
ビシファジン塩酸塩の多形
多形相Aとして呼ばれるビシファジン塩酸塩の既知多形相よりも熱力学的によりいっそう安定している、多形相Bと呼ばれる新規なビシファジン塩酸塩の多結晶形、上記多形相Bを調製するための方法、および上記多形相Bを含む医薬組成物。
【発明の詳細な説明】
【技術分野】
【0001】
ビシファジン塩酸塩の新規多形結晶に関わる。より詳しくは、多形相Aと称するビシファジン塩酸塩の既知の多形相よりも、熱力学的に安定な多形相Bに関する。
【背景技術】
【0002】
以下の式からなる塩酸付加塩であるビシファジン塩酸塩は、非麻薬性鎮痛薬(すなわち、作用がモルヒネ様ではない)である。特許文献1および特許文献2を参照せよ。
【0003】
【化1】

ビシファジン塩酸塩(bicifadine hydrochloride)、その化学名は、(±)−1−(4−メチルフェニル)−3−アザビシクロ[3.1.0]−ヘキサン塩酸塩であり、その同義語は、ラセミ体1−(p−トリル)−3−アザビシクロ[3.1.0]−ヘキサン塩酸塩であり、淡黄褐色プレート状(pale tan plates shaped)結晶として特許文献1の実施例36に記載の通りに製造される。この製品は、1−(p−トリル)−1,2−シクロプロパンジカルボキシミドから生産されていた。1−(p−トリル)−1,2−シクロプロパンジカルボキシミドを有機溶媒に溶かし、アミンに還元して塩酸塩に変換することで、粗ビシファジン塩酸塩の沈殿を生じさせる。粗ビシファジン塩酸塩を濾過によって反応媒体から回収した。次に、粗ビシファジン塩酸塩をアセトニトリル/メタノール混合物から再結晶化して黄褐色プレート形状の結晶を得る。
【0004】
ビシファジン塩酸塩の結晶形は、患者の疼痛に対して錠剤またはカプセル等として種々の経口単位投薬形態で処方されることから、特に重要である。医薬品物質の結晶構造における変化は、医薬品の溶解性、製造性、および安定性、特に固形経口投薬形態の処方で影響を及ぼす可能性がある。したがって、単一の熱力学的に安定な結晶構造からなる純粋な形状でビシファジン塩酸塩を生産することは、重要である。上記の手順に従って生産されるビシファジン塩酸塩結晶構造が、淡黄褐色プレートとして、最も熱力学的に安定な多形相(polymorphic form)でないと判断されている。さらにまた、従来の製造プロセス、例えばグラインディングおよびミリングにかける場合、この多形相(以下、多形相Aという)のビシファジン塩酸塩が、異なる多形相への転換を受けることが示されている。多形相Aがビシファジン塩酸塩で最も熱力学的に安定した形状ではないことから、多形相Aは時間とともに多形転換を起こしうる。したがって、多形相Aは、医薬品への製剤化にとってビシファジン塩酸塩の最適結晶形でない。
【特許文献1】米国特許第4,231,935号
【特許文献2】米国特許第4,196,120号
【発明の開示】
【発明が解決しようとする課題】
【0005】
この発明に従って、我々は結晶ビシファジン塩酸塩に2種類の多形相が存在することを発見し、また米国特許第4,231,935号で生産された結晶形が、淡黄褐色プレートとして、ビシファジン塩酸塩の2種類の多形相のうちの1つであるということを発見した。この多形相は、多形相Aとして表す。さらに、この発明に従って、我々は、従来の製剤加工操作、例えば、グラインディングおよびミリングを受ける場合、結晶がよりいっそう熱力学的に安定していて多形転換を受けない、ビシファジン塩酸塩のもう一つの新しい結晶多形構造が存在するということを発見した。この新しい多形相は、多形相Bと称する。
【0006】
この発明にもとづいて、われわれは多形相Bと称する、ビシファジン塩酸塩の新規多形結晶を発見した。この多形相Bは、多形相Aと称するビシファジン塩酸塩の既知の多形相よりも熱力学的に安定な錠剤である。淡黄褐色プレートの形状である多形相Aの結晶とは異なり、ビシファジン塩酸塩多形相Bの結晶は、白色ないし灰色がかった(off-white)の色を呈するブレード状である。
【課題を解決するための手段】
【0007】
ビシファジン塩酸塩の多形を、赤外線スペクトラムおよび/またはX線粉回折パターンによって特徴づけることが可能である。所定の多形のX線粉回折ピークの相対強度は、パターンの測定に用いる粒度に応じて変化しうる。このことは、試料ホルダー内の結晶の好ましい配向の現象である。しかし、この発明のビシファジン塩酸塩の所定の多形相Bによって、このパターン内に粒度にかかわらず典型的に存在する特定のピークがある。一方で、所定の多形、例えば、この発明のビシファジン塩酸塩の多形相Bの赤外線スペクトラムは、粒度にかかわりなく比較的一定のままである。
【0008】
ラセミ体ビシファジン塩酸塩の多形相AおよびBのX線粉末回折(XRPD)による分析は、CuKα線を用いた島津製作所のXRD−6000粉末回析カメラで実行された。ビシファジンは、結晶性粉末としてその機器に供給された。その機器は、高精度焦点X線管を具備していた。管電圧およびアンペア数をそれぞれ40kVおよび40mVに設定した。ビームの開きおよび散乱スリットを1度に設定し、受け側スリットを0.15mmに設定した。回折された放射線の検出は、ヨウ化ナトリウム・シンチレーション検出器によっておこなった。2.5ないし40°2θの3°/分(0.4秒/0.02°ステップ)シータ−2シータ連続走査を使用した。シリコン標準物質を分析して、機器のアライメントを調べた。データを収集して、XRD−6000バージョン4.1で分析した。
【0009】
ラセミ体ビシファジン塩酸塩の多形相BのX線粉末回折パターンは「d」間隔に換算して与えられ、相対強度(I)は以下の通りであり(s=強、m=中程度、w=弱、v=極めて、d=拡散)、さらにこれらの条件は下記の表1で述べられる。
【0010】
【表1】
【0011】
ビシファジン塩酸塩のA形のX線粉末回折パターンを、以下のように表1の場合と同様の用語で表2に示す。
【0012】
【表2】
【0013】
表1および表2は、それぞれ粒度を下げたビシファジン塩酸塩B形およびA形のピーク位置でのXRPDパターンを表す。これらの表に示す結果は、減少粒度での多形相Aおよび多形相BのXRPDパターン間の差を示す。しかし、このパターンでは重要な主要ピークが所定の角度にあり、所定の角度はビシファジン塩酸塩の所定のB形に特有なもので、その粒度にかかわりなく多形相BのXRPDパターンで、典型的に存在する。2θ(度)として表され、かつCuKα線を用いて多形相Bが特徴づけられる主要ピークが位置づけられているこれらの角度は、5.08、10.07、20.16、25.17、および30.43である。
【0014】
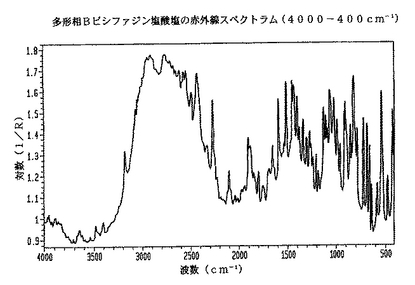
各々の試料について、Ever−Glo 中/遠IR源、拡張範囲臭化カリウム(KBr)ビームスプリッタ、および重水素化硫酸トリグリシン(DTGS)検出器を備えたMagna−IR860(登録商標)フーリエ変換赤外(FT−IR)分光光度計(トーマス・ニコレ(Thomas Nicolet))を用いて、赤外線スペクトルを得た。分光光度計は、所定の波長で試料の各々の赤外光帯の強度を測定した。拡散反射率アクセサリ(コレクター(Collector)(商標)、サーモ・スペクトラ・テク(Thermo SpectraTech))をサンプリングに使用した。各スペクトルは、スペクトラム解像度4cm-1で400から4,000cm-1までの範囲で収集した256回の共付加走査(co−added scans)を表す。試料調製は、多形相Aまたは多形相Bのいずれかである結晶を含む粉末試料を、13mm径カップに入れて、該材料を曇り(frosted)スライドガラスでならすことからなった。バックグラウンド・データ・セットは、所定位置に置かれたアラインメント・ミラーによって得た。反射率Rは、所定の波数における試料の光強度/バックグランド・セットの光強度である。図1は、多形相Bの赤外線スペクトラムを示すもので、横座標は波数cm-1、縦座標はLog(1/R)である。Log 1/R(R=反射率)スペクトルは、各々に対してこれらの2つのデータ・セット(試料およびバックグラウンド光強度)の比を取ることで得た。乾燥結晶粉末としてのラセミ体ビシファジン塩酸塩の多形相Bの赤外線スペクトラムは、表3に示すように、この多形を特徴づける以下のピークを示した。
【0015】
【表3】
【0016】
乾燥結晶粉末状のラセミ体ビシファジン塩酸塩の多形Aの赤外線スペクトラムは、この多形を特徴づける以下の主ピークを示した。
【0017】
【表4】
【0018】
表3および表4は、それぞれビシファジン塩酸塩の多形相Bおよび多形相Aに関して赤外線のピークの位置の完全なパターンを提供する。しかし、特定のキー・ピークがあり、このパターン内では、ビシファジン塩酸塩の多形相Bに特有で、この多形を特徴づけるのに十分である。波数(cm-1)で表すこれらのピークは、2108、891、856、719、および660である。
【0019】
この発明に従って、我々は多形B結晶構造を持つビシファジン塩酸塩を形成する手段を見つけた。ラセミ体ビシファジン塩酸塩を調製する既知の方法は、多形相Aを生産する。なぜなら、それは最初に生産される多形結晶構造であるからである。多形相Aから多形相Bを作るための1つの手段は、通常生産される多形相Aに、運動エネルギーを付与することである。この運動エネルギーは、多形相Aに対して、特に低温度、一般には約−200℃ないし約50℃、好ましくは約−200℃ないし約35℃、より好ましくは−200℃ないし約0℃で印加される。この転換を実行するために、撹拌、グラインディング、またはミリング等の手段によって運動エネルギーを通常のビシファジン塩酸塩多形相Aに印加する方法のいずれも使用することができる。グラインディングまたはミリングを、室温で適用することができる。しかし、この転換は、運動エネルギーを使用するもので、低温でより効率的に実施される。例えば、多形相Aを多形相Bに転換するための運動エネルギー法は、多形Aの固晶を用いて、該結晶に撹拌またはグラインディングを適用する一方で、温度を約35℃以下、一般には約−200℃ないし0℃に保つことである。液体窒素雰囲気下で、グラインディング等によって運動エネルギーを供給している間、低温が利用される。
【0020】
