
ヒトメタボトロピックグルタミン酸受容体、これをコードする核酸およびその用途
【課題】ヒトメタボトロピックグルタミン酸受容体蛋白サブタイプ2および5をコードする核酸、およびこれにコードされる蛋白の提供。
【解決手段】ヒトメタボトロピックグルタミン酸受容体のmGluR2およびmGluR5のサブタイプをコードするメタボトロピックグルタミン酸受容体サブタイプの産生に有用である核酸の提供。関連する受容体サブユニットを同定し単離することを可能にするプローブの提供。さらに新規のメタボトロピックグルタミン酸受容体の機能に影響を与える化合物(例えば、グルタミン酸受容体機能のアゴニスト、アンタゴニスト、およびモジュレーター(活性調節因子))を同定し性状解析するための、このような受容体サブタイプの使用方法の提供。
【解決手段】ヒトメタボトロピックグルタミン酸受容体のmGluR2およびmGluR5のサブタイプをコードするメタボトロピックグルタミン酸受容体サブタイプの産生に有用である核酸の提供。関連する受容体サブユニットを同定し単離することを可能にするプローブの提供。さらに新規のメタボトロピックグルタミン酸受容体の機能に影響を与える化合物(例えば、グルタミン酸受容体機能のアゴニスト、アンタゴニスト、およびモジュレーター(活性調節因子))を同定し性状解析するための、このような受容体サブタイプの使用方法の提供。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、核酸とこれによりコードされる受容体蛋白に関する。本発明の核酸は、新規のヒトメタボトロピック(metabotropic)グルタミン酸受容体サブタイプをコードする核酸に関する。本発明はまたこのような受容体サブタイプの作成方法、およびこのような受容体の機能に影響を与える化合物(例えば、アゴニスト、アンタゴニスト、およびヒトメタボトロピックグルタミン酸受容体のアロステリックモジュレーター)の同定および性状解析のための受容体蛋白の使用方法に関する。
【背景技術】
【0002】
アミノ酸のL−グルタミン酸は、哺乳動物の中枢神経系の主要な興奮性神経伝達物質である。解剖学的、生化学的および電気生理学的分析は、グルタミン酸産生系が広範囲のニューロン作用過程(例えば、急速な興奮性シナプス伝達、神経伝達物質放出の制御、長期増強、学習および記憶、成長性シナプス可塑性、低酸素性虚血傷害およびニューロン細胞の死、てんかん様発作、およびいくつかの神経変性障害の病変性)に関与していることを示唆している。一般的には、モナガン(Monaghan)ら、Ann. Rev. Pharmacol. Toxicol.,29:365−402(1980)を参照。この広範囲の機能(特に学習、神経毒性および神経病理に関する機能)のために、近年グルタミン酸の作用機構を説明し規定しようという研究が始まった。
【0003】
現在グルタミン酸受容体の分類の概要は、薬理学的基準に基づいている。グルタミン酸は、2つの主要な受容体(イオノトロピック(ionotropic)受容体とメタボトロピック(metabotropic)受容体)に分類されている受容体を介してその作用を媒介することが認められている。イオノトロピックグルタミン酸受容体は、完全な陽イオン特異的、リガンドゲート(ligand-gated)のイオンチャネルを含有し、メタボトロピックグルタミン酸受容体は、細胞内第二メッセンジャー系の活性化を介して細胞外シグナルを変換するG蛋白結合受容体である。イオノトロピック受容体は、受容体の薬理学的および機能的性質に基づきさらに少なくとも2つの範疇に分類されている。その2つの主要な型のイオノトロピック受容体は、NMDA(N−メチル−D−アスパラギン酸)受容体とカイニン酸(KA)/AMPA(α−アミノ−3−ヒドロキシ−5−メチル−4−イソキサゾールプロピオン酸)(以前はキスカル酸、すなわちQUISと呼ばれていた)受容体である。メタボトロピック受容体はイオノトロピックグルタミン酸受容体が結合するリガンドと同じリガンドのいくつかに結合するが、メタボトロピック受容体はGTP−結合蛋白および第二メッセンジャー(例えば、サイクリックAMP、サイクリックGMP、ジアシルグリセロール、イノシトール1,4,5−三リン酸およびカルシウム)を介してシナプスの生理機能を変更させる[例えば、グンデルセン(Gundersen)ら、Proc. R. Soc. London Ser. 221:127(1984);スラデツェック(Sladeczek)ら、Nature 317:717(1985);ニコレッティ(Nicoletti)ら、J. Neurosci. 6:1905(1986);スギヤマ(Sugiyama)ら、Nature 325:531(1987)]。
【0004】
メタボトロピックグルタミン酸受容体の電気生理学的および薬理学的性質は、受容体供給源として動物組織や細胞株、および非ヒト組換え受容体を用いて研究されている。ヒトの治療薬開発へのそのような研究の意義は、ヒトでない受容体サブユニットのみしか入手できないことにより限定されている。さらに、メタボトロピックグルタミン酸受容体の特性や構造が分子レベルで研究され出したのはつい最近である。しかしこのような研究は、ヒト以外の種でのみ行われている。メタボトロピックグルタミン酸受容体の生理学的および病理学的重要性の可能性により、グルタミン酸受容体の種々のクラスの代表的メンバーをコードするヒトの配列(すなわち、DNA、RNA、蛋白)が利用(例えば、薬剤スクリーニング測定法のために)できることが必要である。そのようなヒトの配列が入手できれば、ヒトでの受容体の分布の研究、特定の受容体の修飾と種々の疾患の発生の関係の研究も可能になるであろう。
【非特許文献1】グンデルセン(Gundersen)ら、Proc. R. Soc. London Ser. 221:127(1984)
【非特許文献2】スラデツェック(Sladeczek)ら、Nature 317:717(1985)
【非特許文献3】ニコレッティ(Nicoletti)ら、J. Neurosci. 6:1905(1986)
【非特許文献4】スギヤマ(Sugiyama)ら、Nature 325:531(1987)
【発明の開示】
【課題を解決するための手段】
【0005】
(本発明の概要)
本発明は、ヒトメタボトロピックグルタミン酸受容体蛋白サブタイプをコードする新規の核酸、およびこれにコードされる蛋白を開示する。具体的な実施態様において、新規の核酸は、ヒトメタボトロピックグルタミン酸受容体の全長mGluR1、mGluR2、mGluR3およびmGluR5サブタイプまたはその一部をコードする。これらの核酸はメタボトロピックグルタミン酸受容体サブタイプ蛋白の産生に有用である以外に、プローブとしても有用であり、従って当業者は不要な実験をすることなく、関連する受容体サブタイプをコードする核酸を同定し単離することが可能である。
【0006】
本発明は、新規のメタボトロピックグルタミン酸受容体蛋白サブタイプを開示する以外に、このような受容体に影響を与える化合物(例えば、グルタミン酸受容体機能のアゴニスト、アンタゴニスト、およびモジュレーター(活性調節因子))を同定し性状解析するための、このような受容体サブタイプの使用方法を包含する。また本発明は、未知の蛋白がメタボトロピックグルタミン酸受容体サブタイプとして機能するか否かを測定する方法を包含する。
【0007】
(本発明の詳細な説明)
本発明において、ヒトメタボトロピックグルタミン酸受容体サブタイプをコードする単離された核酸が提供される。本発明の1つの面において、mGluR1サブタイプのヒトメタボトロピックグルタミン酸受容体サブタイプをコードする核酸が提供される。別の面において、mGluR2サブタイプのメタボトロピックグルタミン酸受容体の少なくとも1部分をコードする核酸が提供される。さらに別の面において、mGluR3サブタイプのメタボトロピックグルタミン酸受容体をコードする核酸が提供される。さらなる面において、mGluR5サブタイプのメタボトロピックグルタミン酸受容体をコードする核酸が提供される。さらに別の面において、そのような核酸を含有する真核細胞、およびそのような核酸を発現する真核細胞が提供される。
【0008】
また前述の核酸によりコードされる蛋白、および該蛋白に対して産生される抗体が提供される。本発明の別の面において、上記核酸のメタボトロピックグルタミン酸受容体サブタイプ−選択性部分よりなる核酸プローブが提供される。
【0009】
本明細書において、「ヒトメタボトロピックグルタミン酸受容体サブタイプ」とは、グルタミン酸産生リガンドに対する細胞のG−蛋白結合応答に参加する、単離されたおよび/または精製された蛋白を意味する。このような受容体サブタイプは、他のメタボトロピックグルタミン酸受容体サブタイプをコードしない別々の遺伝子により個々にコードされる(すなわち、各サブタイプはユニークな遺伝子によりコードされる)。このような受容体サブタイプの典型的な特徴は、7つの推定されるトランスメンブレンドメインと、その前に大きな推定される細胞外アミノ末端ドメインと、その後に大きな推定される細胞内カルボキシ末端ドメインを有することである。メタボトロピックグルタミン酸受容体は、基本的にメタボトロピックグルタミン酸受容体でない他のG蛋白結合受容体と全くアミノ酸配列相同性を有さない。
【0010】
メタボトロピックグルタミン酸受容体サブタイプの各々の間の関係に関して、mGluR1受容体サブタイプのアミノ酸配列は一般に、他のヒトメタボトロピックグルタミン酸受容体サブタイプのアミノ酸配列との同一性は約70%未満であり、典型的に観察される同一性は45%未満である。mGluR2受容体サブタイプのアミノ酸配列は一般に、他のヒトメタボトロピックグルタミン酸受容体サブタイプのアミノ酸配列との同一性は約60%未満であり、典型的に観察される同一性は45%未満である。mGluR3受容体サブタイプのアミノ酸配列は一般に、他のヒトメタボトロピックグルタミン酸受容体サブタイプのアミノ酸配列との同一性は約60%未満であり、典型的に観察される同一性は45%未満である。mGluR5受容体サブタイプのアミノ酸配列は一般に、他のヒトメタボトロピックグルタミン酸受容体サブタイプのアミノ酸配列との同一性は約70%未満であり、典型的に観察される同一性は45%未満である。
【0011】
上記定義に含まれるものは、一次転写体の別のスプライシングにより産生されるmRNAによりコードされる変種、および上記の生理学的および/または物理的性質の1つまたはそれ以上を有するその断片である。
【0012】
本明細書および請求の範囲において、DNA、RNA、ポリペプチドまたは蛋白の修飾物質としての「単離される」、「純粋な」という用語は、このように呼ばれるDNA、RNA、ポリペプチドまたは蛋白はヒトの手によりそのような形で産生され、従ってその本来のインビボの細胞環境からは分離されていることを示す。このヒトの介入による結果、本発明の組換えDNA、RNA、ポリペプチドおよび蛋白は、天然に存在するDNA、RNA、ポリペプチドまたは蛋白では役に立たないような方法(例えば、選択的薬剤または化合物の同定)において有用である。
【0013】
本発明の受容体蛋白の修飾物質として使用される時、「機能性」とは、グルタミン酸産生関連リガンド(例えばACPDまたはACPD様リガンド、QUIS、AP4など)に受容体蛋白に結合すると、G蛋白との受容体の相互作用を修飾し、これは細胞内第2メッセンジャーのレベルに影響を与え、種々の生理学的作用を引き起こす。言い換えると「機能性」とは、受容体蛋白のアゴニスト活性化の結果として応答が産生されることを意味する。
【0014】
本明細書においてスプライス変種とは、ゲノムDNAの一次転写体の差別的処理により産生され、その結果2つ以上の型のmRNAの産生に至る、変種メタボトロピックグルタミン酸受容体サブタイプをコードする核酸を意味する。差別的に処理された一次転写体から得られるcDNAは、アミノ酸が完全に同一の領域と、異なるアミノ酸配列を有する領域を有する、メタボトロピックグルタミン酸受容体サブタイプをコードするであろう。すなわち同じゲノム配列から、多数の関連するmRNAや蛋白が得られる。得られるmRNAや蛋白はいずれも本明細書では「スプライス変種」と呼ぶ。
【0015】
従って、前記で定義したメタボトロピックグルタミン酸受容体サブタイプをコードするが、遺伝コードの縮重のため特定のハイブリダイゼーション条件下で開示された核酸に必ずしもハイブリダイズしない核酸も、本発明の範囲内にあると考えられる。本明細書に記載の方法または当業者に公知の方法により評価すると、そのようなサブタイプもまた、機能性受容体を形成する。典型的には、メタボトロピックグルタミン酸受容体サブタイプが代替スプライシングにより得られるRNAにコードされない(すなわち、スプライス変種)なら、メタボトロピックグルタミン酸受容体サブタイプをコードする核酸とこれによりコードされるメタボトロピックグルタミン酸受容体蛋白は、本明細書に記載の少なくとも1つのメタボトロピックグルタミン酸受容体サブタイプ核酸(およびこれによりコードされる蛋白)と実質的な配列相同性を有する。スプライス変種をコードするDNAまたはRNAは、本明細書で提供されるDNAまたはRNAとの配列全体の相同性は90%未満であるが、本明細書中のDNA断片とほとんど100%の相同性を有する領域を含有し、開始コドンおよび停止コドンを含むオープンリーディングフレームをコードし、機能性メタボトロピックグルタミン酸受容体サブタイプをコードすることを理解すべきである。
【0016】
ヒトmGluR1サブタイプをコードするDNA配列の例は、配列番号2に記載されるものと実質的に同じアミノ酸配列をコードするヌクレオチドにより示される。現在好適な配列は、配列番号2に示されるアミノ酸配列をコードする。
【0017】
別のDNAの例は、ヒトmGluR1サブタイプをコードし、高緊縮性条件下で配列番号1の実質的に配列全体、またはその大部分(すなわち、典型的には少なくともその25−30個の隣接ヌクレオチド)にハイブリダイズするヌクレオチドであることが特徴である。
【0018】
本明細書において、ハイブリダイゼーションの緊縮性とは、ポリ核酸ハイブリッドが安定である条件を意味する。当業者に公知なように、ハイブリッドの安定性はハイブリッドの融点(Tm)に反映される。
【0019】
Tmは、
式: 81.5℃−16.6(log10[Na+])+0.41(%G+C)−600/l
(式中、lはヌクレオチド中のハイブリッドの長さである)により近似的に表される。
【0020】
配列の相同性が1%減少する毎に、Tmは約1−1.5℃低下する。一般にハイブリッドの安定性は、ナトリウムイオン濃度と温度の関数である。典型的には、ハイブリダイゼーション反応を低緊縮性条件下で行い、次に種々の(ただし、より高い緊縮性の)条件下で洗浄する。ハイブリダイゼーション緊縮性は、このような洗浄条件に関する。
【0021】
すなわち、本明細書において、
(1)断片のハイブリダイゼーションに関して、高緊縮性条件とは、65℃で0.018MのNaCl中で安定なハイブリッドを形成する核酸配列のみのハイブリダイゼーションを可能にする条件を意味する(すなわち、本明細書で企図されるように、もしハイブリッドが65℃で0.018MのNaCl中で安定でない場合、高緊縮性条件下で安定ではないであろう)。高緊縮性条件は、例えば50%ホルムアミド、5×デンハルツ(Denhart's)溶液、5×SSPE、0.2%SDS中で42℃でハイブリダイゼーションし、次に65℃で0.1×SSPE、そして0.1%SDSによる洗浄により提供される。
(2)断片のハイブリダイゼーションに関して、中緊縮性条件とは、42℃で50%ホルムアミド、5×デンハルツ溶液、5×SSPE、0.2%SDS中でハイブリダイゼーションし、次に65℃で0.2×SSPE、そして0.2%SDSによる洗浄と同等の条件を意味する。
(3)断片のハイブリダイゼーションに関して、低緊縮性条件とは、42℃で10%ホルムアミド、5×デンハルツ溶液、6×SSPE、0.2%SDS中でハイブリダイゼーションし、次に50℃で1×SSPE、そして0.2%SDSによる洗浄と同等の条件を意味する。そして、
(4)オリゴヌクレオチド(すなわち、長さが約30ヌクレオチド以下の合成DNA)に関して、高緊縮性条件とは、42℃で10%ホルムアミド、5×デンハルツ溶液、6×SSPE、0.2%SDS中でハイブリダイゼーションし、次に50℃で1×SSPE、そして0.2%SDSによる洗浄に等しい条件を意味する。
【0022】
種々の緩衝液や温度を用いてこれらの条件を再現することができ、これらは必ずしも正確である必要はないことを理解すべきである。
【0023】
デンハルツ溶液およびSSPE(例えば、サムブルーク、フリッチ、およびマニアチス(Sambrook, Fritsch, and Maniatis)、モレキュラークローニング、実験室マニュアル(Molecular Cloning、A Laboratory Manual)、コールド・スプリング・ハーバー・ラボラトリー・プレス(Cold Spring Harbor Laboratory Press)、1989)は、他の適当なハイブリダイゼーション緩衝液と同様に、当業者に公知である。例えば、SSPEはpH7.4のリン酸緩衝化された0.18MのNaClである。SSPEは例えば、175.3gのNaCl、27.6gのNaH2PO4および7.4gのEDTAを800mlの水に溶解しpHを7.4に調整して、次に1リットルになるように水を加えて20×保存溶液として調製することができる。デンハルツ溶液(デンハルト(Denhart)(1966)Biochem. Biophys. Res. Commun.23:641を参照)は、例えば5gのFicoll(タイプ400、ファルマシアLKBバイオテクノロジー社(Pharmacia LKBBiotechnology、INC.)、ピスカタウェイ(Piscataway)、ニュージャージー州)、5gのポリビニルピロリドン、5gのウシ血清アルブミン(第5画分;シグマ(Sigma)、セントルイス、ミズーリ州)、そして水を500mlになるように加え、濾過して粒状物質を除去することにより、50×保存溶液として調製することができる。
【0024】
ヒトmGluR1サブタイプをコードする特に好適な配列は、配列番号1と実質的に同じヌクレオチド配列を有するものであり、配列番号1のコーディング配列と実質的に同じ配列を有するポリ核酸が特に好ましい。
【0025】
本明細書において、「実質的な配列相同性」とは、少なくとも約90%の相同性を有するヌクレオチド配列、そして典型的には95%以上のアミノ酸の同一性を有するヌクレオチド配列を意味する。しかし、スプライス変種であること、または保存的アミノ酸置換(または縮重コドンの置換)により修飾されているため、前記レベルより低い相同性を有する蛋白(およびこのような蛋白をコードするDNAまたはmRNA)は、本発明の範囲内にあることを理解すべきである。
【0026】
本明細書において、「実質的に同じ」とは、本明細書に記載の実際の配列とはわずかに異なるかまたは重大でない配列の変化を有する、DNAのヌクレオチド配列、RNAのリボヌクレオチド配列、または蛋白のアミノ酸配列を意味する。実質的に同じ分子種は、開示された配列と同等であると考えられ、従って本発明の請求の範囲に包含されると考えられる。この点で「わずかに異なるかまたは重大でない配列の変化」は、開示され、特許請求のされているDNA、RNA、または蛋白と実質的に同じ配列が、開示され特許請求のされているヒト由来の配列と機能的に同等であることを意味する。機能的に同等の配列は、実質的に同じ方法で機能して、ヒト由来の核酸および本明細書に開示され特許請求されているアミノ酸組成物と実質的に同じ組成物を産生する。特に機能的に同じDNAは、本明細書に開示されたものと同じであるか、または保存性のアミノ酸変化(例えば非極性残基を別の非極性残基で置換、または荷電残基を類似の荷電残基で置換)を有するものと実質的に同じヒト由来の蛋白をコードする。これらの変化には、当業者に認識される、蛋白の3次元構造を実質的に変化させないものが含まれる。
【0027】
ヒトmGluR2受容体サブタイプの一部をコードするDNA配列の例は、配列番号4に記載のものと実質的に同じアミノ酸配列をコードするヌクレオチド(随時、配列番号13に記載の3’非翻訳配列の343ヌクレオチドの一部またはすべてを含む)、またはクローンMETAB40(1993年5月4日に整理番号75465でアメリカンタイプカルチャーコレクション(ATCC)に寄託された)のヒトmGluR2をコードする部分によりコードされるものと実質的に同じアミノ酸配列をコードするヌクレオチドにより示される。
【0028】
寄託されたクローンは、1993年5月4日に「特許手続上の微生物の寄託の国際的承認に関するブダペスト条約」(the Budapest Treaty on the International Recognition of Deposits of Microorganisms for Purposes of Patent Procedure)の条件と、この条約の名のもとに公布された規則に従い、アメリカンタイプカルチャーコレクション(ATCC)(12301 パークローンドライブ(Parklawn Drive)、ロックヴィル(Rockville)、メリーランド州、アメリカ合衆国 20852)に寄託されている。この条約と規則の条件下で、およびさもなくばアメリカ合衆国の特許法や規則に従い受領する法的資格のある工業所有権事務所および他の個人、または本出願または本出願の優先権を主張する出願が提出されたかまたはこのような出願に対して何らかの特許権が与えられた国または国際組織は、寄託された物質の試料を入手することができる。特にこれに基づきまたはこの優先権を主張する出願またはこれに関する出願を取り込んでいる出願に基づき米国特許の与えられた時点で、本寄託物質の入手に関するすべての規制は永久に取り消されるであろう。
【0029】
ヒトmGluR2受容体サブタイプの一部をコードする好適なポリ核酸配列は、配列番号4に記載のものと同じアミノ酸配列、またはクローンMETAB40(1993年5月4日に整理番号75465でアメリカンタイプカルチャーコレクション(ATCC)に寄託された)のヒトmGluR2をコードする部分によりコードされるものと実質的に同じアミノ酸配列をコードするものである。
【0030】
あるいはDNAの例は、ヒトmGluR2受容体サブタイプをコードするヌクレオチド配列であり、高緊縮性条件下で配列番号3またはその大部分(すなわち、典型的にはその少なくとも25−30個の隣接ヌクレオチド)、またはクローンMETAB40(ATCC整理番号75465)のヒトmGluR2をコードする部分とまたはその大部分とハイブリダイズするヌクレオチド配列として特徴付けられる。ヒトmGluR2受容体サブタイプの部分をコードする特に好適な配列は、配列番号3のコーディング配列と同じヌクレオチド配列、またはクローンMETAB40のヒトmGluR2コーディング部分中のコーディング配列のヌクレオチド配列を有するポリ核酸により示される。
【0031】
ヒトmGluR3受容体サブタイプをコードするDNA配列の例は、配列番号6に記載のアミノ酸配列と実質的に同じアミノ酸配列をコードするヌクレオチドにより示される。好適なポリ核酸配列は、配列番号6と同じ配列をコードするものである。
【0032】
あるいはDNAの例は、ヒトmGluR3受容体サブタイプをコードするヌクレオチド配列であり、高緊縮性条件下で配列番号5またはその大部分(すなわち、典型的にはその少なくとも25−30個の隣接ヌクレオチド)とハイブリダイズするヌクレオチド配列として特徴付けられる。ヒトmGluR3サブタイプの部分をコードする特に好適な配列は、配列番号5のコーディング配列と実質的に同じヌクレオチド配列を有するものであり、配列番号5のコーディング配列と同じヌクレオチド配列を有するポリ核酸が最も好ましい。
【0033】
DNAの例は、ヒトmGluR3受容体サブタイプまたはその部分をコードするヌクレオチド配列であり、配列番号8、10または12のアミノ酸配列と実質的に同じアミノ酸配列をコードするヌクレオチドにより示される。好適なポリ核酸配列は、配列番号8、10または12と同じ配列をコードするものである。
【0034】
あるいはDNAの例は、ヒトmGluR5受容体サブタイプをコードするヌクレオチド配列であり、高緊縮性条件下で配列番号7、9または11の実質的に全配列、またはそれらの大部分(すなわち、典型的には少なくとも25−30個の隣接ヌクレオチド)にハイブリダイズするヌクレオチドとして特徴付けられる。ヒトmGluR5サブタイプをコードする特に好適な配列は、配列番号7、9または11のコーディング配列と実質的に同じヌクレオチド配列を有するものであり、配列番号7、9または11のコーディング配列と同じ配列を有するポリ核酸が最も好ましい。
【0035】
ヒトメタボトロピックグルタミン酸受容体サブタイプをコードするDNAは、本明細書に開示したDNA(配列番号1、3、5、7、9または11の任意のものより得られるヌクレオチドを含む)により、適当なハイブリダイゼーション条件下で適当なヒトcDNAまたはヒトゲノムライブラリーをスクリーニングすることにより単離される。適当なライブラリーは、神経組織試料(例えば、海馬や小脳組織、細胞株など)から調製される。例えばライブラリーは、その実質的に全ての受容体サブタイプコーディング配列を含むDNAの一部を用いてスクリーニングされるか、あるいはそのDNAの一部に基づく適当なオリゴヌクレオチドプローブでスクリーニングされる。
【0036】
本明細書においてプローブは、配列番号1、3、5、7、9または11に記載の任意のものの25またはそれ以上の隣接する塩基と同じであるか少なくとも約25−30個の隣接する塩基(またはその相補物)を含む、ヌクレオチドの配列を有する1本鎖DNAまたはRNAである。プローブを作成するのに好適な領域は、5’および/または3’コーディング配列、トランスメンブレンドメインをコードすると予測される配列、細胞質ループをコードすると予測される配列、シグナル配列、リガンド結合部位などがある。
【0037】
全長cDNAクローン、その断片、またはそのDNAの部分に基づくオリゴヌクレオチドはプローブとして使用され、好ましくは容易に検出するために適当な標識物で標識される。断片がプローブとして使用される時、そのプローブのDNA配列は好ましくはDNAのカルボキシ末端をコードする部分由来であり、最も好ましくはDNA配列の予測されるトランスメンブレンドメインをコードする部分を含む(ドメインは、例えばカイトとドーリトル(Kyte and Doolittle)(1982)、J. Mol. Biol. 第157巻:105の方法を用いて、推定されるアミノ酸配列のハイドロパシー解析に基づき予測される)。これらのプローブは例えば、グルタミン酸受容体ファミリーの追加メンバーの同定と単離のために使用される。
【0038】
本発明の配列の特定の応用として、本発明のヌクレオチド配列をプローブとして使用して遺伝子スクリーニングを実施することができる。すなわち、任意の内因性グルタミン酸受容体に異常が存在するか否かを測定するために、グルタミン酸受容体の任意の1つまたはそれ以上の変化/修飾が関与していることが疑われる神経病理的症状を有する患者の核酸試料を、適当なプローブでスクリーニングすることができる。同様に、グルタミン酸受容体の機能不全に関連する疾患の家族歴がある患者をスクリーニングして、このような疾患に罹りやすい体質であるか否かを決定することができる。
【0039】
本発明の別の実施態様において、ヒトメタボトロピックグルタミン酸受容体蛋白サブタイプをコードするDNAの同定方法が提供され、この方法は、ヒトDNAを上記の核酸プローブと接触させ(使用されるプローブがポリ核酸断片の時は低緊縮性または中緊縮性条件下で接触させ、または使用されるプローブがオリゴヌクレオチドの時は高緊縮性条件下で接触させる)、そして該プローブにハイブリダイズするDNAを同定することよりなる。
【0040】
ライブラリーのスクリーニング後、ハイブリダイゼーションシグナルを検出して陽性クローンを同定する。同定されたクローンを制限酵素マッピングおよび/またはDNA配列解析により性状解析し、次にこれらが完全なメタボトロピックグルタミン酸受容体サブタイプをコードするDNAを含有するか否か(すなわち、これらが翻訳開始コドンおよび停止コドンを含有するか否か)を確認するために本明細書に記載の配列と比較することにより試験する。選択された配列が不完全な場合はこれらを用いて、同じかまたは異なるライブラリーを再スクリーニングして重複するクローンを得ることができる。もしライブラリーがゲノム性の場合は、重複クローンはエクソンとイントロンを含有してもよい。もしライブラリーがcDNAライブラリーの場合は、重複クローンはオープンリーディングフレームを含有するであろう。いずれの場合も完全なクローンは、本明細書で提供されるDNAおよび推定されるアミノ酸配列と比較して同定される。
【0041】
種々のヒトメタボトロピックグルタミン酸受容体サブタイプ(例えば、mGluR1、mGluR2、mGluR3、mGluR5)をコードする、相補的なDNAクローンが単離されている。各サブタイプは、異なる遺伝子にコードされているようである。本明細書に記載されるDNAクローンは、各サブタイプをコードするゲノムクローンを単離し、異なる神経組織から調製されたライブラリーをスクリーニングすることにより、スプライス変種を単離するために使用できる。当業者に公知の核酸増幅法を使用して、ヒトメタボトロピックグルタミン酸受容体サブタイプのスプライス変種をコードするDNAを見つけることができる。これは、公知のまたは予測される互いに異なる配列を取り囲むDNA配列に基づくオリゴヌクレオチドを、ヒトRNAまたはゲノムDNAを増幅するためのプライマーとして用いることにより達成される。増幅生成物のサイズと配列の測定により、スプライス変種の存在が明らかになる。さらにハイブリダイゼーションによるヒトゲノムDNA配列の単離により、ヒトメタボトロピックグルタミン酸受容体サブタイプをコードする転写体の異なるスプライス変種に対応する、イントロンにより分離された多数のエクソンを含有するDNAが得られる。
【0042】
必ずしもすべてのメタボトロピックグルタミン酸受容体サブタイプ(およびその変種)がすべての神経組織または脳のすべての部分に発現されているのではないことは、わかっている。すなわち特定のサブタイプ(またはそのスプライス変種)をコードするcDNAを単離するためには、異なるニューロンまたは神経組織または細胞から調製されるライブラリーをスクリーニングすることが好ましい。各サブユニットをコードするDNAを得るために好適なライブラリーは:ヒトmGluR1をコードするDNAを単離するための小脳;mGluR2をコードするDNAを単離するための海馬;mGluR3をコードするDNAを単離するための海馬と小脳;mGluR5をコードするDNAを単離するための海馬と小脳などがある。
【0043】
特定の受容体サブタイプをコードするDNAが一旦単離されると、そのようなサブタイプ(またはそのスプライス変種)をコードするmRNAを発現する組織を決定するために、リボヌクレアーゼ(RNase)保護測定法が実施される。この測定法は、全細胞性RNAの複雑な混合物中の1つのRNA種を検出し定量するための高感度の手段を与える。サブタイプDNAは標識され、細胞性RNAとハイブリダイズされる。もし細胞性RNA中に相補的なmRNAが存在する場合、DNA−RNAハイブリッドが得られる。次にRNA試料をRNaseで処理する(これは1本鎖RNAを分解する)。RNA−DNAハイブリッドは、RNase分解から保護され、ゲル電気泳動やオートラジオグラフィーにより肉眼で見ることができる。特定のメタボトロピックグルタミン酸受容体サブタイプをコードするmRNAを発現する組織を決定するために、in situ ハイブリダイゼーションを使用することができる。こうして、標識されたサブタイプDNAは異なる脳の領域のスライスとハイブリダイズして、サブタイプmRNA発現が肉眼で見えるようになる。
【0044】
いくつかのヒトのメタボトロピックグルタミン酸受容体サブタイプの発現の分布は、この受容体のラットでの分布とは異なる。例えばラットのmGluR5サブタイプをコードするRNAはラットの海馬に多く存在し、ラットの小脳にはあまり存在しない[例えば、アベ(Abe)ら、J. Biol. Chem.267:13361−13368(1992)を参照]が、ヒトの小脳ライブラリーからは無数のヒトmGluR5をコードするcDNAが得られている。すなわちヒトとラットではいくつかのメタボトロピックグルタミン酸受容体サブタイプの分布が異なっているようである。
【0045】
前述のヌクレオチド配列はベクターに取り込んでさらに操作することができる。本明細書においてベクター(またはプラスミド)とは、異種DNAを細胞に取り込んでそれを発現または複製をさせるのに使用される、独立した成分である。このような担体(vehicle)の選択と使用は、当業者には公知である。
【0046】
発現ベクターとは、DNA断片の発現を制御することができる制御配列(例えばプロモーター領域)に機能的に結合したDNAを発現することができるベクターを含む。すなわち発現ベクターとは、適当な宿主細胞内に取り込まれるとクローン化DNAを発現する、組換えDNAまたはRNA作成体(例えば、プラスミド、ファージ、組換えウイルスまたは他のベクター)を意味する。適当な発現ベクターは当業者に公知であり、真核細胞および/または原核細胞中で複製されるもの、およびエピソームとしてとどまるか宿主細胞ゲノム内に取り込まれるものも含まれる。真核生物の宿主細胞(特に哺乳動物細胞)中での本発明のメタボトロピックグルタミン酸受容体サブタイプの発現のために好適なプラスミドには、例えば、pCMV−T7−2、pCMV−T7−3(図1を参照)、pcDNA1などのサイトメガロウイルス(CMV)プロモーター含有ベクター、そして、SV40プロモーター含有ベクター、およびpMMTVT7(+)またはpMMTVT7(−)(本明細書に記載のように調製されたpMAMneo(クロンテク(Clontek)、パロアルト(Palo Alto)、カリホルニア州)を修飾したもの)などのMMTVLTRプロモーター含有ベクターがある。
【0047】
本明細書において、プロモーター領域とは、そこに機能的に結合したDNAの転写を調節するDNAのセグメントを意味する。プロモーター領域は、RNAポリメラーゼによる認識、結合および転写の開始に充分な特異的配列を含有する。このプロモーター領域の部分をプロモーターと呼ぶ。さらにこのプロモーター領域は、このRNAポリメラーゼの認識、結合および転写開始を調節する配列を含む。これらの配列はcis作用性であるか、またはtrans活性化因子に対して応答する。制御の性質により、プロモーターは構成性(constitutive)であるかまたは制御されている。本発明の実施において使用されるプロモーターの例には、SV40早期プロモーター、サイトメガロウイルス(CMV)プロモーター、マウス乳癌ウイルス(MMTV)ステロイド誘導性プロモーター、モロニー(Moloney)マウス白血病ウイルス(MMLV)プロモーターなどがある。
【0048】
