
ヒトCD16トランスジェニック非ヒト動物とその利用
【課題】抗体医薬の生物活性及び薬理評価を非ヒト動物モデルを用いて行なうことが求められている。
【解決手段】ヒトCD16とそのヒトアクセサリー分子とを、共発現させるようにゲノムを改変した非ヒト動物を取得し、ヒトCD16とそのヒトアクセサリー分子とを共発現する非ヒト動物またはその子孫及びその利用方法を提供する。
【解決手段】ヒトCD16とそのヒトアクセサリー分子とを、共発現させるようにゲノムを改変した非ヒト動物を取得し、ヒトCD16とそのヒトアクセサリー分子とを共発現する非ヒト動物またはその子孫及びその利用方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ヒトCD16とそのヒトアクセサリー分子とを共発現させるようにゲノムが改変された、非ヒト動物またはその子孫及びその利用に関する。
【背景技術】
【0002】
近年の抗体工学の進歩により、ヒトに対する抗原性が低く、臨床応用可能な遺伝子組換え抗体の作製が可能となった。具体的には、ヒト型キメラ抗体やヒト化抗体〔ヒト型相補性決定領域(complementarity determining region; CDR)移植抗体ともいう〕などのヒト化抗体作製技術の開発(非特許文献1−5参照)や、ヒト抗体産生トランスジェニックマウスやファージライブラリーを利用した完全ヒト抗体作製技術の開発(非特許文献6−12参照)が、モノクローナル抗体治療を医薬として用いることを可能にした。その結果、1990年後半からモノクローナル抗体医薬品が次々と認可された。医薬として開発されている抗体のほとんどは分子量約150kDaのヒトIgGであり、そのFc領域に2本のN-グリコシド結合複合型糖鎖が結合する糖蛋白質である。Fc領域はFc受容体や補体などが結合する領域であり、この部分を通じて抗体依存性細胞傷害活性(antibody-dependent cellular cytotoxicity; ADCC)や補体依存性細胞傷害活性(complement-dependent cytotoxicity; CDC)といったエフェクター活性が発揮される。最近の抗体医薬の臨床成績では、乳癌、大腸癌、血液癌といった多くのヒト悪性腫瘍において、延命や病態悪化に至るまでの期間延長といった効果が観察されている(非特許文献13−16参照)。また、癌患者Fc受容体の多型解析から、非ホジキンリンパ腫治療薬抗CD20抗体RituxanR(リツキシマブ)や乳癌治療薬抗Her2抗体HerceptinR(トラスツズマブ)の主たる抗腫瘍メカニズムの一つはADCC活性であることが明らかにされ(非特許文献17−21参照)、医薬開発に応用可能なADCC活性増強技術の開発が次世代抗体技術として注目されている。抗原となるヒトのゲノム情報が完全に解読された今日、抗体医薬は分子標的医薬の代表として熾烈な競争時代に入っており(非特許文献22参照)、現在、世界では、100を超える臨床試験と300を超える前臨床試験が行われていると言われている。
【0003】
しかしながら、現行の抗体医薬の臨床効果は必ずしも充分なものではなく、また、投与量が多いため薬剤費が高いといった課題がある。この原因の一つには、病態モデル動物を用いた非臨床実験において抗体医薬の十分な薬理評価が行なえていないことが挙げられる。これは、種差の違いから、披検薬であるヒト抗体が病態モデル動物のFc受容体ときちんとは結合せずエフェクター活性の十分な評価が難しいこと、Fc受容体の発現分布が必ずしもヒトと一致せずヒトでの効果が想定できないことに起因している。
【0004】
この問題を解決するためには、ヒトの免疫系を再現した疾患モデル動物の作製が欠かせない。ヒトCD16を発現するモデル動物としては、約4.6 kbのヒトCD16プロモーターを含むヒトCD16発現ベクターを用いてNK細胞やマクロファージ上にヒトCD16を発現させたトランスジェニックマウスの作製が報告されている(非特許文献23参照)。しかしながら、本トランスジェニックマウスを用いたヒト抗体のエフェクター活性評価あるいは薬理評価の報告はなく、ヒト抗体の十分な機能評価には成功していない。その後、本トランスジェニックマウスを用いて、C57BL/6JNIcrマウスやC.B17/Icr-scidマウスとの戻し交配(非特許文献24参照)、あるいは、CD16ノックアウトマウスやヒトCD20発現マウスなどの交配(特許文献1参照)などが行われている。
【0005】
【特許文献1】WO2004/060052
【非特許文献1】Proc. Natl. Acad. Sci. U.S.A., 81, 6851 (1984)
【非特許文献2】Nature,312, 643 (1984)
【非特許文献3】Nature, 321, 522 (1986)
【非特許文献4】Nature, 332, 323 (1988)
【非特許文献5】Science, 239, 1534 (1988)
【非特許文献6】Nat. Biotechnol., 14, 845 (1996)
【非特許文献7】Nat. Genet., 15, 146 (1997)
【非特許文献8】Nat. Genet.,16, 133 (1997)
【非特許文献9】Proc. Natl. Acad. Sci. U.S.A.. 97, 722 (2000)
【非特許文献10】J. Mol. Biol., 222, 581 (1991)
【非特許文献11】Biotechnology (N Y), 11, 1145 (1993)
【非特許文献12】Blood 86, 4430 (1995)
【非特許文献13】Semin. Oncol., 30, 1 (2003)
【非特許文献14】Expert Rev. Anticancer Ther., 3, 767 (2003)
【非特許文献15】Breast J., 9, 452 (2003)
【非特許文献16】Trends Mol. Med., 8, S19 (2002)
【非特許文献17】Blood 99, 754 (2002)
【非特許文献18】Cancer Res., 64, 4664 (2004)
【非特許文献19】Arthritis Rheum., 48, 455 (2003)
【非特許文献20】J. Clin. Oncol., 21, 3940 (2003)
【非特許文献21】Clin. Cancer Res., 10, 5650 (2004)
【非特許文献22】Nat. Biotechnol., 23, 1073 (2005)
【非特許文献23】J. Exp. Med., 183, 1259 (1996)
【非特許文献24】Cancer Immunol. Immunother., 48, 443 (1999)
【発明の開示】
【発明が解決しようとする課題】
【0006】
ヒトでは生体防御のために高度に分化した免疫系が構築されており、この免疫系を利用した抗体医薬は、本来、高い薬理効果が期待できる。どのようなヒト抗体が高い薬理効果を発揮するのかをスクリーニングし選択するには、適切なヒト疾患モデルを用いた評価が欠かせない。このようなヒト疾患モデルが構築できる、種差の壁を越えヒトの免疫系を再現しうる非ヒト動物の開発が、有効な抗体医薬開発の観点から望まれる。本発明は、ヒトCD16とそのヒトアクセサリー分子とを共発現させるようにゲノムが改変された、非ヒト動物またはその子孫及びその利用方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明は、以下の(1)〜(16)に関する。
(1)ヒトCD16とそのヒトアクセサリー分子とを共発現させるようにゲノムが改変された、非ヒト動物またはその子孫。
(2)ヒトCD16のヒトアクセサリー分子が、以下の(a)〜(c) からなる群から選ばれる分子である、(1)に記載の非ヒト動物またはその子孫。
(a) ヒトCD3ζ鎖;
(b) ヒトFcε受容体Iγ鎖;
(c) ヒトCD3ζ鎖とヒトFcε受容体Iγ鎖。
(3)ヒトCD16またはそのヒトアクセサリー分子の発現が、ヒトCD16プロモーター領域で制御されるようにゲノムが改変された、(1)または(2)に記載の非ヒト動物またはその子孫。
(4)ヒトCD16が、以下の (a) 〜(d)からなる群から選ばれるDNAがコードする蛋白質である、(1)〜(3)のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号1で表される塩基配列からなるDNA;
(b) 配列番号2で表される塩基配列からなるDNA;
(c) 配列番号1で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒト抗体のFc領域と結合する活性を有する蛋白質をコードするDNA;
(d) 配列番号2で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒト抗体のFc領域と結合する活性を有する蛋白質をコードするDNA。
(5)ヒトCD16が、以下の(a) 〜(f)からなる群から選ばれる蛋白質である、(1)〜(3)のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号3で表されるアミノ酸配列からなる蛋白質;
(b) 配列番号4で表されるアミノ酸配列からなる蛋白質;
(c) 配列番号3で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(d) 配列番号4で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(e) 配列番号3で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(f) 配列番号4で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質。
(6)ヒトCD3ζ鎖が、以下の (a)または(b)であるDNAがコードする蛋白質である、(2)〜(5)のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号5で表される塩基配列からなるDNA;
(b) 配列番号5で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質をコードするDNA。
(7)ヒトCD3ζ鎖が、以下の(a) 〜(c)からなる群から選ばれる蛋白質である、(2)〜(5)のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号6で表されるアミノ酸配列からなる蛋白質;
(b) 配列番号6で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質;
(c) 配列番号6で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質。
(8)ヒトFcε受容体Iγ鎖が、以下の (a)または(b)から選ばれるDNAがコードする蛋白質である、(2)〜(7)のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号7で表される塩基配列からなるDNA;
(b) 配列番号7で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質をコードするDNA。
(9)ヒトFcε受容体Iγ鎖が、以下の(a) 〜(c)からなる群から選ばれる蛋白質である、(2)〜(7)のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号8で表されるアミノ酸配列からなる蛋白質;
(b) 配列番号8で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質;
(c) 配列番号8で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質。
(10)ヒトCD16プロモーター領域が、以下の(a)または(b)から選ばれるDNAである、(3)〜(9)のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号9で表される塩基配列からなるDNA;
(b) 配列番号9で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつナチュラルキラー細胞、マクロファージ、単球または樹状細胞内でヒトCD16またはそのヒトアクセサリー分子を発現させる活性を有するDNA;
(11)非ヒト動物が、ウシ、ヒツジ、ヤギ、ブタ、ウマ、マウス、ラット、ニワトリ、サル及びウサギからなら群から選ばれる動物である、(1)〜(10)のいずれか1項に記載の非ヒト動物またはその子孫。
(12)以下の(1)〜(3)からなる工程を含むことを特徴とする、抗体の抗体依存性細胞傷害活性を測定する方法。
(1) (1)〜(11)のいずれか1項に記載の非ヒト動物またはその子孫より、エフェクター細胞を単離する工程;
(2) 単離したエフェクター細胞と、抗体及び該抗体の標的となる抗原を発現している細胞とを混合し、反応液中でエフェクター細胞と、抗体及び該抗体の標的となる抗原を発現している細胞とを接触させる工程;
(3)反応液中の該標的となる抗原を発現している細胞の生存率を定量する工程。
(13)以下の(1)および(2)からなる工程を含むことを特徴とする、抗体の抗体依存性細胞傷害活性に基づく薬理活性を評価する方法。
(1) (1)〜(11)のいずれか1項に記載の非ヒト動物またはその子孫に、被検物質である抗体を投与する工程;
(2) 該非ヒト動物またはその子孫において被検物質である抗体の抗体依存性細胞傷害活性に基づく薬理活性を評価する工程。
(14)以下の(1)〜(3)からなる工程を含むことを特徴とする、抗体のスクリーニング方法。
(1)(1)〜(11)のいずれか1項に記載の非ヒト動物またはその子孫に被検物質である抗体を投与する工程;
(2) 該非ヒト動物またはその子孫において被検物質である抗体の抗体依存性細胞傷害活性に基づく薬理活性を評価する工程;
(3) 該薬理活性を有する抗体を選択する工程。
(15)抗体が、以下の(a)〜(e)からなる群から選ばれる蛋白質である、(12)〜(14)のいずれか1項に記載の方法。
(a) ヒト抗体;
(b) ヒト化抗体;
(c) ヒト型キメラ抗体
(d) (a)、(b)または(c)のFc領域を含む抗体断片;
(e) (a)、(b)または(c)のFc領域を有する融合蛋白質。
(16)抗体のクラスがIgGである、(15)に記載の方法。
【発明の効果】
【0008】
本発明により、ヒトCD16とそのヒトアクセサリー分子とを共発現させるようにゲノムが改変された、非ヒト動物またはその子孫及びその利用方法が提供される。本発明の非ヒト動物及びその子孫は、ヒト抗体医薬の生物活性及び薬理評価に有用である。
【発明を実施するための最良の形態】
【0009】
本発明の非ヒト動物またはその子孫とは、ヒトCD16とそのヒトアクセサリー分子とを共発現させるように、ゲノムが改変された非ヒト動物またはその子孫であれば、いかなる非ヒト動物またはその子孫も包含される。
本発明において、ヒトCD16とは、ヒト抗体のFc領域と結合する活性を有する蛋白質を意味する。ヒトCD16には遺伝的多型が存在することが知られており、具体的には、ヒトCD16シグナル配列のN末端メチオニンから176番目のアミノ酸残基がフェニルアラニン(以下、ヒトCD16 Pheと称す)である蛋白質と、バリン(以下、ヒトCD16 Valと称す)である蛋白質の存在が知られている。したがって、ヒトにおいては、ヒトCD16 Phe/ヒトCD16 PheあるいはヒトCD16 Val/ヒトCD16 Valのホモ型、ヒトCD16 Phe/ヒトCD16 Valのヘテロ型の3種類の表現型が存在する。本発明において、ヒトCD16とは、いずれの多型も包含する。
【0010】
本発明におけるヒトCD16としては、具体的には、下記(a)、(b)、(c)または(d)のDNAがコードする蛋白質、または下記(e)、(f)、(g)、(h)、(i)または(j)の蛋白質があげられる。
(a) 配列番号1で表される塩基配列からなるDNA;
(b) 配列番号2で表される塩基配列からなるDNA;
(c) 配列番号1で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒト抗体のFc領域と結合する活性を有する蛋白質をコードするDNA;
(d) 配列番号2で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒト抗体のFc領域と結合する活性を有する蛋白質をコードするDNA;
または、
(e) 配列番号3で表されるアミノ酸配列からなる蛋白質;
(f) 配列番号4で表されるアミノ酸配列からなる蛋白質;
(g) 配列番号3で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(h) 配列番号4で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(i) 配列番号3で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(j) 配列番号4で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質。
【0011】
本発明において、ストリンジェントな条件下でハイブリダイズするDNAとは、例えば配列番号1または2で表される塩基配列を有するDNAなどのDNAまたはその一部の断片をプローブとして、コロニー・ハイブリダイゼーション法、プラーク・ハイブリダイゼーション法あるいはサザンブロットハイブリダイゼーション法等を用いることにより得られるDNAを意味し、具体的には、コロニーあるいはプラーク由来のDNAを固定化したフィルターを用いて、0.7〜1.0Mの塩化ナトリウム存在下、65℃でハイブリダイゼーションを行った後、0.1〜2倍濃度のSSC溶液(1倍濃度のSSC溶液の組成は、150mM塩化ナトリウム、15mMクエン酸ナトリウムよりなる)を用い、65℃条件下でフィルターを洗浄することにより同定できるDNAをあげることができる。ハイブリダイゼーションは、Molecular Cloning, A Laboratory Manual, Second Edition, Cold Spring Harbor Laboratory Press, 1989(以下、モレキュラー・クローニング第2版と略す)、Current Protocols in Molecular Biology, John Wiley & Sons, 1987-1997(以下、カレント・プロトコールズ・イン・モレキュラー・バイオロジーと略す)、DNA Cloning 1: Core Techniques, A Practical Approach, Second Edition, Oxford University (1995)等に記載されている方法に準じて行うことができる。ハイブリダイズ可能なDNAとして具体的には、配列番号1または2で表される塩基配列と少なくとも60%以上の相同性を有するDNA、好ましくは70%以上、より好ましくは80%以上、さらに好ましくは90%以上、特に好ましくは95%以上、最も好ましくは98%以上の相同性を有するDNAをあげることができる。
【0012】
本発明において、配列番号3または4で表されるアミノ酸配列において1以上のアミノ酸が欠出、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質とは、モレキュラー・クローニング第2版、カレント・プロトコールズ・イン・モレキュラー・バイオロジー、Nucleic Acids Research, 10, 6487 (1982)、Proc. Natl. Acad. Sci., USA,79, 6409(1982)、Gene, 34, 315 (1985)、Nucleic Acids Research, 13, 4431 (1985)、Proc. Natl. Acad. Sci USA,82, 488 (1985)等に記載の部位特異的変異導入法を用いて、例えば、配列番号3または4で表されるアミノ酸配列を有する蛋白質をコードするDNAに部位特異的変異を導入することにより取得することができる蛋白質を意味する。欠出、置換、挿入および/または付加されるアミノ酸の数は1個以上でありその数は特に限定されないが、上記の部位特異的変異導入法等の周知の技術により、欠失、置換もしくは付加できる程度の数であり、例えば、1〜数十個、好ましくは1〜20個、より好ましくは1〜10個、さらに好ましくは1〜5個である。
【0013】
また、本発明において、配列番号3または4で表されるアミノ酸配列と80%以上の相同性を有し、かつヒト抗体のFc領域と結合する活性を有する蛋白質とは、BLAST〔J. Mol. Biol., 215, 403 (1990)〕やFASTA〔Methods in Enzymology, 183, 63 (1990)〕等の解析ソフトを用いて計算したときに、配列番号3または4に記載のアミノ酸配列を有する蛋白質と少なくとも80%以上、好ましくは85%以上、より好ましくは90%以上、さらに好ましくは95%以上、特に好ましくは97%以上、最も好ましくは99%以上である蛋白質である。
【0014】
本発明において、ヒトCD16のアクセサリー分子とは、ヒト抗体のFc領域が結合したヒトCD16からのシグナルを細胞内に伝達することができる蛋白質を意味する。ヒトCD16からのシグナルを細胞内に伝達することができる蛋白質であればいずれの分子でも包含されるが、具体的には、ヒトCD3ζ鎖、ヒトFcε受容体Iγ鎖またはそれらの組合わせなどが挙げられる。
【0015】
本発明におけるヒトCD3ζ鎖としては、具体的には、下記(a)または(b)のDNAがコードする蛋白質、または下記(c)、(d)または(e)の蛋白質があげられる。
(a) 配列番号5で表される塩基配列からなるDNA;
(b) 配列番号5で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質をコードするDNA;
または、
(c) 配列番号6で表されるアミノ酸配列からなる蛋白質;
(d) 配列番号6で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質;
(e) 配列番号6で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質。
【0016】
本発明におけるヒトFcε受容体Iγ鎖としては、具体的には、下記(a)または(b)のDNAがコードする蛋白質、または下記(c)、(d)または(e)の蛋白質があげられる。
(a) 配列番号7で表される塩基配列からなるDNA;
(b) 配列番号7で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質をコードするDNA;
または、
(c) 配列番号8で表されるアミノ酸配列からなる蛋白質;
(d) 配列番号8で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質;
(e) 配列番号8で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質。
【0017】
本発明において、ストリンジェントな条件下でハイブリダイズするDNAとは、例えば配列番号5または7で表される塩基配列を有するDNAなどのDNAまたはその一部の断片をプローブとして、コロニー・ハイブリダイゼーション法、プラーク・ハイブリダイゼーション法あるいはサザンブロットハイブリダイゼーション法等を用いることにより得られるDNAを意味し、具体的には、コロニーあるいはプラーク由来のDNAを固定化したフィルターを用いて、0.7〜1.0Mの塩化ナトリウム存在下、65℃でハイブリダイゼーションを行った後、0.1〜2倍濃度のSSC溶液(1倍濃度のSSC溶液の組成は、150mM塩化ナトリウム、15mMクエン酸ナトリウムよりなる)を用い、65℃条件下でフィルターを洗浄することにより同定できるDNAをあげることができる。ハイブリダイゼーションは、モレキュラー・クローニング第2版、カレント・プロトコールズ・イン・モレキュラー・バイオロジー、DNA Cloning 1: Core Techniques, A Practical Approach, Second Edition, Oxford University (1995)等に記載されている方法に準じて行うことができる。ハイブリダイズ可能なDNAとして具体的には、配列番号5または7で表される塩基配列と少なくとも60%以上の相同性を有するDNA、好ましくは70%以上、より好ましくは80%以上、さらに好ましくは90%以上、特に好ましくは95%以上、最も好ましくは98%以上の相同性を有するDNAをあげることができる。
【0018】
本発明において、配列番号6または8で表されるアミノ酸配列において1以上のアミノ酸が欠出、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質とは、モレキュラー・クローニング第2版、カレント・プロトコールズ・イン・モレキュラー・バイオロジー、Nucleic Acids Research, 10, 6487 (1982)、Proc. Natl. Acad. Sci., USA,79, 6409(1982)、Gene, 34, 315 (1985)、Nucleic Acids Research, 13, 4431 (1985)、Proc. Natl. Acad. Sci USA,82, 488 (1985)等に記載の部位特異的変異導入法を用いて、例えば、配列番号6または8で表されるアミノ酸配列を有する蛋白質をコードするDNAに部位特異的変異を導入することにより取得することができる蛋白質を意味する。欠出、置換、挿入および/または付加されるアミノ酸の数は1個以上でありその数は特に限定されないが、上記の部位特異的変異導入法等の周知の技術により、欠失、置換もしくは付加できる程度の数であり、例えば、1〜数十個、好ましくは1〜20個、より好ましくは1〜10個、さらに好ましくは1〜5個である。
【0019】
また、本発明において、配列番号6または8で表されるアミノ酸配列と80%以上の相同性を有し、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質とは、BLAST〔J. Mol. Biol., 215, 403 (1990)〕やFASTA〔Methods in Enzymology, 183, 63 (1990)〕等の解析ソフトを用いて計算したときに、配列番号6または8に記載のアミノ酸配列を有する蛋白質と少なくとも80%以上、好ましくは85%以上、より好ましくは90%以上、さらに好ましくは95%以上、特に好ましくは97%以上、最も好ましくは99%以上である蛋白質である。
【0020】
本発明の非ヒト動物またはその子孫としては、ヒトCD16の発現がヒトCD16プロモーター領域で制御されるようにゲノムが改変された、非ヒト動物またはその子孫があげられる。
ここで、ヒトCD16の発現がヒトCD16プロモーター領域で制御されるようにゲノムが改変されたとは、ヒトCD16をコードする遺伝子が、ヒト染色体上のヒトCD16遺伝子の発現を制御するゲノム領域の制御下に位置するように改変することを意味する。ヒトCD16遺伝子の発現を制御するゲノム領域の制御下に位置するように改変するとは、具体的には、ヒト染色体上のヒトCD16遺伝子の発現を制御するゲノム領域の下流に、ヒトCD16をコードする遺伝子が位置するように改変する例が挙げられる。このような非ヒト動物またはその子孫を取得する方法としては、目的とするゲノムの改変を行うことができれば、いずれの手法でも用いることができる。具体的には、目的とした遺伝子改変を施すDNAを受精卵や胚性幹細胞に導入し個体を作製する方法、目的とした遺伝子改変を施した細胞の核を用いたクローン個体の作製の手法などがあげられる。
【0021】
本発明において、ヒトCD16プロモーター領域とは、ヒト染色体上においてヒトCD16遺伝子の組織特異的な発現制御に関与している領域であればいかなる領域も包含される。ヒトCD16は、ナチュラルキラー細胞、マクロファージ、単球及び樹状細胞での発現が観察されており[Nat. Rev. Immunol., 4, 89 (2004); Nat. Rev. Immunol., 2, 580 (2002)]、ヒトCD16プロモーター領域には少なくともこれらの細胞内で制御下にある遺伝子を発現させる活性を有している。
【0022】
本発明におけるヒトCD16プロモーター領域としては、具体的には、下記(a)、(b)または(c)のDNAがあげられる。
(a) 配列番号9で表される塩基配列からなるDNA;
(b) 配列番号9で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒトナチュラルキラー細胞で制御下にある遺伝子を発現させることが可能な活性を有するDNA;
(c) ナチュラルキラー細胞、マクロファージ、単球及び樹状細胞特異的に制御下にある遺伝子を発現させることが可能な活性を有するDNA。
【0023】
本発明において、抗体とは、抗体のFc領域を含む分子であればいかなる分子も包含されるが、ヒト由来の抗体が好ましい。具体的には、ヒト抗体、ヒト化抗体、ヒト型キメラ抗体、それらの抗体のFc領域を含む抗体断片、それらの抗体のFc領域と他の蛋白質とを融合させた該Fc領域を有する融合蛋白質などがあげられる。
ヒト抗体としては、抗原に感作したヒト脾臓細胞より作製したハイブリドーマ細胞が分泌する抗体のほかにも、遺伝子組換え技術により作製されたヒト抗体、すなわち、抗体遺伝子を挿入した抗体発現ベクターを、宿主細胞へ導入することにより取得されたヒト抗体などがあげられる。
【0024】
ヒト化抗体
ヒト型キメラ抗体は、ヒト以外の動物の抗体H鎖V領域(以下、「HV」または「VH」とも称す)および抗体L鎖V領域(以下、「LV」または「VL」とも称す)とヒト抗体のH鎖C領域(以下、「CH」とも称す)およびヒト抗体のL鎖C領域(以下、「CL」とも称す)とからなる抗体を意味する。ヒト以外の動物としては、マウス、ラット、ハムスター、ラビット等、ハイブリドーマを作製することが可能であれば、いかなるものも用いることができる。
【0025】
ヒト型キメラ抗体は、ヒト以外の哺乳動物に抗原を免疫して得られたB細胞と、マウス、ラット等に由来するミエローマ細胞とを細胞融合させて得られる、所望の抗原特異性を有したモノクローナル抗体を生産するハイブリドーマより、VHおよびVLをコードするcDNAを取得し、ヒト抗体CHおよびヒト抗体CLをコードする遺伝子を有する宿主細胞用発現ベクターにそれぞれ挿入してヒト型キメラ抗体発現ベクターを構築し、宿主細胞へ導入することにより発現させ、製造することができる。
【0026】
ヒト型キメラ抗体のCHとしては、ヒトイムノグロブリン(以下、「hIg」と表記する)に属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。また、ヒト型キメラ抗体のCLとしては、hIgに属すればいかなるものでもよく、κクラスあるいはλクラスのものを用いることができる。
【0027】
ヒト化抗体はヒト型CDR移植抗体ともいい、ヒト以外の動物の抗体のVHおよびVLのCDRのアミノ酸配列をヒト抗体のVHおよびVLの適切な位置に移植した抗体をいう。
ヒト化抗体は、ヒト以外の動物の抗体のVHおよびVLのCDR配列を任意のヒト抗体のVHおよびVLのCDR配列に移植したV領域をコードするcDNAを構築し、ヒト抗体のCHおよびヒト抗体のCLをコードする遺伝子を有する宿主細胞用発現ベクターにそれぞれ挿入してヒト化抗体発現ベクターを構築し、該発現ベクターを宿主細胞へ導入することによりヒト化抗体を発現させ、製造することができる。
【0028】
ヒト化抗体のCHとしては、hIgに属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。また、ヒト化抗体のCLとしては、hIgに属すればいかなるものでもよく、κクラスあるいはλクラスのものを用いることができる。
ヒト抗体は、元来、ヒト体内に天然に存在する抗体をいうが、最近の遺伝子工学的、細胞工学的、発生工学的な技術の進歩により作製されたヒト抗体ファージライブラリーならびにヒト抗体トランスジェニック動物あるいはヒト抗体トランスジェニック植物から得られる抗体等も含まれる。
【0029】
ヒト体内に存在する抗体は、例えば、ヒト末梢血リンパ球を単離し、EBウイルス等を感染させ不死化、クローニングすることにより、該抗体を生産するリンパ球を培養でき、培養物中より該抗体を精製することができる。
ヒト抗体ファージライブラリーは、ヒトB細胞から調製した抗体遺伝子をファージ遺伝子に挿入することによりFab、一本鎖抗体等の抗体断片をファージ表面に発現させたライブラリーである。該ライブラリーより、抗原を固定化した基質に対する結合活性を指標として所望の抗原結合活性を有する抗体断片を発現しているファージを回収することができる。該抗体断片は、更に遺伝子工学的手法により、2本の完全なH鎖および2本の完全なL鎖からなるヒト抗体分子へも変換することができる。
【0030】
ヒト抗体トランスジェニック非ヒト動物は、ヒト抗体遺伝子が細胞内に組込まれた動物をいう。具体的には、胚性幹細胞へヒト抗体遺伝子を導入し、該胚性幹細胞を他の初期胚へ移植後、発生させることによりヒト抗体を産生するトランスジェニック非ヒト動物を作製することができる。また、動物の受精卵にヒト抗体遺伝子を導入し、該受精卵を発生させることにヒト抗体を産生するトランスジェニック非ヒト動物を作製することもできる。ヒト抗体を産生するトランスジェニック非ヒト動物からのヒト抗体の作製方法は、通常のヒト以外の哺乳動物で行われているハイブリドーマ作製方法によりヒト抗体ハイブリドーマを得、培養することで培養物中にヒト抗体を蓄積させることができる。
【0031】
トランスジェニック非ヒト動物は、ウシ、ヒツジ、ヤギ、ブタ、ウマ、マウス、ラット、ニワトリ、サル又はウサギ等があげられる。
また、本発明において、抗体は、腫瘍関連抗原を認識する抗体、アレルギーあるいは炎症に関連する抗原を認識する抗体、循環器疾患に関連する抗原を認識する抗体、自己免疫疾患に関連する抗原を認識する抗体、またはウイルスあるいは細菌感染に関連する抗原を認識する抗体であることが好ましく、抗体のクラスはIgGが好ましい。
【0032】
抗体の断片は、上記抗体のFc領域を含んだ断片いう。抗体の断片としては、上記抗体のFc領域を含んだ断片であればいかなるものでもよいが、具体的には、H鎖の単量体、H鎖の2量体などがあげられる。
Fc領域を有する融合蛋白質としては、抗体のFc領域を含んだ抗体あるいは抗体断片と、酵素、サイトカインなどの蛋白質とを融合させた物質であればいかなるものでもよい。
【0033】
以下、本発明の非ヒト動物及びその子孫の作出方法と利用方法について詳細に説明する。
本発明の、ヒトCD16とそのヒトアクセサリー分子とを共発現させるようにゲノムが改変された非ヒト動物またはその子孫は、1)目的とする遺伝子を動物個体に導入するトランスジェニック動物の作製方法、2)標的遺伝子と目的とする導入遺伝子とを入れ換えた遺伝子置換動物の作製方法、3)目的とした遺伝子改変を施した細胞の核を用いたクローン個体の作製方法を用いることにより、作製することができる。以下にそれぞれ具体的に説明する。
1.目的とする遺伝子を動物個体に導入するトランスジェニック動物の作製方法を用いた、本発明の非ヒト動物の作製
(1)ヒトCD16をコードするDNAの取得
ヒトCD16をコードするDNAとしては、ヒトCD16をコードするcDNAおよびゲノムDNAをあげることができる。ゲノムDNAはイントロンを含んでいてもよい。また、コード領域のコドンは、cDNAおよびゲノムDNAにおけるコドンに限られるものでなく、ヒトCD16のアミノ酸配列をコードするコドンであれば、いかなるコドンの組み合わせでもよい。ヒトCD16をコードするDNAの例としては、配列番号1または2に示す塩基配列からなるヒトCD16のcDNAをあげることができる。
【0034】
塩基配列データベースより、ヒトCD16cDNAの配列、またはヒトCD16遺伝子のゲノム配列を検索し、その塩基配列情報を取得する。塩基配列データベースから取得できるヒトCD16遺伝子のゲノム配列としては、例えば、GenBank登録番号NC_000001およびAL_590385で登録されている塩基配列、ヒトCD16cDNAの配列としては、例えば、GenBank登録番号NM_000569で登録されている塩基配列をあげることができる。
【0035】
得られたヒトCD16をコードするDNAの塩基配列情報に基づいて、ヒトCD16をコードする領域を含むcDNAの配列またはゲノム配列の領域を適当に選択し、この領域の5'端20〜40bpの配列を3'端に含むDNAおよび、この領域の3'端20〜40bpの配列と相補的な配列を3'端に含むDNAを作製する。これらのDNAをプライマーとして用い、ヒトcDNAを鋳型としたPCRにより、ヒトCD16をコードするcDNAを、ヒトゲノムDNAを鋳型としたPCRにより、ヒトCD16をコードするゲノムDNAを単離することができる。
【0036】
ヒトcDNAは、ヒトの組織または細胞から調製したRNAから作製できる。組織または細胞から全RNAを調製する方法としては、チオシアン酸グアニジン−トリフルオロ酢酸セシウム法(Methods in Enzymology,154, 3, 1987)、酸性チオシアン酸グアニジン・フェノール・クロロホルム法(Analytical Biochemistry, 162, 156, 1987; 実験医学, 9, 1937, 1991)などがあげられる。また、アブソリュートリーRNA RT-PCRミニプレップ・キット(Absolutely RNA RT-PCR Miniprep Kit、ストラタジーン社製)、RNイージー・ミニ・キット(RNeasy MIni Kit、キアゲン社製)等のキットを用いることによっても調製することができる。全RNAからポリ(A)+ RNAとしてmRNAを調製する方法としては、オリゴ(dT)固定化セルロースカラム法〔Molecular Cloning: A Laboratory Manual, 3rd Edition, Cold Spring Harbor Laboratory Press, 2001(以下、モレキュラー・クローニング第3版と略す)〕等があげられる。さらに、ファーストトラック2.0mRNA分離キット(FastTrack 2.0 mRNA Isolation Kit、インビトロジェン社製)、クィックプレップmRNA精製キット(QuickPrep mRNA Purification Kit、アマシャム・バイオサイエンス社製)等のキットを用いることによりヒト組織や細胞からmRNAを調製することができる。全RNAまたはmRNAからcDNAを合成する方法としてはモレキュラー・クローニング第3版、カレント・プロトコールズ・イン・モレキュラー・バイオロジー等に記載された方法、あるいは市販のキット、例えばRT-PCR用スーパースクリプト第一鎖合成システム(SUPERSCRIPT First-Strand Synthesis System for RT-PCR、インビトロジェン社製)、プロスター第一鎖RT-PCRキット(ProSTAR First Strand RT-PCR Kit、ストラタジーン社製)、PCR−セレクトcDNAサブトラクション・キット(PCR-Select cDNA Subtraction Kit、クロンテック社製)を用いる方法などがあげられる。また、クロンテック社等から市販されているヒトの各種組織のcDNAを用いることもできる。このようにして得られたヒトCD16をコードするDNAの例として、配列番号1または2に示す塩基配列を有するDNAをあげることができる。
【0037】
ヒトゲノムDNAは、ヒトの組織または細胞から、モレキュラー・クローニング第3版に記載の方法により調製することができる。または、イージーDNAキット(Easy-DNA Kit、インビトロジェン社製)、DNA抽出キット(DNA Extraction Kit、ストラタジーン社製)等のキットを用いることにより、調製できる。クロンテック社等から市販されているヒトのゲノムDNAを用いることもできる。
(2)ヒトCD3ζ鎖をコードするDNAの取得
ヒトCD3ζ鎖をコードするDNAとしては、ヒトCD3ζ鎖をコードするcDNAおよびゲノムDNAをあげることができる。ゲノムDNAはイントロンを含んでいてもよい。また、コード領域のコドンは、cDNAおよびゲノムDNAにおけるコドンに限られるものでなく、ヒトCD3ζ鎖のアミノ酸配列をコードするコドンであれば、いかなるコドンの組み合わせでもよい。ヒトCD3ζ鎖をコードするDNAの例としては、配列番号5に示す塩基配列からなるヒトCD3ζ鎖のcDNAをあげることができる。
