
ヒト抗TNFアルファ抗体の安定した高蛋白質濃度製剤
本発明はNaClを含有せず、20mgを上回るポリオールと、少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤を提供する。本発明は長期安定性と皮下投与に有利な特徴をもつ高濃度抗体製剤を提供する。
【発明の詳細な説明】
【背景技術】
【0001】
抗体等の治療用蛋白質の製剤化は経済的及び治療薬として成功するために多数の望ましい特性(例えば安定性、投与適性、濃度)を満たす必要があるため、困難であることが多い。製造、保存及び配送中に、治療用蛋白質は物理的及び化学的劣化を受けることが知られている。これらの不安定性は蛋白質の力価を下げ、患者に有害なイベントが発生する危険を増し、従って、当局の許可に著しく影響する可能性がある(例えばWang,et al.(2007)J Pharm Sci 96:1参照)。従って、治療用蛋白質の成功には安定した蛋白質製剤が不可欠である。
【0002】
有効にするために、多くの治療用蛋白質は高用量を投与する必要があり、高濃度製剤に製剤化することが好ましい。高濃度蛋白質製剤は薬剤を対象に投与する方法(例えば静脈内と皮下のどちらにするか)と頻度に影響を与えることができるので望ましい。
【0003】
高濃度蛋白質製剤は有益であるが、高濃度治療用蛋白質を製剤化するには多くの問題がある。例えば、蛋白質濃度を上げると、蛋白質凝集、溶解度、安定性及び粘度にマイナスの影響を与えることが多い(例えばShire,et al.(2004)J Pharm Sci 93:1390参照)。高蛋白質溶液に非常に一般的な問題である粘度増加は製剤の投与にマイナスの影響(例えば疼痛感及び灼熱痛症候群や、製造、加工、充填−仕上げ及び薬剤送達装置選択の制限)を与える恐れがある(例えばShire,et al.(2004)J Pharm Sci 93:1390参照)。一般的な構造特徴をもつ治療用蛋白質(例えば抗体)でも、今日までに認可されている製剤は成分と濃度範囲が多様であった。例えば、抗CD20抗体であるリツキサンは10mg/mLの濃度で静脈内投与用に製剤化されているが、抗RSV抗体であるシナジスは100mg/mLの濃度で筋肉内投与用に製剤化されている。このように、治療目的に使用することができる高蛋白質製剤、特に抗体製剤には課題が残っている。従って、投薬及び投与上の利点を提供する安定した高濃度蛋白質製剤が必要とされている。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Wang,et al.(2007)J Pharm Sci 96:1
【非特許文献2】Shire,et al.(2004)J Pharm Sci 93:1390
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明は少なくとも部分的にヒト抗TNFα抗体又はその抗原結合フラグメント(例えばアダリムマブ)の新規高濃度製剤の発見に基づく。本発明の製剤は抗体濃度が高いため、多数の驚くべき特徴を提供する。例えば、本発明の製剤は蛋白質濃度が高いにも拘わらず、物理的及び化学的安定性を長期間維持し、皮下投与に適した粘度をもつ。本発明の製剤は少なくとも部分的にヒト抗TNFα抗体又はその抗原結合部分が高濃度(例えば100mg/mL)で可溶性に維持できると共に、注射(例えば皮下投与)に適した粘度を維持しながら非凝集状態に維持できるという驚くべき知見に基づく。本発明の製剤は、高濃度(例えば100mg/mL)のヒト抗TNFα抗体又はその抗原結合部分が可溶性に維持できると共に、広いpH範囲(例えば約pH5.2〜約pH6.0)にわたって非凝集状態で且つ化学的に安定に維持できる(例えば酸化や脱アミド化を生じない)という点も意外である。これらの有益な特徴は安定剤としてNaClを必要とせずに、糖アルコール添加剤を増加することにより達成される。
【課題を解決するための手段】
【0006】
本発明の1側面は、40mgを上回るポリオールと、少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤を提供する。
【0007】
本発明の別の側面は、20mgを上回るポリオールと、少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤を提供する。1態様において、本発明の製剤はNaClを含有しない。
【0008】
本発明は更に、pHが約5.0〜6.4であり、少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤であって、NaClを含有せず、標準24時間撹拌ストレスアッセイ後又は液体として24カ月間の長期保存後の濁度が60NTU未満である前記製剤に関する。
【0009】
本発明は更に、pHが約5.0〜6.4であり、少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤であって、NaClを含有せず、標準48時間撹拌ストレスアッセイ後の濁度が100NTU未満である前記製剤を提供する。
【0010】
本発明の別の側面は、pHが約5.0〜6.4であり、少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤であって、NaClを含有せず、5℃、25℃又は40℃で3カ月間保存の濁度が40NTU未満である前記製剤を包含する。
【0011】
本発明は更に、少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分と、約20mg/mLを上回るポリオールと、0.1〜2.0mg/mLの界面活性剤と、約1.15〜1.45mg/mLのクエン酸・H2Oと、約0.2〜0.4mg/mLのクエン酸ナトリウム脱水和物と、約1.35〜1.75mg/mLのNa2HPO4・2H2Oと、約0.75〜0.95mg/mLのNaH2PO4・2H2Oを含有する液体医薬製剤であって、pHが約4.7〜6.5であり、NaClを含有しない前記製剤を提供する。
【0012】
本発明の製剤は皮下投与に適している。従って、本発明は更に、対象における有害なTNFα活性に関連する疾患の治療における、ヒトTNFα抗体又はその抗原結合部分を含有する本発明の製剤の使用を包含する。
【0013】
1態様において、本発明の製剤は一定濃度のヒトTNFα抗体又はその抗原結合部分と、約3.1〜3.3mPa・sの粘度を有する。
【0014】
1態様において、本発明の製剤は20mgを上回るポリオールを含有する。本発明の製剤に添加することができるポリオールのその他の量は30mgを上回るポリオールである。あるいは、本発明の製剤では40mgを上回るポリオールを使用することもでき、限定されないが、40〜45mg又は約42mgが挙げられる。
【0015】
1態様において、本発明の製剤で使用されるポリオールは糖アルコールであり、限定されないが、マンニトール又はソルビトールが挙げられる。1態様において、製剤は約40〜45mg/mLのマンニトール又はソルビトールを含有する。
【0016】
本発明の製剤では当分野で公知の各種界面活性剤を使用することができる。1態様において、界面活性剤はポリソルベート80である。別の態様において、本発明の製剤では約0.1〜2.0mg/mLのポリソルベート80を使用する。
【0017】
本発明の1態様において、製剤は約1.30〜1.31mg/mLのクエン酸・H2Oを含有する。
【0018】
本発明の別の態様において、製剤は約0.30〜0.31mg/mLのクエン酸ナトリウム脱水和物を含有する。
【0019】
本発明の更に別の態様において、製剤は約1.50〜1.56mg/mLのNa2HPO4・2H2Oを含有する。
【0020】
本発明の別の態様において、製剤は約0.83〜0.89mg/mLのNaH2PO4・2H2Oを含有する。
【0021】
別の態様において、本発明の製剤のpHは約4.8〜約6.4である。例えば、本発明の製剤のpHは約5.0〜約5.4(例えば約5.2)でもよいし、約5.8〜約6.4(例えば約6.0)でもよい。
【0022】
本発明の製剤の1つの利点は、一般に蛋白質濃度の上昇と共に生じる蛋白質凝集の増加を伴わずに高濃度の抗体を提供する点である。1態様において、本発明の製剤は凝集蛋白質含有量が約1%未満である。
【0023】
本願に記載する製剤として、ヒト抗TNFα抗体又はその抗原結合部分の濃度が少なくとも約50mg/mLである製剤も本発明に含むものとする。
【0024】
1態様において、ヒト抗体又はその抗原結合部分は配列番号3に記載のアミノ酸配列を含むCDR3ドメインを含む軽鎖と、配列番号4に記載のアミノ酸配列を含むCDR3ドメインを含む重鎖を含む。
【0025】
本発明の1態様において、抗体は、配列番号3のアミノ酸配列あるいは1、4、5、7もしくは8位の1カ所のアラニン置換又は1、3、4、6、7、8及び/もしくは9位の1〜5カ所の保存アミノ酸置換により配列番号3から変異したアミノ酸配列を含む軽鎖CDR3ドメインと、配列番号4のアミノ酸配列あるいは2、3、4、5、6、8、9、10もしくは11位の1カ所のアラニン置換又は2、3、4、5、6、8、9、10、11及び/もしくは12位の1〜5カ所の保存アミノ酸置換により配列番号4から変異したアミノ酸配列を含む重鎖CDR3ドメインをもつ。
【0026】
本発明の抗体は所定の機能的特徴をもつことができる。例えば、ヒト抗体又はその抗原結合部分は、いずれも表面プラズモン共鳴法により測定した場合に、1×10−8M以下のKdでヒトTNFαから解離することができ、1×10−3s−1以下のKoff速度定数でヒトTNFαから解離することができ、及び/又は標準インビトロL929アッセイにおいて1×10−7M以下のIC50でヒトTNFα細胞傷害作用を中和することができる。
【0027】
1態様において、ヒト抗体又はその抗原結合部分はヒトIgG1κ抗体である。
【0028】
本発明の1態様において、ヒト抗体又はその抗原結合部分の軽鎖は更に配列番号5に記載のアミノ酸配列を含むCDR2ドメインと、配列番号7に記載のアミノ酸配列を含むCDR1ドメインを含み、及び/又はヒト抗体の重鎖は配列番号6に記載のアミノ酸配列を含むCDR2ドメインと、配列番号8に記載のアミノ酸配列を含むCDR1ドメインを含む。別の態様において、ヒト抗体又はその抗原結合部分の軽鎖は配列番号1に記載のアミノ酸配列を含み、ヒト抗体の重鎖は配列番号2に記載のアミノ酸配列を含む。本願に記載する各種配列番号と少なくとも80%、85%、90%、95%、96%、97%、98%又は99%一致するアミノ酸配列をもつヒト抗体又はその抗原結合部分も本発明に包含する。
【0029】
本発明の更に別の態様において、ヒト抗体又はその抗原結合部分はアダリムマブである。
【図面の簡単な説明】
【0030】
【図1】0.1% Solutolを含有する溶液中の高分子量(hmw)蛋白質試料の存在を示すグラフである。MALS(灰色の線)によると、凝集物モル質量はほぼ109g/molまでに等しく、総蛋白質の2.6%に相当する(UV280,黒線)。40℃で12週間保存。
【図2】図2A及び2Bは40℃で保存中に出現する高分子量(hmw)凝集物の初期段階検出を示すグラフである。UV280(黒曲線)では凝集物を検出できなかったが、MALS(灰色曲線)は明白にhmw試料の存在を立証した。1週間保存後(A)と元のサンプル(B)を比較。
【図3】製剤F1〜F6の凍結/解凍サイクルに対する濁度を示すグラフである。
【図4】製剤F1〜F6の凍結/解凍サイクルに対する多分散指数を示すグラフである。
【図5】製剤F1〜F6の凍結/解凍サイクルに対するSECによる凝集物濃度を示すグラフである。
【図6】T0における製剤F1〜F6のDSCによるTm(℃)を示すグラフである。
【図7】製剤F1〜F6の撹拌時間に対するSECによる凝集物濃度を示すグラフである。
【図8】F2、F6及びF7(3種類の代表的なバッチ01032〜0134)の3カ月間保存後の安定性試験で得られた濁度値の比較を示すグラフである。
【図9】F2、F6及びF7(3種類の代表的なバッチ01032〜0134)の3カ月間保存後の安定性試験で得られたDACスコアによる可視粒子値の比較を示すグラフである。
【図10】F2、F6及びF7(3種類の代表的なバッチ01032〜0134)の3カ月間保存後の安定性試験で得られた不可視粒子値(≧10μm)の比較を示すグラフである。
【図11】F2、F6及びF7(3種類の代表的なバッチ01032〜0134)の3カ月間保存後の安定性試験で得られた不可視粒子値(≧25μm)の比較を示すグラフである。
【図12】F2、F6及びF7(3種類の代表的なバッチ01032〜0134)の3カ月間保存後の安定性試験で得られた残留モノマー含有量の比較を示すグラフである。
【図13】F2、F6及びF7(3種類の代表的なバッチ01032〜0134)の3カ月間保存後の安定性試験で得られたリジン変異体合計の比較を示すグラフである。
【図14】24時間後の各種撹拌速度の撹拌ストレスに対する安定性についてF2、F6及びF7を比較した濁度データを示すグラフである。
【図15】24時間後の各種撹拌速度の撹拌ストレスに対する安定性についてF2、F6及びF7を比較したDLSデータ(Z平均値)を示すグラフである。
【図16】数種のポンプサイクル前後のストレスに対する安定性についてF2、F6及びF7を比較した濁度データを示すグラフである。
【図17】数種のポンプサイクル前後の安定性についてF2、F6及びF7を比較したDLSデータ(Z平均)を示すグラフである。
【図18】数種のポンプサイクル前後の安定性についてF2、F6及びF7を比較したSECデータ(凝集物濃度)を示すグラフである。
【図19】蠕動ポンプを使用して充填した100mg/mL製剤の目視スコアを示すグラフである。
【図20】ピストンポンプを使用して充填した100mg/mL製剤の目視スコアを示すグラフである。
【図21】蠕動ポンプを使用して充填した100mg/mL製剤の濁度を示すグラフである。
【図22】ピストンポンプを使用して充填した100mg/mL製剤の濁度を示すグラフである。
【図23】T0及び5℃で4週間保存後における製剤F8〜F11の濁度を示すグラフである。
【図24】T0及び5℃で4週間保存後における製剤F8〜F11のモノマー含有量を示すグラフである。
【図25】T0及び5℃で4週間保存後における製剤F8〜F11の凝集物濃度を示すグラフである。
【図26】T0及び5℃で4週間保存後における製剤F8〜F11の不可視粒子数を示すグラフである。
【発明を実施するための形態】
【0031】
I.定義
本発明を理解し易くするために、先ず所定の用語を定義する。更に、当然のことながら、パラメーターの数値又は数値範囲を記載する場合には、記載する数値の中間の数値及び範囲も常に本発明に含むものとする。
【0032】
「医薬製剤」なる用語は活性成分の生物活性を明白に有効にすることが可能な形態であり、製剤を投与する対象に有意に毒性となるような付加成分を含有しない製剤を意味する。
【0033】
「医薬的に許容可能な担体」なる用語は当業者に公知であり、哺乳動物に投与するのに適した医薬的に許容可能な材料、組成物又はビヒクルを包含する。担体としては、ある臓器又は生体の一部から別の臓器又は生体の一部への主剤の輸送又は移送に関与する液体又は固体増量剤、希釈剤、添加剤、溶媒又は封入材料が挙げられる。各担体は製剤の他の成分と適合可能であり、患者の安全性に有害な作用や影響を与えないという意味で「許容可能」でなければならない。
【0034】
「医薬的に許容可能な添加剤」(ビヒクル、助剤)とは、使用する活性成分の有効用量を提供するために対象哺乳動物に妥当に投与することができる添加剤である。
【0035】
「添加剤」なる用語は例えばバルク特性を改変する所望コンシステンシーを提供するため、安定性を改善するため、及び/又は浸透圧を調節するために製剤に添加することができる物質を意味する。一般に使用される添加剤の例としては、限定されないが、糖質、ポリオール、アミノ酸、界面活性剤及びポリマーが挙げられる。
【0036】
一般に使用される添加剤はポリオールである。本願で使用する「ポリオール」とは複数のヒドロキシル基をもつ物質を意味し、糖質(還元糖及び非還元糖)、糖アルコール及び糖酸を包含する。本願で好ましいポリオールは約600kD未満(例えば約120〜約400kD)の分子量を有する。ポリオールの非限定的な例はフルクトース、マンノース、マルトース、ラクトース、アラビノース、キシロース、リボース、ラムノース、ガラクトース、グルコース、スクロース、トレハロース、ソルボース、メレジトース、ラフィノース、マンニトール、キシリトール、エリスリトール、スレイトール、ソルビトール、グリセロール、L−グルコン酸及びその金属塩である。
【0037】
本願で使用する「緩衝液」とは、その酸−塩基共役成分の作用によりpHの変化を阻止する緩衝溶液を意味する。本発明の緩衝液はpHが約4〜約8、好ましくは約4.5〜約7の範囲であり、最も好ましくはpHが約5.0〜約6.5の範囲である。pHをこの範囲に制御する緩衝液の例としては、リン酸塩、酢酸塩(例えば酢酸ナトリウム)、コハク酸塩(例えばコハク酸ナトリウム)、グルコン酸塩、グルタミン酸塩、ヒスチジン、クエン酸塩及び他の有機酸緩衝液が挙げられる。1態様において、本発明の製剤で使用するのに適した緩衝液はクエン酸リン酸緩衝液である。
【0038】
「界面活性剤」なる用語は一般に空気/溶液界面に誘導されるストレスと溶液/表面に誘導されるストレスから製剤中の蛋白質を保護する物質を包含する。例えば、界面活性剤は蛋白質を凝集から保護することができる。適切な界面活性剤としては、例えばポリソルベート、ポリオキシエチレンアルキルエーテル(例えばBrij 35(登録商標))、又はポロキサマー(例えばTween 20、Tween 80又はポロキサマー188)が挙げられる。好ましい界面活性剤はポロキサマー(例えばポロキサマー188、ポロキサマー407)、ポリオキシエチレンアルキルエーテル(例えばBrij 35(登録商標)、Cremophor A25、Sympatens ALM/230)、及びポリソルベート/Tween類(例えばポリソルベート20、ポリソルベート80、Mirjとポロキサマー類(例えばポロキサマー188)及びTween類(例えばTween 20及びTween 80))である。
【0039】
「安定」した製剤とは製剤中の抗体が製造工程中及び/又は保存後にその物理的安定性及び/又は化学的安定性及び/又は生物活性を本質的に維持する製剤である。蛋白質安定性を測定するための各種分析技術が当分野で利用可能であり、Peptide and Protein Drug Delivery,247−301,Vincent Lee Ed.,Marcel Dekker,Inc.,New York,N.Y.,Pubs.(1991)及びJones,A.(1993)Adv.Drug Delivery Rev.10:29−90に記載されている。例えば、1態様において、蛋白質の安定性は溶液中のモノマー蛋白質の百分率に従って測定され、分解(例えば断片化)及び/又は凝集蛋白質の百分率が低いほうが安定である。製剤は室温(約30℃)もしくは40℃で少なくとも約1カ月間安定であり、及び/又は約2〜8℃で少なくとも約1年間もしくは少なくとも2年間安定であることが好ましい。更に、製剤は製剤の(例えば−70℃まで)凍結及び解凍(以下、「凍結/解凍サイクル」と言う)後に安定であることが好ましい。
【0040】
抗体は色及び/もしくは透明度の目視試験後、又はUV光散乱法もしくはサイズ排除クロマトグラフィーにより測定したときに例えば凝集、沈殿及び/又は変性の徴候を実質的に示さない場合に、製剤中で「その物理的安定性を維持する」。凝集とは、個々の分子又は複合体が共有的又は非共有的に会合して凝集物を形成するプロセスである。凝集は可視沈殿が形成される程度まで進行することができる。
【0041】
製剤の物理的安定性等の安定性はサンプルの見かけの光減衰(吸光度ないし光学密度)の測定を含む当分野で周知の方法により評価することができる。このような光減衰の測定は製剤の濁度に相関する。製剤の濁度は溶液に溶解した蛋白質の固有特性でもあり、一般にネフェロメトリー法により測定され、ネフェロメトリー濁度単位(NTU)で表される。
【0042】
例えば溶液中の成分の1種以上の濃度(例えば蛋白質及び/又は塩濃度)の関数としての濁度を製剤の「乳白」又は「乳白外観」とも言う。濁度は既知濁度の懸濁液を使用して作成した標準曲線を参照することにより計算することができる。医薬組成物の濁度を測定するための参照標準は欧州薬局方基準(European Pharmacopoeia,Fourth Ed.,Directorate for the Quality of Medicine of the Council of Europe(EDQM),Strasbourg,France)に準じることができる。欧州薬局方基準によると、透明溶液は欧州薬局方標準に基づいて濁度約3を有する参照懸濁液と同等以下の濁度を有する溶液として定義される。ネフェロメトリー濁度測定は会合又は非理想効果の不在下で一般に濃度と共に線形変化するレイリー散乱を検出することができる。物理的安定性を評価するための他の方法も当分野で周知である。
【0043】
所定時点の化学的安定性に関して、抗体が以下に定義するようにその生物活性を維持するとみなされるような状況である場合に、抗体は医薬製剤中で「その化学的安定性を維持する」。化学的安定性は例えば抗体の化学的改変形を検出及び定量することにより評価することができる。化学的改変としては、サイズ改変(例えば切り取り)が挙げられ、例えばサイズ排除クロマトグラフィー、SDS−PAGE及び/又はマトリックス支援レーザー脱離イオン化/飛行時間型質量分析法(MALDI/TOF MS)により測定することができる。他の型の化学的改変としては、(例えば脱アミド化又は酸化の結果として生じる)電荷改変が挙げられ、例えばイオン交換クロマトグラフィーにより評価することができる。
【0044】
医薬製剤中の抗体がその所期目的のために生物学的に活性である場合に、抗体は医薬製剤中で「その生物活性を維持する」。例えば、医薬製剤中の抗体の生物活性が(例えば抗原結合アッセイで測定したときに)医薬製剤の製造時に示される生物活性の(アッセイの誤差内で)約30%、約20%又は約10%以内にある場合に、生物活性は維持される。
【0045】
薬理学的な意味で、本発明に関して抗体の「治療有効量」又は「有効量」とは、抗体が治療に有効である疾患の症状の予防又は治療又は緩和に有効な量を意味する。「疾患」とは抗体投与が有効となる任意病態である。これは慢性及び急性疾患ないし疾病を含み、該当疾患の素因を対象に与える病態を含む。
【0046】
「治療」とは治療処置と予防ないし防止措置の両方を意味する。治療を必要とする対象としては、既に疾患に罹患している対象と、疾患を予防すべき対象が挙げられる。
【0047】
本願で使用する「非経口投与」及び「非経口投与する」なる用語は経腸管及び局所投与以外の投与方法を意味し、通常は注射による投与方法であり、限定されないが、静脈内、筋肉内、動脈内、髄腔内、関節包内、眼窩内、心臓内、真皮内、腹腔内、経気管、皮下、表皮下、関節内、関節包下、クモ膜下、頭蓋内、関節内、脊髄内及び胸骨内注射及び輸液が挙げられる。
【0048】
本願で使用する「全身投与」、「全身投与する」、「末梢投与」及び「末梢投与する」なる用語は、化合物、薬剤及び他の材料が患者の生体系に入り、従って、代謝等のプロセスを受けるように、中枢神経系への直接投与以外の方法で投与することを意味する(例えば皮下投与)。
【0049】
本願で使用する「ヒトTNFα」(本願ではhTNFα、TNFα、又は単にhTNFと略称する)なる用語は17kD分泌型及びその生物活性型が非共有的に結合した17kD分子の三量体から構成される26kD膜結合型として存在するヒトサイトカインを意味する。hTNFαの構造は例えばPennica,D.,et al.(1984)Nature 312:724−729;Davis,J.M.,et al.(1987)Biochem 26:1322−1326;及びJones,E.Y.,et al.(1989)Nature 338:225−228に詳細に記載されている。ヒトTNFαなる用語は標準組換え発現法により作製することもできるし、市販品(R & D Systems,Catalog No.210−TA,Minneapolis,Minn.)として購入することもできる組換えヒトTNFα(rhTNFα)を包含するものとする。
【0050】
本願で使用する「抗体」なる用語はジスルフィド結合により相互に結合した2本の重(H)鎖と2本の軽(L)鎖からなる4本のポリペプチド鎖から構成される免疫グロブリン分子を意味する。構造の異なる他の天然抗体(例えばラクダ抗体)もこの定義に含まれる。各重鎖は重鎖可変領域(本願ではHCVR又はVHと略称する)と重鎖定常領域から構成される。重鎖定常領域はCH1、CH2及びCH3の3個のドメインから構成される。各軽鎖は軽鎖可変領域(本願ではLCVR又はVLと略称する)と軽鎖定常領域から構成される。軽鎖定常領域は1個のドメインCLから構成される。VH領域とVL領域はフレームワーク領域(FR)と呼ばれる保存度の高い領域を挟んで相補性決定領域(CDR)と呼ばれる超可変領域に更に分割することができる。各VH及びVLはアミノ末端からカルボキシ末端に向かってFR1、CDR1、FR2、CDR2、FR3、CDR3、FR4の順に配置された3個のCDRと4個のFRから構成される。本発明の1態様において、製剤は各々本願に援用する米国特許第6,090,382号及び6,258,562号に記載されている配列等のCDR1、CDR2及びCDR3配列をもつ抗体を含む。
【0051】
本願で使用する「CDR」なる用語は抗体可変配列内の相補性決定領域を意味する。重鎖と軽鎖の可変領域の各々に3個のCDRが存在し、可変領域の各々についてCDR1、CDR2及びCDR3と呼ぶ。これらのCDRの厳密な境界は種々のシステムに従って種々に定義されている。Kabat(同書)により記載されているシステムは抗体の任意可変領域に適用可能な明白な残基ナンバリングシステムを提供するのみならず、3個のCDRを規定する厳密な残基境界も提供する。これらのCDRをKabat CDRと言う場合もある。ChothiaらはKabat CDR内の所定のサブ部分がアミノ酸配列レベルでは多様性が高いにも拘わらず、ほぼ同一のペプチド骨格構造をとることを見出した(Chothia et al.(1987)Mol.Biol.196:901−917;Chothia et al.(1989)Nature 342:877−883)。これらのサブ部分はL1、L2及びL3又はH1、H2及びH3と呼ばれ、ここで「L」及び「H」は夫々軽鎖領域と重鎖領域を意味する。これらの領域をChothia CDRと言う場合もあり、その境界はKabat CDRとオーバーラップする。Kabat CDRとオーバーラップするCDRを規定する他の境界もPadlan(1995)FASEB J.9:133−139及びMacCallum(1996)J.Mol.Biol.262(5):732−45に記載されている。本願に記載するシステムと厳密に一致しない場合もあるが、Kabat CDRとオーバーラップする更に他のCDR境界定義もあり、特定残基もしくは残基群又はCDR全体が抗原結合にさほど影響しないという予想又は実験結果を踏まえると、CDRを短縮又は延長してもよい。本願で使用する方法はこれらのシステムのいずれかに従って定義されるCDRを利用することができるが、所定の態様はKabat又はChothiaにより定義されるCDRを使用する。
【0052】
本願で使用する抗体の「抗原結合部分」(又は単に「抗体部分」)なる用語は抗原(例えばhTNFα)と特異的に結合する能力を保持する抗体の1個以上のフラグメントを意味する。全長抗体のフラグメントにより抗体の抗原結合機能を実施できることが分かっている。抗体の「抗原結合部分」なる用語に含まれる結合フラグメントの例としては、(i)VL、VH、CL及びCH1ドメインから構成される1価フラグメントであるFabフラグメント;(ii)ヒンジ領域でジスルフィド橋により結合した2個のFabフラグメントから構成される2価フラグメントであるF(ab’)2フラグメント;(iii)VHドメインとCH1ドメインから構成されるFdフラグメント;(iv)抗体のシングルアームのVLドメインとVHドメインから構成されるFvフラグメント;(v)VHドメインから構成されるdAbフラグメント(Ward et al.,(1989)Nature 341:544−546);並びに(vi)単離された相補性決定領域(CDR)が挙げられる。更に、Fvフラグメントの2領域であるVLとVHは別々の遺伝子によりコードされるが、組換え法を使用し、VL領域とVH領域が対合して1価分子(1本鎖Fv(scFv)として知られる;例えばBird et al.(1988)Science 242:423−426;及びHuston et al.(1988)Proc.Natl.Acad.Sci.USA 85:5879−5883参照)を形成する1本の蛋白質鎖とすることが可能な合成リンカーにより結合することができる。このような1本鎖抗体も抗体の「抗原結合部分」なる用語に含むものとする。ダイアボディ等の他の型の1本鎖抗体も包含する。ダイアボディはVHドメインとVLドメインが1本のポリペプチド鎖上で発現される2価の二重特異性抗体であるが、同一鎖上の2領域間を対合させるには短いリンカーを使用することにより、これらの領域を別の鎖の相補領域と対合させ、2個の抗原結合部位を形成する(例えばHolliger,P.,et al.(1993)Proc.Natl.Acad.Sci.USA 90:6444−6448;Poljak,R.J.,et al.(1994)Structure 2:1121−1123参照)。本発明の1態様において、製剤は各々本願に援用する米国特許第6,090,382号及び6,258,562号に記載されている抗原結合部分を含有する。
【0053】
更に、抗体又はその抗原結合部分は抗体又は抗体部分と1個以上の他の蛋白質又はペプチドの共有的又は非共有的会合により形成される大きな免疫接着分子の一部でもよい。このような免疫接着分子の例としては、ストレプトアビジンコア領域を使用して四量体scFv分子を作製する場合(Kipriyanov,S.M.,et al.(1995)Human Antibodies and Hybridomas 6:93−101)や、システイン残基、マーカーペプチド及びC末端ポリヒスチジンタグを使用して2価のビオチン化scFv分子を作製する場合(Kipriyanov,S.M.,et al.(1994)Mol.Immunol.31:1047−1058)が挙げられる。Fab及びF(ab’)2フラグメント等の抗体部分は全長抗体の夫々パパイン又はペプシン消化等の従来技術を使用して全長抗体から作製することができる。更に、抗体、抗体部分及び免疫接着分子は本願に記載するような標準組換えDNAを使用して得ることができる。
【0054】
本願で使用する「ヒト抗体」なる用語はヒト生殖系列免疫グロブリン配列に由来する可変領域と定常領域をもつ抗体を包含する。本発明で使用されるヒト抗体はヒト生殖系列免疫グロブリン配列によりコードされないアミノ酸残基(例えばランダムもしくは部位特異的突然変異誘発によりインビトロ又は体細胞突然変異によりインビボ導入される突然変異)を例えばCDR、特にCDR3に含むことができる。他方、本願で使用する「ヒト抗体」なる用語はマウス等の別の哺乳動物種の生殖系列に由来するCDR配列をヒトフレームワーク配列に移植した抗体を包含しない。
【0055】
本願で使用する「組換えヒト抗体」なる用語は組換え手段により作製、発現、創製又は単離される全ヒト抗体を包含し、例えば宿主細胞にトランスフェクトした組換え発現ベクターを使用して発現される抗体(下記セクションIIに詳述)、組換えコンビナトリアルヒト抗体ライブラリーから単離される抗体(下記セクションIIIに詳述)、ヒト免疫グロブリン遺伝子に対してトランスジェニックな動物(例えばマウス)から単離される抗体(例えばTaylor,L.D.,et al.(1992)Nucl.Acids Res.20:6287−6295参照)又はヒト免疫グロブリン遺伝子配列と他のDNA配列のスプライシングを含む他の任意手段により作製、発現、創製もしくは単離される抗体が挙げられる。このような組換えヒト抗体はヒト生殖系列免疫グロブリン配列に由来する可変領域と定常領域をもつ。他方、所定態様では、このような組換えヒト抗体をインビトロ突然変異誘発(又は、ヒトIg配列に対してトランスジェニックな動物を使用する場合には、インビボ体細胞突然変異誘発)するため、組換え抗体のVH領域とVL領域のアミノ酸配列はヒト生殖系列VH配列及びVL配列に由来及び関連するが、ヒト抗体生殖系列レパートリー内に天然にインビボ存在し得ない配列となる。
【0056】
本願で使用する「単離抗体」とは、異なる抗原特異性をもつ他の抗体を実質的に含まない抗体を意味する(例えば、hTNFαと特異的に結合する単離抗体はhTNFα以外の抗原と特異的に結合する抗体を実質的に含まない)。他方、hTNFαと特異的に結合する単離抗体は他の種に由来するTNFα分子等の他の抗原に対して交差反応性をもつ場合がある。更に、単離抗体は他の細胞材料及び/又は薬品を実質的に排除することができる。
【0057】
本願で使用する「中和抗体」(又は「hTNFα活性を中和した抗体」)とはhTNFαと結合する結果、hTNFαの生物活性を阻害する抗体を意味する。hTNFαの生物活性のこの阻害はhTNFαにより(インビトロ又はインビボで)誘導される細胞傷害作用、hTNFαにより誘導される細胞活性化及びhTNFαとhTNFα受容体の結合等のhTNFα生物活性の1種以上の指標を測定することにより評価することができる。hTNFα生物活性のこれらの指標は各々本願に援用する米国特許第6,090,382号及び6,258,562号に記載されている当分野で公知の数種類の標準インビトロ又はインビボアッセイの1種以上により評価することができる。抗体がhTNFα活性を中和する能力は、hTNFαにより誘導されるL929細胞の細胞傷害作用の阻害により評価することが好ましい。hTNFα活性の付加又は代替パラメーターとして、抗体がHUVEC上でhTNFαにより誘導されるELAM−1の発現を阻害する能力は、hTNFαにより誘導される細胞活性化の指標として評価することができる。
【0058】
本願で使用する「表面プラズモン共鳴法」なる用語は例えばBIAcoreシステム(Pharmacia Biosensor AB,Uppsala,Sweden及びPiscataway,N.J.)を使用してバイオセンサーマトリックス内の蛋白質濃度の変化の検出によりリアルタイム生体特異的相互作用の分析を可能にする光学現象を意味する。詳細については、Jonsson,U.,et al.(1993)Ann.Biol.Clin.51:19−26;Jonsson,U.,et al.(1991)Biotechniques 11:620−627;Johnsson,B.,et al.(1995)J.Mol.Recognit.8:125−131;及びJohnnson,B.,et al.(1991)Anal.Biochem.198:268−277参照。
【0059】
本願で使用する「Kon」なる用語は当分野で公知のように結合性蛋白質(例えば抗体)が抗原と会合して例えば抗体/抗原複合体を形成する会合速度定数を意味する。
【0060】
本願で使用する「Koff」なる用語は抗体が抗体/抗原複合体から解離する解離速度定数を意味する。
【0061】
本願で使用する「Kd」なる用語は特定抗体−抗原相互作用の解離定数を意味し、平衡状態の滴定測定又は解離速度定数(koff)を会合速度定数(kon)で割ることにより得られる値を意味する。
【0062】
以下のサブセクションでは本発明の各種側面について更に詳細に記載する。
【0063】
II.本発明の製剤
本発明は当業者に公知の製剤に比較して特性を改善した液体医薬製剤(例えば抗体製剤)に関する。本発明はNaClを除去し、20mg/mLを上回るポリオール(例えば糖アルコール)を加えることにより、製剤中のヒトTNFα抗体の濃度を約100mg/mLまで上げることができるという驚くべき知見に基づく。抗体濃度が高いにも拘わらず、本発明の製剤は例えば製造、保存及び/もしくは反復凍結/解凍処理工程中又は拡大した気液界面への長期暴露中に溶解度と安定性を維持することが可能である。更に、本発明の製剤は約100mg/mLの抗体濃度でありながら、低レベル蛋白質凝集(即ち1%未満)を維持する。本発明の製剤は更に、驚くべきことに、約100mg/mLの抗体濃度でありながら、皮下注射に適した範囲内の低粘度を維持する。更に、本発明の製剤(例えば高濃度TNFα抗体)は皮下注射に適した低粘度を維持すると共に、ほぼ1のpH範囲(例えばpH5.2〜pH6.0)にわたって安定性を維持する。1態様において、製剤の濁度は標準48時間撹拌ストレスアッセイ後に100NTU未満である。従って、本発明の高濃度抗体製剤は安定性、粘度、濁度及び物理的劣化の問題を含む製剤の多数の公知課題を解決する。
【0064】
本発明の製剤の驚くべき特徴の1つは、高い抗体濃度(例えば100mg/mL以上)でありながらNaClの不在下で製剤の総粘度が低く維持される点である(例えば約3.1〜3.3mPa・s、例えば約3.00、3.05、3.10、3.15、3.20、3.25、3.30、3.35又は約3.40mPa・s)。一般に、粘度は蛋白質濃度の増加と共に増加する(詳細については、Shire et al.(2004)J Pharm Sci 93:1390参照)。このような増加は殆どの場合、イオン性添加剤(例えばNaClやMgCl2)を添加することにより抑制されるが、このような添加剤を添加すると、溶液の濁度も上昇する可能性がある。濁度上昇は不溶性蛋白質凝集物、沈殿又は蛋白質粒子の形成(例えば凝集)と関係していることが多い。従って、本発明の液体医薬製剤はNaClを添加する必要なしに、皮下投与に適した粘度で高い抗体濃度(例えば少なくとも100mg/mL)を実現する。
【0065】
1態様において、本発明の製剤は液体製剤が有意乳白、凝集又は沈殿を示さないような高濃度の蛋白質を含有する。
【0066】
別の態様において、本発明の製剤は(例えば目視アナログスケール(VAS)スコアにより判定した場合に)有意疼痛感覚なしに例えば皮下投与に適するような高濃度の蛋白質を含有する。
【0067】
本発明の製剤は例えば約50mg/mL又は約100mg/mLの蛋白質濃度のヒト抗TNFα抗体又はその抗原結合フラグメントを含む高濃度蛋白質を含有する。従って、下記実施例1に記載するように、本発明の1側面において、液体医薬製剤は約50mg/mLの濃度のヒト抗TNFα抗体を含有する。下記実施例2〜6に記載するように、本発明の別の側面において、液体医薬製剤は約100mg/mLの濃度のヒト抗TNFα抗体を含有する。本発明の更に別の側面において、液体医薬製剤は約150mg/mLの濃度のヒト抗TNFα抗体を含有する。本発明の好ましい態様は高濃度蛋白質を含有する製剤であるが、本発明の製剤が約1mg/mL〜約150mg/mL又は約40mg/mL〜125mg/mLの抗体濃度となり得ることも考えられる。上記濃度の中間の濃度及び範囲(例えば1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、101、102、103、104、105、106、107、108、109、110、111、112、113、114、115、116、117、118、119、120、121、122、123、124、125、126、127、128、129、130、131、132、133、134、135、136、137、138、139、140、141、142、143、144、145、146、147、148、149、150、151、152、153、154、155、156、157、158、159、160、161、162、163、164、165、166、167、168、169、170、171、172、173、174、175、176、177、178、179、180、181、182、183、184、185、186、187、188、189、190、191、192、193、194、195、196、197、198、199又は200mg/mL)も本発明に含むものとする。
【0068】
別の側面において、本発明は例えば約100mg/mLを上回る濃度の治療用抗体(例えばアダリムマブ)を製剤化するために十分な量のポリオール、界面活性剤及び緩衝液系を含有する液体医薬組成物を提供する。1態様において、液体医薬組成物はNaClを含有しない。
【0069】
但し、当然のことながら、本発明の好ましい製剤はNaClを含有しないが、少量(例えば約0.01mM〜約300mM)のNaClが製剤中に存在していてもよい。更に、上記数値の中間の任意量のNaClも含むものとする。
【0070】
1側面において、本発明はヒト抗TNFα抗体又はその抗原結合フラグメント(例えばアダリムマブ)と、NaClの不在下で治療用抗体を製剤化するために十分な量のポリオールを含有する液体医薬組成物を提供する。
【0071】
