
ヒト化K33Nモノクローナル抗体の作製、発現及び解析
本発明は、ヒトα9インテグリンを免疫特異的に認識するヒト化抗体を提供する。これらの抗体のいくつかは、α9インテグリンの生物学的機能を阻害し、それにより、例えば、癌細胞の増殖及び転移等の癌、並びに、例えば、関節リウマチ、変形性関節症、肝炎、気管支喘息、線維症、糖尿病、動脈硬化症、多発性硬化症、肉芽種、炎症性腸疾患(潰瘍性大腸炎及びクローン病)、自己免疫疾患等の炎症性疾患を含む、α9インテグリンと関連する様々な障害又は疾患に対する治療効果を示す。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、免疫特異的にヒトα9インテグリンを認識するヒト化抗体、並びに癌、炎症性疾患、自己免疫疾患、α9インテグリンにより誘導される疾患状態等を含む、α9インテグリンと関連するか又はα9インテグリンが関与する様々な疾患又は障害に対する治療及び診断における該抗体の使用に関する。
【背景技術】
【0002】
細胞は、インテグリンと呼ばれる一群の細胞表面受容体を介して細胞外マトリクス(以下、ECMと略す)に接着している。インテグリンは、α鎖とβ鎖の1:1のヘテロ二量体を形成することによりその機能を果たす。これまでに、少なくとも18種類のα鎖、8種類のβ鎖、及び24種類のαβヘテロ二量体が同定され、確認されている。各々のインテグリンは特異的なリガンドを認識することが知られている。インテグリンは、そのリガンド特異性又は機能によってサブファミリーに分類され、コラーゲン受容体、ラミニン受容体、フィブロネクチンやビトロネクチン等に存在するArg−Gly−Asp(RGD)配列を認識するRGD受容体、白血球にのみ存在する白血球特異的受容体に分けられる(Hynes,R.O.,2002,Integrins:Bidirectional,Allosteric Signaling Machines.Cell 110:673−87;宮坂 昌之,2000,新版 接着分子ハンドブック,秀潤社)。α4インテグリンとα9インテグリンは、これらのタイプのいずれにも属さないサブファミリーのメンバーであり、α4インテグリンサブファミリーと呼ばれる(Elise L.Palmer,Curzio Rfiegg,Ronald Ferrando,Robert Pytela,Sheppard D.,1993,Sequence and Tissue Distribution of the Integrin α9 Subunit,a Novel Partner of β1 That Is Widely Distributed in Epithelia and Muscle.The Journal of Cell Biology,123:1289−97)。一方、しばらくの間ECMは、細胞間を固定する物質として機能するにすぎないと考えられていた。しかし、現在は、インテグリン介在性のECM−細胞相互作用が、細胞の増殖、接着、移動等の調節に明らかに関与し、癌の進行、炎症の悪化等を含む疾患の発症と関連することが明らかになっている。
【0003】
例えば、ECMの1つであるオステオポンチン(以後、OPNと略す)は、分子量約41kDaの、分泌型の酸性リン酸化糖タンパク質であり、母乳、尿、尿細管、破骨細胞、骨芽細胞、マクロファージ、活性化T細胞、腫瘍組織等に広くその発現が認められる分子である。OPNは、その分子の中心に接着配列GRGDS(配列番号1)を有し、ヒトOPNはSVVYGLR(配列番号2)配列又はマウスOPNはSLAYGLR(配列番号3)配列、及びそれらに近接するトロンビン切断部位を有する。OPNは、GRGDS(配列番号1)配列を介してRGDインテグリンと結合し、また、SVVYGLR(配列番号2)配列もしくはSLAYGLR(配列番号3)配列を介してα4(α4β1)インテグリン及びα9(α9β1)インテグリンと結合する。
【0004】
WO02/081522号は、OPNノックアウトマウス又はOPNに対する中和抗体を用いてOPNの機能を阻害することにより、関節リウマチ又は肝炎を治療する効果があることを開示している。また、該文献は、SVVYGLR(配列番号2)配列が、α9インテグリン及びα4インテグリンを認識するため炎症性疾患の発症に不可欠であること、及び、OPNの受容体が免疫担当細胞等で発現して、炎症性疾患と関連していることを開示している。
【0005】
結合プロファイルの違いとして、α4β1がトロンビンで切断されていないOPN(未切断型OPN)とトロンビンで切断されたOPNのN末端断片(切断型OPN)の両方に結合するのに対し、α9β1は切断型OPNにのみ結合することが見出されている(Y.Yokosaki,et al.,(1999) The Journal of Biological Chemistry),274:36328−36334;P.M.Green,et al.,(2001) FEBS Letters,503:75−79;S.T.Barry,et al.,(2000) Experimental Cell Research,258:342−351)。
【0006】
α4インテグリンとα9インテグリンは、OPN以外にも多くのリガンドを共有している。知られているリガンドとしては、フィブロネクチンのEDAドメイン、プロペプチド−フォン・ヴィレブランド因子(pp−vWF)、組織トランスグルタミナーゼ(tTG)、血液凝固第XIII因子、血管細胞接着分子−1(VCAM−1)等を挙げることができる。また、フィブロネクチンのCS−1ドメイン、MadCAM−1(α4β7)等は、α4インテグリンに特異的に認識されるリガンドとして知られている。テネイシン−C、プラスミン等は、α9インテグリンによって特異的に認識されるリガンドとして知られている。
【0007】
インテグリンサブユニットα9、α4及びβ1のアミノ酸配列は公知である。例えば、ヒトα9はNM_002207として、マウスα9はNM_133721として、ヒトα4はNM_000885として、マウスα4はNM_010576として、ヒトβ1はX07979として、及びマウスβ1はNM_010578としてGenBankに登録されている。これらのインテグリンは、種間でアミノ酸配列における高い類似性を有することも知られている。
【発明の概要】
【0008】
現在、癌、炎症性疾患及び自己免疫疾患を治療するための多くの種類の薬物が知られているが、癌、炎症性疾患及び自己免疫疾患に対する薬理効果がより改善された予防剤及び/又は治療剤等の開発が望まれている。本発明は、一部、α9インテグリンに対する特異的阻害抗体に癌抑制効果や抗炎症効果があるという本発明者らによる発見に基づくものである。
【0009】
これまで本発明者らは、免疫特異的にヒトα9インテグリンを認識し、ハイブリドーマクローンK33N(受託番号FERM BP−10830)によって産生されるマウスモノクローナル抗体を単離した。本明細書においては、ハイブリドーマクローンの名称は、このクローンによって産生されるモノクローナル抗体の名称と共通で使用する。該マウス抗ヒトα9インテグリン抗体はIgG1アイソタイプであった。また、該モノクローナル抗体は、ヒト及び/又はマウスのα9インテグリンと、オステオポンチン等のα9インテグリンのリガンドとの結合を阻害する。よって、該抗α9インテグリン抗体はα9インテグリンの機能を阻害し、例えば、癌細胞の増殖又は転移等の癌、並びに、例えば、関節リウマチ、変形性関節炎、肝炎、気管支喘息、線維症、糖尿病、動脈硬化症、多発性硬化症、肉芽種、炎症性腸疾患(潰瘍性大腸炎及びクローン病)、自己免疫疾患、α9インテグリンによって誘導される疾患状態等の炎症性疾患に対する治療効果を示す。
【0010】
さらに、本発明の抗α9インテグリン抗体は、対象におけるα9インテグリン発現の存在とレベルを検出し、それによりα9インテグリンと関係する障害又は疾患を診断するためのインビボでの診断薬として用いることができる。
【0011】
しかし、該モノクローナル抗体はマウス由来であることから、ヒトにおいてはその免疫原性による副作用の可能性があり、このことがヒトにおいて診断用又は治療用として直接適用することを妨げてきた。この免疫原性を低下させるために、本発明者らは、由来するマウス抗α9インテグリン抗体が示す活性と同等の生物学的活性を有するヒト化抗体を調製した。
【0012】
したがって、本発明は、免疫特異的にヒトα9インテグリンを認識するヒト化抗体又はその抗原結合断片であって、一部が非ヒト起源に由来し、一部がヒト起源に由来する抗原結合領域を含む抗体又はその抗原結合断片を提供するものである。具体的な実施形態においては、本発明のヒト化抗体又はその抗原結合断片は、K33Nモノクローナル抗体等の非ヒト源(ドナー)に由来する相補性決定領域(CDR)と、ヒト源(アクセプター)に由来するフレームワーク領域(FR)とを含む。一実施形態では、該ヒト化抗体又はその抗原結合断片は、ヒトα9インテグリンとヒトα9インテグリンのリガンドとの結合を阻害する。
【0013】
具体的な実施形態においては、上述の免疫特異的にヒトα9インテグリンを認識するヒト化抗体又はその抗原結合断片は、(i)ヒトH鎖の可変領域(V領域)に由来する少なくとも1つのH鎖FR(FRH)と、免疫特異的にヒトα9インテグリンを認識する非ヒト抗体K33NのCDRHの少なくとも1つに由来する少なくとも1つのH鎖相補性決定領域(CDRH)とを含む重鎖(H鎖);又は(ii)ヒトL鎖のV領域に由来する少なくとも1つのL鎖FR(FRL)と、免疫特異的にヒトα9インテグリンを認識する非ヒト抗体K33NのCDRLの少なくとも1つに由来する少なくとも1つのL鎖相補性決定領域(CDRL)とを含む軽鎖(L鎖);又は上記の(i)と(ii)の両方を含む。例えば、本発明のヒト化抗体のCDRHの少なくとも1つ及び/又はCDRLの少なくとも1つが由来する該非ヒト抗体は、受託番号FERM BP−10830のハイブリドーマによって産生されるモノクローナル抗体である。
【0014】
好ましい実施形態では、本発明のヒト化抗体又はその抗原結合断片は、(i)ヒトFRHに由来する少なくとも1つのFRHと、配列番号4、5及び6のアミノ酸配列からなる群から選択されるアミノ酸配列を含む少なくとも1つのCDRH;又は(ii)ヒトFRLに由来する少なくとも1つのFRLと、配列番号11、12及び13のアミノ酸配列からなる群から選択されるアミノ酸配列を含む少なくとも1つのCDRL;又は(iii)上記の(i)と(ii)の両方を含む。本発明の該ヒト化抗体又はその抗原結合断片は、それぞれが、配列番号4、5及び6のアミノ酸配列を有する、CDRH1、CDRH2及びCDRH3を含んでいてもよい。あるいは、本発明の該ヒト化抗体又はその抗原結合断片は、それぞれが、配列番号11、12及び13のアミノ酸配列を有する、CDRL1、CDRL2及びCDRL3を含む。好ましい実施形態では、上述の本発明のヒト化抗体又はその抗原結合断片は、それぞれ、配列番号4、5、6、11、12及び13のアミノ酸配列を有する、CDRH1、CDRH2、CDRH3、CDRL1、CDRL2及びCDRL3を含む。あるいは、本発明の該ヒト化抗体又はその抗原結合断片は、GenBankアクセッション番号DA980102(配列番号18)によってコードされるヒトH鎖の可変領域に由来するFRH、又はGenBankアクセッション番号X72441(配列番号23)によってコードされるヒトκ−L鎖の可変領域に由来するFRLを含む。好ましい実施形態では、本発明のヒト化抗体のFRHは、配列番号19、20、21及び22のアミノ酸配列(それぞれ順に、DA980102の対応する部分によってコードされるFRH1、FRH2、FRH3及びFRH4)からなる群から選択される少なくとも1つのアミノ酸配列を含む。別の好ましい実施形態では、本発明のヒト化抗体のFRLは、配列番号24、25、26及び27のアミノ酸配列(それぞれ順に、X72441の対応する部分によってコードされるFRL1、FRL2、FRL3及びFRL4)からなる群から選択される少なくとも1つのアミノ酸配列を含む。より好ましい実施形態では、本発明のヒト化抗体又はその抗原結合断片は、(i)配列番号29のアミノ酸配列を含むH鎖可変領域(VH領域);又は(ii)配列番号31のアミノ酸配列を含むL鎖可変領域(VL領域);又は(iii)上記の(i)と(ii)の両方を含む。最も好ましい実施形態では、本発明のヒト化抗体又はその抗原結合断片は、(i)配列番号37のアミノ酸配列を含むγ1H鎖;又は(ii)配列番号39のアミノ酸配列を含むκL鎖;又は(iii)上記の(i)と(ii)の両方を含む。
【0015】
本発明はさらに、免疫特異的にヒトα9インテグリンを認識する本発明のヒト化抗体又はその抗原結合断片をコードするヌクレオチド配列を含む単離された核酸分子を提供する。具体的には、本発明は、配列番号4、5及び6からなる群から選択される少なくとも1つのアミノ酸配列を含むヒト化H鎖、又は配列番号11、12及び13からなる群から選択される少なくとも1つのアミノ酸配列を含むヒト化L鎖、又は該ヒト化H鎖と該ヒト化L鎖の両方をコードするヌクレオチド配列を含む単離された核酸分子を提供する。好ましい具体的な実施形態では、このような単離された核酸分子は、VH領域をコードする配列番号28のヌクレオチド配列、又は配列番号29のアミノ酸配列をコードするヌクレオチド配列を含む。別の好ましい具体的な実施形態では、このような単離された核酸分子は、VL領域をコードする配列番号30のヌクレオチド配列、又は配列番号31のアミノ酸配列をコードするヌクレオチド配列を含む。さらに別の好ましい具体的な実施形態では、本発明の単離された核酸分子は、配列番号28と30の両方のヌクレオチド配列を含む。好ましい具体的な実施形態では、このような単離された核酸分子は、γ1H鎖をコードする配列番号36のヌクレオチド配列、又は配列番号37のアミノ酸配列をコードするヌクレオチド配列を含む。別の好ましい具体的な実施形態では、このような単離された核酸分子は、κL鎖をコードする配列番号38のヌクレオチド配列、又は配列番号39のアミノ酸配列をコードするヌクレオチド配列を含む。さらに別の好ましい具体的な実施形態では、本発明の単離された核酸分子は、配列番号36と38の両方のヌクレオチド配列を含む。別の好ましい具体的な実施形態では、本発明の単離された核酸分子は、更に、例えば、それぞれ、配列番号10及び17のアミノ酸配列のようなドナー起源のシグナルペプチド又は異種起源のシグナルペプチドをコードするヌクレオチド配列を含む。
【0016】
本発明はさらに、免疫特異的にヒトα9インテグリンを認識する本発明のヒト化抗体又はその抗原結合断片のH鎖又はL鎖又はその両方をコードするヌクレオチド配列を含むベクター(例えば、発現ベクター)を提供する。このようなベクターでは、本発明のヌクレオチド配列は、1以上の調節エレメントに機能的に連結され得る。本発明のヌクレオチド配列は、CDRが由来する非ヒトドナー抗体起源のシグナルペプチド、又は異種起源のシグナルペプチドをコードするヌクレオチド配列を含み得る。
【0017】
さらに、本発明は、本発明の核酸分子(本発明の核酸分子を含むベクターを含む)を含む宿主細胞を提供する。一実施形態では、本発明は、本発明のヒト化H鎖をコードする第1の核酸分子と、本発明のヒト化L鎖をコードする第2の核酸分子とを含む単離された宿主細胞を提供するものであり、該第1及び第2の核酸分子は、本発明の生物学的に機能的なヒト化抗体又はその抗原結合断片が発現されるように、それぞれが調節エレメントに機能的に連結されている。
【0018】
したがって、本発明はさらに、ヒト化抗体が発現されるような条件下で本発明の宿主細胞を培養することと、及び産生されたヒト化抗体を回収することとを含む、本発明のヒト化抗体の調製方法を提供する。
【0019】
本発明はさらに、本発明のヒト化抗体の少なくとも1つを含む組成物を提供する。さらに、本発明は、本発明のヒト化抗体の少なくとも1つと薬学的に許容される担体とを含む、α9インテグリンと関連する障害又は疾患を予防又は治療するための薬学的組成物を提供する。前記のいずれの組成物も、当該障害又は疾患を相加的に又は相乗的に改善することができる別の活性物質をさらに含むことができる。このような活性物質としては、これに限定されるものではないが、抗炎症化合物、化学療法剤等や、抗体又はその抗原結合断片(例えば、ヒトα4インテグリンに免疫特異的に結合することができる抗体)が挙げられる。
【0020】
別の態様では、本発明は、α9インテグリンと関連するか又はα9インテグリンが関与する疾患又は障害を予防又は治療する方法であって、予防に有効な量又は治療に有効な量の本発明のヒト化抗体の少なくとも1つをそれを必要とする対象に投与することを含む方法を提供する。このような使用のために、本発明のヒト化抗体はヒト化抗体の生物学的効果を増強する治療的成分と結合していてもよい。このような治療的成分の例としては、(例えば、二重特異性抗体を形成するための)抗α4抗体等の別の抗体、細胞増殖抑制性もしくは細胞破壊性の細胞毒素、放射性元素、及び/又は抗炎症剤、抗生物質等を含む他の治療剤が挙げられる。
【0021】
さらに別の態様では、本発明は、対象におけるα9インテグリンと関連するか又はα9インテグリンが関与する疾患又は障害を診断する方法であって、診断に有効な量の本発明のヒト化抗体を検査対象に投与することを含む方法を提供する。このような診断への使用のために、本発明のヒト化抗体を放射性元素等の検出可能なマーカーで標識してもよい。
【0022】
3.1.定義
本明細書で使用する場合、「抗体」という用語は、例えば、α9インテグリン等の所望の抗原に免疫特異的に結合可能な抗体分子を指し、完全な抗体分子、又は抗原結合断片を含む抗体分子の断片を包含する。
【0023】
本明細書で使用される「免疫特異的に認識する」という用語は、標的ポリペプチド又はタンパク質、特にヒトα9インテグリンに特異的に結合する、抗体又はその抗原結合断片の能力を指す。このような抗体は、他のポリペプチド又はタンパク質に非特異的に結合しない。しかしながら、標的ポリペプチド又はタンパク質(例えば、ヒトα9インテグリン)に免疫特異的に結合する抗体又はその抗原結合断片は、他の抗原と交差反応してもよい。例えば、ヒトα9インテグリンを免疫特異的に認識する本発明のヒト化抗体又は抗原結合断片は、例えば、他の種のα9インテグリンと交差反応してもよい。好ましくは、ヒトα9インテグリンと免疫特異的に結合する抗体又はその抗原結合断片は、他の抗原と交差反応しない。
【0024】
本明細書で使用される「抗原結合断片」という用語は、標的ポリペプチド又はタンパク質、特に、ヒトα9インテグリン及び/又は非ヒトα9インテグリンと免疫特異的に結合する能力を保持する抗体の任意の断片を指し、単鎖抗体、Fab断片、F(ab’)2断片、ジスルフィド結合Fv、並びに標的ポリペプチド又はタンパク質と特異的に結合する軽鎖(VL)の可変領域及び/又は重鎖(VH)の可変領域又は相補性決定領域(CDR)を含有する断片を含む。したがって、ヒト化抗体のこのような抗原結合断片には、部分的又は全長のヒト定常領域が含まれていても、含まれていなくてもよい。上記の抗体断片を得るための様々な方法が当該技術分野で周知である。
【0025】
本明細書で使用される「ヒト源に由来する」又は「非ヒト源に由来する」という用語は、抗体の一部であってそのアミノ酸配列がヒト抗体又は非ヒト抗体の対応する部分に由来する部分を指す。
【0026】
本明細書で使用される「アクセプター配列」という用語は、通常、非ヒト抗体であるドナー抗体由来のCDRのアクセプターとして働く、ヒト抗体VH領域又はVL領域に由来するフレームワーク領域のヌクレオチド配列又はアミノ酸配列を指す。
【図面の簡単な説明】
【0027】
【図1】抗ヒトα9インテグリン抗体(本発明の2つのクローン(すなわち、K33N及びM35A)、5つの他のクローン(1K11、21C5、24I11、25B6、及び28S1)、並びにY9A2)の細胞接着阻害活性を、ヒトα9インテグリン発現細胞(ヒトメラノーマ細胞G361)とOPN α9インテグリン結合部位ペプチド(SVVYGLR)とを用いて測定した実験の結果を示す図である。ヒトオステオポンチンに対するモノクローナル抗体(5A1)を陰性対照として使用した。
【図2】抗ヒトα9インテグリン抗体(本発明の2つのクローン(すなわち、K33N及びM35A)、5つの他のクローン(1K11、21C5、24I11、25B6、及び28S1)、並びにY9A2)の細胞接着阻害活性を、ヒトα9インテグリン発現細胞(ヒトメラノーマ細胞G361)とテネイシン−C断片のα9インテグリン結合部位ペプチドとを用いて測定した実験の結果を示す図である。ヒトオステオポンチンに対するモノクローナル抗体(5A1)を陰性対照として使用した。
【図3】マウスK33N VH cDNAのヌクレオチド配列(配列番号7)を推定アミノ酸配列(配列番号8)と共に示す図である。アミノ酸残基は一文字表記で示す。シグナルペプチド配列(配列番号10)はイタリック体で示す。成熟VHのN末端アミノ酸残基(Q)は二重下線で示す。Kabatら(Sequences of Proteins of Immunological Interests,第5版,NIH Publication 第91−3242号,U.S.Department of Health and Human Services,1991)の定義によるCDR配列を下線で示す。
【図4】マウスK33N VL cDNAのヌクレオチド配列(配列番号14)を推定アミノ酸配列(配列番号15)と共に示す図である。アミノ酸残基は一文字表記で示す。シグナルペプチド配列(配列番号17)はイタリック体で示す。成熟VLのN末端アミノ酸残基(D)は二重下線で示す。Kabatら(1991)の定義によるCDR配列を下線で示す。
【図5】SpeI部位とHindIII部位(下線)が両側にあるようにデザインされたK33N VH(ChK33N VH)遺伝子のヌクレオチド配列(配列番号32)を推定アミノ酸配列(配列番号8)と共に示す。アミノ酸残基は一文字表記で示す。シグナルペプチド配列(配列番号10)はイタリック体で示す。成熟VHのN末端アミノ酸残基(Q)は二重下線で示す。Kabatら(1991)の定義によるCDR配列を下線で示す。イントロン配列をイタリック体で示す。
【図6】NheI部位とEcoRI部位(下線)が両側にあるようにデザインされたK33N VL(ChK33N VL)遺伝子のヌクレオチド配列(配列番号33)を推定アミノ酸配列(配列番号15)と共に示す。アミノ酸残基は一文字表記で示す。シグナルペプチド配列(配列番号17)はイタリック体で示す。成熟VLのN末端アミノ酸残基(D)は二重下線で示す。Kabatら(1991)の定義によるCDR配列を下線で示す。イントロン配列はイタリック体で示す。
【図7】pChK33N及びpHuK33N(総称して発現ベクターという)の模式的構造を示す図である。プラスミドは、一番上のSalI部位から時計回りに、抗体重鎖遺伝子の転写を開始するためのヒトサイトメガロウイルス(CMV)主要前初期プロモーター及びエンハンサー(CMVプロモーター)から始まる重鎖転写ユニットを含有している。CMVプロモーターに続いて、VHエキソンと、CH1、ヒンジ、CH2及びCH3のエキソンが介在イントロンと共に含まれるヒトγ1重鎖定常領域を含有するゲノム配列とがあり、CH3に続いてmRNAのプロセッシングのためのγ1遺伝子のポリアデニル化部位がある。重鎖遺伝子配列の後、軽鎖転写ユニットがCMVプロモーターから始まり、続いてVLエキソンと、ヒトκ鎖定常領域エキソン(CL)をその前にあるイントロンの一部と共に含有するゲノム配列、及びκ遺伝子のポリAシグナルがある。次に、軽鎖遺伝子に続いて、SV40初期プロモーター(SV40プロモーター)と、大腸菌キサンチングアニンホスホリボシルトランスフェラーゼ遺伝子(gpt)と、SV40ポリアデニル化部位(SV40ポリ(A)部位)を含有する部分とがある。最後に、プラスミドは、細菌の複製起点(pUC ori)とβ−ラクタマーゼ遺伝子(β−ラクタマーゼ)とを含むプラスミドpUC19の一部を含有する。
【図8】K33N VH(配列番号9)、ヒト化K33N(HuK33N)VH(配列番号29)並びにGenBankアクセッション番号DA980102のヌクレオチド配列によってコードされるアミノ酸配列に由来する、ヒトアクセプター配列のFRH1(配列番号19)、FRH2(配列番号20)、FRH3(配列番号21)及びFRH4(配列番号22)のアミノ酸配列のアラインメントを示す図である。アミノ酸残基は一文字表記で示す。配列の上にある数字は、Kabatら(1991)による位置を示す。Kabatら(1991)によって定義されたCDR配列は下線で示す。二重下線が付された残基はCDRと接触すると予想され、ヒト化型においてはこれらの位置のマウス残基は保持された。ヒトVH配列のこの位置では非典型的であるDA980102の82番目のMet(下線)を典型的な残基Leuと置き換えて、潜在的免疫原性を減らした。DA980102中のCDR残基はこの図では省略されている。
【図9】K33N VL(配列番号16)、ヒト化K33N(HuK33N)VL(配列番号31)並びにGenBankアクセッション番号X72441のヌクレオチド配列によってコードされるアミノ酸配列に由来する、ヒトアクセプター配列のFRL1(配列番号24)、FRL2(配列番号25)、FRL3(配列番号26)及びFRL4(配列番号27)のアミノ酸配列のアラインメントを示す図である。アミノ酸残基は一文字表記で示す。配列の上にある数字は、Kabatら(1991)による位置を示す。Kabatら(1991)によって定義されたCDR配列は下線で示す。二重下線が付された残基はCDRと接触すると予想され、ヒト化型においてはこれらの位置のマウス残基は保持された。X72441中のCDR残基はこの図では省略されている。
【図10】HuK33N VH遺伝子の構築に使用されたオリゴヌクレオチドを示す図である。
【図11】HuK33N VL遺伝子の構築に使用されたオリゴヌクレオチドを示す図である。
【図12】HuK33N VH遺伝子の構築に使用されたオリゴヌクレオチドを示す図である。矢印は、各々のオリゴヌクレオチドの位置と方向(5’から3’)を表す。VH領域(配列番号29)のアミノ酸残基は一文字表記で示す。
【図13】HuK33N VL遺伝子の構築に使用されたオリゴヌクレオチドを示す図である。矢印は、各々のオリゴヌクレオチドの位置と方向(5’から3’)を表す。VL領域(配列番号31)のアミノ酸残基は一文字表記で示す。