多形相Aを多形相Bに変換するもう一つの方法は、加熱された溶液から多形相Bを結晶化させ、上記多形Bを形成するのに充分な時間にわたって、上記溶液を冷却する。多形相A結晶は、通常、室温では有機溶剤に十分に溶解しない。しかし、加熱されると、それは可溶性になる。この転換にもとづいて、ビシファジン塩酸塩の多形相A結晶を少なくとも50℃の沸点を持つ有機溶媒と混合し、スラリーを形成させる。この転換を行う際に、少なくとも50℃の沸点を持つ従来の有機溶媒のいずれも利用することができる。つぎに、スラリーが透明な溶液となる温度まで、上記スラリーを加熱する。この溶液を、最大で約35℃、好ましくは約0℃ないし約20℃の温度まで冷やす。冷却された溶液を、多形相Aを含まない純粋な結晶形としての上記多形相Bが形成されるのに十分なしばらくの間、最大で約35℃の温度に保つ。必要に応じて、撹拌しながら冷却をおこなう。冷却している間、混合物から結晶の多形Bが形成されるのに十分な時間をかけるように、モニターしていなければならない。この手順では、冷却するにつれて、多形相Bの「種結晶」を加えることによって転換の時間もまた促進される。
【0021】
また、多形相Bの生産は、ビシファジン塩酸塩A形またはビシファジン塩酸塩A形とB形との混合物から、A形またはA形とB形との混合物のいずれかとして、不活性有機溶媒中にビシファジン塩酸塩のスラリーを形成し、最大で約35℃の温度でスラリーを撹拌することによって、おこなうことができる。スラリーの形成の際に、従来の不活性有機溶媒のいずれかを用いることができる。アジテーションは、ビシファジン塩酸塩を多形相Bに変換するのに十分な時間、実施される。この期間を24時間とすることができるだろう。この時間は、多形相Bの「種」結晶を用いることによって、および/または温度を35℃未満にすることで、短くすることができると考えられる。
【0022】
上記方法のいずれかによって生産された多形相Bは、XRPDまたはInfra−Redによって測定されるように、ビシファジン塩酸塩の他の多形相がなんら含まれない純粋な形状であることができる。このように、ビシファジン塩酸塩の多形相Bの純粋なラセミ体は、ラセミ体ビシファジン塩酸塩のその他の多形相が存在することなく生産される。
【0023】
ビシファジン塩酸塩の多形相Bを、疼痛を抑えるためにヒト患者に投与することができる。これは、多形相Bの結晶構造と不活性担体または希釈剤とを持つビシファジン塩酸塩を含有する組成物による処置を必要とする、上記疼痛で苦しんでいる患者を処置することによって達成され、該組成物は疼痛緩和に有効な量で投与される。この発明によれば、結晶多形相B状態のラセミ体ビシファジン塩酸塩を、疼痛緩和に有効な量で投与する。疼痛緩和に必要とされる任意の有効量のビシファジン塩酸塩を、この組成物で利用することができる。一般に、1日あたり約0.5mg/kgないし約20mg/kgの経口量が用いられる。しかし、投与される経口単位服用量の結晶多形相Bラセミ体ビシファジン塩酸塩の量は、大部分は疼痛の量と患者の体重とに依存し、もちろん医師の判断にゆだねられる。本発明によれば、経口単位投薬形態の結晶多形相Bラセミ体ビシファジン塩酸塩が1日あたり1ないし2回または必要に応じて、25ないし600mgの服用量で投与される。約60kgないし約80kgの患者では、100mgないし600mgを含む単位経口投薬形態を用いることができ、一般に好ましくは200ないし400mgの服用量が用いられる。1日あたり1ないし2回または必要に応じて、経口単位服用量を投与することができる。疼痛が少なく、体重が60kg未満の患者に対しては、約25mgないし約200mgを含む経口単位投薬形態を、患者の要求に応じて1日あたり1ないし2回以上投与する。
【0024】
この発明の組成物では、従来の医薬的に許容される担体または希釈剤のいずれかを使用することができる。経口投与を目的として、医薬組成物は、従来の賦形剤によって調製された従来の経口単位投薬形態、例えば錠剤、カプセル、粉体、溶液、シロップ、または懸濁液のいずれかであってもよい。したがって、組成物の経口投与は、錠剤またはカプセル、ロゼンジ等、従来の方法で処方される形態を取ることが可能である。これらの製剤の各々は、その結晶多形相Bの形態で、ラセミ体ビシファジン塩酸塩を含有する。通常、これらの製剤で結晶多形相A状態にあるビシファジン塩酸塩が全く存在しない状態で、多形相Bビシファジン塩酸塩を用いることが好ましい。多形相Bは、ビシファジン塩酸塩の熱力学的に安定な多形であり、製造の過程および医薬品製剤の予想された商業的な有効期間を超えて、結晶間転換を受けることはない。
【0025】
必要に応じて、その結晶多形相Bのラセミ体ビシファジン塩酸塩は、経口単位投薬形態で親水性徐放高分子(ヒドロキシプロピル・メチルセルロース)の使用によって、徐放多形相で投与されうる。任意の親水性徐放高分子(例えば、約100cpsないし約100,000cpsの範囲の粘度を持つヒドロキシプロピル・メチル・セルロース・ポリマー)を用いることができる。
【0026】
この発明に従って、経口単位投薬形態の構成は、担体を含む可能性がある。薬学的製剤技術に共通の適当な担体として、限定されるものではないが、微結晶性セルロース、ラクトース、スクロース、フルクトース、グルコースデキストロース、または他の糖類、二塩基リン酸カルシウム、硫酸カルシウム、セルロース、メチルセルロース、セルロース誘導体、カオリン、マンニトール、ラクチトール、マルチトール、キシリトール、ソルビトール、又はその他の糖アルコール、乾燥澱粉、デキストリン、マルトデキストリン、または他の多糖類、イノシトール、またはそれらの混合物が挙げられる。
【0027】
この発明用の好ましい単位経口の剤形は、錠剤である。医薬経口単位投薬形態を調製する従来方法のいずれも、この発明の単位剤形を調製する際に利用することができる。医薬経口単位投薬形態(例えば錠剤)は、従来の付加的な配合剤の一つ以上を含む。これらの成分は、医薬製剤技術で既知の幅広く多様な賦形剤から選択される。経口投薬形態の所望の特性によれば、いくつかの成分を、単独で、または組みになって、錠剤としてのそのような投薬形態を調製する際の既知の使用のために、選択することが可能である。そのような成分として、限定されるものではないが、変性剤、流動促進剤、圧縮補助剤(compression aid)、崩壊剤、滑沢剤、バインダー、香料、調味料、甘味料、および防腐剤が挙げられる。
【0028】
適当な滑沢剤として、ステアリン酸、ステアリン酸マグネシウム、タルク、ステアリン酸カルシウム、水素添加された植物油、安息香酸ナトリウム、ロイシン・カーボワックス、マグネシウム・ラウリル硫酸塩、コロイド状二酸化ケイ素、およびモノステアリン酸グリセリンが挙げられる。適当な流動促進剤として、コロイダルシリカ、ヒュームド二酸化珪素、二酸化ケイ素、タルク、ヒュームドシリカ、石膏、およびモノステアリン酸グリセリンが挙げられる。
【0029】
この発明にもとづいて、標準の経口単位投薬形態を調製するための従来手段のいずれかを用いることができる。錠剤を形成する際に、結晶多形相B状態にあるビシファジン塩酸塩から形成された錠剤を生産するために従来の手段によって、配合物を圧縮することができる。本明細書で用いられる「錠剤」という用語は、コーティングまたは非コーティングのいずれでも全てのサイズの圧縮された医薬投薬形態を包含することを意図している。コーティングに用いることができる物質として、ヒドロキシプロピルセルロース、酸化チタン、タルク、甘味料、および着色剤が挙げられる。
【発明を実施するための最良の形態】
【0030】
【実施例1】
【0031】
ラセミ体ビシファジン塩酸塩の調製
300ガロンのリアクターに水(150L)と水酸化ナトリウム(100kg)とを充填し、その溶液を10±5℃に冷却した。第2の300ガロン・リアクターにクロロホルム(203kg)、ベンジルトリエチルアンモニウム・クロリド(8.2kg)、および4−トルアルデヒド(99kg)を充填した。反応混合物を加熱して穏やかに還流し、水酸化ナトリウム溶液を還流が維持する率で加えた。添加は、約6時間かかったが、その後、還流を少なくともさらに3時間連続しておこなった。熱い反応溶液を500Lの冷水(5℃)に添加し、混合物を約15分間にわたって撹拌した。相分離が可能となった。下側の有機層を保持タンクに移した。水層を、未反応の4−トルアルデヒドいっさいを取り除くために、クロロホルム(1x72kg、続いて4x20kg)で洗った。後での再処理用に、混合有機相を保存した。水層を、濃HCl(48kg)でpH2まで酸性化し、4−メチル・マンデル酸を黄色粒状固形分として沈殿させた。スラリーを一晩撹拌した後、産物を濾過によって単離し、水(150L)で洗浄し、60℃で少なくとも24時間真空(in vacuno)乾燥した。このプロセスによって、約80kgの4−メチル・マンデル酸が得られた。上からのクロロホルム抽出物に対して塩化ベンジルトリエチルアンモニウム(約4.5kg)を添加し、混合物を還流させながら加熱する。反応混合物に対して、水酸化ナトリウム(50kg)を含んだ水(75L)からなる溶液を加えた。添加が完了(少なくとも1.5時間)した後、反応混合物を一晩還流させながら加熱した。熱い反応混合物を、250kgの水を含有するリアクターに、5±5℃で添加した。層を分離することが可能であり、下側の有機層を放棄した。水層をクロロホルム(1x36kg、続いて4x10kg)で洗浄した。水層のpHを濃塩酸で0.5ないし1.5に調整した。スラリーを25±5℃で一晩撹拌した。産物を濾過により単離し、水(50L)で洗浄し、少なくとも24時間、真空(in vacuno)中で乾燥させた。このことは、さらに34kgの4−メチルマンデル酸の生産をもたらした。
【0032】
塩化チオニル(179kg)を、ジメチルホルムアミド(730ml)含有トルエン(102kg)に含まれる4−メチルマンデリン酸(110kg)からなる混合物に、2時間にわたって添加した。供給終了後、混合物をさらに3時間、周囲温度で撹拌した。メタノール(362kg)を、ガス放出を制御する率で2時間にわたって添加した。過剰のメタノールを、大気圧で蒸留によって除去した。内部ポット温度が少なくとも85℃に到達するまで、蒸留を続けた。トルエン(577kg)を残留物に供給し、温度を50±5℃に調整した。水(152L)を添加し、混合物を15分間撹拌し、続いてさらに30分間静置させた。水層を捨て、トルエン層を10%の水性重炭酸ナトリウム(186kg)と水(169kg)とで連続的に洗浄した。含水量が0.1%未満となるまで、トルエン溶液を共沸蒸留によって乾燥させた。メチルアクリレート(55kg)を、45±5℃で、ラセミ体メチル2−クロロ−2−(p−トリル)酢酸塩の無水物トルエン溶液に添加した。ナトリウムメトキシド(32.1kg)を、3時間にわたって、等しく約16に分けて添加した。添加終了後、反応混合物をさらに3時間45±5℃で保持した。反応混合物を、5%の塩酸(215L)および水(169L)で連続的に洗浄した。トルエンを、95℃よりも高いポット温度まで、減圧蒸留によって除去した。残留物を50±5℃に冷やし、メタノール(137kg)と水(450L)とで処理した。水酸化カリウム(約39kg)を添加し、混合物を加熱して約6時間にわたって還流した。アルコールの除去は、最大ポット温度98ないし100℃まで、蒸留によっておこなった。結果として生じる溶液を25℃未満に冷やし、濃HCl(86kg)と水(150L)との混合物で、pH 1に酸性化した。混合物を撹拌した。沈殿物を濾過によって回収し、水(36L)でよく洗い、空気乾燥して粗1−(4−メチルフェニル)−1,2−シクロプロプランジカルボキシル酸を得た。
