
ヒト胚性幹細胞の分化
本発明は、多能性幹細胞の分化を促進するための方法を提供する。詳細には、本発明は膵臓内胚葉、膵臓ホルモン発現細胞及び膵臓ホルモン分泌細胞を形成するための改良された方法を提供する。本発明は更に、フィーダー細胞層を使用することなく多能性幹細胞の分化を促進する方法を提供する。
【発明の詳細な説明】
【開示の内容】
【0001】
〔技術分野〕
本発明は、多能性幹細胞の分化を促進するための方法を提供する。詳細には、本発明は膵臓内胚葉、膵臓ホルモン発現細胞及び膵臓ホルモン分泌細胞を形成するための改良された方法を提供する。本発明は更に、フィーダー細胞層を使用することなく多能性幹細胞の分化を促進する方法を提供する。
【0002】
〔背景技術〕
1型糖尿病の細胞置換療法における進歩及び移植可能なランゲルハンス島の不足のため、生着に適したインスリン産生細胞即ち、β細胞の供給源の開発に注目が集まっている。1つの方法として、例えば、胚性幹細胞のような多能性幹細胞から機能性のβ細胞を生成することがある。
【0003】
脊椎動物の胚の発生においては、多能性細胞は原腸胚形成として知られる過程において3つの胚細胞層(外胚葉、中胚葉、内胚葉)を含む細胞群を生ずる。甲状腺、胸腺、膵臓、腸、及び肝臓等の組織は中間段階を経て内胚葉から発生する。この過程における中間段階は、胚体内胚葉の形成である。胚体内胚葉細胞は、HNF−3β、GATA4、Mixl1、CXCR4及びSox−17等の多くのマーカーを発現する。
【0004】
胚体内胚葉が膵臓内胚葉に分化することによって膵臓が形成される。膵臓内胚葉の細胞は膵十二指腸ホメオボックス遺伝子Pdx1を発現する。Pdx1の非存在下では、膵臓の発生は腹側及び背側膵芽が形成された時点で停止してしまう。したがって、Pdx1の発現は膵臓の臓器発生における重要なステップである。成熟した膵臓は異なる細胞種の中でも、特に外分泌組織及び内分泌組織を含んでいる。外分泌及び内分泌組織は、膵臓内胚葉の分化によって生じるものである。
【0005】
島細胞の特徴を有する細胞が、マウスの胚細胞から誘導されたことが報告されている。例えば、ルメルスキー等(Lumelsky et al. Science 292:1389, 2001)は、マウスの胚性幹細胞が膵島に似たインスリン分泌構造に分化したことを報告している。ソリア等(Soria et al. Diabetes 49:157, 2000)は、マウスの胚性幹細胞から誘導されたインスリン分泌細胞が、ストレプトゾトシン誘導糖尿病マウスにおいて血糖症を正常化させたことを報告している。
【0006】
1つの例として、ホリ等(Hori et al. PNAS 99: 16105, 2002)は、マウス胚性幹細胞をホスホイノシチド3−キナーゼ(LY294002)の阻害剤で処理することによって、β細胞に似た細胞が生じたことを開示している。
【0007】
別の例として、ブリスザック等(Blyszczuk et al. PNAS 100:998, 2003)は、Pax4を構成的に発現しているマウス胚性幹細胞からインスリン産生細胞が発生したことを報告している。
【0008】
ミカレフ等(Micallef et al.)は、レチノイン酸が、胚性幹細胞がPdx1陽性膵臓内胚葉を形成する運命決定を調節しうることを報告している。レチノイン酸は、胚の原腸胚形成の終期に対応する期間に胚性幹細胞分化の4日目に細胞培養に添加した場合にPdx1の発現を誘導する効果が最も高くなる(Diabetes 54:301, 2005)。
【0009】
ミヤザキ(Miyazaki)等は、Pdx1を過剰発現するマウス胚性幹細胞を報告している。ミヤザキ等の結果は、外因性のPdx1の発現によって、得られた分化細胞におけるインスリン、ソマトスタチン、グルコキナーゼ、ニューロゲニン3、P48、Pax6、及びHNFの遺伝子の発現が明らかに高められたことを示すものである(Diabetes 53: 1030, 2004)。
【0010】
スコウディー(Skoudy)等は、アクチビンA(TGFβスーパーファミリーのメンバー)がマウス胚性幹細胞における外分泌性膵臓遺伝子(p48及びアミラーゼ)及び内分泌性遺伝子(Pdx1、インスリン及びグルカゴン)の発現をアップレギュレートすることを報告している。1nMのアクチビンAを使用した場合に、最大の効果が認められた。スコウディー等は更に、インスリン及びPdx1のmRNAの発現レベルはレチノイン酸によって影響されなかったが、3nMのFGF7で処理することによってPdx1の転写産物のレベルが増大したことを観察している(Biochem. J. 379: 749, 2004)。
【0011】
シラキ(Shiraki)等は、胚性幹細胞のPdx1陽性細胞への分化を特異的に促進する増殖因子の効果を研究している。シラキ等は、TGFβ2によってPdx陽性細胞が高い比率で再現可能に得られたことを観察している(Genes Cells. 2005 Jun; 10(6): 503-16)。
【0012】
ゴードン(Gordon)等は、血清の非存在下及びアクチビンとWntシグナル伝達の阻害剤の存在下で、マウス胚性幹細胞からブラキュリ(brachyury)+/HNF−3β+内胚葉細胞が誘導されることを示している(米国特許出願公開第2006/00034468(A1)号)。
【0013】
ゴードン(Gordon et al. PNAS, Vol103, p16806, 2006)等は、「Wnt及びTGF−β/ノーダル(nodal)/アクチビンの同時シグナル伝達が前原始線条の形成には必要であった」と述べている。
【0014】
しかしながら胚性幹細胞の発生のマウスモデルは、例えば、ヒト等のより高等な哺乳動物における発生プログラムを正確に模倣していない可能性がある。
【0015】
トムソン(Thomson)等はヒトの胚盤胞から胚性幹細胞を単離した(Science 282:114, 1998)。これと同時に、ギアハート(Gearhart)及び共同研究者等は、胎児の生殖腺組織からヒト胚性生殖(hEG)細胞系を誘導した(Shamblott et al., Proc. Natl. Acad. Sci. USA 95:13726, 1998)。単純に白血病阻害因子(LIF)と培養することによって分化を妨げることが可能なマウス胚性幹細胞と異なり、ヒト胚性幹細胞は極めて特殊な条件下に維持しなければならない(米国特許第6,200,806号、国際特許出願公開第99/20741号、同第01/51616号)。
【0016】
ダムール(D’Amour)等は、高濃度のアクチビン及び低濃度の血清の存在下でヒト胚性幹細胞由来の胚体内胚葉の濃縮された培養物が作製されたことを述べている(Nature Biotechnology 2005)。これらの細胞をマウスの腎臓カプセル下に移植すると、一定の内胚葉性臓器の特徴を有するより成熟した細胞に分化した。ヒト胚性幹細胞由来の胚体内胚葉細胞はFGF−10の添加後、更にPdx1陽性細胞に分化させることができる(米国特許出願第2005/0266554(A1)号)。
【0017】
ダムール等(D’Amour et al. Nature Biotechnology - 24, 1392 - 1401(2006))は、「我々は、ヒト胚性幹細胞(hES)を、インスリン、グルカゴン、ソマトスタチン、膵臓ポリペプチド及びグレリンといった膵臓ホルモンを合成可能な内分泌細胞に転換させる分化プロセスを開発した。このプロセスは、胚体内胚葉、腸管内胚葉、膵臓内胚葉及び内分泌前駆細胞に似た段階を経て、内分泌ホルモンを発現する細胞に細胞を誘導することによりインビボで膵臓の器官形成を模倣するものである。」と述べている。
【0018】
別の例として、フィスク(Fisk)等は、ヒト胚性幹細胞から膵臓島細胞を作製するためのシステムを報告している(米国特許出願第2006/0040387(A1)号)。この場合、分化経路は3つの段階に分けられている。先ず、ヒト胚性幹細胞を、酪酸ナトリウムとアクチビンAの組み合わせを用いて内胚葉に分化させる。次に細胞をノギン等のTGFβアンタゴニストとEGF又はベータセルリンの組み合わせと培養してPdx1陽性細胞を作製する。最後の分化はニコチンアミドによって誘導する。
【0019】
1つの例において、ベンベニストリー(Benvenistry)等は、「我々は、Pdx1の過剰発現が膵臓に多く見られる遺伝子の発現を高めたことを結論付けるものである。インスリン発現の誘導には、インビボでのみ存在する更なるシグナルを必要とする可能性がある。」と述べている(Benvenistry et al, Stem Cells 2006; 24:1923-1930)。
【0020】
〔発明の概要〕
〔発明が解決しようとする課題〕
したがって、膵臓内分泌細胞、膵臓ホルモン発現細胞、又は膵臓ホルモン分泌細胞への分化能を維持する一方で、今日の臨床的要求に応えるように拡張することが可能な多能性細胞系を樹立するための条件を開発することが依然、大きく求められている。本発明者等は、ヒト胚性幹細胞を膵臓内分泌細胞に分化させる効率を高めることを目的として代替的な手法を取ったものである。
【0021】
〔課題を解決するための手段〕
一実施形態において、本発明は、多能性幹細胞を分化させるための方法において、
a.多能性幹細胞を培養する工程と、
b.前記多能性幹細胞を、胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
c.前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
d.前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させる工程と、を含む、方法を提供する。
【0022】
一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞は、以下の方法のいずれか1つによって多能性幹細胞を処理することによって多能性幹細胞から分化させられる。即ち、
a.血清の非存在下、アクチビンAを含む培地中で多能性幹細胞を培養し、次いで細胞をアクチビンA及び血清と培養し、次いで細胞をアクチビンA及び異なる濃度の血清と培養するか、
b.血清の非存在下、アクチビンAを含む培地中で多能性幹細胞を培養し、次いで細胞をアクチビンA及び別の濃度の血清と培養するか、
c.血清の非存在下、アクチビンA及びWntリガンドを含む培地中で多能性幹細胞を培養し、次いでWntリガンドを除去して細胞をアクチビンA及び血清と培養するか、
d.多能性幹細胞を、細胞外基質でコーティングした組織培養基質上で培養し、この多能性幹細胞をアクチビンA及びWntリガンドと培養するか、
e.多能性幹細胞を、細胞外基質でコーティングした組織培養基質上で培養し、次いでこの多能性幹細胞を、血清を含む第1の培地中でアクチビンA及びWntリガンドと培養し、次いでこの多能性幹細胞を、血清を含む第2の培地中でアクチビンAと培養するか、
f.多能性幹細胞を、細胞外基質でコーティングした組織培養基質上で培養し、次いでこの多能性幹細胞を、血清を含む第1の培地中でアクチビンA及びWntリガンドと培養し、次いでこの多能性幹細胞を、異なる濃度の血清を含む第2の培地中でアクチビンA及びWntリガンドと培養する。
【0023】
一実施形態では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞は、以下の方法のいずれか1つによって胚体内胚葉系統に特徴的なマーカーを発現する細胞を処理することによって胚体内胚葉系統に特徴的なマーカーを発現する細胞から分化させられる。即ち、
a.胚体内胚葉系統に特徴的なマーカーを発現する細胞を、繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤で処理し、次いで繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤を含む培地を取り除いた後、レチノイン酸、繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤を含む培地中で細胞を培養するか、
b.胚体内胚葉系統に特徴的なマーカーを発現する細胞を、レチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で処理するか、
c.胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸で処理し、次いでレチノイン酸を除去した後、細胞を少なくとも1種類の繊維芽細胞増殖因子で処理する。
【0024】
一実施形態では、膵臓内分泌系統に特徴的なマーカーを発現する細胞は、以下の方法のいずれか1つによって膵臓内胚葉系統に特徴的なマーカーを発現する細胞を処理することによって膵臓内胚葉系統に特徴的なマーカーを発現する細胞から分化させられる。即ち、
a.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、DAPT及びエキセンジン4を含む培地中で培養し、次いでDAPT及びエキセンジン4を含む培地を除去した後、細胞をエキセンジン1、IGF−1及びHGFを含む培地中で培養するか、
b.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、エキセンジン4を含む培地中で培養し、次いでエキセンジン4を含む培地を除去した後、細胞をエキセンジン1、IGF−1及びHGFを含む培地中で培養するか、
c.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、DAPT及びエキセンジン4を含む培地中で培養するか、
d.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、エキセンジン4を含む培地中で培養するか、
e.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、ノッチシグナル伝達経路を阻害する因子で処理するか、
f.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、約10mM〜約20mMのグルコース及びエキセンジン4を含む培地中で培養する。
【0025】
一実施形態では、本発明は糖尿病を罹患した患者を治療するための方法において、
a.多能性幹細胞を培養する工程と、
b.前記多能性幹細胞を、胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
c.前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
d.前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、β細胞系統の細胞に分化させる工程と、
e.前記β細胞系統の細胞を前記患者に移植する工程と、を含む、方法を提供する。
【図面の簡単な説明】
【0026】
【図1】パネルa)は、100ng/mlのアクチビンAで2日間、5日間、及び8日間処理した後のヒト胚性幹細胞系H9における胚体内胚葉マーカーCXCR4、GATA4、HNF−3β、Mixl1、Sox−17の発現。胚体内胚葉マーカーの発現はmRNAレベルでアッセイされ、非処理のヒト胚性幹細胞における発現レベルに標準化された。パネルbは、100ng/mlのアクチビンAで3日間及び5日間処理した後のヒト胚性幹細胞系H9における前方内胚葉マーカーCerberus、Otx−1及びHex遺伝子の発現。
【図2】100ng/mlのアクチビンAで5日間処理した後のヒト胚性幹細胞系H9における胚体内胚葉マーカーの発現。胚体内胚葉の発現は、免疫組織化学法によって検出した。パネル(a)は、Sox−17の発現。パネル(b)は、HNF−3βの発現を示す。パネル(c)は、Oct3/4の発現。
【図3】段階的分化プロトコールを行った後のヒト胚性幹細胞系H9における胚体内胚葉マーカーの発現。胚体内胚葉マーカーの発現はmRNAレベルでアッセイされ、非処理のヒト胚性幹細胞における発現レベルに標準化された。パネル(a)は、GATA4の発現。パネル(b)は、Sox−17の発現。パネル(c)は、HNF−3βの発現。パネル(d)は、Mixl1の発現。「AA」で示されるデータポイントは、1日間(1d)、3日間(3d)、5日間(5d)又は7日間(7d)にわたったアクチビンAによる処理。「UT」で示されるデータポイントは、1日間(1d)、3日間(3d)、5日間(5d)又は7日間(7d)にわたって培養した非処理のコントロール。
【図4】段階的分化プロトコールを行った後のヒト胚性幹細胞系H9における胚体外内胚葉マーカーの発現。胚体外内胚葉マーカーの発現はmRNAレベルでアッセイされ、非処理のヒト胚性幹細胞における発現レベルに標準化された。パネル(a)は、AFPの発現に対する100ng/mlアクチビンAの影響。パネル(b)は、Sox7の発現に対する100ng/mlアクチビンAの影響。「AA」で示されるデータポイントは、1日間(1d)、3日間(3d)、5日間(5d)又は7日間(7d)にわたったアクチビンAによる処理。「UT」で示されるデータポイントは、1日間(1d)、3日間(3d)、5日間(5d)又は7日間(7d)にわたって培養した非処理のコントロール。
【図5】段階的分化プロトコールを行った後のヒト胚性幹細胞系H9における中胚葉及び内胚葉マーカーの発現。中胚葉及び内胚葉マーカーの発現はmRNAレベルでアッセイされ、非処理のヒト胚性幹細胞における発現レベルに標準化された。パネル(a)は、Brachyuryの発現に対する100ng/mlアクチビンAの影響。パネル(b)は、Zic7の発現に対する100ng/mlアクチビンAの影響。「AA」で示されるデータポイントは、1日間(1d)、3日間(3d)、5日間(5d)又は7日間(7d)にわたったアクチビンAによる処理。「UT」で示されるデータポイントは、1日間(1d)、3日間(3d)、5日間(5d)又は7日間(7d)にわたって培養した非処理のコントロール。
【図6】100ng/mlのアクチビンAで1日間、3日間、5日間、及び7日間処理した後のヒト胚性幹細胞系H7における胚体内胚葉マーカーBrachyury(パネルa)、CXCR4(パネルb)、Mixl1(パネルc)、Sox17(パネルd)、HNF−3beta(パネルe)、Oct4(パネルf)の発現。胚体内胚葉マーカーの発現はmRNAレベルでアッセイされ、非処理のヒト胚性幹細胞における発現レベルに標準化された。
【図7】分化プロトコールを適用した後のヒト胚性幹細胞系H9における胚体内胚葉マーカーの発現。胚体内胚葉の発現は、免疫組織化学法によって検出した。パネル(a)及び(b)は、Sox−17の発現。パネル(c)及び(d)は、HNF−3βの発現。パネル(e)及び(f)は、GATA4の発現。パネル(b)、(d)及び(f)は、DAPIによる核の対比染色。「処理」で示される列は、5日間のアクチビンA処理(100ng/ml)を行ったものである。「非処理」で示される列は、非処理のコントロール。
【図8】第2の分化プロトコールを適用した後のヒト胚性幹細胞系H9における膵臓内胚葉マーカーの発現。膵臓内胚葉マーカーの発現はPCRによってアッセイされ、アクチビンAで処理したヒト胚性幹細胞における発現レベルに標準化された。パネル(a)は、Pdx1の発現。パネル(b)は、GLUT−2の発現。パネル(c)は、PTF1aの発現。
【図9】第2の分化プロトコールを適用した後のヒト胚性幹細胞系H9における膵臓内胚葉マーカーの発現。膵臓内胚葉の発現を、免疫組織化学法によって検出した。パネル(a)は非処理のコントロールにおけるPdx1の発現を示し、パネル(b)は段階的分化プロトコールによって処理した培養中でのPdx1の発現を示す。
【図10】第3の分化プロトコールを適用した後のヒト胚性幹細胞系H9における膵臓内分泌マーカーの発現。膵臓内分泌マーカーの発現はPCRによってアッセイされ、アクチビンAで処理したヒト胚性幹細胞における発現レベルに標準化された。パネル(a)は、NeuroD1の発現。パネル(b)は、Ngn3の発現。パネル(c)は、インスリンの発現。パネル(d)は、Hes−1の発現。発現レベルは膵臓内胚葉細胞に標準化してある。
【図11】分化プロトコールを適用した後のヒト胚性幹細胞系H9における膵臓内胚葉マーカーの発現。膵臓内胚葉マーカーの発現はPCRによってアッセイされ、アクチビンAで処理したヒト胚性幹細胞における発現レベルに標準化された。パネル(a)は、Nkx2.2の発現。パネル(b)は、Pdx1の発現。
【図12】培養の各継代(P0、P1及びP2)の細胞におけるPDX−1の発現。PDX−1の発現はPCRによってアッセイされ、アクチビンAで処理したヒト胚性幹細胞H9における発現レベルに標準化された。
【図13】第3の分化プロトコールを適用した後のヒト胚性幹細胞系H9における肝細胞マーカーの発現。肝細胞マーカーの発現はPCRによってアッセイされ、アクチビンAで処理したヒト胚性幹細胞における発現レベルに標準化された。パネル(a)は、AFPの発現。パネル(b)は、アルブミンの発現。
【図14】ヒト胚性幹細胞系H9における多能性のマーカーの発現。多能性のマーカーの発現は免疫組織化学法によってアッセイされた。パネル(a)は、Oct−4の発現。パネル(b)は、アルカリホスファターゼの発現。
【図15】ヒト胚性幹細胞系H9の核型。核型はマウス胚性繊維芽フィーダー細胞上で培養した継代数P36の細胞で判定した。
【図16】ヒト胚性幹細胞を無フィーダー系で胚体内胚葉に分化させる本発明の分化プロトコールの概要。
【図17】異なる濃度のMATRIGEL上で培養し、(0.5〜2%の)低濃度血清及び高濃度のアクチビンA(100ng/ml)に5日間曝露した継代数44のヒト胚性幹細胞系H9のFACSプロファイル。胚体内胚葉マーカーCXCR4(CD184)の発現をY軸に示し、ESマーカーCD9の発現をX軸に示す。
【図18】希釈率1:10のMATRIGEL(■)、希釈率1:20のMATRIGEL(□(グレーで表示))、又は希釈率1:30のMATRIGEL(□)上で培養し、実施例14で開示する分化プロトコールに供した継代数44のヒト胚性幹細胞系H9の培養から得られた胚体内胚葉のマーカーについてのリアルタイムPCRの結果。発光量比(fold induction)は、マウス胚性繊維芽細胞を使用して調整した培地で培養した継代数44のヒト胚性幹細胞系H9の未分化細胞に対するもの。
【図19】未分化の多能性幹細胞、及び分化している多能性幹細胞から得られた胚体内胚葉細胞における網羅的遺伝子発現の散布図。示されたデータは、マウス胚性繊維芽細胞上で培養した継代数44(右パネル)及びMATRIGEL上で培養した継代数83(左パネル)のヒト胚性幹細胞系H9細胞系の培養から得られたもの。
【図20】マウス胚性繊維芽フィーダー細胞上で培養し、実施例4に開示される胚体内胚葉の分化プロトコールに供したヒト胚性幹細胞系H1(パネルa)、ヒト胚性幹細胞系H7(パネルb)、及びヒト胚性幹細胞系H9(パネルc)について5日目のFACSによるCXCR4の発現。
【図21】マウス胚性繊維芽フィーダー細胞上で培養したヒト胚性幹細胞系H7(パネルa)及びヒト胚性幹細胞系H9(パネルb)の培養における、図に示された胚体内胚葉マーカーの発現のリアルタイムPCRによる結果。結果を、未分化細胞に対する増加倍率として表わす。
【図22】MATRIGEL(希釈率1:30)上で培養し、実施例4に開示される胚体内胚葉の分化プロトコールに供したヒト胚性幹細胞系H1(パネルa)、ヒト胚性幹細胞系H7(パネルb)、及びヒト胚性幹細胞系H9(パネルb)について5日目のFACSによるCXCR4の発現。
【図23】ヒト胚性幹細胞系H7(パネルa)、ヒト胚性幹細胞系H9(パネルb)及びヒト胚性幹細胞系H1(パネルc)の培養における、図に示された胚体内胚葉マーカーの発現のリアルタイムPCRによる結果。結果を、未分化細胞に対する増加倍率として表わす。細胞を実施例4に開示される方法に従って処理した。
【図24】100ng/mlのアクチビンA(パネルa)、又は100ng/mlのアクチビンA+20ng/mlのWnt−3a(パネルb)の存在下で継代数46のヒト胚性幹細胞系H9の培養の位相差顕微鏡画像。細胞を5日間処理した。
【図25】実施例4に開示される方法に従って処理した後の、継代数44のヒト胚性幹細胞系H7(パネルa及びb)及び継代数46のヒト胚性幹細胞系H9(パネルc及びd)の培養におけるFACSによるCXCR4の発現。パネルb及びdは、CXCR4の発現への20ng/mlのWnt−3aの影響。パネルa及びcは、Wnt−3aの非存在下におけるCXCR4の発現。結果は処理後5日目に得られたもの。
【図26】ヒト胚性幹細胞系H7(パネルa)及びH9(パネルb)の培養で認められる遺伝子の発現についてリアルタイムPCRのデータ。培養は、実施例4に開示される分化プロトコールによって処理した。各パネルに示すようにWntアゴニストであるWnt−3a(20ng/ml)、Wnt−5a(20ng/ml)及びWnt−7a(20ng/ml)の影響についても試験を行った。細胞を5日間処理した。結果は、未分化細胞に対する増加倍率として表わす。
【図27】FACSによる、処理後5日目の継代数46のヒト胚性幹細胞系H9の培養におけるCXCR4の発現。パネル(a)は、Wnt−3aの非存在下におけるCXCR4の発現。パネル(b)は、10ng/mlのWnt−3aによる処理後のCXCR4の発現。パネル(c)は、20ng/mlのWnt−3aによる処理後のCXCR4の発現を示し、パネル(d)は、50ng/mlのWnt−3aによる処理後のCXCR4の発現を示す。
【図28】処理後5日目のヒト胚性幹細胞系H9の培養中に認められる胚体内胚葉マーカーの発現。結果は、リアルタイムPCRによって求められる非処理細胞に対する発現の増加倍率として示した。パネル(a)は、図に示される胚体内胚葉マーカー遺伝子の発現に対する10、20、及び50ng/mlのWnt−3aの影響。パネル(b)は、処理後2日目(2d)及び5日目(5d)におけるgoosecoid(■)及びCXCR4(□)の発現に対する1、5、又は10ng/mlのWnt−3a(x軸上で10、5、1として示す)の影響。パネル(c)は、2日目(■)又は5日目(□)における細胞数に対する1、5、又は10ng/mlのWnt−3aの影響。
【図29】実施例4に開示される分化プロトコールによる5日間の処理後の、FACSによるヒト胚性幹細胞系H9の培養におけるCXCR4の発現。細胞を、Wnt−3a又はGSK−3B阻害剤の非存在下(パネルa)、20ng/mlのWnt−3aで5日間の全体(パネルb)、1000nMのGSK−3B阻害剤IXで5日間の全体(パネルc)、500nMのGSK−3B阻害剤IXで5日間の全体(パネルd)、100nMのGSK−3B阻害剤IXで5日間の全体(パネルe)、10nMのGSK−3B阻害剤IXで5日間の全体(パネルf)、100nMのGSK−3B阻害剤IXで1〜2日目(パネルg)、10nMのGSK−3B阻害剤IXで1〜2日目(パネルh)、培養。
【図30】リアルタイプPCRによる胚体内胚葉マーカーの遺伝子発現。結果を、非処理細胞に対する増加倍率として表わす。パネル(a)は、図に示される濃度及び時間でWnt−3a又はGSK−3B阻害剤を含む、実施例4に開示される胚体内胚葉プロトコールに従って処理した、継代数48のヒト胚性幹細胞系H9から得られたデータ。パネル(b)は、図に示される濃度及び時間でWnt−3a又はGSK−3B阻害剤を含む、実施例4で開示する胚体内胚葉プロトコールに従って処理した、継代数46のヒト胚性幹細胞系H9から得られたデータ。
【図31】本発明で使用する胚性幹細胞系についてFACSによるCXCR4の発現。パネル(a〜d)は、継代数49のヒト胚性幹細胞系H9から得られたデータ。パネル(e〜f)は、継代数46のヒト胚性幹細胞系H1から得られたデータ。データは処理後5日目に得た。細胞を以下の条件で処理した。パネル(a):10ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間、パネル(b):100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間、パネル(c):100ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、パネル(d):10ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、パネル(e):100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間、パネル(f):10ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間。
【図32】10、50、又は100ng/mlのアクチビンA及び20ng/mlのWnt−3aで処理した、継代数49のヒト胚性幹細胞系H9の培養についてリアルタイムPCRで調べた胚体内胚葉マーカーの遺伝子発現。パネル(a):AFP、Bry、CXCR4、GSC、HNF−3B、及びPOU5F(Oct−4)の発現、並びに、パネル(b):SOX−17及びGATA4の発現。結果を非処理細胞に対する増加倍率として表わす。
【図33】継代数53の胚性幹細胞系H9についてFACSによるCXCR4の発現。データは処理後5日目に得た。細胞を以下の条件で処理した。パネル(a):100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間及び25ng/mlのBMP−4を3〜5日目、パネル(b):100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間、パネル(c):100ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、パネル(d):20ng/mlのWnt−3a及び25ng/mlのBMP−4を5日間全体、パネル(e):100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3a及び100nMのGSK−3B阻害剤IXを最初の2日間、並びに、パネル(f):100ng/mlのアクチビンA及び25ng/mlのBMP−4を5日間全体。すべてのパネルについてX軸はCD9の発現を表し、Y軸はCXCR4(CD184)の発現を表す。
【図34】10又は100ng/mlのアクチビンA及び20ng/mlのWnt−3a又は100nMのGSK−3B阻害剤で処理した、継代数46のヒト胚性幹細胞系H1の培養についてリアルタイムPCRで調べた胚体内胚葉マーカーの遺伝子発現。パネル(a):AFP、Bry、CXCR4、GSC、及びPOU5F(Oct−4)の発現、並びに、パネル(b):SOX−17、HNF−3B及びGATA4の発現。結果は非処理細胞に対する増加倍率として表わす。
【図35】50又は100ng/mlのアクチビンA及び10又は100nMのGSK−3B阻害剤で処理した、継代数49のヒト胚性幹細胞系H9の培養についてリアルタイムPCRで調べた胚体内胚葉マーカーの遺伝子発現。パネル(a):AFP、Bry、CXCR4、GSC、HNF−3B及びPOU5F(Oct−4)の発現、並びに、パネル(b):SOX−17及びGATA4の発現。結果は非処理細胞に対する増加倍率として表わす。
【図36】アクチビンA、Wnt−3a、GSK−3阻害剤、及びBMP−4の組み合わせで5日間処理した、継代数53のヒト胚性幹細胞系H9の培養についてリアルタイムPCRで調べた胚体内胚葉マーカーの遺伝子発現。パネル(a):AFP、Bry、CXCR4、GSC、HNF−3B及びSOX7の発現、並びに、パネル(b):SOX−17、HNF−3B及びGATA4の発現。
【図37】実施例22に列記した条件で処理したヒト胚性幹細胞系H9の培養において、FACSによって調べたCXCR4の発現の比率。
【図38】フィブロネクチン(パネルa)又はMATRIGEL(商標)(パネルb)上で培養したヒト胚性幹細胞系H9の培養において、FACSによって調べた胚体内胚葉マーカーの発現。
【図39】フィブロネクチン(□)又は希釈率1:10の増殖因子低減MATRIGEL(■)上で培養したヒト胚性幹細胞系H9の培養においてリアルタイムPCRによって求めた胚体内胚葉マーカーの発現。
【図40】低濃度の血清、100ng/mlのアクチビンA及び20ng/mlのWnt−3aの存在下において異なる濃度のMATRIGELがヒト胚性幹細胞の胚体内胚葉への分化に与える影響。細胞を実施例4に開示される方法に従って処理。示した結果はリアルタイプPCRによって調べた、図に示される遺伝子の発現レベル。
【図41−1】MATRIGEL上で維持し、マウス胚性繊維芽細胞上で分化させたヒト胚性幹細胞による胚体内胚葉の形成におけるWnt−3aの役割。パネル(a)〜(d)は、図に示される遺伝子についてリアルタイムPCRのデータ。パネル(e)〜(g)は、図に示す条件についてFACSのデータ。
【図41−2】MATRIGEL上で維持し、マウス胚性繊維芽細胞上で分化させたヒト胚性幹細胞による胚体内胚葉の形成におけるWnt−3aの役割。パネル(a)〜(d)は、図に示される遺伝子についてリアルタイムPCRのデータ。パネル(e)〜(g)は、図に示す条件についてFACSのデータ。
【図41−3】MATRIGEL上で維持し、マウス胚性繊維芽細胞上で分化させたヒト胚性幹細胞による胚体内胚葉の形成におけるWnt−3aの役割。パネル(a)〜(d)は、図に示される遺伝子についてリアルタイムPCRのデータ。パネル(e)〜(g)は、図に示す条件についてFACSのデータ。
【図41−4】MATRIGEL上で維持し、マウス胚性繊維芽細胞上で分化させたヒト胚性幹細胞による胚体内胚葉の形成におけるWnt−3aの役割。パネル(a)〜(d)は、図に示される遺伝子についてリアルタイムPCRのデータ。パネル(e)〜(g)は、図に示す条件についてFACSのデータ。
【図42】Wnt阻害剤DKK−1による処理後の、MATRIGEL(商標)でコーティングされた組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化。示される結果は、20ng/mlのWnt−3A及び100ng/mlのDKK1(DE+DKK1)の存在下、又はDKK1(DE)の非存在下で実施例4に開示される方法に従って処理したH9細胞においてリアルタイムPCRによって調べた、図に示される遺伝子の発現。
【図43】MATRIGELでコーティングした組織培養基質上で培養し、低濃度血清及び100ng/mlのアクチビンAに20ng/mlのWnt−3aを加えないもの(パネルa)又は加えたもの(パネルb)で分化させたヒト胚性幹細胞系H9の培養における胚体内胚葉マーカーの免疫蛍光染色。Ecad=E−カドヘリン、NCAM=N−カドヘリン。
【図44】継代数38のヒト胚性幹細胞系SA002の胚体内胚葉への分化。細胞は図に示した条件で5日間処理し、各パネルに示した遺伝子について遺伝子の発現をリアルタイムPCRで調べた。
【図45】100ng/mlのアクチビンA処理(パネルa)、100ng/mlのアクチビンA+20ng/mlのWnt−3a(パネルb)、又は100ng/mlのアクチビンA+100nMのGSK−3B阻害剤IX(パネルc)による処理後の、継代数38のヒト胚性幹細胞系SA002における、FACSによるCXCR4の発現。細胞を5日間処理。
【図46】ヒト血清でコーティングした組織培養基質上での継代数55のヒト胚性幹細胞系H1の胚体内胚葉への分化。細胞は図に示した条件で処理し、各パネルに示した遺伝子について遺伝子の発現をリアルタイムPCRで調べた。
【図47】MATRIGEL(商標)でコーティングした組織培養基質上での継代数P54のヒト胚性幹細胞系H1の培養の胚体内胚葉への分化。5日間のDEプロトコールの後、異なるGSK−B阻害剤の影響について試験を行った。以下のGSK−3B阻害剤(GSK−3B VIII、IX、XI及びXII)を100nMにて処理の最初の2日間について評価を行った。
【図48】20ng/mlのWnt−3aの存在下で実施例4に開示される方法に従って培養及び処理した継代数49のヒト胚性幹細胞系H9の培養における、処理の最初の2日間のAFP(パネルa)、Pdx−1(パネルb)、Cdx−2及びGlut−2(パネルc)、並びに、HNF−3β、HNF−6及びソマトスタチン(パネルd)の発現。この処理の後、細胞を、2%FBS及び1μMのレチノイン酸、0.1〜1μMのTTNPB(4−[(E)−2−(5,6,7,8−テトラヒドロ−5,5,8,8−テトラメチル−2−ナフタレニル)−1−プロペニル]安息香酸(アロチノイド酸))、又は0.1〜10μMのAM−580(4−[(5,6,7,8−テトラヒドロ−5,5,8,8−テトラメチル−2−ナフタレニル)カルボキサミド]安息香酸)で更に3日間処理。次いで、細胞を2%FBS及び20ng/mlのbFGF中で更に3日間処理。
【図49】図に示した時間及び濃度でアクチビンA及びWnt−1によって処理したヒト胚性幹細胞系H1の培養において、パネルa及びbに示す胚体内胚葉マーカーの発現のリアルタイムPCRによる結果。
【図50】膵臓内胚葉細胞をDMEM/F12又はDMEM/低グルコース中で処理することによって形成された膵臓内分泌細胞の培養中におけるインスリン(パネルa)及びグルカゴン(パネルb)のmRNAの発現。図に示されるデータは、2つの別々の実験で観察されたもの。
【図51】DMDM/低グルコース(パネルa)、DMEM/F12(パネルb)中で処理した細胞において免疫細胞化学法によって調べたインスリンの発現。パネルcは、PDX−1及びインスリンの共染色。
【図52−1】ヒト胚性幹細胞系H9から誘導された膵臓内分泌細胞における遺伝子発現に対するグルコース濃度の影響。各遺伝子はパネル中で特定される。
【図52−2】ヒト胚性幹細胞系H9から誘導された膵臓内分泌細胞における遺伝子発現に対するグルコース濃度の影響。各遺伝子はパネル中で特定される。
【図52−3】ヒト胚性幹細胞系H9から誘導された膵臓内分泌細胞における遺伝子発現に対するグルコース濃度の影響。各遺伝子はパネル中で特定される。
【図53】2、10及び20mMのグルコース中で形成された膵臓内分泌細胞からのcペプチドの放出。細胞をIBMX又は20mMのグルコースで刺激した。
【発明を実施するための形態】
【0027】
開示を分かりやすくするため、限定を目的とすることなく、本発明の詳細な説明を、本発明の特定の特徴、実施形態、又は応用を説明又は図示した以下の小項目に分ける。
【0028】
用語の定義
幹細胞とは、1個の細胞レベルで自己再生し、かつ、自己再生性の前駆細胞、非自己再生性の前駆細胞、及び最終分化細胞のような前駆細胞を生ずるように分化する能力によって定義される未分化細胞のことである。幹細胞はまた、複数の胚細胞層(内胚葉、中胚葉及び外胚葉)から異なる細胞系の機能性細胞へとインビトロで分化する能力、並びに、移植後に複数の胚細胞層の組織を生ずる能力、及び胚盤胞への注入後にすべての組織とまではいかないにしてもほぼ大部分の組織に分化する能力によっても特徴付けられる。
【0029】
幹細胞は、発生上の能力によって、(1)すべての胚性又は胚体外細胞のタイプを生ずる能力を有することを意味する、分化全能性、(2)すべての胚性細胞のタイプを生ずる能力を有することを意味する、分化万能性、(3)細胞系統のサブセットを生ずる能力を有するが、それらがすべて特定の組織、臓器、又は生理学的システムのものであるような、分化多能性(例えば、造血幹細胞(HSC)は、HSC(自己再生性)、血球限定的寡能性前駆細胞、及び、血液の通常の成分であるすべての細胞種及び要素(例、血小板)を生じうる)、(4)多能性幹細胞よりも限定された細胞系統のサブセットを生ずる能力を有することを意味する、分化寡能性、及び(5)単一の細胞系統(例、精原幹細胞)を生ずる能力を有することを意味する、分化単一性に分類される。
【0030】
分化とは、特化していない(「分化決定していない」)か又は完全には特化していない細胞が、例えば、神経細胞や筋細胞のような特化した細胞の特徴を獲得するプロセスである。分化した細胞、又は分化誘導された細胞とは、細胞のその系統内においてより特化した(「分化決定した」)位置を占めるものである。分化のプロセスについて用いられる場合、「分化決定した」なる用語は、通常の条件下で特定の細胞種又は細胞種のサブセットにまで分化し続けるが、通常の条件下では異なる細胞種には分化できず、より分化の未熟な細胞種にも戻ることができない点にまで分化経路を進んだ細胞のことを指して言う。脱分化とは、細胞が細胞の系統においてより特化(又は分化決定)していない位置にまで戻るプロセスのことを指して言う。本明細書で言うところの細胞の系統とは、その細胞の遺伝、即ち、その細胞がどの細胞から生じたものであり、どのような細胞を生じうるか、ということを定義するものである。ある細胞の系統とは、発生及び分化の所定の遺伝スキーム内にその細胞を位置付けるものである。系統特異的マーカーとは、対象とする系統の細胞の表現型と特異的に関連した特徴を指し、分化決定していない細胞の、対象とする系統への分化を評価するために使用することができる。
【0031】
培養中の細胞を述べるために異なる用語が用いられる。「維持」とは、細胞の増殖及び/又は分裂を促進する条件下で増殖培地中に入れられた細胞のことを一般に指して言うものであり、より大きな細胞集団が形成される場合もそうでない場合もある。「継代」とは、1つの培養容器から細胞を取り出して、細胞の増殖及び/又は分裂を促進する条件下でこれらの細胞を第2の培養容器に入れるプロセスのことを指して言う。
【0032】
特定の細胞集団又は細胞系は、しばしば継代された回数によって呼称されるか、又はそれによって特徴付けられる。例えば、10回継代された培養細胞集団は、P10培養と呼ばれる場合がある。初代培養、即ち、組織から細胞を単離した後の最初の培養はP0と指定される。最初の継代培養の後、細胞は2次培養(P1又は継代数1)として述べられる。2回目の継代培養の後では細胞は3次培養(P2又は継代数2)となる、といった具合である。当業者であれば、継代期間中に集団は何度も倍加しうるものであり、したがってある培養の集団倍加の回数は継代数よりも大きいことは理解されるであろう。各継代間の期間における細胞の増殖(即ち、集団倍加数)は、播種密度、基質、培地、培養条件、及び継代間の時間等を含むがこれらに限定されない多くの因子に依存する。
【0033】
「β細胞系統」とは、転写因子PDX−1、並びに以下の転写因子、即ち、NGN−3、Nkx2.2、Nkx6.1、NeuroD、Isl−1、HNF−3β、MAFA、Pax4、及びPax6の内の少なくとも1つについて遺伝子発現が陽性である細胞を指して言う。β細胞系統に特徴的なマーカーを発現する細胞としてはβ細胞が挙げられる。
【0034】
本明細書で言うところの「胚体内胚葉系統に特徴的なマーカーを発現する細胞」とは、以下のマーカー、即ち、SOX−17、GATA−4、HNF−3β、GSC、Cer1、ノーダル(Nodal)、FGF8、ブラキュリ(Brachyury)、Mix様ホメオボックスタンパク質、FGF4 CD48、エオメソダーミン(eomesodermin)(EOMES)、DKK4、FGF17、GATA−6、CXCR4、C−Kit、CD99又はOTX2の内の少なくとも1つを発現する細胞を指して言う。胚体内胚葉系統に特徴的なマーカーを発現する細胞としては、原始線条前駆細胞、原始線条細胞、中内胚葉細胞及び胚体内胚葉細胞が挙げられる。
【0035】
本明細書で言うところの「膵臓内胚葉系統に特徴的なマーカーを発現する細胞」とは、以下のマーカー、即ち、PDX−1、HNF−1β、HNF−3β、PTF−1α、HNF−6、又はHB9の内の少なくとも1つを発現する細胞を指して言う。膵臓内胚葉系統に特徴的なマーカーを発現する細胞としては、膵臓内胚葉細胞が挙げられる。
【0036】
本明細書で言うところの「膵臓内分泌系統に特徴的なマーカーを発現する細胞」とは、以下のマーカー、即ち、NGN−3、NeuroD、Islet−1、PDX−1、NKX6.1、Pax−4、Ngn−3、又はPTF−1αを発現する細胞を指して言う。膵臓内分泌系統に特徴的なマーカーを発現する細胞としては、膵臓内分泌細胞、膵臓ホルモン発現細胞及び膵臓ホルモン分泌細胞、並びにβ細胞系統の細胞が挙げられる。
【0037】
本明細書で言うところの「胚体内胚葉」とは、原腸胚形成時に胚盤葉上層から生ずる細胞の特徴を有し、消化管及びその派生器官を形成する細胞を指して言う。胚体内胚葉細胞は、以下のマーカー、即ち、CXCR4、HNF−3β、GATA−4、SOX−17、ケルベロス(Cerberus)、OTX2、グースコイド(goosecoid)、c−Kit、CD99、及びMixl1を発現する。
【0038】
本明細書で言うところの「胚体外内胚葉」とは、以下のマーカー、即ち、SOX−7、AFP、及びSPARCの内の少なくとも1つを発現する細胞の集団を指して言う。
【0039】
本明細書で言うところの「マーカー」とは、対象とする細胞で発現の仕方が異なる核酸又はポリペプチド分子である。ここで言う異なる発現の仕方とは、陽性のマーカーでは発現レベルの上昇を、陰性のマーカーでは発現レベルの低下を意味する。マーカー核酸又はポリペプチドの検出可能なレベルは、他の細胞と比較して対象とする細胞において充分に高いか又は低いことから、当該技術分野において知られる各種の方法のいずれを用いても対象とする細胞を他の細胞から識別及び区別することが可能である。
【0040】
本明細書で言うところの「中内胚葉細胞」とは、以下のマーカー、即ち、CD48、エオメソダーミン(EOMES)、SOX−17、DKK4、HNF−3β、GSC、FGF17、GATA−6の内の少なくとも1つを発現する細胞を指して言う。
【0041】
本明細書で言うところの「膵臓内分泌細胞」又は「膵臓ホルモン発現細胞」とは、以下のホルモン、即ち、インスリン、グルカゴン、ソマトスタチン、膵臓ポリペプチド、及びグレリンの内の少なくとも1つを発現することが可能な細胞を指して言う。
【0042】
本明細書で言うところの「膵臓ホルモン分泌細胞」とは、以下のホルモン、即ち、インスリン、グルカゴン、ソマトスタチン、及び膵臓ポリペプチドの内の少なくとも1つを分泌することが可能な細胞を指して言う。
【0043】
本明細書で言うところの「前原始線条細胞」とは、以下のマーカー、即ち、ノーダル(Nodal)、又はFGF8の内の少なくとも1つを発現する細胞を指して言う。
【0044】
本明細書で言うところの「原始線条細胞」とは、以下のマーカー、即ち、ブラキュリ(Brachiury)、Mix様ホメオボックスタンパク質、又はFGF4の内の少なくとも1つを発現する細胞を指して言う。
【0045】
多能性幹細胞の単離、増殖及び培養
多能性幹細胞の特徴付け
多能性幹細胞は、発生段階特異的胚性抗原(SSEA)3及び4の1つ以上、及びTra−1−60及びTra−1−81と呼ばれる抗体を用いて検出可能なマーカーを発現しうる(Thomson et al., Science 282:1145, 1998)。インビトロで多能性幹細胞を分化させると、SSEA−4、Tra−1−60、及びTra−1−81の発現が消失し(存在する場合)、SSEA−1の発現が増大する。未分化の多能性幹細胞は通常アルカリホスファターゼ活性を有し、これは、細胞を4%パラホルムアルデヒドで固定し、次いで製造業者(ベクターラボラトリーズ、カリフォルニア州バーリンゲーム所在)によって述べられるようにVectorRedを基質として現像することによって検出することができる。未分化の多能性幹細胞は、RT−PCRによって検出されるOct−4及びTERTを通常更に発現している。
【0046】
増殖させた多能性幹細胞の別の望ましい表現型は、内胚葉、中胚葉、外胚葉組織の3つの胚細胞層のすべての細胞に分化する能力である。多能性幹細胞の多能性を、例えば、細胞を重症複合免疫不全症(SCID)マウスに注入し、形成される奇形腫を4%パラホルムアルデヒドで固定し、次いでこれを3つの胚細胞層からの細胞種の証拠について組織学的に調べることによって確認することができる。また、多能性を、胚様体を形成し、3つの胚細胞層に関連したマーカーの存在について胚様体を評価することによって調べることもできる。
【0047】
増殖させた多能性幹細胞系は、標準的なGバンド法を用いて核型を決定し、対応する霊長類種の公表されている核型と比較することができる。細胞は「正常な核型」を得ることが望ましい。これは、細胞が、ヒトの染色体がすべて揃っていて、かつ目立った変化のない正倍数体であることを意味する。
【0048】
多能性幹細胞の供給源
使用が可能な多能性幹細胞の種類としては、妊娠期間中の任意の時期(必ずしもではないが、通常は妊娠約10〜12週よりも前)に採取した前胚性組織(例えば、胚盤胞等)、胚性組織、胎児組織等の、妊娠後に形成される組織に由来する多能性幹細胞の株化細胞系が含まれる。非限定的な例としては、例えば、ヒト胚性幹細胞系H1、H7及びH9(WiCell)等のヒト胚性幹細胞又はヒト胚性生殖細胞の株化細胞系がある。更に、こうした細胞の初期の株化又は安定化の際に本開示の組成物を使用することも考えられるが、その場合は、供給源となる細胞は供給源組織から直接採取される1次多能性細胞である。フィーダー細胞の非存在下で既に培養された多能性幹細胞集団から得られる細胞も好適である。例えば、BG01v(ブレサジェン社(BresaGen)、ジョージア州アテネ所在)等の変異型ヒト胚性幹細胞系も好適である。
【0049】
一実施形態では、ヒト胚性幹細胞をトムソン等(Thomson et al.)によって述べられるように調製する(米国特許第5,843,780号、Science 282:1145, 1998; Curr. Top. Dev. Biol. 38:133 ff., 1998; Proc. Natl. Acad. Sci. U.S.A. 92:7844, 1995)。
【0050】
多能性幹細胞の培養
一実施形態では、通常、多能性幹細胞を、様々な点で多能性幹細胞を支持するフィーダー細胞の層上で培養する。あるいは、多能性幹細胞を、フィーダー細胞を基本的に含まないにも関わらず、細胞を大きく分化させることなく多能性幹細胞の増殖を支持する培養システム中で培養する。分化をともなわない無フィーダー細胞培養中での多能性幹細胞の増殖は、別の細胞種と予め培養することによって調整した培地を用いることで支持される。また、分化をともなわない無フィーダー細胞培養中での多能性幹細胞の増殖は、合成培地を用いることによっても支持される。
【0051】
例えば、ルービノフ等(Reubinoff et al. Nature Biotechnology 18: 399 - 404(2000))及びトンプソン等(Thompson et al. Science 6 November 1998: Vol. 282. no. 5391, pp. 1145 - 1147)は、マウス胚性繊維芽フィーダー細胞層を用いてヒト胚盤胞からの多能性幹細胞系を培養することについて開示している。
【0052】
リチャード等(Richards et al. Stem Cells 21: 546-556, 2003)は、11種類の異なるヒトの成人、胎児、及び新生児フィーダー細胞層についてヒト多能性幹細胞の培養を支持する能力の評価を行っている。リチャード等は、「成人の皮膚繊維芽フィーダー細胞上で培養したヒト胚性幹細胞系は、ヒト胚性幹細胞の形態を有し、多能性を維持する」と述べている。
【0053】
米国特許出願公開第20020072117号は、無フィーダー細胞培養中で霊長類の多能性幹細胞の増殖を支持する培地を生成する細胞系を開示している。使用される細胞系は、胚性組織から得られるか、あるいは胚性幹細胞から分化した間葉系かつ繊維芽細胞様の細胞系である。米国特許出願公開第20020072117号は、この細胞系の1次フィーダー細胞層としての使用を更に開示している。
【0054】
別の例として、ワン等(Wang et al. Stem Cells 23: 1221-1227, 2005)は、ヒト胚性幹細胞由来のフィーダー細胞層上でヒト多能性幹細胞を長期にわたって増殖させるための方法を開示している。
【0055】
別の例として、ストイコビッチ等(Stojkovic et al. Stem Cells 2005 23: 306-314, 2005)は、ヒト胚性幹細胞の自然分化により誘導されたフィーダー細胞システムを開示している。
【0056】
更なる別の例として、ミヤモト等(Miyamoto et al. Stem Cells 22: 433-440, 2004)は、ヒトの胎盤から得られたフィーダー細胞の供給源を開示している。
【0057】
アミット等(Amit et al. Biol. Reprod 68: 2150-2156, 2003)は、ヒトの包皮由来のフィーダー細胞層を開示している。
【0058】
別の例として、インズンザ等(Inzunza et al. Stem Cells 23: 544-549, 2005)は、ヒトの出生直後産児の包皮繊維芽細胞から得られたフィーダー細胞層を開示している。
【0059】
米国特許第6642048号は、無フィーダー細胞培養中での霊長類の多能性幹(pPS)細胞の増殖を支持する培地、及びこうした培地の製造に有用な細胞系を開示している。米国特許第6642048号は、「本発明は、胚性組織から得られるか、あるいは胚性幹細胞から分化した間葉系かつ繊維芽細胞様の細胞系を含む。本開示では、こうした細胞系を誘導し、培地を調整し、この馴化培地を用いて幹細胞を増殖させるための方法を説明及び図示する」と述べている。
【0060】
別の例として、国際特許出願公開第2005014799号は、哺乳動物細胞の維持、増殖及び分化のための馴化培地を開示している。国際特許出願公開第2005014799号は、「本発明に基づいて製造される培地は、マウス細胞、特にMMH(Metマウス肝細胞)と称される分化及び不死化したトランスジェニック肝細胞の細胞分泌活性によって調整される」と述べている。
【0061】
別の例として、スー等(Xu et al. Stem Cells 22: 972-980, 2004)は、ヒトテロメラーゼ逆転写酵素を過剰発現するように遺伝子改変されたヒト胚性幹細胞由来細胞から得られた馴化培地を開示している。
【0062】
別の例として、米国特許出願公開第20070010011号は、多能性幹細胞の維持のための合成培地を開示している。
【0063】
代替的な培養システムでは、胚性幹細胞の増殖を促進することが可能な増殖因子を添加した無血清培地を使用している。例えば、チェオン等(Cheon et al. BioReprod DOI:10.1095/biolreprod.105.046870, October 19, 2005)は、胚性幹細胞の自己再生を誘発することが可能な異なる増殖因子を添加した非馴化血清補充(SR)培地中に胚性幹細胞が維持された、無フィーダー細胞かつ無血清の培養システムを開示している。
【0064】
別の例として、リーベンシュタイン等(Levenstein et al. Stem Cells 24: 568-574, 2006)は、bFGFを添加した培地を用い、繊維芽細胞又は馴化培地の非存在下でヒト胚性幹細胞を長期にわたって培養するための方法を開示している。
【0065】
別の例として、米国特許出願公開第20050148070号は、血清の非存在下及び繊維芽フィーダー細胞の非存在下において合成培地中でヒト胚性幹細胞を培養する方法を開示している。この方法は、アルブミン、アミノ酸、ビタミン、ミネラル、少なくとも1種類のトランスフェリン又はトランスフェリン代替物、少なくとも1種類のインスリン又はインスリン代替物を含む培地中で幹細胞を培養する工程を含み、前記培地は、哺乳動物胎児血清を基本的に含まず、少なくとも約100ng/mlの、繊維芽細胞増殖因子シグナル伝達受容体を活性化することが可能な繊維芽細胞増殖因子を含み、該増殖因子は、単に繊維芽細胞フィーダー層以外の供給源から供給され、前記培地は、フィーダー細胞又は馴化培地の非存在下で未分化状態の幹細胞の増殖を支持するものである。
【0066】
別の例として、米国特許出願公開第20050233446号は、未分化の霊長類始原幹細胞等の幹細胞を培養するうえで有用な合成培地を開示している。溶液中では、培地は培養される幹細胞と比較して実質的に等張である。与えられた培養中、この特定の培地は基礎培地、並びに始原幹細胞の実質的に未分化状態での増殖を支持するうえで必要とされるbFGF、インスリン、及びアスコルビン酸のそれぞれの所定量を含む。
【0067】
別の例として、米国特許第6800480号は、「一実施形態において、実質的に未分化状態の霊長類由来の始原幹細胞を増殖させるための細胞培地であって、霊長類由来の始原幹細胞の増殖を支持するうえで効果的な低浸透圧、低エンドトキシンの基礎培地を含む細胞培地を提供する。この基礎培地は、霊長類由来の始原幹細胞の増殖を支持するうえで効果的な栄養素血清、並びに、フィーダー細胞、及びフィーダー細胞から誘導される細胞外基質成分からなる群から選択される基質と組み合わされる。培地は更に、非必須アミノ酸、抗酸化剤、並びに、ヌクレオシド及びピルビン酸塩からなる群から選択される第1の増殖因子を含む。」と述べている。
【0068】
別の例では、米国特許出願公開第20050244962号は、「一態様において本発明は、霊長類の胚性幹細胞を培養する方法を提供する。哺乳動物の胎児血清を基本的に含まない(好ましくは、あらゆる動物の血清をも基本的に含まない)培養中で、単に繊維芽フィーダー細胞層以外の供給源から供給される繊維芽細胞増殖因子の存在下で幹細胞を培養する。好ましい一形態では、充分な量の繊維芽増殖因子を添加することによって、幹細胞の培養を維持するために従来必要とされていた繊維芽フィーダー細胞層の必要性がなくなる」と述べている。
【0069】
更なる例として、国際特許出願公開第2005065354号は、基本的に無フィーダー細胞かつ無血清の合成等張培地であって、a.基礎培地、b.実質的に未分化の哺乳動物幹細胞の増殖を支持するうえで充分な量のbFGF、c.実質的に未分化の哺乳動物幹細胞の増殖を支持するうえで充分な量のインスリン、及び、d.実質的に未分化の哺乳動物幹細胞の増殖を支持するうえで充分な量のアスコルビン酸を含む培地を開示している。
【0070】
別の例として、国際特許出願公開第2005086845号は、未分化の幹細胞を維持するための方法であって、幹細胞を未分化状態に維持するのに充分な量の形質転換増殖因子β(transforming growth factor:TGFβ)のタンパク質ファミリーのメンバー、繊維芽細胞増殖因子(FGF)のタンパク質ファミリーのメンバー、又はニコチンアミド(NIC)に、所望の結果を得るのに充分な時間だけ、幹細胞を曝露することを含む方法を開示している。
【0071】
多能性幹細胞は、適当な培養基質上に播種すればよい。一実施形態では、適当な培養基質は例えば、基底膜から誘導されるもの、又は接着分子の受容体/リガンド結合の一部を形成するもののような細胞外基質成分である。一実施形態では、適当な培養基質はMATRIGEL(登録商標)(ベクトン・ディッキンソン社(Becton Dickenson))である。MATRIGEL(登録商標)は、室温でゲル化して再構成基底膜を形成する、エンゲルブレス−ホルム・スワーム(Engelbreth-Holm Swarm)腫瘍細胞から得られる可溶性調製物である。
【0072】
他の細胞外基質成分及び成分混合物も代替物として適している。増殖させられる細胞の種類に応じ、他の細胞外基質成分及び成分混合物は、ラミニン、フィブロネクチン、プロテオグリカン、エンタクチン、硫酸ヘパリン等を、単独又は異なる組み合わせで含んでもよい。
【0073】
多能性幹細胞は、適当な分布で、かつ細胞の生存率、増殖、及び所望の特性の維持を促進する培地の存在下で基質上に播種することができる。これらの特性はいずれも、播種密度に充分な注意を払うことによって利するところがあるものであり、当業者が容易に決定することができるものである。
【0074】
好適な培地は、以下の成分、即ち、例えば、ダルベッコ改変イーグル培地(DMEM)、ギブコ(Gibco)No.11965−092;ノックアウトダルベッコ改変イーグル培地(KO DMEM)、ギブコNo.10829−018;ハムF12/50%DMEM基礎培地;200mM L−グルタミン、ギブコNo.15039−027;非不可欠アミノ酸溶液、ギブコNo.11140−050;β−メルカプトエタノール、シグマ(Sigma)No.7522;ヒト組み換え塩基性繊維芽細胞増殖因子(bFGF)、ギブコNo.13256−029等から調製することができる。
【0075】
膵臓内分泌系統に特徴的なマーカーを発現する細胞への多能性幹細胞の分化
本発明における使用に適した多能性幹細胞としては例えば、胚性幹細胞系H9(NIHコード:WA09)、ヒト胚性幹細胞系H1(NIHコード:WA01)、ヒト胚性幹細胞系H7(NIHコード:WA07)、及びヒト胚性幹細胞系SA002(セラーティス社(Cellartis)、スウェーデン)が挙げられる。多能性細胞に特徴的な以下のマ−カー、即ち、ABCG2、クリプト(cripto)、CD9、FoxD3、コネキシン43、コネキシン45、Oct4、Sox2、ナノグ(Nanog)、hTERT、UTF−1、ZFP42、SSEA−3、SSEA−4、Tra1−60、Tra1−81の内の少なくとも1つを発現する細胞も本発明における使用に適している。
【0076】
胚体内胚葉系統に特徴的なマーカーは、SOX−17、GATA4、Hnf−3β、GSC、Cer1、ノーダル(Nodal)、FGF8、ブラキュリ(Brachyury)、Mix様ホメオボックスタンパク質、FGF4 CD48、エオメソダーミン(eomesodermin)(EOMES)、DKK4、FGF17、GATA6、CXCR4、C−Kit、CD99、及びOTX2からなる群から選択される。胚体内胚葉系統に特徴的なこれらのマーカーの内の少なくとも1つを発現する細胞が本発明における使用に適している。本発明の一態様では、胚体内胚葉系統に特徴的なマーカーを発現する細胞は、原始線条前駆細胞である。代替的な態様では、胚体内胚葉系統に特徴的なマーカーを発現する細胞は、中内胚葉細胞である。代替的な態様では、胚体内胚葉系統に特徴的なマーカーを発現する細胞は、胚体内胚葉細胞である。
【0077】
膵臓内胚葉系統に特徴的なマーカーは、Pdx1、HNF−1β、PTF1a、HNF−6、HB9及びPROX1からなる群から選択される。膵臓内胚葉系統に特徴的なこれらのマーカーの内の少なくとも1つを発現する細胞が本発明における使用に適している。本発明の一態様では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞は、膵臓内胚葉細胞である。
【0078】
膵臓内分泌系統に特徴的なマーカーは、NGN−3、ニューロD(NeuroD)、アイレット−1(Islet-1)、Pdx−1、NKX6.1、Pax−4、Ngn−3、及びPTF−1αからなる群から選択される。一実施形態では、膵臓内分泌細胞は、以下のホルモン、即ち、インスリン、グルカゴン、ソマトスタチン、及び膵臓ポリペプチドの内の少なくとも1つを発現することが可能である。膵臓内分泌系統に特徴的なこれらのマーカーの内の少なくとも1つを発現する細胞が本発明における使用に適している。本発明の一態様では、膵臓内分泌系統に特徴的なマーカーを発現する細胞は、膵臓内分泌細胞である。膵臓内分泌細胞は、膵臓ホルモン発現細胞であってもよい。また、膵臓内分泌細胞は膵臓ホルモン分泌細胞であってもよい。
【0079】
本発明の一態様では、膵臓内分泌細胞は、β細胞系統に特徴的なマーカーを発現する細胞である。β細胞系統に特徴的なマーカーを発現する細胞は、Pdx1、並びに以下の転写因子、即ち、NGN−3、Nkx2.2、Nkx6.1、ニューロD(NeuroD)、Isl−1、HNF−3β、MAFA、Pax4及びPax6の内の少なくとも1つを発現する。本発明の一態様では、β細胞系統に特徴的なマーカーを発現する細胞は、β細胞である。
【0080】