本発明は更に、ヒト抗TNFα抗体又はその抗原結合フラグメントを含有する液体製剤であって、pHが約5.0〜6.4であり、NaClを添加せずに標準24時間撹拌ストレスアッセイ後の濁度が約60NTU未満(例えば約20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37.38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62又は63NTU)である製剤を提供する。別の側面において、本発明はヒト抗TNFα抗体又はその抗原結合フラグメントを含有する液体製剤であって、pHが約5.0〜6.4であり、NaClを添加せずに標準48時間撹拌ストレスアッセイ後の濁度が約100NTU未満(例えば約35、36、37.38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99又は100NTU)である製剤を提供する。更に別の側面において、本発明はヒト抗TNFα抗体又はその抗原結合フラグメントを含有する液体製剤であって、pHが約5.0〜6.4であり、NaClを添加せずに5℃、25℃又は40℃で3カ月間保存の濁度が約40NTU未満(例えば約20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37.38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60NTU)である製剤を提供する。
【0072】
本発明の製剤の1つの特徴は20mg/mLを上回る濃度のポリオール(例えば糖アルコール)を含有する点にある。1態様において、ポリオールはソルビトール又はマンニトールである。なお、蛋白質溶液にソルビトール又はマンニトールを添加しても必ずしも蛋白質安定性が増すわけではない。例えば、熱又は界面ストレス条件下で評価した場合にソルビトールは夫々Tween 20とヒドロキシプロピル−β−シクロデキストリンに比較してブタ成長ホルモンの沈殿に対する効果がなかった(Charman et al.(1993)Pharm Res.10(7):954−62)。
【0073】
1態様において、本発明の製剤で使用するのに適切なポリオールは糖アルコール(例えばマンニトール又はソルビトール)である。ポリオールを含有する本発明の液体製剤は一般に約20mgを上回るポリオールを含有する。1態様において、製剤は約30mg/mLを上回るポリオールを含有する。別の態様において、製剤は約40mg/mLを上回るポリオールを含有する。別の態様において、製剤は約40〜45mg/mL、例えば約35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54又は55mg/mLのポリオールを含有する。更に、上記数値のいずれかの組合せを上限及び/又は下限として使用する数値範囲も含むものとする。
【0074】
本発明の所定態様では、pH緩衝溶液中に抗体を含有する液体製剤を製造する。本発明の緩衝液はpH約4〜約8、好ましくは約4.5〜約7.0、より好ましくは約4.5〜約6.0、更に好ましくは約4.8〜約5.5であり、最も好ましくはpH約5.0〜約6.4である。1態様において、本発明の製剤のpHは約5.2である。別の態様において、本発明の製剤のpHは約6.0である。上記pH値の中間の範囲(例えば4.5、4.6、4.7、4.8、4.9、5.0、5.1、5.2、5.3、5.4、5.5、5.6、5.7、5.8、5.9、6.0、6.1、6.2、6.3、6.4)も本発明に含むものとする。上記数値のいずれかの組合せを上限及び/又は下限として使用する数値範囲(例えば5.2〜5.8)も含むものとする。pHをこの範囲内に制御する緩衝液の例としては、リン酸塩、酢酸塩(例えば酢酸ナトリウム)、コハク酸塩(例えばコハク酸ナトリウム)、グルコン酸塩、グルタミン酸塩、ヒスチジン、クエン酸塩及び他の有機酸緩衝液が挙げられる。
【0075】
本発明の特定態様において、製剤はpHを約5.0〜約6.4の範囲に維持するためにクエン酸塩及び/又はリン酸塩を含む緩衝液系を含有する。1態様において、製剤のpHは約5.2である。別の態様において、製剤のpHは約6.0である。
【0076】
別の好ましい態様において、緩衝液系はクエン酸一水和物、クエン酸ナトリウム、リン酸二ナトリウム二水和物、及び/又はリン酸二水素ナトリウム二水和物を含む。別の好ましい態様において、緩衝液系は約1.15〜1.45(例えば約1.15、1.20、1.25、1.30、1.35、1.40又は1.45)mg/mlのクエン酸と、約0.2〜0.4(例えば約0.2、0.25、0.3、0.35又は0.4)mg/mLのクエン酸ナトリウム脱水和物と、約1.35〜1.75(例えば1.35、1.40、1.45、1.50、1.55、1.60、1.65、1.70又は1.75)mg/mLのリン酸二ナトリウム脱水和物と、約0.75〜0.95(例えば約0.75、0.80、0.85、0.9又は0.95)mg/mLのリン酸二水素ナトリウム脱水和物を含む。
【0077】
上記濃度の中間の数値及び範囲も本発明に含むものとする。更に、上記数値のいずれかの組合せを上限及び/又は下限として使用する数値範囲(例えば1.20〜1.40mg/mL)も含むものとする。
【0078】
他の態様において、緩衝液系は1.3〜1.31mg/mL(例えば約1.305mg/mL)のクエン酸を含む。別の態様において、緩衝液系は約0.27〜0.33mg/mL(例えば約0.305mg/mL)のクエン酸ナトリウム脱水和物を含む。1態様において、緩衝液系は約1.5〜1.56mg/mL(例えば約1.53mg/mL)のリン酸二ナトリウム脱水和物を含む。別の態様において、緩衝液系は約0.83〜0.89mg/mL(例えば約0.86mg/mL)のリン酸二水素ナトリウム二水和物を含む。
【0079】
デタージェントないし界面活性剤も本発明の抗体製剤に添加することができる。典型的な界面活性剤としては、ポリソルベート(例えばポリソルベート20、80等)やポロキサマー(例えばポロキサマー188)等の非イオン性界面活性剤が挙げられる。界面活性剤の添加量は製剤化抗体の凝集を抑え、及び/又は製剤中の粒状物の形成を最小限にし、及び/又は吸着を減らすように選択する。本発明の好ましい1態様において、製剤はポリソルベートである界面活性剤を含有する。本発明の別の好ましい態様において、製剤は界面活性剤としてポリソルベート80を含有する。好ましい1態様において、製剤は約0.1〜約2.0mg/mL(例えば約1mg/mL)のポリソルベート80を含有する。
【0080】
上記濃度の中間の数値及び範囲(例えば0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9)も本発明に含むものとする。更に、上記数値のいずれかの組合せを上限及び/又は下限として使用する数値範囲(例えば0.3〜1.1mg/mL)も含むものとする。
【0081】
1態様において、本発明の製剤は少なくとも約100mg/mLの濃度のヒトTNFα抗体又はその抗原結合部分と、界面活性剤(例えばポリソルベート80)と、ポリオール(例えば20mg/mLを上回るソルビトール又はマンニトール)と、緩衝液系(例えばクエン酸一水和物、クエン酸ナトリウム、リン酸二ナトリウム二水和物及び/又はリン酸二水素ナトリウム二水和物)から本質的に構成され、NaClを含有しない。
【0082】
1態様において、製剤は上記成分を含有し(即ち少なくとも約100mg/mLの濃度の抗体と、緩衝液系と、ポリオールと、界面活性剤を含有し、NaClを含有しない)、ベンジルアルコール、フェノール、m−クレゾール、クロロブタノール及びベンゼトニウムCl等の防腐剤を実質的に含有しない。別の態様では、防腐剤を製剤に加えることができる。製剤の望ましい特性を著しく悪化させない限り、Remington’s Pharmaceutical Sciences 16th edition,Osol,A.Ed.(1980)に記載されているもの等の1種以上の他の医薬的に許容可能な担体、添加剤又は安定剤も製剤に加えることができる。許容可能な担体、添加剤又は安定剤は使用する用量と濃度でレシピエントに非毒性であり、他の緩衝剤;補助溶媒;アスコルビン酸やメチオニン等の酸化防止剤;EDTA等のキレート剤;金属錯体(例えばZn−蛋白質錯体);ポリエステル等の生体適合性ポリマー;及び/又はナトリウム等の塩形成対イオンを含む。
【0083】
本願の製剤は治療する特定適応症の必要に応じて1種以上の他の治療剤、好ましくは製剤の抗体に悪影響を与えない相補活性をもつ治療剤と併用してもよい。このような治療剤は所期目的に有効な量で併存すると適切である。本発明の製剤と併用することができる他の治療剤は各々本願に援用する米国特許第6,090,382号及び6,258,562号に詳細に記載されている。
【0084】
インビボ投与に使用する製剤は無菌でなければならない。これは製剤の製造前又は製造後に滅菌濾過膜で濾過することにより容易に実施される。
【0085】
上記のように、本発明の液体製剤は有利な安定性と保存性をもつ。液体製剤の安定性は保存形態に依存せず、限定されないが、凍結製剤、凍結乾燥製剤、噴霧乾燥製剤、又は活性成分を懸濁した製剤が挙げられる。安定性は選択された温度で選択された期間にわたって測定することができる。本発明の1側面において、液体製剤中の蛋白質は液体形態で少なくとも約3カ月間、少なくとも約4カ月間、少なくとも約5カ月間、少なくとも約6カ月間、少なくとも約12カ月間、少なくとも約18カ月間安定である。上記期間の中間の数値及び範囲(例えば約3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23又は約24カ月間)も本発明に含むものとする。更に、上記数値のいずれかの組合せを上限及び/又は下限として使用する数値範囲も含むものとする。好ましくは、製剤は室温(約30℃)もしくは40℃で少なくとも約1カ月間安定であり、及び/又は約2〜8℃で少なくとも約1年間安定であり、又はより好ましくは約2〜8℃で少なくとも約2年間安定である。更に、製剤は製剤の(例えば−80℃まで)凍結及び解凍(以下、「凍結/解凍サイクル」と言う)後に安定であることが好ましい。
【0086】
液体製剤中の蛋白質の安定性は製剤中の蛋白質のモノマー、凝集物もしくはフラグメント又はその組合せの百分率として表すこともできる。蛋白質は色及び/もしくは透明度の目視試験後、又はUV光散乱法もしくはサイズ排除クロマトグラフィーにより測定したときに凝集、沈殿及び/又は変性の徴候を実質的に示さない場合に、製剤中で「その物理的安定性を維持する」。本発明の1側面において、安定した液体製剤とは製剤中に凝集物として存在する蛋白質が約10%未満、好ましくは約5%未満の製剤である。
【0087】
1態様において、液体製剤の物理的安定性は撹拌ストレスアッセイ(例えば24時間又は48時間撹拌ストレスアッセイ)後に製剤の濁度を測定することにより測定される。例えば、磁気スターラー(例えばマルチポイントHP,550rpm)を取り付けたビーカーに適切な容量の液体製剤を入れ、適切な任意時点(例えばT0〜T48(時間))でアリコートを分取し、アリコートに応じて適切なアッセイを実施することにより、撹拌ストレスアッセイを実施することができる。同一条件下で非撹拌下の製剤のサンプルを対照とする。
【0088】
Hach(ドイツ)製実験室濁度測定システムを使用して濁度測定を実施することができ、ネフェロメトリー単位(NTU)として報告する。
【0089】
本発明の液体製剤は有利な耐容性も有する。耐容性は疼痛視覚的アナログスケール(VAS)を使用して注射部位疼痛を知覚した対象の評価に基づいて評価される。
【0090】
(VAS)は例えば無痛から極限量の疼痛までの一連の値にわたって疼痛を測定する計器である。操作上、VASは長さ約100mmの水平線であり、数値及び/又は語句表記(例えば0もしくは10、又は「無痛」もしくは「耐え難い痛み」)を表示され、場合により極限間の他の語句又は数値表記(例えば、軽度、中度及び重度;又は1〜9)を加えてもよい(例えばLee JS,et al.(2000)Acad Emerg Med 7:550参照)。
【0091】
測定することができる耐容性の他の指標としては、例えばドレイズスケール(出血、点状出血、紅斑、浮腫、痒み)と皮下出血が挙げられる。
【0092】
III.本発明の製剤で使用される抗体
本発明の製剤で使用することができる抗体はヒトTNFα(ないしhTNFα)を含む抗原TNFαに対する抗体である。
【0093】
1態様において、本発明は高い親和性と低い解離速度でヒトTNFαと結合すると共に、高い中和能をもつ単離ヒト抗体又はその抗原結合部分に関する。好ましくは、本発明で使用されるヒト抗体は組換え中和ヒト抗hTNFα抗体である。本発明の最も好ましい組換え中和抗体を本願ではD2E7と言い、HUMIRA(登録商標)ないしアダリムマブとも言う(D2E7 VL領域のアミノ酸配列を配列番号1に示し、D2E7 VH領域のアミノ酸配列を配列番号2に示す)。D2E7(アダリムマブ/HUMIRA(登録商標))の特性は各々本願に援用するSalfeldらの米国特許第6,090,382号、6,258,562号及び6,509,015号に記載されている。
【0094】
1態様において、ヒトTNFα又はその抗原結合部分は、いずれも表面プラズモン共鳴法により測定した場合に、1×10−8M以下のKdと1×10−3s−1以下のKoff速度定数でヒトTNFαから解離し、標準インビトロL929アッセイにおいて1×10−7M以下のIC50でヒトTNFα細胞傷害作用を中和する。より好ましくは、単離ヒト抗体又はその抗原結合部分は5×10−4s−1以下のKoff、更に好ましくは1×10−4s−1以下のKoffでヒトTNFαから解離する。より好ましくは、単離ヒト抗体又はその抗原結合部分は標準インビトロL929アッセイにおいて1×10−8M以下のIC50、更に好ましくは1×10−9M以下のIC50、更に好ましくは1×10−10M以下のIC50でヒトTNFα細胞傷害作用を中和する。好ましい1態様において、抗体は単離ヒト組換え抗体又はその抗原結合部分である。
【0095】
抗体重鎖及び軽鎖CDR3ドメインが抗原に対する抗体の結合特異性/親和性において重要な役割を果たすことは周知である。従って、別の側面において、本発明はhTNFαとの会合の解離速度が遅く、D2E7と構造的に一致又は関連する軽鎖及び重鎖CDR3ドメインをもつヒト抗体を投与することによるクローン病の治療に関する。Koffを実質的に変化させずにD2E7 VL CDR3の9位をAla又はThrで占めることができる。従って、D2E7 VL CDR3のコンセンサスモチーフはアミノ酸配列Q−R−Y−N−R−A−P−Y−(T/A)(配列番号3)を含む。更に、Koffを実質的に変化させずにD2E7 VH CDR3の12位をTyr又はAsnで占めることができる。従って、D2E7 VH CDR3のコンセンサスモチーフはアミノ酸配列V−S−Y−L−S−T−A−S−S−L−D−(Y/N)(配列番号4)を含む。更に、米国特許第6,090,382号の実施例2に実証されているように、Koffを実質的に変化させずにD2E7重鎖及び軽鎖のCDR3ドメイン(VL CDR3内の1、4、5、7もしくは8位又はVH CDR3内の2、3、4、5、6、8、9、10もしくは11位)を1個のアラニン残基で置換することができる。更に、当業者に自明の通り、D2E7 VL及びVH CDR3ドメインをアラニンで置換できるならば、抗体の低い解離速度定数を維持しながらCDR3ドメイン内の他のアミノ酸の置換、特に保存アミノ酸による置換も可能であると思われる。D2E7 VL及び/又はVH CDR3ドメイン内に1〜5カ所以下の保存アミノ酸置換を導入することが好ましい。D2E7 VL及び/又はVH CDR3ドメイン内に1〜3カ所以下の保存アミノ酸置換を導入することがより好ましい。更に、hTNFαとの結合に必須のアミノ酸位置には保存アミノ酸置換を導入すべきでない。D2E7 VL CDR3の2位及び5位と、D2E7 VH CDR3の1位及び7位はhTNFαとの相互作用に必須であると思われるため、これらの位置には保存アミノ酸置換を導入しない(但し、上記のように、D2E7 VL CDR3の5位のアラニン置換は許容可能である)(米国特許第6,090,382号参照)。
【0096】
従って、別の態様において、抗体又はその抗原結合部分は以下の特徴を含むことが好ましい:
a)表面プラズモン共鳴法により測定した場合に、1×10−3s−1以下のKoff速度定数でヒトTNFαから解離する;
b)配列番号3のアミノ酸配列あるいは1、4、5、7もしくは8位の1カ所のアラニン置換又は1、3、4、6、7、8及び/もしくは9位の1〜5カ所の保存アミノ酸置換により配列番号3から変異したアミノ酸配列を含む軽鎖CDR3ドメインをもつ;
c)配列番号4のアミノ酸配列あるいは2、3、4、5、6、8、9、10もしくは11位の1カ所のアラニン置換又は2、3、4、5、6、8、9、10、11及び/もしくは12位の1〜5カ所の保存アミノ酸置換により配列番号4から変異したアミノ酸配列を含む重鎖CDR3ドメインをもつ。
【0097】
より好ましくは、抗体又はその抗原結合部分は5×10−4s−1以下のKoffでヒトTNFαから解離する。更により好ましくは、抗体又はその抗原結合部分は1×10−4s−1以下のKoffでヒトTNFαから解離する。
【0098】
更に別の態様において、抗体又はその抗原結合部分は好ましくは配列番号3のアミノ酸配列又は1、4、5、7もしくは8位の1カ所のアラニン置換により配列番号3から変異したアミノ酸配列を含むCDR3ドメインをもつ軽鎖可変領域(LCVR)と、配列番号4のアミノ酸配列又は2、3、4、5、6、8、9、10もしくは11位の1カ所のアラニン置換により配列番号4から変異したアミノ酸配列を含むCDR3ドメインをもつ重鎖可変領域(HCVR)を含む。好ましくは、LCVRは更に配列番号5のアミノ酸配列を含むCDR2ドメイン(即ちD2E7 VL CDR2)をもち、HCVRは更に配列番号6のアミノ酸配列を含むCDR2ドメイン(即ちD2E7 VH CDR2)をもつ。更により好ましくは、LCVRは更に配列番号7のアミノ酸を含むCDR1ドメイン(即ちD2E7 VL CDR1)をもち、HCVRは配列番号8のアミノ酸配列を含むCDR1ドメイン(即ちD2E7 VH CDR1)をもつ。VLのフレームワーク領域は好ましくはVκIヒト生殖系列ファミリーに由来し、より好ましくはA20ヒト生殖系列Vk遺伝子に由来し、最も好ましくは米国特許第6,090,382号の図1A及び1Bに示されるD2E7 VLフレームワーク配列に由来する。VHのフレームワーク領域は好ましくはVH3ヒト生殖系列ファミリーに由来し、より好ましくはDP−31ヒト生殖系列VH遺伝子に由来し、最も好ましくは米国特許第6,090,382号の図2A及び2Bに示されるD2E7 VHフレームワーク配列に由来する。
【0099】
従って、別の態様において、抗体又はその抗原結合部分は好ましくは配列番号1のアミノ酸配列を含む軽鎖可変領域(LCVR)(即ちD2E7 VL)と、配列番号2のアミノ酸配列を含む重鎖可変領域(HCVR)(即ちD2E7 VH)を含む。所定態様において、抗体はIgG1、IgG2、IgG3、IgG4、IgA、IgE、IgM又はIgD定常領域等の重鎖定常領域を含む。好ましくは、重鎖定常領域はIgG1重鎖定常領域又はIgG4重鎖定常領域である。更に、抗体はκ軽鎖定常領域又はλ軽鎖定常領域である軽鎖定常領域を含むことができる。好ましくは、抗体はκ軽鎖定常領域を含む。あるいは、抗体部分は例えばFabフラグメント又は1本鎖Fvフラグメントとすることができる。
【0100】
更に他の態様において、本発明はD2E7関連VL及びVH CDR3ドメインを含む単離ヒト抗体又はその抗原結合部分の使用を包含する。例えば、配列番号3、配列番号11、配列番号12、配列番号13、配列番号14、配列番号15、配列番号16、配列番号17、配列番号18、配列番号19、配列番号20、配列番号21、配列番号22、配列番号23、配列番号24、配列番号25及び配列番号26から構成される群から選択されるアミノ酸配列を含むCDR3ドメインをもつ軽鎖可変領域(LCVR)と、配列番号4、配列番号27、配列番号28、配列番号29、配列番号30、配列番号31、配列番号32、配列番号33、配列番号34及び配列番号35から構成される群から選択されるアミノ酸配列を含むCDR3ドメインをもつ重鎖可変領域(HCVR)を含む抗体又はその抗原結合部分が挙げられる。
【0101】
本発明の方法及び組成物で使用される抗体又は抗体部分は宿主細胞で免疫グロブリン軽鎖及び重鎖遺伝子を組換え発現させることにより作製することができる。抗体を組換え発現させるためには、抗体の免疫グロブリン軽鎖及び重鎖をコードするDNAフラグメントを組込んだ1個以上の組換え発現ベクターを宿主細胞にトランスフェクトし、軽鎖及び重鎖を宿主細胞で発現させ、好ましくは宿主細胞を培養する培地に分泌させ、培地から抗体を回収することができる。抗体重鎖及び軽鎖遺伝子を獲得し、これらの遺伝子を組換え発現ベクターに組込み、ベクターを宿主細胞に導入するためには、Sambrook,Fritsch and Maniatis(eds),Molecular Cloning;A Laboratory Manual,Second Edition,Cold Spring Harbor,N.Y.,(1989),Ausubel,F.M.et al.(eds.)Current Protocols in Molecular Biology,Greene Publishing Associates,(1989)及びBossらの米国特許第4,816,397号に記載されている方法等の標準組換えDNA法を使用する。
【0102】
アダリムマブ(D2E7)又はアダリムマブ(D2E7)関連抗体を発現させるためには、先ず軽鎖及び重鎖可変領域をコードするDNAフラグメントを得る。これらのDNAはポリメラーゼ連鎖反応(PCR)を使用して生殖系列軽鎖及び重鎖可変配列の増幅と修飾により得ることができる。ヒト重鎖及び軽鎖可変領域遺伝子の生殖系列DNA配列は当分野で公知である(例えば「Vbase」ヒト生殖系列配列データベース参照;更に各々その開示内容を特に本願に援用するKabat,E.A.,et al.(1991)Sequences of Proteins of Immunological Interest,Fifth Edition,U.S.Department of Health and Human Services,NIH Publication No.91−3242;Tomlinson,LM.,et al.(1992)“The Repertoire of Human Germline VH Sequences Reveals about Fifty Groups of VH Segments with Different Hypervariable Loops”J.Mol.Biol.227:776−798;及びCox,J.P.L.et al.(1994)“A Directory of Human Germ−line V78 Segments Reveals a Strong Bias in their Usage”Eur.J.Immunol.24:827−836も参照)。D2E7又はD2E7関連抗体の重鎖可変領域をコードするDNAフラグメントを得るためには、ヒト生殖系列VH遺伝子のVH3ファミリーのメンバーを標準PCRにより増幅する。DP−31 VH生殖系列配列を増幅するのが最も好ましい。D2E7又はD2E7関連抗体の軽鎖可変領域をコードするDNAフラグメントを得るためには、ヒト生殖系列VL遺伝子のVκIファミリーのメンバーを標準PCRにより増幅する。A20 VL生殖系列配列を増幅するのが最も好ましい。DP−31生殖系列VH及びA20生殖系列VL配列の増幅用に適切なPCRプライマーは標準方法を使用して上記引用文献に開示されているヌクレオチド配列に基づいて設計することができる。
【0103】
生殖系列VH及びVLフラグメントが得られたら、本願に開示するD2E7又はD2E7関連アミノ酸配列をコードするように、これらの配列を突然変異させることができる。生殖系列VH及びVL DNA配列によりコードされるアミノ酸配列を先ずD2E7又はD2E7関連VH及びVLアミノ酸配列と比較し、D2E7又はD2E7関連配列のうちで生殖系列と異なるアミノ酸残基を同定する。次に、どのヌクレオチド変異を導入すべきかを決定するために遺伝コードを使用し、突然変異後の生殖系列配列がD2E7又はD2E7関連アミノ酸配列をコードするように、生殖系列DNA配列の適切なヌクレオチドを突然変異させる。(PCR産物が突然変異を含むように、突然変異させたヌクレオチドをPCRプライマーに導入する)PCR突然変異誘発法や部位特異的突然変異誘発法等の標準方法により生殖系列配列の突然変異誘発を実施する。
【0104】
更に、当然のことながら、PCR増幅により得られた「生殖系列」配列が真の生殖系列構成に由来するフレームワーク領域でアミノ酸変異(即ち、例えば体細胞突然変異の結果として、真の生殖系列配列に対する増幅配列の変異)をコードする場合には、これらのアミノ酸変異を真の生殖系列配列に復帰変異(即ちフレームワーク残基を生殖系列構成に「復帰突然変異」)させることが望ましいと思われる。
【0105】
(例えば上記のように生殖系列VH及びVL遺伝子の増幅と突然変異誘発により)D2E7又はD2E7関連VH及びVLセグメントをコードするDNAフラグメントが得られたら、例えば可変領域遺伝子を全長抗体鎖遺伝子、Fabフラグメント遺伝子又はscFv遺伝子に変換するように、これらのDNAフラグメントを標準組換えDNA技術により更に操作することができる。これらの操作では、VL又はVHをコードするDNAフラグメントを、抗体定常領域又はフレキシブルリンカー等の別の蛋白質をコードする別のDNAフラグメントと機能的に連結する。この文脈で使用する「機能的に連結」なる用語は、2つのDNAフラグメントによりコードされるアミノ酸配列がインフレームに維持されるように2つのDNAフラグメントを結合することを意味する。
【0106】
VHをコードするDNAを、重鎖定常領域(CH1、CH2及びCH3)をコードする別のDNA分子と機能的に連結することにより、VH領域をコードする単離DNAを全長重鎖遺伝子に変換することができる。ヒト重鎖定常領域遺伝子の配列は当分野で公知であり(例えばKabat,E.A.,et al.(1991)Sequences of Proteins of Immunological Interest,Fifth Edition,U.S.Department of Health and Human Services,NIH Publication No.91−3242参照)、これらの領域を含むDNAフラグメントは標準PCR増幅法により得ることができる。重鎖定常領域はIgG1、IgG2、IgG3、IgG4、IgA、IgE、IgM又はIgD定常領域とすることができるが、IgG1又はIgG4定常領域が最も好ましい。Fabフラグメント重鎖遺伝子では、VHをコードするDNAを、重鎖CH1定常領域のみをコードする別のDNA分子と機能的に連結することができる。
【0107】
VLをコードするDNAを、軽鎖定常領域CLをコードする別のDNA分子と機能的に連結することにより、VL領域をコードする単離DNAを全長軽鎖遺伝子(及びFab軽鎖遺伝子)に変換することができる。ヒト軽鎖定常領域遺伝子の配列は当分野で公知であり(例えばKabat,E.A.,et al.(1991)Sequences of Proteins of Immunological Interest,Fifth Edition,U.S.Department of Health and Human Services,NIH Publication No.91−3242参照)、これらの領域を含むDNAフラグメントは標準PCR増幅法により得ることができる。軽鎖定常領域はκ又はλ定常領域とすることができるが、κ定常領域が最も好ましい。
【0108】
scFv遺伝子を作製するためには、VL領域とVH領域をフレキシブルリンカーでつないでVH配列とVL配列を連続する1本鎖蛋白質として発現させることができるように、VHとVLをコードするDNAフラグメントを、フレキシブルリンカー、例えばアミノ酸配列(Gly4−Ser)3をコードする別のフラグメントと機能的に連結する(例えばBird et al.(1988)Science 242:423−426;Huston et al.(1988)Proc.Natl.Acad.Sci.USA 85:5879−5883;McCafferty et al.,Nature(1990)348:552−554参照)。
【0109】
本発明で使用される抗体又は抗体部分を発現させるためには、遺伝子を転写及び翻訳制御配列と機能的に連結するように、上記のように得られた部分的又は全長軽鎖及び重鎖をコードするDNAを発現ベクターに挿入する。この文脈で「機能的に連結」なる用語は、ベクター内の転写及び翻訳制御配列が抗体遺伝子の転写及び翻訳を制御するというその所期機能を発揮するように、抗体遺伝子をベクターにライゲーションすることを意味する。発現ベクターと発現制御配列は使用する発現宿主細胞と適合するように選択する。抗体軽鎖遺伝子と抗体重鎖遺伝子を別々のベクターに挿入することもできるが、より一般には、両方の遺伝子を同一の発現ベクターに挿入する。標準方法(例えば抗体遺伝子フラグメントとベクター上の相補的制限部位のライゲーション、又は制限部位が存在しない場合には平滑末端ライゲーション)により抗体遺伝子を発現ベクターに挿入する。D2E7又はD2E7関連軽鎖又は重鎖配列を挿入する前に、発現ベクターに予め定常領域配列を組込んでもよい。例えば、D2E7又はD2E7関連VH及びVL配列を全長抗体遺伝子に変換する1つのアプローチは、VHセグメントをベクター内のCHセグメントと機能的に連結し、VLセグメントをベクター内のCLセグメントと機能的に連結するように、夫々重鎖定常領域と軽鎖定常領域を予めコードする発現ベクターに挿入する方法である。上記の追加又は代替として、組換え発現ベクターは宿主細胞からの抗体鎖の分泌を助長するシグナルペプチドをコードすることができる。シグナルペプチドを抗体鎖遺伝子のアミノ末端とインフレームで連結するように、抗体鎖遺伝子をベクターにクローニングすることができる。シグナルペプチドは免疫グロブリンシグナルペプチド又は異種シグナルペプチド(即ち非免疫グロブリン蛋白質に由来するシグナルペプチド)とすることができる。
【0110】
抗体鎖遺伝子に加え、本発明の組換え発現ベクターは宿主細胞における抗体鎖遺伝子の発現を制御する調節配列をもつ。「調節配列」なる用語はプロモーター、エンハンサー及び抗体鎖遺伝子の転写又は翻訳を制御する他の発現制御エレメント(例えばポリアデニル化シグナル)を包含するものとする。このような調節配列は例えば、Goeddel;Gene Expression Technology:Methods in Enzymology 185,Academic Press,San Diego,CA(1990)に記載されている。当業者に自明の通り、調節配列の選択を含めて発現ベクターの設計は、形質転換させる宿主細胞の選択、所望される蛋白質発現レベル等の因子に依存し得る。哺乳動物宿主細胞発現に好ましい調節配列としては、哺乳動物細胞における高レベルの蛋白質発現を誘導するウイルスエレメントが挙げられ、例えばサイトメガロウイルス(CMV)(例えばCMVプロモーター/エンハンサー)、サルウイルス40(SV40)(例えばSV40プロモーター/エンハンサー)、アデノウイルス(例えばアデノウイルス主要後期プロモーター(AdMLP))及びポリオーマに由来するプロモーター及び/又はエンハンサーが挙げられる。ウイルス調節エレメントとその配列の詳細な説明については、例えばStinskiの米国特許第5,168,062号、Bellらの米国特許第4,510,245号及びSchaffnerらの米国特許第4,968,615号参照。
【0111】
抗体鎖遺伝子と調節配列に加え、本発明で使用される組換え発現ベクターには、宿主細胞におけるベクターの複製を調節する配列(例えば複製起点)や選択マーカー遺伝子等の他の配列を組込むことができる。選択マーカー遺伝子はベクターを導入された宿主細胞の選択を容易にする(例えばいずれもAxelらの米国特許第4,399,216号、4,634,665号及び5,179,017号参照)。例えば、一般に選択マーカー遺伝子はベクターを導入された宿主細胞にG418、ハイグロマイシン又はメトトレキサート等の薬剤に対耐性を付与する。好ましい選択マーカー遺伝子としては、ジヒドロ葉酸レダクターゼ(DHFR)遺伝子(dhfr欠損宿主細胞におけるメトトレキサート選択/増幅用)とneo遺伝子(G418選択用)が挙げられる。
【0112】
軽鎖と重鎖を発現させるために、重鎖と軽鎖をコードする発現ベクターを標準技術により宿主細胞にトランスフェクトする。「トランスフェクション」なる用語の各種語形は外来DNAを原核又は真核宿主細胞に導入するために一般に使用されている多様な技術(例えばエレクトロポレーション、リン酸カルシウム沈殿法、DEAE−デキストラントランスフェクション等)を包含するものとする。理論的には本発明の抗体を原核又は真核宿主細胞のどちらでも発現させることが可能であるが、真核細胞、特に哺乳動物細胞は原核細胞よりも正しく折り畳まれた免疫的に活性な抗体を会合・分泌させるので、真核細胞、最も好ましくは哺乳動物宿主細胞で抗体を発現させるのが最も好ましい。抗体遺伝子の原核細胞発現は活性抗体の高収率生産には無効であることが報告されている(Boss,M.A.and Wood,C.R.(1985)Immunology Today 6:12−13)。
【0113】
本発明の組換え抗体を発現させるために好ましい哺乳動物宿主細胞としては、チャイニーズハムスター卵巣(CHO細胞)(Urlaub and Chasin,(1980)Proc.Natl.Acad.Sci.USA 77:4216−4220に記載されているdhfr欠損CHO細胞と、例えばR.J.Kaufman and P.A.Sharp (1982)Mol.Biol.159:601−621に記載されているようなDHFR選択マーカーの併用を含む)、NS0ミエローマ細胞、COS細胞及びSP2細胞が挙げられる。抗体遺伝子をコードする組換え発現ベクターを哺乳動物宿主細胞に導入する場合には、宿主細胞における抗体の発現、又はより好ましくは宿主細胞を増殖させる培地への抗体の分泌を可能にするために十分な時間にわたって宿主細胞を培養することにより抗体を産生させる。標準蛋白質精製法を使用して培地から抗体を回収することができる。
【0114】
FabフラグメントやscFv分子等の無傷の抗体の部分を作製するために宿主細胞を使用することもできる。当然のことながら、上記手順の変形も本発明の範囲に含まれる。例えば、本発明の抗体の軽鎖又は重鎖のいずれか(両方ではない)をコードするDNAを宿主細胞にトランスフェクトすることが望ましい場合がある。hTNFαとの結合に不要な軽鎖及び重鎖のいずれか又は両方をコードするDNAの一部又は全部を除去するために組換えDNA技術を使用してもよい。このような低分子DNA分子から発現される分子も本発明の抗体に含まれる。更に、標準化学架橋法により本発明の抗体を第2の抗体と架橋させることにより、一方の重鎖と一方の軽鎖が本発明の抗体であり、他方の重鎖と軽鎖がhTNFα以外の抗原に特異的である2価抗体を作製することもできる。
【0115】
本発明の抗体又はその抗原結合部分の組換え発現に好ましいシステムでは、抗体重鎖と抗体軽鎖の両方をコードする組換え発現ベクターをリン酸カルシウムトランスフェクション法によりdhfr欠損CHO細胞に導入する。組換え発現ベクター内で、遺伝子の高レベル転写を誘導するように抗体重鎖及び軽鎖遺伝子を各々CMVエンハンサー/AdMLPプロモーター調節エレメントと機能的に連結する。メトトレキサート選択/増幅を使用してベクターをトランスフェクトされたCHO細胞の選択を可能にするDHFR遺伝子も組換え発現ベクターに組込む。選択された形質転換宿主細胞を培養し、抗体重鎖及び軽鎖を発現させ、無傷の抗体を培地から回収する。標準分子生物学技術を使用して組換え発現ベクターを作製し、宿主細胞にトランスフェクトし、形質転換細胞を選択し、宿主細胞を培養し、培地から抗体を回収する。
【0116】
上記に鑑み、本発明で使用される抗体及び抗体部分の組換え発現に使用することができる核酸、ベクター及び宿主細胞としては、ヒトTNFα抗体アダリムマブ(D2E7)を含む核酸と、前記核酸を組込んだベクターが挙げられる。D2E7軽鎖可変領域をコードするヌクレオチド配列を配列番号36に示す。LCVRのCDR1ドメインはヌクレオチド70〜102を含み、CDR2ドメインはヌクレオチド148〜168を含み、CDR3ドメインはヌクレオチド265〜291を含む。D2E7重鎖可変領域をコードするヌクレオチド配列を配列番号37に示す。HCVRのCDR1ドメインはヌクレオチド91〜105を含み、CDR2ドメインはヌクレオチド148〜198を含み、CDR3ドメインはヌクレオチド295〜330を含む。当業者に自明の通り、D2E7関連抗体又はその部分(例えばCDR3ドメイン等のCDRドメイン)をコードするヌクレオチド配列は、遺伝コード及び標準分子生物学技術を使用してD2E7 LCVR及びHCVRをコードするヌクレオチド配列から誘導することができる。
【0117】
1態様において、液体医薬製剤は抗体アダリムマブに対して生物学的に等価又は生物学的に同等のヒトTNFα抗体又はその抗原結合部分を含有する。1態様において、生物学的に同等の抗体とは参照抗体(例えばアダリムマブ)と比較した場合に臨床的に有意な差を示さない抗体である。生物学的に同等の抗体は参照抗体(例えばアダリムマブ)と等価の安全性、純度及び力価をもつ。
【0118】
IV.本発明の製剤の投与
本発明の製剤の1つの利点は高濃度のヒト抗TNFα抗体又は抗原結合部分(例えばアダリムマブ)を対象に皮下送達するために使用できるという点である。従って、1態様では、本発明の製剤を対象に皮下送達する。1態様において、対象は製剤を自己投与する。
【0119】
1態様では、有効量の製剤を投与する。製剤の「有効量」なる用語はTNFα活性を阻害するため、例えば有害なTNFα活性に関連する状態の各種形態的及び身体的症状を予防するために必要又は十分な量である。別の態様において、製剤の有効量は所望結果を達成するために必要な量である。
【0120】
製剤の有効量の1例は有害なTNFα活性を阻害するため、又はTNFα活性が有害である疾患を治療するために十分な量である。本願で使用する「TNFα活性が有害である疾患」なる用語は疾患に罹患している対象におけるTNFαの存在が前記疾患の病態生理に関与しているか又は前記疾患の増悪に寄与する因子であることが分かっているか又はその疑いのある疾病及び他の疾患を包含するものとする。従って、TNFα活性が有害である疾患とは、TNFα活性の阻害が疾患の症状及び/又は進行を緩和すると予想される疾患である。このような疾患は例えば、疾患に罹患している対象の体液中のTNFα濃度の上昇(例えば対象の血清、血漿、滑液等におけるTNFα濃度上昇)により確認することができ、このような上昇は例えば抗TNFα抗体を使用して検出することができる。
【0121】
下記実施例に記載するように、本発明の製剤の1つの利点は製剤の粘度を増加せずに高濃度の抗体を含有する製剤を製造できる点である。従って、同じく以下に記載するように、新規製剤は従来の市販製剤に比較して少ない容量で多量(例えば有効量)の抗体の投与を可能にすることにより、疼痛を緩和する。
【0122】
1態様において、抗体の有効量は厳密に体重に基づく投与スキーム(例えばmg/kg)に従って決定することもできるし、体重から独立した総用量(固定用量とも言う)でもよい。1例において、製剤の有効量は総用量約80mgの抗体を含有する製剤0.8mL(即ち本発明の100mg/mL抗体製剤0.8mL)である。別の例において、製剤の有効量は総用量約40mgの抗体を含有する本発明の製剤0.4mL(即ち本発明の100mg/mL抗体製剤0.4mL)である。更に別の例において、製剤の有効量は総用量約160mgの抗体を含有する製剤0.8mL×2(即ち本発明の100mg/mL抗体製剤0.8mLずつを含む2単位)である。別の例において、製剤の有効量は総用量約20mgの抗体を含有する本発明の製剤0.2mL(即ち本発明の100mg/mL抗体製剤0.2mL)である。あるいは、体重に基づく固定用量レジメンに従って有効量を決定してもよい(例えば本願に援用するWO2008/154543参照)。
【0123】
本発明は、1態様ではTNFα活性が有害である疾患に罹患している対象におけるTNFα活性を阻害するために使用される貯蔵寿命の長い安定した高濃度製剤を提供し、対象におけるTNFα活性を阻害するように本発明の製剤を対象に投与する。好ましくは、TNFαはヒトTNFαであり、対象はヒト対象である。あるいは、対象は本発明の抗体と交差反応するTNFαを発現する哺乳動物でもよい。更に、対象は(例えばhTNFαの投与又はhTNFαトランスジーンの発現により)hTNFαを導入された哺乳動物でもよい。
【0124】