【図14】SpeI部位とHindIII部位(下線)が両側にある、HuK33N VH遺伝子のヌクレオチド配列(配列番号34)を、シグナルペプチド(配列番号58;イタリック体で示す)及びVH領域(配列番号29)の推定アミノ酸配列と共に示す図である。アミノ酸残基は一文字表記で示す。シグナルペプチド配列はイタリック体で示す。成熟VHのN末端アミノ酸残基(Q)は二重下線で示す。Kabatら(1991)の定義によるCDR配列は下線で示す。イントロン配列はイタリック体で示す。
【図15】NheI部位とEcoRI部位(下線)が両側にある、HuK33N VL遺伝子のヌクレオチド配列(配列番号35)を、シグナルペプチド(配列番号59;イタリック体で示す)及びVL領域(配列番号31)の推定アミノ酸配列と共に示す図である。アミノ酸残基は一文字表記で示す。シグナルペプチド配列はイタリック体で示す。成熟VLのN末端アミノ酸残基(D)は二重下線で示す。Kabatら(1991)の定義によるCDR配列は下線で示す。イントロン配列はイタリック体で示す。
【図16】キメラK33N抗体とヒト化K33N抗体のヒトα9インテグリンに対する親和性の比較を示す。1μg/mL及び0.5μg/mLのキメラK33N及びヒト化K33NのCHO/α9細胞への結合を細胞ELISAで調べた。実験は3重で実施した。吸光度の平均値を標準誤差と共に示す図である。
【図17】HuK33N重鎖及び軽鎖cDNAのPCR増幅及び配列決定に使用したオリゴヌクレオチドの配列(配列番号44〜50)を示す。
【図18】pHuK33N中のHuK33N γ1重鎖のコード領域のヌクレオチド配列(配列番号36)を推定アミノ酸配列(配列番号37)と共に示す図である。アミノ酸残基は一文字表記で示す。終止コドンは「・」で示す。
【図19】pHuK33N中のHuK33N κ軽鎖のコード領域のヌクレオチド配列(配列番号38)を推定アミノ酸配列(配列番号39)と共に示す図である。アミノ酸残基は一文字表記で示す。終止コドンは「・」で示す。
【図20】精製抗体のSDS−PAGE解析の結果を示す図である。6μgのキメラIgG1/κ抗体及びヒト化IgG1/κ抗体(それぞれ、ChK33N及びHuK33Nという)を、Sambrookら(Molecular Cloning,A Laboratory Manual,第2版,1989,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY)に従って、還元条件下、SDS存在下の10%ポリアクリルアミドゲル上で泳動させた。広域タンパク質マーカー(MW;New England Biolabs,Ipswich,MA)をサイズマーカーとして使用した。右側に示す数字は、マーカーのサイズをキロダルトン(kDa)単位で示す。
【図21】ヒトα9インテグリンに対するマウスK33N抗体の結合をFACS解析した結果を示す図である。1.67μg/mLから連続的に3倍希釈した様々な濃度のマウスK33N抗体について、CHO/huα9細胞への結合を試験した。図中、試験した各々の抗体濃度(X軸)における、幾何平均チャンネル蛍光値(MCF;Y軸)がプロットされている。EC50値を、GraphPad Prism(GraphPad Software,San Diego,CA)を用いて算出した。
【図22】ヒトα9インテグリンに対するキメラK33N抗体とヒト化K33N抗体の結合をFACS解析した結果を示す図である。5μg/mlから連続3倍希釈した様々な濃度の各抗体について、CHO/huα9細胞への結合を試験した。図中、試験した各々の抗体濃度(X軸)における、幾何平均チャンネル蛍光値(MCF;Y軸)がプロットされている。EC50値を、GraphPad Prism(GraphPad Software,San Diego,CA)を用いて算出した。
【図23】ヒトα9インテグリンに対するマウスK33N抗体、キメラK33N抗体及びヒト化K33N抗体、並びにマウス抗ヒトα9インテグリン抗体Y9A2の細胞接着阻害率の用量依存性を示す図である。各抗体を5、1、0.2、0.04、0.008及び0.0016μg/mLの濃度で、ヒトα9インテグリン発現細胞株G−361の細胞接着について試験した。実験は4重で実施した。平均阻害率の値を図に示す。
【発明を実施するための形態】
【0028】
5.1.ヒトα9インテグリンに対する抗体の調製
ヒトα9インテグリン又はその任意のエピトープを免疫特異的に認識する抗体は、当該技術分野で公知の任意の好適な方法により作製することができる。
【0029】
本発明において抗原として使用されるα9インテグリンは、(1)α9インテグリンを発現するヒト由来の全ての細胞、もしくはこれらの細胞が存在する全ての組織から得られるタンパク質、(2)α9インテグリンをコードする遺伝子DNA、好ましくはcDNAが、細菌、酵母、細胞株(動物細胞を含む)等に導入され、発現された、組換えタンパク質、又は(3)合成タンパク質であってもよい。
【0030】
α9インテグリンには、ヒトα9インテグリン(配列番号55、ここで、1〜29番目の残基はシグナルペプチドである)のアミノ酸配列と実質的に同一のアミノ酸配列を含むポリペプチドが含まれる。
【0031】
本明細書中、「実質的に同一のアミノ酸配列を含むポリペプチド」という用語は、天然のヒトα9インテグリンと実質的に同等の生物学的特性を有する限り、複数のアミノ酸、好ましくは1〜10個のアミノ酸、より好ましくは1〜数個(例えば、1〜5個)のアミノ酸が置換され、欠失し及び/又は改変されたアミノ酸配列を含む変異体ポリペプチド;並びに複数のアミノ酸、好ましくは1〜10個のアミノ酸、より好ましくは1〜数個(例えば、1〜5個)のアミノ酸が天然のヒトα9インテグリンのアミノ酸配列に付加されたアミノ酸配列を含む変異体ポリペプチドを意味する。さらに、変異体ポリペプチドは、これらのアミノ酸置換、欠失、改変及び付加の複数を有するものであってもよい。
【0032】
本発明における抗原としてのヒトα9インテグリンは、当該技術分野で周知の方法により産生することができ、例えば、遺伝子組換え技術の他、化学合成法、細胞培養法等、又はそれらの変法により産生することができる。
【0033】
変異体ポリペプチドを産生する方法の例としては、合成オリゴヌクレオチド部位特異的突然変異生成(ギャップ二重鎖法)、亜硝酸又は亜硫酸処理によってランダムに点突然変異を導入することを含む点突然変異生成法、Bal31酵素又は他の酵素を用いて欠失突然変異を調製することを含む方法、カセット突然変異生成、リンカースキャニング法、ミス組込み法、ミスマッチプライマー法、DNA断片合成法等が挙げられる。
【0034】
本発明において抗原として使用されるヒトα9インテグリンは、該α9インテグリンの「一部」も含む。本明細書において、「一部」とは、α9インテグリンのリガンド、例えば、OPN、VCAM−1、テネイシン−C等との結合に必要な領域を含む部分;特には、成熟ヒトα9インテグリン(配列番号55の30番目から1035番目までのアミノ酸残基)の、14番目から980番目までのアミノ酸残基を含む部分、及び11番目から981番目までのアミノ酸残基を含む部分を指す。該α9インテグリンの「一部」は、以下に記載する当該技術分野で公知の方法に従って、遺伝子組換えもしくは化学合成、又はそれらの変法によって産生することができ、あるいは、細胞培養法より単離されたヒトα9インテグリンをタンパク質分解的酵素等で適当に消化することによって産生することができる。
【0035】
抗原として、細胞膜上にα9インテグリンを過剰発現する細胞それ自体、又はその膜画分を使用することもできる。ヒトα9インテグリンを過剰発現する細胞は、当該技術分野で周知の組換えDNA技術によって調製することができる。
【0036】
上記のように調製された適当な抗原を用いて、ヒトα9インテグリン又は任意のそのエピトープに特異的な抗体を、当該技術分野で周知の様々な方法によって調製することができる。ヒトα9インテグリンに対するポリクローナル抗体は、当該技術分野で周知の様々な手順によって産生することができる。例えば、目的の抗原を、これに限定するものではないが、ウサギ、マウス、ラット等を含む様々な宿主動物に投与し、抗原に特異的なポリクローナル抗体を含有する抗血清の産生を誘導することができる。宿主の種に応じて、様々なアジュバントを用いて免疫学的応答を増大させてもよい。アジュバントとしては、フロイント(完全及び不完全)アジュバント、水酸化アルミニウム等の鉱物ゲル、リゾレシチン等の表面活性物質、プルロニックポリオール、ポリアニオン、ペプチド、オイルエマルジョン、キーホールリンペットヘモシアニン、ジニトロフェノール、並びに潜在的に有用なヒト用のアジュバント(例えば、BCG(カルメット・ゲラン桿菌)及びコリネバクテリウム・パルブム)が挙げられるが、これらに限定されるものではない。このようなアジュバントも当該技術分野において周知である。
【0037】
モノクローナル抗体は、ハイブリドーマ、組換え体及びファージディスプレイ技術、又はそれらの組合せを用いる方法を含む、当該技術分野で公知の多種多様な技術を用いて調製することができる。例えば、モノクローナル抗体は、当該技術分野で公知であり、また、例えば、Harlowら,Antibodies:A Laboratory Manual,(Cold Spring Harbor Laboratory Press,第2版.1988);Hammerling,et al.,:Monoclonal Antibodies and T−Cell Hybridomas,563−681ページ(Elsevier,N.Y.,1981)(両文献は参照により本明細書に組み込まれる)等に教示されている技術を含む、ハイブリドーマ技術を用いて産生することができる。本明細書で使用される「モノクローナル抗体」という用語は、ハイブリドーマ技術によって産生される抗体に限定されるものではない。「モノクローナル抗体」という用語は、任意の真核生物、原核生物、又はファージクローンを含む、単一のクローンに由来する抗体を指すものであり、それが産生される方法を指すものではない。
【0038】
ハイブリドーマ技術を用いた特異的抗体の産生及びスクリーニングする方法は、当該技術分野においてルーチンで行われておりかつ周知である。非限定的な例としては、マウスを目的の抗原又はそのような抗原を発現する細胞で免疫化することができる。免疫応答が検出されれば、例えば、抗原に特異的な抗体がマウス血清中に検出されれば、マウス脾臓を摘出し、脾細胞を単離する。その後、脾細胞を周知の技術により任意の好適なミエローマ細胞(例えば、P3U1、P3X63−Ag8、P3X63−Ag8−U1、P3NS1−Ag4、SP2/0−Ag14、P3X63−Ag8−653等)に融合させる。ハイブリドーマを選択し、限界希釈によりクローニングする。その後、ハイブリドーマクローンを、当該技術分野で公知の方法により、抗原に結合することができる抗体を分泌する細胞についてアッセイする。通常高レベルの抗体を含有する腹水は、陽性のハイブリドーマクローンをマウスの腹腔内に接種することにより作製することができる。
【0039】
特異的エピトープを認識する抗体断片は、公知の技術により作製することができる。例えば、Fab及びF(ab’)2断片は、免疫グロブリン分子をパパイン(Fab断片を産生するため)又はペプシン(F(ab’)2断片を産生するため)等の酵素を用いてタンパク質分解切断させることにより産生することができる。F(ab’)2断片は、完全な軽鎖と、重鎖の可変領域、CH1領域及びヒンジ領域とを含有する。
【0040】
本発明の抗体又はその抗原結合断片は、抗体の合成について当該技術分野で知られている任意の方法により、具体的には、化学合成又は好ましくは、組換え発現技術により産生することもできる。
【0041】
抗体をコードするヌクレオチド配列は、当業者が利用可能な任意の情報から(すなわち、GenBank、文献から、又は日常的なクローニング及び配列解析により)取得することができる。特定の抗体又はそのエピトープ結合断片をコードする核酸を含有するクローンが入手できず、抗体分子又はそのエピトープ結合断片の配列が知られている場合、免疫グロブリンをコードする核酸は、化学合成、あるいは、抗体配列の5’末端にハイブリダイズ可能な合成プライマーを用いたPCR増幅によって、又は、抗体をコードするcDNAライブラリー由来のcDNAクローン等を同定するための特定の遺伝子配列に特異的なオリゴヌクレオチドプローブを用いたクローニングによって、好適な供給源(例えば、抗体cDNAライブラリー、又は、例えば、抗体を発現する選択されたハイブリドーマ細胞等の抗体を発現する任意の組織もしくは細胞から作製されたcDNA、又はそのような組織若しくは細胞から単離された核酸、好ましくはポリA+ RNA)から取得し得る。その後、PCRで生成した増幅核酸を、当該技術分野で周知の任意の方法を用いて、複製可能なクローニングベクターにクローニングすることができる。
【0042】
5.2.組換え抗体の調製
抗体のヌクレオチド配列決定後、抗体のヌクレオチド配列を、ヌクレオチド配列の操作に関する当該技術分野で周知の方法、例えば、組換えDNA技術、部位特異的突然変異誘発法、PCR等(例えば、Sambrookら.,前記;及びAusubelら.,編,1998,Current Protocols in Molecular Biology,John Wiley & Sons,NYに記載されている技術を参照。両文献とも、その全体が参照により本明細書に組み込まれる。)を用いて操作して、例えば、抗体のエピトープ結合ドメイン領域又は抗体の生物学的活性を増強しもしくは低下させ得る抗体の任意の部分にアミノ酸の置換、欠失、及び/又は挿入を導入することにより、様々なアミノ酸配列を有する抗体を作製することができる。
【0043】
抗体の組換え発現には、抗体をコードするヌクレオチド配列を含有する発現ベクターの構築が必要である。抗体分子又は抗体の重鎖もしくは軽鎖、あるいはそれらの一部をコードするヌクレオチド配列が得られた後、前記節に記載されたように当該技術分野で周知の技術を用いた組換えDNA技術により抗体分子を産生するためのベクターを産生することができる。当業者に周知の方法を用いて、抗体コード配列と適当な転写及び翻訳制御シグナルとを含有する発現ベクターを構築することができる。これらの方法としては、例えば、インビトロ組換えDNA技術、合成技術、及びインビボ遺伝子組換えが挙げられる。抗体の重鎖可変領域、軽鎖可変領域、重鎖と軽鎖の両方の可変領域、重鎖及び/又は軽鎖可変領域のエピトープ結合断片、あるいは1以上の相補性決定領域(CDR)をコードするヌクレオチド配列を、このような発現用ベクターにクローニングすることができる。このような配列を、基となる抗体に由来するシグナルペプチド又は異種シグナルペプチドをコードするポリヌクレオチドと融合させることができる。その後、このように調製された発現ベクターは、抗体を発現させるための適当な宿主細胞に導入することができる。したがって、本発明は、免疫特異的にヒトα9インテグリンを認識するヒト化抗体又はその抗原結合断片をコードするポリヌクレオチドを含有する宿主細胞を含む。
【0044】
宿主細胞に、重鎖由来のポリペプチドをコードする第1のベクターと軽鎖由来のポリペプチドをコードする第2のベクターの本発明の2つの発現ベクターをコトランスフェクトしてもよい。この2つのベクターは、重鎖ポリペプチドと軽鎖ポリペプチドが同等に発現することを可能にする同一の選択マーカーを有していてもよいし、又は両方のプラスミドの維持を確実にするための異なる選択マーカーを含有していてもよい。あるいは、重鎖と軽鎖の両方のポリペプチドをコードし、それらを発現することができる単一のベクターを使用してもよい。重鎖及び軽鎖のコード配列は、cDNA又はゲノムDNAのいずれを含んでいてもよい。
【0045】
別の実施形態では、抗体は、当該技術分野で公知の様々なファージディスプレイ法を用いて作製することもできる。ファージディスプレイ法では、機能的抗体ドメインが、それらをコードするポリヌクレオチド配列を担持するファージ粒子の表面上に提示される。具体的な実施形態では、このようなファージを、レパートリー又はコンビナトリアル抗体ライブラリー(例えば、ヒト又はマウス)により発現される抗原結合ドメイン(例えば、Fab及びFv又はジスルフィド結合安定化Fv)を提示させるために利用することができる。目的の抗原に結合する抗原結合ドメインを発現するファージは、例えば、標識された抗原又は固体表面もしくはビーズに結合もしくは捕捉された抗原を用いて、抗原で選択又は同定することができる。これらの方法で使用されるファージは、典型的には、fd及びM13を含む繊維状ファージである。抗原結合ドメインは、ファージ遺伝子III又は遺伝子VIIIタンパク質のいずれかに融合された組み換えタンパク質として発現される。本発明の免疫グロブリン、又はその断片を作製するために使用することができるファージディスプレイ法の例としては、Brinkmanら,J.Immunol.Methods,182:41−50,1995;Amesら,J.Immunol.Methods,184:177−186,1995;Kettleboroughら,Eur.J.Immunol.,24:952−958,1994;Persicら,Gene,187:9−18,1997;Burtonら,Advances in Immunology,57:191−280,1994;PCT出願PCT/GB91/01134号;PCT公開公報WO90/02809号;WO91/10737号;WO92/01047号;WO92/18619号;WO93/11236号;WO95/15982号;WO95/20401号;並びに米国特許第5,698,426号;第5,223,409号;第5,403,484号;第5,580,717号;第5,427,908号;第5,750,753号;第5,821,047号;第5,571,698号;第5,427,908号;第5,516,637号;第5,780,225号;第5,658,727号;第5,733,743号及び第5,969,108号に開示されているものが挙げられ、これらは各々、その全体が参照により本明細書に組み込まれる。
【0046】
上述の参考文献に記載されているように、ファージ選択の後、ファージ由来の抗体コード領域を単離し、例えば、以下で詳細に記載するように、ヒト抗体を含む完全抗体、又は任意の他の所望の断片を作製するために使用し、哺乳動物細胞、昆虫細胞、植物細胞、酵母、及び細菌を含む任意の所望の宿主で発現させることができる。例えば、Fab、Fab’及びF(ab’)2断片を産生する組換え技術を、例えば、PCT公開公報WO92/22324号;Mullinaxら,BioTechniques,12(6):864−869,1992;及びSawaiら,AJRI,34:26−34,1995;及びBetterら,Science,240:1041−1043,1988に開示された方法(各々、その全体が参照により本明細書に組み込まれる)等の当該技術分野で公知の方法を用いて採用することもできる。単鎖Fv及び抗体を産生することができる技術の例としては、米国特許第4,946,778号及び第5,258,498号;Hustonら,Methods in Enzymology,203:46−88,1991;Shuら,PNAS,90:7995−7999,1993;並びにSkerraら,Science,240:1038−1040,1988に記載されているものが挙げられる。
【0047】
本発明の抗体分子は、上述の任意の方法で産生後、免疫グロブリン分子の精製に関する当該技術分野で公知の任意の方法によって、例えば、クロマトグラフィー(例えば、イオン交換、アフィニティ、具体的には、プロテインAもしくはプロテインG精製後の特異的抗原に対するアフィニティ、及びサイズ分離カラムクロマトグラフィー)、遠心分離、示差溶解度、又はタンパク質の精製のための任意の他の標準的な技術によって精製することができる。また、本発明の抗体又はその断片は、精製を容易にするため、本明細書に記載の又は当該技術分野で公知の異種ポリペプチド配列と融合させてもよい。
【0048】
ヒトでの抗体のインビボ使用及びインビトロ検出アッセイを含む、いくつかの用途のために、キメラ、ヒト化又はヒト抗体を使用することが好ましい場合がある。キメラ抗体及びヒト化抗体は、下記の第5.3節で詳細に説明されている。
【0049】
他の化合物又は異種ポリペプチドと融合しているか又はコンジュゲートしている抗体は、インビトロでのイムノアッセイ、精製方法(例えば、アフィニティクロマトグラフィー)、及びインビボでの治療的又は診断的用途で利用することができる。例えば、PCT公開公報WO93/21232号;欧州特許第439,095号;Naramuraら,Immunol.Lett.,39:91−99,1994;米国特許第5,474,981号;Gilliesら,PNAS,89:1428−1432,1992;及びFellら,J.Immunol.,146:2446−2452,1991を参照(これらは、その全体が参照により本明細書に組み込まれる)。例えば、抗体は、既知の方法又は市販のキット(例えば、ビオチン標識、FITC標識、APC標識)を用いて様々な方法で標識することができる。他の例として、抗体は、インビボでの抗体の生物学的効果を増強する治療成分とコンジュゲートしていてもよい。このような治療成分の例としては、他の抗体、細胞増殖抑制性もしくは細胞破壊性の細胞毒素、放射性元素、及び/又は抗炎症剤、抗生物質等を含む他の治療剤が挙げられる。本発明において、ヒト化抗ヒトα9インテグリンは、抗α4抗体等の別の抗体とコンジュゲートして(例えば、二重特異性抗体を形成して)いてもよい。他の例として、本発明のヒト化抗体は、インビボでの診断的用途のため、例えば、放射性元素等の検出可能なマーカーで標識されていてもよい。
【0050】
5.3.キメラ抗体及びヒト化抗体
キメラ抗体は、マウスモノクローナル抗体に由来する可変領域とヒト免疫グロブリンに由来する定常領域とを有する抗体等の、抗体の異なる部分が異なる動物種に由来する分子である。キメラ抗体を産生する方法は当該技術分野で公知である。例えば、Morrison,Science,229:1202,1985;Oら,BioTechniques,4:214 1986;Gilliesら,J.Immunol.Methods,125:191−202,1989;米国特許第5,807,715号;第4,816,567号;及び第4,816,397号を参照(これらは、その全体が参照により本明細書に組み込まれる)。
【0051】
ヒト化抗体は、所望の抗原に結合する分子であり、非ヒト種に由来する1以上の相補性決定領域(CDR)とヒト免疫グロブリン分子に由来する1以上のフレームワーク領域とを含有する可変領域を含む。非ヒト抗体をヒト化する典型的な方法は、様々な参考文献、例えば、Queenら,1989,Proc.Natl.Acad.Sci USA 86:10029−10033、並びに米国特許第5,585,089号及び第5,693,762号;Riechmannら,Nature,332:323,1988;Tsurushitaら,Methods 36:69−83,2005に記載されており、これらは全て、その全体が参照により本明細書に組み込まれる。例えば、Tsurushitaら(2005,前記;以後、「Tsurushita」という)による参考文献は、もともとQueenら(1989,前記)によって開発された抗体ヒト化方法に基づくマウスモノクローナル抗体のヒト化についての実用的かつ有益なプロトコルを提供している。Tsurushitaに開示されている一般的プロトコルを以下で簡単に説明する。
【0052】
5.3.1.ヒト化抗体を調製するための一般的プロトコル
マウスV遺伝子のクローニング及びシークエンシング
標的マウスモノクローナル抗体のVH領域及びVL領域をコードするcDNAをクローニングするために様々な方法を利用することができる。例えば、SMART RACE cDNA増幅キット(BD Biosciences,CA)又はGeneRacerキット(Invitrogen,CA)を用いた、5’RACE(cDNA末端の迅速な増幅)法が一般に使用されている。5’RACEの遺伝子特異的プライマーは、H鎖とL鎖の各々の可変領域のすぐ下流に結合することができるように、標的モノクローナル抗体のH鎖とL鎖のアイソタイプに基づいて調製することができる。したがって、5’RACEプライマーは、γ1、γ2a、γ2b又はγ3等の、マウスにおける各々のサブタイプに特異的となるように設計してもよい。あるいは、サブタイプ間のコンセンサス領域又は相同性の高い領域に基づいて全てのサブタイプに共通するプライマーを設計してもよい。Tsurushitaでは、以下の5’RACEプライマーが例として開示されている:
(i)5’−GCCAGTGGATAGACTGATGG−(配列番号56)(マウスγ1、γ2a、γ2b及びγ3H鎖のクローニング用)
(ii)5’−GATGGATACAGTTGGTGCAGC−(配列番号57)(マウスκ軽鎖のクローニング用)。
【0053】
PCR増幅したV遺伝子断片は、例えば、Zero Blunt TOPO PCRクローニングキット(Invitrogen)と、決定されたDNA配列とを用いて、プラスミドベクターに直接クローニングすることができる。例えば、得られたDNA配列がコードするアミノ酸配列を、例えば、Model 241タンパク質シークエンサー(Hewlett−Packard,CA)等を用いたN末端アミノ酸シークエンシングによって決定された標的モノクローナル抗体のアミノ酸配列と比較することによって、得られたDNA配列を確認すべきである。典型的には、例えば、エドマン分解等により、標的抗体のN末端の少なくとも15〜20アミノ酸残基を決定すれば、クローニングされたDNA配列の真偽を十分確認することができる。Tsurushitaは、マウスにおいて最も一般的な2つのN末端アミノ酸の1つであるグルタミンがN末端アミノ酸である場合、それがピログルタミンに変換されて、N末端のシークエンシングを阻止することがあることに注意が必要であるとしている。この場合、配列を得るためにN末端のブロックを解除する必要がある。
【0054】
V領域の三次元モデリング