胚体内胚葉系統に特徴的なマーカーを発現する細胞の形成
多能性幹細胞を、当該技術分野のいかなる方法、又は本発明で提案されるいかなる方法によって胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させてもよい。
【0081】
例えば、多能性幹細胞を、ダムール(D’Amour)等により開示される方法に従って胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる(D’Amour et al, Nature Biotechnology 23, 1534 - 1541(2005))。
【0082】
例えば、多能性幹細胞を、シノザキ(Shinozaki)等により開示される方法に従って胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる(Shinozaki et al, Development 131, 1651 - 1662(2004))。
【0083】
例えば、多能性幹細胞を、マクレーン(McLean)等により開示される方法に従って胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる(McLean et al, Stem Cells 25, 29 - 38(2007))。
【0084】
例えば、多能性幹細胞を、ダムール(D’Amour)等により開示される方法に従って胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる(D’Amour et al, Nature Biotechnology 24, 1392 - 1401(2006))。
【0085】
例えば、多能性幹細胞を、アクチビンAを含む培地中、血清の非存在下で多能性幹細胞を培養し、次いで細胞をアクチビンA及び血清と培養し、次いで細胞をアクチビンA及び異なる濃度の血清と培養することによって胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる。この方法の一例は、(Nature Biotechnology 23, 1534 - 1541(2005))に開示されている。
【0086】
例えば、多能性幹細胞を、アクチビンAを含む培地中、血清の非存在下で多能性幹細胞を培養し、次いで細胞をアクチビンA及び血清と培養し、次いで細胞をアクチビンAと、別の濃度の血清の存在下で培養することによって胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる。この方法の一例は、ダムール(D’Amour)等により開示されている(D’Amour et al, Nature Biotechnology, 2005)。
【0087】
例えば、多能性幹細胞を、アクチビンA及びWntリガンドを含む培地中、血清の非存在下で多能性幹細胞を培養し、次いでWntリガンドを除去し、細胞をアクチビンAと、血清の存在下で培養することによって胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる。この方法の一例は、(Nature Biotechnology 24, 1392 - 1401(2006))に記載されている。
【0088】
本発明の一態様では、多能性幹細胞は、細胞外基質でコーティングした組織培養基質上に多能性幹細胞を播種し、次いでこの多能性幹細胞をアクチビンA及びWntリガンドと、血清を含む第1の培地中で所定の時間培養し、次いでこの多能性幹細胞をアクチビンAと、より高い濃度の血清を含む第2の培地中でおおよそ別の所定の時間培養することによって胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる。
【0089】
上記に開示される第1の培地中の血清の濃度は、約0〜約0.5%であってもよく、培養時間は約1〜約3日間であってもよい。上記に開示される第2の培地中の血清の濃度は、約0.5〜約2%であってもよく、培養時間は約1〜約4日間であってもよい。
【0090】
本発明の代替的な一態様では、多能性幹細胞は、細胞外基質でコーティングした組織培養基質上に多能性幹細胞を播種し、次いでこの多能性幹細胞をアクチビンA及びWntリガンドと、血清を含む第1の培地中でおおよそ所定の時間培養し、次いでこの多能性幹細胞をアクチビンA及びWntリガンドと、より高い濃度の血清を含む第2の培地中で別の所定の時間培養することによって胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる。
【0091】
上記に開示される第1の培地中の血清の濃度は、約0〜約0.5%であってもよく、培養時間は約1〜約3日間であってもよい。上記に開示される第2の培地中の血清の濃度は、約0.5〜約2%であってもよく、培養時間は約1〜約4日間であってもよい。
【0092】
一実施形態では、本発明は、胚体内胚葉系統に特徴的なマーカーを発現する多能性幹細胞を分化させる方法であって、
a.細胞外基質でコーティングされた組織培養基質上に多能性幹細胞を播種する工程と、
b.前記多能性幹細胞を、アクチビンA及びWntリガンドと培養する工程と、を含む、方法を提供する。
【0093】
多能性幹細胞をアクチビンA及びWntリガンドと培養する工程は、1個の培地中で行うことが可能である。また、多能性幹細胞をアクチビンA及びWntリガンドと培養する工程は、1個よりも多い培地中で別々に又は同時に行うことも可能である。一実施形態では、多能性幹細胞をアクチビンA及びWntリガンドと培養する工程を2個の培地中で行う。
【0094】
細胞外基質
本発明の一態様では、多能性細胞を、細胞外基質でコーティングされた組織培養基質上で培養して分化させる。細胞外基質は、マウスの肉腫細胞から抽出された可溶化基底膜の調製物(BDバイオサイエンス社(BD Biosciences)よりMATRIGELの商品名で販売されている)であってよい。また、細胞外基質は、増殖因子を低減させたMATRIGELであってもよい。また、細胞外基質はフィブロネクチンであってもよい。代替的な一実施形態では、多能性幹細胞を、ヒト血清でコーティングされた組織培養基質上で培養して分化させる。
【0095】
細胞外基質は、組織培養基質をコーティングするのに先立って希釈してもよい。組織培養基質をコーティングするための細胞外基質を希釈するのに適した方法の例は、クラインマン(Kleinman)等(Kleinman, H.K., et al., Biochemistry 25:312(1986))、及びハドレイ(Hadley)等(Hadley, M.A., et al., J.Cell.Biol. 101:1511(1985))に見ることができる。
【0096】
一実施形態では、細胞外基質はMATRIGELである。一実施形態では、組織培養基質を希釈率が1:10のMATRIGELでコーティングする。代替的な一実施形態では、組織培養基質を希釈率が1:15のMATRIGELでコーティングする。代替的な一実施形態では、組織培養基質を希釈率が1:30のMATRIGELでコーティングする。代替的な一実施形態では、組織培養基質を希釈率が1:60のMATRIGELでコーティングする。
【0097】
一実施形態では、細胞外基質は増殖因子低減MATRIGELである。一実施形態では、組織培養基質を希釈率が1:10の増殖因子低減MATRIGELでコーティングする。代替的な一実施形態では、組織培養基質を希釈率が1:15の増殖因子低減MATRIGELでコーティングする。代替的な一実施形態では、組織培養基質を希釈率が1:30の増殖因子低減MATRIGELでコーティングする。代替的な一実施形態では、組織培養基質を希釈率が1:60の増殖因子低減MATRIGELでコーティングする。
【0098】
1個の培地を使用した、細胞外基質上での、胚体内胚葉系統に特徴的なマーカーを発現する細胞への多能性幹細胞の分化
1個の培地を使用する場合、培地は、例えば(国際特許出願公開第2006020919号に開示されるような)インスリン及びIGF等の特定の因子を、多能性幹細胞が胚体内胚葉に分化できるように充分に低い濃度で含有しなければならない。これは血清濃度を低下させるか、あるいはインスリン及びIGFを含まない合成培地を使用することによって実現することが可能である。合成培地の例はワイルズ(Wiles)等によって開示されている(Wiles et al. Exp Cell Res. 1999 Feb 25; 247(1): 241-8)。
【0099】
前記培地は、約0%〜約10%の範囲の血清濃度を有してもよい。代替的な一実施形態では、濃度は約0%〜約5%の範囲であってもよい。代替的な一実施形態では、濃度は約0%〜約2%の範囲であってもよい。代替的な一実施形態では、濃度は約2%であってもよい。
【0100】
アクチビンA及びWntリガンとの培養時間は約1日〜約7日であってもよい。代替的な一実施形態では、培養時間は約1日〜約3日であってもよい。代替的な一実施形態では、培養時間は約3日であってもよい。
【0101】
アクチビンAは、多能性幹細胞の分化を引き起こすのに適した任意の濃度で使用することができる。濃度は約1pg/ml〜約100μg/mlであってもよい。代替的な一実施形態では、濃度は約1pg/ml〜約1μg/mlであってもよい。別の代替的な一実施形態では、濃度は約1pg/ml〜約100ng/mlであってもよい。別の代替的な一実施形態では、濃度は約50ng/ml〜約100ng/mlであってもよい。別の代替的な一実施形態では、濃度は約100ng/mlであってもよい。
【0102】
Wntリガンドの選択を最適化することによって分化プロセスの効率を高めることが可能である。Wntリガンドは、Wnt−1、Wnt−3a、Wnt−5a及びWnt−7aからなる群から選択することができる。一実施形態では、WntリガンドはWnt−1である。代替的な一実施形態では、WntリガンドはWnt−3aである。
【0103】
Wntリガンドは約1ng/ml〜約1000ng/mlの濃度でもよい。代替的な一実施形態では、濃度は約10ng/ml〜約100ng/mlであってもよい。
【0104】
前記1個の培地は、GSK−3B阻害剤を更に含んでもよい。GSK−3B阻害剤は、GSK−3B阻害剤IX及びGSK−3B阻害剤XIからなる群から選択することができる。一実施形態では、GSK−3B阻害剤はGSK−3B阻害剤IXである。
【0105】
多能性幹細胞をGSK−3B阻害剤と培養する場合、GSK−3B阻害剤の濃度は約1nM〜約1000nMでもよい。代替的な一実施形態では、多能性幹細胞を約10nM〜約100nMの濃度のGSK−3B阻害剤と培養する。
【0106】
前記1個の培地は、胚体内胚葉系統に特徴的なマーカーを発現する細胞の多能性細胞からの形成を促進することが可能な少なくとも1種類の更なる他の因子を含んでもよい。また、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される胚体内胚葉に特徴的なマーカーを発現する細胞の増殖を促進するものであってもよい。更に、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される胚体内胚葉系統に特徴的なマーカーを発現する細胞が、他の細胞種を形成する能力を促進するものであってもよく、あるいは他のいずれかの更なる分化の段階の効率を高めるものであってもよい。
【0107】
前記少なくとも1種類の更なる他の因子は、例えば、ニコチンアミド、TGF−β1、2及び3等のTGFβファミリーのメンバー、血清アルブミン、繊維芽細胞増殖因子ファミリーのメンバー、血小板由来増殖因子−AA及び−BB、血小板濃縮血漿、インスリン様増殖因子(IGF−I、II)、増殖分化因子(GDF−5、−6、−8、−10、−11)、グルカゴン様ペプチド−1及び−II(GLP−1及びII)、GLP−1及びGLP−2模倣体(mimetobody)、エキセンジン−4、レチノイン酸、副甲状腺ホルモン、インスリン、プロゲステロン、アプロチニン、ヒドロコルチゾン、エタノールアミン、βメルカプトエタノール、上皮増殖因子(EGF)、ガストリンI及びII、例えば、トリエチレンペンタミン等の銅キレート化剤、フォルスコリン、酪酸ナトリウム、アクチビン、ベータセルリン、ITS、ノギン、神経突起増殖因子、ノーダル、バルプロ酸、トリコスタチンA、酪酸ナトリウム、肝細胞増殖因子(HGF)、スフィンゴシン1、VEGF、MG132(EMD社、カリフォルニア州)、N2及びB27添加物(ギブコ社(Gibco)、カリフォルニア州)、例えば、シクロパミン(EMD社、カリフォルニア州)等のステロイドアルカロイド、ケラチノサイト増殖因子(KGF)、Dickkopfタンパク質ファミリー、ウシ脳下垂体抽出物、膵島新生関連タンパク質(INGAP)、インディアンヘッジホッグ、ソニックヘッジホッグ、プロテアソーム阻害剤、ノッチ経路阻害剤、ソニックヘッジホッグ阻害剤、又はこれらの組み合わせであってもよい。
【0108】
前記少なくとも1種類の更なる他の因子は、例えば、PANC−1(ATCC No:CRL−1469)、CAPAN−1(ATCC No:HTB−79)、BxPC−3(ATCC No:CRL−1687)、HPAF−II(ATCC No:CRL−1997)等の膵臓細胞系、例えば、HepG2(ATCC No:HTB−8065)等の肝細胞系、例えば、FHs74(ATCC No:CCL−241)等の腸細胞系、及び始原又は形質転換内皮細胞から得られる馴化培地によって供給することが可能である。
【0109】
2個の培地を使用した、細胞外基質上での、胚体内胚葉系統に特徴的なマーカーを発現する細胞への多能性幹細胞の分化
多能性幹細胞の胚体内胚葉系統細胞への分化は、2個の培地を使用して多能性幹細胞をアクチビンA及びWntリガンドと培養することによって実現することが可能である。即ち、多能性幹細胞の分化は以下のようにして実現することが可能である。
a.細胞外基質でコーティングされた組織培養基質上に多能性幹細胞を播種し、
b.前記多能性幹細胞を第1の培地中でアクチビンA及びWntリガンドと培養し、
c.前記多能性幹細胞を第2の培地中でアクチビンAと培養する。
【0110】
第1の培地は低濃度の血清を含んでもよく、第2の培地は第1の培地よりも高濃度の血清を含んでもよい。
【0111】
第2の培地はWntリガンドを含んでもよい。
【0112】
第1の培地:第1の培地は、例えば(国際特許出願公開第2006020919号に開示されるような)インスリン及びIGF等の特定の因子を、多能性幹細胞が胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化できるように充分に低い濃度で含有しなければならない。これは血清濃度を低下させるか、あるいはインスリン及びIGFを含まない合成培地を使用することによって実現することが可能である。合成培地の例はワイルズ(Wiles)等によって開示されている(Wiles et al. Exp Cell Res. 1999 Feb 25; 247(1): 241-8)。
【0113】
第1の培地では、第2の培地と比較してより低濃度の血清が存在してもよい。第2の培地の血清濃度を増大させると細胞の生存率が高まるか、あるいは細胞の増殖が促進される場合がある。第1の培地の血清濃度は約0%〜約10%の範囲であってもよい。また、第1の培地の血清濃度は約0%〜約2%の範囲であってもよい。また、第1の培地の血清濃度は約0%〜約1%の範囲であってもよい。また、第1の培地の血清濃度は約0.5%であってもよい。
【0114】
少なくとも2個の培地を使用して多能性幹細胞をアクチビンA及びWntリガンドと培養する場合、第1の培地中での培養時間は約1日〜約3日であってもよい。
【0115】
アクチビンAは、多能性幹細胞の分化を引き起こすのに適した任意の濃度で使用することができる。濃度は約1pg/ml〜約100μg/mlであってもよい。代替的な一実施形態では、濃度は約1pg/ml〜約1μg/mlであってもよい。別の代替的な一実施形態では、濃度は約1pg/ml〜約100ng/mlであってもよい。別の代替的な一実施形態では、濃度は約50ng/ml〜約100ng/mlであってもよい。別の代替的な一実施形態では、濃度は約100ng/mlであってもよい。
【0116】
Wntリガンドの選択を最適化することによって分化プロセスの効率を高めることが可能である。Wntリガンドを、Wnt−1、Wnt−3a、Wnt−5a及びWnt−7aからなる群から選択することができる。一実施形態では、WntリガンドはWnt−1である。代替的な一実施形態では、WntリガンドはWnt−3aである。
【0117】
Wntリガンドは約1ng/ml〜約1000ng/mlの濃度でもよい。代替的な一実施形態では、濃度は約10ng/ml〜約100ng/mlであってもよい。
【0118】
前記第1の培地は、GSK−3B阻害剤を更に含んでもよい。GSK−3B阻害剤は、第1の培地、第2の培地、又は第1及び第2の培地の両方に添加することができる。
【0119】
GSK−3B阻害剤は、GSK−3B阻害剤IX及びGSK−3B阻害剤XIからなる群から選択することができる。一実施形態では、GSK−3B阻害剤はGSK−3B阻害剤IXである。
【0120】
多能性幹細胞をGSK−3B阻害剤と培養する場合、GSK−3B阻害剤の濃度は約1nM〜約1000nMでもよい。代替的な一実施形態では、多能性幹細胞を約10nM〜約100nMの濃度のGSK−3B阻害剤と培養する。
【0121】
前記第1の培地は、胚体内胚葉系統に特徴的なマーカーを発現する細胞の多能性細胞からの形成を促進することが可能な少なくとも1種類の更なる他の因子を含んでもよい。また、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される胚体内胚葉に特徴的なマーカーを発現する細胞の増殖を促進するものであってもよい。更に、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される胚体内胚葉系統に特徴的なマーカーを発現する細胞が他の細胞種を形成する能力を促進するものであってもよく、あるいは他のいずれかの更なる分化の段階の効率を高めるものであってもよい。
【0122】
前記少なくとも1種類の更なる他の因子は、例えば、ニコチンアミド、TGF−β1、2及び3等のTGFβファミリーのメンバー、血清アルブミン、繊維芽細胞増殖因子ファミリーのメンバー、血小板由来増殖因子−AA及び−BB、血小板濃縮血漿、インスリン様増殖因子(IGF−I、II)、増殖分化因子(GDF−5、−6、−8、−10、−11)、グルカゴン様ペプチド−1及び−II(GLP−1及びII)、GLP−1及びGLP−2模倣体(mimetobody)、エキセンジン−4、レチノイン酸、副甲状腺ホルモン、インスリン、プロゲステロン、アプロチニン、ヒドロコルチゾン、エタノールアミン、βメルカプトエタノール、上皮増殖因子(EGF)、ガストリンI及びII、例えば、トリエチレンペンタミン等の銅キレート化剤、フォルスコリン、酪酸ナトリウム、アクチビン、ベータセルリン、ITS、ノギン、神経突起増殖因子、ノーダル、バルプロ酸、トリコスタチンA、酪酸ナトリウム、肝細胞増殖因子(HGF)、スフィンゴシン1、VEGF、MG132(EMD社、カリフォルニア州)、N2及びB27添加物(ギブコ社(Gibco)、カリフォルニア州)、例えば、シクロパミン(EMD社、カリフォルニア州)等のステロイドアルカロイド、ケラチノサイト増殖因子(KGF)、Dickkopfタンパク質ファミリー、ウシ脳下垂体抽出物、膵島新生関連タンパク質(INGAP)、インディアンヘッジホッグ、ソニックヘッジホッグ、プロテアソーム阻害剤、ノッチ経路阻害剤、ソニックヘッジホッグ阻害剤、又はこれらの組み合わせであってもよい。
【0123】
前記少なくとも1種類の更なる他の因子は、例えば、PANC−1(ATCC No:CRL−1469)、CAPAN−1(ATCC No:HTB−79)、BxPC−3(ATCC No:CRL−1687)、HPAF−II(ATCC No:CRL−1997)等の膵臓細胞系、例えばHepG2(ATCC No:HTB−8065)等の肝細胞系、及び例えば、FHs74(ATCC No:CCL−241)等の腸細胞系から得られる馴化培地によって供給することが可能である。
【0124】
第2の培地:第2の培地は、例えば、(国際特許出願公開第2006020919号に開示されるような)インスリン及びIGF等の特定の因子を培養細胞の生存を促すだけの充分な濃度で含有しなければならない。これは、血清濃度を増大させるか、あるいはインスリン及びIGFの濃度が第1の培地と比較して高い合成培地を使用することによって実現することが可能である。合成培地の例はワイルズ(Wiles)等によって開示されている(Wiles et al. Exp Cell Res. 1999 Feb 25; 247(1): 241-8)。
【0125】
より高い血清濃度を有する第2の培地では、第2の培地の血清濃度は、約0.5%〜約10%の範囲であってもよい。また、第2の培地の血清濃度は約0.5%〜約5%の範囲であってもよい。また、第2の培地の血清濃度は約0.5%〜約2%の範囲であってもよい。また、第2の培地の血清濃度は約2%であってもよい。多能性幹細胞を第2の培地と培養する場合、培養時間は約1日〜約4日であってもよい。
【0126】
第1の培地と同様、アクチビンAは、多能性幹細胞の分化を引き起こすのに適した任意の濃度で使用することができる。濃度は約1pg/ml〜約100μg/mlであってもよい。代替的な一実施形態では、濃度は約1pg/ml〜約1μg/mlであってもよい。別の代替的な一実施形態では、濃度は約1pg/ml〜約100ng/mlであってもよい。別の代替的な一実施形態では、濃度は約50ng/ml〜約100ng/mlであってもよい。別の代替的な一実施形態では、濃度は約100ng/mlであってもよい。
【0127】
Wntリガンドは約1ng/ml〜約1000ng/mlの濃度でもよい。代替的な一実施形態では、濃度は約10ng/ml〜約100ng/mlであってもよい。
【0128】
Wntリガンドは、Wnt−1、Wnt−3a、Wnt−5a及びWnt−7aからなる群から選択することができる。一実施形態では、WntリガンドはWnt−1である。代替的な一実施形態では、WntリガンドはWnt−3aである。
【0129】
前記第2の培地は、GSK−3B阻害剤を更に含んでもよい。GSK−3B阻害剤は、第1の培地、第2の培地、又は第1及び第2の培地の両方に添加することができる。
【0130】
GSK−3B阻害剤は、GSK−3B阻害剤IX及びGSK−3B阻害剤XIからなる群から選択することができる。一実施形態では、GSK−3B阻害剤はGSK−3B阻害剤IXである。
【0131】
多能性幹細胞をGSK−3B阻害剤と培養する場合、GSK−3B阻害剤の濃度は約1nM〜約1000nMでもよい。代替的な一実施形態では、多能性幹細胞を約10nM〜約100nMの濃度のGSK−3B阻害剤と培養する。
【0132】
第1の培地と同様、第2の培地も、胚体内胚葉系統に特徴的なマーカーを発現する細胞の多能性細胞からの形成を促進することが可能な少なくとも1種類の更なる他の因子を含んでもよい。また、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される胚体内胚葉に特徴的なマーカーを発現する細胞の増殖を促進するものであってもよい。更に、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される胚体内胚葉系統に特徴的なマーカーを発現する細胞が他の細胞種を形成する能力を促進するものであってもよく、あるいは他のいずれかの更なる分化の段階の効率を高めるものであってもよい。
【0133】
前記少なくとも1種類の更なる他の因子は、例えば、ニコチンアミド、TGF−β1、2及び3等のTGFβファミリーのメンバー、血清アルブミン、繊維芽細胞増殖因子ファミリーのメンバー、血小板由来増殖因子−AA及び−BB、血小板濃縮血漿、インスリン様増殖因子(IGF−I、II)、増殖分化因子(GDF−5、−6、−8、−10、−11)、グルカゴン様ペプチド−1及び−II(GLP−1及びII)、GLP−1及びGLP−2模倣体(mimetobody)、エキセンジン−4、レチノイン酸、副甲状腺ホルモン、インスリン、プロゲステロン、アプロチニン、ヒドロコルチゾン、エタノールアミン、βメルカプトエタノール、上皮増殖因子(EGF)、ガストリンI及びII、例えば、トリエチレンペンタミン等の銅キレート化剤、フォルスコリン、酪酸ナトリウム、アクチビン、ベータセルリン、ITS、ノギン、神経突起増殖因子、ノーダル、バルプロ酸、トリコスタチンA、酪酸ナトリウム、肝細胞増殖因子(HGF)、スフィンゴシン1、VEGF、MG132(EMD社、カリフォルニア州)、N2及びB27添加物(ギブコ社(Gibco)、カリフォルニア州)、例えば、シクロパミン(EMD社、カリフォルニア州)等のステロイドアルカロイド、ケラチノサイト増殖因子(KGF)、Dickkopfタンパク質ファミリー、ウシ脳下垂体抽出物、膵島新生関連タンパク質(INGAP)、インディアンヘッジホッグ、ソニックヘッジホッグ、プロテアソーム阻害剤、ノッチ経路阻害剤、ソニックヘッジホッグ阻害剤、又はこれらの組み合わせであってもよい。
【0134】
前記少なくとも1種類の更なる他の因子を、例えば、PANC−1(ATCC No:CRL−1469)、CAPAN−1(ATCC No:HTB−79)、BxPC−3(ATCC No:CRL−1687)、HPAF−II(ATCC No:CRL−1997)等の膵臓細胞系、例えばHepG2(ATCC No:HTB−8065)等の肝細胞系、及び例えば、FHs74(ATCC No:CCL−241)等の腸細胞系から得られる馴化培地によって供給することが可能である。
【0135】
胚体内胚葉系統に特徴的なマーカーを発現する細胞の分化
胚体内胚葉系統に特徴的なマーカーを発現する細胞が形成されたことは、特定のプロトコールを行う前後にマーカーの存在について試験を行うことによって判定することができる。多能性幹細胞は通常、こうしたマーカーを発現しない。したがって、多能性細胞の分化は、細胞がマーカーを発現し始めることで検出される。
【0136】
分化の効率は、胚体内胚葉系統に特徴的なマーカーを発現する細胞によって発現されるタンパク質マーカーを特異的に認識する薬剤(抗体等)に、処理した細胞集団を曝露することによって決定することができる。
【0137】
培養細胞又は単離細胞中のタンパク質及び核酸マーカーの発現を評価するための方法は、当該技術分野では標準的なものである。こうした方法には、定量的逆転写ポリメラーゼ連鎖反応(RT−PCR)、ノーザンブロット、in situハイブリダイゼーション(例えば、「分子生物学の最新プロトコール」(Current Protocols in Molecular Biology)オウスベル(Ausubel)等編、2001年度版補遺、を参照)、並びに、切片化材料の免疫組織化学的分析、ウエスタンブロッティング、及び無傷細胞中のアクセシブルなマーカーに対する免疫アッセイ、フローサイトメトリー分析(FACS)(例えば、ハーロー及びレーン(Harlow and Lane)著、「抗体の使用:実験マニュアル」(Using Antibodies: A Laboratory Manual)ニューヨーク州コールドスプリングハーバーラボラトリープレス(1998年)を参照)等の免疫アッセイが含まれる。
【0138】
特定のタンパク質マーカーの検出に有用な抗体の例を表IAに列記する。表IAに列記した抗体によって認識されるものと同じマーカーを標的とする代替的な抗体も入手可能であるか、容易に開発が可能である点は注意を要する。こうした代替的な抗体を、本発明に基づいて単離した細胞におけるマーカーの発現を評価するために用いることもできる。
【0139】
例えば、多能性幹細胞の特徴は当業者にはよく知られたものであり、多能性幹細胞の更なる特徴も次々に特定されている。多能性幹細胞のマーカーとしては、例えば、以下のもの、即ち、ABCG2、クリプト(cripto)、FoxD3、コネキシン43、コネキシン45、Oct4、Sox2、ナノグ(nanog)、hTERT、UTF−1、ZFP42、SSEA−3、SSEA−4、Tra1−60、Tra1−81の内の1つ以上の発現が挙げられる。
【0140】
多能性幹細胞を本発明の方法で処理した後、胚体内胚葉系統に特徴的なマーカーを発現する細胞によって発現されるCXCR4のようなタンパク質マーカーを特異的に認識する薬剤(抗体等)に処理細胞集団を曝露することによって、分化した細胞を精製することができる。
【0141】
膵臓内胚葉系統に特徴的なマーカーを発現する細胞の形成
胚体内胚葉系統に特徴的なマーカーを発現する細胞を、当該技術分野におけるいずれかの方法又は本発明において提案されるいずれかの方法によって膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる。
【0142】
例えば、胚体内胚葉系統に特徴的なマーカーを発現する細胞を、ダムール(D’Amour)等により開示される方法に従って膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる(D’Amour et al, Nature Biotechnology 24, 1392 - 1401(2006))。
【0143】
例えば、胚体内胚葉系統に特徴的なマーカーを発現する細胞を繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤KAAD−シクロパミンで処理し、次いで繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤KAAD−シクロパミンを含む培地を取り除いた後、レチノイン酸、繊維芽細胞増殖因子及びKAAD−シクロパミンを含む培地中で細胞を培養することによって、胚体内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内胚葉系統に特徴的なマーカーを発現する細胞に更に分化させる。この方法の一例は、(Nature Biotechnology 24, 1392 - 1401(2006))に開示されている。
【0144】
本発明の一態様では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で所定の時間処理することによって、胚体内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内胚葉系統に特徴的なマーカーを発現する細胞に更に分化させる。その時間は、約1〜約6日でもよい。
【0145】
本発明の代替的一態様では、胚体内胚葉系統に特徴的なマーカーを発現する細胞を、細胞をレチノイン酸で所定の時間処理することによって膵臓内胚葉系統に特徴的なマーカーを発現する細胞に更に分化させる。その時間は、約1〜約3日でもよい。レチノイン酸を後で取り除き、細胞を少なくとも1種類の繊維芽細胞増殖因子で別の所定の時間処理する。その時間は、約1〜約3日でもよい。
【0146】
一実施形態において、本発明は、胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させるための方法であって、
a.胚体内胚葉系統に特徴的なマーカーを発現する細胞を培養する工程と、
b.胚体内胚葉系統に特徴的なマーカーを発現する前記細胞をレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で処理する工程と、を含む、方法を提供する。
【0147】
胚体内胚葉系統に特徴的なマーカーを発現するいずれの細胞も、この方法を用いて膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させるうえで適している。
【0148】
一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で約1〜約6日間処理する。一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で約6日間処理する。
【0149】
前記少なくとも1種類の繊維芽細胞増殖因子は、FGF−2、FGF−4及びFGF−10からなる群から選択される。
【0150】
胚体内胚葉系統に特徴的なマーカーを発現するいずれの細胞も、この方法を用いて膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させるうえで適している。
【0151】
代替的な一実施形態において、本発明は、胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させるための方法であって、
a.胚体内胚葉系統に特徴的なマーカーを発現する細胞を培養する工程と、
b.胚体内胚葉系統に特徴的なマーカーを発現する前記細胞をレチノイン酸で処理する工程と、
c.レチノイン酸を取り除き、その後、細胞を少なくとも1種類の繊維芽細胞増殖因子で処理する工程と、を含む、方法を提供する。
【0152】
胚体内胚葉系統に特徴的なマーカーを発現するいずれの細胞も、この方法を用いて膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させるうえで適している。
【0153】
一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸で約1〜約3日間処理する。一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸で約3日間処理する。一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞を少なくとも1種類の繊維芽細胞増殖因子で約1〜約3日間処理する。一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞を少なくとも1種類の繊維芽細胞増殖因子で約3日間処理する。
【0154】
前記少なくとも1種類の繊維芽細胞増殖因子は、FGF−2、FGF−4及びFGF−10からなる群から選択される。
【0155】
胚体内胚葉系統に特徴的なマーカーを発現するいずれの細胞も、この方法を用いて膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させるうえで適している。一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸で処理する。あるいは、胚体内胚葉系統に特徴的なマーカーを発現する細胞をFGF−2、又はFGF−4、又はFGF−10で処理する。代替的な一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞を以下の因子、即ち、レチノイン酸、FGF−2、FGF−4、又はFGF−10の内の少なくとも1つで処理する。代替的な一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞を、レチノイン酸と、以下の繊維芽細胞増殖因子、即ち、FGF−2、FGF−4、又はFGF−10の内の少なくとも1つと、で処理する。一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸及びFGF−2で処理する。別の実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸及びFGF−4で処理する。更なる実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸及びFGF−10で処理する。
【0156】
レチノイン酸を約1nM〜約1mMの濃度で使用することができる。一実施形態では、レチノイン酸を1μMの濃度で使用する。
【0157】
FGF−2は約50pg/ml〜約50μg/mlの濃度で使用することができる。一実施形態では、FGF−2を50ng/mlの濃度で使用する。
【0158】
FGF−4は約50pg/ml〜約50μg/mlの濃度で使用することができる。一実施形態では、FGF−4を50ng/mlの濃度で使用する。
【0159】
FGF−10は約50pg/ml〜約50μg/mlの濃度で使用することができる。一実施形態では、FGF−10を50ng/mlの濃度で使用する。
【0160】
胚体内胚葉系統に特徴的なマーカーを発現する細胞は、膵臓内胚葉系統に特徴的なマーカーを発現する細胞の形成を促進することが可能な少なくとも1種類の更なる他の因子で処理してもよい。また、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される膵臓内胚葉系統に特徴的なマーカーを発現する細胞の増殖を促進するものであってもよい。更に、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される膵臓内胚葉系統に特徴的なマーカーを発現する細胞が、他の細胞種を形成する能力を促進するものであってもよく、あるいは他のいずれかの更なる分化の段階の効率を高めるものであってもよい。
【0161】
前記少なくとも1種類の更なる他の因子は、例えば、ニコチンアミド、TGF−β1、2及び3等のTGFβファミリーのメンバー、血清アルブミン、繊維芽細胞増殖因子ファミリーのメンバー、血小板由来増殖因子−AA及び−BB、血小板濃縮血漿、インスリン様増殖因子(IGF−I、II)、増殖分化因子(GDF−5、−6、−8、−10、11)、グルカゴン様ペプチド−1及びII(GLP−1及びII)、GLP−1及びGLP−2模倣体(mimetobody)、エキセンジン−4、レチノイン酸、副甲状腺ホルモン、インスリン、プロゲステロン、アプロチニン、ヒドロコルチゾン、エタノールアミン、βメルカプトエタノール、上皮増殖因子(EGF)、ガストリンI及びII、例えば、トリエチレンペンタミン等の銅キレート化剤、フォルスコリン、酪酸ナトリウム、アクチビン、ベータセルリン、ITS、ノギン、神経突起増殖因子、ノーダル、バルプロ酸、トリコスタチンA、酪酸ナトリウム、肝細胞増殖因子(HGF)、スフィンゴシン1、VEGF、MG132(EMD社、カリフォルニア州)、N2及びB27添加物(ギブコ社(Gibco)、カリフォルニア州)、例えばシクロパミン(EMD社、カリフォルニア州)等のステロイドアルカロイド、ケラチノサイト増殖因子(KGF)、Dickkopfタンパク質ファミリー、ウシ脳下垂体抽出物、膵島新生関連タンパク質(INGAP)、インディアンヘッジホッグ、ソニックヘッジホッグ、プロテアソーム阻害剤、ノッチ経路阻害剤、ソニックヘッジホッグ阻害剤、又はこれらの組み合わせであってもよい。
【0162】
前記少なくとも1種類の更なる他の因子は、例えばPANC−1(ATCC No:CRL−1469)、CAPAN−1(ATCC No:HTB−79)、BxPC−3(ATCC No:CRL−1687)、HPAF−II(ATCC No:CRL−1997)等の膵臓細胞系、例えばHepG2(ATCC No:HTB−8065)等の肝細胞系、及び例えば、FHs74(ATCC No:CCL−241)等の腸細胞系から得られる馴化培地によって供給することが可能である。
【0163】
膵臓内胚葉系統に特徴的なマーカーを発現する細胞の検出
膵臓内胚葉系統に特徴的なマーカーは当業者にはよく知られたものであり、膵臓内胚葉系統に特徴的な更なるマーカーも次々に特定されている。これらのマーカーを用いて、本発明に基づいて処理を行った細胞が、膵臓内胚葉系統に特徴的な性質を獲得するように分化したことを確認することが可能である。膵臓内胚葉系統に特異的なマーカーとしては、例えば、Hlxb9、PTF−1a、PDX−1、HNF−6、HNF−1β等、1以上の転写因子の発現が挙げられる。
【0164】
分化の効率は、膵臓内胚葉系統に特徴的なマーカーを発現する細胞によって発現されるタンパク質マーカーを特異的に認識する薬剤(抗体等)に、処理した細胞集団を曝露することによって決定することができる。
【0165】
培養細胞又は単離細胞中のタンパク質及び核酸マーカーの発現を評価するための方法は、当該技術分野では標準的なものである。こうした方法には、定量的逆転写ポリメラーゼ連鎖反応(RT−PCR)、ノーザンブロット、in situハイブリダイゼーション(例えば、「分子生物学の最新プロトコール」(Current Protocols in Molecular Biology)オウスベル(Ausubel)等編、2001年度版補遺、を参照)、並びに、切片化材料の免疫組織化学的分析、ウエスタンブロッティング、及び無傷細胞中のアクセシブルなマーカーに対する免疫アッセイ、フローサイトメトリー分析(FACS)(例えば、ハーロー及びレーン(Harlow and Lane)著、「抗体の使用:実験マニュアル」(Using Antibodies: A Laboratory Manual)ニューヨーク州コールドスプリングハーバーラボラトリープレス(1998年)を参照)等の免疫アッセイが含まれる。
【0166】
特定のタンパク質マーカーの検出に有用な抗体の例を表IAに列記する。表IAに列記した抗体によって認識されるものと同じマーカーを標的とする代替的な抗体も入手可能であるか、容易に開発が可能である点は注意を要する。こうした代替的な抗体を、本発明に基づいて単離した細胞におけるマーカーの発現を評価するために用いることもできる。
【0167】
膵臓内分泌系統に特徴的なマーカーを発現する細胞の形成
膵臓内胚葉系統に特徴的なマーカーを発現する細胞は、当該技術分野におけるいずれかの方法又は本発明において開示されるいずれかの方法によって膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させることができる。
【0168】
例えば、膵臓内胚葉系統に特徴的なマーカーを発現する細胞は、ダムール(D’Amour)等により開示される方法に従って膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させることができる(D’Amour et al, Nature Biotechnology 24, 1392 - 1401(2006))。
【0169】
例えば、膵臓内胚葉系統に特徴的なマーカーを発現する細胞をDAPT及びエキセンジン4を含む培地中で培養し、次いでDAPT及びエキセンジン4を含む培地を取り除いた後、エキセンジン1、IGF−1及びHGFを含む培地中で細胞を培養することによって、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に更に分化させる。この方法の一例は、(Nature Biotechnology 24, 1392 - 1401(2006))に記載されている。
【0170】
例えば、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、エキセンジン4を含む培地中で培養し、次いでエキセンジン4を含む培地を取り除いた後、エキセンジン1、IGF−1及びHGFを含む培地中で細胞を培養することによって、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に更に分化させる。この方法の一例は、ダムール(D’Amour)等により開示されている(D’Amour et al, Nature Biotechnology, 2006)。
【0171】
例えば、膵臓内胚葉系統に特徴的なマーカーを発現する細胞をDAPT及びエキセンジン4を含む培地中で培養することによって、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に更に分化させる。この方法の一例は、ダムール(D’Amour)等により開示されている(D’Amour et al, Nature Biotechnology, 2006)。
【0172】
例えば、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、エキセンジン4を含む培地中で培養することによって、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に更に分化させる。この方法の一例は、ダムール(D’Amour)等により開示されている(D’Amour et al, Nature Biotechnology, 2006)。
【0173】
本発明の一態様では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、ノッチシグナル伝達経路を阻害する因子で処理することによって、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に更に分化させる。ノッチシグナル伝達経路を阻害する因子は、ノッチ細胞外受容体のアンタゴニストであってもよい。また、この因子は、ノッチ受容体の生物活性を阻害するものであってもよい。また、この因子は、細胞内におけるノッチシグナル伝達経路を阻害するか、ノッチシグナル伝達経路内の特定の要素のアンタゴニストであってもよい。
【0174】
一実施形態では、ノッチシグナル伝達経路を阻害する因子は、γ−セクレターゼ阻害剤である。一実施形態では、γ−セクレターゼ阻害剤は、L−685,458としても知られる、1S−ベンジル−4R−[1−(1S−カルバモイル−2−フェネチルカルバモイル)−1S−3−メチルブチルカルバモイル]−2R−ヒドロキシ−5−フェネチルペンチル]カルバミン酸tert−ブチルエステルである。
【0175】
L−685,458は、約0.1μM〜約100μMの濃度で使用することができる。一実施形態では、L−685,458を約90μMの濃度で使用する。一実施形態では、L−685,458を約80μMの濃度で使用する。一実施形態では、L−685,458を約70μMの濃度で使用する。一実施形態では、L−685,458を約60μMの濃度で使用する。一実施形態では、L−685,458を約50μMの濃度で使用する。一実施形態では、L−685,458を約40μMの濃度で使用する。一実施形態では、L−685,458を約30μMの濃度で使用する。一実施形態では、L−685,458を約20μMの濃度で使用する。一実施形態では、L−685,458を約10μMの濃度で使用する。
【0176】
一実施形態において、本発明は、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させるための方法であって、
a.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を培養する工程と、
b.ノッチシグナル伝達経路を阻害する因子で前記細胞を処理する工程と、を含む、方法を提供する。
【0177】
膵臓内胚葉系統に特徴的なマーカーを発現するいずれの細胞も、この方法を用いて膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させるうえで適している。
【0178】
一実施形態では、ノッチシグナル伝達経路を阻害する因子は、γ−セクレターゼ阻害剤である。一実施形態では、γ−セクレターゼ阻害剤は、L−685,458としても知られる、1S−ベンジル−4R−[1−(1S−カルバモイル−2−フェネチルカルバモイル)−1S−3−メチルブチルカルバモイル]−2R−ヒドロキシ−5−フェネチルペンチル]カルバミン酸tert−ブチルエステルである。
【0179】
膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、ノッチシグナル伝達経路を阻害する因子で約1〜約5日間処理する。あるいは、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、ノッチシグナル伝達経路を阻害する因子で約3〜約5日間処理する。あるいは、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、ノッチシグナル伝達経路を阻害する因子で約5日間処理する。
【0180】
一実施形態では、ノッチシグナル伝達経路を阻害する因子は、γ−セクレターゼ阻害剤である。一実施形態では、γ−セクレターゼ阻害剤は、L−685,458としても知られる、1S−ベンジル−4R−[1−(1S−カルバモイル−2−フェネチルカルバモイル)−1S−3−メチルブチルカルバモイル]−2R−ヒドロキシ−5−フェネチルペンチル]カルバミン酸tert−ブチルエステルである。
【0181】
L−685,458は、約0.1μM〜約100μMの濃度で使用することができる。一実施形態では、L−685,458を約90μMの濃度で使用する。一実施形態では、L−685,458を約80μMの濃度で使用する。一実施形態では、L−685,458を約70μMの濃度で使用する。一実施形態では、L−685,458を約60μMの濃度で使用する。一実施形態では、L−685,458を約50μMの濃度で使用する。一実施形態では、L−685,458を約40μMの濃度で使用する。一実施形態では、L−685,458を約30μMの濃度で使用する。一実施形態では、L−685,458を約20μMの濃度で使用する。一実施形態では、L−685,458を約10μMの濃度で使用する。
【0182】
膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞の形成を促進することが可能な少なくとも1種類の更なる他の因子で処理してもよい。また、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される膵臓内分泌系統に特徴的なマーカーを発現する細胞の増殖を促進するものであってもよい。更に、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される膵臓内分泌系統に特徴的なマーカーを発現する細胞が、他の細胞種を形成する能力を促進するものであってもよく、あるいは他のいずれかの更なる分化の段階の効率を高めるものであってもよい。
【0183】
前記少なくとも1種類の更なる他の因子は、例えば、ニコチンアミド、TGF−β1、2及び3等のTGFβファミリーのメンバー、血清アルブミン、繊維芽細胞増殖因子ファミリーのメンバー、血小板由来増殖因子−AA及び−BB、血小板濃縮血漿、インスリン様増殖因子(IGF−I、II)、増殖分化因子(GDF−5、−6、−8、−10、11)、グルカゴン様ペプチド−1及びII(GLP−1及びII)、GLP−1及びGLP−2模倣体(mimetobody)、エキセンジン−4、レチノイン酸、副甲状腺ホルモン、インスリン、プロゲステロン、アプロチニン、ヒドロコルチゾン、エタノールアミン、βメルカプトエタノール、上皮増殖因子(EGF)、ガストリンI及びII、例えばトリエチレンペンタミン等の銅キレート化剤、フォルスコリン、酪酸ナトリウム、アクチビン、ベータセルリン、ITS、ノギン、神経突起増殖因子、ノーダル、バルプロ酸、トリコスタチンA、酪酸ナトリウム、肝細胞増殖因子(HGF)、スフィンゴシン1、VEGF、MG132(EMD社、カリフォルニア州)、N2及びB27添加物(ギブコ社(Gibco)、カリフォルニア州)、例えば、シクロパミン(EMD社、カリフォルニア州)等のステロイドアルカロイド、ケラチノサイト増殖因子(KGF)、Dickkopfタンパク質ファミリー、ウシ脳下垂体抽出物、膵島新生関連タンパク質(INGAP)、インディアンヘッジホッグ、ソニックヘッジホッグ、プロテアソーム阻害剤、ノッチ経路阻害剤、ソニックヘッジホッグ阻害剤、又はこれらの組み合わせであってもよい。
【0184】
前記少なくとも1種類の更なる他の因子は、例えば、PANC−1(ATCC No:CRL−1469)、CAPAN−1(ATCC No:HTB−79)、BxPC−3(ATCC No:CRL−1687)、HPAF−II(ATCC No:CRL−1997)等の膵臓細胞系、例えばHepG2(ATCC No:HTB−8065)等の肝細胞系、及び例えば、FHs74(ATCC No:CCL−241)等の腸細胞系から得られる馴化培地によって供給することが可能である。
【0185】
一実施形態において、本発明は、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させるための改良された方法であって、
a.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を培養する工程と、
b.グルコースを約10mM〜約20mMの濃度で含む培地中で、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させることが可能な因子で前記細胞を処理する工程と、を含む、方法を提供する。
【0186】
膵臓内胚葉系統に特徴的なマーカーを発現するいずれの細胞も、この方法を用いて膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させるうえで適している。
【0187】
膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させることが可能ないずれの方法も、本発明の改良に適している。
【0188】
一実施形態では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を約10mMの濃度のグルコースを含む培地中で処理する。代替的な一実施形態では、約20mMの濃度のグルコースを含む培地中で細胞を処理する。
【0189】
膵臓内胚葉系統に特徴的なマーカーを発現する細胞を約2〜約30日間処理する。一実施形態では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を約2〜約20日間処理する。一実施形態では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を約2〜約10日間処理する。一実施形態では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を約10日間処理する。一実施形態では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を約4日間処理する。一実施形態では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を約2日間処理する。
【0190】
膵臓内分泌系統に特徴的なマーカーを発現する細胞の検出
膵臓内分泌系統の細胞に特徴的なマーカーは当業者にはよく知られたものであり、膵臓内分泌系統に特徴的な更なるマーカーも次々に特定されている。これらのマーカーを用いて、本発明に基づいて処理を行った細胞が膵臓内分泌系統に特徴的な性質を獲得するように分化したことを確認することが可能である。膵臓内分泌系統に特異的なマーカーとしては、例えば、NGN−3、ニューロD(NeuroD)、Islet−1等、1以上の転写因子の発現が挙げられる。
【0191】
β細胞系統の細胞に特徴的なマーカーは当業者にはよく知られたものであり、β細胞系統に特徴的な更なるマーカーも次々に特定されている。これらのマーカーを用いて、本発明に基づいて処理を行った細胞がβ細胞系統に特徴的な性質を獲得するように分化したことを確認することが可能である。β細胞系統に特徴的な性質としては、例えば、特にPdx1(膵臓及び十二指腸ホメオボックス遺伝子−1)、Nkx2.2、Nkx6.1、Isl1、Pax6、Pax4、ニューロD(NeuroD)、Hnf1b、Hnf−6、Hnf−3β、及びMafA等、1以上の転写因子の発現が挙げられる。これらの転写因子は、当該技術分野では内分泌細胞を同定する目的でよく確立されている。例として、エドルンド(Edlund)による(Nature Reviews Genetics 3: 524-632(2002))等を参照されたい。
【0192】
分化の効率は、膵臓内分泌系統に特徴的なマーカーを発現する細胞によって発現されるタンパク質マーカーを特異的に認識する薬剤(抗体等)に、処理した細胞集団を曝露することによって決定することができる。また、分化の効率は、β細胞系統に特徴的なマーカーを発現する細胞によって発現されるタンパク質マーカーを特異的に認識する薬剤(抗体等)に、処理した細胞集団を曝露することによって決定することもできる。
【0193】
培養細胞又は単離細胞中のタンパク質及び核酸マーカーの発現を評価するための方法は、当該技術分野では標準的なものである。こうした方法には、定量的逆転写ポリメラーゼ連鎖反応(RT−PCR)、ノーザンブロット、in situハイブリダイゼーション(例えば、「分子生物学の最新プロトコール」(Current Protocols in Molecular Biology)オウスベル(Ausubel)等編、2001年度版補遺、を参照)、並びに、切片化材料の免疫組織化学的分析、ウエスタンブロッティング、及び無傷細胞中のアクセシブルなマーカーに対する免疫アッセイ、フローサイトメトリー分析(FACS)(例えば、ハーロー及びレーン(Harlow and Lane)著、「抗体の使用:実験マニュアル」(Using Antibodies: A Laboratory Manual)ニューヨーク州コールドスプリングハーバーラボラトリープレス(1998年)を参照)等の免疫アッセイが含まれる。
【0194】
特定のタンパク質マーカーの検出に有用な抗体の例を表IAに列記する。表IAに列記した抗体によって認識されるものと同じマーカーを標的とする代替的な抗体も入手可能であるか、又は容易に開発が可能である点は注意を要する。こうした代替的な抗体を、本発明に基づいて単離した細胞におけるマーカーの発現を評価するために用いることもできる。
【0195】
治療法
一態様において、本発明は、1型糖尿病を罹患しているかあるいは発症するリスクを有する患者を治療するための方法を提供する。この方法では、多能性幹細胞を培養し、その多能性幹細胞をインビトロでβ細胞系統に分化させ、そのβ細胞系統の細胞を患者に移植することを含む。
【0196】
更に別の一態様において、本発明は、2型糖尿病を罹患しているかあるいは発症するリスクを有する患者を治療するための方法を提供する。この方法では、多能性幹細胞を培養し、培養した細胞をインビトロでβ細胞系統に分化させ、そのβ細胞系統の細胞を患者に移植することを含む。
【0197】
必要に応じて、移植細胞の生存率及び機能を高める医薬薬剤又は生理活性物質で患者を更に治療することができる。こうした薬剤としては例えば、特にインスリン、TGF−β1、2及び3等のTGF−βファミリーのメンバー、骨形態形成タンパク質(BMP−2、−3、−4、−5、−6、−7、−11、−12、及び−13)、繊維芽細胞増殖因子−1及び−2、血小板由来増殖因子−AA及び−BB、血小板濃縮血漿、インスリン様増殖因子(IGF−I、II)、増殖分化因子(GDF−5、−6、−7、−8、−10、−15)、血管内皮細胞由来増殖因子(VEGF)、プレイオトロフィン、エンドセリン等が挙げられる。他の医薬化合物としては例えば、ニコチンアミド、グルカゴン様ペプチド−I(GLP−1)及びII、GLP−1及び2模倣体(mimetibody)、エキセンジン−4、レチノイン酸、副甲状腺ホルモン、例えば、米国特許出願公開第2004/0209901号及び同第2004/0132729号に開示される化合物のようなMAPK阻害剤等が挙げられる。
【0198】
多能性幹細胞は、レシピエントへの移植に先立ってインスリン産生細胞へと分化させることができる。特定の実施形態では、多能性幹細胞をレシピエントへの移植に先立ってβ細胞に完全に分化させる。また、多能性幹細胞は未分化又は部分的に分化した状態でレシピエントに移植してもよい。更なる分化はレシピエントの体内で起こりうる。
【0199】
胚体内胚葉細胞、又は膵臓内胚葉細胞、又はβ細胞は、分散した細胞として移植するか、あるいは肝門脈に注入することが可能な細胞塊に形成することができる。また、細胞を、生体適合性分解性ポリマー支持体、多孔質の非分解性装置中で与えるか、あるいはカプセル化することによってホストの免疫反応から保護することもできる。細胞はレシピエントの適当な部位に移植することができる。移植部位としては、例えば、肝臓、天然膵臓、腎嚢下腔、網、腹膜、漿膜下腔、腸、胃、及び皮下ポケット等が挙げられる。
【0200】
移植細胞の更なる分化、生存又は活性を促進する目的で、増殖因子等の更なる因子、抗酸化剤、又は抗炎症剤を、細胞の投与の前、同時、又は後に投与することができる。特定の実施形態では、増殖因子を用いて投与細胞をインビボで分化させる。これらの因子は、内因性の細胞によって分泌され、投与細胞にインサイチューで曝露されるものでもよい。移植細胞は、当該技術分野で知られる内因性及び外因性の投与された増殖因子の任意の組み合わせによって分化を誘導することができる。
【0201】
移植に用いられる細胞の量は、患者の状態、及び治療に対する患者の応答等の多くの異なる因子に依存し、当業者が決定できるものである。
【0202】
一態様において本発明は、糖尿病を罹患しているか、あるいは発症するリスクを有する患者を治療するための方法を提供する。この方法では、多能性幹細胞を培養し、培養した細胞をインビトロでβ細胞系統に分化させ、その細胞を3次元的な支持体に組み込むことを含む。細胞は、患者への移植に先立ってインビトロでこの支持体上に維持することができる。また、細胞を含んだ支持体を、更なるインビトロ培養を行うことなく患者に直接移植することも可能である。場合により、支持体に、移植細胞の生存率及び機能を高める少なくとも1種類の医薬薬剤を取り込ませてもよい。
【0203】
本発明の目的における使用に適した支持材料としては、組織修復に有用な組織テンプレート、導管、障壁、及びリザーバが挙げられる。特に、生物学的組織を再構築又は再生する目的で、及び組織の増殖を誘導するための走化性物質を送達する目的でインビトロ及びインビボで使用されてきた、発泡材、スポンジ、ゲル、ヒドロゲル、織物及び不織布構造の形態の合成及び天然の材料が、本発明の方法を実施するうえでの使用に適している。例として、米国特許第5,770,417号、同第6,022,743号、同第5,567,612号、同第5,759,830号、同第6,626,950号、同第6,534,084号、同第6,306,424号、同第6,365,149号、同第6,599,323号、同第6,656,488号、米国特許出願公開第2004/0062753(A1)号、米国特許第4,557,264号、及び同第6,333,029号に開示される材料を参照されたい。
【0204】
医薬薬剤を取り込んだ支持体を形成するには、支持体を形成するのに先立って医薬薬剤をポリマー溶液と混合することができる。また、医薬薬剤を、好ましくは医薬用担体の存在下で、製造された支持体上にコーティングしてもよい。医薬薬剤は、液体、微粉化した固体、又は他の任意の適当な物理的形態として存在してもよい。また、支持体に賦形剤を添加することによって医薬薬剤の放出速度を変化させることもできる。代替的な一実施形態では、例えば、米国特許第6,509,369号に開示される化合物のような抗炎症性化合物である少なくとも1種類の医薬化合物を支持体に取り込ませる。
【0205】
例えば、米国特許第6,793,945号に開示される化合物のような抗アポトーシス性化合物である少なくとも1種類の医薬化合物を支持体に取り込ませてもよい。
【0206】
例えば、米国特許第6,331,298号に開示される化合物のような線維症の阻害剤である少なくとも1種類の医薬化合物を支持体に更に取り込ませてもよい。
【0207】
例えば、米国特許出願公開第2004/0220393号及び米国特許出願公開第2004/0209901号に開示される化合物のような血管新生を促進することが可能な化合物である少なくとも1種類の医薬化合物を支持体に更に取り込ませてもよい。
【0208】