本発明の製剤は(以下に詳述する)治療目的でヒト対象に投与することができる。本発明の1態様において、液体医薬製剤は容易に投与可能であり、例えば患者により自己投与される製剤が挙げられる。好ましい1態様において、本発明の製剤は皮下注射により好ましくは単回投与される。更に、本発明の製剤は獣医学的目的のため又はヒト疾患の動物モデルとして抗体と交差反応するTNFαを発現する非ヒト哺乳動物(例えば霊長類、ブタ又はマウス)に投与することもできる。後者に関して、このような動物モデルは本発明の抗体の治療効力の評価(例えば投与用量及び時間コースの試験)に有用であると思われる。
【0125】
1態様では、プレフィルドシリンジ、自己注射ペン又は無針投与装置により本発明の液体医薬製剤を対象に投与することができる。従って、本発明は本発明の液体医薬製剤を収容する自己注射ペン、プレフィルドシリンジ又は無針投与装置にも関する。1態様において、本発明は100mg/mLのヒトTNFα抗体又はその抗原結合部分を含有する製剤の1用量を収容する送達装置に関し、例えば自己注射ペン又はプレフィルドシリンジは約19mg、20、mg、21mg、22mg、23mg、24mg、25mg、26mg、27mg、28mg、29mg、30mg、31mg、32mg、33mg、34mg、35mg、36mg、37mg、38mg、39mg、40mg、41mg、42mg、43mg、44mg、45mg、46mg、47mg、48mg、49mg、50mg、51mg、52mg、53mg、54mg、55mg、56mg、57mg、58mg、59mg、60mg、61mg、62mg、63mg、64mg、65mg、66mg、67mg、68mg、69mg、70mg、71mg、72mg、73mg、74mg、75mg、76mg、77mg、78mg、79mg、80mg、81mg、82mg、83mg、84mg、85mg、86mg、87mg、88mg、89mg、90mg、91mg、92mg、93mg、94mg、95mg、96mg、97mg、98mg、99mg、100mg、101mg、102mg、103mg、104mg、105mg等の製剤の1用量を収容する。
【0126】
好ましくは、本発明の製剤はTNFα活性が有害である疾患を治療するために使用される。本願で使用する「TNFα活性が有害である疾患」なる用語は疾患に罹患している対象におけるTNFαの存在が前記疾患の病態生理に関与しているか又は前記疾患の増悪に寄与する因子であることが分かっているか又はその疑いのある疾病及び他の疾患を包含するものとする。従って、TNFα活性が有害である疾患とは、TNFα活性の阻害が疾患の症状及び/又は進行を緩和すると予想される疾患である。このような疾患は例えば、疾患に罹患している対象の体液中のTNFα濃度の上昇(例えば対象の血清、血漿、滑液等におけるTNFα濃度上昇)により確認することができ、このような上昇は例えば上記のように抗TNFα抗体を使用して検出することができる。
【0127】
TNFα活性が有害である疾患には多数の例が存在する。TNFα活性が有害である疾患の例はいずれもその開示内容全体を本願に援用する米国特許第6,015,557号、6,177,077号、6,379,666号、6,419,934号、6,419,944号、6,423,321号、6,428,787号、及び6,537,549号、並びにPCT公開第WO00/50079号及びWO01/49321号にも記載されている。本発明の製剤はいずれもその開示内容全体を本願に援用する米国特許第6,090,382号、6,258,562号及び米国特許出願公開第US20040126372号に記載されているようなTNFα活性が有害である疾患を治療するために使用することもできる。
【0128】
以下、特定の代表的疾患の治療における本発明の製剤の使用について詳述する。
【0129】
A.敗血症
腫瘍壊死因子は血圧低下、心筋抑制、血管漏出症候群、臓器壊死、毒性二次メディエーターの放出の刺激及び血液凝固カスケードの活性化を含む生物学的効果により、敗血症の病態生理に関与することが知られている(例えばTracey,K.J.and Cerami,A.(1994)Annu.Rev.Med.45:491−503;Russell,D and Thompson,R.C.(1993)Curr.Opin.Biotech.4:714−721参照)。従って、本発明の製剤は敗血症性ショック、内毒素ショック、グラム陰性菌敗血症及び毒素性ショック症候群を含むその臨床背景のいずれかにおいて敗血症を治療するために使用することができる。
【0130】
更に、敗血症を治療するために、インターロイキン1阻害剤(例えばPCT公開第WO92/16221号及びWO92/17583に記載されているもの)、サイトカインであるインターロイキン6(例えばPCT公開第WO93/11793号参照)又は血小板活性化因子のアンタゴニスト(例えば欧州特許出願公開第EP374510号参照)等の敗血症を更に緩和することができる1種以上の他の治療剤と本発明の製剤を併用投与することができる。
【0131】
更に、好ましい1態様では、治療時のIL−6の血清又は血漿濃度が500pg/ml、より好ましくは1000pg/mlを上回る敗血症患者のサブグループ内のヒト対象に本発明の製剤を投与する(PCT公開第WO95/20978号参照)。
【0132】
B.自己免疫疾患
腫瘍壊死因子は各種自己免疫疾患の病態生理に関与すると考えられている。例えば、TNFαは組織炎症を活発にし、関節リウマチにおける関節破壊をもたらすと考えられている(例えばTracey and Cerami,前出;Arend,W.P.and Dayer,J−M.(1995)Arth.Rheum.38:151−160;Fava,R.A.,et al.(1993)Clin.Exp.Immunol.94:261−266参照)。TNFαは膵島細胞の死滅を促進し、糖尿病におけるインスリン抵抗性に関与するとも考えられている(例えばTracey and Cerami,前出;PCT公開第WO94/08609号参照)。TNFαは多発性硬化症において希突起膠細胞に対する細胞傷害作用と炎症性プラークの誘導に関与するとも考えられている(例えばTracey and Cerami,前出参照)。本発明の製剤を使用して治療することができる自己免疫疾患には、若年性特発性関節炎(JIA)(若年性関節リウマチとも言う)も含まれる(Grom et al.(1996)Arthritis Rheum.39:1703;Mangge et al.(1995)Arthritis Rheum.8:211参照)。
【0133】
本発明の製剤は自己免疫疾患、特に炎症を伴うものを治療するために使用することができ、関節リウマチ、リウマチ性脊椎炎(強直性脊椎炎とも言う)、変形性関節症及び通風性関節炎、アレルギー、多発性硬化症、自己免疫性糖尿病、自己免疫性ぶどう膜炎、若年性特発性関節炎(若年性関節リウマチとも言う)、並びにネフローゼ症候群が挙げられる。
【0134】
C.感染症
腫瘍壊死因子は各種感染症に認められる生物学的作用に関与すると考えられている。例えば、TNFαはマラリアにおいて脳炎症と毛細血管血栓及び梗塞に関与すると考えられている(例えばTracey and Cerami,前出参照)。TNFαは髄膜炎において脳炎症に関与し、血液脳関門の破壊を誘導し、敗血症性ショック症候群を誘発し、静脈梗塞を活性化するとも考えられている(例えばTracey and Cerami,前出参照)。TNFαは後天性免疫不全症候群(エイズ)において悪液質を誘導し、ウイルス増殖を刺激し、中枢神経系損傷に関与するとも考えられている(例えばTracey and Cerami,前出参照)。従って、本発明の抗体及び抗体部分は細菌性髄膜炎(例えば欧州特許出願公開第EP585705号参照)、脳マラリア、エイズ及びエイズ関連症候群(ARC)(例えば欧州特許出願公開第EP230574号参照)、並びに移植に伴うサイトメガロウイルス感染(例えばFietze,E.,et al.(1994)Transplantation 58:675−680参照)を含む感染症の治療に使用することができる。本発明の製剤は感染による発熱及び筋肉痛(例えばインフルエンザ)や、感染に伴う(例えばエイズ又はARCに伴う)悪液質を含めて感染症に伴う症状を緩和するために使用することもできる。
【0135】
D.移植
腫瘍壊死因子は同種移植片拒絶反応及び移植片対宿主病(GVHD)の主要なメディエータであり、腎移植の拒絶反応を抑制するためにT細胞受容体CD3複合体に対するラット抗体OKT3を使用する場合に認められている有害反応に関与すると考えられている(例えばTracey and Cerami,前出;Eason,J.D.,et al.(1995)Transplantation 59:300−305;Suthanthiran,M.and Strom,T.B.(1994)New Engl.J.Med.331:365−375参照)。従って、本発明の製剤は同種移植片及び異種移植片の拒絶反応を含む移植拒絶反応を抑制し、GVHDを抑制するために使用することができる。抗体又は抗体部分は単独で使用してもよいが、同種移植片に対する免疫応答を抑制又はGVHDを抑制する1種以上の他の物質と併用することができる。例えば、1態様では、OKT3により誘導される反応を抑制するために本発明の製剤をOKT3と併用する。別の態様では、細胞表面分子CD25(インターロイキン2受容体α)、CD11a(LFA−1)、CD54(ICAM−1)、CD4、CD45、CD28/CTLA4、CD80(B7−1)及び/又はCD86(B7−2)等の免疫応答の調節に関与する他の標的に対する1種以上の抗体と本発明の製剤を併用する。更に別の態様では、シクロスポリンAやFK506等の1種以上の一般的な免疫抑制剤と本発明の製剤を併用する。
【0136】
E.悪性腫瘍
腫瘍壊死因子は悪性腫瘍において悪液質を誘導し、腫瘍増殖を刺激し、転移の可能性を増し、細胞傷害作用に関与すると考えられている(例えばTracey and Cerami,前出参照)。従って、本発明の製剤は腫瘍増殖もしくは転移を抑制し、及び/又は悪性腫瘍に伴う悪液質を緩和するために、悪性腫瘍の治療で使用することができる。
【0137】
F.肺疾患
腫瘍壊死因子は白血球−内皮細胞活性化の刺激、肺細胞に対する細胞傷害作用の誘発及び血管漏出症候群の誘導を含む成人呼吸窮迫症候群の病態生理に関係があると考えられている(例えばTracey and Cerami,前出参照)。従って、本発明の製剤は成人呼吸窮迫症候群(例えばPCT公開第WO91/04054号参照)、ショック肺、慢性肺炎症疾患、肺サルコイドーシス、肺線維症及び珪肺症を含む各種肺疾患を治療するために使用することができる。
【0138】
G.腸疾患
腫瘍壊死因子は炎症性腸疾患の病態生理に関係があると考えられている(例えばTracy,K.J.,et al.(1986)Science 234:470−474;Sun,X−M.,et al.(1988)J.Clin.Invest.81:1328−1331;MacDonald,T.T.,et al.(1990)Clin.Exp.Immunol.81:301−305参照)。キメラマウス抗hTNFα抗体がクローン病の治療用に臨床試験されている(van Dullemen,H.M.,et al.(1995)Gastroenterology 109:129−135)。本発明の製剤はクローン病と潰瘍性大腸炎の2種類の症候群を含む特発性炎症性腸疾患等の腸疾患を治療するために使用することもできる。
【0139】
H.心疾患
本発明の製剤は心筋虚血(例えば欧州特許出願公開第EP453898号参照)と心不全(心筋の弱化)(例えばPCT公開第WO94/20139号参照)を含む各種心疾患を治療するために使用することもできる。
【0140】
I.脊椎関節症
TNFαは脊椎関節症等の炎症性疾患を含む多様な疾患の病態生理に関係があると考えられている(例えばMoeller,A.,et al.(1990)Cytokine 2:162−169;Moellerらの米国特許第5,231,024号;Moeller,Aの欧州特許公開第260610B1号参照)。本発明の製剤により治療することができる脊椎関節症の1例としては乾癬性関節炎が挙げられる。腫瘍壊死因子は乾癬性関節炎の病態生理に関係があると考えられている(Partsch et al.(1998)Ann Rheum Dis.57:691;Ritchlin et al.(1998)J Rheumatol.25:1544)。
【0141】
J.皮膚及び爪疾患
1態様において、本発明の製剤は皮膚及び爪疾患を治療するために使用される。本願で使用する「TNFα活性が有害である皮膚及び爪疾患」なる用語は疾患に罹患している対象におけるTNFαの存在が前記疾患の病態生理に関与しているか又は前記疾患の増悪に寄与する因子であることが分かっているか又はその疑いのある皮膚及び/又は爪疾患と他の疾患(例えば乾癬)を包含するものとする。本発明の製剤を使用して治療することができる皮膚疾患の1例は乾癬である。1態様において、本発明の製剤はプラーク乾癬を治療するために使用される。腫瘍壊死因子は乾癬の病態生理に関係があると考えられている(Takematsu et al.(1989)Arch Dermatol Res.281:398;Victor and Gottlieb(2002)J Drugs Dermatol.1(3):264)。
【0142】
1態様において、本発明の製剤は関節リウマチ、乾癬性関節炎又は強直性脊椎炎を治療するために使用される。単離ヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)を含有する本発明の製剤は関節リウマチ、乾癬性関節炎又は強直性脊椎炎を治療するために有効な投与スキーム及び用量に従ってヒト対象に投与することができる。1態様では、関節リウマチ、乾癬性関節炎又は強直性脊椎炎を治療するために、本発明の製剤中のヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)約40mgの1用量(例えば本発明の100mg/mL製剤0.4mL)を1週間置きにヒト対象に投与する。1態様では、関節リウマチ、強直性脊椎炎又は乾癬性関節炎を治療するために製剤を1週間置き(隔週とも言う、本願に援用するUS20030235585に記載されている投与方法参照)に皮下投与する。
【0143】
1態様において、本発明の製剤はクローン病を治療するために使用される。単離ヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)を含有する本発明の製剤はクローン病を治療するために有効な投与スキーム及び用量に従ってヒト対象に投与することができる。1態様では、クローン病を治療するために、本発明の製剤中のヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)約160mgの1用量(例えば本発明の100mg/mL製剤1.6mL)をヒト対象に約1日目に初期投与した後、2週間後に抗体80mgの後続用量(例えば本発明の100mg/mL製剤0.8mL)を投与し、更にその後、1週間置きに約40mg(例えば本発明の100mg/mL製剤0.4mL)を投与する。1態様では、クローン病を治療するために誘導用量と維持用量を含む多回可変用量レジメン(例えば、各々本願に援用する米国特許公開第US20060009385号及びUS20090317399号参照)に従って製剤を皮下投与する。
【0144】
1態様において、本発明の製剤は乾癬を治療するために使用される。単離ヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)を含有する本発明の製剤は乾癬を治療するために有効な投与スキーム及び用量に従ってヒト対象に投与することができる。1態様では、本発明の製剤中のヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)約80mgの初期用量(例えば本発明の100mg/mL製剤0.8mL)をヒト対象に投与した後、初期投与の1週間後から開始して1週間置きに抗体40mgの後続用量(例えば本発明の100mg/mL製剤0.4mL)を投与する。1態様では、乾癬を治療するために誘導用量と維持用量を含む多回可変用量レジメン(例えば、各々本願に援用するUS20060009385及びWO2007/120823参照)に従って製剤を皮下投与する。
【0145】
1態様において、本発明の製剤は若年性特発性関節炎(JIA)を治療するために使用される。単離ヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)を含有する本発明の製剤はJIAを治療するために有効な投与スキーム及び用量に従ってヒト対象に投与することができる。1態様では、JIAを治療するために、本発明の製剤中のヒトTNFα抗体又はその抗原結合部分20mg(例えば本発明の100mg/mL製剤0.2mL)を体重15kg(約33lb)〜30kg(66lb)未満の対象に1週間置きに投与する。別の態様では、JIAを治療するために、本発明の製剤中のヒトTNFα抗体又はその抗原結合部分40mg(例えば本発明の100mg/mL製剤0.4mL)を体重30kg(66lb)以上の対象に1週間置きに投与する。1態様では、JIAを治療するために、体重に基づく固定用量(例えば、本願に援用する米国特許公開第20090271164号参照)に従って製剤を皮下投与する。
【0146】
1態様では、有害なTNFα活性に関連する疾患を治療するために、抗体を1カ月に一度又は4週間に一度投与するような毎月投与スケジュールに従って単離ヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)をヒト対象に投与することができる。上記のように、毎月投与スケジュールに従って治療することができる疾患の例としては、限定されないが、関節リウマチ、強直性脊椎炎、JIA、乾癬、クローン病又は乾癬性関節炎が挙げられる。従って、単離ヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)を含有する本発明の製剤は有害なTNFα活性に関連する疾患を治療するために毎月投与スケジュールに従ってヒト対象に投与することができる。1態様では、有害なTNFα活性に関連する疾患をもつ対象に本発明の製剤中のヒトTNFα抗体又はその抗原結合部分80mg(例えば本発明の100mg/mL製剤0.8mL)を投与する。
【0147】
本願に記載する投与量は単回用量(例えば0.4mL中に40mg又は0.8mL中に80mgの単回用量)として送達してもよいし、あるいは多回用量(例えば160mg用量を送達するために40mgずつ4回又は80mgずつ2回)として送達してもよい。
【0148】
単離ヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)を含有する本発明の製剤を対象に別の治療剤と併用投与することもできる。1態様では、関節リウマチを治療するために製剤をヒト対象にメトトレキサート又は他の疾患修飾性抗リウマチ薬(DMARD)と併用投与する。別の態様では、JIAを治療するために製剤をヒト対象にメトトレキサート又は他の疾患修飾性抗リウマチ薬(DMARD)と併用投与する。その他の併用療法も米国特許第6,258,562号及び7,541,031号と、米国特許公開第US20040126372号に記載されており、いずれもその開示内容全体を本願に援用する。
【0149】
ヒトTNFα抗体又はその抗原結合部分を含有する本発明の製剤は過去にTNF阻害剤療法の効果がなかった対象、例えばインフリキシマブに応答しなかった対象又は不寛容な対象を治療するために使用することもできる。
【0150】
以下、実施例により本発明を更に例証するが、これらの実施例は限定的であるとみなすべきではない。
【実施例1】
【0151】
ヒト抗TNFα抗体液体医薬製剤の安定性の改善
本実施例は抗体アダリムマブの医薬製剤の安定性の改善を目的とする実験の結果について記載する。
【0152】
材料及び方法
最終薬剤濃度50mg/mLの液体非経口剤形を作製するために、アダリムマブ(サブクラスG1,約47kDa)を修飾医薬製剤に製剤化した。過去の製剤実験によると、リン酸/クエン酸緩衝液系はアダリムマブの蛋白質安定化に関して他の緩衝液系よりも優れていることが分かっている。従って、液体50mg/mL製剤に添加剤を添加することにより安定性の改善を試みた。使用した全添加剤は最高純度(「プロアナリシス」グレード)のものをMerck KGaA,Darmstadt,Germanyから購入した。マンニトールはMallinckrodt Baker B.V.,Deventer,Hollandから入手した。
【0153】
可視粒状物の分析はPh.Eur.2002の規定に従って実施した(§2.9.20 Contamination with particulate matter−visible particles)。不可視粒状物分析は光遮蔽により判定した(SVSS−C40,PAMAS GmbH,Rutesheim,Germany)。Superose TM6 10/30カラム(Amersham Pharmacia Europe GmbH,Freiburg,Germany)をSE−HPLC分析(蛋白質モノマー含有量の評価)に使用し、pH7.5のPBS緩衝液を流速0.5mL/minで流し、オンライン検出のためにUV280分光分析、屈折率検出及びMALSと連動させた。各サンプルの分析を少なくとも3回ずつ実施した。特に指定しない限り、全SE−HPLCデータでSrelは0.13未満であり、全光遮蔽データで2.3未満であった。
【0154】
アダリムマブ濃厚液(〜70mg/mL)を添加剤ストック溶液で希釈することにより個々の蛋白質製剤を調製した。70mg/mLアダリムマブストック溶液は表16に示すようなクエン酸リン酸緩衝液成分の組成(即ちクエン酸・H2O、クエン酸ナトリウム脱水和物、Na2HPO4・2H2O、NaH2PO4・2H2O)を使用して調製した。
【0155】
表16に示すようなクエン酸リン酸緩衝液成分の組成(即ちクエン酸・H2O,クエン酸ナトリウム脱水和物、Na2HPO4・2H2O、NaH2PO4・2H2O)を使用して添加剤をリン酸/クエン酸緩衝液溶媒に溶解することにより添加剤ストック溶液を作製した。滅菌濾過(0.2μm,Minisart(登録商標),Sartorius AG,Goettingen,Germany)前に、緩衝液成分の酸/塩基試料を添加することによりpH調節を実施した。全製剤を少なくとも2本ずつ調製し、無菌空気層流条件下で熱滅菌(180℃,25分)2Rガラスバイアル(Schott Glas,Mainz,Germany)に溶液バッチを最終滅菌濾過することにより作製した。使用前にPh.Eur.に従ってテフロン(登録商標)被覆ブチルラバーストッパーを湿熱(121℃)滅菌した。
【0156】
各種製剤を3種類の異なる温度(5℃、25℃、40℃)で3カ月短期保存した。
【0157】
リン酸/クエン酸緩衝液溶媒を緩衝液交換に使用し、アダリムマブバルク溶液をVivaflow 50 units(カットオフ50kDa,Vivascience G,Hannover,Germany)で透析濾過することによりアダリムマブ濃厚液を準備した。バイオ医薬品溶液の現行の濃縮法と緩衝液交換法はIEX、SE−HPLC、限外/透析濾過及び接線流濾過に基づく(Christy et al.(2002)Desalination,144:133−136)。透析濾過は可変流速で1回の操作で精製、濃縮及び緩衝液交換が可能であるため、蛋白質ストレスが最小限になることから、透析濾過を適用した(表1)。
【0158】
【表1】

実施した各サイクルは総蛋白質の〜0.25%の蛋白質損失をもたらした。一般に、蛋白質損失は濃厚液生成中に7%を超えなかった。
【0159】
1回の透析濾過サイクル以内で、蛋白質濃度は倍増したので、最終濃縮段階以外は元の濃度まで希釈した。従って、存在すべきでない望ましくない溶解物質を有効に除去することができる(例えば透析濾過サイクル10回以内で1.00%濃度を0.00098%まで低下させることができる)。精製及び濃縮後、アダリムマブ濃厚液を遠心した(5℃,3000g,20分間)。
【0160】
pH最適値の評価
最適溶液pH(即ちpH5.2又はpH6.0)を評価するために、pHのみを変化させた2種類の異なるアダリムマブ製剤を分析した。1mg/mL Tween 80を含有する製剤の安定性データを表2A及び2Bに示す。
【0161】
【表2】
【0162】
【表3】
モノマー含有量については、両製剤とも40℃保存時に同等のモノマー損失を示したため、pHによる優劣はないことが分かった。25℃保存条件のデータは40℃データと同様であったが、5℃では、この試験中に分析した全蛋白質溶液はモノマー含有量の有意変化を生じなかった。
【0163】
他方、濁度には差が認められた。保存温度に関係なく、溶液pH6.0では、12週間保存中に不可視粒状物の形成を生じた。粒状物形成の強度は低温に結び付けられるので、粒子の起源は蛋白性ではないと予想される。この点で、深刻な粒状物形成が蛋白質不安定のみに起因したのであるならば、これは保存試験中の高温暴露に関係があると考えられる(Constantino,et al.(1994b)J.Pharm.Sci.83:1662−1669)。
【0164】
Tween 80の代わりに6.16mg/mL NaClを含有する50mg/mLアダリムマブ製剤については、塩添加の結果、1μmよりも大きい粒子の数が両溶液で同程度増加したので、不可視粒子が形成された(表3A及び3B参照)。更に、12週間後に、SE−HPLCデータによると、差は最小限(〜0.3%)であり、25℃結果により確証されなかったが、pH6.0溶液はpH5.2溶液よりもモノマー含有量が多かった。
【0165】
【表4】
【0166】
【表5】
粒子形成はNaCl添加とpH6.0保存により助長され、Tween 80添加と溶液pH5.2により改善されると思われた。従って、NaCl等の塩を含有する溶液中の粒子汚染を軽減することができる成分としてTween 80を提案した(表4A及び4B)。次に6.16mg/mL NaClと1mg/mL Tween 80の両方を含有する溶液を試験した。
【0167】
【表6】
【0168】
【表7】
表4Bに示すように、塩と界面活性剤を含有する製剤では、Tween 80の添加にも拘わらず不可視粒子が明白であったことから、界面活性剤を添加しても不可視粒子形成については影響がなかった。興味深いことに、全サンプルで粒子数は最低保存温度(5℃)で最大であり、粒子起源が潜在的に無機材料によることが示唆された。更に、塩を含有する溶液の目視検査によると、保存温度に関係なく、4週間保存後に若干の濁りが判明した。保存が一時的であっても、低温保存の結果として可視無機成分が沈殿する場合があり、例えばリン酸ナトリウム緩衝液は4℃で比較的不溶性のNa2HPO4・12H2Oを生じる場合がある(Borchert et al.(1986)PDA J.Pharm.Sci.Technol.,40:212−241)。他方、粒状物を評価基準とすると、溶液pH5.2は試験した溶液ではpH6.0よりも有利であった。
【0169】
他方、モノマー含有量については、どちらの溶液pH値も保存中に同一のモノマー含有量となり、NaCl含有製剤(Tween 80不含)の場合、pH6.0では安定性が若干高くなるとさえ思われた。この同様のモノマープロファイルにも拘わらず、中性又は塩基性条件よりのpH値で蛋白質はより多様な潜在的分解メカニズムを受け易いと一般に認められており(Wang(1999)Int.J.Pharm.,185:129−188)、例えば非イオン化蛋白質アミドのカルボニル−アミン反応、(塩基触媒)β脱離及び脱アミド化は各種酸化反応と同様に高pH値により助長される(Akers and DeFelippis,Peptides and proteins as parenteral solutions,in Pharmaceutical formulation development of peptides and proteins,ed.by Frokjaer,S;Hovgaard,L.(2000)145−177)。従って、要約すると、溶液pH5.2はアダリムマブ50mg/mL長期安定性の点で6.0値よりも優れていると判断された。
【0170】
添加剤による安定化:界面活性剤
50mg/mLアダリムマブ製剤に及ぼす界面活性剤の安定化能を調べるために、6.16mg/mL NaClを含有する蛋白質溶液に種々の量のTween 80(0.%、0.03%、0.1%)を加えた。一般に、Tween 80は例えば疎水性表面相互作用により結合することにより蛋白質を安定化させると予想される。蛋白質の表面特性は塩の存在に影響されるので、NaCl不在の影響も調べた(NaCl不含0.1% Tween 80溶液として表5に示す)(Kheirolomoom et al.(1998)Biochem.Eng.J.,2:81−88も参照5.53een)。
【0171】
【表8】
NaClの存在下と不在下でTween 80の量を変化させた結果を表5に示す。この結果から明らかなように、Tween 80はNaClの存在下又は不在下で製剤に安定性を提供することができなかった。0.03% Tween 80/NaClについては、併用の結果、40℃で12週間保存後にモノマー含有量が低下した。一般にTween 80の安定化効果は界面活性剤濃度の増加に相関する(0.001〜1%の範囲で有効)ので、この結果はこの主題に関する大半の論文と相反するものであった(Arakawa et al.(2001)Adv.Drug Deliv.Rev.,46:307−326参照)。
【0172】
NaClの存在下及び不在下で種々のTween 80百分率におけるモノマー濃度に加え、種々の温度で不可視粒子形成も試験した(表6参照)。全保存温度で、Tween 80を加えると、特に0.03%の濃度で不可視粒子数は実質的に増加し、SE−HPLC分析の結果が確証された。興味深いことに、保存温度に関係なく、NaClの不在下では不可視粒子形成は著しく減ることが判明した。
【0173】
【表9】
【0174】
更に凍結/解凍サイクル後の粒状物形成について各種濃度のTween 80を試験した。保存中の溶液に及ぼす僅かな安定化効果とは対照的に、Tween 80は凍結・解凍サイクル中にアダリムマブに対する顕著な安定性を付与することが判明した(表7)。
【0175】
【表10】
【0176】
凍結(−80℃,12時間)と解凍(5℃,12時間)によるストレスを溶液に繰返し付与することにより更にTween 80の影響を検討した。適用した凍結・解凍(凍結/解凍)サイクル数は不可視粒状物の増加に密接に相関した。他方、Tween 80濃度0又は0.03%の溶液に及ぼす5回の凍結/解凍サイクルの影響の結果、粒子汚染(粒子≧1μm)は〜10倍に増加したが、0.1% Tween 80溶液では状況は殆ど変わらなかった。これらの結果はSE−HPLC分析により確証された(表8)。
【0177】
【表11】
他の蛋白質に及ぼす凍結/解凍サイクルの影響について発表されている多数の試験の結果と厳密に一致し、界面活性剤の非添加時に反復凍結/解凍ストレスに暴露すると、50mg/mLアダリムマブの安定性は低下した。逆に、界面活性剤を添加すると、(多角度光散乱法(MALS)を使用して検証した)天然モノマー含有量は変わらなかったため、凍結/解凍に関連する有害なパラメーターに対して蛋白質は保護された。
【0178】
要約すると、0.1% Tween 80をアダリムマブ50mg/mL溶液に添加することが好ましかった。0.1% Tweenは保存液中の蛋白質安定性を僅かに改善したが、凍結・解凍等のプロセス中に安定化効果は十分であった。他方、凍結は蛋白質製剤の製造、保存及び輸送における一般的な単位操作であるため、Tween 80の添加は大きな効果として現れる(Cao et al.(2003)Biotechnol.Bioeng.,82:684−690)。更に、Orthoclone(登録商標)(マウスIgG2a)が1986年という初期にFDAに認可されていることから明らかなように、薬剤中で0.1% Tween 80を使用することは定着している。
【0179】
Tween 80以外に、非イオン性界面活性剤Solutol(登録商標)HS15についてもそのアダリムマブ安定化能を試験した。濃度0.03及び0.1%のSolutol(登録商標)の保護特徴はアビスクミン非経口薬について最近報告されている(Steckel et al.(2003)Int.J.Pharm.,257:181−194)。従って、粒状物形成汚染の形成についてアダリムマブ溶液に及ぼすSolutol(登録商標)の影響を、0.1% Tween 80を含有する蛋白質溶液と比較した(表9)。
【0180】
【表12】
0.03%及び0.1% Solutol(登録商標)を含有する溶液とは対照的に、夫々1% Solutol(登録商標)及び0.1% Tween 80を含有するアダリムマブ溶液は保存中に粒状物の顕著な増加を示した。低濃度Solutol(登録商標)のこのプラスの効果はSE−HPLC分析データには反映されなかった。12週間保存(40℃)後に、Solutol(登録商標)を含有する全溶液は参照(0.1% Tween 80)に比較して〜0.5%のモノマー含有量の低下を示した(図1)。
【0181】
この実験では、高分子量(hmw)蛋白質凝集物の初期段階検出で大きな効果が得られることもMALSにより判明した(図2A及び2B)。大量の分析物で高感度であることから、MALSにより凝集物を検出するには最低濃度で十分であり、例えば1週間保存(40℃)後のhmw凝集物の形成はMALSにより確認することができたが、UV280検出では殆ど検出不能であった。
【0182】
従って、加速貯蔵寿命試験の初期段階で既にhmw凝集物が形成されていることは一般に許容できないため、潜在的安定剤のリストからSolutolを除外した。最少量(<0.1%)の蛋白質でも沈殿に原因となることが知られている(Hoffman,Analytical methods and stability testing of biopharmaceuticals,in Protein formulation and delivery,ed.by McNaIIy,E.J.,3(2000)71−110)。上記結果はより高濃度(>1%)のSolutol(登録商標)HS15がセルピン関連プロテアーゼ阻害剤の溶液を脱安定化し、可視粒状物現象に有効であったという従来の報告を裏付けるものである(例えばWO2006037606参照)。
【0183】
添加剤による安定化:ポリオール
多くの糖質(例えばスクロース、グルコース、ラフィノース、トレハロース)とポリオール(例えばグリセロール、ソルビトール、マンニトール)は蛋白質を安定化する補助溶媒に分類される。これらの物質は主に立体排除メカニズムにより作用すると広く考えられている。例えば、ソルビトール等のポリオールは、例えばMumpsvax(登録商標)、Meruvax(登録商標)II及びAttenuvax(登録商標)等の多数の凍結乾燥ワクチン製剤又はCardene(登録商標)等の静脈内投与可能な溶液で非経口薬を安定化させるために使用されることが多い。
【0184】
界面活性剤等の他の添加剤とは対照的に、糖質とポリオールはその完全な安定化能を発揮するためにはより高濃度(>0.5M)で添加する必要がある。従って、50及び100mg/mLの濃度のソルビトールをアダリムマブ溶液に加え、12週間保存した(表10)。
【0185】
【表13】
【0186】
ソルビトールはソルビトールを添加しない溶液に比較して保存中の粒子形成傾向を低下させた。ソルビトールの添加量は殆ど差を生じなかった。モノマー含有量については、ソルビトールの安定化効果は厳密に濃度依存性であることが判明した。NaClの存在は蛋白質安定性を低下させる(表11)。
【0187】
【表14】
表11によると、100mg/mLソルビトールを添加すると、40℃で12週間保存中にモノマー含有量は〜1.5%増加した。添加剤の量を減らすと、アダリムマブ安定性は低下する。ウマ免疫グロブリンの安定性に関する最近の報告によると、180mg/mLソルビトールは熱ストレスに対する蛋白質安定化に関して90mg/mLの添加よりも優れていることが判明したが、これらの結果はこの報告を裏付けるものである(Rodrigues−Silva et al.,1999 Toxicon 37(1),33−45)。糖質及び糖質由来ポリオールの安定化の濃度依存性は報告されている(Chan et al.(1996)Pharm.Res.,13:756−761;Fatouros et al.(1997b)Pharm.Res.,14:1679−1684)。興味深いことに、表11に示すように、4mg/mL塩を添加すると、ソルビトールの安定化能は著しく低下した(〜0.25%モノマー)。他方、(表11に示すように)0.1% Tween 80を含有するアダリムマブ溶液にNaClを添加しないと、貯蔵寿命実験中にモノマー含有量はごく少量であるが増加した。
【0188】
表12に示すように、ソルビトールの代わりにマンニトールを使用して実験を繰返した。ソルビトールで得られた結果はマンニトールをアダリムマブ溶液に添加することにより確証され、(1)80mg/mLマンニトールを添加した溶液は12週間保存(40℃)後にマンニトール不含溶液よりも蛋白質モノマー含有量が〜1.5%上回り、(2)マンニトールの安定化添加量は濃度依存プロフィルの傾向があり、(3)NaClはマンニトール単独の漸減するモノマー含有量を低下させた。興味深いことに、これらのデータは25℃で実施した同一実験により確証された。
【0189】
【表15】
要約すると、濃度50mg/mLのアダリムマブはソルビトールとマンニトールのどちらでも安定化された。この安定化はNaClにより妨害された。0.1% Tween 80を含有する蛋白質溶液に添加した場合にNaClがアダリムマブ安定性を妨害しないという結果は上記結論と一致した。
【0190】
表13に示すように、各アダリムマブ製剤中の天然モノマーの量はポリオールの添加と添加剤混合物に依存した。これに対応して、凝集物とフラグメントの量も変化した。モノマー損失量における凝集物の割合は添加剤を添加しても添加に関係なく一定であった。換言するならば、アダリムマブ凝集物とフラグメントの比は平衡しており(即ち〜38%凝集物と〜72%フラグメント)、この平衡はポリオールと塩の添加により影響されなかった。ソルビトールとマンニトールが天然状態安定化のみによりアダリムマブ安定性に寄与していたならば、これは凝集物割合の変化に反映されるはずである。そうでなかったことから、ソルビトール/マンニトールによるアダリムマブ安定化には別のメカニズムがあり、その結果、断片化プロセスが妨害されたと考えられる。
【0191】
【表16】
結論として、濃度50mg/mLのアダリムマブはマンニトール又はソルビトールを製剤に加えることにより有効に安定化された。天然状態保護による蛋白質安定性への寄与以外に、マンニトールとソルビトールは別のメカニズムにより蛋白質を安定化し、長期保存中に断片化を抑制した。
【0192】
添加剤による安定化:塩
NaClは蛋白質非経口薬の製剤化で最もよく使用されている塩である。しかし、上記結果によると、50mg/mLのアダリムマブ濃度でNaClはポリオールの存在下でアダリムマブ安定性を妨害し、単独添加剤として蛋白質安定性を増加しなかった。塩の潜在的安定化効果を考慮した場合、ホフマイスター離液順列に従ってその挙動を考察すると、大まかな原則が得られた。従って、ナトリウム塩における対イオンとして塩化物の代わりにアニオン性酢酸塩の使用について検討した。
【0193】
表14に示すように、各溶液(即ち50mg/mLソルビトール/4mg/mL酢酸Na、50mg/mLソルビトール/4mg/mL NaCl、及び50mg/mLソルビトール、塩不含)は異なる蛋白質安定性を示した。NaCl又は酢酸ナトリウムを含有する製剤を比較すると、僅か4週間保存(40℃)後に酢酸ナトリウムを添加したバッチにおけるモノマー含有量はNaClを含有する製剤よりも〜0.25%高くなり、12週間後に差は>0.4%まで拡大したことから、NaClを含有するアダリムマブ溶液は蛋白質安定性に対して不利であった。従って、酢酸ナトリウムは塩化ナトリウムよりもアダリムマブ安定性に大きく寄与した。他方、塩不含製剤はモノマー含有量が同一であったことから、酢酸ナトリウムを添加しても蛋白質安定化は増加しなかった。
【0194】
【表17】
他の2種類の製剤(50mg/mLソルビトールと4mg/mL NaClを含有する製剤と、塩を含有しない製剤)と比較すると、酢酸塩を含有する製剤は1μm超の粒子数が多かった(<6,000個/mLに対して〜180,000個/mL)。
【0195】
緩衝液系も試験し、ソルビトール濃度を変化させてナトリウム緩衝液系とカリウム緩衝液系を比較した。表15に示すように、リン酸カリウム緩衝液に溶解したアダリムマブの安定性はリン酸ナトリウム緩衝液中で測定された安定性と同等であった。これらの結果は25℃で実施した保存試験のデータにより裏付けられた。更に、両者緩衝液系は粒状物汚染も同等であった。従って、液体蛋白質製剤ではリン酸カリウムのほうが好ましいと判断された。
【0196】
【表18】
