
ヒドロコドンとナルトレキソンとの併用医薬
約5〜約20 mgのヒドロコドンまたはその製薬上許容可能な塩と0.055〜約0.56 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物が開示されている。
【発明の詳細な説明】
【技術分野】
【0001】
ヒドロコドン製剤は乱用の対象となることがある。ヒドロコドンは特定量を非経口投与した場合、同量を経口投与するよりも効力が高い場合がある。経口ヒドロコドン製剤の乱用の1形態ドは活性剤を溶液中に入れて注射することを含む。
【背景技術】
【0002】
従来、これらの薬物の非経口乱用を阻止するために、オピオイドアンタゴニストが特定のオピオイドアゴニストと組み合わされてきた。
【0003】
即時放出型ペンタゾシンとナロキソンとの組み合わせが米国で入手可能なタブレットとして利用されており、Sanofi-WinthropからTalwin(登録商標)Nxとして市販されている。Talwin(登録商標)Nxは、50 mg基剤と同等の即時放出型塩酸ペンタゾシンと、0.5 mg基剤と同等の塩酸ナロキソンとを含む。ドイツでは1978年から、疼痛を管理するためにチリジン(50 mg)とナロキソン(4 mg)とを含む固定併用治療が利用可能である(Valoron(登録商標)N, Goedecke)。ニュージーランドでは1991年に、疼痛治療用にブプレノルフィンとナロキソンとの固定併用が導入された(Temgesic(登録商標)Nx, Reckitt & Colman)。
【0004】
Kreekの米国特許第4,769,372号および第4,785,000号は、慢性疼痛または慢性咳に苦しむ患者に対し、腸運動障害を引き起こすことなく治療する方法を記載している。上記方法は、約1.5〜約100 mgのオピオイド鎮痛薬または鎮咳薬と、経口投与した場合に全身性アンタゴニスト活性をほとんど又は全く有しない約1〜約18 mgのオピオイドアンタゴニストとを含む投与単位を1〜2だけ、毎日1〜5回投与することを含む。
【0005】
Crainらの米国特許第5,472,943号は、オピオイドアンタゴニストを含むアゴニストを投与することにより2モードで作用するオピオイドアゴニストの鎮痛性を高める方法を記載している。
【0006】
ヒドロコドンはアセトアミノフェンとの組み合わせで市販されており、疼痛治療に適応される。市販されている商品名は、MallinckrodtのAnexsia(登録商標)、UCB PharmaのLortab(登録商標)、Watson PharmaceuticalsのNorco(登録商標)、AbbottのVicodin(登録商標)、およびEndo LabsのZydone(登録商標)である。
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明の目的及び要旨
本発明の目的は、ヒドロコドンの経口剤形を提供することである。
【0008】
本発明の特定の実施形態の目的は、他の剤形に比べて非経口および/または経口乱用されにくいヒドロコドンの経口剤形を提供することである。
【0009】
本発明の特定の実施形態の目的は、他の剤形に比べて転用されにくいヒドロコドンの経口剤形を提供することである。
【0010】
本発明の特定の実施形態の目的は、ヒドロコドンの経口剤形を用いて、剤形の乱用可能性を低減しながらヒト患者の疼痛を治療する方法を提供することである。
【0011】
本発明の特定の実施形態の目的は、乱用可能性の低い、ヒドロコドンの経口剤形を製造する方法を提供することである。
【課題を解決するための手段】
【0012】
上記および他の目的は以下に述べる本発明によって達成される。本発明は一つには、5〜20 mgのヒドロコドンまたはその製薬上許容可能な塩と、0.055〜0.56 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物であって、前記ナルトレキソンまたはその製薬上許容可能な塩と前記ヒドロコドンまたはその製薬上許容可能な塩とが0.011:1〜0.028:1の割合で含まれる医薬組成物を対象とする。
【0013】
特定の実施形態では、本発明は、約5 mgのヒドロコドンまたはその製薬上許容可能な塩と0.055〜0.14 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物を対象とする。
【0014】
特定の実施形態では、本発明は、約7.5 mgのヒドロコドンまたはその製薬上許容可能な塩と0.0825〜0.21 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物を対象とする。
【0015】
特定の実施形態では、本発明は、約10 mgのヒドロコドンまたはその製薬上許容可能な塩と0.11〜0.28 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物を対象とする。
【0016】
特定の実施形態では、本発明は、約15 mgのヒドロコドンまたはその製薬上許容可能な塩と0.165〜0.42 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物を対象とする。
【0017】
特定の実施形態では、本発明は、約20 mgのヒドロコドンまたはその製薬上許容可能な塩と0.22〜0.56 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物を対象とする。
【0018】
本明細書に開示する本発明の特定の実施形態では、剤形が前記ヒドロコドン、前記ナルトレキソンを徐放するか、または両方を徐放する。
【0019】
本明細書に開示する本発明の特定の実施形態では、剤形が、ヒト患者への定常状態での経口投与後少なくとも12時間に亘って有効に鎮痛を行う。
【0020】
本明細書に開示する本発明の特定の実施形態では、剤形が、ヒト患者への定常状態での経口投与後少なくとも24時間に亘って有効に鎮痛を行う。
【0021】
本明細書に開示する本発明の特定の実施形態では、剤形が、ヒドロコドンまたはその製薬上許容可能な塩と、ナルトレキソンまたはその製薬上許容可能な塩とを含むマトリクスを含み、前記ヒドロコドンまたはその製薬上許容可能な塩および前記ナルトレキソンまたはその製薬上許容可能な塩の両方が徐放性賦形剤中で実質的に互いに分散している。
【0022】
特定の実施形態では、本発明は、ヒドロコドン製剤の経口乱用の可能性を低減する方法であって、本明細書に記載の組成物を調製することを含む方法を対象とする。
【0023】
特定の実施形態では、本発明は、ヒト患者において疼痛を治療する方法であって、患者への定常状態での経口投与後少なくとも12時間に亘って有効に鎮痛を行う、本明細書に記載の医薬組成物を経口投与することを含む方法を対象とする。
【0024】
特定の実施形態では、本発明は、ヒト患者において疼痛を治療する方法であって、患者への定常状態での経口投与後少なくとも24時間に亘って有効に鎮痛を行う、本明細書に記載の医薬組成物を経口投与することを含む方法を対象とする。
【0025】
用語「徐放」は本発明の目的に関して、ヒドロコドンまたはその塩が、血液(例えば血漿)濃度(レベル)が、8〜24時間の期間に亘って、好ましくは1日2回または1日1回の処方を示す期間に亘って、治療範囲内(最小有効鎮痛濃度または「MEAC」より高い)であって且つ毒性レベル未満に維持されるような速度で剤形から放出されることと定義される。
【0026】
用語「非経口」は本明細書において、皮下注射、静脈注射、筋肉内注射、胸骨間注射、点滴技術または当該分野で公知の他の注入方法を含む。
【0027】
用語「ヒドロコドン」は他に特定しない限り、ヒドロコドン塩基を意味する。用語「ナルトレキソン」は他に特定しない限り、ナルトレキソン塩基を意味する。用語「塩」は製薬上許容可能な塩を意味する。
【0028】
用語「定常状態」は、系に到達する薬物の量がその系から出る薬物の量とほぼ等しいことを意味する。従って「定常状態」において、患者の身体は、薬物が血流に吸収されることにより患者の系に使用可能になるのとほぼ同じ速度で薬物を排除している。
【発明を実施するための最良の形態】
【0029】
発明の詳細な説明
本発明の剤形は、約5〜約20 mgのヒドロコドンまたはその製薬上許容可能な塩を含む。ヒドロコドンまたはその塩の特に好ましい投与量は約5 mg、約7.5 mg、約10 mg、約15 mgおよび約20 mgである。特定の実施形態では、ヒドロコドンまたはその塩を適切な製薬上許容可能な賦形剤で処方することにより、ヒドロコドンの徐放を実現する。
【0030】
本発明の剤形は約0.055〜約0.56 mgのナルトレキソンまたはその製薬上許容可能な塩を含む。ナルトレキソンまたはその塩の特に好ましい投与量は約0.0625 mg、約0.09375 mg、約0.125 mg、約0.1875 mgおよび約0.25 mgである。
【0031】
ヒドロコドンまたはその塩およびナルトレキソンまたはその塩は、一方または両方の剤を即時放出するように処方してもよいし、一方または両方の剤を徐放するように適切な製薬上許容可能な賦形剤と組み合わせてもよい。ナルトレキソンまたはその塩の徐放率は、ヒドロコドンまたはその塩の徐放率と同じでもよいし、異なっていてもよい。本発明の特に好ましい実施形態による剤形は、約5 mgのヒドロコドン塩と約0.0625 mgのナルトレキソン塩、約7.5 mgのヒドロコドン塩と約0.09375 mgのナルトレキソン塩、約10 mgのヒドロコドン塩と約0.125 mgのナルトレキソン塩、約15 mgのヒドロコドン塩と約0.1875 mgのナルトレキソン塩、および約20 mgのヒドロコドン塩と約0.25 mgのナルトレキソン塩を含む。ヒドロコドンの重酒石酸塩とナルトレキソンの塩酸塩が特に好ましい。
【0032】
本発明の特定の実施形態では、ナルトレキソンまたはその塩の開示された範囲は、身体的依存性被験者において、製剤の鼻腔内および非経口乱用を阻止するに十分な量であってよい。これは、製剤が、改ざんされ、そして鼻の粘膜に投与され、又は非経口に投与された場合に、ヒドロコドンのオピオイド効果を少なくとも部分的に阻止することにより実現される。好ましくはその量は、身体的依存性が最も高い個体に鼻腔内または非経口投与された結果、オピオイドの突然の中止後に見られる症状に酷似した、中程度から高度の離脱症状が現れるに十分な量である。離脱症状の最も一般的な症状は、瞳孔の拡張、悪寒と極度の発汗とが交互に現れること、腹部の痙攣、吐き気、嘔吐、筋肉の痙攣、過剰被刺激性、流涙、鼻漏、鳥肌、および心拍数の増加を含む。
【0033】
特定の実施形態では、剤形に安定剤を含むことにより、ナルトレキソンまたはその製薬上許容可能な塩の劣化を防止する。特定の実施形態では、剤形に用いられる安定剤は例えば、有機酸、カルボン酸、アミノ酸の酸塩(例えばシステイン、L-システイン、システインハイドロクロライド、グリシンハイドロクロライド、またはシステインジヒドロクロライド)、ソディウムメタビサルファイト(sodium metabisulphite)、アスコルビン酸およびその誘導体、リンゴ酸、イソアスコルビン酸、クエン酸、酒石酸、パルミチン酸、炭酸ナトリウム、炭酸水素ナトリウム、炭酸カルシウム、リン酸水素カルシウム、二酸化硫黄、亜硫酸ナトリウム、重硫酸ナトリウム(sodium bisulphate)、トコフェノール並びにその水溶性および油溶性誘導体(例えばトコフェルソラン(tocofersolan)または酢酸トコフェノール)、亜硫酸塩、重亜硫酸塩(bisulphite)および亜硫酸水素塩またはアルカリ金属、アルカリ土類金属および他の金属、PHBエステル、ガラート、ブチル化ヒドロキシアニソール(BHA)またはブチル化ヒドロキシトルエン(BHT)、および2,6-ジ-t-ブチル-.アルファ.-ジメチルアミノ-p-クレゾール、t-ブチルヒドロキノン、ジ-t-アミルヒドロキノン、ジ-t-ブチルヒドロキノン、ブチルヒドロキシトルエン、ブチルヒドロキシアニソール、ピロカテコール、ピロガロール、プロピル/ガラート、およびノルジヒドログアイアレチン酸(nordihydroguaiaretic acid)、並びに低級脂肪酸、フルーツ酸、リン酸、ソルビン酸および安息香酸並びにこれらの塩、エステル、誘導体および異性体化合物、パルミチン酸アスコルビル、レシチン、モノおよびポリヒドロキシル化ベンゼン誘導体、エチレンジアミン−四酢酸およびその塩、シトラコン酸、コニデンドリン(conidendrine)、炭酸ジエチル、メチレンジオキシフェノール、ケファリン(kephaline)、β,β'-ジチオプロピオン酸、ビフェニルおよび他のフェニル誘導体、これらの製薬上許容可能な塩およびこれらの混合物を含むがこれらに限られない。
【0034】
本発明の経口剤形はヒドロコドンとナルトレキソンとに加えてさらに、これらと共に相乗効果をもたらす又はもたらさない1以上の薬物を含んでもよい。したがって特定の実施形態では、製剤に非オピオイド薬物も含まれる。このような非オピオイド薬物は好ましくはさらに鎮痛をもたらし、例えばアスピリン、非ステロイド性抗炎症剤("NSAIDS";例えばイブプロフェン、ケトプロフェンなど)、N-メチル-D-アスパラギン酸(NMDA)レセプターアンタゴニスト(例えばモルフィナン(例えばデキストロメトルファンまたはデキストロファン)、またはケタミン)、シクロオキシゲナーゼ-II阻害剤("COX-II阻害剤")、および/またはグリシンレセプターアンタゴニストなどが含まれる。
【0035】
本発明の特定の好ましい実施形態は、本発明はさらに非オピオイド鎮痛剤を含むことにより、より低い投与量のヒドロコドンを使用することができる。非オピオイド鎮痛剤は例えば、NSAIDまたはCOX-2阻害剤である。より少ない量の一方または両方の薬物を使用することにより、ヒトにおいて有効な疼痛管理に関連して起こる副作用を低減することができる。
【0036】
適切な非ステロイド性抗炎症剤は、イブプロフェン、ジクロフェナク、ナプロキセン、ベノキサプロフェン、フルルビプロフェン、フェノプロフェン、フルブフェン(flubufen)、ケトプロフェン、インドプロフェン、ピロプロフェン、カルプロフェン、オキサプロジン、プラモプロフェン、ムロプロフェン、トリオキサプロフェン、スプロフェン、アミノプロフェン、チアプロフェン酸、フルプロフェン、ブクロキシン酸(bucloxic acid)、インドメタシン、スリンダク、トルメチン、ゾメピラク、チオピナク、ジドメタシン、アセメタシン、フェンチアザク、クリダナク、オキシピナク、メフェナム酸、メクロフェナム酸、フルフェナム酸、ニフルム酸、トルフェナム酸、ジフルリサル(diflurisal)、フルフェニサル(flufenisal)、ピロキシカム、スドキシカム、イソキシカム、これらの製薬上許容可能な塩、これらの混合物などを含む。これらの薬物の有用な投与量は当業者に周知である。
【0037】
N-メチル-D-アスパラギン酸(NMDA)レセプターアンタゴニストは当該分野で周知であり、例えばモルフィナン(例えばデキストロメトルファンまたはデキストロファン)、ケタミン、d-メタドン、およびこれらの製薬上許容可能な塩を含む。本発明の目的のために、用語「NMDAアンタゴニスト」は、さらにNMDAレセプター活性化の細胞内反応を阻止する薬物、例えばガングリオシド(例えばGM1またはGT1b)、フェノチアジン(例えばトリフルオペラジン)、またはナフタレンスルホンアミド(例えばN-(6-アミノテキシル(aminothexyl))-5-クロロ-1-ナフタレンスルホンアミド)を含むと解釈される。これらの薬物は、Mayerらの米国特許第5,321,012号および第5,556,838号において、習慣性薬物(例えば麻薬鎮痛剤:モルヒネ、コデインなど)に対する耐性および/または依存性の発生を阻害すると記載されている。これらの薬物はまた、Mayerらの米国特許第5,502,058号において、慢性疼痛を治療するとも記載されている。これらの米国特許はすべて参照により本明細書に組み込まれる。NMDAアンタゴニストは単体で含まれてもよいし、Mayerらの上記特許に記載されているように局部麻酔薬(例えばリドカイン)と共に含まれてもよい。
【0038】
グリシンレセプターアンタゴニストを用いて慢性疼痛を治療すること及びこれらの薬物の適応症(identification)は、Weberらの米国特許第5,514,680号に記載されている。
【0039】
COX-2阻害剤は当該分野で報告されており、シクロオキシゲナーゼ-2阻害を引き起こす多くの化学構造が知られている。COX-2阻害剤は例えば、米国特許第5,616,601号、第5,604,260号、第5,593,994号、第5,550,142号、第5,536,752号、第5,521,213号、第5,475,995号、第5,639,780号、第5,604,253号、第5,552,422号、第5,510,368号、第5,436,265号、第5,409,944号、および第5,130,311号に記載されている。これらの米国特許はすべて参照により本明細書に組み込まれる。特定の好ましいCOX-2阻害剤は、セレコシキブ、5-ブロモ-s-(4-フルオロフェニル)-3-[4-(メチルスルホニル)-フェニル]チオフェン、フロスリド(flosulide)、メロキシカム、ロフェコキシブ(rofecoxib)、6-メトキシ-2-ナフチル酢酸、ナブメトン、ニメスリド、N-[2-(シクロヘキシルオキシ)-4-ニトロフェニル]メタンスルホンアミド、1-フルオロ-4-[2-[4-(メチルスホニル)フェニル]-1-シクロペンテン-1-イル]ベンゼン、5-(4-フルオロフェニル)-1-[4-(メチルスホニル)フェニル]-3-トリフルオロメチル 1H-ピラゾール、N-[3-(ホルミルアミノ)-4-オキソ-6-フェノキシ-4H-1-ベンゾピラン-7-イル]メタンスルホンアミド、これらの混合物、およびこららの製薬上許容可能な塩を含む。COX-2阻害剤の投与レベルとしては、オピオイド鎮痛剤と組み合わせた場合、1日に体重1キロ当たり約0.005 mg〜約140 mgのオーダーが治療上有効である。あるいは1人の患者に対して1日に約0.25 mg〜約7 gのCOX-2阻害剤がオピオイド鎮痛剤と組み合わせて投与される。
【0040】
さらなる実施形態では、鎮痛剤以外の所望の効果を提供する非オピオイド薬物、例えば鎮咳剤、去痰薬、鬱血除去剤、抗ヒスタミン薬、局部麻酔薬などを含んでもよい。
【0041】
徐放性剤形
ヒドロコドン(またはヒドロコドン塩)および/またはナルトレキソン(またはナルトレキソン塩)は当業者に公知の任意の適切なタブレット、コーティング済みタブレットまたはマルチ微粒子製剤という形態で徐放性経口製剤として処方することができる。徐放性剤形は、マトリクスに含有させた徐放性材料をヒドロコドンまたはその塩と共に含むが、この場合ナルトレキソンまたはその塩を含んでもよいし含まなくともよい。例えば、ヒドロコドン塩を徐放性マトリクスに含有させ、ナルトレキソン塩をマトリクスとは別にしてもよいしマトリクスに含有させてもよい。
【0042】
特定の実施形態における徐放性剤形は、ヒドロコドンまたはその塩およびナルトレキソンまたはその塩の両方を含む1群の粒子を含んでもよい。他の実施形態では剤形は、ヒドロコドンまたはその塩を含む第1の粒子群とナルトレキソンまたはその塩を含む第2の粒子群とを含んでもよい。1または複数の粒子群を含む実施形態では、粒子の直径は約0.1 mm〜約2.5 mmであってよく、好ましくは約0.5 mm〜約2 mmであるとよい。上記に開示したように、ナルトレキソンまたはナルトレキソン塩は、ヒドロコドンまたはヒドロコドン塩を含む粒子に含有されてもよいし、別の粒子に含有されてもよい。あるいは、ヒドロコドンまたはヒドロコドン塩粒子を含むタブレットまたはカプセルに含有されてもよい。特定の実施形態では、粒子は水性媒質中において活性剤の放出をある徐放率で許容する徐放性材料でコーティングされる。コーティング剤は、記載された他の特性に加えて所望のインビトロ放出率を達成するように選択される。本発明の徐放性コーティング製剤は、平滑で外観が美しく、顔料および他のコーティング添加剤を支持することができ、非毒性、不活性で且つべたつきのない、強靱な連続薄膜を生成することができるべきである。
【0043】
コーティング済みビーズ
本発明の特定の実施形態では、疎水性材料を用いて、活性剤でコーティングされた不活性な薬剤ビーズ(例えばnu pariel 18/20ビーズ)にオーバーコーティングを施す。その後、得られた複数の固体徐放性ビーズを十分な量だけゼラチンカプセル中に入れることにより、周囲の液体(例えば胃液または溶解培地)に吸い込まれたり接触したりする際に有効な徐放量を提供するようにしてもよい。特定の実施形態では、ヒドロコドンまたはヒドロコドン塩を含む徐放性ビーズをナルトレキソンまたはナルトレキソン塩でさらにコーティングしてもよい。あるいはナルトレキソンまたはナルトレキソン塩を、徐放性ヒドロコドンまたはヒドロコドン塩ビーズと共に(例えば粉体混合物として、または別々のビーズに処方された形態で)カプセル中に入れてもよい。
【0044】
本発明の徐放性ビーズ製剤は、例えば胃液に、その後腸液に吸い込まれ曝されると、本発明の活性剤をゆっくりと放出する。本発明の製剤の徐放プロファイルは変更することができるが、その変更は例えばオーバーコーティングに用いる疎水性材料の量を変更すること、疎水性材料に可塑剤を添加する様式を変更すること、疎水性材料に対する可塑剤の量を変更すること、追加の成分または賦形剤を含ませること、製造方法を変更することなどによって行われる。最終生成品の溶解プロファイルもまた改変することができるが、その改変は例えば遅延剤コーティングの厚みを増減することによって行われる。
【0045】
本発明の活性剤でコーティングされたスフェロイドまたはビーズは例えば、活性剤を水に溶解し、次いでWusterインサートを用いて溶液を基質(例えばnu pariel 18/20ビーズ)にスプレーすることによって調製する。活性剤がビーズに結合することを補助するために、および/または溶液に色をつけるためなどに、ビーズをコーティングする前に追加の成分をさらに適宜添加する。例えばヒドロキシプロピルメチルセルロースなどを含み、色素を含むか含まない生成物(例えばColorcon, Inc.から市販されているOpadry(登録商標))を溶液に添加して混合(例えば約1時間)した後に得られた溶液を同じくビーズに塗ってもよい。その後得られたコーティング済み基質(この例ではビーズ)を緩衝剤(barrier agent)で適宜オーバーコーティングすることにより、活性剤を疎水性徐放性コーティング剤から分離してもよい。適切な緩衝剤の一例はヒドロキシプロピルメチルセルロースを含むものであるが、当該分野で公知のいずれの薄膜生成剤も用いることができる。緩衝剤は最終生成物の溶解率に影響を与えないことが好ましい。
【0046】
その後ビーズを疎水性材料の水分散液でオーバーコーティングしてもよい。疎水性材料の水分散液は好ましくはさらに有効量の可塑剤(例えばクエン酸トリエチル)を含む。エチルセルロースの予備処方された水分散液(例えばAquacoat(登録商標)またはSurelease(登録商標))を用いてもよい。Surelease(登録商標)を用いる場合、可塑剤を別途添加する必要はない。あるいはアクリル性ポリマーの予備処方された水分散液(例えばEudragit(登録商標))を用いることもできる。
【0047】
本発明のコーティング溶液は好ましくは薄膜生成剤、可塑剤および溶媒系(即ち水)に加えて、色素を含むことにより、美しく且つ他とは異なる外観を提供する。疎水性材料の水分散液に代えて又はこれに加えて、活性剤の溶液に色を付けてもよい。例えばアルコールまたはプロピレングリコールベースの色分散液、粉砕したアルミニウムレーキおよび乳白剤(例えば二酸化チタン)を用いてAquacoat(登録商標)に色を付けてもよい。これは水溶性ポリマー溶液に剪断力で色を添加し、その後可塑化したAquacoat(登録商標)に低い剪断力を用いることにより行う。あるいは本発明の製剤に色を付ける任意の方法を用いてもよい。アクリル性ポリマーの水分散液を用いた場合に製剤に色を付ける適切な成分は、二酸化チタンおよび色顔料(例えば酸化鉄顔料)を含む。但し、顔料を含有させるとコーティング剤の遅延効果が増す。
【0048】
上記剤を含む基質に、可塑化した疎水性材料を適用してもよい。これは当該分野で公知の適切なスプレー装置を用いてスプレーすることにより行う。好ましい方法によると、Wurster流動化床システムを用いる。これによるとアクリル性ポリマーコーティング剤をスプレーしている間に下から注入された空気流がコア材料を流動化し乾燥させる。コーティングされた基質が水溶液(例えば胃液)に曝されたときに上記剤の所定の徐放性を得るに十分な量の疎水性材料を適用してもよい。疎水性材料でコーティングした後、任意にビーズに薄膜生成剤(例えばOpadry(登録商標))をさらに適用する。このオーバーコーティングは、仮にあったとしても、ビーズの凝集を実質的に低減するために行う。
【0049】
本発明の徐放性製剤からの上記剤の放出には、1以上の放出改変剤を添加すること、またはコーティングにより1以上の通路を提供することにより、さらに影響を与えることができる(即ち所望の速度に調整することができる)。水溶性材料に対する疎水性材料の割合は、他の要因の中で特に、必要な放出率と選択される材料の溶解度特性とによって決定される。
【0050】
孔形成剤として機能する放出改変剤は有機性でも無機性でもよく、使用環境内でコーティング剤から溶解、抽出または浸出させることができる材料を含む。孔形成剤は1以上の疎水性材料(例えばヒドロキシプロピルメチルセルロール)を含んでもよい。
【0051】
放出改変剤はさらに又はこれに代えて、半透性ポリマーを含んでもよい。
【0052】
特定の実施形態では、放出改変剤はヒドロキシプロピルメチルセルロース、ラクトース、金属ステアリン酸塩、およびこれらの任意の混合物から選択される。
【0053】
本発明の徐放性コーティング剤はさらに浸食促進剤(例えばデンプンおよびゴム)を含んでもよい。
【0054】
本発明の徐放性コーティング剤はさらに使用環境内で微細孔性薄膜を生成するに有用な材料(例えば炭酸の線形ポリエステルを含み、ポリマー鎖内で炭酸基が反復するポリカーボネート)を含んでもよい。
【0055】
本発明の徐放性コーティング剤はさらに、少なくとも1つの通路、オリフィスなどを含む排出手段を含んでもよい。通路は米国特許第3,845,770号、第3,916,889号、第4,063,064号、および第4,088,864号に開示されたような方法により形成することができる。通路は円形、三角形、四角形、楕円形、不定形などの任意の形状を有することができる。
【0056】
マトリクス製剤
本発明の他の実施形態では、徐放性製剤は、本明細書に記載する徐放性コーティング剤を任意に有するマトリクスによって得られる。徐放性マトリクスに含まれるのに適した材料はマトリクスを形成するために用いた方法に依存する。
【0057】
例えば、ヒドロコドン(またはヒドロコドン塩)および必要に応じて含まれるナルトレキソン(またはナルトレキソン塩)に添加するマトリクスは以下から選択することができる:(i)親水性および/または疎水性材料、例えばゴム、セルロールエーテル、アクリル性ポリマーまたは樹脂、タンパク質由来の材料、および活性剤を徐放することができ且つ溶融する(または除去するに必要な程度まで軟化する)任意の製薬上許容可能な疎水性材料または親水性材料、(ii)消化可能な長鎖(C8-C50、特にC12-C40)置換または非置換炭化水素(例えば脂肪酸;脂肪性アルコール;脂肪酸のグリセリルエステル;鉱油、植物油および蝋;並びにステアリルアルコール)、および(iii)ポリアルキレングリコール。
【0058】
これらのポリマーのうち、アクリル性ポリマー(特にEudragit(登録商標) RSPO)およびセルロースエーテル(特にヒドロキシアルキルセルロースおよびカルボキシアルキルセルロース)が好ましい。経口剤形は少なくとも1つの親水性または疎水性材料を1%〜80%(重量比)含んでもよい。
【0059】
疎水性材料が炭化水素である場合、炭化水素は好ましくは25℃と90℃との間の融点を有する。長鎖炭化水素材料のうち、脂肪性(脂肪族)アルコールが好ましい。経口剤形は少なくとも1つの消化可能な長鎖炭化水素を最高60%(重量比)含んでもよい。
【0060】
好ましくは経口剤形は少なくとも1つのポリアルキレングリコールを最高60%(重量比)含む。
【0061】
疎水性材料は、アルキルセルロース、アクリル酸およびメタクリル酸ポリマーおよびコポリマー、シェラック、ゼイン、水素化ひまし油、水素化植物油、またはこれらの混合物からなる群より選択されてもよい。本発明の特定の好ましい実施形態では、疎水性材料は、アクリル酸およびメタクリル酸コポリマー、メタクリル酸メチル、メタクリル酸メチルコポリマー、メタクリル酸エトキシエチル、メタクリル酸シアノエチル、メタクリル酸アミノアルキルコポリマー、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミンコポリマー、ポリ(メタクリル酸メチル)、ポリ(メタクリル酸)(無水物)、ポリメタクリル酸、ポリアクリルアミド、ポリ(メタクリル酸無水物)、およびメタクリル酸グリシジルコポリマーなどの材料から選択される製薬上許容可能なアクリル性ポリマーである。他の実施形態では、疎水性材料は、ヒドロキシアルキルセルロース(例えばヒドロキシプロピルメチルセルロース)およびその混合物などの材料から選択される。
【0062】
好ましい疎水性材料は、非水溶性であり、多かれ少なかれ明白な親水性および/または疎水性の傾向を有する。好ましくは、本発明で有用な疎水性材料は約30℃〜約200℃の融点を有し、より好ましくは約45℃〜約90℃の融点を有する。特に、疎水性材料は天然または合成蝋、脂肪性アルコール(例えばラウリル、ミスチリル、ステアリル、セチルまたは好ましくはセトステアリルアルコール)、脂肪酸(脂肪酸エステル、脂肪酸グリセリド(モノグリセリド、ジグリセリドおよびトリグリセリド)を含むがこれらに限られない)、水素化脂肪、炭化水素、ノーマルワックス、ステアリン酸補助剤(stearic aid)、ステアリルアルコール、並びに炭化水素骨格を有する疎水性および親水性材料を含んでもよい。適切な蝋は例えば、蜜蝋、糖蝋(glycowax)、ひまし蝋(castor wax)、カルナウバ蝋を含む。本発明の目的のためには、蝋状物質は、室温では通常固体であり約30℃〜約100℃の融点を有する任意の材料と定義される。
【0063】
本発明に用いることができる適切な疎水性材料は、消化可能な長鎖(C8-C50、特にC12-C40)置換または非置換炭化水素(例えば脂肪酸、脂肪性アルコール、脂肪酸のグリセリルエステル、鉱油、植物油、並びに天然および合成蝋)を含む。約25℃〜約90℃の融点を有する炭化水素が好ましい。特定の実施形態では、長鎖炭化水素材料のうち脂肪性(脂肪族)アルコールが好ましい。経口剤形は少なくとも1つの消化可能な長鎖炭化水素を最高60%(重量比)含んでもよい。
【0064】
好ましくは、マトリクス製剤には2以上の疎水性材料の組み合わせが含まれる。追加の疎水性材料が含まれる場合、その疎水性材料は好ましくは天然および合成蝋、脂肪酸、脂肪性アルコール、およびこれらの混合物から選択される。これらの材料は例えば、蜜蝋、カルナウバ蝋、ステアリン酸、およびステアリルアルコールであるがこれらに限られない。
【0065】
