
ヒドロキシルを末端基とする部分でのカーボンナノチューブの側壁の官能基化
本発明は、側壁が官能基化されたカーボンナノチューブの形成方法を対象とし、ここにおいて、そのような官能基化カーボンナノチューブは、その側壁に共有結合的に付着していてヒドロキシルを末端基とする部分を有する。一般に、そのような方法は、最初にフッ素化されているカーボンナノチューブに関する化学を包含する。いくつかの態様において、フッ素化カーボンナノチューブ(“フルオロナノチューブ”)をジアルコールの一金属塩MO−R−OH[式中、Mは金属であり、Rは炭化水素または他の有機鎖および/もしくは環構造単位である]と反応させる。このような態様では、−O−R−OHがナノチューブ上の−Fと置換し、フッ素はMFとして離れる。一般に、そのような一金属塩は、フルオロナノチューブを分散させてある1種以上のジアルコールにMOHを加えることによりその場で形成される。いくつかの態様では、フルオロナノチューブをアミノアルコール、例えばタイプH2N−R−OHのものと反応させ、ここにおいて、−N(H)−R−OHがナノチューブ上の−Fと置換し、フッ素はHFとして離れる。

【発明の詳細な説明】
【発明の開示】
【0001】
本発明は、Robert A.Welch財団、補助金交付番号C−0109;およびTexas Higher Education Coordinating Board、ATP補助金交付番号003604−0026−2001からの支援によりなされた。
関連出願の相互参照
【0002】
本出願は、それぞれ2003年6月16日および2003年7月28日に提出された米国仮特許出願第60/478936号および第60/490556号に対する優先権を請求するものである。本出願は、“Fabrication of Carbon Nanotube Reinforced Epoxy Polymer Copmposites Using Functionalized Carbon Nanotubes”という名称で本出願と同時に提出される同一に譲渡された特許出願と関連しており、これを本明細書中で参考として援用する。
発明の分野
【0003】
本発明は、一般にカーボンナノチューブに関し、具体的には、カーボンナノチューブを、ヒドロキシルを末端基とする部分で官能基化する方法に関する。
背景
【0004】
複数の同軸シェルを含み多層カーボンナノチューブ(MWNT)とよばれるカーボンナノチューブ(CNT)は、Iijimaにより1991年に発見された[Iijima,S.Nature 1991,354,56]。この発見の後、丸く閉じた単一のグラフェンを含む単層カーボンナノチューブ(SWNT)が、遷移金属でドープされた炭素電極を用いるアーク放電法で合成された[Iijima,S.;Ichihashi,T.Nature 1993,363,603;およびBethune,D.S.,Kiang,C.H.;de Vries,M.S.;Gorman,G.;Savoy,R.;Vasquez,J;Beyers,R.Nature 1993,363,605]。これらのカーボンナノチューブ(特にSWNT)は独特の機械的、電気的および熱的性質を持ち、そのような性質によりそれらは多種多様な用途に関し興味深いものになっている。
【0005】
最近、単層カーボンナノチューブ(SWNT)の化学的処理、特に側壁の官能基化が、基礎的かつ技術的にますます興味深い分野になっている。SWNTの共有結合的および非共有結合的な側壁の化学の両方が報告されており、これは例えば、直接的フッ素化およびこれに続く誘導体化;ラジカル、カルベンおよびニトレンの付加ならびに1,3−双極付加および求電子付加;ならびに芳香族分子またはポリマーとのファンデルワールス相互作用による修飾である。Encyclopedia of Nanoscience and Nanotechnology,S.Nalwa編集,American Scientific Publishers,2004,第1巻、849〜861頁のKhabashesku,V.N.;Margrave,J.L.“Chemistry of Carbon Nanotubes”およびそれの参考文献;Khabashesku,V.N.;Billups,W.E.;Margrave,J.L.Acc.Chem.Res.,2002,35,1087;Bahr,J.L.;Tour,J.M.J.Mater.Chem.2002,12,1952参照。共有結合的に統合されたポリマー複合体の加工用の強化材として[Barrera,E.V.JOM,2000,52,38;Zhu,J.;Kim,J.;Peng,H.;Margrave,J.L.;Khabashesku,V.N.;Barrera,E.V.Nano Lett.2003,3,1107;Zhu,J.;Peng,H.;Rodriguez−Macias,F.;Margrave,J.L.;Khabashesku,V.N.;Imam,M.A.;Lozano,K.;Barrera,E.V.Adv.Funct.Mater,2003近刊]および標的化薬物送達のための賦形剤としての官能基化SWNTの施用が、最近立証されている。Pantarotto,D.;Partidos,C.D.;Graff,R.;Hoebeke,J.;Briand,J.P.;Prato,M.;Bianco,A.J.Am.Chem.Soc.2003,125,6160参照。これらの研究で、共有結合または水素結合の形成により高い結合親和性および選択性を提供することができる有機官能基でSWNTを誘導体化する必要性が確認されている。それらはまた、加工、とりわけ生物医学的用途における加工を改善するためには、ヒドロキシル基のような親水性置換基を末端基とする部分での共有結合的な側壁の官能基化が、もっとも重要でありうることを示唆している。
【0006】
最近の実験的研究[Khabashesku,V.N.;Billups,W.E.;Margrave,J.L.Acc.Chem.Res.,2002,35,1087]で、SWNTの直接的フッ素化により調製したフルオロナノチューブを、フッ素の求核置換により側壁が官能基化されたナノチューブ誘導体の調製に有用な前駆体として用いることができることが示された。フッ素化カーボンナノチューブを中間体として利用してヒドロキシル官能性をCNT、特にSWNTに導入するための簡単な方法は、
とりわけ極性溶媒中でのカーボンナノチューブの分散が必要とされる状況で、非常に有利である。
概要
【0007】
本発明は、側壁が官能基化されたカーボンナノチューブの形成方法を対象とし、ここにおいて、そのような官能基化カーボンナノチューブは、その側壁に共有結合的に付着していてヒドロキシルを末端基とする部分を有する。一般に、そのような方法は、最初にフッ素化されているカーボンナノチューブに関する化学を包含する。
【0008】
いくつかの態様において、フッ素化カーボンナノチューブ(“フルオロナノチューブ”)をジアルコールの一金属塩MO−R−OH[式中、Mは金属であり、Rは炭化水素または他の有機鎖および/もしくは環構造単位である]と反応させる。このような態様では、−O−R−OHがナノチューブ上の−Fと置換し、フッ素はMFとして離れる。一般に、そのような一金属塩は、フルオロナノチューブを分散させてある1種以上のジアルコールにMOHを加えることによりその場で形成される。
【0009】
いくつかの態様では、フルオロナノチューブをアミノアルコール、例えばタイプH2N−R−OHのものと反応させ、ここにおいて、−N(H)−R−OHがナノチューブ上の−Fと置換し、フッ素はHFとして離れる。
【0010】
いくつかの態様では上記化学の変形を用い、そのような態様では、チオール基−SHがジアルコール中の−OH基の一方もしくは両方、および/またはアミノアルコール中の−OH基と置き換わる。
【0011】
ヒドロキシルを末端基とする部分で官能基化されたそのようなナノチューブの用途は広範囲であるが、多くは、極性溶媒中でのその高い分散能力(dispersability)および/または溶解性、ならびに末端ヒドロキシル基で達成することができるさらなる官能基化を十分に利用するものであろうことは疑いない。一例として、カーボンナノチューブ上にありヒドロキシルを末端基とする部分をエピクロロヒドリンと反応させると、側壁にエポキシド基が付着しているカーボンナノチューブを得ることができる。これらエポキシドで官能基化されたカーボンナノチューブをエポキシ樹脂と混合し、適切な硬化剤で硬化すると、カーボンナノチューブ−エポキシ複合体を形成させることができる。
【0012】
上記では、以下の発明の詳細な説明をよりよく理解することができるように、本発明の特徴をいくぶん大まかに概説した。本発明の特許請求の範囲の題目を形成する本発明の追加的な特徴および利点を、以下に説明する。
詳細な説明
【0013】
本発明およびその利点がより完全に理解されるように、ここで添付図面と併せて解釈して以下の説明に触れる。ここにおいて:
【0014】
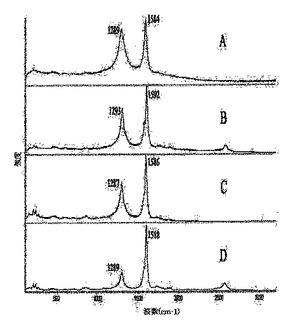
図1は、SWNT材料のラマンスペクトルを表している:フルオロナノチューブ1(A)、ヒドロキシル−ナノチューブ3a(B)、3b(C)、および3bのTGA後の残留物;
【0015】
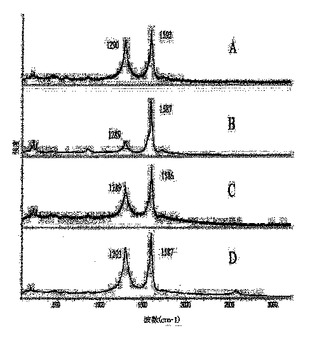
図2は、ヒドロキシル−ナノチューブのラマンスペクトルを表している:(A)3c、(B)3d、(C)3e、(D)3f;
【0016】
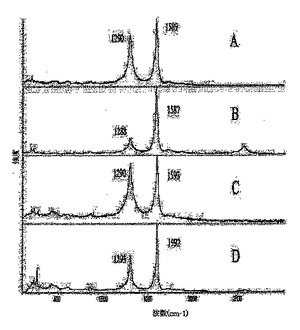
図3は、SWNT材料のラマンスペクトルを表している:(A)3g、(B)3gのTGA後の残留物、(C)3h、(D)3I;そして
【0017】
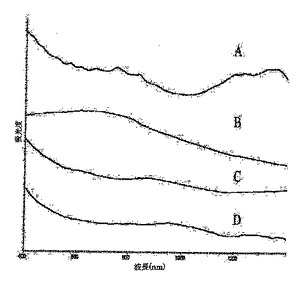
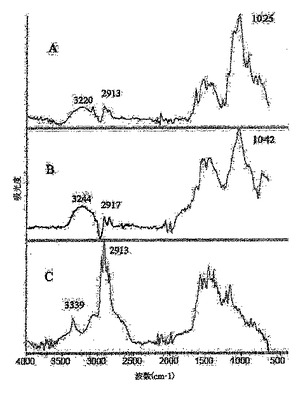
図4は、初期状態のSWNT(A)、フルオロナノチューブ1(B)、ならびにヒドロキシル−ナノチューブ3f(C)および3g(D)のUV−vis−NIRスペクトルを表している;
【0018】
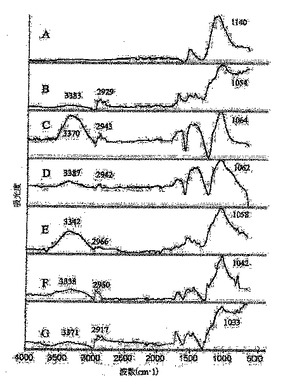
図5は、フルオロナノチューブ1(A)、ならびにヒドロキシル−ナノチューブ(B)3a、(C)3b、(D)3c、(E)3d、(F)3e、(G)3fのATR−FTIRスペクトルを表している;
【0019】
図6は、ヒドロキシル−ナノチューブのATR−FTIRスペクトルを表している:(A)3g、(B)3h、(C)3I;
【0020】
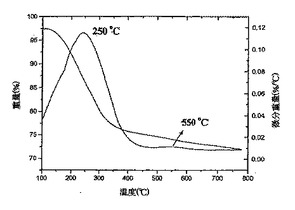
図7は、ヒドロキシル−ナノチューブ3bのTGA−DTAを表している;
【0021】
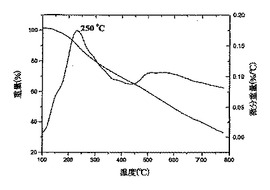
図8は、ヒドロキシル−ナノチューブ3fのTGA−DTAを表している;
【0022】
図9は、ヒドロキシル−ナノチューブ3gのTGA−DTAを表している;
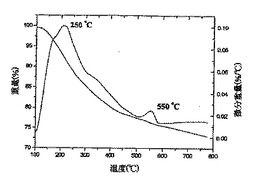
【0023】
図10は、ヒドロキシル−ナノチューブ3fの試験片のTEM像を表しており、ここにおいて、挿入図は単一の官能基化ナノチューブの拡大像を表している;
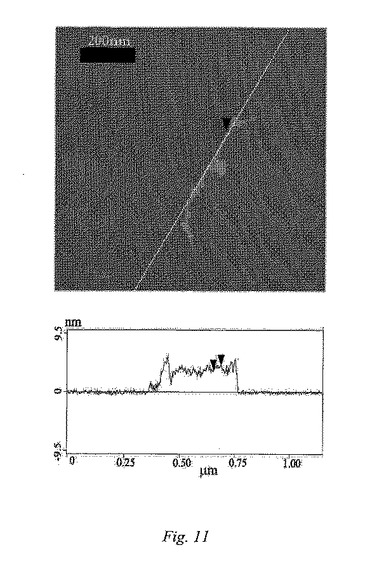
【0024】
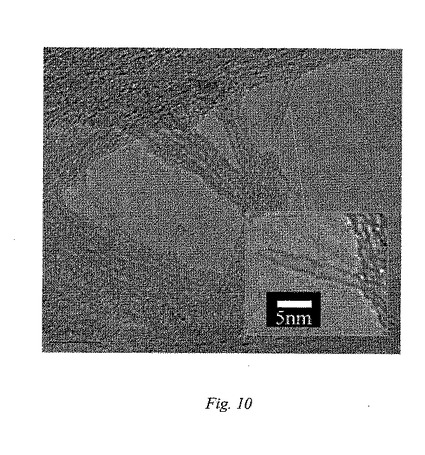
図11は、ヒドロキシル−ナノチューブ3fの束のバックボーンに沿ったAFM像および高さの分析を表しており、ここにおいて、矢印は、側壁の官能基化に起因する0.8nmの高さの差異を指している;そして
【0025】
図12は、エタノール中のSWNT材料の分散液の写真を表している:(A)初期状態のSWNT、および(B)グリセロールで官能基化されたSWNT3f。
【0026】
本発明は、側壁が官能基化されたカーボンナノチューブの形成方法であって、そのような官能基化カーボンナノチューブが、その側壁に共有結合的に付着していてヒドロキシルを末端基とする部分を有する前記形成方法(“ヒドロキシル−ナノチューブ”)、ならびに、そのような方法により作成される組成物および製品を対象とする。一般に、そのような方法は、最初にフッ素化されているカーボンナノチューブに関する化学を包含する。本発明のさまざまな態様の作成および/または使用について以下で検討するが、本発明は多様な具体的状況で具体化することができる多くの施用可能な発明的概念を提供することを、理解すべきである。本明細書中で検討する具体的態様は本発明を作成および/または使用するための具体的方法の例示に過ぎず、本発明の範囲を限定するものではない。
【0027】