本明細書において、「機能的に結合している」とは、DNAとヌクレオチドの制御配列やエフェクター配列(例えばプロモーター、エンハンサー、転写および翻訳停止部位、および他のシグナル配列)との機能的関係を意味する。例えば、DNAとプロモーターの機能的結合は、DNAの転写はプロモーターを特異的に認識してこれに結合し、DNAを転写するRNAポリメラーゼによりプロモーターから開始されるような、DNAとプロモーターの間の物理的および機能的結合を意味する。発現および/またはインビトロの転写を最適化するために、転写または翻訳のレベルで発現を妨害するかまたは低下させる、不適当な代替翻訳開始コドンまたは他の配列を除去するために、クローンの5’および/または3’非翻訳部分を除去、付加または変更することが必要なこともある。あるいはコンセンサスリボゾーム結合部位(例えば、コザック(Kozak))(1991)J. Biol.Chem. 266:19867−19870)を開始コドンの5’のすぐ近くに挿入して、発現を増強させることもできる。同様に、転写を増強するために、メタボトロピックグルタミン酸受容体サブユニットのコーディング配列の代わりに同じアミノ酸をコードする代替コドンを用いることができる(例えば、宿主細胞のコドンの選択性が採用され、G−Cの豊富なドメインを減少させるなど)。さらに両生類の卵母細胞中のメタボトロピックグルタミン酸受容体サブユニットの発現増強の可能性のために、サブユニットコーディング配列を随時発現作成体中に取り込むことができる(ここではコーディング配列の5’末端と3’末端は、それぞれツメガエル(Xenopus)のβ−グロビン遺伝子の5’と3’の非翻訳配列に隣接している)。例えば、メタボトロピックグルタミン酸受容体サブユニットコーディング配列は、pSP64(プロメガ(Promega)、マジソン、ウィスコンシン州、より入手できる)の修飾型であるベクターpSP64Tに取り組むことができる(クリーグとメルトン(Krieg and Melton)(1984)Nucleic Acids Research 12:7057−7070を参照)。このコーディング配列は、β−グロビン遺伝子の5’末端とSP6プロモーターの下流に位置する3’非翻訳配列の間に挿入される。次に得られるベクターから、インビトロの転写体が作成される。このような修飾の好ましい点(またはニーズ)は実験的に決定されている。
本明細書において、発現とはポリ核酸がmRNAに転写され、ペプチド、ポリペプチドまたは蛋白に翻訳される過程を意味する。ポリ核酸がゲノムDNAから得られる場合、適当な真核生物宿主細胞または微生物が選択されるなら、発現にはmRNAのスプライシングを含む。
【0049】
哺乳動物細胞のトランスフェクションのための、ヒトメタボトロピックグルタミン酸受容体をコードするDNAに結合した制御成分を含有する特に好適な基礎ベクターは、サイトメガロウイルス(CMV)プロモーターをベースにしたベクター(例えば、pCMV−T7−2およびpCMV−T7−3(本明細書に記載)、またはpcDNA1(インビトロゲン(Invitrogen)、サンジエゴ、カリホルニア州))、およびMMTVプロモーターをベースにしたベクター(例えば、pMMTVT7(+)またはpMMTVT7(−)(本明細書中に記載))、およびSV40プロモーターをベースにしたベクター(例えば、pSVβ(クロンテック(Clontech)、パロアルト(Palo Alto)、カリホルニア州))である。
【0050】
ヒトメタボトロピックグルタミン酸受容体サブタイプをコードする全長DNAは、ベクターpMMTVT7(+)、pMMTVT7(−)、pCMV−T7−2またはpCMV−T7−3に挿入されている。pCMV−T7−2(およびpCMV−T7−3)は、CMVプロモーター/エンハンサー、プロモーターのすぐ下流に存在するSV40スプライス/ドナー部位、スプライス部位の下流に位置するT7バクテリオファージRNAポリメラーゼプロモーター、その後にSV40ポリアデニル化シグナル、そしてT7プロモーターとポリアデニル化シグナルの間にポリリンカーを有する、pUC19をベースにした哺乳動物細胞発現ベクターである。CMVプロモーターとSV40ポリアデニル化シグナルの間にメタボトロピックグルタミン酸受容体サブタイプを配置させると、この作成体によりトランスフェクションされた哺乳動物の宿主細胞中で外来DNAの構成的発現(constitutive expression)が与えられる。
【0051】
ベクターpMMTVT7(+)とpMMTVT7(−)はベクターpMAMneo(クロンテック(Clontech)、パロアルト(Palo Alto)、カリホルニア州)を修飾することにより調製された。pMAMneoは、デキサメタゾン誘導性マウス乳癌ウイルス(MMTV)−LTRプロモーターに結合した(この後にSV40スプライス部位とポリアデニル化部位が続く)ラウス肉腫ウイルス(RSV)の長い末端反復配列(LTR)を含有する、哺乳動物の発現ベクターである。pMAMneoはまた、大腸菌の増殖のためのβ−ラクタマーゼ遺伝子(アンピシリン耐性を与える蛋白をコードする)と同様に、形質転換体の選択のための大腸菌neo遺伝子を含有する。
【0052】
ベクターpMMTVT7(+)は、pMAMneoからneo遺伝子を除去し、pBluescript(ストラタジーン(Stratagene)、ラホイア(La Jolla)、カリホルニア州)からのマルチプルクローニング部位とT7およびT3プロモーターを挿入することにより作成される。従ってpMMTVT7(+)はMMTV−LTRプロモーターに結合したRSV−LTRエンハンサー、MMTV−LTRプロモーターの下流に位置するT7バクテリオファージRNAポリメラーゼプロモーター、T7プロモーターの下流に位置するポリリンカー、T7プロモーターの下流に位置するT3バクテリオファージRNAポリメラーゼプロモーター、そしてT3プロモーターの下流に位置するSV40スプライシング部位とポリアデニル化部位を含有する。pMAMneoからのβ−ラクタマーゼ遺伝子(アンピシリン耐性を与える蛋白をコードする)はpMMTVT7(+)中で保持されるが、pMAMneo中の配向に対して配向は逆転している。
【0053】
ベクターpMMTVT7(−)はpMMTVT7(+)と同じであるが、T7プロモーターとT3プロモーターの位置が逆転しており、pMMTVT7(−)中のT3プロモーターはpMMTVT7(+)中のT7プロモーターがある場所に位置しており、pMMTVT7(−)中のT7プロモーターはpMMTVT7(+)中のT3プロモーターがある場所に位置している。従ってpMMTVT7(+)とpMMTVT7(−)ベクターは、哺乳動物宿主細胞中で異種DNA(ここで異種DNAはポリリンカーでベクター内に取り込まれている)が発現するのに必要な制御成分のすべてを含有している。さらにT7プロモーターとT3プロモーターはポリリンカーのどちらかの側に位置しているため、これらのプラスミドはポリリンカーでベクター内にサブクローニングされている異種DNAのインビトロ転写体の合成に使用することができる。
【0054】
哺乳動物細胞中のヒトメタボトロピックグルタミン酸受容体サブタイプをコードするDNAの誘導性発現のために、DNAをpMMTVT7(+)またはpMMTVT7(−)のようなプラスミド中に挿入することができる。これらのプラスミドは、機能的に関連する外来DNAのステロイド誘導性発現のためのマウス乳癌ウイルス(MMTV)LTRプロモーターを含有する。宿主細胞が細胞内への糖質コルチコイド(すなわちMMTVLTRプロモーターの誘導物質)の摂取に必要な内因性糖質コルチコイド受容体を発現しない場合は、糖質コルチコイド受容体をコードするDNA(ATCC整理番号67200)で細胞を追加的にトランスフェクションする必要がある。インビトロ転写体の合成のために、ヒトmGluR1、mGluR3、およびmGluR5をコードする全長ヒトDNAクローンを、pIBI24(インターナショナル・バイオテクノロジーズ社(International Biotechnologies, Inc.)、ニューヘーブン、コネチカット州)、pCMV−T7−2、pCMV−T7−3(図1参照)、pMMTVT7(+)、pMMTVT7(−)、pBluescript(ストラタジーン(Stratagene)、ラホイア(La Jolla)、カリホルニア州)またはpGEM7Z(プロメガ(Promega)、マジソン、ウィスコンシン州)などの中にサブクローニングすることもできる。
【0055】
本発明の別の実施態様において、前述のポリ核酸(すなわちDNAまたはmRNA)を含有する細胞が提供される。細菌、酵母および哺乳動物のような宿主細胞は、DNAを複製しメタボトロピックグルタミン酸受容体サブタイプを産生するために使用することができる。本発明に記載の発現ベクターの作成、インビトロ転写体の調製、哺乳動物細胞へのDNAのトランスフェクション、卵母細胞の注入の方法、および受容体の発現と機能の評価のための電気生理的および他の分析方法は、PCT出願PCT/US91/05625とPCT/US92/11090、および同時係属出願米国特許出願第07/563,751号と07/812,254号にも記載されている。これらの文献の主題はその全体が参考のため本明細書中に引用されている。
【0056】
適当な発現ベクターへのクローン化DNAの導入法、1つのプラスミドベクターまたはプラスミドベクターの組合せ(それぞれが1つまたはそれ以上の異なる遺伝子をコードする)または線状DNAによる真核細胞のトランスフェクション、そしてトランスフェクションした細胞の選択法は公知である(例えば、サムブルーク(Sambrook)ら、(1989)分子クローニング:実験室マニュアル(Molecular Cloning:A Laboratory Manual)、第2版、コールド・スプリング・ハーバー・ラボラトリー・プレス(Cold Spring Harbor Laboratory Press)を参照)。異種DNAは、当業者に公知の任意の方法により宿主細胞に導入される:例えば、CaPO4沈殿法(例えば、ウィグラー(Wigler)ら、(1979)、Proc. Natl. Acad. Sci.76:1373−1376)を用いる、異種DNAをコードするベクターによるトランスフェクション。次に組換え細胞は、DNAによりコードされたサブタイプが発現される条件下で培養することができる。好適な細胞は哺乳動物細胞(例えば、HEK293、CHO、およびLtk−細胞)、酵母細胞(例えば、ピキア・パストリス(Pichia Pastoris)のようなメチル親和性(methilotrophic)酵母細胞)、細菌細胞(例えば大腸菌)などがある。
【0057】
本明細書で提供されるDNAは任意の真核細胞(例えば、ピキア・パストリス(Pichia Pastoris)(米国特許第4,882,279号、4,837,148号、4,929,555号および4,855,231号を参照)、サッカロミセス・セレビッシェ(Saccharomyces cerevisiae)、カンジダ・トロピカリス(Candida tropicalis)、ハンゼヌラ・ポリモルファ(Hansenula polymorpha)などの酵母細胞)中で発現されるが、本明細書で提供されるヒトメタボトロピックグルタミン酸受容体サブタイプをコードするDNAの発現のためには、G蛋白を発現する(内因性にまたは組換えで)市販の発現系や当業者に公知の他の発現系を含む哺乳動物発現系が好ましい。PI加水分解/Ca++シグナル経路に結合したヒトメタボトロピック受容体サブタイプをコードするDNAのインビトロmRNA転写体の発現にはツノガエル(Xenopus)卵母細胞が好適である。卵母細胞中の内因性イノシトール3’末端リン酸第二メッセンジャー介在経路は、これらの細胞におけるヒトメタボトロピック受容体の機能性発現を可能にする。組換えヒトメタボトロピック受容体を発現する卵母細胞は、卵母細胞G蛋白結合IP3生成経路を介してアゴニストに応答し、内部の貯蔵状態からCa++を放出し、電圧固定記録法により遅延振動電流として検出される塩素チャネルを活性化すると報告されている。
【0058】
ヒトメタボトロピック受容体の機能性組換え発現のための宿主細胞は、好ましくは内因性または組換えグアニンヌクレオチド結合蛋白(すなわちG蛋白)を発現する。G蛋白はα、βおよびγサブユニットよりなる高度に保存された膜関連蛋白のファミリーの1つである。αサブユニットはGDPとGTPに結合し、異なるG蛋白毎に異なる。結合した対のβおよびγサブユニットはユニークであることもそうでないこともあり、異なるα鎖が同じβγ対または異なる対に結合している[リンダーとギルマン(Linder and Gilman)、Sci. Am.267:56−65(1992)]。G蛋白α鎖をコードする30以上の異なるcDNAがクローン化されている[サイモン(Simon)ら、Science 252:802(1991)]。4つの異なるβポリペプチドが知られている[サイモン(Simon)ら、Science 252:802(1991)]。同定された5つのγcDNAのうち3つがクローン化されている[ハーレー(Hurley)ら、PNAS U.S.A. 81:6948(1984):ガウタム(Gautam)ら、Science 244:971(1989):ガウタム(Gautam)ら、PNAS U.S.A. 87:7973(1990):]。4番目のγcDNA[クレウス(Kleus)ら、Science 259:832(1993)]と5番目のγcDNA[フィッシャーとアロンソン(Fischer and Aronson)、Mol. Cell. Bio. 12:1585(1992)]の配列は確立されており、さらに追加のγサブユニットが存在するかも知れない[タミール(Tamir)ら、Biochemistry 30:3929(1991)]。グアニンヌクレオチド交換とGTP加水分解により、G蛋白は活性状態と不活性状態の間で変化する。不活性G蛋白は、リガンド活性化受容体により刺激されてGDPをGTPに交換する。活性型では、GTPに結合したαサブユニットはβγ複合体から解離し、次にサブユニットは細胞性エフェクター分子と特異的に相互作用して細胞性応答を引き出す。異なるG蛋白は異なるエフェクター系(例えば、ホスホリパーゼC、アデニルサイクラーゼ系)や異なる受容体と相互作用することができるため、異なる組換えヒトメタ受容体サブタイプの発現のための異なる宿主細胞を研究することは有用である。あるいは宿主細胞は、異なるG蛋白の異種発現のためにG蛋白サブユニットをコードするDNAでトランスフェクションすることができる。
【0059】
好適な実施態様において、ヒトメタボトロピックグルタミン酸受容体サブタイプをコードするDNAは、ベクターに結合され、適当な宿主細胞中に導入されて、特異的なヒトメタボトロピックルグルタミン酸受容体サブタイプまたはサブタイプの特異的な組合せを発現する、形質転換された細胞株を産生する。得られる細胞株は次に、公知の薬剤または可能性のある薬剤の受容体機能への影響の再現性のある定量的解析のために大量に産生される。他の実施態様においては、各サブタイプをコードするDNAのインビトロ転写によりmRNAが産生される。このmRNA(単一のサブタイプクローンからであってもまたはクローンの組合せからであっても)は、ツノガエル(Xenopus)卵母細胞に注入され、ここでmRNAはヒトメタボトロピックグルタミン酸受容体サブタイプの合成を指令する。あるいは機能性ヒトメタボトロピックグルタミン酸受容体サブタイプの発現のために、サブタイプをコードするDNAは卵母細胞に直接注入される。次にトランスフェクションされた哺乳動物細胞または注入された卵母細胞は、本明細書に記載の薬剤スクリーニング法に使用される。
【0060】
DNAまたはRNAが導入される真核細胞は、このようなDNAまたはRNAによりトランスフェクションされることができる細胞、またはこのようなDNAまたはRNAを注入されてG蛋白を(内因性にまたは組換えにより)発現することができる細胞である。好適な細胞は、内因性メタボトロピックグルタミン酸受容体を(あったとしても)ほんの少ししか発現せず、かつ一時的または安定にトランスフェクションされ、また本発明のDNAやRNAを発現するものである。最も好適な細胞は、異種DNAによりコードされる1つまたはそれ以上のサブタイプよりなる組換えまたは異種ヒトメタボトロピックグルタミン酸受容体を形成することができるものである。このような細胞は経験的に同定されるか、または容易にトランスフェクションまたは注入されることが知られているものの中から選択される。
【0061】
DNAを導入するための細胞の例は、哺乳動物起源の細胞(例えば、COS細胞、マウスL細胞、チャイニーズハムスター卵巣(CHO)細胞、ヒト胎児性腎(HEK)細胞、アフリカミドリザル(African green monkey)細胞および当業者に公知の他の細胞)、両生類の細胞(例えば、ゼノプス・ラエビス(Xenopus laevis)卵母細胞)、酵母細胞(例えば、サッカロミセス・セレビッシェ(Saccharomyces cerevisiae)、ピキア・パストリス(Pichia Pastoris))などがある。注入されたRNA転写体を発現するための細胞の例は、ゼノプス・ラエビス(Xenopus laevis)卵母細胞がある。DNAのトランスフェクションに好適な細胞は、当業者に公知であるかまたは経験的に同定され、HEK293細胞(整理番号#CRL1573でATCCより入手できる);Ltk−細胞(整理番号#CCL1.3でATCCより入手できる);COS−7細胞(整理番号#CRL1651でATCCより入手できる);CHO細胞(整理番号#CRL9618、CCL61またはCRL9096でATCCより入手できる);DG44細胞(dhfr−CHO細胞;例えば、ウーラウプ(Urlaub)ら(1986)Cell. Molec. Genet. 12:555を参照);そしてBHK細胞(ウェクターとバセルガ(Waechter and Baserga)、PNAS U.S.A. 79:1106−1110(1982);整理番号#CRL10314でATCCより入手できる)を参照)がある。好適な細胞には、CHO細胞とHEK293細胞があり、特に液体窒素中で凍結され次に融解されて再増殖されるHEK293細胞(例えば、ゴーマン(Gorman)の米国特許第5,024,939号(またスティルマン(Stillman)ら、(1985)Mol. Cell. Biol. 5:2051−2060も参照)に記載のもの)、DG44細胞、LTK−細胞などがある。使用される宿主細胞にかかわらず、使用されるリガンドに誘導されるグルタミン酸産生のバックグランドレベルの測定、またはリガンドの非存在下での宿主細胞中に存在するグルタミン酸のバックグランドレベルの測定のために、比較実験を実施すべきであることは、当業者は理解できるであろう。
【0062】
DNAは当業者に公知の方法を用いて細胞内に安定に取り込まれるか、または一時的に発現される。安定にトランスフェクションされる哺乳動物細胞は、選択マーカー遺伝子(例えば、チミジンキナーゼ、ジヒドロ葉酸還元酵素、ネオマイシン耐性などの遺伝子)を有する発現ベクターで細胞をトランスフェクションし、トランスフェクションした細胞をマーカー遺伝子を発現する細胞に選択的な条件下で増殖させることにより調製される。一時的トランスフェクション体を調製するためには、哺乳動物細胞をレポーター遺伝子(例えば大腸菌β−ガラクトシダーゼ遺伝子)でトランスフェクションしてトランスフェクション効率を追跡する。
【0063】
トランスフェクション体は典型的には選択性条件下では増殖せず、普通トランスフェクション後数日以内に分析されるため、選択マーカー遺伝子は一時的トランスフェクション体には含有されない。
【0064】
このような安定トランスフェクション細胞または一時的トランスフェクション細胞を産生するために、細胞を充分な濃度のサブタイプをコードする核酸でトランスフェクションして、異種DNAにコードされるヒトサブタイプを示すヒトメタボトロピックグルタミン酸受容体を形成させる。サブタイプをコードするDNAの正確な量は経験的に決定されるか、または特定のサブタイプ、細胞および測定条件について最適化される。異種DNAまたはRNAのみにコードされるサブタイプを含有するメタボトロピックグルタミン酸受容体を発現する組換え細胞が特に好適である。
【0065】
異種DNAは細胞中でエピソーム成分として維持されるか、または細胞の染色体DNAに取り込まれる。次に得られる組換え細胞を培養し、この培養物またはサブ培養物からサブ培養(または、哺乳動物細胞の場合は継代する)する。組換え細胞のトランスフェクション、注入および培養法は当業者に公知である。同様に当業者に公知の蛋白精製法を用いて、ヒトメタボトロピックグルタミン酸受容体サブタイプが精製される。例えば1つまたはそれ以上のサブタイプに特異的に結合する抗体または他のリガンドが、ヒトメタボトロピックグルタミン酸受容体の親和性精製に使用される。
【0066】
本明細書において、異種DNAまたは外来DNAおよびRNAは、互いに交換して使用することができ、細胞のゲノムの一部としては天然には存在しないか、または天然に存在するものとは異なるゲノム中の位置に見いだされるDNAまたはRNAを意味する。典型的には異種または外来DNAおよびRNAは、宿主細胞に内因性ではなく人工的に細胞に取り込まれたDNAまたはRNAを意味する。異種DNAの例としては、ヒトメタボトロピックグルタミン酸受容体サブタイプをコードするDNA、転写、翻訳または他の制御可能な生化学過程などに影響を与えることにより内因性DNAの発現を媒介または変更するRNAまたは蛋白をコードするDNAなどがある。異種DNAを発現する細胞は、同じであるかまたは異なる発現生成物をコードするDNAを含有する。異種DNAは発現される必要はなく、宿主細胞ゲノム中に取り込まれるかまたはエピソーム的に維持される。
【0067】
機能性mGluRの発現を検出するために使用する種々の測定法を当業者は容易に見いだすことができる。その例としては、PIターンオーバー測定法[例えば、ナカジマ(Nakajima)ら、J. Biol. Chem.267:2437−2442(1992)および実施例3.C.2を参照]、cAMP測定法[例えば、ナカジマ(Nakajima)ら、前述、および実施例3.C.4を参照]、カルシウムイオン流入測定法[例えば、イトウ(Ito)ら、J. Neurochem. 56:531−540(1991)、および実施例3.C.1を参照]、cGMP測定法[例えば、スタイナー(Steiner)ら、J. Biol. Chem.247:1106−1113(1972)]、アラキドン酸放出測定法[例えば、フェルダー(Felder)ら、J. Biol.Chem.264:20356−20362(1989)を参照]などがある。さらに、陽イオンをベースにした測定法(本明細書に記載)を、細胞内サイクリックヌクレオチドレベルの受容体誘導変化を追跡するために使用することができる。そのような測定法は、サイクリックヌクレオチドゲートのイオンチャネルを発現する宿主細胞を使用する。例えば棹状光受容体細胞、嗅覚細胞およびウシ腎細胞中に存在するこれらのチャネル(例えば、カウプ(Kaupp)ら、Nature 342:762−766(1989)、ダレン(Dallen)ら、Nature 347:184−187(1990)、およびビエル(Biel)ら、Proc. Natl. Acad. Sci. USA 91:3505−3509(1994))は、cAMPまたはcGMPの結合による活性化により陽イオンに対して透過性となる。すなわち本発明の測定法において、内因性または組換えサイクリックヌクレオチドゲートのチャネルを発現する宿主細胞は、サイクリックヌクレオチドレベルに影響を与えることが疑われる受容体をコードする核酸(例えば、メタボトロピックグルタミン酸受容体をコードするDNA)でトランスフェクション(または注入)され、次にチャネルのサイクリックヌクレオチド活性化の量の変化を追跡する。チャネルのサイクリックヌクレオチドの活性化の変化を追跡すると、活性化された時cAMPまたはcGMPの変化を引き起こす受容体を機能性であるとして間接的に同定することができる。サイクリックヌクレオチドゲートのチャネルの活性化の量の変化は、チャネルを介するイオンの流入を測定するか、または電流の電気生理学的測定または細胞内陽イオンレベルの変化を測定(例えば、細胞内カルシウムの蛍光測定)することにより、決定することができる。
【0068】
活性化によりサイクリックヌクレオチドの低下を引き起こす受容体(例えば、メタボトロピックグルタミン酸受容体)を発現する細胞の測定法において、受容体活性化化合物を測定物内の細胞に添加する前に、サイクリックヌクレオチドの細胞内レベルを上昇させる物質(例えば、フォルスコリンやIBMX)に細胞を暴露させることが好ましい。
【0069】
上記の測定法での使用に適した宿主細胞は、検討される受容体の発現に適した宿主細胞を含む(例えば、メタボトロピックグルタミン酸受容体の測定のためのL細胞、HEK293細胞、CHO細胞またはツノガエル(Xenopus)卵母細胞)。細胞は、サイクリックヌクレオチドゲートのチャネルをコードする核酸と受容体をコードする核酸で連続的にトランスフェクション(または注入)されるか、または細胞は2つの核酸で同時トランスフェクションされる。実施例3Aと3Bに記載の一時的または安定なトランスフェクションを実施することができる。
【0070】
サイクリックヌクレオチドゲートのチャネルでトランスフェクション(または注入)された細胞は、機能の測定の前にインキュベートされる(典型的には約24−48時間)。チャネルの活性は、トランスフェクションされた細胞からの引き剥がした裏返した膜パッチを用いて測定される(こうして細胞質面に達するcAMPの濃度が調節される)。トランスフェクション体はまた、内部カルシウムレベル([Ca++]i)の単細胞ビデオイメージングにより分析される。この方法は、細胞内カルシウムレベル(これはチャネルのサイクリックヌクレオチド活性化により制御される、チャネルを介するカルシウムの流入量とともに変化する)の測定によるサイクリックヌクレオチドゲートのチャネルの分析を可能にする。イメジーング測定法は、基本的に実施例3.C.4.bに記載のように実施される。
【0071】
本明細書に記載のDNA、mRNA、ベクター、受容体サブタイプおよび細胞により、選択されたメタボトロピックグルタミン酸受容体サブタイプやその受容体サブタイプに対する抗体の産生が可能である。これは、その存在が単一のメタボトロピックグルタミン酸受容体サブタイプの分析を妨害する多くの他の受容体蛋白の汚染が実質的にない、合成または組換え受容体および受容体サブタイプを調製する手段を与える。目的の受容体サブタイプが利用できることが、特定の受容体サブタイプまたはメタボトロピックグルタミン酸受容体サブタイプの組合せへの薬剤の影響を観察し、従ってヒトに特異的でありヒトメタボトロピックグルタミン酸受容体サブタイプまたはメタボトロピックグルタミン酸受容体サブタイプの組合せに特異的な試験系で、薬剤の初期のインビトロスクリーニングを行うことを可能にする。特異的抗体が利用できることは、インビボで発現されるサブタイプの組合せを同定することを可能にする。すなわち、このような特異的組合せは、薬剤スクリーニングの好適な標的として使用される。
【0072】
特異的な受容体組成物への薬剤の影響を測定するためにインビトロで薬剤物質をスクリーニングすることができることは、受容体サブタイプ特異的または疾患特異的薬剤の開発とスクリーニングを可能にするはずである。また単一の受容体サブタイプまたは種々の受容体サブタイプと種々のアゴニストまたはアンタゴニストの可能性のある物質との特定の組合せを試験することにより、個々のサブタイプの機能と活性に関する追加情報が得られ、1つまたはそれ以上の受容体サブタイプと非常に特異的に相互作用することができる化合物の同定と設計を可能にするであろう。得られる薬剤は、種々の受容体サブタイプを発現する細胞を用いてスクリーニングすることにより同定される薬剤より、不要な副作用はより少ないであろう。
【0073】
さらに種々の疾患の薬剤開発と治療法に関して、ヒトメタボトロピックグルタミン酸受容体サブタイプをコードするDNAが利用できることは、ある種の疾患の発症に相関する遺伝子の変化(例えば、突然変異)の同定を可能にする。さらに、このような突然変異を合成DNA配列に特異的に導入し、次にこれを実験室動物に導入するかまたはその効果を測定するためのインビトロ測定系により、そのような疾患の動物モデルの作成が可能になる。
【0074】
別の面において、本発明は本発明のDNAによりコードされる機能性ペプチド断片およびその機能性組合せよりなる。ペプチドがグルタミン酸受容体として機能するのに必須ではない配列中のアミノ酸の一部またはすべてを排除することにより、不要な実験をすることなく当業者はこのような機能性ペプチド断片を産生することができる。グルタミン酸受容体機能に必須のアミノ酸は、例えばペプチドをコードするDNAの体系的消化および/またはDNAへの欠失の導入により決定することができる。修飾(例えば欠失または消化)DNAは、例えばDNAを転写し、次に得られるmRNAをツノガエル(Xenopus)卵母細胞に導入して、mRNAを翻訳させることにより発現することができる。卵母細胞中のこうして発現された蛋白の機能的分析は、グルタミン酸受容体に結合しこれを機能的に活性化することが公知のリガンドに卵母細胞を暴露して、次に内因性チャネルが活性化されるか否かについて卵母細胞を追跡することにより、行われる。電流が検出されるなら、断片はグルタミン酸受容体として機能する。
【0075】
本発明の別の実施態様において、競合的結合測定法で本発明の受容体蛋白を使用することよりなる、メタボトロピックグルタミン酸受容体サブタイプに結合する化合物の同定方法が提供される。この測定法は多数の化合物を迅速にスクリーニングして、どの化合物(もし、あるとすれば)が、特異的に結合した[3H]グルタミン酸を排除することができる(すなわち、メタボトロピックグルタミン酸受容体に結合することができる)かを調べることができる。結合することが見いだされた化合物を用いて、このような化合物が本発明の受容体のモジュレーター、アゴニストまたはアンタゴニストとして作用するのか否かをさらに調べるために、さらに詳しい測定法が実施される。
【0076】
本発明の結合測定法の別の応用は、本発明の受容体の存在の有無に関する試料(例えば生物学的液体)の測定である。例えばグルタミン酸産生経路の機能不全に関連する症状を示す患者の血清を測定して、観察される症状がこのような受容体の型の産生過剰または不足により引き起こされるのかを決めることができる。
【0077】
本発明において企図される結合測定法は、当業者は容易に判定できるように種種の方法で実施することができる。例えばラジオリセプター測定法などの競合的結合測定法を使用することができる。
【0078】
本発明のさらなる実施態様において、本発明のヒトメタボトロピックグルタミン酸受容体サブタイプの活性を調節する化合物を同定するための生物測定法が提供され、該方法は:
(a)ヒトメタボトロピックグルタミン酸受容体サブタイプをコードするDNAを含有する細胞(この細胞は機能性メタボトロピックグルタミン酸受容体を発現する)を、受容体の活性の調節能力を測定すべき少なくとも1つの化合物に暴露させて、
(b)第二メッセンジャー活性の変化について細胞を追跡する
ことよりなる。
【0079】
上記の生物測定法はヒトメタボトロピックグルタミン酸受容体のアゴニストやアンタゴニストおよびアロステリックモジュレーターの同定を可能にする。この方法では組換えメタボトロピックグルタミン酸受容体に「未知の」すなわち試験物質と(アンタゴニスト活性が試験される時は、さらに公知のメタボトロピックグルタミン酸アゴニストの存在下で)接触させ、この「未知の」すなわち試験物質との接触後、公知のグルタミン酸受容体の第二メッセンジャー活性を追跡し、公知のグルタミン酸受容体の活性を追跡し、公知のグルタミン酸受容体の第二メッセンジャー応答を増加または減少させる物質は、ヒトメタボトロピックグルタミン酸受容体の機能性リガンド(すなわち、モジュレーター、アゴニストまたはアンタゴニスト)として同定される。追跡することができる第二メッセンジャー活性には、細胞内カルシウムイオンの濃度、IP3、cAMPレベルの変化、またはアラキドン酸放出またはイオン電流の活性または阻害の追跡(細胞が卵母細胞の場合)がある。
【0080】
本発明の特定の実施態様において、組換えヒトメタボトロピックグルタミン酸受容体を発現する哺乳動物細胞または卵母細胞を試験化合物と接触させて、次に試験化合物がある場合またはない場合のメタボトロピックグルタミン酸受容体介在応答を比較し、化合物の存在に対する試験細胞または対照細胞(すなわち、メタボトロピックグルタミン酸受容体を発現しない細胞)のメタボトロピックグルタミン酸受容体介在応答を比較して、その調節作用を評価することができる。
【0081】
本明細書において、「メタボトロピックグルタミン酸受容体の活性を調節する」化合物またはシグナルとは、メタボトロピックグルタミン酸受容体の活性を変化させ、化合物またはシグナルの存在下ではこれらが存在しない場合とはメタボトロピックグルタミン酸受容体の活性が異なるようになる化合物またはシグナルを意味する。特にこのような化合物またはシグナルはアゴニストやアンタゴニストを含む。アゴニストという用語は、受容体機能を活性化するグルタミン酸またはACPDのような物質またはシグナルを意味し;アンタゴニストという用語は、アゴニストに誘導される受容体活性化を妨害する物質を意味する。アンタゴニストには競合的および非競合的アンタゴニストがある。競合的アンタゴニスト(または競合的ブロッカー)は、アゴニスト(例えば、リガンドまたは神経伝達物質)の近傍または特異的な部位と、その同じ部位または近傍に対して相互作用する。非競合的アンタゴニストまたはブロッカーは、アゴニストと相互作用する部位とは異なる部位と相互作用することにより、受容体の機能を不活性化する。
【0082】
当業者は理解できるように、ヒトメタボトロピックグルタミン酸受容体活性を調節する化合物(例えば、アゴニストおよびアンタゴニスト)の同定方法には、一般に対照との比較が必要である。「対照」細胞または「対照」培養物の1つのタイプは、試験化合物に暴露された細胞または培養物と実質的に同様に処理された細胞または培養物である(ただし、対照培養物は試験化合物に暴露されない)。例えば、電圧固定電気生理学的方法を用いる方法では、細胞を浸漬している外部溶液を単に交換することにより、試験化合物の存在下または非存在下で同じ細胞を試験することができる。別のタイプの「対照」細胞または「対照」培養物は、トランスフェクションされた細胞と同一の細胞または細胞の培養物である(ただし、対照培養物に使用される細胞は、トランスフェクションされた細胞に発現される組換えヒトメタボトロピックグルタミン酸受容体サブタイプを発現しない)。この場合、細胞または各タイプの細胞の培養物が、測定される化合物の存在下と実質的に同じ反応条件下に暴露される時、試験化合物に対する試験細胞の応答は、受容体のない(対照)細胞の試験化合物に対する応答(または応答の欠如)と比較される。
【0083】
本発明のさらに別の実施態様において、ヒトメタボトロピックグルタミン酸受容体の第二メッセンジャー活性は、この受容体を、上記生物測定法により同定される少なくとも1つの化合物の有効量と接触させることにより調節される。
【0084】
本発明のさらに別の実施態様において、上記の受容体蛋白に対して産生された抗体が提供される。この抗体は、受容体組織局在化、サブタイプ組成、機能性ドメインの構造の研究用、受容体の精製、および診断応用治療応用などに使用することができる。好ましくは治療用途では、使用される抗体はモノクローナル抗体である。
【0085】
本発明の受容体蛋白またはその部分を抗体産生の抗原として用いることにより、上記の抗体は当業者に公知の標準的方法を使用して産生することができる。抗ペプチドおよび抗融合蛋白抗体ともに使用することができる[例えば、バホウト(Bahouth)ら、(1991)Trends Parmacol Sci. 第12巻:338−343;Current Protocols in Molecular Biology(アウスベル(Ausubel)ら編、ジョン・ワイリー・アンド・サンズ(John Wiley and Sons)、ニューヨーク(1989)]。免疫原として(合成ペプチドまたは組換え産生細菌融合蛋白として)使用するためのメタボトロピックグルタミン酸受容体サブタイプの部分を選択する場合に考慮すべき因子は、抗原性、入手し易さ(すなわち、細胞外ドメインおよび細胞質ドメイン)、特定のサブタイプに特異的であるかなどである。
【0086】
サブタイプ特異的抗体が利用できることは、(例えば、正常の脳組織対疾患の脳組織中の)種々のサブタイプの分布と発現密度を追跡するのに免疫組織学的方法を使用することを可能にする。このような抗体はまた、診断および治療応用に利用できる。
【0087】
本発明のさらに別の実施態様において、有効量の上記の抗体に本発明の受容体を接触させることにより、該受容体のイオンチャネル活性を調節する方法が提供される。
【0088】
本発明の抗体は、標準的方法(例えば、腹腔内投与、筋肉内投与、静脈内投与、または皮下注射、移植または経皮的投与法など)により、対象者に投与することができる。当業者は、使用される投与法により、投与型、治療処方などを容易に決定することができる。
【0089】
