
ヒト抗体及びタンパク質
【課題】低免疫原性を示す抗体を含む合成タンパク質を提供する。
【解決手段】ヒト用複合抗体を提供し、具体的には、一又はそれ以上のT細胞エピトープを除去するように修飾された抗体を提供する。修飾抗体又は前記修飾抗体の抗原結合フラグメントにおいて、前記修飾抗体又は抗原結合フラグメントの重鎖及び軽鎖可変領域が、各々1又はそれ以上の他の抗体又は抗原結合フラグメントからのアミノ酸配列の2又はそれ以上のセグメントから成り、前記セグメントが全てCDRsでもなくフレームワーク領域でもないことを特徴とする修飾抗体又は前記修飾抗体の抗原結合フラグメント。このようなタンパク質を生成する方法。
【解決手段】ヒト用複合抗体を提供し、具体的には、一又はそれ以上のT細胞エピトープを除去するように修飾された抗体を提供する。修飾抗体又は前記修飾抗体の抗原結合フラグメントにおいて、前記修飾抗体又は抗原結合フラグメントの重鎖及び軽鎖可変領域が、各々1又はそれ以上の他の抗体又は抗原結合フラグメントからのアミノ酸配列の2又はそれ以上のセグメントから成り、前記セグメントが全てCDRsでもなくフレームワーク領域でもないことを特徴とする修飾抗体又は前記修飾抗体の抗原結合フラグメント。このようなタンパク質を生成する方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、最終抗体又はタンパク質分子内で、ヒト抗体又はタンパク質からのアミノ酸配列の2又はそれ以上のセグメントを結合する抗体及びタンパク質の生成に関する。具体的には、本発明は、配列セグメントのこのような結合を提供して、最終抗体又はタンパク質分子中のT細胞エピトープの数を低減又は回避するようにする。本発明は、特に、ヒトの医薬品又はインビボ診断剤として用いる抗体及びタンパク質の生成に関する。
【0002】
ここ20年で、ヒトの潜在的医薬品として用いる組み換え型モノクローナル抗体の生成に大幅な進歩が見られている。ファージ提示法又は遺伝子導入マウスによる、キメラ化技術、ヒト型化技術及びヒト抗体クローニング技術は、非ヒトモノクローナル抗体よりも低い免疫原性を有するヒトに投与される場合に一般的に良好な耐容性を有する抗体を提供している。しかしながら、これらの技術によって生成したいくつかの抗体は、このような抗体の遺伝子起源がヒトである場合でさえ、患者の免疫原性を誘発することを示している。例えば、ヒト抗体Humira(登録商標)は、12%の関節リウマチ患者に免疫原性を誘発し、ヒト化抗体CAMPATH(登録商標)は、約50%の患者に免疫原性を誘発する。このような免疫原性の誘発は、抗体可変領域内での非アミノ酸配列の一部の存在に起因しているようである。この非アミノ酸配列の一部は、いくつかの場合に、免疫原性をもたらすT細胞反応を誘発するT細胞エピトープを作ることができる。従って、高いヒト起源を有するが、改善した技術と、T細胞反応を誘発する配列の作成をできるだけ避ける抗体組成物が求められている。
【0003】
本発明は、方法と、それによって、治療用に、このような抗体(ここでは、「複合抗体」という)が、最終抗体分子内でヒト抗体からのアミノ酸配列の2又はそれ以上のセグメントを結合するような結果物としての抗体組成を提供する。
【0004】
従って、第1の態様では、本発明は修飾抗体、又はこれの抗原結合フラグメントを提供しており、これは修飾抗体又は抗原結合フラグメントの重鎖及び軽鎖可変領域が、1又はそれ以上の別の抗体又は抗原結合フラグメントからのアミノ酸配列の2又はそれ以上のセグメントから成り、これらのセグメントが、全CDRでもフレームワーク領域でもない。
【0005】
本発明のコンテキストでは、用語「セグメント」は、抗体分子内で見つけられる隣接アミノ酸配列をいい、このようなセグメントは、2から125アミノ酸長サイズの範囲であり、好ましくは、2から31アミノ酸長の範囲であり、このようなセグメントは、全CDRでも全フレームワーク領域でもない。治療に用いる場合、本発明の複合抗体は、一般的に、複合抗体の可変領域内で、様々なヒト抗体からのアミノ酸配列の2又はそれ以上のセグメントを結合する。具体的には、本発明は、複合抗体重鎖及び軽鎖可変領域(それぞれ、VH及びVLという)に関し、VHとVLは各々、完全に2又はそれ以上のヒト抗体可変領域からの配列のセグメントから成り、一般的に、各合成VH及びVLは、源ヒト抗体VH及びVL中の位置に相当するヒト可変領域配列位のセグメントを含み、例えば、合成VH配列中のアミノ酸1から10は、ヒト抗体のアミノ酸1から10から導出する。代替的に、複合抗体中のヒトVH又はVL配列のセグメントは、源ヒト抗体VH又はVL中の配列位に関わりなく、どの配列位に位置してもいてもよい。この源ヒト抗体VH及びVLは、例えば、ヒトモノクローナル抗体V領域配列のデータベースに提供されているような、現存のヒト抗体可変(V)領域アミノ酸配列であり、V領域体細胞変異体、及び生殖細胞系とは異なるその他の変異体を有する親和性成熟抗体からの配列、生殖細胞系V領域からの配列、固定V領域フレームワークセットを有するが可変CDRを有する抗体等といった種の抗体からの配列のセグメントから作られた人工的に構成された抗体V領域からの配列、ファージ表示ライブラリ等のヒト抗体ライブラリから選択された配列、及びヒト抗体又は抗体フラグメントをコードするトランスジェニック動物発現遺伝子から導出したヒト抗体の配列を含んでもよい。
【0006】
本発明の好ましい実施例では、治療用の本発明の複合抗体は、最終複合抗体V領域のヒトT細胞エピトープを制限又は回避する組み合わせで多重ヒトVH及びVL配列セグメントを結合することによって構成される。
【0007】
これに関して、ヒトT細胞エピトープは、ヒトMHCクラスII分子に結合することができるアミノ酸配列であり、CD4+T細胞への提示によって、ヘルパーT細胞反応を誘発する。最終複合抗体中のT細胞エピトープを制限、又は回避するヒトVH及びVL配列セグメント及びセグメントの組み合わせを選択できる。これは、ヒト生殖細胞系配列から等のT細胞エピトープを含まないセグメントの使用によって、及び隣接するセグメントを結合して、例えば、2つのセグメントの接合点における非MHC結合配列の生成によって、別のヒト生殖細胞系配列の生成によって、又は非生殖細胞系配列にも関わらず、ヘルパーT細胞反応を誘発しない配列の生成によって、T細胞エピトープを含まない新しい配列を作ることによって達成することができる。
【0008】
本発明の別の好ましい実施例では、更なるアミノ酸配列を加える、又は、1又はそれ以上の制御性T細胞エピトープ(「Trエピトープ」)を規定する複合抗体分子内で作ることができる。本発明の目的のために、Trエピトープは、接触依存性メカニズムと同様に、IL−10及びTGF−β等の阻害性サイトカインの分泌によって免疫反応を調整する能力を有するCD4+ CD25+ T細胞エピトープを刺激するMHC結合ペプチドである。このように、本発明の範囲内では、制御性T細胞エピトープは、ある条件下で、免疫反応の制御に寄与する1又はそれ以上のインビトロ又はインビボ活性を誘発するように示されるペプチドを含むことができる。例えば、制御性T細胞エピトープは、CD4+ CD25+ T細胞を誘発する又は活性化する作用、IL−10及び/又はTGF−β等の阻害性サイトカインの放出を誘発する作用、又はその他の測定可能な免疫抑制関連インビトロ又はインビボ活性を有するペプチドを含む。これらの全ての場合において、これらの作用は、CD4+ CD25+ T細胞の作用に関連する。従って、このようなTrエピトープは、複合抗体の免疫原性を制限又は回避する更なる基準を追加するものである。Trエピトープは、これらのエピトープを含むヒトVH及びVLのセグメントを組み込むことによって、又は2又はそれ以上のヒト配列セグメントの結合を介してこのようなエピトープを生成することによって、又は、例えば、CD4+ CD25+ T細胞の誘発又は活性化のために、例えば、IL−10及び/又はTGF−β等の阻害性サイトカインの放出の測定によって、ヒト抗体又はタンパク質配列のセグメントに相当するペプチドからの新しいTrエピトープをスクリーニングすることによって、複合抗体VH又はVL内に導入することができる(例えば、Hall et al.,Blood,vol.100(2002)p4529−36)。代替的に、公知のTrエピトープは、複合抗体の結合又は機能又は発現を阻害しないVH及び/又はVL内の位置で複合抗体V領域内に組み込むことができ、又は、例えば、VHのN末端に合成VH又はVL配列の一端で組み込むことができる。代替的に、Trエピトープは、複合抗体の機能に干渉しない位置に(例えば、ヒンジ領域内で)、又は、発現の欠如等のいくつかの他の悪影響を引き起こさない位置に、複合抗体の一方又は双方の定常領域内に組み込むことができる。代替的に、抗体融合タンパク質内の合成VH及びVLの一方又は双方について、抗体は、Fab及びFv−タイプ形状(VH及びVLに結合した単鎖抗体(SCAs)を含む)、単一ドメイン抗体、又はホモ二量体型抗体を接合して、Trエピトープを、複合抗体の機能に干渉しない位置に、又は発現の欠如等のいくつかの他の悪影響を引き起こさない位置に組み込むことができる。例えば、SCAsでは、Trエピトープ用に特に好ましい位置は、VHとVLを結合するリンカ領域内である。最も良いのは、Trエピトープが、適切な配列によって側面に位置し、例えば、エンドサイトーシスプロテアーゼの作用に反応する配列を有するエピトープの側面に位置することによって、MHCクラスII分子上の制御性T細胞エピトープの放出及び表示を最適化する。一般的に、抗原の処理中のプロテアーゼの作用を標的にするP−20からP30(P1からP9のように規定されているコアノノメータを有する)を範囲とする位置で側面にある残渣は、必要ならば、ヒト抗体配列の追加のセグメントを用いて導入される。
【0009】
ここで議論されているように、本発明は、修飾抗体又は複合抗体の生成方法も提供している。従って、別の態様では、本発明は;
(1)その他の抗体可変領域の範囲からアミノ酸配列のセグメントを結合することによって抗体可変領域遺伝子を準備して、様々な可変領域遺伝子のライブラリを生成するステップと、
(2)抗体可変領域遺伝子のライブラリを発現ベクタ内へクローニングするステップと、
(3)抗体可変領域のライブラリをスクリーニングして、所望の特性を有するライブラリの員を回復するステップと、
を具える修飾抗体の生成方法を提供する。
【0010】
本発明の第1の好ましい方法「A」では、合成ヒト抗体のライブラリが生成され、特異的抗原に結合する等、所望の特性を有する抗体にスクリーニングされる。この方法は、次の6ステップ;
(1)合成VH及びVL遺伝子を設計するステップと、
(2)合成VH及びVL遺伝子をクローニングするステップと、
(3)合成VH及びVL遺伝子を発現するステップと、
(4)所望の特性を有する複合抗体をスクリーニングして選択するステップと、
(5)リード複合抗体を最適化するステップと、
(6)(選択的に)T細胞エピトープを回避するステップと、
を含む。
【0011】
ステップ(1)については、合成VH及びVL配列のライブラリは、Kabat antibody database(www.bioinf.org.uk/abs/simkab.html)、NCBI database(www.ncbi.nlm.nih.gov)で入手可能なもの等の公知のヒトV領域配列から、及び、UniProt(www.ebi.uniprot.org)及びPRF/SEQDB(www.prf.or.jp)等のタンパク質データベースから、VH及びVL配列のセグメントを選択することによって構成されている。加えて、これらは、1又はそれ以上の個人のドナーから増幅したVH及びVLmRNAの直接配列によるヒトVH及びVL配列の収集によって補充される。配列セグメントの様々な結合は、VH及びVL遺伝子の設計であると考えられる。用いられる1つの方法は、合成VH及びVL配列の長さを固定して、様々なヒトV領域の相当するKabatナンバリング位置からの固定長配列セグメントを用いて、これらを設計することである。
【0012】
例えば、このライブラリは、121及び107アミノ酸VH及びVL領域をそれぞれ具え、例えば、Kabatナンバリングを用いて、VHアミノ酸1−27について様々なセグメントの組み合わせを含む。KabatナンバリングCDR1:30−35、CDR2:50−66、及びCDR:95−106に相当するCDRを有するVHについては、次のKabat位についての配列セグメントが、1つの選択枝:1−27、28−31、32−36、37−42、43−50、51−56、57−60、61−63、64−69、70−82a、82b−96、97−98、99−101、102−117として用いられている。KabatナンバリングCDR1:24−34、CDR2:50−56、CDR3:89−97に相当するCDRを有するVLについて、次のKabat位についての配列セグメントが、1つの選択枝:1−22、23−27、28−30、31−33、34−35、36−47、48−52、53−55、56−59、60−87、88−92、93−94、95−107として用いられている。従って、この例では、合成VHは、14ヒトセグメントから成り、合成VLは、13のヒトセグメントから成る。実際には、コンピュータプログラムを用いて、これらのセグメントの組み合わせを生成する。好ましくは、このプログラムは、例えば、CDRのある種のカノニカル構造を回避する、或いは、VH及び/又はVL折り畳み、又はVH/VL相互作用を分離するあるセグメントの好ましくない組み合わせを回避するアルゴリズムを含む。選択的な追加として、このプログラムは、配列セグメントの組み合わせによって形成したT細胞エピトープの数を制限するアルゴリズムを含んでいてもよい(以下に述べるステップ(6)のコンピュータを利用した方法参照)。
【0013】
ステップ(2)については、合成ヒト配列のライブラリを設計して、好ましくは合成オリゴヌクレオチドを用いて、合成VH及びVL遺伝子が生成される。一般的に、V領域配列のより長いセグメントをコードする合成オリゴヌクレオチドは、V領域配列の2又はそれ以上の連続したセグメントをコードするオリゴヌクレオチドの混合物に結紮される。代替的に、合成V領域は、現存のヒトVH及びVL遺伝子をテンプレートとして用いて、PCRのオーバーラップ又はその他の増幅技術等の他の方法によって結合することができる。例えば、PCRを用いて、V領域の小さなセグメントを個別に増幅し、次いで、PCR反応をオーバーラップさせることによって結合する。
【0014】
その他の方法では、混合合成オリゴヌクレオチドを生成して、好ましくは特異的V領域セグメントをコードする配列を濃縮するドーピング方法を用いて、一連の配列セグメントを作ることができる。大きく変動するヒトV領域セグメント表示を有する合成ヒトVH及びVL遺伝子は、Molecular Cloning:A Laboratory Manual;3rd Ed.,vols.1−3(2001)Cold Spring Harbor Laboratory Pressに記載されているもの等の当業者に知られている技術を用いて、及びOrlandi et al.,Proc Natl Acad Sci USA.,86(1989)3833−3837に記載されているもの等の免疫グロブリンについての標準PCR法を用いて、多くの方法で結合させることができる。
【0015】
ステップ(3)については、合成ヒトVH及びVL遺伝子が生成されると、これらは、完全抗体分子又はFv’s、Fab’s、Fab2、SCA等の抗原結合フラグメント、単一ドメイン抗体(例えば、VHのみを具える)、及びこれらの各々の多重誘導体の生成用の様々な発現ベクタ内にクローンすることができる。代替的に、VH及びVL遺伝子は、その他の分子をコードする遺伝子に融合させて、融合プロテインを生成することができる。また、Fv又はFabの1つの鎖のC末端で、ポリヒスチジンタグ等の検出可能なマーカをコードする配列を含んでいてもよい。発現ベクタは、ほ乳類細胞、細菌細胞、バクテリオファージ、イースト、真菌、及びその他の微生物における発現用ベクタを含む。このようなベクタには、また、トランスジェニック動物からのインビボ発現用ベクタや、リボソーム調製を用いるインビトロ翻訳等のインビトロ系を用いた発現用ベクタも含まれる。
【0016】
ステップ(4)については、合成ヒト抗体のライブラリのスクリーニングは、通常、対象の1又はそれ以上の特異的抗原への結合用である。多くのスクリーニング方法が当業者に知られており、これらの方法の選択は、完全ヒト抗体の発現形状と、抗体分子、即ち、完全ヒト抗体、又はFab、Fv、SCA、単一ドメイン抗体等の組成に依存する。対象の抗原に結合する現存の抗体が入手可能である場合に、この抗体からのVH又はVLは、合成ヒトVL又はVHとそれぞれ組み合わせて、結合を試験することができる。
【0017】
スクリーニング法は、固相上のこのような員のライブラリ又はプールの個々の員を固定することから、個別に又はプール中の対象の抗原を固定することにまで及ぶ。抗体が固定される場合、対象の抗原が追加され、1又はそれ以上の追加試薬の添加によって、直接的、又は間接的に検出することができる。例えば、抗原が融合タンパク質であるか、又はアルカリホスファターゼ等の酵素と結合する場合に、色、蛍光又は化学発光シグナルを生成する幅広い基質から順次添加することによって検出することができる。抗体プールが、1つのロケーション(例えば、マイクロタイター皿)に固定され、シグナルが、抗原の添加に起因する場合、このプールを、このプール又はより小さいプールの個々の員の再スクリーニングに先立って脱複製する(dereplicated)ことができる。対象の抗原が固定される場合、合成抗原ライブラリは、個々の抗体の添加から、特異的ロケーションに固定された対象の抗原まで、抗体のプールの添加まで、全合成ライブラリの添加及び対象の抗原に結合した抗体の次の回復までに及ぶいくつかの方法でスクリーニングすることができる。最後のケースでは、一般的な戦略は、カラム内、又はビード上等の固相上に抗原を固定してライブラリを加え、次いで、例えば、低塩濃度緩衝液でこの固相を洗浄して(ライブラリに緩やかに結びついた員を分離するために)、次いで、例えば高塩濃度緩衝液を用いて抗原に結合する抗体を溶離することである。この目的のためのライブラリの員の発現用の一般的な形式は、ファージ表示、イースト表示、リボソーム表示、及びビード表示である各場合において、合成VH及びVL鎖をコードする核酸が、抗原に結合する合成V領域に付着し続ける。
【0018】
また、スクリーニング方法は、直接抗原結合試験に代わる機能性又は生物学的試験も含み、細胞増殖、細胞増殖阻害、細胞分化、又は細胞移動を含むインビトロ試験、又は、全有機体のレベルでの抗原への反応、例えば、マウスの血球数の変化や移植した腫瘍の増殖阻害を測定するステップを含む、代替的なインビボ試験等で機能性又は生物学的活性が測定される。
【0019】
ステップ(5)については、対象の抗原への結合等の所望の特性を有する1又はそれ以上の「リード」合成ヒト抗体の選択の後、例えば、この抗原の結合親和性を増加させる、又はこの抗体を追加成分に融合させることによって、リード抗体の特性が、選択的に改善されることがある。親和性の増加は、合成可変領域配列の突然変異によって達成され、所望の方法の結合を増加させる又は変更する選択した合成V領域配列の突然変異を選択する。本発明は、リード抗体からの1又はそれ以上の個々のV領域配列セグメントを、1又はそれ以上のヒト抗体配列からの相当する配列セグメントで置換することによる、可変領域配列の突然変異のための新規な方法を含む。具体的には、CDR領域と、又はCDR領域内でオーバーラップするセグメントを、様々な長さのセグメントを含むその他のヒト抗体からの1又はそれ以上の代替セグメントによって置換することができる。本発明の範囲内は、特異的セグメントは、関連する特性を有するヒト抗体から、例えば、同じ抗原に結合する抗体から、又は、関連する特性を有する非ヒト抗体から、又は、関連する配列を有する非ヒト抗体の機能にとって重要である、ある種のキーアミノ酸を保持する配列セグメントを有するヒト抗体から、といった選択したリード抗体まで含む。このような突然変異の影響を受ける1又はそれ以上の合成ヒト抗体をスクリーニングして、特性を改善する。
【0020】
選択的ステップ(6)については、リード合成ヒト抗体の選択後、必要であれば、T細胞エピトープを回避する代替セグメントでT細胞エピトープに寄与、又はT細胞エピトープをコードするV領域セグメントを交換することによって、T細胞エピトープが制限又は回避される。このようなT細胞エピトープは、ある範囲の方法によって検出することができる。例えば、合成V領域配列の1又はそれ以上の座に相当するペプチドを合成して、T細胞アッセイ試験をして、T細胞エピトープの存在を決定することができる。一般的に、このようなペプチドは、長さが15アミノ酸であり、接触領域がより長い配列を試験することが望ましく、12アミノ酸オーバーラップを有する15マー等の配列からオーバーラップペプチドを用いる。T細胞エピトープの検出については、様々なT細胞アッセイの範囲を、サイトカイン放出、増殖(例えば、3H−チミジンの摂取によって)、Ca2+フラックス、表面マーカ発現、遺伝子転写等のCD4+T細胞の活性又は増殖の測定に用いることができる。
【0021】
代替的に、合成V領域配列に相当するオーバーラップペプチドは、インビトロ法又はインシリコ(in silico)法を用いて分析し、ヒトMHCクラスII分子に結合するいずれの場合も、潜在的なT細胞エピトープ、即ち、T細胞反応を誘発するMHC結合ペプチドを決定する。インシリコ(in silico)法は、ペプチド−MHCクラスII結合相互作用のモデリングを含む方法と、MHCクラスIIに結合する一般的なモチーフの同定を含む方法と、ペプチド又は公知のインビトロMHC結合特性を有するペプチド内での特異的アミノ酸のデータベースを用いる方法とを含む。合成V領域配列からのより長いペプチドの生成、又は合成V領域配列を含む全抗体を生成して、例えば、MHC−ペプチドテトラマによって、又は公知のヒトT細胞エピトープのデータベースに提案されている又は構成された配列を調べることによって、T細胞アッセイ中又はMHC結合アッセイ中の合成V領域配列を試験するステップ等のその他の方法を用いることができる。合成ヒトV領域のT細胞エピトープの回避は、MHCクラスII結合モチーフの回避、又はMHCクラスIIへのペプチドの結合を固定する特定のアミノ酸の回避によっても補助することができる。1又はそれ以上のリード合成ヒト抗体からのT細胞エピトープの回避のための好ましい方法では、インシリコ(in silico)法を最初に適用して、潜在的なT細胞エピトープについての合成ヒト抗体V領域を分析し、これらが同定される場合、ヒトVH又はVL配列の新しいセグメントを導入して、これらのエピトープを回避し、新しいT細胞エピトープの導入を回避する。
【0022】
新しいヒトV領域セグメントのこのような導入と、所望の特性について、このように修飾したリード合成ヒト抗体の再スクリーニングの後に、1又はそれ以上の最終リード合成ヒトV領域を、一般的に、15から45アミノ酸の長さのオーバーラップペプチド、例えば、合成ヒト領域配列(全V領域又はこれらの一部分)からの12アミノ酸オーバーラップしている15マーペプチドを試験するステップによって、又は、ヒトT細胞アッセイで、直接的に全合成ヒト抗体を試験するステップによって、ヒトT細胞アッセイで更に試験することができる。全抗体に対してT細胞活性を直接試験できるようにする全合成ヒト抗体を試験するT細胞アッセイを用いた最終分析が好ましい。
【0023】
本発明の第2の好ましい方法「B」では、合成ヒト抗体のライブラリを、所望の特性を有する1又はそれ以上の参照抗体からの所望のアミノ酸を含むように生成する。この方法は、次の7ステップ;
(1)1又はそれ以上の参照抗体の配列を分析するステップと、
(2)合成VH及びVL遺伝子を設計するステップと、
(3)(選択的に)T細胞エピトープを回避するステップと、
(4)合成VH及びVL遺伝子をクローニングするステップと、
(5)合成VH及びVL遺伝子を発現するステップと、
(6)所望の特性を有する複合抗体をスクリーニングして選択するステップと、
(7)T細胞エピトープの最適な回避を含むリード複合抗体を最適化するステップと、
を含む。
【0024】
ステップ(1)では、典型的な参照抗体は、ヒト型の抗体中の所望の特性及び/又は結合特異性を有する齧歯類、特にマウスである。1又はそれ以上の参照抗体V領域配列が入手できる場合、これらの配列を分析してCDRの配列を決定し、結合特性等の抗体の所望の特性に重要であるアミノ酸を同定する。参照抗体については、例えば、参照V領域配列の同種のその他の配列とのアラインメントによってこのような分析を実施する。また、参照抗体が非ヒト、ヒトV領域配列である場合も、このような分析を行う。例えば、プログラムCLUSTAL(Thompson et al.,Nucleic Acids Res.22(1994)p4673−80)を用いて、このようなアラインメントを実施する。このようなアラインメントは、参照抗体のV領域及び相同性v領域ファミリィ中の異常又は微量アミノ酸を同定することができる。加えて、CDRのカノニカル構造等の保存V領域構造を、例えば、Protein Data Bank(Berman et al.:The Protein Data Bank, Nucleic Acids Research,28(2000)235−242)を用いて同定することができる。加えて、構造が知られていない場合、MODELLER(Sali and Blundell,J.Mol.Biol.234(1993)p779−815)等のモデリングソフトウエアを用いて、参照抗体可変領域をモデリングし、いくつかの場合では、抗体−抗原相互作用のモデルを生成することができる。参照抗体V領域のこのような分析は、合成ヒト抗体についてのヒトV領域配列のセグメントの選択を導くのに用いる。
【0025】
ステップ(2)については、合成ヒト抗体の所望の特性に重要であるアミノ酸を決定して、これらのアミノ酸のいくつか又は全てを含むように、ヒトV領域配列のセグメントを選択する。合成ヒト抗体の特性についてのこのようなセグメントの効果が不明確である特定の座に一般的な1又はそれ以上の代替ヒトV領域セグメントを有する選択されたセグメントを含む合成ヒトV領域配列のライブラリがこれによって設計される。このような合成ヒト抗体配列は、その他のヒト抗体配列と保存構造とのアラインメントによって参照抗体で更に分析することができ、加えて、合成ヒト抗体V領域のこの構造の更なるモデリングを行って、合成ヒト抗体に用いられるヒトV領域セグメントの組み合わせを、所望のように精製し、合成V領域内でのタンパク質構造、分子間及び分子内相互作用における欠陥、及び重要なアミノ酸の不正確な構造配向を回避する。
【0026】
選択的ステップ(3)については、セグメント選択の付加的基準として、最終合成ヒトV領域のT細胞エピトープを制限又は回避するこれらのセグメント又はセグメントの組み合わせが選択される。T細胞エピトープをインシリコ(in silico)又はインビトロ法を用いて、好ましくは、合成ヒトV領域配列を設計する段階でインシリコ(in silico)法を用いて、上述した方法A、ステップ(6)に記載した方法によって分析する。
【0027】
ステップ(4)については、合成ヒト配列のライブラリを設計して、次いで、合成VH及びVL遺伝子を、好ましくは合成オリゴヌクレオチドを用いて生成する。典型的には、V領域配列のより長いセグメントをコードする合成オリゴヌクレオチドが、配列の代替セグメントをコードするオリゴヌクレオチドの混合物に結紮して、合成ヒトV領域のライブラリの様々な員を生成する。代替として、合成ヒトV領域のライブラリの各員は、特異的ヒトV領域の配列をコードするオリゴヌクレオチドを部分的に用いて個別に生成する。代替として、合成V領域は、既存のヒトVH及びVL遺伝子をテンプレートとして用いて、又は、テンプレートとして1又はそれ以上の参照抗体V領域遺伝子を用いて、オーバーラップPCR、又は他の増幅技術等のその他の方法によってアセンブルされる。
【0028】
方法Bについてのステップ(5)及び(6)は、方法A、ステップ(4)及び(5)に記載されているのと同様である。
【0029】
選択的なステップ(7)は、方法A、ステップ(6)と同様に用いられ、リード合成ヒト抗体でT細胞エピトープの更なる回避が要求される。全抗体からのT細胞活性について直接試験することを可能にする全合成ヒト抗体を試験するT細胞アッセイを用いた最終分析が好ましい。
【0030】
方法A及びBに加えて、合成ヒト抗体を作成して試験し、このような抗体の特性を最適化するその他の方法があることは、当業者に理解されよう。本発明の合成ヒト抗体は新規であり、V領域の全ヒト起源の結果として、非ヒト配列を含むその他の抗体よりもヒトの免疫原性が低くなくてはならない。また、合成ヒト抗体の追加の選択的特徴、即ち、T細胞エピトープの回避及び/又はTrエピトープの付加は、より低い免疫原性に寄与する。より低い免疫原性の目的は、例えば、源ヒト抗体中の配列位置と異なる複合抗体の配列位置にセグメントを含む合成ヒト抗体、ヒトV領域配列のセグメントの部分的組み込みのみを有する複合抗体、非ヒト配列のセグメントを有する複合抗体、又は、例えば、抗体に対する結合親和性を増加する、又はT細胞エピトープを避けるように突然変異したヒト配列を有する複合抗体等の全ヒト配列セグメントの無いV領域を含む、より好ましくない複合抗体を用いて達成することができることは、当業者に理解されるであろう。
【0031】
合成ヒト抗体内でのV領域配列セグメント及びこれらの組み合わせを選択して、上記のようなT細胞エピトープの選択的回避を含む基準範囲を満たすことが理解されよう。例えば、ヒトV領域配列のセグメント及びこれらの組み合わせは、B細胞エピトープ及びMHCクラスI−制限エピトープ等のその他のエピトープの回避、複合抗体の発現に有害であるアミノ酸配列の回避、N−グリコシル化等の複合抗体の不適切な修飾を導く配列の回避、ヘルパーT細胞エピトープ及び/又はB細胞エピトープ(例えば、ワクチンの使用)の包含等のある種の機能の包含、1又はそれ以上の表面リシン残渣等のその他の部分への連続した結合、及びその他の基準範囲用に選択することができる。
【0032】
また、ヒトに加えて、その他の種由来のV領域セグメント全体又は一部を有する複合抗体を生成することができ、これも本発明の範囲内であると考えるべきであることは、当業者に理解されよう。例えば、マウスの研究用に、マウス起源のV領域配列セグメントの全体又は一部を具える合成マウス抗体を生成することができる。
【0033】
また、本発明は、抗体以外のタンパク質に適用し、治療用途には、このようなタンパク質(ここでは、「合成タンパク質」という)は、最終タンパク質分子内で、ヒトタンパク質からのアミノ酸配列の2又はそれ以上のセグメントを結合する。
【0034】
従って、更なる態様では、本発明は、アミノ酸配列の1又はそれ以上のセグメントの挿入によって改善した免疫原性を有する修飾タンパク質を提供する。
【0035】
タンパク質に関して、用語「セグメント」は、タンパク質分子内で見出される隣接するアミノ酸配列をいい、このようなセグメントは、2から250アミノ酸長のサイズの範囲である。治療用には、本発明の合成タンパク質は、通常、合成タンパク質内で様々なヒトタンパク質からのアミノ酸配列の2又はそれ以上を結合する。特に、本発明は、全体が2又はそれ以上のヒトタンパク質からの配列のセグメントからなる挿入を有する合成タンパク質に関する。ヒトタンパク質が合成タンパク質に対して相同で、又は合成タンパク質の領域に対して相同な領域で存在する場合、源ヒトタンパク質の配列位置に相当する合成タンパク質配列のセグメントを用いることができる。例えば、合成タンパク質配列のアミノ酸1から10は、源ヒトタンパク質のアミノ酸1から10に由来する。代替として、ヒトタンパク質配列中の配列位置にあるヒトタンパク質配列のセグメントは、源ヒトタンパク質の配列位置に関わりない合成タンパク質中のあらゆる配列位置にある合成タンパク質に配置されていてもよい。源ヒトタンパク質は、例えば、ヒトタンパク質配列のデータベースで提供されるようなあらゆる現存のヒトタンパク質アミノ酸配列であり、ヒトタンパク質の天然の突然変異形又は転位形、及び生殖細胞系と異なるその他の変異、人工的に構成されたヒト由来タンパク質からの配列、及び相当するタンパク質が発現するか、或いは発現しないヒト遺伝子又はRNA由来の配列が含まれる。
【0036】
本発明のこの態様の好ましい実施例では、治療用合成タンパク質は、最終合成タンパク質中のヒトT細胞エピトープを制限又は回避する組み合わせのヒトタンパク質配列セグメントを結合又は挿入することによって構成される。合成タンパク質に適用される本発明の好ましい態様は、ヒトタンパク質配列セグメントの挿入によって非ヒトタンパク質等の現存の参照タンパク質を修飾して、最終合成タンパク質のT細胞エピトープを制限又は回避することである。
【0037】
合成タンパク質を生成する本発明の好ましい方法では、合成ヒトタンパク質のライブラリを生成して、T細胞エピトープ不在等の所望の特性を有する1又はそれ以上の参照タンパク質からの所望のアミノ酸を含めるようにしている。この方法は、次の7ステップを含む;
(1)T細胞エピトープの選択的分析を含む1又はそれ以上の参照タンパク質の配列分析をするステップと、