【0033】
粗1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシル酸(約184kg))を300ガロンのリアクター内で、周囲温度で水(900L)にスラリー化した。少なくともpH10が得られるまで、水酸化ナトリウムを部分的に加えた。溶液が得られた後、濃塩酸(19.8kg)を用心深く添加することで、pHを約5.5に調整した。25℃に冷却した後に、塩化メチレン(100kg)を加え、混合物を30分間混合し、さらに下層を捨てた。塩化メチレンを増量(100kg)してこの洗浄手順を繰り返した。つぎに、水溶液を濃塩酸(70kg)で、pH 1まで酸性化した。20〜25℃で約3時間撹拌した後、淡黄色固形分を濾過した。ケークを水(2x100L)で洗浄し、少なくとも24時間、60℃で真空(in vacuo)乾燥することで、約74kgの1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシル酸を得た。
精製された1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシル酸(58kg)、トルエン(465kg)、および尿素(23.8kg)の混合物を加熱して還流して、反応が完了するまで混合物を還流させながら保持した。反応時間が完了すると、溶液を80−90℃に冷やし、水(69L)で洗浄した。下側の水層を破棄した。このようにして、(±)−1−(4−メチルフェニル)−3−アザビシクロ[3.1.0]ヘキサン−2,4−ジオンを、トルエン中溶液として得た。
【0034】
トルエン溶液を還流させながら加熱して含水量が0.1%未満になるまで、共沸的に水を取り除いた。溶液を50±5℃まで冷やし、バイトライド(Vitride)T(186.6kg)をゆっくりと加えて泡立ちを抑えた(約60℃まで反応発熱)。添加終了後、反応が完了するまで、リアクターの内容物を95±5℃まで加熱し、反応が完了するまでその温度に保った。20±10℃まで冷却した後、水酸化ナトリウム(108kg)を含む水(418L)からなる溶液に、ゆっくりと加えた。40±10℃でのアジテーションを短時間行った後、層を分離し、下側の水層を捨てた。有機相を水(2x836L)で洗浄した。下側の水層を捨て、有機層を含水量が0.1%以下になるまで、共沸的に乾燥させた。20℃未満まで冷やした後に、水性抽出物のpHが6.5以下になるまで、乾燥トルエン溶液を無水塩酸で処理した。過剰の塩酸を窒素によるスパージングで取り除き、濾過による単離に先立ってスラリーを約10℃で少なくとも1時間撹拌した。スラリーはろ過された。ケークをアセトン(2x25L)で洗浄し、水分含有量が3%以下になるまで、真空(in vacuo)で乾燥させた。収率は、灰色かかった固晶として31.8kgの粗ビシファジン塩酸塩であった。
【0035】
粗ビシファジン塩酸塩をイソプロピルアルコール(490kg)に添加した。反応混合物を穏やかなに加熱して還流した。溶液を活性炭(2.4kg)とセライト(3.4g)とで処理した。溶液を1ないし2時間還流し、濾過によってセライトと炭素とを取り除き、さらに0.2μフィルターに通した。溶液を20℃以下に冷やして、終夜その温度に保った。産物を濾過によって単離し、冷イソプロピルアルコール(2X23 L)で洗浄し、最終的にアセトン(2x23L)で洗浄した。産物を50℃で真空(in vacuo)乾燥させて水分含有量を0.1%未満にし、白から灰色かかった固晶として、約24kgのビシファジン塩酸塩が得られた。以下、この固形分は実施例7ないし9で用いられた。
【実施例2】
【0036】
ラセミ体ビシファジン塩酸塩の調製
2本の50Lフラスコの各々に、12Lのトルエン、3000gのメチル−α−クロロ−p−トリルアセテート、および1300gのメチルアクリレートを充填した。鉱油中60%の水酸化ナトリウム(725g)を、各々のフラスコに加えた、結果として得られたスラリーを約30分間、室温で撹拌した。反応混合物を−10℃に冷やした。温度を約−10℃に保つ一方で、636mlのメタノールを1.2Lのメチル−t−ブチルエーテルとともに各フラスコに添加した。反応混合物を室温で最低12時間撹拌した。反応混合物が灰色のスラリーから澄んだオレンジ色の溶液となることを観察し、出発原料が5%以下だったことを確認した後に、1.6Lの水を各々のフラスコに加えた。混合物を、さらに10分間撹拌した。両方の反応混合物を一つの40L分液ロートで混合し、水層および有機層を分離した。各々の層を、2つの5ガロン・バケツのうちの1つに排出した。水層を分液ロートに注ぎ戻し、酢酸エチルエステル(2X2 L及び1X1 L)で抽出した。水層を捨てた。複数の有機層を混合し、分液ロートに戻して1Lの水で洗った。有機層を2つの5ガロン・バケツに等量排出した。各々のバケツに、250gの炭と250gの硫酸マグネシウムとを加え、混合物が十分に混ざり合うまで撹拌した。材料を濾過し、酢酸エチルエステル(2x500mL)で洗浄した。反応混合物を、60℃、ブチ・ロタバポア(Buchi Rotavapor)で淡黄色の油に濃縮した。油性溶液を22Lのフラスコに移して、冷浴に入れた。撹拌している間、10Lのエーテルを添加し、固形分が沈殿し始めるまで溶液を0℃に冷やした。つぎに、溶液を−20℃未満に冷やして約1.5時間撹拌した。ポリプロピレン・フイルター・パッドを用い、固形分を18”クロック・フィルター上で濾過し、エチルエーテル(3x2L)で洗浄し、パイレックス乾燥トレイに移した。ジメチル−1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシレートを真空中(in vacuo)25℃で乾燥させた。
【0037】
2本の22Lのフラスコの各々に、9Lの水、2250gのジメチル−1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシレート、および2.25Lのエチルアルコールを充填した。50%の水酸化ナトリウム溶液(1.2L)を各々のフラスコに加えて、反応混合物を加熱して還流し、約1時間15分間、還流状態に保った。ジメチル−1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシレートが5%以下であったことを確認した後に、反応混合物を約75℃で撹拌した。両方の反応混合物を、一本の30ガロン・クロックに移した。氷を添加し、反応混合物を10℃以下に冷やした。4Lの濃塩酸を用いて混合物をpH1まで酸性化させ、最低30分間撹拌した。フィルタ・パッドを用いて固形分をクロック・フィルターで濾過した。濾過ケークを切り離して12Lの酢酸エチルに入れて層の分離をおこなった。有機層を硫酸マグネシウムで乾燥させ、ブフナー漏斗で濾過しながら22Lのフラスコに入れた。ブチ・ロタバポア(Buchi Rotavapor)を用いて、材料を濃縮して8Lの容量にし、10Lのヘキサンで希釈し、撹拌しながら0℃に冷却した。固形分を濾過し、ヘキサン(3x2L)で洗浄した。1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシル酸をパイレックス乾燥トレイに移し、最低12時間、真空中(in vacuno)35℃で乾燥させた。
【0038】
2本の22Lフラスコの各々に、14Lの2−メトキシエチル・エーテル[ダイグライム]、1750gの1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシル酸、および716gの尿素を充填した。反応混合物を、少なくとも12時間、約155℃まで加熱した。出発原料が5%以下であったことを確認した後に、反応混合物を約40Kgの氷を含む55ガロン・クロックに移し、室温に冷却した。この混合物を、総容量100Lになるまで水で希釈し、約4時間撹拌した。混合物を、ポリプロピレン・フィルタ・パッドを用いてクロック・フィルター(18”)で濾過した。固形分を水(3x2L)で洗浄し、5ガロン・バケツに移し、12Lの酢酸エチルエステルに溶解した。溶液を40Lの分液ロートに注ぎ込み、層の分離をおこなった。水層および有機層を各々、2つの5ガロン・バケツの1つに排出し、水溶液を分液ロートに注ぎ戻し、酢酸エチルエステル(2x1L)で抽出した。水層を捨て、複数の有機層は混ぜ合わせた。炭(500g)と硫酸マグネシウム(500g)とを有機層に添加し、結果として生ずる混合物を十分混合されるまで撹拌した。溶液を、ワットマン(Whatman)GFフィルター・パッドを用いてブフナー漏斗(18.5cm)で濾過し、固形分を酢酸エチルエステル(2x500mL)で洗浄した。ブチ・ロタバポア(Buchi Rotavapor)を用いて、濾過液を約60℃の浴槽温度で、最初の結晶が見えるまで、または総容量5Lが達成されるまで濃縮した。この濃縮から得られた残留物を5ガロン・バケツに移して撹拌した。スラリーを12Lのヘキサンで希釈して、室温で2時間撹拌した。固形分を、ポリプロピレン・フィルター・パッドを用いたクロック・フィルター(18”)で濾過し、ヘキサン(3x2L)で洗浄した。1−(4−メチルフェニル)−3−アザビシクロ[3.1.0]ヘキサン−2,4−ジオンをパイレックス乾燥トレイに移し、真空中(in vacuo)35℃で最低12時間乾燥させた。
【0039】
