
ヒトトロンボポエチン受容体に対するアゴニスト抗体の製造方法
【課題】ヒトトロンボポエチン受容体(ヒトc-Mpl)に対するアゴニスト抗体の製造方法を提供する。
【解決手段】抗体の定常領域が、(1)ヒト抗体の重鎖定常領域及び軽鎖定常領域のアミノ酸配列、(2)ヒト抗体サブクラス間で重鎖定常領域のドメインを入れ替えた重鎖定常領域のアミノ酸配列、及び、ヒト抗体軽鎖定常領域のアミノ酸配列、或いは(3)前記(1)又は(2)のアミノ酸配列において1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、のいずれかのアミノ酸配列を有し、且つ抗体の可変領域がヒトトロンボポエチン受容体に結合し該受容体を活性化する能力を有する、並びに、(a)10,000ng/mL以下の濃度でコロニー形成を誘導すること、及び(b)最大活性が、PEG-rHuMGDFの50%以上であり、かつ50%有効濃度(EC50)が100nM以下であるアゴニスト抗体の作製方法。
【解決手段】抗体の定常領域が、(1)ヒト抗体の重鎖定常領域及び軽鎖定常領域のアミノ酸配列、(2)ヒト抗体サブクラス間で重鎖定常領域のドメインを入れ替えた重鎖定常領域のアミノ酸配列、及び、ヒト抗体軽鎖定常領域のアミノ酸配列、或いは(3)前記(1)又は(2)のアミノ酸配列において1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、のいずれかのアミノ酸配列を有し、且つ抗体の可変領域がヒトトロンボポエチン受容体に結合し該受容体を活性化する能力を有する、並びに、(a)10,000ng/mL以下の濃度でコロニー形成を誘導すること、及び(b)最大活性が、PEG-rHuMGDFの50%以上であり、かつ50%有効濃度(EC50)が100nM以下であるアゴニスト抗体の作製方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ヒトトロンボポエチン受容体(別名:ヒトc-Mpl)に対するアゴニスト抗体の製造方法に関する。
【0002】
抗ヒトc-Mplアゴニスト抗体は、臨床上血小板を増多させる必要のある患者/疾患に対する治療剤、特に血小板減少症治療剤の有効成分となりうる。
【背景技術】
【0003】
<TPOとTPO受容体について>
トロンボポエチン(Thrombopoietin:TPO)は、生体内で巨核球および血小板の増殖を促進する造血因子である。ヒトTPOは全長332アミノ酸残基からなる糖タンパク質であり、その活性にはN末端側の配列が重要であることが知られている。細胞膜上のTPO受容体と結合することで機能を発揮する。
【0004】
c-Mplは、現在知られている唯一のTPO受容体である。ヒトc-Mplは、シグナルペプチドを含むと635アミノ酸、成熟型は610アミノ酸からなる膜一回貫通型糖タンパク質であり、I型のサイトカイン受容体ファミリーに属している。ヒトc-MplのメッセンジャーRNAおよびタンパク質の配列は既に報告されている(Genbank: NM_005373, NP_005364参照)。同じファミリーの分子としてエリスロポエチン受容体(EpoR)や、G-CSF受容体(G-CSFR)、インターロイキン3受容体(IL-3R)などを挙げることができる。ヒトc-Mplは細胞外に2つのCRH(Cytokine Receptor Homologue)ドメイン(N末端側からCRH1, CRH2と呼ばれる)を持ち、そのなかにサイトカイン受容体ファミリーに特徴的なWSXWSモチーフを含んでいる。細胞内ドメインにはBox1, Box2と呼ばれる二つの配列があり、これらはシグナル伝達に必須である。TPOはCRH1に結合し、c-Mplを二量体化することでシグナルを入れると考えられているが、詳細な結合および活性化の様式は解明されていない。c-Mplが二量体化すると、細胞内ドメインに結合したシグナル伝達キナーゼが活性化され、リン酸化シグナルが細胞内に伝えられる。TPO-MplシグナルによってJak-STAT, PI3K-Akt, Ras-MAPKの各経路が動かされることが知られている。TPOまたはc-Mplを欠損したマウスは、血小板数が野生型マウスの10-20%程度まで減少することが報告されており、TPO-Mplシステムが血小板数を調節する主要なシステムであることを示している。c-Mplの発現は、巨核球のみならず、さらに未分化な造血前駆細胞や造血幹細胞にも認められる。骨髄中のc-Mpl陽性細胞画分は、c-Mpl陰性画分に比べ、高い骨髄再構築能を持っていることが示されており、また、c-Mpl欠損マウスでは巨核球・血小板だけではなく造血幹細胞も減少していることが判っている (非特許文献1,2参照)。これらの知見から、TPO-Mplシステムが造血系に幹細胞レベルから関与していることが示唆されている。
【0005】
TPOがクローニングされて以来、血小板減少症の治療薬としての利用が期待され、これまでに二種類の組換え型TPOの臨床試験が行われている(非特許文献3参照)。完全長のヒトTPO(rhTPO)と、ヒトTPOの活性部位であるN末端側163アミノ酸のペプチド配列をポリエチレングリコール(PEG)化したPEG-rHuMGDF(Pegylated Recombinant Human Megakaryocyte Growth and Development Factor)である。臨床試験において、これらの組換え型TPOは健常人および特発性血小板減少性紫斑病(Idiopathic Thrombocytopenic Purpura:ITP)患者の血小板を増加させることに成功している。また、骨髄非破壊的な化学療法に伴う血小板減少症を軽減させる効果が示されている。さらに、例数は少ないながらも、再生不良性貧血(Aplastic Anemia:AA)、骨髄異形成症候群(Myelodysplastic Syndrome:MDS)の患者に対しても組換え型TPOの有効性が報告されている(非特許文献4,5)。
【0006】
<c-Mplアゴニスト抗体について>
TPOと同様にc-Mplを介してシグナルを入れる性質を持ちながら、分子としては全く異なる種々のTPOミメティクスが研究されている(非特許文献6,7参照)。ミメティクスには大きく分けて、ペプチド性低分子、非ペプチド性低分子、抗体由来分、アゴニスト抗体などが知られている。
【0007】
公知の抗c-Mplアゴニストヒト抗体として、12B5, 12E10, 12D5(特許文献1参照)を挙げることができる。これらは全長抗体(Whole antibody、 例えばWhole IgG)の形態でプライマリーヒト細胞に対する活性を持たない。なお、本明細書で使用するプライマリーヒト細胞とは、TPOに対して高感受性を有する樹立された特殊な株化細胞やTPO受容体遺伝子を遺伝子組換え技術により導入し該受容体を高発現させた細胞ではなく、ヒト臍帯血やヒト骨髄由来のCD34+細胞等の本来生体内でTPOの作用対象となっている細胞を意味する。
【0008】
また、公知のアゴニストマウス抗体として、BAH-1(特許文献2,非特許文献8参照), VB22B(特許文献3参照)を挙げることが出来るが、マウス抗体はヒト血中において抗原性を示すことが知られており、医薬品としての利用には適さない。また、一般に、アゴニスト抗体を、全長抗体で、例えばCDR grafting等の方法を用いて、その活性を保ったままヒト化することは困難である(特許文献3, 非特許文献9参照)。従って、上記のような公知のアゴニスト抗体が存在するとしても、プライマリー細胞に作用するアゴニストヒト抗体の創出は容易ではない。
【0009】
また前述のTPOミメティクスで述べた抗体由来の低分子も、ある種のアゴニスト抗体であり、抗体の一部を利用改変したDiabody,一本鎖(Fv)2(sc(Fv)2)が報告されている(特許文献1,3参照)。しかしながら、この方法によって作出された改変抗体は、分子の大幅な改変による抗原性が懸念されるうえ、全長抗体に比べ血中半減期が短縮される等、医薬品として利用するためには課題が多い。
【0010】
以上、全長抗体は低抗原性や血中半減期の長さ等、医薬品として有用な性質を備えている一方、上述のように、全長抗体で充分な活性を持つアゴニストヒト抗体を作製するのは容易ではない。
【0011】
そこで、以下に述べるように、本発明者らは、抗体の構造に大幅な改変を加えることなく、充分な活性を持ったアゴニストヒト抗体の取得を試み、その取得に至った。さらに抗体のヒンジ領域に改変を加え、アゴニスト活性を増強することに成功した。本発明によって作出された抗体は血小板減少症治療薬としての利用に適していると考えられる。
【0012】
【非特許文献1】宮崎洋,「トロンボポエチンの将来展望」, Japanese Journal of Transfusion Medicine, 46(3) , 311-316, 2000
【非特許文献2】Murone Mら, Stem Cell 16:1-6, 1998
【非特許文献3】Kuter DJら, Blood 100(10):3457-69, 2002
【非特許文献4】Yonemura Yら, Int J Hemat (82) 307-309, 2005
【非特許文献5】Komatsu Nら, Blood 96 296a, 2000
【非特許文献6】Broudy VCら, Cytokine. 25(2):52-60, 2004
【非特許文献7】Wang Bら, Clin Pharmacol Ther. 76(6):628-38, 2004
【非特許文献8】Deng Bら, Blood 92(6):1981-1988, 1998
【非特許文献9】Ji Hee Sonら, Journal of Immunological Methods 286:187-201, 2004
【特許文献1】国際公開第WO 99/10494号パンフレット
【特許文献2】国際公開第WO 99/03495号パンフレット
【特許文献3】国際公開第WO 2005/056604号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0013】
本発明の目的は、新規な抗ヒトc-Mplアゴニスト抗体を提供することである。
本発明において、抗体とは、これまで全長抗体では困難であった、ヒトc-Mplに対し天然のリガンドであるTPOとほぼ同等のシグナルを入れることができ、プライマリーヒト細胞に対する増殖刺激活性を有する抗体である。
【0014】
本発明の第2の目的は、抗体の断片化を行わずにアゴニスト抗体の活性を高める技術を提供することにより、抗体分子が本来持つ長半減期、低抗原性といった医薬品として望ましい性質を有する新規な抗ヒトc-Mplアゴニスト抗体を提供することである。
【課題を解決するための手段】
【0015】
上記課題を解決するため、本発明者らは、抗ヒトc-Mplアゴニスト抗体の研究に鋭意取り組んだ。その結果、全長抗体で、天然リガンドとほぼ同等のシグナルを生じ、ヒトプライマリー細胞に対する活性を有するヒト抗体を取得した。また、取得したアゴニスト抗体につき更に鋭意検討を行い、抗体の断片化を行わずにそのアゴニスト活性を高める改変方法を見出し、本発明を完成するに至った。
【0016】
すなわち、本発明は以下の特徴を含む。
1.ヒトトロンボポエチン受容体に対するアゴニスト抗体
本発明にかかるヒトトロンボポエチン受容体に対するアゴニスト抗体は、以下の(1)〜(6)に示す抗体を含む。
(1)抗体の定常領域が以下の(i)〜(iii):
(i) ヒト抗体の重鎖定常領域及び軽鎖定常領域のアミノ酸配列、
(ii) ヒト抗体サブクラス間で重鎖定常領域のドメインを入れ替えた重鎖定常領域のアミノ酸配列、及び、ヒト抗体軽鎖定常領域のアミノ酸配列、或いは
(iii) 前記(i)又は(ii)のアミノ酸配列において1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
のいずれかのアミノ酸配列を有し、且つ抗体の可変領域がヒトトロンボポエチン受容体に結合し該受容体を活性化する能力を有する、並びに、以下の(a)及び/又は(b):
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、10,000ng/mL以下の濃度でコロニー形成を誘導すること、
(b) UT7/TPO細胞を用いた増殖能測定法において、活性が下記の構造を有するPEG-rHuMGDFの50%以上であり、かつ50%有効濃度(EC50)が100nM以下であること、
の性質を有する、ヒトトロンボポエチン受容体に対するアゴニスト抗体。
【0017】
本明細書中、ヒト抗体サブクラスは、IgG1, IgG2, IgG3及びIgG4を含む。ヒトイムノグロブリンの定常領域等の配列は、例えばNCBIホームページ(GenBank、UniGeneなど)から入手可能であり、例えばヒトIgG1重鎖定常領域については登録番号J00228、ヒトIgG2重鎖定常領域については登録番号J00230、ヒトIgG3重鎖定常領域については登録番号X03604、ヒトIgG4重鎖定常領域については登録番号K01316、ヒト軽鎖κ定常領域については登録番号V00557, X64135, X64133など、ヒト軽鎖λ定常領域については登録番号X64132, X64134などを含むことができる。
【0018】
本明細書中、ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法とは、後述の実施例6に記載した測定法を意味し、コロニー形成に必要な抗体の濃度は、この測定法に基づき求めることができる。
【0019】
本明細書中、UT7/TPO細胞を用いた増殖能測定法とは、後述の実施例5に記載した測定法を意味し、増殖活性及びEC50はこの測定法に基づき求めることができる。
【0020】
本明細書中、PEG-rHuMGDFとは、ヒトTPOのアミノ末端受容体結合ドメインを含むトランケート化タンパク質をコードするcDNAを含有するプラスミドで形質転換された大腸菌(E. coli)を使用して産生される(Ulichら, Blood 86:971-976 (1995))ポリペプチドを抽出し、リフォールディングし、精製し、そのアミノ末端にポリエチレングリコール(PEG)部分を共有結合させた配列番号1のアミノ酸配列を有する分子であり、以下の構造:
PEG-NH-SPAPPACDLRVLSKLLRDSHVLHSRLSQCPEVHPLPTPVLLPAVDFSLGEWKTQMEETKAQDILGAVTLLLEGVMAARGQLGPTCLSSLLGQLSGQVRLLLGALQSLLGTQLPPQGRTTAHKDPNAIFLSFQHLLRGKVRFLMLVGGSTLCVRRAPPTTAVPS-COOH
を有するものである。
【0021】
本明細書中、ヒトc-Mplを活性化するとは、ヒトc-Mplを発現している細胞においてヒトc-Mplに係る細胞内シグナル伝達を起こすことをいう。
【0022】
本明細書中、数個なる用語は、2〜約10個、例えば2〜9個、2〜8個、2〜7個、2〜6個、2〜5個、2〜4個、又は2〜3個の整数を意味する。
【0023】
(2)上記コロニー形成測定法によりコロニー形成を誘導する活性を有し、及び/又は、上記UT7/TPO細胞を用いた増殖能測定法により細胞増殖活性を有する抗体のうち、コロニー形成活性については10,000ng/mL以下の濃度でコロニー形成を誘導する活性を有する、好ましくは1,000ng/mL以下の濃度で該活性を有する、より好ましくは100ng/mL以下の濃度で該活性を有する上記(1)の抗体。
【0024】
(3)上記細胞増殖活性がPEG-rHuMGDFの50%以上、好ましくは70%以上、より好ましくは90%以上の活性を示し、50%有効濃度(EC50)が、100nM以下, 好ましくは10nM以下、より好ましくは1nM以下のものである上記(1)の抗体。
【0025】
(4)上記コロニー形成測定法及び上記増殖能測定法の両者の測定方法によりそれぞれ以下の活性を示す上記(1)の抗体。
(i) 以下の(a)および(b)の性質を有する、ヒトトロンボポエチン受容体に対するアゴニスト抗体。
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、10,000ng/mL以下の濃度でコロニー形成を誘導する。
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性が下記の構造を有するPEG-rHuMGDFの50%以上であり、かつ50%有効濃度(EC50)が100nM以下である。
(ii) 以下の(a)および(b)の性質を有する、ヒトc-Mplに対するアゴニスト抗体。
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、1,000ng/mL以下の濃度でコロニー形成を誘導する。
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性がPEG-rHuMGDFの70%以上であり、かつEC50が10nM以下である。
(iii) 以下の(a)および(b)の性質を有する、ヒトc-Mplに対するアゴニスト抗体。
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、100ng/mL以下の濃度でコロニー形成を誘導する。
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性がPEG-rHuMGDFの90%以上であり、かつEC50が1nM以下である。
【0026】
(5)以下の(a)〜(h)からなる群から選択される重鎖アミノ酸配列可変領域及び軽鎖アミノ酸配列可変領域を有する上記(1)の抗体。(カッコ内に、各可変領域配列が由来する後述実施例の抗体の名称を示す。)
(a) 配列番号2で示されるアミノ酸配列を含む重鎖可変領域及び配列番号3で示されるアミノ酸配列を含む軽鎖可変領域。(抗体の名称:7-10)
(b) 配列番号4で示されるアミノ酸配列を含む重鎖可変領域及び配列番号5で示されるアミノ酸配列を含む軽鎖可変領域。(抗体の名称:4-49)
(c) 配列番号6で示されるアミノ酸配列を含む重鎖可変領域及び配列番号7で示されるアミノ酸配列を含む軽鎖可変領域。(抗体の名称:6-4-50)
(d) 配列番号8で示されるアミノ酸配列を含む重鎖可変領域及び配列番号9で示されるアミノ酸配列を含む軽鎖可変領域。(抗体の名称:6-5-2)
(e) 配列番号2で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号3で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列
を含む軽鎖可変領域。
(f) 配列番号4で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号5で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域。
(g) 配列番号6で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号7で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域。
(h) 配列番号8で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号9で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域。
【0027】
(6)ヒトc-Mplに対するアゴニスト抗体がヒト抗体である、上記(1)〜(5)に記載の抗体。
【0028】
2.重鎖改変アゴニスト抗体
本発明に係る重鎖改変アゴニスト抗体とは、以下のものを含む。
(1)重鎖定常領域のアッパーヒンジ部が、以下の(a)〜(b)のアミノ酸配列から選択されるいずれか1つのアミノ酸配列を有し、並びに、該重鎖定常領域のミドルヒンジ部以降のC末端側がヒトイムノグロブリンG4のアミノ酸配列或いは該G4のアミノ酸配列においてADCC(抗体依存性細胞障害)活性等に係るアゴニスト抗体として好ましくない性質に関する部位を変異させたアミノ酸配列を有するアゴニスト抗体。
(a) 配列番号10で示されるアミノ酸配列。
(b) 配列番号11で示されるアミノ酸配列。
【0029】
本明細書の中で用いるアッパーヒンジ(Upper hinge)とは、Kabat EU 番号付け(Kabatら、Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institute of Health, Bethesda, Md.(1991))による表記で216位以降、226位よりN末端側の配列を指す。ミドルヒンジ(Middle hinge)とは、同表記で226位以降、231位よりN末端側の配列を指す。ヒトイムノグロブリンG4を含む各サブタイプにつき、アッパーヒンジ、ミドルヒンジ及びその前後のアミノ酸配列を図4Bに示す。図中、CH1はアッパーヒンジに隣接するCH1領域の一部、CH2はCH2領域中のローワーヒンジ(lower hinge)と称される部分である。
【0030】
(2)上記重鎖定常領域のミドルヒンジ部以降のC末端側が、ヒトイムノグロブリンG4のアミノ酸配列において、Kabat EU番号付けにおける228位セリンがプロリンに且つ235位ロイシンがグルタミン酸に置換されたアミノ酸配列を有する重鎖を含む抗体。
【0031】
(3)以下の(i)又は(ii)に示された、ヒトc-Mplに対するアゴニストヒト抗体である上記(2)の重鎖改変抗体。
(i) 重鎖定常領域のアッパーヒンジ部が以下の(a)〜(b)のアミノ酸配列から選択されるいずれか一つのアミノ酸配列を有し、並びに、該重鎖定常領域のミドルヒンジ部以降のC末端側がヒトイムノグロブリンG4のアミノ酸配列或いは該G4のアミノ酸配列においてKabat EU番号付けにおける228位セリンがプロリンに且つ235位ロイシンがグルタミン酸に置換されたアミノ酸配列を有する重鎖を含むヒトc-Mplに対するアゴニスト抗体。
(a) 配列番号10で示されるアミノ酸配列。
(b) 配列番号11で示されるアミノ酸配列。
(ii) 更に好ましい実施形態としては、以下(a)〜(h)からなる群から選択される、上記(i)に記載のヒトc-Mplに対するアゴニスト抗体を挙げることができる。
(a) 配列番号2のアミノ酸配列を含む重鎖と配列番3のアミノ酸配列を含む軽鎖を有する抗体。
(b) 配列番号4のアミノ酸配列を含む重鎖と配列番号5のアミノ酸配列を含む軽鎖を有する抗体。
(c) 配列番号6のアミノ酸配列を含む重鎖と配列番号7のアミノ酸配列を含む軽鎖を有する抗体。
(d) 配列番号8のアミノ酸配列を含む重鎖と配列番号9のアミノ酸配列を含む軽鎖を有する抗体。
(e) 配列番号2のアミノ酸配列を含む重鎖と、配列番号3のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖を有する抗体。
(f) 配列番号4のアミノ酸配列を含む重鎖と、配列番号5のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖を有する抗体。
(g) 配列番号6のアミノ酸配列を含む重鎖と、配列番号7のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖を有する抗体。
(h) 配列番号8のアミノ酸配列を含む重鎖と、配列番号9のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖を有する抗体。
【0032】
3.ヒトc-MPLに対するアゴニスト抗体の医薬用途及び医薬組成物
本発明に係るヒトc-MPLに対するアゴニスト抗体は、c-Mp1受容体に結合しそれを活性化する能力、および/または(インビボおよびインビトロの両方で)血小板の産生を刺激する能力(「血小板生成活性」)および血小板前駆体の産生を刺激する能力(「巨核球生成活性」)を有する。
【0033】
本発明に係るヒトc-Mplに対するアゴニスト抗体を有効成分とする医薬組成物/医薬用途としては、具体的には次のものを挙げることができる。
(1)上記1.(1)〜(6)及び同2.(3)のいずれかの抗体を有効成分とする医薬組成物。
(2)上記1.(1)〜(6)及び同2.(3)のいずれかに記載の抗体を有効成分とする血小板増多剤。
(3)骨髄移植、臍帯血移植時の血小板回復促進用であることを特徴とする上記(2)に記載の血小板増多剤
(4)上記1.(1)〜(6)及び同2.(3)のいずれかの抗体を有効成分とする血小板減少症治療剤。
(5)血小板減少症が、以下の(a)〜(f)のいずれか1つである、上記(4)に記載の血小板減少症治療剤。
(a) 特発性血小板減少性紫斑病(ITP)、
(b) 癌化学療法後の血小板減少症、
(c) 再生不良性貧血、
(d) 骨髄異形性症候群(MDS)、
(e) 肝疾患にともなう血小板減少症、又は
(f) 骨髄移植もしくは臍帯血移植後の血小板減少症。
(6)造血幹細胞移植後の血球回復促進用であるヒトc-Mplアゴニスト抗体を有効成分として含む血球増多剤。
(7)上記1.(1)〜(6)及び同2.(3)のいずれかに記載の抗体を有効成分として含む上記(6)に記載の血球増多剤。
【0034】
4.本発明に係る抗体の製造方法
本発明の抗体は、本発明に係る抗体を産生するハイブリドーマを用いて抗体を製造してもよいし、又は、ハイブリドーマ等の抗体産生細胞からモノクローナル抗体をコードする遺伝子をクローニングし、適当なベクターに組み込んで、遺伝子組換え技術を用いて産生させた組換え型抗体を製造することもできる。
【0035】
本発明の抗体は、次の方法によって製造できる。
ヒトトロンボポエチン受容体(c-Mpl)に対するアゴニスト抗体の製造方法であって、該抗体の定常領域が、以下の(1)〜(3):
(1) ヒト抗体の重鎖定常領域及び軽鎖定常領域のアミノ酸配列、
(2) ヒト抗体サブクラス間で重鎖定常領域のドメインを入れ替えた重鎖定常領域のアミノ酸配列、及び、ヒト抗体軽鎖定常領域のアミノ酸配列、或いは
(3) 前記(1)又は(2)のアミノ酸配列において1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
のいずれかのアミノ酸配列を有し、且つ抗体の可変領域がヒトトロンボポエチン受容体に結合し該受容体を活性化する能力を有する、並びに、以下の(a)及び(b):
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、10,000ng/mL以下の濃度でコロニー形成を誘導すること、及び
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性が、そのN末端がペグ(PEG)化された配列番号1のアミノ酸配列からなる下記の構造:
PEG-NH-SPAPPACDLRVLSKLLRDSHVLHSRLSQCPEVHPLPTPVLLPAVDFSLGEWKTQMEETKAQDILGAVTLLLEGVMAARGQLGPTCLSSLLGQLSGQVRLLLGALQSLLGTQLPPQGRTTAHKDPNAIFLSFQHLLRGKVRFLMLVGGSTLCVRRAPPTTAVPS-COOH
を有するPEG-rHuMGDFの50%以上であり、且つ50%有効濃度(EC50)が100nM以下であること、
の性質を有する前記アゴニスト抗体を構成する重鎖及び軽鎖をそれぞれコードする塩基配列を含むDNAと、それらのDNAの発現を制御する塩基配列を含む1又は複数のDNAとを保持する哺乳動物細胞を作製し、該哺乳動物細胞を培養した培養液から、該重鎖及び軽鎖からなる抗体をコードする塩基配列を含むDNAの発現産物を単離精製することを含む、上記方法。
【0036】
実施形態において、上記抗体が、以下の(a)及び(b):
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、1,000ng/mL以下の濃度でコロニー形成を誘導すること、及び
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性がPEG-rHuMGDFの70%以上であり、且つEC50が10nM以下であること、
の性質を有する。
【0037】
別の実施形態において、上記抗体が、以下の(a)及び(b):
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、100ng/mL以下の濃度でコロニー形成を誘導すること、及び
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性がPEG-rHuMGDFの90%以上であり、かつEC50が1nM以下であること、
の性質を有する。
【0038】
別の実施形態において、上記抗体が、以下の(1)〜(8):
(1) 配列番号2で示されるアミノ酸配列を含む重鎖可変領域及び配列番号3で示されるアミノ酸配列を含む軽鎖可変領域、
(2) 配列番号4で示されるアミノ酸配列を含む重鎖可変領域及び配列番号5で示されるアミノ酸配列を含む軽鎖可変領域、
(3) 配列番号6で示されるアミノ酸配列を含む重鎖可変領域及び配列番号7で示されるアミノ酸配列を含む軽鎖可変領域、
(4) 配列番号8で示されるアミノ酸配列を含む重鎖可変領域及び配列番号9で示されるアミノ酸配列を含む軽鎖可変領域、
(5) 配列番号2で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号3で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、
(6) 配列番号4で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号5で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、
(7) 配列番号6で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号7で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、及び
(8) 配列番号8で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号9で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、
からなる群から選択される重鎖可変領域及び軽鎖可変領域を有する。
【0039】
別の実施形態において、前記抗体が、ヒト抗体である。
【0040】
別の実施形態において、上記抗体の重鎖定常領域のアッパーヒンジ部が、以下の(1)及び(2):
(1) 配列番号10で示されるアミノ酸配列、及び
(2) 配列番号11で示されるアミノ酸配列、
のアミノ酸配列から選択されるいずれか1つのアミノ酸配列を有し、並びに、該重鎖定常領域のミドルヒンジ部以降のC末端側が、ヒトイムノグロブリンG4のアミノ酸配列或いは該G4のアミノ酸配列においてKabat EU番号付けにおける228位セリンがプロリンに且つ235位ロイシンがグルタミン酸に置換されたアミノ酸配列を有する。
【0041】
本発明の抗体の製造方法として、好ましくは、以下の方法が挙げられる。
下記の(a)〜(h):
(a) 配列番号2のアミノ酸配列を含む重鎖と配列番号3のアミノ酸配列を含む軽鎖をコードする塩基配列、
(b) 配列番号4のアミノ酸配列を含む重鎖と配列番号5のアミノ酸配列を含む軽鎖をコードする塩基配列、
(c) 配列番号6のアミノ酸配列を含む重鎖と配列番号7のアミノ酸配列を含む軽鎖をコードする塩基配列、
(d) 配列番号8のアミノ酸配列を含む重鎖と配列番号9のアミノ酸配列を含む軽鎖をコードする塩基配列、
(e) 配列番号2のアミノ酸配列を含む重鎖と、配列番号3のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、
(f) 配列番号4のアミノ酸配列を含む重鎖と、配列番号5のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、
(g) 配列番号6のアミノ酸配列を含む重鎖と、配列番号7のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、及び
(h) 配列番号8のアミノ酸配列を含む重鎖と、配列番号9のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、
からなる群から選択される、重鎖をコードする塩基配列を含むDNA及び軽鎖をコードする塩基配列を含むDNAと、それらのDNAの発現を制御する塩基配列を含む1又は複数のDNAとを保持する哺乳動物細胞を作製し、該哺乳動物細胞を培養した培養液から、該重鎖及び軽鎖からなる抗体をコードするDNAの発現産物を単離精製することを含む、ヒトc-Mplに対するアゴニスト抗体の製造方法。
【0042】
5.本発明に係るDNA
本発明に係るDNAとして、以下のものを挙げることができる。
(1)ヒトMplに対するアゴニスト抗体の重鎖可変領域のアミノ酸配列をコードする塩基配列を含む新規なDNAであって、以下の(a)〜(d)からなる群から選択されるアミノ酸配列をコードする塩基配列を含むDNA:
(a) 配列番号2で示されるアミノ酸配列;
(b) 配列番号4で示されるアミノ酸配列;
(c) 配列番号6で示されるアミノ酸配列;
(d) 配列番号8で示されるアミノ酸配列。
(2)ヒトMplに対するアゴニスト抗体の軽鎖可変領域のアミノ酸配列をコードする塩基配列を含む新規なDNAであって、以下の(a)〜(h)からなる群から選択されるアミノ酸配列をコードする塩基配列を含むDNA:
(a) 配列番号3で示されるアミノ酸配列;
(b) 配列番号5で示されるアミノ酸配列;
(c) 配列番号7で示されるアミノ酸配列;
(d) 配列番号9で示されるアミノ酸配列;
(e) 配列番号3のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列;
(f) 配列番号5のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列;
(g) 配列番号7のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列;
(h) 配列番号9のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列。
(3)上記(1)又は(2)のDNAが、それぞれ可変領域及び定常領域を含む抗体重鎖或いは軽鎖をコードするDNA。
(4)抗体の重鎖定常領域のアッパーヒンジ部が、以下の(a)及び(b)のアミノ酸配列から選択されるいずれか一つのアミノ酸配列を有し、並びに、該重鎖定常領域のミドルヒンジ部以降のC末端側がヒトイムノグロブリンG4のアミノ酸配列或いは該G4のアミノ酸配列においてKabat EU番号付けにおける228位セリンがプロリンに且つ235位ロイシンがグルタミン酸に置換されたアミノ酸配列を有する、上記(3)に記載の抗体重鎖をコードするDNA。
(a) 配列番号10で示されるアミノ酸配列。
(b) 配列番号11で示されるアミノ酸配列。
【0043】
本明細書は本願の優先権の基礎である日本国特許出願2006-81322号、2006-299554号の明細書および/または図面に記載される内容を包含する。
【発明を実施するための最良の形態】
【0044】
以下、本発明を詳細に説明する。
本発明は、プライマリーヒト細胞に作用する抗ヒトc-Mplアゴニストヒト抗体を提供する。
【0045】
本発明における抗体は、ヒト抗体産生マウス(例えば、KMマウスTM(キリンビール社))をヒトMpl組換えタンパク質もしくはヒトMpl発現細胞で免疫し、通常のモノクローナル抗体作製法にて単離することができる。また、ハイブリドーマから抗体遺伝子を単離し、発現ベクターを構築し、発現細胞を作製し、その過程で、種々の定常領域を有する組換え抗体を作製することができる。
【0046】
1.本発明に係る抗体について
本明細書において抗体とは、Fab領域、ヒンジ領域、Fc領域を有する抗体を意味し、天然に存在する抗体、及び、同様の構成を有する範囲において、それ自体公知の方法で得られたモノクローナル抗体産生ハイブリドーマにより産生される、或いは、一旦抗体遺伝子を取得しそれを用いて遺伝子工学的に産生される抗体、更に部位特異的突然変異誘発法により一部改変され遺伝子工学的に産生される抗体を含む。そして本発明に係るヒトcMplに対するアゴニスト抗体及び重鎖改変アゴニスト抗体は前述した通りのものである。
【0047】
一般に、アゴニスト抗体は細胞膜上の標的分子に結合し、複合体を形成させることでシグナルを伝える。ホモ二量体を形成するサイトカイン受容体ファミリー、例えばエリスロポエチン受容体(EpoR)、G-CSF受容体(G-CSFR)、トロンボポエチン受容体(c-Mpl)等に対するアゴニスト抗体は、二価の抗体が、二つの分子と結合することで二量体を形成させると考えられる。多くのアゴニスト抗体がFab断片のみでは活性を示さないこともそれを示唆している。
【0048】
複合体形成には、二つの抗原結合部位が接近し易いことが重要と考えられる。全長抗体では充分な活性を持たない抗体でも、sc(Fv)2等の形態に低分子化することでアゴニスト活性が上昇するのはこのことを示唆している。一方で、低分子化抗体は、分子の大幅な改変による抗原性が懸念されるうえ、血中半減期が短縮される等、医薬品として利用するためには課題が多い。全長抗体の持つ、低抗原性や血中半減期の長さ等、医薬品として有用な性質を生かすためには、抗体の構造に大幅な改変を加えることなく、高い活性を持ったアゴニスト抗体が望まれる。
【0049】
後述の実施例2に示すように、本発明者らは、免疫の方法に工夫をこらし、全長抗体で高い活性を持った抗ヒトc-Mplアゴニスト抗体を取得した。工夫とは、例えば高発現細胞株による免疫や恒常活性型の変異受容体発現細胞を用いた免疫である。該アゴニスト抗体は後述の実施例6に記載のヒト臍帯血CD34+細胞を用いたコロニーアッセイでコロニー形成を誘導することが示されており、医薬品として有用であることが期待される。
【0050】
さらに、本発明者らはヒンジ部分の柔軟性を高めることで、複合体形成の効率を高め、アゴニスト活性を増強することを考案した。柔軟性の高い配列は、例えばグリシンリンカーのような配列を考えることができる。もしくは、ヒトIgGの中でも、最も柔軟性が高いIgG3のヒンジ領域を用いることができる。抗体の低抗原性を損なわないためには、なるべく天然に存在する配列を用いることが望ましい。そこで、IgG3のヒンジ配列がより好ましい。
【0051】
また、遺伝子工学的改変により、細胞傷害活性が低く、かつ、高いヒンジの柔軟性を持った、アゴニスト抗体に最適な定常領域として、ヒトIgG3のアッパーヒンジ領域を持ち、ミドルヒンジ以降C末端側の領域はヒトIgG4の配列とするような抗体の作製も可能である。
【0052】
より具体的には、当業者に周知である遺伝子工学的改変(例えば、欧州特許EP314161公報を参照のこと)により異なるサブクラスのものに変換し、即ち、本発明の抗体の可変領域をコードするDNAを用いて遺伝子工学的手法を用いて元のサブクラスとは異なるサブクラスの抗体に変更することができる。更にヒトIgG4重鎖定常部分のEU番号付け(Sequences of proteins of immunological interest, NIH Publication No.91-3242 を参照)における228番目のセリンをプロリンに変異させることにより、IgG4の分子内架橋(S-S結合)による単量体形成を抑制することが、また、同じく235番目ロイシンをグルタミン酸に変異させることにより、抗体依存性細胞傷害(Antibody-dependent cellular cytotoxicity:ADCC)活性を低減させることができる。上記二つの変異を持ったIgG4をIgG4PEと呼ぶ。
【0053】
上記の事柄を考慮し、本発明者らは細胞傷害活性が低く、かつ、高いヒンジの柔軟性を持った、アゴニスト抗体に最適な定常領域を作製した。本定常領域は、ヒトIgG3のアッパーヒンジ領域を持ち、ミドルヒンジ以降C末端側の領域はヒトIgG4の配列となっている。この定常領域と抗c-Mplアゴニスト抗体の可変領域を組み合わせることにより、安全性と高活性を兼ね備えたアゴニスト抗体を作製することができる。
【0054】
2.本発明に係る抗体の製造方法
本発明の抗体は、種々の方法により生産できるが、まずは本発明抗体産生ハイブリドーマを得る必要がある。後述の実施例1に記載するような本発明の抗原を用い、マウス等を免疫し、特にヒト抗体を得る場合にはヒト抗体産生トランスジェニックマウスなどの非ヒト哺乳動物に免疫する。モノクローナル抗体は、定法に従い、免疫感作動物から得た抗体産生細胞と、自己抗体産生能のない骨髄腫系細胞(ミエローマ細胞)を融合することにより得られるハイブリドーマを培養し、免疫に用いた抗原に対して特異的親和性を示すモノクローナル抗体を産生するクローンを選択することによって取得することができる。取得した抗体群から更にアゴニスト抗体を選択する必要があるが、アゴニスト抗体の作用対象受容体に対するリガンドの活性測定方法として確立した方法を用いればよく、ヒトc-Mplに対するアゴニスト抗体を選択する場合は、後述の実施例5に示したUT7/TPO細胞増殖アッセイ等のTPO活性測定方法として既に確立された方法を用いて適宜行うことができる。
【0055】
本発明に係るヒトc-Mplに対するアゴニスト抗体、特にモノクローナル抗体の製造にあたっては、下記の工程を包含する。すなわち、(1)免疫原として使用する、生体高分子の精製及び/又は抗原タンパク質を細胞表面に過剰に発現している細胞の作製、(2)抗原を動物に注射することにより免疫した後、血液を採取しその抗体価を検定して脾臓等の摘出の時期を決定してから、抗体産生細胞を調製する工程、(3)骨髄腫細胞(ミエローマ)の調製、(4)抗体産生細胞とミエローマとの細胞融合、(5)目的とする抗体を産生するハイブリドーマ群の選別、(6)単一細胞クローンへの分割(クローニング)、(7)場合によっては、モノクローナル抗体を大量に製造するためのハイブリドーマの培養、又はハイブリドーマを移植した動物の飼育、(8)このようにして製造されたモノクローナル抗体の生理活性及びその認識特異性の検討、あるいは標識試薬としての特性の検定、(9)モノクローナル抗体遺伝子のクローニングと組換え抗体作製等である。
【0056】
以下、ヒトc-Mplに対するアゴニストモノクローナル抗体の作製法を上記工程に沿って詳述するが、該抗体の作製法はこれに制限されず、例えば脾細胞以外の抗体産生細胞及びミエローマを使用することもできる。
【0057】
(1) 抗原
一般にヒトc-mpl抗体を得る場合には、ヒトc-Mplタンパク質の一次構造が公知(Genbank: NP_005364参照)であるので、当業者に周知の方法により、c-Mplのアミノ酸配列からペプチドを化学合成し、これを抗原として使用することができ、またc-Mplの細胞膜貫通領域および細胞内領域を欠損した可溶化c-Mpl組換えタンパク質を抗原として用いることもできる。
【0058】
或いは、各種ヒト巨核球系細胞株や強制発現株等のヒトc-Mpl発現細胞株の抗原としての利用が挙げられる。但し、ヒトc-Mpl発現細胞株としては、各種ヒト巨核球系細胞株や強制発現株が知られているが、これらの細胞株のc-Mpl発現量は細胞あたり数千分子と低く、抗原には不向きである。実際、マウス造血系細胞株であるFDCP2にヒトc-Mplを導入した発現株FDCP-hMpl(FEBS Lett. 1996 Oct 21;395(2-3):228-34参照)をヒト抗体産生マウス(KMマウスTM)に免疫した場合、抗体価上昇が不十分であり、hMpl特異的なヒト抗体を得ることができなかった。また、ヒト巨核球系細胞株を抗原として用いる場合、他の膜分子に対する抗体も誘導されるため、c-Mpl特異的な抗体を効率良く誘導するには必ずしも適切でない。従って、目的とするものが単にヒトc-mpl抗体でなくアゴニスト活性を有する抗体の取得に際し抗原タンパク質を発現する細胞株を免疫に用いる場合には、発現量が高い細胞を選択することが望ましい。特に、マウス細胞株、しかも可能な限りMHC適合した細胞株を宿主に、ヒトc-Mplを導入した高発現株を用いるのが望ましい。例として、後述の実施例1に記載の細胞(発現ベクターとしてヒトc-mpl全長遺伝子が担持されたpEF-MPL635或いはpCMV-MPL635を、宿主としてマウス細胞株L929およびFM3Aを用いたもの)を挙げることができる。
【0059】
また、野生型のヒトc-mplに代えて、ヒトc-mplの恒常活性型変異体(例えば、508番目TrpがSerに変異しリガンド非依存性に恒常的にアゴニストシグナルを伝達する変異体;Abe Mら、Leukemia. 2002 Aug;16(8):1500-1506)を同様の方法で強制発現させた細胞株を用いてもよい。このような変異体は野生型とは立体構造が異なることが予想され、そのような恒常活性型変異体に親和性が高い抗体は、強力なアゴニスト活性を示す可能性がある。
【0060】
また、これら強制発現細胞株は、ヒトcMPL或いはその細胞外可溶性領域等と適宜組み合わせて抗原として用いることもできる。
【0061】
(2) 抗体産生細胞の調製工程
上記(1)で得られた抗原と、フロインドの完全若しくは不完全アジュバント、又はカリミョウバンのような助剤とを混合し、免疫原として実験動物に免疫する。実験動物としては、遺伝子改変によってヒト抗体を産生する能力を有するマウス(ヒト抗体産生マウス)が最適である。
【0062】
本発明において用いられたヒト抗体産生マウス(例えばKMマウスTM)は、内在性マウスイムノグロブリン(Ig)重鎖及びマウスκ軽鎖を欠損しており、かつ、ヒトIg重鎖遺伝子を含む14番染色体断片(SC20)及びヒトIgκ鎖トランスジーン(KCo5)を同時に保持する。このマウスはヒトIg重鎖遺伝子座を持つ系統Aのマウスと、ヒトIgκ鎖トランスジーンを持つ系統Bのマウスとの交配により作製される。系統Aは、内因性Ig重鎖及びκ軽鎖破壊の両者についてホモ接合体であり、子孫伝達可能な14番染色体断片(SC20)を保持するマウス系統(Tomizuka. et al., Proc Natl Acad Sci USA., 2000 Vol97:722)である。また、系統Bは内在性マウスIg重鎖及びκ軽鎖欠損の両者についてホモ接合体であり、ヒトIgκ鎖トランスジーン(KCo5)を保持するマウス系統(Nat Biotechnol., 1996 Vol14:845)である。従って、KMマウスはヒト抗体を産生する能力を持ち、マウスIg重鎖およびκ鎖を欠損したマウスである。
【0063】
マウス免疫の際の免疫原投与法は、皮下注射、腹腔内注射、静脈内注射、皮内注射、筋肉内注射、足蹠注射などいずれでもよいが、腹腔内注射、足蹠注射又は静脈内注射が好ましい。
【0064】
免疫は、一回、又は、適当な間隔で(好ましくは2週間から4週間間隔で)複数回繰返し行うことができる。その後、免疫した動物の血清中の抗原に対する抗体価を測定し、抗体価が十分高くなった動物を抗体産生細胞の供給源として用いれば、以後の操作の効果を高めることができる。一般的には、最終免疫後3〜5日後の動物由来の抗体産生細胞を、後の細胞融合に用いることが好ましい。
【0065】
ここで用いられる抗体価の測定法としては、フローサイトメトリー法、放射性同位元素免疫定量法(以下、「RIA法」という)、固相酵素免疫定量法(以下、「ELISA法」という)、蛍光抗体法、受身血球凝集反応法など種々の公知技術があげられるが、検出感度、迅速性、正確性、及び操作の自動化の可能性、等の観点から、フローサイトメトリー法又はELISA法がより好適である。
【0066】
本発明における抗体価の測定は、例えばフローサイトメトリー法によれば、以下に記載するような手順により行うことができる。まず、抗原を発現した細胞とヒト抗体を含む検体(例えばマウス血清,ハイブリドーマの培養上清,精製抗体等)と反応させる。さらに二次抗体として蛍光標識されたヒト抗体に対する抗体を加えてヒト抗体に結合させ、洗浄後、細胞に結合した二次抗体の量を蛍光によって測定することにより、抗体価を算出する。
【0067】
(3) ミエローマの調製工程
ミエローマとしては、マウス、ラット、モルモット、ハムスター、ウサギ又はヒト等の哺乳動物に由来する自己抗体産生能のない細胞を用いることが出来るが、一般的にはマウスから得られた株化細胞、例えば8-アザグアニン耐性マウス(BALB/c由来)ミエローマ株P3X63Ag8U.1(P3-U1)[Yelton, D.E. et al. Current Topics in Microbiology and Immunology, 81, 1-7(1978)]、P3/NSI/1-Ag4-1(NS-1) [Kohler, G. et al. European J. Immunology, 6, 511-519 (1976)]、Sp2/O-Ag14(SP-2)[Shulman, M. et al. Nature, 276, 269-270 (1978)]、P3X63Ag8.653(653)[Kearney, J. F. et al. J. Immunology, 123, 1548-1550(1979)]、P3X63Ag8(X63)[Horibata, K. and Harris, A. W. Nature, 256, 495-497 (1975)]などを用いることが好ましい。これらの細胞株は、適当な培地、例えば8-アザグアニン培地[グルタミン、2-メルカプトエタノール、ゲンタマイシン及びウシ胎児血清(以下、「FCS」という)を加えたRPMI-1640培地に8-アザグアニンを加えた培地]、イスコフ改変ダルベッコ培地(Iscove's Modified Dulbecco's Medium;以下、「IMDM」という)、又はダルベッコ改変イーグル培地(Dulbecco's Modified Eagle Medium;以下、「DMEM」という)で継代培養するが、細胞融合の3〜4日前に正常培地(例えば、10% FCSを含むDMEM培地)で継代培養し、融合当日に2×107以上の細胞数を確保しておく。
【0068】
(4) 細胞融合
抗体産生細胞は、形質細胞、及びその前駆細胞であるリンパ球であり、これは個体のいずれの部位から得てもよく、一般には脾臓、リンパ節、骨髄、扁桃、末梢血、又はこれらを適宜組み合わせたもの等から得ることができるが、脾細胞が最も一般的に用いられる。
【0069】