【0038】
塩基配列データベースより、ヒトCD3ζ鎖cDNAの配列、またはヒトCD3ζ鎖遺伝子のゲノム配列を検索し、その塩基配列情報を取得する。塩基配列データベースから取得できるヒトCD3ζ鎖遺伝子のゲノム配列としては、例えば、GenBank登録番号NC_000001で登録されている塩基配列、ヒトCD3ζ鎖cDNAの配列としては、例えば、GenBank登録番号NM_000734で登録されている塩基配列をあげることができる。
【0039】
得られたヒトCD3ζ鎖をコードするDNAの塩基配列情報に基づいて、ヒトCD3ζ鎖をコードする領域を含むcDNAの配列またはゲノム配列の領域を適当に選択し、この領域の5'端20〜40bpの配列を3'端に含むDNAおよび、この領域の3'端20〜40bpの配列と相補的な配列を3'端に含むDNAを作製する。これらのDNAをプライマーとして用い、ヒトcDNAを鋳型としたPCRにより、ヒトCD3ζ鎖をコードするcDNAを、ヒトゲノムDNAを鋳型としたPCRにより、ヒトCD3ζ鎖をコードするゲノムDNAを単離することができる。
【0040】
ヒトcDNAおよびヒトゲノムDNAは、上述の1.の(1)に記載の方法にしたがって調製することができる。このようにして得られたヒトCD3ζ鎖をコードするDNAの例として、配列番号5に示す塩基配列を有するDNAをあげることができる。
(3)ヒトFcε受容体Iγ鎖をコードするDNAの取得
ヒトFcε受容体Iγ鎖をコードするDNAとしては、ヒトFcε受容体Iγ鎖をコードするcDNAおよびゲノムDNAをあげることができる。ゲノムDNAはイントロンを含んでいてもよい。また、コード領域のコドンは、cDNAおよびゲノムDNAにおけるコドンに限られるものでなく、ヒトFcε受容体Iγ鎖のアミノ酸配列をコードするコドンであれば、いかなるコドンの組み合わせでもよい。ヒトFcε受容体Iγ鎖をコードするDNAの例としては、配列番号7に示す塩基配列からなるヒトFcε受容体Iγ鎖のcDNAをあげることができる。
【0041】
塩基配列データベースより、ヒトFcε受容体Iγ鎖cDNAの配列、またはヒトFcε受容体Iγ鎖遺伝子のゲノム配列を検索し、その塩基配列情報を取得する。塩基配列データベースから取得できるヒトFcε受容体Iγ鎖遺伝子のゲノム配列としては、例えば、GenBank登録番号NC_000001で登録されている塩基配列、ヒトFcε受容体Iγ鎖cDNAの配列としては、例えば、GenBank登録番号NM_004106で登録されている塩基配列をあげることができる。
【0042】
得られたヒトFcε受容体Iγ鎖をコードするDNAの塩基配列情報に基づいて、ヒトFcε受容体Iγ鎖をコードする領域を含むcDNAの配列またはゲノム配列の領域を適当に選択し、この領域の5'端20〜40bpの配列を3'端に含むDNAおよび、この領域の3'端20〜40bpの配列と相補的な配列を3'端に含むDNAを作製する。これらのDNAをプライマーとして用い、ヒトcDNAを鋳型としたPCRにより、ヒトFcε受容体Iγ鎖をコードするcDNAを、ヒトゲノムDNAを鋳型としたPCRにより、ヒトFcε受容体Iγ鎖をコードするゲノムDNAを単離することができる。
【0043】
ヒトcDNAおよびヒトゲノムDNAは、上述の1.の(1)に記載の方法にしたがって調製することができる。このようにして得られたヒトFcε受容体Iγ鎖をコードするDNAの例として、配列番号7に示す塩基配列を有するDNAをあげることができる。
(4)ヒトCD16プロモーター領域の取得
ヒトCD16プロモーター領域としては、ヒト染色体上においてヒトCD16遺伝子の組織特異的な発現制御に関与しているゲノムDNAをあげることができる。具体的には、ヒトCD16遺伝子の組織特異的な発現制御を担う活性を有する、ヒトCD16遺伝子の開始コドンから上流10kb以内の領域のゲノムDNAをあげることができる。ヒトCD16プロモーター領域の例としては、配列番号9に示す塩基配列からなるヒトCD16プロモーター領域をあげることができる。
【0044】
塩基配列データベースより、ヒトCD16cDNAの配列、またはヒトCD16遺伝子のゲノム配列を検索し、その塩基配列情報を取得する。塩基配列データベースから取得できるヒトCD16遺伝子のゲノム配列としては、例えば、GenBank登録番号NC_000001およびAL_590385で登録されている塩基配列、ヒトCD16cDNAの配列としては、例えば、GenBank登録番号NM_000569で登録されている塩基配列をあげることができる。
【0045】
得られたヒトCD16をコードするDNAの塩基配列情報に基づいて、ヒトCD16をコードする領域を含むcDNAの配列またはゲノム配列の領域を適当に選択し、この領域の5'端20〜40bpの配列を3'端に含むDNAおよび、この領域の3'端20〜40bpの配列と相補的な配列を3'端に含むDNAを作製する。これらのDNAをプライマーとして用い、ヒトゲノムDNAを鋳型としたPCRにより、ヒトCD16遺伝子の開始コドンから上流10kb以内の領域のゲノムDNAを数kbずつ増幅し単離することができる。
【0046】
ヒトゲノムDNAは、上述の1.の(1)に記載の方法にしたがって調製することができる。また、ゲノムDNAライブラリースクリーニングシステム(Genome Systems社製)やUniversal GenomeWalkerTM Kits(CLONTECH社製)などを用いることにより、ヒトCD16プロモーター領域を単離することもできる。このようにして得られたヒトCD16プロモーター領域の例として、配列番号9に示す塩基配列を有するDNAをあげることができる。
(5)ヒトCD16とそのヒトアクセサリー分子をコードするDNAを含む導入遺伝子の作製
上述の1.の(1)で得られたヒトCD16をコードするDNAと、上述の1.の(2)及び1.の(3)で得られたヒトCD16のヒトアクセサリー分子をコードするDNAを非ヒト動物に導入する際には、適当なプロモーターの下流に該DNAを連結した発現ベクターを導入遺伝子として用いるのが好ましい。具体的には、ヒトCD16とそのヒトアクセサリー分子のそれぞれを適当なプロモーターの下流に連結した発現ベクターを導入遺伝子として用いても良いし、ヒトCD16とそのヒトアクセサリー分子を配列内リボソーム進入部位(internal ribosome entry site; IRES)で連結し一つのプロモーターの下流に連結した発現ベクターを導入遺伝子として用いても良い。
【0047】
発現ベクターに用いられるプロモーターとしては、ヒトCD16とそのヒトアクセサリー分子をコードするDNAを導入する非ヒト動物の細胞内で転写を開始できるプロモーターであればいずれも用いることができる。例えばウイルス(サイトメガロウイルス、モロニー白血病ウイルス、JCウイルス、乳癌ウイルス、シミアンウイルス、レトロウイルスなど)由来遺伝子のプロモーター、各種哺乳動物(ヒト、ウサギ、イヌ、ネコ、モルモット、ハムスター、ラット、マウスなど)および鳥類(ニワトリなど)由来遺伝子〔例えば、アルブミン、インスリンII、エリスロポエチン、エンドセリン、オステオカルシン、筋クレアチンキナーゼ、血小板由来成長因子β、ケラチンK1、K10およびK14、コラーゲンI型およびII型、心房ナトリウム利尿性因子、ドーパミンβ−水酸化酵素、内皮レセプターチロシンキナーゼ(Tie2)、ナトリウムカリウムアデノシン3リン酸化酵素(Na,K-ATPase)、ニューロフィラメント軽鎖、メタロチオネインIおよびIIA、メタロプロテイナーゼ1組織インヒビター(TIMP1)、MHCクラスI抗原(H-2L)、平滑筋αアクチン、ポリペプチド鎖延長因子1α(EF-1α)、βアクチン、αおよびβミオシン重鎖、ミオシン軽鎖1および2、ミエリン塩基性タンパク、血清アミロイドPコンポーネント、ミオグロビン、レニン、ホスホグリセリン酸キナーゼ(PGK)等の解糖系酵素、ヒートショック蛋白質などの遺伝子〕のプロモーター、上記遺伝子のエンハンサー配列やプロモーター配列を融合させたプロモーター〔例えば、シミアンウイルス40(SV40)の初期遺伝子のプロモーターとヒトT細胞白血病ウイルス1のロング・ターミナル・リピートの一部の配列からなるSRαプロモーター、サイトメガロウイルスの前初期(IE)遺伝子エンハンサーとニワトリβ−アクチンプロモーターからなるCAGプロモーターなど〕などがあげられるが、ヒトCD16遺伝子の組織特異的な発現制御と同じ転写制御活性を有するプロモーターを用いることが好ましく、特にヒトCD16遺伝子の自身のプロモーターを用いることが好ましい。その具体的な例としては、上述の1.の(4)で得られたヒトCD16プロモーター領域があげられる。ヒトCD16遺伝子組織特異的な発現を示すプロモーターを使用することにより、ヒトCD16とそのヒトアクセサリー分子をコードするDNAを導入したトランスジェニック非ヒト動物の特定の組織に発現させることができる。また、Cre-loxP系との組合せにより、導入した外来遺伝子の発現部位、発現時期あるいは発現量等を制御することもできる。発現ベクター中の構造遺伝子をコードするDNAを2つのloxP配列の間に挿入し、該発現ベクターを用いて(6)に後述する方法でトランスジェニック非ヒト動物を作製する。そして、該トランスジェニック非ヒト動物に、アデノウイルスベクターを利用してCreリコンビナーゼを発現させれば、Creリコンビナーゼ発現用のアデノウイルスを感染させた部位または時期に特異的にヒトCD16やそのヒトアクセサリー分子をコードするDNAが欠失し、発現させたい導入遺伝子の発現を抑制することができる。また、(6)に後述する方法でヒトCD16とそのヒトアクセサリー分子の発現ベクターに加えて、例えば組織特異的な発現を示すプロモーターを利用したCreリコンビナーゼ発現ベクターを導入してトランスジェニック非ヒト動物を作製することにより、Creリコンビナーゼが発現する組織でのみ、ヒトCD16やそのヒトアクセサリー分子の発現が抑制されたトランスジェニック非ヒト動物を作製することができる。
【0048】
発現ベクターは、ヒトCD16やそのヒトアクセサリー分子をコードするDNAの下流に、mRNAの3'末端のポリアデニル化に必要なポリアデニル化シグナルを有していることが好ましい。ポリアデニル化シグナルとしては、上記のウィルス由来、各種動物および鳥類由来の各遺伝子に含まれるポリアデニル化シグナル、例えば、SV40の後期遺伝子または初期遺伝子、ウサギβグロビン遺伝子、ウシ成長ホルモン遺伝子、ヒトCD16遺伝子等のポリアデニル化シグナルをあげることができる。
【0049】
その他、ヒトCD16やそのヒトアクセサリー分子をさらに高発現させるために、各遺伝子のスプライシングシグナル、エンハンサー領域、イントロンの一部をプロモーター領域の5'上流、プロモーター領域と翻訳領域間あるいは翻訳領域の3'下流に連結してもよい。以下、プロモーター、ヒトCD16やそのヒトアクセサリー分子をコードするDNAおよびポリアデニル化シグナルからなるDNA構築物をヒトCD16とそのヒトアクセサリー分子発現ユニットと呼ぶ。
【0050】
さらに発現ベクターには、大腸菌(Escherichia coli、以下E. coliと略す)を用いてベクターを調製するため、E. coliで複製可能な複製開始点およびE. coliの形質転換体の選択マーカーとなる内因性プロモーターを含む薬剤耐性遺伝子が必要である。薬剤耐性遺伝子としては、E. coli由来のアンピシリン耐性遺伝子(β−ラクタマーゼ遺伝子)、テトラサイクリン耐性遺伝子、カナマイシン耐性遺伝子等があげられる。E. coliでの複製可能な複製開始点としては、pBR322の複製開始点、colE1の複製開始点等があげられる。
【0051】
レトロウイルスを遺伝子の導入に用いる場合は、レトロウイルスベクターにヒトCD16とそのヒトアクセサリー分子発現ユニットを挿入した組換えレトロウイルスベクターを作製する。該組換えレトロウイルスベクターを、適切なパッケージング細胞に導入することにより、組換えレトロウイルスを生産させることができる。得られた組換えウイルスを受精卵等に感染させることにより、ウイルスベクターを導入することができる。パッケージング細胞への組換えレトロウイルスベクターの導入には、公知の遺伝子導入の手法(例えば、リン酸カルシウム法、電気パルス法、リポフェクション法、凝集法、マイクロインジェクション法、パーティクルガン法、DEAE−デキストラン法等)を用いることができる。
(6)受精卵等への発現ベクターの導入とトランスジェニック非ヒト動物の選択
本発明の非ヒト動物は、上述の1.の(5)で作製したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を対象となる非ヒト動物に導入することによって作製できる。具体的には、該遺伝子を対象となる非ヒト動物の受精卵、胚性幹細胞(以下、ES細胞と略す)、精子または未受精卵へ導入し、これらの細胞を用いて発生させた個体から、ヒトCD16とそのヒトアクセサリー分子をコードするDNAを含む遺伝子が胚芽細胞を含むすべての細胞の染色体上に組み込まれた個体を選択することにより、本発明の非ヒト動物を作出できる。作出したトランスジェニック非ヒト動物の胚芽細胞において該導入遺伝子が存在することは、作出動物の子孫がその胚芽細胞および体細胞の全てに該導入遺伝子を有することで確認することができる。個体の選択は、個体を構成する組織、例えば、血液組織、上皮組織、結合組織、軟骨組織、骨組織、筋組織、口腔内組織または骨格系組織の一部から調製したゲノムDNA上に該導入遺伝子が存在することをDNAレベルで確認することによって行われる。このようにして選択された個体は通常、相同染色体の片方に導入遺伝子を有するヘテロ接合体なので、ヘテロ接合体の個体同士を交配することにより、子孫の中から導入遺伝子を相同染色体の両方に持つホモ接合体動物を取得することができる。このホモ接合体の雌雄の動物を交配することにより、すべての子孫が該遺伝子を安定に保持するホモ接合体となるので、通常の飼育環境で、本発明の非ヒト動物を繁殖継代することができる。
(i)受精卵への遺伝子導入によるトランスジェニック非ヒト動物の作製
受精卵への遺伝子導入によるトランスジェニック非ヒト動物の作製は、Manipulating the Mouse Embryo A Laboratory Manual, Second Edition, Cold Spring Harbor Laboratory Press (1994)(以下、マニピュレーティング・マウス・エンブリオ第2版と略す)、Gene Targeting, A Practical Approach, IRL Press at Oxford University Press (1993)、バイオマニュアルシリーズ8 ジーンターゲッティング, ES細胞を用いた変異マウスの作製,羊土社 (1995)、発生工学実験マニュアル, トランスジェニック・マウスの作り方, 講談社 (1987)等に記載された方法により、作製することができる。
【0052】
ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を導入する受精卵は、同種の雄非ヒト動物と雌非ヒト動物を交配させることによって得られる。例えば、雄マウスと雌マウス、好ましくは近交系の雄マウスと雌マウスを交配することによりマウスの受精卵が得られ、雄ラットと雌ラット、好ましくはWistar系統の雄ラットとWistar系統の雌ラットを交配することによりラットの受精卵が得られる。受精卵は自然交配によっても得られるが、雌非ヒト動物の性周期を人工的に調節した後、雄非ヒト動物と交配させる方法が好ましい。雌非ヒト動物の性周期を人工的に調節する方法としては、例えば初めに卵胞刺激ホルモン、次いで黄体形成ホルモンを例えば腹腔注射などにより投与する方法が好ましいが、好ましいホルモンの投与量、投与間隔は非ヒト動物の種類によりそれぞれ異なる。非ヒト動物がマウスの場合は、雌マウスに卵胞刺激ホルモン投与後、約48時間後に黄体形成ホルモンを投与し、雄マウスと交配させることにより受精卵を得る方法が好ましく、卵胞刺激ホルモンの投与量は2〜20 IU/個体、好ましくは約5IU/個体、黄体形成ホルモンの投与量は0〜10 IU/個体、好ましくは約5IU/個体である。非ヒト動物がラットの場合は、雌ラットに卵胞刺激ホルモン投与後、約48時間後に黄体形成ホルモンを投与し、雄ラットと交配させることにより受精卵を得る方法が好ましく、卵胞刺激ホルモンの投与量は20〜50 IU/個体、好ましくは約30IU/個体、黄体形成ホルモンの投与量は0〜10 IU/個体、好ましくは約5IU/個体である。また、近交系のマウスやWister系統のラットを用いる場合は、約12時間明期条件(例えば7:00-19:00)で約1週間飼育した8週齢以上のものを用いるのが好ましい。
【0053】
得られた受精卵にマイクロインジェクション法(W. J. Gordonら; Proc. Natl. Acad. Sci. USA, 77, 7380, 1980、J. J. Mullinsら; Nature, 344, 541, 1990、R. E. Hammerら; Nature, 315, 680, 1985、V. G. Purselら; Immunol. Immunopathol., 17, 303, 1987)やレトロウイルスを用いた方法(マニピュレーティング・マウス・エンブリオ第2版)等によりヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子が導入された後、該受精卵を雌非ヒト動物に人工的に移植および着床させることによって、導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を組み込んだ染色体DNAを有する非ヒト動物が得られる。
【0054】
マイクロインジェクション法により遺伝子を導入する場合には、受精後約12〜約24時間の雄性前核が出現している受精卵を用いることが好ましい。マイクロインジェクションで導入されるヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子は、環状化状態、直線化状態いずれでも用いることができるが、ヒトCD16とそのヒトアクセサリー分子発現ユニットの構造遺伝子領域およびプロモーター等の発現調節領域を破壊しない形で直線化し導入することが好ましい。レトロウイルスを用いた方法により遺伝子を導入する場合には、桑実胚以前(一般には、8細胞期以前)の発生段階の受精卵を用いることが好ましい。
【0055】
雌非ヒト動物へ受精卵を移植する場合には、精管結紮雄非ヒト動物と交配させることにより、受精能を誘起された偽妊娠雌非ヒト動物に得られた受精卵を人工的に移植および着床させる方法が好ましい。偽妊娠雌非ヒト動物は自然交配によっても得られるが、黄体形成ホルモン放出ホルモン(以下、LHRHと略する)あるいはその類縁体を投与後、雄非ヒト動物と交配させることにより、受精能を誘起された偽妊娠雌非ヒト動物を得ることもできる。LHRHの類縁体としては、例えば[3,5-Dil-Tyr5]-LHRH、[Gln8]-LHRH、[D-Ala6]-LHRH、des-Gly10-[D-His(Bzl)6]-LHRH ethylamide等があげられる。LHRHあるいはその類縁体の投与量ならびにその投与後に雄非ヒト動物と交配させる時期は非ヒト動物の種類によりそれぞれ異なる。非ヒト動物が雌ラットの場合は、通常、LHRHあるいはその類縁体を投与後、約4日目に雄ラットと交配させることが好ましく、LHRHあるいはその類縁体の投与量は、通常、10〜60μg/個体、好ましくは約40μg/個体である。
(ii)ES細胞への遺伝子導入によるトランスジェニック非ヒト動物の作製
ES細胞への遺伝子導入によるトランスジェニック非ヒト動物の作製は、マニピュレーティング・マウス・エンブリオ第2版 、Gene Targeting, A Practical Approach, IRL Press at Oxford University Press (1993)、バイオマニュアルシリーズ8 ジーンターゲッティング, ES細胞を用いた変異マウスの作製,羊土社 (1995)、発生工学実験マニュアル, トランスジェニック・マウスの作り方, 講談社 (1987)等に記載された方法により、作製することができる。
【0056】
非ヒト動物のES細胞にヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を導入後、得られたES細胞を集合キメラ法または注入キメラ法を用いて、非ヒト動物の受精卵に取り込ませ、該受精卵を雌非ヒト動物に人工的に移植および着床させることによって導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を組み込んだ染色体DNAを有する細胞を部分的に有する非ヒトキメラ動物が得られる。
【0057】
非ヒト動物のES細胞は、マウスのES細胞(M. J. Evansら; Nature, 292, 154, 1981 ; G. R. Martin; Proc. Natl. Acad. Sci. USA, 78, 7634, 1981)、ラットのES細胞(P. M. Iannacconeら; Dev. Biol., 163, 288, 1994)ブタのES細胞(M. B. Wheeler; Reprod. Fertil. Dev., 6, 563, 1994)、サルのES細胞(J. A. Thomsonら; Proc. Natl. Acad. Sci. USA, 92, 7844, 1996)の各樹立方法、US5453357またはUS5670372に記載のES細胞の樹立方法に準じて、樹立することができる。マウスのES細胞として、AB2.2〔レキシコン・ジェネティックス(Lexicon Genetics)社製〕等を用いることができる。
【0058】
ES細胞への外来性遺伝子の導入には、公知の遺伝子導入の手法(例えば、リン酸カルシウム法、電気パルス法、リポフェクション法、凝集法、マイクロインジェクション法、パーティクルガン法、DEAE−デキストラン法、ウイルスベクター法等)を用いることができる。導入されるヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子は、環状化状態、直線化状態いずれでも用いることができるが、ヒトCD16とそのヒトアクセサリー分子発現ユニットの構造遺伝子をコードする領域およびプロモーター等の発現調節領域を破壊しない形で直線化し導入することが好ましい。
【0059】
受精卵は、上述の1.の(6)の(i)に記載した方法により取得できる。
ES細胞を集合キメラ法を用いて非ヒト動物の受精卵に取り込ませる場合には、一般に8細胞期以前の発生段階の受精卵を用いることが好ましい。ES細胞を注入キメラ法を用いて非ヒト動物の受精卵に取り込ませる場合には、一般に8細胞期から胚盤胞の発生段階の受精卵を用いることが好ましい。
【0060】
雌非ヒト動物へ受精卵を移植する場合には、精管結紮雄非ヒト動物と交配させることにより、受精能を誘起された偽妊娠雌非ヒト動物に得られた受精卵を人工的に移植および着床させる方法が好ましく、偽妊娠雌非ヒト動物は、上述の1.の(6)の(i)に記載した方法を用いることで得られる。
ES細胞の非ヒト動物と受精卵の非ヒト動物を、同じ種であるが体毛の色等の外見的な表現型が異なる系統にしておけば、非ヒトキメラ動物である個体、およびキメラ率(ES細胞に由来する細胞、すなわち導入した外来性遺伝子組み込んだ染色体DNAを有する細胞の割合)の高い個体を容易に選択することができる。
【0061】
ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を組み込んだ染色体DNAを有する細胞を部分的に有する非ヒトキメラ動物のうちキメラ率の高い個体(好ましくは、雄)と非ヒト動物の個体(好ましくは、雌)を交配する。生まれた仔の組織、例えば、血液組織、上皮組織、結合組織、軟骨組織、骨組織、筋組織、口腔内組織または骨格系組織の一部から調製したゲノムDNA上に導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子が存在するかどうかをサザンハイブリダイゼーション、PCR等により調べ、生まれた全ての仔のゲノムDNA上に導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子が存在する場合、親の非ヒトキメラ動物を生殖系列キメラとして選択する。生殖系列キメラと非ヒト動物の個体を交配することによって、全身の細胞の染色体上に導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を有する個体を仔として得ることができる。この個体は通常、相同染色体の片方にのみ、導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を有するヘテロ接合体である。さらにその個体の雄雌どうしを交配することにより相同染色体の双方に導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子が入ったホモ接合体のトランスジェニック非ヒト動物を得ることができる。
(iii)精子への遺伝子導入によるトランスジェニック非ヒト動物の作製
精子への遺伝子導入によるトランスジェニック非ヒト動物の作製は、Perryら(Science, 284, 1180, 1999) やWakayamaら(Nature Biotechnology, 16, 639, 1998)によって報告された方法を用いて行なうことができる。
【0062】
非ヒト動物の精子に上述の1.の(5)で作製したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を取り込ませた後、得られた精子を顕微受精の方法を用いて未受精卵に受精させ、発生を開始した卵を雌非ヒト動物に人工的に移植および着床させることによって、精子に取り込ませたヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を染色体上に組み込んだ非ヒト動物が得られる。
【0063】
精子に外来性遺伝子を取り込ませる場合には、細胞膜を破壊した精子を、約0.5〜約100ng/mL(好ましくは、5〜10ng/mL)の濃度の外来性遺伝子を含む培地(例えば、CZB培地やNIM培地など)中で数分間(好ましくは、1分間程度)インキュベーションする方法を用いることが好ましい。精子の細胞膜を破壊する方法としては、低濃度(例えば、0.05%)の界面活性剤(例えば、TritonX-100など)で処理する方法、10%の仔ウシ胎児血清(以下、FCSと略す)を含みEDTAを含まない培地(例えば、CZB培地など)中に懸濁した精子を凍結乾燥する方法 、該精子懸濁液を凍結融解する方法のいずれかが好ましい。このような処理をして細胞膜を破壊した精子は、もはや自発的に受精する能力を失っているが、マイクロマニュピレーター等を用いた顕微受精の方法を用いて受精させることで胚の発生を促す。顕微受精を行う場合には、外来性遺伝子を取り込ませた精子の頭部を減数第二分裂中期にある同種の雌非ヒト動物未受精卵の細胞質に注入する方法が好ましい。
(iv)未受精卵への遺伝子導入と体外受精によるトランスジェニック非ヒト動物の作製
未受精卵への遺伝子導入と体外受精によるトランスジェニック非ヒト動物の作製は、Chanら(Proc. Natl. Acad. Sci. USA, 95, 14028, 1998) によって報告された方法を用いて行なうことができる。
【0064】
非ヒト動物の未受精卵に、ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を組み込んだレトロウイルスを感染させることで該遺伝子を導入し、得られた卵を体外受精の方法を用いて発生を開始させ、発生を開始した受精卵を雌非ヒト動物に人工的に移植および着床させることによって、導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を染色体上に組み込んだ非ヒト動物が得られる。
【0065】
ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を組み込んだレトロウイルスは、マニピュレーティング・マウス・エンブリオ第2版等に記載の方法により調製できる。組換えレトロウイルスを未受精卵に感染させる場合には、透明帯を除いた未受精卵を用いることが好ましい。未受精卵の透明帯を除去する方法としては、マイクロマニュピレーター等を用いて物理的に膜を除く方法、化学物質(例えば、酸性タイロードなど)を用いて化学的に膜を溶解して除く方法のいずれかが好ましい。
2.標的遺伝子と目的とする導入遺伝子とを入れ換えた遺伝子置換動物の作製方法を用いた、本発明の非ヒト動物の作製
(1)非動物の標的遺伝子をコードするDNAの取得
上述の1.の(1)に記載の方法で、非ヒト動物の標的遺伝子をコードするDNAを取得する。後述するように、ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子領域の5'端および3'端に非ヒト動物の標的遺伝子の非コード領域を付加するために、5'端および3'端の近傍に適当な制限酵素サイトを合成DNAを用いて付加しておく。
(2)非ヒト動物の標的遺伝子を含むゲノムDNAの取得
非ヒト動物の標的遺伝子を含むゲノムDNAを取得する。該ゲノムDNAには、標的遺伝子をコードする領域およびイントロンの他に、標的遺伝子をコードする領域よりも5'側および3'側の領域も含む必要がある。その長さは、標的遺伝子をコードする領域よりも5'側および3'側へ、少なくとも500bp以上、好ましくは1kbp以上、より好ましくは3kbp以上、最も好ましくは5kbp以上が望ましい。
標的遺伝子を含むゲノムDNAを調製する方法としては、該非ヒト動物のゲノムDNAライブラリーを作製し、プラークハイブリダイゼーションあるいはコロニーハイブリダイゼーションにより、該遺伝子を含むゲノムDNAクローンを単離する方法があげられる。
【0066】
ゲノムDNAライブラリーは、該非ヒト動物の細胞や組織からゲノムDNAを抽出し、制限酵素で部分的に切断した後、適当なベクターに挿入し、ベクターに応じたE. coli等の適当な宿主に導入することにより作製することができる。ゲノムDNAの調製およびゲノムDNAライブラリーは、モレキュラー・クローニング第3版やカレント・プロトコールズ・イン・モレキュラー・バイオロジー等に記載された方法により作製することができる。ベクターとしては、λ EMBL3(ストラタジーン社製)、λ DASH II(ストラタジーン社製)、λ FIX II(ストラタジーン社製)等のλファージベクター、BeloBACII等の細菌人工染色体(BAC)ベクター、pAd10sacBII等のバクテリオファージP1ベクター、pCYPAC1等のP1人工染色体(PAC)ベクター、SuperCos I(ストラタジーン社製)等のコスミドベクター等があげられる。
【0067】
プラークハイブリダイゼーションあるいはコロニーハイブリダイゼーションに用いるプローブとしては、該非ヒト動物の標的となるゲノム遺伝子の部分断片、標的遺伝子のcDNAの部分断片を標識したものを用いることができる。これらの部分断片は、該非ヒト動物の標的遺伝子のゲノム配列またはcDNAの配列を塩基配列データベースから検索し、この塩基配列に基づいて作製したプライマーを用いたPCRにより取得することができる。
【0068】
プラークハイブリダイゼーション、コロニーハイブリダイゼーションおよびプローブの標識と調製は、モレキュラー・クローニング第3版やカレント・プロトコールズ・イン・モレキュラー・バイオロジー等に記載された方法により行なうことができる。
また、該非ヒト動物の標的遺伝子のゲノム配列が塩基配列データベースの検索により得られる場合は、増幅するゲノム配列の領域を適当に選択し、この領域の5'端20〜40bpの配列を3'端に含むDNAおよび、この領域の3'端20〜40bpの配列と相補的な配列を3'端に含むDNAを作製し、このDNAをプライマーとし、該非ヒト動物のゲノムDNAを鋳型としたPCRにより、該非ヒト動物の該標的遺伝子を含むゲノムDNAを単離することができる。
【0069】
また、ゲノムDNAライブラリースクリーニングシステム(Genome Systems社製)やユニバーサル・ゲノムウォーカー・キット(Universal GenomeWalkerTM Kit、クロンテック社製)などのキットを用いることによって、該非ヒト動物の標的遺伝子を含むゲノムDNAを単離することもできる。
上記の方法で得られる非ヒト動物の標的遺伝子を含むゲノムDNAとして、例えば、X染色体上のヒポキサンチンホスホリボシルトランスフェラーゼ(hprt)遺伝子を含むマウスゲノムDNAや、第6染色体上のRosa26遺伝子を含むマウスゲノムDNAをあげることができる。
(3)ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子の両末端に、非ヒト動物の標的遺伝子のゲノム配列の5'非コード領域、3'非コード領域を付加したDNAの作製
上述の1.の(5)で調製したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子の5'端に、上述の2.の(2)で調製した非ヒト動物の標的遺伝子ゲノム配列の開始コドンより5'側の適当な長さの非コード領域を付加し、3'端に該ゲノム配列の終止コドンより3'側の適当な長さの非コード領域を付加したDNA(以下、相同組換え用DNAとも称する)を、標的遺伝子の相同組換えに必要なターゲッティングベクターの構築に用いる。このDNAは、通常の遺伝子工学的な手法を用いれば様々な方法で作製することが可能である。その具体的な例としては、例えば、それぞれの遺伝子断片に、連結のための適当な制限酵素サイトを含む合成DNAをプライマーとして用いたPCRを行うことで調製取得することができる。付加する非コード領域の長さは、3'側、5'側とも500bp以上、より好ましくは1kbp以上、さらに好ましくは3kbp以上、最も好ましくは5kbp以上が望ましい。
(4)ターゲットベクターの作製とES細胞への導入
非ヒト動物標的遺伝子を相同組換えするためのターゲットベクターは、Gene Targeting, A Practical Approach, IRL Press at Oxford University Press (1993)、バイオマニュアルシリーズ8 ジーンターゲッティング, ES細胞を用いた変異マウスの作製(羊土社)(1995)等に記載の方法にしたがって作製することができる。ターゲットベクターは、リプレースメント型、インサーション型いずれでも用いることができる。ターゲットベクターは、上述の2.の(3)で作製した相同組換え用DNA、相同組換え体の選別に必要な遺伝子、および、E. coliを用いたベクターの調製に必要なE. coliで自律複製可能な複製開始点および薬剤耐性遺伝子を有する。相同組換え体の選別に必要な遺伝子としては、ポジティブ選択(選別に用いた遺伝子を含む組換え体を選別する方法)に用いる、hprt遺伝子、ネオマイシン耐性遺伝子等の薬剤耐性遺伝子、ネガティブ選択(選別に用いた遺伝子を含まない組換え体を選別する方法)に用いるジフテリアトキシン(DT)遺伝子等をあげることができる。ポジティブ選択に用いる遺伝子は相同組換え用DNAの非コード領域内に挿入し、ネガティブ選択に用いる遺伝子は相同組換え用DNAとは別の位置に挿入する。ポジティブ選択に用いる遺伝子は、該遺伝子の発現に必要なプロモーターに連結してもよいし、プロモーターに連結せず、相同組換えが起きた場合に、ES細胞の染色体上の標的遺伝子のプロモーターにより発現させるようにしてもよい。ポジティブ選択に用いる遺伝子の両端に2つのloxP配列を付加して挿入すると、相同組換え体の選別を行なった後に、相同組換え体にCreリコンビナーゼ発現ベクターを導入することにより、相同組換え体の染色体からポジティブ選択に用いる遺伝子を除去することができる。
【0070】
具体的なターゲットベクターとしては、GenOway社のプラスミドready-to-use Quick knock-inTM vectorに、相同組換え用DNAを挿入したベクター、例えば、ready-to-use Quick knock-inTM vectorにマウス標的受容体遺伝子を含むゲノムDNAの非コード領域とヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子からなる相同組換え用DNAの発現ユニットを挿入したプラスミドSAT1-HRをあげることができる。
【0071】
ES細胞へのターゲットベクターの導入には、公知の遺伝子導入の手法、例えば、リン酸カルシウム法、電気パルス法、リポフェクション法、凝集法、マイクロインジェクション法、パーティクルガン法、DEAE−デキストラン法、ウイルスベクター法等を用いることができる。導入されるターゲットベクターは、環状、直鎖状いずれの形状でも用いることができるが、相同組換え用DNAおよび相同組換え体の選別に必要な遺伝子を破壊しない形で直線化し導入することが好ましい。
【0072】
相同組換え体を効率的に選別する方法として、例えば、Gene Targeting, A Practical Approach, IRL Press at Oxford University Press (1993)、バイオマニュアルシリーズ8 ジーンターゲッティング, ES細胞を用いた変異マウスの作製(羊土社)(1995)等に記載のポジティブ選択、プロモーター選択、ネガティブ選択、ポリA選択などの方法を用いることができる。例えば、hprt遺伝子を含むターゲットベクターの場合は、hprt遺伝子を欠損したES細胞に導入後、ES細胞をアミノプテリン、ヒポキサンチンおよびチミジンを含む培地で培養し、アミノプテリン耐性の株を選別することにより、hprt遺伝子を含む相同組換え体を選別するポジティブ選択を行なうことができる。ネオマイシン耐性遺伝子を含むターゲットベクターの場合は、ベクターを導入したES細胞をG418を含む培地で培養し、G418耐性の株を選別することにより、ネオマイシン耐性遺伝子を含む相同組換え体を選別するポジティブ選択を行なうことができる。DT遺伝子を含むターゲットベクターの場合は、ベクターを導入したES細胞を培養し、生育してきた株を選別する(相同組換え以外のランダムに染色体に挿入された組換え体は、DT遺伝子が染色体に組み込まれて発現するため、DTの毒性により生育できない)ことにより、DT遺伝子を含まない相同組換え体を選別するネガティブ選択を行なうことができる。選別した細胞株の中から目的とする相同組換え体を選択する方法としては、ゲノムDNAに対するサザンハイブリダイゼーション法(モレキュラー・クローニング第3版)やPCR等があげられる。
(4)受精卵へのES細胞の取り込みと非ヒトトランスジェニック動物の作製
受精卵へのES細胞の取り込みと、受精卵の非ヒト動物への移植は、上述の1.の(6)の(ii)に記載の方法により行なうことができる。
【0073】
得られた仔から上述の1.の(6)の(ii)に記載の方法により、全身の細胞の相同染色体の片方に相同組換えによる遺伝子置換を有するヘテロ接合体を得ることができる。さらにその個体の雄雌どうしを交配することにより相同染色体の双方に相同組換えによる遺伝子置換を有するホモ接合体のトランスジェニック非ヒト動物を得ることができる。
このようにして得られたホモ接合体のトランスジェニック非ヒト動物は、導入した遺伝子がコードするヒトCD16とそのヒトアクセサリー分子を発現し、自己が持っていた標的遺伝子は発現しないトランスジェニック非ヒト動物である。このホモ接合体のトランスジェニック非ヒト動物は、上述の1.の方法で得られるトランスジェニック非ヒト動物よりも、4.および5.に後述する物質の薬理評価に用いる非ヒト動物として、(a)非ヒト動物が本来発現している標的遺伝子を発現していないので、薬理評価に用いる場合に該標的遺伝子を介した薬理効果を考慮する必要がない、(b)導入遺伝子の導入部位に存在する遺伝子の破壊あるいは導入遺伝子による導入部位周辺の遺伝子の活性化等の予測不可能な副次的な表現形の変化がおきる可能性がほとんどない、等の点で好ましい。
3.目的とした遺伝子改変を施した細胞の核を用いたクローン個体の作製方法を用いた、本発明の非ヒト動物の作製
本発明の非ヒト動物は、文献に記載されたクローンヒツジ(I. Wilmutら; Nature, 385, 810, 1997)、クローンウシ(J. B. Cibelliら; Science, 280, 1256, 1998、入谷明; 蛋白核酸酵素, 44, 892, 1999)、クローンヤギ(A. Baguisiら; Nature Biotechnology, 17, 456, 1999)、クローンマウス(T. Wakayamaら; Nature, 394, 369, 1998、T. Wakayamaら; Nature Genetics, 22, 127, 1999)の作製方法を用い、例えば以下のように作製することができる。
