR結合に陽性であった。
【0103】
第二順選択後の抗-IGF-IR Fabクローンの多様性を制限酵素消化パターン(すなわちDNAフィンガープリント)によって分析した。個々のクローンのFab遺伝子挿入は、以下のプライマーを用いるPCR増幅であった:ファージベクター内の独自のFab遺伝子をフランキングする配列に特異的な、リバースPUC19(5'-AGCGGATAACAATTTCACACAGG-3';配列番号31)及びfdtet配列(5'-GTCGTCTTTCCAGACGTTAGT-3';配列番号32)。各増幅産物を通常の切断酵素、BstNIで消化し、3%アガロースゲルで分析した。25個の個々のパターンを全て同定した。各消化パターン由来の代表的クローンのDNA配列をジデオキシヌクレオチド配列決定法によって決定した。
【0104】
IGF-IR及び独自のDNAプロファイルへの正結合を示す個々のクローン由来のプラスミドを使用し、非サプレッサーE. coli宿主HB2151を形質転換した。細胞を1 mMイソプピル-1-β-D-ガラクトピラノシド(IPTG, Sigma)を含む2YTA培地で30℃で培養することによって、HB2151におけるFab断片の発現を誘起した。20% (w/v)ショ糖、200 mM NaCl、1 mM EDTA及び0.1 mM PMSF含む25 mM Tris (pH 7.5)中で細胞ペレットを再懸濁し、次いで4℃で1時間緩やかに攪拌しながらインキュベーションすることによって、細胞のペリプラスム抽出物を調製した。15,000 rpmで15分間遠心分離した後、溶解性Fabタンパク質を、プロティンGカラムを用いるアフィニティークロマトグラフィーによって上清から精製し、製造者のプロトコール(Amersham Pharmacia Biotech)に従った。
【0105】
Fabクローン結合候補化合物を、96ストリップ(strip)-ウェルプレートにコートされた固定IGF-IR(100μg/ml)に対する放射性同位体標識ヒトIGF-Iリガンドの競争的ブロッキングでスクリーニングした。Fab調製物を希釈し、PBS/0.1% BSA中で、室温で0.5〜1時間、IGR-IRプレートでインキュベートした。次いで、40 pMの125I-IGF-Iを加え、プレートを更に90分間インキュベートした。次いで、ウェルを氷冷PBS/0.1% BSAで3回洗浄し、乾燥し、ガンマシンチレーションカウンタで測定した。単一ポイントアッセイにおいて、対照放射性同位体リガンド結合の阻害が30%より高かった候補化合物を選択し、in vitroでのブロッキング力価を決定した。4クローンを同定した。当然のことながら、Fabクローン2G8のみが50%を超えるリガンド結合を阻害し及び約200 nMのIC50を有することが分かった、また完全長IgG1型への変換のために選択した。2F8の重鎖可変領域配列及び翻訳アミノ酸配列をそれぞれ図1及び2に示す。完全長IgG1として設計された2F8重鎖のDNA配列及び翻訳ポリペプチド配列をそれぞれ図3及び4に示す。
【0106】
Fab 2F8塩基配列決定法は、このFabがラムダ軽鎖不変領域にあると決定した。2F8軽鎖のDNA配列及び翻訳アミノ酸配列をそれぞれ図5及び6に示した。完全長ラムダ軽鎖型の配列を図7及び8に示す。結合動的解析をBIAcoreユニットを用いて2F8 IgGで行った。この抗体は0.5〜1 nM (0.5-1 x 10-9M)の親和性でIGF-IRに結合すると決定した。
【0107】
この抗体の親和性を向上させるために、第二世代Fabファージライブラリーを、そこで2F8重鎖が保存され、軽鎖が108種の独自の種類を越える多様性に変化するように作製した。この方法は、軽鎖シャフリングと呼ばれ、所与の標的抗原のための親和性成熟選択抗体に対して首尾良く利用されている(Chamesら, J. Immunol. 169: 1110-18 (2002))。次いで、このライブラリーを上記処理後のヒトIGF-IR(10μg/ml)への結合のためにスクリーニングし、パニング(panning)処理は、高親和性結合Fabを向上させるために低IGF-IR濃度((2μg/ml)を用いて追加の3順を反復した。7クローンを4順後に分析した。7種全ては同一のDNA及び制限消化プロファイルを含んだ。単一単離FabをA12と表わし、ラムダ軽鎖不変領域を有することが明らかになった。軽鎖DNA配列を図9に、アミノ酸配列を図10に示す。完全ラムダ軽鎖配列及び翻訳ポリペプチド配列をそれぞれ図11及び12に示す。2G8及びA12軽鎖のアミノ酸配列比較により、2種の可変領域は全部で11アミノ酸が異なることを決定した(図13及び14に言及)。9種の変化はCDR領域内に存在し、ほとんど(6アミノ酸残基)はCDR3内に生じた。
【0108】
ヒトIGF-IR及びそれらのリガンドのブロッキング活性に対する2抗体(完全IgG)親和性の比較を表3に示した。結合結果は、ヒトIGF-IR ELISAによって決定し、飽和に対して50%結合活性を達成するために必要な抗体力価の濃度を表わす。ブロッキング結果は、固定ヒトIGF-IRに対する125I-IGF-Iリガンドの50%結合を阻害するために必要な抗体レベルを示す。親和性を製造者の使用書(Pharmacia BIACORE 3000)に従うBIAcore分析によって測定した。溶解性IGF-IRをセンサーチップに固定し、抗体結合反応速度を決定した。