(2)合成タンパク質遺伝子を設計するステップと、
(3)T細胞エピトープを(選択的に)回避するステップと、
(4)合成タンパク質遺伝子をクローニングするステップと、
(5)合成タンパク質遺伝子を発現させるステップと、
(6)スクリーニングを行い、所望の特性を有する合成タンパク質を選択するステップと、
(7)T細胞エピトープの選択的回避を含むリード合成タンパク質を最適化するステップ。
【0038】
ステップ(1)では、典型的な参照タンパク質は、合成タンパク質で所望される特性を有する非ヒトである。治療用途には、典型的に、合成タンパク質の免疫原性の低減又は除去が目的となる。1又はそれ以上の参照タンパク質配列が入手可能である場合、これらはタンパク質の所望の特性に重要であるアミノ酸を同定するために分析される。加えて、参照タンパク質のあらゆる公知の構造を分析することができ、又は、代替的に、モデル化ソフトウエアを用いて、構造をモデル化することができる。種間又は種内のどちらかの参照タンパク質の相同体が入手できる場合、時に、これらを用いて、配列差の関係と相同体間の特性の差の関係を決定することができる。タンパク質が別の分子と相互作用する場合、この相互作用のモデルを、生成し、相互作用に重要なアミノ酸を決定することができる。ステップ1への選択的付加として、特に、上記の合成ヒト抗体について詳細に述べているようなインビトロヒトT細胞アッセイを用いて、参照タンパク質中のT細胞エピトープの配列ロケーションが決定される。代替として、T細胞エピトープを分析するためのインシリコ(in silico)法を用いることができる。参照タンパク質のこのような分析を用いて、合成タンパク質用に選択されたヒトタンパク質配列のセグメントに導く。参照タンパク質、特に非ヒトと比較して免疫原性の低減又は除去が目的である合成タンパク質については、一般的に、T細胞エピトープのロケーションに相当する1又はそれ以上のヒト配列セグメントが、T細胞エピトープ無しの他のロケーションからの参照タンパク質からの配列のセグメントと組み合わせて合成タンパク質中に用いられる。
【0039】
ステップ(2)については、合成タンパク質の所望の特性に重要であるアミノ酸を決定して、次いで、これらのアミノ酸のいくつか又は全てを含むように、タンパク質配列のセグメントが選択される。これによって、典型的に、特定の座で、1又はそれ以上の代替ヒトタンパク質セグメントを有する選択されたセグメントを含む合成ヒトタンパク質配列のライブラリが設計される。合成タンパク質の特性のこのようなセグメントの効果は、不明である。このような合成タンパク質配列は、基準タンパク質を伴っていれば、任意の相同体と整列させることによって、又は合成タンパク質の構造のモデル化によって、又は、タンパク質構造の欠陥や重要なアミノ酸の不正確な構造配向を回避するために合成ヒトタンパク質に用いられるヒトタンパク質セグメントの組み合わせを、必要に応じて精製するためにその他の分析を行うことによって、更に分析することができる。
【0040】
選択的ステップ(3)について、セグメントの選択用の追加基準又は基準のみとして、最終合成タンパク質中のT細胞エピトープを制限又は回避するこれらのセグメント、又はセグメントの組み合わせが選択される。T細胞エピトープを、インシリコ(in silico)又はインビトロ法を用いて上記の合成ヒト抗体用に記載された方法によって分析する。
【0041】
ステップ(4)については、合成タンパク質のライブラリを設計して、次いで、合成タンパク質遺伝子を、好ましくは、合成オリゴヌクレオチドを用いて生成する。典型的には、タンパク質配列のより長いセグメントをコードする合成オリゴヌクレオチドが、配列の代替セグメントをコードして合成タンパク質のライブラリの様々な員を生成するオリゴヌクレオチドの混合物に結紮する。代替的に、合成タンパク質のライブラリの各員が、特異的合成タンパク質の配列をコードするオリゴヌクレオチドを用いて部分的に生成される。代替として、合成タンパク質は、オーバーラップしているPCR、又は、現存のヒトタンパク質遺伝子をテンプレートとして用いた、又は、1又はそれ以上の参照タンパク質遺伝子をテンプレートとして用いたその他の増幅技術等の他の方法によってアセンブルすることができる。
【0042】
ステップ(5)では、合成タンパク質のライブラリのスクリーニングは、通常、1又はそれ以上の合成タンパク質の所望の特性用である。多くのスクリーニング方法が当業者に知られており、これらの方法の選択は、合成タンパク質及びタンパク質機能の発現の形に依存する。スクリーニング方法は、固相上のこのような員のライブラリ又はプールの個別の員を固定するステップから、溶液相中のライブラリの員をスクリーニングして、もう1つの分子を同定し、これで合成タンパク質を設計して、個別に、又はプールで結合することによって相互作用させる範囲にまで及ぶ。また、スクリーニング方法は、機能的又は生物学的試験を含み、ここでは、細胞増殖、細胞増殖阻害、細胞分化、又は細胞遊走を含むインビトロ試験、又は全有機体のレベルでの合成タンパク質に対する反応、例えば、マウスの血球数又は移植した腫瘍の増殖阻害の変化を測定するステップを含む代替インビボ試験等の、機能的又は生物学的活性を測定する。
【0043】
ステップ(6)では、所望の特性を有する1又はそれ以上の「リード」合成タンパク質の選択に続いて、選択的に、例えば、酵素の特異的活性を増やすことによって、又はレセプタに対するタンパク質リガンドの結合を増やすことによって、このリードタンパク質の特性を強化することができる。特性の強化は、合成タンパク質配列の突然変異生成によって達成されて、所望の方法で、合成タンパク質の特性を変化させる突然変異が選択される。本発明は、このタンパク質からの1又はそれ以上の個々のタンパク質配列セグメントを1又はそれ以上のヒトタンパク質配列からの配列セグメントで置換することによって、タンパク質配列の突然変異を生成する新規な方法を含む。次いで、このような突然変異生成を行う1又はそれ以上の合成タンパク質をスクリーニングして、特性を強化することができる。
【0044】
選択的ステップ(7)では、リード合成タンパク質の選択に続いて、所望の場合に、T細胞エピトープに寄与する、又はこれをコードするタンパク質配列セグメントをT細胞エピトープを回避する代替セグメントと交換することによって、T細胞エピトープを制限又は回避する。このようなT細胞エピトープは、ある範囲の方法によって検出される。例えば、合成タンパク質中の1又はそれ以上の座に相当するペプチドを合成し、T細胞アッセイで試験してT細胞エピトープの存在を決定することができる。典型的には、このようなペプチドは、長さ15アミノ酸であり、12アミノ酸オーバーラップを有する15マー等の配列からのオーバーラップペプチドを用いてより長い隣接した配列を試験することが望ましい。代替として、潜在的T細胞エピトープ、即ち、T細胞反応を誘発するMHC結合ペプチドを決定する各場合に、合成タンパク質配列に相当するオーバーラップペプチドを、インビトロ法かインシリコ(in silico)法のどちらかを用いて、ヒトMHCクラスII分子への結合について分析する。インシリコ(in silico)法は、ペプチド−MHCクラスII結合相互作用をモデル化するステップを含む方法と、MHCクラスIIへの結合に一般的なモチーフを同定するステップを含む方法と、公知のインビトロMHC結合特性を有するペプチド内のペプチド又は特異的アミノ酸のデータベースを用いる方法を含む。合成タンパク質配列又は全合成タンパク質からより長いペプチドを生成する方法、及び、抗原提示細胞上のT細胞アッセイ、又はMHC結合アッセイでこれらを試験する方法等の他の方法を用いることができる。また、合成タンパク質のT細胞エピトープの回避は、MHCクラスII結合モチーフの回避によって、又はMHCクラスIIへのペプチドの結合をアンカする特異的アミノ酸の回避によって援助される。1又はそれ以上のリード合成タンパク質からT細胞エピトープの回避のための好ましい方法では、先ずインシリコ(in silico)法を適用して、潜在的T細胞エピトープについての合成タンパク質を分析し、これらが決定されると、ヒトタンパク質配列の新しいセグメントを導入して、これらのエピトープを回避して、新しいT細胞エピトープの導入を回避する。所望であれば、T細胞エピトープを回避する新しいヒトセグメントのこのような導入と、所望の特性についての修飾リード合成タンパク質の再スクリーニングに続いて、1又はそれ以上の最終リード合成タンパク質を、典型的に15から45アミノ酸長のオーバーラップペプチド、例えば、合成タンパク質配列(全タンパク質又はこれらの一部分)からの12アミノ酸オーバーラップした15マーペプチドを試験することによって、或いは、ヒトT細胞アッセイで、直接的に、全合成タンパク質を試験することによって、ヒトT細胞アッセイで選択的に試験することができる。全タンパク質からのT細胞活性について直接試験することができる全合成タンパク質を試験するT細胞アッセイを用いた最終分析が好ましい。
【0045】
合成タンパク質を作成して試験し、このようなタンパク質の特性を最適化する他の方法があることは、当業者には理解されよう。本発明の合成タンパク質は新規で、治療目的で用いられる場合は、ヒト起源のいくつかの、又は全てのタンパク質配列セグメントは、その他の比較可能な、又は非ヒト配列を含む非ヒト参照タンパク質より、ヒトの免疫原性が少ない峰性タンパク質を提供するべきである。また、合成タンパク質の付加的な選択的特徴、即ち、T細胞エピトープの回避及び/又はTrエピトープの付加も、より低い免疫原性に寄与する。より低い免疫原性の目的は、全ヒト配列の無い合成タンパク質を用いて達成され、また、突然変異して、参照タンパク質に相同する非ヒトタンパク質のT細胞エピトープ又はセグメントを除去するヒト配列セグメントを有する合成タンパク質を含むことが、当業者には理解される。T細胞エピトープの最適な回避を含む基準の範囲を満足するように合成タンパク質内のタンパク質セグメント及びその組み合わせを選択することが理解されよう。例えば、ヒトタンパク質配列のセグメント及びこれらの組み合わせを、B細胞エピトープ及びMHCクラスI−制限エピトープ等のその他のエピトープの回避と、合成タンパク質の発現に有害であるアミノ酸配列の回避と、N−グリコシル化等の合成タンパク質の不適切な修飾を導く配列の回避と、ヘルパーT細胞エピトープ、及び/又はB細胞エピトープ(例えば、ワクチン適用)の包含等のある種の機能の包含と、その他の部分への連続した結合と、その他の基準の範囲用に選択することができる。
【0046】
また、ヒトに加えて、他の種に由来する配列セグメントの全体又は一部分を有する合成タンパク質を生成することができ、これは、本発明の範囲内であると考えるべきである。例えば、マウスの研究では、マウスタンパク質配列セグメントを含む合成タンパク質を生成することができる。また、合成タンパク質が、同種内の相同タンパク質からのその他のタンパク質配列セグメントと結合した1つの種からのタンパク質配列セグメントを含むことができることが理解される。例えば、本発明は、植物型I RIP(リボソーム阻害タンパク質)の構造を含む。ここでRIPは、入手可能な多数の植物型I RIP配列からの配列セグメントを用いてアセンブルされる。このような合成RIPは、RIP活性を保持するであろう配列セグメントの組み合わせを導入することによってアセンブルされ、ヒトに用いる場合には、最終合成配列中のヒトT細胞エピトープの回避を含むであろう。
【0047】
抗体の場合、本発明は、最終タンパク質の1又はそれ以上のその他の特性に更なる改良を行う合成タンパク質のランダムな、セミランダムな、又は直接的な突然変異生成によって、合成タンパク質配列への更なる修飾の選択枝を含む。本発明は、ヒトに用いられるとき、又は薬学的な用途のタンパク質、又は、アレルギィ反応が本発明の組成物の使用によって制限又は除去される食料、合成洗剤、化粧品、及び他の消費者商品の用途のタンパク質等のヒトによって用いられるときに、低い免疫原性を有するタンパク質生成に特に好適である。本発明は、特に非アレルギィ関連エピトープ(例えば、TH1 T細胞−誘発エピトープ用のTH2)によって、及び/又はアレルギィ患者の免疫反応を抑制するTrエピトープの付加によって除去又は置換されたアレルギィ関連T細胞エピトープを有するタンパク質を生成することによって、ヒト内での低いアレルギィ性を有するタンパク質の生成に特に好適であることが理解されよう。本発明は、特に、非炎症関連エピトープ(例えば、TH2 T細胞誘発エピトープ用のTH1)及び/又は炎症反応を抑制するためのTrエピトープの添加によって除去又は置換された炎症関連T細胞エピトープを有するタンパク質を生成することによって、ヒトの炎症特性を低減するタンパク質を生成するのに特に好適であることが理解されよう。
【0048】
ここで議論されているように、本発明の修飾/合成タンパク質及び抗体は、病気の治療に有益であり、低い免疫原性を示す。従って、更なる態様で、本発明は、選択的に、1又はそれ以上の薬学的に許容できる賦形剤、担体、又は希釈剤と共に、請求項1から18のいずれか1項に記載の修飾抗体、抗体結合性フラグメント、又はタンパク質を具える薬学的調製を提供する。
【0049】
本発明の組成物は、1投与当たり所定量の各活性成分を含む単位投与形で存在する。このような単位は、5−100mg/1日の化合物、好ましくは、5−15mg/1日、10−30mg/1日、25−50mg/1日、40−80mg/1日、又は60−100mg/1日のいずれかを提供するように構成されている。式Iの化合物については、100−1000mg/1日、好ましくは、100−400mg/1日、300−600mg/1日、500−1000mg/1日のいずれかの範囲の投与を提供する。このような投与は、単回投与又は複数回の分散投与で提供することができる。最終的な投与は、もちろん、治療の状態、投与経路、及び患者の年齢、体重、及び状態に依存しており、医師の裁量である。
【0050】
本発明の組成物は、あらゆる適切な経路、例えば、経口(口腔又は舌下を含む)、直腸、鼻腔、局所(口腔、舌下又は経皮を含む)、経腟、又は非経口(皮下、筋内、静脈、又は皮内)を含む経路による投与用に構成されている。このような調製は、医薬の分野で知られているあらゆる方法、例えば、担体や賦形剤と活性成分を関連させることによって調製することができる。
【0051】
経口投与に適した医薬調製物は、カプセル又はタブレット;パウダ又は顆粒;水性又は非水性の液体の溶液又は懸濁液;可食発泡体又はホイップ;又は水中油型液体エマルジョン又は油中水型液体エマルジョン等の分散単位として存在している。
【0052】
経皮投与に適した医薬調製物は、長期間、レシピエントの表皮と密着し続けるように意図した分散パッチとして存在する。例えば、有効成分は、Pharmaceutical Research,3(6),318(1986)に一般的に記載されているような、イオントフォレシスによって、パッチから送り込まれる。
【0053】
局所投与に適した医薬調製物は、軟膏、クリーム、懸濁液、ローション、パウダ、溶液、ペースト、ジェル、スプレィ、エアロゾル、又はオイルとして調製される。
【0054】
眼、又はその他の外部組織である、例えば、口及び皮膚への塗布には、この調製物は、局所軟膏又はクリームとして塗布するのが好ましい。軟膏に調製する場合、有効成分を、パラフィン、又は水混和性軟膏ベースのどちらかと共に用いることができる。代替として、有効成分を、水中油型クリームベース又は油中水型ベースのクリームと共に調製するようにしてもよい。
【0055】
眼への局所投与に適した医薬調製物は、点眼薬を含み、この有効成分を好適な担体、特に水性溶媒に溶解又は懸濁させる。
【0056】
口内局所投与に適した医薬調製物は、薬用キャンディ、トローチ剤、及び洗口液を含む。
【0057】
直腸投与に適した医薬調製物は、座薬又は寛腸剤として存在する。
【0058】
経鼻投与に適合した医薬調製物であって、担体が固体である医薬調製物は、例えば、スナッフで摂取する方法、即ち、鼻腔に近づけたパウダの容器から鼻道を通る素早い吸入によって投与される20から500ミクロンの範囲の粒径を有する粗粉末を含む。スプレィ式点鼻薬として、又は点鼻薬としての投与に好適な調製物であって、担体が液体である調製物は、水性又は油性溶液の有効成分を含む。
【0059】
吸入による投与に適した医薬調製物は、様々なタイプの定量加圧エアロゾル、噴霧器、又は吸入器の手段によって生成する細粒粉末又は噴霧を含む。
【0060】
経腟投与に適した医薬調製物は、ペッサリー、タンポン、クリーム、ジェル、ペースト、発泡体又はスプレィ調製物として存在する。
【0061】
非経口投与に適した医薬調製物は、抗酸化剤、緩衝液、静菌薬及び溶質を含む水性及び非水性滅菌注射液;及び、懸濁剤及び増粘剤を含む対象とするレシピエントの血液を用いる等張の調製を行う水性及び非水性滅菌懸濁液を含む。調製物は、単位投与及び複数投与容器である、例えば、密封アンプル及びバイアルで存在し、また、使用直前に例えば注入用の水といった滅菌液体担体の添加を必要とするのみのフリーズ−ドライ(凍結乾燥)状態で貯蔵されているものでもよい。即時調合の注射液及び懸濁液は、滅菌パウダ、顆粒、及びタブレットから調製される。
【0062】
好ましい単位投与調製物は、上記のような有効成分の日用量、又はサブドーズ(sub−dose)、又はこれらの適切な画分を含むものである。
【0063】
特に上記した成分に加えて、調製物は、問題の調製物のタイプを考慮したこの分野では従来のものである他の薬剤を含んでいてもよい。例えば、経口投与に好適なものは、香料添加剤を含んでいてもよい。
【0064】
次の例は、本発明の範囲を限定するものと考えるべきではない。図及び表は、以下に述べる例に、次のように関連している;
【0065】
表1から表3は、186×9残渣長VH CDR3(表1)、77×8残渣長VL CDR3(表2)、及び153×10残渣長VL CDR3(表3)を具える合成ヒト抗体scFvライブラリに用いられるCDRである。
【0066】
表4は、一次合成ヒト抗TNFαVH及びVL変異株に用いるヒト配列セグメントである。
【0067】
表5は、合成ヒト抗TNFα変異株の活性である。
【0068】
表6は、ブーガニン変異株のブーガニン及び置換ヒトセグメントの免疫原性ペプチド配列である。
【0069】
例1−合成ヒト抗−HER2抗体の構造
【0070】
ヒト可変領域配列セグメントライブラリの作成用に、ある範囲のヒト免疫グロブリンの範囲からのアミノ酸配列を重鎖(VH)及び軽鎖(VL)可変領域配列を含むインシリコ(in silico)ヒト可変領域配列ライブラリを具える単一データベース内に集めた。配列の源としては、NCBI Igblastデータベース(www.ncbi.nih.gov)、Kabatデータベース(Kabat et al.,Sequences of Proteins of Immunological Interest,NIH publication 91−3242,5th ed.(1991)(後にアップデートされた)),Vbase(www.mrc−cpe.cam.ac.uk/imt.doc),Genbank(Benson et al.,Nucl.Acids Res.25(1997)pl−6、又はwww.bioinf.org.uk/absを介した)データベースが挙げられる。用いられた参照抗体可変領域配列は、ハーセプチン(登録商標)として知られているヒト化抗−HER2抗体であった(Carter et al.,Proc.Nat.Acad.Sci.USA,vol 89(1992)p4285,US5821337)。インシリコ(in silico)ヒト可変領域配列ライブラリからのセグメントを、ハーセプチン(登録商標)可変領域配列の相当するアミノ酸に対する同定用に選択し、結合させて、図1及び図2にそれぞれ示されているような合成ヒトVH及びVL配列を生成した。
【0071】
組み換えDNA技術を、この分野で公知の方法と、必要に応じて、これらの方法で用いられる酵素を用いるための供給者の指示を用いて実施した。一般的な方法の源としては、Molecular Cloning,A Laboratory Manual,3rd edition,vols 1−3,eds.Sambrook and Russel(2001)Cold Spring Harbor Laboratory Press、及びCurrent Protocols in Molecular Biology,ed.Ausubel,John Wiley and Sonsが挙げられる。また、詳細な実験室方法は、以下の例7に記載されている。ハーセプチン(登録商標)に相当する合成ヒトVH及びVL配列を、各鎖について、完全合成ヒトVH及びVL配列をコードする長さ30−60アミノ酸の8つの合成ヌクレオチドを用いて作成した。また、同時に、対照試薬として、マウスモノクローナル抗体4D5(Hudziak et al.,Mol.Cell.Biol.,(1989年3月)p1165−1172))のキメラ形を、鎖毎に8つの合成オリゴヌクレオチドを用いて作成した。分離VH及びVLオリゴヌクレオチドを、先ずリン酸化して、等モル比で混合して、サーマルサイクラ内で5分間、94℃に加熱し、次いで65℃に冷却して、65℃で2分間インキュベートした。次いで、45℃で2分間、35℃で2分間、25℃で2分間、4℃で30分間インキュベートを継続した。更に、オリゴヌクレオチドを、T4 DNAリガーゼ(英国ペイズリィ所在のLife Technologies社)を用いて、14℃で18時間、結紮した。
【0072】
Kozak配列、リーダーシグナルペプチド(leader signal peptide)配列及びリーダーイントロン(leader intron)を含む5’フランキング配列、及びスプライス座及びイントロン配列(intron sequence)を含む3’フランキング配列をコードする追加のオリゴヌクレオチドを、VH及びVLオリゴヌクレオチド混合物に上記のように加えて、アニールした。合成ヒトVH及びVKと、生成した4D5発現カセットを、プラスミドベクタpUC19へBamHIフラグメントに対するHindIIIとしてクローニングし、完全なDNA配列を確認した。これらは、ヒトIgG1(VH)又はKappa(VK)定常領域をそれぞれと、ほ乳類細胞の選択用マーカを含む発現ベクタpSVgpt及びpSVhygに転写された。DNA配列を確認して、合成ヒトVH及びVKと、発現ベクタ中の4D5VH及びVKを補正した。
【0073】
抗体発現用主宿細胞株は、NS0である非免疫グロブリン生成マウス骨髄腫であり、英国ポートン所在のEuropean Collection of Animal Cell Cultures(ECACC No85110503)から得た。重鎖及び軽鎖発現ベクタをエレクトロポレーションによって、NS0細胞内に同時遺伝子導入した。gpt遺伝子を発現するコロニィは、10%ウシ胎仔血清、0.8μg/mlミコフェノール酸、及び250μg/mlキサンチンを補充したダルベッコ変法イーグル培地(DMEM)中で選択した。遺伝子導入した細胞クローンは、ヒトIgGについてELISAによってヒト抗体を生成用にスクリーニングした。細胞株分泌抗体を拡張し、最も高い生成株を選択し、液体窒素中に凝固させた。修飾抗体をProsep(登録商標)−A(英国ノースアンバーランド所在のBioprocessing Ltd社)を用いて精製した。ヒトIgGκ抗体についてELISAによって、濃度を決定した。
【0074】
合成ヒト抗体及びキメラ4D5抗体を、Hudziak et al.(同書)によって正確に記載されているように、陰性対照非−Her−2結合ヒトIgG1/Kappa抗体と共に、HER2+ヒト乳癌細胞株SK−BR−3の増殖の阻害について試験した。この結果(図3)は、合成ヒト抗体及びキメラ4D5抗体が、SK−BR−3細胞の増殖を阻害において等価な効果を有することを示す。また、図3は、以下のように生成した代替「エピトープ回避」複合抗体についてのデータを示す。
【0075】
本発明のエピトープ回避オプションを試験するために、合成ヒト重鎖及び軽鎖可変領域の配列を、Peptide Threading(www.csd.abdn.ac.uk/〜gjlk/MHC−thread)を用いて非自己ヒトMHCクラスIIバインダについて分析した。このソフトウエアは、ペプチドのアミノ酸側鎖と、MHCクラスII結合溝内の特定の結合ポケットの間の好適な相互作用を予測する。合成ヒト重鎖及び軽鎖可変配列からの全オーバーラップ13マーは、MHCクラスIIアロタイプのデータベースを通り、MHCクラスII分子との結合、及び、相互作用に基づいて得た。MHCクラスIIを結合すると予想されるペプチドは、VHの残渣16及び67、及びVLの9及び44で始まる132マーであった。結果として、ヒト可変領域配列ライブラリの新しいセグメントが、図1の合成ヒト配列で用いられるセグメントの代わりに選択され、アミノ酸交換VH18L−A/69I−G;VL11L−A、46L−Aを導入した。相当する「エピトープ回避」合成ヒト抗体(「EACHAB」=Epitope Avoided Composite Human AntiBody)は、図1の配列に相当する抗体を作るのに用いられるいくつかのオリゴヌクレオチドを置換することによって作られ、EACHABは上記の方法で作られ、標準合成ヒト抗体と同等であるSK−BR−5の増殖の阻害を示すために試験した(図3)。このデータは、合成ヒト抗体が、対照キメラ抗−HER−2抗体と等しい効果をもって成功裏に構成されて、EACHABバージョンの合成ヒト抗体が、効果の損失すること無く生成され得ることを示している。
【0076】
例2−合成ヒト抗−HER2抗体の免疫原性
【0077】
T細胞増殖アッセイを実施して、合成ヒト抗−HER2抗体、EACHAB変異株及びキメラ4D5抗体の免疫原性を比較した(例1参照)。これらの抗体は、血清の入っていない、動物由来成分の入っていない、タンパク質の入っていない培地、HyQ(登録商標)LS1000脂質補給(Hyclone Cat No:SH30554)及びピルビン酸ナトリウム(Gibco Cat No:11360−039)を補充したHyClone HyQ(登録商標)ADCF−Mab(商標)(Hyclone Cat No:Cat no:SH30349)中で増殖したNS0細胞から調製された。Sephadex G25(PDlOカラム)上の緩衝液を50mMのMESpH6に交換後、抗体は、陽イオン交換カラム(Mono−S 10/10)をそれぞれ通り、塩化ナトリウム勾配(0から0.5M)で溶出した。次いで、抗体含有画分を、PBSのSuperdex 200保存カラム(XK16/60)ランに塗布した。ピーク画分を貯留して、4℃で保存した。抗体濃度は、ヒトIgGについてのELISAによって決定した。
【0078】
健康なヒトのドナー血液から単離し、液体窒素で凍結保存したPBMC(末梢血単核球)を用いて、免疫原性分析を実施した。各ドナーは、Allset(商標)PCRベース組織分類キット(英国Wirral所在のDynal社)を用いて組織分類を行い、20人の健康なドナーを、個人MHCハプロタイプに従って選択した。AIM V(英国ペイズリィ所在のInvitrogen社)の4×106PBMCを含有する2mlバルク培地を試験ペプチド(5μM最終濃度)を有する24ウエル組織培養プレートでインキュベートし、バルク培地を緩やかに再懸濁し、PBMCの3倍の100μlのサンプルをU−底96ウエルプレートに移すことによって、5日、6日、7日、及び8日目に増殖を評価した。Tomtec Mach IIIプレートハーベスタ(英国所在のReceptor Technologies社)を用いて、培地をグラスファイバフィルタマット上に採取し、カウント毎分(cpm)値を、Wallac Microbeta TriLuxプレートリーダ(パララックス高効率計数プロトコルを用いて)を用いてシンチレーション計数によって決定した。各試験抗体について、刺激インデックス(SI)を、試験抗体のカウント毎分(cpm):顕著なT細胞エピトープ反応と考えられるSI>2を有する陰性対照のcpmの率として計算した。この結果は、顕著な増殖性反応キメラ(chimaeric)4D5抗体が、試験された20人の健康的なドナーのうちの5人(25%)の試験された4日間の増殖(2より大きいSI)の少なくとも1日に顕著な増殖性反応を誘発し、合成ヒト抗−HER2抗体が、20人のドナーのうち3人(15%)にSI>2を誘発するが、EACHAB抗−HER2抗体は、20人のドナーのうちの誰にもSI>2を誘発しない(0%)ことを示した。これらの結果は、キメラ4D5>合成ヒト抗−HER2抗体>EACHAB抗−HER2の順番を示し、後者が試験されたいずれのドナー血液サンプルに免疫原性の証拠がないことを示している。
【0079】
例3−合成ヒト抗−Lewis Y抗体の構造
【0080】
シアル化Lewis Y抗原に特異的な合成ヒト抗体を、参照抗体可変領域配列として、ヒト化3S193抗体を用いて、例1で記載されているように構成した(Scott et al.,Cancer Res.,60(2000)p3254−3261,US5874060)。インシリコ(in silico)ヒト可変領域配列ライブラリからのセグメントを、ヒト化3S193可変領域配列中の相当するアミノ酸に対する同定用に選択し、結合して図4及び5それぞれに示されているような合成ヒトVH及びVL配列を生成した。同時に、参照キメラ抗−Lewis Y抗体を、参照V領域配列から作った。ヒトIgG1(VH)及びKappa(VK)定常領域を、合成ヒト抗−Lewis Y抗体と、キメラ参照抗体の双方に用いて、抗体を、US5874060に記載されているような合成Lewis Y−HSA複合体に対するELISAによって試験した。このデータは、US5874060のデータと一致する0.15μg/ml合成ヒト抗体と比較したアッセイの結合シグナルを与えるための0.1μg/mlキメラ抗体の最小濃度を示した。
【0081】
例4−合成ヒト抗−IgE抗体の構造
【0082】
合成ヒト抗−IgE抗体を、参照抗体可変領域配列として、Xolair(登録商標)(Presta et al.,J.Immunol.,151(5)(1993)p2623−2632)として知られているヒト化抗−IgE抗体を用いて、例1に記載されているように構成した。インシリコ(in silico)ヒト可変配列ライブラリからのセグメントを、Xolair(登録商標)可変領域配列の相当する相当するアミノ酸に対する同定用に選択し、結合して図6及び7各々に示されているような合成ヒトVH及びVL配列を生成した。同時に、参照キメラ抗−IgE抗体を、参照V領域配列から作った。ヒトIgG1(VH)及びKappa(VK)定常領域を、合成ヒト抗−IgE抗体とキメラ参照抗体の双方に用いた。
【0083】
Fabの特異性を、表面プラズモン共鳴によって更に特徴付けた(瑞国Uppsala所在のBiacore AB社製BIAcore 2000)。組み換えヒトIgE Fabを、Flicker et al.,J.Immunol.,165(2000)p3849−3859によって記載されている通り生成した。試験抗体を精製し、NHS/EDC kit(Biacore社)を用いてCMチップのフロー電池に固定して、キメラ抗−IgE用の2010RU、及び合成ヒト抗−IgE用の2029RUを得た。Hepes緩衝生理食塩水(10mMのHepes、3.4mMのEDTA、150mMのNaCl、0.05%(v/v)界面活性剤P20、pH7.4)中の10及び25nM組み換えヒトIgE Fabを、10分間5μl/minの流量で試験抗体を除いた。この結果は、10及び25nMのIgE Fabの双方について、等価SPR(表面プラズモン共鳴)曲線が、キメラ抗−IgEについて検出され、合成ヒト抗−IgE抗体は、後者が、参照抗−IgE抗体と等価な結合効果を成功裏に達成することを示した。
【0084】
例5−合成ヒトscFvライブラリの生成及びスクリーニング
【0085】
ヒトscFv(単鎖Fv)の初期構造についての戦略は、Knappik et al.,J.MoI.Biol.,296(2000)57−86に詳細に記載されているように、7人分のヒトVHと、4人分のヒトVL(kappa)遺伝子を構成することであり、ヒト可変領域のデータベースから多数のVH及びVL CDR3セグメントにクローニングすることであった。このCDR3のリストは、VH CDR3については表1に、8つのアミノ酸のVL CDR3については表2に、10のアミノ酸のVL CDR3については表3に示されている。マスターVH及びVL構造については、フレームワーク3の端までのVH及びVLをコードする6つのオーバーラップ合成オリゴヌクレオチドが、Knappik et al.,同書に詳細に記載されているように合成され、繰り返しPCR(Prodromou and Pearl,Protein Engineering,5(1992)827−829)を行った。これらは、EcoRV消化pZero−1ベクタ(英国ペイズリィ所在のInvitrogen社)に結紮された。双方ともが開始時に4D5 CDR3を有するCH1及びC Kappaを添加するため(Carter et al,Bio/Technology,10(1992)163−167)、Knappik et al.,同書のプロトコルを、VH−CH1SapI−EcoRIと、VL−kappa Nsi−ShIフラグメントが、EcoRV消化pZero−1にクローニングされた平滑末端であることを除いて行った。
【0086】
表1

