
ヒトGタンパク質共役型受容体の内因性および非内因性型

【課題】受容体アゴニスト、逆アゴニストまたは部分アゴニストとしての候補化合物の直
接同定のために使用され得る受容体を提供すること。
【解決手段】本発明の特許明細書の中で開示される本発明は、膜貫通受容体に関し、さら
に具体的には内因性リガンドが未知であるヒトGタンパク質共役型受容体(「オーファン
GPCR受容体」)に関し、そして最も具体的には、構成的活性の証拠のためのヒトGP
CRの変異(非内因性)型に関する。
接同定のために使用され得る受容体を提供すること。
【解決手段】本発明の特許明細書の中で開示される本発明は、膜貫通受容体に関し、さら
に具体的には内因性リガンドが未知であるヒトGタンパク質共役型受容体(「オーファン
GPCR受容体」)に関し、そして最も具体的には、構成的活性の証拠のためのヒトGP
CRの変異(非内因性)型に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本特許明細書中に開示される本発明は膜貫通受容体に関し、そしてさらに具体的にはヒ
トGタンパク質共役型受容体に関し、そして特定すると、受容体の構成的活性を確立また
は増強するために改変されたGPCRの非内因性型(version)に特に重点を置いた内因性
ヒトGPCRに関する。好ましくは、改変されたGPCRは、治療薬剤として潜在的な適
用可能性を有する受容体アゴニスト、逆アゴニストまたは部分アゴニストとしての候補化
合物の直接同定のために使用される。
【背景技術】
【0002】
多数の受容体の種類がヒト内に存在するけれども、特に最も数が多くそして治療的に関
連するものはGタンパク質共役型受容体(GPCRまたはGPCRs)の種類により代表
される。ヒトゲノム内に約100,000個の遺伝子が存在すると推定され、これらの中
の約2%、すなわち2,000個の遺伝子がGPCRをコードすると推定される。そのた
めの内因性リガンドが同定されているGPCRを含む受容体は、「既知」受容体と呼ばれ
、一方そのための内因性リガンドが同定されていない受容体は「オーファン」受容体と呼
ばれる。GPCRは薬剤製品の開発のために重要な分野であり、100種の既知GPCR
の中の約20種から、約60%がすべて処方薬で開発されている。
【0003】
GPCRは、共通の構造モチーフを共有している。すべてのこれらの受容体は、7個の
αラセンを形成する22〜24個の疎水性アミノ酸の7個の配列を有し、これらそれぞれ
は膜をスパン(span)する(それぞれのスパンは番号で特定され、すなわち、膜貫通部−1
(TM−1)、膜貫通部−1(TM−1)、などである)膜貫通ラセンは、膜貫通部−2
と膜貫通部−3、膜貫通部−4と膜貫通部−5、および膜貫通部−6と膜貫通部−7との
間のアミノ酸の鎖により細胞膜の外側、すなわち「細胞外」側で結合される(これらは「
細胞外」領域1、2および3(EC−1、EC−2およびEC−3)とそれぞれ呼ばれる
)。膜貫通ラセンは、膜貫通部−1と膜貫通部−2、膜貫通部−3と膜貫通部−4、およ
び膜貫通部−5と膜貫通部−6との間のアミノ酸の鎖により細胞膜の内側、すなわち「細
胞内」側でも結合される(これらは「細胞内」領域1、2および3(IC−1、IC−2
およびIC−3)とそれぞれ呼ばれる)。受容体の「カルボキシ」(”C”)末端は細胞
内部の細胞内空間内にあり、そして受容体の「アミノ」(”N”)末端は細胞外側の細胞
外空間内にある。
【0004】
一般に、内因性リガンドが受容体を結合する場合には(しばしば受容体の「活性化」と
呼ばれる)、細胞内領域と細胞内「Gタンパク質」との間の共役を許容する細胞内領域の
コンホーメーションの変化がある。GPCRはGタンパク質に関して「多数関係」性であ
る、すなわち、GPCRは1個を越えるGタンパク質と相互作用できると報告されている
。非特許文献1参照。他のGタンパク質も存在するけれども、現在、Gq、Gs、Gi、
GzおよびGoが同定されたGタンパク質である。Gタンパク質との内因性リガンド活性
化GPCR共役は、シグナル伝達カスケードプロセスを開始する(「シグナル伝達」と呼
ばれる)。正常条件下で、シグナル伝達は、最終的に細胞活性化または細胞阻害をもたら
す。受容体のIC−3ループならびにカルボキシ末端がGタンパク質と相互作用すると考
えられる。
【0005】
生理的条件下で、GPCRは、2個の異なるコンホーメーション、すなわち「不活性」
状態および「活性」状態の間で平衡して細胞膜内に存在する。不活性状態にある受容体は
、細胞内シグナル伝達経路に連結して生物学的応答を生成できない。活性状態への受容体
コンホーメーションの変化は、シグナル伝達経路への連結を許容し(Gタンパク質を介し
て)そして生物学的反応を生成する。
【0006】
受容体は、内因性リガンドまたは薬剤などの化合物により活性状態に安定化されてもよ
い。受容体のアミノ酸配列の変更を含みこれに限られない最近の発見は、活性状態コンホ
ーメーションにおける受容体を促進および安定化するための内因性リガンドまたは薬剤以
外の手段を提供する。これらの手段は、受容体に結合する内因性リガンドの作用を模擬す
ることにより活性状態にある受容体を効果的に安定化する。このようなリガンド依存手段
による安定化は、「構成的受容体活性化」と呼ばれる。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Kenakin,T., 43 Life Science 1095(1988)
【発明の概要】
【課題を解決するための手段】
【0008】
(発明の概要)
本明細書中には、ヒトGPCRの内因性および非内因性型およびその使用を開示する。
本発明はまた、以下の項目を提供する。
(項目1) 配列番号2のアミノ酸配列によりコードされるGタンパク質共役型受容
体。
(項目2) 項目1のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目3) 配列番号1のベクターおよびcDNAを含んでなるプラスミド。
(項目4) 項目3のプラスミドを含んでなる宿主細胞。
(項目5) 配列番号4のアミノ酸配列によりコードされるGタンパク質共役型受容
体。
(項目6) 項目5のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目7) 配列番号3のベクターおよびcDNAを含んでなるプラスミド。
(項目8) 項目7のプラスミドを含んでなる宿主細胞。
(項目9) 配列番号6のアミノ酸配列によりコードされるGタンパク質共役型受容
体。
(項目10) 項目9のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目11) 配列番号5のベクターおよびcDNAを含んでなるプラスミド。
(項目12) 項目11のプラスミドを含んでなる宿主細胞。
(項目13) 配列番号8のアミノ酸配列によりコードされるGタンパク質共役型受
容体。
(項目14) 項目13のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目15) 配列番号7のベクターおよびcDNAを含んでなるプラスミド。
(項目16) 項目15のプラスミドを含んでなる宿主細胞。
(項目17) 配列番号10のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目18) 項目17のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目19) 配列番号9のベクターおよびcDNAを含んでなるプラスミド。
(項目20) 項目19のプラスミドを含んでなる宿主細胞。
(項目21) 配列番号12のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目22) 配列番号84のアミノ酸配列を含んでなる項目21のGタンパク質共
役型受容体の非内因性、構成的活性化型。
(項目23) 配列番号11のベクターおよびcDNAを含んでなるプラスミド。
(項目24) 項目23のプラスミドを含んでなる宿主細胞。
(項目25) 配列番号14のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目26) 配列番号88のアミノ酸配列を含んでなる項目25のGタンパク質共
役型受容体の非内因性、構成的活性化型。
(項目27) 配列番号13のベクターおよびcDNAを含んでなるプラスミド。
(項目28) 項目27のプラスミドを含んでなる宿主細胞。
(項目29) 配列番号16のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目30) 配列番号92のアミノ酸配列を含んでなる項目29のGタンパク質共
役型受容体の非内因性、構成的活性化型。
(項目31) 配列番号15のベクターおよびcDNAを含んでなるプラスミド。
(項目32) 項目31のプラスミドを含んでなる宿主細胞。
(項目33) 配列番号18のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目34) 項目33のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目35) 配列番号17のベクターおよびcDNAを含んでなるプラスミド。
(項目36) 項目35のプラスミドを含んでなる宿主細胞。
(項目37) 配列番号20のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目38) 項目37のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目39) 配列番号19のベクターおよびcDNAを含んでなるプラスミド。
(項目40) 項目39のプラスミドを含んでなる宿主細胞。
(項目41) 配列番号22のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目42) 項目41のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目43) 配列番号21のベクターおよびcDNAを含んでなるプラスミド。
(項目44) 項目43のプラスミドを含んでなる宿主細胞。
(項目45) 配列番号24のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目46) 項目45のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目47) 配列番号23のベクターおよびcDNAを含んでなるプラスミド。
(項目48) 項目47のプラスミドを含んでなる宿主細胞。
(項目49) 配列番号26のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目50) 項目49のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目51) 配列番号25のベクターおよびcDNAを含んでなるプラスミド。
(項目52) 項目51のプラスミドを含んでなる宿主細胞。
(項目53) 配列番号28のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目54) 項目53のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目55) 配列番号27のベクターおよびcDNAを含んでなるプラスミド。
(項目56) 項目55のプラスミドを含んでなる宿主細胞。
(項目57) 配列番号30のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目58) 項目57のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目59) 配列番号29のベクターおよびcDNAを含んでなるプラスミド。
(項目60) 項目59のプラスミドを含んでなる宿主細胞。
(項目61) 配列番号32のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目62) 配列番号96のアミノ酸配列を含んでなる項目61のGタンパク質共
役型受容体の非内因性、構成的活性化型。
(項目63) 配列番号95のベクターおよびcDNAを含んでなるプラスミド。
(項目64) 項目63のプラスミドを含んでなる宿主細胞。
(項目65) 配列番号34のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目66) 項目65のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目67) 配列番号33のベクターおよびcDNAを含んでなるプラスミド。
(項目68) 項目67のプラスミドを含んでなる宿主細胞。
(項目69) 配列番号36のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目70) 項目69のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目71) 配列番号35のベクターおよびcDNAを含んでなるプラスミド。
(項目72) 項目71のプラスミドを含んでなる宿主細胞。
(項目73) 配列番号38のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目74) 項目73のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目75) 配列番号37のベクターおよびcDNAを含んでなるプラスミド。
(項目76) 項目75のプラスミドを含んでなる宿主細胞。
(項目77) 配列番号40のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目78) 項目77のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目79) 配列番号39のベクターおよびcDNAを含んでなるプラスミド。
(項目80) 項目79のプラスミドを含んでなる宿主細胞。
【図面の簡単な説明】
【0009】
【図1】対照(「CMV」)と比較した内因性型RUP12(「RUP12」)からの第二メッセンジャーIP3 産生の説明である。
【図2】内因性RUP13(「RUP13」)と対照ベクター(「CMV」)との構成的シグナル伝達の比較結果を与える第二メッセンジャー細胞基礎サイクリックAMPアッセイの結果の図示である。
【図3】CMV、内因性RUP13(「RUP13wt」)および非内因性、構成的活性化RUP13(「RUP13(A268K)」)を8XCRE−Lucレポータープラスミドを用いて比較して測定したシグナルの図示である。
【図4】RUP13:「Gs融合タンパク質」(「RUP13−Gs」)と対照ベクター(「CMV」)による構成的シグナル伝達の比較結果を与える〔35S〕GTPγSアッセイの結果の図示である。
【図5】CMV、内因性RUP14(「RUP14wt」)および非内因性、構成的活性化RUP13(「RUP14(L246K)」)を8XCRE−Lucレポータープラスミドを用いて比較して測定したシグナルの図示である。
【図6】CMV、内因性RUP15(「RUP15wt」)および非内因性、構成的活性化RUP15(「RUP15(A398K)」)を8XCRE−Lucレポータープラスミドを用いて比較して測定したシグナルの図示である。
【図7】内因性RUP15(「RUP15wt」)、RUP15の非内因性、構成的活性化型(「RUP15(A398K)」)および対照ベクター(「CMV」)の構成的シグナル伝達の比較結果を与える第二メッセンジャー細胞基礎サイクリックAMPアッセイの結果の図示である。
【図8】RUP15:「Gs融合タンパク質」(「RUP15−Gs」)と対照ベクター(「CMV」)による構成的シグナル伝達の比較結果を与える〔35S〕GTPγSアッセイの結果の図示である。
【図9】対照(「CMV」)と比較した内因性型RUP17(「RUP17」)からの第二メッセンジャーIP3 産生の説明である。
【図10】対照(「CMV」)と比較した内因性型RUP21(「RUP21」)からの第二メッセンジャーIP3 産生の説明である。
【図11】CMV、内因性RUP23(「RUP23wt」)および非内因性、構成的活性化RUP23(「RUP23(W275K)」)を8XCRE−Lucレポータープラスミドを用いて比較して測定したシグナルの図示である。
【図12】RUP13に対する各種の候補化合物の一次スクリーニングからの結果の図示である。「化合物A」の結果はウエルA2に示されそして「化合物B」の結果はウエルG9に示される。
【発明を実施するための形態】
【0010】
(発明の詳細な記述)
受容体に関連して作成された科学文献は、受容体に関して種々の作用を有するリガンド
に関する多数の用語を採用している。明白さおよび統一のために、下記の定義を本特許明
細書中を通じて使用する。これらの定義がこれらの用語のための他の定義と矛盾する場合
には、下記の定義が優先する。
【0011】
「アゴニスト」は、物質が受容体と結合した場合に細胞内反応を活性化するか、または
膜にへのGTP結合を増強する物質(例えばリガンド、候補化合物)を意味するものとす
る。
【0012】
本明細書中に使用される「アミノ酸の略字」を表Aに記載する。
表A
アラニン ALA A
アルギニン ARG R
アスパラギン ASN N
アスパラギン酸 ASP D
システイン CYS C
グルタミン酸 GLU E
グルタミン GLN Q
グリシン GLY G
ヒスチジン HIS H
イソロイシン ILE I
ロイシン LEU L
リシン LYS K
メチオニン MET M
フェニルアラニン PHE F
プロリン PRO P
セリン SER S
トレオニン THR T
トリプトファン TRP W
チロシン TYR Y
バリン VAL V
「部分アゴニスト」は、これらが受容体に結合された場合にアゴニストの作用よりも低
い程度/範囲で細胞内反応を活性化するか、またはアゴニストの作用よりも低い程度/範
囲で膜へのGTP結合を増強する物質(例えばリガンド、候補化合物)を意味するものと
する。
【0013】
「アンタゴニスト」は、アゴニストと同じ部位で受容体に競合的に結合するがしかし受
容体の活性形により開始された細胞内反応を活性化せず、そしてこれによりアゴニストま
たは部分アゴニストによる細胞内反応を阻害できる物質(例えばリガンド、候補化合物)
を意味するものとする。「アンタゴニスト」は、アゴニストまたは部分アゴニストが存在
しない場合に基準細胞内反応を低下させない。
【0014】
「候補化合物」は、スクリーニング技術に適用できる分子(例えば化学化合物であり、
これに限定はされない)を意味するものとする。好ましくは、語句「候補化合物」は、あ
らかじめ間接同定過程により決定されたもの(「間接同定された化合物」)であって、受
容体に対して逆アゴニスト、アゴニストまたはアンタゴニストからなる群から選ばれた化
合物として一般的に知られている化合物を含まず、さらに好ましくは、少なくとも1種の
哺乳動物内において治療効力を有すると以前に決定された間接同定された化合物を含まず
、そして最も好ましくは、ヒトにおいて治療効力を有すると以前に決定された間接同定さ
れた化合物を含まない。
【0015】
「組成物」は、少なくとも1種の成分を含んでなる物質を意味する。「薬剤組成物」は
、組成物の一例である。
【0016】
「化合物効力」は、受容体結合親和性に対抗して、受容体機能性を阻害または刺激する
ための化合物の能力の尺度を意味するものとする。化合物効力を検出するための例示的な
手段は、本明細書の実施例の部分に開示される。
【0017】
「コドン」は、一般に、リン酸基に結合し、そして翻訳されるとアミノ酸をコードする
ヌクレオシド(アデノシン(A)、グアノシン(G)、シチジン(C)、ウリジン(U)
およびチミジン(T))を含んでなる3個のヌクレオチド(またはヌクレオチドの等価基
)の群を意味するものとする。
【0018】
「構成的活性化受容体」は、構成的受容体活性化を受ける受容体を意味するものとする
。構成的活性化受容体は内因性または非内因性であることができる。
【0019】
「構成的受容体活性化」は、受容体とその内因性リガンドまたはその化学的等価物との
結合以外の手段により活性状態にある受容体の安定化を意味するものとする。
【0020】
「接触」または「接触する」は、生体外系または生体内系のいずれでも、少なくとも2
個の部分を一緒にすることを意味するものとする。
【0021】
語句「候補化合物」に関連する「直接同定する」または「直接同定」は、構成的活性化
受容体、好ましくは構成的活性化オーファン受容体、そして最も好ましくは構成的活性化
Gタンパク質共役細胞表面オーファン受容体に対して候補化合物をスクリーニングし、そ
してこの化合物の化合物効力を評価することを意味するものとする。この語句は、いかな
る場合でも、語句「間接同定する」または「間接同定された」により包含されまたはこれ
らを包含するとは解釈または理解されない。
【0022】
「内因性」は、哺乳動物が本来的に産生する物質を意味するものとする。例えば、限定
ではないが用語「受容体」に関する「内因性」は、哺乳動物(例えば、限定ではないがヒ
ト)またはウイルスにより本来的に産生されるものを意味するものとする。反対に、この
範囲内での用語「非内因性」は、哺乳動物(例えば、限定ではないがヒト)またはウイル
スにより本来的には産生されないものを意味するものとする。例えば、限定ではないが、
その内因性形では構成的活性ではないが、しかし操作されると構成的活性となる受容体は
、本明細書内では「非内因性、構成的活性化受容体」と呼ばれるのが最も好ましい。両方
の用語は、「インビボ」および「インビトロ」系の両方を記述するために使用できる。例
えば、限定ではないが、スクリーニング法において、内因性または非内因性受容体は、生
体内スクリーニング系に関連してもよい。別の例であって限定ではないが、哺乳動物のゲ
ノムが非内因性、構成的活性化受容体を含むように操作された場合に、生体内系の手段に
よる候補化合物のスクリーニングが適用可能である。
【0023】
「Gタンパク質共役型受容体融合タンパク質」および「GPCR融合タンパク質」は、
本明細書中で開示する本発明の範囲内で、それぞれ、少なくとも1種のGタンパク質、最
も好ましくはこのようなGタンパク質のアルファ(α)サブユニット(これはGTPを結
合するサブユニットである)に融合された内因性、構成的活性化GPCRまたは非内因性
、構成的活性化GPCRを含んでなる非内因性タンパク質を意味し、ここでGタンパク質
は、好ましくは内因性オーファンGPCRと本来的に共役するGタンパク質と同じ種類で
ある。例えば、限定ではないが、内因性状態において、Gタンパク質”Gsα”がGPC
Rと共役する優勢なGタンパク質である場合に、特定のGPCRに基づく「GPCR融合
タンパク質」はGsαに融合したGPCRを含んでなる非内因性タンパク質であろう。あ
る場合には、以下に記載するように、非−優勢Gタンパク質はGPCRに融合できる。G
タンパク質は、構成的活性GPCRのc−末端に直接融合できるかまたはこれら2者の間
にスペーサーがあってもよい。
【0024】
「宿主細胞」は、その中に「プラスミド」および/または「ベクター」を組み込まれる
ことができる細胞を意味するものとする。原核生物「宿主細胞」の場合に、「プラスミド
」は、典型的には、「宿主細胞」複製物として自律性分子として複製される(従って一般
に、「プラスミド」は、真核生物「宿主細胞」内への導入のために単離される)。真核生
物「宿主細胞」の場合には、「プラスミド」は真核生物「宿主細胞」が複製された場合に
「プラスミド」が複製されるように、「宿主細胞」の細胞DNA中に組み込まれる。好ま
しくは、本明細書中に開示する本発明の目的のためには、「宿主細胞」は真核生物であり
、さらに好ましくは、哺乳動物であり、そして最も好ましくは293、293TおよびC
OS−7細胞からなる群より選ばれる。
【0025】
「間接同定」または「間接同定された」は、内因性受容体に特異性の内因性リガンドの
同定、リガンド−受容体相互作用を妨害および/またはこれと競合するものの決定のため
の受容体に対する候補化合物のスクリーニング、および活性化受容体と関連する少なくと
も1種の第二メッセンジャー経路に影響するための化合物の効力の評価を含む薬剤発見過
程への伝統的な接近方法を意味する。
【0026】
「阻害」または「阻害する」は、用語「反応」に関連して、化合物が存在しない場合の
反対に、化合物が存在する場合には反応が低下または防止されることを意味するものとす
る。
【0027】
「逆アゴニスト」は、受容体の内因性形または受容体の構成的活性化形のいずれかに結
合し、そしてアゴニストまたは部分アゴニストが存在しない場合に観察される正常の活性
基準レベル以下の受容体の活性形により開始される基礎細胞内反応を阻害し、または膜へ
のGTP結合を低下させる物質(例えばリガンド、候補化合物)を意味するものとする。
好ましくは、基礎細胞内反応は、逆アゴニストが存在しない場合の基準反応と比較して、
逆アゴニストの存在においては少なくとも30%、さらに好ましくは少なくとも50%、
そして最も好ましくは少なくとも75%が阻害される。
【0028】
「既知受容体」は、当該受容体に特異性の内因性リガンドが同定されている内因性受容
体を意味するものとする。
【0029】
「リガンド」は、内因性で本来的に存在する受容体に特異性の内因性で本来的に存在す
る分子を意味するものとする。
【0030】
内因性受容体の核酸および/またはアミノ酸配列に関する「変異体」または「変異」は
、内因性、非構成的活性化受容体の変異形が受容体の構成的活性化を明らかにするような
内因性配列への特定の変化または多数の変化を意味するものとする。特定の配列の等価体
に関して、(a)ヒト受容体の次の変異した形の構成的活性化のレベルが受容体の最初の
変異により明らかにされたものと本質的に同じであり、そして(b)受容体の次の変異形
と受容体の最初の変異体との間の配列(アミノ酸および/または核酸)相同性の割合が少
なくとも約80%、さらに好ましくは少なくとも約90%そして最も好ましくは少なくと
も約95%である場合に、ヒト受容体の次の変異形は、ヒト受容体の最初の変異に等価と
考えられる。理想的には、そして構成的活性化を達成するために本明細書中で開示した最
も好ましいカセットがGPCRの内因性と非内因性形との間の単一アミノ酸および/また
はコドン変化を含むという事実のために、配列相同性の割合は少なくとも98%でなけれ
ばならない。
【0031】
「非オーファン受容体」は、受容体へのリガンドの結合が分子内シグナル伝達経路を活
性化する、内因性で本来的に存在するリガンドに特異性の内因性で本来的に存在する分子
を意味するものとする。
【0032】
「オーファン受容体」は、その受容体に特異性の内因性リガンドが同定されていないか
または既知ではない内因性受容体を意味するものとする。
【0033】
「薬剤組成物」は、少なくとも1種の活性成分を含んでなる組成物を意味するものとし
、ここで組成物は哺乳動物(例えば、限定ではないがヒト)において特定の効力的な成果
のための試験が可能である。通常の当該技術分野の熟練者は、活性成分が、熟練者の要求
に基づいて所望の効力的な成果を有するかどうかを決定するために適当な技術を理解しそ
して認めるであろう。
【0034】
「プラスミド」は、「ベクター」およびcDNAの組合せを意味するものとする。一般
に、「プラスミド」は、タンパク質としてのcDNAの複製および/または発現の目的で
「宿主細胞」内に導入される。
【0035】
「第二メッセンジャー」は、受容体活性化の結果として生成した細胞内反応を意味する
ものとする。第二メッセンジャーは、例えば、イノシトール三リン酸(IP3 )、ジアシ
ルグリセロール(DAG)、サイクリックAMP(cAMP)、およびサイクリックGM
P(cGMP)を含むことができる。第二メッセンジャー反応は、受容体活性化の決定の
ために測定できる。さらに、第二メッセンジャー反応は、例えば逆アゴニスト、アゴニス
ト、部分アゴニストおよびアンタゴニストを含む候補化合物の直接同定のために測定でき
る。
【0036】
「刺激」または「刺激する」は、用語「反応」に関連して、反応が化合物の存在下で、
これが存在しない場合の反対に増加することを意味するものとする。
【0037】