本発明によると、カーボンナノチューブ(CNT)としては、単層カーボンナノチューブ(SWNT)、多層カーボンナノチューブ(MWNT)、二層カーボンナノチューブ、バッキーチューブ、フラーレンチューブ、管状フラーレン、黒鉛フィブリル、およびそれらの組合わせが挙げられるが、これらに限定されない。そのようなカーボンナノチューブは、アーク放電[Ebbesen,Annu.Rev.Mater.Sci.1994,24,235−264];レーザーオーブン(laser oven)[Thess et al.,Science 1996,273,483−487];フレーム合成(flame synthesis)[Vander Wal et al.,Chem.Phys.Lett.2001,349,178−184];化学蒸着[米国特許第5374415号]、ここにおいて、担持[Hafner et al.,Chem.Phys.Lett.1998,296,195−202]または非担持[Cheng et al.,Chem.Phys.Lett.1998,289,602−610;Nikolaev et al.,Chem.Phys.Lett.1999,313,91−97]金属触媒を用いてもよい;およびそれらの組合わせを含む任意の公知技術により作成することができるが、これらに限定されない。態様によっては、CNTを、それらをフッ素化する前、またはそれらを本発明のいずれかの化学に付す前に、1以上の加工工程に付すことができる。いくつかの態様では、CNTを、キラリティー、電気伝導率、熱伝導率、直径、長さ、層数およびそれらの組合わせからなる群より選択される性質に基づき分ける。O’Connell et al.,Science 2002,297,593−596;Bachilo et al.,Science 2002,298,2361−2366,;Strano et al.,Science 2003,301,1519−1522参照。いくつかの態様において、CNTは精製されている。代表的精製技術としては、Chiang et al.によるもの[Chiang et al.,J.Phys.Chem.B 2001,105,1157−1161;Chiang et al.,J.Phys.Chem.B 2001,105,8297−8301]が挙げられるが、これに限定されない。いくつかの態様において、CNTは切断工程により切断されている。Liu et al.,Science 1998,280,1253−1256;Gu et al.,Nano Lett.2002,2(9),1009−1013参照。“CNT”および“ナノチューブ”という用語を、本明細書中では同意語として使用する。
【0028】
いくつかの態様において、一般に約C1F0.01〜約C1F1の化学量論を含むフッ素化カーボンナノチューブ(“フルオロナノチューブ”)を、ジアルコールの一金属塩MO−R−OH[式中、Mは金属であり、Rは炭化水素(例えば、−(CH2)n−)または他の有機鎖および/もしくは環構造単位である]と反応させる。このような態様では、−O−R−OHがナノチューブ上の−Fと置換し、フッ素はMFとして離れる。一般に、そのような一金属塩は、フルオロナノチューブを分散させてある1種以上のジアルコールにMOHを加えることによりその場で形成される。
【0029】
上記反応は一般に、約0.5時間〜約3時間の反応時間を必要とする。いくつかの態様では、反応物を加熱手段で加熱する。いくつかの態様では、超音波処理を用いて、ナノチューブを分散させ、および/または反応を促進する。いくつかの態様では、均質化手段を用いて反応物を均質化または混合する。適した均質化手段としては機械的撹拌が挙げられるが、これに限定されない。
【0030】
ジアルコールは、任意のジアルコールであって、フルオロナノチューブをその中に分散させることができ、フルオロナノチューブが適切な条件下でそれと反応するものであることができる。代表的ジアルコールを利用するいくつかの代表的化学経路をスキーム1Aに示す。スキーム1Aにおいて、フルオロナノチューブ1を、ジアルコール2a〜eのいずれかをMOH[式中、MはLi、NaまたはKのいずれかと同等である]と反応させることにより生じたジアルコールの一金属塩と反応させると、官能基化された生成物3a〜eのいずれかが得られる。他の代表的ジアルコールとしてはビスフェノールAが挙げられる。
【0031】
上記化学は、スキーム1Bに示すようにマルチアルコール(multi-alcohol)にも拡大することができる。スキーム1Bにおいて、フルオロナノチューブ1を、マルチアルコール2fをMOH[式中、MはLi、NaまたはKのいずれかと同等でる]と反応させることにより生じるマルチアルコールR(OH)nの一金属塩と反応させると、官能基化された生成物3fが得られる。このように、上記記載は、フルオロナノチューブと一般式MOR(OH)n−1の一金属塩のいずれかとの反応に拡大することができる。Rは同様に、官能基化する部分の主鎖として働くことができる任意の炭化水素または他の有機鎖および/もしくは環構造単位である。
【化1】
【0032】
いくつかの態様では、フルオロナノチューブを最初にジアルコールまたはマルチアルコールに分散して分散液を形成させる。その後、金属水酸化物を同一または異なるジアルコールもしくはマルチアルコールに溶解して溶液を形成させ、その後溶液と分散液を組み合わせて混合物を形成させる。上記のように、超音波処理を利用すると、分散液の形性および/または混合工程を促進することができる。
【0033】
いくつかの態様では、フルオロナノチューブをアミノアルコール、例えばタイプH2N−R−OHのものと反応させ、ここにおいて、−N(H)−R−OHがナノチューブ上の−Fと置換し、フッ素はHFとして離れる。一般に、そのような態様では、フルオロナノチューブを適切なアミノアルコールに分散して反応混合物を形成させ;ピリジン触媒を反応混合物に加え;そして反応混合物+触媒をそのまま反応させて、アミノ(アミン)を末端基とする部分を伴う官能基化カーボンナノチューブを形成させる。いくつかの態様では、超音波処理を用いて、フルオロナノチューブの分散を促進し、混合を誘導する。これらまたは他の態様では、他の混合操作を利用してもよい。反応は一般に、約1時間〜約5時間にわたり約70℃〜約150℃の温度で行われる。
【0034】
アミノアルコールは、任意のアミノアルコールであって、フルオロナノチューブをその中に分散させることができ、フルオロナノチューブが適切な条件下でそれと反応するものであることができる。代表的アミノアルコールを利用するいくつかの代表的化学経路をスキーム2に示す。スキーム2において、フルオロナノチューブ1をアミノアルコール2g〜Iと反応させると、アミノを末端基とする部分が側壁に付着している官能基化カーボンナノチューブ3g〜Iが形成する。
【化2】
【0035】
いくつかの態様では、本発明の方法を、少なくとも部分的に、不活性雰囲気中で実施する。そのような不活性雰囲気としてはAr、Kr、He、Ne、N2、CF4およびそれらの組合わせが挙げられるが、これらに限定されない。
【0036】
上記方法によりヒドロキシ−ナノチューブ生成物が得られる。いくつかの態様において、ヒドロキシ−ナノチューブ生成物は、一般式CNT−[OR(OH)m]x[式中、Rは適切な有機主鎖であり、mは少なくとも1であり、そしてxはナノチューブ炭素原子1000個あたり約1〜約500である]を有する。他の態様において、ヒドロキシ−ナノチューブ生成物は、一般式CNT−[N(Y)R(OH)m]x[式中、Rは適切な有機主鎖であり、Yは水素または他の有機種であり、mは少なくとも1であり、そしてxはナノチューブ炭素原子1000個あたり約1〜約500である]を有する。
【0037】
いくつかの態様では上記化学の変形を用い、そのような態様では、チオール基−SHがジアルコール中の−OH基の一方もしくは両方、および/またはアミノアルコール中の−OH基と置き換わる。
【0038】
理論に拘束される意図はないが、最近のDFTの計算[Kudin,K.N.;Bettinger,H.F.;Scusseria,G.E.Phys.Rev.B,2001,63,45413]は、フルオロナノチューブが裸のカーボンナノチューブより優れた電子受容体であり、したがって強い求核試薬と容易に相互作用しうることを示唆している。これらの反応はまた、フルオロナノチューブ中の弱くなったC−F結合(フッ化アルキルと比較して)により促進され、したがってフッ素がより容易に置換されることが可能になる。アルコールへのフルオロナノチューブの溶解性は、アルコキシドとの反応によりそれらを官能基化する試みを促してきた。本研究に先立ち立証されているこの反応の一例において、ナトリウムメトキシドのメタノール溶液中でフルオロナノチューブ(ほぼC2F)を2時間にわたり音波処理することにより、C4.4F(OCH3)0.25の化学量論を伴い側壁がメトキシル化されたSWNTが生じることが示された。赤外分光学的データおよび可変温度(variable temperature)−質量分析(VTP−MS)データ、ならびに電子マイクロプローブ分析からの高い酸素含有量により、フルオロナノチューブ中のフッ素の部分的置換およびナノチューブ側壁へのメトキシ基の結合が裏付けられた。Mickelson,E.T.Novel Chemistry of Elemental Carbon:Graphite,Fullerenes and Nanotubes,Ph.D.Thesis,Rice大学,Houston,TX,1999;Mickelson,E.T.;Chiang,I.W.;Zimmerman,J.L.;Boul,P.J.;Lozano,J.;Liu,J.;Smalley,R.E.;Hauge,R.H.;Margrave,J.L.J.Phys.Chem.B,1999,103,4318参照。しかしながら、アルコール(メタノール、エタノール、イソ−プロパノール、エタンジオールおよびグリセロール)中でのフルオロナノチューブの音波処理または還流だけではフッ素の著しい置換または脱離は生じないことに留意することが重要である。Shukla,R.;McClain,B.;Khabashesku,V.N.;Margrave,J.L.Rice Quantum Institute 15th Annual Summer Research Colloquium.2001年8月17日、要旨19頁参照。したがって、アルコール種(すなわちジオールおよびグリセロール)を溶媒および試薬の両方として用いると、アルカリ塩基との反応によりヒドロキシルを末端基とするモノアルコキシドを過剰に得ることができる(スキーム1)。
【0039】
これまでの研究において、末端ジアミン、例えばH2N(CH2)nNH2(n=2、3、4、6)はフルオロナノチューブを溶解することができ、高温(90〜150℃)において触媒的量のピリジン存在下でそれらと化学的に反応することが立証されている。該反応はフッ素のほぼ完全な除去および置換をもたらし、側壁への直接的C−N結合付着を作り出すことによりアミノ基を末端基とする官能基化SWNTを生じさせた。Stevens,J.L.;Kiny,V.U.;Huang,A.Y.;Chiang,I.W.;Derrien,G.A.;Khabashesku,V.N.;Margrave,J.L.Proc.NanoTech 2003,第3巻,169−172;Huang,A.Y.;Chiang,I.W.;Khabashesku,V.N.;Margrave,J.L.Rice Quantum Institute 15th Annual Summer Research Colloquium.2001年8月17日、要旨18頁;Stevens,J.L.;Huang,A.Y.;Peng,H.;Chiang,I.W.;Khabashesku,V.N.;Margrave,J.L.NanoLett.2003,3,331;および2003年11月14日提出の同一に譲渡された米国特許出願第10/714187号参照。
【0040】
ヒドロキシルを末端基とする部分で官能基化されたそのようなナノチューブの用途は広範囲であるが、多くは、極性溶媒中でのその高い分散能力および/または溶解性、ならびに末端ヒドロキシル基で達成することができるさらなる官能基化を十分に利用するものであろうことは疑いない。一例として、カーボンナノチューブ上にありヒドロキシルを末端基とする部分をエピクロロヒドリンと反応させると、側壁にエポキシド基が付着しているカーボンナノチューブを得ることができる。これらエポキシドで官能基化されたカーボンナノチューブをエポキシ樹脂と混合し、適切な硬化剤で硬化すると、カーボンナノチューブ−エポキシ複合体を形成させることができる。
【0041】
上記のように、出願人らは、−OH基を末端基とする部分でカーボンナノチューブの側壁を官能基化するための好都合で効率的な方法を開発し、これを“ヒドロキシル−ナノチューブ”と名付けた。これらの官能基は、C−OまたはC−N共有結合(Cはナノチューブ固有の炭素である)のいずれかによりナノチューブの側壁に付着した。このような方法をスキーム1および2に例示する。この方法では、容易に追跡することができる穏やかな反応条件を利用している。このように調製した官能基化カーボンナノチューブの用途は、水素結合能力および側鎖中の末端ヒドロキシル基の化学的反応性に基づくことができる。OH基の化学は非常に富んでいるので、ヒドロキシルナノチューブを用いると、共有結合的に統合されたナノチューブ−強化コポリマーおよびセラミックスならびに生体適合材料を生産することができる。
【0042】
以下の実施例を、本発明の態様のいくつかをより完全に例示するために提供する。以下の実施例で開示する技術は、本発明の実施において十分に機能することが発明者らにより発見され、したがってその実施について代表的なモードを構成すると考えることができる技術を表している。しかしながら、本開示の観点から、当業者は、開示した具体的態様に多くの変更を加えることができ、本発明の精神および範囲から逸脱することなく同様または類似の結果をなお得ることができることを、理解すべきである。
実施例1
【0043】
この実施例は、用いることができる材料のタイプと、本発明のいくつかの態様で使用するためにあるタイプのフルオロナノチューブをどのように調製することができるかを例示するのに役立つ。この実施例ではSWNTを用いたが、他のタイプのCNTを用いてフルオロナノチューブを作成してもよいことに留意されたい。
【0044】