1つの特定の適切なマトリクスは、少なくとも1つの水溶性ヒドロキシアルキルセルロース、少なくとも1つのC12-C36(好ましくはC14-C22)脂肪族アルコール、および必要に応じて少なくとも1つのポリアルキレングリコールを含む。ヒドロキシアルキルセルロースは好ましくはヒドロキシ(C1-C6)アルキルセルロース(例えばヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、またはヒドロキシエチルセルロース)である。本発明の経口剤形中のヒドロキシアルキルセルロースの量は、特にヒドロコドンおよび/またはナルトレキソンに要求される厳密な放出率によって決定される。脂肪族アルコールは例えば、ラウリルアルコール、ミリスチルアルコール、またはステアリルアルコールであってもよい。但し本発明の経口剤形の特に好ましい実施形態では、脂肪族アルコールはセチルアルコールまたはセトステアリルアルコールである。本発明の経口剤形中の脂肪族アルコールの量は、上記のようにヒドロコドンおよび/またはナルトレキソンに要求される厳密な放出率によって決定される。上記量はさらに、経口剤形中に少なくとも1つのポリアルキレングリコールが存在するか否かによっても左右される。少なくとも1つのポリアルキレングリコールが存在しない場合、経口剤形は好ましくは脂肪族アルコールを20%〜50%(重量比)含む。経口剤形中にポリアルキレングリコールが存在する場合、脂肪族アルコールとポリアルキレングリコールとの総重量が総剤形の20%〜50%(重量比)を占めることが好ましい。
【0066】
一実施形態では、脂肪族アルコール/ポリアルキレングリコールに対する例えばヒドロキシアルキルセルロースまたはアクリル性樹脂の割合が、ヒドロコドンおよび/またはナルトレキソンの製剤からの放出率をかなりの度合いで決定する。脂肪族アルコール/ポリアルキレングリコールに対するヒドロキシアルキルセルロースの割合は1:2〜1:4が好ましく、1:3〜1:4が特に好ましい。
【0067】
ポリアルキレングリコールは例えば、ポリプロピレングリコールまたはポリエチレングリコールであってもよい。少なくとも1つのポリアルキレングリコールの数平均分子量は好ましくは1,000〜15,000であり、特に好ましくは1,500〜12,000である。
【0068】
別の適切な徐放性マトリクスは、アルキルセルロース(特にエチルセルロース)、C12〜C36脂肪族アルコール、および必要に応じてポリアルキレングリコールを含む。
【0069】
別の好ましい実施形態では、マトリクスは少なくとも2つの疎水性材料の製薬上許容可能な組み合わせを含む。
【0070】
徐放性マトリクスは上記成分に加えてさらに、適量の他の材料、例えば製薬分野で従来から用いられている希釈剤、滑沢剤、結合剤、粒状化補助剤、色素、芳香剤、および/または流動促進剤を含んでもよい。
【0071】
マトリクス−微粒子
本発明による固体の徐放性経口剤形の調製を促進するためには、当業者に公知のいずれのマトリクス製剤調製方法を用いてもよい。マトリクスへの含有は例えば以下の方法により行われる: 例えば(a)少なくとも1つの水溶性ヒドロキシアルキルセルロース、ヒドロコドン(またはヒドロコドン塩)、および必要に応じてナルトレキソン(またはナルトレキソン塩)を含む顆粒を形成し、(b)得られた顆粒を少なくとも1つのC12-C36脂肪族アルコールと混合し、(c)必要に応じて顆粒を圧縮成形する。好ましくは、ヒドロキシアルキルセルロース顆粒を水で湿粒状化することによって顆粒を形成する。
【0072】
さらに別の実施形態では、ヒドロコドン(またはヒドロコドン塩)および必要に応じて含まれるナルトレキソン(またはナルトレキソン塩)と共にスフェロイド化剤を用いることによりスフェロイドを形成してもよい。微結晶セルロースは好ましいスフェロイド化剤である。適切な微結晶セルロースは例えば、Avicel PH 101(商標、FMC Corporation)として販売されている材料である。この実施形態では、スフェロイドは活性成分およびスフェロイド化剤に加えて結合剤を含んでもよい。適切な結合剤は例えば低粘度の水溶性ポリマーであり、製薬分野の当業者には公知である。しかし、水溶性ヒドロキシ低級アルキルセルロース(例えばヒドロキシプロピルセルロース)が好ましい。スフェロイドはこれに加えて(またはこれに代えて)、非水溶性ポリマー、特にアクリル性ポリマー、アクリル性コポリマー(例えばメタクリル酸−アクリル酸エチルコポリマー)またはエチルセルロースを含んでもよい。この実施形態では、徐放性コーティング剤は概して疎水性材料(例えば(a)蝋単独または蝋と脂肪性アルコールとの混合物または(b)シェラックまたはゼイン)を含む。
【0073】
溶融押出マトリクス
徐放性マトリクスは、溶融粒状化法または溶融押出法によっても調製することができる。溶融粒状化法は概して、通常は固体の疎水性材料(例えば蝋)を溶融することと、粉体化した薬物を疎水性材料中に含有させることとを含む。徐放性剤形を得るためには、追加の疎水性物質(例えばエチルセルロースまたは非水溶性アクリル性ポリマー)を、溶融蝋疎水性材料中に含有させることが必要となる場合もある。溶融粒状化法によって調製された徐放性製剤の例は米国特許第4,861,598号に記載されている。
【0074】
追加の疎水性材料は、1以上の非水溶性蝋状熱可塑性物質を含んでもよい。この非水溶性蝋状熱可塑性物質は、これよりも疎水度の低い別の1以上の蝋状熱可塑性物質と混合されてもよい。定常的放出を実現するためには、製剤中の個々の蝋状物質は初期の放出フェーズにおいて胃腸液に対して実質的に非変性であり不溶性でなければならない。有用な非水溶性蝋状物質は、約1:5,000(w/w)未満の水溶解度を有するものである。
【0075】
徐放性マトリクスは上記成分に加えてさらに、適量の他の材料、例えば製薬分野で従来用いられている希釈剤、滑沢剤、結合剤、粒状化補助剤、色素、芳香剤、および流動促進剤を含んでもよい。これら追加の材料は所望の製剤に所望の影響を与えるに十分な量だけ含まれる。
【0076】
溶融押出マルチ微粒子を含有する徐放性マトリクスは上記成分に加えてさらに、適量の他の材料、例えば製薬分野で従来から用いられている希釈剤、滑沢剤、結合剤、粒状化補助剤、色素、芳香剤、および/または流動促進剤を含んでもよい。
【0077】
経口剤形を処方するために用いることができる製薬上許容可能な担体および賦形剤の特定の例は、Handbook of Pharmaceutical Excipients, American Pharmaceutical Association, 第3版(2000)に記載されている。
【0078】
溶融押出マルチ微粒子
本発明による適切な溶融押出マトリクスの調製は例えば、ヒドロコドン(またはヒドロコドン塩)および/またはナルトレキソン(またはナルトレキソン塩)を少なくとも1つの疎水性材料と混合することにより均質混合物を得る工程を含んでもよい。次いで均質混合物を、少なくとも当該混合物を押出できるようになるまで十分に軟化させるに十分な温度まで加熱する。その後得られた均質混合物を押出してストランドを形成する。好ましくは押出物を当該分野で公知の任意の手段で冷却し、切断してマルチ微粒子にする。ストランドを切断してマルチ微粒子にする。その後マルチ微粒子を単位投与量に分割する。押出物は好ましくは約0.1〜約5 mmの直径を有し、約8〜約24時間の期間に亘って活性剤を徐放する。
【0079】
本発明の溶融押出物を調製するために必要に応じて行われるプロセスは、疎水性材料、ヒドロコドン(またはヒドロコドン塩)および必要に応じて含まれるナルトレキソン(またはナルトレキソン塩)、並びに必要に応じて含まれる結合剤を押出器に直接計量して供給すること、これらの成分を混合加熱して均質混合物を生成すること、均質混合物を押出することによりストランドを形成すること、均質混合物を含むストランドを冷却すること、ストランドを切断して約0.1 mm〜約12 mmのサイズを有する粒子にすること、および粒子を単位投与量に分割することを含む。本発明のこの局面では、比較的連続した製造手順が実現される。
【0080】
押出アパーチャまたは排出ポートの直径を調整することにより、押出されたストランドの厚みを変化させることも可能である。さらに、押出器の排出ポートは円形である必要はなく、楕円形、矩形などであってもよい。排出されてくるストランドは、ホットワイヤカッタ、ギロチンなどを用いて粒子に切断することができる。
【0081】
溶融押出マルチ微粒子系は、押出器の排出ポートに依存して例えば顆粒、スフェロイド、またはペレットの形態であり得る。本発明の目的のためには、用語「溶融押出マルチ微粒子」(MEMS)、および「溶融押出マルチ微粒子系」および「溶融押出粒子」は好ましくは類似のサイズおよび/または形状の範囲内にあり且つ1以上の活性剤と1以上の賦形剤とを含み、好ましくは本明細書に記載する疎水性材料を含む複数の単位を指す。この点に関して、溶融押出マルチ微粒子は長さ約0.1〜約12 mm、直径約0.1〜約5 mmである。さらに溶融押出マルチ微粒子はこのサイズの範囲内であればいずれの形状を有してもよいことが理解される。あるいは押出物を単に所望の長さに切断して治療上活性な薬剤の単位投与量に分割してもよく、この場合スフェロイド化工程は不要である。
【0082】
一つの好ましい実施形態では、経口剤形は、有効量の溶融押出マルチ微粒子をカプセル内に含むように調製される。例えば、複数の溶融押出マルチ微粒子を、摂取され胃液に接触されたときに有効に徐放されるに十分な量でゼラチンカプセル内に入れてもよい。
【0083】
別の好ましい実施形態では、従来のタブレット化装置を用い、標準的な手法を用いて適量のマルチ微粒子押出物を圧縮して経口タブレットにする。タブレット(圧縮され成形された)、カプセル(硬および軟ゼラチン)並びにピルを形成する手法および組成物は、Remington's Pharmaceutical Sciences (Arthur Osol編)、1553〜1593頁(1980)にも記載されている。
【0084】
さらに別の好ましい実施形態では、上記に詳細に記載した米国特許第4,957,681号(Klimeschら)に記載されているように押出物をタブレットに成形してもよい。
【0085】
必要に応じて、例えば上記した徐放性コーティング剤などの徐放性コーティング剤を用いて徐放性溶融押出マルチ微粒子系またはタブレットをコーティングしてもよいし、ゼラチンカプセルをさらにコーティングしてもよい。これらのコーティング剤は好ましくは、約2〜約30%の重量増加レベルを得るに十分な量の疎水性材料を含む。但し、オーバーコーティング剤の量は、特に所望の徐放率によってはこれよりも多い場合もある。
【0086】
本発明の溶融押出単位剤形はカプセル化される前に、溶融押出粒子の組み合わせ(例えばヒドロコドン(またはヒドロコドン塩)を有する一群の粒子とナルトレキソン(またはナルトレキソン塩)を有する一群の粒子)をさらに含んでもよい。単位剤形は即時放出のために、ある量の即時放出型活性剤をさらに含んでもよい。即時放出型剤は、例えばゼラチンカプセル内の分離したペレットとして含有されてもよいし、剤形(例えば徐放性コーティングまたはマトリクスベース)の調製後にマルチ微粒子の表面にコーティングされてもよい。本発明の単位剤形は、所望の効果を達成するために徐放性ビーズとマトリクスマルチ微粒子との組み合わせをさらに含んでもよい。
【0087】
本発明の徐放性製剤は好ましくは、例えば摂取され、胃液、次いで腸液に接触されたときに、ゆっくりと薬剤を放出する。本発明の溶融押出製剤の徐放プロファイルは、例えば遅延剤(すなわち疎水性材料)の量を変更すること、疎水性材料に対する可塑剤の量を変更すること、追加の成分または賦形剤を含ませること、製造方法を変更することなどによって変化させることができる。
【0088】
本発明の別の実施形態では、溶融押出材料を、ヒドロコドン(またはヒドロコドン塩)もナルトレキソン(またはナルトレキソン塩)も含むことなく調製する。ヒドロコドン(またはヒドロコドン塩)とナルトレキソン(またはナルトレキソン塩)とは後に押出物に添加することができる。このような製剤では典型的には、薬剤を押出マトリクス材料と混合し、その後ゆっくりと放出する目的で混合物をタブレット化する。
【0089】
コーティング剤
本発明の剤形は必要に応じて、放出の制御または製剤の保護に適した1以上の材料でコーティングしてもよい。一実施形態では、コーティング剤はpH依存型またはpH独立型放出を可能にするために提供される。pH依存型コーティング剤は、ヒドロコドンおよび/またはナルトレキソンを胃腸(GI)管(例えば胃または小腸)の所望の領域において放出するために役立つ。これにより、患者に少なくとも8時間、好ましくは約12時間、最高約24時間の沈痛を与えることができる吸入プロファイルが提供される。pH独立型コーティング剤が望まれる場合、コーティング剤は、周囲、例えばGI管の液のpH変化にかかわらず最適な放出を達成するように設計される。GI管の1つの所望領域(例えば胃)に投与量の一部を放出し、GI管の別の領域(例えば小腸)に投与量の残りを放出する組成物を処方することも可能である。
【0090】
pH依存型コーティング剤を利用する本発明による製剤はさらに反復作用効果を提供することができる。反復作用効果によると、保護されていない薬物を腸溶性コーティング剤にコーティングして胃で放出し、腸溶性コーティング剤によって保護された残りの部分を胃腸管のさらに下の胃腸管で放出する。pH依存型コーティング剤は、シェラック、酢酸フタル酸セルロース(CAP)、酢酸フタル酸ポリビニル(PVAP)、フタル酸ヒドロキシプロピルメチルセルロース、およびメタクリル酸エステルコポリマー、ゼインなどを含む。
【0091】
特定の好ましい実施形態では、ヒドロコドンまたはその塩および必要に応じてナルトレキソンまたはその塩を含む基質(例えばコーティングされたビーズ、マトリクス粒子)を、(i)アルキルセルロース、(ii)アクリル性ポリマー、(iii)それらの混合物から選択される疎水性材料によってコーティングする。コーティング剤は有機または水溶液または水分散液の形態で適用してもよい。コーティング剤は、所望の徐放プロファイルを得るために基質の約2〜約25%の重量増加を得るように適用してもよい。水分散液由来のコーティング剤は、例えば米国特許第5,273,760号および第5,286,493号に詳細に記載されている。
【0092】
本発明に用いることができる徐放性製剤およびコーティング剤の他の例は、米国特許第5,324,351号、第5,356,467号、および第5,472,712号に記載されているものを含む。
【0093】
アルキルセルロースポリマー
アルキルセルロースを含むセルロース材料およびポリマーは、本発明によるビーズのコーティングに良く適した疎水性材料を提供する。単なる例として1つの好ましいアルキルセルロースポリマーは、エチルセルロースであるが、当業者であれば他のセルロースおよび/またはアルキルセルロースポリマーも単一または任意の組み合わせで、本発明による疎水性コーティング剤の全体または一部として容易に使用可能であることを理解する。
【0094】
エチルセルロースの1つの市販されている水分散液はAquacoat(登録商標) (FMC Corp., Philadelphia, Pennsylvania, U.S.A)である。Aquacoat(登録商標)は、エチルセルロースを水と混合不能な有機溶媒中に溶解し、その後得られたものを水中で界面活性剤および安定剤の存在下で乳化することによって調製される。均質化によりサブミクロンサイズの小滴を生成した後、有機溶媒を真空中で蒸発させて疑似ラテックスを生成する。製造フェーズにおいて可塑剤は疑似ラテックスに含有されない。従って疑似ラテックスをコーティング剤として用いる前に、Aquacoat(登録商標)を適切な可塑剤と混合することが必要である。エチルセルロースの別の水分散液はSurelease(登録商標) (Colorcon, Inc., West Point, Pennsylvania, U.S.A)として市販されている。この製品は、製造プロセスにおいて分散液中に可塑剤を含有させることによって調製される。ポリマーと可塑剤(セバシン酸ジブチル)と安定剤(オレイン酸)とのホットメルトを均質混合物として調製し、その後これをアルカリ溶液で希釈することにより、基質に直接適用することができる水分散液を得る。
【0095】
アクリル性ポリマー
本発明の他の好ましい実施形態では、徐放性コーティング剤を含む疎水性材料は、製薬上許容可能なアクリル性ポリマーであり、これはアクリル酸およびメタクリル酸コポリマー、メタクリル酸メチルコポリマー、メタクリル酸エトキシエチル、メタクリル酸シアノエチル、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミドコポリマー、ポリ(メタクリル酸メチル)、ポリメタクリル酸、ポリ(メタクリル酸メチル)コポリマー、ポリアクリルアミド、メタクリル酸アミノアルキルコポリマー、ポリ(メタクリル酸無水物)、およびメタクリル酸グリシジルコポリマーを含むがこれらに限られない。
【0096】
特定の好ましい実施形態では、アクリル性ポリマーは1以上のメタクリル酸アンモニオ(ammonio)コポリマーを含む。メタクリル酸アンモニオコポリマーは当該分野で周知であり、低含有量の第四級アンモニア基を含むアクリル酸エステルとメタクリル酸エステルとの完全重合コポリマーとしてNational Formulary XVIIに記載されている。
【0097】
所望の溶解プロファイルを得るために、異なる物性(例えば中性(メタ)クリル酸エステルに対する第四級アンモニア基の異なるモル比)を有する2以上のメタクリル酸アンモニオコポリマーを含有させることが必要な場合がある。
【0098】
特定の好ましい実施形態では、アクリル性コーティング剤は、Rohm PharmaからそれぞれEudragit(登録商標) RL30DおよびのEudragit(登録商標) RS30Dの商品名で市販されている2つのアクリル性樹脂ラッカーの混合物を含む。Eudragit(登録商標) RL30DおよびのEudragit(登録商標) RS30Dは、低含有量の第四級アンモニア基を含むアクリル酸エステルとメタクリル酸エステルとのコポリマーである。アンモニア基の、残りの中性(メタ)クリル酸エステルに対するモル比は、Eudragit(登録商標) RL30Dでは1:20であり、Eudragit(登録商標) RS30Dでは1:40である。平均分子量は約150,000である。名称RL(高透過性)およびRS(低透過性)はこれらの剤の透過特性を示す。Eudragit(登録商標) RL/RS混合物は水および消化器液に対して不溶性である。しかし当該混合物から生成されたコーティング剤は水溶液および消化器液中で膨張可能且つ透過可能である。
【0099】
本発明のEudragit( RL/RS分散液は、所望の溶解プロファイルを有する徐放性製剤を最終的に得るために任意の所望の割合で一緒に混合することができる。所望の徐放性製剤は、例えば100%Eudragit(登録商標) RL由来の遅延コーティング剤から、50% Eudragit(登録商標) RLおよび50% Eudragit(登録商標) RSから、または10% Eudragit(登録商標) RLおよびEudragit(登録商標) 90% RSから得ることができる。言うまでもなく当業者であれば他のアクリル性ポリマー(例えばEudragit(登録商標) L)も使用可能であることを認識する。
【0100】
可塑剤
本発明の実施形態では、コーティング剤が疎水性材料の水分散液を含む場合、疎水性材料の水分散液中に有効量の可塑剤を含ませることにより徐放性コーティング剤の物性をさらに高めることができる。例えば、エチルセルロースは比較的高いガラス転移点を有し且つ通常のコーティング条件下では可撓性膜を形成しないため、徐放性コーティング剤を含むエチルセルロースコーティング剤をコーティング材料として用いる前に、これに可塑剤を含有させることが好ましい。概してコーティング溶液中に含まれる可塑剤の量は、薄膜生成剤の濃度(例えば最も多くの場合、薄膜生成剤の約1〜約50%(重量比))に基づく。しかし可塑剤の濃度は、特定のコーティング溶液および適用方法を慎重に実験した後でのみ適切に決定することができる。
【0101】
エチルセルロースに適した可塑剤の例は、非水溶性可塑剤(例えばセバシン酸ジブチル、フタル酸ジエチル、クエン酸トリエチル、クエン酸トリブチル、およびトリアセチン)を含む。本発明で用いられるエチルセルロースの水分散液にとっては、クエン酸トリエチルが特に好ましい可塑剤である。
【0102】
本発明のアクリル性ポリマーに適した可塑剤の例は、クエン酸エステル(例えばクエン酸トリエチルNF XVI、クエン酸トリブチル、フタル酸ジブチル、および1,2-プロピレングリコール)を含むがこれらに限られない。アクリル性薄膜(例えばEudragit(登録商標) RL/RSラッカー溶液)から形成された薄膜の弾性を高めるのに適していることが証明されている他の可塑剤は、ポリエチレングリコール、プロピレングリコール、フタル酸ジエチル、ひまし油、およびトリアセチンを含む。本発明で用いられるアクリル酸ポリマーの水分散液にとっては、クエン酸トリエチルが特に好ましい可塑剤である。
【0103】
少量のタルクを添加することにより処理中に水分散液が粘着する傾向が低減され、タルクは研磨剤として作用することがさらに判明している。
【0104】
徐放性浸透性投与量
本発明による徐放性剤形はさらに浸透性投与製剤として調製してもよい。浸透性剤形は好ましくは、薬物層(ヒドロコドン(またはヒドロコドン塩)および必要に応じてナルトレキソン(またはナルトレキソン層)を含む)と送達または押し出し層(ナルトレキソン(またはナルトレキソン塩))を含んでもよい)とを含む二層コアを含む。二層コアは半透壁により取り囲まれ、半透壁は必要に応じて内部に設けられた少なくとも1つの通路を有する。
【0105】
本発明の目的のために、用語「通路」は、ファイバ、毛細管、多孔性オーバーレイ、多孔性インサート、微孔性部材、または多孔性組成物を介してヒドロコドンまたはヒドロコドン塩(ナルトレキソンまたはナルトレキソン塩を伴うまたは伴わず)を注入、拡散または移送することができるアパーチャ、オリフィス、穴(bore)、孔(pore)、または有孔の要素を含む。通路はさらに、用いられる液体環境中で壁から浸食するか浸出することによって少なくとも1つの通路を形成する化合物を含んでもよい。通路を形成する代表的な化合物は、壁内の浸食性ポリ(グリコール)酸またはポリ(乳)酸、ゼラチン性フィラメント、水除去可能ポリ(ビニルアルコール)、浸出性化合物(例えば液体除去可能孔形成ポリサッカリド、酸、塩または酸化物)を含む。通路は、壁(例えばソルビトール、スクロース、ラクトース、マルトースまたはフルクトース)から化合物を浸出させることにより形成することができ、これにより徐放用寸法を有する孔通路が形成される。通路は、ヒドロコドンまたはヒドロコドン塩を剤形から調節しながら徐放することを補助する任意の形状(例えば円形、三角形、四角形および楕円形)を有することができる。剤形は、その1以上の表面上に1以上の通路を有するように製造することができる。通路および通路を形成する装置は、米国特許第3,845,770号、第3,916,899号、第4,063,064号、および第4,088,864号に開示されている。水を通すことにより形成されて、徐放率を有する放出孔としてのサイズおよび形状を有し当該放出孔として適合された徐放性広がりを有する通路は、米国特許第4,200,098号および第4,285,987号に記載されている。
【0106】
特定の実施形態では、二層コアは、ヒドロコドンまたはその塩を有する薬物層と、ナルトレキソンまたはその塩を含む移動または押し出し層とを含む。特定の実施形態では、薬物層はさらに少なくとも1つのポリマーヒドロゲルを含んでもよい。ポリマーヒドロゲルは約500〜約6,000,000の平均分子量を有してもよい。ポリマーヒドロゲルの例は以下のマルトデキストリンポリマー、ポリ(酸化アルキレン)、アルカリカルボキシアルキルセルロース、およびエチレン−アクリル酸コポリマーを含むがこれらに限られない。マルトデキストリンポリマーは、化学式(C6H12O5)n・H20を含み且つnが3〜7,500であり、500〜1,250,000の数平均分子量を有する。ポリ(酸化アルキレン)は、例えば50,000〜750,000の重量平均分子量を有するポリ(酸化エチレン)およびポリ(酸化プロピレン)に代表され、より好ましくは100,000、200,000、300,000および400,000のうち少なくとも1つの重量平均分子量を有するポリ(酸化エチレン)に代表される。アルカリカルボキシアルキルセルロースは、アルカリがナトリウムまたはカリウムであり、アルキルセルロースが10,000〜175,000の重量平均分子量を有する。エチレン−アクリル酸コポリマーは、10,000〜500,000の数平均分子量を有するメタクリル酸およびエタクリル酸を含む。
【0107】
本発明の特定の実施形態では、送達または押し出し層はオスモポリマーを含む。オスモポリマーの例は、酸化ポリアルキレンとカルボキシアルキルセルロースとからなる群より選択される物質を含むがこれらに限られない。酸化ポリアルキレンは1,000,000〜10,000,000の数平均分子量を有する。酸化ポリアルキレンは、酸化ポリメチレン、酸化ポリエチレン、酸化ポリプロピレン、1,000,000の平均分子量を有する酸化ポリエチレン、5,000,000の平均分子量を有する酸化ポリエチレン、7,000,000の平均分子量を有する酸化ポリエチレン、1,000,000の平均分子量を有する架橋酸化ポリメチレン、および1,200,000の平均分子量を有する酸化ポリプロピレンからなる群より選択される物質であってもよい。典型的なオスモポリマーであるカルボキシアルキルセルロースは、アルカリカルボキシアルキルセルロース、ソディウムカルボキシメチルセルロース、ポタシウムカルボキシメチルセルロース、ソディウムカルボキシエチルセルロース、リチウムカルボキシメチルセルロース、ソディウムカルボキシエチルセルロース、およびカルボキシアルキルヒドロキシアルキルセルロース(例えば、カルボキシメチルヒドロキシエチルセルロース、カルボキシエチルヒドロキシエチルセルロース、およびカルボキシメチルヒドロキシプロピルセルロース)からなる群より選択される物質を含む。移動層に用いられるオスモポリマーは、半透壁に対して浸透圧グラジエントを呈する。オスモポリマーは液体を吸収して剤形になり、それにより浸透性ヒドロゲル(オスモゲルとしても知られる)として膨張および拡張する。これによりオスモポリマーは浸透性剤形からヒドロコドンまたはその製薬上許容可能な塩を押し出す。
【0108】
押し出し層はさらに、オスマゲント(osmagent)または浸透有効溶質としても知られる1以上の浸透有効化合物を含んでもよい。これらは、例えば胃腸管から剤形に周囲の液体を吸収し、移動層の送達速度に寄与する。浸透活性化合物の例は、浸透性塩と浸透性炭水化物とからなる群より選択される物質を含む。特定のオスモゲントの例は、塩化ナトリウム、塩化カリウム、硫酸マグネシウム、リン酸リチウム、塩化リチウム、リン酸ナトリウム、硫酸カリウム、硫酸ナトリウム、リン酸カリウム、グルコース、フルクトース、およびマルトースを含むがこれらに限られない。
【0109】
押し出し層は必要に応じて、9,000〜450,000の数平均分子量を有するヒドロキシプロピルアルキルセルロースを含んでもよい。ヒドロキシプロピルアルキルセルロースは、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルエチルセルロース、ヒドロキシプロピルイソプロピルセルロース、ヒドロキシプロピルブチルセルロース、およびヒドロキシプロピルペンチルセルロースからなる群より選択される物質により代表される。
【0110】
押し出し層は必要に応じて、非毒性色素または染料を含んでもよい。色素または染料の例は、Food and Drug Administration Colorant (FD&C)(例えばFD&C No. 1青染料、FD&C No. 4赤染料、赤酸化第二鉄、黄酸化第二鉄、二酸化チタン、カーボンブラック、およびインディゴ)を含むがこれらに限られない。
【0111】
押し出し層はさらに必要に応じて、成分の酸化を阻止する酸化防止剤を含んでもよい。酸化防止剤のいくつかの例は、アスコルビン酸、パルミチン酸アルコルビル、ブチル化ヒドロキシアニソール、2および3第三級-ブチル-4-ヒドロキシアニソール、ブチル化ヒドロキシトルエン、イソアスコルビン酸ナトリウム、ジヒドログアレチン酸(dihydroguaretic acid)、ソルビン酸カリウム、重硫酸ナトリウム、メタ重硫酸ナトリウム、ソルビン酸、アスコルビン酸カリウム、ビタミンE、4-クロロ-2,6-2第三級(ditertiary)ブチルフェノール、アルファトコフェノール、およびプロピルガラートからなる群より選択される物質を含むがこれらに限られない。
【0112】
特定の別の実施形態では、剤形は均質コアを含み、均質コアは、ヒドロコドンまたはその製薬上許容可能な塩、ナルトレキソンまたはその製薬上許容可能な塩、製薬上許容可能なポリマー(例えば酸化ポリエチレン)、必要に応じて崩壊剤(例えばポリビニルピロリドン)、および必要に応じて吸収促進剤(例えば脂肪酸、界面活性剤、キレート剤、胆汁酸塩など)を含む。均質コアは、ヒドロコドンまたはその製薬上許容可能な塩を放出する通路(上記で定義した)を有する半透壁によって取り囲まれている。
【0113】
特定の実施形態では、半透壁は、セルロースエステルポリマー、セルロースエーテルポリマー、およびセルロースエステル−エーテルポリマーからなる群より選択される物質を含む。代表的な壁ポリマーは、セルロースアシレート、セルロースジアシレート、セルローストリアシレート、セルロースアセテート、セルロースジアセテート、セルローストリアセテート、モノ、ジおよびトリセルロースアルケニレート、並びにモノ、ジおよびトリセルロースアルキニレートからなる群より選択される物質を含む。本発明に用いられるポリ(セルロース)は20,000〜7,500,000の数平均分子量を有する。
【0114】
追加の半透性ポリマーは、アセトアルデヒドジメチルセルロースアセテート、セルロースアセテートエチルカルバメート、セルロースアセテートメチルカルバメート、セルロースジアセテート、プロピルカルバメート、セルロースアセテートジエチルアミノアセテート、半透性ポリアミド、半透性ポリウレタン、半透性スルホン化ポリスチレン、ポリアニオンとポリカチオンとの共沈殿により形成される半透性架橋ポリマー(米国特許第3,173,876号、第3,276,586号、第3,541,005号、第3,541,006号、および第3,546,876号に記載)、米国特許第3,133,132号でLoebおよびSourirajanが開示した半透性ポリマー、半透性架橋ポリスチレン、半透性架橋ポリ(スチレンスルホン酸ナトリウム)、半透性架橋ポリ(塩化ビニルベンジルトリメチルアンモニウム)、および2.5x10-8〜2.5x10-2 (cm2/hr・atm)の液体透過性を有する半透性ポリマーを含む。上記液体透過性は、半透壁の静水圧差または浸透圧差の気圧単位で表している。本発明において有用な他のポリマーは、当該分野で公知であり、米国特許第3,845,770号、第3,916,899号、および第4,160,020号、並びにHandbook of Common Polymers, Scott, J. R.およびW. J. Roff, 1971, CRC Press, Cleveland, Ohioに記載されている。
【0115】
特定の実施形態では、半透壁は好ましくは非毒性且つ不活性であり、薬物の調剤中に亘ってその物理的および化学的完全性を維持する。
【0116】
特定の実施形態では、剤形は結合剤を含む。結合剤の例は、5,000〜350,000の粘度平均分子量を有する治療上許容可能なビニルポリマーを含むがこれに限られない。上記治療上許容可能なビニルポリマーは、酢酸ビニル、ビニルアルコール、塩化ビニル、フッ化ビニル、ブチル酸ビニル、ラウリン酸ビニル、およびステアリン酸ビニルからなる群より選択される物質との、ポリ-n-ビニルアミド、ポリ-n-ビニルアセトアミド、ポリ(ビニルピロリドン)(ポリ-n-ビニルピロリドンとしても知られる)、ポリ-n-ビニルカプロラクトン、ポリ-n-ビニル-5-メチル-2-ピロリドン、およびポリ-n-ビニル-ピロリドンポリマーからなる群より選択される物質に代表される。他の結合剤は例えば、アカシア、デンプン、ゼラチン、および9,200〜250,000の平均分子量を有するヒドロキシプロピルアルキルセルロースを含む。
【0117】