最終免疫後、所定の抗体価が得られたマウスから抗体産生細胞が存在する部位、例えば脾臓を摘出し、抗体産生細胞である脾細胞を調製する。次いで、脾細胞とミエローマを融合させればよい。この脾細胞と工程(3)で得られたミエローマを融合させる手段として現在最も一般的に行われているのは、細胞毒性が比較的少なく融合操作も簡単な、ポリエチレングリコールを用いる方法である。この方法は、例えば以下の手順よりなる。
【0070】
脾細胞とミエローマとを無血清培地(例えば、DMEM)、又はリン酸緩衝生理食塩液(以下、「PBS」という)でよく洗浄し、脾細胞とミエローマの細胞数の比が5:1〜10:1程度になるように混合し、遠心分離する。上清を除去し、沈澱した細胞群をよくほぐした後、撹拌しながら1mLの50%(w/v)ポリエチレングリコール(分子量1000〜4000)を含む無血清培地を滴下する。その後、10mLの無血清培地をゆっくりと加えた後遠心分離する。再び上清を捨て、沈澱した細胞を適量のヒポキサンチン・アミノプテリン・チミジン(以下「HAT」という)液及びヒトインターロイキン-6(以下、「IL-6」という)を含む正常培地(以下、「HAT培地」という)中に懸濁して培養用プレート(以下、「プレート」という)の各ウェルに分注し、5%炭酸ガス存在下、37℃で2週間程度培養する。途中適宜HAT培地を補う。
【0071】
(5) ハイブリドーマ群の選択
上記ミエローマ細胞が、8-アザグアニン耐性株である場合、すなわち、ヒポキサンチン・グアニン・ホスホリボシルトランスフェラーゼ(HGPRT)欠損株である場合、融合しなかった該ミエローマ細胞、及びミエローマ細胞どうしの融合細胞は、HAT含有培地中では生存できない。一方、抗体産生細胞どうしの融合細胞、あるいは、抗体産生細胞とミエローマ細胞とのハイブリドーマは生存することができるが、抗体産生細胞どうしの融合細胞には寿命がある。従って、HAT含有培地中での培養を続けることによって、抗体産生細胞とミエローマ細胞との融合細胞であるハイブリドーマのみが生き残り、結果的にハイブリドーマを選択することができる。コロニー状に生育してきたハイブリドーマについて、HAT培地からアミノプテリンを除いた培地(以下、「HT培地」という)への培地交換を行う。以後、培養上清の一部を採取し、例えば、フローサイトメトリー法により抗ヒトc-Mpl抗体価を測定する。以上、8-アザグアニン耐性の細胞株を用いる方法を例示したが、その他の細胞株もハイブリドーマの選択方法に応じて使用することができ、その場合使用する培地組成も変化する。
【0072】
(6) クローニング工程
前述(2)の記載と同様の方法で抗体価を測定することにより、特異的抗体を産生することが判明したハイブリドーマを、別のプレートに移しクローニングを行う。このクローニング法としては、プレートの1ウェルに1個のハイブリドーマが含まれるように希釈して培養する限界希釈法、軟寒天培地中で培養しコロニーを回収する軟寒天法、マイクロマニュピレーターによって1個ずつの細胞を取り出し培養する方法、セルソーターによって1個の細胞を分離する「ソータクローン」などが挙げられるが、限界希釈法が簡便であり、よく用いられる。
【0073】
抗体価の認められたウェルについて、例えば限界希釈法によるクローニングを2〜4回繰返し、安定して抗体価の認められたものを抗ヒトc-Mplモノクローナル抗体産生ハイブリドーマ株として選択する。
【0074】
(7) アゴニスト抗体の選択
得られた抗ヒトc-Mplモノクローナル抗体産生ハイブリドーマ株の培養上清或いは後記(8)に従って該上清より精製した抗体を、種々のTPO活性測定系にて測定することにより、アゴニスト抗体を選択することができる。スクリーニングとして好適な方法として、哺乳動物細胞にヒトMplを発現させ、細胞増殖アッセイを行う方法が挙げられる。例えばマウス細胞株BaF3にヒトMplを発現させた細胞による増殖アッセイ(Orita et al. Blood. 2005 Jan 15;105(2):562-6.)等も使用し得るが、マウス細胞を用いた場合に、ヒト細胞の反応を反映しているとは限らないことを考慮すると、よりヒト細胞に強い活性を持つ抗体を選抜するにはヒトMplが発現しているヒト細胞を用いる増殖アッセイ方法がより好ましい。ヒト細胞による系として具体的には、後述の実施例5に記載のUT7/TPO細胞を用いた細胞増殖アッセイを挙げることができる。
【0075】
(8) ハイブリドーマ培養によるモノクローナル抗体の調製
クローニングを完了したハイブリドーマは、培地をHT培地から正常培地に換えて培養される。大量培養は、大型培養瓶を用いた回転培養、スピナー培養、あるいはホローファイバーシステム等を用いた培養で行われる。この大量培養における上清を、ゲルろ過等、当業者に周知の方法を用いて精製することにより、抗ヒトc-Mplモノクローナル抗体を得ることができる。また、同系統のマウス(例えばBALB/c)若しくはnu/nuマウス、ラット、モルモット、ハムスター又はウサギ等の腹腔内で該ハイブリドーマを増殖させることにより、抗ヒトc-Mplモノクローナル抗体を大量に含む腹水を得ることができる。精製の簡便な方法としては、市販のモノクローナル抗体精製キット(例えば、MAbTrap GIIキット;アマシャムファルマシアバイオテク社製)等を利用することもできる。かくして得られるモノクローナル抗体は、ヒトc-Mplに対して高い抗原特異性を有する。
【0076】
(9) モノクローナル抗体の検定
かくして得られたモノクローナル抗体のアイソタイプ及びサブクラスの決定は以下のように行うことができる。まず、同定法としてはオクテルロニー(Ouchterlony)法、ELISA法、又はRIA法が挙げられる。オクテルロニー法は簡便ではあるが、モノクローナル抗体の濃度が低い場合には濃縮操作が必要である。一方、ELISA法又はRIA法を用いた場合は、培養上清をそのまま抗原吸着固相と反応させ、さらに二次抗体として各種イムノグロブリンアイソタイプ、サブクラスに対応する抗体を用いることにより、モノクローナル抗体のアイソタイプ、サブクラスを同定することが可能である。さらに、タンパク質の定量は、フォーリンロウリー法、及び280nmにおける吸光度[1.4(OD280)=イムノグロブリン1mg/mL]より算出する方法等により行うことができる。また、ハイブリドーマからモノクローナル抗体をコードする遺伝子をクローニングし、配列を決定することでサブクラスを特定することもできる。
【0077】
(10) モノクローナル抗体をコードする遺伝子のクローニングと組換え型抗体作製
また、ハイブリドーマ等の抗体産生細胞からモノクローナル抗体をコードする遺伝子をクローニングし、適当なベクターに組み込んで、これを宿主(例えば哺乳類細胞細胞株、酵母細胞、昆虫細胞など)に導入し、遺伝子組換え技術を用いて産生させた組換え型抗体を調製することもできる(P.J.Delves., ANTIBODY PRODUCTION ESSENTIAL TECHNIQUES., 1997 WILEY、P.Shepherd and C.Dean., Monoclonal Antibodies., 2000 OXFORD UNIVERSITY PRESS, J.W.Goding., Monoclonal Antibodies:principles and practice., 1993 ACADEMIC PRESS)。
【0078】
本発明は、本発明の抗体を産生するハイブリドーマが保有する抗体の遺伝子配列を含む核酸、特に後述の、本発明のハイブリドーマが産生する抗体の重鎖可変領域及び軽鎖可変領域の核酸も包含する。ここで、核酸にはDNA及びRNAが含まれる。
【0079】
ハイブリドーマからモノクローナル抗体をコードする遺伝子を調製するには、モノクローナル抗体のL鎖V領域、L鎖C領域、H鎖V領域及びH鎖C領域をそれぞれコードするDNAをPCR法等により調製する方法が採用される。プライマーは、抗体遺伝子又はアミノ酸配列から設計したオリゴDNAを、鋳型としてはハイブリドーマから調製したDNAを使用することができる。これらのDNAを1つの適当なベクターに組み込み、これを宿主に導入して発現させるか、あるいはこれらのDNAをそれぞれ適当なベクターに組み込み、共発現させる。
【0080】
ベクターには、宿主微生物で自律的に増殖し得るファージ又はプラスミドが使用される。プラスミド DNAとしては、大腸菌、枯草菌又は酵母由来のプラスミドなどが挙げられ、ファージDNAとしてはλファージが挙げられる。
【0081】
形質転換に使用する宿主としては、抗体の立体構造を正しく構成できることから、真核細胞を挙げることができ、例えば、酵母、動物細胞(COS細胞、CHO細胞等)、昆虫細胞が挙げられる。特に動物細胞宿主を用いる場合、ベクターとしては、例えばN5KG1-Val Larkベクター(IDEC pharmaceuticals: US patent 6001358)を用いることができる。本ベクターは、組換え抗体を動物細胞に発現させる目的で使用される発現ベクターで、2つのCMV プロモーター/エンハンサーを持ち、それぞれの 下流に重鎖および軽鎖可変領域のクローニングサイトを持つ。さらにその下流には、ヒトγ1鎖定常領域とヒトκ鎖定常領域をコードする遺伝子配列をあらかじめ持っている。本ベクターの可変領域クローニングサイトに任意の重鎖および軽鎖可変領域を、読み枠(フレーム)をあわせて組み込むことにより、軽鎖可変領域がヒトκ鎖定常領域と、重鎖可変領域がヒトγ1定常領域と連結された抗体を発現させることができる。本ベクターを導入された動物細胞は培養液中に抗体(ヒトIgG1)を産生する。また、異なる重鎖定常領域遺伝子を含むベクターも使用することができる。例えばN5KG4PEベクター(IDEC pharmaceuticals社製)は定常領域遺伝子として、ヒトγ4に、上述した二つの変異(Ser228Pro, Leu235Glu)が入った配列を持っている。N5KG4PEベクターに任意の重鎖および軽鎖可変領域遺伝子配列を組み込むことで、任意の可変領域を持つIgG4PEを発現させることができる。さらに、重鎖または軽鎖遺伝子に改変を加えることで、様々な定常領域を持った抗体を作製することが可能である。
【0082】
もちろん、本発明に用いる哺乳動物細胞用発現ベクターは上記のものに限定されるものではない。例えば発現を制御する塩基配列として前述CMVプロモーター/エンハンサーを利用した他の発現ベクターを用いてもよいし、これとは異なった公知のプロモーター/エンハンサー(1若しくは複数)を発現制御配列として使ってもよい。プロモーターとしては、例えば、ポリオーマウイルス、鶏痘ウイルス(1989年7月5日公開のUK2211504)、アデノウイルス(例えばアデノウイルス2)、ウシ乳頭腫ウイルス、鳥の肉腫ウイルス、サイトメガロウイルス、レトロウイルス、B型肝炎ウイルスおよび最も好ましくはシミアンウイルス40(SV40)のようなウイルスのゲノムから得られるプロモーター、ヘテロローガスな哺乳動物プロモーター(例えばアクチンプロモーター、免疫グロブリンプロモーター、熱衝撃プロモーター)を挙げることができる。プロモーターに作用し転写を増大させるエンハンサーとしては、公知の哺乳動物遺伝子由来のエンハンサー(グロビン、エラスターゼ、アルブミン、α−フェトプロテイン、およびインシュリン)、および、真核細胞ウイルス由来のエンハンサー(複製起点の後期側のSV40エンハンサー(bp100-270)、複製起点の後期側のポリオーマエンハンサー、およびアデノウイルスエンハンサー)を用いることができる。
【0083】
また、発現ベクターには、転写の終止およびmRNAの安定化に必要な配列を含むことができる。このような配列は普通、真核生物のまたはウイルスのDNAまたはcDNAの5'および時には3'非翻訳領域から取得することができる。
【0084】
宿主への遺伝子の導入方法は公知であり、任意の方法(例えばカルシウムイオンを用いる方法、エレクトロポレーション法、スフェロプラスト法、酢酸リチウム法、リン酸カルシウム法、リポフェクション法等)が挙げられる。また、後述の動物に遺伝子を導入する方法としては、マイクロインジェクション法、ES細胞にエレクトロポレーションやリポフェクション法を使用して遺伝子を導入する方法、核移植法などが挙げられる。
【0085】
本発明において、目的とする抗体は、形質転換体を培養し、その培養上清から採取することにより得ることができる。形質転換体を培養するには、使用する宿主に適した培地を用い、静置培養法、ローラーボトルによる培養法などが採用される。
【0086】
培養後、細胞外に生産された抗体の精製は、培養液をそのまま使用するか、遠心分離等により細胞を除去して行う。その後、タンパク質の単離精製に用いられる各種クロマトグラフィーを用いた一般的な生化学的方法を単独で又は適宜組み合わせて用いることにより、前記培養物中から目的の抗体を単離精製することができる。
【0087】
さらに、トランスジェニック動物作製技術を用いて、目的抗体の遺伝子が内在性遺伝子に組み込まれた動物宿主、例えばトランスジェニックウシ、トランスジェニックヤギ、トランスジェニックヒツジ又はトランスジェニックブタを作製し、そのトランスジェニック動物から分泌されるミルク中からその抗体遺伝子に由来するモノクローナル抗体を大量に取得することも可能である(Wright, G., et al. (1991) Bio/Technology 9, 830-834)。
【0088】
本発明に係るヒトMplに対するアゴニスト抗体の製法として、好ましい方法は、前述の[課題を解決するための手段]の4.にて例示した遺伝子組換え技術による方法であるが、これに限定されるものではない。
【0089】
3.本発明に係るDNA
本発明は、上で述べたように、
(1) ヒトMplに対するアゴニスト抗体の重鎖可変領域のアミノ酸配列をコードする塩基配列であって以下の(a)〜(d)から選択されたアミノ酸配列をコードする塩基配列を含むDNA:
(a) 配列番号2で示されるアミノ酸配列;
(b) 配列番号4で示されるアミノ酸配列;
(c) 配列番号6で示されるアミノ酸配列;
(d) 配列番号8で示されるアミノ酸配列;
及び、
(2) ヒトMplに対するアゴニスト抗体の軽鎖可変領域のアミノ酸配列をコードする塩基配列であって以下の(a)〜(h)から選択されたアミノ酸配列をコードする塩基配列を含むDNA:
(a) 配列番号3で示されるアミノ酸配列;
(b) 配列番号5で示されるアミノ酸配列;
(c) 配列番号7で示されるアミノ酸配列;
(d) 配列番号9で示されるアミノ酸配列;
(e) 配列番号3のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列;
(f) 配列番号5のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列;
(g) 配列番号7のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列;
(h) 配列番号9のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列;
を提供するものであり、上記2.の本発明に係るヒトMplに対するアゴニスト抗体の製法、より具体的には、遺伝子組換え技術を用いた抗体の製造において、用いることができる。
【0090】
これら可変領域のアミノ酸配列(a)〜(d)をコードするDNAは、前述のヒトMplに対するアゴニスト抗体産生ハイブリドーマ取得方法によって得られたハイブリドーマ株より、後述実施例7の通り、定法によりmRNAを抽出し既知である抗体定常領域のアミノ酸配列に基づくプライマーを用いて5’RACE法により取得したものであり、その可変領域をコードする各DNAを含むプラスミドは、独立行政法人産業技術総合研究所特許生物寄託センター(日本国茨城県つくば市東1-1-1 つくばセンター中央第6)に2006年3月14日付にてブダペスト条約に基づく国際寄託がされている。
【0091】
【表1】

【0092】
本発明のアゴニスト抗体を構成する軽鎖の可変領域は、その特定例として、配列番号3、5、7又は9に示されるアミノ酸配列を含むが、これらのアミノ酸配列のフレームワーク領域に1若しくは数個のアミノ酸残基の欠失、置換、付加又は挿入を含んでもよいし、或いはこれらのアミノ酸配列のフレームワーク領域の配列と少なくとも85%、86%、87%、88%又は89%、好ましくは少なくとも90%、92%、93%又は94%、より好ましくは少なくとも95%、96%、97%、98%又は99%の同一性を有する配列を含んでいてもよい。フレームワーク領域は、可変領域において3つのCDR(相補性決定部位;complementarity-determining region)、すなわち配列番号3、5又は7に示されるアミノ酸配列においては、RASQGISS(A又はT)LA(アミノ酸位置24-34)、DASSLES(アミノ酸位置50-56)、及びQQFNSYP(L又はY又はW)T(アミノ酸位置89-97)、を除く領域を指し、配列番号9に示されるアミノ酸領域においては、RASQSVSSSYLA(アミノ酸位置24-35)、DASSRAT(アミノ酸配列51-57)、及びQQYGSSPIT(アミノ酸位置90-98)、を除く領域を指す。後述の実施例17で実証するとおり、フレームワーク領域内にアミノ酸変異が存在しても、本発明の変異型抗体はいずれも、非変異型抗体の場合とほぼ同等のアゴニスト活性、すなわち、FM3A-hMpl細胞などのFM3A-hMpl細胞のヒトトロンボポエチン受容体に結合し該受容体を活性化する能力及び/又はUT-7/TPO細胞の増殖能を有することができる。
【0093】
そのような変異の例は、保存的アミノ酸間での置換である。保存的アミノ酸は、電荷、構造、極性などの性質が類似したアミノ酸群であり、例えば塩基性アミノ酸(Arg、His、Lys)、酸性アミノ酸(Glu、Asp)、非極性アミノ酸(Ala、Leu、Ile、Val、Gly、Pro)、極性アミノ酸(Ser、Thr、Cys、Met、Asn、Gln)、芳香族アミノ酸(Phe、Tyr、Trp)に分類することができる。
【0094】
また、配列同一性は、2つ又はそれ以上の配列を、ギャップを導入して又はギャップを導入しないで整列比較したときのアミノ酸(又はヌクレオチド)配列間の一致率を表わし、一般に全アミノ酸(又はヌクレオチド)数に対する同一アミノ酸(又はヌクレオチド)数のパーセンテージである。配列同一性は、必要に応じてNCBI(米国)などのデータバンクにアクセスし、配列検索用のBLAST、FASTAなどの公知のアルゴリズムを利用して決定することができる。
【0095】
変異の導入は、変異を含まないアミノ酸配列をコードするDNAに対して、例えば部位特異的突然変異誘発法、PCR法(変異を含むプライマーを使用する)などを適用することによって実施することができる。変異導入法は、例えばSambrookら, Molecular Cloning A Laboratory Mannual, Cold Spring Harbor Laboratory Press (1989)などに記載されている。
【0096】
本発明の上記DNAはさらに、可変領域に加え、それぞれ重鎖或いは軽鎖の定常領域をコードする塩基配列を含むものであってもよい。
【0097】
また、本発明に係る抗体の製造方法の項で述べた重鎖定常領域の改変は、寄託したDNA及び公知のヒト抗体定常領域の配列に基づき、周知の遺伝子工学的改変手法により得ることができる。
【0098】
4.ヒトc-Mplに対するアゴニスト抗体の医薬用途及び医薬組成物
本発明に係るヒトc-Mplに対するアゴニスト抗体は、c-MPL受容体に結合しそれを活性化する能力、および/または(インビボおよびインビトロの両方で)血小板の産生を刺激する能力(「血小板生成活性」)および血小板前駆体の産生を刺激する能力(「巨核球生成活性」)を有する。
【0099】
また、ヒトc-Mpl受容体は、巨核球のみならず造血幹細胞においても発現しているとされている。正常動物において、PEG-rHuMGDF投与により骨髄中の赤芽球系や顆粒球/マクロファージ系の前駆細胞が増加したとの報告もある(Stem Cell,14:651-660,1996)。しかし、ヒト臍帯血移植マウスにおいては、PEG-rHuMGDFの投与によりマウスの巨核球系以外の前駆細胞の増殖は見られたが、ヒトの前駆細胞の増殖は認められなかった。一方、ヒトc-Mplに対するアゴニスト抗体においては骨髄中のヒト赤血球および顆粒球/マクロファージ系の前駆細胞の数が有意に高かった(実施例14)。このことは、ヒトc-Mplに対するアゴニスト抗体がヒト細胞に選択的にシグナルを入れ、巨核球系のみならず、他系列の細胞の生着も促進できる可能性を示唆している。
【0100】
本発明に係るヒトc-Mplに対するアゴニスト抗体を有効成分とする医薬組成物により治療される状態は、一般には、存在する巨核球/血小板の欠乏または将来において予期または予想される巨核球/血小板の欠乏(例えば、計画されている手術または血小板供与によるもの)を伴う状態である。そのような状態は、インビボにおける活性Mp1リガンドの(一時的または永久的な)欠乏により生じうる。したがって本発明の組成物は、血小板欠乏状態即ち血小板減少症の治療を要する患者において血小板減少症を予防的または治療的な治療するために利用可能である。さらに、汎血球減少を長期間に渡って伴う造血幹細胞移植(骨髄移植、臍帯血移植、末梢血幹細胞移植)後の血球回復のための治療を要する患者において、汎血球減少症を予防的または治療的な治療をするためにも利用可能である。
【0101】
血小板減少症(血小板欠乏症)は、化学療法および種々の薬物での他の療法、放射線療法、手術、不慮の出血、および他の具体的な病態を含む種々の理由により生じうる。血小板減少症を伴い本発明に従い治療されうる典型的な具体的な病態としては以下のものが挙げられる:無形成貧血;特発性または免疫血小板減少症(ITP)、例えば乳癌に伴う特発性血小板減少性紫斑病;HIVに伴うITPおよびHIVに関連した血栓性血小板減少性紫斑病;血小板減少症を引き起こす転移性腫瘍;全身性エリテマトーデス、例えば新生児ループス症候群脾腫;ファンコーニ症候群;ビタミンB12欠乏症;葉酸欠乏症;メイ-ヘグリンの異常;ウィスコット-アルドリッチ症候群;慢性肝疾患;血小板減少症に関連した骨髄形成異常症候群;発作性夜間ヘモグロビン尿症;C7E3 Fab(Abciximab)療法後の急性深部血小板減少症;同種免疫血小板減少症、例えば母性同種免疫血小板減少症;抗リン脂質抗体および血栓症に関連した血小板減少症;自己免疫血小板減少症;薬物により誘発された免疫血小板減少症、例えばカルボプラチンにより誘発された血小板減少症、ヘパリンにより誘発された血小板減少症;胎児血小板減少症;妊娠血小板減少症;ヒュージー症候群;ルポイド血小板減少症;不慮の及び/又は大量の失血;骨髄増殖性異常症;悪性疾患を有する患者における血小板減少症;血栓性血小板減少紫斑病、例えば癌患者において血栓性血小板減少性紫斑病/溶血性尿毒症性症候群として現れる血栓性細小血管症;自己免疫性溶血性貧血;潜在性空腸憩室穿孔;真正赤血球系無形成症;自己免疫血小板減少症;流行性(epidemica)腎障害;ラファンピシンに関連した急性腎不全;Paris-Trousseau血小板減少症;新生児同種免疫血小板減少症;発作性夜間ヘモグロビン尿症;胃癌における血液学的変化;小児における溶血性尿毒症性症候群;A型関連ウイルスを含むウイルス感染に関連した血液学的徴候およびCMVに関連した血小板減少症。また、ある種のエイズ治療は血小板減少症を引き起こす(例えば、AZT)。また、ある種の創傷治癒障害は、血小板数の増加が有益であろう。なお上記疾患においては、血小板減少のみならず、その他の血球減少を伴う疾患も存在する。
【0102】
予想される血小板の欠乏(例えば、将来の手術によるもの)に対して、血小板が必要となる前に数日から数時間にわたり有効成分としての本発明のアゴニスト抗体を投与することが可能であろう。緊急の状況(例えば、不慮の及び大量の失血)では、血液または精製血小板と共に本発明のアゴニスト抗体を投与することが可能であろう。あるいは、汎血球の欠乏(例えば、臍帯血移植によるもの)に対しても、有効成分としての本発明のアゴニスト抗体を投与することが可能であろう。
【0103】
特に好ましい治療対象としては、(1)特発性血小板減少性紫斑病もしくは肝疾患にともなって起こる血小板減少症、(2)癌化学療法、再生不良性貧血、骨髄異形性症候群(MDS)、骨髄移植もしくは臍帯血移植にともなって起こる血小板減少症および/または汎血球減少症を挙げることができる。
【0104】
本発明のヒトc-MPLに対するアゴニスト抗体はまた、血小板および/または巨核球および関連細胞の生存能または保存寿命を維持するのに有用で有り得る。したがって、そのような細胞を含有する組成物中に有効量を含有させるのが有用であろう。
【0105】
本発明にかかるヒトc-MPLに対するアゴニスト抗体を有効成分とする医薬組成物は、注射用または経口、鼻、経皮または他の投与形態のためのものであってもよい。それらには、例えば、静脈内、皮内、筋肉内、乳房内、腹腔内、鞘内、眼内、延髄後方、肺内(例えば、エアゾール化薬)または皮下注射(長期にわたる放出のためのデポ投与を含む)、舌下、肛門、膣または外科的移植、例えば脾漿膜下、脳または角膜内の埋め込みによる投与が含まれる。該治療は、単回投与または一定期間にわたる複数回の投与よりなるものであってもよい。一般には、本発明にかかるヒトc-MPLに対するアゴニスト抗体の有効量と医薬上許容される希釈剤、保存剤、可溶化剤、乳化剤、佐剤および/または担体とを含む医薬組成物が本発明に含まれる。そのような組成物は、種々のバッファー含有物(例えば、Tris-HCl、酢酸塩、リン酸塩)、pHおよびイオン強度の希釈剤;添加物、例えば界面活性剤および可溶化剤(例えば、Tween 80、Polysorbate 80)、抗酸化剤(例えば、アスコルビン酸、メタ重亜硫酸ナトリウム)、保存剤(例えば、Thimersol、ベンジルアルコール)および増量物質(例えば、ラクトース、マンニトール);ポリ乳酸、ポリグリコール酸などの重合体化合物の粒子状製剤内またはリポソーム内に該物質が封入されたものを含む。所望により、医薬ビヒクル、賦形剤または媒体として働く更に他の医薬上許容される液体、半固体または固体希釈剤を含んでいてもよい。それらには、ポリオキシエチレンソルビタンモノラウラート、ステアリン酸マグネシウム、メチル-およびプロピルヒドロキシベンゾアート、デンプン、スクロース、デキストロース、アラビアゴム、リン酸カルシウム、鉱油、カカオ脂およびテオブロマの油が含まれるが、これらに限定されるものではない。該組成物は、液体形態または乾燥粉末(例えば、凍結乾燥形態)として製造することができる。移植可能な徐放製剤、および経皮製剤も意図される。
【0106】
上記の状態の治療方法にかかわる投与計画は、薬物の作用を改変する種々の要因(例えば、患者の年齢、状態、体重、性別および食事、いずれかの感染の重症度、投与時間ならびに他の臨床的要因)を考慮して担当医師により決定されるであろう。一般には、該用量は、1日当たり本発明抗体100μg〜1mg/kg体重、好ましくは10〜100μg/kg、より好ましくは1〜10μg/kgであり、これらを、1日量で、あるいはより長い又はより短い間隔で(例えば、1日おき、1週間に2回、毎週、または毎日2回または3回)同等量で投与する。
【0107】
また、本発明にかかるヒトc-MPLに対するアゴニスト抗体を有効成分とする医薬組成物は、単独で又は他のサイトカイン、可溶性Mp1受容体、造血因子、インターロイキン、増殖因子と組合せて、血小板の欠乏および他の症状により特徴づけられる病態の治療において使用することができる。造血の一般的な刺激因子(例えば、IL-3またはGM-CSF)と組合せて、いくつかの形態の血小板減少症を治療するのに有用であると予想される。また、他の巨核球刺激因子、すなわちmeg-CSF、幹細胞因子(SCF)、白血病阻害因子(LIF)、オンコスタチンM(OSM)または巨核球刺激活性を有する他の分子を、Mp1リガンドと共に使用することができる。そのような共投与のための追加的な代表的なサイトカインまたは造血因子には、IL-1アルファ、IL-1ベータ、IL-2、IL-3、IL-4、IL5、IL-6、IL-11、コロニー刺激因子-1(CSF-1)、M-CSF、SCF、GM-CSF、顆粒球コロニー刺激因子(G-CSF)、EPO、インターフェロン-アルファ(IFN-アルファ)、コンセンサスインターフェロン、IFN-ベータ、IFN-ガンマ、IL-7、IL-8、IL-9、IL-10、IL-12、IL-13、IL-14、IL-15、IL-16、IL-17、IL-18、トロンボポエチン(TPO)、アンジオポエチン、例えばAng-1、Ang-2、Ang-3、Ang-4、Ang-Y、ヒトアンジオポエチン様ポリペプチド、血管内皮増殖因子(VEGF)、アンジオゲニン、骨形成因子(bone morphogenic protein)-1、骨形成因子-2、骨形成因子-3、骨形成因子-4、骨形成因子-5、骨形成因子-6、骨形成因子-7、骨形成因子-8、骨形成因子-9、骨形成因子-10、骨形成因子-11、骨形成因子-12、骨形成因子-13、骨形成因子-14、骨形成因子-15、骨形成因子受容体IA、骨形成因子受容体IB、脳由来神経栄養因子、毛様体神経栄養因子、毛様体神経栄養因子α、サイトカイン誘導性好中球走化因子1、サイトカイン誘導性好中球走化因子2α、サイトカイン誘導性好中球走化因子2β、β内皮細胞増殖因子、エンドセリン1、上皮増殖因子、上皮由来好中球誘引物質、繊維芽細胞増殖因子4、繊維芽細胞増殖因子5、繊維芽細胞増殖因子6、繊維芽細胞増殖因子7、繊維芽細胞増殖因子8、繊維芽細胞増殖因子8b、繊維芽細胞増殖因子8c、繊維芽細胞増殖因子9、繊維芽細胞増殖因子10、酸性繊維芽細胞増殖因子、塩基性繊維芽細胞増殖因子、グリア細胞株由来神経栄養因子受容体α1、グリア細胞株由来神経栄養因子受容体α2、成長関連タンパク質(growth related protein)、成長関連タンパク質α、成長関連タンパク質β、成長関連タンパク質γ、ヘパリン結合性上皮増殖因子、肝細胞増殖因子、肝細胞増殖因子受容体、インスリン様増殖因子I、インスリン様増殖因子受容体、インスリン様増殖因子II,インスリン様増殖因子結合タンパク質、角質細胞増殖因子、白血病阻害因子、白血病阻害因子受容体α、神経成長因子、神経成長因子受容体、ニューロトロフィン-3、ニューロトロフィン-4、胎盤増殖因子、胎盤増殖因子2、血小板由来内皮細胞増殖因子、血小板由来増殖因子、血小板由来増殖因子A鎖、血小板由来増殖因子AA、血小板由来増殖因子AB、血小板由来増殖因子B鎖、血小板由来増殖因子BB、血小板由来増殖因子受容体α、血小板由来増殖因子受容体β、プレB細胞増殖刺激因子、幹細胞因子受容体、TNF(TFN0、TNF1、TNF2を含む)、トランスフォーミング増殖因子α、トランスフォーミング増殖因子β、トランスフォーミング増殖因子β1、トランスフォーミング増殖因子β1.2、トランスフォーミング増殖因子β2、トランスフォーミング増殖因子β3、トランスフォーミング増殖因子β5、潜在(latent)トランスフォーミング増殖因子β結合タンパク質I、トランスフォーミング増殖因子β結合タンパク質II、トランスフォーミング増殖因子β結合タンパク質III、腫瘍壊死因子受容体I型、腫瘍壊死因子受容体II型、ウロキナーゼ型プラスミノーゲンアクチベーター受容体、血管内皮増殖因子、ならびにそれらのキメラタンパク質が含まれる。
【0108】
したがって、本発明にかかるヒトc-Mplに対するアゴニスト抗体を有効成分とする医薬組成物の投与(成熟巨核球の数を増加させるためのもの)は、血小板の産生を刺激する特に有効な手段であると予想される。さらに造血幹細胞の産生を刺激することにおいても有用な手段であると予想される。前記で列挙した用量は、該治療用組成物中のそのような追加的な成分を補償するように調節されるであろう。治療された患者の経過は、通常の方法によりモニターすることができる。
【0109】
以下、実施例により本発明をさらに詳細に説明するが、本発明の範囲はこれらの実施例に限定されるものではない。
【実施例】
【0110】
[実施例1] 抗原の調製
1-1 ヒトc-Mpl発現細胞の作製
抗原タンパク質を発現する細胞株を免疫に用いる場合、一般的に発現量が高いほど抗体作製には有利である。ヒトc-Mpl発現細胞株としては、各種ヒト巨核球系細胞株や強制発現株が知られているが、これらの細胞株のc-Mpl発現量は細胞あたり数千分子と低く、抗原には不向きである。実際、マウス造血系細胞株であるFDCP2にヒトc-Mplを導入した発現株FDCP-hMpl(FEBS Lett. 1996 Oct 21;395(2-3):228-34.参照)をヒト抗体産生マウス(KMマウスTM)に免疫した場合、抗体価上昇が不十分であり、hMpl特異的なヒト抗体を得ることができなかった。また、ヒト巨核球系細胞株を抗原として用いる場合、他の膜分子に対する抗体も誘導されるため、c-Mpl特異的な抗体を効率良く誘導するためには、マウス細胞株、しかも可能な限りMHC適合した細胞株を宿主に、ヒトc-Mplを導入した高発現株を用いるのが望ましい。そこで、ヒトc-Mpl(hMpl)高発現株を作製するために、hMpl発現ベクターを以下の通り作製し、かつ2種類のマウス細胞株(L929およびFM3A)へ導入を行った。
【0111】
更に、hMplにはリガンド非依存性に恒常的にアゴニストシグナルを伝達する変異型受容体が報告されている(508番目TrpがSerに変換している変異体、Abe Mら、Leukemia. 2002 Aug;16(8):1500-1506)が、このような変異体は野生型とは立体構造が異なることが予想される。このような恒常活性型変異体に親和性が高い抗体は、強力なアゴニスト活性を示す可能性がある。そこで、恒常活性型変異体(以下hMpl-Ser)の発現ベクターも作製し、発現細胞を作製して免疫に用いた。
【0112】
1) 抗ヒトc-Mpl(hMpl)発現ベクターの調製
hMplの全長cDNAを保持するプラスミドDNAであるhumpl-Pas12(Bartley TDら、 Cell 1994 Jul 1;77(7):1117-1124.或はMorita Hら、FEBS Lett. 1996 Oct 21;395(2-3):228-234.)のDNAを鋳型としてhMplのコーディング領域全域を増幅するPCR反応を実施した。プライマーとして、末端に連結のための制限酵素部位(5’末側EcoRI、3’末側XbaI)を付加するようにデザインした、Mpl_F1及びMpl_R2を用いて、KOD-Plus- DNAポリメラーゼ(東洋紡績(株)製、日本)によるPCRで増幅した。以下、実施例中のPCRの反応温度調節は、ジーンアンプPCRシステム9700((株)パーキンエルマー・ジャパン社製)を使用した。反応温度条件としては、94℃の初期温度で5分間の加熱後、98℃/10秒間と68℃/3分間のサイクルを30回反復し、最後に72℃/7分間加熱した。増幅したPCR断片は、エタノール沈殿で回収した後、アガロースゲル電気泳動で分離し、メンブランを用いるDNA精製キットであるQIAquick Gel Extraction Kit(キアゲン社製)にて精製した。精製したDNA断片をpCR4Blunt- TOPO ベクター(東洋紡績(株)製)にサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。DNA塩基配列決定のためのプライマーとして、M13-20FW及びM13RVを用いた。挿入部分のDNA塩基配列解析を行い、hMplの配列(GenBank ACCESSION :M90102)と相違がなく、また、プライマー部分もデザインどおりの配列を有するプラスミドDNAを選択した。次に、hMplの配列を含むプラスミドDNAを精製した後、制限酵素EcoRIとXbaIで消化して、アガロースゲル電気泳動で約2kb弱のDNAを回収し精製した。他方、ヒトEFプロモーターとブラストサイジン(Bsd)選択マーカーを有する発現ベクターであるpEF6/Myc-His(インビトロジェン(株)社製)、及び、CMVプロモーターとネオマイシン(Neo)選択マーカーを有するpEGEP-N1ベクター(ベクトン・ディキンソン・バイオサイエンス・クローンテック社製)についても、同様に制限酵素EcoRI、とXbaIでの消化処理を行った後、脱リン酸化のためにAlkaline Phosphatase (E. coli C75)(タカラバイオ(株)、日本)で処理した後に、アガロースゲル電気泳動とDNA精製キットでDNAを回収した。精製したhMplの全領域DNA断片を、それぞれの発現ベクターDNAにT4 DNA ligase を用いてライゲーションして、大腸菌DH10Bへ導入し、それぞれ形質転換体を得た。インサートDNAを含む形質転換体のプラスミドDNAについてDNA塩基配列を解析して、hMplの全長cDNAが挿入されたpEF-MPL635及びpCMV-MPL635を取得した。
Mpl_F1:5’-AGAGAGAGAG GAATTCGCCA CCATGCCCTC CTGGGCCCTC TT-3’(配列番号12)
Mpl_R2:5’- AGAGAGAGAG CGGCCGCTCA AGGCTGCTGC CAATAGCTTA GTG -3’(配列番号13)
M13-20FW: 5’-GTAAAACGACGGCCAGTG-3’(配列番号14)
M13RV:5’-CAGGAAACAGCTATGAC-3’(配列番号15)
【0113】
2) 恒常活性型ヒトc-Mpl(hMpl-Ser)発現ベクターの調製
TPO非依存性に細胞内のシグナル活性化が報告されているhMpl変異体(508番目TrpがSerに変換している変異体、Abe Mら、Leukemia. 2002 Aug;16(8):1500-1506)の発現ベクターを作製した。508番目のアミノ酸残基をコードするコドンを変更(TGG→TCG)するために、pEF-MPL635のDNAを鋳型として、GeneEditorTM in vitro Site-Directed Mutagenesis System(プロメガ社)を用いた部位特異的変異導入法を実施した。変異導入用オリゴヌクレオチド(5’末端リン酸化済み)としては、Mut_MplSer508を用いた。目的の変異導入用オリゴヌクレオチドと上記キット付属のSelection Oligonucleotideを鋳型DNAとアニーリングさせて変異導入鎖を合成した後、GeneEditorTM Antibiotic Selection Mix存在下では変異体のみが増殖することを利用して変異体を選択した。より具体的には、dsDNAテンプレートをアルカリ条件下(0.2M NaOH、0.2 mMEDTA(最終濃度))室温で5分間インキュベートした後、2M酢酸アンモニウム(pH4.6)を10分の1容量加えて中和してからエタノール沈殿により回収した。アルカリ変性処理した鋳型DNAに、変異導入用オリゴヌクレオチドと新しい抗生物質耐性獲得用Selection Oligonucleotide(5’末端リン酸化)、及び、キット添付のアニーリングバッファーを加えた後、75℃で5分間保温し、37℃にゆっくり下げることによりアニーリングを行なった。次に、変異鎖の合成と連結のために、キット付属のSynthesis 10×buffer、T4 DNA Polymerase、及びT4 DNA ligaseを加えて、37℃で90分反応を行なった。GeneEditorTM Antibiotic Selection Mix存在下でコンピテントセルBMH 71-18 mutSに形質転換して培養した形質転換体大腸菌よりプラスミドDNAを調製し、更にそのDNAによりコンピテントセルJM109を形質転換後、GeneEditorTM Antibiotic Selection Mixを含むLBプレートに播種した。プレートに生じた形質転換体を培養して、プラスミドDNAのDNA塩基配列を解析して、508番目のアミノ酸が変換(TrpからSer)したhMplを発現するpEF-MPL635-Serベクターを取得した。
Mut_MplSer508:5’-CTGCTGCTGC TGAGGTCGCA GTTTCCTGCA CACTAC-3’(配列番号16)
【0114】
3) 全長ヒトc-Mpl発現L929細胞の作製
作製したpEF-MPL635ベクター(1μg)を、Lipofectamine試薬(Invitrogenより購入)および Lipofectamine PLUS(Invitrogenより購入)試薬と混合し、さらに無血清Dulbecco’s Modified Eagle Medium(DMEM)培地と混合した。混合液を1.5×105cells/well で6-well plateに培養したL929細胞に添加し、3時間培養することで細胞にDNAを導入した。10%ウシ胎児血清(FBS)添加DMEM培地にて一晩培養し、翌日より、培地に10μg/mL Blasticidin(Invitrogenより購入)を加えて薬剤耐性細胞を選抜した。その後、抗c-Mpl抗体を用いたFluorescence Activated Cell Sorting(FACS)法にてc-Mpl発現細胞を単離し、全長ヒトc-Mpl発現L929細胞株(以下、L929-hMpl)を樹立した。FACSはFACS-Vantage(Becton Dickinson社製)によって行った。選抜後は5μg/mL Blasticidin, 10%FBS添加DMEM培地にて培養、維持した。
【0115】
4) 全長ヒトc-Mpl発現FM3A細胞の作製
上記 3)と同様の方法でpEF-MPL635ベクターをFM3A細胞に導入し、全長ヒトc-Mpl発現FM3A細胞株(以下、FM3A-hMpl)を樹立した。5μg/mL Blasticidin, 10%FBS添加 Roswell Park Memorial Institute(RPMI)培地にて培養、維持した。
【0116】
5) 恒常活性型ヒトMpl発現FM3A細胞作製
上記、pEF-MPL635-Serベクターを、3)と同様の方法で、FM3A細胞に導入し、hMpl-Ser発現FM3A細胞株(以下、FM3A-hMpl-Ser)を樹立した。5μg/mL Blasticidin, 10%FBS添加RPMI培地にて培養、維持した。
【0117】
1-2 可溶化ヒトc-Mpl組換えタンパク質の作製
ヒトc-Mplの細胞膜貫通領域および細胞内領域を欠失させた、下記配列の可溶化型ヒトc-MplをコードするDNAを発現ベクターpEAK8(EdgeBioSystems社製)に結合し、トランスフェクタム試薬(Promega社より入手可能)にてHek293細胞に導入した。安定発現株を選択後、その培養上清を抗Mpl抗体カラムにて精製し、可溶化型ヒトc-Mpl組換えタンパク質(以下soluble Mpl-x、sMpl-xと略記)を調製した。
NH2-MPSWALFMVTSCLLLAPQNLAQVSSQDVSLLASDSEPLKCFSRTFEDLTCFWDEEEAAPSGTYQLLYAYPREKPRACPLSSQSMPHFGTRYVCQFPDQEEVRLFFPLHLWVKNVFLNQTRTQRVLFVDSVGLPAPPSIIKAMGGSQPGELQISWEEPAPEISDFLRYELRYGPRDPKNSTGPTVIQLIATETCCPALQRPHSASALDQSPCAQPTMPWQDGPKQTSPSREASALTAEGGSCLISGLQPGNSYWLQLRSEPDGISLGGSWGSWSLPVTVDLPGDAVALGLQCFTLDLKNVTCQWQQQDHASSQGFFYHSRARCCPRDRYPIWENCEEEEKTNPGLQTPQFSRCHFKSRNDSIIHILVEVTTAPGTVHSYLGSPFWIHQAVRLPTPNLHWREISSGHLELEWQHPSSWAAQETCYQLRYTGEGHQDWKVLEPPLGARGGTLELRPRSRYRLQLRARLNGPTYQGPWSSWSDPTRVETATETAW-COOH(配列番号17)
【0118】
[実施例2] モノクローナル抗体の作製
本発明の抗体は、遺伝子改変によってヒト抗体を産生する能力を持つヒト抗体産生マウス(KMマウスTM)を免疫し、モノクローナル抗体を作製することで得られた。KMマウスは、内在性マウスイムノグロブリン(Ig)重鎖及びマウスκ軽鎖を欠損しており、かつ、ヒトIg重鎖遺伝子を含む14番染色体断片(SC20)及びヒトIgκ鎖トランスジーン(KCo5)を同時に保持する。即ち、KMマウスはヒト抗体を産生する能力を持ち、マウスIg重鎖およびκ鎖を欠損したマウスである。このマウスはヒトIg重鎖遺伝子座を持つ系統Aのマウスと、ヒトIgκ鎖トランスジーンを持つ系統Bのマウスとの交配により作製される。系統Aは、内因性Ig重鎖及びκ軽鎖破壊の両者についてホモ接合体であり、子孫伝達可能な14番染色体断片(SC20)を保持するマウス系統(Tomizuka. et al., Proc Natl Acad Sci USA., 2000 Vol97:722参照)である。また、系統Bは内在性マウスIg重鎖及びκ軽鎖欠損の両者についてホモ接合体であり、ヒトIgκ鎖トランスジーン(KCo5)を保持するマウス系統(Nat Biotechnol., 1996 Vol14:845参照)である。
【0119】
本実施例におけるモノクローナル抗体の作製は、公知の方法によって行った(単クローン抗体実験操作入門 安東民衛ら著作、講談社(東京、日本)発行 1991参照)。
【0120】
1) 免疫
免疫原としてのヒトc-Mplは、実施例1で作製したL929-hMpl細胞、FM3A-hMpl細胞、恒常活性型c-Mpl発現FM3A-hMpl-Ser細胞、sMpl-x組換えタンパク質を用いた。被免疫動物は、実施例2で作製したヒト免疫グロブリンを産生するヒト抗体産生マウス使用し、下記の免疫方法で行った。
【0121】
免疫方法1: 9週齢のヒト抗体産生マウスに、実施例1で作製したL929-hMpl細胞(5×106細胞)をRibiアジュバンドと混合し、腹腔内に初回免疫した。初回免疫以降、一週間ごとに、同細胞(2×106細胞)を、インターロイキン6(IL-6)(5μg)と同時に、尾静脈経由で7回免疫した。さらに、脾臓及びリンパ節の取得3日前に同細胞を尾静脈経由で最終免疫した。
【0122】
免疫方法2: 9週齢のヒト抗体産生マウスに、実施例1で作製したFM3A-hMpl-Ser細胞(5×106細胞)に紫外線照射し、Ribiアジュバンドを加え腹腔内に初回免疫した。初回免疫から以降、同細胞(5×106細胞)を腹腔内投与で、一週間ごとに7回免疫した。さらに、脾臓及びリンパ節の取得3日前に、実施例1で作製したFM3A-hMpl細胞(2×106細胞)をIL-6(5μg)を加えて、尾静脈経由で最終免疫した。
【0123】
免疫方法3: 9週齢のヒト抗体産生マウスに、実施例1で作製したsMpl-x組換えタンパク質(10μg)を、完全フロイントアジュバンド(CFA)と混合して皮下投与で初回免疫した。2回目から5回目の免疫は、一週間に一度、sMpl-x組換えタンパク質(5μg)を不完全フロイントアジュバンド(IFA)と混合して皮下投与で免疫した。6回目から8回目の免疫では、L929-hMpl細胞(5×106細胞)を腹腔内に免疫した。さらに、脾臓及びリンパ節の取得3日前に、sMpl-x組換えタンパク質(5μg)およびIL-6(5μg)を、尾静脈経由で最終免疫した。
【0124】
2) ハイブリドーマ作製
最終免疫の3日後にマウスから脾臓及び/又はリンパ節を外科的に取得し、350mg/ml 炭酸水素ナトリウム、50単位/ml ペニシリン、50μg/ml ストレプトマイシンを含む無血清DMEM培地 10ml中に入れ、メッシュ(セルストレイナー:ファルコン社製)上でスパーテルを用いてつぶした。メッシュを通過した細胞懸濁液を遠心して細胞を沈澱させた後、この細胞を無血清DMEM培地で2回洗浄してから、無血清DMEM培地に懸濁して細胞数を測定した。一方、10% FCSを含むDMEM培地にて、37℃、5%炭酸ガス存在下で細胞濃度が1×108細胞/mlを越えないように培養したミエローマ細胞 SP2/0(ATCC No.CRL-1581)を同様に無血清DMEM培地で洗浄し、無血清DMEM培地に懸濁して細胞数を測定した。回収した細胞の懸濁液とマウスミエローマ懸濁液とを細胞数5:1で混合し、遠心後、上清を完全に除去した。このペレットに、融合剤として50%(w/v)ポリエチレングリコール1500(ベーリンガーマンハイム社製)1mlを、ピペットの先で撹拌しながらゆっくり添加した後、予め37℃に加温しておいた無血清DMEM培地1mlを2回に分けてゆっくり添加し、さらに7mlの無血清DMEM培地を添加した。遠心後、上清を除去して得られた融合細胞を、以下に記載する限界希釈法によるスクリーニングに供した。ハイブリドーマの選択は、10%のウシ胎児血清(Fetal Calf Serum、FCS)とヒポキサンチン(H)、アミノプテリン(A)、チミジン(T)(以下「HAT」という。:シグマ社製)を含有するDMEM培地中で培養することによって行った。さらに、10% FCSとHT(シグマ社製)とを含有するDMEM培地を用いて限界希釈法によりシングルクローンにした。培養は、96穴マイクロタイタープレート(ベクトンディッキンソン社製)中で行った。抗ヒトc-Mplヒトモノクローナル抗体を産生するハイブリドーマクローンの選択(スクリーニング)及び各々のハイブリドーマが産生するヒトモノクローナル抗体の特徴付けは、実施例4に記載のフローサイトメトリー、あるいは実施例5に記載のUT7/TPO細胞を用いた細胞増殖アッセイによって行った。アゴニスト抗体の活性評価系として、BaF3等のマウス細胞株にヒトMplを発現させ、細胞増殖アッセイを行うことができる(Orita et al. Blood. 2005 Jan 15;105(2):562-6.)が、そのような細胞の反応が、ヒト細胞の反応を反映しているとは限らない。UT7/TPOはヒト由来細胞株であるため、これをスクリーニングに用いることで、よりヒト細胞に強い活性を持つ抗体を選抜し易いと考えられる。
【0125】
スクリーニングの結果、抗ヒトMplアゴニスト抗体産生ハイブリドーマとして、免疫方法1によるハイブリドーマ7-10、免疫方法2によるハイブリドーマ4-49、免疫方法3による ハイブリドーマ6-4-50, 6-5-2の4クローンが選抜された。また、非アゴニスト抗体産生ハイブリドーマとして2-35(免疫方法1により得たもの)をコントロールとして選抜した。
【0126】
[実施例3] ハイブリドーマ培養上清からの精製抗体調製
