
ヒト幹細胞含有組成物の培養方法及び形質転換方法
【解決手段】ヒト造血系由来幹細胞を含有する細胞組成物を、約24〜約48時間の間に1mLの培養あたり1mLの培地で連続的又は周期的に交換される液体培養培地内で培養し、該培養を生理学上許容しうる条件下に保持しながら代謝産生物を除去し、消費した栄養素を補給することを特徴とするエクスビボでヒト造血系由来幹細胞分裂物を得るための培養方法。
【効果】ヒト幹細胞及び/又はヒト造血前駆細胞及び/又はヒト間質細胞を、連続的又は周期的に交替好ましくは潅流される液体培養培地中で培養し、培養を生理学的に許容可能な条件下で保持しながら栄養素を補給することにより、エクスビボでヒト幹細胞が効率良く得られる。
【効果】ヒト幹細胞及び/又はヒト造血前駆細胞及び/又はヒト間質細胞を、連続的又は周期的に交替好ましくは潅流される液体培養培地中で培養し、培養を生理学的に許容可能な条件下で保持しながら栄養素を補給することにより、エクスビボでヒト幹細胞が効率良く得られる。
【発明の詳細な説明】
【技術分野】
【0001】
この発明は培養動物細胞の増殖及び形質転換の為の、特に培養造血細胞の増殖及び形質転換の為の方法、反応器及び組成物に関するものである。
【背景技術】
【0002】
正常成人の循環血液細胞は赤血球、白血球、血小板及びリンパ球を含むがこれらの細胞は骨髄内の細胞に由来する。骨髄内の細胞は非常に未分化な前駆細胞と呼ばれる細胞に由来する。前駆細胞はメチルセルロースや寒天のような半固体の培地中で1〜3週間培養すると分化した血液細胞が集まったコロニーを生ずることより検定出来る。前駆細胞それ自体も一種の前駆細胞である幹細胞と呼ばれる細胞に由来している。幹細胞は分裂時に幹細胞自身の再生と前駆細胞への分化の両方を行なう能力を有する。それ故分裂する幹細胞は未分化の幹細胞といくぶん分化の進んだ前駆細胞を生産する。血液細胞を生産する他に幹細胞は骨芽細胞と破骨細胞をも生産すると考えられ、多分他組織の細胞も幹細胞から派生するであろう。この明細書はヒト造血幹細胞の最初のインビトロ培養を成功させた方法及び組成物を記述する。この方法により造血幹細胞は増殖し、前駆細胞及びより成熟した血液細胞に分化した。
【0003】
1970年代の後半、造血骨髄細胞をインビトロで増殖させる液体培養系が発達した。この培養系は、正常及び白血病の造血の分析及びレトロウイルス媒介による遺伝子導入の様な骨髄の実験的操作に非常に重要な価値を持つ可能性がある。この培養系を用いてマウスの造血の詳細な分析が可能になりマウス系の細部が理解出来る様になった。更に培養マウス骨髄細胞へのレトロウイルス遺伝子導入を可能にした。このことはマウス造血細胞に標識を付けることを可能にし、多機能幹細胞の存在を証明し、白血病発生の過程に於ける種々の遺伝子の研究を可能にした。
【0004】
しかしレトロウイルス遺伝子を培養マウス骨髄細胞に導入することは可能になったが、培養ヒト骨髄細胞に遺伝子導入することは未だ不可能である。なぜなら現在に至るまでヒト骨髄細胞の長期培養は、その培養期間を長く出来ない事と、幹細胞を生存させておき前駆細胞を生産する能力を維持する事が出来ないという制限があるからである。
【0005】
ヒト骨髄細胞の液体培養は最初は造血能力が制限されており、生産される前駆細胞や分化した血液細胞数は減少し、6週から8週の培養で細胞生産は停止した。その後この系に種々の修正がなされたが僅かな改善が見られただけであった。この問題を解決することは非常に価値のあることである。もし成功すればヒト幹細胞及び前駆細胞を増加させることが出来、骨髄移植、化学療法からの保護に使用し、そのような細胞を選択、操作して遺伝子導入に使い、また分化した血液細胞を生産し、輸血療法に用いることも可能になる。
造血とインビトロ骨髄液体培養の研究は付着層内の線維芽細胞と内皮細胞が中心細胞基質の要素であることを明らかにした。これらの細胞は増殖している造血細胞に付着する場を与えると共に前駆細胞の増殖と分化を促進する造血細胞増殖因子を誘導することが出来る。これらの造血細胞増殖因子は、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)及びインターロイキン−6(IL−6)等を含む。
【0006】
しかしそのような付着層上でのヒト骨髄細胞インビトロ培養はたいていはうまく行かなかった。マウスや木ネズミ(tree shrew)の骨髄細胞は液体培養出来るがヒト液体骨髄培養は非付着性の造血前駆細胞或いはクローンを形成する前駆細胞を6〜8週間以上十分な量を生産しない。3〜5カ月保持する培養が報告されたが、幹細胞から前駆細胞を安定して4〜6週以上生産し続ける細胞培養は報告されていない。
【0007】
更に、非付着性細胞及び前駆細胞の生産はこれらの培養の短い生存期間中に典型的に低下するので、幹細胞が生きのびているのか増殖しているのかはっきりしない。分離して調べて見ると刺激を受けない骨髄基質細胞は、検出し得る諸造血増殖因子(HGFs)を殆ど分泌しない。
これらの培養中に安定した前駆細胞及び分化した血液細胞の生産が見られないことから、これらの培養は幹細胞の連続的再生、拡大を支持出来ないものと考えられた。それゆえこれらの培養はある必須の幹細胞刺激因子を欠いているか及び/又は新しい幹細胞阻害因子を含んでいるものと仮定された。HGFを検出できない事と基質細胞が誘導されていない事の説明はなされたが、HGFの検出できない事とヒト骨髄液体培養が他の種の動物のそれと比較して成功しない事を説明する、検証され得る仮説は、インビトロの培養系は付着性骨髄基質細胞がインビボで持つ総合造血支援機能を提供しないと言うものである。
【0008】
骨髄移植のための幹細胞と前駆細胞の増殖はヒト長期骨髄培養に応用出来る可能性がある。ヒトの自己及び同種骨髄移植は現在、白血病、リンパ腫或いは他の生命を脅かす病気の治療法として行なわれている。この治療法に於いて十分な細胞を移植するためには、供与者の骨髄を大量に採集しなければならない。
幹細胞と前駆細胞を増殖させる培養は大量の骨髄供与の必要性を減少させ、小量の骨髄の供与を受け、受血者に輸血する前にインビトロで幹細胞と前駆細胞を増殖させることを可能にする。小量の幹細胞及び前駆細胞が血流中に存在することが知られている。もしこれらの幹細胞と前駆細胞を泳動法により採集し、増殖させる事が可能であれば、移植に必要な数の幹細胞と前駆細胞を末梢血から採集し、骨髄供与の必要性を除くことができる。
【0009】
骨髄移植には患者の体重1kg当り約1×108から2×108の骨髄単核細胞を移植しなければならない。これは体重70kgの受血者は70mlの骨髄供与を必要とすることになる。70mlの骨髄は供与者の骨髄のほんの一部ではあるが、供与過程で供与者に強度の処置を必要とし、多量の血液が失われる。もし幹細胞と前駆細胞を10倍に増やすことが出来れば、供与過程は大巾に軽減し、末梢血から幹細胞と前駆細胞を採集しこれを増殖させて用いることが可能になるかも知れない。
【0010】
前駆細胞の増殖が可能になれば、化学療法の補助として有用であろう。これもまたヒト骨髄長期培養の応用のひとつである。癌治療医のジレンマは殆どの化学療法剤は細胞分裂している全ての細胞を殺すことにより癌細胞を破壊すると言うことである。骨髄は体内で最も増殖の盛んな組織のひとつで、それゆえ最初に化学療法剤で損傷をうける器官であることが多い。その結果血液細胞生産能は化学療法を行なっている期間急速に破壊されるので、化学療法を中止し造血系が血液細胞を回復するのを待たなければ、患者に化学療法を再び行なう事は出来ない。静止幹細胞が活性化して化学療法を再開できるに十分な白血球数に回復するまでは1カ月かそれ以上かかるかも知れない。そして2回目の化学療法中も血球数の降下は繰り返される。化学療法の間隙期間に血球が再生されるが、不幸にして癌細胞もこの期間に増殖し自然淘汰されて化学療法剤に対して耐性を持つようになる可能性がある。
【0011】
化学療法間の間隙時間を短くするために大量の前駆細胞と未分化の血液細胞を患者に戻すことが出来る。この方法により患者の血球数が低くなる時間を大巾に短縮でき、化学療法の再開を早めることが出来る。化学療法の期間が長いほど、また化学療法間の間隙時間が短いほど癌細胞を殺すことに成功する可能性は高くなる。
【0012】
前駆細胞を増殖させるに必要な造血細胞は骨髄回収か或いは末梢血採集に依って得られる。骨髄採集により約4×105CFU−GM前駆細胞を得ることが出来る。5リットルの末梢血から泳動により105CFU−GMが得られるが、供与者をGM−CSFで前処理することにより106CFU−GMに増やすことが可能である。患者の急速な回復には1×108から5×108CFU−GMが必要であるが、これは通常の骨髄供与或いは末梢血供与で得られる量の100倍から1000倍である。それ故、骨髄或いは末梢血のCFU−GMを100倍から1000倍増やすことが出来れば、抗癌剤投与、癌の治療に重要な影響を与えるであろう。
【0013】
遺伝子療法は医学の中で急速に進歩している分野であり、計り知れない臨床応用の可能性を持っている。遺伝子療法は病気の治療に数多くの応用の可能性が考えられ、それに関して広範囲に論評されている。例として下記の参照文献を挙げられる:
Boggs, Int. J. Cell Cloning. (1990) 8:80-96、Kohn et al, Cancer Invest. (1989) 7 (2):179-192、Lehn, Bone Marrow Transp. (1990) 5:287-293、及び Verma, Scientific Amer. (1990) pp. 68-84。
遺伝的形質転換したヒト幹細胞は遺伝子療法の手段として臨床医学に広く応用出来る可能性がある。
【0014】
遺伝子療法は従来の医療から発展してきた臨床療法に新しい方法を加えた。何世紀もかけて進歩してきた最古の医療は粗放な外科処置と天然混合物を医薬として投与することであった。前世紀になって生化学的薬理学が医療の主たる方法として現われた。この範例の下では純粋な生化学的分子が患者に与えられる。一般的にそのような薬剤は毒薬(例えば抗生物質或いは抗癌剤など)、内在の受容体を刺激する生理的模倣剤(例えば阿片やアドレナリン拮抗剤)或いは内在受容体を刺激する生理的拮抗薬(例えば血圧降下剤や麻酔剤)である。
【0015】
遺伝子療法はその定義によれば医療目的で細胞内に遺伝子を挿入することである。遺伝子療法の背景にある原則は薬理学的物質を投与するよりもむしろ、機能する遺伝子を導入して、そのRNA或いは蛋白生産物が目的細胞或いは組織に希望する生化学的効果をもたらすことである。遺伝子療法が古典的な生化学的薬理学に較べて有利である可能性のあるのは次の理由による。第一に、導入された遺伝子は蛋白やRNAを含めて非常に複雑な分子を造ることが出来る。これらの分子自体を投与して目標に到達させるのは非常に困難か不可能である。次に、希望する遺伝子を特定の目標細胞に制御導入すれば特定の組織でその遺伝子生成物の生産を制御出来る。最後に遺伝子療法は、遺伝子が目標細胞とその子孫細胞内で機能し続けるので原則として個体内では永久的である。
【0016】
遺伝子治療を成功に導く為には幾つかの問題を解決しなければならない。第一に、希望する治療遺伝子を選択した細胞に導入することが出来なければならない。第二に、その遺伝子は目標細胞内で十分に発現し、遺伝子の生成物が適当なレベルに達しなくてはならない。最後にRNA或いは蛋白生成物は目標細胞内で適当に処理されて機能を持ち、臨床治療としての意味を持たなければならない。ヒト細胞に遺伝子をインビトロで導入する数種の方法を表1に示してある。
【0017】
【表1】

【0018】
相同組換えの様な他の方法も多くの研究室で開発された。遺伝子療法の研究はここ数年来進行中でインビトロでの数種の細胞タイプに於ける実験から、動物実験に進み最近ヒトの臨床試験の段階に入ったものもある。
【0019】
造血系は遺伝子療法の導入系としては理想的な対象である。造血細胞は骨髄吸引或いは末梢血を採集することにより容易に得ることが出来る。インビトロでの遺伝子導入が成功すれば、処理された細胞は静脈より再注入され骨髄に到達し、そこで増殖する。分化した血液細胞は体内を循環しているので遺伝子を改変された細胞は特定の遺伝子生成物を希望するどの組織にも導入することが出来る。
【0020】
最も重要な事は造血組織は非常に強い(多分無限の)自己再生能力を持つ幹細胞を含んでいる事である。この事はもし遺伝子がこれらの幹細胞に安定して導入されれば造血組織に再注入した際これらの変化した幹細胞は増殖し、骨髄を新しい遺伝子を表現している細胞で再び満たすことが出来る事を意味する。これにより長期間の、多分一生の間の希望する遺伝子生成物の供給を可能にするであろう。同様にして他の組織に存在する幹細胞や胚幹細胞に遺伝子を導入する事に成功すれば同様に長期にわたる、希望する遺伝子生成物の供給が可能になる。
【0021】
造血幹細胞の遺伝子療法は造血系に特有の病気にも他の器官系の病気にも広い応用が可能である。造血系の病気は遺伝性であっても、後天的であっても遺伝子療法での治療が可能である。例えばアルファ及びベータ・タラセミアのようなヘモグロビン異常はグロビンのアルファ又はベータ鎖をコードする遺伝子と高度の組織特異性を与える制御配列を導入する事により治療する事が出来る。(Grossveld et al, Cell (1987) 51:975-986参照)。同様に鎌型赤血球性貧血は胎児グロビン遺伝子を造血幹細胞に導入する事により治すことが出来る。なぜなら制御された胎児ヘモグロビンの高度の発現は鎌状ヘモグロビンの存在にも拘らず赤血球の鎌型化を防ぐに十分であるからである。(Sunshine et al, J. Molec. Biol. (1979) 133:435 参照)。
【0022】
白血球付着不全(LAD)或いは慢性肉芽腫病(CGD)のような、機能的蛋白不全によって引き起こされる好中球の遺伝子病は欠陥遺伝子や欠損遺伝子を高度の組織特異性を与える制御配列と共に造血幹細胞に導入する事により治療する事が出来る(Wilson et al, Science (1990) 248:1413-1416 参照)。フォンウィレンブランツ(von Willebrands')病のような血小板に関する遺伝病はウィレンブランツ(Willebrands)因子をコードする遺伝子とその遺伝子を発現させ、その生成物を分泌させる配列とを導入することにより治す事が出来る。
【0023】
アデノシン・デアミナーゼの欠損による重度複合免疫不全のようなリンパ球免疫不全病を造血幹細胞遺伝子療法で治療するのが特に適していることは明らかである。ADA遺伝子を用いて循環T細胞をレトロウイルス遺伝子療法で治療する方法は、この病気の患者の臨床上の免疫不全経験を減少させるのに成功したが、遺伝子を導入したTリンパ球は生体内では限られた寿命しか無いため効果は一時的なものであった(Kasid et al, Proc. Nat. Acad. Sci. (USA)(1990) 87:473-477、或いはCulver et al, Proc. Nat. Acad. Sci. (USA), (1991) 88:3155-3195 参照)。しかしながら、もし遺伝子を造血幹細胞に導入出来れば、これらの幹細胞から派生するTリンパ球はADA遺伝子を含み発現することであろう。それ故遺伝子導入した幹細胞は患者の生涯生存し増殖を続けるであろうから、T細胞ADA不全は幹細胞の一回の遺伝子療法で永久的に治療出来る(Wilson et al, Proc. Nat. Acad. Sci. (USA), (1990) 87:439-443 参照)。
【0024】
造血素の遺伝的な酵素異常の治療に加えて、幹細胞遺伝子療法は幹細胞とその子孫をウイルスや化学療法剤のような外来作用物から保護するにも有用であるかも知れない。例えばHIVのTATトランス活性化因子のTAR結合位置をコードするDNA配列を導入するとT細胞をHIVウイルスの感染から保護することが示された(Sullenger et al, Cell (1990) 63:601-608 参照)。この配列を造血幹細胞に導入し安定させればこれらの幹細胞から派生したHIVの伝染に比較的あるいは絶対的に抵抗を持つT細胞のプールが出来る。
同様にして多薬剤耐性遺伝子或いはメトトレキセート耐性遺伝子をコードする遺伝子をヒト骨髄幹細胞に導入するのに成功すると癌の化学療法の影響に比較的耐性のある幹細胞を造り出す事が出来る。これらの遺伝子操作された細胞で自己骨髄移植を行なった後は患者は抗癌剤に依って普通引き起こされる深刻な骨髄抑制から幹細胞が保護される事より抗癌剤による化学療法に耐えることが出来る。これにより患者は癌化学療法剤のより効果的な使用量を、より少ない毒性で受ける事が出来る。
【0025】
造血幹細胞遺伝子療法が白血病、リンパ腫及び再生不良性貧血等の後天的造血病に対して有用であることを予想する事はたやすい。一度これらの病気の遺伝的原因が発見されたなら、その遺伝子生成物を投与し細胞内の異常遺伝子の生成物を克服するか、或いは直接遺伝子異常を修正する(多分遺伝子を切り外し、置換することにより)様な遺伝子を導入することにより異常を修正出来るであろう。 しかし更に広いレベルでは造血幹細胞遺伝子療法は造血系以外の病気の治療にも有用である可能性がある。治療効果のある可溶性蛋白をコードするDNA配列の遺伝子導入をすれば、希望する量の、治療効果のある分子を恒久的に分泌する分化した血液細胞を生産することが出来る。例としてはこの方法は、例えばインスリン遺伝子を持つDNA配列とインスリン遺伝子の発現を適切に調節をする(多分血中グルコースのレベルの上昇に反応して)調節遺伝子配列を導入して糖尿病の治療に当てるのに有用である。全身性の高血圧は幹細胞にアンギオテンシン変換酵素、血管平滑筋のカルシウム・チャンネル或いはアドレナリン受容体等の拮抗的阻害剤として機能する分泌ペプチドをコードするDNA配列を導入することにより治療が可能である。アルツハイマー病は中枢神経系内のアミロイドのプラクを破壊する酵素をコードするDNA配列を幹細胞に導入する事により治療可能であるかも知れない。
【0026】
遺伝子療法の多くの応用、特に幹細胞への遺伝子導入はよく知られており、広範に論評されている(Boggs et al, 上記、Kohn et al, 上記、Lehn, 上記、及びVerma et al,上記)。分化したヒト幹細胞への治療的遺伝子導入(例えばTリンパ球への)のある程度の成功例が増加している(Kalsd et al, Proc. Nat. Acad. Sci. (USA) (1990) 87:473-477、Culver et al, Proc. Nat. Acad. Sci. (USA) (1991) 88:3155-3159 参照)。
【0027】
不幸にしてこの発明の以前にはヒト幹細胞への遺伝子導入をして安定化する事に成功した例は無い。幾つかの研究グループがヒト造血細胞へ、ヒトのレトロウイルスを使って遺伝子導入が可能であることを示したが、ヒト未分化造血幹細胞への導入に成功した例は無い。これはレトロウイルス媒介による造血細胞への遺伝子導入がある程度可能であるマウスに於ける実験と対照的である(Wilson et al, Pro. Nat. Acad. Sci. (USA) (1990) 87:439-443 参照)。
【0028】
ヒトの造血幹細胞遺伝子療法が成功しなかった主たる原因は幹細胞が分裂、増殖している状態でヒト造血細胞に遺伝子を導入出来なかった為であった。目標細胞に遺伝子を安定導入するのに成功するにはその目標細胞が少なくとも1回細胞分裂をしなければならない。それ故、導入したい遺伝子の存在下でも幹細胞が分裂していなければ、遺伝子は幹細胞に安定に導入されない。本発明以前は、体外でのヒト幹細胞の分裂と増殖を支持する系は存在せず、ヒト幹細胞の遺伝的形質転換も可能ではなかった。
【0029】
本発明に関連する文献を以下に記述する。米国特許No.4,721,096は造血細胞の増殖用の、基質細胞を含む三次元系を記述している。その特許の中に言及してある文献も参照のこと。グランビル等はマウスのメタロチオネイン−I遺伝子について記述した(Glanville et al, Nature (1981) 292:267-269)。ウオングらはヒトGM−CSFについて記述し(Wong et al, Science (1985) 228:810-815)、レミシュカ等はレトロウイルス媒介の遺伝子導入を造血幹細胞のマーカーとして利用し、これらの幹細胞が生体に移植された後どうなるかを記述している(Lemischka et al, Cell (1986) 45:917-927)。ヤン等はヒトIL−3について(Yang et al, Cell (1986) 47:3-10)、チェン等はプラスミドDNAによる哺乳動物細胞の形質転換について(Chen et al, Mol. Cell. Biol.(1987)7:2745-2752)、グリーヴェス等はヒトCD2遺伝子について(Greaves et al, Cell (1989) 56:975-986)、シヴィン等はCD34抗原について(Civin et al, J. Immunol. (1984) 133:1576)、マーチン等はヒトS−CSFについて(Martin et al, Cell (1990) 63:203-211)、フォレスター等はパラレル・フロー・チャンバーについて(Forrester et al, J. Cell Science (1984), 70:93-110)、またクロンベル等は静止培養に於いてCML細胞が失われる事を(Coulombel et al, J.Clin. Invest.(1986)75:961)それぞれ記述している。
【0030】
それ故ヒト幹細胞の生体外増殖及び安定遺伝的形質転換のための方法及び組成物、並びにヒト造血前駆細胞培養の最適条件を見出す必要がある。特に幹細胞増殖、前駆細胞増殖、また遺伝子療法の重要性を考えた場合、その必要性は更に重大である。不幸にして、今日までそのような目的を達成しようとする試みは期待外れであった。
【発明の開示】
【発明が解決しようとする課題】
【0031】
本発明の目的はヒト幹細胞の生体外増殖と安定遺伝子形質転換にのための、培地の条件、反応器等を包含する新規の方法を提供することである。
本発明の他の目的は(i)ヒト造血前駆細胞培養及び(ii)ヒト造血前駆細胞の安定遺伝子形質転換の最適条件の為の培地条件及び反応器を包含する新規の方法を提供することである。
【課題を解決するための手段】
【0032】
本発明はヒト幹細胞の生体外分裂と安定遺伝子形質転換に関する、及び/又は、ヒト造血前駆細胞培養の最適化に関する、培地の条件、反応器等を包含する新規の方法を本発明者が発見した事に基づいている。これらの方法はヒト幹細胞及び/又はヒト造血前駆細胞を液体培地中で培養する方法である。この方法に於いて、液体培地を24時間から48時間の間に1mlの培養を1mlの培地で連続的に或いは定期的に(好ましくは灌流により)置換し、代謝生成物を除去し、減少した栄養素を補い、培養を生理的に適当な状態に維持する。本発明の特に好ましい実施態様に於いて、上記の培地置換速度は急速に交換される培地に造血増殖因子を添加する条件に関連して決定される。
【0033】
本発明者らは急速に交換される培地に最適量の造血増殖因子を添加し、本発明に従って培地交換速度を増加した場合、驚くべきことに(1)培養中でヒト幹細胞が少なくとも5カ月間の長期間増殖を続け、(2)培養中でヒト造血前駆細胞がヒト幹細胞の分裂と分化により少なくとも5カ月間の長期間生産され、そして(3)ヒト骨髄基質細胞を包含するヒト基質細胞の代謝を活発にし、GM−CSF分泌を活性化する。本発明は培養に於いてヒト幹細胞の生存と増殖を初めて可能にした。
【0034】
本発明はまた、ヒト幹細胞の連続的増殖を支持する体外培養系を提供し、ヒト幹細胞に遺伝子を導入することを可能にし、遺伝的に安定な形質転換したヒト幹細胞を得る事に成功した。この発明の実施態様は組換えレトロウイルス、或いは細胞分裂を必要とする他の遺伝子導入ベクターに組み込ませる事の可能などのような遺伝子の導入にも用いることが出来る。この方法によって製造される遺伝的に変化したヒト幹細胞は、先に述べた様に臨床的な病気に広範に適用する事が可能である。
本発明はまた、ヒト造血前駆細胞への遺伝子導入の効率を高める方法を提供すると同時に遺伝的に安定な形質転換したヒト幹細胞及び/又は遺伝的に安定な形質転換したヒト造血前駆細胞を提供する。
【0035】
本発明は培養中の造血細胞、特に幹細胞を包含する分化の初期段階の細胞の効率的な増殖をさせるための反応器及び組成物を提供する。本方法は通常の方法で形質転換した基質細胞を用いる。その基質細胞は成長因子の本質的な或いは誘導的な生成を提供し、その細胞は物理的に分離していて造血細胞の分離を容易にする。連続的灌流を行い適宜細胞のリサイクルを行なう事により、生存し得る造血細胞を高密度、高収量で得る事が出来る。本反応器は基質細胞に蛋白性の表面を用い、基質細胞と造血細胞の分離を維持する為に表面或いは他の障壁を用いる。
【発明を実施するための最良の形態】
【0036】
本発明の長所は本発明がヒト幹細胞及び/又は前駆細胞液体培養(造血組織培養に用いられる様な)用の標準系に応用された場合いつでも認められる。本発明の方法に従い、補足的に造血増殖因子を最適濃度に培養に添加し、急速培地交換速度を用いた場合、本発明者は、驚いた事に、ヒト造血液体培養の標準系を作る事が出来る事を発見した。その培養は馬、子牛、牛胎児より得られた血清、血漿又はヒト血清の存在下又は非存在下で実施される培養を含み、質的に優れた操作で実施できる。
【0037】
本発明の方法に従って用いられるヒト造血液体培養は1ml当り104から109細胞の細胞密度で、血清アルブミン、コレステロール及び/又はレシチン、セレニウム及び無機塩と共に用いる事の出来る既知の標準培地組成(例えばIMDM, MEM, DMEM, RPMI 1640, Alpha Medium 或いはMcCoy's Medium)を用いて行なう事が出来る。知られている様に、これらの培養は10-4から10-7Mの濃度のハイドロコーチゾンの様なコルチコステロイドを補っても良いし、コーチゾン、デキサメタゾン或いはソルメドロールの様な他のコルチコステロイドを同じ効果を持つ濃度で補っても良い。これらの培養は普通はほぼ生理的なpH,例えば6.9 から7.4で行なわれる。培地は普通4から20容積%の酸素、好ましくは6から8容積%の酸素、を含む気相に曝される。
【0038】
これらの標準培養技術を用いて細胞塊は希望する量に、例えば含まれる幹細胞或いは造血前駆細胞の量を1000倍又はそれ以上に増やす事が出来る。負選択法或いは陽選択法に相当する、異なった既知の方法をこの増大を達成する為に用いる事が出来る。例えば負選択法に相当する例としては分化した細胞は免疫学的技術を用いて除去される。その免疫学的技術とは例えば非前駆、非幹細胞を一群のマウス抗ヒトモノクローナル抗体で標識し、マウス抗体で覆われた細胞をウサギ抗マウスIgでコートしたプラスチックに付着させて除去する技術である(Emerson et al, J. Clin. Invest. (1985) 76:1286-1290)。
【0039】
本発明は上記のすべての条件の下でヒト骨髄液体培養条件の根本的な改良、つまり急速栄養培地交換、に依存するものである。標準培養計画は毎週培地と血清の交換を必要とする。これは1週間に一度培地と血清を交換するか或いは1週間に二度培地と血清を半分ずつ交換することによってなされる。本発明によれば培養の栄養培地は連続的に或いは定期的に、好ましくは灌流法により、1ml当り2×106から1×107の細胞密度の培養に対し24時間から48時間の間に置換される。細胞密度が1ml当り1×104から2×106の場合は同じ培地交換速度が用いられる。細胞密度が1ml当り107以上の場合には培地交換速度は単位時間、細胞当りの培地と血清の流量が一定になるように比例して増加させることが出来る。
【0040】
本発明による栄養培地の交換は、培地を交換する結果を達成できればいかなる方法を用いることも可能である。例えば一定量の消費された培地を除き、一定量の新鮮な培地を加える方法などである。加えられる一定量の培地の流れは重力、ポンプその他の適当な手段による。培養の配列構成と包装に依存して培地はいずれの一方向にも或いは多方向にも流す事が可能である。好ましくは新しい培地は細胞塊に接触する様に加えられるべきである。生体内の灌流を模倣する方法で培養に加えられるのが最も望ましい。即ち少なくとも細胞塊の一部を灌流し、そして全細胞塊まで灌流する方法である。
【0041】
他の任意のしかし重要な本発明の実施態様は合成造血増殖因子を含む、造血増殖因子を急速培地交換培養に添加する事にある。この実施態様の特に好ましい態様は、サイトカインIL−3とGM−CSFの両方を培地に1日当り0.1 から100ng/mlの割合で添加する事である。好ましい割合は1日当り0.5 から10ng/mlであり、更に最も好ましい割合は1日当り1から2ng/mlである。Epoを栄養培地に1日当り0.001から10U/ml、好ましくは0.05から0.15U/ml添加する事が出来る。マスト細胞増殖因子(MCF,C−キットリガンド、スチール因子)を培地に1日当り1から100ng/ml、好ましくは10から50ng/ml添加する事が出来る。IL−1(アルファ又はベータ)を3日から5日の期間に10から100U/ml添加する事が出来る。更にIL−6,G−CSF,基本線維芽細胞増殖因子、IL−7,IL−8,IL−9,IL−10,IL−11,PDGF或いはEGFを1日当り1から100ng/ml添加することもできる。
【0042】
本発明者は、IL−3、GM−CSF及びEpoを上記の様に用いた場合、特異的に赤血球細胞を産生する系統を得る事を発見した。代りにIL−3とGM−CSFを(IL−6或いはG−CSFを加えたり、加えなくとも)使用した場合は、培養は顆粒球を優先的に生産する。本発明の培養で時間と共にT及びBリンパ球が失われる事を本発明者は発見した。
【0043】
培地中の代謝生成物のレベルは通常特定の範囲に維持される。グルコースの濃度は通常5から20mMの範囲に維持される。乳酸塩の濃度は通常35mM以下に維持される。グルタミンの濃度は一般的に1から3mMの範囲に維持される。アンモニウム塩の濃度は通常2.4mM 以下に維持されている。これらの濃度は定期的に或いはオンラインで連続的に既知の方法を用いて監視する事が出来る(Caldwell et al, J. Cell. Physiol. (1991) 147:344-353参照)。
【0044】
本発明に従って培養出来る細胞はあらゆるヒト幹細胞或いはヒト幹細胞を含む細胞塊である。細胞塊はヒト末梢単核細胞、ヒト骨髄細胞、ヒト胎児肝臓細胞、胚幹細胞及び/又はヒト臍帯血細胞を包含する。これらの細胞塊はヒト幹細胞及び/又はヒト造血前駆細胞を含む。ヒト骨髄に見出されるいかなるヒト幹細胞も含めて、ヒトの幹細胞を含む他の細胞塊も本発明に従って使用出来る可能性がある。
【0045】
本発明の好ましい実施態様に於いて、細胞培養は細胞塊中のヒト幹細胞の含有量を増加させる可能性がある。そのような含有量増加は上記の様にしてなされる可能性があるが、本発明に従って用いられた場合、ヒト幹細胞(ヒト骨髄に存在するヒト幹細胞及びヒト骨髄幹細胞を含む)に遺伝子導入による遺伝子療法の為の最初の有用な手段を提供する。ヒト骨髄に存在する幹細胞はヒト骨髄、末梢血、胎児肝臓或いはヒト臍帯血から得る事が出来る。
【0046】
一般にこの実施態様に於いて、安定した遺伝的に形質転換したヒト幹細胞を得る為に、レトロウイルスで感染したパッケジング細胞或いはそのようなパッケジング細胞培養から得られた上清或いは他の遺伝子導入ベクターは本発明に従ってヒト幹細胞培養に加えられる。本発明は幹細胞と造血前駆細胞の分裂のレベルの増加をもたらすが、対照的に従来の培養は造血前駆細胞の低レベルの分裂(つまり減退している培養)を提供するのみであった。本発明の培養系によって初めて培養での細胞の増加が可能になった。このことは細胞にレトロウイルスを感染させるのに必要である。従来の系でレトロウイルス感染を減退細胞培養で行なったが、従来の細胞にはレトロウイルス感染は起こらなかった。本発明は幹細胞のインビトロ・レトロウイルス感染を起こさせる非常に有効な方法を提供する。特別に幹細胞の含有量を増加させた場合、更に特別に合成増殖因子を含む造血増殖因子を用いた場合はレトロウイルス感染は更に有効である。
【0047】
本発明者は組換えレトロウイルスを含む上清を培養に加えるとウイルスとそのウイルスが担う遺伝子がヒト(造血)幹細胞に導入される事を発見した。これらの幹細胞の分裂と分化によって派生した前駆細胞及びこれらの前駆細胞が更に分裂と分化を行なって出来た成熟した血液細胞はインビトロ造血細胞培養の期間中は導入されたDNAを含んでいた。本発明者は次の事を観察した。レトロウイルスが培養の初期にのみ加えられた場合、トランスフェクトされた前駆細胞と成熟した血液細胞が得られたが、それ等の細胞は存在していた幹細胞が培養初期に増殖し、安定な遺伝子導入を受けたものからのみ派生したものである。なぜならこの系ではレトロウイルス感染細胞は隣接した細胞を感染させる事が出来ないからである。希望する遺伝子を含む前駆細胞とより成熟した細胞とは、それ故、初期のレトロウイルス感染期間に形質転換した、より未成熟の幹細胞より遺伝子を受け継いでいる。
【0048】
本発明のこの面でのより好ましい実施態様に於いては、骨髄、末梢血、胎児肝臓或いは臍帯血から分離されたヒト造血細胞は、最初に、より成熟した血液細胞を除去し幹細胞の含有率を増加させる。これは造血細胞を、成熟した血液細胞と、骨髄前駆細胞のエピトープに反応するが幹細胞のエピトープとは反応しないマウス・モノクローナル抗体とをインキュベートし、それから標識された細胞をウサギ抗マウスIg免疫吸着面に免疫吸着させて除去する事により達成される。そのようにして得られた無系統細胞(Lin−)を本発明に従ってレトロウイルス或いは他のベクターと共に培養する。好ましくは、培養はGM−CSF(好ましくは1mg/ml/日)及びIL3(好ましくは1mg/ml/日)の存在下で、また加えても加えなくても良いがIL−1(好ましくは50U/ml/4日)、同じく加えても加えなくても良いがc−キット・リガンド(マスト細胞増殖因子)(好ましくは10μg/ml/日)の存在下で行なう。
【0049】
レトロウイルス感染は培養開始後2から21日、好ましくは10から14日に培地に組換えレトロウイルスで感染したレトロウイルス・パッケジング細胞培養上清を混入して(例えば5から20%容積/容積)行なうか或いはLin−細胞を直接レトロウイルスで感染したレトロウイルス・パッケジング細胞の上で培養して行なうか或いは両方の方法を同時に行なう。
好ましくはレトロウイルス上清を用い、ウイルス存在下でのインキュベートは12日から16日である。また好ましくはパッケジング細胞は集密近くまで増殖させ、培地を交換し、更に12時間から15時間インキュベートする。それから培地を集めてヒト幹細胞のトランスフェクションに用いる。しかしこの過程は必ずしも厳密に行なわれる必要は無く、レトロウイルス・パッケジング細胞のどのような上清も使用して良い。本発明に依ればどのようなレトロウイルス・パッケジング細胞系(既知の)をどのような既知の方法でも用いることが可能である(Wilson et al, Science (1990) 248:1413-1416 及び/又はSullenger et al, Cell (1990) 63:601-608 参照)。実例としてのパッケジング細胞系はNIH3T3細胞及び腎臓癌細胞5637を含む。
【0050】
遺伝子を発現させるのに適当なプロモーター及びエンハンサー要素と共に組換えレトロウイルスに挿入出来るいかなる遺伝子もヒト幹細胞及び造血前駆細胞に導入することが出来る。本発明は培養の中で幹細胞の生存と増殖を可能とし、安定してトランスフェクトされ遺伝的に変えられたヒト造血細胞を培養中で製造する条件を初めて提供した。「安定してトランスフェクトされた」及び「安定して形質転換された」という語は本明細書では外来のDNAがヒト幹細胞染色体に導入される事を意味する。これは本発明が体外で分裂しているヒト幹細胞をそのような外来のDNAに曝すことを可能にした結果出来る様になったのである。
【0051】
本発明に従って、少なくとも5カ月の培養期間中、ヒト造血前駆細胞がヒト幹細胞から分裂と分化によって生産されるような培養方法を得ることが出来る。即ち培養中での幹細胞の生存と増殖を支持する培養方法を得ることが出来る。
本発明者が得たデータは体外ヒト骨髄培養の動向を決定する非常に重要な変数は培地灌流速度であることを示した。培地交換速度を従来の1週一度のデクスター速度から毎日培地交換をする、週当り7倍量に増加した場合体外造血に重要な影響がある事を本発明のデータは示した。本発明者によって行なわれた実験に於ける全ての培養において、かなりの細胞が最初の3〜4週の間に失われた。この後退の後、培養は安定し培地灌流速度の効果がよりはっきりしてきた。
【0052】
