
ビシクロ誘導体
下記一般式(1)で示される優れたDPP-IV阻害活性を有する新規なビシクロ誘導体、または薬理学的に許容されるその塩(具体例:(2S,4S)−1−[[N−(4−メチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル)を提供する。
【化1】

【化1】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ジペプチジルペプチダーゼIV(DPP-IV)阻害活性を有し、II型糖尿病などのDPP-IVが関与する疾患の予防および/または治療に有用なビシクロ誘導体、または薬理学的に許容されるその塩に関する。
【背景技術】
【0002】
ジペプチジルペプチダーゼIV(EC 3.4.14.5、以下、DPP-IVまたはCD26)は、N末端から2番目にプロリンまたはアラニンを有するポリペプチド鎖から、Xaa-ProまたはXaa-Ala(Xaaは任意のアミノ酸を示す)で表されるジペプチドをC末端側で特異的に加水分解するセリンプロテアーゼの1種である。
【0003】
DPP-IVの生体内における機能の1つとして、グルカゴン様ペプチド―1(以下、GLP-1)のN末端にあるHis-Alaのジペプチドを加水分解することによってGLP-1を不活性化することが知られている(非特許文献1)。それに加えて、DPP-IVによって不活性化された不活性型GLP-1がGLP-1受容体に対して拮抗作用を示すことにより、GLP-1の生理的作用がさらに減弱すると考えられている(非特許文献2)。GLP-1は主として小腸腸管上皮に存在する内分泌細胞であるL細胞から分泌されるペプチドホルモンであり、グルコース濃度依存的に膵臓ランゲルハンス島に存在するβ細胞に作用してインスリンの放出を促進することにより、血糖を降下させることが知られている(非特許文献3、4)。またGLP-1は、インスリンの生合成を亢進し、β細胞の増殖も促すことから、β細胞の維持にとっても欠くことのできない因子である(非特許文献5、6)。さらにGLP-1には末梢組織において糖の利用を亢進する作用や、GLP-1の脳室内投与による摂食抑制作用、消化管運動抑制作用が報告されている(非特許文献7−10)。
【0004】
DPP-IVの酵素活性を阻害する物質は、その阻害作用により内在性のGLP-1の分解を抑制することでGLP-1の作用を高め、その結果インスリン分泌を亢進して糖代謝を改善することができると考えられている。そのためDPP-IV阻害剤は、糖尿病、特にII型糖尿病に対する予防および/または治療剤となり得ることが期待されている(非特許文献11、12)。また糖代謝の低下によって惹起、あるいは増悪されるその他の疾患(例えば糖尿病合併症、高インスリン血症、過血糖、脂質代謝異常、肥満など)における予防および/または治療に対する効果も期待されている。
【0005】
GLP-1の不活性化以外にもDPP-IVの生体内における役割や疾病との関係については以下のような報告がある。
(a) DPP-IVの阻害剤またはその抗体が、HIVウイルスの細胞内への侵入を阻害する。HIV-1感染患者由来のT細胞では、CD26の発現が減少している(非特許文献13)。また、HIV-1Tatタンパクは、DPP-IVに結合する(非特許文献14)。
(b) DPP-IVは免疫応答に関与する DPP-IVの阻害剤またはその抗体は、抗原刺激によるT細胞の増殖を抑制する(非特許文献15)。また、抗原刺激によりT細胞でのDPP-IVの発現が増加する(非特許文献16)。DPP-IVは、サイトカイン産生などのT細胞の機能に関与している(非特許文献17)。またDPP-IVは、T細胞表面でアデノシンデアミネース(ADA)と結合する(非特許文献18)。
(c) 慢性関節リウマチ、乾癬および偏平苔蘚患者の皮膚の線維芽細胞において、DPP-IVの発現が増加する(非特許文献19)。
(d) 良性前立腺肥大の患者および前立腺組織のホモジネートにおいて、DPP-IV活性が亢進している(非特許文献20)。肺内皮に存在するDPP-IVは、ラットの肺転移性乳癌および前立腺癌に対して接着分子として作用する(非特許文献21)。
(e)
DPP-IV活性を欠損している変異型F344ラットは、野生型F344ラットと比較して血圧が低いこと、および腎臓でナトリウムの再吸収に重要な役目を担っているタンパクとDPP-IVが相互作用する(特許文献1、2)。
(f) DPP-IV活性を阻害することによって、骨髄抑制性疾患の予防および/または治療が期待でき、DPP-IV活性剤が白血球数増加剤および/または感染症治療剤として期待できる(特許文献3)。
【0006】
これらの知見からDPP-IV阻害剤は、糖尿病(特にII型糖尿病)および/または糖尿病合併症以外のDPP-IVが関与する疾病の予防および/または治療剤となり得ることが期待される。例えば,HIV-1感染に基づくAIDS、臓器・組織移植における拒絶反応、多発性硬化症、慢性関節リウマチ、炎症、アレルギー、骨粗鬆症、乾癬および偏平苔蘚、良性前立腺肥大、乳癌および前立腺癌の肺転移抑制、高血圧、利尿、骨髄抑制の低減、白血球数増加、および感染症などに用いられる薬剤として有用であると考えられる。
【0007】
現在までにDPP-IV阻害剤として、(特許文献4−11)にピロリジン誘導体が、(特許文献12、13)にヘテロ環誘導体が、(特許文献14、15)にβアミノ酸誘導体が開示されている。
【0008】
また、(特許文献16)にDPP-IV阻害活性を有するビシクロ[2.2.2]オクタン誘導体が1化合物のみ開示されているが、本発明は、当該米国特許とは構造、DPP-IV阻害活性の面からも全く異なるものである。また(特許文献17)には、本発明に構造上近似したビシクロ誘導体を示唆する記述が見られるが、その記述内容は具体的に本発明化合物を何ら説明しておらず、また、本発明化合物のいずれをも実施例によって説明しているものではない。
【0009】
これまでに開示されているDPP-IV阻害剤はいずれも、DPP-IV阻害活性、DPP-IV選択性、安定性、毒性、および体内動態において満足できるものではなく、優れたDPP-IV阻害剤が常に求められている。
【非特許文献1】American Journal of Physiology、271巻、E458−E464頁(1996年)
【非特許文献2】European Journal of Pharmacology、318巻、429−435頁(1996年)
【非特許文献3】European Journal Clinical Investigation、22巻、154頁(1992年)
【非特許文献4】Lancet、2巻、1300頁(1987年)
【非特許文献5】Endocrinology、42巻、856頁(1992年)
【非特許文献6】Diabetologia、42巻、856頁(1999年)
【非特許文献7】Endocrinology、135巻、2070頁(1994年)
【非特許文献8】Diabetologia、37巻、1163頁(1994年)
【非特許文献9】Digestion、54巻、392頁(1993年)
【非特許文献10】Dig. Dis. Sci.、43巻、1113頁(1998年)
【非特許文献11】Diabetes、47巻、1663−1670頁(1998年)
【非特許文献12】Diabetologia、42巻、1324−1331頁(1999年)
【非特許文献13】Journal of Immunology、149巻、3073頁(1992年)
【非特許文献14】Journal of Immunology、150巻、2544頁(1993年)
【非特許文献15】Biological Chemistry、305頁(1991年)
【非特許文献16】Scandinavian Journal of Immunology、33巻、737頁(1991年)
【非特許文献17】Scandinavian Journal of Immunology、29巻、127頁(1989年)
【非特許文献18】Science、261巻、466頁(1993年)
【非特許文献19】Journal of Cellular Physiology、151巻、378頁(1992年)
【非特許文献20】European Journal of Clinical Chemistry and Clinical Biochemistry、30巻、333頁(1992年)
【非特許文献21】Journal of Cellular Physiology、121巻、1423頁(1993年)
【特許文献1】WO 03/015775 パンフレット
【特許文献2】WO 03/017936 パンフレット
【特許文献3】WO 03/080633 パンフレット
【特許文献4】WO 95/15309 パンフレット
【特許文献5】WO 98/19998 パンフレット
【特許文献6】WO 00/34241 パンフレット
【特許文献7】WO 02/14271 パンフレット
【特許文献8】WO 02/30890 パンフレット
【特許文献9】WO 02/38541 パンフレット
【特許文献10】WO 03/002553 パンフレット
【特許文献11】US 02/0193390 公報
【特許文献12】WO 02/062764 パンフレット
【特許文献13】WO 03/004496 パンフレット
【特許文献14】WO 03/000180 パンフレット
【特許文献15】WO 03/004498 パンフレット
【特許文献16】US 02/0193390 公報
【特許文献17】WO 02/38541 パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明が解決しようとする問題点は、優れたDPP-IV阻害活性を有する新規な化合物、または薬理学的に許容されるその塩を提供することにある。また、優れたDPP-IV阻害活性を有する新規な化合物、または薬理学的に許容されるその塩を含む医薬組成物、糖尿病およびその合併症の予防および/または治療剤あるいはDPP-IVが関与する疾患に対する予防および/または治療剤を提供することにある。
【課題を解決するための手段】
【0011】
本発明は、優れたDPP-IV阻害活性を有する新規なビシクロ誘導体、または薬理学的に許容されるその塩を提供する。また、優れたDPP-IV阻害活性を有する新規なビシクロ誘導体、または薬理学的に許容されるその塩を含む医薬組成物、糖尿病およびその合併症の予防および/または治療剤あるいはDPP-IVが関与する疾患に対する予防および/または治療剤を提供する。
【0012】
すなわち本発明は、
一般式(1)
【0013】
【化1】
【0014】
[式中、R1は、水素原子、ハロゲン原子、カルボキシル基、水酸基で置換されていてもよいC1〜C4のアルキル基、および置換されていてもよいアリール基を示し、
Xは、CH2、CHF、CF2、CHOH、SおよびOを示し、
nは1、2または3を示す。]
で表されるビシクロ誘導体、または薬理学的に許容されるその塩、前記一般式(1)で表されるビシクロ誘導体、または薬理学的に許容されるその塩を有効成分として含有する医薬、DPP-IV阻害剤、DPP-IVが関与する疾患の治療剤およびDPP-IVが関与する疾患が糖尿病及びその合併症である治療剤に関するものである。
【0015】
ここでC1〜C4のアルキル基とは、メチル基、エチル基、プロピル基、イソプロピル基、およびt−ブチル基などを意味する。置換されていてもよいアリール基とは、ハロゲン原子、C1〜C6のアルキル基、ヒドロキシル基、C1〜C6のアルコキシ基、C1〜C6のアルコキシカルボニル基、C1〜C6のアルキルチオ基、アミノ基、モノまたはジ置換のC1〜C6のアルキルアミノ基、1〜3個のヘテロ原子を含んでいてもよい4〜9員の環状アミノ基、ホルミルアミノ基、C1〜C6のアルキルカルボニルアミノ基、C1〜C6のアルコキシカルボニルアミノ基、ベンジルオキシカルボニルアミノ基、C1〜C6のアルキルスルホニルアミノ基、および置換されていてもよいアリールスルホニルアミノ基などから選ばれた1〜5個の置換基を有していてもよいアリール基を意味する。
【0016】
アリール基とは芳香族炭化水素または芳香族へテロ環(窒素原子、酸素原子、および硫黄原子の中から任意に選ばれた1〜3個のヘテロ原子を含む5員または6員の芳香族単環式複素環、あるいは9員または10員の芳香族縮合複素環)、例えばベンゼン環、ナフタレン環、アントラセン環、ピリジン環、ピリミジン環、ピリダジン環、トリアジン環、キノリン環、ナフチリジン環、キナゾリン環、アクリジン環、ピロール環、フラン環、チオフェン環、イミダゾール環、ピラゾール環、オキサゾール環、イソキサゾール環、チアゾール環、インドール環、ベンゾフラン環、ベンゾチアゾール環、ベンズイミダゾール環、およびベンゾオキサゾール環などを意味する。