ハイブリドーマの培養上清からの抗ヒトc-Mplモノクローナル抗体の精製は以下の方法で行った。抗体を含む培養上清をrmp Protein A(アマシャムファルマシアバイオテク社製)及び0.8×40cm カラム(バイオラッド社製)を用い、吸着緩衝液としてPBS、溶出緩衝液として0.02M グリシン緩衝液(pH 3)を用いてアフィニティー精製した。溶出画分は1M Tris (pH 9.0)を添加してpH7.2付近に調整した。調製された抗体溶液は、透析膜(10000カット、Spectrum Laboratories社製)を用いてPBSに置換し、孔径0.22μm のメンブランフィルターMILLEX-GV(ミリポア社製)でろ過滅菌し、精製抗ヒトc-Mplモノクローナル抗体を得た。精製抗体の濃度は280nmの吸光度を測定し、1mg/ml を1.4 OD として算出した。
【0127】
抗ヒトc-Mplモノクローナル抗体を含む培養上清の調製は以下の方法にて行った。
まず、抗体産生ハイブリドーマを10ng/ml Recombinant Human IL-6(R&D Systems社製)、10% Low IgG Fetal Bovine Serum(HyClone社製)含有eRDF培地(極東製薬社製)に馴化した。この馴化したハイブリドーマを凍結保存した。次に、その一部を、ウシインシュリン(5μg/ml、ギブコビーアールエル社製)、ヒトトランスフェリン(5μg/ml、ギブコビーアールエル社製)、エタノールアミン(0.01mM、シグマ社製)、亜セレン酸ナトリウム(2.5×10-5mM、シグマ社製)、10ng/ml Recombinant Human IL-6(R&D Systems社製)、1% Low IgG Fetal Bovine Serum(HyClone社製)含有eRDF培地(極東製薬社製)に馴化した。フラスコにて培養し、ハイブリドーマの生細胞率が90%になった時点で培養上清を回収した。回収した上清は、10μm と0.2μmのフィルター(ゲルマンサイエンス社製)に供し、きょう雑物を除去した。
【0128】
[実施例4] フローサイトメトリーによる抗ヒトc-Mpl抗体の結合活性評価
ハイブリドーマ培養上清もしくは精製抗体を用いたフローサイトメトリーによって、抗ヒトc-Mpl抗体の結合活性を測定した。手順は以下の通りである。細胞はFM3A-hMpl細胞もしくはヒトMpl発現FDCP2細胞(FDCP-hMpl)(FEBS Lett. 1996 Oct 21;395(2-3):228-34.参照)を用いた。
【0129】
反応あたり4×105個の細胞を50 μLのFACS staining medium(2% FBS, 0.1%NaN3, 1mM EDTA in PBS)に懸濁し、50μLのハイブリドーマの培養上清もしくは精製ヒト抗体(終濃度0.1-1μg/mL)を加え、氷上にて30分間反応させた。FACS staining mediumによる洗浄後、二次抗体であるR-phycoerythrin (RPE)標識ヤギ抗ヒトIgγ F(ab’)抗体(Southern Biotechnology 社製Cat#2043-09)を加え、再び氷上で30分遮光反応させた後、再び洗浄した。細胞をPropidium Iodide(PI)を加えたFACS staining mediumに懸濁し、解析を行った。解析はFACS Calibur(Becton Dickinson社製)にて行った。
【0130】
図1に各抗体の精製抗体を用いたフローサイトメトリーの結果を示す。各抗体はFDCP-hMpl細胞に結合したが、その親細胞であるFDCP2細胞(FDCP parent)には結合しなかった。従って、これらの抗体はヒトMpl特異的に結合していることが示された。
【0131】
[実施例5] UT7/TPO細胞を用いた抗ヒトc-Mpl抗体のアゴニスト活性評価
ハイブリドーマ上清もしくは精製抗体を用いて、UT7/TPO細胞増殖アッセイを行い、アゴニスト活性を評価した。UT7/TPO細胞はTPO依存性のヒト巨核球系細胞株である(Ozaki K et al. Blood. 1998 Dec 15;92(12):4652-62.参照)。通常は10%FBS, 5ng/mL PEG-rHuMGDF添加 Iscove’s Modified Dulbecco’s Medium(IMDM)で培養、維持した。細胞増殖アッセイの手順は以下の通りである。
【0132】
(1) UT7/TPO細胞培養液を50mLチューブに移し、遠心分離(1,500rpm, 5min, 4℃)により細胞をペレットにした。培地を除き、サイトカイン非添加, 10%FBS添加 IMDM培地(以下、増殖アッセイ用培地)でペレットを懸濁した。再び遠心し、新しい増殖アッセイ用培地で懸濁した。遠心、懸濁をもう一度繰り返した。
(2) 上記1.で増殖アッセイ用培地に懸濁した細胞を37℃, 5%CO2条件下で6時間培養した。
(3) 培養後、細胞を遠心してペレットにし、増殖アッセイ用培地にて懸濁した。このとき、細胞濃度を6×105cells/mLにあわせ、細胞懸濁液を、各ウェル50μLで96-well plateに播いた。
(4) 次に、ハイブリドーマ培養上清10μLに、増殖アッセイ用培地40μLを加え、各ウェルに添加する。精製抗体の場合は、増殖アッセイ用培地50μLに検体を終濃度の2倍の濃度で加え、各ウェルに添加した。
(5) 37℃,5%CO2にて、48時間培養する。
(6) WST-8試薬(同仁化学研究所社製)を10μL/wellで添加し、2時間培養する。
(7) 吸光マイクロプレートリーダー(TECAN社製SUNRISE RAINBOW)にて各ウェルの吸光度を測定する。(測定波長450nm, 参照波長600nm以上)
図2に7-10(図2A), 4-49(図2B), 6-4-50(図2C), 6-5-2(図2D)各精製抗体を用いたUT7/TPO細胞増殖アッセイにおける増殖曲線を示す。また、スクリーニングの結果得られた抗ヒトc-Mpl抗体のサブクラス、活性の強さ(UT7/TPO細胞増殖アッセイにおける50%有効濃度(EC50)、最大活性(Max))、各抗体が得られた実施例2に記載の免疫方法を表2に示す。
【0133】
【表2】
【0134】
[実施例6] コロニーアッセイ
ヒト臍帯血由来CD34+ 細胞を用いたCFU-Mkコロニー形成アッセイを行い、精製抗体のヒトプライマリー細胞に対する作用を検討した。アッセイにはMegaCultTM-C(Stem Cell Technologies社製Cat#04972)を使用した。手順は以下の通りである。
【0135】
(1) 検体を含んだ0.15mLのIMDMに、0.85mLのMegaCultTM-C培地を加え1mLとした。
(2) ヒト臍帯血より調製したCD34+細胞を1.1×105cells/mLの濃度で、IMDMに懸濁し、上記1.の培地が入ったチューブに0.05mLずつ添加した。
(3) 細胞を加えた各チューブをボルテックスで攪拌し、0.6mLの氷冷したコラーゲン液を加え、再びボルテックスで攪拌した。
(4) 上記3.までの細胞、検体混合液をチャンバースライドの各ウェルに0.75mLずつ添加した。
(5) チャンバースライドを、100mmペトリディッシュ中に入れた。乾燥を防ぐために、3mLの精製水を入れた35mmペトリディッシュを同じ100mmペトリディッシュ中に入れた。
(6) チャンバースライドが入ったペトリディッシュをインキュベーター中に静置し、37℃,5%CO2条件下で10〜12日間培養した。
(7) 培養後、固定液(メタノール:アセトン=1:3)にて固定した。
(8) 抗ヒトCD41抗体による免疫染色を行い、CFU-Mkコロニーを検出した。顕微鏡下でコロニー数をカウントし、各検体のCFU-Mkコロニー形成能を比較した。
【0136】
図3にコロニーアッセイの結果を示した。7-10_IgG1, 4-49_IgG1によってコロニー形成が誘導された。
【0137】
[実施例7] 抗体遺伝子クローニングと配列決定
組み換え抗体を作製するために、選抜された抗ヒトc-Mplアゴニスト抗体産生ハイブリドーマから抗体遺伝子、具体的には重鎖(H鎖)をコードするヒトIgγ cDNAおよび軽鎖(L鎖)をコードするヒトIgκ cDNAを単離し、配列を決定した。
1)各モノクローナル抗体のcDNA合成
各ハイブリドーマで発現するヒト抗体重鎖、及び軽鎖の抗体の可変領域を含むDNA断片を取得するために、ヒトIgγ、及びヒトIgκの各々の定常領域に特異的なプライマーを用いた5’RACE(5' rapid amplification of cDNA ends)法によるクローニングを行なった。具体的には、BD SMART RACE cDNA Amplification Kit(ベクトン・ディキンソン・バイオサイエンス・クローンテック社製)を用い、添付の説明書にしたがって実施した。
【0138】
cDNA合成の材料としては、ハイブリドーマ7-10, 4-49, 6-4-50, 6-5-2細胞にRNA抽出用試薬であるISOGEN(日本ジーン社製、日本)を添加し、取扱説明書にしたがってTotal RNAを精製した。精製したtotal RNAより各約1μgを鋳型として用いて、1st strand cDNAを作製した。
1st strand cDNA の合成は、
Total RNA 1μg/3μl
5’CDS 1μl
SMART Oligo 1μl
上記組成の反応液を70℃で2分間インキュベートした後、
5×Buffer 2μl
DTT 1μl
DNTP mix 1μl
PowerScript Reverse Transcriptase 1μl
を加え42℃で1.5時間インキュベートした。
さらに、50μlのTricine -EDTA Bufferを加えた後、72℃で7分間インキュベートし、1st strand cDNAを取得した。
【0139】
2)PCRによる重鎖遺伝子、軽鎖遺伝子の増幅と塩基配列の確認
2-1)PCRによる重鎖遺伝子、軽鎖遺伝子の増幅
ヒト抗体遺伝子のcDNAを増幅するために、ヒト抗体特異的配列を有する3’プライマー(具体的な配列は後記)とBD SMART RACE cDNA Amplification Kit で合成されたcDNAの5’末端に付加された配列に特異的にハイブリダイズする5’プライマー(Universal primer A mix)をPCR用のプライマーセットとして、またPCR用酵素としてKOD-Plus- DNAポリメラーゼ(東洋紡績(株)社製)を用いて、下記の反応液を調製してPCRに供した。
【0140】
sterile H2O 28μl
cDNA 2.5μl
KOD-Plus- buffer (10X) 5μl
dNTP Mix (2mM) 5μl
MgSO4 (25mM) 2μl
KOD-Plus- (1 unit/μl) 1μl
Universal primer A mix (UPM) (10X) 5μl
Gene specific primers (GSP)(10μM) 1.5μl
Total volume 50μl
重鎖遺伝子の増幅反応には、SMART RACE cDNA Amplification Kit付属のUPMプライマーとIgG1pプライマーを用い、他方、軽鎖遺伝子の増幅にはUPMプライマーとhk-2プライマーの各セットを使用した。
IgG1pプライマー:5’-TCTTGTCCACCTTGGTGTTGCTGGGCTTGTG-3’(配列番号18)
hk-2:5’-GTT GAA GCT CTT TGT GAC GGG CGA GC -3’(配列番号19)
また反応温度条件は次のとおりである。
94℃ /30 秒間、72℃/3分間のサイクルを5回反復、
94℃ /30 秒間、70℃/30秒間、72℃/3分間のサイクルを5回反復、
94℃ /30 秒間、68℃/30秒間、72℃/3分間のサイクルを25回反復した。
【0141】
さらに、この反応液2μlにTricine -EDTA Buffer 98μlを加えて希釈したもの5μlを鋳型とし、第一PCRよりも内側にプライマーを設定した第二PCR(Nested PCR)を実施した。PCR反応溶液の組成を次に示す。
【0142】
sterile H2O 30μl
第一PCR反応液(50倍希釈) 5μl
KOD-Plus- buffer (10X) 5μl
dNTP Mix (2mM) 5μl
MgSO4 (25mM) 2μl
KOD-Plus- (1 unit/μl) 1μl
Nested Universal primer A (NUP;10μM) 1μl
Gene specific primers (GSP)(10μM) 1μl
Total volume 50μl
上記反応のプライマーセットとして、重鎖遺伝子増幅用の場合は、NUPMプライマー(SMART RACE cDNA amplification Kit付属;ベクトン・ディキンソン・バイオサイエンス・クローンテック社製)とhh2プライマー(4-49、6-4-50、6-5-2の場合)、或はIgG2p_134(7-10の場合)を使用して、また、軽鎖遺伝子の増幅の場合は、UPMプライマーとhk-5プライマーを用いた。反応温度条件としては、94℃の初期温度で1分間の後、94℃/5秒間、68℃/10秒及び72℃/3分間のサイクルを20回反復、最後に72℃/7分間加熱した。
【0143】
2-2)抗体遺伝子の塩基配列決定
上記の方法で増幅した重鎖PCR断片(以下HV[C]と記載)は、重鎖の5’非翻訳領域、リーダー配列(分泌シグナル配列)、可変領域(HV)及び定常領域の一部([C])より構成される。同様に、軽鎖のPCR増幅断片(以下LV[C]と記載)は、軽鎖の5’非翻訳領域、リーダー配列(分泌シグナル配列)、可変領域(LV)及び定常領域の一部([C])より構成される。ここでリーダー配列(分泌シグナル)とは抗体の分泌に必要で、成熟抗体タンパク質からは切り離されるアミノ酸配列である。HV[C]断片およびLV[C]断片は、PCR反応液からエタノール沈殿で回収した後、アガロースゲル電気泳動で分離し、メンブランを用いるDNA精製キットであるQIAquick Gel Extraction Kit (キアゲン社製)にて精製した。精製したHV[C]増幅断片あるいはLV[C]増幅断片は、それぞれZero Blunt TOPO PCR Cloning Kit(インビトロジェン社製)のpCR 4 Blunt- TOPO ベクター(東洋紡績(株)製)にサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。DNA塩基配列決定のためにプライマーとして、M13-20FW及びM13RVを用いた。
hk-5:5’- AGG CAC ACA ACA GAG GCA GTT CCA GAT TTC-3’(配列番号20)
hh2プライマー:5’- GCT GGA GGG CAC GG TCA CCA CGC TG -3’(配列番号21)
IgG2p_134:5’- TGCACGCCGC TGGTCAGGGC GCCTGAGTTC C -3’(配列番号22)
【0144】
アゴニスト抗体7-10の重鎖可変領域、及び軽鎖可変領域をコードするDNA塩基配列、並びに重鎖可変領域及び軽鎖可変領域のアミノ酸配列をそれぞれ以下に示す。
<7-10重鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGAGTTGGGACTGAGCTGGATTTTCCTTTTGGCTATTTTAAAAGGTGTCCAGTGTGAAGTGCAGCTGGTGGAGTCTGGGGGAGGCTTGGTACAGCCTGGCAGGTCCCTGAGACTCTCCTGTGCAGCCTCTGGATTCACCTTTGATGATTATGCCATGCACTGGGTCCGGCAAGCTCCAGGGAAGGGCCTGGAGTGGGTCTCAGGTATTAGTTGGAATAGTGGTAGCATAGGCTATGCGGACTCTGTGAAGGGCCGATTCACCATCTCCAGAGACAACGCCAAGAACTCCCTGTATCTGCAAATGAACAGTCTGAGAGCTGAGGACACGGCCTTGTATTACTGTGCAAAAAATCTATGGTTCGGGGAGTTCCGTTACTGGTACTTCGATCTCTGGGGCCGTGGCACCCTGGTCACTGTCTCCTCA(配列番号23)
<7-10重鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
MELGLSWIFLLAILKGVQCEVQLVESGGGLVQPGRSLRLSCAASGFTFDDYAMHWVRQAPGKGLEWVSGISWNSGSIGYADSVKGRFTISRDNAKNSLYLQMNSLRAEDTALYYCAKNLWFGEFRYWYFD LWGRGTLVTV SS(配列番号24)
<7-10軽鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGACATGAGGGTCCCCGCTCAGCTCCTGGGGCTTCTGCTGCTCTGGCTCCCAGGTGCCAGATGTGCCATCCAGTTGACCCAGTCTCCATCCTCCCTGTCTGCATCTGTAGGAGACAGAGTCACCATCACTTGCCGGGCAAGTCAGGGCATTAGCAGTGCTTTAGCCTGGTATCAGCAGAAACCAGGGAAAGCTCCTAAGCTCCTGATCTATGATGCCTCCAGTTTGGAAAGTGGGGTCCCATCAAGGTTCAGCGGCAGTGGATCTGGGACAGATTTCACTCTCACCATCAGCAGCCTGCAGCCTGAAGATTTTGCAACTTATTACTGTCAACAGTTTAATAGTTACCCGCTCACTTTCGGCGGAGGGACCAAGGTGGAGATCAAA(配列番号25)
<7-10軽鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
MELGLSWIFLLAILKGVQCEVQLVESGGGLVQPGRSLRLSCAASGFTFDDYAMHWVRQAPGKGLEWVSGISWNSGSIGYADSVKGRFTISRDNAKNSLYLQMNSLRAEDTALYYCAKNLWFGEFRYWYFDLWGRGTLVTVSS(配列番号26)
【0145】
アゴニスト抗体4-49の重鎖可変領域、及び軽鎖可変領域をコードするDNA塩基配列、並びに重鎖可変領域及び軽鎖可変領域のアミノ酸配列をそれぞれ以下に示す。
<4-49重鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGAGTTGGGACTGAGCTGGATTTTCCTTGTGGCTATTTTAAAAGGTGTCCAGTGTGAAGAGCAGCTGGTGGAGTCTGGGGGAGGCTTGGTACAGCCTGGCAGGTCCCTGAGACTCTCCTGTACAGCCTCTGGATTCACCTTTGATGATTATGCCATGTACTGGGTCCGGCAAGTTCCAGGGAAGGGCCTGGAGTGGGTCTCAGGTATTAGTTGGAACAGTGGTAGCATAGGCTATGCGGACTCTGTGAAGGGCCGATTCACCGTTTCCAGAGACAACGCCAAGAACTCCCTGTATCTGCAAATGAACAGTCTGAGAGCTGAGGACACGGCCTTATATTACTGTGCAAAAGCCCTATGGTTCGGGGAGTTCCCCCACTACTACGGTATGGACGTCTGGGGCCAAGGGACCACGGTCACCGTCTCCTCA(配列番号27)
<4-49重鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
MELGLSWIFLVAILKGVQCEEQLVESGGGLVQPGRSLRLSCTASGFTFDDYAMYWVRQVPGKGLEWVSGISWNSGSIGYADSVKGRFTVSRDNAKNSLYLQMNSLRAEDTALYYCAKALWFGEFPHYYGMDVWGQGTTVTVSS(配列番号28)
<4-49軽鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGACATGAGGGTCCCCGCTCAGCTCCTGGGGCTTCTGCTGCTCTGGCTCCCAGGTGCCAGATGTGCCATCCAGTTGACCCAGTCTCCATCCTCCCTGTCTGCATCTGTAGGAGACAGAGTCACCATCACTTGCCGGGCAAGTCAGGGCATTAGCAGTACTTTAGCCTGGTATCAGCAGAAACCAGGGAAAGCTCCTAAGCTCCTGATCTATGATGCCTCCAGTTTGGAAAGTGGGGTCCCATCAAGGTTCAGCGGCAGTGGATCTGGGACAGATTTCACTCTCACCATCAGCAGCCTGCAGCCTGAAGATTTTGCAACTTATTACTGTCAACAGTTTAATAGTTACCCGTACACTTTTGGCCAGGGGACCAAGCTGGAGATCAAACGT(配列番号29)
<4-49軽鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
MDMRVPAQLLGLLLLWLPGARCAIQLTQSPSSLSASVGDRVTITCRASQGISSTLAWYQQKPGKAPKLLIYDASSLESGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQFNSYPYTFGQGTKLEIKR(配列番号30)
【0146】
アゴニスト抗体6-4-50の重鎖可変領域、及び軽鎖可変領域をコードするDNA塩基配列、並びに重鎖可変領域及び軽鎖可変領域のアミノ酸配列をそれぞれ以下に示す。
<6-4-50重鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGAATTGGGACTGAGCTGGATTTTCCTTTTGGCTATTTTAAAAGGTGTCCAGTGTGAAGTGCAGCTGGTGGAGTCTGGGGGAGGCTTGGTACAGCCTGGCAGGTCCCTGAGACTCTCCTGTGCAACCTCTGGATTCACCTTTGATAATTATGCCATGTACTGGGTCCGGCAAGCTCCAGGGAAGGGCCTGGAGTGGGTCTCAGGTATTAGTTGGAATAGTGGTGACATAGGCTATGCGGACTCTGTGAAGGGCCGATTCACCATCTCCAGAGACAACGCCAAGAACTCCCTGTATCTGCAAATGAACAGTCTGAGAGCTGAGGACACGGCCTTGTATTACTGTGCAAGGGATGCGGGGTTCGGGGAGTTCCACTACGGTCTGGACGTCTGGGGCCAAGGGACCACGGTCACCGTCTCCTCA(配列番号31)
<6-4-50重鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
MELGLSWIFLLAILKGVQCEVQLVESGGGLVQPGRSLRLSCATSGFTFDNYAMYWVRQAP
GKGLEWVSGISWNSGDIGYADSVKGRFTISRDNAKNSLYLQMNSLRAEDTALYYCARDAG
FGEFHYGLDVWGQGTTVTVSS(配列番号32)
<6-4-50軽鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGACATGAGGGTCCCCGCTCAGCTCCTGGGGCTTCTGCTGCTCTGGCTCCCAGGTGCCAGATGTGCCATCCAGTTGACCCAGTCTCCATCCTCCCTGTCTGCATCTGTAGGAGACAGAGTCACCATCACTTGCCGGGCAAGTCAGGGCATTAGCAGTGCTTTAGCCTGGTATCAGCAGAAACCAGGGAAAGTTCCTAAGCTCCTGATCTATGATGCCTCCAGTTTGGAAAGTGGGGTCCCATCAAGGTTCAGCGGCAGTGGATCTGGGACAGATTTCACTCTCACCATCAGCAGCCTGCAGCCTGAAGATTTTGCAACTTATTACTGTCAACAGTTTAATAGTTACCCGTGGACGTTCGGCCAAGGGACCAAGGTGGAAATCAAACGT(配列番号33)
<6-4-50軽鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
MDMRVPAQLLGLLLLWLPGARCAIQLTQSPSSLSASVGDRVTITCRASQGISSALAWYQQKPGKVPKLLIYDASSLESGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQFNSYPWTFGQGTKVEIKR(配列番号34)
【0147】
アゴニスト抗体6-5-2の重鎖可変領域、及び軽鎖可変領域をコードするDNA塩基配列、並びに重鎖可変領域及び軽鎖可変領域のアミノ酸配列をそれぞれ以下に示す。
<6-5-2重鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGAGTTGGGACTGAGCTGGATTTTCCTTTTGGCTATTTTAAAAGGTGTCCAGTGTGAAGTGCAACTGGTGGAGTGTGGGGGAGGCTTGGTACAGCCTGGCAGGTCCCTGAGACTCTCCTGTGCAGCCTCTGGATTCACCTTTGATGATTATGCCATGCACTGGGTCCGGCAAGCTCCAGGGAAGGGCCTGGAGTGGGTCTCAGGTATTAGTTGGAATAGTGGTAGTATAGGTTATGCGGACTCTGTGAAGGGCCGATTCACCATCTCCAGAGACAACGCCAAGAACTCCCTGTATCTGCAAATGAACAGTCTGAGAGCTGAGGACACGGCCTTGTATTACTGTGCAAAACCTATATGGTTCGGGGAGTGGGGAAACTACTACGGTATGGACGTCTGGGGCCAAGGGACCACGGTCACCGTCTCCTCA(配列番号35)
<6-5-2重鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
MELGLSWIFLLAILKGVQCEVQLVECGGGLVQPGRSLRLSCAASGFTFDDYAMHWVRQAPGKGLEWVSGISWNSGSIGYADSVKGRFTISRDNAKNSLYLQMNSLRAEDTALYYCAKPIWFGEWGNYYGMDVWGQGTTVTVSS(配列番号36)
<6-5-2軽鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGAAACCCCAGCGCAGCTTCTCTTCCTCCTGCTACTCTGGCTCCCAGATACCACCGGAGAAATTGTGTTGACGCAGTCTCCAGGCACCCTGTCTTTGTCTCCAGGGGAAAGAGCCACCCTCTCCTGCAGGGCCAGTCAGAGTGTTAGCAGCAGCTACTTAGCCTGGTACCAGCAGAAACCTGGCCAGGCTCCCAGGCTCCTCATCTATGATGCATCCAGCAGGGCCACTGGCATCCCAGACAGGTTCAGTGGCAGTGGGTCTGGGACAGACTTCACTCTCACCATCAGCAGACTGGAGCCTGAAGATTTTGCAGTGTATTACTGTCAGCAGTATGGTAGCTCACCGATCACCTTCGGCCAAGGGACACGACTGGAGATTAAACGT(配列番号37)
<6-5-2軽鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
METPAQLLFLLLLWLPDTTGEIVLTQSPGTLSLSPGERATLSCRASQSVSSSYLAWYQQKPGQAPRLLIYDASSRATGIPDRFSGSGSGTDFTLTISRLEPEDFAVYYCQQYGSSPITFGQGTRLEIKR(配列番号38)
【0148】
[実施例8] 組換え抗体発現ベクターの構築
上記の方法でハイブリドーマからクローニングした抗体の可変領域を、ヒト抗体発現用ベクターに組み込み、様々な定常領域を持った組換え抗体発現ベクターを作製した。
【0149】
ヒト抗体発現用ベクター、N5KG1-Val Lark(以下N5KG1と略記)(IDEC Pharmaceuticals社製, US patent 6001358参照)は、組換え抗体を動物細胞に発現させる目的で使用されるプラスミドベクターである。N5KG1の構造を図4Aに示した。N5KG1は、2つのCMV プロモーター/エンハンサーを持ち、それぞれの下流に重鎖および軽鎖可変領域遺伝子のクローニングサイトを持つ。さらにその下流には、ヒト重鎖定常領域(γ1)およびヒト軽鎖定常領域(κ)をコードする遺伝子配列をあらかじめ持っている。本ベクターの可変領域クローニングサイトに任意の重鎖および軽鎖可変領域(リーダー配列=分泌シグナル配列を含む)を、読み枠(フレーム)をあわせて組み込むことにより、軽鎖可変領域がヒトκ鎖定常領域と、重鎖可変領域がヒトγ1鎖定常領域と連結された抗体を発現させることができる。従って、本ベクターを導入された動物細胞は培養液中にIgG1抗体を産生する。
【0150】
同様に、発現ベクターN5KG4PE(IDEC Pharmaceuticals社製)は、IgG4PEの重鎖定常領域を持っている。IgG4PEとはIgG4に二つの変異(Ser228Pro, Leu235Glu)が入った配列である。Ser228Pro はIgG4の分子内架橋(S-S結合)による単量体形成を抑制する変異である。また、Leu235Gluは抗体依存性細胞傷害(Antibody-dependent cellular cytotoxicity: ADCC)活性を低減させる変異である。
【0151】
また、N5KG1のIgG1定常領域をIgG3に変換し、N5KG3を作製した。
さらに本実施例では、N5KG1, N5KG3, N5KG4PEをもとに、重鎖定常領域(特にヒンジ領域)に種々の改変を加えた発現ベクターを作製した。
【0152】
本実施例で定常領域に加えた改変は、第一に、抗体ドメイン間のサブクラスの入れ替えによるものである。抗体重鎖定常領域は、N末端側からCH1-ヒンジ-CH2-CH3というドメイン構造を持っている。本実施例では、このドメイン単位で各サブクラスの配列を組み合わせた重鎖定常領域を作製した。例えば、CH1およびヒンジ領域はヒトIgG3の配列であり、CH2およびCH3はヒトIgG1である重鎖定常領域を作製した。そして、このような重鎖定常領域を持つ抗体を、CH1/ヒンジ/CH2/CH3の順にサブクラスを表記し、IgG3/3/1/1と名付けた(以降はIgG3311というように”/”を省略して表記する)。また、例えば、ヒンジ領域はヒトIgG3の配列であり、CH1, CH2およびCH3はヒトIgG4PEである重鎖定常領域を作製した。このような重鎖定常領域を持つ抗体をIgG4344と名付けた。
【0153】
第二に、ヒトIgG3ヒンジ領域の改変体を作製した。抗体のヒンジ領域はアッパー(upper)ヒンジとミドル(middle)ヒンジに分けることができる。アッパーヒンジとはKabat EU numbering(Kabatら、Sequences of Proteins of Immunological Interest, 5thEd. Public Health Service, National Institute of Health, Bethesda, Md.(1991)) による表記で216以降、226よりN末端側の配列を指す。ミドルヒンジとは、同表記で226以降、231よりN末端側の配列を指す。ヒトIgG3のヒンジ領域は、アッパーヒンジが12アミノ酸、ミドルヒンジが50アミノ酸から成る。さらにミドルヒンジは、5アミノ酸と15アミノ酸×3回のリピート配列に分けられる(5+15x3=50)。本実施例では、IgG3ミドルヒンジの繰り返し配列を1回に短縮した変異体を作製した。このようなヒンジをG3h1と名付け、このタイプのヒンジを持つ抗体を、前述のドメイン単位の変異とあわせて、IgGx3xxh1(xは任意)というように表記した。
【0154】
また、IgG3のミドルヒンジ後半の繰り返し配列を無くした重鎖定常領域も作製した。このようなヒンジをG3uh(upper hingeの略)と名付け、IgGx3xxuhというように表記した。
さらにG3uhヒンジにL217SとR228Pの変異を加えた重鎖定常領域を作製した。この変異にはG3uhヒンジをよりIgG4PEの配列に近づける意図がある。これをG3uhm(upper hinge mutationの略)と名付け、これを持つ抗体をIgGx3xxuhmと表記した。
【0155】
図4Bに天然型のヒトイムノグロブリンと、IgG4PE, IgG4344, IgG4344h1, IgG4344uh, IgG4344uhmのヒンジ領域のアミノ酸配列を示す。
【0156】
本実施例では抗Mplアゴニスト抗体の可変領域を用いて、以下の定常領域を持つ抗体の発現ベクターを作製した。
【0157】
IgG1, IgG4PE, IgG3311, IgG3331, IgG3344, IgG3344h1, IgG4344, IgG4344h1, IgG4344uh, IgG4344uhm
【0158】
以下に各発現ベクターの作製法を記載する。
1)IgG1サブクラスの抗c-Mpl抗体発現ベクターの作製
1-1)抗ヒトc-Mpl抗体4-49_IgG1及び7-10_IgG1発現ベクターの作製
7-10と4-49については、N5KG1ベクターに最初に重鎖可変領域、次に、軽鎖可変領域の順に挿入して発現ベクターを作製した。
【0159】
発現ベクター作製の工程を図4Cに示す。7-10及び4-49それぞれのHV[C]およびLV[C]断片(実施例7に記載)を含むプラスミドDNAを鋳型として、末端に連結のための制限酵素部位(5’末側SalI、3’末側NheI)を付加するようにデザインしたプライマーセットを用いて、重鎖および軽鎖のリーダー配列と可変領域のDNAをKOD-Plus- DNAポリメラーゼによるPCRで増幅した。このPCRにより増幅された、重鎖および軽鎖のリーダー配列+可変領域を、それぞれHV断片、LV断片と表す。
【0160】
まず、N5KG1に7-10HV, 4-49HV断片の挿入を行った。HV断片増幅用のプライマーは、以下の通りである。
7-10;
HV断片5’用プライマー:40-3H5Sal
5’- AGAGAGAGAG GTCGACCACC ATGGAGTTGG GACTGAGCTG GATTT -3’(配列番号39)
HV断片3’用プライマー:40-3H3Nhe
5’- AGAGAGAGAG GCTAGCTGAG GAGACAGTGA CCAGGGTGCC A -3’(配列番号40)
4-49;
HV断片5’用プライマー:F24HSal
5’-AGAGAGAGAGGTCGACCACCATGGAGTTGGGACTGAGCTGGATTT -3’(配列番号41)
HV断片3’用プライマー:C15H3Nhe
5’- AGAGAGAGAGGCTAGCTGAGGAGACGGTGACCGTGGT-3’(配列番号42)
【0161】
反応温度条件としては、94℃の初期温度で1分間の加熱後、94℃/5秒間と68℃/45秒間のサイクルを35回反復し、最後に72℃/7分間加熱した。増幅されたDNA断片を制限酵素SalIとNheIで消化して、アガロースゲル電気泳動で約430bpのDNAを回収し精製した。他方、N5KG1ベクターについては制限酵素SalI、NheI処理を順次行った後、脱リン酸化のためにAlkaline Phosphatase (E. coli C75) (宝酒造社製、日本)にて処理した後に、アガロースゲル電気泳動とDNA精製キットで約8.9kbのDNAを回収した。これら2つの断片をT4 DNA ligase を用いてライゲーションして、大腸菌DH10Bへ導入して形質転換体を得た。得られた形質転換体のプラスミドDNAについてDNA塩基配列を解析して、重鎖定常領域の5’上流に、HV断片がフレームを合わせて挿入されたプラスミドDNA、N5KG1_7-10_Hv、及びN5KG1_4-49_Hvを取得した。
【0162】
続いて、これらHV断片が挿入されたプラスミドベクターにLV断片(軽鎖リーダー配列+可変領域)の挿入を行なった。LV[C]断片を含むプラスミドDNAを鋳型として、末端に連結のための制限酵素部位(5’末側BglII、3’末側BsiWI)を付加するようにデザインしたプライマーを用いて、LV断片をPCRで増幅した。LV増幅用プライマーは以下の通りである。
7-10;
LV断片5’用プライマー:165-1B_L18Bgl
5’- AGAGAGAGAGATCTCTCACCATGGACATGAGGGTCCCCGCTC-3’(配列番号43)
LV断片3’用プライマー:165_1B_L18_Bsi
5’- AGAGAGAGAG CGTACGTTTG ATCTCCACCT TGGTCCCTCC -3’(配列番号44)
4-49;
LV断片5’用プライマー:DNP_L1Bglp
5’- AGAGAGAGAGATCTCTCACCATGAGGGTCCCCGCTCAGCTC -3’(配列番号45)
LV断片3’用プライマー:A27_R_N202
5’- AGAGAGAGAGCGTACGTTTGATTTCCACCTTGGTCCCTTGGC-3’(配列番号46)
【0163】
反応は94℃の初期温度で1分間の加熱後、94℃/5秒間と68℃/45秒間のサイクルを35回反復し、最後に72℃/7分間の保温という条件で実施した。精製したLVの増幅DNA断片は、pCR4Blunt- TOPO ベクター(東洋紡績(株)製)にサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。DNA塩基配列決定のためにプライマーとして、M13-20FWとM13RVを用いた。挿入部分のDNA塩基配列解析を行い、鋳型としたLVと相違がなく、また、プライマー部分もデザインどおりの配列を有するプラスミドDNA(TOPO_7-10_Lv、及びTOPO_4-49_Lv)を選択した。次に、それぞれのDNAを制限酵素BglIIとBsiWIで消化して、アガロースゲル電気泳動で約400 bpのDNAを回収し精製した。その精製DNA断片と、制限酵素処理(BglII, BsiWI)及び、脱リン酸化処理を施した7-10或は4-49のHVが挿入されたベクター(約9.3kb)をT4 DNA ligase によりライゲーションした後、大腸菌DH10Bへ導入して形質転換体を得た。形質転換体について、DNA配列解析や制限酵素切断パターンを解析し、目的のプラスミドDNAを含むクローンを選択した。更に、得られた抗体発現プラスミドDNAの大量精製を行い、重鎖全領域と軽鎖全領域、及び、その挿入部位周辺のDNA塩基配列にクローニング工程での変異がないことを確認した。7-10_IgG1および4-49_IgG1発現ベクターは、各々N5KG1_7-10、及びN5KG1_4-49と名付けた。
図4CにN5KG1_7-10、及びN5KG1_4-49の作製工程を図示した。
【0164】
1-2)抗ヒトc-Mpl抗体6-4-50_IgG1及び6-5-2_IgG1抗体発現ベクターの作製
6-4-50、及び、6-5-2については、ヒト抗体発現用ベクターに最初に軽鎖可変領域、次に、重鎖可変領域の順に挿入して発現ベクターを作製した。
【0165】
6-4-50、及び、6-5-2抗体それぞれのLV[C]断片を含むプラスミドDNA(実施例7)を鋳型として、末端に連結のための制限酵素部位(5’末側BglII、3’末側BsiWI)を付加するようにデザインしたプライマーセットを用いて、LV断片(軽鎖のリーダー配列+可変領域)のDNAをKOD-Plus- DNAポリメラーゼによるPCRで増幅した。プライマーは以下の通りである。
6-4-50;
LV断片5’用プライマー:208LF
5’-AGAGAGAGAGATCTCTCACCATGGACATGAGGGTCCCCGCTCAGC -3’(配列番号47)
LV断片3’用プライマー:62LP3Bsi
5’- AGAGAGAGAGCGTACGTTTGATTTCCACCTTGGTCCCTTG -3’(配列番号48)
6-5-2;
LV断片5’用プライマー:A27_F
5’-AGAGAGAGAGATCTCTCACCATGGAAACCCCAGCGCAGCTTCTCTTC -3’(配列番号49)
LV断片3’用プライマー:202LR
5’- AGAGAGAGAGCGTACGTTTAATCTCCAGTCGTGTCCCTTGGC -3’(配列番号50)
【0166】
反応温度条件としては、94℃の初期温度で1分間の加熱後、94℃/5秒間と68℃/45秒間のサイクルを35回反復し、最後に72℃/7分間加熱した。増幅されたDNA断片を制限酵素BglIIとBsiWIで消化して、アガロースゲル電気泳動で約400bpのDNAを回収し精製した。他方、N5KG1ベクターについては制限酵素BglIIとBsiWI処理を順次行った後、脱リン酸化処理としてAlkaline Phosphatase (E. coli C75) (宝酒造社製)にて処理した後に、アガロースゲル電気泳動とDNA精製キットで約8.9kbのDNAを回収した。これら2つの断片をT4 DNA ligase を用いてライゲーションして、大腸菌DH10Bへ導入して形質転換体を得た。インサートDNAを含む形質転換体のプラスミドDNAについてDNA塩基配列を解析して、LV断片がN5KG1のヒト抗体軽鎖定常領域をコードする5’上流にフレームを合わせて挿入されたプラスミドDNA、N5KG1_6-4-50_Lv、及びN5KG1_6-5-2_Lvを取得した。引き続いて、このLV断片が挿入されたプラスミドベクターにHV断片(重鎖のリーダー配列+可変領域)の挿入を行なった。HV[C]を含むプラスミドDNA(実施例7)を鋳型として、末端に連結のための制限酵素部位(5’末側SalI、3’末側NheI)を付加するようにデザインしたプライマーを用いて、HV断片をPCRで増幅した。使用したプライマーは以下の通りである。
6-4-50;
HV断片5’用プライマー:50-5-7Hsal
5’- AGAGAGAGAG GTCGACCACC ATGGAATTGG GACTGAGCTG GATTTT -3’(配列番号51)
HV断片3’用プライマー:C15H3Nhe
5’ -AGAGAGAGAGGCTAGCTGAGGAGACGGTGACCGTGGT-3’(配列番号52)
6-5-2;
HV断片5’用プライマー:F24HSal
5’- AGAGAGAGAGGTCGACCACCATGGAGTTGGGACTGAGCTGGATTT-3’(配列番号53)
HV断片3’用プライマー:L66H3Nhe
5’- AGAGAGAGAGGCTAGCTGAGGAGACGGTGACCGTGGTC-3’(配列番号54)
【0167】
反応は94℃の初期温度で1分間の加熱後、94℃/5秒間と68℃/45秒間のサイクルを35回反復し、最後に72℃/7分間の保温という条件で実施した。精製したHV断片の増幅DNA断片は、一度pCR4Blunt- TOPO ベクター(東洋紡績(株)製)にサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。DNA塩基配列決定のためにプライマーとして、M13-20FWとM13RVを用いた。挿入部分のDNA塩基配列解析を行い、鋳型としたHVと相違がなく、また、プライマー部分もデザインどおりの配列を有するプラスミドDNA(TOPO_6-4-50_Hv、及びTOPO_6-5-2_Hv)を選択した。次に、それぞれのDNAを制限酵素SalIとNheIで消化して、アガロースゲル電気泳動で約430 bpのDNAを回収し精製した。並行して制限酵素処理(SalIとNheI)、及び、脱リン酸化処理した6-4-50或は6-5-2のLV断片が挿入されたベクター(約9.3kb)に、インサートしたいDNA断片をライゲーションした後、大腸菌DH10Bへ導入して、得られた形質転換体から目的のプラスミドDNAを持つクローンを選択した。こうして得られた抗体発現プラスミドDNAの大量精製を行い、重鎖全領域と軽鎖全領域、及び、その挿入部位周辺のDNA塩基配列にクローニング工程での変異がないことを確認した。6-4-50_IgG1及び6-5-2_IgG1抗体発現ベクターを、それぞれN5KG1_6-4-50、及びN5KG1_6-5-2と名付けた。
図4DにN5KG1_6-4-50、及びN5KG1_6-5-2の作製工程を図示した。
【0168】
2)IgG4PEサブクラスの抗ヒトc-Mpl抗体の作製
IgG4PEサブクラス抗体の発現ベクター作製には前述のN5KG4PEベクターを用いた。N5KG4PEのプラスミドDNAを制限酵素NheIとBamHIで切断して、重鎖定常領域を含む断片を精製して、抗c-Mpl抗体であるN5KG1_7-10、及びN5KG1_4-49の同じ制限酵素部位内につなぎ換えることにより、N5KG4PE_7-10、及びN5KG4PE_4-49を作製した。
【0169】
3) N5KG3の作製
ヒトIgG3用発現ベクターN5KG3はN5KG1のIgG1重鎖定常領域を下記配列のIgG3定常領域に置換することで作製した。
IgG3定常領域アミノ酸配列
STKGPSVFPLAPCSRSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYTCNVNHKPSNTKVDKRVELKTPLGDTTHTCPRCPEPKSCDTPPPCPRCPEPKSCDTPPPCPRCPEPKSCDTPPPCPRCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFKWYVDGVEVHNAKTKLREEQYNSTFRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYNTTPPMLDSDGSFFLYSKLTVDKSRWQQGNIFSCSVMHEALHNRYTQKSLSLSPGK*(配列番号55)
IgG3定常領域塩基配列
CTAGCACCAAGGGCCCATCGGTCTTCCCCCTGGCGCCCTGCTCCAGGAGCACCTCTGGGGGCACAGCGGCCCTGGGCTGCCTGGTCAAGGACTACTTCCCCGAACCGGTGACGGTGTCGTGGAACTCAGGCGCCCTGACCAGCGGCGTGCACACCTTCCCGGCTGTCCTACAGTCCTCAGGACTCTACTCCCTCAGCAGCGTGGTGACCGTGCCCTCCAGCAGTTTGGGCACCCAGACCTACACCTGCAACGTGAATCACAAGCCCAGCAACACCAAGGTGGACAAGAGAGTTGAGCTCAAAACCCCACTTGGTGACACAACTCACACATGCCCACGGTGCCCAGAGCCCAAATCTTGTGACACACCTCCCCCGTGCCCACGGTGCCCAGAGCCCAAATCTTGTGACACACCTCCCCCATGCCCACGGTGCCCAGAGCCCAAATCTTGTGACACACCTCCCCCGTGCCCAAGGTGCCCAGCACCTGAACTCCTGGGAGGACCGTCAGTCTTCCTCTTCCCCCCAAAACCCAAGGATACCCTTATGATTTCCCGGACCCCTGAGGTCACGTGCGTGGTGGTGGACGTGAGCCACGAAGACCCCGAGGTCCAGTTCAAGTGGTACGTGGACGGCGTGGAGGTGCATAATGCCAAGACAAAGCTGCGGGAGGAGCAGTACAACAGCACGTTCCGTGTGGTCAGCGTCCTCACCGTCCTGCACCAGGACTGGCTGAACGGCAAGGAGTACAAGTGCAAGGTCTCCAACAAAGCCCTCCCAGCCCCCATCGAGAAAACCATCTCCAAAACCAAAGGACAGCCCCGAGAACCACAGGTGTACACCCTGCCCCCATCCCGGGAGGAGATGACCAAGAACCAGGTCAGCCTGACCTGCCTGGTCAAAGGCTTCTACCCCAGCGACATCGCCGTGGAGTGGGAGAGCAATGGGCAGCCGGAGAACAACTACAACACCACGCCTCCCATGCTGGACTCCGACGGCTCCTTCTTCCTCTACAGCAAGCTCACCGTGGACAAGAGCAGGTGGCAGCAGGGGAACATCTTCTCATGCTCCGTGATGCATGAGGCTCTGCACAACCGCTACACGCAGAAGAGCCTCTCCCTGTCTCCGGGTAAATGA(配列番号56)
【0170】
4) IgG3311発現ベクターの作製
IgG3311発現ベクターは、N5KG3を鋳型として、プライマーlinkH, 13ch1-Rで98℃1秒、60℃30秒、72℃30秒の反応を15回行った。同時に、N5KG1を鋳型として、プライマー13ch1、linkH2を用いて、98℃1秒、60℃30秒、72℃30秒の反応を15回行った。増幅したDNA断片をPCR purification kit で精製し、2つの精製DNA断片を等量混合したのち、98℃1秒、60℃30秒、72℃30秒の反応を5回行い、プライマーlinkH、linkH2を加えて、15回反応した。増幅したDNA断片をNheI, BamHIで切断し、N5KG1ベクターのIgG1定常領域と置き換えた。この発現ベクターをN5KG3311と名づけた。
linkH: GGG TAC GTC CTC ACA TTC AGT GAT CAG(配列番号57)
13ch1-R: GTC TTC GTG GCT CAC GTC CAC CAC CAC GCA(配列番号58)
13ch1: TGC GTG GTG GTG GAC GTG AGC CAC GAA GAC(配列番号59)
linkH2: TGA TCA TAC GTA GAT ATC ACG GC(配列番号60)
【0171】
5) IgG3331発現ベクターの作製
IgG3331発現ベクターは、N5KG3を鋳型として、プライマーlinkH, CH3consRで98℃1秒、60℃30秒、72℃30秒の反応を15回行った。同時に、N5KG1を鋳型として、プライマーCH3cons、linkH2を用いて、98℃1秒、60℃30秒、72℃30秒の反応を15回行った。増幅したDNA断片をPCR purification kit で精製し、2つの精製DNA断片を等量混合したのち、98℃1秒、60℃30秒、72℃30秒の反応を5回行い、プライマーlinkH、linkH2を加えて、15回反応した。増幅したDNA断片をNheI, BamHIで切断し、N5KG1ベクターのIgG1定常領域と置き換えた。この発現ベクターをN5KG3331と名づけた。