cDNAの関連する「ベクター」は、少なくとも1種のcDNAを組み込むことが可能
でそして「宿主細胞」内に組み込むことが可能な環状DNAを意味するものとする。
【0038】
以下の各セクションの順序は、説明の効率のために決定したものであり、下記の開示ま
たは請求の制限を意図せず、そしてそのように解釈してはならない。
A.はじめに
受容体の伝統的な研究は、発見が受容体に影響できるアンタゴニストおよびその他の分
子を発見できるまで進行する前に内因性リガンドが最初に同定されなければならないとい
うアプリオリな仮定(歴史に基づく)から常に前進していた。アンタゴニストが最初に判
明した場合でも、探索は直ちに内因性リガンドの探索に進んだ。この思考の様式は、構成
的活性化受容体の発見の後でも受容体研究で継続された。これまで認められていなかった
ことは、受容体のアゴニスト、部分アゴニスト、および逆アゴニストの発見に最も有用な
ことは受容体の活性状態であることである。過剰活性の受容体または不足活性の受容体か
らもたらされる疾患に対して、治療薬剤に望まれるものは、それぞれ、受容体の活性状態
を低下するかまたは受容体の活性を増強する化合物であり、必ずしも内因性リガンドに対
するアンタゴニストである薬剤ではない。これは、活性受容体状態の活性を低下または増
強する化合物は、内因性リガンドとして同じ部位に結合する必要はないためである。従っ
て、本発明の方法により教示されるように、治療化合物のためのあらゆる探索もリガンド
に依存しない活性状態に対する化合物をスクリーニングすることにから開始すべきである
。
B.ヒトGPCRの同定
ヒトゲノムプロジェクトの研究は、ヒトゲノム内に位置する核酸配列に関する
極めて多数の情報の同定をもたらした。この研究の場合に、ゲノム配列情報は、
特定のゲノム配列がヒトタンパク質を翻訳するオープンリーディングフレーム情
報を含むかまたは含まないかどうかに関する理解または認識を伴わずにに提供さ
れた。ヒトゲノム内の核酸配列を同定する数種の方法は、通常の当該技術分野を
有する熟練者の認識範囲内にある。例えば、限定ではないが、本明細書中で開示
される種々のヒトGPCRは、ジーンバンク(GeneBank)TMデータベースを調査し
て発見された。下記の表Bは、我々が発見した数種類の内因性GPCRを、開示
GPCRに相同である他のGPCRと共に表示する。
【0039】
【表1】

【0040】
受容体相同性は、ヒト身体内の受容体の役割の評価を得るために有用である。本明細書
の記載の進行に応じて、我々は、これらの受容体の非内因性、構成的活性化型を確立する
ためにこれらの受容体を変異するための技術を開示するであろう。
【0041】
本明細書中に開示される技術は、当該技術分野では既知の他のヒト、オーファンGPC
Rにも適用され、これは本明細書の記載の進行に従って明らかとなるであろう。
【0042】
C.受容体スクリーニング
本明細書中で開示されるヒトGPCRの非内因性、構成的活性化型に対する候補化合物
のスクリーニングは、受容体の内因性リガンドの使用を必要としないで、この細胞表面受
容体に作用する候補化合物の直接同定を可能とする。慣用でしばしば商業的に利用できる
技術を用いて、本明細書中で開示されるヒトGPCRの内因性型が発現および/または過
剰発現される身体内の領域を決定できる。受容体の発現および/または過剰発現と関連す
る関連疾患/障害状態を決定するためにこれらの技術を用いることも可能であり、このよ
うな方法は、本特許明細書中に開示される。
【0043】
本明細書中で開示されるヒトGPCRの構成的活性化を明らかにするであろう変異の創
成に関しては、GPCRのTM6内に位置すると想定されるプロリン残基からの距離に基
づいている。このアルゴリズム技術は、2000年4月20日付けWO00/22129
として公開された同時係属および共通して譲渡された特許明細書PCT出願番号PCT/
US99/23938号中に開示されており、これは本明細書中に表示する他の特許文献
と一緒に引用することにより本明細書中に編入される。このアルゴリズム技術は、従来の
配列「整列(alignment) 」に基づいては断定されず、上記のTM6プロリン残基(または
、当然ではあるがこのようなプロリン残基の内因性構成的置換)からの指定の距離により
で決定される。この残基から16アミノ酸残基に位置するアミノ酸(推定すると受容体の
IC3領域内に位置する)を最も好ましいリシン残基に変異することにより、このような
活性化が得られるであろう。その他のアミノ酸残基は、この目的を達成するためにこの位
置における変異に有用であろう。
【0044】
D.疾患/障害同定および/または選択
以下に詳細に記載するように、最も好ましくは非内因性、構成的活性化GPCRに対す
る逆アゴニストおよびアゴニストは、本発明の方法論により同定できる。このような逆ア
ゴニストおよびアゴニストは、本受容体に関連する疾患を治療するための薬剤発見プログ
ラムにおけるリード化合物として理想の候補である。GPCRに対する逆アゴニストを直
接同定するための能力、これによる薬剤組成物の開発を可能とすることにより、GPCR
に関連する疾患および障害の探索が可能である。例えば、GPCRの存在に関する疾患お
よび正常組織試料の両方のスキャニングは、学問的な実験または特定のGPCRへの内因
性リガンドを同定するための経路に沿って追跡する程度を越えてきている。組織スキャン
は、健康および疾患組織の広い範囲にわたって行うことができる。このような組織スキャ
ンは、特定の受容体を疾患および/または障害と関連させる場合の好ましい第一段階を提
供する。
【0045】
好ましくは、ヒトGPCRのDNA配列は、(a)組織−mRNAに対するドット−ブ
ロット分析、および/または(b)組織試料中の受容体の発現のRT−PCT同定のため
のプローブを作製するために使用される。組織採取元内の受容体の存在、または疾患組織
、または正常組織と比較して疾患組織内の高い濃度にある受容体の存在は、限定はされな
いが治療方針とその疾患と関連する疾患との相関を同定するために好んで使用できる。受
容体は、この技術により器官の領域に同様に良く定位できる。受容体が局在する特定組織
の既知機能に基づいて、受容体の推定される機能的役割が推論できる。
【0046】
E.候補化合物のスクリーニング
1.包括的GPCRスクリーニングアッセイ技術
Gタンパク質受容体が構成的活性となった場合に、これはGタンパク質(例えばGq、
Gs、Gi、Gz、Go)に結合しそしてGタンパク質へのGTP結合を刺激する。従っ
てGタンパク質はGTPアーゼとして作用し、そしてゆっくりとGTPをGDPに加水分
解し、これにより正常の条件下で受容体は脱活性化する。しかし構成的活性化受容体は、
GDPをGTPに交換することを続ける。GTPの非加水分解性類似体である〔35S〕G
TPγSは、構成的活性化受容体を発現する膜への増強された結合を測定するために使用
できる。〔35S〕GTPγSが、リガンドが存在しないかまたは存在する場合に膜へのG
タンパク質共役を測定するために使用できることが報告されている。当該技術分野の熟練
者に周知で使用できるその他の例の中でも、この測定の例は、Traynor およびNahorskiに
より1995年に報告された。このアッセイ系の好ましい使用は、候補化合物の最初のス
クリーニングであり、それは、受容体の細胞内ドメインと相互作用する特定のGタンパク
質にかかわらずすべてのGタンパク質共役型受容体に系が包括的に適用できるからである
。
2.特異的GPCRスクリーニングアッセイ技術
候補化合物が「包括的」Gタンパク質共役型受容体アッセイ(すなわち、アゴニスト、
部分アゴニスト、または逆アゴニストである化合物を選択するためのアッセイ)を用いて
同定され、さらに化合物が受容体部位で相互作用したことを確認するためのスクリーニン
グをすることが好ましい。例えば、「包括的」アッセイで同定された化合物は、受容体に
結合しないであろうが、しかし細胞内ドメインからのGタンパク質を単に「脱共役」する
であろう。
【0047】
a.Gs、GzおよびGi
Gsは、酵素アデニル酸シクラーゼを刺激する。Gi(およびGzおよびGo)は、反
対にこの酵素を阻害する。アデニル酸シクラーゼは、ATPからcAMPへの変換に触媒
作用する。従って、Gsタンパク質に共役する構成的活性化GPCRは、cAMPの増加
した細胞レベルと関連する。反対に、Gi(またはGz、Go)タンパク質に共役する構
成的活性化GPCRは、cAMPの低下した細胞レベルと関連する。一般的には「シナプ
ス伝達の間接機序」第8章("Indirect Mechanisms of Synaptic Transmission," Chpt 8,
FromNeuron To Brain(3rd Ed.) Nichols, J.G. et al eds, Sinauer Associates, Inc.(1
992)) 参照。従って、cAMPを検出するアッセイは、候補化合物が例えば受容体に対し
て逆アゴニストであるかどうかを決定するために使用できる(すなわちこのような化合物
はcAMPのレベルを低下するであろう)。cAMPを測定するために当該技術分野で既
知の種々の方法が使用できる。最も好ましい方法は、ELISAに基づくフォーマットで
の抗−cAMP抗体の使用による。使用できる別のアッセイの種類は、全細胞第二メッセ
ンジャーレポーター系アッセイである。遺伝子上のプロモーターは、特定の遺伝子をコー
ドするタンパク質の発現を駆動する。サイクリックAMPは、cAMP反応性DNA結合
タンパク質または転写因子(CREB)の結合を促進して遺伝子発現を駆動し、転写因子
は次いでcAMP反応因子と呼ばれる特定部位でプロモーターに結合しそして遺伝子の発
現を駆動する。レポーター遺伝子、例えばβ−ガラクトシダーゼまたはルシフェラーゼの
前の多重cAMP反応因子を含むプロモーターを有するレポーター系が構築できる。従っ
て、構成体活性化されたGs−連結受容体は、次いで遺伝子を活性化するcAMPの蓄積
およびレポータータンパク質の発現を起こす。レポータータンパク質、例えばβ−ガラク
トシダーゼまたはルシフェラーゼは、次いで標準生物化学アッセイを用いて検出できる(C
henet al. 1995)。
【0048】
b.GoおよびGq
GoおよびGqは、酵素ホスホリパーゼCの活性化と関連し、これは次いでリン脂質P
IP2 を加水分解し、2個の細胞内メッセンジャー、ジアシクログリセロール(diacyclog
lycerol)(DAG)およびイノシトール 1,4,5−トリリン酸(IP3)を遊離する
。IP3 の増加した蓄積は、GqおよびGo関連受容体の活性化と関連する。一般的には
「シナプス伝達の間接機序」第8章("IndirectMechanisms of Synaptic Transmission,"
Chpt 8,From Neuron To Brain(3rdEd.) Nichols, J.G. et al eds, Sinauer Associates,
Inc. (1992)) 参照。IP3蓄積を検出するアッセイは、候補化合物が、例えばGqまた
はGo関連受容体に対する逆アゴニストであるかどうかを決定するために使用できる(す
なわち、このような化合物がIP3のレベルを低下する)。Gq関連受容体は、Gq依存
性ホスホリパーゼCがAP1因子を含む遺伝子の活性化を起こすAP1レポーターアッセ
イを用いてでも試験できる。従って、活性化Gq関連受容体は、この遺伝子の発現の増加
を明らかにし、これによりその逆アゴニストはこの発現の低下を明らかにし、そしてアゴ
ニストはこの発現の増加を明らかにする。このアッセイのために商業的に利用できるアッ
セイが利用できる。
【0049】
3.GPCR融合タンパク質
逆アゴニスト、アゴニストおよび部分アゴニストの直接同定のための候補化合物のスク
リーニングに使用するための内因性、構成的活性オーファンGPCRまたは非内因性、構
成的活性化オーファンGPCRの使用は、その定義により、結合する内因性リガンドが存
在しない場合でも受容体が活性であるという興味あるスクリーニング問題を提供する。従
って、例えば候補化合物の存在における非内因性受容体と、その化合物が存在しない場合
の非内因性受容体との間を区別するために、このような化合物が逆アゴニスト、アゴニス
ト、部分アゴニストであるかどうかまたはこのような受容体に影響しないかどうかに関す
る理解を可能とさせるこのような区別の目的で、このような区別を増強できる方法を使用
することが好ましい。好ましい方法は、「GPCR融合タンパク質」の使用である。
【0050】
一般に、非内因性オーファンGPCRが上記のアッセイ技術(ならびのその他のもの)
を用いて構成的に活性化されたことが一旦決定されると、内因性GPCRと共役する優勢
なGタンパク質を決定できる。GPCRへのGタンパク質の共役は、評価できるシグナル
伝達経路を提供する。スクリーニングが哺乳動物発現系を用いて行われることが最も好ま
しいので、このような系は、その中に内因性Gタンパク質を有すると期待される。従って
、定義により、このような系内で、非内因性、構成的活性化オーファンGPCRは、連続
的にシグナル発生する。この点に関して、例えば受容体への逆アゴニストの存在下でこの
シグナルが増強されることが好ましく、特にはスクリーニングの範囲内で、逆アゴニスト
と接触した受容体の間でさらに容易に区別できる可能性があるようにシグナルが増強され
ることがさらに好ましい。
【0051】
「GPCR融合タンパク質」は、非内因性GPCRと共役したGタンパク質の効力を増
強することを意図する。「GPCR融合タンパク質」は、非内因性、構成的活性化GPC
Rを用いるスクリーニングに好ましく、それは、このような方法がこのスクリーニング技
術で最も好ましく使用される信号を増加するからである。これは、有意の「信号対雑音比
」を容易に得るために重要である。このような有意の比は、本明細書中に開示する候補化
合物のスクリーニングに有利に導入される。
【0052】
「GPCR融合タンパク質」の発現のために有用な構築物の構築は、当該技術分野の通
常の熟練を有する者の認識範囲内である。商業的に利用できる発現ベクターおよび系は、
研究者の特定の要求を満足できる各種の方法を提供する。このような「GPCR融合タン
パク質」構築物のために重要な基準は、内因性GPCR配列およびGタンパク質配列の両
方がインフレームであり(好ましくは、内因性GPCRの配列がGタンパク質配列の上流
にある)そしてGPCRの「終止」コドンが、GPCRの発現の際にGタンパク質も発現
できるように欠失または置換されていなければならないことである。GPCRは、Gタン
パク質に直接連結してもよく、または2個の間のスペーサー残基があることもできる(好
ましくは、約12個を越えず、この数は当該技術分野の通常の熟練者には容易に確認でき
るものである)。我々は、使用されない幾つかの制限部位が、効果的に発現の際にスペー
サーとなるスペーサーの使用を好む(便利さのために)。最も好ましくは、非内因性GP
CRに共役するGタンパク質は、「GPCR融合タンパク質」構築物の創成の前に同定さ
れる。同定されたGタンパク質は僅かしかないので、Gタンパク質の配列を含んでなる構
築物(すなわち汎用Gタンパク質構築物)は、本明細書内の内因性GPCR配列の挿入の
ために利用できる。これは、異なる配列を有する種々の異なる内因性GPCRの大規模ス
クリーニングの範囲内で効率を与える。
【0053】
上記のように、Gi、GzおよびGoに共役する構成的活性化GPCRは、cAMPの
形成を阻害すると期待され、問題のGPCRのこれらの種類に基づくアッセイを興味ある
ものとする(すなわちcAMPシグナルは活性化の際に低下し、従って例えば逆アゴニス
トの直接同定を行う(これはこのシグナルをさらに低下させるであろう))。本明細書中
で開示するように、受容体のこれらの種類に対して、使用可能なシクラーゼに基づくアッ
セイを確立する研究において、内因性GPCRの内因性Gタンパク質には基づかない「G
PCR融合タンパク質」を創成することが可能であることを我々は確認した。従って、例
えば、内因性Gi共役型受容体はGsタンパク質に融合でき、我々はこのような融合構築
物が発現されると、内因性GPCRを、「本来的な」Giタンパク質ではなくて、例えば
Gsと共役するように「駆動」または「強制」し、シクラーゼに基づくアッセイを確立で
きると信じる。このように、Gi、GzおよびGo共役型受容体に対して、我々は、「G
PCR融合タンパク質」が使用されそしてアッセイがアデニル酸シクラーゼ活性の検出に
基づく場合に、融合構築物がGs(または酵素アデニル酸シクラーゼの形成を刺激する等
価Gタンパク質)を用いて確立されることを好む。
【0054】
「Gs、Gi、GzまたはGoタンパク質」と融合した「Gqタンパク質」を用いる「
Gタンパク質融合」構築物も同様に有効である。最も好ましい融合構築物は、「Gqタン
パク質」を用いて達成でき、ここで、Gタンパク質α−サブユニット(「Gαq」)の最
初の6個のアミノ酸が欠失しそしてGαqのC−末端の最後の5個のアミノ酸が、関係す
るGタンパク質のGαの相当するアミノ酸を用いて置換される。例えば、融合構築物は、
「Giタンパク質」と融合したGq(アミノ酸6個欠失)を有するとができ、「Gq/G
i融合構築物」をもたらす。我々は、この融合構築物が内因性Gi共役型受容体を非内因
性Gタンパク質、Gq、に共役させて、第二メッセンジャー、例えばイノシトール三リン
酸またはジアシルグリセロールがcAMP産生の代わりに測定できるように強制すると信
じる。
【0055】
4.シグナルエンハンサーGs共役型GPCRを用いる標的Gi共役型GPCR
のコトランスフェクション(cAMPに基づくアッセイ)
Gi共役型受容体はアデニル酸シクラーゼを阻害するとして知られており、従ってcA
MP産生のレベルを低下し、これはcAMPレベルの評価を興味深くさせることができる
。活性化の際に主としてGiに共役する受容体の構成的活性化の指標としてcAMPの産
生の低下を測定する有効な手段は、シグナルエンハンサー、例えば、Gi連結GPCRを
用いる活性化の際にGsと主として共役する非内因性、構成的活性化受容体(例えば以下
の開示するTSHR−A623I)をコトランスフェクションして達成できる。明らかな
ように、Gs共役型受容体の構成的活性化は、cAMPの産生の増加に基づいて決定でき
る。Gi共役型受容体の構成的活性化は、cAMP産生の低下に導く。従って、コトラン
スフェクション法は、これらの「反対」効果を有利に活用することを意図する。例えば、
非内因性、構成的活性化Gs共役型受容体(「シグナルエンハンサー」)と内因性Gi共
役型受容体(「標的受容体」)とのコトランスフェクションは、基準cAMPシグナルを
与える(すなわち、Gi共役型受容体はcAMPレベルを低下するけれども、この「低下
」は、構成的活性化Gs共役型受容体シグナルエンハンサーにより確立されるcAMPレ
ベルの本質的な増加と関連する)。その際、シグナルエンハンサーと標的受容体の構成的
活性化型とのコトランスフェクションにより、cAMPは、Gi標的の増加した機能的活
性(すなわち、これはcAMPを低下する)によりそれ以上に低下(基準に対する)する
と予想される。
【0056】
cAMPに基づくアッセイを用いる候補化合物のスクリーニングは、次いで2条件下で
達成できる。第一条件は、Gi共役標的受容体に関して、「反対」効果をもたらし、すな
わちGi共役標的受容体の逆アゴニストは測定されるcAMPシグナルを増加し、一方G
i共役標的受容体のアゴニストはこのシグナルを低下する。第二条件は、明らかなように
、この方法を用いて直接同定される候補化合物は、これらがシグナルエンハンシング受容
体を標的としないことを確認するために独立して評価されるべきであることである(これ
はコトランスフェクションした受容体に対するスクリーニングの前または後に行うことが
できる)。
【0057】
F.薬剤化学
必ずではないが一般的には、候補化合物の直接同定は、好ましくは、組合せ化学技術を
介して生成された化合物と関連して行われ、これにより数千の化合物がこの分析のために
ランダムに調製される。一般に、このようなスクリーニングの結果は、独自のコア構造を
有する化合物である。その後、これらの化合物は、好ましくは、これらの薬剤的性質をさ
らに増強するために好ましいコア構造の周囲に追加の化学修飾が行われる。このような技
術は、当該技術分野の熟練者には公知でありそして本明細書中では詳細には取り扱わない
。
【0058】
G.薬剤組成物
さらに開発するために選択された候補化合物は、当該技術分野の熟練者には周知の技術
を用いて薬剤組成物に調剤できる。適当な薬剤的に許容できるキャリヤーは、当該技術分
野の熟練者には利用できる。例えば、「レミントンの薬剤科学」(Remington's Pharmaceu
tical Sciences, 16th. Edition, 1980, MackPublishing Co., (Oslo et al., eds.))参
照。
【0059】
H.その他の効用
本明細書に開示したヒトGPCRの非内因型の好ましい使用は、逆アゴニスト、アゴニ
ストまたは部分アゴニストとしての候補化合物の直接同定のためであるが(好ましくは薬
剤としての使用のため)、ヒトGPCRのこれらの型は、研究設定にも使用できる。例え
ば、GPCRを組み込んだ生体内および生体外系は、正常および疾患の両方のヒトの状態
においてこれら受容体が演じる役割をさらに解明および理解するため、ならびにシグナル
伝達カスケードを理解するめに適用された構成的活性化の役割を理解するために使用でき
る。非内因性ヒトGPCRの価値は、これらの独自の特徴により、非内因性ヒトGPCR
が、内因性リガンドが同定される前にヒト身体内のこれら受容体の役割を理解するために
使用できることにより、研究手段としてのこれらの有用性が高くなることである。開示さ
れた受容体のその他の利用は、なかでも本明細書を検討すると当該技術分野の熟練者には
明らかとなるであろう。
【実施例】
【0060】
以下の実施例は、本発明の説明の目的で提示するものであり、これを制限するものでは
ない。特定の核酸およびアミノ酸配列が本明細書中で開示されるが、当該技術分野の通常
の熟練者は、これらの配列に小さい変更を加えて以下に報告すると同じかまたは実質的に
同様の結果を得る能力を有する。一つの配列から他のものへの配列カセットの適用または
理解の従来の方法(例えば、ラット受容体からヒト受容体へ、またはヒト受容体Aからヒ
ト受容体Bへ)は、一般に配列整列技術に基づいて断定され、その際、配列は、共通の領
域を決定するため研究において整列される。本明細書中に開示する変異法はこの方法には
よらないが、しかしその代わりにアルゴリズム法およびヒトGPCRのTM6領域内に位
置する保存されたプロリン残基からの位置的距離に基づく。この方法が確立されると、当
該技術分野の熟練者は、本明細書中に開示されると本質的に同じ結果(すなわち構成的活
性化)を得るためにこれに小さい変更を加える能力を有すると信じられる。このような変
更方法は、本開示の範囲内にあると考える。
【0061】
実施例1
内因性ヒトGPCR
1.ヒトGPCRの同定
開示される内因性ヒトGPCRは、ジーンバンク(GeneBank)TMデータベース情報の調査
に基づいて同定された。データベースを調査する間に、下記のcDNAクローンが下記に
明らかなように同定された(表C)
【0062】
【表2】
【0063】
2.全長クローニング
a.hRUP8(配列番号1および2)
開示されるヒトRUP8は、ESTデータベース(dbEST)情報の使用に基づいて
同定された。dbESTを調査する間に、アクセッション番号AL121755を有する
cDNAクローンが、新規のGPCRをコードすると同定された。下記のPCRプライマ
ーがヒト精巣マラソン−レディー(Marathon-Ready)cDNA(Clontech)を用いるRT−P
CRのために鋳型として用いられた。
5’−CTTGCAGACATCACCATGGCAGCC−3’(配列番号41、セン
ス)および
5’−GTGATGCTCTGAGTACTGGACTGG−3’(配列番号42、アン
チセンス)
PCRはアドヴァンテージ(Advantage) cDNAポリメラーゼ(Clontech、製造指針に従
う)を用い、50ul反応物中で下記のサイクルにより行った:30秒間94℃、10秒
間94℃、20秒間65℃、1.5分間72℃、および7分間72℃。サイクル2〜4を
35回反復した。
【0064】
1.2kbPCR断片を単離し、そしてpCRII−TOPOベクター(Invitrogen)中
にクローニングし、そしてABIビッグダイターミネーターキット(Big Dye Terminatork
it)(P.E. Biosystem)を用いて配列決定した。配列番号1参照、RUP8の推定アミノ酸
配列を配列番号2に記載する。
【0065】
b.hRUP9(配列番号3および4)
開示されるヒトRUP9は、ジーンバンクデータベース情報の使用に基づいて同定され
た。調査する間に、アクセッション番号AC011375を有するcDNAクローンが、
染色体5からのヒトゲノム配列として同定された。全長RUP9をプライマー
5’−GAAGCTGTGAAGAGTGATGC−3’(配列番号43、センス)、
5’−GTCAGCAATATTGATAAGCAGCAG−3’(配列番号44、アン
チセンス)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョン(Taq Plus Precision)ポリメラーゼ(Stratagene)を5%DMS
Oを用いる100μl反応物中で、下記のサイクルでステップ2〜ステップ4の35回反
復による増幅のために使用した:1分間94℃、30秒間94℃、30秒間56℃、2分
間72℃、および5分間72℃。
【0066】
1.3KbPCR断片を単離し、そしてpCRII−TOPOベクター(Invitrogen)中
に1%アガロースゲルからクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E.Biosystem)を用いて完全に配列決定した。配列番号3参照、RUP8の推定アミ
ノ酸配列を配列番号4に記載する。ヒトゲノムDNAから単離したRUP9クローンの配
列は、データベースから得た配列と一致した。
【0067】
c.hRUP10(配列番号5および6)
開示されるヒトRUP10は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC008754を有するcDNAクローンが
、染色体19からのヒトゲノム配列として同定された。全長RUP10をプライマー
5’−CCATGGGGAACGATTCTGTCAGCTACG−3’(配列番号45
、センス)および
5’−GCTATGCCTGAAGCCAGTCTTGTG−3’(配列番号46、アン
チセンス)
およびヒト白血球マラソンレディーcDNA(Clontech)を鋳型として用いるRT−PCR
によりクローニングした。アドヴァンテージcDNAポリメラーゼ(Clontech)を50μl
反応物中で下記のサイクルでステップ2〜ステップ4の35回反復による増幅のために使
用した:30秒間94℃、10秒間94℃、20秒間62℃、1.5分間72℃、および
7分間72℃。1.0KbPCR断片を単離し、そしてpCRII−TOPOベクター(I
nvitrogen)中にクローニングし、そしてABIビッグダイターミネーターキット(P.E.Bio
system)を用いて完全に配列決定した。新規のヒト受容体RUP10の核酸配列を配列番
号5に記載し、そしてその推定アミノ酸配列を配列番号6に記載する。
【0068】
d.hRUP11(配列番号7および8)
開示されるヒトRUP11は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC013396を有するcDNAクローンが
、染色体2からのヒトゲノム配列として同定された。全長RUP11をプライマー
5’−CCAGGATGTTGTGTCACCGTGGTGGC−3’(配列番号47、
センス)、
5’−CACAGCGCTGCAGCCCTGCAGCTGGC−3’(配列番号48、
アンチセンス)
およびヒトゲノムDNA(Clontech)を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンDNAポリメラーゼ(Stratagene)を50μl反応物中で下記の
サイクルでステップ2〜ステップ4の35回反復による増幅のために使用した:3分間9
4℃、20秒間94℃、20秒間67℃、1.5分間72℃、および7分間72℃。1.