特定の実施形態では、剤形は滑沢剤を含み、滑沢剤は剤形の製造中に金型(die)の壁またはパンチ面に対する粘着を防止するために用いることができる。滑沢剤の例は、ステアリン酸マグネシウム、ステアリン酸ナトリウム、ステアリン酸、ステアリン酸カルシウム、オレイン酸マグネシウム、オレイン酸、オレイン酸カリウム、カプリル酸、ステアリルフマル酸ナトリウム、およびパルミチン酸マグネシウムを含むがこれらに限られない。
【0118】
本発明は特定の好ましい実施形態において、治療用組成物を含み、治療用組成物は、5〜20 mgのヒドロコドンまたはその製薬上許容可能な塩と、25〜500 mgのポリ(酸化アルキレン)(150,000〜500,000の平均分子量を有する)と、1〜50 mgのポリビニルピロリドン(40,000の平均分子量を有する)と、0〜約7.5 mgの滑沢剤とを含む。好ましくは0.05〜0.56 mgのナルトレキソンまたはその製薬上許容可能な塩が薬物層に含まれる。
【0119】
座薬
本発明の徐放性製剤は、直腸投与用の座薬として処方されてもよい。上記座薬は、ヒドロコドン(またはヒドロコドン塩)とナルトレキソン(またはナルトレキソン塩)とを本明細書に開示する投与量で含む。徐放性座薬製剤の調製は、例えば米国特許第5,215,758号に記載されている。
【0120】
薬物は吸収される前に溶液状態でなければならない。座薬の場合、溶液状態にする前に、基剤を溶解しなければならないか、あるいは基剤を溶融しなければならず、次いで基剤から薬物を分離して直腸液にいれなければならない。体内への薬物の吸収は坐薬基剤によって変化し得る。従って特定の薬物と共に用いられる特定の基剤は、薬物の物性を考慮して選択しなければならない。例えば、脂質溶解性薬物は容易に分離して直腸液に流入することができないが、脂質基剤に僅かに溶解する薬物は容易に分離して直腸液に流入することができる。
【0121】
薬物の溶解時間(または放出率)に影響を与える様々な要因は、溶解溶媒媒質に提示される薬物物質の表面積、溶液のpH、特定の溶媒媒質中での物質の溶解度、および溶媒媒質中に溶解した物質の飽和濃度の駆動力などである。直腸投与される座薬からの薬物の吸収に影響を与える要因は概して、座薬ビヒクル、吸収部位pH、薬物pKa、イオン化度、および脂質溶解度を含む。
【0122】
選択される座薬基剤は、本発明の活性剤と適合性を有すべきである。さらに座薬基剤は好ましくは、粘膜に対して非毒性且つ非刺激性であり、直腸液中に溶融または溶解し、保存中安定している。
【0123】
本発明の特定の好ましい実施形態では、水溶性薬物および非水溶性薬物のいずれの場合も、座薬基剤は、C12〜C18の鎖長を有する飽和天然脂肪酸のモノ、ジおよびトリクリセリドからなる群より選択される脂肪酸蝋を含む。
【0124】
本発明の座薬を調製する際に、他の賦形剤を用いてもよい。例えば、直腸ルートの投与に適した形状を形成するために蝋を用いてもよい。この系は、蝋なしで用いてもよいが、その場合は直腸投与および経口投与の両方の場合においてゼラチンカプセルに希釈剤を充填する。
【0125】
適切な市販のモノ、ジおよびトリクリセリドの例は、Novata TM(タイプAB、AB、B、BC、BD、BBC、E、BCF、C、Dおよび299)という商品名(Henkel製)およびWitepsol TM(タイプH5、H12、H15、H175、H185、H19、H32、H35、H39、H42、W25、W31、W35、W45、S55、S58、E75、E76およびE85)という商品名(Dynamit Nobel製)で販売されている、12〜18の炭素原子鎖を有する飽和天然脂肪酸を含む。
【0126】
他の製薬上許容可能な座薬基剤は、全体的にまたは部分的に上記のモノ、ジおよびトリクリセリドと置換してもよい。座薬中の基剤の量は剤形のサイズ(即ち実際の重量)、基剤(例えばアルギン酸塩)の量、および用いられる活性剤によって決定される。座薬基剤の量は概して、座薬の総重量の約20重量%〜約重量90%である。好ましくは座薬中の基剤の量は座薬の総重量の約65重量%〜約80重量%である。
【0127】
他の形態
本明細書に開示する発明は、ヒドロコドンおよびナルトレキソンのすべての製薬上許容可能な塩を用いることを含む。製薬上許容可能な塩は、金属塩(例えば、ナトリウム塩、カリウム塩、セシウム塩など)、アルカリ土類金属(例えばカルシウム塩、マグネシウム塩など)、有機アミン塩(例えばトリエチルアミン塩、ピリジン塩、ピコリン塩、エタノールアミン塩、トリエタノールアミン塩、ジシクロヘキシルアミン塩、N,N'-ジベンジルエチレンジアミン塩など)、無機酸塩(例えば塩酸塩、臭化水素酸塩、硫酸塩、リン酸塩など)、有機酸塩(例えば蟻酸塩、酢酸塩、トリフルオロ酢酸塩、マレイン酸塩、酒石酸塩、重酒石酸塩など)、スルホン酸塩(例えばメタンスルホン酸塩、ベンゼンスルホン酸塩、p-トルエンスルホン酸塩など)、アミノ酸塩(例えばアルギン酸塩、アスパラギン酸塩、グルタミン酸塩など)を含むがこれらに限られない。
【0128】
ヒドロコドン(またはヒドロコドン塩)とナルトレキソン(またはナルトレキソン塩)との組み合わせは、従来の賦形剤、即ち少なくともヒドロコドンまたはその塩を徐放するためのものとして当該分野で公知の、経口投与に適した製薬上許容可能な有機または無機担体物質と混合して用いることができる。適切な製薬上許容可能な担体は、アルコール、アラビアゴム、植物油、ベンジルアルコール、ポリエチレングリコール、ゲル(gelate)、炭水化物(例えばラクトース、アミロースまたはデンプン)、ステアリン酸マグネシウム、タルク、珪酸、粘性パラフィン、香油、脂肪酸モノグリセリドおよびジグリセリド、ペンタエリスリトール脂肪酸エステル、ヒドロキシメチルセルロース、ポリビニルピロリドンなどを含むがこれらに限られない。医薬調製物は滅菌してもよく、所望であれば補助剤(例えば滑沢剤、崩壊剤、保存料、安定剤、湿潤剤、乳化剤、浸透圧緩衝剤に影響を与える塩、色素、芳香剤および/または芳香物質などと混合してもよい。経口投与を意図した組成物は、当該分野で公知の任意の方法を用いて調製することができる。これらの組成物は、タブレットの製造に適した不活性且つ非毒性の製薬上許容可能な賦形剤からなる群より選択される1以上の薬剤を含む。これらの賦形剤は例えば、不活性希釈剤(例えばラクトース)、粒状化および崩壊剤(例えばコーンスターチ)、結合剤(例えばデンプン)、および滑沢剤(例えばステアリン酸マグネシウム)を含む。タブレットはコーティングしなくてもよいし、外見の美しさまたは活性成分の遅延放出とのために公知の手法でコーティングしてもよい。経口投与用の製剤はさらに、硬いゼラチンカプセルとして提示されてもよく、その場合、活性成分を不活性希釈剤と混合する。
【0129】
本発明の経口剤形は、タブレット、トローチ、ロゼンジ(lozenge)、粉剤または顆粒、硬性または軟性カプセル、微粒子(例えばマイクロカプセル、マイクロスフィアなど)、口内タブレット、座薬などの形態とすることができる。ヒドロコドン(またはヒドロコドン塩)とナルトレキソン(またはナルトレキソン塩)とは実質的に互いに分散していてもよい。
【0130】
本発明は特定の実施形態において、上記に開示したヒドロコドン/ナルトレキソン剤形のいずれかを調製することにより、経口ヒドロコドン剤形(またはヒドロコドン塩)の非経口乱用を阻止する方法を提供する。
【0131】
本発明は特定の実施形態において、上記に開示したヒドロコドン/ナルトレキソン剤形のいずれかを調製することを含む経口ヒドロコドン剤形の転用を阻止する方法を提供する。
【0132】
本発明は特定の実施形態において、上記に開示した剤形をヒト患者に投与することにより疼痛を治療する方法を提供する。
【0133】
以下の実施例は本発明の様々な局面を説明するが、請求の範囲を何ら限定するものと解釈されてはならない。
【実施例1】
【0134】
この予言的実施例では、塩酸ナルトレキソンを含む徐放性ヒドロコドン製剤を以下に示す表1の処方で調製する。
【0135】
【表1】

*99.6%アッセイおよび4.2%残基水分に合わせて調整。
【0136】
この実施例では、粒状化プロセスで製剤に塩酸ナルトレキソンを添加する。プロセスは以下の通りである。
【0137】
1.分散:塩酸ナルトレキソンを水中に溶解し、溶液をEudragit/トリアセチン分散液に添加する。
2.粒状化:流体床粒状化器を用いて、Eudragit/トリアセチン分散液を塩酸ヒドロコドン、スプレー乾燥ラクトースおよびポビドンにスプレーする。
3.ミリング(Milling):顆粒を放出して約1 mmの開口(18メッシュスクリーン)を有するミル内を通過させる。
4.蝋づけ:ステアリルアルコールを約50℃で溶融し、高剪断力ミキサを用いてミリングされた顆粒に添加する。トレイまたは流体床上で室温で冷却する。
5.ミリング:冷却した顆粒を約18メッシュスクリーンのミル内を通過させる。
6.潤滑化:ミキサを用いてタルクおよびステアリン酸マグネシウムで顆粒を潤滑化する。
7.圧縮:Kilianタブレットプレスを用いて顆粒を圧縮してタブレットにする。
8.薄膜コーティング:ロータリーパンを用いてタブレットに水性薄膜を適用する。
【実施例2】
【0138】
この予言的実施例では、ヒドロコドン塩/ナルトレキソン塩徐放性浸透性タブレットを以下に示す表2の処方で生成する。
【0139】
【表2】
【0140】
上記処方を有する剤形を以下の手順に従って調製する。
【0141】
まず、無水塩酸ヒドロコドンと塩酸ナルトレキソン二水和物と、200,000の平均分子量を有するポリ(酸化エチレン)と、40,000の平均分子量を有するポリビニルピロリドンとをミキサに加えて10分間混合する。その後混合した材料に変性無水アルコールを添加して10分間混合を続ける。その後湿った顆粒を20メッシュスクリーン内を通過させ、20時間に亘って室温で乾燥し、その後16メッシュスクリーン内を通過させる。次に顆粒をミキサに送り、ステアリン酸マグネシウムと混合し潤滑化する。
【0142】
その後、塩酸ヒドロコドン/塩酸ナルトレキソン組成物を剤形から押し出すための移動または押し出し組成物を以下のように調製する。まず11,200の平均分子量を有する3910 gのヒドロキシプロピルメチルセルロースを45,339 gの水中に溶解する。その後101 gのブチル化ヒドロキシトルエンを650 gの変性無水アルコール中に溶解する。次に2.5 kgのヒドロキシプロピルメチルセルロース水溶液をブチル化ヒドロキシトルエンアルコール溶液に加えながら混合する。その後ブチル化ヒドロキシトルエンアルコール溶液に残りのヒドロキシプロピルメチルセルロース水溶液を加えながら混合することにより、結合剤溶液の調製を完了する。
【0143】
次に21メッシュスクリーンを有するQuadro Comil((登録商標)ミルを用いて、36,000 gの塩化ナトリウムのサイズを一定にする。その後1200 gの酸化第二鉄を40メッシュスクリーン内を通過させる。その後スクリーニングした材料と、7,500,000の平均分子量を有する76,400 gの製薬上許容可能なポリ(酸化エチレン)と、11,200の平均分子量を有する2500 gのヒドロキシプロピルメチルセルロースとを、Glatt(登録商標)流体床粒状化ボウルに加える。ボウルを粒状化器に取り付け、粒状化プロセスを開始して粒状化を実施する。次いで乾燥粉体を空気中に浮遊させて10分間混合する。その後結合剤溶液を3つのノズルから粉体にスプレーする。以下の条件で行うプロセス中、粒状化をモニターする:総溶液スプレー速度:800 g/分、入口温度:43℃、空気流:4300 m3/時間。溶液スプレーの終了時に、得られたコーティング済み粒状化粒子のうち45,033 gを35分間乾燥させる。
【0144】
8メッシュスクリーンを有するQuadro Comil(登録商標)ミルを用いて、コーティング済み顆粒のサイズを一定にする。顆粒をTote(登録商標)タンブラーに送り、281.7 gのステアリン酸マグネシウムと混合し潤滑化する。
【0145】
次いで塩酸ヒドロコドン/塩酸ナルトレキソンを含む薬物組成物と押し出し組成物とを、Kilian(登録商標)タブレットプレス上で圧縮して二層タブレットにする。まず薬物組成物をダイのキャビティに添加して予備圧縮する。その後135 gの押し出し組成物を添加し、3メトリックトンの圧力ヘッドで層を押圧することにより、層が互いに接触する直径11/32インチ(0.873 cm)の配列体とする。
【0146】
二層配列体に半透壁をコーティングする。壁形成組成物は、39.8%のアセチル含有量を有する100%セルロースアセテートを含む。壁形成組成物をアセトン:水(95:5 wt:wt)共溶媒中に溶解することにより、4%固体溶液を生成する。24インチ(60 cm) Vector(登録商標) Hi-Coaterを用いて壁形成組成物を二層構造体の上および周囲にスプレーする。次に半透壁に20ミル(0.508 mm)の出口通路を1つ開けることにより、薬物ヒドロコドン層を剤形の外面部と連結させる。残りの溶媒を、温度45℃、湿度45%で72時間に亘って乾燥することにより除去する。次いで浸透性投与系を45℃で4時間に亘って乾燥させることにより余分な水分を除去する。
【実施例3】
【0147】
この予言的実施例では、ヒドロコドン5 mg/ナルトレキソン0.0625 mgの徐放性カプセルを以下に示す表3の処方で調製する。
【0148】
【表3】
【0149】
上記の処方を以下の手順に従って調製する。
【0150】
1.ステアリン酸アルコールフレークをインパクトミル内を通過させる。
2.塩酸ヒドロコドンと、塩酸ナルトレキソンと、ステアリン酸と、ステアリン酸アルコールと、Eudragit RSPOとを適切なブレンダ/ミキサ中で混合する。
3.混合した材料を高温でツインスクリュー押出器に連続的に送り、得られたストランドをコンベア上に収集する。
4.ストランドをコンベア上で冷却する。
5.ペレタイザを用いてストランドを1 mmのペレットに切断する。
6.微細なペレットおよび大きすぎるペレットに関してペレットをスクリーニングし、約0.8〜1.4 mmの範囲の許容可能なサイズにする。
7.120 mg/カプセルの重さになるようにカプセルを充填する(サイズ2のカプセルに充填する)。
【実施例4】
【0151】
この予言的実施例では、ヒドロコドン5 mg/ナルトレキソン0.0625 mgの徐放性カプセルを以下の手順に従って調製する。
【0152】
まず即時放出型ヒドロコドンビーズを以下に示す表4の処方で調製する。
【0153】
【表4】
【0154】
プロセス
1.薬物層溶液:塩酸ヒドロコドンとOpadryクリアとを水中に溶解する。
2.薬物ローディング:流体床乾燥器内でNuPareilビーズに薬物溶液をスプレーする。
3.コーティング:水中にOpadryバタースコッチを分散し、薬物をロードしたビーズにスプレーする。
その後、徐放性ビーズを以下に示す表5の処方で調製する。
【0155】
【表5】
【0156】
プロセス
1.制御型放出コーティング溶液:水中でクエン酸トリエチルを均質化する。Eudragit(登録商標) RS30DおよびEudragit(登録商標) RL30Dに分散液を添加し、その後混合物にCab-O-Sil(登録商標)を添加する。
2.シールコーティング溶液: Opadry(登録商標)クリアを水中に溶解する。
3.コーティング:流動化床ボトムスプレー法を用いて、制御型放出コーティング溶液、次いでシールコーティング溶液を塩酸ヒドロコドンIRビーズに適用する。
4.硬化:コーティング済みビーズをトレイ上に載置し、45℃で24時間に亘ってオーブンで硬化する。
【0157】
ヒドロコドン/ナルトレキソン徐放性ビーズを生成するために、単位当たり0.0625 mgのナルトレキソンを上記処方に含ませてもよい。ナルトレキソンを塩酸ヒドロコドンと共に純水中に溶解して、その後NuPareilビーズにスプレーしてもよい。
【実施例5】
【0158】
実施例5では、単一施設プラシーボ制御二重盲検無作為9処置3期間交差試験を、オープンレーベルスクリーニングフェーズと共に行った。試験は、経口ナルトレキソン(NTX)を経口即時放出型ヒドロコドン(HYIR)のアゴニスト作用と併用することによる、18歳〜45歳の、体重約45〜100 kgの範囲で且つ理想体重から15%以内の健常な成人男女における換気量に与える影響を評価するために行った。
【0159】
試験は、最長14日間のスクリーニングフェーズと、最長5日間のオープンレーベルHYIR滴定フェーズと、二重盲検フェーズ(1期間1日の処置を3期間行い、各処置期間の間に24時間の洗浄期間を挟む)と、最後の処置期間の後に行う最長14日間の試験終了時の往診から構成された。総試験期間は少なくとも39日間であった。
【0160】
登録前に、試験対象患者基準と除外基準とを用いて各被験者は試験に参加する資格を有すると認められた。各被験者から詳細な医療履歴を得た。オープンレーベルHYIR滴定フェーズを始める前に全被験者が以下のスクリーニング手順を終了した:身体検査、ECG測定、生命徴候、および臨床試験(血液学、化学、尿検査、HIVスクリーニング、肝炎スクリーニング、薬物スクリーニング、血中アルコールテスト、および妊娠テスト)。
【0161】
被験者は参加基準を満たした後、オープンレーベルHYIR滴定フェーズに参加した。オープンレーベルHYIR滴定フェーズは、悪影響を最小に保ちつつ呼吸運動の検出可能な変化を引き起こす、HYIRの最高許容投与量を決定するために計画された。呼吸運動の検出可能な変化は、投与後60分と90分の時点、または投与後90分と120分の時点で20 L/分のMV(換気量)において、投与前からのPETCO2(終末呼気二酸化炭素濃度(Torrで表す))の増加が少なくとも3 Torrであることと定義されている。呼吸運動の検出可能な変化を引き起こしたHYIRの最高許容投与量は、この試験の二重盲検部分で各被験者に投与するHYIRの量として用いた。被験者はCO2再呼吸テストで用いるスピロメータを作動する訓練を受けた。その後各被験者に対して最高3回に分けて滴定セッションを行い、HYIRを15 mg、20 mg、25 mgと順に多くなるように投与した。滴定セッション間には24時間の洗浄期間を設けた。被験者が許容不可能な悪影響を呈することなく25 mgのHYIRを投与されるまで、あるいは投与により許容不可能な悪影響が現れるまで、オープンレーベルフェーズを続けた。被験者にHYIRを投与して許容不可能な悪影響が現れた場合、許容不可能な悪影響が見られなかったHYIRの最高投与量を二重盲検フェーズで用いた。各滴定セッション中に行われたCO2再呼吸テストによって、処置前30分(0時間)並びに投与後30分、60分、90分、120分および180分の時点でのMV値とPETCO2値を得た。MVが変化した被験者に、試験の二重盲検フェーズを行った。
【0162】
33人の被験者がスクリーニングフェーズに登録された。13人が呼吸器のオピオイド感受性またはオピオイド不耐性の欠如のために離脱した。20人の被験者に対し無作為に二重盲検フェーズを行い、18人の被験者が二重盲検フェーズを終了した。
【0163】
試験薬物、投与形態、剤形、単位強度、並びに二重盲検フェーズのテスト処置および基準処置は以下の通りであった。
【0164】
テスト処置
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+NOS(ナルトレキソン経口溶液)プラシーボ
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+0.125 mg NOS
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+0.25 mg NOS
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+0.375 mg NOS
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+0.5 mg NOS
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+0.75 mg NOS
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+1.5 mg NOS
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+3.0 mg NOS
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+8.0 mg NOS
【0165】
基準処置
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+NOSプラシーボ
【0166】
HYIRの5 mgタブレットはAAI Pharma, Wilmington, NCから供給された。
【0167】
NOSを処方するために塩酸ナルトレキソン粉体(Mallinckrodt Chemical Inc., St. Louis, MO)を用いた。ナルトレキソン粉体の必要量を計量し、50 mlの蒸留水および50 mlの単純シロップ中に別々に溶解してNFで最終容量100 mlを得た。これらの濃縮物(concentrations)は各処置期間中に同一容量(10 ml)のNOSが投与されることを可能にした。
【0168】
NOSプラシーボは苦味成分であるビターガード(安息香酸デナトニウム、NF)粉体を含んでいた。NOSプラシーボは、NOSの調製で用いたものと同一のビヒクルを用いて調製した。プラシーボ溶液の外見および味は活性溶液に似ていた。NOSプラシーボの投与容量(10 ml)は活性NOSの投与容量(10 ml)と合致させた。
【0169】
二重盲検フェーズにおいて、被験者に、オープンレーベルフェーズで決定した有効投与量(15、20または25 mg)のHYIRと、9つの考えられるNOS(ナルトレキソン経口溶液)処置(プラシーボ、0.125、0.25、0.375、0.5、0.75、1.5、3.0または8.0 mg)のうちの3つとを3期間に亘る交差臨床試験という形態で投与した。各被験者を6時間に亘って絶食させた後に、HYIRとNOSとを投与し、絶食を投与後3時間続けた。試験薬物投与の少なくとも30前(0時間)並びに投与後30分、60分、90分、120分および180分の時点でCO2再呼吸テストを行った。
【0170】
評価基準
薬力学:CO2再呼吸テストの結果を用いて、換気量に対するHYIRの影響およびHYIRとNOSとの影響を測定した。
【0171】
安全性:有害イベント、臨床試験結果、生命徴候、身体検査、および心電図(ECG)測定結果を用いて安全性を評価した。
【0172】
統計方法
PETCO2に対するMVのプロットから引き出した薬力学変数は、20および30 L/分のMV率(20リットルおよび30リットルのインターセプト値)におけるとMV/PETCO2回帰直線の傾斜とを含んでいた。投与前からの最大変化量(最大可能効果、MPE)を試験のオープンレーベルフェーズ(MPE(OL)=最大呼吸抑制)および二重盲検フェーズ(MPE(DB)=HYIR+NTXによる呼吸抑制)における各変数(MPE20、MPE30およびMPEslope)について計算した。二重盲検フェーズにおける各処置による最大呼吸抑制率(%MPE)を、各変数についてMPE(DB)/MPE(OL)x100という比から計算した。
【0173】
一次薬力学変数は、二重盲検フェーズでの各処置における20および30リットルインターセプトの%MPE (それぞれ%MPE20および%MPE30)であった。二次的薬力学変数は、%MPE並びに二重盲検フェーズのMPE20、MPE30およびMPEslopeの傾斜を含んでいた。これらの値を記述的統計を用いて処置群ごとにまとめ、無作為に選ばれた被験者、固定期間および固定処置というパラメータを伴う混合影響の分散分析(ANOVA)モデルを用いて分析した。さらに線形対照テストを用いて、NOS投与量と%MPE20および%MPE30との投与量−反応関係を調査した。
【0174】
結果
薬力学:
オープンレーベル20リットルおよび30リットルインターセプトMPEおよび傾斜値の効果サイズ分析により、呼吸抑制の最も高感度な(平均値についてのバラツキが最小である)測定値は20リットルおよび30リットルインターセプトMPE値であることが判明した。
【0175】
NTXの投与量が増加すると、呼吸抑制が統計的に有意な程度まで減少する傾向が見られた。これは、全ての処置において、CO2再呼吸テストから引き出された%MPE20、%MPE30、および二重盲検MPE20およびMPE30値に関して見られた。これらのデータは投与量依存的なHYIRにより誘導される呼吸抑制性のアンタゴニスト作用を示している。
【0176】
安全性:
呼吸抑制をもたらすために用いた15、20、25 mgのHYIR投与量、またはこれを0.125〜8.0 mgの範囲のNTX投与量と組み合わせたときに関して新たに同定された安全上の懸念はなかった。
【0177】
結論
0.125〜8.0 mgの範囲の経口NTXは投与量に依存して、15、20または25 mgのHYIRによって誘発される呼吸抑制を阻止した。安全上の新たな懸念も予期せぬ懸念もなかった。
【実施例6】
【0178】
実施例6は、維持量(60〜90 mg)の経口メタドンを毎日投与されている男女の被験者に対して行ったオープンレーベルフェーズと二重盲検処置フェーズからなった。維持量のメタドンは予定された各期間の前日に被験者に与えられた。維持量のメタドンを与えてから16時間後以降かつ22時間後以内に試験薬物を投与した。14人の被験者を試験に登録した(オープンレーベルフェーズ(ナルトレキソンの投与量を徐々に増加させる安全性評価)に2人、二重盲検フェーズに12人)。
【0179】
オープンレーベルフェーズ
オープンレーベルフェーズでは、維持量メタドン治療を受けている被験者に対するプロトコルで計画した2つのナルトレキソン投与量(0.75および2.0 mg)の安全性を評価した。試験のこのフェーズは、試験薬物の投与前に行われる最長14日間のスクリーニング往診と、ナルトレキソン定滴往診とから構成された。ナルトレキソン滴定往診中、2人の被験者に0時間の時点で30 mgのヒドロコドンと0.125 mgのナルトレキソンとを投与し、続く4時間に亘って1時間おきにさらにナルトレキソンを投与し、累積量で最高2.0 mgまで投与した。オープンレーベルフェーズでは、メタドンによる救済が必要となるまで、いずれの被験者にも累積量で1.0 mgを超えるナルトレキソンは投与しなかった。これら2人の被験者に見られた禁断症状の強度のために、試験で用いるナルトレキソンの投与量をプラシーボ0.75 mgおよび2.0 mgからプラシーボ、0.25 mgおよび0.5 mgに変更した。
【0180】
二重盲検フェーズ
二重盲検フェーズは、ナルトレキソン投与とナルトレキソンプラシーボ処置が無作為に選択される3期間に亘って3様式で行う無作為の交差試験として設計された。。試験のこのフェーズは、特定の処置シーケンスが無作為に選択される前に最長14日に亘って行われるスクリーニング往診と、これに続く二重盲検研究対象薬物を投与する3回の往診から構成された。
【0181】
各処置シーケンスは、1期間が4時間の処置を3期間行い、処置期間の間には48時間の洗浄期間が設けられた。各期間では、各被験者に30 mgのヒドロコドンと1〜3つの異なる量のナルトレキソン(プラシーボ、0.25 mgまたは0.5 mg)とを投与した。各被験者が二重盲検フェーズに参加した総期間は約20日間であった。
【0182】
8人の被験者が試験を終了した後、試験内容から0.5 mgのナルトレキソン投与を削除した。残り4人の被験者を登録し、ナルトレキソンを2つの投与量だけ、即ちプラシーボと0.25 mgナルトレキソンとを投与して試験を終了した。元々の無作為スケジュールおよび処置シーケンスは引き続き用いたが、処置シーケンスから0.5 mgのナルトレキソンを投与する期間を削除した。0.5 mgの投与量を削除した後に登録した被験者が試験に参加した期間は約17日間であった。
【0183】
試験の設計は、0.25および0.5 mgのナルトレキソン経口投与と組み合わせて経口投与された30 mgのヒドロコドンが、維持量メタドン治療を受けている被験者におけるいくつかの主観的および客観的測定値に与える影響の時間的経過および大きさを評価するのに適していた。この結論は、以下の試験設計特徴に基づいている。
【0184】
試験の偏りは、2(3x3)ラテン方格(但し0.5 mgのナルトレキソンによる処置は特定の被験者においては終了させた)、試験対象薬物の二重盲検投与、および無作為ナルトレキソン投与量としての試験設計によって制御した。
【0185】
オープンレーベルフェーズによって、この被験者集団が許容することができるナルトレキソン投与量の選択が可能になった。試験のオープンレーベルフェーズで見られた禁断症状の強度のために、試験で用いるナルトレキソンの投与量を0.75および2.0 mgから0.25および0.5 mgに減少した。
【0186】
嗜癖重症度指標を用いて被験者の依存度/常用癖を実証した。
【0187】
薬力学変数は、オピオイドの公知の生理学的および主観的影響の測定基準であった。
【0188】
生理学的薬力学変数は、皮膚温度と瞳孔の直径の測定値であった。オピオイドアゴニストは末梢動脈と静脈の拡張を引き起こすこと、および瞳孔に分布する副交感神経に対する興奮活性のために瞳孔を収縮することが知られている。
【0189】
この試験の主観的および客観的薬力学変数は、オピオイド薬物乱用の可能性および依存度の測定基準である主観的および観察者薬物影響スケール、オピオイド依存症の個人におけるオピオイド離脱症状および維持をモニタするための認知された測定基準である主観的および観察者症状評価スケール、オピオイド依存症の個人における乱用の可能性の主観的測定基準であるストリートバリュー評価アンケート、および薬物の区別および乱用の可能性を測定するために設計されたアンケートである薬物識別アンケートを含んだ。
【0190】
この試験の安全性パラメータは、有害イベント、臨床試験、心電図、および生命徴候であった。
【0191】
この試験のコントロール処置は30 mgのヒドロコドンおよびナルトレキソンプラシーボであった。
【0192】
各被験者は各期間の終了時に1日分のメタドン投与量を受け取ることになっていた。しかし被験者が許容不可能な離脱症状を経験した場合は、期間中のどの時点でも通常投与されている量のメタドンを救済薬として受け取ることができた。この試験の期間に投与される投与量30 mgのヒドロコドンはメタドンの経口維持量60〜90 mgと同等であった。
【0193】
この試験のオープンレーベルフェーズと二重盲検フェーズの両方に登録した被験者は、1日分の経口の維持量(60〜90 mg)のメタドンを受け取ることになっており、その結果物理的にオピオイドに依存することが予想された。
【0194】
薬力学パラメータは、瞳孔の直径、皮膚温度、主観的および観察者薬物影響スケール、並びに主観的および観察者症状評価スケールを含み、パラメータの測定は各テスト処置投与前0.5時間以内(投与前)並びに投与後0.25、0.5、1、2、3および4時間後に行った。ストリートバリュー評価アンケートは投与後0.25、0.5、1、2、3および4時間後に終了させた。薬物識別アンケートは投与後1および3時間後に終了させた。
【0195】
安全性測定は、身体検査、臨床試験(血液学および血液化学)およびECGを含み、スクリーニング往診および試験終了時または早期終了の場合の往診で行った。スクリーニング時、投与前、投与後0.25、0.5、1、2、3および4時間後、並びに試験終了時に生命徴候および酸素飽和を記録した。有害イベントは初日の試験薬物投与時から各被験者を試験から解放するまで収集した。
【0196】
薬力学測定基準
この試験で用いた薬力学測定基準は以下の通りである。
【0197】
薬物影響スケール(主観的および観察者)
主観的薬物影響スケールは、被験者が異なるテスト処置で経験した可能性がある4つの事項を評価した。
【0198】
この感覚が好きだ
悪い効果
気分が悪い
良い効果
【0199】
被験者に、4つの経験に関してどのように感じたかを0〜10の分類別の可視アナログスケール上でランクづけするように依頼した。スコアが高いほどオピオイドアゴニストの影響(陶酔感)が高いことを反映し、スコアが低いほどオピオイドアゴニストの影響が低下していること、またはアンタゴニスト作用(離脱症状)が増加していることを示した。