例えば、米国特許出願公開第2004/017623号に開示される化合物のような免疫抑制化合物である少なくとも1種類の医薬化合物を支持体に更に取り込ませてもよい。
【0209】
例えば、特にTGF−β1、2及び3等のTGF−βファミリー、骨形態形成タンパク質(BMP−2、−3、−4、−5、−6、−7、−11、−12及び−13)、繊維芽細胞増殖因子−1及び−2、血小板由来増殖因子−AA及び−BB、血小板濃縮血漿、インスリン様増殖因子(IGF−I、II)、増殖分化因子(GDF−5、−6、−8、−10、−15)、血管内皮細胞由来増殖因子(VEGF)、プレイオトロフィン、エンドセリン等の増殖因子である少なくとも1種類の医薬化合物を支持体に更に取り込ませてもよい。他の医薬化合物としては、例えば、ニコチンアミド、低酸素症誘導因子1−α、グルカゴン様ペプチド−I(GLP−1)、GLP−1及びGLP−2模倣体、及びII、エキセンジン−4、ノーダル、ノギン、NGF、レチノイン酸、副甲状腺ホルモン、テナシン−C、トロポエラスチン、トロンビン由来ペプチド、カテリシジン、ディフェンシン、ラミニン、フィブロネクチン及びビトロネクチン等の接着細胞外基質タンパク質の細胞及びヘパリン結合ドメインを含む生物学的ペプチド、米国特許出願公開第2004/0209901号及び同第2004/0132729号に開示される化合物等のMAPK阻害剤が挙げられる。
【0210】
本発明の細胞は、足場上に細胞を単純に置くだけで足場中に取り込ませることができる。細胞は単純に拡散によって足場に入り込むことができる(J. Pediatr.Surg. 23(1 Pt 2): 3-9(1988))。細胞の播種効率を高めるための他の幾つかの方法が開発されている。例えば、軟骨細胞をポリグリコール酸の足場上に播種するにはスピナーフラスコが使用される(Biotechnol.Prog. 14(2): 193-202(1998))。細胞を播種するための別の方法は遠心を使用することであり、これにより播種細胞へのストレスが最小に抑えられ、播種効率が高められる。例えば、ヤン(Yang)等は遠心細胞固定法(CCI)と呼ばれる細胞播種法を開発している(J.Biomed.Mater.Res. 55(3): 379-86(2001))。
【0211】
本発明を以下の実施例により更に説明するが、本発明はこれらの実施例によって限定されない。
【0212】
背景
【実施例】
【0213】
(実施例1)
ヒト胚性幹細胞の培養
ヒト胚性幹細胞系H1、H7及びH9をウィセル・リサーチ・インスティテュート社(WiCell Research Institute, Inc.)(ウイスコンシン州マディソン所在)より入手し、当該供給元機関によって与えられる指示に従って培養した。簡単に述べると、20%ノックアウト血清リプレースメント、100nM MEM非必須アミノ酸、0.5mM βメルカプトエタノール、2mM L−グルタミンと4ng/mlヒト塩基性繊維芽細胞増殖因子(bFGF)(すべてインビトロジェン/ギブコ社(Invitrogen/GIBCO)より入手)を添加したDMEM/F12(インビトロジェン/ギブコ社(Invitrogen/GIBCO))からなるES細胞培地中、マウス胚繊維芽(MEF)フィーダー細胞上で細胞を培養した。MEF細胞はE13〜13.5のマウス胚から誘導されたものをチャールズリバー社(Charles River)より購入した。MEF細胞は、10%FBS(ハイクローン社(Hyclone))、2mMグルタミン、及び100mM MEM非必須アミノ酸を添加したDMEM培地で増殖させた。サブコンフルエンスに達したMEF細胞培養を、10μg/mlマイトマイシンC(シグマ社(Sigma)ミズーリ州セントルイス所在)で3時間処理して細胞分裂を停止させた後、トリプシン処理し、0.1%ウシゼラチンコートディッシュに2×104/cm2で播いた。継代数2〜4からのMEF細胞をフィーダー層として使用した。MEF細胞のフィーダー層に播いたヒト胚性幹細胞を、加湿した組織培養インキュベーター内で、5%CO2雰囲気中37℃で培養した。コンフルエンスに達した時点(播種後約5〜7日後)で、ヒト胚性幹細胞を1mg/ml IV型コラゲナーゼ(インビトロジェン/ギブコ社(Invitrogen/GIBCO))で5〜10分間処理した後、5mlピペットを用いて表面から静かに掻き取った。細胞を900rpmで5分間遠心し、得られたペレットを再懸濁して新鮮な培地に1:3〜1:4の細胞比で再び播いた。
【0214】
(実施例2)
胚体内胚葉細胞の形成
胚体内胚葉のマーカーの発現に対するアクチビンAの影響を調べた。アクチビンA(100ng/ml)をマウス胚繊維芽細胞上で培養したヒト胚性幹細胞の集団に添加した。細胞をアクチビンAの存在下で継続的に培養し、示した時間において採取した。胚体内胚葉マーカーの発現レベルをPCR(図1)、FACS(表IIに結果をまとめる)、及び免疫組織化学法(図2)によって調べた。
【0215】
アクチビンAは、H9細胞系においてCXCR4、GATA4、HNF−3β、Mixl1及びSox−17のmRNAの発現を時間依存的に増大させた(図1、パネルa)。前方内胚葉マーカーであるケルベロス(Cerverus)、Otx−1及びHex遺伝子の顕著なアップレギュレーションも観察された(図1、パネルb)。アクチビンAによる処理後ではCXCR4タンパク質の増大がFACSによって観察された。E−カドヘリン及びN−カドヘリンの発現にはアクチビンAによる処理後に変化は見られなかった(表IIA)。CXCR4陽性細胞はC−kit、EPCAM、CD99についても顕著に陽性であり、CD9については陰性であった。これらのマーカーの発現パターンは、検討した3種類のhES細胞系で一貫していた(H7については表IIB、H1については表IIC)。アクチビンAで5日間処理した細胞で行った免疫細胞化学法によって、処理培養中の30〜40%の細胞がSox17及びHNF3βについて陽性であることが明らかとなった。同時に、分化した細胞のほぼ100%が依然Oct4陽性であった(図2)。多能性の表面マーカーの発現の減少を、胚体内胚葉マーカーの発現の増大と考え合わせると、これらのデータは、アクチビンAがヒト胚性幹細胞の胚体内胚葉への分化を促進することを示唆するものである。
【0216】
(実施例3)
膵臓内胚葉細胞の形成
ヒト胚性幹細胞の膵臓内胚葉への分化を誘導することが知られている増殖因子を細胞培養に添加した。具体的には、膵臓内胚葉の形成を誘導することが知られているアクチビンA、bFGF、及びレチノイン酸を細胞培養に添加した。
【0217】
第1の実験群では、0%〜2%の血清及びアクチビンA(100ng/ml)を添加したDMEM/F12中、マウス胚繊維芽細胞上で最大で7日間培養したヒト胚性幹細胞の集団にアクチビンAを添加した。細胞を図3に示した時点で採取し、図に示した遺伝子の発現についてPCRでアッセイされた(図3、4及び5)。図3のPCR分析により、アクチビン処理した細胞は、GATA4(図3、パネルa)、Sox−17(図3、パネルb)、HNF−3β(図3、パネルc)、及びMixl−1(図3、パネルd)を含む、内胚葉の発生に関連した広範な遺伝子を発現していることが示された。しかしながら、Pdx1遺伝子の発現は認められなかった。内胚葉系統マーカーの同じ発現パターンが、アクチビンAで処理したH7細胞で認められた(図6、パネルa〜f)。この段階では、Oct4の発現に有意な減少は認められなかった。
【0218】
アクチビンAは、胚体外内胚葉マーカーであるSox7(図4、パネルa)及びAFP(図4、パネルb)の発現を時間依存的に減少させた。アクチビンAはブラキュリ(Brachyury)の発現を減少させた(図5、パネルa)が、神経マーカーであるZic1の発現には影響を及ぼさなかった(図5、パネルb)。
【0219】
以上を考え合わせると、これらのデータは、Sox−17、Mixl1、Gata4及びHNF−3βの発現の増加が、前方内胚葉マーカーであるOtx1、Cer1及びHex遺伝子がアップレギュレーションされていることと相まって、アクチビンA処理に応じた胚体内胚葉の形成を示していることを示唆するものである。免疫細胞化学法による胚体内胚葉マーカーの分析により、これらの遺伝子のタンパク質発現もmRNA発現に認められた傾向を反映していることが明らかとなった。HNF−3β、Sox−17及びGATA4の発現レベルは、非処理細胞では全細胞の約10〜20%と低かった。5日間のアクチビンA(100ng/ml)処理により、HNF−3β、Sox−17及びGATA4の発現は、全細胞の約50%〜90%にまで増加した(図7)。
【0220】
第2の実験群では、ヒト胚性幹細胞の培養を実施例1に述べた方法に従って2〜3日間未分化条件に維持した。細胞が70〜80%コンフルエンスに達した後、アクチビンAを100ng/mlとなるように加えた0〜2%FBSを含むDMEM/F12に培地を換え、3日、5日、又は7日間にわたってアクチビンAの存在下で培養した。この時間間隔後、図8に示されるようなレチノイン酸とbFGFの組み合わせで細胞を5〜6日間更に処理した。培養物を採取し、mRNAの試料を収集して分析に供した。アクチビンA単独で5日間処理した細胞からなるコントロール培養も含めた。
【0221】
遺伝子発現分析により、アクチビンA又はレチノイン酸単独ではPdx1の発現を誘導しないことが示された。同様の結果が、アクチビンAの存在下でレチノイン酸とFGFの組み合わせにより処理した培養細胞で観察された(図8、パネルa)。しかしながら、アクチビンAの非存在下におけるレチノイン酸とFGFの組み合わせによる細胞の処理により、Pdx1の発現は更に増大した(図8、パネルa)。アクチビンAで3日間処理した後、アクチビンAの非存在下で1μMレチノイン酸及び50ng/ml bFGF(FGF−2としても知られる)で5日間処理した細胞は、アクチビンA単独で5日間処理した試料で観察されたPdx発現レベルよりも約3500倍高いPdx1の発現レベルを示した(図8、パネルa)。免疫細胞化学分析法によれば、全細胞の5〜20%がPdx1を発現していることが示された(図9)。
【0222】
アクチビンAの非存在下での1μMレチノイン酸及びbFGFによる処理は、アクチビンA単独の存在下で処理された細胞では認められなかったGLUT−2及びPTF1aの発現の増大も引き起こした(図8、パネルc)。1μMレチノイン酸及び50ng/ml bFGFで処理した細胞においてGLUT−2及びPTF1aの発現の最大の増加が認められた。これらを考え合わせると、これらのデータは、胚体内胚葉が形成された後に細胞培養からアクチビンAを除去することによって膵臓内胚葉の形成が更に促進されることを示唆するものである。
【0223】
(実施例4)
膵臓内分泌細胞の形成
ヒト胚性幹細胞の培養を実施例1に述べた方法に従って3〜4日間未分化条件に維持した。細胞が50〜60%コンフルエンスに達した後、100ng/mlのアクチビンAを含む、FBSを含まないDMEM/F12に培地を換え、この培地中で細胞を1日間培養した。1日間の培養の後、培地を除去し、100ng/mlのアクチビンAを加えた0.5%FBSを含む培地と交換し、細胞を1日間培養した。この2回目の1日の培養の後、培地を除去し、100ng/mlのアクチビンAを加えた2%FBSを含む培地と交換し、細胞を1日間培養した。この時間間隔後、実施例2に概略を述べたように細胞をレチノイン酸とFGFの組み合わせで6日間処理し、その後、培地を除去し、10μMのγ−セクレターゼ阻害剤L−685,458を含む、2%FBSを含んだDMEM/F12からなる培地と交換し、3日間培養した。細胞を収穫し、mRNAの試料を収集して分析に供した。アクチビンA単独で5日間処理した細胞からなるコントロール培養も含めた。
【0224】
遺伝子発現分析により、アクチビンA単独、又はアクチビンAをレチノイン酸及びFGFと組み合わせた場合には、Ngn3の発現もインスリンの発現も誘導しないことが明らかとなった(図10、パネルa、c)。L−685,458による処理の後ではHes−1の発現の減少も認められた。処理後3日目に最大の阻害が観察された(図10、パネルd)。しかしながら、L−685,458による処理は、アクチビンA単独又はレチノイン酸とFGFとの組み合わせで処理した試料で観察された発現レベルよりも約50倍高いレベルでNgn3の発現を誘導した。γ−セクレターゼ阻害剤で処理した試料ではインスリンの発現に70倍の増大が見られた。ニューロD1(NeuroD1)の発現もL−685,458による処理によって更に増大した(図10、パネルa)。以上を考え合わせると、これらのデータは、膵臓内胚葉が形成された後、レチノイン酸及びFGFを細胞培養から除去し、γ−セクレターゼ阻害剤を添加することによって内分泌細胞の形成が更に促進されることを示唆するものである。
【0225】
(実施例5)
Nkx2.2を発現する膵臓内分泌細胞の形成
実施例2に概略を述べた方法に従って得られた胚体内胚葉細胞を以下のように処理した。即ち、50ng/mlアクチビンA、50ng/ml塩基性FGF及び1μMのレチノイン酸を加えた2%FBS含有DMEM/F12からなる基本培地中で細胞を3〜5日間培養した。1μMのレチノイン酸を単独で又はbFGFとともに含む基本培地中で細胞を更に3〜5日間引き続き培養した。方向付けされた細胞の分化の評価を助けるため、このプロセスに沿った異なる時点で細胞からRNA試料を採取した。更に、この分化プロトコールの全体を通じて培地及び各因子を定期的に除去して補充した。アクチビンAの添加により、アクチビンAを加えない試料と比較してNkx2.2の発現は約35倍に増大した。培養の最初の3日間にアクチビンAで処理した試料では、Pdx1の発現はアクチビンAを含まない試料と同等のレベルに維持された(図11)。以上を考え合わせると、これらのデータは、膵臓内分泌マーカーであるNkx2.2の発現は、レチノイン酸及びbFGFによる処理の最初の3日間にアクチビンAを添加することによって更に促進されることを示唆するものである。
【0226】
(実施例6)
膵臓内胚葉細胞の培養における継代及び増殖
本実施例では、本願でヒト胚性幹細胞より誘導された膵臓内胚葉細胞を細胞培養中で維持し、更に分化させることなく継代することが可能であることを実証する。膵臓内胚葉細胞を低血清DMEM/F12中、100ng/mlのアクチビンAの存在下で分化させた。低血清DMEM/F12は、1日目には0%(v/v)のウシ胎児血清(FBS)、2日目には0.5%(v/v)のFBS、その後は毎日2%(v/v)のFBSを含有するものを使用した。4日間分化させた後、細胞を2%(v/v)FBS、1μMのレチノイン酸、及び50ng/mlのbFGFを含む低血清DMEM/F12中で、全体で6日間更に培養した。6日間の分化の後、細胞を、2%(v/v)FBSを含む低血清DMEM/F12中、50ng/mlのFGF10の存在下で、全体で6日間、培養中で維持した。6日間の培養期間中、膵臓内胚葉細胞を2回継代した。細胞集団の倍加時間はこの6日間の培養期間で約36〜48時間であった。培養の0、3及び6日目において、Q−PCRを用いて膵臓内胚葉を示すマーカー遺伝子の発現を測定した。図12は、50ng/mlのFGF10の存在下で増殖させた細胞が、誘導後の6日間の培養期間中、膵臓内胚葉マーカーであるPdx1の発現を維持したことを示している。
【0227】
(実施例7)
ヒト胚性幹細胞からの肝細胞の誘導
ヒト胚性幹細胞の培養を実施例1で述べた方法に従って未分化培養条件に2〜3日間維持した。細胞が70〜80%コンフルエンスに達した後、100ng/mlのアクチビンAを含む2%FBS含有DMEM/F12に培地を換え、アクチビンAの存在下で細胞を7日間培養した。7日間のアクチビンA処理の後、細胞を図13に示す各条件で5日間処理した。この後、細胞を採取し、mRNAの試料を収集して分析に供した。
【0228】
アクチビンAの非存在下で培養した細胞では、α−フェトタンパク質(AFP)及びアルブミンの発現の増大が認められた(図13、パネルA)。これはレチノイン酸及びFGF−4によって更に増大した(図13、パネルb)。以上を考え合わせると、これらのデータは、ヒト胚性幹細胞の培養が、上記に述べた処理の後、肝細胞マーカーを発現することが可能であることを示唆するものである。更に、ヒト胚性幹細胞を肝細胞に特徴的なマーカーを発現する細胞に分化させることが可能である。
【0229】
(実施例8)
H9ヒト胚性幹細胞系の特徴付け
未分化のES細胞が発現する幾つかのマーカーの発現を評価することにより、H9細胞の性質を経時的に監視した(Carpenter et al., 2001; Reubinoff et al., 2000; Thomson et al., 1998a)。H9細胞では、段階特異的胚抗原の交互発現が見られた(表III)。H9細胞は、いずれも未分化のヒト胚性幹細胞に特徴的なSSEA−3、SSEA−4、Tra−1−60、Tra−1−81、AP及びCD9抗原に対して強力な免疫応答性を示す。
【0230】
例えば、OCT3/4、SOX−2、UTF−1、REX−1、Cx43、Cx45、ABCG−2及びTERT等の胚性幹細胞に特徴的な遺伝子の発現を評価するためにリアルタイムPCRを行ったところ、本実施例で増殖させた細胞は、既に述べられている未分化の胚性幹細胞と似ていることが確認された(表III)。OCT3/4タンパク質の発現及びアルカリホスファターゼ活性(ケミコン社(Chemicon))を免疫染色によって確認した。H9細胞の大部分はOCT3/4及びAPについて陽性であった(図14)。以上をまとめると、これらの結果は、本実施例で使用したH9細胞が、他の研究機関からの報告と比較して、形態、抗原免疫染色、又は多能性マーカーの発現において大きく異ならないものであることを証明するものである。
【0231】
(実施例9)
蛍光活性化細胞選別(FACS)分析
接着した細胞をTrypLE(商標)Express溶液(インビトロジェン社(Invitrogen)カリフォルニア州所在)と5分間インキュベートすることによって培養皿から剥がした。剥がした細胞をヒト胚性幹細胞培地に再懸濁し、遠心して回収した後、洗浄し、この細胞を2%BSA、0.05%アジ化ナトリウムを含むPBSからなる染色緩衝液(シグマ社(Sigma)ミズーリ州)に再懸濁した。必要に応じて、0.1%γグロブリン(シグマ社(Sigma))溶液を用いて15分間、細胞のFc受容体をブロックした。一定分量(約105個の細胞)を表Iに示されるようなフィコエリスリン(PE)若しくはアロフィコシアニン(APC)を結合したモノクローナル抗体(106個の細胞毎に5μLの抗体)、又は非結合一次抗体とインキュベートした。コントロールには、適当なアイソタイプ一致抗体、非染色細胞、及び二次結合抗体のみで染色した細胞を含めた。抗体とのインキュベートをすべて、4℃で30分間行い、その後、細胞を染色緩衝液で洗った。非結合一次抗体で染色した試料を、PE又はAPC結合標識二次抗体と更に30分間、4℃でインキュベートした。使用した二次抗体の一覧については表Iを参照されたい。洗った細胞はペレットとし、染色緩衝液に再懸濁した。細胞表面の分子をFACS Array(BDバイオサイエンス社(BD Biosciences))装置を使用し、少なくとも10,000のイベントを収集して特定した。
【0232】
(実施例10)
免疫細胞化学法
0.1%Matrigel(BD社)でコーティングした培養皿に播種した細胞を室温で20分間、4%パラホルムアルデヒドで固定した。固定した細胞を室温で1時間、PBS/0.1%BSA/10%正常ニワトリ血清/0.5%TritonX−100でブロックした後、PBS/0.1%BSA/10%正常ニワトリ血清中、4℃で一次抗体と一晩インキュベートした。一次抗体及びその実用希釈率を表IBに示す。PBS/0.1%BSA中で3回洗った後、PBS中に1:100の希釈率で希釈した蛍光二次抗体と細胞を室温で1時間インキュベートして結合させた。コントロール試料として、一次抗体を省略するか、一次抗体と同じ濃度の、対応する一致したネガティブコントロールとしての免疫グロブリンで一次抗体を置き換えた反応を含めた。染色した試料はすすいだ。ジアミジノ−2−フェニルインドール二塩酸塩(DAPI)を含むPROLONG(登録商標)(インビトロジェン社(Invitrogen)カリフォルニア州所在)を、核を対比染色し、蛍光退色防止剤として機能するように各試料に1滴ずつ加えた。ニコンコンフォーカルEclipseC−1倒立顕微鏡(ニコン社、日本)及び10〜60倍の対物レンズを使用して画像を得た。
【0233】
(実施例11)
未分化細胞のPCR分析
RNAの抽出、精製及びcDNAの合成:エタノール含有高塩濃度緩衝液の存在下でシリカゲル膜(Rneasy Mini Kit、キアゲン社(Qiagen)カリフォルニア州所在)に結合させた後、洗浄して夾雑物を除去することによりRNA試料を精製した。TURBO DNA−Free Kit(アンビオン社(Ambion, Inc.))を使用してRNAを更に精製し、高品質RNAを水で溶出した。収率及び純度を分光光度計のA260及びA280の指示値によって評価した。ABI(ABI、カリフォルニア州所在)製高容量cDNA Archive Kitを使用して精製したRNAからcDNAコピーを作製した。
【0234】
リアルタイムPCR及び定量分析:特に断らないかぎり、試薬はすべてアプライドバイオシステムズ(Applied Biosystems)社より購入した。リアルタイムPCR反応を、ABI PRISM(登録商標)7900配列検出システムを使用して行った。TAQMAN(登録商標)UNIVERSAL PCR MASTER MIX(登録商標)(ABI社、カリフォルニア州所在)を、20ngの逆転写RNAと全反応容量を20μLとして使用した。各cDNA試料はピペット操作による誤差を補正するために2重で流した。プライマー及びFAM標識TAQMAN(登録商標)プローブは200nMの濃度で使用した。各標的遺伝子の発現レベルは、アプライドバイオシステムズ社(Applied Biosystem)によって以前に開発されているヒトグリセルアルデヒド−3−リン酸デヒドロゲナーゼ(GAPDH)内因性コントロールを使用して標準化された。プライマー及びプローブの組は以下の通りである。Oct3/4(Hs00742896)、SOX−2(Hs00602736)、UTF−1(Hs00747497)、Rex−1(Hs00399279)、コネキシン(Connexin)43(Hs00748445)、コネキシン(Connexin)45(Hs00271416)、ABCG2(Hs00184979)、Tert(Hs00162669)、HNF3β(Hs00232764)、GATA−4(Hs00171403)、Mixl1(Hs00430824)、Sox7(Hs00846731)、AFP(Hs00173490)、ブラキュリ(Brachyury)(Hs00610080)、GSC(Hs00418279_m1)、Pdx−1(Hs00426216)、PTF1a(Hs00603586)、Ngn3(Hs00360700)、ニューロ(Neuro)D1(Hs00159598)、インスリン(Hs00355773)、及びGlu2(Hs00165775)。Sox17プライマーはPRIMERSプログラム(ABI社、カリフォルニア州所在)を使用して設計されたものであり、以下の配列である。Sox17:TGGCGCAGCAGATACCA(配列番号1)、AGCGCCTTCCACGACTTG(配列番号2)、及びCCAGCATCTTGCTCAACTCGGCG(配列番号3)。最初に50℃で2分間、次いで95℃で10分間インキュベーションした後、試料を2段階(95℃で15秒間の変性ステップの後、60℃で1分間のアニーリング/伸長ステップ)で40サイクル反応させた。GENEAMP(登録商標)7000配列検出システムソフトウェアを使用してデータ分析を行った。各プライマー/プローブの組について、増幅が指数関数的に起こる領域の中央において蛍光強度が特定の値に達するサイクル数としてCt値を求めた。比較Ct法を用いて相対的遺伝子発現レベルを計算した。簡単に述べると、各cDNA試料について、内因性コントロールのCt値を対象遺伝子のCt値から差し引くことによってΔCt値(ΔCt)を得た。増幅の効率を100%と仮定すると、標準化された標的の量は2ΔCtとして計算される。最終的なデータは、検量試料に対して表わした。
【0235】
(実施例12)
核型の分析
H9細胞の核型を、標準的なGバンディング核型分析法によって調べた。全部で100個の中期染色体展開像を評価した(アプライドジェネティクスラボラトリー社(Applied Genetics Laboratories, Inc.))。分析した100個の細胞で染色体異常は見られなかった。細胞遺伝子分析により、常染色体の数は正常であり、染色体数(modal chromosome number)は46本であることが示された。図15は、ヒト胚性幹細胞系H9から得られる典型的な核型を示したものである。
【0236】
(実施例13)
細胞外基質でコーティングした組織培養基質上でのヒト胚性幹細胞の培養
ヒト胚性幹細胞系H1、H7及びH9をウィセル・リサーチ・インスティテュート社(WiCell Research Institute, Inc.)(ウイスコンシン州マディソン所在)より入手し、当該供給元機関によって与えられる指示に従って培養した。簡単に述べると、20%ノックアウト血清リプレースメント、100nM MEM非必須アミノ酸、0.5mM βメルカプトエタノール、2mM L−グルタミンと4ng/mlヒト塩基性繊維芽細胞増殖因子(bFGF)を添加したDMEM/F12(インビトロジェン/ギブコ社(Invitrogen/GIBCO))からなるES細胞培地中、マウス胚繊維芽(MEF)フィーダー細胞上で細胞を培養した。MEF細胞はE13〜13.5のマウス胚から誘導されたものをチャールズリバー社(Charles River)より購入した。MEF細胞は、10%FBS(ハイクローン社(Hyclone))、2mMグルタミン、及び100mM MEM非必須アミノ酸を添加したDMEM培地で増殖させた。サブコンフルエンスに達したMEF細胞培養を、10μg/mlマイトマイシンC(シグマ社(Sigma)ミズーリ州セントルイス所在)で3時間処理して細胞分裂を停止させた後、トリプシン処理し、0.1%ウシゼラチンコートディッシュに2×104/cm2で播いた。継代数2〜4からのMEF細胞をフィーダー層として使用した。MEF細胞のフィーダー層に播いたヒト胚性幹細胞を、加湿した組織培養インキュベーター内で、5%CO2雰囲気中37℃で培養した。コンフルエンスに達した時点(播種後約5〜7日後)で、ヒト胚性幹細胞を1mg/ml IV型コラゲナーゼ(インビトロジェン/ギブコ社(Invitrogen/GIBCO))で5〜10分間処理した後、5mlのガラス製ピペットを用いて表面から静かに掻き取った。細胞を900rpmで5分間遠心し、得られたペレットを再懸濁して希釈率が1:30の増殖因子低減MATRIGEL(商標)(BDバイオサイエンス社(BD Biosciences))でコーティングしたプレート上に1:3〜1:4の細胞比で再播種した。この後、細胞を、8ng/mlのbFGF及びコラゲナーゼを添加したMEF馴化培地中で培養し、MATRIGELでコーティングしたプレート上で少なくとも5代にわたって継代した。MATRIGEL(商標)上で培養した細胞を、IV型コラゲナーゼ(インビトロジェン/ギブコ社(Invitrogen/GIBCO))、ディスパーゼ(BDバイオサイエンス社(BD Biosciences))、又はリベラーゼ酵素(ロシュ社(Roche)、インディアナ州)を用いて日常的に継代した。
【0237】
(実施例14)
細胞外基質でコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
Nature Biotechnology 23,1534〜1541(Dec 2005)に既に記載されているようにして胚性幹細胞を胚体内胚葉に分化させた。簡単に述べると、約60〜70%コンフルエンスのH9培養を、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。H9細胞は希釈率1:30〜1:10の増殖因子低減MATRIGELでコーティングしたプレート上又は希釈率1:30〜1:10の通常のMATRIGEL上で培養した。各プレートは室温で1時間、MATRIGELでコーティングした。
【0238】
5日目に、CXCR4、E−カドヘリン、CD9、及びN−カドヘリンの発現についてはFACSによって、SOX−17、SOX−7、α胎児タンパク質(AFP)、CXCR4、ブラキュリ(Brychyury)(Bry)、グースコイド(gooscecoid)(GSC)、HNF−3β、及びGATA4についてはリアルタイムPCRによって培養を分析した。AFP及びSOX−7が臓側内胚葉マーカーとみなされるのに対して、GATA4、HNF−3β及びSOX−17は胚体内胚葉マーカーを代表するものであり、GSC、Bry、及びCXCR4は原始線条のマーカーを代表するものである。図17に、FACSにより分析したCXCR4の発現を示す。低濃度のMATRIGELと比較して、希釈率が1:10のMATRIGELでコーティングしたプレート上で培養した細胞によるCXCR4の発現は顕著に増大した。更に、増殖因子低減MATRIGELは、通常のMATRIGELと比較して胚体内胚葉細胞の形成に有効ではなかった。
【0239】
図18は、リアルタイムPCRの結果を示したものであり、希釈率1:10のMATRIGELでコーティングしたプレート上で培養した細胞では、希釈率1:30のMATRIGEL上で培養した細胞と比較して胚体内胚葉が顕著にアップレギュレートされていることが実証された。
【0240】
(実施例15)
胚体内胚葉形成後のヒト胚性幹細胞における遺伝子発現の変化のマイクロアレイによる分析
RNeasy mini kit(キアゲン社(Qiagen))を使用して以下のヒト胚性幹細胞の培養から全RNAを単離した。即ち、MATRIGELコーティングしたプレート上で培養し、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理したH9P83細胞、及び、MEF細胞上で培養し、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地で更に3日間処理したH9P44細胞。各群のコントロールには、MATRIGELでコーティングした培養皿に播種し、MEF馴化培地中で培養した細胞、又はMEF上に播種し、ES培地中で培養した細胞を含めた。
【0241】
試料の調製、ハイブリダイゼーション、及び画像分析は、アフィメトリックス社(Affymetrix)製Human Genome U133 Plus2.0 Arrayに従って行った。正規化及び対数変換を行った後、OmniViz(登録商標)ソフトウェア(マサチューセッツ州所在)及びGENESIFTER(ビズエックスラブズ社(VizXLabs)、ワシントン州所在)を使用してデータ分析を行った。各処理群内及び異なる処理群間の変動は、ピアソン相関係数を用いて比較した。異なる処理群間の遺伝子発現プロファイルの分散を、グラフ間の相関係数とともに図19に示す。試料間の遺伝子発現における有意差は、分散分析及び0.05以下の調節p値を用いたF検定(Benjamini and Hochberg相関)を用いて評価した。プレゼントコール(present call)が示された遺伝子のみが分析に含まれた。表IVに、異なる試料間で発現量に少なくとも5倍の差が見られた遺伝子を示す。有意に発現された遺伝子の正規化された強度値を、各遺伝子の標準誤差(SEM)とともに示した。
【0242】
(実施例16)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
Nature Biotechnology 23,1534〜1541(Dec 2005)に既に記載されているようにして胚性幹細胞を胚体内胚葉に分化させた。簡単に述べると、約60〜70%コンフルエンスに達した増殖因子低減MATRIGEL(商標)(希釈率1:30)培養上に播いたH9、H7又はH1細胞を、0.5%FBS及び100ng/mlアクチビンA(R&Dシステムズ社(R&D Systems)ミネソタ州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。以下のすべての実施例において、特に断らないかぎり、この処理レジメンを胚体内胚葉(DE)プロトコールと称する。
【0243】
これと並行して、MEFフィーダー上で培養したH9、H7又はH1細胞も上記に述べた同じDEプロトコールに供した。
【0244】
5日目に、CXCR4、E−カドヘリン、CD9、CD99及びN−カドヘリン(CD56)の発現についてはFACSによって、SOX−17、SOX−7、α胎児タンパク質(AFP)、CXCR4、ブラキュリ(Brychyury)(Bry)、グースコイド(gooscecoid)(GSC)、HNF−3β、及びGATA4についてはリアルタイムPCRによって培養を分析した。AFP及びSOX−7が臓側内胚葉マーカーとみなされるのに対して、GATA4、HNF−3β及びSOX−17は胚体内胚葉マーカーを代表するものであり、GSC、Bry、及びCXCR4は原始線条のマーカーを代表するものである。
【0245】
マウスフィーダー細胞上で培養し、DEプロトコールに供したH細胞系は、各胚体内胚葉(DE)マーカーを著明に発現し、FACSによってCXCR4の発現が確認された(図20)。DEプロトコールによる処理後にはE−カドヘリンの発現の顕著な減少も認められた。最後に、CXCR4+の集団はCD117についても陽性に染色された。図21は、非処理のH7(図21、パネルa)及びH9細胞(図21、パネルb)と比較して、各胚体内胚葉マーカーの顕著なアップレギュレーションを示している。
【0246】
MEFフィーダー上で培養したH細胞系と異なり、MATRIGEL(商標)(希釈率1:30)上で培養し、胚体内胚葉プロトコールで処理したH細胞系では、胚体内胚葉マーカーの著明な発現は認められなかった。特に、FACS及びリアルタイムPCRにより示される発現は、マウス胚繊維芽細胞上で培養した細胞と比較して、MATRIGEL(商標)上で培養した細胞において大幅に低かった。胚体内胚葉マーカーの発現は、H1がH9よりも高く、H9がH7よりも高いという一般的な応答パターンにしたがっている(図22及び23)。図22から分かるように、H1細胞はH7及びH9細胞系と比較して、CXCR4の発現に顕著な増大が認められた。いずれの場合においても、CXCR4の発現は、マウス胚繊維芽細胞上で培養した細胞と比較してMATRIGEL(商標)(希釈率1:30)上で培養した細胞では低かった点に注意されたい。図23(パネルa〜c)は、リアルタイムPCRの結果を示したものであり、H7(図23、パネルa)及びH9(図23、パネルb)細胞系において胚体内胚葉マ−カーのアップレギュレーションがある程度増大したことを示している。しかしながら、H1(図23、パネルc)細胞系は、H7及びH9細胞系と比較してより顕著な胚体内胚葉マーカーのアップレギュレーションを示した。
【0247】
(実施例17)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−Wntリガンドの役割
H7P44及びH9P46胚性幹細胞を、MATRIGEL(商標)(希釈度1:10)でコーティングした培養皿上で培養し、0.5%FBS及び100ng/mlアクチビンA(R&Dシステムズ社(R&D Systems)ミネソタ州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。培養の一部のものには、20ng/mlのWnt−3a(カタログNo.1324−WN−002、R&Dシステムズ社(R&D Systems)ミネソタ州所在)、20ng/mlのWnt−5a(カタログNo.654−WN−010、R&Dシステムズ社(R&D Systems)ミネソタ州所在)、25ng/mlのWnt−7a(カタログNo.3008−WN−025、R&Dシステムズ社(R&D Systems)ミネソタ州所在)、又は25ng/mlのWnt−5b(カタログNo.3006−WN−025、R&Dシステムズ社(R&D Systems)ミネソタ州所在)を5日間の処理期間の全体を通じて加えた。図24は、高濃度の(A)AA、又は(B)AA+20ng/mlのWnt−3aの存在下でのH9P46胚体内胚葉培養の位相差顕微鏡画像を示したものである。図25は、MATRIGEL(商標)(希釈率1:30)上で培養し、DEプロトコール+Wnt−3a(図25、パネルb及びd)、及び−Wnt−3a(図25、パネルa及びc)に供したH7P44及びH9P46細胞系について、FACSにより調べた5日目のCXCR4の発現を示したものである。Wnt−3aがDE培養中に存在する場合、低血清及び高濃度のAAで処理したDE培養と比較して、CXCR4(CD184)の著明な発現が見られた。図26は、低血清+AA±Wntリガンドで処理した、A)H7及びB)H9の培養についてリアルタイムPCRのデータを示したものである。いずれのH細胞系においても、Wnt−3aを添加することにより、胚体内胚葉マーカーの顕著なアップレギュレーションが認められた。これに対し、Wnt−5a、Wnt−5b及びWnt−7aは、胚体内胚葉マーカーの発現にほとんど影響を及ぼさなかった。
【0248】
(実施例18)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−Wnt−3aの有効量
H9P46胚性幹細胞を、MATRIGEL(商標)(希釈度1:10)でコーティングした培養皿上で培養し、0.5%FBS、100ng/mlアクチビンA(AA)及び10〜50ng/mlのWnt−3a(R&Dシステムズ社(R&D Systems)ミネソタ州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS、100ng/mlアクチビンA(AA)、及び10〜50ng/mlのWnt−3aを添加したDMEM/F12培地で更に3日間処理した。コントロール培養はWnt−3aで処理しなかった。図27において、パネルa)はWnt−3aの非存在下、b)10ng/mlのWnt−3a、c)20ng/mlのWnt−3a、及びd)50ng/mlのWnt−3aの存在下における5日目のFACSによるCXCR4の発現を示したものである。Wnt−3aの非存在下ではCXCR4の発現は極めて低かった。これに対し、10〜50ng/mlのWnt−3aを添加することにより、CXCR4陽性細胞の数が顕著に増加した。更に、10ng/mlのWnt−3aの添加は、50ng/mlのWnt−3aの添加と同様に効果的であった。リアルタイムPCRの結果によってもこの知見が確認された(図28、パネルa)。
【0249】
別の実験において、H9P52細胞を1:30に希釈した低増殖因子MATRIGEL(商標)上に播種した。DEプロトコールの最初の2日間において、10ng/ml、5ng/ml、1ng/mlの異なる用量のWnt−3aを使用した。図28のパネルbは、処理の5日後のDEマーカーのPCR分析を示したものである。実験終了実施例における細胞数を図28のパネルcに示す。この結果は、高用量のWnt−3aを使用した場合に細胞が増殖することを示している。Wnt−3aによる処理を5日間に延長することによって(5D)、PCRによるDEマーカーの結果にはほとんど影響は見られず、細胞数の大きな増加も認められなかった(図28、パネルc)。これらのデータは、2日間の10ng/mlのWnt−3aによる処理が、細胞の最適な増殖及び胚体内胚葉分化に充分であることを示すものである。
【0250】
(実施例19)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−GSK−3B阻害剤の影響
Wnt−3aの影響がWnt経路を介したものであることを確認するため、GSK−3阻害剤を使用してβカテニン等のWntの下流の標的分子を活性化した。H9P46〜P48胚性幹細胞をMATRIGEL(商標)でコーティングした培養皿(希釈率1:10)上で培養し、0.5%FBS、100ng/mlアクチビンA(AA)、及び10〜1000nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)カリフォルニア州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS、100ng/mlアクチビンA(AA)、及び0〜1000nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)カリフォルニア州所在)を添加したDMEM/F12培地で更に3日間処理した。コントロール培養は、低血清及び高用量のアクチビンA±Wnt−3aで処理した。図29において、パネルaはWnt−3aもGSK−3B阻害剤も加えない場合、b)+20ng/mlのWnt−3a、c)+1000ng/mlのGSK−3B阻害剤IX、d)+500ng/mlのGSK−3B阻害剤IX、e)+100ng/mlのGSK−3B阻害剤IX、f)+10ng/mlのGSK−3B阻害剤IX、g)+100ng/mlのGSK−3B阻害剤IXを1〜2日目、及びh)+10ng/mlのGSK−3B阻害剤IXを1〜2日目に加えた場合の5日目のFACSによるCXCR4の発現を示したものである。
【0251】
Wnt−3aの非存在下又は10nMのGSK−3B阻害剤の存在下では、CXCR4の発現は極めて低かった。これに対し、20ng/mlのWnt−3a又は100〜1000nMのGSK−3B阻害剤の添加により、CXCR4陽性細胞の数は顕著に増大した。更に、100nMのGSK−3B阻害剤を1〜2日目に添加した場合、100nMのGSK−3B阻害剤を5日間の期間全体にわたって添加した場合と同様に効果的であった。図30は、H9P48細胞(パネルa)及びH9P46細胞(パネルb)における胚体内胚葉マーカーの遺伝子発現を示す。
【0252】
図16は、無フィーダー系で胚性幹細胞を胚体内胚葉に分化させる本発明の分化プロトコールの概要を示す。
【0253】
(実施例20)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−GSK−3B阻害剤又はWnt−3aの存在下におけるアクチビンAの有効量
H9P49及びH1P46胚性幹細胞をMATRIGEL(商標)でコーティングした培養皿(希釈率1:10)上で培養し、0.5%FBS、10〜100ng/mlアクチビンA(AA)、及び100nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)、カリフォルニア州所在)又は20ng/mlのWnt−3aを添加したDMEM/F12培地に2日間曝露した後、2%FBS、10〜100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。コントロール培養は、低血清及び100ng/mlのアクチビンAで処理した。図31は、a)10ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間、b)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3Aを最初の2日間、c)100ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、d)10ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、e)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3Aを最初の2日間、並びに、f)10ng/mlのアクチビンA及び20ng/mlのWnt−3Aを最初の2日間加えた場合の、5日目のH9P49及びH1P46におけるFACSによるCXCR4の発現を示したものである。図31のパネルa〜dはH9P49細胞のものであり、パネルE〜FはH1P46細胞のものである。図32は、10、50、又は100ng/mlのアクチビンA及び20ng/mlのWnt−3aで処理したH9P49細胞における胚体内胚葉マーカーの遺伝子発現を示したものであり、パネルaはAFP、Bry、CXCR4、GSC、HNF−3B、及びPOU5F(Oct−4)の発現を示し、パネルbはSOX−17及びGATA4の発現を示す。50ng/mlのAA+20ng/mlのWnt−3A又は100nMのGSK−3B阻害剤IXを使用することによって胚体内胚葉マーカーが著明に発現するようである。アクチビンAを低用量で使用すると胚体外内胚葉が形成される。
【0254】
(実施例16)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−Wnt−3aとGSK−3B阻害剤の組み合わせ
H9P53胚性幹細胞をMATRIGEL(商標)でコーティングした培養皿(希釈率1:30)上で培養し、0.5%FBS、100ng/mlアクチビンA(AA)、及び100nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)、カリフォルニア州所在)を添加し、更に20ng/mlのWnt−3aを加える(+)か、加えない(−)DMEM/F12培地に2日間曝露した後、2%FBS、10〜100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。これと並行して、H9P53培地を、25ng/mlのBMP−4(カタログNo.314−BP−010、R&Dシステムズ社(R&D Systems)ミネソタ州所在)±20ng/mlのWnt−3A±100ng/mlのアクチビンAで処理した。コントロール培養は、低血清及び100ng/mlのアクチビンAで処理した。図33は、a)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間及び25ng/mlのBMP−4を3〜5日目、b)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間、c)100ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、d)20ng/mlのWnt−3a及び25ng/mlのBMP−4を5日間全体、e)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3a及び100nMのGSK−3B阻害剤IXを最初の2日間、並びに、f)100ng/mlのアクチビンA及び25ng/mlのBMP−4を5日間全体にわたって加えた場合の、5日目のFACSによるCXCR4の発現を示したものである。図34は、10又は100ng/mlのアクチビンA及び20ng/mlのWnt−3a又は100nMのGSK−3B阻害剤で処理した、継代数46のヒト胚性幹細胞系H1の培養についてリアルタイムPCRで調べた胚体内胚葉マーカーの遺伝子発現を示す。パネル(a):AFP、Bry、CXCR4、GSC、及びPOU5F(Oct−4)の発現、並びに、パネル(b):SOX−17、HNF−3B及びGATA4の発現。結果は非処理細胞に対する増加倍率として表わす。図35は、50又は100ng/mlのアクチビンA及び10又は100nMのGSK−3B阻害剤で処理した、継代数49のヒト胚性幹細胞系H9の培養についてリアルタイムPCRで調べた胚体内胚葉マーカーの遺伝子発現を示す。パネル(a):AFP、Bry、CXCR4、GSC、HNF−3B及びPOU5F(Oct−4)の発現、並びに、パネル(b):SOX−17及びGATA4の発現。結果は非処理細胞に対する増加倍率として表わす。図36は、アクチビンA、Wnt−3a、GSK−3阻害剤、及びBMP−4の組み合わせで処理したH9P53培養における胚体内胚葉マーカーの遺伝子発現を示す。A)AFP、Bry、CXCR4、GSC、HNF−3B及びSOX7の発現、並びに、B)SOX−17、HNF−3B及びGATA4の発現。DEプロトコールにBMP−4を加えることにより、中胚葉マーカーであるBRYの形成が誘導されるようであるが、Wnt−3AとGSK−4B阻害剤の組み合わせでは、アクチビンAの存在下で各物質を添加した場合と比較して、胚体内胚葉マーカーの顕著なアップレギュレーションは認められなかった。
【0255】
(実施例22)
MEF上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−低血清中でのWnt−3a、アクチビンA、Wnt−5a、BMP−2、BMP−4、BMP−6、BMP−7、IL−4及びSDF−1の組み合わせ
H9P44細胞を、マイトマイシン処理したマウス胚繊維芽細胞(MEF)で予めコーティングした6穴プレート上に播種した。20%ノックアウト血清リプレースメント、100nM MEM非必須アミノ酸、0.5mM βメルカプトエタノール、2mM L−グルタミン(すべてインビトロジェン/ギブコ社(Invitrogen/GIBCO)より入手)、及び8ng/mlヒト塩基性繊維芽細胞増殖因子(bFGF)(R&Dシステムズ社(R&D Systems))を添加したDMEM/F12(インビトロジェン/ギブコ社(Invitrogen/GIBCO))からなるES細胞培地中で70〜80%コンフルエンスに達するまで細胞を培養した。
【0256】
胚体内胚葉(DE)を形成するため、細胞をアクチビンA(100ng/ml)の存在下又は非存在下で、更に下記に詳述する他の増殖因子で処理した。各増殖因子は、下記のレジメンに示されるように段階的に濃度を増加させたFBSに添加した。
0日目:0%のFBSをDMEM/F12に加えた。
1日目:0.5%のFBSをDMEM/F12に加えた。
2日目:2%のFBSをDMEM/F12に加えた。
3日目:細胞を採取してFACS分析及びRT−PCRに供した。
【0257】
増殖因子はすべて、R&Dシステムズ社((R&D Systems)ミネソタ州所在)より購入した。各処理群における増殖因子の詳細な説明及び濃度を下記に示す。
1.コントロール:増殖因子を添加しなかった。
2.アクチビンA(100ng/ml)
3.アクチビンA(100ng/ml)+Wnt−3a(10ng/ml)+Wnt5a(10ng/ml)
4.アクチビンA(100ng/ml)+Wnt−3a(10ng/ml)+Wnt5a(10ng/ml)+BMP2(100ng/ml)
5.アクチビンA(100ng/ml)+BMP−4(100ng/ml)
6.アクチビンA(100ng/ml)+BMP−6(100ng/ml)
7.アクチビンA(100ng/ml)+BMP−7(100ng/ml)
8.アクチビンA(100ng/ml)+BMP−4(100ng/ml)+BMP−6(100ng/ml)+BMP−7(100ng/ml)
9.IL−4(10ng/ml)
10.SDF1a(20ng/ml)
11.アクチビンA(100ng/ml)+IL−4(10ng/ml)+SDF1a(20ng/ml)
12.BMP2(100ng/ml)+BMP−4(100ng.ml)+BMP−6(100ng/ml)+BMP−7(100ng/ml)
13.アクチビンA(100ng/ml)BMP−2(100ng/ml)+BMP−4(100ng.ml)+BMP−6(100ng/ml)+BMP−7(100ng/ml)
14.アクチビンA(100ng/ml)+IL−4(10ng/ml)
15.アクチビンA(100ng/ml)+(SDF1a(20ng/ml)
16.アクチビンA(100ng/ml)+Wnt−3a(10ng/ml)+Wnt−5a(10ng/ml)+Wnt−7a(10ng/ml)
17.アクチビンA(100ng/ml)+IL−4(10ng/ml)+SDF1a(20ng/ml)+BMP−4(100ng/ml)
【0258】
結果
DEプロトコール処理の3日目に細胞を採取した。分析に際しては、処理細胞の一定分量をRT−PCR用のRNAの調製に使用し、残りの細胞をFACS分析に使用した。CXCR4の出現頻度(%)を図37に示す。上記の増殖因子の添加により、CXCR4の発現は、低血清中での100ng/mlのAAによる処理以上には促進されなかった。
【0259】
RT−PCR分析では、選択された一群の胚体内胚葉マーカーの発現について細胞を分析した。示した結果は、基本培地中で増殖させたがアクチビンA又は他の増殖因子のいずれによっても処理しない細胞に対して検量したものである。FACSのデータと一致するように、表Vは、低血清中で高用量のアクチビンAで処理した培養にWnt−3a等の増殖因子を添加しても胚体内胚葉マーカーの顕著なアップレギュレーションが認められなかったことを示している。これは、アクチビンA、WNT3A、及び低血清の存在下で培養したES細胞においてDEマーカーの顕著な増大が認められた上記の実施例とは対照的である。
【0260】
(実施例23)
MATRIGEL又はヒトフィブロネクチンでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
【0261】
H9P55細胞を、ヒトフィブロネクチン又は通常の増殖因子MATRIGEL(商標)(BDバイオサイエンス社(BD Biosciences))上で増殖及び分化させた。1μg/mlのヒトフィブロネクチン(R&Dシステムズ社(R&D Systems))を含む1mlのDMEM/F12(インビトロジェン/ギブコ社(Invitrogen/GIBCO))を組織培養処理した6穴培養皿の各ウェルに加えた。これとは別に、通常の増殖因子MATRIGEL(商標名)をDMEM/F12中で1:10に希釈し、希釈したMATRIGEL(商標名)を組織培養処理した6穴培養皿の各ウェルに1mlずつ加えた。細胞を、コラゲナーゼを用いて継代した。細胞が80%コンフルエンスに達した後、以下のように処理した。即ち、10ng/mlのマウス組み換えWnt3a(R&D)及び100ng/mlのアクチビンA(R&D)を含む0.5%FBS中で2日間処理した。この後、2%FBS及び100ng/mlアクチビンAで3日間処理した。図38のパネルa及びbは、それぞれ、フィブロネクチン及びMATRIGEL上で培養した胚性幹細胞によるCXCR4の発現を示したものである。リアルタイムPCRの結果(図39)により、フィブロネクチンでコーティングしたプレート及びMATRIGEL(商標)でコーティングしたプレートで胚体内胚葉の形成は同等であることが確認された。
【0262】
(実施例24)
異なる濃度のMATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
【0263】
約60〜70%コンフルエンスのH9培養を、0.5%FBS、20ng/mlのWnt−3a及び100ng/mlのアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS、20ng/mlのWnt−3a及び100ng/mlのアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。H9細胞は希釈率1:60〜1:10の通常のMATRIGELでコーティングしたプレート上で培養した。各プレートはMATRIGELで1時間、室温でコーティングした。
【0264】
リアルタイムPCRの結果を図40に示す。低血清、アクチビンA、及びWnt−3aによるヒト胚性幹細胞の処理により、CXCR4、GATA4、グースコイド(Goosecoid)、HNF−3β、及びSOX−17遺伝子の発現が認められ、細胞が胚体内胚葉段階に分化していることが示唆された。しかしながら、Wnt−3aの存在下では、MATRIGEL(商標)コーティングの濃度は分化において重要な役割を果たしてはいないようである。
【0265】
(実施例25)
細胞外基質でコーティングした組織培養基質上で培養した後、MEF上で培養したヒト胚性幹細胞の胚体内胚葉への分化−Wnt−3aの役割
少なくとも5代にわたってMATRIGEL(商標)上で培養したヒト胚性幹細胞系H9から得た細胞をES培地中でMEFフィーダー上に播いた。細胞が約60〜70%コンフルエンスに達した時点で、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露し、その後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。更なる処理群として、20ng/mlのWnt−3aで5日間すべて、及び10〜100ng/mlのアクチビンAで処理した群を置いた。
【0266】
3日目及び5日目に、培養をSOX−17、SOX−7、α胎児タンパク質(AFP)、CXCR4、ブラキュリ(Brychyury)(Bry)、グースコイド(gooscecoid)(GSC)、HNF−3β、GATA4、hTERT及びOct4についてリアルタイムPCRにより分析した。AFP及びSOX−7が臓側内胚葉マーカーとみなされるのに対し、GATA4、HNF−3β及びSOX−17は胚体内胚葉マーカーを代表するものであり、GSC、Bry、及びCXCR4は原始線条のマーカーを代表するものである。hTERT及びOct−4はそれぞれ自己再生能及び多能性のマーカーである。リアルタイムPCRの結果を図41のパネルa〜dに示す。3日目及び5日目にFACS分析も行った。CXCR4及びCD9の発現レベルを分析し、図41のパネルeに示した。
【0267】
Wnt−3aの非存在下では、100ng/mlのアクチビンA中で培養した細胞のAFP発現レベルは非処理のコントロールにおける発現レベルと同様であった。しかしながら、100ng/mlのアクチビンA中で培養した細胞にWnt−3aを加えることによって、AFPの発現に経時的な増加が認められた。低濃度のアクチビンAを使用した場合、Wnt−3aの存在に関わらずAFPの発現は極めて高かった(図41、パネルa)。このことは、細胞の胚体外組織への分化を防止するには高濃度のアクチビンAが必要であることを示唆するものである。
【0268】
FACS分析により、高濃度のアクチビンAで処理したがWnt−3aでは処理しない試料の集団ではCXCR4陽性細胞が32〜42%であったのに対して、3日目に高濃度のアクチビンA及びWnt3aで処理した試料の集団では23〜33%であることが示された(図41、パネルe)。処理5日目までには、高濃度のアクチビンAで処理したがWnt−3aでは処理しなかった細胞の28〜32%がCXCR4を発現したのに対し、高濃度のアクチビンA及びWnt−3aで処理した細胞では43〜51%であった(図41、パネルf)。低濃度のアクチビンAで処理した細胞では、Wnt−3a処理群(3〜4%)と比較してWnt−3aで処理しなかった処理群においてCXCR4陽性細胞がより多かった(11〜20%)(図41、パネルg)。以上まとめると、Wnt−3aは、MEF上で培養されたヒト胚性幹細胞の胚体内胚葉への分化において重要な役割を果たしてはいないようである。このことは、フィーダー層が、アクチビンAによって誘導される胚体内胚葉の形成を促進するうえで充分なWnt−3a又は類似のリガンドを恐らく分泌していることを示唆するものである。
【0269】
(実施例26)
細胞外基質でコーティングした組織培養基質上で培養されたヒト胚性幹細胞の、Wnt阻害剤DKK−1による処理後の胚体内胚葉への分化
Wnt−3aの添加によって分化の増大がもたらされるか否かを調べるため、Wnt−3シグナル伝達の阻害剤を培養に加えた。約60〜70%コンフルエンスのH9培養を、0.5%FBS、20ng/mlのWnt3a、100ng/mlのDikkopf−1(DKK−1)及び100ng/mlのアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlのアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。H9細胞は希釈率1:30の増殖因子低減MATRIGELでコーティングしたプレート上で培養した。各プレートはMATRIGELで1時間、室温でコーティングした。
【0270】
5日目に、培養を、SOX−17、SOX−7、α胎児タンパク質(AFP)、CXCR4、ブラキュリ(Brychyury)(Bry)、グースコイド(gooscecoid)(GSC)、HNF−3β、GATA4、hTERT及びOct4についてリアルタイムPCRにより分析した。AFP及びSOX−7が臓側内胚葉マーカーとみなされるのに対し、GATA4、HNF−3β及びSOX−17は胚体内胚葉マーカーを代表するものであり、GSC、Bry、及びCXCR4は原始線条のマーカーを代表するものである。hTERT及びOct−4はそれぞれ自己再生能及び多能性のマーカーである。結果を図42に示した。
【0271】
Wnt−3aの存在下では、細胞はいずれも胚体内胚葉のマーカーであるCXCR4、GATA4、HNF−3β及びSOX17を発現している。グースコイド(gooscecoid)等の原始線条形成のマーカーも、非処理のコントロールにおいて検出されるよりも高いレベルで検出された。DKK1の添加により、上記の分化のマーカーの発現レベルは非処理の細胞における発現レベルと同等のレベルにまで劇的に減少した。
【0272】
(実施例27)
MATRIGELでコーティングした組織培養基質上で培養し、低血清及びアクチビンA±Wnt−3aで分化させたH9胚性幹細胞におけるDEマーカーの免疫蛍光染色
5日目のH9細胞のDE培養を、実施例10に従ってSOX−17、HNF−3B、GATA−4、N−カドヘリン及びE−カドヘリンについて染色した。すべての核をDAPIで対比染色した。20ng/mlのWnt−3aにより、Wnt−3aの非存在下で分化させた培養と比較して大幅に多数の核がSOX−17、HNF−3β及びGATA−4について陽性に染色された。更に、Wnt−3aの添加により、E−カドヘリンの発現が著しく消失し、N−カドヘリンの発現が昂進した(図43、パネルa及び図43、パネルb)。
【0273】
(実施例28)
MEF又はMATRIGEL上での胚体内胚葉形成後の胚性幹細胞における遺伝子発現の変化のマイクロアレイによる分析
RNeasy mini kit(キアゲン社(Qiagen))を使用して以下の胚性幹細胞の培養から全RNAを単離した。即ち、A)MATRIGEL(商標)コーティングしたプレート(希釈率1:30)上で培養し、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理したH9P33細胞、B)MEF細胞上で培養し、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理したH9P44細胞、及びC)MATRIGEL(商標名)コーティングしたプレート(希釈率1:30)上で培養し、0.5%FBS及び100ng/mlアクチビンA及び20ng/mlのWnt−3aを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理したH9P48細胞。各群のコントロールには、MATRIGELコーティングした培養皿に播種し、MEF馴化培地中で培養した細胞、又はMEF上に播種し、ES培地中で培養した細胞を含めた。すべての群で3つの生物学的複製物を用い、各生物学的複製物を2個の別々の遺伝子チップ上で繰り返し用いた。
【0274】
試料の調製、ハイブリダイゼーション、及び画像分析は、アフィメトリックス社(Affymetrix)製Human Genome U133 Plus2.0 Arrayに従って行った。正規化及び対数変換を行った後、OmniViz(登録商標)ソフトウェア(マサチューセッツ州所在)及びGENESIFTER(ビズエックスラブズ社(VizXLabs)、ワシントン州所在)を使用してデータ分析を行った。試料間の遺伝子発現における有意差は、分散分析及び0.05以下の調節p値を用いたF検定(Benjamini and Hochberg補正)を用いて評価した。少なくとも1つの群においてプレゼントコール(present call)が示された遺伝子のみが分析に含まれた。表VIに、A群、B群及びC群間で少なくとも5倍の差を示す平均正規化、対数変換した遺伝子のシグナル強度を、各遺伝子の調節p値とともに示す。