【0109】
【表3】
【0110】
抗体A12を作製するためにSF8軽鎖中で生じる抗体変化は、2G8よりもIGF-IRに対するA12の顕著に高い親和性に影響をもたらした。これに伴い、この増加は、受容体に対するより高いA12結合能をもたらした。これは、ELISAによって測定され、固定受容体のためのリガンドのブロッキング活性が少なくとも3倍増加した。図15は、受容体ブロッキングアッセイにおける2種の抗-IGF-IR抗体の代表的力価を示す。軽鎖がヒトラムダ類又はカッパ類の不変領域について設計されたか否かに関らず、A12の活性は同一であった。ラムダ類の軽鎖で設計した抗体A12を全ての以下の過程で利用した。このアッセイでは、A12は、寒冷リガンドとの競争よりもIGF-IRへの放射性同位体標識IGF-Iの結合を大幅に阻害した。2F8の活性を寒冷リガントとの競争と比較した。これは、2種の抗体(表3を参照されたい)及びIGF-Iの相対的親和性(0.5〜1 nM)と一致した。
【0111】
Fabクローン由来の完全ヒトIgG1抗-IGF-IR抗体の設計及び発現
Fab 2F8及びFab A12の重及び軽鎖遺伝子をコードするDNA配列を、製造者の教示に従うBoerhinger Mannheim Expandキットを用いるポリメラーゼチェーン反応(PCR)によって増幅した。フォワード及びリバースプライマーは、哺乳動物発現ベクターへのクローニング用の制限エンドヌクレアーゼ部位の配列を含んだ。重鎖用の受容ベクターは、強力な真核生物性プロモーター及び3’ポリアデニル化配列が隣接する、完全ヒトガンマ1不変領域cDNA配列を含んだ。2F8又はA12の完全長ラムダ軽鎖配列をそれぞれ、哺乳動物細胞中の発現の真核性調節要素のみを有する第二ベクターにクローン化した。選択マーカーは、プラスミドの哺乳動物細胞へのトランスフェクション後の安定DNA構成要素の選択用ベクター上にも存在する。フォワードプライマーはまた、発現抗体の適正な分泌のための強力な哺乳動物シグナルペプチド配列をコードする配列を用いて設計した。適正にクローン化された免疫グロブリン遺伝子配列の同定後に、DNAを塩基配列決定し、一時的トランスフェクションにおける発現を試験した。製造者の仕様書に従い、リポフェクチンを用いて一時的トランスフェクションをCOS7霊長類細胞株に行った。24時間又は48時間トランスフェクション後に、完全IgG抗体の発現を抗-ヒト-Fc結合ELISAによって調整培養上清中で検出した。ELISAプレート(96ウェル)を100 ng/ウェルのヤギ抗ヒトFc-特異的ポリクローナル抗体(Sigma)で被覆することによって調製し、4℃で終夜、5%ミルク/PBSでブロックした。次いで、プレートをPBSで5回洗浄した。調整上清をウェルに加え、室温で1.5時間インキュベーションした。結合抗体をヤギ抗ヒトラムダ軽鎖-HRP抗体(Sigma)で検出し、TMB試薬及び上記のマイクロプレートリーダーで視覚化した。抗-IGF-IR抗体の大規模調製を、COS細胞への大規模な一時的トランスフェクションによって、リポフェクション法のスケールアップによって、又は好適な宿主細胞例えばマウス骨髄腫細胞株(NSO, Sp2/0)又はチャイニーズハムスター卵巣細胞株(CHO)への安定トランスフェクションによって達成した。抗-IGF-IR抗体をコードするプラスミドを、エレクトロポレーション法によって宿主細胞へトランスフェクトし、約2週間、好適な薬物選択培地中で選択した。抗-Fc ELISAによる抗体発現及び血清なしの細胞培養培地に拡張された陽性クローンについて、安定選択コロニーをスクリーニングした。最大2週間の期間、スピナーフラスコ又はバイオリアクターの懸濁培養中で、安定選択細胞由来の抗体産生を実行した。一時的又は安定トランスフェクションによって産生した抗体をProAアフィニィークロマトグラフィー(Harlow and Lane. Antibodies. A Laboratory Manual. Cold Spring Harbor Press.1988)で精製し、中性緩衝生理食塩水中へ溶出し、定量した。
【0112】
ヒト腫瘍細胞における抗-IGF-IRモノクローナル抗体のリガンドブロッキング活性の測定
【0113】
次いで、抗-IGF-IR抗体について、ヒト腫瘍細胞を用いて、野生型IGF-IRに対する放射性同位体標識リガンドのブロッキングを試験した。アッセイ条件は、Arteaga and Osborne(Cancer Res. 49: 6237-41 (1989))に若干の変更を加えて行った。MCF7ヒト乳癌細胞を24ウェル皿に播種し、終夜培養した。サブコンフルエント単層を2〜3回、結合緩衝液(Iscove's培地/0.1% BSA)中で洗浄し、結合緩衝液に抗体を添加した。室温で当該抗体で短時間インキュベーションを行った後、40 pM 125I-IGF-I(約40,000 cpm/ウェル)を各ウェルに加え、穏やかに攪拌しながら更に1時間インキュベートした。次いで、ウェルを氷冷PBS/0.1 % BSAで3回洗浄した。次いで、単層を200μl 0.5N NaOHで溶解し、ガンマカウンタでカウントした。結果を図16に図式的に示す。ヒト腫瘍細胞上で、抗体A12は、IC50 3 nM (0.45llg/ml)でIGF-IRへのリガンド結合を阻害した。