要約すると、50mg/mLのアダリムマブ溶液を製剤化する際にはNaClの添加を避けるべきである。例えば浸透圧の理由により塩を添加することが好ましい場合には、酢酸ナトリウムのほうが塩化ナトリウムよりも有利である。同様に、カリウム系リン酸緩衝液系はアダリムマブ安定性に関してリン酸ナトリウム緩衝液系と同等であった。
【0197】
要約すると、約50mg/mLのアダリムマブ溶液には溶液pH5.2と0.1% Tween 80の添加が他の方法よりも有利であった。凍結/解凍試験及び(加速)保存試験後の蛋白質安定性及び粒状物汚染を評価基準として使用した。更に、マンニトールやソルビトール等のポリオールはほぼ同一の効力で蛋白質安定性に実質的に寄与した。蛋白質凝集と断片化のどちらも妨げられたので、天然状態蛋白質における優先的蓄積が唯一の安定化経路ではなかった。NaClはポリオールの存在下で蛋白質安定性を妨害した。酢酸ナトリウムの添加は蛋白質安定性に有害な影響を生じなかった。
【0198】
これらのデータから、pH5.2のリン酸カリウム緩衝液と、0.1% Tween 80と、〜50mg/mLマンニトール又はソルビトールを含有しており、アダリムマブ濃度50mg/mLで最終浸透圧値〜300mosM/kgを目標とする製剤が提案された。
【実施例2】
【0199】
高濃度アダリムマブ製剤
本実施例は抗TNFα抗体アダリムマブを含有する多数の高濃度蛋白質製剤の成分について記載する。驚くべきことに、下記製剤は抗体濃度が高い(即ち約100mg/mL)にも拘わらず、多数の有利な特性があった。
【0200】
濁度を含む製剤(F1〜F6と言う)の多数の特徴を市販50mg/mLアダリムマブ製剤(F7)と比較検討した。溶液の濁度は未希釈溶液の分析により測定した。濁度をNTU値(ネフェロメトリー濁度単位)として報告する。
【0201】
ドイツ医薬品規格(German Drug Codex)に記載されているような目視検査により可視粒子汚染を測定した。USPに準拠する光遮蔽法により不可視粒子をモニターした。希釈溶液の動的光散乱分析を使用し、流体力学的直径を求めた(粒子の粒度分布のDLS測定強度自己相関関数と多分散指数PDIのキュムラント分析により計算した平均又はZ平均寸法として報告する)。
【0202】
フラグメントと凝集物の検出を可能にするSECにより製剤の物理化学的安定性を評価した。化学的安定性をモニターするために、SE−HPLC(フラグメント及び加水分解試料の検出)とCEX−HPLC(カチオン交換HPLC)を使用した。CEX−HPLCは保存中に形成され得る各種リジンアイソフォーム及び分解生成物(例えば脱アミド化及び酸化種)を分離する。
【0203】
試験した製剤はF1〜F6(表16)とし、pH5.2〜pH6.0の各種基剤中に100mg/mLアダリムマブを含有しており、各種ポリオールを添加し、塩化ナトリウムの存在下又は不在下で製剤化した。
【0204】
【表19】
【0205】
下記実施例3〜6に記載するように、総合的な安定性と粘度を決定するために、上記100mg/mL製剤(F1〜F7)を更に試験した。
【0206】
以下、特に代表的溶液F2及びF6に関して、高濃度アダリムマブ製剤の製造方法について説明する。出発溶液は精製抗体を液体緩衝剤(例えば前段階の製造工程段階からの緩衝液)に低濃度(本発明の高濃度よりも低濃度)で溶解した溶液とする。この場合には、pH5.2で界面活性剤を添加しないF7と同一の緩衝液系に約70mg/mLの濃度で溶解したアダリムマブ溶液を準備した。次に、出発溶液を濃縮し、例えば10kDのカットオフで抗体を定量的に保持することが可能な膜を使用して、好ましくは接線流濾過システムで限外濾過により透析濾過する。
【0207】
1例として、透析濾過緩衝液として界面活性剤を添加しない対応する基剤を使用して濃厚液を約50mg/Lまで希釈することにより代表的製剤F2及びF6を製造した。接線流濾過システムを使用して連続緩衝液交換を実施した。一般に少なくとも5容量、又は好ましくは8容量の透析濾過緩衝液を使用して一定の保持液量で透析濾過を実施した。最終段階で、透析濾過溶液を高濃度(例えば150mg/mL以上)まで更に濃縮した。次に管を透析濾過緩衝液でフラッシュすることにより濁った最終保持液を限外濾過システムから回収した。各量のポリソルベート80を添加し、透析濾過緩衝液を使用して目標蛋白質濃度に調整後、透明〜やや乳白色の高濃度液体製剤を得た。0.22μmフィルターで濾過後、溶液は約2〜8℃で保存した場合に少なくとも約12カ月間安定であった。
【実施例3】
【0208】
凍結/解凍ストレスに対する高濃度アダリムマブ製剤の安定性
アダリムマブ製剤が100mg/mL蛋白質濃度で安定であることを実証するために、凍結/解凍ストレス(−80℃で凍結を実施し、25℃で解凍を実施)実験を実施した。
【0209】
粒子形成に対して感受性の一連の分析方法を使用して潜在的な物理的不安定を検出した。コロイドないし可視範囲の粒状凝集物の生成の指標として濁度を測定した。濁度(NTU値として報告する)は4回目の凍結/解凍サイクル後もさほど変化しなかった(図3)。高pH溶液の濁度上昇は蛋白質のpI(アダリムマブ8.5)に近いpHにおける電荷反発の低下による蛋白質−蛋白質相互作用の増大に起因すると考えられる(Wang et al.(2007)J Pharm Sci 96(1)2457−2468)。
【0210】
サブミクロン範囲の粒度を測定するための方法として動的光散乱法を使用した。粒度分布測定中に得られた多分散指数値をコロイドないしマイクロメーター寸法範囲の凝集の別の感受性指標として使用した。濁度データと同様に、試験した製剤のうちで物理的不安定の徴候を示すものは皆無であった(図4)。
【0211】
更に、サイズ排除データを評価した。図5は凝集物濃度を示す。反復凍結/解凍ストレスに対して物理化学的不安定の徴候は全く検出されなかった。
【0212】
凍結/解凍処理により実質的な蛋白質変性と凝集が生じ、その結果、可溶性及び不溶性凝集物形成を生じる場合があることは周知である(Parborji et al.(1994)Pharm Res 11(5)764−771)。本願に記載する全製剤を反復凍結・解凍処理した結果、反復凍結/解凍サイクル(−80℃/25℃)に感受性の製剤は皆無であることが判明した。製剤のpHがアダリムマブのpI(即ち8.5)に近い高値であるにも拘わらず、全製剤はそのpHから独立して同様に安定であった(いずれの場合も初期値に比較して優位変化は認められなかった)。
【0213】
これらの結果は各種緩衝液を比較した別の試験からのデータにより裏付けられた。凍結・解凍後に均質溶液(即ちpH、浸透圧、密度の勾配が最少の溶液)が得られるという点と凍結・解凍中のpH変化が最少であるという点で最も有益な緩衝液系はNaClを添加しない緩衝液組成であることが判明した(実施例1参照)。pH6で製剤化したNaCl不含緩衝液系は評価した全pHレベルのpH変化が最少であることが判明した。
【実施例4】
【0214】
等張化剤として各種ポリオールを含有する100mg/mL製剤の安定性
示差走査熱量測定(DSC)を利用し、全100mg/mLアダリムマブ製剤を安定性全般について試験した。Microcal製VP Capillary DSCを使用してDSCデータを得た。全実験は以下の標準化手順:温度範囲:20℃〜90℃、加熱速度:1K/min、蛋白質濃度1mg/mLを使用する1回の加熱試験で実施した。
【0215】
Tm値が高いほど一般に構造安定性は高い(Singh et al.(2003)AAPS PharmSciTech 4(3)article 42)。図6は100mg/mLアダリムマブ製剤のTm値を示す。これらのデータから明らかなように、全製剤は高いTm値に達する。他方、塩化ナトリウム不含製剤(F2,F3,F5,F6)は有意に高いTm値を示し、これらの製剤の堅牢性を示した。1mg/mLで製剤を試験するので、F1のTmデータはF7のTmと同一であり、塩化ナトリウムの不在下又はpH6.0の100mg/mL製剤の安定性はF7製剤よりも改善されることが確認された。
【0216】
磁気撹拌棒を使用する撹拌ストレスモデルを使用し、新規アダリムマブ製剤の物理化学的不安定を検出した。この周知モデルはアダリムマブを長期気液界面暴露と撹拌関連キャビテーションに供することによりストレスを誘導し、予測可能な方法で可溶性及び不溶性蛋白質凝集物の形成をもたらす。
【0217】
一般に、夫々のpI(アダリムマブpI8.5,低純電荷,最小化静電反発力)の範囲のpH値で製剤化した蛋白質は反発力が弱いため、気液界面に関連する凝集に対する感受性が高い。更に、塩化ナトリウム等のイオン性添加剤はそれらのイオン遮断性により蛋白質凝集に関与する。塩化ナトリウムの存在により疎水性吸引力を低下させ、蛋白質−蛋白質相互作用を抑制し、コロイド安定性を増すことができる(Shire et al.(2004)J Pharm Sci,93(6)1390−1402)。
【0218】
撹拌ストレスにより誘導される凝集物形成を検出するために濁度データを評価した。表17は製剤組成と撹拌時間に対するネフェロメトリー値を示す。(5.2の低pHで製剤化した)F1〜F3の初期濁度値は塩化ナトリウム含有溶液(F1)とNaCl不含溶液(F2,F3)で差を示した。他方、6.0の高pHに調整した溶液(F4〜F6)は濁度が高かった。NaClが撹拌等の機械的ストレス後にmAb溶液の透明度を低下させ得ることは当分野で知られている(例えばFesinmeyer et al.(2009)Pharm Res,26(4)903−913)。
【0219】
【表20】
【0220】
48時間まで撹拌すると、試験した全製剤で濁度値が上昇した。低pHのNaCl不含製剤は撹拌による濁度上昇を最も生じにくかった。驚くべきことに、試験した全100mg/mL製剤は低濃度(50mg/mL)アダリムマブ製剤に比較して撹拌後の濁度が有意に低かった(表18)。
【0221】
一般に、乳白外観は単なるレイリー散乱の結果であり、蛋白質濃度と線形相関する。他方、乳白外観は物理的不安定を生じない(Sukumar et al.(2004)Pharm Res 21(7)1087−1093)。50mg/mLアダリムマブ製剤は24時間撹拌後に63〜130NTU、48時間後に109〜243NTUの濁度を示したが、100mg/mLアダリムマブ製剤は27〜63(24時間)から40〜87(48時間)の数値であった。Treuheit et al.((2002)Pharm Res 19(4)511−516)によると、蛋白質濃度が高いと、10mg/mL未満の範囲のOPC−Fc溶液中で気液界面に誘導される凝集は減少する。同様の結果がKiese et al.((2008)J Pharm Sci 97(10)4347−4366)からも報告されている。意外にも、新規アダリムマブ製剤は100mg/mLという著しく高い蛋白質濃度で撹拌ストレス安定性の増加の特徴を示した。
【0222】
従って、新規製剤は50mg/mL製剤に比較して安定性が増加した。
【0223】
【表21】
【0224】
更に、サイズ排除クロマトグラフィーデータによると、全100mg/mL製剤は48時間撹拌後の凝集物濃度が<1%であり、新規製剤の安定性が裏付けられた(図7)。やはり、低pHと塩化ナトリウムの不在が有益であった。一般には高pHで純電荷が低いと不安定が増すと考えられているが、このデータは新規製剤がアダリムマブのpIに近いpH値であるにも拘わらず安定であり、NaClの不在が有益であるという驚くべき知見を裏付けるものである。
【実施例5】
【0225】
塩化ナトリウムの存在下及び不在下で2種類の異なるポリオールを添加したpH5.2及び6.0の100mg/mLアダリムマブ製剤の長期安定性
新規100mg/mLアダリムマブ製剤を長期保存し、50mg/mL標準製剤と比較して安定性が優れていることを確認した。5℃(市販品の推奨保存温度)で12カ月間の安定性データを評価した。データは実際に、新規製剤が安定性の低下を示さなかったことを示唆している(表19)。
【0226】
SEC及びIEXに関して、モノマー含有量の有意低下又は測定可能な分解は生じなかった。
【0227】
更に、新規アダリムマブ製剤は蛋白質濃度が高いにも拘わらず、50mg/mL市販アダリムマブ製剤の12Mデータに比較して不可視範囲の粒子汚染に関する有意強化が得られた。(凝集、沈殿及び一般物理的不安定現象を示す)不可視粒状物汚染を試験した処、新規アダリムマブ製剤は実際に不可視粒子を含まずに維持されることが判明した。最大値28(≧10)及び最大値3(≧25)の初期粒子数は50mg/mL製剤F7(夫々703と38)よりも有意に少なかった。
【0228】
更に、粒子レベルは12カ月間安定性試験を通して有意に変化せず、F7よりも有意に低レベルに維持された。
【0229】
薬剤製品バッチは試験した全保存条件下でその物理化学的安定性に関してほぼ等価であった。例えば蛋白質濃度が高いほど物理的安定性は低下する傾向があることは広く認められているため、これは意外である(Wang W.(1999)Int J Pharm 185:129−188)。
【0230】
【表22】
【0231】
新規100mg/mL製剤の保存安定性増加の結果を検証するために、2種類の代表的な製剤であるF2及びF6を加速安定性試験(5℃、25℃、40℃で3カ月)に供し、市販50mg/mL製剤(対照試験からの代表的バッチ)と比較した。これらの実験の結果を図8〜13にまとめる。
【0232】
これらのバッチからの濁度データは特に5.2の低pHで100mg/mLのNaCl不含製剤の優れた挙動を実証する。溶液中の蛋白質の濃度が増加すると、乳白が増し、レイリー散乱により濁度読取り値が増すことは一般に知られている(Sukumar et al.(2004)Pharm Res 21(7)1087−1093)。驚くべきことに、塩化ナトリウムを添加しない新規製剤は50mg/mL製剤と同一pHで同等の濁度レベルであることが判明した(図8)。
【0233】
図9〜11は新規製剤の粒状物形成(可視粒子と不可視粒子)の詳細なデータを示す。安定性の増加という驚くべき知見が確証された。実際に、高温で3カ月間保存後も不可視粒子及び可視粒子両方のスコアを下げることが可能であった。
【0234】
図12〜13に示すデータはSEC分析とIEXを使用して試験した化学的安定性の両方で安定性の問題を示さないため、100mg/mL製剤の安定性を更に裏付けた。
【実施例6】
【0235】
50mg/mLアダリムマブ製剤と比較した100mg/mLアダリムマブ製剤の製造適性の強化
本実施例は現在市販されている50mg/mL製剤に比較して新規100mg/mLアダリムマブ製剤(代表的製剤F2及びF6)のプロセス安定性の改善に関するデータをまとめる。
【0236】
ポンピング、濾過、混合、充填−仕上げ工程、輸送又は振盪により生じる機械的ストレスは空気−水界面、材料表面及び剪断力への蛋白質の暴露により、変性とそれに続いて凝集を生じる恐れがある(Mahler at al.(2005)Eur J Pharm Biopharm 59:407−417;Shire et al.(2004)J Pharm Sci,93(6)1390−1402)。
【0237】
先ず蛋白質溶液の処理適性の特徴を表す基本パラメーターとして粘度値を測定した。表20はF1〜F7製剤で得られた粘度データを示す。蛋白質濃度が増加すると、50mg/mL製剤(F7)に比較して粘度が増加した。
【0238】
静電遮断剤であるNaClを除去すると、特にアダリムマブのpIに近いpH値で疎水性蛋白質相互作用が増加し、従って粘度が増加すると予想される。この作用はNaCl濃度<200mMで最も顕著であると報告されている(Shire et al.(2004)J Pharm Sci,93(6)1390−1402)。
【0239】
しかし、意外にも、製剤からNaClを除去しても(F1は〜105mM NaClを含有する)依然として約3.1〜3.3mPa・sという比較的低い粘度値であった(F2、F3、F5及びF6)。これは6.0という高pH値の溶液(F5及びF6)で特に意外であった。
【0240】
要約すると、全製剤は液体充填−仕上げ製造操作に最適な範囲の粘度をもつことを特徴とする。
【0241】
【表23】
無菌製造工程中の滅菌濾過により誘導されるストレスを模倣した実験室モデルで、100mg/mLアダリムマブを含有する2種類の代表的な新規製剤から分析データを得た処、全製剤は濾過に関連する剪断ストレスに対して安定であることが分かった。低レベルの高分子量サブ集団の感受性指標である多分散指数は有意に増加しなかったため、DLSデータは高分子量凝集物の形成の徴候を示さなかった。高分子量種は散乱強度が高い(d6に比例する)ため、ZAve及びZAve粒度分布の指標としての多分散指数に有意に影響しないので、特にDLS測定を使用して粒度分布における少量の高分子量種、例えば凝集物を検出する。更に、濾過により凝集が誘導されないことがSECデータにより確認された。
【0242】
驚くべきことに、100mg/mL製剤でも不安定を示さなかった。最悪の場合のシナリオとして複数回滅菌濾過後でも、蛋白質含有量が高いにも拘わらず、処理適性は高レベルに維持された。
【0243】
【表24】
プロセス関連ストレスに対する新規アダリムマブ製剤の高い安定性を更に実証するために、磁気撹拌棒の撹拌速度を変えてその挙動を比較する撹拌ストレスモデルで製剤を試験した(撹拌ストレスは配合プロセス段階で製造条件下に生じる)。
【0244】
撹拌ストレス耐性を比較した処、100mg/mL蛋白質濃度で濁度の上昇は認められなかった(図14)。塩化ナトリウムを添加せずにポリオール含有量を高くした代表的な100mg/mL製剤はいずれも試験した全撹拌速度でpH5.2の市販製剤と同様に挙動した。撹拌速度を上げると、全製剤は24時間撹拌後に濁度値の僅かな上昇を示したが、100mg/mLで剪断ストレスによる不安定に対する感受性の顕著な増加は検出されなかった。
【0245】
DLS測定により得られる流体力学的直径の変化を比較した処、同様のデータが得られた。蛋白質濃度の高い製剤のほうが撹拌ストレスに対する感受性が強いと考えられるが、どちらの100mg/mL製剤も50mg/mL製剤と同様に挙動した。驚くべきことに、pHが最高の製剤F2は濁度及び流体力学的直径のどちらの分析でも相対増加が最低であることが判明した(図15)。
【0246】
蛋白質濃度が高くてもプロセス安定性は同様であるというこの驚くべき結果はポンピングプロセスにより誘導されるストレスを模倣した機械的ストレスモデルにより更に確証された。製造工程のこの最終段階は蠕動ポンピングによる剪断ストレスを伴い、溶液不安定の危険を増す。この場合も、濁度(図16)とDLS(図17,表22)を使用して得られたデータは、新規100mg/mL製剤が粒子成長反応を生じず、50mg/mL製剤と同様に安定に維持されることを裏付けた。ポンプストレスにより誘導した凝集物形成に対する感受性は検出できなかった。この結果は更にSECデータにより裏付けられ、試験した製剤はポンプサイクルに関して差がないことが分かった(図18)。
【0247】
【表25】
各種充填装置(回転ピストンと蠕動ポンプ)を使用し、100mg/mL製剤の安定性の差を評価した。
【0248】
これらの試験の結果、特にpHの高い塩化ナトリウム含有製剤(F1及びF4)では、ピストンポンプで発生する剪断ストレスが高いほど、可視粒子数が増加した。同様の結果がBausch,Ursula J.から最近報告されている(Impact of filling processes on protein solutions.2008,PhD Thesis,University of Basel,Faculty of Science;http://edoc.unibas.ch/845/1/DissB_8427.pdf)が、これはリツキシマブ溶液の10mg/mLの蛋白質濃度に過ぎない。驚くべきことに、100mg/mLアダリムマブを含有する塩化ナトリウム製剤はピストンポンプを使用する高剪断条件下で処理適性の改善を示した。
【0249】
図19〜22はプロセスストレス増加条件に対するNaCl含有アダリムマブ溶液の感受性の増加を裏付ける粒子数と濁度データを示し、DAC目視スコア法による≧10μm及び≧25μmの粒度範囲の測定は非経口薬に不可欠の品質属性である。従って、NaCl不含製剤における不可視粒子の減少により有意な製剤改善が得られる。
【0250】
図19に示すように、蠕動充填の結果、充填直後(T0)及び保存後に可視粒子形成は生じなかった。他方、ピストン充填の結果、pH6.0で製剤化した溶液ではT0でも有意粒子数が認められた(図20)。塩化ナトリウムを含有するF4で最高値が測定されたが、F5〜F6はスコアが有意に低く、プロセスストレスに対する塩化ナトリウム不含製剤の安定性の改善が確証された。
【0251】
濁度測定により裏付けとなる結果が得られた(図21〜22)。ピストンポンプを使用して充填した溶液の初期値は蠕動充填法を使用して充填した溶液よりも高かった。塩化ナトリウム不含製剤は塩化ナトリウムを含有する製剤よりも濁度が低かった。更に、ピストン充填による剪断ストレスの結果、濁度に関してF4(塩化ナトリウム添加)をF5及びF6(塩化ナトリウム非添加)から識別することができた。
【実施例7】
【0252】
塩化ナトリウム不含製剤中の各種ポリオール濃度の比較
100mg/mLアダリムマブを含有する以下の塩化ナトリウム不含製剤を5℃における短期安定性に及ぼすポリオール濃度の影響について試験した。
【0253】
凝集及び粒子形成傾向に関して不良な条件となるように、製剤をpH6.0に調整した。
【0254】
【表26】
塩化ナトリウム不含溶液の張度要件に見合うように、マンニトール又はソルビトールを42mg/mLの濃度で使用した。データによると、先に使用した12mg/mLの濃度に比較して、どちらのポリオールも溶液の浸透圧に寄与するのみならず、更に蛋白質安定性にも有意影響を与えることが判明した。
【0255】
安定性データによると、ポリオールの種類に関係なく、ポリオール濃度が高いほうが透明度は改善されると思われた。一般に最適とはみなされない条件下(例えばアダリムマブのpIに近いpH6.0)で、ポリオール濃度の高い製剤は5℃で4週間の短期保存後でも透明度の改善を示した。これは数種類の分析法で確認された。
【0256】
図23から明らかなように、試験した製剤の透明度はポリオール濃度を増加することにより有意に低下し、試験期間にわたって低レベルに保つことができた。更に、5℃で4週間後に凝集がやや低下した結果、高いポリオール濃度で高いモノマー含有量が認められた(図24及び25)。ポリオール濃度が高いと、≧10μmの範囲の不可視粒子は(例えばT0で)減少した。
【実施例8】
【0257】
ヒト抗TNFα抗体の安定した高蛋白質濃度製剤
加速安定性試験条件下と推奨保存温度条件における長期保存下の両方でアダリムマブの物理的及び化学的安定性を維持する適性について各種アダリムマブ製剤を試験した(下表1参照)。製剤のpH(pH5.2とpH6)、添加剤条件(例えばマンニトール又はソルビトールの濃度)、塩/イオン強度条件(例えばNaClの濃度)、及び蛋白質濃度(50mg/mLと100mg/mL)を変化させた。
【0258】
【表27】
【0259】
表2はストレス温度とサンプル抽出点の総覧である。製剤F2及びF6はアダリムマブの物理的及び化学的安定性の両方を夫々少なくとも18カ月間及び12カ月間維持する製剤であると認められた。製剤添加剤NaClをマンニトール(製剤F2)とソルビトール(製剤F6)に交換すると、蛋白質濃度の100%増加(製剤F7の50mg/mLから製剤F2及びF6の100mg/mLまで)にも拘わらず、高い安定化能が得られる。驚くべきことに、両製剤の物理的安定性は夫々少なくとも12カ月間及び18カ月維持された。12カ月間保存後も、両製剤のモノマー含有量は99%を上回り(SECデータ)、凝集物濃度は1%未満であった。
【0260】
同様に、リジン変異体合計(L0+L1+L2)を表す安定性は80%を上回ったため、非常に多くの場合に蛋白質薬剤における貯蔵寿命制限因子である化学的安定性も安定性モニター期間にわたって維持された。
【0261】
蛋白質製剤の物理的及び/又は化学的安定性をモニターするのに適切であるとして当分野で認められている他の試験(例えば不可視粒子試験、濁度測定、目視検査、透明度又は色モニター)でも製剤F2及びF6の安定化能が確認された。
【0262】
重要な点として、抗TNF中和試験を示す効力によると、どちらの製剤も完全なサンプル抽出スケジュールにわたってアダリムマブの効力を維持し、データは75〜125%の高品質レベル範囲内にあることが判明した。
【0263】
【表28】
【0264】
【表29】
【0265】
【表30】
【実施例9】
【0266】
高濃度アダリムマブの疼痛試験
皮下注射によりモノクローナル抗体を投与された患者は注射部位に疼痛又は不快感を感じる場合がある(例えばFransson,J.;Espander−Jansson,A.(1996)Journal of Pharmacy and Pharmacology 48(10),1012−1015;Parham,S.M.;Pasieka,J.L.(1996)Can.J.Surg.39,31−35;Moriel E Z;Rajfer J(1993)The Journal of urology 149(5 Pt 2),1299−300参照)。患者の体験を模倣した動物モデルを使用し、疼痛及び耐容性効果を評価すると共に、人体使用前の可能な製剤修飾を評価した。蛋白質製剤の特性を識別する適性について利用可能な動物モデルを評価した。注射時の発声、足フリンチ(注射後0〜10分)、機械的アロディニアの試験、及び熱痛覚過敏(注射後30分)について測定した。患部足をなめたり、振ったりする侵害受容挙動や、注射部位の発赤又は腫脹についても動物を観察した。
【0267】
注射部位疼痛を評価するためにフリンチモデルを選択し、耐容性及び疼痛感覚に及ぼす製剤組成の影響を評価するために使用した。
【0268】
各種アダリムマブ100mg/mL製剤の耐容性を製剤F7(50mg/mLアダリムマブ製剤)と比較した。得られたデータは、50mg/mL製剤(F7)に比較して皮下注射後の注射部位で100mg/mL製剤の耐容性が改善されるという驚くべき結果を裏付けた。
【0269】
注射部位の疼痛等の皮下注射に伴う副作用を抑えるように新規100mg/mL製剤を最適化した。注射部位の疼痛は注射針穿刺に関連する疼痛と、皮下デポーへの溶液注入に関連する感覚の両者を含む。文献から入手可能なデータによると、所定の注射針設計が注射部位の不快感を軽減するために有利であると思われたが、製剤寄与に関する明白なデータは入手できなかった(例えばChan,G.C.F.,et al.(2003)American Journal of Hematology 76(4):398−404参照)。
【0270】
ラット疼痛モデルを使用した本発明者らのデータによると、新規100mg/mL製剤は現在市販されているHumira(登録商標)製剤に比較して同等の治療用量の皮下注射後の注射部位疼痛を軽減するのに有効であると思われた。これは新規100mg/mL製剤の容量注射低下により達成され、患者治療の最適化と患者コンプライアンスの向上という非常に有益な効果を示した。
【0271】
同時に、本発明者らは100mg/mL製剤を製剤化するために許容可能な範囲内の製剤pHが注射部位疼痛に影響を与えないことも確認した。興味深いことに、生理的pH範囲からかけ離れた低いpH値も同様の耐容性で投与可能であった。
【0272】
耐容性試験に適用した方法:
足フリンチ及び侵害防御挙動アッセイ
成体雄性Sprague Dawleyラットを右後足への試験溶液の足裏(s.c.)注射前20〜30分間試験条件に馴化させた。足フリンチ数を記録し、注射後、最初の10分間、侵害防御挙動(足をかばう又はなめる動作)に費やした時間を測定した。特に指定しない限り、全試験溶液は総容量150μLを注射した。実験はコード化し、盲検・無作為法で行った。食塩水とカプサイシン(2.5μg)を夫々陰性及び陽性対照として使用した。
【0273】
容量効果
足フリンチ応答に及ぼす注射容量の影響をプラセボ及び試験製剤F7の両者で試験した。物理的容量を減らすことにより応答を改善できるか否かを調べるために、注射容量の変動(10μl、50μl及び150μlを足裏注射)がフリンチ結果に及ぼす影響を試験した。
【0274】
試験データから容量効果を以下のようにまとめることができる:150μlではプラセボ(32±12)とF7の両者で食塩水(4±2)に比較してフリンチが有意に増加したが、容量を減らすと、食塩水と区別がつかなかった。150μLという高い注射容量では高いフリンチ応答が常に生じたが、低容量(10μL及び50μL)では有意に低い応答となった。
【0275】
この結果から、注射容量を減らすと刺激が低下すると思われ、F2及びF6等の高濃度製剤はF7等の低濃度製剤に比較して耐容性及び疼痛感覚に関して有利であると考えられる。
【0276】
【表31】
【実施例10】
【0277】
耐容性/疼痛に及ぼすアダリムマブ含有溶液のpH効果
アダリムマブを含有する活性溶液で別の実験を実施した。試験した製剤はF2(pH5.2)、生理的条件により近いpH値の対応する製剤であるF5及びF7とした。
【0278】
データによると、pHは足フリンチ応答と侵害防御挙動に費やされた時間を使用して測定した動物応答に影響を与えないと思われた。陽性及び陰性対照データは予想範囲内であった。製剤pHが低い(即ち酸性)ほど、特に皮下注射による非経口投与後に不耐容及び疼痛感覚の危険が増すことは文献から周知である。従って、F2及びF5アダリムマブ製剤では、製剤pHが耐容性及び/又は疼痛感覚に影響を与えないことは意外であった。これは製剤pH、物理的安定性及び凝集物濃度(潜在的に免疫原性の危険に相関する)等の他のパラメーターに製剤化意志決定に関する高い優先性を与えることができるので、非常に有益である。
【0279】
【表32】
【実施例11】
【0280】
アダリムマブ不含溶液の製剤pH効果の影響
製剤組成の影響(例えばリン酸塩等の緩衝液、マンニトール等の添加剤、又はポリソルベート80等の界面活性剤の影響)を試験するために、別の実験を実施し、蛋白質不含製剤で同様のデータを得た。プラセボ溶液のpHを約5〜7の範囲で変動させた処、驚くべきことに、pHの異なる製剤で認められたフリンチ応答は同様であったため、疼痛改善効果はないとも思われた。上記に説明したように、これにより製剤処方者は製剤pH、物理的安定性及び凝集物濃度(潜在的に免疫原性の危険に相関する)等の他のパラメーターに製剤化意志決定に関する高い優先性を与えることができるので、バイオ医薬品製剤開発において非常に有益である。
【0281】
【表33】
要約すると、上記データは耐容性を低下0---)--------------)---------+--及び/又は疼痛感覚を増加せずに一定範囲のpHでこれらの高蛋白質濃度の粘性溶液を低容量で投与できるという100mg/mLアダリムマブ製剤の利点を明白に立証するものである。
【0282】
文献援用
本願の随所に引用する全引用文献(例えば、文献資料、特許、特許出願及びウェブサイトを含む)の内容は任意目的でその全文を特に本願に援用する。本発明の実施は、特に指定しない限り、当分野で周知の蛋白質製剤の従来技術を利用する。
【0283】
等価物
その趣旨又は本質的特徴から離れずに本発明を他の特定形態で具体化することもできる。従って、上記態様は本願に記載する発明を制限するものではなく、あらゆる点で例示とみなすべきである。従って、本発明の範囲は上記記載ではなく、以下の特許請求の範囲により指定され、特許請求の範囲の等価物の意味及び範囲に該当する全変更も本願に含むものとする。
【背景技術】
【0001】
抗体等の治療用蛋白質の製剤化は経済的及び治療薬として成功するために多数の望ましい特性(例えば安定性、投与適性、濃度)を満たす必要があるため、困難であることが多い。製造、保存及び配送中に、治療用蛋白質は物理的及び化学的劣化を受けることが知られている。これらの不安定性は蛋白質の力価を下げ、患者に有害なイベントが発生する危険を増し、従って、当局の許可に著しく影響する可能性がある(例えばWang,et al.(2007)J Pharm Sci 96:1参照)。従って、治療用蛋白質の成功には安定した蛋白質製剤が不可欠である。
【0002】
有効にするために、多くの治療用蛋白質は高用量を投与する必要があり、高濃度製剤に製剤化することが好ましい。高濃度蛋白質製剤は薬剤を対象に投与する方法(例えば静脈内と皮下のどちらにするか)と頻度に影響を与えることができるので望ましい。
【0003】
高濃度蛋白質製剤は有益であるが、高濃度治療用蛋白質を製剤化するには多くの問題がある。例えば、蛋白質濃度を上げると、蛋白質凝集、溶解度、安定性及び粘度にマイナスの影響を与えることが多い(例えばShire,et al.(2004)J Pharm Sci 93:1390参照)。高蛋白質溶液に非常に一般的な問題である粘度増加は製剤の投与にマイナスの影響(例えば疼痛感及び灼熱痛症候群や、製造、加工、充填−仕上げ及び薬剤送達装置選択の制限)を与える恐れがある(例えばShire,et al.(2004)J Pharm Sci 93:1390参照)。一般的な構造特徴をもつ治療用蛋白質(例えば抗体)でも、今日までに認可されている製剤は成分と濃度範囲が多様であった。例えば、抗CD20抗体であるリツキサンは10mg/mLの濃度で静脈内投与用に製剤化されているが、抗RSV抗体であるシナジスは100mg/mLの濃度で筋肉内投与用に製剤化されている。このように、治療目的に使用することができる高蛋白質製剤、特に抗体製剤には課題が残っている。従って、投薬及び投与上の利点を提供する安定した高濃度蛋白質製剤が必要とされている。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Wang,et al.(2007)J Pharm Sci 96:1
【非特許文献2】Shire,et al.(2004)J Pharm Sci 93:1390
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明は少なくとも部分的にヒト抗TNFα抗体又はその抗原結合フラグメント(例えばアダリムマブ)の新規高濃度製剤の発見に基づく。本発明の製剤は抗体濃度が高いため、多数の驚くべき特徴を提供する。例えば、本発明の製剤は蛋白質濃度が高いにも拘わらず、物理的及び化学的安定性を長期間維持し、皮下投与に適した粘度をもつ。本発明の製剤は少なくとも部分的にヒト抗TNFα抗体又はその抗原結合部分が高濃度(例えば100mg/mL)で可溶性に維持できると共に、注射(例えば皮下投与)に適した粘度を維持しながら非凝集状態に維持できるという驚くべき知見に基づく。本発明の製剤は、高濃度(例えば100mg/mL)のヒト抗TNFα抗体又はその抗原結合部分が可溶性に維持できると共に、広いpH範囲(例えば約pH5.2〜約pH6.0)にわたって非凝集状態で且つ化学的に安定に維持できる(例えば酸化や脱アミド化を生じない)という点も意外である。これらの有益な特徴は安定剤としてNaClを必要とせずに、糖アルコール添加剤を増加することにより達成される。
【課題を解決するための手段】
【0006】
本発明の1側面は、40mgを上回るポリオールと、少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤を提供する。
【0007】
本発明の別の側面は、20mgを上回るポリオールと、少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤を提供する。1態様において、本発明の製剤はNaClを含有しない。
【0008】
本発明は更に、pHが約5.0〜6.4であり、少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤であって、NaClを含有せず、標準24時間撹拌ストレスアッセイ後又は液体として24カ月間の長期保存後の濁度が60NTU未満である前記製剤に関する。
【0009】
本発明は更に、pHが約5.0〜6.4であり、少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤であって、NaClを含有せず、標準48時間撹拌ストレスアッセイ後の濁度が100NTU未満である前記製剤を提供する。
【0010】
本発明の別の側面は、pHが約5.0〜6.4であり、少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤であって、NaClを含有せず、5℃、25℃又は40℃で3カ月間保存の濁度が40NTU未満である前記製剤を包含する。
【0011】
本発明は更に、少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分と、約20mg/mLを上回るポリオールと、0.1〜2.0mg/mLの界面活性剤と、約1.15〜1.45mg/mLのクエン酸・H2Oと、約0.2〜0.4mg/mLのクエン酸ナトリウム脱水和物と、約1.35〜1.75mg/mLのNa2HPO4・2H2Oと、約0.75〜0.95mg/mLのNaH2PO4・2H2Oを含有する液体医薬製剤であって、pHが約4.7〜6.5であり、NaClを含有しない前記製剤を提供する。
【0012】
本発明の製剤は皮下投与に適している。従って、本発明は更に、対象における有害なTNFα活性に関連する疾患の治療における、ヒトTNFα抗体又はその抗原結合部分を含有する本発明の製剤の使用を包含する。
【0013】
1態様において、本発明の製剤は一定濃度のヒトTNFα抗体又はその抗原結合部分と、約3.1〜3.3mPa・sの粘度を有する。
【0014】
1態様において、本発明の製剤は20mgを上回るポリオールを含有する。本発明の製剤に添加することができるポリオールのその他の量は30mgを上回るポリオールである。あるいは、本発明の製剤では40mgを上回るポリオールを使用することもでき、限定されないが、40〜45mg又は約42mgが挙げられる。
【0015】
1態様において、本発明の製剤で使用されるポリオールは糖アルコールであり、限定されないが、マンニトール又はソルビトールが挙げられる。1態様において、製剤は約40〜45mg/mLのマンニトール又はソルビトールを含有する。
【0016】
本発明の製剤では当分野で公知の各種界面活性剤を使用することができる。1態様において、界面活性剤はポリソルベート80である。別の態様において、本発明の製剤では約0.1〜2.0mg/mLのポリソルベート80を使用する。
【0017】
本発明の1態様において、製剤は約1.30〜1.31mg/mLのクエン酸・H2Oを含有する。
【0018】
本発明の別の態様において、製剤は約0.30〜0.31mg/mLのクエン酸ナトリウム脱水和物を含有する。
【0019】
本発明の更に別の態様において、製剤は約1.50〜1.56mg/mLのNa2HPO4・2H2Oを含有する。
【0020】
本発明の別の態様において、製剤は約0.83〜0.89mg/mLのNaH2PO4・2H2Oを含有する。
【0021】
別の態様において、本発明の製剤のpHは約4.8〜約6.4である。例えば、本発明の製剤のpHは約5.0〜約5.4(例えば約5.2)でもよいし、約5.8〜約6.4(例えば約6.0)でもよい。
【0022】
本発明の製剤の1つの利点は、一般に蛋白質濃度の上昇と共に生じる蛋白質凝集の増加を伴わずに高濃度の抗体を提供する点である。1態様において、本発明の製剤は凝集蛋白質含有量が約1%未満である。
【0023】
本願に記載する製剤として、ヒト抗TNFα抗体又はその抗原結合部分の濃度が少なくとも約50mg/mLである製剤も本発明に含むものとする。
【0024】
1態様において、ヒト抗体又はその抗原結合部分は配列番号3に記載のアミノ酸配列を含むCDR3ドメインを含む軽鎖と、配列番号4に記載のアミノ酸配列を含むCDR3ドメインを含む重鎖を含む。
【0025】
本発明の1態様において、抗体は、配列番号3のアミノ酸配列あるいは1、4、5、7もしくは8位の1カ所のアラニン置換又は1、3、4、6、7、8及び/もしくは9位の1〜5カ所の保存アミノ酸置換により配列番号3から変異したアミノ酸配列を含む軽鎖CDR3ドメインと、配列番号4のアミノ酸配列あるいは2、3、4、5、6、8、9、10もしくは11位の1カ所のアラニン置換又は2、3、4、5、6、8、9、10、11及び/もしくは12位の1〜5カ所の保存アミノ酸置換により配列番号4から変異したアミノ酸配列を含む重鎖CDR3ドメインをもつ。
【0026】
本発明の抗体は所定の機能的特徴をもつことができる。例えば、ヒト抗体又はその抗原結合部分は、いずれも表面プラズモン共鳴法により測定した場合に、1×10−8M以下のKdでヒトTNFαから解離することができ、1×10−3s−1以下のKoff速度定数でヒトTNFαから解離することができ、及び/又は標準インビトロL929アッセイにおいて1×10−7M以下のIC50でヒトTNFα細胞傷害作用を中和することができる。
【0027】
1態様において、ヒト抗体又はその抗原結合部分はヒトIgG1κ抗体である。
【0028】