【0074】
上述の1.あるいは2.に記載した方法を用い、非ヒト動物の任意の細胞の染色体上に、ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を導入する。得られた細胞の核を初期化する。核を初期化するとは、核を再び発生を繰り返すことができるような状態に戻す操作を行なうことをいう。
初期化した細胞の核を除核した非ヒト動物の未受精卵に注入することによって発生を開始させる。
【0075】
発生を開始した卵を雌非ヒト動物に人工的に移植および着床させることによって導入した遺伝子を発現する、本発明の非ヒト動物を得ることができる。また、上述の2.に記載の方法で、遺伝子置換を行なった細胞の核を用いて得られたトランスジェニック非ヒト動物は、通常相同染色体の片方にヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子が導入されたヘテロ接合体であるため、得られたヘテロ接合体同士を交配することにより、遺伝子置換された自己の標的遺伝子は発現せず、ヒトCD16とそのヒトアクセサリー分子を発現するトランスジェニック非ヒト動物を得ることができる。このホモ接合体のトランスジェニック非ヒト動物も、上述の2.に記載の方法で得られるホモ接合体のトランスジェニック非ヒト動物と同様の点で、薬理評価に用いる非ヒト動物として、上述の1.に記載した方法で得られるトランスジェニック非ヒト動物よりも好ましい。
【0076】
細胞の核を初期化する方法は、非ヒト動物の種類によりそれぞれ異なる。非ヒト動物がヒツジ、ヤギ、ウシなどの場合は、外来性遺伝子を導入した細胞を5〜30%、好ましくは10%のFCSを含む培地(例えば、M2培地)から3〜10日、好ましくは5日間、0〜1%、好ましくは0.5%のFCSを含む貧栄養培地で培養することで細胞周期を休止期状態(G0期もしくはG1期)に誘導し初期化することが好ましい。非ヒト動物がマウスなどの場合は、同種の非ヒト動物の除核した未受精卵に、外来性遺伝子を導入した細胞の核を注入し数時間、好ましくは約1〜6時間培養することで初期化することが好ましい。
【0077】
初期化された核を除核された未受精卵中で発生を開始させる方法は、非ヒト動物の種類によりそれぞれ異なる。非ヒト動物がヒツジ、ヤギ、ウシなどの場合は、細胞周期を休止期状態(G0期もしくはG1期)に誘導し初期化した核を、電気融合法などによって同種の非ヒト動物の除核した未受精卵に移植することで卵子を活性化し発生を開始させることが好ましい。非ヒト動物がマウスなどの場合は、外来性遺伝子を導入した細胞の核を注入した未受精卵を、卵子活性化物質(例えば、ストロンチウムなど)で刺激し細胞分裂の阻害物質(例えば、サイトカラシンBなど)で処理し第二極体の放出を抑制することで発生を開始させることが好ましい。
4.本発明の非ヒト動物またはその子孫から得られる細胞を用いた物質の薬理評価方法
本発明の非ヒト動物またはその子孫から得られる、ナチュラルキラー細胞、マクロファージ、単球及び樹状細胞などFc受容体を発現する細胞(エフェクター細胞)を摘出し、抗体の細胞障害活性等のエフェクター活性を評価することができる。抗体の生物活性を測定する方法としては、モノクローナルアンチボディズ(Monoclonal Antibodies: Principles and Practice, Third Edition, Acad. Press, 1993)、あるいはアンチボディエンジニアリング(Antibody Engineering: A Practical Approach, IRL Press at Oxford University Press, 1996)等に記載の公知の方法を用いることができる。
その具体的な例としては、ヒト抗体の抗原との結合活性、あるいは、ヒト抗体の抗原陽性培養細胞株に対する結合活性は、ELISA法及び蛍光抗体法[キャンサー・イムノロジー・イムノセラピー(Cancer Immunol. Immunother.), 36, 373 (1993)]等により測定できる。抗原陽性培養細胞株に対する細胞障害活性は、CDC活性、ADCC活性等を測定することにより、評価することができる[キャンサー・イムノロジー・イムノセラピー(Cancer Immunol. Immunother.), 36, 373 (1993)]。
また、本発明の非ヒト動物またはその子孫から得られるES細胞を分化誘導することにより、さまざまな種類の細胞を得ることができる。分化の誘導方法としては、ES細胞を同種同系統の動物の皮下に移植することにより、様々な組織が混じりあった奇形腫瘍(テラトーマ)を誘導する方法(マニピュレーティング・マウス・エンブリオ第2版)、適当な条件でインビトロ培養することにより、内胚葉細胞、外胚葉細胞、中胚葉細胞、血液細胞、内皮細胞、軟骨細胞、骨格筋細胞、平滑筋細胞、心筋細胞、神経細胞、グリア細胞、上皮細胞、メラノサイト、ケラチノサイトに分化誘導する方法(Reprod. Fertil. Dev., 10, 31, 1998)をあげることができる。この分化後の細胞と、試験物質とを接触させ、試験物質非存在下での細胞と比較して、細胞内のCa2+濃度の上昇等の種々の細胞の応答や細胞の形態の変化等の薬理作用を調べることにより、試験物質の該細胞に対する薬理評価を行なうことができる。これらの方法によって、ヒトの生体から摘出しにくい細胞や少数しか存在しない細胞などに対する薬理評価を行うことができる。
5.本発明の非ヒト動物またはその子孫を用いた物質の薬理評価方法
本発明の非ヒト動物またはその子孫に試験物質、例えば、抗体を投与し、該試験物質を投与しない該動物と比較して、該動物の血圧、呼吸数、体重等の種々の身体的パラメーターの測定、外見や行動の観察、病理組織学的検討等の薬理作用を調べることにより、該試験物質の薬理評価を行なうことができる。特に、本発明の非ヒト動物またはその子孫は、ADCC活性等のエフェクター活性に基づく薬理評価を行なうのに好ましい。
【0078】
また、本発明の非ヒト動物またはその子孫に各種の疾患を誘発させた病態モデル動物を作製し、該病態モデル動物に試験物質、例えば、抗体を投与し、該病態モデル動物の血圧、呼吸数、体重等の種々の身体的パラメーターの測定、病態や、外見、行動の観察、病理組織学的検討等を、試験物質を投与しない該病態モデル動物と比較して行なうことにより、該試験物質の該疾患に対する有効性および副作用等の薬理評価を行なうことができる。また、この評価に基づき、該疾患の治療薬として好ましい物質を選択することができる。特に、抗体では、抗体のFc領域とのFc受容体との結合に種差が存在しており、通常の病態モデル動物を用いた薬理評価では最適なエフェクター活性等の評価は困難であったが、本発明の非ヒト動物またはその子孫を用いることにより、より適切なADCC活性等のエフェクター活性に基づく抗体の薬理評価を行なうことができる。
本発明の非ヒト動物またはその子孫に誘発させる疾患としては、心疾患(例えば、急性心不全、慢性心不全、心筋炎など)、呼吸器系疾患、関節疾患(例えば、関節リュウマチ、変形性関節症など)、腎疾患(例えば、腎不全、糸球体腎炎、IgA腎症など)、動脈硬化症、乾癬症、高脂血症、アレルギー疾患(例えば、喘息、アレルギー性鼻炎、アトピー性皮膚炎など)、骨疾患(例えば、骨粗鬆症、くる病、骨軟化症、低カルシュウム血症など)、血液疾患、脳血管性傷害、外傷性脳障害、感染症、痴呆症、癌、糖尿病、肝疾患、皮膚疾患、神経変性疾患および慢性炎症性疾患などがあげられる。病態モデル動物の作製は、「マニュアル疾患モデルマウス」〔Molecular Medicine, 31 臨時増刊号, 中山書店 (1994)〕、「図説・薬理学のための病態動物モデル」〔西村書店 (1984)〕、「関節炎モデル動物」〔医歯薬出版 (1985)〕、「神経・筋疾患モデル動物」〔医歯薬出版 (1982)〕、「活性酸素と病態 疾患モデルからベッドサイドへ」〔学会出版センター (1992)〕等に記載の方法を用いることができる。
【0079】
本薬理評価方法、4.及び8.に後述する薬理評価方法に供する試験物質としては、癌、炎症疾患、自己免疫疾患、アレルギーなどの免疫疾患、循環器疾患、またはウィルスあるいは細菌感染をはじめとする各種疾患の予防および治療に用いられるヒト抗体があげられる。
腫瘍関連抗原を認識する抗体、アレルギーあるいは炎症に関連する抗原を認識する抗体、循環器疾患に関連する抗原を認識する抗体、自己免疫疾患に関連する抗原を認識する抗体、またはウイルスあるいは細菌感染に関連する抗原を認識するヒト抗体の具体例を、以下に述べる。
【0080】
腫瘍関連抗原を認識する抗体としては、抗CA125抗体(Immunology Today, 21, 403-410, 2000)、抗17−1A抗体(Immunology Today, 21, 403-410, 2000)、抗インテグリンαvβ3抗体(Immunology Today, 21, 403-410, 2000)、抗CD33抗体(Immunology Today, 21, 403-410, 2000)、抗CD22抗体(Immunology Today, 21, 403-410, 2000)、抗HLA抗体(Immunology Today, 21, 403-410, 2000)、抗HLA−DR抗体(Immunology Today, 21, 403-410, 2000)、抗CD20抗体(Immunology Today, 21, 403-410, 2000)、抗CD19抗体(Immunology Today, 21, 403-410, 2000)、抗EGF受容体抗体(Immunology Today, 21, 403-410, 2000)、抗CD10抗体(American Journal of Clinical Pathology, 113, 374-382, 2000; Proc. Natl. Acad. Sci. USA, 79:4386-4391, 1982)、抗GD2抗体(Anticancer Res., 13, 331-336, 1993)、抗GD3抗体(Cancer Immunol. Immunother., 36, 260-266, 1993)、抗GM2抗体(Cancer Res., 54, 1511-1516, 1994)、抗HER2抗体(Proc. Natl. Acad. Sci. USA, 89, 4285-4289, 1992)、抗CD52抗体(Nature, 332, 323-327, 1988)、抗MAGE抗体(British J. Cancer, 83, 493-497, 2000)、抗HM1.24抗体(Molecular Immunol., 36, 387-395, 1999)、抗副甲状腺ホルモン関連蛋白(PTHrP)抗体(Cancer, 88, 2909-2911, 2000)、抗FGF8抗体(Proc. Natl. Acad. Sci. USA, 86, 9911-9915, 1989)抗塩基性繊維芽細胞増殖因子抗体、抗FGF8受容体抗体(J. Biol. Chem., 265, 16455-16463, 1990)、抗塩基性繊維芽細胞増殖因子受容体抗体、抗インスリン様増殖因子抗体(J. Neurosci. Res., 40, 647-659, 1995)、抗インスリン様増殖因子受容体抗体(J. Neurosci. Res., 40, 647-659, 1995)、抗PMSA抗体(J. Urology, 160, 2396-2401, 1998)、抗血管内皮細胞増殖因子抗体(Cancer Res., 57, 4593-4599, 1997)または抗血管内皮細胞増殖因子受容体抗体(Oncogene, 19, 2138-2146, 2000)などが挙げられる。
【0081】
アレルギーあるいは炎症に関連する抗原を認識する抗体としては、抗IgE抗体(Immunology Today, 21, 403-410, 2000)、抗CD23抗体(Immunology Today, 21, 403-410, 2000)、抗CD11a抗体(Immunology Today, 21, 403-410, 2000)、抗CRTH2抗体(J. Immunol., 162, 1278-1286, 1999)、抗CCR8抗体(WO99/25734)、抗CCR3抗体(US6207155)、抗インターロイキン6抗体(Immunol. Rev., 127, 5-24, 1992)、抗インターロイキン6受容体抗体(Molecular Immunol., 31, 371-381, 1994)、抗インターロイキン5抗体(Immunol. Rev., 127, 5-24, 1992)、抗インターロイキン5受容体抗体、抗インターロイキン4抗体(Cytokine, 3, 562-567, 1991)、抗インターロイキン4受容体抗体(J. Immunol. Meth., 217, 41-50, 1998)、抗腫瘍壊死因子抗体(Hybridoma, 13, 183-190, 1994)、抗腫瘍壊死因子受容体抗体(Molecular Pharmacol., 58, 237-245, 2000)、抗CCR4抗体(Nature, 400, 776-780, 1999)、抗ケモカイン抗体(J. Immunol. Meth., 174, 249-257, 1994)または抗ケモカイン受容体抗体(J. Exp. Med., 186, 1373-1381, 1997)などが挙げられる。
【0082】
循環器疾患に関連する抗原を認識する抗体としては、抗GpIIb/IIIa抗体(J. Immunol., 152, 2968-2976, 1994)、抗血小板由来増殖因子抗体(Science, 253, 1129-1132, 1991)、抗血小板由来増殖因子受容体抗体(J. Biol. Chem., 272, 17400-17404, 1997)または抗血液凝固因子抗体(Circulation, 101, 1158-1164, 2000)などが挙げられる。
【0083】
自己免疫疾患、例えば、乾癬、関節リウマチ、クローン病、潰瘍性大腸炎、全身性エリテマトーデス、多発性硬化症に関連する抗原を認識する抗体としては、抗自己DNA抗体(Immunol. Letters, 72, 61-68, 2000)、抗CD11a抗体(Immunology Today, 21, 403-410, 2000)、抗ICAM3抗体(Immunology Today, 21, 403-410, 2000)、抗CD80抗体(Immunology Today, 21, 403-410, 2000)、抗CD2抗体(Immunology Today, 21, 403-410, 2000)、抗CD3抗体(Immunology Today, 21, 403-410, 2000)、抗CD4抗体(Immunology Today, 21, 403-410, 2000)、抗インテグリンα4β7抗体(Immunology Today, 21, 403-410, 2000)、抗CD40L抗体(Immunology Today, 21, 403-410, 2000)、抗IL−2受容体抗体(Immunology Today, 21, 403-410, 2000)などが挙げられる。
【0084】
ウイルスあるいは細菌感染に関連する抗原を認識する抗体としては、抗gp120抗体(Structure, 8, 385-395, 2000)、抗CD4抗体(J. Rheumatology, 25, 2065-2076, 1998)、抗CCR4抗体または抗ベロ毒素抗体(J. Clin. Microbiol., 37, 396-399, 1999)などが挙げられる。
上記抗体は、ATCC(The American Type Culture Collection)、理化学研究所細胞開発銀行、工業技術院生命工業技術研究所等の公的な機関、あるいは大日本製薬株式会社、R&D SYSTEMS社、PharMingen社、コスモバイオ社、フナコシ株式会社等の民間試薬販売会社から入手することができる。
6.本発明の非ヒト動物またはその子孫のES細胞、卵、精子、核を用いた遺伝子改変動物の作製
本発明の非ヒト動物またはその子孫から得られるES細胞、卵、精子または核を用いて、上述の1.、2.および3.に記載の方法によりヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を有し、かつさらなる染色体上の遺伝子の改変を行った遺伝子改変非ヒト動物またはその子孫を得ることができる。特に、その遺伝子の機能を破壊することである病態が惹起されることが公知になっている遺伝子を上述の2.に記載した相同組換えの手法を利用して欠損させたノックアウト動物や、該遺伝子の機能を阻害するドミナントネガティブ体の遺伝子を上述の1.または3.に記載の方法で導入し発現させたトランスジェニック動物は、病態モデル動物として有用である。
7.本発明の非ヒト動物またはその子孫と同種他系統の動物との交配による遺伝子改変動物の作製と作製された該遺伝子改変動物の利用
本発明の非ヒト動物またはその子孫と同種他系統の動物(例えば、ヒト疾患モデル動物)とを交配させることにより、ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を染色体上に有し、かつある表現系(例えば、ヒト病態と類似の症状)を示す遺伝子改変哺乳動物を得ることができる。交配する動物が、ヒト疾患モデル動物であれば、ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を有し、かつヒト病態と類似の症状を表現系として示す病態モデル動物が得られる。交配の方法としては、自然交配以外に体外受精の方法があげられる。疾患モデル動物としては、先天性および後天性を疾患を問わずいかなる疾患モデル動物も用いることができる。例えば後天性の病態モデル動物は、「マニュアル疾患モデルマウス」〔Molecular Medicine, 31 臨時増刊号, 中山書店 (1994)〕、「図説・薬理学のための病態動物モデル」〔西村書店 (1984)〕、「関節炎モデル動物」〔医歯薬出版 (1985)〕、「神経・筋疾患モデル動物」〔医歯薬出版 (1982)〕、「活性酸素と病態 疾患モデルからベッドサイドへ」〔学会出版センター (1992)〕等に記載の方法により作製することができる。
8.遺伝子改変動物を用いた物質の薬理評価方法
上述の6.および7.に記載の方法で得られた遺伝子改変病態モデル動物に試験物質、例えば、抗体等を投与し、該病態モデル動物の血圧、呼吸数、体重等の種々の身体的パラメーターの測定、病態や、外見、行動の観察、病理組織学的検討等を、試験物質を投与しない該病態モデル動物と比較して行なうことにより、該試験物質の該疾患に対する有効性および副作用等の薬理評価を行なうことができる。また、この評価に基づき、該病態の治療薬として好ましい物質を選択することができる。特に、抗体では、抗体のFc領域とFc受容体との結合に種差が存在しており、通常の病態モデル動物を用いた薬理評価では最適なエフェクター活性等の評価は困難であったが、本発明の非ヒト動物またはその子孫を用いることにより、より適切なADCC活性等のエフェクター活性に基づく薬理評価を行なうことができる。
9.本発明の非ヒト動物またはその子孫を用いる薬理活性を有する物質のスクリーニング方法
本発明の非ヒト動物またはその子孫に試験物質、例えば、抗体を投与し、該試験物質を投与しない該動物と比較して、該動物の血圧、呼吸数、体重等の種々の身体的パラメーターの測定、外見や行動の観察、病理組織学的検討等の薬理作用を調べることにより、該試験物質の薬理評価を行ない、試験物質のなかから、薬理活性を有する試験物質を選択することにより、所望の薬理活性を有する試験物質をスクリーニングすることができる。特に、本発明の非ヒト動物またはその子孫は、抗体を投与し、ADCC活性等のエフェクター活性に基づく薬理活性を評価して、被検抗体から当該エフェクター活性に基づく薬理活性を有する抗体を選択することにより、in vivoにおいて薬理活性を有する抗体をスクリーニングすることができる。
以下に、本発明の実施例を示す。
【実施例1】
【0085】
ヒトCD16発現ベクターの作製
1.ヒトCD16をコードするcDNAの取得
ヒトCD16(別名;FcγRIIIa)のcDNAの塩基配列(GenBank Accession No. NM_000569)(配列番号1)をもとに、翻訳開始コドンを含む領域の配列に制限酵素認識配列を付加したフォワードプライマー(配列番号10)、および翻訳終止コドンを含む領域の配列に制限酵素認識配列を付加したリバースプライマー(配列番号11)を設計した。
【0086】
まず、DNAポリメラーゼKOD(東洋紡社製)を用いて、Human Leukocyte 5'-STRETCH PLUS cDNA Library(クロンテック社製)のlibrary lysate 2μLを含む50μLの反応液 [2.5ユニットのDNAポリメラーゼKOD 、10倍希釈した10×KOD Buffer #1(東洋紡社製)、0.2mmol/L dNTPs、1mmol/L MgCl2、0.4μmol/L上記プライマー(配列番号10および配列番号11)] を調製し、PCRを行った。PCRは、94℃で30秒間、57℃で30秒間、74℃で60秒間からなる反応を1サイクルとして、30サイクル行った。PCR後の反応液から、QIAquick PCR Purification Kit(QIAGEN社製)を用いてDNAを精製し、滅菌水20μLに溶解した。続いて、精製したDNAを制限酵素HindIII(タカラバイオ社製)およびBamHI(タカラバイオ社製)で消化したのち、1.5%アガロースゲル電気泳動に供し、約0.8 kbのPCR増幅断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0087】
次に、プラスミドpBluescript II SK(-) 3μg(Stratagene社製)を制限酵素HindIII(タカラバイオ社製)およびBamHI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約2.9 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
上記で得たHuman Leukocyte 5'-STRETCH PLUS cDNA Library由来のPCR増幅断片と、プラスミドpBluescript II SK (-)由来のDNA断片を混合し、DNA Ligation Kit Ver.2.0(タカラバイオ社製)により連結反応を行った。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のアミノ酸配列のうち、176番目がバリン(以下、ヒトCD16(Val)もしくはFcγRIIIa(V)と表記する)からなるcDNA(配列番号1)を含むプラスミド、および176番目がフェニルアラニン(以下、ヒトCD16(Phe)もしくはhFcγRIIIa(F)と表記する)からなるcDNA(配列番号2)を含むプラスミドを取得し、それぞれをpBshFcγRIIIa(V)、pBshFcγRIIIa(F)と名付けた。
2.ヒトCD3ζをコードするcDNAの取得
ヒトCD3ζのcDNAの塩基配列(GenBank Accession No. NM_000734)(配列番号5)をもとに、翻訳開始コドンを含む領域の配列に制限酵素認識配列を付加したフォワードプライマー(配列番号12)、および翻訳終止コドンを含む領域の配列に制限酵素認識配列を付加したリバースプライマー(配列番号13)を設計した。
【0088】
まず、本項1に記載の方法に準じて、上記プライマー(配列番号12および配列番号13)を用いたPCRを行い、PCR後の反応液から、QIAquick PCR Purification Kit(QIAGEN社製)を用いてDNAを精製し、滅菌水20μLに溶解した。制限酵素EcoRI(タカラバイオ社製)およびKpnI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約0.5 kbのPCR増幅断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0089】
次に、プラスミドpBluescript II SK(-) 3μg(Stratagene社製)を制限酵素EcoRI(タカラバイオ社製)およびKpnI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約2.9 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
上記で得たHuman Leukocyte 5'-STRETCH PLUS cDNA Library由来のPCR増幅断片と、プラスミドpBluescript II SK (-)由来のDNA断片を混合し、DNA Ligation Kit Ver.2.0(タカラバイオ社製)により連結反応を行った。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD3ζのcDNA(配列番号5)を含むプラスミドpBshCD3ζを取得した。
3.ヒトFcεRIγをコードするcDNAの取得
ヒトFcεRIγのcDNAの塩基配列(GenBank Accession No. NM_004106)(配列番号7)をもとに、翻訳開始コドンを含む領域の配列に制限酵素認識配列を付加したフォワードプライマー(配列番号14)、および翻訳終止コドンを含む領域の配列に制限酵素認識配列を付加したリバースプライマー(配列番号15)を設計した。
【0090】
まず、本項1に記載の方法に準じて、上記プライマー(配列番号14および配列番号15)を用いたPCRを行い、PCR後の反応液から、QIAquick PCR Purification Kit(QIAGEN社製)を用いてDNAを精製し、滅菌水20μLに溶解した。制限酵素EcoRI(タカラバイオ社製)およびKpnI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約0.3 kbのPCR増幅断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0091】
上記で得たHuman Leukocyte 5'-STRETCH PLUS cDNA Library由来のPCR増幅断片と、プラスミドpBluescript II SK (-)から調製した本項2に記載の約2.9kbpのDNA断片を混合し、DNA Ligation Kit Ver.2.0(タカラバイオ社製)により連結反応を行った。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトFcεRIγのcDNA(配列番号7)を含むプラスミドpBshFcεRIγを取得した。
4. pKANTEX hFcγRIIIa(V)およびpKANTEX hFcγRIIIa(F)の構築
本項1で得たプラスミドpBshFcγRIIIa(F)とpBshFcγRIIIa(V)、およびWO97/10354に記載のプラスミドpKANTEX93を用いて、以下の手順に従いpKANTEX hFcγRIIIa(V)およびpKANTEX hFcγRIIIa(F)を構築した(図1)。
【0092】
本項1で得たプラスミドpBshFcγRIIIa(V)、pBshFcγRIIIa(F) 各5.0μgを制限酵素NotI(タカラバイオ社製)およびBamHI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約0.8 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いてそれぞれ精製した。
次に、WO97/10354に記載のプラスミドpKANTEX93 10μgを制限酵素NotI(タカラバイオ社製)およびBamHI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約11.7 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0093】
上記で得たヒトCD16(Val)(別名;FcγRIIIa(V))もしくはヒトCD16(Phe)(別名;FcγRIIIa(F))のcDNAを含むDNA断片と、プラスミドpKANTEX93由来のDNA断片を混合し、DNA Ligation Kit Ver.2.0(タカラバイオ社製)により連結反応を行った。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、目的のヒトFcγRIIIa(V)およびヒトFcγRIIIa(F)のcDNAを含むプラスミドpKANTEX hFcγRIIIa(V)およびpKANTEX hFcγRIIIa(F)を取得した。
5.ヒトCD16とそのアクセサリー分子を共発現する発現ベクターの構築
本項2、3で取得したプラスミドpBshCD3ζまたはpBshFcεRIγ、および本項4で得たプラスミドpKANTEX hFcγRIIIa(V)またはpKANTEX hFcγRIIIa(F)を用いて、以下の手順に従い、ヒトCD16(FcγRIIIa)とそのアクセサリー分子を共発現する発現ベクターを構築した(図2)。
【0094】
まず、本項2、3で取得したプラスミドpBshCD3ζおよびpBshFcεRIγの各5.0μgを、制限酵素EcoRI(タカラバイオ社製)およびKpnI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、pBshCD3ζ由来の0.5 kbのDNA断片、およびpBshFcεRIγ由来の約0.3 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いてそれぞれ精製した。
【0095】
次に、本項(4)で取得したプラスミドpKANTEX hFcγRIIIa(V)およびpKANTEX hFcγRIIIa(F)の各2.0μgを、制限酵素EcoRI(タカラバイオ社製)およびNotI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約10.0kbと約2.5kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いてそれぞれ精製した。さらに、得られたpKANTEX hFcγRIIIa(V)由来の約2.5kbのDNA断片1.5μgを制限酵素KpnI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約2.0 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0096】
上記で得たpBshCD3ζ由来の約0.5 kbのDNA断片、pKANTEX hFcγRIIIa(V)由来の約10.0 kbのDNA断片、およびpKANTEX hFcγRIIIa(V)由来の約2.0 kbのDNA断片を混合し、DNA Ligation Kit Ver.2.0(タカラバイオ社製)により連結反応を行った。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、目的のヒトFcγRIIIa(V)およびヒトCD3ζのcDNAを含むヒトFcγRIIIa発現ベクターpKANTEX hFcγRIIIa(V)+hCD3ζを取得した。
【0097】
さらに、pBshFcεRIγ由来の約0.3 kbのDNA断片、pKANTEX hFcγRIIIa(F)由来の約10.0 kbのDNA断片を用い、上記の方法に準じて、hFcγRIIIa(V)とhFcεRIγ、hFcγRIIIa(F)とhCD3ζ、hFcγRIIIa(V)とhFcεRIγの各cDNAを含むhFcγRIIIa発現ベクターを構築した。得られたプラスミドをそれぞれpKANTEX hFcγRIIIa(V)+hFcεRIγ、pKANTEX hFcγRIIIa(F)+hCD3ζ、pKANTEX hFcγRIIIa(F)+hFcεRIγと名付けた。
【実施例2】
【0098】
ヒトCD16プロモーターによって制御されたヒトCD16とヒトCD3ζのcDNAの発現ユニットを相同組換え法によりマウスHPRT遺伝子領域へ挿入するためのプラスミドSAT1-HRの作製
1.プラスミドSAT1-linkerの作製
12種類の制限酵素(KpnI、AscI、BstBI、XbaI、NsiI、PmeI、NruI、SpeI、ClaI、HpaI、SrfI、SacI)の認識配列から成るオリゴDNAセンス鎖SAT1-link1(配列番号16)およびアンチセンス鎖SAT1-link2(配列番号17)をファスマック社にて合成した。SAT1-link1とSAT1-link2をそれぞれ10 pmolずつ含む100μLの蒸留水を調製し、90℃で10分間熱したのち室温までゆっくり戻すことによりSAT1-link1とSAT1-link2をアニーリングさせ、SAT1-linkerを作製した。
【0099】
続いて、5μgのpBluescriptIISK(+) (STRATAGENE社製)をKpnI(タカラバイオ社製)およびSacIで消化したのち、フェノール/クロロホルム抽出処理及びエタノール沈殿を行い、回収したプラスミドを50μLの水に溶解した。
上記で得られたSAT1-linkerを0.2 pmol及びpBluescriptIISK(+)断片(KpnI-SacI)を50 ng含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で2時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、設計したSAT1-linker配列を含むプラスミドSAT1-linkerを得た(図3)。
2.プラスミドSAT1-IRESの作製
5μgのプラスミドG68(GenOway社製)をSmaI(New England Biolabs社製)とNsiI(New England Biolabs社製)にて、また本項1にて作製したプラスミドSAT1-linkerを5μg分PmeI(New England Biolabs社製)とNsiI(New England Biolabs社製)にてそれぞれ消化したのち、プラスミドG68及びプラスミドSAT1-linkerのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約0.6 kb、約3 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0100】
次いでプラスミドG68由来DNA断片(NsiI-SmaI)20ng、プラスミドSAT1-linker由来DNA断片(NsiI-PmeI)80ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、プラスミドG68由来のIRES配列を含むプラスミドSAT1-IRESを得た(図4)。
3.プラスミドSAT1-IRES-polyAの作製
5μgのプラスミドG147(GenOway社製)をSmaI(New England Biolabs社製)とClaI(New England Biolabs社製)にて、また本項2にて作製したプラスミドSAT1-IRESを 5μg分HpaI(New England Biolabs社製)とClaI(New England Biolabs社製)にてそれぞれ消化したのち、プラスミドG147及びプラスミドSAT1-IRESのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約0.8 kb、約3.6 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0101】
次いでプラスミドG147由来DNA断片(ClaI-SmaI)20ng、プラスミドSAT1-IRES由来DNA断片(ClaI-HpaI)80ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、IRES配列の下流にヒト成長ホルモン由来polyA配列が連結されたプラスミドSAT1-IRESを得た(図5)。
4.プラスミドCD3Zeta in pBSの作製
ヒトCD3ζのcDNAの塩基配列(GenBank Accession No. NM_000734)(配列番号5)をもとに、翻訳開始コドンを含む領域の配列に制限酵素認識配列(EcoRI)及びコザック配列を付加したフォワードプライマー(配列番号18)及び翻訳停止コドンを含む領域の配列に制限酵素認識配列(XbaI、SpeI)を付加したリバースプライマー(配列番号19)を作製し、以下のPCRを行なった。即ち、実施例1で作製したpKANTEX hFcγRIIIa(V)+hCD3ζをテンプレートとして含む20μLの反応液 [Pyrobest DNA polymerase(タカラバイオ社製)、10×PCR buffer、0.2mmol/L dNTP mixture、0.5μmol/L上記プライマー(配列番号18および配列番号19)]を調製し、94℃で5分間加熱した後、94℃で1分間、68℃で2分間を1サイクルとした30サイクルの反応でPCRを行なった。PCR反応後の溶液は1.5%アガロースゲル電気泳動に供し、約0.5 kbのPCR増幅断片をQIAquick Gel Extraction kit(QIAGEN社製)を用いて精製した。
【0102】
上記で得られたPCR増幅断片は、Zero Blunt TOPO PCR Cloning Kit for Sequencing(インビトロジェン社製)を用いてプラスミドpCR4Blunt-TOPOと連結させたのち、本プラスミドを用いてheat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD3ζのcDNA配列を含むプラスミドCD3Zeta in pCR4Bluntを得た(図6)。
【0103】
続いて、上記で作製したプラスミドCD3Zeta in pCR4Bluntを5μg分EcoRI(タカラバイオ社製)およびSpeI(タカラバイオ社製)にて、また5μgのプラスミドpBluescriptIISK(+)(STRATAGENE社製)をEcoRI(タカラバイオ社製)とSpeI(タカラバイオ社製)にてそれぞれ消化したのち、プラスミドCD3Zeta in pCR4Blunt及びプラスミドpBluescriptIISK(+)のDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約0.5 kbと約3.0 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。次いでプラスミドCD3Zeta in pCR4Blunt由来DNA断片(EcoRI-SpeI)20ng、プラスミドpBluescriptIISK(+)由来DNA断片(EcoRI-SpeI)80ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD3ζのcDNA配列を含むプラスミドCD3Zeta in pBSを得た(図6)。
5.プラスミドSAT1-IRES-CD3 Zetaの作製
本項3にて作製したプラスミドSAT1-IRES-polyAを 5μg分NruI(New England Biolabs社製)とSpeI(タカラバイオ社製)にて、また本項4で作製したプラスミドCD3Zeta in pBSを5μg分EcoRV(タカラバイオ社製)とSpeI(タカラバイオ社製)にてそれぞれ消化したのち、プラスミドSAT1-IRES-polyA及びプラスミドCD3Zeta in pBSのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約4.4 kb、約0.5 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0104】
次いでプラスミドSAT1-IRES-polyA由来DNA断片(NruI-SpeI)80ng、プラスミドCD3Zeta in pBS由来DNA断片(EcoRV-SpeI)20ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、IRES配列とヒト成長ホルモン由来polyA配列の間にヒトCD3ζのcDNA配列が連結されたプラスミドSAT1-IRES-CD3 Zetaを得た(図7)。
6.プラスミドCD16 in pBSの作製