2本の50Lフラスコの各々に、12Lのトルエンおよび1250gの1−(4−メチルフェニル)−3−アザビシクロ[3.1.0]ヘキサン−2,4−ジオンを充填した。ナトリウム・ビス(2−メトキシエトキシ)アルミニウム・ハイドライド(5700mLのRedAl)を各々のフラスコに添加し、反応混合物を3時間加熱して還流した。出発原料が5%以下であったことを確認した後に、反応混合物を氷/水浴を用いて60℃未満に冷やした。30ガロン・クロックに、氷/水混合物(6.4kg水、4.4kg氷)を2.2Lの水酸化ナトリウム、50%溶液と共に充填し、氷を加えることで温度を約0℃に調整した。この混合物を撹拌して、両反応混合物をそれに注ぎ込んだ。氷を加えることで、温度を20℃未満に下げて、結果として得られる混合物を最低2時間撹拌した。溶液を40L分液漏斗に移した。層を分離し、各層を2つの5ガロン・バケツの1つに排出した。水層を酢酸エチルエステル(3x2L)で抽出して捨て、複数の有機抽出物を混ぜ合わせ、水(3x2L)で洗浄した。水洗浄液を捨てた。6Lの水を含む2ガロンのバケツに、溶液が飽和(塩水)するまで塩化ナトリウムを添加した。有機抽出物を塩水(3x2L)で洗浄し、該水洗浄液を捨てた。有機層を、複数の5ガロン・バケツに等量排出した。250gの炭と250gの硫酸マグネシウムとを各々のバケツに加えて、結果として生じる混合物が十分に混ざり合うまで混合した。これらの溶液をワットマン(Whatman)GFフィルター・パッドを用いてブフナー漏斗(18.5cm)で濾過した。固形分を酢酸エチル(2X500 mL)で洗浄し、濾過液をブチ・ロタバポア(Buchi Rotavapor)で、約60℃の温度で総量約12Lまで濃縮した。20℃未満の温度を保つ一方で、pH2〜3に達するまで反応混合物に無水HClガスをバブリングした。混合物を10℃未満でさらに2時間混合し、その後、12時間以上、フリーザーに置いた。固形分を、ポリプロピレン・フィルター・パッドを用いたクロック・フィルター(18”)で濾過し、ヘキサン(3x2L)、さらにアセトン(3x2L)で洗浄した。粗ビ
シファジン塩酸塩をパイレックス乾燥トレイに移し、最低2時間乾燥させた。
【0040】
4本の22Lフラスコの各々に、約1500gの粗ビシファジン塩酸塩を充填した。イソプロピルアルコール(6−10mL/g)を添加し、材料を撹拌および加熱して還流した。いったん、還流で透明な液体が達成されると、200gの炭を各々のフラスコに添加した。溶液を約10分間撹拌し、GFフィルターが載せられたポリプロピレン・パッドを用いたブフナー漏斗(10”)で濾過して、20Lフィルター・ボトルに入れた。結果として生じる濾過液は、30ガロンPEクロックへ移し、室温で3時間撹拌した。クロックを、最低12時間低温室に置いた。固形分を、ポリプロピレン・フィルタ・パッドを用いたクロック・フィルター(18”)上で濾過し、アセトン(4x2L)で洗浄した。1%未満の乾燥減量が達成されるまで、ステップの産物であるビシファジン塩酸塩を真空中(in vacuo)50℃で乾燥した。この物質を実施例6で使用した。
【実施例3】
【0041】
ビシファジン塩酸塩多形相Bへの転換
多形相Aと多形相Bとの混合物としてのラセミ体ビシファジン塩酸塩を、スラリーを形成するのに十分な量でイソプロピル・アルコールに添加した。スラリーを30℃未満の温度で混合等のアジテーションにかけた。1日後、生成物を濾過によって単離し、乾燥減量が1%未満まで達成されるまで、真空中50℃で乾燥した。生産された材料はビシファジン塩酸塩多形相Bであった。
【実施例4】
【0042】
ビシファジン塩酸塩多形相Bへの転換
多形相AおよびBの混合物としての20グラムのラセミ体ビシファジン塩酸塩を、60mlのイソプロピルアルコールに加えてスラリーにした。このスラリーを約30℃の温度で24時間撹拌した。生成物を濾過して単離し、真空中(in vacuo)乾燥した。生産された材料は、検出可能な多形相Aの存在が認められないビシファジン塩酸塩多形相Bであった。
【実施例5】
【0043】
実施例5において、米国特許第4,231,935号の実施例36で述べられるように、その多形結晶の形状であるビシファジン塩酸塩を、アセトニトリル/メタノールから調製および結晶化した。試料(すなわち、表5の試料1〜5)のビシファジン塩酸塩を以下に説明するように蒸発することで、再結晶化させた。
【0044】
蒸発
以下の容量−容量混合物のアセトニトリル−メタノールを調製した。すなわち、9:1、2:1、1:1、1:2,および1:9.透明な溶液が生ずるまで、計量された量のA形ビシファジン塩酸塩(約90mg)を溶媒混合物の一定分量で処理した。各溶液を0.2ミクロン・ナイロン・シリンジで濾過してきれいなバイアルに入れた。ピンホールによる穿孔が形成されたアルミホイルでバイアルを覆い、ヒューム・フードの下に置いて蒸発を可能にした。溶媒が完全に蒸発すると、固形分が回収された。
【0045】
得られる多形に関する結果を表5に示す。これらの結果は、X線粉末回折技術(XRPD)を用いて得られた。
【0046】
【表5】
【0047】
表5の場合のように、上記手順にもとづく米国特許第4,231,935号の実施例36の製品の単純な再結晶化は、蒸発によってビシファジン塩酸塩多形相Aを生産した。
【実施例6】
【0048】
A形の調製
実施例2で調製したように、ビシファジン塩酸塩(20.1g)を加熱してイソプロピルアルコール(365mL)に撹拌し、80℃で澄んだ溶液が得られた。溶液を55℃で冷やすことで、スラリーが得られた。製品を濾過して回収し、周囲温度で乾燥させることで、A形ビシファジン塩酸塩が得られた。
【実施例7】
【0049】
A形の調製
一定量のエタノール(1mL)をビシファジン塩酸塩(97mg)(実施例1での固晶として生産される)で飽和し、80℃に設定した加熱撹拌プレート上に置かれたバイアルに入れた。溶液を濾過して、−20℃に冷やされた1mLトルエン含有のバイアルに、直接入れた。混合物を15分間、−20℃フリーザーに入れることで、沈殿が生じた。製品を濾過して回収し、周囲温度で減圧下、乾燥することで、A形ビシファジン塩酸塩が得られた。
【実施例8】
【0050】
B形の調製
実施例1で固晶として生産される、A形ビシファジン塩酸塩(3.99g)を加熱して、イソプロピルアルコール(50mL)で撹拌することで、82℃で澄んだ溶液が得られた。溶液を周囲温度に冷やすことで、厚いスラリーが得られた。撹拌機構をシャット・オフし、混合物を2日間、周囲温度に設定した。固形分(白色から灰色かかったブレード)を回収し、空気乾燥することで、B形ビシファジン塩酸塩が得られた。
【実施例9】
【0051】
多形相Bの調製
実施例1で固晶として生産される、A形ビシファジン塩酸塩(約1g)を小さなステンレス・スチール・シリンダーに充填した。ステンレス・スチール棒を加えてシリンダーをキャッピングした。シリンダーを液体窒素が充填されたSPEX/Centriprep 6750型フリーザー・ミルに取り付けた。6分間のグラインディング時間について、試料を一度に2分間挽いて2分間の休憩時間をとった。タンクに液体窒素を満たし、上記周期を繰り返し、グラインディング合計時間を12分とした。プロセスは、A形ビシファジン塩酸塩がまったく検出されることなくB形ビシファジン塩酸塩に対する完全な転換に帰着した。
【実施例10】
【0052】
グラインディングでのA形の不安定性
A形ビシファジン塩酸塩(207mg)を小さなステンレス・スチール・シリンダーに充填した。ステンレス・スチール・ボールを加え、シリンダーをキャッピングし、全体で20分間にわたり周期30/sで5分間の間隔で、レッシュ・ミキサー・ミル(Retsch Mixer Mill)(MM200型)上で上記ユニットを振とうさせた。挽いた材料を除去し、XRPDで分析した。プロセスは、B形ビシファジン塩酸塩への部分的転換をもたらした。
【実施例11】
【0053】
グラインディングでの多形相Bの安定性
B形ビシファジン塩酸塩(111mg)を小さなステンレス・スチール・シリンダーに充填した。ステンレス・スチール・ボールを加え、シリンダーをキャッピングし、全体で20分間にわたり周期30/sで5分間の間隔で、レッシュ・ミキサー・ミル(Retsch Mixer Mill)(MM200型)上で上記ユニットを振とうさせた。挽いた材料を除去し、XRPDで分析した。挽かれた固形分は、B形のままだった。
【図面の簡単な説明】
【0054】
【図1】実施例9で調製される多形相Bラセミ体ビシファジン塩酸塩の赤外線スペクトラムである。
【技術分野】
【0001】
ビシファジン塩酸塩の新規多形結晶に関わる。より詳しくは、多形相Aと称するビシファジン塩酸塩の既知の多形相よりも、熱力学的に安定な多形相Bに関する。
【背景技術】
【0002】
以下の式からなる塩酸付加塩であるビシファジン塩酸塩は、非麻薬性鎮痛薬(すなわち、作用がモルヒネ様ではない)である。特許文献1および特許文献2を参照せよ。
【0003】
【化1】
ビシファジン塩酸塩(bicifadine hydrochloride)、その化学名は、(±)−1−(4−メチルフェニル)−3−アザビシクロ[3.1.0]−ヘキサン塩酸塩であり、その同義語は、ラセミ体1−(p−トリル)−3−アザビシクロ[3.1.0]−ヘキサン塩酸塩であり、淡黄褐色プレート状(pale tan plates shaped)結晶として特許文献1の実施例36に記載の通りに製造される。この製品は、1−(p−トリル)−1,2−シクロプロパンジカルボキシミドから生産されていた。1−(p−トリル)−1,2−シクロプロパンジカルボキシミドを有機溶媒に溶かし、アミンに還元して塩酸塩に変換することで、粗ビシファジン塩酸塩の沈殿を生じさせる。粗ビシファジン塩酸塩を濾過によって反応媒体から回収した。次に、粗ビシファジン塩酸塩をアセトニトリル/メタノール混合物から再結晶化して黄褐色プレート形状の結晶を得る。
【0004】
ビシファジン塩酸塩の結晶形は、患者の疼痛に対して錠剤またはカプセル等として種々の経口単位投薬形態で処方されることから、特に重要である。医薬品物質の結晶構造における変化は、医薬品の溶解性、製造性、および安定性、特に固形経口投薬形態の処方で影響を及ぼす可能性がある。したがって、単一の熱力学的に安定な結晶構造からなる純粋な形状でビシファジン塩酸塩を生産することは、重要である。上記の手順に従って生産されるビシファジン塩酸塩結晶構造が、淡黄褐色プレートとして、最も熱力学的に安定な多形相(polymorphic form)でないと判断されている。さらにまた、従来の製造プロセス、例えばグラインディングおよびミリングにかける場合、この多形相(以下、多形相Aという)のビシファジン塩酸塩が、異なる多形相への転換を受けることが示されている。多形相Aがビシファジン塩酸塩で最も熱力学的に安定した形状ではないことから、多形相Aは時間とともに多形転換を起こしうる。したがって、多形相Aは、医薬品への製剤化にとってビシファジン塩酸塩の最適結晶形でない。
【特許文献1】米国特許第4,231,935号
【特許文献2】米国特許第4,196,120号
【発明の開示】
【発明が解決しようとする課題】
【0005】