本発明の1態様において、ヒト抗体又はその抗原結合部分の軽鎖は更に配列番号5に記載のアミノ酸配列を含むCDR2ドメインと、配列番号7に記載のアミノ酸配列を含むCDR1ドメインを含み、及び/又はヒト抗体の重鎖は配列番号6に記載のアミノ酸配列を含むCDR2ドメインと、配列番号8に記載のアミノ酸配列を含むCDR1ドメインを含む。別の態様において、ヒト抗体又はその抗原結合部分の軽鎖は配列番号1に記載のアミノ酸配列を含み、ヒト抗体の重鎖は配列番号2に記載のアミノ酸配列を含む。本願に記載する各種配列番号と少なくとも80%、85%、90%、95%、96%、97%、98%又は99%一致するアミノ酸配列をもつヒト抗体又はその抗原結合部分も本発明に包含する。
【0029】
本発明の更に別の態様において、ヒト抗体又はその抗原結合部分はアダリムマブである。
【図面の簡単な説明】
【0030】
【図1】0.1% Solutolを含有する溶液中の高分子量(hmw)蛋白質試料の存在を示すグラフである。MALS(灰色の線)によると、凝集物モル質量はほぼ109g/molまでに等しく、総蛋白質の2.6%に相当する(UV280,黒線)。40℃で12週間保存。
【図2】図2A及び2Bは40℃で保存中に出現する高分子量(hmw)凝集物の初期段階検出を示すグラフである。UV280(黒曲線)では凝集物を検出できなかったが、MALS(灰色曲線)は明白にhmw試料の存在を立証した。1週間保存後(A)と元のサンプル(B)を比較。
【図3】製剤F1〜F6の凍結/解凍サイクルに対する濁度を示すグラフである。
【図4】製剤F1〜F6の凍結/解凍サイクルに対する多分散指数を示すグラフである。
【図5】製剤F1〜F6の凍結/解凍サイクルに対するSECによる凝集物濃度を示すグラフである。
【図6】T0における製剤F1〜F6のDSCによるTm(℃)を示すグラフである。
【図7】製剤F1〜F6の撹拌時間に対するSECによる凝集物濃度を示すグラフである。
【図8】F2、F6及びF7(3種類の代表的なバッチ01032〜0134)の3カ月間保存後の安定性試験で得られた濁度値の比較を示すグラフである。
【図9】F2、F6及びF7(3種類の代表的なバッチ01032〜0134)の3カ月間保存後の安定性試験で得られたDACスコアによる可視粒子値の比較を示すグラフである。
【図10】F2、F6及びF7(3種類の代表的なバッチ01032〜0134)の3カ月間保存後の安定性試験で得られた不可視粒子値(≧10μm)の比較を示すグラフである。
【図11】F2、F6及びF7(3種類の代表的なバッチ01032〜0134)の3カ月間保存後の安定性試験で得られた不可視粒子値(≧25μm)の比較を示すグラフである。
【図12】F2、F6及びF7(3種類の代表的なバッチ01032〜0134)の3カ月間保存後の安定性試験で得られた残留モノマー含有量の比較を示すグラフである。
【図13】F2、F6及びF7(3種類の代表的なバッチ01032〜0134)の3カ月間保存後の安定性試験で得られたリジン変異体合計の比較を示すグラフである。
【図14】24時間後の各種撹拌速度の撹拌ストレスに対する安定性についてF2、F6及びF7を比較した濁度データを示すグラフである。
【図15】24時間後の各種撹拌速度の撹拌ストレスに対する安定性についてF2、F6及びF7を比較したDLSデータ(Z平均値)を示すグラフである。
【図16】数種のポンプサイクル前後のストレスに対する安定性についてF2、F6及びF7を比較した濁度データを示すグラフである。
【図17】数種のポンプサイクル前後の安定性についてF2、F6及びF7を比較したDLSデータ(Z平均)を示すグラフである。
【図18】数種のポンプサイクル前後の安定性についてF2、F6及びF7を比較したSECデータ(凝集物濃度)を示すグラフである。
【図19】蠕動ポンプを使用して充填した100mg/mL製剤の目視スコアを示すグラフである。
【図20】ピストンポンプを使用して充填した100mg/mL製剤の目視スコアを示すグラフである。
【図21】蠕動ポンプを使用して充填した100mg/mL製剤の濁度を示すグラフである。
【図22】ピストンポンプを使用して充填した100mg/mL製剤の濁度を示すグラフである。
【図23】T0及び5℃で4週間保存後における製剤F8〜F11の濁度を示すグラフである。
【図24】T0及び5℃で4週間保存後における製剤F8〜F11のモノマー含有量を示すグラフである。
【図25】T0及び5℃で4週間保存後における製剤F8〜F11の凝集物濃度を示すグラフである。
【図26】T0及び5℃で4週間保存後における製剤F8〜F11の不可視粒子数を示すグラフである。
【発明を実施するための形態】
【0031】
I.定義
本発明を理解し易くするために、先ず所定の用語を定義する。更に、当然のことながら、パラメーターの数値又は数値範囲を記載する場合には、記載する数値の中間の数値及び範囲も常に本発明に含むものとする。
【0032】
「医薬製剤」なる用語は活性成分の生物活性を明白に有効にすることが可能な形態であり、製剤を投与する対象に有意に毒性となるような付加成分を含有しない製剤を意味する。
【0033】
「医薬的に許容可能な担体」なる用語は当業者に公知であり、哺乳動物に投与するのに適した医薬的に許容可能な材料、組成物又はビヒクルを包含する。担体としては、ある臓器又は生体の一部から別の臓器又は生体の一部への主剤の輸送又は移送に関与する液体又は固体増量剤、希釈剤、添加剤、溶媒又は封入材料が挙げられる。各担体は製剤の他の成分と適合可能であり、患者の安全性に有害な作用や影響を与えないという意味で「許容可能」でなければならない。
【0034】
「医薬的に許容可能な添加剤」(ビヒクル、助剤)とは、使用する活性成分の有効用量を提供するために対象哺乳動物に妥当に投与することができる添加剤である。
【0035】
「添加剤」なる用語は例えばバルク特性を改変する所望コンシステンシーを提供するため、安定性を改善するため、及び/又は浸透圧を調節するために製剤に添加することができる物質を意味する。一般に使用される添加剤の例としては、限定されないが、糖質、ポリオール、アミノ酸、界面活性剤及びポリマーが挙げられる。
【0036】
一般に使用される添加剤はポリオールである。本願で使用する「ポリオール」とは複数のヒドロキシル基をもつ物質を意味し、糖質(還元糖及び非還元糖)、糖アルコール及び糖酸を包含する。本願で好ましいポリオールは約600kD未満(例えば約120〜約400kD)の分子量を有する。ポリオールの非限定的な例はフルクトース、マンノース、マルトース、ラクトース、アラビノース、キシロース、リボース、ラムノース、ガラクトース、グルコース、スクロース、トレハロース、ソルボース、メレジトース、ラフィノース、マンニトール、キシリトール、エリスリトール、スレイトール、ソルビトール、グリセロール、L−グルコン酸及びその金属塩である。
【0037】
本願で使用する「緩衝液」とは、その酸−塩基共役成分の作用によりpHの変化を阻止する緩衝溶液を意味する。本発明の緩衝液はpHが約4〜約8、好ましくは約4.5〜約7の範囲であり、最も好ましくはpHが約5.0〜約6.5の範囲である。pHをこの範囲に制御する緩衝液の例としては、リン酸塩、酢酸塩(例えば酢酸ナトリウム)、コハク酸塩(例えばコハク酸ナトリウム)、グルコン酸塩、グルタミン酸塩、ヒスチジン、クエン酸塩及び他の有機酸緩衝液が挙げられる。1態様において、本発明の製剤で使用するのに適した緩衝液はクエン酸リン酸緩衝液である。
【0038】
「界面活性剤」なる用語は一般に空気/溶液界面に誘導されるストレスと溶液/表面に誘導されるストレスから製剤中の蛋白質を保護する物質を包含する。例えば、界面活性剤は蛋白質を凝集から保護することができる。適切な界面活性剤としては、例えばポリソルベート、ポリオキシエチレンアルキルエーテル(例えばBrij 35(登録商標))、又はポロキサマー(例えばTween 20、Tween 80又はポロキサマー188)が挙げられる。好ましい界面活性剤はポロキサマー(例えばポロキサマー188、ポロキサマー407)、ポリオキシエチレンアルキルエーテル(例えばBrij 35(登録商標)、Cremophor A25、Sympatens ALM/230)、及びポリソルベート/Tween類(例えばポリソルベート20、ポリソルベート80、Mirjとポロキサマー類(例えばポロキサマー188)及びTween類(例えばTween 20及びTween 80))である。
【0039】
「安定」した製剤とは製剤中の抗体が製造工程中及び/又は保存後にその物理的安定性及び/又は化学的安定性及び/又は生物活性を本質的に維持する製剤である。蛋白質安定性を測定するための各種分析技術が当分野で利用可能であり、Peptide and Protein Drug Delivery,247−301,Vincent Lee Ed.,Marcel Dekker,Inc.,New York,N.Y.,Pubs.(1991)及びJones,A.(1993)Adv.Drug Delivery Rev.10:29−90に記載されている。例えば、1態様において、蛋白質の安定性は溶液中のモノマー蛋白質の百分率に従って測定され、分解(例えば断片化)及び/又は凝集蛋白質の百分率が低いほうが安定である。製剤は室温(約30℃)もしくは40℃で少なくとも約1カ月間安定であり、及び/又は約2〜8℃で少なくとも約1年間もしくは少なくとも2年間安定であることが好ましい。更に、製剤は製剤の(例えば−70℃まで)凍結及び解凍(以下、「凍結/解凍サイクル」と言う)後に安定であることが好ましい。
【0040】
抗体は色及び/もしくは透明度の目視試験後、又はUV光散乱法もしくはサイズ排除クロマトグラフィーにより測定したときに例えば凝集、沈殿及び/又は変性の徴候を実質的に示さない場合に、製剤中で「その物理的安定性を維持する」。凝集とは、個々の分子又は複合体が共有的又は非共有的に会合して凝集物を形成するプロセスである。凝集は可視沈殿が形成される程度まで進行することができる。
【0041】
製剤の物理的安定性等の安定性はサンプルの見かけの光減衰(吸光度ないし光学密度)の測定を含む当分野で周知の方法により評価することができる。このような光減衰の測定は製剤の濁度に相関する。製剤の濁度は溶液に溶解した蛋白質の固有特性でもあり、一般にネフェロメトリー法により測定され、ネフェロメトリー濁度単位(NTU)で表される。
【0042】
例えば溶液中の成分の1種以上の濃度(例えば蛋白質及び/又は塩濃度)の関数としての濁度を製剤の「乳白」又は「乳白外観」とも言う。濁度は既知濁度の懸濁液を使用して作成した標準曲線を参照することにより計算することができる。医薬組成物の濁度を測定するための参照標準は欧州薬局方基準(European Pharmacopoeia,Fourth Ed.,Directorate for the Quality of Medicine of the Council of Europe(EDQM),Strasbourg,France)に準じることができる。欧州薬局方基準によると、透明溶液は欧州薬局方標準に基づいて濁度約3を有する参照懸濁液と同等以下の濁度を有する溶液として定義される。ネフェロメトリー濁度測定は会合又は非理想効果の不在下で一般に濃度と共に線形変化するレイリー散乱を検出することができる。物理的安定性を評価するための他の方法も当分野で周知である。
【0043】
所定時点の化学的安定性に関して、抗体が以下に定義するようにその生物活性を維持するとみなされるような状況である場合に、抗体は医薬製剤中で「その化学的安定性を維持する」。化学的安定性は例えば抗体の化学的改変形を検出及び定量することにより評価することができる。化学的改変としては、サイズ改変(例えば切り取り)が挙げられ、例えばサイズ排除クロマトグラフィー、SDS−PAGE及び/又はマトリックス支援レーザー脱離イオン化/飛行時間型質量分析法(MALDI/TOF MS)により測定することができる。他の型の化学的改変としては、(例えば脱アミド化又は酸化の結果として生じる)電荷改変が挙げられ、例えばイオン交換クロマトグラフィーにより評価することができる。
【0044】
医薬製剤中の抗体がその所期目的のために生物学的に活性である場合に、抗体は医薬製剤中で「その生物活性を維持する」。例えば、医薬製剤中の抗体の生物活性が(例えば抗原結合アッセイで測定したときに)医薬製剤の製造時に示される生物活性の(アッセイの誤差内で)約30%、約20%又は約10%以内にある場合に、生物活性は維持される。
【0045】
薬理学的な意味で、本発明に関して抗体の「治療有効量」又は「有効量」とは、抗体が治療に有効である疾患の症状の予防又は治療又は緩和に有効な量を意味する。「疾患」とは抗体投与が有効となる任意病態である。これは慢性及び急性疾患ないし疾病を含み、該当疾患の素因を対象に与える病態を含む。
【0046】
「治療」とは治療処置と予防ないし防止措置の両方を意味する。治療を必要とする対象としては、既に疾患に罹患している対象と、疾患を予防すべき対象が挙げられる。
【0047】
本願で使用する「非経口投与」及び「非経口投与する」なる用語は経腸管及び局所投与以外の投与方法を意味し、通常は注射による投与方法であり、限定されないが、静脈内、筋肉内、動脈内、髄腔内、関節包内、眼窩内、心臓内、真皮内、腹腔内、経気管、皮下、表皮下、関節内、関節包下、クモ膜下、頭蓋内、関節内、脊髄内及び胸骨内注射及び輸液が挙げられる。
【0048】
本願で使用する「全身投与」、「全身投与する」、「末梢投与」及び「末梢投与する」なる用語は、化合物、薬剤及び他の材料が患者の生体系に入り、従って、代謝等のプロセスを受けるように、中枢神経系への直接投与以外の方法で投与することを意味する(例えば皮下投与)。
【0049】
本願で使用する「ヒトTNFα」(本願ではhTNFα、TNFα、又は単にhTNFと略称する)なる用語は17kD分泌型及びその生物活性型が非共有的に結合した17kD分子の三量体から構成される26kD膜結合型として存在するヒトサイトカインを意味する。hTNFαの構造は例えばPennica,D.,et al.(1984)Nature 312:724−729;Davis,J.M.,et al.(1987)Biochem 26:1322−1326;及びJones,E.Y.,et al.(1989)Nature 338:225−228に詳細に記載されている。ヒトTNFαなる用語は標準組換え発現法により作製することもできるし、市販品(R & D Systems,Catalog No.210−TA,Minneapolis,Minn.)として購入することもできる組換えヒトTNFα(rhTNFα)を包含するものとする。
【0050】
本願で使用する「抗体」なる用語はジスルフィド結合により相互に結合した2本の重(H)鎖と2本の軽(L)鎖からなる4本のポリペプチド鎖から構成される免疫グロブリン分子を意味する。構造の異なる他の天然抗体(例えばラクダ抗体)もこの定義に含まれる。各重鎖は重鎖可変領域(本願ではHCVR又はVHと略称する)と重鎖定常領域から構成される。重鎖定常領域はCH1、CH2及びCH3の3個のドメインから構成される。各軽鎖は軽鎖可変領域(本願ではLCVR又はVLと略称する)と軽鎖定常領域から構成される。軽鎖定常領域は1個のドメインCLから構成される。VH領域とVL領域はフレームワーク領域(FR)と呼ばれる保存度の高い領域を挟んで相補性決定領域(CDR)と呼ばれる超可変領域に更に分割することができる。各VH及びVLはアミノ末端からカルボキシ末端に向かってFR1、CDR1、FR2、CDR2、FR3、CDR3、FR4の順に配置された3個のCDRと4個のFRから構成される。本発明の1態様において、製剤は各々本願に援用する米国特許第6,090,382号及び6,258,562号に記載されている配列等のCDR1、CDR2及びCDR3配列をもつ抗体を含む。
【0051】
本願で使用する「CDR」なる用語は抗体可変配列内の相補性決定領域を意味する。重鎖と軽鎖の可変領域の各々に3個のCDRが存在し、可変領域の各々についてCDR1、CDR2及びCDR3と呼ぶ。これらのCDRの厳密な境界は種々のシステムに従って種々に定義されている。Kabat(同書)により記載されているシステムは抗体の任意可変領域に適用可能な明白な残基ナンバリングシステムを提供するのみならず、3個のCDRを規定する厳密な残基境界も提供する。これらのCDRをKabat CDRと言う場合もある。ChothiaらはKabat CDR内の所定のサブ部分がアミノ酸配列レベルでは多様性が高いにも拘わらず、ほぼ同一のペプチド骨格構造をとることを見出した(Chothia et al.(1987)Mol.Biol.196:901−917;Chothia et al.(1989)Nature 342:877−883)。これらのサブ部分はL1、L2及びL3又はH1、H2及びH3と呼ばれ、ここで「L」及び「H」は夫々軽鎖領域と重鎖領域を意味する。これらの領域をChothia CDRと言う場合もあり、その境界はKabat CDRとオーバーラップする。Kabat CDRとオーバーラップするCDRを規定する他の境界もPadlan(1995)FASEB J.9:133−139及びMacCallum(1996)J.Mol.Biol.262(5):732−45に記載されている。本願に記載するシステムと厳密に一致しない場合もあるが、Kabat CDRとオーバーラップする更に他のCDR境界定義もあり、特定残基もしくは残基群又はCDR全体が抗原結合にさほど影響しないという予想又は実験結果を踏まえると、CDRを短縮又は延長してもよい。本願で使用する方法はこれらのシステムのいずれかに従って定義されるCDRを利用することができるが、所定の態様はKabat又はChothiaにより定義されるCDRを使用する。
【0052】
本願で使用する抗体の「抗原結合部分」(又は単に「抗体部分」)なる用語は抗原(例えばhTNFα)と特異的に結合する能力を保持する抗体の1個以上のフラグメントを意味する。全長抗体のフラグメントにより抗体の抗原結合機能を実施できることが分かっている。抗体の「抗原結合部分」なる用語に含まれる結合フラグメントの例としては、(i)VL、VH、CL及びCH1ドメインから構成される1価フラグメントであるFabフラグメント;(ii)ヒンジ領域でジスルフィド橋により結合した2個のFabフラグメントから構成される2価フラグメントであるF(ab’)2フラグメント;(iii)VHドメインとCH1ドメインから構成されるFdフラグメント;(iv)抗体のシングルアームのVLドメインとVHドメインから構成されるFvフラグメント;(v)VHドメインから構成されるdAbフラグメント(Ward et al.,(1989)Nature 341:544−546);並びに(vi)単離された相補性決定領域(CDR)が挙げられる。更に、Fvフラグメントの2領域であるVLとVHは別々の遺伝子によりコードされるが、組換え法を使用し、VL領域とVH領域が対合して1価分子(1本鎖Fv(scFv)として知られる;例えばBird et al.(1988)Science 242:423−426;及びHuston et al.(1988)Proc.Natl.Acad.Sci.USA 85:5879−5883参照)を形成する1本の蛋白質鎖とすることが可能な合成リンカーにより結合することができる。このような1本鎖抗体も抗体の「抗原結合部分」なる用語に含むものとする。ダイアボディ等の他の型の1本鎖抗体も包含する。ダイアボディはVHドメインとVLドメインが1本のポリペプチド鎖上で発現される2価の二重特異性抗体であるが、同一鎖上の2領域間を対合させるには短いリンカーを使用することにより、これらの領域を別の鎖の相補領域と対合させ、2個の抗原結合部位を形成する(例えばHolliger,P.,et al.(1993)Proc.Natl.Acad.Sci.USA 90:6444−6448;Poljak,R.J.,et al.(1994)Structure 2:1121−1123参照)。本発明の1態様において、製剤は各々本願に援用する米国特許第6,090,382号及び6,258,562号に記載されている抗原結合部分を含有する。
【0053】
更に、抗体又はその抗原結合部分は抗体又は抗体部分と1個以上の他の蛋白質又はペプチドの共有的又は非共有的会合により形成される大きな免疫接着分子の一部でもよい。このような免疫接着分子の例としては、ストレプトアビジンコア領域を使用して四量体scFv分子を作製する場合(Kipriyanov,S.M.,et al.(1995)Human Antibodies and Hybridomas 6:93−101)や、システイン残基、マーカーペプチド及びC末端ポリヒスチジンタグを使用して2価のビオチン化scFv分子を作製する場合(Kipriyanov,S.M.,et al.(1994)Mol.Immunol.31:1047−1058)が挙げられる。Fab及びF(ab’)2フラグメント等の抗体部分は全長抗体の夫々パパイン又はペプシン消化等の従来技術を使用して全長抗体から作製することができる。更に、抗体、抗体部分及び免疫接着分子は本願に記載するような標準組換えDNAを使用して得ることができる。
【0054】
本願で使用する「ヒト抗体」なる用語はヒト生殖系列免疫グロブリン配列に由来する可変領域と定常領域をもつ抗体を包含する。本発明で使用されるヒト抗体はヒト生殖系列免疫グロブリン配列によりコードされないアミノ酸残基(例えばランダムもしくは部位特異的突然変異誘発によりインビトロ又は体細胞突然変異によりインビボ導入される突然変異)を例えばCDR、特にCDR3に含むことができる。他方、本願で使用する「ヒト抗体」なる用語はマウス等の別の哺乳動物種の生殖系列に由来するCDR配列をヒトフレームワーク配列に移植した抗体を包含しない。
【0055】
本願で使用する「組換えヒト抗体」なる用語は組換え手段により作製、発現、創製又は単離される全ヒト抗体を包含し、例えば宿主細胞にトランスフェクトした組換え発現ベクターを使用して発現される抗体(下記セクションIIに詳述)、組換えコンビナトリアルヒト抗体ライブラリーから単離される抗体(下記セクションIIIに詳述)、ヒト免疫グロブリン遺伝子に対してトランスジェニックな動物(例えばマウス)から単離される抗体(例えばTaylor,L.D.,et al.(1992)Nucl.Acids Res.20:6287−6295参照)又はヒト免疫グロブリン遺伝子配列と他のDNA配列のスプライシングを含む他の任意手段により作製、発現、創製もしくは単離される抗体が挙げられる。このような組換えヒト抗体はヒト生殖系列免疫グロブリン配列に由来する可変領域と定常領域をもつ。他方、所定態様では、このような組換えヒト抗体をインビトロ突然変異誘発(又は、ヒトIg配列に対してトランスジェニックな動物を使用する場合には、インビボ体細胞突然変異誘発)するため、組換え抗体のVH領域とVL領域のアミノ酸配列はヒト生殖系列VH配列及びVL配列に由来及び関連するが、ヒト抗体生殖系列レパートリー内に天然にインビボ存在し得ない配列となる。
【0056】
本願で使用する「単離抗体」とは、異なる抗原特異性をもつ他の抗体を実質的に含まない抗体を意味する(例えば、hTNFαと特異的に結合する単離抗体はhTNFα以外の抗原と特異的に結合する抗体を実質的に含まない)。他方、hTNFαと特異的に結合する単離抗体は他の種に由来するTNFα分子等の他の抗原に対して交差反応性をもつ場合がある。更に、単離抗体は他の細胞材料及び/又は薬品を実質的に排除することができる。
【0057】
本願で使用する「中和抗体」(又は「hTNFα活性を中和した抗体」)とはhTNFαと結合する結果、hTNFαの生物活性を阻害する抗体を意味する。hTNFαの生物活性のこの阻害はhTNFαにより(インビトロ又はインビボで)誘導される細胞傷害作用、hTNFαにより誘導される細胞活性化及びhTNFαとhTNFα受容体の結合等のhTNFα生物活性の1種以上の指標を測定することにより評価することができる。hTNFα生物活性のこれらの指標は各々本願に援用する米国特許第6,090,382号及び6,258,562号に記載されている当分野で公知の数種類の標準インビトロ又はインビボアッセイの1種以上により評価することができる。抗体がhTNFα活性を中和する能力は、hTNFαにより誘導されるL929細胞の細胞傷害作用の阻害により評価することが好ましい。hTNFα活性の付加又は代替パラメーターとして、抗体がHUVEC上でhTNFαにより誘導されるELAM−1の発現を阻害する能力は、hTNFαにより誘導される細胞活性化の指標として評価することができる。
【0058】
本願で使用する「表面プラズモン共鳴法」なる用語は例えばBIAcoreシステム(Pharmacia Biosensor AB,Uppsala,Sweden及びPiscataway,N.J.)を使用してバイオセンサーマトリックス内の蛋白質濃度の変化の検出によりリアルタイム生体特異的相互作用の分析を可能にする光学現象を意味する。詳細については、Jonsson,U.,et al.(1993)Ann.Biol.Clin.51:19−26;Jonsson,U.,et al.(1991)Biotechniques 11:620−627;Johnsson,B.,et al.(1995)J.Mol.Recognit.8:125−131;及びJohnnson,B.,et al.(1991)Anal.Biochem.198:268−277参照。
【0059】
本願で使用する「Kon」なる用語は当分野で公知のように結合性蛋白質(例えば抗体)が抗原と会合して例えば抗体/抗原複合体を形成する会合速度定数を意味する。
【0060】
本願で使用する「Koff」なる用語は抗体が抗体/抗原複合体から解離する解離速度定数を意味する。
【0061】
本願で使用する「Kd」なる用語は特定抗体−抗原相互作用の解離定数を意味し、平衡状態の滴定測定又は解離速度定数(koff)を会合速度定数(kon)で割ることにより得られる値を意味する。
【0062】
以下のサブセクションでは本発明の各種側面について更に詳細に記載する。
【0063】
II.本発明の製剤
本発明は当業者に公知の製剤に比較して特性を改善した液体医薬製剤(例えば抗体製剤)に関する。本発明はNaClを除去し、20mg/mLを上回るポリオール(例えば糖アルコール)を加えることにより、製剤中のヒトTNFα抗体の濃度を約100mg/mLまで上げることができるという驚くべき知見に基づく。抗体濃度が高いにも拘わらず、本発明の製剤は例えば製造、保存及び/もしくは反復凍結/解凍処理工程中又は拡大した気液界面への長期暴露中に溶解度と安定性を維持することが可能である。更に、本発明の製剤は約100mg/mLの抗体濃度でありながら、低レベル蛋白質凝集(即ち1%未満)を維持する。本発明の製剤は更に、驚くべきことに、約100mg/mLの抗体濃度でありながら、皮下注射に適した範囲内の低粘度を維持する。更に、本発明の製剤(例えば高濃度TNFα抗体)は皮下注射に適した低粘度を維持すると共に、ほぼ1のpH範囲(例えばpH5.2〜pH6.0)にわたって安定性を維持する。1態様において、製剤の濁度は標準48時間撹拌ストレスアッセイ後に100NTU未満である。従って、本発明の高濃度抗体製剤は安定性、粘度、濁度及び物理的劣化の問題を含む製剤の多数の公知課題を解決する。
【0064】
本発明の製剤の驚くべき特徴の1つは、高い抗体濃度(例えば100mg/mL以上)でありながらNaClの不在下で製剤の総粘度が低く維持される点である(例えば約3.1〜3.3mPa・s、例えば約3.00、3.05、3.10、3.15、3.20、3.25、3.30、3.35又は約3.40mPa・s)。一般に、粘度は蛋白質濃度の増加と共に増加する(詳細については、Shire et al.(2004)J Pharm Sci 93:1390参照)。このような増加は殆どの場合、イオン性添加剤(例えばNaClやMgCl2)を添加することにより抑制されるが、このような添加剤を添加すると、溶液の濁度も上昇する可能性がある。濁度上昇は不溶性蛋白質凝集物、沈殿又は蛋白質粒子の形成(例えば凝集)と関係していることが多い。従って、本発明の液体医薬製剤はNaClを添加する必要なしに、皮下投与に適した粘度で高い抗体濃度(例えば少なくとも100mg/mL)を実現する。
【0065】
1態様において、本発明の製剤は液体製剤が有意乳白、凝集又は沈殿を示さないような高濃度の蛋白質を含有する。
【0066】
別の態様において、本発明の製剤は(例えば目視アナログスケール(VAS)スコアにより判定した場合に)有意疼痛感覚なしに例えば皮下投与に適するような高濃度の蛋白質を含有する。
【0067】
本発明の製剤は例えば約50mg/mL又は約100mg/mLの蛋白質濃度のヒト抗TNFα抗体又はその抗原結合フラグメントを含む高濃度蛋白質を含有する。従って、下記実施例1に記載するように、本発明の1側面において、液体医薬製剤は約50mg/mLの濃度のヒト抗TNFα抗体を含有する。下記実施例2〜6に記載するように、本発明の別の側面において、液体医薬製剤は約100mg/mLの濃度のヒト抗TNFα抗体を含有する。本発明の更に別の側面において、液体医薬製剤は約150mg/mLの濃度のヒト抗TNFα抗体を含有する。本発明の好ましい態様は高濃度蛋白質を含有する製剤であるが、本発明の製剤が約1mg/mL〜約150mg/mL又は約40mg/mL〜125mg/mLの抗体濃度となり得ることも考えられる。上記濃度の中間の濃度及び範囲(例えば1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、101、102、103、104、105、106、107、108、109、110、111、112、113、114、115、116、117、118、119、120、121、122、123、124、125、126、127、128、129、130、131、132、133、134、135、136、137、138、139、140、141、142、143、144、145、146、147、148、149、150、151、152、153、154、155、156、157、158、159、160、161、162、163、164、165、166、167、168、169、170、171、172、173、174、175、176、177、178、179、180、181、182、183、184、185、186、187、188、189、190、191、192、193、194、195、196、197、198、199又は200mg/mL)も本発明に含むものとする。
【0068】
別の側面において、本発明は例えば約100mg/mLを上回る濃度の治療用抗体(例えばアダリムマブ)を製剤化するために十分な量のポリオール、界面活性剤及び緩衝液系を含有する液体医薬組成物を提供する。1態様において、液体医薬組成物はNaClを含有しない。
【0069】
但し、当然のことながら、本発明の好ましい製剤はNaClを含有しないが、少量(例えば約0.01mM〜約300mM)のNaClが製剤中に存在していてもよい。更に、上記数値の中間の任意量のNaClも含むものとする。
【0070】
1側面において、本発明はヒト抗TNFα抗体又はその抗原結合フラグメント(例えばアダリムマブ)と、NaClの不在下で治療用抗体を製剤化するために十分な量のポリオールを含有する液体医薬組成物を提供する。
【0071】
本発明は更に、ヒト抗TNFα抗体又はその抗原結合フラグメントを含有する液体製剤であって、pHが約5.0〜6.4であり、NaClを添加せずに標準24時間撹拌ストレスアッセイ後の濁度が約60NTU未満(例えば約20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37.38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62又は63NTU)である製剤を提供する。別の側面において、本発明はヒト抗TNFα抗体又はその抗原結合フラグメントを含有する液体製剤であって、pHが約5.0〜6.4であり、NaClを添加せずに標準48時間撹拌ストレスアッセイ後の濁度が約100NTU未満(例えば約35、36、37.38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99又は100NTU)である製剤を提供する。更に別の側面において、本発明はヒト抗TNFα抗体又はその抗原結合フラグメントを含有する液体製剤であって、pHが約5.0〜6.4であり、NaClを添加せずに5℃、25℃又は40℃で3カ月間保存の濁度が約40NTU未満(例えば約20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37.38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60NTU)である製剤を提供する。
【0072】
本発明の製剤の1つの特徴は20mg/mLを上回る濃度のポリオール(例えば糖アルコール)を含有する点にある。1態様において、ポリオールはソルビトール又はマンニトールである。なお、蛋白質溶液にソルビトール又はマンニトールを添加しても必ずしも蛋白質安定性が増すわけではない。例えば、熱又は界面ストレス条件下で評価した場合にソルビトールは夫々Tween 20とヒドロキシプロピル−β−シクロデキストリンに比較してブタ成長ホルモンの沈殿に対する効果がなかった(Charman et al.(1993)Pharm Res.10(7):954−62)。
【0073】
1態様において、本発明の製剤で使用するのに適切なポリオールは糖アルコール(例えばマンニトール又はソルビトール)である。ポリオールを含有する本発明の液体製剤は一般に約20mgを上回るポリオールを含有する。1態様において、製剤は約30mg/mLを上回るポリオールを含有する。別の態様において、製剤は約40mg/mLを上回るポリオールを含有する。別の態様において、製剤は約40〜45mg/mL、例えば約35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54又は55mg/mLのポリオールを含有する。更に、上記数値のいずれかの組合せを上限及び/又は下限として使用する数値範囲も含むものとする。
【0074】
本発明の所定態様では、pH緩衝溶液中に抗体を含有する液体製剤を製造する。本発明の緩衝液はpH約4〜約8、好ましくは約4.5〜約7.0、より好ましくは約4.5〜約6.0、更に好ましくは約4.8〜約5.5であり、最も好ましくはpH約5.0〜約6.4である。1態様において、本発明の製剤のpHは約5.2である。別の態様において、本発明の製剤のpHは約6.0である。上記pH値の中間の範囲(例えば4.5、4.6、4.7、4.8、4.9、5.0、5.1、5.2、5.3、5.4、5.5、5.6、5.7、5.8、5.9、6.0、6.1、6.2、6.3、6.4)も本発明に含むものとする。上記数値のいずれかの組合せを上限及び/又は下限として使用する数値範囲(例えば5.2〜5.8)も含むものとする。pHをこの範囲内に制御する緩衝液の例としては、リン酸塩、酢酸塩(例えば酢酸ナトリウム)、コハク酸塩(例えばコハク酸ナトリウム)、グルコン酸塩、グルタミン酸塩、ヒスチジン、クエン酸塩及び他の有機酸緩衝液が挙げられる。
【0075】
本発明の特定態様において、製剤はpHを約5.0〜約6.4の範囲に維持するためにクエン酸塩及び/又はリン酸塩を含む緩衝液系を含有する。1態様において、製剤のpHは約5.2である。別の態様において、製剤のpHは約6.0である。
【0076】
別の好ましい態様において、緩衝液系はクエン酸一水和物、クエン酸ナトリウム、リン酸二ナトリウム二水和物、及び/又はリン酸二水素ナトリウム二水和物を含む。別の好ましい態様において、緩衝液系は約1.15〜1.45(例えば約1.15、1.20、1.25、1.30、1.35、1.40又は1.45)mg/mlのクエン酸と、約0.2〜0.4(例えば約0.2、0.25、0.3、0.35又は0.4)mg/mLのクエン酸ナトリウム脱水和物と、約1.35〜1.75(例えば1.35、1.40、1.45、1.50、1.55、1.60、1.65、1.70又は1.75)mg/mLのリン酸二ナトリウム脱水和物と、約0.75〜0.95(例えば約0.75、0.80、0.85、0.9又は0.95)mg/mLのリン酸二水素ナトリウム脱水和物を含む。
【0077】
上記濃度の中間の数値及び範囲も本発明に含むものとする。更に、上記数値のいずれかの組合せを上限及び/又は下限として使用する数値範囲(例えば1.20〜1.40mg/mL)も含むものとする。
【0078】
他の態様において、緩衝液系は1.3〜1.31mg/mL(例えば約1.305mg/mL)のクエン酸を含む。別の態様において、緩衝液系は約0.27〜0.33mg/mL(例えば約0.305mg/mL)のクエン酸ナトリウム脱水和物を含む。1態様において、緩衝液系は約1.5〜1.56mg/mL(例えば約1.53mg/mL)のリン酸二ナトリウム脱水和物を含む。別の態様において、緩衝液系は約0.83〜0.89mg/mL(例えば約0.86mg/mL)のリン酸二水素ナトリウム二水和物を含む。
【0079】
デタージェントないし界面活性剤も本発明の抗体製剤に添加することができる。典型的な界面活性剤としては、ポリソルベート(例えばポリソルベート20、80等)やポロキサマー(例えばポロキサマー188)等の非イオン性界面活性剤が挙げられる。界面活性剤の添加量は製剤化抗体の凝集を抑え、及び/又は製剤中の粒状物の形成を最小限にし、及び/又は吸着を減らすように選択する。本発明の好ましい1態様において、製剤はポリソルベートである界面活性剤を含有する。本発明の別の好ましい態様において、製剤は界面活性剤としてポリソルベート80を含有する。好ましい1態様において、製剤は約0.1〜約2.0mg/mL(例えば約1mg/mL)のポリソルベート80を含有する。
【0080】
上記濃度の中間の数値及び範囲(例えば0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9)も本発明に含むものとする。更に、上記数値のいずれかの組合せを上限及び/又は下限として使用する数値範囲(例えば0.3〜1.1mg/mL)も含むものとする。
【0081】
1態様において、本発明の製剤は少なくとも約100mg/mLの濃度のヒトTNFα抗体又はその抗原結合部分と、界面活性剤(例えばポリソルベート80)と、ポリオール(例えば20mg/mLを上回るソルビトール又はマンニトール)と、緩衝液系(例えばクエン酸一水和物、クエン酸ナトリウム、リン酸二ナトリウム二水和物及び/又はリン酸二水素ナトリウム二水和物)から本質的に構成され、NaClを含有しない。
【0082】
1態様において、製剤は上記成分を含有し(即ち少なくとも約100mg/mLの濃度の抗体と、緩衝液系と、ポリオールと、界面活性剤を含有し、NaClを含有しない)、ベンジルアルコール、フェノール、m−クレゾール、クロロブタノール及びベンゼトニウムCl等の防腐剤を実質的に含有しない。別の態様では、防腐剤を製剤に加えることができる。製剤の望ましい特性を著しく悪化させない限り、Remington’s Pharmaceutical Sciences 16th edition,Osol,A.Ed.(1980)に記載されているもの等の1種以上の他の医薬的に許容可能な担体、添加剤又は安定剤も製剤に加えることができる。許容可能な担体、添加剤又は安定剤は使用する用量と濃度でレシピエントに非毒性であり、他の緩衝剤;補助溶媒;アスコルビン酸やメチオニン等の酸化防止剤;EDTA等のキレート剤;金属錯体(例えばZn−蛋白質錯体);ポリエステル等の生体適合性ポリマー;及び/又はナトリウム等の塩形成対イオンを含む。
【0083】
本願の製剤は治療する特定適応症の必要に応じて1種以上の他の治療剤、好ましくは製剤の抗体に悪影響を与えない相補活性をもつ治療剤と併用してもよい。このような治療剤は所期目的に有効な量で併存すると適切である。本発明の製剤と併用することができる他の治療剤は各々本願に援用する米国特許第6,090,382号及び6,258,562号に詳細に記載されている。
【0084】
インビボ投与に使用する製剤は無菌でなければならない。これは製剤の製造前又は製造後に滅菌濾過膜で濾過することにより容易に実施される。
【0085】
上記のように、本発明の液体製剤は有利な安定性と保存性をもつ。液体製剤の安定性は保存形態に依存せず、限定されないが、凍結製剤、凍結乾燥製剤、噴霧乾燥製剤、又は活性成分を懸濁した製剤が挙げられる。安定性は選択された温度で選択された期間にわたって測定することができる。本発明の1側面において、液体製剤中の蛋白質は液体形態で少なくとも約3カ月間、少なくとも約4カ月間、少なくとも約5カ月間、少なくとも約6カ月間、少なくとも約12カ月間、少なくとも約18カ月間安定である。上記期間の中間の数値及び範囲(例えば約3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23又は約24カ月間)も本発明に含むものとする。更に、上記数値のいずれかの組合せを上限及び/又は下限として使用する数値範囲も含むものとする。好ましくは、製剤は室温(約30℃)もしくは40℃で少なくとも約1カ月間安定であり、及び/又は約2〜8℃で少なくとも約1年間安定であり、又はより好ましくは約2〜8℃で少なくとも約2年間安定である。更に、製剤は製剤の(例えば−80℃まで)凍結及び解凍(以下、「凍結/解凍サイクル」と言う)後に安定であることが好ましい。