この実施例では、Rice大学のCarbon Nanotechnology研究所でHiPco法により調製された粗製SWNTを、先に記載したように完全に精製して鉄および他の不純物を除去した。Chiang,I.W.;Brinson,B.E.;Huang,A.Y.;Willis,P.A.;Bronikowski,M.J.;Margrave,J.L.;Smalley,R.E.;Hauge,R.H.J.Phys.Chem.B,2001,105,8297参照。精製後、SWNT中の鉄含有量は1重量%を超えていなかった。Carbon Nanotechnologies Inc.,Houston,TXにより供給されるもののような粉末形の精製SWNTを用いることもできる。この実施例では、ほぼC2.5Fの化学量論のフルオロナノチューブ1を、われわれのグループが以前報告した手順に従って、150℃で精製SWNTを直接フッ素化することにより調製した。Mickelson E.T.;Huffman,C.B.;Rinzler,A.G.;Smalley,R.E.;Hauge,R.H.;Margrave,J.L.Chem.Phys.Lett.1998,296,188参照。他のすべての化学物質、例えば、ヒドロキシル−ナノチューブを生産するためのさらなる加工工程で用いられるアルコール2a〜fおよびアミノアルコール2g〜iは、Aldrich Chemical Co.,Milwaukee,WIから購入した。
実施例2
【0045】
この実施例は、スキーム1に対応する本発明の方法のための合成手順を例示するのに役立つ。
【0046】
この方法(スキーム1)によるヒドロキシル−ナノチューブの調製では、10〜15mgのフルオロナノチューブ1を10mLの対応するジオールまたはトリオール2a〜fと一緒にバイアルに入れ、完全な分散液を達成するために80〜90℃で30分間音波処理(17W/55kHz Cole Palmer製浴)した。別のバイアルにおいて、60〜80mgのLiOH(またはNaOHまたはKOH)を、10mLの対応するアルカノール中で完全に溶解するまで30分間音波処理した。ジオール2a〜hの場合この手順を室温で実施したが、より粘性であるグリセロール2fの場合、高温(80〜90℃)での音波処理が必要であった。次の工程で、両バイアルからの溶液を組合わせ、得られた混合物を約1時間音波処理した。その後、反応混合物を孔径1ミクロンのCole Palmer製TEFLON膜に通して濾過し、大量のエタノールおよび水で洗浄して、LiF(またはNaFまたはKF)およびLiOH(またはNaOHまたはKOH)副生物の完全な除去を確実にした。ヒドロキシル−ナノチューブ3a〜fの黒色フィルムとして膜に密着している沈殿生成物を剥がし、70℃の真空オーブンで一晩乾燥した。エネルギー分散型X線分析(EDAX)元素分析は、3a〜f誘導体の試料中の残留フッ素含有量が3〜5at.%であることを示した。
実施例3
【0047】
この実施例は、スキーム2に対応する本発明の方法のための合成手順を例示するのに役立つ。
【0048】
この実施例(スキーム2)では、フルオロナノチューブ1(10〜15mg)を30mLのアミノアルコール2g〜i中で3分間音波処理した。これによりフルオロナノチューブの完全な分散液が得られ、黒色溶液が形成した。その後、5滴のピリジン(Py)を触媒として溶液に加え、反応混合物を窒素雰囲気下、80〜90℃で3時間撹拌した。その後、反応混合物を大量のエタノールと一緒に孔径1ミクロンのCole Palmer製TEFLON膜に通して濾過し、未反応アミノアルコールおよび好ましくない反応副生物の完全な除去を確実にした。官能基化SWNT3g〜iをフィルター膜から取り外し、70℃の真空オーブンで一晩乾燥した。EDAX分析は、3g〜i中の残留フッ素含有量が11〜13at.%であることを示した。
実施例4
【0049】
この実施例は、上記方法により生産された生成物の特性をどのように決定することができるかを例示するのに役立つ。
【0050】
ラマン分光法、減衰全反射−フーリエ変換赤外(ATR−FTIR)分光法、紫外−可視−近赤外(UV−vis−NIR)分光法、熱重量分析/示差熱分析法(TGA/DTA)、走査型電子顕微鏡法/エネルギー分散型X線分析法(SEM/EDAX)、原子間力顕微鏡法(AFM)および透過型電子顕微鏡法(TEM)のすべてを、初期状態のSWNT、フルオロナノチューブ、ならびに実施例2および3で調製したヒドロキシル−ナノチューブ3a〜iの特性決定に使用した。標準的な顕微鏡用スライドの上面に置いた試料についてのラマンスペクトルは、AlGaAsダイオード780−nmレーザー源で操作するRenishaw 1000マイクロラマンシステムで収集した。ATR−FTIRスペクトル測定に関しては、ATRアクセサリーを備えるThermal Nicolet Nexus 870 FTIRシステムを利用した。UV−vis−NIR範囲中のスペクトルは、Shimadzu 3101 PC UV/vis/NIR分光計を用いて測定した。熱分解分析は、TA−SDT−2960 TGA/DTA分析計で実施した。走査型電子顕微鏡法(SEM)は、エネルギー分散型X線(EDAX)分析計を装備したPhillips XL−30電界放出電子顕微鏡を用いて30kVのビームエネルギーで実施した。モデル2570JV−Zスキャナーを備えるDigital Instruments MultiMode走査型プローブ顕微鏡(SPM)を、タッピングモード原子間力顕微鏡分析(AFM)に使用した。レース状炭素(lacey carbon)をコーティングした銅グリッド(サイズ200メッシュ)上に置いた試験片の透過型電子顕微鏡法(TEM)の写真像は、100kVの加速電圧で操作するJEOL JEM−2010電子顕微鏡を用いて得た。
a.光学的分光法
【0051】
ラマン分光法は、ナノチューブの共有結合的な側壁修飾の迅速な評価を提供する。実施例2および3のSWNT誘導体について収集したラマンスペクトルを、図1〜3に示す。1285〜1300cm−1領域におけるピークの観察は炭素のsp3状態に関連付けられており、付着した官能基によりナノチューブ側壁上のπ−電子の芳香族系が崩壊した証拠として通常用いられる。フルオロナノチューブ1のラマンスペクトル(図1A)において、1293cm−1で観察された高強度ピークは、実施例2および3で調製したすべての官能基化SWNTのうちsp3−混成側壁炭素の含有量がもっとも高い(約40%)ことを反映している。1におけるこの高度な側壁修飾は、初期状態のSWNTにおいて200〜260cm−1で見られるSWNTブリージング(breathing)モードのピークの完全な消失、ならびに裸のナノチューブでの1594cm−1から1での1584cm−1まで赤側にシフトしている接線(tangential)モードのピークのブロードニングおよび弱化を引き起こす。ヒドロキシル−ナノチューブ3a〜Iのラマンスペクトルで観察されるように、1287〜1293cm−1の範囲におけるSP3炭素のピークはしたがって、共有結合的官能基化を示す。フルオロナノチューブ1のラマンスペクトルに比べこれらのピークの相対的強度が低いということは、研究した反応(スキーム1、2)における1からのフッ素の側壁での脱離の進行(フッ素の置換に加えて)により説明することができ、この脱離はsp3炭素状態の数を減少させ、ナノチューブ側壁上のsp2−結合を部分的に復活させる。1とは異なり、200〜260cm−1でのブリージングモードのピークは3a〜iのスペクトルにおいて可視的になり、最も低い程度で官能基化されているSWNT誘導体3aおよび3dについてはより高い強度を示している(図1Bおよび2F)。このモードは、側壁に付着している基をより多く持つ誘導体3b、c、e〜iではより弱くなる−これらの基は、ナノチューブの放射状のブリージング振動を妨害すると思われる。Encyclopedia of Nanoscience and Nanotechnology、S.Nalwa編集、American Scientific Publishers、2004年、第1巻、849〜861頁のKhabashesku,V.N.;Margrave,J.L.Chemistry of Carbon Nanotubesおよびそれの参考文献;Khabashesku,V.N.;Billups,W.E.;Margrave,J.L.Acc.Chem.Res.,2002,35,1087;Bahr,J.L.;Tour,J.M.J.Mater.Chem.2002,12,1952参照。
【0052】
UV−vis−NIR分光法は、SWNT側壁の官能基化の他の分光学的精査として役立つ。この場合、電子構造の改変により、初期状態のナノチューブのスペクトルに通常観察されるvan Hove転移特性の喪失がもたらされる。この実施例では、この主張を、初期状態のSWNTのUV−vis−NIRスペクトルと、ジメチルホルムアミド(DMF)溶液中のフルオロナノチューブ1およびヒドロキシル−ナノチューブ3f、gについて測定したものとを、図4で比較することにより例示する。裸の(例えば初期状態の)SWNTとは異なり、van Hove特異点は、高度に官能基化された誘導体1のスペクトル中にまったく存在しない。3f、gで観察されるvan Hove特異点の強度における劇的な低下により、それらのUV−vis−NIRスペクトルは側壁が官能基化されたSWNTに典型的なものに見えるようになり、これにより化学修飾の発生の重要な証拠を提供する。Encyclopedia of Nanoscience and Nanotechnology、S.Nalwa編集、American Scientific Publishers、2004年、第1巻、849〜861頁のKhabashesku,V.N.;Margrave,J.L.Chemistry of Carbon Nanotubesおよびそれの参考文献;Khabashesku,V.N.;Billups,W.E.;Margrave,J.L.Acc.Chem.Res.,2002,35,1087;Bahr,J.L.;Tour,J.M.J.Mater.Chem.2002,12,1952;Stevens,J.L.;Kiny,V.U.;Huang,A.Y.;Chiang,I.W.;Derrien,G.A.;Khabashesku,V.N.;Margrave,J.L.Proc.NanoTech2003,第3巻,169−172参照。
【0053】
図5および6に示したATR−FTIRスペクトルを用いて、SWNTの側壁に共有結合していてヒドロキシル基を末端基とする部分を同定した。フルオロナノチューブ1中のC−F結合の伸縮に特徴的な約1140cm−1の強いピーク(図5A)は、ジオール、トリオールおよびアミノアルコールとの反応後に消失している。このピークは、ヒドロキシル−ナノチューブ3a〜iのスペクトルでは、ナノチューブ−O−CおよびC−OH単位のC−O結合の伸縮に起因する1020〜1070cm−1領域中のピークにより置き換わられた。3000〜3600cm−1の範囲における新しい非常に幅広いバンドはO−H伸縮に帰属するが、2800〜3000cm−1および1360〜1460cm−1領域中のピークは、それぞれC−H伸縮および変形モードに帰属する。誘導体3g,hおよび3i中のナノチューブ−N(H)−Cまたはナノチューブ−N(C)−C構造単位のC−N伸縮モードは、1120〜1210cm−1のスペクトル範囲で観察されており(図6A〜C)、これは、それぞれ第二級および第三級アミンでのC−Nモードに特徴的である。Lin−Vien,D.;Colthup,N.B.;Fatelley,W.G.;Grasselli,J.G.The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules;Academic Press Inc.:San Diego,CA,1991,299頁参照。1および3a〜i中の活性化C=C伸縮モードのピークは、1540〜1580cm−1領域で観察された。
b.熱分解の研究
【0054】
これらの研究はさらに、ナノチューブの共有結合的な側壁の誘導体化に関する証拠を提供した。試料3b、3fおよび3gについて得られたTGA−DTAデータプロット(図7〜9)は、微分プロット上で250℃に主要ピークを示している。そのような高温におけるこれらのピークの出現は、重量減少が、ナノチューブからの物理吸着種の脱着によるのではなく、OH基を末端基とする部分の分離およびフラグメンテーションによって引き起こされることを示している。これらのSWNT誘導体についてのDTAプロット上で約550℃において観察される第2のピークは、残留C−F結合の脱離に起因し[Khabashesku,V.N.;Billups,W.E.;Margrave,J.L.Acc Chem.Res.,2002,35,1087;Mickelson,E.T.Novel Chemistry of Elemental Carbon:Graphite,Fullerenes and Nanotubes,Ph.D.Thesis,Rice大学,Houston,TX,1999]、EDAXで測定した残留フッ素含有量と相関している。これらのピークの主要ではない特徴は、フルオロナノチューブ1中のC−F結合の大部分が、反応の過程(スキーム1、2)でOH基を末端基とする部分により効率的に置き換わられたことを示している。3b、3fおよび3gのTGAプロット上で250℃において観察される主要ピークは、それぞれ20%、35%および22%の平均重量減少を示している。この重量減少がOH基を末端基とする部分の脱離に起因すると仮定すると、これらの誘導体における側壁の官能基化の程度は、大体3bで1/25、3fで1/16、および3gで1/20と概算することができる。
【0055】