【0275】
(実施例29)
MATRIGELでコーティングした組織培養基質上で培養されたSA002 ES細胞系の胚体内胚葉への分化
8ng/mlのbFGFを添加したMEF−CM中、MATRIGELコーティングしたプレート(希釈率1:30)上で少なくとも3代にわたって予め培養したSA002 P38細胞(セラーティス社(Cellartis)スウェーデン)を、0.5%FBS、及び100ng/mlアクチビンA(R&Dシステムズ社(R&D Systems)ミネソタ州所在)±20ng/mlのWnt−3a又は100nMのGSK−3BIX阻害剤を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。リアルタイムPCRの結果を図44のパネルa及びbに示す。H1、H7及びH9細胞系と同様、SA002細胞系も、DEマーカーの顕著な発現にはWnt−3Aの添加をやはり必要とした。図45にCXCR4の発現を示す。a)AAで処理、b)AA+Wnt−3a、c)AA+GSK−3B阻害剤。
【0276】
(実施例25)
ヒト血清でコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
継代数55のヒト胚性幹細胞系H1の培養をヒト血清(シグマ社(Sigma)No.H1388、ミズーリ州所在)でコーティングしたプレート上で増殖及び分化させた。0.5mlのヒト血清を組織培養処理した6穴培養皿の各ウェルに加え、室温で1時間インキュベートし、ヒト胚性幹細胞を加える前に吸引した。細胞が80%コンフルエンスに達した後、以下のように処理した。即ち、10ng/mlのマウス組み換えWnt3a(R&D)又は100nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)、カリフォルニア州所在)及び100ng/mlのアクチビンA(R&D)を含む0.5%FBS中で2日間処理した。この後、2%FBS及び100ng/mlアクチビンAで3日間処理した。この後、培養をリアルタイムPCRで分析した(図46、パネルa及びb)。アクチビンAのみで処理した細胞と比較して、アクチビンA+GSK−3B阻害剤又はWnt−3Aで処理した細胞では胚体内胚葉マーカーの著明な発現が認められた。これらの所見は、MATRIGEL(商標)又はヒトフィブロネクチンでコーティングしたプレート上で培養したヒト胚性幹細胞における我々の発見に対応したものである。
【0277】
(実施例31)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−異なるGSK−3B阻害剤の評価
ヒト胚性幹細胞からの胚体内胚葉(DE)の形成における市販の多くのGSK−3B阻害剤の有効性を評価した。以下のGSK−3B阻害剤を100nMで評価した。即ち、GSK−3B阻害剤VIII(カタログNo.361549、カルバイオケム社(Calbiochem)、カリフォルニア州所在)、GSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)、カリフォルニア州所在)、GSK−3B阻害剤XI(カタログNo.361553、カルバイオケム社(Calbiochem)、カリフォルニア州所在)、GSK−3B阻害剤XII(カタログNo.361554、カルバイオケム社(Calbiochem)、カリフォルニア州所在)。H1P54 ES細胞を、MATRIGEL(商標名)(希釈度1:30)でコーティングした培養皿上で培養し、0.5%FBS、100ng/mlアクチビンA(AA)±各種GSK−3B阻害剤を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。コントロール培養を低血清及び高用量のAAで処理した。図47のパネルa及びbは、5日目における胚体内胚葉マーカーの遺伝子発現を示したものである。GSK−3B阻害剤IX及びXIは、GSK−3B阻害剤VIII及びXIIと比較してDEの形成を誘導するうえでいずれも有効であった。
【0278】
(実施例32)
無フィーダー条件下で培養されたヒト胚性幹細胞による膵臓内胚葉の形成−レチノイン酸類似体の評価
H9P49胚性幹細胞を、MATRIGEL(商標)(希釈度1:30)でコーティングした培養皿上で培養し、0.5%FBS、20ng/mlのWnt−3a(カタログNo.1324−WN−002、R&Dシステムズ社(R&D Systems)ミネソタ州所在)、及び100ng/mlアクチビンA(R&Dシステムズ社(R&D Systems)ミネソタ州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。5日目に細胞を回収してFACS及びリアルタイムPCRによる評価を行った。上記の実施例において示したように、このプロトコールによりCXCR4及びSOX−17のような胚体内胚葉マーカーが顕著にアップレギュレートされた。得られた5日目の胚体内胚葉細胞を以下の培地条件に曝露して膵臓内胚葉の形成を誘導した。即ち、2%FBS及び1μMのオールトランスレチノイン酸(RA)(カタログNo.R2625、シグマ社(Sigma)、ミズーリ州所在)、又は0.1〜10μMのAM−580(4−[(5,6,7,8−テトラヒドロ−5,5,8,8−テトラメチル−2−ナフタレニル)カルボキサミド]安息香酸、カタログNo.A8843、シグマ社(Sigma)、ミズーリ州所在)、又は0.1〜1μMのTTNPB(4−[(E)−2−(5,6,7,8−テトラヒドロ−5,5,8,8−テトラメチル−2−ナフタレニル)−1−プロペニル]安息香酸(アロチノイド酸)、カタログNo.T3757、シグマ社(Sigma)、ミズーリ州所在)を添加したDMEM/F12培地中で3日間培養した。AM−580及びTTNPBは、レチノイン酸受容体に対する親和性を有するレチノイン酸類似体である。RA処理の後、2%FBS及び20〜50ng/mlのbFGF(カタログNo.F0291、シグマ社(Sigma)、ミズーリ州所在)を添加したDMEM/F12培地中で更に3日間処理した。細胞を収穫し、mRNAの試料を採取して分析に供した。
【0279】
遺伝子発現分析(図48パネルa〜d)により、1μMのRAの添加後にbFGFに曝露することにより、PDX−1等の膵臓内胚葉マーカーが顕著にアップレギュレートされることが示された。更にこのプロトコールにより、CDX−1及びAFP等の前腸内胚葉マーカーの著明な発現が認められた。1μMの濃度では、RA類似体の添加により、同等の膵臓内胚葉及び前腸内胚葉マーカーの発現が見られた。しかしながら、1μMのRA類似体の添加により、オールトランスレチノイン酸と比較してより顕著なAFPの発現が認められた。しかしながら、10μMのAM−580の添加によってPDX−1の発現レベルは高く維持されたままでAFP及びCDX−2の発現が抑制された。
【0280】
(実施例34)
ヒト胚性幹細胞におけるサイトカイン発現に対するWnt−3a処理の影響
Wnt−3aによる処理がサイトカイン発現に与える影響についてタンパク質アレイを用いて分析を行った。ヒト胚性幹細胞系H9の細胞を実施例15で述べた方法に従って培養した。継代数54において、0.5%FBSを含むDMEM/F12中、100ng/mlのアクチビンA±10ng/mlのWnt−3aの存在下で細胞を2日間分化させた。この後、100ng/mlのアクチビンA及び2%FBSを含むDMEM/F12中で細胞を更に3日間培養した。5日目の終わりに、CXCR4の発現を各処理群についてFACSにより調べた。アクチビンAのみで処理した細胞ではCXCR4を発現した細胞は1%に過ぎなかった。アクチビンA及びWnt3aで処理した細胞では、73%の細胞がCXCR4の発現について陽性であった。
【0281】
哺乳動物細胞溶解キット(シグマ・アルドリッチ社(Sigma-Aldrich)、ミズーリ州所在)を用いて各処理群の細胞から細胞溶解物を調製した。各処理群からの馴化培地を回収して濃縮した。レイ・バイオテック社((RayBiotech)、ジョージア州所在)(http://www.raybiotech.com/)によって提供されるCytokine Arrayパネルを使用してサイトカインアレイ分析を行った。表VIIに、サイトカイン、増殖因子、並びにデータの正規化及びバックグランド除去後の受容体の発現を示す。各パネルに対して、ポジティブ及びネガティブコントロールも含まれた。示したデータは、細胞処理群ごとに2つの独立した試料(1、2)のものである。
【0282】
Wnt−3a処理細胞で馴化した培地では、アンジオゲニン、IGFBP−1及びEGFの著明なアップレギュレーションが認められた。IGFBP−1、TGFβ−1及びTGFβ−3等の多くのタンパク質がWnt−3a処理細胞の溶解物中でアップレギュレートされた。これらのアップレギュレートされたタンパク質を分化培地に再び添加することにより、胚体内胚葉形成に対するWnt−3aの効果を補充又は促進することが可能である。
【0283】
(実施例35)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化:Wnt1の役割
H1P55 ES細胞を、MATRIGEL(商標)(希釈度1:30)でコーティングした培養皿上で培養し、0.5%FBS及び100ng/mlのアクチビンA±10〜20ng/mlのWnt−1(ペプロテック社(PeproTech)、ニュージャージー州所在、カタログNo.120−17)を添加したDMEM/F12培地に2日間曝露した後、2%FBS、100ng/mlアクチビンA(AA)±10又は20ng/mlのWnt−1を添加したDMEM/F12培地で更に3日間処理した。以下のWnt1+AAの組み合わせについて試験を行った。
【0284】
a)0.5%FBS+DM−F12中、20ng/mlのWNT1+100ng/mlのAAで1〜2日目の後、2%FBS+DM−F12+100ng/mlのAAで3日目、b)0.5%FBS+DM−F12中、20ng/mlのWNT1+100ng/mlのAAで1〜2日目の後、2%FBS+DM−F12+100ng/mlのAAで3〜5日目、c)0.5%FBS+DM−F12中、10ng/mlのWNT1+100ng/mlのAAで1〜2日目の後、2%FBS+DM−F12+100ng/mlのAAで3日目、d)0.5%FBS+DM−F12中、10ng/mlのWNT1+100ng/mlのAAで1〜2日目の後、2%FBS+DM−F12+100ng/mlのAAで3〜5日目、e)0.5%FBS+DM−F12中、20ng/mlのWNT1+100ng/mlのAAで1〜2日目の後、2%FBS+DM−F12+100ng/ml+のAA+20ng/mlのWNT1で3日目、f)0.5%FBS+DM−F12中、20ng/mlのWNT1+100ng/mlのAAで1〜2日目の後、2%FBS+DM−F12+100ng/mlのAA+20ng/mlのWNT1で3〜5日目。図49のパネルa及びbに、H1細胞を低血清、AA及びWNT1で処理した後の胚体内胚葉マーカーについてのリアルタイムPCRのデータを示す。100ng/mlのAAの存在下で20ng/mlのWNT1を添加することにより、胚体内胚葉マーカー(Bry、CXCR4、GSC、SOX17、HNF−3B及びGATA−4)の顕著なアップレギュレーションが認められた。
【0285】
(実施例36)
膵臓内分泌分化に対するグルコースの影響
膵臓内胚葉細胞の膵臓内分泌細胞への分化の効率は、例えば基本培地の選択、又はグルコースの濃度等の多くの因子に依存する。胚性幹細胞から誘導された膵臓内胚葉細胞の膵臓内分泌細胞への分化に対するグルコース濃度の影響を調べた。
【0286】
基本培地を変更することによるグルコース濃度の変化:未分化のヒト胚性幹細胞(H1及びH9)の培養物を、膵臓内胚葉細胞への分化に先立って、実施例1に述べた方法に従って培養した。胚性幹細胞を、血清の非存在下、100ng/mlのアクチビンAを含むRPMI中で1日培養することによって膵臓内胚葉細胞に分化させた。この期間の後、100ng/mlのアクチビンA及び0.2%FBSを含むRPMI中で細胞を更に2日間培養した。この処理の後、2%FBS、FGF10(50ng/ml)及びKAAD−シクロパミン(250nM)を含むRPMIで培地を置き換えた。この培地中で細胞を4日間培養した。この期間の後、1xB27を添加した、オールトランスレチノイン酸(2μM)、FGF10(50ng/ml)及びKAAD−シクロパミン(0.25μM)を含む培地に4日間置き換えて膵臓内胚葉細胞の形成を誘導した。膵臓内胚葉細胞の収率は、低グルコースDMEM又はDMEM/F12で処理した培養で有意な差は見られなかった。
【0287】
膵臓内胚葉細胞をエキセンジン4及びHGFで処理することによって膵臓内分泌細胞に分化させた。エキセンジン4(50ng/ml)及びHGF(50ng/ml)は低グルコースDMEM又はDMEM/F12に10日間加えた。両培地に1xB27を添加した。培養を収穫し、mRNAの試料を採取して分析に供した。試料は、(Nature Biotechnology 24, 1392 - 1401(2006))に開示される方法に従って得られた膵臓内胚葉に対して標準化された。
【0288】
インスリンの発現をリアルタイムPCRにより分析した。図50のパネルa及びbに示されるように、インスリン及びグルカゴン遺伝子の発現は、DMEM−低グルコース中で処理した細胞と比較してDMEM/F12中で処理した細胞においていずれも大きく増大した。インスリンの発現を更に免疫組織化学法によって分析した(図51)。DMEM/F12中での処理により、DMEM−低グルコースと比較してインスリン陽性細胞の比率が大きくなった(図51、パネルa及びb)。インスリン陽性細胞はPDX−1についても陽性であった(パネルc)。
【0289】
グルコース濃度の変化:未分化のヒト胚性幹細胞(H1及びH9)の培養物を、膵臓内胚葉細胞への分化に先立って、実施例1に述べた方法に従って培養した。胚性幹細胞を、血清の非存在下、100ng/mlのアクチビンAを含むRPMI中で1日培養することによって膵臓内胚葉細胞に分化させた。この期間の後、100ng/mlのアクチビンA及び0.2%FBSを含むRPMI中で細胞を更に2日間培養した。この処理の後、2%FBS、FGF10(50ng/ml)及びKAAD−シクロパミン(250nM)を含むRPMIで培地を置き換えた。この培地中で細胞を4日間培養した。この期間の後、1xB27を添加した、オールトランスレチノイン酸(2μM)、FGF10(50ng/ml)及びKAAD−シクロパミン(0.25μM)を含むCMRLに4日間置き換えて膵臓内胚葉細胞の形成を誘導した。この培地に5、10又は20mMのグルコースを添加した。膵臓内胚葉細胞の収率は、5、10又は20mMのグルコースで処理したH9胚性幹細胞から誘導された培養で有意な差は見られなかった(図52、パネルa)。
【0290】
膵臓内胚葉細胞を、1xB27、エキセンジン4(50ng/ml)及びHGF(50ng/ml)を添加したCMRLで、5、10又は20mMグルコース中、2、4又は10日間細胞を処理することによって膵臓内分泌細胞に分化させた。細胞を収穫し、mRNAの試料を採取して分析に供した。試料は、(Nature Biotechnology 24, 1392-1401(2006))に開示される方法に従って得られた膵臓内胚葉に対して標準化された。
【0291】
図52のパネルb〜gは、ヒト胚性幹細胞系H9から誘導された細胞におけるNgn−3、NeuroD−1、Nkx2.2、Pax−4、インスリン及びグルカゴンの発現に対するグルコースの影響を示したものである。Ngn3は、膵臓の内分泌細胞の運命決定に関与する最初の転写因子であり、NeuroD1はNgn3の直接的な標的である。グルコース刺激はNgn3及びNeuroD1の両方のmRNAレベルを用量依存的に増大させた。別の2つの重要な膵臓マーカーであるNkx2.2及びPax−4もやはり同様の発現パターンを示した(図52、パネルd及びe)。10mMのグルコースで10日間処理した細胞において最適なインスリン及びグルカゴンの発現が観察された(図52、パネルf及びg)。
【0292】
Ngn−3、NeuroD−1、Nkx2.2、Pax−4について同様の結果が、ヒト胚性幹細胞系H1から誘導された培養において観察された(表VIII)。しかしながら、20mMのグルコースで10日間処理した細胞において最適なインスリン及びシナプトフィシンの発現が観察された(表VIII)。
【0293】
本発明の方法によって形成されたインスリン発現細胞から放出されるC−ペプチド:グルコース介在C−ペプチド放出を、2、10又は20mMのグルコースで処理したH1細胞から誘導されたインスリン陽性細胞で観察した。C−ペプチド放出を引き起こすため、細胞をまず、重炭酸塩及びHEPESを含むクレブス・リンゲル液(KRBH;129mM NaCl、4.8mM KCl、2.5mM CaCl2、1.2mM KH2PO4、1.2mM MgSO4、5mM NaHCO3、10mM HEPES、0.1%BSA)と1時間インキュベートした。培地を捨て、2mMのD−グルコースを含むクレブス・リンゲル液で置き換えた。細胞を20mMグルコース又は0.5mMのIBMX(すべてシグマ(Sigma)社より購入)で1時間刺激した。刺激上清中のC−ペプチド濃度を基本上清中のC−ペプチド濃度で割ることによって各培養について刺激倍数(fold stimulation)を計算した。
【0294】
BMXは、C−ペプチドの放出を1.2〜3倍に刺激した(図53)。20mMグルコースはC−ペプチド放出を刺激しなかった。2、10及び20mMグルコース中で形成されたインスリン陽性細胞間で、C−ペプチド分泌に有意な差は認められなかった。
【0295】
以上まとめると、我々のデータは、グルコースが内分泌マーカーであるNgn3及びNeuroD1の用量依存的なアップレギュレーションを誘導することを示唆するものであり、このことは、グルコースが、ヒト胚細胞の膵臓内分泌細胞への用量依存的な分化を誘導することを示唆している。インスリンの発現もまた、グルコースによって用量依存的に調節される。
【0296】
本文書の全体を通じて引用した刊行物はそれらの全容を本明細書に援用するものである。本発明の様々な態様を実施例及び好適な実施形態を参照することで上記に説明したが、本発明の範囲は上記の記載によって規定されるものではなく、以下の特許請求の範囲を特許法の原則に則って適切に解釈することによって定義されるものであることは認識されるであろう。
【0297】
〔実施態様〕
(1) 膵臓内分泌系統に特徴的なマーカーを発現する細胞を生成するための方法において、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、約10mM〜約20mMの濃度のグルコースを添加した培地中で、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって処理することによって、前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させること、を含む、方法。
(2) 前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞が、多能性幹細胞から分化させられる、実施態様1に記載の方法。
(3) 前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞が、胚体内胚葉系統に特徴的なマーカーを発現する細胞から誘導される、実施態様1に記載の方法。
(4) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞が、多能性幹細胞から誘導される、実施態様1に記載の方法。
(5) 前記多能性幹細胞が、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に更に分化させられる前に胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させられる、実施態様2に記載の方法。
(6) 前記グルコースが約10mMの濃度で使用される、実施態様1に記載の方法。
(7) 前記グルコースが約20mMの濃度で使用される、実施態様1に記載の方法。
(8) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約30日間処理する、実施態様1に記載の方法。
(9) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約20日間処理する、実施態様1に記載の方法。
(10) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約10日間処理する、実施態様1に記載の方法。
【0298】
(11) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約10日間処理する、実施態様1に記載の方法。
(12) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約4日間処理する、実施態様1に記載の方法。
(13) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間処理する、実施態様1に記載の方法。
(14) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、実施態様6に記載の方法。
(15) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、実施態様7に記載の方法。
(16) 多能性幹細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させるための方法において、
a.前記多能性幹細胞を培養する工程と、
b.前記多能性細胞をアクチビンAで処理することによって、前記多能性幹細胞を、胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
c.前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、少なくとも1種類の繊維芽細胞増殖因子、又はレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で処理することによって、前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
d.前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、約10mM〜約20mMの濃度のグルコースを添加した培地中で、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって処理することによって、前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させる工程と、を含む、方法。
(17) 前記グルコースが約10mMの濃度で使用される、実施態様16に記載の方法。
(18) 前記グルコースが約20mMの濃度で使用される、実施態様16に記載の方法。
(19) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約30日間処理する、実施態様16に記載の方法。
(20) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約20日間処理する、実施態様16に記載の方法。
【0299】
(21) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約10日間処理する、実施態様16に記載の方法。
(22) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約10日間処理する、実施態様16に記載の方法。
(23) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約4日間処理する、実施態様16に記載の方法。
(24) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間処理する、実施態様16に記載の方法。
(25) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、実施態様21に記載の方法。
(26) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、実施態様22に記載の方法。
(27) 糖尿病を有する患者に実施態様1に記載の細胞を移植することによって前記患者を治療するための方法。
(28) 糖尿病を有する患者に実施態様16に記載の細胞を移植することによって前記患者を治療するための方法。
(29) 前記多能性幹細胞が胚性幹細胞である、実施態様16に記載の方法。
(30) 前記胚性幹細胞がヒト胚性幹細胞である、実施態様29に記載の方法。
【0300】
(31) 前記ヒト胚性幹細胞が、H1及びH9からなる群の細胞系から誘導される、実施態様30に記載の方法。
【開示の内容】
【0001】
〔技術分野〕
本発明は、多能性幹細胞の分化を促進するための方法を提供する。詳細には、本発明は膵臓内胚葉、膵臓ホルモン発現細胞及び膵臓ホルモン分泌細胞を形成するための改良された方法を提供する。本発明は更に、フィーダー細胞層を使用することなく多能性幹細胞の分化を促進する方法を提供する。
【0002】
〔背景技術〕
1型糖尿病の細胞置換療法における進歩及び移植可能なランゲルハンス島の不足のため、生着に適したインスリン産生細胞即ち、β細胞の供給源の開発に注目が集まっている。1つの方法として、例えば、胚性幹細胞のような多能性幹細胞から機能性のβ細胞を生成することがある。
【0003】
脊椎動物の胚の発生においては、多能性細胞は原腸胚形成として知られる過程において3つの胚細胞層(外胚葉、中胚葉、内胚葉)を含む細胞群を生ずる。甲状腺、胸腺、膵臓、腸、及び肝臓等の組織は中間段階を経て内胚葉から発生する。この過程における中間段階は、胚体内胚葉の形成である。胚体内胚葉細胞は、HNF−3β、GATA4、Mixl1、CXCR4及びSox−17等の多くのマーカーを発現する。
【0004】
胚体内胚葉が膵臓内胚葉に分化することによって膵臓が形成される。膵臓内胚葉の細胞は膵十二指腸ホメオボックス遺伝子Pdx1を発現する。Pdx1の非存在下では、膵臓の発生は腹側及び背側膵芽が形成された時点で停止してしまう。したがって、Pdx1の発現は膵臓の臓器発生における重要なステップである。成熟した膵臓は異なる細胞種の中でも、特に外分泌組織及び内分泌組織を含んでいる。外分泌及び内分泌組織は、膵臓内胚葉の分化によって生じるものである。
【0005】
島細胞の特徴を有する細胞が、マウスの胚細胞から誘導されたことが報告されている。例えば、ルメルスキー等(Lumelsky et al. Science 292:1389, 2001)は、マウスの胚性幹細胞が膵島に似たインスリン分泌構造に分化したことを報告している。ソリア等(Soria et al. Diabetes 49:157, 2000)は、マウスの胚性幹細胞から誘導されたインスリン分泌細胞が、ストレプトゾトシン誘導糖尿病マウスにおいて血糖症を正常化させたことを報告している。
【0006】
1つの例として、ホリ等(Hori et al. PNAS 99: 16105, 2002)は、マウス胚性幹細胞をホスホイノシチド3−キナーゼ(LY294002)の阻害剤で処理することによって、β細胞に似た細胞が生じたことを開示している。
【0007】
別の例として、ブリスザック等(Blyszczuk et al. PNAS 100:998, 2003)は、Pax4を構成的に発現しているマウス胚性幹細胞からインスリン産生細胞が発生したことを報告している。
【0008】
ミカレフ等(Micallef et al.)は、レチノイン酸が、胚性幹細胞がPdx1陽性膵臓内胚葉を形成する運命決定を調節しうることを報告している。レチノイン酸は、胚の原腸胚形成の終期に対応する期間に胚性幹細胞分化の4日目に細胞培養に添加した場合にPdx1の発現を誘導する効果が最も高くなる(Diabetes 54:301, 2005)。
【0009】
ミヤザキ(Miyazaki)等は、Pdx1を過剰発現するマウス胚性幹細胞を報告している。ミヤザキ等の結果は、外因性のPdx1の発現によって、得られた分化細胞におけるインスリン、ソマトスタチン、グルコキナーゼ、ニューロゲニン3、P48、Pax6、及びHNFの遺伝子の発現が明らかに高められたことを示すものである(Diabetes 53: 1030, 2004)。
【0010】
スコウディー(Skoudy)等は、アクチビンA(TGFβスーパーファミリーのメンバー)がマウス胚性幹細胞における外分泌性膵臓遺伝子(p48及びアミラーゼ)及び内分泌性遺伝子(Pdx1、インスリン及びグルカゴン)の発現をアップレギュレートすることを報告している。1nMのアクチビンAを使用した場合に、最大の効果が認められた。スコウディー等は更に、インスリン及びPdx1のmRNAの発現レベルはレチノイン酸によって影響されなかったが、3nMのFGF7で処理することによってPdx1の転写産物のレベルが増大したことを観察している(Biochem. J. 379: 749, 2004)。
【0011】
シラキ(Shiraki)等は、胚性幹細胞のPdx1陽性細胞への分化を特異的に促進する増殖因子の効果を研究している。シラキ等は、TGFβ2によってPdx陽性細胞が高い比率で再現可能に得られたことを観察している(Genes Cells. 2005 Jun; 10(6): 503-16)。
【0012】
ゴードン(Gordon)等は、血清の非存在下及びアクチビンとWntシグナル伝達の阻害剤の存在下で、マウス胚性幹細胞からブラキュリ(brachyury)+/HNF−3β+内胚葉細胞が誘導されることを示している(米国特許出願公開第2006/00034468(A1)号)。
【0013】
ゴードン(Gordon et al. PNAS, Vol103, p16806, 2006)等は、「Wnt及びTGF−β/ノーダル(nodal)/アクチビンの同時シグナル伝達が前原始線条の形成には必要であった」と述べている。
【0014】
しかしながら胚性幹細胞の発生のマウスモデルは、例えば、ヒト等のより高等な哺乳動物における発生プログラムを正確に模倣していない可能性がある。
【0015】
トムソン(Thomson)等はヒトの胚盤胞から胚性幹細胞を単離した(Science 282:114, 1998)。これと同時に、ギアハート(Gearhart)及び共同研究者等は、胎児の生殖腺組織からヒト胚性生殖(hEG)細胞系を誘導した(Shamblott et al., Proc. Natl. Acad. Sci. USA 95:13726, 1998)。単純に白血病阻害因子(LIF)と培養することによって分化を妨げることが可能なマウス胚性幹細胞と異なり、ヒト胚性幹細胞は極めて特殊な条件下に維持しなければならない(米国特許第6,200,806号、国際特許出願公開第99/20741号、同第01/51616号)。
【0016】
ダムール(D’Amour)等は、高濃度のアクチビン及び低濃度の血清の存在下でヒト胚性幹細胞由来の胚体内胚葉の濃縮された培養物が作製されたことを述べている(Nature Biotechnology 2005)。これらの細胞をマウスの腎臓カプセル下に移植すると、一定の内胚葉性臓器の特徴を有するより成熟した細胞に分化した。ヒト胚性幹細胞由来の胚体内胚葉細胞はFGF−10の添加後、更にPdx1陽性細胞に分化させることができる(米国特許出願第2005/0266554(A1)号)。
【0017】
ダムール等(D’Amour et al. Nature Biotechnology - 24, 1392 - 1401(2006))は、「我々は、ヒト胚性幹細胞(hES)を、インスリン、グルカゴン、ソマトスタチン、膵臓ポリペプチド及びグレリンといった膵臓ホルモンを合成可能な内分泌細胞に転換させる分化プロセスを開発した。このプロセスは、胚体内胚葉、腸管内胚葉、膵臓内胚葉及び内分泌前駆細胞に似た段階を経て、内分泌ホルモンを発現する細胞に細胞を誘導することによりインビボで膵臓の器官形成を模倣するものである。」と述べている。
【0018】
別の例として、フィスク(Fisk)等は、ヒト胚性幹細胞から膵臓島細胞を作製するためのシステムを報告している(米国特許出願第2006/0040387(A1)号)。この場合、分化経路は3つの段階に分けられている。先ず、ヒト胚性幹細胞を、酪酸ナトリウムとアクチビンAの組み合わせを用いて内胚葉に分化させる。次に細胞をノギン等のTGFβアンタゴニストとEGF又はベータセルリンの組み合わせと培養してPdx1陽性細胞を作製する。最後の分化はニコチンアミドによって誘導する。
【0019】
1つの例において、ベンベニストリー(Benvenistry)等は、「我々は、Pdx1の過剰発現が膵臓に多く見られる遺伝子の発現を高めたことを結論付けるものである。インスリン発現の誘導には、インビボでのみ存在する更なるシグナルを必要とする可能性がある。」と述べている(Benvenistry et al, Stem Cells 2006; 24:1923-1930)。
【0020】
〔発明の概要〕
〔発明が解決しようとする課題〕
したがって、膵臓内分泌細胞、膵臓ホルモン発現細胞、又は膵臓ホルモン分泌細胞への分化能を維持する一方で、今日の臨床的要求に応えるように拡張することが可能な多能性細胞系を樹立するための条件を開発することが依然、大きく求められている。本発明者等は、ヒト胚性幹細胞を膵臓内分泌細胞に分化させる効率を高めることを目的として代替的な手法を取ったものである。
【0021】
〔課題を解決するための手段〕
一実施形態において、本発明は、多能性幹細胞を分化させるための方法において、
a.多能性幹細胞を培養する工程と、
b.前記多能性幹細胞を、胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
c.前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
d.前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させる工程と、を含む、方法を提供する。
【0022】
一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞は、以下の方法のいずれか1つによって多能性幹細胞を処理することによって多能性幹細胞から分化させられる。即ち、
a.血清の非存在下、アクチビンAを含む培地中で多能性幹細胞を培養し、次いで細胞をアクチビンA及び血清と培養し、次いで細胞をアクチビンA及び異なる濃度の血清と培養するか、
b.血清の非存在下、アクチビンAを含む培地中で多能性幹細胞を培養し、次いで細胞をアクチビンA及び別の濃度の血清と培養するか、
c.血清の非存在下、アクチビンA及びWntリガンドを含む培地中で多能性幹細胞を培養し、次いでWntリガンドを除去して細胞をアクチビンA及び血清と培養するか、
d.多能性幹細胞を、細胞外基質でコーティングした組織培養基質上で培養し、この多能性幹細胞をアクチビンA及びWntリガンドと培養するか、
e.多能性幹細胞を、細胞外基質でコーティングした組織培養基質上で培養し、次いでこの多能性幹細胞を、血清を含む第1の培地中でアクチビンA及びWntリガンドと培養し、次いでこの多能性幹細胞を、血清を含む第2の培地中でアクチビンAと培養するか、
f.多能性幹細胞を、細胞外基質でコーティングした組織培養基質上で培養し、次いでこの多能性幹細胞を、血清を含む第1の培地中でアクチビンA及びWntリガンドと培養し、次いでこの多能性幹細胞を、異なる濃度の血清を含む第2の培地中でアクチビンA及びWntリガンドと培養する。
【0023】
一実施形態では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞は、以下の方法のいずれか1つによって胚体内胚葉系統に特徴的なマーカーを発現する細胞を処理することによって胚体内胚葉系統に特徴的なマーカーを発現する細胞から分化させられる。即ち、
a.胚体内胚葉系統に特徴的なマーカーを発現する細胞を、繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤で処理し、次いで繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤を含む培地を取り除いた後、レチノイン酸、繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤を含む培地中で細胞を培養するか、
b.胚体内胚葉系統に特徴的なマーカーを発現する細胞を、レチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で処理するか、
c.胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸で処理し、次いでレチノイン酸を除去した後、細胞を少なくとも1種類の繊維芽細胞増殖因子で処理する。
【0024】
一実施形態では、膵臓内分泌系統に特徴的なマーカーを発現する細胞は、以下の方法のいずれか1つによって膵臓内胚葉系統に特徴的なマーカーを発現する細胞を処理することによって膵臓内胚葉系統に特徴的なマーカーを発現する細胞から分化させられる。即ち、
a.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、DAPT及びエキセンジン4を含む培地中で培養し、次いでDAPT及びエキセンジン4を含む培地を除去した後、細胞をエキセンジン1、IGF−1及びHGFを含む培地中で培養するか、
b.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、エキセンジン4を含む培地中で培養し、次いでエキセンジン4を含む培地を除去した後、細胞をエキセンジン1、IGF−1及びHGFを含む培地中で培養するか、
c.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、DAPT及びエキセンジン4を含む培地中で培養するか、
d.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、エキセンジン4を含む培地中で培養するか、
e.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、ノッチシグナル伝達経路を阻害する因子で処理するか、
f.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、約10mM〜約20mMのグルコース及びエキセンジン4を含む培地中で培養する。
【0025】
一実施形態では、本発明は糖尿病を罹患した患者を治療するための方法において、
a.多能性幹細胞を培養する工程と、
b.前記多能性幹細胞を、胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
c.前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
d.前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、β細胞系統の細胞に分化させる工程と、
e.前記β細胞系統の細胞を前記患者に移植する工程と、を含む、方法を提供する。
【図面の簡単な説明】
【0026】
【図1】パネルa)は、100ng/mlのアクチビンAで2日間、5日間、及び8日間処理した後のヒト胚性幹細胞系H9における胚体内胚葉マーカーCXCR4、GATA4、HNF−3β、Mixl1、Sox−17の発現。胚体内胚葉マーカーの発現はmRNAレベルでアッセイされ、非処理のヒト胚性幹細胞における発現レベルに標準化された。パネルbは、100ng/mlのアクチビンAで3日間及び5日間処理した後のヒト胚性幹細胞系H9における前方内胚葉マーカーCerberus、Otx−1及びHex遺伝子の発現。
【図2】100ng/mlのアクチビンAで5日間処理した後のヒト胚性幹細胞系H9における胚体内胚葉マーカーの発現。胚体内胚葉の発現は、免疫組織化学法によって検出した。パネル(a)は、Sox−17の発現。パネル(b)は、HNF−3βの発現を示す。パネル(c)は、Oct3/4の発現。
【図3】段階的分化プロトコールを行った後のヒト胚性幹細胞系H9における胚体内胚葉マーカーの発現。胚体内胚葉マーカーの発現はmRNAレベルでアッセイされ、非処理のヒト胚性幹細胞における発現レベルに標準化された。パネル(a)は、GATA4の発現。パネル(b)は、Sox−17の発現。パネル(c)は、HNF−3βの発現。パネル(d)は、Mixl1の発現。「AA」で示されるデータポイントは、1日間(1d)、3日間(3d)、5日間(5d)又は7日間(7d)にわたったアクチビンAによる処理。「UT」で示されるデータポイントは、1日間(1d)、3日間(3d)、5日間(5d)又は7日間(7d)にわたって培養した非処理のコントロール。
【図4】段階的分化プロトコールを行った後のヒト胚性幹細胞系H9における胚体外内胚葉マーカーの発現。胚体外内胚葉マーカーの発現はmRNAレベルでアッセイされ、非処理のヒト胚性幹細胞における発現レベルに標準化された。パネル(a)は、AFPの発現に対する100ng/mlアクチビンAの影響。パネル(b)は、Sox7の発現に対する100ng/mlアクチビンAの影響。「AA」で示されるデータポイントは、1日間(1d)、3日間(3d)、5日間(5d)又は7日間(7d)にわたったアクチビンAによる処理。「UT」で示されるデータポイントは、1日間(1d)、3日間(3d)、5日間(5d)又は7日間(7d)にわたって培養した非処理のコントロール。
【図5】段階的分化プロトコールを行った後のヒト胚性幹細胞系H9における中胚葉及び内胚葉マーカーの発現。中胚葉及び内胚葉マーカーの発現はmRNAレベルでアッセイされ、非処理のヒト胚性幹細胞における発現レベルに標準化された。パネル(a)は、Brachyuryの発現に対する100ng/mlアクチビンAの影響。パネル(b)は、Zic7の発現に対する100ng/mlアクチビンAの影響。「AA」で示されるデータポイントは、1日間(1d)、3日間(3d)、5日間(5d)又は7日間(7d)にわたったアクチビンAによる処理。「UT」で示されるデータポイントは、1日間(1d)、3日間(3d)、5日間(5d)又は7日間(7d)にわたって培養した非処理のコントロール。
【図6】100ng/mlのアクチビンAで1日間、3日間、5日間、及び7日間処理した後のヒト胚性幹細胞系H7における胚体内胚葉マーカーBrachyury(パネルa)、CXCR4(パネルb)、Mixl1(パネルc)、Sox17(パネルd)、HNF−3beta(パネルe)、Oct4(パネルf)の発現。胚体内胚葉マーカーの発現はmRNAレベルでアッセイされ、非処理のヒト胚性幹細胞における発現レベルに標準化された。
【図7】分化プロトコールを適用した後のヒト胚性幹細胞系H9における胚体内胚葉マーカーの発現。胚体内胚葉の発現は、免疫組織化学法によって検出した。パネル(a)及び(b)は、Sox−17の発現。パネル(c)及び(d)は、HNF−3βの発現。パネル(e)及び(f)は、GATA4の発現。パネル(b)、(d)及び(f)は、DAPIによる核の対比染色。「処理」で示される列は、5日間のアクチビンA処理(100ng/ml)を行ったものである。「非処理」で示される列は、非処理のコントロール。
【図8】第2の分化プロトコールを適用した後のヒト胚性幹細胞系H9における膵臓内胚葉マーカーの発現。膵臓内胚葉マーカーの発現はPCRによってアッセイされ、アクチビンAで処理したヒト胚性幹細胞における発現レベルに標準化された。パネル(a)は、Pdx1の発現。パネル(b)は、GLUT−2の発現。パネル(c)は、PTF1aの発現。
【図9】第2の分化プロトコールを適用した後のヒト胚性幹細胞系H9における膵臓内胚葉マーカーの発現。膵臓内胚葉の発現を、免疫組織化学法によって検出した。パネル(a)は非処理のコントロールにおけるPdx1の発現を示し、パネル(b)は段階的分化プロトコールによって処理した培養中でのPdx1の発現を示す。
【図10】第3の分化プロトコールを適用した後のヒト胚性幹細胞系H9における膵臓内分泌マーカーの発現。膵臓内分泌マーカーの発現はPCRによってアッセイされ、アクチビンAで処理したヒト胚性幹細胞における発現レベルに標準化された。パネル(a)は、NeuroD1の発現。パネル(b)は、Ngn3の発現。パネル(c)は、インスリンの発現。パネル(d)は、Hes−1の発現。発現レベルは膵臓内胚葉細胞に標準化してある。
【図11】分化プロトコールを適用した後のヒト胚性幹細胞系H9における膵臓内胚葉マーカーの発現。膵臓内胚葉マーカーの発現はPCRによってアッセイされ、アクチビンAで処理したヒト胚性幹細胞における発現レベルに標準化された。パネル(a)は、Nkx2.2の発現。パネル(b)は、Pdx1の発現。
【図12】培養の各継代(P0、P1及びP2)の細胞におけるPDX−1の発現。PDX−1の発現はPCRによってアッセイされ、アクチビンAで処理したヒト胚性幹細胞H9における発現レベルに標準化された。
【図13】第3の分化プロトコールを適用した後のヒト胚性幹細胞系H9における肝細胞マーカーの発現。肝細胞マーカーの発現はPCRによってアッセイされ、アクチビンAで処理したヒト胚性幹細胞における発現レベルに標準化された。パネル(a)は、AFPの発現。パネル(b)は、アルブミンの発現。
【図14】ヒト胚性幹細胞系H9における多能性のマーカーの発現。多能性のマーカーの発現は免疫組織化学法によってアッセイされた。パネル(a)は、Oct−4の発現。パネル(b)は、アルカリホスファターゼの発現。
【図15】ヒト胚性幹細胞系H9の核型。核型はマウス胚性繊維芽フィーダー細胞上で培養した継代数P36の細胞で判定した。
【図16】ヒト胚性幹細胞を無フィーダー系で胚体内胚葉に分化させる本発明の分化プロトコールの概要。
【図17】異なる濃度のMATRIGEL上で培養し、(0.5〜2%の)低濃度血清及び高濃度のアクチビンA(100ng/ml)に5日間曝露した継代数44のヒト胚性幹細胞系H9のFACSプロファイル。胚体内胚葉マーカーCXCR4(CD184)の発現をY軸に示し、ESマーカーCD9の発現をX軸に示す。
【図18】希釈率1:10のMATRIGEL(■)、希釈率1:20のMATRIGEL(□(グレーで表示))、又は希釈率1:30のMATRIGEL(□)上で培養し、実施例14で開示する分化プロトコールに供した継代数44のヒト胚性幹細胞系H9の培養から得られた胚体内胚葉のマーカーについてのリアルタイムPCRの結果。発光量比(fold induction)は、マウス胚性繊維芽細胞を使用して調整した培地で培養した継代数44のヒト胚性幹細胞系H9の未分化細胞に対するもの。
【図19】未分化の多能性幹細胞、及び分化している多能性幹細胞から得られた胚体内胚葉細胞における網羅的遺伝子発現の散布図。示されたデータは、マウス胚性繊維芽細胞上で培養した継代数44(右パネル)及びMATRIGEL上で培養した継代数83(左パネル)のヒト胚性幹細胞系H9細胞系の培養から得られたもの。
【図20】マウス胚性繊維芽フィーダー細胞上で培養し、実施例4に開示される胚体内胚葉の分化プロトコールに供したヒト胚性幹細胞系H1(パネルa)、ヒト胚性幹細胞系H7(パネルb)、及びヒト胚性幹細胞系H9(パネルc)について5日目のFACSによるCXCR4の発現。
【図21】マウス胚性繊維芽フィーダー細胞上で培養したヒト胚性幹細胞系H7(パネルa)及びヒト胚性幹細胞系H9(パネルb)の培養における、図に示された胚体内胚葉マーカーの発現のリアルタイムPCRによる結果。結果を、未分化細胞に対する増加倍率として表わす。
【図22】MATRIGEL(希釈率1:30)上で培養し、実施例4に開示される胚体内胚葉の分化プロトコールに供したヒト胚性幹細胞系H1(パネルa)、ヒト胚性幹細胞系H7(パネルb)、及びヒト胚性幹細胞系H9(パネルb)について5日目のFACSによるCXCR4の発現。
【図23】ヒト胚性幹細胞系H7(パネルa)、ヒト胚性幹細胞系H9(パネルb)及びヒト胚性幹細胞系H1(パネルc)の培養における、図に示された胚体内胚葉マーカーの発現のリアルタイムPCRによる結果。結果を、未分化細胞に対する増加倍率として表わす。細胞を実施例4に開示される方法に従って処理した。
【図24】100ng/mlのアクチビンA(パネルa)、又は100ng/mlのアクチビンA+20ng/mlのWnt−3a(パネルb)の存在下で継代数46のヒト胚性幹細胞系H9の培養の位相差顕微鏡画像。細胞を5日間処理した。
【図25】実施例4に開示される方法に従って処理した後の、継代数44のヒト胚性幹細胞系H7(パネルa及びb)及び継代数46のヒト胚性幹細胞系H9(パネルc及びd)の培養におけるFACSによるCXCR4の発現。パネルb及びdは、CXCR4の発現への20ng/mlのWnt−3aの影響。パネルa及びcは、Wnt−3aの非存在下におけるCXCR4の発現。結果は処理後5日目に得られたもの。
【図26】ヒト胚性幹細胞系H7(パネルa)及びH9(パネルb)の培養で認められる遺伝子の発現についてリアルタイムPCRのデータ。培養は、実施例4に開示される分化プロトコールによって処理した。各パネルに示すようにWntアゴニストであるWnt−3a(20ng/ml)、Wnt−5a(20ng/ml)及びWnt−7a(20ng/ml)の影響についても試験を行った。細胞を5日間処理した。結果は、未分化細胞に対する増加倍率として表わす。
【図27】FACSによる、処理後5日目の継代数46のヒト胚性幹細胞系H9の培養におけるCXCR4の発現。パネル(a)は、Wnt−3aの非存在下におけるCXCR4の発現。パネル(b)は、10ng/mlのWnt−3aによる処理後のCXCR4の発現。パネル(c)は、20ng/mlのWnt−3aによる処理後のCXCR4の発現を示し、パネル(d)は、50ng/mlのWnt−3aによる処理後のCXCR4の発現を示す。
【図28】処理後5日目のヒト胚性幹細胞系H9の培養中に認められる胚体内胚葉マーカーの発現。結果は、リアルタイムPCRによって求められる非処理細胞に対する発現の増加倍率として示した。パネル(a)は、図に示される胚体内胚葉マーカー遺伝子の発現に対する10、20、及び50ng/mlのWnt−3aの影響。パネル(b)は、処理後2日目(2d)及び5日目(5d)におけるgoosecoid(■)及びCXCR4(□)の発現に対する1、5、又は10ng/mlのWnt−3a(x軸上で10、5、1として示す)の影響。パネル(c)は、2日目(■)又は5日目(□)における細胞数に対する1、5、又は10ng/mlのWnt−3aの影響。
【図29】実施例4に開示される分化プロトコールによる5日間の処理後の、FACSによるヒト胚性幹細胞系H9の培養におけるCXCR4の発現。細胞を、Wnt−3a又はGSK−3B阻害剤の非存在下(パネルa)、20ng/mlのWnt−3aで5日間の全体(パネルb)、1000nMのGSK−3B阻害剤IXで5日間の全体(パネルc)、500nMのGSK−3B阻害剤IXで5日間の全体(パネルd)、100nMのGSK−3B阻害剤IXで5日間の全体(パネルe)、10nMのGSK−3B阻害剤IXで5日間の全体(パネルf)、100nMのGSK−3B阻害剤IXで1〜2日目(パネルg)、10nMのGSK−3B阻害剤IXで1〜2日目(パネルh)、培養。
【図30】リアルタイプPCRによる胚体内胚葉マーカーの遺伝子発現。結果を、非処理細胞に対する増加倍率として表わす。パネル(a)は、図に示される濃度及び時間でWnt−3a又はGSK−3B阻害剤を含む、実施例4に開示される胚体内胚葉プロトコールに従って処理した、継代数48のヒト胚性幹細胞系H9から得られたデータ。パネル(b)は、図に示される濃度及び時間でWnt−3a又はGSK−3B阻害剤を含む、実施例4で開示する胚体内胚葉プロトコールに従って処理した、継代数46のヒト胚性幹細胞系H9から得られたデータ。
【図31】本発明で使用する胚性幹細胞系についてFACSによるCXCR4の発現。パネル(a〜d)は、継代数49のヒト胚性幹細胞系H9から得られたデータ。パネル(e〜f)は、継代数46のヒト胚性幹細胞系H1から得られたデータ。データは処理後5日目に得た。細胞を以下の条件で処理した。パネル(a):10ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間、パネル(b):100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間、パネル(c):100ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、パネル(d):10ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、パネル(e):100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間、パネル(f):10ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間。
【図32】10、50、又は100ng/mlのアクチビンA及び20ng/mlのWnt−3aで処理した、継代数49のヒト胚性幹細胞系H9の培養についてリアルタイムPCRで調べた胚体内胚葉マーカーの遺伝子発現。パネル(a):AFP、Bry、CXCR4、GSC、HNF−3B、及びPOU5F(Oct−4)の発現、並びに、パネル(b):SOX−17及びGATA4の発現。結果を非処理細胞に対する増加倍率として表わす。
【図33】継代数53の胚性幹細胞系H9についてFACSによるCXCR4の発現。データは処理後5日目に得た。細胞を以下の条件で処理した。パネル(a):100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間及び25ng/mlのBMP−4を3〜5日目、パネル(b):100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間、パネル(c):100ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、パネル(d):20ng/mlのWnt−3a及び25ng/mlのBMP−4を5日間全体、パネル(e):100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3a及び100nMのGSK−3B阻害剤IXを最初の2日間、並びに、パネル(f):100ng/mlのアクチビンA及び25ng/mlのBMP−4を5日間全体。すべてのパネルについてX軸はCD9の発現を表し、Y軸はCXCR4(CD184)の発現を表す。
【図34】10又は100ng/mlのアクチビンA及び20ng/mlのWnt−3a又は100nMのGSK−3B阻害剤で処理した、継代数46のヒト胚性幹細胞系H1の培養についてリアルタイムPCRで調べた胚体内胚葉マーカーの遺伝子発現。パネル(a):AFP、Bry、CXCR4、GSC、及びPOU5F(Oct−4)の発現、並びに、パネル(b):SOX−17、HNF−3B及びGATA4の発現。結果は非処理細胞に対する増加倍率として表わす。
【図35】50又は100ng/mlのアクチビンA及び10又は100nMのGSK−3B阻害剤で処理した、継代数49のヒト胚性幹細胞系H9の培養についてリアルタイムPCRで調べた胚体内胚葉マーカーの遺伝子発現。パネル(a):AFP、Bry、CXCR4、GSC、HNF−3B及びPOU5F(Oct−4)の発現、並びに、パネル(b):SOX−17及びGATA4の発現。結果は非処理細胞に対する増加倍率として表わす。
【図36】アクチビンA、Wnt−3a、GSK−3阻害剤、及びBMP−4の組み合わせで5日間処理した、継代数53のヒト胚性幹細胞系H9の培養についてリアルタイムPCRで調べた胚体内胚葉マーカーの遺伝子発現。パネル(a):AFP、Bry、CXCR4、GSC、HNF−3B及びSOX7の発現、並びに、パネル(b):SOX−17、HNF−3B及びGATA4の発現。
【図37】実施例22に列記した条件で処理したヒト胚性幹細胞系H9の培養において、FACSによって調べたCXCR4の発現の比率。
【図38】フィブロネクチン(パネルa)又はMATRIGEL(商標)(パネルb)上で培養したヒト胚性幹細胞系H9の培養において、FACSによって調べた胚体内胚葉マーカーの発現。
【図39】フィブロネクチン(□)又は希釈率1:10の増殖因子低減MATRIGEL(■)上で培養したヒト胚性幹細胞系H9の培養においてリアルタイムPCRによって求めた胚体内胚葉マーカーの発現。
【図40】低濃度の血清、100ng/mlのアクチビンA及び20ng/mlのWnt−3aの存在下において異なる濃度のMATRIGELがヒト胚性幹細胞の胚体内胚葉への分化に与える影響。細胞を実施例4に開示される方法に従って処理。示した結果はリアルタイプPCRによって調べた、図に示される遺伝子の発現レベル。
【図41−1】MATRIGEL上で維持し、マウス胚性繊維芽細胞上で分化させたヒト胚性幹細胞による胚体内胚葉の形成におけるWnt−3aの役割。パネル(a)〜(d)は、図に示される遺伝子についてリアルタイムPCRのデータ。パネル(e)〜(g)は、図に示す条件についてFACSのデータ。
【図41−2】MATRIGEL上で維持し、マウス胚性繊維芽細胞上で分化させたヒト胚性幹細胞による胚体内胚葉の形成におけるWnt−3aの役割。パネル(a)〜(d)は、図に示される遺伝子についてリアルタイムPCRのデータ。パネル(e)〜(g)は、図に示す条件についてFACSのデータ。
【図41−3】MATRIGEL上で維持し、マウス胚性繊維芽細胞上で分化させたヒト胚性幹細胞による胚体内胚葉の形成におけるWnt−3aの役割。パネル(a)〜(d)は、図に示される遺伝子についてリアルタイムPCRのデータ。パネル(e)〜(g)は、図に示す条件についてFACSのデータ。
【図41−4】MATRIGEL上で維持し、マウス胚性繊維芽細胞上で分化させたヒト胚性幹細胞による胚体内胚葉の形成におけるWnt−3aの役割。パネル(a)〜(d)は、図に示される遺伝子についてリアルタイムPCRのデータ。パネル(e)〜(g)は、図に示す条件についてFACSのデータ。
【図42】Wnt阻害剤DKK−1による処理後の、MATRIGEL(商標)でコーティングされた組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化。示される結果は、20ng/mlのWnt−3A及び100ng/mlのDKK1(DE+DKK1)の存在下、又はDKK1(DE)の非存在下で実施例4に開示される方法に従って処理したH9細胞においてリアルタイムPCRによって調べた、図に示される遺伝子の発現。
【図43】MATRIGELでコーティングした組織培養基質上で培養し、低濃度血清及び100ng/mlのアクチビンAに20ng/mlのWnt−3aを加えないもの(パネルa)又は加えたもの(パネルb)で分化させたヒト胚性幹細胞系H9の培養における胚体内胚葉マーカーの免疫蛍光染色。Ecad=E−カドヘリン、NCAM=N−カドヘリン。
【図44】継代数38のヒト胚性幹細胞系SA002の胚体内胚葉への分化。細胞は図に示した条件で5日間処理し、各パネルに示した遺伝子について遺伝子の発現をリアルタイムPCRで調べた。
【図45】100ng/mlのアクチビンA処理(パネルa)、100ng/mlのアクチビンA+20ng/mlのWnt−3a(パネルb)、又は100ng/mlのアクチビンA+100nMのGSK−3B阻害剤IX(パネルc)による処理後の、継代数38のヒト胚性幹細胞系SA002における、FACSによるCXCR4の発現。細胞を5日間処理。
【図46】ヒト血清でコーティングした組織培養基質上での継代数55のヒト胚性幹細胞系H1の胚体内胚葉への分化。細胞は図に示した条件で処理し、各パネルに示した遺伝子について遺伝子の発現をリアルタイムPCRで調べた。
【図47】MATRIGEL(商標)でコーティングした組織培養基質上での継代数P54のヒト胚性幹細胞系H1の培養の胚体内胚葉への分化。5日間のDEプロトコールの後、異なるGSK−B阻害剤の影響について試験を行った。以下のGSK−3B阻害剤(GSK−3B VIII、IX、XI及びXII)を100nMにて処理の最初の2日間について評価を行った。
【図48】20ng/mlのWnt−3aの存在下で実施例4に開示される方法に従って培養及び処理した継代数49のヒト胚性幹細胞系H9の培養における、処理の最初の2日間のAFP(パネルa)、Pdx−1(パネルb)、Cdx−2及びGlut−2(パネルc)、並びに、HNF−3β、HNF−6及びソマトスタチン(パネルd)の発現。この処理の後、細胞を、2%FBS及び1μMのレチノイン酸、0.1〜1μMのTTNPB(4−[(E)−2−(5,6,7,8−テトラヒドロ−5,5,8,8−テトラメチル−2−ナフタレニル)−1−プロペニル]安息香酸(アロチノイド酸))、又は0.1〜10μMのAM−580(4−[(5,6,7,8−テトラヒドロ−5,5,8,8−テトラメチル−2−ナフタレニル)カルボキサミド]安息香酸)で更に3日間処理。次いで、細胞を2%FBS及び20ng/mlのbFGF中で更に3日間処理。
【図49】図に示した時間及び濃度でアクチビンA及びWnt−1によって処理したヒト胚性幹細胞系H1の培養において、パネルa及びbに示す胚体内胚葉マーカーの発現のリアルタイムPCRによる結果。
【図50】膵臓内胚葉細胞をDMEM/F12又はDMEM/低グルコース中で処理することによって形成された膵臓内分泌細胞の培養中におけるインスリン(パネルa)及びグルカゴン(パネルb)のmRNAの発現。図に示されるデータは、2つの別々の実験で観察されたもの。
【図51】DMDM/低グルコース(パネルa)、DMEM/F12(パネルb)中で処理した細胞において免疫細胞化学法によって調べたインスリンの発現。パネルcは、PDX−1及びインスリンの共染色。
【図52−1】ヒト胚性幹細胞系H9から誘導された膵臓内分泌細胞における遺伝子発現に対するグルコース濃度の影響。各遺伝子はパネル中で特定される。
【図52−2】ヒト胚性幹細胞系H9から誘導された膵臓内分泌細胞における遺伝子発現に対するグルコース濃度の影響。各遺伝子はパネル中で特定される。
【図52−3】ヒト胚性幹細胞系H9から誘導された膵臓内分泌細胞における遺伝子発現に対するグルコース濃度の影響。各遺伝子はパネル中で特定される。
【図53】2、10及び20mMのグルコース中で形成された膵臓内分泌細胞からのcペプチドの放出。細胞をIBMX又は20mMのグルコースで刺激した。
【発明を実施するための形態】
【0027】
開示を分かりやすくするため、限定を目的とすることなく、本発明の詳細な説明を、本発明の特定の特徴、実施形態、又は応用を説明又は図示した以下の小項目に分ける。
【0028】
用語の定義
幹細胞とは、1個の細胞レベルで自己再生し、かつ、自己再生性の前駆細胞、非自己再生性の前駆細胞、及び最終分化細胞のような前駆細胞を生ずるように分化する能力によって定義される未分化細胞のことである。幹細胞はまた、複数の胚細胞層(内胚葉、中胚葉及び外胚葉)から異なる細胞系の機能性細胞へとインビトロで分化する能力、並びに、移植後に複数の胚細胞層の組織を生ずる能力、及び胚盤胞への注入後にすべての組織とまではいかないにしてもほぼ大部分の組織に分化する能力によっても特徴付けられる。