本発明のさらなる実施態様において、細胞内サイクリックヌクレオチドレベルの受容体に誘導される変化を追跡するための、陽イオンベースの生物測定法が提供され、該生物測定法は:
内因性または組換えサイクリックヌクレオチドゲートのチャネルを発現する宿主細胞に、細胞内サイクリックヌクレオチドレベルに影響を与えることが疑われる受容体をコードする核酸を導入し、そして
細胞内サイクリックヌクレオチドレベルに影響を与えることが疑われる、該受容体に対するリガンドの存在下または非存在下で、サイクリックヌクレオチドゲートのチャネルのサイクリックヌクレオチド活性化の量の変化を追跡することよりなる。
【0090】
以下の非限定的実施例を参照して本発明をさらに詳細に説明する。
【実施例1】
【0091】
ヒトメタボトロピックグルタミン酸受容体をコードするDNAの単離
A.mGluR5受容体cDNA
cDNAライブラリースクリーニング
【0092】
ヒト海馬組織から単離したRNAを、標準的方法[例えば、グブラーとホフマン(Gubler and Hoffman)、(1983)Gene 25:263−269]に従い、オリゴdT−プライムした1本鎖cDNAの合成のための鋳型として使用した。1本鎖cDNAを2本鎖cDNAに変換し、そのcDNAの末端にEcoRI/SnaBI/XhoIアダプターを付け加えた。アガロースゲル電気泳動を用いてサイズによりcDNAを分離し、>2.5kbのものをEcoRI消化したλgt10バクテリオファージベクターに結合させた。得られる一次ヒト海馬cDNAライブラリー(約2×105組換え体)を、ラットmGluR1受容体(ヌクレオチド1から1723+5’非翻訳配列;マス(Masu)ら、(1991)Nature 349:760−765を参照)をコードするDNAの断片に対するハイブリダイゼーションについてスクリーニングした。ハイブリダイゼーションは、42℃で5×SSPE、5×デンハルツ(Denhart's)溶液、50%ホルムアミド、0.2%SDSおよび200μg/mlの変性した超音波処理したニシン精子DNA中で行ない、洗浄は65℃で1×SSPEと0.2%SDS中で行なった。3273塩基対を含有するハイブリダイズする1つのプラーク(METAB1)を同定した。
【0093】
追加のヒトmGluR5コーディングクローンを得るために、METAB1を放射標識し、以下のように調製した2つのヒト小脳cDNAライブラリーをスクリーニングするのに使用した。ランダムプライマーを使用して、ヒト小脳組織から単離したRNAから最初のcDNA合成をプライムしてcDNAを合成した。長さを基準にしてcDNAをプールし、2つのライブラリーを作成した:1つは2.8kb以上の長さの挿入体を有し(すなわち、大挿入体ライブラリー)、もう1つは1−2.8kbの長さの挿入体を有する(すなわち、中挿入体ライブラリー)。ラットmGluR1DNA断片へのハイブリダイゼーションに対して海馬ライブラリーをスクリーニングするために使用したのと同じハイブリダイゼーション条件を用いて、METAB1に対するハイブリダイゼーションについてライブラリーをスクリーニングした。洗浄は55℃で1×SSPE、0.2%SDS中で行なった。大挿入体ライブラリー中に1つのハイブリダイズするプラーク(METAB2)を同定し、中挿入体ライブラリー中に4つのハイブリダイズするプラーク(METAB3−METAB6)を同定した。
【0094】
ヒトmGluR5をコードするDNAの別のスクリーニングで、METAB1により大挿入体または中挿入体小脳ライブラリーをスクリーニングするのに使用した条件と同じ条件を用いて、サイズが1−2kbの範囲の挿入体を含有するランダムにプライムしたヒト海馬cDNAライブラリー(2×106組換え体)および中挿入体小脳cDNAライブラリーを、放射能標識したMETAB5へのハイブリダイゼーションについてスクリーニングした。海馬ライブラリーに3つのハイブリダイズするプラーク(METAB10−METAB12)が同定され、小脳ライブラリーの別の一次スクリーニングで5つの追加のハイブリダイズするプラーク(METAB13−METAB17)が同定された。選択されたプラークを精製した。
【0095】
単離されたクローンの性状解析
制限酵素マッピングとDNA配列解析による精製したプラークの挿入体の性状解析から、単離されたクローンによりヒトmGluR5転写体の少なくとも3つの明らかなスプライス変種が表されていることが判明した。METAB1の分析により、これは翻訳開始コドンを含有するが翻訳停止コドンは含有しないことが示された。推測されるアミノ酸配列は、ラットのmGluR1の推測されるアミノ酸配列に約70%同一であり、ラットのmGluR5の推測されるアミノ酸配列とは>90%の同一性を有している[アベ(Abe)ら、(1992)J. Biol. Bhem. 267:13361−13368]。
【0096】
METAB5のDNA配列解析は、METAB5は5’末端でMETAB1の3’末端と重複し、3’方向へ追加の343ヌクレオチドが続いていることを示した。METAB1とMETAB5の重複領域を比較すると、METAB1はMETAB5に存在しない96ヌクレオチドを有していた(すなわち、METAB1はMETAB5に対して96ヌクレオチド挿入を含有する)。METAB5はまた、翻訳停止コドンを持っていない。METAB12挿入体は5’末端でMETAB5の3’末端と重複するが、さらに3’末端方向に延長して翻訳停止コドンを有する。
【0097】
METAB2のDNA配列解析により、5’末端の最初の869ヌクレオチドはMETAB1の3’末端の一部と重複し、同じであった。しかし、METAB1とMETAB2の配列はMETAB1の96ヌクレオチド挿入の最初で分岐していた。METAB2は3’末端方向に約2700ヌクレオチド延長し、METAB1との分岐点の4ヌクレオチドだけ3’方向に推定翻訳停止コドンを有する。
【0098】
METAB4の部分的DNA配列解析は、これが別のヒトメタボトロピック受容体であるmGluR1の一部を含有することを示していた(実施例1.B.を参照)。
【0099】
全長mGluR5 cDNA作成体の調製
cDNAのインビトロ転写体の調製および/または哺乳動物細胞でのcDNAの発現に使用するために、ヒトmGluR5転写体の3つの推定スプライス変種を表す全長作成体(mGluR5a、mGluR5bそしてmGluR5cと呼ぶ)を作成し、発現ベクター中に取り込むことができる。典型的に使用される基礎となる発現ベクターは、pCMV−T7−3またはpCMV−T7−2である(図1を参照)。プラスミドpCMV−T7−3はpUC19ベースのベクターであり、CMV(CMV)プロモーター/エンハンサー、プロモーターのすぐ下流に位置するSV40スプライスドナー/スプライスアクセプター部位、SV40スプライス部位のすぐ下流に位置するT7バクテリオファージRNAポリメラーゼプロモーター、T7プロモーターの下流のSV40ポリアデニル化シグナル、およびT7プロモーターとポリアデニル化シグナルの間のポリリンカーを含有する。すなわちこのベクターは、哺乳動物宿主細胞中での異種DNAの発現に必要なすべての制御成分を含有する(異種DNAはポリリンカーでベクターに取り込まれる)。さらにT7プロモーターはポリリンカーのすぐ上流に位置するため、このプラスミドは、ポリリンカーでベクターにサブクローニングされた異種DNAのインビトロ転写体の合成に使用される。pCMV−T7−3とpCMV−T7−2は、ポリリンカー中の制限酵素部位の配向のみが異なる。
【0100】
全長mGluR5a作成体(配列番号7を参照)を調製するために、クローンMETAB1、METAB5およびMETAB12の部分を一緒に結合させた。まず操作を容易にするために、METAB1、METAB5およびMETAB12の挿入体をEcoRI断片としてλgt10から別々に、EcoRI消化pGEM−7Zf(プロメガ(Promega)、マジソン、ウィスコンシン州)に移した。METAB1挿入体を含有するpGEM−7ZfベクターをScaI/NheIで消化して、アンピシリン耐性遺伝子の5’半分とMETAB1挿入体の5’部分(配列番号7のヌクレオチド1−2724)を含有する3.8kb断片を放出させた。METAB5の挿入体を含有するpGEM−7ZfベクターをScaI/NheIで消化して、アンピシリン耐性遺伝子の3’半分とMETAB5挿入体の3’部分(配列番号7のヌクレオチド2725−3469)を含有する2.6kb断片を放出させ、この断片を、METAB1を含有するpGEM−7Zfベクターからの3.8kb断片に結合させてpGEM−METAB1+5を作成した。pGEM−METAB1+5をScaI/NheIで消化して、アンピシリン耐性遺伝子の5’半分と配列番号7のヌクレオチド1−3316を含有する4.4kb断片を放出させた。次にこの4.4kb断片を、METAB12挿入体を含有するpGEM−7ZfベクターのScaI/NheI(部分)消化により得られた2.6kb断片[この2.6kb断片はアンピシリン耐性遺伝子の3’半分とMETAB12の3’部分(配列番号7のヌクレオチド3317−4085)を含有する]に結合させた。得られたベクターはpGEM−7Zf中に完全なmGluR5aコーディング配列を含有していた。全長mGluR5a cDNAを、Aat II(平滑末端)−Hind III断片としてベクターから単離し、NotI(平滑末端)/Hind III消化pCMV−T7−3にサブクローニングして、作成体mGluR5a1を作成した。
【0101】
要約すると、作成体mGluR5a1は、METAB1からの5’非翻訳配列の369塩基対(配列番号7のヌクレオチド1−369)と、mGluR5受容体のmGluR5a変種の完全なコーディング配列(配列番号7のヌクレオチド370−3912)、および3’非翻訳配列の173塩基対(配列番号7のヌクレオチド3913−4085)を含有する。mGluR5aをコードする配列は、哺乳動物宿主細胞中で受容体を発現するために、そしてツノガエル(Xenopus)卵母細胞で発現されるDNAのインビトロ転写体を作成するのに使用するために、pCMV−T7−3中の制御成分に機能的に結合している。
【0102】
2つの追加のmGluR5a作成体(mGluR5a2とmGluR5a3)を、最初のmGluR5a作成体の5’非翻訳領域を修飾して調製した。上記のmGluR5a作成体は、提唱されている翻訳開始コドン(配列番号7のヌクレオチド370から372)に先行する5’非翻訳領域中に7つのおそらく不適切なATGコドンを含有する。mGluR5a1作成体をBAl31で消化して、以下を行う:(1)配列の255ヌクレオチド(配列番号7のヌクレオチド1−255であり、上流のATGトリプレット7つのうち6つを含有する)を除去して、mGluR5a2を作成する、そして(2)配列の348ヌクレオチド(配列番号7のヌクレオチド1−348であり、上流のATGトリプレットのすべてを含有する)を除去して、mGluR5a3を作成する。すなわち、mGluR5a2は、5’非翻訳配列の一部が欠如していること以外はmGluR5a1と同じであり、従って提唱される翻訳開始コドンの上流に1つのみのATGトリプレットを含有する。同様に、mGluR5a3は、提唱される翻訳開始コドンの上流のすべてのATGトリプレットが欠如していること以外はmGluR5a1と同じであり、5’非翻訳配列の21ヌクレオチドのみを含有する。
【0103】
3つ目のmGluR5a作成体(MMTV−hmGluR5a)は、以下のようにmGluR5aのMMTVプロモーター制御発現に使用するために調製した。mGluR5a3をXbaIで消化した。SV40スプライス部位、全長mGluR5aコーディング配列(および5’非翻訳配列の21ヌクレオチドと3’非翻訳配列の173ヌクレオチド)、およびポリアデニル化シグナルを含有する4.1kb断片を単離し、平滑末端とし、pBR322の2kbのEcoRI−NdeI(平滑末端)断片に結合させて、pBR−hmGluR5を作成した。MMTVLTRプロモーターとアンピシリンおよびネオマイシン耐性遺伝子を含有するベクターpMAMneo(クロンテック(Clontech)、パロアルト(Palo Alto)、カリホルニア州)をBamHIで消化して、ネオマイシン耐性遺伝子を除去し、再結合させた。次にこのベクターをEcoRIで消化し、アンピシリン耐性遺伝子を含有する断片を逆配向で大きい方のベクター断片に再結合させてpMAMneo ampoppを作成した。このベクターをPstI/NheIで消化して、アンピシリン耐性遺伝子の5’部分とMMTV−LTRを含有する2.3kbの断片を単離した。プラスミドpBR−hmGluR5をPstI/XbaIで消化し、アンピシリン耐性遺伝子の3’部分とmGluR5a配列(SV40スプライス部位とポリアデニル化シグナルとともに)を含有する5.3kbの断片を、pMAMneo ampoppの2.3kbのPst/NheI断片に結合させてMMTV−hmGluR5aを作成した。
【0104】
すなわち、pMMTV−hmGluR5aは、MMTV−LTRを含有し、mGluR5a DNA(配列番号7のヌクレオチド349−4085を含有する)と機能的に結合したSV40スプライス部位が続き、この後にポリアデニル化シグナルが続く。
【0105】
4つ目のmGluR5a作成体(pSV−hmGluR5)は、以下のようにmGluR5aのSV40プロモーター制御発現に使用するために調製した。mGluR5a3をXhoIで部分的に消化し、クレノウ(Klenow)で処理し、自身と再結合させ、こうしてmGluR5a DNAの3’に位置するXhoI部位を破壊した。次にプラスミドをScaI/XhoIで消化し、SV40スプライス部位、全長mGluR5aコーディング配列(および5’非翻訳配列の21ヌクレオチドと3’非翻訳配列の173ヌクレオチド)、ポリアデニル化シグナルおよびアンピシリン耐性遺伝子の3’部分を含有する断片を作成した。プラスミドpSVβ(クロンテック(Clontech)、パロアルト(Palo Alto)、カリホルニア州)をScaI/XhoIで消化し、アンピシリン耐性遺伝子とSV40早期プロモーターを含有する断片を、mGluR5a DNAを含有するScaI/XhoI断片に結合させて、pSV−hmGluR5を作成した。すなわち、pSV−hmGluR5は、SV40早期プロモーターを含有し、mGluR5a DNA(配列番号7のヌクレオチド349−4085を含有する)と機能的に結合したSV40スプライス部位が続き、この後にポリアデニル化シグナルが続く。
【0106】
全長mGluR5b作成体を調製するために、mGluR5a作成体
(mGluR1a1、mGluR5a2またはmGluR5a3)をNheI/PmlIで消化し、配列番号7のヌクレオチド2725−3020を含有する断片を放出させる。次に残りのベクター断片を、METAB1からの単離したNheI/PmlI断片に結合させる。得られるベクター(mGluR5b)は、配列番号7のヌクレオチド2999と3000の間に位置する96塩基対の挿入体(配列番号9のヌクレオチド3000−3095)を含有する以外は、これが得られたmGluR5a作成体と同じである。配列番号9は、ベクターmGluR5a1から調製した全長mGluR5b cDNAの完全なヌクレオチド配列である。
【0107】
全長mGluR5c作成体を調製するために、mGluR5a作成体
(mGluR1a1、mGluR5a2またはmGluR5a3)をNheI/HindIII(Hind III部位は、mGluR5aベクターのpCMV−T7−3部分のポリリンカー内に存在する)で消化し、配列番号7のヌクレオチド2725−4085を含有する断片を放出させる。次に残りのベクター断片を、METAB2から単離したNheI/Hind III断片に結合させる。得られる全長cDNA(mGluR5c)(配列番号11)は、コーディング配列の最初の2630ヌクレオチドについては、これが調製されたmGluR5a作成体と同じであるが、コーディング配列のヌクレオチド2631で、mGluR5cとmGluR5aのコーディング配列は分岐(例えば、配列番号7のヌクレオチド3000で始まる)し、mGluR5cコーディング配列はコーディング配列のヌクレオチド2631としてグアニンヌクレオチドを有し、その後に翻訳停止コドン(配列番号11のヌクレオチド3001−3003)が続く。
【0108】
B.mGluR1受容体cDNA
cDNAライブラリースクリーニング
中挿入体小脳ライブラリーを、ラットのmGluR1受容体(ヌクレオチド1から3031および5’非翻訳配列:マス(Masu)ら、(1991)Nature 349:760−765)をコードするDNAの断片へのハイブリダイゼーションについてスクリーニングした。ハイブリダイゼーションは、42℃で5×SSPE、5×デンハルツ(Denhart's)溶液、50%ホルムアルミド、0.2%SDSおよび200μg/mlの変性した超音波処理したニシン精子DNA中で行ない、洗浄は55℃で1×SSPEと0.2%SDS中で行なった。3つのハイブリダイズするクローン(METAB7−METAB9)が同定された。
【0109】
次のスクリーニングで、ヒト中挿入体小脳cDNAライブラリーの1×106組換え体の各平板を、追加のヒトmGluR1クローンとプローブ結合させた。この平板をまず、ラットのmGluR1 cDNAのヌクレオチド1−1256(および5’非翻訳配列)を含有するDNA断片(すなわち、5’プローブ)とハイブリダイゼーションさせ、次にクローンMETAB7−METAB9を同定したこの前のスクリーニングで使用したものと同じハイブリダイゼーション条件と洗浄条件を用いて、ラットmGluR1a cDNAのヌクレオチド2075−3310を含有するDNA断片(すなわち、3’プローブ)に連続してハイブリダイズさせた。5’プローブへのハイブリダイゼーションにより3つのクローン(METAB18、METAB21およびMETAB22)が同定され、そして3’プローブへのハイブリダイゼーションにより4つのクローン(METAB14、METAB20、METAB32およびMETAB35)が同定された。
【0110】
このラットのmGluR1断片をプローブとして使用して、さらなるmGluR1クローンのために大挿入体ヒト小脳cDNAライブラリーをスクリーニングした。ハイブリダイゼーションと洗浄条件は、中挿入体小脳ライブラリーから6つのmGluR1クローンを単離した時に使用したものと同じである(例えば、ハイブリダイゼーション溶液では20%ホルムアルデヒドを使用した)。3つのプラーク(METAB58、METAB59およびMETAB60)がプローブにハイブリダイズした。
【0111】
単離されたクローンの性状解析
精製したプラークの挿入体を制限酵素マッピングとDNA配列解析により性状解析した。METAB58は、約2.8kbであり、5’非翻訳配列、翻訳開始コドンおよび約2.3kbのコーディング配列を含有する。METAB58の3’末端は、METAB14の5’末端と重複する。METAB14は3’方向へ約700塩基対延長し、翻訳停止コドンを含有する。すなわち、METAB58とMETAB14は重複して、全長mGluR1受容体をコードする(配列番号1を参照)。他のクローンも、翻訳開始コドンと翻訳停止コドンの間に位置するmGluR1コーディング配列の部分からのヌクレオチド配列を含有する、部分mGluR1 cDNAである。
【0112】
ヒトmGluR1転写体の3’末端をコードする追加のクローンがヒトcDNAライブラリー中に存在するか否かを測定するために、海馬/脳幹神経節および小脳ライブラリーからのcDNAの核酸増幅を行なった。5’プライマーは配列番号1のヌクレオチド2218から2240よりなり、3’プライマーは、ラットのmGluR1aコーディング配列のアミノ酸890−897に基づく縮重オリゴヌクレオチドであった(ピン(Pin)ら、(1992)Neurobiology 89:10331−10335を参照)。増幅生成物をゲル電気泳動で解析した。海馬/脳幹神経節中にのみ1つの生成物(すなわち、500塩基対断片)が検出された。
【0113】
mGluR1転写体の3’末端を示す追加のクローンを得るために、海馬および小脳cDNAライブラリーを、前述のヒトmGluR1 cDNAを得るのに使用した条件と同様の条件を使用してラットのmGluR1a cDNAの3’末端からの断片(例えば、ラットのmGluR1a cDNAの約2kbのNcoI/ClaI断片)でスクリーニングすることができる。このプローブは、択一的にスプライスされるようではないmGluR1 cDNAの3’部分に対応する。次にハイブリダイズするクローンを制限酵素マッピングとDNA配列決定により解析して、異なる3’末端が示されるか否かを決定する。
【0114】
全長mGluR1 cDNA作成体の調製
ヒトmGluR1受容体のB型をコードする全長作成体を調製するために、クローンMETAB58とMETAB14の一部を結合させた。METAB58をEcoRI/AccIで消化し、配列番号1のヌクレオチド154−2612を含有する2459塩基対の断片を単離する。METAB14の704塩基対断片(配列番号1のヌクレオチド2613−3321を含有する)を、METAB14のAccI/XhoI消化により単離する。次にこの断片をMETAB58の2459塩基対に結合させて、EcoRI/SalI消化ベクターpCMV−T7−3に結合させる。ヒトmGluR1Bをコードする得られた作成体は、5’非翻訳配列の234ヌクレオチド(配列番号1のヌクレオチド154−387)、全mGluR1Bコーディング配列(配列番号1のヌクレオチド388−3108)、および3’非翻訳配列の213ヌクレオチド(配列番号1のヌクレオチド3109−3321)を含有する。mGluR1Bをコードする配列は、哺乳動物細胞中での発現のためのpCMV−T7−3中の制御成分に機能的に結合している。
【0115】
種々のヒト組織中で実際に発現されるmGluR5およびmGluR1受容体を決定するために、種々の方法が使用できる。例えば、本明細書に記載のmGluR転写体の挿入/欠失(すなわち、分岐の領域)の5’と3’に位置するヌクレオチド配列に特異的なオリゴヌクレオチドを用いて、種々の組織から単離されたRNAおよび/または種々の組織から調製されたcDNAライブラリーの核酸増幅をプライムすることができる。増幅生成物の有無および生成物のサイズは、組織中で発現される変種を示す。生成物はまた、DNA配列解析により詳細に性状解析される。
【0116】
RNase保護測定法を用いて、種々の組織中で発現される変種転写体を決定することもできる。これらの測定法は、総細胞性RNAの複雑な混合物中のRNA種を検出し定量するための感度の良い方法である。mGluRDNAの一部を標識し、細胞性RNAとハイブリダイズさせる。細胞性RNA中に相補的なmRNAが存在する場合は、DNA−RNAハイブリッドが形成する。次にこのRNA試料をRNaseで処理して、1本鎖RNAを分解させる。RNA−DNAはRNase分解から保護され、ゲル電気泳動やオートラジオグラフィーにより目視できるようにされる。
【0117】
例えばヒトmGluR cDNAへのハイブリダイゼーションによるヒトメタボトロピック受容体をコードする配列を含有するゲノムクローンの単離と、引き続くクローンの性状解析は、mGluR一次転写体の可能なスプライス変種に関してさらなる情報を与える。
【0118】
C.mGluR3受容体cDNA
cDNAライブラリースクリーニング
ヒト海馬cDNAライブラリー(ランダムプライマーを用いてcDNA合成をプライムさせ、次にλgt10ベクターへの結合について1.0−2.8kbのcDNAを選択することにより作成された)を、ラットmGluR2 cDNAの500塩基対のSmaI/XbaI断片とラットmGluR3 cDNAの3kbのAccI/BamHI断片へのハイブリダイゼーションについて選択した[タナベ(Tanabe)ら、(1992)Neuron 8:169−179]。ハイブリダイゼーションは、42℃で5×SSPE、5×デンハルツ(Denhart's)溶液、50%ホルムアミド、0.2%SDSおよび200μg/mlの変性した超音波処理したニシン精子DNA中で行ない、洗浄は65℃で0.5×SSPEと0.2%SDS中で行なった。3つのハイブリダイズするクローン(METAB40、METAB41、およびMETAB45)が同定された。
【0119】
次にMETAB45の5’末端の一部(すなわち、最初の244塩基対;配列番号5のヌクレオチド2634−2877)を用いて、増幅した小脳ライブラリー(ランダムプライマーを用いてcDNA合成をプライムし、次にλgt10ベクターへの結合について>2.8kbのcDNAを選択することにより作成される)、そして増幅した海馬cDNAライブラリー(ランダムプライマーを用いてcDNA合成をプライムし、次にλgt10ベクターへの結合について>2.0kbのcDNAを選択することにより作成される)を追加のmGluR3についてスクリーニングした。各ライブラリーから100万のクローンがスクリーニングされた。ハイブリダイゼーション条件と洗浄条件は、海馬ライブラリーからのMETAB40、METAB41およびMETAB45を単離するのに使用したものと同じである。3つのハイブリダイズするクローンが各ライブラリーで同定された(小脳ライブラリーではMETAB46、METAB49およびMETAB50、そして海馬ライブラリーではMETAB47、METAB48およびMETAB51B)。
【0120】
単離したクローンの性状解析
精製したプラークの挿入体を、制限酵素マッピングとDNA配列解析により性状解析した。単離したクローンのおのおのは、ヒトmGluR2受容体の一部をコードするクローンメタボトロピック40(例えば実施例1.D.を参照)を除いて、ヒトmGluR3受容体の一部である。クローンMETAB41、METAB45、METAB47−49は、mGluR3コーディング配列の3’からの配列および翻訳停止コドンを含有する。クローンMETAB46、METAB50およびMETAB51Bは、mGluR3 cDNAの5’末端からの配列(翻訳開始コドンと種々の量の5’非翻訳配列を含む)を含有する。
【0121】
全長mGluR3 cDNA作成体の調製
METAB48とMETAB46またはMETAB51Bの一部を結合させて、全長ヒトmGluR3コーディング配列を含有する4つの作成体を調製した。この全長コーディング配列は配列番号5(ヌクレオチド1064−3703)に与えられる。クローンMETAB46とMETAB51Bの挿入体は、EcoRI断片としてpCMV−T7−3中に別々にサブクローニングされた。クローンMETAB48の挿入体は、EcoRI断片としてpCMV−T7−2にサブクローニングされた。
【0122】
作成体mGluR3Bを作成するために、METAB51B挿入体を含有するpCMV−T7−3プラスミドをScaI/Bgl IIで消化し、アンピシリン耐性遺伝子の5’半分とMETAB51B挿入体の5’部分(配列番号5のヌクレオチド748−1671)を含有する2.6kbの断片を単離した。この断片を、METAB48の挿入体を有するpCMV−T7−2プラスミドのScaI/Bgl II消化物から単離した4.3kbの断片[この4.3kbの断片はアンピシリン耐性遺伝子の3’半分とMETAB48の3’部分を有する(配列番号5のヌクレオチド1672−3919)]に結合させた。得られた作成体(mGluR3B)は、316ヌクレオチドの5’非翻訳配列(配列番号5のヌクレオチド748−1063)、全mGluR3コーディング配列(配列番号5のヌクレオチド1064−3703)、および216ヌクレオチドの3’非翻訳配列(配列番号5のヌクレオチド3704−3919)を含有する。mGluR3Bをコードする配列は、哺乳動物細胞中での発現のためにベクターpCMV−T7−3とpCMV−T7−2からの制御成分に機能的に結合している。
【0123】
作成体mGluR3Cを作成するために、METAB46の挿入体を有するpCMV−T7−3プラスミドを、ScaI/Bgl IIで消化し、アンピシリン耐性遺伝子の5’半分とMETAB46の5’部分を含有する3.4kbの断片(配列番号5のヌクレオチド1−1671)を単離した。この断片を、METAB48の作成体mGluR3Bで使用したものと同じScaI/Bgl II断片に結合させた。得られた作成体(mGluR3C)は、5’非翻訳配列の1063ヌクレオチド(配列番号5のヌクレオチド1−1063)、全mGluR3コーディング配列(配列番号5のヌクレオチド1064−3703)、および3’非翻訳配列の216ヌクレオチド(配列番号5のヌクレオチド3704−3919)を含有する。このmGluR3Cをコードする配列は、哺乳動物細胞中での発現のためにベクターpCMV−T7−2とpCMV−T7−3からの制御成分に機能的に結合している。
【0124】
作成体mGluR3Aは、mGluR3CをEcoRVとNotIで消化して、配列番号5のヌクレオチド1−1035を含有する断片を除去して、NotI部位を平滑末端として、次に大きい方のベクター断片を再結合させることにより作成した。作成体mGluR3Aは、28ヌクレオチドの5’非翻訳配列(配列番号5のヌクレオチド1036−1063)、全mGluR3コーディング配列(配列番号5のヌクレオチド1064−3703)、および216ヌクレオチドの3’非翻訳配列(配列番号5のヌクレオチド3704−3919)を含有する。このmGluR3Aをコードする配列は、哺乳動物細胞中での発現のためにベクターpCMV−T7−3とpCMV−T7−2からの制御成分に機能的に結合している。
【0125】
作成体pSV−hmGluR3C(mGluR3のSV40プロモーター制御発現に使用するため)を作成するために、METAB46の挿入体を有するpCMV−T7−3プラスミドをScaI/NotIで消化し、アンピシリン耐性遺伝子の3’部分と全METAB46挿入体を含有する断片を単離した。プラスミドpSVβをScaI/NotIで消化し、アンピシリン耐性遺伝子5’部分とSV40早期プロモーターおよびスプライス部位を含有する断片を、METAB46を有するpCMV−T7−3ベクターからのScaI/NotI断片に結合させて、pSV−METAB46を作成した。プラスミドpSV−METAB46をScaI/Bgl IIで消化し、アンピシリン耐性遺伝子の5’部分、SV40早期プロモーターおよびスプライス部位そしてMETAB46の5’部分(配列番号5のヌクレオチド1−1671)を含有する断片を単離した。この断片を、作成体mGluR3BやmGluR3Cで使用したものと同じMETAB48のScaI/Bgl II断片に結合させた。得られた作成体(pSV−hmGluR3C)は、SV40プロモーターを含有し、mGluR3 DNA(配列番号5のヌクレオチド1−3919を含有する)と機能的に結合したSV40スプライス部位が続き、この後にポリアデニル化シグナルが続く。
【0126】
D.mGluR2受容体cDNA
実施例1.C.に記載したようにヒト海馬cDNAライブラリーからクローンMETAB40を単離した。このMETAB40の挿入体cDNAは長さが1100塩基対であり、ヒトmGluR2受容体の3’末端(翻訳停止コドンと3’末端非翻訳配列を含む)をコードする。METAB40の最初の355ヌクレオチドは配列番号3に提供され、METAB40の最後の343ヌクレオチド(すべて3’非翻訳配列からである)は配列番号13に提供される。
【0127】
mGluR2転写体の5’部分を示すDNAを含有するクローンを単離するために、METAB40の5’末端に対応するオリゴヌクレオチドへのハイブリダイゼーションについて、ヒト海馬cDNAライブラリーをスクリーニングすることができる。ハイブリダイズするクローンを精製し、DNA配列解析により性状解析する。METAB40に重複し翻訳開始コドンを含有するクローンを、適当な制限部位でMETAB40に結合させて、全長mGluR2をコードするcDNA作成体を作成することができる。
【実施例2】
【0128】
卵母細胞中の組換えヒトメタボトロピックグルタミン酸受容体の発現
ヒトメタボトロピック受容体をコードするDNAを含有する作成体から調製したインビトロ転写体を、ツノガエル(Xenopus)卵母細胞に注入した。2電極の電位固定法(例えば、スツマー(Stuhmer)(1992)Meth. Enzymol. 207:319−339を参照)を用いて、卵母細胞トランスメンブレン電流の電気生理的測定を行なった。
【0129】
A.インビトロ転写体の調製
作成体mGluR5a3中に含有されるメタボトロピック受容体cDNAのキャップされた組換え転写体を、メガスクリプト(Megascript)キット(カタログ番号#1334、アンビオン社(Ambion Inc.)、オースチン、テキサス州)を用いて、線状化したプラスミドから合成した。各合成転写体の質量を紫外吸光度法により測定し、各転写体の完全性をアガロースゲルの電気泳動により測定した。
【0130】
B.電気生理学
卵母細胞1つにつき、10−50ngのメタボトロピック受容体転写体を、ツノガエル(Xenopus)卵母細胞に注入した。卵母細胞の調製と注入は、ダスカル(Dascal)の方法[(1987)Crit. Rev. Biochem. 22:317−387]に従って行なった。mRNA注入の2から6日後、2電極電位固定法を用いて卵母細胞を試験した。細胞はリンゲル溶液(115mM NaCl、2.5mM KCl、1.8mM CaCl2、10mM HEPES、pH7.3)浴に浸漬し、膜電位は−80から−100mVに固定した。薬剤含有溶液の60μlずつをピペットで直接浴に入れた。アクソテープ(AXOTAPE)バージョン1.2ソフトウェア(アクソン・インスツルメンツ(Axon Instruments)、フォスターシティ、カリホルニア州)を用いてPC−386中のラボマスター(Labmaster)データ採取ボードを用いて2−5Hzでデータをサンプリングした。データをレーザープリンターに取り出すか、またはシグマプロット(Sigmaplot)バージョン5.0を用いてプロットした。
【0131】
メタボトロピック受容体調節化合物(すなわち、0.001−0.1μMキスカル酸、0.1−10μMグルタミン酸および0.1−300μM 1S,3R−ACPD(1−アミノ−シクロペンチル−1,3−ジカルボン酸)を浴に適用し、トランスメンブレン電流を記録した。化合物の適用後相当の電流が検出された。種々の量の各化合物を適用後の電流を比較する用量応答試験で、電流の大きさは各化合物の濃度の上昇に伴い増加することが明らかになった。これらのデータを解析した結果、各化合物のEC50を計算することができ、これを化合物の相対電位の測定に使用した。
【実施例3】
【0132】
哺乳動物細胞におけるヒトメタボトロピックグルタミン酸受容体サブユニットの組換え発現
ヒト胎児性腎(HEK)細胞とチャイニーズハムスター卵巣(CHO)細胞(すなわち、DG44細胞;ウーラウプ(Urlaub)ら(1986)Som. Cell. Molec. Genet. 12:555を参照)を、ヒトメタボトロピック受容体をコードするDNAでトランスフェクションさせた。種々の方法(例えば、イノシオトールリン酸(IP1)測定法、Ca++−感受性蛍光指示薬ベースの測定法、および[3H]−グルタミン酸結合測定法)を用いて、メタボトロピック受容体の発現について解析した。
【0133】
A.HEK293細胞の一時的トランスフェクション
HEK293細胞を、mGluR5a(作成体mGluR5a2とmGluR5a3および作成体MMTV−hmGluR5a)受容体をコードするDNAで一時的トランスフェクションを行なった。約2×106のHEK293細胞を標準的CaPO4トランスフェクション法[ウィグラー(Wigler)ら、(1979)Proc. Natl. Acad. Sci. U.S.A 76:1373−1376を参照]に従い、目的のプラスミドの5−18μg(あるトランスフェクション体においては0.18μg、実施例3.C.2.を参照)で一時的にトランスフェクションした。さらにCMVプロモーターに融合した大腸菌β−ガラクトシダーゼ遺伝子を含有する、プラスミドpCMVβgal(クロンテック・ラボラトリーズ(Clontech Laboratories)、パロアルト(Palo Alto)、カリホルニア州)0.5−2μgを、レポーター遺伝子として同時トランスフェクションして、トランスフェクションの効率を追跡した。β−ガラクトシダーゼとX−gal基質を含む反応の生成物の直接染色により、β−ガラクトシダーゼ発現についてトランスフェクション体を解析した[ジョーンズ(Jones)(1986)EMBO 5:3133−3142]。トランスフェクション体はまた、β−ガラクトシダーゼ活性の測定によるβ−ガラクトシダーゼ発現について解析することもできる[ミラー(Miller)(1972)分子遺伝子学実験(Experiments in Molecular Genetics)のPP. 352−355, コールド・スプリング・ハーバー・プレス(Cold Spring Harbor Press)]。