3KbPCR断片を単離し、そしてpCRII−TOPOベクター(Invitrogen)中にクロ
ーニングし、そしてABIビッグダイターミネーターキット(P.E.Biosystem)を用いて完
全に配列決定した。新規のヒト受容体RUP11の核酸配列を配列番号7に記載し、そし
てその推定アミノ酸配列を配列番号8に記載する。
【0069】
e.hRUP12(配列番号9および10)
開示されるヒトRUP12は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AP000808を有するcDNAクローンが
、ラットRTAおよびヒトmas1腫瘍遺伝子GPCRと著しい相同性を有する新規のG
PCRをコードすると同定された。全長RUP12をプライマー
5’−CTTCCTCTCGTAGGGATGAACCAGAC−3’(配列番号49、
センス)
5’−CTCGCACAGGTGGGAAGCACCTGTGG−3’(配列番号50、
アンチセンス)
およびヒトゲノムDNA(Clontech)を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンDNAポリメラーゼ(Stratagene)を下記のサイクルでステップ
2〜ステップ4の35回反復による増幅のために使用した:3分間94℃、20秒間94
℃、20秒間65℃、2分間72℃、および7分間72℃。1.0kbPCR断片を単離
し、そしてpCRII−TOPOベクター(Invitrogen)中にクローニングし、そしてAB
Iビッグダイターミネーターキット(P.E.Biosystem)を用いて完全に配列決定した(核酸
配列に関しては配列番号9そして推論したアミノ酸配列に関しては配列番号10参照)。
【0070】
f.hRUP13(配列番号11および12)
開示されるヒトRUP13は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC011780を有するcDNAクローンが
、GPCRサカナGRPX−ORYLAと著しい相同性を有する新規のGPCRをコード
すると同定された。全長RUP13をプライマー
5’−GCCTGTGACAGGAGGTACCCTGG−3’(配列番号51、センス
)
5’−CATATCCCTCCGAGTGTCCAGCGGC−3’(配列番号52、ア
ンチセンス)
およびヒトゲノムDNA(Clontech)を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンDNAポリメラーゼ(Stratagene)を下記のサイクルでステップ
2〜ステップ4の35回反復による増幅のために使用した:3分間94℃、20秒間94
℃、20秒間65℃、2分間72℃、および7分間72℃。1.35kbPCR断片を単
離し、そしてpCRII−TOPOベクター(Invitrogen)中にクローニングし、そしてA
BIビッグダイターミネーターキット(P.E.Biosystem)を用いて完全に配列決定した(核
酸配列に関しては配列番号11そして推論したアミノ酸配列に関しては配列番号12参照
)。
g.hRUP14(配列番号13および14)
開示されるヒトRUP14は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AL137188を有するcDNAクローンが
、染色体13からのヒトゲノム配列として同定された。全長RUP14をプライマー
5’−GCATGGAGAGAAAATTTATGTCCTTGCAACC−3’(配列
番号53、センス)
5’−CAAGAACAGGTCTCATCTAAGAGCTCC−3’(配列番号54
、アンチセンス)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンポリメラーゼ(Stratagene)および5%DMSOを下記のサイク
ルでステップ2およびステップ3の35回反復による増幅のために使用した:3分間94
℃、20秒間94℃、2分間58℃、10分間72℃。
【0071】
1.1KbPCR断片を単離し、そしてpCRII−TOPOベクター(Invitrogen)中
にクローニングし、そしてABIビッグダイターミネーターキット(P.E. Biosystem)を用
いて完全に配列決定した(核酸配列に関しては配列番号13そして推論したアミノ酸配列
に関しては配列番号14参照)。ヒトゲノムDNAから単離したRUP14クローンの配
列は、データベースから得た配列と一致した。
【0072】
h.hRUP15(配列番号15および16)
開示されるヒトRUP15は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC016468を有するcDNAクローンが
、ヒトゲノム配列として同定された。全長RUP15をプライマー
5’−GCTGTTGCCATGACGTCCACCTGCAC−3’(配列番号55、
センス)
5’−GGACAGTTCAAGGTTTGCCTTAGAAC−3’(配列番号56、
アンチセンス)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンポリメラーゼ(Stratagene)を下記のサイクルでステップ2〜4
の35回反復による増幅のために使用した:3分間94℃、20秒間94℃、20秒間6
5℃、2分間72℃および7分間72℃。
【0073】
1.5KbPCR断片を単離し、そしてpCRII−TOPOベクター(Invitrogen)中
にクローニングし、そしてABIビッグダイターミネーターキット(P.E. Biosystem)を用
いて完全に配列決定した。核酸配列に関しては配列番号15そして推論したアミノ酸配列
に関しては配列番号16参照。ヒトゲノムDNAから単離したRUP15クローンの配列
は、データベースから得た配列と一致した。
【0074】
i.hRUP16(配列番号17および18)
開示されるヒトRUP16は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AL136106を有するcDNAクローンが
、染色体13からのヒトゲノム配列として同定された。全長RUP16をプライマー
5’−CTTTCGATACTGCTCCTATGCTC−3’(配列番号57、センス
、開始コドンの5’)
5’−GTAGTCCACTGAAAGTCCAGTGATCC−3’(配列番号58、
アンチセンス、終止コドンの3’)
およびヒト骨格筋マラソンレディーcDNA(Clontech)を鋳型として用いるPCRにより
クローニングした。アドヴァンテージcDNAポリメラーゼ(Clontech)を50ul反応物
中で下記のサイクルでステップ2〜4の35回反復による増幅のために使用した:30秒
間94℃、5秒間94℃、15秒間69℃、1分間72℃および5分間72℃。
【0075】
1.1KbPCR断片を単離し、そしてpCRII−TOPOベクター(Invitrogen)中
にクローニングし、そしてT7シーケナーゼキット(sequenase kit)(Amersham) を用いて
完全に配列決定した。核酸配列に関しては配列番号17そして推論したアミノ酸配列に関
しては配列番号18参照。RUP16クローンの配列は、AL136106の4個の不規
則断片と一致し、これはRUP16cDNAが4個のエキソンからなることを示す。
【0076】
j.hRUP17(配列番号19および20)
開示されるヒトRUP17は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC023078を有するcDNAクローンが
、染色体11からのヒトゲノム配列として同定された。全長RUP17をプライマー
5’−TTTCTGAGCATGGATCCAACCATCTC−3’(配列番号59、
センス、開始コドンを含む)
5’−CTGTCTGACAGGGCAGAGGCTCTTC−3’(配列番号60、ア
ンチセンス、終止コドンの3’)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
アドヴァンテージcDNAポリメラーゼミックス(Clontech)を5%DMSOを含む100
ul反応物中で下記のサイクルでステップ2〜4の30回反復による増幅のために使用し
た:1分間94℃、15秒間94℃、20秒間67℃、1分30秒間72℃および5分間
72℃。
【0077】
970bpPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号19そし
て推論したアミノ酸配列に関しては配列番号20参照。
【0078】
k.hRUP18(配列番号21および22)
開示されるヒトRUP18は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC008547を有するcDNAクローンが
、染色体5からのヒトゲノム配列として同定された。全長RUP18をプライマー
5’−GGAACTCGTATAGACCCAGCGTCGCTCC−3’(配列番号6
1、センス、開始コドンの5’)、
5’−GGAGGTTGCGCCTTAGCGACAGATGACC−3’(配列番号6
2、アンチセンス、終止コドンの3’)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンDNAポリメラーゼ(Stratagene)を5%DMSOを含む100
ul反応物中で下記のサイクルでステップ2〜4の35回反復による増幅のために使用し
た:5分間95℃、30秒間95℃、30秒間65℃、2分間72℃および5分間72℃
。
【0079】
1.3kbPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号21そし
て推論したアミノ酸配列に関しては配列番号22参照。
【0080】
l.hRUP19(配列番号23および24)
開示されるヒトRUP19は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC026331を有するcDNAクローンが
、染色体12からのヒトゲノム配列として同定された。全長RUP19をプライマー
5’−CTGCACCCGGACACTTGCTCTG−3’(配列番号63、センス、
開始コドンの5’)
5’−GTCTGCTTGTTCAGTGCCACTCAAC−3’(配列番号64、ア
ンチセンス、終止コドンを含む)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンDNAポリメラーゼ(Stratagene)を5%DMSOを用い下記の
サイクルでステップ2〜4の35回反復による増幅のために使用した:1分間94℃、1
5秒間94℃、20秒間70℃、1分30秒間72℃および5分間72℃。
【0081】
1.1kbPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号23そし
て推論したアミノ酸配列に関しては配列番号24参照。
【0082】
m.hRUP20(配列番号25および26)
開示されるヒトRUP20は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AL161458を有するcDNAクローンが
、染色体1からのヒトゲノム配列として同定された。全長RUP20をプライマー
5’−TATCTGCAATTCTATTCTAGCTCCTG−3’(配列番号65、
センス、開始コドンの5’)、
5’−TGTCCCTAATAAAGTCACATGAATGC−3’(配列番号66、
アンチセンス、終止コドンの3’)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
アドヴァンテージcDNAポリメラーゼミックス(Clontech)を5%DMSOを用い下記の
サイクルでステップ2〜4の35回反復による増幅のために使用した:1分間95℃、1
5秒間95℃、20秒間60℃、1分30秒間72℃および5分間72℃。
【0083】
1.0kbPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号25そし
て推論したアミノ酸配列に関しては配列番号26参照。
【0084】
n.hRUP21(配列番号27および28)
開示されるヒトRUP21は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC026756を有するcDNAクローンが
、染色体13からのヒトゲノム配列として同定された。全長RUP21をプライマー
5’−GGAGACAACCATGAATGAGCCAC−3’(配列番号67、センス
)
5’−TATTTCAAGGGTTGTTTGAGTAAC−3’(配列番号68、アン
チセンス)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンポリメラーゼ(Stratagene)を5%DMSOを含む100ul反
応物中で下記のサイクルでステップ2〜4の30回反復による増幅のために使用した:1
分間94℃、15秒間94℃、20秒間55℃、1分30秒間72℃および5分間72℃
。
【0085】
1,014bpPCR断片を1%アガロースゲルから単離し、そしてpCRII−TO
POベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーター
キット(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号27
そして推論したアミノ酸配列に関しては配列番号28参照。
【0086】
o.hRUP22(配列番号29および30)
開示されるヒトRUP22は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC027026を有するcDNAクローンが
、染色体11からのヒトゲノム配列として同定された。全長RUP22をプライマー
5’−GGCACCAGTGGAGGTTTTCTGAGCATG−3’(配列番号69
、センス、開始コドンを含む)
5’−CTGATGGAAGTAGAGGCTGTCCATCTC−3’(配列番号70
、アンチセンス、終止コドンの3’)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンDNAポリメラーゼ(Stratagene)を5%DMSOを含む100
ul反応物中で下記のサイクルでステップ2〜4の30回反復による増幅のために使用し
た:94℃、1分間94℃、15秒間55℃、20秒間72℃、1.5分間72℃、5分
間。
【0087】
970bpPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号29そし
て推論したアミノ酸配列に関しては配列番号30参照。
【0088】
p.hRUP23(配列番号31および32)
開示されるヒトRUP23は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC007104を有するcDNAクローンが
、染色体4からのヒトゲノム配列として同定された。全長RUP23をプライマー
5’−CCAGGCGAGCCGCTAGCGCCATG−3’(配列番号71、センス
、開始コドンとしてのATC)
5’−ATGAGCCCTGCCAGGCCCTCAGT−3’(配列番号72、アンチ
センス、終止コドンとしてのTCA)
およびヒト胎盤マラソンレディーDNA(Clontech)を鋳型として用いるPCRによりクロ
ーニングした。アドヴァンテージcDNAポリメラーゼ(Clontech)を50ul反応物中で
下記のサイクルでステップ2〜4の35回反復による増幅のために使用した:30秒間9
5℃、15秒間95℃、20秒間66℃、1分20秒間72℃および5分間72℃。
【0089】
1.0kbPCR断片を単離し、そしてpCRII−TOPOベクター(Invitrogen)中
にクローニングし、そしてABIビッグダイターミネーターキット(P.E. Biosystem)を用
いて完全に配列決定した。核酸配列に関しては配列番号31そして推論したアミノ酸配列
に関しては配列番号32参照。
【0090】
q.hRUP24(配列番号33および34)
開示されるヒトRUP25は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC026331を有するcDNAクローンが
、染色体12からのヒトゲノム配列として同定された。全長RUP25をプライマー
5’−GCTGGAGCATTCACTAGGCGAG−3’(配列番号73、センス、
開始コドンの5’)
5’−AGATCCTGGTTCTTGGTGACAATG−3’(配列番号74、アン
チセンス、終止コドンの3’)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
アドヴァンテージcDNAポリメラーゼミックス(Clontech)を5%DMSOを用い、下記
のサイクルでステップ2〜4の35回反復による増幅のために使用した:1分間94℃、
15秒間94℃、20秒間56℃、1分30秒間72℃および5分間72℃。
【0091】
1.2kbPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号33そし
て推論したアミノ酸配列に関しては配列番号34参照。
【0092】
r.hRUP25(配列番号35および36)
開示されるヒトRUP25は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC026331を有するcDNAクローンが
、染色体12からのヒトゲノム配列として同定された。全長RUP25をプライマー
5’−GCTGGAGCATTCACTAGGCGAG−3’(配列番号75、センス、
開始コドンの5’)
5’−AGATCCTGGTTCTTGGTGACAATG−3’(配列番号76、アン
チセンス、終止コドンの3’)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
アドヴァンテージcDNAポリメラーゼミックス(Clontech)を5%DMSOを用い、下記
のサイクルでステップ2〜4の35回反復による増幅のために使用した:1分間94℃、
15秒間94℃、20秒間56℃、1分30秒間72℃および5分間72℃。
【0093】
1.2kbPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号35そし
て推論したアミノ酸配列に関しては配列番号36参照。
【0094】
s.hRUP26(配列番号37および38)
開示されるヒトRUP26は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC023040を有するcDNAクローンが
、染色体2からのヒトゲノム配列として同定された。全長RUP26をRUP26特異性
プライマー
5’−AGCCATCCCTGCCAGGAAGCATGG−3’(配列番号77、セン
ス、開始コドンを含む)
5’−CCAGACTGTGGACTCAAGAACTCTAGG−3’(配列番号78
、アンチセンス、終止コドンを含む)
およびヒト胎盤マラソンレディーcDNA(Clontech)を鋳型として用いるRT−PCRに
よりクローニングした。アドヴァンテージcDNAポリメラーゼミックス(Clontech)を5
%DMSOを含む100ul反応物中で下記のサイクルでステップ2〜4の35回反復に
よる増幅のために使用した:5分間94℃、30秒間95℃、30秒間65℃、2分間7
2℃および5分間72℃。
【0095】
1.1kbPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号37そし
て推論したアミノ酸配列に関しては配列番号38参照。
【0096】
t.hRUP27(配列番号39および40)
開示されるヒトRUP27は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC027643を有するcDNAクローンが
、染色体12からのヒトゲノム配列として同定された。全長RUP27をRUP17特異
性プライマー
5’−AGTCCACGAACAATGAATCCATTTCATG−3’(配列番号7
9、センス、開始コドンを含む)
5’−ATCATGTCTAGACTCATGGTGATCC−3’(配列番号80、ア
ンチセンス、終止コドンの3’)
およびヒト成人脳マラソンレディーcDNA(Clontech)を鋳型として用いるPCRにより
クローニングした。アドヴァンテージcDNAポリメラーゼミックス(Clontech)を5%D
MSOを含む50ul反応物中で下記のサイクルでステップ2〜4の35回反復による増
幅のために使用した:1分間94℃、10秒間94℃、20秒間58℃、1分30秒間7
2℃および5分間72℃。
【0097】
1.1kbPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号35そし
て推論したアミノ酸配列に関しては配列番号36参照。ヒト脳から単離されたRUP27
cDNAクローンの配列は,AC027643の5個の不規則断片と一致することが決定
され、RUP27cDNAが5個のエキソンからなることを示す。
【0098】
実施例2
非内因性、構成的活性化GPCRの調製
当該技術分野の熟練者は、核酸配列の変異のための技術を選択する能力を有すると信じ
られる。下記は、以上に開示した数種のヒトGPCRの非内因性型を創成するために使用
される方法である。以下に開示される変異は、保存されるプロリン(またはこれの内因性
の保存性置換基)残基(TM6/IC3界面近くのGPCRのTM6領域内に位置する)
から16番目のアミノ酸(GPCRのIC3領域内に位置する)が、好ましくはアラニン
、ヒスチジン、アルギニンまたはリシンアミノ酸残基に、最も好ましくはリシンアミノ酸
残基に変異されるアルゴリスム法に基づく。
【0099】
1.トランスフォーマーサイトディレクテッドTM変異誘発
非内因性ヒトGPCRの調製は、製造者の指針に従って、トランスフォーマーサイトデ
ィレクテッド(Transformer Site-Directed) TM変異誘発キット(Clontech)を用いて、ヒト
GPCRについて行ってもよい。2個の変異誘発プライマーを使用し、最も好ましくはリ
シン変異を創成するリシン変異誘発オリゴヌクレオチド、および選択マーカーオリゴヌク
レオチドを用いる。便宜のために、ヒトGPCR内に組み込まれるべきコドン変異も標準
形で記録する(表D)。
【0100】
【表3】
【0101】
2.クイクチェンジTMサイトディレクテッドTM変異誘発
非内因性ヒトGPCRの調製は、クイクチェンジ(QuikChange)TMサイトディレクテッド
(Site-Directed) TM変異誘発キット(Stratagene、製造者の指針に従う)によっても行う
ことができる。内因性GPCRが好ましい鋳型として使用され、そして2個の変異誘発プ
ライマーを用い、ならびに最も好ましくはリシン変異誘発オリゴヌクレオチドおよび選択
マーカーオリゴヌクレオチド(キット内に含まれる)が使用される。便宜のために、新規
のヒトGPCR内に組み込まれたコドン変異およびそれぞれのオリゴヌクレオチドを標準
形で記録する(表E)
【0102】
【表4】
【0103】
非内因性ヒトGPCRを次いで配列決定しそして誘導および確認された核酸およびアミ
ノ酸配列を本特許明細書に添付した「配列表」に表示し、これを下記の表Fに要約した。
【0104】
【表5】
【0105】
実施例3
受容体発現
種々の細胞がタンパク質の発現のために当該技術分野で利用できるが、哺乳動物細胞の
使用が最も好ましい。この主要な理由は、実用性により定められ、すなわち例えばGPC
Rの発現のための酵母細胞の利用は、可能な限り、哺乳動物系のために発展された受容体
共役、遺伝機序および分泌経路を含まない(確かに酵母の場合には含まない)非哺乳動物
細胞をプロトコール中に導入し、これにより非哺乳動物細胞で得られた結果は、利用可能
な限り、哺乳動物細胞から得られたほどは好ましくない。使用される特定の哺乳動物細胞
は熟練者の特定の要求により決定できるけれども、哺乳動物細胞のなかでは、COS−7
、293および293T細胞が特に好ましい。
【0106】
a.一過性トランスフェクション
第一日目に、6x106 /(293細胞のウエル10cm皿)をプレートアウトした。
第二日目に、2本の反応管を調製した(それぞれの管についての下記の比率はプレートあ
たりである)。管Aは、4μgDNA(例えばpCMVベクター、受容体cDNAを含む
pCMVベクターなど)を0.5mlの血清を含まないDMEM(GibcoBRL) 中で混合し
て調製した。管Bは、24μlリポフェクタミン(Gibco BRL) を0.5mlの血清を含ま
ないDMEM中で混合して調製した。管AおよびBを倒置(数回)により混合し、次いで
室温で30〜45分間インキュベーションした。この混合物を「トランスフェクション混
合物」と呼ぶ。プレーティングした293細胞を1XPBSを用いて洗浄し、次いで0.
5mlの血清を含まないDMEMを加えた。トランスフェクション混合物1mlを細胞に
加え、次いで37℃/5%CO2で4時間インキュベーションした。トランスフェクショ
ン混合物を吸引して取り出し、次いでDMEM/10%ウシ胎児血清の10mlを加えた
。細胞を37℃/5%CO2でインキュベーションした。48時間インキュベーションし
た後、細胞を採取しそして分析に使用した。
【0107】
b.安定細胞株:Gs融合タンパク質
約12x106 293細胞を15cm組織培養プレート上にプレーティングした。10
%ウシ胎児血清および1%ピルビン酸ナトリウム、L−グルタミン、および抗生物質を含
むDMEハイ・グルコース媒体(HighGlucose Medium) 中で培養した。プレーティングし
た24時間後に293細胞は約80%集密度となり、細胞をDNA12μgを用いてトラ
ンスフェクションした。DNA12μgを、リポフェクタミン60ulおよび血清を含ま
ないDMEハイ・グルコース媒体2mLと混合した。媒体をプレートから吸引しそして細
胞を血清を含まない媒体を用いて一回洗浄した。DNA、リポフェクタミン、および媒体
混合物を血清を含まない媒体10mLと一緒にプレートに加えた。37℃で4〜5時間の
インキュベーションの後に、媒体を吸引し、そして血清を含む媒体25mlを加えた。ト
ランスフェクションの24時間後に媒体を再度吸引し、そして血清を含む新しい媒体を加
えた。トランスフェクションの48時間後に、媒体を吸引しそして血清を含みゲネチシン
(G418薬剤)を含む媒体を加え、最終濃度500μg/mLまで加えた。トランスフ
ェクションした細胞は、ここでG418耐性遺伝子を含む陽性にトランスフェクションさ
れた細胞に関して選択する。選択が起きる度に媒体を4〜5日毎に交換した。選択の間に
、細胞は安定なプールを創成するために増殖するか、または安定クローン選択のためにス
プリットする。
【0108】
実施例4 非内因性GPCRの構成的活性の決定のためのアッセイ
種々の方法が非内因性ヒトGPCRの構成的活性の評価のために利用される。以下は例
示である。当該技術分野の通常の熟練者は、熟練者の要求に優先的に役立つこのような技
術を決定する能力を有すると信じられる。
1.膜結合アッセイ:〔35S〕GTPγSアッセイ
Gタンパク質共役型受容体がその活性状態にある場合に、リガンド結合または構成的活
性化のいずれかの結果として、受容体はGタンパク質に結合しそしてGDPの遊離および
その後のGタンパク質へのGTPの結合を刺激する。Gタンパク質共役型受容体複合体の
αサブユニットは、GTPアーゼとして作用しそしてGTPをGDPにゆっくりと加水分
解し、その時点で受容体は通常は脱活される。構成的活性化受容体はGTPをGDPへ交
換することを続ける。非−加水分解性GTP類似体である〔35S〕GTPγSは、構成的
活性化受容体を発現する膜への〔35S〕GTPγSの増強された結合を証明するために使
用できる。構成的活性化を測定するために〔35S〕GTPγS結合を用いる利点は、(a
)これがすべてのGタンパク質共役型受容体に包括的に適用される、(b)これが、膜表
面の近くで細胞内カスケードに影響する分子を取り上げることを困難とすることである。
【0109】
このアッセイは、関係する受容体を発現する膜に結合する〔35S〕GTPγSを刺激す
るGタンパク質共役型受容体の能力を利用する。従って、このアッセイは、既知のオーフ
ァンおよび構成的活性化Gタンパク質共役型受容体への候補化合物をスクリーニングする
ための直接同定法に使用できる。アッセイは包括的であり、そしてすべてのGタンパク質
共役型受容体における薬剤発見に適用される。
【0110】
〔35S〕GTPγSアッセイは、20mM HEPESおよび1〜約20mM MgC
l2 (この量は、20mMが好ましいけれども、結果の最適化のために調節できる)pH
7.4、約0.3〜約1.2nM 〔35S〕GTPγS(この量は、1.2が好ましいけ
れども、結果の最適化のために調節できる)を含む結合緩衝液および12.5〜75μg
膜タンパク質(例えばGs融合タンパク質を発現する293細胞、この量は最適化のため
に調節できる)および10μM GDP(この量は最適化のために調節できる)中で1時
間インキュベーションした。次いでコムギ胚芽凝集素ビーズ(25μl、Amersham)を加
えそして混合物をさらに30分間、室温でインキュベーションした。次いで管を1500
x gで5分間、室温で遠心分離し次いでシンチレーションカウンターでカウントした
。
【0111】
2.アデニル酸シクラーゼ
細胞に基づくアッセイのために設計されたフラッシュ・プレート(Flash Plate) TMアデ
ニル酸シクラーゼキット(New England Nuclear;カタログ番号SMP004A)は、粗血漿膜と
共に用いるために変更できる。フラッシュ・プレートウエルは、cAMPを認識する特定
の抗体も含むシンチラント(scintillant)コーティングを含むことができる。ウエル内で
産生されたcAMPはcAMP抗体に対する放射性cAMPトレーサーの結合に関する直
接競合により定量できる。以下の記載は、受容体を発現する全細胞内のcAMPレベル内
の変化の測定のための簡単なプロトコールとして役立つ。
【0112】
トランスフェクションされた細胞を一過性トランスフェクションの約24時間後に採取
した。媒体を注意して吸引して取り出し廃棄した。PBS10mlを細胞のそれぞれの皿
におだやかに加え次いで注意して吸引した。Sigma 細胞分離緩衝液1mlおよびPBS3
mlをそれぞれのプレートに加えた。細胞を皿からピペットで取り出しそして細胞懸濁液
を50ml円錐型遠心分離管内に集めた。次いで細胞を室温、1,100rpmで5分間
遠心分離した。細胞ペレットを注意して適当な体積のPBS(約3ml/プレート)内に
再懸濁した。次いで細胞を血球計数器を用いて計数しそして追加のPBSを加えて細胞の
概略数を与えた(最終体積約50μl/ウエル)。
【0113】
cAMP標準および「検出緩衝液」(「検出緩衝液」11mlにトレーサー〔125 Ic
AMP(50μl〕の1μCiを含んでなる)を調製し、そして製造者の指示に従って維
持した。「アッセイ緩衝液」をスクリーニングのために新たに調製しそしてこれは「刺激
緩衝液」50μl、試験化合物3ul(12uMの最終アッセイ濃度)および細胞50μ
lを含み、「アッセイ緩衝液」は使用するまで氷上に保管した。アッセイは、適当なウエ
ルにcAMP標準50μlの添加から始まり、次いでウエルH−11およびH−12へP
BSA50ulを加えた。「刺激緩衝液」50μlをすべてのウエルに加えた。DMSO
(または選択した候補化合物)を化合物溶液3μlの供給ができるピン器具を用いて適当
なウエルに加え、12μM試験化合物の最終アッセイ濃度および全アッセイ体積100μ
lとした。次いで細胞をウエルに加えそして60分間、室温でインキュベーションした。
次いでトレーサーcAMPを含む「検出混合物」100μlをウエルに加えた。次いでプ
レートをさらに2時間インキュベーションし、次いでウオラック・マイクロベータ(Walla