【0200】
観察者薬物影響スケールは、被験者が異なるテスト処置で示した可能性がある4つの事項を評価した。
【0201】
楽しい
不快な
具合が悪い
陶酔感
【0202】
観察者に、被験者が4つの経験の各々に関してどのように感じたと思うかを0〜10の分類別の可視アナログスケール上でランクづけするように依頼した。スコアが高いほどオピオイドアゴニストの影響(陶酔感)が高いことを反映し、スコアが低いほどオピオイドアゴニストの影響が低下していること、またはアンタゴニスト作用(離脱症状)が増加していることを示した。
【0203】
症状評価スケール(主観的および観察者)
主観的症状評価スケールは、オピオイドレセプター活性または禁断症状という症状を、アンタゴニスト項目およびその強度レベルに関して評価するために被験者によって用いられた。
【0204】
【0205】
これらの症状は1〜3のスケールで評価された。
【0206】
全く感じない。
ある程度感じる。
大いに感じる。
【0207】
スコアが高いほどオピオイドアゴニストまたはアンタゴニスト症状が増加していることを反映し、スコアが低いほどオピオイドアゴニストまたはアンタゴニスト症状が低下していることを示した。
【0208】
観察者症状評価スケールアンケートは、被験者が示したオピオイドレセプターアゴニストまたはアンタゴニスト活性およびその強度レベルを示す可能性のある徴候を評価するために用いられた。
【0209】
【0210】
これらの症状は1〜4のスケールで評価された。
【0211】
全くない。
相対的に目立たないが良く観察すると気づく。
かなり明らかである。細かく観察しなくても気づく。
非常に明らかである。持続的であるか、または被験者にとって煩わしく見える。
【0212】
スコアが高いほどオピオイドアゴニストまたはアンタゴニスト徴候が増加していることを反映し、スコアが低いほどオピオイドアゴニストまたはアンタゴニスト徴候が低下していることを示した。
【0213】
瞳孔の直径
2倍レンズを取り付けたPolaroid(Cambridge, MA)カメラを用いて常に周囲光がある状態で被験者の目を撮影した。各写真からの瞳孔の直径を測径器を用いてミリメートル単位で測定した。各期間で同じ目を用いて全ての決定を行った。測定に用いた目を記録した。
【0214】
薬物識別アンケート
薬物識別アンケートは、10の薬物カテゴリーのリストからなり、オピオイド乱用集団になじみのある文言が用いられた。被験者は、試験薬物がどのカテゴリーに最も近いかを選択した。アンケートには以下のカテゴリーをリストアップした。
【0215】
ブランクまたはブラシーボ
アヘン剤(例えばモルヒネ、ヘロイン、コデイン、メタドン)
アヘンアンタゴニスト(例えば、ナロキソン、ナルトレキソン)
抗精神病薬または神経弛緩薬(例えば、ハルドール、ステラジン)
バルビツール酸系催眠薬および睡眠薬(例えば、クアールード、ペントバルビタール、セコナール)
抗鬱薬(例えば、エラビル、イミプラミン)
PCPまたは幻覚剤(例えば、LSD、メスカリン、MDA、STP)
ベンゾジアゼピン(例えば、バリウム、リブリウム、アチバン、キサナックス)
コカインまたは興奮剤(例えば、アンフェタミン、デキセドリン、リタリン)
その他
【0216】
ストリートバリュー評価
被験者に「街でこの薬物を買うのにいくら払いますか」と質問する。
【0217】
被験者はその後薬物の価格はいくらだと思うかを直接CRFに記録する。
【0218】
皮膚温度
使い捨て温度プローブ付きのデュアルチャネル、デュアルディスプレイ電子温度計を用いて皮膚温度を測定した。温度を摂氏で記録した。
有害イベント
【0219】
有害イベントとは、医薬品と関係があると考えられるか否かにかかわらず一時的にその医薬品の使用と関連づけられる、好ましくなく意図しない何らかの徴候(実験室での異常の発見を含む)、症状、または疾病であった。自発的に報告されたか調査員が観察したかにかかわらず、試験薬物を最初に投与してから被験者を試験から解放するまでの間に起こったすべての有害イベントを、有害イベントの形式で報告した。医学的干渉を必要とする有害イベントが起こった場合、適切な資格を有しライセンスを有する医療担当者が適切なサポートおよび/または信頼できる治療を提供した。
【0220】
全体的結論
この試験は、維持量メタドン治療を受けている被験者において、30 mgのHYIRの経口投与と組み合わせてある範囲の量のNTXを経口投与することが、オピオイドアゴニストおよびアンタゴニスト活性の様々な主観的および生理学的測定値に与える影響を特徴づけるために設計された。
この被験者集団では、30 mgのHYIRの経口投与は有意な主観的または生理学的オピオイドアゴニスト活性を引き起こさなかった。HYIRおよびNTXプラシーボによる処置の後、全ての薬力学変数の投与前からの変化は最小であった。この処置を受けた12人の被験者のうち4人が救済用のメタドン投与を必要とした。これは突然禁断が現れている可能性を示している。
【0221】
この試験の一次薬力学変数は、「この感覚が好きだ」「良い効果」「悪い効果」および「気分が悪い」という質問に対する平均PDmax(最大投与前スコア)値であった。主観的薬物影響スケールにおける4つの質問のすべてにおいて、平均PDmax値に対してNTX投与量に関係する影響が見られた。NTXの投与量を0.25から0.5 mgに増加した結果、各質問に対する投与前スコアからの最大変化がますます否定的になった。このことはすべての場合において、オピオイドアゴニストの影響に対する、NTX投与量に関係するアンタゴニスムを示している。NTXプラシーボ処置とNTXの投与量0.25 mgとの間には、「この感覚が好きだ」および「悪い効果」という質問に関して統計的に有意な差異があった。NTXプラシーボ処置とNTXの投与量0.5 mgとの間には、「気分が悪い」以外の全ての質問に関して統計的に有意な差異があった。
【0222】
主観的および観察者症状評価スケールでは、アゴニストの合計値以外の二次薬力学変数に対して、NTXの投与量に関係する影響があった。但しこれは必ずしも統計的に有意ではない。NTX投与量0.25 mgは、感情が否定的になる傾向(オピオイドアゴニスト影響の低下)とアンタゴニスト活性の増加(禁断症状の傾向)の閾値投与量であった。NTX投与量0.5 mgは、主観的および観察者薬物影響スケールにおいて示されたNTXプラシーボ処置に対する統計的に有意な差異、主観的および観察者症状評価スケールから得られたアンタゴニスト合計値、および瞳孔の直径により、禁断症状の強力な証拠を提示した。0.25および0.5 mgのNTXを投与された被験者のそれぞれ約60%および90%が救済用のメタドン投与を必要とした。
【0223】
オピオイド薬物の数々の測定のPDmaxおよびAUCに対する、3期間の各々における処置手段を示す図面(図1〜図12)は、HYIR+NTXプラシーボによる処置とHYIR+0.25 mgのNTXによる処置とHYIR+0.5 mgのNTXによる処置との間で3期間中の変化の傾向が異なることを示した。観察された手段に見られる傾向は、HYIR+NTXプラシーボ投与による処置後の3期間に亘って主観的および生理学的オピオイドアゴニスト影響が増加し、且つHYIR+0.25 mgのNTXまたはHYIR+0.5 mgのNTX投与による処置後の3期間に亘ってオピオイドアンタゴニスト効果が増加するという仮定に合致する。
【0224】
0.25または0.5 mgのNTXの経口投与を30 mgのHYIR投与と共に投与することの安全性の測定を行ったが、この被験者集団については新しいまたは予期しない安全上の懸念は示唆されなかった。NTX投与量が増加するにつれて、各被験者のオピオイド離脱症状に共通して関連づけられる、処置に伴う有害イベントの数は増えたが、処置に伴う有害イベントのほとんどは軽度か中程度であった。1被験者のみが臨床的に顕著な異常実験値を呈したが、これはこの被験者の過去の医療履歴にリストアップされた状態によるものであった。臨床的に顕著な生命徴候異常の発生は被験者および処置の両方に関して隔離された事項であった。
【0225】
結論として、0.25および0.5 mgのNTX経口投与量は、維持量メタドンを投与されている被験者において有害な影響を引き起こすことが判明した。NTXは投与量に依存して、否定的な感情の状態および禁断症状を増加させる。経口NTX(0.25または0.5 mg)と経口HYIR(30 mg)の組み合わせは、新しいまたは予期しない安全上の懸念をもたらさなかった。実際、HYIRに低量のNTXを添加すると、魅力が低減し、そのため身体的にオピオイドに依存している被験者においてHYIRの乱用傾向の可能性が低減した。
【実施例7】
【0226】
実施例7は、プラシーボ制御型二重盲検無作為4処置4期間交差試験として行った単一施設試験からなった。これは単一盲検フェーズを含んだ。各処置シーケンスは、少なくとも5日間の洗浄期間により分けられた、1期間が4時間である4処置期間からなった。各処置期間において、各被験者に15 mgの経口HYIRおよび、プラシーボ、0.25、0.5または1.0 mgのNTXのいずれかを投与した。この試験の総時間はスクリーニングを含めて約52日間であった。
【0227】
スクリーニングフェーズ
無作為化によりこの試験の二重盲検部分を行う前に、スクリーニングフェーズを最長21日間に亘って行った。被験者は熱不快度テストの訓練セッションに参加した。この訓練は、43℃、46℃および49℃に加熱した銅塊を順に前腕の指定された部位に5秒以内の時間に亘って適用することを含んだ。各適用後、被験者は100 mmの可視アナログスケール(VAS)を用いて疼痛の強度を評価した。調査員の裁量で被験者が常に信頼できるVASスコアを生成できるようになるまで、この手順を新しい肌部位で繰り返した。満足にスクリーニングフェーズを終了することができなかった被験者を試験から除いた。
【0228】
単一盲検フェーズ
スクリーニングフェーズの終了後、以下のように単一盲検フェーズで熱不快度評価を行った。
【0229】
前腕の部位を選択し洗浄可能マーカーでマーキングした。ベースラインの生命徴候を記録した。前腕の所定の部位に局部麻酔薬(EMLA(登録商標)クリーム、AstraZeneca, Wilmington, DE)を塗った。約1.5時間後(麻酔薬が効果を呈した後)に、クリームを除去し、52℃に加熱した銅塊を用いて3分間に亘って熱刺激を前腕の部位に適用した。局部麻酔から感覚が回復するまで約1時間待った。その後各被験者にプラシーボとしての7.5 mg HYIRタブレット2個とプラシーボとしてのNTXタブレット2個とを経口投与した。
【0230】
投与後1.5時間の時点で、生命徴候を記録し熱不快度テストを行った。熱不快度テストは、43℃、46℃および49℃に加熱した銅塊を順に部位に5秒間適用することから構成された。各適用後、被験者は100 mmのVASスケールを用いて疼痛の強度を評価した。これらの測定から得られたVASスコアを加算し、60 mm以上の合計値を出した被験者のみをスクリーニングプロセスに進ませた。
【0231】
スクリーニングに進んだ被験者に、7.5 mgのHYIRタブレット2個とプラシーボとしてのNTXタブレット2個とを投与した。HYIRの投与後1.5時間の時点で、3つの温度の各々についてテスト測定を繰り返した。VASの疼痛スコアの合計値が前回のテストセッションに比べて少なくとも20 mm減少した被験者を、この試験の二重盲検部分に参加するに適していた。
【0232】
二重盲検フェーズ
少なくとも5日後、単一盲検フェーズで成功裡に終了した被験者を無作為に試験の二重盲検フェーズに選択した。健常な男女志願者をこの試験に登録した。各被験者は交差計画の4処置期間に参加し、15 mgのHYIRをNTXの4つの投与量(プラシーボ、0.25、0.5および1.0 mg)の各々と共に投与された。この二重盲検試験は、HYIRの鎮痛効果に対するNTXの影響を評価するために設計された。試験は、痛覚過敏症である健常な志願者における、15 mgのHYIRの鎮痛効果に対する0.25、0.5および1.0 mgの経口NTXの影響を評価した。この試験ではNTXの3つの異なる投与量(0.25、0.5および1.0 mg)を用い、プラシーボNTXと比較した。
【0233】
4試験期間の各々において処置シーケンス毎に同じ手順を続けた。被験者は単一盲検フェーズまたは前回の交差期間5日以上後に各処置期間を開始した。被験者は最初の処置期間の開始時に痛覚計(疼痛潜伏テストで用いる)を使用する訓練を受けた。各被験者は処置当日、少なくとも6時間絶食した後に施設に入った。生命徴候を記録し、尿による薬物スクリーニング、アルコールスクリーニング、および尿による妊娠スクリーニング(女性の被験者)を行った。被験者がテストを続けるには、全ての薬物スクリーニングの結果が試験薬物以外の全ての薬物について陰性である必要があった。
【0234】
各前腕においてテスト部位およびコントロール部位を選択し、洗浄可能マーカーでマーキングした。次いで、テスト部位に局部麻酔薬を塗った。約1.5時間後(麻酔薬が効果を呈した後)にクリームを除去し、52℃に加熱した銅塊を用いて3分間に亘って熱刺激をテスト部位に適用した。感覚が回復するまで約1時間待った後、以下のベースライン測定を行った:生命徴候、瞳孔計測、熱不快度(43℃、46℃および49℃において)、疼痛潜伏(放射型熱刺激適用後の潜伏)、症状評価スケール、薬物評価アンケート、およびオピオイド誘導型薬物影響アンケート。
【0235】
ベースライン評価が終了すると即座に、各被験者に15 mgの経口HYIRと、プラシーボ、0.25、0.5および1.0 mgのNTXのいずれかとを無作為コードに従って投与した。その後、ベースラインで行ったものと同じテスト測定を、投与後0.5、1、2、3および4時間の時点で行った。各時点でコントロール部位(熱刺激を受けていない方の前腕上の領域)での熱不快度テストおよび疼痛潜伏テストを行った。コントロール部位でのテストは、各時点でテスト部位でのテストより前に行った。各被験者は休息および回復のために5日以上待ってから次の期間のテストに戻った。追跡フェーズでは、被験者は試験の終了後7日以内にクリニックに戻ってテスト部位の最終チェックと被験者の実験室データの検討を受け、その後試験から正式に解放された。
【0236】
薬力学的測定
各テストセッションにおいて、投与前30分以内並びに投与後0.5、1、2、3および4時間の時点で以下の薬力学パラメータを記録した。
【0237】
熱不快度テスト(3つの温度において)
疼痛潜伏テスト
瞳孔の直径
症状評価スケールアンケート
オピオイド誘導型薬物影響アンケート
薬物評価アンケート
【0238】
グレード付き熱刺激を用いた熱不快度テスト
熱不快度テストは、直径1インチ(1”)の銅塊(Uniformed Services University of the Health Sciences, Bethesda, MD)に5秒間接触した後の被験者の不快感の感知を測定するために設計された。加熱(52℃)した銅塊を被験者の前腕に3分間適用することにより温熱損傷を引き起こした。その後テストは、被験者を3つの異なる温度、即ち43℃、46℃および49℃に加熱した銅塊に曝すことから構成された。各銅塊の必要温度は、Isotemp Dry Bath(Fisher Scientific, Indiana, PA)内に載置した加熱ブロック(モデル145および147、Fisher Scientific, Indiana, PA)に銅塊を挿入することにより得た。各測定時点で適用された銅塊の温度シーケンスは、試験の二重盲検部分を開始したときに各被験者に対して無作為に選択した。熱不快度テストはコントロール部位と実験皮膚部位との両方で行った。被験者は、100 mmのVASを用いて自分の不快度を評価した。スケールの左端は「全く不快感なし」となっており、右端は「私が耐えられる最大の不快感」となっていた。各被験者は、0 mmと100 mmとの間の水平スケール上に垂直の線でマーキングすることにより回答した。左端から垂直マークまでの距離を測定し、熱不快度の量的測定値として用いた。
【0239】
疼痛潜伏テスト
疼痛潜伏テストは、放射型熱刺激の付与から疼痛の開始(放射型熱刺激の自己終了により実証される)までの潜伏期間を秒で把握するために設計された。放射型熱刺激はモデル33テイルフリック鎮痛メータ(IITC Inc, Woodland Hills, CA)を用いてコントロール部位と実験皮膚部位との両方に適用した。各被験者は、最初の二重盲検期間の開始時にこの痛覚計を用いる訓練を受けた。痛覚計は被験者の皮膚から4インチという固定された距離に置かれ、選択された皮膚部位に向けて高強度の光を発した。調査員が痛覚計をオンにし、被験者は疼痛が始まったときにストップボタンを押すことによりテストを終了させた(痛覚計をオフにした)。被験者が高強度の光に曝されていた総時間を適切なCRFページに記録した。
【0240】
瞳孔計測
瞳孔計測は、瞳孔の直径に対する試験処置の影響を測定するために行った。改変された倍率2倍の接眼レンズ(John Hopkins University, Baltimore, MD)を有するPolaroidワンステップクローズアップカメラ(Polaroid Corporation, Cambridge, MA)とPolaroid600カラーフィルム(Polaroid Corporation, Cambridge, MA)とを用いて瞳孔を撮影した。実験室の背景照明は、モデルL-246 sekonic LUXメータ(Sekonic Co., Tokyo, Japan)を用いて測定した。被験者の眼窩にカメラを位置づけ、虹彩をレンズアダプタの開口部中央に整合させた。モデルCD-6C Mitutoyoデジタル測径器(Judge Tool Sales, Southport, CT)を用いて写真から瞳孔の直径をミリメートル単位で測定した。各被験者について常に同じ目を測定した。
【0241】
症状評価スケールアンケート
症状評価スケールアンケートは25項目から構成された。被験者には、各項目について「今どのように感じているか」を示すように指示した。各項目は3ポイントスケール、即ち「全くそう感じない」「ある程度そう感じる」または「大いにそう感じる」により評価した。12項目はアゴニスト項目に分類され、13項目はアンタゴニスト項目に分類された。アゴニスト項目はオピオイド投与に関連づけられる症状であった。アンタゴニスト項目はオピオイド離脱症状に関連づけられる症状であった。12のアゴニスト項目は、口数が多い、エネルギッシュである、身体が重い/動きがのろい、無頓着である、肌が痒い、幸せである、そわそわしている、安心している、頷き、リラックスしている、愉快である、および成り行きまかせである、であった。13のアンタゴニスト項目は、落ち着かない、吐き気がする、短気である、緊張している、神経過敏である、ほてりまたは冷え、肌がひんやりまたはじっとりとしている、赤面、あくびが出る、涙目、鼻水が出る、寒気/鳥肌、および発汗であった。
【0242】
オピオイド誘導型薬物影響アンケート
左端に「全くそうでない」とあり右端に「大いにそうである」となっている0〜100 mmのVASスケールを用いて、7つの薬物影響を評価した。これらの影響は、吐き気、嘔吐、めまい、嗜眠状態、便秘、掻痒、および口が渇くことを含んでいた。被験者は、その瞬間に薬物によってどのように感じたかを最もよく表す水平線上の距離に垂直のマークを記入した。
【0243】
薬物評価アンケート
左端に「全くそうでない」とあり右端に「大いにそうである」となっている0〜100 mmのVASスケールを用いて、3つの薬物に関する質問について評価した。これらの質問は、「今薬物の影響を感じていますか」「今感じている薬物の影響が好きですか」そして「今感じている薬物の影響が嫌いですか」であった。被験者は、その瞬間に薬物によってどのように感じたかを最もよく表す水平線上の距離に垂直のマークを記入した。
【0244】
有害イベント
AE(有害イベント)を、医薬品と関係があると考えられるか否かにかかわらず一時的にその医薬品の使用と関連づけられる、好ましくなく意図しない何らかの徴候(実験室での異常の発見を含む)、症状、または疾病と定義した。AEは、試験に登録した被験者に試験薬物を最初に投与した後に起こった場合にのみ、TEAE(処置による有害イベント)と分類した。TEAEの観察期間は、試験薬物を最初に投与してから期間4の終了後試験から解放されるまで、またはそれより早い中止時までであった。試験期間中に被験者が報告したか、または調査員/試験スタッフが観察したAEはすべて完全に記録した。医学的干渉を必要とするAEが起こった場合、適切な資格を有しライセンスを有する医療担当者が適切なサポートおよび/または信頼できる治療を提供した。研究の終了時/中止時にAEが解決しなかった場合は、解決するまで、これ以上の向上が望めないと調査員が決定するまで、または被験者に連絡がつかなくなるまで被験者をフォローした。
【0245】
薬力学的結果
熱不快度の平均VASスコアおよび疼痛潜伏、並びに痛覚過敏の誘導型薬力学測定値については、HYIR+プラシーボNTX処置とHYIR+0.25、0.5または1.0 mgのNTX処置との間で統計的に有意な差異はなかった。
【0246】
HYIR+0.5 mgのNTXおよびHYIR+1.0 mgのNTXの投与後には、HYIR+プラシーボNTXの投与後に比べて、瞳孔の直径AUC(投与前からの変化)に統計的に有意な増加があったが、PDmaxにはなかった。
【0247】
HYIRの主観的オピオイドアゴニスト影響に関して、処置間、またはNTXの投与量に関係する一貫した傾向の間で統計的に有意な差異は概してなかった。これは主観的症状評価スケールおよび主観的薬物評価アンケートによって評価された通りである。
【0248】
全体的結論
健常な志願者に対するこの試験の結果から以下の結論が導かれた。
【0249】
この試験で用いたNTXの投与量(0.25、0.5または1.0 mg)はいずれも、15 mgの経口HYIRによる鎮痛に対して統計的に有意な低下をもたらさなかった。これは熱不快度VASスコアおよび疼痛潜伏テストによって測定された通りである。
【0250】
15 mg投与量のHYIRの生理学的オピオイドアゴニスト活性に対して、NTX投与量に関係する一貫した影響はなかった。但しすべてのNTX投与量はHYIR誘導型瞳孔収縮を低減した。
【0251】
15 mg投与量のHYIRの主観的オピオイドアゴニスト活性に対して、NTX投与量に関係する一貫した影響はなかった。
【0252】
健常な志願者を、15 mgの経口HYIRと0.25、0.5または1.0 mgのNTXとの組み合わせで処置することに関連する安全上の懸念はなかった。
【0253】
一般的にオピオイドの使用と関連づけられる報告済みTEAEはの数は、NTX投与量の増加に伴って減少した。
【0254】
当業者には本発明の多くの他の改変が明らかであり、これらの改変も添付の請求の範囲の範囲に含まれるものとする。
【図面の簡単な説明】
【0255】
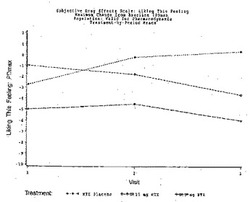
【図1】図1は実施例6の3処置期間の各々における、「この感覚が好きだ」という主観的薬物効果の、ベースラインからの最大変化(PDmax)を示す図である。
【図2】図2は実施例6の3処置期間の各々における、「この感覚が好きだ」という主観的薬物効果のPDmaxの曲線下面積(AUC)を示す図である。
【図3】図3は実施例6の3処置期間の各々における、「良い効果」という主観的薬物効果の、ベースラインからの最大変化(PDmax)を示す図である。
【図4】図4は実施例6の3処置期間の各々における、「良い効果」という主観的薬物効果のPDmaxの曲線下面積(AUC)を示す図である。
【図5】図5は実施例6の3処置期間の各々における、「気分が悪い」という主観的薬物効果の、ベースラインからの最大変化(PDmax)を示す図である。
【図6】図6は実施例6の3処置期間の各々における、「気分が悪い」という主観的薬物効果のPDmaxの曲線下面積(AUC)を示す図である。
【図7】図7は実施例6の3処置期間の各々における、「悪い効果」という主観的薬物効果の、ベースラインからの最大変化(PDmax)を示す図である。
【図8】図8は実施例6の3処置期間の各々における、「悪い効果」という主観的薬物効果のPDmaxの曲線下面積(AUC)を示す図である。
【図9】図9は実施例6の3処置期間の各々における、主観的「アンタゴニスト全症状スコア値」の、ベースラインからの最大変化(PDmax)を示す図である。
【図10】図10は実施例6の3処置期間の各々における、主観的「アンタゴニスト全症状スコア値」のPDmaxの曲線下面積(AUC)を示す図である。
【図11】図11は実施例6の3処置期間の各々における、瞳孔の直径の、ベースラインからの最大変化(PDmax)を示す図である。
【図12】図12は実施例6の3処置期間の各々における、瞳孔の直径のPDmaxの曲線下領域(AUC)を示す図である。
【技術分野】
【0001】
ヒドロコドン製剤は乱用の対象となることがある。ヒドロコドンは特定量を非経口投与した場合、同量を経口投与するよりも効力が高い場合がある。経口ヒドロコドン製剤の乱用の1形態ドは活性剤を溶液中に入れて注射することを含む。
【背景技術】
【0002】
従来、これらの薬物の非経口乱用を阻止するために、オピオイドアンタゴニストが特定のオピオイドアゴニストと組み合わされてきた。
【0003】
即時放出型ペンタゾシンとナロキソンとの組み合わせが米国で入手可能なタブレットとして利用されており、Sanofi-WinthropからTalwin(登録商標)Nxとして市販されている。Talwin(登録商標)Nxは、50 mg基剤と同等の即時放出型塩酸ペンタゾシンと、0.5 mg基剤と同等の塩酸ナロキソンとを含む。ドイツでは1978年から、疼痛を管理するためにチリジン(50 mg)とナロキソン(4 mg)とを含む固定併用治療が利用可能である(Valoron(登録商標)N, Goedecke)。ニュージーランドでは1991年に、疼痛治療用にブプレノルフィンとナロキソンとの固定併用が導入された(Temgesic(登録商標)Nx, Reckitt & Colman)。
【0004】
Kreekの米国特許第4,769,372号および第4,785,000号は、慢性疼痛または慢性咳に苦しむ患者に対し、腸運動障害を引き起こすことなく治療する方法を記載している。上記方法は、約1.5〜約100 mgのオピオイド鎮痛薬または鎮咳薬と、経口投与した場合に全身性アンタゴニスト活性をほとんど又は全く有しない約1〜約18 mgのオピオイドアンタゴニストとを含む投与単位を1〜2だけ、毎日1〜5回投与することを含む。
【0005】
Crainらの米国特許第5,472,943号は、オピオイドアンタゴニストを含むアゴニストを投与することにより2モードで作用するオピオイドアゴニストの鎮痛性を高める方法を記載している。
【0006】
ヒドロコドンはアセトアミノフェンとの組み合わせで市販されており、疼痛治療に適応される。市販されている商品名は、MallinckrodtのAnexsia(登録商標)、UCB PharmaのLortab(登録商標)、Watson PharmaceuticalsのNorco(登録商標)、AbbottのVicodin(登録商標)、およびEndo LabsのZydone(登録商標)である。
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明の目的及び要旨
本発明の目的は、ヒドロコドンの経口剤形を提供することである。
【0008】
本発明の特定の実施形態の目的は、他の剤形に比べて非経口および/または経口乱用されにくいヒドロコドンの経口剤形を提供することである。
【0009】
本発明の特定の実施形態の目的は、他の剤形に比べて転用されにくいヒドロコドンの経口剤形を提供することである。
【0010】
本発明の特定の実施形態の目的は、ヒドロコドンの経口剤形を用いて、剤形の乱用可能性を低減しながらヒト患者の疼痛を治療する方法を提供することである。
【0011】
本発明の特定の実施形態の目的は、乱用可能性の低い、ヒドロコドンの経口剤形を製造する方法を提供することである。
【課題を解決するための手段】
【0012】
上記および他の目的は以下に述べる本発明によって達成される。本発明は一つには、5〜20 mgのヒドロコドンまたはその製薬上許容可能な塩と、0.055〜0.56 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物であって、前記ナルトレキソンまたはその製薬上許容可能な塩と前記ヒドロコドンまたはその製薬上許容可能な塩とが0.011:1〜0.028:1の割合で含まれる医薬組成物を対象とする。
【0013】
特定の実施形態では、本発明は、約5 mgのヒドロコドンまたはその製薬上許容可能な塩と0.055〜0.14 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物を対象とする。
【0014】
特定の実施形態では、本発明は、約7.5 mgのヒドロコドンまたはその製薬上許容可能な塩と0.0825〜0.21 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物を対象とする。
【0015】
特定の実施形態では、本発明は、約10 mgのヒドロコドンまたはその製薬上許容可能な塩と0.11〜0.28 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物を対象とする。
【0016】
特定の実施形態では、本発明は、約15 mgのヒドロコドンまたはその製薬上許容可能な塩と0.165〜0.42 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物を対象とする。
【0017】
特定の実施形態では、本発明は、約20 mgのヒドロコドンまたはその製薬上許容可能な塩と0.22〜0.56 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物を対象とする。
【0018】
本明細書に開示する本発明の特定の実施形態では、剤形が前記ヒドロコドン、前記ナルトレキソンを徐放するか、または両方を徐放する。
【0019】
本明細書に開示する本発明の特定の実施形態では、剤形が、ヒト患者への定常状態での経口投与後少なくとも12時間に亘って有効に鎮痛を行う。
【0020】
本明細書に開示する本発明の特定の実施形態では、剤形が、ヒト患者への定常状態での経口投与後少なくとも24時間に亘って有効に鎮痛を行う。
【0021】
本明細書に開示する本発明の特定の実施形態では、剤形が、ヒドロコドンまたはその製薬上許容可能な塩と、ナルトレキソンまたはその製薬上許容可能な塩とを含むマトリクスを含み、前記ヒドロコドンまたはその製薬上許容可能な塩および前記ナルトレキソンまたはその製薬上許容可能な塩の両方が徐放性賦形剤中で実質的に互いに分散している。
【0022】
特定の実施形態では、本発明は、ヒドロコドン製剤の経口乱用の可能性を低減する方法であって、本明細書に記載の組成物を調製することを含む方法を対象とする。
【0023】
特定の実施形態では、本発明は、ヒト患者において疼痛を治療する方法であって、患者への定常状態での経口投与後少なくとも12時間に亘って有効に鎮痛を行う、本明細書に記載の医薬組成物を経口投与することを含む方法を対象とする。
【0024】
特定の実施形態では、本発明は、ヒト患者において疼痛を治療する方法であって、患者への定常状態での経口投与後少なくとも24時間に亘って有効に鎮痛を行う、本明細書に記載の医薬組成物を経口投与することを含む方法を対象とする。
【0025】
用語「徐放」は本発明の目的に関して、ヒドロコドンまたはその塩が、血液(例えば血漿)濃度(レベル)が、8〜24時間の期間に亘って、好ましくは1日2回または1日1回の処方を示す期間に亘って、治療範囲内(最小有効鎮痛濃度または「MEAC」より高い)であって且つ毒性レベル未満に維持されるような速度で剤形から放出されることと定義される。
【0026】