【0017】
ここでハロゲン原子とは、フッ素原子、塩素原子、臭素原子、およびヨウ素原子を意味する。
【0018】
本発明の好ましい化合物としては、
(2S,4S)−1−[[N−(4−カルボキシビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル、
(2S)−1−[[N−(4−カルボキシビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリル、
(2S,4S)−1−[[N−(4−メチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル、
(2S)−1−[[N−(4−メチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリル、
(2S,4S)−1−[[N−(4−ヒドロキシメチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル、
(2S)−1−[[N−(4−ヒドロキシメチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリル、
(2S,4S)−1−[[N−(4−フルオロビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル、
(2S)−1−[[N−(4−フルオロビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリル、
などが例示できる。
【発明の効果】
【0019】
本発明化合物は優れたDPP-IV阻害活性を有する新規な化合物であり、糖尿病およびその合併症の予防および/または治療剤あるいはDPP-IVが関与する疾患に対する予防および/または治療剤を提供する。
【図面の簡単な説明】
【0020】
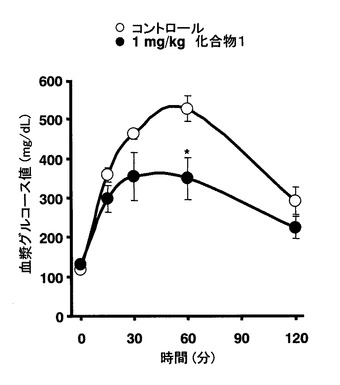
【図1】正常マウスにおける経口糖負荷試験時の血漿グルコース値に対する化合物1の効果を示すグラフ。各データは4例の平均値 ± 標準誤差で示した。*P< 0.05 vs. control (t検定)
【発明を実施するための最良の形態】
【0021】
本発明化合物が薬理学上許容な塩を形成する場合、塩酸、臭化水素酸、硫酸、硝酸、および燐酸などの無機酸、または酢酸、マレイン酸、フマル酸、コハク酸、乳酸、リンゴ酸、酒石酸、クエン酸、メタンスルホン酸、p−トルエンスルホン酸、ベンゼンスルホン酸、サリチル酸、ステアリン酸、パルミチン酸、およびトリフルオロ酢酸などの有機酸との塩、ナトリウム塩、カリウム塩、カルシウム塩、マグネシウム塩、アルミニウム塩、および亜鉛塩などの金属塩、アンモニウム塩およびテトラメチルアンモニウム塩などのアンモニウム塩、モルホリン、およびピペリジンなどとの有機アミン塩、およびグリシン、リジン、アルギニン、フェニルアラニンおよびプロリンなどのアミノ酸との付加塩が例示できる。
【0022】
上記一般式(1)で表される本発明化合物またはその塩には、1個または2個以上の不斉中心に基づく複数の光学異性体が存在し得るが、本発明はこれらの光学異性体もしくはジアステレオ異性体のいずれをも含み、またそれらの任意の比率を示す混合物またはラセミ体をも含むものである。また、上記一般式(1)で表される本発明化合物またはその塩に二重結合を含む場合には、その配置はZまたはEのいずれであってもよく、これらの任意の比率を示す混合物をも本発明に含まれる。さらには、上記一般式(1)で表される本発明化合物またはその塩の中には互変異性体や回転異性体が存在し得るものがあるが、それぞれの異性体およびそれらの任意の比率を示す混合物をも本発明に含まれる。
【0023】
上記一般式(1)で表される本発明化合物またはその塩は、分子内塩や付加物、それらの溶媒和物あるいは水和物などのいずれも含むものである。
【0024】
上記一般式(1)で表される本発明化合物またはその塩は、単独で、または一種以上の製剤上許容される補助剤と共に医薬組成物として用いることができ、薬理学上許容される担体、賦形剤(例えば、デンプン、乳糖、リン酸カルシウム、または炭酸カルシウムなど)、滑沢剤(例えば、ステアリン酸マグネシウム、ステアリン酸カルシウムタルク、またはステアリン酸など)、結合剤(例えば、デンプン、結晶セルロース、カルボキシメチルセルロース、アラビアゴム、ポリビニルピロリドン、またはアルギン酸など)、崩壊剤(例えば、タルク、またはカルボキシメチルセルロースカルシウムなど)、希釈剤(例えば、生理食塩水、グルコース、マンニトール、またはラクトースなどの水溶液など)などと混合し、通常の方法により錠剤、カプセル剤、顆粒剤、散剤、細粒剤、アンプル剤または注射剤などの形態で経口的または非経口的に投与することができる。投与量は上記一般式(1)で表される本発明化合物またはその塩の種類、投与方法、患者の年齢、体重、症状などにより異なるが、通常、人を含む哺乳動物に対して上記一般式(1)で表される本発明化合物またはその塩として0.0001〜1000mg/kg/日である。投与は例えば1日1回または数回に分割して投与する。
【0025】
上記一般式(1)で表される本発明化合物またはその塩は、必要であれば一種以上のDPP-IV阻害剤以外の糖尿病治療剤と併用することができる。本発明化合物またはその塩と併用される糖尿病治療剤としては、インスリンやその誘導体、GLP-1やその誘導体、その他の経口糖尿病治療剤が挙げられる。経口糖尿病治療剤としては、スルホニルウレア系糖尿病治療剤、非スルホニルウレア系インスリン分泌促進剤、ビグアナイド系糖尿病治療剤、α−グリコシダーゼ阻害剤、グルカゴンアンタゴニスト、GLP-1アゴニスト、PPARアゴニスト、β3アゴニスト、SGLT阻害剤、PKC阻害剤、グルカゴンシンテースキナーゼ-3(GSK-3)阻害剤、プロテインチロシンホスファターゼ-1B(PTP-1B)阻害剤、カリウムチャネルオープナー、インスリン増感剤、グルコース取込み調節剤、脂質代謝作用剤、食欲抑制剤などがあげられる。
【0026】
これらのうちGLP-1やその誘導体としては、ベタトロピン、またはNN-2211などが挙げられ、スルホニルウレア系糖尿病治療剤としては、トルブタミド、グリベンクラミド、グリクラジド、グリメピリド、またはグリピジドなどが挙げられ、非スルホニルウレア系インスリン分泌促進剤としては、ナテグリニド、レパグリニド、ミチグリニド、またはJTT-608などが挙げられ、ビグアナイド系糖尿病治療剤としては、メトホルミンなどが挙げられ、α−グリコシダーゼ阻害剤としては、ボグリボースまたはミグリトールなどが挙げられ、PPARアゴニストとしては、トログリタゾン、ロシグリタゾン、ピオグリタゾン、シグリタゾン、KRP-297(MK-767)、イサグリタゾン、GI-262570、JTT-501などが挙げられ、β3アゴニストとしては、AJ-9677、YM-178、またはN-5984などが挙げられる。
【0027】
本発明化合物(1)は、種々の合成法によって製造することができる。本発明化合物(1)は通常の分離手段(例えば抽出、再結晶、蒸留、クロマトグラフィー等)によって単離、精製することができる。また、得られた化合物が塩を形成する様な場合には、通常の方法あるいはそれに準ずる方法(例えば中和等)によって各種の塩を製造することができる。
【0028】
次に、本発明化合物およびその塩の代表的な製造工程について説明する。
【0029】
A法
【0030】
【化2】
【0031】
A法第一工程
本工程は、一般式(3)(式中、R1およびnは前記に同じ)で表されるビシクロアミン誘導体に、一般式(4)(式中、Y1はClまたはBrを表す。
Xは前記に同じ)で表されるハロ酢酸誘導体を反応させて、一般式(1)(式中、R1、n、およびXは前記に同じ) で表されるビシクロ誘導体を製造する工程である。本反応は、塩基の存在下または非存在下に行われる。本反応に塩基を用いる場合には、水酸化ナトリウム、水酸化カリウム、炭酸水素ナトリウム、炭酸水素カリウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウムなどの無機塩基や、トリエチルアミン、ジイソプロピルエチルアミン、N,N,N,N−テトラメチルエチレンジアミン、ジアザビシクロ[5.4.0]−7−ウンデセン、ジアザビシクロ[4.3.0]−5−ノネン、ホスファゼンベースまたはペンタイソプロピルグアニジンなどの有機塩基が例示できる。本反応に触媒を用いる場合には、テトラブチルアンモニウムブロミド、テトラブチルアンモニウムヨージド、ベンジルトリエチルアンモニウムブロミド、臭化リチウム、ヨウ化リチウム、ヨウ化ナトリウム、臭化カリウム、ヨウ化カリウム、臭化セシウム、ヨウ化セシウムなどの相関移動触媒または無機塩が例示できる。本反応に用いられる溶媒としては、反応に関与しない不活性な溶媒、例えばアセトン、エタノール、トルエン、アセトニトリル、テトラヒドロフラン、ジオキサン、エチルエーテル、t−ブチルメチルエーテル、ジメトキシエタン、酢酸エチル、ジクロロメタン、N,N−ジメチルホルムアミド、ジメチルスルホキシド、N−メチル−2−ピロリドンなどが用いられる。反応は0〜150℃で円滑に進行する。
【0032】
B法
【0033】
【化3】
【0034】
B法第一工程
本工程は、一般式(5)(式中、nは前記に同じ、THPはテトラヒドロピラニル基)で表されるビシクロアミン誘導体に、一般式(4)(式中、XおよびY1は前記に同じ)で表されるハロ酢酸誘導体を反応させて、一般式(6)(式中、nおよびXは前記に同じ)
で表されるビシクロ誘導体を製造する工程である。本反応は、塩基の存在下または非存在下に行われる。本反応に塩基を用いる場合には、水酸化ナトリウム、水酸化カリウム、炭酸水素ナトリウム、炭酸水素カリウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウムなどの無機塩基や、トリエチルアミン、ジイソプロピルエチルアミン、N,N,N,N−テトラメチルエチレンジアミン、ジアザビシクロ[5.4.0]−7−ウンデセン、ジアザビシクロ[4.3.0]−5−ノネン、ホスファゼンベースまたはペンタイソプロピルグアニジンなどの有機塩基が例示できる。本反応に触媒を用いる場合には、テトラブチルアンモニウムブロミド、テトラブチルアンモニウムヨージド、ベンジルトリエチルアンモニウムブロミド、臭化リチウム、ヨウ化リチウム、ヨウ化ナトリウム、臭化カリウム、ヨウ化カリウム、臭化セシウム、ヨウ化セシウムなどの相関移動触媒または無機塩が例示できる。本反応に用いられる溶媒としては、反応に関与しない不活性な溶媒、例えばアセトン、エタノール、トルエン、アセトニトリル、テトラヒドロフラン、ジオキサン、エチルエーテル、t−ブチルメチルエーテル、ジメトキシエタン、酢酸エチル、ジクロロメタン、N,N−ジメチルホルムアミド、ジメチルスルホキシド、N−メチル−2−ピロリドンなどが用いられる。反応は0〜150℃で円滑に進行する。
【0035】
B法第二工程
本工程は、一般式(6)(式中、nおよびXは前記に同じ) で表されるビシクロ誘導体のテトラヒドロピラニル基を除去して、一般式(2)(式中、nおよびXは前記に同じ)
で表されるビシクロ誘導体を製造する工程である。テトラヒドロピラニル基の除去は、公知の方法に従って酢酸やp−トルエンスルホン酸、または塩酸などにより、容易に除去することができる。
【0036】
以下の試験例および実施例により本発明の有用性を示すが本発明は試験例および実施例に限定されるものではない。
【0037】
<参考例1>
4−アミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニルの合成
第一工程:
4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸メチルの合成
ビシクロ[2.2.2]オクタン−1、4−ジカルボン酸水素メチル (25.0 g)、アジ化ジフェニルホスホリル (32.5 g)、トリエチルアミン (17.3
mL) およびトルエン (500 mL) を混合して室温で2時間撹拌し、次いで2時間加熱還流した。反応混合物にベンジルアルコール (122 mL) を加えて、さらに17時間加熱還流した。冷後、反応混合物を10%クエン酸水溶液、飽和炭酸水素ナトリウム水溶液、および飽和食塩水の順で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:ヘキサン:酢酸エチル=2:1)にて精製し、4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸メチル
(32.2 g) を得た。
MS (FAB+) m/z: 318 (MH+).