培地交換速度、週当り3.5 回が培養の増殖を最も高め、また前駆細胞の生産面から培養最長の寿命をもたらした。特に注意すべき点は4から10週目にかけて2週毎の非付着性細胞の生産数は恒常的であるか又は増加した。
培養の全期間に亙って、3.5 週以降に生産された細胞の累積数は、従来のデクスター法で生産された場合の殆ど3倍であった。更に前駆細胞の安定生産は18週まで維持されていた。
ヒト骨髄にあるような基質細胞は、本発明の培養中に存在してもよいし、存在しなくてもよい。典型的な培養に於いては基質細胞は細胞培養中に1000分の1から10分の1(基質細胞数/全細胞数)存在している。
【0053】
本発明の他の一面に於いて、本発明者は驚いたことには、本発明の培養はヒト骨髄基質細胞の代謝活性を促進し、GM−CSF及びIL−6の分泌を高める事を発見した。GM−CSFはヒト骨髄基質細胞上清中には検出されないが、本発明に従った急速培地交換はヒト骨髄基質細胞を刺激して300 セントグラム/ml/日から200ピコグラム/ml/日のGM−CSFを分泌させた。ヒト骨髄基質細胞によるIL−6の分泌もまた本発明に従った急速培地交換により1〜2ng/ml/日から2〜4ng/ml/日増加した。この増加は本発明の急速培地交換速度を用いた時にも、またその急速培地交換速度を用いると共に造血増殖因子を添加した場合のいずれにも観察された。本発明者によって得られたデータに基づいて本発明の急速培地交換速度のヒト基質細胞サイトカイン生産能に及ぼす影響は、どのような複合組織培養中の基質細胞においても観察されるはずである。
【0054】
実例として、本発明に従って用いられる培地は3つの基本成分よりなる。第1の成分はIMDM,MEM、DMEM,RPMI1640,アルファ培地或いはマッコイの培地或いは既知の培養培地成分である。第2は血清成分である。これは少なくとも、馬血清、ヒト血清を含み、更に随意的に牛胎児血清、牛新生児血清及び/又は子牛血清から成る。第3の成分はハイドロコーチゾン、コーチゾン、デキサメサゾン、ソルメドロール或いはこれらの組み合わせ、好ましくはハイドロコーチゾンの様なコルチコステロイドである。
使用出来る種々の培地の成分を以下に示す。
【0055】
【表2】
【0056】
【表3】
【0057】
【表4】
【0058】
【表5】
【0059】
【表6】
【0060】
【表7】
【0061】
【表8】
【0062】
【表9】
【0063】
【表10】
【0064】
【表11】
【0065】
【表12】
【0066】
【表13】
【0067】
血清成分は培地中に少なくとも1%(V/V)から50%(V/V)存在し得る。血清濃度は好ましくは15から30%(V/V)付近である。血清濃度を高める場合は交換速度を比例して増加させる。第3の成分は10-7Mから104M存在し得るが、好ましくは5×10-6から5×10-5Mである。培地の成分は3つの成分を加えた時に100 %になる様に構成されている。選択的に、血清成分は数種存在する標準的な代用血清混合物のどれとも交換できる。典型的な代用血清混合物はインスリン、アルブミン、及びレシチンかコレステロールを含む。以下の文献を参照:Migliaccio et al, Exp. Hematol.(1990) 18:1049-1055、Iscove et al, Exp. Cell Res.(1980) 126:121-126、及びDainiak et al, J. Clin. Invest. (1985) 76:1237-1242。
【0068】
実例として、ヒト造血幹細胞の細胞濃度は次のようにして増殖させる。吸引採集された骨髄から赤血球をフィコール:ハイパーク密度勾配遠心により除去する。その後、単核細胞を分化した血液成分を認識する抗体の「カクテル」と共にインキュベートする。ここで、分化した血液成分とは、赤血球、顆粒球、マクロファージ及び分化したリンパ球(B−及びT−細胞両方)を含む。更に決定前駆細胞を認識する抗体(抗CD33を含む)を加える。分化した細胞はパニング、磁気ビーズ、或いは蛍光活性セルソーター(FACS)等を含む種々の方法の1つにより除去される。分化した細胞を除去する事により造血幹細胞の組変えウイルスによる感染の機会、それ故希望する遺伝子の導入の機会は非常に改善される。
【0069】
他の実施態様に於いて、培養中で造血細胞の増殖の為の方法が提供される。その方法は効果的な増殖環境を維持する為に、増殖因子を提供する為の通常に形質転換した線維芽細胞を用い、蛋白成分を線維芽細胞と造血細胞の混合物に加え、実質的な連続灌流を行い、随意的に培地の再利用を行なう。
【0070】
それ故この実施態様の方法の詳細を、灌流条件、反応器とその内部構造及び形質転換線維芽細胞の3つに分けて説明する。
反応器には、必要な細胞分散、栄養素と酸素の取り入れ、代謝老廃物の除去、随意的な造血細胞のリサイクル、基質細胞の交換及び造血細胞の採取が出来るあらゆる形の容器が含まれる。反応器は実質的に骨に於ける灌流を模倣する状態を作り出す事が出来るものとする。生体内では骨髄1mlあたり毎分約0.08mlの血清が灌流している。これは106細胞1日当り0.3mlの血清量に相当する。それ故、代謝産物を増殖制限しないレベルに維持する為に、培地は24時間の間に、平均少なくとも50%、好ましくは100%交換する必要がある。経験的に生体内灌流速度を模倣すれば、交換速度は一般的に1日106細胞当り約0.5 から1.0ml灌流培地である。
【0071】
バイオリアクター内の灌流速度は反応器内の細胞密度に依存して変化する。2〜10×106細胞/mlの細胞培養ではこの速度は24〜48時間当り1mlの反応器の体積に対して1mlの培地に相当する。この場合使用された培地は20%の血清(10%牛胎児血清と10%ウマ血清の混合物か或いは20%牛胎児血清)を含む。細胞密度がより高い場合には灌流速度は細胞数当りと時間当りの血清流が一定になる様に、比例して増加する。それ故細胞が5×108細胞/mlで培養される場合は、灌流速度は反応器体積1ml当り毎分0.1mlである。この流速は細胞密度に相当する血清と培地の流量に一致し、培養内の正常ヒト骨髄基質細胞からの造血増殖因子の内部生産を刺激する。この血清と培地の流速で誘導される造血増殖因子にはGM−CSFが含まれ、さらにS−CSF,IL−6、G−CSF及び他の造血増殖因子が含まれていてもよい。幹細胞と前駆細胞が基質細胞と付着している場所で受ける縦方向流れによるせん断応力が1.0から5.0dyne/cm2の間になるように、これらの流速はバイオリアクター内で決定されるであろう。
【0072】
種々の培地を造血及び基質細胞の増殖の為に用いる事が出来る。実例としての培地はMEM、IMDM、及びRPMIを含み、これら培地は5〜20%牛胎児血清、5〜20%子牛血清及び0〜15%ウマ血清の組み合わせで補う事が出来るし及び/又は血清なしでPDGF,EGF、FGF,HGF,或いは基質細胞や幹細胞を刺激するための他の増殖因子等で補う事が出来る。特殊な系統のある細胞だけを特に得たい場合、形質転換した線維芽細胞によって提供される増殖因子を補う為に更に増殖因子を灌流培地に加える事が出来る。基質細胞の分泌により或いは添加により灌流培地に加える事の出来る増殖因子はGM−CSF,G−CSF,M−CSF,インターロイキン1−7(特に1、3、6及び7)、TGF−アルファ或いはベータ、エリスロポエチン、或いは同様の物質、特にヒト因子等で
ある。特に興味があるのは0.5 〜2ng/ml、好ましくは1ng/mlのGM−CSF、
及び最終濃度0.1〜2U/ml/日のエリスロポエチン、100〜300ng/ml/日のG−CS
F及び約1〜10ng/ml/日の幹細胞増殖因子(S−CSF,マスト細胞増殖因子或
いはキット・リガンドとも呼ばれる)等である。ひとつ或いはそれ以上、好ましくは少なくともふたつの増殖因子が形質転換した細胞から分泌され、灌流培地中には増殖因子の希望するレベルを維持するに十分な量の形質転換した細胞が存在することが分かっている。
【0073】
反応器内では生理的温度、つまり37℃に維持されている。33℃を含めて低い温度で行なう事も可能であるが通常は25℃以下ではない。湿度は一般に100%で気相は5%の二酸化炭素を含んでいる。灌流培地は反応器外で酸素添加しても良いし、反応器内でしてもよい。反応器内で酸素添加する種々の手段がある。反応器内酸素添加はハローファイバー、焼結ガラス・ディスク、シリコン管、或いは適当な有孔性と疎水性を持つ他の膜を用いて行う事が出来る。栄養素のレベルと代謝生成物のレベルは比較的狭い範囲に維持される。グルコースのレベルは約5から20mMの範囲で通常10から20mMであり、乳酸のレベルは通常35mM以下に維持しているが20mM以上であることも可能である。グルタミンの濃度は一般的には1から3mM の範囲に、通常は1.5から2.5mM の範囲に維持している。アンモニアの濃度は通常2.5mM 以下に維持しているが、好ましくは2.0mM 以下である。
【0074】
液体の流れは重力、ポンプ或いは他の方法によってなされても良い。流れの方向は反応器内のパッキングの性質に依存して、どの1方向でも構わないし多方向でも差し支えない。望ましくは流れの方向が反応器を横切って実質的に水平であるラミナー・フローを用いても良いし、流れが反応器の底から上方へ向かう垂直流を用いても良い。
【0075】
ヒト造血細胞が腫瘍細胞、例えば白血病、リンパ腫或いは癌を含むと疑われる場合は、灌流速度を正常前駆細胞と腫瘍性の造血細胞とを分離する様に調節出来る。正常の造血前駆細胞は約1.5〜2.0dyne/cm2の縦方向液体流によるストレスに耐える親和力で基質及び実質蛋白に付着している。対照的に腫瘍細胞とそれ等の前駆細胞は基質に対する親和力がかなり弱まっており、約0.05〜1.2dyne/cm2の範囲である。しかし、正常と腫瘍前駆細胞が耐える事の出来るシアー・ストレスの中間のシアー・ストレス、通常1dyne/cm2以上を与える灌流流速を用いれば、腫瘍前駆細胞を正常前駆細胞から分離出来る。分離を達成するには一般的には灌流を少なくとも2日維持し、好ましくは少なくとも5日、更に好ましくは7日或いはそれ以上維持する。このようにして患者からの正常造血細胞を拡大し、同時に適当な流速を採用する事により腫瘍細胞を分離出来る。この方法により腫瘍を持つ患者からの自己造血細胞を化学療法或いはX線療法の間正常造血細胞を拡大して、患者に返し、患者の造血及び免疫系を回復させる。
【0076】
せん断応力を用いて造血腫瘍細胞を正常造血細胞とを分離する実例としては慢性骨髄性白血病(CML)の状況がある。CML細胞のせん断応力耐性は0.05〜1.2dyne/cm2の範囲である。この差が個々の骨髄標品について効率の良いCML細胞除去を可能にした。せん断応力約1.2〜1.5dyne/cm2、好ましくは1.3dyne/cm2を用いて、CML細胞は効率良く分離出来る。
【0077】
個々の骨髄細胞内のせん断応力耐性は先細放射状フローチャンバー(a tapered radial flow chamber)を用いて決定出来る。放射状フローチャンバーに於いては、細胞がうけるせん断応力はチャンバーの始まりからの距離dの関数1/dとして減少する。せん断応力のバンドを細胞集団について分析し、希望する細胞集団を残存させるようにせん断応力をセットする。
【0078】
白血病幹細胞を除去するために、白血病患者の骨髄標品の前駆細胞と幹細胞は最初に放射状フローチャンバーに入れられる。放射状フローチャンバーはポリカーボネートかガラスよりなる2枚のプレートより成り、下方のプレートに骨髄基質細胞が付着出来る様になっている。最初の測定は1)造血細胞の注入の前に骨髄基質細胞の集密的細胞単層を形成しておき12〜24時間後に液体を流し始めるか、或いは2)患者の骨髄を基質細胞の単層なしに直接フローチャンバーに植え、3〜4日待ってから液体を流す(通常0.05〜1.0cc/分)。プレートの端をゴムのガスケットを用いてシールし、調節ねじで一緒にする。チャンバーの狭い注入口に貯蔵タンクから注射器型の定圧ポンプによって管を通して液体をチャンバー内に入れる。広い捕集端で液体と除去された細胞は別の管を通り捕集される(図3及び図3b参照)。一定期間の灌流の後(通常3〜7日)、付着していない細胞を除去し、プレートを分離し3〜5領域のそれぞれから細胞を別々に吸引とゴムのポリスマンで取り、各々の分画を標準的な方法で白血病細胞の有無を調べる(通常は染色体バンディングによる核型分析)。各々の分画での白血病細胞の分析はどの分画(つまりどのくらいのせん断応力)で白血病細胞が基質細胞に付着出来なくなり、除去されたかを示す。このチャンバーでは細胞にかかるせん断応力は入り口からの距離が遠のくにつれて対数的に減少する(図3c参照)。典型的に、付着しない細胞は全て或いは殆ど全て白血病であるが、最も狭い、チャンバーの半分に付着しているのは殆ど全て正常である。
【0079】
これらの測定の結果に基づいて、一連の平行、矩形のチャンバーを設置し、その中に液体を一定の流速で流し、下面にせん断応力をかける。その流速は先細りチャンバーの中で基質細胞から白血病細胞は除去するが全部の正常細胞は除去しないせん断応力に相当するものである。慢性骨髄白血病患者の骨髄の場合、このせん断応力は典型的には0.01〜0.05dyne/cm2である。実際の流速はチャンバーの大きさと形状に依存する。患者からの骨髄細胞をこれらの矩形チャンバーの中で5×106/mlから50×106/mlの濃度で、イスコブの改変ダルベッコの培地に5〜20%(典型的には10%)の牛胎児血清と0〜15%(典型的には10%)のウマ血清を加え、更に10-6Mハイドロコーチゾンを加えた(加えない場合もある)培地で培養する。骨髄細胞を12〜24時間液体の流れなしで培養し、その後流れを開始する。細胞を3〜7日間培養し、非付着性の細胞は捨てられる。付着細胞を矩形プレートから吸引と機械的振動で回収し、集める。これらの細胞は直接患者に戻すか後で用いるために標準技術で液体窒素中で保存する。
【0080】
造血系の細胞以外の細胞でもせん断応力に対する耐性の差による分離が可能である。細胞の複合体の中に区別し得る細胞集団がある場合は、上記の方法を用いて皮膚、肝臓、筋肉、神経或いは上皮細胞に由来する細胞懸濁の中に興味のある細胞を分離する事も可能である。特に興味のある事は正常細胞の集団中に存在する腫瘍細胞を分離することである。分離されるべき細胞集団を後記の様な適当な基質、例えば目的とする細胞が付着し得る精製された蛋白或いは細胞成分の様な基質に接触させる。各々の付着した細胞の小集団についてせん断応力耐性を上記の方法で決める。そして目的とする細胞を上記のようにして集める。
【0081】
反応器の中で種々のパッキングを用いて細胞の付着増殖の場を提供し、一方では基質細胞と造血細胞の間のある程度の物理的な隔離を保持し、また一方では基質細胞と造血細胞の間のある程度の接触或いは接近を維持する。この様にして基質細胞によって分泌された因子がたやすく造血細胞に取り込まれ、造血細胞の増殖と、適当であれば分化と成熟化を助長する。
【0082】
細胞を支持する蛋白マトリックスは切削されたコラーゲン粒子の形を取っても良い。例えば多孔性のコラーゲンのスポンジ或いはビーズ;骨髄からの細胞外の骨マトリックス蛋白より成るスポンジ或いはビーズ;或いは蛋白でコートされた膜等が挙げられる。ただしここで蛋白はコラーゲン、フィブロネクチン、ヘモネクチン、RGDペプチド、混合した骨髄マトリックス蛋白或いは似た物でも良い。膜孔サイズは物理的な隔離を維持しながら異なったタイプの細胞の相互作用を維持する為に、一般的には1〜5μの範囲にある。
【0083】
蛋白でコートした膜を用いてもよい。種々の膜の材料を用いることが出来る。例えばポリプロピレン、ポリエチレン、ポリカーボネート、ポリスルフォネート等である。種々の蛋白、特にコラーゲン或いは既に示された他の蛋白質を用いる事が出来る。膜孔は十分小さく形質転換した細胞は膜を通り抜けてはならないが、その膜の1面に細胞を増殖させ、集密的細胞単層を形成させ細胞膜の一部を孔の中へ伸ばす様にする。一般に孔のサイズは1〜5μの範囲にある。このようにして造血幹細胞は膜の反対側の面で増殖し、形質転換細胞と相互作用を持ち、ここで種々の因子は形質転換細胞から造血前駆細胞へ直接伝達される。前駆細胞、幹細胞は孔を通って侵入してきた細胞質突起に付着する事が出来る。幹細胞からの造血分化は膜の片側で起こり分化した子孫細胞は、基質細胞が集密単層を形成した際、孔は既に塞がれているので、孔を通り抜けることは出来ない。例えば複数のチャンバーを提供し基質細胞を増殖させ、集密状態に達していないチャンバーに造血細胞を移動させる事が出来る。かくして、可動の障壁をチャンバーの間に設け、基質細胞が集密状態に近ずいた時(一般に8〜12週)チャンバー間の障壁を開くか除去するかして基質細胞が新しいチャンバーに移動するようにし、造血細胞が集密状態に達していない基質細胞と接触するようにする。その間集密状態に達していない基質細胞は種々の因子を造血細胞を含むチャンバーに与える(図5a及び図5b)。造血細胞の移動は適当な流速か又は他の適当な方法で達成する事が出来る。適当な壁で分けられたチャンバーに種々のウエルを作ることが出来る。ひとつのウエルに細胞を植えて集密になると細胞は隣のウエルに移動し隣のウエルを集密以下の状態で細胞を植えた事になる。本システムの他の変更は8〜12週の培養の後、造血細胞は新鮮な増殖している基質細胞に曝される事である。この新基質細胞に曝す事は幾つかの方法のうちのひとつにより達成される。第1の技術では、培養をEDTAに3〜5分曝し基質細胞から造血幹細胞を分離する。分離された細胞はそこで新しい培養容器に移される。その培養容器は3〜7日以前に植えられた骨髄基質細胞を含んでいる。この過程を8〜12週毎に繰り返す。他の異なった方法は8〜12週後に培養の体積を増やし培養にコラーゲンのビーズを加えて表面積を増やす方法である。最後に小有機物分子又は蛋白、特に血小板由来の増殖因子(100〜500ng/ml)のようなホルモン、インターロイキン1α、腫瘍壊死因子α、又は塩基性線維芽細胞増殖因子又は他の線維芽細胞のマイトジェニック分子を3〜7日毎に培養に加える。基質細胞をマイトジェンに曝すと骨髄基質細胞の連続した増殖と造血増殖因子の生産が促進される。かくて基質細胞を連続して集密以下の状態に置くことが出来る。
【0084】
連続した液体の流れは骨髄細胞集団の中の正常細胞と癌細胞を選択的に分離するのにも用いる事が出来る。この方法に於いては、放射状フローチャンバーを最初に用いて正常及び癌細胞の特異的基質付着性を決定し、癌細胞を除去するせん断応力を与える流速のフローチャンバーで手術前に正常細胞と癌細胞を分離する。
【0085】
本方法と器具は灌流培地の流れによって失われた幹細胞を再使用する機会も提供する。膜表面蛋白マーカーCD34は実質的に未熟の造血細胞を成熟した造血細胞から分離する。かくして、CD34+である細胞を捕集し、再使用する事により培地中の幹細胞が不足することを防止できる。
【0086】
種々の技術を用いて細胞の未熟分画を捕集し反応器へ戻す事が出来る。例えばCD34に特異的な抗体で細胞を標識し、それからその抗体に対する抗体を用いてCD34+細胞を集め反応器に戻す事によって行うことが出来る。正選択法に代わって、負選択法を用いて成熟した細胞特有のマーカーに対する抗体で成熟細胞を除去することができる。成熟細胞のマーカーとは糖蛋白A、CD33,MO1,OKT3,OKT8,OKT11,OKT16,OKM1,OKM5,Leu7,Leu9,LeuM1等である。種々の造血細胞系統の成熟細胞(リンパ球、骨髄細胞、赤血球)の特異的なマーカーに対する種々の抗体を得る事が出来、これらの抗体を用いて反応器からの流出液から成熟細胞を除去し、残りの細胞を集めて反応器に戻す事が出来る。この方法で、幹細胞を失う事による培養の衰退を防ぐ事が出来、幹細胞の無制限のインビトロ生存を維持する事が出来る。
【0087】
抗体マーカーを用いての分離は種々の方法によって達成する事が出来る。パニング、蛍光活性化セルソーティング、種々の表面、例えばポリスチレン表面、金属小球及び磁性体等に抗体を結合する様な標準技術を単一に或いは複合して用いる事が出来る。抗体を固相表面に結合させて、付着、非付着細胞間の分離をすることができる。或いは抗体を標識し、直接に或いは間接に標識された細胞と標識されない細胞の分離も可能である。
【0088】
本技法を用いれば造血細胞のインビトロ増殖の期間を延長する事が出来る。一般的に生体外でのヒトの造血機能は少なくとも6カ月維持され、顆粒球形成機能は少なくとも4カ月、赤血球形成機能は少なくとも3カ月維持される。更に加えるに、造血前駆細胞は培養期間中連続して生産され、投入細胞の10倍以上の前駆細胞の純収率を得る。
【0089】
更に加えるに、本技法によれば幹細胞の細胞分裂の速度を大きく増大させ、レトロウイルス媒介の遺伝子導入を効率良く行なわせることができる。初期2週間の感染期間に適当なレトロウイルスによって導入された遺伝子はその後4カ月にわたる培養期間に生産された全ての前駆細胞の10〜30%の中で発現させる事が出来る。本技法はそれ故高度に増殖しているヒト造血幹細胞に遺伝子を導入することを可能にする。
【0090】
図1は灌流チャンバーの概略図である。反応器10、蓋プレート12及び床プレート14はボルト16で組み立てられ、ウィングナット18で固定される。歪みを避ける為に3本のボルトが用いられる。チャンバー20は3区画があり、中区画22は基質細胞の為の支持マトリックス、基質細胞床、及び骨髄細胞を含む。中区画22は上区画24と下区画26から膜或いは網の28及び30で分けられている。好適には、ポリスルフォン膜を用いることが出来るし或いは細胞をチャンバーの中区画にとどめておける細かい網目を持つステンレススチールの網を用いる事も出来る。分離間層を、分離膜に機械的な支持を与える為に区分してある内筒27を用いて、チャンバーに置く。上区画及び下区画は同一である必要はなく、液体培地或いはガス交換のための管や膜が通る。ガスは疎水性の管、例えばシリコン製管を通して交換される。管の長さ(それによるガス/液体接触領域)は、中区画で代謝している細胞集団の要する十分なガスの流れができる様に変える事が出来る。培地は上区画或いは下区画からポート32を通じて注入、取り出し可能であり、供給管34を通じて加えることも可能である。
【0091】
希望するなら、上区画及び下区画は外部酸素添加器を用いて除く事が出来る。この場合、分離膜は円筒溝プレート12と14に適合するガラス筒36の下に置かれ、円筒溝の内側領域は膜を通じての流れの分布を良くする為に切り込みを入れてある。この構造は有限数の導入ポートからの液体が混合し、放射状の圧力が平衡化し、分離膜を通しての液体の流れを均一にする。この機構は酸素添加が限界に達しない様な、比較的小数の細胞を培養するチャンバーに適当である。
【0092】
図2に示してあるのは灌流チャンバーを横培地チャンバー、酸素添加器、センサー・チャンバー及び取り出し/注入ポートとをつなぐループの概略図である。
【0093】
外部新鮮培地源50はポンプ52により配管56を通して培地槽に供給し、使用された培地は配管58を通して槽54からポンプ52により使用済み培地容器60に導かれ更に処理される。2番目のポンプ62は培地を培地槽52から配管64を通してホローファイバー酸素添加器66に注入する。培地は配管68を通ってバイオリアクター70の第1チャンバーに導かれる。適宜に培地成分の注入装置82を装着し、培地成分を配管68を通して培地によってバイオリアクター70の第1チャンバーに供給する。培地成分は試験成分、添加因子等である。バイオリアクター70からの培地は中チャンバー72を通ってバイオリアクターの第2チャンバー74に導かれる。そこから培地は配管76により培地組成の変化を検出する為のインライン・センサー78に導かれる。
【0094】
例えばグルタミン:グルコース比は使用する細胞系に依存して約1:5〜8の範囲にあるのが望ましく、例えば形質転換3T3細胞は1:8であるのが好ましい。更にアンモニアの濃度は好ましくは2.0mM以下、乳酸の濃度は40mM以下であることが望ましい。バイオリアクターからの流出液を監視する事により、バイオリアクターに供給される培地を変更する事が出来る。酸素分圧を変え、ガス流量を変化させ、種々の成分を強化し、灌流速度を増加或いは減少する事が出来る。
【0095】
センサー78から培地は配管80を通ってポンプ62により槽54に導かれる。
上記の流れ径路により槽中の培地は別のポンプを用いてゆっくりと交換される。この機構は培地交換速度(外ポンプによる)と酸素添加器と灌流チャンバーを通る流速との別々の制御を可能にする。前者は培地成分と灌流の長期の交換の制御に用いられ、後者は可溶酸素分圧の制御とチャンバー内の流れパターンの制御に用いられる。小メッシュ生体適合膜の使用はチャンバー内でのプラグ(ピストン)フローを可能にし造血細胞や基質細胞にに対し、非常に正確な量を加えることが望まれる増殖因子や他の特別な化合物の供給の正確な制御が出来るようにした。
【0096】
チャンバーとループの構成要素を高圧蒸気減菌した後、反応器は無菌状態で組み立てられる。培地は横ループとチャンバーを通して数日循環し汚染の徴候を監視する。無菌組み立てが完成したならば、チャンバーの中区画へ細胞外マトリックスのみ或いは基質細胞を含む既に接種してある細胞外マトリックスを入れる。基質細胞をそれから:1)チャンバー内に数日間とどめ代謝活動及び/又は増殖因子に対する反応性を監視する。もし結果が満足するものであれば、骨髄を接種する;或いは2)骨髄をただちに接種する。どちらの場合も細胞層は灌流チャンバーの中区画の底に保持される。細胞は追加の細胞外マトリックス下におき、細胞層は分離膜に付着する。この時チャンバーをひっくり返し細胞層を中区画の天井に持ってくる事も可能である。この構成では成熟細胞は基質細胞に対する付着力を失うに連れて、中チャンバーの底にたまる。この特色は成熟細胞が基質細胞及び/又は成熟度の低い造血細胞に損傷を与えるのを防ぐ為に重要である。この特色は成熟細胞の連続除去を容易にする。
【0097】
これらの細胞は細胞を注射器で回収し或いは灌流培地の圧力によりチャンバーより細胞を取り出し管を通して流出させる。
基質細胞は、大部分については、必要な造血増殖因子を提供するひとつ或いはそれ以上の遺伝子で形質転換された線維芽細胞である。同じ或いは異なった細胞を宿主細胞の特異的な選択方式により種々の遺伝子で形質転換出来る。複数の遺伝子に対して、同じ或いは異なった細胞を用いる事が出来る。
【0098】
非常に多くの正常細胞或いは安定細胞系統を用いる事が出来る。しかし、ある系統の細胞の形質転換はその細胞の過剰な増殖をもたらす事があるので、全ての細胞系統を使用出来るわけではない。使用される細胞系は癌性のものではなく、支持体に付着するものであるのが望ましい。哺乳動物細胞はヒト由来のものである必要は無く、サルである必要も無い。種々の形質転換していない細胞も付着細胞層に加えても良い。それ等の細胞は正常ヒト骨髄付着細胞、正常ヒト脾臓付着細胞及び正常ヒト胸腺上皮細胞である。
【0099】
線維芽を含む哺乳動物細胞を形質転換する方法はよく知られており多くの文献があるがその内の少数の参考文献は前記した。構成物はプロモーター、適当であればエンハンサーより成る天然の転写開始制御領域を用いても良いし、定常或いは誘導性の異なった転写開始領域を用いても良い。
数多くの転写開始領域を得る事が出来、それらは誘導性であり、又は定常性である。天然にあるエンハンサーがある場合もあるし、エンハンサーを加えても良い。ある特定の細胞にのみ誘導出来るし、数種の細胞にも全部の細胞にも機能を持たせる事も出来る。転写開始領域はウイルス、天然の遺伝子、合成或いはこれらの組み合わせから得る事が出来る。
【0100】
入手可能で、使用可能なプロモーターは染色体プロモーターを含み、それ等はマウス又はヒトメタロチオネイン−I或いは−IIプロモーター、アクチン・プロモーター等であり、ウイルス・プロモーターとしてはSV40初期遺伝子プロモーター、CMVプロモーター、アデノウイルス・プロモーター、レトロウイルスLTRのプロモーター等である。これらのプロモーターは手に入れる事が出来、転写開始領域及び目的遺伝子を挿入する為のポリリンカーを持つ適当なベクターにたやすく挿入する事が出来る。他の場合、転写開始領域と転写終了領域の間にポリリンカーがあり、メッセンジャーRNAの翻訳のための処理に関係ある種々のシグナル、例えばキャップの位置、ポリアデニレーション・シグナルを提供する発現ベクターを手に入れることが出来る。制御領域と構造遺伝子より成る発現カセットを構成するにはひとつ又はそれ以上の制限酵素、アダプター、ポリリンカー、インビトロ突然変異、プライマーリペアー、切除等を使用する。
【0101】
発現カセットは通常マーカーとひとつ又はそれ以上の複製系を含むベクターの一部である。マーカーは発現カセットとマーカーが導入された細胞の検出及び/又は選択を可能にするものである。種々のマーカーを用いる事が出来るが、トキシン、特に抗生物質に対する抵抗性を与えるマーカーが特に好ましく用いられる。好ましくは宿主として用いられる哺乳動物細胞にG418に対する耐性を与えるゲンタマイシン耐性が用いられる。複製系は原核生物複製系より成り、それにより発現カセットの種々の成分を組み立てる種々の段階でのクローニングを可能にする。宿主細胞に於いてエピゾーム要素を維持する為に他の複製系を用いる事も可能である。しかし複製系の大部分は発現カセットが宿主の染色体に組み込まれるようにさせる性質のものから選択される。
【0102】
発現カセットを宿主に導入する為に通常用いられる技術のどれを用いても良い。それ等はカルシウム沈澱DNA、トランスフェクション、感染、エレクトロポレーション、誘導粒子等である。一度宿主細胞が形質転換されたら、それ等のマーカーを持つ細胞を選択薬剤を含む適当な栄養培地中で選択、増大させる事が可能である。生存した細胞は更に増大させる事が可能である。
使用可能な宿主細胞はアフリカミドリサル細胞系CV1,マウス細胞NIH−3T3,正常ヒト骨髄線維芽細胞、ヒト脾臓線維芽細胞、正常マウス骨髄線維芽細胞及び正常マウス脾臓線維芽細胞等を含む。時によるとベクターと細胞系統の選択によっては細胞が癌化する事があるので注意が必要である。得られた形質転換細胞が付着能を持っていて蛋白スポンジ、蛋白コート膜等の支持体への結合を維持出来る事が重要である。
【0103】
一度適当な増殖因子を発現するベクターが作成されたら、適当な方法で細胞を形質転換させるのに用いる事が出来る。得られた形質転換細胞を先に記述した支持体にシードする事が出来る。これらの支持体は反応器に導入されるか或いはシードする時に既に反応器中に入っている。細胞は十分な時間増殖させ、細胞が生存して必要な増殖因子を生産する事が出来るようにする。
次に、反応器に適当な時期に造血細胞をシードする。造血細胞は、実質的に純粋な幹細胞、ひとつ或いはそれ以上の系統の成熟した造血細胞が実質的に存在しない造血細胞の混合物、或いは造血系の全ての又は実質的に全ての系統の、種々の段階の成熟度の細胞より成る混合物を含む。
【0104】
反応器を通る実質的に連続な灌流を行い、そして種々の栄養素と関連する種々の因子を監視しながら、細胞を増殖させる。大抵の場合、主要な因子は基質細胞から供給されるので、通常は増殖因子の濃度の平衡状態が達成される。調節された上清は造血細胞の増殖に効果的である事が見出されているので、これを用いて反応器内の増殖因子を適当な濃度レベルに維持するような基質細胞と造血細胞の比を与える事が出来る。
【0105】
トランスフェクトした基質細胞はヒト幹細胞への遺伝子導入を助ける事ができる。マウスに於いては、レトロウイルス媒介の幹細胞への遺伝子導入はマウスを5−FUで前処理し、IL−3及びGM−CSFを含むWEHI調節培地で採集した骨髄細胞を増殖させることにより可能になった(Lemischka, Cell (1986)45:917)。興味あるレトロウイルスを分泌しているレトロウイルス・パッケジング細胞と共に増殖している、人工基質が遺伝子をヒト幹細胞に効率良く導入する為に用いる事が出来る。例えばヒトT細胞は幹細胞をCD2制御配列の支配下にあるHIVのアンチセンス配列を含むレトロウイルス・ベクターで感染させる事によりHIV感染に抵抗性を持つように成る(Greaves, Cell (1989) 56:979-986)。レトロウイルスパッケジング細胞によって提供される、レトロウイルスの複製に必須の因子がある。この因子は目標となる造血細胞には存在しない。ウイルスが一度造血目標細胞に導入されれば、ウイルスは複製出来ない。
【0106】
図3aとbに於いて、取り入れ口102、取り出し口104、25のチャンバー106を持つ放射状フローチャンバー100が示されている。矢印108は流れの方向を示す。造血細胞110はチャンバー内の基質層112の上にシードされ増殖する。流速がどの細胞が付着出来るかを決定し、付着出来ない細胞114は取り出し口104を通って外へ出る。
図4aと4bに於いて、取り入れ口122と取り出し口124のある増殖チャンバー120が示されている。図4bに於いては取り入れ口122はマニフォールド128より成る。マニフォールドは細胞110と基質112を含む個々のチャンバー126に入る。そこで細胞は増殖し分離される。
図5aと5bに於いて、増殖チャンバーとその中で障壁134、136、138が培養中除かれている状態を図示している。障壁134は約8〜10週目に;障壁136は約18〜20週目に;障壁138は約28〜32週目に除かれる。
【0107】
本発明の一般的な説明をした後、更に理解を深めるために、次に特定の実施例を挙げて説明をする。ここで提供する実施例は例示のみを目的とし、特記した場合を除いて本発明を制限するものではない。
【0108】
細胞分離及び染色方法
フィコールによる骨髄細胞の分離
1.骨髄試料を室温に保ったI−MDM(イスコブの改変ダルベッコ培地;GIBCO;カタログNo.430-2200)で1:4の比で希釈する。
2.室温で、50mlの遠心管の中で希釈した骨髄試料35mlを注意深く15mlのフィコール−パク(Ficoll-Paque)(比重1.077g/cc;Pharmacia;カタログNo.17-0840-02)にのせる。
3.700×g(1800rpm、Beckman遠心機)で30分、室温(20℃)で遠心分離する。
4.遠心分離後、上層の大部分を除き(間層の上約5mlを残す)、間層(骨髄細胞)を回収し、氷冷したI−MDMで次の様に3回洗浄する:
第1洗浄:1400rpm/15分/4℃
第2洗浄:1200rpm/10分/4℃
第3洗浄:1200rpm/10分/4℃
5.3回目の洗浄の後、細胞は培地か或いは平衡塩類溶液等(細胞の使用目的に依存)に懸濁し、細胞を酢酸液で1:10に希釈した後に(細胞懸濁液10μl=2%酢酸を含むPBS90μl)細胞数をカウントする。この方法では赤血球は酢酸中で溶血するので、白血球のみをカウントすることになる。
6.次に、適当な培地中に細胞を希望する最終濃度に懸濁する(培地については各々の応用参照)。
【0109】
MY−10陽性の骨髄細胞の蛍光染色法:
試薬
標準緩衝液:
粉末バクト乾燥DIFCO緩衝液
(Baxter;カタログNo.2314-15GB): 200g
10%NaN3(アジ化ナトリウム) 20g
非動化牛胎児血清(56℃、30分) 200ml
2回蒸留水で20リットルに希釈;pH7.15〜7.25;
4℃で保存
1ヵ月有効
2%パラフォルムアルデヒド溶液
パラフォルムアルデヒド: 10g
2回蒸留水 500
10N NaOH(ドラフト内で) 8〜20滴
粉末バクト乾燥DIFCO緩衝液 5g
ドラフト内で2回蒸留水を500mlフラスコに入れ、ホットプレート上で撹拌 して60℃にする。
パラフォルムアルデヒド10gを加える。
NaOHを溶液が透明になるまで滴下する。
DIFCO5gを加える。
冷却し、2N塩酸でpHを7.35〜7.45にする。
1.細胞数を数えた後、標準緩衝液で1回洗浄する(100rpm,5分,4℃)。
2.次に、細胞を標準緩衝液に2×105細胞/mlの濃度に懸濁する。
3.2つの15ml遠心管に、それぞれ50μlの細胞懸濁液を入れる。
4.ひとつの遠心管には、1:5に希釈した50μlの抗HPCA−1を加える(抗ヒト前駆細胞抗原;Becton Dickinson; カタログNo.7660;標準緩衝液で1:5に希釈する)。他の遠心管には、1:5に希釈した50μlのMIgを加える(マウスIgG1コントロール;Becton Dickinson;カタログNo.9040;標準緩衝液で1:5に希釈する)。
5.両方の遠心管を氷中で1/2時間インキュベートする。
6.インキュベーション後、細胞を5mlの標準緩衝液で2回洗浄する(1000rpm,5分,4℃)。
7.2回目の洗浄後細胞ペレットは1:40に希釈したGAM−FITC(アフィニティーで分離された抗マウスIgG及び抗IgMヤギF(ab’)2、ヒトIg吸収、フルオレシン結合;TAGO;カタログNo.4353;標準緩衝液で1:40に希釈する)50μlに再懸濁する。
8.細胞を氷冷下、暗闇で1/2時間インキュベートする。