ヒトCD16のcDNAの塩基配列(GenBank Accession No. NM_000569)(配列番号1)をもとに、5’非翻訳領域及び翻訳開始コドンを含む配列よりなるフォワードプライマー(配列番号20)及び翻訳停止コドンを含む領域の配列に制限酵素認識配列(XbaI、SpeI)を付加したリバースプライマー(配列番号21)を作製し、以下のPCRを行なった。即ち、実施例1で作製したpKANTEX hFcγRIIIa(V)+hCD3ζをテンプレートとして含む20μLの反応液 [Pyrobest DNA polymerase(タカラバイオ社製)、10×PCR buffer、0.2mmol/L dNTP mixture、0.5μmol/L上記プライマー(配列番号20および配列番号21)]を調製し、94℃で5分間加熱した後、94℃で1分間、68℃で2分間を1サイクルとした30サイクルの反応でPCRを行なった。PCR反応後の溶液は1.5%アガロースゲル電気泳動に供し、約0.8 kbのPCR増幅断片をQIAquick Gel Extraction kit(QIAGEN社製)を用いて精製した。
【0105】
上記で得られたPCR増幅断片は、Zero Blunt TOPO PCR Cloning Kit for Sequencing(インビトロジェン社製)を用いてプラスミドpCR4Blunt-TOPOと連結させたのち、本プラスミドを用いてheat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD3ζのcDNA配列を含むプラスミドCD16 in pCR4Bluntを得た(図8)。
【0106】
続いて、上記で作製したプラスミドCD16 in pCR4Bluntを5μg分EcoRI(タカラバイオ社製)とSpeI(タカラバイオ社製)にて、また5μgのプラスミドpBluescriptIISK(+)(STRATAGENE社製)をEcoRI(タカラバイオ社製)とSpeI(タカラバイオ社製)にてそれぞれ消化したのち、上記で得られたプラスミドCD16 in pCR4Blunt及びプラスミドpBluescriptIISK(+)のDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約0.8 kb、3.0 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0107】
次いでプラスミドCD16 in pCR4Blunt由来DNA断片(EcoRI-SpeI)20ng、プラスミドpBluescriptIISK(+)由来DNA断片(EcoRI-SpeI)80ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のcDNA配列を含むプラスミドCD16 in pBSを得た(図8)。
7.プラスミドCD16Pmoの作製
ヒトCD16のプロモーター配列(GenBank Accession No. AL_590385)(配列番号9)をもとに、フォワードプライマー(配列番号22)及びリバースプライマー(配列番号23)を作製し、以下のPCRを行なった。即ち、Human Genomic DNA(Novagen社製)をテンプレートとして含む20μLの反応液 [Pyrobest DNA polymerase(タカラバイオ社製)、10×PCR buffer、0.2mmol/L dNTP mixture、0.5μmol/L上記プライマー(配列番号22および配列番号23)]を調製し、94℃で5分間加熱した後、94℃で1分間、68℃で2分間を1サイクルとした30サイクルの反応でPCRを行なった。PCR反応後の溶液は1.5%アガロースゲル電気泳動に供し、約1.1 kbのPCR増幅断片をQIAquick Gel Extraction kit(QIAGEN社製)を用いて精製した。
【0108】
上記で得られたPCR増幅断片は、Zero Blunt TOPO PCR Cloning Kit for Sequencing(インビトロジェン社製)を用いてプラスミドpCR4Blunt-TOPOと連結させたのち、本プラスミドを用いてheat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のプロモーター配列を含むプラスミドCD16Pmo-Nを得た(図9)。
【0109】
次に、ヒトCD16のプロモーター配列(GenBank Accession No. AL_590385)(配列番号9)をもとに、フォワードプライマー(配列番号24)及びリバースプライマー(配列番号25)を作製し、以下のPCRを行なった。即ち、Human Genomic DNA(Novagen社製)をテンプレートとして含む20μLの反応液 [Pyrobest DNA polymerase(タカラバイオ社製)、10×PCR buffer、0.2mmol/L dNTP mixture、0.5μmol/L上記プライマー(配列番号24および配列番号25)]を調製し、94℃で5分間加熱した後、94℃で1分間、68℃で2分間を1サイクルとした30サイクルの反応でPCRを行なった。PCR反応後の溶液は1.5%アガロースゲル電気泳動に供し、約0.8 kbのPCR増幅断片をQIAquick Gel Extraction kit(QIAGEN社製)を用いて精製した。
【0110】
上記で得られたPCR増幅断片は、Zero Blunt TOPO PCR Cloning Kit for Sequencing(インビトロジェン社製)を用いてプラスミドpCR4Blunt-TOPOと連結させたのち、本プラスミドを用いてheat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のプロモーター配列を含むプラスミドCD16Pmo-Cを得た(図10)。
【0111】
続いて、上記で作製したプラスミドCD16Pmo-Nを5μg分HindIII(タカラバイオ社製)とApaI(タカラバイオ社製)にて、また同じく上記で作製したプラスミドCD16Pmo-Cを5μg分HindIII(タカラバイオ社製)とApaI(タカラバイオ社製)にてそれぞれ消化したのち、上記で得られたプラスミドCD16Pmo-N及びプラスミドCD16Pmo-CのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約1.1 kbと約4.8 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。次いでプラスミドCD16Pmo-N由来DNA断片(HindIII-ApaI)20ng、プラスミドCD16Pmo-C由来DNA断片(HindIII-ApaI)80ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、約1.8 kbのヒトCD16のプロモーター配列を含むプラスミドCD16Pmoを得た(図11)。
8.プラスミドCD16Pmo+cDNAの作製
本項6で作製したプラスミドCD16 in pBSを5μg分BspEI(New England Biolabs社製)とHindIII(タカラバイオ社製)にて、また本項7で作製したプラスミドCD16Pmoを5μg分BspEI(New England Biolabs社製)とHindIII(タカラバイオ社製)にてそれぞれ消化したのち、上記で得られたプラスミドCD16 in pBS及びプラスミドCD16PmoのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約3.8 kb、1.8 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。次いでプラスミドCD16 in pBS由来DNA断片(HindIII-BspEI)80ng、プラスミドCD16Pmo由来DNA断片(HindIII-BspEI)20ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のプロモーター配列およびcDNA配列が連結されたプラスミドCD16Pmo+cDNAを得た(図12)。
9.プラスミドSAT1-Tgの作製
本項5で作製したプラスミドSAT1-IRES-CD3 Zetaを5μg分BstBI(New England Biolabs社製)とXbaI(タカラバイオ社製)にて、また本項8で作製したプラスミドCD16Pmo+cDNAを5μg分ClaI(New England Biolabs社製)とXbaI(タカラバイオ社製)にてそれぞれ消化したのち、上記で得られたプラスミドSAT1-IRES-CD3 Zeta及びプラスミドCD16Pmo+cDNAのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約4.9 kbと約2.6 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。次いでプラスミドSAT1-IRES-CD3 Zeta由来DNA断片(BstBI-XbaI)80ng、プラスミドCD16Pmo+cDNA由来DNA断片(ClaI-XbaI)20ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のプロモーター配列下流にIRES配列で連結されたヒトCD16のcDNAとヒトCD3ζのcDNA配列が配置され、さらにその下流にヒト成長ホルモン由来polyA配列が付加されたプラスミドSAT1-Tgを得た(図13)。
10.プラスミドSAT1-HRの作製
本項9で作製したプラスミドSAT1-Tgを制限酵素AscI及びSrfIで切断することにより生じる約4.6 kbのDNA断片を回収し、AscI及びSwaIで切断したプラスミドready-to-use Quick knock-inTM vector(GenOway社製)とLigase酵素を用いて連結反応を行った。得られた連結済プラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ネガティブセレクションマーカーとしてジフテリア毒素発現ユニット(DTA)およびポジティブセレクションマーカーとしてネオマイシン耐性遺伝子発現ユニットを有し、またマウスHPRT遺伝子領域に相同組換えを起こさせるための5’側アームとしてマウスHPRT遺伝子の第1エクソン上流の塩基配列、同じく3’側アームとしてマウスHPRT遺伝子の第3エクソンおよび第2、3イントロンの塩基配列を含むready-to-use Quick knock-inTM vectorに、ヒトCD16のプロモーター配列下流にIRES配列で連結されたヒトCD16のcDNAとヒトCD3ζのcDNA配列が配置され、さらにその下流にヒト成長ホルモン由来polyA配列が付加された遺伝子発現構成体が導入された、プラスミドSAT1-HRを得た(図14)。
【実施例3】
【0112】
ヒトCD16プロモーター制御下においてヒトCD16及びヒトCD3ζを発現している遺伝子改変マウスの作製
1.遺伝子改変ES細胞株の樹立
以下の方法を用いて、マウスES細胞のHPRT遺伝子のエクソン3上流に、ヒトCD16プロモーターによって制御されたヒトCD16のcDNA及びヒトCD3ζの発現ユニットを挿入した。遺伝子導入は公知のエレクトロポレーション法 [サイトテクノロジー (Cytotechnology), 3, 133 (1990)] により以下の手順で行った。
【0113】
実施例2で作製したプラスミドSAT1-HRをPmeI(New England Biolabs社製)で切断した後、40 μg分の線状化プラスミドを5×106細胞の129Olaマウス由来E14 ES細胞(GenOway社製)へパルス電圧260 V、電気容量500μFの条件で遺伝子導入を行った。なお、129Olaマウスとは、129系統マウスよりHPRT遺伝子のエクソン1およびその周辺の染色体領域を欠失させることにより作製したマウスである。2日後、200μg/mlのG418入り培地でES細胞を選抜培養し、G418耐性ES細胞株を400クローン単離した。400クローンはそれぞれ96穴プレートでコンフルエントまで培養した後、96穴プレート2枚(細胞培養用とレプリカのゲノムDNA取得用)へそれぞれ継代した。
【0114】
ゲノムDNA取得用の96穴プレートより公知の方法(WO03/085107(山根さんのFUT8KO株特許))でゲノムDNAを取得した後、相同組換え法により目的のHPRT遺伝子領域にヒトCD16プロモーターによって制御されたヒトCD16のcDNA及びヒトCD3ζの発現ユニットが挿入されている相同組換えクローンを、以下に示すゲノムPCR法を用いて検出した。
相同組換え領域の5’側の確認は、PCR用プライマーGW496(配列番号26)およびPCR用プライマーGW497(配列番号27)を用いて行った(図15)。100 ngのゲノムDNAをテンプレートとして含む50μLの反応液 [2.6 unitsのExpand High Fidelity polymerase(Roche diagnostics社製)、10×PCR buffer、0.5 mmol/L dNTP mixture、15 pmolずつのプライマーGW496(配列番号26)およびGW497(配列番号27)]を調製し、94℃で2分間加熱した後、94℃で30秒間、62℃で1分間、68℃で4分間を1サイクルとした35サイクルの反応でPCRを行なった。PCR反応終了溶液はアガロースゲル電気泳動に供し、4529 bpのPCR増幅断片が検出されたES細胞クローン株をクローニングした(図16)。その結果、G418耐性を有する400クローン中、37クローンにおいて4529 bpのPCR増幅断片が認められた。この37クローン中20クローンについて、次に示す3’側のゲノムPCR解析を行った。
【0115】
相同組換え領域の3’側の確認は、PCR用プライマーGX2922(配列番号28)およびPCR用プライマーGX0025(配列番号29)を用いて行った(図17)。100 ngのゲノムDNAをテンプレートとして含む50μLの反応液 [2.6 unitsのExpand High Fidelity polymerase(Roche diagnostics社製)、10×PCR buffer、0.5 mmol/L dNTP mixture、15 pmolずつのプライマーGX2922(配列番号28)およびGX0025(配列番号29)]を調製し、94℃で2分間加熱した後、94℃で30秒間、62℃で1分間、68℃で4分間を1サイクルとした35サイクルの反応でPCRを行なった。PCR反応終了溶液はアガロースゲル電気泳動に供し、5733 bpのPCR増幅断片が検出されたES細胞クローン株を同定した(図17)。その結果、5’側の相同組換えが確認された20クローン中17クローンにおいて、5733 bpのPCR増幅断片が認められた。
【0116】
5’側および3’側のPCR解析により特異的バンドが検出された17クローンについて、次に示すサザンブロット解析を行った(図18)。各クローンのゲノムDNAをPstI(New England Biolabs社製)で16時間切断させたのち、アガロースゲル電気泳動に供し、既存の方法(J. Mol. Biol., 98, 503, 1975)を用いてナイロンフィルターへDNAを転写した。65℃のプレハイブリ溶液(4×SSC、1% SDS、65℃0.5% スキムミルク、20 mM EDTA、100 μg/mLのサケ精子DNA)に上記で作製したフィルターを浸したのち、さらに32P標識体やDIG標識体で標識した3’側プローブ(配列番号30)を添加し、65℃で18時間ハイブリダイゼーションを行った。なお、3’側プローブはマウスHPRT遺伝子の第3エクソン配列(GenBank Accession No. K01509)より設計した。ハイブリダイゼーション後、3×SSCと1%SDSを含有する溶液を用いて65℃で15分間の洗浄を2回行ったのち、さらに0.5×SSCと1%SDSを含有する溶液を用いて65℃で15分間の洗浄を2回行った。X線フィルムを洗浄後のフィルターで3日間感光させたのち現像を行い、目的通りの相同組換えが起こっているES細胞クローン株(約6.1 kbのバンドが検出される)を同定した(図18、19)。その結果、#1C8、#2A8、#12A2、#15A9の4クローン株において約6.1 kbのバンドが観察され、これらクローン株では正常に相同組換えが行われていることが確認された。
【0117】
上記のESクローン株(#1C8、#2A8、#12A2、#15A9)をC57BL/6マウス由来の胚盤胞へとインジェクションしたのち、偽妊娠させたOF1マウスの卵管へと移植し、産仔を得た。その結果、#1C8、#12A2、#15A9のES細胞をインジェクションした胚盤胞より、95〜100%のキメラ率(体表の毛の色がアグチである割合をキメラ率として表記する)を有するオスのキメラ体を計8匹得た。この8匹より、#1C8由来のキメラ体2匹、#12A2由来のキメラ体1匹、#15A9由来のキメラ体1匹をそれぞれC57BL/6マウスのメスと交配させ、産まれたメスのF1体について、以下に示すPCR解析およびサザンブロット解析を行った。
【0118】
相同組換え領域の5’側の確認は、PCR用プライマーGW496(GenOway社製(配列番号26))およびPCR用プライマーGW497(GenOway社製(配列番号27))を用いて行った(図15)。メスのF1体の尾より取得した100 ngのゲノムDNAをテンプレートとして含む50μLの反応液 [2.6 unitsのExpand High Fidelity polymerase(Roche diagnostics社製)、10×PCR buffer、0.5 mmol/L dNTP mixture、15 pmolずつのプライマーGW496(配列番号26)およびGW497(配列番号27)]を調製し、94℃で2分間加熱した後、94℃で30秒間、62℃で1分間、68℃で4分間を1サイクルとした35サイクルの反応でPCRを行なった。PCR反応終了溶液はアガロースゲル電気泳動に供し、4529 bpのPCR増幅断片が検出されるかどうか確認した(図15、20)。その結果、#1C8、#12A2および#15A9由来のキメラ体より産まれたメスのF1体より、4529 bpのPCR増幅断片が検出された。
【0119】
続いて、相同組換え領域の3’側の確認はサザンブロット解析にて行った。メスのF1体の尾より取得したゲノムDNAをPstI(New England Biolabs社製)で16時間切断させたのち、アガロースゲル電気泳動に供し、既存の方法(J. Mol. Biol., 98, 503, 1975)を用いてナイロンフィルターへDNAを転写した。65℃のプレハイブリ溶液(4×SSC、1% SDS、65℃0.5% スキムミルク、20 mM EDTA、100 μg/mLのサケ精子DNA)に上記で作製したフィルターを浸したのち、さらに32P標識体やDIG標識体で標識した3’側プローブ(配列番号30)を添加し、65℃で18時間ハイブリダイゼーションを行った。なお、3’側プローブはマウスHPRT遺伝子の第3エクソン配列(GenBank Accession No. K01509)より設計した。ハイブリダイゼーション後、3×SSCと1%SDSを含有する溶液を用いて65℃で15分間の洗浄を2回行ったのち、さらに0.5×SSCと1%SDSを含有する溶液を用いて65℃で15分間の洗浄を2回行った。X線フィルムを洗浄後のフィルターで3日間感光させたのち現像を行い、目的通りの相同組換えが起こっているF1体(約6.1 kbのバンドが検出される)を同定した(図21、22)。その結果、4529 bpのPCR増幅断片が検出されたF1体において、C57Bl/6マウスの染色体由来バンド(約7.8 kb)および約6.1 kbの相同組換え体由来バンドが検出された。
【0120】
以上の結果、オスの#1C8、#12A2および#15A9由来キメラ体の生殖系列は目的通りの相同組換えを起こしたES細胞株由来であり、このキメラ体とメスの野生型マウスを掛け合わせることにより、片方のX染色体のHPRT遺伝子領域にはヒトCD16プロモーター下流にヒトCD16とヒトCD3ζのcDNAをIRES配列で連結させた遺伝子発現ユニットが挿入されたF1体が産まれることが確認された。
【実施例4】
【0121】
ヒトCD16プロモーター制御下においてヒトFcεRIγ及びGFPを発現している遺伝子改変マウスの作製
1.プラスミドEpsilon in pCR4Bluntの作製
ヒトFcεRIγのcDNAの塩基配列(GenBank Accession No. NM_004106)(配列番号7)をもとに、翻訳開始コドンを含む領域の配列に制限酵素認識配列(BspEI)及びヒトCD16プロモーター配列を付加したフォワードプライマー(配列番号31)及び翻訳停止コドンを含む領域の配列に制限酵素認識配列(SpeI)を付加したリバースプライマー(配列番号32)を作製し、以下のPCRを行なった。即ち、実施例1で作製したpKANTEX hFcγRIIIa(V)+hFcεRIγをテンプレートとして含む20μLの反応液 [Pyrobest DNA polymerase(タカラバイオ社製)、10×PCR buffer、0.2mmol/L dNTP mixture、0.5μmol/L上記プライマー(配列番号31および配列番号32)]を調製し、94℃で5分間加熱した後、94℃で1分間、68℃で2分間を1サイクルとした30サイクルの反応でPCRを行なった。PCR反応後の溶液は1.5%アガロースゲル電気泳動に供し、約0.3 kbのPCR増幅断片をQIAquick Gel Extraction kit(QIAGEN社製)を用いて精製した。
【0122】
上記で得られたPCR増幅断片は、Zero Blunt TOPO PCR Cloning Kit for Sequencing(インビトロジェン社製)を用いてプラスミドpCR4Blunt-TOPOと連結させたのち、本プラスミドを用いてheat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトFcεRIγのcDNA配列を含むプラスミドEpsilon in pCR4Bluntを得た(図23)。
2.プラスミドGFP in pCR4Bluntの作製
公知のGreen fluorescent protein(GFP)cDNA配列(GenBank Accession No. M62653)をもとに、より強い発光が観察される改変型GFPのcDNA配列を設計し(配列番号33)、合成オリゴDNAを用いて本配列を含むDNA鎖を作製した。
【0123】
続いて、翻訳開始コドンを含む領域の配列に制限酵素認識配列(EcoRI)及びコザック配列を付加したフォワードプライマー(配列番号34)及び翻訳停止コドンを含む領域の配列に制限酵素認識配列(SpeI)を付加したリバースプライマー(配列番号35)を作製し、以下のPCRを行なった。即ち、改変型GFPのcDNA配列(配列番号33)をテンプレートとして含む20μLの反応液 [Pyrobest DNA polymerase(タカラバイオ社製)、10×PCR buffer、0.2mmol/L dNTP mixture、0.5μmol/L上記プライマー(配列番号34および配列番号35)]を調製し、94℃で5分間加熱した後、94℃で1分間、68℃で2分間を1サイクルとした30サイクルの反応でPCRを行なった。PCR反応後の溶液は1.5%アガロースゲル電気泳動に供し、約0.8 kbのPCR増幅断片をQIAquick Gel Extraction kit(QIAGEN社製)を用いて精製した。
【0124】
上記で得られたPCR増幅断片は、Zero Blunt TOPO PCR Cloning Kit for Sequencing(インビトロジェン社製)を用いてプラスミドpCR4Blunt-TOPOと連結させたのち、本プラスミドを用いてheat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、改変型GFPのcDNA配列を含むプラスミドGFP in pCR4Bluntを得た(図24)。
3.プラスミドPmo+epsilonの作製
実施例2で作製したプラスミドCD16Pmo+cDNAを5μg分BspEI(New England Biolabs社製)とSpeI(タカラバイオ社製)にて、また本項1にて作製したプラスミドEpsilon in pCR4Bluntを 5μg分BspEI(New England Biolabs社製)とSpeI(タカラバイオ社製)にてそれぞれ消化したのち、上記で得られたプラスミドCD16Pmo+cDNA及びプラスミドEpsilon in pCR4BluntのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約3.8 kb、約0.3 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0125】
次いでプラスミドCD16Pmo+cDNA由来DNA断片(BspEI-SpeI)50ng、プラスミドEpsilon in pCR4Blunt由来DNA断片(BspEI-SpeI)50ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のプロモーター配列下流にヒトFcεRIγのcDNA配列が連結されたプラスミドPmo+epsilonを得た(図25)。
4.プラスミドSAT1-IRES-GFPの作製
実施例2で作製したプラスミドSAT1-IRES-polyAを5μg分NruI(New England Biolabs社製)とSpeI(タカラバイオ社製)にて、また本項2にて作製したプラスミドGFP in pCR4Bluntを 5μg分EcoRV(タカラバイオ社製)とSpeI(タカラバイオ社製)にてそれぞれ消化したのち、上記で得られたプラスミドSAT1-IRES-polyA及びプラスミドGFP in pCR4BluntのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約4.4 kb、0.8 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。次いでプラスミドSAT1-IRES-polyA由来DNA断片(NruI-SpeI)50ng、プラスミドGFP in pCR4Blunt由来DNA断片(EcoRV-SpeI)50ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、IRES配列とヒト成長ホルモン由来polyA配列の間に改変型GFPのcDNA配列が連結されたプラスミドSAT1-IRES-GFPを得た(図26)。
5.プラスミドEpsGFP-Tgの作製
本項4で作製したプラスミドSAT1-IRES-GFPを5μg分BstBI(New England Biolabs社製)とXbaI(タカラバイオ社製)にて、また本項3で作製したプラスミドPmo+epsilonを5μg分ClaI(New England Biolabs社製)とXbaI(タカラバイオ社製)、10μLの10×Mバッファー(タカラバイオ社製)にてそれぞれ消化したのち、上記で得られたプラスミドSAT1-IRES-GFP及びプラスミドPmo+epsilonのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約5.0 kb、約2.1 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。次いでプラスミドSAT1-IRES-GFP由来DNA断片(BstBI-XbaI)80ng、プラスミドPmo+epsilon由来DNA断片(ClaI-XbaI)20ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のプロモーター配列下流にIRES配列で連結されたヒトFcεRIγのcDNAと改変型GFPのcDNA配列が配置され、さらにその下流にヒト成長ホルモン由来polyA配列が付加されたプラスミドEpsGFP-Tgを得た(図27)。
6.ヒトCD16プロモーター制御下においてヒトFcεRIγ及びGFPを発現している遺伝子改変マウスの作製
本項5で作製したプラスミドEpsGFP-Tgを線状化もしくはマウス染色体の特定の遺伝子領域へ挿入するためのプラスミドに挿入したのち、マウスの受精卵もしくはES細胞へと導入し、ヒトCD16プロモーター制御下においてヒトFcεRIγ及びGFPを発現している遺伝子改変マウスを作製した。さらに、本マウスを実施例3に記載のヒトCD16プロモーター制御下においてヒトCD16及びヒトCD3ζを発現している遺伝子改変マウスと掛け合わせることにより、ヒトCD16プロモーター制御下においてヒトCD16、ヒトCD3ζ、ヒトFcεRIγ、および改変型GFPを発現している遺伝子改変マウスを作製した。
【0126】
また、本項5で作製したプラスミドEpsGFP-Tgを実施例3に記載のヒトCD16プロモーター制御下においてヒトCD16及びヒトCD3ζを発現している遺伝子改変マウスの受精卵もしくはES細胞へと導入し、ヒトCD16プロモーター制御下においてヒトCD16、ヒトCD3ζ、ヒトFcεRIγ、および改変型GFPを発現している遺伝子改変マウスを作製した。
【図面の簡単な説明】
【0127】
【図1】プラスミドpKANTEXhFcγRIIIa(V)およびpKANTEXhFcγRIIIa(F)の作製方法を示す。
【図2】プラスミドpKANTEXhFcγRIIIa(V)+hCD3ζ、pKANTEXhFcγRIIIa(F)+hCD3ζ、pKANTEXhFcγRIIIa(V)+hFcεRIγ、およびpKANTEXhFcγRIIIa(F)+hFcεRIγの作製方法を示す。
【図3】プラスミドSAT1-linkerの作製方法を示す。
【図4】プラスミドSAT1-IRESの作製方法を示す。
【図5】プラスミドSAT1-IRES-polyAの作製方法を示す。
【図6】プラスミドCD3Zeta in pBSの作製方法を示す。
【図7】プラスミドSAT1-IRES-CD3Zetaの作製方法を示す。
【図8】プラスミドCD16 in pBSの作製方法を示す。
【図9】プラスミドCD16Pmo-Nの作製方法を示す。
【図10】プラスミドCD16Pmo-Cの作製方法を示す。
【図11】プラスミドCD16Pmoの作製方法を示す。
【図12】プラスミドCD16Pmo+cDNAの作製方法を示す。
【図13】プラスミドSAT1-Tgの作製方法を示す。
【図14】プラスミドSAT1-HRの作製方法を示す。
【図15】予想される相同組換え体の染色体構造(HPRT遺伝子領域)及び相同組換え体を検出するための5’側および3’側のPCRプライマーの位置を模式的に示す。
【図16】5’側のPCRによって検出される、相同組換え体ES細胞株のゲノムDNA由来の増幅バンドを示す。
【図17】3’側のPCRによって検出される、相同組換え体ES細胞株のゲノムDNA由来の増幅バンドを示す。
【図18】予想される相同組換え体の染色体構造(HPRT遺伝子領域)及び相同組換え体を検出するためのサザンブロット解析に用いる3’側プローブの位置を模式的に示す。
【図19】3’側プローブを用いたサザンブロット解析によって検出される、宿主株のE14 ES細胞株及び相同組換え体ES細胞株のゲノムDNA由来のバンドを示す。
【図20】5’側のPCRによって検出される、野生型及びF1体マウスのゲノムDNA由来の増幅バンドを示す。
【図21】親株のE14 ES細胞株、予想される相同組換え体、ならびに野生型マウスの染色体構造(HPRT遺伝子領域)及びサザンブロット解析に用いる3’側プローブの位置を模式的に示す。
【図22】3’側プローブを用いたサザンブロット解析によって検出される、宿主株のE14 ES細胞株、野生型マウス、及びF1体のゲノムDNA由来のバンドを示す。
【図23】プラスミドEpsilon in pCR4Bluntの作製方法を示す。
【図24】プラスミドGFP in pCR4Bluntの作製方法を示す。
【図25】プラスミドPmo+epsilonの作製方法を示す。
【図26】プラスミドSAT1-IRES-GFPの作製方法を示す。
【図27】プラスミドEpsGFP-Tgの作製方法を示す。
【産業上の利用可能性】
【0128】
本発明により、ヒトCD16とそのヒトアクセサリー分子とを共発現させるようにゲノムが改変された、非ヒト動物またはその子孫及びその利用方法が提供される。本発明の非ヒト動物及びその子孫は、抗体の生物活性及び薬理評価に有用である。
【技術分野】
【0001】
本発明は、ヒトCD16とそのヒトアクセサリー分子とを共発現させるようにゲノムが改変された、非ヒト動物またはその子孫及びその利用に関する。
【背景技術】
【0002】
近年の抗体工学の進歩により、ヒトに対する抗原性が低く、臨床応用可能な遺伝子組換え抗体の作製が可能となった。具体的には、ヒト型キメラ抗体やヒト化抗体〔ヒト型相補性決定領域(complementarity determining region; CDR)移植抗体ともいう〕などのヒト化抗体作製技術の開発(非特許文献1−5参照)や、ヒト抗体産生トランスジェニックマウスやファージライブラリーを利用した完全ヒト抗体作製技術の開発(非特許文献6−12参照)が、モノクローナル抗体治療を医薬として用いることを可能にした。その結果、1990年後半からモノクローナル抗体医薬品が次々と認可された。医薬として開発されている抗体のほとんどは分子量約150kDaのヒトIgGであり、そのFc領域に2本のN-グリコシド結合複合型糖鎖が結合する糖蛋白質である。Fc領域はFc受容体や補体などが結合する領域であり、この部分を通じて抗体依存性細胞傷害活性(antibody-dependent cellular cytotoxicity; ADCC)や補体依存性細胞傷害活性(complement-dependent cytotoxicity; CDC)といったエフェクター活性が発揮される。最近の抗体医薬の臨床成績では、乳癌、大腸癌、血液癌といった多くのヒト悪性腫瘍において、延命や病態悪化に至るまでの期間延長といった効果が観察されている(非特許文献13−16参照)。また、癌患者Fc受容体の多型解析から、非ホジキンリンパ腫治療薬抗CD20抗体RituxanR(リツキシマブ)や乳癌治療薬抗Her2抗体HerceptinR(トラスツズマブ)の主たる抗腫瘍メカニズムの一つはADCC活性であることが明らかにされ(非特許文献17−21参照)、医薬開発に応用可能なADCC活性増強技術の開発が次世代抗体技術として注目されている。抗原となるヒトのゲノム情報が完全に解読された今日、抗体医薬は分子標的医薬の代表として熾烈な競争時代に入っており(非特許文献22参照)、現在、世界では、100を超える臨床試験と300を超える前臨床試験が行われていると言われている。
【0003】
しかしながら、現行の抗体医薬の臨床効果は必ずしも充分なものではなく、また、投与量が多いため薬剤費が高いといった課題がある。この原因の一つには、病態モデル動物を用いた非臨床実験において抗体医薬の十分な薬理評価が行なえていないことが挙げられる。これは、種差の違いから、披検薬であるヒト抗体が病態モデル動物のFc受容体ときちんとは結合せずエフェクター活性の十分な評価が難しいこと、Fc受容体の発現分布が必ずしもヒトと一致せずヒトでの効果が想定できないことに起因している。
【0004】
この問題を解決するためには、ヒトの免疫系を再現した疾患モデル動物の作製が欠かせない。ヒトCD16を発現するモデル動物としては、約4.6 kbのヒトCD16プロモーターを含むヒトCD16発現ベクターを用いてNK細胞やマクロファージ上にヒトCD16を発現させたトランスジェニックマウスの作製が報告されている(非特許文献23参照)。しかしながら、本トランスジェニックマウスを用いたヒト抗体のエフェクター活性評価あるいは薬理評価の報告はなく、ヒト抗体の十分な機能評価には成功していない。その後、本トランスジェニックマウスを用いて、C57BL/6JNIcrマウスやC.B17/Icr-scidマウスとの戻し交配(非特許文献24参照)、あるいは、CD16ノックアウトマウスやヒトCD20発現マウスなどの交配(特許文献1参照)などが行われている。
【0005】
【特許文献1】WO2004/060052
【非特許文献1】Proc. Natl. Acad. Sci. U.S.A., 81, 6851 (1984)
【非特許文献2】Nature,312, 643 (1984)
【非特許文献3】Nature, 321, 522 (1986)
【非特許文献4】Nature, 332, 323 (1988)
【非特許文献5】Science, 239, 1534 (1988)
【非特許文献6】Nat. Biotechnol., 14, 845 (1996)
【非特許文献7】Nat. Genet., 15, 146 (1997)
【非特許文献8】Nat. Genet.,16, 133 (1997)
【非特許文献9】Proc. Natl. Acad. Sci. U.S.A.. 97, 722 (2000)
【非特許文献10】J. Mol. Biol., 222, 581 (1991)
【非特許文献11】Biotechnology (N Y), 11, 1145 (1993)
【非特許文献12】Blood 86, 4430 (1995)
【非特許文献13】Semin. Oncol., 30, 1 (2003)
【非特許文献14】Expert Rev. Anticancer Ther., 3, 767 (2003)
【非特許文献15】Breast J., 9, 452 (2003)
【非特許文献16】Trends Mol. Med., 8, S19 (2002)
【非特許文献17】Blood 99, 754 (2002)
【非特許文献18】Cancer Res., 64, 4664 (2004)
【非特許文献19】Arthritis Rheum., 48, 455 (2003)
【非特許文献20】J. Clin. Oncol., 21, 3940 (2003)
【非特許文献21】Clin. Cancer Res., 10, 5650 (2004)
【非特許文献22】Nat. Biotechnol., 23, 1073 (2005)
【非特許文献23】J. Exp. Med., 183, 1259 (1996)
【非特許文献24】Cancer Immunol. Immunother., 48, 443 (1999)
【発明の開示】
【発明が解決しようとする課題】
【0006】
ヒトでは生体防御のために高度に分化した免疫系が構築されており、この免疫系を利用した抗体医薬は、本来、高い薬理効果が期待できる。どのようなヒト抗体が高い薬理効果を発揮するのかをスクリーニングし選択するには、適切なヒト疾患モデルを用いた評価が欠かせない。このようなヒト疾患モデルが構築できる、種差の壁を越えヒトの免疫系を再現しうる非ヒト動物の開発が、有効な抗体医薬開発の観点から望まれる。本発明は、ヒトCD16とそのヒトアクセサリー分子とを共発現させるようにゲノムが改変された、非ヒト動物またはその子孫及びその利用方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明は、以下の(1)〜(16)に関する。
(1)ヒトCD16とそのヒトアクセサリー分子とを共発現させるようにゲノムが改変された、非ヒト動物またはその子孫。
(2)ヒトCD16のヒトアクセサリー分子が、以下の(a)〜(c) からなる群から選ばれる分子である、(1)に記載の非ヒト動物またはその子孫。
(a) ヒトCD3ζ鎖;
(b) ヒトFcε受容体Iγ鎖;
(c) ヒトCD3ζ鎖とヒトFcε受容体Iγ鎖。
(3)ヒトCD16またはそのヒトアクセサリー分子の発現が、ヒトCD16プロモーター領域で制御されるようにゲノムが改変された、(1)または(2)に記載の非ヒト動物またはその子孫。
(4)ヒトCD16が、以下の (a) 〜(d)からなる群から選ばれるDNAがコードする蛋白質である、(1)〜(3)のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号1で表される塩基配列からなるDNA;
(b) 配列番号2で表される塩基配列からなるDNA;
(c) 配列番号1で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒト抗体のFc領域と結合する活性を有する蛋白質をコードするDNA;
(d) 配列番号2で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒト抗体のFc領域と結合する活性を有する蛋白質をコードするDNA。
(5)ヒトCD16が、以下の(a) 〜(f)からなる群から選ばれる蛋白質である、(1)〜(3)のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号3で表されるアミノ酸配列からなる蛋白質;
(b) 配列番号4で表されるアミノ酸配列からなる蛋白質;
(c) 配列番号3で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(d) 配列番号4で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(e) 配列番号3で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(f) 配列番号4で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質。