cMicroBeta)シンチレーションカウンター内で計数した。次いでcAMP/ウエルの値を
それぞれのアッセイプレート内に含まれる標準cAMP曲線から外挿した。
【0114】
3.Gi共役型標的GPCRのための細胞に基づくcAMP
TSHRは、活性化の際にcAMPの蓄積を起こすGs共役型GPCRである。TSH
Rは、アミノ酸残基623を変異すること(すなわち、アラニン残基をイソロイシン残基
に変化する)により構成的に活性化される。Gi共役型受容体は、アデニル酸シクラーゼ
を阻害すると予想され、そして、そのために、cAMP産生のレベルを低下し、これはc
AMPレベルの問題の評価を可能とする。Gi共役型受容体の構成的活性化の指標として
のcAMPの産生の低下を測定するために有効な技術は、cAMPの基準レベルを確立す
るためのGi連結標的GPCRを用いる「シグナルエンハンサー」として、最も好ましく
は非内因性、構成的活性化TSHR(TSHR−A6231)(または内因性、構成的活
性Gs共役型受容体)を同時トランスフェクションすることにより達成できる。Gi共役
型受容体の非内因性型が創成されると、次いで標的GPCRのこの非内因性型は、シグナ
ルエンハンサーと一緒に同時トランスフェクションされ、そしてスクリーニングに使用で
きるのはこの物質である。我々は、cAMPアッセイを使用する場合にシグナルを効率的
に発生するためにこの方法を使用した。この方法は、好ましくはGi共役型受容体に対す
る候補化合物の直接同定に使用される。この方法を使用した場合に、Gi共役型受容体に
とって標的GPCRの逆アゴニストはcAMPシグナルを増加しそしてアゴニストはcA
MPシグナルを低下するであろう。
【0115】
第一日目に、2X104 293および293細胞/ウエルをプレートアウトする。第二
日目に、2個の反応管を調製する(それぞれの管についての下記の比率はプレートあたり
である)。管Aは、哺乳動物細胞内にトランスフェクションされたそれぞれの受容体の2
μgDNAを、1.21の血清を含まないDMEM(IrvineScientific, Irvine, CA) 中
で全体で4μgDNA(例えばpCMVベクター、変異THSR(THSR−A6231
I)を含むpCMVベクター、THSR−A6231IおよびGPCRなど)と混合して
調製する。管Bは、120μlリポフェクタミン(GibcoBRL) を1.2mlの血清を含ま
ないDMEM中で混合して調製する。次いで、管AおよびBを倒置(数回)してよく混合
し、次いで室温で30〜45分間インキュベーションした。混合物を「トランスフェクシ
ョン混合物」と呼ぶ。プレーティングした293細胞を1XPBSを用いて洗浄し、次い
で血清を含まないDMEM10mlを加える。次いでトランスフェクション混合物2.4
mlを細胞に加え、次いで4時間、37℃/5%CO2でインキュベーションする。次い
でトランスフェクション混合物を吸引して取り出し、次いでDMEM/10%ウシ胎児血
清25mlを加える。次いで細胞を37℃/5%CO2でインキュベーションする。24
時間インキュベーションの後、細胞を採取しそして分析に使用する。
【0116】
しかし、細胞に基づくアッセイのために設計されたフラッシュ・プレートTMアデニル酸
シクラーゼキット(New England Nuclear;カタログ番号SMP004A)は、熟練者の要求に従
って粗血漿膜と用いるために変更できる。フラッシュ・プレートウエルは、cAMPを認
識する特定の抗体も含むシンチラントコーティングを含むことができる。ウエル内で産生
されたcAMPはcAMP抗体に対する放射性cAMPトレーサーの結合に関する直接競
合により定量できる。以下の記載は、受容体を発現する全細胞内のcAMPレベル内の変
化の測定のための簡単なプロトコールとして役立つ。
【0117】
トランスフェクションされた細胞を一過性トランスフェクションの約24時間後に採取
する。媒体を注意して吸引して取り出して廃棄する。PBS10mlを細胞のそれぞれの
皿におだやかに加え次いで注意して吸引する。Sigma細胞分離緩衝液1mlおよびP
BS3mlをそれぞれのプレートに加える。細胞を皿からピペットで取り出しそして細胞
懸濁液を50ml円錐型遠心分離管内に集める。次いで細胞を室温、1,100rpmで
5分間遠心分離する。細胞ペレットを注意して適当な体積のPBS(約3ml/プレート
)内に再懸濁する。次いで細胞を血球計数器を用いて計数しそして追加のPBSを加えて
細胞の概略数を与える(最終体積約50μl/ウエル)。
【0118】
cAMP標準および「検出緩衝液」(「検出緩衝液」11mlにトレーサー〔125 Ic
AMP(50μl〕の1μCiを含んでなる)を調製し、そして製造者の指示に従って維
持する。「アッセイ緩衝液」をスクリーニングのために新たに調製しなればならずそして
これは「刺激緩衝液」50μl、試験化合物3ul(12uM最終アッセイ濃度)および
細胞50μlを含み、「アッセイ緩衝液」は使用するまで氷上で保管できる。アッセイは
、適当なウエルにcAMP標準50μlの添加から始めることができ、次いでウエルH−
11およびH−12へPBSA50ulを加える。刺激緩衝液50μlをウエルに加える
。選択した化合物(例えばTSH)を化合物溶液3μlの供給ができるピン器具を用いて
適当なウエルに加え、12μM試験化合物の最終アッセイ濃度および全アッセイ体積10
0μlとする。次いで細胞をウエルに加えそして60分間、室温でインキュベーションす
る。次いでトレーサーcAMPを含む「検出混合物」100μlをウエルに加える。次い
でプレートをさらに2時間インキュベーションし、次いでウオラック・マイクロベータシ
ンチレーションカウンター内で計数する。次いでcAMP/ウエルの値をそれぞれのアッ
セイプレート内に含まれる標準cAMP曲線から外挿する。
【0119】
4.レポーターに基づくアッセイ
a.Gre−Lucレポーターアッセイ(Gs−関連受容体)
293および293t細胞をウエルあたりに2x104 細胞の密度で96ウエルプレー
ト上にプレートアウトし、そして製造者の指示に従って次の日にリポフェクタミン試薬(B
RL)を用いてトランスフェクションした。DNA/脂質混合物をそれぞれ6ウエルトラン
スフェクション物について次のように調製した:DMEM100μl中のプラスミドDN
A260ngをDMEM100μl中の脂質2μlとおだやかに混合した(8xCRE−
Lucレポータープラスミド200ng、内因性受容体または非内因性受容体を含んでな
るpCMVまたはpCMV単独の50ng、およびGPRS発現プラスミド(pcDNA
3中のGPRS(Invitrogen))10ngから成るプラスミドDNA260ng)。8XC
RE−Lucレポータープラスミドは、以下のようにして調製した:ベクターSRIF−
β−galは、pβgal−ベーシックベクター(BasicVector)(Clontech)中のBglV
−HindIII部位でラット・ソマトスタチンプロモーター(−71/+51)をクロ
ーニングして得られた。cAMP反応因子の8個のコピーは、アデノウイルス鋳型Adp
CF126CCRE8(7Human Gene Therapy 1883(1996)参照)からPCRにより得られ
そしてKpn−BglV部位でSRIF−β−galベクター内にクローニングすると、
8xCRE−β−galレポーターベクターが得られた。8xCRE−Lucレポタープ
ラスミドは、8xCRE−β−galレポーターベクター中のベータ−ガラクトシダーゼ
遺伝子をHindIII−BamHI部位におけるpGL3−ベーシックベクター(Prome
ga)から得たルシフェラーゼ遺伝子と置換して生成した。30分、室温でのインキュベー
ションの後に、DNA/脂質混合物をDMEM400μlを用いて希釈し、そして希釈混
合物100μlをそれぞれのウエルに加えた。10%FCSを含むDMEM100μlを
、細胞カルチャーインキュベーター中での4時間のインキュベーションの後にそれぞれの
ウエルに加えた。次の日に、トランスフェクションした細胞を10%FCSを含むDME
M200μl/ウエルに交換した。8日後に、PBSを用いる洗浄1回の後にフェノール
レッドを含まないDMEM100μl/ウエルにウエルを交換した。ルシフェラーゼ活性
は、ルクライト(LucLite)TMレポーター遺伝子アッセイキット(Packard)を用い製造者の指
示に従いそして1450マイクロベータ(MicroBeta) TMシンチレーションおよび蛍光カウ
ンター(Wallac)を用いて読み取って次の日に測定した。
【0120】
b.AP1レポーターアッセイ(Gq−関連受容体)
Gq刺激を検出するための方法は、これらのプロモーター中にAP1因子を含む遺伝子
の活性化を起こすGq−依存性ホスホリパーゼCの既知の性質に依存する。パスデテクト
(Pathdetect) TM AP−1 cis−レポーティングシステム(Stratagene 、カタログ番
号#219073)が、CREBレポーターアッセイに関して以上に記載したプロトコールにした
がって使用できるが、リン酸カルシウム沈降物の成分が410ng pAP1−Luc、
80ng pCMV−受容体発現プラスミド、および20ng CMV−SEAPであっ
た点が異なる。
c.Srf−Lucレポーターアッセイ(Gq−関連受容体)
Gq刺激を検出するための一つの方法は、そのプロモーター中に血清反応因子を含む遺
伝子の活性化を起こすGq依存性ホスホリパーゼCの既知の性質に依存する。パスデテク
トTMSRF−Luc−レポーティングシステム(Stratagene)が、例えばCOS7細胞中の
Gq共役活性のアッセイに使用できる。細胞を系のプラスミド成分を用いてトランスフェ
クションしそして哺乳動物トランスフェクション(MannmalianTransfection) TMキット(St
ratagene 、カタログ番号#200285)を製造者の指示に従って使用して内因性または非内因
性GPCRをコードする発現プラスミドを示す。要約すると、410ng SRF−Lu
c、80ng pCMV−受容体発現プラスミド、および20ng CMV−SEAP(
分泌されたアルカリホスファターゼ発現プラスミド。アルカリホスファターゼ活性は試料
間のトランスフェクション効果における変動を制御するためにトランスフェクションされ
た細胞の媒体中で測定する)を、製造者の指示に従ってリン酸カルシウム沈降物中に混和
する。沈降物の半分を96−ウエルプレート中の3ウエル上に均等に分布させ、血清を含
まない媒体中の細胞上に24時間保持する。最後の5時間に、指示された場合に、細胞を
1μMアンギオテンシンを用いてインキュベーションする。次いで、細胞を溶解しそして
ルシライト(Lucilite)TMキット(Packard、カタログ番号#6016911)および「トリラックス(
Trilux)1450マイクロベータ(Micrebeta) 」液体シンチレーションおよび蛍光カウン
ター(Wallac)を用いて、製造者の指示に従ってルシフェラーゼ活性をアッセイする。デー
タはグラフパッドプリズム(GraphPadPrism)TM2.0a(GraphPad Software Inc.)を用いて分
析できる。
【0121】
d.細胞内IP3 蓄積アッセイ(Gw−関連受容体)
第一日目に、受容体(内因性および/または非内因性)を含んでなる細胞を24ウエル
プレート上に、通常は1x105 細胞/ウエルでプレーティングできる(この数は最適化
できるが)。第二日目に、細胞を最初、血清を含まないDMEM50μl/ウエル中の0
.25μgDNAおよび血清を含まないDMEM50μl/ウエル中の2μlリポフェタ
ミンと混合してトランスフェクションできる。溶液をおだやかに攪拌しそして15〜30
分間、室温でインキュベーションする。細胞を0.5ml PBSを用いて洗浄し、血清
を含まない媒体400μlをトランスフェクション媒体と混合しそして細胞に加える。次
いで細胞を3〜4時間、37℃/5%CO2でインキュベーションし次いでトランスフェ
クション媒体を除きそして通常の増殖媒体1ml/ウエルで置き換えた。第三日目に、細
胞を 3H−ミオ−イノシトールを用いて標識する。要約すると、媒体を除きそして細胞を
0.5mlPBSを用いて洗浄する。次いで、0.5mlイノシトール不含/血清不含媒
体(GIBCOBRL) を、ウエルあたりに 3H−ミオ−イノシトールの0.25μCiと一緒に
ウエルあたりに加えそして細胞を16〜18時間、37℃/5%CO2でインキュベーシ
ョンする。第四日目に、細胞を0.5mlPBSで洗浄し、そしてイノシトール不含/血
清不含媒体10μMパルジリン10mM塩化リチウムを含むアッセイ媒体0.45mlも
しくはアッセイ媒体0.4mlおよび10xケタンセリン(ketanserin、ket)50μ
lを最終濃度10μMとしたものを加える。次いで細胞を30分間37℃でインキュベー
ションする。次いで細胞をPBS0.5mlで洗浄しそして新規/氷冷却停止溶液(1M
KOH;18mMホウ酸Na;3.8mM EDTA)200μlをウエルあたりに加
える。溶液を氷上で5〜10分間、または細胞が溶解するまで保持し、次いで新規/氷冷
中和溶液(7.5%HCL)200μlにより中和する。次いで1.5mlエッペンドル
フ(eppendorf)管内に溶解物を移しそしてクロロホルム/メタノール(1:2)1mlを
管当たりに加える。溶液を15秒間ウズ攪拌し、上相をバイオラド(Biorad)AG1−X8
TMアニオン交換樹脂(100〜200メッシュ)に加える。最初に樹脂を1:1.25W
/Vの水を用いて洗浄しそして上相0.9mlをカラム上に加える。カラムを5mMミオ
−イノシトール10mlおよび5mM ホウ酸Na/60mMギ酸Na 10mlを用い
て洗浄する。イノシトール・三リン酸が0.1Mギ酸/1Mギ酸アンモニウムの2mlを
含むシンチレーションカクテル10mlを含むシンチレーションバイアル内で溶離する。
カラムを0.1Mギ酸/3Mギ酸アンモニウムの10mlを用いて洗浄しそして2回、d
dH2Oを用いてリンスしそして4℃水中で保管する。
【0122】
例示的な結果を以下の表Gに示す。
【0123】
【表6】
【0124】
上記の実施例4(1)で開示した構成的活性化を検出するためのGTPγSアッセイの
例示的な結果は、ヒトRUP13およびRUP15上での「Gs:融合タンパク質構築物
」を用いて達成された。下記の表Hは、このアッセイから生成されたシグナルおよび示し
たようなシグナルの相違を表示する。
【0125】
【表7】
【0126】
実施例5
融合タンパク質調製
a.GPCR:Gs融合構築物
構成的活性化GPCR−Gタンパク質融合構築物の設計は、以下のように行った:ラッ
トGタンパク質Gsα(長形;Itoh et al., 83 PNAS 3776 (1986))の5’および3’末
端の両方を、この上にHindIII(5’−AAGCTT−3’)配列を含むように操
作した。正しい配列(近接する(flanking)HindIII配列を含む)の確認の後に、全
配列を、そのベクターのHindIII制限部位を用いてサブクローニングしてpcDN
A3.1(−)(Invitrogenカタログ番号V795-20)中にシャトル(shuttle) した。Gsα
配列の正しい方向は、pcDNA3.1(−)中へのサブクローニングの後に決定した。
HindIII配列で変更したラットGsα遺伝子を含むpcDNA3.1(−)を次い
で確認した。これでこのベクターは、「汎用」Gsαタンパク質ベクターとして利用でき
た。pcDNA3.1(−)ベクターは種々の周知の制限部位をHindIII部位の上
流に含み、従ってGsタンパク質の上流に、内因性、構成的活性GPCRのコーディング
配列を挿入する能力を有利に与える。この同じ方法は、他の「汎用」G−タンパク質ベク
ターを創成するためにも利用でき、そして当然ながら、その他の商業的に利用可能、また
は熟練者に既知の独自のベクターも使用可能であり、その際、重要な基準は、GPCRの
配列がGタンパク質のものの上流でそしてインフレームであることである。
【0127】
RUP13は、Gsを介して共役する。下記の例示的「GPCR融合タンパク質」につ
いて、Gsαへの融合が達成された。
【0128】
RUP13−「Gsα融合タンパク質」構築物を以下のようにして作製した。
プライマーは以下のように設計した。
5’−gatc〔TCTAGAAT〕GGAGTCCTCACCCATCCCCCAG−
3’(配列番号97、センス)
5’−gatc〔GATATC〕CGTGACTCCAGCCGGGGTGAGGCGG
C−3’(配列番号98、アンチセンス)
小文字のヌクレオチドはGタンパク質とRUP13との間の制限部位(括弧内に示す)内
のスペーサーとして含まれる。センスおよびアンチセンスプライマーは、スペーサー(制
限部位に帰属する)がGタンパク質とRUP15との間に存在するように、それぞれXb
aIとEcoRVのための制限部位を含んでいた。
【0129】
次いで、上記に開示したGsα汎用ベクター内の融合ためのそれぞれの受容体配列を確
保するためにPCRを使用し、それぞれ下記のプロトコールを用いた。RUP15のため
の100ng cDNAを、それぞれプライマー(センスおよびアンチ−センス)2μl
、10mM dNTP3μL、10XタックプラスTMプレシジョン緩衝液(Precision Buf
fer)10μL、タックプラスTMプレシジョンポリメラーゼ(Stratagene:#600211) 1μL
、および水80μLを含む別々の管に加えた。RUP15のための反応温度およびサイク
ル時間は、サイクルステップ2〜4の35回反復を含んで下記であった。1分間94℃、
30秒間94℃、20秒間62℃、1分40秒間72℃、および5分間72℃。PCR産
物を1%アガロースゲル上を流し次いで精製した(データは記載しない)。精製した産物
をXbaIおよびEcoRVを用いて消化しそして所望の内挿物を精製しそしてそれぞれ
の制限部位でGs汎用ベクター内に連結した。陽性クローンを形質転換の後に単離しそし
て制限酵素消化により決定した。293細胞を用いる発現が、本明細書内に記載したプロ
トコールを用いて達成された。RUP15−「Gs融合タンパク質」のそれぞれの陽性ク
ローンを正確さを確認するために配列決定した。(核酸配列に関しては配列番号99、そ
してアミノ酸配列に関しては配列番号100参照)。
【0130】
RUP15は、Gsを介して共役する。下記の例示的「GPCR融合タンパク質」に
ついて、Gsαへの融合が達成された。
【0131】
RUP15−「Gsα融合タンパク質」構築物を以下のようにして作製した。プライマ
ーは以下のように設計した。
5’−TCTAGAATGACGTCCACCTGCACCAACAGC−3’(配列番
号101、センス)
5’−gatatcGCAGGAAAAGTAGCAGAATCGTAGGAAG−3’
(配列番号102、アンチセンス)
小文字のヌクレオチドはGタンパク質とRUP15との間の制限部位内のスペーサーと
して含まれる。センスおよびアンチセンスプライマーは、スペーサー(制限部位に帰属す
る)がGタンパク質とRUP15との間に存在するように、それぞれEcoRVとXba
Iのための制限部位を含んでいた。
【0132】
次いで、上記に開示したGsα汎用ベクター内の融合ためのそれぞれの受容体配列を確
保するためにPCRを使用し、それぞれ下記のプロトコールを用いた。RUP15のため
の100ng cDNAを、それぞれプライマー(センスおよびアンチ−センス)2μl
、10mM dNTP3μL、10XタックプラスTMプレシジョン緩衝液10μL、タッ
クプラスTMプレシジョンポリメラーゼ(Stratagene: #600211) 1μL、および水80μL
を含む別々の管に加えた。RUP15のための反応温度およびサイクル時間は、サイクル
ステップ2〜4の35回反復を含んで下記であった。1分間94℃、30秒間94℃、2
0秒間62℃、1分40秒間72℃、および5分間72℃。PCR産物を1%アガロース
ゲル上を流し次いで精製した(データは記載しない)。精製した産物を消化した。)精製
した産物をEcoRVおよびXbaIを用いて消化しそして所望の内挿物を精製しそして
それぞれの制限部位でGs汎用ベクター内に連結した。陽性クローンを形質転換の後に単
離しそして制限酵素消化により決定した。293細胞を用いる発現が、本明細書内に記載
したプロトコールを用いて達成された。RUP15−「Gs融合タンパク質」のそれぞれ
の陽性クローンを正確さを確認するために配列決定した。(核酸配列に関しては配列番号
103、そしてアミノ酸配列に関しては配列番号104参照)。
【0133】
b.Gq(6アミノ酸欠失)/Gi融合構築物
Gq(欠失)/Gi融合構築物の設計は、以下のようにして達成できた。N−末端の6
個のアミノ酸(TELSIMの配列(配列番号129)Gαq−サブユニットを有する2
〜7のアミノ酸を欠失しそして配列EYNLB(配列番号130)を有するC−末端の5
個のアミノ酸を、配列DCGLF(配列番号131)を有する「GαIタンパク質」の相
当するアミノ酸と置き換える。この融合構築物は、下記のプライマー
5’−gatcaagcttcCATGGCGTGCTGCCTGAGCGAGGAG−
3’(配列番号132)および
5’−gatcggatccTTAGAACAGGCCGCAGTCCTTCAGGTT
CAGCTGCAGGATGGTG−3’(配列番号133)
およびマウスGαq−野生型を含むプラスミド63313を用い、鋳型として血球凝集素
タグを用いるPCRにより得られる。小文字のヌクレオチドはスペーサーとして含まれる
。
【0134】
タックプラスプレシジョンDNAポリメラーゼ(Stratagene)は、ステップ2〜4の35
回反復を含む下記のサイクルによる複製に使用される。2分間95℃、20秒間95℃、
20秒間56℃、2分間72℃、および7分間72℃。PCR産物をpCRII−TOP
Oベクター(Invitrogen)内にクローニングしそしてABIビッグダイターミネーターキッ
ト(P.E.Biosystems) を用いて配列決定した。融合構築物の配列を含むTOPOクローン
からの挿入物を発現ベクターpcDNA3.1(+)中に、HindIII/BamHI
部位で2段クローニング法によりシャトルした。
【0135】
実施例6
開示したヒトGPCRの組織分布:RT−PCR
数種の新規のヒトGPCRの発現を確認しそして組織分布を決定するためにRT−PC
Rを適用した。使用したオリゴヌクレオチドは、鋳型としてGPCR特異性そしてヒト多
重組織cDNAパネル(MTC、Clontech) であった。タック DNAポリメラーゼ(Str
atagene)を製造者の指示に従う40μl反応物内の増幅のために使用した。反応物20μ
lを1.5%アガロースゲル上に加えてRT−PCR産物を分析した。下記の表Jは、受
容体、サイクル条件および使用したプライマーを表示する。
【0136】
【表8】
【0137】
【表9】
【0138】
実施例7
プロトコール:逆アゴニストおよびアゴニストの直接同定
A.〔35S〕GTPγSアッセイ
我々は内因性、構成的活性GPCRを候補化合物、例えば逆アゴニストの直接同定のた
めに完全には理解されていない理由から使用したけれども、アッセイ内の変動が大きくな
ることがある。次いで好ましくは、以上に公開したように「GPCR融合タンパク質」も
非内因性、構成的活性化GPCRと一緒に使用される。我々は、このようなタンパク質を
使用する場合にアッセイ内変動が本質的に安定化され、これにより効果的な信号対騒音比
が得られるように見えることを認めた。これは候補化合物のさらに強固な同定を可能とす
る有利な結果を有する。このように、直接同定のために「GPCR融合タンパク質」が使
用されそして利用する場合には下記のアッセイプロトコールを利用することが望ましい。
【0139】
1.膜調製
関係する構成的活性オーファン「GPCR融合タンパク質」を含んでなりそして逆アゴ
ニスト、アゴニストまたは部分アゴニストとしての候補化合物の直接同定に使用するため
の膜は、好ましくは下記のように調製される。
【0140】
a.材料
「膜スクレープ緩衝液」は20mM HEPESおよび10mM EDTA、pH7.
4を含んでなる。「膜洗浄緩衝液」は20mM HEPESおよび0.1mM EDTA
、pH7.4を含んでなる。「結合緩衝液」は20mM HEPES、100mM Na
Cl、および10mM MgCl2 、pH7.4を含んでなる。
【0141】
b.操作
操作の間を通じてすべての材料を氷上に保持する。第一に、媒体を細胞の集密単層から
吸引し、次いで10ml冷PBSを用いてリンスし、次いで吸引する。その後、「膜スク
レープ緩衝液」5mlをスクレープ細胞に加える、これに続いて、50ml遠心分離管内
に細胞抽出物を移す(20,000rpmで17分間、4℃で遠心分離する)。その後、
上清を吸引しそしてペレットを膜「洗浄緩衝液」30ml中に再懸濁し、次いで20,0
00rpmで17分間、4℃で遠心分離する。次いで、上清を吸引しそしてペレットを「
結合緩衝液」内に再懸濁する。次いでこれをブリンクマン・ポリトロン(Brinkman polytr
on) TMホモジナイザーを用いてホモジナイジングする(すべての材料が懸濁するまで15
〜20秒間バースト)。これを本明細書中では「膜タンパク質」と呼ぶ。
【0142】
2.ブラドフォードタンパク質アッセイ
ホモジナイゼーションに次いで、膜のタンパク質濃度をブラドフォード(Bradford)タン
パク質アッセイを用いて決定する(タンパク質は約1.5mg/mlまで希釈し、アリコ
ートに入れ、以後の使用のために冷凍(−80℃)できる。冷凍した場合に、使用のため
のプロトコールは以下である:アッセイの日に、冷凍した「膜タンパク質」を室温で解凍
し、次いでウズ攪拌しそして約12x1.000rpmで約5〜10秒間ポリトロンを用
いてホモジナイズする。複数の調製のためには、異なる調製物のホモジナイゼーションの
間でホモジナイザーを完全に清浄化することに注意すること)
【0143】
a.材料
「結合緩衝液」(上記の通り)、「ブラドフォード染色試薬」、「ブラドフォードタン
パク質標準」を製造者の指示に従って使用する(Biorad カタログ番号500-0006) 。
【0144】
b.操作
2本の管を調製し、一方は膜を含みそして他方は対照の「ブランク」とする。それぞれ
は「結合緩衝液」800ulを含む。その後、「ブラドフォードタンパク質標準」(1m
g/ml)10μlをそれぞれの管に加え、次いで膜「タンパク質」10μlを一本の管
(ブランク以外)を除いて加える。その後、「ブラドフォード染色試薬」200μlをそ
れぞれの管に加え、次いでそれぞれウズ攪拌する。5分後に、管を再びウズ攪拌しそして
その中の材料をキュベットに移す。次いでキュベットをCECIL3041分光光度計を
用いて波長595で読み取る。
【0145】
3.直接同定アッセイ
a.材料
「GDP緩衝液」は、37.5ml「結合緩衝液」および2mg GDP(Sigmaカタロ
グ番号G-7127) から成り、次いで「結合緩衝液」中の一連の希釈により0.2μM GD
Pが得られる(それぞれのウエル内のGDPの最終濃度は、0.1μM GDP)。候補
化合物を含んでなるそれぞれのウエルは100μl「DPG緩衝液」(最終濃度、0.1
μM GDP)、「結合緩衝液」中の「膜タンパク質」50ul、および「結合緩衝液」
中の〔35S〕GTPγS(0.6nM)50μl(10ml「結合緩衝液」あたり2.5
μl〔35S〕GTPγS)から成る200ulの最終体積を有する。
【0146】
b.操作
候補化合物は、好ましくは96ウエルプレートフォーマットを用いてスクリーニングさ
れる(これらは−80℃で冷凍できる)。「膜タンパク質」(または対照として「GPC
R融合タンパク質」を含まない発現ベクターを有する膜)を懸濁するまで短時間ホモジナ
イズする。次いで、タンパク質濃度を上記の「ブラドフォードタンパク質アッセイ」セッ
トを用いて決定する。次いで「膜タンパク質」(および対照)を「結合緩衝液」中で0.
25mg/mlまで希釈する(最終アッセイ濃度、12.5μgウエル)。その後、「G
DP緩衝液」100μlをウオラックシンチストリップ(Wallac Scintistrip) TM (Walla
c)のそれぞれのウエルに加えた。次いで候補化合物5ulをこのウエル内に移すために5
ulピン器具を用いる(すなわち200μlの全アッセイ体積中の5ulは、候補化合物
の最終スクリーニング濃度が10μMであるような1:40の比である)。再度、汚染を
防ぐために、それぞれの移動ステップの後、水(1X)、エタノール(1X)および水(
2X)を含んでなる3基の容器内でピン器具を洗浄すること。過剰の液体をそれぞれのリ
ンスの後に器具から振り払いそして紙およびキムワイプ(kimwipe)を用いて乾燥すること
。その後、「膜タンパク質」50μlをそれぞれのウエルに加え(「GPCR融合タンパ
ク質」を含まない膜を含んでなる対照ウエルも使用する)、そして5〜10分間、室温で
予備インキュベーションする。その後、「結合緩衝液」中の〔35S〕GTPγS(0.6
nM)50μlをそれぞれのウエルに加え、次いで振とう器内で60分間、室温でインキ
ュベーションする(この実施例でもプレートをフォイルで覆う)。次いで、プレートを4
00rpmで15分間、22℃で回転してアッセイを停止する。次いでプレートを8チャ
ンネルマニホルドを用いて吸引しそしてプレートカバーを用いてシールする。次いで、ワ
ラック1450で設定「Prot.$37」を用いてプレートを読み取る(製造者の指定
に従う)
【0147】
B.サイクリックAMPアッセイ
候補化合物を直接同定するための別のアッセイ方法は、シクラーゼに基づくアッセイを
用いて達成される。直接同定に加えて、このアッセイ法は、上記のような〔35S〕GTP
γS法からの結果の確認を与えるための独立した方法としても使用できる。
【0148】
変更したフラッシュプレートTMアデニル酸シクラーゼ・キット(New England Nuclear,
カタログ番号SMP004A)が、下記のプロトコールに従う構成的活性化オーファンGPCRに
対する逆アゴニストおよびアゴニストとしての候補化合物の直接同定のために好ましくは
使用された。
【0149】
トランスフェクションされた細胞をトランスフェクションの約3日後に採取した。20
mM HEPES、pH7.4および10mM MgCl2 を含む緩衝液中に懸濁した細
胞のホモジナイゼーションにより膜を調製した。ホモジナイゼーションは、ブリンクマン
ポリトロンTMを約10秒間使用して氷上で行った。得られたホモジネートを49,000
Xgで15分間、4℃で遠心分離する。次いで得られたペレットを20mM HEPES
、pH7.4および0.1mM EDTAを含む緩衝液中に再懸濁し、10秒間ホモジナ
イズし、次いで49,000Xgで15分間、4℃で遠心分離した。次いで得られたペレ
ットを−80℃で、使用するまで保管した。直接同定スクリーニングの日に、膜ペレット
を室温でゆっくりと解凍し、20mM HEPES、pH7.4および10mM MgC
l2を含む緩衝液中に再懸濁すると、0.60mg/mlの最終タンパク質濃度が得られ
た(再懸濁した膜は使用まで氷上に置く)。
【0150】
cAMP標準および「検出緩衝液」(「検出緩衝液」11mlにトレーサー〔12IcA
MP(100μl〕の2μCiを含んでなる)を調製しそして製造者の指示に従って維持
した。「アッセイ緩衝液」をスクリーニングのために調製し、これは20mM HEPE
S、pH7.4、10mM MgCl2 、20mMホスホクレアチン(Sigma) 、0.1単
位/mlクレアチンホスホキナーゼ(Sigma)、50μM GTP(Sigma)、および0.2m
M ATP(Sigma) を含んでいた。次いで「アッセイ緩衝液」を使用まで氷上で保管した
。
【0151】
上記のようにして同定された候補化合物(冷凍された場合には、室温で解凍)を、40
μl「膜タンパク質」(30μg/ウエル)および「アッセイ緩衝液」50μlと一緒に
、好ましくは96プレートウエルに加えた(3μl/ウエル、12μM最終アッセイ濃度
)。次いでこの混合物を30分間、室温で穏やかに振とうしてインキュベーションした。
【0152】