用語「非経口」は本明細書において、皮下注射、静脈注射、筋肉内注射、胸骨間注射、点滴技術または当該分野で公知の他の注入方法を含む。
【0027】
用語「ヒドロコドン」は他に特定しない限り、ヒドロコドン塩基を意味する。用語「ナルトレキソン」は他に特定しない限り、ナルトレキソン塩基を意味する。用語「塩」は製薬上許容可能な塩を意味する。
【0028】
用語「定常状態」は、系に到達する薬物の量がその系から出る薬物の量とほぼ等しいことを意味する。従って「定常状態」において、患者の身体は、薬物が血流に吸収されることにより患者の系に使用可能になるのとほぼ同じ速度で薬物を排除している。
【発明を実施するための最良の形態】
【0029】
発明の詳細な説明
本発明の剤形は、約5〜約20 mgのヒドロコドンまたはその製薬上許容可能な塩を含む。ヒドロコドンまたはその塩の特に好ましい投与量は約5 mg、約7.5 mg、約10 mg、約15 mgおよび約20 mgである。特定の実施形態では、ヒドロコドンまたはその塩を適切な製薬上許容可能な賦形剤で処方することにより、ヒドロコドンの徐放を実現する。
【0030】
本発明の剤形は約0.055〜約0.56 mgのナルトレキソンまたはその製薬上許容可能な塩を含む。ナルトレキソンまたはその塩の特に好ましい投与量は約0.0625 mg、約0.09375 mg、約0.125 mg、約0.1875 mgおよび約0.25 mgである。
【0031】
ヒドロコドンまたはその塩およびナルトレキソンまたはその塩は、一方または両方の剤を即時放出するように処方してもよいし、一方または両方の剤を徐放するように適切な製薬上許容可能な賦形剤と組み合わせてもよい。ナルトレキソンまたはその塩の徐放率は、ヒドロコドンまたはその塩の徐放率と同じでもよいし、異なっていてもよい。本発明の特に好ましい実施形態による剤形は、約5 mgのヒドロコドン塩と約0.0625 mgのナルトレキソン塩、約7.5 mgのヒドロコドン塩と約0.09375 mgのナルトレキソン塩、約10 mgのヒドロコドン塩と約0.125 mgのナルトレキソン塩、約15 mgのヒドロコドン塩と約0.1875 mgのナルトレキソン塩、および約20 mgのヒドロコドン塩と約0.25 mgのナルトレキソン塩を含む。ヒドロコドンの重酒石酸塩とナルトレキソンの塩酸塩が特に好ましい。
【0032】
本発明の特定の実施形態では、ナルトレキソンまたはその塩の開示された範囲は、身体的依存性被験者において、製剤の鼻腔内および非経口乱用を阻止するに十分な量であってよい。これは、製剤が、改ざんされ、そして鼻の粘膜に投与され、又は非経口に投与された場合に、ヒドロコドンのオピオイド効果を少なくとも部分的に阻止することにより実現される。好ましくはその量は、身体的依存性が最も高い個体に鼻腔内または非経口投与された結果、オピオイドの突然の中止後に見られる症状に酷似した、中程度から高度の離脱症状が現れるに十分な量である。離脱症状の最も一般的な症状は、瞳孔の拡張、悪寒と極度の発汗とが交互に現れること、腹部の痙攣、吐き気、嘔吐、筋肉の痙攣、過剰被刺激性、流涙、鼻漏、鳥肌、および心拍数の増加を含む。
【0033】
特定の実施形態では、剤形に安定剤を含むことにより、ナルトレキソンまたはその製薬上許容可能な塩の劣化を防止する。特定の実施形態では、剤形に用いられる安定剤は例えば、有機酸、カルボン酸、アミノ酸の酸塩(例えばシステイン、L-システイン、システインハイドロクロライド、グリシンハイドロクロライド、またはシステインジヒドロクロライド)、ソディウムメタビサルファイト(sodium metabisulphite)、アスコルビン酸およびその誘導体、リンゴ酸、イソアスコルビン酸、クエン酸、酒石酸、パルミチン酸、炭酸ナトリウム、炭酸水素ナトリウム、炭酸カルシウム、リン酸水素カルシウム、二酸化硫黄、亜硫酸ナトリウム、重硫酸ナトリウム(sodium bisulphate)、トコフェノール並びにその水溶性および油溶性誘導体(例えばトコフェルソラン(tocofersolan)または酢酸トコフェノール)、亜硫酸塩、重亜硫酸塩(bisulphite)および亜硫酸水素塩またはアルカリ金属、アルカリ土類金属および他の金属、PHBエステル、ガラート、ブチル化ヒドロキシアニソール(BHA)またはブチル化ヒドロキシトルエン(BHT)、および2,6-ジ-t-ブチル-.アルファ.-ジメチルアミノ-p-クレゾール、t-ブチルヒドロキノン、ジ-t-アミルヒドロキノン、ジ-t-ブチルヒドロキノン、ブチルヒドロキシトルエン、ブチルヒドロキシアニソール、ピロカテコール、ピロガロール、プロピル/ガラート、およびノルジヒドログアイアレチン酸(nordihydroguaiaretic acid)、並びに低級脂肪酸、フルーツ酸、リン酸、ソルビン酸および安息香酸並びにこれらの塩、エステル、誘導体および異性体化合物、パルミチン酸アスコルビル、レシチン、モノおよびポリヒドロキシル化ベンゼン誘導体、エチレンジアミン−四酢酸およびその塩、シトラコン酸、コニデンドリン(conidendrine)、炭酸ジエチル、メチレンジオキシフェノール、ケファリン(kephaline)、β,β'-ジチオプロピオン酸、ビフェニルおよび他のフェニル誘導体、これらの製薬上許容可能な塩およびこれらの混合物を含むがこれらに限られない。
【0034】
本発明の経口剤形はヒドロコドンとナルトレキソンとに加えてさらに、これらと共に相乗効果をもたらす又はもたらさない1以上の薬物を含んでもよい。したがって特定の実施形態では、製剤に非オピオイド薬物も含まれる。このような非オピオイド薬物は好ましくはさらに鎮痛をもたらし、例えばアスピリン、非ステロイド性抗炎症剤("NSAIDS";例えばイブプロフェン、ケトプロフェンなど)、N-メチル-D-アスパラギン酸(NMDA)レセプターアンタゴニスト(例えばモルフィナン(例えばデキストロメトルファンまたはデキストロファン)、またはケタミン)、シクロオキシゲナーゼ-II阻害剤("COX-II阻害剤")、および/またはグリシンレセプターアンタゴニストなどが含まれる。
【0035】
本発明の特定の好ましい実施形態は、本発明はさらに非オピオイド鎮痛剤を含むことにより、より低い投与量のヒドロコドンを使用することができる。非オピオイド鎮痛剤は例えば、NSAIDまたはCOX-2阻害剤である。より少ない量の一方または両方の薬物を使用することにより、ヒトにおいて有効な疼痛管理に関連して起こる副作用を低減することができる。
【0036】
適切な非ステロイド性抗炎症剤は、イブプロフェン、ジクロフェナク、ナプロキセン、ベノキサプロフェン、フルルビプロフェン、フェノプロフェン、フルブフェン(flubufen)、ケトプロフェン、インドプロフェン、ピロプロフェン、カルプロフェン、オキサプロジン、プラモプロフェン、ムロプロフェン、トリオキサプロフェン、スプロフェン、アミノプロフェン、チアプロフェン酸、フルプロフェン、ブクロキシン酸(bucloxic acid)、インドメタシン、スリンダク、トルメチン、ゾメピラク、チオピナク、ジドメタシン、アセメタシン、フェンチアザク、クリダナク、オキシピナク、メフェナム酸、メクロフェナム酸、フルフェナム酸、ニフルム酸、トルフェナム酸、ジフルリサル(diflurisal)、フルフェニサル(flufenisal)、ピロキシカム、スドキシカム、イソキシカム、これらの製薬上許容可能な塩、これらの混合物などを含む。これらの薬物の有用な投与量は当業者に周知である。
【0037】
N-メチル-D-アスパラギン酸(NMDA)レセプターアンタゴニストは当該分野で周知であり、例えばモルフィナン(例えばデキストロメトルファンまたはデキストロファン)、ケタミン、d-メタドン、およびこれらの製薬上許容可能な塩を含む。本発明の目的のために、用語「NMDAアンタゴニスト」は、さらにNMDAレセプター活性化の細胞内反応を阻止する薬物、例えばガングリオシド(例えばGM1またはGT1b)、フェノチアジン(例えばトリフルオペラジン)、またはナフタレンスルホンアミド(例えばN-(6-アミノテキシル(aminothexyl))-5-クロロ-1-ナフタレンスルホンアミド)を含むと解釈される。これらの薬物は、Mayerらの米国特許第5,321,012号および第5,556,838号において、習慣性薬物(例えば麻薬鎮痛剤:モルヒネ、コデインなど)に対する耐性および/または依存性の発生を阻害すると記載されている。これらの薬物はまた、Mayerらの米国特許第5,502,058号において、慢性疼痛を治療するとも記載されている。これらの米国特許はすべて参照により本明細書に組み込まれる。NMDAアンタゴニストは単体で含まれてもよいし、Mayerらの上記特許に記載されているように局部麻酔薬(例えばリドカイン)と共に含まれてもよい。
【0038】
グリシンレセプターアンタゴニストを用いて慢性疼痛を治療すること及びこれらの薬物の適応症(identification)は、Weberらの米国特許第5,514,680号に記載されている。
【0039】
COX-2阻害剤は当該分野で報告されており、シクロオキシゲナーゼ-2阻害を引き起こす多くの化学構造が知られている。COX-2阻害剤は例えば、米国特許第5,616,601号、第5,604,260号、第5,593,994号、第5,550,142号、第5,536,752号、第5,521,213号、第5,475,995号、第5,639,780号、第5,604,253号、第5,552,422号、第5,510,368号、第5,436,265号、第5,409,944号、および第5,130,311号に記載されている。これらの米国特許はすべて参照により本明細書に組み込まれる。特定の好ましいCOX-2阻害剤は、セレコシキブ、5-ブロモ-s-(4-フルオロフェニル)-3-[4-(メチルスルホニル)-フェニル]チオフェン、フロスリド(flosulide)、メロキシカム、ロフェコキシブ(rofecoxib)、6-メトキシ-2-ナフチル酢酸、ナブメトン、ニメスリド、N-[2-(シクロヘキシルオキシ)-4-ニトロフェニル]メタンスルホンアミド、1-フルオロ-4-[2-[4-(メチルスホニル)フェニル]-1-シクロペンテン-1-イル]ベンゼン、5-(4-フルオロフェニル)-1-[4-(メチルスホニル)フェニル]-3-トリフルオロメチル 1H-ピラゾール、N-[3-(ホルミルアミノ)-4-オキソ-6-フェノキシ-4H-1-ベンゾピラン-7-イル]メタンスルホンアミド、これらの混合物、およびこららの製薬上許容可能な塩を含む。COX-2阻害剤の投与レベルとしては、オピオイド鎮痛剤と組み合わせた場合、1日に体重1キロ当たり約0.005 mg〜約140 mgのオーダーが治療上有効である。あるいは1人の患者に対して1日に約0.25 mg〜約7 gのCOX-2阻害剤がオピオイド鎮痛剤と組み合わせて投与される。
【0040】
さらなる実施形態では、鎮痛剤以外の所望の効果を提供する非オピオイド薬物、例えば鎮咳剤、去痰薬、鬱血除去剤、抗ヒスタミン薬、局部麻酔薬などを含んでもよい。
【0041】
徐放性剤形
ヒドロコドン(またはヒドロコドン塩)および/またはナルトレキソン(またはナルトレキソン塩)は当業者に公知の任意の適切なタブレット、コーティング済みタブレットまたはマルチ微粒子製剤という形態で徐放性経口製剤として処方することができる。徐放性剤形は、マトリクスに含有させた徐放性材料をヒドロコドンまたはその塩と共に含むが、この場合ナルトレキソンまたはその塩を含んでもよいし含まなくともよい。例えば、ヒドロコドン塩を徐放性マトリクスに含有させ、ナルトレキソン塩をマトリクスとは別にしてもよいしマトリクスに含有させてもよい。
【0042】
特定の実施形態における徐放性剤形は、ヒドロコドンまたはその塩およびナルトレキソンまたはその塩の両方を含む1群の粒子を含んでもよい。他の実施形態では剤形は、ヒドロコドンまたはその塩を含む第1の粒子群とナルトレキソンまたはその塩を含む第2の粒子群とを含んでもよい。1または複数の粒子群を含む実施形態では、粒子の直径は約0.1 mm〜約2.5 mmであってよく、好ましくは約0.5 mm〜約2 mmであるとよい。上記に開示したように、ナルトレキソンまたはナルトレキソン塩は、ヒドロコドンまたはヒドロコドン塩を含む粒子に含有されてもよいし、別の粒子に含有されてもよい。あるいは、ヒドロコドンまたはヒドロコドン塩粒子を含むタブレットまたはカプセルに含有されてもよい。特定の実施形態では、粒子は水性媒質中において活性剤の放出をある徐放率で許容する徐放性材料でコーティングされる。コーティング剤は、記載された他の特性に加えて所望のインビトロ放出率を達成するように選択される。本発明の徐放性コーティング製剤は、平滑で外観が美しく、顔料および他のコーティング添加剤を支持することができ、非毒性、不活性で且つべたつきのない、強靱な連続薄膜を生成することができるべきである。
【0043】
コーティング済みビーズ
本発明の特定の実施形態では、疎水性材料を用いて、活性剤でコーティングされた不活性な薬剤ビーズ(例えばnu pariel 18/20ビーズ)にオーバーコーティングを施す。その後、得られた複数の固体徐放性ビーズを十分な量だけゼラチンカプセル中に入れることにより、周囲の液体(例えば胃液または溶解培地)に吸い込まれたり接触したりする際に有効な徐放量を提供するようにしてもよい。特定の実施形態では、ヒドロコドンまたはヒドロコドン塩を含む徐放性ビーズをナルトレキソンまたはナルトレキソン塩でさらにコーティングしてもよい。あるいはナルトレキソンまたはナルトレキソン塩を、徐放性ヒドロコドンまたはヒドロコドン塩ビーズと共に(例えば粉体混合物として、または別々のビーズに処方された形態で)カプセル中に入れてもよい。
【0044】
本発明の徐放性ビーズ製剤は、例えば胃液に、その後腸液に吸い込まれ曝されると、本発明の活性剤をゆっくりと放出する。本発明の製剤の徐放プロファイルは変更することができるが、その変更は例えばオーバーコーティングに用いる疎水性材料の量を変更すること、疎水性材料に可塑剤を添加する様式を変更すること、疎水性材料に対する可塑剤の量を変更すること、追加の成分または賦形剤を含ませること、製造方法を変更することなどによって行われる。最終生成品の溶解プロファイルもまた改変することができるが、その改変は例えば遅延剤コーティングの厚みを増減することによって行われる。
【0045】
本発明の活性剤でコーティングされたスフェロイドまたはビーズは例えば、活性剤を水に溶解し、次いでWusterインサートを用いて溶液を基質(例えばnu pariel 18/20ビーズ)にスプレーすることによって調製する。活性剤がビーズに結合することを補助するために、および/または溶液に色をつけるためなどに、ビーズをコーティングする前に追加の成分をさらに適宜添加する。例えばヒドロキシプロピルメチルセルロースなどを含み、色素を含むか含まない生成物(例えばColorcon, Inc.から市販されているOpadry(登録商標))を溶液に添加して混合(例えば約1時間)した後に得られた溶液を同じくビーズに塗ってもよい。その後得られたコーティング済み基質(この例ではビーズ)を緩衝剤(barrier agent)で適宜オーバーコーティングすることにより、活性剤を疎水性徐放性コーティング剤から分離してもよい。適切な緩衝剤の一例はヒドロキシプロピルメチルセルロースを含むものであるが、当該分野で公知のいずれの薄膜生成剤も用いることができる。緩衝剤は最終生成物の溶解率に影響を与えないことが好ましい。
【0046】
その後ビーズを疎水性材料の水分散液でオーバーコーティングしてもよい。疎水性材料の水分散液は好ましくはさらに有効量の可塑剤(例えばクエン酸トリエチル)を含む。エチルセルロースの予備処方された水分散液(例えばAquacoat(登録商標)またはSurelease(登録商標))を用いてもよい。Surelease(登録商標)を用いる場合、可塑剤を別途添加する必要はない。あるいはアクリル性ポリマーの予備処方された水分散液(例えばEudragit(登録商標))を用いることもできる。
【0047】
本発明のコーティング溶液は好ましくは薄膜生成剤、可塑剤および溶媒系(即ち水)に加えて、色素を含むことにより、美しく且つ他とは異なる外観を提供する。疎水性材料の水分散液に代えて又はこれに加えて、活性剤の溶液に色を付けてもよい。例えばアルコールまたはプロピレングリコールベースの色分散液、粉砕したアルミニウムレーキおよび乳白剤(例えば二酸化チタン)を用いてAquacoat(登録商標)に色を付けてもよい。これは水溶性ポリマー溶液に剪断力で色を添加し、その後可塑化したAquacoat(登録商標)に低い剪断力を用いることにより行う。あるいは本発明の製剤に色を付ける任意の方法を用いてもよい。アクリル性ポリマーの水分散液を用いた場合に製剤に色を付ける適切な成分は、二酸化チタンおよび色顔料(例えば酸化鉄顔料)を含む。但し、顔料を含有させるとコーティング剤の遅延効果が増す。
【0048】
上記剤を含む基質に、可塑化した疎水性材料を適用してもよい。これは当該分野で公知の適切なスプレー装置を用いてスプレーすることにより行う。好ましい方法によると、Wurster流動化床システムを用いる。これによるとアクリル性ポリマーコーティング剤をスプレーしている間に下から注入された空気流がコア材料を流動化し乾燥させる。コーティングされた基質が水溶液(例えば胃液)に曝されたときに上記剤の所定の徐放性を得るに十分な量の疎水性材料を適用してもよい。疎水性材料でコーティングした後、任意にビーズに薄膜生成剤(例えばOpadry(登録商標))をさらに適用する。このオーバーコーティングは、仮にあったとしても、ビーズの凝集を実質的に低減するために行う。
【0049】
本発明の徐放性製剤からの上記剤の放出には、1以上の放出改変剤を添加すること、またはコーティングにより1以上の通路を提供することにより、さらに影響を与えることができる(即ち所望の速度に調整することができる)。水溶性材料に対する疎水性材料の割合は、他の要因の中で特に、必要な放出率と選択される材料の溶解度特性とによって決定される。
【0050】
孔形成剤として機能する放出改変剤は有機性でも無機性でもよく、使用環境内でコーティング剤から溶解、抽出または浸出させることができる材料を含む。孔形成剤は1以上の疎水性材料(例えばヒドロキシプロピルメチルセルロール)を含んでもよい。
【0051】
放出改変剤はさらに又はこれに代えて、半透性ポリマーを含んでもよい。
【0052】
特定の実施形態では、放出改変剤はヒドロキシプロピルメチルセルロース、ラクトース、金属ステアリン酸塩、およびこれらの任意の混合物から選択される。
【0053】
本発明の徐放性コーティング剤はさらに浸食促進剤(例えばデンプンおよびゴム)を含んでもよい。
【0054】
本発明の徐放性コーティング剤はさらに使用環境内で微細孔性薄膜を生成するに有用な材料(例えば炭酸の線形ポリエステルを含み、ポリマー鎖内で炭酸基が反復するポリカーボネート)を含んでもよい。
【0055】
本発明の徐放性コーティング剤はさらに、少なくとも1つの通路、オリフィスなどを含む排出手段を含んでもよい。通路は米国特許第3,845,770号、第3,916,889号、第4,063,064号、および第4,088,864号に開示されたような方法により形成することができる。通路は円形、三角形、四角形、楕円形、不定形などの任意の形状を有することができる。
【0056】
マトリクス製剤
本発明の他の実施形態では、徐放性製剤は、本明細書に記載する徐放性コーティング剤を任意に有するマトリクスによって得られる。徐放性マトリクスに含まれるのに適した材料はマトリクスを形成するために用いた方法に依存する。
【0057】
例えば、ヒドロコドン(またはヒドロコドン塩)および必要に応じて含まれるナルトレキソン(またはナルトレキソン塩)に添加するマトリクスは以下から選択することができる:(i)親水性および/または疎水性材料、例えばゴム、セルロールエーテル、アクリル性ポリマーまたは樹脂、タンパク質由来の材料、および活性剤を徐放することができ且つ溶融する(または除去するに必要な程度まで軟化する)任意の製薬上許容可能な疎水性材料または親水性材料、(ii)消化可能な長鎖(C8-C50、特にC12-C40)置換または非置換炭化水素(例えば脂肪酸;脂肪性アルコール;脂肪酸のグリセリルエステル;鉱油、植物油および蝋;並びにステアリルアルコール)、および(iii)ポリアルキレングリコール。
【0058】
これらのポリマーのうち、アクリル性ポリマー(特にEudragit(登録商標) RSPO)およびセルロースエーテル(特にヒドロキシアルキルセルロースおよびカルボキシアルキルセルロース)が好ましい。経口剤形は少なくとも1つの親水性または疎水性材料を1%〜80%(重量比)含んでもよい。
【0059】
疎水性材料が炭化水素である場合、炭化水素は好ましくは25℃と90℃との間の融点を有する。長鎖炭化水素材料のうち、脂肪性(脂肪族)アルコールが好ましい。経口剤形は少なくとも1つの消化可能な長鎖炭化水素を最高60%(重量比)含んでもよい。
【0060】
好ましくは経口剤形は少なくとも1つのポリアルキレングリコールを最高60%(重量比)含む。
【0061】
疎水性材料は、アルキルセルロース、アクリル酸およびメタクリル酸ポリマーおよびコポリマー、シェラック、ゼイン、水素化ひまし油、水素化植物油、またはこれらの混合物からなる群より選択されてもよい。本発明の特定の好ましい実施形態では、疎水性材料は、アクリル酸およびメタクリル酸コポリマー、メタクリル酸メチル、メタクリル酸メチルコポリマー、メタクリル酸エトキシエチル、メタクリル酸シアノエチル、メタクリル酸アミノアルキルコポリマー、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミンコポリマー、ポリ(メタクリル酸メチル)、ポリ(メタクリル酸)(無水物)、ポリメタクリル酸、ポリアクリルアミド、ポリ(メタクリル酸無水物)、およびメタクリル酸グリシジルコポリマーなどの材料から選択される製薬上許容可能なアクリル性ポリマーである。他の実施形態では、疎水性材料は、ヒドロキシアルキルセルロース(例えばヒドロキシプロピルメチルセルロース)およびその混合物などの材料から選択される。
【0062】
好ましい疎水性材料は、非水溶性であり、多かれ少なかれ明白な親水性および/または疎水性の傾向を有する。好ましくは、本発明で有用な疎水性材料は約30℃〜約200℃の融点を有し、より好ましくは約45℃〜約90℃の融点を有する。特に、疎水性材料は天然または合成蝋、脂肪性アルコール(例えばラウリル、ミスチリル、ステアリル、セチルまたは好ましくはセトステアリルアルコール)、脂肪酸(脂肪酸エステル、脂肪酸グリセリド(モノグリセリド、ジグリセリドおよびトリグリセリド)を含むがこれらに限られない)、水素化脂肪、炭化水素、ノーマルワックス、ステアリン酸補助剤(stearic aid)、ステアリルアルコール、並びに炭化水素骨格を有する疎水性および親水性材料を含んでもよい。適切な蝋は例えば、蜜蝋、糖蝋(glycowax)、ひまし蝋(castor wax)、カルナウバ蝋を含む。本発明の目的のためには、蝋状物質は、室温では通常固体であり約30℃〜約100℃の融点を有する任意の材料と定義される。
【0063】
本発明に用いることができる適切な疎水性材料は、消化可能な長鎖(C8-C50、特にC12-C40)置換または非置換炭化水素(例えば脂肪酸、脂肪性アルコール、脂肪酸のグリセリルエステル、鉱油、植物油、並びに天然および合成蝋)を含む。約25℃〜約90℃の融点を有する炭化水素が好ましい。特定の実施形態では、長鎖炭化水素材料のうち脂肪性(脂肪族)アルコールが好ましい。経口剤形は少なくとも1つの消化可能な長鎖炭化水素を最高60%(重量比)含んでもよい。
【0064】
好ましくは、マトリクス製剤には2以上の疎水性材料の組み合わせが含まれる。追加の疎水性材料が含まれる場合、その疎水性材料は好ましくは天然および合成蝋、脂肪酸、脂肪性アルコール、およびこれらの混合物から選択される。これらの材料は例えば、蜜蝋、カルナウバ蝋、ステアリン酸、およびステアリルアルコールであるがこれらに限られない。
【0065】
1つの特定の適切なマトリクスは、少なくとも1つの水溶性ヒドロキシアルキルセルロース、少なくとも1つのC12-C36(好ましくはC14-C22)脂肪族アルコール、および必要に応じて少なくとも1つのポリアルキレングリコールを含む。ヒドロキシアルキルセルロースは好ましくはヒドロキシ(C1-C6)アルキルセルロース(例えばヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、またはヒドロキシエチルセルロース)である。本発明の経口剤形中のヒドロキシアルキルセルロースの量は、特にヒドロコドンおよび/またはナルトレキソンに要求される厳密な放出率によって決定される。脂肪族アルコールは例えば、ラウリルアルコール、ミリスチルアルコール、またはステアリルアルコールであってもよい。但し本発明の経口剤形の特に好ましい実施形態では、脂肪族アルコールはセチルアルコールまたはセトステアリルアルコールである。本発明の経口剤形中の脂肪族アルコールの量は、上記のようにヒドロコドンおよび/またはナルトレキソンに要求される厳密な放出率によって決定される。上記量はさらに、経口剤形中に少なくとも1つのポリアルキレングリコールが存在するか否かによっても左右される。少なくとも1つのポリアルキレングリコールが存在しない場合、経口剤形は好ましくは脂肪族アルコールを20%〜50%(重量比)含む。経口剤形中にポリアルキレングリコールが存在する場合、脂肪族アルコールとポリアルキレングリコールとの総重量が総剤形の20%〜50%(重量比)を占めることが好ましい。
【0066】
一実施形態では、脂肪族アルコール/ポリアルキレングリコールに対する例えばヒドロキシアルキルセルロースまたはアクリル性樹脂の割合が、ヒドロコドンおよび/またはナルトレキソンの製剤からの放出率をかなりの度合いで決定する。脂肪族アルコール/ポリアルキレングリコールに対するヒドロキシアルキルセルロースの割合は1:2〜1:4が好ましく、1:3〜1:4が特に好ましい。
【0067】
ポリアルキレングリコールは例えば、ポリプロピレングリコールまたはポリエチレングリコールであってもよい。少なくとも1つのポリアルキレングリコールの数平均分子量は好ましくは1,000〜15,000であり、特に好ましくは1,500〜12,000である。
【0068】
別の適切な徐放性マトリクスは、アルキルセルロース(特にエチルセルロース)、C12〜C36脂肪族アルコール、および必要に応じてポリアルキレングリコールを含む。
【0069】
別の好ましい実施形態では、マトリクスは少なくとも2つの疎水性材料の製薬上許容可能な組み合わせを含む。
【0070】
徐放性マトリクスは上記成分に加えてさらに、適量の他の材料、例えば製薬分野で従来から用いられている希釈剤、滑沢剤、結合剤、粒状化補助剤、色素、芳香剤、および/または流動促進剤を含んでもよい。
【0071】
マトリクス−微粒子
本発明による固体の徐放性経口剤形の調製を促進するためには、当業者に公知のいずれのマトリクス製剤調製方法を用いてもよい。マトリクスへの含有は例えば以下の方法により行われる: 例えば(a)少なくとも1つの水溶性ヒドロキシアルキルセルロース、ヒドロコドン(またはヒドロコドン塩)、および必要に応じてナルトレキソン(またはナルトレキソン塩)を含む顆粒を形成し、(b)得られた顆粒を少なくとも1つのC12-C36脂肪族アルコールと混合し、(c)必要に応じて顆粒を圧縮成形する。好ましくは、ヒドロキシアルキルセルロース顆粒を水で湿粒状化することによって顆粒を形成する。
【0072】
さらに別の実施形態では、ヒドロコドン(またはヒドロコドン塩)および必要に応じて含まれるナルトレキソン(またはナルトレキソン塩)と共にスフェロイド化剤を用いることによりスフェロイドを形成してもよい。微結晶セルロースは好ましいスフェロイド化剤である。適切な微結晶セルロースは例えば、Avicel PH 101(商標、FMC Corporation)として販売されている材料である。この実施形態では、スフェロイドは活性成分およびスフェロイド化剤に加えて結合剤を含んでもよい。適切な結合剤は例えば低粘度の水溶性ポリマーであり、製薬分野の当業者には公知である。しかし、水溶性ヒドロキシ低級アルキルセルロース(例えばヒドロキシプロピルセルロース)が好ましい。スフェロイドはこれに加えて(またはこれに代えて)、非水溶性ポリマー、特にアクリル性ポリマー、アクリル性コポリマー(例えばメタクリル酸−アクリル酸エチルコポリマー)またはエチルセルロースを含んでもよい。この実施形態では、徐放性コーティング剤は概して疎水性材料(例えば(a)蝋単独または蝋と脂肪性アルコールとの混合物または(b)シェラックまたはゼイン)を含む。
【0073】
溶融押出マトリクス
徐放性マトリクスは、溶融粒状化法または溶融押出法によっても調製することができる。溶融粒状化法は概して、通常は固体の疎水性材料(例えば蝋)を溶融することと、粉体化した薬物を疎水性材料中に含有させることとを含む。徐放性剤形を得るためには、追加の疎水性物質(例えばエチルセルロースまたは非水溶性アクリル性ポリマー)を、溶融蝋疎水性材料中に含有させることが必要となる場合もある。溶融粒状化法によって調製された徐放性製剤の例は米国特許第4,861,598号に記載されている。
【0074】
追加の疎水性材料は、1以上の非水溶性蝋状熱可塑性物質を含んでもよい。この非水溶性蝋状熱可塑性物質は、これよりも疎水度の低い別の1以上の蝋状熱可塑性物質と混合されてもよい。定常的放出を実現するためには、製剤中の個々の蝋状物質は初期の放出フェーズにおいて胃腸液に対して実質的に非変性であり不溶性でなければならない。有用な非水溶性蝋状物質は、約1:5,000(w/w)未満の水溶解度を有するものである。
【0075】
徐放性マトリクスは上記成分に加えてさらに、適量の他の材料、例えば製薬分野で従来用いられている希釈剤、滑沢剤、結合剤、粒状化補助剤、色素、芳香剤、および流動促進剤を含んでもよい。これら追加の材料は所望の製剤に所望の影響を与えるに十分な量だけ含まれる。
【0076】
溶融押出マルチ微粒子を含有する徐放性マトリクスは上記成分に加えてさらに、適量の他の材料、例えば製薬分野で従来から用いられている希釈剤、滑沢剤、結合剤、粒状化補助剤、色素、芳香剤、および/または流動促進剤を含んでもよい。
【0077】
経口剤形を処方するために用いることができる製薬上許容可能な担体および賦形剤の特定の例は、Handbook of Pharmaceutical Excipients, American Pharmaceutical Association, 第3版(2000)に記載されている。
【0078】
溶融押出マルチ微粒子
本発明による適切な溶融押出マトリクスの調製は例えば、ヒドロコドン(またはヒドロコドン塩)および/またはナルトレキソン(またはナルトレキソン塩)を少なくとも1つの疎水性材料と混合することにより均質混合物を得る工程を含んでもよい。次いで均質混合物を、少なくとも当該混合物を押出できるようになるまで十分に軟化させるに十分な温度まで加熱する。その後得られた均質混合物を押出してストランドを形成する。好ましくは押出物を当該分野で公知の任意の手段で冷却し、切断してマルチ微粒子にする。ストランドを切断してマルチ微粒子にする。その後マルチ微粒子を単位投与量に分割する。押出物は好ましくは約0.1〜約5 mmの直径を有し、約8〜約24時間の期間に亘って活性剤を徐放する。
【0079】
本発明の溶融押出物を調製するために必要に応じて行われるプロセスは、疎水性材料、ヒドロコドン(またはヒドロコドン塩)および必要に応じて含まれるナルトレキソン(またはナルトレキソン塩)、並びに必要に応じて含まれる結合剤を押出器に直接計量して供給すること、これらの成分を混合加熱して均質混合物を生成すること、均質混合物を押出することによりストランドを形成すること、均質混合物を含むストランドを冷却すること、ストランドを切断して約0.1 mm〜約12 mmのサイズを有する粒子にすること、および粒子を単位投与量に分割することを含む。本発明のこの局面では、比較的連続した製造手順が実現される。
【0080】