9.インキュベーション後、細胞を5mlの標準緩衝液で2回洗浄し(100rpm,5分,4℃)、各々のペレットは、標準緩衝液100μlと2%パラフォルムアルデヒド100μlの溶液に再懸濁する。
10.次に、フローサイトメーターで細胞を蛍光分析する。%蛍光陽性は抗HPCA−1標品の%蛍光からMIg標品の%蛍光を引いたもの。
【0110】
成熟前駆細胞を選別するための骨髄細胞の蛍光染色法:
目的:
この染色法の目的は、成熟細胞集団を除去し、フローサイトメーター又は磁気ビーズを用いることにより、造血幹細胞(最も原始的な幹細胞)を濃縮することである。選別の結果と比較し濃縮程度を求めるために、いくつかの細胞を(フィコール(Ficoll)分離後)全骨髄細胞として保持して、MY−10陽性細胞を染色するすることは、常に良い方法であると考えられる。
1.細胞は前記したようにフィコール−パク(Ficoll-Paque)で分離される(0.5×106細胞を除去し、2つに分け、抗−HPCA−1/GAM−FITC及びMIg/GAM−FITCで染色する)。
2.3回目の洗浄後、細胞ペレットをモノクローナル抗体混合液(この混合液の作り方の記載部分を参照)に107細胞あたり該混合液1mlを用いて懸濁し、細胞を氷上で1時間インキュベートする。
3.その後、細胞を過剰の氷冷I−MDMで3回洗浄する(1000rpm,5分,4℃)。
4.3回目の洗浄後、細胞を1:40に希釈したGAM−FITC(標準緩衝液ではなく、I−MDMで希釈したもの)に0.25×106細胞当たり50μlの比率で懸濁し、氷冷下、暗闇で1/2時間インキュベートする。
5.インキュベーション後、細胞を氷冷I−MDM中で3回洗浄し、最後の洗浄後、2〜4mlの氷冷I−MDMに懸濁し、選別まで氷冷して保持する。
6.その後、蛍光に基づくフローサイトメーターで細胞を選別し、蛍光ヒストグラムの上部85%を除く。より良く濃縮するために、この選別を二度繰り返してもよい。
7.選別後、細胞をカウントし、洗浄し、アリコートはMY−10陽性細胞の染色を(上記のようにして)行ない、全骨髄細胞の染色アリコートと比較して濃縮程度を求める。
【0111】
磁気抗体を用いた未熟細胞の選択:
1.成熟細胞を選別するために、骨髄細胞の蛍光染色法操作の工程1〜3に従う。 注:アジ化ナトリウムはいずれの緩衝液にも含まれない。
2.その間に、適当量の磁気ヤギ抗−マウスIg(Biomag; Collaborative Re
search; カタログNo.74340-50;1mg/ml;5×108粒子/ml)を氷冷I−MDM中、1500rpm、5分、4℃で3回洗浄する(防腐剤として用いられているアジ化ナトリウムを洗い出すため)。
3.「工程1」における3回目の洗浄後に得られた細胞ペレットを、バイオマグ中に50粒子バイオマグ/細胞の割合で再懸濁する(例えば、1×106細胞に対して5×107粒子を用いる。従って、0.1mlのバイオマグである)。
4.細胞を(細胞数により)T−25又はT−75組織培養フラスコに入れ、断続的に攪拌しながら氷冷下、1/2時間インキュベートする。
5.インキュベートした後、該フラスコ(バイオマグが入っている)を平らな磁石の上に置き、輪ゴム又はテープでしっかり留め、4℃で10〜15分インキュベートする。
6.該磁石及びフラスコを垂直に立て、上清を集める。
7.工程4〜6を2回以上繰り返す。
8.細胞をカウントし、氷冷I−MDM中で1回洗浄し、アリコートを除去してMY−10陽性に対して染色を行ない、適当な培地に再懸濁してさらに使用する。
【0112】
I.培地交換
材料及び方法:
細胞: ヒト骨髄細胞は、通知を受け承諾した個人の腸骨稜からの吸引物にヘパリンを加えたものから得た。フィコール−パク(Pharmacia,No.17-0840-02)密度勾配遠心分離により骨髄を分離し、低密度細胞(<1.077gm/cm3)を集め、イスコブの改変ダルベッコ培地(IMDM)で3回洗浄した。細胞を2回目と3回目の洗浄の間にカウントした。その後、細胞を2連又は3連の24ウェル組織培養プレート(Costar, No.3524)に322μl/ウェル、1、2、及び5×106細胞/mlで播種した。
【0113】
長期培養条件: 低密度細胞を湿潤5%CO2/95%空気雰囲気下、10%ウシ胎児血清(Hyclone Laboratories)、10%ウマ血清(Hyclone Laboratories)、1%ペニシリン/ストレプトマイシン(Sigma, 10,000U/mlペニシリンG及び10mg/mlストレプトマイシン,カタログNo.P3539)及び10-5Mヒドロコルチゾン(17- ヒドロキシコルチコステロン,Sigma,カタログNo.H0888)を補給したIMDM中でインキュベートした。培養を、3種の培地交換スケジュール、すなわち100 %毎日培地交換(7/週)、50%毎日培地交換(3.5/週)、又は50%週2回培地交換(1/週)のうちの一つで処理した。培地交換の期間中、週2回、非付着性細胞の50%を各培養ウェルから除去し、ヘモサイトメーターを用いてカウントした。
【0114】
細胞をカウントするために除去した時(2回/週)、3.5/週及び1/週培養の栄養補給の間に除去された培地は、細胞カウント用に全部とっておき、新規培地をウェルに戻した。7/週培養は細胞カウント用に除去された培地の1/2をとっておけばよく、残りの1/2中の非付着性細胞は遠心分離して戻した。その後、新規培地を各ウェルに添加し、細胞カウントのために除去された培地を補充した。細胞がカウントのために除去されなった日は、7/週及び3.5/週培養ウェルから、ぞれぞれ培地の100%又は50%の培地を除去した。細胞を遠心分離し、さらに新規培地とともに元のウェルに戻した。
【0115】
メチルセルロース及び形態学的アッセイ: 一週間毎に、細胞カウント用に除去された非付着性細胞をエリトロポエチン(erythropoietin)、GM−CSF及びIL−3の存在下、メチルセルロースにプレートし、顆粒球マクロファージ−コロニー形成ユニット(CFU−GM)を数えた。除去細胞のアリコートを細胞遠心分離し、ライト−ギムザ(Wright-Giemsa)で染色し、差分細胞カウントを行なった。
【0116】
統計学的解析: 週2回の細胞産生の結果は、同型培養から平均±SEMとして表わされる。培養グループ間に有意差の生じる確率は、一対のt−検定を用いて、急速交換培養(7/週及び3.5/週)とそれに対抗するコントロール培養(1/週)との標準化累積細胞産生値の比較により求めた。統計学的有意性は5%レベルで判定した。
【0117】
結果:
非付着性細胞産生の速度論: 非付着性細胞産生は、接種細胞密度の関数(1〜5×106細胞/mlの範囲にわたっての)及び培地交換速度の両者を試験した。培地交換速度は、1培地体積交換/週、すなわち伝統的なデクスター(Dexter)培養速度から、7培地体積交換/週まで、様々であった。集められた週2回の細胞数は、培養当たりの接種細胞数で割ることにより標準化した。
【0118】
各培地交換速度において、標準化した細胞コレクションカーブは接種密度でそれ程変化はしなかった。7/週、3.5/週及び1/週の3つの培地潅流速度で保持された培養の細胞産生は、培養当たりの接種細胞数に標準化した場合、同様であった。接種密度間の最終累積細胞産生の比較から、3つの培地交換速度のいずれにおいても顕著な差異は全く示されなかった(サンプルの全組の一対のt−検定によりp> .20)。
【0119】
反対に、培地交換速度はこれらの培養における細胞産生の速度及び寿命に大きく影響した。1/週(コントロール)、3.5/週及び7/週で交換された培地の細胞産生は全て初めの数週間にわたり減少した。しかしながら、培養産生性の差異は培養の第3週以降に明らかになった。第3〜10週の間に、細胞産生は7/週培養においては一定であり、1/週培養においては低レベルで一定であったが、3.5/週培養においては指数関数的に増加した。第10〜12週以後には、全ての培養において細胞産生は培養が終結するまで減退した。
【0120】
1/週交換培養の結果は、伝統的なヒト・デクスター培養の様々な系において共通して見られる結果と同じであったが、一方、3.5/週及び7/週の急速交換培養では、前の最適培養方法に比べて細胞産生性の増加が見られた。毎日培地の1/2が交換される培養(3.5/週)では、コントロール(1/週)又は毎日完全交換(7/週)培地よりも実質的に長期間細胞産生の増加が維持された。第3週及び第9週の間、3.5/週交換培養から集められた非付着性細胞の数は指数対数的に増加し、2.1週間毎に2倍になった。
【0121】
3.5/週及び1/週プロトコールでの細胞産生は、培養の産生のパーセンテージとして3.5/週交換速度下での細胞産生をプロットすることにより、1/週の交換速度と直接比較することができる。この比較により、最初の減少相では、2つのプロトコールでの細胞産生は同じであることが示される。しかし、第3.5週及び第18週の間では、3.5/週交換速度下での細胞産生は常に高い。
したがって、培養物の増殖能は、初めの減少後の細胞の産生能により測定する
ことができる。第3週以後の標準化累積細胞産生(Σ(i=7)n,Ci/Co)は7/週、3.5/週の培地交換速度に対する細胞接種密度には依存しなかった。同様の培地交換速度での培養から得られる細胞産生データは、質的にも統計学的にも類似しており、従って、密度は平均化され、組合せて、大きな統計サンプルを得た(下表)。第3.5週及び第20週の間の密度平均化累積細胞産生は:7/週培養では0.22;3.5週培養では0.40;及び1/週培養では0.15であった。したがって、1/週から7/週への培地交換速度の増加は、細胞産生を代表的なデクスター培養培地交換スケジュールよりも約60%程増加させた。3.5/週交換速度は、1/週デクスタープロトコールと比較してほとんどその3倍の累積細胞産生増加となった。これらのデータの一対のt−検定を用いた統計学的解析は、7/週対1/週及び3.5/週対1/週において共に5%程度の有意性で著しい差異があることを証明した。したがって、3.5/週の培地交換速度は、1/週の伝統的なデクスタープロトコールより細胞産生速度を改善する。
【0122】
顆粒球−マクロファージ前駆細胞産生: 顆粒球−マクロファージ前駆細胞アッセイは、或る培地潅流スケジュール及び接種密度の反復実験から行なわれた(表14)。培地潅流速度は、産生される顆粒球−マクロファージ前駆細胞の数に著しい影響を示した。3.5/週培地交換培養では、前駆細胞産生の寿命が最も長かった。これらの培養物では、第4週と第18週の間に前駆体が安定な速度で産生された。
【0123】
前駆細胞産生についての最適条件は、週当たり3.5 回交換され、かつ5×106細胞/mlで接種された培養である。これらの培養は、第20週までに相当数の前駆細胞を産生した。一対のt−検定を用いた統計学的解析により、3.5/週の最適培地交換速度培養は、3種の接種密度全てにおいて第8週以後に7/週及び1/週培養よりも1%レベルの有意性でかなり多くの顆粒球−マクロファージ前駆細胞を産生することが示された。産生された前駆細胞数は幹細胞再生の非直接的な尺度なので、重要である。前駆細胞は、培養中に依然として存在する初期細胞、おそらくは幹細胞から分化することにより、培養の数週間後に存在することが可能である。したがって、これらのデータから、より生理学的で急速な培地/血清交換速度及びより高い細胞密度が、5ケ月間の幹細胞再生の幾分かを支持する条件を提供したであろうことが示唆される。
【0124】
非付着性細胞形態: 3.5/週培養により支持される延長化造血機能がそれ以外の培養物では性質上異なるか否かを決定するために、第10週と第19週の間に集められた非付着性細胞を染色し、形態学的に分類した。1/週及び7/週の交換速度において、産生された細胞の大部分は、第15週付近及びそれ以降ではマクロファージであり(表15)、それは他の研究機関における研究で得られた結果と同様であった。それとは反対に、週当たり3.5培地体積の速度で潅流され、5×106細胞/mlで播種された培養物は、19週間を通じてマクロファージだけでなく顆粒球をも産生した。したがって、この培地交換速度及び接種密度はインビトロにおいてより効果的に顆粒球機能を再構築したと考えられる。
【0125】
【表14】
【0126】
この結果は、長期ヒトデクスター培養条件が次善であり、かつインビボ造血環境に近いより優れたインビトロ培養として、骨髄エクスビボのより効果的な再構築が達成できることを支持するものである。
【0127】
物理的外観: 培地交換速度は、培養物の物理的外観に著しく影響した。培養の10週付近で、7/週培養では多数の脂肪細胞が間質に生じ、一方3.5/週培養ではわずかに脂肪細胞が生じ、1/週培養では脂肪細胞は全く生じなかった。26週で培養が終結した時、7/週培養の間質はおよそ20〜30%の脂肪細胞からなるが、3.5/週培養ではわずかな脂肪細胞を有するだけであった。付着性コロニー分布も、培地潅流速度の異なる培養間ではそれぞれ異なる。3.5/週培養における付着性コロニーは、7/週及び1/週培養のものよりも長期間存続した。
【0128】
【表15】
【0129】
II.造血細胞成長因子を有する培地の補給を組合せた培地交換
材料及び方法:
細胞: ヒト骨髄細胞は、通知承諾に従い、腸骨稜からの吸引物にヘパリンを加えたものから、ユニバーシティ・オブ・ミシガン・ヒューマン・インベスティゲーション・コミッティー(University of Michigan Human Invenstigation Committee)から提供されたプロトコールに基づいて得た。骨髄をフィコール−パ
ク(Pharmacia)密度勾配遠心分離により分離し、低密度細胞(<1.077gm/cm3)
を集め、IMDMで3回洗浄した。細胞を2回目と3回目の洗浄の間にカウントした。その後、細胞を6−ウェル組織培養プレート(Costar No.3406)又はコラーゲン被覆6−ウェルプレート(ウサギ尾タイプ1コラーゲン,Biocoat. Collaborative Research Inc. カタログNo.40400)上に1.5ml/ウェルで、5×106細胞/mlで2連に播種した。
【0130】
培養培地: 使用した培地は、10%ウシ胎児血清(Hyclone Laboratories)、10%ウマ血清(Hyclone Laboratories)、1%ペニシリン/ストレプトマイシン(
Sigma,10,000U/mlペニシリンG及び10mg/mlストレプトマイシン,カタログNo.P
3539)及び10-5Mヒドロコルチゾン(17-ヒドロキシコルチコステロン,Sigma, カタログNo.H0888)を含有するIMDM(Gibco Laboratories, カタログNo.430-2200)である。
【0131】
造血成長因子(HGH): 急速培地交換による頻繁培地補給により、造血成長因子はクローナルアッセイ4において最大限のコロニー形成を促進することがわかっているおよそ1/20の濃度で培地に添加した。使用された濃度はIL−3が1ng/ml、GM−CSF(Amgen Biologicals, カタログNo.13050)が1ng/ml、Epo(Terry Fox Labs. Vancouver, Canada)が0.1U/mlであった。
【0132】
造血前駆細胞アッセイ: 培地から除去された非付着性造血細胞をカウントし、1×105細胞/ml又はそれ以下の細胞をメチルセルロースにプレートした。GM−CSF及びEpoを該メチルセルロースにそれぞれ20ng/ml及び2U/mlで添加した。細胞を24−ウェルプレートに0.25ml/ウェルでプレートし、37℃で14日間インキュベートした。その後、コロニーを倒立顕微鏡下でカウントし、50細胞より大きいコロニーはGM−コロニー形成ユニット(CFU−GM)、赤芽球コロニー群−形成ユニット(BFU−E)、又は顆粒球赤芽球巨核球マクロファージ−コロニー形成ユニット(CFU−GEMM)として記録した。
【0133】
LTBMC条件: 培養は湿潤5%CO2/95%空気雰囲気下、37℃でインキュベートし、50%毎日培地交換の速度で潅流(培地交換)した。培養の第1週の間、毎日培地交換の間に除去された細胞は全て遠心分離して元のウェルに戻した。培養1週間後、全非付着性細胞の50%を、培地交換の間培養物から週2回除去し、単核化細胞をカウントし、新規培地をウェルに戻した。細胞をカウントしない残りの5日/週は、培地の50%を各培養ウェルから除去し、新規培地で置き換えた。除去した培地は遠心分離し、培地を細胞ペレットからデカントし、細胞を元のウェルに戻した。
【0134】
統計学的解析: 培養グループ間に相当な差異が生じる確率は、一対のt検定を用いて、造血成長因子を補給した急速潅流培養物と対する未処理コントロール培養物との標準化累積細胞産生値の比較により決定した。統計学的有意性は5%レベルで判定した。組織培養プラスティック上で培養された対抗する急速潅流LTBMCとI型ラット尾コラーゲンには5%レベルで統計的な差異はなかった。したがって、プラスティック及びコラーゲンマトリックスについてのデータは、ここにおける図及びそれ以外の図を提示するために組合せ、この組合せのデータについて統計学的解析を行なった。
【0135】
結果:
急速交換された成長因子補給LTBMCにおける細胞産生の速度論: 長期骨髄培養(LTBMC)の寿命と産生能が不十分なHGFの産生により制限される、という仮説の第1のテストとして、我々は、IL−3又はEpoが補給された急速交換エクスビボ骨髄培養を保持した。これらの培養物においては、培地の50%が毎日除去され、IL−3又はEpoが補給された同体積の新規培地で置き換えられた。その後、除去細胞を遠心分離し、培地をデカントして捨て、細胞を再懸濁し、元の培養物に戻した。IL−3及びEpoはそれぞれ急速交換LTBMCの細胞産生能を促進した。初めからEpoのみを含有する培養物は、実質的に終結した赤血球の分化により高い細胞産生速度を示した。しかし、4週付近で、赤血球形成は終了し、細胞産生速度はコントロール培養のレベルまで低下した。IL−3及びEpoは、非付着性細胞産生において、18週間を通して平均的な増加を誘発し、コントロールに対して、それぞれ175%及び173%であった。
【0136】
成長因子の組合せは、非付着性細胞産生速度を増加させることにおいて、より効果的であることが明らかになった。細胞産生の最高速度は、IL−3+GM−CSF+Epoの組合せにおいて観察された。これらの培養は、培養において最初の6週間に週2回接種された細胞数のおよそ25%を産生し、非付着性細胞産生が第2〜8週の間にコントロールに対して平均4.8倍増加した。IL−3+GM−CSFの組合せでは、8週通して非付着性細胞がコントロールに比較して平均3.5倍増加した。分離実験において、IL−3+GM−CSF+Epoの組み合わせにIL−6及びG−CSFのいずれも添加しない場合には、非付着性細胞産生速度は改善されたが、その代わり、結果的に細胞産生速度が、IL−3+GM−CSFの組合せを含有する培養と区別できなくなった。いずれの場合も、HGF添加により誘発される細胞産生における刺激効果は0〜8週の間で最大となったが、細胞産生は培養全般を通じてコントロールよりも高かった。
【0137】
HGFを組合せることにより、急速交換LTBMCにおいて非常に多数の非付着性細胞が産生された。培養物の産生能は、接種細胞数(CO)に対する経時的産生細胞累積数(Σi=1n,Ci,Ciは時間iにおいて集められた非付着性細胞の数)を、時間の関数としてその割合(Σ(i=7)n,Ci,Co)をプロットして比較することにより示すことができる。この比率が1を越える場合、培養物は接種された細胞よりも多く細胞を産生し、この培養物では細胞数が増大する。
【0138】
IL−3+GM−CSF+Epoの組合せにより、接種細胞数よりも3倍以上の累積細胞産生が誘発された。この細胞産生速度は、培養における初めの6週間で最高であり、この間、培養は2週間毎に接種された細胞数とおよそ同数の細胞を産生した。この最高細胞産生速度は、推定インビボ骨髄細胞産生速度の15%であり、そこでは骨髄細胞塊の50%が毎日発生する。培養第3〜7週の間において、IL−3+GM−CSFの組合せは、IL−3+GM−CSF+Epoの組合せと同等の速度で細胞数は2倍以上増大した。HGFが補給されない未処理の急速交換(50%毎日培地交換)及び遅交換(50%培地交換週2回)コントロール培養は、それぞれ18週以降に接種された細胞数の約1及び0.37倍を産生した。より重要なことは、これらの非補給培養物から除去された全細胞の半分以上が最初の2つのサンプリングに由来していたことであり、さらにこれらの細胞の多くが初めの接種物からのものであること、及び培養物にHGFを補給することが前駆体と幹細胞の重要なサイクリングを誘発するために必要であることを示したことである。
【0139】
非付着性細胞の形態学的解析: 複数のHGFの添加もまた、培養物で産生される骨髄細胞の多様性を増加させた。コントロール培養は、培養の第3週後に主にマクロファージである非付着性細胞を産生した。赤血球系細胞の産生は急に減少し、第5週後にはわずかな赤血球系細胞しか検出されなかった。Epoを含有する培養(Epo単独,IL−3+Epo,及びIL−3+GM−CSF+Epo)では、赤血球系細胞の産生は一時的に増加し、高パーセンテージ(55〜75%)の赤血球系である非付着性細胞が3週を通じて産生された。IL−3+Epo±GM−CSFが存在する場合、培養は続き、培養16週を通じて、赤血球系として分類された約5〜15%の非付着性細胞と共に、赤血球系細胞を産生した。したがって、IL−3+Epoの存在下では、赤血球形成は始終活性であった。
【0140】
IL−3±Epoでは、第5週に、主に(60〜70%)後期顆粒球(LG)である非付着性細胞集団が生じた。LGのパーセンテージは着々と減少し、第18週で約20%までになった。それに対応してマクロファージの産生が増加した。GM−CSFをIL−3±Epoに添加した場合は、高パーセンテージのLGが18週を通じて存続した。したがって、IL−3+GM−CSFの組合せにより、培養の18週に活性な顆粒球形成が起こり、同様にEpoを添加することにより赤血球形成も保持された。培養5.5週でのコントロール及びIL−3+GM−CSF+Epoを補給した培養の顕微鏡写真からは、培養密度及び産生細胞の多様性がめざましく向上したことがわかる。
【0141】
非付着性前駆細胞産生の速度論: 前駆細胞産は、複数のHGFを添加するにしたがって増加した。未処理のコントロールにおける顆粒球マクロファージコロニー形成ユニット(CFU−GMs)の産生は、18週間以上にわたり長く安定であり、それは急速に潅流されたHGF無添加のLTBMCを用いて得られた初期の結果と一致する。IL−3+GM−CSF及びIL−3+Epo±GM−CSF培養中で産生されたCFU−GMは、第3〜5週の間ではコントロールのおよそ10倍であった。
【0142】
ヒトLTBMCにおける赤芽球コロニー群形成ユニット(BFU−E)の産生は低く、すぐに終結してしまうことが報告されている(Coutinho et al, Blood (1990) 75(11): 2118-2129)。急速交換で未処理のコントロールは、BFU−E産生が急速に減少したが、培養17週を通じて低レベルのBFU−Eが産生された。Epoだけを添加しても、産生されるBFU−Eの数はそれほど大きな影響を受けなかった。IL−3だけを添加すると、第3〜5週においてBFU−E産生の穏やかな短い刺激が誘発された。一方、IL−3にEpo又はGM−CSFのいずれかを加えたものは、培養第3〜5週の間に非付着性BFU−Eレベルがコントロールに比べて10〜20倍増加した。
【0143】
III. ヒト幹細胞の形質転換
材料及び方法
細胞 ヒト骨髄細胞は通知承諾に従い、腸骨稜からの吸引物にヘパリンを加えたものから、ユニバーシティ・オブ・ミシガン・ヒューマン・インベスティゲーション・コミッティー(University of Michigan Human Investigation Committee)から提供されたプロトコールに基づいて得た。骨髄はフィコール−パク(Pharmacia)密度勾配遠心分離により分離し、低密度細胞(<1.077gm/cm3)を集め、IMDMで3回洗浄した。細胞を2回目と3回目の洗浄の間にカウントした。CD18遺伝子伝達実験を行なうために、骨髄は通告承諾に従い、CD18欠失患者提供者から得た。
【0144】
骨髄細胞の系統陰性(Lin-)選択 IMDMでの3回目の洗浄後、成熟単核細胞は、モノクローナル抗体(MAb)混合物と共にインキュベートすることにより、上記細胞調製物から除去した。107細胞を1mlのMAb混合液中、氷上で10〜15分毎に静かに混ぜながら1時間インキュベートした。用いたMAb混合液を表16に示す。
【0145】
【表16】
【0146】
細胞を過剰の氷冷IMDMで3回洗浄し、4℃で遠心分離した。適当量の磁気ヤギ抗−マウスIg(Biomag; Collaborative Research Corp., カタログNo.743
40-50,1mg/ml,5×108粒子/ml)を氷冷IMDMで3回洗浄し、遠心分離した。細胞を50粒子/細胞でバイオマグに再懸濁し、T−25又はT−75組織培養フラスコに入れ、氷上で1/2時間、断続的に攪拌しながらインキュベートした。インキュベート後、フラスコを平らな磁石の上に置き、磁石をフラスコにしっかり留め、4℃で10〜15分間インキュベートした。この磁石とフラスコを垂直に立て、上清を集めた。遮光、磁石の添加、垂直に立てること、及び上清の収集を伴うインキュベートを2回以上繰り返した。この細胞をカウントし、6−ウェル組織培養プレート(Costar No.3406)に接種した。
【0147】
培養培地 使用した培地は、10%ウシ胎児血清(Hyclone Laboratories)、10%ウマ血清(Hyclone Laboratories)、1%ペニシリン/ストレプトマイシン(Sigma,10,000U/mlペニシリンG及び10mg/mlストレプトマイシン,カタログNo.P3539)及び10-5Mヒドロコルチゾン(17-ヒドロキシコルチコステロン,Sigma, カタログNo.H0888)を含有するIMDM(Gibco Laboratories, カタログNo.430-2200)である。
【0148】
造血生育因子 造血成長因子は最適なものを上記した。用いた濃度は1ng/ml又は0.4U/mlのIL−3(Genetics Institute, Cambridge, MA からの寄贈)、1ng/mlのGM−CSF(Genetics Institute, Cambridge, MA からの寄贈)、50U/mlのIL−1a(Genzyme Corp.)、0.1U/mlのEpo(Terry Fox Labs, Vancouver Canada)、10ng/ml のMGF(マスト細胞増殖因子,c−kitリガンド,Immunex Corp., Seattle, WA)、及び2.0ng/mlのヒブリカイン([PIXY321]Immunex Corp., Seattle, WA)であった。
【0149】
造血前駆細胞アッセイ 毎週のサンプリングで培養物から除去された非付着性造血細胞をカウントし、1〜105細胞/ml 又はそれ以下の細胞をメチルセルロースにプレートした。MGF、GM−CSF及びEpoを該メチルセルロースにそれぞれ50ng/ml、20ng/ml及び2U/ml添加した。細胞を24−ウェルのプレートに0.25ml/ウェルでプレートし、37℃で14日間インキュベートした。その後、コロニーを倒立顕微鏡下でカウントし、50細胞以上のコロニーをGM−コロニー形成ユニット(CFU−GM)、赤芽球コロニー群−形成ユニット(BFU−E)、又は顆粒球赤血球巨核球マクロファージ−コロニー形成ユニット(CFU−GEMM)として記録した。
【0150】
レトロウイルス産生細胞系 2つのレトロウイルス産生細胞系をメモリアル・スローン・ケターリング・キャンサー・センター(Memorial Sloan Kettering Cancer Center, New York, NY)のドクター・エリー・ギルボア(Dr. Eli Gilboa)の研究室から入手した。この細胞系は、哺乳動物ネオマイシンアナログG418耐性を付与するネオマイシンフォスフォトランスフェラーゼ産生NEO遺伝子を含有するアンホトロフィックなウイルス性粒子を産生する。また、この両細胞系は、レトロウイルスに感染した細胞がそれ自身感染性のウイルスを産生しないように、必要なレトロウイルス遺伝子が欠損しているレトロウイルス粒子を産生する。
【0151】
パッケージング細胞系を含有するSAXは、改変モロニー・ムリン・ロイケミア・ウイルス(Moloney Murine Leukemia Virus) (MoMuLV)を含有する3T3系細胞系である。SAXプロウイルスはNEO遺伝子、SV−40促進化アデノシンデアミナーゼ遺伝子をXhol制限部位に含有する。また、このSAXプロウイルスはφパッケージング領域を含有するが、gag(コア蛋白質)、pol(リバーストランスクリプターゼ)及びenv(エンベロープ蛋白質)遺伝子が欠損している。この第2のレトロウイルス粒子は外来DNAのダブルコピーを含有しており、このレトロウイルス粒子はDC−29(ダブルコピー−29番目クローン)と表わされる。DC−29プロウイルスは、NEO遺伝子及びその他のレトロウイルス性及び外来のDNAの2つのコピーを3T3細胞系に含有している。
【0152】
CD18試験を行なうために、ヒトCD19 cDNA全長を含有するレトロウイルスベクターに感染したアンホトロフィックなパッケージング細胞Psi−Cripを用いた(Wilson et al, Science (1990) 248:1413-1416)。このレトロウイルスにおいて、ヒトCD18のcDNA全長は、BA−CD18と呼ばれるトリβ−アクチン遺伝子の5′領域をスキャンしている異種配列から組換え遺伝子を発現するベクターのBamH1部位にクローンされた。ヒト・サイトメガロウイルスの5′方向から即時型初期(IE)遺伝子までの配列をPUC19にサブクローンし、IE促進配列を含有する部分をXho1上で(ポリリンカーから)Nco1(IE遺伝子の−220)フラグメントまで除去した。合成リンカーを用いてNco1部位をXho1部位に変換し、この改変フラグメントを5′からβ−アクチンプロモーター方向に配置したBA−CD18のXho1部位にクローンした。この新しいベクターをCMV−BA−CD18と呼んだ。
【0153】
レトロウイルス粒子産生 SAXレトロウイルス粒子はドクター・エリー・ギルボア(Dr. Eli Gilboa)の研究室のドクター・クレイ・スミス(Dr. Clay Smith)より、凍結して-80℃で保存されたウイルスの上清溶液として提供された。DC−29及びCD18レトロウイルス粒子は、DC−29及びCD18ウイルスパッケージング系をT−75フラスコ内でほとんど集密するまで増殖させ、培地を全て交換し、細胞を12〜15時間インキュベートし、その後ウイルス粒子を含有する培地を集めることにより産生させた。その後、この上清を含有するウイルスを遠心分離してウイルスパッケージング細胞を除去し、培地を除去して、アリコート中、-80℃で凍結した。
【0154】
SAXレトロウイルス、DC29レトロウイルス又はCD18レトロウイルス添加上清含有LTBMC 培養物を湿潤5%CO2/95%空気雰囲気下、37℃でインキュベートした。培養の最初の2週間は毎日、培地の2/3(1ml)を各培養ウェルから除去し、培地をHGF(0.85ml)及びウイルス細胞産生上清(0.15ml)を含有する同体積の新規培地で置き換えた。レトロウイルス上清を含有する培地を使用直前に解凍し、使用する必要がない場合には冷凍庫の氷上で保存した。培養物から除去された培地を遠心分離し、培地をデカントし、細胞を元のウェルに戻した。
【0155】
SAXレトロウイルスパッケージング細胞系と共培養したLTBMC SAXレトロウイルスパッケージング細胞系をT−25フラスコ(Costar, No.3056)内でおよそ10%集密するまで生育させ、それから2000ラドの放射線にさらした。上記のようにして調製された造血細胞を照射ウイルス産生細胞に添加し、50%毎日培地交換して全細胞をウェルに戻しながら2.5 週間培養した。培養2.5 週目に、EDTAの0.5mM溶液をフラスコに添加し、間質は残したまま造血細胞を除去した。除去された造血細胞を、ウェル当たり1000細胞の新たにトリプシンを添加した骨髄線維芽細胞とともに6−ウェルプレートの3ウェルに添加した。
【0156】
感染LTBMCのサンプリング 培養2週目の初めに、レトロウイルス添加が終了又は共培養が終了した後で、解析するために交換培地中の非付着性細胞を週に一度除去しながら、培養物を一日当たり50%の培地交換した。毎日培地交換を行なっている間、非付着性細胞を培養物から除去し、単核細胞をカウントし、新規培地をウェルに戻した。細胞をカウントしない残りの6日/週には、培地の50%を各培養ウェルから除去し、新規培地で置き換え、除去した培地を遠心分離し、培地を細胞ペレットからデカントして、細胞を元のウェルに戻した。
【0157】
レトロウイルス感染の解析 初めの骨髄接種物を、0、0.4、0.8、1.2、1.6及び2.0mg/ml のG418にプレートし、ポスト感染骨髄細胞をプレートするためのG418の濃度決定用死滅曲線(kill curve)を得た。培養物から除去された細胞は0.0、0.8、及び1.6mg/mlのG418を含有するメチルセルロースにプレートした。2週間後、前駆細胞のコロニー数をメチルセルロース中で数えた。その後、各コロニーをメチルセルロースから掻き取り、ポリメラーゼ・チェーン・リアクション(PCR)によりレトロウイルスDNAをアッセイした。
【0158】
統計学的解析 培養グループ間に有意差が生じる確率は、一対のt検定を用いて、試験サンプルと対するコントロール培養物との累積細胞産生値を比較することにより決定した。統計学的な有意性は5%レベルで判定した。
【0159】
結果
SAXレトロウイルスを用いたレトロウイルス感染
SAXレトロウイルス感染LTBMCにおける細胞産生の速度論 レトロウイルス感染した培地における細胞産生はレトロウイルス感染の可能性の指標であり、したがって、測定の有用なパラメーターである。ターゲット細胞ゲノ厶へのレトロウイルスの組込みは、唯一、細胞分裂の間に起こると考えられる。この理由により、増大する培養産生性は幹細胞有糸分裂の可能性を高め、したがってレトロウイルス感染の可能性を高めるものである。最高の細胞産生は、培養の4週間を通じて産生細胞数を増加させるIL−3+GM−CSF及びIL−3+GM−CSF+IL−1αを補給した上清ウイルス添加培養物において起こる。SAXウイルスパッケージング細胞と共培養されたLTBMCは、2週目に上清添加培地よりも多くの細胞を産生したが、2週目以後では細胞産生は減少した。
【0160】
上清SAXウイルス含有LTBMCにおけるレトロウイルス感染の解析 高[G418]中で生存している前駆細胞のパーセンテージは、IL−3+GM−CSF又はIL−3+GM−CSF+IL−1α補給培養物では、培養の初めの4〜6週において2〜50%と様々であった。培養第10週付近(8週間後にはウイルスの添加は終了していた)では、造血コロニーにクローン可能な前駆細胞数の43%がG418にさらされて生存していた。このことから、レトロウイルスによって培養物中に存在する幹細胞に転移されたG418耐性遺伝子を含有するウイルスにより、最初の感染14日間で、これらの前駆細胞がG418耐性になっていたことが示される。急速に潅流されたHGF無補給培養物は、第8週と第11週の間に、平均12%の高[G418]中生存前駆細胞を有していた。11週で培養が終了した時、IL−3+GM−CSF補給培養の間質層にはトリプリシンが添加され、間質に付着した前駆細胞の17%が高[G418]中で生存していた。これは、相当なパーセンテージの付着前駆細胞もSAXウイルスに感染したことを示唆するものである。
【0161】
照射SAXウイルスパッケージング細胞と共培養されたLTBMCにおけるレトロウイルス感染の解析 照射を受けたSAX細胞と共培養した場合、G418中で生存している前駆細胞のパーセンテージは、0〜36%と様々であった。HGFが補給されない培養は、高[G418]中で第4〜7週の間のみ生存するCFU−GMを産生した。第7週以後、高[G418]中で生存するCFU−GMは、これらの培養では全く産生されず、これは、幹細胞の感染がほとんど、あるいは全く起こらなかったことを示唆する。IL−3+GM−CSFが補給され照射SAX細胞と2.5週間共培養されたLTBMCは、0.8mg/ml G418中で第4、5及び8週において生存するCFU−GMを高パーセンテージ産生した。しかし、第10週で、これらの培養はG418に耐性のCFU−GMを産生しなくなった。このことは、これらの培養では幹細胞の感染がほとんど、あるいは全く起こらなかったか、又は幹細胞が分化あるいは死滅しただろうことを示唆するものである。