異なる方法により調製された(スキーム1、2)3bおよび3g誘導体のTGA後の残留材料について測定したラマンスペクトル(図1Dおよび3B)は、両方ともsp3炭素モードの強度における劇的な低下を示している。このデータは、ナノチューブ側壁からの官能基の分離が、他の共有結合的に官能基化されたSWNTにおける先の熱分解の観察結果に一致して起こることを示している。Encyclopedia of Nanoscience and Nanotechnology、S.Nalwa編集、American Scientific Publishers、2004年、第1巻、849〜861頁のKhabashesku,V.N.;Margrave,J.L.Chemistry of Carbon Nanotubesおよびそれの参考文献;Khabashesku,V.N.;Billups,W.E.;Margrave,J.L.Acc.Chem.Res.,2002,35,1087;Bahr,J.L.;Tour,J.M.J.Mater.Chem.2002,12,1952;Stevens,J.L.;Huang,A.Y.;Peng,H.;Chiang,I.W.;Khabashesku,V.N.;Margrave,J.L.NanoLett.2003,3,331;Peng,H.;Reverdy,P.;Khabashesku,V.N.;Margrave,J.L.Chem.Comm.2003,362;Peng,H.;Alemany,L.B.;Margrave,J.L.;Khabashesku、V.N.J.Am.Chem.Soc.2003,125,15174−15182参照。
【0056】
SWNT誘導体の熱分解中に発生する揮発性種は、可変温度熱分解−質量分析法(VTP−MS)により分析した。3a〜fについてVTP−MSにより得られたデータは、300〜550℃の温度範囲での真空条件下における付着基のフラグメンテーションを示しており、これは、質量スペクトルにおいて、3aおよび3d〜fではm/z44(C2H4O)、29(HCO)のピーク、ならびに3bおよび3cではそれぞれm/z58、57、56、55(C3H6O〜C3H2O)およびm/z72、71、70(C4H8O〜C4H6O)のピークの追加的群により検出される。誘導体3iにおける側壁のC−N結合基の存在は、250〜400℃の温度でのジエタノールアミンの分離に起因するm/z105の主要ピークの出現をもたらす。
c.顕微鏡分析
【0057】
TEMにより、ヒドロキシルナノチューブにおける側壁修飾の直接的画像化が可能になる。図10は、レース状炭素をコーティングした銅グリッド上に置いたグリセロールで官能基化したSWNT3f試験片のTEM像を示している。挿入図は、単一ナノチューブの“でこぼこのある”表面をはっきり示しており、これは、側壁上の炭素−炭素結合フラクションが、連結を形成している炭素でより短いsp2からより長いsp3状態へ、共有結合的に改変することに起因する。
【0058】
3f誘導体のAFM研究(図11)は、側壁の官能基化に起因する、初期状態のSWNTナノチューブと比較して著しく低減した束のサイズを示している。初期状態のSWNTは、直径で数十〜100ナノメートルの束に凝集することが知られている。3fの束の平均サイズは、直径でわずか3〜6nmであることが測定された。これらの束の内部において、個々のヒドロキシル−ナノチューブは、側鎖からの末端OH基により形成される水素結合によって一緒に連結していると考えられる。官能基化SWNTの束のバックボーンプロフィルのタッピングモードでの分析は、高さの平均が4.4nmであることを示している。非晶質炭素粒子不純物を含まないバックボーン区域に沿って測定した高さの差異(約0.8nm)は、図10の挿入図でのTEM像により示されるように、“伸縮した”様式でナノチューブ側壁に付着しているOCH2CH(OH)CH2OH鎖の大体の長さに関連していると思われる。
実施例5
【0059】
この実施例は、非官能基化CNTと比較して、ヒドロキシルを末端基とする部分で官能基化されたカーボンナノチューブが極性溶媒中で有する改善された分散能力または溶解性を例示するのに役立つ。
【0060】
実施例2および3で調製したすべてのヒドロキシル−ナノチューブSWNT誘導体は、初期状態のSWNTと比較して極性溶媒への改善された溶解性を示した(図12A)。もっとも安定な溶液はグリセロールで誘導したSWNT材料3fから得られた。これは、それらがナノチューブ側鎖中に最高含有量のヒドロキシル基を持つためと思われる。水への3fの溶解性(約40mg/L)は数日間安定であったが、3f濃度がより高い(約80mg/L)エタノール溶液では(図12B)、数カ月後でもわずかに沈殿が見られた。
【0061】
本明細書中で参照したすべての特許および出版物を、本明細書中で参考として援用する。上記構造、作用、および上記態様の操作のいくつかは、本発明を実施するのに必須ではなく、代表的態様または複数の態様を完結させるためにのみ説明中に挙げられていることは、理解されるであろう。これに加えて、上記の参照した特許および出版物に記載されている具体的な構造、作用および操作は、本発明と併せて実施することができるが、その実施に不可欠なものではないことは、理解されるであろう。したがって、本発明は、添付の特許請求の範囲により定義されるような本発明の精神および範囲から実質的に逸脱することなく、具体的に記載したこととは異なるように実施してもよいことを、理解すべきである。
【図面の簡単な説明】
【0062】
【図1】SWNT材料のラマンスペクトルを表す図である:フルオロナノチューブ1(A)、ヒドロキシル−ナノチューブ3a(B)、3b(C)、および3bのTGA後の残留物。
【図2】ヒドロキシル−ナノチューブのラマンスペクトルを表す図である:(A)3c、(B)3d、(C)3e、(D)3f。
【図3】SWNT材料のラマンスペクトルを表す図である:(A)3g、(B)3gのTGA後の残留物、(C)3h、(D)3I。
【図4】初期状態のSWNT(A)、フルオロナノチューブ1(B)、ならびにヒドロキシル−ナノチューブ3f(C)および3g(D)のUV−vis−NIRスペクトルを表す図である。
【図5】フルオロナノチューブ1(A)、ならびにヒドロキシル−ナノチューブ(B)3a、(C)3b、(D)3c、(E)3d、(F)3e、(G)3fのATR−FTIRスペクトルを表す図である。
【図6】ヒドロキシル−ナノチューブのATR−FTIRスペクトルを表す図である:(A)3g、(B)3h、(C)3I。
【図7】ヒドロキシル−ナノチューブ3bのTGA−DTAを表す図である。
【図8】ヒドロキシル−ナノチューブ3fのTGA−DTAを表す図である。
【図9】ヒドロキシル−ナノチューブ3gのTGA−DTAを表す図である。
【図10】ヒドロキシル−ナノチューブ3fの試験片のTEM像を表す図であり、ここにおいて、挿入図は単一の官能基化ナノチューブの拡大像を表す。
【図11】ヒドロキシル−ナノチューブ3fの束のバックボーンに沿ったAFM像および高さの分析を表す図であり、ここにおいて、矢印は、側壁の官能基化に起因する0.8nmの高さの差異を指している。
【図12】エタノール中のSWNT材料の分散液の写真を表す図である:(A)初期状態のSWNT、および(B)グリセロールで官能基化したSWNT3f。
【発明の開示】
【0001】
本発明は、Robert A.Welch財団、補助金交付番号C−0109;およびTexas Higher Education Coordinating Board、ATP補助金交付番号003604−0026−2001からの支援によりなされた。
関連出願の相互参照
【0002】
本出願は、それぞれ2003年6月16日および2003年7月28日に提出された米国仮特許出願第60/478936号および第60/490556号に対する優先権を請求するものである。本出願は、“Fabrication of Carbon Nanotube Reinforced Epoxy Polymer Copmposites Using Functionalized Carbon Nanotubes”という名称で本出願と同時に提出される同一に譲渡された特許出願と関連しており、これを本明細書中で参考として援用する。
発明の分野
【0003】
本発明は、一般にカーボンナノチューブに関し、具体的には、カーボンナノチューブを、ヒドロキシルを末端基とする部分で官能基化する方法に関する。
背景
【0004】
複数の同軸シェルを含み多層カーボンナノチューブ(MWNT)とよばれるカーボンナノチューブ(CNT)は、Iijimaにより1991年に発見された[Iijima,S.Nature 1991,354,56]。この発見の後、丸く閉じた単一のグラフェンを含む単層カーボンナノチューブ(SWNT)が、遷移金属でドープされた炭素電極を用いるアーク放電法で合成された[Iijima,S.;Ichihashi,T.Nature 1993,363,603;およびBethune,D.S.,Kiang,C.H.;de Vries,M.S.;Gorman,G.;Savoy,R.;Vasquez,J;Beyers,R.Nature 1993,363,605]。これらのカーボンナノチューブ(特にSWNT)は独特の機械的、電気的および熱的性質を持ち、そのような性質によりそれらは多種多様な用途に関し興味深いものになっている。
【0005】
最近、単層カーボンナノチューブ(SWNT)の化学的処理、特に側壁の官能基化が、基礎的かつ技術的にますます興味深い分野になっている。SWNTの共有結合的および非共有結合的な側壁の化学の両方が報告されており、これは例えば、直接的フッ素化およびこれに続く誘導体化;ラジカル、カルベンおよびニトレンの付加ならびに1,3−双極付加および求電子付加;ならびに芳香族分子またはポリマーとのファンデルワールス相互作用による修飾である。Encyclopedia of Nanoscience and Nanotechnology,S.Nalwa編集,American Scientific Publishers,2004,第1巻、849〜861頁のKhabashesku,V.N.;Margrave,J.L.“Chemistry of Carbon Nanotubes”およびそれの参考文献;Khabashesku,V.N.;Billups,W.E.;Margrave,J.L.Acc.Chem.Res.,2002,35,1087;Bahr,J.L.;Tour,J.M.J.Mater.Chem.2002,12,1952参照。共有結合的に統合されたポリマー複合体の加工用の強化材として[Barrera,E.V.JOM,2000,52,38;Zhu,J.;Kim,J.;Peng,H.;Margrave,J.L.;Khabashesku,V.N.;Barrera,E.V.Nano Lett.2003,3,1107;Zhu,J.;Peng,H.;Rodriguez−Macias,F.;Margrave,J.L.;Khabashesku,V.N.;Imam,M.A.;Lozano,K.;Barrera,E.V.Adv.Funct.Mater,2003近刊]および標的化薬物送達のための賦形剤としての官能基化SWNTの施用が、最近立証されている。Pantarotto,D.;Partidos,C.D.;Graff,R.;Hoebeke,J.;Briand,J.P.;Prato,M.;Bianco,A.J.Am.Chem.Soc.2003,125,6160参照。これらの研究で、共有結合または水素結合の形成により高い結合親和性および選択性を提供することができる有機官能基でSWNTを誘導体化する必要性が確認されている。それらはまた、加工、とりわけ生物医学的用途における加工を改善するためには、ヒドロキシル基のような親水性置換基を末端基とする部分での共有結合的な側壁の官能基化が、もっとも重要でありうることを示唆している。
【0006】
最近の実験的研究[Khabashesku,V.N.;Billups,W.E.;Margrave,J.L.Acc.Chem.Res.,2002,35,1087]で、SWNTの直接的フッ素化により調製したフルオロナノチューブを、フッ素の求核置換により側壁が官能基化されたナノチューブ誘導体の調製に有用な前駆体として用いることができることが示された。フッ素化カーボンナノチューブを中間体として利用してヒドロキシル官能性をCNT、特にSWNTに導入するための簡単な方法は、
とりわけ極性溶媒中でのカーボンナノチューブの分散が必要とされる状況で、非常に有利である。
概要
【0007】
本発明は、側壁が官能基化されたカーボンナノチューブの形成方法を対象とし、ここにおいて、そのような官能基化カーボンナノチューブは、その側壁に共有結合的に付着していてヒドロキシルを末端基とする部分を有する。一般に、そのような方法は、最初にフッ素化されているカーボンナノチューブに関する化学を包含する。
【0008】
いくつかの態様において、フッ素化カーボンナノチューブ(“フルオロナノチューブ”)をジアルコールの一金属塩MO−R−OH[式中、Mは金属であり、Rは炭化水素または他の有機鎖および/もしくは環構造単位である]と反応させる。このような態様では、−O−R−OHがナノチューブ上の−Fと置換し、フッ素はMFとして離れる。一般に、そのような一金属塩は、フルオロナノチューブを分散させてある1種以上のジアルコールにMOHを加えることによりその場で形成される。
【0009】
いくつかの態様では、フルオロナノチューブをアミノアルコール、例えばタイプH2N−R−OHのものと反応させ、ここにおいて、−N(H)−R−OHがナノチューブ上の−Fと置換し、フッ素はHFとして離れる。
【0010】
いくつかの態様では上記化学の変形を用い、そのような態様では、チオール基−SHがジアルコール中の−OH基の一方もしくは両方、および/またはアミノアルコール中の−OH基と置き換わる。
【0011】