【0087】
表2
【0088】
表3
【0089】
CDR3の挿入について、プラス鎖から表Hの各CDR3をコードする単一オリゴヌクレオチドを、コンセンサスVH及びVL遺伝子にアニーリングするために各端に加えられた12つの相同性ヌクレオチドで合成した。これらのCDR配列に加えて、抗体E25からのCDR(例4参照)が含まれていた。これらのプライマを拡張し、二次プライマを加えて、VHについて5’NotI−3’XbaI座、VLについて5’SpeI−3’BamHIを、VH及びVL遺伝子(C領域のない)のN及びC末端のすぐ隣に導入した。クローニングに先立って、更なる相補プライマ対を用いて、VHとVLの間にリンカ配列(Gly4Ser)3を挿入し、一方、XbaI及びSpeI座は維持した。フルサイズのVH−リンカ−VLフラグメントは、NotI及びBamHIで消化され、NotI−BamHI消化pBluescript II KS(+/−)(蘭国アムステルダム所在のStratagene社)にクローニングした。
【0090】
個々のBluescriptクローンを採取し、国際公開番号第WO99/11777号に記載されているように、プラスミドDNAを精製して96ウエルプレート内にロボット制御で分注した。次いで、国際公開番号第WO99/11777号に記載されているように、DNAを、tRNA−ビオチニル−リシンを含有するIVTTにさらし、ストレプトアビジン表面に更にロボット制御で配置した。次いで、10,000の独立したクローンの固定開始scFvライブラリを組み換えヒトIgE Fabでインキュベートすることによってスクリーニングした(例4参照)。ウエルを室温で1時間、PBS/3%BSAでブロックし、PBSで3回洗浄して、PBS/3%BSAの5μg/mlヒトIgE Fabで1時間、処理した。次いで、ウエルを、PBSで更に3回洗浄し、PBS/3%BSAの5μg/mlアルカリ性ホスフォターゼ同定キメラ抗−IgE(例4)で1時間半、処理した。ウエルを、PBSで5回、更に洗浄し、可視化用に基質5−ブロモ−1−クロロ−3−インドリルホスファート及びニトロブルーテトラゾリウム(Roche Molecular)を用いて染色現像した。9600ウエルに1つの頻度で観察される強いシグナルは、E25 CDR3’sを含有するVHとVLの対双方から誘導されることを示した。
【0091】
例6−合成マウス抗−TNFα抗体の構造
【0092】
マウス可変領域配列ライブラリを、NCBI Igblast、Kabat及びGenbankデータベースを用いてヒトライブラリについて、例1に記載されているように作成した。用いた参照抗体可変領域配列は、マウスcA2抗体の可変領域を用いるRemicade(登録商標)(Le et al.,US6277969)として知られているキメラ抗−TNFα抗体であった。インシリコ(in silico)マウス可変領域配列ライブラリからのセグメントは、Remicade(登録商標)可変領域の部分的に相当するアミノ酸から選択されるが、例1で測定される非−自己ヒトMHCクラスIIバインダの形の配列のヒトT細胞エピトープを回避するように設計された変異体を含んでいた。キメラ抗体に用いた配列と比較した合成マウスVH及びVL配列を図8に示し、これは、合成マウス抗体のエピトープ回避のためのセグメント選択の結果として、2つの抗体間のVHとVLそれぞれの9及び16アミノ酸の差を示す。
【0093】
合成マウス及びキメラ抗−TNFα抗体を、例1に記載されているように生成した。標準ELISAの固定ヒトTNFαに結合する精製抗体の比較(国際公開番号第WO03/042247A2号に記載されている)は、合成マウス抗体が、キメラ抗−TNFα抗体の完全な結合能力を保持することを示した(図9)。次いで、これらの抗体の免疫原性を、T細胞アッセイ用の24HLA−DR型ヒト血液サンプルを用いて、例2で記載されているように比較した。この結果は、24人のドナーの誰もSI>2を誘発しない(0%)合成マウス抗−TNFα抗体と比較して、キメラ抗−TNFα抗体が、試験した24人の健康なドナーのうち9人(37.5%)に顕著な増殖反応(SIが2より大きい)を誘発することを示した。これらの結果は、ヒトT細胞エピトープを回避するためのこのようなセグメントの選択でマウスV領域から完全に取り出される可変領域配列のセグメントを具える合成マウス抗体が、なんらエピトープ回避方法を用いることなく、相当するキメラ抗体によって表示されるヒトT細胞アッセイの免疫原性を除去することができることを示した。
【0094】
例7:合成ヒト抗−TNFα抗体の構造
【0095】
ヒトTNFαに対する抗体A2の参照マウス可変領域重鎖及び軽鎖配列を、米国特許第5656272号から得た(図10、各SEQ.ID No1及びNo.2)。構造モデルを、マウス参照可変領域から作成し、CDR確認に欠かせないアミノ酸とCDRに近いパッキングを、CDR(<3Å)からの距離に基づいて同定した。重要ではあるが、余り気にかけなくてもよい残渣中、CDR(>3Å <6Å)からの距離と、CDRに近いより重要な残渣のパッキングの影響に基づいて同定した。更なる残渣セットを、マウス抗体配列、即ち、1%より少ない頻度で特定の座で見出されるアミノ酸の生成頻度に基づいて同定した。
【0096】
これらの残渣のできるだけ多くに含まれるヒトV領域配列セグメントを選択して(表4)、完全な長さのVH及びVL配列を作成した。同定された構造上重要な全ての残渣を含むようにこれらの配列に代替を作成し、エピトープ回避及び合成ヒト抗体設計用のテンプレートとしての機能する配列を作成した。各合成VH及びVLについての好ましい配列を、参照マウス抗体からの重要な残渣を含むように設計した。これらの可変重鎖及び軽鎖アミノ酸配列を、図11及び図12のSEQ ID No.3及びNo.4にそれぞれ示す。
【0097】
表4
(a)重鎖
【0098】
(b)軽鎖
【0099】
合成重鎖及び軽鎖可変領域配列を、様々なインシリコ(in silico)法(例えば、Propred[http://imtech.res.in/raghava/propred/index.html]、Peptide Threading[www.csd.abdn.ac.uk/〜gjlk/MHC−thread]、SYFPEITHI(www.syfpeithi.de)、MHCpred(www.jenner.ac.uk/MHCPred/)を用いてスキャンして、潜在的T細胞エピトープの存在を探し、参照マウスCDRに関連して同種ヒト生殖細胞フレームワーク領域配列と比較した。
【0100】
次の重鎖可変領域変異体を作成した(図11参照):
SEQ.ID.No.5は、SEQ.ID.No.3に対して、次の変化を含む:
T82aN+R83K
【0101】
SEQ.ID.No.6は、SEQ.ID.No.3に対して、次の変化を含む:
T82aN+R83K+D82bS
【0102】
SEQ.ID.No.7は、SEQ.ID.No.3に対して、次の変化を含む:
T82aN+R83K+D82bS+V32A
【0103】
SEQ.ID.No.8は、SEQ.ID.No.3に対して、次の変化を含む:
T82aN+R83K+D82bS+V32A+V78A
【0104】
次の軽鎖可変領域変異体を作成した(図12参照):
SEQ.ID.No.9は、SEQ.ID.No.4に対して、次の変化を含む:
I10T+N103R
【0105】
SEQ.ID.No.10は、SEQ.ID.No.4に対して、次の変化を含む:
I10T+N103R+S80A
【0106】
SEQ.ID.No.11は、SEQ.ID.No.4に対して、次の変化を含む:
I10T+N103R+S80AN41D
【0107】
対照キメラ抗体の構造について、SEQ.ID.No.1及びSEQ.ID.No.2を与えるように翻訳するヌクレオチド配列を、一連のオーバーラップ40マー合成オリゴヌクレオチドを用いて構成した。V領域配列の側面には、追加の5’及び3’配列が位置し、ほ乳類発現ベクタへのクローニングを容易にしている。オリゴヌクレオチドの配列を図13及び図14に示す。
【0108】
オリゴヌクレオチドを、Sigma−Genosys社(英国プール所在)から購入し、100μMの濃度で再懸濁した。ほとんどの5’オリゴヌクレオチドを除く、1μlの各重鎖有意DNAオリゴヌクレオチドを共に混合して、1.5μl(ほぼ、1μg)の混合物を:2μl 10× PNK緩衝液、2μl 10mMのATP、14μl H2O、0.5μl(5単位)PNKを更に含む20μlの反応液中でポリヌクレオチドキナーゼ(PNK、英国ペイズリー所在のInvitrogen社)を用いて処理した。この反応液を37℃で30分間インキュベートし、70℃で20分間加熱することによって、酵素を不活化した。重鎖アンチセンス、軽鎖センス、及びアンチセンスオリゴヌクレオチドを、同様にリン酸化した。各セットからの5’オリゴヌクレオチドを、H2Oで9倍に希釈し、適切な反応混合物に1.5μlを添加した。次いで、各反応液を0.5mlに希釈して、体積が最高で44μlになるまで、8000rpmで90分間、Amicon microcon YM3濃縮器で回転透析した。
【0109】
重鎖についてのセンス及びアンチセンス混合物と、軽鎖についてのセンス及びアンチセンス混合物を混合して、H2Oで88μlにした。10μlの10× リガーゼ連鎖反応(LCR)緩衝液、及び2μlのPfu リガーゼ(8単位、英国ケンブリッジ所在のStratagene社)を、各反応液に添加し、プログラム可能な加熱ブロック(heating block)中で、次のようにインキュベートした:94℃で4分間、次いで、1サイクルにつき60℃で3分間、その後、94℃で39秒間、次いで60℃で3分間を20サイクル。最後に反応液を、60℃で5分間インキュベートした。各LCR10μlを、臭化エチジウムで染色した1%のアガロースゲルに通し、1Kbラダーマーカ(Invitrogen社)と比較した。ほぼ400bpの微かな特異的バンド(faint specific band)の周囲に、結紮DNAの塗抹標本を各レーンで観察した。
【0110】
重鎖についてSEQ.ID.No.12が、22、及び軽鎖についてSEQ.ID.No.33及び43をプライマとして用いて、重鎖及び軽鎖LCRをPCRを介して増幅した。各反応液には、次のものが含まれていた:5μlのLCR、5μlの10× エクスパンドHiFi緩衝液(英国ルイス所在のRoche社)、lμlの10mM NTP混合物、0.25μlの各プライマ(l00μMのストックから)、0.5μlのエクスパンドHiFiポリメラーゼ(3単位、Roche社)、及び38μlのH2O。この反応液を次のように循環させた:94℃で2分間、次いで94℃で30秒間を20サイクル、60℃で30分間、及び72℃で30秒間。最後に、反応液を72℃で5分間インキュベートした。反応液の収量及び特性を、上記のようなアガロースゲル電解質によって確認した。ほぼ400bpで特異的でシャープなバンドが、各反応液について観察された。
【0111】
Qiagen PCR精製キットを用いて、反応生成物を精製し、各々を30μlのH2Oで溶離した。重鎖生成物を、制限酵素Mlu I及びHind IIIを用いて標準反応で消化させ、軽鎖生成物を、BssH II及びBamH Iを用いて消化させた。Qiagen PCR精製キットを用いて、反応生成物を再び精製し、各々を30μlのH2Oで溶離した。
【0112】
軽鎖発現ベクタpANT08は、pAT153バックボーンに基づいており:CMV即時/アーリーエンハンサプロモータ−590から+7、高発現マウス抗体軽鎖RNA由来の30nt 5’UTR、可変領域開始コドン付近のBssH II制限座を組み込むマウスコンセンサス軽鎖シグナル配列、可変領域スプライス座からBamH I制限座への33ntを含むヒト合成イントロンに対するショートリンカ(可変領域の代用)、次いで、ヒト定常Kappa(CK)領域遺伝子に先立つ343ntのイントロンを含むヒト遺伝子DNAのフラグメント、CK遺伝子及びCKポリAをこの順に含む。
【0113】
重鎖発現ベクタpANT09は、プロモータ領域を通るpANT08と類似しており、この後に:上記の重鎖発現ベクタ由来の62nt5’UTRと、可変領域開始コドン付近のMlu I座を組み込むマウス重鎖コンセンサスシグナル配列と、可変領域スプライス座に対するショートリンカ(可変領域の代用)、その直後にCH領域ポリA座の端に対するCH1遺伝子のイントロン211ntの上流に位置したHind III制限座からのヒトゲノムDNAと、が続く。このフラグメントは、CH1、ヒンジ、CH2及びCH3イントロン、及びヒトIgG1のエクソンが含まれる。また、このベクタも、メトトレキサートに対する耐性について、SV40プロモータとポリAシグナルによって制御される、ジヒドロ葉酸還元酵素用の遺伝子を含んでいた。
【0114】
各ベクタ2μgを、全体積30μgの標準的反応液中で関連制限酵素で消化させた。各反応液は、上記のような1%アガロースゲルを通り、ベクタ特異バンド(6.0Kbp重鎖及び4.2Kbp軽鎖)をゲルから切り取り、Qiagenゲル抽出キットを用いて精製し、30μlH2O中で溶離した。
【0115】
各消化ベクタ1μlを、Ligafastキット(英国サウサンプトン所在のPromega社)を用いて、3μgの相当する消化可変遺伝子PCR生成物に結紮した。各結紮反応液2.5μlを、製造者によって指示されているように、サブクローニング効率適格(efficiency competent)XLl−blue(Stratagene社)に形質転換し、100μg/mlのアンピシリンを含有するLBアガープレート上に置き、37℃で夜通しインキュベートした。各結紮からの10の細菌コロニィを、100μg/mlアンピシリンを含有する10mlの2× YT培養液内に植菌し、振盪しながら37℃で夜通し培養した。プラスミドを、Qiagenプラスミド調製キットを用いて、1.5mlの各一夜培養物から精製し、各々を50μlのH2Oで溶離した。プラスミドを、契約しているシークエンシング施設に送り、正確なV領域遺伝子配列を同定する標準CMVプロモータプライマ及びクローンで配列した。
【0116】
合成ヒト抗体の構造について、SEQ.ID.No.3及びSEQ.ID.No.4を与えるように翻訳するヌクレオチド配列を、一連のオーバーラップ40マー合成オリゴヌクレオチドを用いて構成した。オリゴヌクレオチドの配列を、図15及び図16に示す。SEQ.ID.No.5を与えるように翻訳するヌクレオチド配列を、オリゴヌクレオチドプライマSEQ.ID.No.94及び95(図17)を、オリゴヌクレオチドSEQ.ID.No.53及び63とプライマリ合成ヒト抗体重鎖変異体のプラスミドDNAと共にテンプレートとして用いて、オーバーラップPCRを介して構成した。2つのPCR反応を、SEQ.ID.No.53及び95、又はSEQ.ID.No.94及び63のプライマ対として用いて実施した。各反応液中には、次のものが含まれていた:1μl(100ng)のプラスミドテンプレート、5μlの10× エクスパンドHiFi緩衝液(Roche社)、1μlの10mM NTP混合物、0.25μlの各プライマ(100μMストックから)、0.5μlのエクスパンドHiFiポリメラーゼ(3単位、Roche社)、及び42μlのH2O。これらの反応液を次のように循環させた:94℃で2分間、次いで、94℃で30秒間を20サイクル、60℃で30秒間、及び72℃で30秒間。最後に、この反応液を72℃で5分間インキュベートした。全反応液を1%のアガロースゲルによって電気泳動させ、295bp及び126bpの特異的バンドを除去し、Qiagenゲル抽出キットを用いて精製した。DNAを30μlのH2Oで溶離した。
【0117】
テンプレートとして、1μlの295bp生成物及び1μlの126bp生成物を用いたことを除いて、2つの精製フラグメントを、上述のPCR条件でオリゴヌクレオチドプライマSEQ.ID.No.53及び63を用いて、PCR反応液中で結合した。従って、H2Oの量は、41μlに減少した。396bpの結合したPCR生成物を、QiagenPCR精製キットを用いて精製し、30μlのH2Oで溶離した。
【0118】
SEQ.ID.No.6を与えるように翻訳するヌクレオチド配列を、オリゴヌクレオチドプライマSEQ.ID.No.96及び97(図17)を、オリゴヌクレオチドSEQ.ID.No.53及び63とプライマリ合成ヒト抗体重鎖変異体のプラスミドDNAと共にテンプレートとして用いて、オーバーラップPCRを介して構成した。2つのPCR反応を、SEQ.ID.No.53及び97、又はSEQ.ID.No.96及び63のプライマ対として用いて実施した。第1段階のPCRを上記のように実施して、295bp及び126bpのフラグメントを得た。これらのフラグメントを、上記のように精製し、共に結合させて、再精製した。
【0119】
SEQ.ID.No.7を与えるように翻訳するヌクレオチド配列を、オリゴヌクレオチドプライマSEQ.ID.No.98及び99(図17)を、オリゴヌクレオチドSEQ.ID.No.53及び63とSEQ.ID.No.6についてのPCR生成物と共にテンプレートとして用いて、オーバーラップPCRを介して構成した。2つのPCR反応を、SEQ.ID.No.53及び99又はオリゴヌクレオチドSEQ.ID.No.98及び63のプライマ対のいずれかを用いて実施した第1段階のPCRを上記のように実施して、98bp及び318bpのフラグメントを得た。これらのフラグメントを、上記のように精製し、共に結合させて、再精製した。
【0120】
SEQ.ID.No.8を与えるように翻訳するヌクレオチド配列を、オリゴヌクレオチドプライマSEQ.ID.No.100及び101(図17)を、オリゴヌクレオチドSEQ.ID.No.53及び63とSEQ.ID.No.7についてのPCR生成物と共にテンプレートとして用いて、オーバーラップPCRを介して構成した。2つのPCR反応をSEQ.ID.No.53及び101、又はSEQ.ID.No.100及び63のどちらかをプライマ対として用いて実施した。第1段階PCRを上記のように実施して、270bp及び155bpのフラグメントを得た。これらのフラグメントを、上記のように精製し、共に結合させて、再精製した。
【0121】
上記のPCR生成物のそれぞれを、Mlu I及びHind IIIで消化させ、同様に消化させたpANT09内に結紮した。この結紮を、形質転換して配置し、コロニィを選定し、プラスミドを調製して、全て配列した。
【0122】
SEQ.ID.No.9を与えるように翻訳するヌクレオチド配列を、テンプレートとして、オリゴヌクレオチドプライマSEQ.ID.No.102及び103(図17)と、プライマリ合成ヒト抗体軽鎖変異体のプラスミドDNAとを用いて、PCRを介して構成した。重鎖変異体について記載されているように、PCR単反応を実施し、383bpの生成物を得た。完全な反応液を1%アガロースゲルによって電気泳動し、特異的なバンドを除去し、Qiagenゲル抽出キットを用いて精製した。DNAを30μlのH2Oで溶離した。
【0123】
SEQ.ID.No.10を与えるように翻訳するヌクレオチド配列を、テンプレートとして、オリゴヌクレオチドSEQ.ID.No.74及び84と共にオリゴヌクレオチドプライマSEQ.ID.No.104及び105(図17)と、SEQ.ID.No.9についてのPCR生成物とを用いて、オーバーラップPCRを介して構成した。2つのPCR反応を、SEQ.ID.No.74及び105、又はSEQ.ID.No.104及び84のプライマ対として用いて実施した。第1段階のPCRを重鎖変異体についての上記のように実施して、265bp及び139bpのフラグメントを得た。これらのフラグメントを、重鎖変異体についての上記のように精製し、共に結合させて383bpの生成物を作成し、再精製した。
【0124】
SEQ.ID.No.11を与えるように翻訳するヌクレオチド配列を、テンプレートとして、オリゴヌクレオチドSEQ.ID.No.74及び84と共にオリゴヌクレオチドプライマSEQ.ID.No.106及び107(図17)と、SEQ.ID.No.10についてのPCR生成物とを用いて、オーバーラップPCRを介して構成した。2つのPCR反応を、SEQ.ID.No.74及び107、又はSEQ.ID.No.106及び84のプライマ対として用いて実施した。第1段階のPCRを重鎖変異体についての上記のように実施して、148bp及び256bpのフラグメントを得た。これらのフラグメントを、重鎖変異体についての上記のように精製し、共に結合させて383bpの生成物を作成し、再精製した。
【0125】
上記のPCR生成物のそれぞれは、BssH II及びBamH Iで消化され、同様に消化されたpANT08内に結紮される。この結紮を上記のように、形質転換して配置し、コロニィを選定し、プラスミドを調製して、上述した通り全て配列した。
【0126】
CHO−K1細胞(ATCC#CCL−61)を、10%FCS、L−グルタミン、ピルビン酸ナトリウム、及びL−プロリンを含む高グルコースDMEM中で増殖させた。近融合性培地を、製造業者(Invitrogen)によって指示されているように、リポフェクトアミン2000(Lipofectamine 2000)を用いてトランスフェクション用に採取した。トランスフェクションは、0.3μgの重鎖作成物及び0.2μgの軽鎖作成物を具える0.5μgのプラスミドDNAの全てを用いて、3×105セル/mlで200μlの細胞を播種した48ウエルプレート中で実施した。
【0127】
上澄みを採取する前に、トランスフェクションを、37℃/5%CO2で48から72時間インキュベートした。抗体発現は:マウスモノクローナル抗−ヒトIgGキャプチャ抗体と、ヒトIgG1/Kappaスタンダードと、HRP複合ヤギ抗−ヒトKappa軽鎖と、を検出抗体として用いてELISAによって定量化した(全ての試薬はSigma製)。
【0128】
重鎖及び軽鎖の全ての組み合わせを遺伝子導入した(即ち、6重鎖×5軽鎖=30トランスフェクション)。抗体発現レベルは一般的に、0.5から2.0μg/mlの範囲内であるが、重鎖SEQ.ID.No.8では、発現は観察されなかった。
【0129】
発現抗体を、TNF−感受性WEHI−164細胞を用いて、ヒトTNFαの活性を無毒化する能力について試験した(Espevik et al.,J.Immunol.Methods 1986,95,99−105)。細胞を、3−4時間、96−ウエルマイクロタイタープレートに、1ウエル当たり5×104個の細胞で、1μg/mlのアクチノミシンDに置いた。細胞を40pMヒトTNFα、及び様々な濃度のキメラ抗体(1ng/mlから500ng/mlの範囲)に曝露させて、検量線を作成した。重鎖及び軽鎖の様々な組み合わせを、25ng/mlの単一濃度点で試験した。この濃度は、キメラ抗体のED50として既に決定してある。ED50のキメラ抗体として以前決定した全アッセイを3回実施した。
【0130】
混合物を37℃で夜通しインキュベートした。最終濃度0.5mg/mlに、3−[4,5−ジメチル−チアゾール−2−イル]−2,5−ジフェニルテトラゾリウムブロミド染料(MTT)を添加し、37℃で4時間、インキュベートし、0.1MのHCl、0.1% SDS中で細胞を溶解し、550nmの波長で吸光度を測定して、細胞生存率を決定した。
【0131】
重鎖及び軽鎖の組み合わせからの吸光度を用いて、検量線から見掛けの抗体の濃度を計算した。キメラの見掛けの濃度は、変異体の各組み合わせの濃度によって分けられて、倍の差分値を与えた。キメラの値よりも低い値は、これらの組み合わせが、TNFα細胞毒性から細胞を保護する点でより効果的であることを示すが、より高い値は、これらの効果がより低いことを示した。全ての組み合わせについての値を表5に示す。
【0132】
表5
キメラ抗体と比較した合成ヒト抗体変異体の活性率
【0133】
次の合成ヒト抗体重鎖及び軽鎖の組み合わせは、1.0に近い倍の差分値を与えた:SEQ.ID.No.5/10、SEQ.ID.No.7/4、SEQ.ID.No.7/9、SEQ.ID.No.7/10。これらの組み合わせを更なる研究用に選択した。
【0134】
上記で選択された配列を運搬する発現プラスミドをNS0細胞内に遺伝子導入した(ECACC番号第85110503号)。これらの細胞を、L−グルタミン、ピルビン酸ナトリウム、5%超低IgG FCS及びペン/ストレプ(pen/strep)を含む高グルコースDMEMで培養した。細胞を、対数増殖期中に採取し、遠沈して新しい増殖培地内で5×106セル/mlで再懸濁した。750μlの細胞を全量30μgの各プラスミド対で混合し、これを、50μlのH2O内でSsp Iで線形化した。細胞/プラスミド混合物を4mmギャップのキュベットに移し、250V、1500μF、無限抵抗で、Equibio Easyject Plusを用いてエレクトロポレートした。このエレクトロポレートは、25mlの予熱した増殖培地に即時移し、100μl/ウエルで、5×96ウエル平底プレートに置かれた。このプレートを37℃/5%CO2でインキュベートした。48時間の後エレクトロポレーションを行い、300nMメトトレキサートを含有する50μl培地を、各ウエルに加えて、100nMの最終濃度を与えた。7日間の後エレクトロポレーションを行い、100nMメトトレキサートを含有する更なる50μlの培地を、各ウエルに加えた。
【0135】
ほぼ2週間後のエレクトロポレーションを行うと、いくつかのウエルの培地は、黄色になり始め、遺伝子導入されたコロニィ増殖を示した。これらのウエルからの培地を、抗−ヒトIgG Fcキャプチャ/抗−ヒトIg Kappa軽鎖HRP複合検出ELISAを用いて、抗体発現について試験した。試験サンプルを、ヒトIgG1/Kappa標準及び確立されている抗体発現レベルと比較した。抗体のコロニィ発現有効量を、200nMメトトレキサートを含む培地で増やした。
【0136】
抗体をタンパク質A親和性クロマトグラフィを介して500ml培養培地から精製し、Sephacryl S200を用いて、サイズ排除クロマトグラフィを実施した。精製した抗体を、280nmでのUV吸光度によって定量化し、OD2801=1.4mg/mlを仮定した。
【0137】
精製キメラ及び複合抗体を、上記の例4に記載されているWEHI−164保護アッセイを介して活性について試験した。各抗体を、検量線を作成するのに先に用いた全濃度範囲に渡って試験した(図18参照)。合成ヒト抗体7/10(即ち、SEQ.ID.No.7及び10を含む)は、最も活性な変異体であり、キメラ抗体と同じ活性を有することが分かった。合成ヒト抗体7/9及び5/10は、キメラと比較して僅かに少ない同様の活性を有し、合成ヒト抗体7/4は、最も活性がなかった。
【0138】
従って、合成ヒト抗体7/10が、最も好ましいMHCクラスII結合プロファイルを有すると予測され、最も活性な変異体であるので、これを、経時変化T細胞増殖アッセイでの試験用に選択した。ヒトPBMCを、2回のFicoll密度遠心分離を介して、ヒト血由来の軟膜から調製した。調製PBMCを、90%ヒトAB血清/10%DMSO中の1mlアリコートで3×107セル/mlの密度で再懸濁し、液体窒素化で貯蔵した。PBMCをDynal Allset(登録商標)PCRタイピングキットを用いて組織分類した。
【0139】
リード合成ヒト抗体を、20人の健康なドナーからのヒトPBMCを用いて、全タンパク質T細胞アッセイ中のキメラ抗体と比較した。各ドナーからのPBMCを解凍し、洗浄して、AIM V血清フリーリンパ球増殖培地で再懸濁した。第1日目に、50μgのタンパク質を、24ウエルプレートの4×106のPBMC2mlのバルク培地に加え、第6日目から第9日目に、3重に100μlアリコートを除去して、96ウエルプレートに移した。採取して放射能の取り込みを測定する前に、各アリコートを、1μCiトリチウム化チミジンを含有する75μl培地で24時間パルスした。結果を刺激インデックス(SI)の計算によって標準化した。広範囲のHLA DRアロタイプのガレージは、個々のMHCハプロタイプによってドナーを選択することによって達成される。
【0140】
経時アッセイの結果を、図19で示す。これらの結果は、キメラ抗体(図19(a))が、20人のドナー中10人に少なくとも1日で、T細胞反応(SI>=2)を導くことを示す。逆に、合成ヒト抗体(図19(b))は、あらゆる時点であらゆるドナーの反応も誘導することができなかった。従って、非免疫原性合成ヒト抗体は、マウス抗−TNFα抗体(A2)を参照として用いて、ヒト抗体のセグメントから成功裏に構成された。
【0141】
例8:合成タイプIリボソーム阻害タンパク質の構造
【0142】
プラントタイプIリボソーム阻害タンパク質(RIP)ブーガニン(ブーゲンビレアスペクタビリス(Bougainvillea spectabilis)に由来する)の合成変異体を、国際公開番号第WO2005090579号で記載されている方法を用いて生成した。ブーガニン中のT細胞エピトープのロケーションを、国際公開番号第WO2005090579号に記載のようにオーバーラップ15マーペプチドの分析によって試験し、表6のSEQ ID11−13のペプチド(残渣121−135、130−144、及び148−162に相当する)をエピトープとして同定した。ブーガニンを、ブーゲンビレアスペクタビリス(Bougainvillea spectabilis)植物の葉の組織からクローニングした。製造業者によって指示されているように100mg組織からポリA Tract System 1000キット(Promega社)を用いてmRNAを抽出した。cDNAを、以下のプライマ:ATGTACAACACTGTGTCATTTAAC及びTTATTTGGAGCTTTTAAACTTAAGGATACCを有するAccessQuick RT−PCR system(Promega社)を用いて、mRNAテンプレートから合成した。第1のプライマは、ATG開始コドンを更に含み、第2のプライマは、TAA停止コドンを更に含む。PCR生成物を、T/Aクローニングシステム(pGEM T Easy、Promega社)を用いてクローニングし、いくつかのクローンをベクタ内に含まれるT7プロモータの転写方向に配向する修正クローンを同定するように配列した。