これは、寒冷IGF-I リガンドの阻害活性(IC50 = 1 nM)よりも少し低かったが、寒冷IGF-IIの阻害活性(IC50 = 9 nM)よりも高かった。2種のIGFリガンドで観察された相違は、リガンドIGF-IよりもIGF-IRについてのIGF-IIの低い結合反応速度に起因するらしい(Janssonら, J Biol Chem. 272: 8189-97 (1997))。抗体2F8のIC50は30 nM (4.5 g/ml)であると決定した。我々は、続いて、いくつかの異なったヒト腫瘍型におけるA12抗体のIGF-Iリガンドブロッキング活性を測定した。結果を表4に示す。抗体A12は内生的細胞IGF-IRに対する結合に効果的であり、及び乳癌、膵臓癌及び結腸癌組織を含むヒト腫瘍型の範囲に結合するリガンドの阻害に効果的であった。
【0114】
【表4】
【0115】
IGF-IRは、インシュリン受容体(IR)に対してかなりの相同性を共有する。抗-IGF-IR抗体がこのIGF-IRに特異的であり、インシュリン結合をブロックしないか否かを決定するために、細胞型ブロッキングアッセイをヒトZR-75I乳癌細胞で行った。インシュリンは、僅かであるがIRよりも3オーダー低くIGF-IRに結合することができるため、我々は、MCF7細胞と比べてIR対IGF-IR比が高いヒト乳癌細胞株ZR-75Iを利用した。この細胞株の利用によって、我々は、細胞に対するインシュリン結合が特異的IR結合のよりよい指標となると結論した。アッセイはMCF7細胞について上記の方法で行い、結果を表17に示す。寒冷インシュリンは放射性同位体標識インシュリンの細胞への結合を滴定することができるが、2F8も高親和性抗体A12抗体も、200 nM抗体濃度でさえもインシュリン結合をブロックせず、IGF-IRへのこれらの抗体の選択的結合と一致し、IRへのこれらの抗体の選択的結合とは一致しなかった。
【0116】
リガンド-依存的細胞有糸分裂生起の抗体-介在阻害
IGF-IRへのIGF-I結合のブロッキングが阻害細胞増殖を阻害するか否かを決定するために、マイトジェンアッセイをMCF7乳癌細胞、BxPC-3膵臓癌細胞及びHT-29結腸癌細胞で行った。アッセイは、若干の変更加えて、Pragerら(Proc. Natl. Acad. Sci. U. S. A. 91: 2181-85 (1994)に従って行った。細胞を96-ウェル組織培養プレートに5000〜10000細胞/ウェルで播き、終夜吸着させた。次いで、培地を血清なしの培地で交換し、37℃で終夜インキュベートした。次いで、細胞を抗体A12の存在又は非存在下でIGF-Iでインキュベートし、更に37℃で終夜インキュベートした。次いで、0.25μCi [3H]チミジンを各ウェル加え、37℃で5時間インキュベートした。上清を吸引し、細胞をトリプシン処理で5分間懸濁した。次いで、細胞をフィルターで回収し、セル・ハーベスターを用いて水で3回洗浄した。乾燥後、フィルターをシンチレーションカウンタで読むために処理した。結果を図18A、B及びCに示す(それぞれ、MCF7、BxPC-3及びHT-29)。IGF-Iデータポイントは最大のマイトジェン反応を達成するために必要な量を決定するリガンドの滴定を示す。様々な癌細胞型に対する抗体A12の活性の測定では、IGF-Iを5nMの濃度で添加し、抗体は200 nM〜0.05 nMの力価を示した。抗体A12は、用量依存的にIGF-Iリガンドに反応して、IC50 6nMで、MCF7有糸分裂生起を阻害した。
【0117】
次いで、いくつかの追加のヒト腫瘍細胞株に対するマイトジェン阻害を抗体A12について試験した。結果を表5に示す。抗体A12は、乳癌、結腸癌及び多発性骨髄腫を含む多数のヒト腫瘍細胞株のIGF-Iリガンド-介在有糸分裂生起の阻害に効果的であった。
【0118】
【表5】
【0119】
IGF-I指向受容体ホスホリル化及び下流シグナル伝達の抗体-介在阻害
IGF-Iシグナル伝達に対する抗−IGF-IR抗体の阻害効果を視覚化するために、抗体A12又は2F8の存在又は非存在下で、受容体自己ホスホリル化及び下流エフェクター分子ホスホリル化分析を行った。MCF7ヒト乳癌細胞株をその高IGF-IR多様性に起因する使用のために選択した。細胞を10 cm又は6ウェル培養皿に播き、70〜80%コンフルエンスになるまで培養した。次いで、単層をPBSで2回洗浄し、血清なしの培地で終夜培養した。抗-IGF-IR抗体を新鮮な血清なし培地(100 nM〜10 nM)で加え、リガンド(10 nM)の添加前に30分間インキュベートした。細胞を10分間リガンドと共にインキュベートし、その後氷上に置き、氷冷PBSで洗浄した。細胞を溶解液(50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% TritonX-100, 1 mM EDTA, 1 mM PMSF, 0.5 mM Na3V04, 1 /ml ロイペプチン, 1μg/ml ペプスタチン, 及び1μg/ml アプロチニン)を添加して溶解し、氷上の遠心管に15分間ですくい取った。次いで溶解物を4℃で遠心して清浄した。次いで、その溶解物から溶解IGF-IRを免疫沈降させた(IP)。1μg/mlの抗体3B7(Santa Cruz)又はA12を、4℃で400μlの溶解物と共にインキュベートした。