【0029】
幹細胞は、発生上の能力によって、(1)すべての胚性又は胚体外細胞のタイプを生ずる能力を有することを意味する、分化全能性、(2)すべての胚性細胞のタイプを生ずる能力を有することを意味する、分化万能性、(3)細胞系統のサブセットを生ずる能力を有するが、それらがすべて特定の組織、臓器、又は生理学的システムのものであるような、分化多能性(例えば、造血幹細胞(HSC)は、HSC(自己再生性)、血球限定的寡能性前駆細胞、及び、血液の通常の成分であるすべての細胞種及び要素(例、血小板)を生じうる)、(4)多能性幹細胞よりも限定された細胞系統のサブセットを生ずる能力を有することを意味する、分化寡能性、及び(5)単一の細胞系統(例、精原幹細胞)を生ずる能力を有することを意味する、分化単一性に分類される。
【0030】
分化とは、特化していない(「分化決定していない」)か又は完全には特化していない細胞が、例えば、神経細胞や筋細胞のような特化した細胞の特徴を獲得するプロセスである。分化した細胞、又は分化誘導された細胞とは、細胞のその系統内においてより特化した(「分化決定した」)位置を占めるものである。分化のプロセスについて用いられる場合、「分化決定した」なる用語は、通常の条件下で特定の細胞種又は細胞種のサブセットにまで分化し続けるが、通常の条件下では異なる細胞種には分化できず、より分化の未熟な細胞種にも戻ることができない点にまで分化経路を進んだ細胞のことを指して言う。脱分化とは、細胞が細胞の系統においてより特化(又は分化決定)していない位置にまで戻るプロセスのことを指して言う。本明細書で言うところの細胞の系統とは、その細胞の遺伝、即ち、その細胞がどの細胞から生じたものであり、どのような細胞を生じうるか、ということを定義するものである。ある細胞の系統とは、発生及び分化の所定の遺伝スキーム内にその細胞を位置付けるものである。系統特異的マーカーとは、対象とする系統の細胞の表現型と特異的に関連した特徴を指し、分化決定していない細胞の、対象とする系統への分化を評価するために使用することができる。
【0031】
培養中の細胞を述べるために異なる用語が用いられる。「維持」とは、細胞の増殖及び/又は分裂を促進する条件下で増殖培地中に入れられた細胞のことを一般に指して言うものであり、より大きな細胞集団が形成される場合もそうでない場合もある。「継代」とは、1つの培養容器から細胞を取り出して、細胞の増殖及び/又は分裂を促進する条件下でこれらの細胞を第2の培養容器に入れるプロセスのことを指して言う。
【0032】
特定の細胞集団又は細胞系は、しばしば継代された回数によって呼称されるか、又はそれによって特徴付けられる。例えば、10回継代された培養細胞集団は、P10培養と呼ばれる場合がある。初代培養、即ち、組織から細胞を単離した後の最初の培養はP0と指定される。最初の継代培養の後、細胞は2次培養(P1又は継代数1)として述べられる。2回目の継代培養の後では細胞は3次培養(P2又は継代数2)となる、といった具合である。当業者であれば、継代期間中に集団は何度も倍加しうるものであり、したがってある培養の集団倍加の回数は継代数よりも大きいことは理解されるであろう。各継代間の期間における細胞の増殖(即ち、集団倍加数)は、播種密度、基質、培地、培養条件、及び継代間の時間等を含むがこれらに限定されない多くの因子に依存する。
【0033】
「β細胞系統」とは、転写因子PDX−1、並びに以下の転写因子、即ち、NGN−3、Nkx2.2、Nkx6.1、NeuroD、Isl−1、HNF−3β、MAFA、Pax4、及びPax6の内の少なくとも1つについて遺伝子発現が陽性である細胞を指して言う。β細胞系統に特徴的なマーカーを発現する細胞としてはβ細胞が挙げられる。
【0034】
本明細書で言うところの「胚体内胚葉系統に特徴的なマーカーを発現する細胞」とは、以下のマーカー、即ち、SOX−17、GATA−4、HNF−3β、GSC、Cer1、ノーダル(Nodal)、FGF8、ブラキュリ(Brachyury)、Mix様ホメオボックスタンパク質、FGF4 CD48、エオメソダーミン(eomesodermin)(EOMES)、DKK4、FGF17、GATA−6、CXCR4、C−Kit、CD99又はOTX2の内の少なくとも1つを発現する細胞を指して言う。胚体内胚葉系統に特徴的なマーカーを発現する細胞としては、原始線条前駆細胞、原始線条細胞、中内胚葉細胞及び胚体内胚葉細胞が挙げられる。
【0035】
本明細書で言うところの「膵臓内胚葉系統に特徴的なマーカーを発現する細胞」とは、以下のマーカー、即ち、PDX−1、HNF−1β、HNF−3β、PTF−1α、HNF−6、又はHB9の内の少なくとも1つを発現する細胞を指して言う。膵臓内胚葉系統に特徴的なマーカーを発現する細胞としては、膵臓内胚葉細胞が挙げられる。
【0036】
本明細書で言うところの「膵臓内分泌系統に特徴的なマーカーを発現する細胞」とは、以下のマーカー、即ち、NGN−3、NeuroD、Islet−1、PDX−1、NKX6.1、Pax−4、Ngn−3、又はPTF−1αを発現する細胞を指して言う。膵臓内分泌系統に特徴的なマーカーを発現する細胞としては、膵臓内分泌細胞、膵臓ホルモン発現細胞及び膵臓ホルモン分泌細胞、並びにβ細胞系統の細胞が挙げられる。
【0037】
本明細書で言うところの「胚体内胚葉」とは、原腸胚形成時に胚盤葉上層から生ずる細胞の特徴を有し、消化管及びその派生器官を形成する細胞を指して言う。胚体内胚葉細胞は、以下のマーカー、即ち、CXCR4、HNF−3β、GATA−4、SOX−17、ケルベロス(Cerberus)、OTX2、グースコイド(goosecoid)、c−Kit、CD99、及びMixl1を発現する。
【0038】
本明細書で言うところの「胚体外内胚葉」とは、以下のマーカー、即ち、SOX−7、AFP、及びSPARCの内の少なくとも1つを発現する細胞の集団を指して言う。
【0039】
本明細書で言うところの「マーカー」とは、対象とする細胞で発現の仕方が異なる核酸又はポリペプチド分子である。ここで言う異なる発現の仕方とは、陽性のマーカーでは発現レベルの上昇を、陰性のマーカーでは発現レベルの低下を意味する。マーカー核酸又はポリペプチドの検出可能なレベルは、他の細胞と比較して対象とする細胞において充分に高いか又は低いことから、当該技術分野において知られる各種の方法のいずれを用いても対象とする細胞を他の細胞から識別及び区別することが可能である。
【0040】
本明細書で言うところの「中内胚葉細胞」とは、以下のマーカー、即ち、CD48、エオメソダーミン(EOMES)、SOX−17、DKK4、HNF−3β、GSC、FGF17、GATA−6の内の少なくとも1つを発現する細胞を指して言う。
【0041】
本明細書で言うところの「膵臓内分泌細胞」又は「膵臓ホルモン発現細胞」とは、以下のホルモン、即ち、インスリン、グルカゴン、ソマトスタチン、膵臓ポリペプチド、及びグレリンの内の少なくとも1つを発現することが可能な細胞を指して言う。
【0042】
本明細書で言うところの「膵臓ホルモン分泌細胞」とは、以下のホルモン、即ち、インスリン、グルカゴン、ソマトスタチン、及び膵臓ポリペプチドの内の少なくとも1つを分泌することが可能な細胞を指して言う。
【0043】
本明細書で言うところの「前原始線条細胞」とは、以下のマーカー、即ち、ノーダル(Nodal)、又はFGF8の内の少なくとも1つを発現する細胞を指して言う。
【0044】
本明細書で言うところの「原始線条細胞」とは、以下のマーカー、即ち、ブラキュリ(Brachiury)、Mix様ホメオボックスタンパク質、又はFGF4の内の少なくとも1つを発現する細胞を指して言う。
【0045】
多能性幹細胞の単離、増殖及び培養
多能性幹細胞の特徴付け
多能性幹細胞は、発生段階特異的胚性抗原(SSEA)3及び4の1つ以上、及びTra−1−60及びTra−1−81と呼ばれる抗体を用いて検出可能なマーカーを発現しうる(Thomson et al., Science 282:1145, 1998)。インビトロで多能性幹細胞を分化させると、SSEA−4、Tra−1−60、及びTra−1−81の発現が消失し(存在する場合)、SSEA−1の発現が増大する。未分化の多能性幹細胞は通常アルカリホスファターゼ活性を有し、これは、細胞を4%パラホルムアルデヒドで固定し、次いで製造業者(ベクターラボラトリーズ、カリフォルニア州バーリンゲーム所在)によって述べられるようにVectorRedを基質として現像することによって検出することができる。未分化の多能性幹細胞は、RT−PCRによって検出されるOct−4及びTERTを通常更に発現している。
【0046】
増殖させた多能性幹細胞の別の望ましい表現型は、内胚葉、中胚葉、外胚葉組織の3つの胚細胞層のすべての細胞に分化する能力である。多能性幹細胞の多能性を、例えば、細胞を重症複合免疫不全症(SCID)マウスに注入し、形成される奇形腫を4%パラホルムアルデヒドで固定し、次いでこれを3つの胚細胞層からの細胞種の証拠について組織学的に調べることによって確認することができる。また、多能性を、胚様体を形成し、3つの胚細胞層に関連したマーカーの存在について胚様体を評価することによって調べることもできる。
【0047】
増殖させた多能性幹細胞系は、標準的なGバンド法を用いて核型を決定し、対応する霊長類種の公表されている核型と比較することができる。細胞は「正常な核型」を得ることが望ましい。これは、細胞が、ヒトの染色体がすべて揃っていて、かつ目立った変化のない正倍数体であることを意味する。
【0048】
多能性幹細胞の供給源
使用が可能な多能性幹細胞の種類としては、妊娠期間中の任意の時期(必ずしもではないが、通常は妊娠約10〜12週よりも前)に採取した前胚性組織(例えば、胚盤胞等)、胚性組織、胎児組織等の、妊娠後に形成される組織に由来する多能性幹細胞の株化細胞系が含まれる。非限定的な例としては、例えば、ヒト胚性幹細胞系H1、H7及びH9(WiCell)等のヒト胚性幹細胞又はヒト胚性生殖細胞の株化細胞系がある。更に、こうした細胞の初期の株化又は安定化の際に本開示の組成物を使用することも考えられるが、その場合は、供給源となる細胞は供給源組織から直接採取される1次多能性細胞である。フィーダー細胞の非存在下で既に培養された多能性幹細胞集団から得られる細胞も好適である。例えば、BG01v(ブレサジェン社(BresaGen)、ジョージア州アテネ所在)等の変異型ヒト胚性幹細胞系も好適である。
【0049】
一実施形態では、ヒト胚性幹細胞をトムソン等(Thomson et al.)によって述べられるように調製する(米国特許第5,843,780号、Science 282:1145, 1998; Curr. Top. Dev. Biol. 38:133 ff., 1998; Proc. Natl. Acad. Sci. U.S.A. 92:7844, 1995)。
【0050】
多能性幹細胞の培養
一実施形態では、通常、多能性幹細胞を、様々な点で多能性幹細胞を支持するフィーダー細胞の層上で培養する。あるいは、多能性幹細胞を、フィーダー細胞を基本的に含まないにも関わらず、細胞を大きく分化させることなく多能性幹細胞の増殖を支持する培養システム中で培養する。分化をともなわない無フィーダー細胞培養中での多能性幹細胞の増殖は、別の細胞種と予め培養することによって調整した培地を用いることで支持される。また、分化をともなわない無フィーダー細胞培養中での多能性幹細胞の増殖は、合成培地を用いることによっても支持される。
【0051】
例えば、ルービノフ等(Reubinoff et al. Nature Biotechnology 18: 399 - 404(2000))及びトンプソン等(Thompson et al. Science 6 November 1998: Vol. 282. no. 5391, pp. 1145 - 1147)は、マウス胚性繊維芽フィーダー細胞層を用いてヒト胚盤胞からの多能性幹細胞系を培養することについて開示している。
【0052】
リチャード等(Richards et al. Stem Cells 21: 546-556, 2003)は、11種類の異なるヒトの成人、胎児、及び新生児フィーダー細胞層についてヒト多能性幹細胞の培養を支持する能力の評価を行っている。リチャード等は、「成人の皮膚繊維芽フィーダー細胞上で培養したヒト胚性幹細胞系は、ヒト胚性幹細胞の形態を有し、多能性を維持する」と述べている。
【0053】
米国特許出願公開第20020072117号は、無フィーダー細胞培養中で霊長類の多能性幹細胞の増殖を支持する培地を生成する細胞系を開示している。使用される細胞系は、胚性組織から得られるか、あるいは胚性幹細胞から分化した間葉系かつ繊維芽細胞様の細胞系である。米国特許出願公開第20020072117号は、この細胞系の1次フィーダー細胞層としての使用を更に開示している。
【0054】
別の例として、ワン等(Wang et al. Stem Cells 23: 1221-1227, 2005)は、ヒト胚性幹細胞由来のフィーダー細胞層上でヒト多能性幹細胞を長期にわたって増殖させるための方法を開示している。
【0055】
別の例として、ストイコビッチ等(Stojkovic et al. Stem Cells 2005 23: 306-314, 2005)は、ヒト胚性幹細胞の自然分化により誘導されたフィーダー細胞システムを開示している。
【0056】
更なる別の例として、ミヤモト等(Miyamoto et al. Stem Cells 22: 433-440, 2004)は、ヒトの胎盤から得られたフィーダー細胞の供給源を開示している。
【0057】
アミット等(Amit et al. Biol. Reprod 68: 2150-2156, 2003)は、ヒトの包皮由来のフィーダー細胞層を開示している。
【0058】
別の例として、インズンザ等(Inzunza et al. Stem Cells 23: 544-549, 2005)は、ヒトの出生直後産児の包皮繊維芽細胞から得られたフィーダー細胞層を開示している。
【0059】
米国特許第6642048号は、無フィーダー細胞培養中での霊長類の多能性幹(pPS)細胞の増殖を支持する培地、及びこうした培地の製造に有用な細胞系を開示している。米国特許第6642048号は、「本発明は、胚性組織から得られるか、あるいは胚性幹細胞から分化した間葉系かつ繊維芽細胞様の細胞系を含む。本開示では、こうした細胞系を誘導し、培地を調整し、この馴化培地を用いて幹細胞を増殖させるための方法を説明及び図示する」と述べている。
【0060】
別の例として、国際特許出願公開第2005014799号は、哺乳動物細胞の維持、増殖及び分化のための馴化培地を開示している。国際特許出願公開第2005014799号は、「本発明に基づいて製造される培地は、マウス細胞、特にMMH(Metマウス肝細胞)と称される分化及び不死化したトランスジェニック肝細胞の細胞分泌活性によって調整される」と述べている。
【0061】
別の例として、スー等(Xu et al. Stem Cells 22: 972-980, 2004)は、ヒトテロメラーゼ逆転写酵素を過剰発現するように遺伝子改変されたヒト胚性幹細胞由来細胞から得られた馴化培地を開示している。
【0062】
別の例として、米国特許出願公開第20070010011号は、多能性幹細胞の維持のための合成培地を開示している。
【0063】
代替的な培養システムでは、胚性幹細胞の増殖を促進することが可能な増殖因子を添加した無血清培地を使用している。例えば、チェオン等(Cheon et al. BioReprod DOI:10.1095/biolreprod.105.046870, October 19, 2005)は、胚性幹細胞の自己再生を誘発することが可能な異なる増殖因子を添加した非馴化血清補充(SR)培地中に胚性幹細胞が維持された、無フィーダー細胞かつ無血清の培養システムを開示している。
【0064】
別の例として、リーベンシュタイン等(Levenstein et al. Stem Cells 24: 568-574, 2006)は、bFGFを添加した培地を用い、繊維芽細胞又は馴化培地の非存在下でヒト胚性幹細胞を長期にわたって培養するための方法を開示している。
【0065】
別の例として、米国特許出願公開第20050148070号は、血清の非存在下及び繊維芽フィーダー細胞の非存在下において合成培地中でヒト胚性幹細胞を培養する方法を開示している。この方法は、アルブミン、アミノ酸、ビタミン、ミネラル、少なくとも1種類のトランスフェリン又はトランスフェリン代替物、少なくとも1種類のインスリン又はインスリン代替物を含む培地中で幹細胞を培養する工程を含み、前記培地は、哺乳動物胎児血清を基本的に含まず、少なくとも約100ng/mlの、繊維芽細胞増殖因子シグナル伝達受容体を活性化することが可能な繊維芽細胞増殖因子を含み、該増殖因子は、単に繊維芽細胞フィーダー層以外の供給源から供給され、前記培地は、フィーダー細胞又は馴化培地の非存在下で未分化状態の幹細胞の増殖を支持するものである。
【0066】
別の例として、米国特許出願公開第20050233446号は、未分化の霊長類始原幹細胞等の幹細胞を培養するうえで有用な合成培地を開示している。溶液中では、培地は培養される幹細胞と比較して実質的に等張である。与えられた培養中、この特定の培地は基礎培地、並びに始原幹細胞の実質的に未分化状態での増殖を支持するうえで必要とされるbFGF、インスリン、及びアスコルビン酸のそれぞれの所定量を含む。
【0067】
別の例として、米国特許第6800480号は、「一実施形態において、実質的に未分化状態の霊長類由来の始原幹細胞を増殖させるための細胞培地であって、霊長類由来の始原幹細胞の増殖を支持するうえで効果的な低浸透圧、低エンドトキシンの基礎培地を含む細胞培地を提供する。この基礎培地は、霊長類由来の始原幹細胞の増殖を支持するうえで効果的な栄養素血清、並びに、フィーダー細胞、及びフィーダー細胞から誘導される細胞外基質成分からなる群から選択される基質と組み合わされる。培地は更に、非必須アミノ酸、抗酸化剤、並びに、ヌクレオシド及びピルビン酸塩からなる群から選択される第1の増殖因子を含む。」と述べている。
【0068】
別の例では、米国特許出願公開第20050244962号は、「一態様において本発明は、霊長類の胚性幹細胞を培養する方法を提供する。哺乳動物の胎児血清を基本的に含まない(好ましくは、あらゆる動物の血清をも基本的に含まない)培養中で、単に繊維芽フィーダー細胞層以外の供給源から供給される繊維芽細胞増殖因子の存在下で幹細胞を培養する。好ましい一形態では、充分な量の繊維芽増殖因子を添加することによって、幹細胞の培養を維持するために従来必要とされていた繊維芽フィーダー細胞層の必要性がなくなる」と述べている。
【0069】
更なる例として、国際特許出願公開第2005065354号は、基本的に無フィーダー細胞かつ無血清の合成等張培地であって、a.基礎培地、b.実質的に未分化の哺乳動物幹細胞の増殖を支持するうえで充分な量のbFGF、c.実質的に未分化の哺乳動物幹細胞の増殖を支持するうえで充分な量のインスリン、及び、d.実質的に未分化の哺乳動物幹細胞の増殖を支持するうえで充分な量のアスコルビン酸を含む培地を開示している。
【0070】
別の例として、国際特許出願公開第2005086845号は、未分化の幹細胞を維持するための方法であって、幹細胞を未分化状態に維持するのに充分な量の形質転換増殖因子β(transforming growth factor:TGFβ)のタンパク質ファミリーのメンバー、繊維芽細胞増殖因子(FGF)のタンパク質ファミリーのメンバー、又はニコチンアミド(NIC)に、所望の結果を得るのに充分な時間だけ、幹細胞を曝露することを含む方法を開示している。
【0071】
多能性幹細胞は、適当な培養基質上に播種すればよい。一実施形態では、適当な培養基質は例えば、基底膜から誘導されるもの、又は接着分子の受容体/リガンド結合の一部を形成するもののような細胞外基質成分である。一実施形態では、適当な培養基質はMATRIGEL(登録商標)(ベクトン・ディッキンソン社(Becton Dickenson))である。MATRIGEL(登録商標)は、室温でゲル化して再構成基底膜を形成する、エンゲルブレス−ホルム・スワーム(Engelbreth-Holm Swarm)腫瘍細胞から得られる可溶性調製物である。
【0072】
他の細胞外基質成分及び成分混合物も代替物として適している。増殖させられる細胞の種類に応じ、他の細胞外基質成分及び成分混合物は、ラミニン、フィブロネクチン、プロテオグリカン、エンタクチン、硫酸ヘパリン等を、単独又は異なる組み合わせで含んでもよい。
【0073】
多能性幹細胞は、適当な分布で、かつ細胞の生存率、増殖、及び所望の特性の維持を促進する培地の存在下で基質上に播種することができる。これらの特性はいずれも、播種密度に充分な注意を払うことによって利するところがあるものであり、当業者が容易に決定することができるものである。
【0074】
好適な培地は、以下の成分、即ち、例えば、ダルベッコ改変イーグル培地(DMEM)、ギブコ(Gibco)No.11965−092;ノックアウトダルベッコ改変イーグル培地(KO DMEM)、ギブコNo.10829−018;ハムF12/50%DMEM基礎培地;200mM L−グルタミン、ギブコNo.15039−027;非不可欠アミノ酸溶液、ギブコNo.11140−050;β−メルカプトエタノール、シグマ(Sigma)No.7522;ヒト組み換え塩基性繊維芽細胞増殖因子(bFGF)、ギブコNo.13256−029等から調製することができる。
【0075】
膵臓内分泌系統に特徴的なマーカーを発現する細胞への多能性幹細胞の分化
本発明における使用に適した多能性幹細胞としては例えば、胚性幹細胞系H9(NIHコード:WA09)、ヒト胚性幹細胞系H1(NIHコード:WA01)、ヒト胚性幹細胞系H7(NIHコード:WA07)、及びヒト胚性幹細胞系SA002(セラーティス社(Cellartis)、スウェーデン)が挙げられる。多能性細胞に特徴的な以下のマ−カー、即ち、ABCG2、クリプト(cripto)、CD9、FoxD3、コネキシン43、コネキシン45、Oct4、Sox2、ナノグ(Nanog)、hTERT、UTF−1、ZFP42、SSEA−3、SSEA−4、Tra1−60、Tra1−81の内の少なくとも1つを発現する細胞も本発明における使用に適している。
【0076】
胚体内胚葉系統に特徴的なマーカーは、SOX−17、GATA4、Hnf−3β、GSC、Cer1、ノーダル(Nodal)、FGF8、ブラキュリ(Brachyury)、Mix様ホメオボックスタンパク質、FGF4 CD48、エオメソダーミン(eomesodermin)(EOMES)、DKK4、FGF17、GATA6、CXCR4、C−Kit、CD99、及びOTX2からなる群から選択される。胚体内胚葉系統に特徴的なこれらのマーカーの内の少なくとも1つを発現する細胞が本発明における使用に適している。本発明の一態様では、胚体内胚葉系統に特徴的なマーカーを発現する細胞は、原始線条前駆細胞である。代替的な態様では、胚体内胚葉系統に特徴的なマーカーを発現する細胞は、中内胚葉細胞である。代替的な態様では、胚体内胚葉系統に特徴的なマーカーを発現する細胞は、胚体内胚葉細胞である。
【0077】
膵臓内胚葉系統に特徴的なマーカーは、Pdx1、HNF−1β、PTF1a、HNF−6、HB9及びPROX1からなる群から選択される。膵臓内胚葉系統に特徴的なこれらのマーカーの内の少なくとも1つを発現する細胞が本発明における使用に適している。本発明の一態様では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞は、膵臓内胚葉細胞である。
【0078】
膵臓内分泌系統に特徴的なマーカーは、NGN−3、ニューロD(NeuroD)、アイレット−1(Islet-1)、Pdx−1、NKX6.1、Pax−4、Ngn−3、及びPTF−1αからなる群から選択される。一実施形態では、膵臓内分泌細胞は、以下のホルモン、即ち、インスリン、グルカゴン、ソマトスタチン、及び膵臓ポリペプチドの内の少なくとも1つを発現することが可能である。膵臓内分泌系統に特徴的なこれらのマーカーの内の少なくとも1つを発現する細胞が本発明における使用に適している。本発明の一態様では、膵臓内分泌系統に特徴的なマーカーを発現する細胞は、膵臓内分泌細胞である。膵臓内分泌細胞は、膵臓ホルモン発現細胞であってもよい。また、膵臓内分泌細胞は膵臓ホルモン分泌細胞であってもよい。
【0079】
本発明の一態様では、膵臓内分泌細胞は、β細胞系統に特徴的なマーカーを発現する細胞である。β細胞系統に特徴的なマーカーを発現する細胞は、Pdx1、並びに以下の転写因子、即ち、NGN−3、Nkx2.2、Nkx6.1、ニューロD(NeuroD)、Isl−1、HNF−3β、MAFA、Pax4及びPax6の内の少なくとも1つを発現する。本発明の一態様では、β細胞系統に特徴的なマーカーを発現する細胞は、β細胞である。
【0080】
胚体内胚葉系統に特徴的なマーカーを発現する細胞の形成
多能性幹細胞を、当該技術分野のいかなる方法、又は本発明で提案されるいかなる方法によって胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させてもよい。
【0081】
例えば、多能性幹細胞を、ダムール(D’Amour)等により開示される方法に従って胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる(D’Amour et al, Nature Biotechnology 23, 1534 - 1541(2005))。
【0082】
例えば、多能性幹細胞を、シノザキ(Shinozaki)等により開示される方法に従って胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる(Shinozaki et al, Development 131, 1651 - 1662(2004))。
【0083】
例えば、多能性幹細胞を、マクレーン(McLean)等により開示される方法に従って胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる(McLean et al, Stem Cells 25, 29 - 38(2007))。
【0084】
例えば、多能性幹細胞を、ダムール(D’Amour)等により開示される方法に従って胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる(D’Amour et al, Nature Biotechnology 24, 1392 - 1401(2006))。
【0085】
例えば、多能性幹細胞を、アクチビンAを含む培地中、血清の非存在下で多能性幹細胞を培養し、次いで細胞をアクチビンA及び血清と培養し、次いで細胞をアクチビンA及び異なる濃度の血清と培養することによって胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる。この方法の一例は、(Nature Biotechnology 23, 1534 - 1541(2005))に開示されている。
【0086】
例えば、多能性幹細胞を、アクチビンAを含む培地中、血清の非存在下で多能性幹細胞を培養し、次いで細胞をアクチビンA及び血清と培養し、次いで細胞をアクチビンAと、別の濃度の血清の存在下で培養することによって胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる。この方法の一例は、ダムール(D’Amour)等により開示されている(D’Amour et al, Nature Biotechnology, 2005)。
【0087】
例えば、多能性幹細胞を、アクチビンA及びWntリガンドを含む培地中、血清の非存在下で多能性幹細胞を培養し、次いでWntリガンドを除去し、細胞をアクチビンAと、血清の存在下で培養することによって胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる。この方法の一例は、(Nature Biotechnology 24, 1392 - 1401(2006))に記載されている。
【0088】
本発明の一態様では、多能性幹細胞は、細胞外基質でコーティングした組織培養基質上に多能性幹細胞を播種し、次いでこの多能性幹細胞をアクチビンA及びWntリガンドと、血清を含む第1の培地中で所定の時間培養し、次いでこの多能性幹細胞をアクチビンAと、より高い濃度の血清を含む第2の培地中でおおよそ別の所定の時間培養することによって胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる。
【0089】
上記に開示される第1の培地中の血清の濃度は、約0〜約0.5%であってもよく、培養時間は約1〜約3日間であってもよい。上記に開示される第2の培地中の血清の濃度は、約0.5〜約2%であってもよく、培養時間は約1〜約4日間であってもよい。
【0090】
本発明の代替的な一態様では、多能性幹細胞は、細胞外基質でコーティングした組織培養基質上に多能性幹細胞を播種し、次いでこの多能性幹細胞をアクチビンA及びWntリガンドと、血清を含む第1の培地中でおおよそ所定の時間培養し、次いでこの多能性幹細胞をアクチビンA及びWntリガンドと、より高い濃度の血清を含む第2の培地中で別の所定の時間培養することによって胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる。
【0091】
上記に開示される第1の培地中の血清の濃度は、約0〜約0.5%であってもよく、培養時間は約1〜約3日間であってもよい。上記に開示される第2の培地中の血清の濃度は、約0.5〜約2%であってもよく、培養時間は約1〜約4日間であってもよい。
【0092】
一実施形態では、本発明は、胚体内胚葉系統に特徴的なマーカーを発現する多能性幹細胞を分化させる方法であって、
a.細胞外基質でコーティングされた組織培養基質上に多能性幹細胞を播種する工程と、
b.前記多能性幹細胞を、アクチビンA及びWntリガンドと培養する工程と、を含む、方法を提供する。
【0093】
多能性幹細胞をアクチビンA及びWntリガンドと培養する工程は、1個の培地中で行うことが可能である。また、多能性幹細胞をアクチビンA及びWntリガンドと培養する工程は、1個よりも多い培地中で別々に又は同時に行うことも可能である。一実施形態では、多能性幹細胞をアクチビンA及びWntリガンドと培養する工程を2個の培地中で行う。
【0094】
細胞外基質
本発明の一態様では、多能性細胞を、細胞外基質でコーティングされた組織培養基質上で培養して分化させる。細胞外基質は、マウスの肉腫細胞から抽出された可溶化基底膜の調製物(BDバイオサイエンス社(BD Biosciences)よりMATRIGELの商品名で販売されている)であってよい。また、細胞外基質は、増殖因子を低減させたMATRIGELであってもよい。また、細胞外基質はフィブロネクチンであってもよい。代替的な一実施形態では、多能性幹細胞を、ヒト血清でコーティングされた組織培養基質上で培養して分化させる。
【0095】
細胞外基質は、組織培養基質をコーティングするのに先立って希釈してもよい。組織培養基質をコーティングするための細胞外基質を希釈するのに適した方法の例は、クラインマン(Kleinman)等(Kleinman, H.K., et al., Biochemistry 25:312(1986))、及びハドレイ(Hadley)等(Hadley, M.A., et al., J.Cell.Biol. 101:1511(1985))に見ることができる。
【0096】
一実施形態では、細胞外基質はMATRIGELである。一実施形態では、組織培養基質を希釈率が1:10のMATRIGELでコーティングする。代替的な一実施形態では、組織培養基質を希釈率が1:15のMATRIGELでコーティングする。代替的な一実施形態では、組織培養基質を希釈率が1:30のMATRIGELでコーティングする。代替的な一実施形態では、組織培養基質を希釈率が1:60のMATRIGELでコーティングする。
【0097】
一実施形態では、細胞外基質は増殖因子低減MATRIGELである。一実施形態では、組織培養基質を希釈率が1:10の増殖因子低減MATRIGELでコーティングする。代替的な一実施形態では、組織培養基質を希釈率が1:15の増殖因子低減MATRIGELでコーティングする。代替的な一実施形態では、組織培養基質を希釈率が1:30の増殖因子低減MATRIGELでコーティングする。代替的な一実施形態では、組織培養基質を希釈率が1:60の増殖因子低減MATRIGELでコーティングする。
【0098】
1個の培地を使用した、細胞外基質上での、胚体内胚葉系統に特徴的なマーカーを発現する細胞への多能性幹細胞の分化
1個の培地を使用する場合、培地は、例えば(国際特許出願公開第2006020919号に開示されるような)インスリン及びIGF等の特定の因子を、多能性幹細胞が胚体内胚葉に分化できるように充分に低い濃度で含有しなければならない。これは血清濃度を低下させるか、あるいはインスリン及びIGFを含まない合成培地を使用することによって実現することが可能である。合成培地の例はワイルズ(Wiles)等によって開示されている(Wiles et al. Exp Cell Res. 1999 Feb 25; 247(1): 241-8)。
【0099】
前記培地は、約0%〜約10%の範囲の血清濃度を有してもよい。代替的な一実施形態では、濃度は約0%〜約5%の範囲であってもよい。代替的な一実施形態では、濃度は約0%〜約2%の範囲であってもよい。代替的な一実施形態では、濃度は約2%であってもよい。
【0100】
アクチビンA及びWntリガンとの培養時間は約1日〜約7日であってもよい。代替的な一実施形態では、培養時間は約1日〜約3日であってもよい。代替的な一実施形態では、培養時間は約3日であってもよい。
【0101】
アクチビンAは、多能性幹細胞の分化を引き起こすのに適した任意の濃度で使用することができる。濃度は約1pg/ml〜約100μg/mlであってもよい。代替的な一実施形態では、濃度は約1pg/ml〜約1μg/mlであってもよい。別の代替的な一実施形態では、濃度は約1pg/ml〜約100ng/mlであってもよい。別の代替的な一実施形態では、濃度は約50ng/ml〜約100ng/mlであってもよい。別の代替的な一実施形態では、濃度は約100ng/mlであってもよい。
【0102】
Wntリガンドの選択を最適化することによって分化プロセスの効率を高めることが可能である。Wntリガンドは、Wnt−1、Wnt−3a、Wnt−5a及びWnt−7aからなる群から選択することができる。一実施形態では、WntリガンドはWnt−1である。代替的な一実施形態では、WntリガンドはWnt−3aである。
【0103】
Wntリガンドは約1ng/ml〜約1000ng/mlの濃度でもよい。代替的な一実施形態では、濃度は約10ng/ml〜約100ng/mlであってもよい。
【0104】
前記1個の培地は、GSK−3B阻害剤を更に含んでもよい。GSK−3B阻害剤は、GSK−3B阻害剤IX及びGSK−3B阻害剤XIからなる群から選択することができる。一実施形態では、GSK−3B阻害剤はGSK−3B阻害剤IXである。
【0105】
多能性幹細胞をGSK−3B阻害剤と培養する場合、GSK−3B阻害剤の濃度は約1nM〜約1000nMでもよい。代替的な一実施形態では、多能性幹細胞を約10nM〜約100nMの濃度のGSK−3B阻害剤と培養する。
【0106】
前記1個の培地は、胚体内胚葉系統に特徴的なマーカーを発現する細胞の多能性細胞からの形成を促進することが可能な少なくとも1種類の更なる他の因子を含んでもよい。また、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される胚体内胚葉に特徴的なマーカーを発現する細胞の増殖を促進するものであってもよい。更に、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される胚体内胚葉系統に特徴的なマーカーを発現する細胞が、他の細胞種を形成する能力を促進するものであってもよく、あるいは他のいずれかの更なる分化の段階の効率を高めるものであってもよい。
【0107】
前記少なくとも1種類の更なる他の因子は、例えば、ニコチンアミド、TGF−β1、2及び3等のTGFβファミリーのメンバー、血清アルブミン、繊維芽細胞増殖因子ファミリーのメンバー、血小板由来増殖因子−AA及び−BB、血小板濃縮血漿、インスリン様増殖因子(IGF−I、II)、増殖分化因子(GDF−5、−6、−8、−10、−11)、グルカゴン様ペプチド−1及び−II(GLP−1及びII)、GLP−1及びGLP−2模倣体(mimetobody)、エキセンジン−4、レチノイン酸、副甲状腺ホルモン、インスリン、プロゲステロン、アプロチニン、ヒドロコルチゾン、エタノールアミン、βメルカプトエタノール、上皮増殖因子(EGF)、ガストリンI及びII、例えば、トリエチレンペンタミン等の銅キレート化剤、フォルスコリン、酪酸ナトリウム、アクチビン、ベータセルリン、ITS、ノギン、神経突起増殖因子、ノーダル、バルプロ酸、トリコスタチンA、酪酸ナトリウム、肝細胞増殖因子(HGF)、スフィンゴシン1、VEGF、MG132(EMD社、カリフォルニア州)、N2及びB27添加物(ギブコ社(Gibco)、カリフォルニア州)、例えば、シクロパミン(EMD社、カリフォルニア州)等のステロイドアルカロイド、ケラチノサイト増殖因子(KGF)、Dickkopfタンパク質ファミリー、ウシ脳下垂体抽出物、膵島新生関連タンパク質(INGAP)、インディアンヘッジホッグ、ソニックヘッジホッグ、プロテアソーム阻害剤、ノッチ経路阻害剤、ソニックヘッジホッグ阻害剤、又はこれらの組み合わせであってもよい。
【0108】
前記少なくとも1種類の更なる他の因子は、例えば、PANC−1(ATCC No:CRL−1469)、CAPAN−1(ATCC No:HTB−79)、BxPC−3(ATCC No:CRL−1687)、HPAF−II(ATCC No:CRL−1997)等の膵臓細胞系、例えば、HepG2(ATCC No:HTB−8065)等の肝細胞系、例えば、FHs74(ATCC No:CCL−241)等の腸細胞系、及び始原又は形質転換内皮細胞から得られる馴化培地によって供給することが可能である。
【0109】
2個の培地を使用した、細胞外基質上での、胚体内胚葉系統に特徴的なマーカーを発現する細胞への多能性幹細胞の分化
多能性幹細胞の胚体内胚葉系統細胞への分化は、2個の培地を使用して多能性幹細胞をアクチビンA及びWntリガンドと培養することによって実現することが可能である。即ち、多能性幹細胞の分化は以下のようにして実現することが可能である。
a.細胞外基質でコーティングされた組織培養基質上に多能性幹細胞を播種し、
b.前記多能性幹細胞を第1の培地中でアクチビンA及びWntリガンドと培養し、
c.前記多能性幹細胞を第2の培地中でアクチビンAと培養する。
【0110】
第1の培地は低濃度の血清を含んでもよく、第2の培地は第1の培地よりも高濃度の血清を含んでもよい。
【0111】
第2の培地はWntリガンドを含んでもよい。
【0112】
第1の培地:第1の培地は、例えば(国際特許出願公開第2006020919号に開示されるような)インスリン及びIGF等の特定の因子を、多能性幹細胞が胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化できるように充分に低い濃度で含有しなければならない。これは血清濃度を低下させるか、あるいはインスリン及びIGFを含まない合成培地を使用することによって実現することが可能である。合成培地の例はワイルズ(Wiles)等によって開示されている(Wiles et al. Exp Cell Res. 1999 Feb 25; 247(1): 241-8)。
【0113】
第1の培地では、第2の培地と比較してより低濃度の血清が存在してもよい。第2の培地の血清濃度を増大させると細胞の生存率が高まるか、あるいは細胞の増殖が促進される場合がある。第1の培地の血清濃度は約0%〜約10%の範囲であってもよい。また、第1の培地の血清濃度は約0%〜約2%の範囲であってもよい。また、第1の培地の血清濃度は約0%〜約1%の範囲であってもよい。また、第1の培地の血清濃度は約0.5%であってもよい。
【0114】
少なくとも2個の培地を使用して多能性幹細胞をアクチビンA及びWntリガンドと培養する場合、第1の培地中での培養時間は約1日〜約3日であってもよい。
【0115】
アクチビンAは、多能性幹細胞の分化を引き起こすのに適した任意の濃度で使用することができる。濃度は約1pg/ml〜約100μg/mlであってもよい。代替的な一実施形態では、濃度は約1pg/ml〜約1μg/mlであってもよい。別の代替的な一実施形態では、濃度は約1pg/ml〜約100ng/mlであってもよい。別の代替的な一実施形態では、濃度は約50ng/ml〜約100ng/mlであってもよい。別の代替的な一実施形態では、濃度は約100ng/mlであってもよい。
【0116】
Wntリガンドの選択を最適化することによって分化プロセスの効率を高めることが可能である。Wntリガンドを、Wnt−1、Wnt−3a、Wnt−5a及びWnt−7aからなる群から選択することができる。一実施形態では、WntリガンドはWnt−1である。代替的な一実施形態では、WntリガンドはWnt−3aである。
【0117】
Wntリガンドは約1ng/ml〜約1000ng/mlの濃度でもよい。代替的な一実施形態では、濃度は約10ng/ml〜約100ng/mlであってもよい。
【0118】
前記第1の培地は、GSK−3B阻害剤を更に含んでもよい。GSK−3B阻害剤は、第1の培地、第2の培地、又は第1及び第2の培地の両方に添加することができる。
【0119】
GSK−3B阻害剤は、GSK−3B阻害剤IX及びGSK−3B阻害剤XIからなる群から選択することができる。一実施形態では、GSK−3B阻害剤はGSK−3B阻害剤IXである。
【0120】
多能性幹細胞をGSK−3B阻害剤と培養する場合、GSK−3B阻害剤の濃度は約1nM〜約1000nMでもよい。代替的な一実施形態では、多能性幹細胞を約10nM〜約100nMの濃度のGSK−3B阻害剤と培養する。
【0121】
前記第1の培地は、胚体内胚葉系統に特徴的なマーカーを発現する細胞の多能性細胞からの形成を促進することが可能な少なくとも1種類の更なる他の因子を含んでもよい。また、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される胚体内胚葉に特徴的なマーカーを発現する細胞の増殖を促進するものであってもよい。更に、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される胚体内胚葉系統に特徴的なマーカーを発現する細胞が他の細胞種を形成する能力を促進するものであってもよく、あるいは他のいずれかの更なる分化の段階の効率を高めるものであってもよい。
【0122】
前記少なくとも1種類の更なる他の因子は、例えば、ニコチンアミド、TGF−β1、2及び3等のTGFβファミリーのメンバー、血清アルブミン、繊維芽細胞増殖因子ファミリーのメンバー、血小板由来増殖因子−AA及び−BB、血小板濃縮血漿、インスリン様増殖因子(IGF−I、II)、増殖分化因子(GDF−5、−6、−8、−10、−11)、グルカゴン様ペプチド−1及び−II(GLP−1及びII)、GLP−1及びGLP−2模倣体(mimetobody)、エキセンジン−4、レチノイン酸、副甲状腺ホルモン、インスリン、プロゲステロン、アプロチニン、ヒドロコルチゾン、エタノールアミン、βメルカプトエタノール、上皮増殖因子(EGF)、ガストリンI及びII、例えば、トリエチレンペンタミン等の銅キレート化剤、フォルスコリン、酪酸ナトリウム、アクチビン、ベータセルリン、ITS、ノギン、神経突起増殖因子、ノーダル、バルプロ酸、トリコスタチンA、酪酸ナトリウム、肝細胞増殖因子(HGF)、スフィンゴシン1、VEGF、MG132(EMD社、カリフォルニア州)、N2及びB27添加物(ギブコ社(Gibco)、カリフォルニア州)、例えば、シクロパミン(EMD社、カリフォルニア州)等のステロイドアルカロイド、ケラチノサイト増殖因子(KGF)、Dickkopfタンパク質ファミリー、ウシ脳下垂体抽出物、膵島新生関連タンパク質(INGAP)、インディアンヘッジホッグ、ソニックヘッジホッグ、プロテアソーム阻害剤、ノッチ経路阻害剤、ソニックヘッジホッグ阻害剤、又はこれらの組み合わせであってもよい。
【0123】
前記少なくとも1種類の更なる他の因子は、例えば、PANC−1(ATCC No:CRL−1469)、CAPAN−1(ATCC No:HTB−79)、BxPC−3(ATCC No:CRL−1687)、HPAF−II(ATCC No:CRL−1997)等の膵臓細胞系、例えばHepG2(ATCC No:HTB−8065)等の肝細胞系、及び例えば、FHs74(ATCC No:CCL−241)等の腸細胞系から得られる馴化培地によって供給することが可能である。
【0124】
第2の培地:第2の培地は、例えば、(国際特許出願公開第2006020919号に開示されるような)インスリン及びIGF等の特定の因子を培養細胞の生存を促すだけの充分な濃度で含有しなければならない。これは、血清濃度を増大させるか、あるいはインスリン及びIGFの濃度が第1の培地と比較して高い合成培地を使用することによって実現することが可能である。合成培地の例はワイルズ(Wiles)等によって開示されている(Wiles et al. Exp Cell Res. 1999 Feb 25; 247(1): 241-8)。
【0125】
より高い血清濃度を有する第2の培地では、第2の培地の血清濃度は、約0.5%〜約10%の範囲であってもよい。また、第2の培地の血清濃度は約0.5%〜約5%の範囲であってもよい。また、第2の培地の血清濃度は約0.5%〜約2%の範囲であってもよい。また、第2の培地の血清濃度は約2%であってもよい。多能性幹細胞を第2の培地と培養する場合、培養時間は約1日〜約4日であってもよい。
【0126】
第1の培地と同様、アクチビンAは、多能性幹細胞の分化を引き起こすのに適した任意の濃度で使用することができる。濃度は約1pg/ml〜約100μg/mlであってもよい。代替的な一実施形態では、濃度は約1pg/ml〜約1μg/mlであってもよい。別の代替的な一実施形態では、濃度は約1pg/ml〜約100ng/mlであってもよい。別の代替的な一実施形態では、濃度は約50ng/ml〜約100ng/mlであってもよい。別の代替的な一実施形態では、濃度は約100ng/mlであってもよい。
【0127】
Wntリガンドは約1ng/ml〜約1000ng/mlの濃度でもよい。代替的な一実施形態では、濃度は約10ng/ml〜約100ng/mlであってもよい。
【0128】
Wntリガンドは、Wnt−1、Wnt−3a、Wnt−5a及びWnt−7aからなる群から選択することができる。一実施形態では、WntリガンドはWnt−1である。代替的な一実施形態では、WntリガンドはWnt−3aである。
【0129】
前記第2の培地は、GSK−3B阻害剤を更に含んでもよい。GSK−3B阻害剤は、第1の培地、第2の培地、又は第1及び第2の培地の両方に添加することができる。
【0130】
GSK−3B阻害剤は、GSK−3B阻害剤IX及びGSK−3B阻害剤XIからなる群から選択することができる。一実施形態では、GSK−3B阻害剤はGSK−3B阻害剤IXである。
【0131】
多能性幹細胞をGSK−3B阻害剤と培養する場合、GSK−3B阻害剤の濃度は約1nM〜約1000nMでもよい。代替的な一実施形態では、多能性幹細胞を約10nM〜約100nMの濃度のGSK−3B阻害剤と培養する。
【0132】
第1の培地と同様、第2の培地も、胚体内胚葉系統に特徴的なマーカーを発現する細胞の多能性細胞からの形成を促進することが可能な少なくとも1種類の更なる他の因子を含んでもよい。また、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される胚体内胚葉に特徴的なマーカーを発現する細胞の増殖を促進するものであってもよい。更に、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される胚体内胚葉系統に特徴的なマーカーを発現する細胞が他の細胞種を形成する能力を促進するものであってもよく、あるいは他のいずれかの更なる分化の段階の効率を高めるものであってもよい。
【0133】
前記少なくとも1種類の更なる他の因子は、例えば、ニコチンアミド、TGF−β1、2及び3等のTGFβファミリーのメンバー、血清アルブミン、繊維芽細胞増殖因子ファミリーのメンバー、血小板由来増殖因子−AA及び−BB、血小板濃縮血漿、インスリン様増殖因子(IGF−I、II)、増殖分化因子(GDF−5、−6、−8、−10、−11)、グルカゴン様ペプチド−1及び−II(GLP−1及びII)、GLP−1及びGLP−2模倣体(mimetobody)、エキセンジン−4、レチノイン酸、副甲状腺ホルモン、インスリン、プロゲステロン、アプロチニン、ヒドロコルチゾン、エタノールアミン、βメルカプトエタノール、上皮増殖因子(EGF)、ガストリンI及びII、例えば、トリエチレンペンタミン等の銅キレート化剤、フォルスコリン、酪酸ナトリウム、アクチビン、ベータセルリン、ITS、ノギン、神経突起増殖因子、ノーダル、バルプロ酸、トリコスタチンA、酪酸ナトリウム、肝細胞増殖因子(HGF)、スフィンゴシン1、VEGF、MG132(EMD社、カリフォルニア州)、N2及びB27添加物(ギブコ社(Gibco)、カリフォルニア州)、例えば、シクロパミン(EMD社、カリフォルニア州)等のステロイドアルカロイド、ケラチノサイト増殖因子(KGF)、Dickkopfタンパク質ファミリー、ウシ脳下垂体抽出物、膵島新生関連タンパク質(INGAP)、インディアンヘッジホッグ、ソニックヘッジホッグ、プロテアソーム阻害剤、ノッチ経路阻害剤、ソニックヘッジホッグ阻害剤、又はこれらの組み合わせであってもよい。
【0134】
前記少なくとも1種類の更なる他の因子を、例えば、PANC−1(ATCC No:CRL−1469)、CAPAN−1(ATCC No:HTB−79)、BxPC−3(ATCC No:CRL−1687)、HPAF−II(ATCC No:CRL−1997)等の膵臓細胞系、例えばHepG2(ATCC No:HTB−8065)等の肝細胞系、及び例えば、FHs74(ATCC No:CCL−241)等の腸細胞系から得られる馴化培地によって供給することが可能である。
【0135】
胚体内胚葉系統に特徴的なマーカーを発現する細胞の分化
胚体内胚葉系統に特徴的なマーカーを発現する細胞が形成されたことは、特定のプロトコールを行う前後にマーカーの存在について試験を行うことによって判定することができる。多能性幹細胞は通常、こうしたマーカーを発現しない。したがって、多能性細胞の分化は、細胞がマーカーを発現し始めることで検出される。
【0136】
分化の効率は、胚体内胚葉系統に特徴的なマーカーを発現する細胞によって発現されるタンパク質マーカーを特異的に認識する薬剤(抗体等)に、処理した細胞集団を曝露することによって決定することができる。
【0137】
培養細胞又は単離細胞中のタンパク質及び核酸マーカーの発現を評価するための方法は、当該技術分野では標準的なものである。こうした方法には、定量的逆転写ポリメラーゼ連鎖反応(RT−PCR)、ノーザンブロット、in situハイブリダイゼーション(例えば、「分子生物学の最新プロトコール」(Current Protocols in Molecular Biology)オウスベル(Ausubel)等編、2001年度版補遺、を参照)、並びに、切片化材料の免疫組織化学的分析、ウエスタンブロッティング、及び無傷細胞中のアクセシブルなマーカーに対する免疫アッセイ、フローサイトメトリー分析(FACS)(例えば、ハーロー及びレーン(Harlow and Lane)著、「抗体の使用:実験マニュアル」(Using Antibodies: A Laboratory Manual)ニューヨーク州コールドスプリングハーバーラボラトリープレス(1998年)を参照)等の免疫アッセイが含まれる。
【0138】
特定のタンパク質マーカーの検出に有用な抗体の例を表IAに列記する。表IAに列記した抗体によって認識されるものと同じマーカーを標的とする代替的な抗体も入手可能であるか、容易に開発が可能である点は注意を要する。こうした代替的な抗体を、本発明に基づいて単離した細胞におけるマーカーの発現を評価するために用いることもできる。
【0139】
例えば、多能性幹細胞の特徴は当業者にはよく知られたものであり、多能性幹細胞の更なる特徴も次々に特定されている。多能性幹細胞のマーカーとしては、例えば、以下のもの、即ち、ABCG2、クリプト(cripto)、FoxD3、コネキシン43、コネキシン45、Oct4、Sox2、ナノグ(nanog)、hTERT、UTF−1、ZFP42、SSEA−3、SSEA−4、Tra1−60、Tra1−81の内の1つ以上の発現が挙げられる。
【0140】
多能性幹細胞を本発明の方法で処理した後、胚体内胚葉系統に特徴的なマーカーを発現する細胞によって発現されるCXCR4のようなタンパク質マーカーを特異的に認識する薬剤(抗体等)に処理細胞集団を曝露することによって、分化した細胞を精製することができる。
【0141】
膵臓内胚葉系統に特徴的なマーカーを発現する細胞の形成
胚体内胚葉系統に特徴的なマーカーを発現する細胞を、当該技術分野におけるいずれかの方法又は本発明において提案されるいずれかの方法によって膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる。
【0142】
例えば、胚体内胚葉系統に特徴的なマーカーを発現する細胞を、ダムール(D’Amour)等により開示される方法に従って膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる(D’Amour et al, Nature Biotechnology 24, 1392 - 1401(2006))。
【0143】
例えば、胚体内胚葉系統に特徴的なマーカーを発現する細胞を繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤KAAD−シクロパミンで処理し、次いで繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤KAAD−シクロパミンを含む培地を取り除いた後、レチノイン酸、繊維芽細胞増殖因子及びKAAD−シクロパミンを含む培地中で細胞を培養することによって、胚体内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内胚葉系統に特徴的なマーカーを発現する細胞に更に分化させる。この方法の一例は、(Nature Biotechnology 24, 1392 - 1401(2006))に開示されている。
【0144】
本発明の一態様では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で所定の時間処理することによって、胚体内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内胚葉系統に特徴的なマーカーを発現する細胞に更に分化させる。その時間は、約1〜約6日でもよい。
【0145】
本発明の代替的一態様では、胚体内胚葉系統に特徴的なマーカーを発現する細胞を、細胞をレチノイン酸で所定の時間処理することによって膵臓内胚葉系統に特徴的なマーカーを発現する細胞に更に分化させる。その時間は、約1〜約3日でもよい。レチノイン酸を後で取り除き、細胞を少なくとも1種類の繊維芽細胞増殖因子で別の所定の時間処理する。その時間は、約1〜約3日でもよい。
【0146】
一実施形態において、本発明は、胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させるための方法であって、
a.胚体内胚葉系統に特徴的なマーカーを発現する細胞を培養する工程と、
b.胚体内胚葉系統に特徴的なマーカーを発現する前記細胞をレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で処理する工程と、を含む、方法を提供する。
【0147】
胚体内胚葉系統に特徴的なマーカーを発現するいずれの細胞も、この方法を用いて膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させるうえで適している。
【0148】
一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で約1〜約6日間処理する。一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で約6日間処理する。
【0149】
前記少なくとも1種類の繊維芽細胞増殖因子は、FGF−2、FGF−4及びFGF−10からなる群から選択される。
【0150】
胚体内胚葉系統に特徴的なマーカーを発現するいずれの細胞も、この方法を用いて膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させるうえで適している。
【0151】
代替的な一実施形態において、本発明は、胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させるための方法であって、
a.胚体内胚葉系統に特徴的なマーカーを発現する細胞を培養する工程と、
b.胚体内胚葉系統に特徴的なマーカーを発現する前記細胞をレチノイン酸で処理する工程と、
c.レチノイン酸を取り除き、その後、細胞を少なくとも1種類の繊維芽細胞増殖因子で処理する工程と、を含む、方法を提供する。
【0152】
胚体内胚葉系統に特徴的なマーカーを発現するいずれの細胞も、この方法を用いて膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させるうえで適している。
【0153】
一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸で約1〜約3日間処理する。一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸で約3日間処理する。一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞を少なくとも1種類の繊維芽細胞増殖因子で約1〜約3日間処理する。一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞を少なくとも1種類の繊維芽細胞増殖因子で約3日間処理する。
【0154】
前記少なくとも1種類の繊維芽細胞増殖因子は、FGF−2、FGF−4及びFGF−10からなる群から選択される。
【0155】
胚体内胚葉系統に特徴的なマーカーを発現するいずれの細胞も、この方法を用いて膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させるうえで適している。一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸で処理する。あるいは、胚体内胚葉系統に特徴的なマーカーを発現する細胞をFGF−2、又はFGF−4、又はFGF−10で処理する。代替的な一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞を以下の因子、即ち、レチノイン酸、FGF−2、FGF−4、又はFGF−10の内の少なくとも1つで処理する。代替的な一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞を、レチノイン酸と、以下の繊維芽細胞増殖因子、即ち、FGF−2、FGF−4、又はFGF−10の内の少なくとも1つと、で処理する。一実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸及びFGF−2で処理する。別の実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸及びFGF−4で処理する。更なる実施形態では、胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸及びFGF−10で処理する。
【0156】
レチノイン酸を約1nM〜約1mMの濃度で使用することができる。一実施形態では、レチノイン酸を1μMの濃度で使用する。
【0157】
FGF−2は約50pg/ml〜約50μg/mlの濃度で使用することができる。一実施形態では、FGF−2を50ng/mlの濃度で使用する。
【0158】
FGF−4は約50pg/ml〜約50μg/mlの濃度で使用することができる。一実施形態では、FGF−4を50ng/mlの濃度で使用する。
【0159】
FGF−10は約50pg/ml〜約50μg/mlの濃度で使用することができる。一実施形態では、FGF−10を50ng/mlの濃度で使用する。
【0160】
胚体内胚葉系統に特徴的なマーカーを発現する細胞は、膵臓内胚葉系統に特徴的なマーカーを発現する細胞の形成を促進することが可能な少なくとも1種類の更なる他の因子で処理してもよい。また、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される膵臓内胚葉系統に特徴的なマーカーを発現する細胞の増殖を促進するものであってもよい。更に、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される膵臓内胚葉系統に特徴的なマーカーを発現する細胞が、他の細胞種を形成する能力を促進するものであってもよく、あるいは他のいずれかの更なる分化の段階の効率を高めるものであってもよい。
【0161】
前記少なくとも1種類の更なる他の因子は、例えば、ニコチンアミド、TGF−β1、2及び3等のTGFβファミリーのメンバー、血清アルブミン、繊維芽細胞増殖因子ファミリーのメンバー、血小板由来増殖因子−AA及び−BB、血小板濃縮血漿、インスリン様増殖因子(IGF−I、II)、増殖分化因子(GDF−5、−6、−8、−10、11)、グルカゴン様ペプチド−1及びII(GLP−1及びII)、GLP−1及びGLP−2模倣体(mimetobody)、エキセンジン−4、レチノイン酸、副甲状腺ホルモン、インスリン、プロゲステロン、アプロチニン、ヒドロコルチゾン、エタノールアミン、βメルカプトエタノール、上皮増殖因子(EGF)、ガストリンI及びII、例えば、トリエチレンペンタミン等の銅キレート化剤、フォルスコリン、酪酸ナトリウム、アクチビン、ベータセルリン、ITS、ノギン、神経突起増殖因子、ノーダル、バルプロ酸、トリコスタチンA、酪酸ナトリウム、肝細胞増殖因子(HGF)、スフィンゴシン1、VEGF、MG132(EMD社、カリフォルニア州)、N2及びB27添加物(ギブコ社(Gibco)、カリフォルニア州)、例えばシクロパミン(EMD社、カリフォルニア州)等のステロイドアルカロイド、ケラチノサイト増殖因子(KGF)、Dickkopfタンパク質ファミリー、ウシ脳下垂体抽出物、膵島新生関連タンパク質(INGAP)、インディアンヘッジホッグ、ソニックヘッジホッグ、プロテアソーム阻害剤、ノッチ経路阻害剤、ソニックヘッジホッグ阻害剤、又はこれらの組み合わせであってもよい。
【0162】