【0038】
第二工程:
4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸の合成
4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸メチル (64.3 g) をエタノール (1100 mL) に溶解し、1mol/L水酸化ナトリウム水溶液
(1000 mL) を加え、50℃で1時間撹拌した。反応液中のエタノールを減圧留去し、残渣をジエチルエーテル (500 mL) で洗浄した後、濃塩酸で酸性(pH1)とした。析出した結晶を濾取し、水洗後、減圧乾燥して4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸
(56.1 g) を得た。
MS (FAB+) m/z: 304 (MH+).
【0039】
第三工程:
4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニルの合成
4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸 (1.00 g) をジクロロメタン (10 mL) に懸濁して、3,4−ジヒドロ−2H−ピラン
(1.20 mL)、次いでp−トルエンスルホン酸・1水和物 (6.3 mg) を加え、室温で30分間撹拌した。反応混合物を飽和炭酸水素ナトリウム水溶液、次いで水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:ヘキサン:酢酸エチル=4:1)で精製し、4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニル
(1.18 g) を得た。
【0040】
1H NMR (CDCl3) δ 1.53-1.95
(m, 18H), 3.67-3.71 (m, 1H), 3.82-3.89 (m, 1H), 4.59 (br, 1H), 5.03 (s, 2H),
5.95 (br, 1H), 7.29-7.38 (m, 5H).
【0041】
第四工程
4−アミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニルの合成
4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニル (548 mg) を用いて、参考例1の第四工程と同様に反応を行い、4−アミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニル
(357 mg) を得た。
MS (EI+) m/z: 253 (M+).
【0042】
<参考例2>
4−アミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタンの合成
第一工程:
4−ベンジルオキシカルボニルアミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタンの合成
4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸 (500 mg) をテトラヒドロフラン (8 mL) に溶解して、食塩−氷浴上で冷却しながらN−メチルモルホリン
(0.18 mL) を加えた後、クロロ炭酸エチル (0.16 mL) を滴下し、さらに10分間撹拌した。次いで反応混合物に水素化ホウ素ナトリウム (187
mg) を加えた後、メタノール (15 mL) を加えて、0℃以下で1時間撹拌した。反応混合物を減圧濃縮して、残渣に水を加え、酢酸エチルで抽出した。酢酸エチル層を1mol/L塩酸、次いで水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:ヘキサン:酢酸エチル=1:1)で精製し、4−ベンジルオキシカルボニルアミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタン
(453 mg) を得た。
MS (EI+) m/z: 289 (M+).
【0043】
第二工程:
4−アミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタンの合成
4−ベンジルオキシカルボニルアミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタン (448 mg) を用いて、参考例1の第四工程と同様に反応を行い、4−アミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタン
(185 mg) を得た。
MS (EI+) m/z: 155 (M+).
【0044】
<参考例3>
1−アミノ−4−フルオロビシクロ[2.2.2]オクタンの合成
第一工程:
1−ベンジルオキシカルボニルアミノ−4−フルオロビシクロ[2.2.2]オクタンの合成
4−フルオロビシクロ[2.2.2]オクタン−1−カルボン酸 (265 mg) を用いて、参考例1の第一工程と同様に反応を行い、1−ベンジルオキシカルボニルアミノ−4−フルオロビシクロ[2.2.2]オクタン
(365 mg) を得た。
MS (EI+) m/z: 277 (M+).
【0045】
第二工程:
1−アミノ−4−フルオロビシクロ[2.2.2]オクタンの合成
1−ベンジルオキシカルボニルアミノ−4−フルオロビシクロ[2.2.2]オクタン
(350 mg) を用いて、参考例1の第四工程と同様に反応を行い、1−アミノ−4−フルオロビシクロ[2.2.2]オクタン (144 mg) を得た。
MS (EI+) m/z: 143 (M+).
【0046】
<参考例4>
(2S、4S)−1−(2−クロロアセチル)−4−フルオロピロリジン−2−カルボニトリルの合成
文献記載(WO02/38541 パンフレット)の(2S、4S)−1−(2−ブロモアセチル)−4−フルオロピロリジン−2−カルボニトリルの製法に準じて、(2S、4S)−4−フルオロピロリジン−2−カルボキサミド塩酸塩
(5.00 g) およびクロロアセチルクロリド (2.60 mL) から、(2S、4S)−1−(2−クロロアセチル)−4−フルオロピロリジン−2−カルボニトリル
(4.96 g) を得た。
MS (EI+) m/z: 190 (M+).
HRMS (EI+) for C7H8ClFN2O(M+):
calcd, 190.0309; found, 190.0283.
【実施例1】
【0047】
【化4】
【0048】
(2S,4S)−1−[[N−(4−カルボキシビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリルの合成
第一工程
(2S,4S)−1−[[N−[4−(2−テトラヒドロピラニル)オキシカルボニルビシクロ[2.2.2]オクト−1−イル]アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリルの合成
4−アミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニル (62.9 mg) をアセトニトリル (1 mL) に懸濁して、ジイソプロピルエチルアミン
(47 μL) を加え、氷冷下で(2S、4S)−1−(2−ブロモアセチル)−4−フルオロピロリジン−2−カルボニトリル (53.1 mg) のアセトニトリル
(0.8 mL) 溶液を加えて4時間撹拌した。反応液を濃縮した残渣に酢酸エチルおよび水を加えて溶解し、炭酸水素ナトリウム水溶液を用いてアルカリ性とした後に抽出した。酢酸エチル層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:ジクロロメタン:メタノール=10:1)で精製し、(2S,4S)−1−[[N−[4−(2−テトラヒドロピラニル)オキシカルボニルビシクロ[2.2.2]オクト−1−イル]アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル
(73.3 mg) を得た。
MS (FAB+) m/z: 408 (MH+).
HRMS (FAB+) for C21H31FN3O4 (MH+):
calcd, 408.2299; found, 408.2295.
【0049】
第二工程
(2S,4S)−1−[[N−(4−カルボキシビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリルの合成
(2S,4S)−1−[[N−[4−(2−テトラヒドロピラニル)オキシカルボニルビシクロ[2.2.2]オクト−1−イル]アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル
(71.6 mg) を酢酸 (4 mL) に溶解し、室温で6時間撹拌した。反応混合物を減圧濃縮して、残渣をジクロロメタンで懸濁して濾取し、(2S,4S)−1−[[N−(4−カルボキシビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル
(31.4 mg) を得た。
MS (EI+) m/z: 323 (M+).
【実施例2】
【0050】
【化5】
【0051】
(2S)−1−[[N−(4−カルボキシビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリルの合成
第一工程
(2S)−1−[[N−[4−(2−テトラヒドロピラニル)オキシカルボニルビシクロ[2.2.2]オクト−1−イル]アミノ]アセチル]ピロリジン−2−カルボニトリルの合成
4−アミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニル (90.0 mg) および(2S)−1−(2−ブロモアセチル)ピロリジン−2−カルボニトリル
(70.0 mg)を用いて、実施例1と同様に反応を行い、(2S)−1−[[N−[4−(2−テトラヒドロピラニル)オキシカルボニルビシクロ[2.2.2]オクト−1−イル]アミノ]アセチル]ピロリジン−2−カルボニトリル
(85.2 mg) を得た。
MS (EI+) m/z: 383 (M+).
HRMS (EI+) for C21H31N3O4 (M+):
calcd, 383.2315; found, 383.2296.
【0052】
第二工程
(2S)−1−[[N−(4−カルボキシビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリルの合成
(2S)−1−[[N−[4−(2−テトラヒドロピラニル)オキシカルボニルビシクロ[2.2.2]オクト−1−イル]アミノ]アセチル]ピロリジン−2−カルボニトリル
(73.0 mg) を用いて、実施例1と同様に反応を行い、(2S)−1−[[N−[4−カルボキシビシクロ[2.2.2]オクト−1−イル]アミノ]アセチル]ピロリジン−2−カルボニトリル
(56.0 mg) を得た。
MS (EI+) m/z: 305 (M+).
HRMS (EI+) for C16H23N3O3 (M+):
calcd, 305.1739; found, 305.1736.
【実施例3】
【0053】
【化6】
【0054】
(2S,4S)−1−[[N−(4−メチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリルの合成
4−アミノ−1−メチルビシクロ[2.2.2]オクタン(56.0 mg) および(2S、4S)−1−(2−ブロモアセチル)−4−フルオロピロリジン−2−カルボニトリル
(95.0 mg) を用いて、実施例1と同様に反応を行い、(2S,4S)−1−[[N−(4−メチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル
(89.2 mg) を得た。
MS (EI+) m/z: 293 (M+).