CH3consR: GGTGTACACCTGTGGCTCTCGGGGCTGCCC(配列番号61)
CH3cons: GGGCAGCCCCGAGAGCCACAGGTGTACACC(配列番号62)
以下に、IgG3344, IgG3344h1, IgG4344, IgG4344h1, IgG4344uh, IgG4344uhmの作製法を記す。これらについては、各定常領域をPCR法によって増幅し、それをクローニングしたプラスミドを作製した。ついで、それら改変定常領域をN5KG1_7-10等のIgG1定常領域と置換した。
【0172】
6) IgG3344, IgG3344h1定常領域の作製
IgG3344発現ベクターは、N5KG3331およびN5KG4PEを鋳型として、PCRを用いた変異導入(Overlap Extention法による部位特異的変異導入法)によって以下の手順で行った。
【0173】
N5KG3331を鋳型として、プライマーとしてG3G4_P1_FとG3G4_P2_Rを用いて94℃の初期温度で1分間の加熱後、94℃/15秒間、55℃/10秒間、そして68℃/1分間のサイクルを35回反復し、最後に72℃/7分間加熱した。同時に、前述発現ベクターN5KG4PEを鋳型として、プライマーとしてG3G4_P3_FとG3G4_P4_Rを用いて同条件にてPCRを実施した。増幅したDNA断片をアガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kit (キアゲン社製)にて精製した。これらの精製DNA断片を等量混合した後、最初の5サイクルについては、2DNA断片のオーバーラップ部分をアニーリングさせて94℃の初期温度で1分間の加熱後、94℃/10秒間、55℃/10秒間、そして68℃/1.5分間という5サイクルの伸長反応を行なった後、全長を増幅させる為に反応液にプライマーとしてG3G4_P1_FとG3G4_P4_Rを加えて、更に94℃/5秒間と68℃/2分間のサイクルを20回反復し、最後に72℃/7分間の加熱を行なった。G3G4_P1_FとG3G4_P4_Rのプライマーは、ヒト抗体定常領域のコード部分を切りだして、抗体の発現ベクターの該当する部分に変更可能とするために制限酵素部位(G3G4_P1_FにNheI、G3G4_P4_RにBamHI部位)が存在する。増幅したPCR断片は、アガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kitにて精製した。精製した増幅断片は、Zero Blunt TOPO PCR Cloning Kit(インビトロジェン社製)のpCR 4 Blunt- TOPO ベクターにサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。塩基配列の解析よりIgG3344およびIgG3344h1定常領域を持つクローンを選択した。
G3G4_P1_F:5’-AGAGAGGCTA GCACCAAGGG CCCATCG-3’(配列番号63)
G3G4_P2_R:5’-GAACTCAGGT GCTGGGCACC TTGGGCACG-3’(配列番号64)
G3G4_P3_F:5’- CCAAGGTGCC CAGCACCTGA GTTCGAGGGG GGA -3’(配列番号65)
G3G4_P4_R:5’- AGAGAGGGAT CCTCATTTAC CCAGAGACAG GGA -3’(配列番号66)
【0174】
7) IgG4344定常領域の作製
IgG4344発現ベクターの作製においては、N5KG3331を鋳型として、プライマーとしてG434_P5_FとG434_P6_Rを用いて94℃の初期温度で1分間の加熱後、94℃/15秒間、55℃/10秒間、そして68℃/1分間のサイクルを35回反復し、最後に72℃/7分間加熱した。同時に、N5KG4PEを鋳型として、プライマーとしてG434_P7_FとG3G4_P2_Rを用いて同条件にてPCRを実施した。増幅したDNA断片をアガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kit (キアゲン社製)にて精製した。これらの精製2DNA断片と、前述したN5KG4PEを鋳型として、G3G4_P3_FとG3G4_P4_Rをプライマーとして増幅・精製しておいたDNA断片の計3種類のOverlap Extention反応を実施した。すなわち、3種類のDNA断片のオーバーラップ部分をアニーリングさせて94℃の初期温度で1分間の加熱後、94℃/10秒間、55℃/10秒間68℃/1.5分間のサイクルを5回反復して伸長した後、全長を増幅させる為に反応液にプライマーとしてG434_P5_FとG3G4_P4_Rを加えて、94℃/5秒間と68℃/2分間のサイクルを20回反復し、最後に72℃/7分間の加熱を行なった。増幅したPCR断片はQIAquick Gel Extraction Kitにて精製した後、pCR 4 Blunt- TOPO ベクターにサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。塩基配列の解析よりIgG4344定常領域を持つクローンを選択した。
G434_P5_F:5’- AGAGAGGCTA GCACCAAGGG GCCATCC -3’(配列番号67)
G434_P6_R:5’- GGTTTTGAGC TCAACTCTCT TGTCCACCTT GGTGTTGC -3’(配列番号68)
G434_P7_F:5’- GTGGACAAGA GAGTTGAGCT CAAAACCCCA CTTGGTGACA C -3’(配列番号69)
【0175】
8) IgG4344h1定常領域の作製
IgG4344h1発現ベクターの作製は、N5KG4344を鋳型として、プライマーとしてG434_P5_F, G434_P6_Rを用いて98℃の初期温度で10秒間の加熱後、98℃/10秒間、55℃/30秒間、そして72℃/1分間のサイクルを7回反復し、次いで98℃/10秒間、68℃/1分間のサイクルを30回反復して、最後に72℃/7分間加熱した。PCR用酵素としては、Pyrobest DNA Polymerase(タカラバイオ株式会社)を使用した。同時に、N5KG3344h1を鋳型として、プライマーとしてG434_P7_FとG3G4_P4_Rを用いて同条件にてPCRを実施した。増幅したDNA断片をアガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kit (キアゲン社製)にて精製した。これらの精製DNA断片を等量混合した後、2DNA断片のオーバーラップ部分をアニーリングさせて、98℃の初期温度で10秒間の加熱後、98℃/10秒間、55℃/30秒間、そして72℃/1分間のサイクルを7回反復して伸長した後、全長を増幅させる為に反応液にプライマーとしてG434_P5_FとG3G4_P4_Rを加えて、更に98℃/10秒間と68℃/1分間のサイクルを30回反復し、最後に72℃/7分間の加熱を行なった。増幅したPCR断片は、アガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kitにて精製した。精製した増幅断片は、pCR 4 Blunt- TOPO ベクターにサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。塩基配列の解析よりG4344h1定常領域持つクローンを選択した。
【0176】
9) IgG4344uh定常領域の作製
G4344uhの作製は、N5KG4344を鋳型として、プライマーとしてG434_P5_Fと17-1Rを用いて98℃の初期温度で10秒間の加熱後、98℃/10秒間、50℃/30秒間、そして72℃/1分間のサイクルを5回反復し、次いで98℃/10秒間、55℃/30秒間、そして72℃/1分間のサイクルを5回反復し、98℃/10秒間、68℃/1分間のサイクルを25回反復して、最後に72℃/7分間加熱した。PCR用酵素としては、Pyrobest DNA Polymerase(タカラバイオ株式会社)を使用した。並行して、N5KG3344h1を鋳型として、プライマーとして17-2FとG3G4_P4_Rを用いて同条件にてPCRを実施した。増幅したDNA断片をアガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kitにて精製した。これらの精製DNA断片を等量混合した後、2DNA断片のオーバーラップ部分をアニーリングさせて、98℃の初期温度で10秒間の加熱後、98℃/10秒間、68℃/1分間のサイクルを5回、さらに98℃/10秒間、55℃/30秒間、そして72℃/1分間のサイクルを5回反復して伸長した後、全長を増幅させる為に反応液にプライマーとしてG434_P5_FとG3G4_P4_Rを加えて、更に94℃/30秒間と68℃/1分間のサイクルを30回反復し、最後に72℃/7分間の加熱を行なった。増幅したPCR断片は、アガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kitにて精製した。精製した増幅断片は、pCR 4 Blunt- TOPO ベクターにサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。塩基配列の解析よりIgG4344uh定常領域を持つクローンを選択した。
17-1R:5’- AGGTGCTGGG CACCGTGGGC ATGTGTGAGT TGT -3’(配列番号70)
17-2F:5’- CACACATGCC CACGGTGCCC AGCACCTGAG TTC -3’(配列番号71)
【0177】
10) IgG4344uhm定常領域の作製
IgG4344uhm発現ベクターの作製は、N5KG4PEを鋳型として、プライマーとしてG434_P5_Fと17m-1Rを用いて98℃の初期温度で10秒間の加熱後、98℃/10秒間、50℃/30秒間、そして72℃/1分間のサイクルを5回反復し、次いで98℃/10秒間、55℃/30秒間、そして72℃/1分間のサイクルを5回反復し、98℃/10秒間、68℃/1分間のサイクルを25回反復して、最後に72℃/7分間加熱した。PCR用酵素としては、Pyrobest DNA Polymeraseを使用した。並行して、N5KG4PEを鋳型として、プライマーとして17m-2FとG3G4_P4_Rを用いて同条件にてPCRを実施した。増幅したDNA断片をアガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kitにて精製した。これらの精製DNA断片を等量混合した後、2DNA断片のオーバーラップ部分をアニーリングさせて、94℃/30秒間、55℃/30秒間、そして72℃/1分間のサイクルを7回反復して伸長した後、全長を増幅させる為に反応液にプライマーとしてG434_P5_FとG3G4_P4_Rを加えて、更に94℃/30秒間と68℃/1分間のサイクルを30回反復し、最後に72℃/7分間の加熱を行なった。増幅したPCR断片は、アガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kitにて精製した。精製した増幅断片は、pCR 4 Blunt- TOPO ベクターにサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。塩基配列の解析よりIgG4344uhm定常領域を持つクローンを選択した。
17m-1R:5’- TGTGTGAGTT GTGTCACCAA GTGGGGTTTT GGACTCAACT CTCTTGTCCA CCTTGGT -3’(配列番号72)
17m-2F:5’- ACCCCACTTG GTGACACAAC TCACACATGC CCACCATGCC CAGCACCTGA GTTCGAG -3’(配列番号73)
図4Eに各種改変重鎖のアミノ酸配列を示した。
【0178】
11) 各種改変重鎖定常領域を持った抗体発現ベクターの作製
作製した各種改変重鎖定常領域を持つプラスミドDNAを、制限酵素NheI, BamHIで切断後、定常領域配列を精製分離した。続いて、抗ヒトc-Mpl抗体発現ベクターN5KG1_7-10, N5KG1_4-49, N5KG1_6-4-50, N5KG1_6-5-2を同酵素で処理し、定常領域を置換した。
図4Fに7-10_IgG4344uhmの重鎖配列を示した。
図4Gに7-10_IgG4344uhmの軽鎖配列を示した。
【0179】
[実施例9] 抗ヒトc-Mpl抗体の293F細胞での一過性発現、及び精製
実施例8で作製した発現ベクターDNAをEndoFree Plasmid Kit(キアゲン社)にて調製し、FreeStyleTM 293 Expression System(インビトロジェンライフテクノロジー社)を用いて浮遊性293細胞(インビトロジェンライフテクノロジー社)に導入して、一過性発現により各抗体を含む培養上清を得た。孔径0.22μm のメンブランフィルター(MILLIPORE 製)で濾過した培養上清(IgGとして約500μg)を抗体精製用アフニティーカラムであるHiTrap rProtein A FF(カラム体積1ml)(アマシャムバイオサイエンス社)にチャージし、PBS (-)で洗浄後20 mMクエン酸バッファー(pH3.4)により溶出し、200 mMリン酸バッファー(pH7.0)を含むチューブに回収した。
【0180】
[実施例10] 組換え型抗体の作製
構築した抗体発現ベクターを宿主細胞に導入して、抗体発現細胞を作製した。発現のための宿主細胞には、dhfr欠損のCHO DG44 細胞(IDEC Pharmaceuticals Corporation)を無血清培地であるEX-CELL325PF(JRH)に馴化した細胞株を用いた。宿主細胞へのベクターの導入はエレクトロポレーションにより実施した。エレクトロポレーションは抗体発現ベクター約2μgを制限酵素AscIで線状化し、Bio-Rad electrophoreterをもちいて350V、500μFの条件で、4×106個のCHO細胞に遺伝子を導入し、96well culture plateに播種した。ベクターの導入処理後、G418を添加して培養を継続した。コロニーを確認した後、抗体発現株を選別した。選択したCHO細胞株をEX-CELL325-PF培地(JRH)(2 mM glutamine、100 units/ml penicillin、100 μg/ml streptomycin、 hypoxanthine and thymidine (HT) サプリメント(1:100) (Invitrogen)を含む)で5% CO2条件下で培養した。培養上清をMabselect Protein A カラム(Amersham Pharmacia Biotech, Co., Ltd.)に吸着後、PBSで洗浄して、20mMクエン酸-Na、50mM NaCl (pH3.4)バッファーで溶出した。溶出液は50mM Phosphate-Na, pH7.0 にて中和した。Milli-Q水にて、約1.5倍に希釈してConductivityを4.0ms/cm以下に調製した。次に、Q-Sepharose(Hitrap Q HP)(Amersham Pharmacia Biotech, Co., Ltd.)と、SP-Sepharose(HiTrap SP FF)(Amersham Pharmacia Biotech, Co., Ltd.)を連結したカラムに、サンプルをチャージして吸着後、20mMリン酸ナトリウム緩衝液(pH5.0)にて洗浄後、1×PBSバッファーにて溶出した。調製された抗体溶液は、孔径0.22μmのメンブランフィルターMILLEX-GV (ミリポア社製)でろ過滅菌した。精製した抗体の濃度は280nmの吸光度を測定し、1mg/mL を1.4 ODとして算出した。
【0181】
組換え型の改変抗体を用いて、UT7/TPOアッセイ(実施例5)における活性を測定した。4-49_IgG1に比べ、IgG3311とIgG3331は活性の増強が見られた(図5A)。7-10_IgG4344uhmおよび4-49_IgG4344uhmはPEG-rHuMGDFと同等の活性を示した。
【0182】
表3に各種改変抗体の活性をまとめた。全てのアゴニスト抗体に関して、定常領域改変による活性の増強が見られた。7-10, 4-49に関しては、IgG1とIgG4PEは同等の活性であったが、IgG4344uhmはIgG4PEに比べ高活性であった。IgG4344uhmは、IgG4PEのアッパーヒンジ部7アミノ酸配列中のC末側4−7位がIgG3のアッパーヒンジ12アミノ酸配列中の4−12位の配列に置き換わっている(図4B参照)。従って、この部分が活性の増強に重要であると考えられる。
【0183】
【表3】
【0184】
[実施例11] アゴニスト抗体によるシグナル伝達
TPOが受容体であるc-Mplに結合すると、細胞内タンパク質のリン酸化が起こる。TPOによって活性化する主要な経路は、Jak-STAT, Ras-MAPK, PI3K-Aktの3つが知られている。アゴニスト抗体によるc-Mpl下流のリン酸化シグナル伝達の解析を行った。解析は、リン酸化タンパク質特異的な抗体を用いたウェスタンブロット法で行った。以下に使用した抗体を列記する。抗STAT5(Cell Signaling社製, Cat#9352), 抗phospho-STAT5(Cell Signaling社製, Cat#9351L), 抗JAK2(Upstate社製, Cat#06-255), 抗phospho-JAK2(Upstate社製, Cat#07-606), 抗Erk1/2(Cell Signaling社製, Cat#9272), 抗phospho-Erk1/2(Cell Signaling社製, Cat#9271L), 抗Akt(Cell Signaling社製, Cat#9102), 抗phospho-Akt(Cell Signaling社製, Cat#9101S)。
【0185】
これら抗体を使用し、以下の手順でアッセイを行った。
1) UT7/TPO細胞をサイトカイン非添加IMDM培地で洗浄し、6時間培養した。
2) 培養後、細胞を1×106cells/mLに調製し、6-wellプレートに2mL/wellで播いた。
3) ウェルに、アゴニスト抗体または陽性コントロールとしてPEG-rHuMGDFを添加し、細胞を刺激した。
4) 最短5分〜最長2時間の刺激時間の後に細胞を回収し、氷冷PBSにて細胞を洗浄した。
5) 遠心分離で細胞をペレットにし、上清を除去後、PhosphoSafeTM Extraction Reagent(Novagen社製, Cat#71296)でペレットを溶解したのち、再度遠心して、上清(細胞抽出液)を回収した。
6) 上記5.の細胞抽出液を用いて、ウェスタンブロット法でリン酸化タンパク質を検出した。
【0186】
結果を図6に示す。アゴニスト抗体7-10G4344uhmおよび4-49G4344uhmによって、TPOシグナルと同様の経路のリン酸化が見られた(図6A)。抗体6-5-2については、IgG1はJak2, STAT5のリン酸化が観察されなかったが、IgG3344はJak2, STAT5のリン酸化が見られた(図6B)。
【0187】
[実施例12] ヒト血小板に対するプライミング作用検討
TPOはそれ自体では血小板凝集を起こさないが、ADP等の凝集惹起物質による血小板凝集を促進する作用(プライミング作用)がある。アゴニスト抗体によるヒト血小板プライミング作用を以下の手順で調べた。
【0188】
1) 抗凝固剤として、3.1%(w/v)クエン酸三ナトリウムを10分の1容量加えた、健常人の末梢血を遠心分離(140g, 15min)し、多血小板血漿(Platelet Rich Plasma、以下 PRP)を調製した。
2) さらに遠心分離(2500g, 15min)して血球成分を沈殿させ、血漿を採取した。
3) PRPに含まれる血小板数を測定し、血漿を用いて血小板数を3×105/μLにあわせた。
4) 上記3.で調製した血小板浮遊液100μLに検体を加え、攪拌しながら3分間インキュベーションした。
5) 30μM ADP(SIGMA社製)を5μL添加し、血小板凝集による濁度の低下を測定した。測定にはMCメディカル社製, Hematracer801を用いた。
【0189】
結果を図7に示す。ADP添加条件下で、アゴニスト抗体によるプライミング作用が観察された。抗体単独(ADPなし)では、血小板凝集は起こらなかった。
【0190】
[実施例13] カニクイザル投与
アゴニスト抗体をカニクイザルに投与し、血小板数の変動を解析した。使用個体のTPOに対する反応性を確かめるため、初日(Day0)にPEG-rHuMGDF(10μg/kg)を静脈内投与し3週間経過を観察した後、初回投与後21日目に精製アゴニスト抗体7-10G4PE(個体A)7-10G3344h1(個体B)を用量1mg/kgで静脈内投与した。
【0191】
結果を図8に示す。A,B両方の個体でPEG-rHuMGDFによる一過性の血小板上昇が見られた。個体Bではアゴニスト抗体7-10G3344h1投与後に、血小板数の上昇が見られた。また、抗体投与による重篤な毒性は見られなかった。
【0192】
[実施例14] ヒト臍帯血移植モデルにおける作用検討
実施例10で作製したアゴニスト抗体が、ヒト臍帯血移植モデルにおけるヒト造血系の構築を促進することを確認するため、以下の手順で実験を行った。
【0193】
・ NOG(NOD/SCID/IL2-γR KO)マウス(実験動物中央研究所(日本国神奈川県川崎市)より購入)に移植前処置として放射線照射(2グレイ)を行い、ヒト臍帯血由来CD34+細胞を1,000ないし10,000個、尾静脈より注入移植した。
・ 移植後一日目に最初の被検物質投与を行い、その後、週に一回投与を実施した。群構成と各々の被検物質および投与量は下記のとおりである。匹数は各群6匹、投与は腹腔内投与で行った。また、毎週の投与時に体重測定を行った。
【0194】
<群構成,被検物質,投与量>
I: 移植数10,000, PBS(コントロール)投与
II: 移植数1,000, PBS投与
III: 移植数10,000, 抗体7-10G4344uhm投与, 100μg/head/week
IV: 移植数1,000, 抗体7-10G4344uhm投与, 100μg/head/week
V: 移植数10,000, TPO (PEG-rHuMGDF)投与, 5μg/head/week
VI: 移植数1,000, TPO (PEG-rHuMGDF)投与, 5μg/head/week
・ 移植前1日と移植後2、4、6週に末梢血の解析を行った。末梢血解析の手順は下記のとおりである。
【0195】
<末梢血解析手順>
・ キャピラリ−を用いて、マウス眼窩静脈より末梢血(約70μL)を採取した。
・ KX-21自動血球解析システム(Sysmex社製)を用いて血球数を測定した。
・ ヒト血小板および白血球のキメラ率を調べるため、以下のAおよびBに挙げた各抗体の組み合わせで染色し、FACS Caliburで解析した。A(血小板解析用):PE標識−抗ヒトCD41抗体(Dako社製 R7058)+FITC標識−抗マウスCD41抗体(BD Pharmingen社製#553848)、B(白血球解析用): APC標識−抗ヒトCD45抗体(ベックマン・コールター社製IM2473)+FITC標識−抗マウスCD45抗体(BD Pharmingen社製#553080)。また、解析の際に定量用の蛍光ビーズ(フローカウントビーズ)を加え、一定量の血液を解析できるようにした。
・ 血小板・白血球ともに、ヒト細胞数/(ヒト細胞数+マウス細胞数)×100(%)、という式でキメラ率を算出した。末梢血中の全血小板数とキメラ率を乗じることにより、ヒト血小板数を算出した。
・ 6週目にマウスを屠殺し、大腿骨から骨髄細胞を採取した。それを用いてコロニーアッセイを行い、ヒトの巨核球系(MK)および赤血球系(E)、顆粒球・マクロファージ系(GM)の前駆細胞の数を測定した。巨核球系前駆細胞(CFU-Mk)を検出するコロニーアッセイは、培養中にTPO(50ng/mL)とSCF(100ng/mL)を加えて行った。37℃、5%CO2条件下で12日間の培養を行った。コロニーの検出は実施例6と同様に抗ヒトCD41抗体を用いて行った。赤血球系、顆粒球・マクロファージ系の前駆細胞を検出するコロニーアッセイは、Methocult system(Stem Cell Technologies社製)を用いて、培養中にEPO(4IU/mL), SCF(100ng/mL), IL-3(20ng/mL), GM-CSF(10ng/mL)を加えて行った。37℃, 5%CO2, 5%O2条件下で14日間の培養を行った。培養後、顕微鏡下でコロニーのカウントを行った。
【0196】
図9A、9B及び9Cに本実験の結果を図示した。
移植後6週の時点において、抗体投与群では、他群に比べ、末梢血ヒト血小板数が有意に高かった(図9A)。これにより、アゴニスト抗体7-10G4344uhmは臍帯血移植時の血小板回復を促進することが示唆された。さらに抗体投与群では、骨髄中のヒト赤血球系および顆粒球・マクロファージ系前駆細胞の数が有意に高かった(図9B)。またマウス白血球に対するヒト白血球の割を示すCD45キメラ率においても有意に高かったことから、抗体投与群においてヒト白血球が増加していることがわかる(図9C)。このことは7-10G4344uhmが巨核球系のみならず、他系列の細胞の生着も促進できる可能性を示唆している。
【0197】
これらの知見から、アゴニスト抗体は、血球が巨核球系・赤血球系・顆粒球・マクロファージ系に分かれるより上流の細胞に作用していることが示唆された。Mplが造血幹細胞に発現しているという知見とあわせて考えると、アゴニスト抗体は造血幹細胞の増殖を促進している可能性が高い。
【0198】
ちなみに本実験ではTPO投与群では同様の効果が見られなかった。これに対しては、TPOがマウス造血細胞にも作用するため、TPO投与群では骨髄中でヒト細胞とマウス細胞の競合が起こり、純粋にヒト細胞に対する作用を観察できていない可能性を考慮しなければならない。本アゴニスト抗体は、ヒトMplにしか作用しないことを特徴とすることから、Mplを介したシグナルがヒト臍帯血造血幹細胞の増幅に効果があることを、in vivoで初めて明らかにすることができた。
【0199】
[実施例15] ヒンジ改変抗体の抗原性解析
本発明のアゴニスト抗体はヒンジ部分の改変によって活性を増強していることを特徴としているが、改変による抗原性の上昇が懸念された。そこで、ヒンジ改変体である7-10G4344uhmのアミノ酸配列をもとに、コンピュータ上での抗原性予測を実施した。
【0200】
体内に投与された外来タンパク質は樹状細胞、マクロファージ等の抗原提示細胞(APC)に取り込まれ、分解された後、ペプチドが主要組織適合抗原複合体(MHC)クラスII分子(ヒトの場合HLA class II, HLA-DR, DQ, DP)によって抗原提示される。APCによって提示されたペプチドはT細胞受容体(TCR)によって認識され、T細胞を活性化させる。活性化されたT細胞(ヘルパーT細胞)は同抗原を認識する抗体を発現するB細胞を活性化し、外来タンパク質に対する抗体が産生される。この機構の中で、ペプチドとMHCクラスII分子との親和性が抗原性を規定する大きな要因となっている。ヒトのMHCクラスII分子には多くの型(多型)があり、同じペプチドであってもクラスII分子の型によって親和性は大きく異なることが知られている。
【0201】
そこで、7-10G4344uhmとIgG4PEの定常領域を持つ別のヒト抗体のアミノ酸配列を、様々な型のヒトHLA-DR, DQ, DP分子との親和性を解析した(HLA分子のデータベースおよび、解析のアルゴリズムはAlgoNomics社より提供された)。
【0202】
その結果、ヒンジ改変による新たなエピトープ出現はなかった。本改変抗体を医薬品として利用する際に抗原性の問題はないことが示唆された。
【0203】
[実施例16] ヒトMplトランスジェニックマウスへの抗体投与
本発明の抗体はマウスMplには交叉しないため、薬効を測定するため、外来遺伝子としてヒトMplを導入したトランスジェニック(Tg)マウスを作製し、抗体を投与する実験を行った。まず、マウスMplのプロモータ領域5.5kbをPCRで増幅し、pBluescriptプラスミドベクターにクローニングした。次に、ヒトMplの翻訳領域および3’側非翻訳領域をPCRで増幅し、マウスMplプロモーター下流に連結した。このコンストラクトをC57BL/6マウス受精卵に注入し、受精卵を仮親に戻し出産させた。生後3週経過した時点で尻尾からゲノムDNAを抽出し、PCRでTgマウスを選別した。得られたTgマウス個体をC57BL/6と交配し、系統化した。骨髄でのヒトMplの発現解析を行った。
【0204】
その結果、複数のヒトMplを持つTgマウス系統が得られた。そのうち、39L系統の骨髄ではヒトMplが発現していることがRT-PCRで確認された。39L系統のマウスを用いて抗体の薬効を確認した。
【0205】
アゴニスト抗体7-10G4344uhmを単回投与(3又は10μg/mL)し、末梢血中の血小板数の推移を、KX-21自動血球解析装置を用いて調べた。末梢血の採取は眼窩静脈より行い、毎週測定を行った。陽性コントロールとしてTPO(PEG-rHuMGDF)を用いた。群構成は以下のとおりである(各群6匹)。
【0206】
I: 7-10G4344uhm 10μg投与群
II: 7-10G4344uhm 3μg投与群
III: TPO 3μg投与群
IV: PBS投与群
VI: 野生型マウス 7-10G4344uhm 10μg投与群
結果を図10に示す。抗体投与群、TPO投与群で血小板が増加した。TPO投与群は投与後2週間でほぼベースラインに戻った。これに対して抗体投与群は投与後一ヶ月を経過しても血小板数は上昇したままであった。この結果から、アゴニスト抗体は血中で非常に安定であり、単回投与で長期にわたって血小板造血を促進できることが示唆された。このことから、特にアゴニスト抗体は慢性血小板減少症の治療に適していることが示唆された。
【0207】
[実施例17] 7-10G4344uhm軽鎖変異体の活性評価
アゴニスト抗体7-10の軽鎖可変領域(7-10VL)のフレームワーク領域に変異を導入し、結合活性及びアゴニスト活性に与える影響を調べた。軽鎖の変異体はアゴニスト抗体4-49の軽鎖(V104L)、アゴニスト6-4-50の軽鎖のアミノ酸を1つ置換したもの(A43V,G100Q)の3種である。これら変異軽鎖と7−10G4344uhm重鎖を組み合わせた抗体を作製したところ、全て、結合活性、アゴニスト活性ともにオリジナルの7-10G4344uhmと同等であった。一方で、アゴニスト抗体7-10の軽鎖可変領域の相補性決定領域(CDR領域)に変異(Y94F)を導入した場合、結合活性、アゴニスト活性ともに10分の1程度に低下した。この結果から軽鎖アミノ酸配列には、ある程度の自由度があることが示された。
各変異体の軽鎖アミノ酸配列及び7-10VLのアミノ酸配列は、以下のとおりである。なお、変異部は太字と下線で示した。
【0208】
【化1】
【0209】
結合活性解析:各抗体の濃度を1, 0.1, 0.01μg/mLに調製し、FM3A-hMpl細胞を用いたフローサイトメトリーを行った。実験方法は実施例4に記載の方法で行った。コントロールに抗DNP(ジニトロフェノール)抗体(サブクラスIgG4;ヒト抗体)を用いた。軽鎖変異抗体は7-10G4344uhmと同等の結合活性を示した(図11)。
アゴニスト活性解析: 実施例5に記載の方法で、UT-7/TPO細胞を用いた細胞増殖アッセイを行った。軽鎖変異抗体は7-10G4344uhmと同等のアゴニスト活性を示した(図12)。
【産業上の利用可能性】
【0210】
本発明により、種々の血小板減少症に対する治療薬として利用できる、抗ヒトcMplアゴニストヒト抗体が提供された。また他のアゴニスト抗体にも応用し得る、高い安全性と薬効を期待し得る、抗体の定常領域が提供された。
【0211】
本発明により、全長抗体でヒトトロンボポエチン受容体(c-Mpl)を活性化しうるヒトc-Mplに対するアゴニスト抗体が提供される。このアゴニスト抗体は、種々の血小板減少症に対する治療薬として使用可能であり、医療産業上多大な寄与が期待される。
【0212】
本明細書で引用した全ての刊行物、特許および特許出願をそのまま参考として本明細書にとり入れるものとする。
【図面の簡単な説明】
【0213】
【図1】図1は、アゴニスト抗体結合活性を示す。FDCP-hMpl細胞とFDCP2細胞(FDCP parent)を用いたフローサイトメトリー(実施例2)で、図示した各抗体の結合活性を調べた。各抗体はヒトc-Mpl特異的に結合することが示された。
【図2】図2は、UT7/TPOアッセイ結果を示す。図A〜Dに示した各精製抗体(IgG1)のUT7/TPO細胞増殖アッセイ(実施例5)における各抗体の増殖曲線を示した。
【図3】図3は、CFU-Mkアッセイ結果を示す。これは、ヒト臍帯血CD34+細胞を用いたコロニー形成アッセイ(実施例6)の結果である。
【図4A】図4Aは、組換え抗体作製に関するN5KG1ベクターの構造を示す。図中、Cはサイトメガロウイルスのプロモーター/エンハンサー、Bはウシ成長ホルモンポリアデニル化領域、N1はネオマイシンホスホトランスフェラーゼのエクソン1、Kはヒトイムノグロブリンκ定常領域、G1はヒトイムノグロブリンγ1定常領域、BTはマウスβグロブリン主要プロモーター、N2はネオマイシンホスホトランスフェラーゼのエクソン2、Dはジヒドロ葉酸レダクターゼ、VHは重鎖可変領域、VLは軽鎖可変領域をそれぞれ表す。
【図4B】図4Bは、組換え抗体作製に関する、天然型のヒトイムノグロブリンと、IgG4PE, IgG4344, IgG4344h1, IgG4344uh, IgG4344uhmのCH1領域とヒンジ領域(アッパーヒンジ及びミドルヒンジ)のアミノ酸配列を示す。
【図4C−1】図4C(すなわち、図4C-1〜図4C-3)は、組換え抗体作製に関する、N5KG1_7-10, N5KG1_4-49発現ベクター作製工程図を示す。
【図4C−2】図4C(すなわち、図4C-1〜図4C-3)は、組換え抗体作製に関する、N5KG1_7-10, N5KG1_4-49発現ベクター作製工程図を示す。
【図4C−3】図4C(すなわち、図4C-1〜図4C-3)は、組換え抗体作製に関する、N5KG1_7-10, N5KG1_4-49発現ベクター作製工程図を示す。
【図4D−1】図4D(すなわち、図4D-1〜図4D-3)は、組換え抗体作製に関する、N5KG1_6-4-50, N5KG1_6-5-2発現ベクター作製工程を示す。
【図4D−2】図4D(すなわち、図4D-1〜図4D-3)は、組換え抗体作製に関する、N5KG1_6-4-50, N5KG1_6-5-2発現ベクター作製工程を示す。
【図4D−3】図4D(すなわち、図4D-1〜図4D-3)は、組換え抗体作製に関する、N5KG1_6-4-50, N5KG1_6-5-2発現ベクター作製工程を示す。
【図4E】図4Eは、組換え抗体作製に関する、図示した各種改変重鎖の定常領域の配列を示す。
【図4F−1】図4F(すなわち、図4F-1〜図4F-2)は、組換え抗体作製に関する、7-10G4344uhm重鎖の核酸およびアミノ酸配列を示す。
【図4F−2】図4F(すなわち、図4F-1〜図4F-2)は、組換え抗体作製に関する、7-10G4344uhm重鎖の核酸およびアミノ酸配列を示す。
【図4G】図4Gは、組換え抗体作製に関する、7-10G4344uhm軽鎖の核酸およびアミノ酸配列を示す。
【図5】図5は、ヒンジ改変抗体の活性を示す。A:UT7/TPO細胞増殖アッセイにおける4-49G1, 4-49G3311, 4-49G3331の活性。B:UT7/TPO細胞増殖アッセイにおける7-10G4344uhm, 4-49G4344uhmの活性。
【図6A】図6Aは、シグナル伝達解析(実施例11)における、アゴニスト抗体7-10G4344uhm, 4-49G4344uhmのシグナル伝達解析結果を示す。
【図6B】図6Bは、シグナル伝達解析(実施例11)における、アゴニスト抗体6-5-2G1, 6-5-2G3344のシグナル伝達解析結果を示す。
【図7】図7は、ヒト血小板プライミング作用を示す。これは、実施例12記載の試験の結果を示す。アゴニスト抗体7-10G3311, 4-49G3311によるヒト血小板プライミング作用が示された。また、アゴニスト抗体のみ(ADP非添加)では血小板の凝集は起こらないことが示された。
【図8】図8は、カニクイザルへのアゴニスト抗体投与による血小板数の変化を示したグラフである。実施例13に記載のように、カニクイザルにアゴニスト抗体を投与し、血小板数をモニターした。矢印は一回目(PEG-rHuMGDF)および二回目(アゴニスト抗体)の投与日を示す。
【図9A】図9Aは、NOG臍帯血移植モデルマウスにおいてCD34+細胞を1,000個(右図)又は10,000個(左図)を移植した後に被検物質を投与したときの末梢血ヒト血小板数の経時的推移を示す。図中、Preは投与前の血小板数を示す。
【図9B】図9Bは、NOG臍帯血移植モデルマウスにおいてCD34+細胞を1,000個(右図)又は10,000個(左図)を移植した後に被検物質を投与したときの、移植後6週における骨髄ヒト前駆細胞数(colony count; GM+E+GEM)を示す。前駆細胞数は、巨核球系以外の合計細胞数であり、GMは顆粒球・マクロファージ系、Eは赤血球系、GEMはColony-forming-unit-granulocyte-macrophage-erythroidを表わす。結果は、平均±標準偏差(mean±SD)で表わしている。また、ベヒクル(Vehicle)はコントロールとしてのPBS(リン酸緩衝塩水)を表わし、また、NTは非処理(Non-treated)を表わす。
【図9C】図9Cは、NOG臍帯血移植モデルマウスにおいてCD34+細胞を1,000個(右図)又は10,000個(左図)を移植した後に被検物質を投与したときの、移植後6週における末梢血ヒト細胞キメラ率を示す。ベヒクル(Vehicle)は対照としてのPBS(リン酸緩衝塩水)を表わし、また、NTは非処理(Non-treated)を表わす。
【図10】図10は、ヒトMpl Tgマウスへのアゴニスト抗体投与後の血小板数の経日推移を示す。対照として、該TgマウスへのTPO又はベヒクル(PBS)の投与、並びに、非Tg(野生型;Non-Tg)マウスへの7-10G4344uhm(10μg)の投与実験を行い、その結果も示した。結果は、平均±SEMで表わした。
【図11】図11は、アゴニスト抗体7-10G4344uhmの軽鎖変異抗体のFM3A-hMpl細胞への結合性を示す。
【図12】図12は、アゴニスト抗体7-10G4344uhmの軽鎖変異抗体に関するUT-7/TPO細胞を用いた細胞増殖アッセイの結果を示す。
【配列表フリーテキスト】
【0214】
配列番号11:変異ヒンジドメインUH2G3uhm
配列番号12〜16:プライマー
配列番号18〜22:プライマー
配列番号39〜54:プライマー
配列番号57〜73:プライマー
配列番号74:G3344h1
配列番号75:G3344
配列番号76:G4344
配列番号77:G4344h1
配列番号78:G4344uh
配列番号79:G4344uhm
配列番号80:G4PE
配列番号81:7-10G4344uhm H鎖
配列番号82:7-10G4344uhm H鎖
配列番号83:7-10G4344uhm L鎖
配列番号84:7-10G4344uhm L鎖
配列番号85:7-10VL_V104L(変異体)
配列番号86:7-10VL_G100Q(変異体)
配列番号87:7-10VL_A43V(変異体)
【技術分野】
【0001】
本発明は、ヒトトロンボポエチン受容体(別名:ヒトc-Mpl)に対するアゴニスト抗体の製造方法に関する。
【0002】
抗ヒトc-Mplアゴニスト抗体は、臨床上血小板を増多させる必要のある患者/疾患に対する治療剤、特に血小板減少症治療剤の有効成分となりうる。
【背景技術】
【0003】
<TPOとTPO受容体について>
トロンボポエチン(Thrombopoietin:TPO)は、生体内で巨核球および血小板の増殖を促進する造血因子である。ヒトTPOは全長332アミノ酸残基からなる糖タンパク質であり、その活性にはN末端側の配列が重要であることが知られている。細胞膜上のTPO受容体と結合することで機能を発揮する。
【0004】
c-Mplは、現在知られている唯一のTPO受容体である。ヒトc-Mplは、シグナルペプチドを含むと635アミノ酸、成熟型は610アミノ酸からなる膜一回貫通型糖タンパク質であり、I型のサイトカイン受容体ファミリーに属している。ヒトc-MplのメッセンジャーRNAおよびタンパク質の配列は既に報告されている(Genbank: NM_005373, NP_005364参照)。同じファミリーの分子としてエリスロポエチン受容体(EpoR)や、G-CSF受容体(G-CSFR)、インターロイキン3受容体(IL-3R)などを挙げることができる。ヒトc-Mplは細胞外に2つのCRH(Cytokine Receptor Homologue)ドメイン(N末端側からCRH1, CRH2と呼ばれる)を持ち、そのなかにサイトカイン受容体ファミリーに特徴的なWSXWSモチーフを含んでいる。細胞内ドメインにはBox1, Box2と呼ばれる二つの配列があり、これらはシグナル伝達に必須である。TPOはCRH1に結合し、c-Mplを二量体化することでシグナルを入れると考えられているが、詳細な結合および活性化の様式は解明されていない。c-Mplが二量体化すると、細胞内ドメインに結合したシグナル伝達キナーゼが活性化され、リン酸化シグナルが細胞内に伝えられる。TPO-MplシグナルによってJak-STAT, PI3K-Akt, Ras-MAPKの各経路が動かされることが知られている。TPOまたはc-Mplを欠損したマウスは、血小板数が野生型マウスの10-20%程度まで減少することが報告されており、TPO-Mplシステムが血小板数を調節する主要なシステムであることを示している。c-Mplの発現は、巨核球のみならず、さらに未分化な造血前駆細胞や造血幹細胞にも認められる。骨髄中のc-Mpl陽性細胞画分は、c-Mpl陰性画分に比べ、高い骨髄再構築能を持っていることが示されており、また、c-Mpl欠損マウスでは巨核球・血小板だけではなく造血幹細胞も減少していることが判っている (非特許文献1,2参照)。これらの知見から、TPO-Mplシステムが造血系に幹細胞レベルから関与していることが示唆されている。
【0005】
TPOがクローニングされて以来、血小板減少症の治療薬としての利用が期待され、これまでに二種類の組換え型TPOの臨床試験が行われている(非特許文献3参照)。完全長のヒトTPO(rhTPO)と、ヒトTPOの活性部位であるN末端側163アミノ酸のペプチド配列をポリエチレングリコール(PEG)化したPEG-rHuMGDF(Pegylated Recombinant Human Megakaryocyte Growth and Development Factor)である。臨床試験において、これらの組換え型TPOは健常人および特発性血小板減少性紫斑病(Idiopathic Thrombocytopenic Purpura:ITP)患者の血小板を増加させることに成功している。また、骨髄非破壊的な化学療法に伴う血小板減少症を軽減させる効果が示されている。さらに、例数は少ないながらも、再生不良性貧血(Aplastic Anemia:AA)、骨髄異形成症候群(Myelodysplastic Syndrome:MDS)の患者に対しても組換え型TPOの有効性が報告されている(非特許文献4,5)。
【0006】
<c-Mplアゴニスト抗体について>
TPOと同様にc-Mplを介してシグナルを入れる性質を持ちながら、分子としては全く異なる種々のTPOミメティクスが研究されている(非特許文献6,7参照)。ミメティクスには大きく分けて、ペプチド性低分子、非ペプチド性低分子、抗体由来分、アゴニスト抗体などが知られている。
【0007】
公知の抗c-Mplアゴニストヒト抗体として、12B5, 12E10, 12D5(特許文献1参照)を挙げることができる。これらは全長抗体(Whole antibody、 例えばWhole IgG)の形態でプライマリーヒト細胞に対する活性を持たない。なお、本明細書で使用するプライマリーヒト細胞とは、TPOに対して高感受性を有する樹立された特殊な株化細胞やTPO受容体遺伝子を遺伝子組換え技術により導入し該受容体を高発現させた細胞ではなく、ヒト臍帯血やヒト骨髄由来のCD34+細胞等の本来生体内でTPOの作用対象となっている細胞を意味する。
【0008】
また、公知のアゴニストマウス抗体として、BAH-1(特許文献2,非特許文献8参照), VB22B(特許文献3参照)を挙げることが出来るが、マウス抗体はヒト血中において抗原性を示すことが知られており、医薬品としての利用には適さない。また、一般に、アゴニスト抗体を、全長抗体で、例えばCDR grafting等の方法を用いて、その活性を保ったままヒト化することは困難である(特許文献3, 非特許文献9参照)。従って、上記のような公知のアゴニスト抗体が存在するとしても、プライマリー細胞に作用するアゴニストヒト抗体の創出は容易ではない。
【0009】
また前述のTPOミメティクスで述べた抗体由来の低分子も、ある種のアゴニスト抗体であり、抗体の一部を利用改変したDiabody,一本鎖(Fv)2(sc(Fv)2)が報告されている(特許文献1,3参照)。しかしながら、この方法によって作出された改変抗体は、分子の大幅な改変による抗原性が懸念されるうえ、全長抗体に比べ血中半減期が短縮される等、医薬品として利用するためには課題が多い。
【0010】
以上、全長抗体は低抗原性や血中半減期の長さ等、医薬品として有用な性質を備えている一方、上述のように、全長抗体で充分な活性を持つアゴニストヒト抗体を作製するのは容易ではない。
【0011】
そこで、以下に述べるように、本発明者らは、抗体の構造に大幅な改変を加えることなく、充分な活性を持ったアゴニストヒト抗体の取得を試み、その取得に至った。さらに抗体のヒンジ領域に改変を加え、アゴニスト活性を増強することに成功した。本発明によって作出された抗体は血小板減少症治療薬としての利用に適していると考えられる。
【0012】
【非特許文献1】宮崎洋,「トロンボポエチンの将来展望」, Japanese Journal of Transfusion Medicine, 46(3) , 311-316, 2000
【非特許文献2】Murone Mら, Stem Cell 16:1-6, 1998
【非特許文献3】Kuter DJら, Blood 100(10):3457-69, 2002
【非特許文献4】Yonemura Yら, Int J Hemat (82) 307-309, 2005
【非特許文献5】Komatsu Nら, Blood 96 296a, 2000
【非特許文献6】Broudy VCら, Cytokine. 25(2):52-60, 2004
【非特許文献7】Wang Bら, Clin Pharmacol Ther. 76(6):628-38, 2004
【非特許文献8】Deng Bら, Blood 92(6):1981-1988, 1998
【非特許文献9】Ji Hee Sonら, Journal of Immunological Methods 286:187-201, 2004
【特許文献1】国際公開第WO 99/10494号パンフレット
【特許文献2】国際公開第WO 99/03495号パンフレット
【特許文献3】国際公開第WO 2005/056604号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0013】
本発明の目的は、新規な抗ヒトc-Mplアゴニスト抗体を提供することである。
本発明において、抗体とは、これまで全長抗体では困難であった、ヒトc-Mplに対し天然のリガンドであるTPOとほぼ同等のシグナルを入れることができ、プライマリーヒト細胞に対する増殖刺激活性を有する抗体である。
【0014】
本発明の第2の目的は、抗体の断片化を行わずにアゴニスト抗体の活性を高める技術を提供することにより、抗体分子が本来持つ長半減期、低抗原性といった医薬品として望ましい性質を有する新規な抗ヒトc-Mplアゴニスト抗体を提供することである。
【課題を解決するための手段】
【0015】
上記課題を解決するため、本発明者らは、抗ヒトc-Mplアゴニスト抗体の研究に鋭意取り組んだ。その結果、全長抗体で、天然リガンドとほぼ同等のシグナルを生じ、ヒトプライマリー細胞に対する活性を有するヒト抗体を取得した。また、取得したアゴニスト抗体につき更に鋭意検討を行い、抗体の断片化を行わずにそのアゴニスト活性を高める改変方法を見出し、本発明を完成するに至った。
【0016】
すなわち、本発明は以下の特徴を含む。
1.ヒトトロンボポエチン受容体に対するアゴニスト抗体
本発明にかかるヒトトロンボポエチン受容体に対するアゴニスト抗体は、以下の(1)〜(6)に示す抗体を含む。
(1)抗体の定常領域が以下の(i)〜(iii):
(i) ヒト抗体の重鎖定常領域及び軽鎖定常領域のアミノ酸配列、
(ii) ヒト抗体サブクラス間で重鎖定常領域のドメインを入れ替えた重鎖定常領域のアミノ酸配列、及び、ヒト抗体軽鎖定常領域のアミノ酸配列、或いは
(iii) 前記(i)又は(ii)のアミノ酸配列において1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
のいずれかのアミノ酸配列を有し、且つ抗体の可変領域がヒトトロンボポエチン受容体に結合し該受容体を活性化する能力を有する、並びに、以下の(a)及び/又は(b):
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、10,000ng/mL以下の濃度でコロニー形成を誘導すること、
(b) UT7/TPO細胞を用いた増殖能測定法において、活性が下記の構造を有するPEG-rHuMGDFの50%以上であり、かつ50%有効濃度(EC50)が100nM以下であること、
の性質を有する、ヒトトロンボポエチン受容体に対するアゴニスト抗体。
【0017】