インキュベーションの後、「検出緩衝液」100μlをそれぞれのウエルに加え、次い
で2〜24時間インキュベーションした。次いでプレートをワラック・マイクロベータTM
プレートリーダーで「Prot#31」を用いて計数した(製造者の指示に従う)。
【0153】
代表的なスクリーニングアッセイプレート(96ウエルフォーマット)結果を図12に
示す。それぞれの棒は、それぞれのウエル内の種々の化合物と、上記の実施例5(a)で
調製したRUP13−GαA融合タンパク質に対する結果を示す。図12に示した代表的
な結果は、それぞれのプレートの平均結果(「m」)に基づいた標準偏差および平均も与
え、そして一次スクリーンから「リード」としての逆アゴニストの選択のための平均プラ
ス2個の任意選択は、少なくとも平均プレート反応による反応百分率から標準偏差の二倍
を差し引いて減らした候補化合物の選択を含む。反対に、一次スクリーンから「リード」
としてのアゴニストの選択のため任意選択は、少なくとも平均プレート反応による反応百
分率に標準偏差の二倍を加えて増加した候補化合物の選択を含む。これらの選択手順に基
づいて、以下のウエル内の候補化合物は、ウエルA2およびG9それぞれの中のRUP1
3に対する推定逆アゴニスト(化合物A)およびアゴニスト(化合物B)として直接同定
された。図12参照。明確化のために以下を記す。これらの化合物はこのGPCRのため
の内因性リガンドのいかなる知識も持たないで直接同定された。受容体機能に基づくアッ
セイ技術を中心とし、そして化合物結合親和性ではなくて、我々はこの受容体の機能的活
性を低下でき(化合物A)ならびに受容体の機能的活性を増加できる(化合物B)化合物
を確認できる。肺組織内のこれら受容体の位置に基づいて(例えば実施例6のhRUP1
3およびhRUP21参照)、薬剤が肺ガンの可能な治療処置のために開発できる。
【0154】
本明細書内で引用された文献は、同時係属および関連特許を含み、特に断らない限り、
引用することにより全体を本明細書中に編入される。熟練者の判断の範囲内にある開示さ
れた本発明の変更および延長は、上記の開示および特許請求範囲の範囲内に包含される。
【0155】
種々の発現ベクターが当該技術分野の熟練者により利用可能であるが、内因性および非
内因性ヒトGPCRの両者のための使用の目的で、使用されるベクターがpCMVである
ことが最も好ましい。このベクターは、1998年10月13日付けで、特許手続上の微
生物の寄託の国際的承認に関するブダペスト条約の規定に基づいてアメリカンタイプカル
チャーコレクション(ATCC)に寄託された(10801 University Blvd., Manassas, VA
20110-2209 USA) 。DNAはATCCDにより試験されそして利用可能と決定された。A
TCCは、pCMVに下記の寄託番号を割り当てた:ATCC#203351。
【技術分野】
【0001】
本特許明細書中に開示される本発明は膜貫通受容体に関し、そしてさらに具体的にはヒ
トGタンパク質共役型受容体に関し、そして特定すると、受容体の構成的活性を確立また
は増強するために改変されたGPCRの非内因性型(version)に特に重点を置いた内因性
ヒトGPCRに関する。好ましくは、改変されたGPCRは、治療薬剤として潜在的な適
用可能性を有する受容体アゴニスト、逆アゴニストまたは部分アゴニストとしての候補化
合物の直接同定のために使用される。
【背景技術】
【0002】
多数の受容体の種類がヒト内に存在するけれども、特に最も数が多くそして治療的に関
連するものはGタンパク質共役型受容体(GPCRまたはGPCRs)の種類により代表
される。ヒトゲノム内に約100,000個の遺伝子が存在すると推定され、これらの中
の約2%、すなわち2,000個の遺伝子がGPCRをコードすると推定される。そのた
めの内因性リガンドが同定されているGPCRを含む受容体は、「既知」受容体と呼ばれ
、一方そのための内因性リガンドが同定されていない受容体は「オーファン」受容体と呼
ばれる。GPCRは薬剤製品の開発のために重要な分野であり、100種の既知GPCR
の中の約20種から、約60%がすべて処方薬で開発されている。
【0003】
GPCRは、共通の構造モチーフを共有している。すべてのこれらの受容体は、7個の
αラセンを形成する22〜24個の疎水性アミノ酸の7個の配列を有し、これらそれぞれ
は膜をスパン(span)する(それぞれのスパンは番号で特定され、すなわち、膜貫通部−1
(TM−1)、膜貫通部−1(TM−1)、などである)膜貫通ラセンは、膜貫通部−2
と膜貫通部−3、膜貫通部−4と膜貫通部−5、および膜貫通部−6と膜貫通部−7との
間のアミノ酸の鎖により細胞膜の外側、すなわち「細胞外」側で結合される(これらは「
細胞外」領域1、2および3(EC−1、EC−2およびEC−3)とそれぞれ呼ばれる
)。膜貫通ラセンは、膜貫通部−1と膜貫通部−2、膜貫通部−3と膜貫通部−4、およ
び膜貫通部−5と膜貫通部−6との間のアミノ酸の鎖により細胞膜の内側、すなわち「細
胞内」側でも結合される(これらは「細胞内」領域1、2および3(IC−1、IC−2
およびIC−3)とそれぞれ呼ばれる)。受容体の「カルボキシ」(”C”)末端は細胞
内部の細胞内空間内にあり、そして受容体の「アミノ」(”N”)末端は細胞外側の細胞
外空間内にある。
【0004】
一般に、内因性リガンドが受容体を結合する場合には(しばしば受容体の「活性化」と
呼ばれる)、細胞内領域と細胞内「Gタンパク質」との間の共役を許容する細胞内領域の
コンホーメーションの変化がある。GPCRはGタンパク質に関して「多数関係」性であ
る、すなわち、GPCRは1個を越えるGタンパク質と相互作用できると報告されている
。非特許文献1参照。他のGタンパク質も存在するけれども、現在、Gq、Gs、Gi、
GzおよびGoが同定されたGタンパク質である。Gタンパク質との内因性リガンド活性
化GPCR共役は、シグナル伝達カスケードプロセスを開始する(「シグナル伝達」と呼
ばれる)。正常条件下で、シグナル伝達は、最終的に細胞活性化または細胞阻害をもたら
す。受容体のIC−3ループならびにカルボキシ末端がGタンパク質と相互作用すると考
えられる。
【0005】
生理的条件下で、GPCRは、2個の異なるコンホーメーション、すなわち「不活性」
状態および「活性」状態の間で平衡して細胞膜内に存在する。不活性状態にある受容体は
、細胞内シグナル伝達経路に連結して生物学的応答を生成できない。活性状態への受容体
コンホーメーションの変化は、シグナル伝達経路への連結を許容し(Gタンパク質を介し
て)そして生物学的反応を生成する。
【0006】
受容体は、内因性リガンドまたは薬剤などの化合物により活性状態に安定化されてもよ
い。受容体のアミノ酸配列の変更を含みこれに限られない最近の発見は、活性状態コンホ
ーメーションにおける受容体を促進および安定化するための内因性リガンドまたは薬剤以
外の手段を提供する。これらの手段は、受容体に結合する内因性リガンドの作用を模擬す
ることにより活性状態にある受容体を効果的に安定化する。このようなリガンド依存手段
による安定化は、「構成的受容体活性化」と呼ばれる。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Kenakin,T., 43 Life Science 1095(1988)
【発明の概要】
【課題を解決するための手段】
【0008】
(発明の概要)
本明細書中には、ヒトGPCRの内因性および非内因性型およびその使用を開示する。
本発明はまた、以下の項目を提供する。
(項目1) 配列番号2のアミノ酸配列によりコードされるGタンパク質共役型受容
体。
(項目2) 項目1のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目3) 配列番号1のベクターおよびcDNAを含んでなるプラスミド。
(項目4) 項目3のプラスミドを含んでなる宿主細胞。
(項目5) 配列番号4のアミノ酸配列によりコードされるGタンパク質共役型受容
体。
(項目6) 項目5のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目7) 配列番号3のベクターおよびcDNAを含んでなるプラスミド。
(項目8) 項目7のプラスミドを含んでなる宿主細胞。
(項目9) 配列番号6のアミノ酸配列によりコードされるGタンパク質共役型受容
体。
(項目10) 項目9のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目11) 配列番号5のベクターおよびcDNAを含んでなるプラスミド。
(項目12) 項目11のプラスミドを含んでなる宿主細胞。
(項目13) 配列番号8のアミノ酸配列によりコードされるGタンパク質共役型受
容体。
(項目14) 項目13のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目15) 配列番号7のベクターおよびcDNAを含んでなるプラスミド。
(項目16) 項目15のプラスミドを含んでなる宿主細胞。
(項目17) 配列番号10のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目18) 項目17のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目19) 配列番号9のベクターおよびcDNAを含んでなるプラスミド。
(項目20) 項目19のプラスミドを含んでなる宿主細胞。
(項目21) 配列番号12のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目22) 配列番号84のアミノ酸配列を含んでなる項目21のGタンパク質共
役型受容体の非内因性、構成的活性化型。
(項目23) 配列番号11のベクターおよびcDNAを含んでなるプラスミド。
(項目24) 項目23のプラスミドを含んでなる宿主細胞。
(項目25) 配列番号14のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目26) 配列番号88のアミノ酸配列を含んでなる項目25のGタンパク質共
役型受容体の非内因性、構成的活性化型。
(項目27) 配列番号13のベクターおよびcDNAを含んでなるプラスミド。
(項目28) 項目27のプラスミドを含んでなる宿主細胞。
(項目29) 配列番号16のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目30) 配列番号92のアミノ酸配列を含んでなる項目29のGタンパク質共
役型受容体の非内因性、構成的活性化型。
(項目31) 配列番号15のベクターおよびcDNAを含んでなるプラスミド。
(項目32) 項目31のプラスミドを含んでなる宿主細胞。
(項目33) 配列番号18のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目34) 項目33のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目35) 配列番号17のベクターおよびcDNAを含んでなるプラスミド。
(項目36) 項目35のプラスミドを含んでなる宿主細胞。
(項目37) 配列番号20のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目38) 項目37のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目39) 配列番号19のベクターおよびcDNAを含んでなるプラスミド。
(項目40) 項目39のプラスミドを含んでなる宿主細胞。
(項目41) 配列番号22のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目42) 項目41のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目43) 配列番号21のベクターおよびcDNAを含んでなるプラスミド。
(項目44) 項目43のプラスミドを含んでなる宿主細胞。
(項目45) 配列番号24のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目46) 項目45のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目47) 配列番号23のベクターおよびcDNAを含んでなるプラスミド。
(項目48) 項目47のプラスミドを含んでなる宿主細胞。
(項目49) 配列番号26のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目50) 項目49のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目51) 配列番号25のベクターおよびcDNAを含んでなるプラスミド。
(項目52) 項目51のプラスミドを含んでなる宿主細胞。
(項目53) 配列番号28のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目54) 項目53のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目55) 配列番号27のベクターおよびcDNAを含んでなるプラスミド。
(項目56) 項目55のプラスミドを含んでなる宿主細胞。
(項目57) 配列番号30のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目58) 項目57のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目59) 配列番号29のベクターおよびcDNAを含んでなるプラスミド。
(項目60) 項目59のプラスミドを含んでなる宿主細胞。
(項目61) 配列番号32のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目62) 配列番号96のアミノ酸配列を含んでなる項目61のGタンパク質共
役型受容体の非内因性、構成的活性化型。
(項目63) 配列番号95のベクターおよびcDNAを含んでなるプラスミド。
(項目64) 項目63のプラスミドを含んでなる宿主細胞。
(項目65) 配列番号34のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目66) 項目65のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目67) 配列番号33のベクターおよびcDNAを含んでなるプラスミド。
(項目68) 項目67のプラスミドを含んでなる宿主細胞。
(項目69) 配列番号36のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目70) 項目69のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目71) 配列番号35のベクターおよびcDNAを含んでなるプラスミド。
(項目72) 項目71のプラスミドを含んでなる宿主細胞。
(項目73) 配列番号38のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目74) 項目73のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目75) 配列番号37のベクターおよびcDNAを含んでなるプラスミド。
(項目76) 項目75のプラスミドを含んでなる宿主細胞。
(項目77) 配列番号40のアミノ酸配列によりコードされるGタンパク質共役型
受容体。
(項目78) 項目77のGタンパク質共役型受容体の非内因性、構成的活性化型。
(項目79) 配列番号39のベクターおよびcDNAを含んでなるプラスミド。
(項目80) 項目79のプラスミドを含んでなる宿主細胞。
【図面の簡単な説明】
【0009】
【図1】対照(「CMV」)と比較した内因性型RUP12(「RUP12」)からの第二メッセンジャーIP3 産生の説明である。
【図2】内因性RUP13(「RUP13」)と対照ベクター(「CMV」)との構成的シグナル伝達の比較結果を与える第二メッセンジャー細胞基礎サイクリックAMPアッセイの結果の図示である。
【図3】CMV、内因性RUP13(「RUP13wt」)および非内因性、構成的活性化RUP13(「RUP13(A268K)」)を8XCRE−Lucレポータープラスミドを用いて比較して測定したシグナルの図示である。
【図4】RUP13:「Gs融合タンパク質」(「RUP13−Gs」)と対照ベクター(「CMV」)による構成的シグナル伝達の比較結果を与える〔35S〕GTPγSアッセイの結果の図示である。
【図5】CMV、内因性RUP14(「RUP14wt」)および非内因性、構成的活性化RUP13(「RUP14(L246K)」)を8XCRE−Lucレポータープラスミドを用いて比較して測定したシグナルの図示である。
【図6】CMV、内因性RUP15(「RUP15wt」)および非内因性、構成的活性化RUP15(「RUP15(A398K)」)を8XCRE−Lucレポータープラスミドを用いて比較して測定したシグナルの図示である。
【図7】内因性RUP15(「RUP15wt」)、RUP15の非内因性、構成的活性化型(「RUP15(A398K)」)および対照ベクター(「CMV」)の構成的シグナル伝達の比較結果を与える第二メッセンジャー細胞基礎サイクリックAMPアッセイの結果の図示である。
【図8】RUP15:「Gs融合タンパク質」(「RUP15−Gs」)と対照ベクター(「CMV」)による構成的シグナル伝達の比較結果を与える〔35S〕GTPγSアッセイの結果の図示である。
【図9】対照(「CMV」)と比較した内因性型RUP17(「RUP17」)からの第二メッセンジャーIP3 産生の説明である。
【図10】対照(「CMV」)と比較した内因性型RUP21(「RUP21」)からの第二メッセンジャーIP3 産生の説明である。
【図11】CMV、内因性RUP23(「RUP23wt」)および非内因性、構成的活性化RUP23(「RUP23(W275K)」)を8XCRE−Lucレポータープラスミドを用いて比較して測定したシグナルの図示である。
【図12】RUP13に対する各種の候補化合物の一次スクリーニングからの結果の図示である。「化合物A」の結果はウエルA2に示されそして「化合物B」の結果はウエルG9に示される。
【発明を実施するための形態】
【0010】
(発明の詳細な記述)
受容体に関連して作成された科学文献は、受容体に関して種々の作用を有するリガンド
に関する多数の用語を採用している。明白さおよび統一のために、下記の定義を本特許明
細書中を通じて使用する。これらの定義がこれらの用語のための他の定義と矛盾する場合
には、下記の定義が優先する。
【0011】
「アゴニスト」は、物質が受容体と結合した場合に細胞内反応を活性化するか、または
膜にへのGTP結合を増強する物質(例えばリガンド、候補化合物)を意味するものとす
る。
【0012】
本明細書中に使用される「アミノ酸の略字」を表Aに記載する。
表A
アラニン ALA A
アルギニン ARG R
アスパラギン ASN N
アスパラギン酸 ASP D
システイン CYS C
グルタミン酸 GLU E
グルタミン GLN Q
グリシン GLY G
ヒスチジン HIS H
イソロイシン ILE I
ロイシン LEU L
リシン LYS K
メチオニン MET M
フェニルアラニン PHE F
プロリン PRO P
セリン SER S
トレオニン THR T
トリプトファン TRP W
チロシン TYR Y
バリン VAL V
「部分アゴニスト」は、これらが受容体に結合された場合にアゴニストの作用よりも低
い程度/範囲で細胞内反応を活性化するか、またはアゴニストの作用よりも低い程度/範
囲で膜へのGTP結合を増強する物質(例えばリガンド、候補化合物)を意味するものと
する。
【0013】
「アンタゴニスト」は、アゴニストと同じ部位で受容体に競合的に結合するがしかし受
容体の活性形により開始された細胞内反応を活性化せず、そしてこれによりアゴニストま
たは部分アゴニストによる細胞内反応を阻害できる物質(例えばリガンド、候補化合物)
を意味するものとする。「アンタゴニスト」は、アゴニストまたは部分アゴニストが存在
しない場合に基準細胞内反応を低下させない。
【0014】
「候補化合物」は、スクリーニング技術に適用できる分子(例えば化学化合物であり、
これに限定はされない)を意味するものとする。好ましくは、語句「候補化合物」は、あ
らかじめ間接同定過程により決定されたもの(「間接同定された化合物」)であって、受
容体に対して逆アゴニスト、アゴニストまたはアンタゴニストからなる群から選ばれた化
合物として一般的に知られている化合物を含まず、さらに好ましくは、少なくとも1種の
哺乳動物内において治療効力を有すると以前に決定された間接同定された化合物を含まず
、そして最も好ましくは、ヒトにおいて治療効力を有すると以前に決定された間接同定さ
れた化合物を含まない。
【0015】
「組成物」は、少なくとも1種の成分を含んでなる物質を意味する。「薬剤組成物」は
、組成物の一例である。
【0016】
「化合物効力」は、受容体結合親和性に対抗して、受容体機能性を阻害または刺激する
ための化合物の能力の尺度を意味するものとする。化合物効力を検出するための例示的な
手段は、本明細書の実施例の部分に開示される。
【0017】
「コドン」は、一般に、リン酸基に結合し、そして翻訳されるとアミノ酸をコードする
ヌクレオシド(アデノシン(A)、グアノシン(G)、シチジン(C)、ウリジン(U)
およびチミジン(T))を含んでなる3個のヌクレオチド(またはヌクレオチドの等価基
)の群を意味するものとする。
【0018】
「構成的活性化受容体」は、構成的受容体活性化を受ける受容体を意味するものとする
。構成的活性化受容体は内因性または非内因性であることができる。
【0019】
「構成的受容体活性化」は、受容体とその内因性リガンドまたはその化学的等価物との
結合以外の手段により活性状態にある受容体の安定化を意味するものとする。
【0020】
「接触」または「接触する」は、生体外系または生体内系のいずれでも、少なくとも2
個の部分を一緒にすることを意味するものとする。
【0021】
語句「候補化合物」に関連する「直接同定する」または「直接同定」は、構成的活性化
受容体、好ましくは構成的活性化オーファン受容体、そして最も好ましくは構成的活性化
Gタンパク質共役細胞表面オーファン受容体に対して候補化合物をスクリーニングし、そ
してこの化合物の化合物効力を評価することを意味するものとする。この語句は、いかな
る場合でも、語句「間接同定する」または「間接同定された」により包含されまたはこれ
らを包含するとは解釈または理解されない。
【0022】
「内因性」は、哺乳動物が本来的に産生する物質を意味するものとする。例えば、限定
ではないが用語「受容体」に関する「内因性」は、哺乳動物(例えば、限定ではないがヒ
ト)またはウイルスにより本来的に産生されるものを意味するものとする。反対に、この
範囲内での用語「非内因性」は、哺乳動物(例えば、限定ではないがヒト)またはウイル
スにより本来的には産生されないものを意味するものとする。例えば、限定ではないが、
その内因性形では構成的活性ではないが、しかし操作されると構成的活性となる受容体は
、本明細書内では「非内因性、構成的活性化受容体」と呼ばれるのが最も好ましい。両方
の用語は、「インビボ」および「インビトロ」系の両方を記述するために使用できる。例
えば、限定ではないが、スクリーニング法において、内因性または非内因性受容体は、生
体内スクリーニング系に関連してもよい。別の例であって限定ではないが、哺乳動物のゲ
ノムが非内因性、構成的活性化受容体を含むように操作された場合に、生体内系の手段に
よる候補化合物のスクリーニングが適用可能である。
【0023】
「Gタンパク質共役型受容体融合タンパク質」および「GPCR融合タンパク質」は、
本明細書中で開示する本発明の範囲内で、それぞれ、少なくとも1種のGタンパク質、最
も好ましくはこのようなGタンパク質のアルファ(α)サブユニット(これはGTPを結
合するサブユニットである)に融合された内因性、構成的活性化GPCRまたは非内因性
、構成的活性化GPCRを含んでなる非内因性タンパク質を意味し、ここでGタンパク質
は、好ましくは内因性オーファンGPCRと本来的に共役するGタンパク質と同じ種類で
ある。例えば、限定ではないが、内因性状態において、Gタンパク質”Gsα”がGPC
Rと共役する優勢なGタンパク質である場合に、特定のGPCRに基づく「GPCR融合
タンパク質」はGsαに融合したGPCRを含んでなる非内因性タンパク質であろう。あ
る場合には、以下に記載するように、非−優勢Gタンパク質はGPCRに融合できる。G
タンパク質は、構成的活性GPCRのc−末端に直接融合できるかまたはこれら2者の間
にスペーサーがあってもよい。
【0024】
「宿主細胞」は、その中に「プラスミド」および/または「ベクター」を組み込まれる
ことができる細胞を意味するものとする。原核生物「宿主細胞」の場合に、「プラスミド
」は、典型的には、「宿主細胞」複製物として自律性分子として複製される(従って一般
に、「プラスミド」は、真核生物「宿主細胞」内への導入のために単離される)。真核生
物「宿主細胞」の場合には、「プラスミド」は真核生物「宿主細胞」が複製された場合に
「プラスミド」が複製されるように、「宿主細胞」の細胞DNA中に組み込まれる。好ま
しくは、本明細書中に開示する本発明の目的のためには、「宿主細胞」は真核生物であり
、さらに好ましくは、哺乳動物であり、そして最も好ましくは293、293TおよびC
OS−7細胞からなる群より選ばれる。
【0025】
「間接同定」または「間接同定された」は、内因性受容体に特異性の内因性リガンドの
同定、リガンド−受容体相互作用を妨害および/またはこれと競合するものの決定のため
の受容体に対する候補化合物のスクリーニング、および活性化受容体と関連する少なくと
も1種の第二メッセンジャー経路に影響するための化合物の効力の評価を含む薬剤発見過
程への伝統的な接近方法を意味する。
【0026】
「阻害」または「阻害する」は、用語「反応」に関連して、化合物が存在しない場合の
反対に、化合物が存在する場合には反応が低下または防止されることを意味するものとす
る。
【0027】
「逆アゴニスト」は、受容体の内因性形または受容体の構成的活性化形のいずれかに結
合し、そしてアゴニストまたは部分アゴニストが存在しない場合に観察される正常の活性
基準レベル以下の受容体の活性形により開始される基礎細胞内反応を阻害し、または膜へ
のGTP結合を低下させる物質(例えばリガンド、候補化合物)を意味するものとする。
好ましくは、基礎細胞内反応は、逆アゴニストが存在しない場合の基準反応と比較して、
逆アゴニストの存在においては少なくとも30%、さらに好ましくは少なくとも50%、
そして最も好ましくは少なくとも75%が阻害される。
【0028】
「既知受容体」は、当該受容体に特異性の内因性リガンドが同定されている内因性受容
体を意味するものとする。
【0029】
「リガンド」は、内因性で本来的に存在する受容体に特異性の内因性で本来的に存在す
る分子を意味するものとする。
【0030】
内因性受容体の核酸および/またはアミノ酸配列に関する「変異体」または「変異」は
、内因性、非構成的活性化受容体の変異形が受容体の構成的活性化を明らかにするような
内因性配列への特定の変化または多数の変化を意味するものとする。特定の配列の等価体
に関して、(a)ヒト受容体の次の変異した形の構成的活性化のレベルが受容体の最初の
変異により明らかにされたものと本質的に同じであり、そして(b)受容体の次の変異形
と受容体の最初の変異体との間の配列(アミノ酸および/または核酸)相同性の割合が少
なくとも約80%、さらに好ましくは少なくとも約90%そして最も好ましくは少なくと
も約95%である場合に、ヒト受容体の次の変異形は、ヒト受容体の最初の変異に等価と
考えられる。理想的には、そして構成的活性化を達成するために本明細書中で開示した最
も好ましいカセットがGPCRの内因性と非内因性形との間の単一アミノ酸および/また
はコドン変化を含むという事実のために、配列相同性の割合は少なくとも98%でなけれ
ばならない。
【0031】
「非オーファン受容体」は、受容体へのリガンドの結合が分子内シグナル伝達経路を活
性化する、内因性で本来的に存在するリガンドに特異性の内因性で本来的に存在する分子
を意味するものとする。
【0032】
「オーファン受容体」は、その受容体に特異性の内因性リガンドが同定されていないか
または既知ではない内因性受容体を意味するものとする。
【0033】
「薬剤組成物」は、少なくとも1種の活性成分を含んでなる組成物を意味するものとし
、ここで組成物は哺乳動物(例えば、限定ではないがヒト)において特定の効力的な成果
のための試験が可能である。通常の当該技術分野の熟練者は、活性成分が、熟練者の要求
に基づいて所望の効力的な成果を有するかどうかを決定するために適当な技術を理解しそ
して認めるであろう。
【0034】
「プラスミド」は、「ベクター」およびcDNAの組合せを意味するものとする。一般
に、「プラスミド」は、タンパク質としてのcDNAの複製および/または発現の目的で
「宿主細胞」内に導入される。
【0035】
「第二メッセンジャー」は、受容体活性化の結果として生成した細胞内反応を意味する
ものとする。第二メッセンジャーは、例えば、イノシトール三リン酸(IP3 )、ジアシ
ルグリセロール(DAG)、サイクリックAMP(cAMP)、およびサイクリックGM
P(cGMP)を含むことができる。第二メッセンジャー反応は、受容体活性化の決定の
ために測定できる。さらに、第二メッセンジャー反応は、例えば逆アゴニスト、アゴニス
ト、部分アゴニストおよびアンタゴニストを含む候補化合物の直接同定のために測定でき
る。
【0036】
「刺激」または「刺激する」は、用語「反応」に関連して、反応が化合物の存在下で、
これが存在しない場合の反対に増加することを意味するものとする。
【0037】
cDNAの関連する「ベクター」は、少なくとも1種のcDNAを組み込むことが可能
でそして「宿主細胞」内に組み込むことが可能な環状DNAを意味するものとする。
【0038】
以下の各セクションの順序は、説明の効率のために決定したものであり、下記の開示ま
たは請求の制限を意図せず、そしてそのように解釈してはならない。
A.はじめに
受容体の伝統的な研究は、発見が受容体に影響できるアンタゴニストおよびその他の分
子を発見できるまで進行する前に内因性リガンドが最初に同定されなければならないとい
うアプリオリな仮定(歴史に基づく)から常に前進していた。アンタゴニストが最初に判
明した場合でも、探索は直ちに内因性リガンドの探索に進んだ。この思考の様式は、構成
的活性化受容体の発見の後でも受容体研究で継続された。これまで認められていなかった
ことは、受容体のアゴニスト、部分アゴニスト、および逆アゴニストの発見に最も有用な
ことは受容体の活性状態であることである。過剰活性の受容体または不足活性の受容体か
らもたらされる疾患に対して、治療薬剤に望まれるものは、それぞれ、受容体の活性状態
を低下するかまたは受容体の活性を増強する化合物であり、必ずしも内因性リガンドに対
するアンタゴニストである薬剤ではない。これは、活性受容体状態の活性を低下または増
強する化合物は、内因性リガンドとして同じ部位に結合する必要はないためである。従っ
て、本発明の方法により教示されるように、治療化合物のためのあらゆる探索もリガンド
に依存しない活性状態に対する化合物をスクリーニングすることにから開始すべきである
。
B.ヒトGPCRの同定
ヒトゲノムプロジェクトの研究は、ヒトゲノム内に位置する核酸配列に関する
極めて多数の情報の同定をもたらした。この研究の場合に、ゲノム配列情報は、
特定のゲノム配列がヒトタンパク質を翻訳するオープンリーディングフレーム情
報を含むかまたは含まないかどうかに関する理解または認識を伴わずにに提供さ