【0162】
DC−29レトロウイルスを用いたレトロウイルス感染
DC−29レトロウイルス感染したLTBMCにおける細胞産生の速度論
DC−29レトロウイルス上清に感染した培養中で産生される細胞数はHGF依存性であった。IL−3+GM−CSF+Epo補給培養では、培養の10週間を通じて週毎に1.5〜4×106細胞が産生された。IL−3+GM−CSF+Epo+MGF補給培養ではさらに多いが、ヒブリカイン+Epo補給培養では最も高い細胞産生が見られた。興味深いことは、IL−3+GM−CSF+Epoは補給されているがDC−29レトロウイルス上清が添加されていないコントロール培養は、DC−29レトロウイルス上清が添加されている類似の培養よりもそれほど多くなかったことである。IL−3+GM−CSF+Epo、IL−3+GM−CSF+Epo+MGF及びヒブリカイン+Epo培養(ウイルス添加)における細胞産生は、それぞれ5%、1%及び1%の有意性において、コントロール培養(IL−3+GM−CSF+Epo,ウイルス無添加)よりも著しく高かった。レトロウイルス上清添加培養における産生の増加の一部は、3T3系パッケージング細胞系により産生されることが知られているMGF(c−kitリガンド)などの成長因子の存在によるものであろう。
【0163】
上清DC−29ウイルス添加LTBMCにおけるレトロウイルス感染の解析
レトロウイルス感染の効率は、1.6mg/ml G418中のCFU−GM生存、レトロウイルス感染前に全骨髄細胞を死滅させる濃度により評価した。第8週(6週間後に感染)におけるCFU−GM生存の平均パーセンテージは全ての感染培養において高いものであった(表17参照)。
【0164】
【表17】
【0165】
IL−3+GM−CSF+Epoの組合せにMGFを添加すると、培養の第8〜10週における感染効率が増大し、培養物の一つは7.7%のG418耐性コロニーを含有していると考えられる。第8週におけるデータからは、ヒブリカイン+Epo補給培養物が高い感染効率を有していたことが示唆されるが、第10週のデータでは感染が全く起こらなかったことが示唆される。第10週では、いくつかの培養物から除去されたCFU−GMの数は減少し、およそ10%感染において、CFU−GMはほとんど、あるいは全く見られないと予想されたであろうことに注目されたい。さらに、1.5mg/ml G418は極めて高い抗生物質処方量であり、わずかに次善の最適レベルでさえトランスフェクトされたG418耐性遺伝子の効能を封じ込めるにおそらく十分である。したがって、これらの培養における造血幹細胞への遺伝子転移の効率は、いくつかのサンプルにおいては少なくとも7.7%であるが、おそらく45%程度又はそれ以上であろう。
【0166】
非付着性前駆細胞産生の速度論
これらの試験におけるレトロウイルス感染の解析は、前駆細胞をアッセイして幹細胞の存在、感染及びサイクリングを推察することによるので、培養におけるHGFの前駆細胞産生への効果を調べることは重要である。また、このレトロウイルス試験において、MGF及びヒブリカインは他のHGFとの組合せで用いられるものである。従来MGF及びヒブリカインは共にLTBMCが補給された急速潅流化HGFには用いられておらず、したがってそれらの造血形成への効果を調べることが必要である。
【0167】
前駆細胞産生は、HGF補給に大きく依存した。培養物から除去されたCFU−GMの数を表18に示す。
【0168】
【表18】
【0169】
レトロウイルス上清の添加により、前駆細胞除去はレトロウイルス上清無添加の場合の2.2倍に増加した。レトロウイルス上清及びIL−3+GM−CSF+Epo+MGF又はヒブリカイン+Epo補給培養物において除去されたCFU−GMの数の増加は、それぞれ非感染コントロールの4.2 及び3.8 倍大きいものであった。CFU−GMの除去は、接種されたCFU−GMの数と比較した時、全てのウイルス補給培養において1%レベルの有意性で統計学上重要であった。
【0170】
培養における前駆細胞産生
CFU−GM区分における集団バランスは、MGF又はヒブリカインのIL−3+GM−CSF+Epo補給培養への添加が、CFU−GMプールにおいてかなりの正の効果を有するものであることを示す。IL−3+GM−CSF+Epoの組合せにMGFを添加することは、MGFが補給されていない同様の培養と比較して、CFU−GM除去を1.9 倍、CFU−GM分化を0.5 倍増大させた(表19参照)。
【0171】
【表19】
【0172】
ヒブリカイン+Epoの組合せは、CFU−GMの産生及び分化において極めて著しい効果を有していた。ヒブリカイン+Epoは、IL−3+GM−CSF+Epoが補給された従来の最適培養と比較して、CFU−GM除去において1.8倍、CFU−GM分化において3倍以上増大させた。また、ヒブリカイン+Epoは、IL−3+GM−CSF+Epo+MGFの組合せのほとんど2倍の数の顆粒球及びマクロファージの産生を誘発した。このことは、ヒブリカインが顆粒球マクロファージ系の強力な誘発剤であることを示している。
【0173】
レトロウイルスをコードするCD18に感染した幹細胞から産生される好中球の解析 CD18欠損骨髄は、上記のようにして初期造血細胞を濃縮し、それから1.0ng/ml/日 GM−CSF及び1.0ng/ml/日 IL−3及び40U/ml/日 IL1α及びCD18レトロウイルス産生系上清を補給して、50%毎日培地交換を行ないながら14日間培養した。第15日目から、レトロウイルス上清を添加しないこと以外は同様の条件下で該細胞を培養した。非付着性細胞を培養物から毎週除去し、標準法によりビオチン添加した抗CD18モノクローナル抗体を用いたフローサイトメトリーで細胞表面CD18の存在を調べた(Updyke et al Meth. Enzymol. (1986) 121: 717-725)。このアッセイにより、CD18欠損骨髄細胞はいかなる細胞表面CD18も発現しなかったが、レトロウイルス感染培地から生じた好中球及び単核細胞は細胞表面CD18をまさに発現した。3連の培地において、細胞表面CD18の発現は、6週間で3.5%/5%/2%であり、11週間で11%/28%/3%であった。11週間目に培地中に存在する好中球及び単核細胞は、CFU−GM前駆細胞から10〜14日だけ早く発生したので、これらのデータは、培養の初めの2週間で、ヒト造血幹細胞が組換えレトロウイルスと、うまく安定にトランスフェクトされたことを示す。
【0174】
この結果の解釈において重要なのは、数種のグループはヒト造血前駆細胞へのレトロウイルス仲介遺伝子の転移を実証しているが、ヒト造血幹細胞への遺伝子転移は示されていないことである。アッセイに必要な前駆細胞が存在しなかったので、急速な細胞産生の減衰、ゆっくりと潅流されたヒトLTBMCにより、感染分析は制限されていた。
【0175】
本研究において、レトロウイルスの感染は、初めに哺乳動物細胞抗生物質G418の存在下、メチルセルロース中のLTBMCから除去された増殖細胞により評価した。SAX又はDC−29レトロウイルスに感染したNEO産生物を発現する前駆細胞は、高濃度のG418中で生存しコロニーを形成できるであろうが、感染していない細胞は高濃度のG418中で死滅するであろう。また、ウイルス添加終了後6週間以上高濃度のG418中で生存する前駆細胞の産生においては、それらの細胞が少し前により原始的な細胞(幹細胞)から分化していたことが必要であり、したがって、幹細胞が感染していることが示唆される。同様に、好中球及び単核細胞の表面に発現し、かつ培養の第11週の間に産生されるCD18では、感染期間に存在する全ての成熟細胞、前駆体及びクロノジェニック(Clonogenic)な前駆体が培養中4〜5週間で死滅してしまうので、原始的な造血幹細胞は培養の初めの2週間の間に感染していることが必要である。
【0176】
ここにおいて用いられるレトロウイルス感染に関する解析では、NEO遺伝子産生物が十分に発現しないので、レトロウイルスに感染した細胞のパーセンテージを軽視することができる。転移された遺伝子の産生物の発現の低さは、従来、ヒト及び霊長類モデルにおける問題とされてきた。したがって、本研究における感染前駆細胞のパーセンテージは、おそらく保守的な評価である。
【0177】
高[G418]中で生存していた前駆細胞のパーセンテージは、SAXレトロウイルス上清で感染させ、かつIL−3+GM−CSF±IL−1αを補給した培養の10週目において、およそ40%であった。これらの結果は、高パーセンテージの幹細胞が、培養の最初の2週間でレトロウイルス上清を補給した培養中で感染したことを示す。
【0178】
DC−29レトロウイルスで感染した前駆細胞のパーセンテージは、ウイルス添加終了後初めの4週間においてIL−3+GM−CSF+Epo+MGF及びヒブリカイン+Epo培地中では高いものであった(0〜21%)。この高レベル前駆細胞感染は、おそらくレトロウイルスによる前駆体及び原始細胞の直接感染によるものであった。高G418濃度中で生存している前駆細胞のパーセンテージは、ウイルス添加終了後4週間で減少したが、2週間後には高G418濃度中での生存は0〜22%に戻った。
【0179】
このDC−29試験におけるG418耐性前駆細胞の産生は、実際にDC−29レトロウイルスに感染した前駆細胞のパーセンテージを軽視できるであろう。感染したコロニーを選択するために用いられる高濃度のG418(1.6mg/mlのG418)は、SAX感染試験で用いられたものの2倍であり、生存するためには、NEO遺伝子産生物、ネオマイシンホスフォトランスフェラーゼの高い発現レベルが必要である。
【0180】
興味深いことに、上清を含有するSAX又はDC−29レトロウイルス、HGF IL−3+GM−CSF±Epo、IL−1α、又はMGF、又はヒブリカイン+Epoの組合せのいずれかを補給したLTBMCにおいて、G418中で生存している前駆細胞のパーセンテージは、ウイルス添加終了後6〜8時間で増加した。G418生存前駆細胞のパーセンテージが培養の後の段階になるに従って増加してゆくそのメカニズムはわかっていないが、一つの可能性としては、培養の初めの2週間に感染した幹細胞が、培養が進行するに従ってより活性になったということである。これはG418中で生存している細胞のパーセンテージを効果的に増加させるであろう。もう一つの可能性としては、最初の感染培養期間中に直接トランスフェクトされた前駆細胞とは異なる、より発現可能な組込み部位を有する幹細胞からの分化により、NEO遺伝子の発現が、培養中の後期に産生された前駆細胞において増大したのだろうということである。したがって、おそらくいくつかの原因によると考えられるが、培養におけるこの高レベルの前駆細胞生存率は、幹細胞がこれらのLTBMC中で感染したことを強く示唆するものである。
【0181】
さらに、これらのデータは、本発明において開示された培養条件下で造血幹細胞が培養中に増殖しており、レトロウイルスで転移された遺伝性物質をこれらの細胞へ組込むことができることを証明するものである。前駆体はこれらの幹細胞から連続的かつ活発に産生され、これら前駆体の多くはトランスフェクトされた遺伝子を含有し、発現した。これらのデータは、遺伝学的に改変されたヒト造血幹細胞が存在し、これらの培養中で増殖していたことを示すものである。
【0182】
IV.実験
I.形質転換細胞の形成
成長因子ヒトGM−CSF(Wong, Science (1984) 228:810-815)を原核性発現ベクターに挿入した。hGM−CSF cDNA(EcoRI〜AhaIII,およそ700bpフラグメント)をpSP65のEcoRI〜PstIフラグメントにクローン化した(Melton, Nucl, Acids Res. (1984) 2: 7035-7056)。得られたプラスミドをSP65GM−CSFとした。マウス メタロチオネインプロモーター(Glanville, Nature, (1981) 292: 267-269)をEcoRI及びBglIIで消化し、該プロモーターを含有するおよそ2kbのフラグメントをpSP65のEcoRI〜BamHIフラグメントに挿入してp65MTを作製した。その後、pSP65GM−CSFをEcoRIで消化し、DNAポリメラーゼIのクレノウ(Klenow)フラグメントを突出部に充填し、得られた連結されたDNAをHindIII で消化してGM−CSFのコード領域からなる700bpフラグメントを単離することにより、プラスミドpMT GM−CSFを構築した。このフラグメントをp65MTのSalI充填/HindIII 部位にサブクローンした。その後、メタロチオネインプロモーター及びGM−CSFコード領域を含む2.7kbフラグメントを単離し、SV−40プロモーターが除去されたpSV2neo(Southern and Berg, J. Mol. Appl. Genet (1982) 1: 327)においた。この結果、GM−CSFコード配列の下流にSV−40ポリAシグナルが存在するものが得られた。
【0183】
抗生物質ゲンタマイシン(G418)への耐性を付与するネオマイシン耐性遺伝子は、およそ3kbPvuII〜EcoRIフラグメントを単離し、EcoRIリンカーをPvuII部位におくことにより、pSV2neoから取り出した。EcoRI末端を有するneo耐性遺伝子をGM−CSF発現プラスミドのEcoRI部位にサブクローン化してプラスミドMTGM−CSFneoを作製した。
【0184】
単独で、及びSV−40プロモーター及びポリA部位のコントロール下、ギボンエイプ(gibbon ape)IL−3遺伝子をコードするプラスミドと共にトランスフェクションするもの(Yang, Cell (1986) 47:3-10)としてのプラスミドMTGM−CSFneoを、線形化DNAの電気穿孔法によりアフリカミドリサル細胞系CV1及びマウス細胞系NIH 3T3細胞へトランスフェクトした。形質転換細胞を、500mg/mlのG418を含有する培地選択により選択し、単離し、AML−193細胞(Adams et al, Leukemia (1989) 3: 314)を用いる上清のバイオアッセイにより、GM−CSF又はIL−3の産生でスクリーンした。その後、陽性の系のうちいくつかをでデクスター培養におけるヒト骨髄細胞用ストローマとして使用した。
さらに、オカヤマのカルシウム/リン酸法(Chen, Mol. Cell. Biol.(1987) 7: 2745-2752)を用いて正常マウス骨髄細胞を上記プラスミドとトランスフェクトし、誘導遺伝子を効果的に発現することがわかった。
【0185】
トランスフェクトされた線維芽細胞によるGM−CSF及びIL−3分泌を調べた。無血清72時間培養上清をNIH−3T3細胞より得、ウサギ抗GM−CSF又は抗IL−3抗体を中和することにより阻害可能なターゲット細胞における3Hの取り込みによってhGF分泌をアッセイした。20mg/ml GM−CSFにより誘発された増殖を100ユニットGM−CSFとし、10ng/ml IL−3により誘発された増殖を100ユニットIL−3とした。共にトランスフェクトされた細胞は、約35U/mlのGM−CSF及び約57U/mlのIL−3を産生した。
【0186】
II.潅流チャンバー
潅流チャンバーは、分解や生体融合することなく加圧滅菌できるようにデルリン(Delrin)キャップが付いたガラス製のシリンダーである。キャップには円柱状のグルーブがあり、そこへガラスシリンダーがフィットする。グルーブの底部にはチャンバーのルーメン(lumen)をシールするためのOリングがある。キャップにはいくつかの穴があり、そこにはルアー(Luer Lok)フィッティングがあり、さらにそのルアーフィッティングには付着性及び/又は非付着性細胞をサンプリングするために、チャンバー中央部へ伸びるチューブだけでなく、培地及びガス供給ラインも配置されている。このキャップは、120°間隔でガラスシリンダーの外側に配置されている3つの長いボルトで取付けてあり、組立をしっかりさせるために蝶ナット及びワッシャーが用いられている。
【0187】
チャンバーはサイドの貯蔵槽に掛ける。ループにはポンプ、オンラインセンサーのチャンバー、酸素供給器、及びサンプル注入孔、さらにサイド培地貯蔵槽がある。そして、サイド貯蔵槽中の培地は、セパレートポンプを用いてゆっくり交換される。この構成は、酸素供給器及び潅流チャンバーを通じて培地交換速度及び流速の独立制御を可能にする。前者は培地組成の長期変化を制御し潅流するために用いられ、後者はチャンバー中の溶解酸素の膨張及びフローパターンを制御するために用いることができる。小さなメッシュのポリスルフォネート膜を用いれば、チャンバー内でのプラグフロー及び非常に正確な量の成長因子及びバイオリアクターに導入させたい他の特別な成分の供給を正確に制御することが可能になる。
【0188】
トランスフェクトされた間質細胞は、寸断したコラーゲンスポンジ層の上に接種されるか、あるいは該間質細胞はコラーゲンでプレコートされた5μ孔のポリカーボネートフィルターの片側に置かれ、間質細胞が長時間フィルターに付着できるようになっている。細胞は、細胞質突出物を孔を通じて送りながら、細胞が片側に集密するまで適当な栄養培地中で生育できるようになっている。その後、骨髄細胞を膜のもう一方側に接種し、孔を通過して入り込んできた細胞質突出物に幹細胞を付着させる。
【0189】
ループのチャンバー及び成分を加熱滅菌した後、滅菌環境下でリアクターを組み立てる。そして雑菌混入のサインをモニターしながら、培地をサイドループ及びチャンバーを通じて数日間循環させる。そしてバイオリアクターの中央部に、細胞外マトリックスのみ、もしくは間質細胞を含有する前接種された細胞外マトリックス支持体を接種する。その代謝挙動及び/又は成長因子の反応性をモニターしながら、間質細胞をその後数日間チャンバー内に保持してもよく、結果が満足できるものであれば、骨髄細胞を接種するか、或いはすぐに骨髄細胞を接種する。いずれの場合においても、細胞層は潅流チャンバーの中央底部に保持される。
【0190】
細胞にはさらに細胞外マトリックスを接種し、細胞層は支持体に付着する。膜が使用される場合、チャンバーは倒置させてもよく、その場合、細胞層は中央部の天井に位置する。この配置において、間質細胞への付着がゆるむと、成熟している細胞は中央チャンバーの底部に沈む。そして、非付着細胞は、培地潅流圧力によって作動する出管の方への一定の細胞の流れにより採取される。
【0191】
代表的な実験においては、第1日目にチャンバーでは、寸断されたコラーゲンスポンジ支持体上にNIH−3T3細胞が接種される。最初の40日間は、潅流速度及び他の操作変数を調整した。40日目にはかなり安定した状態になり、それは約20日間持続した。64日目に、チャンバーに33×106ヒト骨髄細胞を接種した。初めの10日間で、採取された細胞の数は減少し、3日毎に約7〜8×105細胞が産生される安定した状態に落ち着いた。フローサイトメトリー解析は、一定のフラクション、すなわち採取細胞の約20%はHLA−DR陽性であることを示した。90日目には、ポンプの故障が起こり、pHは一夜で6.9以下に下がった。潅流速度を元に戻すと、非付着性細胞の産生は回復し、雑細菌混入が起こる前の安定した状態の産生速度に近づいた。この時点で本研究を終了した。
【0192】
上記の結果は、潅流チャンバーでは生体外造血形成を行なうことができ、pH下落後造血形成を生体外で元に戻すことができるであろうことを証明し、またグルコース濃度のデータは、酸素添加が制限されることを示す乳酸塩濃度の増加が起こらずに接種後にグルコース濃度が低下するので、おもに造血細胞はグルコース上で好気的に生育することを示した。グルコース/乳酸塩(嫌気性)代謝は、主にNIH−3T3間質層によると考えられる。同様に、一度造血細胞数がレベルダウンすると、グルタミン及びアンモニア濃度は接種前のレベルになり、これは骨髄細胞によるグルタミン消費が間質層と同レベルであったことを意味する。
【0193】
III.代謝生成物のモニタリング
グルタミンとアンモニアのみならずグルコースと乳酸塩の消費及び形成速度もトランスフェクトしたNIH−3T3細胞について求めた。(培地はIMDM+20%FCSであった。)グルコース消費の増加は毎日栄養補給したT−フラスコについて観察されただけであったが、低頻度栄養供給培地では全てが、同じゆるやかに減少するグルコース摂取速度パターンに従う。毎日50%交換した培養を、第18日目に毎日100%交換スケジュールに変更したところ、グルコース消費はただちに増加し、続いて第1日目から毎日100%交換した培地において観察されたものと同じ傾向を示した。グルコースにおける乳酸塩の収率が本質的に一定である場合、乳酸塩産生速度は同様のパターンに従う(0.9乳酸塩/グルコース;主に嫌気性間質代謝であることを示している。)
【0194】
グルタミン及びアンモニア濃度は、グルコース/乳酸塩代謝に類似したパターンを示す。37℃におけるグルタミンの化学的分解に対して補正された値を用いると、相対摂取速度を表わすグルタミン消費速度対グルコース消費速度は一定で、約1:8 グルタミン:グルコースである。予測される最適比率は酸素摂取速度に伴って変化し、最適摂取速度が増加するに従って、比率は下がる。
【0195】
同じようなことが、正常骨髄間質線維芽細胞から得られるグルコース/乳酸塩代謝データからも支持された。低頻度培地交換の条件下においては、培養は根本的には嫌気性であり、高い安定したレベルの乳酸塩の生成がすぐに達成され、かつ持続した。より頻度の高い培地交換では、細胞代謝はより急速になり、グルコース消費及び乳酸塩産生は増加した。自然な化学的分解のデータを補正した後で、グルコース消費が全く検出されないことが観察された。3T3細胞及び正常ヒト骨髄細胞の両者にたいして、上記の血清/培地交換速度である場合、細胞は分割し続け、ぎっしりと詰まり、それは臨界的な交換スケジュールである。
【0196】
さらに血清の潅流速度対栄養素の還流速度の相対的な重要性を確認するために、以下の実験を行なった:1)毎日交換される20%血清含有培地の入った一組のT−フラスコ;2)2組のT−フラスコ,一組には20%血清及び隔日交換される培地が入っており、もう一組には10%血清及び毎日交換される培地が入っている;3)2組のT−フラスコ、一組は10%血清及び隔日交換される培地が入っており、もう一組には5%血清及び毎日交換される培地が入っている;4)2組のT−フラスコ、一組には5%血清及び隔日交換される培地が入っており、もう一組には2.5%血清及び毎日交換される培地が入っている。血清交換速度は各グループで同じであるが、培地を含有する栄養素の交換速度は様々である。これらの実験の結果から、血清の交換速度が臨界的であることが示される。実験1)については、グルコース消費が増加し、第4日付近で実質的に安定して約9.5ミリモル/日の速度になったが、それ以外の全ての場合には、2倍量の血清を用いて培地を隔日に交換、又は半量の血清を用いて培地を毎日交換したこととは関係なく、グルコース消費はグループIの最初のグルコース消費以下からスタートして減少し、実質的に直線となった。このことは、間質細胞の代謝生育挙動に影響する血清又は一つ以上の血清成分の臨界的な潅流速度が必要であることを支持する。
【0197】
上記の結果から、効果的な方法でバイオリアクター内で造血細胞を生育させることができるであろうことは明確である。間質細胞は類似又は異種の提供源から提供することができ、そこにおいて間質細胞は重要な成長因子を提供するための遺伝子でトランスフェクトされている。この方法では、細胞の生育を補助するために、血清を培地に添加する必要はない。造血細胞を間質細胞から分離することを可能にする方法で支持体に付着する間質細胞を提供することにより、造血細胞は連続的に採取されて利用される。因子の組合せを適当に選択することにより、造血細胞の特異的な系統が生育されるであろう。さらに、所望により、遺伝子を造血細胞へ導入するためのトランスフェクトウイルスの貯蔵槽として間質細胞を提供してもよい。
【0198】
本明細書中で引用されている全ての刊行物及び特許出願は、ここにおいて引例として加入するものとし、各刊行物及び特許出願を明確に独立して引例に加入されるものとして示した。
明らかに、上記の技術から鑑みて本発明の多くの改変及び変形が可能である。したがって、添付する請求の範囲内で、本発明はここにおいて特に記載した以外の方法でも実施できることが理解される。
【図面の簡単な説明】
【0199】
【図1】灌流室の概略を示す。
【図2】灌流培地通路の概略及びフロー・ダイアグラムを図示している。
【図3】3aは細胞分離のせん断応力を測定する為のフローチャンバーの概略を示す。 3bは3aのフローチャンバーの側面を示す。 3cは造血細胞のせん断応力・プロフィールのグラフである。
【図4】4a及び4bは造血細胞の増殖、分離用のフローチャンバーの上面及び側面図である。
【図5】5a及び5bは基質細胞の連続増殖を可能にするために障壁を順番に取り除いたフローチャンバーの概略を示す。
【技術分野】
【0001】
この発明は培養動物細胞の増殖及び形質転換の為の、特に培養造血細胞の増殖及び形質転換の為の方法、反応器及び組成物に関するものである。
【背景技術】
【0002】
正常成人の循環血液細胞は赤血球、白血球、血小板及びリンパ球を含むがこれらの細胞は骨髄内の細胞に由来する。骨髄内の細胞は非常に未分化な前駆細胞と呼ばれる細胞に由来する。前駆細胞はメチルセルロースや寒天のような半固体の培地中で1〜3週間培養すると分化した血液細胞が集まったコロニーを生ずることより検定出来る。前駆細胞それ自体も一種の前駆細胞である幹細胞と呼ばれる細胞に由来している。幹細胞は分裂時に幹細胞自身の再生と前駆細胞への分化の両方を行なう能力を有する。それ故分裂する幹細胞は未分化の幹細胞といくぶん分化の進んだ前駆細胞を生産する。血液細胞を生産する他に幹細胞は骨芽細胞と破骨細胞をも生産すると考えられ、多分他組織の細胞も幹細胞から派生するであろう。この明細書はヒト造血幹細胞の最初のインビトロ培養を成功させた方法及び組成物を記述する。この方法により造血幹細胞は増殖し、前駆細胞及びより成熟した血液細胞に分化した。
【0003】
1970年代の後半、造血骨髄細胞をインビトロで増殖させる液体培養系が発達した。この培養系は、正常及び白血病の造血の分析及びレトロウイルス媒介による遺伝子導入の様な骨髄の実験的操作に非常に重要な価値を持つ可能性がある。この培養系を用いてマウスの造血の詳細な分析が可能になりマウス系の細部が理解出来る様になった。更に培養マウス骨髄細胞へのレトロウイルス遺伝子導入を可能にした。このことはマウス造血細胞に標識を付けることを可能にし、多機能幹細胞の存在を証明し、白血病発生の過程に於ける種々の遺伝子の研究を可能にした。
【0004】
しかしレトロウイルス遺伝子を培養マウス骨髄細胞に導入することは可能になったが、培養ヒト骨髄細胞に遺伝子導入することは未だ不可能である。なぜなら現在に至るまでヒト骨髄細胞の長期培養は、その培養期間を長く出来ない事と、幹細胞を生存させておき前駆細胞を生産する能力を維持する事が出来ないという制限があるからである。
【0005】
ヒト骨髄細胞の液体培養は最初は造血能力が制限されており、生産される前駆細胞や分化した血液細胞数は減少し、6週から8週の培養で細胞生産は停止した。その後この系に種々の修正がなされたが僅かな改善が見られただけであった。この問題を解決することは非常に価値のあることである。もし成功すればヒト幹細胞及び前駆細胞を増加させることが出来、骨髄移植、化学療法からの保護に使用し、そのような細胞を選択、操作して遺伝子導入に使い、また分化した血液細胞を生産し、輸血療法に用いることも可能になる。
造血とインビトロ骨髄液体培養の研究は付着層内の線維芽細胞と内皮細胞が中心細胞基質の要素であることを明らかにした。これらの細胞は増殖している造血細胞に付着する場を与えると共に前駆細胞の増殖と分化を促進する造血細胞増殖因子を誘導することが出来る。これらの造血細胞増殖因子は、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)及びインターロイキン−6(IL−6)等を含む。
【0006】
しかしそのような付着層上でのヒト骨髄細胞インビトロ培養はたいていはうまく行かなかった。マウスや木ネズミ(tree shrew)の骨髄細胞は液体培養出来るがヒト液体骨髄培養は非付着性の造血前駆細胞或いはクローンを形成する前駆細胞を6〜8週間以上十分な量を生産しない。3〜5カ月保持する培養が報告されたが、幹細胞から前駆細胞を安定して4〜6週以上生産し続ける細胞培養は報告されていない。
【0007】
更に、非付着性細胞及び前駆細胞の生産はこれらの培養の短い生存期間中に典型的に低下するので、幹細胞が生きのびているのか増殖しているのかはっきりしない。分離して調べて見ると刺激を受けない骨髄基質細胞は、検出し得る諸造血増殖因子(HGFs)を殆ど分泌しない。
これらの培養中に安定した前駆細胞及び分化した血液細胞の生産が見られないことから、これらの培養は幹細胞の連続的再生、拡大を支持出来ないものと考えられた。それゆえこれらの培養はある必須の幹細胞刺激因子を欠いているか及び/又は新しい幹細胞阻害因子を含んでいるものと仮定された。HGFを検出できない事と基質細胞が誘導されていない事の説明はなされたが、HGFの検出できない事とヒト骨髄液体培養が他の種の動物のそれと比較して成功しない事を説明する、検証され得る仮説は、インビトロの培養系は付着性骨髄基質細胞がインビボで持つ総合造血支援機能を提供しないと言うものである。
【0008】
骨髄移植のための幹細胞と前駆細胞の増殖はヒト長期骨髄培養に応用出来る可能性がある。ヒトの自己及び同種骨髄移植は現在、白血病、リンパ腫或いは他の生命を脅かす病気の治療法として行なわれている。この治療法に於いて十分な細胞を移植するためには、供与者の骨髄を大量に採集しなければならない。
幹細胞と前駆細胞を増殖させる培養は大量の骨髄供与の必要性を減少させ、小量の骨髄の供与を受け、受血者に輸血する前にインビトロで幹細胞と前駆細胞を増殖させることを可能にする。小量の幹細胞及び前駆細胞が血流中に存在することが知られている。もしこれらの幹細胞と前駆細胞を泳動法により採集し、増殖させる事が可能であれば、移植に必要な数の幹細胞と前駆細胞を末梢血から採集し、骨髄供与の必要性を除くことができる。
【0009】
骨髄移植には患者の体重1kg当り約1×108から2×108の骨髄単核細胞を移植しなければならない。これは体重70kgの受血者は70mlの骨髄供与を必要とすることになる。70mlの骨髄は供与者の骨髄のほんの一部ではあるが、供与過程で供与者に強度の処置を必要とし、多量の血液が失われる。もし幹細胞と前駆細胞を10倍に増やすことが出来れば、供与過程は大巾に軽減し、末梢血から幹細胞と前駆細胞を採集しこれを増殖させて用いることが可能になるかも知れない。
【0010】
前駆細胞の増殖が可能になれば、化学療法の補助として有用であろう。これもまたヒト骨髄長期培養の応用のひとつである。癌治療医のジレンマは殆どの化学療法剤は細胞分裂している全ての細胞を殺すことにより癌細胞を破壊すると言うことである。骨髄は体内で最も増殖の盛んな組織のひとつで、それゆえ最初に化学療法剤で損傷をうける器官であることが多い。その結果血液細胞生産能は化学療法を行なっている期間急速に破壊されるので、化学療法を中止し造血系が血液細胞を回復するのを待たなければ、患者に化学療法を再び行なう事は出来ない。静止幹細胞が活性化して化学療法を再開できるに十分な白血球数に回復するまでは1カ月かそれ以上かかるかも知れない。そして2回目の化学療法中も血球数の降下は繰り返される。化学療法の間隙期間に血球が再生されるが、不幸にして癌細胞もこの期間に増殖し自然淘汰されて化学療法剤に対して耐性を持つようになる可能性がある。
【0011】
化学療法間の間隙時間を短くするために大量の前駆細胞と未分化の血液細胞を患者に戻すことが出来る。この方法により患者の血球数が低くなる時間を大巾に短縮でき、化学療法の再開を早めることが出来る。化学療法の期間が長いほど、また化学療法間の間隙時間が短いほど癌細胞を殺すことに成功する可能性は高くなる。
【0012】
前駆細胞を増殖させるに必要な造血細胞は骨髄回収か或いは末梢血採集に依って得られる。骨髄採集により約4×105CFU−GM前駆細胞を得ることが出来る。5リットルの末梢血から泳動により105CFU−GMが得られるが、供与者をGM−CSFで前処理することにより106CFU−GMに増やすことが可能である。患者の急速な回復には1×108から5×108CFU−GMが必要であるが、これは通常の骨髄供与或いは末梢血供与で得られる量の100倍から1000倍である。それ故、骨髄或いは末梢血のCFU−GMを100倍から1000倍増やすことが出来れば、抗癌剤投与、癌の治療に重要な影響を与えるであろう。
【0013】
遺伝子療法は医学の中で急速に進歩している分野であり、計り知れない臨床応用の可能性を持っている。遺伝子療法は病気の治療に数多くの応用の可能性が考えられ、それに関して広範囲に論評されている。例として下記の参照文献を挙げられる:
Boggs, Int. J. Cell Cloning. (1990) 8:80-96、Kohn et al, Cancer Invest. (1989) 7 (2):179-192、Lehn, Bone Marrow Transp. (1990) 5:287-293、及び Verma, Scientific Amer. (1990) pp. 68-84。
遺伝的形質転換したヒト幹細胞は遺伝子療法の手段として臨床医学に広く応用出来る可能性がある。
【0014】
遺伝子療法は従来の医療から発展してきた臨床療法に新しい方法を加えた。何世紀もかけて進歩してきた最古の医療は粗放な外科処置と天然混合物を医薬として投与することであった。前世紀になって生化学的薬理学が医療の主たる方法として現われた。この範例の下では純粋な生化学的分子が患者に与えられる。一般的にそのような薬剤は毒薬(例えば抗生物質或いは抗癌剤など)、内在の受容体を刺激する生理的模倣剤(例えば阿片やアドレナリン拮抗剤)或いは内在受容体を刺激する生理的拮抗薬(例えば血圧降下剤や麻酔剤)である。
【0015】
遺伝子療法はその定義によれば医療目的で細胞内に遺伝子を挿入することである。遺伝子療法の背景にある原則は薬理学的物質を投与するよりもむしろ、機能する遺伝子を導入して、そのRNA或いは蛋白生産物が目的細胞或いは組織に希望する生化学的効果をもたらすことである。遺伝子療法が古典的な生化学的薬理学に較べて有利である可能性のあるのは次の理由による。第一に、導入された遺伝子は蛋白やRNAを含めて非常に複雑な分子を造ることが出来る。これらの分子自体を投与して目標に到達させるのは非常に困難か不可能である。次に、希望する遺伝子を特定の目標細胞に制御導入すれば特定の組織でその遺伝子生成物の生産を制御出来る。最後に遺伝子療法は、遺伝子が目標細胞とその子孫細胞内で機能し続けるので原則として個体内では永久的である。
【0016】
遺伝子治療を成功に導く為には幾つかの問題を解決しなければならない。第一に、希望する治療遺伝子を選択した細胞に導入することが出来なければならない。第二に、その遺伝子は目標細胞内で十分に発現し、遺伝子の生成物が適当なレベルに達しなくてはならない。最後にRNA或いは蛋白生成物は目標細胞内で適当に処理されて機能を持ち、臨床治療としての意味を持たなければならない。ヒト細胞に遺伝子をインビトロで導入する数種の方法を表1に示してある。
【0017】
【表1】
【0018】
相同組換えの様な他の方法も多くの研究室で開発された。遺伝子療法の研究はここ数年来進行中でインビトロでの数種の細胞タイプに於ける実験から、動物実験に進み最近ヒトの臨床試験の段階に入ったものもある。
【0019】
造血系は遺伝子療法の導入系としては理想的な対象である。造血細胞は骨髄吸引或いは末梢血を採集することにより容易に得ることが出来る。インビトロでの遺伝子導入が成功すれば、処理された細胞は静脈より再注入され骨髄に到達し、そこで増殖する。分化した血液細胞は体内を循環しているので遺伝子を改変された細胞は特定の遺伝子生成物を希望するどの組織にも導入することが出来る。
【0020】
最も重要な事は造血組織は非常に強い(多分無限の)自己再生能力を持つ幹細胞を含んでいる事である。この事はもし遺伝子がこれらの幹細胞に安定して導入されれば造血組織に再注入した際これらの変化した幹細胞は増殖し、骨髄を新しい遺伝子を表現している細胞で再び満たすことが出来る事を意味する。これにより長期間の、多分一生の間の希望する遺伝子生成物の供給を可能にするであろう。同様にして他の組織に存在する幹細胞や胚幹細胞に遺伝子を導入する事に成功すれば同様に長期にわたる、希望する遺伝子生成物の供給が可能になる。
【0021】