【0143】
表6
ブーガニン(bouganin)の免疫原性ペプチド配列及び置換ヒト配列セグメント
【0144】
表6に示されるように同定されたヒト配列セグメントを含む一連の変異体を作成した。これらは、高品質ポリメラーゼ(エクスパンドHiFi、Roche社)を有するオーバーラップPCRを用いて構成した。5’及び3’プライマは上述のものであり、PCR生成物を上述の通りT/Aクローニングベクタにクローニングし、T7プロモータの転写方向に配向した修正クローンを同定した。クローンを、発光酵素遺伝子を発現する対照DNA(Luciferase T7 Control、Promega社)を含む共役転写及び転写反応中の活性について分析した。ブーガニンはリボソーム不活化タンパク質であるので、発光酵素遺伝子の転写のレベルを著しく下げる。この減少は、従来Steady−Glo(Promega社)等の発光酵素検出システムを用いて分析されている。精製野生型又は突然変異体ブーニガンプラスミドを、Not Iで線形化し、10ng/mlに希釈した。Luciferase T7 Control DNAを125ng/μlに希釈した。1μlの各DNAを、10μlのTnT混合物(Promega社)と、0.25μlのメチオニンと、0.25μlのヌクレアーゼ自由水と混合した(TnTキットで供給される)。対照は、野生型(wt)ブーニガンとLuciferase T7 Controlのみであった。反応を3回実施し、30℃で1時間インキュベートした。5μlの各反応液を、黒い壁付の96ウエル照度計プレートに移し、45μlの水と、50μlのSteady−Glo試薬を混合した。Wallac Microbeta Trilux照度計で発光を読んだ。活性は、Luciferase T7 Control DNAのみで観察された蛍光の割合として表した。
【0145】
図20は、多数の異なる変異体の活性プロファイルを示す。これは、最も活性な変異体が:ペプチド41中のV123T;ペプチド44中のV132P/Y133Q;ペプチド44中のI152Q;であることを示す。これらの4つの突然変異を含む複合変異体を作成し、活性アッセイで再試験した。この変異体の活性は、図20のCOMBによって示され、wtタンパク質の活性のほぼ75%を維持する。
【0146】
残渣121−135、130−144、及び148−162に相当する活性COMB変異体内のヒト配列セグメントを含むペプチドを合成し、例7に記載したように、20人の健康なドナーからのヒトPBMCを有する経時T細胞アッセイ中の相当する野生型ペプチドと比較した。この結果は、ヒト配列セグメントを含むペプチドが、どの時点でも、どのドナーのT細胞増殖も誘発しないが、各野生型ペプチドは、少なくとも1の時点で全ドナーの>10%のSI>2を有する増殖を誘発することを示した。
【0147】
例9:合成ヒルジンの構造
【0148】
トロンビン阻害ヒルジン(医用蛭(Hirudo medicinalis)に由来する)の合成変異体を野生型として表7のSEQ ID No 14を有するタンパク質を用いて、国際公開番号第WO2004113386号に記載されている方法で生成した。ヒルジンのT細胞エピトープのロケーションを、国際公開番号第WO2004113386号に記載されているように、オーバーラップ15マーペプチドの分析によって試験し、ペプチド27−41CILGSDGEKNQCVTGが、20人の健康なドナーからのヒトPBMCで有意なT細胞反応を与えることが示された。ヒトメラノーマ関連抗原(AAN40505.1)からのヒト配列セグメントKCRHを、例8のオーバーラップPCRを用いて、26−29でヒルジン残渣を置換するのに使用し、国際公開番号第WO2004113386号に記載されているアッセイを用いる野生型ヒルジンの完全活性化を維持した28/29RHに変わる28/29ILを有する変異体ヒルジン分子を得た。修飾ペプチド27−41CRHGSDGEKNQCVTGを、この修飾ペプチド中のT細胞エピトープ活性の損失を示す例8に示すようなT細胞アッセイ中の野生型ペプチド27−41CILGSDGEKNQCVTGと共に試験した。
【0149】
例10:Trエピトープを有する合成ヒト抗−IgE抗体の構造
【0150】
例4の合成ヒト抗−IgE抗体からのVH及びVL遺伝子を、同書内のOrlandi et al.からの標準ポリメラーゼ連鎖反応(PCR)法によって、オリゴヌクレオチドプライマ対を、VL−及びVH−特異PCR用のテンプレートとして用いて、分離プラスミドベクタにクローニングした。オーバーラップする相補配列を、続く融合PCR中に結合したPCR生成物内に導入し、20アミノ酸(G4S1)4リンカ、又は代替的に、2つのアスパラギン残渣及びGGSトリプレットによって各側に並んだC型肝炎コアタンパク質(P19,MacDonald et al.,Journal of Infectious Diseases,185(2002)p720−727)からの10アミノ酸Trエピトープを含む20アミノ酸配列GGSNNLSCLTIPASANNGGSのどちらかのコード配列を形成した。この最終増幅ステップを、制限酵素EcoRV及びBspE1を有する続く開裂用のプライマ対で実施し、bluescriptKS vector(Stratagene社)にクローニングした。次いで、合成ヒト抗−IgE単鎖抗体(scFvs)の二量体形を、Mack et al.,Proc Natl Acad Sci USA.,92(1995)p7021−7025の方法によって構成した。二量体VL−リンカ−VH−VL−リンカ−VHフラグメントを、発現ベクタpEF−DHFR(Mack et al.,同書)のEcoR1/Sal1座にサブクローニングし、エレクトロポレーションによって、DHFR−欠損中国ハムスタ卵巣(CHO)細胞に遺伝子導入した。選択、遺伝子増幅、タンパク質生成を、Mach et al.,同書によって記載されているように実施した。二量体scFvを、ニッケル−ニトリロ三酢酸(Ni−NTA)カラム(Qiagen社)上の親和性クロマトグラフィよってC−末端ヒスチジンテールを介して精製して、CHABIgEG4S 1×4((VLとVHの間のG4SO4リンカ)とCHABIgEHCVP19(VLとVHの間のHCV Trエピトープ)を指定した二量体Fvsを与えた。
【0151】
二量体scFvsを、20人の健康なドナーからのPBMCを用いてHall et al.,Blood 100(2002)p4529−4536に記載された通りに正確に50μg/mlのヒトT細胞アッセイで順次試験した。これらの結果は、CHABIgEG4S 1×4、又はCHABIgEHCVP19のどちらかについてT細胞の有意な増殖を示していないが、CHABIgEG4S 1×4ではなくCHABIgEHCVP19で刺激した20人のドナーのうちの4人からのIL−10生成の有意なレベル(SI>2)を示した(20人のドナー中0人のSI>2)。これは、免疫抑制サイトカインIL−10の誘発についての抗体分子内に含まれるTrエピトープの効果を示している。
【図面の簡単な説明】
【0152】
【図1−1】図1は、合成ヒト抗HER−2抗体に用いられるVH遺伝子の配列である。
【図1−2】図1は、合成ヒト抗HER−2抗体に用いられるVH遺伝子の配列である。
【図1−3】図1は、合成ヒト抗HER−2抗体に用いられるVH遺伝子の配列である。
【図2−1】図2は、合成ヒト抗HER−2抗体に用いられるVL遺伝子の配列である。
【図2−2】図2は、合成ヒト抗HER−2抗体に用いられるVL遺伝子の配列である。
【図2−3】図2は、合成ヒト抗HER−2抗体に用いられるVL遺伝子の配列である。
【図3】図3は、キメラ4D5IgG1/kappaで8日間、インキュベーションした後のヒトSK−BR−3細胞と、合成ヒト抗−HER2抗体と、キメラ抗−IgE対照で抗−HER2「EACHAB」を回避したエピトープの増殖の阻害を示す(例4参照)。
【図4−1】図4は、合成ヒト抗−Lewis Y抗体を用いたVH遺伝子の配列である。
【図4−2】図4は、合成ヒト抗−Lewis Y抗体を用いたVH遺伝子の配列である。
【図4−3】図4は、合成ヒト抗−Lewis Y抗体を用いたVH遺伝子の配列である。
【図5−1】図5は、合成ヒト抗−Lewis Y抗体を用いたVL遺伝子の配列である。
【図5−2】図5は、合成ヒト抗−Lewis Y抗体を用いたVL遺伝子の配列である。
【図5−3】図5は、合成ヒト抗−Lewis Y抗体を用いたVL遺伝子の配列である。
【図6−1】図6は、合成ヒト抗ヒトIgE抗体を用いたVH遺伝子の配列である。
【図6−2】図6は、合成ヒト抗ヒトIgE抗体を用いたVH遺伝子の配列である。
【図6−3】図6は、合成ヒト抗ヒトIgE抗体を用いたVH遺伝子の配列である。
【図7−1】図7は、合成ヒト抗ヒトIgE抗体を用いたVL遺伝子の配列である。
【図7−2】図7は、合成ヒト抗ヒトIgE抗体を用いたVL遺伝子の配列である。
【図7−3】図7は、合成ヒト抗ヒトIgE抗体を用いたVL遺伝子の配列である。
【図8】図8は、ヒトT細胞エピトープの回避を含む合成マウス抗ヒトTNFα抗体を用いたVH及びVL遺伝子の配列である。
【図9】図9は、合成マウス及びキメラ抗ヒトTNFα抗体によるヒトTNFαへの結合用ELISAである。
【図10】図10は、抗TNFα抗体A2のV領域配列である。
【図11】図11は、合成ヒト抗TNFαVH変異株の配列である。
【図12】図12は、合成ヒト抗TNFαVL変異株の配列である。
【図13】図13は、キメラマウス:ヒト抗TNFαVHの構成用オリゴヌクレオチドである。
【図14】図14は、キメラマウス:ヒト抗TNFαVLの構成用オリゴヌクレオチドである。
【図15】図15は、一次合成ヒト抗TNFαVH(SEQ ID No.3 図11に相当する)の構成用オリゴヌクレオチドである。
【図16】図16は、一次合成ヒト抗TNFαVL(SEQ ID No.4 図12に相当する)の構成用オリゴヌクレオチドである。
【図17】図17は、二次合成ヒト抗TNFαVH及びVL変異株の構成用オリゴヌクレオチドである。
【図18】図18は、合成ヒト抗TNFα抗体用WEHI−164保護アッセイである。
【図19】図19は、リード合成ヒト抗TNFαの経時ヒトT細胞アッセイである。
【図20】図20は、挿入されたヒト配列セグメントを有する合成ブーガニン分子の活性である。
【技術分野】
【0001】
本発明は、最終抗体又はタンパク質分子内で、ヒト抗体又はタンパク質からのアミノ酸配列の2又はそれ以上のセグメントを結合する抗体及びタンパク質の生成に関する。具体的には、本発明は、配列セグメントのこのような結合を提供して、最終抗体又はタンパク質分子中のT細胞エピトープの数を低減又は回避するようにする。本発明は、特に、ヒトの医薬品又はインビボ診断剤として用いる抗体及びタンパク質の生成に関する。
【0002】
ここ20年で、ヒトの潜在的医薬品として用いる組み換え型モノクローナル抗体の生成に大幅な進歩が見られている。ファージ提示法又は遺伝子導入マウスによる、キメラ化技術、ヒト型化技術及びヒト抗体クローニング技術は、非ヒトモノクローナル抗体よりも低い免疫原性を有するヒトに投与される場合に一般的に良好な耐容性を有する抗体を提供している。しかしながら、これらの技術によって生成したいくつかの抗体は、このような抗体の遺伝子起源がヒトである場合でさえ、患者の免疫原性を誘発することを示している。例えば、ヒト抗体Humira(登録商標)は、12%の関節リウマチ患者に免疫原性を誘発し、ヒト化抗体CAMPATH(登録商標)は、約50%の患者に免疫原性を誘発する。このような免疫原性の誘発は、抗体可変領域内での非アミノ酸配列の一部の存在に起因しているようである。この非アミノ酸配列の一部は、いくつかの場合に、免疫原性をもたらすT細胞反応を誘発するT細胞エピトープを作ることができる。従って、高いヒト起源を有するが、改善した技術と、T細胞反応を誘発する配列の作成をできるだけ避ける抗体組成物が求められている。
【0003】
本発明は、方法と、それによって、治療用に、このような抗体(ここでは、「複合抗体」という)が、最終抗体分子内でヒト抗体からのアミノ酸配列の2又はそれ以上のセグメントを結合するような結果物としての抗体組成を提供する。
【0004】
従って、第1の態様では、本発明は修飾抗体、又はこれの抗原結合フラグメントを提供しており、これは修飾抗体又は抗原結合フラグメントの重鎖及び軽鎖可変領域が、1又はそれ以上の別の抗体又は抗原結合フラグメントからのアミノ酸配列の2又はそれ以上のセグメントから成り、これらのセグメントが、全CDRでもフレームワーク領域でもない。
【0005】
本発明のコンテキストでは、用語「セグメント」は、抗体分子内で見つけられる隣接アミノ酸配列をいい、このようなセグメントは、2から125アミノ酸長サイズの範囲であり、好ましくは、2から31アミノ酸長の範囲であり、このようなセグメントは、全CDRでも全フレームワーク領域でもない。治療に用いる場合、本発明の複合抗体は、一般的に、複合抗体の可変領域内で、様々なヒト抗体からのアミノ酸配列の2又はそれ以上のセグメントを結合する。具体的には、本発明は、複合抗体重鎖及び軽鎖可変領域(それぞれ、VH及びVLという)に関し、VHとVLは各々、完全に2又はそれ以上のヒト抗体可変領域からの配列のセグメントから成り、一般的に、各合成VH及びVLは、源ヒト抗体VH及びVL中の位置に相当するヒト可変領域配列位のセグメントを含み、例えば、合成VH配列中のアミノ酸1から10は、ヒト抗体のアミノ酸1から10から導出する。代替的に、複合抗体中のヒトVH又はVL配列のセグメントは、源ヒト抗体VH又はVL中の配列位に関わりなく、どの配列位に位置してもいてもよい。この源ヒト抗体VH及びVLは、例えば、ヒトモノクローナル抗体V領域配列のデータベースに提供されているような、現存のヒト抗体可変(V)領域アミノ酸配列であり、V領域体細胞変異体、及び生殖細胞系とは異なるその他の変異体を有する親和性成熟抗体からの配列、生殖細胞系V領域からの配列、固定V領域フレームワークセットを有するが可変CDRを有する抗体等といった種の抗体からの配列のセグメントから作られた人工的に構成された抗体V領域からの配列、ファージ表示ライブラリ等のヒト抗体ライブラリから選択された配列、及びヒト抗体又は抗体フラグメントをコードするトランスジェニック動物発現遺伝子から導出したヒト抗体の配列を含んでもよい。
【0006】
本発明の好ましい実施例では、治療用の本発明の複合抗体は、最終複合抗体V領域のヒトT細胞エピトープを制限又は回避する組み合わせで多重ヒトVH及びVL配列セグメントを結合することによって構成される。
【0007】
これに関して、ヒトT細胞エピトープは、ヒトMHCクラスII分子に結合することができるアミノ酸配列であり、CD4+T細胞への提示によって、ヘルパーT細胞反応を誘発する。最終複合抗体中のT細胞エピトープを制限、又は回避するヒトVH及びVL配列セグメント及びセグメントの組み合わせを選択できる。これは、ヒト生殖細胞系配列から等のT細胞エピトープを含まないセグメントの使用によって、及び隣接するセグメントを結合して、例えば、2つのセグメントの接合点における非MHC結合配列の生成によって、別のヒト生殖細胞系配列の生成によって、又は非生殖細胞系配列にも関わらず、ヘルパーT細胞反応を誘発しない配列の生成によって、T細胞エピトープを含まない新しい配列を作ることによって達成することができる。
【0008】
本発明の別の好ましい実施例では、更なるアミノ酸配列を加える、又は、1又はそれ以上の制御性T細胞エピトープ(「Trエピトープ」)を規定する複合抗体分子内で作ることができる。本発明の目的のために、Trエピトープは、接触依存性メカニズムと同様に、IL−10及びTGF−β等の阻害性サイトカインの分泌によって免疫反応を調整する能力を有するCD4+ CD25+ T細胞エピトープを刺激するMHC結合ペプチドである。このように、本発明の範囲内では、制御性T細胞エピトープは、ある条件下で、免疫反応の制御に寄与する1又はそれ以上のインビトロ又はインビボ活性を誘発するように示されるペプチドを含むことができる。例えば、制御性T細胞エピトープは、CD4+ CD25+ T細胞を誘発する又は活性化する作用、IL−10及び/又はTGF−β等の阻害性サイトカインの放出を誘発する作用、又はその他の測定可能な免疫抑制関連インビトロ又はインビボ活性を有するペプチドを含む。これらの全ての場合において、これらの作用は、CD4+ CD25+ T細胞の作用に関連する。従って、このようなTrエピトープは、複合抗体の免疫原性を制限又は回避する更なる基準を追加するものである。Trエピトープは、これらのエピトープを含むヒトVH及びVLのセグメントを組み込むことによって、又は2又はそれ以上のヒト配列セグメントの結合を介してこのようなエピトープを生成することによって、又は、例えば、CD4+ CD25+ T細胞の誘発又は活性化のために、例えば、IL−10及び/又はTGF−β等の阻害性サイトカインの放出の測定によって、ヒト抗体又はタンパク質配列のセグメントに相当するペプチドからの新しいTrエピトープをスクリーニングすることによって、複合抗体VH又はVL内に導入することができる(例えば、Hall et al.,Blood,vol.100(2002)p4529−36)。代替的に、公知のTrエピトープは、複合抗体の結合又は機能又は発現を阻害しないVH及び/又はVL内の位置で複合抗体V領域内に組み込むことができ、又は、例えば、VHのN末端に合成VH又はVL配列の一端で組み込むことができる。代替的に、Trエピトープは、複合抗体の機能に干渉しない位置に(例えば、ヒンジ領域内で)、又は、発現の欠如等のいくつかの他の悪影響を引き起こさない位置に、複合抗体の一方又は双方の定常領域内に組み込むことができる。代替的に、抗体融合タンパク質内の合成VH及びVLの一方又は双方について、抗体は、Fab及びFv−タイプ形状(VH及びVLに結合した単鎖抗体(SCAs)を含む)、単一ドメイン抗体、又はホモ二量体型抗体を接合して、Trエピトープを、複合抗体の機能に干渉しない位置に、又は発現の欠如等のいくつかの他の悪影響を引き起こさない位置に組み込むことができる。例えば、SCAsでは、Trエピトープ用に特に好ましい位置は、VHとVLを結合するリンカ領域内である。最も良いのは、Trエピトープが、適切な配列によって側面に位置し、例えば、エンドサイトーシスプロテアーゼの作用に反応する配列を有するエピトープの側面に位置することによって、MHCクラスII分子上の制御性T細胞エピトープの放出及び表示を最適化する。一般的に、抗原の処理中のプロテアーゼの作用を標的にするP−20からP30(P1からP9のように規定されているコアノノメータを有する)を範囲とする位置で側面にある残渣は、必要ならば、ヒト抗体配列の追加のセグメントを用いて導入される。
【0009】
ここで議論されているように、本発明は、修飾抗体又は複合抗体の生成方法も提供している。従って、別の態様では、本発明は;
(1)その他の抗体可変領域の範囲からアミノ酸配列のセグメントを結合することによって抗体可変領域遺伝子を準備して、様々な可変領域遺伝子のライブラリを生成するステップと、
(2)抗体可変領域遺伝子のライブラリを発現ベクタ内へクローニングするステップと、
(3)抗体可変領域のライブラリをスクリーニングして、所望の特性を有するライブラリの員を回復するステップと、
を具える修飾抗体の生成方法を提供する。
【0010】
本発明の第1の好ましい方法「A」では、合成ヒト抗体のライブラリが生成され、特異的抗原に結合する等、所望の特性を有する抗体にスクリーニングされる。この方法は、次の6ステップ;
(1)合成VH及びVL遺伝子を設計するステップと、
(2)合成VH及びVL遺伝子をクローニングするステップと、
(3)合成VH及びVL遺伝子を発現するステップと、
(4)所望の特性を有する複合抗体をスクリーニングして選択するステップと、
(5)リード複合抗体を最適化するステップと、
(6)(選択的に)T細胞エピトープを回避するステップと、
を含む。
【0011】
ステップ(1)については、合成VH及びVL配列のライブラリは、Kabat antibody database(www.bioinf.org.uk/abs/simkab.html)、NCBI database(www.ncbi.nlm.nih.gov)で入手可能なもの等の公知のヒトV領域配列から、及び、UniProt(www.ebi.uniprot.org)及びPRF/SEQDB(www.prf.or.jp)等のタンパク質データベースから、VH及びVL配列のセグメントを選択することによって構成されている。加えて、これらは、1又はそれ以上の個人のドナーから増幅したVH及びVLmRNAの直接配列によるヒトVH及びVL配列の収集によって補充される。配列セグメントの様々な結合は、VH及びVL遺伝子の設計であると考えられる。用いられる1つの方法は、合成VH及びVL配列の長さを固定して、様々なヒトV領域の相当するKabatナンバリング位置からの固定長配列セグメントを用いて、これらを設計することである。
【0012】
例えば、このライブラリは、121及び107アミノ酸VH及びVL領域をそれぞれ具え、例えば、Kabatナンバリングを用いて、VHアミノ酸1−27について様々なセグメントの組み合わせを含む。KabatナンバリングCDR1:30−35、CDR2:50−66、及びCDR:95−106に相当するCDRを有するVHについては、次のKabat位についての配列セグメントが、1つの選択枝:1−27、28−31、32−36、37−42、43−50、51−56、57−60、61−63、64−69、70−82a、82b−96、97−98、99−101、102−117として用いられている。KabatナンバリングCDR1:24−34、CDR2:50−56、CDR3:89−97に相当するCDRを有するVLについて、次のKabat位についての配列セグメントが、1つの選択枝:1−22、23−27、28−30、31−33、34−35、36−47、48−52、53−55、56−59、60−87、88−92、93−94、95−107として用いられている。従って、この例では、合成VHは、14ヒトセグメントから成り、合成VLは、13のヒトセグメントから成る。実際には、コンピュータプログラムを用いて、これらのセグメントの組み合わせを生成する。好ましくは、このプログラムは、例えば、CDRのある種のカノニカル構造を回避する、或いは、VH及び/又はVL折り畳み、又はVH/VL相互作用を分離するあるセグメントの好ましくない組み合わせを回避するアルゴリズムを含む。選択的な追加として、このプログラムは、配列セグメントの組み合わせによって形成したT細胞エピトープの数を制限するアルゴリズムを含んでいてもよい(以下に述べるステップ(6)のコンピュータを利用した方法参照)。
【0013】
ステップ(2)については、合成ヒト配列のライブラリを設計して、好ましくは合成オリゴヌクレオチドを用いて、合成VH及びVL遺伝子が生成される。一般的に、V領域配列のより長いセグメントをコードする合成オリゴヌクレオチドは、V領域配列の2又はそれ以上の連続したセグメントをコードするオリゴヌクレオチドの混合物に結紮される。代替的に、合成V領域は、現存のヒトVH及びVL遺伝子をテンプレートとして用いて、PCRのオーバーラップ又はその他の増幅技術等の他の方法によって結合することができる。例えば、PCRを用いて、V領域の小さなセグメントを個別に増幅し、次いで、PCR反応をオーバーラップさせることによって結合する。
【0014】
その他の方法では、混合合成オリゴヌクレオチドを生成して、好ましくは特異的V領域セグメントをコードする配列を濃縮するドーピング方法を用いて、一連の配列セグメントを作ることができる。大きく変動するヒトV領域セグメント表示を有する合成ヒトVH及びVL遺伝子は、Molecular Cloning:A Laboratory Manual;3rd Ed.,vols.1−3(2001)Cold Spring Harbor Laboratory Pressに記載されているもの等の当業者に知られている技術を用いて、及びOrlandi et al.,Proc Natl Acad Sci USA.,86(1989)3833−3837に記載されているもの等の免疫グロブリンについての標準PCR法を用いて、多くの方法で結合させることができる。
【0015】
ステップ(3)については、合成ヒトVH及びVL遺伝子が生成されると、これらは、完全抗体分子又はFv’s、Fab’s、Fab2、SCA等の抗原結合フラグメント、単一ドメイン抗体(例えば、VHのみを具える)、及びこれらの各々の多重誘導体の生成用の様々な発現ベクタ内にクローンすることができる。代替的に、VH及びVL遺伝子は、その他の分子をコードする遺伝子に融合させて、融合プロテインを生成することができる。また、Fv又はFabの1つの鎖のC末端で、ポリヒスチジンタグ等の検出可能なマーカをコードする配列を含んでいてもよい。発現ベクタは、ほ乳類細胞、細菌細胞、バクテリオファージ、イースト、真菌、及びその他の微生物における発現用ベクタを含む。このようなベクタには、また、トランスジェニック動物からのインビボ発現用ベクタや、リボソーム調製を用いるインビトロ翻訳等のインビトロ系を用いた発現用ベクタも含まれる。
【0016】
ステップ(4)については、合成ヒト抗体のライブラリのスクリーニングは、通常、対象の1又はそれ以上の特異的抗原への結合用である。多くのスクリーニング方法が当業者に知られており、これらの方法の選択は、完全ヒト抗体の発現形状と、抗体分子、即ち、完全ヒト抗体、又はFab、Fv、SCA、単一ドメイン抗体等の組成に依存する。対象の抗原に結合する現存の抗体が入手可能である場合に、この抗体からのVH又はVLは、合成ヒトVL又はVHとそれぞれ組み合わせて、結合を試験することができる。
【0017】
スクリーニング法は、固相上のこのような員のライブラリ又はプールの個々の員を固定することから、個別に又はプール中の対象の抗原を固定することにまで及ぶ。抗体が固定される場合、対象の抗原が追加され、1又はそれ以上の追加試薬の添加によって、直接的、又は間接的に検出することができる。例えば、抗原が融合タンパク質であるか、又はアルカリホスファターゼ等の酵素と結合する場合に、色、蛍光又は化学発光シグナルを生成する幅広い基質から順次添加することによって検出することができる。抗体プールが、1つのロケーション(例えば、マイクロタイター皿)に固定され、シグナルが、抗原の添加に起因する場合、このプールを、このプール又はより小さいプールの個々の員の再スクリーニングに先立って脱複製する(dereplicated)ことができる。対象の抗原が固定される場合、合成抗原ライブラリは、個々の抗体の添加から、特異的ロケーションに固定された対象の抗原まで、抗体のプールの添加まで、全合成ライブラリの添加及び対象の抗原に結合した抗体の次の回復までに及ぶいくつかの方法でスクリーニングすることができる。最後のケースでは、一般的な戦略は、カラム内、又はビード上等の固相上に抗原を固定してライブラリを加え、次いで、例えば、低塩濃度緩衝液でこの固相を洗浄して(ライブラリに緩やかに結びついた員を分離するために)、次いで、例えば高塩濃度緩衝液を用いて抗原に結合する抗体を溶離することである。この目的のためのライブラリの員の発現用の一般的な形式は、ファージ表示、イースト表示、リボソーム表示、及びビード表示である各場合において、合成VH及びVL鎖をコードする核酸が、抗原に結合する合成V領域に付着し続ける。
【0018】
また、スクリーニング方法は、直接抗原結合試験に代わる機能性又は生物学的試験も含み、細胞増殖、細胞増殖阻害、細胞分化、又は細胞移動を含むインビトロ試験、又は、全有機体のレベルでの抗原への反応、例えば、マウスの血球数の変化や移植した腫瘍の増殖阻害を測定するステップを含む、代替的なインビボ試験等で機能性又は生物学的活性が測定される。
【0019】
ステップ(5)については、対象の抗原への結合等の所望の特性を有する1又はそれ以上の「リード」合成ヒト抗体の選択の後、例えば、この抗原の結合親和性を増加させる、又はこの抗体を追加成分に融合させることによって、リード抗体の特性が、選択的に改善されることがある。親和性の増加は、合成可変領域配列の突然変異によって達成され、所望の方法の結合を増加させる又は変更する選択した合成V領域配列の突然変異を選択する。本発明は、リード抗体からの1又はそれ以上の個々のV領域配列セグメントを、1又はそれ以上のヒト抗体配列からの相当する配列セグメントで置換することによる、可変領域配列の突然変異のための新規な方法を含む。具体的には、CDR領域と、又はCDR領域内でオーバーラップするセグメントを、様々な長さのセグメントを含むその他のヒト抗体からの1又はそれ以上の代替セグメントによって置換することができる。本発明の範囲内は、特異的セグメントは、関連する特性を有するヒト抗体から、例えば、同じ抗原に結合する抗体から、又は、関連する特性を有する非ヒト抗体から、又は、関連する配列を有する非ヒト抗体の機能にとって重要である、ある種のキーアミノ酸を保持する配列セグメントを有するヒト抗体から、といった選択したリード抗体まで含む。このような突然変異の影響を受ける1又はそれ以上の合成ヒト抗体をスクリーニングして、特性を改善する。