れた。ヒトゲノム内の核酸配列を同定する数種の方法は、通常の当該技術分野を
有する熟練者の認識範囲内にある。例えば、限定ではないが、本明細書中で開示
される種々のヒトGPCRは、ジーンバンク(GeneBank)TMデータベースを調査し
て発見された。下記の表Bは、我々が発見した数種類の内因性GPCRを、開示
GPCRに相同である他のGPCRと共に表示する。
【0039】
【表1】
【0040】
受容体相同性は、ヒト身体内の受容体の役割の評価を得るために有用である。本明細書
の記載の進行に応じて、我々は、これらの受容体の非内因性、構成的活性化型を確立する
ためにこれらの受容体を変異するための技術を開示するであろう。
【0041】
本明細書中に開示される技術は、当該技術分野では既知の他のヒト、オーファンGPC
Rにも適用され、これは本明細書の記載の進行に従って明らかとなるであろう。
【0042】
C.受容体スクリーニング
本明細書中で開示されるヒトGPCRの非内因性、構成的活性化型に対する候補化合物
のスクリーニングは、受容体の内因性リガンドの使用を必要としないで、この細胞表面受
容体に作用する候補化合物の直接同定を可能とする。慣用でしばしば商業的に利用できる
技術を用いて、本明細書中で開示されるヒトGPCRの内因性型が発現および/または過
剰発現される身体内の領域を決定できる。受容体の発現および/または過剰発現と関連す
る関連疾患/障害状態を決定するためにこれらの技術を用いることも可能であり、このよ
うな方法は、本特許明細書中に開示される。
【0043】
本明細書中で開示されるヒトGPCRの構成的活性化を明らかにするであろう変異の創
成に関しては、GPCRのTM6内に位置すると想定されるプロリン残基からの距離に基
づいている。このアルゴリズム技術は、2000年4月20日付けWO00/22129
として公開された同時係属および共通して譲渡された特許明細書PCT出願番号PCT/
US99/23938号中に開示されており、これは本明細書中に表示する他の特許文献
と一緒に引用することにより本明細書中に編入される。このアルゴリズム技術は、従来の
配列「整列(alignment) 」に基づいては断定されず、上記のTM6プロリン残基(または
、当然ではあるがこのようなプロリン残基の内因性構成的置換)からの指定の距離により
で決定される。この残基から16アミノ酸残基に位置するアミノ酸(推定すると受容体の
IC3領域内に位置する)を最も好ましいリシン残基に変異することにより、このような
活性化が得られるであろう。その他のアミノ酸残基は、この目的を達成するためにこの位
置における変異に有用であろう。
【0044】
D.疾患/障害同定および/または選択
以下に詳細に記載するように、最も好ましくは非内因性、構成的活性化GPCRに対す
る逆アゴニストおよびアゴニストは、本発明の方法論により同定できる。このような逆ア
ゴニストおよびアゴニストは、本受容体に関連する疾患を治療するための薬剤発見プログ
ラムにおけるリード化合物として理想の候補である。GPCRに対する逆アゴニストを直
接同定するための能力、これによる薬剤組成物の開発を可能とすることにより、GPCR
に関連する疾患および障害の探索が可能である。例えば、GPCRの存在に関する疾患お
よび正常組織試料の両方のスキャニングは、学問的な実験または特定のGPCRへの内因
性リガンドを同定するための経路に沿って追跡する程度を越えてきている。組織スキャン
は、健康および疾患組織の広い範囲にわたって行うことができる。このような組織スキャ
ンは、特定の受容体を疾患および/または障害と関連させる場合の好ましい第一段階を提
供する。
【0045】
好ましくは、ヒトGPCRのDNA配列は、(a)組織−mRNAに対するドット−ブ
ロット分析、および/または(b)組織試料中の受容体の発現のRT−PCT同定のため
のプローブを作製するために使用される。組織採取元内の受容体の存在、または疾患組織
、または正常組織と比較して疾患組織内の高い濃度にある受容体の存在は、限定はされな
いが治療方針とその疾患と関連する疾患との相関を同定するために好んで使用できる。受
容体は、この技術により器官の領域に同様に良く定位できる。受容体が局在する特定組織
の既知機能に基づいて、受容体の推定される機能的役割が推論できる。
【0046】
E.候補化合物のスクリーニング
1.包括的GPCRスクリーニングアッセイ技術
Gタンパク質受容体が構成的活性となった場合に、これはGタンパク質(例えばGq、
Gs、Gi、Gz、Go)に結合しそしてGタンパク質へのGTP結合を刺激する。従っ
てGタンパク質はGTPアーゼとして作用し、そしてゆっくりとGTPをGDPに加水分
解し、これにより正常の条件下で受容体は脱活性化する。しかし構成的活性化受容体は、
GDPをGTPに交換することを続ける。GTPの非加水分解性類似体である〔35S〕G
TPγSは、構成的活性化受容体を発現する膜への増強された結合を測定するために使用
できる。〔35S〕GTPγSが、リガンドが存在しないかまたは存在する場合に膜へのG
タンパク質共役を測定するために使用できることが報告されている。当該技術分野の熟練
者に周知で使用できるその他の例の中でも、この測定の例は、Traynor およびNahorskiに
より1995年に報告された。このアッセイ系の好ましい使用は、候補化合物の最初のス
クリーニングであり、それは、受容体の細胞内ドメインと相互作用する特定のGタンパク
質にかかわらずすべてのGタンパク質共役型受容体に系が包括的に適用できるからである
。
2.特異的GPCRスクリーニングアッセイ技術
候補化合物が「包括的」Gタンパク質共役型受容体アッセイ(すなわち、アゴニスト、
部分アゴニスト、または逆アゴニストである化合物を選択するためのアッセイ)を用いて
同定され、さらに化合物が受容体部位で相互作用したことを確認するためのスクリーニン
グをすることが好ましい。例えば、「包括的」アッセイで同定された化合物は、受容体に
結合しないであろうが、しかし細胞内ドメインからのGタンパク質を単に「脱共役」する
であろう。
【0047】
a.Gs、GzおよびGi
Gsは、酵素アデニル酸シクラーゼを刺激する。Gi(およびGzおよびGo)は、反
対にこの酵素を阻害する。アデニル酸シクラーゼは、ATPからcAMPへの変換に触媒
作用する。従って、Gsタンパク質に共役する構成的活性化GPCRは、cAMPの増加
した細胞レベルと関連する。反対に、Gi(またはGz、Go)タンパク質に共役する構
成的活性化GPCRは、cAMPの低下した細胞レベルと関連する。一般的には「シナプ
ス伝達の間接機序」第8章("Indirect Mechanisms of Synaptic Transmission," Chpt 8,
FromNeuron To Brain(3rd Ed.) Nichols, J.G. et al eds, Sinauer Associates, Inc.(1
992)) 参照。従って、cAMPを検出するアッセイは、候補化合物が例えば受容体に対し
て逆アゴニストであるかどうかを決定するために使用できる(すなわちこのような化合物
はcAMPのレベルを低下するであろう)。cAMPを測定するために当該技術分野で既
知の種々の方法が使用できる。最も好ましい方法は、ELISAに基づくフォーマットで
の抗−cAMP抗体の使用による。使用できる別のアッセイの種類は、全細胞第二メッセ
ンジャーレポーター系アッセイである。遺伝子上のプロモーターは、特定の遺伝子をコー
ドするタンパク質の発現を駆動する。サイクリックAMPは、cAMP反応性DNA結合
タンパク質または転写因子(CREB)の結合を促進して遺伝子発現を駆動し、転写因子
は次いでcAMP反応因子と呼ばれる特定部位でプロモーターに結合しそして遺伝子の発
現を駆動する。レポーター遺伝子、例えばβ−ガラクトシダーゼまたはルシフェラーゼの
前の多重cAMP反応因子を含むプロモーターを有するレポーター系が構築できる。従っ
て、構成体活性化されたGs−連結受容体は、次いで遺伝子を活性化するcAMPの蓄積
およびレポータータンパク質の発現を起こす。レポータータンパク質、例えばβ−ガラク
トシダーゼまたはルシフェラーゼは、次いで標準生物化学アッセイを用いて検出できる(C
henet al. 1995)。
【0048】
b.GoおよびGq
GoおよびGqは、酵素ホスホリパーゼCの活性化と関連し、これは次いでリン脂質P
IP2 を加水分解し、2個の細胞内メッセンジャー、ジアシクログリセロール(diacyclog
lycerol)(DAG)およびイノシトール 1,4,5−トリリン酸(IP3)を遊離する
。IP3 の増加した蓄積は、GqおよびGo関連受容体の活性化と関連する。一般的には
「シナプス伝達の間接機序」第8章("IndirectMechanisms of Synaptic Transmission,"
Chpt 8,From Neuron To Brain(3rdEd.) Nichols, J.G. et al eds, Sinauer Associates,
Inc. (1992)) 参照。IP3蓄積を検出するアッセイは、候補化合物が、例えばGqまた
はGo関連受容体に対する逆アゴニストであるかどうかを決定するために使用できる(す
なわち、このような化合物がIP3のレベルを低下する)。Gq関連受容体は、Gq依存
性ホスホリパーゼCがAP1因子を含む遺伝子の活性化を起こすAP1レポーターアッセ
イを用いてでも試験できる。従って、活性化Gq関連受容体は、この遺伝子の発現の増加
を明らかにし、これによりその逆アゴニストはこの発現の低下を明らかにし、そしてアゴ
ニストはこの発現の増加を明らかにする。このアッセイのために商業的に利用できるアッ
セイが利用できる。
【0049】
3.GPCR融合タンパク質
逆アゴニスト、アゴニストおよび部分アゴニストの直接同定のための候補化合物のスク
リーニングに使用するための内因性、構成的活性オーファンGPCRまたは非内因性、構
成的活性化オーファンGPCRの使用は、その定義により、結合する内因性リガンドが存
在しない場合でも受容体が活性であるという興味あるスクリーニング問題を提供する。従
って、例えば候補化合物の存在における非内因性受容体と、その化合物が存在しない場合
の非内因性受容体との間を区別するために、このような化合物が逆アゴニスト、アゴニス
ト、部分アゴニストであるかどうかまたはこのような受容体に影響しないかどうかに関す
る理解を可能とさせるこのような区別の目的で、このような区別を増強できる方法を使用
することが好ましい。好ましい方法は、「GPCR融合タンパク質」の使用である。
【0050】
一般に、非内因性オーファンGPCRが上記のアッセイ技術(ならびのその他のもの)
を用いて構成的に活性化されたことが一旦決定されると、内因性GPCRと共役する優勢
なGタンパク質を決定できる。GPCRへのGタンパク質の共役は、評価できるシグナル
伝達経路を提供する。スクリーニングが哺乳動物発現系を用いて行われることが最も好ま
しいので、このような系は、その中に内因性Gタンパク質を有すると期待される。従って
、定義により、このような系内で、非内因性、構成的活性化オーファンGPCRは、連続
的にシグナル発生する。この点に関して、例えば受容体への逆アゴニストの存在下でこの
シグナルが増強されることが好ましく、特にはスクリーニングの範囲内で、逆アゴニスト
と接触した受容体の間でさらに容易に区別できる可能性があるようにシグナルが増強され
ることがさらに好ましい。
【0051】
「GPCR融合タンパク質」は、非内因性GPCRと共役したGタンパク質の効力を増
強することを意図する。「GPCR融合タンパク質」は、非内因性、構成的活性化GPC
Rを用いるスクリーニングに好ましく、それは、このような方法がこのスクリーニング技
術で最も好ましく使用される信号を増加するからである。これは、有意の「信号対雑音比
」を容易に得るために重要である。このような有意の比は、本明細書中に開示する候補化
合物のスクリーニングに有利に導入される。
【0052】
「GPCR融合タンパク質」の発現のために有用な構築物の構築は、当該技術分野の通
常の熟練を有する者の認識範囲内である。商業的に利用できる発現ベクターおよび系は、
研究者の特定の要求を満足できる各種の方法を提供する。このような「GPCR融合タン
パク質」構築物のために重要な基準は、内因性GPCR配列およびGタンパク質配列の両
方がインフレームであり(好ましくは、内因性GPCRの配列がGタンパク質配列の上流
にある)そしてGPCRの「終止」コドンが、GPCRの発現の際にGタンパク質も発現
できるように欠失または置換されていなければならないことである。GPCRは、Gタン
パク質に直接連結してもよく、または2個の間のスペーサー残基があることもできる(好
ましくは、約12個を越えず、この数は当該技術分野の通常の熟練者には容易に確認でき
るものである)。我々は、使用されない幾つかの制限部位が、効果的に発現の際にスペー
サーとなるスペーサーの使用を好む(便利さのために)。最も好ましくは、非内因性GP
CRに共役するGタンパク質は、「GPCR融合タンパク質」構築物の創成の前に同定さ
れる。同定されたGタンパク質は僅かしかないので、Gタンパク質の配列を含んでなる構
築物(すなわち汎用Gタンパク質構築物)は、本明細書内の内因性GPCR配列の挿入の
ために利用できる。これは、異なる配列を有する種々の異なる内因性GPCRの大規模ス
クリーニングの範囲内で効率を与える。
【0053】
上記のように、Gi、GzおよびGoに共役する構成的活性化GPCRは、cAMPの
形成を阻害すると期待され、問題のGPCRのこれらの種類に基づくアッセイを興味ある
ものとする(すなわちcAMPシグナルは活性化の際に低下し、従って例えば逆アゴニス
トの直接同定を行う(これはこのシグナルをさらに低下させるであろう))。本明細書中
で開示するように、受容体のこれらの種類に対して、使用可能なシクラーゼに基づくアッ
セイを確立する研究において、内因性GPCRの内因性Gタンパク質には基づかない「G
PCR融合タンパク質」を創成することが可能であることを我々は確認した。従って、例
えば、内因性Gi共役型受容体はGsタンパク質に融合でき、我々はこのような融合構築
物が発現されると、内因性GPCRを、「本来的な」Giタンパク質ではなくて、例えば
Gsと共役するように「駆動」または「強制」し、シクラーゼに基づくアッセイを確立で
きると信じる。このように、Gi、GzおよびGo共役型受容体に対して、我々は、「G
PCR融合タンパク質」が使用されそしてアッセイがアデニル酸シクラーゼ活性の検出に
基づく場合に、融合構築物がGs(または酵素アデニル酸シクラーゼの形成を刺激する等
価Gタンパク質)を用いて確立されることを好む。
【0054】
「Gs、Gi、GzまたはGoタンパク質」と融合した「Gqタンパク質」を用いる「
Gタンパク質融合」構築物も同様に有効である。最も好ましい融合構築物は、「Gqタン
パク質」を用いて達成でき、ここで、Gタンパク質α−サブユニット(「Gαq」)の最
初の6個のアミノ酸が欠失しそしてGαqのC−末端の最後の5個のアミノ酸が、関係す
るGタンパク質のGαの相当するアミノ酸を用いて置換される。例えば、融合構築物は、
「Giタンパク質」と融合したGq(アミノ酸6個欠失)を有するとができ、「Gq/G
i融合構築物」をもたらす。我々は、この融合構築物が内因性Gi共役型受容体を非内因
性Gタンパク質、Gq、に共役させて、第二メッセンジャー、例えばイノシトール三リン
酸またはジアシルグリセロールがcAMP産生の代わりに測定できるように強制すると信
じる。
【0055】
4.シグナルエンハンサーGs共役型GPCRを用いる標的Gi共役型GPCR
のコトランスフェクション(cAMPに基づくアッセイ)
Gi共役型受容体はアデニル酸シクラーゼを阻害するとして知られており、従ってcA
MP産生のレベルを低下し、これはcAMPレベルの評価を興味深くさせることができる
。活性化の際に主としてGiに共役する受容体の構成的活性化の指標としてcAMPの産
生の低下を測定する有効な手段は、シグナルエンハンサー、例えば、Gi連結GPCRを
用いる活性化の際にGsと主として共役する非内因性、構成的活性化受容体(例えば以下
の開示するTSHR−A623I)をコトランスフェクションして達成できる。明らかな
ように、Gs共役型受容体の構成的活性化は、cAMPの産生の増加に基づいて決定でき
る。Gi共役型受容体の構成的活性化は、cAMP産生の低下に導く。従って、コトラン
スフェクション法は、これらの「反対」効果を有利に活用することを意図する。例えば、
非内因性、構成的活性化Gs共役型受容体(「シグナルエンハンサー」)と内因性Gi共
役型受容体(「標的受容体」)とのコトランスフェクションは、基準cAMPシグナルを
与える(すなわち、Gi共役型受容体はcAMPレベルを低下するけれども、この「低下
」は、構成的活性化Gs共役型受容体シグナルエンハンサーにより確立されるcAMPレ
ベルの本質的な増加と関連する)。その際、シグナルエンハンサーと標的受容体の構成的
活性化型とのコトランスフェクションにより、cAMPは、Gi標的の増加した機能的活
性(すなわち、これはcAMPを低下する)によりそれ以上に低下(基準に対する)する
と予想される。
【0056】
cAMPに基づくアッセイを用いる候補化合物のスクリーニングは、次いで2条件下で
達成できる。第一条件は、Gi共役標的受容体に関して、「反対」効果をもたらし、すな
わちGi共役標的受容体の逆アゴニストは測定されるcAMPシグナルを増加し、一方G
i共役標的受容体のアゴニストはこのシグナルを低下する。第二条件は、明らかなように
、この方法を用いて直接同定される候補化合物は、これらがシグナルエンハンシング受容
体を標的としないことを確認するために独立して評価されるべきであることである(これ
はコトランスフェクションした受容体に対するスクリーニングの前または後に行うことが
できる)。
【0057】
F.薬剤化学
必ずではないが一般的には、候補化合物の直接同定は、好ましくは、組合せ化学技術を
介して生成された化合物と関連して行われ、これにより数千の化合物がこの分析のために
ランダムに調製される。一般に、このようなスクリーニングの結果は、独自のコア構造を
有する化合物である。その後、これらの化合物は、好ましくは、これらの薬剤的性質をさ
らに増強するために好ましいコア構造の周囲に追加の化学修飾が行われる。このような技
術は、当該技術分野の熟練者には公知でありそして本明細書中では詳細には取り扱わない
。
【0058】
G.薬剤組成物
さらに開発するために選択された候補化合物は、当該技術分野の熟練者には周知の技術
を用いて薬剤組成物に調剤できる。適当な薬剤的に許容できるキャリヤーは、当該技術分
野の熟練者には利用できる。例えば、「レミントンの薬剤科学」(Remington's Pharmaceu
tical Sciences, 16th. Edition, 1980, MackPublishing Co., (Oslo et al., eds.))参
照。
【0059】
H.その他の効用
本明細書に開示したヒトGPCRの非内因型の好ましい使用は、逆アゴニスト、アゴニ
ストまたは部分アゴニストとしての候補化合物の直接同定のためであるが(好ましくは薬
剤としての使用のため)、ヒトGPCRのこれらの型は、研究設定にも使用できる。例え
ば、GPCRを組み込んだ生体内および生体外系は、正常および疾患の両方のヒトの状態
においてこれら受容体が演じる役割をさらに解明および理解するため、ならびにシグナル
伝達カスケードを理解するめに適用された構成的活性化の役割を理解するために使用でき
る。非内因性ヒトGPCRの価値は、これらの独自の特徴により、非内因性ヒトGPCR
が、内因性リガンドが同定される前にヒト身体内のこれら受容体の役割を理解するために
使用できることにより、研究手段としてのこれらの有用性が高くなることである。開示さ
れた受容体のその他の利用は、なかでも本明細書を検討すると当該技術分野の熟練者には
明らかとなるであろう。
【実施例】
【0060】
以下の実施例は、本発明の説明の目的で提示するものであり、これを制限するものでは
ない。特定の核酸およびアミノ酸配列が本明細書中で開示されるが、当該技術分野の通常
の熟練者は、これらの配列に小さい変更を加えて以下に報告すると同じかまたは実質的に
同様の結果を得る能力を有する。一つの配列から他のものへの配列カセットの適用または
理解の従来の方法(例えば、ラット受容体からヒト受容体へ、またはヒト受容体Aからヒ
ト受容体Bへ)は、一般に配列整列技術に基づいて断定され、その際、配列は、共通の領
域を決定するため研究において整列される。本明細書中に開示する変異法はこの方法には
よらないが、しかしその代わりにアルゴリズム法およびヒトGPCRのTM6領域内に位
置する保存されたプロリン残基からの位置的距離に基づく。この方法が確立されると、当
該技術分野の熟練者は、本明細書中に開示されると本質的に同じ結果(すなわち構成的活
性化)を得るためにこれに小さい変更を加える能力を有すると信じられる。このような変
更方法は、本開示の範囲内にあると考える。
【0061】
実施例1
内因性ヒトGPCR
1.ヒトGPCRの同定
開示される内因性ヒトGPCRは、ジーンバンク(GeneBank)TMデータベース情報の調査
に基づいて同定された。データベースを調査する間に、下記のcDNAクローンが下記に
明らかなように同定された(表C)
【0062】
【表2】
【0063】
2.全長クローニング
a.hRUP8(配列番号1および2)
開示されるヒトRUP8は、ESTデータベース(dbEST)情報の使用に基づいて
同定された。dbESTを調査する間に、アクセッション番号AL121755を有する
cDNAクローンが、新規のGPCRをコードすると同定された。下記のPCRプライマ
ーがヒト精巣マラソン−レディー(Marathon-Ready)cDNA(Clontech)を用いるRT−P
CRのために鋳型として用いられた。
5’−CTTGCAGACATCACCATGGCAGCC−3’(配列番号41、セン
ス)および
5’−GTGATGCTCTGAGTACTGGACTGG−3’(配列番号42、アン
チセンス)
PCRはアドヴァンテージ(Advantage) cDNAポリメラーゼ(Clontech、製造指針に従
う)を用い、50ul反応物中で下記のサイクルにより行った:30秒間94℃、10秒
間94℃、20秒間65℃、1.5分間72℃、および7分間72℃。サイクル2〜4を
35回反復した。
【0064】
1.2kbPCR断片を単離し、そしてpCRII−TOPOベクター(Invitrogen)中
にクローニングし、そしてABIビッグダイターミネーターキット(Big Dye Terminatork
it)(P.E. Biosystem)を用いて配列決定した。配列番号1参照、RUP8の推定アミノ酸
配列を配列番号2に記載する。
【0065】
b.hRUP9(配列番号3および4)
開示されるヒトRUP9は、ジーンバンクデータベース情報の使用に基づいて同定され
た。調査する間に、アクセッション番号AC011375を有するcDNAクローンが、
染色体5からのヒトゲノム配列として同定された。全長RUP9をプライマー
5’−GAAGCTGTGAAGAGTGATGC−3’(配列番号43、センス)、
5’−GTCAGCAATATTGATAAGCAGCAG−3’(配列番号44、アン
チセンス)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョン(Taq Plus Precision)ポリメラーゼ(Stratagene)を5%DMS
Oを用いる100μl反応物中で、下記のサイクルでステップ2〜ステップ4の35回反
復による増幅のために使用した:1分間94℃、30秒間94℃、30秒間56℃、2分
間72℃、および5分間72℃。
【0066】
1.3KbPCR断片を単離し、そしてpCRII−TOPOベクター(Invitrogen)中
に1%アガロースゲルからクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E.Biosystem)を用いて完全に配列決定した。配列番号3参照、RUP8の推定アミ
ノ酸配列を配列番号4に記載する。ヒトゲノムDNAから単離したRUP9クローンの配
列は、データベースから得た配列と一致した。
【0067】
c.hRUP10(配列番号5および6)
開示されるヒトRUP10は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC008754を有するcDNAクローンが
、染色体19からのヒトゲノム配列として同定された。全長RUP10をプライマー
5’−CCATGGGGAACGATTCTGTCAGCTACG−3’(配列番号45
、センス)および
5’−GCTATGCCTGAAGCCAGTCTTGTG−3’(配列番号46、アン
チセンス)
およびヒト白血球マラソンレディーcDNA(Clontech)を鋳型として用いるRT−PCR
によりクローニングした。アドヴァンテージcDNAポリメラーゼ(Clontech)を50μl
反応物中で下記のサイクルでステップ2〜ステップ4の35回反復による増幅のために使
用した:30秒間94℃、10秒間94℃、20秒間62℃、1.5分間72℃、および
7分間72℃。1.0KbPCR断片を単離し、そしてpCRII−TOPOベクター(I
nvitrogen)中にクローニングし、そしてABIビッグダイターミネーターキット(P.E.Bio
system)を用いて完全に配列決定した。新規のヒト受容体RUP10の核酸配列を配列番
号5に記載し、そしてその推定アミノ酸配列を配列番号6に記載する。
【0068】
d.hRUP11(配列番号7および8)
開示されるヒトRUP11は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC013396を有するcDNAクローンが
、染色体2からのヒトゲノム配列として同定された。全長RUP11をプライマー
5’−CCAGGATGTTGTGTCACCGTGGTGGC−3’(配列番号47、
センス)、
5’−CACAGCGCTGCAGCCCTGCAGCTGGC−3’(配列番号48、
アンチセンス)
およびヒトゲノムDNA(Clontech)を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンDNAポリメラーゼ(Stratagene)を50μl反応物中で下記の
サイクルでステップ2〜ステップ4の35回反復による増幅のために使用した:3分間9
4℃、20秒間94℃、20秒間67℃、1.5分間72℃、および7分間72℃。1.
3KbPCR断片を単離し、そしてpCRII−TOPOベクター(Invitrogen)中にクロ
ーニングし、そしてABIビッグダイターミネーターキット(P.E.Biosystem)を用いて完
全に配列決定した。新規のヒト受容体RUP11の核酸配列を配列番号7に記載し、そし
てその推定アミノ酸配列を配列番号8に記載する。
【0069】
e.hRUP12(配列番号9および10)
開示されるヒトRUP12は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AP000808を有するcDNAクローンが
、ラットRTAおよびヒトmas1腫瘍遺伝子GPCRと著しい相同性を有する新規のG
PCRをコードすると同定された。全長RUP12をプライマー
5’−CTTCCTCTCGTAGGGATGAACCAGAC−3’(配列番号49、
センス)
5’−CTCGCACAGGTGGGAAGCACCTGTGG−3’(配列番号50、
アンチセンス)
およびヒトゲノムDNA(Clontech)を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンDNAポリメラーゼ(Stratagene)を下記のサイクルでステップ
2〜ステップ4の35回反復による増幅のために使用した:3分間94℃、20秒間94
℃、20秒間65℃、2分間72℃、および7分間72℃。1.0kbPCR断片を単離
し、そしてpCRII−TOPOベクター(Invitrogen)中にクローニングし、そしてAB
Iビッグダイターミネーターキット(P.E.Biosystem)を用いて完全に配列決定した(核酸
配列に関しては配列番号9そして推論したアミノ酸配列に関しては配列番号10参照)。
【0070】
f.hRUP13(配列番号11および12)
開示されるヒトRUP13は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC011780を有するcDNAクローンが
、GPCRサカナGRPX−ORYLAと著しい相同性を有する新規のGPCRをコード
すると同定された。全長RUP13をプライマー
5’−GCCTGTGACAGGAGGTACCCTGG−3’(配列番号51、センス
)
5’−CATATCCCTCCGAGTGTCCAGCGGC−3’(配列番号52、ア
ンチセンス)
およびヒトゲノムDNA(Clontech)を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンDNAポリメラーゼ(Stratagene)を下記のサイクルでステップ
2〜ステップ4の35回反復による増幅のために使用した:3分間94℃、20秒間94
℃、20秒間65℃、2分間72℃、および7分間72℃。1.35kbPCR断片を単
離し、そしてpCRII−TOPOベクター(Invitrogen)中にクローニングし、そしてA
BIビッグダイターミネーターキット(P.E.Biosystem)を用いて完全に配列決定した(核
酸配列に関しては配列番号11そして推論したアミノ酸配列に関しては配列番号12参照
)。
g.hRUP14(配列番号13および14)
開示されるヒトRUP14は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AL137188を有するcDNAクローンが
、染色体13からのヒトゲノム配列として同定された。全長RUP14をプライマー
5’−GCATGGAGAGAAAATTTATGTCCTTGCAACC−3’(配列
番号53、センス)
5’−CAAGAACAGGTCTCATCTAAGAGCTCC−3’(配列番号54
、アンチセンス)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンポリメラーゼ(Stratagene)および5%DMSOを下記のサイク
ルでステップ2およびステップ3の35回反復による増幅のために使用した:3分間94
℃、20秒間94℃、2分間58℃、10分間72℃。
【0071】
1.1KbPCR断片を単離し、そしてpCRII−TOPOベクター(Invitrogen)中
にクローニングし、そしてABIビッグダイターミネーターキット(P.E. Biosystem)を用
いて完全に配列決定した(核酸配列に関しては配列番号13そして推論したアミノ酸配列
に関しては配列番号14参照)。ヒトゲノムDNAから単離したRUP14クローンの配
列は、データベースから得た配列と一致した。
【0072】
h.hRUP15(配列番号15および16)
開示されるヒトRUP15は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC016468を有するcDNAクローンが
、ヒトゲノム配列として同定された。全長RUP15をプライマー
5’−GCTGTTGCCATGACGTCCACCTGCAC−3’(配列番号55、
センス)
5’−GGACAGTTCAAGGTTTGCCTTAGAAC−3’(配列番号56、
アンチセンス)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンポリメラーゼ(Stratagene)を下記のサイクルでステップ2〜4
の35回反復による増幅のために使用した:3分間94℃、20秒間94℃、20秒間6
5℃、2分間72℃および7分間72℃。
【0073】
1.5KbPCR断片を単離し、そしてpCRII−TOPOベクター(Invitrogen)中
にクローニングし、そしてABIビッグダイターミネーターキット(P.E. Biosystem)を用