【0134】
5μgのMMTV−hmGluR5Aで一時的トランスフェクションを行なったHEK293細胞は、ラウス肉腫ウイルス(RSV)LTRプロモーターに機能的に結合した糖質コルチコイド受容体をコードするDNAを含有するpRShGR(ATCC整理番号67200)5μgと同時トランスフェクションをした。これらの細胞における糖質コルチコイド受容体の同時トランスフェクションは、細胞への糖質コルチコイド(例えば、デキサメタゾン)の添加によるMMTVプロモーター−mGluR5aDNAの発現の誘導を確実にする。
【0135】
HEK細胞のこれらのトランスフェクションの効率は、標準的効率に典型的なものであった(すなわち、約50%)。
【0136】
B.哺乳動物細胞の安定なトランスフェクション
HEK293細胞、Ltk−細胞およびCHO細胞のような哺乳動物細胞は、リン酸カルシウムトランスフェクション法を用いて安定にトランスフェクションすることができる[Current Protocols in Molecular Biology, Vol.1,ワイリー・インターサイエンス(Wiley Inter-Science)、増刊14号、単元9.1.1.−9.1.9(1990)]。CHO細胞を宿主として使用するときは、ヒトメタボトロピック受容体をコードするcDNAの発現を制御するにはSV40プロモーターの使用が一般に好ましい。それぞれ1−2×106細胞を含有する10cmのプレートを、メタボトロピック受容体をコードするDNA約5−10μgと選択マーカー(例えば、ネオマイシン−耐性遺伝子(すなわち、HEK293形質転換体の選択にはネオマイシン耐性遺伝子(すなわち、pSV2neo)、Ltk−細胞トランスフェクション体にはチミジンキナーゼ遺伝子、またはDG44細胞形質転換体の選択にはジヒドロ葉酸還元酵素(dhfr)遺伝子))をコードするDNA 0.5−1μgを含有するDNA/リン酸カルシウム沈殿物でトランスフェクションする。適当な培地中で約14日間の増殖の後、コロニーが形成され、クローニングシリンダーを用いて個々に単離される。次に単離物を限界希釈を行いスクリーニングして、例えば後述の方法を用いてメタボトロピック受容体を発現するものを同定する。
【0137】
C.トランスフェクション体の解析
1.蛍光指示薬に基づく測定
アゴニストによるG蛋白結合メタボトロピック受容体の活性化により、ホスファチジルイノシトール(PI)加水分解/細胞内Ca++シグナル化経路および/または阻害性cAMPカスケードが刺激される。細胞内カルシウム濃度の一時的上昇の検出法は、PI加水分解/Ca++動員(mobilization)経路またはPI加水分解/Ca++動員(mobilization)経路と阻害性cAMPカスケードの両方にカップリングしたメタボトロピック受容体の機能性発現の解析に適用できる。細胞内カルシウムレベルを測定する1つの方法は、カルシウム感受性蛍光指示薬を用いる。
【0138】
フルオ−3(fluo−3)やフラ−2(fura−2)(モレキュラー・プローブズ社(Molecular Probes Inc.)、ユージーン(Eugene)、オレゴン州)のようなカルシウム感受性指示薬は、膜透過性であるアセトキシメチルエステルとして入手できる。アセトキシメチルエステル型の指示薬が細胞内に入ると、サイトゾル性エステラーゼによりエステル基は除去され、このため遊離の指示薬はサイトゾル内に捕捉される。遊離の指示薬とカルシウムが相互作用すると、指示薬の蛍光強度が増加する。従って、指示薬を含有する細胞の細胞内Ca++濃度の上昇は直接蛍光の増強(または、フラ−2を使用する時は、2つの波長での蛍光の比率の増強)として発現される。メタボトロピック受容体を測定するための自動蛍光検出装置が、本出願人の係属中の米国特許出願第07/812,254号およびその対応するPCT特許出願US92/11090号(参考のためその全体が本明細書中に引用される)に記載されている。さらに細胞内Ca++振動を目視できるようにするために蛍光イメージング法を使用することができる。
【0139】
ヒトmGluR5a受容体をコードするDNAで一時的トランスフェクションを行なったHEK細胞を、自動蛍光指示薬ベースの測定法と蛍光イメージング測定法を用いて、機能性組換えメタボトロピック受容体の発現について解析した。同様に、メタボトロピック受容体DNAで安定にトランスフェクションした細胞も、これらの測定法系を用いて機能性メタボトロピック受容体について解析した。
【0140】
a.自動蛍光測定法
2×105細胞/ウェルの濃度でポリ−L−リジンでプレコーティングし、20μMフルオ−3、0.2%プルロニック(Pluronic)F−127をHBS(125mM NaCl、5mM KCl、1.8mM CaCl2、0.62mM MgCl2、20mMグルコース、20mM HEPES、pH7.4)中に含有する培地中で、20℃で2時間インキュベートしてフルオ−3を充填した96ウェルマイクロタイタープレート(ヌンク(Nunc)、カタログ番号1−6708、アラメダ・インダストリーズ(Alameda Industries)、エスコンジド(Escondido)、カリホルニア州)に、トランスフェクションしていないHEK293細胞(またはpCMV−T7−3で一時的にトランスフェクションしたHEK293細胞)およびmGluR5aをコードするDNAでトランスフェクションしたHEK293細胞を広げた。次に細胞を測定緩衝液(すなわち、HBS)で洗浄した。次にマイクロタイタープレートを蛍光プレートリーダー(例えば、フルオロスカン(Fluoroskan)II、ラボ・プロダクツ・インターナショナル社(Lab Products International, Ltd.)、ラレー(Raleigh)、ノースカロライナ州)に入れ、各ウェルの基礎蛍光を測定し記録してから、ウェルにキスカル酸、グルタミン酸、トランス−ACPD(1−アミノ−シクロペンタン−1,3−ジカルボン酸)、1S,3R−ACPD、AP3(2−アミノ−3−ホスホノプロピオン酸)、AP5(2−アミノ−5−ホスホノペンタノン酸)、およびCNQX(6−シアノ−7−ニトロキノキサリン−2,3−ジオン)を添加した。アゴニストの添加後ウェルの蛍光を繰り返し(0.63秒間隔で75回読む)追跡する。
【0141】
一般に、トランスフェクションしていないHEK293細胞の蛍光は、これらの化合物のどれを添加しても変化しなかった。mGluR5a3またはMMTV−hmGluR5a作成体のいずれかで一時的にトランスフェクションしたHEK293細胞の蛍光は、グルタミン酸、キスカル酸、トランス−ACPD、または1S,3R−ACPDの添加に応答して上昇した。蛍光はピーク値まで増加し、次に時間とともに化合物の添加前の細胞の基礎レベルまで低下した。mGluR5a2でトランスフェクションした細胞について、グルタミン酸、キスカル酸またはトランス−ACPD刺激蛍光の増強に対するAP3、AP5またはCNQXの影響を調べた。これらの化合物(AP3、AP5、またはCNQX)のいずれも、これらの細胞中のアゴニスト誘導蛍光増強を阻害しなかった。
【0142】
種々の量のグルタミン酸、キスカル酸または1S,3R−ACPDを添加後に測定したピーク蛍光値を比較する用量応答試験で、ピーク蛍光の大きさは各化合物の濃度の上昇に伴い増加することが明らかになった。これらのデータを解析した結果、各化合物のEC50を計算することができ、これを化合物の相対電位の測定に使用した。
【0143】
MMTV−hmGluR5aおよびpRShgGR(糖質コルチコイド受容体作成体)で一時的に同時トランスフェクションしたHEK293細胞も、この蛍光測定法で解析した。これらの細胞の蛍光は100μMのキスカル酸に応答して増加し、測定前に細胞をデキサメタゾン(約1M)で37℃で16時間プレインキュベートするとピーク応答は大きかった。
【0144】
b.蛍光イメージング測定法
細胞内Ca++濃度のメタボトロピック受容体介在変化を目視できるようにするために、mGluR5a3で一時的にトランスフェクションしたHEK293細胞とトランスフェクションしていないHEK293細胞(対照)を、デジタルビデオイメージングで解析した。暗所中で室温で25分間細胞を1μMのフラ−2(アセトキシメチルエステル)に暴露することにより、トランスフェクション体(ガラス挿入底を有する35mm培養皿あたり4×105細胞)をフラ−2で充填した。次に細胞をDMEMで3回そしてリンゲル溶液(160mM NaCl、5mM KCl、2mM CaCl2、1mM MgCl2、11mMグルコース、5mM HEPES、pH7.3)で4回洗浄した。
【0145】
次にトランスフェクション体と非トランスフェクション細胞を、紫外光源として150Wのキセノンランプを有するアキシオベルト(Axiovert)100TV倒立顕微鏡(ツァイス(Zeiss)、オーベルコフレン(Oberkochren)、ドイツ)のステージの上に広げた。40×1.3N.A.油浸対物レンズを介して細胞を340nmと380nmで交互励起するのに、イメージ1フルオル(Fluor)システム(ユニバーサル・イメージング(Univeersal Imaging)、ウェストチェスター、ペンシルバニア州)を使用した。510nmより長い波長で出る光を、CCD72強化CCDカメラ(エム・ティー・アイ ダーゲ(MTI Dage)、ミシガン・シティ、インディアナ州)により集め、デジタル化した。340nmと380nmの励起イメージからバックグランドの蛍光を差し引いた。補正値を使用して340/380強度比を計算した。較正していないフラ−2比は、細胞内Ca++濃度の変化の信頼できる指標である。
【0146】
較正していないフラ−2比を用いて、休止細胞内Ca++濃度(約100nM)を紫色で、高細胞内Ca++濃度(約1μM)を赤色で疑色イメージを作成した。実験の各比率のイメージについて、定量解析のために、各細胞の12×12ピクセル領域内の平均比をソフトウェアにより計算し、さらに解析しグラフを描くために表に取り込んだ。
【0147】
HEK293細胞は、PI加水分解/Ca++動員経路の受容体介在活性化に必要な細胞内成分を発現することを証明するために、トランスフェクション体と非トランスフェクション細胞(内因性G蛋白結合ムスカリン性アセチルコリン受容体を発現する)を1mMのカルバミルコリン(CCh;ムスカリン性アセチルコリン受容体アゴニスト)に暴露し、細胞内Ca++濃度の上昇を追跡した。典型的には、イメージング試験でCChの添加に応答して、大多数の細胞の細胞内Ca++濃度の上昇が検出された。
【0148】
トランスフェクションしたHEK293細胞およびトランスフェクションしていないHEK293細胞を、100μMキスカル酸に応答して細胞内Ca++濃度が上昇するか否かについて追跡した。平均すると、非トランスフェクション細胞の細胞内Ca++濃度は、キスカル酸への暴露後増加しない。これに対して、トランスフェクションした細胞の26.7±22.3%のCa++濃度は100μMのキスカル酸の添加に応答して増加した。
【0149】
2.ホスファチジルイノシトール加水分解(IP1)測定法
G蛋白結合メタボトロピック受容体のアゴニストによる活性化により、ホスファチジルイノシトール(PI)加水分解経路が刺激されるため、PI加水分解の生成物(例えば、IP3、IP2またはIP1)の増加を検出する方法は、PI加水分解/Ca++動員経路またはPI加水分解/Ca++動員経路と阻害性cAMPカスケードにカップリングしたメタボトロピック受容体の機能性発現の解析に応用することができる。PIの加水分解により生成されるIP1および/またはIP2および/またはIP3を測定する1つの方法は、細胞膜リン脂質内への[3H]−ミオ−イノシトールを取り込み、次に[3H]−IP1、[3H]−IP2および[3H]−IP3を分離し、以下のように各画分の放射能を定量する。
mGluR5a3で一時的にトランスフェクションしたHEK293細胞を、8×105細胞/ウェルで24ウェルのマイクロタイタープレートに広げた。細胞を数時間沈降させ、プレートの底に付着させた後、2μCiの[3H]−ミオ−イノシトール(アマーシャム(Amersham)、カタログ番号#PT6−271、アーリントンハイツ(Arlington Heights)、イリノイ州;比活性=17.7Ci/mmol)を各ウェルに添加し37℃で一晩インキュベートした。翌日、ニコンダイアフォト(Nikon Diaphot)倒立顕微鏡下で細胞を観察し、細胞の形態の健常性を評価し、ウェルがコンフルエント(confluent)な細胞層を含有するか否かを測定した。次に培地を吸引し、細胞を0.5mlのクレブス重炭酸緩衝液[117.9mM NaCl、4.72mM KCl、2.54mM CaCl2、1.18mM MgSO4、1.19mM KH2PO4、25mM NaHCO3、11.1mMデキストロース(95% O2、5% CO2、pH7.4で平衡化されている)]で2回洗浄した。細胞を室温で45分間インキュベートした。次に各ウェルから緩衝液を吸引し、細胞を洗浄し、0.5ml/ウェルで室温で45分間インキュベートした。次に緩衝液を各ウェルから吸引し、次に10mM NaClの代わりに10mMのLiClを含有する450μlのクレブス重炭酸緩衝液(IP1のイノシトールと無機リンへの加水分解を防ぐために)とともに細胞を37℃で20分間インキュベートした。
【0150】
メタボトロピック受容体調節化合物による細胞の処理を始める前に、50μlのクレブス重炭酸緩衝液(対照)または化合物の最終濃度の10×のものを各ウェルに添加し、40分間インキュベートを続けた。各ウェルに1mlの氷冷メタノールを加えてインキュベートを停止させた。
【0151】
細胞からIP1を単離するために、プラスチックピペットのチップでプレートの細胞を掻き取って、細胞懸濁物を12×75mmガラス試験管に移した。試験管を完全にボルテックス混合し、各反応混合物の150μl分(すなわち、総量の10分の1)を別の試験管に移して蛋白を測定した。1mlのクロロホルムによる抽出により、放射標識膜リン脂質から水溶性リン酸イノシトールを分離した。試験管を室温で30分間インキュベートした後、4℃で500×gで5分間遠心分離した。[3H]−リン酸イノシトールを含有する水(上)層を、アクセルQMAセパク(Accell QMA SEP−PAK)カラム(ミリポア(Millipore)、カリホルニア州)に接続した10mlのシリンジに移した(これは、20mlのシンチレーションバイアルに集められるように変更したアマーシャム・スーパーセパレーター(Amersham Superseparator)装置に結合してある)。カートリッジに水(10ml)を入れて[3H]−イノシトール前駆体を除き、次に4mlの0.02Mトリエチルアンモニウム水素炭酸化緩衝液(TEAB、フルカ(Fluka);ニューヨーク)を加えた。カートリッジから[3H]−IP1、[3H]−IP2そして[3H]−IP3を別々に除去するために、4mlの0.1M TEAB、4mlの0.3M TEAB、そして4mlの0.4M TEABをカートリッジに連続的に添加して、画分を溶出させ、大きいシンチレーションバイアルに採取した。エコルメ・カクテル(Ecolume cocktail)(15ml;ICN;カリホルニア州)を各バイアルに加え、次にシンチレーション計測して別々の画分の各IPの量を測定した。蛋白濃度は、バイオラッド(Bio−Rad)蛋白マイクロ測定法(バイオラッド(Bio−Rad)、リッチモンド、カリホルニア州)を用いて測定した。
【0152】
この測定法で測定した時、18μgのmGluR5a3で一時的にトランスフェクションしたHEK293細胞は、比較的高い基礎レベルのIP1を示した。しかし、0.18μgのmGluR5a3で一時的にトランスフェクションしたHEK293細胞は、低い基礎IP1レベルを示し、1mMのグルタミン酸、1mMのキスカル酸または1mMの1S,3R−ACPDで処理した時はIP1レベルの検出できる増加を示した。IP1レベルのキスカル酸に誘導される増加は、1mMのAP3により影響を受けなかった。
【0153】
mGluR5a3でトランスフェクションした細胞に、種々の濃度のグルタミン酸、キスカル酸または1S,3R−ACPDの添加後測定したIP1レベルを比較した用量応答試験では、各化合物の濃度の上昇とともにIP1レベルが増加することを示していた。これらのデータの解析により、各化合物のEC50を計算することができ、これを化合物の相対電位の測定に使用した。
【0154】
3.メタボトロピック受容体リガンド結合測定法
mGluR5a3またはpUC19(陰性対照)で一時的にトランスフェクションしたHEK細胞を、[3H]−グルタミン酸結合について解析した。陽性対照として結合測定法にラット脳の膜を加えた。
【0155】
a.膜の調製
i.ラット前脳膜
シェープ(Schoepp)ら[(1992)Neurosci. Lett. 145:100]が記載したように、ラット前脳膜を調製した。簡単に説明すると、10個のラット脳からの基本的に脳皮質、線条体と海馬よりなる前脳を、2.5mM CaCl2、pH7.6を含有する50部の30mMの氷冷トリス−塩酸中で、ポリトロン(Polytron)(ブリンクマン(Brinkman)、ウェストベリー(Westbury)、ニューヨーク州))を用いてホモゲナイズした。ホモゲネートを4℃で30、000×gで15分間遠心分離した。上澄液を捨て、ポリトロンを用いてペレットを50部の緩衝液に再懸濁し、懸濁物を30、000×gで15分間遠心分離した。この工程を2回繰り返した。ペレットを緩衝液に再懸濁し、37℃で30分間インキュベートした。次に懸濁物を4℃で30、000×gで15分間遠心分離した。この工程を3回繰り返した。最終ペレットを15部の50mM トリス−塩酸、pH7.6に再懸濁し、分注し、急速に凍結し、−70℃で保存した。
【0156】
ii.トランスフェクションしたHEK293細胞およびトランスフェクションしないHEK293細胞の膜
mGluR5aをコードするDNAまたはpUC19(陰性対照)でトランスフェクションしたHEK293細胞から膜を調製するために、組織培養プレートから細胞を掻き取り、プレートを5mlのPBS(リン酸緩衝化生理食塩水:137mM NaCl、2.7mM KCl、10mM Na2HPO4、1.7mM KH2PO4)で洗浄した。卓上型遠心分離機で細胞を低速度で遠心分離し、細胞ペレットをPBSで洗浄した。0.5mM PMSF、pH7.6を含有する50mMトリス−塩酸20部に、細胞ペレットを再懸濁した。ダウンス(Dounce)(テフロン(登録商標)/ガラス)ホモゲナイザー中で10−20ストロークを用いて、細胞を氷の上でホモゲナイズした。ホモゲネートを4℃で120、000×gで30分間遠心分離した。最終の膜ペレットを、0.5mM PMSF、pH7.6を含有する50mMトリス−塩酸再懸濁した。膜調製物を分注し、急速に凍結し、−701℃で保存した。蛋白濃度は、ブラッドフォード(Bradford)[(1976)Anal. Biochem.72:248]の方法で測定した。
【0157】
b.[3H]−グルタミン酸結合測定法
ラット前脳の膜中のメタボトロピック受容体への[3H]−グルタミン酸の特異的結合は、基本的にシェープ(Schoepp)ら(前述)が記載したように調製した。測定の日に凍結ホモゲネートを融解し、50mMトリス−塩酸、pH7.6で3回洗浄した。最終ペレットを50mMトリス−塩酸、pH7.6に再懸濁した。蛋白濃度は、ブラッドフォード(Bradford)[(1976)Anal. Biochem.72:248]の方法で測定した。懸濁物を30、000×gで15分間遠心分離し、ペレットを1mg/mlの濃度で測定緩衝液(50mMトリス−塩酸、0.5mM PMSF、0.1%BSA、pH7.6)に再懸濁した。膜懸濁物を、総量0.5mlの測定緩衝液[イオノトロピックグルタミン酸受容体への[3H]−グルタミン酸の結合を組織するために、100μM NMDA(シグマ(Sigma)、セントルイス、ミズーリ州)、100μM AMPAおよび100μMカイニン酸(リサーチ・バイオケミカルズ社(Research Biochemiclas Inc)、ナチック(Natick)、マサチューセッツ州)を含有し、塩化物依存性摂取部位への[3H]−グルタミン酸の結合を阻害するために、100μM SITS(シグマ(Sigma)、セントルイス、ミズーリ州)を含有する]中で、3重測定で10または100nMの[3H]−グルタミン酸(ニュー・イングランド・ニュクレア(New England Nuclear)、ボストン、マサチューセッツ州;カタログ番号NET−490、比活性=57.4Ci/mmol)と、氷上で45分間インキュベートした。SM−24ローター(ソーバル(Sorvall)、ウィルミントン(Wilmington)、デラウェア(Delaware))中で4℃で20、000×gで5分間遠心分離して、結合した放射能を遊離の放射能から分離した。ペレットを5−6mlの氷冷50mMトリス−塩酸、pH7.6で2回洗浄した。ペレットを5mlのエコルメシンチレーションカクテル(Ecolume scintillation cocktail)中でボルテックス混合して可溶化させた。ベックマン(Beckman)シンチレーション計測機で放射能を測定した。総結合量から、1mMのグルタミン酸の存在下で得られた非特異的結合量を引いて、特異的結合量を求めた。
【0158】
mGluR5aをコードするDNAまたはpUC19でトランスフェクションしたHEK293細胞から調製した膜への[3H]−グルタミン酸の特異的結合を、基本的にラット脳の膜への結合の測定で記載したように測定したが、若干の修飾を行なった。測定の日に凍結ホモゲネートを融解し、MR−150高速冷却マイクロ遠心分離機(ペニンスラ・ラボラトリーズ社(Peninsula Laboratories Inc.)、ベルモント(Belmont)、カリホルニア州)で遠心分離した。ペレットを測定緩衝液(50mMトリス−塩酸、0.5mM PMSF、0.1%BSA、pH7.6)で2回洗浄し、最終ペレットを1mg/mlの濃度で測定緩衝液に再懸濁した。NMDA、AMPAおよびカイニン酸を測定混合物から除きながら、HEK293細胞を[3H]−グルタミン酸への結合について解析した。
【0159】
200μgの膜と100nMの[3H]−グルタミン酸を用いて、ラット脳の膜への[3H]−グルタミン酸の特異的結合を測定した。総結合量対非特異結合量の比は約2:1であった。
【0160】
mGluR5a3またはpUC19でトランスフェクションしたHEK293細胞から調製した膜への[3H]−グルタミン酸の特異的結合を、200μgの膜と100nMの[3H]−グルタミン酸を用いて測定した。mGluR5a3でトランスフェクションしたHEK293細胞から調製した膜への特異的結合量は、pUC19でトランスフェクションしたHEK293細胞から調製した膜への結合量より有意に高かった。競合結合試験を行い、種々の濃度の非標識グルタミン酸の存在下でmGluR5a3でトランスフェクションしたHEK293細胞から調製した膜への[3H]−グルタミン酸の特異的結合の量を測定した。これらの試験で得られたデータからIC50値を求めた。
【0161】
4.サイクリックAMP(cAMP)測定
a.RIAベースの測定
ある種のG蛋白結合受容体の活性化は、cAMPの(増加ではなく)低下を引き起こすため、細胞内cAMPレベルの測定法は、哺乳動物宿主細胞中で発現される組換えヒトメタボトロピック受容体を評価するのに使用される。ヒトメタボトロピック受容体をコードするDNAまたはpUC19(陰性対照)で一時的にまたは安定にトランスフェクションした哺乳動物細胞を、5×105細胞/ウェルの濃度で24ウェルのマイクロタイタープレートに広げ、一晩インキュベートさせる。翌日、ニコンダイアフォト(Nikon Diaphot)倒立顕微鏡下で細胞を観察し、細胞の形態の健常性を評価し、ウェルはコンフルエントな細胞層を含有するか否かを測定した。次に培地を吸引し、細胞を、1mMのIBMX(3−イソブチル−1−メチルキサンチン;シグマ(Sigma)、セントルイス、ミズーリ州)と0.1%BSAを含有するクレブス重炭酸緩衝液0.5ml(PI加水分解測定法で使用したものと同じ緩衝液:実施例3.C.2を参照)で2回洗浄した。あるいはクレブス重炭酸緩衝液の代わりに1×PBSを使用することもできる。各洗浄の後に37℃で30分間インキュベートを行う。各ウェルから緩衝液を吸引し、次に細胞を、1mMのIBMXと0.1%BSAを含有する0.2mlのクレブス重炭酸緩衝液で37℃で20分間インキュベートする。
【0162】
メタボトロピック受容体調節化合物による細胞の処理を始める前に、50μlのクレブス重炭酸緩衝液(最終濃度の5×のフォルスコリンの有りまたは無し)を、一部の細胞(基礎対照)に加え、一部の細胞(試験細胞)に最小濃度の5×の化合物および最終濃度の5×のフォルスコリンを加え、37℃で15分間インキュベートする。15分間のインキュベートの最後に、1%トリトンX−100溶液25μlを加えて反応を停止させ、インキュベートをさらに10分間続ける。溶解した細胞および細胞懸濁物を、プラスチックピペットのチップで12×75mmのポリプロピレン試験管に移す。各ウェルを、1mMのIBMXと0.1%BSAを含有するクレブス重炭酸緩衝液75μlで洗浄する。洗浄物を細胞溶解物を一緒にする。細胞溶解懸濁物を2300×gで5分間遠心分離し、RIAキット(アマーシャム・ライフサイエンシーズ(Amersham Life Sciences)カタログ#TRK432;アーリントンハイツ(Arlington Heights)、イリノイ州)を用いて、上澄液のcAMPレベルを測定する。
【0163】
b.サイクリックヌクレオチドゲートのチャネルに基づく測定法
5%の規定補足ウシ血清(ハイクローン(Hyclone))を含有するダルベッコー改変イーグル培地(DMEM;ギブコ(GIBCO))(100U/mlのペニシリンと100μg/mlの硫酸ストレプトマイシンを含む)中で、HEK293細胞を単層で増殖させた(10cmのポリ−D−リジン被覆プレート当たり約2×106細胞)。CMVプロモーターに結合した5μgのpCMV−OCNA(嗅覚性サイクリックヌクレオチドゲートのチャネル(ダレン(Dhallen)らを参照、前述)、2μgのpCMV−βgal(クロンテック(Clontech)、パロアルト(Palo Alto)、カリホルニア州)、および対照プラスミドとしての13μgのpUC19を用いて、細胞をリン酸カルシウム法(アウスベル(Ausubel)ら、前述、pp9.1.1−9.1.7)で一時的にトランスフェクションした。ベクターpCMV−OCNAは、pBluescriptKSからの約3.0kbのEcoRI断片として嗅覚性サイクリックヌクレオチドゲートのチャネルをコードするDNAを単離し、得られた断片をEcoRI消化pCMV−T7−3に結合させることにより、作成した。トランスフェクションの6時間後、リン酸カルシウム沈殿物を洗い流し、10%透析胎児牛血清(ハイクローン(Hyclone))、100U/mlのペニシリン、100μg/mlのストレプトマイシン、および2mMグルタミンが補足されたDMEMを細胞に与えた。β−ガラクトシダーゼ活性の測定によるトランスフェクション効率は、50−70%であった。
【0164】
嗅覚性サイクリックヌクレオチドゲートのチャネルDNAでトランスフェクションしたHEK細胞を、24−48時間インキュベートしてから、機能を試験した。まず、細胞質の面に達するcAMPの濃度が調節できるように(例えば、単一チャネル記録(Single Channel Recording)、サクマンとネヘル(Sakman and Neher)編、プレヌム・プレス(Plenum Press)、ニューヨーク、(1983))、トランスフェクションした細胞から引っ張った裏返した膜パッチを用いて、電気生理学的にチャネルの活性を測定した。パッチの両面を、Ca++/Mg++を含まないリンゲル液に接触させた。1つのパッチでは、1mMのcAMPの存在下で膜電位を−100から+100mVへ2秒で勾配をつけて電流を発生させた。この結果は、チャネルが機能的に発現されていることを示唆していた。
【0165】
トランスフェクション体もまた、細胞内カルシウムレベル([Ca++])の単細胞ビデオイメージングにより解析した。この方法は、細胞内カルシウムレベルの測定(これはチャネルのサイクリックヌクレオチド活性化により制御される、チャネルを介するカルシウムの流入量とともに変化する)によるサイクリックヌクレオチドゲートのチャネルの分析を可能にする。イメジーング測定法は、基本的に実施例3.C.1.bに記載のように実施したが、若干の修飾を行なった。色素を充填後、細胞をツァイスアクシオヴェルト(Zeiss Axiovert)顕微鏡と100Wの水銀ランプ、デージ(Dage)増強CCDカメラ、そしてイメージ−1(Image−1)ハードウェアとイメージ処理のソフトウェアを用いて観察した。このソフトウェアは、20×1.3N.A.油浸対物レンズを介して細胞を350nmと385nmの交互励起を調節した(典型的には5秒毎)。510nmより長い波長で出る光を、CCDカメラにより集め、デジタル化し、350nmと385nmの励起イメージからバックグランドの発光を差し引いた後、350/385強度比を計算した。
【0166】
定量的解析のために、実験の各比率イメージについてソフトウェアにより各細胞の12×12ピクセル領域内の350/385平均比を計算し、さらに解析しグラフを描くために表に取り込んだ。フラ−2シグナルを未変性の細胞で較正し、細胞をリンゲル液(10μMイオノマイシン、10mM EGTAを含有し、およびCa++の添加無し)に接触させてRminを得た。次に細胞をリンゲル液(10μMイオノマイシン、10mM Ca++を含有する)に接触させて3回洗浄してRmaxを得た。生きている細胞中のフラ−2のKd250nMとグリンキーウイックツ(Grynkiewicz)ら(J. Biol. Chem.260:3440(1985))を使用して、静止[Ca++]iは典型的には100nMであった。
【0167】
これらの実験で、細胞内cAMPレベルを上昇させる物質にHEK293細胞トランスフェクション体を暴露し、以後の[Ca++]iの変化を追跡した。100μMのフォルスコリン(アデニルシクラーゼのアクチベータ)の添加に応答して、64個の細胞の平均した結果と各細胞で、[Ca++]iにわずかな増加が見られた。1mMのIBMX(cAMPホスホジエステラーゼの阻害剤)の添加後、さらに有意な増加が見られた。対照実験では、64個の非トランスフェクションHEK293細胞中の1つのみが、細胞内cAMPレベルの上昇に対応して、[Ca++]iの増加を示した。この応答は一時的であり、サイクリックヌクレオチドゲートのチャネルDNAでトランスフェクションしたHEK293細胞中で見られた持続性応答とは明らかに異なっていた。
【0168】
これらの結果は、サイクリックヌクレオチドゲートのチャネルを発現するHEK細胞は、活性化された時細胞内サイクリックヌクレオチドレベルに変化を引き起こす受容体(例えば、メタボトロピック受容体)の測定において宿主細胞として使用できることを証明している。
【0169】
5.ノーザンブロットハイブリダイゼーション解析
ヒトメタボトロピック受容体をコードするDNAでトランスフェクションした細胞はまた、ノーザンブロット解析により対応する転写体の発現について解析することができる。ヒトメタボトロピック受容体をコードするDNAでトランスフェクションした約1×107個の細胞から総RNAを単離し、10−15μgのRNAをノーザンハイブリダイゼーション解析に使用した。ヒトメタボトロピック受容体をコードするプラスミドからの挿入体をニックトランスレーションし、プローブとして使用した。ノーザンブロットハイブリダイゼーションの典型的な条件は以下の通りである:
ハイブリダイゼーションは、42℃で5×SSPE、5×デンハルツ(Denhart's)溶液、50%ホルムアルミド中で行ない、洗浄は65℃で0.2×SSPEと0.1%SDS中で行なった。
【0170】
本発明をいくつかの好適な実施態様を参照して詳細に説明したが、ここに記載され特許請求のされている本発明の精神と範囲を逸脱することなくその修飾や変更が可能であることが理解されるであろう。
【0171】
(配列の要約)
配列番号1は、本発明のメタボトロピックグルタミン酸受容体サブタイプ(mGluR1B)をコードするDNAの核酸配列(およびその推定アミノ酸配列)である。
配列番号2は、配列番号1のヌクレオチド配列の推定アミノ酸配列である。
配列番号3は、ヒトmGluR2受容体サブタイプの一部をコードする部分クローンのヌクレオチド配列(および推定アミノ酸配列)である。
配列番号4は、配列番号3のヌクレオチド配列によりコードされるヒトmGluR2受容体サブタイプの一部のアミノ酸配列である。
配列番号5は、本発明のメタボトロピックグルタミン酸受容体サブタイプ(mGluR3)をコードするDNAの核酸配列(および推定アミノ酸配列)である。
配列番号6は、配列番号5のヌクレオチド配列の推定アミノ酸配列である。
配列番号7は、本発明のメタボトロピックグルタミン酸受容体(mGluR5a1)をコードするDNAの核酸配列(および推定アミノ酸配列)である。
配列番号8は、配列番号7のヌクレオチド配列の推定アミノ酸配列である。
配列番号9は、本発明のmGluR5変種メタボトロピックグルタミン酸受容体(mGluR5b)をコードするDNAの核酸配列(および推定アミノ酸配列)である。
配列番号10は、配列番号9のヌクレオチド配列の推定アミノ酸配列である。
配列番号11は、本発明のmGluR5変種メタボトロピックグルタミン酸受容体(mGluR5c)をコードするDNAの核酸配列(および推定アミノ酸配列)である。
配列番号12は、配列番号11のヌクレオチド配列の推定アミノ酸配列である。
配列番号13は、ヒトmGluR2受容体サブタイプの3’非翻訳配列の343ヌクレオチドである。
【0172】
(配列表)
(1)一般情報:
(i) 出願人:ダゲット,ロリー(Daggett, Lorrie)
エリス,スティーブン・ビー(Ellis, Steven B.)
ルー,チェン(Lu, Chen)
ポンツラー,アーロン(Pontsler, Aaron)
ジョンソン,エドウィン・シー(Johnson, Edwin C.)
ヘス,ステファン・ディー(Hess, Stephen D.)
(ii)発明の名称:ヒトメタボトロピックグルタミン酸受容体、これをコードする核酸およびその用途
(iii)配列の数:13
(iv)連絡住所:
(A)宛名:プリティー、シュレーダー、ブルーゲマンおよびクラーク(Pretty, Schroeder, Brueggemann & Clark)
(B)通り:444サウス・フラワー通り、スイート2000(444 South Flower Street, Suite 2000 )
(C)市:ロサンゼルス
(D)州:カリホルニア
(E)国:米国
(F)郵便番号:90071
(v) コンピューターで読める形式:
(A)媒体の型:フロッピー(登録商標)ディスク
(B)コンピューター:IBM PCコンパチブル
(C)オペレーティング・システム:PC−DOS/MS−DOS
(D)ソフトウェア:パテントイン・リリース#1.0(PatentIn Release #1.0)、バージョン#1.25
(vi)現行出願のデータ:
(A)出願番号:
(B)出願日:1994年6月2日
(C)分類:
(vii)先行出願のデータ:
(A)出願番号:US08/072、574
(B)出願日:1993年6月4日
(viii)弁理士/代理人情報:
(A)氏名:ライター,ステファン・イー(Reiter, Stephen E.)