【0020】
選択的ステップ(6)については、リード合成ヒト抗体の選択後、必要であれば、T細胞エピトープを回避する代替セグメントでT細胞エピトープに寄与、又はT細胞エピトープをコードするV領域セグメントを交換することによって、T細胞エピトープが制限又は回避される。このようなT細胞エピトープは、ある範囲の方法によって検出することができる。例えば、合成V領域配列の1又はそれ以上の座に相当するペプチドを合成して、T細胞アッセイ試験をして、T細胞エピトープの存在を決定することができる。一般的に、このようなペプチドは、長さが15アミノ酸であり、接触領域がより長い配列を試験することが望ましく、12アミノ酸オーバーラップを有する15マー等の配列からオーバーラップペプチドを用いる。T細胞エピトープの検出については、様々なT細胞アッセイの範囲を、サイトカイン放出、増殖(例えば、3H−チミジンの摂取によって)、Ca2+フラックス、表面マーカ発現、遺伝子転写等のCD4+T細胞の活性又は増殖の測定に用いることができる。
【0021】
代替的に、合成V領域配列に相当するオーバーラップペプチドは、インビトロ法又はインシリコ(in silico)法を用いて分析し、ヒトMHCクラスII分子に結合するいずれの場合も、潜在的なT細胞エピトープ、即ち、T細胞反応を誘発するMHC結合ペプチドを決定する。インシリコ(in silico)法は、ペプチド−MHCクラスII結合相互作用のモデリングを含む方法と、MHCクラスIIに結合する一般的なモチーフの同定を含む方法と、ペプチド又は公知のインビトロMHC結合特性を有するペプチド内での特異的アミノ酸のデータベースを用いる方法とを含む。合成V領域配列からのより長いペプチドの生成、又は合成V領域配列を含む全抗体を生成して、例えば、MHC−ペプチドテトラマによって、又は公知のヒトT細胞エピトープのデータベースに提案されている又は構成された配列を調べることによって、T細胞アッセイ中又はMHC結合アッセイ中の合成V領域配列を試験するステップ等のその他の方法を用いることができる。合成ヒトV領域のT細胞エピトープの回避は、MHCクラスII結合モチーフの回避、又はMHCクラスIIへのペプチドの結合を固定する特定のアミノ酸の回避によっても補助することができる。1又はそれ以上のリード合成ヒト抗体からのT細胞エピトープの回避のための好ましい方法では、インシリコ(in silico)法を最初に適用して、潜在的なT細胞エピトープについての合成ヒト抗体V領域を分析し、これらが同定される場合、ヒトVH又はVL配列の新しいセグメントを導入して、これらのエピトープを回避し、新しいT細胞エピトープの導入を回避する。
【0022】
新しいヒトV領域セグメントのこのような導入と、所望の特性について、このように修飾したリード合成ヒト抗体の再スクリーニングの後に、1又はそれ以上の最終リード合成ヒトV領域を、一般的に、15から45アミノ酸の長さのオーバーラップペプチド、例えば、合成ヒト領域配列(全V領域又はこれらの一部分)からの12アミノ酸オーバーラップしている15マーペプチドを試験するステップによって、又は、ヒトT細胞アッセイで、直接的に全合成ヒト抗体を試験するステップによって、ヒトT細胞アッセイで更に試験することができる。全抗体に対してT細胞活性を直接試験できるようにする全合成ヒト抗体を試験するT細胞アッセイを用いた最終分析が好ましい。
【0023】
本発明の第2の好ましい方法「B」では、合成ヒト抗体のライブラリを、所望の特性を有する1又はそれ以上の参照抗体からの所望のアミノ酸を含むように生成する。この方法は、次の7ステップ;
(1)1又はそれ以上の参照抗体の配列を分析するステップと、
(2)合成VH及びVL遺伝子を設計するステップと、
(3)(選択的に)T細胞エピトープを回避するステップと、
(4)合成VH及びVL遺伝子をクローニングするステップと、
(5)合成VH及びVL遺伝子を発現するステップと、
(6)所望の特性を有する複合抗体をスクリーニングして選択するステップと、
(7)T細胞エピトープの最適な回避を含むリード複合抗体を最適化するステップと、
を含む。
【0024】
ステップ(1)では、典型的な参照抗体は、ヒト型の抗体中の所望の特性及び/又は結合特異性を有する齧歯類、特にマウスである。1又はそれ以上の参照抗体V領域配列が入手できる場合、これらの配列を分析してCDRの配列を決定し、結合特性等の抗体の所望の特性に重要であるアミノ酸を同定する。参照抗体については、例えば、参照V領域配列の同種のその他の配列とのアラインメントによってこのような分析を実施する。また、参照抗体が非ヒト、ヒトV領域配列である場合も、このような分析を行う。例えば、プログラムCLUSTAL(Thompson et al.,Nucleic Acids Res.22(1994)p4673−80)を用いて、このようなアラインメントを実施する。このようなアラインメントは、参照抗体のV領域及び相同性v領域ファミリィ中の異常又は微量アミノ酸を同定することができる。加えて、CDRのカノニカル構造等の保存V領域構造を、例えば、Protein Data Bank(Berman et al.:The Protein Data Bank, Nucleic Acids Research,28(2000)235−242)を用いて同定することができる。加えて、構造が知られていない場合、MODELLER(Sali and Blundell,J.Mol.Biol.234(1993)p779−815)等のモデリングソフトウエアを用いて、参照抗体可変領域をモデリングし、いくつかの場合では、抗体−抗原相互作用のモデルを生成することができる。参照抗体V領域のこのような分析は、合成ヒト抗体についてのヒトV領域配列のセグメントの選択を導くのに用いる。
【0025】
ステップ(2)については、合成ヒト抗体の所望の特性に重要であるアミノ酸を決定して、これらのアミノ酸のいくつか又は全てを含むように、ヒトV領域配列のセグメントを選択する。合成ヒト抗体の特性についてのこのようなセグメントの効果が不明確である特定の座に一般的な1又はそれ以上の代替ヒトV領域セグメントを有する選択されたセグメントを含む合成ヒトV領域配列のライブラリがこれによって設計される。このような合成ヒト抗体配列は、その他のヒト抗体配列と保存構造とのアラインメントによって参照抗体で更に分析することができ、加えて、合成ヒト抗体V領域のこの構造の更なるモデリングを行って、合成ヒト抗体に用いられるヒトV領域セグメントの組み合わせを、所望のように精製し、合成V領域内でのタンパク質構造、分子間及び分子内相互作用における欠陥、及び重要なアミノ酸の不正確な構造配向を回避する。
【0026】
選択的ステップ(3)については、セグメント選択の付加的基準として、最終合成ヒトV領域のT細胞エピトープを制限又は回避するこれらのセグメント又はセグメントの組み合わせが選択される。T細胞エピトープをインシリコ(in silico)又はインビトロ法を用いて、好ましくは、合成ヒトV領域配列を設計する段階でインシリコ(in silico)法を用いて、上述した方法A、ステップ(6)に記載した方法によって分析する。
【0027】
ステップ(4)については、合成ヒト配列のライブラリを設計して、次いで、合成VH及びVL遺伝子を、好ましくは合成オリゴヌクレオチドを用いて生成する。典型的には、V領域配列のより長いセグメントをコードする合成オリゴヌクレオチドが、配列の代替セグメントをコードするオリゴヌクレオチドの混合物に結紮して、合成ヒトV領域のライブラリの様々な員を生成する。代替として、合成ヒトV領域のライブラリの各員は、特異的ヒトV領域の配列をコードするオリゴヌクレオチドを部分的に用いて個別に生成する。代替として、合成V領域は、既存のヒトVH及びVL遺伝子をテンプレートとして用いて、又は、テンプレートとして1又はそれ以上の参照抗体V領域遺伝子を用いて、オーバーラップPCR、又は他の増幅技術等のその他の方法によってアセンブルされる。
【0028】
方法Bについてのステップ(5)及び(6)は、方法A、ステップ(4)及び(5)に記載されているのと同様である。
【0029】
選択的なステップ(7)は、方法A、ステップ(6)と同様に用いられ、リード合成ヒト抗体でT細胞エピトープの更なる回避が要求される。全抗体からのT細胞活性について直接試験することを可能にする全合成ヒト抗体を試験するT細胞アッセイを用いた最終分析が好ましい。
【0030】
方法A及びBに加えて、合成ヒト抗体を作成して試験し、このような抗体の特性を最適化するその他の方法があることは、当業者に理解されよう。本発明の合成ヒト抗体は新規であり、V領域の全ヒト起源の結果として、非ヒト配列を含むその他の抗体よりもヒトの免疫原性が低くなくてはならない。また、合成ヒト抗体の追加の選択的特徴、即ち、T細胞エピトープの回避及び/又はTrエピトープの付加は、より低い免疫原性に寄与する。より低い免疫原性の目的は、例えば、源ヒト抗体中の配列位置と異なる複合抗体の配列位置にセグメントを含む合成ヒト抗体、ヒトV領域配列のセグメントの部分的組み込みのみを有する複合抗体、非ヒト配列のセグメントを有する複合抗体、又は、例えば、抗体に対する結合親和性を増加する、又はT細胞エピトープを避けるように突然変異したヒト配列を有する複合抗体等の全ヒト配列セグメントの無いV領域を含む、より好ましくない複合抗体を用いて達成することができることは、当業者に理解されるであろう。
【0031】
合成ヒト抗体内でのV領域配列セグメント及びこれらの組み合わせを選択して、上記のようなT細胞エピトープの選択的回避を含む基準範囲を満たすことが理解されよう。例えば、ヒトV領域配列のセグメント及びこれらの組み合わせは、B細胞エピトープ及びMHCクラスI−制限エピトープ等のその他のエピトープの回避、複合抗体の発現に有害であるアミノ酸配列の回避、N−グリコシル化等の複合抗体の不適切な修飾を導く配列の回避、ヘルパーT細胞エピトープ及び/又はB細胞エピトープ(例えば、ワクチンの使用)の包含等のある種の機能の包含、1又はそれ以上の表面リシン残渣等のその他の部分への連続した結合、及びその他の基準範囲用に選択することができる。
【0032】
また、ヒトに加えて、その他の種由来のV領域セグメント全体又は一部を有する複合抗体を生成することができ、これも本発明の範囲内であると考えるべきであることは、当業者に理解されよう。例えば、マウスの研究用に、マウス起源のV領域配列セグメントの全体又は一部を具える合成マウス抗体を生成することができる。
【0033】
また、本発明は、抗体以外のタンパク質に適用し、治療用途には、このようなタンパク質(ここでは、「合成タンパク質」という)は、最終タンパク質分子内で、ヒトタンパク質からのアミノ酸配列の2又はそれ以上のセグメントを結合する。
【0034】
従って、更なる態様では、本発明は、アミノ酸配列の1又はそれ以上のセグメントの挿入によって改善した免疫原性を有する修飾タンパク質を提供する。
【0035】
タンパク質に関して、用語「セグメント」は、タンパク質分子内で見出される隣接するアミノ酸配列をいい、このようなセグメントは、2から250アミノ酸長のサイズの範囲である。治療用には、本発明の合成タンパク質は、通常、合成タンパク質内で様々なヒトタンパク質からのアミノ酸配列の2又はそれ以上を結合する。特に、本発明は、全体が2又はそれ以上のヒトタンパク質からの配列のセグメントからなる挿入を有する合成タンパク質に関する。ヒトタンパク質が合成タンパク質に対して相同で、又は合成タンパク質の領域に対して相同な領域で存在する場合、源ヒトタンパク質の配列位置に相当する合成タンパク質配列のセグメントを用いることができる。例えば、合成タンパク質配列のアミノ酸1から10は、源ヒトタンパク質のアミノ酸1から10に由来する。代替として、ヒトタンパク質配列中の配列位置にあるヒトタンパク質配列のセグメントは、源ヒトタンパク質の配列位置に関わりない合成タンパク質中のあらゆる配列位置にある合成タンパク質に配置されていてもよい。源ヒトタンパク質は、例えば、ヒトタンパク質配列のデータベースで提供されるようなあらゆる現存のヒトタンパク質アミノ酸配列であり、ヒトタンパク質の天然の突然変異形又は転位形、及び生殖細胞系と異なるその他の変異、人工的に構成されたヒト由来タンパク質からの配列、及び相当するタンパク質が発現するか、或いは発現しないヒト遺伝子又はRNA由来の配列が含まれる。
【0036】
本発明のこの態様の好ましい実施例では、治療用合成タンパク質は、最終合成タンパク質中のヒトT細胞エピトープを制限又は回避する組み合わせのヒトタンパク質配列セグメントを結合又は挿入することによって構成される。合成タンパク質に適用される本発明の好ましい態様は、ヒトタンパク質配列セグメントの挿入によって非ヒトタンパク質等の現存の参照タンパク質を修飾して、最終合成タンパク質のT細胞エピトープを制限又は回避することである。
【0037】
合成タンパク質を生成する本発明の好ましい方法では、合成ヒトタンパク質のライブラリを生成して、T細胞エピトープ不在等の所望の特性を有する1又はそれ以上の参照タンパク質からの所望のアミノ酸を含めるようにしている。この方法は、次の7ステップを含む;
(1)T細胞エピトープの選択的分析を含む1又はそれ以上の参照タンパク質の配列分析をするステップと、
(2)合成タンパク質遺伝子を設計するステップと、
(3)T細胞エピトープを(選択的に)回避するステップと、
(4)合成タンパク質遺伝子をクローニングするステップと、
(5)合成タンパク質遺伝子を発現させるステップと、
(6)スクリーニングを行い、所望の特性を有する合成タンパク質を選択するステップと、
(7)T細胞エピトープの選択的回避を含むリード合成タンパク質を最適化するステップ。
【0038】
ステップ(1)では、典型的な参照タンパク質は、合成タンパク質で所望される特性を有する非ヒトである。治療用途には、典型的に、合成タンパク質の免疫原性の低減又は除去が目的となる。1又はそれ以上の参照タンパク質配列が入手可能である場合、これらはタンパク質の所望の特性に重要であるアミノ酸を同定するために分析される。加えて、参照タンパク質のあらゆる公知の構造を分析することができ、又は、代替的に、モデル化ソフトウエアを用いて、構造をモデル化することができる。種間又は種内のどちらかの参照タンパク質の相同体が入手できる場合、時に、これらを用いて、配列差の関係と相同体間の特性の差の関係を決定することができる。タンパク質が別の分子と相互作用する場合、この相互作用のモデルを、生成し、相互作用に重要なアミノ酸を決定することができる。ステップ1への選択的付加として、特に、上記の合成ヒト抗体について詳細に述べているようなインビトロヒトT細胞アッセイを用いて、参照タンパク質中のT細胞エピトープの配列ロケーションが決定される。代替として、T細胞エピトープを分析するためのインシリコ(in silico)法を用いることができる。参照タンパク質のこのような分析を用いて、合成タンパク質用に選択されたヒトタンパク質配列のセグメントに導く。参照タンパク質、特に非ヒトと比較して免疫原性の低減又は除去が目的である合成タンパク質については、一般的に、T細胞エピトープのロケーションに相当する1又はそれ以上のヒト配列セグメントが、T細胞エピトープ無しの他のロケーションからの参照タンパク質からの配列のセグメントと組み合わせて合成タンパク質中に用いられる。
【0039】
ステップ(2)については、合成タンパク質の所望の特性に重要であるアミノ酸を決定して、次いで、これらのアミノ酸のいくつか又は全てを含むように、タンパク質配列のセグメントが選択される。これによって、典型的に、特定の座で、1又はそれ以上の代替ヒトタンパク質セグメントを有する選択されたセグメントを含む合成ヒトタンパク質配列のライブラリが設計される。合成タンパク質の特性のこのようなセグメントの効果は、不明である。このような合成タンパク質配列は、基準タンパク質を伴っていれば、任意の相同体と整列させることによって、又は合成タンパク質の構造のモデル化によって、又は、タンパク質構造の欠陥や重要なアミノ酸の不正確な構造配向を回避するために合成ヒトタンパク質に用いられるヒトタンパク質セグメントの組み合わせを、必要に応じて精製するためにその他の分析を行うことによって、更に分析することができる。
【0040】
選択的ステップ(3)について、セグメントの選択用の追加基準又は基準のみとして、最終合成タンパク質中のT細胞エピトープを制限又は回避するこれらのセグメント、又はセグメントの組み合わせが選択される。T細胞エピトープを、インシリコ(in silico)又はインビトロ法を用いて上記の合成ヒト抗体用に記載された方法によって分析する。
【0041】
ステップ(4)については、合成タンパク質のライブラリを設計して、次いで、合成タンパク質遺伝子を、好ましくは、合成オリゴヌクレオチドを用いて生成する。典型的には、タンパク質配列のより長いセグメントをコードする合成オリゴヌクレオチドが、配列の代替セグメントをコードして合成タンパク質のライブラリの様々な員を生成するオリゴヌクレオチドの混合物に結紮する。代替的に、合成タンパク質のライブラリの各員が、特異的合成タンパク質の配列をコードするオリゴヌクレオチドを用いて部分的に生成される。代替として、合成タンパク質は、オーバーラップしているPCR、又は、現存のヒトタンパク質遺伝子をテンプレートとして用いた、又は、1又はそれ以上の参照タンパク質遺伝子をテンプレートとして用いたその他の増幅技術等の他の方法によってアセンブルすることができる。
【0042】
ステップ(5)では、合成タンパク質のライブラリのスクリーニングは、通常、1又はそれ以上の合成タンパク質の所望の特性用である。多くのスクリーニング方法が当業者に知られており、これらの方法の選択は、合成タンパク質及びタンパク質機能の発現の形に依存する。スクリーニング方法は、固相上のこのような員のライブラリ又はプールの個別の員を固定するステップから、溶液相中のライブラリの員をスクリーニングして、もう1つの分子を同定し、これで合成タンパク質を設計して、個別に、又はプールで結合することによって相互作用させる範囲にまで及ぶ。また、スクリーニング方法は、機能的又は生物学的試験を含み、ここでは、細胞増殖、細胞増殖阻害、細胞分化、又は細胞遊走を含むインビトロ試験、又は全有機体のレベルでの合成タンパク質に対する反応、例えば、マウスの血球数又は移植した腫瘍の増殖阻害の変化を測定するステップを含む代替インビボ試験等の、機能的又は生物学的活性を測定する。
【0043】
ステップ(6)では、所望の特性を有する1又はそれ以上の「リード」合成タンパク質の選択に続いて、選択的に、例えば、酵素の特異的活性を増やすことによって、又はレセプタに対するタンパク質リガンドの結合を増やすことによって、このリードタンパク質の特性を強化することができる。特性の強化は、合成タンパク質配列の突然変異生成によって達成されて、所望の方法で、合成タンパク質の特性を変化させる突然変異が選択される。本発明は、このタンパク質からの1又はそれ以上の個々のタンパク質配列セグメントを1又はそれ以上のヒトタンパク質配列からの配列セグメントで置換することによって、タンパク質配列の突然変異を生成する新規な方法を含む。次いで、このような突然変異生成を行う1又はそれ以上の合成タンパク質をスクリーニングして、特性を強化することができる。
【0044】
選択的ステップ(7)では、リード合成タンパク質の選択に続いて、所望の場合に、T細胞エピトープに寄与する、又はこれをコードするタンパク質配列セグメントをT細胞エピトープを回避する代替セグメントと交換することによって、T細胞エピトープを制限又は回避する。このようなT細胞エピトープは、ある範囲の方法によって検出される。例えば、合成タンパク質中の1又はそれ以上の座に相当するペプチドを合成し、T細胞アッセイで試験してT細胞エピトープの存在を決定することができる。典型的には、このようなペプチドは、長さ15アミノ酸であり、12アミノ酸オーバーラップを有する15マー等の配列からのオーバーラップペプチドを用いてより長い隣接した配列を試験することが望ましい。代替として、潜在的T細胞エピトープ、即ち、T細胞反応を誘発するMHC結合ペプチドを決定する各場合に、合成タンパク質配列に相当するオーバーラップペプチドを、インビトロ法かインシリコ(in silico)法のどちらかを用いて、ヒトMHCクラスII分子への結合について分析する。インシリコ(in silico)法は、ペプチド−MHCクラスII結合相互作用をモデル化するステップを含む方法と、MHCクラスIIへの結合に一般的なモチーフを同定するステップを含む方法と、公知のインビトロMHC結合特性を有するペプチド内のペプチド又は特異的アミノ酸のデータベースを用いる方法を含む。合成タンパク質配列又は全合成タンパク質からより長いペプチドを生成する方法、及び、抗原提示細胞上のT細胞アッセイ、又はMHC結合アッセイでこれらを試験する方法等の他の方法を用いることができる。また、合成タンパク質のT細胞エピトープの回避は、MHCクラスII結合モチーフの回避によって、又はMHCクラスIIへのペプチドの結合をアンカする特異的アミノ酸の回避によって援助される。1又はそれ以上のリード合成タンパク質からT細胞エピトープの回避のための好ましい方法では、先ずインシリコ(in silico)法を適用して、潜在的T細胞エピトープについての合成タンパク質を分析し、これらが決定されると、ヒトタンパク質配列の新しいセグメントを導入して、これらのエピトープを回避して、新しいT細胞エピトープの導入を回避する。所望であれば、T細胞エピトープを回避する新しいヒトセグメントのこのような導入と、所望の特性についての修飾リード合成タンパク質の再スクリーニングに続いて、1又はそれ以上の最終リード合成タンパク質を、典型的に15から45アミノ酸長のオーバーラップペプチド、例えば、合成タンパク質配列(全タンパク質又はこれらの一部分)からの12アミノ酸オーバーラップした15マーペプチドを試験することによって、或いは、ヒトT細胞アッセイで、直接的に、全合成タンパク質を試験することによって、ヒトT細胞アッセイで選択的に試験することができる。全タンパク質からのT細胞活性について直接試験することができる全合成タンパク質を試験するT細胞アッセイを用いた最終分析が好ましい。
【0045】
合成タンパク質を作成して試験し、このようなタンパク質の特性を最適化する他の方法があることは、当業者には理解されよう。本発明の合成タンパク質は新規で、治療目的で用いられる場合は、ヒト起源のいくつかの、又は全てのタンパク質配列セグメントは、その他の比較可能な、又は非ヒト配列を含む非ヒト参照タンパク質より、ヒトの免疫原性が少ない峰性タンパク質を提供するべきである。また、合成タンパク質の付加的な選択的特徴、即ち、T細胞エピトープの回避及び/又はTrエピトープの付加も、より低い免疫原性に寄与する。より低い免疫原性の目的は、全ヒト配列の無い合成タンパク質を用いて達成され、また、突然変異して、参照タンパク質に相同する非ヒトタンパク質のT細胞エピトープ又はセグメントを除去するヒト配列セグメントを有する合成タンパク質を含むことが、当業者には理解される。T細胞エピトープの最適な回避を含む基準の範囲を満足するように合成タンパク質内のタンパク質セグメント及びその組み合わせを選択することが理解されよう。例えば、ヒトタンパク質配列のセグメント及びこれらの組み合わせを、B細胞エピトープ及びMHCクラスI−制限エピトープ等のその他のエピトープの回避と、合成タンパク質の発現に有害であるアミノ酸配列の回避と、N−グリコシル化等の合成タンパク質の不適切な修飾を導く配列の回避と、ヘルパーT細胞エピトープ、及び/又はB細胞エピトープ(例えば、ワクチン適用)の包含等のある種の機能の包含と、その他の部分への連続した結合と、その他の基準の範囲用に選択することができる。
【0046】
また、ヒトに加えて、他の種に由来する配列セグメントの全体又は一部分を有する合成タンパク質を生成することができ、これは、本発明の範囲内であると考えるべきである。例えば、マウスの研究では、マウスタンパク質配列セグメントを含む合成タンパク質を生成することができる。また、合成タンパク質が、同種内の相同タンパク質からのその他のタンパク質配列セグメントと結合した1つの種からのタンパク質配列セグメントを含むことができることが理解される。例えば、本発明は、植物型I RIP(リボソーム阻害タンパク質)の構造を含む。ここでRIPは、入手可能な多数の植物型I RIP配列からの配列セグメントを用いてアセンブルされる。このような合成RIPは、RIP活性を保持するであろう配列セグメントの組み合わせを導入することによってアセンブルされ、ヒトに用いる場合には、最終合成配列中のヒトT細胞エピトープの回避を含むであろう。
【0047】
抗体の場合、本発明は、最終タンパク質の1又はそれ以上のその他の特性に更なる改良を行う合成タンパク質のランダムな、セミランダムな、又は直接的な突然変異生成によって、合成タンパク質配列への更なる修飾の選択枝を含む。本発明は、ヒトに用いられるとき、又は薬学的な用途のタンパク質、又は、アレルギィ反応が本発明の組成物の使用によって制限又は除去される食料、合成洗剤、化粧品、及び他の消費者商品の用途のタンパク質等のヒトによって用いられるときに、低い免疫原性を有するタンパク質生成に特に好適である。本発明は、特に非アレルギィ関連エピトープ(例えば、TH1 T細胞−誘発エピトープ用のTH2)によって、及び/又はアレルギィ患者の免疫反応を抑制するTrエピトープの付加によって除去又は置換されたアレルギィ関連T細胞エピトープを有するタンパク質を生成することによって、ヒト内での低いアレルギィ性を有するタンパク質の生成に特に好適であることが理解されよう。本発明は、特に、非炎症関連エピトープ(例えば、TH2 T細胞誘発エピトープ用のTH1)及び/又は炎症反応を抑制するためのTrエピトープの添加によって除去又は置換された炎症関連T細胞エピトープを有するタンパク質を生成することによって、ヒトの炎症特性を低減するタンパク質を生成するのに特に好適であることが理解されよう。
【0048】
ここで議論されているように、本発明の修飾/合成タンパク質及び抗体は、病気の治療に有益であり、低い免疫原性を示す。従って、更なる態様で、本発明は、選択的に、1又はそれ以上の薬学的に許容できる賦形剤、担体、又は希釈剤と共に、請求項1から18のいずれか1項に記載の修飾抗体、抗体結合性フラグメント、又はタンパク質を具える薬学的調製を提供する。
【0049】
本発明の組成物は、1投与当たり所定量の各活性成分を含む単位投与形で存在する。このような単位は、5−100mg/1日の化合物、好ましくは、5−15mg/1日、10−30mg/1日、25−50mg/1日、40−80mg/1日、又は60−100mg/1日のいずれかを提供するように構成されている。式Iの化合物については、100−1000mg/1日、好ましくは、100−400mg/1日、300−600mg/1日、500−1000mg/1日のいずれかの範囲の投与を提供する。このような投与は、単回投与又は複数回の分散投与で提供することができる。最終的な投与は、もちろん、治療の状態、投与経路、及び患者の年齢、体重、及び状態に依存しており、医師の裁量である。
【0050】
本発明の組成物は、あらゆる適切な経路、例えば、経口(口腔又は舌下を含む)、直腸、鼻腔、局所(口腔、舌下又は経皮を含む)、経腟、又は非経口(皮下、筋内、静脈、又は皮内)を含む経路による投与用に構成されている。このような調製は、医薬の分野で知られているあらゆる方法、例えば、担体や賦形剤と活性成分を関連させることによって調製することができる。
【0051】
経口投与に適した医薬調製物は、カプセル又はタブレット;パウダ又は顆粒;水性又は非水性の液体の溶液又は懸濁液;可食発泡体又はホイップ;又は水中油型液体エマルジョン又は油中水型液体エマルジョン等の分散単位として存在している。
【0052】
経皮投与に適した医薬調製物は、長期間、レシピエントの表皮と密着し続けるように意図した分散パッチとして存在する。例えば、有効成分は、Pharmaceutical Research,3(6),318(1986)に一般的に記載されているような、イオントフォレシスによって、パッチから送り込まれる。
【0053】
局所投与に適した医薬調製物は、軟膏、クリーム、懸濁液、ローション、パウダ、溶液、ペースト、ジェル、スプレィ、エアロゾル、又はオイルとして調製される。
【0054】
眼、又はその他の外部組織である、例えば、口及び皮膚への塗布には、この調製物は、局所軟膏又はクリームとして塗布するのが好ましい。軟膏に調製する場合、有効成分を、パラフィン、又は水混和性軟膏ベースのどちらかと共に用いることができる。代替として、有効成分を、水中油型クリームベース又は油中水型ベースのクリームと共に調製するようにしてもよい。
【0055】
眼への局所投与に適した医薬調製物は、点眼薬を含み、この有効成分を好適な担体、特に水性溶媒に溶解又は懸濁させる。
【0056】
口内局所投与に適した医薬調製物は、薬用キャンディ、トローチ剤、及び洗口液を含む。
【0057】
直腸投与に適した医薬調製物は、座薬又は寛腸剤として存在する。
【0058】