HRMS (EI+) for C16H24FN3O(M+):
calcd, 293.1903; found, 293.1881.
【実施例4】
【0055】
【化7】
【0056】
(2S)−1−[[N−(4−メチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリルの合成
4−アミノ−1−メチルビシクロ[2.2.2]オクタン(75.0 mg) および(2S)−1−(2−ブロモアセチル)ピロリジン−2−カルボニトリル (110
mg) を用いて、実施例1と同様に反応を行い、(2S)−1−[[N−(4−メチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリル
(84.0 mg) を得た。
MS (EI+) m/z: 275 (M+).
HRMS (EI+) for C16H25N3O(M+):
calcd, 275.1998; found, 275.1981.
【実施例5】
【0057】
【化8】
【0058】
(2S,4S)−1−[[N−(4−ヒドロキシメチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリルの合成
4−アミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタン (50.0 mg) および(2S、4S)−1−(2−ブロモアセチル)−4−フルオロピロリジン−2−カルボニトリル
(75.7 mg) を用いて、実施例5と同様に反応を行い、(2S,4S)−1−[[N−(4−ヒドロキシメチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル
(93.0 mg) を得た。
MS (FAB+) m/z: 310 (MH+).
HRMS (FAB+) for C16H25FN3O2 (MH+):
calcd, 310.1931; found, 310.1942.
【実施例6】
【0059】
【化9】
【0060】
(2S)−1−[[N−(4−ヒドロキシメチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリルの合成
4−アミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタン (50.5 mg) および(2S)−1−(2−ブロモアセチル)ピロリジン−2−カルボニトリル
(61.7 mg) を用いて、実施例5と同様に反応を行い、(2S)−1−[[N−(4−ヒドロキシメチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリル
(57.0 mg) を得た。
MS (FAB+) m/z: 292 (MH+).
HRMS (FAB+) for C16H25N3O2 (MH+):
calcd, 292.2025; found, 292.2025.
【実施例7】
【0061】
【化10】
【0062】
(2S,4S)−1−[[N−(4−フルオロビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリルの合成
4−アミノ−1−フルオロビシクロ[2.2.2]オクタン(50.0 mg) および(2S、4S)−1−(2−ブロモアセチル)−4−フルオロピロリジン−2−カルボニトリル
(82.1 mg) を用いて、実施例10と同様に反応を行い、(2S,4S)−1−[[N−(4−フルオロビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル
(93.1 mg) を得た。
MS (EI+) m/z: 297 (M+).
HRMS (EI+) for C15H21F2N3O(M+):
calcd, 297.1653; found, 297.1628.
【実施例8】
【0063】
【化11】
【0064】
(2S)−1−[[N−(4−フルオロビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリルの合成
4−アミノ−1−フルオロビシクロ[2.2.2]オクタン(50.0 mg) および(2S)−1−(2−ブロモアセチル)ピロリジン−2−カルボニトリル
(73.5 mg) を用いて、実施例10と同様に反応を行い、(2S)−1−[[N−(4−フルオロビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリル
(72.3mg) を得た。
MS (EI+) m/z: 279 (M+).
HRMS (EI+) for C15H22FN3O(M+):
calcd, 279.1747; found, 279.1766.
【0065】
<試験例1>[ジペプチジルペプチダーゼ IV活性阻害試験]
基質であるH−Gly−Pro−AMC (7−アミノ−4−メチル−クマリン)・HBrが血漿ジペプチジルペプチダーゼ IVにより分解されて遊離するAMC濃度を蛍光強度により測定した。
方法
平底96穴プレートを用いて、生理食塩水で8倍希釈した血漿20μLに化合物を溶解させた緩衝液(25mmol/L ヘペス、140 mmol/ L 塩化ナトリウム、1%
ウシ血清アルブミン、80mmol/ L 塩化マグネシウム・6水和物、pH7.4)20μLを添加し室温で5分間放置した後、0.1mmol/LのH−Gly−Pro−AMC・HBr溶液10μLを添加して反応を開始した。遮光下室温で20分間放置した後、25%酢酸溶液20μLを添加して反応を停止させた。遊離したAMC濃度を、蛍光プレートリーダーを用いて355nmで励起させた時の460nmの蛍光強度を測定した。得られた結果から50%阻害濃度(IC50値)をプリズム3.02
(グラフパッド ソフトウェア) を用いて算出した。結果を表1に記載した。
【0066】
【表1】
【0067】
化合物A:(2S)−1−[[(3−ヒドロキシ−1−アダマンチル)アミノ]アセチル]−2−シアノピロリジン(LAF-237)
【0068】
<試験例2>[経口投与におけるマウスのジペプチジルペプチダーゼ IV活性阻害試験]
0.3% カルボキシメチルセルロースナトリウム塩を用いて化合物を0.1 mg/mLの濃度で懸濁し、8週齢の雄性ICRマウス (日本チャールスリハ゛ー) に10mL/kgで経口投与した。投与前および投与後30分にEDTA・2K処理毛細管を用いて尾静脈から採血を行い、採取した各血液を6000回転で2分間遠心分離して血漿を得た。試験例1と同様の方法を用いて、酵素活性を測定した。投与前の酵素活性値からの減少率を阻害率として算出した[阻害率={(投与前値−投与後値)÷投与前値}×100]。結果を表2に記載した。
【0069】
【表2】
【0070】
化合物A:(2S)−1−[[(3−ヒドロキシ−1−アダマンチル)アミノ]アセチル
]−2−シアノピロリジン(LAF−237)
【0071】
<試験例3>[経口投与におけるマウス耐糖能試験]
実施例5の本発明化合物(以下化合物1)を0.3%カルボキシメチル-セルロース ナトリウム塩 (CMC-Na, シグマ) で懸濁した。7週齢の雄性ICRマウス(日本チャールスリバー)を1週間予備飼育した。この時、標準食(CE-2,
日本クレア) および水は自由摂取させた。8週齢のICRマウスを16時間絶食し、0.3%CMC-Na (10 mL/kg) または化合物1(1 0 mg/kg,
10 mL/kg) を経口投与した。投与30分後にグルコース溶液を5 g/kgの用量で経口投与した。採血はEDTA-2K処理毛細管を用いて、グルコース溶液投与前および投与15,
30, 60および120分後に尾静脈から行った。血漿グルコース値の測定にはグルコース Bテストワコー (和光純薬工業) を用いた。結果は平均値 ± 標準誤差で示した。統計解析はt検定を用いて、有意水準は5%未満とした。結果を図1に記した。
【0072】
<試験例4>[薬剤性白血球減少症に対する薬効評価試験]
本発明化合物の薬剤性白血球減少症に対する薬効評価実験をOkabeらの方法(薬理と治療、19巻、6号、55頁、1991年)に準じて行った。
8週齢の雄性ICR系マウス(日本チャールスリハ゛ー)を用いて、Day0にシクロホスファミド(200mg/kg)を単回腹腔内投与した。翌日から対照群には生理食塩水を投与し、薬物投与群には本発明化合物(1〜200mg/kg)を1日1〜2回、5日間経口投与した。試験開始から2,4,6,および8日後にそれぞれ採血を行い、白血球数を経時的に測定し、シクロホスファミド投与前の白血球数をコントロールとすることによって、本発明化合物の薬剤性白血球減少症に対する薬効を評価した。コントロールと比較して、本発明化合物は白血球の減少を有意に抑制した。
【0073】
<試験例5>[血中G−CSF濃度の増加作用試験]
7週齢の雄性ICR系マウス(日本チャールスリハ゛ー)を用いて、対照群には生理食塩水を投与し、薬物投与群には本発明化合物(1〜200mg/kg)を1日1〜2回、5日間経口投与した。投与終了翌日に麻酔下で採血し、マウスG−CSF ELISA測定キット(R&D SYSTEM社)を用いて血漿中のG−CSF濃度を測定した。コントロールと比較して、本発明化合物は血漿中のG−CSF濃度を有意に増加させた。
【産業上の利用可能性】
【0074】
本願化合物は、優れたDPP-IV阻害活性を有する新規なビシクロ誘導体、または薬理学的に許容されるその塩である。本願化合物を有効成分として含有する医薬組成物は、糖尿病およびその合併症の予防および/または治療剤あるいはDPP-IVが関与する疾患に対する予
防および/または治療剤として有用である。
【技術分野】
【0001】
本発明は、ジペプチジルペプチダーゼIV(DPP-IV)阻害活性を有し、II型糖尿病などのDPP-IVが関与する疾患の予防および/または治療に有用なビシクロ誘導体、または薬理学的に許容されるその塩に関する。
【背景技術】
【0002】
ジペプチジルペプチダーゼIV(EC 3.4.14.5、以下、DPP-IVまたはCD26)は、N末端から2番目にプロリンまたはアラニンを有するポリペプチド鎖から、Xaa-ProまたはXaa-Ala(Xaaは任意のアミノ酸を示す)で表されるジペプチドをC末端側で特異的に加水分解するセリンプロテアーゼの1種である。
【0003】
DPP-IVの生体内における機能の1つとして、グルカゴン様ペプチド―1(以下、GLP-1)のN末端にあるHis-Alaのジペプチドを加水分解することによってGLP-1を不活性化することが知られている(非特許文献1)。それに加えて、DPP-IVによって不活性化された不活性型GLP-1がGLP-1受容体に対して拮抗作用を示すことにより、GLP-1の生理的作用がさらに減弱すると考えられている(非特許文献2)。GLP-1は主として小腸腸管上皮に存在する内分泌細胞であるL細胞から分泌されるペプチドホルモンであり、グルコース濃度依存的に膵臓ランゲルハンス島に存在するβ細胞に作用してインスリンの放出を促進することにより、血糖を降下させることが知られている(非特許文献3、4)。またGLP-1は、インスリンの生合成を亢進し、β細胞の増殖も促すことから、β細胞の維持にとっても欠くことのできない因子である(非特許文献5、6)。さらにGLP-1には末梢組織において糖の利用を亢進する作用や、GLP-1の脳室内投与による摂食抑制作用、消化管運動抑制作用が報告されている(非特許文献7−10)。
【0004】
DPP-IVの酵素活性を阻害する物質は、その阻害作用により内在性のGLP-1の分解を抑制することでGLP-1の作用を高め、その結果インスリン分泌を亢進して糖代謝を改善することができると考えられている。そのためDPP-IV阻害剤は、糖尿病、特にII型糖尿病に対する予防および/または治療剤となり得ることが期待されている(非特許文献11、12)。また糖代謝の低下によって惹起、あるいは増悪されるその他の疾患(例えば糖尿病合併症、高インスリン血症、過血糖、脂質代謝異常、肥満など)における予防および/または治療に対する効果も期待されている。