押出アパーチャまたは排出ポートの直径を調整することにより、押出されたストランドの厚みを変化させることも可能である。さらに、押出器の排出ポートは円形である必要はなく、楕円形、矩形などであってもよい。排出されてくるストランドは、ホットワイヤカッタ、ギロチンなどを用いて粒子に切断することができる。
【0081】
溶融押出マルチ微粒子系は、押出器の排出ポートに依存して例えば顆粒、スフェロイド、またはペレットの形態であり得る。本発明の目的のためには、用語「溶融押出マルチ微粒子」(MEMS)、および「溶融押出マルチ微粒子系」および「溶融押出粒子」は好ましくは類似のサイズおよび/または形状の範囲内にあり且つ1以上の活性剤と1以上の賦形剤とを含み、好ましくは本明細書に記載する疎水性材料を含む複数の単位を指す。この点に関して、溶融押出マルチ微粒子は長さ約0.1〜約12 mm、直径約0.1〜約5 mmである。さらに溶融押出マルチ微粒子はこのサイズの範囲内であればいずれの形状を有してもよいことが理解される。あるいは押出物を単に所望の長さに切断して治療上活性な薬剤の単位投与量に分割してもよく、この場合スフェロイド化工程は不要である。
【0082】
一つの好ましい実施形態では、経口剤形は、有効量の溶融押出マルチ微粒子をカプセル内に含むように調製される。例えば、複数の溶融押出マルチ微粒子を、摂取され胃液に接触されたときに有効に徐放されるに十分な量でゼラチンカプセル内に入れてもよい。
【0083】
別の好ましい実施形態では、従来のタブレット化装置を用い、標準的な手法を用いて適量のマルチ微粒子押出物を圧縮して経口タブレットにする。タブレット(圧縮され成形された)、カプセル(硬および軟ゼラチン)並びにピルを形成する手法および組成物は、Remington's Pharmaceutical Sciences (Arthur Osol編)、1553〜1593頁(1980)にも記載されている。
【0084】
さらに別の好ましい実施形態では、上記に詳細に記載した米国特許第4,957,681号(Klimeschら)に記載されているように押出物をタブレットに成形してもよい。
【0085】
必要に応じて、例えば上記した徐放性コーティング剤などの徐放性コーティング剤を用いて徐放性溶融押出マルチ微粒子系またはタブレットをコーティングしてもよいし、ゼラチンカプセルをさらにコーティングしてもよい。これらのコーティング剤は好ましくは、約2〜約30%の重量増加レベルを得るに十分な量の疎水性材料を含む。但し、オーバーコーティング剤の量は、特に所望の徐放率によってはこれよりも多い場合もある。
【0086】
本発明の溶融押出単位剤形はカプセル化される前に、溶融押出粒子の組み合わせ(例えばヒドロコドン(またはヒドロコドン塩)を有する一群の粒子とナルトレキソン(またはナルトレキソン塩)を有する一群の粒子)をさらに含んでもよい。単位剤形は即時放出のために、ある量の即時放出型活性剤をさらに含んでもよい。即時放出型剤は、例えばゼラチンカプセル内の分離したペレットとして含有されてもよいし、剤形(例えば徐放性コーティングまたはマトリクスベース)の調製後にマルチ微粒子の表面にコーティングされてもよい。本発明の単位剤形は、所望の効果を達成するために徐放性ビーズとマトリクスマルチ微粒子との組み合わせをさらに含んでもよい。
【0087】
本発明の徐放性製剤は好ましくは、例えば摂取され、胃液、次いで腸液に接触されたときに、ゆっくりと薬剤を放出する。本発明の溶融押出製剤の徐放プロファイルは、例えば遅延剤(すなわち疎水性材料)の量を変更すること、疎水性材料に対する可塑剤の量を変更すること、追加の成分または賦形剤を含ませること、製造方法を変更することなどによって変化させることができる。
【0088】
本発明の別の実施形態では、溶融押出材料を、ヒドロコドン(またはヒドロコドン塩)もナルトレキソン(またはナルトレキソン塩)も含むことなく調製する。ヒドロコドン(またはヒドロコドン塩)とナルトレキソン(またはナルトレキソン塩)とは後に押出物に添加することができる。このような製剤では典型的には、薬剤を押出マトリクス材料と混合し、その後ゆっくりと放出する目的で混合物をタブレット化する。
【0089】
コーティング剤
本発明の剤形は必要に応じて、放出の制御または製剤の保護に適した1以上の材料でコーティングしてもよい。一実施形態では、コーティング剤はpH依存型またはpH独立型放出を可能にするために提供される。pH依存型コーティング剤は、ヒドロコドンおよび/またはナルトレキソンを胃腸(GI)管(例えば胃または小腸)の所望の領域において放出するために役立つ。これにより、患者に少なくとも8時間、好ましくは約12時間、最高約24時間の沈痛を与えることができる吸入プロファイルが提供される。pH独立型コーティング剤が望まれる場合、コーティング剤は、周囲、例えばGI管の液のpH変化にかかわらず最適な放出を達成するように設計される。GI管の1つの所望領域(例えば胃)に投与量の一部を放出し、GI管の別の領域(例えば小腸)に投与量の残りを放出する組成物を処方することも可能である。
【0090】
pH依存型コーティング剤を利用する本発明による製剤はさらに反復作用効果を提供することができる。反復作用効果によると、保護されていない薬物を腸溶性コーティング剤にコーティングして胃で放出し、腸溶性コーティング剤によって保護された残りの部分を胃腸管のさらに下の胃腸管で放出する。pH依存型コーティング剤は、シェラック、酢酸フタル酸セルロース(CAP)、酢酸フタル酸ポリビニル(PVAP)、フタル酸ヒドロキシプロピルメチルセルロース、およびメタクリル酸エステルコポリマー、ゼインなどを含む。
【0091】
特定の好ましい実施形態では、ヒドロコドンまたはその塩および必要に応じてナルトレキソンまたはその塩を含む基質(例えばコーティングされたビーズ、マトリクス粒子)を、(i)アルキルセルロース、(ii)アクリル性ポリマー、(iii)それらの混合物から選択される疎水性材料によってコーティングする。コーティング剤は有機または水溶液または水分散液の形態で適用してもよい。コーティング剤は、所望の徐放プロファイルを得るために基質の約2〜約25%の重量増加を得るように適用してもよい。水分散液由来のコーティング剤は、例えば米国特許第5,273,760号および第5,286,493号に詳細に記載されている。
【0092】
本発明に用いることができる徐放性製剤およびコーティング剤の他の例は、米国特許第5,324,351号、第5,356,467号、および第5,472,712号に記載されているものを含む。
【0093】
アルキルセルロースポリマー
アルキルセルロースを含むセルロース材料およびポリマーは、本発明によるビーズのコーティングに良く適した疎水性材料を提供する。単なる例として1つの好ましいアルキルセルロースポリマーは、エチルセルロースであるが、当業者であれば他のセルロースおよび/またはアルキルセルロースポリマーも単一または任意の組み合わせで、本発明による疎水性コーティング剤の全体または一部として容易に使用可能であることを理解する。
【0094】
エチルセルロースの1つの市販されている水分散液はAquacoat(登録商標) (FMC Corp., Philadelphia, Pennsylvania, U.S.A)である。Aquacoat(登録商標)は、エチルセルロースを水と混合不能な有機溶媒中に溶解し、その後得られたものを水中で界面活性剤および安定剤の存在下で乳化することによって調製される。均質化によりサブミクロンサイズの小滴を生成した後、有機溶媒を真空中で蒸発させて疑似ラテックスを生成する。製造フェーズにおいて可塑剤は疑似ラテックスに含有されない。従って疑似ラテックスをコーティング剤として用いる前に、Aquacoat(登録商標)を適切な可塑剤と混合することが必要である。エチルセルロースの別の水分散液はSurelease(登録商標) (Colorcon, Inc., West Point, Pennsylvania, U.S.A)として市販されている。この製品は、製造プロセスにおいて分散液中に可塑剤を含有させることによって調製される。ポリマーと可塑剤(セバシン酸ジブチル)と安定剤(オレイン酸)とのホットメルトを均質混合物として調製し、その後これをアルカリ溶液で希釈することにより、基質に直接適用することができる水分散液を得る。
【0095】
アクリル性ポリマー
本発明の他の好ましい実施形態では、徐放性コーティング剤を含む疎水性材料は、製薬上許容可能なアクリル性ポリマーであり、これはアクリル酸およびメタクリル酸コポリマー、メタクリル酸メチルコポリマー、メタクリル酸エトキシエチル、メタクリル酸シアノエチル、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミドコポリマー、ポリ(メタクリル酸メチル)、ポリメタクリル酸、ポリ(メタクリル酸メチル)コポリマー、ポリアクリルアミド、メタクリル酸アミノアルキルコポリマー、ポリ(メタクリル酸無水物)、およびメタクリル酸グリシジルコポリマーを含むがこれらに限られない。
【0096】
特定の好ましい実施形態では、アクリル性ポリマーは1以上のメタクリル酸アンモニオ(ammonio)コポリマーを含む。メタクリル酸アンモニオコポリマーは当該分野で周知であり、低含有量の第四級アンモニア基を含むアクリル酸エステルとメタクリル酸エステルとの完全重合コポリマーとしてNational Formulary XVIIに記載されている。
【0097】
所望の溶解プロファイルを得るために、異なる物性(例えば中性(メタ)クリル酸エステルに対する第四級アンモニア基の異なるモル比)を有する2以上のメタクリル酸アンモニオコポリマーを含有させることが必要な場合がある。
【0098】
特定の好ましい実施形態では、アクリル性コーティング剤は、Rohm PharmaからそれぞれEudragit(登録商標) RL30DおよびのEudragit(登録商標) RS30Dの商品名で市販されている2つのアクリル性樹脂ラッカーの混合物を含む。Eudragit(登録商標) RL30DおよびのEudragit(登録商標) RS30Dは、低含有量の第四級アンモニア基を含むアクリル酸エステルとメタクリル酸エステルとのコポリマーである。アンモニア基の、残りの中性(メタ)クリル酸エステルに対するモル比は、Eudragit(登録商標) RL30Dでは1:20であり、Eudragit(登録商標) RS30Dでは1:40である。平均分子量は約150,000である。名称RL(高透過性)およびRS(低透過性)はこれらの剤の透過特性を示す。Eudragit(登録商標) RL/RS混合物は水および消化器液に対して不溶性である。しかし当該混合物から生成されたコーティング剤は水溶液および消化器液中で膨張可能且つ透過可能である。
【0099】
本発明のEudragit( RL/RS分散液は、所望の溶解プロファイルを有する徐放性製剤を最終的に得るために任意の所望の割合で一緒に混合することができる。所望の徐放性製剤は、例えば100%Eudragit(登録商標) RL由来の遅延コーティング剤から、50% Eudragit(登録商標) RLおよび50% Eudragit(登録商標) RSから、または10% Eudragit(登録商標) RLおよびEudragit(登録商標) 90% RSから得ることができる。言うまでもなく当業者であれば他のアクリル性ポリマー(例えばEudragit(登録商標) L)も使用可能であることを認識する。
【0100】
可塑剤
本発明の実施形態では、コーティング剤が疎水性材料の水分散液を含む場合、疎水性材料の水分散液中に有効量の可塑剤を含ませることにより徐放性コーティング剤の物性をさらに高めることができる。例えば、エチルセルロースは比較的高いガラス転移点を有し且つ通常のコーティング条件下では可撓性膜を形成しないため、徐放性コーティング剤を含むエチルセルロースコーティング剤をコーティング材料として用いる前に、これに可塑剤を含有させることが好ましい。概してコーティング溶液中に含まれる可塑剤の量は、薄膜生成剤の濃度(例えば最も多くの場合、薄膜生成剤の約1〜約50%(重量比))に基づく。しかし可塑剤の濃度は、特定のコーティング溶液および適用方法を慎重に実験した後でのみ適切に決定することができる。
【0101】
エチルセルロースに適した可塑剤の例は、非水溶性可塑剤(例えばセバシン酸ジブチル、フタル酸ジエチル、クエン酸トリエチル、クエン酸トリブチル、およびトリアセチン)を含む。本発明で用いられるエチルセルロースの水分散液にとっては、クエン酸トリエチルが特に好ましい可塑剤である。
【0102】
本発明のアクリル性ポリマーに適した可塑剤の例は、クエン酸エステル(例えばクエン酸トリエチルNF XVI、クエン酸トリブチル、フタル酸ジブチル、および1,2-プロピレングリコール)を含むがこれらに限られない。アクリル性薄膜(例えばEudragit(登録商標) RL/RSラッカー溶液)から形成された薄膜の弾性を高めるのに適していることが証明されている他の可塑剤は、ポリエチレングリコール、プロピレングリコール、フタル酸ジエチル、ひまし油、およびトリアセチンを含む。本発明で用いられるアクリル酸ポリマーの水分散液にとっては、クエン酸トリエチルが特に好ましい可塑剤である。
【0103】
少量のタルクを添加することにより処理中に水分散液が粘着する傾向が低減され、タルクは研磨剤として作用することがさらに判明している。
【0104】
徐放性浸透性投与量
本発明による徐放性剤形はさらに浸透性投与製剤として調製してもよい。浸透性剤形は好ましくは、薬物層(ヒドロコドン(またはヒドロコドン塩)および必要に応じてナルトレキソン(またはナルトレキソン層)を含む)と送達または押し出し層(ナルトレキソン(またはナルトレキソン塩))を含んでもよい)とを含む二層コアを含む。二層コアは半透壁により取り囲まれ、半透壁は必要に応じて内部に設けられた少なくとも1つの通路を有する。
【0105】
本発明の目的のために、用語「通路」は、ファイバ、毛細管、多孔性オーバーレイ、多孔性インサート、微孔性部材、または多孔性組成物を介してヒドロコドンまたはヒドロコドン塩(ナルトレキソンまたはナルトレキソン塩を伴うまたは伴わず)を注入、拡散または移送することができるアパーチャ、オリフィス、穴(bore)、孔(pore)、または有孔の要素を含む。通路はさらに、用いられる液体環境中で壁から浸食するか浸出することによって少なくとも1つの通路を形成する化合物を含んでもよい。通路を形成する代表的な化合物は、壁内の浸食性ポリ(グリコール)酸またはポリ(乳)酸、ゼラチン性フィラメント、水除去可能ポリ(ビニルアルコール)、浸出性化合物(例えば液体除去可能孔形成ポリサッカリド、酸、塩または酸化物)を含む。通路は、壁(例えばソルビトール、スクロース、ラクトース、マルトースまたはフルクトース)から化合物を浸出させることにより形成することができ、これにより徐放用寸法を有する孔通路が形成される。通路は、ヒドロコドンまたはヒドロコドン塩を剤形から調節しながら徐放することを補助する任意の形状(例えば円形、三角形、四角形および楕円形)を有することができる。剤形は、その1以上の表面上に1以上の通路を有するように製造することができる。通路および通路を形成する装置は、米国特許第3,845,770号、第3,916,899号、第4,063,064号、および第4,088,864号に開示されている。水を通すことにより形成されて、徐放率を有する放出孔としてのサイズおよび形状を有し当該放出孔として適合された徐放性広がりを有する通路は、米国特許第4,200,098号および第4,285,987号に記載されている。
【0106】
特定の実施形態では、二層コアは、ヒドロコドンまたはその塩を有する薬物層と、ナルトレキソンまたはその塩を含む移動または押し出し層とを含む。特定の実施形態では、薬物層はさらに少なくとも1つのポリマーヒドロゲルを含んでもよい。ポリマーヒドロゲルは約500〜約6,000,000の平均分子量を有してもよい。ポリマーヒドロゲルの例は以下のマルトデキストリンポリマー、ポリ(酸化アルキレン)、アルカリカルボキシアルキルセルロース、およびエチレン−アクリル酸コポリマーを含むがこれらに限られない。マルトデキストリンポリマーは、化学式(C6H12O5)n・H20を含み且つnが3〜7,500であり、500〜1,250,000の数平均分子量を有する。ポリ(酸化アルキレン)は、例えば50,000〜750,000の重量平均分子量を有するポリ(酸化エチレン)およびポリ(酸化プロピレン)に代表され、より好ましくは100,000、200,000、300,000および400,000のうち少なくとも1つの重量平均分子量を有するポリ(酸化エチレン)に代表される。アルカリカルボキシアルキルセルロースは、アルカリがナトリウムまたはカリウムであり、アルキルセルロースが10,000〜175,000の重量平均分子量を有する。エチレン−アクリル酸コポリマーは、10,000〜500,000の数平均分子量を有するメタクリル酸およびエタクリル酸を含む。
【0107】
本発明の特定の実施形態では、送達または押し出し層はオスモポリマーを含む。オスモポリマーの例は、酸化ポリアルキレンとカルボキシアルキルセルロースとからなる群より選択される物質を含むがこれらに限られない。酸化ポリアルキレンは1,000,000〜10,000,000の数平均分子量を有する。酸化ポリアルキレンは、酸化ポリメチレン、酸化ポリエチレン、酸化ポリプロピレン、1,000,000の平均分子量を有する酸化ポリエチレン、5,000,000の平均分子量を有する酸化ポリエチレン、7,000,000の平均分子量を有する酸化ポリエチレン、1,000,000の平均分子量を有する架橋酸化ポリメチレン、および1,200,000の平均分子量を有する酸化ポリプロピレンからなる群より選択される物質であってもよい。典型的なオスモポリマーであるカルボキシアルキルセルロースは、アルカリカルボキシアルキルセルロース、ソディウムカルボキシメチルセルロース、ポタシウムカルボキシメチルセルロース、ソディウムカルボキシエチルセルロース、リチウムカルボキシメチルセルロース、ソディウムカルボキシエチルセルロース、およびカルボキシアルキルヒドロキシアルキルセルロース(例えば、カルボキシメチルヒドロキシエチルセルロース、カルボキシエチルヒドロキシエチルセルロース、およびカルボキシメチルヒドロキシプロピルセルロース)からなる群より選択される物質を含む。移動層に用いられるオスモポリマーは、半透壁に対して浸透圧グラジエントを呈する。オスモポリマーは液体を吸収して剤形になり、それにより浸透性ヒドロゲル(オスモゲルとしても知られる)として膨張および拡張する。これによりオスモポリマーは浸透性剤形からヒドロコドンまたはその製薬上許容可能な塩を押し出す。
【0108】
押し出し層はさらに、オスマゲント(osmagent)または浸透有効溶質としても知られる1以上の浸透有効化合物を含んでもよい。これらは、例えば胃腸管から剤形に周囲の液体を吸収し、移動層の送達速度に寄与する。浸透活性化合物の例は、浸透性塩と浸透性炭水化物とからなる群より選択される物質を含む。特定のオスモゲントの例は、塩化ナトリウム、塩化カリウム、硫酸マグネシウム、リン酸リチウム、塩化リチウム、リン酸ナトリウム、硫酸カリウム、硫酸ナトリウム、リン酸カリウム、グルコース、フルクトース、およびマルトースを含むがこれらに限られない。
【0109】
押し出し層は必要に応じて、9,000〜450,000の数平均分子量を有するヒドロキシプロピルアルキルセルロースを含んでもよい。ヒドロキシプロピルアルキルセルロースは、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルエチルセルロース、ヒドロキシプロピルイソプロピルセルロース、ヒドロキシプロピルブチルセルロース、およびヒドロキシプロピルペンチルセルロースからなる群より選択される物質により代表される。
【0110】
押し出し層は必要に応じて、非毒性色素または染料を含んでもよい。色素または染料の例は、Food and Drug Administration Colorant (FD&C)(例えばFD&C No. 1青染料、FD&C No. 4赤染料、赤酸化第二鉄、黄酸化第二鉄、二酸化チタン、カーボンブラック、およびインディゴ)を含むがこれらに限られない。
【0111】
押し出し層はさらに必要に応じて、成分の酸化を阻止する酸化防止剤を含んでもよい。酸化防止剤のいくつかの例は、アスコルビン酸、パルミチン酸アルコルビル、ブチル化ヒドロキシアニソール、2および3第三級-ブチル-4-ヒドロキシアニソール、ブチル化ヒドロキシトルエン、イソアスコルビン酸ナトリウム、ジヒドログアレチン酸(dihydroguaretic acid)、ソルビン酸カリウム、重硫酸ナトリウム、メタ重硫酸ナトリウム、ソルビン酸、アスコルビン酸カリウム、ビタミンE、4-クロロ-2,6-2第三級(ditertiary)ブチルフェノール、アルファトコフェノール、およびプロピルガラートからなる群より選択される物質を含むがこれらに限られない。
【0112】
特定の別の実施形態では、剤形は均質コアを含み、均質コアは、ヒドロコドンまたはその製薬上許容可能な塩、ナルトレキソンまたはその製薬上許容可能な塩、製薬上許容可能なポリマー(例えば酸化ポリエチレン)、必要に応じて崩壊剤(例えばポリビニルピロリドン)、および必要に応じて吸収促進剤(例えば脂肪酸、界面活性剤、キレート剤、胆汁酸塩など)を含む。均質コアは、ヒドロコドンまたはその製薬上許容可能な塩を放出する通路(上記で定義した)を有する半透壁によって取り囲まれている。
【0113】
特定の実施形態では、半透壁は、セルロースエステルポリマー、セルロースエーテルポリマー、およびセルロースエステル−エーテルポリマーからなる群より選択される物質を含む。代表的な壁ポリマーは、セルロースアシレート、セルロースジアシレート、セルローストリアシレート、セルロースアセテート、セルロースジアセテート、セルローストリアセテート、モノ、ジおよびトリセルロースアルケニレート、並びにモノ、ジおよびトリセルロースアルキニレートからなる群より選択される物質を含む。本発明に用いられるポリ(セルロース)は20,000〜7,500,000の数平均分子量を有する。
【0114】
追加の半透性ポリマーは、アセトアルデヒドジメチルセルロースアセテート、セルロースアセテートエチルカルバメート、セルロースアセテートメチルカルバメート、セルロースジアセテート、プロピルカルバメート、セルロースアセテートジエチルアミノアセテート、半透性ポリアミド、半透性ポリウレタン、半透性スルホン化ポリスチレン、ポリアニオンとポリカチオンとの共沈殿により形成される半透性架橋ポリマー(米国特許第3,173,876号、第3,276,586号、第3,541,005号、第3,541,006号、および第3,546,876号に記載)、米国特許第3,133,132号でLoebおよびSourirajanが開示した半透性ポリマー、半透性架橋ポリスチレン、半透性架橋ポリ(スチレンスルホン酸ナトリウム)、半透性架橋ポリ(塩化ビニルベンジルトリメチルアンモニウム)、および2.5x10-8〜2.5x10-2 (cm2/hr・atm)の液体透過性を有する半透性ポリマーを含む。上記液体透過性は、半透壁の静水圧差または浸透圧差の気圧単位で表している。本発明において有用な他のポリマーは、当該分野で公知であり、米国特許第3,845,770号、第3,916,899号、および第4,160,020号、並びにHandbook of Common Polymers, Scott, J. R.およびW. J. Roff, 1971, CRC Press, Cleveland, Ohioに記載されている。
【0115】
特定の実施形態では、半透壁は好ましくは非毒性且つ不活性であり、薬物の調剤中に亘ってその物理的および化学的完全性を維持する。
【0116】
特定の実施形態では、剤形は結合剤を含む。結合剤の例は、5,000〜350,000の粘度平均分子量を有する治療上許容可能なビニルポリマーを含むがこれに限られない。上記治療上許容可能なビニルポリマーは、酢酸ビニル、ビニルアルコール、塩化ビニル、フッ化ビニル、ブチル酸ビニル、ラウリン酸ビニル、およびステアリン酸ビニルからなる群より選択される物質との、ポリ-n-ビニルアミド、ポリ-n-ビニルアセトアミド、ポリ(ビニルピロリドン)(ポリ-n-ビニルピロリドンとしても知られる)、ポリ-n-ビニルカプロラクトン、ポリ-n-ビニル-5-メチル-2-ピロリドン、およびポリ-n-ビニル-ピロリドンポリマーからなる群より選択される物質に代表される。他の結合剤は例えば、アカシア、デンプン、ゼラチン、および9,200〜250,000の平均分子量を有するヒドロキシプロピルアルキルセルロースを含む。
【0117】
特定の実施形態では、剤形は滑沢剤を含み、滑沢剤は剤形の製造中に金型(die)の壁またはパンチ面に対する粘着を防止するために用いることができる。滑沢剤の例は、ステアリン酸マグネシウム、ステアリン酸ナトリウム、ステアリン酸、ステアリン酸カルシウム、オレイン酸マグネシウム、オレイン酸、オレイン酸カリウム、カプリル酸、ステアリルフマル酸ナトリウム、およびパルミチン酸マグネシウムを含むがこれらに限られない。
【0118】
本発明は特定の好ましい実施形態において、治療用組成物を含み、治療用組成物は、5〜20 mgのヒドロコドンまたはその製薬上許容可能な塩と、25〜500 mgのポリ(酸化アルキレン)(150,000〜500,000の平均分子量を有する)と、1〜50 mgのポリビニルピロリドン(40,000の平均分子量を有する)と、0〜約7.5 mgの滑沢剤とを含む。好ましくは0.05〜0.56 mgのナルトレキソンまたはその製薬上許容可能な塩が薬物層に含まれる。
【0119】
座薬
本発明の徐放性製剤は、直腸投与用の座薬として処方されてもよい。上記座薬は、ヒドロコドン(またはヒドロコドン塩)とナルトレキソン(またはナルトレキソン塩)とを本明細書に開示する投与量で含む。徐放性座薬製剤の調製は、例えば米国特許第5,215,758号に記載されている。
【0120】
薬物は吸収される前に溶液状態でなければならない。座薬の場合、溶液状態にする前に、基剤を溶解しなければならないか、あるいは基剤を溶融しなければならず、次いで基剤から薬物を分離して直腸液にいれなければならない。体内への薬物の吸収は坐薬基剤によって変化し得る。従って特定の薬物と共に用いられる特定の基剤は、薬物の物性を考慮して選択しなければならない。例えば、脂質溶解性薬物は容易に分離して直腸液に流入することができないが、脂質基剤に僅かに溶解する薬物は容易に分離して直腸液に流入することができる。
【0121】
薬物の溶解時間(または放出率)に影響を与える様々な要因は、溶解溶媒媒質に提示される薬物物質の表面積、溶液のpH、特定の溶媒媒質中での物質の溶解度、および溶媒媒質中に溶解した物質の飽和濃度の駆動力などである。直腸投与される座薬からの薬物の吸収に影響を与える要因は概して、座薬ビヒクル、吸収部位pH、薬物pKa、イオン化度、および脂質溶解度を含む。
【0122】
選択される座薬基剤は、本発明の活性剤と適合性を有すべきである。さらに座薬基剤は好ましくは、粘膜に対して非毒性且つ非刺激性であり、直腸液中に溶融または溶解し、保存中安定している。
【0123】
本発明の特定の好ましい実施形態では、水溶性薬物および非水溶性薬物のいずれの場合も、座薬基剤は、C12〜C18の鎖長を有する飽和天然脂肪酸のモノ、ジおよびトリクリセリドからなる群より選択される脂肪酸蝋を含む。
【0124】
本発明の座薬を調製する際に、他の賦形剤を用いてもよい。例えば、直腸ルートの投与に適した形状を形成するために蝋を用いてもよい。この系は、蝋なしで用いてもよいが、その場合は直腸投与および経口投与の両方の場合においてゼラチンカプセルに希釈剤を充填する。
【0125】
適切な市販のモノ、ジおよびトリクリセリドの例は、Novata TM(タイプAB、AB、B、BC、BD、BBC、E、BCF、C、Dおよび299)という商品名(Henkel製)およびWitepsol TM(タイプH5、H12、H15、H175、H185、H19、H32、H35、H39、H42、W25、W31、W35、W45、S55、S58、E75、E76およびE85)という商品名(Dynamit Nobel製)で販売されている、12〜18の炭素原子鎖を有する飽和天然脂肪酸を含む。
【0126】
他の製薬上許容可能な座薬基剤は、全体的にまたは部分的に上記のモノ、ジおよびトリクリセリドと置換してもよい。座薬中の基剤の量は剤形のサイズ(即ち実際の重量)、基剤(例えばアルギン酸塩)の量、および用いられる活性剤によって決定される。座薬基剤の量は概して、座薬の総重量の約20重量%〜約重量90%である。好ましくは座薬中の基剤の量は座薬の総重量の約65重量%〜約80重量%である。
【0127】
他の形態
本明細書に開示する発明は、ヒドロコドンおよびナルトレキソンのすべての製薬上許容可能な塩を用いることを含む。製薬上許容可能な塩は、金属塩(例えば、ナトリウム塩、カリウム塩、セシウム塩など)、アルカリ土類金属(例えばカルシウム塩、マグネシウム塩など)、有機アミン塩(例えばトリエチルアミン塩、ピリジン塩、ピコリン塩、エタノールアミン塩、トリエタノールアミン塩、ジシクロヘキシルアミン塩、N,N'-ジベンジルエチレンジアミン塩など)、無機酸塩(例えば塩酸塩、臭化水素酸塩、硫酸塩、リン酸塩など)、有機酸塩(例えば蟻酸塩、酢酸塩、トリフルオロ酢酸塩、マレイン酸塩、酒石酸塩、重酒石酸塩など)、スルホン酸塩(例えばメタンスルホン酸塩、ベンゼンスルホン酸塩、p-トルエンスルホン酸塩など)、アミノ酸塩(例えばアルギン酸塩、アスパラギン酸塩、グルタミン酸塩など)を含むがこれらに限られない。
【0128】
ヒドロコドン(またはヒドロコドン塩)とナルトレキソン(またはナルトレキソン塩)との組み合わせは、従来の賦形剤、即ち少なくともヒドロコドンまたはその塩を徐放するためのものとして当該分野で公知の、経口投与に適した製薬上許容可能な有機または無機担体物質と混合して用いることができる。適切な製薬上許容可能な担体は、アルコール、アラビアゴム、植物油、ベンジルアルコール、ポリエチレングリコール、ゲル(gelate)、炭水化物(例えばラクトース、アミロースまたはデンプン)、ステアリン酸マグネシウム、タルク、珪酸、粘性パラフィン、香油、脂肪酸モノグリセリドおよびジグリセリド、ペンタエリスリトール脂肪酸エステル、ヒドロキシメチルセルロース、ポリビニルピロリドンなどを含むがこれらに限られない。医薬調製物は滅菌してもよく、所望であれば補助剤(例えば滑沢剤、崩壊剤、保存料、安定剤、湿潤剤、乳化剤、浸透圧緩衝剤に影響を与える塩、色素、芳香剤および/または芳香物質などと混合してもよい。経口投与を意図した組成物は、当該分野で公知の任意の方法を用いて調製することができる。これらの組成物は、タブレットの製造に適した不活性且つ非毒性の製薬上許容可能な賦形剤からなる群より選択される1以上の薬剤を含む。これらの賦形剤は例えば、不活性希釈剤(例えばラクトース)、粒状化および崩壊剤(例えばコーンスターチ)、結合剤(例えばデンプン)、および滑沢剤(例えばステアリン酸マグネシウム)を含む。タブレットはコーティングしなくてもよいし、外見の美しさまたは活性成分の遅延放出とのために公知の手法でコーティングしてもよい。経口投与用の製剤はさらに、硬いゼラチンカプセルとして提示されてもよく、その場合、活性成分を不活性希釈剤と混合する。
【0129】
本発明の経口剤形は、タブレット、トローチ、ロゼンジ(lozenge)、粉剤または顆粒、硬性または軟性カプセル、微粒子(例えばマイクロカプセル、マイクロスフィアなど)、口内タブレット、座薬などの形態とすることができる。ヒドロコドン(またはヒドロコドン塩)とナルトレキソン(またはナルトレキソン塩)とは実質的に互いに分散していてもよい。
【0130】
本発明は特定の実施形態において、上記に開示したヒドロコドン/ナルトレキソン剤形のいずれかを調製することにより、経口ヒドロコドン剤形(またはヒドロコドン塩)の非経口乱用を阻止する方法を提供する。
【0131】
本発明は特定の実施形態において、上記に開示したヒドロコドン/ナルトレキソン剤形のいずれかを調製することを含む経口ヒドロコドン剤形の転用を阻止する方法を提供する。
【0132】
本発明は特定の実施形態において、上記に開示した剤形をヒト患者に投与することにより疼痛を治療する方法を提供する。