VH領域及びVL領域の配列に基づいて、CDRの立体構造を維持するために潜在的に重要な標的抗体のフレームワーク残基を、例えば、R.Levyら,1989,Biochemistry 28:7168−7175;及びB.Zilberら,1990,Biochemistry 29:10032−10041に記載されている方法によってまず同定する。典型的には、VH領域及びVL領域の各々は、14個の構造的に意味のある部分に分けられ、これらの部分は、免疫グロブリンスーパーファミリーのドメイン構造を含むβ鎖及びループ様構造である。標的抗体由来の各々の部位のアミノ酸配列と、PDBデータベース中の構造が既知の抗体の対応する部分の配列とを比較する(H.M.Bermanら,2000,Nucleic Acids Res.28:235−342を参照)。複数の配列を比較することにより、各々の標的部位との配列の相同性が最も高い対応する部分を選択し、V領域の三次元モデルを構築する。構造を最適化するために、このモデルを、(例えば、ENCADを用いて、又はPressら,1990,“Numerical Recipes”,Cambridge University Press,Cambridgeの記載に従って;Weinerら,1981,J.Comp.Chem.2:287−303に記載されたAMBER;英国がん研究所が運営するBio Molecular Modelling、又は“BMM”ウェブサイトで入手可能な3D−JIG−SAW;又はジェノバにあるスイスバイオインフォマティクス研究所が運営するExPASy Proteomics Serverウェブサイトで入手可能なSWISS−MODELによって)、複数回、共役勾配法によりエネルギー最小化を行った。
【0055】
ヒトフレームワークの選択
V領域の構造のモデリングと並行して、マウスVH領域及びVL領域のcDNAクローニングから推定されるアミノ酸配列を、それぞれ、例えば、Kabatデータベース(Johnsonら,2000,Nucleic Acids Res.28:214−218を参照)、GenBank等におけるヒトV領域配列と比較する。配列全体でマウス配列と少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、又は少なくとも95%の相同性のあるヒトフレームワーク領域は、例えば、Smith−Watermanアルゴリズム(“Algorithms on Strings,Trees,and Sequences”,Cambridge University Press,Cambridgeにおける、Gusfield,1997)、又はBLAST(Karlinら,1990,Proc.Natl.Acad.Sci USA 87:2264−2268)等を用いて、検索することができる。これらのヒト配列は、cDNAに基づく配列及びタンパク質由来の配列のいずれに基づくものであってもよいが、多くの場合、生殖系列の使用がcDNAに基づく配列、タンパク質由来の配列における体細胞超突然変異と関連する潜在的免疫原性を消失させるのに有用であることから、生殖細胞系列の使用が好ましい。あるいは、Queenら(1989,前記)に記載されているように、コンセンサスフレームワーク配列を用いて、cDNAに基づく配列又はタンパク質由来の配列から得られたフレームワーク中のそのような超突然変異残基を同定し、除去することもできる。生殖細胞系列のVH部分をアクセプターフレームワークとして使用する場合、第14番染色体上のVH部分のみが機能的なVH領域を生じさせるので、第15番及び第16番染色体ではなく、第14番染色体上にコードされるVH部分を使用する。
【0056】
ヒト化V領域の設計
Queenら(1989,前記)によれば、CDRの約4〜6Åの範囲内にあるフレームワークアミノ酸は、正確なCDR構造を支持する潜在的に重要なフレームワーク残基であると考えられることから、これらのフレームワークアミノ酸を同定することが必要である。このプロセスは、例えば、米国国立科学財団(NSF)が支援するMolecular Visualization Freewareウェブサイトで入手可能なRASMOL等の原子座標から原子間距離を計算するコンピュータプログラムを用いるか、又は手作業でコンピュータモデルを検査することによって行うことができる。重要なフレームワーク位置のアミノ酸が、マウスドナー配列とヒトアクセプター配列の間で異なる場合、通常ヒト残基をマウスドナー配列で置き換える。しかしながら、このような残基のCDR構造を支持する上での寄与がわずかである場合、典型的には対応するヒト残基が用いられる。また、選択されたヒトアクセプターが、V領域配列の約10〜20%未満で「非典型的な」アミノ酸を含有する場合、それらは親和性成熟の間の体細胞超突然変異の結果であり得ることから、ヒトにおける潜在的免疫原性を回避するためにドナー残基と置き換えるべきである。
【0057】
さらに、ヒト化V領域を設計するために、潜在的なN結合型グリコシル化シグナルの残基等の他の因子を注意深く考慮する必要がある(詳細についてはTsurushitaを参照)。
【0058】
ヒト化抗体は、治療目的で必要とされ又は消失させられるべきエフェクター機能に応じて、ヒトκもしくはλ軽鎖由来のヒト定常領域もしくはその部分、及び/又はヒト抗体のγ1、γ2、γ3、γ4、μ、α1、α2、δ、もしくはε重鎖、あるいはそれらの変異体を含有していてもよい。例えば、突然変異を有する定常領域のFc部分を、抗体のFc受容体への結合を低下させるため、及び/又は補体を固定する能力を低下させるために、本発明のキメラ抗体又はヒト化抗体の可変領域と融合させてもよい(例えば、Winterら、ドイツ特許第2,209,757号;Morrisonら、WO89/07142号、Morganら、WO94/29351号を参照)。このような抗体分子の操作は、第5.2節に記載の組換えDNA技術により実施することができる。
【0059】
好ましくは得られたキメラ抗体又はヒト化抗体は、非ヒトドナー抗体と同じ特異性及び非ヒトドナー抗体の親和性と同等か、又は少なくとも約1/3、少なくとも約1/2、もしくは少なくとも約2/3の親和性を有する。別の態様では、得られたキメラ抗体又はヒト化抗体は、少なくとも約1×107M−1、好ましくは少なくとも約1×108M−1、最も好ましくは少なくとも約1×109M−1の親和定数を有する。
【0060】
上述の一般的プロトコルに加えて、抗体は、当該技術分野で公知の種々の技術を用いてヒト化することができ、例えば、CDRグラフティング(欧州特許第239,400号;PCT公開公報WO91/09967号;米国特許第5,225,539号;第5,530,101号及び第5,585,089号)、ベニアリング(veneering)又はリサーフェイシング(resurfacing)(欧州特許第592,106号;欧州特許第519,596号;Padlan,Molecular Immunology,28(4/5):489−498,1991;Studnickaら,Protein Engineering,7(6):805−814,1994;Roguskaら,Proc Natl.Acad.Sci.USA,91:969−973,1994)、並びに鎖シャッフリング(米国特許第5,565,332号)が挙げられ、これらは全てその全体が参照により本明細書に組み込まれる。
【0061】
5.3.2.ヒト化抗体を医薬品として調製するために更に考慮すべき事項
ヒト化抗体を医薬品として提供するために、効率的で安定的な産生系を調製する必要がある。例えば、H鎖及びL鎖配列を挿入することによってヒト化抗体用の適切な発現ベクターを調製し、この発現ベクターがトランスフェクトされた生産性の高い細胞株をマスター細胞バンク(MCB)用の種細胞として得ることができる。マスター細胞バンク(MCB)は、ワーキング細胞バンク(WCB)の安定的かつ半永久的な供給源として利用される。その後、WCB由来のワーキング細胞を培養し、その培養培地を回収することにより、ヒト化抗体を調製することができる。
【0062】
適当な調節遺伝子を含む様々な発現ベクターを、そのような産生細胞株の調製に用いることができる。哺乳動物タンパク質を発現するために一般に用いられる細胞を宿主細胞としてヒト化抗体の発現に用いることができる。このような宿主細胞としては、これに限定されるものではないが、チャイニーズハムスター卵巣(CHO)細胞、SP2/0−Ag14.19細胞、NSO細胞等が挙げられる。発現ベクターと宿主細胞の最良の組合せを選択することにより、ヒト化抗体の生産性を最大化することができる。さらに、宿主細胞によるヒト化抗体の発現を最適化するように好適な培地を選択するため、培養培地の組成を様々な無血清培地及び添加物から検討するべきである。
【0063】
効率と最終的な収率に基づいて、宿主細胞によって産生されたヒト化抗体を、アフィニティクロマトグラフィー、イオン交換クロマトグラフィー、疎水性相互作用クロマトグラフィー等を含む、当該技術分野で周知の様々な方法を用いて培養上清から精製することができる。
【0064】
5.4.医薬組成物及び治療用途
本発明は、免疫特異的にヒトα9インテグリンを認識する、上述のヒト化抗体又はその抗原結合断片を含む医薬組成物を提供する。本発明のヒト化抗体を有効成分として含有する医薬組成物は、これに限定されるものではないが、例えば、癌細胞の増殖又は転移等の癌、及び、例えば、関節リウマチ、変形性関節症、肝炎、気管支喘息、線維症、真性糖尿病、動脈硬化症、多発性硬化症、肉芽腫、炎症性腸疾患(潰瘍性大腸炎及びクローン病)、自己免疫疾患等の炎症性疾患を含む、α9インテグリンと関連する障害又は疾患を予防及び/又は治療するための薬剤として用いることができる。
【0065】
本発明のヒト化抗体を含有する医薬組成物は、臓器移植後の慢性的拒絶、及び、例えば、全身性自己免疫疾患、エリテマトーデス、ブドウ膜炎、ベーチェット病、多発性筋炎、増殖性糸球体腎炎、サルコイドーシス、α9インテグリンによって誘導される疾患状態等の自己免疫疾患を治療するためにも用いることができる。
【0066】
上述の障害又は疾患を予防又は治療するための、本発明のヒト化抗体を含む予防用及び/又は治療用薬剤は毒性が低く、好適な溶媒中で混合することにより液体調製物として直接、又は適当な剤形の医薬組成物として、ヒトに経口投与又は非経口投与することができる。
【0067】
上述の投与に用いられる医薬組成物は、前述の抗体又はその塩と薬学的に許容される担体、希釈剤又は賦形剤とを含有する。このような組成物は、経口投与又は非経口投与に好適な剤形で提供される。
【0068】
投与量は、投与されるべき対象の年齢及び大きさ、標的疾患、状態、投与経路等によって様々に変わり得る。抗体を、例えば、成人患者の関節リウマチ等の予防及び/又は治療に用いる場合、本発明の抗体を、一回当たり通常約0.01〜約20mg/体重kg、好ましくは約0.1〜約10mg/体重kg、より好ましくは約0.1〜約5mg/体重kgの用量で、1日におよそ1〜5回、好ましくは1日におよそ1〜3回静脈内投与することが有効である。他の非経口投与及び経口投与では、抗体を前記用量に対応する用量で投与することができる。状態が特に重篤である場合は、その状態によって用量を増加させてもよい。
【0069】
例えば、リポソーム、マイクロ粒子、マイクロカプセルへの封入、突然変異体ウイルスを発現することができる組換え細胞、受容体を介するエンドサイトーシス等の様々な送達系が知られており、本発明の医薬組成物を投与するために用いることができる(例えば、Wu and Wu,1987,J.Biol.Chem.262:4429 4432を参照)。投与経路としては、これに限定されるものではないが、皮内、筋肉内、腹腔内、静脈内、皮下、鼻腔内、硬膜外、及び経口経路が挙げられる。化合物は、任意の利用しやすい経路、例えば、注入又はボーラス注射、あるいは上皮又は皮膚粘膜内層(例えば、口腔粘膜、直腸及び腸粘膜等)からの吸収によって投与してもよく、また、他の生物学的に活性のある薬剤と共に投与してもよい。投与は全身性又は局所性のいずれでもよい。例えば、吸入器又はネブライザーとエアロゾル化剤を用いて、経肺投与を採用してもよい。
【0070】
特定の実施形態では、本発明の医薬組成物を、治療を必要とする部位に局所的に投与することが望ましい場合がある。このような投与は、例えば、これに限定されるものではないが、手術時の局所注入、手術後の創傷包帯と組み合わせ等による局所塗布、注射、カテーテル、坐剤、鼻スプレー、又はインプラントによって行うことができる。前記インプラントは、多孔性、非多孔性、又はゼラチン様の物質からなり、シラスティック膜等の膜、又は繊維を含む。一実施形態では、感染した組織部位(又は、かつて感染組織部位であった部位)への直接注射により投与してもよい。
【0071】
別の実施形態では、医薬組成物は、小胞、特にリポソーム内で送達することができる(Langer,1990,Science 249:1527−1533;Liposomes in the Therapy of Infectious Disease and Cancer,Lopez Berestein and Fidler(編),Liss,New York,353−365頁(1989)のTreatらの稿;同書,317−327頁のLopez−Beresteinの稿を参照;一般には同書を参照)。
【0072】
更に別の実施形態では、医薬組成物は、放出制御系で送達することができる。一実施形態では、ポンプを用いてもよい(Langer,前記;Sefton,1987,CRC Crit.Ref.Biomed.Eng.14:201;Buchwaldら,1980,Surgery 88:507;及びSaudekら,1989,N.Engl.J.Med.321:574を参照)。別の実施形態では、ポリマー材料を用いることができる(Medical Applications of Controlled Release,Langer and Wise(編),CRC Pres.,Boca Raton,Florida(1974);Controlled Drug Bioavailability,Drug Product Design and Performance,Smolen and Ball(編),Wiley,New York(1984);Ranger and Peppas,J.Macromol.Sci.Rev.Macromol.Chem.23:61(1983)を参照;また、Levyら,1985,Science 228:190;Duringら.,1989,Ann.Neurol.25:351;Howardら.,1989,J.Neurosurg.71:105も参照)。さらに別の実施形態では、放出制御系を組成物の標的の近くに置くことができ、それにより全身投与量の一部しか必要としないようにすることができる(例えば、Medical Applications of Controlled Release,前記,第2巻,115−138頁(1984)のGoodsonの稿を参照)。他の放出制御系は、Langer(Science 249:1527−1533(1990))による総説に記載されている。
【0073】
経口投与用の組成物の例としては、固体又は液体剤形、特に、錠剤(糖衣錠及びフィルムコート錠を含む)、丸薬、顆粒、粉末調製物、カプセル(ソフトカプセルを含む)、シロップ、エマルジョン、懸濁液等が挙げられる。このような組成物は、公知の方法により製造され、薬学的調製の分野で従来から用いられているビヒクル、希釈剤又は賦形剤を含有する。錠剤用のビヒクル又は賦形剤の例としては、ラクトース、デンプン、スクロース、ステアリン酸マグネシウム等が挙げられる。
【0074】
注射用調製物は、静脈内、皮下、皮内及び筋肉内注射、点滴等のための剤形を含んでいてもよい。これらの注射用調製物は、公知の方法により調製することができる。注射用調製物は、例えば、上述の抗体又はその塩を、従来から注射用に用いられている滅菌水性媒体又は油性媒体中で溶解、懸濁又は乳化させることによって調製してもよい。注射用の水性媒体としては、例えば、生理的食塩水、グルコース及び他の助剤を含有する等張溶液等が挙げられ、これらは、例えば、アルコール(例えば、エタノール)、多価アルコール(例えば、プロピレングリコール、ポリエチレングリコール)、非イオン性界面活性剤[例えば、ポリソルベート80、HCO−50(水素化ヒマシ油のポリオキシエチレン(50モル)付加物)]等の適切な可溶化剤と組み合わせて用いることができる。油性媒体としては、例えば、ゴマ油、大豆油等を利用することができ、これらは、例えば、安息香酸ベンジル、ベンジルアルコール等の可溶化剤と組み合わせて用いることができる。このように調製された注射液は、好ましくは適当なアンプルに充填される。直腸投与に用いられる坐剤は、前述の抗体又はその塩を従来の坐剤用基剤と混合することにより調製することができる。
【0075】
有効な手段としては、上述の経口又は非経口で使用するための医薬組成物は、有効成分の投与量に合わせるのに適した単位用量からなる剤形で調製する。このような単位投与量からなる剤形としては、例えば、錠剤、丸薬、カプセル、注射液(アンプル)、坐剤等が挙げられる。含有する前述の抗体の量は、通常、単位投与量からなる剤形当たり約5〜500mgであり;特に、注射液の形態では、前述の抗体を約5〜100mg含有することが好ましく、また、他の剤形の場合、約10〜250mg含有することが好ましい。
【0076】
上述の各々の組成物は、製剤が上述の抗体との何らかの有害な相互作用を生じさせない限り、他の有効成分をさらに含有していてもよい。
【0077】
本発明はまた、α9インテグリン結合性機能分子(例えば、OPN、VCAM−1、テネイシン−C、フィブロネクチン、pp−vWF、tTG等)を有効成分として含有する細胞及び/又は組織再構築の阻害剤及び/又は促進剤;並びにα9インテグリン発現細胞及び/又は組織(例えば、腫瘍細胞、好中球、平滑筋等)をα9インテグリン結合性機能分子と接触させることを含む、細胞及び/又は組織再構築の阻害方法及び/又は促進方法に関する。このような治療剤中の有効成分の投与量、投与方法、薬学的調製方法等は、本発明のヒト化抗体を含む医薬品に関する前述の記載を参照して適切に決定することができる。
【0078】
上述のように、本発明は更にα9インテグリンと関連するか又はα9インテグリンが関与する障害又は疾患を予防又は治療する方法であって、有効量の本発明のヒト化抗体の少なくとも1つをそれを必要とする対象に投与することを含む方法を提供する。
【0079】
5.5.診断的使用
本発明のヒト化抗体を含む医薬組成物は、例えば、癌細胞の増殖又は転移等の癌、及び、例えば、関節リウマチ、変形性関節症、肝炎、気管支喘息、線維症、真性糖尿病、癌転移、動脈硬化症、多発性硬化症、肉芽腫等の炎症性疾患のための診断薬として、又は臓器移植後の慢性的拒絶、及び、例えば、全身性自己免疫疾患、エリテマトーデス、ブドウ膜炎、ベーチェット病、多発性筋炎、増殖性糸球体腎炎、サルコイドーシス、α9インテグリンによって誘導される疾患状態等の自己免疫疾患、並びにその他の疾患ための診断薬として用いることができる。本発明のヒト化抗体は、α9インテグリンを特異的に認識することができることから、被験液中のα9インテグリンの定量に用いることができ、特に、サンドイッチイムノアッセイ、競合アッセイ、イムノメトリー、ネフロメトリー等、免疫染色、又は同様の方法による定量に用いることができる。これらの免疫学的方法を本発明のアッセイ方法に適用する場合には、特別な条件、手順等の説明は必要なく、当該技術分野における通常の技術的検討を従来の条件及び手順に加えてアッセイ系を構築することで十分である。これらの一般的技術手段の詳細は、総説、教科書等を参照することができる。
【0080】
上述のように、α9インテグリンは、本発明の抗体を用いることにより高感度に定量することができる。本発明のヒト化抗体は、α9インテグリンの定量をインビボで行うことによりα9インテグリンと関連する様々な疾患を診断する上で特に有用である。例えば、α9インテグリンの発現レベルの増加又は減少が検出される場合、対象はα9インテグリンと関連する疾患、例えば、癌もしくは炎症性疾患を患っている可能性が極めて高く、又は将来においてこれらの疾患を患う可能性が極めて高いと診断することができる。したがって、本発明はまた、対象におけるα9インテグリンと関連するか又はα9インテグリンが関与する障害又は疾患を診断する方法であって、有効量の本発明のヒト化抗体の少なくとも1つ又は両方を、それを必要とする対象に投与することを含む方法を提供する。このようなインビボ診断に必要な投与量は、治療用途に必要な投与量よりも少なくてもよく、日常的な手順によって当業者が決定することができる。
【0081】
本発明のヒト化抗体は、体液等の被験液、組織等に存在するα9インテグリンを特異的に検出するために用いることもできる。ヒト化抗体は、α9インテグリンの精製用の抗体カラムを調製し、精製時の各画分に含まれるα9インテグリンを検出し、又は被験細胞中のα9インテグリンの挙動を解析するために用いることもできる。
【実施例】
【0082】
6.実施例
以下の実施例では、ヒト及び/又はマウスのα9インテグリンを免疫特異的に認識するモノクローナル抗体の調製、これらのモノクローナル抗体の可変領域の配列決定、及びこれらの抗体の他の特徴、そのような抗体のキメラ化及びヒト化、並びに得られたキメラ抗体及びヒト化抗体の特徴を説明する。これらの実施例は、これに限定するものと解釈されるものではない。
【0083】
6.1.ヒトα9インテグリンに対するマウス抗体の調製
ヒトα9インテグリンに対するマウスモノクローナル抗体は、サブトラクティブ免疫化法(Williams C.V.ら,1992,Biotechniques 12:842−847による)に従って調製した。簡単には、3匹のBalb/cマウスに、マウス1匹当たり3×106個の、ヒトα9インテグリンを発現するNIH−3T3細胞(ヒトα9/NIH−3T3細胞)を腹腔内注射した。注射の1週間後及び2週間後に、マウスに、3×106細胞/マウスのヒトα9/NIH−3T3細胞を腹腔内注射し、次いで1週間後に、同じ細胞を2×106細胞/マウスでさらに静脈内注射した。ハイブリドーマを当該技術分野で周知の方法により調製した(例えば、Harlowら,Antibodies:A Laboratory Manual,(Cold Spring Harbor Laboratory Press,第2版.1988);Monoclonal Antibodies and T−CeIl Hybridomas,563−681頁のHammerlingらの稿(Elsevier,N.Y.,1981)を参照)。ヒトα9インテグリンを発現するヒトα9/CHO−K1細胞及びヒトα9インテグリンを内在性に発現するヒトメラノーマ細胞とは免疫特異的に反応するが、ヒトα4インテグリンを発現するCHO−K1細胞とは免疫特異的に反応しないモノクローナル抗体を産生するハイブリドーマクローンを樹立し、ヒトα9インテグリンを免疫特異的に認識するモノクローナル抗体を産生するハイブリドーマクローン(すなわち、K33N)を単離した。
【0084】
6.2.抗ヒトα9インテグリン抗体のCDR解析
モノクローナル抗体(すなわち、K33N)のCDRのアミノ酸配列を、対応するハイブリドーマから抽出したmRNAを逆転写して、cDNAを調製することにより決定した。このcDNAを鋳型として用いて、H鎖とL鎖の可変領域を、ScFv−クローニングプライマー(Light Primer Mix and Heavy Primer Mix;Amersham Biosciences Corp.,IL製)を用いてPCRで伸長させ、増幅させた。このPCR産物をpCRII TOPOベクターにクローニングし、シークエンシングしてアミノ酸配列を決定した。このプロセスを3回繰り返した。結果を表1に示す。
【0085】
【表1】

【0086】
6.3.細胞接着阻害活性
(1)細胞接着は、α9インテグリンのそのリガンド、すなわち、OPN、フィブロネクチン、テネイシン−C、VCAM−1等を含む様々なECM、への結合を伴うことが知られていることから、ヒトα9インテグリンを発現する細胞(ヒトメラノーマG361細胞)のリガンドへの結合を用いることにより、単離された抗ヒトα9インテグリン抗体の細胞接着阻害活性について調べた。
【0087】
簡単には、OPNペプチドを、ウシ血清アルブミン(BSA)−融合SVVYGLR(配列番号2)ペプチドとして調製した。TN−C fn3(RAA)は、ヒトテネイシン−C中のフィブロネクチンタイプIII反復の3番目の領域として調製する。このペプチドのGRD配列は、大腸菌宿主細胞で発現させることによりRAA配列に置き換えられている。
【0088】
OPNペプチド及びテネイシン−C断片(TN−C fn3(RAA))を、96穴プレートに5μg/mLで添加し、37℃で1時間インキュベートしてプレートにコーティングし、その後、このプレートをブロッキング溶液(0.5%BSA/PBS)でブロッキングした。ヒトメラノーマG361細胞を0.25%BSA/DMEMに懸濁し(1×105細胞/mL)、各濃度の抗ヒトα9インテグリン抗体をこの細胞懸濁液に添加した。0.25%BSA/DMEM中のヒトメラノーマG361細胞(1×105細胞/mL)と抗体の混合物を、96穴プレートに200μL/穴で添加し、5%CO2、37℃で1時間インキュベートした。非接着細胞をPBSで洗い流し、接着細胞を固定し、0.5%クリスタルバイオレット(和光純薬工業,大阪,日本製)/20%メタノールで染色した。染色された細胞を蒸留水で3回洗浄し、次に、20%酢酸溶液をそれに添加して、溶解させた。590nmの波長におけるODを測定して、接着活性を定量した。陰性対照として抗ヒトOPNモノクローナル抗体(5A1)、陽性対照として以前に調製した抗ヒトα9インテグリン抗体(1K11,21C5、24I11、25B6及び28S1)を用いた。
【0089】
抗ヒトα9インテグリン抗体による、G361細胞のOPNペプチドへの結合に対する効果を図1に示し、テネイシン−C断片への結合に対する効果を図2に示す。図1に示すように、OPNが関与する細胞接着は、陽性対照の21C5及び24I11と比べて低い濃度のK33Nにより阻害され、かつ、Y9A2と同じ濃度のK33Nにより阻害された。テネイシン−Cが関与する細胞接着は、低濃度のK33N、かつ、Y9A2と同じ濃度のK33Nによって阻害され、この阻害効果は、陽性対照の21C5及び24I11よりも著しく強かった。