【0005】
GLP-1の不活性化以外にもDPP-IVの生体内における役割や疾病との関係については以下のような報告がある。
(a) DPP-IVの阻害剤またはその抗体が、HIVウイルスの細胞内への侵入を阻害する。HIV-1感染患者由来のT細胞では、CD26の発現が減少している(非特許文献13)。また、HIV-1Tatタンパクは、DPP-IVに結合する(非特許文献14)。
(b) DPP-IVは免疫応答に関与する DPP-IVの阻害剤またはその抗体は、抗原刺激によるT細胞の増殖を抑制する(非特許文献15)。また、抗原刺激によりT細胞でのDPP-IVの発現が増加する(非特許文献16)。DPP-IVは、サイトカイン産生などのT細胞の機能に関与している(非特許文献17)。またDPP-IVは、T細胞表面でアデノシンデアミネース(ADA)と結合する(非特許文献18)。
(c) 慢性関節リウマチ、乾癬および偏平苔蘚患者の皮膚の線維芽細胞において、DPP-IVの発現が増加する(非特許文献19)。
(d) 良性前立腺肥大の患者および前立腺組織のホモジネートにおいて、DPP-IV活性が亢進している(非特許文献20)。肺内皮に存在するDPP-IVは、ラットの肺転移性乳癌および前立腺癌に対して接着分子として作用する(非特許文献21)。
(e)
DPP-IV活性を欠損している変異型F344ラットは、野生型F344ラットと比較して血圧が低いこと、および腎臓でナトリウムの再吸収に重要な役目を担っているタンパクとDPP-IVが相互作用する(特許文献1、2)。
(f) DPP-IV活性を阻害することによって、骨髄抑制性疾患の予防および/または治療が期待でき、DPP-IV活性剤が白血球数増加剤および/または感染症治療剤として期待できる(特許文献3)。
【0006】
これらの知見からDPP-IV阻害剤は、糖尿病(特にII型糖尿病)および/または糖尿病合併症以外のDPP-IVが関与する疾病の予防および/または治療剤となり得ることが期待される。例えば,HIV-1感染に基づくAIDS、臓器・組織移植における拒絶反応、多発性硬化症、慢性関節リウマチ、炎症、アレルギー、骨粗鬆症、乾癬および偏平苔蘚、良性前立腺肥大、乳癌および前立腺癌の肺転移抑制、高血圧、利尿、骨髄抑制の低減、白血球数増加、および感染症などに用いられる薬剤として有用であると考えられる。
【0007】
現在までにDPP-IV阻害剤として、(特許文献4−11)にピロリジン誘導体が、(特許文献12、13)にヘテロ環誘導体が、(特許文献14、15)にβアミノ酸誘導体が開示されている。
【0008】
また、(特許文献16)にDPP-IV阻害活性を有するビシクロ[2.2.2]オクタン誘導体が1化合物のみ開示されているが、本発明は、当該米国特許とは構造、DPP-IV阻害活性の面からも全く異なるものである。また(特許文献17)には、本発明に構造上近似したビシクロ誘導体を示唆する記述が見られるが、その記述内容は具体的に本発明化合物を何ら説明しておらず、また、本発明化合物のいずれをも実施例によって説明しているものではない。
【0009】
これまでに開示されているDPP-IV阻害剤はいずれも、DPP-IV阻害活性、DPP-IV選択性、安定性、毒性、および体内動態において満足できるものではなく、優れたDPP-IV阻害剤が常に求められている。
【非特許文献1】American Journal of Physiology、271巻、E458−E464頁(1996年)
【非特許文献2】European Journal of Pharmacology、318巻、429−435頁(1996年)
【非特許文献3】European Journal Clinical Investigation、22巻、154頁(1992年)
【非特許文献4】Lancet、2巻、1300頁(1987年)
【非特許文献5】Endocrinology、42巻、856頁(1992年)
【非特許文献6】Diabetologia、42巻、856頁(1999年)
【非特許文献7】Endocrinology、135巻、2070頁(1994年)
【非特許文献8】Diabetologia、37巻、1163頁(1994年)
【非特許文献9】Digestion、54巻、392頁(1993年)
【非特許文献10】Dig. Dis. Sci.、43巻、1113頁(1998年)
【非特許文献11】Diabetes、47巻、1663−1670頁(1998年)
【非特許文献12】Diabetologia、42巻、1324−1331頁(1999年)
【非特許文献13】Journal of Immunology、149巻、3073頁(1992年)
【非特許文献14】Journal of Immunology、150巻、2544頁(1993年)
【非特許文献15】Biological Chemistry、305頁(1991年)
【非特許文献16】Scandinavian Journal of Immunology、33巻、737頁(1991年)
【非特許文献17】Scandinavian Journal of Immunology、29巻、127頁(1989年)
【非特許文献18】Science、261巻、466頁(1993年)
【非特許文献19】Journal of Cellular Physiology、151巻、378頁(1992年)
【非特許文献20】European Journal of Clinical Chemistry and Clinical Biochemistry、30巻、333頁(1992年)
【非特許文献21】Journal of Cellular Physiology、121巻、1423頁(1993年)
【特許文献1】WO 03/015775 パンフレット
【特許文献2】WO 03/017936 パンフレット
【特許文献3】WO 03/080633 パンフレット
【特許文献4】WO 95/15309 パンフレット
【特許文献5】WO 98/19998 パンフレット
【特許文献6】WO 00/34241 パンフレット
【特許文献7】WO 02/14271 パンフレット
【特許文献8】WO 02/30890 パンフレット
【特許文献9】WO 02/38541 パンフレット
【特許文献10】WO 03/002553 パンフレット
【特許文献11】US 02/0193390 公報
【特許文献12】WO 02/062764 パンフレット
【特許文献13】WO 03/004496 パンフレット
【特許文献14】WO 03/000180 パンフレット
【特許文献15】WO 03/004498 パンフレット
【特許文献16】US 02/0193390 公報
【特許文献17】WO 02/38541 パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明が解決しようとする問題点は、優れたDPP-IV阻害活性を有する新規な化合物、または薬理学的に許容されるその塩を提供することにある。また、優れたDPP-IV阻害活性を有する新規な化合物、または薬理学的に許容されるその塩を含む医薬組成物、糖尿病およびその合併症の予防および/または治療剤あるいはDPP-IVが関与する疾患に対する予防および/または治療剤を提供することにある。
【課題を解決するための手段】
【0011】
本発明は、優れたDPP-IV阻害活性を有する新規なビシクロ誘導体、または薬理学的に許容されるその塩を提供する。また、優れたDPP-IV阻害活性を有する新規なビシクロ誘導体、または薬理学的に許容されるその塩を含む医薬組成物、糖尿病およびその合併症の予防および/または治療剤あるいはDPP-IVが関与する疾患に対する予防および/または治療剤を提供する。
【0012】
すなわち本発明は、
一般式(1)
【0013】
【化1】
【0014】
[式中、R1は、水素原子、ハロゲン原子、カルボキシル基、水酸基で置換されていてもよいC1〜C4のアルキル基、および置換されていてもよいアリール基を示し、
Xは、CH2、CHF、CF2、CHOH、SおよびOを示し、
nは1、2または3を示す。]
で表されるビシクロ誘導体、または薬理学的に許容されるその塩、前記一般式(1)で表されるビシクロ誘導体、または薬理学的に許容されるその塩を有効成分として含有する医薬、DPP-IV阻害剤、DPP-IVが関与する疾患の治療剤およびDPP-IVが関与する疾患が糖尿病及びその合併症である治療剤に関するものである。
【0015】
ここでC1〜C4のアルキル基とは、メチル基、エチル基、プロピル基、イソプロピル基、およびt−ブチル基などを意味する。置換されていてもよいアリール基とは、ハロゲン原子、C1〜C6のアルキル基、ヒドロキシル基、C1〜C6のアルコキシ基、C1〜C6のアルコキシカルボニル基、C1〜C6のアルキルチオ基、アミノ基、モノまたはジ置換のC1〜C6のアルキルアミノ基、1〜3個のヘテロ原子を含んでいてもよい4〜9員の環状アミノ基、ホルミルアミノ基、C1〜C6のアルキルカルボニルアミノ基、C1〜C6のアルコキシカルボニルアミノ基、ベンジルオキシカルボニルアミノ基、C1〜C6のアルキルスルホニルアミノ基、および置換されていてもよいアリールスルホニルアミノ基などから選ばれた1〜5個の置換基を有していてもよいアリール基を意味する。
【0016】
アリール基とは芳香族炭化水素または芳香族へテロ環(窒素原子、酸素原子、および硫黄原子の中から任意に選ばれた1〜3個のヘテロ原子を含む5員または6員の芳香族単環式複素環、あるいは9員または10員の芳香族縮合複素環)、例えばベンゼン環、ナフタレン環、アントラセン環、ピリジン環、ピリミジン環、ピリダジン環、トリアジン環、キノリン環、ナフチリジン環、キナゾリン環、アクリジン環、ピロール環、フラン環、チオフェン環、イミダゾール環、ピラゾール環、オキサゾール環、イソキサゾール環、チアゾール環、インドール環、ベンゾフラン環、ベンゾチアゾール環、ベンズイミダゾール環、およびベンゾオキサゾール環などを意味する。
【0017】
ここでハロゲン原子とは、フッ素原子、塩素原子、臭素原子、およびヨウ素原子を意味する。
【0018】
本発明の好ましい化合物としては、
(2S,4S)−1−[[N−(4−カルボキシビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル、
(2S)−1−[[N−(4−カルボキシビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリル、
(2S,4S)−1−[[N−(4−メチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル、
(2S)−1−[[N−(4−メチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリル、
(2S,4S)−1−[[N−(4−ヒドロキシメチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル、
(2S)−1−[[N−(4−ヒドロキシメチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリル、
(2S,4S)−1−[[N−(4−フルオロビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル、
(2S)−1−[[N−(4−フルオロビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリル、
などが例示できる。
【発明の効果】
【0019】