本明細書中、ヒト抗体サブクラスは、IgG1, IgG2, IgG3及びIgG4を含む。ヒトイムノグロブリンの定常領域等の配列は、例えばNCBIホームページ(GenBank、UniGeneなど)から入手可能であり、例えばヒトIgG1重鎖定常領域については登録番号J00228、ヒトIgG2重鎖定常領域については登録番号J00230、ヒトIgG3重鎖定常領域については登録番号X03604、ヒトIgG4重鎖定常領域については登録番号K01316、ヒト軽鎖κ定常領域については登録番号V00557, X64135, X64133など、ヒト軽鎖λ定常領域については登録番号X64132, X64134などを含むことができる。
【0018】
本明細書中、ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法とは、後述の実施例6に記載した測定法を意味し、コロニー形成に必要な抗体の濃度は、この測定法に基づき求めることができる。
【0019】
本明細書中、UT7/TPO細胞を用いた増殖能測定法とは、後述の実施例5に記載した測定法を意味し、増殖活性及びEC50はこの測定法に基づき求めることができる。
【0020】
本明細書中、PEG-rHuMGDFとは、ヒトTPOのアミノ末端受容体結合ドメインを含むトランケート化タンパク質をコードするcDNAを含有するプラスミドで形質転換された大腸菌(E. coli)を使用して産生される(Ulichら, Blood 86:971-976 (1995))ポリペプチドを抽出し、リフォールディングし、精製し、そのアミノ末端にポリエチレングリコール(PEG)部分を共有結合させた配列番号1のアミノ酸配列を有する分子であり、以下の構造:
PEG-NH-SPAPPACDLRVLSKLLRDSHVLHSRLSQCPEVHPLPTPVLLPAVDFSLGEWKTQMEETKAQDILGAVTLLLEGVMAARGQLGPTCLSSLLGQLSGQVRLLLGALQSLLGTQLPPQGRTTAHKDPNAIFLSFQHLLRGKVRFLMLVGGSTLCVRRAPPTTAVPS-COOH
を有するものである。
【0021】
本明細書中、ヒトc-Mplを活性化するとは、ヒトc-Mplを発現している細胞においてヒトc-Mplに係る細胞内シグナル伝達を起こすことをいう。
【0022】
本明細書中、数個なる用語は、2〜約10個、例えば2〜9個、2〜8個、2〜7個、2〜6個、2〜5個、2〜4個、又は2〜3個の整数を意味する。
【0023】
(2)上記コロニー形成測定法によりコロニー形成を誘導する活性を有し、及び/又は、上記UT7/TPO細胞を用いた増殖能測定法により細胞増殖活性を有する抗体のうち、コロニー形成活性については10,000ng/mL以下の濃度でコロニー形成を誘導する活性を有する、好ましくは1,000ng/mL以下の濃度で該活性を有する、より好ましくは100ng/mL以下の濃度で該活性を有する上記(1)の抗体。
【0024】
(3)上記細胞増殖活性がPEG-rHuMGDFの50%以上、好ましくは70%以上、より好ましくは90%以上の活性を示し、50%有効濃度(EC50)が、100nM以下, 好ましくは10nM以下、より好ましくは1nM以下のものである上記(1)の抗体。
【0025】
(4)上記コロニー形成測定法及び上記増殖能測定法の両者の測定方法によりそれぞれ以下の活性を示す上記(1)の抗体。
(i) 以下の(a)および(b)の性質を有する、ヒトトロンボポエチン受容体に対するアゴニスト抗体。
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、10,000ng/mL以下の濃度でコロニー形成を誘導する。
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性が下記の構造を有するPEG-rHuMGDFの50%以上であり、かつ50%有効濃度(EC50)が100nM以下である。
(ii) 以下の(a)および(b)の性質を有する、ヒトc-Mplに対するアゴニスト抗体。
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、1,000ng/mL以下の濃度でコロニー形成を誘導する。
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性がPEG-rHuMGDFの70%以上であり、かつEC50が10nM以下である。
(iii) 以下の(a)および(b)の性質を有する、ヒトc-Mplに対するアゴニスト抗体。
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、100ng/mL以下の濃度でコロニー形成を誘導する。
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性がPEG-rHuMGDFの90%以上であり、かつEC50が1nM以下である。
【0026】
(5)以下の(a)〜(h)からなる群から選択される重鎖アミノ酸配列可変領域及び軽鎖アミノ酸配列可変領域を有する上記(1)の抗体。(カッコ内に、各可変領域配列が由来する後述実施例の抗体の名称を示す。)
(a) 配列番号2で示されるアミノ酸配列を含む重鎖可変領域及び配列番号3で示されるアミノ酸配列を含む軽鎖可変領域。(抗体の名称:7-10)
(b) 配列番号4で示されるアミノ酸配列を含む重鎖可変領域及び配列番号5で示されるアミノ酸配列を含む軽鎖可変領域。(抗体の名称:4-49)
(c) 配列番号6で示されるアミノ酸配列を含む重鎖可変領域及び配列番号7で示されるアミノ酸配列を含む軽鎖可変領域。(抗体の名称:6-4-50)
(d) 配列番号8で示されるアミノ酸配列を含む重鎖可変領域及び配列番号9で示されるアミノ酸配列を含む軽鎖可変領域。(抗体の名称:6-5-2)
(e) 配列番号2で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号3で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列
を含む軽鎖可変領域。
(f) 配列番号4で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号5で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域。
(g) 配列番号6で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号7で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域。
(h) 配列番号8で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号9で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域。
【0027】
(6)ヒトc-Mplに対するアゴニスト抗体がヒト抗体である、上記(1)〜(5)に記載の抗体。
【0028】
2.重鎖改変アゴニスト抗体
本発明に係る重鎖改変アゴニスト抗体とは、以下のものを含む。
(1)重鎖定常領域のアッパーヒンジ部が、以下の(a)〜(b)のアミノ酸配列から選択されるいずれか1つのアミノ酸配列を有し、並びに、該重鎖定常領域のミドルヒンジ部以降のC末端側がヒトイムノグロブリンG4のアミノ酸配列或いは該G4のアミノ酸配列においてADCC(抗体依存性細胞障害)活性等に係るアゴニスト抗体として好ましくない性質に関する部位を変異させたアミノ酸配列を有するアゴニスト抗体。
(a) 配列番号10で示されるアミノ酸配列。
(b) 配列番号11で示されるアミノ酸配列。
【0029】
本明細書の中で用いるアッパーヒンジ(Upper hinge)とは、Kabat EU 番号付け(Kabatら、Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institute of Health, Bethesda, Md.(1991))による表記で216位以降、226位よりN末端側の配列を指す。ミドルヒンジ(Middle hinge)とは、同表記で226位以降、231位よりN末端側の配列を指す。ヒトイムノグロブリンG4を含む各サブタイプにつき、アッパーヒンジ、ミドルヒンジ及びその前後のアミノ酸配列を図4Bに示す。図中、CH1はアッパーヒンジに隣接するCH1領域の一部、CH2はCH2領域中のローワーヒンジ(lower hinge)と称される部分である。
【0030】
(2)上記重鎖定常領域のミドルヒンジ部以降のC末端側が、ヒトイムノグロブリンG4のアミノ酸配列において、Kabat EU番号付けにおける228位セリンがプロリンに且つ235位ロイシンがグルタミン酸に置換されたアミノ酸配列を有する重鎖を含む抗体。
【0031】
(3)以下の(i)又は(ii)に示された、ヒトc-Mplに対するアゴニストヒト抗体である上記(2)の重鎖改変抗体。
(i) 重鎖定常領域のアッパーヒンジ部が以下の(a)〜(b)のアミノ酸配列から選択されるいずれか一つのアミノ酸配列を有し、並びに、該重鎖定常領域のミドルヒンジ部以降のC末端側がヒトイムノグロブリンG4のアミノ酸配列或いは該G4のアミノ酸配列においてKabat EU番号付けにおける228位セリンがプロリンに且つ235位ロイシンがグルタミン酸に置換されたアミノ酸配列を有する重鎖を含むヒトc-Mplに対するアゴニスト抗体。
(a) 配列番号10で示されるアミノ酸配列。
(b) 配列番号11で示されるアミノ酸配列。
(ii) 更に好ましい実施形態としては、以下(a)〜(h)からなる群から選択される、上記(i)に記載のヒトc-Mplに対するアゴニスト抗体を挙げることができる。
(a) 配列番号2のアミノ酸配列を含む重鎖と配列番3のアミノ酸配列を含む軽鎖を有する抗体。
(b) 配列番号4のアミノ酸配列を含む重鎖と配列番号5のアミノ酸配列を含む軽鎖を有する抗体。
(c) 配列番号6のアミノ酸配列を含む重鎖と配列番号7のアミノ酸配列を含む軽鎖を有する抗体。
(d) 配列番号8のアミノ酸配列を含む重鎖と配列番号9のアミノ酸配列を含む軽鎖を有する抗体。
(e) 配列番号2のアミノ酸配列を含む重鎖と、配列番号3のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖を有する抗体。
(f) 配列番号4のアミノ酸配列を含む重鎖と、配列番号5のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖を有する抗体。
(g) 配列番号6のアミノ酸配列を含む重鎖と、配列番号7のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖を有する抗体。
(h) 配列番号8のアミノ酸配列を含む重鎖と、配列番号9のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖を有する抗体。
【0032】
3.ヒトc-MPLに対するアゴニスト抗体の医薬用途及び医薬組成物
本発明に係るヒトc-MPLに対するアゴニスト抗体は、c-Mp1受容体に結合しそれを活性化する能力、および/または(インビボおよびインビトロの両方で)血小板の産生を刺激する能力(「血小板生成活性」)および血小板前駆体の産生を刺激する能力(「巨核球生成活性」)を有する。
【0033】
本発明に係るヒトc-Mplに対するアゴニスト抗体を有効成分とする医薬組成物/医薬用途としては、具体的には次のものを挙げることができる。
(1)上記1.(1)〜(6)及び同2.(3)のいずれかの抗体を有効成分とする医薬組成物。
(2)上記1.(1)〜(6)及び同2.(3)のいずれかに記載の抗体を有効成分とする血小板増多剤。
(3)骨髄移植、臍帯血移植時の血小板回復促進用であることを特徴とする上記(2)に記載の血小板増多剤
(4)上記1.(1)〜(6)及び同2.(3)のいずれかの抗体を有効成分とする血小板減少症治療剤。
(5)血小板減少症が、以下の(a)〜(f)のいずれか1つである、上記(4)に記載の血小板減少症治療剤。
(a) 特発性血小板減少性紫斑病(ITP)、
(b) 癌化学療法後の血小板減少症、
(c) 再生不良性貧血、
(d) 骨髄異形性症候群(MDS)、
(e) 肝疾患にともなう血小板減少症、又は
(f) 骨髄移植もしくは臍帯血移植後の血小板減少症。
(6)造血幹細胞移植後の血球回復促進用であるヒトc-Mplアゴニスト抗体を有効成分として含む血球増多剤。
(7)上記1.(1)〜(6)及び同2.(3)のいずれかに記載の抗体を有効成分として含む上記(6)に記載の血球増多剤。
【0034】
4.本発明に係る抗体の製造方法
本発明の抗体は、本発明に係る抗体を産生するハイブリドーマを用いて抗体を製造してもよいし、又は、ハイブリドーマ等の抗体産生細胞からモノクローナル抗体をコードする遺伝子をクローニングし、適当なベクターに組み込んで、遺伝子組換え技術を用いて産生させた組換え型抗体を製造することもできる。
【0035】
本発明の抗体は、次の方法によって製造できる。
ヒトトロンボポエチン受容体(c-Mpl)に対するアゴニスト抗体の製造方法であって、該抗体の定常領域が、以下の(1)〜(3):
(1) ヒト抗体の重鎖定常領域及び軽鎖定常領域のアミノ酸配列、
(2) ヒト抗体サブクラス間で重鎖定常領域のドメインを入れ替えた重鎖定常領域のアミノ酸配列、及び、ヒト抗体軽鎖定常領域のアミノ酸配列、或いは
(3) 前記(1)又は(2)のアミノ酸配列において1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
のいずれかのアミノ酸配列を有し、且つ抗体の可変領域がヒトトロンボポエチン受容体に結合し該受容体を活性化する能力を有する、並びに、以下の(a)及び(b):
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、10,000ng/mL以下の濃度でコロニー形成を誘導すること、及び
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性が、そのN末端がペグ(PEG)化された配列番号1のアミノ酸配列からなる下記の構造:
PEG-NH-SPAPPACDLRVLSKLLRDSHVLHSRLSQCPEVHPLPTPVLLPAVDFSLGEWKTQMEETKAQDILGAVTLLLEGVMAARGQLGPTCLSSLLGQLSGQVRLLLGALQSLLGTQLPPQGRTTAHKDPNAIFLSFQHLLRGKVRFLMLVGGSTLCVRRAPPTTAVPS-COOH
を有するPEG-rHuMGDFの50%以上であり、且つ50%有効濃度(EC50)が100nM以下であること、
の性質を有する前記アゴニスト抗体を構成する重鎖及び軽鎖をそれぞれコードする塩基配列を含むDNAと、それらのDNAの発現を制御する塩基配列を含む1又は複数のDNAとを保持する哺乳動物細胞を作製し、該哺乳動物細胞を培養した培養液から、該重鎖及び軽鎖からなる抗体をコードする塩基配列を含むDNAの発現産物を単離精製することを含む、上記方法。
【0036】
実施形態において、上記抗体が、以下の(a)及び(b):
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、1,000ng/mL以下の濃度でコロニー形成を誘導すること、及び
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性がPEG-rHuMGDFの70%以上であり、且つEC50が10nM以下であること、
の性質を有する。
【0037】
別の実施形態において、上記抗体が、以下の(a)及び(b):
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、100ng/mL以下の濃度でコロニー形成を誘導すること、及び
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性がPEG-rHuMGDFの90%以上であり、かつEC50が1nM以下であること、
の性質を有する。
【0038】
別の実施形態において、上記抗体が、以下の(1)〜(8):
(1) 配列番号2で示されるアミノ酸配列を含む重鎖可変領域及び配列番号3で示されるアミノ酸配列を含む軽鎖可変領域、
(2) 配列番号4で示されるアミノ酸配列を含む重鎖可変領域及び配列番号5で示されるアミノ酸配列を含む軽鎖可変領域、
(3) 配列番号6で示されるアミノ酸配列を含む重鎖可変領域及び配列番号7で示されるアミノ酸配列を含む軽鎖可変領域、
(4) 配列番号8で示されるアミノ酸配列を含む重鎖可変領域及び配列番号9で示されるアミノ酸配列を含む軽鎖可変領域、
(5) 配列番号2で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号3で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、
(6) 配列番号4で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号5で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、
(7) 配列番号6で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号7で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、及び
(8) 配列番号8で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号9で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、
からなる群から選択される重鎖可変領域及び軽鎖可変領域を有する。
【0039】
別の実施形態において、前記抗体が、ヒト抗体である。
【0040】
別の実施形態において、上記抗体の重鎖定常領域のアッパーヒンジ部が、以下の(1)及び(2):
(1) 配列番号10で示されるアミノ酸配列、及び
(2) 配列番号11で示されるアミノ酸配列、
のアミノ酸配列から選択されるいずれか1つのアミノ酸配列を有し、並びに、該重鎖定常領域のミドルヒンジ部以降のC末端側が、ヒトイムノグロブリンG4のアミノ酸配列或いは該G4のアミノ酸配列においてKabat EU番号付けにおける228位セリンがプロリンに且つ235位ロイシンがグルタミン酸に置換されたアミノ酸配列を有する。
【0041】
本発明の抗体の製造方法として、好ましくは、以下の方法が挙げられる。
下記の(a)〜(h):
(a) 配列番号2のアミノ酸配列を含む重鎖と配列番号3のアミノ酸配列を含む軽鎖をコードする塩基配列、
(b) 配列番号4のアミノ酸配列を含む重鎖と配列番号5のアミノ酸配列を含む軽鎖をコードする塩基配列、
(c) 配列番号6のアミノ酸配列を含む重鎖と配列番号7のアミノ酸配列を含む軽鎖をコードする塩基配列、
(d) 配列番号8のアミノ酸配列を含む重鎖と配列番号9のアミノ酸配列を含む軽鎖をコードする塩基配列、
(e) 配列番号2のアミノ酸配列を含む重鎖と、配列番号3のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、
(f) 配列番号4のアミノ酸配列を含む重鎖と、配列番号5のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、
(g) 配列番号6のアミノ酸配列を含む重鎖と、配列番号7のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、及び
(h) 配列番号8のアミノ酸配列を含む重鎖と、配列番号9のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、
からなる群から選択される、重鎖をコードする塩基配列を含むDNA及び軽鎖をコードする塩基配列を含むDNAと、それらのDNAの発現を制御する塩基配列を含む1又は複数のDNAとを保持する哺乳動物細胞を作製し、該哺乳動物細胞を培養した培養液から、該重鎖及び軽鎖からなる抗体をコードするDNAの発現産物を単離精製することを含む、ヒトc-Mplに対するアゴニスト抗体の製造方法。
【0042】
5.本発明に係るDNA
本発明に係るDNAとして、以下のものを挙げることができる。
(1)ヒトMplに対するアゴニスト抗体の重鎖可変領域のアミノ酸配列をコードする塩基配列を含む新規なDNAであって、以下の(a)〜(d)からなる群から選択されるアミノ酸配列をコードする塩基配列を含むDNA:
(a) 配列番号2で示されるアミノ酸配列;
(b) 配列番号4で示されるアミノ酸配列;
(c) 配列番号6で示されるアミノ酸配列;
(d) 配列番号8で示されるアミノ酸配列。
(2)ヒトMplに対するアゴニスト抗体の軽鎖可変領域のアミノ酸配列をコードする塩基配列を含む新規なDNAであって、以下の(a)〜(h)からなる群から選択されるアミノ酸配列をコードする塩基配列を含むDNA:
(a) 配列番号3で示されるアミノ酸配列;
(b) 配列番号5で示されるアミノ酸配列;
(c) 配列番号7で示されるアミノ酸配列;
(d) 配列番号9で示されるアミノ酸配列;
(e) 配列番号3のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列;
(f) 配列番号5のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列;
(g) 配列番号7のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列;
(h) 配列番号9のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列。
(3)上記(1)又は(2)のDNAが、それぞれ可変領域及び定常領域を含む抗体重鎖或いは軽鎖をコードするDNA。
(4)抗体の重鎖定常領域のアッパーヒンジ部が、以下の(a)及び(b)のアミノ酸配列から選択されるいずれか一つのアミノ酸配列を有し、並びに、該重鎖定常領域のミドルヒンジ部以降のC末端側がヒトイムノグロブリンG4のアミノ酸配列或いは該G4のアミノ酸配列においてKabat EU番号付けにおける228位セリンがプロリンに且つ235位ロイシンがグルタミン酸に置換されたアミノ酸配列を有する、上記(3)に記載の抗体重鎖をコードするDNA。
(a) 配列番号10で示されるアミノ酸配列。
(b) 配列番号11で示されるアミノ酸配列。
【0043】
本明細書は本願の優先権の基礎である日本国特許出願2006-81322号、2006-299554号の明細書および/または図面に記載される内容を包含する。
【発明を実施するための最良の形態】
【0044】
以下、本発明を詳細に説明する。
本発明は、プライマリーヒト細胞に作用する抗ヒトc-Mplアゴニストヒト抗体を提供する。
【0045】
本発明における抗体は、ヒト抗体産生マウス(例えば、KMマウスTM(キリンビール社))をヒトMpl組換えタンパク質もしくはヒトMpl発現細胞で免疫し、通常のモノクローナル抗体作製法にて単離することができる。また、ハイブリドーマから抗体遺伝子を単離し、発現ベクターを構築し、発現細胞を作製し、その過程で、種々の定常領域を有する組換え抗体を作製することができる。
【0046】
1.本発明に係る抗体について
本明細書において抗体とは、Fab領域、ヒンジ領域、Fc領域を有する抗体を意味し、天然に存在する抗体、及び、同様の構成を有する範囲において、それ自体公知の方法で得られたモノクローナル抗体産生ハイブリドーマにより産生される、或いは、一旦抗体遺伝子を取得しそれを用いて遺伝子工学的に産生される抗体、更に部位特異的突然変異誘発法により一部改変され遺伝子工学的に産生される抗体を含む。そして本発明に係るヒトcMplに対するアゴニスト抗体及び重鎖改変アゴニスト抗体は前述した通りのものである。
【0047】
一般に、アゴニスト抗体は細胞膜上の標的分子に結合し、複合体を形成させることでシグナルを伝える。ホモ二量体を形成するサイトカイン受容体ファミリー、例えばエリスロポエチン受容体(EpoR)、G-CSF受容体(G-CSFR)、トロンボポエチン受容体(c-Mpl)等に対するアゴニスト抗体は、二価の抗体が、二つの分子と結合することで二量体を形成させると考えられる。多くのアゴニスト抗体がFab断片のみでは活性を示さないこともそれを示唆している。
【0048】
複合体形成には、二つの抗原結合部位が接近し易いことが重要と考えられる。全長抗体では充分な活性を持たない抗体でも、sc(Fv)2等の形態に低分子化することでアゴニスト活性が上昇するのはこのことを示唆している。一方で、低分子化抗体は、分子の大幅な改変による抗原性が懸念されるうえ、血中半減期が短縮される等、医薬品として利用するためには課題が多い。全長抗体の持つ、低抗原性や血中半減期の長さ等、医薬品として有用な性質を生かすためには、抗体の構造に大幅な改変を加えることなく、高い活性を持ったアゴニスト抗体が望まれる。
【0049】
後述の実施例2に示すように、本発明者らは、免疫の方法に工夫をこらし、全長抗体で高い活性を持った抗ヒトc-Mplアゴニスト抗体を取得した。工夫とは、例えば高発現細胞株による免疫や恒常活性型の変異受容体発現細胞を用いた免疫である。該アゴニスト抗体は後述の実施例6に記載のヒト臍帯血CD34+細胞を用いたコロニーアッセイでコロニー形成を誘導することが示されており、医薬品として有用であることが期待される。
【0050】
さらに、本発明者らはヒンジ部分の柔軟性を高めることで、複合体形成の効率を高め、アゴニスト活性を増強することを考案した。柔軟性の高い配列は、例えばグリシンリンカーのような配列を考えることができる。もしくは、ヒトIgGの中でも、最も柔軟性が高いIgG3のヒンジ領域を用いることができる。抗体の低抗原性を損なわないためには、なるべく天然に存在する配列を用いることが望ましい。そこで、IgG3のヒンジ配列がより好ましい。
【0051】
また、遺伝子工学的改変により、細胞傷害活性が低く、かつ、高いヒンジの柔軟性を持った、アゴニスト抗体に最適な定常領域として、ヒトIgG3のアッパーヒンジ領域を持ち、ミドルヒンジ以降C末端側の領域はヒトIgG4の配列とするような抗体の作製も可能である。
【0052】
より具体的には、当業者に周知である遺伝子工学的改変(例えば、欧州特許EP314161公報を参照のこと)により異なるサブクラスのものに変換し、即ち、本発明の抗体の可変領域をコードするDNAを用いて遺伝子工学的手法を用いて元のサブクラスとは異なるサブクラスの抗体に変更することができる。更にヒトIgG4重鎖定常部分のEU番号付け(Sequences of proteins of immunological interest, NIH Publication No.91-3242 を参照)における228番目のセリンをプロリンに変異させることにより、IgG4の分子内架橋(S-S結合)による単量体形成を抑制することが、また、同じく235番目ロイシンをグルタミン酸に変異させることにより、抗体依存性細胞傷害(Antibody-dependent cellular cytotoxicity:ADCC)活性を低減させることができる。上記二つの変異を持ったIgG4をIgG4PEと呼ぶ。
【0053】
上記の事柄を考慮し、本発明者らは細胞傷害活性が低く、かつ、高いヒンジの柔軟性を持った、アゴニスト抗体に最適な定常領域を作製した。本定常領域は、ヒトIgG3のアッパーヒンジ領域を持ち、ミドルヒンジ以降C末端側の領域はヒトIgG4の配列となっている。この定常領域と抗c-Mplアゴニスト抗体の可変領域を組み合わせることにより、安全性と高活性を兼ね備えたアゴニスト抗体を作製することができる。
【0054】
2.本発明に係る抗体の製造方法
本発明の抗体は、種々の方法により生産できるが、まずは本発明抗体産生ハイブリドーマを得る必要がある。後述の実施例1に記載するような本発明の抗原を用い、マウス等を免疫し、特にヒト抗体を得る場合にはヒト抗体産生トランスジェニックマウスなどの非ヒト哺乳動物に免疫する。モノクローナル抗体は、定法に従い、免疫感作動物から得た抗体産生細胞と、自己抗体産生能のない骨髄腫系細胞(ミエローマ細胞)を融合することにより得られるハイブリドーマを培養し、免疫に用いた抗原に対して特異的親和性を示すモノクローナル抗体を産生するクローンを選択することによって取得することができる。取得した抗体群から更にアゴニスト抗体を選択する必要があるが、アゴニスト抗体の作用対象受容体に対するリガンドの活性測定方法として確立した方法を用いればよく、ヒトc-Mplに対するアゴニスト抗体を選択する場合は、後述の実施例5に示したUT7/TPO細胞増殖アッセイ等のTPO活性測定方法として既に確立された方法を用いて適宜行うことができる。
【0055】
本発明に係るヒトc-Mplに対するアゴニスト抗体、特にモノクローナル抗体の製造にあたっては、下記の工程を包含する。すなわち、(1)免疫原として使用する、生体高分子の精製及び/又は抗原タンパク質を細胞表面に過剰に発現している細胞の作製、(2)抗原を動物に注射することにより免疫した後、血液を採取しその抗体価を検定して脾臓等の摘出の時期を決定してから、抗体産生細胞を調製する工程、(3)骨髄腫細胞(ミエローマ)の調製、(4)抗体産生細胞とミエローマとの細胞融合、(5)目的とする抗体を産生するハイブリドーマ群の選別、(6)単一細胞クローンへの分割(クローニング)、(7)場合によっては、モノクローナル抗体を大量に製造するためのハイブリドーマの培養、又はハイブリドーマを移植した動物の飼育、(8)このようにして製造されたモノクローナル抗体の生理活性及びその認識特異性の検討、あるいは標識試薬としての特性の検定、(9)モノクローナル抗体遺伝子のクローニングと組換え抗体作製等である。
【0056】
以下、ヒトc-Mplに対するアゴニストモノクローナル抗体の作製法を上記工程に沿って詳述するが、該抗体の作製法はこれに制限されず、例えば脾細胞以外の抗体産生細胞及びミエローマを使用することもできる。
【0057】
(1) 抗原
一般にヒトc-mpl抗体を得る場合には、ヒトc-Mplタンパク質の一次構造が公知(Genbank: NP_005364参照)であるので、当業者に周知の方法により、c-Mplのアミノ酸配列からペプチドを化学合成し、これを抗原として使用することができ、またc-Mplの細胞膜貫通領域および細胞内領域を欠損した可溶化c-Mpl組換えタンパク質を抗原として用いることもできる。
【0058】
或いは、各種ヒト巨核球系細胞株や強制発現株等のヒトc-Mpl発現細胞株の抗原としての利用が挙げられる。但し、ヒトc-Mpl発現細胞株としては、各種ヒト巨核球系細胞株や強制発現株が知られているが、これらの細胞株のc-Mpl発現量は細胞あたり数千分子と低く、抗原には不向きである。実際、マウス造血系細胞株であるFDCP2にヒトc-Mplを導入した発現株FDCP-hMpl(FEBS Lett. 1996 Oct 21;395(2-3):228-34参照)をヒト抗体産生マウス(KMマウスTM)に免疫した場合、抗体価上昇が不十分であり、hMpl特異的なヒト抗体を得ることができなかった。また、ヒト巨核球系細胞株を抗原として用いる場合、他の膜分子に対する抗体も誘導されるため、c-Mpl特異的な抗体を効率良く誘導するには必ずしも適切でない。従って、目的とするものが単にヒトc-mpl抗体でなくアゴニスト活性を有する抗体の取得に際し抗原タンパク質を発現する細胞株を免疫に用いる場合には、発現量が高い細胞を選択することが望ましい。特に、マウス細胞株、しかも可能な限りMHC適合した細胞株を宿主に、ヒトc-Mplを導入した高発現株を用いるのが望ましい。例として、後述の実施例1に記載の細胞(発現ベクターとしてヒトc-mpl全長遺伝子が担持されたpEF-MPL635或いはpCMV-MPL635を、宿主としてマウス細胞株L929およびFM3Aを用いたもの)を挙げることができる。
【0059】
また、野生型のヒトc-mplに代えて、ヒトc-mplの恒常活性型変異体(例えば、508番目TrpがSerに変異しリガンド非依存性に恒常的にアゴニストシグナルを伝達する変異体;Abe Mら、Leukemia. 2002 Aug;16(8):1500-1506)を同様の方法で強制発現させた細胞株を用いてもよい。このような変異体は野生型とは立体構造が異なることが予想され、そのような恒常活性型変異体に親和性が高い抗体は、強力なアゴニスト活性を示す可能性がある。
【0060】
また、これら強制発現細胞株は、ヒトcMPL或いはその細胞外可溶性領域等と適宜組み合わせて抗原として用いることもできる。
【0061】
(2) 抗体産生細胞の調製工程
上記(1)で得られた抗原と、フロインドの完全若しくは不完全アジュバント、又はカリミョウバンのような助剤とを混合し、免疫原として実験動物に免疫する。実験動物としては、遺伝子改変によってヒト抗体を産生する能力を有するマウス(ヒト抗体産生マウス)が最適である。
【0062】
本発明において用いられたヒト抗体産生マウス(例えばKMマウスTM)は、内在性マウスイムノグロブリン(Ig)重鎖及びマウスκ軽鎖を欠損しており、かつ、ヒトIg重鎖遺伝子を含む14番染色体断片(SC20)及びヒトIgκ鎖トランスジーン(KCo5)を同時に保持する。このマウスはヒトIg重鎖遺伝子座を持つ系統Aのマウスと、ヒトIgκ鎖トランスジーンを持つ系統Bのマウスとの交配により作製される。系統Aは、内因性Ig重鎖及びκ軽鎖破壊の両者についてホモ接合体であり、子孫伝達可能な14番染色体断片(SC20)を保持するマウス系統(Tomizuka. et al., Proc Natl Acad Sci USA., 2000 Vol97:722)である。また、系統Bは内在性マウスIg重鎖及びκ軽鎖欠損の両者についてホモ接合体であり、ヒトIgκ鎖トランスジーン(KCo5)を保持するマウス系統(Nat Biotechnol., 1996 Vol14:845)である。従って、KMマウスはヒト抗体を産生する能力を持ち、マウスIg重鎖およびκ鎖を欠損したマウスである。
【0063】
マウス免疫の際の免疫原投与法は、皮下注射、腹腔内注射、静脈内注射、皮内注射、筋肉内注射、足蹠注射などいずれでもよいが、腹腔内注射、足蹠注射又は静脈内注射が好ましい。
【0064】
免疫は、一回、又は、適当な間隔で(好ましくは2週間から4週間間隔で)複数回繰返し行うことができる。その後、免疫した動物の血清中の抗原に対する抗体価を測定し、抗体価が十分高くなった動物を抗体産生細胞の供給源として用いれば、以後の操作の効果を高めることができる。一般的には、最終免疫後3〜5日後の動物由来の抗体産生細胞を、後の細胞融合に用いることが好ましい。
【0065】
ここで用いられる抗体価の測定法としては、フローサイトメトリー法、放射性同位元素免疫定量法(以下、「RIA法」という)、固相酵素免疫定量法(以下、「ELISA法」という)、蛍光抗体法、受身血球凝集反応法など種々の公知技術があげられるが、検出感度、迅速性、正確性、及び操作の自動化の可能性、等の観点から、フローサイトメトリー法又はELISA法がより好適である。
【0066】
本発明における抗体価の測定は、例えばフローサイトメトリー法によれば、以下に記載するような手順により行うことができる。まず、抗原を発現した細胞とヒト抗体を含む検体(例えばマウス血清,ハイブリドーマの培養上清,精製抗体等)と反応させる。さらに二次抗体として蛍光標識されたヒト抗体に対する抗体を加えてヒト抗体に結合させ、洗浄後、細胞に結合した二次抗体の量を蛍光によって測定することにより、抗体価を算出する。
【0067】
(3) ミエローマの調製工程
ミエローマとしては、マウス、ラット、モルモット、ハムスター、ウサギ又はヒト等の哺乳動物に由来する自己抗体産生能のない細胞を用いることが出来るが、一般的にはマウスから得られた株化細胞、例えば8-アザグアニン耐性マウス(BALB/c由来)ミエローマ株P3X63Ag8U.1(P3-U1)[Yelton, D.E. et al. Current Topics in Microbiology and Immunology, 81, 1-7(1978)]、P3/NSI/1-Ag4-1(NS-1) [Kohler, G. et al. European J. Immunology, 6, 511-519 (1976)]、Sp2/O-Ag14(SP-2)[Shulman, M. et al. Nature, 276, 269-270 (1978)]、P3X63Ag8.653(653)[Kearney, J. F. et al. J. Immunology, 123, 1548-1550(1979)]、P3X63Ag8(X63)[Horibata, K. and Harris, A. W. Nature, 256, 495-497 (1975)]などを用いることが好ましい。これらの細胞株は、適当な培地、例えば8-アザグアニン培地[グルタミン、2-メルカプトエタノール、ゲンタマイシン及びウシ胎児血清(以下、「FCS」という)を加えたRPMI-1640培地に8-アザグアニンを加えた培地]、イスコフ改変ダルベッコ培地(Iscove's Modified Dulbecco's Medium;以下、「IMDM」という)、又はダルベッコ改変イーグル培地(Dulbecco's Modified Eagle Medium;以下、「DMEM」という)で継代培養するが、細胞融合の3〜4日前に正常培地(例えば、10% FCSを含むDMEM培地)で継代培養し、融合当日に2×107以上の細胞数を確保しておく。
【0068】
(4) 細胞融合
抗体産生細胞は、形質細胞、及びその前駆細胞であるリンパ球であり、これは個体のいずれの部位から得てもよく、一般には脾臓、リンパ節、骨髄、扁桃、末梢血、又はこれらを適宜組み合わせたもの等から得ることができるが、脾細胞が最も一般的に用いられる。
【0069】
最終免疫後、所定の抗体価が得られたマウスから抗体産生細胞が存在する部位、例えば脾臓を摘出し、抗体産生細胞である脾細胞を調製する。次いで、脾細胞とミエローマを融合させればよい。この脾細胞と工程(3)で得られたミエローマを融合させる手段として現在最も一般的に行われているのは、細胞毒性が比較的少なく融合操作も簡単な、ポリエチレングリコールを用いる方法である。この方法は、例えば以下の手順よりなる。
【0070】
脾細胞とミエローマとを無血清培地(例えば、DMEM)、又はリン酸緩衝生理食塩液(以下、「PBS」という)でよく洗浄し、脾細胞とミエローマの細胞数の比が5:1〜10:1程度になるように混合し、遠心分離する。上清を除去し、沈澱した細胞群をよくほぐした後、撹拌しながら1mLの50%(w/v)ポリエチレングリコール(分子量1000〜4000)を含む無血清培地を滴下する。その後、10mLの無血清培地をゆっくりと加えた後遠心分離する。再び上清を捨て、沈澱した細胞を適量のヒポキサンチン・アミノプテリン・チミジン(以下「HAT」という)液及びヒトインターロイキン-6(以下、「IL-6」という)を含む正常培地(以下、「HAT培地」という)中に懸濁して培養用プレート(以下、「プレート」という)の各ウェルに分注し、5%炭酸ガス存在下、37℃で2週間程度培養する。途中適宜HAT培地を補う。
【0071】
(5) ハイブリドーマ群の選択
上記ミエローマ細胞が、8-アザグアニン耐性株である場合、すなわち、ヒポキサンチン・グアニン・ホスホリボシルトランスフェラーゼ(HGPRT)欠損株である場合、融合しなかった該ミエローマ細胞、及びミエローマ細胞どうしの融合細胞は、HAT含有培地中では生存できない。一方、抗体産生細胞どうしの融合細胞、あるいは、抗体産生細胞とミエローマ細胞とのハイブリドーマは生存することができるが、抗体産生細胞どうしの融合細胞には寿命がある。従って、HAT含有培地中での培養を続けることによって、抗体産生細胞とミエローマ細胞との融合細胞であるハイブリドーマのみが生き残り、結果的にハイブリドーマを選択することができる。コロニー状に生育してきたハイブリドーマについて、HAT培地からアミノプテリンを除いた培地(以下、「HT培地」という)への培地交換を行う。以後、培養上清の一部を採取し、例えば、フローサイトメトリー法により抗ヒトc-Mpl抗体価を測定する。以上、8-アザグアニン耐性の細胞株を用いる方法を例示したが、その他の細胞株もハイブリドーマの選択方法に応じて使用することができ、その場合使用する培地組成も変化する。
【0072】
(6) クローニング工程
前述(2)の記載と同様の方法で抗体価を測定することにより、特異的抗体を産生することが判明したハイブリドーマを、別のプレートに移しクローニングを行う。このクローニング法としては、プレートの1ウェルに1個のハイブリドーマが含まれるように希釈して培養する限界希釈法、軟寒天培地中で培養しコロニーを回収する軟寒天法、マイクロマニュピレーターによって1個ずつの細胞を取り出し培養する方法、セルソーターによって1個の細胞を分離する「ソータクローン」などが挙げられるが、限界希釈法が簡便であり、よく用いられる。
【0073】
抗体価の認められたウェルについて、例えば限界希釈法によるクローニングを2〜4回繰返し、安定して抗体価の認められたものを抗ヒトc-Mplモノクローナル抗体産生ハイブリドーマ株として選択する。
【0074】
(7) アゴニスト抗体の選択
得られた抗ヒトc-Mplモノクローナル抗体産生ハイブリドーマ株の培養上清或いは後記(8)に従って該上清より精製した抗体を、種々のTPO活性測定系にて測定することにより、アゴニスト抗体を選択することができる。スクリーニングとして好適な方法として、哺乳動物細胞にヒトMplを発現させ、細胞増殖アッセイを行う方法が挙げられる。例えばマウス細胞株BaF3にヒトMplを発現させた細胞による増殖アッセイ(Orita et al. Blood. 2005 Jan 15;105(2):562-6.)等も使用し得るが、マウス細胞を用いた場合に、ヒト細胞の反応を反映しているとは限らないことを考慮すると、よりヒト細胞に強い活性を持つ抗体を選抜するにはヒトMplが発現しているヒト細胞を用いる増殖アッセイ方法がより好ましい。ヒト細胞による系として具体的には、後述の実施例5に記載のUT7/TPO細胞を用いた細胞増殖アッセイを挙げることができる。
【0075】
(8) ハイブリドーマ培養によるモノクローナル抗体の調製
クローニングを完了したハイブリドーマは、培地をHT培地から正常培地に換えて培養される。大量培養は、大型培養瓶を用いた回転培養、スピナー培養、あるいはホローファイバーシステム等を用いた培養で行われる。この大量培養における上清を、ゲルろ過等、当業者に周知の方法を用いて精製することにより、抗ヒトc-Mplモノクローナル抗体を得ることができる。また、同系統のマウス(例えばBALB/c)若しくはnu/nuマウス、ラット、モルモット、ハムスター又はウサギ等の腹腔内で該ハイブリドーマを増殖させることにより、抗ヒトc-Mplモノクローナル抗体を大量に含む腹水を得ることができる。精製の簡便な方法としては、市販のモノクローナル抗体精製キット(例えば、MAbTrap GIIキット;アマシャムファルマシアバイオテク社製)等を利用することもできる。かくして得られるモノクローナル抗体は、ヒトc-Mplに対して高い抗原特異性を有する。
【0076】
(9) モノクローナル抗体の検定
かくして得られたモノクローナル抗体のアイソタイプ及びサブクラスの決定は以下のように行うことができる。まず、同定法としてはオクテルロニー(Ouchterlony)法、ELISA法、又はRIA法が挙げられる。オクテルロニー法は簡便ではあるが、モノクローナル抗体の濃度が低い場合には濃縮操作が必要である。一方、ELISA法又はRIA法を用いた場合は、培養上清をそのまま抗原吸着固相と反応させ、さらに二次抗体として各種イムノグロブリンアイソタイプ、サブクラスに対応する抗体を用いることにより、モノクローナル抗体のアイソタイプ、サブクラスを同定することが可能である。さらに、タンパク質の定量は、フォーリンロウリー法、及び280nmにおける吸光度[1.4(OD280)=イムノグロブリン1mg/mL]より算出する方法等により行うことができる。また、ハイブリドーマからモノクローナル抗体をコードする遺伝子をクローニングし、配列を決定することでサブクラスを特定することもできる。
【0077】
(10) モノクローナル抗体をコードする遺伝子のクローニングと組換え型抗体作製
また、ハイブリドーマ等の抗体産生細胞からモノクローナル抗体をコードする遺伝子をクローニングし、適当なベクターに組み込んで、これを宿主(例えば哺乳類細胞細胞株、酵母細胞、昆虫細胞など)に導入し、遺伝子組換え技術を用いて産生させた組換え型抗体を調製することもできる(P.J.Delves., ANTIBODY PRODUCTION ESSENTIAL TECHNIQUES., 1997 WILEY、P.Shepherd and C.Dean., Monoclonal Antibodies., 2000 OXFORD UNIVERSITY PRESS, J.W.Goding., Monoclonal Antibodies:principles and practice., 1993 ACADEMIC PRESS)。
【0078】
本発明は、本発明の抗体を産生するハイブリドーマが保有する抗体の遺伝子配列を含む核酸、特に後述の、本発明のハイブリドーマが産生する抗体の重鎖可変領域及び軽鎖可変領域の核酸も包含する。ここで、核酸にはDNA及びRNAが含まれる。
【0079】
ハイブリドーマからモノクローナル抗体をコードする遺伝子を調製するには、モノクローナル抗体のL鎖V領域、L鎖C領域、H鎖V領域及びH鎖C領域をそれぞれコードするDNAをPCR法等により調製する方法が採用される。プライマーは、抗体遺伝子又はアミノ酸配列から設計したオリゴDNAを、鋳型としてはハイブリドーマから調製したDNAを使用することができる。これらのDNAを1つの適当なベクターに組み込み、これを宿主に導入して発現させるか、あるいはこれらのDNAをそれぞれ適当なベクターに組み込み、共発現させる。
【0080】
ベクターには、宿主微生物で自律的に増殖し得るファージ又はプラスミドが使用される。プラスミド DNAとしては、大腸菌、枯草菌又は酵母由来のプラスミドなどが挙げられ、ファージDNAとしてはλファージが挙げられる。
【0081】
形質転換に使用する宿主としては、抗体の立体構造を正しく構成できることから、真核細胞を挙げることができ、例えば、酵母、動物細胞(COS細胞、CHO細胞等)、昆虫細胞が挙げられる。特に動物細胞宿主を用いる場合、ベクターとしては、例えばN5KG1-Val Larkベクター(IDEC pharmaceuticals: US patent 6001358)を用いることができる。本ベクターは、組換え抗体を動物細胞に発現させる目的で使用される発現ベクターで、2つのCMV プロモーター/エンハンサーを持ち、それぞれの 下流に重鎖および軽鎖可変領域のクローニングサイトを持つ。さらにその下流には、ヒトγ1鎖定常領域とヒトκ鎖定常領域をコードする遺伝子配列をあらかじめ持っている。本ベクターの可変領域クローニングサイトに任意の重鎖および軽鎖可変領域を、読み枠(フレーム)をあわせて組み込むことにより、軽鎖可変領域がヒトκ鎖定常領域と、重鎖可変領域がヒトγ1定常領域と連結された抗体を発現させることができる。本ベクターを導入された動物細胞は培養液中に抗体(ヒトIgG1)を産生する。また、異なる重鎖定常領域遺伝子を含むベクターも使用することができる。例えばN5KG4PEベクター(IDEC pharmaceuticals社製)は定常領域遺伝子として、ヒトγ4に、上述した二つの変異(Ser228Pro, Leu235Glu)が入った配列を持っている。N5KG4PEベクターに任意の重鎖および軽鎖可変領域遺伝子配列を組み込むことで、任意の可変領域を持つIgG4PEを発現させることができる。さらに、重鎖または軽鎖遺伝子に改変を加えることで、様々な定常領域を持った抗体を作製することが可能である。
【0082】
もちろん、本発明に用いる哺乳動物細胞用発現ベクターは上記のものに限定されるものではない。例えば発現を制御する塩基配列として前述CMVプロモーター/エンハンサーを利用した他の発現ベクターを用いてもよいし、これとは異なった公知のプロモーター/エンハンサー(1若しくは複数)を発現制御配列として使ってもよい。プロモーターとしては、例えば、ポリオーマウイルス、鶏痘ウイルス(1989年7月5日公開のUK2211504)、アデノウイルス(例えばアデノウイルス2)、ウシ乳頭腫ウイルス、鳥の肉腫ウイルス、サイトメガロウイルス、レトロウイルス、B型肝炎ウイルスおよび最も好ましくはシミアンウイルス40(SV40)のようなウイルスのゲノムから得られるプロモーター、ヘテロローガスな哺乳動物プロモーター(例えばアクチンプロモーター、免疫グロブリンプロモーター、熱衝撃プロモーター)を挙げることができる。プロモーターに作用し転写を増大させるエンハンサーとしては、公知の哺乳動物遺伝子由来のエンハンサー(グロビン、エラスターゼ、アルブミン、α−フェトプロテイン、およびインシュリン)、および、真核細胞ウイルス由来のエンハンサー(複製起点の後期側のSV40エンハンサー(bp100-270)、複製起点の後期側のポリオーマエンハンサー、およびアデノウイルスエンハンサー)を用いることができる。
【0083】
また、発現ベクターには、転写の終止およびmRNAの安定化に必要な配列を含むことができる。このような配列は普通、真核生物のまたはウイルスのDNAまたはcDNAの5'および時には3'非翻訳領域から取得することができる。
【0084】
宿主への遺伝子の導入方法は公知であり、任意の方法(例えばカルシウムイオンを用いる方法、エレクトロポレーション法、スフェロプラスト法、酢酸リチウム法、リン酸カルシウム法、リポフェクション法等)が挙げられる。また、後述の動物に遺伝子を導入する方法としては、マイクロインジェクション法、ES細胞にエレクトロポレーションやリポフェクション法を使用して遺伝子を導入する方法、核移植法などが挙げられる。
【0085】
本発明において、目的とする抗体は、形質転換体を培養し、その培養上清から採取することにより得ることができる。形質転換体を培養するには、使用する宿主に適した培地を用い、静置培養法、ローラーボトルによる培養法などが採用される。
【0086】
培養後、細胞外に生産された抗体の精製は、培養液をそのまま使用するか、遠心分離等により細胞を除去して行う。その後、タンパク質の単離精製に用いられる各種クロマトグラフィーを用いた一般的な生化学的方法を単独で又は適宜組み合わせて用いることにより、前記培養物中から目的の抗体を単離精製することができる。
【0087】