次いで、4℃で2時間、免疫複合体をProA-セファロースビーズの添加によって沈殿させ、ペレット化し、溶解緩衝液で3回洗浄した。ProAビーズに結合したIPをすくい取り、変性ゲルランニング緩衝液に加えた。溶解物又はIPを変性ゲル電気泳動のために処理し、4〜12%アクリルアミドゲルで泳動し、Towbinら (Biotechnology 24: 145-9 (1992))に従うウェスタンブロットによってナイロン又はニトロセルロース膜にブロットした。チロシンホスホリル化タンパク質を抗-p-チロシン抗体(Cell Signaling #9411)及び抗-マウス-HRP第二抗体を用いてブロットで検出した。IGF-IRをモノクローナル抗体C-20(Santa Cruz)で検出した。Aktホスホリル化のために、リン酸-Aktを抗体#559029で検出し、総Aktを#559028(BDPhanningen)で検出した。MARKホスホリル化のために、リン酸-p44/42を#9101で、総p44/42を#9102(Cell Signaling Tech.)で検出した。バンドは、X-線フィルムを用いECL試薬で視覚化した。
【0120】
図19Aから明らかなように、MCF7細胞におけるIGF-IRの自己-ホスホリル化は血清除去後に停止され、2F8又はA12のいずれかの添加は、受容体ホスホリル化を誘起しなかった。これは、検出可能なアゴニスト活性の欠落を証明した。10nM IGF-Iを添加した場合に、IGF-IRホスホリル化は強く誘起された。抗体2F8は、IGF-IRホスホリル化を約50%低減させた、一方、高親和性抗体A12はほとんど完全にホスホリル化を阻害した。同様に、抗体A12は、HT-29結腸癌細胞及びBxPC-3膵臓癌細胞においてIGF-IRの自己ホスホリル化を阻害した(図19B)。
【0121】
IGF-Iに応答する下流エフェクターシグナル伝達もまた抗-IGF-IR抗体によって阻害された(図20)。MAPKホスホリル化は、2F8及びA12によってかなりの程度阻害された。抗-アポトーシス分子Aktのホスホリル化は、2F8を有するIGF-IR抗体遮断に対して感受性が低かった。それは、Aktホスホリル化をいくらか減少させたにすぎなかた。A12は、濃度lO nMでさえも顕著にAktホスホリル化を阻害した。抗体A12は同等に、市販の抗体3B7として可溶化IGF-IRの免疫沈降に優れたが、A12は、ウェスタンブロット転移後のナイロン膜上に固定された変性IGF-IRを検出することができなかった。
【0122】
モノクローナル抗体A12の腫瘍細胞株に対するFACS結合解析
A12は内生IGF-IRを免疫沈降することができるので、我々は、A12を蛍光活性細胞選別法(FACS)に検出抗体として用いることができるか否かを決定する点に興味を持った。ヒト腫瘍細胞株を培地で培養し、氷冷PBSですくい取り、測定した。一次抗体A12(0.5μg)を250μl PBS/5% FBS中の約5百万細胞に加え、氷上で1時間インキュベートした。次いで、細胞をPBS/5% FBSで3 mlに希釈し、ペレット化し、上清を吸引した。次いで、二次フィコエリトリン(PE)-標識ヤギ抗-ヒトIgG F(ab)2断片を250μl PBS/5% FBSに1:200で加え、氷上で60分間インキュベートした。その後、既に述べたように、細胞を再度希釈し、ペレット化した後、500μl PBS/5% FBSに再懸濁した。次いで、Epics XLユニット(Coulter)でFACS解析を行った。図21から明らかなように、抗体A12は完全にヒト乳癌細胞株MCF7及びヒト白血病細胞数HELをシフトした。IGF-IR陰性マウス胚繊維芽細胞(R-細胞) (R. Basera, Thomas Jefferson University, Philadelphia, PAから入手)は負の対照として働いた。A12はこれらの細胞に結合できず、IGF-IRに対する抗体結合特異性の指標である。しかしながら、A12はマウス腫瘍細胞ルイス肺腫瘍に結合し、当該腫瘍を部分的にシフトした。これは、この抗-ヒトIGF-IR抗体がマウスIGF-IRに対していくらか交差反応性を有することを示唆するものである。
【0123】
抗体A12の結合後のIGF-I受容体内在化
抗体A12は、高親和性を有するヒト腫瘍細胞に野生型IGF-IRを結合することが明らかとなった。抗体A12は、製造者の教示に従うIODO-ビーズ(Pierce)を用いて125ヨウ素で放射標識した。MCF7ヒト乳癌細胞を6-ウェルプレートに播き、50%コンフルエンスになるまで終夜培養した。125I-A12の1μgを各ウェルに加え、37℃でインキュベートするか又は氷上で4℃に維持した。プレートを30分間、90分間又は180分間インキュベートし、各時点について三点で行った。4℃での培養を180分間維持した。各時点で、ウェルを1 x w/PBSで洗浄した後、100 mMグリシン-HCl、2M尿素で5分間すくい取った。すくい取った材料は膜結合抗体を示し、氷上においてカウントした。次いで、ウェルをPBSで3回洗浄し、細胞を1N NaOH/1% TritonX100で溶解した。溶解画分は内在化抗体を示した。次いで、すくい取られた溶解画分をガンマカウンタで読み、標準偏差を用いてプロットした。図22に示すように、内在的放射活性のレベルは、37℃で培養された細胞中で時間と共に増加したが、膜輸送をおそらく遅らせる4℃に維持された細胞中では、取り込みはほとんど観察されなかった。これは、IGF-IRへの結合の際に、抗体A12は迅速に内在化され、強力に表面結合受容体の枯渇をもたらす、ことを証明した。