【0090】
6.4.非ヒト抗体のヒト化
6.4.1.マウスK33NのV遺伝子のクローニング及びシークエンシング
マウスK33Nハイブリドーマ細胞を、7.5%CO2インキュベーター内、37℃で、10%胎仔ウシ血清(FBS;HyClone,Logan,UT)を含有するTIL培地I(免疫生物研究所,群馬,日本製)中で増殖させた。TRIzol試薬(Invitrogen,Carlsbad,CA)を用いて製造者のプロトコルに従い、約107個のハイブリドーマ細胞から全RNAを抽出した。オリゴdTでプライミングされたcDNAを、GeneRacerキット(Invitrogen)を用いて製造者のプロトコルに従い合成した。K33Nの重鎖及び軽鎖の可変領域cDNAは、それぞれ、マウスγ1鎖及びκ鎖の定常領域にアニールする3’プライマーと、GeneRacerキットに含まれるGeneRacer 5’プライマー(5’−CGACTGGAGCACGAGGACACTGA−3’)(配列番号51)とを用いて、Phusion DNAポリメラーゼ(New England Biolabs,Beverly,MA)によるポリメラーゼ連鎖反応(PCR)により増幅させた。重鎖可変領域(VH)のPCR増幅には、5’−GCCAGTGGATAGACAGATGG−3’(配列番号52)を有する3’プライマーを用いた。軽鎖可変領域(VL)のPCR増幅には、5’−GATGGATACAGTTGGTGCAGC−3’(配列番号53)配列を有する3’プライマーを用いた。増幅されたVHとVLのcDNAを、配列決定のためにpCR4Blunt−TOPOベクター(Invitrogen)にサブクローニングした。可変領域のDNAシークエンシングはTocore(Menlo Park,CA)で実施した。いくつかの重鎖及び軽鎖クローンをシークエンシングし、典型的なマウス重鎖及び軽鎖可変領域に相同なユニーク配列を同定した。コンセンサスcDNA配列を、K33NのVH及びVLの推定アミノ酸配列と共に、それぞれ、図3及び4に示す。
【0091】
6.4.2.キメラK33N IgG1/κ抗体の構築
K33N VHをコードする遺伝子を、K33N VHのcDNAを鋳型として、5’−GGGACTAGTACCACCATGGGATGGAGCTGGATCTTTCTC−3’(下線はSpeI部位)(配列番号40)を5’プライマーとして、5’−GGGAAGCTTGTTTTAAGGACTCACCTGAGGAGACTCTGAGACTGGTGCC−3’(配列番号41)(下線はHindIII部位)を3’プライマーとして用いてPCRを行うことにより、スプライスドナーシグナルと適当な側面制限酵素部位とを含むエキソンとして生成した(図5)。同様に、K33N VLをコードする遺伝子を、K33N VLのcDNAを鋳型として、5’−GGGGCTAGCACCACCATGAGTGTGCCCACTCAACTCCTG−3’(配列番号42)(下線はNheI部位)を5’プライマーとして、5’−GGGGAATTCTGAGAAGACTACTTACGTTTTATTTCCAGCTTGGTCCCCCC−3’(配列番号43)(下線はEcoRI部位)を3’プライマーとして用いてPCRを行うことにより、スプライスドナーシグナルと適当な側面制限酵素部位とを含むエキソンとして生成した(図6)。K33NのVHエキソンとVLエキソンのスプライスドナーシグナルは、それぞれ、マウス生殖系列JH4配列とJκ2配列から得た。PCR増幅断片は、QIAquickゲル抽出キット(Qiagen,Valencia,CA)を用いてゲル精製し、SpeIとHindIII(VHの場合)又はNheIとEcoRI(VLの場合)で消化し、キメラK33N IgG1/κ抗体を産生するために、ヒトγ1定常領域とκ定常領域とを担持する哺乳動物発現ベクターにクローニングした。得られた発現ベクター、pChK33Nの模式的構造を図7に示す。
【0092】
6.4.3.ヒト化K33N V遺伝子の作製
K33N可変領域のヒト化を、Queenら(Proc.Natl.Acad.Sci.USA 86:10029−10033,1989)に概説されているように実施した。まず、コンピュータプログラムを活用してK33N可変領域の分子モデルを構築した。次に、ヒト可変領域配列に対する相同性検索に基づいて、DA980102(GenBankアクセッション番号)に由来するヒトアミノ酸配列であって、K33N VHに対する高い相同性を有する配列を、ヒト化K33N VHのフレームワークを提供するアクセプターとして選択した。マウスK33NのVH領域とDA980102のVH領域のフレームワークにおけるアミノ酸の同一性は、74.7%(65/87)である。同様に、X72441(GenBankアクセッション番号)のヒトアミノ酸配列を、K33N VLのヒト化のためのアクセプターとして選択した。マウスK33NとX72441のVL領域のフレームワークにおけるアミノ酸の同一性は、76.3%(61/80)である。
【0093】
コンピュータモデルによって相補性決定領域(CDR)との顕著な接触が示唆されたフレームワーク位置においては、K33N可変領域由来のアミノ酸をヒトフレームワークアミノ酸の代わりに用いた。28、48、66、67及び71番目においてこの操作を行ない、ヒト化K33N(HuK33N)VHを作製した(図8)。さらに、ヒトDA980102アクセプターの82番目のMetは、ヒトVH配列の同じサブグループでは非典型的であることが判明したことから、最も一般的なアミノ酸残基(Leu)に置き換えて、潜在的免疫原性を減らした。軽鎖については、70番目と71番目の残基で置換を行ない、HuK33N VLを作製した(図9)。K33N、デザインしたHuK33N、及びヒトアクセプターアミノ酸配列のアラインメントを、VHについては図8に、VLについては図9に示す。
【0094】
HuK33NのVHとVLの各々をコードする遺伝子を、シグナルペプチドと、スプライスドナーシグナルと、後に哺乳動物発現ベクターにクローニングするための適当な制限酵素部位とを含むエキソンとして設計した。HuK33NのVHエキソンとVLエキソンのスプライスドナーシグナルは、それぞれ、ヒト生殖系列JH4配列とJκ1配列から得た。マウスK33N VH遺伝子のシグナルペプチド配列は、SIG−Pred シグナルペプチド予測ソフトウェア(http://bmbpcu36.leeds.ac.uk/prot_analysis/Signal.html)によって、正確な切断に準最適であることが示された。そのため、SIG−Predソフトウェアで効率的かつ正確に切断されると予測された、マウス抗ヒトα9インテグリンモノクローナル抗体24I11(ジーンテクノサイエンス社)のVH遺伝子のシグナルペプチド配列を、HuK33N VH遺伝子で用いた。ヒト化K33N VLエキソン中のシグナルペプチド配列は、対応するマウスK33N VL配列から得た。SIG−Predソフトウェアでは、HuK33N VL遺伝子のシグナルペプチドは、効率的かつ正確に切断されると予測された。
【0095】
HuK33NのVH遺伝子とVL遺伝子を、Heら(J.Immunol.160:1029−1035,1998)に概説されているように、ThermalAce DNAポリメラーゼ(Invitrogen)を用いて、いくつかの重複する合成オリゴヌクレオチドプライマーの伸長とPCR増幅によって構築した。HuK33NのVH遺伝子とVL遺伝子の構築に用いたオリゴヌクレオチドを、ぞれぞれ、図10と図11に示す。HuK33NのVH遺伝子とVL遺伝子におけるこれらのオリゴヌクレオチドの位置を、それぞれ、図12と図13に示す。PCR増幅断片をQIAquickゲル抽出キット(Qiagen)を用いてゲル精製し、配列決定のためにpCR4Blunt−TOPOベクターにクローニングした。SpeIとHindIII(VHの場合)又はNheIとEcoRI(VLの場合)で消化した後、HuK33NのVH遺伝子とVL遺伝子を、ヒトIgG1/κ形態で産生するための哺乳動物発現ベクター内の対応する部位にサブクローニングした。得られた発現ベクター、pHuK33Nの模式的構造を図7に示す。得られたHuK33NのVH遺伝子とVL遺伝子のヌクレオチド配列を、推定アミノ酸配列と共に、ぞれぞれ、図14と図15に示す。
【0096】
6.4.4.キメラ及びヒト化K33N IgG1/κの一過性発現
キメラ及びヒト化K33N IgG1/κ抗体は、それぞれ、pChK33N及びpHuK33NのプラスミドDNAを、Durocherら(Nucl.Acids Res.30:e9,2002)に従ってポリエチレンイミンを用いてHEK293細胞にトランスフェクトすることにより一過性に発現させた。一過性にトランスフェクトされたHEK293細胞は、7.5%CO2インキュベーター内、37℃で、10%FBSを含有するDMEM中で4日間維持した。培養上清中のChK33N IgG1/κ抗体及びHuK33N IgG1/κ抗体の各々の発現レベルをサンドイッチELISAで測定した。ELISAプレートは、PBS中1/2,000希釈したヤギ抗ヒトIgG Fcγ鎖特異的ポリクローナル抗体(SouthernBiotech,Birmingham,AL)を100μL/穴で用いて、4℃で一晩コーティングし、洗浄緩衝液(0.05%Tween20を含有するPBS)で洗浄し、ブロッキング緩衝液(2%スキムミルク及び0.05%Tween20を含有するPBS)を300μL/穴で用いて、室温で1時間ブロッキングした。洗浄緩衝液で洗浄した後、ELISA緩衝液(1%スキムミルク及び0.025%Tween 20を含有するPBS)で適当に希釈した試料を100μL/穴でELISAプレートに添加した。ヒトミエローマ血清(SouthernBiotech)から精製したヒトIgG1/κ抗体を標準品として用いた。ELISAプレートを室温で2時間インキュベートし、洗浄緩衝液で洗浄した後、1/2,000希釈したHRPコンジュゲートヤギ抗ヒトκ鎖ポリクローナル抗体(SouthernBiotech)を100μL/穴で用いて、結合した抗体を検出した。室温で1時間インキュベートし、洗浄緩衝液で洗浄した後、100μL/穴のABTS基質(bio WORLD,Dublin,OH)を添加して、発色を行なった。100μL/穴の2%シュウ酸を添加して、発色を停止させた。吸光度を405nmで測定した。
【0097】
一過性に発現されたChK33N抗体とHuK33N抗体のヒトα9インテグリンへの結合を細胞ELISAで調べた。表面に組換えヒトα9インテグリンを発現するCHO−K1安定トランスフェクタント(CHO/huα9;ジーンテクノサイエンス社提供)を、10%FBSを含有する50μLのF12/DMEM(HyClone)中2×105細胞/穴で96穴組織培養プレートに播種し、7.5%CO2インキュベーター内で、37℃で一晩増殖させた。ヒトα9インテグリンへの結合を検討するために、10%FBSを含有するF12/DMEM中のChK33N、HuK33N又は無関係なヒトIgG1/κミエローマ抗体(SouthernBiotech)50μLを各穴に添加した。4℃で1時間インキュベートし、細胞を氷冷PBSで2回洗浄した後、100μLの1/1,000希釈したHRPコンジュゲートヤギ抗ヒトIgGポリクローナル抗体(SouthernBiotech)を各穴に添加した。4℃で1時間インキュベートした後、細胞を氷冷PBSで3回洗浄した。発色させるために、100μLのABTS基質を添加した。100μLの2%シュウ酸を添加して、発色を停止させた。吸光度を405nmで測定した。結果から、0.5μg/mLと1μg/mLの両方において、ChK33N抗体のヒトα9インテグリンへの結合が、HuK33N抗体のヒトα9インテグリンへの結合とほぼ同じであることが示された(図16)。
【0098】
6.4.5.ChK33N及びHuK33N IgG1/κ抗体の各々を産生するNS0安定トランスフェクタントの作製
ChK33N IgG1/κ抗体及びHuK33N IgG1/κ抗体を安定的に産生する細胞株を得るために、それぞれ、発現ベクターpChK33N及びpHuK33Nを、マウスミエローマ細胞株NS0(European Collection of Animal Cell Cultures,Salisbury,Wiltshire,UK)の染色体に導入した。NS0細胞を、7.5%CO2インキュベーター内、37℃で、10%FBSを含有するDME培地中で増殖させた。Bebbingtonら(Bio/Technology 10:169−175,1992)に記載されているように、エレクトロポレーションによりNS0への安定トランスフェクションを行なった。トランスフェクションの前に、発現ベクターを、FspIを用いて線状化した。約107個の細胞を10μgの線状化プラスミドでトランスフェクトし、10%FBSを含有するDME培地に懸濁し、いくつかの96穴プレートに播種した。24時間後、選択培地(10%FBS、HT培地サプリメント(Sigma,St.Louis,MO)、0.25mg/mLキサンチン及び1μg/mLミコフェノール酸を含有するDME培地)を添加した。選択を開始してから約10日後、培養上清をアッセイして抗体産生を調べた。
【0099】
ChK33N IgG1/κ抗体とHuK33N IgG1/κ抗体の発現を、基本的には第6.4.3節に記載した手順に従って、サンドイッチELISAで測定した。高レベルのChK33N抗体を産生するNS0安定トランスフェクタント(NS0−ChK33N 3D11)を、Hybridoma SFM(Invitrogen)を用いて無血清培地中での増殖に適応させた。高レベルのHuK33N抗体を産生するNS0安定トランスフェクタントは、更に、限界希釈によって96穴プレートでサブクローニングした。これらのサブクローンのうちの1つ(NS0−HuK33N 8G8−11)をHybridoma−SFM中での増殖に適応させた。PCRマイコプラズマ検出セット(Takara Bio USA,Madison,WI)を用いた検査により、NS0−ChK33N 3D11とNS0−HuK33N 8G8−11の両方について、マイコプラズマの存在が陰性であることが示された。
【0100】
NS0−HuK33N 6D5−11により産生されたHuK33Nの重鎖と軽鎖が正しいかどうかをcDNAシークエンシングにより確認した。全RNAを、TRIzol試薬(Invitrogen)を用いてNS0−HuK33N 6D5−11細胞から抽出し、オリゴdTでプライミングされたcDNAを、GeneRacerキット(Invitrogen)を用いて製造者のプロトコルに従い合成した。γ1重鎖のコード領域を、CMV2及びJNT098(図17)をプライマーとして用い、Phusionポリメラーゼ(New England Biolabs)を用いてPCRにより増幅した。PCR断片をゲル精製し、CMV2、JNT082、JNT097及びJNT098をプライマーとして用いてシークエンシングした(図17)。同様に、κ軽鎖のコード領域を、CMV2とJNT026とを用いて増幅した(図17)。ゲル精製したDNA断片を、CMV2、JNT026、JNT080及びJNT084をプライマーとして用いてシークエンシングした(図17)。HuK33Nの重鎖と軽鎖の各々のコード領域の得られたヌクレオチド配列は、pHuK33Nベクター中の対応する配列と完全に一致した(それぞれ、図18及び図19)。
【0101】
6.4.6.ChK33N抗体とHuK33N抗体の精製
NS0−ChK33N 3D11細胞及びNS0−HuK33N 8G8−11細胞をローラーボトル中、Hybridoma−SFMで枯渇するまで増殖させた。遠心分離及び濾過後、培養上清をプロテイン−Aセファロースカラム(GE Healthcare,Piscataway,NJ)にかけた。カラムをPBSで洗浄後、抗体を0.1Mのグリシン−HCl(pH3.0)で溶出させた。1MのTris−HCl(pH8)で中和後、溶出した抗体の緩衝液を透析によってPBSに変えた。抗体濃度を、280nmにおける吸光度を測定して決定した(1mg/mL=1.4OD)。抗体発現レベルは、NS0−ChK33N 3D11細胞では50μg/mL、NS0−HuK33N 8G8−11細胞では12μg/mLであった。
【0102】
精製したChK33N抗体とHuK33N抗体を、標準的な手順に従ったSDS−PAGEにより解析した。還元条件下での解析から、ChK33N抗体及びHuK33N抗体は、各々、分子量約50kDaの重鎖と分子量約25kDaの軽鎖から構成されていることが示された(図20)。各抗体の純度は95%を上回るようであった。
【0103】
6.4.7.ChK33N抗体とHuK33N抗体の特徴付け
マウスK33N抗体、キメラK33N抗体、及びヒト化K33N抗体のヒトα9インテグリンへの結合を、CHO/huα9細胞を用いたFACS結合アッセイで調べた。約5×105個のCHO/huα9細胞/試験を、FACS結合緩衝液(0.5%BSA及び0.05%NaN3を含有するPBS)で洗浄し、様々な量の試験抗体を含有する200μLのFACS結合緩衝液に懸濁した。氷上で30分置いた後、細胞をFACS結合緩衝液で洗浄した。次に、マウスK33Nで染色した細胞を、1/200希釈したFITC標識ヤギ抗マウスIgGポリクローナル抗体(SouthernBiotech)を含むFACS結合緩衝液100μLに懸濁した。キメラK33N又はヒト化K33Nで染色した細胞は、1/200希釈したFITC標識ヤギ抗ヒトIgGポリクローナル抗体(SouthernBiotech)を含むFACS結合緩衝液100μLに懸濁した。氷上に30分置いた後、細胞をFACS結合緩衝液で洗浄し、200μLのFACS結合緩衝液に懸濁し、FACScanフローサイトメータ(BD Biosciences,Franklin Lakes,NJ)を用いて解析した。マウスK33N抗体のCHO/huα9細胞への結合のEC50値は104ng/mLであった(図21)。ChK33N抗体とHuK33N抗体の間で、結合パターンは互いによく似ていた(図22)。さらに、ChK33N抗体とHuK33N抗体のEC50値は互いにほぼ同じであった(ChK33N抗体は143ng/mL、HuK33N抗体は151ng/mL)。この結果は、抗原結合親和性を失わせずに、マウスK33N抗体をヒト化することに成功したことを示している。
【0104】
6.4.8.細胞接着アッセイ
96穴平底マイクロタイタープレート(Nunc)を、50μLの5μg/mL hTNC(AEIDGIEL)−BSA溶液で、CO2インキュベーター内、37℃で1時間コーティングした。対照穴を、50μLの5μg/mL BSA溶液でコーティングした。コーティング反応の後、穴中の溶液を200μLの0.5w/v%BSA(ブロッキング)溶液と交換し、室温で1時間インキュベートした。ブロッキング反応後、穴をPBSで洗浄した。0.25w/v%BSAを含有するTIL培地に懸濁した2.5×105細胞/mLのG−361細胞150μLと、試験抗体溶液(0.25w/v%BSA/TIL)50μLとをこれらの穴に添加し、37℃のCO2インキュベーター内で1時間インキュベートした。インキュベート後、非接着細胞を除去するために、各穴をPBS溶液で3回注意深く洗浄した。50μLの0.5w/v%クリスタルバイオレット溶液を添加して、接着細胞を固定及び染色した。30分後、穴一杯の水で3回洗浄して、余分な色素を除去し、穴中の色素を50μLの20v/v%酢酸で可溶化させた。各穴の595nmにおける吸光度を、吸光度計を用いて測定した。細胞接着阻害率は、以下の式;(1−(A−B)/(C−B))×100(%)により算出した。A:抗体が存在し、hTNC−BSAでコーティングした穴の吸光度、B:いずれの試験抗体も存在しない、BSAでコーティングした穴の吸光度、C:正常ヒトIgG存在下のhTNC−BSAでコーティングした穴の吸光度(陰性対照)(図23)。
【0105】
結果
Y9A2のIC50は0.053μg/mL(95%CI:0.032〜0.089)であった。K33N、ChK33N、及びHuK33NのIC50は、それぞれ、0.075μM(0.053〜0.106)、0.090μg/mL(0.063〜0.128)、0.084μg/mL(0.055〜0.129)であり、試験抗体のIC50値がY9A2のIC50値と同じレベルにあることを示した。
【0106】
7.寄託
マウス抗ヒトα9インテグリンモノクローナル抗体を産生する、本明細書でK33Nと表されているハイブリドーマは、2007年5月29日に、微生物の寄託に関するブダペスト条約に準拠して、(郵便番号305−8566)茨城県つくば市東1−1−1中央第6にある、産業技術総合研究所特許生物寄託センターに寄託され、アクセッション番号FERM BP−10830が与えられた。これは、その全体が参照により本明細書に組み込まれる。
【0107】
8.産業上の適用可能性
本発明のヒト化モノクローナル抗体は、α9インテグリンの機能を阻害して、例えば、癌細胞の増殖又は転移等の癌、及び、例えば、関節リウマチ、変形性関節症、肝炎、気管支喘息、線維症、真性糖尿病、癌転移、動脈硬化症、多発性硬化症、肉芽腫、炎症性腸疾患(潰瘍性大腸炎及びクローン病)、自己免疫疾患等の炎症性疾患に対する治療効果を示す。
【0108】
9.配列表
本明細書の全体を通じて言及されている配列を以下にまとめる。
【表2】
【技術分野】
【0001】
本発明は、免疫特異的にヒトα9インテグリンを認識するヒト化抗体、並びに癌、炎症性疾患、自己免疫疾患、α9インテグリンにより誘導される疾患状態等を含む、α9インテグリンと関連するか又はα9インテグリンが関与する様々な疾患又は障害に対する治療及び診断における該抗体の使用に関する。
【背景技術】
【0002】
細胞は、インテグリンと呼ばれる一群の細胞表面受容体を介して細胞外マトリクス(以下、ECMと略す)に接着している。インテグリンは、α鎖とβ鎖の1:1のヘテロ二量体を形成することによりその機能を果たす。これまでに、少なくとも18種類のα鎖、8種類のβ鎖、及び24種類のαβヘテロ二量体が同定され、確認されている。各々のインテグリンは特異的なリガンドを認識することが知られている。インテグリンは、そのリガンド特異性又は機能によってサブファミリーに分類され、コラーゲン受容体、ラミニン受容体、フィブロネクチンやビトロネクチン等に存在するArg−Gly−Asp(RGD)配列を認識するRGD受容体、白血球にのみ存在する白血球特異的受容体に分けられる(Hynes,R.O.,2002,Integrins:Bidirectional,Allosteric Signaling Machines.Cell 110:673−87;宮坂 昌之,2000,新版 接着分子ハンドブック,秀潤社)。α4インテグリンとα9インテグリンは、これらのタイプのいずれにも属さないサブファミリーのメンバーであり、α4インテグリンサブファミリーと呼ばれる(Elise L.Palmer,Curzio Rfiegg,Ronald Ferrando,Robert Pytela,Sheppard D.,1993,Sequence and Tissue Distribution of the Integrin α9 Subunit,a Novel Partner of β1 That Is Widely Distributed in Epithelia and Muscle.The Journal of Cell Biology,123:1289−97)。一方、しばらくの間ECMは、細胞間を固定する物質として機能するにすぎないと考えられていた。しかし、現在は、インテグリン介在性のECM−細胞相互作用が、細胞の増殖、接着、移動等の調節に明らかに関与し、癌の進行、炎症の悪化等を含む疾患の発症と関連することが明らかになっている。
【0003】
例えば、ECMの1つであるオステオポンチン(以後、OPNと略す)は、分子量約41kDaの、分泌型の酸性リン酸化糖タンパク質であり、母乳、尿、尿細管、破骨細胞、骨芽細胞、マクロファージ、活性化T細胞、腫瘍組織等に広くその発現が認められる分子である。OPNは、その分子の中心に接着配列GRGDS(配列番号1)を有し、ヒトOPNはSVVYGLR(配列番号2)配列又はマウスOPNはSLAYGLR(配列番号3)配列、及びそれらに近接するトロンビン切断部位を有する。OPNは、GRGDS(配列番号1)配列を介してRGDインテグリンと結合し、また、SVVYGLR(配列番号2)配列もしくはSLAYGLR(配列番号3)配列を介してα4(α4β1)インテグリン及びα9(α9β1)インテグリンと結合する。
【0004】
WO02/081522号は、OPNノックアウトマウス又はOPNに対する中和抗体を用いてOPNの機能を阻害することにより、関節リウマチ又は肝炎を治療する効果があることを開示している。また、該文献は、SVVYGLR(配列番号2)配列が、α9インテグリン及びα4インテグリンを認識するため炎症性疾患の発症に不可欠であること、及び、OPNの受容体が免疫担当細胞等で発現して、炎症性疾患と関連していることを開示している。
【0005】
結合プロファイルの違いとして、α4β1がトロンビンで切断されていないOPN(未切断型OPN)とトロンビンで切断されたOPNのN末端断片(切断型OPN)の両方に結合するのに対し、α9β1は切断型OPNにのみ結合することが見出されている(Y.Yokosaki,et al.,(1999) The Journal of Biological Chemistry),274:36328−36334;P.M.Green,et al.,(2001) FEBS Letters,503:75−79;S.T.Barry,et al.,(2000) Experimental Cell Research,258:342−351)。
【0006】
α4インテグリンとα9インテグリンは、OPN以外にも多くのリガンドを共有している。知られているリガンドとしては、フィブロネクチンのEDAドメイン、プロペプチド−フォン・ヴィレブランド因子(pp−vWF)、組織トランスグルタミナーゼ(tTG)、血液凝固第XIII因子、血管細胞接着分子−1(VCAM−1)等を挙げることができる。また、フィブロネクチンのCS−1ドメイン、MadCAM−1(α4β7)等は、α4インテグリンに特異的に認識されるリガンドとして知られている。テネイシン−C、プラスミン等は、α9インテグリンによって特異的に認識されるリガンドとして知られている。