経鼻投与に適合した医薬調製物であって、担体が固体である医薬調製物は、例えば、スナッフで摂取する方法、即ち、鼻腔に近づけたパウダの容器から鼻道を通る素早い吸入によって投与される20から500ミクロンの範囲の粒径を有する粗粉末を含む。スプレィ式点鼻薬として、又は点鼻薬としての投与に好適な調製物であって、担体が液体である調製物は、水性又は油性溶液の有効成分を含む。
【0059】
吸入による投与に適した医薬調製物は、様々なタイプの定量加圧エアロゾル、噴霧器、又は吸入器の手段によって生成する細粒粉末又は噴霧を含む。
【0060】
経腟投与に適した医薬調製物は、ペッサリー、タンポン、クリーム、ジェル、ペースト、発泡体又はスプレィ調製物として存在する。
【0061】
非経口投与に適した医薬調製物は、抗酸化剤、緩衝液、静菌薬及び溶質を含む水性及び非水性滅菌注射液;及び、懸濁剤及び増粘剤を含む対象とするレシピエントの血液を用いる等張の調製を行う水性及び非水性滅菌懸濁液を含む。調製物は、単位投与及び複数投与容器である、例えば、密封アンプル及びバイアルで存在し、また、使用直前に例えば注入用の水といった滅菌液体担体の添加を必要とするのみのフリーズ−ドライ(凍結乾燥)状態で貯蔵されているものでもよい。即時調合の注射液及び懸濁液は、滅菌パウダ、顆粒、及びタブレットから調製される。
【0062】
好ましい単位投与調製物は、上記のような有効成分の日用量、又はサブドーズ(sub−dose)、又はこれらの適切な画分を含むものである。
【0063】
特に上記した成分に加えて、調製物は、問題の調製物のタイプを考慮したこの分野では従来のものである他の薬剤を含んでいてもよい。例えば、経口投与に好適なものは、香料添加剤を含んでいてもよい。
【0064】
次の例は、本発明の範囲を限定するものと考えるべきではない。図及び表は、以下に述べる例に、次のように関連している;
【0065】
表1から表3は、186×9残渣長VH CDR3(表1)、77×8残渣長VL CDR3(表2)、及び153×10残渣長VL CDR3(表3)を具える合成ヒト抗体scFvライブラリに用いられるCDRである。
【0066】
表4は、一次合成ヒト抗TNFαVH及びVL変異株に用いるヒト配列セグメントである。
【0067】
表5は、合成ヒト抗TNFα変異株の活性である。
【0068】
表6は、ブーガニン変異株のブーガニン及び置換ヒトセグメントの免疫原性ペプチド配列である。
【0069】
例1−合成ヒト抗−HER2抗体の構造
【0070】
ヒト可変領域配列セグメントライブラリの作成用に、ある範囲のヒト免疫グロブリンの範囲からのアミノ酸配列を重鎖(VH)及び軽鎖(VL)可変領域配列を含むインシリコ(in silico)ヒト可変領域配列ライブラリを具える単一データベース内に集めた。配列の源としては、NCBI Igblastデータベース(www.ncbi.nih.gov)、Kabatデータベース(Kabat et al.,Sequences of Proteins of Immunological Interest,NIH publication 91−3242,5th ed.(1991)(後にアップデートされた)),Vbase(www.mrc−cpe.cam.ac.uk/imt.doc),Genbank(Benson et al.,Nucl.Acids Res.25(1997)pl−6、又はwww.bioinf.org.uk/absを介した)データベースが挙げられる。用いられた参照抗体可変領域配列は、ハーセプチン(登録商標)として知られているヒト化抗−HER2抗体であった(Carter et al.,Proc.Nat.Acad.Sci.USA,vol 89(1992)p4285,US5821337)。インシリコ(in silico)ヒト可変領域配列ライブラリからのセグメントを、ハーセプチン(登録商標)可変領域配列の相当するアミノ酸に対する同定用に選択し、結合させて、図1及び図2にそれぞれ示されているような合成ヒトVH及びVL配列を生成した。
【0071】
組み換えDNA技術を、この分野で公知の方法と、必要に応じて、これらの方法で用いられる酵素を用いるための供給者の指示を用いて実施した。一般的な方法の源としては、Molecular Cloning,A Laboratory Manual,3rd edition,vols 1−3,eds.Sambrook and Russel(2001)Cold Spring Harbor Laboratory Press、及びCurrent Protocols in Molecular Biology,ed.Ausubel,John Wiley and Sonsが挙げられる。また、詳細な実験室方法は、以下の例7に記載されている。ハーセプチン(登録商標)に相当する合成ヒトVH及びVL配列を、各鎖について、完全合成ヒトVH及びVL配列をコードする長さ30−60アミノ酸の8つの合成ヌクレオチドを用いて作成した。また、同時に、対照試薬として、マウスモノクローナル抗体4D5(Hudziak et al.,Mol.Cell.Biol.,(1989年3月)p1165−1172))のキメラ形を、鎖毎に8つの合成オリゴヌクレオチドを用いて作成した。分離VH及びVLオリゴヌクレオチドを、先ずリン酸化して、等モル比で混合して、サーマルサイクラ内で5分間、94℃に加熱し、次いで65℃に冷却して、65℃で2分間インキュベートした。次いで、45℃で2分間、35℃で2分間、25℃で2分間、4℃で30分間インキュベートを継続した。更に、オリゴヌクレオチドを、T4 DNAリガーゼ(英国ペイズリィ所在のLife Technologies社)を用いて、14℃で18時間、結紮した。
【0072】
Kozak配列、リーダーシグナルペプチド(leader signal peptide)配列及びリーダーイントロン(leader intron)を含む5’フランキング配列、及びスプライス座及びイントロン配列(intron sequence)を含む3’フランキング配列をコードする追加のオリゴヌクレオチドを、VH及びVLオリゴヌクレオチド混合物に上記のように加えて、アニールした。合成ヒトVH及びVKと、生成した4D5発現カセットを、プラスミドベクタpUC19へBamHIフラグメントに対するHindIIIとしてクローニングし、完全なDNA配列を確認した。これらは、ヒトIgG1(VH)又はKappa(VK)定常領域をそれぞれと、ほ乳類細胞の選択用マーカを含む発現ベクタpSVgpt及びpSVhygに転写された。DNA配列を確認して、合成ヒトVH及びVKと、発現ベクタ中の4D5VH及びVKを補正した。
【0073】
抗体発現用主宿細胞株は、NS0である非免疫グロブリン生成マウス骨髄腫であり、英国ポートン所在のEuropean Collection of Animal Cell Cultures(ECACC No85110503)から得た。重鎖及び軽鎖発現ベクタをエレクトロポレーションによって、NS0細胞内に同時遺伝子導入した。gpt遺伝子を発現するコロニィは、10%ウシ胎仔血清、0.8μg/mlミコフェノール酸、及び250μg/mlキサンチンを補充したダルベッコ変法イーグル培地(DMEM)中で選択した。遺伝子導入した細胞クローンは、ヒトIgGについてELISAによってヒト抗体を生成用にスクリーニングした。細胞株分泌抗体を拡張し、最も高い生成株を選択し、液体窒素中に凝固させた。修飾抗体をProsep(登録商標)−A(英国ノースアンバーランド所在のBioprocessing Ltd社)を用いて精製した。ヒトIgGκ抗体についてELISAによって、濃度を決定した。
【0074】
合成ヒト抗体及びキメラ4D5抗体を、Hudziak et al.(同書)によって正確に記載されているように、陰性対照非−Her−2結合ヒトIgG1/Kappa抗体と共に、HER2+ヒト乳癌細胞株SK−BR−3の増殖の阻害について試験した。この結果(図3)は、合成ヒト抗体及びキメラ4D5抗体が、SK−BR−3細胞の増殖を阻害において等価な効果を有することを示す。また、図3は、以下のように生成した代替「エピトープ回避」複合抗体についてのデータを示す。
【0075】
本発明のエピトープ回避オプションを試験するために、合成ヒト重鎖及び軽鎖可変領域の配列を、Peptide Threading(www.csd.abdn.ac.uk/〜gjlk/MHC−thread)を用いて非自己ヒトMHCクラスIIバインダについて分析した。このソフトウエアは、ペプチドのアミノ酸側鎖と、MHCクラスII結合溝内の特定の結合ポケットの間の好適な相互作用を予測する。合成ヒト重鎖及び軽鎖可変配列からの全オーバーラップ13マーは、MHCクラスIIアロタイプのデータベースを通り、MHCクラスII分子との結合、及び、相互作用に基づいて得た。MHCクラスIIを結合すると予想されるペプチドは、VHの残渣16及び67、及びVLの9及び44で始まる132マーであった。結果として、ヒト可変領域配列ライブラリの新しいセグメントが、図1の合成ヒト配列で用いられるセグメントの代わりに選択され、アミノ酸交換VH18L−A/69I−G;VL11L−A、46L−Aを導入した。相当する「エピトープ回避」合成ヒト抗体(「EACHAB」=Epitope Avoided Composite Human AntiBody)は、図1の配列に相当する抗体を作るのに用いられるいくつかのオリゴヌクレオチドを置換することによって作られ、EACHABは上記の方法で作られ、標準合成ヒト抗体と同等であるSK−BR−5の増殖の阻害を示すために試験した(図3)。このデータは、合成ヒト抗体が、対照キメラ抗−HER−2抗体と等しい効果をもって成功裏に構成されて、EACHABバージョンの合成ヒト抗体が、効果の損失すること無く生成され得ることを示している。
【0076】
例2−合成ヒト抗−HER2抗体の免疫原性
【0077】
T細胞増殖アッセイを実施して、合成ヒト抗−HER2抗体、EACHAB変異株及びキメラ4D5抗体の免疫原性を比較した(例1参照)。これらの抗体は、血清の入っていない、動物由来成分の入っていない、タンパク質の入っていない培地、HyQ(登録商標)LS1000脂質補給(Hyclone Cat No:SH30554)及びピルビン酸ナトリウム(Gibco Cat No:11360−039)を補充したHyClone HyQ(登録商標)ADCF−Mab(商標)(Hyclone Cat No:Cat no:SH30349)中で増殖したNS0細胞から調製された。Sephadex G25(PDlOカラム)上の緩衝液を50mMのMESpH6に交換後、抗体は、陽イオン交換カラム(Mono−S 10/10)をそれぞれ通り、塩化ナトリウム勾配(0から0.5M)で溶出した。次いで、抗体含有画分を、PBSのSuperdex 200保存カラム(XK16/60)ランに塗布した。ピーク画分を貯留して、4℃で保存した。抗体濃度は、ヒトIgGについてのELISAによって決定した。
【0078】
健康なヒトのドナー血液から単離し、液体窒素で凍結保存したPBMC(末梢血単核球)を用いて、免疫原性分析を実施した。各ドナーは、Allset(商標)PCRベース組織分類キット(英国Wirral所在のDynal社)を用いて組織分類を行い、20人の健康なドナーを、個人MHCハプロタイプに従って選択した。AIM V(英国ペイズリィ所在のInvitrogen社)の4×106PBMCを含有する2mlバルク培地を試験ペプチド(5μM最終濃度)を有する24ウエル組織培養プレートでインキュベートし、バルク培地を緩やかに再懸濁し、PBMCの3倍の100μlのサンプルをU−底96ウエルプレートに移すことによって、5日、6日、7日、及び8日目に増殖を評価した。Tomtec Mach IIIプレートハーベスタ(英国所在のReceptor Technologies社)を用いて、培地をグラスファイバフィルタマット上に採取し、カウント毎分(cpm)値を、Wallac Microbeta TriLuxプレートリーダ(パララックス高効率計数プロトコルを用いて)を用いてシンチレーション計数によって決定した。各試験抗体について、刺激インデックス(SI)を、試験抗体のカウント毎分(cpm):顕著なT細胞エピトープ反応と考えられるSI>2を有する陰性対照のcpmの率として計算した。この結果は、顕著な増殖性反応キメラ(chimaeric)4D5抗体が、試験された20人の健康的なドナーのうちの5人(25%)の試験された4日間の増殖(2より大きいSI)の少なくとも1日に顕著な増殖性反応を誘発し、合成ヒト抗−HER2抗体が、20人のドナーのうち3人(15%)にSI>2を誘発するが、EACHAB抗−HER2抗体は、20人のドナーのうちの誰にもSI>2を誘発しない(0%)ことを示した。これらの結果は、キメラ4D5>合成ヒト抗−HER2抗体>EACHAB抗−HER2の順番を示し、後者が試験されたいずれのドナー血液サンプルに免疫原性の証拠がないことを示している。
【0079】
例3−合成ヒト抗−Lewis Y抗体の構造
【0080】
シアル化Lewis Y抗原に特異的な合成ヒト抗体を、参照抗体可変領域配列として、ヒト化3S193抗体を用いて、例1で記載されているように構成した(Scott et al.,Cancer Res.,60(2000)p3254−3261,US5874060)。インシリコ(in silico)ヒト可変領域配列ライブラリからのセグメントを、ヒト化3S193可変領域配列中の相当するアミノ酸に対する同定用に選択し、結合して図4及び5それぞれに示されているような合成ヒトVH及びVL配列を生成した。同時に、参照キメラ抗−Lewis Y抗体を、参照V領域配列から作った。ヒトIgG1(VH)及びKappa(VK)定常領域を、合成ヒト抗−Lewis Y抗体と、キメラ参照抗体の双方に用いて、抗体を、US5874060に記載されているような合成Lewis Y−HSA複合体に対するELISAによって試験した。このデータは、US5874060のデータと一致する0.15μg/ml合成ヒト抗体と比較したアッセイの結合シグナルを与えるための0.1μg/mlキメラ抗体の最小濃度を示した。
【0081】
例4−合成ヒト抗−IgE抗体の構造
【0082】
合成ヒト抗−IgE抗体を、参照抗体可変領域配列として、Xolair(登録商標)(Presta et al.,J.Immunol.,151(5)(1993)p2623−2632)として知られているヒト化抗−IgE抗体を用いて、例1に記載されているように構成した。インシリコ(in silico)ヒト可変配列ライブラリからのセグメントを、Xolair(登録商標)可変領域配列の相当する相当するアミノ酸に対する同定用に選択し、結合して図6及び7各々に示されているような合成ヒトVH及びVL配列を生成した。同時に、参照キメラ抗−IgE抗体を、参照V領域配列から作った。ヒトIgG1(VH)及びKappa(VK)定常領域を、合成ヒト抗−IgE抗体とキメラ参照抗体の双方に用いた。
【0083】
Fabの特異性を、表面プラズモン共鳴によって更に特徴付けた(瑞国Uppsala所在のBiacore AB社製BIAcore 2000)。組み換えヒトIgE Fabを、Flicker et al.,J.Immunol.,165(2000)p3849−3859によって記載されている通り生成した。試験抗体を精製し、NHS/EDC kit(Biacore社)を用いてCMチップのフロー電池に固定して、キメラ抗−IgE用の2010RU、及び合成ヒト抗−IgE用の2029RUを得た。Hepes緩衝生理食塩水(10mMのHepes、3.4mMのEDTA、150mMのNaCl、0.05%(v/v)界面活性剤P20、pH7.4)中の10及び25nM組み換えヒトIgE Fabを、10分間5μl/minの流量で試験抗体を除いた。この結果は、10及び25nMのIgE Fabの双方について、等価SPR(表面プラズモン共鳴)曲線が、キメラ抗−IgEについて検出され、合成ヒト抗−IgE抗体は、後者が、参照抗−IgE抗体と等価な結合効果を成功裏に達成することを示した。
【0084】
例5−合成ヒトscFvライブラリの生成及びスクリーニング
【0085】
ヒトscFv(単鎖Fv)の初期構造についての戦略は、Knappik et al.,J.MoI.Biol.,296(2000)57−86に詳細に記載されているように、7人分のヒトVHと、4人分のヒトVL(kappa)遺伝子を構成することであり、ヒト可変領域のデータベースから多数のVH及びVL CDR3セグメントにクローニングすることであった。このCDR3のリストは、VH CDR3については表1に、8つのアミノ酸のVL CDR3については表2に、10のアミノ酸のVL CDR3については表3に示されている。マスターVH及びVL構造については、フレームワーク3の端までのVH及びVLをコードする6つのオーバーラップ合成オリゴヌクレオチドが、Knappik et al.,同書に詳細に記載されているように合成され、繰り返しPCR(Prodromou and Pearl,Protein Engineering,5(1992)827−829)を行った。これらは、EcoRV消化pZero−1ベクタ(英国ペイズリィ所在のInvitrogen社)に結紮された。双方ともが開始時に4D5 CDR3を有するCH1及びC Kappaを添加するため(Carter et al,Bio/Technology,10(1992)163−167)、Knappik et al.,同書のプロトコルを、VH−CH1SapI−EcoRIと、VL−kappa Nsi−ShIフラグメントが、EcoRV消化pZero−1にクローニングされた平滑末端であることを除いて行った。
【0086】
表1
【0087】
表2
【0088】
表3
【0089】
CDR3の挿入について、プラス鎖から表Hの各CDR3をコードする単一オリゴヌクレオチドを、コンセンサスVH及びVL遺伝子にアニーリングするために各端に加えられた12つの相同性ヌクレオチドで合成した。これらのCDR配列に加えて、抗体E25からのCDR(例4参照)が含まれていた。これらのプライマを拡張し、二次プライマを加えて、VHについて5’NotI−3’XbaI座、VLについて5’SpeI−3’BamHIを、VH及びVL遺伝子(C領域のない)のN及びC末端のすぐ隣に導入した。クローニングに先立って、更なる相補プライマ対を用いて、VHとVLの間にリンカ配列(Gly4Ser)3を挿入し、一方、XbaI及びSpeI座は維持した。フルサイズのVH−リンカ−VLフラグメントは、NotI及びBamHIで消化され、NotI−BamHI消化pBluescript II KS(+/−)(蘭国アムステルダム所在のStratagene社)にクローニングした。
【0090】
個々のBluescriptクローンを採取し、国際公開番号第WO99/11777号に記載されているように、プラスミドDNAを精製して96ウエルプレート内にロボット制御で分注した。次いで、国際公開番号第WO99/11777号に記載されているように、DNAを、tRNA−ビオチニル−リシンを含有するIVTTにさらし、ストレプトアビジン表面に更にロボット制御で配置した。次いで、10,000の独立したクローンの固定開始scFvライブラリを組み換えヒトIgE Fabでインキュベートすることによってスクリーニングした(例4参照)。ウエルを室温で1時間、PBS/3%BSAでブロックし、PBSで3回洗浄して、PBS/3%BSAの5μg/mlヒトIgE Fabで1時間、処理した。次いで、ウエルを、PBSで更に3回洗浄し、PBS/3%BSAの5μg/mlアルカリ性ホスフォターゼ同定キメラ抗−IgE(例4)で1時間半、処理した。ウエルを、PBSで5回、更に洗浄し、可視化用に基質5−ブロモ−1−クロロ−3−インドリルホスファート及びニトロブルーテトラゾリウム(Roche Molecular)を用いて染色現像した。9600ウエルに1つの頻度で観察される強いシグナルは、E25 CDR3’sを含有するVHとVLの対双方から誘導されることを示した。
【0091】
例6−合成マウス抗−TNFα抗体の構造
【0092】
マウス可変領域配列ライブラリを、NCBI Igblast、Kabat及びGenbankデータベースを用いてヒトライブラリについて、例1に記載されているように作成した。用いた参照抗体可変領域配列は、マウスcA2抗体の可変領域を用いるRemicade(登録商標)(Le et al.,US6277969)として知られているキメラ抗−TNFα抗体であった。インシリコ(in silico)マウス可変領域配列ライブラリからのセグメントは、Remicade(登録商標)可変領域の部分的に相当するアミノ酸から選択されるが、例1で測定される非−自己ヒトMHCクラスIIバインダの形の配列のヒトT細胞エピトープを回避するように設計された変異体を含んでいた。キメラ抗体に用いた配列と比較した合成マウスVH及びVL配列を図8に示し、これは、合成マウス抗体のエピトープ回避のためのセグメント選択の結果として、2つの抗体間のVHとVLそれぞれの9及び16アミノ酸の差を示す。
【0093】
合成マウス及びキメラ抗−TNFα抗体を、例1に記載されているように生成した。標準ELISAの固定ヒトTNFαに結合する精製抗体の比較(国際公開番号第WO03/042247A2号に記載されている)は、合成マウス抗体が、キメラ抗−TNFα抗体の完全な結合能力を保持することを示した(図9)。次いで、これらの抗体の免疫原性を、T細胞アッセイ用の24HLA−DR型ヒト血液サンプルを用いて、例2で記載されているように比較した。この結果は、24人のドナーの誰もSI>2を誘発しない(0%)合成マウス抗−TNFα抗体と比較して、キメラ抗−TNFα抗体が、試験した24人の健康なドナーのうち9人(37.5%)に顕著な増殖反応(SIが2より大きい)を誘発することを示した。これらの結果は、ヒトT細胞エピトープを回避するためのこのようなセグメントの選択でマウスV領域から完全に取り出される可変領域配列のセグメントを具える合成マウス抗体が、なんらエピトープ回避方法を用いることなく、相当するキメラ抗体によって表示されるヒトT細胞アッセイの免疫原性を除去することができることを示した。
【0094】
例7:合成ヒト抗−TNFα抗体の構造
【0095】
ヒトTNFαに対する抗体A2の参照マウス可変領域重鎖及び軽鎖配列を、米国特許第5656272号から得た(図10、各SEQ.ID No1及びNo.2)。構造モデルを、マウス参照可変領域から作成し、CDR確認に欠かせないアミノ酸とCDRに近いパッキングを、CDR(<3Å)からの距離に基づいて同定した。重要ではあるが、余り気にかけなくてもよい残渣中、CDR(>3Å <6Å)からの距離と、CDRに近いより重要な残渣のパッキングの影響に基づいて同定した。更なる残渣セットを、マウス抗体配列、即ち、1%より少ない頻度で特定の座で見出されるアミノ酸の生成頻度に基づいて同定した。
【0096】
これらの残渣のできるだけ多くに含まれるヒトV領域配列セグメントを選択して(表4)、完全な長さのVH及びVL配列を作成した。同定された構造上重要な全ての残渣を含むようにこれらの配列に代替を作成し、エピトープ回避及び合成ヒト抗体設計用のテンプレートとしての機能する配列を作成した。各合成VH及びVLについての好ましい配列を、参照マウス抗体からの重要な残渣を含むように設計した。これらの可変重鎖及び軽鎖アミノ酸配列を、図11及び図12のSEQ ID No.3及びNo.4にそれぞれ示す。
【0097】
表4
(a)重鎖
【0098】
(b)軽鎖
【0099】
合成重鎖及び軽鎖可変領域配列を、様々なインシリコ(in silico)法(例えば、Propred[http://imtech.res.in/raghava/propred/index.html]、Peptide Threading[www.csd.abdn.ac.uk/〜gjlk/MHC−thread]、SYFPEITHI(www.syfpeithi.de)、MHCpred(www.jenner.ac.uk/MHCPred/)を用いてスキャンして、潜在的T細胞エピトープの存在を探し、参照マウスCDRに関連して同種ヒト生殖細胞フレームワーク領域配列と比較した。
【0100】
次の重鎖可変領域変異体を作成した(図11参照):
SEQ.ID.No.5は、SEQ.ID.No.3に対して、次の変化を含む:
T82aN+R83K
【0101】
SEQ.ID.No.6は、SEQ.ID.No.3に対して、次の変化を含む:
T82aN+R83K+D82bS
【0102】
SEQ.ID.No.7は、SEQ.ID.No.3に対して、次の変化を含む:
T82aN+R83K+D82bS+V32A
【0103】
SEQ.ID.No.8は、SEQ.ID.No.3に対して、次の変化を含む:
T82aN+R83K+D82bS+V32A+V78A
【0104】
次の軽鎖可変領域変異体を作成した(図12参照):
SEQ.ID.No.9は、SEQ.ID.No.4に対して、次の変化を含む:
I10T+N103R
【0105】
SEQ.ID.No.10は、SEQ.ID.No.4に対して、次の変化を含む:
I10T+N103R+S80A
【0106】
SEQ.ID.No.11は、SEQ.ID.No.4に対して、次の変化を含む:
I10T+N103R+S80AN41D
【0107】
対照キメラ抗体の構造について、SEQ.ID.No.1及びSEQ.ID.No.2を与えるように翻訳するヌクレオチド配列を、一連のオーバーラップ40マー合成オリゴヌクレオチドを用いて構成した。V領域配列の側面には、追加の5’及び3’配列が位置し、ほ乳類発現ベクタへのクローニングを容易にしている。オリゴヌクレオチドの配列を図13及び図14に示す。
【0108】
オリゴヌクレオチドを、Sigma−Genosys社(英国プール所在)から購入し、100μMの濃度で再懸濁した。ほとんどの5’オリゴヌクレオチドを除く、1μlの各重鎖有意DNAオリゴヌクレオチドを共に混合して、1.5μl(ほぼ、1μg)の混合物を:2μl 10× PNK緩衝液、2μl 10mMのATP、14μl H2O、0.5μl(5単位)PNKを更に含む20μlの反応液中でポリヌクレオチドキナーゼ(PNK、英国ペイズリー所在のInvitrogen社)を用いて処理した。この反応液を37℃で30分間インキュベートし、70℃で20分間加熱することによって、酵素を不活化した。重鎖アンチセンス、軽鎖センス、及びアンチセンスオリゴヌクレオチドを、同様にリン酸化した。各セットからの5’オリゴヌクレオチドを、H2Oで9倍に希釈し、適切な反応混合物に1.5μlを添加した。次いで、各反応液を0.5mlに希釈して、体積が最高で44μlになるまで、8000rpmで90分間、Amicon microcon YM3濃縮器で回転透析した。
【0109】
重鎖についてのセンス及びアンチセンス混合物と、軽鎖についてのセンス及びアンチセンス混合物を混合して、H2Oで88μlにした。10μlの10× リガーゼ連鎖反応(LCR)緩衝液、及び2μlのPfu リガーゼ(8単位、英国ケンブリッジ所在のStratagene社)を、各反応液に添加し、プログラム可能な加熱ブロック(heating block)中で、次のようにインキュベートした:94℃で4分間、次いで、1サイクルにつき60℃で3分間、その後、94℃で39秒間、次いで60℃で3分間を20サイクル。最後に反応液を、60℃で5分間インキュベートした。各LCR10μlを、臭化エチジウムで染色した1%のアガロースゲルに通し、1Kbラダーマーカ(Invitrogen社)と比較した。ほぼ400bpの微かな特異的バンド(faint specific band)の周囲に、結紮DNAの塗抹標本を各レーンで観察した。
【0110】
重鎖についてSEQ.ID.No.12が、22、及び軽鎖についてSEQ.ID.No.33及び43をプライマとして用いて、重鎖及び軽鎖LCRをPCRを介して増幅した。各反応液には、次のものが含まれていた:5μlのLCR、5μlの10× エクスパンドHiFi緩衝液(英国ルイス所在のRoche社)、lμlの10mM NTP混合物、0.25μlの各プライマ(l00μMのストックから)、0.5μlのエクスパンドHiFiポリメラーゼ(3単位、Roche社)、及び38μlのH2O。この反応液を次のように循環させた:94℃で2分間、次いで94℃で30秒間を20サイクル、60℃で30分間、及び72℃で30秒間。最後に、反応液を72℃で5分間インキュベートした。反応液の収量及び特性を、上記のようなアガロースゲル電解質によって確認した。ほぼ400bpで特異的でシャープなバンドが、各反応液について観察された。
【0111】
Qiagen PCR精製キットを用いて、反応生成物を精製し、各々を30μlのH2Oで溶離した。重鎖生成物を、制限酵素Mlu I及びHind IIIを用いて標準反応で消化させ、軽鎖生成物を、BssH II及びBamH Iを用いて消化させた。Qiagen PCR精製キットを用いて、反応生成物を再び精製し、各々を30μlのH2Oで溶離した。
【0112】
軽鎖発現ベクタpANT08は、pAT153バックボーンに基づいており:CMV即時/アーリーエンハンサプロモータ−590から+7、高発現マウス抗体軽鎖RNA由来の30nt 5’UTR、可変領域開始コドン付近のBssH II制限座を組み込むマウスコンセンサス軽鎖シグナル配列、可変領域スプライス座からBamH I制限座への33ntを含むヒト合成イントロンに対するショートリンカ(可変領域の代用)、次いで、ヒト定常Kappa(CK)領域遺伝子に先立つ343ntのイントロンを含むヒト遺伝子DNAのフラグメント、CK遺伝子及びCKポリAをこの順に含む。