(B)登録番号:31,192
(C)参照/整理番号:FP41 9772
(ix)電話連絡先情報:
(A)電話:619−546−4737
(B)ファックス:619−546−9392
(2)配列番号:1の情報:
(i) 配列の特色:
(A)長さ:3321塩基対
(B)型:核酸
(C)鎖の数:両形態
(D)トポロジー:両形態
(ii)分子の型:cDNA
(ix)配列の特徴:
(A)名称/記号:CDS
(B)存在位置:388..3108
(D)他の情報:/生成物=“ヒトmGluR1B”
(xi)配列:配列番号:1:

【0173】
(2)配列番号:2の情報:
(i) 配列の特色:
(A)長さ:906アミノ酸
(B)型:アミノ酸
(D)トポロジー:直鎖状
(ii)分子の型:蛋白
(xi)配列:配列番号:2:
【0174】
(2)配列番号:3の情報:
(i) 配列の特色:
(A)長さ:355塩基対
(B)型:核酸
(C)鎖の数:両形態
(D)トポロジー:両形態
(ii)分子の型:cDNA
(ix)配列の特徴:
(A)名称/記号:CDS
(B)存在位置:1..354
(D)他の情報:/生成物=“ヒトmGluR2断片”
(xi)配列:配列番号:3:
【0175】
(2)配列番号:4の情報:
(i) 配列の特色:
(A)長さ:118アミノ酸
(B)型:アミノ酸
(D)トポロジー:直鎖状
(ii)分子の型:蛋白
(xi)配列:配列番号:4:
【0176】
(2)配列番号:5の情報:
(i) 配列の特色:
(A)長さ:3919塩基対
(B)型:核酸
(C)鎖の数:両形態
(D)トポロジー:両形態
(ii)分子の型:cDNA
(ix)配列の特徴:
(A)名称/記号:CDS
(B)存在位置:1064..3703
(D)他の情報:/生成物=“ヒトmGluR3”
(xi)配列:配列番号:5:
【0177】
(2)配列番号:6の情報:
(i) 配列の特色:
(A)長さ:879アミノ酸
(B)型:アミノ酸
(D)トポロジー:直鎖状
(ii)分子の型:蛋白
(xi)配列:配列番号:6:
【0178】
(2)配列番号:7の情報:
(i) 配列の特色:
(A)長さ:4085塩基対
(B)型:核酸
(C)鎖の数:両形態
(D)トポロジー:両形態
(ii)分子の型:cDNA
(ix)配列の特徴:
(A)名称/記号:CDS
(B)存在位置:370..3912
(D)他の情報:/生成物=“ヒトmGluR5A”
(xi)配列:配列番号:7:
【0179】
(2)配列番号:8の情報:
(i) 配列の特色:
(A)長さ:1180アミノ酸
(B)型:アミノ酸
(D)トポロジー:直鎖状
(ii)分子の型:蛋白
(xi)配列:配列番号:8:
【0180】
(2)配列番号:9の情報:
(i) 配列の特色:
(A)長さ:4181塩基対
(B)型:核酸
(C)鎖の数:両形態
(D)トポロジー:両形態
(ii)分子の型:cDNA
(ix)配列の特徴:
(A)名称/記号:CDS
(B)存在位置:370..4008
(D)他の情報:/生成物=“ヒトmGluR5B”/注=“ヌクレオチド2998と2999の間に96塩基対が挿入されたmGluR5A変種”
(xi)配列:配列番号:9:
【0181】
(2)配列番号:10の情報:
(i) 配列の特色:
(A)長さ:1212アミノ酸
(B)型:アミノ酸
(D)トポロジー:直鎖状
(ii)分子の型:蛋白
(xi)配列:配列番号:10:
【0182】
(2)配列番号:11の情報:
(i) 配列の特色:
(A)長さ:3282塩基対
(B)型:核酸
(C)鎖の数:両形態
(D)トポロジー:両形態
(ii)分子の型:cDNA
(ix)配列の特徴:
(A)名称/記号:CDS
(B)存在位置:370..3003
(D)他の情報:/生成物=“ヒトmGluR5C”/注=“3’末端が切り取られたmGluR5aの変種”
(xi)配列:配列番号:11:
【0183】
(2)配列番号:12の情報:
(i) 配列の特色:
(A)長さ:877アミノ酸
(B)型:アミノ酸
(D)トポロジー:直鎖状
(ii)分子の型:蛋白
(xi)配列:配列番号:12:
【0184】
(i) 配列の特色:
(A)長さ:343塩基対
(B)型:核酸
(C)鎖の数:両形態
(D)トポロジー:両形態
(ii)分子の型:cDNA
(ix)配列の特徴:
(A)名称/記号:種々の特徴
(B)存在位置:1..343
(D)他の情報:/注=“mGluR2の部分配列−3’非翻訳配列”
(xi)配列:配列番号:13:
【図面の簡単な説明】
【0185】
【図1】図1は、CMVプロモーターをベースにしたベクターであるpCMV−T7−2とpCMV−T7−3を示す。
【技術分野】
【0001】
本発明は、核酸とこれによりコードされる受容体蛋白に関する。本発明の核酸は、新規のヒトメタボトロピック(metabotropic)グルタミン酸受容体サブタイプをコードする核酸に関する。本発明はまたこのような受容体サブタイプの作成方法、およびこのような受容体の機能に影響を与える化合物(例えば、アゴニスト、アンタゴニスト、およびヒトメタボトロピックグルタミン酸受容体のアロステリックモジュレーター)の同定および性状解析のための受容体蛋白の使用方法に関する。
【背景技術】
【0002】
アミノ酸のL−グルタミン酸は、哺乳動物の中枢神経系の主要な興奮性神経伝達物質である。解剖学的、生化学的および電気生理学的分析は、グルタミン酸産生系が広範囲のニューロン作用過程(例えば、急速な興奮性シナプス伝達、神経伝達物質放出の制御、長期増強、学習および記憶、成長性シナプス可塑性、低酸素性虚血傷害およびニューロン細胞の死、てんかん様発作、およびいくつかの神経変性障害の病変性)に関与していることを示唆している。一般的には、モナガン(Monaghan)ら、Ann. Rev. Pharmacol. Toxicol.,29:365−402(1980)を参照。この広範囲の機能(特に学習、神経毒性および神経病理に関する機能)のために、近年グルタミン酸の作用機構を説明し規定しようという研究が始まった。
【0003】
現在グルタミン酸受容体の分類の概要は、薬理学的基準に基づいている。グルタミン酸は、2つの主要な受容体(イオノトロピック(ionotropic)受容体とメタボトロピック(metabotropic)受容体)に分類されている受容体を介してその作用を媒介することが認められている。イオノトロピックグルタミン酸受容体は、完全な陽イオン特異的、リガンドゲート(ligand-gated)のイオンチャネルを含有し、メタボトロピックグルタミン酸受容体は、細胞内第二メッセンジャー系の活性化を介して細胞外シグナルを変換するG蛋白結合受容体である。イオノトロピック受容体は、受容体の薬理学的および機能的性質に基づきさらに少なくとも2つの範疇に分類されている。その2つの主要な型のイオノトロピック受容体は、NMDA(N−メチル−D−アスパラギン酸)受容体とカイニン酸(KA)/AMPA(α−アミノ−3−ヒドロキシ−5−メチル−4−イソキサゾールプロピオン酸)(以前はキスカル酸、すなわちQUISと呼ばれていた)受容体である。メタボトロピック受容体はイオノトロピックグルタミン酸受容体が結合するリガンドと同じリガンドのいくつかに結合するが、メタボトロピック受容体はGTP−結合蛋白および第二メッセンジャー(例えば、サイクリックAMP、サイクリックGMP、ジアシルグリセロール、イノシトール1,4,5−三リン酸およびカルシウム)を介してシナプスの生理機能を変更させる[例えば、グンデルセン(Gundersen)ら、Proc. R. Soc. London Ser. 221:127(1984);スラデツェック(Sladeczek)ら、Nature 317:717(1985);ニコレッティ(Nicoletti)ら、J. Neurosci. 6:1905(1986);スギヤマ(Sugiyama)ら、Nature 325:531(1987)]。
【0004】
メタボトロピックグルタミン酸受容体の電気生理学的および薬理学的性質は、受容体供給源として動物組織や細胞株、および非ヒト組換え受容体を用いて研究されている。ヒトの治療薬開発へのそのような研究の意義は、ヒトでない受容体サブユニットのみしか入手できないことにより限定されている。さらに、メタボトロピックグルタミン酸受容体の特性や構造が分子レベルで研究され出したのはつい最近である。しかしこのような研究は、ヒト以外の種でのみ行われている。メタボトロピックグルタミン酸受容体の生理学的および病理学的重要性の可能性により、グルタミン酸受容体の種々のクラスの代表的メンバーをコードするヒトの配列(すなわち、DNA、RNA、蛋白)が利用(例えば、薬剤スクリーニング測定法のために)できることが必要である。そのようなヒトの配列が入手できれば、ヒトでの受容体の分布の研究、特定の受容体の修飾と種々の疾患の発生の関係の研究も可能になるであろう。
【非特許文献1】グンデルセン(Gundersen)ら、Proc. R. Soc. London Ser. 221:127(1984)
【非特許文献2】スラデツェック(Sladeczek)ら、Nature 317:717(1985)
【非特許文献3】ニコレッティ(Nicoletti)ら、J. Neurosci. 6:1905(1986)
【非特許文献4】スギヤマ(Sugiyama)ら、Nature 325:531(1987)
【発明の開示】
【課題を解決するための手段】
【0005】
(本発明の概要)
本発明は、ヒトメタボトロピックグルタミン酸受容体蛋白サブタイプをコードする新規の核酸、およびこれにコードされる蛋白を開示する。具体的な実施態様において、新規の核酸は、ヒトメタボトロピックグルタミン酸受容体の全長mGluR1、mGluR2、mGluR3およびmGluR5サブタイプまたはその一部をコードする。これらの核酸はメタボトロピックグルタミン酸受容体サブタイプ蛋白の産生に有用である以外に、プローブとしても有用であり、従って当業者は不要な実験をすることなく、関連する受容体サブタイプをコードする核酸を同定し単離することが可能である。
【0006】
本発明は、新規のメタボトロピックグルタミン酸受容体蛋白サブタイプを開示する以外に、このような受容体に影響を与える化合物(例えば、グルタミン酸受容体機能のアゴニスト、アンタゴニスト、およびモジュレーター(活性調節因子))を同定し性状解析するための、このような受容体サブタイプの使用方法を包含する。また本発明は、未知の蛋白がメタボトロピックグルタミン酸受容体サブタイプとして機能するか否かを測定する方法を包含する。
【0007】
(本発明の詳細な説明)
本発明において、ヒトメタボトロピックグルタミン酸受容体サブタイプをコードする単離された核酸が提供される。本発明の1つの面において、mGluR1サブタイプのヒトメタボトロピックグルタミン酸受容体サブタイプをコードする核酸が提供される。別の面において、mGluR2サブタイプのメタボトロピックグルタミン酸受容体の少なくとも1部分をコードする核酸が提供される。さらに別の面において、mGluR3サブタイプのメタボトロピックグルタミン酸受容体をコードする核酸が提供される。さらなる面において、mGluR5サブタイプのメタボトロピックグルタミン酸受容体をコードする核酸が提供される。さらに別の面において、そのような核酸を含有する真核細胞、およびそのような核酸を発現する真核細胞が提供される。
【0008】
また前述の核酸によりコードされる蛋白、および該蛋白に対して産生される抗体が提供される。本発明の別の面において、上記核酸のメタボトロピックグルタミン酸受容体サブタイプ−選択性部分よりなる核酸プローブが提供される。
【0009】
本明細書において、「ヒトメタボトロピックグルタミン酸受容体サブタイプ」とは、グルタミン酸産生リガンドに対する細胞のG−蛋白結合応答に参加する、単離されたおよび/または精製された蛋白を意味する。このような受容体サブタイプは、他のメタボトロピックグルタミン酸受容体サブタイプをコードしない別々の遺伝子により個々にコードされる(すなわち、各サブタイプはユニークな遺伝子によりコードされる)。このような受容体サブタイプの典型的な特徴は、7つの推定されるトランスメンブレンドメインと、その前に大きな推定される細胞外アミノ末端ドメインと、その後に大きな推定される細胞内カルボキシ末端ドメインを有することである。メタボトロピックグルタミン酸受容体は、基本的にメタボトロピックグルタミン酸受容体でない他のG蛋白結合受容体と全くアミノ酸配列相同性を有さない。
【0010】
メタボトロピックグルタミン酸受容体サブタイプの各々の間の関係に関して、mGluR1受容体サブタイプのアミノ酸配列は一般に、他のヒトメタボトロピックグルタミン酸受容体サブタイプのアミノ酸配列との同一性は約70%未満であり、典型的に観察される同一性は45%未満である。mGluR2受容体サブタイプのアミノ酸配列は一般に、他のヒトメタボトロピックグルタミン酸受容体サブタイプのアミノ酸配列との同一性は約60%未満であり、典型的に観察される同一性は45%未満である。mGluR3受容体サブタイプのアミノ酸配列は一般に、他のヒトメタボトロピックグルタミン酸受容体サブタイプのアミノ酸配列との同一性は約60%未満であり、典型的に観察される同一性は45%未満である。mGluR5受容体サブタイプのアミノ酸配列は一般に、他のヒトメタボトロピックグルタミン酸受容体サブタイプのアミノ酸配列との同一性は約70%未満であり、典型的に観察される同一性は45%未満である。
【0011】
上記定義に含まれるものは、一次転写体の別のスプライシングにより産生されるmRNAによりコードされる変種、および上記の生理学的および/または物理的性質の1つまたはそれ以上を有するその断片である。
【0012】
本明細書および請求の範囲において、DNA、RNA、ポリペプチドまたは蛋白の修飾物質としての「単離される」、「純粋な」という用語は、このように呼ばれるDNA、RNA、ポリペプチドまたは蛋白はヒトの手によりそのような形で産生され、従ってその本来のインビボの細胞環境からは分離されていることを示す。このヒトの介入による結果、本発明の組換えDNA、RNA、ポリペプチドおよび蛋白は、天然に存在するDNA、RNA、ポリペプチドまたは蛋白では役に立たないような方法(例えば、選択的薬剤または化合物の同定)において有用である。
【0013】
本発明の受容体蛋白の修飾物質として使用される時、「機能性」とは、グルタミン酸産生関連リガンド(例えばACPDまたはACPD様リガンド、QUIS、AP4など)に受容体蛋白に結合すると、G蛋白との受容体の相互作用を修飾し、これは細胞内第2メッセンジャーのレベルに影響を与え、種々の生理学的作用を引き起こす。言い換えると「機能性」とは、受容体蛋白のアゴニスト活性化の結果として応答が産生されることを意味する。
【0014】
本明細書においてスプライス変種とは、ゲノムDNAの一次転写体の差別的処理により産生され、その結果2つ以上の型のmRNAの産生に至る、変種メタボトロピックグルタミン酸受容体サブタイプをコードする核酸を意味する。差別的に処理された一次転写体から得られるcDNAは、アミノ酸が完全に同一の領域と、異なるアミノ酸配列を有する領域を有する、メタボトロピックグルタミン酸受容体サブタイプをコードするであろう。すなわち同じゲノム配列から、多数の関連するmRNAや蛋白が得られる。得られるmRNAや蛋白はいずれも本明細書では「スプライス変種」と呼ぶ。
【0015】
従って、前記で定義したメタボトロピックグルタミン酸受容体サブタイプをコードするが、遺伝コードの縮重のため特定のハイブリダイゼーション条件下で開示された核酸に必ずしもハイブリダイズしない核酸も、本発明の範囲内にあると考えられる。本明細書に記載の方法または当業者に公知の方法により評価すると、そのようなサブタイプもまた、機能性受容体を形成する。典型的には、メタボトロピックグルタミン酸受容体サブタイプが代替スプライシングにより得られるRNAにコードされない(すなわち、スプライス変種)なら、メタボトロピックグルタミン酸受容体サブタイプをコードする核酸とこれによりコードされるメタボトロピックグルタミン酸受容体蛋白は、本明細書に記載の少なくとも1つのメタボトロピックグルタミン酸受容体サブタイプ核酸(およびこれによりコードされる蛋白)と実質的な配列相同性を有する。スプライス変種をコードするDNAまたはRNAは、本明細書で提供されるDNAまたはRNAとの配列全体の相同性は90%未満であるが、本明細書中のDNA断片とほとんど100%の相同性を有する領域を含有し、開始コドンおよび停止コドンを含むオープンリーディングフレームをコードし、機能性メタボトロピックグルタミン酸受容体サブタイプをコードすることを理解すべきである。
【0016】
ヒトmGluR1サブタイプをコードするDNA配列の例は、配列番号2に記載されるものと実質的に同じアミノ酸配列をコードするヌクレオチドにより示される。現在好適な配列は、配列番号2に示されるアミノ酸配列をコードする。
【0017】
別のDNAの例は、ヒトmGluR1サブタイプをコードし、高緊縮性条件下で配列番号1の実質的に配列全体、またはその大部分(すなわち、典型的には少なくともその25−30個の隣接ヌクレオチド)にハイブリダイズするヌクレオチドであることが特徴である。
【0018】
本明細書において、ハイブリダイゼーションの緊縮性とは、ポリ核酸ハイブリッドが安定である条件を意味する。当業者に公知なように、ハイブリッドの安定性はハイブリッドの融点(Tm)に反映される。
【0019】
Tmは、
式: 81.5℃−16.6(log10[Na+])+0.41(%G+C)−600/l
(式中、lはヌクレオチド中のハイブリッドの長さである)により近似的に表される。
【0020】
配列の相同性が1%減少する毎に、Tmは約1−1.5℃低下する。一般にハイブリッドの安定性は、ナトリウムイオン濃度と温度の関数である。典型的には、ハイブリダイゼーション反応を低緊縮性条件下で行い、次に種々の(ただし、より高い緊縮性の)条件下で洗浄する。ハイブリダイゼーション緊縮性は、このような洗浄条件に関する。
【0021】
すなわち、本明細書において、
(1)断片のハイブリダイゼーションに関して、高緊縮性条件とは、65℃で0.018MのNaCl中で安定なハイブリッドを形成する核酸配列のみのハイブリダイゼーションを可能にする条件を意味する(すなわち、本明細書で企図されるように、もしハイブリッドが65℃で0.018MのNaCl中で安定でない場合、高緊縮性条件下で安定ではないであろう)。高緊縮性条件は、例えば50%ホルムアミド、5×デンハルツ(Denhart's)溶液、5×SSPE、0.2%SDS中で42℃でハイブリダイゼーションし、次に65℃で0.1×SSPE、そして0.1%SDSによる洗浄により提供される。
(2)断片のハイブリダイゼーションに関して、中緊縮性条件とは、42℃で50%ホルムアミド、5×デンハルツ溶液、5×SSPE、0.2%SDS中でハイブリダイゼーションし、次に65℃で0.2×SSPE、そして0.2%SDSによる洗浄と同等の条件を意味する。
(3)断片のハイブリダイゼーションに関して、低緊縮性条件とは、42℃で10%ホルムアミド、5×デンハルツ溶液、6×SSPE、0.2%SDS中でハイブリダイゼーションし、次に50℃で1×SSPE、そして0.2%SDSによる洗浄と同等の条件を意味する。そして、
(4)オリゴヌクレオチド(すなわち、長さが約30ヌクレオチド以下の合成DNA)に関して、高緊縮性条件とは、42℃で10%ホルムアミド、5×デンハルツ溶液、6×SSPE、0.2%SDS中でハイブリダイゼーションし、次に50℃で1×SSPE、そして0.2%SDSによる洗浄に等しい条件を意味する。
【0022】
種々の緩衝液や温度を用いてこれらの条件を再現することができ、これらは必ずしも正確である必要はないことを理解すべきである。
【0023】
デンハルツ溶液およびSSPE(例えば、サムブルーク、フリッチ、およびマニアチス(Sambrook, Fritsch, and Maniatis)、モレキュラークローニング、実験室マニュアル(Molecular Cloning、A Laboratory Manual)、コールド・スプリング・ハーバー・ラボラトリー・プレス(Cold Spring Harbor Laboratory Press)、1989)は、他の適当なハイブリダイゼーション緩衝液と同様に、当業者に公知である。例えば、SSPEはpH7.4のリン酸緩衝化された0.18MのNaClである。SSPEは例えば、175.3gのNaCl、27.6gのNaH2PO4および7.4gのEDTAを800mlの水に溶解しpHを7.4に調整して、次に1リットルになるように水を加えて20×保存溶液として調製することができる。デンハルツ溶液(デンハルト(Denhart)(1966)Biochem. Biophys. Res. Commun.23:641を参照)は、例えば5gのFicoll(タイプ400、ファルマシアLKBバイオテクノロジー社(Pharmacia LKBBiotechnology、INC.)、ピスカタウェイ(Piscataway)、ニュージャージー州)、5gのポリビニルピロリドン、5gのウシ血清アルブミン(第5画分;シグマ(Sigma)、セントルイス、ミズーリ州)、そして水を500mlになるように加え、濾過して粒状物質を除去することにより、50×保存溶液として調製することができる。
【0024】
ヒトmGluR1サブタイプをコードする特に好適な配列は、配列番号1と実質的に同じヌクレオチド配列を有するものであり、配列番号1のコーディング配列と実質的に同じ配列を有するポリ核酸が特に好ましい。
【0025】
本明細書において、「実質的な配列相同性」とは、少なくとも約90%の相同性を有するヌクレオチド配列、そして典型的には95%以上のアミノ酸の同一性を有するヌクレオチド配列を意味する。しかし、スプライス変種であること、または保存的アミノ酸置換(または縮重コドンの置換)により修飾されているため、前記レベルより低い相同性を有する蛋白(およびこのような蛋白をコードするDNAまたはmRNA)は、本発明の範囲内にあることを理解すべきである。
【0026】
本明細書において、「実質的に同じ」とは、本明細書に記載の実際の配列とはわずかに異なるかまたは重大でない配列の変化を有する、DNAのヌクレオチド配列、RNAのリボヌクレオチド配列、または蛋白のアミノ酸配列を意味する。実質的に同じ分子種は、開示された配列と同等であると考えられ、従って本発明の請求の範囲に包含されると考えられる。この点で「わずかに異なるかまたは重大でない配列の変化」は、開示され、特許請求のされているDNA、RNA、または蛋白と実質的に同じ配列が、開示され特許請求のされているヒト由来の配列と機能的に同等であることを意味する。機能的に同等の配列は、実質的に同じ方法で機能して、ヒト由来の核酸および本明細書に開示され特許請求されているアミノ酸組成物と実質的に同じ組成物を産生する。特に機能的に同じDNAは、本明細書に開示されたものと同じであるか、または保存性のアミノ酸変化(例えば非極性残基を別の非極性残基で置換、または荷電残基を類似の荷電残基で置換)を有するものと実質的に同じヒト由来の蛋白をコードする。これらの変化には、当業者に認識される、蛋白の3次元構造を実質的に変化させないものが含まれる。
【0027】
ヒトmGluR2受容体サブタイプの一部をコードするDNA配列の例は、配列番号4に記載のものと実質的に同じアミノ酸配列をコードするヌクレオチド(随時、配列番号13に記載の3’非翻訳配列の343ヌクレオチドの一部またはすべてを含む)、またはクローンMETAB40(1993年5月4日に整理番号75465でアメリカンタイプカルチャーコレクション(ATCC)に寄託された)のヒトmGluR2をコードする部分によりコードされるものと実質的に同じアミノ酸配列をコードするヌクレオチドにより示される。
【0028】
寄託されたクローンは、1993年5月4日に「特許手続上の微生物の寄託の国際的承認に関するブダペスト条約」(the Budapest Treaty on the International Recognition of Deposits of Microorganisms for Purposes of Patent Procedure)の条件と、この条約の名のもとに公布された規則に従い、アメリカンタイプカルチャーコレクション(ATCC)(12301 パークローンドライブ(Parklawn Drive)、ロックヴィル(Rockville)、メリーランド州、アメリカ合衆国 20852)に寄託されている。この条約と規則の条件下で、およびさもなくばアメリカ合衆国の特許法や規則に従い受領する法的資格のある工業所有権事務所および他の個人、または本出願または本出願の優先権を主張する出願が提出されたかまたはこのような出願に対して何らかの特許権が与えられた国または国際組織は、寄託された物質の試料を入手することができる。特にこれに基づきまたはこの優先権を主張する出願またはこれに関する出願を取り込んでいる出願に基づき米国特許の与えられた時点で、本寄託物質の入手に関するすべての規制は永久に取り消されるであろう。
【0029】
ヒトmGluR2受容体サブタイプの一部をコードする好適なポリ核酸配列は、配列番号4に記載のものと同じアミノ酸配列、またはクローンMETAB40(1993年5月4日に整理番号75465でアメリカンタイプカルチャーコレクション(ATCC)に寄託された)のヒトmGluR2をコードする部分によりコードされるものと実質的に同じアミノ酸配列をコードするものである。
【0030】
あるいはDNAの例は、ヒトmGluR2受容体サブタイプをコードするヌクレオチド配列であり、高緊縮性条件下で配列番号3またはその大部分(すなわち、典型的にはその少なくとも25−30個の隣接ヌクレオチド)、またはクローンMETAB40(ATCC整理番号75465)のヒトmGluR2をコードする部分とまたはその大部分とハイブリダイズするヌクレオチド配列として特徴付けられる。ヒトmGluR2受容体サブタイプの部分をコードする特に好適な配列は、配列番号3のコーディング配列と同じヌクレオチド配列、またはクローンMETAB40のヒトmGluR2コーディング部分中のコーディング配列のヌクレオチド配列を有するポリ核酸により示される。
【0031】
ヒトmGluR3受容体サブタイプをコードするDNA配列の例は、配列番号6に記載のアミノ酸配列と実質的に同じアミノ酸配列をコードするヌクレオチドにより示される。好適なポリ核酸配列は、配列番号6と同じ配列をコードするものである。
【0032】
あるいはDNAの例は、ヒトmGluR3受容体サブタイプをコードするヌクレオチド配列であり、高緊縮性条件下で配列番号5またはその大部分(すなわち、典型的にはその少なくとも25−30個の隣接ヌクレオチド)とハイブリダイズするヌクレオチド配列として特徴付けられる。ヒトmGluR3サブタイプの部分をコードする特に好適な配列は、配列番号5のコーディング配列と実質的に同じヌクレオチド配列を有するものであり、配列番号5のコーディング配列と同じヌクレオチド配列を有するポリ核酸が最も好ましい。
【0033】
DNAの例は、ヒトmGluR3受容体サブタイプまたはその部分をコードするヌクレオチド配列であり、配列番号8、10または12のアミノ酸配列と実質的に同じアミノ酸配列をコードするヌクレオチドにより示される。好適なポリ核酸配列は、配列番号8、10または12と同じ配列をコードするものである。
【0034】
あるいはDNAの例は、ヒトmGluR5受容体サブタイプをコードするヌクレオチド配列であり、高緊縮性条件下で配列番号7、9または11の実質的に全配列、またはそれらの大部分(すなわち、典型的には少なくとも25−30個の隣接ヌクレオチド)にハイブリダイズするヌクレオチドとして特徴付けられる。ヒトmGluR5サブタイプをコードする特に好適な配列は、配列番号7、9または11のコーディング配列と実質的に同じヌクレオチド配列を有するものであり、配列番号7、9または11のコーディング配列と同じ配列を有するポリ核酸が最も好ましい。
【0035】
ヒトメタボトロピックグルタミン酸受容体サブタイプをコードするDNAは、本明細書に開示したDNA(配列番号1、3、5、7、9または11の任意のものより得られるヌクレオチドを含む)により、適当なハイブリダイゼーション条件下で適当なヒトcDNAまたはヒトゲノムライブラリーをスクリーニングすることにより単離される。適当なライブラリーは、神経組織試料(例えば、海馬や小脳組織、細胞株など)から調製される。例えばライブラリーは、その実質的に全ての受容体サブタイプコーディング配列を含むDNAの一部を用いてスクリーニングされるか、あるいはそのDNAの一部に基づく適当なオリゴヌクレオチドプローブでスクリーニングされる。
【0036】
本明細書においてプローブは、配列番号1、3、5、7、9または11に記載の任意のものの25またはそれ以上の隣接する塩基と同じであるか少なくとも約25−30個の隣接する塩基(またはその相補物)を含む、ヌクレオチドの配列を有する1本鎖DNAまたはRNAである。プローブを作成するのに好適な領域は、5’および/または3’コーディング配列、トランスメンブレンドメインをコードすると予測される配列、細胞質ループをコードすると予測される配列、シグナル配列、リガンド結合部位などがある。
【0037】
全長cDNAクローン、その断片、またはそのDNAの部分に基づくオリゴヌクレオチドはプローブとして使用され、好ましくは容易に検出するために適当な標識物で標識される。断片がプローブとして使用される時、そのプローブのDNA配列は好ましくはDNAのカルボキシ末端をコードする部分由来であり、最も好ましくはDNA配列の予測されるトランスメンブレンドメインをコードする部分を含む(ドメインは、例えばカイトとドーリトル(Kyte and Doolittle)(1982)、J. Mol. Biol. 第157巻:105の方法を用いて、推定されるアミノ酸配列のハイドロパシー解析に基づき予測される)。これらのプローブは例えば、グルタミン酸受容体ファミリーの追加メンバーの同定と単離のために使用される。
【0038】
本発明の配列の特定の応用として、本発明のヌクレオチド配列をプローブとして使用して遺伝子スクリーニングを実施することができる。すなわち、任意の内因性グルタミン酸受容体に異常が存在するか否かを測定するために、グルタミン酸受容体の任意の1つまたはそれ以上の変化/修飾が関与していることが疑われる神経病理的症状を有する患者の核酸試料を、適当なプローブでスクリーニングすることができる。同様に、グルタミン酸受容体の機能不全に関連する疾患の家族歴がある患者をスクリーニングして、このような疾患に罹りやすい体質であるか否かを決定することができる。
【0039】
本発明の別の実施態様において、ヒトメタボトロピックグルタミン酸受容体蛋白サブタイプをコードするDNAの同定方法が提供され、この方法は、ヒトDNAを上記の核酸プローブと接触させ(使用されるプローブがポリ核酸断片の時は低緊縮性または中緊縮性条件下で接触させ、または使用されるプローブがオリゴヌクレオチドの時は高緊縮性条件下で接触させる)、そして該プローブにハイブリダイズするDNAを同定することよりなる。
【0040】
ライブラリーのスクリーニング後、ハイブリダイゼーションシグナルを検出して陽性クローンを同定する。同定されたクローンを制限酵素マッピングおよび/またはDNA配列解析により性状解析し、次にこれらが完全なメタボトロピックグルタミン酸受容体サブタイプをコードするDNAを含有するか否か(すなわち、これらが翻訳開始コドンおよび停止コドンを含有するか否か)を確認するために本明細書に記載の配列と比較することにより試験する。選択された配列が不完全な場合はこれらを用いて、同じかまたは異なるライブラリーを再スクリーニングして重複するクローンを得ることができる。もしライブラリーがゲノム性の場合は、重複クローンはエクソンとイントロンを含有してもよい。もしライブラリーがcDNAライブラリーの場合は、重複クローンはオープンリーディングフレームを含有するであろう。いずれの場合も完全なクローンは、本明細書で提供されるDNAおよび推定されるアミノ酸配列と比較して同定される。
【0041】
種々のヒトメタボトロピックグルタミン酸受容体サブタイプ(例えば、mGluR1、mGluR2、mGluR3、mGluR5)をコードする、相補的なDNAクローンが単離されている。各サブタイプは、異なる遺伝子にコードされているようである。本明細書に記載されるDNAクローンは、各サブタイプをコードするゲノムクローンを単離し、異なる神経組織から調製されたライブラリーをスクリーニングすることにより、スプライス変種を単離するために使用できる。当業者に公知の核酸増幅法を使用して、ヒトメタボトロピックグルタミン酸受容体サブタイプのスプライス変種をコードするDNAを見つけることができる。これは、公知のまたは予測される互いに異なる配列を取り囲むDNA配列に基づくオリゴヌクレオチドを、ヒトRNAまたはゲノムDNAを増幅するためのプライマーとして用いることにより達成される。増幅生成物のサイズと配列の測定により、スプライス変種の存在が明らかになる。さらにハイブリダイゼーションによるヒトゲノムDNA配列の単離により、ヒトメタボトロピックグルタミン酸受容体サブタイプをコードする転写体の異なるスプライス変種に対応する、イントロンにより分離された多数のエクソンを含有するDNAが得られる。
【0042】
必ずしもすべてのメタボトロピックグルタミン酸受容体サブタイプ(およびその変種)がすべての神経組織または脳のすべての部分に発現されているのではないことは、わかっている。すなわち特定のサブタイプ(またはそのスプライス変種)をコードするcDNAを単離するためには、異なるニューロンまたは神経組織または細胞から調製されるライブラリーをスクリーニングすることが好ましい。各サブユニットをコードするDNAを得るために好適なライブラリーは:ヒトmGluR1をコードするDNAを単離するための小脳;mGluR2をコードするDNAを単離するための海馬;mGluR3をコードするDNAを単離するための海馬と小脳;mGluR5をコードするDNAを単離するための海馬と小脳などがある。
【0043】
特定の受容体サブタイプをコードするDNAが一旦単離されると、そのようなサブタイプ(またはそのスプライス変種)をコードするmRNAを発現する組織を決定するために、リボヌクレアーゼ(RNase)保護測定法が実施される。この測定法は、全細胞性RNAの複雑な混合物中の1つのRNA種を検出し定量するための高感度の手段を与える。サブタイプDNAは標識され、細胞性RNAとハイブリダイズされる。もし細胞性RNA中に相補的なmRNAが存在する場合、DNA−RNAハイブリッドが得られる。次にRNA試料をRNaseで処理する(これは1本鎖RNAを分解する)。RNA−DNAハイブリッドは、RNase分解から保護され、ゲル電気泳動やオートラジオグラフィーにより肉眼で見ることができる。特定のメタボトロピックグルタミン酸受容体サブタイプをコードするmRNAを発現する組織を決定するために、in situ ハイブリダイゼーションを使用することができる。こうして、標識されたサブタイプDNAは異なる脳の領域のスライスとハイブリダイズして、サブタイプmRNA発現が肉眼で見えるようになる。
【0044】
いくつかのヒトのメタボトロピックグルタミン酸受容体サブタイプの発現の分布は、この受容体のラットでの分布とは異なる。例えばラットのmGluR5サブタイプをコードするRNAはラットの海馬に多く存在し、ラットの小脳にはあまり存在しない[例えば、アベ(Abe)ら、J. Biol. Chem.267:13361−13368(1992)を参照]が、ヒトの小脳ライブラリーからは無数のヒトmGluR5をコードするcDNAが得られている。すなわちヒトとラットではいくつかのメタボトロピックグルタミン酸受容体サブタイプの分布が異なっているようである。
【0045】
前述のヌクレオチド配列はベクターに取り込んでさらに操作することができる。本明細書においてベクター(またはプラスミド)とは、異種DNAを細胞に取り込んでそれを発現または複製をさせるのに使用される、独立した成分である。このような担体(vehicle)の選択と使用は、当業者には公知である。
【0046】
発現ベクターとは、DNA断片の発現を制御することができる制御配列(例えばプロモーター領域)に機能的に結合したDNAを発現することができるベクターを含む。すなわち発現ベクターとは、適当な宿主細胞内に取り込まれるとクローン化DNAを発現する、組換えDNAまたはRNA作成体(例えば、プラスミド、ファージ、組換えウイルスまたは他のベクター)を意味する。適当な発現ベクターは当業者に公知であり、真核細胞および/または原核細胞中で複製されるもの、およびエピソームとしてとどまるか宿主細胞ゲノム内に取り込まれるものも含まれる。真核生物の宿主細胞(特に哺乳動物細胞)中での本発明のメタボトロピックグルタミン酸受容体サブタイプの発現のために好適なプラスミドには、例えば、pCMV−T7−2、pCMV−T7−3(図1を参照)、pcDNA1などのサイトメガロウイルス(CMV)プロモーター含有ベクター、そして、SV40プロモーター含有ベクター、およびpMMTVT7(+)またはpMMTVT7(−)(本明細書に記載のように調製されたpMAMneo(クロンテク(Clontek)、パロアルト(Palo Alto)、カリホルニア州)を修飾したもの)などのMMTVLTRプロモーター含有ベクターがある。
【0047】
本明細書において、プロモーター領域とは、そこに機能的に結合したDNAの転写を調節するDNAのセグメントを意味する。プロモーター領域は、RNAポリメラーゼによる認識、結合および転写の開始に充分な特異的配列を含有する。このプロモーター領域の部分をプロモーターと呼ぶ。さらにこのプロモーター領域は、このRNAポリメラーゼの認識、結合および転写開始を調節する配列を含む。これらの配列はcis作用性であるか、またはtrans活性化因子に対して応答する。制御の性質により、プロモーターは構成性(constitutive)であるかまたは制御されている。本発明の実施において使用されるプロモーターの例には、SV40早期プロモーター、サイトメガロウイルス(CMV)プロモーター、マウス乳癌ウイルス(MMTV)ステロイド誘導性プロモーター、モロニー(Moloney)マウス白血病ウイルス(MMLV)プロモーターなどがある。
【0048】
本明細書において、「機能的に結合している」とは、DNAとヌクレオチドの制御配列やエフェクター配列(例えばプロモーター、エンハンサー、転写および翻訳停止部位、および他のシグナル配列)との機能的関係を意味する。例えば、DNAとプロモーターの機能的結合は、DNAの転写はプロモーターを特異的に認識してこれに結合し、DNAを転写するRNAポリメラーゼによりプロモーターから開始されるような、DNAとプロモーターの間の物理的および機能的結合を意味する。発現および/またはインビトロの転写を最適化するために、転写または翻訳のレベルで発現を妨害するかまたは低下させる、不適当な代替翻訳開始コドンまたは他の配列を除去するために、クローンの5’および/または3’非翻訳部分を除去、付加または変更することが必要なこともある。あるいはコンセンサスリボゾーム結合部位(例えば、コザック(Kozak))(1991)J. Biol.Chem. 266:19867−19870)を開始コドンの5’のすぐ近くに挿入して、発現を増強させることもできる。同様に、転写を増強するために、メタボトロピックグルタミン酸受容体サブユニットのコーディング配列の代わりに同じアミノ酸をコードする代替コドンを用いることができる(例えば、宿主細胞のコドンの選択性が採用され、G−Cの豊富なドメインを減少させるなど)。さらに両生類の卵母細胞中のメタボトロピックグルタミン酸受容体サブユニットの発現増強の可能性のために、サブユニットコーディング配列を随時発現作成体中に取り込むことができる(ここではコーディング配列の5’末端と3’末端は、それぞれツメガエル(Xenopus)のβ−グロビン遺伝子の5’と3’の非翻訳配列に隣接している)。例えば、メタボトロピックグルタミン酸受容体サブユニットコーディング配列は、pSP64(プロメガ(Promega)、マジソン、ウィスコンシン州、より入手できる)の修飾型であるベクターpSP64Tに取り組むことができる(クリーグとメルトン(Krieg and Melton)(1984)Nucleic Acids Research 12:7057−7070を参照)。このコーディング配列は、β−グロビン遺伝子の5’末端とSP6プロモーターの下流に位置する3’非翻訳配列の間に挿入される。次に得られるベクターから、インビトロの転写体が作成される。このような修飾の好ましい点(またはニーズ)は実験的に決定されている。