【0124】
IGF-IR表面受容体多様性をFACSで決定した。接着性MCF7細胞を37℃で4時間、50 nMのIGF-Iの抗体A12で処理した。細胞を氷冷PBS/5% BSAで2回洗浄し、1 x 106 細胞を染色チューブに等分し、氷上に置いた。次いで、非ブロッキングマウスモノクローナル抗体(Ab-1, NeoMarkers, Fremont, CA)に特異的なIGF-Iアルファ-ポリペプチドを4℃で2時間、細胞でインキュベートした。PBS/BSA洗浄後、細胞を氷上で1時間、抗-マウスIgGフィコエリトリン-複合化第二抗体(PharMingen, BD Biosciences)でインキュベートした。PBS/BSA洗浄後、FACSvantage SEフローサイトメトリー(BD Bioscience)を用いる蛍光活性化細胞選別アッセイによって、細胞を分析した。図22bから明らかなように、A12への暴露は、MCF7細胞の蛍光強度に顕著な減少をもたらした。これは、内在化に起因する表面受容体低調節の指標である。計算した平均蛍光強度比は、A12によるインキュベーション後に90%のIGF-IR表面染色の減少を示した。4℃でA12で細胞をインキュベートした場合には、このシフトは見られず、エネルギー依存性抗体-介在受容体内在化処理と一致した。IGI-Iへの細胞の暴露は、表面IGF-IR蛍光強度における顕著な変化を起こさず、IGF-IR分解にリガンドの効果をほとんど及ぼさないことを示すウェスタンブロッティング解析と一致した。
【0125】
全細胞IGF-IRは、IGF-I又はA12による処理に応答するHT-29細胞及びBxPC-3細胞について決定した。図22cに示すように、培地へのIGF-Iの添加はIGF-IR発現に何の影響を及ぼさなかった。反対に、抗体A12の添加は、3〜6時間後の細胞のIGF-IRレベルの重大な枯渇をもたらした。
【0126】
単独又はイリノテカン(CPT-11)と併用したヒト結腸癌の成長阻害
抗-IGF-IR抗体がin vivoでヌードマウス異種移植モデルにおいてヒト腫瘍成長を阻害することができるか否かを決定することに興味を持った。細胞培養中の2〜3百万個の生存HT-29ヒト結腸癌細胞の皮下投与によって、腫瘍を、3〜4週齢の胸腺欠損マウス(nu/nu)に誘発させた。腫瘍を樹立し、腫瘍体積が200 mm3に達した時に抗体処置を開始した。10動物に1処理群当たりの腫瘍細胞を注射した。抗体を0.5 ml TBS中の1 mg又は0.5 mgで3日間ごとに腹腔内投与(IP)した。薬物イリノテカン(CPT-11) (LKT Laboratories)を抗体処置の開始から4週に1回IP (100 mg/kg)投与した。対照動物には、クラスが一致した無関係なヒトIgG抗体を与えた。高さ、幅及び長さを測定するVernierキャリパーを用いて一定の間隔で腫瘍測定を行い、計算し、総腫瘍体積を決定した。対照腫瘍が3000mm3に達した時に試験を終了した。図23で示されるように、毎3日間の0.5 mg又は1 mgの抗体2F8用量は、このモデルにおいて顕著な阻害効果を示した(P<0.05)。0.5又は1 mgの2F8での処置群の腫瘍サイズ間には全く統計上の誤差はなく、その反応はCPT-11単独で処置しものと同様であった。2F8及びCPT-11を一緒に与えた場合には、より高い腫瘍成長阻害(72%減少)を示した。これは、抗-IGF-IR抗体療法が腫瘍成長に与える化学療法剤CPT-11の抗腫瘍活性を亢進することができたことを証明するものである。
【0127】
ヒト結腸癌に対するin vivoでの抗体A12の抗腫瘍活性
抗体A12は、抗体2F8よりもIGF-IRに対して10倍より高い親和性を有する。顕著な腫瘍阻害が抗体2F8を用いてin vivoで観察されたので、我々は、マウス異種移植モデルにおけるヒト結腸癌株H-29の腫瘍成長に与えるA12の活性を試験した。腫瘍を先述のようにして誘発させ、腫瘍樹立後(大きさ200 mm3)直ちに抗体処置を開始した。次いで、抗体処置を実験期間中毎3日間、1 mg、100μg又は10μgの濃度で行った。1処置群につき10動物を使用し、対照動物にはクラス一致IgG対照抗体を与えた。図24に示すように、抗体A12は、対照に比べて腫瘍成長が74%減少した(P<0.05)。これは、A12がこの異種移植モデルにおいて結腸癌成長の阻害に単一治療剤として有効である、ことを証明した。この実験では、明確な用量-応答効果が認められた。100μg A12の用量で抗腫瘍活性も観察された。
【0128】
異種腫瘍モデルにおけるin vivoでのヒト乳癌に対する抗体A12活性