さらに、トランスジェニック動物作製技術を用いて、目的抗体の遺伝子が内在性遺伝子に組み込まれた動物宿主、例えばトランスジェニックウシ、トランスジェニックヤギ、トランスジェニックヒツジ又はトランスジェニックブタを作製し、そのトランスジェニック動物から分泌されるミルク中からその抗体遺伝子に由来するモノクローナル抗体を大量に取得することも可能である(Wright, G., et al. (1991) Bio/Technology 9, 830-834)。
【0088】
本発明に係るヒトMplに対するアゴニスト抗体の製法として、好ましい方法は、前述の[課題を解決するための手段]の4.にて例示した遺伝子組換え技術による方法であるが、これに限定されるものではない。
【0089】
3.本発明に係るDNA
本発明は、上で述べたように、
(1) ヒトMplに対するアゴニスト抗体の重鎖可変領域のアミノ酸配列をコードする塩基配列であって以下の(a)〜(d)から選択されたアミノ酸配列をコードする塩基配列を含むDNA:
(a) 配列番号2で示されるアミノ酸配列;
(b) 配列番号4で示されるアミノ酸配列;
(c) 配列番号6で示されるアミノ酸配列;
(d) 配列番号8で示されるアミノ酸配列;
及び、
(2) ヒトMplに対するアゴニスト抗体の軽鎖可変領域のアミノ酸配列をコードする塩基配列であって以下の(a)〜(h)から選択されたアミノ酸配列をコードする塩基配列を含むDNA:
(a) 配列番号3で示されるアミノ酸配列;
(b) 配列番号5で示されるアミノ酸配列;
(c) 配列番号7で示されるアミノ酸配列;
(d) 配列番号9で示されるアミノ酸配列;
(e) 配列番号3のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列;
(f) 配列番号5のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列;
(g) 配列番号7のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列;
(h) 配列番号9のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列;
を提供するものであり、上記2.の本発明に係るヒトMplに対するアゴニスト抗体の製法、より具体的には、遺伝子組換え技術を用いた抗体の製造において、用いることができる。
【0090】
これら可変領域のアミノ酸配列(a)〜(d)をコードするDNAは、前述のヒトMplに対するアゴニスト抗体産生ハイブリドーマ取得方法によって得られたハイブリドーマ株より、後述実施例7の通り、定法によりmRNAを抽出し既知である抗体定常領域のアミノ酸配列に基づくプライマーを用いて5’RACE法により取得したものであり、その可変領域をコードする各DNAを含むプラスミドは、独立行政法人産業技術総合研究所特許生物寄託センター(日本国茨城県つくば市東1-1-1 つくばセンター中央第6)に2006年3月14日付にてブダペスト条約に基づく国際寄託がされている。
【0091】
【表1】
【0092】
本発明のアゴニスト抗体を構成する軽鎖の可変領域は、その特定例として、配列番号3、5、7又は9に示されるアミノ酸配列を含むが、これらのアミノ酸配列のフレームワーク領域に1若しくは数個のアミノ酸残基の欠失、置換、付加又は挿入を含んでもよいし、或いはこれらのアミノ酸配列のフレームワーク領域の配列と少なくとも85%、86%、87%、88%又は89%、好ましくは少なくとも90%、92%、93%又は94%、より好ましくは少なくとも95%、96%、97%、98%又は99%の同一性を有する配列を含んでいてもよい。フレームワーク領域は、可変領域において3つのCDR(相補性決定部位;complementarity-determining region)、すなわち配列番号3、5又は7に示されるアミノ酸配列においては、RASQGISS(A又はT)LA(アミノ酸位置24-34)、DASSLES(アミノ酸位置50-56)、及びQQFNSYP(L又はY又はW)T(アミノ酸位置89-97)、を除く領域を指し、配列番号9に示されるアミノ酸領域においては、RASQSVSSSYLA(アミノ酸位置24-35)、DASSRAT(アミノ酸配列51-57)、及びQQYGSSPIT(アミノ酸位置90-98)、を除く領域を指す。後述の実施例17で実証するとおり、フレームワーク領域内にアミノ酸変異が存在しても、本発明の変異型抗体はいずれも、非変異型抗体の場合とほぼ同等のアゴニスト活性、すなわち、FM3A-hMpl細胞などのFM3A-hMpl細胞のヒトトロンボポエチン受容体に結合し該受容体を活性化する能力及び/又はUT-7/TPO細胞の増殖能を有することができる。
【0093】
そのような変異の例は、保存的アミノ酸間での置換である。保存的アミノ酸は、電荷、構造、極性などの性質が類似したアミノ酸群であり、例えば塩基性アミノ酸(Arg、His、Lys)、酸性アミノ酸(Glu、Asp)、非極性アミノ酸(Ala、Leu、Ile、Val、Gly、Pro)、極性アミノ酸(Ser、Thr、Cys、Met、Asn、Gln)、芳香族アミノ酸(Phe、Tyr、Trp)に分類することができる。
【0094】
また、配列同一性は、2つ又はそれ以上の配列を、ギャップを導入して又はギャップを導入しないで整列比較したときのアミノ酸(又はヌクレオチド)配列間の一致率を表わし、一般に全アミノ酸(又はヌクレオチド)数に対する同一アミノ酸(又はヌクレオチド)数のパーセンテージである。配列同一性は、必要に応じてNCBI(米国)などのデータバンクにアクセスし、配列検索用のBLAST、FASTAなどの公知のアルゴリズムを利用して決定することができる。
【0095】
変異の導入は、変異を含まないアミノ酸配列をコードするDNAに対して、例えば部位特異的突然変異誘発法、PCR法(変異を含むプライマーを使用する)などを適用することによって実施することができる。変異導入法は、例えばSambrookら, Molecular Cloning A Laboratory Mannual, Cold Spring Harbor Laboratory Press (1989)などに記載されている。
【0096】
本発明の上記DNAはさらに、可変領域に加え、それぞれ重鎖或いは軽鎖の定常領域をコードする塩基配列を含むものであってもよい。
【0097】
また、本発明に係る抗体の製造方法の項で述べた重鎖定常領域の改変は、寄託したDNA及び公知のヒト抗体定常領域の配列に基づき、周知の遺伝子工学的改変手法により得ることができる。
【0098】
4.ヒトc-Mplに対するアゴニスト抗体の医薬用途及び医薬組成物
本発明に係るヒトc-Mplに対するアゴニスト抗体は、c-MPL受容体に結合しそれを活性化する能力、および/または(インビボおよびインビトロの両方で)血小板の産生を刺激する能力(「血小板生成活性」)および血小板前駆体の産生を刺激する能力(「巨核球生成活性」)を有する。
【0099】
また、ヒトc-Mpl受容体は、巨核球のみならず造血幹細胞においても発現しているとされている。正常動物において、PEG-rHuMGDF投与により骨髄中の赤芽球系や顆粒球/マクロファージ系の前駆細胞が増加したとの報告もある(Stem Cell,14:651-660,1996)。しかし、ヒト臍帯血移植マウスにおいては、PEG-rHuMGDFの投与によりマウスの巨核球系以外の前駆細胞の増殖は見られたが、ヒトの前駆細胞の増殖は認められなかった。一方、ヒトc-Mplに対するアゴニスト抗体においては骨髄中のヒト赤血球および顆粒球/マクロファージ系の前駆細胞の数が有意に高かった(実施例14)。このことは、ヒトc-Mplに対するアゴニスト抗体がヒト細胞に選択的にシグナルを入れ、巨核球系のみならず、他系列の細胞の生着も促進できる可能性を示唆している。
【0100】
本発明に係るヒトc-Mplに対するアゴニスト抗体を有効成分とする医薬組成物により治療される状態は、一般には、存在する巨核球/血小板の欠乏または将来において予期または予想される巨核球/血小板の欠乏(例えば、計画されている手術または血小板供与によるもの)を伴う状態である。そのような状態は、インビボにおける活性Mp1リガンドの(一時的または永久的な)欠乏により生じうる。したがって本発明の組成物は、血小板欠乏状態即ち血小板減少症の治療を要する患者において血小板減少症を予防的または治療的な治療するために利用可能である。さらに、汎血球減少を長期間に渡って伴う造血幹細胞移植(骨髄移植、臍帯血移植、末梢血幹細胞移植)後の血球回復のための治療を要する患者において、汎血球減少症を予防的または治療的な治療をするためにも利用可能である。
【0101】
血小板減少症(血小板欠乏症)は、化学療法および種々の薬物での他の療法、放射線療法、手術、不慮の出血、および他の具体的な病態を含む種々の理由により生じうる。血小板減少症を伴い本発明に従い治療されうる典型的な具体的な病態としては以下のものが挙げられる:無形成貧血;特発性または免疫血小板減少症(ITP)、例えば乳癌に伴う特発性血小板減少性紫斑病;HIVに伴うITPおよびHIVに関連した血栓性血小板減少性紫斑病;血小板減少症を引き起こす転移性腫瘍;全身性エリテマトーデス、例えば新生児ループス症候群脾腫;ファンコーニ症候群;ビタミンB12欠乏症;葉酸欠乏症;メイ-ヘグリンの異常;ウィスコット-アルドリッチ症候群;慢性肝疾患;血小板減少症に関連した骨髄形成異常症候群;発作性夜間ヘモグロビン尿症;C7E3 Fab(Abciximab)療法後の急性深部血小板減少症;同種免疫血小板減少症、例えば母性同種免疫血小板減少症;抗リン脂質抗体および血栓症に関連した血小板減少症;自己免疫血小板減少症;薬物により誘発された免疫血小板減少症、例えばカルボプラチンにより誘発された血小板減少症、ヘパリンにより誘発された血小板減少症;胎児血小板減少症;妊娠血小板減少症;ヒュージー症候群;ルポイド血小板減少症;不慮の及び/又は大量の失血;骨髄増殖性異常症;悪性疾患を有する患者における血小板減少症;血栓性血小板減少紫斑病、例えば癌患者において血栓性血小板減少性紫斑病/溶血性尿毒症性症候群として現れる血栓性細小血管症;自己免疫性溶血性貧血;潜在性空腸憩室穿孔;真正赤血球系無形成症;自己免疫血小板減少症;流行性(epidemica)腎障害;ラファンピシンに関連した急性腎不全;Paris-Trousseau血小板減少症;新生児同種免疫血小板減少症;発作性夜間ヘモグロビン尿症;胃癌における血液学的変化;小児における溶血性尿毒症性症候群;A型関連ウイルスを含むウイルス感染に関連した血液学的徴候およびCMVに関連した血小板減少症。また、ある種のエイズ治療は血小板減少症を引き起こす(例えば、AZT)。また、ある種の創傷治癒障害は、血小板数の増加が有益であろう。なお上記疾患においては、血小板減少のみならず、その他の血球減少を伴う疾患も存在する。
【0102】
予想される血小板の欠乏(例えば、将来の手術によるもの)に対して、血小板が必要となる前に数日から数時間にわたり有効成分としての本発明のアゴニスト抗体を投与することが可能であろう。緊急の状況(例えば、不慮の及び大量の失血)では、血液または精製血小板と共に本発明のアゴニスト抗体を投与することが可能であろう。あるいは、汎血球の欠乏(例えば、臍帯血移植によるもの)に対しても、有効成分としての本発明のアゴニスト抗体を投与することが可能であろう。
【0103】
特に好ましい治療対象としては、(1)特発性血小板減少性紫斑病もしくは肝疾患にともなって起こる血小板減少症、(2)癌化学療法、再生不良性貧血、骨髄異形性症候群(MDS)、骨髄移植もしくは臍帯血移植にともなって起こる血小板減少症および/または汎血球減少症を挙げることができる。
【0104】
本発明のヒトc-MPLに対するアゴニスト抗体はまた、血小板および/または巨核球および関連細胞の生存能または保存寿命を維持するのに有用で有り得る。したがって、そのような細胞を含有する組成物中に有効量を含有させるのが有用であろう。
【0105】
本発明にかかるヒトc-MPLに対するアゴニスト抗体を有効成分とする医薬組成物は、注射用または経口、鼻、経皮または他の投与形態のためのものであってもよい。それらには、例えば、静脈内、皮内、筋肉内、乳房内、腹腔内、鞘内、眼内、延髄後方、肺内(例えば、エアゾール化薬)または皮下注射(長期にわたる放出のためのデポ投与を含む)、舌下、肛門、膣または外科的移植、例えば脾漿膜下、脳または角膜内の埋め込みによる投与が含まれる。該治療は、単回投与または一定期間にわたる複数回の投与よりなるものであってもよい。一般には、本発明にかかるヒトc-MPLに対するアゴニスト抗体の有効量と医薬上許容される希釈剤、保存剤、可溶化剤、乳化剤、佐剤および/または担体とを含む医薬組成物が本発明に含まれる。そのような組成物は、種々のバッファー含有物(例えば、Tris-HCl、酢酸塩、リン酸塩)、pHおよびイオン強度の希釈剤;添加物、例えば界面活性剤および可溶化剤(例えば、Tween 80、Polysorbate 80)、抗酸化剤(例えば、アスコルビン酸、メタ重亜硫酸ナトリウム)、保存剤(例えば、Thimersol、ベンジルアルコール)および増量物質(例えば、ラクトース、マンニトール);ポリ乳酸、ポリグリコール酸などの重合体化合物の粒子状製剤内またはリポソーム内に該物質が封入されたものを含む。所望により、医薬ビヒクル、賦形剤または媒体として働く更に他の医薬上許容される液体、半固体または固体希釈剤を含んでいてもよい。それらには、ポリオキシエチレンソルビタンモノラウラート、ステアリン酸マグネシウム、メチル-およびプロピルヒドロキシベンゾアート、デンプン、スクロース、デキストロース、アラビアゴム、リン酸カルシウム、鉱油、カカオ脂およびテオブロマの油が含まれるが、これらに限定されるものではない。該組成物は、液体形態または乾燥粉末(例えば、凍結乾燥形態)として製造することができる。移植可能な徐放製剤、および経皮製剤も意図される。
【0106】
上記の状態の治療方法にかかわる投与計画は、薬物の作用を改変する種々の要因(例えば、患者の年齢、状態、体重、性別および食事、いずれかの感染の重症度、投与時間ならびに他の臨床的要因)を考慮して担当医師により決定されるであろう。一般には、該用量は、1日当たり本発明抗体100μg〜1mg/kg体重、好ましくは10〜100μg/kg、より好ましくは1〜10μg/kgであり、これらを、1日量で、あるいはより長い又はより短い間隔で(例えば、1日おき、1週間に2回、毎週、または毎日2回または3回)同等量で投与する。
【0107】
また、本発明にかかるヒトc-MPLに対するアゴニスト抗体を有効成分とする医薬組成物は、単独で又は他のサイトカイン、可溶性Mp1受容体、造血因子、インターロイキン、増殖因子と組合せて、血小板の欠乏および他の症状により特徴づけられる病態の治療において使用することができる。造血の一般的な刺激因子(例えば、IL-3またはGM-CSF)と組合せて、いくつかの形態の血小板減少症を治療するのに有用であると予想される。また、他の巨核球刺激因子、すなわちmeg-CSF、幹細胞因子(SCF)、白血病阻害因子(LIF)、オンコスタチンM(OSM)または巨核球刺激活性を有する他の分子を、Mp1リガンドと共に使用することができる。そのような共投与のための追加的な代表的なサイトカインまたは造血因子には、IL-1アルファ、IL-1ベータ、IL-2、IL-3、IL-4、IL5、IL-6、IL-11、コロニー刺激因子-1(CSF-1)、M-CSF、SCF、GM-CSF、顆粒球コロニー刺激因子(G-CSF)、EPO、インターフェロン-アルファ(IFN-アルファ)、コンセンサスインターフェロン、IFN-ベータ、IFN-ガンマ、IL-7、IL-8、IL-9、IL-10、IL-12、IL-13、IL-14、IL-15、IL-16、IL-17、IL-18、トロンボポエチン(TPO)、アンジオポエチン、例えばAng-1、Ang-2、Ang-3、Ang-4、Ang-Y、ヒトアンジオポエチン様ポリペプチド、血管内皮増殖因子(VEGF)、アンジオゲニン、骨形成因子(bone morphogenic protein)-1、骨形成因子-2、骨形成因子-3、骨形成因子-4、骨形成因子-5、骨形成因子-6、骨形成因子-7、骨形成因子-8、骨形成因子-9、骨形成因子-10、骨形成因子-11、骨形成因子-12、骨形成因子-13、骨形成因子-14、骨形成因子-15、骨形成因子受容体IA、骨形成因子受容体IB、脳由来神経栄養因子、毛様体神経栄養因子、毛様体神経栄養因子α、サイトカイン誘導性好中球走化因子1、サイトカイン誘導性好中球走化因子2α、サイトカイン誘導性好中球走化因子2β、β内皮細胞増殖因子、エンドセリン1、上皮増殖因子、上皮由来好中球誘引物質、繊維芽細胞増殖因子4、繊維芽細胞増殖因子5、繊維芽細胞増殖因子6、繊維芽細胞増殖因子7、繊維芽細胞増殖因子8、繊維芽細胞増殖因子8b、繊維芽細胞増殖因子8c、繊維芽細胞増殖因子9、繊維芽細胞増殖因子10、酸性繊維芽細胞増殖因子、塩基性繊維芽細胞増殖因子、グリア細胞株由来神経栄養因子受容体α1、グリア細胞株由来神経栄養因子受容体α2、成長関連タンパク質(growth related protein)、成長関連タンパク質α、成長関連タンパク質β、成長関連タンパク質γ、ヘパリン結合性上皮増殖因子、肝細胞増殖因子、肝細胞増殖因子受容体、インスリン様増殖因子I、インスリン様増殖因子受容体、インスリン様増殖因子II,インスリン様増殖因子結合タンパク質、角質細胞増殖因子、白血病阻害因子、白血病阻害因子受容体α、神経成長因子、神経成長因子受容体、ニューロトロフィン-3、ニューロトロフィン-4、胎盤増殖因子、胎盤増殖因子2、血小板由来内皮細胞増殖因子、血小板由来増殖因子、血小板由来増殖因子A鎖、血小板由来増殖因子AA、血小板由来増殖因子AB、血小板由来増殖因子B鎖、血小板由来増殖因子BB、血小板由来増殖因子受容体α、血小板由来増殖因子受容体β、プレB細胞増殖刺激因子、幹細胞因子受容体、TNF(TFN0、TNF1、TNF2を含む)、トランスフォーミング増殖因子α、トランスフォーミング増殖因子β、トランスフォーミング増殖因子β1、トランスフォーミング増殖因子β1.2、トランスフォーミング増殖因子β2、トランスフォーミング増殖因子β3、トランスフォーミング増殖因子β5、潜在(latent)トランスフォーミング増殖因子β結合タンパク質I、トランスフォーミング増殖因子β結合タンパク質II、トランスフォーミング増殖因子β結合タンパク質III、腫瘍壊死因子受容体I型、腫瘍壊死因子受容体II型、ウロキナーゼ型プラスミノーゲンアクチベーター受容体、血管内皮増殖因子、ならびにそれらのキメラタンパク質が含まれる。
【0108】
したがって、本発明にかかるヒトc-Mplに対するアゴニスト抗体を有効成分とする医薬組成物の投与(成熟巨核球の数を増加させるためのもの)は、血小板の産生を刺激する特に有効な手段であると予想される。さらに造血幹細胞の産生を刺激することにおいても有用な手段であると予想される。前記で列挙した用量は、該治療用組成物中のそのような追加的な成分を補償するように調節されるであろう。治療された患者の経過は、通常の方法によりモニターすることができる。
【0109】
以下、実施例により本発明をさらに詳細に説明するが、本発明の範囲はこれらの実施例に限定されるものではない。
【実施例】
【0110】
[実施例1] 抗原の調製
1-1 ヒトc-Mpl発現細胞の作製
抗原タンパク質を発現する細胞株を免疫に用いる場合、一般的に発現量が高いほど抗体作製には有利である。ヒトc-Mpl発現細胞株としては、各種ヒト巨核球系細胞株や強制発現株が知られているが、これらの細胞株のc-Mpl発現量は細胞あたり数千分子と低く、抗原には不向きである。実際、マウス造血系細胞株であるFDCP2にヒトc-Mplを導入した発現株FDCP-hMpl(FEBS Lett. 1996 Oct 21;395(2-3):228-34.参照)をヒト抗体産生マウス(KMマウスTM)に免疫した場合、抗体価上昇が不十分であり、hMpl特異的なヒト抗体を得ることができなかった。また、ヒト巨核球系細胞株を抗原として用いる場合、他の膜分子に対する抗体も誘導されるため、c-Mpl特異的な抗体を効率良く誘導するためには、マウス細胞株、しかも可能な限りMHC適合した細胞株を宿主に、ヒトc-Mplを導入した高発現株を用いるのが望ましい。そこで、ヒトc-Mpl(hMpl)高発現株を作製するために、hMpl発現ベクターを以下の通り作製し、かつ2種類のマウス細胞株(L929およびFM3A)へ導入を行った。
【0111】
更に、hMplにはリガンド非依存性に恒常的にアゴニストシグナルを伝達する変異型受容体が報告されている(508番目TrpがSerに変換している変異体、Abe Mら、Leukemia. 2002 Aug;16(8):1500-1506)が、このような変異体は野生型とは立体構造が異なることが予想される。このような恒常活性型変異体に親和性が高い抗体は、強力なアゴニスト活性を示す可能性がある。そこで、恒常活性型変異体(以下hMpl-Ser)の発現ベクターも作製し、発現細胞を作製して免疫に用いた。
【0112】
1) 抗ヒトc-Mpl(hMpl)発現ベクターの調製
hMplの全長cDNAを保持するプラスミドDNAであるhumpl-Pas12(Bartley TDら、 Cell 1994 Jul 1;77(7):1117-1124.或はMorita Hら、FEBS Lett. 1996 Oct 21;395(2-3):228-234.)のDNAを鋳型としてhMplのコーディング領域全域を増幅するPCR反応を実施した。プライマーとして、末端に連結のための制限酵素部位(5’末側EcoRI、3’末側XbaI)を付加するようにデザインした、Mpl_F1及びMpl_R2を用いて、KOD-Plus- DNAポリメラーゼ(東洋紡績(株)製、日本)によるPCRで増幅した。以下、実施例中のPCRの反応温度調節は、ジーンアンプPCRシステム9700((株)パーキンエルマー・ジャパン社製)を使用した。反応温度条件としては、94℃の初期温度で5分間の加熱後、98℃/10秒間と68℃/3分間のサイクルを30回反復し、最後に72℃/7分間加熱した。増幅したPCR断片は、エタノール沈殿で回収した後、アガロースゲル電気泳動で分離し、メンブランを用いるDNA精製キットであるQIAquick Gel Extraction Kit(キアゲン社製)にて精製した。精製したDNA断片をpCR4Blunt- TOPO ベクター(東洋紡績(株)製)にサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。DNA塩基配列決定のためのプライマーとして、M13-20FW及びM13RVを用いた。挿入部分のDNA塩基配列解析を行い、hMplの配列(GenBank ACCESSION :M90102)と相違がなく、また、プライマー部分もデザインどおりの配列を有するプラスミドDNAを選択した。次に、hMplの配列を含むプラスミドDNAを精製した後、制限酵素EcoRIとXbaIで消化して、アガロースゲル電気泳動で約2kb弱のDNAを回収し精製した。他方、ヒトEFプロモーターとブラストサイジン(Bsd)選択マーカーを有する発現ベクターであるpEF6/Myc-His(インビトロジェン(株)社製)、及び、CMVプロモーターとネオマイシン(Neo)選択マーカーを有するpEGEP-N1ベクター(ベクトン・ディキンソン・バイオサイエンス・クローンテック社製)についても、同様に制限酵素EcoRI、とXbaIでの消化処理を行った後、脱リン酸化のためにAlkaline Phosphatase (E. coli C75)(タカラバイオ(株)、日本)で処理した後に、アガロースゲル電気泳動とDNA精製キットでDNAを回収した。精製したhMplの全領域DNA断片を、それぞれの発現ベクターDNAにT4 DNA ligase を用いてライゲーションして、大腸菌DH10Bへ導入し、それぞれ形質転換体を得た。インサートDNAを含む形質転換体のプラスミドDNAについてDNA塩基配列を解析して、hMplの全長cDNAが挿入されたpEF-MPL635及びpCMV-MPL635を取得した。
Mpl_F1:5’-AGAGAGAGAG GAATTCGCCA CCATGCCCTC CTGGGCCCTC TT-3’(配列番号12)
Mpl_R2:5’- AGAGAGAGAG CGGCCGCTCA AGGCTGCTGC CAATAGCTTA GTG -3’(配列番号13)
M13-20FW: 5’-GTAAAACGACGGCCAGTG-3’(配列番号14)
M13RV:5’-CAGGAAACAGCTATGAC-3’(配列番号15)
【0113】
2) 恒常活性型ヒトc-Mpl(hMpl-Ser)発現ベクターの調製
TPO非依存性に細胞内のシグナル活性化が報告されているhMpl変異体(508番目TrpがSerに変換している変異体、Abe Mら、Leukemia. 2002 Aug;16(8):1500-1506)の発現ベクターを作製した。508番目のアミノ酸残基をコードするコドンを変更(TGG→TCG)するために、pEF-MPL635のDNAを鋳型として、GeneEditorTM in vitro Site-Directed Mutagenesis System(プロメガ社)を用いた部位特異的変異導入法を実施した。変異導入用オリゴヌクレオチド(5’末端リン酸化済み)としては、Mut_MplSer508を用いた。目的の変異導入用オリゴヌクレオチドと上記キット付属のSelection Oligonucleotideを鋳型DNAとアニーリングさせて変異導入鎖を合成した後、GeneEditorTM Antibiotic Selection Mix存在下では変異体のみが増殖することを利用して変異体を選択した。より具体的には、dsDNAテンプレートをアルカリ条件下(0.2M NaOH、0.2 mMEDTA(最終濃度))室温で5分間インキュベートした後、2M酢酸アンモニウム(pH4.6)を10分の1容量加えて中和してからエタノール沈殿により回収した。アルカリ変性処理した鋳型DNAに、変異導入用オリゴヌクレオチドと新しい抗生物質耐性獲得用Selection Oligonucleotide(5’末端リン酸化)、及び、キット添付のアニーリングバッファーを加えた後、75℃で5分間保温し、37℃にゆっくり下げることによりアニーリングを行なった。次に、変異鎖の合成と連結のために、キット付属のSynthesis 10×buffer、T4 DNA Polymerase、及びT4 DNA ligaseを加えて、37℃で90分反応を行なった。GeneEditorTM Antibiotic Selection Mix存在下でコンピテントセルBMH 71-18 mutSに形質転換して培養した形質転換体大腸菌よりプラスミドDNAを調製し、更にそのDNAによりコンピテントセルJM109を形質転換後、GeneEditorTM Antibiotic Selection Mixを含むLBプレートに播種した。プレートに生じた形質転換体を培養して、プラスミドDNAのDNA塩基配列を解析して、508番目のアミノ酸が変換(TrpからSer)したhMplを発現するpEF-MPL635-Serベクターを取得した。
Mut_MplSer508:5’-CTGCTGCTGC TGAGGTCGCA GTTTCCTGCA CACTAC-3’(配列番号16)
【0114】
3) 全長ヒトc-Mpl発現L929細胞の作製
作製したpEF-MPL635ベクター(1μg)を、Lipofectamine試薬(Invitrogenより購入)および Lipofectamine PLUS(Invitrogenより購入)試薬と混合し、さらに無血清Dulbecco’s Modified Eagle Medium(DMEM)培地と混合した。混合液を1.5×105cells/well で6-well plateに培養したL929細胞に添加し、3時間培養することで細胞にDNAを導入した。10%ウシ胎児血清(FBS)添加DMEM培地にて一晩培養し、翌日より、培地に10μg/mL Blasticidin(Invitrogenより購入)を加えて薬剤耐性細胞を選抜した。その後、抗c-Mpl抗体を用いたFluorescence Activated Cell Sorting(FACS)法にてc-Mpl発現細胞を単離し、全長ヒトc-Mpl発現L929細胞株(以下、L929-hMpl)を樹立した。FACSはFACS-Vantage(Becton Dickinson社製)によって行った。選抜後は5μg/mL Blasticidin, 10%FBS添加DMEM培地にて培養、維持した。
【0115】
4) 全長ヒトc-Mpl発現FM3A細胞の作製
上記 3)と同様の方法でpEF-MPL635ベクターをFM3A細胞に導入し、全長ヒトc-Mpl発現FM3A細胞株(以下、FM3A-hMpl)を樹立した。5μg/mL Blasticidin, 10%FBS添加 Roswell Park Memorial Institute(RPMI)培地にて培養、維持した。
【0116】
5) 恒常活性型ヒトMpl発現FM3A細胞作製
上記、pEF-MPL635-Serベクターを、3)と同様の方法で、FM3A細胞に導入し、hMpl-Ser発現FM3A細胞株(以下、FM3A-hMpl-Ser)を樹立した。5μg/mL Blasticidin, 10%FBS添加RPMI培地にて培養、維持した。
【0117】
1-2 可溶化ヒトc-Mpl組換えタンパク質の作製
ヒトc-Mplの細胞膜貫通領域および細胞内領域を欠失させた、下記配列の可溶化型ヒトc-MplをコードするDNAを発現ベクターpEAK8(EdgeBioSystems社製)に結合し、トランスフェクタム試薬(Promega社より入手可能)にてHek293細胞に導入した。安定発現株を選択後、その培養上清を抗Mpl抗体カラムにて精製し、可溶化型ヒトc-Mpl組換えタンパク質(以下soluble Mpl-x、sMpl-xと略記)を調製した。
NH2-MPSWALFMVTSCLLLAPQNLAQVSSQDVSLLASDSEPLKCFSRTFEDLTCFWDEEEAAPSGTYQLLYAYPREKPRACPLSSQSMPHFGTRYVCQFPDQEEVRLFFPLHLWVKNVFLNQTRTQRVLFVDSVGLPAPPSIIKAMGGSQPGELQISWEEPAPEISDFLRYELRYGPRDPKNSTGPTVIQLIATETCCPALQRPHSASALDQSPCAQPTMPWQDGPKQTSPSREASALTAEGGSCLISGLQPGNSYWLQLRSEPDGISLGGSWGSWSLPVTVDLPGDAVALGLQCFTLDLKNVTCQWQQQDHASSQGFFYHSRARCCPRDRYPIWENCEEEEKTNPGLQTPQFSRCHFKSRNDSIIHILVEVTTAPGTVHSYLGSPFWIHQAVRLPTPNLHWREISSGHLELEWQHPSSWAAQETCYQLRYTGEGHQDWKVLEPPLGARGGTLELRPRSRYRLQLRARLNGPTYQGPWSSWSDPTRVETATETAW-COOH(配列番号17)
【0118】
[実施例2] モノクローナル抗体の作製
本発明の抗体は、遺伝子改変によってヒト抗体を産生する能力を持つヒト抗体産生マウス(KMマウスTM)を免疫し、モノクローナル抗体を作製することで得られた。KMマウスは、内在性マウスイムノグロブリン(Ig)重鎖及びマウスκ軽鎖を欠損しており、かつ、ヒトIg重鎖遺伝子を含む14番染色体断片(SC20)及びヒトIgκ鎖トランスジーン(KCo5)を同時に保持する。即ち、KMマウスはヒト抗体を産生する能力を持ち、マウスIg重鎖およびκ鎖を欠損したマウスである。このマウスはヒトIg重鎖遺伝子座を持つ系統Aのマウスと、ヒトIgκ鎖トランスジーンを持つ系統Bのマウスとの交配により作製される。系統Aは、内因性Ig重鎖及びκ軽鎖破壊の両者についてホモ接合体であり、子孫伝達可能な14番染色体断片(SC20)を保持するマウス系統(Tomizuka. et al., Proc Natl Acad Sci USA., 2000 Vol97:722参照)である。また、系統Bは内在性マウスIg重鎖及びκ軽鎖欠損の両者についてホモ接合体であり、ヒトIgκ鎖トランスジーン(KCo5)を保持するマウス系統(Nat Biotechnol., 1996 Vol14:845参照)である。
【0119】
本実施例におけるモノクローナル抗体の作製は、公知の方法によって行った(単クローン抗体実験操作入門 安東民衛ら著作、講談社(東京、日本)発行 1991参照)。
【0120】
1) 免疫
免疫原としてのヒトc-Mplは、実施例1で作製したL929-hMpl細胞、FM3A-hMpl細胞、恒常活性型c-Mpl発現FM3A-hMpl-Ser細胞、sMpl-x組換えタンパク質を用いた。被免疫動物は、実施例2で作製したヒト免疫グロブリンを産生するヒト抗体産生マウス使用し、下記の免疫方法で行った。
【0121】
免疫方法1: 9週齢のヒト抗体産生マウスに、実施例1で作製したL929-hMpl細胞(5×106細胞)をRibiアジュバンドと混合し、腹腔内に初回免疫した。初回免疫以降、一週間ごとに、同細胞(2×106細胞)を、インターロイキン6(IL-6)(5μg)と同時に、尾静脈経由で7回免疫した。さらに、脾臓及びリンパ節の取得3日前に同細胞を尾静脈経由で最終免疫した。
【0122】
免疫方法2: 9週齢のヒト抗体産生マウスに、実施例1で作製したFM3A-hMpl-Ser細胞(5×106細胞)に紫外線照射し、Ribiアジュバンドを加え腹腔内に初回免疫した。初回免疫から以降、同細胞(5×106細胞)を腹腔内投与で、一週間ごとに7回免疫した。さらに、脾臓及びリンパ節の取得3日前に、実施例1で作製したFM3A-hMpl細胞(2×106細胞)をIL-6(5μg)を加えて、尾静脈経由で最終免疫した。
【0123】
免疫方法3: 9週齢のヒト抗体産生マウスに、実施例1で作製したsMpl-x組換えタンパク質(10μg)を、完全フロイントアジュバンド(CFA)と混合して皮下投与で初回免疫した。2回目から5回目の免疫は、一週間に一度、sMpl-x組換えタンパク質(5μg)を不完全フロイントアジュバンド(IFA)と混合して皮下投与で免疫した。6回目から8回目の免疫では、L929-hMpl細胞(5×106細胞)を腹腔内に免疫した。さらに、脾臓及びリンパ節の取得3日前に、sMpl-x組換えタンパク質(5μg)およびIL-6(5μg)を、尾静脈経由で最終免疫した。
【0124】
2) ハイブリドーマ作製
最終免疫の3日後にマウスから脾臓及び/又はリンパ節を外科的に取得し、350mg/ml 炭酸水素ナトリウム、50単位/ml ペニシリン、50μg/ml ストレプトマイシンを含む無血清DMEM培地 10ml中に入れ、メッシュ(セルストレイナー:ファルコン社製)上でスパーテルを用いてつぶした。メッシュを通過した細胞懸濁液を遠心して細胞を沈澱させた後、この細胞を無血清DMEM培地で2回洗浄してから、無血清DMEM培地に懸濁して細胞数を測定した。一方、10% FCSを含むDMEM培地にて、37℃、5%炭酸ガス存在下で細胞濃度が1×108細胞/mlを越えないように培養したミエローマ細胞 SP2/0(ATCC No.CRL-1581)を同様に無血清DMEM培地で洗浄し、無血清DMEM培地に懸濁して細胞数を測定した。回収した細胞の懸濁液とマウスミエローマ懸濁液とを細胞数5:1で混合し、遠心後、上清を完全に除去した。このペレットに、融合剤として50%(w/v)ポリエチレングリコール1500(ベーリンガーマンハイム社製)1mlを、ピペットの先で撹拌しながらゆっくり添加した後、予め37℃に加温しておいた無血清DMEM培地1mlを2回に分けてゆっくり添加し、さらに7mlの無血清DMEM培地を添加した。遠心後、上清を除去して得られた融合細胞を、以下に記載する限界希釈法によるスクリーニングに供した。ハイブリドーマの選択は、10%のウシ胎児血清(Fetal Calf Serum、FCS)とヒポキサンチン(H)、アミノプテリン(A)、チミジン(T)(以下「HAT」という。:シグマ社製)を含有するDMEM培地中で培養することによって行った。さらに、10% FCSとHT(シグマ社製)とを含有するDMEM培地を用いて限界希釈法によりシングルクローンにした。培養は、96穴マイクロタイタープレート(ベクトンディッキンソン社製)中で行った。抗ヒトc-Mplヒトモノクローナル抗体を産生するハイブリドーマクローンの選択(スクリーニング)及び各々のハイブリドーマが産生するヒトモノクローナル抗体の特徴付けは、実施例4に記載のフローサイトメトリー、あるいは実施例5に記載のUT7/TPO細胞を用いた細胞増殖アッセイによって行った。アゴニスト抗体の活性評価系として、BaF3等のマウス細胞株にヒトMplを発現させ、細胞増殖アッセイを行うことができる(Orita et al. Blood. 2005 Jan 15;105(2):562-6.)が、そのような細胞の反応が、ヒト細胞の反応を反映しているとは限らない。UT7/TPOはヒト由来細胞株であるため、これをスクリーニングに用いることで、よりヒト細胞に強い活性を持つ抗体を選抜し易いと考えられる。
【0125】
スクリーニングの結果、抗ヒトMplアゴニスト抗体産生ハイブリドーマとして、免疫方法1によるハイブリドーマ7-10、免疫方法2によるハイブリドーマ4-49、免疫方法3による ハイブリドーマ6-4-50, 6-5-2の4クローンが選抜された。また、非アゴニスト抗体産生ハイブリドーマとして2-35(免疫方法1により得たもの)をコントロールとして選抜した。
【0126】
[実施例3] ハイブリドーマ培養上清からの精製抗体調製
ハイブリドーマの培養上清からの抗ヒトc-Mplモノクローナル抗体の精製は以下の方法で行った。抗体を含む培養上清をrmp Protein A(アマシャムファルマシアバイオテク社製)及び0.8×40cm カラム(バイオラッド社製)を用い、吸着緩衝液としてPBS、溶出緩衝液として0.02M グリシン緩衝液(pH 3)を用いてアフィニティー精製した。溶出画分は1M Tris (pH 9.0)を添加してpH7.2付近に調整した。調製された抗体溶液は、透析膜(10000カット、Spectrum Laboratories社製)を用いてPBSに置換し、孔径0.22μm のメンブランフィルターMILLEX-GV(ミリポア社製)でろ過滅菌し、精製抗ヒトc-Mplモノクローナル抗体を得た。精製抗体の濃度は280nmの吸光度を測定し、1mg/ml を1.4 OD として算出した。
【0127】
抗ヒトc-Mplモノクローナル抗体を含む培養上清の調製は以下の方法にて行った。
まず、抗体産生ハイブリドーマを10ng/ml Recombinant Human IL-6(R&D Systems社製)、10% Low IgG Fetal Bovine Serum(HyClone社製)含有eRDF培地(極東製薬社製)に馴化した。この馴化したハイブリドーマを凍結保存した。次に、その一部を、ウシインシュリン(5μg/ml、ギブコビーアールエル社製)、ヒトトランスフェリン(5μg/ml、ギブコビーアールエル社製)、エタノールアミン(0.01mM、シグマ社製)、亜セレン酸ナトリウム(2.5×10-5mM、シグマ社製)、10ng/ml Recombinant Human IL-6(R&D Systems社製)、1% Low IgG Fetal Bovine Serum(HyClone社製)含有eRDF培地(極東製薬社製)に馴化した。フラスコにて培養し、ハイブリドーマの生細胞率が90%になった時点で培養上清を回収した。回収した上清は、10μm と0.2μmのフィルター(ゲルマンサイエンス社製)に供し、きょう雑物を除去した。
【0128】
[実施例4] フローサイトメトリーによる抗ヒトc-Mpl抗体の結合活性評価
ハイブリドーマ培養上清もしくは精製抗体を用いたフローサイトメトリーによって、抗ヒトc-Mpl抗体の結合活性を測定した。手順は以下の通りである。細胞はFM3A-hMpl細胞もしくはヒトMpl発現FDCP2細胞(FDCP-hMpl)(FEBS Lett. 1996 Oct 21;395(2-3):228-34.参照)を用いた。
【0129】
反応あたり4×105個の細胞を50 μLのFACS staining medium(2% FBS, 0.1%NaN3, 1mM EDTA in PBS)に懸濁し、50μLのハイブリドーマの培養上清もしくは精製ヒト抗体(終濃度0.1-1μg/mL)を加え、氷上にて30分間反応させた。FACS staining mediumによる洗浄後、二次抗体であるR-phycoerythrin (RPE)標識ヤギ抗ヒトIgγ F(ab’)抗体(Southern Biotechnology 社製Cat#2043-09)を加え、再び氷上で30分遮光反応させた後、再び洗浄した。細胞をPropidium Iodide(PI)を加えたFACS staining mediumに懸濁し、解析を行った。解析はFACS Calibur(Becton Dickinson社製)にて行った。
【0130】
図1に各抗体の精製抗体を用いたフローサイトメトリーの結果を示す。各抗体はFDCP-hMpl細胞に結合したが、その親細胞であるFDCP2細胞(FDCP parent)には結合しなかった。従って、これらの抗体はヒトMpl特異的に結合していることが示された。
【0131】
[実施例5] UT7/TPO細胞を用いた抗ヒトc-Mpl抗体のアゴニスト活性評価
ハイブリドーマ上清もしくは精製抗体を用いて、UT7/TPO細胞増殖アッセイを行い、アゴニスト活性を評価した。UT7/TPO細胞はTPO依存性のヒト巨核球系細胞株である(Ozaki K et al. Blood. 1998 Dec 15;92(12):4652-62.参照)。通常は10%FBS, 5ng/mL PEG-rHuMGDF添加 Iscove’s Modified Dulbecco’s Medium(IMDM)で培養、維持した。細胞増殖アッセイの手順は以下の通りである。
【0132】
(1) UT7/TPO細胞培養液を50mLチューブに移し、遠心分離(1,500rpm, 5min, 4℃)により細胞をペレットにした。培地を除き、サイトカイン非添加, 10%FBS添加 IMDM培地(以下、増殖アッセイ用培地)でペレットを懸濁した。再び遠心し、新しい増殖アッセイ用培地で懸濁した。遠心、懸濁をもう一度繰り返した。
(2) 上記1.で増殖アッセイ用培地に懸濁した細胞を37℃, 5%CO2条件下で6時間培養した。
(3) 培養後、細胞を遠心してペレットにし、増殖アッセイ用培地にて懸濁した。このとき、細胞濃度を6×105cells/mLにあわせ、細胞懸濁液を、各ウェル50μLで96-well plateに播いた。
(4) 次に、ハイブリドーマ培養上清10μLに、増殖アッセイ用培地40μLを加え、各ウェルに添加する。精製抗体の場合は、増殖アッセイ用培地50μLに検体を終濃度の2倍の濃度で加え、各ウェルに添加した。
(5) 37℃,5%CO2にて、48時間培養する。
(6) WST-8試薬(同仁化学研究所社製)を10μL/wellで添加し、2時間培養する。
(7) 吸光マイクロプレートリーダー(TECAN社製SUNRISE RAINBOW)にて各ウェルの吸光度を測定する。(測定波長450nm, 参照波長600nm以上)
図2に7-10(図2A), 4-49(図2B), 6-4-50(図2C), 6-5-2(図2D)各精製抗体を用いたUT7/TPO細胞増殖アッセイにおける増殖曲線を示す。また、スクリーニングの結果得られた抗ヒトc-Mpl抗体のサブクラス、活性の強さ(UT7/TPO細胞増殖アッセイにおける50%有効濃度(EC50)、最大活性(Max))、各抗体が得られた実施例2に記載の免疫方法を表2に示す。
【0133】
【表2】
【0134】
[実施例6] コロニーアッセイ
ヒト臍帯血由来CD34+ 細胞を用いたCFU-Mkコロニー形成アッセイを行い、精製抗体のヒトプライマリー細胞に対する作用を検討した。アッセイにはMegaCultTM-C(Stem Cell Technologies社製Cat#04972)を使用した。手順は以下の通りである。
【0135】
(1) 検体を含んだ0.15mLのIMDMに、0.85mLのMegaCultTM-C培地を加え1mLとした。
(2) ヒト臍帯血より調製したCD34+細胞を1.1×105cells/mLの濃度で、IMDMに懸濁し、上記1.の培地が入ったチューブに0.05mLずつ添加した。
(3) 細胞を加えた各チューブをボルテックスで攪拌し、0.6mLの氷冷したコラーゲン液を加え、再びボルテックスで攪拌した。
(4) 上記3.までの細胞、検体混合液をチャンバースライドの各ウェルに0.75mLずつ添加した。
(5) チャンバースライドを、100mmペトリディッシュ中に入れた。乾燥を防ぐために、3mLの精製水を入れた35mmペトリディッシュを同じ100mmペトリディッシュ中に入れた。
(6) チャンバースライドが入ったペトリディッシュをインキュベーター中に静置し、37℃,5%CO2条件下で10〜12日間培養した。
(7) 培養後、固定液(メタノール:アセトン=1:3)にて固定した。
(8) 抗ヒトCD41抗体による免疫染色を行い、CFU-Mkコロニーを検出した。顕微鏡下でコロニー数をカウントし、各検体のCFU-Mkコロニー形成能を比較した。
【0136】
図3にコロニーアッセイの結果を示した。7-10_IgG1, 4-49_IgG1によってコロニー形成が誘導された。
【0137】
[実施例7] 抗体遺伝子クローニングと配列決定
組み換え抗体を作製するために、選抜された抗ヒトc-Mplアゴニスト抗体産生ハイブリドーマから抗体遺伝子、具体的には重鎖(H鎖)をコードするヒトIgγ cDNAおよび軽鎖(L鎖)をコードするヒトIgκ cDNAを単離し、配列を決定した。
1)各モノクローナル抗体のcDNA合成
各ハイブリドーマで発現するヒト抗体重鎖、及び軽鎖の抗体の可変領域を含むDNA断片を取得するために、ヒトIgγ、及びヒトIgκの各々の定常領域に特異的なプライマーを用いた5’RACE(5' rapid amplification of cDNA ends)法によるクローニングを行なった。具体的には、BD SMART RACE cDNA Amplification Kit(ベクトン・ディキンソン・バイオサイエンス・クローンテック社製)を用い、添付の説明書にしたがって実施した。
【0138】
cDNA合成の材料としては、ハイブリドーマ7-10, 4-49, 6-4-50, 6-5-2細胞にRNA抽出用試薬であるISOGEN(日本ジーン社製、日本)を添加し、取扱説明書にしたがってTotal RNAを精製した。精製したtotal RNAより各約1μgを鋳型として用いて、1st strand cDNAを作製した。
1st strand cDNA の合成は、
Total RNA 1μg/3μl
5’CDS 1μl
SMART Oligo 1μl
上記組成の反応液を70℃で2分間インキュベートした後、
5×Buffer 2μl
DTT 1μl
DNTP mix 1μl
PowerScript Reverse Transcriptase 1μl
を加え42℃で1.5時間インキュベートした。
さらに、50μlのTricine -EDTA Bufferを加えた後、72℃で7分間インキュベートし、1st strand cDNAを取得した。
【0139】
2)PCRによる重鎖遺伝子、軽鎖遺伝子の増幅と塩基配列の確認
2-1)PCRによる重鎖遺伝子、軽鎖遺伝子の増幅
ヒト抗体遺伝子のcDNAを増幅するために、ヒト抗体特異的配列を有する3’プライマー(具体的な配列は後記)とBD SMART RACE cDNA Amplification Kit で合成されたcDNAの5’末端に付加された配列に特異的にハイブリダイズする5’プライマー(Universal primer A mix)をPCR用のプライマーセットとして、またPCR用酵素としてKOD-Plus- DNAポリメラーゼ(東洋紡績(株)社製)を用いて、下記の反応液を調製してPCRに供した。
【0140】
sterile H2O 28μl
cDNA 2.5μl
KOD-Plus- buffer (10X) 5μl
dNTP Mix (2mM) 5μl
MgSO4 (25mM) 2μl
KOD-Plus- (1 unit/μl) 1μl
Universal primer A mix (UPM) (10X) 5μl
Gene specific primers (GSP)(10μM) 1.5μl
Total volume 50μl
重鎖遺伝子の増幅反応には、SMART RACE cDNA Amplification Kit付属のUPMプライマーとIgG1pプライマーを用い、他方、軽鎖遺伝子の増幅にはUPMプライマーとhk-2プライマーの各セットを使用した。
IgG1pプライマー:5’-TCTTGTCCACCTTGGTGTTGCTGGGCTTGTG-3’(配列番号18)
hk-2:5’-GTT GAA GCT CTT TGT GAC GGG CGA GC -3’(配列番号19)
また反応温度条件は次のとおりである。
94℃ /30 秒間、72℃/3分間のサイクルを5回反復、
94℃ /30 秒間、70℃/30秒間、72℃/3分間のサイクルを5回反復、
94℃ /30 秒間、68℃/30秒間、72℃/3分間のサイクルを25回反復した。
【0141】
さらに、この反応液2μlにTricine -EDTA Buffer 98μlを加えて希釈したもの5μlを鋳型とし、第一PCRよりも内側にプライマーを設定した第二PCR(Nested PCR)を実施した。PCR反応溶液の組成を次に示す。
【0142】
sterile H2O 30μl
第一PCR反応液(50倍希釈) 5μl
KOD-Plus- buffer (10X) 5μl
dNTP Mix (2mM) 5μl
MgSO4 (25mM) 2μl
KOD-Plus- (1 unit/μl) 1μl
Nested Universal primer A (NUP;10μM) 1μl
Gene specific primers (GSP)(10μM) 1μl
Total volume 50μl
上記反応のプライマーセットとして、重鎖遺伝子増幅用の場合は、NUPMプライマー(SMART RACE cDNA amplification Kit付属;ベクトン・ディキンソン・バイオサイエンス・クローンテック社製)とhh2プライマー(4-49、6-4-50、6-5-2の場合)、或はIgG2p_134(7-10の場合)を使用して、また、軽鎖遺伝子の増幅の場合は、UPMプライマーとhk-5プライマーを用いた。反応温度条件としては、94℃の初期温度で1分間の後、94℃/5秒間、68℃/10秒及び72℃/3分間のサイクルを20回反復、最後に72℃/7分間加熱した。