【0113】
重鎖発現ベクタpANT09は、プロモータ領域を通るpANT08と類似しており、この後に:上記の重鎖発現ベクタ由来の62nt5’UTRと、可変領域開始コドン付近のMlu I座を組み込むマウス重鎖コンセンサスシグナル配列と、可変領域スプライス座に対するショートリンカ(可変領域の代用)、その直後にCH領域ポリA座の端に対するCH1遺伝子のイントロン211ntの上流に位置したHind III制限座からのヒトゲノムDNAと、が続く。このフラグメントは、CH1、ヒンジ、CH2及びCH3イントロン、及びヒトIgG1のエクソンが含まれる。また、このベクタも、メトトレキサートに対する耐性について、SV40プロモータとポリAシグナルによって制御される、ジヒドロ葉酸還元酵素用の遺伝子を含んでいた。
【0114】
各ベクタ2μgを、全体積30μgの標準的反応液中で関連制限酵素で消化させた。各反応液は、上記のような1%アガロースゲルを通り、ベクタ特異バンド(6.0Kbp重鎖及び4.2Kbp軽鎖)をゲルから切り取り、Qiagenゲル抽出キットを用いて精製し、30μlH2O中で溶離した。
【0115】
各消化ベクタ1μlを、Ligafastキット(英国サウサンプトン所在のPromega社)を用いて、3μgの相当する消化可変遺伝子PCR生成物に結紮した。各結紮反応液2.5μlを、製造者によって指示されているように、サブクローニング効率適格(efficiency competent)XLl−blue(Stratagene社)に形質転換し、100μg/mlのアンピシリンを含有するLBアガープレート上に置き、37℃で夜通しインキュベートした。各結紮からの10の細菌コロニィを、100μg/mlアンピシリンを含有する10mlの2× YT培養液内に植菌し、振盪しながら37℃で夜通し培養した。プラスミドを、Qiagenプラスミド調製キットを用いて、1.5mlの各一夜培養物から精製し、各々を50μlのH2Oで溶離した。プラスミドを、契約しているシークエンシング施設に送り、正確なV領域遺伝子配列を同定する標準CMVプロモータプライマ及びクローンで配列した。
【0116】
合成ヒト抗体の構造について、SEQ.ID.No.3及びSEQ.ID.No.4を与えるように翻訳するヌクレオチド配列を、一連のオーバーラップ40マー合成オリゴヌクレオチドを用いて構成した。オリゴヌクレオチドの配列を、図15及び図16に示す。SEQ.ID.No.5を与えるように翻訳するヌクレオチド配列を、オリゴヌクレオチドプライマSEQ.ID.No.94及び95(図17)を、オリゴヌクレオチドSEQ.ID.No.53及び63とプライマリ合成ヒト抗体重鎖変異体のプラスミドDNAと共にテンプレートとして用いて、オーバーラップPCRを介して構成した。2つのPCR反応を、SEQ.ID.No.53及び95、又はSEQ.ID.No.94及び63のプライマ対として用いて実施した。各反応液中には、次のものが含まれていた:1μl(100ng)のプラスミドテンプレート、5μlの10× エクスパンドHiFi緩衝液(Roche社)、1μlの10mM NTP混合物、0.25μlの各プライマ(100μMストックから)、0.5μlのエクスパンドHiFiポリメラーゼ(3単位、Roche社)、及び42μlのH2O。これらの反応液を次のように循環させた:94℃で2分間、次いで、94℃で30秒間を20サイクル、60℃で30秒間、及び72℃で30秒間。最後に、この反応液を72℃で5分間インキュベートした。全反応液を1%のアガロースゲルによって電気泳動させ、295bp及び126bpの特異的バンドを除去し、Qiagenゲル抽出キットを用いて精製した。DNAを30μlのH2Oで溶離した。
【0117】
テンプレートとして、1μlの295bp生成物及び1μlの126bp生成物を用いたことを除いて、2つの精製フラグメントを、上述のPCR条件でオリゴヌクレオチドプライマSEQ.ID.No.53及び63を用いて、PCR反応液中で結合した。従って、H2Oの量は、41μlに減少した。396bpの結合したPCR生成物を、QiagenPCR精製キットを用いて精製し、30μlのH2Oで溶離した。
【0118】
SEQ.ID.No.6を与えるように翻訳するヌクレオチド配列を、オリゴヌクレオチドプライマSEQ.ID.No.96及び97(図17)を、オリゴヌクレオチドSEQ.ID.No.53及び63とプライマリ合成ヒト抗体重鎖変異体のプラスミドDNAと共にテンプレートとして用いて、オーバーラップPCRを介して構成した。2つのPCR反応を、SEQ.ID.No.53及び97、又はSEQ.ID.No.96及び63のプライマ対として用いて実施した。第1段階のPCRを上記のように実施して、295bp及び126bpのフラグメントを得た。これらのフラグメントを、上記のように精製し、共に結合させて、再精製した。
【0119】
SEQ.ID.No.7を与えるように翻訳するヌクレオチド配列を、オリゴヌクレオチドプライマSEQ.ID.No.98及び99(図17)を、オリゴヌクレオチドSEQ.ID.No.53及び63とSEQ.ID.No.6についてのPCR生成物と共にテンプレートとして用いて、オーバーラップPCRを介して構成した。2つのPCR反応を、SEQ.ID.No.53及び99又はオリゴヌクレオチドSEQ.ID.No.98及び63のプライマ対のいずれかを用いて実施した第1段階のPCRを上記のように実施して、98bp及び318bpのフラグメントを得た。これらのフラグメントを、上記のように精製し、共に結合させて、再精製した。
【0120】
SEQ.ID.No.8を与えるように翻訳するヌクレオチド配列を、オリゴヌクレオチドプライマSEQ.ID.No.100及び101(図17)を、オリゴヌクレオチドSEQ.ID.No.53及び63とSEQ.ID.No.7についてのPCR生成物と共にテンプレートとして用いて、オーバーラップPCRを介して構成した。2つのPCR反応をSEQ.ID.No.53及び101、又はSEQ.ID.No.100及び63のどちらかをプライマ対として用いて実施した。第1段階PCRを上記のように実施して、270bp及び155bpのフラグメントを得た。これらのフラグメントを、上記のように精製し、共に結合させて、再精製した。
【0121】
上記のPCR生成物のそれぞれを、Mlu I及びHind IIIで消化させ、同様に消化させたpANT09内に結紮した。この結紮を、形質転換して配置し、コロニィを選定し、プラスミドを調製して、全て配列した。
【0122】
SEQ.ID.No.9を与えるように翻訳するヌクレオチド配列を、テンプレートとして、オリゴヌクレオチドプライマSEQ.ID.No.102及び103(図17)と、プライマリ合成ヒト抗体軽鎖変異体のプラスミドDNAとを用いて、PCRを介して構成した。重鎖変異体について記載されているように、PCR単反応を実施し、383bpの生成物を得た。完全な反応液を1%アガロースゲルによって電気泳動し、特異的なバンドを除去し、Qiagenゲル抽出キットを用いて精製した。DNAを30μlのH2Oで溶離した。
【0123】
SEQ.ID.No.10を与えるように翻訳するヌクレオチド配列を、テンプレートとして、オリゴヌクレオチドSEQ.ID.No.74及び84と共にオリゴヌクレオチドプライマSEQ.ID.No.104及び105(図17)と、SEQ.ID.No.9についてのPCR生成物とを用いて、オーバーラップPCRを介して構成した。2つのPCR反応を、SEQ.ID.No.74及び105、又はSEQ.ID.No.104及び84のプライマ対として用いて実施した。第1段階のPCRを重鎖変異体についての上記のように実施して、265bp及び139bpのフラグメントを得た。これらのフラグメントを、重鎖変異体についての上記のように精製し、共に結合させて383bpの生成物を作成し、再精製した。
【0124】
SEQ.ID.No.11を与えるように翻訳するヌクレオチド配列を、テンプレートとして、オリゴヌクレオチドSEQ.ID.No.74及び84と共にオリゴヌクレオチドプライマSEQ.ID.No.106及び107(図17)と、SEQ.ID.No.10についてのPCR生成物とを用いて、オーバーラップPCRを介して構成した。2つのPCR反応を、SEQ.ID.No.74及び107、又はSEQ.ID.No.106及び84のプライマ対として用いて実施した。第1段階のPCRを重鎖変異体についての上記のように実施して、148bp及び256bpのフラグメントを得た。これらのフラグメントを、重鎖変異体についての上記のように精製し、共に結合させて383bpの生成物を作成し、再精製した。
【0125】
上記のPCR生成物のそれぞれは、BssH II及びBamH Iで消化され、同様に消化されたpANT08内に結紮される。この結紮を上記のように、形質転換して配置し、コロニィを選定し、プラスミドを調製して、上述した通り全て配列した。
【0126】
CHO−K1細胞(ATCC#CCL−61)を、10%FCS、L−グルタミン、ピルビン酸ナトリウム、及びL−プロリンを含む高グルコースDMEM中で増殖させた。近融合性培地を、製造業者(Invitrogen)によって指示されているように、リポフェクトアミン2000(Lipofectamine 2000)を用いてトランスフェクション用に採取した。トランスフェクションは、0.3μgの重鎖作成物及び0.2μgの軽鎖作成物を具える0.5μgのプラスミドDNAの全てを用いて、3×105セル/mlで200μlの細胞を播種した48ウエルプレート中で実施した。
【0127】
上澄みを採取する前に、トランスフェクションを、37℃/5%CO2で48から72時間インキュベートした。抗体発現は:マウスモノクローナル抗−ヒトIgGキャプチャ抗体と、ヒトIgG1/Kappaスタンダードと、HRP複合ヤギ抗−ヒトKappa軽鎖と、を検出抗体として用いてELISAによって定量化した(全ての試薬はSigma製)。
【0128】
重鎖及び軽鎖の全ての組み合わせを遺伝子導入した(即ち、6重鎖×5軽鎖=30トランスフェクション)。抗体発現レベルは一般的に、0.5から2.0μg/mlの範囲内であるが、重鎖SEQ.ID.No.8では、発現は観察されなかった。
【0129】
発現抗体を、TNF−感受性WEHI−164細胞を用いて、ヒトTNFαの活性を無毒化する能力について試験した(Espevik et al.,J.Immunol.Methods 1986,95,99−105)。細胞を、3−4時間、96−ウエルマイクロタイタープレートに、1ウエル当たり5×104個の細胞で、1μg/mlのアクチノミシンDに置いた。細胞を40pMヒトTNFα、及び様々な濃度のキメラ抗体(1ng/mlから500ng/mlの範囲)に曝露させて、検量線を作成した。重鎖及び軽鎖の様々な組み合わせを、25ng/mlの単一濃度点で試験した。この濃度は、キメラ抗体のED50として既に決定してある。ED50のキメラ抗体として以前決定した全アッセイを3回実施した。
【0130】
混合物を37℃で夜通しインキュベートした。最終濃度0.5mg/mlに、3−[4,5−ジメチル−チアゾール−2−イル]−2,5−ジフェニルテトラゾリウムブロミド染料(MTT)を添加し、37℃で4時間、インキュベートし、0.1MのHCl、0.1% SDS中で細胞を溶解し、550nmの波長で吸光度を測定して、細胞生存率を決定した。
【0131】
重鎖及び軽鎖の組み合わせからの吸光度を用いて、検量線から見掛けの抗体の濃度を計算した。キメラの見掛けの濃度は、変異体の各組み合わせの濃度によって分けられて、倍の差分値を与えた。キメラの値よりも低い値は、これらの組み合わせが、TNFα細胞毒性から細胞を保護する点でより効果的であることを示すが、より高い値は、これらの効果がより低いことを示した。全ての組み合わせについての値を表5に示す。
【0132】
表5
キメラ抗体と比較した合成ヒト抗体変異体の活性率
【0133】
次の合成ヒト抗体重鎖及び軽鎖の組み合わせは、1.0に近い倍の差分値を与えた:SEQ.ID.No.5/10、SEQ.ID.No.7/4、SEQ.ID.No.7/9、SEQ.ID.No.7/10。これらの組み合わせを更なる研究用に選択した。
【0134】
上記で選択された配列を運搬する発現プラスミドをNS0細胞内に遺伝子導入した(ECACC番号第85110503号)。これらの細胞を、L−グルタミン、ピルビン酸ナトリウム、5%超低IgG FCS及びペン/ストレプ(pen/strep)を含む高グルコースDMEMで培養した。細胞を、対数増殖期中に採取し、遠沈して新しい増殖培地内で5×106セル/mlで再懸濁した。750μlの細胞を全量30μgの各プラスミド対で混合し、これを、50μlのH2O内でSsp Iで線形化した。細胞/プラスミド混合物を4mmギャップのキュベットに移し、250V、1500μF、無限抵抗で、Equibio Easyject Plusを用いてエレクトロポレートした。このエレクトロポレートは、25mlの予熱した増殖培地に即時移し、100μl/ウエルで、5×96ウエル平底プレートに置かれた。このプレートを37℃/5%CO2でインキュベートした。48時間の後エレクトロポレーションを行い、300nMメトトレキサートを含有する50μl培地を、各ウエルに加えて、100nMの最終濃度を与えた。7日間の後エレクトロポレーションを行い、100nMメトトレキサートを含有する更なる50μlの培地を、各ウエルに加えた。
【0135】
ほぼ2週間後のエレクトロポレーションを行うと、いくつかのウエルの培地は、黄色になり始め、遺伝子導入されたコロニィ増殖を示した。これらのウエルからの培地を、抗−ヒトIgG Fcキャプチャ/抗−ヒトIg Kappa軽鎖HRP複合検出ELISAを用いて、抗体発現について試験した。試験サンプルを、ヒトIgG1/Kappa標準及び確立されている抗体発現レベルと比較した。抗体のコロニィ発現有効量を、200nMメトトレキサートを含む培地で増やした。
【0136】
抗体をタンパク質A親和性クロマトグラフィを介して500ml培養培地から精製し、Sephacryl S200を用いて、サイズ排除クロマトグラフィを実施した。精製した抗体を、280nmでのUV吸光度によって定量化し、OD2801=1.4mg/mlを仮定した。
【0137】
精製キメラ及び複合抗体を、上記の例4に記載されているWEHI−164保護アッセイを介して活性について試験した。各抗体を、検量線を作成するのに先に用いた全濃度範囲に渡って試験した(図18参照)。合成ヒト抗体7/10(即ち、SEQ.ID.No.7及び10を含む)は、最も活性な変異体であり、キメラ抗体と同じ活性を有することが分かった。合成ヒト抗体7/9及び5/10は、キメラと比較して僅かに少ない同様の活性を有し、合成ヒト抗体7/4は、最も活性がなかった。
【0138】
従って、合成ヒト抗体7/10が、最も好ましいMHCクラスII結合プロファイルを有すると予測され、最も活性な変異体であるので、これを、経時変化T細胞増殖アッセイでの試験用に選択した。ヒトPBMCを、2回のFicoll密度遠心分離を介して、ヒト血由来の軟膜から調製した。調製PBMCを、90%ヒトAB血清/10%DMSO中の1mlアリコートで3×107セル/mlの密度で再懸濁し、液体窒素化で貯蔵した。PBMCをDynal Allset(登録商標)PCRタイピングキットを用いて組織分類した。
【0139】
リード合成ヒト抗体を、20人の健康なドナーからのヒトPBMCを用いて、全タンパク質T細胞アッセイ中のキメラ抗体と比較した。各ドナーからのPBMCを解凍し、洗浄して、AIM V血清フリーリンパ球増殖培地で再懸濁した。第1日目に、50μgのタンパク質を、24ウエルプレートの4×106のPBMC2mlのバルク培地に加え、第6日目から第9日目に、3重に100μlアリコートを除去して、96ウエルプレートに移した。採取して放射能の取り込みを測定する前に、各アリコートを、1μCiトリチウム化チミジンを含有する75μl培地で24時間パルスした。結果を刺激インデックス(SI)の計算によって標準化した。広範囲のHLA DRアロタイプのガレージは、個々のMHCハプロタイプによってドナーを選択することによって達成される。
【0140】
経時アッセイの結果を、図19で示す。これらの結果は、キメラ抗体(図19(a))が、20人のドナー中10人に少なくとも1日で、T細胞反応(SI>=2)を導くことを示す。逆に、合成ヒト抗体(図19(b))は、あらゆる時点であらゆるドナーの反応も誘導することができなかった。従って、非免疫原性合成ヒト抗体は、マウス抗−TNFα抗体(A2)を参照として用いて、ヒト抗体のセグメントから成功裏に構成された。
【0141】
例8:合成タイプIリボソーム阻害タンパク質の構造
【0142】
プラントタイプIリボソーム阻害タンパク質(RIP)ブーガニン(ブーゲンビレアスペクタビリス(Bougainvillea spectabilis)に由来する)の合成変異体を、国際公開番号第WO2005090579号で記載されている方法を用いて生成した。ブーガニン中のT細胞エピトープのロケーションを、国際公開番号第WO2005090579号に記載のようにオーバーラップ15マーペプチドの分析によって試験し、表6のSEQ ID11−13のペプチド(残渣121−135、130−144、及び148−162に相当する)をエピトープとして同定した。ブーガニンを、ブーゲンビレアスペクタビリス(Bougainvillea spectabilis)植物の葉の組織からクローニングした。製造業者によって指示されているように100mg組織からポリA Tract System 1000キット(Promega社)を用いてmRNAを抽出した。cDNAを、以下のプライマ:ATGTACAACACTGTGTCATTTAAC及びTTATTTGGAGCTTTTAAACTTAAGGATACCを有するAccessQuick RT−PCR system(Promega社)を用いて、mRNAテンプレートから合成した。第1のプライマは、ATG開始コドンを更に含み、第2のプライマは、TAA停止コドンを更に含む。PCR生成物を、T/Aクローニングシステム(pGEM T Easy、Promega社)を用いてクローニングし、いくつかのクローンをベクタ内に含まれるT7プロモータの転写方向に配向する修正クローンを同定するように配列した。
【0143】
表6
ブーガニン(bouganin)の免疫原性ペプチド配列及び置換ヒト配列セグメント
【0144】
表6に示されるように同定されたヒト配列セグメントを含む一連の変異体を作成した。これらは、高品質ポリメラーゼ(エクスパンドHiFi、Roche社)を有するオーバーラップPCRを用いて構成した。5’及び3’プライマは上述のものであり、PCR生成物を上述の通りT/Aクローニングベクタにクローニングし、T7プロモータの転写方向に配向した修正クローンを同定した。クローンを、発光酵素遺伝子を発現する対照DNA(Luciferase T7 Control、Promega社)を含む共役転写及び転写反応中の活性について分析した。ブーガニンはリボソーム不活化タンパク質であるので、発光酵素遺伝子の転写のレベルを著しく下げる。この減少は、従来Steady−Glo(Promega社)等の発光酵素検出システムを用いて分析されている。精製野生型又は突然変異体ブーニガンプラスミドを、Not Iで線形化し、10ng/mlに希釈した。Luciferase T7 Control DNAを125ng/μlに希釈した。1μlの各DNAを、10μlのTnT混合物(Promega社)と、0.25μlのメチオニンと、0.25μlのヌクレアーゼ自由水と混合した(TnTキットで供給される)。対照は、野生型(wt)ブーニガンとLuciferase T7 Controlのみであった。反応を3回実施し、30℃で1時間インキュベートした。5μlの各反応液を、黒い壁付の96ウエル照度計プレートに移し、45μlの水と、50μlのSteady−Glo試薬を混合した。Wallac Microbeta Trilux照度計で発光を読んだ。活性は、Luciferase T7 Control DNAのみで観察された蛍光の割合として表した。
【0145】
図20は、多数の異なる変異体の活性プロファイルを示す。これは、最も活性な変異体が:ペプチド41中のV123T;ペプチド44中のV132P/Y133Q;ペプチド44中のI152Q;であることを示す。これらの4つの突然変異を含む複合変異体を作成し、活性アッセイで再試験した。この変異体の活性は、図20のCOMBによって示され、wtタンパク質の活性のほぼ75%を維持する。
【0146】
残渣121−135、130−144、及び148−162に相当する活性COMB変異体内のヒト配列セグメントを含むペプチドを合成し、例7に記載したように、20人の健康なドナーからのヒトPBMCを有する経時T細胞アッセイ中の相当する野生型ペプチドと比較した。この結果は、ヒト配列セグメントを含むペプチドが、どの時点でも、どのドナーのT細胞増殖も誘発しないが、各野生型ペプチドは、少なくとも1の時点で全ドナーの>10%のSI>2を有する増殖を誘発することを示した。
【0147】
例9:合成ヒルジンの構造
【0148】
トロンビン阻害ヒルジン(医用蛭(Hirudo medicinalis)に由来する)の合成変異体を野生型として表7のSEQ ID No 14を有するタンパク質を用いて、国際公開番号第WO2004113386号に記載されている方法で生成した。ヒルジンのT細胞エピトープのロケーションを、国際公開番号第WO2004113386号に記載されているように、オーバーラップ15マーペプチドの分析によって試験し、ペプチド27−41CILGSDGEKNQCVTGが、20人の健康なドナーからのヒトPBMCで有意なT細胞反応を与えることが示された。ヒトメラノーマ関連抗原(AAN40505.1)からのヒト配列セグメントKCRHを、例8のオーバーラップPCRを用いて、26−29でヒルジン残渣を置換するのに使用し、国際公開番号第WO2004113386号に記載されているアッセイを用いる野生型ヒルジンの完全活性化を維持した28/29RHに変わる28/29ILを有する変異体ヒルジン分子を得た。修飾ペプチド27−41CRHGSDGEKNQCVTGを、この修飾ペプチド中のT細胞エピトープ活性の損失を示す例8に示すようなT細胞アッセイ中の野生型ペプチド27−41CILGSDGEKNQCVTGと共に試験した。
【0149】
例10:Trエピトープを有する合成ヒト抗−IgE抗体の構造
【0150】
例4の合成ヒト抗−IgE抗体からのVH及びVL遺伝子を、同書内のOrlandi et al.からの標準ポリメラーゼ連鎖反応(PCR)法によって、オリゴヌクレオチドプライマ対を、VL−及びVH−特異PCR用のテンプレートとして用いて、分離プラスミドベクタにクローニングした。オーバーラップする相補配列を、続く融合PCR中に結合したPCR生成物内に導入し、20アミノ酸(G4S1)4リンカ、又は代替的に、2つのアスパラギン残渣及びGGSトリプレットによって各側に並んだC型肝炎コアタンパク質(P19,MacDonald et al.,Journal of Infectious Diseases,185(2002)p720−727)からの10アミノ酸Trエピトープを含む20アミノ酸配列GGSNNLSCLTIPASANNGGSのどちらかのコード配列を形成した。この最終増幅ステップを、制限酵素EcoRV及びBspE1を有する続く開裂用のプライマ対で実施し、bluescriptKS vector(Stratagene社)にクローニングした。次いで、合成ヒト抗−IgE単鎖抗体(scFvs)の二量体形を、Mack et al.,Proc Natl Acad Sci USA.,92(1995)p7021−7025の方法によって構成した。二量体VL−リンカ−VH−VL−リンカ−VHフラグメントを、発現ベクタpEF−DHFR(Mack et al.,同書)のEcoR1/Sal1座にサブクローニングし、エレクトロポレーションによって、DHFR−欠損中国ハムスタ卵巣(CHO)細胞に遺伝子導入した。選択、遺伝子増幅、タンパク質生成を、Mach et al.,同書によって記載されているように実施した。二量体scFvを、ニッケル−ニトリロ三酢酸(Ni−NTA)カラム(Qiagen社)上の親和性クロマトグラフィよってC−末端ヒスチジンテールを介して精製して、CHABIgEG4S 1×4((VLとVHの間のG4SO4リンカ)とCHABIgEHCVP19(VLとVHの間のHCV Trエピトープ)を指定した二量体Fvsを与えた。
【0151】
二量体scFvsを、20人の健康なドナーからのPBMCを用いてHall et al.,Blood 100(2002)p4529−4536に記載された通りに正確に50μg/mlのヒトT細胞アッセイで順次試験した。これらの結果は、CHABIgEG4S 1×4、又はCHABIgEHCVP19のどちらかについてT細胞の有意な増殖を示していないが、CHABIgEG4S 1×4ではなくCHABIgEHCVP19で刺激した20人のドナーのうちの4人からのIL−10生成の有意なレベル(SI>2)を示した(20人のドナー中0人のSI>2)。これは、免疫抑制サイトカインIL−10の誘発についての抗体分子内に含まれるTrエピトープの効果を示している。
【図面の簡単な説明】
【0152】
【図1−1】図1は、合成ヒト抗HER−2抗体に用いられるVH遺伝子の配列である。
【図1−2】図1は、合成ヒト抗HER−2抗体に用いられるVH遺伝子の配列である。
【図1−3】図1は、合成ヒト抗HER−2抗体に用いられるVH遺伝子の配列である。
【図2−1】図2は、合成ヒト抗HER−2抗体に用いられるVL遺伝子の配列である。
【図2−2】図2は、合成ヒト抗HER−2抗体に用いられるVL遺伝子の配列である。
【図2−3】図2は、合成ヒト抗HER−2抗体に用いられるVL遺伝子の配列である。
【図3】図3は、キメラ4D5IgG1/kappaで8日間、インキュベーションした後のヒトSK−BR−3細胞と、合成ヒト抗−HER2抗体と、キメラ抗−IgE対照で抗−HER2「EACHAB」を回避したエピトープの増殖の阻害を示す(例4参照)。
【図4−1】図4は、合成ヒト抗−Lewis Y抗体を用いたVH遺伝子の配列である。
【図4−2】図4は、合成ヒト抗−Lewis Y抗体を用いたVH遺伝子の配列である。
【図4−3】図4は、合成ヒト抗−Lewis Y抗体を用いたVH遺伝子の配列である。
【図5−1】図5は、合成ヒト抗−Lewis Y抗体を用いたVL遺伝子の配列である。
【図5−2】図5は、合成ヒト抗−Lewis Y抗体を用いたVL遺伝子の配列である。
【図5−3】図5は、合成ヒト抗−Lewis Y抗体を用いたVL遺伝子の配列である。
【図6−1】図6は、合成ヒト抗ヒトIgE抗体を用いたVH遺伝子の配列である。
【図6−2】図6は、合成ヒト抗ヒトIgE抗体を用いたVH遺伝子の配列である。
【図6−3】図6は、合成ヒト抗ヒトIgE抗体を用いたVH遺伝子の配列である。
【図7−1】図7は、合成ヒト抗ヒトIgE抗体を用いたVL遺伝子の配列である。
【図7−2】図7は、合成ヒト抗ヒトIgE抗体を用いたVL遺伝子の配列である。
【図7−3】図7は、合成ヒト抗ヒトIgE抗体を用いたVL遺伝子の配列である。
【図8】図8は、ヒトT細胞エピトープの回避を含む合成マウス抗ヒトTNFα抗体を用いたVH及びVL遺伝子の配列である。
【図9】図9は、合成マウス及びキメラ抗ヒトTNFα抗体によるヒトTNFαへの結合用ELISAである。
【図10】図10は、抗TNFα抗体A2のV領域配列である。
【図11】図11は、合成ヒト抗TNFαVH変異株の配列である。
【図12】図12は、合成ヒト抗TNFαVL変異株の配列である。
【図13】図13は、キメラマウス:ヒト抗TNFαVHの構成用オリゴヌクレオチドである。
【図14】図14は、キメラマウス:ヒト抗TNFαVLの構成用オリゴヌクレオチドである。
【図15】図15は、一次合成ヒト抗TNFαVH(SEQ ID No.3 図11に相当する)の構成用オリゴヌクレオチドである。
【図16】図16は、一次合成ヒト抗TNFαVL(SEQ ID No.4 図12に相当する)の構成用オリゴヌクレオチドである。
【図17】図17は、二次合成ヒト抗TNFαVH及びVL変異株の構成用オリゴヌクレオチドである。
【図18】図18は、合成ヒト抗TNFα抗体用WEHI−164保護アッセイである。
【図19】図19は、リード合成ヒト抗TNFαの経時ヒトT細胞アッセイである。
【図20】図20は、挿入されたヒト配列セグメントを有する合成ブーガニン分子の活性である。
【特許請求の範囲】
【請求項1】
修飾抗体又は前記修飾抗体の抗原結合フラグメントにおいて、前記修飾抗体又は抗原結合フラグメントの重鎖及び軽鎖可変領域が、各々1又はそれ以上の他の抗体又は抗原結合フラグメントからのアミノ酸配列の2又はそれ以上のセグメントから成り、前記セグメントが全てCDRsでもなくフレームワーク領域でもないことを特徴とする修飾抗体又は前記修飾抗体の抗原結合フラグメント。
【請求項1】
修飾抗体又は前記修飾抗体の抗原結合フラグメントにおいて、前記修飾抗体又は抗原結合フラグメントの重鎖及び軽鎖可変領域が、各々1又はそれ以上の他の抗体又は抗原結合フラグメントからのアミノ酸配列の2又はそれ以上のセグメントから成り、前記セグメントが全てCDRsでもなくフレームワーク領域でもないことを特徴とする修飾抗体又は前記修飾抗体の抗原結合フラグメント。
【図1−1】