前記少なくとも1種類の更なる他の因子は、例えばPANC−1(ATCC No:CRL−1469)、CAPAN−1(ATCC No:HTB−79)、BxPC−3(ATCC No:CRL−1687)、HPAF−II(ATCC No:CRL−1997)等の膵臓細胞系、例えばHepG2(ATCC No:HTB−8065)等の肝細胞系、及び例えば、FHs74(ATCC No:CCL−241)等の腸細胞系から得られる馴化培地によって供給することが可能である。
【0163】
膵臓内胚葉系統に特徴的なマーカーを発現する細胞の検出
膵臓内胚葉系統に特徴的なマーカーは当業者にはよく知られたものであり、膵臓内胚葉系統に特徴的な更なるマーカーも次々に特定されている。これらのマーカーを用いて、本発明に基づいて処理を行った細胞が、膵臓内胚葉系統に特徴的な性質を獲得するように分化したことを確認することが可能である。膵臓内胚葉系統に特異的なマーカーとしては、例えば、Hlxb9、PTF−1a、PDX−1、HNF−6、HNF−1β等、1以上の転写因子の発現が挙げられる。
【0164】
分化の効率は、膵臓内胚葉系統に特徴的なマーカーを発現する細胞によって発現されるタンパク質マーカーを特異的に認識する薬剤(抗体等)に、処理した細胞集団を曝露することによって決定することができる。
【0165】
培養細胞又は単離細胞中のタンパク質及び核酸マーカーの発現を評価するための方法は、当該技術分野では標準的なものである。こうした方法には、定量的逆転写ポリメラーゼ連鎖反応(RT−PCR)、ノーザンブロット、in situハイブリダイゼーション(例えば、「分子生物学の最新プロトコール」(Current Protocols in Molecular Biology)オウスベル(Ausubel)等編、2001年度版補遺、を参照)、並びに、切片化材料の免疫組織化学的分析、ウエスタンブロッティング、及び無傷細胞中のアクセシブルなマーカーに対する免疫アッセイ、フローサイトメトリー分析(FACS)(例えば、ハーロー及びレーン(Harlow and Lane)著、「抗体の使用:実験マニュアル」(Using Antibodies: A Laboratory Manual)ニューヨーク州コールドスプリングハーバーラボラトリープレス(1998年)を参照)等の免疫アッセイが含まれる。
【0166】
特定のタンパク質マーカーの検出に有用な抗体の例を表IAに列記する。表IAに列記した抗体によって認識されるものと同じマーカーを標的とする代替的な抗体も入手可能であるか、容易に開発が可能である点は注意を要する。こうした代替的な抗体を、本発明に基づいて単離した細胞におけるマーカーの発現を評価するために用いることもできる。
【0167】
膵臓内分泌系統に特徴的なマーカーを発現する細胞の形成
膵臓内胚葉系統に特徴的なマーカーを発現する細胞は、当該技術分野におけるいずれかの方法又は本発明において開示されるいずれかの方法によって膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させることができる。
【0168】
例えば、膵臓内胚葉系統に特徴的なマーカーを発現する細胞は、ダムール(D’Amour)等により開示される方法に従って膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させることができる(D’Amour et al, Nature Biotechnology 24, 1392 - 1401(2006))。
【0169】
例えば、膵臓内胚葉系統に特徴的なマーカーを発現する細胞をDAPT及びエキセンジン4を含む培地中で培養し、次いでDAPT及びエキセンジン4を含む培地を取り除いた後、エキセンジン1、IGF−1及びHGFを含む培地中で細胞を培養することによって、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に更に分化させる。この方法の一例は、(Nature Biotechnology 24, 1392 - 1401(2006))に記載されている。
【0170】
例えば、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、エキセンジン4を含む培地中で培養し、次いでエキセンジン4を含む培地を取り除いた後、エキセンジン1、IGF−1及びHGFを含む培地中で細胞を培養することによって、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に更に分化させる。この方法の一例は、ダムール(D’Amour)等により開示されている(D’Amour et al, Nature Biotechnology, 2006)。
【0171】
例えば、膵臓内胚葉系統に特徴的なマーカーを発現する細胞をDAPT及びエキセンジン4を含む培地中で培養することによって、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に更に分化させる。この方法の一例は、ダムール(D’Amour)等により開示されている(D’Amour et al, Nature Biotechnology, 2006)。
【0172】
例えば、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、エキセンジン4を含む培地中で培養することによって、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に更に分化させる。この方法の一例は、ダムール(D’Amour)等により開示されている(D’Amour et al, Nature Biotechnology, 2006)。
【0173】
本発明の一態様では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、ノッチシグナル伝達経路を阻害する因子で処理することによって、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に更に分化させる。ノッチシグナル伝達経路を阻害する因子は、ノッチ細胞外受容体のアンタゴニストであってもよい。また、この因子は、ノッチ受容体の生物活性を阻害するものであってもよい。また、この因子は、細胞内におけるノッチシグナル伝達経路を阻害するか、ノッチシグナル伝達経路内の特定の要素のアンタゴニストであってもよい。
【0174】
一実施形態では、ノッチシグナル伝達経路を阻害する因子は、γ−セクレターゼ阻害剤である。一実施形態では、γ−セクレターゼ阻害剤は、L−685,458としても知られる、1S−ベンジル−4R−[1−(1S−カルバモイル−2−フェネチルカルバモイル)−1S−3−メチルブチルカルバモイル]−2R−ヒドロキシ−5−フェネチルペンチル]カルバミン酸tert−ブチルエステルである。
【0175】
L−685,458は、約0.1μM〜約100μMの濃度で使用することができる。一実施形態では、L−685,458を約90μMの濃度で使用する。一実施形態では、L−685,458を約80μMの濃度で使用する。一実施形態では、L−685,458を約70μMの濃度で使用する。一実施形態では、L−685,458を約60μMの濃度で使用する。一実施形態では、L−685,458を約50μMの濃度で使用する。一実施形態では、L−685,458を約40μMの濃度で使用する。一実施形態では、L−685,458を約30μMの濃度で使用する。一実施形態では、L−685,458を約20μMの濃度で使用する。一実施形態では、L−685,458を約10μMの濃度で使用する。
【0176】
一実施形態において、本発明は、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させるための方法であって、
a.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を培養する工程と、
b.ノッチシグナル伝達経路を阻害する因子で前記細胞を処理する工程と、を含む、方法を提供する。
【0177】
膵臓内胚葉系統に特徴的なマーカーを発現するいずれの細胞も、この方法を用いて膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させるうえで適している。
【0178】
一実施形態では、ノッチシグナル伝達経路を阻害する因子は、γ−セクレターゼ阻害剤である。一実施形態では、γ−セクレターゼ阻害剤は、L−685,458としても知られる、1S−ベンジル−4R−[1−(1S−カルバモイル−2−フェネチルカルバモイル)−1S−3−メチルブチルカルバモイル]−2R−ヒドロキシ−5−フェネチルペンチル]カルバミン酸tert−ブチルエステルである。
【0179】
膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、ノッチシグナル伝達経路を阻害する因子で約1〜約5日間処理する。あるいは、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、ノッチシグナル伝達経路を阻害する因子で約3〜約5日間処理する。あるいは、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、ノッチシグナル伝達経路を阻害する因子で約5日間処理する。
【0180】
一実施形態では、ノッチシグナル伝達経路を阻害する因子は、γ−セクレターゼ阻害剤である。一実施形態では、γ−セクレターゼ阻害剤は、L−685,458としても知られる、1S−ベンジル−4R−[1−(1S−カルバモイル−2−フェネチルカルバモイル)−1S−3−メチルブチルカルバモイル]−2R−ヒドロキシ−5−フェネチルペンチル]カルバミン酸tert−ブチルエステルである。
【0181】
L−685,458は、約0.1μM〜約100μMの濃度で使用することができる。一実施形態では、L−685,458を約90μMの濃度で使用する。一実施形態では、L−685,458を約80μMの濃度で使用する。一実施形態では、L−685,458を約70μMの濃度で使用する。一実施形態では、L−685,458を約60μMの濃度で使用する。一実施形態では、L−685,458を約50μMの濃度で使用する。一実施形態では、L−685,458を約40μMの濃度で使用する。一実施形態では、L−685,458を約30μMの濃度で使用する。一実施形態では、L−685,458を約20μMの濃度で使用する。一実施形態では、L−685,458を約10μMの濃度で使用する。
【0182】
膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞の形成を促進することが可能な少なくとも1種類の更なる他の因子で処理してもよい。また、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される膵臓内分泌系統に特徴的なマーカーを発現する細胞の増殖を促進するものであってもよい。更に、前記少なくとも1種類の更なる他の因子は、本発明の方法によって形成される膵臓内分泌系統に特徴的なマーカーを発現する細胞が、他の細胞種を形成する能力を促進するものであってもよく、あるいは他のいずれかの更なる分化の段階の効率を高めるものであってもよい。
【0183】
前記少なくとも1種類の更なる他の因子は、例えば、ニコチンアミド、TGF−β1、2及び3等のTGFβファミリーのメンバー、血清アルブミン、繊維芽細胞増殖因子ファミリーのメンバー、血小板由来増殖因子−AA及び−BB、血小板濃縮血漿、インスリン様増殖因子(IGF−I、II)、増殖分化因子(GDF−5、−6、−8、−10、11)、グルカゴン様ペプチド−1及びII(GLP−1及びII)、GLP−1及びGLP−2模倣体(mimetobody)、エキセンジン−4、レチノイン酸、副甲状腺ホルモン、インスリン、プロゲステロン、アプロチニン、ヒドロコルチゾン、エタノールアミン、βメルカプトエタノール、上皮増殖因子(EGF)、ガストリンI及びII、例えばトリエチレンペンタミン等の銅キレート化剤、フォルスコリン、酪酸ナトリウム、アクチビン、ベータセルリン、ITS、ノギン、神経突起増殖因子、ノーダル、バルプロ酸、トリコスタチンA、酪酸ナトリウム、肝細胞増殖因子(HGF)、スフィンゴシン1、VEGF、MG132(EMD社、カリフォルニア州)、N2及びB27添加物(ギブコ社(Gibco)、カリフォルニア州)、例えば、シクロパミン(EMD社、カリフォルニア州)等のステロイドアルカロイド、ケラチノサイト増殖因子(KGF)、Dickkopfタンパク質ファミリー、ウシ脳下垂体抽出物、膵島新生関連タンパク質(INGAP)、インディアンヘッジホッグ、ソニックヘッジホッグ、プロテアソーム阻害剤、ノッチ経路阻害剤、ソニックヘッジホッグ阻害剤、又はこれらの組み合わせであってもよい。
【0184】
前記少なくとも1種類の更なる他の因子は、例えば、PANC−1(ATCC No:CRL−1469)、CAPAN−1(ATCC No:HTB−79)、BxPC−3(ATCC No:CRL−1687)、HPAF−II(ATCC No:CRL−1997)等の膵臓細胞系、例えばHepG2(ATCC No:HTB−8065)等の肝細胞系、及び例えば、FHs74(ATCC No:CCL−241)等の腸細胞系から得られる馴化培地によって供給することが可能である。
【0185】
一実施形態において、本発明は、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させるための改良された方法であって、
a.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を培養する工程と、
b.グルコースを約10mM〜約20mMの濃度で含む培地中で、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させることが可能な因子で前記細胞を処理する工程と、を含む、方法を提供する。
【0186】
膵臓内胚葉系統に特徴的なマーカーを発現するいずれの細胞も、この方法を用いて膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させるうえで適している。
【0187】
膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させることが可能ないずれの方法も、本発明の改良に適している。
【0188】
一実施形態では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を約10mMの濃度のグルコースを含む培地中で処理する。代替的な一実施形態では、約20mMの濃度のグルコースを含む培地中で細胞を処理する。
【0189】
膵臓内胚葉系統に特徴的なマーカーを発現する細胞を約2〜約30日間処理する。一実施形態では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を約2〜約20日間処理する。一実施形態では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を約2〜約10日間処理する。一実施形態では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を約10日間処理する。一実施形態では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を約4日間処理する。一実施形態では、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を約2日間処理する。
【0190】
膵臓内分泌系統に特徴的なマーカーを発現する細胞の検出
膵臓内分泌系統の細胞に特徴的なマーカーは当業者にはよく知られたものであり、膵臓内分泌系統に特徴的な更なるマーカーも次々に特定されている。これらのマーカーを用いて、本発明に基づいて処理を行った細胞が膵臓内分泌系統に特徴的な性質を獲得するように分化したことを確認することが可能である。膵臓内分泌系統に特異的なマーカーとしては、例えば、NGN−3、ニューロD(NeuroD)、Islet−1等、1以上の転写因子の発現が挙げられる。
【0191】
β細胞系統の細胞に特徴的なマーカーは当業者にはよく知られたものであり、β細胞系統に特徴的な更なるマーカーも次々に特定されている。これらのマーカーを用いて、本発明に基づいて処理を行った細胞がβ細胞系統に特徴的な性質を獲得するように分化したことを確認することが可能である。β細胞系統に特徴的な性質としては、例えば、特にPdx1(膵臓及び十二指腸ホメオボックス遺伝子−1)、Nkx2.2、Nkx6.1、Isl1、Pax6、Pax4、ニューロD(NeuroD)、Hnf1b、Hnf−6、Hnf−3β、及びMafA等、1以上の転写因子の発現が挙げられる。これらの転写因子は、当該技術分野では内分泌細胞を同定する目的でよく確立されている。例として、エドルンド(Edlund)による(Nature Reviews Genetics 3: 524-632(2002))等を参照されたい。
【0192】
分化の効率は、膵臓内分泌系統に特徴的なマーカーを発現する細胞によって発現されるタンパク質マーカーを特異的に認識する薬剤(抗体等)に、処理した細胞集団を曝露することによって決定することができる。また、分化の効率は、β細胞系統に特徴的なマーカーを発現する細胞によって発現されるタンパク質マーカーを特異的に認識する薬剤(抗体等)に、処理した細胞集団を曝露することによって決定することもできる。
【0193】
培養細胞又は単離細胞中のタンパク質及び核酸マーカーの発現を評価するための方法は、当該技術分野では標準的なものである。こうした方法には、定量的逆転写ポリメラーゼ連鎖反応(RT−PCR)、ノーザンブロット、in situハイブリダイゼーション(例えば、「分子生物学の最新プロトコール」(Current Protocols in Molecular Biology)オウスベル(Ausubel)等編、2001年度版補遺、を参照)、並びに、切片化材料の免疫組織化学的分析、ウエスタンブロッティング、及び無傷細胞中のアクセシブルなマーカーに対する免疫アッセイ、フローサイトメトリー分析(FACS)(例えば、ハーロー及びレーン(Harlow and Lane)著、「抗体の使用:実験マニュアル」(Using Antibodies: A Laboratory Manual)ニューヨーク州コールドスプリングハーバーラボラトリープレス(1998年)を参照)等の免疫アッセイが含まれる。
【0194】
特定のタンパク質マーカーの検出に有用な抗体の例を表IAに列記する。表IAに列記した抗体によって認識されるものと同じマーカーを標的とする代替的な抗体も入手可能であるか、又は容易に開発が可能である点は注意を要する。こうした代替的な抗体を、本発明に基づいて単離した細胞におけるマーカーの発現を評価するために用いることもできる。
【0195】
治療法
一態様において、本発明は、1型糖尿病を罹患しているかあるいは発症するリスクを有する患者を治療するための方法を提供する。この方法では、多能性幹細胞を培養し、その多能性幹細胞をインビトロでβ細胞系統に分化させ、そのβ細胞系統の細胞を患者に移植することを含む。
【0196】
更に別の一態様において、本発明は、2型糖尿病を罹患しているかあるいは発症するリスクを有する患者を治療するための方法を提供する。この方法では、多能性幹細胞を培養し、培養した細胞をインビトロでβ細胞系統に分化させ、そのβ細胞系統の細胞を患者に移植することを含む。
【0197】
必要に応じて、移植細胞の生存率及び機能を高める医薬薬剤又は生理活性物質で患者を更に治療することができる。こうした薬剤としては例えば、特にインスリン、TGF−β1、2及び3等のTGF−βファミリーのメンバー、骨形態形成タンパク質(BMP−2、−3、−4、−5、−6、−7、−11、−12、及び−13)、繊維芽細胞増殖因子−1及び−2、血小板由来増殖因子−AA及び−BB、血小板濃縮血漿、インスリン様増殖因子(IGF−I、II)、増殖分化因子(GDF−5、−6、−7、−8、−10、−15)、血管内皮細胞由来増殖因子(VEGF)、プレイオトロフィン、エンドセリン等が挙げられる。他の医薬化合物としては例えば、ニコチンアミド、グルカゴン様ペプチド−I(GLP−1)及びII、GLP−1及び2模倣体(mimetibody)、エキセンジン−4、レチノイン酸、副甲状腺ホルモン、例えば、米国特許出願公開第2004/0209901号及び同第2004/0132729号に開示される化合物のようなMAPK阻害剤等が挙げられる。
【0198】
多能性幹細胞は、レシピエントへの移植に先立ってインスリン産生細胞へと分化させることができる。特定の実施形態では、多能性幹細胞をレシピエントへの移植に先立ってβ細胞に完全に分化させる。また、多能性幹細胞は未分化又は部分的に分化した状態でレシピエントに移植してもよい。更なる分化はレシピエントの体内で起こりうる。
【0199】
胚体内胚葉細胞、又は膵臓内胚葉細胞、又はβ細胞は、分散した細胞として移植するか、あるいは肝門脈に注入することが可能な細胞塊に形成することができる。また、細胞を、生体適合性分解性ポリマー支持体、多孔質の非分解性装置中で与えるか、あるいはカプセル化することによってホストの免疫反応から保護することもできる。細胞はレシピエントの適当な部位に移植することができる。移植部位としては、例えば、肝臓、天然膵臓、腎嚢下腔、網、腹膜、漿膜下腔、腸、胃、及び皮下ポケット等が挙げられる。
【0200】
移植細胞の更なる分化、生存又は活性を促進する目的で、増殖因子等の更なる因子、抗酸化剤、又は抗炎症剤を、細胞の投与の前、同時、又は後に投与することができる。特定の実施形態では、増殖因子を用いて投与細胞をインビボで分化させる。これらの因子は、内因性の細胞によって分泌され、投与細胞にインサイチューで曝露されるものでもよい。移植細胞は、当該技術分野で知られる内因性及び外因性の投与された増殖因子の任意の組み合わせによって分化を誘導することができる。
【0201】
移植に用いられる細胞の量は、患者の状態、及び治療に対する患者の応答等の多くの異なる因子に依存し、当業者が決定できるものである。
【0202】
一態様において本発明は、糖尿病を罹患しているか、あるいは発症するリスクを有する患者を治療するための方法を提供する。この方法では、多能性幹細胞を培養し、培養した細胞をインビトロでβ細胞系統に分化させ、その細胞を3次元的な支持体に組み込むことを含む。細胞は、患者への移植に先立ってインビトロでこの支持体上に維持することができる。また、細胞を含んだ支持体を、更なるインビトロ培養を行うことなく患者に直接移植することも可能である。場合により、支持体に、移植細胞の生存率及び機能を高める少なくとも1種類の医薬薬剤を取り込ませてもよい。
【0203】
本発明の目的における使用に適した支持材料としては、組織修復に有用な組織テンプレート、導管、障壁、及びリザーバが挙げられる。特に、生物学的組織を再構築又は再生する目的で、及び組織の増殖を誘導するための走化性物質を送達する目的でインビトロ及びインビボで使用されてきた、発泡材、スポンジ、ゲル、ヒドロゲル、織物及び不織布構造の形態の合成及び天然の材料が、本発明の方法を実施するうえでの使用に適している。例として、米国特許第5,770,417号、同第6,022,743号、同第5,567,612号、同第5,759,830号、同第6,626,950号、同第6,534,084号、同第6,306,424号、同第6,365,149号、同第6,599,323号、同第6,656,488号、米国特許出願公開第2004/0062753(A1)号、米国特許第4,557,264号、及び同第6,333,029号に開示される材料を参照されたい。
【0204】
医薬薬剤を取り込んだ支持体を形成するには、支持体を形成するのに先立って医薬薬剤をポリマー溶液と混合することができる。また、医薬薬剤を、好ましくは医薬用担体の存在下で、製造された支持体上にコーティングしてもよい。医薬薬剤は、液体、微粉化した固体、又は他の任意の適当な物理的形態として存在してもよい。また、支持体に賦形剤を添加することによって医薬薬剤の放出速度を変化させることもできる。代替的な一実施形態では、例えば、米国特許第6,509,369号に開示される化合物のような抗炎症性化合物である少なくとも1種類の医薬化合物を支持体に取り込ませる。
【0205】
例えば、米国特許第6,793,945号に開示される化合物のような抗アポトーシス性化合物である少なくとも1種類の医薬化合物を支持体に取り込ませてもよい。
【0206】
例えば、米国特許第6,331,298号に開示される化合物のような線維症の阻害剤である少なくとも1種類の医薬化合物を支持体に更に取り込ませてもよい。
【0207】
例えば、米国特許出願公開第2004/0220393号及び米国特許出願公開第2004/0209901号に開示される化合物のような血管新生を促進することが可能な化合物である少なくとも1種類の医薬化合物を支持体に更に取り込ませてもよい。
【0208】
例えば、米国特許出願公開第2004/017623号に開示される化合物のような免疫抑制化合物である少なくとも1種類の医薬化合物を支持体に更に取り込ませてもよい。
【0209】
例えば、特にTGF−β1、2及び3等のTGF−βファミリー、骨形態形成タンパク質(BMP−2、−3、−4、−5、−6、−7、−11、−12及び−13)、繊維芽細胞増殖因子−1及び−2、血小板由来増殖因子−AA及び−BB、血小板濃縮血漿、インスリン様増殖因子(IGF−I、II)、増殖分化因子(GDF−5、−6、−8、−10、−15)、血管内皮細胞由来増殖因子(VEGF)、プレイオトロフィン、エンドセリン等の増殖因子である少なくとも1種類の医薬化合物を支持体に更に取り込ませてもよい。他の医薬化合物としては、例えば、ニコチンアミド、低酸素症誘導因子1−α、グルカゴン様ペプチド−I(GLP−1)、GLP−1及びGLP−2模倣体、及びII、エキセンジン−4、ノーダル、ノギン、NGF、レチノイン酸、副甲状腺ホルモン、テナシン−C、トロポエラスチン、トロンビン由来ペプチド、カテリシジン、ディフェンシン、ラミニン、フィブロネクチン及びビトロネクチン等の接着細胞外基質タンパク質の細胞及びヘパリン結合ドメインを含む生物学的ペプチド、米国特許出願公開第2004/0209901号及び同第2004/0132729号に開示される化合物等のMAPK阻害剤が挙げられる。
【0210】
本発明の細胞は、足場上に細胞を単純に置くだけで足場中に取り込ませることができる。細胞は単純に拡散によって足場に入り込むことができる(J. Pediatr.Surg. 23(1 Pt 2): 3-9(1988))。細胞の播種効率を高めるための他の幾つかの方法が開発されている。例えば、軟骨細胞をポリグリコール酸の足場上に播種するにはスピナーフラスコが使用される(Biotechnol.Prog. 14(2): 193-202(1998))。細胞を播種するための別の方法は遠心を使用することであり、これにより播種細胞へのストレスが最小に抑えられ、播種効率が高められる。例えば、ヤン(Yang)等は遠心細胞固定法(CCI)と呼ばれる細胞播種法を開発している(J.Biomed.Mater.Res. 55(3): 379-86(2001))。
【0211】
本発明を以下の実施例により更に説明するが、本発明はこれらの実施例によって限定されない。
【0212】
背景
【実施例】
【0213】
(実施例1)
ヒト胚性幹細胞の培養
ヒト胚性幹細胞系H1、H7及びH9をウィセル・リサーチ・インスティテュート社(WiCell Research Institute, Inc.)(ウイスコンシン州マディソン所在)より入手し、当該供給元機関によって与えられる指示に従って培養した。簡単に述べると、20%ノックアウト血清リプレースメント、100nM MEM非必須アミノ酸、0.5mM βメルカプトエタノール、2mM L−グルタミンと4ng/mlヒト塩基性繊維芽細胞増殖因子(bFGF)(すべてインビトロジェン/ギブコ社(Invitrogen/GIBCO)より入手)を添加したDMEM/F12(インビトロジェン/ギブコ社(Invitrogen/GIBCO))からなるES細胞培地中、マウス胚繊維芽(MEF)フィーダー細胞上で細胞を培養した。MEF細胞はE13〜13.5のマウス胚から誘導されたものをチャールズリバー社(Charles River)より購入した。MEF細胞は、10%FBS(ハイクローン社(Hyclone))、2mMグルタミン、及び100mM MEM非必須アミノ酸を添加したDMEM培地で増殖させた。サブコンフルエンスに達したMEF細胞培養を、10μg/mlマイトマイシンC(シグマ社(Sigma)ミズーリ州セントルイス所在)で3時間処理して細胞分裂を停止させた後、トリプシン処理し、0.1%ウシゼラチンコートディッシュに2×104/cm2で播いた。継代数2〜4からのMEF細胞をフィーダー層として使用した。MEF細胞のフィーダー層に播いたヒト胚性幹細胞を、加湿した組織培養インキュベーター内で、5%CO2雰囲気中37℃で培養した。コンフルエンスに達した時点(播種後約5〜7日後)で、ヒト胚性幹細胞を1mg/ml IV型コラゲナーゼ(インビトロジェン/ギブコ社(Invitrogen/GIBCO))で5〜10分間処理した後、5mlピペットを用いて表面から静かに掻き取った。細胞を900rpmで5分間遠心し、得られたペレットを再懸濁して新鮮な培地に1:3〜1:4の細胞比で再び播いた。
【0214】
(実施例2)
胚体内胚葉細胞の形成
胚体内胚葉のマーカーの発現に対するアクチビンAの影響を調べた。アクチビンA(100ng/ml)をマウス胚繊維芽細胞上で培養したヒト胚性幹細胞の集団に添加した。細胞をアクチビンAの存在下で継続的に培養し、示した時間において採取した。胚体内胚葉マーカーの発現レベルをPCR(図1)、FACS(表IIに結果をまとめる)、及び免疫組織化学法(図2)によって調べた。
【0215】
アクチビンAは、H9細胞系においてCXCR4、GATA4、HNF−3β、Mixl1及びSox−17のmRNAの発現を時間依存的に増大させた(図1、パネルa)。前方内胚葉マーカーであるケルベロス(Cerverus)、Otx−1及びHex遺伝子の顕著なアップレギュレーションも観察された(図1、パネルb)。アクチビンAによる処理後ではCXCR4タンパク質の増大がFACSによって観察された。E−カドヘリン及びN−カドヘリンの発現にはアクチビンAによる処理後に変化は見られなかった(表IIA)。CXCR4陽性細胞はC−kit、EPCAM、CD99についても顕著に陽性であり、CD9については陰性であった。これらのマーカーの発現パターンは、検討した3種類のhES細胞系で一貫していた(H7については表IIB、H1については表IIC)。アクチビンAで5日間処理した細胞で行った免疫細胞化学法によって、処理培養中の30〜40%の細胞がSox17及びHNF3βについて陽性であることが明らかとなった。同時に、分化した細胞のほぼ100%が依然Oct4陽性であった(図2)。多能性の表面マーカーの発現の減少を、胚体内胚葉マーカーの発現の増大と考え合わせると、これらのデータは、アクチビンAがヒト胚性幹細胞の胚体内胚葉への分化を促進することを示唆するものである。
【0216】
(実施例3)
膵臓内胚葉細胞の形成
ヒト胚性幹細胞の膵臓内胚葉への分化を誘導することが知られている増殖因子を細胞培養に添加した。具体的には、膵臓内胚葉の形成を誘導することが知られているアクチビンA、bFGF、及びレチノイン酸を細胞培養に添加した。
【0217】
第1の実験群では、0%〜2%の血清及びアクチビンA(100ng/ml)を添加したDMEM/F12中、マウス胚繊維芽細胞上で最大で7日間培養したヒト胚性幹細胞の集団にアクチビンAを添加した。細胞を図3に示した時点で採取し、図に示した遺伝子の発現についてPCRでアッセイされた(図3、4及び5)。図3のPCR分析により、アクチビン処理した細胞は、GATA4(図3、パネルa)、Sox−17(図3、パネルb)、HNF−3β(図3、パネルc)、及びMixl−1(図3、パネルd)を含む、内胚葉の発生に関連した広範な遺伝子を発現していることが示された。しかしながら、Pdx1遺伝子の発現は認められなかった。内胚葉系統マーカーの同じ発現パターンが、アクチビンAで処理したH7細胞で認められた(図6、パネルa〜f)。この段階では、Oct4の発現に有意な減少は認められなかった。
【0218】
アクチビンAは、胚体外内胚葉マーカーであるSox7(図4、パネルa)及びAFP(図4、パネルb)の発現を時間依存的に減少させた。アクチビンAはブラキュリ(Brachyury)の発現を減少させた(図5、パネルa)が、神経マーカーであるZic1の発現には影響を及ぼさなかった(図5、パネルb)。
【0219】
以上を考え合わせると、これらのデータは、Sox−17、Mixl1、Gata4及びHNF−3βの発現の増加が、前方内胚葉マーカーであるOtx1、Cer1及びHex遺伝子がアップレギュレーションされていることと相まって、アクチビンA処理に応じた胚体内胚葉の形成を示していることを示唆するものである。免疫細胞化学法による胚体内胚葉マーカーの分析により、これらの遺伝子のタンパク質発現もmRNA発現に認められた傾向を反映していることが明らかとなった。HNF−3β、Sox−17及びGATA4の発現レベルは、非処理細胞では全細胞の約10〜20%と低かった。5日間のアクチビンA(100ng/ml)処理により、HNF−3β、Sox−17及びGATA4の発現は、全細胞の約50%〜90%にまで増加した(図7)。
【0220】
第2の実験群では、ヒト胚性幹細胞の培養を実施例1に述べた方法に従って2〜3日間未分化条件に維持した。細胞が70〜80%コンフルエンスに達した後、アクチビンAを100ng/mlとなるように加えた0〜2%FBSを含むDMEM/F12に培地を換え、3日、5日、又は7日間にわたってアクチビンAの存在下で培養した。この時間間隔後、図8に示されるようなレチノイン酸とbFGFの組み合わせで細胞を5〜6日間更に処理した。培養物を採取し、mRNAの試料を収集して分析に供した。アクチビンA単独で5日間処理した細胞からなるコントロール培養も含めた。
【0221】
遺伝子発現分析により、アクチビンA又はレチノイン酸単独ではPdx1の発現を誘導しないことが示された。同様の結果が、アクチビンAの存在下でレチノイン酸とFGFの組み合わせにより処理した培養細胞で観察された(図8、パネルa)。しかしながら、アクチビンAの非存在下におけるレチノイン酸とFGFの組み合わせによる細胞の処理により、Pdx1の発現は更に増大した(図8、パネルa)。アクチビンAで3日間処理した後、アクチビンAの非存在下で1μMレチノイン酸及び50ng/ml bFGF(FGF−2としても知られる)で5日間処理した細胞は、アクチビンA単独で5日間処理した試料で観察されたPdx発現レベルよりも約3500倍高いPdx1の発現レベルを示した(図8、パネルa)。免疫細胞化学分析法によれば、全細胞の5〜20%がPdx1を発現していることが示された(図9)。
【0222】
アクチビンAの非存在下での1μMレチノイン酸及びbFGFによる処理は、アクチビンA単独の存在下で処理された細胞では認められなかったGLUT−2及びPTF1aの発現の増大も引き起こした(図8、パネルc)。1μMレチノイン酸及び50ng/ml bFGFで処理した細胞においてGLUT−2及びPTF1aの発現の最大の増加が認められた。これらを考え合わせると、これらのデータは、胚体内胚葉が形成された後に細胞培養からアクチビンAを除去することによって膵臓内胚葉の形成が更に促進されることを示唆するものである。
【0223】
(実施例4)
膵臓内分泌細胞の形成
ヒト胚性幹細胞の培養を実施例1に述べた方法に従って3〜4日間未分化条件に維持した。細胞が50〜60%コンフルエンスに達した後、100ng/mlのアクチビンAを含む、FBSを含まないDMEM/F12に培地を換え、この培地中で細胞を1日間培養した。1日間の培養の後、培地を除去し、100ng/mlのアクチビンAを加えた0.5%FBSを含む培地と交換し、細胞を1日間培養した。この2回目の1日の培養の後、培地を除去し、100ng/mlのアクチビンAを加えた2%FBSを含む培地と交換し、細胞を1日間培養した。この時間間隔後、実施例2に概略を述べたように細胞をレチノイン酸とFGFの組み合わせで6日間処理し、その後、培地を除去し、10μMのγ−セクレターゼ阻害剤L−685,458を含む、2%FBSを含んだDMEM/F12からなる培地と交換し、3日間培養した。細胞を収穫し、mRNAの試料を収集して分析に供した。アクチビンA単独で5日間処理した細胞からなるコントロール培養も含めた。
【0224】
遺伝子発現分析により、アクチビンA単独、又はアクチビンAをレチノイン酸及びFGFと組み合わせた場合には、Ngn3の発現もインスリンの発現も誘導しないことが明らかとなった(図10、パネルa、c)。L−685,458による処理の後ではHes−1の発現の減少も認められた。処理後3日目に最大の阻害が観察された(図10、パネルd)。しかしながら、L−685,458による処理は、アクチビンA単独又はレチノイン酸とFGFとの組み合わせで処理した試料で観察された発現レベルよりも約50倍高いレベルでNgn3の発現を誘導した。γ−セクレターゼ阻害剤で処理した試料ではインスリンの発現に70倍の増大が見られた。ニューロD1(NeuroD1)の発現もL−685,458による処理によって更に増大した(図10、パネルa)。以上を考え合わせると、これらのデータは、膵臓内胚葉が形成された後、レチノイン酸及びFGFを細胞培養から除去し、γ−セクレターゼ阻害剤を添加することによって内分泌細胞の形成が更に促進されることを示唆するものである。
【0225】
(実施例5)
Nkx2.2を発現する膵臓内分泌細胞の形成
実施例2に概略を述べた方法に従って得られた胚体内胚葉細胞を以下のように処理した。即ち、50ng/mlアクチビンA、50ng/ml塩基性FGF及び1μMのレチノイン酸を加えた2%FBS含有DMEM/F12からなる基本培地中で細胞を3〜5日間培養した。1μMのレチノイン酸を単独で又はbFGFとともに含む基本培地中で細胞を更に3〜5日間引き続き培養した。方向付けされた細胞の分化の評価を助けるため、このプロセスに沿った異なる時点で細胞からRNA試料を採取した。更に、この分化プロトコールの全体を通じて培地及び各因子を定期的に除去して補充した。アクチビンAの添加により、アクチビンAを加えない試料と比較してNkx2.2の発現は約35倍に増大した。培養の最初の3日間にアクチビンAで処理した試料では、Pdx1の発現はアクチビンAを含まない試料と同等のレベルに維持された(図11)。以上を考え合わせると、これらのデータは、膵臓内分泌マーカーであるNkx2.2の発現は、レチノイン酸及びbFGFによる処理の最初の3日間にアクチビンAを添加することによって更に促進されることを示唆するものである。
【0226】
(実施例6)
膵臓内胚葉細胞の培養における継代及び増殖
本実施例では、本願でヒト胚性幹細胞より誘導された膵臓内胚葉細胞を細胞培養中で維持し、更に分化させることなく継代することが可能であることを実証する。膵臓内胚葉細胞を低血清DMEM/F12中、100ng/mlのアクチビンAの存在下で分化させた。低血清DMEM/F12は、1日目には0%(v/v)のウシ胎児血清(FBS)、2日目には0.5%(v/v)のFBS、その後は毎日2%(v/v)のFBSを含有するものを使用した。4日間分化させた後、細胞を2%(v/v)FBS、1μMのレチノイン酸、及び50ng/mlのbFGFを含む低血清DMEM/F12中で、全体で6日間更に培養した。6日間の分化の後、細胞を、2%(v/v)FBSを含む低血清DMEM/F12中、50ng/mlのFGF10の存在下で、全体で6日間、培養中で維持した。6日間の培養期間中、膵臓内胚葉細胞を2回継代した。細胞集団の倍加時間はこの6日間の培養期間で約36〜48時間であった。培養の0、3及び6日目において、Q−PCRを用いて膵臓内胚葉を示すマーカー遺伝子の発現を測定した。図12は、50ng/mlのFGF10の存在下で増殖させた細胞が、誘導後の6日間の培養期間中、膵臓内胚葉マーカーであるPdx1の発現を維持したことを示している。
【0227】
(実施例7)
ヒト胚性幹細胞からの肝細胞の誘導
ヒト胚性幹細胞の培養を実施例1で述べた方法に従って未分化培養条件に2〜3日間維持した。細胞が70〜80%コンフルエンスに達した後、100ng/mlのアクチビンAを含む2%FBS含有DMEM/F12に培地を換え、アクチビンAの存在下で細胞を7日間培養した。7日間のアクチビンA処理の後、細胞を図13に示す各条件で5日間処理した。この後、細胞を採取し、mRNAの試料を収集して分析に供した。
【0228】
アクチビンAの非存在下で培養した細胞では、α−フェトタンパク質(AFP)及びアルブミンの発現の増大が認められた(図13、パネルA)。これはレチノイン酸及びFGF−4によって更に増大した(図13、パネルb)。以上を考え合わせると、これらのデータは、ヒト胚性幹細胞の培養が、上記に述べた処理の後、肝細胞マーカーを発現することが可能であることを示唆するものである。更に、ヒト胚性幹細胞を肝細胞に特徴的なマーカーを発現する細胞に分化させることが可能である。
【0229】
(実施例8)
H9ヒト胚性幹細胞系の特徴付け
未分化のES細胞が発現する幾つかのマーカーの発現を評価することにより、H9細胞の性質を経時的に監視した(Carpenter et al., 2001; Reubinoff et al., 2000; Thomson et al., 1998a)。H9細胞では、段階特異的胚抗原の交互発現が見られた(表III)。H9細胞は、いずれも未分化のヒト胚性幹細胞に特徴的なSSEA−3、SSEA−4、Tra−1−60、Tra−1−81、AP及びCD9抗原に対して強力な免疫応答性を示す。
【0230】
例えば、OCT3/4、SOX−2、UTF−1、REX−1、Cx43、Cx45、ABCG−2及びTERT等の胚性幹細胞に特徴的な遺伝子の発現を評価するためにリアルタイムPCRを行ったところ、本実施例で増殖させた細胞は、既に述べられている未分化の胚性幹細胞と似ていることが確認された(表III)。OCT3/4タンパク質の発現及びアルカリホスファターゼ活性(ケミコン社(Chemicon))を免疫染色によって確認した。H9細胞の大部分はOCT3/4及びAPについて陽性であった(図14)。以上をまとめると、これらの結果は、本実施例で使用したH9細胞が、他の研究機関からの報告と比較して、形態、抗原免疫染色、又は多能性マーカーの発現において大きく異ならないものであることを証明するものである。
【0231】
(実施例9)
蛍光活性化細胞選別(FACS)分析
接着した細胞をTrypLE(商標)Express溶液(インビトロジェン社(Invitrogen)カリフォルニア州所在)と5分間インキュベートすることによって培養皿から剥がした。剥がした細胞をヒト胚性幹細胞培地に再懸濁し、遠心して回収した後、洗浄し、この細胞を2%BSA、0.05%アジ化ナトリウムを含むPBSからなる染色緩衝液(シグマ社(Sigma)ミズーリ州)に再懸濁した。必要に応じて、0.1%γグロブリン(シグマ社(Sigma))溶液を用いて15分間、細胞のFc受容体をブロックした。一定分量(約105個の細胞)を表Iに示されるようなフィコエリスリン(PE)若しくはアロフィコシアニン(APC)を結合したモノクローナル抗体(106個の細胞毎に5μLの抗体)、又は非結合一次抗体とインキュベートした。コントロールには、適当なアイソタイプ一致抗体、非染色細胞、及び二次結合抗体のみで染色した細胞を含めた。抗体とのインキュベートをすべて、4℃で30分間行い、その後、細胞を染色緩衝液で洗った。非結合一次抗体で染色した試料を、PE又はAPC結合標識二次抗体と更に30分間、4℃でインキュベートした。使用した二次抗体の一覧については表Iを参照されたい。洗った細胞はペレットとし、染色緩衝液に再懸濁した。細胞表面の分子をFACS Array(BDバイオサイエンス社(BD Biosciences))装置を使用し、少なくとも10,000のイベントを収集して特定した。
【0232】
(実施例10)
免疫細胞化学法
0.1%Matrigel(BD社)でコーティングした培養皿に播種した細胞を室温で20分間、4%パラホルムアルデヒドで固定した。固定した細胞を室温で1時間、PBS/0.1%BSA/10%正常ニワトリ血清/0.5%TritonX−100でブロックした後、PBS/0.1%BSA/10%正常ニワトリ血清中、4℃で一次抗体と一晩インキュベートした。一次抗体及びその実用希釈率を表IBに示す。PBS/0.1%BSA中で3回洗った後、PBS中に1:100の希釈率で希釈した蛍光二次抗体と細胞を室温で1時間インキュベートして結合させた。コントロール試料として、一次抗体を省略するか、一次抗体と同じ濃度の、対応する一致したネガティブコントロールとしての免疫グロブリンで一次抗体を置き換えた反応を含めた。染色した試料はすすいだ。ジアミジノ−2−フェニルインドール二塩酸塩(DAPI)を含むPROLONG(登録商標)(インビトロジェン社(Invitrogen)カリフォルニア州所在)を、核を対比染色し、蛍光退色防止剤として機能するように各試料に1滴ずつ加えた。ニコンコンフォーカルEclipseC−1倒立顕微鏡(ニコン社、日本)及び10〜60倍の対物レンズを使用して画像を得た。
【0233】
(実施例11)
未分化細胞のPCR分析
RNAの抽出、精製及びcDNAの合成:エタノール含有高塩濃度緩衝液の存在下でシリカゲル膜(Rneasy Mini Kit、キアゲン社(Qiagen)カリフォルニア州所在)に結合させた後、洗浄して夾雑物を除去することによりRNA試料を精製した。TURBO DNA−Free Kit(アンビオン社(Ambion, Inc.))を使用してRNAを更に精製し、高品質RNAを水で溶出した。収率及び純度を分光光度計のA260及びA280の指示値によって評価した。ABI(ABI、カリフォルニア州所在)製高容量cDNA Archive Kitを使用して精製したRNAからcDNAコピーを作製した。
【0234】
リアルタイムPCR及び定量分析:特に断らないかぎり、試薬はすべてアプライドバイオシステムズ(Applied Biosystems)社より購入した。リアルタイムPCR反応を、ABI PRISM(登録商標)7900配列検出システムを使用して行った。TAQMAN(登録商標)UNIVERSAL PCR MASTER MIX(登録商標)(ABI社、カリフォルニア州所在)を、20ngの逆転写RNAと全反応容量を20μLとして使用した。各cDNA試料はピペット操作による誤差を補正するために2重で流した。プライマー及びFAM標識TAQMAN(登録商標)プローブは200nMの濃度で使用した。各標的遺伝子の発現レベルは、アプライドバイオシステムズ社(Applied Biosystem)によって以前に開発されているヒトグリセルアルデヒド−3−リン酸デヒドロゲナーゼ(GAPDH)内因性コントロールを使用して標準化された。プライマー及びプローブの組は以下の通りである。Oct3/4(Hs00742896)、SOX−2(Hs00602736)、UTF−1(Hs00747497)、Rex−1(Hs00399279)、コネキシン(Connexin)43(Hs00748445)、コネキシン(Connexin)45(Hs00271416)、ABCG2(Hs00184979)、Tert(Hs00162669)、HNF3β(Hs00232764)、GATA−4(Hs00171403)、Mixl1(Hs00430824)、Sox7(Hs00846731)、AFP(Hs00173490)、ブラキュリ(Brachyury)(Hs00610080)、GSC(Hs00418279_m1)、Pdx−1(Hs00426216)、PTF1a(Hs00603586)、Ngn3(Hs00360700)、ニューロ(Neuro)D1(Hs00159598)、インスリン(Hs00355773)、及びGlu2(Hs00165775)。Sox17プライマーはPRIMERSプログラム(ABI社、カリフォルニア州所在)を使用して設計されたものであり、以下の配列である。Sox17:TGGCGCAGCAGATACCA(配列番号1)、AGCGCCTTCCACGACTTG(配列番号2)、及びCCAGCATCTTGCTCAACTCGGCG(配列番号3)。最初に50℃で2分間、次いで95℃で10分間インキュベーションした後、試料を2段階(95℃で15秒間の変性ステップの後、60℃で1分間のアニーリング/伸長ステップ)で40サイクル反応させた。GENEAMP(登録商標)7000配列検出システムソフトウェアを使用してデータ分析を行った。各プライマー/プローブの組について、増幅が指数関数的に起こる領域の中央において蛍光強度が特定の値に達するサイクル数としてCt値を求めた。比較Ct法を用いて相対的遺伝子発現レベルを計算した。簡単に述べると、各cDNA試料について、内因性コントロールのCt値を対象遺伝子のCt値から差し引くことによってΔCt値(ΔCt)を得た。増幅の効率を100%と仮定すると、標準化された標的の量は2ΔCtとして計算される。最終的なデータは、検量試料に対して表わした。
【0235】
(実施例12)
核型の分析
H9細胞の核型を、標準的なGバンディング核型分析法によって調べた。全部で100個の中期染色体展開像を評価した(アプライドジェネティクスラボラトリー社(Applied Genetics Laboratories, Inc.))。分析した100個の細胞で染色体異常は見られなかった。細胞遺伝子分析により、常染色体の数は正常であり、染色体数(modal chromosome number)は46本であることが示された。図15は、ヒト胚性幹細胞系H9から得られる典型的な核型を示したものである。
【0236】
(実施例13)
細胞外基質でコーティングした組織培養基質上でのヒト胚性幹細胞の培養
ヒト胚性幹細胞系H1、H7及びH9をウィセル・リサーチ・インスティテュート社(WiCell Research Institute, Inc.)(ウイスコンシン州マディソン所在)より入手し、当該供給元機関によって与えられる指示に従って培養した。簡単に述べると、20%ノックアウト血清リプレースメント、100nM MEM非必須アミノ酸、0.5mM βメルカプトエタノール、2mM L−グルタミンと4ng/mlヒト塩基性繊維芽細胞増殖因子(bFGF)を添加したDMEM/F12(インビトロジェン/ギブコ社(Invitrogen/GIBCO))からなるES細胞培地中、マウス胚繊維芽(MEF)フィーダー細胞上で細胞を培養した。MEF細胞はE13〜13.5のマウス胚から誘導されたものをチャールズリバー社(Charles River)より購入した。MEF細胞は、10%FBS(ハイクローン社(Hyclone))、2mMグルタミン、及び100mM MEM非必須アミノ酸を添加したDMEM培地で増殖させた。サブコンフルエンスに達したMEF細胞培養を、10μg/mlマイトマイシンC(シグマ社(Sigma)ミズーリ州セントルイス所在)で3時間処理して細胞分裂を停止させた後、トリプシン処理し、0.1%ウシゼラチンコートディッシュに2×104/cm2で播いた。継代数2〜4からのMEF細胞をフィーダー層として使用した。MEF細胞のフィーダー層に播いたヒト胚性幹細胞を、加湿した組織培養インキュベーター内で、5%CO2雰囲気中37℃で培養した。コンフルエンスに達した時点(播種後約5〜7日後)で、ヒト胚性幹細胞を1mg/ml IV型コラゲナーゼ(インビトロジェン/ギブコ社(Invitrogen/GIBCO))で5〜10分間処理した後、5mlのガラス製ピペットを用いて表面から静かに掻き取った。細胞を900rpmで5分間遠心し、得られたペレットを再懸濁して希釈率が1:30の増殖因子低減MATRIGEL(商標)(BDバイオサイエンス社(BD Biosciences))でコーティングしたプレート上に1:3〜1:4の細胞比で再播種した。この後、細胞を、8ng/mlのbFGF及びコラゲナーゼを添加したMEF馴化培地中で培養し、MATRIGELでコーティングしたプレート上で少なくとも5代にわたって継代した。MATRIGEL(商標)上で培養した細胞を、IV型コラゲナーゼ(インビトロジェン/ギブコ社(Invitrogen/GIBCO))、ディスパーゼ(BDバイオサイエンス社(BD Biosciences))、又はリベラーゼ酵素(ロシュ社(Roche)、インディアナ州)を用いて日常的に継代した。
【0237】
(実施例14)
細胞外基質でコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
Nature Biotechnology 23,1534〜1541(Dec 2005)に既に記載されているようにして胚性幹細胞を胚体内胚葉に分化させた。簡単に述べると、約60〜70%コンフルエンスのH9培養を、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。H9細胞は希釈率1:30〜1:10の増殖因子低減MATRIGELでコーティングしたプレート上又は希釈率1:30〜1:10の通常のMATRIGEL上で培養した。各プレートは室温で1時間、MATRIGELでコーティングした。
【0238】
5日目に、CXCR4、E−カドヘリン、CD9、及びN−カドヘリンの発現についてはFACSによって、SOX−17、SOX−7、α胎児タンパク質(AFP)、CXCR4、ブラキュリ(Brychyury)(Bry)、グースコイド(gooscecoid)(GSC)、HNF−3β、及びGATA4についてはリアルタイムPCRによって培養を分析した。AFP及びSOX−7が臓側内胚葉マーカーとみなされるのに対して、GATA4、HNF−3β及びSOX−17は胚体内胚葉マーカーを代表するものであり、GSC、Bry、及びCXCR4は原始線条のマーカーを代表するものである。図17に、FACSにより分析したCXCR4の発現を示す。低濃度のMATRIGELと比較して、希釈率が1:10のMATRIGELでコーティングしたプレート上で培養した細胞によるCXCR4の発現は顕著に増大した。更に、増殖因子低減MATRIGELは、通常のMATRIGELと比較して胚体内胚葉細胞の形成に有効ではなかった。
【0239】
図18は、リアルタイムPCRの結果を示したものであり、希釈率1:10のMATRIGELでコーティングしたプレート上で培養した細胞では、希釈率1:30のMATRIGEL上で培養した細胞と比較して胚体内胚葉が顕著にアップレギュレートされていることが実証された。
【0240】
(実施例15)
胚体内胚葉形成後のヒト胚性幹細胞における遺伝子発現の変化のマイクロアレイによる分析
RNeasy mini kit(キアゲン社(Qiagen))を使用して以下のヒト胚性幹細胞の培養から全RNAを単離した。即ち、MATRIGELコーティングしたプレート上で培養し、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理したH9P83細胞、及び、MEF細胞上で培養し、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地で更に3日間処理したH9P44細胞。各群のコントロールには、MATRIGELでコーティングした培養皿に播種し、MEF馴化培地中で培養した細胞、又はMEF上に播種し、ES培地中で培養した細胞を含めた。
【0241】
試料の調製、ハイブリダイゼーション、及び画像分析は、アフィメトリックス社(Affymetrix)製Human Genome U133 Plus2.0 Arrayに従って行った。正規化及び対数変換を行った後、OmniViz(登録商標)ソフトウェア(マサチューセッツ州所在)及びGENESIFTER(ビズエックスラブズ社(VizXLabs)、ワシントン州所在)を使用してデータ分析を行った。各処理群内及び異なる処理群間の変動は、ピアソン相関係数を用いて比較した。異なる処理群間の遺伝子発現プロファイルの分散を、グラフ間の相関係数とともに図19に示す。試料間の遺伝子発現における有意差は、分散分析及び0.05以下の調節p値を用いたF検定(Benjamini and Hochberg相関)を用いて評価した。プレゼントコール(present call)が示された遺伝子のみが分析に含まれた。表IVに、異なる試料間で発現量に少なくとも5倍の差が見られた遺伝子を示す。有意に発現された遺伝子の正規化された強度値を、各遺伝子の標準誤差(SEM)とともに示した。
【0242】
(実施例16)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
Nature Biotechnology 23,1534〜1541(Dec 2005)に既に記載されているようにして胚性幹細胞を胚体内胚葉に分化させた。簡単に述べると、約60〜70%コンフルエンスに達した増殖因子低減MATRIGEL(商標)(希釈率1:30)培養上に播いたH9、H7又はH1細胞を、0.5%FBS及び100ng/mlアクチビンA(R&Dシステムズ社(R&D Systems)ミネソタ州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。以下のすべての実施例において、特に断らないかぎり、この処理レジメンを胚体内胚葉(DE)プロトコールと称する。
【0243】
これと並行して、MEFフィーダー上で培養したH9、H7又はH1細胞も上記に述べた同じDEプロトコールに供した。
【0244】
5日目に、CXCR4、E−カドヘリン、CD9、CD99及びN−カドヘリン(CD56)の発現についてはFACSによって、SOX−17、SOX−7、α胎児タンパク質(AFP)、CXCR4、ブラキュリ(Brychyury)(Bry)、グースコイド(gooscecoid)(GSC)、HNF−3β、及びGATA4についてはリアルタイムPCRによって培養を分析した。AFP及びSOX−7が臓側内胚葉マーカーとみなされるのに対して、GATA4、HNF−3β及びSOX−17は胚体内胚葉マーカーを代表するものであり、GSC、Bry、及びCXCR4は原始線条のマーカーを代表するものである。
【0245】
マウスフィーダー細胞上で培養し、DEプロトコールに供したH細胞系は、各胚体内胚葉(DE)マーカーを著明に発現し、FACSによってCXCR4の発現が確認された(図20)。DEプロトコールによる処理後にはE−カドヘリンの発現の顕著な減少も認められた。最後に、CXCR4+の集団はCD117についても陽性に染色された。図21は、非処理のH7(図21、パネルa)及びH9細胞(図21、パネルb)と比較して、各胚体内胚葉マーカーの顕著なアップレギュレーションを示している。
【0246】
MEFフィーダー上で培養したH細胞系と異なり、MATRIGEL(商標)(希釈率1:30)上で培養し、胚体内胚葉プロトコールで処理したH細胞系では、胚体内胚葉マーカーの著明な発現は認められなかった。特に、FACS及びリアルタイムPCRにより示される発現は、マウス胚繊維芽細胞上で培養した細胞と比較して、MATRIGEL(商標)上で培養した細胞において大幅に低かった。胚体内胚葉マーカーの発現は、H1がH9よりも高く、H9がH7よりも高いという一般的な応答パターンにしたがっている(図22及び23)。図22から分かるように、H1細胞はH7及びH9細胞系と比較して、CXCR4の発現に顕著な増大が認められた。いずれの場合においても、CXCR4の発現は、マウス胚繊維芽細胞上で培養した細胞と比較してMATRIGEL(商標)(希釈率1:30)上で培養した細胞では低かった点に注意されたい。図23(パネルa〜c)は、リアルタイムPCRの結果を示したものであり、H7(図23、パネルa)及びH9(図23、パネルb)細胞系において胚体内胚葉マ−カーのアップレギュレーションがある程度増大したことを示している。しかしながら、H1(図23、パネルc)細胞系は、H7及びH9細胞系と比較してより顕著な胚体内胚葉マーカーのアップレギュレーションを示した。
【0247】
(実施例17)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−Wntリガンドの役割
H7P44及びH9P46胚性幹細胞を、MATRIGEL(商標)(希釈度1:10)でコーティングした培養皿上で培養し、0.5%FBS及び100ng/mlアクチビンA(R&Dシステムズ社(R&D Systems)ミネソタ州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。培養の一部のものには、20ng/mlのWnt−3a(カタログNo.1324−WN−002、R&Dシステムズ社(R&D Systems)ミネソタ州所在)、20ng/mlのWnt−5a(カタログNo.654−WN−010、R&Dシステムズ社(R&D Systems)ミネソタ州所在)、25ng/mlのWnt−7a(カタログNo.3008−WN−025、R&Dシステムズ社(R&D Systems)ミネソタ州所在)、又は25ng/mlのWnt−5b(カタログNo.3006−WN−025、R&Dシステムズ社(R&D Systems)ミネソタ州所在)を5日間の処理期間の全体を通じて加えた。図24は、高濃度の(A)AA、又は(B)AA+20ng/mlのWnt−3aの存在下でのH9P46胚体内胚葉培養の位相差顕微鏡画像を示したものである。図25は、MATRIGEL(商標)(希釈率1:30)上で培養し、DEプロトコール+Wnt−3a(図25、パネルb及びd)、及び−Wnt−3a(図25、パネルa及びc)に供したH7P44及びH9P46細胞系について、FACSにより調べた5日目のCXCR4の発現を示したものである。Wnt−3aがDE培養中に存在する場合、低血清及び高濃度のAAで処理したDE培養と比較して、CXCR4(CD184)の著明な発現が見られた。図26は、低血清+AA±Wntリガンドで処理した、A)H7及びB)H9の培養についてリアルタイムPCRのデータを示したものである。いずれのH細胞系においても、Wnt−3aを添加することにより、胚体内胚葉マーカーの顕著なアップレギュレーションが認められた。これに対し、Wnt−5a、Wnt−5b及びWnt−7aは、胚体内胚葉マーカーの発現にほとんど影響を及ぼさなかった。
【0248】
(実施例18)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−Wnt−3aの有効量
H9P46胚性幹細胞を、MATRIGEL(商標)(希釈度1:10)でコーティングした培養皿上で培養し、0.5%FBS、100ng/mlアクチビンA(AA)及び10〜50ng/mlのWnt−3a(R&Dシステムズ社(R&D Systems)ミネソタ州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS、100ng/mlアクチビンA(AA)、及び10〜50ng/mlのWnt−3aを添加したDMEM/F12培地で更に3日間処理した。コントロール培養はWnt−3aで処理しなかった。図27において、パネルa)はWnt−3aの非存在下、b)10ng/mlのWnt−3a、c)20ng/mlのWnt−3a、及びd)50ng/mlのWnt−3aの存在下における5日目のFACSによるCXCR4の発現を示したものである。Wnt−3aの非存在下ではCXCR4の発現は極めて低かった。これに対し、10〜50ng/mlのWnt−3aを添加することにより、CXCR4陽性細胞の数が顕著に増加した。更に、10ng/mlのWnt−3aの添加は、50ng/mlのWnt−3aの添加と同様に効果的であった。リアルタイムPCRの結果によってもこの知見が確認された(図28、パネルa)。
【0249】