【0143】
2-2)抗体遺伝子の塩基配列決定
上記の方法で増幅した重鎖PCR断片(以下HV[C]と記載)は、重鎖の5’非翻訳領域、リーダー配列(分泌シグナル配列)、可変領域(HV)及び定常領域の一部([C])より構成される。同様に、軽鎖のPCR増幅断片(以下LV[C]と記載)は、軽鎖の5’非翻訳領域、リーダー配列(分泌シグナル配列)、可変領域(LV)及び定常領域の一部([C])より構成される。ここでリーダー配列(分泌シグナル)とは抗体の分泌に必要で、成熟抗体タンパク質からは切り離されるアミノ酸配列である。HV[C]断片およびLV[C]断片は、PCR反応液からエタノール沈殿で回収した後、アガロースゲル電気泳動で分離し、メンブランを用いるDNA精製キットであるQIAquick Gel Extraction Kit (キアゲン社製)にて精製した。精製したHV[C]増幅断片あるいはLV[C]増幅断片は、それぞれZero Blunt TOPO PCR Cloning Kit(インビトロジェン社製)のpCR 4 Blunt- TOPO ベクター(東洋紡績(株)製)にサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。DNA塩基配列決定のためにプライマーとして、M13-20FW及びM13RVを用いた。
hk-5:5’- AGG CAC ACA ACA GAG GCA GTT CCA GAT TTC-3’(配列番号20)
hh2プライマー:5’- GCT GGA GGG CAC GG TCA CCA CGC TG -3’(配列番号21)
IgG2p_134:5’- TGCACGCCGC TGGTCAGGGC GCCTGAGTTC C -3’(配列番号22)
【0144】
アゴニスト抗体7-10の重鎖可変領域、及び軽鎖可変領域をコードするDNA塩基配列、並びに重鎖可変領域及び軽鎖可変領域のアミノ酸配列をそれぞれ以下に示す。
<7-10重鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGAGTTGGGACTGAGCTGGATTTTCCTTTTGGCTATTTTAAAAGGTGTCCAGTGTGAAGTGCAGCTGGTGGAGTCTGGGGGAGGCTTGGTACAGCCTGGCAGGTCCCTGAGACTCTCCTGTGCAGCCTCTGGATTCACCTTTGATGATTATGCCATGCACTGGGTCCGGCAAGCTCCAGGGAAGGGCCTGGAGTGGGTCTCAGGTATTAGTTGGAATAGTGGTAGCATAGGCTATGCGGACTCTGTGAAGGGCCGATTCACCATCTCCAGAGACAACGCCAAGAACTCCCTGTATCTGCAAATGAACAGTCTGAGAGCTGAGGACACGGCCTTGTATTACTGTGCAAAAAATCTATGGTTCGGGGAGTTCCGTTACTGGTACTTCGATCTCTGGGGCCGTGGCACCCTGGTCACTGTCTCCTCA(配列番号23)
<7-10重鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
MELGLSWIFLLAILKGVQCEVQLVESGGGLVQPGRSLRLSCAASGFTFDDYAMHWVRQAPGKGLEWVSGISWNSGSIGYADSVKGRFTISRDNAKNSLYLQMNSLRAEDTALYYCAKNLWFGEFRYWYFD LWGRGTLVTV SS(配列番号24)
<7-10軽鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGACATGAGGGTCCCCGCTCAGCTCCTGGGGCTTCTGCTGCTCTGGCTCCCAGGTGCCAGATGTGCCATCCAGTTGACCCAGTCTCCATCCTCCCTGTCTGCATCTGTAGGAGACAGAGTCACCATCACTTGCCGGGCAAGTCAGGGCATTAGCAGTGCTTTAGCCTGGTATCAGCAGAAACCAGGGAAAGCTCCTAAGCTCCTGATCTATGATGCCTCCAGTTTGGAAAGTGGGGTCCCATCAAGGTTCAGCGGCAGTGGATCTGGGACAGATTTCACTCTCACCATCAGCAGCCTGCAGCCTGAAGATTTTGCAACTTATTACTGTCAACAGTTTAATAGTTACCCGCTCACTTTCGGCGGAGGGACCAAGGTGGAGATCAAA(配列番号25)
<7-10軽鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
MELGLSWIFLLAILKGVQCEVQLVESGGGLVQPGRSLRLSCAASGFTFDDYAMHWVRQAPGKGLEWVSGISWNSGSIGYADSVKGRFTISRDNAKNSLYLQMNSLRAEDTALYYCAKNLWFGEFRYWYFDLWGRGTLVTVSS(配列番号26)
【0145】
アゴニスト抗体4-49の重鎖可変領域、及び軽鎖可変領域をコードするDNA塩基配列、並びに重鎖可変領域及び軽鎖可変領域のアミノ酸配列をそれぞれ以下に示す。
<4-49重鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGAGTTGGGACTGAGCTGGATTTTCCTTGTGGCTATTTTAAAAGGTGTCCAGTGTGAAGAGCAGCTGGTGGAGTCTGGGGGAGGCTTGGTACAGCCTGGCAGGTCCCTGAGACTCTCCTGTACAGCCTCTGGATTCACCTTTGATGATTATGCCATGTACTGGGTCCGGCAAGTTCCAGGGAAGGGCCTGGAGTGGGTCTCAGGTATTAGTTGGAACAGTGGTAGCATAGGCTATGCGGACTCTGTGAAGGGCCGATTCACCGTTTCCAGAGACAACGCCAAGAACTCCCTGTATCTGCAAATGAACAGTCTGAGAGCTGAGGACACGGCCTTATATTACTGTGCAAAAGCCCTATGGTTCGGGGAGTTCCCCCACTACTACGGTATGGACGTCTGGGGCCAAGGGACCACGGTCACCGTCTCCTCA(配列番号27)
<4-49重鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
MELGLSWIFLVAILKGVQCEEQLVESGGGLVQPGRSLRLSCTASGFTFDDYAMYWVRQVPGKGLEWVSGISWNSGSIGYADSVKGRFTVSRDNAKNSLYLQMNSLRAEDTALYYCAKALWFGEFPHYYGMDVWGQGTTVTVSS(配列番号28)
<4-49軽鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGACATGAGGGTCCCCGCTCAGCTCCTGGGGCTTCTGCTGCTCTGGCTCCCAGGTGCCAGATGTGCCATCCAGTTGACCCAGTCTCCATCCTCCCTGTCTGCATCTGTAGGAGACAGAGTCACCATCACTTGCCGGGCAAGTCAGGGCATTAGCAGTACTTTAGCCTGGTATCAGCAGAAACCAGGGAAAGCTCCTAAGCTCCTGATCTATGATGCCTCCAGTTTGGAAAGTGGGGTCCCATCAAGGTTCAGCGGCAGTGGATCTGGGACAGATTTCACTCTCACCATCAGCAGCCTGCAGCCTGAAGATTTTGCAACTTATTACTGTCAACAGTTTAATAGTTACCCGTACACTTTTGGCCAGGGGACCAAGCTGGAGATCAAACGT(配列番号29)
<4-49軽鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
MDMRVPAQLLGLLLLWLPGARCAIQLTQSPSSLSASVGDRVTITCRASQGISSTLAWYQQKPGKAPKLLIYDASSLESGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQFNSYPYTFGQGTKLEIKR(配列番号30)
【0146】
アゴニスト抗体6-4-50の重鎖可変領域、及び軽鎖可変領域をコードするDNA塩基配列、並びに重鎖可変領域及び軽鎖可変領域のアミノ酸配列をそれぞれ以下に示す。
<6-4-50重鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGAATTGGGACTGAGCTGGATTTTCCTTTTGGCTATTTTAAAAGGTGTCCAGTGTGAAGTGCAGCTGGTGGAGTCTGGGGGAGGCTTGGTACAGCCTGGCAGGTCCCTGAGACTCTCCTGTGCAACCTCTGGATTCACCTTTGATAATTATGCCATGTACTGGGTCCGGCAAGCTCCAGGGAAGGGCCTGGAGTGGGTCTCAGGTATTAGTTGGAATAGTGGTGACATAGGCTATGCGGACTCTGTGAAGGGCCGATTCACCATCTCCAGAGACAACGCCAAGAACTCCCTGTATCTGCAAATGAACAGTCTGAGAGCTGAGGACACGGCCTTGTATTACTGTGCAAGGGATGCGGGGTTCGGGGAGTTCCACTACGGTCTGGACGTCTGGGGCCAAGGGACCACGGTCACCGTCTCCTCA(配列番号31)
<6-4-50重鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
MELGLSWIFLLAILKGVQCEVQLVESGGGLVQPGRSLRLSCATSGFTFDNYAMYWVRQAP
GKGLEWVSGISWNSGDIGYADSVKGRFTISRDNAKNSLYLQMNSLRAEDTALYYCARDAG
FGEFHYGLDVWGQGTTVTVSS(配列番号32)
<6-4-50軽鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGACATGAGGGTCCCCGCTCAGCTCCTGGGGCTTCTGCTGCTCTGGCTCCCAGGTGCCAGATGTGCCATCCAGTTGACCCAGTCTCCATCCTCCCTGTCTGCATCTGTAGGAGACAGAGTCACCATCACTTGCCGGGCAAGTCAGGGCATTAGCAGTGCTTTAGCCTGGTATCAGCAGAAACCAGGGAAAGTTCCTAAGCTCCTGATCTATGATGCCTCCAGTTTGGAAAGTGGGGTCCCATCAAGGTTCAGCGGCAGTGGATCTGGGACAGATTTCACTCTCACCATCAGCAGCCTGCAGCCTGAAGATTTTGCAACTTATTACTGTCAACAGTTTAATAGTTACCCGTGGACGTTCGGCCAAGGGACCAAGGTGGAAATCAAACGT(配列番号33)
<6-4-50軽鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
MDMRVPAQLLGLLLLWLPGARCAIQLTQSPSSLSASVGDRVTITCRASQGISSALAWYQQKPGKVPKLLIYDASSLESGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQFNSYPWTFGQGTKVEIKR(配列番号34)
【0147】
アゴニスト抗体6-5-2の重鎖可変領域、及び軽鎖可変領域をコードするDNA塩基配列、並びに重鎖可変領域及び軽鎖可変領域のアミノ酸配列をそれぞれ以下に示す。
<6-5-2重鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGAGTTGGGACTGAGCTGGATTTTCCTTTTGGCTATTTTAAAAGGTGTCCAGTGTGAAGTGCAACTGGTGGAGTGTGGGGGAGGCTTGGTACAGCCTGGCAGGTCCCTGAGACTCTCCTGTGCAGCCTCTGGATTCACCTTTGATGATTATGCCATGCACTGGGTCCGGCAAGCTCCAGGGAAGGGCCTGGAGTGGGTCTCAGGTATTAGTTGGAATAGTGGTAGTATAGGTTATGCGGACTCTGTGAAGGGCCGATTCACCATCTCCAGAGACAACGCCAAGAACTCCCTGTATCTGCAAATGAACAGTCTGAGAGCTGAGGACACGGCCTTGTATTACTGTGCAAAACCTATATGGTTCGGGGAGTGGGGAAACTACTACGGTATGGACGTCTGGGGCCAAGGGACCACGGTCACCGTCTCCTCA(配列番号35)
<6-5-2重鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
MELGLSWIFLLAILKGVQCEVQLVECGGGLVQPGRSLRLSCAASGFTFDDYAMHWVRQAPGKGLEWVSGISWNSGSIGYADSVKGRFTISRDNAKNSLYLQMNSLRAEDTALYYCAKPIWFGEWGNYYGMDVWGQGTTVTVSS(配列番号36)
<6-5-2軽鎖核酸配列>(ATG開始コドンより可変領域C末端アミノ酸残基をコードするDNA配列まで)
ATGGAAACCCCAGCGCAGCTTCTCTTCCTCCTGCTACTCTGGCTCCCAGATACCACCGGAGAAATTGTGTTGACGCAGTCTCCAGGCACCCTGTCTTTGTCTCCAGGGGAAAGAGCCACCCTCTCCTGCAGGGCCAGTCAGAGTGTTAGCAGCAGCTACTTAGCCTGGTACCAGCAGAAACCTGGCCAGGCTCCCAGGCTCCTCATCTATGATGCATCCAGCAGGGCCACTGGCATCCCAGACAGGTTCAGTGGCAGTGGGTCTGGGACAGACTTCACTCTCACCATCAGCAGACTGGAGCCTGAAGATTTTGCAGTGTATTACTGTCAGCAGTATGGTAGCTCACCGATCACCTTCGGCCAAGGGACACGACTGGAGATTAAACGT(配列番号37)
<6-5-2軽鎖アミノ酸配列>(リーダー配列及び可変領域まで)
(下線で示すアミノ酸残基は分泌シグナルとなるリーダー配列を示す)
METPAQLLFLLLLWLPDTTGEIVLTQSPGTLSLSPGERATLSCRASQSVSSSYLAWYQQKPGQAPRLLIYDASSRATGIPDRFSGSGSGTDFTLTISRLEPEDFAVYYCQQYGSSPITFGQGTRLEIKR(配列番号38)
【0148】
[実施例8] 組換え抗体発現ベクターの構築
上記の方法でハイブリドーマからクローニングした抗体の可変領域を、ヒト抗体発現用ベクターに組み込み、様々な定常領域を持った組換え抗体発現ベクターを作製した。
【0149】
ヒト抗体発現用ベクター、N5KG1-Val Lark(以下N5KG1と略記)(IDEC Pharmaceuticals社製, US patent 6001358参照)は、組換え抗体を動物細胞に発現させる目的で使用されるプラスミドベクターである。N5KG1の構造を図4Aに示した。N5KG1は、2つのCMV プロモーター/エンハンサーを持ち、それぞれの下流に重鎖および軽鎖可変領域遺伝子のクローニングサイトを持つ。さらにその下流には、ヒト重鎖定常領域(γ1)およびヒト軽鎖定常領域(κ)をコードする遺伝子配列をあらかじめ持っている。本ベクターの可変領域クローニングサイトに任意の重鎖および軽鎖可変領域(リーダー配列=分泌シグナル配列を含む)を、読み枠(フレーム)をあわせて組み込むことにより、軽鎖可変領域がヒトκ鎖定常領域と、重鎖可変領域がヒトγ1鎖定常領域と連結された抗体を発現させることができる。従って、本ベクターを導入された動物細胞は培養液中にIgG1抗体を産生する。
【0150】
同様に、発現ベクターN5KG4PE(IDEC Pharmaceuticals社製)は、IgG4PEの重鎖定常領域を持っている。IgG4PEとはIgG4に二つの変異(Ser228Pro, Leu235Glu)が入った配列である。Ser228Pro はIgG4の分子内架橋(S-S結合)による単量体形成を抑制する変異である。また、Leu235Gluは抗体依存性細胞傷害(Antibody-dependent cellular cytotoxicity: ADCC)活性を低減させる変異である。
【0151】
また、N5KG1のIgG1定常領域をIgG3に変換し、N5KG3を作製した。
さらに本実施例では、N5KG1, N5KG3, N5KG4PEをもとに、重鎖定常領域(特にヒンジ領域)に種々の改変を加えた発現ベクターを作製した。
【0152】
本実施例で定常領域に加えた改変は、第一に、抗体ドメイン間のサブクラスの入れ替えによるものである。抗体重鎖定常領域は、N末端側からCH1-ヒンジ-CH2-CH3というドメイン構造を持っている。本実施例では、このドメイン単位で各サブクラスの配列を組み合わせた重鎖定常領域を作製した。例えば、CH1およびヒンジ領域はヒトIgG3の配列であり、CH2およびCH3はヒトIgG1である重鎖定常領域を作製した。そして、このような重鎖定常領域を持つ抗体を、CH1/ヒンジ/CH2/CH3の順にサブクラスを表記し、IgG3/3/1/1と名付けた(以降はIgG3311というように”/”を省略して表記する)。また、例えば、ヒンジ領域はヒトIgG3の配列であり、CH1, CH2およびCH3はヒトIgG4PEである重鎖定常領域を作製した。このような重鎖定常領域を持つ抗体をIgG4344と名付けた。
【0153】
第二に、ヒトIgG3ヒンジ領域の改変体を作製した。抗体のヒンジ領域はアッパー(upper)ヒンジとミドル(middle)ヒンジに分けることができる。アッパーヒンジとはKabat EU numbering(Kabatら、Sequences of Proteins of Immunological Interest, 5thEd. Public Health Service, National Institute of Health, Bethesda, Md.(1991)) による表記で216以降、226よりN末端側の配列を指す。ミドルヒンジとは、同表記で226以降、231よりN末端側の配列を指す。ヒトIgG3のヒンジ領域は、アッパーヒンジが12アミノ酸、ミドルヒンジが50アミノ酸から成る。さらにミドルヒンジは、5アミノ酸と15アミノ酸×3回のリピート配列に分けられる(5+15x3=50)。本実施例では、IgG3ミドルヒンジの繰り返し配列を1回に短縮した変異体を作製した。このようなヒンジをG3h1と名付け、このタイプのヒンジを持つ抗体を、前述のドメイン単位の変異とあわせて、IgGx3xxh1(xは任意)というように表記した。
【0154】
また、IgG3のミドルヒンジ後半の繰り返し配列を無くした重鎖定常領域も作製した。このようなヒンジをG3uh(upper hingeの略)と名付け、IgGx3xxuhというように表記した。
さらにG3uhヒンジにL217SとR228Pの変異を加えた重鎖定常領域を作製した。この変異にはG3uhヒンジをよりIgG4PEの配列に近づける意図がある。これをG3uhm(upper hinge mutationの略)と名付け、これを持つ抗体をIgGx3xxuhmと表記した。
【0155】
図4Bに天然型のヒトイムノグロブリンと、IgG4PE, IgG4344, IgG4344h1, IgG4344uh, IgG4344uhmのヒンジ領域のアミノ酸配列を示す。
【0156】
本実施例では抗Mplアゴニスト抗体の可変領域を用いて、以下の定常領域を持つ抗体の発現ベクターを作製した。
【0157】
IgG1, IgG4PE, IgG3311, IgG3331, IgG3344, IgG3344h1, IgG4344, IgG4344h1, IgG4344uh, IgG4344uhm
【0158】
以下に各発現ベクターの作製法を記載する。
1)IgG1サブクラスの抗c-Mpl抗体発現ベクターの作製
1-1)抗ヒトc-Mpl抗体4-49_IgG1及び7-10_IgG1発現ベクターの作製
7-10と4-49については、N5KG1ベクターに最初に重鎖可変領域、次に、軽鎖可変領域の順に挿入して発現ベクターを作製した。
【0159】
発現ベクター作製の工程を図4Cに示す。7-10及び4-49それぞれのHV[C]およびLV[C]断片(実施例7に記載)を含むプラスミドDNAを鋳型として、末端に連結のための制限酵素部位(5’末側SalI、3’末側NheI)を付加するようにデザインしたプライマーセットを用いて、重鎖および軽鎖のリーダー配列と可変領域のDNAをKOD-Plus- DNAポリメラーゼによるPCRで増幅した。このPCRにより増幅された、重鎖および軽鎖のリーダー配列+可変領域を、それぞれHV断片、LV断片と表す。
【0160】
まず、N5KG1に7-10HV, 4-49HV断片の挿入を行った。HV断片増幅用のプライマーは、以下の通りである。
7-10;
HV断片5’用プライマー:40-3H5Sal
5’- AGAGAGAGAG GTCGACCACC ATGGAGTTGG GACTGAGCTG GATTT -3’(配列番号39)
HV断片3’用プライマー:40-3H3Nhe
5’- AGAGAGAGAG GCTAGCTGAG GAGACAGTGA CCAGGGTGCC A -3’(配列番号40)
4-49;
HV断片5’用プライマー:F24HSal
5’-AGAGAGAGAGGTCGACCACCATGGAGTTGGGACTGAGCTGGATTT -3’(配列番号41)
HV断片3’用プライマー:C15H3Nhe
5’- AGAGAGAGAGGCTAGCTGAGGAGACGGTGACCGTGGT-3’(配列番号42)
【0161】
反応温度条件としては、94℃の初期温度で1分間の加熱後、94℃/5秒間と68℃/45秒間のサイクルを35回反復し、最後に72℃/7分間加熱した。増幅されたDNA断片を制限酵素SalIとNheIで消化して、アガロースゲル電気泳動で約430bpのDNAを回収し精製した。他方、N5KG1ベクターについては制限酵素SalI、NheI処理を順次行った後、脱リン酸化のためにAlkaline Phosphatase (E. coli C75) (宝酒造社製、日本)にて処理した後に、アガロースゲル電気泳動とDNA精製キットで約8.9kbのDNAを回収した。これら2つの断片をT4 DNA ligase を用いてライゲーションして、大腸菌DH10Bへ導入して形質転換体を得た。得られた形質転換体のプラスミドDNAについてDNA塩基配列を解析して、重鎖定常領域の5’上流に、HV断片がフレームを合わせて挿入されたプラスミドDNA、N5KG1_7-10_Hv、及びN5KG1_4-49_Hvを取得した。
【0162】
続いて、これらHV断片が挿入されたプラスミドベクターにLV断片(軽鎖リーダー配列+可変領域)の挿入を行なった。LV[C]断片を含むプラスミドDNAを鋳型として、末端に連結のための制限酵素部位(5’末側BglII、3’末側BsiWI)を付加するようにデザインしたプライマーを用いて、LV断片をPCRで増幅した。LV増幅用プライマーは以下の通りである。
7-10;
LV断片5’用プライマー:165-1B_L18Bgl
5’- AGAGAGAGAGATCTCTCACCATGGACATGAGGGTCCCCGCTC-3’(配列番号43)
LV断片3’用プライマー:165_1B_L18_Bsi
5’- AGAGAGAGAG CGTACGTTTG ATCTCCACCT TGGTCCCTCC -3’(配列番号44)
4-49;
LV断片5’用プライマー:DNP_L1Bglp
5’- AGAGAGAGAGATCTCTCACCATGAGGGTCCCCGCTCAGCTC -3’(配列番号45)
LV断片3’用プライマー:A27_R_N202
5’- AGAGAGAGAGCGTACGTTTGATTTCCACCTTGGTCCCTTGGC-3’(配列番号46)
【0163】
反応は94℃の初期温度で1分間の加熱後、94℃/5秒間と68℃/45秒間のサイクルを35回反復し、最後に72℃/7分間の保温という条件で実施した。精製したLVの増幅DNA断片は、pCR4Blunt- TOPO ベクター(東洋紡績(株)製)にサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。DNA塩基配列決定のためにプライマーとして、M13-20FWとM13RVを用いた。挿入部分のDNA塩基配列解析を行い、鋳型としたLVと相違がなく、また、プライマー部分もデザインどおりの配列を有するプラスミドDNA(TOPO_7-10_Lv、及びTOPO_4-49_Lv)を選択した。次に、それぞれのDNAを制限酵素BglIIとBsiWIで消化して、アガロースゲル電気泳動で約400 bpのDNAを回収し精製した。その精製DNA断片と、制限酵素処理(BglII, BsiWI)及び、脱リン酸化処理を施した7-10或は4-49のHVが挿入されたベクター(約9.3kb)をT4 DNA ligase によりライゲーションした後、大腸菌DH10Bへ導入して形質転換体を得た。形質転換体について、DNA配列解析や制限酵素切断パターンを解析し、目的のプラスミドDNAを含むクローンを選択した。更に、得られた抗体発現プラスミドDNAの大量精製を行い、重鎖全領域と軽鎖全領域、及び、その挿入部位周辺のDNA塩基配列にクローニング工程での変異がないことを確認した。7-10_IgG1および4-49_IgG1発現ベクターは、各々N5KG1_7-10、及びN5KG1_4-49と名付けた。
図4CにN5KG1_7-10、及びN5KG1_4-49の作製工程を図示した。
【0164】
1-2)抗ヒトc-Mpl抗体6-4-50_IgG1及び6-5-2_IgG1抗体発現ベクターの作製
6-4-50、及び、6-5-2については、ヒト抗体発現用ベクターに最初に軽鎖可変領域、次に、重鎖可変領域の順に挿入して発現ベクターを作製した。
【0165】
6-4-50、及び、6-5-2抗体それぞれのLV[C]断片を含むプラスミドDNA(実施例7)を鋳型として、末端に連結のための制限酵素部位(5’末側BglII、3’末側BsiWI)を付加するようにデザインしたプライマーセットを用いて、LV断片(軽鎖のリーダー配列+可変領域)のDNAをKOD-Plus- DNAポリメラーゼによるPCRで増幅した。プライマーは以下の通りである。
6-4-50;
LV断片5’用プライマー:208LF
5’-AGAGAGAGAGATCTCTCACCATGGACATGAGGGTCCCCGCTCAGC -3’(配列番号47)
LV断片3’用プライマー:62LP3Bsi
5’- AGAGAGAGAGCGTACGTTTGATTTCCACCTTGGTCCCTTG -3’(配列番号48)
6-5-2;
LV断片5’用プライマー:A27_F
5’-AGAGAGAGAGATCTCTCACCATGGAAACCCCAGCGCAGCTTCTCTTC -3’(配列番号49)
LV断片3’用プライマー:202LR
5’- AGAGAGAGAGCGTACGTTTAATCTCCAGTCGTGTCCCTTGGC -3’(配列番号50)
【0166】
反応温度条件としては、94℃の初期温度で1分間の加熱後、94℃/5秒間と68℃/45秒間のサイクルを35回反復し、最後に72℃/7分間加熱した。増幅されたDNA断片を制限酵素BglIIとBsiWIで消化して、アガロースゲル電気泳動で約400bpのDNAを回収し精製した。他方、N5KG1ベクターについては制限酵素BglIIとBsiWI処理を順次行った後、脱リン酸化処理としてAlkaline Phosphatase (E. coli C75) (宝酒造社製)にて処理した後に、アガロースゲル電気泳動とDNA精製キットで約8.9kbのDNAを回収した。これら2つの断片をT4 DNA ligase を用いてライゲーションして、大腸菌DH10Bへ導入して形質転換体を得た。インサートDNAを含む形質転換体のプラスミドDNAについてDNA塩基配列を解析して、LV断片がN5KG1のヒト抗体軽鎖定常領域をコードする5’上流にフレームを合わせて挿入されたプラスミドDNA、N5KG1_6-4-50_Lv、及びN5KG1_6-5-2_Lvを取得した。引き続いて、このLV断片が挿入されたプラスミドベクターにHV断片(重鎖のリーダー配列+可変領域)の挿入を行なった。HV[C]を含むプラスミドDNA(実施例7)を鋳型として、末端に連結のための制限酵素部位(5’末側SalI、3’末側NheI)を付加するようにデザインしたプライマーを用いて、HV断片をPCRで増幅した。使用したプライマーは以下の通りである。
6-4-50;
HV断片5’用プライマー:50-5-7Hsal
5’- AGAGAGAGAG GTCGACCACC ATGGAATTGG GACTGAGCTG GATTTT -3’(配列番号51)
HV断片3’用プライマー:C15H3Nhe
5’ -AGAGAGAGAGGCTAGCTGAGGAGACGGTGACCGTGGT-3’(配列番号52)
6-5-2;
HV断片5’用プライマー:F24HSal
5’- AGAGAGAGAGGTCGACCACCATGGAGTTGGGACTGAGCTGGATTT-3’(配列番号53)
HV断片3’用プライマー:L66H3Nhe
5’- AGAGAGAGAGGCTAGCTGAGGAGACGGTGACCGTGGTC-3’(配列番号54)
【0167】
反応は94℃の初期温度で1分間の加熱後、94℃/5秒間と68℃/45秒間のサイクルを35回反復し、最後に72℃/7分間の保温という条件で実施した。精製したHV断片の増幅DNA断片は、一度pCR4Blunt- TOPO ベクター(東洋紡績(株)製)にサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。DNA塩基配列決定のためにプライマーとして、M13-20FWとM13RVを用いた。挿入部分のDNA塩基配列解析を行い、鋳型としたHVと相違がなく、また、プライマー部分もデザインどおりの配列を有するプラスミドDNA(TOPO_6-4-50_Hv、及びTOPO_6-5-2_Hv)を選択した。次に、それぞれのDNAを制限酵素SalIとNheIで消化して、アガロースゲル電気泳動で約430 bpのDNAを回収し精製した。並行して制限酵素処理(SalIとNheI)、及び、脱リン酸化処理した6-4-50或は6-5-2のLV断片が挿入されたベクター(約9.3kb)に、インサートしたいDNA断片をライゲーションした後、大腸菌DH10Bへ導入して、得られた形質転換体から目的のプラスミドDNAを持つクローンを選択した。こうして得られた抗体発現プラスミドDNAの大量精製を行い、重鎖全領域と軽鎖全領域、及び、その挿入部位周辺のDNA塩基配列にクローニング工程での変異がないことを確認した。6-4-50_IgG1及び6-5-2_IgG1抗体発現ベクターを、それぞれN5KG1_6-4-50、及びN5KG1_6-5-2と名付けた。
図4DにN5KG1_6-4-50、及びN5KG1_6-5-2の作製工程を図示した。
【0168】
2)IgG4PEサブクラスの抗ヒトc-Mpl抗体の作製
IgG4PEサブクラス抗体の発現ベクター作製には前述のN5KG4PEベクターを用いた。N5KG4PEのプラスミドDNAを制限酵素NheIとBamHIで切断して、重鎖定常領域を含む断片を精製して、抗c-Mpl抗体であるN5KG1_7-10、及びN5KG1_4-49の同じ制限酵素部位内につなぎ換えることにより、N5KG4PE_7-10、及びN5KG4PE_4-49を作製した。
【0169】
3) N5KG3の作製
ヒトIgG3用発現ベクターN5KG3はN5KG1のIgG1重鎖定常領域を下記配列のIgG3定常領域に置換することで作製した。
IgG3定常領域アミノ酸配列
STKGPSVFPLAPCSRSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYTCNVNHKPSNTKVDKRVELKTPLGDTTHTCPRCPEPKSCDTPPPCPRCPEPKSCDTPPPCPRCPEPKSCDTPPPCPRCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFKWYVDGVEVHNAKTKLREEQYNSTFRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYNTTPPMLDSDGSFFLYSKLTVDKSRWQQGNIFSCSVMHEALHNRYTQKSLSLSPGK*(配列番号55)
IgG3定常領域塩基配列
CTAGCACCAAGGGCCCATCGGTCTTCCCCCTGGCGCCCTGCTCCAGGAGCACCTCTGGGGGCACAGCGGCCCTGGGCTGCCTGGTCAAGGACTACTTCCCCGAACCGGTGACGGTGTCGTGGAACTCAGGCGCCCTGACCAGCGGCGTGCACACCTTCCCGGCTGTCCTACAGTCCTCAGGACTCTACTCCCTCAGCAGCGTGGTGACCGTGCCCTCCAGCAGTTTGGGCACCCAGACCTACACCTGCAACGTGAATCACAAGCCCAGCAACACCAAGGTGGACAAGAGAGTTGAGCTCAAAACCCCACTTGGTGACACAACTCACACATGCCCACGGTGCCCAGAGCCCAAATCTTGTGACACACCTCCCCCGTGCCCACGGTGCCCAGAGCCCAAATCTTGTGACACACCTCCCCCATGCCCACGGTGCCCAGAGCCCAAATCTTGTGACACACCTCCCCCGTGCCCAAGGTGCCCAGCACCTGAACTCCTGGGAGGACCGTCAGTCTTCCTCTTCCCCCCAAAACCCAAGGATACCCTTATGATTTCCCGGACCCCTGAGGTCACGTGCGTGGTGGTGGACGTGAGCCACGAAGACCCCGAGGTCCAGTTCAAGTGGTACGTGGACGGCGTGGAGGTGCATAATGCCAAGACAAAGCTGCGGGAGGAGCAGTACAACAGCACGTTCCGTGTGGTCAGCGTCCTCACCGTCCTGCACCAGGACTGGCTGAACGGCAAGGAGTACAAGTGCAAGGTCTCCAACAAAGCCCTCCCAGCCCCCATCGAGAAAACCATCTCCAAAACCAAAGGACAGCCCCGAGAACCACAGGTGTACACCCTGCCCCCATCCCGGGAGGAGATGACCAAGAACCAGGTCAGCCTGACCTGCCTGGTCAAAGGCTTCTACCCCAGCGACATCGCCGTGGAGTGGGAGAGCAATGGGCAGCCGGAGAACAACTACAACACCACGCCTCCCATGCTGGACTCCGACGGCTCCTTCTTCCTCTACAGCAAGCTCACCGTGGACAAGAGCAGGTGGCAGCAGGGGAACATCTTCTCATGCTCCGTGATGCATGAGGCTCTGCACAACCGCTACACGCAGAAGAGCCTCTCCCTGTCTCCGGGTAAATGA(配列番号56)
【0170】
4) IgG3311発現ベクターの作製
IgG3311発現ベクターは、N5KG3を鋳型として、プライマーlinkH, 13ch1-Rで98℃1秒、60℃30秒、72℃30秒の反応を15回行った。同時に、N5KG1を鋳型として、プライマー13ch1、linkH2を用いて、98℃1秒、60℃30秒、72℃30秒の反応を15回行った。増幅したDNA断片をPCR purification kit で精製し、2つの精製DNA断片を等量混合したのち、98℃1秒、60℃30秒、72℃30秒の反応を5回行い、プライマーlinkH、linkH2を加えて、15回反応した。増幅したDNA断片をNheI, BamHIで切断し、N5KG1ベクターのIgG1定常領域と置き換えた。この発現ベクターをN5KG3311と名づけた。
linkH: GGG TAC GTC CTC ACA TTC AGT GAT CAG(配列番号57)
13ch1-R: GTC TTC GTG GCT CAC GTC CAC CAC CAC GCA(配列番号58)
13ch1: TGC GTG GTG GTG GAC GTG AGC CAC GAA GAC(配列番号59)
linkH2: TGA TCA TAC GTA GAT ATC ACG GC(配列番号60)
【0171】
5) IgG3331発現ベクターの作製
IgG3331発現ベクターは、N5KG3を鋳型として、プライマーlinkH, CH3consRで98℃1秒、60℃30秒、72℃30秒の反応を15回行った。同時に、N5KG1を鋳型として、プライマーCH3cons、linkH2を用いて、98℃1秒、60℃30秒、72℃30秒の反応を15回行った。増幅したDNA断片をPCR purification kit で精製し、2つの精製DNA断片を等量混合したのち、98℃1秒、60℃30秒、72℃30秒の反応を5回行い、プライマーlinkH、linkH2を加えて、15回反応した。増幅したDNA断片をNheI, BamHIで切断し、N5KG1ベクターのIgG1定常領域と置き換えた。この発現ベクターをN5KG3331と名づけた。
CH3consR: GGTGTACACCTGTGGCTCTCGGGGCTGCCC(配列番号61)
CH3cons: GGGCAGCCCCGAGAGCCACAGGTGTACACC(配列番号62)
以下に、IgG3344, IgG3344h1, IgG4344, IgG4344h1, IgG4344uh, IgG4344uhmの作製法を記す。これらについては、各定常領域をPCR法によって増幅し、それをクローニングしたプラスミドを作製した。ついで、それら改変定常領域をN5KG1_7-10等のIgG1定常領域と置換した。
【0172】
6) IgG3344, IgG3344h1定常領域の作製
IgG3344発現ベクターは、N5KG3331およびN5KG4PEを鋳型として、PCRを用いた変異導入(Overlap Extention法による部位特異的変異導入法)によって以下の手順で行った。
【0173】
N5KG3331を鋳型として、プライマーとしてG3G4_P1_FとG3G4_P2_Rを用いて94℃の初期温度で1分間の加熱後、94℃/15秒間、55℃/10秒間、そして68℃/1分間のサイクルを35回反復し、最後に72℃/7分間加熱した。同時に、前述発現ベクターN5KG4PEを鋳型として、プライマーとしてG3G4_P3_FとG3G4_P4_Rを用いて同条件にてPCRを実施した。増幅したDNA断片をアガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kit (キアゲン社製)にて精製した。これらの精製DNA断片を等量混合した後、最初の5サイクルについては、2DNA断片のオーバーラップ部分をアニーリングさせて94℃の初期温度で1分間の加熱後、94℃/10秒間、55℃/10秒間、そして68℃/1.5分間という5サイクルの伸長反応を行なった後、全長を増幅させる為に反応液にプライマーとしてG3G4_P1_FとG3G4_P4_Rを加えて、更に94℃/5秒間と68℃/2分間のサイクルを20回反復し、最後に72℃/7分間の加熱を行なった。G3G4_P1_FとG3G4_P4_Rのプライマーは、ヒト抗体定常領域のコード部分を切りだして、抗体の発現ベクターの該当する部分に変更可能とするために制限酵素部位(G3G4_P1_FにNheI、G3G4_P4_RにBamHI部位)が存在する。増幅したPCR断片は、アガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kitにて精製した。精製した増幅断片は、Zero Blunt TOPO PCR Cloning Kit(インビトロジェン社製)のpCR 4 Blunt- TOPO ベクターにサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。塩基配列の解析よりIgG3344およびIgG3344h1定常領域を持つクローンを選択した。
G3G4_P1_F:5’-AGAGAGGCTA GCACCAAGGG CCCATCG-3’(配列番号63)
G3G4_P2_R:5’-GAACTCAGGT GCTGGGCACC TTGGGCACG-3’(配列番号64)
G3G4_P3_F:5’- CCAAGGTGCC CAGCACCTGA GTTCGAGGGG GGA -3’(配列番号65)
G3G4_P4_R:5’- AGAGAGGGAT CCTCATTTAC CCAGAGACAG GGA -3’(配列番号66)
【0174】
7) IgG4344定常領域の作製
IgG4344発現ベクターの作製においては、N5KG3331を鋳型として、プライマーとしてG434_P5_FとG434_P6_Rを用いて94℃の初期温度で1分間の加熱後、94℃/15秒間、55℃/10秒間、そして68℃/1分間のサイクルを35回反復し、最後に72℃/7分間加熱した。同時に、N5KG4PEを鋳型として、プライマーとしてG434_P7_FとG3G4_P2_Rを用いて同条件にてPCRを実施した。増幅したDNA断片をアガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kit (キアゲン社製)にて精製した。これらの精製2DNA断片と、前述したN5KG4PEを鋳型として、G3G4_P3_FとG3G4_P4_Rをプライマーとして増幅・精製しておいたDNA断片の計3種類のOverlap Extention反応を実施した。すなわち、3種類のDNA断片のオーバーラップ部分をアニーリングさせて94℃の初期温度で1分間の加熱後、94℃/10秒間、55℃/10秒間68℃/1.5分間のサイクルを5回反復して伸長した後、全長を増幅させる為に反応液にプライマーとしてG434_P5_FとG3G4_P4_Rを加えて、94℃/5秒間と68℃/2分間のサイクルを20回反復し、最後に72℃/7分間の加熱を行なった。増幅したPCR断片はQIAquick Gel Extraction Kitにて精製した後、pCR 4 Blunt- TOPO ベクターにサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。塩基配列の解析よりIgG4344定常領域を持つクローンを選択した。
G434_P5_F:5’- AGAGAGGCTA GCACCAAGGG GCCATCC -3’(配列番号67)
G434_P6_R:5’- GGTTTTGAGC TCAACTCTCT TGTCCACCTT GGTGTTGC -3’(配列番号68)
G434_P7_F:5’- GTGGACAAGA GAGTTGAGCT CAAAACCCCA CTTGGTGACA C -3’(配列番号69)
【0175】
8) IgG4344h1定常領域の作製
IgG4344h1発現ベクターの作製は、N5KG4344を鋳型として、プライマーとしてG434_P5_F, G434_P6_Rを用いて98℃の初期温度で10秒間の加熱後、98℃/10秒間、55℃/30秒間、そして72℃/1分間のサイクルを7回反復し、次いで98℃/10秒間、68℃/1分間のサイクルを30回反復して、最後に72℃/7分間加熱した。PCR用酵素としては、Pyrobest DNA Polymerase(タカラバイオ株式会社)を使用した。同時に、N5KG3344h1を鋳型として、プライマーとしてG434_P7_FとG3G4_P4_Rを用いて同条件にてPCRを実施した。増幅したDNA断片をアガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kit (キアゲン社製)にて精製した。これらの精製DNA断片を等量混合した後、2DNA断片のオーバーラップ部分をアニーリングさせて、98℃の初期温度で10秒間の加熱後、98℃/10秒間、55℃/30秒間、そして72℃/1分間のサイクルを7回反復して伸長した後、全長を増幅させる為に反応液にプライマーとしてG434_P5_FとG3G4_P4_Rを加えて、更に98℃/10秒間と68℃/1分間のサイクルを30回反復し、最後に72℃/7分間の加熱を行なった。増幅したPCR断片は、アガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kitにて精製した。精製した増幅断片は、pCR 4 Blunt- TOPO ベクターにサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。塩基配列の解析よりG4344h1定常領域持つクローンを選択した。
【0176】
9) IgG4344uh定常領域の作製
G4344uhの作製は、N5KG4344を鋳型として、プライマーとしてG434_P5_Fと17-1Rを用いて98℃の初期温度で10秒間の加熱後、98℃/10秒間、50℃/30秒間、そして72℃/1分間のサイクルを5回反復し、次いで98℃/10秒間、55℃/30秒間、そして72℃/1分間のサイクルを5回反復し、98℃/10秒間、68℃/1分間のサイクルを25回反復して、最後に72℃/7分間加熱した。PCR用酵素としては、Pyrobest DNA Polymerase(タカラバイオ株式会社)を使用した。並行して、N5KG3344h1を鋳型として、プライマーとして17-2FとG3G4_P4_Rを用いて同条件にてPCRを実施した。増幅したDNA断片をアガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kitにて精製した。これらの精製DNA断片を等量混合した後、2DNA断片のオーバーラップ部分をアニーリングさせて、98℃の初期温度で10秒間の加熱後、98℃/10秒間、68℃/1分間のサイクルを5回、さらに98℃/10秒間、55℃/30秒間、そして72℃/1分間のサイクルを5回反復して伸長した後、全長を増幅させる為に反応液にプライマーとしてG434_P5_FとG3G4_P4_Rを加えて、更に94℃/30秒間と68℃/1分間のサイクルを30回反復し、最後に72℃/7分間の加熱を行なった。増幅したPCR断片は、アガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kitにて精製した。精製した増幅断片は、pCR 4 Blunt- TOPO ベクターにサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。塩基配列の解析よりIgG4344uh定常領域を持つクローンを選択した。
17-1R:5’- AGGTGCTGGG CACCGTGGGC ATGTGTGAGT TGT -3’(配列番号70)
17-2F:5’- CACACATGCC CACGGTGCCC AGCACCTGAG TTC -3’(配列番号71)
【0177】
10) IgG4344uhm定常領域の作製
IgG4344uhm発現ベクターの作製は、N5KG4PEを鋳型として、プライマーとしてG434_P5_Fと17m-1Rを用いて98℃の初期温度で10秒間の加熱後、98℃/10秒間、50℃/30秒間、そして72℃/1分間のサイクルを5回反復し、次いで98℃/10秒間、55℃/30秒間、そして72℃/1分間のサイクルを5回反復し、98℃/10秒間、68℃/1分間のサイクルを25回反復して、最後に72℃/7分間加熱した。PCR用酵素としては、Pyrobest DNA Polymeraseを使用した。並行して、N5KG4PEを鋳型として、プライマーとして17m-2FとG3G4_P4_Rを用いて同条件にてPCRを実施した。増幅したDNA断片をアガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kitにて精製した。これらの精製DNA断片を等量混合した後、2DNA断片のオーバーラップ部分をアニーリングさせて、94℃/30秒間、55℃/30秒間、そして72℃/1分間のサイクルを7回反復して伸長した後、全長を増幅させる為に反応液にプライマーとしてG434_P5_FとG3G4_P4_Rを加えて、更に94℃/30秒間と68℃/1分間のサイクルを30回反復し、最後に72℃/7分間の加熱を行なった。増幅したPCR断片は、アガロースゲル電気泳動で回収し、QIAquick Gel Extraction Kitにて精製した。精製した増幅断片は、pCR 4 Blunt- TOPO ベクターにサブクローニングを行い、得られたクローンのプラスミドDNAについてインサートDNAの塩基配列を解析した。塩基配列の解析よりIgG4344uhm定常領域を持つクローンを選択した。
17m-1R:5’- TGTGTGAGTT GTGTCACCAA GTGGGGTTTT GGACTCAACT CTCTTGTCCA CCTTGGT -3’(配列番号72)
17m-2F:5’- ACCCCACTTG GTGACACAAC TCACACATGC CCACCATGCC CAGCACCTGA GTTCGAG -3’(配列番号73)
図4Eに各種改変重鎖のアミノ酸配列を示した。
【0178】
11) 各種改変重鎖定常領域を持った抗体発現ベクターの作製
作製した各種改変重鎖定常領域を持つプラスミドDNAを、制限酵素NheI, BamHIで切断後、定常領域配列を精製分離した。続いて、抗ヒトc-Mpl抗体発現ベクターN5KG1_7-10, N5KG1_4-49, N5KG1_6-4-50, N5KG1_6-5-2を同酵素で処理し、定常領域を置換した。
図4Fに7-10_IgG4344uhmの重鎖配列を示した。
図4Gに7-10_IgG4344uhmの軽鎖配列を示した。
【0179】
[実施例9] 抗ヒトc-Mpl抗体の293F細胞での一過性発現、及び精製