【0133】
以下の実施例は本発明の様々な局面を説明するが、請求の範囲を何ら限定するものと解釈されてはならない。
【実施例1】
【0134】
この予言的実施例では、塩酸ナルトレキソンを含む徐放性ヒドロコドン製剤を以下に示す表1の処方で調製する。
【0135】
【表1】
*99.6%アッセイおよび4.2%残基水分に合わせて調整。
【0136】
この実施例では、粒状化プロセスで製剤に塩酸ナルトレキソンを添加する。プロセスは以下の通りである。
【0137】
1.分散:塩酸ナルトレキソンを水中に溶解し、溶液をEudragit/トリアセチン分散液に添加する。
2.粒状化:流体床粒状化器を用いて、Eudragit/トリアセチン分散液を塩酸ヒドロコドン、スプレー乾燥ラクトースおよびポビドンにスプレーする。
3.ミリング(Milling):顆粒を放出して約1 mmの開口(18メッシュスクリーン)を有するミル内を通過させる。
4.蝋づけ:ステアリルアルコールを約50℃で溶融し、高剪断力ミキサを用いてミリングされた顆粒に添加する。トレイまたは流体床上で室温で冷却する。
5.ミリング:冷却した顆粒を約18メッシュスクリーンのミル内を通過させる。
6.潤滑化:ミキサを用いてタルクおよびステアリン酸マグネシウムで顆粒を潤滑化する。
7.圧縮:Kilianタブレットプレスを用いて顆粒を圧縮してタブレットにする。
8.薄膜コーティング:ロータリーパンを用いてタブレットに水性薄膜を適用する。
【実施例2】
【0138】
この予言的実施例では、ヒドロコドン塩/ナルトレキソン塩徐放性浸透性タブレットを以下に示す表2の処方で生成する。
【0139】
【表2】
【0140】
上記処方を有する剤形を以下の手順に従って調製する。
【0141】
まず、無水塩酸ヒドロコドンと塩酸ナルトレキソン二水和物と、200,000の平均分子量を有するポリ(酸化エチレン)と、40,000の平均分子量を有するポリビニルピロリドンとをミキサに加えて10分間混合する。その後混合した材料に変性無水アルコールを添加して10分間混合を続ける。その後湿った顆粒を20メッシュスクリーン内を通過させ、20時間に亘って室温で乾燥し、その後16メッシュスクリーン内を通過させる。次に顆粒をミキサに送り、ステアリン酸マグネシウムと混合し潤滑化する。
【0142】
その後、塩酸ヒドロコドン/塩酸ナルトレキソン組成物を剤形から押し出すための移動または押し出し組成物を以下のように調製する。まず11,200の平均分子量を有する3910 gのヒドロキシプロピルメチルセルロースを45,339 gの水中に溶解する。その後101 gのブチル化ヒドロキシトルエンを650 gの変性無水アルコール中に溶解する。次に2.5 kgのヒドロキシプロピルメチルセルロース水溶液をブチル化ヒドロキシトルエンアルコール溶液に加えながら混合する。その後ブチル化ヒドロキシトルエンアルコール溶液に残りのヒドロキシプロピルメチルセルロース水溶液を加えながら混合することにより、結合剤溶液の調製を完了する。
【0143】
次に21メッシュスクリーンを有するQuadro Comil((登録商標)ミルを用いて、36,000 gの塩化ナトリウムのサイズを一定にする。その後1200 gの酸化第二鉄を40メッシュスクリーン内を通過させる。その後スクリーニングした材料と、7,500,000の平均分子量を有する76,400 gの製薬上許容可能なポリ(酸化エチレン)と、11,200の平均分子量を有する2500 gのヒドロキシプロピルメチルセルロースとを、Glatt(登録商標)流体床粒状化ボウルに加える。ボウルを粒状化器に取り付け、粒状化プロセスを開始して粒状化を実施する。次いで乾燥粉体を空気中に浮遊させて10分間混合する。その後結合剤溶液を3つのノズルから粉体にスプレーする。以下の条件で行うプロセス中、粒状化をモニターする:総溶液スプレー速度:800 g/分、入口温度:43℃、空気流:4300 m3/時間。溶液スプレーの終了時に、得られたコーティング済み粒状化粒子のうち45,033 gを35分間乾燥させる。
【0144】
8メッシュスクリーンを有するQuadro Comil(登録商標)ミルを用いて、コーティング済み顆粒のサイズを一定にする。顆粒をTote(登録商標)タンブラーに送り、281.7 gのステアリン酸マグネシウムと混合し潤滑化する。
【0145】
次いで塩酸ヒドロコドン/塩酸ナルトレキソンを含む薬物組成物と押し出し組成物とを、Kilian(登録商標)タブレットプレス上で圧縮して二層タブレットにする。まず薬物組成物をダイのキャビティに添加して予備圧縮する。その後135 gの押し出し組成物を添加し、3メトリックトンの圧力ヘッドで層を押圧することにより、層が互いに接触する直径11/32インチ(0.873 cm)の配列体とする。
【0146】
二層配列体に半透壁をコーティングする。壁形成組成物は、39.8%のアセチル含有量を有する100%セルロースアセテートを含む。壁形成組成物をアセトン:水(95:5 wt:wt)共溶媒中に溶解することにより、4%固体溶液を生成する。24インチ(60 cm) Vector(登録商標) Hi-Coaterを用いて壁形成組成物を二層構造体の上および周囲にスプレーする。次に半透壁に20ミル(0.508 mm)の出口通路を1つ開けることにより、薬物ヒドロコドン層を剤形の外面部と連結させる。残りの溶媒を、温度45℃、湿度45%で72時間に亘って乾燥することにより除去する。次いで浸透性投与系を45℃で4時間に亘って乾燥させることにより余分な水分を除去する。
【実施例3】
【0147】
この予言的実施例では、ヒドロコドン5 mg/ナルトレキソン0.0625 mgの徐放性カプセルを以下に示す表3の処方で調製する。
【0148】
【表3】
【0149】
上記の処方を以下の手順に従って調製する。
【0150】
1.ステアリン酸アルコールフレークをインパクトミル内を通過させる。
2.塩酸ヒドロコドンと、塩酸ナルトレキソンと、ステアリン酸と、ステアリン酸アルコールと、Eudragit RSPOとを適切なブレンダ/ミキサ中で混合する。
3.混合した材料を高温でツインスクリュー押出器に連続的に送り、得られたストランドをコンベア上に収集する。
4.ストランドをコンベア上で冷却する。
5.ペレタイザを用いてストランドを1 mmのペレットに切断する。
6.微細なペレットおよび大きすぎるペレットに関してペレットをスクリーニングし、約0.8〜1.4 mmの範囲の許容可能なサイズにする。
7.120 mg/カプセルの重さになるようにカプセルを充填する(サイズ2のカプセルに充填する)。
【実施例4】
【0151】
この予言的実施例では、ヒドロコドン5 mg/ナルトレキソン0.0625 mgの徐放性カプセルを以下の手順に従って調製する。
【0152】
まず即時放出型ヒドロコドンビーズを以下に示す表4の処方で調製する。
【0153】
【表4】
【0154】
プロセス
1.薬物層溶液:塩酸ヒドロコドンとOpadryクリアとを水中に溶解する。
2.薬物ローディング:流体床乾燥器内でNuPareilビーズに薬物溶液をスプレーする。
3.コーティング:水中にOpadryバタースコッチを分散し、薬物をロードしたビーズにスプレーする。
その後、徐放性ビーズを以下に示す表5の処方で調製する。
【0155】
【表5】
【0156】
プロセス
1.制御型放出コーティング溶液:水中でクエン酸トリエチルを均質化する。Eudragit(登録商標) RS30DおよびEudragit(登録商標) RL30Dに分散液を添加し、その後混合物にCab-O-Sil(登録商標)を添加する。
2.シールコーティング溶液: Opadry(登録商標)クリアを水中に溶解する。
3.コーティング:流動化床ボトムスプレー法を用いて、制御型放出コーティング溶液、次いでシールコーティング溶液を塩酸ヒドロコドンIRビーズに適用する。
4.硬化:コーティング済みビーズをトレイ上に載置し、45℃で24時間に亘ってオーブンで硬化する。
【0157】
ヒドロコドン/ナルトレキソン徐放性ビーズを生成するために、単位当たり0.0625 mgのナルトレキソンを上記処方に含ませてもよい。ナルトレキソンを塩酸ヒドロコドンと共に純水中に溶解して、その後NuPareilビーズにスプレーしてもよい。
【実施例5】
【0158】
実施例5では、単一施設プラシーボ制御二重盲検無作為9処置3期間交差試験を、オープンレーベルスクリーニングフェーズと共に行った。試験は、経口ナルトレキソン(NTX)を経口即時放出型ヒドロコドン(HYIR)のアゴニスト作用と併用することによる、18歳〜45歳の、体重約45〜100 kgの範囲で且つ理想体重から15%以内の健常な成人男女における換気量に与える影響を評価するために行った。
【0159】
試験は、最長14日間のスクリーニングフェーズと、最長5日間のオープンレーベルHYIR滴定フェーズと、二重盲検フェーズ(1期間1日の処置を3期間行い、各処置期間の間に24時間の洗浄期間を挟む)と、最後の処置期間の後に行う最長14日間の試験終了時の往診から構成された。総試験期間は少なくとも39日間であった。
【0160】
登録前に、試験対象患者基準と除外基準とを用いて各被験者は試験に参加する資格を有すると認められた。各被験者から詳細な医療履歴を得た。オープンレーベルHYIR滴定フェーズを始める前に全被験者が以下のスクリーニング手順を終了した:身体検査、ECG測定、生命徴候、および臨床試験(血液学、化学、尿検査、HIVスクリーニング、肝炎スクリーニング、薬物スクリーニング、血中アルコールテスト、および妊娠テスト)。
【0161】
被験者は参加基準を満たした後、オープンレーベルHYIR滴定フェーズに参加した。オープンレーベルHYIR滴定フェーズは、悪影響を最小に保ちつつ呼吸運動の検出可能な変化を引き起こす、HYIRの最高許容投与量を決定するために計画された。呼吸運動の検出可能な変化は、投与後60分と90分の時点、または投与後90分と120分の時点で20 L/分のMV(換気量)において、投与前からのPETCO2(終末呼気二酸化炭素濃度(Torrで表す))の増加が少なくとも3 Torrであることと定義されている。呼吸運動の検出可能な変化を引き起こしたHYIRの最高許容投与量は、この試験の二重盲検部分で各被験者に投与するHYIRの量として用いた。被験者はCO2再呼吸テストで用いるスピロメータを作動する訓練を受けた。その後各被験者に対して最高3回に分けて滴定セッションを行い、HYIRを15 mg、20 mg、25 mgと順に多くなるように投与した。滴定セッション間には24時間の洗浄期間を設けた。被験者が許容不可能な悪影響を呈することなく25 mgのHYIRを投与されるまで、あるいは投与により許容不可能な悪影響が現れるまで、オープンレーベルフェーズを続けた。被験者にHYIRを投与して許容不可能な悪影響が現れた場合、許容不可能な悪影響が見られなかったHYIRの最高投与量を二重盲検フェーズで用いた。各滴定セッション中に行われたCO2再呼吸テストによって、処置前30分(0時間)並びに投与後30分、60分、90分、120分および180分の時点でのMV値とPETCO2値を得た。MVが変化した被験者に、試験の二重盲検フェーズを行った。
【0162】
33人の被験者がスクリーニングフェーズに登録された。13人が呼吸器のオピオイド感受性またはオピオイド不耐性の欠如のために離脱した。20人の被験者に対し無作為に二重盲検フェーズを行い、18人の被験者が二重盲検フェーズを終了した。
【0163】
試験薬物、投与形態、剤形、単位強度、並びに二重盲検フェーズのテスト処置および基準処置は以下の通りであった。
【0164】
テスト処置
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+NOS(ナルトレキソン経口溶液)プラシーボ
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+0.125 mg NOS
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+0.25 mg NOS
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+0.375 mg NOS
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+0.5 mg NOS
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+0.75 mg NOS
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+1.5 mg NOS
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+3.0 mg NOS
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+8.0 mg NOS
【0165】
基準処置
HYIR(15、20または25 mg; 3、4または5x5 mgタブレット)+NOSプラシーボ
【0166】
HYIRの5 mgタブレットはAAI Pharma, Wilmington, NCから供給された。
【0167】
NOSを処方するために塩酸ナルトレキソン粉体(Mallinckrodt Chemical Inc., St. Louis, MO)を用いた。ナルトレキソン粉体の必要量を計量し、50 mlの蒸留水および50 mlの単純シロップ中に別々に溶解してNFで最終容量100 mlを得た。これらの濃縮物(concentrations)は各処置期間中に同一容量(10 ml)のNOSが投与されることを可能にした。
【0168】
NOSプラシーボは苦味成分であるビターガード(安息香酸デナトニウム、NF)粉体を含んでいた。NOSプラシーボは、NOSの調製で用いたものと同一のビヒクルを用いて調製した。プラシーボ溶液の外見および味は活性溶液に似ていた。NOSプラシーボの投与容量(10 ml)は活性NOSの投与容量(10 ml)と合致させた。
【0169】
二重盲検フェーズにおいて、被験者に、オープンレーベルフェーズで決定した有効投与量(15、20または25 mg)のHYIRと、9つの考えられるNOS(ナルトレキソン経口溶液)処置(プラシーボ、0.125、0.25、0.375、0.5、0.75、1.5、3.0または8.0 mg)のうちの3つとを3期間に亘る交差臨床試験という形態で投与した。各被験者を6時間に亘って絶食させた後に、HYIRとNOSとを投与し、絶食を投与後3時間続けた。試験薬物投与の少なくとも30前(0時間)並びに投与後30分、60分、90分、120分および180分の時点でCO2再呼吸テストを行った。
【0170】
評価基準
薬力学:CO2再呼吸テストの結果を用いて、換気量に対するHYIRの影響およびHYIRとNOSとの影響を測定した。
【0171】
安全性:有害イベント、臨床試験結果、生命徴候、身体検査、および心電図(ECG)測定結果を用いて安全性を評価した。
【0172】
統計方法
PETCO2に対するMVのプロットから引き出した薬力学変数は、20および30 L/分のMV率(20リットルおよび30リットルのインターセプト値)におけるとMV/PETCO2回帰直線の傾斜とを含んでいた。投与前からの最大変化量(最大可能効果、MPE)を試験のオープンレーベルフェーズ(MPE(OL)=最大呼吸抑制)および二重盲検フェーズ(MPE(DB)=HYIR+NTXによる呼吸抑制)における各変数(MPE20、MPE30およびMPEslope)について計算した。二重盲検フェーズにおける各処置による最大呼吸抑制率(%MPE)を、各変数についてMPE(DB)/MPE(OL)x100という比から計算した。
【0173】
一次薬力学変数は、二重盲検フェーズでの各処置における20および30リットルインターセプトの%MPE (それぞれ%MPE20および%MPE30)であった。二次的薬力学変数は、%MPE並びに二重盲検フェーズのMPE20、MPE30およびMPEslopeの傾斜を含んでいた。これらの値を記述的統計を用いて処置群ごとにまとめ、無作為に選ばれた被験者、固定期間および固定処置というパラメータを伴う混合影響の分散分析(ANOVA)モデルを用いて分析した。さらに線形対照テストを用いて、NOS投与量と%MPE20および%MPE30との投与量−反応関係を調査した。
【0174】
結果
薬力学:
オープンレーベル20リットルおよび30リットルインターセプトMPEおよび傾斜値の効果サイズ分析により、呼吸抑制の最も高感度な(平均値についてのバラツキが最小である)測定値は20リットルおよび30リットルインターセプトMPE値であることが判明した。
【0175】
NTXの投与量が増加すると、呼吸抑制が統計的に有意な程度まで減少する傾向が見られた。これは、全ての処置において、CO2再呼吸テストから引き出された%MPE20、%MPE30、および二重盲検MPE20およびMPE30値に関して見られた。これらのデータは投与量依存的なHYIRにより誘導される呼吸抑制性のアンタゴニスト作用を示している。
【0176】
安全性:
呼吸抑制をもたらすために用いた15、20、25 mgのHYIR投与量、またはこれを0.125〜8.0 mgの範囲のNTX投与量と組み合わせたときに関して新たに同定された安全上の懸念はなかった。
【0177】
結論
0.125〜8.0 mgの範囲の経口NTXは投与量に依存して、15、20または25 mgのHYIRによって誘発される呼吸抑制を阻止した。安全上の新たな懸念も予期せぬ懸念もなかった。
【実施例6】
【0178】
実施例6は、維持量(60〜90 mg)の経口メタドンを毎日投与されている男女の被験者に対して行ったオープンレーベルフェーズと二重盲検処置フェーズからなった。維持量のメタドンは予定された各期間の前日に被験者に与えられた。維持量のメタドンを与えてから16時間後以降かつ22時間後以内に試験薬物を投与した。14人の被験者を試験に登録した(オープンレーベルフェーズ(ナルトレキソンの投与量を徐々に増加させる安全性評価)に2人、二重盲検フェーズに12人)。
【0179】
オープンレーベルフェーズ
オープンレーベルフェーズでは、維持量メタドン治療を受けている被験者に対するプロトコルで計画した2つのナルトレキソン投与量(0.75および2.0 mg)の安全性を評価した。試験のこのフェーズは、試験薬物の投与前に行われる最長14日間のスクリーニング往診と、ナルトレキソン定滴往診とから構成された。ナルトレキソン滴定往診中、2人の被験者に0時間の時点で30 mgのヒドロコドンと0.125 mgのナルトレキソンとを投与し、続く4時間に亘って1時間おきにさらにナルトレキソンを投与し、累積量で最高2.0 mgまで投与した。オープンレーベルフェーズでは、メタドンによる救済が必要となるまで、いずれの被験者にも累積量で1.0 mgを超えるナルトレキソンは投与しなかった。これら2人の被験者に見られた禁断症状の強度のために、試験で用いるナルトレキソンの投与量をプラシーボ0.75 mgおよび2.0 mgからプラシーボ、0.25 mgおよび0.5 mgに変更した。
【0180】
二重盲検フェーズ
二重盲検フェーズは、ナルトレキソン投与とナルトレキソンプラシーボ処置が無作為に選択される3期間に亘って3様式で行う無作為の交差試験として設計された。。試験のこのフェーズは、特定の処置シーケンスが無作為に選択される前に最長14日に亘って行われるスクリーニング往診と、これに続く二重盲検研究対象薬物を投与する3回の往診から構成された。
【0181】
各処置シーケンスは、1期間が4時間の処置を3期間行い、処置期間の間には48時間の洗浄期間が設けられた。各期間では、各被験者に30 mgのヒドロコドンと1〜3つの異なる量のナルトレキソン(プラシーボ、0.25 mgまたは0.5 mg)とを投与した。各被験者が二重盲検フェーズに参加した総期間は約20日間であった。
【0182】
8人の被験者が試験を終了した後、試験内容から0.5 mgのナルトレキソン投与を削除した。残り4人の被験者を登録し、ナルトレキソンを2つの投与量だけ、即ちプラシーボと0.25 mgナルトレキソンとを投与して試験を終了した。元々の無作為スケジュールおよび処置シーケンスは引き続き用いたが、処置シーケンスから0.5 mgのナルトレキソンを投与する期間を削除した。0.5 mgの投与量を削除した後に登録した被験者が試験に参加した期間は約17日間であった。
【0183】
試験の設計は、0.25および0.5 mgのナルトレキソン経口投与と組み合わせて経口投与された30 mgのヒドロコドンが、維持量メタドン治療を受けている被験者におけるいくつかの主観的および客観的測定値に与える影響の時間的経過および大きさを評価するのに適していた。この結論は、以下の試験設計特徴に基づいている。
【0184】
試験の偏りは、2(3x3)ラテン方格(但し0.5 mgのナルトレキソンによる処置は特定の被験者においては終了させた)、試験対象薬物の二重盲検投与、および無作為ナルトレキソン投与量としての試験設計によって制御した。
【0185】
オープンレーベルフェーズによって、この被験者集団が許容することができるナルトレキソン投与量の選択が可能になった。試験のオープンレーベルフェーズで見られた禁断症状の強度のために、試験で用いるナルトレキソンの投与量を0.75および2.0 mgから0.25および0.5 mgに減少した。
【0186】
嗜癖重症度指標を用いて被験者の依存度/常用癖を実証した。
【0187】
薬力学変数は、オピオイドの公知の生理学的および主観的影響の測定基準であった。
【0188】
生理学的薬力学変数は、皮膚温度と瞳孔の直径の測定値であった。オピオイドアゴニストは末梢動脈と静脈の拡張を引き起こすこと、および瞳孔に分布する副交感神経に対する興奮活性のために瞳孔を収縮することが知られている。
【0189】
この試験の主観的および客観的薬力学変数は、オピオイド薬物乱用の可能性および依存度の測定基準である主観的および観察者薬物影響スケール、オピオイド依存症の個人におけるオピオイド離脱症状および維持をモニタするための認知された測定基準である主観的および観察者症状評価スケール、オピオイド依存症の個人における乱用の可能性の主観的測定基準であるストリートバリュー評価アンケート、および薬物の区別および乱用の可能性を測定するために設計されたアンケートである薬物識別アンケートを含んだ。
【0190】
この試験の安全性パラメータは、有害イベント、臨床試験、心電図、および生命徴候であった。
【0191】
この試験のコントロール処置は30 mgのヒドロコドンおよびナルトレキソンプラシーボであった。
【0192】
各被験者は各期間の終了時に1日分のメタドン投与量を受け取ることになっていた。しかし被験者が許容不可能な離脱症状を経験した場合は、期間中のどの時点でも通常投与されている量のメタドンを救済薬として受け取ることができた。この試験の期間に投与される投与量30 mgのヒドロコドンはメタドンの経口維持量60〜90 mgと同等であった。
【0193】
この試験のオープンレーベルフェーズと二重盲検フェーズの両方に登録した被験者は、1日分の経口の維持量(60〜90 mg)のメタドンを受け取ることになっており、その結果物理的にオピオイドに依存することが予想された。
【0194】
薬力学パラメータは、瞳孔の直径、皮膚温度、主観的および観察者薬物影響スケール、並びに主観的および観察者症状評価スケールを含み、パラメータの測定は各テスト処置投与前0.5時間以内(投与前)並びに投与後0.25、0.5、1、2、3および4時間後に行った。ストリートバリュー評価アンケートは投与後0.25、0.5、1、2、3および4時間後に終了させた。薬物識別アンケートは投与後1および3時間後に終了させた。
【0195】
安全性測定は、身体検査、臨床試験(血液学および血液化学)およびECGを含み、スクリーニング往診および試験終了時または早期終了の場合の往診で行った。スクリーニング時、投与前、投与後0.25、0.5、1、2、3および4時間後、並びに試験終了時に生命徴候および酸素飽和を記録した。有害イベントは初日の試験薬物投与時から各被験者を試験から解放するまで収集した。
【0196】
薬力学測定基準
この試験で用いた薬力学測定基準は以下の通りである。
【0197】
薬物影響スケール(主観的および観察者)
主観的薬物影響スケールは、被験者が異なるテスト処置で経験した可能性がある4つの事項を評価した。
【0198】
この感覚が好きだ
悪い効果
気分が悪い
良い効果
【0199】
被験者に、4つの経験に関してどのように感じたかを0〜10の分類別の可視アナログスケール上でランクづけするように依頼した。スコアが高いほどオピオイドアゴニストの影響(陶酔感)が高いことを反映し、スコアが低いほどオピオイドアゴニストの影響が低下していること、またはアンタゴニスト作用(離脱症状)が増加していることを示した。
【0200】
観察者薬物影響スケールは、被験者が異なるテスト処置で示した可能性がある4つの事項を評価した。
【0201】
楽しい
不快な
具合が悪い
陶酔感
【0202】
観察者に、被験者が4つの経験の各々に関してどのように感じたと思うかを0〜10の分類別の可視アナログスケール上でランクづけするように依頼した。スコアが高いほどオピオイドアゴニストの影響(陶酔感)が高いことを反映し、スコアが低いほどオピオイドアゴニストの影響が低下していること、またはアンタゴニスト作用(離脱症状)が増加していることを示した。
【0203】
症状評価スケール(主観的および観察者)
主観的症状評価スケールは、オピオイドレセプター活性または禁断症状という症状を、アンタゴニスト項目およびその強度レベルに関して評価するために被験者によって用いられた。
【0204】
【0205】
これらの症状は1〜3のスケールで評価された。
【0206】
全く感じない。
ある程度感じる。
大いに感じる。
【0207】
スコアが高いほどオピオイドアゴニストまたはアンタゴニスト症状が増加していることを反映し、スコアが低いほどオピオイドアゴニストまたはアンタゴニスト症状が低下していることを示した。
【0208】
観察者症状評価スケールアンケートは、被験者が示したオピオイドレセプターアゴニストまたはアンタゴニスト活性およびその強度レベルを示す可能性のある徴候を評価するために用いられた。
【0209】
【0210】
これらの症状は1〜4のスケールで評価された。
【0211】
全くない。
相対的に目立たないが良く観察すると気づく。
かなり明らかである。細かく観察しなくても気づく。
非常に明らかである。持続的であるか、または被験者にとって煩わしく見える。
【0212】
スコアが高いほどオピオイドアゴニストまたはアンタゴニスト徴候が増加していることを反映し、スコアが低いほどオピオイドアゴニストまたはアンタゴニスト徴候が低下していることを示した。
【0213】
瞳孔の直径
2倍レンズを取り付けたPolaroid(Cambridge, MA)カメラを用いて常に周囲光がある状態で被験者の目を撮影した。各写真からの瞳孔の直径を測径器を用いてミリメートル単位で測定した。各期間で同じ目を用いて全ての決定を行った。測定に用いた目を記録した。
【0214】
薬物識別アンケート
薬物識別アンケートは、10の薬物カテゴリーのリストからなり、オピオイド乱用集団になじみのある文言が用いられた。被験者は、試験薬物がどのカテゴリーに最も近いかを選択した。アンケートには以下のカテゴリーをリストアップした。
【0215】
ブランクまたはブラシーボ
アヘン剤(例えばモルヒネ、ヘロイン、コデイン、メタドン)
アヘンアンタゴニスト(例えば、ナロキソン、ナルトレキソン)
抗精神病薬または神経弛緩薬(例えば、ハルドール、ステラジン)
バルビツール酸系催眠薬および睡眠薬(例えば、クアールード、ペントバルビタール、セコナール)
抗鬱薬(例えば、エラビル、イミプラミン)
PCPまたは幻覚剤(例えば、LSD、メスカリン、MDA、STP)
ベンゾジアゼピン(例えば、バリウム、リブリウム、アチバン、キサナックス)
コカインまたは興奮剤(例えば、アンフェタミン、デキセドリン、リタリン)
その他
【0216】
ストリートバリュー評価
被験者に「街でこの薬物を買うのにいくら払いますか」と質問する。
【0217】
被験者はその後薬物の価格はいくらだと思うかを直接CRFに記録する。
【0218】
皮膚温度
使い捨て温度プローブ付きのデュアルチャネル、デュアルディスプレイ電子温度計を用いて皮膚温度を測定した。温度を摂氏で記録した。
有害イベント
【0219】
有害イベントとは、医薬品と関係があると考えられるか否かにかかわらず一時的にその医薬品の使用と関連づけられる、好ましくなく意図しない何らかの徴候(実験室での異常の発見を含む)、症状、または疾病であった。自発的に報告されたか調査員が観察したかにかかわらず、試験薬物を最初に投与してから被験者を試験から解放するまでの間に起こったすべての有害イベントを、有害イベントの形式で報告した。医学的干渉を必要とする有害イベントが起こった場合、適切な資格を有しライセンスを有する医療担当者が適切なサポートおよび/または信頼できる治療を提供した。
【0220】
全体的結論
この試験は、維持量メタドン治療を受けている被験者において、30 mgのHYIRの経口投与と組み合わせてある範囲の量のNTXを経口投与することが、オピオイドアゴニストおよびアンタゴニスト活性の様々な主観的および生理学的測定値に与える影響を特徴づけるために設計された。
この被験者集団では、30 mgのHYIRの経口投与は有意な主観的または生理学的オピオイドアゴニスト活性を引き起こさなかった。HYIRおよびNTXプラシーボによる処置の後、全ての薬力学変数の投与前からの変化は最小であった。この処置を受けた12人の被験者のうち4人が救済用のメタドン投与を必要とした。これは突然禁断が現れている可能性を示している。
【0221】
この試験の一次薬力学変数は、「この感覚が好きだ」「良い効果」「悪い効果」および「気分が悪い」という質問に対する平均PDmax(最大投与前スコア)値であった。主観的薬物影響スケールにおける4つの質問のすべてにおいて、平均PDmax値に対してNTX投与量に関係する影響が見られた。NTXの投与量を0.25から0.5 mgに増加した結果、各質問に対する投与前スコアからの最大変化がますます否定的になった。このことはすべての場合において、オピオイドアゴニストの影響に対する、NTX投与量に関係するアンタゴニスムを示している。NTXプラシーボ処置とNTXの投与量0.25 mgとの間には、「この感覚が好きだ」および「悪い効果」という質問に関して統計的に有意な差異があった。NTXプラシーボ処置とNTXの投与量0.5 mgとの間には、「気分が悪い」以外の全ての質問に関して統計的に有意な差異があった。
【0222】
主観的および観察者症状評価スケールでは、アゴニストの合計値以外の二次薬力学変数に対して、NTXの投与量に関係する影響があった。但しこれは必ずしも統計的に有意ではない。NTX投与量0.25 mgは、感情が否定的になる傾向(オピオイドアゴニスト影響の低下)とアンタゴニスト活性の増加(禁断症状の傾向)の閾値投与量であった。NTX投与量0.5 mgは、主観的および観察者薬物影響スケールにおいて示されたNTXプラシーボ処置に対する統計的に有意な差異、主観的および観察者症状評価スケールから得られたアンタゴニスト合計値、および瞳孔の直径により、禁断症状の強力な証拠を提示した。0.25および0.5 mgのNTXを投与された被験者のそれぞれ約60%および90%が救済用のメタドン投与を必要とした。
【0223】
オピオイド薬物の数々の測定のPDmaxおよびAUCに対する、3期間の各々における処置手段を示す図面(図1〜図12)は、HYIR+NTXプラシーボによる処置とHYIR+0.25 mgのNTXによる処置とHYIR+0.5 mgのNTXによる処置との間で3期間中の変化の傾向が異なることを示した。観察された手段に見られる傾向は、HYIR+NTXプラシーボ投与による処置後の3期間に亘って主観的および生理学的オピオイドアゴニスト影響が増加し、且つHYIR+0.25 mgのNTXまたはHYIR+0.5 mgのNTX投与による処置後の3期間に亘ってオピオイドアンタゴニスト効果が増加するという仮定に合致する。
【0224】