【0007】
インテグリンサブユニットα9、α4及びβ1のアミノ酸配列は公知である。例えば、ヒトα9はNM_002207として、マウスα9はNM_133721として、ヒトα4はNM_000885として、マウスα4はNM_010576として、ヒトβ1はX07979として、及びマウスβ1はNM_010578としてGenBankに登録されている。これらのインテグリンは、種間でアミノ酸配列における高い類似性を有することも知られている。
【発明の概要】
【0008】
現在、癌、炎症性疾患及び自己免疫疾患を治療するための多くの種類の薬物が知られているが、癌、炎症性疾患及び自己免疫疾患に対する薬理効果がより改善された予防剤及び/又は治療剤等の開発が望まれている。本発明は、一部、α9インテグリンに対する特異的阻害抗体に癌抑制効果や抗炎症効果があるという本発明者らによる発見に基づくものである。
【0009】
これまで本発明者らは、免疫特異的にヒトα9インテグリンを認識し、ハイブリドーマクローンK33N(受託番号FERM BP−10830)によって産生されるマウスモノクローナル抗体を単離した。本明細書においては、ハイブリドーマクローンの名称は、このクローンによって産生されるモノクローナル抗体の名称と共通で使用する。該マウス抗ヒトα9インテグリン抗体はIgG1アイソタイプであった。また、該モノクローナル抗体は、ヒト及び/又はマウスのα9インテグリンと、オステオポンチン等のα9インテグリンのリガンドとの結合を阻害する。よって、該抗α9インテグリン抗体はα9インテグリンの機能を阻害し、例えば、癌細胞の増殖又は転移等の癌、並びに、例えば、関節リウマチ、変形性関節炎、肝炎、気管支喘息、線維症、糖尿病、動脈硬化症、多発性硬化症、肉芽種、炎症性腸疾患(潰瘍性大腸炎及びクローン病)、自己免疫疾患、α9インテグリンによって誘導される疾患状態等の炎症性疾患に対する治療効果を示す。
【0010】
さらに、本発明の抗α9インテグリン抗体は、対象におけるα9インテグリン発現の存在とレベルを検出し、それによりα9インテグリンと関係する障害又は疾患を診断するためのインビボでの診断薬として用いることができる。
【0011】
しかし、該モノクローナル抗体はマウス由来であることから、ヒトにおいてはその免疫原性による副作用の可能性があり、このことがヒトにおいて診断用又は治療用として直接適用することを妨げてきた。この免疫原性を低下させるために、本発明者らは、由来するマウス抗α9インテグリン抗体が示す活性と同等の生物学的活性を有するヒト化抗体を調製した。
【0012】
したがって、本発明は、免疫特異的にヒトα9インテグリンを認識するヒト化抗体又はその抗原結合断片であって、一部が非ヒト起源に由来し、一部がヒト起源に由来する抗原結合領域を含む抗体又はその抗原結合断片を提供するものである。具体的な実施形態においては、本発明のヒト化抗体又はその抗原結合断片は、K33Nモノクローナル抗体等の非ヒト源(ドナー)に由来する相補性決定領域(CDR)と、ヒト源(アクセプター)に由来するフレームワーク領域(FR)とを含む。一実施形態では、該ヒト化抗体又はその抗原結合断片は、ヒトα9インテグリンとヒトα9インテグリンのリガンドとの結合を阻害する。
【0013】
具体的な実施形態においては、上述の免疫特異的にヒトα9インテグリンを認識するヒト化抗体又はその抗原結合断片は、(i)ヒトH鎖の可変領域(V領域)に由来する少なくとも1つのH鎖FR(FRH)と、免疫特異的にヒトα9インテグリンを認識する非ヒト抗体K33NのCDRHの少なくとも1つに由来する少なくとも1つのH鎖相補性決定領域(CDRH)とを含む重鎖(H鎖);又は(ii)ヒトL鎖のV領域に由来する少なくとも1つのL鎖FR(FRL)と、免疫特異的にヒトα9インテグリンを認識する非ヒト抗体K33NのCDRLの少なくとも1つに由来する少なくとも1つのL鎖相補性決定領域(CDRL)とを含む軽鎖(L鎖);又は上記の(i)と(ii)の両方を含む。例えば、本発明のヒト化抗体のCDRHの少なくとも1つ及び/又はCDRLの少なくとも1つが由来する該非ヒト抗体は、受託番号FERM BP−10830のハイブリドーマによって産生されるモノクローナル抗体である。
【0014】
好ましい実施形態では、本発明のヒト化抗体又はその抗原結合断片は、(i)ヒトFRHに由来する少なくとも1つのFRHと、配列番号4、5及び6のアミノ酸配列からなる群から選択されるアミノ酸配列を含む少なくとも1つのCDRH;又は(ii)ヒトFRLに由来する少なくとも1つのFRLと、配列番号11、12及び13のアミノ酸配列からなる群から選択されるアミノ酸配列を含む少なくとも1つのCDRL;又は(iii)上記の(i)と(ii)の両方を含む。本発明の該ヒト化抗体又はその抗原結合断片は、それぞれが、配列番号4、5及び6のアミノ酸配列を有する、CDRH1、CDRH2及びCDRH3を含んでいてもよい。あるいは、本発明の該ヒト化抗体又はその抗原結合断片は、それぞれが、配列番号11、12及び13のアミノ酸配列を有する、CDRL1、CDRL2及びCDRL3を含む。好ましい実施形態では、上述の本発明のヒト化抗体又はその抗原結合断片は、それぞれ、配列番号4、5、6、11、12及び13のアミノ酸配列を有する、CDRH1、CDRH2、CDRH3、CDRL1、CDRL2及びCDRL3を含む。あるいは、本発明の該ヒト化抗体又はその抗原結合断片は、GenBankアクセッション番号DA980102(配列番号18)によってコードされるヒトH鎖の可変領域に由来するFRH、又はGenBankアクセッション番号X72441(配列番号23)によってコードされるヒトκ−L鎖の可変領域に由来するFRLを含む。好ましい実施形態では、本発明のヒト化抗体のFRHは、配列番号19、20、21及び22のアミノ酸配列(それぞれ順に、DA980102の対応する部分によってコードされるFRH1、FRH2、FRH3及びFRH4)からなる群から選択される少なくとも1つのアミノ酸配列を含む。別の好ましい実施形態では、本発明のヒト化抗体のFRLは、配列番号24、25、26及び27のアミノ酸配列(それぞれ順に、X72441の対応する部分によってコードされるFRL1、FRL2、FRL3及びFRL4)からなる群から選択される少なくとも1つのアミノ酸配列を含む。より好ましい実施形態では、本発明のヒト化抗体又はその抗原結合断片は、(i)配列番号29のアミノ酸配列を含むH鎖可変領域(VH領域);又は(ii)配列番号31のアミノ酸配列を含むL鎖可変領域(VL領域);又は(iii)上記の(i)と(ii)の両方を含む。最も好ましい実施形態では、本発明のヒト化抗体又はその抗原結合断片は、(i)配列番号37のアミノ酸配列を含むγ1H鎖;又は(ii)配列番号39のアミノ酸配列を含むκL鎖;又は(iii)上記の(i)と(ii)の両方を含む。
【0015】
本発明はさらに、免疫特異的にヒトα9インテグリンを認識する本発明のヒト化抗体又はその抗原結合断片をコードするヌクレオチド配列を含む単離された核酸分子を提供する。具体的には、本発明は、配列番号4、5及び6からなる群から選択される少なくとも1つのアミノ酸配列を含むヒト化H鎖、又は配列番号11、12及び13からなる群から選択される少なくとも1つのアミノ酸配列を含むヒト化L鎖、又は該ヒト化H鎖と該ヒト化L鎖の両方をコードするヌクレオチド配列を含む単離された核酸分子を提供する。好ましい具体的な実施形態では、このような単離された核酸分子は、VH領域をコードする配列番号28のヌクレオチド配列、又は配列番号29のアミノ酸配列をコードするヌクレオチド配列を含む。別の好ましい具体的な実施形態では、このような単離された核酸分子は、VL領域をコードする配列番号30のヌクレオチド配列、又は配列番号31のアミノ酸配列をコードするヌクレオチド配列を含む。さらに別の好ましい具体的な実施形態では、本発明の単離された核酸分子は、配列番号28と30の両方のヌクレオチド配列を含む。好ましい具体的な実施形態では、このような単離された核酸分子は、γ1H鎖をコードする配列番号36のヌクレオチド配列、又は配列番号37のアミノ酸配列をコードするヌクレオチド配列を含む。別の好ましい具体的な実施形態では、このような単離された核酸分子は、κL鎖をコードする配列番号38のヌクレオチド配列、又は配列番号39のアミノ酸配列をコードするヌクレオチド配列を含む。さらに別の好ましい具体的な実施形態では、本発明の単離された核酸分子は、配列番号36と38の両方のヌクレオチド配列を含む。別の好ましい具体的な実施形態では、本発明の単離された核酸分子は、更に、例えば、それぞれ、配列番号10及び17のアミノ酸配列のようなドナー起源のシグナルペプチド又は異種起源のシグナルペプチドをコードするヌクレオチド配列を含む。
【0016】
本発明はさらに、免疫特異的にヒトα9インテグリンを認識する本発明のヒト化抗体又はその抗原結合断片のH鎖又はL鎖又はその両方をコードするヌクレオチド配列を含むベクター(例えば、発現ベクター)を提供する。このようなベクターでは、本発明のヌクレオチド配列は、1以上の調節エレメントに機能的に連結され得る。本発明のヌクレオチド配列は、CDRが由来する非ヒトドナー抗体起源のシグナルペプチド、又は異種起源のシグナルペプチドをコードするヌクレオチド配列を含み得る。
【0017】
さらに、本発明は、本発明の核酸分子(本発明の核酸分子を含むベクターを含む)を含む宿主細胞を提供する。一実施形態では、本発明は、本発明のヒト化H鎖をコードする第1の核酸分子と、本発明のヒト化L鎖をコードする第2の核酸分子とを含む単離された宿主細胞を提供するものであり、該第1及び第2の核酸分子は、本発明の生物学的に機能的なヒト化抗体又はその抗原結合断片が発現されるように、それぞれが調節エレメントに機能的に連結されている。
【0018】
したがって、本発明はさらに、ヒト化抗体が発現されるような条件下で本発明の宿主細胞を培養することと、及び産生されたヒト化抗体を回収することとを含む、本発明のヒト化抗体の調製方法を提供する。
【0019】
本発明はさらに、本発明のヒト化抗体の少なくとも1つを含む組成物を提供する。さらに、本発明は、本発明のヒト化抗体の少なくとも1つと薬学的に許容される担体とを含む、α9インテグリンと関連する障害又は疾患を予防又は治療するための薬学的組成物を提供する。前記のいずれの組成物も、当該障害又は疾患を相加的に又は相乗的に改善することができる別の活性物質をさらに含むことができる。このような活性物質としては、これに限定されるものではないが、抗炎症化合物、化学療法剤等や、抗体又はその抗原結合断片(例えば、ヒトα4インテグリンに免疫特異的に結合することができる抗体)が挙げられる。
【0020】
別の態様では、本発明は、α9インテグリンと関連するか又はα9インテグリンが関与する疾患又は障害を予防又は治療する方法であって、予防に有効な量又は治療に有効な量の本発明のヒト化抗体の少なくとも1つをそれを必要とする対象に投与することを含む方法を提供する。このような使用のために、本発明のヒト化抗体はヒト化抗体の生物学的効果を増強する治療的成分と結合していてもよい。このような治療的成分の例としては、(例えば、二重特異性抗体を形成するための)抗α4抗体等の別の抗体、細胞増殖抑制性もしくは細胞破壊性の細胞毒素、放射性元素、及び/又は抗炎症剤、抗生物質等を含む他の治療剤が挙げられる。
【0021】
さらに別の態様では、本発明は、対象におけるα9インテグリンと関連するか又はα9インテグリンが関与する疾患又は障害を診断する方法であって、診断に有効な量の本発明のヒト化抗体を検査対象に投与することを含む方法を提供する。このような診断への使用のために、本発明のヒト化抗体を放射性元素等の検出可能なマーカーで標識してもよい。
【0022】
3.1.定義
本明細書で使用する場合、「抗体」という用語は、例えば、α9インテグリン等の所望の抗原に免疫特異的に結合可能な抗体分子を指し、完全な抗体分子、又は抗原結合断片を含む抗体分子の断片を包含する。
【0023】
本明細書で使用される「免疫特異的に認識する」という用語は、標的ポリペプチド又はタンパク質、特にヒトα9インテグリンに特異的に結合する、抗体又はその抗原結合断片の能力を指す。このような抗体は、他のポリペプチド又はタンパク質に非特異的に結合しない。しかしながら、標的ポリペプチド又はタンパク質(例えば、ヒトα9インテグリン)に免疫特異的に結合する抗体又はその抗原結合断片は、他の抗原と交差反応してもよい。例えば、ヒトα9インテグリンを免疫特異的に認識する本発明のヒト化抗体又は抗原結合断片は、例えば、他の種のα9インテグリンと交差反応してもよい。好ましくは、ヒトα9インテグリンと免疫特異的に結合する抗体又はその抗原結合断片は、他の抗原と交差反応しない。
【0024】
本明細書で使用される「抗原結合断片」という用語は、標的ポリペプチド又はタンパク質、特に、ヒトα9インテグリン及び/又は非ヒトα9インテグリンと免疫特異的に結合する能力を保持する抗体の任意の断片を指し、単鎖抗体、Fab断片、F(ab’)2断片、ジスルフィド結合Fv、並びに標的ポリペプチド又はタンパク質と特異的に結合する軽鎖(VL)の可変領域及び/又は重鎖(VH)の可変領域又は相補性決定領域(CDR)を含有する断片を含む。したがって、ヒト化抗体のこのような抗原結合断片には、部分的又は全長のヒト定常領域が含まれていても、含まれていなくてもよい。上記の抗体断片を得るための様々な方法が当該技術分野で周知である。
【0025】
本明細書で使用される「ヒト源に由来する」又は「非ヒト源に由来する」という用語は、抗体の一部であってそのアミノ酸配列がヒト抗体又は非ヒト抗体の対応する部分に由来する部分を指す。
【0026】
本明細書で使用される「アクセプター配列」という用語は、通常、非ヒト抗体であるドナー抗体由来のCDRのアクセプターとして働く、ヒト抗体VH領域又はVL領域に由来するフレームワーク領域のヌクレオチド配列又はアミノ酸配列を指す。
【図面の簡単な説明】
【0027】
【図1】抗ヒトα9インテグリン抗体(本発明の2つのクローン(すなわち、K33N及びM35A)、5つの他のクローン(1K11、21C5、24I11、25B6、及び28S1)、並びにY9A2)の細胞接着阻害活性を、ヒトα9インテグリン発現細胞(ヒトメラノーマ細胞G361)とOPN α9インテグリン結合部位ペプチド(SVVYGLR)とを用いて測定した実験の結果を示す図である。ヒトオステオポンチンに対するモノクローナル抗体(5A1)を陰性対照として使用した。
【図2】抗ヒトα9インテグリン抗体(本発明の2つのクローン(すなわち、K33N及びM35A)、5つの他のクローン(1K11、21C5、24I11、25B6、及び28S1)、並びにY9A2)の細胞接着阻害活性を、ヒトα9インテグリン発現細胞(ヒトメラノーマ細胞G361)とテネイシン−C断片のα9インテグリン結合部位ペプチドとを用いて測定した実験の結果を示す図である。ヒトオステオポンチンに対するモノクローナル抗体(5A1)を陰性対照として使用した。
【図3】マウスK33N VH cDNAのヌクレオチド配列(配列番号7)を推定アミノ酸配列(配列番号8)と共に示す図である。アミノ酸残基は一文字表記で示す。シグナルペプチド配列(配列番号10)はイタリック体で示す。成熟VHのN末端アミノ酸残基(Q)は二重下線で示す。Kabatら(Sequences of Proteins of Immunological Interests,第5版,NIH Publication 第91−3242号,U.S.Department of Health and Human Services,1991)の定義によるCDR配列を下線で示す。
【図4】マウスK33N VL cDNAのヌクレオチド配列(配列番号14)を推定アミノ酸配列(配列番号15)と共に示す図である。アミノ酸残基は一文字表記で示す。シグナルペプチド配列(配列番号17)はイタリック体で示す。成熟VLのN末端アミノ酸残基(D)は二重下線で示す。Kabatら(1991)の定義によるCDR配列を下線で示す。
【図5】SpeI部位とHindIII部位(下線)が両側にあるようにデザインされたK33N VH(ChK33N VH)遺伝子のヌクレオチド配列(配列番号32)を推定アミノ酸配列(配列番号8)と共に示す。アミノ酸残基は一文字表記で示す。シグナルペプチド配列(配列番号10)はイタリック体で示す。成熟VHのN末端アミノ酸残基(Q)は二重下線で示す。Kabatら(1991)の定義によるCDR配列を下線で示す。イントロン配列をイタリック体で示す。
【図6】NheI部位とEcoRI部位(下線)が両側にあるようにデザインされたK33N VL(ChK33N VL)遺伝子のヌクレオチド配列(配列番号33)を推定アミノ酸配列(配列番号15)と共に示す。アミノ酸残基は一文字表記で示す。シグナルペプチド配列(配列番号17)はイタリック体で示す。成熟VLのN末端アミノ酸残基(D)は二重下線で示す。Kabatら(1991)の定義によるCDR配列を下線で示す。イントロン配列はイタリック体で示す。
【図7】pChK33N及びpHuK33N(総称して発現ベクターという)の模式的構造を示す図である。プラスミドは、一番上のSalI部位から時計回りに、抗体重鎖遺伝子の転写を開始するためのヒトサイトメガロウイルス(CMV)主要前初期プロモーター及びエンハンサー(CMVプロモーター)から始まる重鎖転写ユニットを含有している。CMVプロモーターに続いて、VHエキソンと、CH1、ヒンジ、CH2及びCH3のエキソンが介在イントロンと共に含まれるヒトγ1重鎖定常領域を含有するゲノム配列とがあり、CH3に続いてmRNAのプロセッシングのためのγ1遺伝子のポリアデニル化部位がある。重鎖遺伝子配列の後、軽鎖転写ユニットがCMVプロモーターから始まり、続いてVLエキソンと、ヒトκ鎖定常領域エキソン(CL)をその前にあるイントロンの一部と共に含有するゲノム配列、及びκ遺伝子のポリAシグナルがある。次に、軽鎖遺伝子に続いて、SV40初期プロモーター(SV40プロモーター)と、大腸菌キサンチングアニンホスホリボシルトランスフェラーゼ遺伝子(gpt)と、SV40ポリアデニル化部位(SV40ポリ(A)部位)を含有する部分とがある。最後に、プラスミドは、細菌の複製起点(pUC ori)とβ−ラクタマーゼ遺伝子(β−ラクタマーゼ)とを含むプラスミドpUC19の一部を含有する。
【図8】K33N VH(配列番号9)、ヒト化K33N(HuK33N)VH(配列番号29)並びにGenBankアクセッション番号DA980102のヌクレオチド配列によってコードされるアミノ酸配列に由来する、ヒトアクセプター配列のFRH1(配列番号19)、FRH2(配列番号20)、FRH3(配列番号21)及びFRH4(配列番号22)のアミノ酸配列のアラインメントを示す図である。アミノ酸残基は一文字表記で示す。配列の上にある数字は、Kabatら(1991)による位置を示す。Kabatら(1991)によって定義されたCDR配列は下線で示す。二重下線が付された残基はCDRと接触すると予想され、ヒト化型においてはこれらの位置のマウス残基は保持された。ヒトVH配列のこの位置では非典型的であるDA980102の82番目のMet(下線)を典型的な残基Leuと置き換えて、潜在的免疫原性を減らした。DA980102中のCDR残基はこの図では省略されている。
【図9】K33N VL(配列番号16)、ヒト化K33N(HuK33N)VL(配列番号31)並びにGenBankアクセッション番号X72441のヌクレオチド配列によってコードされるアミノ酸配列に由来する、ヒトアクセプター配列のFRL1(配列番号24)、FRL2(配列番号25)、FRL3(配列番号26)及びFRL4(配列番号27)のアミノ酸配列のアラインメントを示す図である。アミノ酸残基は一文字表記で示す。配列の上にある数字は、Kabatら(1991)による位置を示す。Kabatら(1991)によって定義されたCDR配列は下線で示す。二重下線が付された残基はCDRと接触すると予想され、ヒト化型においてはこれらの位置のマウス残基は保持された。X72441中のCDR残基はこの図では省略されている。
【図10】HuK33N VH遺伝子の構築に使用されたオリゴヌクレオチドを示す図である。
【図11】HuK33N VL遺伝子の構築に使用されたオリゴヌクレオチドを示す図である。
【図12】HuK33N VH遺伝子の構築に使用されたオリゴヌクレオチドを示す図である。矢印は、各々のオリゴヌクレオチドの位置と方向(5’から3’)を表す。VH領域(配列番号29)のアミノ酸残基は一文字表記で示す。
【図13】HuK33N VL遺伝子の構築に使用されたオリゴヌクレオチドを示す図である。矢印は、各々のオリゴヌクレオチドの位置と方向(5’から3’)を表す。VL領域(配列番号31)のアミノ酸残基は一文字表記で示す。
【図14】SpeI部位とHindIII部位(下線)が両側にある、HuK33N VH遺伝子のヌクレオチド配列(配列番号34)を、シグナルペプチド(配列番号58;イタリック体で示す)及びVH領域(配列番号29)の推定アミノ酸配列と共に示す図である。アミノ酸残基は一文字表記で示す。シグナルペプチド配列はイタリック体で示す。成熟VHのN末端アミノ酸残基(Q)は二重下線で示す。Kabatら(1991)の定義によるCDR配列は下線で示す。イントロン配列はイタリック体で示す。
【図15】NheI部位とEcoRI部位(下線)が両側にある、HuK33N VL遺伝子のヌクレオチド配列(配列番号35)を、シグナルペプチド(配列番号59;イタリック体で示す)及びVL領域(配列番号31)の推定アミノ酸配列と共に示す図である。アミノ酸残基は一文字表記で示す。シグナルペプチド配列はイタリック体で示す。成熟VLのN末端アミノ酸残基(D)は二重下線で示す。Kabatら(1991)の定義によるCDR配列は下線で示す。イントロン配列はイタリック体で示す。
【図16】キメラK33N抗体とヒト化K33N抗体のヒトα9インテグリンに対する親和性の比較を示す。1μg/mL及び0.5μg/mLのキメラK33N及びヒト化K33NのCHO/α9細胞への結合を細胞ELISAで調べた。実験は3重で実施した。吸光度の平均値を標準誤差と共に示す図である。
【図17】HuK33N重鎖及び軽鎖cDNAのPCR増幅及び配列決定に使用したオリゴヌクレオチドの配列(配列番号44〜50)を示す。
【図18】pHuK33N中のHuK33N γ1重鎖のコード領域のヌクレオチド配列(配列番号36)を推定アミノ酸配列(配列番号37)と共に示す図である。アミノ酸残基は一文字表記で示す。終止コドンは「・」で示す。
【図19】pHuK33N中のHuK33N κ軽鎖のコード領域のヌクレオチド配列(配列番号38)を推定アミノ酸配列(配列番号39)と共に示す図である。アミノ酸残基は一文字表記で示す。終止コドンは「・」で示す。
【図20】精製抗体のSDS−PAGE解析の結果を示す図である。6μgのキメラIgG1/κ抗体及びヒト化IgG1/κ抗体(それぞれ、ChK33N及びHuK33Nという)を、Sambrookら(Molecular Cloning,A Laboratory Manual,第2版,1989,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY)に従って、還元条件下、SDS存在下の10%ポリアクリルアミドゲル上で泳動させた。広域タンパク質マーカー(MW;New England Biolabs,Ipswich,MA)をサイズマーカーとして使用した。右側に示す数字は、マーカーのサイズをキロダルトン(kDa)単位で示す。
【図21】ヒトα9インテグリンに対するマウスK33N抗体の結合をFACS解析した結果を示す図である。1.67μg/mLから連続的に3倍希釈した様々な濃度のマウスK33N抗体について、CHO/huα9細胞への結合を試験した。図中、試験した各々の抗体濃度(X軸)における、幾何平均チャンネル蛍光値(MCF;Y軸)がプロットされている。EC50値を、GraphPad Prism(GraphPad Software,San Diego,CA)を用いて算出した。
【図22】ヒトα9インテグリンに対するキメラK33N抗体とヒト化K33N抗体の結合をFACS解析した結果を示す図である。5μg/mlから連続3倍希釈した様々な濃度の各抗体について、CHO/huα9細胞への結合を試験した。図中、試験した各々の抗体濃度(X軸)における、幾何平均チャンネル蛍光値(MCF;Y軸)がプロットされている。EC50値を、GraphPad Prism(GraphPad Software,San Diego,CA)を用いて算出した。
【図23】ヒトα9インテグリンに対するマウスK33N抗体、キメラK33N抗体及びヒト化K33N抗体、並びにマウス抗ヒトα9インテグリン抗体Y9A2の細胞接着阻害率の用量依存性を示す図である。各抗体を5、1、0.2、0.04、0.008及び0.0016μg/mLの濃度で、ヒトα9インテグリン発現細胞株G−361の細胞接着について試験した。実験は4重で実施した。平均阻害率の値を図に示す。
【発明を実施するための形態】
【0028】
5.1.ヒトα9インテグリンに対する抗体の調製
ヒトα9インテグリン又はその任意のエピトープを免疫特異的に認識する抗体は、当該技術分野で公知の任意の好適な方法により作製することができる。
【0029】
本発明において抗原として使用されるα9インテグリンは、(1)α9インテグリンを発現するヒト由来の全ての細胞、もしくはこれらの細胞が存在する全ての組織から得られるタンパク質、(2)α9インテグリンをコードする遺伝子DNA、好ましくはcDNAが、細菌、酵母、細胞株(動物細胞を含む)等に導入され、発現された、組換えタンパク質、又は(3)合成タンパク質であってもよい。
【0030】
α9インテグリンには、ヒトα9インテグリン(配列番号55、ここで、1〜29番目の残基はシグナルペプチドである)のアミノ酸配列と実質的に同一のアミノ酸配列を含むポリペプチドが含まれる。
【0031】
本明細書中、「実質的に同一のアミノ酸配列を含むポリペプチド」という用語は、天然のヒトα9インテグリンと実質的に同等の生物学的特性を有する限り、複数のアミノ酸、好ましくは1〜10個のアミノ酸、より好ましくは1〜数個(例えば、1〜5個)のアミノ酸が置換され、欠失し及び/又は改変されたアミノ酸配列を含む変異体ポリペプチド;並びに複数のアミノ酸、好ましくは1〜10個のアミノ酸、より好ましくは1〜数個(例えば、1〜5個)のアミノ酸が天然のヒトα9インテグリンのアミノ酸配列に付加されたアミノ酸配列を含む変異体ポリペプチドを意味する。さらに、変異体ポリペプチドは、これらのアミノ酸置換、欠失、改変及び付加の複数を有するものであってもよい。