ヒドロキシルを末端基とする部分で官能基化されたそのようなナノチューブの用途は広範囲であるが、多くは、極性溶媒中でのその高い分散能力(dispersability)および/または溶解性、ならびに末端ヒドロキシル基で達成することができるさらなる官能基化を十分に利用するものであろうことは疑いない。一例として、カーボンナノチューブ上にありヒドロキシルを末端基とする部分をエピクロロヒドリンと反応させると、側壁にエポキシド基が付着しているカーボンナノチューブを得ることができる。これらエポキシドで官能基化されたカーボンナノチューブをエポキシ樹脂と混合し、適切な硬化剤で硬化すると、カーボンナノチューブ−エポキシ複合体を形成させることができる。
【0012】
上記では、以下の発明の詳細な説明をよりよく理解することができるように、本発明の特徴をいくぶん大まかに概説した。本発明の特許請求の範囲の題目を形成する本発明の追加的な特徴および利点を、以下に説明する。
詳細な説明
【0013】
本発明およびその利点がより完全に理解されるように、ここで添付図面と併せて解釈して以下の説明に触れる。ここにおいて:
【0014】
図1は、SWNT材料のラマンスペクトルを表している:フルオロナノチューブ1(A)、ヒドロキシル−ナノチューブ3a(B)、3b(C)、および3bのTGA後の残留物;
【0015】
図2は、ヒドロキシル−ナノチューブのラマンスペクトルを表している:(A)3c、(B)3d、(C)3e、(D)3f;
【0016】
図3は、SWNT材料のラマンスペクトルを表している:(A)3g、(B)3gのTGA後の残留物、(C)3h、(D)3I;そして
【0017】
図4は、初期状態のSWNT(A)、フルオロナノチューブ1(B)、ならびにヒドロキシル−ナノチューブ3f(C)および3g(D)のUV−vis−NIRスペクトルを表している;
【0018】
図5は、フルオロナノチューブ1(A)、ならびにヒドロキシル−ナノチューブ(B)3a、(C)3b、(D)3c、(E)3d、(F)3e、(G)3fのATR−FTIRスペクトルを表している;
【0019】
図6は、ヒドロキシル−ナノチューブのATR−FTIRスペクトルを表している:(A)3g、(B)3h、(C)3I;
【0020】
図7は、ヒドロキシル−ナノチューブ3bのTGA−DTAを表している;
【0021】
図8は、ヒドロキシル−ナノチューブ3fのTGA−DTAを表している;
【0022】
図9は、ヒドロキシル−ナノチューブ3gのTGA−DTAを表している;
【0023】
図10は、ヒドロキシル−ナノチューブ3fの試験片のTEM像を表しており、ここにおいて、挿入図は単一の官能基化ナノチューブの拡大像を表している;
【0024】
図11は、ヒドロキシル−ナノチューブ3fの束のバックボーンに沿ったAFM像および高さの分析を表しており、ここにおいて、矢印は、側壁の官能基化に起因する0.8nmの高さの差異を指している;そして
【0025】
図12は、エタノール中のSWNT材料の分散液の写真を表している:(A)初期状態のSWNT、および(B)グリセロールで官能基化されたSWNT3f。
【0026】
本発明は、側壁が官能基化されたカーボンナノチューブの形成方法であって、そのような官能基化カーボンナノチューブが、その側壁に共有結合的に付着していてヒドロキシルを末端基とする部分を有する前記形成方法(“ヒドロキシル−ナノチューブ”)、ならびに、そのような方法により作成される組成物および製品を対象とする。一般に、そのような方法は、最初にフッ素化されているカーボンナノチューブに関する化学を包含する。本発明のさまざまな態様の作成および/または使用について以下で検討するが、本発明は多様な具体的状況で具体化することができる多くの施用可能な発明的概念を提供することを、理解すべきである。本明細書中で検討する具体的態様は本発明を作成および/または使用するための具体的方法の例示に過ぎず、本発明の範囲を限定するものではない。
【0027】
本発明によると、カーボンナノチューブ(CNT)としては、単層カーボンナノチューブ(SWNT)、多層カーボンナノチューブ(MWNT)、二層カーボンナノチューブ、バッキーチューブ、フラーレンチューブ、管状フラーレン、黒鉛フィブリル、およびそれらの組合わせが挙げられるが、これらに限定されない。そのようなカーボンナノチューブは、アーク放電[Ebbesen,Annu.Rev.Mater.Sci.1994,24,235−264];レーザーオーブン(laser oven)[Thess et al.,Science 1996,273,483−487];フレーム合成(flame synthesis)[Vander Wal et al.,Chem.Phys.Lett.2001,349,178−184];化学蒸着[米国特許第5374415号]、ここにおいて、担持[Hafner et al.,Chem.Phys.Lett.1998,296,195−202]または非担持[Cheng et al.,Chem.Phys.Lett.1998,289,602−610;Nikolaev et al.,Chem.Phys.Lett.1999,313,91−97]金属触媒を用いてもよい;およびそれらの組合わせを含む任意の公知技術により作成することができるが、これらに限定されない。態様によっては、CNTを、それらをフッ素化する前、またはそれらを本発明のいずれかの化学に付す前に、1以上の加工工程に付すことができる。いくつかの態様では、CNTを、キラリティー、電気伝導率、熱伝導率、直径、長さ、層数およびそれらの組合わせからなる群より選択される性質に基づき分ける。O’Connell et al.,Science 2002,297,593−596;Bachilo et al.,Science 2002,298,2361−2366,;Strano et al.,Science 2003,301,1519−1522参照。いくつかの態様において、CNTは精製されている。代表的精製技術としては、Chiang et al.によるもの[Chiang et al.,J.Phys.Chem.B 2001,105,1157−1161;Chiang et al.,J.Phys.Chem.B 2001,105,8297−8301]が挙げられるが、これに限定されない。いくつかの態様において、CNTは切断工程により切断されている。Liu et al.,Science 1998,280,1253−1256;Gu et al.,Nano Lett.2002,2(9),1009−1013参照。“CNT”および“ナノチューブ”という用語を、本明細書中では同意語として使用する。
【0028】
いくつかの態様において、一般に約C1F0.01〜約C1F1の化学量論を含むフッ素化カーボンナノチューブ(“フルオロナノチューブ”)を、ジアルコールの一金属塩MO−R−OH[式中、Mは金属であり、Rは炭化水素(例えば、−(CH2)n−)または他の有機鎖および/もしくは環構造単位である]と反応させる。このような態様では、−O−R−OHがナノチューブ上の−Fと置換し、フッ素はMFとして離れる。一般に、そのような一金属塩は、フルオロナノチューブを分散させてある1種以上のジアルコールにMOHを加えることによりその場で形成される。
【0029】
上記反応は一般に、約0.5時間〜約3時間の反応時間を必要とする。いくつかの態様では、反応物を加熱手段で加熱する。いくつかの態様では、超音波処理を用いて、ナノチューブを分散させ、および/または反応を促進する。いくつかの態様では、均質化手段を用いて反応物を均質化または混合する。適した均質化手段としては機械的撹拌が挙げられるが、これに限定されない。
【0030】
ジアルコールは、任意のジアルコールであって、フルオロナノチューブをその中に分散させることができ、フルオロナノチューブが適切な条件下でそれと反応するものであることができる。代表的ジアルコールを利用するいくつかの代表的化学経路をスキーム1Aに示す。スキーム1Aにおいて、フルオロナノチューブ1を、ジアルコール2a〜eのいずれかをMOH[式中、MはLi、NaまたはKのいずれかと同等である]と反応させることにより生じたジアルコールの一金属塩と反応させると、官能基化された生成物3a〜eのいずれかが得られる。他の代表的ジアルコールとしてはビスフェノールAが挙げられる。
【0031】
上記化学は、スキーム1Bに示すようにマルチアルコール(multi-alcohol)にも拡大することができる。スキーム1Bにおいて、フルオロナノチューブ1を、マルチアルコール2fをMOH[式中、MはLi、NaまたはKのいずれかと同等でる]と反応させることにより生じるマルチアルコールR(OH)nの一金属塩と反応させると、官能基化された生成物3fが得られる。このように、上記記載は、フルオロナノチューブと一般式MOR(OH)n−1の一金属塩のいずれかとの反応に拡大することができる。Rは同様に、官能基化する部分の主鎖として働くことができる任意の炭化水素または他の有機鎖および/もしくは環構造単位である。
【化1】
【0032】
いくつかの態様では、フルオロナノチューブを最初にジアルコールまたはマルチアルコールに分散して分散液を形成させる。その後、金属水酸化物を同一または異なるジアルコールもしくはマルチアルコールに溶解して溶液を形成させ、その後溶液と分散液を組み合わせて混合物を形成させる。上記のように、超音波処理を利用すると、分散液の形性および/または混合工程を促進することができる。
【0033】
いくつかの態様では、フルオロナノチューブをアミノアルコール、例えばタイプH2N−R−OHのものと反応させ、ここにおいて、−N(H)−R−OHがナノチューブ上の−Fと置換し、フッ素はHFとして離れる。一般に、そのような態様では、フルオロナノチューブを適切なアミノアルコールに分散して反応混合物を形成させ;ピリジン触媒を反応混合物に加え;そして反応混合物+触媒をそのまま反応させて、アミノ(アミン)を末端基とする部分を伴う官能基化カーボンナノチューブを形成させる。いくつかの態様では、超音波処理を用いて、フルオロナノチューブの分散を促進し、混合を誘導する。これらまたは他の態様では、他の混合操作を利用してもよい。反応は一般に、約1時間〜約5時間にわたり約70℃〜約150℃の温度で行われる。
【0034】
アミノアルコールは、任意のアミノアルコールであって、フルオロナノチューブをその中に分散させることができ、フルオロナノチューブが適切な条件下でそれと反応するものであることができる。代表的アミノアルコールを利用するいくつかの代表的化学経路をスキーム2に示す。スキーム2において、フルオロナノチューブ1をアミノアルコール2g〜Iと反応させると、アミノを末端基とする部分が側壁に付着している官能基化カーボンナノチューブ3g〜Iが形成する。
【化2】
【0035】
いくつかの態様では、本発明の方法を、少なくとも部分的に、不活性雰囲気中で実施する。そのような不活性雰囲気としてはAr、Kr、He、Ne、N2、CF4およびそれらの組合わせが挙げられるが、これらに限定されない。
【0036】
上記方法によりヒドロキシ−ナノチューブ生成物が得られる。いくつかの態様において、ヒドロキシ−ナノチューブ生成物は、一般式CNT−[OR(OH)m]x[式中、Rは適切な有機主鎖であり、mは少なくとも1であり、そしてxはナノチューブ炭素原子1000個あたり約1〜約500である]を有する。他の態様において、ヒドロキシ−ナノチューブ生成物は、一般式CNT−[N(Y)R(OH)m]x[式中、Rは適切な有機主鎖であり、Yは水素または他の有機種であり、mは少なくとも1であり、そしてxはナノチューブ炭素原子1000個あたり約1〜約500である]を有する。
【0037】
いくつかの態様では上記化学の変形を用い、そのような態様では、チオール基−SHがジアルコール中の−OH基の一方もしくは両方、および/またはアミノアルコール中の−OH基と置き換わる。
【0038】
理論に拘束される意図はないが、最近のDFTの計算[Kudin,K.N.;Bettinger,H.F.;Scusseria,G.E.Phys.Rev.B,2001,63,45413]は、フルオロナノチューブが裸のカーボンナノチューブより優れた電子受容体であり、したがって強い求核試薬と容易に相互作用しうることを示唆している。これらの反応はまた、フルオロナノチューブ中の弱くなったC−F結合(フッ化アルキルと比較して)により促進され、したがってフッ素がより容易に置換されることが可能になる。アルコールへのフルオロナノチューブの溶解性は、アルコキシドとの反応によりそれらを官能基化する試みを促してきた。本研究に先立ち立証されているこの反応の一例において、ナトリウムメトキシドのメタノール溶液中でフルオロナノチューブ(ほぼC2F)を2時間にわたり音波処理することにより、C4.4F(OCH3)0.25の化学量論を伴い側壁がメトキシル化されたSWNTが生じることが示された。赤外分光学的データおよび可変温度(variable temperature)−質量分析(VTP−MS)データ、ならびに電子マイクロプローブ分析からの高い酸素含有量により、フルオロナノチューブ中のフッ素の部分的置換およびナノチューブ側壁へのメトキシ基の結合が裏付けられた。Mickelson,E.T.Novel Chemistry of Elemental Carbon:Graphite,Fullerenes and Nanotubes,Ph.D.Thesis,Rice大学,Houston,TX,1999;Mickelson,E.T.;Chiang,I.W.;Zimmerman,J.L.;Boul,P.J.;Lozano,J.;Liu,J.;Smalley,R.E.;Hauge,R.H.;Margrave,J.L.J.Phys.Chem.B,1999,103,4318参照。しかしながら、アルコール(メタノール、エタノール、イソ−プロパノール、エタンジオールおよびグリセロール)中でのフルオロナノチューブの音波処理または還流だけではフッ素の著しい置換または脱離は生じないことに留意することが重要である。Shukla,R.;McClain,B.;Khabashesku,V.N.;Margrave,J.L.Rice Quantum Institute 15th Annual Summer Research Colloquium.2001年8月17日、要旨19頁参照。したがって、アルコール種(すなわちジオールおよびグリセロール)を溶媒および試薬の両方として用いると、アルカリ塩基との反応によりヒドロキシルを末端基とするモノアルコキシドを過剰に得ることができる(スキーム1)。