造血幹細胞の遺伝子療法は造血系に特有の病気にも他の器官系の病気にも広い応用が可能である。造血系の病気は遺伝性であっても、後天的であっても遺伝子療法での治療が可能である。例えばアルファ及びベータ・タラセミアのようなヘモグロビン異常はグロビンのアルファ又はベータ鎖をコードする遺伝子と高度の組織特異性を与える制御配列を導入する事により治療する事が出来る。(Grossveld et al, Cell (1987) 51:975-986参照)。同様に鎌型赤血球性貧血は胎児グロビン遺伝子を造血幹細胞に導入する事により治すことが出来る。なぜなら制御された胎児ヘモグロビンの高度の発現は鎌状ヘモグロビンの存在にも拘らず赤血球の鎌型化を防ぐに十分であるからである。(Sunshine et al, J. Molec. Biol. (1979) 133:435 参照)。
【0022】
白血球付着不全(LAD)或いは慢性肉芽腫病(CGD)のような、機能的蛋白不全によって引き起こされる好中球の遺伝子病は欠陥遺伝子や欠損遺伝子を高度の組織特異性を与える制御配列と共に造血幹細胞に導入する事により治療する事が出来る(Wilson et al, Science (1990) 248:1413-1416 参照)。フォンウィレンブランツ(von Willebrands')病のような血小板に関する遺伝病はウィレンブランツ(Willebrands)因子をコードする遺伝子とその遺伝子を発現させ、その生成物を分泌させる配列とを導入することにより治す事が出来る。
【0023】
アデノシン・デアミナーゼの欠損による重度複合免疫不全のようなリンパ球免疫不全病を造血幹細胞遺伝子療法で治療するのが特に適していることは明らかである。ADA遺伝子を用いて循環T細胞をレトロウイルス遺伝子療法で治療する方法は、この病気の患者の臨床上の免疫不全経験を減少させるのに成功したが、遺伝子を導入したTリンパ球は生体内では限られた寿命しか無いため効果は一時的なものであった(Kasid et al, Proc. Nat. Acad. Sci. (USA)(1990) 87:473-477、或いはCulver et al, Proc. Nat. Acad. Sci. (USA), (1991) 88:3155-3195 参照)。しかしながら、もし遺伝子を造血幹細胞に導入出来れば、これらの幹細胞から派生するTリンパ球はADA遺伝子を含み発現することであろう。それ故遺伝子導入した幹細胞は患者の生涯生存し増殖を続けるであろうから、T細胞ADA不全は幹細胞の一回の遺伝子療法で永久的に治療出来る(Wilson et al, Proc. Nat. Acad. Sci. (USA), (1990) 87:439-443 参照)。
【0024】
造血素の遺伝的な酵素異常の治療に加えて、幹細胞遺伝子療法は幹細胞とその子孫をウイルスや化学療法剤のような外来作用物から保護するにも有用であるかも知れない。例えばHIVのTATトランス活性化因子のTAR結合位置をコードするDNA配列を導入するとT細胞をHIVウイルスの感染から保護することが示された(Sullenger et al, Cell (1990) 63:601-608 参照)。この配列を造血幹細胞に導入し安定させればこれらの幹細胞から派生したHIVの伝染に比較的あるいは絶対的に抵抗を持つT細胞のプールが出来る。
同様にして多薬剤耐性遺伝子或いはメトトレキセート耐性遺伝子をコードする遺伝子をヒト骨髄幹細胞に導入するのに成功すると癌の化学療法の影響に比較的耐性のある幹細胞を造り出す事が出来る。これらの遺伝子操作された細胞で自己骨髄移植を行なった後は患者は抗癌剤に依って普通引き起こされる深刻な骨髄抑制から幹細胞が保護される事より抗癌剤による化学療法に耐えることが出来る。これにより患者は癌化学療法剤のより効果的な使用量を、より少ない毒性で受ける事が出来る。
【0025】
造血幹細胞遺伝子療法が白血病、リンパ腫及び再生不良性貧血等の後天的造血病に対して有用であることを予想する事はたやすい。一度これらの病気の遺伝的原因が発見されたなら、その遺伝子生成物を投与し細胞内の異常遺伝子の生成物を克服するか、或いは直接遺伝子異常を修正する(多分遺伝子を切り外し、置換することにより)様な遺伝子を導入することにより異常を修正出来るであろう。 しかし更に広いレベルでは造血幹細胞遺伝子療法は造血系以外の病気の治療にも有用である可能性がある。治療効果のある可溶性蛋白をコードするDNA配列の遺伝子導入をすれば、希望する量の、治療効果のある分子を恒久的に分泌する分化した血液細胞を生産することが出来る。例としてはこの方法は、例えばインスリン遺伝子を持つDNA配列とインスリン遺伝子の発現を適切に調節をする(多分血中グルコースのレベルの上昇に反応して)調節遺伝子配列を導入して糖尿病の治療に当てるのに有用である。全身性の高血圧は幹細胞にアンギオテンシン変換酵素、血管平滑筋のカルシウム・チャンネル或いはアドレナリン受容体等の拮抗的阻害剤として機能する分泌ペプチドをコードするDNA配列を導入することにより治療が可能である。アルツハイマー病は中枢神経系内のアミロイドのプラクを破壊する酵素をコードするDNA配列を幹細胞に導入する事により治療可能であるかも知れない。
【0026】
遺伝子療法の多くの応用、特に幹細胞への遺伝子導入はよく知られており、広範に論評されている(Boggs et al, 上記、Kohn et al, 上記、Lehn, 上記、及びVerma et al,上記)。分化したヒト幹細胞への治療的遺伝子導入(例えばTリンパ球への)のある程度の成功例が増加している(Kalsd et al, Proc. Nat. Acad. Sci. (USA) (1990) 87:473-477、Culver et al, Proc. Nat. Acad. Sci. (USA) (1991) 88:3155-3159 参照)。
【0027】
不幸にしてこの発明の以前にはヒト幹細胞への遺伝子導入をして安定化する事に成功した例は無い。幾つかの研究グループがヒト造血細胞へ、ヒトのレトロウイルスを使って遺伝子導入が可能であることを示したが、ヒト未分化造血幹細胞への導入に成功した例は無い。これはレトロウイルス媒介による造血細胞への遺伝子導入がある程度可能であるマウスに於ける実験と対照的である(Wilson et al, Pro. Nat. Acad. Sci. (USA) (1990) 87:439-443 参照)。
【0028】
ヒトの造血幹細胞遺伝子療法が成功しなかった主たる原因は幹細胞が分裂、増殖している状態でヒト造血細胞に遺伝子を導入出来なかった為であった。目標細胞に遺伝子を安定導入するのに成功するにはその目標細胞が少なくとも1回細胞分裂をしなければならない。それ故、導入したい遺伝子の存在下でも幹細胞が分裂していなければ、遺伝子は幹細胞に安定に導入されない。本発明以前は、体外でのヒト幹細胞の分裂と増殖を支持する系は存在せず、ヒト幹細胞の遺伝的形質転換も可能ではなかった。
【0029】
本発明に関連する文献を以下に記述する。米国特許No.4,721,096は造血細胞の増殖用の、基質細胞を含む三次元系を記述している。その特許の中に言及してある文献も参照のこと。グランビル等はマウスのメタロチオネイン−I遺伝子について記述した(Glanville et al, Nature (1981) 292:267-269)。ウオングらはヒトGM−CSFについて記述し(Wong et al, Science (1985) 228:810-815)、レミシュカ等はレトロウイルス媒介の遺伝子導入を造血幹細胞のマーカーとして利用し、これらの幹細胞が生体に移植された後どうなるかを記述している(Lemischka et al, Cell (1986) 45:917-927)。ヤン等はヒトIL−3について(Yang et al, Cell (1986) 47:3-10)、チェン等はプラスミドDNAによる哺乳動物細胞の形質転換について(Chen et al, Mol. Cell. Biol.(1987)7:2745-2752)、グリーヴェス等はヒトCD2遺伝子について(Greaves et al, Cell (1989) 56:975-986)、シヴィン等はCD34抗原について(Civin et al, J. Immunol. (1984) 133:1576)、マーチン等はヒトS−CSFについて(Martin et al, Cell (1990) 63:203-211)、フォレスター等はパラレル・フロー・チャンバーについて(Forrester et al, J. Cell Science (1984), 70:93-110)、またクロンベル等は静止培養に於いてCML細胞が失われる事を(Coulombel et al, J.Clin. Invest.(1986)75:961)それぞれ記述している。
【0030】
それ故ヒト幹細胞の生体外増殖及び安定遺伝的形質転換のための方法及び組成物、並びにヒト造血前駆細胞培養の最適条件を見出す必要がある。特に幹細胞増殖、前駆細胞増殖、また遺伝子療法の重要性を考えた場合、その必要性は更に重大である。不幸にして、今日までそのような目的を達成しようとする試みは期待外れであった。
【発明の開示】
【発明が解決しようとする課題】
【0031】
本発明の目的はヒト幹細胞の生体外増殖と安定遺伝子形質転換にのための、培地の条件、反応器等を包含する新規の方法を提供することである。
本発明の他の目的は(i)ヒト造血前駆細胞培養及び(ii)ヒト造血前駆細胞の安定遺伝子形質転換の最適条件の為の培地条件及び反応器を包含する新規の方法を提供することである。
【課題を解決するための手段】
【0032】
本発明はヒト幹細胞の生体外分裂と安定遺伝子形質転換に関する、及び/又は、ヒト造血前駆細胞培養の最適化に関する、培地の条件、反応器等を包含する新規の方法を本発明者が発見した事に基づいている。これらの方法はヒト幹細胞及び/又はヒト造血前駆細胞を液体培地中で培養する方法である。この方法に於いて、液体培地を24時間から48時間の間に1mlの培養を1mlの培地で連続的に或いは定期的に(好ましくは灌流により)置換し、代謝生成物を除去し、減少した栄養素を補い、培養を生理的に適当な状態に維持する。本発明の特に好ましい実施態様に於いて、上記の培地置換速度は急速に交換される培地に造血増殖因子を添加する条件に関連して決定される。
【0033】
本発明者らは急速に交換される培地に最適量の造血増殖因子を添加し、本発明に従って培地交換速度を増加した場合、驚くべきことに(1)培養中でヒト幹細胞が少なくとも5カ月間の長期間増殖を続け、(2)培養中でヒト造血前駆細胞がヒト幹細胞の分裂と分化により少なくとも5カ月間の長期間生産され、そして(3)ヒト骨髄基質細胞を包含するヒト基質細胞の代謝を活発にし、GM−CSF分泌を活性化する。本発明は培養に於いてヒト幹細胞の生存と増殖を初めて可能にした。
【0034】
本発明はまた、ヒト幹細胞の連続的増殖を支持する体外培養系を提供し、ヒト幹細胞に遺伝子を導入することを可能にし、遺伝的に安定な形質転換したヒト幹細胞を得る事に成功した。この発明の実施態様は組換えレトロウイルス、或いは細胞分裂を必要とする他の遺伝子導入ベクターに組み込ませる事の可能などのような遺伝子の導入にも用いることが出来る。この方法によって製造される遺伝的に変化したヒト幹細胞は、先に述べた様に臨床的な病気に広範に適用する事が可能である。
本発明はまた、ヒト造血前駆細胞への遺伝子導入の効率を高める方法を提供すると同時に遺伝的に安定な形質転換したヒト幹細胞及び/又は遺伝的に安定な形質転換したヒト造血前駆細胞を提供する。
【0035】
本発明は培養中の造血細胞、特に幹細胞を包含する分化の初期段階の細胞の効率的な増殖をさせるための反応器及び組成物を提供する。本方法は通常の方法で形質転換した基質細胞を用いる。その基質細胞は成長因子の本質的な或いは誘導的な生成を提供し、その細胞は物理的に分離していて造血細胞の分離を容易にする。連続的灌流を行い適宜細胞のリサイクルを行なう事により、生存し得る造血細胞を高密度、高収量で得る事が出来る。本反応器は基質細胞に蛋白性の表面を用い、基質細胞と造血細胞の分離を維持する為に表面或いは他の障壁を用いる。
【発明を実施するための最良の形態】
【0036】
本発明の長所は本発明がヒト幹細胞及び/又は前駆細胞液体培養(造血組織培養に用いられる様な)用の標準系に応用された場合いつでも認められる。本発明の方法に従い、補足的に造血増殖因子を最適濃度に培養に添加し、急速培地交換速度を用いた場合、本発明者は、驚いた事に、ヒト造血液体培養の標準系を作る事が出来る事を発見した。その培養は馬、子牛、牛胎児より得られた血清、血漿又はヒト血清の存在下又は非存在下で実施される培養を含み、質的に優れた操作で実施できる。
【0037】
本発明の方法に従って用いられるヒト造血液体培養は1ml当り104から109細胞の細胞密度で、血清アルブミン、コレステロール及び/又はレシチン、セレニウム及び無機塩と共に用いる事の出来る既知の標準培地組成(例えばIMDM, MEM, DMEM, RPMI 1640, Alpha Medium 或いはMcCoy's Medium)を用いて行なう事が出来る。知られている様に、これらの培養は10-4から10-7Mの濃度のハイドロコーチゾンの様なコルチコステロイドを補っても良いし、コーチゾン、デキサメタゾン或いはソルメドロールの様な他のコルチコステロイドを同じ効果を持つ濃度で補っても良い。これらの培養は普通はほぼ生理的なpH,例えば6.9 から7.4で行なわれる。培地は普通4から20容積%の酸素、好ましくは6から8容積%の酸素、を含む気相に曝される。
【0038】
これらの標準培養技術を用いて細胞塊は希望する量に、例えば含まれる幹細胞或いは造血前駆細胞の量を1000倍又はそれ以上に増やす事が出来る。負選択法或いは陽選択法に相当する、異なった既知の方法をこの増大を達成する為に用いる事が出来る。例えば負選択法に相当する例としては分化した細胞は免疫学的技術を用いて除去される。その免疫学的技術とは例えば非前駆、非幹細胞を一群のマウス抗ヒトモノクローナル抗体で標識し、マウス抗体で覆われた細胞をウサギ抗マウスIgでコートしたプラスチックに付着させて除去する技術である(Emerson et al, J. Clin. Invest. (1985) 76:1286-1290)。
【0039】
本発明は上記のすべての条件の下でヒト骨髄液体培養条件の根本的な改良、つまり急速栄養培地交換、に依存するものである。標準培養計画は毎週培地と血清の交換を必要とする。これは1週間に一度培地と血清を交換するか或いは1週間に二度培地と血清を半分ずつ交換することによってなされる。本発明によれば培養の栄養培地は連続的に或いは定期的に、好ましくは灌流法により、1ml当り2×106から1×107の細胞密度の培養に対し24時間から48時間の間に置換される。細胞密度が1ml当り1×104から2×106の場合は同じ培地交換速度が用いられる。細胞密度が1ml当り107以上の場合には培地交換速度は単位時間、細胞当りの培地と血清の流量が一定になるように比例して増加させることが出来る。
【0040】
本発明による栄養培地の交換は、培地を交換する結果を達成できればいかなる方法を用いることも可能である。例えば一定量の消費された培地を除き、一定量の新鮮な培地を加える方法などである。加えられる一定量の培地の流れは重力、ポンプその他の適当な手段による。培養の配列構成と包装に依存して培地はいずれの一方向にも或いは多方向にも流す事が可能である。好ましくは新しい培地は細胞塊に接触する様に加えられるべきである。生体内の灌流を模倣する方法で培養に加えられるのが最も望ましい。即ち少なくとも細胞塊の一部を灌流し、そして全細胞塊まで灌流する方法である。
【0041】
他の任意のしかし重要な本発明の実施態様は合成造血増殖因子を含む、造血増殖因子を急速培地交換培養に添加する事にある。この実施態様の特に好ましい態様は、サイトカインIL−3とGM−CSFの両方を培地に1日当り0.1 から100ng/mlの割合で添加する事である。好ましい割合は1日当り0.5 から10ng/mlであり、更に最も好ましい割合は1日当り1から2ng/mlである。Epoを栄養培地に1日当り0.001から10U/ml、好ましくは0.05から0.15U/ml添加する事が出来る。マスト細胞増殖因子(MCF,C−キットリガンド、スチール因子)を培地に1日当り1から100ng/ml、好ましくは10から50ng/ml添加する事が出来る。IL−1(アルファ又はベータ)を3日から5日の期間に10から100U/ml添加する事が出来る。更にIL−6,G−CSF,基本線維芽細胞増殖因子、IL−7,IL−8,IL−9,IL−10,IL−11,PDGF或いはEGFを1日当り1から100ng/ml添加することもできる。
【0042】
本発明者は、IL−3、GM−CSF及びEpoを上記の様に用いた場合、特異的に赤血球細胞を産生する系統を得る事を発見した。代りにIL−3とGM−CSFを(IL−6或いはG−CSFを加えたり、加えなくとも)使用した場合は、培養は顆粒球を優先的に生産する。本発明の培養で時間と共にT及びBリンパ球が失われる事を本発明者は発見した。
【0043】
培地中の代謝生成物のレベルは通常特定の範囲に維持される。グルコースの濃度は通常5から20mMの範囲に維持される。乳酸塩の濃度は通常35mM以下に維持される。グルタミンの濃度は一般的に1から3mMの範囲に維持される。アンモニウム塩の濃度は通常2.4mM 以下に維持されている。これらの濃度は定期的に或いはオンラインで連続的に既知の方法を用いて監視する事が出来る(Caldwell et al, J. Cell. Physiol. (1991) 147:344-353参照)。
【0044】
本発明に従って培養出来る細胞はあらゆるヒト幹細胞或いはヒト幹細胞を含む細胞塊である。細胞塊はヒト末梢単核細胞、ヒト骨髄細胞、ヒト胎児肝臓細胞、胚幹細胞及び/又はヒト臍帯血細胞を包含する。これらの細胞塊はヒト幹細胞及び/又はヒト造血前駆細胞を含む。ヒト骨髄に見出されるいかなるヒト幹細胞も含めて、ヒトの幹細胞を含む他の細胞塊も本発明に従って使用出来る可能性がある。
【0045】
本発明の好ましい実施態様に於いて、細胞培養は細胞塊中のヒト幹細胞の含有量を増加させる可能性がある。そのような含有量増加は上記の様にしてなされる可能性があるが、本発明に従って用いられた場合、ヒト幹細胞(ヒト骨髄に存在するヒト幹細胞及びヒト骨髄幹細胞を含む)に遺伝子導入による遺伝子療法の為の最初の有用な手段を提供する。ヒト骨髄に存在する幹細胞はヒト骨髄、末梢血、胎児肝臓或いはヒト臍帯血から得る事が出来る。
【0046】
一般にこの実施態様に於いて、安定した遺伝的に形質転換したヒト幹細胞を得る為に、レトロウイルスで感染したパッケジング細胞或いはそのようなパッケジング細胞培養から得られた上清或いは他の遺伝子導入ベクターは本発明に従ってヒト幹細胞培養に加えられる。本発明は幹細胞と造血前駆細胞の分裂のレベルの増加をもたらすが、対照的に従来の培養は造血前駆細胞の低レベルの分裂(つまり減退している培養)を提供するのみであった。本発明の培養系によって初めて培養での細胞の増加が可能になった。このことは細胞にレトロウイルスを感染させるのに必要である。従来の系でレトロウイルス感染を減退細胞培養で行なったが、従来の細胞にはレトロウイルス感染は起こらなかった。本発明は幹細胞のインビトロ・レトロウイルス感染を起こさせる非常に有効な方法を提供する。特別に幹細胞の含有量を増加させた場合、更に特別に合成増殖因子を含む造血増殖因子を用いた場合はレトロウイルス感染は更に有効である。
【0047】
本発明者は組換えレトロウイルスを含む上清を培養に加えるとウイルスとそのウイルスが担う遺伝子がヒト(造血)幹細胞に導入される事を発見した。これらの幹細胞の分裂と分化によって派生した前駆細胞及びこれらの前駆細胞が更に分裂と分化を行なって出来た成熟した血液細胞はインビトロ造血細胞培養の期間中は導入されたDNAを含んでいた。本発明者は次の事を観察した。レトロウイルスが培養の初期にのみ加えられた場合、トランスフェクトされた前駆細胞と成熟した血液細胞が得られたが、それ等の細胞は存在していた幹細胞が培養初期に増殖し、安定な遺伝子導入を受けたものからのみ派生したものである。なぜならこの系ではレトロウイルス感染細胞は隣接した細胞を感染させる事が出来ないからである。希望する遺伝子を含む前駆細胞とより成熟した細胞とは、それ故、初期のレトロウイルス感染期間に形質転換した、より未成熟の幹細胞より遺伝子を受け継いでいる。
【0048】
本発明のこの面でのより好ましい実施態様に於いては、骨髄、末梢血、胎児肝臓或いは臍帯血から分離されたヒト造血細胞は、最初に、より成熟した血液細胞を除去し幹細胞の含有率を増加させる。これは造血細胞を、成熟した血液細胞と、骨髄前駆細胞のエピトープに反応するが幹細胞のエピトープとは反応しないマウス・モノクローナル抗体とをインキュベートし、それから標識された細胞をウサギ抗マウスIg免疫吸着面に免疫吸着させて除去する事により達成される。そのようにして得られた無系統細胞(Lin−)を本発明に従ってレトロウイルス或いは他のベクターと共に培養する。好ましくは、培養はGM−CSF(好ましくは1mg/ml/日)及びIL3(好ましくは1mg/ml/日)の存在下で、また加えても加えなくても良いがIL−1(好ましくは50U/ml/4日)、同じく加えても加えなくても良いがc−キット・リガンド(マスト細胞増殖因子)(好ましくは10μg/ml/日)の存在下で行なう。
【0049】
レトロウイルス感染は培養開始後2から21日、好ましくは10から14日に培地に組換えレトロウイルスで感染したレトロウイルス・パッケジング細胞培養上清を混入して(例えば5から20%容積/容積)行なうか或いはLin−細胞を直接レトロウイルスで感染したレトロウイルス・パッケジング細胞の上で培養して行なうか或いは両方の方法を同時に行なう。
好ましくはレトロウイルス上清を用い、ウイルス存在下でのインキュベートは12日から16日である。また好ましくはパッケジング細胞は集密近くまで増殖させ、培地を交換し、更に12時間から15時間インキュベートする。それから培地を集めてヒト幹細胞のトランスフェクションに用いる。しかしこの過程は必ずしも厳密に行なわれる必要は無く、レトロウイルス・パッケジング細胞のどのような上清も使用して良い。本発明に依ればどのようなレトロウイルス・パッケジング細胞系(既知の)をどのような既知の方法でも用いることが可能である(Wilson et al, Science (1990) 248:1413-1416 及び/又はSullenger et al, Cell (1990) 63:601-608 参照)。実例としてのパッケジング細胞系はNIH3T3細胞及び腎臓癌細胞5637を含む。
【0050】
遺伝子を発現させるのに適当なプロモーター及びエンハンサー要素と共に組換えレトロウイルスに挿入出来るいかなる遺伝子もヒト幹細胞及び造血前駆細胞に導入することが出来る。本発明は培養の中で幹細胞の生存と増殖を可能とし、安定してトランスフェクトされ遺伝的に変えられたヒト造血細胞を培養中で製造する条件を初めて提供した。「安定してトランスフェクトされた」及び「安定して形質転換された」という語は本明細書では外来のDNAがヒト幹細胞染色体に導入される事を意味する。これは本発明が体外で分裂しているヒト幹細胞をそのような外来のDNAに曝すことを可能にした結果出来る様になったのである。
【0051】
本発明に従って、少なくとも5カ月の培養期間中、ヒト造血前駆細胞がヒト幹細胞から分裂と分化によって生産されるような培養方法を得ることが出来る。即ち培養中での幹細胞の生存と増殖を支持する培養方法を得ることが出来る。
本発明者が得たデータは体外ヒト骨髄培養の動向を決定する非常に重要な変数は培地灌流速度であることを示した。培地交換速度を従来の1週一度のデクスター速度から毎日培地交換をする、週当り7倍量に増加した場合体外造血に重要な影響がある事を本発明のデータは示した。本発明者によって行なわれた実験に於ける全ての培養において、かなりの細胞が最初の3〜4週の間に失われた。この後退の後、培養は安定し培地灌流速度の効果がよりはっきりしてきた。
【0052】
培地交換速度、週当り3.5 回が培養の増殖を最も高め、また前駆細胞の生産面から培養最長の寿命をもたらした。特に注意すべき点は4から10週目にかけて2週毎の非付着性細胞の生産数は恒常的であるか又は増加した。
培養の全期間に亙って、3.5 週以降に生産された細胞の累積数は、従来のデクスター法で生産された場合の殆ど3倍であった。更に前駆細胞の安定生産は18週まで維持されていた。
ヒト骨髄にあるような基質細胞は、本発明の培養中に存在してもよいし、存在しなくてもよい。典型的な培養に於いては基質細胞は細胞培養中に1000分の1から10分の1(基質細胞数/全細胞数)存在している。
【0053】
本発明の他の一面に於いて、本発明者は驚いたことには、本発明の培養はヒト骨髄基質細胞の代謝活性を促進し、GM−CSF及びIL−6の分泌を高める事を発見した。GM−CSFはヒト骨髄基質細胞上清中には検出されないが、本発明に従った急速培地交換はヒト骨髄基質細胞を刺激して300 セントグラム/ml/日から200ピコグラム/ml/日のGM−CSFを分泌させた。ヒト骨髄基質細胞によるIL−6の分泌もまた本発明に従った急速培地交換により1〜2ng/ml/日から2〜4ng/ml/日増加した。この増加は本発明の急速培地交換速度を用いた時にも、またその急速培地交換速度を用いると共に造血増殖因子を添加した場合のいずれにも観察された。本発明者によって得られたデータに基づいて本発明の急速培地交換速度のヒト基質細胞サイトカイン生産能に及ぼす影響は、どのような複合組織培養中の基質細胞においても観察されるはずである。
【0054】
実例として、本発明に従って用いられる培地は3つの基本成分よりなる。第1の成分はIMDM,MEM、DMEM,RPMI1640,アルファ培地或いはマッコイの培地或いは既知の培養培地成分である。第2は血清成分である。これは少なくとも、馬血清、ヒト血清を含み、更に随意的に牛胎児血清、牛新生児血清及び/又は子牛血清から成る。第3の成分はハイドロコーチゾン、コーチゾン、デキサメサゾン、ソルメドロール或いはこれらの組み合わせ、好ましくはハイドロコーチゾンの様なコルチコステロイドである。
使用出来る種々の培地の成分を以下に示す。
【0055】
【表2】
【0056】
【表3】
【0057】
【表4】
【0058】
【表5】
【0059】
【表6】
【0060】
【表7】
【0061】
【表8】
【0062】
【表9】
【0063】
【表10】
【0064】
【表11】
【0065】
【表12】
【0066】
【表13】
【0067】
血清成分は培地中に少なくとも1%(V/V)から50%(V/V)存在し得る。血清濃度は好ましくは15から30%(V/V)付近である。血清濃度を高める場合は交換速度を比例して増加させる。第3の成分は10-7Mから104M存在し得るが、好ましくは5×10-6から5×10-5Mである。培地の成分は3つの成分を加えた時に100 %になる様に構成されている。選択的に、血清成分は数種存在する標準的な代用血清混合物のどれとも交換できる。典型的な代用血清混合物はインスリン、アルブミン、及びレシチンかコレステロールを含む。以下の文献を参照:Migliaccio et al, Exp. Hematol.(1990) 18:1049-1055、Iscove et al, Exp. Cell Res.(1980) 126:121-126、及びDainiak et al, J. Clin. Invest. (1985) 76:1237-1242。
【0068】
実例として、ヒト造血幹細胞の細胞濃度は次のようにして増殖させる。吸引採集された骨髄から赤血球をフィコール:ハイパーク密度勾配遠心により除去する。その後、単核細胞を分化した血液成分を認識する抗体の「カクテル」と共にインキュベートする。ここで、分化した血液成分とは、赤血球、顆粒球、マクロファージ及び分化したリンパ球(B−及びT−細胞両方)を含む。更に決定前駆細胞を認識する抗体(抗CD33を含む)を加える。分化した細胞はパニング、磁気ビーズ、或いは蛍光活性セルソーター(FACS)等を含む種々の方法の1つにより除去される。分化した細胞を除去する事により造血幹細胞の組変えウイルスによる感染の機会、それ故希望する遺伝子の導入の機会は非常に改善される。
【0069】
他の実施態様に於いて、培養中で造血細胞の増殖の為の方法が提供される。その方法は効果的な増殖環境を維持する為に、増殖因子を提供する為の通常に形質転換した線維芽細胞を用い、蛋白成分を線維芽細胞と造血細胞の混合物に加え、実質的な連続灌流を行い、随意的に培地の再利用を行なう。
【0070】
それ故この実施態様の方法の詳細を、灌流条件、反応器とその内部構造及び形質転換線維芽細胞の3つに分けて説明する。
反応器には、必要な細胞分散、栄養素と酸素の取り入れ、代謝老廃物の除去、随意的な造血細胞のリサイクル、基質細胞の交換及び造血細胞の採取が出来るあらゆる形の容器が含まれる。反応器は実質的に骨に於ける灌流を模倣する状態を作り出す事が出来るものとする。生体内では骨髄1mlあたり毎分約0.08mlの血清が灌流している。これは106細胞1日当り0.3mlの血清量に相当する。それ故、代謝産物を増殖制限しないレベルに維持する為に、培地は24時間の間に、平均少なくとも50%、好ましくは100%交換する必要がある。経験的に生体内灌流速度を模倣すれば、交換速度は一般的に1日106細胞当り約0.5 から1.0ml灌流培地である。
【0071】
バイオリアクター内の灌流速度は反応器内の細胞密度に依存して変化する。2〜10×106細胞/mlの細胞培養ではこの速度は24〜48時間当り1mlの反応器の体積に対して1mlの培地に相当する。この場合使用された培地は20%の血清(10%牛胎児血清と10%ウマ血清の混合物か或いは20%牛胎児血清)を含む。細胞密度がより高い場合には灌流速度は細胞数当りと時間当りの血清流が一定になる様に、比例して増加する。それ故細胞が5×108細胞/mlで培養される場合は、灌流速度は反応器体積1ml当り毎分0.1mlである。この流速は細胞密度に相当する血清と培地の流量に一致し、培養内の正常ヒト骨髄基質細胞からの造血増殖因子の内部生産を刺激する。この血清と培地の流速で誘導される造血増殖因子にはGM−CSFが含まれ、さらにS−CSF,IL−6、G−CSF及び他の造血増殖因子が含まれていてもよい。幹細胞と前駆細胞が基質細胞と付着している場所で受ける縦方向流れによるせん断応力が1.0から5.0dyne/cm2の間になるように、これらの流速はバイオリアクター内で決定されるであろう。
【0072】
種々の培地を造血及び基質細胞の増殖の為に用いる事が出来る。実例としての培地はMEM、IMDM、及びRPMIを含み、これら培地は5〜20%牛胎児血清、5〜20%子牛血清及び0〜15%ウマ血清の組み合わせで補う事が出来るし及び/又は血清なしでPDGF,EGF、FGF,HGF,或いは基質細胞や幹細胞を刺激するための他の増殖因子等で補う事が出来る。特殊な系統のある細胞だけを特に得たい場合、形質転換した線維芽細胞によって提供される増殖因子を補う為に更に増殖因子を灌流培地に加える事が出来る。基質細胞の分泌により或いは添加により灌流培地に加える事の出来る増殖因子はGM−CSF,G−CSF,M−CSF,インターロイキン1−7(特に1、3、6及び7)、TGF−アルファ或いはベータ、エリスロポエチン、或いは同様の物質、特にヒト因子等で
ある。特に興味があるのは0.5 〜2ng/ml、好ましくは1ng/mlのGM−CSF、
及び最終濃度0.1〜2U/ml/日のエリスロポエチン、100〜300ng/ml/日のG−CS
F及び約1〜10ng/ml/日の幹細胞増殖因子(S−CSF,マスト細胞増殖因子或
いはキット・リガンドとも呼ばれる)等である。ひとつ或いはそれ以上、好ましくは少なくともふたつの増殖因子が形質転換した細胞から分泌され、灌流培地中には増殖因子の希望するレベルを維持するに十分な量の形質転換した細胞が存在することが分かっている。
【0073】
反応器内では生理的温度、つまり37℃に維持されている。33℃を含めて低い温度で行なう事も可能であるが通常は25℃以下ではない。湿度は一般に100%で気相は5%の二酸化炭素を含んでいる。灌流培地は反応器外で酸素添加しても良いし、反応器内でしてもよい。反応器内で酸素添加する種々の手段がある。反応器内酸素添加はハローファイバー、焼結ガラス・ディスク、シリコン管、或いは適当な有孔性と疎水性を持つ他の膜を用いて行う事が出来る。栄養素のレベルと代謝生成物のレベルは比較的狭い範囲に維持される。グルコースのレベルは約5から20mMの範囲で通常10から20mMであり、乳酸のレベルは通常35mM以下に維持しているが20mM以上であることも可能である。グルタミンの濃度は一般的には1から3mM の範囲に、通常は1.5から2.5mM の範囲に維持している。アンモニアの濃度は通常2.5mM 以下に維持しているが、好ましくは2.0mM 以下である。
【0074】
液体の流れは重力、ポンプ或いは他の方法によってなされても良い。流れの方向は反応器内のパッキングの性質に依存して、どの1方向でも構わないし多方向でも差し支えない。望ましくは流れの方向が反応器を横切って実質的に水平であるラミナー・フローを用いても良いし、流れが反応器の底から上方へ向かう垂直流を用いても良い。
【0075】
ヒト造血細胞が腫瘍細胞、例えば白血病、リンパ腫或いは癌を含むと疑われる場合は、灌流速度を正常前駆細胞と腫瘍性の造血細胞とを分離する様に調節出来る。正常の造血前駆細胞は約1.5〜2.0dyne/cm2の縦方向液体流によるストレスに耐える親和力で基質及び実質蛋白に付着している。対照的に腫瘍細胞とそれ等の前駆細胞は基質に対する親和力がかなり弱まっており、約0.05〜1.2dyne/cm2の範囲である。しかし、正常と腫瘍前駆細胞が耐える事の出来るシアー・ストレスの中間のシアー・ストレス、通常1dyne/cm2以上を与える灌流流速を用いれば、腫瘍前駆細胞を正常前駆細胞から分離出来る。分離を達成するには一般的には灌流を少なくとも2日維持し、好ましくは少なくとも5日、更に好ましくは7日或いはそれ以上維持する。このようにして患者からの正常造血細胞を拡大し、同時に適当な流速を採用する事により腫瘍細胞を分離出来る。この方法により腫瘍を持つ患者からの自己造血細胞を化学療法或いはX線療法の間正常造血細胞を拡大して、患者に返し、患者の造血及び免疫系を回復させる。
【0076】
せん断応力を用いて造血腫瘍細胞を正常造血細胞とを分離する実例としては慢性骨髄性白血病(CML)の状況がある。CML細胞のせん断応力耐性は0.05〜1.2dyne/cm2の範囲である。この差が個々の骨髄標品について効率の良いCML細胞除去を可能にした。せん断応力約1.2〜1.5dyne/cm2、好ましくは1.3dyne/cm2を用いて、CML細胞は効率良く分離出来る。
【0077】
個々の骨髄細胞内のせん断応力耐性は先細放射状フローチャンバー(a tapered radial flow chamber)を用いて決定出来る。放射状フローチャンバーに於いては、細胞がうけるせん断応力はチャンバーの始まりからの距離dの関数1/dとして減少する。せん断応力のバンドを細胞集団について分析し、希望する細胞集団を残存させるようにせん断応力をセットする。
【0078】
白血病幹細胞を除去するために、白血病患者の骨髄標品の前駆細胞と幹細胞は最初に放射状フローチャンバーに入れられる。放射状フローチャンバーはポリカーボネートかガラスよりなる2枚のプレートより成り、下方のプレートに骨髄基質細胞が付着出来る様になっている。最初の測定は1)造血細胞の注入の前に骨髄基質細胞の集密的細胞単層を形成しておき12〜24時間後に液体を流し始めるか、或いは2)患者の骨髄を基質細胞の単層なしに直接フローチャンバーに植え、3〜4日待ってから液体を流す(通常0.05〜1.0cc/分)。プレートの端をゴムのガスケットを用いてシールし、調節ねじで一緒にする。チャンバーの狭い注入口に貯蔵タンクから注射器型の定圧ポンプによって管を通して液体をチャンバー内に入れる。広い捕集端で液体と除去された細胞は別の管を通り捕集される(図3及び図3b参照)。一定期間の灌流の後(通常3〜7日)、付着していない細胞を除去し、プレートを分離し3〜5領域のそれぞれから細胞を別々に吸引とゴムのポリスマンで取り、各々の分画を標準的な方法で白血病細胞の有無を調べる(通常は染色体バンディングによる核型分析)。各々の分画での白血病細胞の分析はどの分画(つまりどのくらいのせん断応力)で白血病細胞が基質細胞に付着出来なくなり、除去されたかを示す。このチャンバーでは細胞にかかるせん断応力は入り口からの距離が遠のくにつれて対数的に減少する(図3c参照)。典型的に、付着しない細胞は全て或いは殆ど全て白血病であるが、最も狭い、チャンバーの半分に付着しているのは殆ど全て正常である。
【0079】