【0032】
本発明における抗原としてのヒトα9インテグリンは、当該技術分野で周知の方法により産生することができ、例えば、遺伝子組換え技術の他、化学合成法、細胞培養法等、又はそれらの変法により産生することができる。
【0033】
変異体ポリペプチドを産生する方法の例としては、合成オリゴヌクレオチド部位特異的突然変異生成(ギャップ二重鎖法)、亜硝酸又は亜硫酸処理によってランダムに点突然変異を導入することを含む点突然変異生成法、Bal31酵素又は他の酵素を用いて欠失突然変異を調製することを含む方法、カセット突然変異生成、リンカースキャニング法、ミス組込み法、ミスマッチプライマー法、DNA断片合成法等が挙げられる。
【0034】
本発明において抗原として使用されるヒトα9インテグリンは、該α9インテグリンの「一部」も含む。本明細書において、「一部」とは、α9インテグリンのリガンド、例えば、OPN、VCAM−1、テネイシン−C等との結合に必要な領域を含む部分;特には、成熟ヒトα9インテグリン(配列番号55の30番目から1035番目までのアミノ酸残基)の、14番目から980番目までのアミノ酸残基を含む部分、及び11番目から981番目までのアミノ酸残基を含む部分を指す。該α9インテグリンの「一部」は、以下に記載する当該技術分野で公知の方法に従って、遺伝子組換えもしくは化学合成、又はそれらの変法によって産生することができ、あるいは、細胞培養法より単離されたヒトα9インテグリンをタンパク質分解的酵素等で適当に消化することによって産生することができる。
【0035】
抗原として、細胞膜上にα9インテグリンを過剰発現する細胞それ自体、又はその膜画分を使用することもできる。ヒトα9インテグリンを過剰発現する細胞は、当該技術分野で周知の組換えDNA技術によって調製することができる。
【0036】
上記のように調製された適当な抗原を用いて、ヒトα9インテグリン又は任意のそのエピトープに特異的な抗体を、当該技術分野で周知の様々な方法によって調製することができる。ヒトα9インテグリンに対するポリクローナル抗体は、当該技術分野で周知の様々な手順によって産生することができる。例えば、目的の抗原を、これに限定するものではないが、ウサギ、マウス、ラット等を含む様々な宿主動物に投与し、抗原に特異的なポリクローナル抗体を含有する抗血清の産生を誘導することができる。宿主の種に応じて、様々なアジュバントを用いて免疫学的応答を増大させてもよい。アジュバントとしては、フロイント(完全及び不完全)アジュバント、水酸化アルミニウム等の鉱物ゲル、リゾレシチン等の表面活性物質、プルロニックポリオール、ポリアニオン、ペプチド、オイルエマルジョン、キーホールリンペットヘモシアニン、ジニトロフェノール、並びに潜在的に有用なヒト用のアジュバント(例えば、BCG(カルメット・ゲラン桿菌)及びコリネバクテリウム・パルブム)が挙げられるが、これらに限定されるものではない。このようなアジュバントも当該技術分野において周知である。
【0037】
モノクローナル抗体は、ハイブリドーマ、組換え体及びファージディスプレイ技術、又はそれらの組合せを用いる方法を含む、当該技術分野で公知の多種多様な技術を用いて調製することができる。例えば、モノクローナル抗体は、当該技術分野で公知であり、また、例えば、Harlowら,Antibodies:A Laboratory Manual,(Cold Spring Harbor Laboratory Press,第2版.1988);Hammerling,et al.,:Monoclonal Antibodies and T−Cell Hybridomas,563−681ページ(Elsevier,N.Y.,1981)(両文献は参照により本明細書に組み込まれる)等に教示されている技術を含む、ハイブリドーマ技術を用いて産生することができる。本明細書で使用される「モノクローナル抗体」という用語は、ハイブリドーマ技術によって産生される抗体に限定されるものではない。「モノクローナル抗体」という用語は、任意の真核生物、原核生物、又はファージクローンを含む、単一のクローンに由来する抗体を指すものであり、それが産生される方法を指すものではない。
【0038】
ハイブリドーマ技術を用いた特異的抗体の産生及びスクリーニングする方法は、当該技術分野においてルーチンで行われておりかつ周知である。非限定的な例としては、マウスを目的の抗原又はそのような抗原を発現する細胞で免疫化することができる。免疫応答が検出されれば、例えば、抗原に特異的な抗体がマウス血清中に検出されれば、マウス脾臓を摘出し、脾細胞を単離する。その後、脾細胞を周知の技術により任意の好適なミエローマ細胞(例えば、P3U1、P3X63−Ag8、P3X63−Ag8−U1、P3NS1−Ag4、SP2/0−Ag14、P3X63−Ag8−653等)に融合させる。ハイブリドーマを選択し、限界希釈によりクローニングする。その後、ハイブリドーマクローンを、当該技術分野で公知の方法により、抗原に結合することができる抗体を分泌する細胞についてアッセイする。通常高レベルの抗体を含有する腹水は、陽性のハイブリドーマクローンをマウスの腹腔内に接種することにより作製することができる。
【0039】
特異的エピトープを認識する抗体断片は、公知の技術により作製することができる。例えば、Fab及びF(ab’)2断片は、免疫グロブリン分子をパパイン(Fab断片を産生するため)又はペプシン(F(ab’)2断片を産生するため)等の酵素を用いてタンパク質分解切断させることにより産生することができる。F(ab’)2断片は、完全な軽鎖と、重鎖の可変領域、CH1領域及びヒンジ領域とを含有する。
【0040】
本発明の抗体又はその抗原結合断片は、抗体の合成について当該技術分野で知られている任意の方法により、具体的には、化学合成又は好ましくは、組換え発現技術により産生することもできる。
【0041】
抗体をコードするヌクレオチド配列は、当業者が利用可能な任意の情報から(すなわち、GenBank、文献から、又は日常的なクローニング及び配列解析により)取得することができる。特定の抗体又はそのエピトープ結合断片をコードする核酸を含有するクローンが入手できず、抗体分子又はそのエピトープ結合断片の配列が知られている場合、免疫グロブリンをコードする核酸は、化学合成、あるいは、抗体配列の5’末端にハイブリダイズ可能な合成プライマーを用いたPCR増幅によって、又は、抗体をコードするcDNAライブラリー由来のcDNAクローン等を同定するための特定の遺伝子配列に特異的なオリゴヌクレオチドプローブを用いたクローニングによって、好適な供給源(例えば、抗体cDNAライブラリー、又は、例えば、抗体を発現する選択されたハイブリドーマ細胞等の抗体を発現する任意の組織もしくは細胞から作製されたcDNA、又はそのような組織若しくは細胞から単離された核酸、好ましくはポリA+ RNA)から取得し得る。その後、PCRで生成した増幅核酸を、当該技術分野で周知の任意の方法を用いて、複製可能なクローニングベクターにクローニングすることができる。
【0042】
5.2.組換え抗体の調製
抗体のヌクレオチド配列決定後、抗体のヌクレオチド配列を、ヌクレオチド配列の操作に関する当該技術分野で周知の方法、例えば、組換えDNA技術、部位特異的突然変異誘発法、PCR等(例えば、Sambrookら.,前記;及びAusubelら.,編,1998,Current Protocols in Molecular Biology,John Wiley & Sons,NYに記載されている技術を参照。両文献とも、その全体が参照により本明細書に組み込まれる。)を用いて操作して、例えば、抗体のエピトープ結合ドメイン領域又は抗体の生物学的活性を増強しもしくは低下させ得る抗体の任意の部分にアミノ酸の置換、欠失、及び/又は挿入を導入することにより、様々なアミノ酸配列を有する抗体を作製することができる。
【0043】
抗体の組換え発現には、抗体をコードするヌクレオチド配列を含有する発現ベクターの構築が必要である。抗体分子又は抗体の重鎖もしくは軽鎖、あるいはそれらの一部をコードするヌクレオチド配列が得られた後、前記節に記載されたように当該技術分野で周知の技術を用いた組換えDNA技術により抗体分子を産生するためのベクターを産生することができる。当業者に周知の方法を用いて、抗体コード配列と適当な転写及び翻訳制御シグナルとを含有する発現ベクターを構築することができる。これらの方法としては、例えば、インビトロ組換えDNA技術、合成技術、及びインビボ遺伝子組換えが挙げられる。抗体の重鎖可変領域、軽鎖可変領域、重鎖と軽鎖の両方の可変領域、重鎖及び/又は軽鎖可変領域のエピトープ結合断片、あるいは1以上の相補性決定領域(CDR)をコードするヌクレオチド配列を、このような発現用ベクターにクローニングすることができる。このような配列を、基となる抗体に由来するシグナルペプチド又は異種シグナルペプチドをコードするポリヌクレオチドと融合させることができる。その後、このように調製された発現ベクターは、抗体を発現させるための適当な宿主細胞に導入することができる。したがって、本発明は、免疫特異的にヒトα9インテグリンを認識するヒト化抗体又はその抗原結合断片をコードするポリヌクレオチドを含有する宿主細胞を含む。
【0044】
宿主細胞に、重鎖由来のポリペプチドをコードする第1のベクターと軽鎖由来のポリペプチドをコードする第2のベクターの本発明の2つの発現ベクターをコトランスフェクトしてもよい。この2つのベクターは、重鎖ポリペプチドと軽鎖ポリペプチドが同等に発現することを可能にする同一の選択マーカーを有していてもよいし、又は両方のプラスミドの維持を確実にするための異なる選択マーカーを含有していてもよい。あるいは、重鎖と軽鎖の両方のポリペプチドをコードし、それらを発現することができる単一のベクターを使用してもよい。重鎖及び軽鎖のコード配列は、cDNA又はゲノムDNAのいずれを含んでいてもよい。
【0045】
別の実施形態では、抗体は、当該技術分野で公知の様々なファージディスプレイ法を用いて作製することもできる。ファージディスプレイ法では、機能的抗体ドメインが、それらをコードするポリヌクレオチド配列を担持するファージ粒子の表面上に提示される。具体的な実施形態では、このようなファージを、レパートリー又はコンビナトリアル抗体ライブラリー(例えば、ヒト又はマウス)により発現される抗原結合ドメイン(例えば、Fab及びFv又はジスルフィド結合安定化Fv)を提示させるために利用することができる。目的の抗原に結合する抗原結合ドメインを発現するファージは、例えば、標識された抗原又は固体表面もしくはビーズに結合もしくは捕捉された抗原を用いて、抗原で選択又は同定することができる。これらの方法で使用されるファージは、典型的には、fd及びM13を含む繊維状ファージである。抗原結合ドメインは、ファージ遺伝子III又は遺伝子VIIIタンパク質のいずれかに融合された組み換えタンパク質として発現される。本発明の免疫グロブリン、又はその断片を作製するために使用することができるファージディスプレイ法の例としては、Brinkmanら,J.Immunol.Methods,182:41−50,1995;Amesら,J.Immunol.Methods,184:177−186,1995;Kettleboroughら,Eur.J.Immunol.,24:952−958,1994;Persicら,Gene,187:9−18,1997;Burtonら,Advances in Immunology,57:191−280,1994;PCT出願PCT/GB91/01134号;PCT公開公報WO90/02809号;WO91/10737号;WO92/01047号;WO92/18619号;WO93/11236号;WO95/15982号;WO95/20401号;並びに米国特許第5,698,426号;第5,223,409号;第5,403,484号;第5,580,717号;第5,427,908号;第5,750,753号;第5,821,047号;第5,571,698号;第5,427,908号;第5,516,637号;第5,780,225号;第5,658,727号;第5,733,743号及び第5,969,108号に開示されているものが挙げられ、これらは各々、その全体が参照により本明細書に組み込まれる。
【0046】
上述の参考文献に記載されているように、ファージ選択の後、ファージ由来の抗体コード領域を単離し、例えば、以下で詳細に記載するように、ヒト抗体を含む完全抗体、又は任意の他の所望の断片を作製するために使用し、哺乳動物細胞、昆虫細胞、植物細胞、酵母、及び細菌を含む任意の所望の宿主で発現させることができる。例えば、Fab、Fab’及びF(ab’)2断片を産生する組換え技術を、例えば、PCT公開公報WO92/22324号;Mullinaxら,BioTechniques,12(6):864−869,1992;及びSawaiら,AJRI,34:26−34,1995;及びBetterら,Science,240:1041−1043,1988に開示された方法(各々、その全体が参照により本明細書に組み込まれる)等の当該技術分野で公知の方法を用いて採用することもできる。単鎖Fv及び抗体を産生することができる技術の例としては、米国特許第4,946,778号及び第5,258,498号;Hustonら,Methods in Enzymology,203:46−88,1991;Shuら,PNAS,90:7995−7999,1993;並びにSkerraら,Science,240:1038−1040,1988に記載されているものが挙げられる。
【0047】
本発明の抗体分子は、上述の任意の方法で産生後、免疫グロブリン分子の精製に関する当該技術分野で公知の任意の方法によって、例えば、クロマトグラフィー(例えば、イオン交換、アフィニティ、具体的には、プロテインAもしくはプロテインG精製後の特異的抗原に対するアフィニティ、及びサイズ分離カラムクロマトグラフィー)、遠心分離、示差溶解度、又はタンパク質の精製のための任意の他の標準的な技術によって精製することができる。また、本発明の抗体又はその断片は、精製を容易にするため、本明細書に記載の又は当該技術分野で公知の異種ポリペプチド配列と融合させてもよい。
【0048】
ヒトでの抗体のインビボ使用及びインビトロ検出アッセイを含む、いくつかの用途のために、キメラ、ヒト化又はヒト抗体を使用することが好ましい場合がある。キメラ抗体及びヒト化抗体は、下記の第5.3節で詳細に説明されている。
【0049】
他の化合物又は異種ポリペプチドと融合しているか又はコンジュゲートしている抗体は、インビトロでのイムノアッセイ、精製方法(例えば、アフィニティクロマトグラフィー)、及びインビボでの治療的又は診断的用途で利用することができる。例えば、PCT公開公報WO93/21232号;欧州特許第439,095号;Naramuraら,Immunol.Lett.,39:91−99,1994;米国特許第5,474,981号;Gilliesら,PNAS,89:1428−1432,1992;及びFellら,J.Immunol.,146:2446−2452,1991を参照(これらは、その全体が参照により本明細書に組み込まれる)。例えば、抗体は、既知の方法又は市販のキット(例えば、ビオチン標識、FITC標識、APC標識)を用いて様々な方法で標識することができる。他の例として、抗体は、インビボでの抗体の生物学的効果を増強する治療成分とコンジュゲートしていてもよい。このような治療成分の例としては、他の抗体、細胞増殖抑制性もしくは細胞破壊性の細胞毒素、放射性元素、及び/又は抗炎症剤、抗生物質等を含む他の治療剤が挙げられる。本発明において、ヒト化抗ヒトα9インテグリンは、抗α4抗体等の別の抗体とコンジュゲートして(例えば、二重特異性抗体を形成して)いてもよい。他の例として、本発明のヒト化抗体は、インビボでの診断的用途のため、例えば、放射性元素等の検出可能なマーカーで標識されていてもよい。
【0050】
5.3.キメラ抗体及びヒト化抗体
キメラ抗体は、マウスモノクローナル抗体に由来する可変領域とヒト免疫グロブリンに由来する定常領域とを有する抗体等の、抗体の異なる部分が異なる動物種に由来する分子である。キメラ抗体を産生する方法は当該技術分野で公知である。例えば、Morrison,Science,229:1202,1985;Oら,BioTechniques,4:214 1986;Gilliesら,J.Immunol.Methods,125:191−202,1989;米国特許第5,807,715号;第4,816,567号;及び第4,816,397号を参照(これらは、その全体が参照により本明細書に組み込まれる)。
【0051】
ヒト化抗体は、所望の抗原に結合する分子であり、非ヒト種に由来する1以上の相補性決定領域(CDR)とヒト免疫グロブリン分子に由来する1以上のフレームワーク領域とを含有する可変領域を含む。非ヒト抗体をヒト化する典型的な方法は、様々な参考文献、例えば、Queenら,1989,Proc.Natl.Acad.Sci USA 86:10029−10033、並びに米国特許第5,585,089号及び第5,693,762号;Riechmannら,Nature,332:323,1988;Tsurushitaら,Methods 36:69−83,2005に記載されており、これらは全て、その全体が参照により本明細書に組み込まれる。例えば、Tsurushitaら(2005,前記;以後、「Tsurushita」という)による参考文献は、もともとQueenら(1989,前記)によって開発された抗体ヒト化方法に基づくマウスモノクローナル抗体のヒト化についての実用的かつ有益なプロトコルを提供している。Tsurushitaに開示されている一般的プロトコルを以下で簡単に説明する。
【0052】
5.3.1.ヒト化抗体を調製するための一般的プロトコル
マウスV遺伝子のクローニング及びシークエンシング
標的マウスモノクローナル抗体のVH領域及びVL領域をコードするcDNAをクローニングするために様々な方法を利用することができる。例えば、SMART RACE cDNA増幅キット(BD Biosciences,CA)又はGeneRacerキット(Invitrogen,CA)を用いた、5’RACE(cDNA末端の迅速な増幅)法が一般に使用されている。5’RACEの遺伝子特異的プライマーは、H鎖とL鎖の各々の可変領域のすぐ下流に結合することができるように、標的モノクローナル抗体のH鎖とL鎖のアイソタイプに基づいて調製することができる。したがって、5’RACEプライマーは、γ1、γ2a、γ2b又はγ3等の、マウスにおける各々のサブタイプに特異的となるように設計してもよい。あるいは、サブタイプ間のコンセンサス領域又は相同性の高い領域に基づいて全てのサブタイプに共通するプライマーを設計してもよい。Tsurushitaでは、以下の5’RACEプライマーが例として開示されている:
(i)5’−GCCAGTGGATAGACTGATGG−(配列番号56)(マウスγ1、γ2a、γ2b及びγ3H鎖のクローニング用)
(ii)5’−GATGGATACAGTTGGTGCAGC−(配列番号57)(マウスκ軽鎖のクローニング用)。
【0053】
PCR増幅したV遺伝子断片は、例えば、Zero Blunt TOPO PCRクローニングキット(Invitrogen)と、決定されたDNA配列とを用いて、プラスミドベクターに直接クローニングすることができる。例えば、得られたDNA配列がコードするアミノ酸配列を、例えば、Model 241タンパク質シークエンサー(Hewlett−Packard,CA)等を用いたN末端アミノ酸シークエンシングによって決定された標的モノクローナル抗体のアミノ酸配列と比較することによって、得られたDNA配列を確認すべきである。典型的には、例えば、エドマン分解等により、標的抗体のN末端の少なくとも15〜20アミノ酸残基を決定すれば、クローニングされたDNA配列の真偽を十分確認することができる。Tsurushitaは、マウスにおいて最も一般的な2つのN末端アミノ酸の1つであるグルタミンがN末端アミノ酸である場合、それがピログルタミンに変換されて、N末端のシークエンシングを阻止することがあることに注意が必要であるとしている。この場合、配列を得るためにN末端のブロックを解除する必要がある。
【0054】
V領域の三次元モデリング
VH領域及びVL領域の配列に基づいて、CDRの立体構造を維持するために潜在的に重要な標的抗体のフレームワーク残基を、例えば、R.Levyら,1989,Biochemistry 28:7168−7175;及びB.Zilberら,1990,Biochemistry 29:10032−10041に記載されている方法によってまず同定する。典型的には、VH領域及びVL領域の各々は、14個の構造的に意味のある部分に分けられ、これらの部分は、免疫グロブリンスーパーファミリーのドメイン構造を含むβ鎖及びループ様構造である。標的抗体由来の各々の部位のアミノ酸配列と、PDBデータベース中の構造が既知の抗体の対応する部分の配列とを比較する(H.M.Bermanら,2000,Nucleic Acids Res.28:235−342を参照)。複数の配列を比較することにより、各々の標的部位との配列の相同性が最も高い対応する部分を選択し、V領域の三次元モデルを構築する。構造を最適化するために、このモデルを、(例えば、ENCADを用いて、又はPressら,1990,“Numerical Recipes”,Cambridge University Press,Cambridgeの記載に従って;Weinerら,1981,J.Comp.Chem.2:287−303に記載されたAMBER;英国がん研究所が運営するBio Molecular Modelling、又は“BMM”ウェブサイトで入手可能な3D−JIG−SAW;又はジェノバにあるスイスバイオインフォマティクス研究所が運営するExPASy Proteomics Serverウェブサイトで入手可能なSWISS−MODELによって)、複数回、共役勾配法によりエネルギー最小化を行った。
【0055】
ヒトフレームワークの選択
V領域の構造のモデリングと並行して、マウスVH領域及びVL領域のcDNAクローニングから推定されるアミノ酸配列を、それぞれ、例えば、Kabatデータベース(Johnsonら,2000,Nucleic Acids Res.28:214−218を参照)、GenBank等におけるヒトV領域配列と比較する。配列全体でマウス配列と少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、又は少なくとも95%の相同性のあるヒトフレームワーク領域は、例えば、Smith−Watermanアルゴリズム(“Algorithms on Strings,Trees,and Sequences”,Cambridge University Press,Cambridgeにおける、Gusfield,1997)、又はBLAST(Karlinら,1990,Proc.Natl.Acad.Sci USA 87:2264−2268)等を用いて、検索することができる。これらのヒト配列は、cDNAに基づく配列及びタンパク質由来の配列のいずれに基づくものであってもよいが、多くの場合、生殖系列の使用がcDNAに基づく配列、タンパク質由来の配列における体細胞超突然変異と関連する潜在的免疫原性を消失させるのに有用であることから、生殖細胞系列の使用が好ましい。あるいは、Queenら(1989,前記)に記載されているように、コンセンサスフレームワーク配列を用いて、cDNAに基づく配列又はタンパク質由来の配列から得られたフレームワーク中のそのような超突然変異残基を同定し、除去することもできる。生殖細胞系列のVH部分をアクセプターフレームワークとして使用する場合、第14番染色体上のVH部分のみが機能的なVH領域を生じさせるので、第15番及び第16番染色体ではなく、第14番染色体上にコードされるVH部分を使用する。
【0056】
ヒト化V領域の設計
Queenら(1989,前記)によれば、CDRの約4〜6Åの範囲内にあるフレームワークアミノ酸は、正確なCDR構造を支持する潜在的に重要なフレームワーク残基であると考えられることから、これらのフレームワークアミノ酸を同定することが必要である。このプロセスは、例えば、米国国立科学財団(NSF)が支援するMolecular Visualization Freewareウェブサイトで入手可能なRASMOL等の原子座標から原子間距離を計算するコンピュータプログラムを用いるか、又は手作業でコンピュータモデルを検査することによって行うことができる。重要なフレームワーク位置のアミノ酸が、マウスドナー配列とヒトアクセプター配列の間で異なる場合、通常ヒト残基をマウスドナー配列で置き換える。しかしながら、このような残基のCDR構造を支持する上での寄与がわずかである場合、典型的には対応するヒト残基が用いられる。また、選択されたヒトアクセプターが、V領域配列の約10〜20%未満で「非典型的な」アミノ酸を含有する場合、それらは親和性成熟の間の体細胞超突然変異の結果であり得ることから、ヒトにおける潜在的免疫原性を回避するためにドナー残基と置き換えるべきである。
【0057】
さらに、ヒト化V領域を設計するために、潜在的なN結合型グリコシル化シグナルの残基等の他の因子を注意深く考慮する必要がある(詳細についてはTsurushitaを参照)。
【0058】
ヒト化抗体は、治療目的で必要とされ又は消失させられるべきエフェクター機能に応じて、ヒトκもしくはλ軽鎖由来のヒト定常領域もしくはその部分、及び/又はヒト抗体のγ1、γ2、γ3、γ4、μ、α1、α2、δ、もしくはε重鎖、あるいはそれらの変異体を含有していてもよい。例えば、突然変異を有する定常領域のFc部分を、抗体のFc受容体への結合を低下させるため、及び/又は補体を固定する能力を低下させるために、本発明のキメラ抗体又はヒト化抗体の可変領域と融合させてもよい(例えば、Winterら、ドイツ特許第2,209,757号;Morrisonら、WO89/07142号、Morganら、WO94/29351号を参照)。このような抗体分子の操作は、第5.2節に記載の組換えDNA技術により実施することができる。