本発明化合物は優れたDPP-IV阻害活性を有する新規な化合物であり、糖尿病およびその合併症の予防および/または治療剤あるいはDPP-IVが関与する疾患に対する予防および/または治療剤を提供する。
【図面の簡単な説明】
【0020】
【図1】正常マウスにおける経口糖負荷試験時の血漿グルコース値に対する化合物1の効果を示すグラフ。各データは4例の平均値 ± 標準誤差で示した。*P< 0.05 vs. control (t検定)
【発明を実施するための最良の形態】
【0021】
本発明化合物が薬理学上許容な塩を形成する場合、塩酸、臭化水素酸、硫酸、硝酸、および燐酸などの無機酸、または酢酸、マレイン酸、フマル酸、コハク酸、乳酸、リンゴ酸、酒石酸、クエン酸、メタンスルホン酸、p−トルエンスルホン酸、ベンゼンスルホン酸、サリチル酸、ステアリン酸、パルミチン酸、およびトリフルオロ酢酸などの有機酸との塩、ナトリウム塩、カリウム塩、カルシウム塩、マグネシウム塩、アルミニウム塩、および亜鉛塩などの金属塩、アンモニウム塩およびテトラメチルアンモニウム塩などのアンモニウム塩、モルホリン、およびピペリジンなどとの有機アミン塩、およびグリシン、リジン、アルギニン、フェニルアラニンおよびプロリンなどのアミノ酸との付加塩が例示できる。
【0022】
上記一般式(1)で表される本発明化合物またはその塩には、1個または2個以上の不斉中心に基づく複数の光学異性体が存在し得るが、本発明はこれらの光学異性体もしくはジアステレオ異性体のいずれをも含み、またそれらの任意の比率を示す混合物またはラセミ体をも含むものである。また、上記一般式(1)で表される本発明化合物またはその塩に二重結合を含む場合には、その配置はZまたはEのいずれであってもよく、これらの任意の比率を示す混合物をも本発明に含まれる。さらには、上記一般式(1)で表される本発明化合物またはその塩の中には互変異性体や回転異性体が存在し得るものがあるが、それぞれの異性体およびそれらの任意の比率を示す混合物をも本発明に含まれる。
【0023】
上記一般式(1)で表される本発明化合物またはその塩は、分子内塩や付加物、それらの溶媒和物あるいは水和物などのいずれも含むものである。
【0024】
上記一般式(1)で表される本発明化合物またはその塩は、単独で、または一種以上の製剤上許容される補助剤と共に医薬組成物として用いることができ、薬理学上許容される担体、賦形剤(例えば、デンプン、乳糖、リン酸カルシウム、または炭酸カルシウムなど)、滑沢剤(例えば、ステアリン酸マグネシウム、ステアリン酸カルシウムタルク、またはステアリン酸など)、結合剤(例えば、デンプン、結晶セルロース、カルボキシメチルセルロース、アラビアゴム、ポリビニルピロリドン、またはアルギン酸など)、崩壊剤(例えば、タルク、またはカルボキシメチルセルロースカルシウムなど)、希釈剤(例えば、生理食塩水、グルコース、マンニトール、またはラクトースなどの水溶液など)などと混合し、通常の方法により錠剤、カプセル剤、顆粒剤、散剤、細粒剤、アンプル剤または注射剤などの形態で経口的または非経口的に投与することができる。投与量は上記一般式(1)で表される本発明化合物またはその塩の種類、投与方法、患者の年齢、体重、症状などにより異なるが、通常、人を含む哺乳動物に対して上記一般式(1)で表される本発明化合物またはその塩として0.0001〜1000mg/kg/日である。投与は例えば1日1回または数回に分割して投与する。
【0025】
上記一般式(1)で表される本発明化合物またはその塩は、必要であれば一種以上のDPP-IV阻害剤以外の糖尿病治療剤と併用することができる。本発明化合物またはその塩と併用される糖尿病治療剤としては、インスリンやその誘導体、GLP-1やその誘導体、その他の経口糖尿病治療剤が挙げられる。経口糖尿病治療剤としては、スルホニルウレア系糖尿病治療剤、非スルホニルウレア系インスリン分泌促進剤、ビグアナイド系糖尿病治療剤、α−グリコシダーゼ阻害剤、グルカゴンアンタゴニスト、GLP-1アゴニスト、PPARアゴニスト、β3アゴニスト、SGLT阻害剤、PKC阻害剤、グルカゴンシンテースキナーゼ-3(GSK-3)阻害剤、プロテインチロシンホスファターゼ-1B(PTP-1B)阻害剤、カリウムチャネルオープナー、インスリン増感剤、グルコース取込み調節剤、脂質代謝作用剤、食欲抑制剤などがあげられる。
【0026】
これらのうちGLP-1やその誘導体としては、ベタトロピン、またはNN-2211などが挙げられ、スルホニルウレア系糖尿病治療剤としては、トルブタミド、グリベンクラミド、グリクラジド、グリメピリド、またはグリピジドなどが挙げられ、非スルホニルウレア系インスリン分泌促進剤としては、ナテグリニド、レパグリニド、ミチグリニド、またはJTT-608などが挙げられ、ビグアナイド系糖尿病治療剤としては、メトホルミンなどが挙げられ、α−グリコシダーゼ阻害剤としては、ボグリボースまたはミグリトールなどが挙げられ、PPARアゴニストとしては、トログリタゾン、ロシグリタゾン、ピオグリタゾン、シグリタゾン、KRP-297(MK-767)、イサグリタゾン、GI-262570、JTT-501などが挙げられ、β3アゴニストとしては、AJ-9677、YM-178、またはN-5984などが挙げられる。
【0027】
本発明化合物(1)は、種々の合成法によって製造することができる。本発明化合物(1)は通常の分離手段(例えば抽出、再結晶、蒸留、クロマトグラフィー等)によって単離、精製することができる。また、得られた化合物が塩を形成する様な場合には、通常の方法あるいはそれに準ずる方法(例えば中和等)によって各種の塩を製造することができる。
【0028】
次に、本発明化合物およびその塩の代表的な製造工程について説明する。
【0029】
A法
【0030】
【化2】
【0031】
A法第一工程
本工程は、一般式(3)(式中、R1およびnは前記に同じ)で表されるビシクロアミン誘導体に、一般式(4)(式中、Y1はClまたはBrを表す。
Xは前記に同じ)で表されるハロ酢酸誘導体を反応させて、一般式(1)(式中、R1、n、およびXは前記に同じ) で表されるビシクロ誘導体を製造する工程である。本反応は、塩基の存在下または非存在下に行われる。本反応に塩基を用いる場合には、水酸化ナトリウム、水酸化カリウム、炭酸水素ナトリウム、炭酸水素カリウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウムなどの無機塩基や、トリエチルアミン、ジイソプロピルエチルアミン、N,N,N,N−テトラメチルエチレンジアミン、ジアザビシクロ[5.4.0]−7−ウンデセン、ジアザビシクロ[4.3.0]−5−ノネン、ホスファゼンベースまたはペンタイソプロピルグアニジンなどの有機塩基が例示できる。本反応に触媒を用いる場合には、テトラブチルアンモニウムブロミド、テトラブチルアンモニウムヨージド、ベンジルトリエチルアンモニウムブロミド、臭化リチウム、ヨウ化リチウム、ヨウ化ナトリウム、臭化カリウム、ヨウ化カリウム、臭化セシウム、ヨウ化セシウムなどの相関移動触媒または無機塩が例示できる。本反応に用いられる溶媒としては、反応に関与しない不活性な溶媒、例えばアセトン、エタノール、トルエン、アセトニトリル、テトラヒドロフラン、ジオキサン、エチルエーテル、t−ブチルメチルエーテル、ジメトキシエタン、酢酸エチル、ジクロロメタン、N,N−ジメチルホルムアミド、ジメチルスルホキシド、N−メチル−2−ピロリドンなどが用いられる。反応は0〜150℃で円滑に進行する。
【0032】
B法
【0033】
【化3】
【0034】
B法第一工程
本工程は、一般式(5)(式中、nは前記に同じ、THPはテトラヒドロピラニル基)で表されるビシクロアミン誘導体に、一般式(4)(式中、XおよびY1は前記に同じ)で表されるハロ酢酸誘導体を反応させて、一般式(6)(式中、nおよびXは前記に同じ)
で表されるビシクロ誘導体を製造する工程である。本反応は、塩基の存在下または非存在下に行われる。本反応に塩基を用いる場合には、水酸化ナトリウム、水酸化カリウム、炭酸水素ナトリウム、炭酸水素カリウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウムなどの無機塩基や、トリエチルアミン、ジイソプロピルエチルアミン、N,N,N,N−テトラメチルエチレンジアミン、ジアザビシクロ[5.4.0]−7−ウンデセン、ジアザビシクロ[4.3.0]−5−ノネン、ホスファゼンベースまたはペンタイソプロピルグアニジンなどの有機塩基が例示できる。本反応に触媒を用いる場合には、テトラブチルアンモニウムブロミド、テトラブチルアンモニウムヨージド、ベンジルトリエチルアンモニウムブロミド、臭化リチウム、ヨウ化リチウム、ヨウ化ナトリウム、臭化カリウム、ヨウ化カリウム、臭化セシウム、ヨウ化セシウムなどの相関移動触媒または無機塩が例示できる。本反応に用いられる溶媒としては、反応に関与しない不活性な溶媒、例えばアセトン、エタノール、トルエン、アセトニトリル、テトラヒドロフラン、ジオキサン、エチルエーテル、t−ブチルメチルエーテル、ジメトキシエタン、酢酸エチル、ジクロロメタン、N,N−ジメチルホルムアミド、ジメチルスルホキシド、N−メチル−2−ピロリドンなどが用いられる。反応は0〜150℃で円滑に進行する。
【0035】
B法第二工程
本工程は、一般式(6)(式中、nおよびXは前記に同じ) で表されるビシクロ誘導体のテトラヒドロピラニル基を除去して、一般式(2)(式中、nおよびXは前記に同じ)
で表されるビシクロ誘導体を製造する工程である。テトラヒドロピラニル基の除去は、公知の方法に従って酢酸やp−トルエンスルホン酸、または塩酸などにより、容易に除去することができる。
【0036】
以下の試験例および実施例により本発明の有用性を示すが本発明は試験例および実施例に限定されるものではない。
【0037】
<参考例1>
4−アミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニルの合成
第一工程:
4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸メチルの合成
ビシクロ[2.2.2]オクタン−1、4−ジカルボン酸水素メチル (25.0 g)、アジ化ジフェニルホスホリル (32.5 g)、トリエチルアミン (17.3
mL) およびトルエン (500 mL) を混合して室温で2時間撹拌し、次いで2時間加熱還流した。反応混合物にベンジルアルコール (122 mL) を加えて、さらに17時間加熱還流した。冷後、反応混合物を10%クエン酸水溶液、飽和炭酸水素ナトリウム水溶液、および飽和食塩水の順で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:ヘキサン:酢酸エチル=2:1)にて精製し、4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸メチル
(32.2 g) を得た。
MS (FAB+) m/z: 318 (MH+).
【0038】
第二工程:
4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸の合成
4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸メチル (64.3 g) をエタノール (1100 mL) に溶解し、1mol/L水酸化ナトリウム水溶液
(1000 mL) を加え、50℃で1時間撹拌した。反応液中のエタノールを減圧留去し、残渣をジエチルエーテル (500 mL) で洗浄した後、濃塩酸で酸性(pH1)とした。析出した結晶を濾取し、水洗後、減圧乾燥して4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸
(56.1 g) を得た。
MS (FAB+) m/z: 304 (MH+).