【0086】
液体製剤中の蛋白質の安定性は製剤中の蛋白質のモノマー、凝集物もしくはフラグメント又はその組合せの百分率として表すこともできる。蛋白質は色及び/もしくは透明度の目視試験後、又はUV光散乱法もしくはサイズ排除クロマトグラフィーにより測定したときに凝集、沈殿及び/又は変性の徴候を実質的に示さない場合に、製剤中で「その物理的安定性を維持する」。本発明の1側面において、安定した液体製剤とは製剤中に凝集物として存在する蛋白質が約10%未満、好ましくは約5%未満の製剤である。
【0087】
1態様において、液体製剤の物理的安定性は撹拌ストレスアッセイ(例えば24時間又は48時間撹拌ストレスアッセイ)後に製剤の濁度を測定することにより測定される。例えば、磁気スターラー(例えばマルチポイントHP,550rpm)を取り付けたビーカーに適切な容量の液体製剤を入れ、適切な任意時点(例えばT0〜T48(時間))でアリコートを分取し、アリコートに応じて適切なアッセイを実施することにより、撹拌ストレスアッセイを実施することができる。同一条件下で非撹拌下の製剤のサンプルを対照とする。
【0088】
Hach(ドイツ)製実験室濁度測定システムを使用して濁度測定を実施することができ、ネフェロメトリー単位(NTU)として報告する。
【0089】
本発明の液体製剤は有利な耐容性も有する。耐容性は疼痛視覚的アナログスケール(VAS)を使用して注射部位疼痛を知覚した対象の評価に基づいて評価される。
【0090】
(VAS)は例えば無痛から極限量の疼痛までの一連の値にわたって疼痛を測定する計器である。操作上、VASは長さ約100mmの水平線であり、数値及び/又は語句表記(例えば0もしくは10、又は「無痛」もしくは「耐え難い痛み」)を表示され、場合により極限間の他の語句又は数値表記(例えば、軽度、中度及び重度;又は1〜9)を加えてもよい(例えばLee JS,et al.(2000)Acad Emerg Med 7:550参照)。
【0091】
測定することができる耐容性の他の指標としては、例えばドレイズスケール(出血、点状出血、紅斑、浮腫、痒み)と皮下出血が挙げられる。
【0092】
III.本発明の製剤で使用される抗体
本発明の製剤で使用することができる抗体はヒトTNFα(ないしhTNFα)を含む抗原TNFαに対する抗体である。
【0093】
1態様において、本発明は高い親和性と低い解離速度でヒトTNFαと結合すると共に、高い中和能をもつ単離ヒト抗体又はその抗原結合部分に関する。好ましくは、本発明で使用されるヒト抗体は組換え中和ヒト抗hTNFα抗体である。本発明の最も好ましい組換え中和抗体を本願ではD2E7と言い、HUMIRA(登録商標)ないしアダリムマブとも言う(D2E7 VL領域のアミノ酸配列を配列番号1に示し、D2E7 VH領域のアミノ酸配列を配列番号2に示す)。D2E7(アダリムマブ/HUMIRA(登録商標))の特性は各々本願に援用するSalfeldらの米国特許第6,090,382号、6,258,562号及び6,509,015号に記載されている。
【0094】
1態様において、ヒトTNFα又はその抗原結合部分は、いずれも表面プラズモン共鳴法により測定した場合に、1×10−8M以下のKdと1×10−3s−1以下のKoff速度定数でヒトTNFαから解離し、標準インビトロL929アッセイにおいて1×10−7M以下のIC50でヒトTNFα細胞傷害作用を中和する。より好ましくは、単離ヒト抗体又はその抗原結合部分は5×10−4s−1以下のKoff、更に好ましくは1×10−4s−1以下のKoffでヒトTNFαから解離する。より好ましくは、単離ヒト抗体又はその抗原結合部分は標準インビトロL929アッセイにおいて1×10−8M以下のIC50、更に好ましくは1×10−9M以下のIC50、更に好ましくは1×10−10M以下のIC50でヒトTNFα細胞傷害作用を中和する。好ましい1態様において、抗体は単離ヒト組換え抗体又はその抗原結合部分である。
【0095】
抗体重鎖及び軽鎖CDR3ドメインが抗原に対する抗体の結合特異性/親和性において重要な役割を果たすことは周知である。従って、別の側面において、本発明はhTNFαとの会合の解離速度が遅く、D2E7と構造的に一致又は関連する軽鎖及び重鎖CDR3ドメインをもつヒト抗体を投与することによるクローン病の治療に関する。Koffを実質的に変化させずにD2E7 VL CDR3の9位をAla又はThrで占めることができる。従って、D2E7 VL CDR3のコンセンサスモチーフはアミノ酸配列Q−R−Y−N−R−A−P−Y−(T/A)(配列番号3)を含む。更に、Koffを実質的に変化させずにD2E7 VH CDR3の12位をTyr又はAsnで占めることができる。従って、D2E7 VH CDR3のコンセンサスモチーフはアミノ酸配列V−S−Y−L−S−T−A−S−S−L−D−(Y/N)(配列番号4)を含む。更に、米国特許第6,090,382号の実施例2に実証されているように、Koffを実質的に変化させずにD2E7重鎖及び軽鎖のCDR3ドメイン(VL CDR3内の1、4、5、7もしくは8位又はVH CDR3内の2、3、4、5、6、8、9、10もしくは11位)を1個のアラニン残基で置換することができる。更に、当業者に自明の通り、D2E7 VL及びVH CDR3ドメインをアラニンで置換できるならば、抗体の低い解離速度定数を維持しながらCDR3ドメイン内の他のアミノ酸の置換、特に保存アミノ酸による置換も可能であると思われる。D2E7 VL及び/又はVH CDR3ドメイン内に1〜5カ所以下の保存アミノ酸置換を導入することが好ましい。D2E7 VL及び/又はVH CDR3ドメイン内に1〜3カ所以下の保存アミノ酸置換を導入することがより好ましい。更に、hTNFαとの結合に必須のアミノ酸位置には保存アミノ酸置換を導入すべきでない。D2E7 VL CDR3の2位及び5位と、D2E7 VH CDR3の1位及び7位はhTNFαとの相互作用に必須であると思われるため、これらの位置には保存アミノ酸置換を導入しない(但し、上記のように、D2E7 VL CDR3の5位のアラニン置換は許容可能である)(米国特許第6,090,382号参照)。
【0096】
従って、別の態様において、抗体又はその抗原結合部分は以下の特徴を含むことが好ましい:
a)表面プラズモン共鳴法により測定した場合に、1×10−3s−1以下のKoff速度定数でヒトTNFαから解離する;
b)配列番号3のアミノ酸配列あるいは1、4、5、7もしくは8位の1カ所のアラニン置換又は1、3、4、6、7、8及び/もしくは9位の1〜5カ所の保存アミノ酸置換により配列番号3から変異したアミノ酸配列を含む軽鎖CDR3ドメインをもつ;
c)配列番号4のアミノ酸配列あるいは2、3、4、5、6、8、9、10もしくは11位の1カ所のアラニン置換又は2、3、4、5、6、8、9、10、11及び/もしくは12位の1〜5カ所の保存アミノ酸置換により配列番号4から変異したアミノ酸配列を含む重鎖CDR3ドメインをもつ。
【0097】
より好ましくは、抗体又はその抗原結合部分は5×10−4s−1以下のKoffでヒトTNFαから解離する。更により好ましくは、抗体又はその抗原結合部分は1×10−4s−1以下のKoffでヒトTNFαから解離する。
【0098】
更に別の態様において、抗体又はその抗原結合部分は好ましくは配列番号3のアミノ酸配列又は1、4、5、7もしくは8位の1カ所のアラニン置換により配列番号3から変異したアミノ酸配列を含むCDR3ドメインをもつ軽鎖可変領域(LCVR)と、配列番号4のアミノ酸配列又は2、3、4、5、6、8、9、10もしくは11位の1カ所のアラニン置換により配列番号4から変異したアミノ酸配列を含むCDR3ドメインをもつ重鎖可変領域(HCVR)を含む。好ましくは、LCVRは更に配列番号5のアミノ酸配列を含むCDR2ドメイン(即ちD2E7 VL CDR2)をもち、HCVRは更に配列番号6のアミノ酸配列を含むCDR2ドメイン(即ちD2E7 VH CDR2)をもつ。更により好ましくは、LCVRは更に配列番号7のアミノ酸を含むCDR1ドメイン(即ちD2E7 VL CDR1)をもち、HCVRは配列番号8のアミノ酸配列を含むCDR1ドメイン(即ちD2E7 VH CDR1)をもつ。VLのフレームワーク領域は好ましくはVκIヒト生殖系列ファミリーに由来し、より好ましくはA20ヒト生殖系列Vk遺伝子に由来し、最も好ましくは米国特許第6,090,382号の図1A及び1Bに示されるD2E7 VLフレームワーク配列に由来する。VHのフレームワーク領域は好ましくはVH3ヒト生殖系列ファミリーに由来し、より好ましくはDP−31ヒト生殖系列VH遺伝子に由来し、最も好ましくは米国特許第6,090,382号の図2A及び2Bに示されるD2E7 VHフレームワーク配列に由来する。
【0099】
従って、別の態様において、抗体又はその抗原結合部分は好ましくは配列番号1のアミノ酸配列を含む軽鎖可変領域(LCVR)(即ちD2E7 VL)と、配列番号2のアミノ酸配列を含む重鎖可変領域(HCVR)(即ちD2E7 VH)を含む。所定態様において、抗体はIgG1、IgG2、IgG3、IgG4、IgA、IgE、IgM又はIgD定常領域等の重鎖定常領域を含む。好ましくは、重鎖定常領域はIgG1重鎖定常領域又はIgG4重鎖定常領域である。更に、抗体はκ軽鎖定常領域又はλ軽鎖定常領域である軽鎖定常領域を含むことができる。好ましくは、抗体はκ軽鎖定常領域を含む。あるいは、抗体部分は例えばFabフラグメント又は1本鎖Fvフラグメントとすることができる。
【0100】
更に他の態様において、本発明はD2E7関連VL及びVH CDR3ドメインを含む単離ヒト抗体又はその抗原結合部分の使用を包含する。例えば、配列番号3、配列番号11、配列番号12、配列番号13、配列番号14、配列番号15、配列番号16、配列番号17、配列番号18、配列番号19、配列番号20、配列番号21、配列番号22、配列番号23、配列番号24、配列番号25及び配列番号26から構成される群から選択されるアミノ酸配列を含むCDR3ドメインをもつ軽鎖可変領域(LCVR)と、配列番号4、配列番号27、配列番号28、配列番号29、配列番号30、配列番号31、配列番号32、配列番号33、配列番号34及び配列番号35から構成される群から選択されるアミノ酸配列を含むCDR3ドメインをもつ重鎖可変領域(HCVR)を含む抗体又はその抗原結合部分が挙げられる。
【0101】
本発明の方法及び組成物で使用される抗体又は抗体部分は宿主細胞で免疫グロブリン軽鎖及び重鎖遺伝子を組換え発現させることにより作製することができる。抗体を組換え発現させるためには、抗体の免疫グロブリン軽鎖及び重鎖をコードするDNAフラグメントを組込んだ1個以上の組換え発現ベクターを宿主細胞にトランスフェクトし、軽鎖及び重鎖を宿主細胞で発現させ、好ましくは宿主細胞を培養する培地に分泌させ、培地から抗体を回収することができる。抗体重鎖及び軽鎖遺伝子を獲得し、これらの遺伝子を組換え発現ベクターに組込み、ベクターを宿主細胞に導入するためには、Sambrook,Fritsch and Maniatis(eds),Molecular Cloning;A Laboratory Manual,Second Edition,Cold Spring Harbor,N.Y.,(1989),Ausubel,F.M.et al.(eds.)Current Protocols in Molecular Biology,Greene Publishing Associates,(1989)及びBossらの米国特許第4,816,397号に記載されている方法等の標準組換えDNA法を使用する。
【0102】
アダリムマブ(D2E7)又はアダリムマブ(D2E7)関連抗体を発現させるためには、先ず軽鎖及び重鎖可変領域をコードするDNAフラグメントを得る。これらのDNAはポリメラーゼ連鎖反応(PCR)を使用して生殖系列軽鎖及び重鎖可変配列の増幅と修飾により得ることができる。ヒト重鎖及び軽鎖可変領域遺伝子の生殖系列DNA配列は当分野で公知である(例えば「Vbase」ヒト生殖系列配列データベース参照;更に各々その開示内容を特に本願に援用するKabat,E.A.,et al.(1991)Sequences of Proteins of Immunological Interest,Fifth Edition,U.S.Department of Health and Human Services,NIH Publication No.91−3242;Tomlinson,LM.,et al.(1992)“The Repertoire of Human Germline VH Sequences Reveals about Fifty Groups of VH Segments with Different Hypervariable Loops”J.Mol.Biol.227:776−798;及びCox,J.P.L.et al.(1994)“A Directory of Human Germ−line V78 Segments Reveals a Strong Bias in their Usage”Eur.J.Immunol.24:827−836も参照)。D2E7又はD2E7関連抗体の重鎖可変領域をコードするDNAフラグメントを得るためには、ヒト生殖系列VH遺伝子のVH3ファミリーのメンバーを標準PCRにより増幅する。DP−31 VH生殖系列配列を増幅するのが最も好ましい。D2E7又はD2E7関連抗体の軽鎖可変領域をコードするDNAフラグメントを得るためには、ヒト生殖系列VL遺伝子のVκIファミリーのメンバーを標準PCRにより増幅する。A20 VL生殖系列配列を増幅するのが最も好ましい。DP−31生殖系列VH及びA20生殖系列VL配列の増幅用に適切なPCRプライマーは標準方法を使用して上記引用文献に開示されているヌクレオチド配列に基づいて設計することができる。
【0103】
生殖系列VH及びVLフラグメントが得られたら、本願に開示するD2E7又はD2E7関連アミノ酸配列をコードするように、これらの配列を突然変異させることができる。生殖系列VH及びVL DNA配列によりコードされるアミノ酸配列を先ずD2E7又はD2E7関連VH及びVLアミノ酸配列と比較し、D2E7又はD2E7関連配列のうちで生殖系列と異なるアミノ酸残基を同定する。次に、どのヌクレオチド変異を導入すべきかを決定するために遺伝コードを使用し、突然変異後の生殖系列配列がD2E7又はD2E7関連アミノ酸配列をコードするように、生殖系列DNA配列の適切なヌクレオチドを突然変異させる。(PCR産物が突然変異を含むように、突然変異させたヌクレオチドをPCRプライマーに導入する)PCR突然変異誘発法や部位特異的突然変異誘発法等の標準方法により生殖系列配列の突然変異誘発を実施する。
【0104】
更に、当然のことながら、PCR増幅により得られた「生殖系列」配列が真の生殖系列構成に由来するフレームワーク領域でアミノ酸変異(即ち、例えば体細胞突然変異の結果として、真の生殖系列配列に対する増幅配列の変異)をコードする場合には、これらのアミノ酸変異を真の生殖系列配列に復帰変異(即ちフレームワーク残基を生殖系列構成に「復帰突然変異」)させることが望ましいと思われる。
【0105】
(例えば上記のように生殖系列VH及びVL遺伝子の増幅と突然変異誘発により)D2E7又はD2E7関連VH及びVLセグメントをコードするDNAフラグメントが得られたら、例えば可変領域遺伝子を全長抗体鎖遺伝子、Fabフラグメント遺伝子又はscFv遺伝子に変換するように、これらのDNAフラグメントを標準組換えDNA技術により更に操作することができる。これらの操作では、VL又はVHをコードするDNAフラグメントを、抗体定常領域又はフレキシブルリンカー等の別の蛋白質をコードする別のDNAフラグメントと機能的に連結する。この文脈で使用する「機能的に連結」なる用語は、2つのDNAフラグメントによりコードされるアミノ酸配列がインフレームに維持されるように2つのDNAフラグメントを結合することを意味する。
【0106】
VHをコードするDNAを、重鎖定常領域(CH1、CH2及びCH3)をコードする別のDNA分子と機能的に連結することにより、VH領域をコードする単離DNAを全長重鎖遺伝子に変換することができる。ヒト重鎖定常領域遺伝子の配列は当分野で公知であり(例えばKabat,E.A.,et al.(1991)Sequences of Proteins of Immunological Interest,Fifth Edition,U.S.Department of Health and Human Services,NIH Publication No.91−3242参照)、これらの領域を含むDNAフラグメントは標準PCR増幅法により得ることができる。重鎖定常領域はIgG1、IgG2、IgG3、IgG4、IgA、IgE、IgM又はIgD定常領域とすることができるが、IgG1又はIgG4定常領域が最も好ましい。Fabフラグメント重鎖遺伝子では、VHをコードするDNAを、重鎖CH1定常領域のみをコードする別のDNA分子と機能的に連結することができる。
【0107】
VLをコードするDNAを、軽鎖定常領域CLをコードする別のDNA分子と機能的に連結することにより、VL領域をコードする単離DNAを全長軽鎖遺伝子(及びFab軽鎖遺伝子)に変換することができる。ヒト軽鎖定常領域遺伝子の配列は当分野で公知であり(例えばKabat,E.A.,et al.(1991)Sequences of Proteins of Immunological Interest,Fifth Edition,U.S.Department of Health and Human Services,NIH Publication No.91−3242参照)、これらの領域を含むDNAフラグメントは標準PCR増幅法により得ることができる。軽鎖定常領域はκ又はλ定常領域とすることができるが、κ定常領域が最も好ましい。
【0108】
scFv遺伝子を作製するためには、VL領域とVH領域をフレキシブルリンカーでつないでVH配列とVL配列を連続する1本鎖蛋白質として発現させることができるように、VHとVLをコードするDNAフラグメントを、フレキシブルリンカー、例えばアミノ酸配列(Gly4−Ser)3をコードする別のフラグメントと機能的に連結する(例えばBird et al.(1988)Science 242:423−426;Huston et al.(1988)Proc.Natl.Acad.Sci.USA 85:5879−5883;McCafferty et al.,Nature(1990)348:552−554参照)。
【0109】
本発明で使用される抗体又は抗体部分を発現させるためには、遺伝子を転写及び翻訳制御配列と機能的に連結するように、上記のように得られた部分的又は全長軽鎖及び重鎖をコードするDNAを発現ベクターに挿入する。この文脈で「機能的に連結」なる用語は、ベクター内の転写及び翻訳制御配列が抗体遺伝子の転写及び翻訳を制御するというその所期機能を発揮するように、抗体遺伝子をベクターにライゲーションすることを意味する。発現ベクターと発現制御配列は使用する発現宿主細胞と適合するように選択する。抗体軽鎖遺伝子と抗体重鎖遺伝子を別々のベクターに挿入することもできるが、より一般には、両方の遺伝子を同一の発現ベクターに挿入する。標準方法(例えば抗体遺伝子フラグメントとベクター上の相補的制限部位のライゲーション、又は制限部位が存在しない場合には平滑末端ライゲーション)により抗体遺伝子を発現ベクターに挿入する。D2E7又はD2E7関連軽鎖又は重鎖配列を挿入する前に、発現ベクターに予め定常領域配列を組込んでもよい。例えば、D2E7又はD2E7関連VH及びVL配列を全長抗体遺伝子に変換する1つのアプローチは、VHセグメントをベクター内のCHセグメントと機能的に連結し、VLセグメントをベクター内のCLセグメントと機能的に連結するように、夫々重鎖定常領域と軽鎖定常領域を予めコードする発現ベクターに挿入する方法である。上記の追加又は代替として、組換え発現ベクターは宿主細胞からの抗体鎖の分泌を助長するシグナルペプチドをコードすることができる。シグナルペプチドを抗体鎖遺伝子のアミノ末端とインフレームで連結するように、抗体鎖遺伝子をベクターにクローニングすることができる。シグナルペプチドは免疫グロブリンシグナルペプチド又は異種シグナルペプチド(即ち非免疫グロブリン蛋白質に由来するシグナルペプチド)とすることができる。
【0110】
抗体鎖遺伝子に加え、本発明の組換え発現ベクターは宿主細胞における抗体鎖遺伝子の発現を制御する調節配列をもつ。「調節配列」なる用語はプロモーター、エンハンサー及び抗体鎖遺伝子の転写又は翻訳を制御する他の発現制御エレメント(例えばポリアデニル化シグナル)を包含するものとする。このような調節配列は例えば、Goeddel;Gene Expression Technology:Methods in Enzymology 185,Academic Press,San Diego,CA(1990)に記載されている。当業者に自明の通り、調節配列の選択を含めて発現ベクターの設計は、形質転換させる宿主細胞の選択、所望される蛋白質発現レベル等の因子に依存し得る。哺乳動物宿主細胞発現に好ましい調節配列としては、哺乳動物細胞における高レベルの蛋白質発現を誘導するウイルスエレメントが挙げられ、例えばサイトメガロウイルス(CMV)(例えばCMVプロモーター/エンハンサー)、サルウイルス40(SV40)(例えばSV40プロモーター/エンハンサー)、アデノウイルス(例えばアデノウイルス主要後期プロモーター(AdMLP))及びポリオーマに由来するプロモーター及び/又はエンハンサーが挙げられる。ウイルス調節エレメントとその配列の詳細な説明については、例えばStinskiの米国特許第5,168,062号、Bellらの米国特許第4,510,245号及びSchaffnerらの米国特許第4,968,615号参照。
【0111】
抗体鎖遺伝子と調節配列に加え、本発明で使用される組換え発現ベクターには、宿主細胞におけるベクターの複製を調節する配列(例えば複製起点)や選択マーカー遺伝子等の他の配列を組込むことができる。選択マーカー遺伝子はベクターを導入された宿主細胞の選択を容易にする(例えばいずれもAxelらの米国特許第4,399,216号、4,634,665号及び5,179,017号参照)。例えば、一般に選択マーカー遺伝子はベクターを導入された宿主細胞にG418、ハイグロマイシン又はメトトレキサート等の薬剤に対耐性を付与する。好ましい選択マーカー遺伝子としては、ジヒドロ葉酸レダクターゼ(DHFR)遺伝子(dhfr欠損宿主細胞におけるメトトレキサート選択/増幅用)とneo遺伝子(G418選択用)が挙げられる。
【0112】
軽鎖と重鎖を発現させるために、重鎖と軽鎖をコードする発現ベクターを標準技術により宿主細胞にトランスフェクトする。「トランスフェクション」なる用語の各種語形は外来DNAを原核又は真核宿主細胞に導入するために一般に使用されている多様な技術(例えばエレクトロポレーション、リン酸カルシウム沈殿法、DEAE−デキストラントランスフェクション等)を包含するものとする。理論的には本発明の抗体を原核又は真核宿主細胞のどちらでも発現させることが可能であるが、真核細胞、特に哺乳動物細胞は原核細胞よりも正しく折り畳まれた免疫的に活性な抗体を会合・分泌させるので、真核細胞、最も好ましくは哺乳動物宿主細胞で抗体を発現させるのが最も好ましい。抗体遺伝子の原核細胞発現は活性抗体の高収率生産には無効であることが報告されている(Boss,M.A.and Wood,C.R.(1985)Immunology Today 6:12−13)。
【0113】
本発明の組換え抗体を発現させるために好ましい哺乳動物宿主細胞としては、チャイニーズハムスター卵巣(CHO細胞)(Urlaub and Chasin,(1980)Proc.Natl.Acad.Sci.USA 77:4216−4220に記載されているdhfr欠損CHO細胞と、例えばR.J.Kaufman and P.A.Sharp (1982)Mol.Biol.159:601−621に記載されているようなDHFR選択マーカーの併用を含む)、NS0ミエローマ細胞、COS細胞及びSP2細胞が挙げられる。抗体遺伝子をコードする組換え発現ベクターを哺乳動物宿主細胞に導入する場合には、宿主細胞における抗体の発現、又はより好ましくは宿主細胞を増殖させる培地への抗体の分泌を可能にするために十分な時間にわたって宿主細胞を培養することにより抗体を産生させる。標準蛋白質精製法を使用して培地から抗体を回収することができる。
【0114】
FabフラグメントやscFv分子等の無傷の抗体の部分を作製するために宿主細胞を使用することもできる。当然のことながら、上記手順の変形も本発明の範囲に含まれる。例えば、本発明の抗体の軽鎖又は重鎖のいずれか(両方ではない)をコードするDNAを宿主細胞にトランスフェクトすることが望ましい場合がある。hTNFαとの結合に不要な軽鎖及び重鎖のいずれか又は両方をコードするDNAの一部又は全部を除去するために組換えDNA技術を使用してもよい。このような低分子DNA分子から発現される分子も本発明の抗体に含まれる。更に、標準化学架橋法により本発明の抗体を第2の抗体と架橋させることにより、一方の重鎖と一方の軽鎖が本発明の抗体であり、他方の重鎖と軽鎖がhTNFα以外の抗原に特異的である2価抗体を作製することもできる。
【0115】
本発明の抗体又はその抗原結合部分の組換え発現に好ましいシステムでは、抗体重鎖と抗体軽鎖の両方をコードする組換え発現ベクターをリン酸カルシウムトランスフェクション法によりdhfr欠損CHO細胞に導入する。組換え発現ベクター内で、遺伝子の高レベル転写を誘導するように抗体重鎖及び軽鎖遺伝子を各々CMVエンハンサー/AdMLPプロモーター調節エレメントと機能的に連結する。メトトレキサート選択/増幅を使用してベクターをトランスフェクトされたCHO細胞の選択を可能にするDHFR遺伝子も組換え発現ベクターに組込む。選択された形質転換宿主細胞を培養し、抗体重鎖及び軽鎖を発現させ、無傷の抗体を培地から回収する。標準分子生物学技術を使用して組換え発現ベクターを作製し、宿主細胞にトランスフェクトし、形質転換細胞を選択し、宿主細胞を培養し、培地から抗体を回収する。
【0116】
上記に鑑み、本発明で使用される抗体及び抗体部分の組換え発現に使用することができる核酸、ベクター及び宿主細胞としては、ヒトTNFα抗体アダリムマブ(D2E7)を含む核酸と、前記核酸を組込んだベクターが挙げられる。D2E7軽鎖可変領域をコードするヌクレオチド配列を配列番号36に示す。LCVRのCDR1ドメインはヌクレオチド70〜102を含み、CDR2ドメインはヌクレオチド148〜168を含み、CDR3ドメインはヌクレオチド265〜291を含む。D2E7重鎖可変領域をコードするヌクレオチド配列を配列番号37に示す。HCVRのCDR1ドメインはヌクレオチド91〜105を含み、CDR2ドメインはヌクレオチド148〜198を含み、CDR3ドメインはヌクレオチド295〜330を含む。当業者に自明の通り、D2E7関連抗体又はその部分(例えばCDR3ドメイン等のCDRドメイン)をコードするヌクレオチド配列は、遺伝コード及び標準分子生物学技術を使用してD2E7 LCVR及びHCVRをコードするヌクレオチド配列から誘導することができる。
【0117】
1態様において、液体医薬製剤は抗体アダリムマブに対して生物学的に等価又は生物学的に同等のヒトTNFα抗体又はその抗原結合部分を含有する。1態様において、生物学的に同等の抗体とは参照抗体(例えばアダリムマブ)と比較した場合に臨床的に有意な差を示さない抗体である。生物学的に同等の抗体は参照抗体(例えばアダリムマブ)と等価の安全性、純度及び力価をもつ。
【0118】
IV.本発明の製剤の投与
本発明の製剤の1つの利点は高濃度のヒト抗TNFα抗体又は抗原結合部分(例えばアダリムマブ)を対象に皮下送達するために使用できるという点である。従って、1態様では、本発明の製剤を対象に皮下送達する。1態様において、対象は製剤を自己投与する。
【0119】
1態様では、有効量の製剤を投与する。製剤の「有効量」なる用語はTNFα活性を阻害するため、例えば有害なTNFα活性に関連する状態の各種形態的及び身体的症状を予防するために必要又は十分な量である。別の態様において、製剤の有効量は所望結果を達成するために必要な量である。
【0120】
製剤の有効量の1例は有害なTNFα活性を阻害するため、又はTNFα活性が有害である疾患を治療するために十分な量である。本願で使用する「TNFα活性が有害である疾患」なる用語は疾患に罹患している対象におけるTNFαの存在が前記疾患の病態生理に関与しているか又は前記疾患の増悪に寄与する因子であることが分かっているか又はその疑いのある疾病及び他の疾患を包含するものとする。従って、TNFα活性が有害である疾患とは、TNFα活性の阻害が疾患の症状及び/又は進行を緩和すると予想される疾患である。このような疾患は例えば、疾患に罹患している対象の体液中のTNFα濃度の上昇(例えば対象の血清、血漿、滑液等におけるTNFα濃度上昇)により確認することができ、このような上昇は例えば抗TNFα抗体を使用して検出することができる。
【0121】
下記実施例に記載するように、本発明の製剤の1つの利点は製剤の粘度を増加せずに高濃度の抗体を含有する製剤を製造できる点である。従って、同じく以下に記載するように、新規製剤は従来の市販製剤に比較して少ない容量で多量(例えば有効量)の抗体の投与を可能にすることにより、疼痛を緩和する。
【0122】
1態様において、抗体の有効量は厳密に体重に基づく投与スキーム(例えばmg/kg)に従って決定することもできるし、体重から独立した総用量(固定用量とも言う)でもよい。1例において、製剤の有効量は総用量約80mgの抗体を含有する製剤0.8mL(即ち本発明の100mg/mL抗体製剤0.8mL)である。別の例において、製剤の有効量は総用量約40mgの抗体を含有する本発明の製剤0.4mL(即ち本発明の100mg/mL抗体製剤0.4mL)である。更に別の例において、製剤の有効量は総用量約160mgの抗体を含有する製剤0.8mL×2(即ち本発明の100mg/mL抗体製剤0.8mLずつを含む2単位)である。別の例において、製剤の有効量は総用量約20mgの抗体を含有する本発明の製剤0.2mL(即ち本発明の100mg/mL抗体製剤0.2mL)である。あるいは、体重に基づく固定用量レジメンに従って有効量を決定してもよい(例えば本願に援用するWO2008/154543参照)。
【0123】
本発明は、1態様ではTNFα活性が有害である疾患に罹患している対象におけるTNFα活性を阻害するために使用される貯蔵寿命の長い安定した高濃度製剤を提供し、対象におけるTNFα活性を阻害するように本発明の製剤を対象に投与する。好ましくは、TNFαはヒトTNFαであり、対象はヒト対象である。あるいは、対象は本発明の抗体と交差反応するTNFαを発現する哺乳動物でもよい。更に、対象は(例えばhTNFαの投与又はhTNFαトランスジーンの発現により)hTNFαを導入された哺乳動物でもよい。
【0124】
本発明の製剤は(以下に詳述する)治療目的でヒト対象に投与することができる。本発明の1態様において、液体医薬製剤は容易に投与可能であり、例えば患者により自己投与される製剤が挙げられる。好ましい1態様において、本発明の製剤は皮下注射により好ましくは単回投与される。更に、本発明の製剤は獣医学的目的のため又はヒト疾患の動物モデルとして抗体と交差反応するTNFαを発現する非ヒト哺乳動物(例えば霊長類、ブタ又はマウス)に投与することもできる。後者に関して、このような動物モデルは本発明の抗体の治療効力の評価(例えば投与用量及び時間コースの試験)に有用であると思われる。
【0125】
1態様では、プレフィルドシリンジ、自己注射ペン又は無針投与装置により本発明の液体医薬製剤を対象に投与することができる。従って、本発明は本発明の液体医薬製剤を収容する自己注射ペン、プレフィルドシリンジ又は無針投与装置にも関する。1態様において、本発明は100mg/mLのヒトTNFα抗体又はその抗原結合部分を含有する製剤の1用量を収容する送達装置に関し、例えば自己注射ペン又はプレフィルドシリンジは約19mg、20、mg、21mg、22mg、23mg、24mg、25mg、26mg、27mg、28mg、29mg、30mg、31mg、32mg、33mg、34mg、35mg、36mg、37mg、38mg、39mg、40mg、41mg、42mg、43mg、44mg、45mg、46mg、47mg、48mg、49mg、50mg、51mg、52mg、53mg、54mg、55mg、56mg、57mg、58mg、59mg、60mg、61mg、62mg、63mg、64mg、65mg、66mg、67mg、68mg、69mg、70mg、71mg、72mg、73mg、74mg、75mg、76mg、77mg、78mg、79mg、80mg、81mg、82mg、83mg、84mg、85mg、86mg、87mg、88mg、89mg、90mg、91mg、92mg、93mg、94mg、95mg、96mg、97mg、98mg、99mg、100mg、101mg、102mg、103mg、104mg、105mg等の製剤の1用量を収容する。
【0126】
好ましくは、本発明の製剤はTNFα活性が有害である疾患を治療するために使用される。本願で使用する「TNFα活性が有害である疾患」なる用語は疾患に罹患している対象におけるTNFαの存在が前記疾患の病態生理に関与しているか又は前記疾患の増悪に寄与する因子であることが分かっているか又はその疑いのある疾病及び他の疾患を包含するものとする。従って、TNFα活性が有害である疾患とは、TNFα活性の阻害が疾患の症状及び/又は進行を緩和すると予想される疾患である。このような疾患は例えば、疾患に罹患している対象の体液中のTNFα濃度の上昇(例えば対象の血清、血漿、滑液等におけるTNFα濃度上昇)により確認することができ、このような上昇は例えば上記のように抗TNFα抗体を使用して検出することができる。
【0127】
TNFα活性が有害である疾患には多数の例が存在する。TNFα活性が有害である疾患の例はいずれもその開示内容全体を本願に援用する米国特許第6,015,557号、6,177,077号、6,379,666号、6,419,934号、6,419,944号、6,423,321号、6,428,787号、及び6,537,549号、並びにPCT公開第WO00/50079号及びWO01/49321号にも記載されている。本発明の製剤はいずれもその開示内容全体を本願に援用する米国特許第6,090,382号、6,258,562号及び米国特許出願公開第US20040126372号に記載されているようなTNFα活性が有害である疾患を治療するために使用することもできる。
【0128】
以下、特定の代表的疾患の治療における本発明の製剤の使用について詳述する。
【0129】
A.敗血症
腫瘍壊死因子は血圧低下、心筋抑制、血管漏出症候群、臓器壊死、毒性二次メディエーターの放出の刺激及び血液凝固カスケードの活性化を含む生物学的効果により、敗血症の病態生理に関与することが知られている(例えばTracey,K.J.and Cerami,A.(1994)Annu.Rev.Med.45:491−503;Russell,D and Thompson,R.C.(1993)Curr.Opin.Biotech.4:714−721参照)。従って、本発明の製剤は敗血症性ショック、内毒素ショック、グラム陰性菌敗血症及び毒素性ショック症候群を含むその臨床背景のいずれかにおいて敗血症を治療するために使用することができる。
【0130】
更に、敗血症を治療するために、インターロイキン1阻害剤(例えばPCT公開第WO92/16221号及びWO92/17583に記載されているもの)、サイトカインであるインターロイキン6(例えばPCT公開第WO93/11793号参照)又は血小板活性化因子のアンタゴニスト(例えば欧州特許出願公開第EP374510号参照)等の敗血症を更に緩和することができる1種以上の他の治療剤と本発明の製剤を併用投与することができる。
【0131】
更に、好ましい1態様では、治療時のIL−6の血清又は血漿濃度が500pg/ml、より好ましくは1000pg/mlを上回る敗血症患者のサブグループ内のヒト対象に本発明の製剤を投与する(PCT公開第WO95/20978号参照)。
【0132】
B.自己免疫疾患
腫瘍壊死因子は各種自己免疫疾患の病態生理に関与すると考えられている。例えば、TNFαは組織炎症を活発にし、関節リウマチにおける関節破壊をもたらすと考えられている(例えばTracey and Cerami,前出;Arend,W.P.and Dayer,J−M.(1995)Arth.Rheum.38:151−160;Fava,R.A.,et al.(1993)Clin.Exp.Immunol.94:261−266参照)。TNFαは膵島細胞の死滅を促進し、糖尿病におけるインスリン抵抗性に関与するとも考えられている(例えばTracey and Cerami,前出;PCT公開第WO94/08609号参照)。TNFαは多発性硬化症において希突起膠細胞に対する細胞傷害作用と炎症性プラークの誘導に関与するとも考えられている(例えばTracey and Cerami,前出参照)。本発明の製剤を使用して治療することができる自己免疫疾患には、若年性特発性関節炎(JIA)(若年性関節リウマチとも言う)も含まれる(Grom et al.(1996)Arthritis Rheum.39:1703;Mangge et al.(1995)Arthritis Rheum.8:211参照)。
【0133】
本発明の製剤は自己免疫疾患、特に炎症を伴うものを治療するために使用することができ、関節リウマチ、リウマチ性脊椎炎(強直性脊椎炎とも言う)、変形性関節症及び通風性関節炎、アレルギー、多発性硬化症、自己免疫性糖尿病、自己免疫性ぶどう膜炎、若年性特発性関節炎(若年性関節リウマチとも言う)、並びにネフローゼ症候群が挙げられる。
【0134】
C.感染症
腫瘍壊死因子は各種感染症に認められる生物学的作用に関与すると考えられている。例えば、TNFαはマラリアにおいて脳炎症と毛細血管血栓及び梗塞に関与すると考えられている(例えばTracey and Cerami,前出参照)。TNFαは髄膜炎において脳炎症に関与し、血液脳関門の破壊を誘導し、敗血症性ショック症候群を誘発し、静脈梗塞を活性化するとも考えられている(例えばTracey and Cerami,前出参照)。TNFαは後天性免疫不全症候群(エイズ)において悪液質を誘導し、ウイルス増殖を刺激し、中枢神経系損傷に関与するとも考えられている(例えばTracey and Cerami,前出参照)。従って、本発明の抗体及び抗体部分は細菌性髄膜炎(例えば欧州特許出願公開第EP585705号参照)、脳マラリア、エイズ及びエイズ関連症候群(ARC)(例えば欧州特許出願公開第EP230574号参照)、並びに移植に伴うサイトメガロウイルス感染(例えばFietze,E.,et al.(1994)Transplantation 58:675−680参照)を含む感染症の治療に使用することができる。本発明の製剤は感染による発熱及び筋肉痛(例えばインフルエンザ)や、感染に伴う(例えばエイズ又はARCに伴う)悪液質を含めて感染症に伴う症状を緩和するために使用することもできる。
【0135】
D.移植
腫瘍壊死因子は同種移植片拒絶反応及び移植片対宿主病(GVHD)の主要なメディエータであり、腎移植の拒絶反応を抑制するためにT細胞受容体CD3複合体に対するラット抗体OKT3を使用する場合に認められている有害反応に関与すると考えられている(例えばTracey and Cerami,前出;Eason,J.D.,et al.(1995)Transplantation 59:300−305;Suthanthiran,M.and Strom,T.B.(1994)New Engl.J.Med.331:365−375参照)。従って、本発明の製剤は同種移植片及び異種移植片の拒絶反応を含む移植拒絶反応を抑制し、GVHDを抑制するために使用することができる。抗体又は抗体部分は単独で使用してもよいが、同種移植片に対する免疫応答を抑制又はGVHDを抑制する1種以上の他の物質と併用することができる。例えば、1態様では、OKT3により誘導される反応を抑制するために本発明の製剤をOKT3と併用する。別の態様では、細胞表面分子CD25(インターロイキン2受容体α)、CD11a(LFA−1)、CD54(ICAM−1)、CD4、CD45、CD28/CTLA4、CD80(B7−1)及び/又はCD86(B7−2)等の免疫応答の調節に関与する他の標的に対する1種以上の抗体と本発明の製剤を併用する。更に別の態様では、シクロスポリンAやFK506等の1種以上の一般的な免疫抑制剤と本発明の製剤を併用する。
【0136】
E.悪性腫瘍
腫瘍壊死因子は悪性腫瘍において悪液質を誘導し、腫瘍増殖を刺激し、転移の可能性を増し、細胞傷害作用に関与すると考えられている(例えばTracey and Cerami,前出参照)。従って、本発明の製剤は腫瘍増殖もしくは転移を抑制し、及び/又は悪性腫瘍に伴う悪液質を緩和するために、悪性腫瘍の治療で使用することができる。
【0137】
F.肺疾患