(6)ヒトCD3ζ鎖が、以下の (a)または(b)であるDNAがコードする蛋白質である、(2)〜(5)のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号5で表される塩基配列からなるDNA;
(b) 配列番号5で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質をコードするDNA。
(7)ヒトCD3ζ鎖が、以下の(a) 〜(c)からなる群から選ばれる蛋白質である、(2)〜(5)のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号6で表されるアミノ酸配列からなる蛋白質;
(b) 配列番号6で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質;
(c) 配列番号6で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質。
(8)ヒトFcε受容体Iγ鎖が、以下の (a)または(b)から選ばれるDNAがコードする蛋白質である、(2)〜(7)のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号7で表される塩基配列からなるDNA;
(b) 配列番号7で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質をコードするDNA。
(9)ヒトFcε受容体Iγ鎖が、以下の(a) 〜(c)からなる群から選ばれる蛋白質である、(2)〜(7)のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号8で表されるアミノ酸配列からなる蛋白質;
(b) 配列番号8で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質;
(c) 配列番号8で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質。
(10)ヒトCD16プロモーター領域が、以下の(a)または(b)から選ばれるDNAである、(3)〜(9)のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号9で表される塩基配列からなるDNA;
(b) 配列番号9で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつナチュラルキラー細胞、マクロファージ、単球または樹状細胞内でヒトCD16またはそのヒトアクセサリー分子を発現させる活性を有するDNA;
(11)非ヒト動物が、ウシ、ヒツジ、ヤギ、ブタ、ウマ、マウス、ラット、ニワトリ、サル及びウサギからなら群から選ばれる動物である、(1)〜(10)のいずれか1項に記載の非ヒト動物またはその子孫。
(12)以下の(1)〜(3)からなる工程を含むことを特徴とする、抗体の抗体依存性細胞傷害活性を測定する方法。
(1) (1)〜(11)のいずれか1項に記載の非ヒト動物またはその子孫より、エフェクター細胞を単離する工程;
(2) 単離したエフェクター細胞と、抗体及び該抗体の標的となる抗原を発現している細胞とを混合し、反応液中でエフェクター細胞と、抗体及び該抗体の標的となる抗原を発現している細胞とを接触させる工程;
(3)反応液中の該標的となる抗原を発現している細胞の生存率を定量する工程。
(13)以下の(1)および(2)からなる工程を含むことを特徴とする、抗体の抗体依存性細胞傷害活性に基づく薬理活性を評価する方法。
(1) (1)〜(11)のいずれか1項に記載の非ヒト動物またはその子孫に、被検物質である抗体を投与する工程;
(2) 該非ヒト動物またはその子孫において被検物質である抗体の抗体依存性細胞傷害活性に基づく薬理活性を評価する工程。
(14)以下の(1)〜(3)からなる工程を含むことを特徴とする、抗体のスクリーニング方法。
(1)(1)〜(11)のいずれか1項に記載の非ヒト動物またはその子孫に被検物質である抗体を投与する工程;
(2) 該非ヒト動物またはその子孫において被検物質である抗体の抗体依存性細胞傷害活性に基づく薬理活性を評価する工程;
(3) 該薬理活性を有する抗体を選択する工程。
(15)抗体が、以下の(a)〜(e)からなる群から選ばれる蛋白質である、(12)〜(14)のいずれか1項に記載の方法。
(a) ヒト抗体;
(b) ヒト化抗体;
(c) ヒト型キメラ抗体
(d) (a)、(b)または(c)のFc領域を含む抗体断片;
(e) (a)、(b)または(c)のFc領域を有する融合蛋白質。
(16)抗体のクラスがIgGである、(15)に記載の方法。
【発明の効果】
【0008】
本発明により、ヒトCD16とそのヒトアクセサリー分子とを共発現させるようにゲノムが改変された、非ヒト動物またはその子孫及びその利用方法が提供される。本発明の非ヒト動物及びその子孫は、ヒト抗体医薬の生物活性及び薬理評価に有用である。
【発明を実施するための最良の形態】
【0009】
本発明の非ヒト動物またはその子孫とは、ヒトCD16とそのヒトアクセサリー分子とを共発現させるように、ゲノムが改変された非ヒト動物またはその子孫であれば、いかなる非ヒト動物またはその子孫も包含される。
本発明において、ヒトCD16とは、ヒト抗体のFc領域と結合する活性を有する蛋白質を意味する。ヒトCD16には遺伝的多型が存在することが知られており、具体的には、ヒトCD16シグナル配列のN末端メチオニンから176番目のアミノ酸残基がフェニルアラニン(以下、ヒトCD16 Pheと称す)である蛋白質と、バリン(以下、ヒトCD16 Valと称す)である蛋白質の存在が知られている。したがって、ヒトにおいては、ヒトCD16 Phe/ヒトCD16 PheあるいはヒトCD16 Val/ヒトCD16 Valのホモ型、ヒトCD16 Phe/ヒトCD16 Valのヘテロ型の3種類の表現型が存在する。本発明において、ヒトCD16とは、いずれの多型も包含する。
【0010】
本発明におけるヒトCD16としては、具体的には、下記(a)、(b)、(c)または(d)のDNAがコードする蛋白質、または下記(e)、(f)、(g)、(h)、(i)または(j)の蛋白質があげられる。
(a) 配列番号1で表される塩基配列からなるDNA;
(b) 配列番号2で表される塩基配列からなるDNA;
(c) 配列番号1で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒト抗体のFc領域と結合する活性を有する蛋白質をコードするDNA;
(d) 配列番号2で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒト抗体のFc領域と結合する活性を有する蛋白質をコードするDNA;
または、
(e) 配列番号3で表されるアミノ酸配列からなる蛋白質;
(f) 配列番号4で表されるアミノ酸配列からなる蛋白質;
(g) 配列番号3で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(h) 配列番号4で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(i) 配列番号3で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(j) 配列番号4で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質。
【0011】
本発明において、ストリンジェントな条件下でハイブリダイズするDNAとは、例えば配列番号1または2で表される塩基配列を有するDNAなどのDNAまたはその一部の断片をプローブとして、コロニー・ハイブリダイゼーション法、プラーク・ハイブリダイゼーション法あるいはサザンブロットハイブリダイゼーション法等を用いることにより得られるDNAを意味し、具体的には、コロニーあるいはプラーク由来のDNAを固定化したフィルターを用いて、0.7〜1.0Mの塩化ナトリウム存在下、65℃でハイブリダイゼーションを行った後、0.1〜2倍濃度のSSC溶液(1倍濃度のSSC溶液の組成は、150mM塩化ナトリウム、15mMクエン酸ナトリウムよりなる)を用い、65℃条件下でフィルターを洗浄することにより同定できるDNAをあげることができる。ハイブリダイゼーションは、Molecular Cloning, A Laboratory Manual, Second Edition, Cold Spring Harbor Laboratory Press, 1989(以下、モレキュラー・クローニング第2版と略す)、Current Protocols in Molecular Biology, John Wiley & Sons, 1987-1997(以下、カレント・プロトコールズ・イン・モレキュラー・バイオロジーと略す)、DNA Cloning 1: Core Techniques, A Practical Approach, Second Edition, Oxford University (1995)等に記載されている方法に準じて行うことができる。ハイブリダイズ可能なDNAとして具体的には、配列番号1または2で表される塩基配列と少なくとも60%以上の相同性を有するDNA、好ましくは70%以上、より好ましくは80%以上、さらに好ましくは90%以上、特に好ましくは95%以上、最も好ましくは98%以上の相同性を有するDNAをあげることができる。
【0012】
本発明において、配列番号3または4で表されるアミノ酸配列において1以上のアミノ酸が欠出、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質とは、モレキュラー・クローニング第2版、カレント・プロトコールズ・イン・モレキュラー・バイオロジー、Nucleic Acids Research, 10, 6487 (1982)、Proc. Natl. Acad. Sci., USA,79, 6409(1982)、Gene, 34, 315 (1985)、Nucleic Acids Research, 13, 4431 (1985)、Proc. Natl. Acad. Sci USA,82, 488 (1985)等に記載の部位特異的変異導入法を用いて、例えば、配列番号3または4で表されるアミノ酸配列を有する蛋白質をコードするDNAに部位特異的変異を導入することにより取得することができる蛋白質を意味する。欠出、置換、挿入および/または付加されるアミノ酸の数は1個以上でありその数は特に限定されないが、上記の部位特異的変異導入法等の周知の技術により、欠失、置換もしくは付加できる程度の数であり、例えば、1〜数十個、好ましくは1〜20個、より好ましくは1〜10個、さらに好ましくは1〜5個である。
【0013】
また、本発明において、配列番号3または4で表されるアミノ酸配列と80%以上の相同性を有し、かつヒト抗体のFc領域と結合する活性を有する蛋白質とは、BLAST〔J. Mol. Biol., 215, 403 (1990)〕やFASTA〔Methods in Enzymology, 183, 63 (1990)〕等の解析ソフトを用いて計算したときに、配列番号3または4に記載のアミノ酸配列を有する蛋白質と少なくとも80%以上、好ましくは85%以上、より好ましくは90%以上、さらに好ましくは95%以上、特に好ましくは97%以上、最も好ましくは99%以上である蛋白質である。
【0014】
本発明において、ヒトCD16のアクセサリー分子とは、ヒト抗体のFc領域が結合したヒトCD16からのシグナルを細胞内に伝達することができる蛋白質を意味する。ヒトCD16からのシグナルを細胞内に伝達することができる蛋白質であればいずれの分子でも包含されるが、具体的には、ヒトCD3ζ鎖、ヒトFcε受容体Iγ鎖またはそれらの組合わせなどが挙げられる。
【0015】
本発明におけるヒトCD3ζ鎖としては、具体的には、下記(a)または(b)のDNAがコードする蛋白質、または下記(c)、(d)または(e)の蛋白質があげられる。
(a) 配列番号5で表される塩基配列からなるDNA;
(b) 配列番号5で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質をコードするDNA;
または、
(c) 配列番号6で表されるアミノ酸配列からなる蛋白質;
(d) 配列番号6で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質;
(e) 配列番号6で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質。
【0016】
本発明におけるヒトFcε受容体Iγ鎖としては、具体的には、下記(a)または(b)のDNAがコードする蛋白質、または下記(c)、(d)または(e)の蛋白質があげられる。
(a) 配列番号7で表される塩基配列からなるDNA;
(b) 配列番号7で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質をコードするDNA;
または、
(c) 配列番号8で表されるアミノ酸配列からなる蛋白質;
(d) 配列番号8で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質;
(e) 配列番号8で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質。
【0017】
本発明において、ストリンジェントな条件下でハイブリダイズするDNAとは、例えば配列番号5または7で表される塩基配列を有するDNAなどのDNAまたはその一部の断片をプローブとして、コロニー・ハイブリダイゼーション法、プラーク・ハイブリダイゼーション法あるいはサザンブロットハイブリダイゼーション法等を用いることにより得られるDNAを意味し、具体的には、コロニーあるいはプラーク由来のDNAを固定化したフィルターを用いて、0.7〜1.0Mの塩化ナトリウム存在下、65℃でハイブリダイゼーションを行った後、0.1〜2倍濃度のSSC溶液(1倍濃度のSSC溶液の組成は、150mM塩化ナトリウム、15mMクエン酸ナトリウムよりなる)を用い、65℃条件下でフィルターを洗浄することにより同定できるDNAをあげることができる。ハイブリダイゼーションは、モレキュラー・クローニング第2版、カレント・プロトコールズ・イン・モレキュラー・バイオロジー、DNA Cloning 1: Core Techniques, A Practical Approach, Second Edition, Oxford University (1995)等に記載されている方法に準じて行うことができる。ハイブリダイズ可能なDNAとして具体的には、配列番号5または7で表される塩基配列と少なくとも60%以上の相同性を有するDNA、好ましくは70%以上、より好ましくは80%以上、さらに好ましくは90%以上、特に好ましくは95%以上、最も好ましくは98%以上の相同性を有するDNAをあげることができる。
【0018】
本発明において、配列番号6または8で表されるアミノ酸配列において1以上のアミノ酸が欠出、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質とは、モレキュラー・クローニング第2版、カレント・プロトコールズ・イン・モレキュラー・バイオロジー、Nucleic Acids Research, 10, 6487 (1982)、Proc. Natl. Acad. Sci., USA,79, 6409(1982)、Gene, 34, 315 (1985)、Nucleic Acids Research, 13, 4431 (1985)、Proc. Natl. Acad. Sci USA,82, 488 (1985)等に記載の部位特異的変異導入法を用いて、例えば、配列番号6または8で表されるアミノ酸配列を有する蛋白質をコードするDNAに部位特異的変異を導入することにより取得することができる蛋白質を意味する。欠出、置換、挿入および/または付加されるアミノ酸の数は1個以上でありその数は特に限定されないが、上記の部位特異的変異導入法等の周知の技術により、欠失、置換もしくは付加できる程度の数であり、例えば、1〜数十個、好ましくは1〜20個、より好ましくは1〜10個、さらに好ましくは1〜5個である。
【0019】
また、本発明において、配列番号6または8で表されるアミノ酸配列と80%以上の相同性を有し、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質とは、BLAST〔J. Mol. Biol., 215, 403 (1990)〕やFASTA〔Methods in Enzymology, 183, 63 (1990)〕等の解析ソフトを用いて計算したときに、配列番号6または8に記載のアミノ酸配列を有する蛋白質と少なくとも80%以上、好ましくは85%以上、より好ましくは90%以上、さらに好ましくは95%以上、特に好ましくは97%以上、最も好ましくは99%以上である蛋白質である。
【0020】
本発明の非ヒト動物またはその子孫としては、ヒトCD16の発現がヒトCD16プロモーター領域で制御されるようにゲノムが改変された、非ヒト動物またはその子孫があげられる。
ここで、ヒトCD16の発現がヒトCD16プロモーター領域で制御されるようにゲノムが改変されたとは、ヒトCD16をコードする遺伝子が、ヒト染色体上のヒトCD16遺伝子の発現を制御するゲノム領域の制御下に位置するように改変することを意味する。ヒトCD16遺伝子の発現を制御するゲノム領域の制御下に位置するように改変するとは、具体的には、ヒト染色体上のヒトCD16遺伝子の発現を制御するゲノム領域の下流に、ヒトCD16をコードする遺伝子が位置するように改変する例が挙げられる。このような非ヒト動物またはその子孫を取得する方法としては、目的とするゲノムの改変を行うことができれば、いずれの手法でも用いることができる。具体的には、目的とした遺伝子改変を施すDNAを受精卵や胚性幹細胞に導入し個体を作製する方法、目的とした遺伝子改変を施した細胞の核を用いたクローン個体の作製の手法などがあげられる。
【0021】
本発明において、ヒトCD16プロモーター領域とは、ヒト染色体上においてヒトCD16遺伝子の組織特異的な発現制御に関与している領域であればいかなる領域も包含される。ヒトCD16は、ナチュラルキラー細胞、マクロファージ、単球及び樹状細胞での発現が観察されており[Nat. Rev. Immunol., 4, 89 (2004); Nat. Rev. Immunol., 2, 580 (2002)]、ヒトCD16プロモーター領域には少なくともこれらの細胞内で制御下にある遺伝子を発現させる活性を有している。
【0022】
本発明におけるヒトCD16プロモーター領域としては、具体的には、下記(a)、(b)または(c)のDNAがあげられる。
(a) 配列番号9で表される塩基配列からなるDNA;
(b) 配列番号9で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒトナチュラルキラー細胞で制御下にある遺伝子を発現させることが可能な活性を有するDNA;
(c) ナチュラルキラー細胞、マクロファージ、単球及び樹状細胞特異的に制御下にある遺伝子を発現させることが可能な活性を有するDNA。
【0023】
本発明において、抗体とは、抗体のFc領域を含む分子であればいかなる分子も包含されるが、ヒト由来の抗体が好ましい。具体的には、ヒト抗体、ヒト化抗体、ヒト型キメラ抗体、それらの抗体のFc領域を含む抗体断片、それらの抗体のFc領域と他の蛋白質とを融合させた該Fc領域を有する融合蛋白質などがあげられる。
ヒト抗体としては、抗原に感作したヒト脾臓細胞より作製したハイブリドーマ細胞が分泌する抗体のほかにも、遺伝子組換え技術により作製されたヒト抗体、すなわち、抗体遺伝子を挿入した抗体発現ベクターを、宿主細胞へ導入することにより取得されたヒト抗体などがあげられる。
【0024】
ヒト化抗体
ヒト型キメラ抗体は、ヒト以外の動物の抗体H鎖V領域(以下、「HV」または「VH」とも称す)および抗体L鎖V領域(以下、「LV」または「VL」とも称す)とヒト抗体のH鎖C領域(以下、「CH」とも称す)およびヒト抗体のL鎖C領域(以下、「CL」とも称す)とからなる抗体を意味する。ヒト以外の動物としては、マウス、ラット、ハムスター、ラビット等、ハイブリドーマを作製することが可能であれば、いかなるものも用いることができる。
【0025】
ヒト型キメラ抗体は、ヒト以外の哺乳動物に抗原を免疫して得られたB細胞と、マウス、ラット等に由来するミエローマ細胞とを細胞融合させて得られる、所望の抗原特異性を有したモノクローナル抗体を生産するハイブリドーマより、VHおよびVLをコードするcDNAを取得し、ヒト抗体CHおよびヒト抗体CLをコードする遺伝子を有する宿主細胞用発現ベクターにそれぞれ挿入してヒト型キメラ抗体発現ベクターを構築し、宿主細胞へ導入することにより発現させ、製造することができる。
【0026】
ヒト型キメラ抗体のCHとしては、ヒトイムノグロブリン(以下、「hIg」と表記する)に属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。また、ヒト型キメラ抗体のCLとしては、hIgに属すればいかなるものでもよく、κクラスあるいはλクラスのものを用いることができる。
【0027】
ヒト化抗体はヒト型CDR移植抗体ともいい、ヒト以外の動物の抗体のVHおよびVLのCDRのアミノ酸配列をヒト抗体のVHおよびVLの適切な位置に移植した抗体をいう。
ヒト化抗体は、ヒト以外の動物の抗体のVHおよびVLのCDR配列を任意のヒト抗体のVHおよびVLのCDR配列に移植したV領域をコードするcDNAを構築し、ヒト抗体のCHおよびヒト抗体のCLをコードする遺伝子を有する宿主細胞用発現ベクターにそれぞれ挿入してヒト化抗体発現ベクターを構築し、該発現ベクターを宿主細胞へ導入することによりヒト化抗体を発現させ、製造することができる。
【0028】
ヒト化抗体のCHとしては、hIgに属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。また、ヒト化抗体のCLとしては、hIgに属すればいかなるものでもよく、κクラスあるいはλクラスのものを用いることができる。
ヒト抗体は、元来、ヒト体内に天然に存在する抗体をいうが、最近の遺伝子工学的、細胞工学的、発生工学的な技術の進歩により作製されたヒト抗体ファージライブラリーならびにヒト抗体トランスジェニック動物あるいはヒト抗体トランスジェニック植物から得られる抗体等も含まれる。
【0029】
ヒト体内に存在する抗体は、例えば、ヒト末梢血リンパ球を単離し、EBウイルス等を感染させ不死化、クローニングすることにより、該抗体を生産するリンパ球を培養でき、培養物中より該抗体を精製することができる。
ヒト抗体ファージライブラリーは、ヒトB細胞から調製した抗体遺伝子をファージ遺伝子に挿入することによりFab、一本鎖抗体等の抗体断片をファージ表面に発現させたライブラリーである。該ライブラリーより、抗原を固定化した基質に対する結合活性を指標として所望の抗原結合活性を有する抗体断片を発現しているファージを回収することができる。該抗体断片は、更に遺伝子工学的手法により、2本の完全なH鎖および2本の完全なL鎖からなるヒト抗体分子へも変換することができる。
【0030】
ヒト抗体トランスジェニック非ヒト動物は、ヒト抗体遺伝子が細胞内に組込まれた動物をいう。具体的には、胚性幹細胞へヒト抗体遺伝子を導入し、該胚性幹細胞を他の初期胚へ移植後、発生させることによりヒト抗体を産生するトランスジェニック非ヒト動物を作製することができる。また、動物の受精卵にヒト抗体遺伝子を導入し、該受精卵を発生させることにヒト抗体を産生するトランスジェニック非ヒト動物を作製することもできる。ヒト抗体を産生するトランスジェニック非ヒト動物からのヒト抗体の作製方法は、通常のヒト以外の哺乳動物で行われているハイブリドーマ作製方法によりヒト抗体ハイブリドーマを得、培養することで培養物中にヒト抗体を蓄積させることができる。
【0031】
トランスジェニック非ヒト動物は、ウシ、ヒツジ、ヤギ、ブタ、ウマ、マウス、ラット、ニワトリ、サル又はウサギ等があげられる。
また、本発明において、抗体は、腫瘍関連抗原を認識する抗体、アレルギーあるいは炎症に関連する抗原を認識する抗体、循環器疾患に関連する抗原を認識する抗体、自己免疫疾患に関連する抗原を認識する抗体、またはウイルスあるいは細菌感染に関連する抗原を認識する抗体であることが好ましく、抗体のクラスはIgGが好ましい。
【0032】
抗体の断片は、上記抗体のFc領域を含んだ断片いう。抗体の断片としては、上記抗体のFc領域を含んだ断片であればいかなるものでもよいが、具体的には、H鎖の単量体、H鎖の2量体などがあげられる。
Fc領域を有する融合蛋白質としては、抗体のFc領域を含んだ抗体あるいは抗体断片と、酵素、サイトカインなどの蛋白質とを融合させた物質であればいかなるものでもよい。
【0033】
以下、本発明の非ヒト動物及びその子孫の作出方法と利用方法について詳細に説明する。
本発明の、ヒトCD16とそのヒトアクセサリー分子とを共発現させるようにゲノムが改変された非ヒト動物またはその子孫は、1)目的とする遺伝子を動物個体に導入するトランスジェニック動物の作製方法、2)標的遺伝子と目的とする導入遺伝子とを入れ換えた遺伝子置換動物の作製方法、3)目的とした遺伝子改変を施した細胞の核を用いたクローン個体の作製方法を用いることにより、作製することができる。以下にそれぞれ具体的に説明する。
1.目的とする遺伝子を動物個体に導入するトランスジェニック動物の作製方法を用いた、本発明の非ヒト動物の作製
(1)ヒトCD16をコードするDNAの取得
ヒトCD16をコードするDNAとしては、ヒトCD16をコードするcDNAおよびゲノムDNAをあげることができる。ゲノムDNAはイントロンを含んでいてもよい。また、コード領域のコドンは、cDNAおよびゲノムDNAにおけるコドンに限られるものでなく、ヒトCD16のアミノ酸配列をコードするコドンであれば、いかなるコドンの組み合わせでもよい。ヒトCD16をコードするDNAの例としては、配列番号1または2に示す塩基配列からなるヒトCD16のcDNAをあげることができる。
【0034】
塩基配列データベースより、ヒトCD16cDNAの配列、またはヒトCD16遺伝子のゲノム配列を検索し、その塩基配列情報を取得する。塩基配列データベースから取得できるヒトCD16遺伝子のゲノム配列としては、例えば、GenBank登録番号NC_000001およびAL_590385で登録されている塩基配列、ヒトCD16cDNAの配列としては、例えば、GenBank登録番号NM_000569で登録されている塩基配列をあげることができる。
【0035】
得られたヒトCD16をコードするDNAの塩基配列情報に基づいて、ヒトCD16をコードする領域を含むcDNAの配列またはゲノム配列の領域を適当に選択し、この領域の5'端20〜40bpの配列を3'端に含むDNAおよび、この領域の3'端20〜40bpの配列と相補的な配列を3'端に含むDNAを作製する。これらのDNAをプライマーとして用い、ヒトcDNAを鋳型としたPCRにより、ヒトCD16をコードするcDNAを、ヒトゲノムDNAを鋳型としたPCRにより、ヒトCD16をコードするゲノムDNAを単離することができる。
【0036】
ヒトcDNAは、ヒトの組織または細胞から調製したRNAから作製できる。組織または細胞から全RNAを調製する方法としては、チオシアン酸グアニジン−トリフルオロ酢酸セシウム法(Methods in Enzymology,154, 3, 1987)、酸性チオシアン酸グアニジン・フェノール・クロロホルム法(Analytical Biochemistry, 162, 156, 1987; 実験医学, 9, 1937, 1991)などがあげられる。また、アブソリュートリーRNA RT-PCRミニプレップ・キット(Absolutely RNA RT-PCR Miniprep Kit、ストラタジーン社製)、RNイージー・ミニ・キット(RNeasy MIni Kit、キアゲン社製)等のキットを用いることによっても調製することができる。全RNAからポリ(A)+ RNAとしてmRNAを調製する方法としては、オリゴ(dT)固定化セルロースカラム法〔Molecular Cloning: A Laboratory Manual, 3rd Edition, Cold Spring Harbor Laboratory Press, 2001(以下、モレキュラー・クローニング第3版と略す)〕等があげられる。さらに、ファーストトラック2.0mRNA分離キット(FastTrack 2.0 mRNA Isolation Kit、インビトロジェン社製)、クィックプレップmRNA精製キット(QuickPrep mRNA Purification Kit、アマシャム・バイオサイエンス社製)等のキットを用いることによりヒト組織や細胞からmRNAを調製することができる。全RNAまたはmRNAからcDNAを合成する方法としてはモレキュラー・クローニング第3版、カレント・プロトコールズ・イン・モレキュラー・バイオロジー等に記載された方法、あるいは市販のキット、例えばRT-PCR用スーパースクリプト第一鎖合成システム(SUPERSCRIPT First-Strand Synthesis System for RT-PCR、インビトロジェン社製)、プロスター第一鎖RT-PCRキット(ProSTAR First Strand RT-PCR Kit、ストラタジーン社製)、PCR−セレクトcDNAサブトラクション・キット(PCR-Select cDNA Subtraction Kit、クロンテック社製)を用いる方法などがあげられる。また、クロンテック社等から市販されているヒトの各種組織のcDNAを用いることもできる。このようにして得られたヒトCD16をコードするDNAの例として、配列番号1または2に示す塩基配列を有するDNAをあげることができる。
【0037】
ヒトゲノムDNAは、ヒトの組織または細胞から、モレキュラー・クローニング第3版に記載の方法により調製することができる。または、イージーDNAキット(Easy-DNA Kit、インビトロジェン社製)、DNA抽出キット(DNA Extraction Kit、ストラタジーン社製)等のキットを用いることにより、調製できる。クロンテック社等から市販されているヒトのゲノムDNAを用いることもできる。
(2)ヒトCD3ζ鎖をコードするDNAの取得
ヒトCD3ζ鎖をコードするDNAとしては、ヒトCD3ζ鎖をコードするcDNAおよびゲノムDNAをあげることができる。ゲノムDNAはイントロンを含んでいてもよい。また、コード領域のコドンは、cDNAおよびゲノムDNAにおけるコドンに限られるものでなく、ヒトCD3ζ鎖のアミノ酸配列をコードするコドンであれば、いかなるコドンの組み合わせでもよい。ヒトCD3ζ鎖をコードするDNAの例としては、配列番号5に示す塩基配列からなるヒトCD3ζ鎖のcDNAをあげることができる。
【0038】
塩基配列データベースより、ヒトCD3ζ鎖cDNAの配列、またはヒトCD3ζ鎖遺伝子のゲノム配列を検索し、その塩基配列情報を取得する。塩基配列データベースから取得できるヒトCD3ζ鎖遺伝子のゲノム配列としては、例えば、GenBank登録番号NC_000001で登録されている塩基配列、ヒトCD3ζ鎖cDNAの配列としては、例えば、GenBank登録番号NM_000734で登録されている塩基配列をあげることができる。
【0039】
得られたヒトCD3ζ鎖をコードするDNAの塩基配列情報に基づいて、ヒトCD3ζ鎖をコードする領域を含むcDNAの配列またはゲノム配列の領域を適当に選択し、この領域の5'端20〜40bpの配列を3'端に含むDNAおよび、この領域の3'端20〜40bpの配列と相補的な配列を3'端に含むDNAを作製する。これらのDNAをプライマーとして用い、ヒトcDNAを鋳型としたPCRにより、ヒトCD3ζ鎖をコードするcDNAを、ヒトゲノムDNAを鋳型としたPCRにより、ヒトCD3ζ鎖をコードするゲノムDNAを単離することができる。
【0040】
ヒトcDNAおよびヒトゲノムDNAは、上述の1.の(1)に記載の方法にしたがって調製することができる。このようにして得られたヒトCD3ζ鎖をコードするDNAの例として、配列番号5に示す塩基配列を有するDNAをあげることができる。
(3)ヒトFcε受容体Iγ鎖をコードするDNAの取得
ヒトFcε受容体Iγ鎖をコードするDNAとしては、ヒトFcε受容体Iγ鎖をコードするcDNAおよびゲノムDNAをあげることができる。ゲノムDNAはイントロンを含んでいてもよい。また、コード領域のコドンは、cDNAおよびゲノムDNAにおけるコドンに限られるものでなく、ヒトFcε受容体Iγ鎖のアミノ酸配列をコードするコドンであれば、いかなるコドンの組み合わせでもよい。ヒトFcε受容体Iγ鎖をコードするDNAの例としては、配列番号7に示す塩基配列からなるヒトFcε受容体Iγ鎖のcDNAをあげることができる。
【0041】
塩基配列データベースより、ヒトFcε受容体Iγ鎖cDNAの配列、またはヒトFcε受容体Iγ鎖遺伝子のゲノム配列を検索し、その塩基配列情報を取得する。塩基配列データベースから取得できるヒトFcε受容体Iγ鎖遺伝子のゲノム配列としては、例えば、GenBank登録番号NC_000001で登録されている塩基配列、ヒトFcε受容体Iγ鎖cDNAの配列としては、例えば、GenBank登録番号NM_004106で登録されている塩基配列をあげることができる。
【0042】
得られたヒトFcε受容体Iγ鎖をコードするDNAの塩基配列情報に基づいて、ヒトFcε受容体Iγ鎖をコードする領域を含むcDNAの配列またはゲノム配列の領域を適当に選択し、この領域の5'端20〜40bpの配列を3'端に含むDNAおよび、この領域の3'端20〜40bpの配列と相補的な配列を3'端に含むDNAを作製する。これらのDNAをプライマーとして用い、ヒトcDNAを鋳型としたPCRにより、ヒトFcε受容体Iγ鎖をコードするcDNAを、ヒトゲノムDNAを鋳型としたPCRにより、ヒトFcε受容体Iγ鎖をコードするゲノムDNAを単離することができる。
【0043】
ヒトcDNAおよびヒトゲノムDNAは、上述の1.の(1)に記載の方法にしたがって調製することができる。このようにして得られたヒトFcε受容体Iγ鎖をコードするDNAの例として、配列番号7に示す塩基配列を有するDNAをあげることができる。
(4)ヒトCD16プロモーター領域の取得
ヒトCD16プロモーター領域としては、ヒト染色体上においてヒトCD16遺伝子の組織特異的な発現制御に関与しているゲノムDNAをあげることができる。具体的には、ヒトCD16遺伝子の組織特異的な発現制御を担う活性を有する、ヒトCD16遺伝子の開始コドンから上流10kb以内の領域のゲノムDNAをあげることができる。ヒトCD16プロモーター領域の例としては、配列番号9に示す塩基配列からなるヒトCD16プロモーター領域をあげることができる。
【0044】
塩基配列データベースより、ヒトCD16cDNAの配列、またはヒトCD16遺伝子のゲノム配列を検索し、その塩基配列情報を取得する。塩基配列データベースから取得できるヒトCD16遺伝子のゲノム配列としては、例えば、GenBank登録番号NC_000001およびAL_590385で登録されている塩基配列、ヒトCD16cDNAの配列としては、例えば、GenBank登録番号NM_000569で登録されている塩基配列をあげることができる。
【0045】
得られたヒトCD16をコードするDNAの塩基配列情報に基づいて、ヒトCD16をコードする領域を含むcDNAの配列またはゲノム配列の領域を適当に選択し、この領域の5'端20〜40bpの配列を3'端に含むDNAおよび、この領域の3'端20〜40bpの配列と相補的な配列を3'端に含むDNAを作製する。これらのDNAをプライマーとして用い、ヒトゲノムDNAを鋳型としたPCRにより、ヒトCD16遺伝子の開始コドンから上流10kb以内の領域のゲノムDNAを数kbずつ増幅し単離することができる。
【0046】
ヒトゲノムDNAは、上述の1.の(1)に記載の方法にしたがって調製することができる。また、ゲノムDNAライブラリースクリーニングシステム(Genome Systems社製)やUniversal GenomeWalkerTM Kits(CLONTECH社製)などを用いることにより、ヒトCD16プロモーター領域を単離することもできる。このようにして得られたヒトCD16プロモーター領域の例として、配列番号9に示す塩基配列を有するDNAをあげることができる。
(5)ヒトCD16とそのヒトアクセサリー分子をコードするDNAを含む導入遺伝子の作製
上述の1.の(1)で得られたヒトCD16をコードするDNAと、上述の1.の(2)及び1.の(3)で得られたヒトCD16のヒトアクセサリー分子をコードするDNAを非ヒト動物に導入する際には、適当なプロモーターの下流に該DNAを連結した発現ベクターを導入遺伝子として用いるのが好ましい。具体的には、ヒトCD16とそのヒトアクセサリー分子のそれぞれを適当なプロモーターの下流に連結した発現ベクターを導入遺伝子として用いても良いし、ヒトCD16とそのヒトアクセサリー分子を配列内リボソーム進入部位(internal ribosome entry site; IRES)で連結し一つのプロモーターの下流に連結した発現ベクターを導入遺伝子として用いても良い。
【0047】
発現ベクターに用いられるプロモーターとしては、ヒトCD16とそのヒトアクセサリー分子をコードするDNAを導入する非ヒト動物の細胞内で転写を開始できるプロモーターであればいずれも用いることができる。例えばウイルス(サイトメガロウイルス、モロニー白血病ウイルス、JCウイルス、乳癌ウイルス、シミアンウイルス、レトロウイルスなど)由来遺伝子のプロモーター、各種哺乳動物(ヒト、ウサギ、イヌ、ネコ、モルモット、ハムスター、ラット、マウスなど)および鳥類(ニワトリなど)由来遺伝子〔例えば、アルブミン、インスリンII、エリスロポエチン、エンドセリン、オステオカルシン、筋クレアチンキナーゼ、血小板由来成長因子β、ケラチンK1、K10およびK14、コラーゲンI型およびII型、心房ナトリウム利尿性因子、ドーパミンβ−水酸化酵素、内皮レセプターチロシンキナーゼ(Tie2)、ナトリウムカリウムアデノシン3リン酸化酵素(Na,K-ATPase)、ニューロフィラメント軽鎖、メタロチオネインIおよびIIA、メタロプロテイナーゼ1組織インヒビター(TIMP1)、MHCクラスI抗原(H-2L)、平滑筋αアクチン、ポリペプチド鎖延長因子1α(EF-1α)、βアクチン、αおよびβミオシン重鎖、ミオシン軽鎖1および2、ミエリン塩基性タンパク、血清アミロイドPコンポーネント、ミオグロビン、レニン、ホスホグリセリン酸キナーゼ(PGK)等の解糖系酵素、ヒートショック蛋白質などの遺伝子〕のプロモーター、上記遺伝子のエンハンサー配列やプロモーター配列を融合させたプロモーター〔例えば、シミアンウイルス40(SV40)の初期遺伝子のプロモーターとヒトT細胞白血病ウイルス1のロング・ターミナル・リピートの一部の配列からなるSRαプロモーター、サイトメガロウイルスの前初期(IE)遺伝子エンハンサーとニワトリβ−アクチンプロモーターからなるCAGプロモーターなど〕などがあげられるが、ヒトCD16遺伝子の組織特異的な発現制御と同じ転写制御活性を有するプロモーターを用いることが好ましく、特にヒトCD16遺伝子の自身のプロモーターを用いることが好ましい。その具体的な例としては、上述の1.の(4)で得られたヒトCD16プロモーター領域があげられる。ヒトCD16遺伝子組織特異的な発現を示すプロモーターを使用することにより、ヒトCD16とそのヒトアクセサリー分子をコードするDNAを導入したトランスジェニック非ヒト動物の特定の組織に発現させることができる。また、Cre-loxP系との組合せにより、導入した外来遺伝子の発現部位、発現時期あるいは発現量等を制御することもできる。発現ベクター中の構造遺伝子をコードするDNAを2つのloxP配列の間に挿入し、該発現ベクターを用いて(6)に後述する方法でトランスジェニック非ヒト動物を作製する。そして、該トランスジェニック非ヒト動物に、アデノウイルスベクターを利用してCreリコンビナーゼを発現させれば、Creリコンビナーゼ発現用のアデノウイルスを感染させた部位または時期に特異的にヒトCD16やそのヒトアクセサリー分子をコードするDNAが欠失し、発現させたい導入遺伝子の発現を抑制することができる。また、(6)に後述する方法でヒトCD16とそのヒトアクセサリー分子の発現ベクターに加えて、例えば組織特異的な発現を示すプロモーターを利用したCreリコンビナーゼ発現ベクターを導入してトランスジェニック非ヒト動物を作製することにより、Creリコンビナーゼが発現する組織でのみ、ヒトCD16やそのヒトアクセサリー分子の発現が抑制されたトランスジェニック非ヒト動物を作製することができる。