この発明に従って、我々は結晶ビシファジン塩酸塩に2種類の多形相が存在することを発見し、また米国特許第4,231,935号で生産された結晶形が、淡黄褐色プレートとして、ビシファジン塩酸塩の2種類の多形相のうちの1つであるということを発見した。この多形相は、多形相Aとして表す。さらに、この発明に従って、我々は、従来の製剤加工操作、例えば、グラインディングおよびミリングを受ける場合、結晶がよりいっそう熱力学的に安定していて多形転換を受けない、ビシファジン塩酸塩のもう一つの新しい結晶多形構造が存在するということを発見した。この新しい多形相は、多形相Bと称する。
【0006】
この発明にもとづいて、われわれは多形相Bと称する、ビシファジン塩酸塩の新規多形結晶を発見した。この多形相Bは、多形相Aと称するビシファジン塩酸塩の既知の多形相よりも熱力学的に安定な錠剤である。淡黄褐色プレートの形状である多形相Aの結晶とは異なり、ビシファジン塩酸塩多形相Bの結晶は、白色ないし灰色がかった(off-white)の色を呈するブレード状である。
【課題を解決するための手段】
【0007】
ビシファジン塩酸塩の多形を、赤外線スペクトラムおよび/またはX線粉回折パターンによって特徴づけることが可能である。所定の多形のX線粉回折ピークの相対強度は、パターンの測定に用いる粒度に応じて変化しうる。このことは、試料ホルダー内の結晶の好ましい配向の現象である。しかし、この発明のビシファジン塩酸塩の所定の多形相Bによって、このパターン内に粒度にかかわらず典型的に存在する特定のピークがある。一方で、所定の多形、例えば、この発明のビシファジン塩酸塩の多形相Bの赤外線スペクトラムは、粒度にかかわりなく比較的一定のままである。
【0008】
ラセミ体ビシファジン塩酸塩の多形相AおよびBのX線粉末回折(XRPD)による分析は、CuKα線を用いた島津製作所のXRD−6000粉末回析カメラで実行された。ビシファジンは、結晶性粉末としてその機器に供給された。その機器は、高精度焦点X線管を具備していた。管電圧およびアンペア数をそれぞれ40kVおよび40mVに設定した。ビームの開きおよび散乱スリットを1度に設定し、受け側スリットを0.15mmに設定した。回折された放射線の検出は、ヨウ化ナトリウム・シンチレーション検出器によっておこなった。2.5ないし40°2θの3°/分(0.4秒/0.02°ステップ)シータ−2シータ連続走査を使用した。シリコン標準物質を分析して、機器のアライメントを調べた。データを収集して、XRD−6000バージョン4.1で分析した。
【0009】
ラセミ体ビシファジン塩酸塩の多形相BのX線粉末回折パターンは「d」間隔に換算して与えられ、相対強度(I)は以下の通りであり(s=強、m=中程度、w=弱、v=極めて、d=拡散)、さらにこれらの条件は下記の表1で述べられる。
【0010】
【表1】
【0011】
ビシファジン塩酸塩のA形のX線粉末回折パターンを、以下のように表1の場合と同様の用語で表2に示す。
【0012】
【表2】
【0013】
表1および表2は、それぞれ粒度を下げたビシファジン塩酸塩B形およびA形のピーク位置でのXRPDパターンを表す。これらの表に示す結果は、減少粒度での多形相Aおよび多形相BのXRPDパターン間の差を示す。しかし、このパターンでは重要な主要ピークが所定の角度にあり、所定の角度はビシファジン塩酸塩の所定のB形に特有なもので、その粒度にかかわりなく多形相BのXRPDパターンで、典型的に存在する。2θ(度)として表され、かつCuKα線を用いて多形相Bが特徴づけられる主要ピークが位置づけられているこれらの角度は、5.08、10.07、20.16、25.17、および30.43である。
【0014】
各々の試料について、Ever−Glo 中/遠IR源、拡張範囲臭化カリウム(KBr)ビームスプリッタ、および重水素化硫酸トリグリシン(DTGS)検出器を備えたMagna−IR860(登録商標)フーリエ変換赤外(FT−IR)分光光度計(トーマス・ニコレ(Thomas Nicolet))を用いて、赤外線スペクトルを得た。分光光度計は、所定の波長で試料の各々の赤外光帯の強度を測定した。拡散反射率アクセサリ(コレクター(Collector)(商標)、サーモ・スペクトラ・テク(Thermo SpectraTech))をサンプリングに使用した。各スペクトルは、スペクトラム解像度4cm-1で400から4,000cm-1までの範囲で収集した256回の共付加走査(co−added scans)を表す。試料調製は、多形相Aまたは多形相Bのいずれかである結晶を含む粉末試料を、13mm径カップに入れて、該材料を曇り(frosted)スライドガラスでならすことからなった。バックグラウンド・データ・セットは、所定位置に置かれたアラインメント・ミラーによって得た。反射率Rは、所定の波数における試料の光強度/バックグランド・セットの光強度である。図1は、多形相Bの赤外線スペクトラムを示すもので、横座標は波数cm-1、縦座標はLog(1/R)である。Log 1/R(R=反射率)スペクトルは、各々に対してこれらの2つのデータ・セット(試料およびバックグラウンド光強度)の比を取ることで得た。乾燥結晶粉末としてのラセミ体ビシファジン塩酸塩の多形相Bの赤外線スペクトラムは、表3に示すように、この多形を特徴づける以下のピークを示した。
【0015】
【表3】
【0016】
乾燥結晶粉末状のラセミ体ビシファジン塩酸塩の多形Aの赤外線スペクトラムは、この多形を特徴づける以下の主ピークを示した。
【0017】
【表4】
【0018】
表3および表4は、それぞれビシファジン塩酸塩の多形相Bおよび多形相Aに関して赤外線のピークの位置の完全なパターンを提供する。しかし、特定のキー・ピークがあり、このパターン内では、ビシファジン塩酸塩の多形相Bに特有で、この多形を特徴づけるのに十分である。波数(cm-1)で表すこれらのピークは、2108、891、856、719、および660である。
【0019】
この発明に従って、我々は多形B結晶構造を持つビシファジン塩酸塩を形成する手段を見つけた。ラセミ体ビシファジン塩酸塩を調製する既知の方法は、多形相Aを生産する。なぜなら、それは最初に生産される多形結晶構造であるからである。多形相Aから多形相Bを作るための1つの手段は、通常生産される多形相Aに、運動エネルギーを付与することである。この運動エネルギーは、多形相Aに対して、特に低温度、一般には約−200℃ないし約50℃、好ましくは約−200℃ないし約35℃、より好ましくは−200℃ないし約0℃で印加される。この転換を実行するために、撹拌、グラインディング、またはミリング等の手段によって運動エネルギーを通常のビシファジン塩酸塩多形相Aに印加する方法のいずれも使用することができる。グラインディングまたはミリングを、室温で適用することができる。しかし、この転換は、運動エネルギーを使用するもので、低温でより効率的に実施される。例えば、多形相Aを多形相Bに転換するための運動エネルギー法は、多形Aの固晶を用いて、該結晶に撹拌またはグラインディングを適用する一方で、温度を約35℃以下、一般には約−200℃ないし0℃に保つことである。液体窒素雰囲気下で、グラインディング等によって運動エネルギーを供給している間、低温が利用される。
【0020】
多形相Aを多形相Bに変換するもう一つの方法は、加熱された溶液から多形相Bを結晶化させ、上記多形Bを形成するのに充分な時間にわたって、上記溶液を冷却する。多形相A結晶は、通常、室温では有機溶剤に十分に溶解しない。しかし、加熱されると、それは可溶性になる。この転換にもとづいて、ビシファジン塩酸塩の多形相A結晶を少なくとも50℃の沸点を持つ有機溶媒と混合し、スラリーを形成させる。この転換を行う際に、少なくとも50℃の沸点を持つ従来の有機溶媒のいずれも利用することができる。つぎに、スラリーが透明な溶液となる温度まで、上記スラリーを加熱する。この溶液を、最大で約35℃、好ましくは約0℃ないし約20℃の温度まで冷やす。冷却された溶液を、多形相Aを含まない純粋な結晶形としての上記多形相Bが形成されるのに十分なしばらくの間、最大で約35℃の温度に保つ。必要に応じて、撹拌しながら冷却をおこなう。冷却している間、混合物から結晶の多形Bが形成されるのに十分な時間をかけるように、モニターしていなければならない。この手順では、冷却するにつれて、多形相Bの「種結晶」を加えることによって転換の時間もまた促進される。
【0021】
また、多形相Bの生産は、ビシファジン塩酸塩A形またはビシファジン塩酸塩A形とB形との混合物から、A形またはA形とB形との混合物のいずれかとして、不活性有機溶媒中にビシファジン塩酸塩のスラリーを形成し、最大で約35℃の温度でスラリーを撹拌することによって、おこなうことができる。スラリーの形成の際に、従来の不活性有機溶媒のいずれかを用いることができる。アジテーションは、ビシファジン塩酸塩を多形相Bに変換するのに十分な時間、実施される。この期間を24時間とすることができるだろう。この時間は、多形相Bの「種」結晶を用いることによって、および/または温度を35℃未満にすることで、短くすることができると考えられる。
【0022】
上記方法のいずれかによって生産された多形相Bは、XRPDまたはInfra−Redによって測定されるように、ビシファジン塩酸塩の他の多形相がなんら含まれない純粋な形状であることができる。このように、ビシファジン塩酸塩の多形相Bの純粋なラセミ体は、ラセミ体ビシファジン塩酸塩のその他の多形相が存在することなく生産される。
【0023】
ビシファジン塩酸塩の多形相Bを、疼痛を抑えるためにヒト患者に投与することができる。これは、多形相Bの結晶構造と不活性担体または希釈剤とを持つビシファジン塩酸塩を含有する組成物による処置を必要とする、上記疼痛で苦しんでいる患者を処置することによって達成され、該組成物は疼痛緩和に有効な量で投与される。この発明によれば、結晶多形相B状態のラセミ体ビシファジン塩酸塩を、疼痛緩和に有効な量で投与する。疼痛緩和に必要とされる任意の有効量のビシファジン塩酸塩を、この組成物で利用することができる。一般に、1日あたり約0.5mg/kgないし約20mg/kgの経口量が用いられる。しかし、投与される経口単位服用量の結晶多形相Bラセミ体ビシファジン塩酸塩の量は、大部分は疼痛の量と患者の体重とに依存し、もちろん医師の判断にゆだねられる。本発明によれば、経口単位投薬形態の結晶多形相Bラセミ体ビシファジン塩酸塩が1日あたり1ないし2回または必要に応じて、25ないし600mgの服用量で投与される。約60kgないし約80kgの患者では、100mgないし600mgを含む単位経口投薬形態を用いることができ、一般に好ましくは200ないし400mgの服用量が用いられる。1日あたり1ないし2回または必要に応じて、経口単位服用量を投与することができる。疼痛が少なく、体重が60kg未満の患者に対しては、約25mgないし約200mgを含む経口単位投薬形態を、患者の要求に応じて1日あたり1ないし2回以上投与する。