【図1−2】
【図1−3】
【図2−1】

【図2−2】

【図2−3】
【図3】
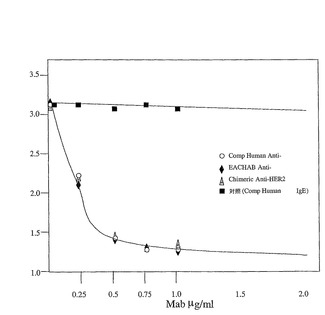
【図4−1】
【図4−2】
【図4−3】

【図5−1】
【図5−2】

【図5−3】
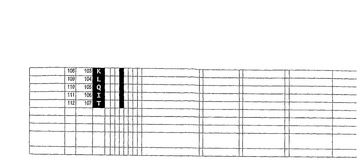
【図6−1】
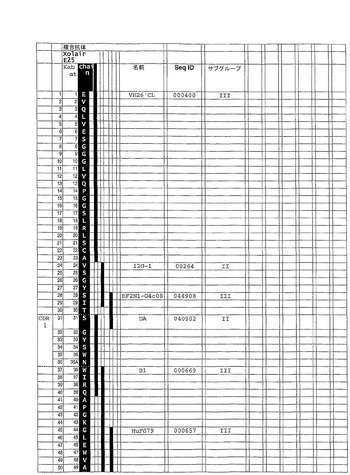
【図6−2】
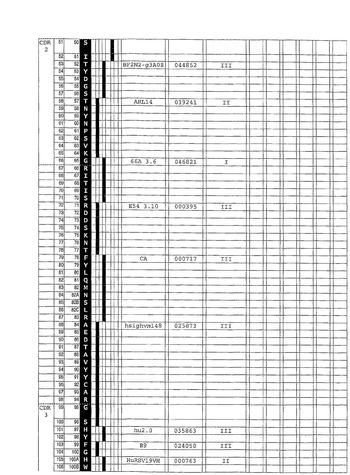
【図6−3】
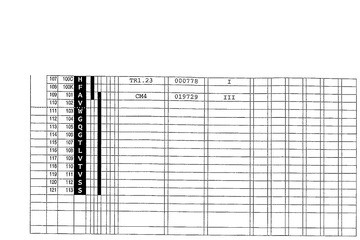
【図7−1】
【図7−2】

【図7−3】
【図8】

【図9】
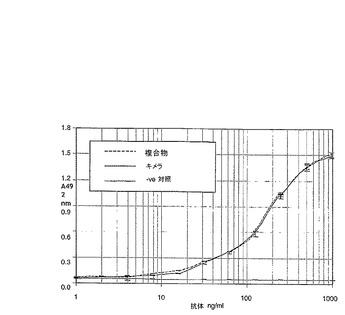
【図10】

【図11】

【図12】

【図13】

【図14】

【図15】

【図16】

【図17】

【図18】
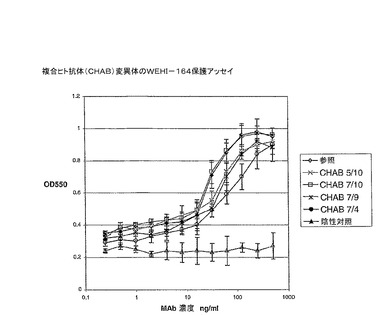
【図19】
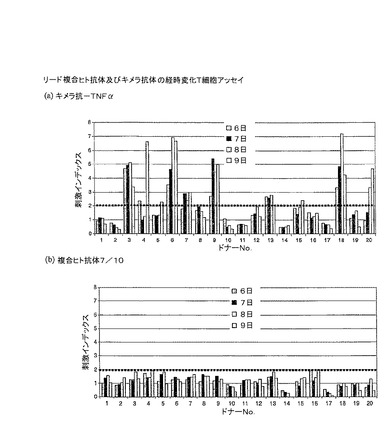
【図20】
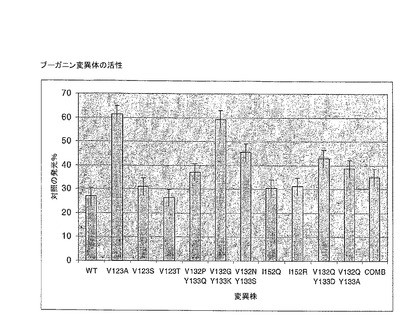
【図1−2】
【図1−3】
【図2−1】
【図2−2】
【図2−3】
【図3】
【図4−1】
【図4−2】
【図4−3】
【図5−1】
【図5−2】
【図5−3】
【図6−1】
【図6−2】
【図6−3】
【図7−1】
【図7−2】
【図7−3】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【公開番号】特開2013−28616(P2013−28616A)
【公開日】平成25年2月7日(2013.2.7)
【国際特許分類】
【出願番号】特願2012−198433(P2012−198433)
【出願日】平成24年9月10日(2012.9.10)
【分割の表示】特願2007−553693(P2007−553693)の分割
【原出願日】平成18年2月3日(2006.2.3)
【出願人】(507165486)アンチトープ リミテッド (5)
【Fターム(参考)】
【公開日】平成25年2月7日(2013.2.7)
【国際特許分類】
【出願日】平成24年9月10日(2012.9.10)
【分割の表示】特願2007−553693(P2007−553693)の分割
【原出願日】平成18年2月3日(2006.2.3)
【出願人】(507165486)アンチトープ リミテッド (5)
【Fターム(参考)】
[ Back to top ]