これらの測定の結果に基づいて、一連の平行、矩形のチャンバーを設置し、その中に液体を一定の流速で流し、下面にせん断応力をかける。その流速は先細りチャンバーの中で基質細胞から白血病細胞は除去するが全部の正常細胞は除去しないせん断応力に相当するものである。慢性骨髄白血病患者の骨髄の場合、このせん断応力は典型的には0.01〜0.05dyne/cm2である。実際の流速はチャンバーの大きさと形状に依存する。患者からの骨髄細胞をこれらの矩形チャンバーの中で5×106/mlから50×106/mlの濃度で、イスコブの改変ダルベッコの培地に5〜20%(典型的には10%)の牛胎児血清と0〜15%(典型的には10%)のウマ血清を加え、更に10-6Mハイドロコーチゾンを加えた(加えない場合もある)培地で培養する。骨髄細胞を12〜24時間液体の流れなしで培養し、その後流れを開始する。細胞を3〜7日間培養し、非付着性の細胞は捨てられる。付着細胞を矩形プレートから吸引と機械的振動で回収し、集める。これらの細胞は直接患者に戻すか後で用いるために標準技術で液体窒素中で保存する。
【0080】
造血系の細胞以外の細胞でもせん断応力に対する耐性の差による分離が可能である。細胞の複合体の中に区別し得る細胞集団がある場合は、上記の方法を用いて皮膚、肝臓、筋肉、神経或いは上皮細胞に由来する細胞懸濁の中に興味のある細胞を分離する事も可能である。特に興味のある事は正常細胞の集団中に存在する腫瘍細胞を分離することである。分離されるべき細胞集団を後記の様な適当な基質、例えば目的とする細胞が付着し得る精製された蛋白或いは細胞成分の様な基質に接触させる。各々の付着した細胞の小集団についてせん断応力耐性を上記の方法で決める。そして目的とする細胞を上記のようにして集める。
【0081】
反応器の中で種々のパッキングを用いて細胞の付着増殖の場を提供し、一方では基質細胞と造血細胞の間のある程度の物理的な隔離を保持し、また一方では基質細胞と造血細胞の間のある程度の接触或いは接近を維持する。この様にして基質細胞によって分泌された因子がたやすく造血細胞に取り込まれ、造血細胞の増殖と、適当であれば分化と成熟化を助長する。
【0082】
細胞を支持する蛋白マトリックスは切削されたコラーゲン粒子の形を取っても良い。例えば多孔性のコラーゲンのスポンジ或いはビーズ;骨髄からの細胞外の骨マトリックス蛋白より成るスポンジ或いはビーズ;或いは蛋白でコートされた膜等が挙げられる。ただしここで蛋白はコラーゲン、フィブロネクチン、ヘモネクチン、RGDペプチド、混合した骨髄マトリックス蛋白或いは似た物でも良い。膜孔サイズは物理的な隔離を維持しながら異なったタイプの細胞の相互作用を維持する為に、一般的には1〜5μの範囲にある。
【0083】
蛋白でコートした膜を用いてもよい。種々の膜の材料を用いることが出来る。例えばポリプロピレン、ポリエチレン、ポリカーボネート、ポリスルフォネート等である。種々の蛋白、特にコラーゲン或いは既に示された他の蛋白質を用いる事が出来る。膜孔は十分小さく形質転換した細胞は膜を通り抜けてはならないが、その膜の1面に細胞を増殖させ、集密的細胞単層を形成させ細胞膜の一部を孔の中へ伸ばす様にする。一般に孔のサイズは1〜5μの範囲にある。このようにして造血幹細胞は膜の反対側の面で増殖し、形質転換細胞と相互作用を持ち、ここで種々の因子は形質転換細胞から造血前駆細胞へ直接伝達される。前駆細胞、幹細胞は孔を通って侵入してきた細胞質突起に付着する事が出来る。幹細胞からの造血分化は膜の片側で起こり分化した子孫細胞は、基質細胞が集密単層を形成した際、孔は既に塞がれているので、孔を通り抜けることは出来ない。例えば複数のチャンバーを提供し基質細胞を増殖させ、集密状態に達していないチャンバーに造血細胞を移動させる事が出来る。かくして、可動の障壁をチャンバーの間に設け、基質細胞が集密状態に近ずいた時(一般に8〜12週)チャンバー間の障壁を開くか除去するかして基質細胞が新しいチャンバーに移動するようにし、造血細胞が集密状態に達していない基質細胞と接触するようにする。その間集密状態に達していない基質細胞は種々の因子を造血細胞を含むチャンバーに与える(図5a及び図5b)。造血細胞の移動は適当な流速か又は他の適当な方法で達成する事が出来る。適当な壁で分けられたチャンバーに種々のウエルを作ることが出来る。ひとつのウエルに細胞を植えて集密になると細胞は隣のウエルに移動し隣のウエルを集密以下の状態で細胞を植えた事になる。本システムの他の変更は8〜12週の培養の後、造血細胞は新鮮な増殖している基質細胞に曝される事である。この新基質細胞に曝す事は幾つかの方法のうちのひとつにより達成される。第1の技術では、培養をEDTAに3〜5分曝し基質細胞から造血幹細胞を分離する。分離された細胞はそこで新しい培養容器に移される。その培養容器は3〜7日以前に植えられた骨髄基質細胞を含んでいる。この過程を8〜12週毎に繰り返す。他の異なった方法は8〜12週後に培養の体積を増やし培養にコラーゲンのビーズを加えて表面積を増やす方法である。最後に小有機物分子又は蛋白、特に血小板由来の増殖因子(100〜500ng/ml)のようなホルモン、インターロイキン1α、腫瘍壊死因子α、又は塩基性線維芽細胞増殖因子又は他の線維芽細胞のマイトジェニック分子を3〜7日毎に培養に加える。基質細胞をマイトジェンに曝すと骨髄基質細胞の連続した増殖と造血増殖因子の生産が促進される。かくて基質細胞を連続して集密以下の状態に置くことが出来る。
【0084】
連続した液体の流れは骨髄細胞集団の中の正常細胞と癌細胞を選択的に分離するのにも用いる事が出来る。この方法に於いては、放射状フローチャンバーを最初に用いて正常及び癌細胞の特異的基質付着性を決定し、癌細胞を除去するせん断応力を与える流速のフローチャンバーで手術前に正常細胞と癌細胞を分離する。
【0085】
本方法と器具は灌流培地の流れによって失われた幹細胞を再使用する機会も提供する。膜表面蛋白マーカーCD34は実質的に未熟の造血細胞を成熟した造血細胞から分離する。かくして、CD34+である細胞を捕集し、再使用する事により培地中の幹細胞が不足することを防止できる。
【0086】
種々の技術を用いて細胞の未熟分画を捕集し反応器へ戻す事が出来る。例えばCD34に特異的な抗体で細胞を標識し、それからその抗体に対する抗体を用いてCD34+細胞を集め反応器に戻す事によって行うことが出来る。正選択法に代わって、負選択法を用いて成熟した細胞特有のマーカーに対する抗体で成熟細胞を除去することができる。成熟細胞のマーカーとは糖蛋白A、CD33,MO1,OKT3,OKT8,OKT11,OKT16,OKM1,OKM5,Leu7,Leu9,LeuM1等である。種々の造血細胞系統の成熟細胞(リンパ球、骨髄細胞、赤血球)の特異的なマーカーに対する種々の抗体を得る事が出来、これらの抗体を用いて反応器からの流出液から成熟細胞を除去し、残りの細胞を集めて反応器に戻す事が出来る。この方法で、幹細胞を失う事による培養の衰退を防ぐ事が出来、幹細胞の無制限のインビトロ生存を維持する事が出来る。
【0087】
抗体マーカーを用いての分離は種々の方法によって達成する事が出来る。パニング、蛍光活性化セルソーティング、種々の表面、例えばポリスチレン表面、金属小球及び磁性体等に抗体を結合する様な標準技術を単一に或いは複合して用いる事が出来る。抗体を固相表面に結合させて、付着、非付着細胞間の分離をすることができる。或いは抗体を標識し、直接に或いは間接に標識された細胞と標識されない細胞の分離も可能である。
【0088】
本技法を用いれば造血細胞のインビトロ増殖の期間を延長する事が出来る。一般的に生体外でのヒトの造血機能は少なくとも6カ月維持され、顆粒球形成機能は少なくとも4カ月、赤血球形成機能は少なくとも3カ月維持される。更に加えるに、造血前駆細胞は培養期間中連続して生産され、投入細胞の10倍以上の前駆細胞の純収率を得る。
【0089】
更に加えるに、本技法によれば幹細胞の細胞分裂の速度を大きく増大させ、レトロウイルス媒介の遺伝子導入を効率良く行なわせることができる。初期2週間の感染期間に適当なレトロウイルスによって導入された遺伝子はその後4カ月にわたる培養期間に生産された全ての前駆細胞の10〜30%の中で発現させる事が出来る。本技法はそれ故高度に増殖しているヒト造血幹細胞に遺伝子を導入することを可能にする。
【0090】
図1は灌流チャンバーの概略図である。反応器10、蓋プレート12及び床プレート14はボルト16で組み立てられ、ウィングナット18で固定される。歪みを避ける為に3本のボルトが用いられる。チャンバー20は3区画があり、中区画22は基質細胞の為の支持マトリックス、基質細胞床、及び骨髄細胞を含む。中区画22は上区画24と下区画26から膜或いは網の28及び30で分けられている。好適には、ポリスルフォン膜を用いることが出来るし或いは細胞をチャンバーの中区画にとどめておける細かい網目を持つステンレススチールの網を用いる事も出来る。分離間層を、分離膜に機械的な支持を与える為に区分してある内筒27を用いて、チャンバーに置く。上区画及び下区画は同一である必要はなく、液体培地或いはガス交換のための管や膜が通る。ガスは疎水性の管、例えばシリコン製管を通して交換される。管の長さ(それによるガス/液体接触領域)は、中区画で代謝している細胞集団の要する十分なガスの流れができる様に変える事が出来る。培地は上区画或いは下区画からポート32を通じて注入、取り出し可能であり、供給管34を通じて加えることも可能である。
【0091】
希望するなら、上区画及び下区画は外部酸素添加器を用いて除く事が出来る。この場合、分離膜は円筒溝プレート12と14に適合するガラス筒36の下に置かれ、円筒溝の内側領域は膜を通じての流れの分布を良くする為に切り込みを入れてある。この構造は有限数の導入ポートからの液体が混合し、放射状の圧力が平衡化し、分離膜を通しての液体の流れを均一にする。この機構は酸素添加が限界に達しない様な、比較的小数の細胞を培養するチャンバーに適当である。
【0092】
図2に示してあるのは灌流チャンバーを横培地チャンバー、酸素添加器、センサー・チャンバー及び取り出し/注入ポートとをつなぐループの概略図である。
【0093】
外部新鮮培地源50はポンプ52により配管56を通して培地槽に供給し、使用された培地は配管58を通して槽54からポンプ52により使用済み培地容器60に導かれ更に処理される。2番目のポンプ62は培地を培地槽52から配管64を通してホローファイバー酸素添加器66に注入する。培地は配管68を通ってバイオリアクター70の第1チャンバーに導かれる。適宜に培地成分の注入装置82を装着し、培地成分を配管68を通して培地によってバイオリアクター70の第1チャンバーに供給する。培地成分は試験成分、添加因子等である。バイオリアクター70からの培地は中チャンバー72を通ってバイオリアクターの第2チャンバー74に導かれる。そこから培地は配管76により培地組成の変化を検出する為のインライン・センサー78に導かれる。
【0094】
例えばグルタミン:グルコース比は使用する細胞系に依存して約1:5〜8の範囲にあるのが望ましく、例えば形質転換3T3細胞は1:8であるのが好ましい。更にアンモニアの濃度は好ましくは2.0mM以下、乳酸の濃度は40mM以下であることが望ましい。バイオリアクターからの流出液を監視する事により、バイオリアクターに供給される培地を変更する事が出来る。酸素分圧を変え、ガス流量を変化させ、種々の成分を強化し、灌流速度を増加或いは減少する事が出来る。
【0095】
センサー78から培地は配管80を通ってポンプ62により槽54に導かれる。
上記の流れ径路により槽中の培地は別のポンプを用いてゆっくりと交換される。この機構は培地交換速度(外ポンプによる)と酸素添加器と灌流チャンバーを通る流速との別々の制御を可能にする。前者は培地成分と灌流の長期の交換の制御に用いられ、後者は可溶酸素分圧の制御とチャンバー内の流れパターンの制御に用いられる。小メッシュ生体適合膜の使用はチャンバー内でのプラグ(ピストン)フローを可能にし造血細胞や基質細胞にに対し、非常に正確な量を加えることが望まれる増殖因子や他の特別な化合物の供給の正確な制御が出来るようにした。
【0096】
チャンバーとループの構成要素を高圧蒸気減菌した後、反応器は無菌状態で組み立てられる。培地は横ループとチャンバーを通して数日循環し汚染の徴候を監視する。無菌組み立てが完成したならば、チャンバーの中区画へ細胞外マトリックスのみ或いは基質細胞を含む既に接種してある細胞外マトリックスを入れる。基質細胞をそれから:1)チャンバー内に数日間とどめ代謝活動及び/又は増殖因子に対する反応性を監視する。もし結果が満足するものであれば、骨髄を接種する;或いは2)骨髄をただちに接種する。どちらの場合も細胞層は灌流チャンバーの中区画の底に保持される。細胞は追加の細胞外マトリックス下におき、細胞層は分離膜に付着する。この時チャンバーをひっくり返し細胞層を中区画の天井に持ってくる事も可能である。この構成では成熟細胞は基質細胞に対する付着力を失うに連れて、中チャンバーの底にたまる。この特色は成熟細胞が基質細胞及び/又は成熟度の低い造血細胞に損傷を与えるのを防ぐ為に重要である。この特色は成熟細胞の連続除去を容易にする。
【0097】
これらの細胞は細胞を注射器で回収し或いは灌流培地の圧力によりチャンバーより細胞を取り出し管を通して流出させる。
基質細胞は、大部分については、必要な造血増殖因子を提供するひとつ或いはそれ以上の遺伝子で形質転換された線維芽細胞である。同じ或いは異なった細胞を宿主細胞の特異的な選択方式により種々の遺伝子で形質転換出来る。複数の遺伝子に対して、同じ或いは異なった細胞を用いる事が出来る。
【0098】
非常に多くの正常細胞或いは安定細胞系統を用いる事が出来る。しかし、ある系統の細胞の形質転換はその細胞の過剰な増殖をもたらす事があるので、全ての細胞系統を使用出来るわけではない。使用される細胞系は癌性のものではなく、支持体に付着するものであるのが望ましい。哺乳動物細胞はヒト由来のものである必要は無く、サルである必要も無い。種々の形質転換していない細胞も付着細胞層に加えても良い。それ等の細胞は正常ヒト骨髄付着細胞、正常ヒト脾臓付着細胞及び正常ヒト胸腺上皮細胞である。
【0099】
線維芽を含む哺乳動物細胞を形質転換する方法はよく知られており多くの文献があるがその内の少数の参考文献は前記した。構成物はプロモーター、適当であればエンハンサーより成る天然の転写開始制御領域を用いても良いし、定常或いは誘導性の異なった転写開始領域を用いても良い。
数多くの転写開始領域を得る事が出来、それらは誘導性であり、又は定常性である。天然にあるエンハンサーがある場合もあるし、エンハンサーを加えても良い。ある特定の細胞にのみ誘導出来るし、数種の細胞にも全部の細胞にも機能を持たせる事も出来る。転写開始領域はウイルス、天然の遺伝子、合成或いはこれらの組み合わせから得る事が出来る。
【0100】
入手可能で、使用可能なプロモーターは染色体プロモーターを含み、それ等はマウス又はヒトメタロチオネイン−I或いは−IIプロモーター、アクチン・プロモーター等であり、ウイルス・プロモーターとしてはSV40初期遺伝子プロモーター、CMVプロモーター、アデノウイルス・プロモーター、レトロウイルスLTRのプロモーター等である。これらのプロモーターは手に入れる事が出来、転写開始領域及び目的遺伝子を挿入する為のポリリンカーを持つ適当なベクターにたやすく挿入する事が出来る。他の場合、転写開始領域と転写終了領域の間にポリリンカーがあり、メッセンジャーRNAの翻訳のための処理に関係ある種々のシグナル、例えばキャップの位置、ポリアデニレーション・シグナルを提供する発現ベクターを手に入れることが出来る。制御領域と構造遺伝子より成る発現カセットを構成するにはひとつ又はそれ以上の制限酵素、アダプター、ポリリンカー、インビトロ突然変異、プライマーリペアー、切除等を使用する。
【0101】
発現カセットは通常マーカーとひとつ又はそれ以上の複製系を含むベクターの一部である。マーカーは発現カセットとマーカーが導入された細胞の検出及び/又は選択を可能にするものである。種々のマーカーを用いる事が出来るが、トキシン、特に抗生物質に対する抵抗性を与えるマーカーが特に好ましく用いられる。好ましくは宿主として用いられる哺乳動物細胞にG418に対する耐性を与えるゲンタマイシン耐性が用いられる。複製系は原核生物複製系より成り、それにより発現カセットの種々の成分を組み立てる種々の段階でのクローニングを可能にする。宿主細胞に於いてエピゾーム要素を維持する為に他の複製系を用いる事も可能である。しかし複製系の大部分は発現カセットが宿主の染色体に組み込まれるようにさせる性質のものから選択される。
【0102】
発現カセットを宿主に導入する為に通常用いられる技術のどれを用いても良い。それ等はカルシウム沈澱DNA、トランスフェクション、感染、エレクトロポレーション、誘導粒子等である。一度宿主細胞が形質転換されたら、それ等のマーカーを持つ細胞を選択薬剤を含む適当な栄養培地中で選択、増大させる事が可能である。生存した細胞は更に増大させる事が可能である。
使用可能な宿主細胞はアフリカミドリサル細胞系CV1,マウス細胞NIH−3T3,正常ヒト骨髄線維芽細胞、ヒト脾臓線維芽細胞、正常マウス骨髄線維芽細胞及び正常マウス脾臓線維芽細胞等を含む。時によるとベクターと細胞系統の選択によっては細胞が癌化する事があるので注意が必要である。得られた形質転換細胞が付着能を持っていて蛋白スポンジ、蛋白コート膜等の支持体への結合を維持出来る事が重要である。
【0103】
一度適当な増殖因子を発現するベクターが作成されたら、適当な方法で細胞を形質転換させるのに用いる事が出来る。得られた形質転換細胞を先に記述した支持体にシードする事が出来る。これらの支持体は反応器に導入されるか或いはシードする時に既に反応器中に入っている。細胞は十分な時間増殖させ、細胞が生存して必要な増殖因子を生産する事が出来るようにする。
次に、反応器に適当な時期に造血細胞をシードする。造血細胞は、実質的に純粋な幹細胞、ひとつ或いはそれ以上の系統の成熟した造血細胞が実質的に存在しない造血細胞の混合物、或いは造血系の全ての又は実質的に全ての系統の、種々の段階の成熟度の細胞より成る混合物を含む。
【0104】
反応器を通る実質的に連続な灌流を行い、そして種々の栄養素と関連する種々の因子を監視しながら、細胞を増殖させる。大抵の場合、主要な因子は基質細胞から供給されるので、通常は増殖因子の濃度の平衡状態が達成される。調節された上清は造血細胞の増殖に効果的である事が見出されているので、これを用いて反応器内の増殖因子を適当な濃度レベルに維持するような基質細胞と造血細胞の比を与える事が出来る。
【0105】
トランスフェクトした基質細胞はヒト幹細胞への遺伝子導入を助ける事ができる。マウスに於いては、レトロウイルス媒介の幹細胞への遺伝子導入はマウスを5−FUで前処理し、IL−3及びGM−CSFを含むWEHI調節培地で採集した骨髄細胞を増殖させることにより可能になった(Lemischka, Cell (1986)45:917)。興味あるレトロウイルスを分泌しているレトロウイルス・パッケジング細胞と共に増殖している、人工基質が遺伝子をヒト幹細胞に効率良く導入する為に用いる事が出来る。例えばヒトT細胞は幹細胞をCD2制御配列の支配下にあるHIVのアンチセンス配列を含むレトロウイルス・ベクターで感染させる事によりHIV感染に抵抗性を持つように成る(Greaves, Cell (1989) 56:979-986)。レトロウイルスパッケジング細胞によって提供される、レトロウイルスの複製に必須の因子がある。この因子は目標となる造血細胞には存在しない。ウイルスが一度造血目標細胞に導入されれば、ウイルスは複製出来ない。
【0106】
図3aとbに於いて、取り入れ口102、取り出し口104、25のチャンバー106を持つ放射状フローチャンバー100が示されている。矢印108は流れの方向を示す。造血細胞110はチャンバー内の基質層112の上にシードされ増殖する。流速がどの細胞が付着出来るかを決定し、付着出来ない細胞114は取り出し口104を通って外へ出る。
図4aと4bに於いて、取り入れ口122と取り出し口124のある増殖チャンバー120が示されている。図4bに於いては取り入れ口122はマニフォールド128より成る。マニフォールドは細胞110と基質112を含む個々のチャンバー126に入る。そこで細胞は増殖し分離される。
図5aと5bに於いて、増殖チャンバーとその中で障壁134、136、138が培養中除かれている状態を図示している。障壁134は約8〜10週目に;障壁136は約18〜20週目に;障壁138は約28〜32週目に除かれる。
【0107】
本発明の一般的な説明をした後、更に理解を深めるために、次に特定の実施例を挙げて説明をする。ここで提供する実施例は例示のみを目的とし、特記した場合を除いて本発明を制限するものではない。
【0108】
細胞分離及び染色方法
フィコールによる骨髄細胞の分離
1.骨髄試料を室温に保ったI−MDM(イスコブの改変ダルベッコ培地;GIBCO;カタログNo.430-2200)で1:4の比で希釈する。
2.室温で、50mlの遠心管の中で希釈した骨髄試料35mlを注意深く15mlのフィコール−パク(Ficoll-Paque)(比重1.077g/cc;Pharmacia;カタログNo.17-0840-02)にのせる。
3.700×g(1800rpm、Beckman遠心機)で30分、室温(20℃)で遠心分離する。
4.遠心分離後、上層の大部分を除き(間層の上約5mlを残す)、間層(骨髄細胞)を回収し、氷冷したI−MDMで次の様に3回洗浄する:
第1洗浄:1400rpm/15分/4℃
第2洗浄:1200rpm/10分/4℃
第3洗浄:1200rpm/10分/4℃
5.3回目の洗浄の後、細胞は培地か或いは平衡塩類溶液等(細胞の使用目的に依存)に懸濁し、細胞を酢酸液で1:10に希釈した後に(細胞懸濁液10μl=2%酢酸を含むPBS90μl)細胞数をカウントする。この方法では赤血球は酢酸中で溶血するので、白血球のみをカウントすることになる。
6.次に、適当な培地中に細胞を希望する最終濃度に懸濁する(培地については各々の応用参照)。
【0109】
MY−10陽性の骨髄細胞の蛍光染色法:
試薬
標準緩衝液:
粉末バクト乾燥DIFCO緩衝液
(Baxter;カタログNo.2314-15GB): 200g
10%NaN3(アジ化ナトリウム) 20g
非動化牛胎児血清(56℃、30分) 200ml
2回蒸留水で20リットルに希釈;pH7.15〜7.25;
4℃で保存
1ヵ月有効
2%パラフォルムアルデヒド溶液
パラフォルムアルデヒド: 10g
2回蒸留水 500
10N NaOH(ドラフト内で) 8〜20滴
粉末バクト乾燥DIFCO緩衝液 5g
ドラフト内で2回蒸留水を500mlフラスコに入れ、ホットプレート上で撹拌 して60℃にする。
パラフォルムアルデヒド10gを加える。
NaOHを溶液が透明になるまで滴下する。
DIFCO5gを加える。
冷却し、2N塩酸でpHを7.35〜7.45にする。
1.細胞数を数えた後、標準緩衝液で1回洗浄する(100rpm,5分,4℃)。
2.次に、細胞を標準緩衝液に2×105細胞/mlの濃度に懸濁する。
3.2つの15ml遠心管に、それぞれ50μlの細胞懸濁液を入れる。
4.ひとつの遠心管には、1:5に希釈した50μlの抗HPCA−1を加える(抗ヒト前駆細胞抗原;Becton Dickinson; カタログNo.7660;標準緩衝液で1:5に希釈する)。他の遠心管には、1:5に希釈した50μlのMIgを加える(マウスIgG1コントロール;Becton Dickinson;カタログNo.9040;標準緩衝液で1:5に希釈する)。
5.両方の遠心管を氷中で1/2時間インキュベートする。
6.インキュベーション後、細胞を5mlの標準緩衝液で2回洗浄する(1000rpm,5分,4℃)。
7.2回目の洗浄後細胞ペレットは1:40に希釈したGAM−FITC(アフィニティーで分離された抗マウスIgG及び抗IgMヤギF(ab’)2、ヒトIg吸収、フルオレシン結合;TAGO;カタログNo.4353;標準緩衝液で1:40に希釈する)50μlに再懸濁する。
8.細胞を氷冷下、暗闇で1/2時間インキュベートする。
9.インキュベーション後、細胞を5mlの標準緩衝液で2回洗浄し(100rpm,5分,4℃)、各々のペレットは、標準緩衝液100μlと2%パラフォルムアルデヒド100μlの溶液に再懸濁する。
10.次に、フローサイトメーターで細胞を蛍光分析する。%蛍光陽性は抗HPCA−1標品の%蛍光からMIg標品の%蛍光を引いたもの。
【0110】
成熟前駆細胞を選別するための骨髄細胞の蛍光染色法:
目的:
この染色法の目的は、成熟細胞集団を除去し、フローサイトメーター又は磁気ビーズを用いることにより、造血幹細胞(最も原始的な幹細胞)を濃縮することである。選別の結果と比較し濃縮程度を求めるために、いくつかの細胞を(フィコール(Ficoll)分離後)全骨髄細胞として保持して、MY−10陽性細胞を染色するすることは、常に良い方法であると考えられる。
1.細胞は前記したようにフィコール−パク(Ficoll-Paque)で分離される(0.5×106細胞を除去し、2つに分け、抗−HPCA−1/GAM−FITC及びMIg/GAM−FITCで染色する)。
2.3回目の洗浄後、細胞ペレットをモノクローナル抗体混合液(この混合液の作り方の記載部分を参照)に107細胞あたり該混合液1mlを用いて懸濁し、細胞を氷上で1時間インキュベートする。
3.その後、細胞を過剰の氷冷I−MDMで3回洗浄する(1000rpm,5分,4℃)。
4.3回目の洗浄後、細胞を1:40に希釈したGAM−FITC(標準緩衝液ではなく、I−MDMで希釈したもの)に0.25×106細胞当たり50μlの比率で懸濁し、氷冷下、暗闇で1/2時間インキュベートする。
5.インキュベーション後、細胞を氷冷I−MDM中で3回洗浄し、最後の洗浄後、2〜4mlの氷冷I−MDMに懸濁し、選別まで氷冷して保持する。
6.その後、蛍光に基づくフローサイトメーターで細胞を選別し、蛍光ヒストグラムの上部85%を除く。より良く濃縮するために、この選別を二度繰り返してもよい。
7.選別後、細胞をカウントし、洗浄し、アリコートはMY−10陽性細胞の染色を(上記のようにして)行ない、全骨髄細胞の染色アリコートと比較して濃縮程度を求める。
【0111】
磁気抗体を用いた未熟細胞の選択:
1.成熟細胞を選別するために、骨髄細胞の蛍光染色法操作の工程1〜3に従う。 注:アジ化ナトリウムはいずれの緩衝液にも含まれない。
2.その間に、適当量の磁気ヤギ抗−マウスIg(Biomag; Collaborative Re
search; カタログNo.74340-50;1mg/ml;5×108粒子/ml)を氷冷I−MDM中、1500rpm、5分、4℃で3回洗浄する(防腐剤として用いられているアジ化ナトリウムを洗い出すため)。
3.「工程1」における3回目の洗浄後に得られた細胞ペレットを、バイオマグ中に50粒子バイオマグ/細胞の割合で再懸濁する(例えば、1×106細胞に対して5×107粒子を用いる。従って、0.1mlのバイオマグである)。
4.細胞を(細胞数により)T−25又はT−75組織培養フラスコに入れ、断続的に攪拌しながら氷冷下、1/2時間インキュベートする。
5.インキュベートした後、該フラスコ(バイオマグが入っている)を平らな磁石の上に置き、輪ゴム又はテープでしっかり留め、4℃で10〜15分インキュベートする。
6.該磁石及びフラスコを垂直に立て、上清を集める。
7.工程4〜6を2回以上繰り返す。
8.細胞をカウントし、氷冷I−MDM中で1回洗浄し、アリコートを除去してMY−10陽性に対して染色を行ない、適当な培地に再懸濁してさらに使用する。
【0112】
I.培地交換
材料及び方法:
細胞: ヒト骨髄細胞は、通知を受け承諾した個人の腸骨稜からの吸引物にヘパリンを加えたものから得た。フィコール−パク(Pharmacia,No.17-0840-02)密度勾配遠心分離により骨髄を分離し、低密度細胞(<1.077gm/cm3)を集め、イスコブの改変ダルベッコ培地(IMDM)で3回洗浄した。細胞を2回目と3回目の洗浄の間にカウントした。その後、細胞を2連又は3連の24ウェル組織培養プレート(Costar, No.3524)に322μl/ウェル、1、2、及び5×106細胞/mlで播種した。
【0113】
長期培養条件: 低密度細胞を湿潤5%CO2/95%空気雰囲気下、10%ウシ胎児血清(Hyclone Laboratories)、10%ウマ血清(Hyclone Laboratories)、1%ペニシリン/ストレプトマイシン(Sigma, 10,000U/mlペニシリンG及び10mg/mlストレプトマイシン,カタログNo.P3539)及び10-5Mヒドロコルチゾン(17- ヒドロキシコルチコステロン,Sigma,カタログNo.H0888)を補給したIMDM中でインキュベートした。培養を、3種の培地交換スケジュール、すなわち100 %毎日培地交換(7/週)、50%毎日培地交換(3.5/週)、又は50%週2回培地交換(1/週)のうちの一つで処理した。培地交換の期間中、週2回、非付着性細胞の50%を各培養ウェルから除去し、ヘモサイトメーターを用いてカウントした。
【0114】
細胞をカウントするために除去した時(2回/週)、3.5/週及び1/週培養の栄養補給の間に除去された培地は、細胞カウント用に全部とっておき、新規培地をウェルに戻した。7/週培養は細胞カウント用に除去された培地の1/2をとっておけばよく、残りの1/2中の非付着性細胞は遠心分離して戻した。その後、新規培地を各ウェルに添加し、細胞カウントのために除去された培地を補充した。細胞がカウントのために除去されなった日は、7/週及び3.5/週培養ウェルから、ぞれぞれ培地の100%又は50%の培地を除去した。細胞を遠心分離し、さらに新規培地とともに元のウェルに戻した。
【0115】
メチルセルロース及び形態学的アッセイ: 一週間毎に、細胞カウント用に除去された非付着性細胞をエリトロポエチン(erythropoietin)、GM−CSF及びIL−3の存在下、メチルセルロースにプレートし、顆粒球マクロファージ−コロニー形成ユニット(CFU−GM)を数えた。除去細胞のアリコートを細胞遠心分離し、ライト−ギムザ(Wright-Giemsa)で染色し、差分細胞カウントを行なった。
【0116】
統計学的解析: 週2回の細胞産生の結果は、同型培養から平均±SEMとして表わされる。培養グループ間に有意差の生じる確率は、一対のt−検定を用いて、急速交換培養(7/週及び3.5/週)とそれに対抗するコントロール培養(1/週)との標準化累積細胞産生値の比較により求めた。統計学的有意性は5%レベルで判定した。
【0117】
結果:
非付着性細胞産生の速度論: 非付着性細胞産生は、接種細胞密度の関数(1〜5×106細胞/mlの範囲にわたっての)及び培地交換速度の両者を試験した。培地交換速度は、1培地体積交換/週、すなわち伝統的なデクスター(Dexter)培養速度から、7培地体積交換/週まで、様々であった。集められた週2回の細胞数は、培養当たりの接種細胞数で割ることにより標準化した。
【0118】
各培地交換速度において、標準化した細胞コレクションカーブは接種密度でそれ程変化はしなかった。7/週、3.5/週及び1/週の3つの培地潅流速度で保持された培養の細胞産生は、培養当たりの接種細胞数に標準化した場合、同様であった。接種密度間の最終累積細胞産生の比較から、3つの培地交換速度のいずれにおいても顕著な差異は全く示されなかった(サンプルの全組の一対のt−検定によりp> .20)。
【0119】
反対に、培地交換速度はこれらの培養における細胞産生の速度及び寿命に大きく影響した。1/週(コントロール)、3.5/週及び7/週で交換された培地の細胞産生は全て初めの数週間にわたり減少した。しかしながら、培養産生性の差異は培養の第3週以降に明らかになった。第3〜10週の間に、細胞産生は7/週培養においては一定であり、1/週培養においては低レベルで一定であったが、3.5/週培養においては指数関数的に増加した。第10〜12週以後には、全ての培養において細胞産生は培養が終結するまで減退した。
【0120】
1/週交換培養の結果は、伝統的なヒト・デクスター培養の様々な系において共通して見られる結果と同じであったが、一方、3.5/週及び7/週の急速交換培養では、前の最適培養方法に比べて細胞産生性の増加が見られた。毎日培地の1/2が交換される培養(3.5/週)では、コントロール(1/週)又は毎日完全交換(7/週)培地よりも実質的に長期間細胞産生の増加が維持された。第3週及び第9週の間、3.5/週交換培養から集められた非付着性細胞の数は指数対数的に増加し、2.1週間毎に2倍になった。
【0121】
3.5/週及び1/週プロトコールでの細胞産生は、培養の産生のパーセンテージとして3.5/週交換速度下での細胞産生をプロットすることにより、1/週の交換速度と直接比較することができる。この比較により、最初の減少相では、2つのプロトコールでの細胞産生は同じであることが示される。しかし、第3.5週及び第18週の間では、3.5/週交換速度下での細胞産生は常に高い。
したがって、培養物の増殖能は、初めの減少後の細胞の産生能により測定する
ことができる。第3週以後の標準化累積細胞産生(Σ(i=7)n,Ci/Co)は7/週、3.5/週の培地交換速度に対する細胞接種密度には依存しなかった。同様の培地交換速度での培養から得られる細胞産生データは、質的にも統計学的にも類似しており、従って、密度は平均化され、組合せて、大きな統計サンプルを得た(下表)。第3.5週及び第20週の間の密度平均化累積細胞産生は:7/週培養では0.22;3.5週培養では0.40;及び1/週培養では0.15であった。したがって、1/週から7/週への培地交換速度の増加は、細胞産生を代表的なデクスター培養培地交換スケジュールよりも約60%程増加させた。3.5/週交換速度は、1/週デクスタープロトコールと比較してほとんどその3倍の累積細胞産生増加となった。これらのデータの一対のt−検定を用いた統計学的解析は、7/週対1/週及び3.5/週対1/週において共に5%程度の有意性で著しい差異があることを証明した。したがって、3.5/週の培地交換速度は、1/週の伝統的なデクスタープロトコールより細胞産生速度を改善する。
【0122】
顆粒球−マクロファージ前駆細胞産生: 顆粒球−マクロファージ前駆細胞アッセイは、或る培地潅流スケジュール及び接種密度の反復実験から行なわれた(表14)。培地潅流速度は、産生される顆粒球−マクロファージ前駆細胞の数に著しい影響を示した。3.5/週培地交換培養では、前駆細胞産生の寿命が最も長かった。これらの培養物では、第4週と第18週の間に前駆体が安定な速度で産生された。
【0123】
前駆細胞産生についての最適条件は、週当たり3.5 回交換され、かつ5×106細胞/mlで接種された培養である。これらの培養は、第20週までに相当数の前駆細胞を産生した。一対のt−検定を用いた統計学的解析により、3.5/週の最適培地交換速度培養は、3種の接種密度全てにおいて第8週以後に7/週及び1/週培養よりも1%レベルの有意性でかなり多くの顆粒球−マクロファージ前駆細胞を産生することが示された。産生された前駆細胞数は幹細胞再生の非直接的な尺度なので、重要である。前駆細胞は、培養中に依然として存在する初期細胞、おそらくは幹細胞から分化することにより、培養の数週間後に存在することが可能である。したがって、これらのデータから、より生理学的で急速な培地/血清交換速度及びより高い細胞密度が、5ケ月間の幹細胞再生の幾分かを支持する条件を提供したであろうことが示唆される。
【0124】
非付着性細胞形態: 3.5/週培養により支持される延長化造血機能がそれ以外の培養物では性質上異なるか否かを決定するために、第10週と第19週の間に集められた非付着性細胞を染色し、形態学的に分類した。1/週及び7/週の交換速度において、産生された細胞の大部分は、第15週付近及びそれ以降ではマクロファージであり(表15)、それは他の研究機関における研究で得られた結果と同様であった。それとは反対に、週当たり3.5培地体積の速度で潅流され、5×106細胞/mlで播種された培養物は、19週間を通じてマクロファージだけでなく顆粒球をも産生した。したがって、この培地交換速度及び接種密度はインビトロにおいてより効果的に顆粒球機能を再構築したと考えられる。
【0125】
【表14】
【0126】
この結果は、長期ヒトデクスター培養条件が次善であり、かつインビボ造血環境に近いより優れたインビトロ培養として、骨髄エクスビボのより効果的な再構築が達成できることを支持するものである。
【0127】
物理的外観: 培地交換速度は、培養物の物理的外観に著しく影響した。培養の10週付近で、7/週培養では多数の脂肪細胞が間質に生じ、一方3.5/週培養ではわずかに脂肪細胞が生じ、1/週培養では脂肪細胞は全く生じなかった。26週で培養が終結した時、7/週培養の間質はおよそ20〜30%の脂肪細胞からなるが、3.5/週培養ではわずかな脂肪細胞を有するだけであった。付着性コロニー分布も、培地潅流速度の異なる培養間ではそれぞれ異なる。3.5/週培養における付着性コロニーは、7/週及び1/週培養のものよりも長期間存続した。