【0024】
この発明の組成物では、従来の医薬的に許容される担体または希釈剤のいずれかを使用することができる。経口投与を目的として、医薬組成物は、従来の賦形剤によって調製された従来の経口単位投薬形態、例えば錠剤、カプセル、粉体、溶液、シロップ、または懸濁液のいずれかであってもよい。したがって、組成物の経口投与は、錠剤またはカプセル、ロゼンジ等、従来の方法で処方される形態を取ることが可能である。これらの製剤の各々は、その結晶多形相Bの形態で、ラセミ体ビシファジン塩酸塩を含有する。通常、これらの製剤で結晶多形相A状態にあるビシファジン塩酸塩が全く存在しない状態で、多形相Bビシファジン塩酸塩を用いることが好ましい。多形相Bは、ビシファジン塩酸塩の熱力学的に安定な多形であり、製造の過程および医薬品製剤の予想された商業的な有効期間を超えて、結晶間転換を受けることはない。
【0025】
必要に応じて、その結晶多形相Bのラセミ体ビシファジン塩酸塩は、経口単位投薬形態で親水性徐放高分子(ヒドロキシプロピル・メチルセルロース)の使用によって、徐放多形相で投与されうる。任意の親水性徐放高分子(例えば、約100cpsないし約100,000cpsの範囲の粘度を持つヒドロキシプロピル・メチル・セルロース・ポリマー)を用いることができる。
【0026】
この発明に従って、経口単位投薬形態の構成は、担体を含む可能性がある。薬学的製剤技術に共通の適当な担体として、限定されるものではないが、微結晶性セルロース、ラクトース、スクロース、フルクトース、グルコースデキストロース、または他の糖類、二塩基リン酸カルシウム、硫酸カルシウム、セルロース、メチルセルロース、セルロース誘導体、カオリン、マンニトール、ラクチトール、マルチトール、キシリトール、ソルビトール、又はその他の糖アルコール、乾燥澱粉、デキストリン、マルトデキストリン、または他の多糖類、イノシトール、またはそれらの混合物が挙げられる。
【0027】
この発明用の好ましい単位経口の剤形は、錠剤である。医薬経口単位投薬形態を調製する従来方法のいずれも、この発明の単位剤形を調製する際に利用することができる。医薬経口単位投薬形態(例えば錠剤)は、従来の付加的な配合剤の一つ以上を含む。これらの成分は、医薬製剤技術で既知の幅広く多様な賦形剤から選択される。経口投薬形態の所望の特性によれば、いくつかの成分を、単独で、または組みになって、錠剤としてのそのような投薬形態を調製する際の既知の使用のために、選択することが可能である。そのような成分として、限定されるものではないが、変性剤、流動促進剤、圧縮補助剤(compression aid)、崩壊剤、滑沢剤、バインダー、香料、調味料、甘味料、および防腐剤が挙げられる。
【0028】
適当な滑沢剤として、ステアリン酸、ステアリン酸マグネシウム、タルク、ステアリン酸カルシウム、水素添加された植物油、安息香酸ナトリウム、ロイシン・カーボワックス、マグネシウム・ラウリル硫酸塩、コロイド状二酸化ケイ素、およびモノステアリン酸グリセリンが挙げられる。適当な流動促進剤として、コロイダルシリカ、ヒュームド二酸化珪素、二酸化ケイ素、タルク、ヒュームドシリカ、石膏、およびモノステアリン酸グリセリンが挙げられる。
【0029】
この発明にもとづいて、標準の経口単位投薬形態を調製するための従来手段のいずれかを用いることができる。錠剤を形成する際に、結晶多形相B状態にあるビシファジン塩酸塩から形成された錠剤を生産するために従来の手段によって、配合物を圧縮することができる。本明細書で用いられる「錠剤」という用語は、コーティングまたは非コーティングのいずれでも全てのサイズの圧縮された医薬投薬形態を包含することを意図している。コーティングに用いることができる物質として、ヒドロキシプロピルセルロース、酸化チタン、タルク、甘味料、および着色剤が挙げられる。
【発明を実施するための最良の形態】
【0030】
【実施例1】
【0031】
ラセミ体ビシファジン塩酸塩の調製
300ガロンのリアクターに水(150L)と水酸化ナトリウム(100kg)とを充填し、その溶液を10±5℃に冷却した。第2の300ガロン・リアクターにクロロホルム(203kg)、ベンジルトリエチルアンモニウム・クロリド(8.2kg)、および4−トルアルデヒド(99kg)を充填した。反応混合物を加熱して穏やかに還流し、水酸化ナトリウム溶液を還流が維持する率で加えた。添加は、約6時間かかったが、その後、還流を少なくともさらに3時間連続しておこなった。熱い反応溶液を500Lの冷水(5℃)に添加し、混合物を約15分間にわたって撹拌した。相分離が可能となった。下側の有機層を保持タンクに移した。水層を、未反応の4−トルアルデヒドいっさいを取り除くために、クロロホルム(1x72kg、続いて4x20kg)で洗った。後での再処理用に、混合有機相を保存した。水層を、濃HCl(48kg)でpH2まで酸性化し、4−メチル・マンデル酸を黄色粒状固形分として沈殿させた。スラリーを一晩撹拌した後、産物を濾過によって単離し、水(150L)で洗浄し、60℃で少なくとも24時間真空(in vacuno)乾燥した。このプロセスによって、約80kgの4−メチル・マンデル酸が得られた。上からのクロロホルム抽出物に対して塩化ベンジルトリエチルアンモニウム(約4.5kg)を添加し、混合物を還流させながら加熱する。反応混合物に対して、水酸化ナトリウム(50kg)を含んだ水(75L)からなる溶液を加えた。添加が完了(少なくとも1.5時間)した後、反応混合物を一晩還流させながら加熱した。熱い反応混合物を、250kgの水を含有するリアクターに、5±5℃で添加した。層を分離することが可能であり、下側の有機層を放棄した。水層をクロロホルム(1x36kg、続いて4x10kg)で洗浄した。水層のpHを濃塩酸で0.5ないし1.5に調整した。スラリーを25±5℃で一晩撹拌した。産物を濾過により単離し、水(50L)で洗浄し、少なくとも24時間、真空(in vacuno)中で乾燥させた。このことは、さらに34kgの4−メチルマンデル酸の生産をもたらした。
【0032】
塩化チオニル(179kg)を、ジメチルホルムアミド(730ml)含有トルエン(102kg)に含まれる4−メチルマンデリン酸(110kg)からなる混合物に、2時間にわたって添加した。供給終了後、混合物をさらに3時間、周囲温度で撹拌した。メタノール(362kg)を、ガス放出を制御する率で2時間にわたって添加した。過剰のメタノールを、大気圧で蒸留によって除去した。内部ポット温度が少なくとも85℃に到達するまで、蒸留を続けた。トルエン(577kg)を残留物に供給し、温度を50±5℃に調整した。水(152L)を添加し、混合物を15分間撹拌し、続いてさらに30分間静置させた。水層を捨て、トルエン層を10%の水性重炭酸ナトリウム(186kg)と水(169kg)とで連続的に洗浄した。含水量が0.1%未満となるまで、トルエン溶液を共沸蒸留によって乾燥させた。メチルアクリレート(55kg)を、45±5℃で、ラセミ体メチル2−クロロ−2−(p−トリル)酢酸塩の無水物トルエン溶液に添加した。ナトリウムメトキシド(32.1kg)を、3時間にわたって、等しく約16に分けて添加した。添加終了後、反応混合物をさらに3時間45±5℃で保持した。反応混合物を、5%の塩酸(215L)および水(169L)で連続的に洗浄した。トルエンを、95℃よりも高いポット温度まで、減圧蒸留によって除去した。残留物を50±5℃に冷やし、メタノール(137kg)と水(450L)とで処理した。水酸化カリウム(約39kg)を添加し、混合物を加熱して約6時間にわたって還流した。アルコールの除去は、最大ポット温度98ないし100℃まで、蒸留によっておこなった。結果として生じる溶液を25℃未満に冷やし、濃HCl(86kg)と水(150L)との混合物で、pH 1に酸性化した。混合物を撹拌した。沈殿物を濾過によって回収し、水(36L)でよく洗い、空気乾燥して粗1−(4−メチルフェニル)−1,2−シクロプロプランジカルボキシル酸を得た。
【0033】
粗1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシル酸(約184kg))を300ガロンのリアクター内で、周囲温度で水(900L)にスラリー化した。少なくともpH10が得られるまで、水酸化ナトリウムを部分的に加えた。溶液が得られた後、濃塩酸(19.8kg)を用心深く添加することで、pHを約5.5に調整した。25℃に冷却した後に、塩化メチレン(100kg)を加え、混合物を30分間混合し、さらに下層を捨てた。塩化メチレンを増量(100kg)してこの洗浄手順を繰り返した。つぎに、水溶液を濃塩酸(70kg)で、pH 1まで酸性化した。20〜25℃で約3時間撹拌した後、淡黄色固形分を濾過した。ケークを水(2x100L)で洗浄し、少なくとも24時間、60℃で真空(in vacuo)乾燥することで、約74kgの1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシル酸を得た。
精製された1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシル酸(58kg)、トルエン(465kg)、および尿素(23.8kg)の混合物を加熱して還流して、反応が完了するまで混合物を還流させながら保持した。反応時間が完了すると、溶液を80−90℃に冷やし、水(69L)で洗浄した。下側の水層を破棄した。このようにして、(±)−1−(4−メチルフェニル)−3−アザビシクロ[3.1.0]ヘキサン−2,4−ジオンを、トルエン中溶液として得た。
【0034】
トルエン溶液を還流させながら加熱して含水量が0.1%未満になるまで、共沸的に水を取り除いた。溶液を50±5℃まで冷やし、バイトライド(Vitride)T(186.6kg)をゆっくりと加えて泡立ちを抑えた(約60℃まで反応発熱)。添加終了後、反応が完了するまで、リアクターの内容物を95±5℃まで加熱し、反応が完了するまでその温度に保った。20±10℃まで冷却した後、水酸化ナトリウム(108kg)を含む水(418L)からなる溶液に、ゆっくりと加えた。40±10℃でのアジテーションを短時間行った後、層を分離し、下側の水層を捨てた。有機相を水(2x836L)で洗浄した。下側の水層を捨て、有機層を含水量が0.1%以下になるまで、共沸的に乾燥させた。20℃未満まで冷やした後に、水性抽出物のpHが6.5以下になるまで、乾燥トルエン溶液を無水塩酸で処理した。過剰の塩酸を窒素によるスパージングで取り除き、濾過による単離に先立ってスラリーを約10℃で少なくとも1時間撹拌した。スラリーはろ過された。ケークをアセトン(2x25L)で洗浄し、水分含有量が3%以下になるまで、真空(in vacuo)で乾燥させた。収率は、灰色かかった固晶として31.8kgの粗ビシファジン塩酸塩であった。