腫瘍壊死因子は白血球−内皮細胞活性化の刺激、肺細胞に対する細胞傷害作用の誘発及び血管漏出症候群の誘導を含む成人呼吸窮迫症候群の病態生理に関係があると考えられている(例えばTracey and Cerami,前出参照)。従って、本発明の製剤は成人呼吸窮迫症候群(例えばPCT公開第WO91/04054号参照)、ショック肺、慢性肺炎症疾患、肺サルコイドーシス、肺線維症及び珪肺症を含む各種肺疾患を治療するために使用することができる。
【0138】
G.腸疾患
腫瘍壊死因子は炎症性腸疾患の病態生理に関係があると考えられている(例えばTracy,K.J.,et al.(1986)Science 234:470−474;Sun,X−M.,et al.(1988)J.Clin.Invest.81:1328−1331;MacDonald,T.T.,et al.(1990)Clin.Exp.Immunol.81:301−305参照)。キメラマウス抗hTNFα抗体がクローン病の治療用に臨床試験されている(van Dullemen,H.M.,et al.(1995)Gastroenterology 109:129−135)。本発明の製剤はクローン病と潰瘍性大腸炎の2種類の症候群を含む特発性炎症性腸疾患等の腸疾患を治療するために使用することもできる。
【0139】
H.心疾患
本発明の製剤は心筋虚血(例えば欧州特許出願公開第EP453898号参照)と心不全(心筋の弱化)(例えばPCT公開第WO94/20139号参照)を含む各種心疾患を治療するために使用することもできる。
【0140】
I.脊椎関節症
TNFαは脊椎関節症等の炎症性疾患を含む多様な疾患の病態生理に関係があると考えられている(例えばMoeller,A.,et al.(1990)Cytokine 2:162−169;Moellerらの米国特許第5,231,024号;Moeller,Aの欧州特許公開第260610B1号参照)。本発明の製剤により治療することができる脊椎関節症の1例としては乾癬性関節炎が挙げられる。腫瘍壊死因子は乾癬性関節炎の病態生理に関係があると考えられている(Partsch et al.(1998)Ann Rheum Dis.57:691;Ritchlin et al.(1998)J Rheumatol.25:1544)。
【0141】
J.皮膚及び爪疾患
1態様において、本発明の製剤は皮膚及び爪疾患を治療するために使用される。本願で使用する「TNFα活性が有害である皮膚及び爪疾患」なる用語は疾患に罹患している対象におけるTNFαの存在が前記疾患の病態生理に関与しているか又は前記疾患の増悪に寄与する因子であることが分かっているか又はその疑いのある皮膚及び/又は爪疾患と他の疾患(例えば乾癬)を包含するものとする。本発明の製剤を使用して治療することができる皮膚疾患の1例は乾癬である。1態様において、本発明の製剤はプラーク乾癬を治療するために使用される。腫瘍壊死因子は乾癬の病態生理に関係があると考えられている(Takematsu et al.(1989)Arch Dermatol Res.281:398;Victor and Gottlieb(2002)J Drugs Dermatol.1(3):264)。
【0142】
1態様において、本発明の製剤は関節リウマチ、乾癬性関節炎又は強直性脊椎炎を治療するために使用される。単離ヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)を含有する本発明の製剤は関節リウマチ、乾癬性関節炎又は強直性脊椎炎を治療するために有効な投与スキーム及び用量に従ってヒト対象に投与することができる。1態様では、関節リウマチ、乾癬性関節炎又は強直性脊椎炎を治療するために、本発明の製剤中のヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)約40mgの1用量(例えば本発明の100mg/mL製剤0.4mL)を1週間置きにヒト対象に投与する。1態様では、関節リウマチ、強直性脊椎炎又は乾癬性関節炎を治療するために製剤を1週間置き(隔週とも言う、本願に援用するUS20030235585に記載されている投与方法参照)に皮下投与する。
【0143】
1態様において、本発明の製剤はクローン病を治療するために使用される。単離ヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)を含有する本発明の製剤はクローン病を治療するために有効な投与スキーム及び用量に従ってヒト対象に投与することができる。1態様では、クローン病を治療するために、本発明の製剤中のヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)約160mgの1用量(例えば本発明の100mg/mL製剤1.6mL)をヒト対象に約1日目に初期投与した後、2週間後に抗体80mgの後続用量(例えば本発明の100mg/mL製剤0.8mL)を投与し、更にその後、1週間置きに約40mg(例えば本発明の100mg/mL製剤0.4mL)を投与する。1態様では、クローン病を治療するために誘導用量と維持用量を含む多回可変用量レジメン(例えば、各々本願に援用する米国特許公開第US20060009385号及びUS20090317399号参照)に従って製剤を皮下投与する。
【0144】
1態様において、本発明の製剤は乾癬を治療するために使用される。単離ヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)を含有する本発明の製剤は乾癬を治療するために有効な投与スキーム及び用量に従ってヒト対象に投与することができる。1態様では、本発明の製剤中のヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)約80mgの初期用量(例えば本発明の100mg/mL製剤0.8mL)をヒト対象に投与した後、初期投与の1週間後から開始して1週間置きに抗体40mgの後続用量(例えば本発明の100mg/mL製剤0.4mL)を投与する。1態様では、乾癬を治療するために誘導用量と維持用量を含む多回可変用量レジメン(例えば、各々本願に援用するUS20060009385及びWO2007/120823参照)に従って製剤を皮下投与する。
【0145】
1態様において、本発明の製剤は若年性特発性関節炎(JIA)を治療するために使用される。単離ヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)を含有する本発明の製剤はJIAを治療するために有効な投与スキーム及び用量に従ってヒト対象に投与することができる。1態様では、JIAを治療するために、本発明の製剤中のヒトTNFα抗体又はその抗原結合部分20mg(例えば本発明の100mg/mL製剤0.2mL)を体重15kg(約33lb)〜30kg(66lb)未満の対象に1週間置きに投与する。別の態様では、JIAを治療するために、本発明の製剤中のヒトTNFα抗体又はその抗原結合部分40mg(例えば本発明の100mg/mL製剤0.4mL)を体重30kg(66lb)以上の対象に1週間置きに投与する。1態様では、JIAを治療するために、体重に基づく固定用量(例えば、本願に援用する米国特許公開第20090271164号参照)に従って製剤を皮下投与する。
【0146】
1態様では、有害なTNFα活性に関連する疾患を治療するために、抗体を1カ月に一度又は4週間に一度投与するような毎月投与スケジュールに従って単離ヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)をヒト対象に投与することができる。上記のように、毎月投与スケジュールに従って治療することができる疾患の例としては、限定されないが、関節リウマチ、強直性脊椎炎、JIA、乾癬、クローン病又は乾癬性関節炎が挙げられる。従って、単離ヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)を含有する本発明の製剤は有害なTNFα活性に関連する疾患を治療するために毎月投与スケジュールに従ってヒト対象に投与することができる。1態様では、有害なTNFα活性に関連する疾患をもつ対象に本発明の製剤中のヒトTNFα抗体又はその抗原結合部分80mg(例えば本発明の100mg/mL製剤0.8mL)を投与する。
【0147】
本願に記載する投与量は単回用量(例えば0.4mL中に40mg又は0.8mL中に80mgの単回用量)として送達してもよいし、あるいは多回用量(例えば160mg用量を送達するために40mgずつ4回又は80mgずつ2回)として送達してもよい。
【0148】
単離ヒトTNFα抗体又はその抗原結合部分(例えばアダリムマブ)を含有する本発明の製剤を対象に別の治療剤と併用投与することもできる。1態様では、関節リウマチを治療するために製剤をヒト対象にメトトレキサート又は他の疾患修飾性抗リウマチ薬(DMARD)と併用投与する。別の態様では、JIAを治療するために製剤をヒト対象にメトトレキサート又は他の疾患修飾性抗リウマチ薬(DMARD)と併用投与する。その他の併用療法も米国特許第6,258,562号及び7,541,031号と、米国特許公開第US20040126372号に記載されており、いずれもその開示内容全体を本願に援用する。
【0149】
ヒトTNFα抗体又はその抗原結合部分を含有する本発明の製剤は過去にTNF阻害剤療法の効果がなかった対象、例えばインフリキシマブに応答しなかった対象又は不寛容な対象を治療するために使用することもできる。
【0150】
以下、実施例により本発明を更に例証するが、これらの実施例は限定的であるとみなすべきではない。
【実施例1】
【0151】
ヒト抗TNFα抗体液体医薬製剤の安定性の改善
本実施例は抗体アダリムマブの医薬製剤の安定性の改善を目的とする実験の結果について記載する。
【0152】
材料及び方法
最終薬剤濃度50mg/mLの液体非経口剤形を作製するために、アダリムマブ(サブクラスG1,約47kDa)を修飾医薬製剤に製剤化した。過去の製剤実験によると、リン酸/クエン酸緩衝液系はアダリムマブの蛋白質安定化に関して他の緩衝液系よりも優れていることが分かっている。従って、液体50mg/mL製剤に添加剤を添加することにより安定性の改善を試みた。使用した全添加剤は最高純度(「プロアナリシス」グレード)のものをMerck KGaA,Darmstadt,Germanyから購入した。マンニトールはMallinckrodt Baker B.V.,Deventer,Hollandから入手した。
【0153】
可視粒状物の分析はPh.Eur.2002の規定に従って実施した(§2.9.20 Contamination with particulate matter−visible particles)。不可視粒状物分析は光遮蔽により判定した(SVSS−C40,PAMAS GmbH,Rutesheim,Germany)。Superose TM6 10/30カラム(Amersham Pharmacia Europe GmbH,Freiburg,Germany)をSE−HPLC分析(蛋白質モノマー含有量の評価)に使用し、pH7.5のPBS緩衝液を流速0.5mL/minで流し、オンライン検出のためにUV280分光分析、屈折率検出及びMALSと連動させた。各サンプルの分析を少なくとも3回ずつ実施した。特に指定しない限り、全SE−HPLCデータでSrelは0.13未満であり、全光遮蔽データで2.3未満であった。
【0154】
アダリムマブ濃厚液(〜70mg/mL)を添加剤ストック溶液で希釈することにより個々の蛋白質製剤を調製した。70mg/mLアダリムマブストック溶液は表16に示すようなクエン酸リン酸緩衝液成分の組成(即ちクエン酸・H2O、クエン酸ナトリウム脱水和物、Na2HPO4・2H2O、NaH2PO4・2H2O)を使用して調製した。
【0155】
表16に示すようなクエン酸リン酸緩衝液成分の組成(即ちクエン酸・H2O,クエン酸ナトリウム脱水和物、Na2HPO4・2H2O、NaH2PO4・2H2O)を使用して添加剤をリン酸/クエン酸緩衝液溶媒に溶解することにより添加剤ストック溶液を作製した。滅菌濾過(0.2μm,Minisart(登録商標),Sartorius AG,Goettingen,Germany)前に、緩衝液成分の酸/塩基試料を添加することによりpH調節を実施した。全製剤を少なくとも2本ずつ調製し、無菌空気層流条件下で熱滅菌(180℃,25分)2Rガラスバイアル(Schott Glas,Mainz,Germany)に溶液バッチを最終滅菌濾過することにより作製した。使用前にPh.Eur.に従ってテフロン(登録商標)被覆ブチルラバーストッパーを湿熱(121℃)滅菌した。
【0156】
各種製剤を3種類の異なる温度(5℃、25℃、40℃)で3カ月短期保存した。
【0157】
リン酸/クエン酸緩衝液溶媒を緩衝液交換に使用し、アダリムマブバルク溶液をVivaflow 50 units(カットオフ50kDa,Vivascience G,Hannover,Germany)で透析濾過することによりアダリムマブ濃厚液を準備した。バイオ医薬品溶液の現行の濃縮法と緩衝液交換法はIEX、SE−HPLC、限外/透析濾過及び接線流濾過に基づく(Christy et al.(2002)Desalination,144:133−136)。透析濾過は可変流速で1回の操作で精製、濃縮及び緩衝液交換が可能であるため、蛋白質ストレスが最小限になることから、透析濾過を適用した(表1)。
【0158】
【表1】
実施した各サイクルは総蛋白質の〜0.25%の蛋白質損失をもたらした。一般に、蛋白質損失は濃厚液生成中に7%を超えなかった。
【0159】
1回の透析濾過サイクル以内で、蛋白質濃度は倍増したので、最終濃縮段階以外は元の濃度まで希釈した。従って、存在すべきでない望ましくない溶解物質を有効に除去することができる(例えば透析濾過サイクル10回以内で1.00%濃度を0.00098%まで低下させることができる)。精製及び濃縮後、アダリムマブ濃厚液を遠心した(5℃,3000g,20分間)。
【0160】
pH最適値の評価
最適溶液pH(即ちpH5.2又はpH6.0)を評価するために、pHのみを変化させた2種類の異なるアダリムマブ製剤を分析した。1mg/mL Tween 80を含有する製剤の安定性データを表2A及び2Bに示す。
【0161】
【表2】
【0162】
【表3】
モノマー含有量については、両製剤とも40℃保存時に同等のモノマー損失を示したため、pHによる優劣はないことが分かった。25℃保存条件のデータは40℃データと同様であったが、5℃では、この試験中に分析した全蛋白質溶液はモノマー含有量の有意変化を生じなかった。
【0163】
他方、濁度には差が認められた。保存温度に関係なく、溶液pH6.0では、12週間保存中に不可視粒状物の形成を生じた。粒状物形成の強度は低温に結び付けられるので、粒子の起源は蛋白性ではないと予想される。この点で、深刻な粒状物形成が蛋白質不安定のみに起因したのであるならば、これは保存試験中の高温暴露に関係があると考えられる(Constantino,et al.(1994b)J.Pharm.Sci.83:1662−1669)。
【0164】
Tween 80の代わりに6.16mg/mL NaClを含有する50mg/mLアダリムマブ製剤については、塩添加の結果、1μmよりも大きい粒子の数が両溶液で同程度増加したので、不可視粒子が形成された(表3A及び3B参照)。更に、12週間後に、SE−HPLCデータによると、差は最小限(〜0.3%)であり、25℃結果により確証されなかったが、pH6.0溶液はpH5.2溶液よりもモノマー含有量が多かった。
【0165】
【表4】
【0166】
【表5】
粒子形成はNaCl添加とpH6.0保存により助長され、Tween 80添加と溶液pH5.2により改善されると思われた。従って、NaCl等の塩を含有する溶液中の粒子汚染を軽減することができる成分としてTween 80を提案した(表4A及び4B)。次に6.16mg/mL NaClと1mg/mL Tween 80の両方を含有する溶液を試験した。
【0167】
【表6】
【0168】
【表7】
表4Bに示すように、塩と界面活性剤を含有する製剤では、Tween 80の添加にも拘わらず不可視粒子が明白であったことから、界面活性剤を添加しても不可視粒子形成については影響がなかった。興味深いことに、全サンプルで粒子数は最低保存温度(5℃)で最大であり、粒子起源が潜在的に無機材料によることが示唆された。更に、塩を含有する溶液の目視検査によると、保存温度に関係なく、4週間保存後に若干の濁りが判明した。保存が一時的であっても、低温保存の結果として可視無機成分が沈殿する場合があり、例えばリン酸ナトリウム緩衝液は4℃で比較的不溶性のNa2HPO4・12H2Oを生じる場合がある(Borchert et al.(1986)PDA J.Pharm.Sci.Technol.,40:212−241)。他方、粒状物を評価基準とすると、溶液pH5.2は試験した溶液ではpH6.0よりも有利であった。
【0169】
他方、モノマー含有量については、どちらの溶液pH値も保存中に同一のモノマー含有量となり、NaCl含有製剤(Tween 80不含)の場合、pH6.0では安定性が若干高くなるとさえ思われた。この同様のモノマープロファイルにも拘わらず、中性又は塩基性条件よりのpH値で蛋白質はより多様な潜在的分解メカニズムを受け易いと一般に認められており(Wang(1999)Int.J.Pharm.,185:129−188)、例えば非イオン化蛋白質アミドのカルボニル−アミン反応、(塩基触媒)β脱離及び脱アミド化は各種酸化反応と同様に高pH値により助長される(Akers and DeFelippis,Peptides and proteins as parenteral solutions,in Pharmaceutical formulation development of peptides and proteins,ed.by Frokjaer,S;Hovgaard,L.(2000)145−177)。従って、要約すると、溶液pH5.2はアダリムマブ50mg/mL長期安定性の点で6.0値よりも優れていると判断された。
【0170】
添加剤による安定化:界面活性剤
50mg/mLアダリムマブ製剤に及ぼす界面活性剤の安定化能を調べるために、6.16mg/mL NaClを含有する蛋白質溶液に種々の量のTween 80(0.%、0.03%、0.1%)を加えた。一般に、Tween 80は例えば疎水性表面相互作用により結合することにより蛋白質を安定化させると予想される。蛋白質の表面特性は塩の存在に影響されるので、NaCl不在の影響も調べた(NaCl不含0.1% Tween 80溶液として表5に示す)(Kheirolomoom et al.(1998)Biochem.Eng.J.,2:81−88も参照5.53een)。
【0171】
【表8】
NaClの存在下と不在下でTween 80の量を変化させた結果を表5に示す。この結果から明らかなように、Tween 80はNaClの存在下又は不在下で製剤に安定性を提供することができなかった。0.03% Tween 80/NaClについては、併用の結果、40℃で12週間保存後にモノマー含有量が低下した。一般にTween 80の安定化効果は界面活性剤濃度の増加に相関する(0.001〜1%の範囲で有効)ので、この結果はこの主題に関する大半の論文と相反するものであった(Arakawa et al.(2001)Adv.Drug Deliv.Rev.,46:307−326参照)。
【0172】
NaClの存在下及び不在下で種々のTween 80百分率におけるモノマー濃度に加え、種々の温度で不可視粒子形成も試験した(表6参照)。全保存温度で、Tween 80を加えると、特に0.03%の濃度で不可視粒子数は実質的に増加し、SE−HPLC分析の結果が確証された。興味深いことに、保存温度に関係なく、NaClの不在下では不可視粒子形成は著しく減ることが判明した。
【0173】
【表9】
【0174】
更に凍結/解凍サイクル後の粒状物形成について各種濃度のTween 80を試験した。保存中の溶液に及ぼす僅かな安定化効果とは対照的に、Tween 80は凍結・解凍サイクル中にアダリムマブに対する顕著な安定性を付与することが判明した(表7)。
【0175】
【表10】
【0176】
凍結(−80℃,12時間)と解凍(5℃,12時間)によるストレスを溶液に繰返し付与することにより更にTween 80の影響を検討した。適用した凍結・解凍(凍結/解凍)サイクル数は不可視粒状物の増加に密接に相関した。他方、Tween 80濃度0又は0.03%の溶液に及ぼす5回の凍結/解凍サイクルの影響の結果、粒子汚染(粒子≧1μm)は〜10倍に増加したが、0.1% Tween 80溶液では状況は殆ど変わらなかった。これらの結果はSE−HPLC分析により確証された(表8)。
【0177】
【表11】
他の蛋白質に及ぼす凍結/解凍サイクルの影響について発表されている多数の試験の結果と厳密に一致し、界面活性剤の非添加時に反復凍結/解凍ストレスに暴露すると、50mg/mLアダリムマブの安定性は低下した。逆に、界面活性剤を添加すると、(多角度光散乱法(MALS)を使用して検証した)天然モノマー含有量は変わらなかったため、凍結/解凍に関連する有害なパラメーターに対して蛋白質は保護された。
【0178】
要約すると、0.1% Tween 80をアダリムマブ50mg/mL溶液に添加することが好ましかった。0.1% Tweenは保存液中の蛋白質安定性を僅かに改善したが、凍結・解凍等のプロセス中に安定化効果は十分であった。他方、凍結は蛋白質製剤の製造、保存及び輸送における一般的な単位操作であるため、Tween 80の添加は大きな効果として現れる(Cao et al.(2003)Biotechnol.Bioeng.,82:684−690)。更に、Orthoclone(登録商標)(マウスIgG2a)が1986年という初期にFDAに認可されていることから明らかなように、薬剤中で0.1% Tween 80を使用することは定着している。
【0179】
Tween 80以外に、非イオン性界面活性剤Solutol(登録商標)HS15についてもそのアダリムマブ安定化能を試験した。濃度0.03及び0.1%のSolutol(登録商標)の保護特徴はアビスクミン非経口薬について最近報告されている(Steckel et al.(2003)Int.J.Pharm.,257:181−194)。従って、粒状物形成汚染の形成についてアダリムマブ溶液に及ぼすSolutol(登録商標)の影響を、0.1% Tween 80を含有する蛋白質溶液と比較した(表9)。
【0180】
【表12】
0.03%及び0.1% Solutol(登録商標)を含有する溶液とは対照的に、夫々1% Solutol(登録商標)及び0.1% Tween 80を含有するアダリムマブ溶液は保存中に粒状物の顕著な増加を示した。低濃度Solutol(登録商標)のこのプラスの効果はSE−HPLC分析データには反映されなかった。12週間保存(40℃)後に、Solutol(登録商標)を含有する全溶液は参照(0.1% Tween 80)に比較して〜0.5%のモノマー含有量の低下を示した(図1)。
【0181】
この実験では、高分子量(hmw)蛋白質凝集物の初期段階検出で大きな効果が得られることもMALSにより判明した(図2A及び2B)。大量の分析物で高感度であることから、MALSにより凝集物を検出するには最低濃度で十分であり、例えば1週間保存(40℃)後のhmw凝集物の形成はMALSにより確認することができたが、UV280検出では殆ど検出不能であった。
【0182】
従って、加速貯蔵寿命試験の初期段階で既にhmw凝集物が形成されていることは一般に許容できないため、潜在的安定剤のリストからSolutolを除外した。最少量(<0.1%)の蛋白質でも沈殿に原因となることが知られている(Hoffman,Analytical methods and stability testing of biopharmaceuticals,in Protein formulation and delivery,ed.by McNaIIy,E.J.,3(2000)71−110)。上記結果はより高濃度(>1%)のSolutol(登録商標)HS15がセルピン関連プロテアーゼ阻害剤の溶液を脱安定化し、可視粒状物現象に有効であったという従来の報告を裏付けるものである(例えばWO2006037606参照)。
【0183】
添加剤による安定化:ポリオール
多くの糖質(例えばスクロース、グルコース、ラフィノース、トレハロース)とポリオール(例えばグリセロール、ソルビトール、マンニトール)は蛋白質を安定化する補助溶媒に分類される。これらの物質は主に立体排除メカニズムにより作用すると広く考えられている。例えば、ソルビトール等のポリオールは、例えばMumpsvax(登録商標)、Meruvax(登録商標)II及びAttenuvax(登録商標)等の多数の凍結乾燥ワクチン製剤又はCardene(登録商標)等の静脈内投与可能な溶液で非経口薬を安定化させるために使用されることが多い。
【0184】
界面活性剤等の他の添加剤とは対照的に、糖質とポリオールはその完全な安定化能を発揮するためにはより高濃度(>0.5M)で添加する必要がある。従って、50及び100mg/mLの濃度のソルビトールをアダリムマブ溶液に加え、12週間保存した(表10)。
【0185】
【表13】
【0186】
ソルビトールはソルビトールを添加しない溶液に比較して保存中の粒子形成傾向を低下させた。ソルビトールの添加量は殆ど差を生じなかった。モノマー含有量については、ソルビトールの安定化効果は厳密に濃度依存性であることが判明した。NaClの存在は蛋白質安定性を低下させる(表11)。
【0187】
【表14】
表11によると、100mg/mLソルビトールを添加すると、40℃で12週間保存中にモノマー含有量は〜1.5%増加した。添加剤の量を減らすと、アダリムマブ安定性は低下する。ウマ免疫グロブリンの安定性に関する最近の報告によると、180mg/mLソルビトールは熱ストレスに対する蛋白質安定化に関して90mg/mLの添加よりも優れていることが判明したが、これらの結果はこの報告を裏付けるものである(Rodrigues−Silva et al.,1999 Toxicon 37(1),33−45)。糖質及び糖質由来ポリオールの安定化の濃度依存性は報告されている(Chan et al.(1996)Pharm.Res.,13:756−761;Fatouros et al.(1997b)Pharm.Res.,14:1679−1684)。興味深いことに、表11に示すように、4mg/mL塩を添加すると、ソルビトールの安定化能は著しく低下した(〜0.25%モノマー)。他方、(表11に示すように)0.1% Tween 80を含有するアダリムマブ溶液にNaClを添加しないと、貯蔵寿命実験中にモノマー含有量はごく少量であるが増加した。
【0188】
表12に示すように、ソルビトールの代わりにマンニトールを使用して実験を繰返した。ソルビトールで得られた結果はマンニトールをアダリムマブ溶液に添加することにより確証され、(1)80mg/mLマンニトールを添加した溶液は12週間保存(40℃)後にマンニトール不含溶液よりも蛋白質モノマー含有量が〜1.5%上回り、(2)マンニトールの安定化添加量は濃度依存プロフィルの傾向があり、(3)NaClはマンニトール単独の漸減するモノマー含有量を低下させた。興味深いことに、これらのデータは25℃で実施した同一実験により確証された。
【0189】
【表15】
要約すると、濃度50mg/mLのアダリムマブはソルビトールとマンニトールのどちらでも安定化された。この安定化はNaClにより妨害された。0.1% Tween 80を含有する蛋白質溶液に添加した場合にNaClがアダリムマブ安定性を妨害しないという結果は上記結論と一致した。
【0190】
表13に示すように、各アダリムマブ製剤中の天然モノマーの量はポリオールの添加と添加剤混合物に依存した。これに対応して、凝集物とフラグメントの量も変化した。モノマー損失量における凝集物の割合は添加剤を添加しても添加に関係なく一定であった。換言するならば、アダリムマブ凝集物とフラグメントの比は平衡しており(即ち〜38%凝集物と〜72%フラグメント)、この平衡はポリオールと塩の添加により影響されなかった。ソルビトールとマンニトールが天然状態安定化のみによりアダリムマブ安定性に寄与していたならば、これは凝集物割合の変化に反映されるはずである。そうでなかったことから、ソルビトール/マンニトールによるアダリムマブ安定化には別のメカニズムがあり、その結果、断片化プロセスが妨害されたと考えられる。
【0191】
【表16】
結論として、濃度50mg/mLのアダリムマブはマンニトール又はソルビトールを製剤に加えることにより有効に安定化された。天然状態保護による蛋白質安定性への寄与以外に、マンニトールとソルビトールは別のメカニズムにより蛋白質を安定化し、長期保存中に断片化を抑制した。
【0192】
添加剤による安定化:塩
NaClは蛋白質非経口薬の製剤化で最もよく使用されている塩である。しかし、上記結果によると、50mg/mLのアダリムマブ濃度でNaClはポリオールの存在下でアダリムマブ安定性を妨害し、単独添加剤として蛋白質安定性を増加しなかった。塩の潜在的安定化効果を考慮した場合、ホフマイスター離液順列に従ってその挙動を考察すると、大まかな原則が得られた。従って、ナトリウム塩における対イオンとして塩化物の代わりにアニオン性酢酸塩の使用について検討した。
【0193】
表14に示すように、各溶液(即ち50mg/mLソルビトール/4mg/mL酢酸Na、50mg/mLソルビトール/4mg/mL NaCl、及び50mg/mLソルビトール、塩不含)は異なる蛋白質安定性を示した。NaCl又は酢酸ナトリウムを含有する製剤を比較すると、僅か4週間保存(40℃)後に酢酸ナトリウムを添加したバッチにおけるモノマー含有量はNaClを含有する製剤よりも〜0.25%高くなり、12週間後に差は>0.4%まで拡大したことから、NaClを含有するアダリムマブ溶液は蛋白質安定性に対して不利であった。従って、酢酸ナトリウムは塩化ナトリウムよりもアダリムマブ安定性に大きく寄与した。他方、塩不含製剤はモノマー含有量が同一であったことから、酢酸ナトリウムを添加しても蛋白質安定化は増加しなかった。
【0194】
【表17】
他の2種類の製剤(50mg/mLソルビトールと4mg/mL NaClを含有する製剤と、塩を含有しない製剤)と比較すると、酢酸塩を含有する製剤は1μm超の粒子数が多かった(<6,000個/mLに対して〜180,000個/mL)。
【0195】
緩衝液系も試験し、ソルビトール濃度を変化させてナトリウム緩衝液系とカリウム緩衝液系を比較した。表15に示すように、リン酸カリウム緩衝液に溶解したアダリムマブの安定性はリン酸ナトリウム緩衝液中で測定された安定性と同等であった。これらの結果は25℃で実施した保存試験のデータにより裏付けられた。更に、両者緩衝液系は粒状物汚染も同等であった。従って、液体蛋白質製剤ではリン酸カリウムのほうが好ましいと判断された。
【0196】
【表18】
要約すると、50mg/mLのアダリムマブ溶液を製剤化する際にはNaClの添加を避けるべきである。例えば浸透圧の理由により塩を添加することが好ましい場合には、酢酸ナトリウムのほうが塩化ナトリウムよりも有利である。同様に、カリウム系リン酸緩衝液系はアダリムマブ安定性に関してリン酸ナトリウム緩衝液系と同等であった。
【0197】
要約すると、約50mg/mLのアダリムマブ溶液には溶液pH5.2と0.1% Tween 80の添加が他の方法よりも有利であった。凍結/解凍試験及び(加速)保存試験後の蛋白質安定性及び粒状物汚染を評価基準として使用した。更に、マンニトールやソルビトール等のポリオールはほぼ同一の効力で蛋白質安定性に実質的に寄与した。蛋白質凝集と断片化のどちらも妨げられたので、天然状態蛋白質における優先的蓄積が唯一の安定化経路ではなかった。NaClはポリオールの存在下で蛋白質安定性を妨害した。酢酸ナトリウムの添加は蛋白質安定性に有害な影響を生じなかった。
【0198】
これらのデータから、pH5.2のリン酸カリウム緩衝液と、0.1% Tween 80と、〜50mg/mLマンニトール又はソルビトールを含有しており、アダリムマブ濃度50mg/mLで最終浸透圧値〜300mosM/kgを目標とする製剤が提案された。
【実施例2】
【0199】
高濃度アダリムマブ製剤
本実施例は抗TNFα抗体アダリムマブを含有する多数の高濃度蛋白質製剤の成分について記載する。驚くべきことに、下記製剤は抗体濃度が高い(即ち約100mg/mL)にも拘わらず、多数の有利な特性があった。
【0200】
濁度を含む製剤(F1〜F6と言う)の多数の特徴を市販50mg/mLアダリムマブ製剤(F7)と比較検討した。溶液の濁度は未希釈溶液の分析により測定した。濁度をNTU値(ネフェロメトリー濁度単位)として報告する。
【0201】
ドイツ医薬品規格(German Drug Codex)に記載されているような目視検査により可視粒子汚染を測定した。USPに準拠する光遮蔽法により不可視粒子をモニターした。希釈溶液の動的光散乱分析を使用し、流体力学的直径を求めた(粒子の粒度分布のDLS測定強度自己相関関数と多分散指数PDIのキュムラント分析により計算した平均又はZ平均寸法として報告する)。
【0202】
フラグメントと凝集物の検出を可能にするSECにより製剤の物理化学的安定性を評価した。化学的安定性をモニターするために、SE−HPLC(フラグメント及び加水分解試料の検出)とCEX−HPLC(カチオン交換HPLC)を使用した。CEX−HPLCは保存中に形成され得る各種リジンアイソフォーム及び分解生成物(例えば脱アミド化及び酸化種)を分離する。
【0203】
試験した製剤はF1〜F6(表16)とし、pH5.2〜pH6.0の各種基剤中に100mg/mLアダリムマブを含有しており、各種ポリオールを添加し、塩化ナトリウムの存在下又は不在下で製剤化した。
【0204】
【表19】
【0205】
下記実施例3〜6に記載するように、総合的な安定性と粘度を決定するために、上記100mg/mL製剤(F1〜F7)を更に試験した。
【0206】
以下、特に代表的溶液F2及びF6に関して、高濃度アダリムマブ製剤の製造方法について説明する。出発溶液は精製抗体を液体緩衝剤(例えば前段階の製造工程段階からの緩衝液)に低濃度(本発明の高濃度よりも低濃度)で溶解した溶液とする。この場合には、pH5.2で界面活性剤を添加しないF7と同一の緩衝液系に約70mg/mLの濃度で溶解したアダリムマブ溶液を準備した。次に、出発溶液を濃縮し、例えば10kDのカットオフで抗体を定量的に保持することが可能な膜を使用して、好ましくは接線流濾過システムで限外濾過により透析濾過する。
【0207】
1例として、透析濾過緩衝液として界面活性剤を添加しない対応する基剤を使用して濃厚液を約50mg/Lまで希釈することにより代表的製剤F2及びF6を製造した。接線流濾過システムを使用して連続緩衝液交換を実施した。一般に少なくとも5容量、又は好ましくは8容量の透析濾過緩衝液を使用して一定の保持液量で透析濾過を実施した。最終段階で、透析濾過溶液を高濃度(例えば150mg/mL以上)まで更に濃縮した。次に管を透析濾過緩衝液でフラッシュすることにより濁った最終保持液を限外濾過システムから回収した。各量のポリソルベート80を添加し、透析濾過緩衝液を使用して目標蛋白質濃度に調整後、透明〜やや乳白色の高濃度液体製剤を得た。0.22μmフィルターで濾過後、溶液は約2〜8℃で保存した場合に少なくとも約12カ月間安定であった。
【実施例3】
【0208】
凍結/解凍ストレスに対する高濃度アダリムマブ製剤の安定性
アダリムマブ製剤が100mg/mL蛋白質濃度で安定であることを実証するために、凍結/解凍ストレス(−80℃で凍結を実施し、25℃で解凍を実施)実験を実施した。
【0209】
粒子形成に対して感受性の一連の分析方法を使用して潜在的な物理的不安定を検出した。コロイドないし可視範囲の粒状凝集物の生成の指標として濁度を測定した。濁度(NTU値として報告する)は4回目の凍結/解凍サイクル後もさほど変化しなかった(図3)。高pH溶液の濁度上昇は蛋白質のpI(アダリムマブ8.5)に近いpHにおける電荷反発の低下による蛋白質−蛋白質相互作用の増大に起因すると考えられる(Wang et al.(2007)J Pharm Sci 96(1)2457−2468)。
【0210】
サブミクロン範囲の粒度を測定するための方法として動的光散乱法を使用した。粒度分布測定中に得られた多分散指数値をコロイドないしマイクロメーター寸法範囲の凝集の別の感受性指標として使用した。濁度データと同様に、試験した製剤のうちで物理的不安定の徴候を示すものは皆無であった(図4)。
【0211】
更に、サイズ排除データを評価した。図5は凝集物濃度を示す。反復凍結/解凍ストレスに対して物理化学的不安定の徴候は全く検出されなかった。
【0212】
凍結/解凍処理により実質的な蛋白質変性と凝集が生じ、その結果、可溶性及び不溶性凝集物形成を生じる場合があることは周知である(Parborji et al.(1994)Pharm Res 11(5)764−771)。本願に記載する全製剤を反復凍結・解凍処理した結果、反復凍結/解凍サイクル(−80℃/25℃)に感受性の製剤は皆無であることが判明した。製剤のpHがアダリムマブのpI(即ち8.5)に近い高値であるにも拘わらず、全製剤はそのpHから独立して同様に安定であった(いずれの場合も初期値に比較して優位変化は認められなかった)。
【0213】
これらの結果は各種緩衝液を比較した別の試験からのデータにより裏付けられた。凍結・解凍後に均質溶液(即ちpH、浸透圧、密度の勾配が最少の溶液)が得られるという点と凍結・解凍中のpH変化が最少であるという点で最も有益な緩衝液系はNaClを添加しない緩衝液組成であることが判明した(実施例1参照)。pH6で製剤化したNaCl不含緩衝液系は評価した全pHレベルのpH変化が最少であることが判明した。
【実施例4】
【0214】
等張化剤として各種ポリオールを含有する100mg/mL製剤の安定性
示差走査熱量測定(DSC)を利用し、全100mg/mLアダリムマブ製剤を安定性全般について試験した。Microcal製VP Capillary DSCを使用してDSCデータを得た。全実験は以下の標準化手順:温度範囲:20℃〜90℃、加熱速度:1K/min、蛋白質濃度1mg/mLを使用する1回の加熱試験で実施した。
【0215】
Tm値が高いほど一般に構造安定性は高い(Singh et al.(2003)AAPS PharmSciTech 4(3)article 42)。図6は100mg/mLアダリムマブ製剤のTm値を示す。これらのデータから明らかなように、全製剤は高いTm値に達する。他方、塩化ナトリウム不含製剤(F2,F3,F5,F6)は有意に高いTm値を示し、これらの製剤の堅牢性を示した。1mg/mLで製剤を試験するので、F1のTmデータはF7のTmと同一であり、塩化ナトリウムの不在下又はpH6.0の100mg/mL製剤の安定性はF7製剤よりも改善されることが確認された。
【0216】
磁気撹拌棒を使用する撹拌ストレスモデルを使用し、新規アダリムマブ製剤の物理化学的不安定を検出した。この周知モデルはアダリムマブを長期気液界面暴露と撹拌関連キャビテーションに供することによりストレスを誘導し、予測可能な方法で可溶性及び不溶性蛋白質凝集物の形成をもたらす。
【0217】
一般に、夫々のpI(アダリムマブpI8.5,低純電荷,最小化静電反発力)の範囲のpH値で製剤化した蛋白質は反発力が弱いため、気液界面に関連する凝集に対する感受性が高い。更に、塩化ナトリウム等のイオン性添加剤はそれらのイオン遮断性により蛋白質凝集に関与する。塩化ナトリウムの存在により疎水性吸引力を低下させ、蛋白質−蛋白質相互作用を抑制し、コロイド安定性を増すことができる(Shire et al.(2004)J Pharm Sci,93(6)1390−1402)。
【0218】
撹拌ストレスにより誘導される凝集物形成を検出するために濁度データを評価した。表17は製剤組成と撹拌時間に対するネフェロメトリー値を示す。(5.2の低pHで製剤化した)F1〜F3の初期濁度値は塩化ナトリウム含有溶液(F1)とNaCl不含溶液(F2,F3)で差を示した。他方、6.0の高pHに調整した溶液(F4〜F6)は濁度が高かった。NaClが撹拌等の機械的ストレス後にmAb溶液の透明度を低下させ得ることは当分野で知られている(例えばFesinmeyer et al.(2009)Pharm Res,26(4)903−913)。
【0219】
【表20】
【0220】
48時間まで撹拌すると、試験した全製剤で濁度値が上昇した。低pHのNaCl不含製剤は撹拌による濁度上昇を最も生じにくかった。驚くべきことに、試験した全100mg/mL製剤は低濃度(50mg/mL)アダリムマブ製剤に比較して撹拌後の濁度が有意に低かった(表18)。
【0221】