いて完全に配列決定した。核酸配列に関しては配列番号15そして推論したアミノ酸配列
に関しては配列番号16参照。ヒトゲノムDNAから単離したRUP15クローンの配列
は、データベースから得た配列と一致した。
【0074】
i.hRUP16(配列番号17および18)
開示されるヒトRUP16は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AL136106を有するcDNAクローンが
、染色体13からのヒトゲノム配列として同定された。全長RUP16をプライマー
5’−CTTTCGATACTGCTCCTATGCTC−3’(配列番号57、センス
、開始コドンの5’)
5’−GTAGTCCACTGAAAGTCCAGTGATCC−3’(配列番号58、
アンチセンス、終止コドンの3’)
およびヒト骨格筋マラソンレディーcDNA(Clontech)を鋳型として用いるPCRにより
クローニングした。アドヴァンテージcDNAポリメラーゼ(Clontech)を50ul反応物
中で下記のサイクルでステップ2〜4の35回反復による増幅のために使用した:30秒
間94℃、5秒間94℃、15秒間69℃、1分間72℃および5分間72℃。
【0075】
1.1KbPCR断片を単離し、そしてpCRII−TOPOベクター(Invitrogen)中
にクローニングし、そしてT7シーケナーゼキット(sequenase kit)(Amersham) を用いて
完全に配列決定した。核酸配列に関しては配列番号17そして推論したアミノ酸配列に関
しては配列番号18参照。RUP16クローンの配列は、AL136106の4個の不規
則断片と一致し、これはRUP16cDNAが4個のエキソンからなることを示す。
【0076】
j.hRUP17(配列番号19および20)
開示されるヒトRUP17は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC023078を有するcDNAクローンが
、染色体11からのヒトゲノム配列として同定された。全長RUP17をプライマー
5’−TTTCTGAGCATGGATCCAACCATCTC−3’(配列番号59、
センス、開始コドンを含む)
5’−CTGTCTGACAGGGCAGAGGCTCTTC−3’(配列番号60、ア
ンチセンス、終止コドンの3’)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
アドヴァンテージcDNAポリメラーゼミックス(Clontech)を5%DMSOを含む100
ul反応物中で下記のサイクルでステップ2〜4の30回反復による増幅のために使用し
た:1分間94℃、15秒間94℃、20秒間67℃、1分30秒間72℃および5分間
72℃。
【0077】
970bpPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号19そし
て推論したアミノ酸配列に関しては配列番号20参照。
【0078】
k.hRUP18(配列番号21および22)
開示されるヒトRUP18は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC008547を有するcDNAクローンが
、染色体5からのヒトゲノム配列として同定された。全長RUP18をプライマー
5’−GGAACTCGTATAGACCCAGCGTCGCTCC−3’(配列番号6
1、センス、開始コドンの5’)、
5’−GGAGGTTGCGCCTTAGCGACAGATGACC−3’(配列番号6
2、アンチセンス、終止コドンの3’)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンDNAポリメラーゼ(Stratagene)を5%DMSOを含む100
ul反応物中で下記のサイクルでステップ2〜4の35回反復による増幅のために使用し
た:5分間95℃、30秒間95℃、30秒間65℃、2分間72℃および5分間72℃
。
【0079】
1.3kbPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号21そし
て推論したアミノ酸配列に関しては配列番号22参照。
【0080】
l.hRUP19(配列番号23および24)
開示されるヒトRUP19は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC026331を有するcDNAクローンが
、染色体12からのヒトゲノム配列として同定された。全長RUP19をプライマー
5’−CTGCACCCGGACACTTGCTCTG−3’(配列番号63、センス、
開始コドンの5’)
5’−GTCTGCTTGTTCAGTGCCACTCAAC−3’(配列番号64、ア
ンチセンス、終止コドンを含む)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンDNAポリメラーゼ(Stratagene)を5%DMSOを用い下記の
サイクルでステップ2〜4の35回反復による増幅のために使用した:1分間94℃、1
5秒間94℃、20秒間70℃、1分30秒間72℃および5分間72℃。
【0081】
1.1kbPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号23そし
て推論したアミノ酸配列に関しては配列番号24参照。
【0082】
m.hRUP20(配列番号25および26)
開示されるヒトRUP20は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AL161458を有するcDNAクローンが
、染色体1からのヒトゲノム配列として同定された。全長RUP20をプライマー
5’−TATCTGCAATTCTATTCTAGCTCCTG−3’(配列番号65、
センス、開始コドンの5’)、
5’−TGTCCCTAATAAAGTCACATGAATGC−3’(配列番号66、
アンチセンス、終止コドンの3’)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
アドヴァンテージcDNAポリメラーゼミックス(Clontech)を5%DMSOを用い下記の
サイクルでステップ2〜4の35回反復による増幅のために使用した:1分間95℃、1
5秒間95℃、20秒間60℃、1分30秒間72℃および5分間72℃。
【0083】
1.0kbPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号25そし
て推論したアミノ酸配列に関しては配列番号26参照。
【0084】
n.hRUP21(配列番号27および28)
開示されるヒトRUP21は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC026756を有するcDNAクローンが
、染色体13からのヒトゲノム配列として同定された。全長RUP21をプライマー
5’−GGAGACAACCATGAATGAGCCAC−3’(配列番号67、センス
)
5’−TATTTCAAGGGTTGTTTGAGTAAC−3’(配列番号68、アン
チセンス)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンポリメラーゼ(Stratagene)を5%DMSOを含む100ul反
応物中で下記のサイクルでステップ2〜4の30回反復による増幅のために使用した:1
分間94℃、15秒間94℃、20秒間55℃、1分30秒間72℃および5分間72℃
。
【0085】
1,014bpPCR断片を1%アガロースゲルから単離し、そしてpCRII−TO
POベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーター
キット(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号27
そして推論したアミノ酸配列に関しては配列番号28参照。
【0086】
o.hRUP22(配列番号29および30)
開示されるヒトRUP22は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC027026を有するcDNAクローンが
、染色体11からのヒトゲノム配列として同定された。全長RUP22をプライマー
5’−GGCACCAGTGGAGGTTTTCTGAGCATG−3’(配列番号69
、センス、開始コドンを含む)
5’−CTGATGGAAGTAGAGGCTGTCCATCTC−3’(配列番号70
、アンチセンス、終止コドンの3’)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
タックプラスプレシジョンDNAポリメラーゼ(Stratagene)を5%DMSOを含む100
ul反応物中で下記のサイクルでステップ2〜4の30回反復による増幅のために使用し
た:94℃、1分間94℃、15秒間55℃、20秒間72℃、1.5分間72℃、5分
間。
【0087】
970bpPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号29そし
て推論したアミノ酸配列に関しては配列番号30参照。
【0088】
p.hRUP23(配列番号31および32)
開示されるヒトRUP23は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC007104を有するcDNAクローンが
、染色体4からのヒトゲノム配列として同定された。全長RUP23をプライマー
5’−CCAGGCGAGCCGCTAGCGCCATG−3’(配列番号71、センス
、開始コドンとしてのATC)
5’−ATGAGCCCTGCCAGGCCCTCAGT−3’(配列番号72、アンチ
センス、終止コドンとしてのTCA)
およびヒト胎盤マラソンレディーDNA(Clontech)を鋳型として用いるPCRによりクロ
ーニングした。アドヴァンテージcDNAポリメラーゼ(Clontech)を50ul反応物中で
下記のサイクルでステップ2〜4の35回反復による増幅のために使用した:30秒間9
5℃、15秒間95℃、20秒間66℃、1分20秒間72℃および5分間72℃。
【0089】
1.0kbPCR断片を単離し、そしてpCRII−TOPOベクター(Invitrogen)中
にクローニングし、そしてABIビッグダイターミネーターキット(P.E. Biosystem)を用
いて完全に配列決定した。核酸配列に関しては配列番号31そして推論したアミノ酸配列
に関しては配列番号32参照。
【0090】
q.hRUP24(配列番号33および34)
開示されるヒトRUP25は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC026331を有するcDNAクローンが
、染色体12からのヒトゲノム配列として同定された。全長RUP25をプライマー
5’−GCTGGAGCATTCACTAGGCGAG−3’(配列番号73、センス、
開始コドンの5’)
5’−AGATCCTGGTTCTTGGTGACAATG−3’(配列番号74、アン
チセンス、終止コドンの3’)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
アドヴァンテージcDNAポリメラーゼミックス(Clontech)を5%DMSOを用い、下記
のサイクルでステップ2〜4の35回反復による増幅のために使用した:1分間94℃、
15秒間94℃、20秒間56℃、1分30秒間72℃および5分間72℃。
【0091】
1.2kbPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号33そし
て推論したアミノ酸配列に関しては配列番号34参照。
【0092】
r.hRUP25(配列番号35および36)
開示されるヒトRUP25は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC026331を有するcDNAクローンが
、染色体12からのヒトゲノム配列として同定された。全長RUP25をプライマー
5’−GCTGGAGCATTCACTAGGCGAG−3’(配列番号75、センス、
開始コドンの5’)
5’−AGATCCTGGTTCTTGGTGACAATG−3’(配列番号76、アン
チセンス、終止コドンの3’)
およびヒトゲノムDNA(Promega) を鋳型として用いるPCRによりクローニングした。
アドヴァンテージcDNAポリメラーゼミックス(Clontech)を5%DMSOを用い、下記
のサイクルでステップ2〜4の35回反復による増幅のために使用した:1分間94℃、
15秒間94℃、20秒間56℃、1分30秒間72℃および5分間72℃。
【0093】
1.2kbPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号35そし
て推論したアミノ酸配列に関しては配列番号36参照。
【0094】
s.hRUP26(配列番号37および38)
開示されるヒトRUP26は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC023040を有するcDNAクローンが
、染色体2からのヒトゲノム配列として同定された。全長RUP26をRUP26特異性
プライマー
5’−AGCCATCCCTGCCAGGAAGCATGG−3’(配列番号77、セン
ス、開始コドンを含む)
5’−CCAGACTGTGGACTCAAGAACTCTAGG−3’(配列番号78
、アンチセンス、終止コドンを含む)
およびヒト胎盤マラソンレディーcDNA(Clontech)を鋳型として用いるRT−PCRに
よりクローニングした。アドヴァンテージcDNAポリメラーゼミックス(Clontech)を5
%DMSOを含む100ul反応物中で下記のサイクルでステップ2〜4の35回反復に
よる増幅のために使用した:5分間94℃、30秒間95℃、30秒間65℃、2分間7
2℃および5分間72℃。
【0095】
1.1kbPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号37そし
て推論したアミノ酸配列に関しては配列番号38参照。
【0096】
t.hRUP27(配列番号39および40)
開示されるヒトRUP27は、ジーンバンクデータベース情報の使用に基づいて同定さ
れた。調査する間に、アクセッション番号AC027643を有するcDNAクローンが
、染色体12からのヒトゲノム配列として同定された。全長RUP27をRUP17特異
性プライマー
5’−AGTCCACGAACAATGAATCCATTTCATG−3’(配列番号7
9、センス、開始コドンを含む)
5’−ATCATGTCTAGACTCATGGTGATCC−3’(配列番号80、ア
ンチセンス、終止コドンの3’)
およびヒト成人脳マラソンレディーcDNA(Clontech)を鋳型として用いるPCRにより
クローニングした。アドヴァンテージcDNAポリメラーゼミックス(Clontech)を5%D
MSOを含む50ul反応物中で下記のサイクルでステップ2〜4の35回反復による増
幅のために使用した:1分間94℃、10秒間94℃、20秒間58℃、1分30秒間7
2℃および5分間72℃。
【0097】
1.1kbPCR断片を1%アガロースゲルから単離し、そしてpCRII−TOPO
ベクター(Invitrogen)中にクローニングし、そしてABIビッグダイターミネーターキッ
ト(P.E. Biosystem)を用いて完全に配列決定した。核酸配列に関しては配列番号35そし
て推論したアミノ酸配列に関しては配列番号36参照。ヒト脳から単離されたRUP27
cDNAクローンの配列は,AC027643の5個の不規則断片と一致することが決定
され、RUP27cDNAが5個のエキソンからなることを示す。
【0098】
実施例2
非内因性、構成的活性化GPCRの調製
当該技術分野の熟練者は、核酸配列の変異のための技術を選択する能力を有すると信じ
られる。下記は、以上に開示した数種のヒトGPCRの非内因性型を創成するために使用
される方法である。以下に開示される変異は、保存されるプロリン(またはこれの内因性
の保存性置換基)残基(TM6/IC3界面近くのGPCRのTM6領域内に位置する)
から16番目のアミノ酸(GPCRのIC3領域内に位置する)が、好ましくはアラニン
、ヒスチジン、アルギニンまたはリシンアミノ酸残基に、最も好ましくはリシンアミノ酸
残基に変異されるアルゴリスム法に基づく。
【0099】
1.トランスフォーマーサイトディレクテッドTM変異誘発
非内因性ヒトGPCRの調製は、製造者の指針に従って、トランスフォーマーサイトデ
ィレクテッド(Transformer Site-Directed) TM変異誘発キット(Clontech)を用いて、ヒト
GPCRについて行ってもよい。2個の変異誘発プライマーを使用し、最も好ましくはリ
シン変異を創成するリシン変異誘発オリゴヌクレオチド、および選択マーカーオリゴヌク
レオチドを用いる。便宜のために、ヒトGPCR内に組み込まれるべきコドン変異も標準
形で記録する(表D)。
【0100】
【表3】
【0101】
2.クイクチェンジTMサイトディレクテッドTM変異誘発
非内因性ヒトGPCRの調製は、クイクチェンジ(QuikChange)TMサイトディレクテッド
(Site-Directed) TM変異誘発キット(Stratagene、製造者の指針に従う)によっても行う
ことができる。内因性GPCRが好ましい鋳型として使用され、そして2個の変異誘発プ
ライマーを用い、ならびに最も好ましくはリシン変異誘発オリゴヌクレオチドおよび選択
マーカーオリゴヌクレオチド(キット内に含まれる)が使用される。便宜のために、新規
のヒトGPCR内に組み込まれたコドン変異およびそれぞれのオリゴヌクレオチドを標準
形で記録する(表E)
【0102】
【表4】
【0103】
非内因性ヒトGPCRを次いで配列決定しそして誘導および確認された核酸およびアミ
ノ酸配列を本特許明細書に添付した「配列表」に表示し、これを下記の表Fに要約した。
【0104】
【表5】
【0105】
実施例3
受容体発現
種々の細胞がタンパク質の発現のために当該技術分野で利用できるが、哺乳動物細胞の
使用が最も好ましい。この主要な理由は、実用性により定められ、すなわち例えばGPC
Rの発現のための酵母細胞の利用は、可能な限り、哺乳動物系のために発展された受容体
共役、遺伝機序および分泌経路を含まない(確かに酵母の場合には含まない)非哺乳動物
細胞をプロトコール中に導入し、これにより非哺乳動物細胞で得られた結果は、利用可能
な限り、哺乳動物細胞から得られたほどは好ましくない。使用される特定の哺乳動物細胞
は熟練者の特定の要求により決定できるけれども、哺乳動物細胞のなかでは、COS−7
、293および293T細胞が特に好ましい。
【0106】
a.一過性トランスフェクション
第一日目に、6x106 /(293細胞のウエル10cm皿)をプレートアウトした。
第二日目に、2本の反応管を調製した(それぞれの管についての下記の比率はプレートあ
たりである)。管Aは、4μgDNA(例えばpCMVベクター、受容体cDNAを含む
pCMVベクターなど)を0.5mlの血清を含まないDMEM(GibcoBRL) 中で混合し
て調製した。管Bは、24μlリポフェクタミン(Gibco BRL) を0.5mlの血清を含ま
ないDMEM中で混合して調製した。管AおよびBを倒置(数回)により混合し、次いで
室温で30〜45分間インキュベーションした。この混合物を「トランスフェクション混
合物」と呼ぶ。プレーティングした293細胞を1XPBSを用いて洗浄し、次いで0.
5mlの血清を含まないDMEMを加えた。トランスフェクション混合物1mlを細胞に
加え、次いで37℃/5%CO2で4時間インキュベーションした。トランスフェクショ
ン混合物を吸引して取り出し、次いでDMEM/10%ウシ胎児血清の10mlを加えた
。細胞を37℃/5%CO2でインキュベーションした。48時間インキュベーションし
た後、細胞を採取しそして分析に使用した。
【0107】
b.安定細胞株:Gs融合タンパク質
約12x106 293細胞を15cm組織培養プレート上にプレーティングした。10
%ウシ胎児血清および1%ピルビン酸ナトリウム、L−グルタミン、および抗生物質を含
むDMEハイ・グルコース媒体(HighGlucose Medium) 中で培養した。プレーティングし
た24時間後に293細胞は約80%集密度となり、細胞をDNA12μgを用いてトラ
ンスフェクションした。DNA12μgを、リポフェクタミン60ulおよび血清を含ま
ないDMEハイ・グルコース媒体2mLと混合した。媒体をプレートから吸引しそして細
胞を血清を含まない媒体を用いて一回洗浄した。DNA、リポフェクタミン、および媒体
混合物を血清を含まない媒体10mLと一緒にプレートに加えた。37℃で4〜5時間の
インキュベーションの後に、媒体を吸引し、そして血清を含む媒体25mlを加えた。ト
ランスフェクションの24時間後に媒体を再度吸引し、そして血清を含む新しい媒体を加
えた。トランスフェクションの48時間後に、媒体を吸引しそして血清を含みゲネチシン
(G418薬剤)を含む媒体を加え、最終濃度500μg/mLまで加えた。トランスフ
ェクションした細胞は、ここでG418耐性遺伝子を含む陽性にトランスフェクションさ
れた細胞に関して選択する。選択が起きる度に媒体を4〜5日毎に交換した。選択の間に
、細胞は安定なプールを創成するために増殖するか、または安定クローン選択のためにス
プリットする。
【0108】
実施例4 非内因性GPCRの構成的活性の決定のためのアッセイ
種々の方法が非内因性ヒトGPCRの構成的活性の評価のために利用される。以下は例
示である。当該技術分野の通常の熟練者は、熟練者の要求に優先的に役立つこのような技
術を決定する能力を有すると信じられる。
1.膜結合アッセイ:〔35S〕GTPγSアッセイ
Gタンパク質共役型受容体がその活性状態にある場合に、リガンド結合または構成的活
性化のいずれかの結果として、受容体はGタンパク質に結合しそしてGDPの遊離および
その後のGタンパク質へのGTPの結合を刺激する。Gタンパク質共役型受容体複合体の
αサブユニットは、GTPアーゼとして作用しそしてGTPをGDPにゆっくりと加水分
解し、その時点で受容体は通常は脱活される。構成的活性化受容体はGTPをGDPへ交
換することを続ける。非−加水分解性GTP類似体である〔35S〕GTPγSは、構成的
活性化受容体を発現する膜への〔35S〕GTPγSの増強された結合を証明するために使
用できる。構成的活性化を測定するために〔35S〕GTPγS結合を用いる利点は、(a
)これがすべてのGタンパク質共役型受容体に包括的に適用される、(b)これが、膜表
面の近くで細胞内カスケードに影響する分子を取り上げることを困難とすることである。
【0109】
このアッセイは、関係する受容体を発現する膜に結合する〔35S〕GTPγSを刺激す
るGタンパク質共役型受容体の能力を利用する。従って、このアッセイは、既知のオーフ
ァンおよび構成的活性化Gタンパク質共役型受容体への候補化合物をスクリーニングする
ための直接同定法に使用できる。アッセイは包括的であり、そしてすべてのGタンパク質
共役型受容体における薬剤発見に適用される。
【0110】
〔35S〕GTPγSアッセイは、20mM HEPESおよび1〜約20mM MgC
l2 (この量は、20mMが好ましいけれども、結果の最適化のために調節できる)pH
7.4、約0.3〜約1.2nM 〔35S〕GTPγS(この量は、1.2が好ましいけ
れども、結果の最適化のために調節できる)を含む結合緩衝液および12.5〜75μg
膜タンパク質(例えばGs融合タンパク質を発現する293細胞、この量は最適化のため
に調節できる)および10μM GDP(この量は最適化のために調節できる)中で1時
間インキュベーションした。次いでコムギ胚芽凝集素ビーズ(25μl、Amersham)を加
えそして混合物をさらに30分間、室温でインキュベーションした。次いで管を1500
x gで5分間、室温で遠心分離し次いでシンチレーションカウンターでカウントした
。
【0111】
2.アデニル酸シクラーゼ
細胞に基づくアッセイのために設計されたフラッシュ・プレート(Flash Plate) TMアデ
ニル酸シクラーゼキット(New England Nuclear;カタログ番号SMP004A)は、粗血漿膜と
共に用いるために変更できる。フラッシュ・プレートウエルは、cAMPを認識する特定
の抗体も含むシンチラント(scintillant)コーティングを含むことができる。ウエル内で
産生されたcAMPはcAMP抗体に対する放射性cAMPトレーサーの結合に関する直
接競合により定量できる。以下の記載は、受容体を発現する全細胞内のcAMPレベル内
の変化の測定のための簡単なプロトコールとして役立つ。
【0112】
トランスフェクションされた細胞を一過性トランスフェクションの約24時間後に採取
した。媒体を注意して吸引して取り出し廃棄した。PBS10mlを細胞のそれぞれの皿
におだやかに加え次いで注意して吸引した。Sigma 細胞分離緩衝液1mlおよびPBS3
mlをそれぞれのプレートに加えた。細胞を皿からピペットで取り出しそして細胞懸濁液
を50ml円錐型遠心分離管内に集めた。次いで細胞を室温、1,100rpmで5分間
遠心分離した。細胞ペレットを注意して適当な体積のPBS(約3ml/プレート)内に
再懸濁した。次いで細胞を血球計数器を用いて計数しそして追加のPBSを加えて細胞の
概略数を与えた(最終体積約50μl/ウエル)。
【0113】
cAMP標準および「検出緩衝液」(「検出緩衝液」11mlにトレーサー〔125 Ic
AMP(50μl〕の1μCiを含んでなる)を調製し、そして製造者の指示に従って維
持した。「アッセイ緩衝液」をスクリーニングのために新たに調製しそしてこれは「刺激
緩衝液」50μl、試験化合物3ul(12uMの最終アッセイ濃度)および細胞50μ
lを含み、「アッセイ緩衝液」は使用するまで氷上に保管した。アッセイは、適当なウエ
ルにcAMP標準50μlの添加から始まり、次いでウエルH−11およびH−12へP
BSA50ulを加えた。「刺激緩衝液」50μlをすべてのウエルに加えた。DMSO
(または選択した候補化合物)を化合物溶液3μlの供給ができるピン器具を用いて適当
なウエルに加え、12μM試験化合物の最終アッセイ濃度および全アッセイ体積100μ
lとした。次いで細胞をウエルに加えそして60分間、室温でインキュベーションした。
次いでトレーサーcAMPを含む「検出混合物」100μlをウエルに加えた。次いでプ
レートをさらに2時間インキュベーションし、次いでウオラック・マイクロベータ(Walla
cMicroBeta)シンチレーションカウンター内で計数した。次いでcAMP/ウエルの値を
それぞれのアッセイプレート内に含まれる標準cAMP曲線から外挿した。
【0114】
3.Gi共役型標的GPCRのための細胞に基づくcAMP
TSHRは、活性化の際にcAMPの蓄積を起こすGs共役型GPCRである。TSH
Rは、アミノ酸残基623を変異すること(すなわち、アラニン残基をイソロイシン残基
に変化する)により構成的に活性化される。Gi共役型受容体は、アデニル酸シクラーゼ
を阻害すると予想され、そして、そのために、cAMP産生のレベルを低下し、これはc
AMPレベルの問題の評価を可能とする。Gi共役型受容体の構成的活性化の指標として
のcAMPの産生の低下を測定するために有効な技術は、cAMPの基準レベルを確立す
るためのGi連結標的GPCRを用いる「シグナルエンハンサー」として、最も好ましく
は非内因性、構成的活性化TSHR(TSHR−A6231)(または内因性、構成的活
性Gs共役型受容体)を同時トランスフェクションすることにより達成できる。Gi共役
型受容体の非内因性型が創成されると、次いで標的GPCRのこの非内因性型は、シグナ
ルエンハンサーと一緒に同時トランスフェクションされ、そしてスクリーニングに使用で
きるのはこの物質である。我々は、cAMPアッセイを使用する場合にシグナルを効率的
に発生するためにこの方法を使用した。この方法は、好ましくはGi共役型受容体に対す
る候補化合物の直接同定に使用される。この方法を使用した場合に、Gi共役型受容体に
とって標的GPCRの逆アゴニストはcAMPシグナルを増加しそしてアゴニストはcA
MPシグナルを低下するであろう。
【0115】
第一日目に、2X104 293および293細胞/ウエルをプレートアウトする。第二
日目に、2個の反応管を調製する(それぞれの管についての下記の比率はプレートあたり
である)。管Aは、哺乳動物細胞内にトランスフェクションされたそれぞれの受容体の2
μgDNAを、1.21の血清を含まないDMEM(IrvineScientific, Irvine, CA) 中
で全体で4μgDNA(例えばpCMVベクター、変異THSR(THSR−A6231
I)を含むpCMVベクター、THSR−A6231IおよびGPCRなど)と混合して
調製する。管Bは、120μlリポフェクタミン(GibcoBRL) を1.2mlの血清を含ま
ないDMEM中で混合して調製する。次いで、管AおよびBを倒置(数回)してよく混合
し、次いで室温で30〜45分間インキュベーションした。混合物を「トランスフェクシ
ョン混合物」と呼ぶ。プレーティングした293細胞を1XPBSを用いて洗浄し、次い
で血清を含まないDMEM10mlを加える。次いでトランスフェクション混合物2.4
mlを細胞に加え、次いで4時間、37℃/5%CO2でインキュベーションする。次い
でトランスフェクション混合物を吸引して取り出し、次いでDMEM/10%ウシ胎児血
清25mlを加える。次いで細胞を37℃/5%CO2でインキュベーションする。24
時間インキュベーションの後、細胞を採取しそして分析に使用する。
【0116】
しかし、細胞に基づくアッセイのために設計されたフラッシュ・プレートTMアデニル酸
シクラーゼキット(New England Nuclear;カタログ番号SMP004A)は、熟練者の要求に従
って粗血漿膜と用いるために変更できる。フラッシュ・プレートウエルは、cAMPを認
識する特定の抗体も含むシンチラントコーティングを含むことができる。ウエル内で産生
されたcAMPはcAMP抗体に対する放射性cAMPトレーサーの結合に関する直接競
合により定量できる。以下の記載は、受容体を発現する全細胞内のcAMPレベル内の変
化の測定のための簡単なプロトコールとして役立つ。
【0117】
トランスフェクションされた細胞を一過性トランスフェクションの約24時間後に採取
する。媒体を注意して吸引して取り出して廃棄する。PBS10mlを細胞のそれぞれの
皿におだやかに加え次いで注意して吸引する。Sigma細胞分離緩衝液1mlおよびP
BS3mlをそれぞれのプレートに加える。細胞を皿からピペットで取り出しそして細胞
懸濁液を50ml円錐型遠心分離管内に集める。次いで細胞を室温、1,100rpmで
5分間遠心分離する。細胞ペレットを注意して適当な体積のPBS(約3ml/プレート
)内に再懸濁する。次いで細胞を血球計数器を用いて計数しそして追加のPBSを加えて
細胞の概略数を与える(最終体積約50μl/ウエル)。
【0118】
cAMP標準および「検出緩衝液」(「検出緩衝液」11mlにトレーサー〔125 Ic
AMP(50μl〕の1μCiを含んでなる)を調製し、そして製造者の指示に従って維
持する。「アッセイ緩衝液」をスクリーニングのために新たに調製しなればならずそして
これは「刺激緩衝液」50μl、試験化合物3ul(12uM最終アッセイ濃度)および
細胞50μlを含み、「アッセイ緩衝液」は使用するまで氷上で保管できる。アッセイは
、適当なウエルにcAMP標準50μlの添加から始めることができ、次いでウエルH−
11およびH−12へPBSA50ulを加える。刺激緩衝液50μlをウエルに加える
。選択した化合物(例えばTSH)を化合物溶液3μlの供給ができるピン器具を用いて
適当なウエルに加え、12μM試験化合物の最終アッセイ濃度および全アッセイ体積10
0μlとする。次いで細胞をウエルに加えそして60分間、室温でインキュベーションす