【0035】
粗ビシファジン塩酸塩をイソプロピルアルコール(490kg)に添加した。反応混合物を穏やかなに加熱して還流した。溶液を活性炭(2.4kg)とセライト(3.4g)とで処理した。溶液を1ないし2時間還流し、濾過によってセライトと炭素とを取り除き、さらに0.2μフィルターに通した。溶液を20℃以下に冷やして、終夜その温度に保った。産物を濾過によって単離し、冷イソプロピルアルコール(2X23 L)で洗浄し、最終的にアセトン(2x23L)で洗浄した。産物を50℃で真空(in vacuo)乾燥させて水分含有量を0.1%未満にし、白から灰色かかった固晶として、約24kgのビシファジン塩酸塩が得られた。以下、この固形分は実施例7ないし9で用いられた。
【実施例2】
【0036】
ラセミ体ビシファジン塩酸塩の調製
2本の50Lフラスコの各々に、12Lのトルエン、3000gのメチル−α−クロロ−p−トリルアセテート、および1300gのメチルアクリレートを充填した。鉱油中60%の水酸化ナトリウム(725g)を、各々のフラスコに加えた、結果として得られたスラリーを約30分間、室温で撹拌した。反応混合物を−10℃に冷やした。温度を約−10℃に保つ一方で、636mlのメタノールを1.2Lのメチル−t−ブチルエーテルとともに各フラスコに添加した。反応混合物を室温で最低12時間撹拌した。反応混合物が灰色のスラリーから澄んだオレンジ色の溶液となることを観察し、出発原料が5%以下だったことを確認した後に、1.6Lの水を各々のフラスコに加えた。混合物を、さらに10分間撹拌した。両方の反応混合物を一つの40L分液ロートで混合し、水層および有機層を分離した。各々の層を、2つの5ガロン・バケツのうちの1つに排出した。水層を分液ロートに注ぎ戻し、酢酸エチルエステル(2X2 L及び1X1 L)で抽出した。水層を捨てた。複数の有機層を混合し、分液ロートに戻して1Lの水で洗った。有機層を2つの5ガロン・バケツに等量排出した。各々のバケツに、250gの炭と250gの硫酸マグネシウムとを加え、混合物が十分に混ざり合うまで撹拌した。材料を濾過し、酢酸エチルエステル(2x500mL)で洗浄した。反応混合物を、60℃、ブチ・ロタバポア(Buchi Rotavapor)で淡黄色の油に濃縮した。油性溶液を22Lのフラスコに移して、冷浴に入れた。撹拌している間、10Lのエーテルを添加し、固形分が沈殿し始めるまで溶液を0℃に冷やした。つぎに、溶液を−20℃未満に冷やして約1.5時間撹拌した。ポリプロピレン・フイルター・パッドを用い、固形分を18”クロック・フィルター上で濾過し、エチルエーテル(3x2L)で洗浄し、パイレックス乾燥トレイに移した。ジメチル−1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシレートを真空中(in vacuo)25℃で乾燥させた。
【0037】
2本の22Lのフラスコの各々に、9Lの水、2250gのジメチル−1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシレート、および2.25Lのエチルアルコールを充填した。50%の水酸化ナトリウム溶液(1.2L)を各々のフラスコに加えて、反応混合物を加熱して還流し、約1時間15分間、還流状態に保った。ジメチル−1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシレートが5%以下であったことを確認した後に、反応混合物を約75℃で撹拌した。両方の反応混合物を、一本の30ガロン・クロックに移した。氷を添加し、反応混合物を10℃以下に冷やした。4Lの濃塩酸を用いて混合物をpH1まで酸性化させ、最低30分間撹拌した。フィルタ・パッドを用いて固形分をクロック・フィルターで濾過した。濾過ケークを切り離して12Lの酢酸エチルに入れて層の分離をおこなった。有機層を硫酸マグネシウムで乾燥させ、ブフナー漏斗で濾過しながら22Lのフラスコに入れた。ブチ・ロタバポア(Buchi Rotavapor)を用いて、材料を濃縮して8Lの容量にし、10Lのヘキサンで希釈し、撹拌しながら0℃に冷却した。固形分を濾過し、ヘキサン(3x2L)で洗浄した。1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシル酸をパイレックス乾燥トレイに移し、最低12時間、真空中(in vacuno)35℃で乾燥させた。
【0038】
2本の22Lフラスコの各々に、14Lの2−メトキシエチル・エーテル[ダイグライム]、1750gの1−(4−メチルフェニル)−1,2−シクロプロパンジカルボキシル酸、および716gの尿素を充填した。反応混合物を、少なくとも12時間、約155℃まで加熱した。出発原料が5%以下であったことを確認した後に、反応混合物を約40Kgの氷を含む55ガロン・クロックに移し、室温に冷却した。この混合物を、総容量100Lになるまで水で希釈し、約4時間撹拌した。混合物を、ポリプロピレン・フィルタ・パッドを用いてクロック・フィルター(18”)で濾過した。固形分を水(3x2L)で洗浄し、5ガロン・バケツに移し、12Lの酢酸エチルエステルに溶解した。溶液を40Lの分液ロートに注ぎ込み、層の分離をおこなった。水層および有機層を各々、2つの5ガロン・バケツの1つに排出し、水溶液を分液ロートに注ぎ戻し、酢酸エチルエステル(2x1L)で抽出した。水層を捨て、複数の有機層は混ぜ合わせた。炭(500g)と硫酸マグネシウム(500g)とを有機層に添加し、結果として生ずる混合物を十分混合されるまで撹拌した。溶液を、ワットマン(Whatman)GFフィルター・パッドを用いてブフナー漏斗(18.5cm)で濾過し、固形分を酢酸エチルエステル(2x500mL)で洗浄した。ブチ・ロタバポア(Buchi Rotavapor)を用いて、濾過液を約60℃の浴槽温度で、最初の結晶が見えるまで、または総容量5Lが達成されるまで濃縮した。この濃縮から得られた残留物を5ガロン・バケツに移して撹拌した。スラリーを12Lのヘキサンで希釈して、室温で2時間撹拌した。固形分を、ポリプロピレン・フィルター・パッドを用いたクロック・フィルター(18”)で濾過し、ヘキサン(3x2L)で洗浄した。1−(4−メチルフェニル)−3−アザビシクロ[3.1.0]ヘキサン−2,4−ジオンをパイレックス乾燥トレイに移し、真空中(in vacuo)35℃で最低12時間乾燥させた。
【0039】
2本の50Lフラスコの各々に、12Lのトルエンおよび1250gの1−(4−メチルフェニル)−3−アザビシクロ[3.1.0]ヘキサン−2,4−ジオンを充填した。ナトリウム・ビス(2−メトキシエトキシ)アルミニウム・ハイドライド(5700mLのRedAl)を各々のフラスコに添加し、反応混合物を3時間加熱して還流した。出発原料が5%以下であったことを確認した後に、反応混合物を氷/水浴を用いて60℃未満に冷やした。30ガロン・クロックに、氷/水混合物(6.4kg水、4.4kg氷)を2.2Lの水酸化ナトリウム、50%溶液と共に充填し、氷を加えることで温度を約0℃に調整した。この混合物を撹拌して、両反応混合物をそれに注ぎ込んだ。氷を加えることで、温度を20℃未満に下げて、結果として得られる混合物を最低2時間撹拌した。溶液を40L分液漏斗に移した。層を分離し、各層を2つの5ガロン・バケツの1つに排出した。水層を酢酸エチルエステル(3x2L)で抽出して捨て、複数の有機抽出物を混ぜ合わせ、水(3x2L)で洗浄した。水洗浄液を捨てた。6Lの水を含む2ガロンのバケツに、溶液が飽和(塩水)するまで塩化ナトリウムを添加した。有機抽出物を塩水(3x2L)で洗浄し、該水洗浄液を捨てた。有機層を、複数の5ガロン・バケツに等量排出した。250gの炭と250gの硫酸マグネシウムとを各々のバケツに加えて、結果として生じる混合物が十分に混ざり合うまで混合した。これらの溶液をワットマン(Whatman)GFフィルター・パッドを用いてブフナー漏斗(18.5cm)で濾過した。固形分を酢酸エチル(2X500 mL)で洗浄し、濾過液をブチ・ロタバポア(Buchi Rotavapor)で、約60℃の温度で総量約12Lまで濃縮した。20℃未満の温度を保つ一方で、pH2〜3に達するまで反応混合物に無水HClガスをバブリングした。混合物を10℃未満でさらに2時間混合し、その後、12時間以上、フリーザーに置いた。固形分を、ポリプロピレン・フィルター・パッドを用いたクロック・フィルター(18”)で濾過し、ヘキサン(3x2L)、さらにアセトン(3x2L)で洗浄した。粗ビ
シファジン塩酸塩をパイレックス乾燥トレイに移し、最低2時間乾燥させた。
【0040】
4本の22Lフラスコの各々に、約1500gの粗ビシファジン塩酸塩を充填した。イソプロピルアルコール(6−10mL/g)を添加し、材料を撹拌および加熱して還流した。いったん、還流で透明な液体が達成されると、200gの炭を各々のフラスコに添加した。溶液を約10分間撹拌し、GFフィルターが載せられたポリプロピレン・パッドを用いたブフナー漏斗(10”)で濾過して、20Lフィルター・ボトルに入れた。結果として生じる濾過液は、30ガロンPEクロックへ移し、室温で3時間撹拌した。クロックを、最低12時間低温室に置いた。固形分を、ポリプロピレン・フィルタ・パッドを用いたクロック・フィルター(18”)上で濾過し、アセトン(4x2L)で洗浄した。1%未満の乾燥減量が達成されるまで、ステップの産物であるビシファジン塩酸塩を真空中(in vacuo)50℃で乾燥した。この物質を実施例6で使用した。
【実施例3】
【0041】
ビシファジン塩酸塩多形相Bへの転換
多形相Aと多形相Bとの混合物としてのラセミ体ビシファジン塩酸塩を、スラリーを形成するのに十分な量でイソプロピル・アルコールに添加した。スラリーを30℃未満の温度で混合等のアジテーションにかけた。1日後、生成物を濾過によって単離し、乾燥減量が1%未満まで達成されるまで、真空中50℃で乾燥した。生産された材料はビシファジン塩酸塩多形相Bであった。
【実施例4】
【0042】
ビシファジン塩酸塩多形相Bへの転換
多形相AおよびBの混合物としての20グラムのラセミ体ビシファジン塩酸塩を、60mlのイソプロピルアルコールに加えてスラリーにした。このスラリーを約30℃の温度で24時間撹拌した。生成物を濾過して単離し、真空中(in vacuo)乾燥した。生産された材料は、検出可能な多形相Aの存在が認められないビシファジン塩酸塩多形相Bであった。