0.25または0.5 mgのNTXの経口投与を30 mgのHYIR投与と共に投与することの安全性の測定を行ったが、この被験者集団については新しいまたは予期しない安全上の懸念は示唆されなかった。NTX投与量が増加するにつれて、各被験者のオピオイド離脱症状に共通して関連づけられる、処置に伴う有害イベントの数は増えたが、処置に伴う有害イベントのほとんどは軽度か中程度であった。1被験者のみが臨床的に顕著な異常実験値を呈したが、これはこの被験者の過去の医療履歴にリストアップされた状態によるものであった。臨床的に顕著な生命徴候異常の発生は被験者および処置の両方に関して隔離された事項であった。
【0225】
結論として、0.25および0.5 mgのNTX経口投与量は、維持量メタドンを投与されている被験者において有害な影響を引き起こすことが判明した。NTXは投与量に依存して、否定的な感情の状態および禁断症状を増加させる。経口NTX(0.25または0.5 mg)と経口HYIR(30 mg)の組み合わせは、新しいまたは予期しない安全上の懸念をもたらさなかった。実際、HYIRに低量のNTXを添加すると、魅力が低減し、そのため身体的にオピオイドに依存している被験者においてHYIRの乱用傾向の可能性が低減した。
【実施例7】
【0226】
実施例7は、プラシーボ制御型二重盲検無作為4処置4期間交差試験として行った単一施設試験からなった。これは単一盲検フェーズを含んだ。各処置シーケンスは、少なくとも5日間の洗浄期間により分けられた、1期間が4時間である4処置期間からなった。各処置期間において、各被験者に15 mgの経口HYIRおよび、プラシーボ、0.25、0.5または1.0 mgのNTXのいずれかを投与した。この試験の総時間はスクリーニングを含めて約52日間であった。
【0227】
スクリーニングフェーズ
無作為化によりこの試験の二重盲検部分を行う前に、スクリーニングフェーズを最長21日間に亘って行った。被験者は熱不快度テストの訓練セッションに参加した。この訓練は、43℃、46℃および49℃に加熱した銅塊を順に前腕の指定された部位に5秒以内の時間に亘って適用することを含んだ。各適用後、被験者は100 mmの可視アナログスケール(VAS)を用いて疼痛の強度を評価した。調査員の裁量で被験者が常に信頼できるVASスコアを生成できるようになるまで、この手順を新しい肌部位で繰り返した。満足にスクリーニングフェーズを終了することができなかった被験者を試験から除いた。
【0228】
単一盲検フェーズ
スクリーニングフェーズの終了後、以下のように単一盲検フェーズで熱不快度評価を行った。
【0229】
前腕の部位を選択し洗浄可能マーカーでマーキングした。ベースラインの生命徴候を記録した。前腕の所定の部位に局部麻酔薬(EMLA(登録商標)クリーム、AstraZeneca, Wilmington, DE)を塗った。約1.5時間後(麻酔薬が効果を呈した後)に、クリームを除去し、52℃に加熱した銅塊を用いて3分間に亘って熱刺激を前腕の部位に適用した。局部麻酔から感覚が回復するまで約1時間待った。その後各被験者にプラシーボとしての7.5 mg HYIRタブレット2個とプラシーボとしてのNTXタブレット2個とを経口投与した。
【0230】
投与後1.5時間の時点で、生命徴候を記録し熱不快度テストを行った。熱不快度テストは、43℃、46℃および49℃に加熱した銅塊を順に部位に5秒間適用することから構成された。各適用後、被験者は100 mmのVASスケールを用いて疼痛の強度を評価した。これらの測定から得られたVASスコアを加算し、60 mm以上の合計値を出した被験者のみをスクリーニングプロセスに進ませた。
【0231】
スクリーニングに進んだ被験者に、7.5 mgのHYIRタブレット2個とプラシーボとしてのNTXタブレット2個とを投与した。HYIRの投与後1.5時間の時点で、3つの温度の各々についてテスト測定を繰り返した。VASの疼痛スコアの合計値が前回のテストセッションに比べて少なくとも20 mm減少した被験者を、この試験の二重盲検部分に参加するに適していた。
【0232】
二重盲検フェーズ
少なくとも5日後、単一盲検フェーズで成功裡に終了した被験者を無作為に試験の二重盲検フェーズに選択した。健常な男女志願者をこの試験に登録した。各被験者は交差計画の4処置期間に参加し、15 mgのHYIRをNTXの4つの投与量(プラシーボ、0.25、0.5および1.0 mg)の各々と共に投与された。この二重盲検試験は、HYIRの鎮痛効果に対するNTXの影響を評価するために設計された。試験は、痛覚過敏症である健常な志願者における、15 mgのHYIRの鎮痛効果に対する0.25、0.5および1.0 mgの経口NTXの影響を評価した。この試験ではNTXの3つの異なる投与量(0.25、0.5および1.0 mg)を用い、プラシーボNTXと比較した。
【0233】
4試験期間の各々において処置シーケンス毎に同じ手順を続けた。被験者は単一盲検フェーズまたは前回の交差期間5日以上後に各処置期間を開始した。被験者は最初の処置期間の開始時に痛覚計(疼痛潜伏テストで用いる)を使用する訓練を受けた。各被験者は処置当日、少なくとも6時間絶食した後に施設に入った。生命徴候を記録し、尿による薬物スクリーニング、アルコールスクリーニング、および尿による妊娠スクリーニング(女性の被験者)を行った。被験者がテストを続けるには、全ての薬物スクリーニングの結果が試験薬物以外の全ての薬物について陰性である必要があった。
【0234】
各前腕においてテスト部位およびコントロール部位を選択し、洗浄可能マーカーでマーキングした。次いで、テスト部位に局部麻酔薬を塗った。約1.5時間後(麻酔薬が効果を呈した後)にクリームを除去し、52℃に加熱した銅塊を用いて3分間に亘って熱刺激をテスト部位に適用した。感覚が回復するまで約1時間待った後、以下のベースライン測定を行った:生命徴候、瞳孔計測、熱不快度(43℃、46℃および49℃において)、疼痛潜伏(放射型熱刺激適用後の潜伏)、症状評価スケール、薬物評価アンケート、およびオピオイド誘導型薬物影響アンケート。
【0235】
ベースライン評価が終了すると即座に、各被験者に15 mgの経口HYIRと、プラシーボ、0.25、0.5および1.0 mgのNTXのいずれかとを無作為コードに従って投与した。その後、ベースラインで行ったものと同じテスト測定を、投与後0.5、1、2、3および4時間の時点で行った。各時点でコントロール部位(熱刺激を受けていない方の前腕上の領域)での熱不快度テストおよび疼痛潜伏テストを行った。コントロール部位でのテストは、各時点でテスト部位でのテストより前に行った。各被験者は休息および回復のために5日以上待ってから次の期間のテストに戻った。追跡フェーズでは、被験者は試験の終了後7日以内にクリニックに戻ってテスト部位の最終チェックと被験者の実験室データの検討を受け、その後試験から正式に解放された。
【0236】
薬力学的測定
各テストセッションにおいて、投与前30分以内並びに投与後0.5、1、2、3および4時間の時点で以下の薬力学パラメータを記録した。
【0237】
熱不快度テスト(3つの温度において)
疼痛潜伏テスト
瞳孔の直径
症状評価スケールアンケート
オピオイド誘導型薬物影響アンケート
薬物評価アンケート
【0238】
グレード付き熱刺激を用いた熱不快度テスト
熱不快度テストは、直径1インチ(1”)の銅塊(Uniformed Services University of the Health Sciences, Bethesda, MD)に5秒間接触した後の被験者の不快感の感知を測定するために設計された。加熱(52℃)した銅塊を被験者の前腕に3分間適用することにより温熱損傷を引き起こした。その後テストは、被験者を3つの異なる温度、即ち43℃、46℃および49℃に加熱した銅塊に曝すことから構成された。各銅塊の必要温度は、Isotemp Dry Bath(Fisher Scientific, Indiana, PA)内に載置した加熱ブロック(モデル145および147、Fisher Scientific, Indiana, PA)に銅塊を挿入することにより得た。各測定時点で適用された銅塊の温度シーケンスは、試験の二重盲検部分を開始したときに各被験者に対して無作為に選択した。熱不快度テストはコントロール部位と実験皮膚部位との両方で行った。被験者は、100 mmのVASを用いて自分の不快度を評価した。スケールの左端は「全く不快感なし」となっており、右端は「私が耐えられる最大の不快感」となっていた。各被験者は、0 mmと100 mmとの間の水平スケール上に垂直の線でマーキングすることにより回答した。左端から垂直マークまでの距離を測定し、熱不快度の量的測定値として用いた。
【0239】
疼痛潜伏テスト
疼痛潜伏テストは、放射型熱刺激の付与から疼痛の開始(放射型熱刺激の自己終了により実証される)までの潜伏期間を秒で把握するために設計された。放射型熱刺激はモデル33テイルフリック鎮痛メータ(IITC Inc, Woodland Hills, CA)を用いてコントロール部位と実験皮膚部位との両方に適用した。各被験者は、最初の二重盲検期間の開始時にこの痛覚計を用いる訓練を受けた。痛覚計は被験者の皮膚から4インチという固定された距離に置かれ、選択された皮膚部位に向けて高強度の光を発した。調査員が痛覚計をオンにし、被験者は疼痛が始まったときにストップボタンを押すことによりテストを終了させた(痛覚計をオフにした)。被験者が高強度の光に曝されていた総時間を適切なCRFページに記録した。
【0240】
瞳孔計測
瞳孔計測は、瞳孔の直径に対する試験処置の影響を測定するために行った。改変された倍率2倍の接眼レンズ(John Hopkins University, Baltimore, MD)を有するPolaroidワンステップクローズアップカメラ(Polaroid Corporation, Cambridge, MA)とPolaroid600カラーフィルム(Polaroid Corporation, Cambridge, MA)とを用いて瞳孔を撮影した。実験室の背景照明は、モデルL-246 sekonic LUXメータ(Sekonic Co., Tokyo, Japan)を用いて測定した。被験者の眼窩にカメラを位置づけ、虹彩をレンズアダプタの開口部中央に整合させた。モデルCD-6C Mitutoyoデジタル測径器(Judge Tool Sales, Southport, CT)を用いて写真から瞳孔の直径をミリメートル単位で測定した。各被験者について常に同じ目を測定した。
【0241】
症状評価スケールアンケート
症状評価スケールアンケートは25項目から構成された。被験者には、各項目について「今どのように感じているか」を示すように指示した。各項目は3ポイントスケール、即ち「全くそう感じない」「ある程度そう感じる」または「大いにそう感じる」により評価した。12項目はアゴニスト項目に分類され、13項目はアンタゴニスト項目に分類された。アゴニスト項目はオピオイド投与に関連づけられる症状であった。アンタゴニスト項目はオピオイド離脱症状に関連づけられる症状であった。12のアゴニスト項目は、口数が多い、エネルギッシュである、身体が重い/動きがのろい、無頓着である、肌が痒い、幸せである、そわそわしている、安心している、頷き、リラックスしている、愉快である、および成り行きまかせである、であった。13のアンタゴニスト項目は、落ち着かない、吐き気がする、短気である、緊張している、神経過敏である、ほてりまたは冷え、肌がひんやりまたはじっとりとしている、赤面、あくびが出る、涙目、鼻水が出る、寒気/鳥肌、および発汗であった。
【0242】
オピオイド誘導型薬物影響アンケート
左端に「全くそうでない」とあり右端に「大いにそうである」となっている0〜100 mmのVASスケールを用いて、7つの薬物影響を評価した。これらの影響は、吐き気、嘔吐、めまい、嗜眠状態、便秘、掻痒、および口が渇くことを含んでいた。被験者は、その瞬間に薬物によってどのように感じたかを最もよく表す水平線上の距離に垂直のマークを記入した。
【0243】
薬物評価アンケート
左端に「全くそうでない」とあり右端に「大いにそうである」となっている0〜100 mmのVASスケールを用いて、3つの薬物に関する質問について評価した。これらの質問は、「今薬物の影響を感じていますか」「今感じている薬物の影響が好きですか」そして「今感じている薬物の影響が嫌いですか」であった。被験者は、その瞬間に薬物によってどのように感じたかを最もよく表す水平線上の距離に垂直のマークを記入した。
【0244】
有害イベント
AE(有害イベント)を、医薬品と関係があると考えられるか否かにかかわらず一時的にその医薬品の使用と関連づけられる、好ましくなく意図しない何らかの徴候(実験室での異常の発見を含む)、症状、または疾病と定義した。AEは、試験に登録した被験者に試験薬物を最初に投与した後に起こった場合にのみ、TEAE(処置による有害イベント)と分類した。TEAEの観察期間は、試験薬物を最初に投与してから期間4の終了後試験から解放されるまで、またはそれより早い中止時までであった。試験期間中に被験者が報告したか、または調査員/試験スタッフが観察したAEはすべて完全に記録した。医学的干渉を必要とするAEが起こった場合、適切な資格を有しライセンスを有する医療担当者が適切なサポートおよび/または信頼できる治療を提供した。研究の終了時/中止時にAEが解決しなかった場合は、解決するまで、これ以上の向上が望めないと調査員が決定するまで、または被験者に連絡がつかなくなるまで被験者をフォローした。
【0245】
薬力学的結果
熱不快度の平均VASスコアおよび疼痛潜伏、並びに痛覚過敏の誘導型薬力学測定値については、HYIR+プラシーボNTX処置とHYIR+0.25、0.5または1.0 mgのNTX処置との間で統計的に有意な差異はなかった。
【0246】
HYIR+0.5 mgのNTXおよびHYIR+1.0 mgのNTXの投与後には、HYIR+プラシーボNTXの投与後に比べて、瞳孔の直径AUC(投与前からの変化)に統計的に有意な増加があったが、PDmaxにはなかった。
【0247】
HYIRの主観的オピオイドアゴニスト影響に関して、処置間、またはNTXの投与量に関係する一貫した傾向の間で統計的に有意な差異は概してなかった。これは主観的症状評価スケールおよび主観的薬物評価アンケートによって評価された通りである。
【0248】
全体的結論
健常な志願者に対するこの試験の結果から以下の結論が導かれた。
【0249】
この試験で用いたNTXの投与量(0.25、0.5または1.0 mg)はいずれも、15 mgの経口HYIRによる鎮痛に対して統計的に有意な低下をもたらさなかった。これは熱不快度VASスコアおよび疼痛潜伏テストによって測定された通りである。
【0250】
15 mg投与量のHYIRの生理学的オピオイドアゴニスト活性に対して、NTX投与量に関係する一貫した影響はなかった。但しすべてのNTX投与量はHYIR誘導型瞳孔収縮を低減した。
【0251】
15 mg投与量のHYIRの主観的オピオイドアゴニスト活性に対して、NTX投与量に関係する一貫した影響はなかった。
【0252】
健常な志願者を、15 mgの経口HYIRと0.25、0.5または1.0 mgのNTXとの組み合わせで処置することに関連する安全上の懸念はなかった。
【0253】
一般的にオピオイドの使用と関連づけられる報告済みTEAEはの数は、NTX投与量の増加に伴って減少した。
【0254】
当業者には本発明の多くの他の改変が明らかであり、これらの改変も添付の請求の範囲の範囲に含まれるものとする。
【図面の簡単な説明】
【0255】
【図1】図1は実施例6の3処置期間の各々における、「この感覚が好きだ」という主観的薬物効果の、ベースラインからの最大変化(PDmax)を示す図である。
【図2】図2は実施例6の3処置期間の各々における、「この感覚が好きだ」という主観的薬物効果のPDmaxの曲線下面積(AUC)を示す図である。
【図3】図3は実施例6の3処置期間の各々における、「良い効果」という主観的薬物効果の、ベースラインからの最大変化(PDmax)を示す図である。
【図4】図4は実施例6の3処置期間の各々における、「良い効果」という主観的薬物効果のPDmaxの曲線下面積(AUC)を示す図である。
【図5】図5は実施例6の3処置期間の各々における、「気分が悪い」という主観的薬物効果の、ベースラインからの最大変化(PDmax)を示す図である。
【図6】図6は実施例6の3処置期間の各々における、「気分が悪い」という主観的薬物効果のPDmaxの曲線下面積(AUC)を示す図である。
【図7】図7は実施例6の3処置期間の各々における、「悪い効果」という主観的薬物効果の、ベースラインからの最大変化(PDmax)を示す図である。
【図8】図8は実施例6の3処置期間の各々における、「悪い効果」という主観的薬物効果のPDmaxの曲線下面積(AUC)を示す図である。
【図9】図9は実施例6の3処置期間の各々における、主観的「アンタゴニスト全症状スコア値」の、ベースラインからの最大変化(PDmax)を示す図である。
【図10】図10は実施例6の3処置期間の各々における、主観的「アンタゴニスト全症状スコア値」のPDmaxの曲線下面積(AUC)を示す図である。
【図11】図11は実施例6の3処置期間の各々における、瞳孔の直径の、ベースラインからの最大変化(PDmax)を示す図である。
【図12】図12は実施例6の3処置期間の各々における、瞳孔の直径のPDmaxの曲線下領域(AUC)を示す図である。
【特許請求の範囲】
【請求項1】
約5〜約20 mgのヒドロコドンまたはその製薬上許容可能な塩と、0.055〜0.56 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物であって、前記ナルトレキソンまたはその製薬上許容可能な塩と前記ヒドロコドンまたはその製薬上許容可能な塩とが0.011:1〜0.028:1の割合で含まれる医薬組成物。
【請求項2】
約5 mgのヒドロコドンまたはその製薬上許容可能な塩と0.055 mg〜0.14 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項3】
約7.5 mgのヒドロコドンまたはその製薬上許容可能な塩と0.0825 mg〜0.21 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項4】
約10 mgのヒドロコドンまたはその製薬上許容可能な塩と0.11 mg〜0.28 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項5】
約15 mgのヒドロコドンまたはその製薬上許容可能な塩と0.165 mg〜0.42 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項6】
約20 mgのヒドロコドンまたはその製薬上許容可能な塩と0.22 mg〜0.56 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項7】
約5 mgのヒドロコドンまたはその製薬上許容可能な塩と0.0625 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項8】
約7.5 mgのヒドロコドンまたはその製薬上許容可能な塩と0.09375 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項9】
約10 mgのヒドロコドンまたはその製薬上許容可能な塩と0.125 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項10】
約15 mgのヒドロコドンまたはその製薬上許容可能な塩と0.1875 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項11】
約20 mgのヒドロコドンまたはその製薬上許容可能な塩と0.25 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項12】
前記ヒドロコドンまたはその製薬上許容可能な塩を徐放する徐放性賦形剤をさらに含む、請求項1に記載の医薬組成物。
【請求項13】
前記ナルトレキソンまたはその製薬上許容可能な塩を徐放する徐放性賦形剤をさらに含む、請求項1に記載の医薬組成物。
【請求項14】
前記ヒドロコドンまたはその製薬上許容可能な塩と、前記ナルトレキソンまたはその製薬上許容可能な塩とを徐放する徐放性賦形剤をさらに含む、請求項1に記載の医薬組成物。
【請求項15】
剤形がヒト患者への定常状態での経口投与後少なくとも12時間に亘って有効に鎮痛を行う、請求項12および14に記載の医薬組成物。
【請求項16】
剤形がヒト患者への定常状態での経口投与後少なくとも24時間に亘って有効に鎮痛を行う、請求項12および14に記載の医薬組成物。
【請求項17】
前記ヒドロコドンまたはその製薬上許容可能な塩と前記ナルトレキソンまたはその製薬上許容可能な塩とが前記徐放性賦形剤中で実質的に相互分散(interdispersed)する、請求項14に記載の医薬組成物。
【請求項18】
前記ヒドロコドンが重酒石酸塩(bitartratre salt)の形態にある、請求項1〜11に記載の医薬組成物。
【請求項19】
前記ナルトレキソンが塩酸塩の形態にある、請求項1〜11に記載の医薬組成物。
【請求項20】
イブプロフェン、ジクロフェナク、ナプロキセン、ベノキサプロフェン、フルルビプロフェン、フェノプロフェン、フルブフェン(flubufen)、ケトプロフェン、インドプロフェン、ピロプロフェン、カルプロフェン、オキサプロジン、プラモプロフェン、ムロプロフェン、トリオキサプロフェン、スプロフェン、アミノプロフェン、チアプロフェン酸(tiaprofenic acid)、フルプロフェン、ブクロキシン酸(bucloxic acid)、インドメタシン、スリンダク、トルメチン、ゾメピラク、チオピナク、ジドメタシン、アセメタシン、フェンチアザク、クリダナク、オキシピナク、メフェナム酸、メクロフェナム酸、フルフェナム酸、ニフルム酸、トルフェナム酸、ジフルリサル(diflurisal)、フルフェニサル(flufenisal)、ピロキシカム、スドキシカム、イソキシカム、これらの製薬上許容可能な塩およびそれらの混合物からなる群より選択される非ステロイド性抗炎症剤をさらに含む、請求項1〜19に記載の医薬組成物。
【請求項21】
請求項1〜20に記載の医薬組成物を経口投与することを含む、ヒト患者の疼痛を治療する方法。
【請求項22】
約5〜約20 mgのヒドロコドンまたはその製薬上許容可能な塩と0.055〜0.56 mgのナルトレキソンまたはその製薬上許容可能な塩とを組み合わせて経口剤形にすることを含む、医薬組成物の調製方法であって、前記ナルトレキソンまたはその製薬上許容可能な塩と前記ヒドロコドンまたはその製薬上許容可能な塩とを0.011:1〜0.028:1の割合で含ませる前記方法。
【請求項23】
請求項1〜20に記載の医薬製剤を調製することを含む、ヒドロコドン製剤の乱用を阻止する方法。
【請求項24】
請求項1〜20のいずれかに記載の剤形の調製における、ヒドロコドンまたはその製薬上許容可能な塩の使用。
【請求項25】
請求項1〜20のいずれかに記載の剤形の調製における、ナルトレキソンまたはその製薬上許容可能な塩の使用。
【請求項26】
請求項1〜20のいずれかに記載の剤形の調製における、ヒドロコドンまたはその製薬上許容可能な塩およびナルトレキソンまたはその製薬上許容可能な塩の使用。
【請求項1】
約5〜約20 mgのヒドロコドンまたはその製薬上許容可能な塩と、0.055〜0.56 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む医薬組成物であって、前記ナルトレキソンまたはその製薬上許容可能な塩と前記ヒドロコドンまたはその製薬上許容可能な塩とが0.011:1〜0.028:1の割合で含まれる医薬組成物。
【請求項2】
約5 mgのヒドロコドンまたはその製薬上許容可能な塩と0.055 mg〜0.14 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項3】
約7.5 mgのヒドロコドンまたはその製薬上許容可能な塩と0.0825 mg〜0.21 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項4】
約10 mgのヒドロコドンまたはその製薬上許容可能な塩と0.11 mg〜0.28 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項5】
約15 mgのヒドロコドンまたはその製薬上許容可能な塩と0.165 mg〜0.42 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項6】
約20 mgのヒドロコドンまたはその製薬上許容可能な塩と0.22 mg〜0.56 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項7】
約5 mgのヒドロコドンまたはその製薬上許容可能な塩と0.0625 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項8】
約7.5 mgのヒドロコドンまたはその製薬上許容可能な塩と0.09375 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項9】
約10 mgのヒドロコドンまたはその製薬上許容可能な塩と0.125 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項10】
約15 mgのヒドロコドンまたはその製薬上許容可能な塩と0.1875 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項11】
約20 mgのヒドロコドンまたはその製薬上許容可能な塩と0.25 mgのナルトレキソンまたはその製薬上許容可能な塩とを含む、請求項1に記載の医薬組成物。
【請求項12】
前記ヒドロコドンまたはその製薬上許容可能な塩を徐放する徐放性賦形剤をさらに含む、請求項1に記載の医薬組成物。
【請求項13】
前記ナルトレキソンまたはその製薬上許容可能な塩を徐放する徐放性賦形剤をさらに含む、請求項1に記載の医薬組成物。
【請求項14】
前記ヒドロコドンまたはその製薬上許容可能な塩と、前記ナルトレキソンまたはその製薬上許容可能な塩とを徐放する徐放性賦形剤をさらに含む、請求項1に記載の医薬組成物。
【請求項15】
剤形がヒト患者への定常状態での経口投与後少なくとも12時間に亘って有効に鎮痛を行う、請求項12および14に記載の医薬組成物。
【請求項16】
剤形がヒト患者への定常状態での経口投与後少なくとも24時間に亘って有効に鎮痛を行う、請求項12および14に記載の医薬組成物。
【請求項17】
前記ヒドロコドンまたはその製薬上許容可能な塩と前記ナルトレキソンまたはその製薬上許容可能な塩とが前記徐放性賦形剤中で実質的に相互分散(interdispersed)する、請求項14に記載の医薬組成物。
【請求項18】
前記ヒドロコドンが重酒石酸塩(bitartratre salt)の形態にある、請求項1〜11に記載の医薬組成物。
【請求項19】
前記ナルトレキソンが塩酸塩の形態にある、請求項1〜11に記載の医薬組成物。
【請求項20】
イブプロフェン、ジクロフェナク、ナプロキセン、ベノキサプロフェン、フルルビプロフェン、フェノプロフェン、フルブフェン(flubufen)、ケトプロフェン、インドプロフェン、ピロプロフェン、カルプロフェン、オキサプロジン、プラモプロフェン、ムロプロフェン、トリオキサプロフェン、スプロフェン、アミノプロフェン、チアプロフェン酸(tiaprofenic acid)、フルプロフェン、ブクロキシン酸(bucloxic acid)、インドメタシン、スリンダク、トルメチン、ゾメピラク、チオピナク、ジドメタシン、アセメタシン、フェンチアザク、クリダナク、オキシピナク、メフェナム酸、メクロフェナム酸、フルフェナム酸、ニフルム酸、トルフェナム酸、ジフルリサル(diflurisal)、フルフェニサル(flufenisal)、ピロキシカム、スドキシカム、イソキシカム、これらの製薬上許容可能な塩およびそれらの混合物からなる群より選択される非ステロイド性抗炎症剤をさらに含む、請求項1〜19に記載の医薬組成物。
【請求項21】
請求項1〜20に記載の医薬組成物を経口投与することを含む、ヒト患者の疼痛を治療する方法。
【請求項22】
約5〜約20 mgのヒドロコドンまたはその製薬上許容可能な塩と0.055〜0.56 mgのナルトレキソンまたはその製薬上許容可能な塩とを組み合わせて経口剤形にすることを含む、医薬組成物の調製方法であって、前記ナルトレキソンまたはその製薬上許容可能な塩と前記ヒドロコドンまたはその製薬上許容可能な塩とを0.011:1〜0.028:1の割合で含ませる前記方法。
【請求項23】
請求項1〜20に記載の医薬製剤を調製することを含む、ヒドロコドン製剤の乱用を阻止する方法。
【請求項24】
請求項1〜20のいずれかに記載の剤形の調製における、ヒドロコドンまたはその製薬上許容可能な塩の使用。
【請求項25】
請求項1〜20のいずれかに記載の剤形の調製における、ナルトレキソンまたはその製薬上許容可能な塩の使用。
【請求項26】
請求項1〜20のいずれかに記載の剤形の調製における、ヒドロコドンまたはその製薬上許容可能な塩およびナルトレキソンまたはその製薬上許容可能な塩の使用。
【図1】


【図2】
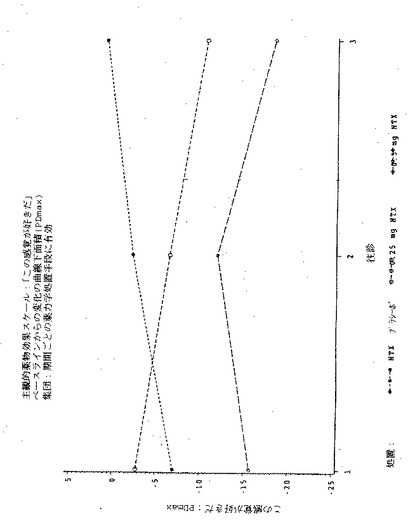
【図3】
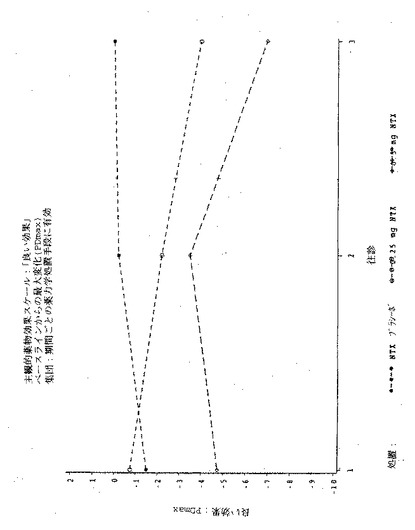
【図4】
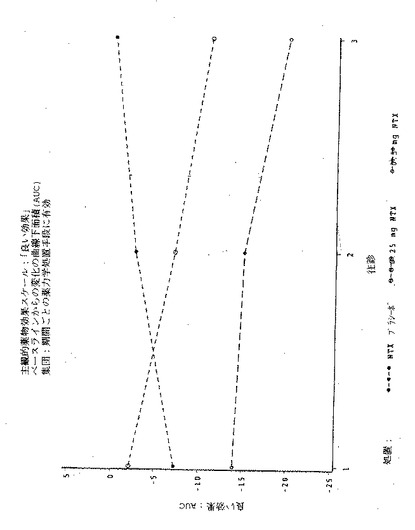
【図5】
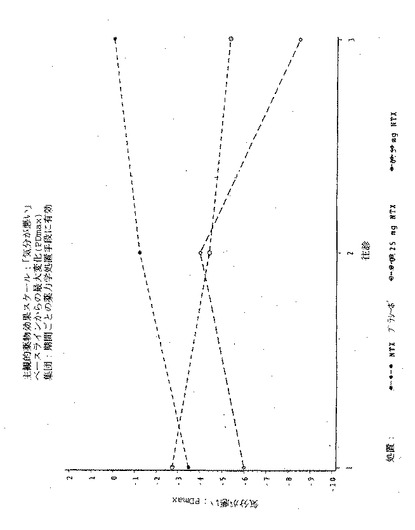
【図6】
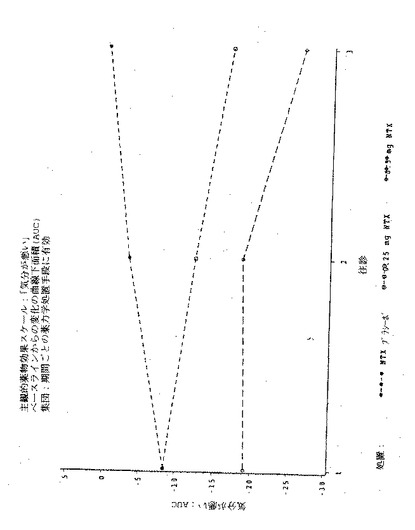
【図7】
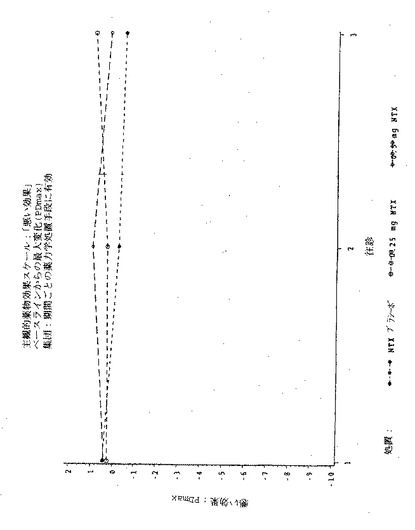
【図8】
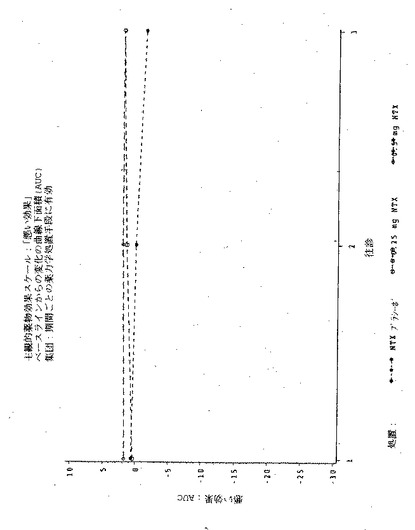
【図9】
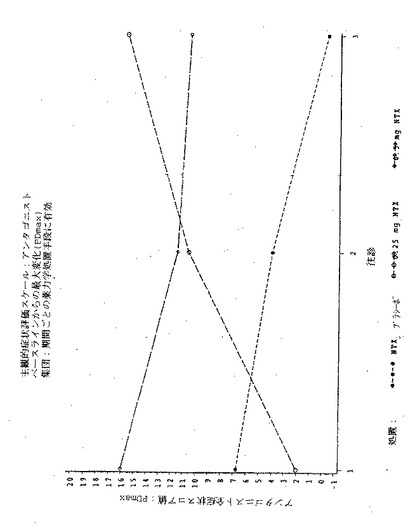
【図10】
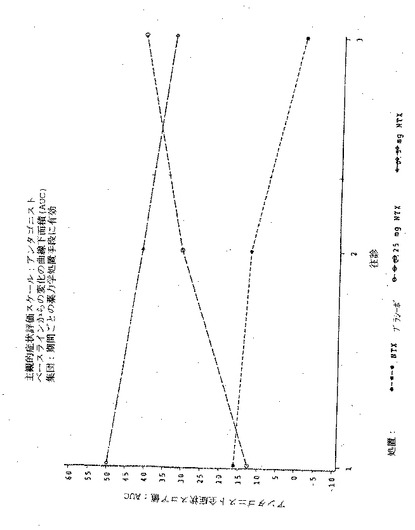
【図11】
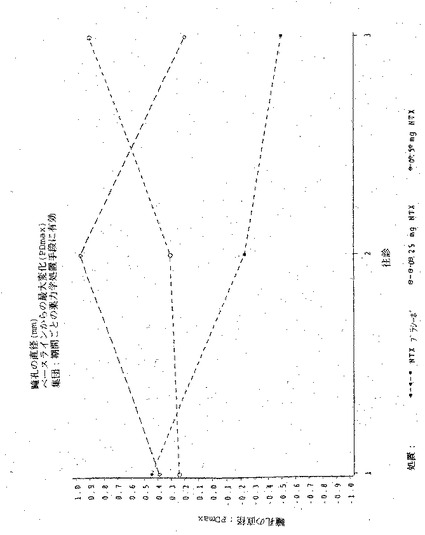
【図12】
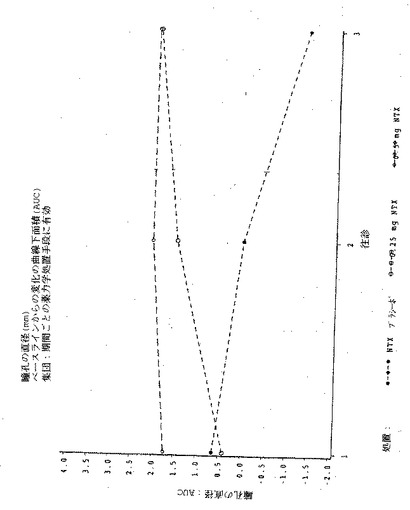
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公表番号】特表2007−506738(P2007−506738A)
【公表日】平成19年3月22日(2007.3.22)
【国際特許分類】
【出願番号】特願2006−528039(P2006−528039)
【出願日】平成16年9月9日(2004.9.9)
【国際出願番号】PCT/US2004/029521
【国際公開番号】WO2005/032555
【国際公開日】平成17年4月14日(2005.4.14)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.POLAROID
【出願人】(599108792)ユーロ−セルティーク エス.エイ. (134)
【Fターム(参考)】
【公表日】平成19年3月22日(2007.3.22)
【国際特許分類】
【出願日】平成16年9月9日(2004.9.9)
【国際出願番号】PCT/US2004/029521
【国際公開番号】WO2005/032555
【国際公開日】平成17年4月14日(2005.4.14)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.POLAROID
【出願人】(599108792)ユーロ−セルティーク エス.エイ. (134)
【Fターム(参考)】
[ Back to top ]