別の実験において、H9P52細胞を1:30に希釈した低増殖因子MATRIGEL(商標)上に播種した。DEプロトコールの最初の2日間において、10ng/ml、5ng/ml、1ng/mlの異なる用量のWnt−3aを使用した。図28のパネルbは、処理の5日後のDEマーカーのPCR分析を示したものである。実験終了実施例における細胞数を図28のパネルcに示す。この結果は、高用量のWnt−3aを使用した場合に細胞が増殖することを示している。Wnt−3aによる処理を5日間に延長することによって(5D)、PCRによるDEマーカーの結果にはほとんど影響は見られず、細胞数の大きな増加も認められなかった(図28、パネルc)。これらのデータは、2日間の10ng/mlのWnt−3aによる処理が、細胞の最適な増殖及び胚体内胚葉分化に充分であることを示すものである。
【0250】
(実施例19)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−GSK−3B阻害剤の影響
Wnt−3aの影響がWnt経路を介したものであることを確認するため、GSK−3阻害剤を使用してβカテニン等のWntの下流の標的分子を活性化した。H9P46〜P48胚性幹細胞をMATRIGEL(商標)でコーティングした培養皿(希釈率1:10)上で培養し、0.5%FBS、100ng/mlアクチビンA(AA)、及び10〜1000nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)カリフォルニア州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS、100ng/mlアクチビンA(AA)、及び0〜1000nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)カリフォルニア州所在)を添加したDMEM/F12培地で更に3日間処理した。コントロール培養は、低血清及び高用量のアクチビンA±Wnt−3aで処理した。図29において、パネルaはWnt−3aもGSK−3B阻害剤も加えない場合、b)+20ng/mlのWnt−3a、c)+1000ng/mlのGSK−3B阻害剤IX、d)+500ng/mlのGSK−3B阻害剤IX、e)+100ng/mlのGSK−3B阻害剤IX、f)+10ng/mlのGSK−3B阻害剤IX、g)+100ng/mlのGSK−3B阻害剤IXを1〜2日目、及びh)+10ng/mlのGSK−3B阻害剤IXを1〜2日目に加えた場合の5日目のFACSによるCXCR4の発現を示したものである。
【0251】
Wnt−3aの非存在下又は10nMのGSK−3B阻害剤の存在下では、CXCR4の発現は極めて低かった。これに対し、20ng/mlのWnt−3a又は100〜1000nMのGSK−3B阻害剤の添加により、CXCR4陽性細胞の数は顕著に増大した。更に、100nMのGSK−3B阻害剤を1〜2日目に添加した場合、100nMのGSK−3B阻害剤を5日間の期間全体にわたって添加した場合と同様に効果的であった。図30は、H9P48細胞(パネルa)及びH9P46細胞(パネルb)における胚体内胚葉マーカーの遺伝子発現を示す。
【0252】
図16は、無フィーダー系で胚性幹細胞を胚体内胚葉に分化させる本発明の分化プロトコールの概要を示す。
【0253】
(実施例20)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−GSK−3B阻害剤又はWnt−3aの存在下におけるアクチビンAの有効量
H9P49及びH1P46胚性幹細胞をMATRIGEL(商標)でコーティングした培養皿(希釈率1:10)上で培養し、0.5%FBS、10〜100ng/mlアクチビンA(AA)、及び100nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)、カリフォルニア州所在)又は20ng/mlのWnt−3aを添加したDMEM/F12培地に2日間曝露した後、2%FBS、10〜100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。コントロール培養は、低血清及び100ng/mlのアクチビンAで処理した。図31は、a)10ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間、b)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3Aを最初の2日間、c)100ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、d)10ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、e)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3Aを最初の2日間、並びに、f)10ng/mlのアクチビンA及び20ng/mlのWnt−3Aを最初の2日間加えた場合の、5日目のH9P49及びH1P46におけるFACSによるCXCR4の発現を示したものである。図31のパネルa〜dはH9P49細胞のものであり、パネルE〜FはH1P46細胞のものである。図32は、10、50、又は100ng/mlのアクチビンA及び20ng/mlのWnt−3aで処理したH9P49細胞における胚体内胚葉マーカーの遺伝子発現を示したものであり、パネルaはAFP、Bry、CXCR4、GSC、HNF−3B、及びPOU5F(Oct−4)の発現を示し、パネルbはSOX−17及びGATA4の発現を示す。50ng/mlのAA+20ng/mlのWnt−3A又は100nMのGSK−3B阻害剤IXを使用することによって胚体内胚葉マーカーが著明に発現するようである。アクチビンAを低用量で使用すると胚体外内胚葉が形成される。
【0254】
(実施例16)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−Wnt−3aとGSK−3B阻害剤の組み合わせ
H9P53胚性幹細胞をMATRIGEL(商標)でコーティングした培養皿(希釈率1:30)上で培養し、0.5%FBS、100ng/mlアクチビンA(AA)、及び100nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)、カリフォルニア州所在)を添加し、更に20ng/mlのWnt−3aを加える(+)か、加えない(−)DMEM/F12培地に2日間曝露した後、2%FBS、10〜100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。これと並行して、H9P53培地を、25ng/mlのBMP−4(カタログNo.314−BP−010、R&Dシステムズ社(R&D Systems)ミネソタ州所在)±20ng/mlのWnt−3A±100ng/mlのアクチビンAで処理した。コントロール培養は、低血清及び100ng/mlのアクチビンAで処理した。図33は、a)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間及び25ng/mlのBMP−4を3〜5日目、b)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間、c)100ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、d)20ng/mlのWnt−3a及び25ng/mlのBMP−4を5日間全体、e)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3a及び100nMのGSK−3B阻害剤IXを最初の2日間、並びに、f)100ng/mlのアクチビンA及び25ng/mlのBMP−4を5日間全体にわたって加えた場合の、5日目のFACSによるCXCR4の発現を示したものである。図34は、10又は100ng/mlのアクチビンA及び20ng/mlのWnt−3a又は100nMのGSK−3B阻害剤で処理した、継代数46のヒト胚性幹細胞系H1の培養についてリアルタイムPCRで調べた胚体内胚葉マーカーの遺伝子発現を示す。パネル(a):AFP、Bry、CXCR4、GSC、及びPOU5F(Oct−4)の発現、並びに、パネル(b):SOX−17、HNF−3B及びGATA4の発現。結果は非処理細胞に対する増加倍率として表わす。図35は、50又は100ng/mlのアクチビンA及び10又は100nMのGSK−3B阻害剤で処理した、継代数49のヒト胚性幹細胞系H9の培養についてリアルタイムPCRで調べた胚体内胚葉マーカーの遺伝子発現を示す。パネル(a):AFP、Bry、CXCR4、GSC、HNF−3B及びPOU5F(Oct−4)の発現、並びに、パネル(b):SOX−17及びGATA4の発現。結果は非処理細胞に対する増加倍率として表わす。図36は、アクチビンA、Wnt−3a、GSK−3阻害剤、及びBMP−4の組み合わせで処理したH9P53培養における胚体内胚葉マーカーの遺伝子発現を示す。A)AFP、Bry、CXCR4、GSC、HNF−3B及びSOX7の発現、並びに、B)SOX−17、HNF−3B及びGATA4の発現。DEプロトコールにBMP−4を加えることにより、中胚葉マーカーであるBRYの形成が誘導されるようであるが、Wnt−3AとGSK−4B阻害剤の組み合わせでは、アクチビンAの存在下で各物質を添加した場合と比較して、胚体内胚葉マーカーの顕著なアップレギュレーションは認められなかった。
【0255】
(実施例22)
MEF上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−低血清中でのWnt−3a、アクチビンA、Wnt−5a、BMP−2、BMP−4、BMP−6、BMP−7、IL−4及びSDF−1の組み合わせ
H9P44細胞を、マイトマイシン処理したマウス胚繊維芽細胞(MEF)で予めコーティングした6穴プレート上に播種した。20%ノックアウト血清リプレースメント、100nM MEM非必須アミノ酸、0.5mM βメルカプトエタノール、2mM L−グルタミン(すべてインビトロジェン/ギブコ社(Invitrogen/GIBCO)より入手)、及び8ng/mlヒト塩基性繊維芽細胞増殖因子(bFGF)(R&Dシステムズ社(R&D Systems))を添加したDMEM/F12(インビトロジェン/ギブコ社(Invitrogen/GIBCO))からなるES細胞培地中で70〜80%コンフルエンスに達するまで細胞を培養した。
【0256】
胚体内胚葉(DE)を形成するため、細胞をアクチビンA(100ng/ml)の存在下又は非存在下で、更に下記に詳述する他の増殖因子で処理した。各増殖因子は、下記のレジメンに示されるように段階的に濃度を増加させたFBSに添加した。
0日目:0%のFBSをDMEM/F12に加えた。
1日目:0.5%のFBSをDMEM/F12に加えた。
2日目:2%のFBSをDMEM/F12に加えた。
3日目:細胞を採取してFACS分析及びRT−PCRに供した。
【0257】
増殖因子はすべて、R&Dシステムズ社((R&D Systems)ミネソタ州所在)より購入した。各処理群における増殖因子の詳細な説明及び濃度を下記に示す。
1.コントロール:増殖因子を添加しなかった。
2.アクチビンA(100ng/ml)
3.アクチビンA(100ng/ml)+Wnt−3a(10ng/ml)+Wnt5a(10ng/ml)
4.アクチビンA(100ng/ml)+Wnt−3a(10ng/ml)+Wnt5a(10ng/ml)+BMP2(100ng/ml)
5.アクチビンA(100ng/ml)+BMP−4(100ng/ml)
6.アクチビンA(100ng/ml)+BMP−6(100ng/ml)
7.アクチビンA(100ng/ml)+BMP−7(100ng/ml)
8.アクチビンA(100ng/ml)+BMP−4(100ng/ml)+BMP−6(100ng/ml)+BMP−7(100ng/ml)
9.IL−4(10ng/ml)
10.SDF1a(20ng/ml)
11.アクチビンA(100ng/ml)+IL−4(10ng/ml)+SDF1a(20ng/ml)
12.BMP2(100ng/ml)+BMP−4(100ng.ml)+BMP−6(100ng/ml)+BMP−7(100ng/ml)
13.アクチビンA(100ng/ml)BMP−2(100ng/ml)+BMP−4(100ng.ml)+BMP−6(100ng/ml)+BMP−7(100ng/ml)
14.アクチビンA(100ng/ml)+IL−4(10ng/ml)
15.アクチビンA(100ng/ml)+(SDF1a(20ng/ml)
16.アクチビンA(100ng/ml)+Wnt−3a(10ng/ml)+Wnt−5a(10ng/ml)+Wnt−7a(10ng/ml)
17.アクチビンA(100ng/ml)+IL−4(10ng/ml)+SDF1a(20ng/ml)+BMP−4(100ng/ml)
【0258】
結果
DEプロトコール処理の3日目に細胞を採取した。分析に際しては、処理細胞の一定分量をRT−PCR用のRNAの調製に使用し、残りの細胞をFACS分析に使用した。CXCR4の出現頻度(%)を図37に示す。上記の増殖因子の添加により、CXCR4の発現は、低血清中での100ng/mlのAAによる処理以上には促進されなかった。
【0259】
RT−PCR分析では、選択された一群の胚体内胚葉マーカーの発現について細胞を分析した。示した結果は、基本培地中で増殖させたがアクチビンA又は他の増殖因子のいずれによっても処理しない細胞に対して検量したものである。FACSのデータと一致するように、表Vは、低血清中で高用量のアクチビンAで処理した培養にWnt−3a等の増殖因子を添加しても胚体内胚葉マーカーの顕著なアップレギュレーションが認められなかったことを示している。これは、アクチビンA、WNT3A、及び低血清の存在下で培養したES細胞においてDEマーカーの顕著な増大が認められた上記の実施例とは対照的である。
【0260】
(実施例23)
MATRIGEL又はヒトフィブロネクチンでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
【0261】
H9P55細胞を、ヒトフィブロネクチン又は通常の増殖因子MATRIGEL(商標)(BDバイオサイエンス社(BD Biosciences))上で増殖及び分化させた。1μg/mlのヒトフィブロネクチン(R&Dシステムズ社(R&D Systems))を含む1mlのDMEM/F12(インビトロジェン/ギブコ社(Invitrogen/GIBCO))を組織培養処理した6穴培養皿の各ウェルに加えた。これとは別に、通常の増殖因子MATRIGEL(商標名)をDMEM/F12中で1:10に希釈し、希釈したMATRIGEL(商標名)を組織培養処理した6穴培養皿の各ウェルに1mlずつ加えた。細胞を、コラゲナーゼを用いて継代した。細胞が80%コンフルエンスに達した後、以下のように処理した。即ち、10ng/mlのマウス組み換えWnt3a(R&D)及び100ng/mlのアクチビンA(R&D)を含む0.5%FBS中で2日間処理した。この後、2%FBS及び100ng/mlアクチビンAで3日間処理した。図38のパネルa及びbは、それぞれ、フィブロネクチン及びMATRIGEL上で培養した胚性幹細胞によるCXCR4の発現を示したものである。リアルタイムPCRの結果(図39)により、フィブロネクチンでコーティングしたプレート及びMATRIGEL(商標)でコーティングしたプレートで胚体内胚葉の形成は同等であることが確認された。
【0262】
(実施例24)
異なる濃度のMATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
【0263】
約60〜70%コンフルエンスのH9培養を、0.5%FBS、20ng/mlのWnt−3a及び100ng/mlのアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS、20ng/mlのWnt−3a及び100ng/mlのアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。H9細胞は希釈率1:60〜1:10の通常のMATRIGELでコーティングしたプレート上で培養した。各プレートはMATRIGELで1時間、室温でコーティングした。
【0264】
リアルタイムPCRの結果を図40に示す。低血清、アクチビンA、及びWnt−3aによるヒト胚性幹細胞の処理により、CXCR4、GATA4、グースコイド(Goosecoid)、HNF−3β、及びSOX−17遺伝子の発現が認められ、細胞が胚体内胚葉段階に分化していることが示唆された。しかしながら、Wnt−3aの存在下では、MATRIGEL(商標)コーティングの濃度は分化において重要な役割を果たしてはいないようである。
【0265】
(実施例25)
細胞外基質でコーティングした組織培養基質上で培養した後、MEF上で培養したヒト胚性幹細胞の胚体内胚葉への分化−Wnt−3aの役割
少なくとも5代にわたってMATRIGEL(商標)上で培養したヒト胚性幹細胞系H9から得た細胞をES培地中でMEFフィーダー上に播いた。細胞が約60〜70%コンフルエンスに達した時点で、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露し、その後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。更なる処理群として、20ng/mlのWnt−3aで5日間すべて、及び10〜100ng/mlのアクチビンAで処理した群を置いた。
【0266】
3日目及び5日目に、培養をSOX−17、SOX−7、α胎児タンパク質(AFP)、CXCR4、ブラキュリ(Brychyury)(Bry)、グースコイド(gooscecoid)(GSC)、HNF−3β、GATA4、hTERT及びOct4についてリアルタイムPCRにより分析した。AFP及びSOX−7が臓側内胚葉マーカーとみなされるのに対し、GATA4、HNF−3β及びSOX−17は胚体内胚葉マーカーを代表するものであり、GSC、Bry、及びCXCR4は原始線条のマーカーを代表するものである。hTERT及びOct−4はそれぞれ自己再生能及び多能性のマーカーである。リアルタイムPCRの結果を図41のパネルa〜dに示す。3日目及び5日目にFACS分析も行った。CXCR4及びCD9の発現レベルを分析し、図41のパネルeに示した。
【0267】
Wnt−3aの非存在下では、100ng/mlのアクチビンA中で培養した細胞のAFP発現レベルは非処理のコントロールにおける発現レベルと同様であった。しかしながら、100ng/mlのアクチビンA中で培養した細胞にWnt−3aを加えることによって、AFPの発現に経時的な増加が認められた。低濃度のアクチビンAを使用した場合、Wnt−3aの存在に関わらずAFPの発現は極めて高かった(図41、パネルa)。このことは、細胞の胚体外組織への分化を防止するには高濃度のアクチビンAが必要であることを示唆するものである。
【0268】
FACS分析により、高濃度のアクチビンAで処理したがWnt−3aでは処理しない試料の集団ではCXCR4陽性細胞が32〜42%であったのに対して、3日目に高濃度のアクチビンA及びWnt3aで処理した試料の集団では23〜33%であることが示された(図41、パネルe)。処理5日目までには、高濃度のアクチビンAで処理したがWnt−3aでは処理しなかった細胞の28〜32%がCXCR4を発現したのに対し、高濃度のアクチビンA及びWnt−3aで処理した細胞では43〜51%であった(図41、パネルf)。低濃度のアクチビンAで処理した細胞では、Wnt−3a処理群(3〜4%)と比較してWnt−3aで処理しなかった処理群においてCXCR4陽性細胞がより多かった(11〜20%)(図41、パネルg)。以上まとめると、Wnt−3aは、MEF上で培養されたヒト胚性幹細胞の胚体内胚葉への分化において重要な役割を果たしてはいないようである。このことは、フィーダー層が、アクチビンAによって誘導される胚体内胚葉の形成を促進するうえで充分なWnt−3a又は類似のリガンドを恐らく分泌していることを示唆するものである。
【0269】
(実施例26)
細胞外基質でコーティングした組織培養基質上で培養されたヒト胚性幹細胞の、Wnt阻害剤DKK−1による処理後の胚体内胚葉への分化
Wnt−3aの添加によって分化の増大がもたらされるか否かを調べるため、Wnt−3シグナル伝達の阻害剤を培養に加えた。約60〜70%コンフルエンスのH9培養を、0.5%FBS、20ng/mlのWnt3a、100ng/mlのDikkopf−1(DKK−1)及び100ng/mlのアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlのアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。H9細胞は希釈率1:30の増殖因子低減MATRIGELでコーティングしたプレート上で培養した。各プレートはMATRIGELで1時間、室温でコーティングした。
【0270】
5日目に、培養を、SOX−17、SOX−7、α胎児タンパク質(AFP)、CXCR4、ブラキュリ(Brychyury)(Bry)、グースコイド(gooscecoid)(GSC)、HNF−3β、GATA4、hTERT及びOct4についてリアルタイムPCRにより分析した。AFP及びSOX−7が臓側内胚葉マーカーとみなされるのに対し、GATA4、HNF−3β及びSOX−17は胚体内胚葉マーカーを代表するものであり、GSC、Bry、及びCXCR4は原始線条のマーカーを代表するものである。hTERT及びOct−4はそれぞれ自己再生能及び多能性のマーカーである。結果を図42に示した。
【0271】
Wnt−3aの存在下では、細胞はいずれも胚体内胚葉のマーカーであるCXCR4、GATA4、HNF−3β及びSOX17を発現している。グースコイド(gooscecoid)等の原始線条形成のマーカーも、非処理のコントロールにおいて検出されるよりも高いレベルで検出された。DKK1の添加により、上記の分化のマーカーの発現レベルは非処理の細胞における発現レベルと同等のレベルにまで劇的に減少した。
【0272】
(実施例27)
MATRIGELでコーティングした組織培養基質上で培養し、低血清及びアクチビンA±Wnt−3aで分化させたH9胚性幹細胞におけるDEマーカーの免疫蛍光染色
5日目のH9細胞のDE培養を、実施例10に従ってSOX−17、HNF−3B、GATA−4、N−カドヘリン及びE−カドヘリンについて染色した。すべての核をDAPIで対比染色した。20ng/mlのWnt−3aにより、Wnt−3aの非存在下で分化させた培養と比較して大幅に多数の核がSOX−17、HNF−3β及びGATA−4について陽性に染色された。更に、Wnt−3aの添加により、E−カドヘリンの発現が著しく消失し、N−カドヘリンの発現が昂進した(図43、パネルa及び図43、パネルb)。
【0273】
(実施例28)
MEF又はMATRIGEL上での胚体内胚葉形成後の胚性幹細胞における遺伝子発現の変化のマイクロアレイによる分析
RNeasy mini kit(キアゲン社(Qiagen))を使用して以下の胚性幹細胞の培養から全RNAを単離した。即ち、A)MATRIGEL(商標)コーティングしたプレート(希釈率1:30)上で培養し、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理したH9P33細胞、B)MEF細胞上で培養し、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理したH9P44細胞、及びC)MATRIGEL(商標名)コーティングしたプレート(希釈率1:30)上で培養し、0.5%FBS及び100ng/mlアクチビンA及び20ng/mlのWnt−3aを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理したH9P48細胞。各群のコントロールには、MATRIGELコーティングした培養皿に播種し、MEF馴化培地中で培養した細胞、又はMEF上に播種し、ES培地中で培養した細胞を含めた。すべての群で3つの生物学的複製物を用い、各生物学的複製物を2個の別々の遺伝子チップ上で繰り返し用いた。
【0274】
試料の調製、ハイブリダイゼーション、及び画像分析は、アフィメトリックス社(Affymetrix)製Human Genome U133 Plus2.0 Arrayに従って行った。正規化及び対数変換を行った後、OmniViz(登録商標)ソフトウェア(マサチューセッツ州所在)及びGENESIFTER(ビズエックスラブズ社(VizXLabs)、ワシントン州所在)を使用してデータ分析を行った。試料間の遺伝子発現における有意差は、分散分析及び0.05以下の調節p値を用いたF検定(Benjamini and Hochberg補正)を用いて評価した。少なくとも1つの群においてプレゼントコール(present call)が示された遺伝子のみが分析に含まれた。表VIに、A群、B群及びC群間で少なくとも5倍の差を示す平均正規化、対数変換した遺伝子のシグナル強度を、各遺伝子の調節p値とともに示す。
【0275】
(実施例29)
MATRIGELでコーティングした組織培養基質上で培養されたSA002 ES細胞系の胚体内胚葉への分化
8ng/mlのbFGFを添加したMEF−CM中、MATRIGELコーティングしたプレート(希釈率1:30)上で少なくとも3代にわたって予め培養したSA002 P38細胞(セラーティス社(Cellartis)スウェーデン)を、0.5%FBS、及び100ng/mlアクチビンA(R&Dシステムズ社(R&D Systems)ミネソタ州所在)±20ng/mlのWnt−3a又は100nMのGSK−3BIX阻害剤を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。リアルタイムPCRの結果を図44のパネルa及びbに示す。H1、H7及びH9細胞系と同様、SA002細胞系も、DEマーカーの顕著な発現にはWnt−3Aの添加をやはり必要とした。図45にCXCR4の発現を示す。a)AAで処理、b)AA+Wnt−3a、c)AA+GSK−3B阻害剤。
【0276】
(実施例25)
ヒト血清でコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
継代数55のヒト胚性幹細胞系H1の培養をヒト血清(シグマ社(Sigma)No.H1388、ミズーリ州所在)でコーティングしたプレート上で増殖及び分化させた。0.5mlのヒト血清を組織培養処理した6穴培養皿の各ウェルに加え、室温で1時間インキュベートし、ヒト胚性幹細胞を加える前に吸引した。細胞が80%コンフルエンスに達した後、以下のように処理した。即ち、10ng/mlのマウス組み換えWnt3a(R&D)又は100nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)、カリフォルニア州所在)及び100ng/mlのアクチビンA(R&D)を含む0.5%FBS中で2日間処理した。この後、2%FBS及び100ng/mlアクチビンAで3日間処理した。この後、培養をリアルタイムPCRで分析した(図46、パネルa及びb)。アクチビンAのみで処理した細胞と比較して、アクチビンA+GSK−3B阻害剤又はWnt−3Aで処理した細胞では胚体内胚葉マーカーの著明な発現が認められた。これらの所見は、MATRIGEL(商標)又はヒトフィブロネクチンでコーティングしたプレート上で培養したヒト胚性幹細胞における我々の発見に対応したものである。
【0277】
(実施例31)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−異なるGSK−3B阻害剤の評価
ヒト胚性幹細胞からの胚体内胚葉(DE)の形成における市販の多くのGSK−3B阻害剤の有効性を評価した。以下のGSK−3B阻害剤を100nMで評価した。即ち、GSK−3B阻害剤VIII(カタログNo.361549、カルバイオケム社(Calbiochem)、カリフォルニア州所在)、GSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)、カリフォルニア州所在)、GSK−3B阻害剤XI(カタログNo.361553、カルバイオケム社(Calbiochem)、カリフォルニア州所在)、GSK−3B阻害剤XII(カタログNo.361554、カルバイオケム社(Calbiochem)、カリフォルニア州所在)。H1P54 ES細胞を、MATRIGEL(商標名)(希釈度1:30)でコーティングした培養皿上で培養し、0.5%FBS、100ng/mlアクチビンA(AA)±各種GSK−3B阻害剤を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。コントロール培養を低血清及び高用量のAAで処理した。図47のパネルa及びbは、5日目における胚体内胚葉マーカーの遺伝子発現を示したものである。GSK−3B阻害剤IX及びXIは、GSK−3B阻害剤VIII及びXIIと比較してDEの形成を誘導するうえでいずれも有効であった。
【0278】
(実施例32)
無フィーダー条件下で培養されたヒト胚性幹細胞による膵臓内胚葉の形成−レチノイン酸類似体の評価
H9P49胚性幹細胞を、MATRIGEL(商標)(希釈度1:30)でコーティングした培養皿上で培養し、0.5%FBS、20ng/mlのWnt−3a(カタログNo.1324−WN−002、R&Dシステムズ社(R&D Systems)ミネソタ州所在)、及び100ng/mlアクチビンA(R&Dシステムズ社(R&D Systems)ミネソタ州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。5日目に細胞を回収してFACS及びリアルタイムPCRによる評価を行った。上記の実施例において示したように、このプロトコールによりCXCR4及びSOX−17のような胚体内胚葉マーカーが顕著にアップレギュレートされた。得られた5日目の胚体内胚葉細胞を以下の培地条件に曝露して膵臓内胚葉の形成を誘導した。即ち、2%FBS及び1μMのオールトランスレチノイン酸(RA)(カタログNo.R2625、シグマ社(Sigma)、ミズーリ州所在)、又は0.1〜10μMのAM−580(4−[(5,6,7,8−テトラヒドロ−5,5,8,8−テトラメチル−2−ナフタレニル)カルボキサミド]安息香酸、カタログNo.A8843、シグマ社(Sigma)、ミズーリ州所在)、又は0.1〜1μMのTTNPB(4−[(E)−2−(5,6,7,8−テトラヒドロ−5,5,8,8−テトラメチル−2−ナフタレニル)−1−プロペニル]安息香酸(アロチノイド酸)、カタログNo.T3757、シグマ社(Sigma)、ミズーリ州所在)を添加したDMEM/F12培地中で3日間培養した。AM−580及びTTNPBは、レチノイン酸受容体に対する親和性を有するレチノイン酸類似体である。RA処理の後、2%FBS及び20〜50ng/mlのbFGF(カタログNo.F0291、シグマ社(Sigma)、ミズーリ州所在)を添加したDMEM/F12培地中で更に3日間処理した。細胞を収穫し、mRNAの試料を採取して分析に供した。
【0279】
遺伝子発現分析(図48パネルa〜d)により、1μMのRAの添加後にbFGFに曝露することにより、PDX−1等の膵臓内胚葉マーカーが顕著にアップレギュレートされることが示された。更にこのプロトコールにより、CDX−1及びAFP等の前腸内胚葉マーカーの著明な発現が認められた。1μMの濃度では、RA類似体の添加により、同等の膵臓内胚葉及び前腸内胚葉マーカーの発現が見られた。しかしながら、1μMのRA類似体の添加により、オールトランスレチノイン酸と比較してより顕著なAFPの発現が認められた。しかしながら、10μMのAM−580の添加によってPDX−1の発現レベルは高く維持されたままでAFP及びCDX−2の発現が抑制された。
【0280】
(実施例34)
ヒト胚性幹細胞におけるサイトカイン発現に対するWnt−3a処理の影響
Wnt−3aによる処理がサイトカイン発現に与える影響についてタンパク質アレイを用いて分析を行った。ヒト胚性幹細胞系H9の細胞を実施例15で述べた方法に従って培養した。継代数54において、0.5%FBSを含むDMEM/F12中、100ng/mlのアクチビンA±10ng/mlのWnt−3aの存在下で細胞を2日間分化させた。この後、100ng/mlのアクチビンA及び2%FBSを含むDMEM/F12中で細胞を更に3日間培養した。5日目の終わりに、CXCR4の発現を各処理群についてFACSにより調べた。アクチビンAのみで処理した細胞ではCXCR4を発現した細胞は1%に過ぎなかった。アクチビンA及びWnt3aで処理した細胞では、73%の細胞がCXCR4の発現について陽性であった。
【0281】
哺乳動物細胞溶解キット(シグマ・アルドリッチ社(Sigma-Aldrich)、ミズーリ州所在)を用いて各処理群の細胞から細胞溶解物を調製した。各処理群からの馴化培地を回収して濃縮した。レイ・バイオテック社((RayBiotech)、ジョージア州所在)(http://www.raybiotech.com/)によって提供されるCytokine Arrayパネルを使用してサイトカインアレイ分析を行った。表VIIに、サイトカイン、増殖因子、並びにデータの正規化及びバックグランド除去後の受容体の発現を示す。各パネルに対して、ポジティブ及びネガティブコントロールも含まれた。示したデータは、細胞処理群ごとに2つの独立した試料(1、2)のものである。
【0282】
Wnt−3a処理細胞で馴化した培地では、アンジオゲニン、IGFBP−1及びEGFの著明なアップレギュレーションが認められた。IGFBP−1、TGFβ−1及びTGFβ−3等の多くのタンパク質がWnt−3a処理細胞の溶解物中でアップレギュレートされた。これらのアップレギュレートされたタンパク質を分化培地に再び添加することにより、胚体内胚葉形成に対するWnt−3aの効果を補充又は促進することが可能である。
【0283】
(実施例35)
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化:Wnt1の役割
H1P55 ES細胞を、MATRIGEL(商標)(希釈度1:30)でコーティングした培養皿上で培養し、0.5%FBS及び100ng/mlのアクチビンA±10〜20ng/mlのWnt−1(ペプロテック社(PeproTech)、ニュージャージー州所在、カタログNo.120−17)を添加したDMEM/F12培地に2日間曝露した後、2%FBS、100ng/mlアクチビンA(AA)±10又は20ng/mlのWnt−1を添加したDMEM/F12培地で更に3日間処理した。以下のWnt1+AAの組み合わせについて試験を行った。
【0284】
a)0.5%FBS+DM−F12中、20ng/mlのWNT1+100ng/mlのAAで1〜2日目の後、2%FBS+DM−F12+100ng/mlのAAで3日目、b)0.5%FBS+DM−F12中、20ng/mlのWNT1+100ng/mlのAAで1〜2日目の後、2%FBS+DM−F12+100ng/mlのAAで3〜5日目、c)0.5%FBS+DM−F12中、10ng/mlのWNT1+100ng/mlのAAで1〜2日目の後、2%FBS+DM−F12+100ng/mlのAAで3日目、d)0.5%FBS+DM−F12中、10ng/mlのWNT1+100ng/mlのAAで1〜2日目の後、2%FBS+DM−F12+100ng/mlのAAで3〜5日目、e)0.5%FBS+DM−F12中、20ng/mlのWNT1+100ng/mlのAAで1〜2日目の後、2%FBS+DM−F12+100ng/ml+のAA+20ng/mlのWNT1で3日目、f)0.5%FBS+DM−F12中、20ng/mlのWNT1+100ng/mlのAAで1〜2日目の後、2%FBS+DM−F12+100ng/mlのAA+20ng/mlのWNT1で3〜5日目。図49のパネルa及びbに、H1細胞を低血清、AA及びWNT1で処理した後の胚体内胚葉マーカーについてのリアルタイムPCRのデータを示す。100ng/mlのAAの存在下で20ng/mlのWNT1を添加することにより、胚体内胚葉マーカー(Bry、CXCR4、GSC、SOX17、HNF−3B及びGATA−4)の顕著なアップレギュレーションが認められた。
【0285】
(実施例36)
膵臓内分泌分化に対するグルコースの影響
膵臓内胚葉細胞の膵臓内分泌細胞への分化の効率は、例えば基本培地の選択、又はグルコースの濃度等の多くの因子に依存する。胚性幹細胞から誘導された膵臓内胚葉細胞の膵臓内分泌細胞への分化に対するグルコース濃度の影響を調べた。
【0286】
基本培地を変更することによるグルコース濃度の変化:未分化のヒト胚性幹細胞(H1及びH9)の培養物を、膵臓内胚葉細胞への分化に先立って、実施例1に述べた方法に従って培養した。胚性幹細胞を、血清の非存在下、100ng/mlのアクチビンAを含むRPMI中で1日培養することによって膵臓内胚葉細胞に分化させた。この期間の後、100ng/mlのアクチビンA及び0.2%FBSを含むRPMI中で細胞を更に2日間培養した。この処理の後、2%FBS、FGF10(50ng/ml)及びKAAD−シクロパミン(250nM)を含むRPMIで培地を置き換えた。この培地中で細胞を4日間培養した。この期間の後、1xB27を添加した、オールトランスレチノイン酸(2μM)、FGF10(50ng/ml)及びKAAD−シクロパミン(0.25μM)を含む培地に4日間置き換えて膵臓内胚葉細胞の形成を誘導した。膵臓内胚葉細胞の収率は、低グルコースDMEM又はDMEM/F12で処理した培養で有意な差は見られなかった。
【0287】
膵臓内胚葉細胞をエキセンジン4及びHGFで処理することによって膵臓内分泌細胞に分化させた。エキセンジン4(50ng/ml)及びHGF(50ng/ml)は低グルコースDMEM又はDMEM/F12に10日間加えた。両培地に1xB27を添加した。培養を収穫し、mRNAの試料を採取して分析に供した。試料は、(Nature Biotechnology 24, 1392 - 1401(2006))に開示される方法に従って得られた膵臓内胚葉に対して標準化された。
【0288】
インスリンの発現をリアルタイムPCRにより分析した。図50のパネルa及びbに示されるように、インスリン及びグルカゴン遺伝子の発現は、DMEM−低グルコース中で処理した細胞と比較してDMEM/F12中で処理した細胞においていずれも大きく増大した。インスリンの発現を更に免疫組織化学法によって分析した(図51)。DMEM/F12中での処理により、DMEM−低グルコースと比較してインスリン陽性細胞の比率が大きくなった(図51、パネルa及びb)。インスリン陽性細胞はPDX−1についても陽性であった(パネルc)。
【0289】
グルコース濃度の変化:未分化のヒト胚性幹細胞(H1及びH9)の培養物を、膵臓内胚葉細胞への分化に先立って、実施例1に述べた方法に従って培養した。胚性幹細胞を、血清の非存在下、100ng/mlのアクチビンAを含むRPMI中で1日培養することによって膵臓内胚葉細胞に分化させた。この期間の後、100ng/mlのアクチビンA及び0.2%FBSを含むRPMI中で細胞を更に2日間培養した。この処理の後、2%FBS、FGF10(50ng/ml)及びKAAD−シクロパミン(250nM)を含むRPMIで培地を置き換えた。この培地中で細胞を4日間培養した。この期間の後、1xB27を添加した、オールトランスレチノイン酸(2μM)、FGF10(50ng/ml)及びKAAD−シクロパミン(0.25μM)を含むCMRLに4日間置き換えて膵臓内胚葉細胞の形成を誘導した。この培地に5、10又は20mMのグルコースを添加した。膵臓内胚葉細胞の収率は、5、10又は20mMのグルコースで処理したH9胚性幹細胞から誘導された培養で有意な差は見られなかった(図52、パネルa)。
【0290】
膵臓内胚葉細胞を、1xB27、エキセンジン4(50ng/ml)及びHGF(50ng/ml)を添加したCMRLで、5、10又は20mMグルコース中、2、4又は10日間細胞を処理することによって膵臓内分泌細胞に分化させた。細胞を収穫し、mRNAの試料を採取して分析に供した。試料は、(Nature Biotechnology 24, 1392-1401(2006))に開示される方法に従って得られた膵臓内胚葉に対して標準化された。
【0291】
図52のパネルb〜gは、ヒト胚性幹細胞系H9から誘導された細胞におけるNgn−3、NeuroD−1、Nkx2.2、Pax−4、インスリン及びグルカゴンの発現に対するグルコースの影響を示したものである。Ngn3は、膵臓の内分泌細胞の運命決定に関与する最初の転写因子であり、NeuroD1はNgn3の直接的な標的である。グルコース刺激はNgn3及びNeuroD1の両方のmRNAレベルを用量依存的に増大させた。別の2つの重要な膵臓マーカーであるNkx2.2及びPax−4もやはり同様の発現パターンを示した(図52、パネルd及びe)。10mMのグルコースで10日間処理した細胞において最適なインスリン及びグルカゴンの発現が観察された(図52、パネルf及びg)。
【0292】
Ngn−3、NeuroD−1、Nkx2.2、Pax−4について同様の結果が、ヒト胚性幹細胞系H1から誘導された培養において観察された(表VIII)。しかしながら、20mMのグルコースで10日間処理した細胞において最適なインスリン及びシナプトフィシンの発現が観察された(表VIII)。
【0293】
本発明の方法によって形成されたインスリン発現細胞から放出されるC−ペプチド:グルコース介在C−ペプチド放出を、2、10又は20mMのグルコースで処理したH1細胞から誘導されたインスリン陽性細胞で観察した。C−ペプチド放出を引き起こすため、細胞をまず、重炭酸塩及びHEPESを含むクレブス・リンゲル液(KRBH;129mM NaCl、4.8mM KCl、2.5mM CaCl2、1.2mM KH2PO4、1.2mM MgSO4、5mM NaHCO3、10mM HEPES、0.1%BSA)と1時間インキュベートした。培地を捨て、2mMのD−グルコースを含むクレブス・リンゲル液で置き換えた。細胞を20mMグルコース又は0.5mMのIBMX(すべてシグマ(Sigma)社より購入)で1時間刺激した。刺激上清中のC−ペプチド濃度を基本上清中のC−ペプチド濃度で割ることによって各培養について刺激倍数(fold stimulation)を計算した。
【0294】
BMXは、C−ペプチドの放出を1.2〜3倍に刺激した(図53)。20mMグルコースはC−ペプチド放出を刺激しなかった。2、10及び20mMグルコース中で形成されたインスリン陽性細胞間で、C−ペプチド分泌に有意な差は認められなかった。
【0295】
以上まとめると、我々のデータは、グルコースが内分泌マーカーであるNgn3及びNeuroD1の用量依存的なアップレギュレーションを誘導することを示唆するものであり、このことは、グルコースが、ヒト胚細胞の膵臓内分泌細胞への用量依存的な分化を誘導することを示唆している。インスリンの発現もまた、グルコースによって用量依存的に調節される。
【0296】
本文書の全体を通じて引用した刊行物はそれらの全容を本明細書に援用するものである。本発明の様々な態様を実施例及び好適な実施形態を参照することで上記に説明したが、本発明の範囲は上記の記載によって規定されるものではなく、以下の特許請求の範囲を特許法の原則に則って適切に解釈することによって定義されるものであることは認識されるであろう。
【0297】
〔実施態様〕
(1) 膵臓内分泌系統に特徴的なマーカーを発現する細胞を生成するための方法において、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、約10mM〜約20mMの濃度のグルコースを添加した培地中で、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって処理することによって、前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させること、を含む、方法。
(2) 前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞が、多能性幹細胞から分化させられる、実施態様1に記載の方法。
(3) 前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞が、胚体内胚葉系統に特徴的なマーカーを発現する細胞から誘導される、実施態様1に記載の方法。
(4) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞が、多能性幹細胞から誘導される、実施態様1に記載の方法。
(5) 前記多能性幹細胞が、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に更に分化させられる前に胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させられる、実施態様2に記載の方法。
(6) 前記グルコースが約10mMの濃度で使用される、実施態様1に記載の方法。
(7) 前記グルコースが約20mMの濃度で使用される、実施態様1に記載の方法。
(8) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約30日間処理する、実施態様1に記載の方法。
(9) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約20日間処理する、実施態様1に記載の方法。
(10) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約10日間処理する、実施態様1に記載の方法。
【0298】
(11) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約10日間処理する、実施態様1に記載の方法。
(12) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約4日間処理する、実施態様1に記載の方法。
(13) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間処理する、実施態様1に記載の方法。
(14) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、実施態様6に記載の方法。
(15) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、実施態様7に記載の方法。
(16) 多能性幹細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させるための方法において、
a.前記多能性幹細胞を培養する工程と、
b.前記多能性細胞をアクチビンAで処理することによって、前記多能性幹細胞を、胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
c.前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、少なくとも1種類の繊維芽細胞増殖因子、又はレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で処理することによって、前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
d.前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、約10mM〜約20mMの濃度のグルコースを添加した培地中で、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって処理することによって、前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させる工程と、を含む、方法。
(17) 前記グルコースが約10mMの濃度で使用される、実施態様16に記載の方法。
(18) 前記グルコースが約20mMの濃度で使用される、実施態様16に記載の方法。
(19) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約30日間処理する、実施態様16に記載の方法。
(20) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約20日間処理する、実施態様16に記載の方法。
【0299】
(21) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約10日間処理する、実施態様16に記載の方法。
(22) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約10日間処理する、実施態様16に記載の方法。
(23) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約4日間処理する、実施態様16に記載の方法。
(24) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間処理する、実施態様16に記載の方法。
(25) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、実施態様21に記載の方法。
(26) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、実施態様22に記載の方法。
(27) 糖尿病を有する患者に実施態様1に記載の細胞を移植することによって前記患者を治療するための方法。
(28) 糖尿病を有する患者に実施態様16に記載の細胞を移植することによって前記患者を治療するための方法。
(29) 前記多能性幹細胞が胚性幹細胞である、実施態様16に記載の方法。
(30) 前記胚性幹細胞がヒト胚性幹細胞である、実施態様29に記載の方法。
【0300】
(31) 前記ヒト胚性幹細胞が、H1及びH9からなる群の細胞系から誘導される、実施態様30に記載の方法。
【特許請求の範囲】
【請求項1】
膵臓内分泌系統に特徴的なマーカーを発現する細胞を生成するための方法において、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、約10mM〜約20mMの濃度のグルコースを添加した培地中で、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって処理することによって、前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させること、を含む、方法。
【請求項2】
前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞が、多能性幹細胞から分化させられる、請求項1に記載の方法。
【請求項3】
前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞が、胚体内胚葉系統に特徴的なマーカーを発現する細胞から誘導される、請求項1に記載の方法。
【請求項4】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞が、多能性幹細胞から誘導される、請求項1に記載の方法。
【請求項5】
前記多能性幹細胞が、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に更に分化させられる前に胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させられる、請求項2に記載の方法。
【請求項6】
前記グルコースが約10mMの濃度で使用される、請求項1に記載の方法。
【請求項7】
前記グルコースが約20mMの濃度で使用される、請求項1に記載の方法。
【請求項8】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約30日間処理する、請求項1に記載の方法。
【請求項9】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約20日間処理する、請求項1に記載の方法。
【請求項10】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約10日間処理する、請求項1に記載の方法。
【請求項11】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約10日間処理する、請求項1に記載の方法。
【請求項12】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約4日間処理する、請求項1に記載の方法。
【請求項13】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間処理する、請求項1に記載の方法。
【請求項14】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、請求項6に記載の方法。
【請求項15】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、請求項7に記載の方法。
【請求項16】
多能性幹細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させるための方法において、
a.前記多能性幹細胞を培養する工程と、
b.前記多能性細胞をアクチビンAで処理することによって、前記多能性幹細胞を、胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
c.前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、少なくとも1種類の繊維芽細胞増殖因子、又はレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で処理することによって、前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
d.前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、約10mM〜約20mMの濃度のグルコースを添加した培地中で、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって処理することによって、前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させる工程と、を含む、方法。
【請求項17】
前記グルコースが約10mMの濃度で使用される、請求項16に記載の方法。
【請求項18】
前記グルコースが約20mMの濃度で使用される、請求項16に記載の方法。
【請求項19】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約30日間処理する、請求項16に記載の方法。
【請求項20】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約20日間処理する、請求項16に記載の方法。
【請求項21】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約10日間処理する、請求項16に記載の方法。
【請求項22】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約10日間処理する、請求項16に記載の方法。
【請求項23】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約4日間処理する、請求項16に記載の方法。
【請求項24】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間処理する、請求項16に記載の方法。
【請求項25】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、請求項21に記載の方法。
【請求項26】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、請求項22に記載の方法。
【請求項27】
糖尿病を有する患者に請求項1に記載の細胞を移植することによって前記患者を治療するための方法。
【請求項28】
糖尿病を有する患者に請求項16に記載の細胞を移植することによって前記患者を治療するための方法。
【請求項29】
前記多能性幹細胞が胚性幹細胞である、請求項16に記載の方法。
【請求項30】
前記胚性幹細胞がヒト胚性幹細胞である、請求項29に記載の方法。
【請求項31】
前記ヒト胚性幹細胞が、H1及びH9からなる群の細胞系から誘導される、請求項30に記載の方法。
【請求項1】
膵臓内分泌系統に特徴的なマーカーを発現する細胞を生成するための方法において、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、約10mM〜約20mMの濃度のグルコースを添加した培地中で、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって処理することによって、前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させること、を含む、方法。
【請求項2】
前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞が、多能性幹細胞から分化させられる、請求項1に記載の方法。
【請求項3】
前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞が、胚体内胚葉系統に特徴的なマーカーを発現する細胞から誘導される、請求項1に記載の方法。
【請求項4】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞が、多能性幹細胞から誘導される、請求項1に記載の方法。
【請求項5】
前記多能性幹細胞が、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に更に分化させられる前に胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させられる、請求項2に記載の方法。
【請求項6】
前記グルコースが約10mMの濃度で使用される、請求項1に記載の方法。
【請求項7】
前記グルコースが約20mMの濃度で使用される、請求項1に記載の方法。
【請求項8】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約30日間処理する、請求項1に記載の方法。
【請求項9】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約20日間処理する、請求項1に記載の方法。
【請求項10】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約10日間処理する、請求項1に記載の方法。
【請求項11】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約10日間処理する、請求項1に記載の方法。
【請求項12】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約4日間処理する、請求項1に記載の方法。
【請求項13】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間処理する、請求項1に記載の方法。
【請求項14】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、請求項6に記載の方法。
【請求項15】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、請求項7に記載の方法。
【請求項16】
多能性幹細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させるための方法において、
a.前記多能性幹細胞を培養する工程と、
b.前記多能性細胞をアクチビンAで処理することによって、前記多能性幹細胞を、胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
c.前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、少なくとも1種類の繊維芽細胞増殖因子、又はレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で処理することによって、前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
d.前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、約10mM〜約20mMの濃度のグルコースを添加した培地中で、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって処理することによって、前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させる工程と、を含む、方法。
【請求項17】
前記グルコースが約10mMの濃度で使用される、請求項16に記載の方法。
【請求項18】
前記グルコースが約20mMの濃度で使用される、請求項16に記載の方法。
【請求項19】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約30日間処理する、請求項16に記載の方法。
【請求項20】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約20日間処理する、請求項16に記載の方法。
【請求項21】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約10日間処理する、請求項16に記載の方法。
【請求項22】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約10日間処理する、請求項16に記載の方法。
【請求項23】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約4日間処理する、請求項16に記載の方法。
【請求項24】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間処理する、請求項16に記載の方法。
【請求項25】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、請求項21に記載の方法。
【請求項26】
前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、請求項22に記載の方法。
【請求項27】
糖尿病を有する患者に請求項1に記載の細胞を移植することによって前記患者を治療するための方法。
【請求項28】
糖尿病を有する患者に請求項16に記載の細胞を移植することによって前記患者を治療するための方法。
【請求項29】
前記多能性幹細胞が胚性幹細胞である、請求項16に記載の方法。
【請求項30】
前記胚性幹細胞がヒト胚性幹細胞である、請求項29に記載の方法。
【請求項31】
前記ヒト胚性幹細胞が、H1及びH9からなる群の細胞系から誘導される、請求項30に記載の方法。
【図1】