【0059】
好ましくは得られたキメラ抗体又はヒト化抗体は、非ヒトドナー抗体と同じ特異性及び非ヒトドナー抗体の親和性と同等か、又は少なくとも約1/3、少なくとも約1/2、もしくは少なくとも約2/3の親和性を有する。別の態様では、得られたキメラ抗体又はヒト化抗体は、少なくとも約1×107M−1、好ましくは少なくとも約1×108M−1、最も好ましくは少なくとも約1×109M−1の親和定数を有する。
【0060】
上述の一般的プロトコルに加えて、抗体は、当該技術分野で公知の種々の技術を用いてヒト化することができ、例えば、CDRグラフティング(欧州特許第239,400号;PCT公開公報WO91/09967号;米国特許第5,225,539号;第5,530,101号及び第5,585,089号)、ベニアリング(veneering)又はリサーフェイシング(resurfacing)(欧州特許第592,106号;欧州特許第519,596号;Padlan,Molecular Immunology,28(4/5):489−498,1991;Studnickaら,Protein Engineering,7(6):805−814,1994;Roguskaら,Proc Natl.Acad.Sci.USA,91:969−973,1994)、並びに鎖シャッフリング(米国特許第5,565,332号)が挙げられ、これらは全てその全体が参照により本明細書に組み込まれる。
【0061】
5.3.2.ヒト化抗体を医薬品として調製するために更に考慮すべき事項
ヒト化抗体を医薬品として提供するために、効率的で安定的な産生系を調製する必要がある。例えば、H鎖及びL鎖配列を挿入することによってヒト化抗体用の適切な発現ベクターを調製し、この発現ベクターがトランスフェクトされた生産性の高い細胞株をマスター細胞バンク(MCB)用の種細胞として得ることができる。マスター細胞バンク(MCB)は、ワーキング細胞バンク(WCB)の安定的かつ半永久的な供給源として利用される。その後、WCB由来のワーキング細胞を培養し、その培養培地を回収することにより、ヒト化抗体を調製することができる。
【0062】
適当な調節遺伝子を含む様々な発現ベクターを、そのような産生細胞株の調製に用いることができる。哺乳動物タンパク質を発現するために一般に用いられる細胞を宿主細胞としてヒト化抗体の発現に用いることができる。このような宿主細胞としては、これに限定されるものではないが、チャイニーズハムスター卵巣(CHO)細胞、SP2/0−Ag14.19細胞、NSO細胞等が挙げられる。発現ベクターと宿主細胞の最良の組合せを選択することにより、ヒト化抗体の生産性を最大化することができる。さらに、宿主細胞によるヒト化抗体の発現を最適化するように好適な培地を選択するため、培養培地の組成を様々な無血清培地及び添加物から検討するべきである。
【0063】
効率と最終的な収率に基づいて、宿主細胞によって産生されたヒト化抗体を、アフィニティクロマトグラフィー、イオン交換クロマトグラフィー、疎水性相互作用クロマトグラフィー等を含む、当該技術分野で周知の様々な方法を用いて培養上清から精製することができる。
【0064】
5.4.医薬組成物及び治療用途
本発明は、免疫特異的にヒトα9インテグリンを認識する、上述のヒト化抗体又はその抗原結合断片を含む医薬組成物を提供する。本発明のヒト化抗体を有効成分として含有する医薬組成物は、これに限定されるものではないが、例えば、癌細胞の増殖又は転移等の癌、及び、例えば、関節リウマチ、変形性関節症、肝炎、気管支喘息、線維症、真性糖尿病、動脈硬化症、多発性硬化症、肉芽腫、炎症性腸疾患(潰瘍性大腸炎及びクローン病)、自己免疫疾患等の炎症性疾患を含む、α9インテグリンと関連する障害又は疾患を予防及び/又は治療するための薬剤として用いることができる。
【0065】
本発明のヒト化抗体を含有する医薬組成物は、臓器移植後の慢性的拒絶、及び、例えば、全身性自己免疫疾患、エリテマトーデス、ブドウ膜炎、ベーチェット病、多発性筋炎、増殖性糸球体腎炎、サルコイドーシス、α9インテグリンによって誘導される疾患状態等の自己免疫疾患を治療するためにも用いることができる。
【0066】
上述の障害又は疾患を予防又は治療するための、本発明のヒト化抗体を含む予防用及び/又は治療用薬剤は毒性が低く、好適な溶媒中で混合することにより液体調製物として直接、又は適当な剤形の医薬組成物として、ヒトに経口投与又は非経口投与することができる。
【0067】
上述の投与に用いられる医薬組成物は、前述の抗体又はその塩と薬学的に許容される担体、希釈剤又は賦形剤とを含有する。このような組成物は、経口投与又は非経口投与に好適な剤形で提供される。
【0068】
投与量は、投与されるべき対象の年齢及び大きさ、標的疾患、状態、投与経路等によって様々に変わり得る。抗体を、例えば、成人患者の関節リウマチ等の予防及び/又は治療に用いる場合、本発明の抗体を、一回当たり通常約0.01〜約20mg/体重kg、好ましくは約0.1〜約10mg/体重kg、より好ましくは約0.1〜約5mg/体重kgの用量で、1日におよそ1〜5回、好ましくは1日におよそ1〜3回静脈内投与することが有効である。他の非経口投与及び経口投与では、抗体を前記用量に対応する用量で投与することができる。状態が特に重篤である場合は、その状態によって用量を増加させてもよい。
【0069】
例えば、リポソーム、マイクロ粒子、マイクロカプセルへの封入、突然変異体ウイルスを発現することができる組換え細胞、受容体を介するエンドサイトーシス等の様々な送達系が知られており、本発明の医薬組成物を投与するために用いることができる(例えば、Wu and Wu,1987,J.Biol.Chem.262:4429 4432を参照)。投与経路としては、これに限定されるものではないが、皮内、筋肉内、腹腔内、静脈内、皮下、鼻腔内、硬膜外、及び経口経路が挙げられる。化合物は、任意の利用しやすい経路、例えば、注入又はボーラス注射、あるいは上皮又は皮膚粘膜内層(例えば、口腔粘膜、直腸及び腸粘膜等)からの吸収によって投与してもよく、また、他の生物学的に活性のある薬剤と共に投与してもよい。投与は全身性又は局所性のいずれでもよい。例えば、吸入器又はネブライザーとエアロゾル化剤を用いて、経肺投与を採用してもよい。
【0070】
特定の実施形態では、本発明の医薬組成物を、治療を必要とする部位に局所的に投与することが望ましい場合がある。このような投与は、例えば、これに限定されるものではないが、手術時の局所注入、手術後の創傷包帯と組み合わせ等による局所塗布、注射、カテーテル、坐剤、鼻スプレー、又はインプラントによって行うことができる。前記インプラントは、多孔性、非多孔性、又はゼラチン様の物質からなり、シラスティック膜等の膜、又は繊維を含む。一実施形態では、感染した組織部位(又は、かつて感染組織部位であった部位)への直接注射により投与してもよい。
【0071】
別の実施形態では、医薬組成物は、小胞、特にリポソーム内で送達することができる(Langer,1990,Science 249:1527−1533;Liposomes in the Therapy of Infectious Disease and Cancer,Lopez Berestein and Fidler(編),Liss,New York,353−365頁(1989)のTreatらの稿;同書,317−327頁のLopez−Beresteinの稿を参照;一般には同書を参照)。
【0072】
更に別の実施形態では、医薬組成物は、放出制御系で送達することができる。一実施形態では、ポンプを用いてもよい(Langer,前記;Sefton,1987,CRC Crit.Ref.Biomed.Eng.14:201;Buchwaldら,1980,Surgery 88:507;及びSaudekら,1989,N.Engl.J.Med.321:574を参照)。別の実施形態では、ポリマー材料を用いることができる(Medical Applications of Controlled Release,Langer and Wise(編),CRC Pres.,Boca Raton,Florida(1974);Controlled Drug Bioavailability,Drug Product Design and Performance,Smolen and Ball(編),Wiley,New York(1984);Ranger and Peppas,J.Macromol.Sci.Rev.Macromol.Chem.23:61(1983)を参照;また、Levyら,1985,Science 228:190;Duringら.,1989,Ann.Neurol.25:351;Howardら.,1989,J.Neurosurg.71:105も参照)。さらに別の実施形態では、放出制御系を組成物の標的の近くに置くことができ、それにより全身投与量の一部しか必要としないようにすることができる(例えば、Medical Applications of Controlled Release,前記,第2巻,115−138頁(1984)のGoodsonの稿を参照)。他の放出制御系は、Langer(Science 249:1527−1533(1990))による総説に記載されている。
【0073】
経口投与用の組成物の例としては、固体又は液体剤形、特に、錠剤(糖衣錠及びフィルムコート錠を含む)、丸薬、顆粒、粉末調製物、カプセル(ソフトカプセルを含む)、シロップ、エマルジョン、懸濁液等が挙げられる。このような組成物は、公知の方法により製造され、薬学的調製の分野で従来から用いられているビヒクル、希釈剤又は賦形剤を含有する。錠剤用のビヒクル又は賦形剤の例としては、ラクトース、デンプン、スクロース、ステアリン酸マグネシウム等が挙げられる。
【0074】
注射用調製物は、静脈内、皮下、皮内及び筋肉内注射、点滴等のための剤形を含んでいてもよい。これらの注射用調製物は、公知の方法により調製することができる。注射用調製物は、例えば、上述の抗体又はその塩を、従来から注射用に用いられている滅菌水性媒体又は油性媒体中で溶解、懸濁又は乳化させることによって調製してもよい。注射用の水性媒体としては、例えば、生理的食塩水、グルコース及び他の助剤を含有する等張溶液等が挙げられ、これらは、例えば、アルコール(例えば、エタノール)、多価アルコール(例えば、プロピレングリコール、ポリエチレングリコール)、非イオン性界面活性剤[例えば、ポリソルベート80、HCO−50(水素化ヒマシ油のポリオキシエチレン(50モル)付加物)]等の適切な可溶化剤と組み合わせて用いることができる。油性媒体としては、例えば、ゴマ油、大豆油等を利用することができ、これらは、例えば、安息香酸ベンジル、ベンジルアルコール等の可溶化剤と組み合わせて用いることができる。このように調製された注射液は、好ましくは適当なアンプルに充填される。直腸投与に用いられる坐剤は、前述の抗体又はその塩を従来の坐剤用基剤と混合することにより調製することができる。
【0075】
有効な手段としては、上述の経口又は非経口で使用するための医薬組成物は、有効成分の投与量に合わせるのに適した単位用量からなる剤形で調製する。このような単位投与量からなる剤形としては、例えば、錠剤、丸薬、カプセル、注射液(アンプル)、坐剤等が挙げられる。含有する前述の抗体の量は、通常、単位投与量からなる剤形当たり約5〜500mgであり;特に、注射液の形態では、前述の抗体を約5〜100mg含有することが好ましく、また、他の剤形の場合、約10〜250mg含有することが好ましい。
【0076】
上述の各々の組成物は、製剤が上述の抗体との何らかの有害な相互作用を生じさせない限り、他の有効成分をさらに含有していてもよい。
【0077】
本発明はまた、α9インテグリン結合性機能分子(例えば、OPN、VCAM−1、テネイシン−C、フィブロネクチン、pp−vWF、tTG等)を有効成分として含有する細胞及び/又は組織再構築の阻害剤及び/又は促進剤;並びにα9インテグリン発現細胞及び/又は組織(例えば、腫瘍細胞、好中球、平滑筋等)をα9インテグリン結合性機能分子と接触させることを含む、細胞及び/又は組織再構築の阻害方法及び/又は促進方法に関する。このような治療剤中の有効成分の投与量、投与方法、薬学的調製方法等は、本発明のヒト化抗体を含む医薬品に関する前述の記載を参照して適切に決定することができる。
【0078】
上述のように、本発明は更にα9インテグリンと関連するか又はα9インテグリンが関与する障害又は疾患を予防又は治療する方法であって、有効量の本発明のヒト化抗体の少なくとも1つをそれを必要とする対象に投与することを含む方法を提供する。
【0079】
5.5.診断的使用
本発明のヒト化抗体を含む医薬組成物は、例えば、癌細胞の増殖又は転移等の癌、及び、例えば、関節リウマチ、変形性関節症、肝炎、気管支喘息、線維症、真性糖尿病、癌転移、動脈硬化症、多発性硬化症、肉芽腫等の炎症性疾患のための診断薬として、又は臓器移植後の慢性的拒絶、及び、例えば、全身性自己免疫疾患、エリテマトーデス、ブドウ膜炎、ベーチェット病、多発性筋炎、増殖性糸球体腎炎、サルコイドーシス、α9インテグリンによって誘導される疾患状態等の自己免疫疾患、並びにその他の疾患ための診断薬として用いることができる。本発明のヒト化抗体は、α9インテグリンを特異的に認識することができることから、被験液中のα9インテグリンの定量に用いることができ、特に、サンドイッチイムノアッセイ、競合アッセイ、イムノメトリー、ネフロメトリー等、免疫染色、又は同様の方法による定量に用いることができる。これらの免疫学的方法を本発明のアッセイ方法に適用する場合には、特別な条件、手順等の説明は必要なく、当該技術分野における通常の技術的検討を従来の条件及び手順に加えてアッセイ系を構築することで十分である。これらの一般的技術手段の詳細は、総説、教科書等を参照することができる。
【0080】
上述のように、α9インテグリンは、本発明の抗体を用いることにより高感度に定量することができる。本発明のヒト化抗体は、α9インテグリンの定量をインビボで行うことによりα9インテグリンと関連する様々な疾患を診断する上で特に有用である。例えば、α9インテグリンの発現レベルの増加又は減少が検出される場合、対象はα9インテグリンと関連する疾患、例えば、癌もしくは炎症性疾患を患っている可能性が極めて高く、又は将来においてこれらの疾患を患う可能性が極めて高いと診断することができる。したがって、本発明はまた、対象におけるα9インテグリンと関連するか又はα9インテグリンが関与する障害又は疾患を診断する方法であって、有効量の本発明のヒト化抗体の少なくとも1つ又は両方を、それを必要とする対象に投与することを含む方法を提供する。このようなインビボ診断に必要な投与量は、治療用途に必要な投与量よりも少なくてもよく、日常的な手順によって当業者が決定することができる。
【0081】
本発明のヒト化抗体は、体液等の被験液、組織等に存在するα9インテグリンを特異的に検出するために用いることもできる。ヒト化抗体は、α9インテグリンの精製用の抗体カラムを調製し、精製時の各画分に含まれるα9インテグリンを検出し、又は被験細胞中のα9インテグリンの挙動を解析するために用いることもできる。
【実施例】
【0082】
6.実施例
以下の実施例では、ヒト及び/又はマウスのα9インテグリンを免疫特異的に認識するモノクローナル抗体の調製、これらのモノクローナル抗体の可変領域の配列決定、及びこれらの抗体の他の特徴、そのような抗体のキメラ化及びヒト化、並びに得られたキメラ抗体及びヒト化抗体の特徴を説明する。これらの実施例は、これに限定するものと解釈されるものではない。
【0083】
6.1.ヒトα9インテグリンに対するマウス抗体の調製
ヒトα9インテグリンに対するマウスモノクローナル抗体は、サブトラクティブ免疫化法(Williams C.V.ら,1992,Biotechniques 12:842−847による)に従って調製した。簡単には、3匹のBalb/cマウスに、マウス1匹当たり3×106個の、ヒトα9インテグリンを発現するNIH−3T3細胞(ヒトα9/NIH−3T3細胞)を腹腔内注射した。注射の1週間後及び2週間後に、マウスに、3×106細胞/マウスのヒトα9/NIH−3T3細胞を腹腔内注射し、次いで1週間後に、同じ細胞を2×106細胞/マウスでさらに静脈内注射した。ハイブリドーマを当該技術分野で周知の方法により調製した(例えば、Harlowら,Antibodies:A Laboratory Manual,(Cold Spring Harbor Laboratory Press,第2版.1988);Monoclonal Antibodies and T−CeIl Hybridomas,563−681頁のHammerlingらの稿(Elsevier,N.Y.,1981)を参照)。ヒトα9インテグリンを発現するヒトα9/CHO−K1細胞及びヒトα9インテグリンを内在性に発現するヒトメラノーマ細胞とは免疫特異的に反応するが、ヒトα4インテグリンを発現するCHO−K1細胞とは免疫特異的に反応しないモノクローナル抗体を産生するハイブリドーマクローンを樹立し、ヒトα9インテグリンを免疫特異的に認識するモノクローナル抗体を産生するハイブリドーマクローン(すなわち、K33N)を単離した。
【0084】
6.2.抗ヒトα9インテグリン抗体のCDR解析
モノクローナル抗体(すなわち、K33N)のCDRのアミノ酸配列を、対応するハイブリドーマから抽出したmRNAを逆転写して、cDNAを調製することにより決定した。このcDNAを鋳型として用いて、H鎖とL鎖の可変領域を、ScFv−クローニングプライマー(Light Primer Mix and Heavy Primer Mix;Amersham Biosciences Corp.,IL製)を用いてPCRで伸長させ、増幅させた。このPCR産物をpCRII TOPOベクターにクローニングし、シークエンシングしてアミノ酸配列を決定した。このプロセスを3回繰り返した。結果を表1に示す。
【0085】
【表1】
【0086】
6.3.細胞接着阻害活性
(1)細胞接着は、α9インテグリンのそのリガンド、すなわち、OPN、フィブロネクチン、テネイシン−C、VCAM−1等を含む様々なECM、への結合を伴うことが知られていることから、ヒトα9インテグリンを発現する細胞(ヒトメラノーマG361細胞)のリガンドへの結合を用いることにより、単離された抗ヒトα9インテグリン抗体の細胞接着阻害活性について調べた。
【0087】
簡単には、OPNペプチドを、ウシ血清アルブミン(BSA)−融合SVVYGLR(配列番号2)ペプチドとして調製した。TN−C fn3(RAA)は、ヒトテネイシン−C中のフィブロネクチンタイプIII反復の3番目の領域として調製する。このペプチドのGRD配列は、大腸菌宿主細胞で発現させることによりRAA配列に置き換えられている。
【0088】
OPNペプチド及びテネイシン−C断片(TN−C fn3(RAA))を、96穴プレートに5μg/mLで添加し、37℃で1時間インキュベートしてプレートにコーティングし、その後、このプレートをブロッキング溶液(0.5%BSA/PBS)でブロッキングした。ヒトメラノーマG361細胞を0.25%BSA/DMEMに懸濁し(1×105細胞/mL)、各濃度の抗ヒトα9インテグリン抗体をこの細胞懸濁液に添加した。0.25%BSA/DMEM中のヒトメラノーマG361細胞(1×105細胞/mL)と抗体の混合物を、96穴プレートに200μL/穴で添加し、5%CO2、37℃で1時間インキュベートした。非接着細胞をPBSで洗い流し、接着細胞を固定し、0.5%クリスタルバイオレット(和光純薬工業,大阪,日本製)/20%メタノールで染色した。染色された細胞を蒸留水で3回洗浄し、次に、20%酢酸溶液をそれに添加して、溶解させた。590nmの波長におけるODを測定して、接着活性を定量した。陰性対照として抗ヒトOPNモノクローナル抗体(5A1)、陽性対照として以前に調製した抗ヒトα9インテグリン抗体(1K11,21C5、24I11、25B6及び28S1)を用いた。
【0089】
抗ヒトα9インテグリン抗体による、G361細胞のOPNペプチドへの結合に対する効果を図1に示し、テネイシン−C断片への結合に対する効果を図2に示す。図1に示すように、OPNが関与する細胞接着は、陽性対照の21C5及び24I11と比べて低い濃度のK33Nにより阻害され、かつ、Y9A2と同じ濃度のK33Nにより阻害された。テネイシン−Cが関与する細胞接着は、低濃度のK33N、かつ、Y9A2と同じ濃度のK33Nによって阻害され、この阻害効果は、陽性対照の21C5及び24I11よりも著しく強かった。
【0090】
6.4.非ヒト抗体のヒト化
6.4.1.マウスK33NのV遺伝子のクローニング及びシークエンシング
マウスK33Nハイブリドーマ細胞を、7.5%CO2インキュベーター内、37℃で、10%胎仔ウシ血清(FBS;HyClone,Logan,UT)を含有するTIL培地I(免疫生物研究所,群馬,日本製)中で増殖させた。TRIzol試薬(Invitrogen,Carlsbad,CA)を用いて製造者のプロトコルに従い、約107個のハイブリドーマ細胞から全RNAを抽出した。オリゴdTでプライミングされたcDNAを、GeneRacerキット(Invitrogen)を用いて製造者のプロトコルに従い合成した。K33Nの重鎖及び軽鎖の可変領域cDNAは、それぞれ、マウスγ1鎖及びκ鎖の定常領域にアニールする3’プライマーと、GeneRacerキットに含まれるGeneRacer 5’プライマー(5’−CGACTGGAGCACGAGGACACTGA−3’)(配列番号51)とを用いて、Phusion DNAポリメラーゼ(New England Biolabs,Beverly,MA)によるポリメラーゼ連鎖反応(PCR)により増幅させた。重鎖可変領域(VH)のPCR増幅には、5’−GCCAGTGGATAGACAGATGG−3’(配列番号52)を有する3’プライマーを用いた。軽鎖可変領域(VL)のPCR増幅には、5’−GATGGATACAGTTGGTGCAGC−3’(配列番号53)配列を有する3’プライマーを用いた。増幅されたVHとVLのcDNAを、配列決定のためにpCR4Blunt−TOPOベクター(Invitrogen)にサブクローニングした。可変領域のDNAシークエンシングはTocore(Menlo Park,CA)で実施した。いくつかの重鎖及び軽鎖クローンをシークエンシングし、典型的なマウス重鎖及び軽鎖可変領域に相同なユニーク配列を同定した。コンセンサスcDNA配列を、K33NのVH及びVLの推定アミノ酸配列と共に、それぞれ、図3及び4に示す。
【0091】
6.4.2.キメラK33N IgG1/κ抗体の構築
K33N VHをコードする遺伝子を、K33N VHのcDNAを鋳型として、5’−GGGACTAGTACCACCATGGGATGGAGCTGGATCTTTCTC−3’(下線はSpeI部位)(配列番号40)を5’プライマーとして、5’−GGGAAGCTTGTTTTAAGGACTCACCTGAGGAGACTCTGAGACTGGTGCC−3’(配列番号41)(下線はHindIII部位)を3’プライマーとして用いてPCRを行うことにより、スプライスドナーシグナルと適当な側面制限酵素部位とを含むエキソンとして生成した(図5)。同様に、K33N VLをコードする遺伝子を、K33N VLのcDNAを鋳型として、5’−GGGGCTAGCACCACCATGAGTGTGCCCACTCAACTCCTG−3’(配列番号42)(下線はNheI部位)を5’プライマーとして、5’−GGGGAATTCTGAGAAGACTACTTACGTTTTATTTCCAGCTTGGTCCCCCC−3’(配列番号43)(下線はEcoRI部位)を3’プライマーとして用いてPCRを行うことにより、スプライスドナーシグナルと適当な側面制限酵素部位とを含むエキソンとして生成した(図6)。K33NのVHエキソンとVLエキソンのスプライスドナーシグナルは、それぞれ、マウス生殖系列JH4配列とJκ2配列から得た。PCR増幅断片は、QIAquickゲル抽出キット(Qiagen,Valencia,CA)を用いてゲル精製し、SpeIとHindIII(VHの場合)又はNheIとEcoRI(VLの場合)で消化し、キメラK33N IgG1/κ抗体を産生するために、ヒトγ1定常領域とκ定常領域とを担持する哺乳動物発現ベクターにクローニングした。得られた発現ベクター、pChK33Nの模式的構造を図7に示す。
【0092】
6.4.3.ヒト化K33N V遺伝子の作製
K33N可変領域のヒト化を、Queenら(Proc.Natl.Acad.Sci.USA 86:10029−10033,1989)に概説されているように実施した。まず、コンピュータプログラムを活用してK33N可変領域の分子モデルを構築した。次に、ヒト可変領域配列に対する相同性検索に基づいて、DA980102(GenBankアクセッション番号)に由来するヒトアミノ酸配列であって、K33N VHに対する高い相同性を有する配列を、ヒト化K33N VHのフレームワークを提供するアクセプターとして選択した。マウスK33NのVH領域とDA980102のVH領域のフレームワークにおけるアミノ酸の同一性は、74.7%(65/87)である。同様に、X72441(GenBankアクセッション番号)のヒトアミノ酸配列を、K33N VLのヒト化のためのアクセプターとして選択した。マウスK33NとX72441のVL領域のフレームワークにおけるアミノ酸の同一性は、76.3%(61/80)である。
【0093】
コンピュータモデルによって相補性決定領域(CDR)との顕著な接触が示唆されたフレームワーク位置においては、K33N可変領域由来のアミノ酸をヒトフレームワークアミノ酸の代わりに用いた。28、48、66、67及び71番目においてこの操作を行ない、ヒト化K33N(HuK33N)VHを作製した(図8)。さらに、ヒトDA980102アクセプターの82番目のMetは、ヒトVH配列の同じサブグループでは非典型的であることが判明したことから、最も一般的なアミノ酸残基(Leu)に置き換えて、潜在的免疫原性を減らした。軽鎖については、70番目と71番目の残基で置換を行ない、HuK33N VLを作製した(図9)。K33N、デザインしたHuK33N、及びヒトアクセプターアミノ酸配列のアラインメントを、VHについては図8に、VLについては図9に示す。