本明細書において、発現とはポリ核酸がmRNAに転写され、ペプチド、ポリペプチドまたは蛋白に翻訳される過程を意味する。ポリ核酸がゲノムDNAから得られる場合、適当な真核生物宿主細胞または微生物が選択されるなら、発現にはmRNAのスプライシングを含む。
【0049】
哺乳動物細胞のトランスフェクションのための、ヒトメタボトロピックグルタミン酸受容体をコードするDNAに結合した制御成分を含有する特に好適な基礎ベクターは、サイトメガロウイルス(CMV)プロモーターをベースにしたベクター(例えば、pCMV−T7−2およびpCMV−T7−3(本明細書に記載)、またはpcDNA1(インビトロゲン(Invitrogen)、サンジエゴ、カリホルニア州))、およびMMTVプロモーターをベースにしたベクター(例えば、pMMTVT7(+)またはpMMTVT7(−)(本明細書中に記載))、およびSV40プロモーターをベースにしたベクター(例えば、pSVβ(クロンテック(Clontech)、パロアルト(Palo Alto)、カリホルニア州))である。
【0050】
ヒトメタボトロピックグルタミン酸受容体サブタイプをコードする全長DNAは、ベクターpMMTVT7(+)、pMMTVT7(−)、pCMV−T7−2またはpCMV−T7−3に挿入されている。pCMV−T7−2(およびpCMV−T7−3)は、CMVプロモーター/エンハンサー、プロモーターのすぐ下流に存在するSV40スプライス/ドナー部位、スプライス部位の下流に位置するT7バクテリオファージRNAポリメラーゼプロモーター、その後にSV40ポリアデニル化シグナル、そしてT7プロモーターとポリアデニル化シグナルの間にポリリンカーを有する、pUC19をベースにした哺乳動物細胞発現ベクターである。CMVプロモーターとSV40ポリアデニル化シグナルの間にメタボトロピックグルタミン酸受容体サブタイプを配置させると、この作成体によりトランスフェクションされた哺乳動物の宿主細胞中で外来DNAの構成的発現(constitutive expression)が与えられる。
【0051】
ベクターpMMTVT7(+)とpMMTVT7(−)はベクターpMAMneo(クロンテック(Clontech)、パロアルト(Palo Alto)、カリホルニア州)を修飾することにより調製された。pMAMneoは、デキサメタゾン誘導性マウス乳癌ウイルス(MMTV)−LTRプロモーターに結合した(この後にSV40スプライス部位とポリアデニル化部位が続く)ラウス肉腫ウイルス(RSV)の長い末端反復配列(LTR)を含有する、哺乳動物の発現ベクターである。pMAMneoはまた、大腸菌の増殖のためのβ−ラクタマーゼ遺伝子(アンピシリン耐性を与える蛋白をコードする)と同様に、形質転換体の選択のための大腸菌neo遺伝子を含有する。
【0052】
ベクターpMMTVT7(+)は、pMAMneoからneo遺伝子を除去し、pBluescript(ストラタジーン(Stratagene)、ラホイア(La Jolla)、カリホルニア州)からのマルチプルクローニング部位とT7およびT3プロモーターを挿入することにより作成される。従ってpMMTVT7(+)はMMTV−LTRプロモーターに結合したRSV−LTRエンハンサー、MMTV−LTRプロモーターの下流に位置するT7バクテリオファージRNAポリメラーゼプロモーター、T7プロモーターの下流に位置するポリリンカー、T7プロモーターの下流に位置するT3バクテリオファージRNAポリメラーゼプロモーター、そしてT3プロモーターの下流に位置するSV40スプライシング部位とポリアデニル化部位を含有する。pMAMneoからのβ−ラクタマーゼ遺伝子(アンピシリン耐性を与える蛋白をコードする)はpMMTVT7(+)中で保持されるが、pMAMneo中の配向に対して配向は逆転している。
【0053】
ベクターpMMTVT7(−)はpMMTVT7(+)と同じであるが、T7プロモーターとT3プロモーターの位置が逆転しており、pMMTVT7(−)中のT3プロモーターはpMMTVT7(+)中のT7プロモーターがある場所に位置しており、pMMTVT7(−)中のT7プロモーターはpMMTVT7(+)中のT3プロモーターがある場所に位置している。従ってpMMTVT7(+)とpMMTVT7(−)ベクターは、哺乳動物宿主細胞中で異種DNA(ここで異種DNAはポリリンカーでベクター内に取り込まれている)が発現するのに必要な制御成分のすべてを含有している。さらにT7プロモーターとT3プロモーターはポリリンカーのどちらかの側に位置しているため、これらのプラスミドはポリリンカーでベクター内にサブクローニングされている異種DNAのインビトロ転写体の合成に使用することができる。
【0054】
哺乳動物細胞中のヒトメタボトロピックグルタミン酸受容体サブタイプをコードするDNAの誘導性発現のために、DNAをpMMTVT7(+)またはpMMTVT7(−)のようなプラスミド中に挿入することができる。これらのプラスミドは、機能的に関連する外来DNAのステロイド誘導性発現のためのマウス乳癌ウイルス(MMTV)LTRプロモーターを含有する。宿主細胞が細胞内への糖質コルチコイド(すなわちMMTVLTRプロモーターの誘導物質)の摂取に必要な内因性糖質コルチコイド受容体を発現しない場合は、糖質コルチコイド受容体をコードするDNA(ATCC整理番号67200)で細胞を追加的にトランスフェクションする必要がある。インビトロ転写体の合成のために、ヒトmGluR1、mGluR3、およびmGluR5をコードする全長ヒトDNAクローンを、pIBI24(インターナショナル・バイオテクノロジーズ社(International Biotechnologies, Inc.)、ニューヘーブン、コネチカット州)、pCMV−T7−2、pCMV−T7−3(図1参照)、pMMTVT7(+)、pMMTVT7(−)、pBluescript(ストラタジーン(Stratagene)、ラホイア(La Jolla)、カリホルニア州)またはpGEM7Z(プロメガ(Promega)、マジソン、ウィスコンシン州)などの中にサブクローニングすることもできる。
【0055】
本発明の別の実施態様において、前述のポリ核酸(すなわちDNAまたはmRNA)を含有する細胞が提供される。細菌、酵母および哺乳動物のような宿主細胞は、DNAを複製しメタボトロピックグルタミン酸受容体サブタイプを産生するために使用することができる。本発明に記載の発現ベクターの作成、インビトロ転写体の調製、哺乳動物細胞へのDNAのトランスフェクション、卵母細胞の注入の方法、および受容体の発現と機能の評価のための電気生理的および他の分析方法は、PCT出願PCT/US91/05625とPCT/US92/11090、および同時係属出願米国特許出願第07/563,751号と07/812,254号にも記載されている。これらの文献の主題はその全体が参考のため本明細書中に引用されている。
【0056】
適当な発現ベクターへのクローン化DNAの導入法、1つのプラスミドベクターまたはプラスミドベクターの組合せ(それぞれが1つまたはそれ以上の異なる遺伝子をコードする)または線状DNAによる真核細胞のトランスフェクション、そしてトランスフェクションした細胞の選択法は公知である(例えば、サムブルーク(Sambrook)ら、(1989)分子クローニング:実験室マニュアル(Molecular Cloning:A Laboratory Manual)、第2版、コールド・スプリング・ハーバー・ラボラトリー・プレス(Cold Spring Harbor Laboratory Press)を参照)。異種DNAは、当業者に公知の任意の方法により宿主細胞に導入される:例えば、CaPO4沈殿法(例えば、ウィグラー(Wigler)ら、(1979)、Proc. Natl. Acad. Sci.76:1373−1376)を用いる、異種DNAをコードするベクターによるトランスフェクション。次に組換え細胞は、DNAによりコードされたサブタイプが発現される条件下で培養することができる。好適な細胞は哺乳動物細胞(例えば、HEK293、CHO、およびLtk−細胞)、酵母細胞(例えば、ピキア・パストリス(Pichia Pastoris)のようなメチル親和性(methilotrophic)酵母細胞)、細菌細胞(例えば大腸菌)などがある。
【0057】
本明細書で提供されるDNAは任意の真核細胞(例えば、ピキア・パストリス(Pichia Pastoris)(米国特許第4,882,279号、4,837,148号、4,929,555号および4,855,231号を参照)、サッカロミセス・セレビッシェ(Saccharomyces cerevisiae)、カンジダ・トロピカリス(Candida tropicalis)、ハンゼヌラ・ポリモルファ(Hansenula polymorpha)などの酵母細胞)中で発現されるが、本明細書で提供されるヒトメタボトロピックグルタミン酸受容体サブタイプをコードするDNAの発現のためには、G蛋白を発現する(内因性にまたは組換えで)市販の発現系や当業者に公知の他の発現系を含む哺乳動物発現系が好ましい。PI加水分解/Ca++シグナル経路に結合したヒトメタボトロピック受容体サブタイプをコードするDNAのインビトロmRNA転写体の発現にはツノガエル(Xenopus)卵母細胞が好適である。卵母細胞中の内因性イノシトール3’末端リン酸第二メッセンジャー介在経路は、これらの細胞におけるヒトメタボトロピック受容体の機能性発現を可能にする。組換えヒトメタボトロピック受容体を発現する卵母細胞は、卵母細胞G蛋白結合IP3生成経路を介してアゴニストに応答し、内部の貯蔵状態からCa++を放出し、電圧固定記録法により遅延振動電流として検出される塩素チャネルを活性化すると報告されている。
【0058】
ヒトメタボトロピック受容体の機能性組換え発現のための宿主細胞は、好ましくは内因性または組換えグアニンヌクレオチド結合蛋白(すなわちG蛋白)を発現する。G蛋白はα、βおよびγサブユニットよりなる高度に保存された膜関連蛋白のファミリーの1つである。αサブユニットはGDPとGTPに結合し、異なるG蛋白毎に異なる。結合した対のβおよびγサブユニットはユニークであることもそうでないこともあり、異なるα鎖が同じβγ対または異なる対に結合している[リンダーとギルマン(Linder and Gilman)、Sci. Am.267:56−65(1992)]。G蛋白α鎖をコードする30以上の異なるcDNAがクローン化されている[サイモン(Simon)ら、Science 252:802(1991)]。4つの異なるβポリペプチドが知られている[サイモン(Simon)ら、Science 252:802(1991)]。同定された5つのγcDNAのうち3つがクローン化されている[ハーレー(Hurley)ら、PNAS U.S.A. 81:6948(1984):ガウタム(Gautam)ら、Science 244:971(1989):ガウタム(Gautam)ら、PNAS U.S.A. 87:7973(1990):]。4番目のγcDNA[クレウス(Kleus)ら、Science 259:832(1993)]と5番目のγcDNA[フィッシャーとアロンソン(Fischer and Aronson)、Mol. Cell. Bio. 12:1585(1992)]の配列は確立されており、さらに追加のγサブユニットが存在するかも知れない[タミール(Tamir)ら、Biochemistry 30:3929(1991)]。グアニンヌクレオチド交換とGTP加水分解により、G蛋白は活性状態と不活性状態の間で変化する。不活性G蛋白は、リガンド活性化受容体により刺激されてGDPをGTPに交換する。活性型では、GTPに結合したαサブユニットはβγ複合体から解離し、次にサブユニットは細胞性エフェクター分子と特異的に相互作用して細胞性応答を引き出す。異なるG蛋白は異なるエフェクター系(例えば、ホスホリパーゼC、アデニルサイクラーゼ系)や異なる受容体と相互作用することができるため、異なる組換えヒトメタ受容体サブタイプの発現のための異なる宿主細胞を研究することは有用である。あるいは宿主細胞は、異なるG蛋白の異種発現のためにG蛋白サブユニットをコードするDNAでトランスフェクションすることができる。
【0059】
好適な実施態様において、ヒトメタボトロピックグルタミン酸受容体サブタイプをコードするDNAは、ベクターに結合され、適当な宿主細胞中に導入されて、特異的なヒトメタボトロピックルグルタミン酸受容体サブタイプまたはサブタイプの特異的な組合せを発現する、形質転換された細胞株を産生する。得られる細胞株は次に、公知の薬剤または可能性のある薬剤の受容体機能への影響の再現性のある定量的解析のために大量に産生される。他の実施態様においては、各サブタイプをコードするDNAのインビトロ転写によりmRNAが産生される。このmRNA(単一のサブタイプクローンからであってもまたはクローンの組合せからであっても)は、ツノガエル(Xenopus)卵母細胞に注入され、ここでmRNAはヒトメタボトロピックグルタミン酸受容体サブタイプの合成を指令する。あるいは機能性ヒトメタボトロピックグルタミン酸受容体サブタイプの発現のために、サブタイプをコードするDNAは卵母細胞に直接注入される。次にトランスフェクションされた哺乳動物細胞または注入された卵母細胞は、本明細書に記載の薬剤スクリーニング法に使用される。
【0060】
DNAまたはRNAが導入される真核細胞は、このようなDNAまたはRNAによりトランスフェクションされることができる細胞、またはこのようなDNAまたはRNAを注入されてG蛋白を(内因性にまたは組換えにより)発現することができる細胞である。好適な細胞は、内因性メタボトロピックグルタミン酸受容体を(あったとしても)ほんの少ししか発現せず、かつ一時的または安定にトランスフェクションされ、また本発明のDNAやRNAを発現するものである。最も好適な細胞は、異種DNAによりコードされる1つまたはそれ以上のサブタイプよりなる組換えまたは異種ヒトメタボトロピックグルタミン酸受容体を形成することができるものである。このような細胞は経験的に同定されるか、または容易にトランスフェクションまたは注入されることが知られているものの中から選択される。
【0061】
DNAを導入するための細胞の例は、哺乳動物起源の細胞(例えば、COS細胞、マウスL細胞、チャイニーズハムスター卵巣(CHO)細胞、ヒト胎児性腎(HEK)細胞、アフリカミドリザル(African green monkey)細胞および当業者に公知の他の細胞)、両生類の細胞(例えば、ゼノプス・ラエビス(Xenopus laevis)卵母細胞)、酵母細胞(例えば、サッカロミセス・セレビッシェ(Saccharomyces cerevisiae)、ピキア・パストリス(Pichia Pastoris))などがある。注入されたRNA転写体を発現するための細胞の例は、ゼノプス・ラエビス(Xenopus laevis)卵母細胞がある。DNAのトランスフェクションに好適な細胞は、当業者に公知であるかまたは経験的に同定され、HEK293細胞(整理番号#CRL1573でATCCより入手できる);Ltk−細胞(整理番号#CCL1.3でATCCより入手できる);COS−7細胞(整理番号#CRL1651でATCCより入手できる);CHO細胞(整理番号#CRL9618、CCL61またはCRL9096でATCCより入手できる);DG44細胞(dhfr−CHO細胞;例えば、ウーラウプ(Urlaub)ら(1986)Cell. Molec. Genet. 12:555を参照);そしてBHK細胞(ウェクターとバセルガ(Waechter and Baserga)、PNAS U.S.A. 79:1106−1110(1982);整理番号#CRL10314でATCCより入手できる)を参照)がある。好適な細胞には、CHO細胞とHEK293細胞があり、特に液体窒素中で凍結され次に融解されて再増殖されるHEK293細胞(例えば、ゴーマン(Gorman)の米国特許第5,024,939号(またスティルマン(Stillman)ら、(1985)Mol. Cell. Biol. 5:2051−2060も参照)に記載のもの)、DG44細胞、LTK−細胞などがある。使用される宿主細胞にかかわらず、使用されるリガンドに誘導されるグルタミン酸産生のバックグランドレベルの測定、またはリガンドの非存在下での宿主細胞中に存在するグルタミン酸のバックグランドレベルの測定のために、比較実験を実施すべきであることは、当業者は理解できるであろう。
【0062】
DNAは当業者に公知の方法を用いて細胞内に安定に取り込まれるか、または一時的に発現される。安定にトランスフェクションされる哺乳動物細胞は、選択マーカー遺伝子(例えば、チミジンキナーゼ、ジヒドロ葉酸還元酵素、ネオマイシン耐性などの遺伝子)を有する発現ベクターで細胞をトランスフェクションし、トランスフェクションした細胞をマーカー遺伝子を発現する細胞に選択的な条件下で増殖させることにより調製される。一時的トランスフェクション体を調製するためには、哺乳動物細胞をレポーター遺伝子(例えば大腸菌β−ガラクトシダーゼ遺伝子)でトランスフェクションしてトランスフェクション効率を追跡する。
【0063】
トランスフェクション体は典型的には選択性条件下では増殖せず、普通トランスフェクション後数日以内に分析されるため、選択マーカー遺伝子は一時的トランスフェクション体には含有されない。
【0064】
このような安定トランスフェクション細胞または一時的トランスフェクション細胞を産生するために、細胞を充分な濃度のサブタイプをコードする核酸でトランスフェクションして、異種DNAにコードされるヒトサブタイプを示すヒトメタボトロピックグルタミン酸受容体を形成させる。サブタイプをコードするDNAの正確な量は経験的に決定されるか、または特定のサブタイプ、細胞および測定条件について最適化される。異種DNAまたはRNAのみにコードされるサブタイプを含有するメタボトロピックグルタミン酸受容体を発現する組換え細胞が特に好適である。
【0065】
異種DNAは細胞中でエピソーム成分として維持されるか、または細胞の染色体DNAに取り込まれる。次に得られる組換え細胞を培養し、この培養物またはサブ培養物からサブ培養(または、哺乳動物細胞の場合は継代する)する。組換え細胞のトランスフェクション、注入および培養法は当業者に公知である。同様に当業者に公知の蛋白精製法を用いて、ヒトメタボトロピックグルタミン酸受容体サブタイプが精製される。例えば1つまたはそれ以上のサブタイプに特異的に結合する抗体または他のリガンドが、ヒトメタボトロピックグルタミン酸受容体の親和性精製に使用される。
【0066】
本明細書において、異種DNAまたは外来DNAおよびRNAは、互いに交換して使用することができ、細胞のゲノムの一部としては天然には存在しないか、または天然に存在するものとは異なるゲノム中の位置に見いだされるDNAまたはRNAを意味する。典型的には異種または外来DNAおよびRNAは、宿主細胞に内因性ではなく人工的に細胞に取り込まれたDNAまたはRNAを意味する。異種DNAの例としては、ヒトメタボトロピックグルタミン酸受容体サブタイプをコードするDNA、転写、翻訳または他の制御可能な生化学過程などに影響を与えることにより内因性DNAの発現を媒介または変更するRNAまたは蛋白をコードするDNAなどがある。異種DNAを発現する細胞は、同じであるかまたは異なる発現生成物をコードするDNAを含有する。異種DNAは発現される必要はなく、宿主細胞ゲノム中に取り込まれるかまたはエピソーム的に維持される。
【0067】
機能性mGluRの発現を検出するために使用する種々の測定法を当業者は容易に見いだすことができる。その例としては、PIターンオーバー測定法[例えば、ナカジマ(Nakajima)ら、J. Biol. Chem.267:2437−2442(1992)および実施例3.C.2を参照]、cAMP測定法[例えば、ナカジマ(Nakajima)ら、前述、および実施例3.C.4を参照]、カルシウムイオン流入測定法[例えば、イトウ(Ito)ら、J. Neurochem. 56:531−540(1991)、および実施例3.C.1を参照]、cGMP測定法[例えば、スタイナー(Steiner)ら、J. Biol. Chem.247:1106−1113(1972)]、アラキドン酸放出測定法[例えば、フェルダー(Felder)ら、J. Biol.Chem.264:20356−20362(1989)を参照]などがある。さらに、陽イオンをベースにした測定法(本明細書に記載)を、細胞内サイクリックヌクレオチドレベルの受容体誘導変化を追跡するために使用することができる。そのような測定法は、サイクリックヌクレオチドゲートのイオンチャネルを発現する宿主細胞を使用する。例えば棹状光受容体細胞、嗅覚細胞およびウシ腎細胞中に存在するこれらのチャネル(例えば、カウプ(Kaupp)ら、Nature 342:762−766(1989)、ダレン(Dallen)ら、Nature 347:184−187(1990)、およびビエル(Biel)ら、Proc. Natl. Acad. Sci. USA 91:3505−3509(1994))は、cAMPまたはcGMPの結合による活性化により陽イオンに対して透過性となる。すなわち本発明の測定法において、内因性または組換えサイクリックヌクレオチドゲートのチャネルを発現する宿主細胞は、サイクリックヌクレオチドレベルに影響を与えることが疑われる受容体をコードする核酸(例えば、メタボトロピックグルタミン酸受容体をコードするDNA)でトランスフェクション(または注入)され、次にチャネルのサイクリックヌクレオチド活性化の量の変化を追跡する。チャネルのサイクリックヌクレオチドの活性化の変化を追跡すると、活性化された時cAMPまたはcGMPの変化を引き起こす受容体を機能性であるとして間接的に同定することができる。サイクリックヌクレオチドゲートのチャネルの活性化の量の変化は、チャネルを介するイオンの流入を測定するか、または電流の電気生理学的測定または細胞内陽イオンレベルの変化を測定(例えば、細胞内カルシウムの蛍光測定)することにより、決定することができる。
【0068】
活性化によりサイクリックヌクレオチドの低下を引き起こす受容体(例えば、メタボトロピックグルタミン酸受容体)を発現する細胞の測定法において、受容体活性化化合物を測定物内の細胞に添加する前に、サイクリックヌクレオチドの細胞内レベルを上昇させる物質(例えば、フォルスコリンやIBMX)に細胞を暴露させることが好ましい。
【0069】
上記の測定法での使用に適した宿主細胞は、検討される受容体の発現に適した宿主細胞を含む(例えば、メタボトロピックグルタミン酸受容体の測定のためのL細胞、HEK293細胞、CHO細胞またはツノガエル(Xenopus)卵母細胞)。細胞は、サイクリックヌクレオチドゲートのチャネルをコードする核酸と受容体をコードする核酸で連続的にトランスフェクション(または注入)されるか、または細胞は2つの核酸で同時トランスフェクションされる。実施例3Aと3Bに記載の一時的または安定なトランスフェクションを実施することができる。
【0070】
サイクリックヌクレオチドゲートのチャネルでトランスフェクション(または注入)された細胞は、機能の測定の前にインキュベートされる(典型的には約24−48時間)。チャネルの活性は、トランスフェクションされた細胞からの引き剥がした裏返した膜パッチを用いて測定される(こうして細胞質面に達するcAMPの濃度が調節される)。トランスフェクション体はまた、内部カルシウムレベル([Ca++]i)の単細胞ビデオイメージングにより分析される。この方法は、細胞内カルシウムレベル(これはチャネルのサイクリックヌクレオチド活性化により制御される、チャネルを介するカルシウムの流入量とともに変化する)の測定によるサイクリックヌクレオチドゲートのチャネルの分析を可能にする。イメジーング測定法は、基本的に実施例3.C.4.bに記載のように実施される。
【0071】
本明細書に記載のDNA、mRNA、ベクター、受容体サブタイプおよび細胞により、選択されたメタボトロピックグルタミン酸受容体サブタイプやその受容体サブタイプに対する抗体の産生が可能である。これは、その存在が単一のメタボトロピックグルタミン酸受容体サブタイプの分析を妨害する多くの他の受容体蛋白の汚染が実質的にない、合成または組換え受容体および受容体サブタイプを調製する手段を与える。目的の受容体サブタイプが利用できることが、特定の受容体サブタイプまたはメタボトロピックグルタミン酸受容体サブタイプの組合せへの薬剤の影響を観察し、従ってヒトに特異的でありヒトメタボトロピックグルタミン酸受容体サブタイプまたはメタボトロピックグルタミン酸受容体サブタイプの組合せに特異的な試験系で、薬剤の初期のインビトロスクリーニングを行うことを可能にする。特異的抗体が利用できることは、インビボで発現されるサブタイプの組合せを同定することを可能にする。すなわち、このような特異的組合せは、薬剤スクリーニングの好適な標的として使用される。
【0072】
特異的な受容体組成物への薬剤の影響を測定するためにインビトロで薬剤物質をスクリーニングすることができることは、受容体サブタイプ特異的または疾患特異的薬剤の開発とスクリーニングを可能にするはずである。また単一の受容体サブタイプまたは種々の受容体サブタイプと種々のアゴニストまたはアンタゴニストの可能性のある物質との特定の組合せを試験することにより、個々のサブタイプの機能と活性に関する追加情報が得られ、1つまたはそれ以上の受容体サブタイプと非常に特異的に相互作用することができる化合物の同定と設計を可能にするであろう。得られる薬剤は、種々の受容体サブタイプを発現する細胞を用いてスクリーニングすることにより同定される薬剤より、不要な副作用はより少ないであろう。
【0073】
さらに種々の疾患の薬剤開発と治療法に関して、ヒトメタボトロピックグルタミン酸受容体サブタイプをコードするDNAが利用できることは、ある種の疾患の発症に相関する遺伝子の変化(例えば、突然変異)の同定を可能にする。さらに、このような突然変異を合成DNA配列に特異的に導入し、次にこれを実験室動物に導入するかまたはその効果を測定するためのインビトロ測定系により、そのような疾患の動物モデルの作成が可能になる。
【0074】
別の面において、本発明は本発明のDNAによりコードされる機能性ペプチド断片およびその機能性組合せよりなる。ペプチドがグルタミン酸受容体として機能するのに必須ではない配列中のアミノ酸の一部またはすべてを排除することにより、不要な実験をすることなく当業者はこのような機能性ペプチド断片を産生することができる。グルタミン酸受容体機能に必須のアミノ酸は、例えばペプチドをコードするDNAの体系的消化および/またはDNAへの欠失の導入により決定することができる。修飾(例えば欠失または消化)DNAは、例えばDNAを転写し、次に得られるmRNAをツノガエル(Xenopus)卵母細胞に導入して、mRNAを翻訳させることにより発現することができる。卵母細胞中のこうして発現された蛋白の機能的分析は、グルタミン酸受容体に結合しこれを機能的に活性化することが公知のリガンドに卵母細胞を暴露して、次に内因性チャネルが活性化されるか否かについて卵母細胞を追跡することにより、行われる。電流が検出されるなら、断片はグルタミン酸受容体として機能する。
【0075】
本発明の別の実施態様において、競合的結合測定法で本発明の受容体蛋白を使用することよりなる、メタボトロピックグルタミン酸受容体サブタイプに結合する化合物の同定方法が提供される。この測定法は多数の化合物を迅速にスクリーニングして、どの化合物(もし、あるとすれば)が、特異的に結合した[3H]グルタミン酸を排除することができる(すなわち、メタボトロピックグルタミン酸受容体に結合することができる)かを調べることができる。結合することが見いだされた化合物を用いて、このような化合物が本発明の受容体のモジュレーター、アゴニストまたはアンタゴニストとして作用するのか否かをさらに調べるために、さらに詳しい測定法が実施される。
【0076】
本発明の結合測定法の別の応用は、本発明の受容体の存在の有無に関する試料(例えば生物学的液体)の測定である。例えばグルタミン酸産生経路の機能不全に関連する症状を示す患者の血清を測定して、観察される症状がこのような受容体の型の産生過剰または不足により引き起こされるのかを決めることができる。
【0077】
本発明において企図される結合測定法は、当業者は容易に判定できるように種種の方法で実施することができる。例えばラジオリセプター測定法などの競合的結合測定法を使用することができる。
【0078】
本発明のさらなる実施態様において、本発明のヒトメタボトロピックグルタミン酸受容体サブタイプの活性を調節する化合物を同定するための生物測定法が提供され、該方法は:
(a)ヒトメタボトロピックグルタミン酸受容体サブタイプをコードするDNAを含有する細胞(この細胞は機能性メタボトロピックグルタミン酸受容体を発現する)を、受容体の活性の調節能力を測定すべき少なくとも1つの化合物に暴露させて、
(b)第二メッセンジャー活性の変化について細胞を追跡する
ことよりなる。
【0079】
上記の生物測定法はヒトメタボトロピックグルタミン酸受容体のアゴニストやアンタゴニストおよびアロステリックモジュレーターの同定を可能にする。この方法では組換えメタボトロピックグルタミン酸受容体に「未知の」すなわち試験物質と(アンタゴニスト活性が試験される時は、さらに公知のメタボトロピックグルタミン酸アゴニストの存在下で)接触させ、この「未知の」すなわち試験物質との接触後、公知のグルタミン酸受容体の第二メッセンジャー活性を追跡し、公知のグルタミン酸受容体の活性を追跡し、公知のグルタミン酸受容体の第二メッセンジャー応答を増加または減少させる物質は、ヒトメタボトロピックグルタミン酸受容体の機能性リガンド(すなわち、モジュレーター、アゴニストまたはアンタゴニスト)として同定される。追跡することができる第二メッセンジャー活性には、細胞内カルシウムイオンの濃度、IP3、cAMPレベルの変化、またはアラキドン酸放出またはイオン電流の活性または阻害の追跡(細胞が卵母細胞の場合)がある。
【0080】
本発明の特定の実施態様において、組換えヒトメタボトロピックグルタミン酸受容体を発現する哺乳動物細胞または卵母細胞を試験化合物と接触させて、次に試験化合物がある場合またはない場合のメタボトロピックグルタミン酸受容体介在応答を比較し、化合物の存在に対する試験細胞または対照細胞(すなわち、メタボトロピックグルタミン酸受容体を発現しない細胞)のメタボトロピックグルタミン酸受容体介在応答を比較して、その調節作用を評価することができる。
【0081】
本明細書において、「メタボトロピックグルタミン酸受容体の活性を調節する」化合物またはシグナルとは、メタボトロピックグルタミン酸受容体の活性を変化させ、化合物またはシグナルの存在下ではこれらが存在しない場合とはメタボトロピックグルタミン酸受容体の活性が異なるようになる化合物またはシグナルを意味する。特にこのような化合物またはシグナルはアゴニストやアンタゴニストを含む。アゴニストという用語は、受容体機能を活性化するグルタミン酸またはACPDのような物質またはシグナルを意味し;アンタゴニストという用語は、アゴニストに誘導される受容体活性化を妨害する物質を意味する。アンタゴニストには競合的および非競合的アンタゴニストがある。競合的アンタゴニスト(または競合的ブロッカー)は、アゴニスト(例えば、リガンドまたは神経伝達物質)の近傍または特異的な部位と、その同じ部位または近傍に対して相互作用する。非競合的アンタゴニストまたはブロッカーは、アゴニストと相互作用する部位とは異なる部位と相互作用することにより、受容体の機能を不活性化する。
【0082】
当業者は理解できるように、ヒトメタボトロピックグルタミン酸受容体活性を調節する化合物(例えば、アゴニストおよびアンタゴニスト)の同定方法には、一般に対照との比較が必要である。「対照」細胞または「対照」培養物の1つのタイプは、試験化合物に暴露された細胞または培養物と実質的に同様に処理された細胞または培養物である(ただし、対照培養物は試験化合物に暴露されない)。例えば、電圧固定電気生理学的方法を用いる方法では、細胞を浸漬している外部溶液を単に交換することにより、試験化合物の存在下または非存在下で同じ細胞を試験することができる。別のタイプの「対照」細胞または「対照」培養物は、トランスフェクションされた細胞と同一の細胞または細胞の培養物である(ただし、対照培養物に使用される細胞は、トランスフェクションされた細胞に発現される組換えヒトメタボトロピックグルタミン酸受容体サブタイプを発現しない)。この場合、細胞または各タイプの細胞の培養物が、測定される化合物の存在下と実質的に同じ反応条件下に暴露される時、試験化合物に対する試験細胞の応答は、受容体のない(対照)細胞の試験化合物に対する応答(または応答の欠如)と比較される。
【0083】
本発明のさらに別の実施態様において、ヒトメタボトロピックグルタミン酸受容体の第二メッセンジャー活性は、この受容体を、上記生物測定法により同定される少なくとも1つの化合物の有効量と接触させることにより調節される。
【0084】
本発明のさらに別の実施態様において、上記の受容体蛋白に対して産生された抗体が提供される。この抗体は、受容体組織局在化、サブタイプ組成、機能性ドメインの構造の研究用、受容体の精製、および診断応用治療応用などに使用することができる。好ましくは治療用途では、使用される抗体はモノクローナル抗体である。
【0085】
本発明の受容体蛋白またはその部分を抗体産生の抗原として用いることにより、上記の抗体は当業者に公知の標準的方法を使用して産生することができる。抗ペプチドおよび抗融合蛋白抗体ともに使用することができる[例えば、バホウト(Bahouth)ら、(1991)Trends Parmacol Sci. 第12巻:338−343;Current Protocols in Molecular Biology(アウスベル(Ausubel)ら編、ジョン・ワイリー・アンド・サンズ(John Wiley and Sons)、ニューヨーク(1989)]。免疫原として(合成ペプチドまたは組換え産生細菌融合蛋白として)使用するためのメタボトロピックグルタミン酸受容体サブタイプの部分を選択する場合に考慮すべき因子は、抗原性、入手し易さ(すなわち、細胞外ドメインおよび細胞質ドメイン)、特定のサブタイプに特異的であるかなどである。
【0086】
サブタイプ特異的抗体が利用できることは、(例えば、正常の脳組織対疾患の脳組織中の)種々のサブタイプの分布と発現密度を追跡するのに免疫組織学的方法を使用することを可能にする。このような抗体はまた、診断および治療応用に利用できる。
【0087】
本発明のさらに別の実施態様において、有効量の上記の抗体に本発明の受容体を接触させることにより、該受容体のイオンチャネル活性を調節する方法が提供される。
【0088】
本発明の抗体は、標準的方法(例えば、腹腔内投与、筋肉内投与、静脈内投与、または皮下注射、移植または経皮的投与法など)により、対象者に投与することができる。当業者は、使用される投与法により、投与型、治療処方などを容易に決定することができる。
【0089】
本発明のさらなる実施態様において、細胞内サイクリックヌクレオチドレベルの受容体に誘導される変化を追跡するための、陽イオンベースの生物測定法が提供され、該生物測定法は:
内因性または組換えサイクリックヌクレオチドゲートのチャネルを発現する宿主細胞に、細胞内サイクリックヌクレオチドレベルに影響を与えることが疑われる受容体をコードする核酸を導入し、そして
細胞内サイクリックヌクレオチドレベルに影響を与えることが疑われる、該受容体に対するリガンドの存在下または非存在下で、サイクリックヌクレオチドゲートのチャネルのサイクリックヌクレオチド活性化の量の変化を追跡することよりなる。
【0090】
以下の非限定的実施例を参照して本発明をさらに詳細に説明する。
【実施例1】
【0091】
ヒトメタボトロピックグルタミン酸受容体をコードするDNAの単離
A.mGluR5受容体cDNA
cDNAライブラリースクリーニング
【0092】
ヒト海馬組織から単離したRNAを、標準的方法[例えば、グブラーとホフマン(Gubler and Hoffman)、(1983)Gene 25:263−269]に従い、オリゴdT−プライムした1本鎖cDNAの合成のための鋳型として使用した。1本鎖cDNAを2本鎖cDNAに変換し、そのcDNAの末端にEcoRI/SnaBI/XhoIアダプターを付け加えた。アガロースゲル電気泳動を用いてサイズによりcDNAを分離し、>2.5kbのものをEcoRI消化したλgt10バクテリオファージベクターに結合させた。得られる一次ヒト海馬cDNAライブラリー(約2×105組換え体)を、ラットmGluR1受容体(ヌクレオチド1から1723+5’非翻訳配列;マス(Masu)ら、(1991)Nature 349:760−765を参照)をコードするDNAの断片に対するハイブリダイゼーションについてスクリーニングした。ハイブリダイゼーションは、42℃で5×SSPE、5×デンハルツ(Denhart's)溶液、50%ホルムアミド、0.2%SDSおよび200μg/mlの変性した超音波処理したニシン精子DNA中で行ない、洗浄は65℃で1×SSPEと0.2%SDS中で行なった。3273塩基対を含有するハイブリダイズする1つのプラーク(METAB1)を同定した。
【0093】
追加のヒトmGluR5コーディングクローンを得るために、METAB1を放射標識し、以下のように調製した2つのヒト小脳cDNAライブラリーをスクリーニングするのに使用した。ランダムプライマーを使用して、ヒト小脳組織から単離したRNAから最初のcDNA合成をプライムしてcDNAを合成した。長さを基準にしてcDNAをプールし、2つのライブラリーを作成した:1つは2.8kb以上の長さの挿入体を有し(すなわち、大挿入体ライブラリー)、もう1つは1−2.8kbの長さの挿入体を有する(すなわち、中挿入体ライブラリー)。ラットmGluR1DNA断片へのハイブリダイゼーションに対して海馬ライブラリーをスクリーニングするために使用したのと同じハイブリダイゼーション条件を用いて、METAB1に対するハイブリダイゼーションについてライブラリーをスクリーニングした。洗浄は55℃で1×SSPE、0.2%SDS中で行なった。大挿入体ライブラリー中に1つのハイブリダイズするプラーク(METAB2)を同定し、中挿入体ライブラリー中に4つのハイブリダイズするプラーク(METAB3−METAB6)を同定した。
【0094】
ヒトmGluR5をコードするDNAの別のスクリーニングで、METAB1により大挿入体または中挿入体小脳ライブラリーをスクリーニングするのに使用した条件と同じ条件を用いて、サイズが1−2kbの範囲の挿入体を含有するランダムにプライムしたヒト海馬cDNAライブラリー(2×106組換え体)および中挿入体小脳cDNAライブラリーを、放射能標識したMETAB5へのハイブリダイゼーションについてスクリーニングした。海馬ライブラリーに3つのハイブリダイズするプラーク(METAB10−METAB12)が同定され、小脳ライブラリーの別の一次スクリーニングで5つの追加のハイブリダイズするプラーク(METAB13−METAB17)が同定された。選択されたプラークを精製した。
【0095】
単離されたクローンの性状解析
制限酵素マッピングとDNA配列解析による精製したプラークの挿入体の性状解析から、単離されたクローンによりヒトmGluR5転写体の少なくとも3つの明らかなスプライス変種が表されていることが判明した。METAB1の分析により、これは翻訳開始コドンを含有するが翻訳停止コドンは含有しないことが示された。推測されるアミノ酸配列は、ラットのmGluR1の推測されるアミノ酸配列に約70%同一であり、ラットのmGluR5の推測されるアミノ酸配列とは>90%の同一性を有している[アベ(Abe)ら、(1992)J. Biol. Bhem. 267:13361−13368]。
【0096】