【0039】
これまでの研究において、末端ジアミン、例えばH2N(CH2)nNH2(n=2、3、4、6)はフルオロナノチューブを溶解することができ、高温(90〜150℃)において触媒的量のピリジン存在下でそれらと化学的に反応することが立証されている。該反応はフッ素のほぼ完全な除去および置換をもたらし、側壁への直接的C−N結合付着を作り出すことによりアミノ基を末端基とする官能基化SWNTを生じさせた。Stevens,J.L.;Kiny,V.U.;Huang,A.Y.;Chiang,I.W.;Derrien,G.A.;Khabashesku,V.N.;Margrave,J.L.Proc.NanoTech 2003,第3巻,169−172;Huang,A.Y.;Chiang,I.W.;Khabashesku,V.N.;Margrave,J.L.Rice Quantum Institute 15th Annual Summer Research Colloquium.2001年8月17日、要旨18頁;Stevens,J.L.;Huang,A.Y.;Peng,H.;Chiang,I.W.;Khabashesku,V.N.;Margrave,J.L.NanoLett.2003,3,331;および2003年11月14日提出の同一に譲渡された米国特許出願第10/714187号参照。
【0040】
ヒドロキシルを末端基とする部分で官能基化されたそのようなナノチューブの用途は広範囲であるが、多くは、極性溶媒中でのその高い分散能力および/または溶解性、ならびに末端ヒドロキシル基で達成することができるさらなる官能基化を十分に利用するものであろうことは疑いない。一例として、カーボンナノチューブ上にありヒドロキシルを末端基とする部分をエピクロロヒドリンと反応させると、側壁にエポキシド基が付着しているカーボンナノチューブを得ることができる。これらエポキシドで官能基化されたカーボンナノチューブをエポキシ樹脂と混合し、適切な硬化剤で硬化すると、カーボンナノチューブ−エポキシ複合体を形成させることができる。
【0041】
上記のように、出願人らは、−OH基を末端基とする部分でカーボンナノチューブの側壁を官能基化するための好都合で効率的な方法を開発し、これを“ヒドロキシル−ナノチューブ”と名付けた。これらの官能基は、C−OまたはC−N共有結合(Cはナノチューブ固有の炭素である)のいずれかによりナノチューブの側壁に付着した。このような方法をスキーム1および2に例示する。この方法では、容易に追跡することができる穏やかな反応条件を利用している。このように調製した官能基化カーボンナノチューブの用途は、水素結合能力および側鎖中の末端ヒドロキシル基の化学的反応性に基づくことができる。OH基の化学は非常に富んでいるので、ヒドロキシルナノチューブを用いると、共有結合的に統合されたナノチューブ−強化コポリマーおよびセラミックスならびに生体適合材料を生産することができる。
【0042】
以下の実施例を、本発明の態様のいくつかをより完全に例示するために提供する。以下の実施例で開示する技術は、本発明の実施において十分に機能することが発明者らにより発見され、したがってその実施について代表的なモードを構成すると考えることができる技術を表している。しかしながら、本開示の観点から、当業者は、開示した具体的態様に多くの変更を加えることができ、本発明の精神および範囲から逸脱することなく同様または類似の結果をなお得ることができることを、理解すべきである。
実施例1
【0043】
この実施例は、用いることができる材料のタイプと、本発明のいくつかの態様で使用するためにあるタイプのフルオロナノチューブをどのように調製することができるかを例示するのに役立つ。この実施例ではSWNTを用いたが、他のタイプのCNTを用いてフルオロナノチューブを作成してもよいことに留意されたい。
【0044】
この実施例では、Rice大学のCarbon Nanotechnology研究所でHiPco法により調製された粗製SWNTを、先に記載したように完全に精製して鉄および他の不純物を除去した。Chiang,I.W.;Brinson,B.E.;Huang,A.Y.;Willis,P.A.;Bronikowski,M.J.;Margrave,J.L.;Smalley,R.E.;Hauge,R.H.J.Phys.Chem.B,2001,105,8297参照。精製後、SWNT中の鉄含有量は1重量%を超えていなかった。Carbon Nanotechnologies Inc.,Houston,TXにより供給されるもののような粉末形の精製SWNTを用いることもできる。この実施例では、ほぼC2.5Fの化学量論のフルオロナノチューブ1を、われわれのグループが以前報告した手順に従って、150℃で精製SWNTを直接フッ素化することにより調製した。Mickelson E.T.;Huffman,C.B.;Rinzler,A.G.;Smalley,R.E.;Hauge,R.H.;Margrave,J.L.Chem.Phys.Lett.1998,296,188参照。他のすべての化学物質、例えば、ヒドロキシル−ナノチューブを生産するためのさらなる加工工程で用いられるアルコール2a〜fおよびアミノアルコール2g〜iは、Aldrich Chemical Co.,Milwaukee,WIから購入した。
実施例2
【0045】
この実施例は、スキーム1に対応する本発明の方法のための合成手順を例示するのに役立つ。
【0046】
この方法(スキーム1)によるヒドロキシル−ナノチューブの調製では、10〜15mgのフルオロナノチューブ1を10mLの対応するジオールまたはトリオール2a〜fと一緒にバイアルに入れ、完全な分散液を達成するために80〜90℃で30分間音波処理(17W/55kHz Cole Palmer製浴)した。別のバイアルにおいて、60〜80mgのLiOH(またはNaOHまたはKOH)を、10mLの対応するアルカノール中で完全に溶解するまで30分間音波処理した。ジオール2a〜hの場合この手順を室温で実施したが、より粘性であるグリセロール2fの場合、高温(80〜90℃)での音波処理が必要であった。次の工程で、両バイアルからの溶液を組合わせ、得られた混合物を約1時間音波処理した。その後、反応混合物を孔径1ミクロンのCole Palmer製TEFLON膜に通して濾過し、大量のエタノールおよび水で洗浄して、LiF(またはNaFまたはKF)およびLiOH(またはNaOHまたはKOH)副生物の完全な除去を確実にした。ヒドロキシル−ナノチューブ3a〜fの黒色フィルムとして膜に密着している沈殿生成物を剥がし、70℃の真空オーブンで一晩乾燥した。エネルギー分散型X線分析(EDAX)元素分析は、3a〜f誘導体の試料中の残留フッ素含有量が3〜5at.%であることを示した。
実施例3
【0047】
この実施例は、スキーム2に対応する本発明の方法のための合成手順を例示するのに役立つ。
【0048】
この実施例(スキーム2)では、フルオロナノチューブ1(10〜15mg)を30mLのアミノアルコール2g〜i中で3分間音波処理した。これによりフルオロナノチューブの完全な分散液が得られ、黒色溶液が形成した。その後、5滴のピリジン(Py)を触媒として溶液に加え、反応混合物を窒素雰囲気下、80〜90℃で3時間撹拌した。その後、反応混合物を大量のエタノールと一緒に孔径1ミクロンのCole Palmer製TEFLON膜に通して濾過し、未反応アミノアルコールおよび好ましくない反応副生物の完全な除去を確実にした。官能基化SWNT3g〜iをフィルター膜から取り外し、70℃の真空オーブンで一晩乾燥した。EDAX分析は、3g〜i中の残留フッ素含有量が11〜13at.%であることを示した。
実施例4
【0049】
この実施例は、上記方法により生産された生成物の特性をどのように決定することができるかを例示するのに役立つ。
【0050】
ラマン分光法、減衰全反射−フーリエ変換赤外(ATR−FTIR)分光法、紫外−可視−近赤外(UV−vis−NIR)分光法、熱重量分析/示差熱分析法(TGA/DTA)、走査型電子顕微鏡法/エネルギー分散型X線分析法(SEM/EDAX)、原子間力顕微鏡法(AFM)および透過型電子顕微鏡法(TEM)のすべてを、初期状態のSWNT、フルオロナノチューブ、ならびに実施例2および3で調製したヒドロキシル−ナノチューブ3a〜iの特性決定に使用した。標準的な顕微鏡用スライドの上面に置いた試料についてのラマンスペクトルは、AlGaAsダイオード780−nmレーザー源で操作するRenishaw 1000マイクロラマンシステムで収集した。ATR−FTIRスペクトル測定に関しては、ATRアクセサリーを備えるThermal Nicolet Nexus 870 FTIRシステムを利用した。UV−vis−NIR範囲中のスペクトルは、Shimadzu 3101 PC UV/vis/NIR分光計を用いて測定した。熱分解分析は、TA−SDT−2960 TGA/DTA分析計で実施した。走査型電子顕微鏡法(SEM)は、エネルギー分散型X線(EDAX)分析計を装備したPhillips XL−30電界放出電子顕微鏡を用いて30kVのビームエネルギーで実施した。モデル2570JV−Zスキャナーを備えるDigital Instruments MultiMode走査型プローブ顕微鏡(SPM)を、タッピングモード原子間力顕微鏡分析(AFM)に使用した。レース状炭素(lacey carbon)をコーティングした銅グリッド(サイズ200メッシュ)上に置いた試験片の透過型電子顕微鏡法(TEM)の写真像は、100kVの加速電圧で操作するJEOL JEM−2010電子顕微鏡を用いて得た。
a.光学的分光法
【0051】
ラマン分光法は、ナノチューブの共有結合的な側壁修飾の迅速な評価を提供する。実施例2および3のSWNT誘導体について収集したラマンスペクトルを、図1〜3に示す。1285〜1300cm−1領域におけるピークの観察は炭素のsp3状態に関連付けられており、付着した官能基によりナノチューブ側壁上のπ−電子の芳香族系が崩壊した証拠として通常用いられる。フルオロナノチューブ1のラマンスペクトル(図1A)において、1293cm−1で観察された高強度ピークは、実施例2および3で調製したすべての官能基化SWNTのうちsp3−混成側壁炭素の含有量がもっとも高い(約40%)ことを反映している。1におけるこの高度な側壁修飾は、初期状態のSWNTにおいて200〜260cm−1で見られるSWNTブリージング(breathing)モードのピークの完全な消失、ならびに裸のナノチューブでの1594cm−1から1での1584cm−1まで赤側にシフトしている接線(tangential)モードのピークのブロードニングおよび弱化を引き起こす。ヒドロキシル−ナノチューブ3a〜Iのラマンスペクトルで観察されるように、1287〜1293cm−1の範囲におけるSP3炭素のピークはしたがって、共有結合的官能基化を示す。フルオロナノチューブ1のラマンスペクトルに比べこれらのピークの相対的強度が低いということは、研究した反応(スキーム1、2)における1からのフッ素の側壁での脱離の進行(フッ素の置換に加えて)により説明することができ、この脱離はsp3炭素状態の数を減少させ、ナノチューブ側壁上のsp2−結合を部分的に復活させる。1とは異なり、200〜260cm−1でのブリージングモードのピークは3a〜iのスペクトルにおいて可視的になり、最も低い程度で官能基化されているSWNT誘導体3aおよび3dについてはより高い強度を示している(図1Bおよび2F)。このモードは、側壁に付着している基をより多く持つ誘導体3b、c、e〜iではより弱くなる−これらの基は、ナノチューブの放射状のブリージング振動を妨害すると思われる。Encyclopedia of Nanoscience and Nanotechnology、S.Nalwa編集、American Scientific Publishers、2004年、第1巻、849〜861頁のKhabashesku,V.N.;Margrave,J.L.Chemistry of Carbon Nanotubesおよびそれの参考文献;Khabashesku,V.N.;Billups,W.E.;Margrave,J.L.Acc.Chem.Res.,2002,35,1087;Bahr,J.L.;Tour,J.M.J.Mater.Chem.2002,12,1952参照。
【0052】