抗体A12は、in vitroで、MCF7細胞のIGF-依存的細胞分裂誘起刺激及び増幅を示した。In vivoでのMCF7腫瘍成長に対するその活性を利用するために、マウス異種移植腫瘍モデルを利用した。MCF7細胞をエストロゲン依存的ヒト腫瘍から最初に単離し、in vivoでの維持及び成長のためにエストロゲンを外部から添加した。ヌードマウスに生分解性エストロゲンペレットを移植した(0.72 mg 17-β-エストラジオール/ペレット, 60日間放出)。加えて、皮下腫瘍細胞注射時には、マウスにはまた、ゴマ種油の50μl懸濁液中の0.5 mgエストラジオールで皮下的に右脇腹に注射した。抗体処置開始前に腫瘍を約150 mm3のサイズに樹立した。抗体を毎3日間、1 mg、100μg又は10μgで投与し、実験期間中、継続した。29日目に、1 mgのA12による動物処置は腫瘍成長が89%減少した(図25)。この処置群では、明らかに樹立腫瘍が最小限であった。当該モデルでは、用量依存的反応がA12処置について認められた。この試験は、A12がin vivoでのヒト乳癌細胞の成長を顕著に減少させる点で有効である、ことを証明した。更に、1 mg/用量で抗体のみで処置すると、腫瘍退縮が観察され、試験終了(50日間)まで続いた。
【0129】
BxPC-3膵臓癌異種移植における、A12のCPT-11又はゲムシタビンとの併用効果
胸腺nu/nuマウスに、Matrigelと1:1混合した2x106 BxPC-3ヒト膵臓癌細胞皮下に投与した。20日後、腫瘍が約200〜300 mm3に達した時に、マウスを無作為化し、処置群に分類した:1) TBS対照; 2) mAb A12 1mg/用量、3回/週; 3) 2 mg イリノテカン、毎7日に1回; 4) mAb A12及びイリノテカン; 5) 2.5 mg ゲムシタビン、毎7日に1回; 6) mAb A12及びゲンシタビン。全ての処置を腹腔内注射によって投与した。式:体積 =(π/σ)lxw2を使って、腫瘍測定を週に2回記録した。
【0130】
18日目に、1群3動物を屠殺し、腫瘍を中間組織的評価のために切除した。TBS群をのぞいて44日目まで残りの動物について処置及び腫瘍測定を継続した。潰瘍形成及び壊死のため、当該対照群を37日目に屠殺した。試験終了時に、最終腫瘍測定を記録し、動物を屠殺し、1群4動物の腫瘍を最終組織的評価のために切除した。
【0131】
BxPC-3腫瘍はmAb A12に対し非常に反応性が高く、8動物中の2動物は処置の5週間後に部分的に腫瘍退縮した。BxPC-3腫瘍のイリノテカン又はゲムシタビンのみへの反応は、抗体と比較できるものであったが、腫瘍退縮は見られなかった。抗体A12をイリノテカン又はゲムシタビンと併用した場合には、薬剤を単独で使用した場合に比べてより効果的であった(図26)。イリノテカン及びMab A12はより効果的な組み合わせである。A12及びイリノテカンの組み合わせについては、9動物中の3動物が部分的に腫瘍退縮を示した。しかしながら、イリノテカンを与えた動物では、腫瘍退縮はなかった。
【0132】
【表6】
【0133】
HT-29結腸癌異種移植におけるCPT-11又はパクリタキセルと併用したA12の効果
雌性胸腺nu/nuマウスに、Matrigelとの0.4 ml混合1:1中に5 x 106 細胞を含むHT-29ヒト結腸癌細胞懸濁液を皮下に投与した。腫瘍が約200 mm3に達した時に、マウスを無作為化し、処置群に分類した:1) TBS対照; 2) mAb A12 1mg/用量、3回/週; 3) 2 mg イリノテカン、毎7日に1回; 4) mAb A12及びイリノテカン; 5) 121μg パクリタキセルの0.2 ml、毎7日に1回; 6) mAb A12及びパクリタキセル。全ての処置を腹腔内注射によって投与した。
【0134】
処置を6週間継続し、式:体積 =(π/σ)lxw2を使って、腫瘍測定を週に2回記録した。
21日目に、1群4動物を屠殺し、腫瘍を組織的評価のために切除した。40日まで残りの動物について処置及び腫瘍測定を継続した。その際に最終測定を記録し、動物を屠殺し、1群4匹の腫瘍を最終組織的評価のために切除した。
【0135】
単剤mAb A12、CPT-11又はパクリタキセルは顕著に(P<0.02)、TBS対照群に比べてHT-29異種移植片の成長を阻害した(図27)。mAb A12とCPT-11又はパクリタキセルのいずれかとの併用療法は、いずれかの単独処置に比べて腫瘍成長において顕著な阻害を示した(P<0.003)。
【0136】
単一療法群では腫瘍退縮は観察されなかった。対照腫瘍はそのままで、40日目での単一療法群と同様に22日目に高度に血管新生を示した。しかしながら、CPT-11及びmAbA12を用いる併用療法は、8動物中6動物において部分的に腫瘍退縮が起こり、パクリタキセル及びmAbA12を用いる併用療法は、8動物中3動物において腫瘍退縮が起こった。
【0137】
【表7】
【0138】
本明細書に開示される本発明の原理におけるバリエーションが当業者によってなされることは理解および予想され、かかる修飾が本発明の範囲に含まれるものであることが意図される。
【特許請求の範囲】
【請求項1】
インシュリン様成長因子−I受容体(IGF−IR)に特異的に結合し、かつ以下:
(i)IGF−I又はIGF−IIからIGF−IRへの結合を阻害する;
(ii)IGF−I又はIGF−IIによってIGF−IRの活性化を中和する;
(iii)IGF−IR表面受容体を少なくとも約80%減少させる;及び
(iv)1×10−10M−1以下のKdでIGF−IRに結合する、
からなる群より選ばれる少なくとも1つの性質を有する、単離ヒト抗体又はその断片であって、VHCDR1に配列番号14、VHCDR2に配列番号16、VHCDR3に配列番号18、VLCDR1に配列番号26、VLCDR2に配列番号28及びVLCDR3に配列番号30で表されるアミノ酸配列を有する相補性決定部位(CDR)を含む単離ヒト抗体又はその断片。