【0094】
HuK33NのVHとVLの各々をコードする遺伝子を、シグナルペプチドと、スプライスドナーシグナルと、後に哺乳動物発現ベクターにクローニングするための適当な制限酵素部位とを含むエキソンとして設計した。HuK33NのVHエキソンとVLエキソンのスプライスドナーシグナルは、それぞれ、ヒト生殖系列JH4配列とJκ1配列から得た。マウスK33N VH遺伝子のシグナルペプチド配列は、SIG−Pred シグナルペプチド予測ソフトウェア(http://bmbpcu36.leeds.ac.uk/prot_analysis/Signal.html)によって、正確な切断に準最適であることが示された。そのため、SIG−Predソフトウェアで効率的かつ正確に切断されると予測された、マウス抗ヒトα9インテグリンモノクローナル抗体24I11(ジーンテクノサイエンス社)のVH遺伝子のシグナルペプチド配列を、HuK33N VH遺伝子で用いた。ヒト化K33N VLエキソン中のシグナルペプチド配列は、対応するマウスK33N VL配列から得た。SIG−Predソフトウェアでは、HuK33N VL遺伝子のシグナルペプチドは、効率的かつ正確に切断されると予測された。
【0095】
HuK33NのVH遺伝子とVL遺伝子を、Heら(J.Immunol.160:1029−1035,1998)に概説されているように、ThermalAce DNAポリメラーゼ(Invitrogen)を用いて、いくつかの重複する合成オリゴヌクレオチドプライマーの伸長とPCR増幅によって構築した。HuK33NのVH遺伝子とVL遺伝子の構築に用いたオリゴヌクレオチドを、ぞれぞれ、図10と図11に示す。HuK33NのVH遺伝子とVL遺伝子におけるこれらのオリゴヌクレオチドの位置を、それぞれ、図12と図13に示す。PCR増幅断片をQIAquickゲル抽出キット(Qiagen)を用いてゲル精製し、配列決定のためにpCR4Blunt−TOPOベクターにクローニングした。SpeIとHindIII(VHの場合)又はNheIとEcoRI(VLの場合)で消化した後、HuK33NのVH遺伝子とVL遺伝子を、ヒトIgG1/κ形態で産生するための哺乳動物発現ベクター内の対応する部位にサブクローニングした。得られた発現ベクター、pHuK33Nの模式的構造を図7に示す。得られたHuK33NのVH遺伝子とVL遺伝子のヌクレオチド配列を、推定アミノ酸配列と共に、ぞれぞれ、図14と図15に示す。
【0096】
6.4.4.キメラ及びヒト化K33N IgG1/κの一過性発現
キメラ及びヒト化K33N IgG1/κ抗体は、それぞれ、pChK33N及びpHuK33NのプラスミドDNAを、Durocherら(Nucl.Acids Res.30:e9,2002)に従ってポリエチレンイミンを用いてHEK293細胞にトランスフェクトすることにより一過性に発現させた。一過性にトランスフェクトされたHEK293細胞は、7.5%CO2インキュベーター内、37℃で、10%FBSを含有するDMEM中で4日間維持した。培養上清中のChK33N IgG1/κ抗体及びHuK33N IgG1/κ抗体の各々の発現レベルをサンドイッチELISAで測定した。ELISAプレートは、PBS中1/2,000希釈したヤギ抗ヒトIgG Fcγ鎖特異的ポリクローナル抗体(SouthernBiotech,Birmingham,AL)を100μL/穴で用いて、4℃で一晩コーティングし、洗浄緩衝液(0.05%Tween20を含有するPBS)で洗浄し、ブロッキング緩衝液(2%スキムミルク及び0.05%Tween20を含有するPBS)を300μL/穴で用いて、室温で1時間ブロッキングした。洗浄緩衝液で洗浄した後、ELISA緩衝液(1%スキムミルク及び0.025%Tween 20を含有するPBS)で適当に希釈した試料を100μL/穴でELISAプレートに添加した。ヒトミエローマ血清(SouthernBiotech)から精製したヒトIgG1/κ抗体を標準品として用いた。ELISAプレートを室温で2時間インキュベートし、洗浄緩衝液で洗浄した後、1/2,000希釈したHRPコンジュゲートヤギ抗ヒトκ鎖ポリクローナル抗体(SouthernBiotech)を100μL/穴で用いて、結合した抗体を検出した。室温で1時間インキュベートし、洗浄緩衝液で洗浄した後、100μL/穴のABTS基質(bio WORLD,Dublin,OH)を添加して、発色を行なった。100μL/穴の2%シュウ酸を添加して、発色を停止させた。吸光度を405nmで測定した。
【0097】
一過性に発現されたChK33N抗体とHuK33N抗体のヒトα9インテグリンへの結合を細胞ELISAで調べた。表面に組換えヒトα9インテグリンを発現するCHO−K1安定トランスフェクタント(CHO/huα9;ジーンテクノサイエンス社提供)を、10%FBSを含有する50μLのF12/DMEM(HyClone)中2×105細胞/穴で96穴組織培養プレートに播種し、7.5%CO2インキュベーター内で、37℃で一晩増殖させた。ヒトα9インテグリンへの結合を検討するために、10%FBSを含有するF12/DMEM中のChK33N、HuK33N又は無関係なヒトIgG1/κミエローマ抗体(SouthernBiotech)50μLを各穴に添加した。4℃で1時間インキュベートし、細胞を氷冷PBSで2回洗浄した後、100μLの1/1,000希釈したHRPコンジュゲートヤギ抗ヒトIgGポリクローナル抗体(SouthernBiotech)を各穴に添加した。4℃で1時間インキュベートした後、細胞を氷冷PBSで3回洗浄した。発色させるために、100μLのABTS基質を添加した。100μLの2%シュウ酸を添加して、発色を停止させた。吸光度を405nmで測定した。結果から、0.5μg/mLと1μg/mLの両方において、ChK33N抗体のヒトα9インテグリンへの結合が、HuK33N抗体のヒトα9インテグリンへの結合とほぼ同じであることが示された(図16)。
【0098】
6.4.5.ChK33N及びHuK33N IgG1/κ抗体の各々を産生するNS0安定トランスフェクタントの作製
ChK33N IgG1/κ抗体及びHuK33N IgG1/κ抗体を安定的に産生する細胞株を得るために、それぞれ、発現ベクターpChK33N及びpHuK33Nを、マウスミエローマ細胞株NS0(European Collection of Animal Cell Cultures,Salisbury,Wiltshire,UK)の染色体に導入した。NS0細胞を、7.5%CO2インキュベーター内、37℃で、10%FBSを含有するDME培地中で増殖させた。Bebbingtonら(Bio/Technology 10:169−175,1992)に記載されているように、エレクトロポレーションによりNS0への安定トランスフェクションを行なった。トランスフェクションの前に、発現ベクターを、FspIを用いて線状化した。約107個の細胞を10μgの線状化プラスミドでトランスフェクトし、10%FBSを含有するDME培地に懸濁し、いくつかの96穴プレートに播種した。24時間後、選択培地(10%FBS、HT培地サプリメント(Sigma,St.Louis,MO)、0.25mg/mLキサンチン及び1μg/mLミコフェノール酸を含有するDME培地)を添加した。選択を開始してから約10日後、培養上清をアッセイして抗体産生を調べた。
【0099】
ChK33N IgG1/κ抗体とHuK33N IgG1/κ抗体の発現を、基本的には第6.4.3節に記載した手順に従って、サンドイッチELISAで測定した。高レベルのChK33N抗体を産生するNS0安定トランスフェクタント(NS0−ChK33N 3D11)を、Hybridoma SFM(Invitrogen)を用いて無血清培地中での増殖に適応させた。高レベルのHuK33N抗体を産生するNS0安定トランスフェクタントは、更に、限界希釈によって96穴プレートでサブクローニングした。これらのサブクローンのうちの1つ(NS0−HuK33N 8G8−11)をHybridoma−SFM中での増殖に適応させた。PCRマイコプラズマ検出セット(Takara Bio USA,Madison,WI)を用いた検査により、NS0−ChK33N 3D11とNS0−HuK33N 8G8−11の両方について、マイコプラズマの存在が陰性であることが示された。
【0100】
NS0−HuK33N 6D5−11により産生されたHuK33Nの重鎖と軽鎖が正しいかどうかをcDNAシークエンシングにより確認した。全RNAを、TRIzol試薬(Invitrogen)を用いてNS0−HuK33N 6D5−11細胞から抽出し、オリゴdTでプライミングされたcDNAを、GeneRacerキット(Invitrogen)を用いて製造者のプロトコルに従い合成した。γ1重鎖のコード領域を、CMV2及びJNT098(図17)をプライマーとして用い、Phusionポリメラーゼ(New England Biolabs)を用いてPCRにより増幅した。PCR断片をゲル精製し、CMV2、JNT082、JNT097及びJNT098をプライマーとして用いてシークエンシングした(図17)。同様に、κ軽鎖のコード領域を、CMV2とJNT026とを用いて増幅した(図17)。ゲル精製したDNA断片を、CMV2、JNT026、JNT080及びJNT084をプライマーとして用いてシークエンシングした(図17)。HuK33Nの重鎖と軽鎖の各々のコード領域の得られたヌクレオチド配列は、pHuK33Nベクター中の対応する配列と完全に一致した(それぞれ、図18及び図19)。
【0101】
6.4.6.ChK33N抗体とHuK33N抗体の精製
NS0−ChK33N 3D11細胞及びNS0−HuK33N 8G8−11細胞をローラーボトル中、Hybridoma−SFMで枯渇するまで増殖させた。遠心分離及び濾過後、培養上清をプロテイン−Aセファロースカラム(GE Healthcare,Piscataway,NJ)にかけた。カラムをPBSで洗浄後、抗体を0.1Mのグリシン−HCl(pH3.0)で溶出させた。1MのTris−HCl(pH8)で中和後、溶出した抗体の緩衝液を透析によってPBSに変えた。抗体濃度を、280nmにおける吸光度を測定して決定した(1mg/mL=1.4OD)。抗体発現レベルは、NS0−ChK33N 3D11細胞では50μg/mL、NS0−HuK33N 8G8−11細胞では12μg/mLであった。
【0102】
精製したChK33N抗体とHuK33N抗体を、標準的な手順に従ったSDS−PAGEにより解析した。還元条件下での解析から、ChK33N抗体及びHuK33N抗体は、各々、分子量約50kDaの重鎖と分子量約25kDaの軽鎖から構成されていることが示された(図20)。各抗体の純度は95%を上回るようであった。
【0103】
6.4.7.ChK33N抗体とHuK33N抗体の特徴付け
マウスK33N抗体、キメラK33N抗体、及びヒト化K33N抗体のヒトα9インテグリンへの結合を、CHO/huα9細胞を用いたFACS結合アッセイで調べた。約5×105個のCHO/huα9細胞/試験を、FACS結合緩衝液(0.5%BSA及び0.05%NaN3を含有するPBS)で洗浄し、様々な量の試験抗体を含有する200μLのFACS結合緩衝液に懸濁した。氷上で30分置いた後、細胞をFACS結合緩衝液で洗浄した。次に、マウスK33Nで染色した細胞を、1/200希釈したFITC標識ヤギ抗マウスIgGポリクローナル抗体(SouthernBiotech)を含むFACS結合緩衝液100μLに懸濁した。キメラK33N又はヒト化K33Nで染色した細胞は、1/200希釈したFITC標識ヤギ抗ヒトIgGポリクローナル抗体(SouthernBiotech)を含むFACS結合緩衝液100μLに懸濁した。氷上に30分置いた後、細胞をFACS結合緩衝液で洗浄し、200μLのFACS結合緩衝液に懸濁し、FACScanフローサイトメータ(BD Biosciences,Franklin Lakes,NJ)を用いて解析した。マウスK33N抗体のCHO/huα9細胞への結合のEC50値は104ng/mLであった(図21)。ChK33N抗体とHuK33N抗体の間で、結合パターンは互いによく似ていた(図22)。さらに、ChK33N抗体とHuK33N抗体のEC50値は互いにほぼ同じであった(ChK33N抗体は143ng/mL、HuK33N抗体は151ng/mL)。この結果は、抗原結合親和性を失わせずに、マウスK33N抗体をヒト化することに成功したことを示している。
【0104】
6.4.8.細胞接着アッセイ
96穴平底マイクロタイタープレート(Nunc)を、50μLの5μg/mL hTNC(AEIDGIEL)−BSA溶液で、CO2インキュベーター内、37℃で1時間コーティングした。対照穴を、50μLの5μg/mL BSA溶液でコーティングした。コーティング反応の後、穴中の溶液を200μLの0.5w/v%BSA(ブロッキング)溶液と交換し、室温で1時間インキュベートした。ブロッキング反応後、穴をPBSで洗浄した。0.25w/v%BSAを含有するTIL培地に懸濁した2.5×105細胞/mLのG−361細胞150μLと、試験抗体溶液(0.25w/v%BSA/TIL)50μLとをこれらの穴に添加し、37℃のCO2インキュベーター内で1時間インキュベートした。インキュベート後、非接着細胞を除去するために、各穴をPBS溶液で3回注意深く洗浄した。50μLの0.5w/v%クリスタルバイオレット溶液を添加して、接着細胞を固定及び染色した。30分後、穴一杯の水で3回洗浄して、余分な色素を除去し、穴中の色素を50μLの20v/v%酢酸で可溶化させた。各穴の595nmにおける吸光度を、吸光度計を用いて測定した。細胞接着阻害率は、以下の式;(1−(A−B)/(C−B))×100(%)により算出した。A:抗体が存在し、hTNC−BSAでコーティングした穴の吸光度、B:いずれの試験抗体も存在しない、BSAでコーティングした穴の吸光度、C:正常ヒトIgG存在下のhTNC−BSAでコーティングした穴の吸光度(陰性対照)(図23)。
【0105】
結果
Y9A2のIC50は0.053μg/mL(95%CI:0.032〜0.089)であった。K33N、ChK33N、及びHuK33NのIC50は、それぞれ、0.075μM(0.053〜0.106)、0.090μg/mL(0.063〜0.128)、0.084μg/mL(0.055〜0.129)であり、試験抗体のIC50値がY9A2のIC50値と同じレベルにあることを示した。
【0106】
7.寄託
マウス抗ヒトα9インテグリンモノクローナル抗体を産生する、本明細書でK33Nと表されているハイブリドーマは、2007年5月29日に、微生物の寄託に関するブダペスト条約に準拠して、(郵便番号305−8566)茨城県つくば市東1−1−1中央第6にある、産業技術総合研究所特許生物寄託センターに寄託され、アクセッション番号FERM BP−10830が与えられた。これは、その全体が参照により本明細書に組み込まれる。
【0107】
8.産業上の適用可能性
本発明のヒト化モノクローナル抗体は、α9インテグリンの機能を阻害して、例えば、癌細胞の増殖又は転移等の癌、及び、例えば、関節リウマチ、変形性関節症、肝炎、気管支喘息、線維症、真性糖尿病、癌転移、動脈硬化症、多発性硬化症、肉芽腫、炎症性腸疾患(潰瘍性大腸炎及びクローン病)、自己免疫疾患等の炎症性疾患に対する治療効果を示す。
【0108】
9.配列表
本明細書の全体を通じて言及されている配列を以下にまとめる。
【表2】
【特許請求の範囲】
【請求項1】
免疫特異的にヒトα9インテグリンを認識するヒト化抗体又はその抗原結合断片であって、
(i)ヒト抗体のVH領域に由来する少なくとも1つのFRHと、免疫特異的にヒトα9インテグリンを認識する非ヒト抗体のCDRHの少なくとも1つに由来する少なくとも1つのCDRHとを含むH鎖;又は
(ii)ヒト抗体のVL領域に由来する少なくとも1つのFRLと、免疫特異的にヒトα9インテグリンを認識する非ヒト抗体のCDRLの少なくとも1つに由来する少なくとも1つのCDRLとを含むL鎖;又は
(iii)上述の(i)と(ii)の両方
を含む抗体。
【請求項2】
前記非ヒト抗体が、受託番号FERM BP−10830のハイブリドーマにより産生されるマウスモノクローナル抗体である、請求項1に記載のヒト化抗体又はその抗原結合断片。
【請求項3】
前記ヒト化抗体の少なくとも1つのCDRHが配列番号4、5及び6のアミノ酸配列からなる群から選択されるアミノ酸配列を含み、かつ、前記ヒト化抗体の少なくとも1つのCDRLが配列番号11、12及び13のアミノ酸配列からなる群から選択されるアミノ酸配列を含む、請求項1に記載のヒト化抗体又はその抗原結合断片。
【請求項4】
前記ヒト抗体のVH領域が、GenBankアクセッション番号DA980102(配列番号18)のヌクレオチド配列によってコードされるアミノ酸配列に由来するアミノ酸配列を含み、かつ、前記ヒト抗体のVL領域が、GenBankアクセッション番号X72441(配列番号23)のヌクレオチド配列によってコードされるアミノ酸配列に由来するアミノ酸配列を含む、請求項2又は3に記載のヒト化抗体又はその抗原結合断片。
【請求項5】
配列番号60のアミノ酸配列を含むH鎖、及び配列番号61のアミノ酸配列を含むL鎖を含む、免疫特異的にヒトα9インテグリンを認識するヒト化抗体又はその抗原結合断片。
【請求項6】
配列番号60のアミノ酸配列をコードするヌクレオチド配列を含む、単離された核酸分子。
【請求項7】
前記ヌクレオチド配列が配列番号62のヌクレオチド配列を有する、請求項6に記載の核酸分子。
【請求項8】
シグナルペプチドをコードするヌクレオチド配列をさらに含む、請求項6に記載の核酸分子。
【請求項9】
前記シグナルペプチドが配列番号58のアミノ酸配列を含む、請求項8に記載の核酸分子。
【請求項10】
配列番号61のアミノ酸配列をコードするヌクレオチド配列を含む、単離された核酸分子。
【請求項11】
前記ヌクレオチド配列が配列番号63のヌクレオチド配列を有する、請求項10に記載の核酸分子。
【請求項12】
シグナルペプチドをコードするヌクレオチド配列をさらに含む、請求項10に記載の核酸分子。
【請求項13】
前記シグナルペプチドが配列番号59のアミノ酸配列を含む、請求項12に記載の核酸分子。
【請求項14】
請求項6に記載の核酸分子、又は請求項10に記載の核酸分子、あるいはその両方を含むベクターであって、前記核酸分子が1以上の調節エレメントに機能的に連結されている、ベクター。
【請求項15】
請求項14に記載のベクターを含む、単離された宿主細胞。
【請求項16】
ヒト化抗体又はその抗原結合断片を製造する方法であって、前記ヒト化抗体又はその抗原結合断片が発現されるような条件下で請求項15に記載の宿主細胞を培養することと、発現されたヒト化抗体を回収することとを含む方法。
【請求項17】
請求項1、2又は5のいずれか一項に記載のヒト化抗体又はその抗原結合断片、及び薬学的に許容される担体を含む医薬組成物。
【請求項18】
α9インテグリンと関連する障害又は疾患を予防又は治療する方法であって、有効量の請求項5に記載のヒト化抗体又はその抗原結合断片をそれを必要とする対象に投与することを含む方法。
【請求項19】
α9インテグリンと関連する障害又は疾患をインビボで診断する方法であって、有効量の請求項1又は2に記載のヒト化抗体又はその抗原結合断片を検査対象に投与することを含む方法。
【請求項1】
免疫特異的にヒトα9インテグリンを認識するヒト化抗体又はその抗原結合断片であって、
(i)ヒト抗体のVH領域に由来する少なくとも1つのFRHと、免疫特異的にヒトα9インテグリンを認識する非ヒト抗体のCDRHの少なくとも1つに由来する少なくとも1つのCDRHとを含むH鎖;又は
(ii)ヒト抗体のVL領域に由来する少なくとも1つのFRLと、免疫特異的にヒトα9インテグリンを認識する非ヒト抗体のCDRLの少なくとも1つに由来する少なくとも1つのCDRLとを含むL鎖;又は
(iii)上述の(i)と(ii)の両方
を含む抗体。
【請求項2】
前記非ヒト抗体が、受託番号FERM BP−10830のハイブリドーマにより産生されるマウスモノクローナル抗体である、請求項1に記載のヒト化抗体又はその抗原結合断片。
【請求項3】
前記ヒト化抗体の少なくとも1つのCDRHが配列番号4、5及び6のアミノ酸配列からなる群から選択されるアミノ酸配列を含み、かつ、前記ヒト化抗体の少なくとも1つのCDRLが配列番号11、12及び13のアミノ酸配列からなる群から選択されるアミノ酸配列を含む、請求項1に記載のヒト化抗体又はその抗原結合断片。
【請求項4】
前記ヒト抗体のVH領域が、GenBankアクセッション番号DA980102(配列番号18)のヌクレオチド配列によってコードされるアミノ酸配列に由来するアミノ酸配列を含み、かつ、前記ヒト抗体のVL領域が、GenBankアクセッション番号X72441(配列番号23)のヌクレオチド配列によってコードされるアミノ酸配列に由来するアミノ酸配列を含む、請求項2又は3に記載のヒト化抗体又はその抗原結合断片。
【請求項5】
配列番号60のアミノ酸配列を含むH鎖、及び配列番号61のアミノ酸配列を含むL鎖を含む、免疫特異的にヒトα9インテグリンを認識するヒト化抗体又はその抗原結合断片。
【請求項6】
配列番号60のアミノ酸配列をコードするヌクレオチド配列を含む、単離された核酸分子。
【請求項7】
前記ヌクレオチド配列が配列番号62のヌクレオチド配列を有する、請求項6に記載の核酸分子。
【請求項8】
シグナルペプチドをコードするヌクレオチド配列をさらに含む、請求項6に記載の核酸分子。
【請求項9】
前記シグナルペプチドが配列番号58のアミノ酸配列を含む、請求項8に記載の核酸分子。
【請求項10】
配列番号61のアミノ酸配列をコードするヌクレオチド配列を含む、単離された核酸分子。
【請求項11】
前記ヌクレオチド配列が配列番号63のヌクレオチド配列を有する、請求項10に記載の核酸分子。
【請求項12】
シグナルペプチドをコードするヌクレオチド配列をさらに含む、請求項10に記載の核酸分子。
【請求項13】
前記シグナルペプチドが配列番号59のアミノ酸配列を含む、請求項12に記載の核酸分子。
【請求項14】
請求項6に記載の核酸分子、又は請求項10に記載の核酸分子、あるいはその両方を含むベクターであって、前記核酸分子が1以上の調節エレメントに機能的に連結されている、ベクター。
【請求項15】
請求項14に記載のベクターを含む、単離された宿主細胞。
【請求項16】
ヒト化抗体又はその抗原結合断片を製造する方法であって、前記ヒト化抗体又はその抗原結合断片が発現されるような条件下で請求項15に記載の宿主細胞を培養することと、発現されたヒト化抗体を回収することとを含む方法。
【請求項17】
請求項1、2又は5のいずれか一項に記載のヒト化抗体又はその抗原結合断片、及び薬学的に許容される担体を含む医薬組成物。
【請求項18】
α9インテグリンと関連する障害又は疾患を予防又は治療する方法であって、有効量の請求項5に記載のヒト化抗体又はその抗原結合断片をそれを必要とする対象に投与することを含む方法。
【請求項19】
α9インテグリンと関連する障害又は疾患をインビボで診断する方法であって、有効量の請求項1又は2に記載のヒト化抗体又はその抗原結合断片を検査対象に投与することを含む方法。
【図1】

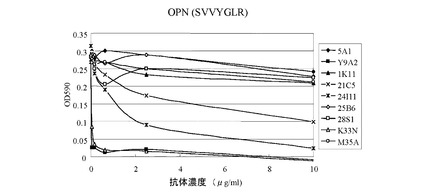
【図2】
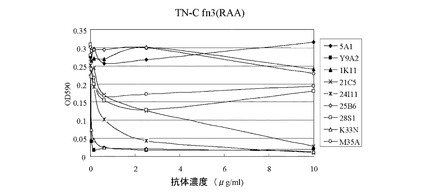
【図3】
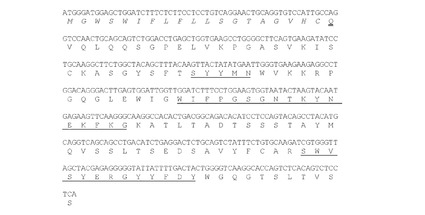
【図4】

【図5】
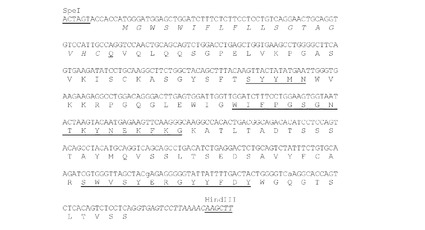
【図6】
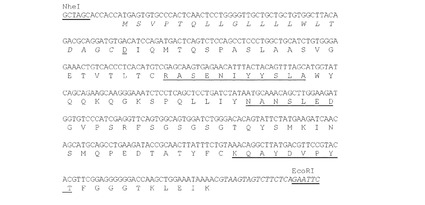
【図7】
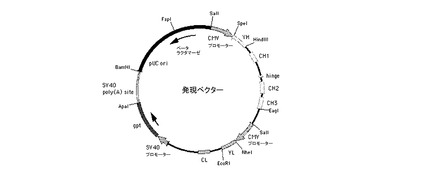
【図8】

【図9】

【図10】

【図11】

【図12】

【図13】
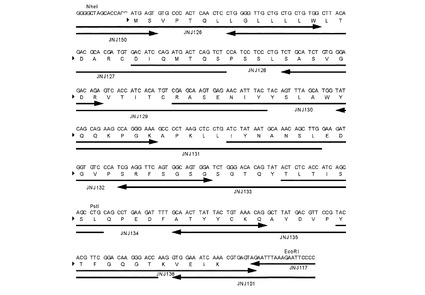
【図14】
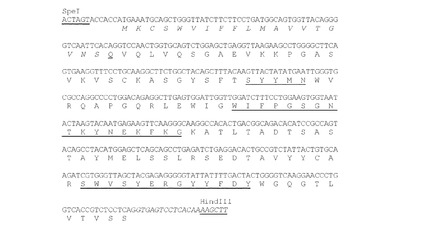
【図15】
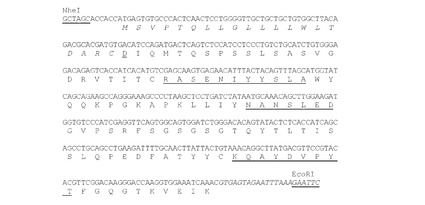
【図16】
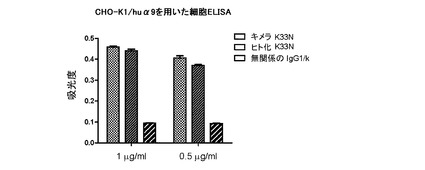
【図17】

【図18】

【図19】

【図21】
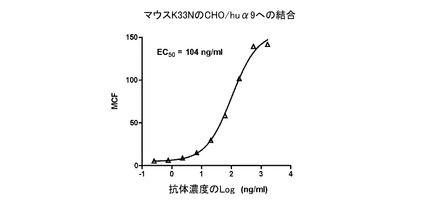
【図22】
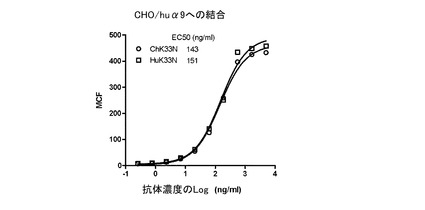
【図23】
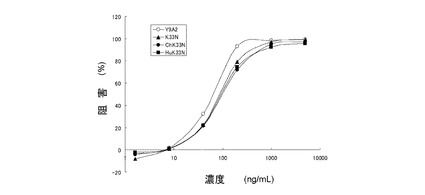
【図20】
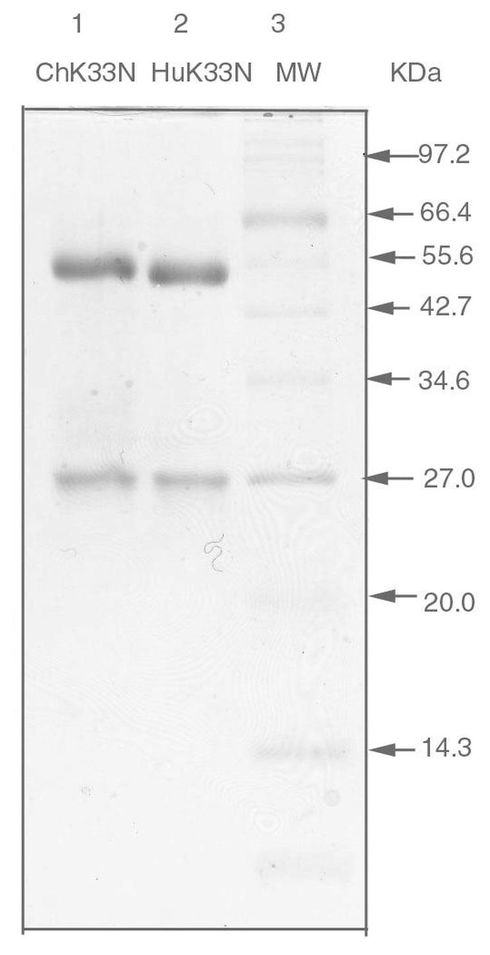
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図21】
【図22】
【図23】
【図20】
【公表番号】特表2012−526518(P2012−526518A)
【公表日】平成24年11月1日(2012.11.1)
【国際特許分類】
【出願番号】特願2011−533480(P2011−533480)
【出願日】平成22年3月10日(2010.3.10)
【国際出願番号】PCT/JP2010/054483
【国際公開番号】WO2010/104208
【国際公開日】平成22年9月16日(2010.9.16)
【出願人】(501416243)株式会社ジーンテクノサイエンス (9)
【出願人】(000124269)科研製薬株式会社 (18)
【Fターム(参考)】
【公表日】平成24年11月1日(2012.11.1)
【国際特許分類】
【出願日】平成22年3月10日(2010.3.10)
【国際出願番号】PCT/JP2010/054483
【国際公開番号】WO2010/104208
【国際公開日】平成22年9月16日(2010.9.16)
【出願人】(501416243)株式会社ジーンテクノサイエンス (9)
【出願人】(000124269)科研製薬株式会社 (18)
【Fターム(参考)】
[ Back to top ]