UV−vis−NIR分光法は、SWNT側壁の官能基化の他の分光学的精査として役立つ。この場合、電子構造の改変により、初期状態のナノチューブのスペクトルに通常観察されるvan Hove転移特性の喪失がもたらされる。この実施例では、この主張を、初期状態のSWNTのUV−vis−NIRスペクトルと、ジメチルホルムアミド(DMF)溶液中のフルオロナノチューブ1およびヒドロキシル−ナノチューブ3f、gについて測定したものとを、図4で比較することにより例示する。裸の(例えば初期状態の)SWNTとは異なり、van Hove特異点は、高度に官能基化された誘導体1のスペクトル中にまったく存在しない。3f、gで観察されるvan Hove特異点の強度における劇的な低下により、それらのUV−vis−NIRスペクトルは側壁が官能基化されたSWNTに典型的なものに見えるようになり、これにより化学修飾の発生の重要な証拠を提供する。Encyclopedia of Nanoscience and Nanotechnology、S.Nalwa編集、American Scientific Publishers、2004年、第1巻、849〜861頁のKhabashesku,V.N.;Margrave,J.L.Chemistry of Carbon Nanotubesおよびそれの参考文献;Khabashesku,V.N.;Billups,W.E.;Margrave,J.L.Acc.Chem.Res.,2002,35,1087;Bahr,J.L.;Tour,J.M.J.Mater.Chem.2002,12,1952;Stevens,J.L.;Kiny,V.U.;Huang,A.Y.;Chiang,I.W.;Derrien,G.A.;Khabashesku,V.N.;Margrave,J.L.Proc.NanoTech2003,第3巻,169−172参照。
【0053】
図5および6に示したATR−FTIRスペクトルを用いて、SWNTの側壁に共有結合していてヒドロキシル基を末端基とする部分を同定した。フルオロナノチューブ1中のC−F結合の伸縮に特徴的な約1140cm−1の強いピーク(図5A)は、ジオール、トリオールおよびアミノアルコールとの反応後に消失している。このピークは、ヒドロキシル−ナノチューブ3a〜iのスペクトルでは、ナノチューブ−O−CおよびC−OH単位のC−O結合の伸縮に起因する1020〜1070cm−1領域中のピークにより置き換わられた。3000〜3600cm−1の範囲における新しい非常に幅広いバンドはO−H伸縮に帰属するが、2800〜3000cm−1および1360〜1460cm−1領域中のピークは、それぞれC−H伸縮および変形モードに帰属する。誘導体3g,hおよび3i中のナノチューブ−N(H)−Cまたはナノチューブ−N(C)−C構造単位のC−N伸縮モードは、1120〜1210cm−1のスペクトル範囲で観察されており(図6A〜C)、これは、それぞれ第二級および第三級アミンでのC−Nモードに特徴的である。Lin−Vien,D.;Colthup,N.B.;Fatelley,W.G.;Grasselli,J.G.The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules;Academic Press Inc.:San Diego,CA,1991,299頁参照。1および3a〜i中の活性化C=C伸縮モードのピークは、1540〜1580cm−1領域で観察された。
b.熱分解の研究
【0054】
これらの研究はさらに、ナノチューブの共有結合的な側壁の誘導体化に関する証拠を提供した。試料3b、3fおよび3gについて得られたTGA−DTAデータプロット(図7〜9)は、微分プロット上で250℃に主要ピークを示している。そのような高温におけるこれらのピークの出現は、重量減少が、ナノチューブからの物理吸着種の脱着によるのではなく、OH基を末端基とする部分の分離およびフラグメンテーションによって引き起こされることを示している。これらのSWNT誘導体についてのDTAプロット上で約550℃において観察される第2のピークは、残留C−F結合の脱離に起因し[Khabashesku,V.N.;Billups,W.E.;Margrave,J.L.Acc Chem.Res.,2002,35,1087;Mickelson,E.T.Novel Chemistry of Elemental Carbon:Graphite,Fullerenes and Nanotubes,Ph.D.Thesis,Rice大学,Houston,TX,1999]、EDAXで測定した残留フッ素含有量と相関している。これらのピークの主要ではない特徴は、フルオロナノチューブ1中のC−F結合の大部分が、反応の過程(スキーム1、2)でOH基を末端基とする部分により効率的に置き換わられたことを示している。3b、3fおよび3gのTGAプロット上で250℃において観察される主要ピークは、それぞれ20%、35%および22%の平均重量減少を示している。この重量減少がOH基を末端基とする部分の脱離に起因すると仮定すると、これらの誘導体における側壁の官能基化の程度は、大体3bで1/25、3fで1/16、および3gで1/20と概算することができる。
【0055】
異なる方法により調製された(スキーム1、2)3bおよび3g誘導体のTGA後の残留材料について測定したラマンスペクトル(図1Dおよび3B)は、両方ともsp3炭素モードの強度における劇的な低下を示している。このデータは、ナノチューブ側壁からの官能基の分離が、他の共有結合的に官能基化されたSWNTにおける先の熱分解の観察結果に一致して起こることを示している。Encyclopedia of Nanoscience and Nanotechnology、S.Nalwa編集、American Scientific Publishers、2004年、第1巻、849〜861頁のKhabashesku,V.N.;Margrave,J.L.Chemistry of Carbon Nanotubesおよびそれの参考文献;Khabashesku,V.N.;Billups,W.E.;Margrave,J.L.Acc.Chem.Res.,2002,35,1087;Bahr,J.L.;Tour,J.M.J.Mater.Chem.2002,12,1952;Stevens,J.L.;Huang,A.Y.;Peng,H.;Chiang,I.W.;Khabashesku,V.N.;Margrave,J.L.NanoLett.2003,3,331;Peng,H.;Reverdy,P.;Khabashesku,V.N.;Margrave,J.L.Chem.Comm.2003,362;Peng,H.;Alemany,L.B.;Margrave,J.L.;Khabashesku、V.N.J.Am.Chem.Soc.2003,125,15174−15182参照。
【0056】
SWNT誘導体の熱分解中に発生する揮発性種は、可変温度熱分解−質量分析法(VTP−MS)により分析した。3a〜fについてVTP−MSにより得られたデータは、300〜550℃の温度範囲での真空条件下における付着基のフラグメンテーションを示しており、これは、質量スペクトルにおいて、3aおよび3d〜fではm/z44(C2H4O)、29(HCO)のピーク、ならびに3bおよび3cではそれぞれm/z58、57、56、55(C3H6O〜C3H2O)およびm/z72、71、70(C4H8O〜C4H6O)のピークの追加的群により検出される。誘導体3iにおける側壁のC−N結合基の存在は、250〜400℃の温度でのジエタノールアミンの分離に起因するm/z105の主要ピークの出現をもたらす。
c.顕微鏡分析
【0057】
TEMにより、ヒドロキシルナノチューブにおける側壁修飾の直接的画像化が可能になる。図10は、レース状炭素をコーティングした銅グリッド上に置いたグリセロールで官能基化したSWNT3f試験片のTEM像を示している。挿入図は、単一ナノチューブの“でこぼこのある”表面をはっきり示しており、これは、側壁上の炭素−炭素結合フラクションが、連結を形成している炭素でより短いsp2からより長いsp3状態へ、共有結合的に改変することに起因する。
【0058】
3f誘導体のAFM研究(図11)は、側壁の官能基化に起因する、初期状態のSWNTナノチューブと比較して著しく低減した束のサイズを示している。初期状態のSWNTは、直径で数十〜100ナノメートルの束に凝集することが知られている。3fの束の平均サイズは、直径でわずか3〜6nmであることが測定された。これらの束の内部において、個々のヒドロキシル−ナノチューブは、側鎖からの末端OH基により形成される水素結合によって一緒に連結していると考えられる。官能基化SWNTの束のバックボーンプロフィルのタッピングモードでの分析は、高さの平均が4.4nmであることを示している。非晶質炭素粒子不純物を含まないバックボーン区域に沿って測定した高さの差異(約0.8nm)は、図10の挿入図でのTEM像により示されるように、“伸縮した”様式でナノチューブ側壁に付着しているOCH2CH(OH)CH2OH鎖の大体の長さに関連していると思われる。
実施例5
【0059】
この実施例は、非官能基化CNTと比較して、ヒドロキシルを末端基とする部分で官能基化されたカーボンナノチューブが極性溶媒中で有する改善された分散能力または溶解性を例示するのに役立つ。
【0060】
実施例2および3で調製したすべてのヒドロキシル−ナノチューブSWNT誘導体は、初期状態のSWNTと比較して極性溶媒への改善された溶解性を示した(図12A)。もっとも安定な溶液はグリセロールで誘導したSWNT材料3fから得られた。これは、それらがナノチューブ側鎖中に最高含有量のヒドロキシル基を持つためと思われる。水への3fの溶解性(約40mg/L)は数日間安定であったが、3f濃度がより高い(約80mg/L)エタノール溶液では(図12B)、数カ月後でもわずかに沈殿が見られた。
【0061】
本明細書中で参照したすべての特許および出版物を、本明細書中で参考として援用する。上記構造、作用、および上記態様の操作のいくつかは、本発明を実施するのに必須ではなく、代表的態様または複数の態様を完結させるためにのみ説明中に挙げられていることは、理解されるであろう。これに加えて、上記の参照した特許および出版物に記載されている具体的な構造、作用および操作は、本発明と併せて実施することができるが、その実施に不可欠なものではないことは、理解されるであろう。したがって、本発明は、添付の特許請求の範囲により定義されるような本発明の精神および範囲から実質的に逸脱することなく、具体的に記載したこととは異なるように実施してもよいことを、理解すべきである。
【図面の簡単な説明】
【0062】
【図1】SWNT材料のラマンスペクトルを表す図である:フルオロナノチューブ1(A)、ヒドロキシル−ナノチューブ3a(B)、3b(C)、および3bのTGA後の残留物。
【図2】ヒドロキシル−ナノチューブのラマンスペクトルを表す図である:(A)3c、(B)3d、(C)3e、(D)3f。
【図3】SWNT材料のラマンスペクトルを表す図である:(A)3g、(B)3gのTGA後の残留物、(C)3h、(D)3I。
【図4】初期状態のSWNT(A)、フルオロナノチューブ1(B)、ならびにヒドロキシル−ナノチューブ3f(C)および3g(D)のUV−vis−NIRスペクトルを表す図である。
【図5】フルオロナノチューブ1(A)、ならびにヒドロキシル−ナノチューブ(B)3a、(C)3b、(D)3c、(E)3d、(F)3e、(G)3fのATR−FTIRスペクトルを表す図である。
【図6】ヒドロキシル−ナノチューブのATR−FTIRスペクトルを表す図である:(A)3g、(B)3h、(C)3I。
【図7】ヒドロキシル−ナノチューブ3bのTGA−DTAを表す図である。
【図8】ヒドロキシル−ナノチューブ3fのTGA−DTAを表す図である。
【図9】ヒドロキシル−ナノチューブ3gのTGA−DTAを表す図である。
【図10】ヒドロキシル−ナノチューブ3fの試験片のTEM像を表す図であり、ここにおいて、挿入図は単一の官能基化ナノチューブの拡大像を表す。
【図11】ヒドロキシル−ナノチューブ3fの束のバックボーンに沿ったAFM像および高さの分析を表す図であり、ここにおいて、矢印は、側壁の官能基化に起因する0.8nmの高さの差異を指している。
【図12】エタノール中のSWNT材料の分散液の写真を表す図である:(A)初期状態のSWNT、および(B)グリセロールで官能基化したSWNT3f。
【特許請求の範囲】
【請求項1】
以下の工程:
a)i)フッ素化カーボンナノチューブ;
ii)一定量の金属水酸化物種;および
iii)一定量のアルコール種、ここにおいて、該種は少なくとも2個のヒドロキシル基を含む、
を含む混合物を提供する工程;および
b)該混合物を反応させて、ヒドロキシルを末端基とする部分が側壁に付着している官能基化カーボンナノチューブを得る工程、
を含む方法。
【請求項2】
フッ素化カーボンナノチューブを、フッ素を、単層カーボンナノチューブ、多層カーボンナノチューブ、二層カーボンナノチューブ、バッキーチューブ、フラーレンチューブ、管状フラーレン、黒鉛フィブリル、およびそれらの組み合わせからなる群より選択されるカーボンナノチューブと接触させることを含む方法により作成する、請求項1に記載の方法。
【請求項3】
フッ素化カーボンナノチューブが約C1F0.01〜約C2Fの化学量論を有する、請求項1に記載の方法。
【請求項4】
金属水酸化物が、LiOH、NaOH、KOHおよびそれらの組み合わせからなる群より選択される、請求項1に記載の方法。
【請求項5】
アルコール種が一般式R(OH)n[式中、nは少なくとも2であり、Rは有機主鎖である]のものである、請求項1に記載の方法。
【請求項6】
金属水酸化物と少なくとも数種のアルコール種を反応させて、アルコール種の一金属塩MOR(OH)n−1を形成させる、請求項5に記載の方法。
【請求項7】
さらに、フッ素化カーボンナノチューブを一定量のアルコール種に分散させる工程を含む、請求項1に記載の方法。
【請求項8】
反応の工程が加熱を包含する、請求項1に記載の方法。
【請求項9】
反応の工程が混合を包含する、請求項1に記載の方法。
【請求項10】
反応の工程が超音波処理を包含する、請求項1に記載の方法。
【請求項11】
さらに、ヒドロキシルを末端基とする部分が側壁に付着している官能基化カーボンナノチューブを含む濾過生成物を収集するために濾過する工程を含む、請求項1に記載の方法。