【0128】
【表15】
【0129】
II.造血細胞成長因子を有する培地の補給を組合せた培地交換
材料及び方法:
細胞: ヒト骨髄細胞は、通知承諾に従い、腸骨稜からの吸引物にヘパリンを加えたものから、ユニバーシティ・オブ・ミシガン・ヒューマン・インベスティゲーション・コミッティー(University of Michigan Human Invenstigation Committee)から提供されたプロトコールに基づいて得た。骨髄をフィコール−パ
ク(Pharmacia)密度勾配遠心分離により分離し、低密度細胞(<1.077gm/cm3)
を集め、IMDMで3回洗浄した。細胞を2回目と3回目の洗浄の間にカウントした。その後、細胞を6−ウェル組織培養プレート(Costar No.3406)又はコラーゲン被覆6−ウェルプレート(ウサギ尾タイプ1コラーゲン,Biocoat. Collaborative Research Inc. カタログNo.40400)上に1.5ml/ウェルで、5×106細胞/mlで2連に播種した。
【0130】
培養培地: 使用した培地は、10%ウシ胎児血清(Hyclone Laboratories)、10%ウマ血清(Hyclone Laboratories)、1%ペニシリン/ストレプトマイシン(
Sigma,10,000U/mlペニシリンG及び10mg/mlストレプトマイシン,カタログNo.P
3539)及び10-5Mヒドロコルチゾン(17-ヒドロキシコルチコステロン,Sigma, カタログNo.H0888)を含有するIMDM(Gibco Laboratories, カタログNo.430-2200)である。
【0131】
造血成長因子(HGH): 急速培地交換による頻繁培地補給により、造血成長因子はクローナルアッセイ4において最大限のコロニー形成を促進することがわかっているおよそ1/20の濃度で培地に添加した。使用された濃度はIL−3が1ng/ml、GM−CSF(Amgen Biologicals, カタログNo.13050)が1ng/ml、Epo(Terry Fox Labs. Vancouver, Canada)が0.1U/mlであった。
【0132】
造血前駆細胞アッセイ: 培地から除去された非付着性造血細胞をカウントし、1×105細胞/ml又はそれ以下の細胞をメチルセルロースにプレートした。GM−CSF及びEpoを該メチルセルロースにそれぞれ20ng/ml及び2U/mlで添加した。細胞を24−ウェルプレートに0.25ml/ウェルでプレートし、37℃で14日間インキュベートした。その後、コロニーを倒立顕微鏡下でカウントし、50細胞より大きいコロニーはGM−コロニー形成ユニット(CFU−GM)、赤芽球コロニー群−形成ユニット(BFU−E)、又は顆粒球赤芽球巨核球マクロファージ−コロニー形成ユニット(CFU−GEMM)として記録した。
【0133】
LTBMC条件: 培養は湿潤5%CO2/95%空気雰囲気下、37℃でインキュベートし、50%毎日培地交換の速度で潅流(培地交換)した。培養の第1週の間、毎日培地交換の間に除去された細胞は全て遠心分離して元のウェルに戻した。培養1週間後、全非付着性細胞の50%を、培地交換の間培養物から週2回除去し、単核化細胞をカウントし、新規培地をウェルに戻した。細胞をカウントしない残りの5日/週は、培地の50%を各培養ウェルから除去し、新規培地で置き換えた。除去した培地は遠心分離し、培地を細胞ペレットからデカントし、細胞を元のウェルに戻した。
【0134】
統計学的解析: 培養グループ間に相当な差異が生じる確率は、一対のt検定を用いて、造血成長因子を補給した急速潅流培養物と対する未処理コントロール培養物との標準化累積細胞産生値の比較により決定した。統計学的有意性は5%レベルで判定した。組織培養プラスティック上で培養された対抗する急速潅流LTBMCとI型ラット尾コラーゲンには5%レベルで統計的な差異はなかった。したがって、プラスティック及びコラーゲンマトリックスについてのデータは、ここにおける図及びそれ以外の図を提示するために組合せ、この組合せのデータについて統計学的解析を行なった。
【0135】
結果:
急速交換された成長因子補給LTBMCにおける細胞産生の速度論: 長期骨髄培養(LTBMC)の寿命と産生能が不十分なHGFの産生により制限される、という仮説の第1のテストとして、我々は、IL−3又はEpoが補給された急速交換エクスビボ骨髄培養を保持した。これらの培養物においては、培地の50%が毎日除去され、IL−3又はEpoが補給された同体積の新規培地で置き換えられた。その後、除去細胞を遠心分離し、培地をデカントして捨て、細胞を再懸濁し、元の培養物に戻した。IL−3及びEpoはそれぞれ急速交換LTBMCの細胞産生能を促進した。初めからEpoのみを含有する培養物は、実質的に終結した赤血球の分化により高い細胞産生速度を示した。しかし、4週付近で、赤血球形成は終了し、細胞産生速度はコントロール培養のレベルまで低下した。IL−3及びEpoは、非付着性細胞産生において、18週間を通して平均的な増加を誘発し、コントロールに対して、それぞれ175%及び173%であった。
【0136】
成長因子の組合せは、非付着性細胞産生速度を増加させることにおいて、より効果的であることが明らかになった。細胞産生の最高速度は、IL−3+GM−CSF+Epoの組合せにおいて観察された。これらの培養は、培養において最初の6週間に週2回接種された細胞数のおよそ25%を産生し、非付着性細胞産生が第2〜8週の間にコントロールに対して平均4.8倍増加した。IL−3+GM−CSFの組合せでは、8週通して非付着性細胞がコントロールに比較して平均3.5倍増加した。分離実験において、IL−3+GM−CSF+Epoの組み合わせにIL−6及びG−CSFのいずれも添加しない場合には、非付着性細胞産生速度は改善されたが、その代わり、結果的に細胞産生速度が、IL−3+GM−CSFの組合せを含有する培養と区別できなくなった。いずれの場合も、HGF添加により誘発される細胞産生における刺激効果は0〜8週の間で最大となったが、細胞産生は培養全般を通じてコントロールよりも高かった。
【0137】
HGFを組合せることにより、急速交換LTBMCにおいて非常に多数の非付着性細胞が産生された。培養物の産生能は、接種細胞数(CO)に対する経時的産生細胞累積数(Σi=1n,Ci,Ciは時間iにおいて集められた非付着性細胞の数)を、時間の関数としてその割合(Σ(i=7)n,Ci,Co)をプロットして比較することにより示すことができる。この比率が1を越える場合、培養物は接種された細胞よりも多く細胞を産生し、この培養物では細胞数が増大する。
【0138】
IL−3+GM−CSF+Epoの組合せにより、接種細胞数よりも3倍以上の累積細胞産生が誘発された。この細胞産生速度は、培養における初めの6週間で最高であり、この間、培養は2週間毎に接種された細胞数とおよそ同数の細胞を産生した。この最高細胞産生速度は、推定インビボ骨髄細胞産生速度の15%であり、そこでは骨髄細胞塊の50%が毎日発生する。培養第3〜7週の間において、IL−3+GM−CSFの組合せは、IL−3+GM−CSF+Epoの組合せと同等の速度で細胞数は2倍以上増大した。HGFが補給されない未処理の急速交換(50%毎日培地交換)及び遅交換(50%培地交換週2回)コントロール培養は、それぞれ18週以降に接種された細胞数の約1及び0.37倍を産生した。より重要なことは、これらの非補給培養物から除去された全細胞の半分以上が最初の2つのサンプリングに由来していたことであり、さらにこれらの細胞の多くが初めの接種物からのものであること、及び培養物にHGFを補給することが前駆体と幹細胞の重要なサイクリングを誘発するために必要であることを示したことである。
【0139】
非付着性細胞の形態学的解析: 複数のHGFの添加もまた、培養物で産生される骨髄細胞の多様性を増加させた。コントロール培養は、培養の第3週後に主にマクロファージである非付着性細胞を産生した。赤血球系細胞の産生は急に減少し、第5週後にはわずかな赤血球系細胞しか検出されなかった。Epoを含有する培養(Epo単独,IL−3+Epo,及びIL−3+GM−CSF+Epo)では、赤血球系細胞の産生は一時的に増加し、高パーセンテージ(55〜75%)の赤血球系である非付着性細胞が3週を通じて産生された。IL−3+Epo±GM−CSFが存在する場合、培養は続き、培養16週を通じて、赤血球系として分類された約5〜15%の非付着性細胞と共に、赤血球系細胞を産生した。したがって、IL−3+Epoの存在下では、赤血球形成は始終活性であった。
【0140】
IL−3±Epoでは、第5週に、主に(60〜70%)後期顆粒球(LG)である非付着性細胞集団が生じた。LGのパーセンテージは着々と減少し、第18週で約20%までになった。それに対応してマクロファージの産生が増加した。GM−CSFをIL−3±Epoに添加した場合は、高パーセンテージのLGが18週を通じて存続した。したがって、IL−3+GM−CSFの組合せにより、培養の18週に活性な顆粒球形成が起こり、同様にEpoを添加することにより赤血球形成も保持された。培養5.5週でのコントロール及びIL−3+GM−CSF+Epoを補給した培養の顕微鏡写真からは、培養密度及び産生細胞の多様性がめざましく向上したことがわかる。
【0141】
非付着性前駆細胞産生の速度論: 前駆細胞産は、複数のHGFを添加するにしたがって増加した。未処理のコントロールにおける顆粒球マクロファージコロニー形成ユニット(CFU−GMs)の産生は、18週間以上にわたり長く安定であり、それは急速に潅流されたHGF無添加のLTBMCを用いて得られた初期の結果と一致する。IL−3+GM−CSF及びIL−3+Epo±GM−CSF培養中で産生されたCFU−GMは、第3〜5週の間ではコントロールのおよそ10倍であった。
【0142】
ヒトLTBMCにおける赤芽球コロニー群形成ユニット(BFU−E)の産生は低く、すぐに終結してしまうことが報告されている(Coutinho et al, Blood (1990) 75(11): 2118-2129)。急速交換で未処理のコントロールは、BFU−E産生が急速に減少したが、培養17週を通じて低レベルのBFU−Eが産生された。Epoだけを添加しても、産生されるBFU−Eの数はそれほど大きな影響を受けなかった。IL−3だけを添加すると、第3〜5週においてBFU−E産生の穏やかな短い刺激が誘発された。一方、IL−3にEpo又はGM−CSFのいずれかを加えたものは、培養第3〜5週の間に非付着性BFU−Eレベルがコントロールに比べて10〜20倍増加した。
【0143】
III. ヒト幹細胞の形質転換
材料及び方法
細胞 ヒト骨髄細胞は通知承諾に従い、腸骨稜からの吸引物にヘパリンを加えたものから、ユニバーシティ・オブ・ミシガン・ヒューマン・インベスティゲーション・コミッティー(University of Michigan Human Investigation Committee)から提供されたプロトコールに基づいて得た。骨髄はフィコール−パク(Pharmacia)密度勾配遠心分離により分離し、低密度細胞(<1.077gm/cm3)を集め、IMDMで3回洗浄した。細胞を2回目と3回目の洗浄の間にカウントした。CD18遺伝子伝達実験を行なうために、骨髄は通告承諾に従い、CD18欠失患者提供者から得た。
【0144】
骨髄細胞の系統陰性(Lin-)選択 IMDMでの3回目の洗浄後、成熟単核細胞は、モノクローナル抗体(MAb)混合物と共にインキュベートすることにより、上記細胞調製物から除去した。107細胞を1mlのMAb混合液中、氷上で10〜15分毎に静かに混ぜながら1時間インキュベートした。用いたMAb混合液を表16に示す。
【0145】
【表16】
【0146】
細胞を過剰の氷冷IMDMで3回洗浄し、4℃で遠心分離した。適当量の磁気ヤギ抗−マウスIg(Biomag; Collaborative Research Corp., カタログNo.743
40-50,1mg/ml,5×108粒子/ml)を氷冷IMDMで3回洗浄し、遠心分離した。細胞を50粒子/細胞でバイオマグに再懸濁し、T−25又はT−75組織培養フラスコに入れ、氷上で1/2時間、断続的に攪拌しながらインキュベートした。インキュベート後、フラスコを平らな磁石の上に置き、磁石をフラスコにしっかり留め、4℃で10〜15分間インキュベートした。この磁石とフラスコを垂直に立て、上清を集めた。遮光、磁石の添加、垂直に立てること、及び上清の収集を伴うインキュベートを2回以上繰り返した。この細胞をカウントし、6−ウェル組織培養プレート(Costar No.3406)に接種した。
【0147】
培養培地 使用した培地は、10%ウシ胎児血清(Hyclone Laboratories)、10%ウマ血清(Hyclone Laboratories)、1%ペニシリン/ストレプトマイシン(Sigma,10,000U/mlペニシリンG及び10mg/mlストレプトマイシン,カタログNo.P3539)及び10-5Mヒドロコルチゾン(17-ヒドロキシコルチコステロン,Sigma, カタログNo.H0888)を含有するIMDM(Gibco Laboratories, カタログNo.430-2200)である。
【0148】
造血生育因子 造血成長因子は最適なものを上記した。用いた濃度は1ng/ml又は0.4U/mlのIL−3(Genetics Institute, Cambridge, MA からの寄贈)、1ng/mlのGM−CSF(Genetics Institute, Cambridge, MA からの寄贈)、50U/mlのIL−1a(Genzyme Corp.)、0.1U/mlのEpo(Terry Fox Labs, Vancouver Canada)、10ng/ml のMGF(マスト細胞増殖因子,c−kitリガンド,Immunex Corp., Seattle, WA)、及び2.0ng/mlのヒブリカイン([PIXY321]Immunex Corp., Seattle, WA)であった。
【0149】
造血前駆細胞アッセイ 毎週のサンプリングで培養物から除去された非付着性造血細胞をカウントし、1〜105細胞/ml 又はそれ以下の細胞をメチルセルロースにプレートした。MGF、GM−CSF及びEpoを該メチルセルロースにそれぞれ50ng/ml、20ng/ml及び2U/ml添加した。細胞を24−ウェルのプレートに0.25ml/ウェルでプレートし、37℃で14日間インキュベートした。その後、コロニーを倒立顕微鏡下でカウントし、50細胞以上のコロニーをGM−コロニー形成ユニット(CFU−GM)、赤芽球コロニー群−形成ユニット(BFU−E)、又は顆粒球赤血球巨核球マクロファージ−コロニー形成ユニット(CFU−GEMM)として記録した。
【0150】
レトロウイルス産生細胞系 2つのレトロウイルス産生細胞系をメモリアル・スローン・ケターリング・キャンサー・センター(Memorial Sloan Kettering Cancer Center, New York, NY)のドクター・エリー・ギルボア(Dr. Eli Gilboa)の研究室から入手した。この細胞系は、哺乳動物ネオマイシンアナログG418耐性を付与するネオマイシンフォスフォトランスフェラーゼ産生NEO遺伝子を含有するアンホトロフィックなウイルス性粒子を産生する。また、この両細胞系は、レトロウイルスに感染した細胞がそれ自身感染性のウイルスを産生しないように、必要なレトロウイルス遺伝子が欠損しているレトロウイルス粒子を産生する。
【0151】
パッケージング細胞系を含有するSAXは、改変モロニー・ムリン・ロイケミア・ウイルス(Moloney Murine Leukemia Virus) (MoMuLV)を含有する3T3系細胞系である。SAXプロウイルスはNEO遺伝子、SV−40促進化アデノシンデアミナーゼ遺伝子をXhol制限部位に含有する。また、このSAXプロウイルスはφパッケージング領域を含有するが、gag(コア蛋白質)、pol(リバーストランスクリプターゼ)及びenv(エンベロープ蛋白質)遺伝子が欠損している。この第2のレトロウイルス粒子は外来DNAのダブルコピーを含有しており、このレトロウイルス粒子はDC−29(ダブルコピー−29番目クローン)と表わされる。DC−29プロウイルスは、NEO遺伝子及びその他のレトロウイルス性及び外来のDNAの2つのコピーを3T3細胞系に含有している。
【0152】
CD18試験を行なうために、ヒトCD19 cDNA全長を含有するレトロウイルスベクターに感染したアンホトロフィックなパッケージング細胞Psi−Cripを用いた(Wilson et al, Science (1990) 248:1413-1416)。このレトロウイルスにおいて、ヒトCD18のcDNA全長は、BA−CD18と呼ばれるトリβ−アクチン遺伝子の5′領域をスキャンしている異種配列から組換え遺伝子を発現するベクターのBamH1部位にクローンされた。ヒト・サイトメガロウイルスの5′方向から即時型初期(IE)遺伝子までの配列をPUC19にサブクローンし、IE促進配列を含有する部分をXho1上で(ポリリンカーから)Nco1(IE遺伝子の−220)フラグメントまで除去した。合成リンカーを用いてNco1部位をXho1部位に変換し、この改変フラグメントを5′からβ−アクチンプロモーター方向に配置したBA−CD18のXho1部位にクローンした。この新しいベクターをCMV−BA−CD18と呼んだ。
【0153】
レトロウイルス粒子産生 SAXレトロウイルス粒子はドクター・エリー・ギルボア(Dr. Eli Gilboa)の研究室のドクター・クレイ・スミス(Dr. Clay Smith)より、凍結して-80℃で保存されたウイルスの上清溶液として提供された。DC−29及びCD18レトロウイルス粒子は、DC−29及びCD18ウイルスパッケージング系をT−75フラスコ内でほとんど集密するまで増殖させ、培地を全て交換し、細胞を12〜15時間インキュベートし、その後ウイルス粒子を含有する培地を集めることにより産生させた。その後、この上清を含有するウイルスを遠心分離してウイルスパッケージング細胞を除去し、培地を除去して、アリコート中、-80℃で凍結した。
【0154】
SAXレトロウイルス、DC29レトロウイルス又はCD18レトロウイルス添加上清含有LTBMC 培養物を湿潤5%CO2/95%空気雰囲気下、37℃でインキュベートした。培養の最初の2週間は毎日、培地の2/3(1ml)を各培養ウェルから除去し、培地をHGF(0.85ml)及びウイルス細胞産生上清(0.15ml)を含有する同体積の新規培地で置き換えた。レトロウイルス上清を含有する培地を使用直前に解凍し、使用する必要がない場合には冷凍庫の氷上で保存した。培養物から除去された培地を遠心分離し、培地をデカントし、細胞を元のウェルに戻した。
【0155】
SAXレトロウイルスパッケージング細胞系と共培養したLTBMC SAXレトロウイルスパッケージング細胞系をT−25フラスコ(Costar, No.3056)内でおよそ10%集密するまで生育させ、それから2000ラドの放射線にさらした。上記のようにして調製された造血細胞を照射ウイルス産生細胞に添加し、50%毎日培地交換して全細胞をウェルに戻しながら2.5 週間培養した。培養2.5 週目に、EDTAの0.5mM溶液をフラスコに添加し、間質は残したまま造血細胞を除去した。除去された造血細胞を、ウェル当たり1000細胞の新たにトリプシンを添加した骨髄線維芽細胞とともに6−ウェルプレートの3ウェルに添加した。
【0156】
感染LTBMCのサンプリング 培養2週目の初めに、レトロウイルス添加が終了又は共培養が終了した後で、解析するために交換培地中の非付着性細胞を週に一度除去しながら、培養物を一日当たり50%の培地交換した。毎日培地交換を行なっている間、非付着性細胞を培養物から除去し、単核細胞をカウントし、新規培地をウェルに戻した。細胞をカウントしない残りの6日/週には、培地の50%を各培養ウェルから除去し、新規培地で置き換え、除去した培地を遠心分離し、培地を細胞ペレットからデカントして、細胞を元のウェルに戻した。
【0157】
レトロウイルス感染の解析 初めの骨髄接種物を、0、0.4、0.8、1.2、1.6及び2.0mg/ml のG418にプレートし、ポスト感染骨髄細胞をプレートするためのG418の濃度決定用死滅曲線(kill curve)を得た。培養物から除去された細胞は0.0、0.8、及び1.6mg/mlのG418を含有するメチルセルロースにプレートした。2週間後、前駆細胞のコロニー数をメチルセルロース中で数えた。その後、各コロニーをメチルセルロースから掻き取り、ポリメラーゼ・チェーン・リアクション(PCR)によりレトロウイルスDNAをアッセイした。
【0158】
統計学的解析 培養グループ間に有意差が生じる確率は、一対のt検定を用いて、試験サンプルと対するコントロール培養物との累積細胞産生値を比較することにより決定した。統計学的な有意性は5%レベルで判定した。
【0159】
結果
SAXレトロウイルスを用いたレトロウイルス感染
SAXレトロウイルス感染LTBMCにおける細胞産生の速度論 レトロウイルス感染した培地における細胞産生はレトロウイルス感染の可能性の指標であり、したがって、測定の有用なパラメーターである。ターゲット細胞ゲノ厶へのレトロウイルスの組込みは、唯一、細胞分裂の間に起こると考えられる。この理由により、増大する培養産生性は幹細胞有糸分裂の可能性を高め、したがってレトロウイルス感染の可能性を高めるものである。最高の細胞産生は、培養の4週間を通じて産生細胞数を増加させるIL−3+GM−CSF及びIL−3+GM−CSF+IL−1αを補給した上清ウイルス添加培養物において起こる。SAXウイルスパッケージング細胞と共培養されたLTBMCは、2週目に上清添加培地よりも多くの細胞を産生したが、2週目以後では細胞産生は減少した。
【0160】
上清SAXウイルス含有LTBMCにおけるレトロウイルス感染の解析 高[G418]中で生存している前駆細胞のパーセンテージは、IL−3+GM−CSF又はIL−3+GM−CSF+IL−1α補給培養物では、培養の初めの4〜6週において2〜50%と様々であった。培養第10週付近(8週間後にはウイルスの添加は終了していた)では、造血コロニーにクローン可能な前駆細胞数の43%がG418にさらされて生存していた。このことから、レトロウイルスによって培養物中に存在する幹細胞に転移されたG418耐性遺伝子を含有するウイルスにより、最初の感染14日間で、これらの前駆細胞がG418耐性になっていたことが示される。急速に潅流されたHGF無補給培養物は、第8週と第11週の間に、平均12%の高[G418]中生存前駆細胞を有していた。11週で培養が終了した時、IL−3+GM−CSF補給培養の間質層にはトリプリシンが添加され、間質に付着した前駆細胞の17%が高[G418]中で生存していた。これは、相当なパーセンテージの付着前駆細胞もSAXウイルスに感染したことを示唆するものである。
【0161】
照射SAXウイルスパッケージング細胞と共培養されたLTBMCにおけるレトロウイルス感染の解析 照射を受けたSAX細胞と共培養した場合、G418中で生存している前駆細胞のパーセンテージは、0〜36%と様々であった。HGFが補給されない培養は、高[G418]中で第4〜7週の間のみ生存するCFU−GMを産生した。第7週以後、高[G418]中で生存するCFU−GMは、これらの培養では全く産生されず、これは、幹細胞の感染がほとんど、あるいは全く起こらなかったことを示唆する。IL−3+GM−CSFが補給され照射SAX細胞と2.5週間共培養されたLTBMCは、0.8mg/ml G418中で第4、5及び8週において生存するCFU−GMを高パーセンテージ産生した。しかし、第10週で、これらの培養はG418に耐性のCFU−GMを産生しなくなった。このことは、これらの培養では幹細胞の感染がほとんど、あるいは全く起こらなかったか、又は幹細胞が分化あるいは死滅しただろうことを示唆するものである。
【0162】
DC−29レトロウイルスを用いたレトロウイルス感染
DC−29レトロウイルス感染したLTBMCにおける細胞産生の速度論
DC−29レトロウイルス上清に感染した培養中で産生される細胞数はHGF依存性であった。IL−3+GM−CSF+Epo補給培養では、培養の10週間を通じて週毎に1.5〜4×106細胞が産生された。IL−3+GM−CSF+Epo+MGF補給培養ではさらに多いが、ヒブリカイン+Epo補給培養では最も高い細胞産生が見られた。興味深いことは、IL−3+GM−CSF+Epoは補給されているがDC−29レトロウイルス上清が添加されていないコントロール培養は、DC−29レトロウイルス上清が添加されている類似の培養よりもそれほど多くなかったことである。IL−3+GM−CSF+Epo、IL−3+GM−CSF+Epo+MGF及びヒブリカイン+Epo培養(ウイルス添加)における細胞産生は、それぞれ5%、1%及び1%の有意性において、コントロール培養(IL−3+GM−CSF+Epo,ウイルス無添加)よりも著しく高かった。レトロウイルス上清添加培養における産生の増加の一部は、3T3系パッケージング細胞系により産生されることが知られているMGF(c−kitリガンド)などの成長因子の存在によるものであろう。
【0163】
上清DC−29ウイルス添加LTBMCにおけるレトロウイルス感染の解析
レトロウイルス感染の効率は、1.6mg/ml G418中のCFU−GM生存、レトロウイルス感染前に全骨髄細胞を死滅させる濃度により評価した。第8週(6週間後に感染)におけるCFU−GM生存の平均パーセンテージは全ての感染培養において高いものであった(表17参照)。
【0164】
【表17】
【0165】
IL−3+GM−CSF+Epoの組合せにMGFを添加すると、培養の第8〜10週における感染効率が増大し、培養物の一つは7.7%のG418耐性コロニーを含有していると考えられる。第8週におけるデータからは、ヒブリカイン+Epo補給培養物が高い感染効率を有していたことが示唆されるが、第10週のデータでは感染が全く起こらなかったことが示唆される。第10週では、いくつかの培養物から除去されたCFU−GMの数は減少し、およそ10%感染において、CFU−GMはほとんど、あるいは全く見られないと予想されたであろうことに注目されたい。さらに、1.5mg/ml G418は極めて高い抗生物質処方量であり、わずかに次善の最適レベルでさえトランスフェクトされたG418耐性遺伝子の効能を封じ込めるにおそらく十分である。したがって、これらの培養における造血幹細胞への遺伝子転移の効率は、いくつかのサンプルにおいては少なくとも7.7%であるが、おそらく45%程度又はそれ以上であろう。
【0166】
非付着性前駆細胞産生の速度論
これらの試験におけるレトロウイルス感染の解析は、前駆細胞をアッセイして幹細胞の存在、感染及びサイクリングを推察することによるので、培養におけるHGFの前駆細胞産生への効果を調べることは重要である。また、このレトロウイルス試験において、MGF及びヒブリカインは他のHGFとの組合せで用いられるものである。従来MGF及びヒブリカインは共にLTBMCが補給された急速潅流化HGFには用いられておらず、したがってそれらの造血形成への効果を調べることが必要である。
【0167】
前駆細胞産生は、HGF補給に大きく依存した。培養物から除去されたCFU−GMの数を表18に示す。
【0168】
【表18】
【0169】
レトロウイルス上清の添加により、前駆細胞除去はレトロウイルス上清無添加の場合の2.2倍に増加した。レトロウイルス上清及びIL−3+GM−CSF+Epo+MGF又はヒブリカイン+Epo補給培養物において除去されたCFU−GMの数の増加は、それぞれ非感染コントロールの4.2 及び3.8 倍大きいものであった。CFU−GMの除去は、接種されたCFU−GMの数と比較した時、全てのウイルス補給培養において1%レベルの有意性で統計学上重要であった。
【0170】
培養における前駆細胞産生
CFU−GM区分における集団バランスは、MGF又はヒブリカインのIL−3+GM−CSF+Epo補給培養への添加が、CFU−GMプールにおいてかなりの正の効果を有するものであることを示す。IL−3+GM−CSF+Epoの組合せにMGFを添加することは、MGFが補給されていない同様の培養と比較して、CFU−GM除去を1.9 倍、CFU−GM分化を0.5 倍増大させた(表19参照)。
【0171】
【表19】
【0172】
ヒブリカイン+Epoの組合せは、CFU−GMの産生及び分化において極めて著しい効果を有していた。ヒブリカイン+Epoは、IL−3+GM−CSF+Epoが補給された従来の最適培養と比較して、CFU−GM除去において1.8倍、CFU−GM分化において3倍以上増大させた。また、ヒブリカイン+Epoは、IL−3+GM−CSF+Epo+MGFの組合せのほとんど2倍の数の顆粒球及びマクロファージの産生を誘発した。このことは、ヒブリカインが顆粒球マクロファージ系の強力な誘発剤であることを示している。
【0173】
レトロウイルスをコードするCD18に感染した幹細胞から産生される好中球の解析 CD18欠損骨髄は、上記のようにして初期造血細胞を濃縮し、それから1.0ng/ml/日 GM−CSF及び1.0ng/ml/日 IL−3及び40U/ml/日 IL1α及びCD18レトロウイルス産生系上清を補給して、50%毎日培地交換を行ないながら14日間培養した。第15日目から、レトロウイルス上清を添加しないこと以外は同様の条件下で該細胞を培養した。非付着性細胞を培養物から毎週除去し、標準法によりビオチン添加した抗CD18モノクローナル抗体を用いたフローサイトメトリーで細胞表面CD18の存在を調べた(Updyke et al Meth. Enzymol. (1986) 121: 717-725)。このアッセイにより、CD18欠損骨髄細胞はいかなる細胞表面CD18も発現しなかったが、レトロウイルス感染培地から生じた好中球及び単核細胞は細胞表面CD18をまさに発現した。3連の培地において、細胞表面CD18の発現は、6週間で3.5%/5%/2%であり、11週間で11%/28%/3%であった。11週間目に培地中に存在する好中球及び単核細胞は、CFU−GM前駆細胞から10〜14日だけ早く発生したので、これらのデータは、培養の初めの2週間で、ヒト造血幹細胞が組換えレトロウイルスと、うまく安定にトランスフェクトされたことを示す。
【0174】
この結果の解釈において重要なのは、数種のグループはヒト造血前駆細胞へのレトロウイルス仲介遺伝子の転移を実証しているが、ヒト造血幹細胞への遺伝子転移は示されていないことである。アッセイに必要な前駆細胞が存在しなかったので、急速な細胞産生の減衰、ゆっくりと潅流されたヒトLTBMCにより、感染分析は制限されていた。
【0175】
本研究において、レトロウイルスの感染は、初めに哺乳動物細胞抗生物質G418の存在下、メチルセルロース中のLTBMCから除去された増殖細胞により評価した。SAX又はDC−29レトロウイルスに感染したNEO産生物を発現する前駆細胞は、高濃度のG418中で生存しコロニーを形成できるであろうが、感染していない細胞は高濃度のG418中で死滅するであろう。また、ウイルス添加終了後6週間以上高濃度のG418中で生存する前駆細胞の産生においては、それらの細胞が少し前により原始的な細胞(幹細胞)から分化していたことが必要であり、したがって、幹細胞が感染していることが示唆される。同様に、好中球及び単核細胞の表面に発現し、かつ培養の第11週の間に産生されるCD18では、感染期間に存在する全ての成熟細胞、前駆体及びクロノジェニック(Clonogenic)な前駆体が培養中4〜5週間で死滅してしまうので、原始的な造血幹細胞は培養の初めの2週間の間に感染していることが必要である。
【0176】
ここにおいて用いられるレトロウイルス感染に関する解析では、NEO遺伝子産生物が十分に発現しないので、レトロウイルスに感染した細胞のパーセンテージを軽視することができる。転移された遺伝子の産生物の発現の低さは、従来、ヒト及び霊長類モデルにおける問題とされてきた。したがって、本研究における感染前駆細胞のパーセンテージは、おそらく保守的な評価である。
【0177】
高[G418]中で生存していた前駆細胞のパーセンテージは、SAXレトロウイルス上清で感染させ、かつIL−3+GM−CSF±IL−1αを補給した培養の10週目において、およそ40%であった。これらの結果は、高パーセンテージの幹細胞が、培養の最初の2週間でレトロウイルス上清を補給した培養中で感染したことを示す。
【0178】
DC−29レトロウイルスで感染した前駆細胞のパーセンテージは、ウイルス添加終了後初めの4週間においてIL−3+GM−CSF+Epo+MGF及びヒブリカイン+Epo培地中では高いものであった(0〜21%)。この高レベル前駆細胞感染は、おそらくレトロウイルスによる前駆体及び原始細胞の直接感染によるものであった。高G418濃度中で生存している前駆細胞のパーセンテージは、ウイルス添加終了後4週間で減少したが、2週間後には高G418濃度中での生存は0〜22%に戻った。
【0179】
このDC−29試験におけるG418耐性前駆細胞の産生は、実際にDC−29レトロウイルスに感染した前駆細胞のパーセンテージを軽視できるであろう。感染したコロニーを選択するために用いられる高濃度のG418(1.6mg/mlのG418)は、SAX感染試験で用いられたものの2倍であり、生存するためには、NEO遺伝子産生物、ネオマイシンホスフォトランスフェラーゼの高い発現レベルが必要である。
【0180】
興味深いことに、上清を含有するSAX又はDC−29レトロウイルス、HGF IL−3+GM−CSF±Epo、IL−1α、又はMGF、又はヒブリカイン+Epoの組合せのいずれかを補給したLTBMCにおいて、G418中で生存している前駆細胞のパーセンテージは、ウイルス添加終了後6〜8時間で増加した。G418生存前駆細胞のパーセンテージが培養の後の段階になるに従って増加してゆくそのメカニズムはわかっていないが、一つの可能性としては、培養の初めの2週間に感染した幹細胞が、培養が進行するに従ってより活性になったということである。これはG418中で生存している細胞のパーセンテージを効果的に増加させるであろう。もう一つの可能性としては、最初の感染培養期間中に直接トランスフェクトされた前駆細胞とは異なる、より発現可能な組込み部位を有する幹細胞からの分化により、NEO遺伝子の発現が、培養中の後期に産生された前駆細胞において増大したのだろうということである。したがって、おそらくいくつかの原因によると考えられるが、培養におけるこの高レベルの前駆細胞生存率は、幹細胞がこれらのLTBMC中で感染したことを強く示唆するものである。
【0181】
さらに、これらのデータは、本発明において開示された培養条件下で造血幹細胞が培養中に増殖しており、レトロウイルスで転移された遺伝性物質をこれらの細胞へ組込むことができることを証明するものである。前駆体はこれらの幹細胞から連続的かつ活発に産生され、これら前駆体の多くはトランスフェクトされた遺伝子を含有し、発現した。これらのデータは、遺伝学的に改変されたヒト造血幹細胞が存在し、これらの培養中で増殖していたことを示すものである。
【0182】
IV.実験
I.形質転換細胞の形成
成長因子ヒトGM−CSF(Wong, Science (1984) 228:810-815)を原核性発現ベクターに挿入した。hGM−CSF cDNA(EcoRI〜AhaIII,およそ700bpフラグメント)をpSP65のEcoRI〜PstIフラグメントにクローン化した(Melton, Nucl, Acids Res. (1984) 2: 7035-7056)。得られたプラスミドをSP65GM−CSFとした。マウス メタロチオネインプロモーター(Glanville, Nature, (1981) 292: 267-269)をEcoRI及びBglIIで消化し、該プロモーターを含有するおよそ2kbのフラグメントをpSP65のEcoRI〜BamHIフラグメントに挿入してp65MTを作製した。その後、pSP65GM−CSFをEcoRIで消化し、DNAポリメラーゼIのクレノウ(Klenow)フラグメントを突出部に充填し、得られた連結されたDNAをHindIII で消化してGM−CSFのコード領域からなる700bpフラグメントを単離することにより、プラスミドpMT GM−CSFを構築した。このフラグメントをp65MTのSalI充填/HindIII 部位にサブクローンした。その後、メタロチオネインプロモーター及びGM−CSFコード領域を含む2.7kbフラグメントを単離し、SV−40プロモーターが除去されたpSV2neo(Southern and Berg, J. Mol. Appl. Genet (1982) 1: 327)においた。この結果、GM−CSFコード配列の下流にSV−40ポリAシグナルが存在するものが得られた。