る。次いでトレーサーcAMPを含む「検出混合物」100μlをウエルに加える。次い
でプレートをさらに2時間インキュベーションし、次いでウオラック・マイクロベータシ
ンチレーションカウンター内で計数する。次いでcAMP/ウエルの値をそれぞれのアッ
セイプレート内に含まれる標準cAMP曲線から外挿する。
【0119】
4.レポーターに基づくアッセイ
a.Gre−Lucレポーターアッセイ(Gs−関連受容体)
293および293t細胞をウエルあたりに2x104 細胞の密度で96ウエルプレー
ト上にプレートアウトし、そして製造者の指示に従って次の日にリポフェクタミン試薬(B
RL)を用いてトランスフェクションした。DNA/脂質混合物をそれぞれ6ウエルトラン
スフェクション物について次のように調製した:DMEM100μl中のプラスミドDN
A260ngをDMEM100μl中の脂質2μlとおだやかに混合した(8xCRE−
Lucレポータープラスミド200ng、内因性受容体または非内因性受容体を含んでな
るpCMVまたはpCMV単独の50ng、およびGPRS発現プラスミド(pcDNA
3中のGPRS(Invitrogen))10ngから成るプラスミドDNA260ng)。8XC
RE−Lucレポータープラスミドは、以下のようにして調製した:ベクターSRIF−
β−galは、pβgal−ベーシックベクター(BasicVector)(Clontech)中のBglV
−HindIII部位でラット・ソマトスタチンプロモーター(−71/+51)をクロ
ーニングして得られた。cAMP反応因子の8個のコピーは、アデノウイルス鋳型Adp
CF126CCRE8(7Human Gene Therapy 1883(1996)参照)からPCRにより得られ
そしてKpn−BglV部位でSRIF−β−galベクター内にクローニングすると、
8xCRE−β−galレポーターベクターが得られた。8xCRE−Lucレポタープ
ラスミドは、8xCRE−β−galレポーターベクター中のベータ−ガラクトシダーゼ
遺伝子をHindIII−BamHI部位におけるpGL3−ベーシックベクター(Prome
ga)から得たルシフェラーゼ遺伝子と置換して生成した。30分、室温でのインキュベー
ションの後に、DNA/脂質混合物をDMEM400μlを用いて希釈し、そして希釈混
合物100μlをそれぞれのウエルに加えた。10%FCSを含むDMEM100μlを
、細胞カルチャーインキュベーター中での4時間のインキュベーションの後にそれぞれの
ウエルに加えた。次の日に、トランスフェクションした細胞を10%FCSを含むDME
M200μl/ウエルに交換した。8日後に、PBSを用いる洗浄1回の後にフェノール
レッドを含まないDMEM100μl/ウエルにウエルを交換した。ルシフェラーゼ活性
は、ルクライト(LucLite)TMレポーター遺伝子アッセイキット(Packard)を用い製造者の指
示に従いそして1450マイクロベータ(MicroBeta) TMシンチレーションおよび蛍光カウ
ンター(Wallac)を用いて読み取って次の日に測定した。
【0120】
b.AP1レポーターアッセイ(Gq−関連受容体)
Gq刺激を検出するための方法は、これらのプロモーター中にAP1因子を含む遺伝子
の活性化を起こすGq−依存性ホスホリパーゼCの既知の性質に依存する。パスデテクト
(Pathdetect) TM AP−1 cis−レポーティングシステム(Stratagene 、カタログ番
号#219073)が、CREBレポーターアッセイに関して以上に記載したプロトコールにした
がって使用できるが、リン酸カルシウム沈降物の成分が410ng pAP1−Luc、
80ng pCMV−受容体発現プラスミド、および20ng CMV−SEAPであっ
た点が異なる。
c.Srf−Lucレポーターアッセイ(Gq−関連受容体)
Gq刺激を検出するための一つの方法は、そのプロモーター中に血清反応因子を含む遺
伝子の活性化を起こすGq依存性ホスホリパーゼCの既知の性質に依存する。パスデテク
トTMSRF−Luc−レポーティングシステム(Stratagene)が、例えばCOS7細胞中の
Gq共役活性のアッセイに使用できる。細胞を系のプラスミド成分を用いてトランスフェ
クションしそして哺乳動物トランスフェクション(MannmalianTransfection) TMキット(St
ratagene 、カタログ番号#200285)を製造者の指示に従って使用して内因性または非内因
性GPCRをコードする発現プラスミドを示す。要約すると、410ng SRF−Lu
c、80ng pCMV−受容体発現プラスミド、および20ng CMV−SEAP(
分泌されたアルカリホスファターゼ発現プラスミド。アルカリホスファターゼ活性は試料
間のトランスフェクション効果における変動を制御するためにトランスフェクションされ
た細胞の媒体中で測定する)を、製造者の指示に従ってリン酸カルシウム沈降物中に混和
する。沈降物の半分を96−ウエルプレート中の3ウエル上に均等に分布させ、血清を含
まない媒体中の細胞上に24時間保持する。最後の5時間に、指示された場合に、細胞を
1μMアンギオテンシンを用いてインキュベーションする。次いで、細胞を溶解しそして
ルシライト(Lucilite)TMキット(Packard、カタログ番号#6016911)および「トリラックス(
Trilux)1450マイクロベータ(Micrebeta) 」液体シンチレーションおよび蛍光カウン
ター(Wallac)を用いて、製造者の指示に従ってルシフェラーゼ活性をアッセイする。デー
タはグラフパッドプリズム(GraphPadPrism)TM2.0a(GraphPad Software Inc.)を用いて分
析できる。
【0121】
d.細胞内IP3 蓄積アッセイ(Gw−関連受容体)
第一日目に、受容体(内因性および/または非内因性)を含んでなる細胞を24ウエル
プレート上に、通常は1x105 細胞/ウエルでプレーティングできる(この数は最適化
できるが)。第二日目に、細胞を最初、血清を含まないDMEM50μl/ウエル中の0
.25μgDNAおよび血清を含まないDMEM50μl/ウエル中の2μlリポフェタ
ミンと混合してトランスフェクションできる。溶液をおだやかに攪拌しそして15〜30
分間、室温でインキュベーションする。細胞を0.5ml PBSを用いて洗浄し、血清
を含まない媒体400μlをトランスフェクション媒体と混合しそして細胞に加える。次
いで細胞を3〜4時間、37℃/5%CO2でインキュベーションし次いでトランスフェ
クション媒体を除きそして通常の増殖媒体1ml/ウエルで置き換えた。第三日目に、細
胞を 3H−ミオ−イノシトールを用いて標識する。要約すると、媒体を除きそして細胞を
0.5mlPBSを用いて洗浄する。次いで、0.5mlイノシトール不含/血清不含媒
体(GIBCOBRL) を、ウエルあたりに 3H−ミオ−イノシトールの0.25μCiと一緒に
ウエルあたりに加えそして細胞を16〜18時間、37℃/5%CO2でインキュベーシ
ョンする。第四日目に、細胞を0.5mlPBSで洗浄し、そしてイノシトール不含/血
清不含媒体10μMパルジリン10mM塩化リチウムを含むアッセイ媒体0.45mlも
しくはアッセイ媒体0.4mlおよび10xケタンセリン(ketanserin、ket)50μ
lを最終濃度10μMとしたものを加える。次いで細胞を30分間37℃でインキュベー
ションする。次いで細胞をPBS0.5mlで洗浄しそして新規/氷冷却停止溶液(1M
KOH;18mMホウ酸Na;3.8mM EDTA)200μlをウエルあたりに加
える。溶液を氷上で5〜10分間、または細胞が溶解するまで保持し、次いで新規/氷冷
中和溶液(7.5%HCL)200μlにより中和する。次いで1.5mlエッペンドル
フ(eppendorf)管内に溶解物を移しそしてクロロホルム/メタノール(1:2)1mlを
管当たりに加える。溶液を15秒間ウズ攪拌し、上相をバイオラド(Biorad)AG1−X8
TMアニオン交換樹脂(100〜200メッシュ)に加える。最初に樹脂を1:1.25W
/Vの水を用いて洗浄しそして上相0.9mlをカラム上に加える。カラムを5mMミオ
−イノシトール10mlおよび5mM ホウ酸Na/60mMギ酸Na 10mlを用い
て洗浄する。イノシトール・三リン酸が0.1Mギ酸/1Mギ酸アンモニウムの2mlを
含むシンチレーションカクテル10mlを含むシンチレーションバイアル内で溶離する。
カラムを0.1Mギ酸/3Mギ酸アンモニウムの10mlを用いて洗浄しそして2回、d
dH2Oを用いてリンスしそして4℃水中で保管する。
【0122】
例示的な結果を以下の表Gに示す。
【0123】
【表6】
【0124】
上記の実施例4(1)で開示した構成的活性化を検出するためのGTPγSアッセイの
例示的な結果は、ヒトRUP13およびRUP15上での「Gs:融合タンパク質構築物
」を用いて達成された。下記の表Hは、このアッセイから生成されたシグナルおよび示し
たようなシグナルの相違を表示する。
【0125】
【表7】
【0126】
実施例5
融合タンパク質調製
a.GPCR:Gs融合構築物
構成的活性化GPCR−Gタンパク質融合構築物の設計は、以下のように行った:ラッ
トGタンパク質Gsα(長形;Itoh et al., 83 PNAS 3776 (1986))の5’および3’末
端の両方を、この上にHindIII(5’−AAGCTT−3’)配列を含むように操
作した。正しい配列(近接する(flanking)HindIII配列を含む)の確認の後に、全
配列を、そのベクターのHindIII制限部位を用いてサブクローニングしてpcDN
A3.1(−)(Invitrogenカタログ番号V795-20)中にシャトル(shuttle) した。Gsα
配列の正しい方向は、pcDNA3.1(−)中へのサブクローニングの後に決定した。
HindIII配列で変更したラットGsα遺伝子を含むpcDNA3.1(−)を次い
で確認した。これでこのベクターは、「汎用」Gsαタンパク質ベクターとして利用でき
た。pcDNA3.1(−)ベクターは種々の周知の制限部位をHindIII部位の上
流に含み、従ってGsタンパク質の上流に、内因性、構成的活性GPCRのコーディング
配列を挿入する能力を有利に与える。この同じ方法は、他の「汎用」G−タンパク質ベク
ターを創成するためにも利用でき、そして当然ながら、その他の商業的に利用可能、また
は熟練者に既知の独自のベクターも使用可能であり、その際、重要な基準は、GPCRの
配列がGタンパク質のものの上流でそしてインフレームであることである。
【0127】
RUP13は、Gsを介して共役する。下記の例示的「GPCR融合タンパク質」につ
いて、Gsαへの融合が達成された。
【0128】
RUP13−「Gsα融合タンパク質」構築物を以下のようにして作製した。
プライマーは以下のように設計した。
5’−gatc〔TCTAGAAT〕GGAGTCCTCACCCATCCCCCAG−
3’(配列番号97、センス)
5’−gatc〔GATATC〕CGTGACTCCAGCCGGGGTGAGGCGG
C−3’(配列番号98、アンチセンス)
小文字のヌクレオチドはGタンパク質とRUP13との間の制限部位(括弧内に示す)内
のスペーサーとして含まれる。センスおよびアンチセンスプライマーは、スペーサー(制
限部位に帰属する)がGタンパク質とRUP15との間に存在するように、それぞれXb
aIとEcoRVのための制限部位を含んでいた。
【0129】
次いで、上記に開示したGsα汎用ベクター内の融合ためのそれぞれの受容体配列を確
保するためにPCRを使用し、それぞれ下記のプロトコールを用いた。RUP15のため
の100ng cDNAを、それぞれプライマー(センスおよびアンチ−センス)2μl
、10mM dNTP3μL、10XタックプラスTMプレシジョン緩衝液(Precision Buf
fer)10μL、タックプラスTMプレシジョンポリメラーゼ(Stratagene:#600211) 1μL
、および水80μLを含む別々の管に加えた。RUP15のための反応温度およびサイク
ル時間は、サイクルステップ2〜4の35回反復を含んで下記であった。1分間94℃、
30秒間94℃、20秒間62℃、1分40秒間72℃、および5分間72℃。PCR産
物を1%アガロースゲル上を流し次いで精製した(データは記載しない)。精製した産物
をXbaIおよびEcoRVを用いて消化しそして所望の内挿物を精製しそしてそれぞれ
の制限部位でGs汎用ベクター内に連結した。陽性クローンを形質転換の後に単離しそし
て制限酵素消化により決定した。293細胞を用いる発現が、本明細書内に記載したプロ
トコールを用いて達成された。RUP15−「Gs融合タンパク質」のそれぞれの陽性ク
ローンを正確さを確認するために配列決定した。(核酸配列に関しては配列番号99、そ
してアミノ酸配列に関しては配列番号100参照)。
【0130】
RUP15は、Gsを介して共役する。下記の例示的「GPCR融合タンパク質」に
ついて、Gsαへの融合が達成された。
【0131】
RUP15−「Gsα融合タンパク質」構築物を以下のようにして作製した。プライマ
ーは以下のように設計した。
5’−TCTAGAATGACGTCCACCTGCACCAACAGC−3’(配列番
号101、センス)
5’−gatatcGCAGGAAAAGTAGCAGAATCGTAGGAAG−3’
(配列番号102、アンチセンス)
小文字のヌクレオチドはGタンパク質とRUP15との間の制限部位内のスペーサーと
して含まれる。センスおよびアンチセンスプライマーは、スペーサー(制限部位に帰属す
る)がGタンパク質とRUP15との間に存在するように、それぞれEcoRVとXba
Iのための制限部位を含んでいた。
【0132】
次いで、上記に開示したGsα汎用ベクター内の融合ためのそれぞれの受容体配列を確
保するためにPCRを使用し、それぞれ下記のプロトコールを用いた。RUP15のため
の100ng cDNAを、それぞれプライマー(センスおよびアンチ−センス)2μl
、10mM dNTP3μL、10XタックプラスTMプレシジョン緩衝液10μL、タッ
クプラスTMプレシジョンポリメラーゼ(Stratagene: #600211) 1μL、および水80μL
を含む別々の管に加えた。RUP15のための反応温度およびサイクル時間は、サイクル
ステップ2〜4の35回反復を含んで下記であった。1分間94℃、30秒間94℃、2
0秒間62℃、1分40秒間72℃、および5分間72℃。PCR産物を1%アガロース
ゲル上を流し次いで精製した(データは記載しない)。精製した産物を消化した。)精製
した産物をEcoRVおよびXbaIを用いて消化しそして所望の内挿物を精製しそして
それぞれの制限部位でGs汎用ベクター内に連結した。陽性クローンを形質転換の後に単
離しそして制限酵素消化により決定した。293細胞を用いる発現が、本明細書内に記載
したプロトコールを用いて達成された。RUP15−「Gs融合タンパク質」のそれぞれ
の陽性クローンを正確さを確認するために配列決定した。(核酸配列に関しては配列番号
103、そしてアミノ酸配列に関しては配列番号104参照)。
【0133】
b.Gq(6アミノ酸欠失)/Gi融合構築物
Gq(欠失)/Gi融合構築物の設計は、以下のようにして達成できた。N−末端の6
個のアミノ酸(TELSIMの配列(配列番号129)Gαq−サブユニットを有する2
〜7のアミノ酸を欠失しそして配列EYNLB(配列番号130)を有するC−末端の5
個のアミノ酸を、配列DCGLF(配列番号131)を有する「GαIタンパク質」の相
当するアミノ酸と置き換える。この融合構築物は、下記のプライマー
5’−gatcaagcttcCATGGCGTGCTGCCTGAGCGAGGAG−
3’(配列番号132)および
5’−gatcggatccTTAGAACAGGCCGCAGTCCTTCAGGTT
CAGCTGCAGGATGGTG−3’(配列番号133)
およびマウスGαq−野生型を含むプラスミド63313を用い、鋳型として血球凝集素
タグを用いるPCRにより得られる。小文字のヌクレオチドはスペーサーとして含まれる
。
【0134】
タックプラスプレシジョンDNAポリメラーゼ(Stratagene)は、ステップ2〜4の35
回反復を含む下記のサイクルによる複製に使用される。2分間95℃、20秒間95℃、
20秒間56℃、2分間72℃、および7分間72℃。PCR産物をpCRII−TOP
Oベクター(Invitrogen)内にクローニングしそしてABIビッグダイターミネーターキッ
ト(P.E.Biosystems) を用いて配列決定した。融合構築物の配列を含むTOPOクローン
からの挿入物を発現ベクターpcDNA3.1(+)中に、HindIII/BamHI
部位で2段クローニング法によりシャトルした。
【0135】
実施例6
開示したヒトGPCRの組織分布:RT−PCR
数種の新規のヒトGPCRの発現を確認しそして組織分布を決定するためにRT−PC
Rを適用した。使用したオリゴヌクレオチドは、鋳型としてGPCR特異性そしてヒト多
重組織cDNAパネル(MTC、Clontech) であった。タック DNAポリメラーゼ(Str
atagene)を製造者の指示に従う40μl反応物内の増幅のために使用した。反応物20μ
lを1.5%アガロースゲル上に加えてRT−PCR産物を分析した。下記の表Jは、受
容体、サイクル条件および使用したプライマーを表示する。
【0136】
【表8】
【0137】
【表9】
【0138】
実施例7
プロトコール:逆アゴニストおよびアゴニストの直接同定
A.〔35S〕GTPγSアッセイ
我々は内因性、構成的活性GPCRを候補化合物、例えば逆アゴニストの直接同定のた
めに完全には理解されていない理由から使用したけれども、アッセイ内の変動が大きくな
ることがある。次いで好ましくは、以上に公開したように「GPCR融合タンパク質」も
非内因性、構成的活性化GPCRと一緒に使用される。我々は、このようなタンパク質を
使用する場合にアッセイ内変動が本質的に安定化され、これにより効果的な信号対騒音比
が得られるように見えることを認めた。これは候補化合物のさらに強固な同定を可能とす
る有利な結果を有する。このように、直接同定のために「GPCR融合タンパク質」が使
用されそして利用する場合には下記のアッセイプロトコールを利用することが望ましい。
【0139】
1.膜調製
関係する構成的活性オーファン「GPCR融合タンパク質」を含んでなりそして逆アゴ
ニスト、アゴニストまたは部分アゴニストとしての候補化合物の直接同定に使用するため
の膜は、好ましくは下記のように調製される。
【0140】
a.材料
「膜スクレープ緩衝液」は20mM HEPESおよび10mM EDTA、pH7.
4を含んでなる。「膜洗浄緩衝液」は20mM HEPESおよび0.1mM EDTA
、pH7.4を含んでなる。「結合緩衝液」は20mM HEPES、100mM Na
Cl、および10mM MgCl2 、pH7.4を含んでなる。
【0141】
b.操作
操作の間を通じてすべての材料を氷上に保持する。第一に、媒体を細胞の集密単層から
吸引し、次いで10ml冷PBSを用いてリンスし、次いで吸引する。その後、「膜スク
レープ緩衝液」5mlをスクレープ細胞に加える、これに続いて、50ml遠心分離管内
に細胞抽出物を移す(20,000rpmで17分間、4℃で遠心分離する)。その後、
上清を吸引しそしてペレットを膜「洗浄緩衝液」30ml中に再懸濁し、次いで20,0
00rpmで17分間、4℃で遠心分離する。次いで、上清を吸引しそしてペレットを「
結合緩衝液」内に再懸濁する。次いでこれをブリンクマン・ポリトロン(Brinkman polytr
on) TMホモジナイザーを用いてホモジナイジングする(すべての材料が懸濁するまで15
〜20秒間バースト)。これを本明細書中では「膜タンパク質」と呼ぶ。
【0142】
2.ブラドフォードタンパク質アッセイ
ホモジナイゼーションに次いで、膜のタンパク質濃度をブラドフォード(Bradford)タン
パク質アッセイを用いて決定する(タンパク質は約1.5mg/mlまで希釈し、アリコ
ートに入れ、以後の使用のために冷凍(−80℃)できる。冷凍した場合に、使用のため
のプロトコールは以下である:アッセイの日に、冷凍した「膜タンパク質」を室温で解凍
し、次いでウズ攪拌しそして約12x1.000rpmで約5〜10秒間ポリトロンを用
いてホモジナイズする。複数の調製のためには、異なる調製物のホモジナイゼーションの
間でホモジナイザーを完全に清浄化することに注意すること)
【0143】
a.材料
「結合緩衝液」(上記の通り)、「ブラドフォード染色試薬」、「ブラドフォードタン
パク質標準」を製造者の指示に従って使用する(Biorad カタログ番号500-0006) 。
【0144】
b.操作
2本の管を調製し、一方は膜を含みそして他方は対照の「ブランク」とする。それぞれ
は「結合緩衝液」800ulを含む。その後、「ブラドフォードタンパク質標準」(1m
g/ml)10μlをそれぞれの管に加え、次いで膜「タンパク質」10μlを一本の管
(ブランク以外)を除いて加える。その後、「ブラドフォード染色試薬」200μlをそ
れぞれの管に加え、次いでそれぞれウズ攪拌する。5分後に、管を再びウズ攪拌しそして
その中の材料をキュベットに移す。次いでキュベットをCECIL3041分光光度計を
用いて波長595で読み取る。
【0145】
3.直接同定アッセイ
a.材料
「GDP緩衝液」は、37.5ml「結合緩衝液」および2mg GDP(Sigmaカタロ
グ番号G-7127) から成り、次いで「結合緩衝液」中の一連の希釈により0.2μM GD
Pが得られる(それぞれのウエル内のGDPの最終濃度は、0.1μM GDP)。候補
化合物を含んでなるそれぞれのウエルは100μl「DPG緩衝液」(最終濃度、0.1
μM GDP)、「結合緩衝液」中の「膜タンパク質」50ul、および「結合緩衝液」
中の〔35S〕GTPγS(0.6nM)50μl(10ml「結合緩衝液」あたり2.5
μl〔35S〕GTPγS)から成る200ulの最終体積を有する。
【0146】
b.操作
候補化合物は、好ましくは96ウエルプレートフォーマットを用いてスクリーニングさ
れる(これらは−80℃で冷凍できる)。「膜タンパク質」(または対照として「GPC
R融合タンパク質」を含まない発現ベクターを有する膜)を懸濁するまで短時間ホモジナ
イズする。次いで、タンパク質濃度を上記の「ブラドフォードタンパク質アッセイ」セッ
トを用いて決定する。次いで「膜タンパク質」(および対照)を「結合緩衝液」中で0.
25mg/mlまで希釈する(最終アッセイ濃度、12.5μgウエル)。その後、「G
DP緩衝液」100μlをウオラックシンチストリップ(Wallac Scintistrip) TM (Walla
c)のそれぞれのウエルに加えた。次いで候補化合物5ulをこのウエル内に移すために5
ulピン器具を用いる(すなわち200μlの全アッセイ体積中の5ulは、候補化合物
の最終スクリーニング濃度が10μMであるような1:40の比である)。再度、汚染を
防ぐために、それぞれの移動ステップの後、水(1X)、エタノール(1X)および水(
2X)を含んでなる3基の容器内でピン器具を洗浄すること。過剰の液体をそれぞれのリ
ンスの後に器具から振り払いそして紙およびキムワイプ(kimwipe)を用いて乾燥すること
。その後、「膜タンパク質」50μlをそれぞれのウエルに加え(「GPCR融合タンパ
ク質」を含まない膜を含んでなる対照ウエルも使用する)、そして5〜10分間、室温で
予備インキュベーションする。その後、「結合緩衝液」中の〔35S〕GTPγS(0.6
nM)50μlをそれぞれのウエルに加え、次いで振とう器内で60分間、室温でインキ
ュベーションする(この実施例でもプレートをフォイルで覆う)。次いで、プレートを4
00rpmで15分間、22℃で回転してアッセイを停止する。次いでプレートを8チャ
ンネルマニホルドを用いて吸引しそしてプレートカバーを用いてシールする。次いで、ワ
ラック1450で設定「Prot.$37」を用いてプレートを読み取る(製造者の指定
に従う)
【0147】
B.サイクリックAMPアッセイ
候補化合物を直接同定するための別のアッセイ方法は、シクラーゼに基づくアッセイを
用いて達成される。直接同定に加えて、このアッセイ法は、上記のような〔35S〕GTP
γS法からの結果の確認を与えるための独立した方法としても使用できる。
【0148】
変更したフラッシュプレートTMアデニル酸シクラーゼ・キット(New England Nuclear,
カタログ番号SMP004A)が、下記のプロトコールに従う構成的活性化オーファンGPCRに
対する逆アゴニストおよびアゴニストとしての候補化合物の直接同定のために好ましくは
使用された。
【0149】
トランスフェクションされた細胞をトランスフェクションの約3日後に採取した。20
mM HEPES、pH7.4および10mM MgCl2 を含む緩衝液中に懸濁した細
胞のホモジナイゼーションにより膜を調製した。ホモジナイゼーションは、ブリンクマン
ポリトロンTMを約10秒間使用して氷上で行った。得られたホモジネートを49,000
Xgで15分間、4℃で遠心分離する。次いで得られたペレットを20mM HEPES
、pH7.4および0.1mM EDTAを含む緩衝液中に再懸濁し、10秒間ホモジナ
イズし、次いで49,000Xgで15分間、4℃で遠心分離した。次いで得られたペレ
ットを−80℃で、使用するまで保管した。直接同定スクリーニングの日に、膜ペレット
を室温でゆっくりと解凍し、20mM HEPES、pH7.4および10mM MgC
l2を含む緩衝液中に再懸濁すると、0.60mg/mlの最終タンパク質濃度が得られ
た(再懸濁した膜は使用まで氷上に置く)。
【0150】
cAMP標準および「検出緩衝液」(「検出緩衝液」11mlにトレーサー〔12IcA
MP(100μl〕の2μCiを含んでなる)を調製しそして製造者の指示に従って維持
した。「アッセイ緩衝液」をスクリーニングのために調製し、これは20mM HEPE
S、pH7.4、10mM MgCl2 、20mMホスホクレアチン(Sigma) 、0.1単
位/mlクレアチンホスホキナーゼ(Sigma)、50μM GTP(Sigma)、および0.2m
M ATP(Sigma) を含んでいた。次いで「アッセイ緩衝液」を使用まで氷上で保管した
。
【0151】
上記のようにして同定された候補化合物(冷凍された場合には、室温で解凍)を、40
μl「膜タンパク質」(30μg/ウエル)および「アッセイ緩衝液」50μlと一緒に
、好ましくは96プレートウエルに加えた(3μl/ウエル、12μM最終アッセイ濃度
)。次いでこの混合物を30分間、室温で穏やかに振とうしてインキュベーションした。
【0152】
インキュベーションの後、「検出緩衝液」100μlをそれぞれのウエルに加え、次い
で2〜24時間インキュベーションした。次いでプレートをワラック・マイクロベータTM
プレートリーダーで「Prot#31」を用いて計数した(製造者の指示に従う)。
【0153】
代表的なスクリーニングアッセイプレート(96ウエルフォーマット)結果を図12に
示す。それぞれの棒は、それぞれのウエル内の種々の化合物と、上記の実施例5(a)で
調製したRUP13−GαA融合タンパク質に対する結果を示す。図12に示した代表的
な結果は、それぞれのプレートの平均結果(「m」)に基づいた標準偏差および平均も与
え、そして一次スクリーンから「リード」としての逆アゴニストの選択のための平均プラ
ス2個の任意選択は、少なくとも平均プレート反応による反応百分率から標準偏差の二倍
を差し引いて減らした候補化合物の選択を含む。反対に、一次スクリーンから「リード」
としてのアゴニストの選択のため任意選択は、少なくとも平均プレート反応による反応百
分率に標準偏差の二倍を加えて増加した候補化合物の選択を含む。これらの選択手順に基
づいて、以下のウエル内の候補化合物は、ウエルA2およびG9それぞれの中のRUP1
3に対する推定逆アゴニスト(化合物A)およびアゴニスト(化合物B)として直接同定
された。図12参照。明確化のために以下を記す。これらの化合物はこのGPCRのため
の内因性リガンドのいかなる知識も持たないで直接同定された。受容体機能に基づくアッ
セイ技術を中心とし、そして化合物結合親和性ではなくて、我々はこの受容体の機能的活
性を低下でき(化合物A)ならびに受容体の機能的活性を増加できる(化合物B)化合物
を確認できる。肺組織内のこれら受容体の位置に基づいて(例えば実施例6のhRUP1
3およびhRUP21参照)、薬剤が肺ガンの可能な治療処置のために開発できる。
【0154】
本明細書内で引用された文献は、同時係属および関連特許を含み、特に断らない限り、
引用することにより全体を本明細書中に編入される。熟練者の判断の範囲内にある開示さ
れた本発明の変更および延長は、上記の開示および特許請求範囲の範囲内に包含される。
【0155】
種々の発現ベクターが当該技術分野の熟練者により利用可能であるが、内因性および非
内因性ヒトGPCRの両者のための使用の目的で、使用されるベクターがpCMVである
ことが最も好ましい。このベクターは、1998年10月13日付けで、特許手続上の微
生物の寄託の国際的承認に関するブダペスト条約の規定に基づいてアメリカンタイプカル
チャーコレクション(ATCC)に寄託された(10801 University Blvd., Manassas, VA
20110-2209 USA) 。DNAはATCCDにより試験されそして利用可能と決定された。A
TCCは、pCMVに下記の寄託番号を割り当てた:ATCC#203351。
【特許請求の範囲】
【請求項1】
明細書中に記載の発明。
【請求項1】
明細書中に記載の発明。
【図1】

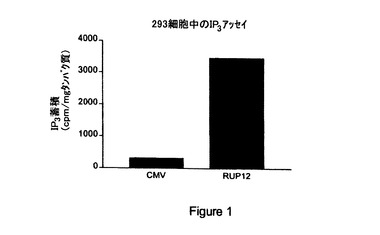
【図2】
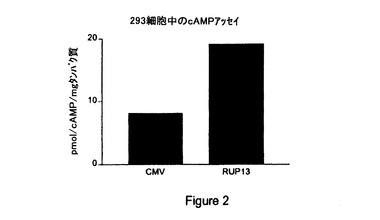
【図3】
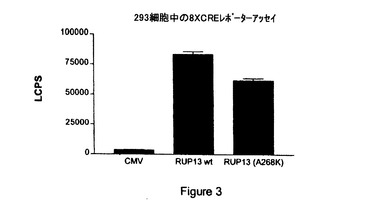
【図4】
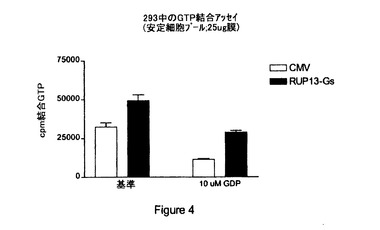
【図5】
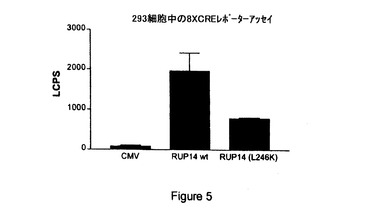
【図6】
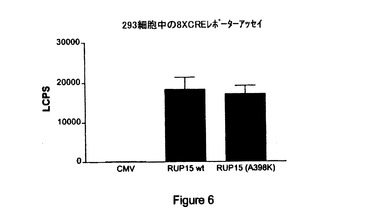
【図7】
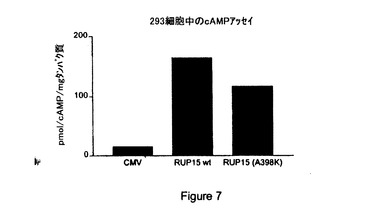
【図8】
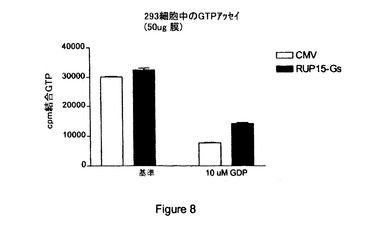
【図9】
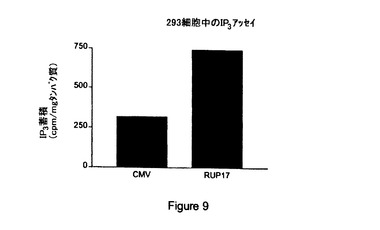
【図10】
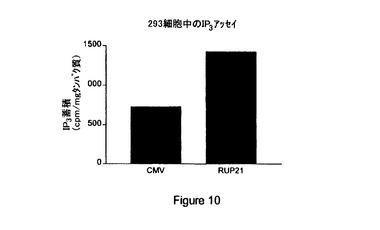
【図11】
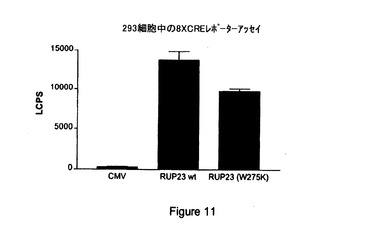
【図12】
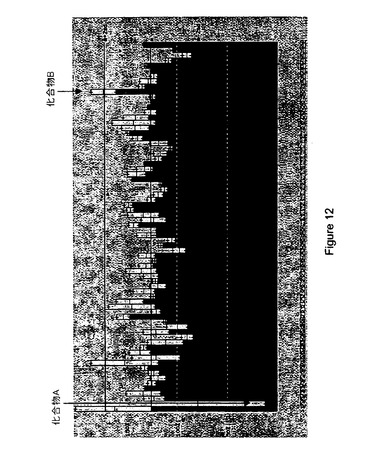
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公開番号】特開2011−147461(P2011−147461A)
【公開日】平成23年8月4日(2011.8.4)
【国際特許分類】
【外国語出願】
【出願番号】特願2011−105725(P2011−105725)
【出願日】平成23年5月10日(2011.5.10)
【分割の表示】特願2001−538960(P2001−538960)の分割
【原出願日】平成12年11月16日(2000.11.16)
【出願人】(500478097)アリーナ ファーマシューティカルズ, インコーポレイテッド (97)
【Fターム(参考)】
【公開日】平成23年8月4日(2011.8.4)
【国際特許分類】
【出願番号】特願2011−105725(P2011−105725)
【出願日】平成23年5月10日(2011.5.10)
【分割の表示】特願2001−538960(P2001−538960)の分割
【原出願日】平成12年11月16日(2000.11.16)
【出願人】(500478097)アリーナ ファーマシューティカルズ, インコーポレイテッド (97)
【Fターム(参考)】
[ Back to top ]