【0048】
発現ベクターは、ヒトCD16やそのヒトアクセサリー分子をコードするDNAの下流に、mRNAの3'末端のポリアデニル化に必要なポリアデニル化シグナルを有していることが好ましい。ポリアデニル化シグナルとしては、上記のウィルス由来、各種動物および鳥類由来の各遺伝子に含まれるポリアデニル化シグナル、例えば、SV40の後期遺伝子または初期遺伝子、ウサギβグロビン遺伝子、ウシ成長ホルモン遺伝子、ヒトCD16遺伝子等のポリアデニル化シグナルをあげることができる。
【0049】
その他、ヒトCD16やそのヒトアクセサリー分子をさらに高発現させるために、各遺伝子のスプライシングシグナル、エンハンサー領域、イントロンの一部をプロモーター領域の5'上流、プロモーター領域と翻訳領域間あるいは翻訳領域の3'下流に連結してもよい。以下、プロモーター、ヒトCD16やそのヒトアクセサリー分子をコードするDNAおよびポリアデニル化シグナルからなるDNA構築物をヒトCD16とそのヒトアクセサリー分子発現ユニットと呼ぶ。
【0050】
さらに発現ベクターには、大腸菌(Escherichia coli、以下E. coliと略す)を用いてベクターを調製するため、E. coliで複製可能な複製開始点およびE. coliの形質転換体の選択マーカーとなる内因性プロモーターを含む薬剤耐性遺伝子が必要である。薬剤耐性遺伝子としては、E. coli由来のアンピシリン耐性遺伝子(β−ラクタマーゼ遺伝子)、テトラサイクリン耐性遺伝子、カナマイシン耐性遺伝子等があげられる。E. coliでの複製可能な複製開始点としては、pBR322の複製開始点、colE1の複製開始点等があげられる。
【0051】
レトロウイルスを遺伝子の導入に用いる場合は、レトロウイルスベクターにヒトCD16とそのヒトアクセサリー分子発現ユニットを挿入した組換えレトロウイルスベクターを作製する。該組換えレトロウイルスベクターを、適切なパッケージング細胞に導入することにより、組換えレトロウイルスを生産させることができる。得られた組換えウイルスを受精卵等に感染させることにより、ウイルスベクターを導入することができる。パッケージング細胞への組換えレトロウイルスベクターの導入には、公知の遺伝子導入の手法(例えば、リン酸カルシウム法、電気パルス法、リポフェクション法、凝集法、マイクロインジェクション法、パーティクルガン法、DEAE−デキストラン法等)を用いることができる。
(6)受精卵等への発現ベクターの導入とトランスジェニック非ヒト動物の選択
本発明の非ヒト動物は、上述の1.の(5)で作製したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を対象となる非ヒト動物に導入することによって作製できる。具体的には、該遺伝子を対象となる非ヒト動物の受精卵、胚性幹細胞(以下、ES細胞と略す)、精子または未受精卵へ導入し、これらの細胞を用いて発生させた個体から、ヒトCD16とそのヒトアクセサリー分子をコードするDNAを含む遺伝子が胚芽細胞を含むすべての細胞の染色体上に組み込まれた個体を選択することにより、本発明の非ヒト動物を作出できる。作出したトランスジェニック非ヒト動物の胚芽細胞において該導入遺伝子が存在することは、作出動物の子孫がその胚芽細胞および体細胞の全てに該導入遺伝子を有することで確認することができる。個体の選択は、個体を構成する組織、例えば、血液組織、上皮組織、結合組織、軟骨組織、骨組織、筋組織、口腔内組織または骨格系組織の一部から調製したゲノムDNA上に該導入遺伝子が存在することをDNAレベルで確認することによって行われる。このようにして選択された個体は通常、相同染色体の片方に導入遺伝子を有するヘテロ接合体なので、ヘテロ接合体の個体同士を交配することにより、子孫の中から導入遺伝子を相同染色体の両方に持つホモ接合体動物を取得することができる。このホモ接合体の雌雄の動物を交配することにより、すべての子孫が該遺伝子を安定に保持するホモ接合体となるので、通常の飼育環境で、本発明の非ヒト動物を繁殖継代することができる。
(i)受精卵への遺伝子導入によるトランスジェニック非ヒト動物の作製
受精卵への遺伝子導入によるトランスジェニック非ヒト動物の作製は、Manipulating the Mouse Embryo A Laboratory Manual, Second Edition, Cold Spring Harbor Laboratory Press (1994)(以下、マニピュレーティング・マウス・エンブリオ第2版と略す)、Gene Targeting, A Practical Approach, IRL Press at Oxford University Press (1993)、バイオマニュアルシリーズ8 ジーンターゲッティング, ES細胞を用いた変異マウスの作製,羊土社 (1995)、発生工学実験マニュアル, トランスジェニック・マウスの作り方, 講談社 (1987)等に記載された方法により、作製することができる。
【0052】
ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を導入する受精卵は、同種の雄非ヒト動物と雌非ヒト動物を交配させることによって得られる。例えば、雄マウスと雌マウス、好ましくは近交系の雄マウスと雌マウスを交配することによりマウスの受精卵が得られ、雄ラットと雌ラット、好ましくはWistar系統の雄ラットとWistar系統の雌ラットを交配することによりラットの受精卵が得られる。受精卵は自然交配によっても得られるが、雌非ヒト動物の性周期を人工的に調節した後、雄非ヒト動物と交配させる方法が好ましい。雌非ヒト動物の性周期を人工的に調節する方法としては、例えば初めに卵胞刺激ホルモン、次いで黄体形成ホルモンを例えば腹腔注射などにより投与する方法が好ましいが、好ましいホルモンの投与量、投与間隔は非ヒト動物の種類によりそれぞれ異なる。非ヒト動物がマウスの場合は、雌マウスに卵胞刺激ホルモン投与後、約48時間後に黄体形成ホルモンを投与し、雄マウスと交配させることにより受精卵を得る方法が好ましく、卵胞刺激ホルモンの投与量は2〜20 IU/個体、好ましくは約5IU/個体、黄体形成ホルモンの投与量は0〜10 IU/個体、好ましくは約5IU/個体である。非ヒト動物がラットの場合は、雌ラットに卵胞刺激ホルモン投与後、約48時間後に黄体形成ホルモンを投与し、雄ラットと交配させることにより受精卵を得る方法が好ましく、卵胞刺激ホルモンの投与量は20〜50 IU/個体、好ましくは約30IU/個体、黄体形成ホルモンの投与量は0〜10 IU/個体、好ましくは約5IU/個体である。また、近交系のマウスやWister系統のラットを用いる場合は、約12時間明期条件(例えば7:00-19:00)で約1週間飼育した8週齢以上のものを用いるのが好ましい。
【0053】
得られた受精卵にマイクロインジェクション法(W. J. Gordonら; Proc. Natl. Acad. Sci. USA, 77, 7380, 1980、J. J. Mullinsら; Nature, 344, 541, 1990、R. E. Hammerら; Nature, 315, 680, 1985、V. G. Purselら; Immunol. Immunopathol., 17, 303, 1987)やレトロウイルスを用いた方法(マニピュレーティング・マウス・エンブリオ第2版)等によりヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子が導入された後、該受精卵を雌非ヒト動物に人工的に移植および着床させることによって、導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を組み込んだ染色体DNAを有する非ヒト動物が得られる。
【0054】
マイクロインジェクション法により遺伝子を導入する場合には、受精後約12〜約24時間の雄性前核が出現している受精卵を用いることが好ましい。マイクロインジェクションで導入されるヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子は、環状化状態、直線化状態いずれでも用いることができるが、ヒトCD16とそのヒトアクセサリー分子発現ユニットの構造遺伝子領域およびプロモーター等の発現調節領域を破壊しない形で直線化し導入することが好ましい。レトロウイルスを用いた方法により遺伝子を導入する場合には、桑実胚以前(一般には、8細胞期以前)の発生段階の受精卵を用いることが好ましい。
【0055】
雌非ヒト動物へ受精卵を移植する場合には、精管結紮雄非ヒト動物と交配させることにより、受精能を誘起された偽妊娠雌非ヒト動物に得られた受精卵を人工的に移植および着床させる方法が好ましい。偽妊娠雌非ヒト動物は自然交配によっても得られるが、黄体形成ホルモン放出ホルモン(以下、LHRHと略する)あるいはその類縁体を投与後、雄非ヒト動物と交配させることにより、受精能を誘起された偽妊娠雌非ヒト動物を得ることもできる。LHRHの類縁体としては、例えば[3,5-Dil-Tyr5]-LHRH、[Gln8]-LHRH、[D-Ala6]-LHRH、des-Gly10-[D-His(Bzl)6]-LHRH ethylamide等があげられる。LHRHあるいはその類縁体の投与量ならびにその投与後に雄非ヒト動物と交配させる時期は非ヒト動物の種類によりそれぞれ異なる。非ヒト動物が雌ラットの場合は、通常、LHRHあるいはその類縁体を投与後、約4日目に雄ラットと交配させることが好ましく、LHRHあるいはその類縁体の投与量は、通常、10〜60μg/個体、好ましくは約40μg/個体である。
(ii)ES細胞への遺伝子導入によるトランスジェニック非ヒト動物の作製
ES細胞への遺伝子導入によるトランスジェニック非ヒト動物の作製は、マニピュレーティング・マウス・エンブリオ第2版 、Gene Targeting, A Practical Approach, IRL Press at Oxford University Press (1993)、バイオマニュアルシリーズ8 ジーンターゲッティング, ES細胞を用いた変異マウスの作製,羊土社 (1995)、発生工学実験マニュアル, トランスジェニック・マウスの作り方, 講談社 (1987)等に記載された方法により、作製することができる。
【0056】
非ヒト動物のES細胞にヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を導入後、得られたES細胞を集合キメラ法または注入キメラ法を用いて、非ヒト動物の受精卵に取り込ませ、該受精卵を雌非ヒト動物に人工的に移植および着床させることによって導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を組み込んだ染色体DNAを有する細胞を部分的に有する非ヒトキメラ動物が得られる。
【0057】
非ヒト動物のES細胞は、マウスのES細胞(M. J. Evansら; Nature, 292, 154, 1981 ; G. R. Martin; Proc. Natl. Acad. Sci. USA, 78, 7634, 1981)、ラットのES細胞(P. M. Iannacconeら; Dev. Biol., 163, 288, 1994)ブタのES細胞(M. B. Wheeler; Reprod. Fertil. Dev., 6, 563, 1994)、サルのES細胞(J. A. Thomsonら; Proc. Natl. Acad. Sci. USA, 92, 7844, 1996)の各樹立方法、US5453357またはUS5670372に記載のES細胞の樹立方法に準じて、樹立することができる。マウスのES細胞として、AB2.2〔レキシコン・ジェネティックス(Lexicon Genetics)社製〕等を用いることができる。
【0058】
ES細胞への外来性遺伝子の導入には、公知の遺伝子導入の手法(例えば、リン酸カルシウム法、電気パルス法、リポフェクション法、凝集法、マイクロインジェクション法、パーティクルガン法、DEAE−デキストラン法、ウイルスベクター法等)を用いることができる。導入されるヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子は、環状化状態、直線化状態いずれでも用いることができるが、ヒトCD16とそのヒトアクセサリー分子発現ユニットの構造遺伝子をコードする領域およびプロモーター等の発現調節領域を破壊しない形で直線化し導入することが好ましい。
【0059】
受精卵は、上述の1.の(6)の(i)に記載した方法により取得できる。
ES細胞を集合キメラ法を用いて非ヒト動物の受精卵に取り込ませる場合には、一般に8細胞期以前の発生段階の受精卵を用いることが好ましい。ES細胞を注入キメラ法を用いて非ヒト動物の受精卵に取り込ませる場合には、一般に8細胞期から胚盤胞の発生段階の受精卵を用いることが好ましい。
【0060】
雌非ヒト動物へ受精卵を移植する場合には、精管結紮雄非ヒト動物と交配させることにより、受精能を誘起された偽妊娠雌非ヒト動物に得られた受精卵を人工的に移植および着床させる方法が好ましく、偽妊娠雌非ヒト動物は、上述の1.の(6)の(i)に記載した方法を用いることで得られる。
ES細胞の非ヒト動物と受精卵の非ヒト動物を、同じ種であるが体毛の色等の外見的な表現型が異なる系統にしておけば、非ヒトキメラ動物である個体、およびキメラ率(ES細胞に由来する細胞、すなわち導入した外来性遺伝子組み込んだ染色体DNAを有する細胞の割合)の高い個体を容易に選択することができる。
【0061】
ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を組み込んだ染色体DNAを有する細胞を部分的に有する非ヒトキメラ動物のうちキメラ率の高い個体(好ましくは、雄)と非ヒト動物の個体(好ましくは、雌)を交配する。生まれた仔の組織、例えば、血液組織、上皮組織、結合組織、軟骨組織、骨組織、筋組織、口腔内組織または骨格系組織の一部から調製したゲノムDNA上に導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子が存在するかどうかをサザンハイブリダイゼーション、PCR等により調べ、生まれた全ての仔のゲノムDNA上に導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子が存在する場合、親の非ヒトキメラ動物を生殖系列キメラとして選択する。生殖系列キメラと非ヒト動物の個体を交配することによって、全身の細胞の染色体上に導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を有する個体を仔として得ることができる。この個体は通常、相同染色体の片方にのみ、導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を有するヘテロ接合体である。さらにその個体の雄雌どうしを交配することにより相同染色体の双方に導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子が入ったホモ接合体のトランスジェニック非ヒト動物を得ることができる。
(iii)精子への遺伝子導入によるトランスジェニック非ヒト動物の作製
精子への遺伝子導入によるトランスジェニック非ヒト動物の作製は、Perryら(Science, 284, 1180, 1999) やWakayamaら(Nature Biotechnology, 16, 639, 1998)によって報告された方法を用いて行なうことができる。
【0062】
非ヒト動物の精子に上述の1.の(5)で作製したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を取り込ませた後、得られた精子を顕微受精の方法を用いて未受精卵に受精させ、発生を開始した卵を雌非ヒト動物に人工的に移植および着床させることによって、精子に取り込ませたヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を染色体上に組み込んだ非ヒト動物が得られる。
【0063】
精子に外来性遺伝子を取り込ませる場合には、細胞膜を破壊した精子を、約0.5〜約100ng/mL(好ましくは、5〜10ng/mL)の濃度の外来性遺伝子を含む培地(例えば、CZB培地やNIM培地など)中で数分間(好ましくは、1分間程度)インキュベーションする方法を用いることが好ましい。精子の細胞膜を破壊する方法としては、低濃度(例えば、0.05%)の界面活性剤(例えば、TritonX-100など)で処理する方法、10%の仔ウシ胎児血清(以下、FCSと略す)を含みEDTAを含まない培地(例えば、CZB培地など)中に懸濁した精子を凍結乾燥する方法 、該精子懸濁液を凍結融解する方法のいずれかが好ましい。このような処理をして細胞膜を破壊した精子は、もはや自発的に受精する能力を失っているが、マイクロマニュピレーター等を用いた顕微受精の方法を用いて受精させることで胚の発生を促す。顕微受精を行う場合には、外来性遺伝子を取り込ませた精子の頭部を減数第二分裂中期にある同種の雌非ヒト動物未受精卵の細胞質に注入する方法が好ましい。
(iv)未受精卵への遺伝子導入と体外受精によるトランスジェニック非ヒト動物の作製
未受精卵への遺伝子導入と体外受精によるトランスジェニック非ヒト動物の作製は、Chanら(Proc. Natl. Acad. Sci. USA, 95, 14028, 1998) によって報告された方法を用いて行なうことができる。
【0064】
非ヒト動物の未受精卵に、ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を組み込んだレトロウイルスを感染させることで該遺伝子を導入し、得られた卵を体外受精の方法を用いて発生を開始させ、発生を開始した受精卵を雌非ヒト動物に人工的に移植および着床させることによって、導入したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を染色体上に組み込んだ非ヒト動物が得られる。
【0065】
ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を組み込んだレトロウイルスは、マニピュレーティング・マウス・エンブリオ第2版等に記載の方法により調製できる。組換えレトロウイルスを未受精卵に感染させる場合には、透明帯を除いた未受精卵を用いることが好ましい。未受精卵の透明帯を除去する方法としては、マイクロマニュピレーター等を用いて物理的に膜を除く方法、化学物質(例えば、酸性タイロードなど)を用いて化学的に膜を溶解して除く方法のいずれかが好ましい。
2.標的遺伝子と目的とする導入遺伝子とを入れ換えた遺伝子置換動物の作製方法を用いた、本発明の非ヒト動物の作製
(1)非動物の標的遺伝子をコードするDNAの取得
上述の1.の(1)に記載の方法で、非ヒト動物の標的遺伝子をコードするDNAを取得する。後述するように、ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子領域の5'端および3'端に非ヒト動物の標的遺伝子の非コード領域を付加するために、5'端および3'端の近傍に適当な制限酵素サイトを合成DNAを用いて付加しておく。
(2)非ヒト動物の標的遺伝子を含むゲノムDNAの取得
非ヒト動物の標的遺伝子を含むゲノムDNAを取得する。該ゲノムDNAには、標的遺伝子をコードする領域およびイントロンの他に、標的遺伝子をコードする領域よりも5'側および3'側の領域も含む必要がある。その長さは、標的遺伝子をコードする領域よりも5'側および3'側へ、少なくとも500bp以上、好ましくは1kbp以上、より好ましくは3kbp以上、最も好ましくは5kbp以上が望ましい。
標的遺伝子を含むゲノムDNAを調製する方法としては、該非ヒト動物のゲノムDNAライブラリーを作製し、プラークハイブリダイゼーションあるいはコロニーハイブリダイゼーションにより、該遺伝子を含むゲノムDNAクローンを単離する方法があげられる。
【0066】
ゲノムDNAライブラリーは、該非ヒト動物の細胞や組織からゲノムDNAを抽出し、制限酵素で部分的に切断した後、適当なベクターに挿入し、ベクターに応じたE. coli等の適当な宿主に導入することにより作製することができる。ゲノムDNAの調製およびゲノムDNAライブラリーは、モレキュラー・クローニング第3版やカレント・プロトコールズ・イン・モレキュラー・バイオロジー等に記載された方法により作製することができる。ベクターとしては、λ EMBL3(ストラタジーン社製)、λ DASH II(ストラタジーン社製)、λ FIX II(ストラタジーン社製)等のλファージベクター、BeloBACII等の細菌人工染色体(BAC)ベクター、pAd10sacBII等のバクテリオファージP1ベクター、pCYPAC1等のP1人工染色体(PAC)ベクター、SuperCos I(ストラタジーン社製)等のコスミドベクター等があげられる。
【0067】
プラークハイブリダイゼーションあるいはコロニーハイブリダイゼーションに用いるプローブとしては、該非ヒト動物の標的となるゲノム遺伝子の部分断片、標的遺伝子のcDNAの部分断片を標識したものを用いることができる。これらの部分断片は、該非ヒト動物の標的遺伝子のゲノム配列またはcDNAの配列を塩基配列データベースから検索し、この塩基配列に基づいて作製したプライマーを用いたPCRにより取得することができる。
【0068】
プラークハイブリダイゼーション、コロニーハイブリダイゼーションおよびプローブの標識と調製は、モレキュラー・クローニング第3版やカレント・プロトコールズ・イン・モレキュラー・バイオロジー等に記載された方法により行なうことができる。
また、該非ヒト動物の標的遺伝子のゲノム配列が塩基配列データベースの検索により得られる場合は、増幅するゲノム配列の領域を適当に選択し、この領域の5'端20〜40bpの配列を3'端に含むDNAおよび、この領域の3'端20〜40bpの配列と相補的な配列を3'端に含むDNAを作製し、このDNAをプライマーとし、該非ヒト動物のゲノムDNAを鋳型としたPCRにより、該非ヒト動物の該標的遺伝子を含むゲノムDNAを単離することができる。
【0069】
また、ゲノムDNAライブラリースクリーニングシステム(Genome Systems社製)やユニバーサル・ゲノムウォーカー・キット(Universal GenomeWalkerTM Kit、クロンテック社製)などのキットを用いることによって、該非ヒト動物の標的遺伝子を含むゲノムDNAを単離することもできる。
上記の方法で得られる非ヒト動物の標的遺伝子を含むゲノムDNAとして、例えば、X染色体上のヒポキサンチンホスホリボシルトランスフェラーゼ(hprt)遺伝子を含むマウスゲノムDNAや、第6染色体上のRosa26遺伝子を含むマウスゲノムDNAをあげることができる。
(3)ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子の両末端に、非ヒト動物の標的遺伝子のゲノム配列の5'非コード領域、3'非コード領域を付加したDNAの作製
上述の1.の(5)で調製したヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子の5'端に、上述の2.の(2)で調製した非ヒト動物の標的遺伝子ゲノム配列の開始コドンより5'側の適当な長さの非コード領域を付加し、3'端に該ゲノム配列の終止コドンより3'側の適当な長さの非コード領域を付加したDNA(以下、相同組換え用DNAとも称する)を、標的遺伝子の相同組換えに必要なターゲッティングベクターの構築に用いる。このDNAは、通常の遺伝子工学的な手法を用いれば様々な方法で作製することが可能である。その具体的な例としては、例えば、それぞれの遺伝子断片に、連結のための適当な制限酵素サイトを含む合成DNAをプライマーとして用いたPCRを行うことで調製取得することができる。付加する非コード領域の長さは、3'側、5'側とも500bp以上、より好ましくは1kbp以上、さらに好ましくは3kbp以上、最も好ましくは5kbp以上が望ましい。
(4)ターゲットベクターの作製とES細胞への導入
非ヒト動物標的遺伝子を相同組換えするためのターゲットベクターは、Gene Targeting, A Practical Approach, IRL Press at Oxford University Press (1993)、バイオマニュアルシリーズ8 ジーンターゲッティング, ES細胞を用いた変異マウスの作製(羊土社)(1995)等に記載の方法にしたがって作製することができる。ターゲットベクターは、リプレースメント型、インサーション型いずれでも用いることができる。ターゲットベクターは、上述の2.の(3)で作製した相同組換え用DNA、相同組換え体の選別に必要な遺伝子、および、E. coliを用いたベクターの調製に必要なE. coliで自律複製可能な複製開始点および薬剤耐性遺伝子を有する。相同組換え体の選別に必要な遺伝子としては、ポジティブ選択(選別に用いた遺伝子を含む組換え体を選別する方法)に用いる、hprt遺伝子、ネオマイシン耐性遺伝子等の薬剤耐性遺伝子、ネガティブ選択(選別に用いた遺伝子を含まない組換え体を選別する方法)に用いるジフテリアトキシン(DT)遺伝子等をあげることができる。ポジティブ選択に用いる遺伝子は相同組換え用DNAの非コード領域内に挿入し、ネガティブ選択に用いる遺伝子は相同組換え用DNAとは別の位置に挿入する。ポジティブ選択に用いる遺伝子は、該遺伝子の発現に必要なプロモーターに連結してもよいし、プロモーターに連結せず、相同組換えが起きた場合に、ES細胞の染色体上の標的遺伝子のプロモーターにより発現させるようにしてもよい。ポジティブ選択に用いる遺伝子の両端に2つのloxP配列を付加して挿入すると、相同組換え体の選別を行なった後に、相同組換え体にCreリコンビナーゼ発現ベクターを導入することにより、相同組換え体の染色体からポジティブ選択に用いる遺伝子を除去することができる。
【0070】
具体的なターゲットベクターとしては、GenOway社のプラスミドready-to-use Quick knock-inTM vectorに、相同組換え用DNAを挿入したベクター、例えば、ready-to-use Quick knock-inTM vectorにマウス標的受容体遺伝子を含むゲノムDNAの非コード領域とヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子からなる相同組換え用DNAの発現ユニットを挿入したプラスミドSAT1-HRをあげることができる。
【0071】
ES細胞へのターゲットベクターの導入には、公知の遺伝子導入の手法、例えば、リン酸カルシウム法、電気パルス法、リポフェクション法、凝集法、マイクロインジェクション法、パーティクルガン法、DEAE−デキストラン法、ウイルスベクター法等を用いることができる。導入されるターゲットベクターは、環状、直鎖状いずれの形状でも用いることができるが、相同組換え用DNAおよび相同組換え体の選別に必要な遺伝子を破壊しない形で直線化し導入することが好ましい。
【0072】
相同組換え体を効率的に選別する方法として、例えば、Gene Targeting, A Practical Approach, IRL Press at Oxford University Press (1993)、バイオマニュアルシリーズ8 ジーンターゲッティング, ES細胞を用いた変異マウスの作製(羊土社)(1995)等に記載のポジティブ選択、プロモーター選択、ネガティブ選択、ポリA選択などの方法を用いることができる。例えば、hprt遺伝子を含むターゲットベクターの場合は、hprt遺伝子を欠損したES細胞に導入後、ES細胞をアミノプテリン、ヒポキサンチンおよびチミジンを含む培地で培養し、アミノプテリン耐性の株を選別することにより、hprt遺伝子を含む相同組換え体を選別するポジティブ選択を行なうことができる。ネオマイシン耐性遺伝子を含むターゲットベクターの場合は、ベクターを導入したES細胞をG418を含む培地で培養し、G418耐性の株を選別することにより、ネオマイシン耐性遺伝子を含む相同組換え体を選別するポジティブ選択を行なうことができる。DT遺伝子を含むターゲットベクターの場合は、ベクターを導入したES細胞を培養し、生育してきた株を選別する(相同組換え以外のランダムに染色体に挿入された組換え体は、DT遺伝子が染色体に組み込まれて発現するため、DTの毒性により生育できない)ことにより、DT遺伝子を含まない相同組換え体を選別するネガティブ選択を行なうことができる。選別した細胞株の中から目的とする相同組換え体を選択する方法としては、ゲノムDNAに対するサザンハイブリダイゼーション法(モレキュラー・クローニング第3版)やPCR等があげられる。
(4)受精卵へのES細胞の取り込みと非ヒトトランスジェニック動物の作製
受精卵へのES細胞の取り込みと、受精卵の非ヒト動物への移植は、上述の1.の(6)の(ii)に記載の方法により行なうことができる。
【0073】
得られた仔から上述の1.の(6)の(ii)に記載の方法により、全身の細胞の相同染色体の片方に相同組換えによる遺伝子置換を有するヘテロ接合体を得ることができる。さらにその個体の雄雌どうしを交配することにより相同染色体の双方に相同組換えによる遺伝子置換を有するホモ接合体のトランスジェニック非ヒト動物を得ることができる。
このようにして得られたホモ接合体のトランスジェニック非ヒト動物は、導入した遺伝子がコードするヒトCD16とそのヒトアクセサリー分子を発現し、自己が持っていた標的遺伝子は発現しないトランスジェニック非ヒト動物である。このホモ接合体のトランスジェニック非ヒト動物は、上述の1.の方法で得られるトランスジェニック非ヒト動物よりも、4.および5.に後述する物質の薬理評価に用いる非ヒト動物として、(a)非ヒト動物が本来発現している標的遺伝子を発現していないので、薬理評価に用いる場合に該標的遺伝子を介した薬理効果を考慮する必要がない、(b)導入遺伝子の導入部位に存在する遺伝子の破壊あるいは導入遺伝子による導入部位周辺の遺伝子の活性化等の予測不可能な副次的な表現形の変化がおきる可能性がほとんどない、等の点で好ましい。
3.目的とした遺伝子改変を施した細胞の核を用いたクローン個体の作製方法を用いた、本発明の非ヒト動物の作製
本発明の非ヒト動物は、文献に記載されたクローンヒツジ(I. Wilmutら; Nature, 385, 810, 1997)、クローンウシ(J. B. Cibelliら; Science, 280, 1256, 1998、入谷明; 蛋白核酸酵素, 44, 892, 1999)、クローンヤギ(A. Baguisiら; Nature Biotechnology, 17, 456, 1999)、クローンマウス(T. Wakayamaら; Nature, 394, 369, 1998、T. Wakayamaら; Nature Genetics, 22, 127, 1999)の作製方法を用い、例えば以下のように作製することができる。
【0074】
上述の1.あるいは2.に記載した方法を用い、非ヒト動物の任意の細胞の染色体上に、ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を導入する。得られた細胞の核を初期化する。核を初期化するとは、核を再び発生を繰り返すことができるような状態に戻す操作を行なうことをいう。
初期化した細胞の核を除核した非ヒト動物の未受精卵に注入することによって発生を開始させる。
【0075】
発生を開始した卵を雌非ヒト動物に人工的に移植および着床させることによって導入した遺伝子を発現する、本発明の非ヒト動物を得ることができる。また、上述の2.に記載の方法で、遺伝子置換を行なった細胞の核を用いて得られたトランスジェニック非ヒト動物は、通常相同染色体の片方にヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子が導入されたヘテロ接合体であるため、得られたヘテロ接合体同士を交配することにより、遺伝子置換された自己の標的遺伝子は発現せず、ヒトCD16とそのヒトアクセサリー分子を発現するトランスジェニック非ヒト動物を得ることができる。このホモ接合体のトランスジェニック非ヒト動物も、上述の2.に記載の方法で得られるホモ接合体のトランスジェニック非ヒト動物と同様の点で、薬理評価に用いる非ヒト動物として、上述の1.に記載した方法で得られるトランスジェニック非ヒト動物よりも好ましい。
【0076】
細胞の核を初期化する方法は、非ヒト動物の種類によりそれぞれ異なる。非ヒト動物がヒツジ、ヤギ、ウシなどの場合は、外来性遺伝子を導入した細胞を5〜30%、好ましくは10%のFCSを含む培地(例えば、M2培地)から3〜10日、好ましくは5日間、0〜1%、好ましくは0.5%のFCSを含む貧栄養培地で培養することで細胞周期を休止期状態(G0期もしくはG1期)に誘導し初期化することが好ましい。非ヒト動物がマウスなどの場合は、同種の非ヒト動物の除核した未受精卵に、外来性遺伝子を導入した細胞の核を注入し数時間、好ましくは約1〜6時間培養することで初期化することが好ましい。
【0077】
初期化された核を除核された未受精卵中で発生を開始させる方法は、非ヒト動物の種類によりそれぞれ異なる。非ヒト動物がヒツジ、ヤギ、ウシなどの場合は、細胞周期を休止期状態(G0期もしくはG1期)に誘導し初期化した核を、電気融合法などによって同種の非ヒト動物の除核した未受精卵に移植することで卵子を活性化し発生を開始させることが好ましい。非ヒト動物がマウスなどの場合は、外来性遺伝子を導入した細胞の核を注入した未受精卵を、卵子活性化物質(例えば、ストロンチウムなど)で刺激し細胞分裂の阻害物質(例えば、サイトカラシンBなど)で処理し第二極体の放出を抑制することで発生を開始させることが好ましい。
4.本発明の非ヒト動物またはその子孫から得られる細胞を用いた物質の薬理評価方法
本発明の非ヒト動物またはその子孫から得られる、ナチュラルキラー細胞、マクロファージ、単球及び樹状細胞などFc受容体を発現する細胞(エフェクター細胞)を摘出し、抗体の細胞障害活性等のエフェクター活性を評価することができる。抗体の生物活性を測定する方法としては、モノクローナルアンチボディズ(Monoclonal Antibodies: Principles and Practice, Third Edition, Acad. Press, 1993)、あるいはアンチボディエンジニアリング(Antibody Engineering: A Practical Approach, IRL Press at Oxford University Press, 1996)等に記載の公知の方法を用いることができる。
その具体的な例としては、ヒト抗体の抗原との結合活性、あるいは、ヒト抗体の抗原陽性培養細胞株に対する結合活性は、ELISA法及び蛍光抗体法[キャンサー・イムノロジー・イムノセラピー(Cancer Immunol. Immunother.), 36, 373 (1993)]等により測定できる。抗原陽性培養細胞株に対する細胞障害活性は、CDC活性、ADCC活性等を測定することにより、評価することができる[キャンサー・イムノロジー・イムノセラピー(Cancer Immunol. Immunother.), 36, 373 (1993)]。
また、本発明の非ヒト動物またはその子孫から得られるES細胞を分化誘導することにより、さまざまな種類の細胞を得ることができる。分化の誘導方法としては、ES細胞を同種同系統の動物の皮下に移植することにより、様々な組織が混じりあった奇形腫瘍(テラトーマ)を誘導する方法(マニピュレーティング・マウス・エンブリオ第2版)、適当な条件でインビトロ培養することにより、内胚葉細胞、外胚葉細胞、中胚葉細胞、血液細胞、内皮細胞、軟骨細胞、骨格筋細胞、平滑筋細胞、心筋細胞、神経細胞、グリア細胞、上皮細胞、メラノサイト、ケラチノサイトに分化誘導する方法(Reprod. Fertil. Dev., 10, 31, 1998)をあげることができる。この分化後の細胞と、試験物質とを接触させ、試験物質非存在下での細胞と比較して、細胞内のCa2+濃度の上昇等の種々の細胞の応答や細胞の形態の変化等の薬理作用を調べることにより、試験物質の該細胞に対する薬理評価を行なうことができる。これらの方法によって、ヒトの生体から摘出しにくい細胞や少数しか存在しない細胞などに対する薬理評価を行うことができる。
5.本発明の非ヒト動物またはその子孫を用いた物質の薬理評価方法
本発明の非ヒト動物またはその子孫に試験物質、例えば、抗体を投与し、該試験物質を投与しない該動物と比較して、該動物の血圧、呼吸数、体重等の種々の身体的パラメーターの測定、外見や行動の観察、病理組織学的検討等の薬理作用を調べることにより、該試験物質の薬理評価を行なうことができる。特に、本発明の非ヒト動物またはその子孫は、ADCC活性等のエフェクター活性に基づく薬理評価を行なうのに好ましい。
【0078】
また、本発明の非ヒト動物またはその子孫に各種の疾患を誘発させた病態モデル動物を作製し、該病態モデル動物に試験物質、例えば、抗体を投与し、該病態モデル動物の血圧、呼吸数、体重等の種々の身体的パラメーターの測定、病態や、外見、行動の観察、病理組織学的検討等を、試験物質を投与しない該病態モデル動物と比較して行なうことにより、該試験物質の該疾患に対する有効性および副作用等の薬理評価を行なうことができる。また、この評価に基づき、該疾患の治療薬として好ましい物質を選択することができる。特に、抗体では、抗体のFc領域とのFc受容体との結合に種差が存在しており、通常の病態モデル動物を用いた薬理評価では最適なエフェクター活性等の評価は困難であったが、本発明の非ヒト動物またはその子孫を用いることにより、より適切なADCC活性等のエフェクター活性に基づく抗体の薬理評価を行なうことができる。
本発明の非ヒト動物またはその子孫に誘発させる疾患としては、心疾患(例えば、急性心不全、慢性心不全、心筋炎など)、呼吸器系疾患、関節疾患(例えば、関節リュウマチ、変形性関節症など)、腎疾患(例えば、腎不全、糸球体腎炎、IgA腎症など)、動脈硬化症、乾癬症、高脂血症、アレルギー疾患(例えば、喘息、アレルギー性鼻炎、アトピー性皮膚炎など)、骨疾患(例えば、骨粗鬆症、くる病、骨軟化症、低カルシュウム血症など)、血液疾患、脳血管性傷害、外傷性脳障害、感染症、痴呆症、癌、糖尿病、肝疾患、皮膚疾患、神経変性疾患および慢性炎症性疾患などがあげられる。病態モデル動物の作製は、「マニュアル疾患モデルマウス」〔Molecular Medicine, 31 臨時増刊号, 中山書店 (1994)〕、「図説・薬理学のための病態動物モデル」〔西村書店 (1984)〕、「関節炎モデル動物」〔医歯薬出版 (1985)〕、「神経・筋疾患モデル動物」〔医歯薬出版 (1982)〕、「活性酸素と病態 疾患モデルからベッドサイドへ」〔学会出版センター (1992)〕等に記載の方法を用いることができる。
【0079】
本薬理評価方法、4.及び8.に後述する薬理評価方法に供する試験物質としては、癌、炎症疾患、自己免疫疾患、アレルギーなどの免疫疾患、循環器疾患、またはウィルスあるいは細菌感染をはじめとする各種疾患の予防および治療に用いられるヒト抗体があげられる。
腫瘍関連抗原を認識する抗体、アレルギーあるいは炎症に関連する抗原を認識する抗体、循環器疾患に関連する抗原を認識する抗体、自己免疫疾患に関連する抗原を認識する抗体、またはウイルスあるいは細菌感染に関連する抗原を認識するヒト抗体の具体例を、以下に述べる。
【0080】