【0183】
抗生物質ゲンタマイシン(G418)への耐性を付与するネオマイシン耐性遺伝子は、およそ3kbPvuII〜EcoRIフラグメントを単離し、EcoRIリンカーをPvuII部位におくことにより、pSV2neoから取り出した。EcoRI末端を有するneo耐性遺伝子をGM−CSF発現プラスミドのEcoRI部位にサブクローン化してプラスミドMTGM−CSFneoを作製した。
【0184】
単独で、及びSV−40プロモーター及びポリA部位のコントロール下、ギボンエイプ(gibbon ape)IL−3遺伝子をコードするプラスミドと共にトランスフェクションするもの(Yang, Cell (1986) 47:3-10)としてのプラスミドMTGM−CSFneoを、線形化DNAの電気穿孔法によりアフリカミドリサル細胞系CV1及びマウス細胞系NIH 3T3細胞へトランスフェクトした。形質転換細胞を、500mg/mlのG418を含有する培地選択により選択し、単離し、AML−193細胞(Adams et al, Leukemia (1989) 3: 314)を用いる上清のバイオアッセイにより、GM−CSF又はIL−3の産生でスクリーンした。その後、陽性の系のうちいくつかをでデクスター培養におけるヒト骨髄細胞用ストローマとして使用した。
さらに、オカヤマのカルシウム/リン酸法(Chen, Mol. Cell. Biol.(1987) 7: 2745-2752)を用いて正常マウス骨髄細胞を上記プラスミドとトランスフェクトし、誘導遺伝子を効果的に発現することがわかった。
【0185】
トランスフェクトされた線維芽細胞によるGM−CSF及びIL−3分泌を調べた。無血清72時間培養上清をNIH−3T3細胞より得、ウサギ抗GM−CSF又は抗IL−3抗体を中和することにより阻害可能なターゲット細胞における3Hの取り込みによってhGF分泌をアッセイした。20mg/ml GM−CSFにより誘発された増殖を100ユニットGM−CSFとし、10ng/ml IL−3により誘発された増殖を100ユニットIL−3とした。共にトランスフェクトされた細胞は、約35U/mlのGM−CSF及び約57U/mlのIL−3を産生した。
【0186】
II.潅流チャンバー
潅流チャンバーは、分解や生体融合することなく加圧滅菌できるようにデルリン(Delrin)キャップが付いたガラス製のシリンダーである。キャップには円柱状のグルーブがあり、そこへガラスシリンダーがフィットする。グルーブの底部にはチャンバーのルーメン(lumen)をシールするためのOリングがある。キャップにはいくつかの穴があり、そこにはルアー(Luer Lok)フィッティングがあり、さらにそのルアーフィッティングには付着性及び/又は非付着性細胞をサンプリングするために、チャンバー中央部へ伸びるチューブだけでなく、培地及びガス供給ラインも配置されている。このキャップは、120°間隔でガラスシリンダーの外側に配置されている3つの長いボルトで取付けてあり、組立をしっかりさせるために蝶ナット及びワッシャーが用いられている。
【0187】
チャンバーはサイドの貯蔵槽に掛ける。ループにはポンプ、オンラインセンサーのチャンバー、酸素供給器、及びサンプル注入孔、さらにサイド培地貯蔵槽がある。そして、サイド貯蔵槽中の培地は、セパレートポンプを用いてゆっくり交換される。この構成は、酸素供給器及び潅流チャンバーを通じて培地交換速度及び流速の独立制御を可能にする。前者は培地組成の長期変化を制御し潅流するために用いられ、後者はチャンバー中の溶解酸素の膨張及びフローパターンを制御するために用いることができる。小さなメッシュのポリスルフォネート膜を用いれば、チャンバー内でのプラグフロー及び非常に正確な量の成長因子及びバイオリアクターに導入させたい他の特別な成分の供給を正確に制御することが可能になる。
【0188】
トランスフェクトされた間質細胞は、寸断したコラーゲンスポンジ層の上に接種されるか、あるいは該間質細胞はコラーゲンでプレコートされた5μ孔のポリカーボネートフィルターの片側に置かれ、間質細胞が長時間フィルターに付着できるようになっている。細胞は、細胞質突出物を孔を通じて送りながら、細胞が片側に集密するまで適当な栄養培地中で生育できるようになっている。その後、骨髄細胞を膜のもう一方側に接種し、孔を通過して入り込んできた細胞質突出物に幹細胞を付着させる。
【0189】
ループのチャンバー及び成分を加熱滅菌した後、滅菌環境下でリアクターを組み立てる。そして雑菌混入のサインをモニターしながら、培地をサイドループ及びチャンバーを通じて数日間循環させる。そしてバイオリアクターの中央部に、細胞外マトリックスのみ、もしくは間質細胞を含有する前接種された細胞外マトリックス支持体を接種する。その代謝挙動及び/又は成長因子の反応性をモニターしながら、間質細胞をその後数日間チャンバー内に保持してもよく、結果が満足できるものであれば、骨髄細胞を接種するか、或いはすぐに骨髄細胞を接種する。いずれの場合においても、細胞層は潅流チャンバーの中央底部に保持される。
【0190】
細胞にはさらに細胞外マトリックスを接種し、細胞層は支持体に付着する。膜が使用される場合、チャンバーは倒置させてもよく、その場合、細胞層は中央部の天井に位置する。この配置において、間質細胞への付着がゆるむと、成熟している細胞は中央チャンバーの底部に沈む。そして、非付着細胞は、培地潅流圧力によって作動する出管の方への一定の細胞の流れにより採取される。
【0191】
代表的な実験においては、第1日目にチャンバーでは、寸断されたコラーゲンスポンジ支持体上にNIH−3T3細胞が接種される。最初の40日間は、潅流速度及び他の操作変数を調整した。40日目にはかなり安定した状態になり、それは約20日間持続した。64日目に、チャンバーに33×106ヒト骨髄細胞を接種した。初めの10日間で、採取された細胞の数は減少し、3日毎に約7〜8×105細胞が産生される安定した状態に落ち着いた。フローサイトメトリー解析は、一定のフラクション、すなわち採取細胞の約20%はHLA−DR陽性であることを示した。90日目には、ポンプの故障が起こり、pHは一夜で6.9以下に下がった。潅流速度を元に戻すと、非付着性細胞の産生は回復し、雑細菌混入が起こる前の安定した状態の産生速度に近づいた。この時点で本研究を終了した。
【0192】
上記の結果は、潅流チャンバーでは生体外造血形成を行なうことができ、pH下落後造血形成を生体外で元に戻すことができるであろうことを証明し、またグルコース濃度のデータは、酸素添加が制限されることを示す乳酸塩濃度の増加が起こらずに接種後にグルコース濃度が低下するので、おもに造血細胞はグルコース上で好気的に生育することを示した。グルコース/乳酸塩(嫌気性)代謝は、主にNIH−3T3間質層によると考えられる。同様に、一度造血細胞数がレベルダウンすると、グルタミン及びアンモニア濃度は接種前のレベルになり、これは骨髄細胞によるグルタミン消費が間質層と同レベルであったことを意味する。
【0193】
III.代謝生成物のモニタリング
グルタミンとアンモニアのみならずグルコースと乳酸塩の消費及び形成速度もトランスフェクトしたNIH−3T3細胞について求めた。(培地はIMDM+20%FCSであった。)グルコース消費の増加は毎日栄養補給したT−フラスコについて観察されただけであったが、低頻度栄養供給培地では全てが、同じゆるやかに減少するグルコース摂取速度パターンに従う。毎日50%交換した培養を、第18日目に毎日100%交換スケジュールに変更したところ、グルコース消費はただちに増加し、続いて第1日目から毎日100%交換した培地において観察されたものと同じ傾向を示した。グルコースにおける乳酸塩の収率が本質的に一定である場合、乳酸塩産生速度は同様のパターンに従う(0.9乳酸塩/グルコース;主に嫌気性間質代謝であることを示している。)
【0194】
グルタミン及びアンモニア濃度は、グルコース/乳酸塩代謝に類似したパターンを示す。37℃におけるグルタミンの化学的分解に対して補正された値を用いると、相対摂取速度を表わすグルタミン消費速度対グルコース消費速度は一定で、約1:8 グルタミン:グルコースである。予測される最適比率は酸素摂取速度に伴って変化し、最適摂取速度が増加するに従って、比率は下がる。
【0195】
同じようなことが、正常骨髄間質線維芽細胞から得られるグルコース/乳酸塩代謝データからも支持された。低頻度培地交換の条件下においては、培養は根本的には嫌気性であり、高い安定したレベルの乳酸塩の生成がすぐに達成され、かつ持続した。より頻度の高い培地交換では、細胞代謝はより急速になり、グルコース消費及び乳酸塩産生は増加した。自然な化学的分解のデータを補正した後で、グルコース消費が全く検出されないことが観察された。3T3細胞及び正常ヒト骨髄細胞の両者にたいして、上記の血清/培地交換速度である場合、細胞は分割し続け、ぎっしりと詰まり、それは臨界的な交換スケジュールである。
【0196】
さらに血清の潅流速度対栄養素の還流速度の相対的な重要性を確認するために、以下の実験を行なった:1)毎日交換される20%血清含有培地の入った一組のT−フラスコ;2)2組のT−フラスコ,一組には20%血清及び隔日交換される培地が入っており、もう一組には10%血清及び毎日交換される培地が入っている;3)2組のT−フラスコ、一組は10%血清及び隔日交換される培地が入っており、もう一組には5%血清及び毎日交換される培地が入っている;4)2組のT−フラスコ、一組には5%血清及び隔日交換される培地が入っており、もう一組には2.5%血清及び毎日交換される培地が入っている。血清交換速度は各グループで同じであるが、培地を含有する栄養素の交換速度は様々である。これらの実験の結果から、血清の交換速度が臨界的であることが示される。実験1)については、グルコース消費が増加し、第4日付近で実質的に安定して約9.5ミリモル/日の速度になったが、それ以外の全ての場合には、2倍量の血清を用いて培地を隔日に交換、又は半量の血清を用いて培地を毎日交換したこととは関係なく、グルコース消費はグループIの最初のグルコース消費以下からスタートして減少し、実質的に直線となった。このことは、間質細胞の代謝生育挙動に影響する血清又は一つ以上の血清成分の臨界的な潅流速度が必要であることを支持する。
【0197】
上記の結果から、効果的な方法でバイオリアクター内で造血細胞を生育させることができるであろうことは明確である。間質細胞は類似又は異種の提供源から提供することができ、そこにおいて間質細胞は重要な成長因子を提供するための遺伝子でトランスフェクトされている。この方法では、細胞の生育を補助するために、血清を培地に添加する必要はない。造血細胞を間質細胞から分離することを可能にする方法で支持体に付着する間質細胞を提供することにより、造血細胞は連続的に採取されて利用される。因子の組合せを適当に選択することにより、造血細胞の特異的な系統が生育されるであろう。さらに、所望により、遺伝子を造血細胞へ導入するためのトランスフェクトウイルスの貯蔵槽として間質細胞を提供してもよい。
【0198】
本明細書中で引用されている全ての刊行物及び特許出願は、ここにおいて引例として加入するものとし、各刊行物及び特許出願を明確に独立して引例に加入されるものとして示した。
明らかに、上記の技術から鑑みて本発明の多くの改変及び変形が可能である。したがって、添付する請求の範囲内で、本発明はここにおいて特に記載した以外の方法でも実施できることが理解される。
【図面の簡単な説明】
【0199】
【図1】灌流室の概略を示す。
【図2】灌流培地通路の概略及びフロー・ダイアグラムを図示している。
【図3】3aは細胞分離のせん断応力を測定する為のフローチャンバーの概略を示す。 3bは3aのフローチャンバーの側面を示す。 3cは造血細胞のせん断応力・プロフィールのグラフである。
【図4】4a及び4bは造血細胞の増殖、分離用のフローチャンバーの上面及び側面図である。
【図5】5a及び5bは基質細胞の連続増殖を可能にするために障壁を順番に取り除いたフローチャンバーの概略を示す。
【特許請求の範囲】
【請求項1】
ヒト造血系由来幹細胞を含有する細胞組成物を、約24〜約48時間の間に1mLの培養あたり1mLの培地で連続的又は周期的に交換される液体培養培地内で培養し、該培養を生理学上許容しうる条件下に保持しながら代謝産生物を除去し、消費した栄養素を補給することを特徴とするエクスビボでヒト造血系由来幹細胞分裂物を得るための培養方法。
【請求項2】
培地が連続的に交換される、請求項1記載の方法。
【請求項3】
培地の交換が、新鮮培地をヒト造血系由来幹細胞塊の少なくとも一部に接触するように加えて潅流されることからなる、請求項2記載の方法。
【請求項4】
培地が周期的に交換される、請求項1記載の方法。
【請求項5】
培地の交換が、新鮮培地をヒト造血系由来幹細胞塊の少なくとも一部に接触するように加えて潅流することにより行われる請求項4記載の方法。
【請求項6】
培地が造血培養培地であり、ヒト末梢血単核細胞、ヒト骨髄細胞、ヒト胎児肝細胞及びヒト臍帯血細胞から選ばれる少なくとも一つが培養される、請求項1記載の方法。
【請求項7】
パッキングに付着している血管間質細胞と造血系由来幹細胞を含有するヒト造血細胞とを反応容器内で混合し、そこにおいて該間質細胞の少なくとも一部は形質転換されて、当該造血細胞の増殖、分化、あるいはその両者を制御する成長因子の少なくとも一つを分泌することができ;
該反応容器を生理学上許容可能な条件下に保持したまま、代謝産生物を除去し消費栄養素を補給しながら実質上連続的に該細胞を栄養培地とともに該反応容器に潅流し;さらに
造血細胞を該反応器から採取するが、該反応容器内に接種された該ヒト造血細胞が悪性細胞からなる疑いがある場合には、該潅流が、該間質細胞に対する悪性細胞の親和力よりも大きくかつ正常造血細胞の親和力よりも小さな力を与えるような速度で行なわれることからなる、請求項1記載の方法。
【請求項8】
造血細胞表面の剪断応力が約1.0dyne/cm2より大きくなるような潅流速度を用いることからなる、請求項7記載の方法。
【請求項9】
遺伝子伝達ベクター存在下で細胞組成物を培養すること、及び安定かつ遺伝学的に形質転換されたヒト造血系由来幹細胞を得ることからなる、請求項1記載の方法。
【請求項10】
培地が連続的に交換される、請求項9記載の方法。
【請求項11】
培地の交換がヒト造血系由来幹細胞の塊の少なくとも一部に接触するように新鮮培地を潅流することからなる、請求項10記載の方法。
【請求項12】
培地が周期的に交換されることからなる、請求項9記載の方法。
【請求項13】
培地の交換がヒト造血系由来幹細胞の塊の少なくとも一部に接触するように新鮮培地を潅流することからなる、請求項12記載の方法。
【請求項14】
培地が造血培養培地であり、ヒト末梢血単核細胞、ヒト骨髄細胞、ヒト胎児肝細胞、胎児性幹細胞及びヒト臍帯血細胞から選ばれる少なくとも一つが培養される、請求項9記載の方法。
【請求項15】
細胞組成物がヒト造血系由来幹細胞に関して濃縮されている、請求項9記載の方法。
【請求項16】
培養ヒト造血系由来幹細胞が、安定かつ遺伝学的に形質転換されたヒト前駆細胞を産生する、請求項9記載の方法。
【請求項17】
培養ヒト造血系由来幹細胞が、安定かつ遺伝学的に形質転換されたヒト造血前駆細胞を産生する、請求項9記載の方法。
【請求項18】
IL−3及びGM−CSFを、各々0.1〜100ng・mL-1・日-1の速度で培地に連続的に添加する、請求項9記載の方法。
【請求項19】
マスト細胞成長因子が1〜100ng・mL-1・日-1の速度で培地に添加されるか、又はIL−1αが3〜5日間に10〜100U・mL-1の速度で培地に添加される、請求項18記載の方法。
【請求項20】
マスト細胞成長因子が培地に添加される、請求項19記載の方法。
【請求項21】
IL−1αが培地に添加される、請求項19記載の方法。
【請求項22】
マスト細胞成長因子及びIL−αの両者が培地に添加される、請求項19記載の方法。
【請求項23】
培地が動物の血清又は血漿を含む、請求項9記載の方法。
【請求項24】
培地がコルチコステロイドを含む、請求項9記載の方法。
【請求項25】
培地中のグルコース濃度が5〜20mMの範囲内に、培地中の乳酸塩濃度が約35mM以下に、培地中のグルタミン濃度が1〜3mMの範囲内に、かつ培地中のアンモニア濃度が2.5mM以下に保持されることからなる、請求項9記載の方法。
【請求項26】
培養が10-3〜10-1(間質細胞/全細胞)のヒト間質細胞を含む条件で行われる請求項14記載の方法。
【請求項27】
遺伝子伝達ベクターがレトロウイルスである、請求項9記載の方法。
【請求項28】
ヒト造血系由来幹細胞をレトロウイルスパッケージング細胞系上清の存在下で培養することからなる、請求項9記載の方法。
【請求項29】
ヒト造血系由来幹細胞をレトロウイルスパッケージング細胞系の存在下で培養することからなる、請求項9記載の方法。
【請求項30】
IL−3及びGM−CSFが各々0.1〜100ng・mL-1・日-1の速度で連続的に培地に添加される、請求項1記載の方法。
【請求項31】
Epoが培地に0.001〜10U・mL-1・日-1の速度で添加されるか、マスト細胞成長因子が培地に1〜100ng・mL-1・日-1の速度で添加されるか、又はIL−1(α又はβ)が培地に3〜5日間当たり10〜100U・mL-1の速度で添加される、請求項30記載の方法。
【請求項32】
Epoが培地に0.001〜10U・mL-1・日-1の速度で添加される、請求項30記載の方法。
【請求項33】
さらに、G−CSF、塩基性線維芽細胞成長因子、IL−6、IL−7、IL−8、IL−9、IL−10、IL−11、PDGF及びEGFから選ばれる少なくとも一つが培地に添加される、請求項32記載の方法。
【請求項34】
培地が動物の血清又は血漿を含む、請求項1記載の方法。
【請求項35】
培地が、コルチコステロイドからなる、請求項1記載の方法。
【請求項36】
培地中のグルコース濃度が5〜20mMの範囲内に、培地中の乳酸塩濃度が約35mM以下に、培地中のグルタミン濃度が1〜3mMの範囲内に、かつ培地中のアンモニア濃度が2.5mM以下に保持されることからなる、請求項1記載の方法。
【請求項37】
培養が10-3〜10-1(間質細胞/全細胞)のヒト間質細胞を含む条件で行われる、請求項6記載の方法。
【請求項38】
増殖したヒト造血系由来幹細胞プールを得ることからなる、請求項1記載の方法。
【請求項39】
増殖したヒト造血幹細胞プールを得ることからなる、請求項38記載の方法。
【請求項40】
ヒト造血前駆細胞を培養することからなる、請求項1記載の方法。
【請求項41】
細胞組成物がヒト造血前駆細胞に関して濃縮されている、請求項40記載の方法。
【請求項42】
IL−3及びGM−CSFが各々0.1〜100ng・mL-1・日-1の速度で連続的に培地に添加される、請求項40記載の方法。
【請求項43】
Epoが培地に0.001〜10U・mL-1・日-1の速度で添加されるか、マスト細胞成長因子が培地に1〜100ng・mL-1・日-1の速度で添加されるか、又はIL−1(α又はβ)が培地に3〜5日間当たり10〜100U・mL-1の速度で添加される、請求項42記載の方法。
【請求項44】
赤血球を含有する細胞培養が得られる、請求項6記載の方法。
【請求項45】
顆粒球を含有する細胞培養が得られる、請求項6記載の方法。
【請求項46】
ヒト間質細胞の代謝、GM−CSF分泌又はIL−6分泌が増加する、請求項37記載の方法。
【請求項47】
ヒト間質細胞がヒト骨髄間質細胞である、請求項46記載の方法。
【請求項48】
成熟細胞を培養から除去する、請求項1記載の方法。
【請求項49】
成熟細胞を培養から除去する、請求項39記載の方法。
【請求項50】
リアクターチャンバー;並びに
栄養培地を該リアクターチャンバーから約24〜約48時間の間に1mLの培養あたり1mLの培地の速度で導入及び除去するための手段及び当該リアクターチャンバーからの溶出物をモニターするための手段を有し;
該リアクターチャンバーにおいて、間質細胞が幹細胞を含むヒト造血細胞とともにパッキングに付着しており、間質細胞の少なくとも一部が形質転換され、幹細胞の増殖、分化、又はその両者を制御する成長因子のうち少なくとも一つを分泌できるバイオリアクター。
【請求項51】
ヒト造血系由来幹細胞組成物を、エクスビボでヒト造血系由来幹細胞分裂物を得るのに充分な速度で交換される液体培養培地中で、当該培地を生理学的に許容し得る条件に保持しながら培養する方法であって、培地の交換速度が細胞密度に関連する速度で交換される方法であり、当該速度が1×104〜1×107細胞/mL培地の細胞密度となるように1日あたり50〜100%交換する速度であることを特徴とするエクスビボで当該ヒト幹細胞分裂物を得るための培養方法。
【請求項52】
ヒト造血系由来幹細胞が、ヒト骨髄中に見出されるヒト幹細胞である請求項51記載の培養方法。
【請求項53】
ヒト造血系由来幹細胞組成物を、少なくとも1日あたり50%交換する速度で交換される液体培養培地中で培養するものである請求項51記載の培養方法。
【請求項54】
IL−3、GM−CSF、スチール因子、Epo、IL−1α、IL−1β、G−CSF、塩基性線維芽細胞増殖因子、IL−6、IL−7、IL−8、IL−9、IL−10、IL−11、PDGF又はEFGを含む培地を用いる請求項51記載の培養方法。
【請求項55】
ヒト造血系由来幹細胞が、ヒト造血幹細胞である請求項51記載の培養方法。
【請求項56】
ヒト造血系由来幹細胞が、ヒト造血幹細胞である請求項53記載の培養方法。
【請求項57】
ヒト造血系由来幹細胞が、ヒト造血幹細胞である請求項51記載の培養方法。
【請求項58】
遺伝子伝達ベクターの存在下に、培養すること、及び安定で遺伝学的に形質転換されたヒト造血系由来幹細胞を得ることからなる請求項51記載の方法。
【請求項59】
ヒト造血系由来幹細胞が、ヒト造血幹細胞である請求項58記載の方法。
【請求項60】
ヒト造血系由来幹細胞が、ヒト末梢血単核球、ヒト骨髄細胞、ヒト胎児肝細胞、ヒト胚幹細胞又はヒト膵帯血細胞である請求項58記載の方法。
【請求項61】
培養されたヒト造血幹細胞が、安定な遺伝学的に形質転換されたヒト造血前駆細胞を生産する請求項59記載の方法。
【請求項62】
IL−3及びGM−CSFを培地に連続的に添加する請求項58記載の方法。
【請求項63】
スチール因子又はIL−1αを培地に添加する請求項58記載の方法。
【請求項64】
スチール因子を培地に添加する請求項58記載の方法。
【請求項65】
スチール因子とIL−1αの両者を培地に添加する請求項58記載の方法。
【請求項66】
遺伝子伝達ベクターがレトロウイルスである請求項58記載の方法。
【請求項67】
ヒト造血幹細胞をレトロウイルスパッケージング細胞系上清の存在下で培養するものである請求項58記載の方法。
【請求項68】
ヒト造血幹細胞を含有するヒト造血細胞を、灌流された液体培養培地中で、マトリックスに接着し、ヒト造血細胞の増殖、分化又はその両者に作用する少なくとも1つの増殖因子を排出し得る形質転換間質細胞の存在下で、細胞を約24〜約48時間毎に液体培養培地を交換することにより灌流し、生理学的に許容される条件下に保持しながら培養し、エクスビボでヒト造血幹細胞分裂物を得ることを特徴とするエクスビボでヒト造血幹細胞分裂物を得る方法。
【請求項69】
ヒト造血細胞が骨髄細胞であり、液体培養培地と間質細胞の灌流がヒト骨髄間質細胞の分裂を促し、その結果ヒト骨髄幹細胞が反応器内に産生され、さらにそのヒト骨髄幹細胞を形質移入することを特徴とする請求項68記載の方法。
【請求項1】
ヒト造血系由来幹細胞を含有する細胞組成物を、約24〜約48時間の間に1mLの培養あたり1mLの培地で連続的又は周期的に交換される液体培養培地内で培養し、該培養を生理学上許容しうる条件下に保持しながら代謝産生物を除去し、消費した栄養素を補給することを特徴とするエクスビボでヒト造血系由来幹細胞分裂物を得るための培養方法。
【請求項2】
培地が連続的に交換される、請求項1記載の方法。
【請求項3】
培地の交換が、新鮮培地をヒト造血系由来幹細胞塊の少なくとも一部に接触するように加えて潅流されることからなる、請求項2記載の方法。
【請求項4】
培地が周期的に交換される、請求項1記載の方法。
【請求項5】
培地の交換が、新鮮培地をヒト造血系由来幹細胞塊の少なくとも一部に接触するように加えて潅流することにより行われる請求項4記載の方法。
【請求項6】
培地が造血培養培地であり、ヒト末梢血単核細胞、ヒト骨髄細胞、ヒト胎児肝細胞及びヒト臍帯血細胞から選ばれる少なくとも一つが培養される、請求項1記載の方法。
【請求項7】
パッキングに付着している血管間質細胞と造血系由来幹細胞を含有するヒト造血細胞とを反応容器内で混合し、そこにおいて該間質細胞の少なくとも一部は形質転換されて、当該造血細胞の増殖、分化、あるいはその両者を制御する成長因子の少なくとも一つを分泌することができ;
該反応容器を生理学上許容可能な条件下に保持したまま、代謝産生物を除去し消費栄養素を補給しながら実質上連続的に該細胞を栄養培地とともに該反応容器に潅流し;さらに
造血細胞を該反応器から採取するが、該反応容器内に接種された該ヒト造血細胞が悪性細胞からなる疑いがある場合には、該潅流が、該間質細胞に対する悪性細胞の親和力よりも大きくかつ正常造血細胞の親和力よりも小さな力を与えるような速度で行なわれることからなる、請求項1記載の方法。
【請求項8】
造血細胞表面の剪断応力が約1.0dyne/cm2より大きくなるような潅流速度を用いることからなる、請求項7記載の方法。
【請求項9】
遺伝子伝達ベクター存在下で細胞組成物を培養すること、及び安定かつ遺伝学的に形質転換されたヒト造血系由来幹細胞を得ることからなる、請求項1記載の方法。
【請求項10】
培地が連続的に交換される、請求項9記載の方法。
【請求項11】
培地の交換がヒト造血系由来幹細胞の塊の少なくとも一部に接触するように新鮮培地を潅流することからなる、請求項10記載の方法。
【請求項12】
培地が周期的に交換されることからなる、請求項9記載の方法。
【請求項13】
培地の交換がヒト造血系由来幹細胞の塊の少なくとも一部に接触するように新鮮培地を潅流することからなる、請求項12記載の方法。
【請求項14】
培地が造血培養培地であり、ヒト末梢血単核細胞、ヒト骨髄細胞、ヒト胎児肝細胞、胎児性幹細胞及びヒト臍帯血細胞から選ばれる少なくとも一つが培養される、請求項9記載の方法。
【請求項15】
細胞組成物がヒト造血系由来幹細胞に関して濃縮されている、請求項9記載の方法。
【請求項16】
培養ヒト造血系由来幹細胞が、安定かつ遺伝学的に形質転換されたヒト前駆細胞を産生する、請求項9記載の方法。
【請求項17】
培養ヒト造血系由来幹細胞が、安定かつ遺伝学的に形質転換されたヒト造血前駆細胞を産生する、請求項9記載の方法。
【請求項18】
IL−3及びGM−CSFを、各々0.1〜100ng・mL-1・日-1の速度で培地に連続的に添加する、請求項9記載の方法。
【請求項19】
マスト細胞成長因子が1〜100ng・mL-1・日-1の速度で培地に添加されるか、又はIL−1αが3〜5日間に10〜100U・mL-1の速度で培地に添加される、請求項18記載の方法。
【請求項20】
マスト細胞成長因子が培地に添加される、請求項19記載の方法。
【請求項21】
IL−1αが培地に添加される、請求項19記載の方法。
【請求項22】
マスト細胞成長因子及びIL−αの両者が培地に添加される、請求項19記載の方法。
【請求項23】
培地が動物の血清又は血漿を含む、請求項9記載の方法。
【請求項24】
培地がコルチコステロイドを含む、請求項9記載の方法。
【請求項25】
培地中のグルコース濃度が5〜20mMの範囲内に、培地中の乳酸塩濃度が約35mM以下に、培地中のグルタミン濃度が1〜3mMの範囲内に、かつ培地中のアンモニア濃度が2.5mM以下に保持されることからなる、請求項9記載の方法。
【請求項26】
培養が10-3〜10-1(間質細胞/全細胞)のヒト間質細胞を含む条件で行われる請求項14記載の方法。
【請求項27】
遺伝子伝達ベクターがレトロウイルスである、請求項9記載の方法。
【請求項28】
ヒト造血系由来幹細胞をレトロウイルスパッケージング細胞系上清の存在下で培養することからなる、請求項9記載の方法。
【請求項29】
ヒト造血系由来幹細胞をレトロウイルスパッケージング細胞系の存在下で培養することからなる、請求項9記載の方法。
【請求項30】
IL−3及びGM−CSFが各々0.1〜100ng・mL-1・日-1の速度で連続的に培地に添加される、請求項1記載の方法。
【請求項31】
Epoが培地に0.001〜10U・mL-1・日-1の速度で添加されるか、マスト細胞成長因子が培地に1〜100ng・mL-1・日-1の速度で添加されるか、又はIL−1(α又はβ)が培地に3〜5日間当たり10〜100U・mL-1の速度で添加される、請求項30記載の方法。
【請求項32】
Epoが培地に0.001〜10U・mL-1・日-1の速度で添加される、請求項30記載の方法。
【請求項33】
さらに、G−CSF、塩基性線維芽細胞成長因子、IL−6、IL−7、IL−8、IL−9、IL−10、IL−11、PDGF及びEGFから選ばれる少なくとも一つが培地に添加される、請求項32記載の方法。
【請求項34】
培地が動物の血清又は血漿を含む、請求項1記載の方法。
【請求項35】
培地が、コルチコステロイドからなる、請求項1記載の方法。
【請求項36】
培地中のグルコース濃度が5〜20mMの範囲内に、培地中の乳酸塩濃度が約35mM以下に、培地中のグルタミン濃度が1〜3mMの範囲内に、かつ培地中のアンモニア濃度が2.5mM以下に保持されることからなる、請求項1記載の方法。
【請求項37】
培養が10-3〜10-1(間質細胞/全細胞)のヒト間質細胞を含む条件で行われる、請求項6記載の方法。
【請求項38】
増殖したヒト造血系由来幹細胞プールを得ることからなる、請求項1記載の方法。
【請求項39】
増殖したヒト造血幹細胞プールを得ることからなる、請求項38記載の方法。
【請求項40】
ヒト造血前駆細胞を培養することからなる、請求項1記載の方法。
【請求項41】
細胞組成物がヒト造血前駆細胞に関して濃縮されている、請求項40記載の方法。
【請求項42】
IL−3及びGM−CSFが各々0.1〜100ng・mL-1・日-1の速度で連続的に培地に添加される、請求項40記載の方法。
【請求項43】
Epoが培地に0.001〜10U・mL-1・日-1の速度で添加されるか、マスト細胞成長因子が培地に1〜100ng・mL-1・日-1の速度で添加されるか、又はIL−1(α又はβ)が培地に3〜5日間当たり10〜100U・mL-1の速度で添加される、請求項42記載の方法。
【請求項44】
赤血球を含有する細胞培養が得られる、請求項6記載の方法。
【請求項45】
顆粒球を含有する細胞培養が得られる、請求項6記載の方法。
【請求項46】
ヒト間質細胞の代謝、GM−CSF分泌又はIL−6分泌が増加する、請求項37記載の方法。
【請求項47】
ヒト間質細胞がヒト骨髄間質細胞である、請求項46記載の方法。
【請求項48】
成熟細胞を培養から除去する、請求項1記載の方法。
【請求項49】
成熟細胞を培養から除去する、請求項39記載の方法。
【請求項50】
リアクターチャンバー;並びに
栄養培地を該リアクターチャンバーから約24〜約48時間の間に1mLの培養あたり1mLの培地の速度で導入及び除去するための手段及び当該リアクターチャンバーからの溶出物をモニターするための手段を有し;
該リアクターチャンバーにおいて、間質細胞が幹細胞を含むヒト造血細胞とともにパッキングに付着しており、間質細胞の少なくとも一部が形質転換され、幹細胞の増殖、分化、又はその両者を制御する成長因子のうち少なくとも一つを分泌できるバイオリアクター。
【請求項51】
ヒト造血系由来幹細胞組成物を、エクスビボでヒト造血系由来幹細胞分裂物を得るのに充分な速度で交換される液体培養培地中で、当該培地を生理学的に許容し得る条件に保持しながら培養する方法であって、培地の交換速度が細胞密度に関連する速度で交換される方法であり、当該速度が1×104〜1×107細胞/mL培地の細胞密度となるように1日あたり50〜100%交換する速度であることを特徴とするエクスビボで当該ヒト幹細胞分裂物を得るための培養方法。
【請求項52】
ヒト造血系由来幹細胞が、ヒト骨髄中に見出されるヒト幹細胞である請求項51記載の培養方法。
【請求項53】
ヒト造血系由来幹細胞組成物を、少なくとも1日あたり50%交換する速度で交換される液体培養培地中で培養するものである請求項51記載の培養方法。
【請求項54】
IL−3、GM−CSF、スチール因子、Epo、IL−1α、IL−1β、G−CSF、塩基性線維芽細胞増殖因子、IL−6、IL−7、IL−8、IL−9、IL−10、IL−11、PDGF又はEFGを含む培地を用いる請求項51記載の培養方法。
【請求項55】
ヒト造血系由来幹細胞が、ヒト造血幹細胞である請求項51記載の培養方法。
【請求項56】
ヒト造血系由来幹細胞が、ヒト造血幹細胞である請求項53記載の培養方法。
【請求項57】
ヒト造血系由来幹細胞が、ヒト造血幹細胞である請求項51記載の培養方法。
【請求項58】
遺伝子伝達ベクターの存在下に、培養すること、及び安定で遺伝学的に形質転換されたヒト造血系由来幹細胞を得ることからなる請求項51記載の方法。
【請求項59】
ヒト造血系由来幹細胞が、ヒト造血幹細胞である請求項58記載の方法。
【請求項60】
ヒト造血系由来幹細胞が、ヒト末梢血単核球、ヒト骨髄細胞、ヒト胎児肝細胞、ヒト胚幹細胞又はヒト膵帯血細胞である請求項58記載の方法。
【請求項61】
培養されたヒト造血幹細胞が、安定な遺伝学的に形質転換されたヒト造血前駆細胞を生産する請求項59記載の方法。
【請求項62】
IL−3及びGM−CSFを培地に連続的に添加する請求項58記載の方法。
【請求項63】
スチール因子又はIL−1αを培地に添加する請求項58記載の方法。
【請求項64】
スチール因子を培地に添加する請求項58記載の方法。
【請求項65】
スチール因子とIL−1αの両者を培地に添加する請求項58記載の方法。
【請求項66】
遺伝子伝達ベクターがレトロウイルスである請求項58記載の方法。
【請求項67】
ヒト造血幹細胞をレトロウイルスパッケージング細胞系上清の存在下で培養するものである請求項58記載の方法。
【請求項68】
ヒト造血幹細胞を含有するヒト造血細胞を、灌流された液体培養培地中で、マトリックスに接着し、ヒト造血細胞の増殖、分化又はその両者に作用する少なくとも1つの増殖因子を排出し得る形質転換間質細胞の存在下で、細胞を約24〜約48時間毎に液体培養培地を交換することにより灌流し、生理学的に許容される条件下に保持しながら培養し、エクスビボでヒト造血幹細胞分裂物を得ることを特徴とするエクスビボでヒト造血幹細胞分裂物を得る方法。
【請求項69】
ヒト造血細胞が骨髄細胞であり、液体培養培地と間質細胞の灌流がヒト骨髄間質細胞の分裂を促し、その結果ヒト骨髄幹細胞が反応器内に産生され、さらにそのヒト骨髄幹細胞を形質移入することを特徴とする請求項68記載の方法。
【図1】

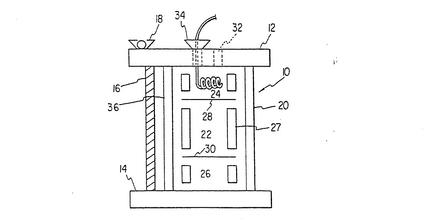
【図2】
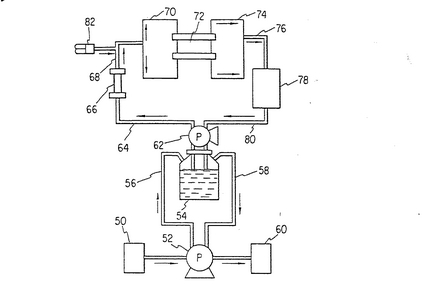
【図3】
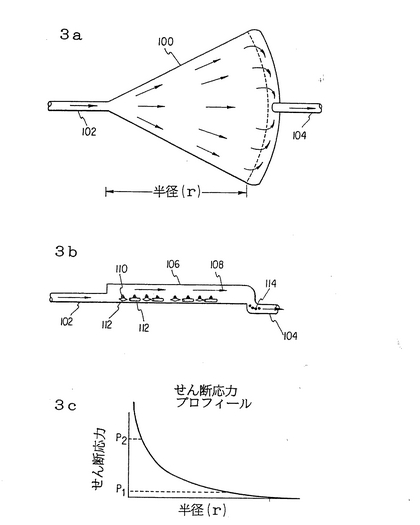
【図4】
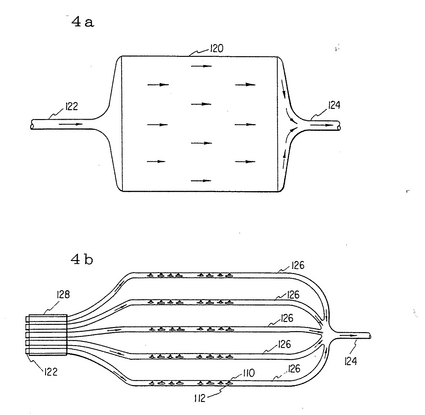
【図5】
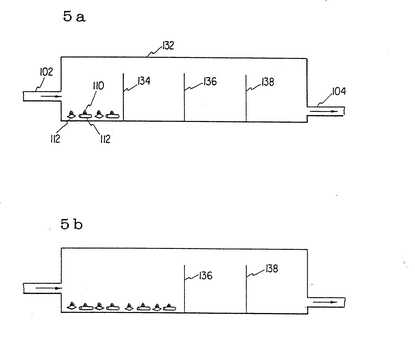
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2006−158401(P2006−158401A)
【公開日】平成18年6月22日(2006.6.22)
【国際特許分類】
【出願番号】特願2006−956(P2006−956)
【出願日】平成18年1月5日(2006.1.5)
【分割の表示】特願平4−504303の分割
【原出願日】平成3年12月17日(1991.12.17)
【出願人】(598166032)リージェンツ オブ ザ ユニバーシティ オブ ミシガン (1)
【氏名又は名称原語表記】REGENTS OF THE UNIVERSITY OF MICHIGAN
【Fターム(参考)】
【公開日】平成18年6月22日(2006.6.22)
【国際特許分類】
【出願日】平成18年1月5日(2006.1.5)
【分割の表示】特願平4−504303の分割
【原出願日】平成3年12月17日(1991.12.17)
【出願人】(598166032)リージェンツ オブ ザ ユニバーシティ オブ ミシガン (1)
【氏名又は名称原語表記】REGENTS OF THE UNIVERSITY OF MICHIGAN
【Fターム(参考)】
[ Back to top ]