一般に、乳白外観は単なるレイリー散乱の結果であり、蛋白質濃度と線形相関する。他方、乳白外観は物理的不安定を生じない(Sukumar et al.(2004)Pharm Res 21(7)1087−1093)。50mg/mLアダリムマブ製剤は24時間撹拌後に63〜130NTU、48時間後に109〜243NTUの濁度を示したが、100mg/mLアダリムマブ製剤は27〜63(24時間)から40〜87(48時間)の数値であった。Treuheit et al.((2002)Pharm Res 19(4)511−516)によると、蛋白質濃度が高いと、10mg/mL未満の範囲のOPC−Fc溶液中で気液界面に誘導される凝集は減少する。同様の結果がKiese et al.((2008)J Pharm Sci 97(10)4347−4366)からも報告されている。意外にも、新規アダリムマブ製剤は100mg/mLという著しく高い蛋白質濃度で撹拌ストレス安定性の増加の特徴を示した。
【0222】
従って、新規製剤は50mg/mL製剤に比較して安定性が増加した。
【0223】
【表21】
【0224】
更に、サイズ排除クロマトグラフィーデータによると、全100mg/mL製剤は48時間撹拌後の凝集物濃度が<1%であり、新規製剤の安定性が裏付けられた(図7)。やはり、低pHと塩化ナトリウムの不在が有益であった。一般には高pHで純電荷が低いと不安定が増すと考えられているが、このデータは新規製剤がアダリムマブのpIに近いpH値であるにも拘わらず安定であり、NaClの不在が有益であるという驚くべき知見を裏付けるものである。
【実施例5】
【0225】
塩化ナトリウムの存在下及び不在下で2種類の異なるポリオールを添加したpH5.2及び6.0の100mg/mLアダリムマブ製剤の長期安定性
新規100mg/mLアダリムマブ製剤を長期保存し、50mg/mL標準製剤と比較して安定性が優れていることを確認した。5℃(市販品の推奨保存温度)で12カ月間の安定性データを評価した。データは実際に、新規製剤が安定性の低下を示さなかったことを示唆している(表19)。
【0226】
SEC及びIEXに関して、モノマー含有量の有意低下又は測定可能な分解は生じなかった。
【0227】
更に、新規アダリムマブ製剤は蛋白質濃度が高いにも拘わらず、50mg/mL市販アダリムマブ製剤の12Mデータに比較して不可視範囲の粒子汚染に関する有意強化が得られた。(凝集、沈殿及び一般物理的不安定現象を示す)不可視粒状物汚染を試験した処、新規アダリムマブ製剤は実際に不可視粒子を含まずに維持されることが判明した。最大値28(≧10)及び最大値3(≧25)の初期粒子数は50mg/mL製剤F7(夫々703と38)よりも有意に少なかった。
【0228】
更に、粒子レベルは12カ月間安定性試験を通して有意に変化せず、F7よりも有意に低レベルに維持された。
【0229】
薬剤製品バッチは試験した全保存条件下でその物理化学的安定性に関してほぼ等価であった。例えば蛋白質濃度が高いほど物理的安定性は低下する傾向があることは広く認められているため、これは意外である(Wang W.(1999)Int J Pharm 185:129−188)。
【0230】
【表22】
【0231】
新規100mg/mL製剤の保存安定性増加の結果を検証するために、2種類の代表的な製剤であるF2及びF6を加速安定性試験(5℃、25℃、40℃で3カ月)に供し、市販50mg/mL製剤(対照試験からの代表的バッチ)と比較した。これらの実験の結果を図8〜13にまとめる。
【0232】
これらのバッチからの濁度データは特に5.2の低pHで100mg/mLのNaCl不含製剤の優れた挙動を実証する。溶液中の蛋白質の濃度が増加すると、乳白が増し、レイリー散乱により濁度読取り値が増すことは一般に知られている(Sukumar et al.(2004)Pharm Res 21(7)1087−1093)。驚くべきことに、塩化ナトリウムを添加しない新規製剤は50mg/mL製剤と同一pHで同等の濁度レベルであることが判明した(図8)。
【0233】
図9〜11は新規製剤の粒状物形成(可視粒子と不可視粒子)の詳細なデータを示す。安定性の増加という驚くべき知見が確証された。実際に、高温で3カ月間保存後も不可視粒子及び可視粒子両方のスコアを下げることが可能であった。
【0234】
図12〜13に示すデータはSEC分析とIEXを使用して試験した化学的安定性の両方で安定性の問題を示さないため、100mg/mL製剤の安定性を更に裏付けた。
【実施例6】
【0235】
50mg/mLアダリムマブ製剤と比較した100mg/mLアダリムマブ製剤の製造適性の強化
本実施例は現在市販されている50mg/mL製剤に比較して新規100mg/mLアダリムマブ製剤(代表的製剤F2及びF6)のプロセス安定性の改善に関するデータをまとめる。
【0236】
ポンピング、濾過、混合、充填−仕上げ工程、輸送又は振盪により生じる機械的ストレスは空気−水界面、材料表面及び剪断力への蛋白質の暴露により、変性とそれに続いて凝集を生じる恐れがある(Mahler at al.(2005)Eur J Pharm Biopharm 59:407−417;Shire et al.(2004)J Pharm Sci,93(6)1390−1402)。
【0237】
先ず蛋白質溶液の処理適性の特徴を表す基本パラメーターとして粘度値を測定した。表20はF1〜F7製剤で得られた粘度データを示す。蛋白質濃度が増加すると、50mg/mL製剤(F7)に比較して粘度が増加した。
【0238】
静電遮断剤であるNaClを除去すると、特にアダリムマブのpIに近いpH値で疎水性蛋白質相互作用が増加し、従って粘度が増加すると予想される。この作用はNaCl濃度<200mMで最も顕著であると報告されている(Shire et al.(2004)J Pharm Sci,93(6)1390−1402)。
【0239】
しかし、意外にも、製剤からNaClを除去しても(F1は〜105mM NaClを含有する)依然として約3.1〜3.3mPa・sという比較的低い粘度値であった(F2、F3、F5及びF6)。これは6.0という高pH値の溶液(F5及びF6)で特に意外であった。
【0240】
要約すると、全製剤は液体充填−仕上げ製造操作に最適な範囲の粘度をもつことを特徴とする。
【0241】
【表23】
無菌製造工程中の滅菌濾過により誘導されるストレスを模倣した実験室モデルで、100mg/mLアダリムマブを含有する2種類の代表的な新規製剤から分析データを得た処、全製剤は濾過に関連する剪断ストレスに対して安定であることが分かった。低レベルの高分子量サブ集団の感受性指標である多分散指数は有意に増加しなかったため、DLSデータは高分子量凝集物の形成の徴候を示さなかった。高分子量種は散乱強度が高い(d6に比例する)ため、ZAve及びZAve粒度分布の指標としての多分散指数に有意に影響しないので、特にDLS測定を使用して粒度分布における少量の高分子量種、例えば凝集物を検出する。更に、濾過により凝集が誘導されないことがSECデータにより確認された。
【0242】
驚くべきことに、100mg/mL製剤でも不安定を示さなかった。最悪の場合のシナリオとして複数回滅菌濾過後でも、蛋白質含有量が高いにも拘わらず、処理適性は高レベルに維持された。
【0243】
【表24】
プロセス関連ストレスに対する新規アダリムマブ製剤の高い安定性を更に実証するために、磁気撹拌棒の撹拌速度を変えてその挙動を比較する撹拌ストレスモデルで製剤を試験した(撹拌ストレスは配合プロセス段階で製造条件下に生じる)。
【0244】
撹拌ストレス耐性を比較した処、100mg/mL蛋白質濃度で濁度の上昇は認められなかった(図14)。塩化ナトリウムを添加せずにポリオール含有量を高くした代表的な100mg/mL製剤はいずれも試験した全撹拌速度でpH5.2の市販製剤と同様に挙動した。撹拌速度を上げると、全製剤は24時間撹拌後に濁度値の僅かな上昇を示したが、100mg/mLで剪断ストレスによる不安定に対する感受性の顕著な増加は検出されなかった。
【0245】
DLS測定により得られる流体力学的直径の変化を比較した処、同様のデータが得られた。蛋白質濃度の高い製剤のほうが撹拌ストレスに対する感受性が強いと考えられるが、どちらの100mg/mL製剤も50mg/mL製剤と同様に挙動した。驚くべきことに、pHが最高の製剤F2は濁度及び流体力学的直径のどちらの分析でも相対増加が最低であることが判明した(図15)。
【0246】
蛋白質濃度が高くてもプロセス安定性は同様であるというこの驚くべき結果はポンピングプロセスにより誘導されるストレスを模倣した機械的ストレスモデルにより更に確証された。製造工程のこの最終段階は蠕動ポンピングによる剪断ストレスを伴い、溶液不安定の危険を増す。この場合も、濁度(図16)とDLS(図17,表22)を使用して得られたデータは、新規100mg/mL製剤が粒子成長反応を生じず、50mg/mL製剤と同様に安定に維持されることを裏付けた。ポンプストレスにより誘導した凝集物形成に対する感受性は検出できなかった。この結果は更にSECデータにより裏付けられ、試験した製剤はポンプサイクルに関して差がないことが分かった(図18)。
【0247】
【表25】
各種充填装置(回転ピストンと蠕動ポンプ)を使用し、100mg/mL製剤の安定性の差を評価した。
【0248】
これらの試験の結果、特にpHの高い塩化ナトリウム含有製剤(F1及びF4)では、ピストンポンプで発生する剪断ストレスが高いほど、可視粒子数が増加した。同様の結果がBausch,Ursula J.から最近報告されている(Impact of filling processes on protein solutions.2008,PhD Thesis,University of Basel,Faculty of Science;http://edoc.unibas.ch/845/1/DissB_8427.pdf)が、これはリツキシマブ溶液の10mg/mLの蛋白質濃度に過ぎない。驚くべきことに、100mg/mLアダリムマブを含有する塩化ナトリウム製剤はピストンポンプを使用する高剪断条件下で処理適性の改善を示した。
【0249】
図19〜22はプロセスストレス増加条件に対するNaCl含有アダリムマブ溶液の感受性の増加を裏付ける粒子数と濁度データを示し、DAC目視スコア法による≧10μm及び≧25μmの粒度範囲の測定は非経口薬に不可欠の品質属性である。従って、NaCl不含製剤における不可視粒子の減少により有意な製剤改善が得られる。
【0250】
図19に示すように、蠕動充填の結果、充填直後(T0)及び保存後に可視粒子形成は生じなかった。他方、ピストン充填の結果、pH6.0で製剤化した溶液ではT0でも有意粒子数が認められた(図20)。塩化ナトリウムを含有するF4で最高値が測定されたが、F5〜F6はスコアが有意に低く、プロセスストレスに対する塩化ナトリウム不含製剤の安定性の改善が確証された。
【0251】
濁度測定により裏付けとなる結果が得られた(図21〜22)。ピストンポンプを使用して充填した溶液の初期値は蠕動充填法を使用して充填した溶液よりも高かった。塩化ナトリウム不含製剤は塩化ナトリウムを含有する製剤よりも濁度が低かった。更に、ピストン充填による剪断ストレスの結果、濁度に関してF4(塩化ナトリウム添加)をF5及びF6(塩化ナトリウム非添加)から識別することができた。
【実施例7】
【0252】
塩化ナトリウム不含製剤中の各種ポリオール濃度の比較
100mg/mLアダリムマブを含有する以下の塩化ナトリウム不含製剤を5℃における短期安定性に及ぼすポリオール濃度の影響について試験した。
【0253】
凝集及び粒子形成傾向に関して不良な条件となるように、製剤をpH6.0に調整した。
【0254】
【表26】
塩化ナトリウム不含溶液の張度要件に見合うように、マンニトール又はソルビトールを42mg/mLの濃度で使用した。データによると、先に使用した12mg/mLの濃度に比較して、どちらのポリオールも溶液の浸透圧に寄与するのみならず、更に蛋白質安定性にも有意影響を与えることが判明した。
【0255】
安定性データによると、ポリオールの種類に関係なく、ポリオール濃度が高いほうが透明度は改善されると思われた。一般に最適とはみなされない条件下(例えばアダリムマブのpIに近いpH6.0)で、ポリオール濃度の高い製剤は5℃で4週間の短期保存後でも透明度の改善を示した。これは数種類の分析法で確認された。
【0256】
図23から明らかなように、試験した製剤の透明度はポリオール濃度を増加することにより有意に低下し、試験期間にわたって低レベルに保つことができた。更に、5℃で4週間後に凝集がやや低下した結果、高いポリオール濃度で高いモノマー含有量が認められた(図24及び25)。ポリオール濃度が高いと、≧10μmの範囲の不可視粒子は(例えばT0で)減少した。
【実施例8】
【0257】
ヒト抗TNFα抗体の安定した高蛋白質濃度製剤
加速安定性試験条件下と推奨保存温度条件における長期保存下の両方でアダリムマブの物理的及び化学的安定性を維持する適性について各種アダリムマブ製剤を試験した(下表1参照)。製剤のpH(pH5.2とpH6)、添加剤条件(例えばマンニトール又はソルビトールの濃度)、塩/イオン強度条件(例えばNaClの濃度)、及び蛋白質濃度(50mg/mLと100mg/mL)を変化させた。
【0258】
【表27】
【0259】
表2はストレス温度とサンプル抽出点の総覧である。製剤F2及びF6はアダリムマブの物理的及び化学的安定性の両方を夫々少なくとも18カ月間及び12カ月間維持する製剤であると認められた。製剤添加剤NaClをマンニトール(製剤F2)とソルビトール(製剤F6)に交換すると、蛋白質濃度の100%増加(製剤F7の50mg/mLから製剤F2及びF6の100mg/mLまで)にも拘わらず、高い安定化能が得られる。驚くべきことに、両製剤の物理的安定性は夫々少なくとも12カ月間及び18カ月維持された。12カ月間保存後も、両製剤のモノマー含有量は99%を上回り(SECデータ)、凝集物濃度は1%未満であった。
【0260】
同様に、リジン変異体合計(L0+L1+L2)を表す安定性は80%を上回ったため、非常に多くの場合に蛋白質薬剤における貯蔵寿命制限因子である化学的安定性も安定性モニター期間にわたって維持された。
【0261】
蛋白質製剤の物理的及び/又は化学的安定性をモニターするのに適切であるとして当分野で認められている他の試験(例えば不可視粒子試験、濁度測定、目視検査、透明度又は色モニター)でも製剤F2及びF6の安定化能が確認された。
【0262】
重要な点として、抗TNF中和試験を示す効力によると、どちらの製剤も完全なサンプル抽出スケジュールにわたってアダリムマブの効力を維持し、データは75〜125%の高品質レベル範囲内にあることが判明した。
【0263】
【表28】
【0264】
【表29】
【0265】
【表30】
【実施例9】
【0266】
高濃度アダリムマブの疼痛試験
皮下注射によりモノクローナル抗体を投与された患者は注射部位に疼痛又は不快感を感じる場合がある(例えばFransson,J.;Espander−Jansson,A.(1996)Journal of Pharmacy and Pharmacology 48(10),1012−1015;Parham,S.M.;Pasieka,J.L.(1996)Can.J.Surg.39,31−35;Moriel E Z;Rajfer J(1993)The Journal of urology 149(5 Pt 2),1299−300参照)。患者の体験を模倣した動物モデルを使用し、疼痛及び耐容性効果を評価すると共に、人体使用前の可能な製剤修飾を評価した。蛋白質製剤の特性を識別する適性について利用可能な動物モデルを評価した。注射時の発声、足フリンチ(注射後0〜10分)、機械的アロディニアの試験、及び熱痛覚過敏(注射後30分)について測定した。患部足をなめたり、振ったりする侵害受容挙動や、注射部位の発赤又は腫脹についても動物を観察した。
【0267】
注射部位疼痛を評価するためにフリンチモデルを選択し、耐容性及び疼痛感覚に及ぼす製剤組成の影響を評価するために使用した。
【0268】
各種アダリムマブ100mg/mL製剤の耐容性を製剤F7(50mg/mLアダリムマブ製剤)と比較した。得られたデータは、50mg/mL製剤(F7)に比較して皮下注射後の注射部位で100mg/mL製剤の耐容性が改善されるという驚くべき結果を裏付けた。
【0269】
注射部位の疼痛等の皮下注射に伴う副作用を抑えるように新規100mg/mL製剤を最適化した。注射部位の疼痛は注射針穿刺に関連する疼痛と、皮下デポーへの溶液注入に関連する感覚の両者を含む。文献から入手可能なデータによると、所定の注射針設計が注射部位の不快感を軽減するために有利であると思われたが、製剤寄与に関する明白なデータは入手できなかった(例えばChan,G.C.F.,et al.(2003)American Journal of Hematology 76(4):398−404参照)。
【0270】
ラット疼痛モデルを使用した本発明者らのデータによると、新規100mg/mL製剤は現在市販されているHumira(登録商標)製剤に比較して同等の治療用量の皮下注射後の注射部位疼痛を軽減するのに有効であると思われた。これは新規100mg/mL製剤の容量注射低下により達成され、患者治療の最適化と患者コンプライアンスの向上という非常に有益な効果を示した。
【0271】
同時に、本発明者らは100mg/mL製剤を製剤化するために許容可能な範囲内の製剤pHが注射部位疼痛に影響を与えないことも確認した。興味深いことに、生理的pH範囲からかけ離れた低いpH値も同様の耐容性で投与可能であった。
【0272】
耐容性試験に適用した方法:
足フリンチ及び侵害防御挙動アッセイ
成体雄性Sprague Dawleyラットを右後足への試験溶液の足裏(s.c.)注射前20〜30分間試験条件に馴化させた。足フリンチ数を記録し、注射後、最初の10分間、侵害防御挙動(足をかばう又はなめる動作)に費やした時間を測定した。特に指定しない限り、全試験溶液は総容量150μLを注射した。実験はコード化し、盲検・無作為法で行った。食塩水とカプサイシン(2.5μg)を夫々陰性及び陽性対照として使用した。
【0273】
容量効果
足フリンチ応答に及ぼす注射容量の影響をプラセボ及び試験製剤F7の両者で試験した。物理的容量を減らすことにより応答を改善できるか否かを調べるために、注射容量の変動(10μl、50μl及び150μlを足裏注射)がフリンチ結果に及ぼす影響を試験した。
【0274】
試験データから容量効果を以下のようにまとめることができる:150μlではプラセボ(32±12)とF7の両者で食塩水(4±2)に比較してフリンチが有意に増加したが、容量を減らすと、食塩水と区別がつかなかった。150μLという高い注射容量では高いフリンチ応答が常に生じたが、低容量(10μL及び50μL)では有意に低い応答となった。
【0275】
この結果から、注射容量を減らすと刺激が低下すると思われ、F2及びF6等の高濃度製剤はF7等の低濃度製剤に比較して耐容性及び疼痛感覚に関して有利であると考えられる。
【0276】
【表31】
【実施例10】
【0277】
耐容性/疼痛に及ぼすアダリムマブ含有溶液のpH効果
アダリムマブを含有する活性溶液で別の実験を実施した。試験した製剤はF2(pH5.2)、生理的条件により近いpH値の対応する製剤であるF5及びF7とした。
【0278】
データによると、pHは足フリンチ応答と侵害防御挙動に費やされた時間を使用して測定した動物応答に影響を与えないと思われた。陽性及び陰性対照データは予想範囲内であった。製剤pHが低い(即ち酸性)ほど、特に皮下注射による非経口投与後に不耐容及び疼痛感覚の危険が増すことは文献から周知である。従って、F2及びF5アダリムマブ製剤では、製剤pHが耐容性及び/又は疼痛感覚に影響を与えないことは意外であった。これは製剤pH、物理的安定性及び凝集物濃度(潜在的に免疫原性の危険に相関する)等の他のパラメーターに製剤化意志決定に関する高い優先性を与えることができるので、非常に有益である。
【0279】
【表32】
【実施例11】
【0280】
アダリムマブ不含溶液の製剤pH効果の影響
製剤組成の影響(例えばリン酸塩等の緩衝液、マンニトール等の添加剤、又はポリソルベート80等の界面活性剤の影響)を試験するために、別の実験を実施し、蛋白質不含製剤で同様のデータを得た。プラセボ溶液のpHを約5〜7の範囲で変動させた処、驚くべきことに、pHの異なる製剤で認められたフリンチ応答は同様であったため、疼痛改善効果はないとも思われた。上記に説明したように、これにより製剤処方者は製剤pH、物理的安定性及び凝集物濃度(潜在的に免疫原性の危険に相関する)等の他のパラメーターに製剤化意志決定に関する高い優先性を与えることができるので、バイオ医薬品製剤開発において非常に有益である。
【0281】
【表33】
要約すると、上記データは耐容性を低下0---)--------------)---------+--及び/又は疼痛感覚を増加せずに一定範囲のpHでこれらの高蛋白質濃度の粘性溶液を低容量で投与できるという100mg/mLアダリムマブ製剤の利点を明白に立証するものである。
【0282】
文献援用
本願の随所に引用する全引用文献(例えば、文献資料、特許、特許出願及びウェブサイトを含む)の内容は任意目的でその全文を特に本願に援用する。本発明の実施は、特に指定しない限り、当分野で周知の蛋白質製剤の従来技術を利用する。
【0283】
等価物
その趣旨又は本質的特徴から離れずに本発明を他の特定形態で具体化することもできる。従って、上記態様は本願に記載する発明を制限するものではなく、あらゆる点で例示とみなすべきである。従って、本発明の範囲は上記記載ではなく、以下の特許請求の範囲により指定され、特許請求の範囲の等価物の意味及び範囲に該当する全変更も本願に含むものとする。
【特許請求の範囲】
【請求項1】
約20mgを上回るポリオールと、配列番号3に記載のアミノ酸配列あるいは1、4、5、7もしくは8位の1カ所のアラニン置換又は1、3、4、6、7、8及び/もしくは9位の1〜5カ所の保存アミノ酸置換により配列番号3から変異したアミノ酸配列を含むCDR3ドメインを含む軽鎖と、配列番号4に記載のアミノ酸配列あるいは2、3、4、5、6、8、9、10もしくは11位の1カ所のアラニン置換又は2、3、4、5、6、8、9、10、11及び/もしくは12位の1〜5カ所の保存アミノ酸置換により配列番号4から変異したアミノ酸配列を含むCDR3ドメインを含む重鎖を含む少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤であって、前記製剤が添加剤NaClを含有しない前記製剤。
【請求項2】
製剤が約30mgを上回るポリオールを含有する請求項1に記載の製剤。
【請求項3】
製剤が約40mgを上回るポリオールを含有する請求項1に記載の製剤。
【請求項4】
約40〜45mgのポリオールを含有する請求項1に記載の製剤。
【請求項5】
ポリオールが糖アルコールである請求項1から4のいずれか一項に記載の製剤。
【請求項6】
糖アルコールがマンニトール又はソルビトールである請求項5に記載の製剤。
【請求項7】
ヒト抗体がヒトIgG1κ抗体である請求項1から6のいずれか一項に記載の製剤。
【請求項8】
ヒト抗体の軽鎖が更に配列番号5に記載のアミノ酸配列を含むCDR2ドメインと、配列番号7に記載のアミノ酸配列を含むCDR1ドメインを含み、及び/又はヒト抗体の重鎖が配列番号6に記載のアミノ酸配列を含むCDR2ドメインと、配列番号8に記載のアミノ酸配列を含むCDR1ドメインを含む請求項1から6のいずれか一項に記載の製剤。
【請求項9】
ヒト抗体の軽鎖が配列番号1に記載のアミノ酸配列を含み、ヒト抗体の重鎖が配列番号2に記載のアミノ酸配列を含む請求項1から6のいずれか一項に記載の製剤。
【請求項10】
抗体がアダリムマブである請求項1から6のいずれか一項に記載の製剤。
【請求項11】
pHが約5.0〜6.4であり、配列番号3に記載のアミノ酸配列を含むCDR3ドメインを含む軽鎖と、配列番号4に記載のアミノ酸配列を含むCDR3ドメインを含む重鎖を含む少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤であって、前記製剤がNaClを含有せず、標準24時間撹拌ストレスアッセイ後の濁度が約60NTU未満である前記製剤。
【請求項12】
pHが約5.0〜6.4であり、配列番号3に記載のアミノ酸配列を含むCDR3ドメインを含む軽鎖と、配列番号4に記載のアミノ酸配列を含むCDR3ドメインを含む重鎖を含む少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤であって、前記製剤がNaClを含有せず、標準48時間撹拌ストレスアッセイ後の濁度が約100NTU未満である前記製剤。
【請求項13】
pHが約5.0〜6.4であり、配列番号3に記載のアミノ酸配列を含むCDR3ドメインを含む軽鎖と、配列番号4に記載のアミノ酸配列を含むCDR3ドメインを含む重鎖を含む少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤であって、前記製剤がNaClを含有せず、5℃、25℃又は40℃で3カ月間保存後の濁度が約40NTU未満である前記製剤。
【請求項14】
更に約20mgを上回るポリオールを含有する請求項11から13のいずれか一項に記載の製剤。
【請求項15】
更に約30mgを上回るポリオールを含有する請求項11から13のいずれか一項に記載の製剤。
【請求項16】
更に約40mgを上回るポリオールを含有する請求項11から13のいずれか一項に記載の製剤。
【請求項17】
更に約40〜45mgのポリオールを含有する請求項11から13のいずれか一項に記載の製剤。
【請求項18】
ポリオールが糖アルコールである請求項13から17のいずれか一項に記載の製剤。
【請求項19】
糖アルコールがマンニトール又はソルビトールである請求項18に記載の製剤。
【請求項20】
pHが約5.0〜5.4又は約5.8〜6.4である請求項11から13のいずれか一項に記載の製剤。
【請求項21】
凝集蛋白質が約1%未満である請求項11から20のいずれか一項に記載の製剤。
【請求項22】
ヒト抗体がヒトIgG1κ抗体である請求項11から21のいずれか一項に記載の製剤。
【請求項23】
ヒト抗体の軽鎖が更に配列番号5に記載のアミノ酸配列を含むCDR2ドメインと、配列番号7に記載のアミノ酸配列を含むCDR1ドメインを含み、及び/又はヒト抗体の重鎖が配列番号6に記載のアミノ酸配列を含むCDR2ドメインと、配列番号8に記載のアミノ酸配列を含むCDR1ドメインを含む請求項11から21のいずれか一項に記載の製剤。
【請求項24】
ヒト抗体の軽鎖が配列番号1に記載のアミノ酸配列を含み、ヒト抗体の重鎖が配列番号2に記載のアミノ酸配列を含む請求項11から21のいずれか一項に記載の製剤。
【請求項25】
抗体がアダリムマブである請求項11から21のいずれか一項に記載の製剤。
【請求項26】
配列番号3に記載のアミノ酸配列を含むCDR3ドメインを含む軽鎖と、配列番号4に記載のアミノ酸配列を含むCDR3ドメインを含む重鎖を含む少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分と;
約20mg/mLを上回る糖アルコールと;
約0.1〜2.0mg/mLの界面活性剤と;
約1.15〜1.45mg/mLのクエン酸・H2Oと;
約0.2〜0.4mg/mLのクエン酸ナトリウム脱水和物と;
約1.35〜1.75mg/mLのNa2HPO4・2H2Oと;
約0.75〜0.95mg/mLのNaH2PO4・2H2O
を含有する液体医薬製剤であって、
前記製剤がpH約4.7〜6.5であり、NaClを含有しない前記製剤。
【請求項27】
糖アルコールがマンニトール又はソルビトールである請求項26に記載の製剤。
【請求項28】
約40〜45mg/mLのマンニトール又はソルビトールを含有する請求項27に記載の製剤。
【請求項29】
界面活性剤がポリソルベート80である請求項26に記載の製剤。
【請求項30】
約1mg/mLのポリソルベート80を含有する請求項29に記載の製剤。
【請求項31】
約1.30〜1.31mg/mLのクエン酸・H2Oを含有する請求項26に記載の製剤。
【請求項32】
約0.30〜0.31mg/mLのクエン酸ナトリウム脱水和物を含有する請求項26に記載の製剤。
【請求項33】
約1.50〜1.56mg/mLのNa2HPO4・2H2Oを含有する請求項26に記載の製剤。
【請求項34】
約0.83〜0.89mg/mLのNaH2PO4・2H2Oを含有する請求項26に記載の製剤。
【請求項35】
pHが約5.2である請求項26に記載の製剤。
【請求項36】
pHが約6.0である請求項26に記載の製剤。
【請求項37】
ヒト抗体がヒトIgGlκ抗体である請求項26から36のいずれか一項に記載の製剤。
【請求項38】
ヒト抗体の軽鎖が更に配列番号5に記載のアミノ酸配列を含むCDR2ドメインと、配列番号7に記載のアミノ酸配列を含むCDR1ドメインを含み、及び/又はヒト抗体の重鎖が配列番号6に記載のアミノ酸配列を含むCDR2ドメインと、配列番号8に記載のアミノ酸配列を含むCDR1ドメインを含む請求項26から36のいずれか一項に記載の製剤。
【請求項39】
ヒト抗体の軽鎖が配列番号1に記載のアミノ酸配列を含み、ヒト抗体の重鎖が配列番号2に記載のアミノ酸配列を含む請求項26から36のいずれか一項に記載の製剤。
【請求項40】
抗体がアダリムマブである請求項26から36のいずれか一項に記載の製剤。
【請求項41】
皮下投与に適している請求項1から40のいずれか一項に記載の製剤。
【請求項42】
対象における有害なTNFα活性に関連する疾患の治療方法であって、請求項1から41のいずれか一項に記載の製剤を前記対象に投与する段階を含む前記方法。
【請求項1】
約20mgを上回るポリオールと、配列番号3に記載のアミノ酸配列あるいは1、4、5、7もしくは8位の1カ所のアラニン置換又は1、3、4、6、7、8及び/もしくは9位の1〜5カ所の保存アミノ酸置換により配列番号3から変異したアミノ酸配列を含むCDR3ドメインを含む軽鎖と、配列番号4に記載のアミノ酸配列あるいは2、3、4、5、6、8、9、10もしくは11位の1カ所のアラニン置換又は2、3、4、5、6、8、9、10、11及び/もしくは12位の1〜5カ所の保存アミノ酸置換により配列番号4から変異したアミノ酸配列を含むCDR3ドメインを含む重鎖を含む少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤であって、前記製剤が添加剤NaClを含有しない前記製剤。
【請求項2】
製剤が約30mgを上回るポリオールを含有する請求項1に記載の製剤。
【請求項3】
製剤が約40mgを上回るポリオールを含有する請求項1に記載の製剤。
【請求項4】
約40〜45mgのポリオールを含有する請求項1に記載の製剤。
【請求項5】
ポリオールが糖アルコールである請求項1から4のいずれか一項に記載の製剤。
【請求項6】
糖アルコールがマンニトール又はソルビトールである請求項5に記載の製剤。
【請求項7】
ヒト抗体がヒトIgG1κ抗体である請求項1から6のいずれか一項に記載の製剤。
【請求項8】
ヒト抗体の軽鎖が更に配列番号5に記載のアミノ酸配列を含むCDR2ドメインと、配列番号7に記載のアミノ酸配列を含むCDR1ドメインを含み、及び/又はヒト抗体の重鎖が配列番号6に記載のアミノ酸配列を含むCDR2ドメインと、配列番号8に記載のアミノ酸配列を含むCDR1ドメインを含む請求項1から6のいずれか一項に記載の製剤。
【請求項9】
ヒト抗体の軽鎖が配列番号1に記載のアミノ酸配列を含み、ヒト抗体の重鎖が配列番号2に記載のアミノ酸配列を含む請求項1から6のいずれか一項に記載の製剤。
【請求項10】
抗体がアダリムマブである請求項1から6のいずれか一項に記載の製剤。
【請求項11】
pHが約5.0〜6.4であり、配列番号3に記載のアミノ酸配列を含むCDR3ドメインを含む軽鎖と、配列番号4に記載のアミノ酸配列を含むCDR3ドメインを含む重鎖を含む少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤であって、前記製剤がNaClを含有せず、標準24時間撹拌ストレスアッセイ後の濁度が約60NTU未満である前記製剤。
【請求項12】
pHが約5.0〜6.4であり、配列番号3に記載のアミノ酸配列を含むCDR3ドメインを含む軽鎖と、配列番号4に記載のアミノ酸配列を含むCDR3ドメインを含む重鎖を含む少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤であって、前記製剤がNaClを含有せず、標準48時間撹拌ストレスアッセイ後の濁度が約100NTU未満である前記製剤。
【請求項13】
pHが約5.0〜6.4であり、配列番号3に記載のアミノ酸配列を含むCDR3ドメインを含む軽鎖と、配列番号4に記載のアミノ酸配列を含むCDR3ドメインを含む重鎖を含む少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分を含有する液体医薬製剤であって、前記製剤がNaClを含有せず、5℃、25℃又は40℃で3カ月間保存後の濁度が約40NTU未満である前記製剤。
【請求項14】
更に約20mgを上回るポリオールを含有する請求項11から13のいずれか一項に記載の製剤。
【請求項15】
更に約30mgを上回るポリオールを含有する請求項11から13のいずれか一項に記載の製剤。
【請求項16】
更に約40mgを上回るポリオールを含有する請求項11から13のいずれか一項に記載の製剤。
【請求項17】
更に約40〜45mgのポリオールを含有する請求項11から13のいずれか一項に記載の製剤。
【請求項18】
ポリオールが糖アルコールである請求項13から17のいずれか一項に記載の製剤。
【請求項19】
糖アルコールがマンニトール又はソルビトールである請求項18に記載の製剤。
【請求項20】
pHが約5.0〜5.4又は約5.8〜6.4である請求項11から13のいずれか一項に記載の製剤。
【請求項21】
凝集蛋白質が約1%未満である請求項11から20のいずれか一項に記載の製剤。
【請求項22】
ヒト抗体がヒトIgG1κ抗体である請求項11から21のいずれか一項に記載の製剤。
【請求項23】
ヒト抗体の軽鎖が更に配列番号5に記載のアミノ酸配列を含むCDR2ドメインと、配列番号7に記載のアミノ酸配列を含むCDR1ドメインを含み、及び/又はヒト抗体の重鎖が配列番号6に記載のアミノ酸配列を含むCDR2ドメインと、配列番号8に記載のアミノ酸配列を含むCDR1ドメインを含む請求項11から21のいずれか一項に記載の製剤。
【請求項24】
ヒト抗体の軽鎖が配列番号1に記載のアミノ酸配列を含み、ヒト抗体の重鎖が配列番号2に記載のアミノ酸配列を含む請求項11から21のいずれか一項に記載の製剤。
【請求項25】
抗体がアダリムマブである請求項11から21のいずれか一項に記載の製剤。
【請求項26】
配列番号3に記載のアミノ酸配列を含むCDR3ドメインを含む軽鎖と、配列番号4に記載のアミノ酸配列を含むCDR3ドメインを含む重鎖を含む少なくとも約100mg/mLのヒト抗TNFα抗体又はその抗原結合部分と;
約20mg/mLを上回る糖アルコールと;
約0.1〜2.0mg/mLの界面活性剤と;
約1.15〜1.45mg/mLのクエン酸・H2Oと;
約0.2〜0.4mg/mLのクエン酸ナトリウム脱水和物と;
約1.35〜1.75mg/mLのNa2HPO4・2H2Oと;
約0.75〜0.95mg/mLのNaH2PO4・2H2O
を含有する液体医薬製剤であって、
前記製剤がpH約4.7〜6.5であり、NaClを含有しない前記製剤。
【請求項27】
糖アルコールがマンニトール又はソルビトールである請求項26に記載の製剤。
【請求項28】
約40〜45mg/mLのマンニトール又はソルビトールを含有する請求項27に記載の製剤。
【請求項29】
界面活性剤がポリソルベート80である請求項26に記載の製剤。
【請求項30】
約1mg/mLのポリソルベート80を含有する請求項29に記載の製剤。
【請求項31】
約1.30〜1.31mg/mLのクエン酸・H2Oを含有する請求項26に記載の製剤。
【請求項32】
約0.30〜0.31mg/mLのクエン酸ナトリウム脱水和物を含有する請求項26に記載の製剤。
【請求項33】
約1.50〜1.56mg/mLのNa2HPO4・2H2Oを含有する請求項26に記載の製剤。
【請求項34】
約0.83〜0.89mg/mLのNaH2PO4・2H2Oを含有する請求項26に記載の製剤。
【請求項35】
pHが約5.2である請求項26に記載の製剤。
【請求項36】
pHが約6.0である請求項26に記載の製剤。
【請求項37】
ヒト抗体がヒトIgGlκ抗体である請求項26から36のいずれか一項に記載の製剤。
【請求項38】
ヒト抗体の軽鎖が更に配列番号5に記載のアミノ酸配列を含むCDR2ドメインと、配列番号7に記載のアミノ酸配列を含むCDR1ドメインを含み、及び/又はヒト抗体の重鎖が配列番号6に記載のアミノ酸配列を含むCDR2ドメインと、配列番号8に記載のアミノ酸配列を含むCDR1ドメインを含む請求項26から36のいずれか一項に記載の製剤。
【請求項39】
ヒト抗体の軽鎖が配列番号1に記載のアミノ酸配列を含み、ヒト抗体の重鎖が配列番号2に記載のアミノ酸配列を含む請求項26から36のいずれか一項に記載の製剤。
【請求項40】
抗体がアダリムマブである請求項26から36のいずれか一項に記載の製剤。
【請求項41】
皮下投与に適している請求項1から40のいずれか一項に記載の製剤。
【請求項42】
対象における有害なTNFα活性に関連する疾患の治療方法であって、請求項1から41のいずれか一項に記載の製剤を前記対象に投与する段階を含む前記方法。
【図1】

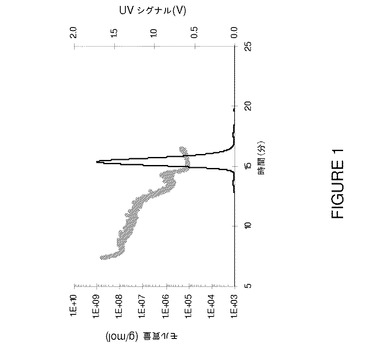
【図2】
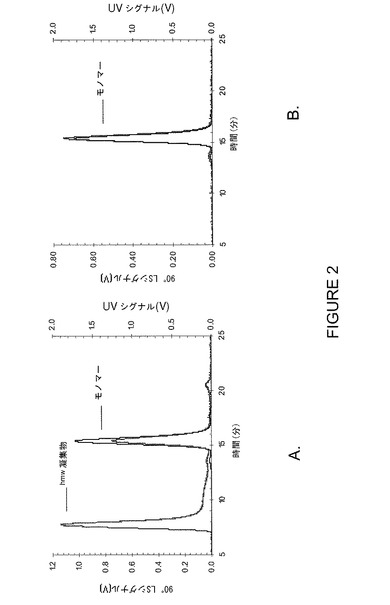
【図3】
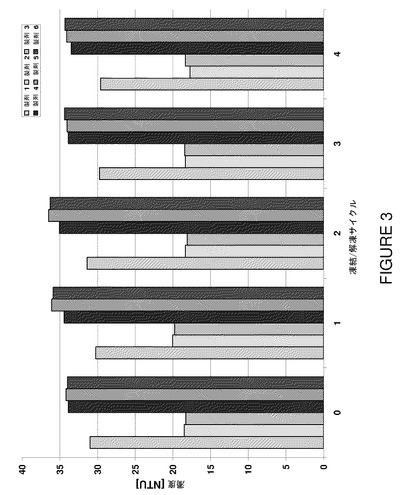
【図4】
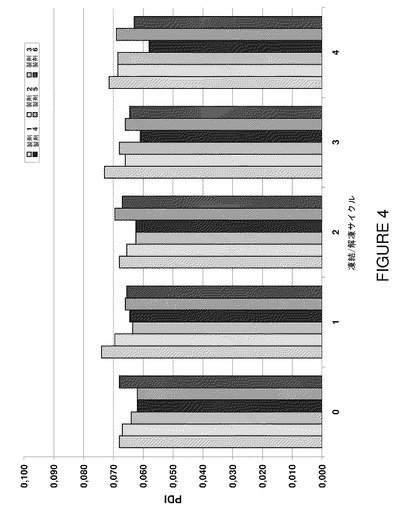
【図5】
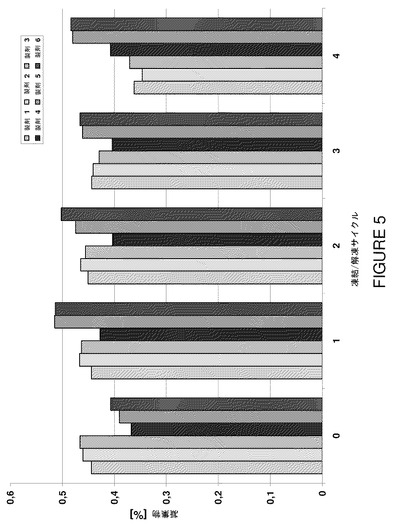
【図6】
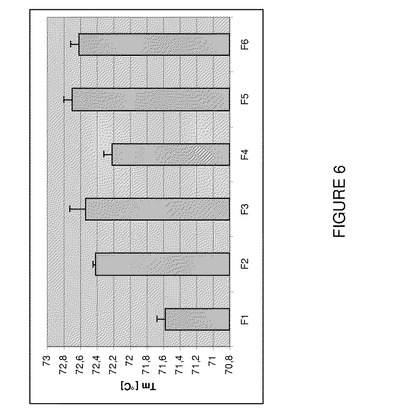
【図7】
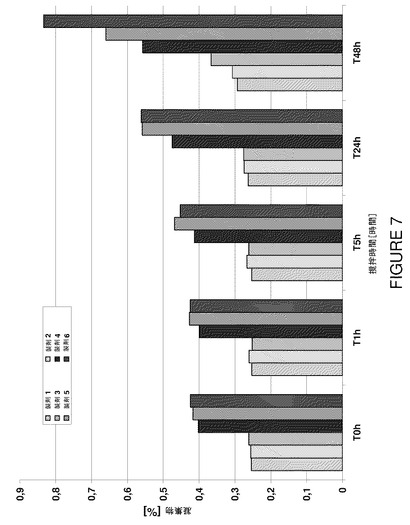
【図8】
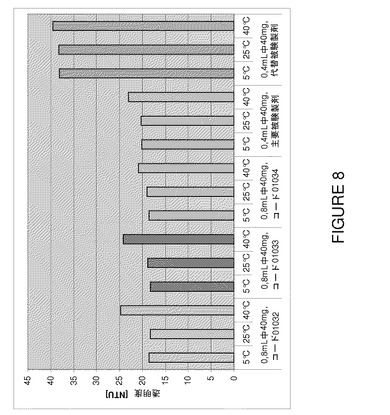
【図9】
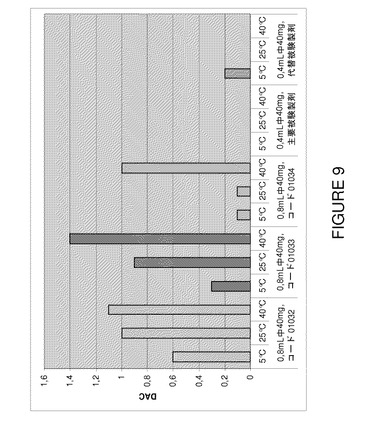
【図10】
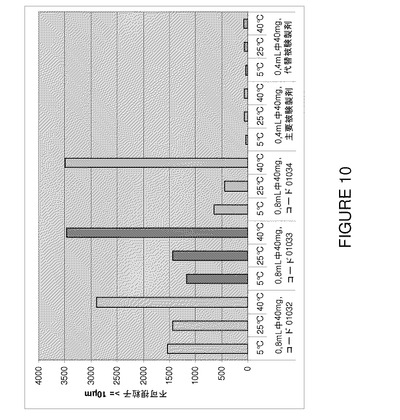
【図11】
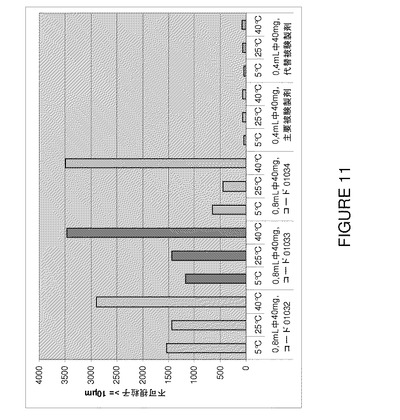
【図12】
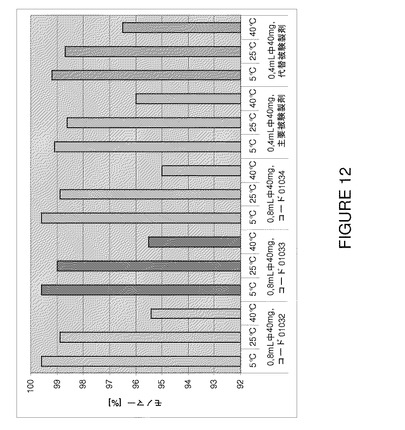
【図13】
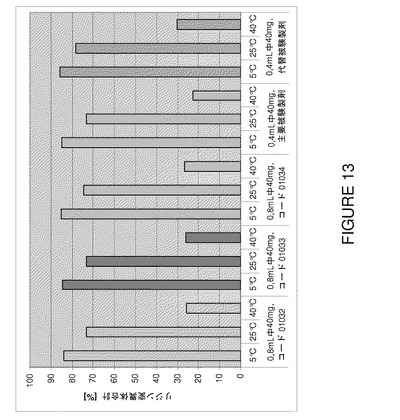
【図14】
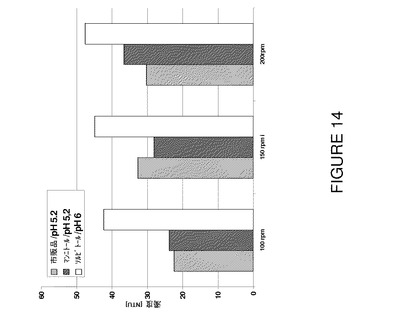
【図15】
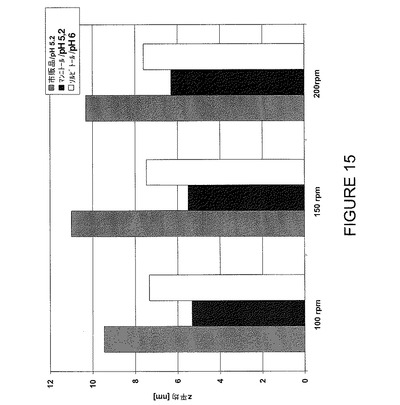
【図16】
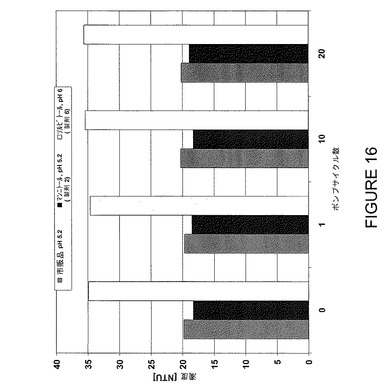
【図17】
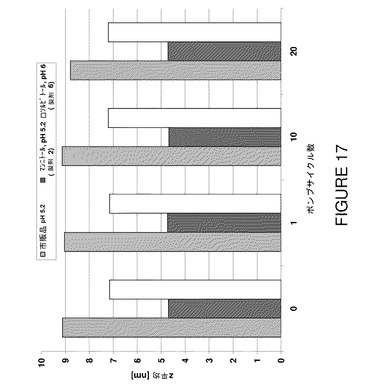
【図18】
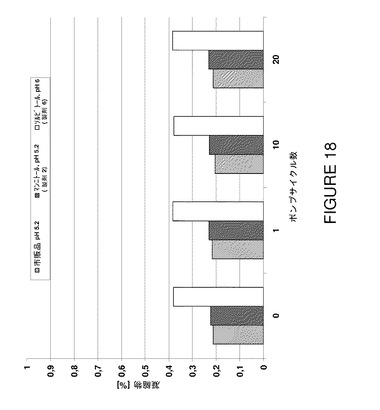
【図19】

【図20】
【図21】
【図22】
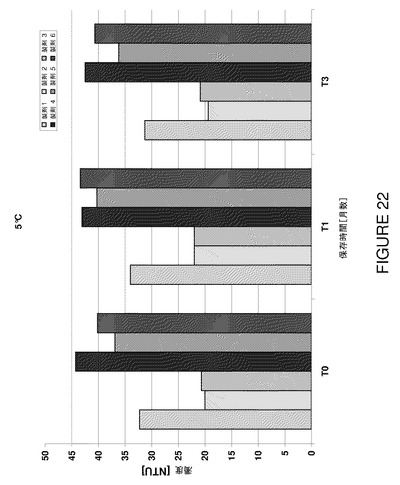
【図23】
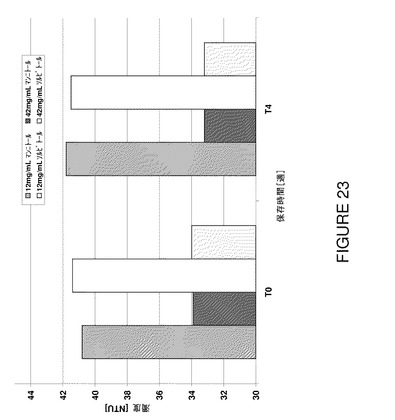
【図24】
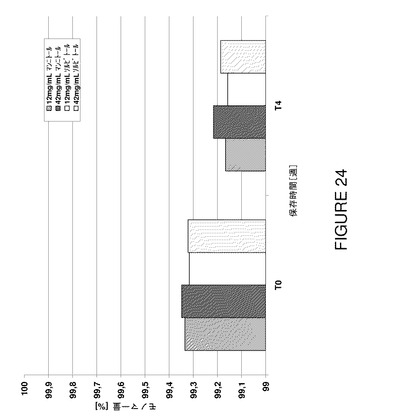
【図25】
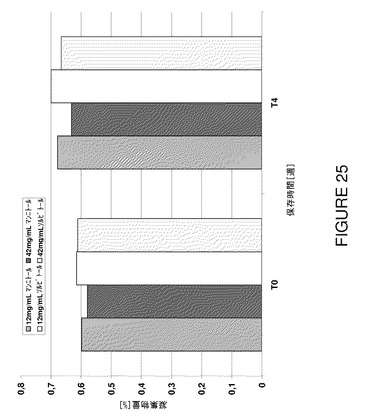
【図26】
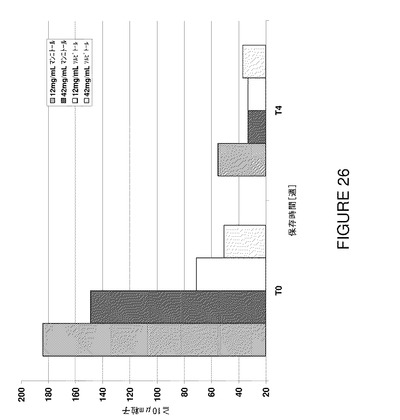
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【公表番号】特表2012−526121(P2012−526121A)
【公表日】平成24年10月25日(2012.10.25)
【国際特許分類】
【出願番号】特願2012−509873(P2012−509873)
【出願日】平成22年5月3日(2010.5.3)
【国際出願番号】PCT/US2010/033387
【国際公開番号】WO2010/129469
【国際公開日】平成22年11月11日(2010.11.11)
【出願人】(503448572)アボツト・バイオテクノロジー・リミテツド (30)
【Fターム(参考)】
【公表日】平成24年10月25日(2012.10.25)
【国際特許分類】
【出願日】平成22年5月3日(2010.5.3)
【国際出願番号】PCT/US2010/033387
【国際公開番号】WO2010/129469
【国際公開日】平成22年11月11日(2010.11.11)
【出願人】(503448572)アボツト・バイオテクノロジー・リミテツド (30)
【Fターム(参考)】
[ Back to top ]