METAB5のDNA配列解析は、METAB5は5’末端でMETAB1の3’末端と重複し、3’方向へ追加の343ヌクレオチドが続いていることを示した。METAB1とMETAB5の重複領域を比較すると、METAB1はMETAB5に存在しない96ヌクレオチドを有していた(すなわち、METAB1はMETAB5に対して96ヌクレオチド挿入を含有する)。METAB5はまた、翻訳停止コドンを持っていない。METAB12挿入体は5’末端でMETAB5の3’末端と重複するが、さらに3’末端方向に延長して翻訳停止コドンを有する。
【0097】
METAB2のDNA配列解析により、5’末端の最初の869ヌクレオチドはMETAB1の3’末端の一部と重複し、同じであった。しかし、METAB1とMETAB2の配列はMETAB1の96ヌクレオチド挿入の最初で分岐していた。METAB2は3’末端方向に約2700ヌクレオチド延長し、METAB1との分岐点の4ヌクレオチドだけ3’方向に推定翻訳停止コドンを有する。
【0098】
METAB4の部分的DNA配列解析は、これが別のヒトメタボトロピック受容体であるmGluR1の一部を含有することを示していた(実施例1.B.を参照)。
【0099】
全長mGluR5 cDNA作成体の調製
cDNAのインビトロ転写体の調製および/または哺乳動物細胞でのcDNAの発現に使用するために、ヒトmGluR5転写体の3つの推定スプライス変種を表す全長作成体(mGluR5a、mGluR5bそしてmGluR5cと呼ぶ)を作成し、発現ベクター中に取り込むことができる。典型的に使用される基礎となる発現ベクターは、pCMV−T7−3またはpCMV−T7−2である(図1を参照)。プラスミドpCMV−T7−3はpUC19ベースのベクターであり、CMV(CMV)プロモーター/エンハンサー、プロモーターのすぐ下流に位置するSV40スプライスドナー/スプライスアクセプター部位、SV40スプライス部位のすぐ下流に位置するT7バクテリオファージRNAポリメラーゼプロモーター、T7プロモーターの下流のSV40ポリアデニル化シグナル、およびT7プロモーターとポリアデニル化シグナルの間のポリリンカーを含有する。すなわちこのベクターは、哺乳動物宿主細胞中での異種DNAの発現に必要なすべての制御成分を含有する(異種DNAはポリリンカーでベクターに取り込まれる)。さらにT7プロモーターはポリリンカーのすぐ上流に位置するため、このプラスミドは、ポリリンカーでベクターにサブクローニングされた異種DNAのインビトロ転写体の合成に使用される。pCMV−T7−3とpCMV−T7−2は、ポリリンカー中の制限酵素部位の配向のみが異なる。
【0100】
全長mGluR5a作成体(配列番号7を参照)を調製するために、クローンMETAB1、METAB5およびMETAB12の部分を一緒に結合させた。まず操作を容易にするために、METAB1、METAB5およびMETAB12の挿入体をEcoRI断片としてλgt10から別々に、EcoRI消化pGEM−7Zf(プロメガ(Promega)、マジソン、ウィスコンシン州)に移した。METAB1挿入体を含有するpGEM−7ZfベクターをScaI/NheIで消化して、アンピシリン耐性遺伝子の5’半分とMETAB1挿入体の5’部分(配列番号7のヌクレオチド1−2724)を含有する3.8kb断片を放出させた。METAB5の挿入体を含有するpGEM−7ZfベクターをScaI/NheIで消化して、アンピシリン耐性遺伝子の3’半分とMETAB5挿入体の3’部分(配列番号7のヌクレオチド2725−3469)を含有する2.6kb断片を放出させ、この断片を、METAB1を含有するpGEM−7Zfベクターからの3.8kb断片に結合させてpGEM−METAB1+5を作成した。pGEM−METAB1+5をScaI/NheIで消化して、アンピシリン耐性遺伝子の5’半分と配列番号7のヌクレオチド1−3316を含有する4.4kb断片を放出させた。次にこの4.4kb断片を、METAB12挿入体を含有するpGEM−7ZfベクターのScaI/NheI(部分)消化により得られた2.6kb断片[この2.6kb断片はアンピシリン耐性遺伝子の3’半分とMETAB12の3’部分(配列番号7のヌクレオチド3317−4085)を含有する]に結合させた。得られたベクターはpGEM−7Zf中に完全なmGluR5aコーディング配列を含有していた。全長mGluR5a cDNAを、Aat II(平滑末端)−Hind III断片としてベクターから単離し、NotI(平滑末端)/Hind III消化pCMV−T7−3にサブクローニングして、作成体mGluR5a1を作成した。
【0101】
要約すると、作成体mGluR5a1は、METAB1からの5’非翻訳配列の369塩基対(配列番号7のヌクレオチド1−369)と、mGluR5受容体のmGluR5a変種の完全なコーディング配列(配列番号7のヌクレオチド370−3912)、および3’非翻訳配列の173塩基対(配列番号7のヌクレオチド3913−4085)を含有する。mGluR5aをコードする配列は、哺乳動物宿主細胞中で受容体を発現するために、そしてツノガエル(Xenopus)卵母細胞で発現されるDNAのインビトロ転写体を作成するのに使用するために、pCMV−T7−3中の制御成分に機能的に結合している。
【0102】
2つの追加のmGluR5a作成体(mGluR5a2とmGluR5a3)を、最初のmGluR5a作成体の5’非翻訳領域を修飾して調製した。上記のmGluR5a作成体は、提唱されている翻訳開始コドン(配列番号7のヌクレオチド370から372)に先行する5’非翻訳領域中に7つのおそらく不適切なATGコドンを含有する。mGluR5a1作成体をBAl31で消化して、以下を行う:(1)配列の255ヌクレオチド(配列番号7のヌクレオチド1−255であり、上流のATGトリプレット7つのうち6つを含有する)を除去して、mGluR5a2を作成する、そして(2)配列の348ヌクレオチド(配列番号7のヌクレオチド1−348であり、上流のATGトリプレットのすべてを含有する)を除去して、mGluR5a3を作成する。すなわち、mGluR5a2は、5’非翻訳配列の一部が欠如していること以外はmGluR5a1と同じであり、従って提唱される翻訳開始コドンの上流に1つのみのATGトリプレットを含有する。同様に、mGluR5a3は、提唱される翻訳開始コドンの上流のすべてのATGトリプレットが欠如していること以外はmGluR5a1と同じであり、5’非翻訳配列の21ヌクレオチドのみを含有する。
【0103】
3つ目のmGluR5a作成体(MMTV−hmGluR5a)は、以下のようにmGluR5aのMMTVプロモーター制御発現に使用するために調製した。mGluR5a3をXbaIで消化した。SV40スプライス部位、全長mGluR5aコーディング配列(および5’非翻訳配列の21ヌクレオチドと3’非翻訳配列の173ヌクレオチド)、およびポリアデニル化シグナルを含有する4.1kb断片を単離し、平滑末端とし、pBR322の2kbのEcoRI−NdeI(平滑末端)断片に結合させて、pBR−hmGluR5を作成した。MMTVLTRプロモーターとアンピシリンおよびネオマイシン耐性遺伝子を含有するベクターpMAMneo(クロンテック(Clontech)、パロアルト(Palo Alto)、カリホルニア州)をBamHIで消化して、ネオマイシン耐性遺伝子を除去し、再結合させた。次にこのベクターをEcoRIで消化し、アンピシリン耐性遺伝子を含有する断片を逆配向で大きい方のベクター断片に再結合させてpMAMneo ampoppを作成した。このベクターをPstI/NheIで消化して、アンピシリン耐性遺伝子の5’部分とMMTV−LTRを含有する2.3kbの断片を単離した。プラスミドpBR−hmGluR5をPstI/XbaIで消化し、アンピシリン耐性遺伝子の3’部分とmGluR5a配列(SV40スプライス部位とポリアデニル化シグナルとともに)を含有する5.3kbの断片を、pMAMneo ampoppの2.3kbのPst/NheI断片に結合させてMMTV−hmGluR5aを作成した。
【0104】
すなわち、pMMTV−hmGluR5aは、MMTV−LTRを含有し、mGluR5a DNA(配列番号7のヌクレオチド349−4085を含有する)と機能的に結合したSV40スプライス部位が続き、この後にポリアデニル化シグナルが続く。
【0105】
4つ目のmGluR5a作成体(pSV−hmGluR5)は、以下のようにmGluR5aのSV40プロモーター制御発現に使用するために調製した。mGluR5a3をXhoIで部分的に消化し、クレノウ(Klenow)で処理し、自身と再結合させ、こうしてmGluR5a DNAの3’に位置するXhoI部位を破壊した。次にプラスミドをScaI/XhoIで消化し、SV40スプライス部位、全長mGluR5aコーディング配列(および5’非翻訳配列の21ヌクレオチドと3’非翻訳配列の173ヌクレオチド)、ポリアデニル化シグナルおよびアンピシリン耐性遺伝子の3’部分を含有する断片を作成した。プラスミドpSVβ(クロンテック(Clontech)、パロアルト(Palo Alto)、カリホルニア州)をScaI/XhoIで消化し、アンピシリン耐性遺伝子とSV40早期プロモーターを含有する断片を、mGluR5a DNAを含有するScaI/XhoI断片に結合させて、pSV−hmGluR5を作成した。すなわち、pSV−hmGluR5は、SV40早期プロモーターを含有し、mGluR5a DNA(配列番号7のヌクレオチド349−4085を含有する)と機能的に結合したSV40スプライス部位が続き、この後にポリアデニル化シグナルが続く。
【0106】
全長mGluR5b作成体を調製するために、mGluR5a作成体
(mGluR1a1、mGluR5a2またはmGluR5a3)をNheI/PmlIで消化し、配列番号7のヌクレオチド2725−3020を含有する断片を放出させる。次に残りのベクター断片を、METAB1からの単離したNheI/PmlI断片に結合させる。得られるベクター(mGluR5b)は、配列番号7のヌクレオチド2999と3000の間に位置する96塩基対の挿入体(配列番号9のヌクレオチド3000−3095)を含有する以外は、これが得られたmGluR5a作成体と同じである。配列番号9は、ベクターmGluR5a1から調製した全長mGluR5b cDNAの完全なヌクレオチド配列である。
【0107】
全長mGluR5c作成体を調製するために、mGluR5a作成体
(mGluR1a1、mGluR5a2またはmGluR5a3)をNheI/HindIII(Hind III部位は、mGluR5aベクターのpCMV−T7−3部分のポリリンカー内に存在する)で消化し、配列番号7のヌクレオチド2725−4085を含有する断片を放出させる。次に残りのベクター断片を、METAB2から単離したNheI/Hind III断片に結合させる。得られる全長cDNA(mGluR5c)(配列番号11)は、コーディング配列の最初の2630ヌクレオチドについては、これが調製されたmGluR5a作成体と同じであるが、コーディング配列のヌクレオチド2631で、mGluR5cとmGluR5aのコーディング配列は分岐(例えば、配列番号7のヌクレオチド3000で始まる)し、mGluR5cコーディング配列はコーディング配列のヌクレオチド2631としてグアニンヌクレオチドを有し、その後に翻訳停止コドン(配列番号11のヌクレオチド3001−3003)が続く。
【0108】
B.mGluR1受容体cDNA
cDNAライブラリースクリーニング
中挿入体小脳ライブラリーを、ラットのmGluR1受容体(ヌクレオチド1から3031および5’非翻訳配列:マス(Masu)ら、(1991)Nature 349:760−765)をコードするDNAの断片へのハイブリダイゼーションについてスクリーニングした。ハイブリダイゼーションは、42℃で5×SSPE、5×デンハルツ(Denhart's)溶液、50%ホルムアルミド、0.2%SDSおよび200μg/mlの変性した超音波処理したニシン精子DNA中で行ない、洗浄は55℃で1×SSPEと0.2%SDS中で行なった。3つのハイブリダイズするクローン(METAB7−METAB9)が同定された。
【0109】
次のスクリーニングで、ヒト中挿入体小脳cDNAライブラリーの1×106組換え体の各平板を、追加のヒトmGluR1クローンとプローブ結合させた。この平板をまず、ラットのmGluR1 cDNAのヌクレオチド1−1256(および5’非翻訳配列)を含有するDNA断片(すなわち、5’プローブ)とハイブリダイゼーションさせ、次にクローンMETAB7−METAB9を同定したこの前のスクリーニングで使用したものと同じハイブリダイゼーション条件と洗浄条件を用いて、ラットmGluR1a cDNAのヌクレオチド2075−3310を含有するDNA断片(すなわち、3’プローブ)に連続してハイブリダイズさせた。5’プローブへのハイブリダイゼーションにより3つのクローン(METAB18、METAB21およびMETAB22)が同定され、そして3’プローブへのハイブリダイゼーションにより4つのクローン(METAB14、METAB20、METAB32およびMETAB35)が同定された。
【0110】
このラットのmGluR1断片をプローブとして使用して、さらなるmGluR1クローンのために大挿入体ヒト小脳cDNAライブラリーをスクリーニングした。ハイブリダイゼーションと洗浄条件は、中挿入体小脳ライブラリーから6つのmGluR1クローンを単離した時に使用したものと同じである(例えば、ハイブリダイゼーション溶液では20%ホルムアルデヒドを使用した)。3つのプラーク(METAB58、METAB59およびMETAB60)がプローブにハイブリダイズした。
【0111】
単離されたクローンの性状解析
精製したプラークの挿入体を制限酵素マッピングとDNA配列解析により性状解析した。METAB58は、約2.8kbであり、5’非翻訳配列、翻訳開始コドンおよび約2.3kbのコーディング配列を含有する。METAB58の3’末端は、METAB14の5’末端と重複する。METAB14は3’方向へ約700塩基対延長し、翻訳停止コドンを含有する。すなわち、METAB58とMETAB14は重複して、全長mGluR1受容体をコードする(配列番号1を参照)。他のクローンも、翻訳開始コドンと翻訳停止コドンの間に位置するmGluR1コーディング配列の部分からのヌクレオチド配列を含有する、部分mGluR1 cDNAである。
【0112】
ヒトmGluR1転写体の3’末端をコードする追加のクローンがヒトcDNAライブラリー中に存在するか否かを測定するために、海馬/脳幹神経節および小脳ライブラリーからのcDNAの核酸増幅を行なった。5’プライマーは配列番号1のヌクレオチド2218から2240よりなり、3’プライマーは、ラットのmGluR1aコーディング配列のアミノ酸890−897に基づく縮重オリゴヌクレオチドであった(ピン(Pin)ら、(1992)Neurobiology 89:10331−10335を参照)。増幅生成物をゲル電気泳動で解析した。海馬/脳幹神経節中にのみ1つの生成物(すなわち、500塩基対断片)が検出された。
【0113】
mGluR1転写体の3’末端を示す追加のクローンを得るために、海馬および小脳cDNAライブラリーを、前述のヒトmGluR1 cDNAを得るのに使用した条件と同様の条件を使用してラットのmGluR1a cDNAの3’末端からの断片(例えば、ラットのmGluR1a cDNAの約2kbのNcoI/ClaI断片)でスクリーニングすることができる。このプローブは、択一的にスプライスされるようではないmGluR1 cDNAの3’部分に対応する。次にハイブリダイズするクローンを制限酵素マッピングとDNA配列決定により解析して、異なる3’末端が示されるか否かを決定する。
【0114】
全長mGluR1 cDNA作成体の調製
ヒトmGluR1受容体のB型をコードする全長作成体を調製するために、クローンMETAB58とMETAB14の一部を結合させた。METAB58をEcoRI/AccIで消化し、配列番号1のヌクレオチド154−2612を含有する2459塩基対の断片を単離する。METAB14の704塩基対断片(配列番号1のヌクレオチド2613−3321を含有する)を、METAB14のAccI/XhoI消化により単離する。次にこの断片をMETAB58の2459塩基対に結合させて、EcoRI/SalI消化ベクターpCMV−T7−3に結合させる。ヒトmGluR1Bをコードする得られた作成体は、5’非翻訳配列の234ヌクレオチド(配列番号1のヌクレオチド154−387)、全mGluR1Bコーディング配列(配列番号1のヌクレオチド388−3108)、および3’非翻訳配列の213ヌクレオチド(配列番号1のヌクレオチド3109−3321)を含有する。mGluR1Bをコードする配列は、哺乳動物細胞中での発現のためのpCMV−T7−3中の制御成分に機能的に結合している。
【0115】
種々のヒト組織中で実際に発現されるmGluR5およびmGluR1受容体を決定するために、種々の方法が使用できる。例えば、本明細書に記載のmGluR転写体の挿入/欠失(すなわち、分岐の領域)の5’と3’に位置するヌクレオチド配列に特異的なオリゴヌクレオチドを用いて、種々の組織から単離されたRNAおよび/または種々の組織から調製されたcDNAライブラリーの核酸増幅をプライムすることができる。増幅生成物の有無および生成物のサイズは、組織中で発現される変種を示す。生成物はまた、DNA配列解析により詳細に性状解析される。
【0116】
RNase保護測定法を用いて、種々の組織中で発現される変種転写体を決定することもできる。これらの測定法は、総細胞性RNAの複雑な混合物中のRNA種を検出し定量するための感度の良い方法である。mGluRDNAの一部を標識し、細胞性RNAとハイブリダイズさせる。細胞性RNA中に相補的なmRNAが存在する場合は、DNA−RNAハイブリッドが形成する。次にこのRNA試料をRNaseで処理して、1本鎖RNAを分解させる。RNA−DNAはRNase分解から保護され、ゲル電気泳動やオートラジオグラフィーにより目視できるようにされる。
【0117】
例えばヒトmGluR cDNAへのハイブリダイゼーションによるヒトメタボトロピック受容体をコードする配列を含有するゲノムクローンの単離と、引き続くクローンの性状解析は、mGluR一次転写体の可能なスプライス変種に関してさらなる情報を与える。
【0118】
C.mGluR3受容体cDNA
cDNAライブラリースクリーニング
ヒト海馬cDNAライブラリー(ランダムプライマーを用いてcDNA合成をプライムさせ、次にλgt10ベクターへの結合について1.0−2.8kbのcDNAを選択することにより作成された)を、ラットmGluR2 cDNAの500塩基対のSmaI/XbaI断片とラットmGluR3 cDNAの3kbのAccI/BamHI断片へのハイブリダイゼーションについて選択した[タナベ(Tanabe)ら、(1992)Neuron 8:169−179]。ハイブリダイゼーションは、42℃で5×SSPE、5×デンハルツ(Denhart's)溶液、50%ホルムアミド、0.2%SDSおよび200μg/mlの変性した超音波処理したニシン精子DNA中で行ない、洗浄は65℃で0.5×SSPEと0.2%SDS中で行なった。3つのハイブリダイズするクローン(METAB40、METAB41、およびMETAB45)が同定された。
【0119】
次にMETAB45の5’末端の一部(すなわち、最初の244塩基対;配列番号5のヌクレオチド2634−2877)を用いて、増幅した小脳ライブラリー(ランダムプライマーを用いてcDNA合成をプライムし、次にλgt10ベクターへの結合について>2.8kbのcDNAを選択することにより作成される)、そして増幅した海馬cDNAライブラリー(ランダムプライマーを用いてcDNA合成をプライムし、次にλgt10ベクターへの結合について>2.0kbのcDNAを選択することにより作成される)を追加のmGluR3についてスクリーニングした。各ライブラリーから100万のクローンがスクリーニングされた。ハイブリダイゼーション条件と洗浄条件は、海馬ライブラリーからのMETAB40、METAB41およびMETAB45を単離するのに使用したものと同じである。3つのハイブリダイズするクローンが各ライブラリーで同定された(小脳ライブラリーではMETAB46、METAB49およびMETAB50、そして海馬ライブラリーではMETAB47、METAB48およびMETAB51B)。
【0120】
単離したクローンの性状解析
精製したプラークの挿入体を、制限酵素マッピングとDNA配列解析により性状解析した。単離したクローンのおのおのは、ヒトmGluR2受容体の一部をコードするクローンメタボトロピック40(例えば実施例1.D.を参照)を除いて、ヒトmGluR3受容体の一部である。クローンMETAB41、METAB45、METAB47−49は、mGluR3コーディング配列の3’からの配列および翻訳停止コドンを含有する。クローンMETAB46、METAB50およびMETAB51Bは、mGluR3 cDNAの5’末端からの配列(翻訳開始コドンと種々の量の5’非翻訳配列を含む)を含有する。
【0121】
全長mGluR3 cDNA作成体の調製
METAB48とMETAB46またはMETAB51Bの一部を結合させて、全長ヒトmGluR3コーディング配列を含有する4つの作成体を調製した。この全長コーディング配列は配列番号5(ヌクレオチド1064−3703)に与えられる。クローンMETAB46とMETAB51Bの挿入体は、EcoRI断片としてpCMV−T7−3中に別々にサブクローニングされた。クローンMETAB48の挿入体は、EcoRI断片としてpCMV−T7−2にサブクローニングされた。
【0122】
作成体mGluR3Bを作成するために、METAB51B挿入体を含有するpCMV−T7−3プラスミドをScaI/Bgl IIで消化し、アンピシリン耐性遺伝子の5’半分とMETAB51B挿入体の5’部分(配列番号5のヌクレオチド748−1671)を含有する2.6kbの断片を単離した。この断片を、METAB48の挿入体を有するpCMV−T7−2プラスミドのScaI/Bgl II消化物から単離した4.3kbの断片[この4.3kbの断片はアンピシリン耐性遺伝子の3’半分とMETAB48の3’部分を有する(配列番号5のヌクレオチド1672−3919)]に結合させた。得られた作成体(mGluR3B)は、316ヌクレオチドの5’非翻訳配列(配列番号5のヌクレオチド748−1063)、全mGluR3コーディング配列(配列番号5のヌクレオチド1064−3703)、および216ヌクレオチドの3’非翻訳配列(配列番号5のヌクレオチド3704−3919)を含有する。mGluR3Bをコードする配列は、哺乳動物細胞中での発現のためにベクターpCMV−T7−3とpCMV−T7−2からの制御成分に機能的に結合している。
【0123】
作成体mGluR3Cを作成するために、METAB46の挿入体を有するpCMV−T7−3プラスミドを、ScaI/Bgl IIで消化し、アンピシリン耐性遺伝子の5’半分とMETAB46の5’部分を含有する3.4kbの断片(配列番号5のヌクレオチド1−1671)を単離した。この断片を、METAB48の作成体mGluR3Bで使用したものと同じScaI/Bgl II断片に結合させた。得られた作成体(mGluR3C)は、5’非翻訳配列の1063ヌクレオチド(配列番号5のヌクレオチド1−1063)、全mGluR3コーディング配列(配列番号5のヌクレオチド1064−3703)、および3’非翻訳配列の216ヌクレオチド(配列番号5のヌクレオチド3704−3919)を含有する。このmGluR3Cをコードする配列は、哺乳動物細胞中での発現のためにベクターpCMV−T7−2とpCMV−T7−3からの制御成分に機能的に結合している。
【0124】
作成体mGluR3Aは、mGluR3CをEcoRVとNotIで消化して、配列番号5のヌクレオチド1−1035を含有する断片を除去して、NotI部位を平滑末端として、次に大きい方のベクター断片を再結合させることにより作成した。作成体mGluR3Aは、28ヌクレオチドの5’非翻訳配列(配列番号5のヌクレオチド1036−1063)、全mGluR3コーディング配列(配列番号5のヌクレオチド1064−3703)、および216ヌクレオチドの3’非翻訳配列(配列番号5のヌクレオチド3704−3919)を含有する。このmGluR3Aをコードする配列は、哺乳動物細胞中での発現のためにベクターpCMV−T7−3とpCMV−T7−2からの制御成分に機能的に結合している。
【0125】
作成体pSV−hmGluR3C(mGluR3のSV40プロモーター制御発現に使用するため)を作成するために、METAB46の挿入体を有するpCMV−T7−3プラスミドをScaI/NotIで消化し、アンピシリン耐性遺伝子の3’部分と全METAB46挿入体を含有する断片を単離した。プラスミドpSVβをScaI/NotIで消化し、アンピシリン耐性遺伝子5’部分とSV40早期プロモーターおよびスプライス部位を含有する断片を、METAB46を有するpCMV−T7−3ベクターからのScaI/NotI断片に結合させて、pSV−METAB46を作成した。プラスミドpSV−METAB46をScaI/Bgl IIで消化し、アンピシリン耐性遺伝子の5’部分、SV40早期プロモーターおよびスプライス部位そしてMETAB46の5’部分(配列番号5のヌクレオチド1−1671)を含有する断片を単離した。この断片を、作成体mGluR3BやmGluR3Cで使用したものと同じMETAB48のScaI/Bgl II断片に結合させた。得られた作成体(pSV−hmGluR3C)は、SV40プロモーターを含有し、mGluR3 DNA(配列番号5のヌクレオチド1−3919を含有する)と機能的に結合したSV40スプライス部位が続き、この後にポリアデニル化シグナルが続く。
【0126】
D.mGluR2受容体cDNA
実施例1.C.に記載したようにヒト海馬cDNAライブラリーからクローンMETAB40を単離した。このMETAB40の挿入体cDNAは長さが1100塩基対であり、ヒトmGluR2受容体の3’末端(翻訳停止コドンと3’末端非翻訳配列を含む)をコードする。METAB40の最初の355ヌクレオチドは配列番号3に提供され、METAB40の最後の343ヌクレオチド(すべて3’非翻訳配列からである)は配列番号13に提供される。
【0127】
mGluR2転写体の5’部分を示すDNAを含有するクローンを単離するために、METAB40の5’末端に対応するオリゴヌクレオチドへのハイブリダイゼーションについて、ヒト海馬cDNAライブラリーをスクリーニングすることができる。ハイブリダイズするクローンを精製し、DNA配列解析により性状解析する。METAB40に重複し翻訳開始コドンを含有するクローンを、適当な制限部位でMETAB40に結合させて、全長mGluR2をコードするcDNA作成体を作成することができる。
【実施例2】
【0128】
卵母細胞中の組換えヒトメタボトロピックグルタミン酸受容体の発現
ヒトメタボトロピック受容体をコードするDNAを含有する作成体から調製したインビトロ転写体を、ツノガエル(Xenopus)卵母細胞に注入した。2電極の電位固定法(例えば、スツマー(Stuhmer)(1992)Meth. Enzymol. 207:319−339を参照)を用いて、卵母細胞トランスメンブレン電流の電気生理的測定を行なった。
【0129】
A.インビトロ転写体の調製
作成体mGluR5a3中に含有されるメタボトロピック受容体cDNAのキャップされた組換え転写体を、メガスクリプト(Megascript)キット(カタログ番号#1334、アンビオン社(Ambion Inc.)、オースチン、テキサス州)を用いて、線状化したプラスミドから合成した。各合成転写体の質量を紫外吸光度法により測定し、各転写体の完全性をアガロースゲルの電気泳動により測定した。
【0130】
B.電気生理学
卵母細胞1つにつき、10−50ngのメタボトロピック受容体転写体を、ツノガエル(Xenopus)卵母細胞に注入した。卵母細胞の調製と注入は、ダスカル(Dascal)の方法[(1987)Crit. Rev. Biochem. 22:317−387]に従って行なった。mRNA注入の2から6日後、2電極電位固定法を用いて卵母細胞を試験した。細胞はリンゲル溶液(115mM NaCl、2.5mM KCl、1.8mM CaCl2、10mM HEPES、pH7.3)浴に浸漬し、膜電位は−80から−100mVに固定した。薬剤含有溶液の60μlずつをピペットで直接浴に入れた。アクソテープ(AXOTAPE)バージョン1.2ソフトウェア(アクソン・インスツルメンツ(Axon Instruments)、フォスターシティ、カリホルニア州)を用いてPC−386中のラボマスター(Labmaster)データ採取ボードを用いて2−5Hzでデータをサンプリングした。データをレーザープリンターに取り出すか、またはシグマプロット(Sigmaplot)バージョン5.0を用いてプロットした。
【0131】
メタボトロピック受容体調節化合物(すなわち、0.001−0.1μMキスカル酸、0.1−10μMグルタミン酸および0.1−300μM 1S,3R−ACPD(1−アミノ−シクロペンチル−1,3−ジカルボン酸)を浴に適用し、トランスメンブレン電流を記録した。化合物の適用後相当の電流が検出された。種々の量の各化合物を適用後の電流を比較する用量応答試験で、電流の大きさは各化合物の濃度の上昇に伴い増加することが明らかになった。これらのデータを解析した結果、各化合物のEC50を計算することができ、これを化合物の相対電位の測定に使用した。
【実施例3】
【0132】
哺乳動物細胞におけるヒトメタボトロピックグルタミン酸受容体サブユニットの組換え発現
ヒト胎児性腎(HEK)細胞とチャイニーズハムスター卵巣(CHO)細胞(すなわち、DG44細胞;ウーラウプ(Urlaub)ら(1986)Som. Cell. Molec. Genet. 12:555を参照)を、ヒトメタボトロピック受容体をコードするDNAでトランスフェクションさせた。種々の方法(例えば、イノシオトールリン酸(IP1)測定法、Ca++−感受性蛍光指示薬ベースの測定法、および[3H]−グルタミン酸結合測定法)を用いて、メタボトロピック受容体の発現について解析した。
【0133】
A.HEK293細胞の一時的トランスフェクション
HEK293細胞を、mGluR5a(作成体mGluR5a2とmGluR5a3および作成体MMTV−hmGluR5a)受容体をコードするDNAで一時的トランスフェクションを行なった。約2×106のHEK293細胞を標準的CaPO4トランスフェクション法[ウィグラー(Wigler)ら、(1979)Proc. Natl. Acad. Sci. U.S.A 76:1373−1376を参照]に従い、目的のプラスミドの5−18μg(あるトランスフェクション体においては0.18μg、実施例3.C.2.を参照)で一時的にトランスフェクションした。さらにCMVプロモーターに融合した大腸菌β−ガラクトシダーゼ遺伝子を含有する、プラスミドpCMVβgal(クロンテック・ラボラトリーズ(Clontech Laboratories)、パロアルト(Palo Alto)、カリホルニア州)0.5−2μgを、レポーター遺伝子として同時トランスフェクションして、トランスフェクションの効率を追跡した。β−ガラクトシダーゼとX−gal基質を含む反応の生成物の直接染色により、β−ガラクトシダーゼ発現についてトランスフェクション体を解析した[ジョーンズ(Jones)(1986)EMBO 5:3133−3142]。トランスフェクション体はまた、β−ガラクトシダーゼ活性の測定によるβ−ガラクトシダーゼ発現について解析することもできる[ミラー(Miller)(1972)分子遺伝子学実験(Experiments in Molecular Genetics)のPP. 352−355, コールド・スプリング・ハーバー・プレス(Cold Spring Harbor Press)]。
【0134】
5μgのMMTV−hmGluR5Aで一時的トランスフェクションを行なったHEK293細胞は、ラウス肉腫ウイルス(RSV)LTRプロモーターに機能的に結合した糖質コルチコイド受容体をコードするDNAを含有するpRShGR(ATCC整理番号67200)5μgと同時トランスフェクションをした。これらの細胞における糖質コルチコイド受容体の同時トランスフェクションは、細胞への糖質コルチコイド(例えば、デキサメタゾン)の添加によるMMTVプロモーター−mGluR5aDNAの発現の誘導を確実にする。
【0135】
HEK細胞のこれらのトランスフェクションの効率は、標準的効率に典型的なものであった(すなわち、約50%)。
【0136】
B.哺乳動物細胞の安定なトランスフェクション
HEK293細胞、Ltk−細胞およびCHO細胞のような哺乳動物細胞は、リン酸カルシウムトランスフェクション法を用いて安定にトランスフェクションすることができる[Current Protocols in Molecular Biology, Vol.1,ワイリー・インターサイエンス(Wiley Inter-Science)、増刊14号、単元9.1.1.−9.1.9(1990)]。CHO細胞を宿主として使用するときは、ヒトメタボトロピック受容体をコードするcDNAの発現を制御するにはSV40プロモーターの使用が一般に好ましい。それぞれ1−2×106細胞を含有する10cmのプレートを、メタボトロピック受容体をコードするDNA約5−10μgと選択マーカー(例えば、ネオマイシン−耐性遺伝子(すなわち、HEK293形質転換体の選択にはネオマイシン耐性遺伝子(すなわち、pSV2neo)、Ltk−細胞トランスフェクション体にはチミジンキナーゼ遺伝子、またはDG44細胞形質転換体の選択にはジヒドロ葉酸還元酵素(dhfr)遺伝子))をコードするDNA 0.5−1μgを含有するDNA/リン酸カルシウム沈殿物でトランスフェクションする。適当な培地中で約14日間の増殖の後、コロニーが形成され、クローニングシリンダーを用いて個々に単離される。次に単離物を限界希釈を行いスクリーニングして、例えば後述の方法を用いてメタボトロピック受容体を発現するものを同定する。
【0137】
C.トランスフェクション体の解析
1.蛍光指示薬に基づく測定
アゴニストによるG蛋白結合メタボトロピック受容体の活性化により、ホスファチジルイノシトール(PI)加水分解/細胞内Ca++シグナル化経路および/または阻害性cAMPカスケードが刺激される。細胞内カルシウム濃度の一時的上昇の検出法は、PI加水分解/Ca++動員(mobilization)経路またはPI加水分解/Ca++動員(mobilization)経路と阻害性cAMPカスケードの両方にカップリングしたメタボトロピック受容体の機能性発現の解析に適用できる。細胞内カルシウムレベルを測定する1つの方法は、カルシウム感受性蛍光指示薬を用いる。
【0138】
フルオ−3(fluo−3)やフラ−2(fura−2)(モレキュラー・プローブズ社(Molecular Probes Inc.)、ユージーン(Eugene)、オレゴン州)のようなカルシウム感受性指示薬は、膜透過性であるアセトキシメチルエステルとして入手できる。アセトキシメチルエステル型の指示薬が細胞内に入ると、サイトゾル性エステラーゼによりエステル基は除去され、このため遊離の指示薬はサイトゾル内に捕捉される。遊離の指示薬とカルシウムが相互作用すると、指示薬の蛍光強度が増加する。従って、指示薬を含有する細胞の細胞内Ca++濃度の上昇は直接蛍光の増強(または、フラ−2を使用する時は、2つの波長での蛍光の比率の増強)として発現される。メタボトロピック受容体を測定するための自動蛍光検出装置が、本出願人の係属中の米国特許出願第07/812,254号およびその対応するPCT特許出願US92/11090号(参考のためその全体が本明細書中に引用される)に記載されている。さらに細胞内Ca++振動を目視できるようにするために蛍光イメージング法を使用することができる。
【0139】
ヒトmGluR5a受容体をコードするDNAで一時的トランスフェクションを行なったHEK細胞を、自動蛍光指示薬ベースの測定法と蛍光イメージング測定法を用いて、機能性組換えメタボトロピック受容体の発現について解析した。同様に、メタボトロピック受容体DNAで安定にトランスフェクションした細胞も、これらの測定法系を用いて機能性メタボトロピック受容体について解析した。
【0140】
a.自動蛍光測定法
2×105細胞/ウェルの濃度でポリ−L−リジンでプレコーティングし、20μMフルオ−3、0.2%プルロニック(Pluronic)F−127をHBS(125mM NaCl、5mM KCl、1.8mM CaCl2、0.62mM MgCl2、20mMグルコース、20mM HEPES、pH7.4)中に含有する培地中で、20℃で2時間インキュベートしてフルオ−3を充填した96ウェルマイクロタイタープレート(ヌンク(Nunc)、カタログ番号1−6708、アラメダ・インダストリーズ(Alameda Industries)、エスコンジド(Escondido)、カリホルニア州)に、トランスフェクションしていないHEK293細胞(またはpCMV−T7−3で一時的にトランスフェクションしたHEK293細胞)およびmGluR5aをコードするDNAでトランスフェクションしたHEK293細胞を広げた。次に細胞を測定緩衝液(すなわち、HBS)で洗浄した。次にマイクロタイタープレートを蛍光プレートリーダー(例えば、フルオロスカン(Fluoroskan)II、ラボ・プロダクツ・インターナショナル社(Lab Products International, Ltd.)、ラレー(Raleigh)、ノースカロライナ州)に入れ、各ウェルの基礎蛍光を測定し記録してから、ウェルにキスカル酸、グルタミン酸、トランス−ACPD(1−アミノ−シクロペンタン−1,3−ジカルボン酸)、1S,3R−ACPD、AP3(2−アミノ−3−ホスホノプロピオン酸)、AP5(2−アミノ−5−ホスホノペンタノン酸)、およびCNQX(6−シアノ−7−ニトロキノキサリン−2,3−ジオン)を添加した。アゴニストの添加後ウェルの蛍光を繰り返し(0.63秒間隔で75回読む)追跡する。
【0141】
一般に、トランスフェクションしていないHEK293細胞の蛍光は、これらの化合物のどれを添加しても変化しなかった。mGluR5a3またはMMTV−hmGluR5a作成体のいずれかで一時的にトランスフェクションしたHEK293細胞の蛍光は、グルタミン酸、キスカル酸、トランス−ACPD、または1S,3R−ACPDの添加に応答して上昇した。蛍光はピーク値まで増加し、次に時間とともに化合物の添加前の細胞の基礎レベルまで低下した。mGluR5a2でトランスフェクションした細胞について、グルタミン酸、キスカル酸またはトランス−ACPD刺激蛍光の増強に対するAP3、AP5またはCNQXの影響を調べた。これらの化合物(AP3、AP5、またはCNQX)のいずれも、これらの細胞中のアゴニスト誘導蛍光増強を阻害しなかった。
【0142】
種々の量のグルタミン酸、キスカル酸または1S,3R−ACPDを添加後に測定したピーク蛍光値を比較する用量応答試験で、ピーク蛍光の大きさは各化合物の濃度の上昇に伴い増加することが明らかになった。これらのデータを解析した結果、各化合物のEC50を計算することができ、これを化合物の相対電位の測定に使用した。
【0143】
MMTV−hmGluR5aおよびpRShgGR(糖質コルチコイド受容体作成体)で一時的に同時トランスフェクションしたHEK293細胞も、この蛍光測定法で解析した。これらの細胞の蛍光は100μMのキスカル酸に応答して増加し、測定前に細胞をデキサメタゾン(約1M)で37℃で16時間プレインキュベートするとピーク応答は大きかった。
【0144】
b.蛍光イメージング測定法
細胞内Ca++濃度のメタボトロピック受容体介在変化を目視できるようにするために、mGluR5a3で一時的にトランスフェクションしたHEK293細胞とトランスフェクションしていないHEK293細胞(対照)を、デジタルビデオイメージングで解析した。暗所中で室温で25分間細胞を1μMのフラ−2(アセトキシメチルエステル)に暴露することにより、トランスフェクション体(ガラス挿入底を有する35mm培養皿あたり4×105細胞)をフラ−2で充填した。次に細胞をDMEMで3回そしてリンゲル溶液(160mM NaCl、5mM KCl、2mM CaCl2、1mM MgCl2、11mMグルコース、5mM HEPES、pH7.3)で4回洗浄した。
【0145】