【0039】
第三工程:
4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニルの合成
4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸 (1.00 g) をジクロロメタン (10 mL) に懸濁して、3,4−ジヒドロ−2H−ピラン
(1.20 mL)、次いでp−トルエンスルホン酸・1水和物 (6.3 mg) を加え、室温で30分間撹拌した。反応混合物を飽和炭酸水素ナトリウム水溶液、次いで水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:ヘキサン:酢酸エチル=4:1)で精製し、4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニル
(1.18 g) を得た。
【0040】
1H NMR (CDCl3) δ 1.53-1.95
(m, 18H), 3.67-3.71 (m, 1H), 3.82-3.89 (m, 1H), 4.59 (br, 1H), 5.03 (s, 2H),
5.95 (br, 1H), 7.29-7.38 (m, 5H).
【0041】
第四工程
4−アミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニルの合成
4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニル (548 mg) を用いて、参考例1の第四工程と同様に反応を行い、4−アミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニル
(357 mg) を得た。
MS (EI+) m/z: 253 (M+).
【0042】
<参考例2>
4−アミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタンの合成
第一工程:
4−ベンジルオキシカルボニルアミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタンの合成
4−ベンジルオキシカルボニルアミノビシクロ[2.2.2]オクタン−1−カルボン酸 (500 mg) をテトラヒドロフラン (8 mL) に溶解して、食塩−氷浴上で冷却しながらN−メチルモルホリン
(0.18 mL) を加えた後、クロロ炭酸エチル (0.16 mL) を滴下し、さらに10分間撹拌した。次いで反応混合物に水素化ホウ素ナトリウム (187
mg) を加えた後、メタノール (15 mL) を加えて、0℃以下で1時間撹拌した。反応混合物を減圧濃縮して、残渣に水を加え、酢酸エチルで抽出した。酢酸エチル層を1mol/L塩酸、次いで水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:ヘキサン:酢酸エチル=1:1)で精製し、4−ベンジルオキシカルボニルアミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタン
(453 mg) を得た。
MS (EI+) m/z: 289 (M+).
【0043】
第二工程:
4−アミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタンの合成
4−ベンジルオキシカルボニルアミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタン (448 mg) を用いて、参考例1の第四工程と同様に反応を行い、4−アミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタン
(185 mg) を得た。
MS (EI+) m/z: 155 (M+).
【0044】
<参考例3>
1−アミノ−4−フルオロビシクロ[2.2.2]オクタンの合成
第一工程:
1−ベンジルオキシカルボニルアミノ−4−フルオロビシクロ[2.2.2]オクタンの合成
4−フルオロビシクロ[2.2.2]オクタン−1−カルボン酸 (265 mg) を用いて、参考例1の第一工程と同様に反応を行い、1−ベンジルオキシカルボニルアミノ−4−フルオロビシクロ[2.2.2]オクタン
(365 mg) を得た。
MS (EI+) m/z: 277 (M+).
【0045】
第二工程:
1−アミノ−4−フルオロビシクロ[2.2.2]オクタンの合成
1−ベンジルオキシカルボニルアミノ−4−フルオロビシクロ[2.2.2]オクタン
(350 mg) を用いて、参考例1の第四工程と同様に反応を行い、1−アミノ−4−フルオロビシクロ[2.2.2]オクタン (144 mg) を得た。
MS (EI+) m/z: 143 (M+).
【0046】
<参考例4>
(2S、4S)−1−(2−クロロアセチル)−4−フルオロピロリジン−2−カルボニトリルの合成
文献記載(WO02/38541 パンフレット)の(2S、4S)−1−(2−ブロモアセチル)−4−フルオロピロリジン−2−カルボニトリルの製法に準じて、(2S、4S)−4−フルオロピロリジン−2−カルボキサミド塩酸塩
(5.00 g) およびクロロアセチルクロリド (2.60 mL) から、(2S、4S)−1−(2−クロロアセチル)−4−フルオロピロリジン−2−カルボニトリル
(4.96 g) を得た。
MS (EI+) m/z: 190 (M+).
HRMS (EI+) for C7H8ClFN2O(M+):
calcd, 190.0309; found, 190.0283.
【実施例1】
【0047】
【化4】
【0048】
(2S,4S)−1−[[N−(4−カルボキシビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリルの合成
第一工程
(2S,4S)−1−[[N−[4−(2−テトラヒドロピラニル)オキシカルボニルビシクロ[2.2.2]オクト−1−イル]アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリルの合成
4−アミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニル (62.9 mg) をアセトニトリル (1 mL) に懸濁して、ジイソプロピルエチルアミン
(47 μL) を加え、氷冷下で(2S、4S)−1−(2−ブロモアセチル)−4−フルオロピロリジン−2−カルボニトリル (53.1 mg) のアセトニトリル
(0.8 mL) 溶液を加えて4時間撹拌した。反応液を濃縮した残渣に酢酸エチルおよび水を加えて溶解し、炭酸水素ナトリウム水溶液を用いてアルカリ性とした後に抽出した。酢酸エチル層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出溶媒:ジクロロメタン:メタノール=10:1)で精製し、(2S,4S)−1−[[N−[4−(2−テトラヒドロピラニル)オキシカルボニルビシクロ[2.2.2]オクト−1−イル]アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル
(73.3 mg) を得た。
MS (FAB+) m/z: 408 (MH+).
HRMS (FAB+) for C21H31FN3O4 (MH+):
calcd, 408.2299; found, 408.2295.
【0049】
第二工程
(2S,4S)−1−[[N−(4−カルボキシビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリルの合成
(2S,4S)−1−[[N−[4−(2−テトラヒドロピラニル)オキシカルボニルビシクロ[2.2.2]オクト−1−イル]アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル
(71.6 mg) を酢酸 (4 mL) に溶解し、室温で6時間撹拌した。反応混合物を減圧濃縮して、残渣をジクロロメタンで懸濁して濾取し、(2S,4S)−1−[[N−(4−カルボキシビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル
(31.4 mg) を得た。
MS (EI+) m/z: 323 (M+).
【実施例2】
【0050】
【化5】
【0051】
(2S)−1−[[N−(4−カルボキシビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリルの合成
第一工程
(2S)−1−[[N−[4−(2−テトラヒドロピラニル)オキシカルボニルビシクロ[2.2.2]オクト−1−イル]アミノ]アセチル]ピロリジン−2−カルボニトリルの合成
4−アミノビシクロ[2.2.2]オクタン−1−カルボン酸2−テトラヒドロピラニル (90.0 mg) および(2S)−1−(2−ブロモアセチル)ピロリジン−2−カルボニトリル
(70.0 mg)を用いて、実施例1と同様に反応を行い、(2S)−1−[[N−[4−(2−テトラヒドロピラニル)オキシカルボニルビシクロ[2.2.2]オクト−1−イル]アミノ]アセチル]ピロリジン−2−カルボニトリル
(85.2 mg) を得た。
MS (EI+) m/z: 383 (M+).
HRMS (EI+) for C21H31N3O4 (M+):
calcd, 383.2315; found, 383.2296.
【0052】
第二工程
(2S)−1−[[N−(4−カルボキシビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリルの合成
(2S)−1−[[N−[4−(2−テトラヒドロピラニル)オキシカルボニルビシクロ[2.2.2]オクト−1−イル]アミノ]アセチル]ピロリジン−2−カルボニトリル
(73.0 mg) を用いて、実施例1と同様に反応を行い、(2S)−1−[[N−[4−カルボキシビシクロ[2.2.2]オクト−1−イル]アミノ]アセチル]ピロリジン−2−カルボニトリル
(56.0 mg) を得た。
MS (EI+) m/z: 305 (M+).
HRMS (EI+) for C16H23N3O3 (M+):
calcd, 305.1739; found, 305.1736.
【実施例3】
【0053】
【化6】
【0054】
(2S,4S)−1−[[N−(4−メチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリルの合成
4−アミノ−1−メチルビシクロ[2.2.2]オクタン(56.0 mg) および(2S、4S)−1−(2−ブロモアセチル)−4−フルオロピロリジン−2−カルボニトリル
(95.0 mg) を用いて、実施例1と同様に反応を行い、(2S,4S)−1−[[N−(4−メチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル
(89.2 mg) を得た。
MS (EI+) m/z: 293 (M+).
HRMS (EI+) for C16H24FN3O(M+):
calcd, 293.1903; found, 293.1881.
【実施例4】
【0055】
【化7】
【0056】
(2S)−1−[[N−(4−メチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリルの合成
4−アミノ−1−メチルビシクロ[2.2.2]オクタン(75.0 mg) および(2S)−1−(2−ブロモアセチル)ピロリジン−2−カルボニトリル (110
mg) を用いて、実施例1と同様に反応を行い、(2S)−1−[[N−(4−メチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリル
(84.0 mg) を得た。
MS (EI+) m/z: 275 (M+).
HRMS (EI+) for C16H25N3O(M+):
calcd, 275.1998; found, 275.1981.
【実施例5】
【0057】
【化8】
【0058】
(2S,4S)−1−[[N−(4−ヒドロキシメチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリルの合成
4−アミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタン (50.0 mg) および(2S、4S)−1−(2−ブロモアセチル)−4−フルオロピロリジン−2−カルボニトリル
(75.7 mg) を用いて、実施例5と同様に反応を行い、(2S,4S)−1−[[N−(4−ヒドロキシメチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル
(93.0 mg) を得た。
MS (FAB+) m/z: 310 (MH+).
HRMS (FAB+) for C16H25FN3O2 (MH+):
calcd, 310.1931; found, 310.1942.
【実施例6】
【0059】
【化9】
【0060】
(2S)−1−[[N−(4−ヒドロキシメチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリルの合成
4−アミノ−1−ヒドロキシメチルビシクロ[2.2.2]オクタン (50.5 mg) および(2S)−1−(2−ブロモアセチル)ピロリジン−2−カルボニトリル
(61.7 mg) を用いて、実施例5と同様に反応を行い、(2S)−1−[[N−(4−ヒドロキシメチルビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリル
(57.0 mg) を得た。
MS (FAB+) m/z: 292 (MH+).
HRMS (FAB+) for C16H25N3O2 (MH+):
calcd, 292.2025; found, 292.2025.
【実施例7】
【0061】
【化10】
【0062】
(2S,4S)−1−[[N−(4−フルオロビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリルの合成
4−アミノ−1−フルオロビシクロ[2.2.2]オクタン(50.0 mg) および(2S、4S)−1−(2−ブロモアセチル)−4−フルオロピロリジン−2−カルボニトリル
(82.1 mg) を用いて、実施例10と同様に反応を行い、(2S,4S)−1−[[N−(4−フルオロビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]−4−フルオロピロリジン−2−カルボニトリル
(93.1 mg) を得た。
MS (EI+) m/z: 297 (M+).
HRMS (EI+) for C15H21F2N3O(M+):
calcd, 297.1653; found, 297.1628.