腫瘍関連抗原を認識する抗体としては、抗CA125抗体(Immunology Today, 21, 403-410, 2000)、抗17−1A抗体(Immunology Today, 21, 403-410, 2000)、抗インテグリンαvβ3抗体(Immunology Today, 21, 403-410, 2000)、抗CD33抗体(Immunology Today, 21, 403-410, 2000)、抗CD22抗体(Immunology Today, 21, 403-410, 2000)、抗HLA抗体(Immunology Today, 21, 403-410, 2000)、抗HLA−DR抗体(Immunology Today, 21, 403-410, 2000)、抗CD20抗体(Immunology Today, 21, 403-410, 2000)、抗CD19抗体(Immunology Today, 21, 403-410, 2000)、抗EGF受容体抗体(Immunology Today, 21, 403-410, 2000)、抗CD10抗体(American Journal of Clinical Pathology, 113, 374-382, 2000; Proc. Natl. Acad. Sci. USA, 79:4386-4391, 1982)、抗GD2抗体(Anticancer Res., 13, 331-336, 1993)、抗GD3抗体(Cancer Immunol. Immunother., 36, 260-266, 1993)、抗GM2抗体(Cancer Res., 54, 1511-1516, 1994)、抗HER2抗体(Proc. Natl. Acad. Sci. USA, 89, 4285-4289, 1992)、抗CD52抗体(Nature, 332, 323-327, 1988)、抗MAGE抗体(British J. Cancer, 83, 493-497, 2000)、抗HM1.24抗体(Molecular Immunol., 36, 387-395, 1999)、抗副甲状腺ホルモン関連蛋白(PTHrP)抗体(Cancer, 88, 2909-2911, 2000)、抗FGF8抗体(Proc. Natl. Acad. Sci. USA, 86, 9911-9915, 1989)抗塩基性繊維芽細胞増殖因子抗体、抗FGF8受容体抗体(J. Biol. Chem., 265, 16455-16463, 1990)、抗塩基性繊維芽細胞増殖因子受容体抗体、抗インスリン様増殖因子抗体(J. Neurosci. Res., 40, 647-659, 1995)、抗インスリン様増殖因子受容体抗体(J. Neurosci. Res., 40, 647-659, 1995)、抗PMSA抗体(J. Urology, 160, 2396-2401, 1998)、抗血管内皮細胞増殖因子抗体(Cancer Res., 57, 4593-4599, 1997)または抗血管内皮細胞増殖因子受容体抗体(Oncogene, 19, 2138-2146, 2000)などが挙げられる。
【0081】
アレルギーあるいは炎症に関連する抗原を認識する抗体としては、抗IgE抗体(Immunology Today, 21, 403-410, 2000)、抗CD23抗体(Immunology Today, 21, 403-410, 2000)、抗CD11a抗体(Immunology Today, 21, 403-410, 2000)、抗CRTH2抗体(J. Immunol., 162, 1278-1286, 1999)、抗CCR8抗体(WO99/25734)、抗CCR3抗体(US6207155)、抗インターロイキン6抗体(Immunol. Rev., 127, 5-24, 1992)、抗インターロイキン6受容体抗体(Molecular Immunol., 31, 371-381, 1994)、抗インターロイキン5抗体(Immunol. Rev., 127, 5-24, 1992)、抗インターロイキン5受容体抗体、抗インターロイキン4抗体(Cytokine, 3, 562-567, 1991)、抗インターロイキン4受容体抗体(J. Immunol. Meth., 217, 41-50, 1998)、抗腫瘍壊死因子抗体(Hybridoma, 13, 183-190, 1994)、抗腫瘍壊死因子受容体抗体(Molecular Pharmacol., 58, 237-245, 2000)、抗CCR4抗体(Nature, 400, 776-780, 1999)、抗ケモカイン抗体(J. Immunol. Meth., 174, 249-257, 1994)または抗ケモカイン受容体抗体(J. Exp. Med., 186, 1373-1381, 1997)などが挙げられる。
【0082】
循環器疾患に関連する抗原を認識する抗体としては、抗GpIIb/IIIa抗体(J. Immunol., 152, 2968-2976, 1994)、抗血小板由来増殖因子抗体(Science, 253, 1129-1132, 1991)、抗血小板由来増殖因子受容体抗体(J. Biol. Chem., 272, 17400-17404, 1997)または抗血液凝固因子抗体(Circulation, 101, 1158-1164, 2000)などが挙げられる。
【0083】
自己免疫疾患、例えば、乾癬、関節リウマチ、クローン病、潰瘍性大腸炎、全身性エリテマトーデス、多発性硬化症に関連する抗原を認識する抗体としては、抗自己DNA抗体(Immunol. Letters, 72, 61-68, 2000)、抗CD11a抗体(Immunology Today, 21, 403-410, 2000)、抗ICAM3抗体(Immunology Today, 21, 403-410, 2000)、抗CD80抗体(Immunology Today, 21, 403-410, 2000)、抗CD2抗体(Immunology Today, 21, 403-410, 2000)、抗CD3抗体(Immunology Today, 21, 403-410, 2000)、抗CD4抗体(Immunology Today, 21, 403-410, 2000)、抗インテグリンα4β7抗体(Immunology Today, 21, 403-410, 2000)、抗CD40L抗体(Immunology Today, 21, 403-410, 2000)、抗IL−2受容体抗体(Immunology Today, 21, 403-410, 2000)などが挙げられる。
【0084】
ウイルスあるいは細菌感染に関連する抗原を認識する抗体としては、抗gp120抗体(Structure, 8, 385-395, 2000)、抗CD4抗体(J. Rheumatology, 25, 2065-2076, 1998)、抗CCR4抗体または抗ベロ毒素抗体(J. Clin. Microbiol., 37, 396-399, 1999)などが挙げられる。
上記抗体は、ATCC(The American Type Culture Collection)、理化学研究所細胞開発銀行、工業技術院生命工業技術研究所等の公的な機関、あるいは大日本製薬株式会社、R&D SYSTEMS社、PharMingen社、コスモバイオ社、フナコシ株式会社等の民間試薬販売会社から入手することができる。
6.本発明の非ヒト動物またはその子孫のES細胞、卵、精子、核を用いた遺伝子改変動物の作製
本発明の非ヒト動物またはその子孫から得られるES細胞、卵、精子または核を用いて、上述の1.、2.および3.に記載の方法によりヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を有し、かつさらなる染色体上の遺伝子の改変を行った遺伝子改変非ヒト動物またはその子孫を得ることができる。特に、その遺伝子の機能を破壊することである病態が惹起されることが公知になっている遺伝子を上述の2.に記載した相同組換えの手法を利用して欠損させたノックアウト動物や、該遺伝子の機能を阻害するドミナントネガティブ体の遺伝子を上述の1.または3.に記載の方法で導入し発現させたトランスジェニック動物は、病態モデル動物として有用である。
7.本発明の非ヒト動物またはその子孫と同種他系統の動物との交配による遺伝子改変動物の作製と作製された該遺伝子改変動物の利用
本発明の非ヒト動物またはその子孫と同種他系統の動物(例えば、ヒト疾患モデル動物)とを交配させることにより、ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を染色体上に有し、かつある表現系(例えば、ヒト病態と類似の症状)を示す遺伝子改変哺乳動物を得ることができる。交配する動物が、ヒト疾患モデル動物であれば、ヒトCD16とそのヒトアクセサリー分子発現ユニットを含む遺伝子を有し、かつヒト病態と類似の症状を表現系として示す病態モデル動物が得られる。交配の方法としては、自然交配以外に体外受精の方法があげられる。疾患モデル動物としては、先天性および後天性を疾患を問わずいかなる疾患モデル動物も用いることができる。例えば後天性の病態モデル動物は、「マニュアル疾患モデルマウス」〔Molecular Medicine, 31 臨時増刊号, 中山書店 (1994)〕、「図説・薬理学のための病態動物モデル」〔西村書店 (1984)〕、「関節炎モデル動物」〔医歯薬出版 (1985)〕、「神経・筋疾患モデル動物」〔医歯薬出版 (1982)〕、「活性酸素と病態 疾患モデルからベッドサイドへ」〔学会出版センター (1992)〕等に記載の方法により作製することができる。
8.遺伝子改変動物を用いた物質の薬理評価方法
上述の6.および7.に記載の方法で得られた遺伝子改変病態モデル動物に試験物質、例えば、抗体等を投与し、該病態モデル動物の血圧、呼吸数、体重等の種々の身体的パラメーターの測定、病態や、外見、行動の観察、病理組織学的検討等を、試験物質を投与しない該病態モデル動物と比較して行なうことにより、該試験物質の該疾患に対する有効性および副作用等の薬理評価を行なうことができる。また、この評価に基づき、該病態の治療薬として好ましい物質を選択することができる。特に、抗体では、抗体のFc領域とFc受容体との結合に種差が存在しており、通常の病態モデル動物を用いた薬理評価では最適なエフェクター活性等の評価は困難であったが、本発明の非ヒト動物またはその子孫を用いることにより、より適切なADCC活性等のエフェクター活性に基づく薬理評価を行なうことができる。
9.本発明の非ヒト動物またはその子孫を用いる薬理活性を有する物質のスクリーニング方法
本発明の非ヒト動物またはその子孫に試験物質、例えば、抗体を投与し、該試験物質を投与しない該動物と比較して、該動物の血圧、呼吸数、体重等の種々の身体的パラメーターの測定、外見や行動の観察、病理組織学的検討等の薬理作用を調べることにより、該試験物質の薬理評価を行ない、試験物質のなかから、薬理活性を有する試験物質を選択することにより、所望の薬理活性を有する試験物質をスクリーニングすることができる。特に、本発明の非ヒト動物またはその子孫は、抗体を投与し、ADCC活性等のエフェクター活性に基づく薬理活性を評価して、被検抗体から当該エフェクター活性に基づく薬理活性を有する抗体を選択することにより、in vivoにおいて薬理活性を有する抗体をスクリーニングすることができる。
以下に、本発明の実施例を示す。
【実施例1】
【0085】
ヒトCD16発現ベクターの作製
1.ヒトCD16をコードするcDNAの取得
ヒトCD16(別名;FcγRIIIa)のcDNAの塩基配列(GenBank Accession No. NM_000569)(配列番号1)をもとに、翻訳開始コドンを含む領域の配列に制限酵素認識配列を付加したフォワードプライマー(配列番号10)、および翻訳終止コドンを含む領域の配列に制限酵素認識配列を付加したリバースプライマー(配列番号11)を設計した。
【0086】
まず、DNAポリメラーゼKOD(東洋紡社製)を用いて、Human Leukocyte 5'-STRETCH PLUS cDNA Library(クロンテック社製)のlibrary lysate 2μLを含む50μLの反応液 [2.5ユニットのDNAポリメラーゼKOD 、10倍希釈した10×KOD Buffer #1(東洋紡社製)、0.2mmol/L dNTPs、1mmol/L MgCl2、0.4μmol/L上記プライマー(配列番号10および配列番号11)] を調製し、PCRを行った。PCRは、94℃で30秒間、57℃で30秒間、74℃で60秒間からなる反応を1サイクルとして、30サイクル行った。PCR後の反応液から、QIAquick PCR Purification Kit(QIAGEN社製)を用いてDNAを精製し、滅菌水20μLに溶解した。続いて、精製したDNAを制限酵素HindIII(タカラバイオ社製)およびBamHI(タカラバイオ社製)で消化したのち、1.5%アガロースゲル電気泳動に供し、約0.8 kbのPCR増幅断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0087】
次に、プラスミドpBluescript II SK(-) 3μg(Stratagene社製)を制限酵素HindIII(タカラバイオ社製)およびBamHI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約2.9 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
上記で得たHuman Leukocyte 5'-STRETCH PLUS cDNA Library由来のPCR増幅断片と、プラスミドpBluescript II SK (-)由来のDNA断片を混合し、DNA Ligation Kit Ver.2.0(タカラバイオ社製)により連結反応を行った。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のアミノ酸配列のうち、176番目がバリン(以下、ヒトCD16(Val)もしくはFcγRIIIa(V)と表記する)からなるcDNA(配列番号1)を含むプラスミド、および176番目がフェニルアラニン(以下、ヒトCD16(Phe)もしくはhFcγRIIIa(F)と表記する)からなるcDNA(配列番号2)を含むプラスミドを取得し、それぞれをpBshFcγRIIIa(V)、pBshFcγRIIIa(F)と名付けた。
2.ヒトCD3ζをコードするcDNAの取得
ヒトCD3ζのcDNAの塩基配列(GenBank Accession No. NM_000734)(配列番号5)をもとに、翻訳開始コドンを含む領域の配列に制限酵素認識配列を付加したフォワードプライマー(配列番号12)、および翻訳終止コドンを含む領域の配列に制限酵素認識配列を付加したリバースプライマー(配列番号13)を設計した。
【0088】
まず、本項1に記載の方法に準じて、上記プライマー(配列番号12および配列番号13)を用いたPCRを行い、PCR後の反応液から、QIAquick PCR Purification Kit(QIAGEN社製)を用いてDNAを精製し、滅菌水20μLに溶解した。制限酵素EcoRI(タカラバイオ社製)およびKpnI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約0.5 kbのPCR増幅断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0089】
次に、プラスミドpBluescript II SK(-) 3μg(Stratagene社製)を制限酵素EcoRI(タカラバイオ社製)およびKpnI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約2.9 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
上記で得たHuman Leukocyte 5'-STRETCH PLUS cDNA Library由来のPCR増幅断片と、プラスミドpBluescript II SK (-)由来のDNA断片を混合し、DNA Ligation Kit Ver.2.0(タカラバイオ社製)により連結反応を行った。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD3ζのcDNA(配列番号5)を含むプラスミドpBshCD3ζを取得した。
3.ヒトFcεRIγをコードするcDNAの取得
ヒトFcεRIγのcDNAの塩基配列(GenBank Accession No. NM_004106)(配列番号7)をもとに、翻訳開始コドンを含む領域の配列に制限酵素認識配列を付加したフォワードプライマー(配列番号14)、および翻訳終止コドンを含む領域の配列に制限酵素認識配列を付加したリバースプライマー(配列番号15)を設計した。
【0090】
まず、本項1に記載の方法に準じて、上記プライマー(配列番号14および配列番号15)を用いたPCRを行い、PCR後の反応液から、QIAquick PCR Purification Kit(QIAGEN社製)を用いてDNAを精製し、滅菌水20μLに溶解した。制限酵素EcoRI(タカラバイオ社製)およびKpnI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約0.3 kbのPCR増幅断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0091】
上記で得たHuman Leukocyte 5'-STRETCH PLUS cDNA Library由来のPCR増幅断片と、プラスミドpBluescript II SK (-)から調製した本項2に記載の約2.9kbpのDNA断片を混合し、DNA Ligation Kit Ver.2.0(タカラバイオ社製)により連結反応を行った。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトFcεRIγのcDNA(配列番号7)を含むプラスミドpBshFcεRIγを取得した。
4. pKANTEX hFcγRIIIa(V)およびpKANTEX hFcγRIIIa(F)の構築
本項1で得たプラスミドpBshFcγRIIIa(F)とpBshFcγRIIIa(V)、およびWO97/10354に記載のプラスミドpKANTEX93を用いて、以下の手順に従いpKANTEX hFcγRIIIa(V)およびpKANTEX hFcγRIIIa(F)を構築した(図1)。
【0092】
本項1で得たプラスミドpBshFcγRIIIa(V)、pBshFcγRIIIa(F) 各5.0μgを制限酵素NotI(タカラバイオ社製)およびBamHI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約0.8 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いてそれぞれ精製した。
次に、WO97/10354に記載のプラスミドpKANTEX93 10μgを制限酵素NotI(タカラバイオ社製)およびBamHI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約11.7 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0093】
上記で得たヒトCD16(Val)(別名;FcγRIIIa(V))もしくはヒトCD16(Phe)(別名;FcγRIIIa(F))のcDNAを含むDNA断片と、プラスミドpKANTEX93由来のDNA断片を混合し、DNA Ligation Kit Ver.2.0(タカラバイオ社製)により連結反応を行った。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、目的のヒトFcγRIIIa(V)およびヒトFcγRIIIa(F)のcDNAを含むプラスミドpKANTEX hFcγRIIIa(V)およびpKANTEX hFcγRIIIa(F)を取得した。
5.ヒトCD16とそのアクセサリー分子を共発現する発現ベクターの構築
本項2、3で取得したプラスミドpBshCD3ζまたはpBshFcεRIγ、および本項4で得たプラスミドpKANTEX hFcγRIIIa(V)またはpKANTEX hFcγRIIIa(F)を用いて、以下の手順に従い、ヒトCD16(FcγRIIIa)とそのアクセサリー分子を共発現する発現ベクターを構築した(図2)。
【0094】
まず、本項2、3で取得したプラスミドpBshCD3ζおよびpBshFcεRIγの各5.0μgを、制限酵素EcoRI(タカラバイオ社製)およびKpnI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、pBshCD3ζ由来の0.5 kbのDNA断片、およびpBshFcεRIγ由来の約0.3 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いてそれぞれ精製した。
【0095】
次に、本項(4)で取得したプラスミドpKANTEX hFcγRIIIa(V)およびpKANTEX hFcγRIIIa(F)の各2.0μgを、制限酵素EcoRI(タカラバイオ社製)およびNotI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約10.0kbと約2.5kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いてそれぞれ精製した。さらに、得られたpKANTEX hFcγRIIIa(V)由来の約2.5kbのDNA断片1.5μgを制限酵素KpnI(タカラバイオ社製)で消化後、1.5%アガロースゲル電気泳動に供し、約2.0 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0096】
上記で得たpBshCD3ζ由来の約0.5 kbのDNA断片、pKANTEX hFcγRIIIa(V)由来の約10.0 kbのDNA断片、およびpKANTEX hFcγRIIIa(V)由来の約2.0 kbのDNA断片を混合し、DNA Ligation Kit Ver.2.0(タカラバイオ社製)により連結反応を行った。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、目的のヒトFcγRIIIa(V)およびヒトCD3ζのcDNAを含むヒトFcγRIIIa発現ベクターpKANTEX hFcγRIIIa(V)+hCD3ζを取得した。
【0097】
さらに、pBshFcεRIγ由来の約0.3 kbのDNA断片、pKANTEX hFcγRIIIa(F)由来の約10.0 kbのDNA断片を用い、上記の方法に準じて、hFcγRIIIa(V)とhFcεRIγ、hFcγRIIIa(F)とhCD3ζ、hFcγRIIIa(V)とhFcεRIγの各cDNAを含むhFcγRIIIa発現ベクターを構築した。得られたプラスミドをそれぞれpKANTEX hFcγRIIIa(V)+hFcεRIγ、pKANTEX hFcγRIIIa(F)+hCD3ζ、pKANTEX hFcγRIIIa(F)+hFcεRIγと名付けた。
【実施例2】
【0098】
ヒトCD16プロモーターによって制御されたヒトCD16とヒトCD3ζのcDNAの発現ユニットを相同組換え法によりマウスHPRT遺伝子領域へ挿入するためのプラスミドSAT1-HRの作製
1.プラスミドSAT1-linkerの作製
12種類の制限酵素(KpnI、AscI、BstBI、XbaI、NsiI、PmeI、NruI、SpeI、ClaI、HpaI、SrfI、SacI)の認識配列から成るオリゴDNAセンス鎖SAT1-link1(配列番号16)およびアンチセンス鎖SAT1-link2(配列番号17)をファスマック社にて合成した。SAT1-link1とSAT1-link2をそれぞれ10 pmolずつ含む100μLの蒸留水を調製し、90℃で10分間熱したのち室温までゆっくり戻すことによりSAT1-link1とSAT1-link2をアニーリングさせ、SAT1-linkerを作製した。
【0099】
続いて、5μgのpBluescriptIISK(+) (STRATAGENE社製)をKpnI(タカラバイオ社製)およびSacIで消化したのち、フェノール/クロロホルム抽出処理及びエタノール沈殿を行い、回収したプラスミドを50μLの水に溶解した。
上記で得られたSAT1-linkerを0.2 pmol及びpBluescriptIISK(+)断片(KpnI-SacI)を50 ng含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で2時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、設計したSAT1-linker配列を含むプラスミドSAT1-linkerを得た(図3)。
2.プラスミドSAT1-IRESの作製
5μgのプラスミドG68(GenOway社製)をSmaI(New England Biolabs社製)とNsiI(New England Biolabs社製)にて、また本項1にて作製したプラスミドSAT1-linkerを5μg分PmeI(New England Biolabs社製)とNsiI(New England Biolabs社製)にてそれぞれ消化したのち、プラスミドG68及びプラスミドSAT1-linkerのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約0.6 kb、約3 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0100】
次いでプラスミドG68由来DNA断片(NsiI-SmaI)20ng、プラスミドSAT1-linker由来DNA断片(NsiI-PmeI)80ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、プラスミドG68由来のIRES配列を含むプラスミドSAT1-IRESを得た(図4)。
3.プラスミドSAT1-IRES-polyAの作製
5μgのプラスミドG147(GenOway社製)をSmaI(New England Biolabs社製)とClaI(New England Biolabs社製)にて、また本項2にて作製したプラスミドSAT1-IRESを 5μg分HpaI(New England Biolabs社製)とClaI(New England Biolabs社製)にてそれぞれ消化したのち、プラスミドG147及びプラスミドSAT1-IRESのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約0.8 kb、約3.6 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0101】
次いでプラスミドG147由来DNA断片(ClaI-SmaI)20ng、プラスミドSAT1-IRES由来DNA断片(ClaI-HpaI)80ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、IRES配列の下流にヒト成長ホルモン由来polyA配列が連結されたプラスミドSAT1-IRESを得た(図5)。
4.プラスミドCD3Zeta in pBSの作製
ヒトCD3ζのcDNAの塩基配列(GenBank Accession No. NM_000734)(配列番号5)をもとに、翻訳開始コドンを含む領域の配列に制限酵素認識配列(EcoRI)及びコザック配列を付加したフォワードプライマー(配列番号18)及び翻訳停止コドンを含む領域の配列に制限酵素認識配列(XbaI、SpeI)を付加したリバースプライマー(配列番号19)を作製し、以下のPCRを行なった。即ち、実施例1で作製したpKANTEX hFcγRIIIa(V)+hCD3ζをテンプレートとして含む20μLの反応液 [Pyrobest DNA polymerase(タカラバイオ社製)、10×PCR buffer、0.2mmol/L dNTP mixture、0.5μmol/L上記プライマー(配列番号18および配列番号19)]を調製し、94℃で5分間加熱した後、94℃で1分間、68℃で2分間を1サイクルとした30サイクルの反応でPCRを行なった。PCR反応後の溶液は1.5%アガロースゲル電気泳動に供し、約0.5 kbのPCR増幅断片をQIAquick Gel Extraction kit(QIAGEN社製)を用いて精製した。
【0102】
上記で得られたPCR増幅断片は、Zero Blunt TOPO PCR Cloning Kit for Sequencing(インビトロジェン社製)を用いてプラスミドpCR4Blunt-TOPOと連結させたのち、本プラスミドを用いてheat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD3ζのcDNA配列を含むプラスミドCD3Zeta in pCR4Bluntを得た(図6)。
【0103】
続いて、上記で作製したプラスミドCD3Zeta in pCR4Bluntを5μg分EcoRI(タカラバイオ社製)およびSpeI(タカラバイオ社製)にて、また5μgのプラスミドpBluescriptIISK(+)(STRATAGENE社製)をEcoRI(タカラバイオ社製)とSpeI(タカラバイオ社製)にてそれぞれ消化したのち、プラスミドCD3Zeta in pCR4Blunt及びプラスミドpBluescriptIISK(+)のDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約0.5 kbと約3.0 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。次いでプラスミドCD3Zeta in pCR4Blunt由来DNA断片(EcoRI-SpeI)20ng、プラスミドpBluescriptIISK(+)由来DNA断片(EcoRI-SpeI)80ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD3ζのcDNA配列を含むプラスミドCD3Zeta in pBSを得た(図6)。
5.プラスミドSAT1-IRES-CD3 Zetaの作製
本項3にて作製したプラスミドSAT1-IRES-polyAを 5μg分NruI(New England Biolabs社製)とSpeI(タカラバイオ社製)にて、また本項4で作製したプラスミドCD3Zeta in pBSを5μg分EcoRV(タカラバイオ社製)とSpeI(タカラバイオ社製)にてそれぞれ消化したのち、プラスミドSAT1-IRES-polyA及びプラスミドCD3Zeta in pBSのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約4.4 kb、約0.5 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0104】
次いでプラスミドSAT1-IRES-polyA由来DNA断片(NruI-SpeI)80ng、プラスミドCD3Zeta in pBS由来DNA断片(EcoRV-SpeI)20ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、IRES配列とヒト成長ホルモン由来polyA配列の間にヒトCD3ζのcDNA配列が連結されたプラスミドSAT1-IRES-CD3 Zetaを得た(図7)。
6.プラスミドCD16 in pBSの作製
ヒトCD16のcDNAの塩基配列(GenBank Accession No. NM_000569)(配列番号1)をもとに、5’非翻訳領域及び翻訳開始コドンを含む配列よりなるフォワードプライマー(配列番号20)及び翻訳停止コドンを含む領域の配列に制限酵素認識配列(XbaI、SpeI)を付加したリバースプライマー(配列番号21)を作製し、以下のPCRを行なった。即ち、実施例1で作製したpKANTEX hFcγRIIIa(V)+hCD3ζをテンプレートとして含む20μLの反応液 [Pyrobest DNA polymerase(タカラバイオ社製)、10×PCR buffer、0.2mmol/L dNTP mixture、0.5μmol/L上記プライマー(配列番号20および配列番号21)]を調製し、94℃で5分間加熱した後、94℃で1分間、68℃で2分間を1サイクルとした30サイクルの反応でPCRを行なった。PCR反応後の溶液は1.5%アガロースゲル電気泳動に供し、約0.8 kbのPCR増幅断片をQIAquick Gel Extraction kit(QIAGEN社製)を用いて精製した。
【0105】
上記で得られたPCR増幅断片は、Zero Blunt TOPO PCR Cloning Kit for Sequencing(インビトロジェン社製)を用いてプラスミドpCR4Blunt-TOPOと連結させたのち、本プラスミドを用いてheat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD3ζのcDNA配列を含むプラスミドCD16 in pCR4Bluntを得た(図8)。
【0106】
続いて、上記で作製したプラスミドCD16 in pCR4Bluntを5μg分EcoRI(タカラバイオ社製)とSpeI(タカラバイオ社製)にて、また5μgのプラスミドpBluescriptIISK(+)(STRATAGENE社製)をEcoRI(タカラバイオ社製)とSpeI(タカラバイオ社製)にてそれぞれ消化したのち、上記で得られたプラスミドCD16 in pCR4Blunt及びプラスミドpBluescriptIISK(+)のDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約0.8 kb、3.0 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0107】
次いでプラスミドCD16 in pCR4Blunt由来DNA断片(EcoRI-SpeI)20ng、プラスミドpBluescriptIISK(+)由来DNA断片(EcoRI-SpeI)80ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のcDNA配列を含むプラスミドCD16 in pBSを得た(図8)。
7.プラスミドCD16Pmoの作製
ヒトCD16のプロモーター配列(GenBank Accession No. AL_590385)(配列番号9)をもとに、フォワードプライマー(配列番号22)及びリバースプライマー(配列番号23)を作製し、以下のPCRを行なった。即ち、Human Genomic DNA(Novagen社製)をテンプレートとして含む20μLの反応液 [Pyrobest DNA polymerase(タカラバイオ社製)、10×PCR buffer、0.2mmol/L dNTP mixture、0.5μmol/L上記プライマー(配列番号22および配列番号23)]を調製し、94℃で5分間加熱した後、94℃で1分間、68℃で2分間を1サイクルとした30サイクルの反応でPCRを行なった。PCR反応後の溶液は1.5%アガロースゲル電気泳動に供し、約1.1 kbのPCR増幅断片をQIAquick Gel Extraction kit(QIAGEN社製)を用いて精製した。
【0108】
上記で得られたPCR増幅断片は、Zero Blunt TOPO PCR Cloning Kit for Sequencing(インビトロジェン社製)を用いてプラスミドpCR4Blunt-TOPOと連結させたのち、本プラスミドを用いてheat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のプロモーター配列を含むプラスミドCD16Pmo-Nを得た(図9)。
【0109】
次に、ヒトCD16のプロモーター配列(GenBank Accession No. AL_590385)(配列番号9)をもとに、フォワードプライマー(配列番号24)及びリバースプライマー(配列番号25)を作製し、以下のPCRを行なった。即ち、Human Genomic DNA(Novagen社製)をテンプレートとして含む20μLの反応液 [Pyrobest DNA polymerase(タカラバイオ社製)、10×PCR buffer、0.2mmol/L dNTP mixture、0.5μmol/L上記プライマー(配列番号24および配列番号25)]を調製し、94℃で5分間加熱した後、94℃で1分間、68℃で2分間を1サイクルとした30サイクルの反応でPCRを行なった。PCR反応後の溶液は1.5%アガロースゲル電気泳動に供し、約0.8 kbのPCR増幅断片をQIAquick Gel Extraction kit(QIAGEN社製)を用いて精製した。
【0110】
上記で得られたPCR増幅断片は、Zero Blunt TOPO PCR Cloning Kit for Sequencing(インビトロジェン社製)を用いてプラスミドpCR4Blunt-TOPOと連結させたのち、本プラスミドを用いてheat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のプロモーター配列を含むプラスミドCD16Pmo-Cを得た(図10)。
【0111】
続いて、上記で作製したプラスミドCD16Pmo-Nを5μg分HindIII(タカラバイオ社製)とApaI(タカラバイオ社製)にて、また同じく上記で作製したプラスミドCD16Pmo-Cを5μg分HindIII(タカラバイオ社製)とApaI(タカラバイオ社製)にてそれぞれ消化したのち、上記で得られたプラスミドCD16Pmo-N及びプラスミドCD16Pmo-CのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約1.1 kbと約4.8 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。次いでプラスミドCD16Pmo-N由来DNA断片(HindIII-ApaI)20ng、プラスミドCD16Pmo-C由来DNA断片(HindIII-ApaI)80ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、約1.8 kbのヒトCD16のプロモーター配列を含むプラスミドCD16Pmoを得た(図11)。
8.プラスミドCD16Pmo+cDNAの作製
本項6で作製したプラスミドCD16 in pBSを5μg分BspEI(New England Biolabs社製)とHindIII(タカラバイオ社製)にて、また本項7で作製したプラスミドCD16Pmoを5μg分BspEI(New England Biolabs社製)とHindIII(タカラバイオ社製)にてそれぞれ消化したのち、上記で得られたプラスミドCD16 in pBS及びプラスミドCD16PmoのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約3.8 kb、1.8 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。次いでプラスミドCD16 in pBS由来DNA断片(HindIII-BspEI)80ng、プラスミドCD16Pmo由来DNA断片(HindIII-BspEI)20ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のプロモーター配列およびcDNA配列が連結されたプラスミドCD16Pmo+cDNAを得た(図12)。
9.プラスミドSAT1-Tgの作製
本項5で作製したプラスミドSAT1-IRES-CD3 Zetaを5μg分BstBI(New England Biolabs社製)とXbaI(タカラバイオ社製)にて、また本項8で作製したプラスミドCD16Pmo+cDNAを5μg分ClaI(New England Biolabs社製)とXbaI(タカラバイオ社製)にてそれぞれ消化したのち、上記で得られたプラスミドSAT1-IRES-CD3 Zeta及びプラスミドCD16Pmo+cDNAのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約4.9 kbと約2.6 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。次いでプラスミドSAT1-IRES-CD3 Zeta由来DNA断片(BstBI-XbaI)80ng、プラスミドCD16Pmo+cDNA由来DNA断片(ClaI-XbaI)20ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のプロモーター配列下流にIRES配列で連結されたヒトCD16のcDNAとヒトCD3ζのcDNA配列が配置され、さらにその下流にヒト成長ホルモン由来polyA配列が付加されたプラスミドSAT1-Tgを得た(図13)。
10.プラスミドSAT1-HRの作製