【請求項2】
インシュリン様成長因子−I受容体(IGF−IR)に特異的に結合し、かつ以下:
(i)IGF−I又はIGF−IIからIGF−IRへの結合を阻害する;
(ii)IGF−I又はIGF−IIによってIGF−IRの活性化を中和する;
(iii)IGF−IR表面受容体を少なくとも約80%減少させる;及び
(iv)1×10−10M−1以下のKdでIGF−IRに結合する、
からなる群より選ばれる少なくとも1つの性質を有する、単離ヒト抗体又はその断片であって、配列番号2で表わされる重鎖可変ドメイン及び配列番号10で表わされる軽鎖可変ドメインを含む単離ヒト抗体又はその断片。
【請求項3】
インシュリン様成長因子−I受容体(IGF−IR)に特異的に結合し、かつ以下:
(i)IGF−I又はIGF−IIからIGF−IRへの結合を阻害する;
(ii)IGF−I又はIGF−IIによってIGF−IRの活性化を中和する;
(iii)IGF−IR表面受容体を少なくとも約80%減少させる;及び
(iv)1×10−10M−1以下のKdでIGF−IRに結合する、
からなる群より選ばれる少なくとも1つの性質を有する、単離ヒト抗体又はその断片であって、配列番号2で表わされる重鎖可変ドメイン、配列番号10で表わされる軽鎖可変ドメイン、ヒトIgG1アイソタイプ重鎖不変領域及びラムダ軽鎖不変領域を含み、VHCDR1に配列番号14、VHCDR2に配列番号16、VHCDR3に配列番号18、VLCDR1に配列番号26、VLCDR2に配列番号28及びVLCDR3に配列番号30で表されるアミノ酸配列を有する相補性決定部位(CDR)を含む単離ヒト抗体又はその断片。
【請求項4】
1×10−10M−1以下のKdでIGF−IRに結合する、請求項1〜3のいずれか1項記載の単離ヒト抗体又はその断片。
【請求項5】
3×10−11M−1以下のKdでIGF−IRに結合する、請求項4記載の単離ヒト抗体又はその断片。
【請求項6】
4×10−11M−1のKdでIGF−IRに結合する、請求項4記載の単離ヒト抗体又はその断片。
【請求項7】
請求項1〜6のいずれか1項記載の抗体又はその断片を含む医薬組成物。
【請求項8】
抗新形成剤を更に含む、請求項7記載の治療組成物。
【請求項9】
前記抗新形成剤が、イリノテカン、カンプトテカン及びエトポシドからなる群より選ば
れる、請求項8記載の治療組成物。
【請求項10】
腫瘍成長を低減させるための医薬品としての使用のための、請求項7〜9のいずれか1
項記載の医薬組成物。
【請求項11】
前記腫瘍が、乳癌、結腸直腸癌、膵癌、卵巣癌、肺癌、前立腺癌、骨組織もしくは軟部
組織肉腫又は骨髄腫からなる群より選択される、請求項10記載の医薬組成物。
【請求項1】
インシュリン様成長因子−I受容体(IGF−IR)に特異的に結合し、かつ以下:
(i)IGF−I又はIGF−IIからIGF−IRへの結合を阻害する;
(ii)IGF−I又はIGF−IIによってIGF−IRの活性化を中和する;
(iii)IGF−IR表面受容体を少なくとも約80%減少させる;及び
(iv)1×10−10M−1以下のKdでIGF−IRに結合する、
からなる群より選ばれる少なくとも1つの性質を有する、単離ヒト抗体又はその断片であって、VHCDR1に配列番号14、VHCDR2に配列番号16、VHCDR3に配列番号18、VLCDR1に配列番号26、VLCDR2に配列番号28及びVLCDR3に配列番号30で表されるアミノ酸配列を有する相補性決定部位(CDR)を含む単離ヒト抗体又はその断片。
【請求項2】
インシュリン様成長因子−I受容体(IGF−IR)に特異的に結合し、かつ以下:
(i)IGF−I又はIGF−IIからIGF−IRへの結合を阻害する;
(ii)IGF−I又はIGF−IIによってIGF−IRの活性化を中和する;
(iii)IGF−IR表面受容体を少なくとも約80%減少させる;及び
(iv)1×10−10M−1以下のKdでIGF−IRに結合する、
からなる群より選ばれる少なくとも1つの性質を有する、単離ヒト抗体又はその断片であって、配列番号2で表わされる重鎖可変ドメイン及び配列番号10で表わされる軽鎖可変ドメインを含む単離ヒト抗体又はその断片。
【請求項3】
インシュリン様成長因子−I受容体(IGF−IR)に特異的に結合し、かつ以下:
(i)IGF−I又はIGF−IIからIGF−IRへの結合を阻害する;
(ii)IGF−I又はIGF−IIによってIGF−IRの活性化を中和する;
(iii)IGF−IR表面受容体を少なくとも約80%減少させる;及び
(iv)1×10−10M−1以下のKdでIGF−IRに結合する、
からなる群より選ばれる少なくとも1つの性質を有する、単離ヒト抗体又はその断片であって、配列番号2で表わされる重鎖可変ドメイン、配列番号10で表わされる軽鎖可変ドメイン、ヒトIgG1アイソタイプ重鎖不変領域及びラムダ軽鎖不変領域を含み、VHCDR1に配列番号14、VHCDR2に配列番号16、VHCDR3に配列番号18、VLCDR1に配列番号26、VLCDR2に配列番号28及びVLCDR3に配列番号30で表されるアミノ酸配列を有する相補性決定部位(CDR)を含む単離ヒト抗体又はその断片。
【請求項4】