【請求項12】
さらに、濾過生成物を洗浄および乾燥することを含む、請求項11に記載の方法。
【請求項13】
以下の工程:
a)i)フッ素化カーボンナノチューブ;
ii)該フッ素化カーボンナノチューブと混合されている一定量のアルコール種の金属塩、ここにおいて、該アルコール種は少なくとも2個のヒドロキシル基を含む、
を含む混合物を提供する工程;および
b)該混合物を反応させて、ヒドロキシルを末端基とする部分が側壁に付着している官能基化カーボンナノチューブを得る工程、
を含む方法。
【請求項14】
フッ素化カーボンナノチューブを、フッ素を、単層カーボンナノチューブ、多層カーボンナノチューブ、二層カーボンナノチューブ、バッキーチューブ、フラーレンチューブ、管状フラーレン、黒鉛フィブリル、およびそれらの組合わせからなる群より選択されるカーボンナノチューブと接触させることを含む方法により作成する、請求項13に記載の方法。
【請求項15】
フッ素化カーボンナノチューブが約C1F0.01〜約C2Fの化学量論を有する、請求項13に記載の方法。
【請求項16】
アルコール種が一般式R(OH)n[式中、nは少なくとも2であり、Rは有機主鎖である]のものである、請求項13に記載の方法。
【請求項17】
アルコール種の金属塩が一般式MOR(OH)n−1のものである、請求項16に記載の方法。
【請求項18】
さらに、フッ素化カーボンナノチューブを一定量のアルコール種に分散させる工程を含む、請求項1に記載の方法。
【請求項19】
以下の工程:
a)i)フッ素化カーボンナノチューブ;
ii)該フッ素化カーボンナノチューブと混合されている一定量のアルコール種の金属塩、ここにおいて、該アルコール種は少なくとも2個のヒドロキシル基を含む、
を含む混合物を提供する工程;および
b)該混合物を反応させて、ヒドロキシルを末端基とする部分が側壁に付着している官能基化カーボンナノチューブを得る工程、
を含む方法により作成される官能基化カーボンナノチューブ。
【請求項20】
フッ素化カーボンナノチューブを、フッ素を、単層カーボンナノチューブ、多層カーボンナノチューブ、二層カーボンナノチューブ、バッキーチューブ、フラーレンチューブ、管状フラーレン、黒鉛フィブリル、およびそれらの組合わせからなる群より選択されるカーボンナノチューブと接触させることを含む方法により作成する、請求項19に記載の官能基化カーボンナノチューブ。
【請求項21】
フッ素化カーボンナノチューブが約C1F0.01〜約C2Fの化学量論を有する、請求項19に記載の官能基化カーボンナノチューブ。
【請求項22】
アルコール種が一般式R(OH)n[式中、nは少なくとも2であり、Rは有機主鎖である]のものである、請求項19に記載の官能基化カーボンナノチューブ。
【請求項23】
アルコール種がビスフェノールAである、請求項22に記載の官能基化カーボンナノチューブ。
【請求項24】
アルコール種の金属塩が一般式MOR(OH)n−1のものである、請求項22に記載の官能基化カーボンナノチューブ。
【請求項25】
さらに、フッ素化カーボンナノチューブを一定量のアルコール種に分散させる工程を含む、請求項19に記載の官能基化カーボンナノチューブ。
【請求項26】
一般式CNT−[OR(OH)m]x[式中、Rは有機主鎖であり、mは少なくとも1であり、そしてxはナノチューブ炭素原子1000個あたり約1〜約500である]を有する、請求項19に記載の官能基化カーボンナノチューブ。
【請求項27】
官能基化カーボンナノチューブがその端において追加的に官能基化されている、請求項19に記載の官能基化カーボンナノチューブ。
【請求項28】
さらに、官能基化カーボンナノチューブをエピクロロヒドリンと反応させて、エポキシドを末端基とする部分で側壁において官能基化されているカーボンナノチューブを形成させる、請求項23に記載の官能基化カーボンナノチューブ。
【請求項29】
複数のカーボンナノチューブを含む官能基化カーボンナノチューブであって、該複数のカーボンナノチューブが、該カーボンナノチューブの側壁に付着している官能基を有し、
a)官能基が形−OR(OH)mのものであり;
b)Rが有機主鎖であり;そして
c)mが少なくとも1である、
前記官能基化カーボンナノチューブ。
【請求項30】
カーボンナノチューブの側壁に付着している官能基がナノチューブ炭素原子1000個あたり約1〜約500個である、請求項29に記載の官能基化カーボンナノチューブ。
【請求項31】
官能基化カーボンナノチューブがその端において官能基で追加的に官能基化されている、請求項29に記載の官能基化カーボンナノチューブ。
【請求項32】
複数のカーボンナノチューブを含む官能基化カーボンナノチューブであって、該複数のカーボンナノチューブが、該カーボンナノチューブの側壁に付着しているエポキシド部分を含む官能基を有し、
a)官能基が形
【化1】
のものであり;そして
b)Rが有機主鎖である、
前記官能基化カーボンナノチューブ。
【請求項33】
カーボンナノチューブの側壁に付着している官能基がナノチューブ炭素原子1000個あたり約1〜約500個である、請求項32に記載の官能基化カーボンナノチューブ。
【請求項1】
以下の工程:
a)i)フッ素化カーボンナノチューブ;
ii)一定量の金属水酸化物種;および
iii)一定量のアルコール種、ここにおいて、該種は少なくとも2個のヒドロキシル基を含む、
を含む混合物を提供する工程;および
b)該混合物を反応させて、ヒドロキシルを末端基とする部分が側壁に付着している官能基化カーボンナノチューブを得る工程、
を含む方法。
【請求項2】
フッ素化カーボンナノチューブを、フッ素を、単層カーボンナノチューブ、多層カーボンナノチューブ、二層カーボンナノチューブ、バッキーチューブ、フラーレンチューブ、管状フラーレン、黒鉛フィブリル、およびそれらの組み合わせからなる群より選択されるカーボンナノチューブと接触させることを含む方法により作成する、請求項1に記載の方法。
【請求項3】
フッ素化カーボンナノチューブが約C1F0.01〜約C2Fの化学量論を有する、請求項1に記載の方法。
【請求項4】
金属水酸化物が、LiOH、NaOH、KOHおよびそれらの組み合わせからなる群より選択される、請求項1に記載の方法。
【請求項5】
アルコール種が一般式R(OH)n[式中、nは少なくとも2であり、Rは有機主鎖である]のものである、請求項1に記載の方法。
【請求項6】
金属水酸化物と少なくとも数種のアルコール種を反応させて、アルコール種の一金属塩MOR(OH)n−1を形成させる、請求項5に記載の方法。
【請求項7】
さらに、フッ素化カーボンナノチューブを一定量のアルコール種に分散させる工程を含む、請求項1に記載の方法。
【請求項8】
反応の工程が加熱を包含する、請求項1に記載の方法。
【請求項9】
反応の工程が混合を包含する、請求項1に記載の方法。
【請求項10】
反応の工程が超音波処理を包含する、請求項1に記載の方法。
【請求項11】
さらに、ヒドロキシルを末端基とする部分が側壁に付着している官能基化カーボンナノチューブを含む濾過生成物を収集するために濾過する工程を含む、請求項1に記載の方法。
【請求項12】
さらに、濾過生成物を洗浄および乾燥することを含む、請求項11に記載の方法。
【請求項13】
以下の工程:
a)i)フッ素化カーボンナノチューブ;
ii)該フッ素化カーボンナノチューブと混合されている一定量のアルコール種の金属塩、ここにおいて、該アルコール種は少なくとも2個のヒドロキシル基を含む、
を含む混合物を提供する工程;および
b)該混合物を反応させて、ヒドロキシルを末端基とする部分が側壁に付着している官能基化カーボンナノチューブを得る工程、
を含む方法。
【請求項14】
フッ素化カーボンナノチューブを、フッ素を、単層カーボンナノチューブ、多層カーボンナノチューブ、二層カーボンナノチューブ、バッキーチューブ、フラーレンチューブ、管状フラーレン、黒鉛フィブリル、およびそれらの組合わせからなる群より選択されるカーボンナノチューブと接触させることを含む方法により作成する、請求項13に記載の方法。
【請求項15】
フッ素化カーボンナノチューブが約C1F0.01〜約C2Fの化学量論を有する、請求項13に記載の方法。
【請求項16】
アルコール種が一般式R(OH)n[式中、nは少なくとも2であり、Rは有機主鎖である]のものである、請求項13に記載の方法。
【請求項17】
アルコール種の金属塩が一般式MOR(OH)n−1のものである、請求項16に記載の方法。
【請求項18】
さらに、フッ素化カーボンナノチューブを一定量のアルコール種に分散させる工程を含む、請求項1に記載の方法。
【請求項19】
以下の工程:
a)i)フッ素化カーボンナノチューブ;
ii)該フッ素化カーボンナノチューブと混合されている一定量のアルコール種の金属塩、ここにおいて、該アルコール種は少なくとも2個のヒドロキシル基を含む、
を含む混合物を提供する工程;および
b)該混合物を反応させて、ヒドロキシルを末端基とする部分が側壁に付着している官能基化カーボンナノチューブを得る工程、
を含む方法により作成される官能基化カーボンナノチューブ。
【請求項20】
フッ素化カーボンナノチューブを、フッ素を、単層カーボンナノチューブ、多層カーボンナノチューブ、二層カーボンナノチューブ、バッキーチューブ、フラーレンチューブ、管状フラーレン、黒鉛フィブリル、およびそれらの組合わせからなる群より選択されるカーボンナノチューブと接触させることを含む方法により作成する、請求項19に記載の官能基化カーボンナノチューブ。
【請求項21】
フッ素化カーボンナノチューブが約C1F0.01〜約C2Fの化学量論を有する、請求項19に記載の官能基化カーボンナノチューブ。
【請求項22】
アルコール種が一般式R(OH)n[式中、nは少なくとも2であり、Rは有機主鎖である]のものである、請求項19に記載の官能基化カーボンナノチューブ。
【請求項23】
アルコール種がビスフェノールAである、請求項22に記載の官能基化カーボンナノチューブ。
【請求項24】
アルコール種の金属塩が一般式MOR(OH)n−1のものである、請求項22に記載の官能基化カーボンナノチューブ。
【請求項25】
さらに、フッ素化カーボンナノチューブを一定量のアルコール種に分散させる工程を含む、請求項19に記載の官能基化カーボンナノチューブ。
【請求項26】
一般式CNT−[OR(OH)m]x[式中、Rは有機主鎖であり、mは少なくとも1であり、そしてxはナノチューブ炭素原子1000個あたり約1〜約500である]を有する、請求項19に記載の官能基化カーボンナノチューブ。
【請求項27】
官能基化カーボンナノチューブがその端において追加的に官能基化されている、請求項19に記載の官能基化カーボンナノチューブ。
【請求項28】
さらに、官能基化カーボンナノチューブをエピクロロヒドリンと反応させて、エポキシドを末端基とする部分で側壁において官能基化されているカーボンナノチューブを形成させる、請求項23に記載の官能基化カーボンナノチューブ。
【請求項29】
複数のカーボンナノチューブを含む官能基化カーボンナノチューブであって、該複数のカーボンナノチューブが、該カーボンナノチューブの側壁に付着している官能基を有し、
a)官能基が形−OR(OH)mのものであり;
b)Rが有機主鎖であり;そして
c)mが少なくとも1である、
前記官能基化カーボンナノチューブ。
【請求項30】
カーボンナノチューブの側壁に付着している官能基がナノチューブ炭素原子1000個あたり約1〜約500個である、請求項29に記載の官能基化カーボンナノチューブ。
【請求項31】
官能基化カーボンナノチューブがその端において官能基で追加的に官能基化されている、請求項29に記載の官能基化カーボンナノチューブ。
【請求項32】
複数のカーボンナノチューブを含む官能基化カーボンナノチューブであって、該複数のカーボンナノチューブが、該カーボンナノチューブの側壁に付着しているエポキシド部分を含む官能基を有し、
a)官能基が形
【化1】
のものであり;そして
b)Rが有機主鎖である、
前記官能基化カーボンナノチューブ。
【請求項33】
カーボンナノチューブの側壁に付着している官能基がナノチューブ炭素原子1000個あたり約1〜約500個である、請求項32に記載の官能基化カーボンナノチューブ。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公表番号】特表2007−523818(P2007−523818A)
【公表日】平成19年8月23日(2007.8.23)
【国際特許分類】
【出願番号】特願2006−517284(P2006−517284)
【出願日】平成16年6月16日(2004.6.16)
【国際出願番号】PCT/US2004/019015
【国際公開番号】WO2005/028740
【国際公開日】平成17年3月31日(2005.3.31)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.TEFLON
【出願人】(501105635)ウィリアム・マーシュ・ライス・ユニバーシティ (26)
【Fターム(参考)】
【公表日】平成19年8月23日(2007.8.23)
【国際特許分類】
【出願日】平成16年6月16日(2004.6.16)
【国際出願番号】PCT/US2004/019015
【国際公開番号】WO2005/028740
【国際公開日】平成17年3月31日(2005.3.31)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.TEFLON
【出願人】(501105635)ウィリアム・マーシュ・ライス・ユニバーシティ (26)
【Fターム(参考)】
[ Back to top ]