【図2a)】

【図2b)】

【図2c)】

【図3】
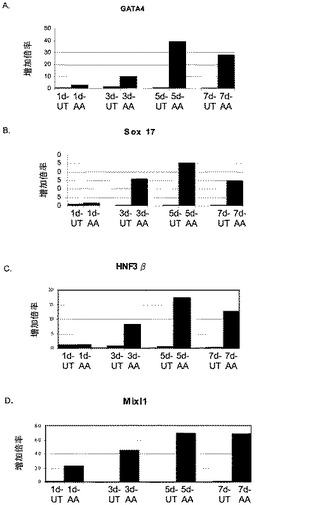
【図4】
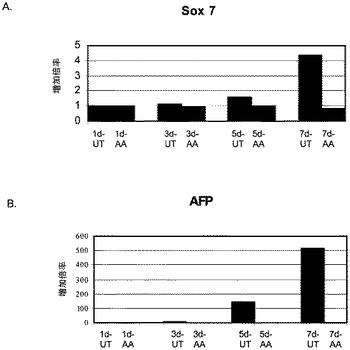
【図5】
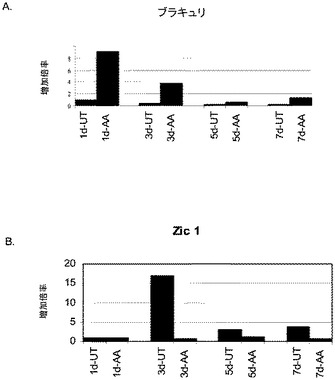
【図6】
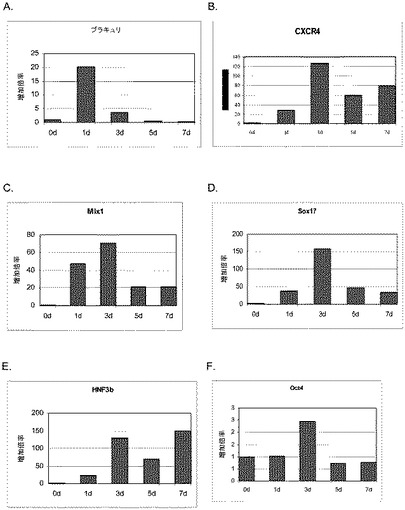
【図7】

【図8】
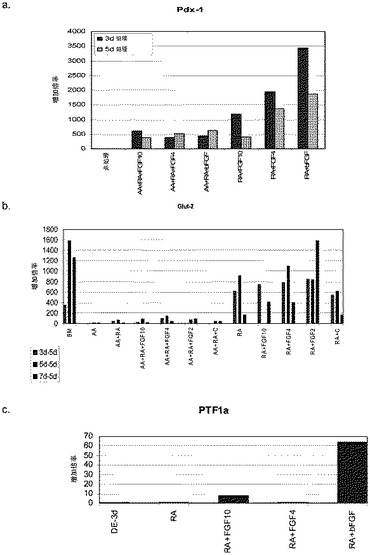
【図9】

【図10】
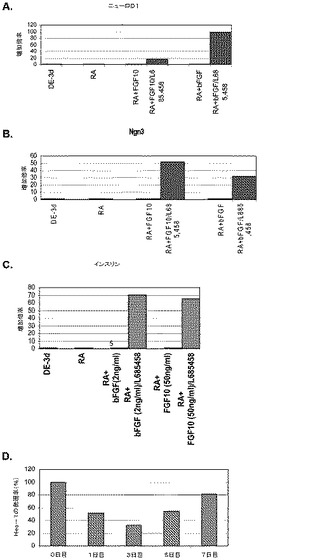
【図11】

【図12】

【図13】
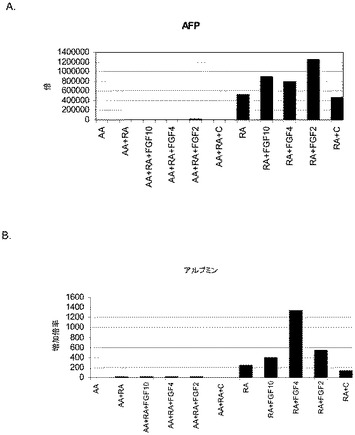
【図14】

【図15】
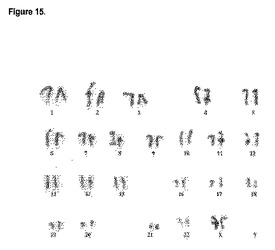
【図16】
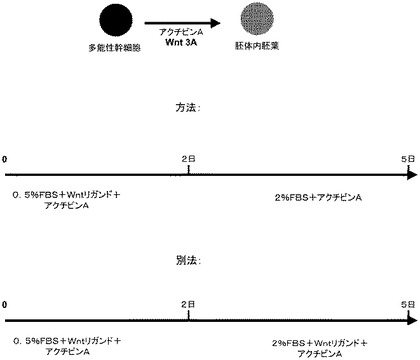
【図17】
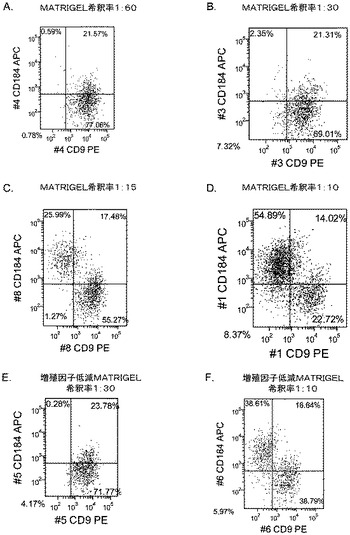
【図18】
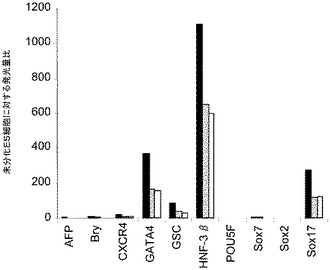
【図19】
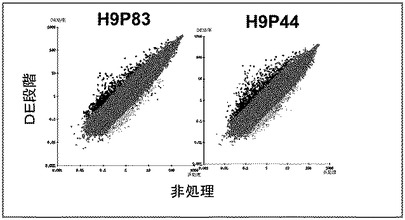
【図20】

【図21】
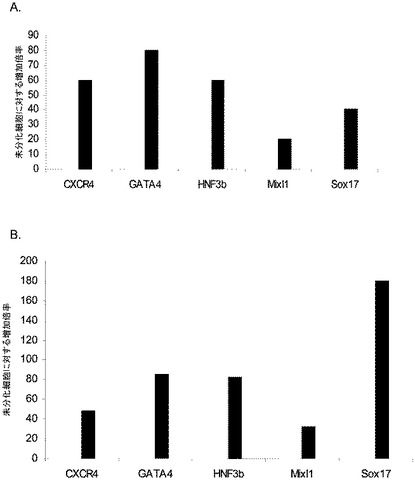
【図22A】
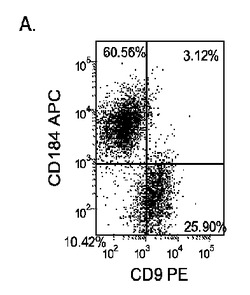
【図22B】

【図22C】
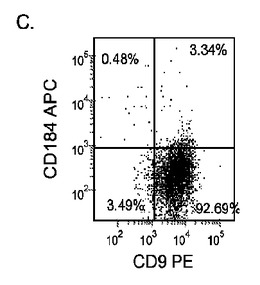
【図23】
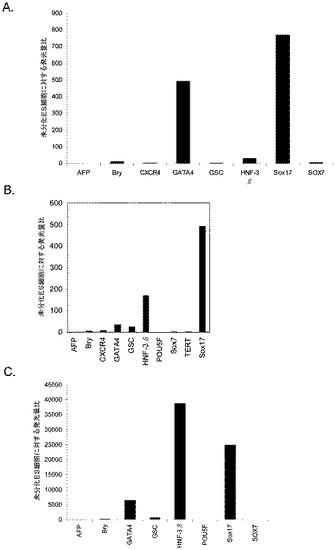
【図24A】

【図24B】

【図25A】
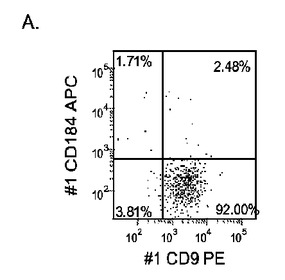
【図25B】
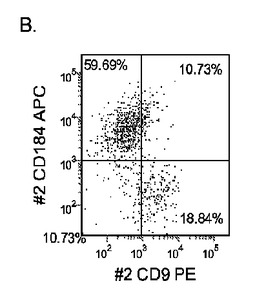
【図25C】

【図25D】
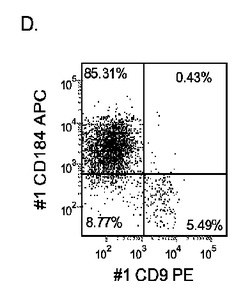
【図26】

【図27A】
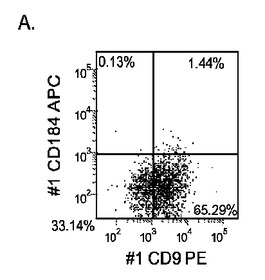
【図27B】
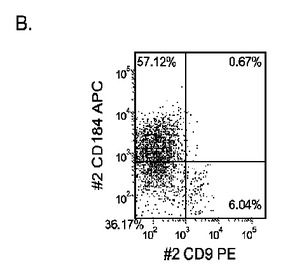
【図27C】
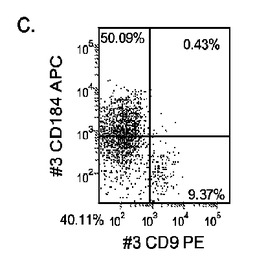
【図27D】

【図28】
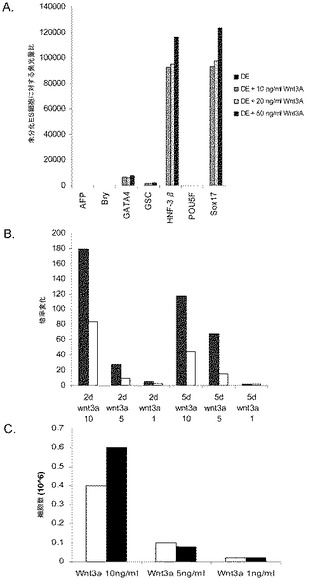
【図29A】
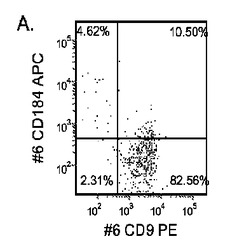
【図29B】
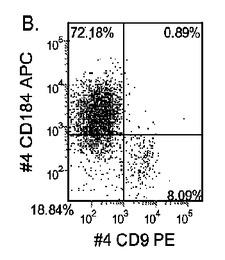
【図29C】
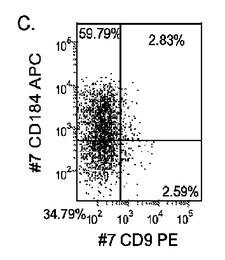
【図29D】

【図29E】
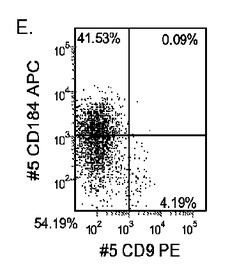
【図29F】

【図29G】
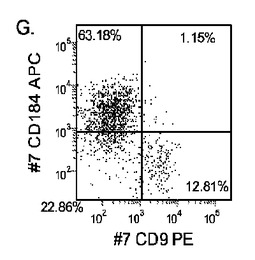
【図29H】
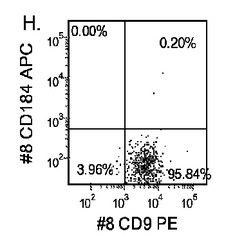
【図30】
【図31A】
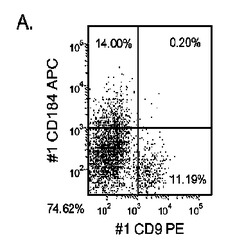
【図31B】
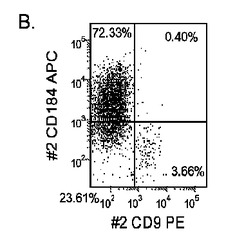
【図31C】
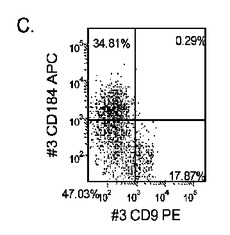
【図31D】
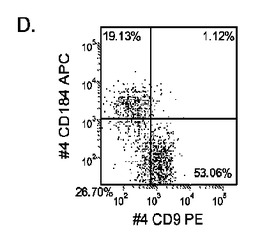
【図31E】
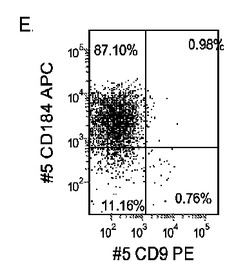
【図31F】

【図32】

【図33A】

【図33B】
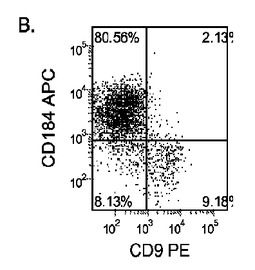
【図33C】
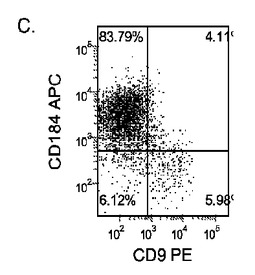
【図33D】
【図33E】
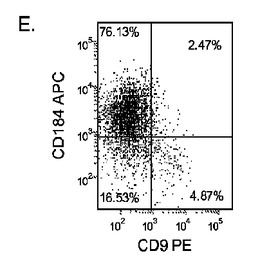
【図33F】
【図34】

【図35】
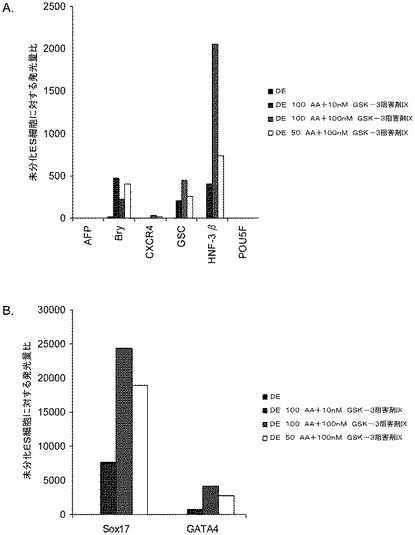
【図36】
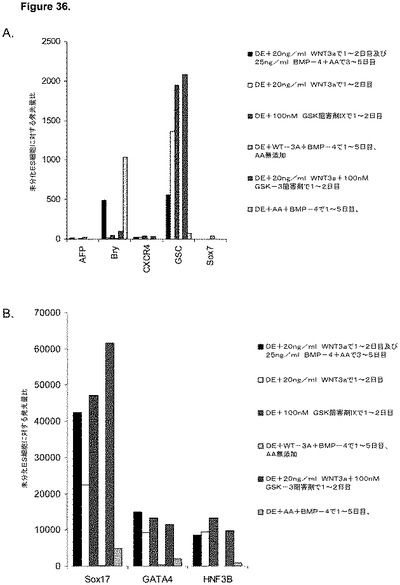
【図37】

【図38A】
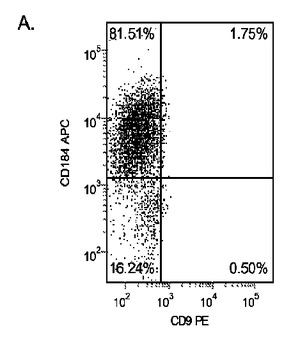
【図38B】
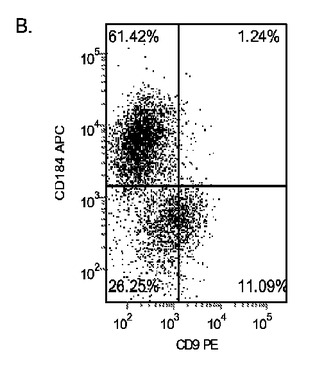
【図39】
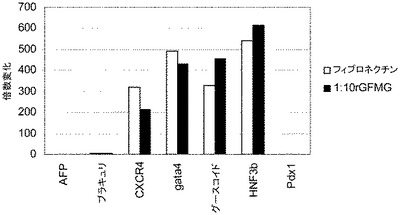
【図40】
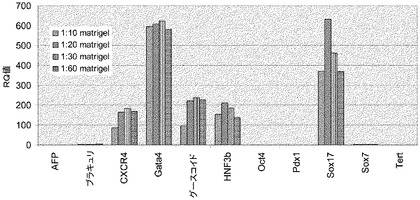
【図41−1】
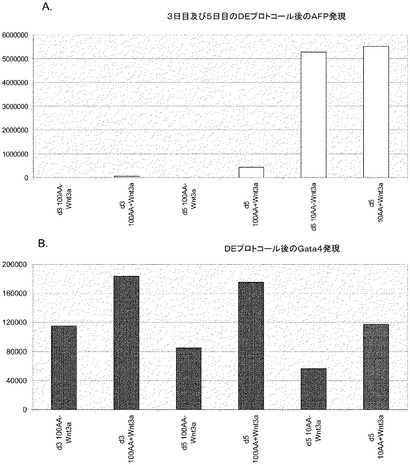
【図41−2】

【図41−3】
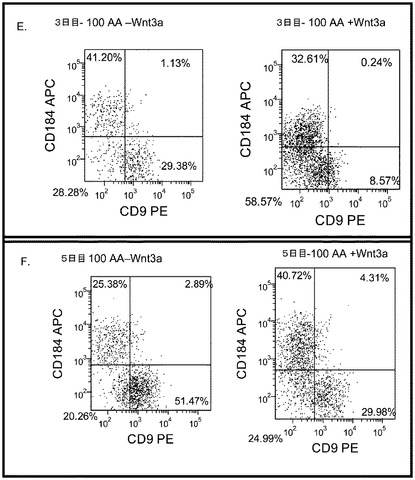
【図41−4】
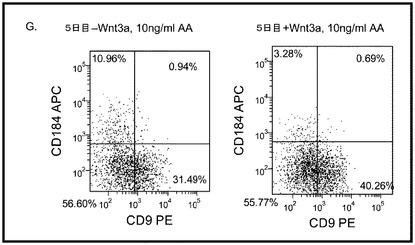
【図42】
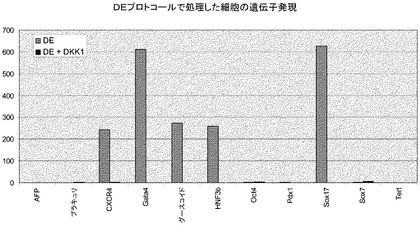
【図43A】

【図43B】

【図44】
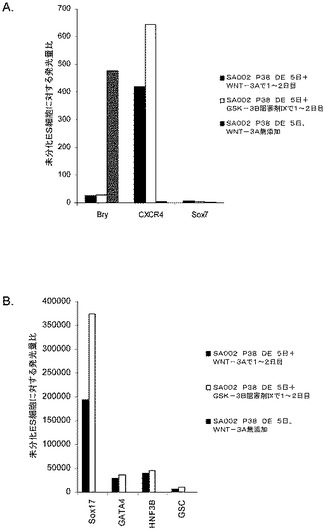
【図45A】

【図45B】
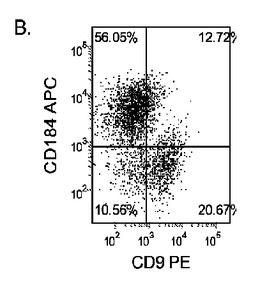
【図45C】
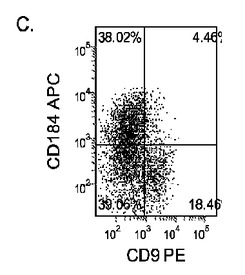
【図46】
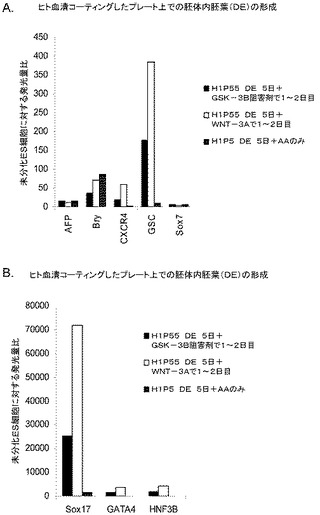
【図47】
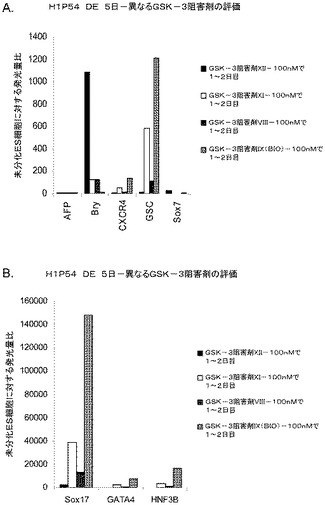
【図48】
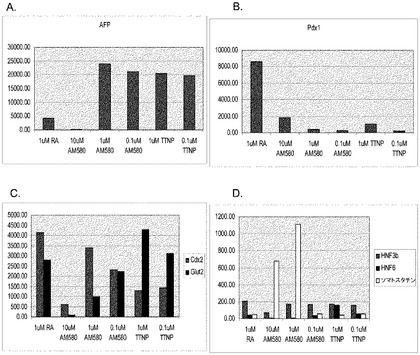
【図49】
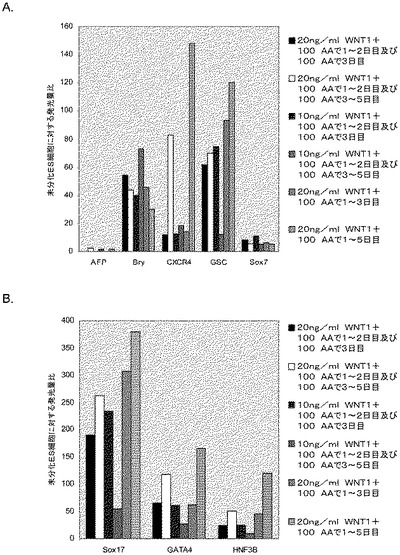
【図50】
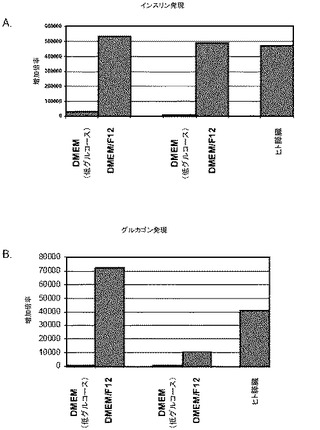
【図51】

【図52−1】
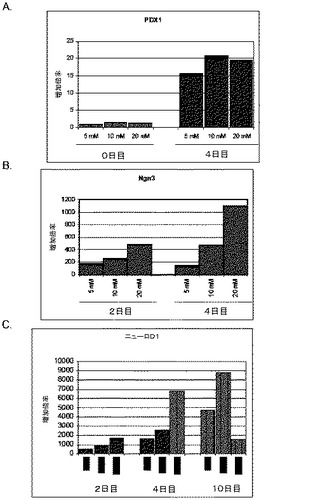
【図52−2】

【図52−3】

【図53】
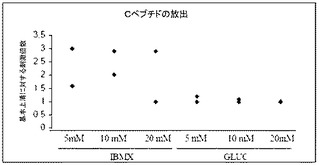
【図2a)】
【図2b)】
【図2c)】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22A】
【図22B】
【図22C】
【図23】
【図24A】
【図24B】
【図25A】
【図25B】
【図25C】
【図25D】
【図26】
【図27A】
【図27B】
【図27C】
【図27D】
【図28】
【図29A】
【図29B】
【図29C】
【図29D】
【図29E】
【図29F】
【図29G】
【図29H】
【図30】
【図31A】
【図31B】
【図31C】
【図31D】
【図31E】
【図31F】
【図32】
【図33A】
【図33B】
【図33C】
【図33D】
【図33E】
【図33F】
【図34】
【図35】
【図36】
【図37】
【図38A】
【図38B】
【図39】
【図40】
【図41−1】
【図41−2】
【図41−3】
【図41−4】
【図42】
【図43A】
【図43B】
【図44】
【図45A】
【図45B】
【図45C】
【図46】
【図47】
【図48】
【図49】
【図50】
【図51】
【図52−1】
【図52−2】
【図52−3】
【図53】
【公表番号】特表2010−535036(P2010−535036A)
【公表日】平成22年11月18日(2010.11.18)
【国際特許分類】
【出願番号】特願2010−520194(P2010−520194)
【出願日】平成20年7月31日(2008.7.31)
【国際出願番号】PCT/US2008/071782
【国際公開番号】WO2009/018453
【国際公開日】平成21年2月5日(2009.2.5)
【出願人】(596159500)ライフスキャン・インコーポレイテッド (100)
【氏名又は名称原語表記】Lifescan,Inc.
【住所又は居所原語表記】1000 Gibraltar Drive,Milpitas,California 95035,United States of America
【Fターム(参考)】
【公表日】平成22年11月18日(2010.11.18)
【国際特許分類】
【出願日】平成20年7月31日(2008.7.31)
【国際出願番号】PCT/US2008/071782
【国際公開番号】WO2009/018453
【国際公開日】平成21年2月5日(2009.2.5)
【出願人】(596159500)ライフスキャン・インコーポレイテッド (100)
【氏名又は名称原語表記】Lifescan,Inc.
【住所又は居所原語表記】1000 Gibraltar Drive,Milpitas,California 95035,United States of America
【Fターム(参考)】
[ Back to top ]