本項9で作製したプラスミドSAT1-Tgを制限酵素AscI及びSrfIで切断することにより生じる約4.6 kbのDNA断片を回収し、AscI及びSwaIで切断したプラスミドready-to-use Quick knock-inTM vector(GenOway社製)とLigase酵素を用いて連結反応を行った。得られた連結済プラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ネガティブセレクションマーカーとしてジフテリア毒素発現ユニット(DTA)およびポジティブセレクションマーカーとしてネオマイシン耐性遺伝子発現ユニットを有し、またマウスHPRT遺伝子領域に相同組換えを起こさせるための5’側アームとしてマウスHPRT遺伝子の第1エクソン上流の塩基配列、同じく3’側アームとしてマウスHPRT遺伝子の第3エクソンおよび第2、3イントロンの塩基配列を含むready-to-use Quick knock-inTM vectorに、ヒトCD16のプロモーター配列下流にIRES配列で連結されたヒトCD16のcDNAとヒトCD3ζのcDNA配列が配置され、さらにその下流にヒト成長ホルモン由来polyA配列が付加された遺伝子発現構成体が導入された、プラスミドSAT1-HRを得た(図14)。
【実施例3】
【0112】
ヒトCD16プロモーター制御下においてヒトCD16及びヒトCD3ζを発現している遺伝子改変マウスの作製
1.遺伝子改変ES細胞株の樹立
以下の方法を用いて、マウスES細胞のHPRT遺伝子のエクソン3上流に、ヒトCD16プロモーターによって制御されたヒトCD16のcDNA及びヒトCD3ζの発現ユニットを挿入した。遺伝子導入は公知のエレクトロポレーション法 [サイトテクノロジー (Cytotechnology), 3, 133 (1990)] により以下の手順で行った。
【0113】
実施例2で作製したプラスミドSAT1-HRをPmeI(New England Biolabs社製)で切断した後、40 μg分の線状化プラスミドを5×106細胞の129Olaマウス由来E14 ES細胞(GenOway社製)へパルス電圧260 V、電気容量500μFの条件で遺伝子導入を行った。なお、129Olaマウスとは、129系統マウスよりHPRT遺伝子のエクソン1およびその周辺の染色体領域を欠失させることにより作製したマウスである。2日後、200μg/mlのG418入り培地でES細胞を選抜培養し、G418耐性ES細胞株を400クローン単離した。400クローンはそれぞれ96穴プレートでコンフルエントまで培養した後、96穴プレート2枚(細胞培養用とレプリカのゲノムDNA取得用)へそれぞれ継代した。
【0114】
ゲノムDNA取得用の96穴プレートより公知の方法(WO03/085107(山根さんのFUT8KO株特許))でゲノムDNAを取得した後、相同組換え法により目的のHPRT遺伝子領域にヒトCD16プロモーターによって制御されたヒトCD16のcDNA及びヒトCD3ζの発現ユニットが挿入されている相同組換えクローンを、以下に示すゲノムPCR法を用いて検出した。
相同組換え領域の5’側の確認は、PCR用プライマーGW496(配列番号26)およびPCR用プライマーGW497(配列番号27)を用いて行った(図15)。100 ngのゲノムDNAをテンプレートとして含む50μLの反応液 [2.6 unitsのExpand High Fidelity polymerase(Roche diagnostics社製)、10×PCR buffer、0.5 mmol/L dNTP mixture、15 pmolずつのプライマーGW496(配列番号26)およびGW497(配列番号27)]を調製し、94℃で2分間加熱した後、94℃で30秒間、62℃で1分間、68℃で4分間を1サイクルとした35サイクルの反応でPCRを行なった。PCR反応終了溶液はアガロースゲル電気泳動に供し、4529 bpのPCR増幅断片が検出されたES細胞クローン株をクローニングした(図16)。その結果、G418耐性を有する400クローン中、37クローンにおいて4529 bpのPCR増幅断片が認められた。この37クローン中20クローンについて、次に示す3’側のゲノムPCR解析を行った。
【0115】
相同組換え領域の3’側の確認は、PCR用プライマーGX2922(配列番号28)およびPCR用プライマーGX0025(配列番号29)を用いて行った(図17)。100 ngのゲノムDNAをテンプレートとして含む50μLの反応液 [2.6 unitsのExpand High Fidelity polymerase(Roche diagnostics社製)、10×PCR buffer、0.5 mmol/L dNTP mixture、15 pmolずつのプライマーGX2922(配列番号28)およびGX0025(配列番号29)]を調製し、94℃で2分間加熱した後、94℃で30秒間、62℃で1分間、68℃で4分間を1サイクルとした35サイクルの反応でPCRを行なった。PCR反応終了溶液はアガロースゲル電気泳動に供し、5733 bpのPCR増幅断片が検出されたES細胞クローン株を同定した(図17)。その結果、5’側の相同組換えが確認された20クローン中17クローンにおいて、5733 bpのPCR増幅断片が認められた。
【0116】
5’側および3’側のPCR解析により特異的バンドが検出された17クローンについて、次に示すサザンブロット解析を行った(図18)。各クローンのゲノムDNAをPstI(New England Biolabs社製)で16時間切断させたのち、アガロースゲル電気泳動に供し、既存の方法(J. Mol. Biol., 98, 503, 1975)を用いてナイロンフィルターへDNAを転写した。65℃のプレハイブリ溶液(4×SSC、1% SDS、65℃0.5% スキムミルク、20 mM EDTA、100 μg/mLのサケ精子DNA)に上記で作製したフィルターを浸したのち、さらに32P標識体やDIG標識体で標識した3’側プローブ(配列番号30)を添加し、65℃で18時間ハイブリダイゼーションを行った。なお、3’側プローブはマウスHPRT遺伝子の第3エクソン配列(GenBank Accession No. K01509)より設計した。ハイブリダイゼーション後、3×SSCと1%SDSを含有する溶液を用いて65℃で15分間の洗浄を2回行ったのち、さらに0.5×SSCと1%SDSを含有する溶液を用いて65℃で15分間の洗浄を2回行った。X線フィルムを洗浄後のフィルターで3日間感光させたのち現像を行い、目的通りの相同組換えが起こっているES細胞クローン株(約6.1 kbのバンドが検出される)を同定した(図18、19)。その結果、#1C8、#2A8、#12A2、#15A9の4クローン株において約6.1 kbのバンドが観察され、これらクローン株では正常に相同組換えが行われていることが確認された。
【0117】
上記のESクローン株(#1C8、#2A8、#12A2、#15A9)をC57BL/6マウス由来の胚盤胞へとインジェクションしたのち、偽妊娠させたOF1マウスの卵管へと移植し、産仔を得た。その結果、#1C8、#12A2、#15A9のES細胞をインジェクションした胚盤胞より、95〜100%のキメラ率(体表の毛の色がアグチである割合をキメラ率として表記する)を有するオスのキメラ体を計8匹得た。この8匹より、#1C8由来のキメラ体2匹、#12A2由来のキメラ体1匹、#15A9由来のキメラ体1匹をそれぞれC57BL/6マウスのメスと交配させ、産まれたメスのF1体について、以下に示すPCR解析およびサザンブロット解析を行った。
【0118】
相同組換え領域の5’側の確認は、PCR用プライマーGW496(GenOway社製(配列番号26))およびPCR用プライマーGW497(GenOway社製(配列番号27))を用いて行った(図15)。メスのF1体の尾より取得した100 ngのゲノムDNAをテンプレートとして含む50μLの反応液 [2.6 unitsのExpand High Fidelity polymerase(Roche diagnostics社製)、10×PCR buffer、0.5 mmol/L dNTP mixture、15 pmolずつのプライマーGW496(配列番号26)およびGW497(配列番号27)]を調製し、94℃で2分間加熱した後、94℃で30秒間、62℃で1分間、68℃で4分間を1サイクルとした35サイクルの反応でPCRを行なった。PCR反応終了溶液はアガロースゲル電気泳動に供し、4529 bpのPCR増幅断片が検出されるかどうか確認した(図15、20)。その結果、#1C8、#12A2および#15A9由来のキメラ体より産まれたメスのF1体より、4529 bpのPCR増幅断片が検出された。
【0119】
続いて、相同組換え領域の3’側の確認はサザンブロット解析にて行った。メスのF1体の尾より取得したゲノムDNAをPstI(New England Biolabs社製)で16時間切断させたのち、アガロースゲル電気泳動に供し、既存の方法(J. Mol. Biol., 98, 503, 1975)を用いてナイロンフィルターへDNAを転写した。65℃のプレハイブリ溶液(4×SSC、1% SDS、65℃0.5% スキムミルク、20 mM EDTA、100 μg/mLのサケ精子DNA)に上記で作製したフィルターを浸したのち、さらに32P標識体やDIG標識体で標識した3’側プローブ(配列番号30)を添加し、65℃で18時間ハイブリダイゼーションを行った。なお、3’側プローブはマウスHPRT遺伝子の第3エクソン配列(GenBank Accession No. K01509)より設計した。ハイブリダイゼーション後、3×SSCと1%SDSを含有する溶液を用いて65℃で15分間の洗浄を2回行ったのち、さらに0.5×SSCと1%SDSを含有する溶液を用いて65℃で15分間の洗浄を2回行った。X線フィルムを洗浄後のフィルターで3日間感光させたのち現像を行い、目的通りの相同組換えが起こっているF1体(約6.1 kbのバンドが検出される)を同定した(図21、22)。その結果、4529 bpのPCR増幅断片が検出されたF1体において、C57Bl/6マウスの染色体由来バンド(約7.8 kb)および約6.1 kbの相同組換え体由来バンドが検出された。
【0120】
以上の結果、オスの#1C8、#12A2および#15A9由来キメラ体の生殖系列は目的通りの相同組換えを起こしたES細胞株由来であり、このキメラ体とメスの野生型マウスを掛け合わせることにより、片方のX染色体のHPRT遺伝子領域にはヒトCD16プロモーター下流にヒトCD16とヒトCD3ζのcDNAをIRES配列で連結させた遺伝子発現ユニットが挿入されたF1体が産まれることが確認された。
【実施例4】
【0121】
ヒトCD16プロモーター制御下においてヒトFcεRIγ及びGFPを発現している遺伝子改変マウスの作製
1.プラスミドEpsilon in pCR4Bluntの作製
ヒトFcεRIγのcDNAの塩基配列(GenBank Accession No. NM_004106)(配列番号7)をもとに、翻訳開始コドンを含む領域の配列に制限酵素認識配列(BspEI)及びヒトCD16プロモーター配列を付加したフォワードプライマー(配列番号31)及び翻訳停止コドンを含む領域の配列に制限酵素認識配列(SpeI)を付加したリバースプライマー(配列番号32)を作製し、以下のPCRを行なった。即ち、実施例1で作製したpKANTEX hFcγRIIIa(V)+hFcεRIγをテンプレートとして含む20μLの反応液 [Pyrobest DNA polymerase(タカラバイオ社製)、10×PCR buffer、0.2mmol/L dNTP mixture、0.5μmol/L上記プライマー(配列番号31および配列番号32)]を調製し、94℃で5分間加熱した後、94℃で1分間、68℃で2分間を1サイクルとした30サイクルの反応でPCRを行なった。PCR反応後の溶液は1.5%アガロースゲル電気泳動に供し、約0.3 kbのPCR増幅断片をQIAquick Gel Extraction kit(QIAGEN社製)を用いて精製した。
【0122】
上記で得られたPCR増幅断片は、Zero Blunt TOPO PCR Cloning Kit for Sequencing(インビトロジェン社製)を用いてプラスミドpCR4Blunt-TOPOと連結させたのち、本プラスミドを用いてheat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトFcεRIγのcDNA配列を含むプラスミドEpsilon in pCR4Bluntを得た(図23)。
2.プラスミドGFP in pCR4Bluntの作製
公知のGreen fluorescent protein(GFP)cDNA配列(GenBank Accession No. M62653)をもとに、より強い発光が観察される改変型GFPのcDNA配列を設計し(配列番号33)、合成オリゴDNAを用いて本配列を含むDNA鎖を作製した。
【0123】
続いて、翻訳開始コドンを含む領域の配列に制限酵素認識配列(EcoRI)及びコザック配列を付加したフォワードプライマー(配列番号34)及び翻訳停止コドンを含む領域の配列に制限酵素認識配列(SpeI)を付加したリバースプライマー(配列番号35)を作製し、以下のPCRを行なった。即ち、改変型GFPのcDNA配列(配列番号33)をテンプレートとして含む20μLの反応液 [Pyrobest DNA polymerase(タカラバイオ社製)、10×PCR buffer、0.2mmol/L dNTP mixture、0.5μmol/L上記プライマー(配列番号34および配列番号35)]を調製し、94℃で5分間加熱した後、94℃で1分間、68℃で2分間を1サイクルとした30サイクルの反応でPCRを行なった。PCR反応後の溶液は1.5%アガロースゲル電気泳動に供し、約0.8 kbのPCR増幅断片をQIAquick Gel Extraction kit(QIAGEN社製)を用いて精製した。
【0124】
上記で得られたPCR増幅断片は、Zero Blunt TOPO PCR Cloning Kit for Sequencing(インビトロジェン社製)を用いてプラスミドpCR4Blunt-TOPOと連結させたのち、本プラスミドを用いてheat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、改変型GFPのcDNA配列を含むプラスミドGFP in pCR4Bluntを得た(図24)。
3.プラスミドPmo+epsilonの作製
実施例2で作製したプラスミドCD16Pmo+cDNAを5μg分BspEI(New England Biolabs社製)とSpeI(タカラバイオ社製)にて、また本項1にて作製したプラスミドEpsilon in pCR4Bluntを 5μg分BspEI(New England Biolabs社製)とSpeI(タカラバイオ社製)にてそれぞれ消化したのち、上記で得られたプラスミドCD16Pmo+cDNA及びプラスミドEpsilon in pCR4BluntのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約3.8 kb、約0.3 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。
【0125】
次いでプラスミドCD16Pmo+cDNA由来DNA断片(BspEI-SpeI)50ng、プラスミドEpsilon in pCR4Blunt由来DNA断片(BspEI-SpeI)50ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のプロモーター配列下流にヒトFcεRIγのcDNA配列が連結されたプラスミドPmo+epsilonを得た(図25)。
4.プラスミドSAT1-IRES-GFPの作製
実施例2で作製したプラスミドSAT1-IRES-polyAを5μg分NruI(New England Biolabs社製)とSpeI(タカラバイオ社製)にて、また本項2にて作製したプラスミドGFP in pCR4Bluntを 5μg分EcoRV(タカラバイオ社製)とSpeI(タカラバイオ社製)にてそれぞれ消化したのち、上記で得られたプラスミドSAT1-IRES-polyA及びプラスミドGFP in pCR4BluntのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約4.4 kb、0.8 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。次いでプラスミドSAT1-IRES-polyA由来DNA断片(NruI-SpeI)50ng、プラスミドGFP in pCR4Blunt由来DNA断片(EcoRV-SpeI)50ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、IRES配列とヒト成長ホルモン由来polyA配列の間に改変型GFPのcDNA配列が連結されたプラスミドSAT1-IRES-GFPを得た(図26)。
5.プラスミドEpsGFP-Tgの作製
本項4で作製したプラスミドSAT1-IRES-GFPを5μg分BstBI(New England Biolabs社製)とXbaI(タカラバイオ社製)にて、また本項3で作製したプラスミドPmo+epsilonを5μg分ClaI(New England Biolabs社製)とXbaI(タカラバイオ社製)、10μLの10×Mバッファー(タカラバイオ社製)にてそれぞれ消化したのち、上記で得られたプラスミドSAT1-IRES-GFP及びプラスミドPmo+epsilonのDNA断片を1.0%(W/V)アガロースゲル電気泳動に供し、それぞれ約5.0 kb、約2.1 kbのDNA断片をQIAquick Gel Extraction Kit(QIAGEN社製)を用いて精製した。次いでプラスミドSAT1-IRES-GFP由来DNA断片(BstBI-XbaI)80ng、プラスミドPmo+epsilon由来DNA断片(ClaI-XbaI)20ngを含む反応液10μLを調製したのちLigation High(東洋紡社製)を5μL加え、16℃で16時間連結反応を行なった。得られたプラスミドDNAを用い、heat shock法により大腸菌DH5α株(東洋紡社製)を形質転換した。形質転換株よりQIAprep(R) Spin Miniprep Kit(QIAGEN社製)を用いてプラスミドDNAを調製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit v2.0(QIAGEN社製)とDNAシーケンサABI PRISM 377(Applied Biosystems社製)を用いて塩基配列を解析した。その結果、ヒトCD16のプロモーター配列下流にIRES配列で連結されたヒトFcεRIγのcDNAと改変型GFPのcDNA配列が配置され、さらにその下流にヒト成長ホルモン由来polyA配列が付加されたプラスミドEpsGFP-Tgを得た(図27)。
6.ヒトCD16プロモーター制御下においてヒトFcεRIγ及びGFPを発現している遺伝子改変マウスの作製
本項5で作製したプラスミドEpsGFP-Tgを線状化もしくはマウス染色体の特定の遺伝子領域へ挿入するためのプラスミドに挿入したのち、マウスの受精卵もしくはES細胞へと導入し、ヒトCD16プロモーター制御下においてヒトFcεRIγ及びGFPを発現している遺伝子改変マウスを作製した。さらに、本マウスを実施例3に記載のヒトCD16プロモーター制御下においてヒトCD16及びヒトCD3ζを発現している遺伝子改変マウスと掛け合わせることにより、ヒトCD16プロモーター制御下においてヒトCD16、ヒトCD3ζ、ヒトFcεRIγ、および改変型GFPを発現している遺伝子改変マウスを作製した。
【0126】
また、本項5で作製したプラスミドEpsGFP-Tgを実施例3に記載のヒトCD16プロモーター制御下においてヒトCD16及びヒトCD3ζを発現している遺伝子改変マウスの受精卵もしくはES細胞へと導入し、ヒトCD16プロモーター制御下においてヒトCD16、ヒトCD3ζ、ヒトFcεRIγ、および改変型GFPを発現している遺伝子改変マウスを作製した。
【図面の簡単な説明】
【0127】
【図1】プラスミドpKANTEXhFcγRIIIa(V)およびpKANTEXhFcγRIIIa(F)の作製方法を示す。
【図2】プラスミドpKANTEXhFcγRIIIa(V)+hCD3ζ、pKANTEXhFcγRIIIa(F)+hCD3ζ、pKANTEXhFcγRIIIa(V)+hFcεRIγ、およびpKANTEXhFcγRIIIa(F)+hFcεRIγの作製方法を示す。
【図3】プラスミドSAT1-linkerの作製方法を示す。
【図4】プラスミドSAT1-IRESの作製方法を示す。
【図5】プラスミドSAT1-IRES-polyAの作製方法を示す。
【図6】プラスミドCD3Zeta in pBSの作製方法を示す。
【図7】プラスミドSAT1-IRES-CD3Zetaの作製方法を示す。
【図8】プラスミドCD16 in pBSの作製方法を示す。
【図9】プラスミドCD16Pmo-Nの作製方法を示す。
【図10】プラスミドCD16Pmo-Cの作製方法を示す。
【図11】プラスミドCD16Pmoの作製方法を示す。
【図12】プラスミドCD16Pmo+cDNAの作製方法を示す。
【図13】プラスミドSAT1-Tgの作製方法を示す。
【図14】プラスミドSAT1-HRの作製方法を示す。
【図15】予想される相同組換え体の染色体構造(HPRT遺伝子領域)及び相同組換え体を検出するための5’側および3’側のPCRプライマーの位置を模式的に示す。
【図16】5’側のPCRによって検出される、相同組換え体ES細胞株のゲノムDNA由来の増幅バンドを示す。
【図17】3’側のPCRによって検出される、相同組換え体ES細胞株のゲノムDNA由来の増幅バンドを示す。
【図18】予想される相同組換え体の染色体構造(HPRT遺伝子領域)及び相同組換え体を検出するためのサザンブロット解析に用いる3’側プローブの位置を模式的に示す。
【図19】3’側プローブを用いたサザンブロット解析によって検出される、宿主株のE14 ES細胞株及び相同組換え体ES細胞株のゲノムDNA由来のバンドを示す。
【図20】5’側のPCRによって検出される、野生型及びF1体マウスのゲノムDNA由来の増幅バンドを示す。
【図21】親株のE14 ES細胞株、予想される相同組換え体、ならびに野生型マウスの染色体構造(HPRT遺伝子領域)及びサザンブロット解析に用いる3’側プローブの位置を模式的に示す。
【図22】3’側プローブを用いたサザンブロット解析によって検出される、宿主株のE14 ES細胞株、野生型マウス、及びF1体のゲノムDNA由来のバンドを示す。
【図23】プラスミドEpsilon in pCR4Bluntの作製方法を示す。
【図24】プラスミドGFP in pCR4Bluntの作製方法を示す。
【図25】プラスミドPmo+epsilonの作製方法を示す。
【図26】プラスミドSAT1-IRES-GFPの作製方法を示す。
【図27】プラスミドEpsGFP-Tgの作製方法を示す。
【産業上の利用可能性】
【0128】
本発明により、ヒトCD16とそのヒトアクセサリー分子とを共発現させるようにゲノムが改変された、非ヒト動物またはその子孫及びその利用方法が提供される。本発明の非ヒト動物及びその子孫は、抗体の生物活性及び薬理評価に有用である。
【特許請求の範囲】
【請求項1】
ヒトCD16とそのヒトアクセサリー分子とを共発現させるようにゲノムが改変された、非ヒト動物またはその子孫。
【請求項2】
ヒトCD16のヒトアクセサリー分子が、以下の(a)〜(c) からなる群から選ばれる分子である、請求項1に記載の非ヒト動物またはその子孫。
(a) ヒトCD3ζ鎖;
(b) ヒトFcε受容体Iγ鎖;
(c) ヒトCD3ζ鎖とヒトFcε受容体Iγ鎖。
【請求項3】
ヒトCD16またはそのヒトアクセサリー分子の発現が、ヒトCD16プロモーター領域で制御されるようにゲノムが改変された、請求項1または2に記載の非ヒト動物またはその子孫。
【請求項4】
ヒトCD16が、以下の (a) 〜(d)からなる群から選ばれるDNAがコードする蛋白質である、請求項1〜3のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号1で表される塩基配列からなるDNA;
(b) 配列番号2で表される塩基配列からなるDNA;
(c) 配列番号1で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒト抗体のFc領域と結合する活性を有する蛋白質をコードするDNA;
(d) 配列番号2で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒト抗体のFc領域と結合する活性を有する蛋白質をコードするDNA。
【請求項5】
ヒトCD16が、以下の(a) 〜(f)からなる群から選ばれる蛋白質である、請求項1〜3のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号3で表されるアミノ酸配列からなる蛋白質;
(b) 配列番号4で表されるアミノ酸配列からなる蛋白質;
(c) 配列番号3で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(d) 配列番号4で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(e) 配列番号3で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(f) 配列番号4で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質。
【請求項6】
ヒトCD3ζ鎖が、以下の (a)または(b)であるDNAがコードする蛋白質である、請求項2〜5のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号5で表される塩基配列からなるDNA;
(b) 配列番号5で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質をコードするDNA。
【請求項7】
ヒトCD3ζ鎖が、以下の(a) 〜(c)からなる群から選ばれる蛋白質である、請求項2〜5のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号6で表されるアミノ酸配列からなる蛋白質;
(b) 配列番号6で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質;
(c) 配列番号6で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質。
【請求項8】
ヒトFcε受容体Iγ鎖が、以下の (a)または(b)から選ばれるDNAがコードする蛋白質である、請求項2〜7のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号7で表される塩基配列からなるDNA;
(b) 配列番号7で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質をコードするDNA。
【請求項9】
ヒトFcε受容体Iγ鎖が、以下の(a) 〜(c)からなる群から選ばれる蛋白質である、請求項2〜7のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号8で表されるアミノ酸配列からなる蛋白質;
(b) 配列番号8で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質;
(c) 配列番号8で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質。
【請求項10】
ヒトCD16プロモーター領域が、以下の(a)または(b)から選ばれるDNAである、請求項3〜9のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号9で表される塩基配列からなるDNA;
(b) 配列番号9で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつナチュラルキラー細胞、マクロファージ、単球または樹状細胞内でヒトCD16またはそのヒトアクセサリー分子を発現させる活性を有するDNA;
【請求項11】
非ヒト動物が、ウシ、ヒツジ、ヤギ、ブタ、ウマ、マウス、ラット、ニワトリ、サル及びウサギからなら群から選ばれる動物である、請求項1〜10のいずれか1項に記載の非ヒト動物またはその子孫。
【請求項12】
以下の(1)〜(3)からなる工程を含むことを特徴とする、抗体の抗体依存性細胞傷害活性を測定する方法。
(1) 請求項1〜11のいずれか1項に記載の非ヒト動物またはその子孫より、エフェクター細胞を単離する工程;
(2) 単離したエフェクター細胞と、抗体及び該抗体の標的となる抗原を発現している細胞とを混合し、反応液中でエフェクター細胞と、抗体及び該抗体の標的となる抗原を発現している細胞とを接触させる工程;
(3) 反応液中の該標的となる抗原を発現している細胞の生存率を定量する工程。
【請求項13】
以下の(1)および(2)からなる工程を含むことを特徴とする、抗体の抗体依存性細胞傷害活性に基づく薬理活性を評価する方法。
(1) 請求項1〜11のいずれか1項に記載の非ヒト動物またはその子孫に、被検物質である抗体を投与する工程;
(2) 該非ヒト動物またはその子孫において
被検物質である抗体の抗体依存性細胞傷害活性に基づく薬理活性を評価する工程。
【請求項14】
以下の(1)〜(3)からなる工程を含むことを特徴とする、抗体のスクリーニング方法。
(1)請求項1〜11のいずれか1項に記載の非ヒト動物またはその子孫に被検物質である抗体を投与する工程;
(2) 該非ヒト動物またはその子孫において被検物質である抗体の抗体依存性細胞傷害活性に基づく薬理活性を評価する工程;
(3)該薬理活性を有する抗体を選択する工程。
【請求項15】
抗体が、以下の(a)〜(e)からなる群から選ばれる蛋白質である、請求項12〜14のいずれか1項に記載の方法。
(a) ヒト抗体;
(b) ヒト化抗体;
(c) ヒト型キメラ抗体
(d) (a)、(b)または(c)のFc領域を含む抗体断片;
(e) (a)、(b)または(c)のFc領域を有する融合蛋白質。
【請求項16】
抗体のクラスがIgGである、請求項15に記載の方法。
【請求項1】
ヒトCD16とそのヒトアクセサリー分子とを共発現させるようにゲノムが改変された、非ヒト動物またはその子孫。
【請求項2】
ヒトCD16のヒトアクセサリー分子が、以下の(a)〜(c) からなる群から選ばれる分子である、請求項1に記載の非ヒト動物またはその子孫。
(a) ヒトCD3ζ鎖;
(b) ヒトFcε受容体Iγ鎖;
(c) ヒトCD3ζ鎖とヒトFcε受容体Iγ鎖。
【請求項3】
ヒトCD16またはそのヒトアクセサリー分子の発現が、ヒトCD16プロモーター領域で制御されるようにゲノムが改変された、請求項1または2に記載の非ヒト動物またはその子孫。
【請求項4】
ヒトCD16が、以下の (a) 〜(d)からなる群から選ばれるDNAがコードする蛋白質である、請求項1〜3のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号1で表される塩基配列からなるDNA;
(b) 配列番号2で表される塩基配列からなるDNA;
(c) 配列番号1で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒト抗体のFc領域と結合する活性を有する蛋白質をコードするDNA;
(d) 配列番号2で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒト抗体のFc領域と結合する活性を有する蛋白質をコードするDNA。
【請求項5】
ヒトCD16が、以下の(a) 〜(f)からなる群から選ばれる蛋白質である、請求項1〜3のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号3で表されるアミノ酸配列からなる蛋白質;
(b) 配列番号4で表されるアミノ酸配列からなる蛋白質;
(c) 配列番号3で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(d) 配列番号4で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(e) 配列番号3で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質;
(f) 配列番号4で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒト抗体のFc領域と結合する活性を有する蛋白質。
【請求項6】
ヒトCD3ζ鎖が、以下の (a)または(b)であるDNAがコードする蛋白質である、請求項2〜5のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号5で表される塩基配列からなるDNA;
(b) 配列番号5で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質をコードするDNA。
【請求項7】
ヒトCD3ζ鎖が、以下の(a) 〜(c)からなる群から選ばれる蛋白質である、請求項2〜5のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号6で表されるアミノ酸配列からなる蛋白質;
(b) 配列番号6で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質;
(c) 配列番号6で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質。
【請求項8】
ヒトFcε受容体Iγ鎖が、以下の (a)または(b)から選ばれるDNAがコードする蛋白質である、請求項2〜7のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号7で表される塩基配列からなるDNA;
(b) 配列番号7で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質をコードするDNA。
【請求項9】
ヒトFcε受容体Iγ鎖が、以下の(a) 〜(c)からなる群から選ばれる蛋白質である、請求項2〜7のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号8で表されるアミノ酸配列からなる蛋白質;
(b) 配列番号8で表されるアミノ酸配列において、1以上のアミノ酸が欠失、置換、挿入および/または付加されたアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質;
(c) 配列番号8で表されるアミノ酸配列と80%以上の相同性を有するアミノ酸配列からなり、かつヒトCD16と会合し細胞内情報伝達機能を有する蛋白質。
【請求項10】
ヒトCD16プロモーター領域が、以下の(a)または(b)から選ばれるDNAである、請求項3〜9のいずれか1項に記載の非ヒト動物またはその子孫。
(a) 配列番号9で表される塩基配列からなるDNA;
(b) 配列番号9で表される塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつナチュラルキラー細胞、マクロファージ、単球または樹状細胞内でヒトCD16またはそのヒトアクセサリー分子を発現させる活性を有するDNA;
【請求項11】
非ヒト動物が、ウシ、ヒツジ、ヤギ、ブタ、ウマ、マウス、ラット、ニワトリ、サル及びウサギからなら群から選ばれる動物である、請求項1〜10のいずれか1項に記載の非ヒト動物またはその子孫。
【請求項12】
以下の(1)〜(3)からなる工程を含むことを特徴とする、抗体の抗体依存性細胞傷害活性を測定する方法。
(1) 請求項1〜11のいずれか1項に記載の非ヒト動物またはその子孫より、エフェクター細胞を単離する工程;
(2) 単離したエフェクター細胞と、抗体及び該抗体の標的となる抗原を発現している細胞とを混合し、反応液中でエフェクター細胞と、抗体及び該抗体の標的となる抗原を発現している細胞とを接触させる工程;
(3) 反応液中の該標的となる抗原を発現している細胞の生存率を定量する工程。
【請求項13】
以下の(1)および(2)からなる工程を含むことを特徴とする、抗体の抗体依存性細胞傷害活性に基づく薬理活性を評価する方法。
(1) 請求項1〜11のいずれか1項に記載の非ヒト動物またはその子孫に、被検物質である抗体を投与する工程;
(2) 該非ヒト動物またはその子孫において
被検物質である抗体の抗体依存性細胞傷害活性に基づく薬理活性を評価する工程。
【請求項14】
以下の(1)〜(3)からなる工程を含むことを特徴とする、抗体のスクリーニング方法。
(1)請求項1〜11のいずれか1項に記載の非ヒト動物またはその子孫に被検物質である抗体を投与する工程;
(2) 該非ヒト動物またはその子孫において被検物質である抗体の抗体依存性細胞傷害活性に基づく薬理活性を評価する工程;
(3)該薬理活性を有する抗体を選択する工程。
【請求項15】
抗体が、以下の(a)〜(e)からなる群から選ばれる蛋白質である、請求項12〜14のいずれか1項に記載の方法。
(a) ヒト抗体;
(b) ヒト化抗体;
(c) ヒト型キメラ抗体
(d) (a)、(b)または(c)のFc領域を含む抗体断片;
(e) (a)、(b)または(c)のFc領域を有する融合蛋白質。
【請求項16】
抗体のクラスがIgGである、請求項15に記載の方法。
【図1】


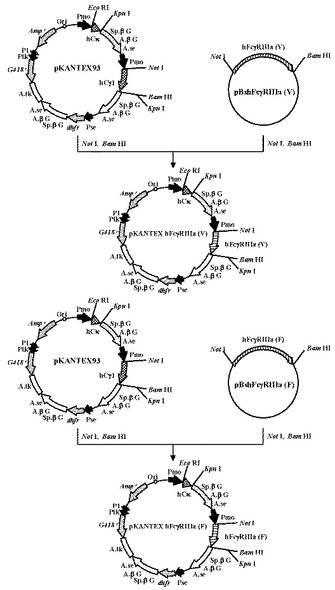
【図2】
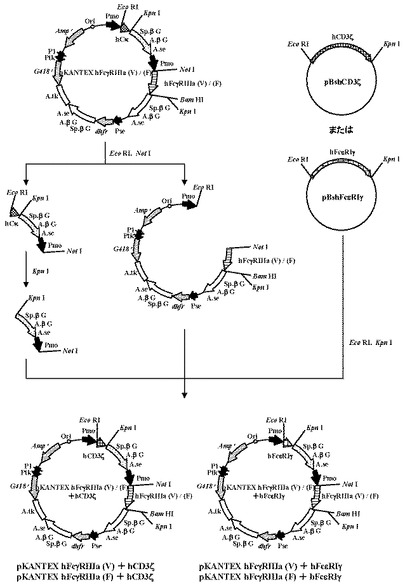
【図3】
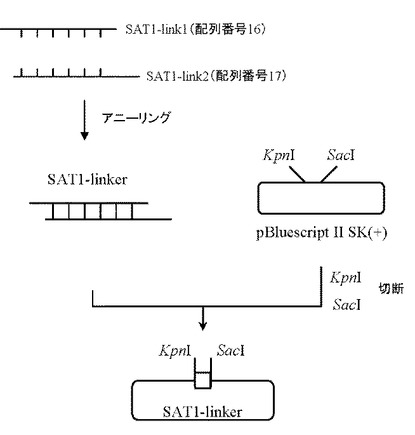
【図4】
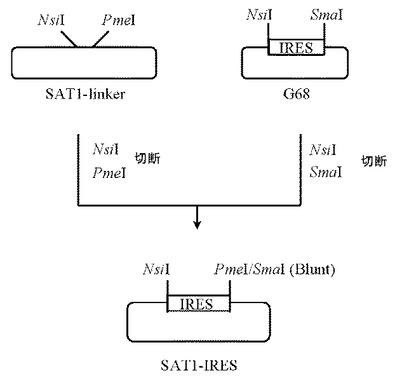
【図5】
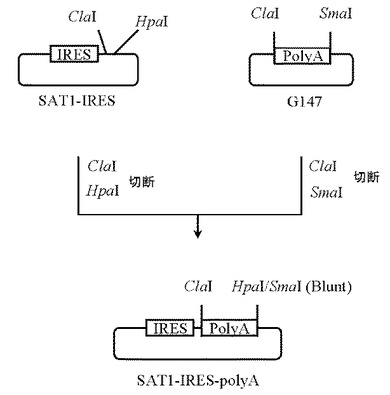
【図6】
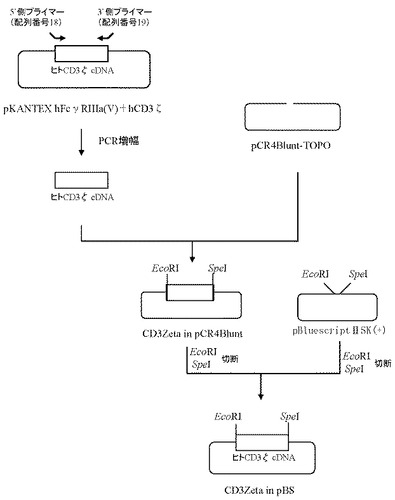
【図7】
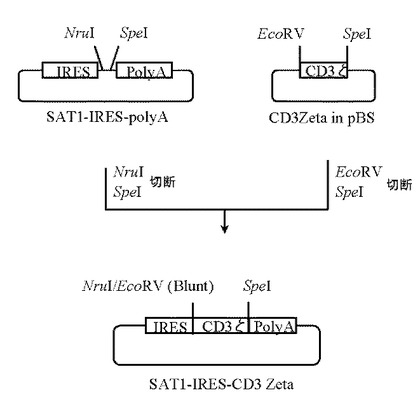
【図8】
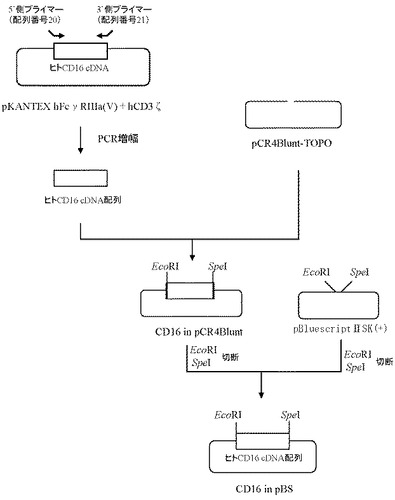
【図9】
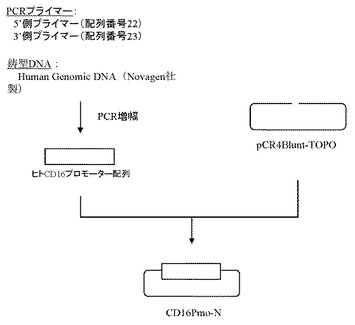
【図10】
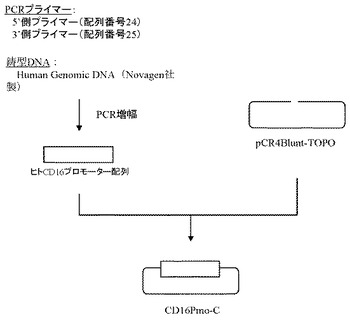
【図11】
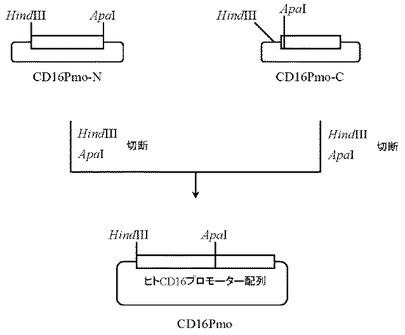
【図12】
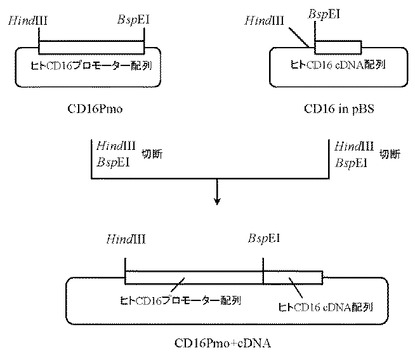
【図13】
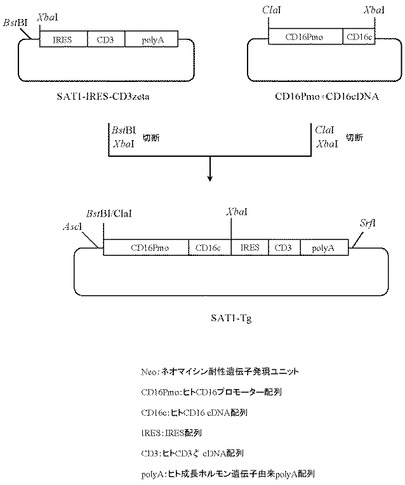
【図14】
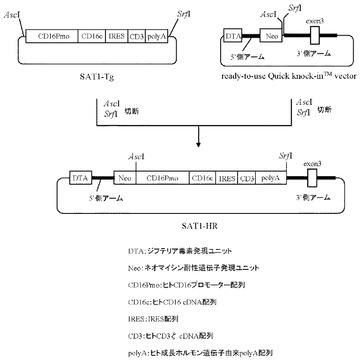
【図15】
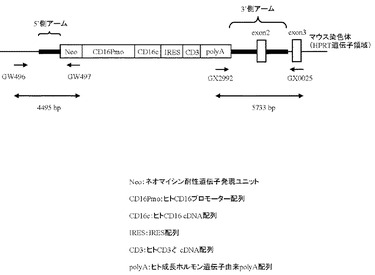
【図16】
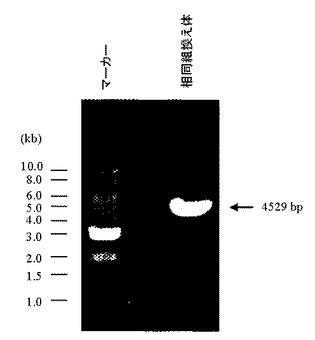
【図17】
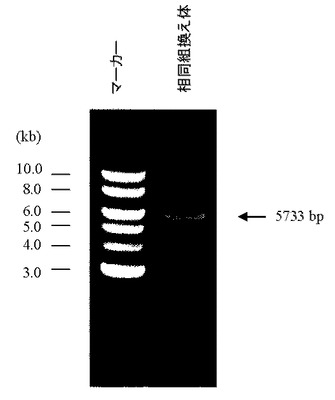
【図18】
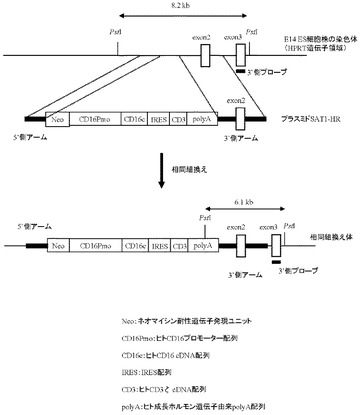
【図19】
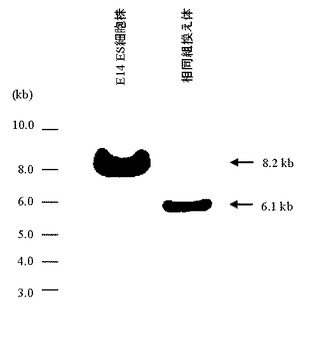
【図20】
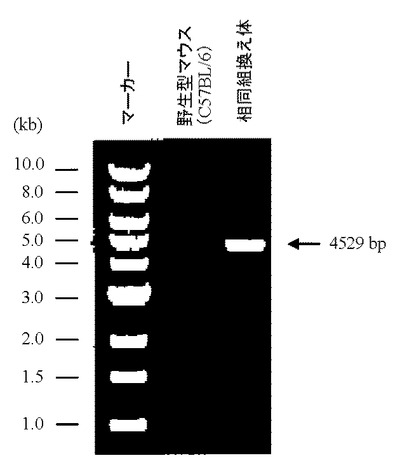
【図21】
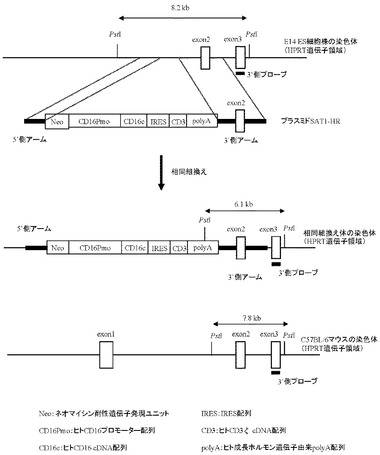
【図22】
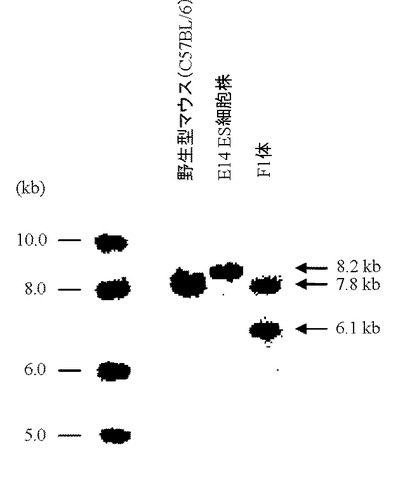
【図23】
【図24】
【図25】
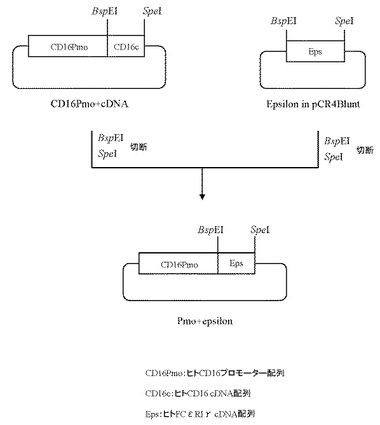
【図26】
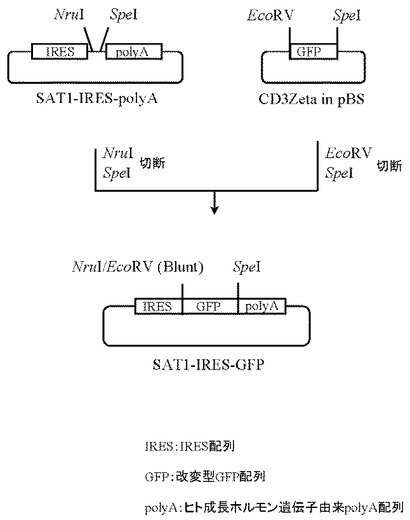
【図27】
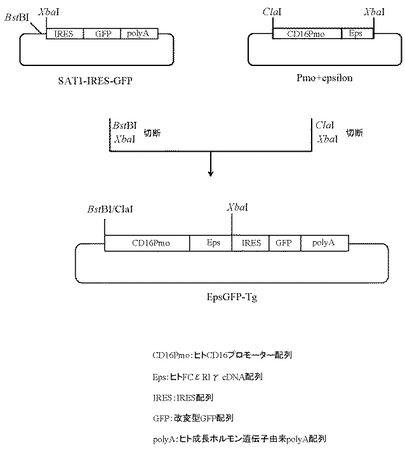
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【公開番号】特開2008−154468(P2008−154468A)
【公開日】平成20年7月10日(2008.7.10)
【国際特許分類】
【出願番号】特願2006−344071(P2006−344071)
【出願日】平成18年12月21日(2006.12.21)
【出願人】(000001029)協和醗酵工業株式会社 (276)
【Fターム(参考)】
【公開日】平成20年7月10日(2008.7.10)
【国際特許分類】
【出願日】平成18年12月21日(2006.12.21)
【出願人】(000001029)協和醗酵工業株式会社 (276)
【Fターム(参考)】
[ Back to top ]