次にトランスフェクション体と非トランスフェクション細胞を、紫外光源として150Wのキセノンランプを有するアキシオベルト(Axiovert)100TV倒立顕微鏡(ツァイス(Zeiss)、オーベルコフレン(Oberkochren)、ドイツ)のステージの上に広げた。40×1.3N.A.油浸対物レンズを介して細胞を340nmと380nmで交互励起するのに、イメージ1フルオル(Fluor)システム(ユニバーサル・イメージング(Univeersal Imaging)、ウェストチェスター、ペンシルバニア州)を使用した。510nmより長い波長で出る光を、CCD72強化CCDカメラ(エム・ティー・アイ ダーゲ(MTI Dage)、ミシガン・シティ、インディアナ州)により集め、デジタル化した。340nmと380nmの励起イメージからバックグランドの蛍光を差し引いた。補正値を使用して340/380強度比を計算した。較正していないフラ−2比は、細胞内Ca++濃度の変化の信頼できる指標である。
【0146】
較正していないフラ−2比を用いて、休止細胞内Ca++濃度(約100nM)を紫色で、高細胞内Ca++濃度(約1μM)を赤色で疑色イメージを作成した。実験の各比率のイメージについて、定量解析のために、各細胞の12×12ピクセル領域内の平均比をソフトウェアにより計算し、さらに解析しグラフを描くために表に取り込んだ。
【0147】
HEK293細胞は、PI加水分解/Ca++動員経路の受容体介在活性化に必要な細胞内成分を発現することを証明するために、トランスフェクション体と非トランスフェクション細胞(内因性G蛋白結合ムスカリン性アセチルコリン受容体を発現する)を1mMのカルバミルコリン(CCh;ムスカリン性アセチルコリン受容体アゴニスト)に暴露し、細胞内Ca++濃度の上昇を追跡した。典型的には、イメージング試験でCChの添加に応答して、大多数の細胞の細胞内Ca++濃度の上昇が検出された。
【0148】
トランスフェクションしたHEK293細胞およびトランスフェクションしていないHEK293細胞を、100μMキスカル酸に応答して細胞内Ca++濃度が上昇するか否かについて追跡した。平均すると、非トランスフェクション細胞の細胞内Ca++濃度は、キスカル酸への暴露後増加しない。これに対して、トランスフェクションした細胞の26.7±22.3%のCa++濃度は100μMのキスカル酸の添加に応答して増加した。
【0149】
2.ホスファチジルイノシトール加水分解(IP1)測定法
G蛋白結合メタボトロピック受容体のアゴニストによる活性化により、ホスファチジルイノシトール(PI)加水分解経路が刺激されるため、PI加水分解の生成物(例えば、IP3、IP2またはIP1)の増加を検出する方法は、PI加水分解/Ca++動員経路またはPI加水分解/Ca++動員経路と阻害性cAMPカスケードにカップリングしたメタボトロピック受容体の機能性発現の解析に応用することができる。PIの加水分解により生成されるIP1および/またはIP2および/またはIP3を測定する1つの方法は、細胞膜リン脂質内への[3H]−ミオ−イノシトールを取り込み、次に[3H]−IP1、[3H]−IP2および[3H]−IP3を分離し、以下のように各画分の放射能を定量する。
mGluR5a3で一時的にトランスフェクションしたHEK293細胞を、8×105細胞/ウェルで24ウェルのマイクロタイタープレートに広げた。細胞を数時間沈降させ、プレートの底に付着させた後、2μCiの[3H]−ミオ−イノシトール(アマーシャム(Amersham)、カタログ番号#PT6−271、アーリントンハイツ(Arlington Heights)、イリノイ州;比活性=17.7Ci/mmol)を各ウェルに添加し37℃で一晩インキュベートした。翌日、ニコンダイアフォト(Nikon Diaphot)倒立顕微鏡下で細胞を観察し、細胞の形態の健常性を評価し、ウェルがコンフルエント(confluent)な細胞層を含有するか否かを測定した。次に培地を吸引し、細胞を0.5mlのクレブス重炭酸緩衝液[117.9mM NaCl、4.72mM KCl、2.54mM CaCl2、1.18mM MgSO4、1.19mM KH2PO4、25mM NaHCO3、11.1mMデキストロース(95% O2、5% CO2、pH7.4で平衡化されている)]で2回洗浄した。細胞を室温で45分間インキュベートした。次に各ウェルから緩衝液を吸引し、細胞を洗浄し、0.5ml/ウェルで室温で45分間インキュベートした。次に緩衝液を各ウェルから吸引し、次に10mM NaClの代わりに10mMのLiClを含有する450μlのクレブス重炭酸緩衝液(IP1のイノシトールと無機リンへの加水分解を防ぐために)とともに細胞を37℃で20分間インキュベートした。
【0150】
メタボトロピック受容体調節化合物による細胞の処理を始める前に、50μlのクレブス重炭酸緩衝液(対照)または化合物の最終濃度の10×のものを各ウェルに添加し、40分間インキュベートを続けた。各ウェルに1mlの氷冷メタノールを加えてインキュベートを停止させた。
【0151】
細胞からIP1を単離するために、プラスチックピペットのチップでプレートの細胞を掻き取って、細胞懸濁物を12×75mmガラス試験管に移した。試験管を完全にボルテックス混合し、各反応混合物の150μl分(すなわち、総量の10分の1)を別の試験管に移して蛋白を測定した。1mlのクロロホルムによる抽出により、放射標識膜リン脂質から水溶性リン酸イノシトールを分離した。試験管を室温で30分間インキュベートした後、4℃で500×gで5分間遠心分離した。[3H]−リン酸イノシトールを含有する水(上)層を、アクセルQMAセパク(Accell QMA SEP−PAK)カラム(ミリポア(Millipore)、カリホルニア州)に接続した10mlのシリンジに移した(これは、20mlのシンチレーションバイアルに集められるように変更したアマーシャム・スーパーセパレーター(Amersham Superseparator)装置に結合してある)。カートリッジに水(10ml)を入れて[3H]−イノシトール前駆体を除き、次に4mlの0.02Mトリエチルアンモニウム水素炭酸化緩衝液(TEAB、フルカ(Fluka);ニューヨーク)を加えた。カートリッジから[3H]−IP1、[3H]−IP2そして[3H]−IP3を別々に除去するために、4mlの0.1M TEAB、4mlの0.3M TEAB、そして4mlの0.4M TEABをカートリッジに連続的に添加して、画分を溶出させ、大きいシンチレーションバイアルに採取した。エコルメ・カクテル(Ecolume cocktail)(15ml;ICN;カリホルニア州)を各バイアルに加え、次にシンチレーション計測して別々の画分の各IPの量を測定した。蛋白濃度は、バイオラッド(Bio−Rad)蛋白マイクロ測定法(バイオラッド(Bio−Rad)、リッチモンド、カリホルニア州)を用いて測定した。
【0152】
この測定法で測定した時、18μgのmGluR5a3で一時的にトランスフェクションしたHEK293細胞は、比較的高い基礎レベルのIP1を示した。しかし、0.18μgのmGluR5a3で一時的にトランスフェクションしたHEK293細胞は、低い基礎IP1レベルを示し、1mMのグルタミン酸、1mMのキスカル酸または1mMの1S,3R−ACPDで処理した時はIP1レベルの検出できる増加を示した。IP1レベルのキスカル酸に誘導される増加は、1mMのAP3により影響を受けなかった。
【0153】
mGluR5a3でトランスフェクションした細胞に、種々の濃度のグルタミン酸、キスカル酸または1S,3R−ACPDの添加後測定したIP1レベルを比較した用量応答試験では、各化合物の濃度の上昇とともにIP1レベルが増加することを示していた。これらのデータの解析により、各化合物のEC50を計算することができ、これを化合物の相対電位の測定に使用した。
【0154】
3.メタボトロピック受容体リガンド結合測定法
mGluR5a3またはpUC19(陰性対照)で一時的にトランスフェクションしたHEK細胞を、[3H]−グルタミン酸結合について解析した。陽性対照として結合測定法にラット脳の膜を加えた。
【0155】
a.膜の調製
i.ラット前脳膜
シェープ(Schoepp)ら[(1992)Neurosci. Lett. 145:100]が記載したように、ラット前脳膜を調製した。簡単に説明すると、10個のラット脳からの基本的に脳皮質、線条体と海馬よりなる前脳を、2.5mM CaCl2、pH7.6を含有する50部の30mMの氷冷トリス−塩酸中で、ポリトロン(Polytron)(ブリンクマン(Brinkman)、ウェストベリー(Westbury)、ニューヨーク州))を用いてホモゲナイズした。ホモゲネートを4℃で30、000×gで15分間遠心分離した。上澄液を捨て、ポリトロンを用いてペレットを50部の緩衝液に再懸濁し、懸濁物を30、000×gで15分間遠心分離した。この工程を2回繰り返した。ペレットを緩衝液に再懸濁し、37℃で30分間インキュベートした。次に懸濁物を4℃で30、000×gで15分間遠心分離した。この工程を3回繰り返した。最終ペレットを15部の50mM トリス−塩酸、pH7.6に再懸濁し、分注し、急速に凍結し、−70℃で保存した。
【0156】
ii.トランスフェクションしたHEK293細胞およびトランスフェクションしないHEK293細胞の膜
mGluR5aをコードするDNAまたはpUC19(陰性対照)でトランスフェクションしたHEK293細胞から膜を調製するために、組織培養プレートから細胞を掻き取り、プレートを5mlのPBS(リン酸緩衝化生理食塩水:137mM NaCl、2.7mM KCl、10mM Na2HPO4、1.7mM KH2PO4)で洗浄した。卓上型遠心分離機で細胞を低速度で遠心分離し、細胞ペレットをPBSで洗浄した。0.5mM PMSF、pH7.6を含有する50mMトリス−塩酸20部に、細胞ペレットを再懸濁した。ダウンス(Dounce)(テフロン(登録商標)/ガラス)ホモゲナイザー中で10−20ストロークを用いて、細胞を氷の上でホモゲナイズした。ホモゲネートを4℃で120、000×gで30分間遠心分離した。最終の膜ペレットを、0.5mM PMSF、pH7.6を含有する50mMトリス−塩酸再懸濁した。膜調製物を分注し、急速に凍結し、−701℃で保存した。蛋白濃度は、ブラッドフォード(Bradford)[(1976)Anal. Biochem.72:248]の方法で測定した。
【0157】
b.[3H]−グルタミン酸結合測定法
ラット前脳の膜中のメタボトロピック受容体への[3H]−グルタミン酸の特異的結合は、基本的にシェープ(Schoepp)ら(前述)が記載したように調製した。測定の日に凍結ホモゲネートを融解し、50mMトリス−塩酸、pH7.6で3回洗浄した。最終ペレットを50mMトリス−塩酸、pH7.6に再懸濁した。蛋白濃度は、ブラッドフォード(Bradford)[(1976)Anal. Biochem.72:248]の方法で測定した。懸濁物を30、000×gで15分間遠心分離し、ペレットを1mg/mlの濃度で測定緩衝液(50mMトリス−塩酸、0.5mM PMSF、0.1%BSA、pH7.6)に再懸濁した。膜懸濁物を、総量0.5mlの測定緩衝液[イオノトロピックグルタミン酸受容体への[3H]−グルタミン酸の結合を組織するために、100μM NMDA(シグマ(Sigma)、セントルイス、ミズーリ州)、100μM AMPAおよび100μMカイニン酸(リサーチ・バイオケミカルズ社(Research Biochemiclas Inc)、ナチック(Natick)、マサチューセッツ州)を含有し、塩化物依存性摂取部位への[3H]−グルタミン酸の結合を阻害するために、100μM SITS(シグマ(Sigma)、セントルイス、ミズーリ州)を含有する]中で、3重測定で10または100nMの[3H]−グルタミン酸(ニュー・イングランド・ニュクレア(New England Nuclear)、ボストン、マサチューセッツ州;カタログ番号NET−490、比活性=57.4Ci/mmol)と、氷上で45分間インキュベートした。SM−24ローター(ソーバル(Sorvall)、ウィルミントン(Wilmington)、デラウェア(Delaware))中で4℃で20、000×gで5分間遠心分離して、結合した放射能を遊離の放射能から分離した。ペレットを5−6mlの氷冷50mMトリス−塩酸、pH7.6で2回洗浄した。ペレットを5mlのエコルメシンチレーションカクテル(Ecolume scintillation cocktail)中でボルテックス混合して可溶化させた。ベックマン(Beckman)シンチレーション計測機で放射能を測定した。総結合量から、1mMのグルタミン酸の存在下で得られた非特異的結合量を引いて、特異的結合量を求めた。
【0158】
mGluR5aをコードするDNAまたはpUC19でトランスフェクションしたHEK293細胞から調製した膜への[3H]−グルタミン酸の特異的結合を、基本的にラット脳の膜への結合の測定で記載したように測定したが、若干の修飾を行なった。測定の日に凍結ホモゲネートを融解し、MR−150高速冷却マイクロ遠心分離機(ペニンスラ・ラボラトリーズ社(Peninsula Laboratories Inc.)、ベルモント(Belmont)、カリホルニア州)で遠心分離した。ペレットを測定緩衝液(50mMトリス−塩酸、0.5mM PMSF、0.1%BSA、pH7.6)で2回洗浄し、最終ペレットを1mg/mlの濃度で測定緩衝液に再懸濁した。NMDA、AMPAおよびカイニン酸を測定混合物から除きながら、HEK293細胞を[3H]−グルタミン酸への結合について解析した。
【0159】
200μgの膜と100nMの[3H]−グルタミン酸を用いて、ラット脳の膜への[3H]−グルタミン酸の特異的結合を測定した。総結合量対非特異結合量の比は約2:1であった。
【0160】
mGluR5a3またはpUC19でトランスフェクションしたHEK293細胞から調製した膜への[3H]−グルタミン酸の特異的結合を、200μgの膜と100nMの[3H]−グルタミン酸を用いて測定した。mGluR5a3でトランスフェクションしたHEK293細胞から調製した膜への特異的結合量は、pUC19でトランスフェクションしたHEK293細胞から調製した膜への結合量より有意に高かった。競合結合試験を行い、種々の濃度の非標識グルタミン酸の存在下でmGluR5a3でトランスフェクションしたHEK293細胞から調製した膜への[3H]−グルタミン酸の特異的結合の量を測定した。これらの試験で得られたデータからIC50値を求めた。
【0161】
4.サイクリックAMP(cAMP)測定
a.RIAベースの測定
ある種のG蛋白結合受容体の活性化は、cAMPの(増加ではなく)低下を引き起こすため、細胞内cAMPレベルの測定法は、哺乳動物宿主細胞中で発現される組換えヒトメタボトロピック受容体を評価するのに使用される。ヒトメタボトロピック受容体をコードするDNAまたはpUC19(陰性対照)で一時的にまたは安定にトランスフェクションした哺乳動物細胞を、5×105細胞/ウェルの濃度で24ウェルのマイクロタイタープレートに広げ、一晩インキュベートさせる。翌日、ニコンダイアフォト(Nikon Diaphot)倒立顕微鏡下で細胞を観察し、細胞の形態の健常性を評価し、ウェルはコンフルエントな細胞層を含有するか否かを測定した。次に培地を吸引し、細胞を、1mMのIBMX(3−イソブチル−1−メチルキサンチン;シグマ(Sigma)、セントルイス、ミズーリ州)と0.1%BSAを含有するクレブス重炭酸緩衝液0.5ml(PI加水分解測定法で使用したものと同じ緩衝液:実施例3.C.2を参照)で2回洗浄した。あるいはクレブス重炭酸緩衝液の代わりに1×PBSを使用することもできる。各洗浄の後に37℃で30分間インキュベートを行う。各ウェルから緩衝液を吸引し、次に細胞を、1mMのIBMXと0.1%BSAを含有する0.2mlのクレブス重炭酸緩衝液で37℃で20分間インキュベートする。
【0162】
メタボトロピック受容体調節化合物による細胞の処理を始める前に、50μlのクレブス重炭酸緩衝液(最終濃度の5×のフォルスコリンの有りまたは無し)を、一部の細胞(基礎対照)に加え、一部の細胞(試験細胞)に最小濃度の5×の化合物および最終濃度の5×のフォルスコリンを加え、37℃で15分間インキュベートする。15分間のインキュベートの最後に、1%トリトンX−100溶液25μlを加えて反応を停止させ、インキュベートをさらに10分間続ける。溶解した細胞および細胞懸濁物を、プラスチックピペットのチップで12×75mmのポリプロピレン試験管に移す。各ウェルを、1mMのIBMXと0.1%BSAを含有するクレブス重炭酸緩衝液75μlで洗浄する。洗浄物を細胞溶解物を一緒にする。細胞溶解懸濁物を2300×gで5分間遠心分離し、RIAキット(アマーシャム・ライフサイエンシーズ(Amersham Life Sciences)カタログ#TRK432;アーリントンハイツ(Arlington Heights)、イリノイ州)を用いて、上澄液のcAMPレベルを測定する。
【0163】
b.サイクリックヌクレオチドゲートのチャネルに基づく測定法
5%の規定補足ウシ血清(ハイクローン(Hyclone))を含有するダルベッコー改変イーグル培地(DMEM;ギブコ(GIBCO))(100U/mlのペニシリンと100μg/mlの硫酸ストレプトマイシンを含む)中で、HEK293細胞を単層で増殖させた(10cmのポリ−D−リジン被覆プレート当たり約2×106細胞)。CMVプロモーターに結合した5μgのpCMV−OCNA(嗅覚性サイクリックヌクレオチドゲートのチャネル(ダレン(Dhallen)らを参照、前述)、2μgのpCMV−βgal(クロンテック(Clontech)、パロアルト(Palo Alto)、カリホルニア州)、および対照プラスミドとしての13μgのpUC19を用いて、細胞をリン酸カルシウム法(アウスベル(Ausubel)ら、前述、pp9.1.1−9.1.7)で一時的にトランスフェクションした。ベクターpCMV−OCNAは、pBluescriptKSからの約3.0kbのEcoRI断片として嗅覚性サイクリックヌクレオチドゲートのチャネルをコードするDNAを単離し、得られた断片をEcoRI消化pCMV−T7−3に結合させることにより、作成した。トランスフェクションの6時間後、リン酸カルシウム沈殿物を洗い流し、10%透析胎児牛血清(ハイクローン(Hyclone))、100U/mlのペニシリン、100μg/mlのストレプトマイシン、および2mMグルタミンが補足されたDMEMを細胞に与えた。β−ガラクトシダーゼ活性の測定によるトランスフェクション効率は、50−70%であった。
【0164】
嗅覚性サイクリックヌクレオチドゲートのチャネルDNAでトランスフェクションしたHEK細胞を、24−48時間インキュベートしてから、機能を試験した。まず、細胞質の面に達するcAMPの濃度が調節できるように(例えば、単一チャネル記録(Single Channel Recording)、サクマンとネヘル(Sakman and Neher)編、プレヌム・プレス(Plenum Press)、ニューヨーク、(1983))、トランスフェクションした細胞から引っ張った裏返した膜パッチを用いて、電気生理学的にチャネルの活性を測定した。パッチの両面を、Ca++/Mg++を含まないリンゲル液に接触させた。1つのパッチでは、1mMのcAMPの存在下で膜電位を−100から+100mVへ2秒で勾配をつけて電流を発生させた。この結果は、チャネルが機能的に発現されていることを示唆していた。
【0165】
トランスフェクション体もまた、細胞内カルシウムレベル([Ca++])の単細胞ビデオイメージングにより解析した。この方法は、細胞内カルシウムレベルの測定(これはチャネルのサイクリックヌクレオチド活性化により制御される、チャネルを介するカルシウムの流入量とともに変化する)によるサイクリックヌクレオチドゲートのチャネルの分析を可能にする。イメジーング測定法は、基本的に実施例3.C.1.bに記載のように実施したが、若干の修飾を行なった。色素を充填後、細胞をツァイスアクシオヴェルト(Zeiss Axiovert)顕微鏡と100Wの水銀ランプ、デージ(Dage)増強CCDカメラ、そしてイメージ−1(Image−1)ハードウェアとイメージ処理のソフトウェアを用いて観察した。このソフトウェアは、20×1.3N.A.油浸対物レンズを介して細胞を350nmと385nmの交互励起を調節した(典型的には5秒毎)。510nmより長い波長で出る光を、CCDカメラにより集め、デジタル化し、350nmと385nmの励起イメージからバックグランドの発光を差し引いた後、350/385強度比を計算した。
【0166】
定量的解析のために、実験の各比率イメージについてソフトウェアにより各細胞の12×12ピクセル領域内の350/385平均比を計算し、さらに解析しグラフを描くために表に取り込んだ。フラ−2シグナルを未変性の細胞で較正し、細胞をリンゲル液(10μMイオノマイシン、10mM EGTAを含有し、およびCa++の添加無し)に接触させてRminを得た。次に細胞をリンゲル液(10μMイオノマイシン、10mM Ca++を含有する)に接触させて3回洗浄してRmaxを得た。生きている細胞中のフラ−2のKd250nMとグリンキーウイックツ(Grynkiewicz)ら(J. Biol. Chem.260:3440(1985))を使用して、静止[Ca++]iは典型的には100nMであった。
【0167】
これらの実験で、細胞内cAMPレベルを上昇させる物質にHEK293細胞トランスフェクション体を暴露し、以後の[Ca++]iの変化を追跡した。100μMのフォルスコリン(アデニルシクラーゼのアクチベータ)の添加に応答して、64個の細胞の平均した結果と各細胞で、[Ca++]iにわずかな増加が見られた。1mMのIBMX(cAMPホスホジエステラーゼの阻害剤)の添加後、さらに有意な増加が見られた。対照実験では、64個の非トランスフェクションHEK293細胞中の1つのみが、細胞内cAMPレベルの上昇に対応して、[Ca++]iの増加を示した。この応答は一時的であり、サイクリックヌクレオチドゲートのチャネルDNAでトランスフェクションしたHEK293細胞中で見られた持続性応答とは明らかに異なっていた。
【0168】
これらの結果は、サイクリックヌクレオチドゲートのチャネルを発現するHEK細胞は、活性化された時細胞内サイクリックヌクレオチドレベルに変化を引き起こす受容体(例えば、メタボトロピック受容体)の測定において宿主細胞として使用できることを証明している。
【0169】
5.ノーザンブロットハイブリダイゼーション解析
ヒトメタボトロピック受容体をコードするDNAでトランスフェクションした細胞はまた、ノーザンブロット解析により対応する転写体の発現について解析することができる。ヒトメタボトロピック受容体をコードするDNAでトランスフェクションした約1×107個の細胞から総RNAを単離し、10−15μgのRNAをノーザンハイブリダイゼーション解析に使用した。ヒトメタボトロピック受容体をコードするプラスミドからの挿入体をニックトランスレーションし、プローブとして使用した。ノーザンブロットハイブリダイゼーションの典型的な条件は以下の通りである:
ハイブリダイゼーションは、42℃で5×SSPE、5×デンハルツ(Denhart's)溶液、50%ホルムアルミド中で行ない、洗浄は65℃で0.2×SSPEと0.1%SDS中で行なった。
【0170】
本発明をいくつかの好適な実施態様を参照して詳細に説明したが、ここに記載され特許請求のされている本発明の精神と範囲を逸脱することなくその修飾や変更が可能であることが理解されるであろう。
【0171】
(配列の要約)
配列番号1は、本発明のメタボトロピックグルタミン酸受容体サブタイプ(mGluR1B)をコードするDNAの核酸配列(およびその推定アミノ酸配列)である。
配列番号2は、配列番号1のヌクレオチド配列の推定アミノ酸配列である。
配列番号3は、ヒトmGluR2受容体サブタイプの一部をコードする部分クローンのヌクレオチド配列(および推定アミノ酸配列)である。
配列番号4は、配列番号3のヌクレオチド配列によりコードされるヒトmGluR2受容体サブタイプの一部のアミノ酸配列である。
配列番号5は、本発明のメタボトロピックグルタミン酸受容体サブタイプ(mGluR3)をコードするDNAの核酸配列(および推定アミノ酸配列)である。
配列番号6は、配列番号5のヌクレオチド配列の推定アミノ酸配列である。
配列番号7は、本発明のメタボトロピックグルタミン酸受容体(mGluR5a1)をコードするDNAの核酸配列(および推定アミノ酸配列)である。
配列番号8は、配列番号7のヌクレオチド配列の推定アミノ酸配列である。
配列番号9は、本発明のmGluR5変種メタボトロピックグルタミン酸受容体(mGluR5b)をコードするDNAの核酸配列(および推定アミノ酸配列)である。
配列番号10は、配列番号9のヌクレオチド配列の推定アミノ酸配列である。
配列番号11は、本発明のmGluR5変種メタボトロピックグルタミン酸受容体(mGluR5c)をコードするDNAの核酸配列(および推定アミノ酸配列)である。
配列番号12は、配列番号11のヌクレオチド配列の推定アミノ酸配列である。
配列番号13は、ヒトmGluR2受容体サブタイプの3’非翻訳配列の343ヌクレオチドである。
【0172】
(配列表)
(1)一般情報:
(i) 出願人:ダゲット,ロリー(Daggett, Lorrie)
エリス,スティーブン・ビー(Ellis, Steven B.)
ルー,チェン(Lu, Chen)
ポンツラー,アーロン(Pontsler, Aaron)
ジョンソン,エドウィン・シー(Johnson, Edwin C.)
ヘス,ステファン・ディー(Hess, Stephen D.)
(ii)発明の名称:ヒトメタボトロピックグルタミン酸受容体、これをコードする核酸およびその用途
(iii)配列の数:13
(iv)連絡住所:
(A)宛名:プリティー、シュレーダー、ブルーゲマンおよびクラーク(Pretty, Schroeder, Brueggemann & Clark)
(B)通り:444サウス・フラワー通り、スイート2000(444 South Flower Street, Suite 2000 )
(C)市:ロサンゼルス
(D)州:カリホルニア
(E)国:米国
(F)郵便番号:90071
(v) コンピューターで読める形式:
(A)媒体の型:フロッピー(登録商標)ディスク
(B)コンピューター:IBM PCコンパチブル
(C)オペレーティング・システム:PC−DOS/MS−DOS
(D)ソフトウェア:パテントイン・リリース#1.0(PatentIn Release #1.0)、バージョン#1.25
(vi)現行出願のデータ:
(A)出願番号:
(B)出願日:1994年6月2日
(C)分類:
(vii)先行出願のデータ:
(A)出願番号:US08/072、574
(B)出願日:1993年6月4日
(viii)弁理士/代理人情報:
(A)氏名:ライター,ステファン・イー(Reiter, Stephen E.)
(B)登録番号:31,192
(C)参照/整理番号:FP41 9772
(ix)電話連絡先情報:
(A)電話:619−546−4737
(B)ファックス:619−546−9392
(2)配列番号:1の情報:
(i) 配列の特色:
(A)長さ:3321塩基対
(B)型:核酸
(C)鎖の数:両形態
(D)トポロジー:両形態
(ii)分子の型:cDNA
(ix)配列の特徴:
(A)名称/記号:CDS
(B)存在位置:388..3108
(D)他の情報:/生成物=“ヒトmGluR1B”
(xi)配列:配列番号:1:
【0173】
(2)配列番号:2の情報:
(i) 配列の特色:
(A)長さ:906アミノ酸
(B)型:アミノ酸
(D)トポロジー:直鎖状
(ii)分子の型:蛋白
(xi)配列:配列番号:2:
【0174】
(2)配列番号:3の情報:
(i) 配列の特色:
(A)長さ:355塩基対
(B)型:核酸
(C)鎖の数:両形態
(D)トポロジー:両形態
(ii)分子の型:cDNA
(ix)配列の特徴:
(A)名称/記号:CDS
(B)存在位置:1..354
(D)他の情報:/生成物=“ヒトmGluR2断片”
(xi)配列:配列番号:3:
【0175】
(2)配列番号:4の情報:
(i) 配列の特色:
(A)長さ:118アミノ酸
(B)型:アミノ酸
(D)トポロジー:直鎖状
(ii)分子の型:蛋白
(xi)配列:配列番号:4:
【0176】
(2)配列番号:5の情報:
(i) 配列の特色:
(A)長さ:3919塩基対
(B)型:核酸
(C)鎖の数:両形態
(D)トポロジー:両形態
(ii)分子の型:cDNA
(ix)配列の特徴:
(A)名称/記号:CDS
(B)存在位置:1064..3703
(D)他の情報:/生成物=“ヒトmGluR3”
(xi)配列:配列番号:5:
【0177】
(2)配列番号:6の情報:
(i) 配列の特色:
(A)長さ:879アミノ酸
(B)型:アミノ酸
(D)トポロジー:直鎖状
(ii)分子の型:蛋白
(xi)配列:配列番号:6:
【0178】
(2)配列番号:7の情報:
(i) 配列の特色:
(A)長さ:4085塩基対
(B)型:核酸
(C)鎖の数:両形態
(D)トポロジー:両形態
(ii)分子の型:cDNA
(ix)配列の特徴:
(A)名称/記号:CDS
(B)存在位置:370..3912
(D)他の情報:/生成物=“ヒトmGluR5A”
(xi)配列:配列番号:7:
【0179】
(2)配列番号:8の情報:
(i) 配列の特色:
(A)長さ:1180アミノ酸
(B)型:アミノ酸
(D)トポロジー:直鎖状
(ii)分子の型:蛋白
(xi)配列:配列番号:8:
【0180】
(2)配列番号:9の情報:
(i) 配列の特色:
(A)長さ:4181塩基対
(B)型:核酸
(C)鎖の数:両形態
(D)トポロジー:両形態
(ii)分子の型:cDNA
(ix)配列の特徴:
(A)名称/記号:CDS
(B)存在位置:370..4008
(D)他の情報:/生成物=“ヒトmGluR5B”/注=“ヌクレオチド2998と2999の間に96塩基対が挿入されたmGluR5A変種”
(xi)配列:配列番号:9:
【0181】
(2)配列番号:10の情報:
(i) 配列の特色:
(A)長さ:1212アミノ酸
(B)型:アミノ酸
(D)トポロジー:直鎖状
(ii)分子の型:蛋白
(xi)配列:配列番号:10:
【0182】
(2)配列番号:11の情報:
(i) 配列の特色:
(A)長さ:3282塩基対
(B)型:核酸
(C)鎖の数:両形態
(D)トポロジー:両形態
(ii)分子の型:cDNA
(ix)配列の特徴:
(A)名称/記号:CDS
(B)存在位置:370..3003
(D)他の情報:/生成物=“ヒトmGluR5C”/注=“3’末端が切り取られたmGluR5aの変種”
(xi)配列:配列番号:11:
【0183】
(2)配列番号:12の情報:
(i) 配列の特色:
(A)長さ:877アミノ酸
(B)型:アミノ酸
(D)トポロジー:直鎖状
(ii)分子の型:蛋白
(xi)配列:配列番号:12:
【0184】
(i) 配列の特色:
(A)長さ:343塩基対
(B)型:核酸
(C)鎖の数:両形態
(D)トポロジー:両形態
(ii)分子の型:cDNA
(ix)配列の特徴:
(A)名称/記号:種々の特徴
(B)存在位置:1..343
(D)他の情報:/注=“mGluR2の部分配列−3’非翻訳配列”
(xi)配列:配列番号:13:
【図面の簡単な説明】
【0185】
【図1】図1は、CMVプロモーターをベースにしたベクターであるpCMV−T7−2とpCMV−T7−3を示す。
【特許請求の範囲】
【請求項1】
以下の(a)または(b)から選択されることを特徴とする単離されたDNA:
(a)配列番号3に記載の全コード領域を有するDNA、
(b)配列番号3に記載の全コード領域を有するDNAと相補的な塩基配列からなるDNAと、高緊縮性条件下でハイブリダイズし、かつヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)としての活性を有する蛋白をコードするDNA。
【請求項2】
以下の(a)または(b)の蛋白をコードすることを特徴とする単離されたDNA:
(a)配列番号4に記載のアミノ酸配列を有する蛋白、
(b)配列番号4に記載のアミノ酸配列において、1または数個のアミノ酸を欠失、置換および/または付加して(a)の蛋白から得られ、ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)としての活性を有する蛋白。
【請求項3】
請求項1又は2に記載のDNAによりコードされる単離された蛋白。
【請求項4】
請求項1又は2に記載のDNAのうち、少なくとも14の連続する塩基、またはその相補的な塩基よりなる、核酸プローブ。
【請求項5】
請求項1又は2に記載のDNAに相補的な単離されたmRNA。
【請求項6】
請求項1又は2に記載のDNAを含有する真核細胞。
【請求項7】
請求項1又は2に記載のDNAを発現する真核細胞。
【請求項8】
請求項5に記載のmRNAを発現する両生類卵母細胞。
【請求項9】
ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)をコードするDNAの同定方法であって、
請求項4に記載の核酸プローブと、ヒトDNAを、該核酸プローブがポリ核酸断片の時は低緊縮性ハイブリダイゼーション条件から中緊縮性ハイブリダイゼーション条件下で、該核酸プローブがオリゴヌクレオチドの時は高緊縮性ハイブリダイゼーション条件下で、接触させて、該プローブにハイブリダイズするDNAを同定することを特徴とするDNAの同定方法。
【請求項10】
競合的結合測定法において、請求項3に記載の蛋白を用いることを特徴とする、ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)に結合する化合物の同定方法。
【請求項11】
ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)の活性を調節する化合物を同定するための生物学的測定法であって、
(a)請求項7に記載の真核細胞を、受容体サブタイプの第二メッセンジャー活性の調節能力を測定すべき少なくとも1つの化合物に暴露させ、
(b)その後、該第二メッセンジャー活性の変化について該真核細胞を追跡することを特徴とする生物学的測定法。
【請求項12】
ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)を、請求の範囲第11項に記載の生物測定法により同定される少なくとも1つの化合物に、該化合物が有効量存在する状態で接触させることを特徴とする、ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)の第二メッセンジャー活性の調節方法。
【請求項13】
請求項3に記載の蛋白またはその免疫原性を発現する部位に対する抗体。
【請求項14】
該抗体はモノクローナル抗体である、請求項13に記載の抗体。
【請求項15】
ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)を、請求項14項に記載のモノクロナール抗体に、該抗体が有効量存在する状態で接触させることよりなる、ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)の第二メッセンジャー活性の調節方法。
【請求項1】
以下の(a)または(b)から選択されることを特徴とする単離されたDNA:
(a)配列番号3に記載の全コード領域を有するDNA、
(b)配列番号3に記載の全コード領域を有するDNAと相補的な塩基配列からなるDNAと、高緊縮性条件下でハイブリダイズし、かつヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)としての活性を有する蛋白をコードするDNA。
【請求項2】
以下の(a)または(b)の蛋白をコードすることを特徴とする単離されたDNA:
(a)配列番号4に記載のアミノ酸配列を有する蛋白、
(b)配列番号4に記載のアミノ酸配列において、1または数個のアミノ酸を欠失、置換および/または付加して(a)の蛋白から得られ、ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)としての活性を有する蛋白。
【請求項3】
請求項1又は2に記載のDNAによりコードされる単離された蛋白。
【請求項4】
請求項1又は2に記載のDNAのうち、少なくとも14の連続する塩基、またはその相補的な塩基よりなる、核酸プローブ。
【請求項5】
請求項1又は2に記載のDNAに相補的な単離されたmRNA。
【請求項6】
請求項1又は2に記載のDNAを含有する真核細胞。
【請求項7】
請求項1又は2に記載のDNAを発現する真核細胞。
【請求項8】
請求項5に記載のmRNAを発現する両生類卵母細胞。
【請求項9】
ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)をコードするDNAの同定方法であって、
請求項4に記載の核酸プローブと、ヒトDNAを、該核酸プローブがポリ核酸断片の時は低緊縮性ハイブリダイゼーション条件から中緊縮性ハイブリダイゼーション条件下で、該核酸プローブがオリゴヌクレオチドの時は高緊縮性ハイブリダイゼーション条件下で、接触させて、該プローブにハイブリダイズするDNAを同定することを特徴とするDNAの同定方法。
【請求項10】
競合的結合測定法において、請求項3に記載の蛋白を用いることを特徴とする、ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)に結合する化合物の同定方法。
【請求項11】
ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)の活性を調節する化合物を同定するための生物学的測定法であって、
(a)請求項7に記載の真核細胞を、受容体サブタイプの第二メッセンジャー活性の調節能力を測定すべき少なくとも1つの化合物に暴露させ、
(b)その後、該第二メッセンジャー活性の変化について該真核細胞を追跡することを特徴とする生物学的測定法。
【請求項12】
ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)を、請求の範囲第11項に記載の生物測定法により同定される少なくとも1つの化合物に、該化合物が有効量存在する状態で接触させることを特徴とする、ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)の第二メッセンジャー活性の調節方法。
【請求項13】
請求項3に記載の蛋白またはその免疫原性を発現する部位に対する抗体。
【請求項14】
該抗体はモノクローナル抗体である、請求項13に記載の抗体。
【請求項15】
ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)を、請求項14項に記載のモノクロナール抗体に、該抗体が有効量存在する状態で接触させることよりなる、ヒトメタボトロピックグルタミン酸受容体サブタイプ2(ヒトmGluR2)の第二メッセンジャー活性の調節方法。
【図1】

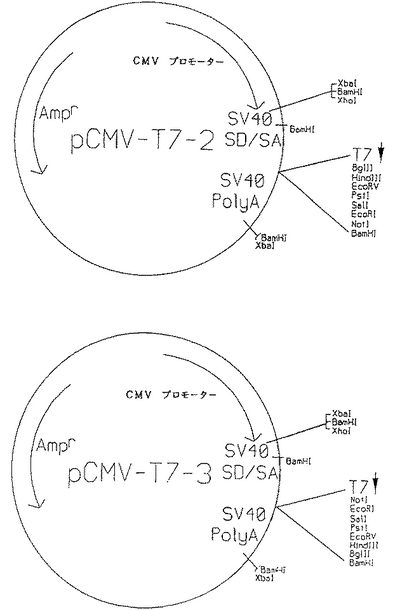
【公開番号】特開2006−254915(P2006−254915A)
【公開日】平成18年9月28日(2006.9.28)
【国際特許分類】
【出願番号】特願2006−85289(P2006−85289)
【出願日】平成18年3月27日(2006.3.27)
【分割の表示】特願2004−340504(P2004−340504)の分割
【原出願日】平成6年6月3日(1994.6.3)
【出願人】(500019948)シビア・ニューロサイエンシーズ・インコーポレイテッド (1)
【Fターム(参考)】
【公開日】平成18年9月28日(2006.9.28)
【国際特許分類】
【出願日】平成18年3月27日(2006.3.27)
【分割の表示】特願2004−340504(P2004−340504)の分割
【原出願日】平成6年6月3日(1994.6.3)
【出願人】(500019948)シビア・ニューロサイエンシーズ・インコーポレイテッド (1)
【Fターム(参考)】
[ Back to top ]