【実施例8】
【0063】
【化11】
【0064】
(2S)−1−[[N−(4−フルオロビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリルの合成
4−アミノ−1−フルオロビシクロ[2.2.2]オクタン(50.0 mg) および(2S)−1−(2−ブロモアセチル)ピロリジン−2−カルボニトリル
(73.5 mg) を用いて、実施例10と同様に反応を行い、(2S)−1−[[N−(4−フルオロビシクロ[2.2.2]オクト−1−イル)アミノ]アセチル]ピロリジン−2−カルボニトリル
(72.3mg) を得た。
MS (EI+) m/z: 279 (M+).
HRMS (EI+) for C15H22FN3O(M+):
calcd, 279.1747; found, 279.1766.
【0065】
<試験例1>[ジペプチジルペプチダーゼ IV活性阻害試験]
基質であるH−Gly−Pro−AMC (7−アミノ−4−メチル−クマリン)・HBrが血漿ジペプチジルペプチダーゼ IVにより分解されて遊離するAMC濃度を蛍光強度により測定した。
方法
平底96穴プレートを用いて、生理食塩水で8倍希釈した血漿20μLに化合物を溶解させた緩衝液(25mmol/L ヘペス、140 mmol/ L 塩化ナトリウム、1%
ウシ血清アルブミン、80mmol/ L 塩化マグネシウム・6水和物、pH7.4)20μLを添加し室温で5分間放置した後、0.1mmol/LのH−Gly−Pro−AMC・HBr溶液10μLを添加して反応を開始した。遮光下室温で20分間放置した後、25%酢酸溶液20μLを添加して反応を停止させた。遊離したAMC濃度を、蛍光プレートリーダーを用いて355nmで励起させた時の460nmの蛍光強度を測定した。得られた結果から50%阻害濃度(IC50値)をプリズム3.02
(グラフパッド ソフトウェア) を用いて算出した。結果を表1に記載した。
【0066】
【表1】
【0067】
化合物A:(2S)−1−[[(3−ヒドロキシ−1−アダマンチル)アミノ]アセチル]−2−シアノピロリジン(LAF-237)
【0068】
<試験例2>[経口投与におけるマウスのジペプチジルペプチダーゼ IV活性阻害試験]
0.3% カルボキシメチルセルロースナトリウム塩を用いて化合物を0.1 mg/mLの濃度で懸濁し、8週齢の雄性ICRマウス (日本チャールスリハ゛ー) に10mL/kgで経口投与した。投与前および投与後30分にEDTA・2K処理毛細管を用いて尾静脈から採血を行い、採取した各血液を6000回転で2分間遠心分離して血漿を得た。試験例1と同様の方法を用いて、酵素活性を測定した。投与前の酵素活性値からの減少率を阻害率として算出した[阻害率={(投与前値−投与後値)÷投与前値}×100]。結果を表2に記載した。
【0069】
【表2】
【0070】
化合物A:(2S)−1−[[(3−ヒドロキシ−1−アダマンチル)アミノ]アセチル
]−2−シアノピロリジン(LAF−237)
【0071】
<試験例3>[経口投与におけるマウス耐糖能試験]
実施例5の本発明化合物(以下化合物1)を0.3%カルボキシメチル-セルロース ナトリウム塩 (CMC-Na, シグマ) で懸濁した。7週齢の雄性ICRマウス(日本チャールスリバー)を1週間予備飼育した。この時、標準食(CE-2,
日本クレア) および水は自由摂取させた。8週齢のICRマウスを16時間絶食し、0.3%CMC-Na (10 mL/kg) または化合物1(1 0 mg/kg,
10 mL/kg) を経口投与した。投与30分後にグルコース溶液を5 g/kgの用量で経口投与した。採血はEDTA-2K処理毛細管を用いて、グルコース溶液投与前および投与15,
30, 60および120分後に尾静脈から行った。血漿グルコース値の測定にはグルコース Bテストワコー (和光純薬工業) を用いた。結果は平均値 ± 標準誤差で示した。統計解析はt検定を用いて、有意水準は5%未満とした。結果を図1に記した。
【0072】
<試験例4>[薬剤性白血球減少症に対する薬効評価試験]
本発明化合物の薬剤性白血球減少症に対する薬効評価実験をOkabeらの方法(薬理と治療、19巻、6号、55頁、1991年)に準じて行った。
8週齢の雄性ICR系マウス(日本チャールスリハ゛ー)を用いて、Day0にシクロホスファミド(200mg/kg)を単回腹腔内投与した。翌日から対照群には生理食塩水を投与し、薬物投与群には本発明化合物(1〜200mg/kg)を1日1〜2回、5日間経口投与した。試験開始から2,4,6,および8日後にそれぞれ採血を行い、白血球数を経時的に測定し、シクロホスファミド投与前の白血球数をコントロールとすることによって、本発明化合物の薬剤性白血球減少症に対する薬効を評価した。コントロールと比較して、本発明化合物は白血球の減少を有意に抑制した。
【0073】
<試験例5>[血中G−CSF濃度の増加作用試験]
7週齢の雄性ICR系マウス(日本チャールスリハ゛ー)を用いて、対照群には生理食塩水を投与し、薬物投与群には本発明化合物(1〜200mg/kg)を1日1〜2回、5日間経口投与した。投与終了翌日に麻酔下で採血し、マウスG−CSF ELISA測定キット(R&D SYSTEM社)を用いて血漿中のG−CSF濃度を測定した。コントロールと比較して、本発明化合物は血漿中のG−CSF濃度を有意に増加させた。
【産業上の利用可能性】
【0074】
本願化合物は、優れたDPP-IV阻害活性を有する新規なビシクロ誘導体、または薬理学的に許容されるその塩である。本願化合物を有効成分として含有する医薬組成物は、糖尿病およびその合併症の予防および/または治療剤あるいはDPP-IVが関与する疾患に対する予
防および/または治療剤として有用である。
【特許請求の範囲】
【請求項1】
一般式(1)
【化1】
[式中、R1は、水素原子、ハロゲン原子、カルボキシル基、水酸基で置換されていてもよいC1〜C4のアルキル基または置換されていてもよいアリール基を示し、
Xは、CH2、CHF、CF2、CHOH、SまたはOを示し、
nは1ないし3の整数を示す。]
で表されるビシクロ誘導体、または薬理学的に許容されるその塩。
【請求項2】
前記一般式(1)において、R1は水素原子を示し、Xは、CH2、CHF、CF2、CHOH、SまたはOを示し、nは1ないし3の整数を示す化合物であることを特徴とする請求項1記載のビシクロ誘導体、または薬理学的に許容されるその塩。
【請求項3】
前記一般式(1)において、R1はハロゲン原子を示し、Xは、CH2、CHF、CF2、CHOH、SまたはOを示し、nは1ないし3の整数を示す化合物であることを特徴とする請求項1記載のビシクロ誘導体、または薬理学的に許容されるその塩。
【請求項4】
前記一般式(1)で表される化合物が、一般式(2)
【化2】
[式中、Xは、CH2、CHF、CF2、CHOH、SまたはOを示し、
nは1ないし3の整数を示す。]
で表される化合物であることを特徴とする請求項1記載のビシクロ誘導体、または薬理学的に許容されるその塩。
【請求項5】
前記一般式(1)において、R1は水酸基で置換されていてもよいC1〜C4のアルキル基を示し、Xは、CH2、CHF、CF2、CHOH、SまたはOを示し、nは1ないし3の整数を示す化合物であることを特徴とする請求項1記載のビシクロ誘導体、または薬理学的に許容されるその塩。
【請求項6】
請求項1記載のビシクロ誘導体、または薬理学的に許容されるその塩を有効成分として含有する医薬。
【請求項7】
請求項1記載のビシクロ誘導体、または薬理学的に許容されるその塩を有効成分として含有するDPP-IV阻害剤。
【請求項8】
請求項1記載のビシクロ誘導体、または薬理学的に許容されるその塩を有効成分とするDPP-IVが関与する疾患の治療剤。
【請求項9】
DPP-IVが関与する疾患が糖尿病及びその合併症である請求項8記載の治療剤。
【請求項1】
一般式(1)
【化1】
[式中、R1は、水素原子、ハロゲン原子、カルボキシル基、水酸基で置換されていてもよいC1〜C4のアルキル基または置換されていてもよいアリール基を示し、
Xは、CH2、CHF、CF2、CHOH、SまたはOを示し、
nは1ないし3の整数を示す。]
で表されるビシクロ誘導体、または薬理学的に許容されるその塩。
【請求項2】
前記一般式(1)において、R1は水素原子を示し、Xは、CH2、CHF、CF2、CHOH、SまたはOを示し、nは1ないし3の整数を示す化合物であることを特徴とする請求項1記載のビシクロ誘導体、または薬理学的に許容されるその塩。
【請求項3】
前記一般式(1)において、R1はハロゲン原子を示し、Xは、CH2、CHF、CF2、CHOH、SまたはOを示し、nは1ないし3の整数を示す化合物であることを特徴とする請求項1記載のビシクロ誘導体、または薬理学的に許容されるその塩。
【請求項4】
前記一般式(1)で表される化合物が、一般式(2)
【化2】
[式中、Xは、CH2、CHF、CF2、CHOH、SまたはOを示し、
nは1ないし3の整数を示す。]
で表される化合物であることを特徴とする請求項1記載のビシクロ誘導体、または薬理学的に許容されるその塩。
【請求項5】
前記一般式(1)において、R1は水酸基で置換されていてもよいC1〜C4のアルキル基を示し、Xは、CH2、CHF、CF2、CHOH、SまたはOを示し、nは1ないし3の整数を示す化合物であることを特徴とする請求項1記載のビシクロ誘導体、または薬理学的に許容されるその塩。
【請求項6】
請求項1記載のビシクロ誘導体、または薬理学的に許容されるその塩を有効成分として含有する医薬。
【請求項7】
請求項1記載のビシクロ誘導体、または薬理学的に許容されるその塩を有効成分として含有するDPP-IV阻害剤。
【請求項8】
請求項1記載のビシクロ誘導体、または薬理学的に許容されるその塩を有効成分とするDPP-IVが関与する疾患の治療剤。
【請求項9】
DPP-IVが関与する疾患が糖尿病及びその合併症である請求項8記載の治療剤。
【図1】

【国際公開番号】WO2005/082847
【国際公開日】平成17年9月9日(2005.9.9)
【発行日】平成19年10月25日(2007.10.25)
【国際特許分類】
【出願番号】特願2006−510421(P2006−510421)
【国際出願番号】PCT/JP2005/002806
【国際出願日】平成17年2月22日(2005.2.22)
【出願人】(000001395)杏林製薬株式会社 (120)
【Fターム(参考)】
【国際公開日】平成17年9月9日(2005.9.9)
【発行日】平成19年10月25日(2007.10.25)
【国際特許分類】
【国際出願番号】PCT/JP2005/002806
【国際出願日】平成17年2月22日(2005.2.22)
【出願人】(000001395)杏林製薬株式会社 (120)
【Fターム(参考)】
[ Back to top ]