1×10−10M−1以下のKdでIGF−IRに結合する、請求項1〜3のいずれか1項記載の単離ヒト抗体又はその断片。
【請求項5】
3×10−11M−1以下のKdでIGF−IRに結合する、請求項4記載の単離ヒト抗体又はその断片。
【請求項6】
4×10−11M−1のKdでIGF−IRに結合する、請求項4記載の単離ヒト抗体又はその断片。
【請求項7】
請求項1〜6のいずれか1項記載の抗体又はその断片を含む医薬組成物。
【請求項8】
抗新形成剤を更に含む、請求項7記載の治療組成物。
【請求項9】
前記抗新形成剤が、イリノテカン、カンプトテカン及びエトポシドからなる群より選ば
れる、請求項8記載の治療組成物。
【請求項10】
腫瘍成長を低減させるための医薬品としての使用のための、請求項7〜9のいずれか1
項記載の医薬組成物。
【請求項11】
前記腫瘍が、乳癌、結腸直腸癌、膵癌、卵巣癌、肺癌、前立腺癌、骨組織もしくは軟部
組織肉腫又は骨髄腫からなる群より選択される、請求項10記載の医薬組成物。
【図1】


【図2】

【図3】

【図4】

【図5】
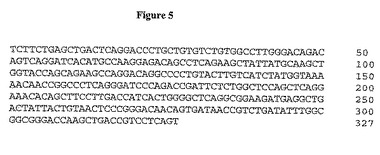
【図6】

【図7】

【図8】

【図9】
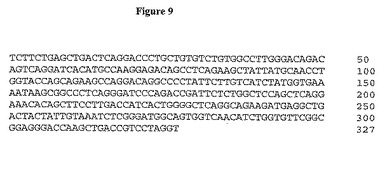
【図10】

【図11】
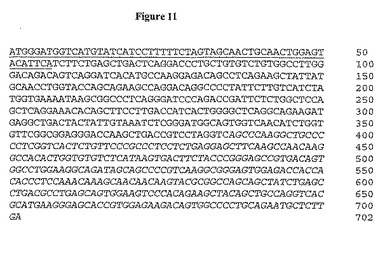
【図12】

【図13】
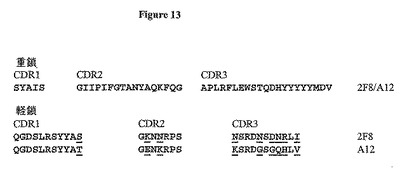
【図14】
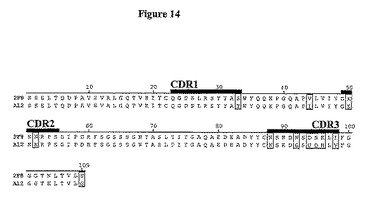
【図15】
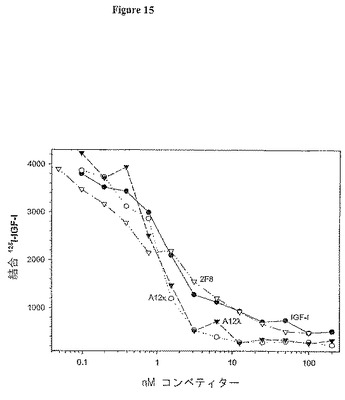
【図16】
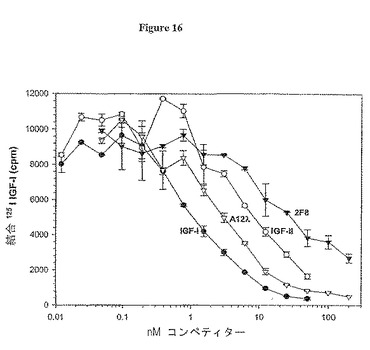
【図17】
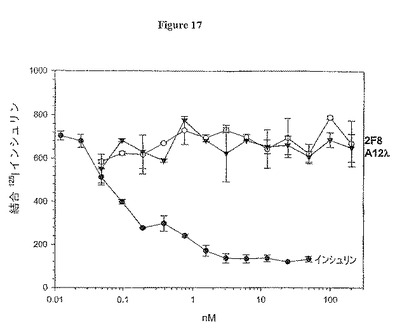
【図18A】
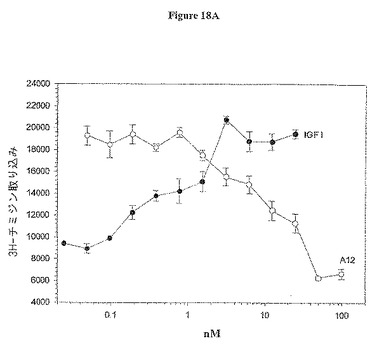
【図18B】
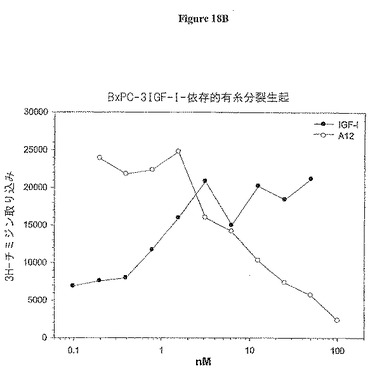
【図18C】
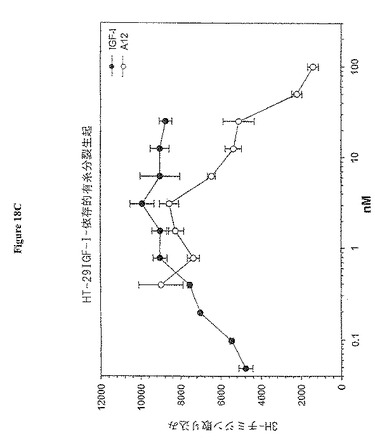
【図19A】
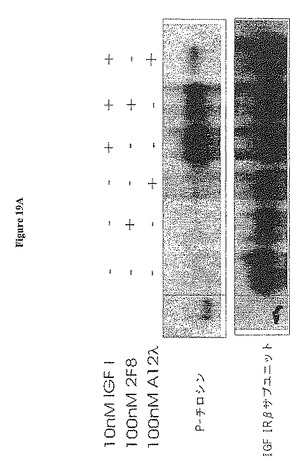
【図19B】
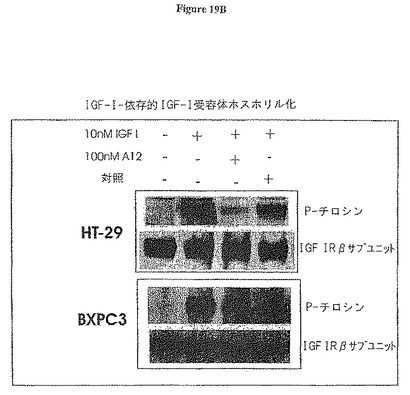
【図20A】
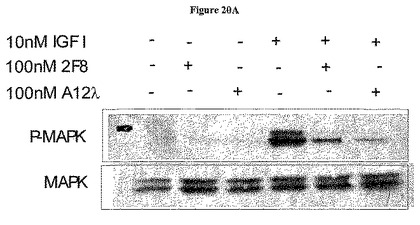
【図20B】
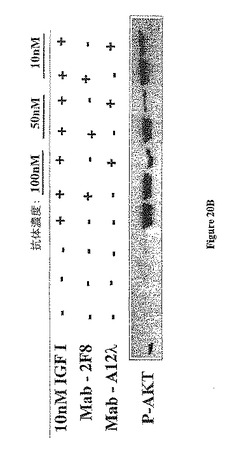
【図21】
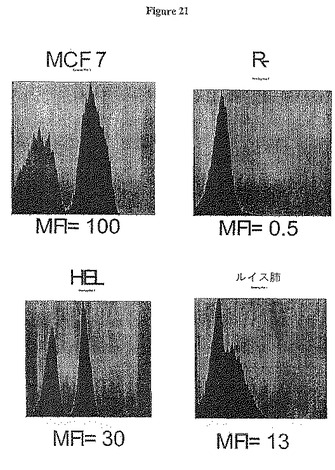
【図22A】
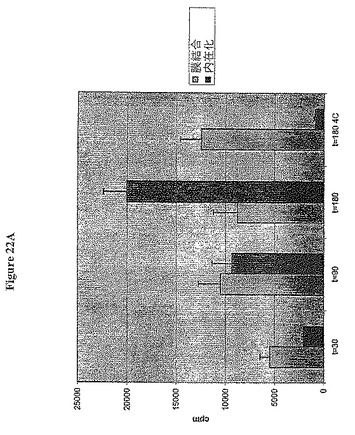
【図22B】
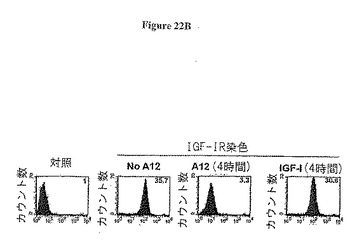
【図22C】
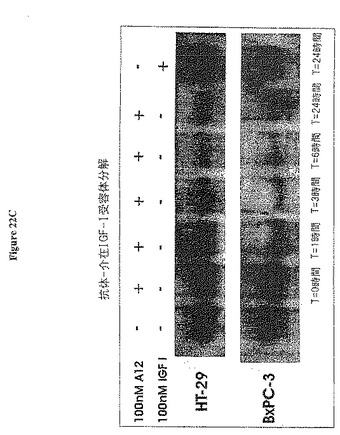
【図23】
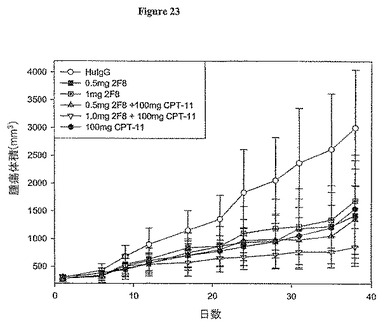
【図24】
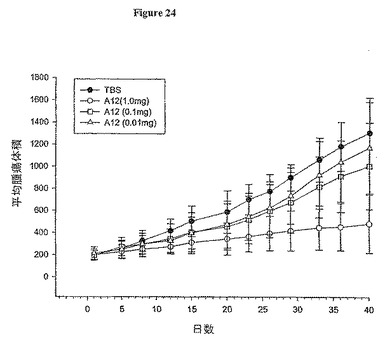
【図25】
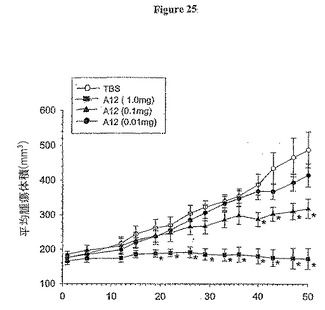
【図26】
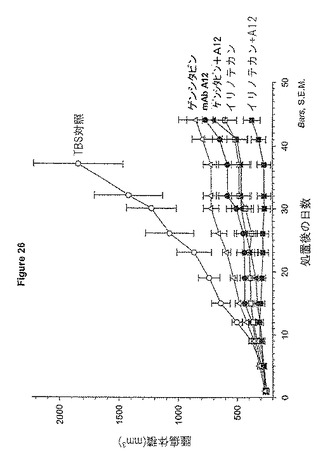
【図27】
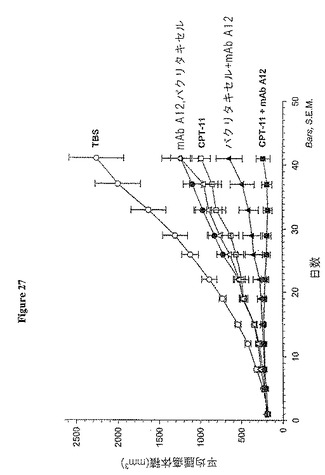
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18A】
【図18B】
【図18C】
【図19A】
【図19B】
【図20A】
【図20B】
【図21】
【図22A】
【図22B】
【図22C】
【図23】
【図24】
【図25】
【図26】
【図27】
【公開番号】特開2011−184440(P2011−184440A)
【公開日】平成23年9月22日(2011.9.22)
【国際特許分類】
【外国語出願】
【出願番号】特願2011−58405(P2011−58405)
【出願日】平成23年3月16日(2011.3.16)
【分割の表示】特願2006−514278(P2006−514278)の分割
【原出願日】平成16年5月3日(2004.5.3)
【出願人】(508188662)イムクローン・リミテッド・ライアビリティ・カンパニー (23)
【氏名又は名称原語表記】Imclone LLC
【Fターム(参考)】
【公開日】平成23年9月22日(2011.9.22)
【国際特許分類】
【出願番号】特願2011−58405(P2011−58405)
【出願日】平成23年3月16日(2011.3.16)
【分割の表示】特願2006−514278(P2006−514278)の分割
【原出願日】平成16年5月3日(2004.5.3)
【出願人】(508188662)イムクローン・リミテッド・ライアビリティ・カンパニー (23)
【氏名又は名称原語表記】Imclone LLC
【Fターム(参考)】
[ Back to top ]