実施例8で作製した発現ベクターDNAをEndoFree Plasmid Kit(キアゲン社)にて調製し、FreeStyleTM 293 Expression System(インビトロジェンライフテクノロジー社)を用いて浮遊性293細胞(インビトロジェンライフテクノロジー社)に導入して、一過性発現により各抗体を含む培養上清を得た。孔径0.22μm のメンブランフィルター(MILLIPORE 製)で濾過した培養上清(IgGとして約500μg)を抗体精製用アフニティーカラムであるHiTrap rProtein A FF(カラム体積1ml)(アマシャムバイオサイエンス社)にチャージし、PBS (-)で洗浄後20 mMクエン酸バッファー(pH3.4)により溶出し、200 mMリン酸バッファー(pH7.0)を含むチューブに回収した。
【0180】
[実施例10] 組換え型抗体の作製
構築した抗体発現ベクターを宿主細胞に導入して、抗体発現細胞を作製した。発現のための宿主細胞には、dhfr欠損のCHO DG44 細胞(IDEC Pharmaceuticals Corporation)を無血清培地であるEX-CELL325PF(JRH)に馴化した細胞株を用いた。宿主細胞へのベクターの導入はエレクトロポレーションにより実施した。エレクトロポレーションは抗体発現ベクター約2μgを制限酵素AscIで線状化し、Bio-Rad electrophoreterをもちいて350V、500μFの条件で、4×106個のCHO細胞に遺伝子を導入し、96well culture plateに播種した。ベクターの導入処理後、G418を添加して培養を継続した。コロニーを確認した後、抗体発現株を選別した。選択したCHO細胞株をEX-CELL325-PF培地(JRH)(2 mM glutamine、100 units/ml penicillin、100 μg/ml streptomycin、 hypoxanthine and thymidine (HT) サプリメント(1:100) (Invitrogen)を含む)で5% CO2条件下で培養した。培養上清をMabselect Protein A カラム(Amersham Pharmacia Biotech, Co., Ltd.)に吸着後、PBSで洗浄して、20mMクエン酸-Na、50mM NaCl (pH3.4)バッファーで溶出した。溶出液は50mM Phosphate-Na, pH7.0 にて中和した。Milli-Q水にて、約1.5倍に希釈してConductivityを4.0ms/cm以下に調製した。次に、Q-Sepharose(Hitrap Q HP)(Amersham Pharmacia Biotech, Co., Ltd.)と、SP-Sepharose(HiTrap SP FF)(Amersham Pharmacia Biotech, Co., Ltd.)を連結したカラムに、サンプルをチャージして吸着後、20mMリン酸ナトリウム緩衝液(pH5.0)にて洗浄後、1×PBSバッファーにて溶出した。調製された抗体溶液は、孔径0.22μmのメンブランフィルターMILLEX-GV (ミリポア社製)でろ過滅菌した。精製した抗体の濃度は280nmの吸光度を測定し、1mg/mL を1.4 ODとして算出した。
【0181】
組換え型の改変抗体を用いて、UT7/TPOアッセイ(実施例5)における活性を測定した。4-49_IgG1に比べ、IgG3311とIgG3331は活性の増強が見られた(図5A)。7-10_IgG4344uhmおよび4-49_IgG4344uhmはPEG-rHuMGDFと同等の活性を示した。
【0182】
表3に各種改変抗体の活性をまとめた。全てのアゴニスト抗体に関して、定常領域改変による活性の増強が見られた。7-10, 4-49に関しては、IgG1とIgG4PEは同等の活性であったが、IgG4344uhmはIgG4PEに比べ高活性であった。IgG4344uhmは、IgG4PEのアッパーヒンジ部7アミノ酸配列中のC末側4−7位がIgG3のアッパーヒンジ12アミノ酸配列中の4−12位の配列に置き換わっている(図4B参照)。従って、この部分が活性の増強に重要であると考えられる。
【0183】
【表3】
【0184】
[実施例11] アゴニスト抗体によるシグナル伝達
TPOが受容体であるc-Mplに結合すると、細胞内タンパク質のリン酸化が起こる。TPOによって活性化する主要な経路は、Jak-STAT, Ras-MAPK, PI3K-Aktの3つが知られている。アゴニスト抗体によるc-Mpl下流のリン酸化シグナル伝達の解析を行った。解析は、リン酸化タンパク質特異的な抗体を用いたウェスタンブロット法で行った。以下に使用した抗体を列記する。抗STAT5(Cell Signaling社製, Cat#9352), 抗phospho-STAT5(Cell Signaling社製, Cat#9351L), 抗JAK2(Upstate社製, Cat#06-255), 抗phospho-JAK2(Upstate社製, Cat#07-606), 抗Erk1/2(Cell Signaling社製, Cat#9272), 抗phospho-Erk1/2(Cell Signaling社製, Cat#9271L), 抗Akt(Cell Signaling社製, Cat#9102), 抗phospho-Akt(Cell Signaling社製, Cat#9101S)。
【0185】
これら抗体を使用し、以下の手順でアッセイを行った。
1) UT7/TPO細胞をサイトカイン非添加IMDM培地で洗浄し、6時間培養した。
2) 培養後、細胞を1×106cells/mLに調製し、6-wellプレートに2mL/wellで播いた。
3) ウェルに、アゴニスト抗体または陽性コントロールとしてPEG-rHuMGDFを添加し、細胞を刺激した。
4) 最短5分〜最長2時間の刺激時間の後に細胞を回収し、氷冷PBSにて細胞を洗浄した。
5) 遠心分離で細胞をペレットにし、上清を除去後、PhosphoSafeTM Extraction Reagent(Novagen社製, Cat#71296)でペレットを溶解したのち、再度遠心して、上清(細胞抽出液)を回収した。
6) 上記5.の細胞抽出液を用いて、ウェスタンブロット法でリン酸化タンパク質を検出した。
【0186】
結果を図6に示す。アゴニスト抗体7-10G4344uhmおよび4-49G4344uhmによって、TPOシグナルと同様の経路のリン酸化が見られた(図6A)。抗体6-5-2については、IgG1はJak2, STAT5のリン酸化が観察されなかったが、IgG3344はJak2, STAT5のリン酸化が見られた(図6B)。
【0187】
[実施例12] ヒト血小板に対するプライミング作用検討
TPOはそれ自体では血小板凝集を起こさないが、ADP等の凝集惹起物質による血小板凝集を促進する作用(プライミング作用)がある。アゴニスト抗体によるヒト血小板プライミング作用を以下の手順で調べた。
【0188】
1) 抗凝固剤として、3.1%(w/v)クエン酸三ナトリウムを10分の1容量加えた、健常人の末梢血を遠心分離(140g, 15min)し、多血小板血漿(Platelet Rich Plasma、以下 PRP)を調製した。
2) さらに遠心分離(2500g, 15min)して血球成分を沈殿させ、血漿を採取した。
3) PRPに含まれる血小板数を測定し、血漿を用いて血小板数を3×105/μLにあわせた。
4) 上記3.で調製した血小板浮遊液100μLに検体を加え、攪拌しながら3分間インキュベーションした。
5) 30μM ADP(SIGMA社製)を5μL添加し、血小板凝集による濁度の低下を測定した。測定にはMCメディカル社製, Hematracer801を用いた。
【0189】
結果を図7に示す。ADP添加条件下で、アゴニスト抗体によるプライミング作用が観察された。抗体単独(ADPなし)では、血小板凝集は起こらなかった。
【0190】
[実施例13] カニクイザル投与
アゴニスト抗体をカニクイザルに投与し、血小板数の変動を解析した。使用個体のTPOに対する反応性を確かめるため、初日(Day0)にPEG-rHuMGDF(10μg/kg)を静脈内投与し3週間経過を観察した後、初回投与後21日目に精製アゴニスト抗体7-10G4PE(個体A)7-10G3344h1(個体B)を用量1mg/kgで静脈内投与した。
【0191】
結果を図8に示す。A,B両方の個体でPEG-rHuMGDFによる一過性の血小板上昇が見られた。個体Bではアゴニスト抗体7-10G3344h1投与後に、血小板数の上昇が見られた。また、抗体投与による重篤な毒性は見られなかった。
【0192】
[実施例14] ヒト臍帯血移植モデルにおける作用検討
実施例10で作製したアゴニスト抗体が、ヒト臍帯血移植モデルにおけるヒト造血系の構築を促進することを確認するため、以下の手順で実験を行った。
【0193】
・ NOG(NOD/SCID/IL2-γR KO)マウス(実験動物中央研究所(日本国神奈川県川崎市)より購入)に移植前処置として放射線照射(2グレイ)を行い、ヒト臍帯血由来CD34+細胞を1,000ないし10,000個、尾静脈より注入移植した。
・ 移植後一日目に最初の被検物質投与を行い、その後、週に一回投与を実施した。群構成と各々の被検物質および投与量は下記のとおりである。匹数は各群6匹、投与は腹腔内投与で行った。また、毎週の投与時に体重測定を行った。
【0194】
<群構成,被検物質,投与量>
I: 移植数10,000, PBS(コントロール)投与
II: 移植数1,000, PBS投与
III: 移植数10,000, 抗体7-10G4344uhm投与, 100μg/head/week
IV: 移植数1,000, 抗体7-10G4344uhm投与, 100μg/head/week
V: 移植数10,000, TPO (PEG-rHuMGDF)投与, 5μg/head/week
VI: 移植数1,000, TPO (PEG-rHuMGDF)投与, 5μg/head/week
・ 移植前1日と移植後2、4、6週に末梢血の解析を行った。末梢血解析の手順は下記のとおりである。
【0195】
<末梢血解析手順>
・ キャピラリ−を用いて、マウス眼窩静脈より末梢血(約70μL)を採取した。
・ KX-21自動血球解析システム(Sysmex社製)を用いて血球数を測定した。
・ ヒト血小板および白血球のキメラ率を調べるため、以下のAおよびBに挙げた各抗体の組み合わせで染色し、FACS Caliburで解析した。A(血小板解析用):PE標識−抗ヒトCD41抗体(Dako社製 R7058)+FITC標識−抗マウスCD41抗体(BD Pharmingen社製#553848)、B(白血球解析用): APC標識−抗ヒトCD45抗体(ベックマン・コールター社製IM2473)+FITC標識−抗マウスCD45抗体(BD Pharmingen社製#553080)。また、解析の際に定量用の蛍光ビーズ(フローカウントビーズ)を加え、一定量の血液を解析できるようにした。
・ 血小板・白血球ともに、ヒト細胞数/(ヒト細胞数+マウス細胞数)×100(%)、という式でキメラ率を算出した。末梢血中の全血小板数とキメラ率を乗じることにより、ヒト血小板数を算出した。
・ 6週目にマウスを屠殺し、大腿骨から骨髄細胞を採取した。それを用いてコロニーアッセイを行い、ヒトの巨核球系(MK)および赤血球系(E)、顆粒球・マクロファージ系(GM)の前駆細胞の数を測定した。巨核球系前駆細胞(CFU-Mk)を検出するコロニーアッセイは、培養中にTPO(50ng/mL)とSCF(100ng/mL)を加えて行った。37℃、5%CO2条件下で12日間の培養を行った。コロニーの検出は実施例6と同様に抗ヒトCD41抗体を用いて行った。赤血球系、顆粒球・マクロファージ系の前駆細胞を検出するコロニーアッセイは、Methocult system(Stem Cell Technologies社製)を用いて、培養中にEPO(4IU/mL), SCF(100ng/mL), IL-3(20ng/mL), GM-CSF(10ng/mL)を加えて行った。37℃, 5%CO2, 5%O2条件下で14日間の培養を行った。培養後、顕微鏡下でコロニーのカウントを行った。
【0196】
図9A、9B及び9Cに本実験の結果を図示した。
移植後6週の時点において、抗体投与群では、他群に比べ、末梢血ヒト血小板数が有意に高かった(図9A)。これにより、アゴニスト抗体7-10G4344uhmは臍帯血移植時の血小板回復を促進することが示唆された。さらに抗体投与群では、骨髄中のヒト赤血球系および顆粒球・マクロファージ系前駆細胞の数が有意に高かった(図9B)。またマウス白血球に対するヒト白血球の割を示すCD45キメラ率においても有意に高かったことから、抗体投与群においてヒト白血球が増加していることがわかる(図9C)。このことは7-10G4344uhmが巨核球系のみならず、他系列の細胞の生着も促進できる可能性を示唆している。
【0197】
これらの知見から、アゴニスト抗体は、血球が巨核球系・赤血球系・顆粒球・マクロファージ系に分かれるより上流の細胞に作用していることが示唆された。Mplが造血幹細胞に発現しているという知見とあわせて考えると、アゴニスト抗体は造血幹細胞の増殖を促進している可能性が高い。
【0198】
ちなみに本実験ではTPO投与群では同様の効果が見られなかった。これに対しては、TPOがマウス造血細胞にも作用するため、TPO投与群では骨髄中でヒト細胞とマウス細胞の競合が起こり、純粋にヒト細胞に対する作用を観察できていない可能性を考慮しなければならない。本アゴニスト抗体は、ヒトMplにしか作用しないことを特徴とすることから、Mplを介したシグナルがヒト臍帯血造血幹細胞の増幅に効果があることを、in vivoで初めて明らかにすることができた。
【0199】
[実施例15] ヒンジ改変抗体の抗原性解析
本発明のアゴニスト抗体はヒンジ部分の改変によって活性を増強していることを特徴としているが、改変による抗原性の上昇が懸念された。そこで、ヒンジ改変体である7-10G4344uhmのアミノ酸配列をもとに、コンピュータ上での抗原性予測を実施した。
【0200】
体内に投与された外来タンパク質は樹状細胞、マクロファージ等の抗原提示細胞(APC)に取り込まれ、分解された後、ペプチドが主要組織適合抗原複合体(MHC)クラスII分子(ヒトの場合HLA class II, HLA-DR, DQ, DP)によって抗原提示される。APCによって提示されたペプチドはT細胞受容体(TCR)によって認識され、T細胞を活性化させる。活性化されたT細胞(ヘルパーT細胞)は同抗原を認識する抗体を発現するB細胞を活性化し、外来タンパク質に対する抗体が産生される。この機構の中で、ペプチドとMHCクラスII分子との親和性が抗原性を規定する大きな要因となっている。ヒトのMHCクラスII分子には多くの型(多型)があり、同じペプチドであってもクラスII分子の型によって親和性は大きく異なることが知られている。
【0201】
そこで、7-10G4344uhmとIgG4PEの定常領域を持つ別のヒト抗体のアミノ酸配列を、様々な型のヒトHLA-DR, DQ, DP分子との親和性を解析した(HLA分子のデータベースおよび、解析のアルゴリズムはAlgoNomics社より提供された)。
【0202】
その結果、ヒンジ改変による新たなエピトープ出現はなかった。本改変抗体を医薬品として利用する際に抗原性の問題はないことが示唆された。
【0203】
[実施例16] ヒトMplトランスジェニックマウスへの抗体投与
本発明の抗体はマウスMplには交叉しないため、薬効を測定するため、外来遺伝子としてヒトMplを導入したトランスジェニック(Tg)マウスを作製し、抗体を投与する実験を行った。まず、マウスMplのプロモータ領域5.5kbをPCRで増幅し、pBluescriptプラスミドベクターにクローニングした。次に、ヒトMplの翻訳領域および3’側非翻訳領域をPCRで増幅し、マウスMplプロモーター下流に連結した。このコンストラクトをC57BL/6マウス受精卵に注入し、受精卵を仮親に戻し出産させた。生後3週経過した時点で尻尾からゲノムDNAを抽出し、PCRでTgマウスを選別した。得られたTgマウス個体をC57BL/6と交配し、系統化した。骨髄でのヒトMplの発現解析を行った。
【0204】
その結果、複数のヒトMplを持つTgマウス系統が得られた。そのうち、39L系統の骨髄ではヒトMplが発現していることがRT-PCRで確認された。39L系統のマウスを用いて抗体の薬効を確認した。
【0205】
アゴニスト抗体7-10G4344uhmを単回投与(3又は10μg/mL)し、末梢血中の血小板数の推移を、KX-21自動血球解析装置を用いて調べた。末梢血の採取は眼窩静脈より行い、毎週測定を行った。陽性コントロールとしてTPO(PEG-rHuMGDF)を用いた。群構成は以下のとおりである(各群6匹)。
【0206】
I: 7-10G4344uhm 10μg投与群
II: 7-10G4344uhm 3μg投与群
III: TPO 3μg投与群
IV: PBS投与群
VI: 野生型マウス 7-10G4344uhm 10μg投与群
結果を図10に示す。抗体投与群、TPO投与群で血小板が増加した。TPO投与群は投与後2週間でほぼベースラインに戻った。これに対して抗体投与群は投与後一ヶ月を経過しても血小板数は上昇したままであった。この結果から、アゴニスト抗体は血中で非常に安定であり、単回投与で長期にわたって血小板造血を促進できることが示唆された。このことから、特にアゴニスト抗体は慢性血小板減少症の治療に適していることが示唆された。
【0207】
[実施例17] 7-10G4344uhm軽鎖変異体の活性評価
アゴニスト抗体7-10の軽鎖可変領域(7-10VL)のフレームワーク領域に変異を導入し、結合活性及びアゴニスト活性に与える影響を調べた。軽鎖の変異体はアゴニスト抗体4-49の軽鎖(V104L)、アゴニスト6-4-50の軽鎖のアミノ酸を1つ置換したもの(A43V,G100Q)の3種である。これら変異軽鎖と7−10G4344uhm重鎖を組み合わせた抗体を作製したところ、全て、結合活性、アゴニスト活性ともにオリジナルの7-10G4344uhmと同等であった。一方で、アゴニスト抗体7-10の軽鎖可変領域の相補性決定領域(CDR領域)に変異(Y94F)を導入した場合、結合活性、アゴニスト活性ともに10分の1程度に低下した。この結果から軽鎖アミノ酸配列には、ある程度の自由度があることが示された。
各変異体の軽鎖アミノ酸配列及び7-10VLのアミノ酸配列は、以下のとおりである。なお、変異部は太字と下線で示した。
【0208】
【化1】
【0209】
結合活性解析:各抗体の濃度を1, 0.1, 0.01μg/mLに調製し、FM3A-hMpl細胞を用いたフローサイトメトリーを行った。実験方法は実施例4に記載の方法で行った。コントロールに抗DNP(ジニトロフェノール)抗体(サブクラスIgG4;ヒト抗体)を用いた。軽鎖変異抗体は7-10G4344uhmと同等の結合活性を示した(図11)。
アゴニスト活性解析: 実施例5に記載の方法で、UT-7/TPO細胞を用いた細胞増殖アッセイを行った。軽鎖変異抗体は7-10G4344uhmと同等のアゴニスト活性を示した(図12)。
【産業上の利用可能性】
【0210】
本発明により、種々の血小板減少症に対する治療薬として利用できる、抗ヒトcMplアゴニストヒト抗体が提供された。また他のアゴニスト抗体にも応用し得る、高い安全性と薬効を期待し得る、抗体の定常領域が提供された。
【0211】
本発明により、全長抗体でヒトトロンボポエチン受容体(c-Mpl)を活性化しうるヒトc-Mplに対するアゴニスト抗体が提供される。このアゴニスト抗体は、種々の血小板減少症に対する治療薬として使用可能であり、医療産業上多大な寄与が期待される。
【0212】
本明細書で引用した全ての刊行物、特許および特許出願をそのまま参考として本明細書にとり入れるものとする。
【図面の簡単な説明】
【0213】
【図1】図1は、アゴニスト抗体結合活性を示す。FDCP-hMpl細胞とFDCP2細胞(FDCP parent)を用いたフローサイトメトリー(実施例2)で、図示した各抗体の結合活性を調べた。各抗体はヒトc-Mpl特異的に結合することが示された。
【図2】図2は、UT7/TPOアッセイ結果を示す。図A〜Dに示した各精製抗体(IgG1)のUT7/TPO細胞増殖アッセイ(実施例5)における各抗体の増殖曲線を示した。
【図3】図3は、CFU-Mkアッセイ結果を示す。これは、ヒト臍帯血CD34+細胞を用いたコロニー形成アッセイ(実施例6)の結果である。
【図4A】図4Aは、組換え抗体作製に関するN5KG1ベクターの構造を示す。図中、Cはサイトメガロウイルスのプロモーター/エンハンサー、Bはウシ成長ホルモンポリアデニル化領域、N1はネオマイシンホスホトランスフェラーゼのエクソン1、Kはヒトイムノグロブリンκ定常領域、G1はヒトイムノグロブリンγ1定常領域、BTはマウスβグロブリン主要プロモーター、N2はネオマイシンホスホトランスフェラーゼのエクソン2、Dはジヒドロ葉酸レダクターゼ、VHは重鎖可変領域、VLは軽鎖可変領域をそれぞれ表す。
【図4B】図4Bは、組換え抗体作製に関する、天然型のヒトイムノグロブリンと、IgG4PE, IgG4344, IgG4344h1, IgG4344uh, IgG4344uhmのCH1領域とヒンジ領域(アッパーヒンジ及びミドルヒンジ)のアミノ酸配列を示す。
【図4C−1】図4C(すなわち、図4C-1〜図4C-3)は、組換え抗体作製に関する、N5KG1_7-10, N5KG1_4-49発現ベクター作製工程図を示す。
【図4C−2】図4C(すなわち、図4C-1〜図4C-3)は、組換え抗体作製に関する、N5KG1_7-10, N5KG1_4-49発現ベクター作製工程図を示す。
【図4C−3】図4C(すなわち、図4C-1〜図4C-3)は、組換え抗体作製に関する、N5KG1_7-10, N5KG1_4-49発現ベクター作製工程図を示す。
【図4D−1】図4D(すなわち、図4D-1〜図4D-3)は、組換え抗体作製に関する、N5KG1_6-4-50, N5KG1_6-5-2発現ベクター作製工程を示す。
【図4D−2】図4D(すなわち、図4D-1〜図4D-3)は、組換え抗体作製に関する、N5KG1_6-4-50, N5KG1_6-5-2発現ベクター作製工程を示す。
【図4D−3】図4D(すなわち、図4D-1〜図4D-3)は、組換え抗体作製に関する、N5KG1_6-4-50, N5KG1_6-5-2発現ベクター作製工程を示す。
【図4E】図4Eは、組換え抗体作製に関する、図示した各種改変重鎖の定常領域の配列を示す。
【図4F−1】図4F(すなわち、図4F-1〜図4F-2)は、組換え抗体作製に関する、7-10G4344uhm重鎖の核酸およびアミノ酸配列を示す。
【図4F−2】図4F(すなわち、図4F-1〜図4F-2)は、組換え抗体作製に関する、7-10G4344uhm重鎖の核酸およびアミノ酸配列を示す。
【図4G】図4Gは、組換え抗体作製に関する、7-10G4344uhm軽鎖の核酸およびアミノ酸配列を示す。
【図5】図5は、ヒンジ改変抗体の活性を示す。A:UT7/TPO細胞増殖アッセイにおける4-49G1, 4-49G3311, 4-49G3331の活性。B:UT7/TPO細胞増殖アッセイにおける7-10G4344uhm, 4-49G4344uhmの活性。
【図6A】図6Aは、シグナル伝達解析(実施例11)における、アゴニスト抗体7-10G4344uhm, 4-49G4344uhmのシグナル伝達解析結果を示す。
【図6B】図6Bは、シグナル伝達解析(実施例11)における、アゴニスト抗体6-5-2G1, 6-5-2G3344のシグナル伝達解析結果を示す。
【図7】図7は、ヒト血小板プライミング作用を示す。これは、実施例12記載の試験の結果を示す。アゴニスト抗体7-10G3311, 4-49G3311によるヒト血小板プライミング作用が示された。また、アゴニスト抗体のみ(ADP非添加)では血小板の凝集は起こらないことが示された。
【図8】図8は、カニクイザルへのアゴニスト抗体投与による血小板数の変化を示したグラフである。実施例13に記載のように、カニクイザルにアゴニスト抗体を投与し、血小板数をモニターした。矢印は一回目(PEG-rHuMGDF)および二回目(アゴニスト抗体)の投与日を示す。
【図9A】図9Aは、NOG臍帯血移植モデルマウスにおいてCD34+細胞を1,000個(右図)又は10,000個(左図)を移植した後に被検物質を投与したときの末梢血ヒト血小板数の経時的推移を示す。図中、Preは投与前の血小板数を示す。
【図9B】図9Bは、NOG臍帯血移植モデルマウスにおいてCD34+細胞を1,000個(右図)又は10,000個(左図)を移植した後に被検物質を投与したときの、移植後6週における骨髄ヒト前駆細胞数(colony count; GM+E+GEM)を示す。前駆細胞数は、巨核球系以外の合計細胞数であり、GMは顆粒球・マクロファージ系、Eは赤血球系、GEMはColony-forming-unit-granulocyte-macrophage-erythroidを表わす。結果は、平均±標準偏差(mean±SD)で表わしている。また、ベヒクル(Vehicle)はコントロールとしてのPBS(リン酸緩衝塩水)を表わし、また、NTは非処理(Non-treated)を表わす。
【図9C】図9Cは、NOG臍帯血移植モデルマウスにおいてCD34+細胞を1,000個(右図)又は10,000個(左図)を移植した後に被検物質を投与したときの、移植後6週における末梢血ヒト細胞キメラ率を示す。ベヒクル(Vehicle)は対照としてのPBS(リン酸緩衝塩水)を表わし、また、NTは非処理(Non-treated)を表わす。
【図10】図10は、ヒトMpl Tgマウスへのアゴニスト抗体投与後の血小板数の経日推移を示す。対照として、該TgマウスへのTPO又はベヒクル(PBS)の投与、並びに、非Tg(野生型;Non-Tg)マウスへの7-10G4344uhm(10μg)の投与実験を行い、その結果も示した。結果は、平均±SEMで表わした。
【図11】図11は、アゴニスト抗体7-10G4344uhmの軽鎖変異抗体のFM3A-hMpl細胞への結合性を示す。
【図12】図12は、アゴニスト抗体7-10G4344uhmの軽鎖変異抗体に関するUT-7/TPO細胞を用いた細胞増殖アッセイの結果を示す。
【配列表フリーテキスト】
【0214】
配列番号11:変異ヒンジドメインUH2G3uhm
配列番号12〜16:プライマー
配列番号18〜22:プライマー
配列番号39〜54:プライマー
配列番号57〜73:プライマー
配列番号74:G3344h1
配列番号75:G3344
配列番号76:G4344
配列番号77:G4344h1
配列番号78:G4344uh
配列番号79:G4344uhm
配列番号80:G4PE
配列番号81:7-10G4344uhm H鎖
配列番号82:7-10G4344uhm H鎖
配列番号83:7-10G4344uhm L鎖
配列番号84:7-10G4344uhm L鎖
配列番号85:7-10VL_V104L(変異体)
配列番号86:7-10VL_G100Q(変異体)
配列番号87:7-10VL_A43V(変異体)
【特許請求の範囲】
【請求項1】
ヒトトロンボポエチン受容体(c-Mpl)に対するアゴニスト抗体の製造方法であって、該抗体の定常領域が、以下の(1)〜(3):
(1) ヒト抗体の重鎖定常領域及び軽鎖定常領域のアミノ酸配列、
(2) ヒト抗体サブクラス間で重鎖定常領域のドメインを入れ替えた重鎖定常領域のアミノ酸配列、及び、ヒト抗体軽鎖定常領域のアミノ酸配列、或いは
(3) 前記(1)又は(2)のアミノ酸配列において1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
のいずれかのアミノ酸配列を有し、且つ抗体の可変領域がヒトトロンボポエチン受容体に結合し該受容体を活性化する能力を有する、並びに、以下の(a)及び(b):
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、10,000ng/mL以下の濃度でコロニー形成を誘導すること、及び
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性が、そのN末端がペグ(PEG)化された配列番号1のアミノ酸配列からなる下記の構造:
PEG-NH-SPAPPACDLRVLSKLLRDSHVLHSRLSQCPEVHPLPTPVLLPAVDFSLGEWKTQMEETKAQDILGAVTLLLEGVMAARGQLGPTCLSSLLGQLSGQVRLLLGALQSLLGTQLPPQGRTTAHKDPNAIFLSFQHLLRGKVRFLMLVGGSTLCVRRAPPTTAVPS-COOH
を有するPEG-rHuMGDFの50%以上であり、且つ50%有効濃度(EC50)が100nM以下であること、
の性質を有する前記アゴニスト抗体を構成する重鎖及び軽鎖をそれぞれコードする塩基配列を含むDNAと、それらのDNAの発現を制御する塩基配列を含む1又は複数のDNAとを保持する哺乳動物細胞を作製し、該哺乳動物細胞を培養した培養液から、該重鎖及び軽鎖からなる抗体をコードする塩基配列を含むDNAの発現産物を単離精製することを含む、前記方法。
【請求項2】
前記抗体が、以下の(a)及び(b):
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、1,000ng/mL以下の濃度でコロニー形成を誘導すること、及び
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性がPEG-rHuMGDFの70%以上であり、且つEC50が10nM以下であること、
の性質を有する、請求項1に記載の方法。
【請求項3】
前記抗体が、以下の(a)及び(b):
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、100ng/mL以下の濃度でコロニー形成を誘導すること、及び
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性がPEG-rHuMGDFの90%以上であり、かつEC50が1nM以下であること、
の性質を有する、請求項1に記載の方法。
【請求項4】
前記抗体が、以下の(1)〜(8):
(1) 配列番号2で示されるアミノ酸配列を含む重鎖可変領域及び配列番号3で示されるアミノ酸配列を含む軽鎖可変領域、
(2) 配列番号4で示されるアミノ酸配列を含む重鎖可変領域及び配列番号5で示されるアミノ酸配列を含む軽鎖可変領域、
(3) 配列番号6で示されるアミノ酸配列を含む重鎖可変領域及び配列番号7で示されるアミノ酸配列を含む軽鎖可変領域、
(4) 配列番号8で示されるアミノ酸配列を含む重鎖可変領域及び配列番号9で示されるアミノ酸配列を含む軽鎖可変領域、
(5) 配列番号2で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号3で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、
(6) 配列番号4で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号5で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、
(7) 配列番号6で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号7で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、及び
(8) 配列番号8で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号9で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、
からなる群から選択される重鎖可変領域及び軽鎖可変領域を有する、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
前記抗体が、ヒトc-Mplに対するアゴニスト抗体がヒト抗体である、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記抗体の重鎖定常領域のアッパーヒンジ部が、以下の(1)及び(2):
(1) 配列番号10で示されるアミノ酸配列、及び
(2) 配列番号11で示されるアミノ酸配列、
のアミノ酸配列から選択されるいずれか1つのアミノ酸配列を有し、並びに、該重鎖定常領域のミドルヒンジ部以降のC末端側が、ヒトイムノグロブリンG4のアミノ酸配列或いは該G4のアミノ酸配列においてKabat EU番号付けにおける228位セリンがプロリンに且つ235位ロイシンがグルタミン酸に置換されたアミノ酸配列を有する、請求項4に記載の方法。
【請求項7】
前記重鎖及び軽鎖をコードする塩基配列が、以下の(1)〜(8):
(1) 配列番号2のアミノ酸配列を含む重鎖と配列番号3のアミノ酸配列を含む軽鎖をコードする塩基配列、
(2) 配列番号4のアミノ酸配列を含む重鎖と配列番号5のアミノ酸配列を含む軽鎖をコードする塩基配列、
(3) 配列番号6のアミノ酸配列を含む重鎖と配列番号7のアミノ酸配列を含む軽鎖をコードする塩基配列、及び
(4) 配列番号8のアミノ酸配列を含む重鎖と配列番号9のアミノ酸配列を含む軽鎖をコードする塩基配列、
(5) 配列番号2のアミノ酸配列を含む重鎖と、配列番号3のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、
(6) 配列番号4のアミノ酸配列を含む重鎖と、配列番号5のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、
(7) 配列番号6のアミノ酸配列を含む重鎖と、配列番号7のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、及び
(8) 配列番号8のアミノ酸配列を含む重鎖と、配列番号9のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、
からなる群から選択される、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
以下の(1)〜(4):
(1) 配列番号2で示されるアミノ酸配列、
(2) 配列番号4で示されるアミノ酸配列、
(3) 配列番号6で示されるアミノ酸配列、及び
(4) 配列番号8で示されるアミノ酸配列、
からなる群から選択されるアミノ酸配列をコードする塩基配列を含むDNA。
【請求項9】
以下の(1)〜(4):
(1) 配列番号2で示されるアミノ酸配列、
(2) 配列番号4で示されるアミノ酸配列、
(3) 配列番号6で示されるアミノ酸配列、及び
(4) 配列番号8で示されるアミノ酸配列、
からなる群から選択されるアミノ酸配列を含む抗体重鎖をコードするDNA。
【請求項10】
抗体重鎖のアッパーヒンジ部が、以下の(1)及び(2):
(1) 配列番号10で示されるアミノ酸配列、及び
(2) 配列番号11で示されるアミノ酸配列、
のアミノ酸配列から選択されるいずれか1つのアミノ酸配列であり、並びに、該抗体重鎖のミドルヒンジ部以降のC末端側が、ヒトイムノグロブリンG4のアミノ酸配列或いは該G4のアミノ酸配列においてKabat EU番号付けにおける228位セリンがプロリンに且つ235位ロイシンがグルタミン酸に置換されたアミノ酸配列を有する、請求項9に記載のDNA。
【請求項11】
以下の(1)〜(8):
(1) 配列番号3で示されるアミノ酸配列、
(2) 配列番号5で示されるアミノ酸配列、
(3) 配列番号7で示されるアミノ酸配列、
(4) 配列番号9で示されるアミノ酸配列、
(5) 配列番号3のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
(6) 配列番号5のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
(7) 配列番号7のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、及び
(8) 配列番号9のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
からなる群から選択されるアミノ酸配列をコードする塩基配列を含むDNA。
【請求項12】
以下の(1)〜(8):
(1) 配列番号3で示されるアミノ酸配列、
(2) 配列番号5で示されるアミノ酸配列、
(3) 配列番号7で示されるアミノ酸配列、
(4) 配列番号9で示されるアミノ酸配列、
(5) 配列番号3のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
(6) 配列番号5のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
(7) 配列番号7のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、及び
(8) 配列番号9のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
からなる群から選択されるアミノ酸配列を含む抗体軽鎖をコードするDNA。
【請求項1】
ヒトトロンボポエチン受容体(c-Mpl)に対するアゴニスト抗体の製造方法であって、該抗体の定常領域が、以下の(1)〜(3):
(1) ヒト抗体の重鎖定常領域及び軽鎖定常領域のアミノ酸配列、
(2) ヒト抗体サブクラス間で重鎖定常領域のドメインを入れ替えた重鎖定常領域のアミノ酸配列、及び、ヒト抗体軽鎖定常領域のアミノ酸配列、或いは
(3) 前記(1)又は(2)のアミノ酸配列において1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
のいずれかのアミノ酸配列を有し、且つ抗体の可変領域がヒトトロンボポエチン受容体に結合し該受容体を活性化する能力を有する、並びに、以下の(a)及び(b):
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、10,000ng/mL以下の濃度でコロニー形成を誘導すること、及び
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性が、そのN末端がペグ(PEG)化された配列番号1のアミノ酸配列からなる下記の構造:
PEG-NH-SPAPPACDLRVLSKLLRDSHVLHSRLSQCPEVHPLPTPVLLPAVDFSLGEWKTQMEETKAQDILGAVTLLLEGVMAARGQLGPTCLSSLLGQLSGQVRLLLGALQSLLGTQLPPQGRTTAHKDPNAIFLSFQHLLRGKVRFLMLVGGSTLCVRRAPPTTAVPS-COOH
を有するPEG-rHuMGDFの50%以上であり、且つ50%有効濃度(EC50)が100nM以下であること、
の性質を有する前記アゴニスト抗体を構成する重鎖及び軽鎖をそれぞれコードする塩基配列を含むDNAと、それらのDNAの発現を制御する塩基配列を含む1又は複数のDNAとを保持する哺乳動物細胞を作製し、該哺乳動物細胞を培養した培養液から、該重鎖及び軽鎖からなる抗体をコードする塩基配列を含むDNAの発現産物を単離精製することを含む、前記方法。
【請求項2】
前記抗体が、以下の(a)及び(b):
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、1,000ng/mL以下の濃度でコロニー形成を誘導すること、及び
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性がPEG-rHuMGDFの70%以上であり、且つEC50が10nM以下であること、
の性質を有する、請求項1に記載の方法。
【請求項3】
前記抗体が、以下の(a)及び(b):
(a) ヒト臍帯血CD34+細胞を用いたCFU-MKコロニー形成測定法において、100ng/mL以下の濃度でコロニー形成を誘導すること、及び
(b) UT7/TPO細胞を用いた増殖能測定法において、最大活性がPEG-rHuMGDFの90%以上であり、かつEC50が1nM以下であること、
の性質を有する、請求項1に記載の方法。
【請求項4】
前記抗体が、以下の(1)〜(8):
(1) 配列番号2で示されるアミノ酸配列を含む重鎖可変領域及び配列番号3で示されるアミノ酸配列を含む軽鎖可変領域、
(2) 配列番号4で示されるアミノ酸配列を含む重鎖可変領域及び配列番号5で示されるアミノ酸配列を含む軽鎖可変領域、
(3) 配列番号6で示されるアミノ酸配列を含む重鎖可変領域及び配列番号7で示されるアミノ酸配列を含む軽鎖可変領域、
(4) 配列番号8で示されるアミノ酸配列を含む重鎖可変領域及び配列番号9で示されるアミノ酸配列を含む軽鎖可変領域、
(5) 配列番号2で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号3で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、
(6) 配列番号4で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号5で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、
(7) 配列番号6で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号7で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、及び
(8) 配列番号8で示されるアミノ酸配列を含む重鎖可変領域、及び配列番号9で示されるアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖可変領域、
からなる群から選択される重鎖可変領域及び軽鎖可変領域を有する、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
前記抗体が、ヒトc-Mplに対するアゴニスト抗体がヒト抗体である、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記抗体の重鎖定常領域のアッパーヒンジ部が、以下の(1)及び(2):
(1) 配列番号10で示されるアミノ酸配列、及び
(2) 配列番号11で示されるアミノ酸配列、
のアミノ酸配列から選択されるいずれか1つのアミノ酸配列を有し、並びに、該重鎖定常領域のミドルヒンジ部以降のC末端側が、ヒトイムノグロブリンG4のアミノ酸配列或いは該G4のアミノ酸配列においてKabat EU番号付けにおける228位セリンがプロリンに且つ235位ロイシンがグルタミン酸に置換されたアミノ酸配列を有する、請求項4に記載の方法。
【請求項7】
前記重鎖及び軽鎖をコードする塩基配列が、以下の(1)〜(8):
(1) 配列番号2のアミノ酸配列を含む重鎖と配列番号3のアミノ酸配列を含む軽鎖をコードする塩基配列、
(2) 配列番号4のアミノ酸配列を含む重鎖と配列番号5のアミノ酸配列を含む軽鎖をコードする塩基配列、
(3) 配列番号6のアミノ酸配列を含む重鎖と配列番号7のアミノ酸配列を含む軽鎖をコードする塩基配列、及び
(4) 配列番号8のアミノ酸配列を含む重鎖と配列番号9のアミノ酸配列を含む軽鎖をコードする塩基配列、
(5) 配列番号2のアミノ酸配列を含む重鎖と、配列番号3のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、
(6) 配列番号4のアミノ酸配列を含む重鎖と、配列番号5のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、
(7) 配列番号6のアミノ酸配列を含む重鎖と、配列番号7のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、及び
(8) 配列番号8のアミノ酸配列を含む重鎖と、配列番号9のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列を含む軽鎖をコードする塩基配列、
からなる群から選択される、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
以下の(1)〜(4):
(1) 配列番号2で示されるアミノ酸配列、
(2) 配列番号4で示されるアミノ酸配列、
(3) 配列番号6で示されるアミノ酸配列、及び
(4) 配列番号8で示されるアミノ酸配列、
からなる群から選択されるアミノ酸配列をコードする塩基配列を含むDNA。
【請求項9】
以下の(1)〜(4):
(1) 配列番号2で示されるアミノ酸配列、
(2) 配列番号4で示されるアミノ酸配列、
(3) 配列番号6で示されるアミノ酸配列、及び
(4) 配列番号8で示されるアミノ酸配列、
からなる群から選択されるアミノ酸配列を含む抗体重鎖をコードするDNA。
【請求項10】
抗体重鎖のアッパーヒンジ部が、以下の(1)及び(2):
(1) 配列番号10で示されるアミノ酸配列、及び
(2) 配列番号11で示されるアミノ酸配列、
のアミノ酸配列から選択されるいずれか1つのアミノ酸配列であり、並びに、該抗体重鎖のミドルヒンジ部以降のC末端側が、ヒトイムノグロブリンG4のアミノ酸配列或いは該G4のアミノ酸配列においてKabat EU番号付けにおける228位セリンがプロリンに且つ235位ロイシンがグルタミン酸に置換されたアミノ酸配列を有する、請求項9に記載のDNA。
【請求項11】
以下の(1)〜(8):
(1) 配列番号3で示されるアミノ酸配列、
(2) 配列番号5で示されるアミノ酸配列、
(3) 配列番号7で示されるアミノ酸配列、
(4) 配列番号9で示されるアミノ酸配列、
(5) 配列番号3のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
(6) 配列番号5のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
(7) 配列番号7のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、及び
(8) 配列番号9のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
からなる群から選択されるアミノ酸配列をコードする塩基配列を含むDNA。
【請求項12】
以下の(1)〜(8):
(1) 配列番号3で示されるアミノ酸配列、
(2) 配列番号5で示されるアミノ酸配列、
(3) 配列番号7で示されるアミノ酸配列、
(4) 配列番号9で示されるアミノ酸配列、
(5) 配列番号3のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
(6) 配列番号5のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
(7) 配列番号7のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、及び
(8) 配列番号9のアミノ酸配列においてフレームワーク領域に1若しくは数個のアミノ酸残基が欠失、置換、付加又は挿入されたアミノ酸配列、
からなる群から選択されるアミノ酸配列を含む抗体軽鎖をコードするDNA。
【図1】

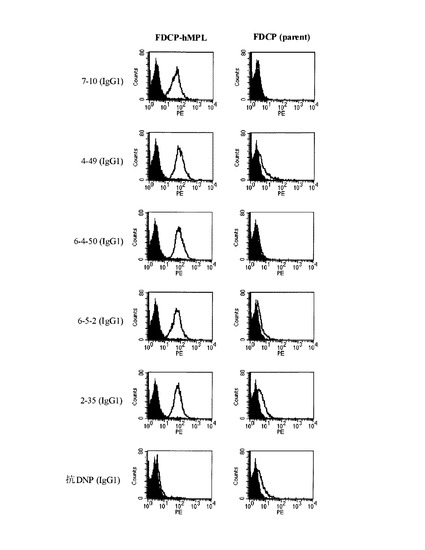
【図2】
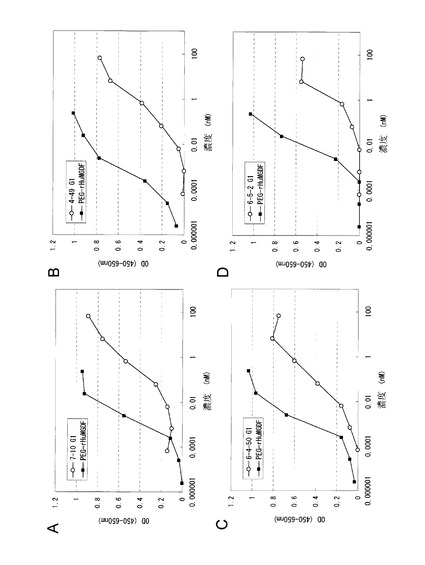
【図3】
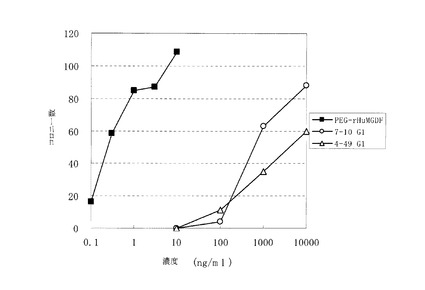
【図4A】
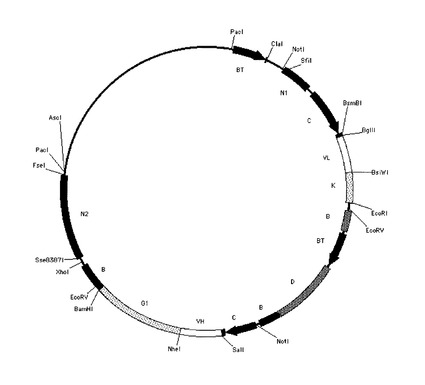
【図4B】
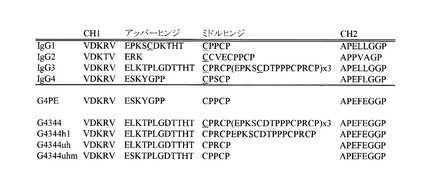
【図4C−1】
【図4C−2】
【図4C−3】
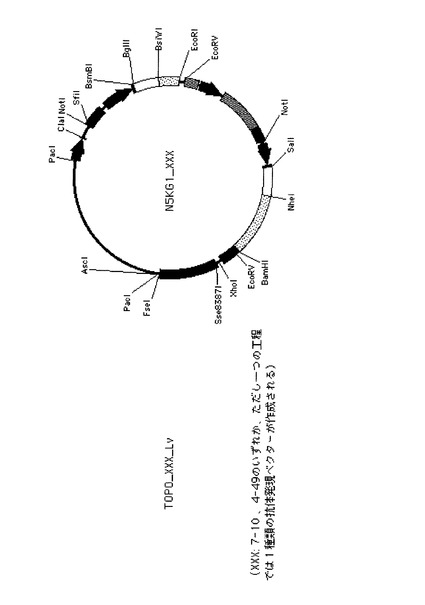
【図4D−1】
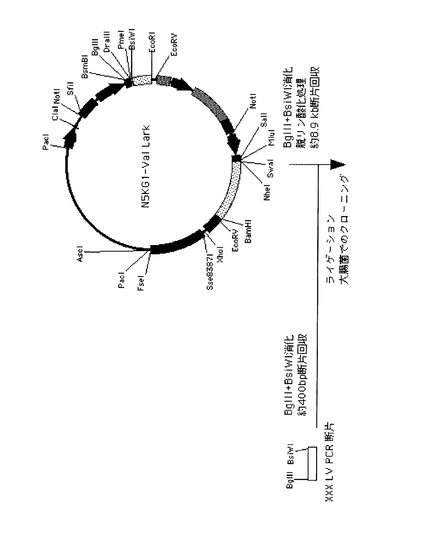
【図4D−2】
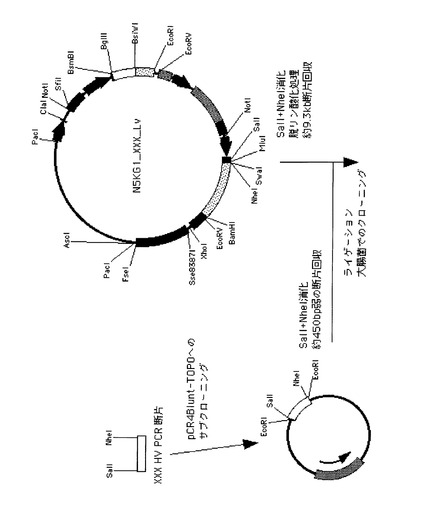
【図4D−3】
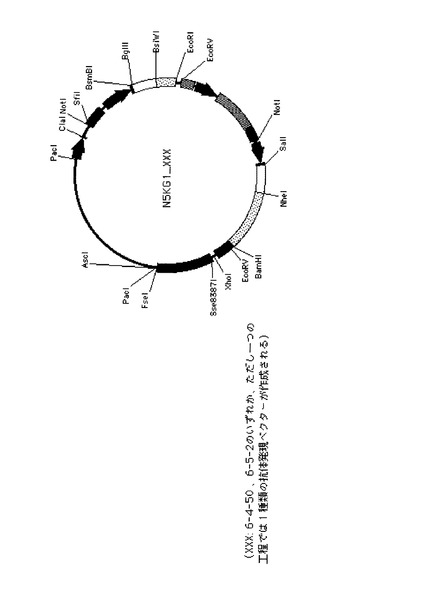
【図4E】

【図4F−1】

【図4F−2】

【図4G】

【図5】
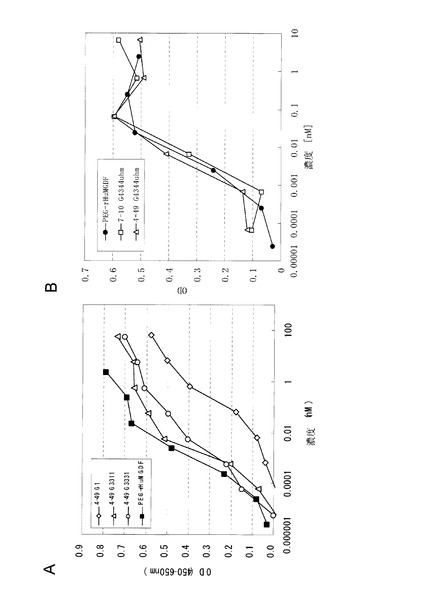
【図6A】
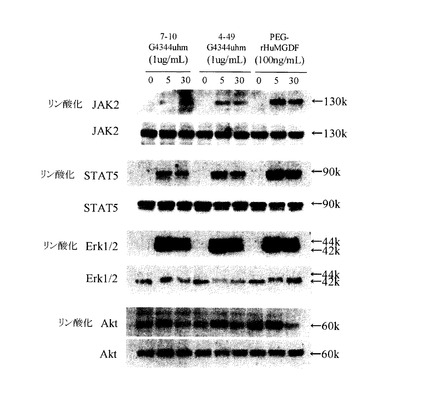
【図6B】
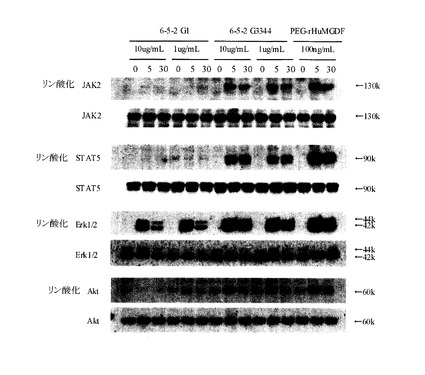
【図7】
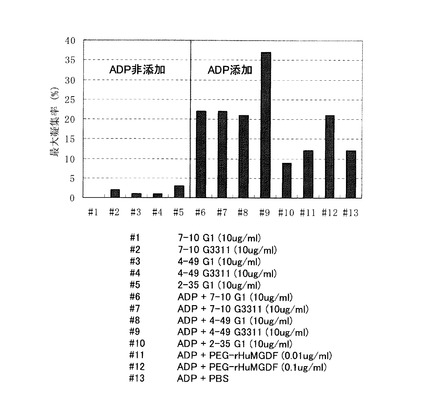
【図8】
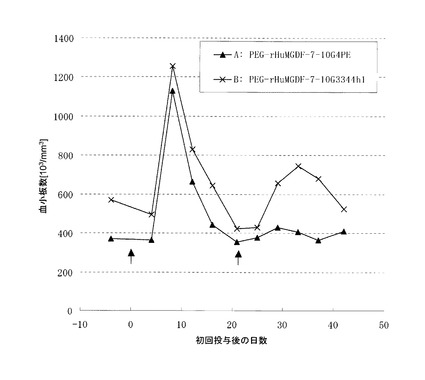
【図9A】
【図9B】
【図9C】
【図10】
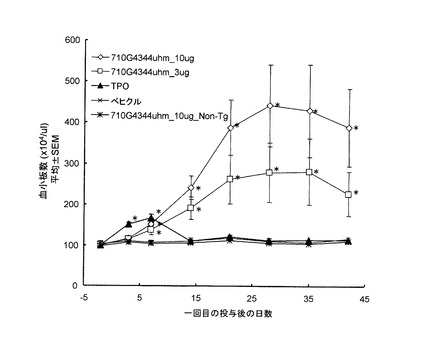
【図11】
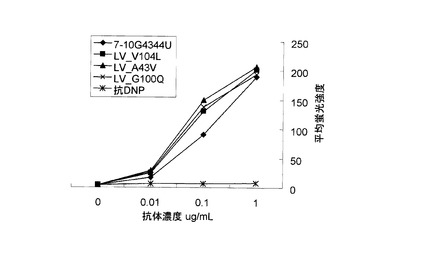
【図12】
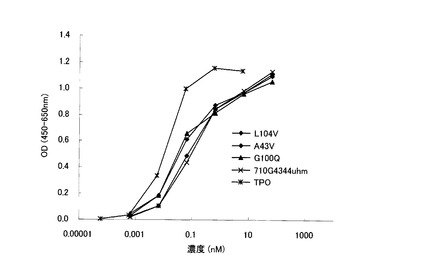
【図2】
【図3】
【図4A】
【図4B】
【図4C−1】
【図4C−2】
【図4C−3】
【図4D−1】
【図4D−2】
【図4D−3】
【図4E】
【図4F−1】
【図4F−2】
【図4G】
【図5】
【図6A】
【図6B】
【図7】
【図8】
【図9A】
【図9B】
【図9C】
【図10】
【図11】
【図12】
【公開番号】特開2009−17881(P2009−17881A)
【公開日】平成21年1月29日(2009.1.29)
【国際特許分類】
【出願番号】特願2008−194088(P2008−194088)
【出願日】平成20年7月28日(2008.7.28)
【分割の表示】特願2008−506356(P2008−506356)の分割
【原出願日】平成19年3月20日(2007.3.20)
【出願人】(000001029)協和発酵キリン株式会社 (276)
【Fターム(参考)】
【公開日】平成21年1月29日(2009.1.29)
【国際特許分類】
【出願日】平成20年7月28日(2008.7.28)
【分割の表示】特願2008−506356(P2008−506356)の分割
【原出願日】平成19年3月20日(2007.3.20)
【出願人】(000001029)協和発酵キリン株式会社 (276)
【Fターム(参考)】
[ Back to top ]