【実施例5】
【0043】
実施例5において、米国特許第4,231,935号の実施例36で述べられるように、その多形結晶の形状であるビシファジン塩酸塩を、アセトニトリル/メタノールから調製および結晶化した。試料(すなわち、表5の試料1〜5)のビシファジン塩酸塩を以下に説明するように蒸発することで、再結晶化させた。
【0044】
蒸発
以下の容量−容量混合物のアセトニトリル−メタノールを調製した。すなわち、9:1、2:1、1:1、1:2,および1:9.透明な溶液が生ずるまで、計量された量のA形ビシファジン塩酸塩(約90mg)を溶媒混合物の一定分量で処理した。各溶液を0.2ミクロン・ナイロン・シリンジで濾過してきれいなバイアルに入れた。ピンホールによる穿孔が形成されたアルミホイルでバイアルを覆い、ヒューム・フードの下に置いて蒸発を可能にした。溶媒が完全に蒸発すると、固形分が回収された。
【0045】
得られる多形に関する結果を表5に示す。これらの結果は、X線粉末回折技術(XRPD)を用いて得られた。
【0046】
【表5】
【0047】
表5の場合のように、上記手順にもとづく米国特許第4,231,935号の実施例36の製品の単純な再結晶化は、蒸発によってビシファジン塩酸塩多形相Aを生産した。
【実施例6】
【0048】
A形の調製
実施例2で調製したように、ビシファジン塩酸塩(20.1g)を加熱してイソプロピルアルコール(365mL)に撹拌し、80℃で澄んだ溶液が得られた。溶液を55℃で冷やすことで、スラリーが得られた。製品を濾過して回収し、周囲温度で乾燥させることで、A形ビシファジン塩酸塩が得られた。
【実施例7】
【0049】
A形の調製
一定量のエタノール(1mL)をビシファジン塩酸塩(97mg)(実施例1での固晶として生産される)で飽和し、80℃に設定した加熱撹拌プレート上に置かれたバイアルに入れた。溶液を濾過して、−20℃に冷やされた1mLトルエン含有のバイアルに、直接入れた。混合物を15分間、−20℃フリーザーに入れることで、沈殿が生じた。製品を濾過して回収し、周囲温度で減圧下、乾燥することで、A形ビシファジン塩酸塩が得られた。
【実施例8】
【0050】
B形の調製
実施例1で固晶として生産される、A形ビシファジン塩酸塩(3.99g)を加熱して、イソプロピルアルコール(50mL)で撹拌することで、82℃で澄んだ溶液が得られた。溶液を周囲温度に冷やすことで、厚いスラリーが得られた。撹拌機構をシャット・オフし、混合物を2日間、周囲温度に設定した。固形分(白色から灰色かかったブレード)を回収し、空気乾燥することで、B形ビシファジン塩酸塩が得られた。
【実施例9】
【0051】
多形相Bの調製
実施例1で固晶として生産される、A形ビシファジン塩酸塩(約1g)を小さなステンレス・スチール・シリンダーに充填した。ステンレス・スチール棒を加えてシリンダーをキャッピングした。シリンダーを液体窒素が充填されたSPEX/Centriprep 6750型フリーザー・ミルに取り付けた。6分間のグラインディング時間について、試料を一度に2分間挽いて2分間の休憩時間をとった。タンクに液体窒素を満たし、上記周期を繰り返し、グラインディング合計時間を12分とした。プロセスは、A形ビシファジン塩酸塩がまったく検出されることなくB形ビシファジン塩酸塩に対する完全な転換に帰着した。
【実施例10】
【0052】
グラインディングでのA形の不安定性
A形ビシファジン塩酸塩(207mg)を小さなステンレス・スチール・シリンダーに充填した。ステンレス・スチール・ボールを加え、シリンダーをキャッピングし、全体で20分間にわたり周期30/sで5分間の間隔で、レッシュ・ミキサー・ミル(Retsch Mixer Mill)(MM200型)上で上記ユニットを振とうさせた。挽いた材料を除去し、XRPDで分析した。プロセスは、B形ビシファジン塩酸塩への部分的転換をもたらした。
【実施例11】
【0053】
グラインディングでの多形相Bの安定性
B形ビシファジン塩酸塩(111mg)を小さなステンレス・スチール・シリンダーに充填した。ステンレス・スチール・ボールを加え、シリンダーをキャッピングし、全体で20分間にわたり周期30/sで5分間の間隔で、レッシュ・ミキサー・ミル(Retsch Mixer Mill)(MM200型)上で上記ユニットを振とうさせた。挽いた材料を除去し、XRPDで分析した。挽かれた固形分は、B形のままだった。
【図面の簡単な説明】
【0054】
【図1】実施例9で調製される多形相Bラセミ体ビシファジン塩酸塩の赤外線スペクトラムである。
【特許請求の範囲】
【請求項1】
多形相Bを持つ固晶としてのビシファジン塩酸塩。
【請求項2】
ビシファジン塩酸塩多形相Bであって、前記多形の固晶の主赤外線スペクトラム・ピークが波数(cm-1)で、2108、891、856、719、および660である、ビシファジン塩酸塩多形相B。
【請求項3】
前記結晶は、以下の波数(cm-1)で表されるピークを持つ固晶としての赤外線スペクトラムによって、特徴づけられる、請求項2に記載の多形相Bのビシファジン塩酸塩。
【表1】
【請求項4】
ビシファジン塩酸塩多形相Bであって、前記多形の固晶は、2θ(度)が5.08、 10.07、20.16、25.17、および 30.43で、CuKα線を用いた粉末X線回折法によって測定される主ピークを持つことによって特徴づけられる、ビシファジン塩酸塩多形相B。
【請求項5】
「d」線間距離および相対強度I(s=強、m=中程度、w=弱、v=極めて、d=拡散)の表現で表した以下のX線回折を持つ、請求項4に記載のビシファジン塩酸塩多形相B。
【表2】
【請求項6】
多形相B固晶としてビシファジン塩酸塩を生産する方法であって、
有機溶媒にビシファジン塩酸塩を含み、沸点が少なくとも約50℃のスラリーを提供するステップと、
前記スラリーが透明な溶液になる温度で、前記スラリーを加熱するステップと、
前記溶液を最大で約35℃の温度まで冷やすステップと、
前記冷却された溶液を、十分な時間、前記最大で約35℃の温度に保ち、前記多形相Bを前記溶液から結晶の形状に結晶化させる、方法。
【請求項7】
前記冷却は、前記溶液をアジテーションしている間に実施される、請求項6に記載の方法。
【請求項8】
前記冷却された溶液は、アジテーションされる一方で最大で35℃の温度に保たれる、請求項6に記載の方法。
【請求項9】
前記加熱された溶液を、約−200℃ないし0℃の温度に冷やす、請求項6に記載の方法。
【請求項10】
ビシファジンを多形相B固晶として生産する方法であって、
ビシファジン塩酸塩の多形相A固晶を提供するステップと、
約−200℃ないし50℃の温度で前記結晶をアジテーションすることで、前記多形相A固晶を多形相B結晶に変換するステップと、を含む方法。
【請求項11】
前記アジテーションをグラインディングによって実行する、請求項10の方法。
【請求項12】
前記アジテーションは、約−200℃ないし約35℃の温度で実行される、請求項10に記載の方法。
【請求項13】
前記アジテーションは、約−200℃ないし0℃の温度で実行される、請求項12に記載の方法。
【請求項14】
経口単位投薬形態である医薬組成物であって、ビシファジン塩酸塩の多形相B固晶と、医薬的に許容される不活性の担体または希釈剤とを含む、医薬組成物。
【請求項15】
多形相B結晶状態の前記ビシファジン塩酸塩が25mgないし約600mgの量で前記経口単位投薬形態で存在する、請求項14の経口単位投薬形態。
【請求項16】
前記経口単位投薬形態は、約25mgないし600mgの前記多形相B結晶を含有する、請求項15に記載の医薬組成物。
【請求項17】
前記経口単位投薬形態は、錠剤またはカプセルである、請求項15に記載の組成物。
【請求項18】
前記処置を必要としている患者の痛みを抑える方法であって、
前記患者に対して、多形相Bの結晶構造を持つビシファジン塩酸塩と不活性担体または希釈剤とを含む組成物を投与するステップを含み、前記組成物は前記痛みを緩和するのに有効な量で投与される、方法。
【請求項19】
多形相Bの結晶形態を持つ前記ビシファジン塩酸塩は、1日あたり0.5mg/kgないし約20mg/kgの量で投与される、請求項18に記載の組成物。
【請求項20】
前記ビシファジンは、約25ないし600mg含む全ての単位投薬形態で投与される、請求項19に記載の方法。
【請求項21】
多形相Aまたは多形相AからBまでの混合物のいずれかに含まれるビシファジン塩酸塩から純粋な多形相B結晶を生産する方法であって、
多形相Aまたは多形相AからBまでの混合物のいずれかに含まれるビシファジン塩酸塩を、有機溶媒に添加してスラリーを形成するステップと、
最大で約35℃の温度で、またはビシファジン塩酸塩の多形相B結晶を形成するのに十分な時間を少なくとも時間として、前記スラリーに運動エネルギーを与える、方法。
【請求項22】
前記運動エネルギーは、アジテーションによって与えられる請求項21に記載の方法。
【請求項23】
前記アジテーションは、撹拌によって実行される、請求項22に記載の方法。
【請求項1】
多形相Bを持つ固晶としてのビシファジン塩酸塩。
【請求項2】
ビシファジン塩酸塩多形相Bであって、前記多形の固晶の主赤外線スペクトラム・ピークが波数(cm-1)で、2108、891、856、719、および660である、ビシファジン塩酸塩多形相B。
【請求項3】
前記結晶は、以下の波数(cm-1)で表されるピークを持つ固晶としての赤外線スペクトラムによって、特徴づけられる、請求項2に記載の多形相Bのビシファジン塩酸塩。
【表1】
【請求項4】
ビシファジン塩酸塩多形相Bであって、前記多形の固晶は、2θ(度)が5.08、 10.07、20.16、25.17、および 30.43で、CuKα線を用いた粉末X線回折法によって測定される主ピークを持つことによって特徴づけられる、ビシファジン塩酸塩多形相B。
【請求項5】
「d」線間距離および相対強度I(s=強、m=中程度、w=弱、v=極めて、d=拡散)の表現で表した以下のX線回折を持つ、請求項4に記載のビシファジン塩酸塩多形相B。
【表2】
【請求項6】
多形相B固晶としてビシファジン塩酸塩を生産する方法であって、
有機溶媒にビシファジン塩酸塩を含み、沸点が少なくとも約50℃のスラリーを提供するステップと、
前記スラリーが透明な溶液になる温度で、前記スラリーを加熱するステップと、
前記溶液を最大で約35℃の温度まで冷やすステップと、
前記冷却された溶液を、十分な時間、前記最大で約35℃の温度に保ち、前記多形相Bを前記溶液から結晶の形状に結晶化させる、方法。
【請求項7】
前記冷却は、前記溶液をアジテーションしている間に実施される、請求項6に記載の方法。
【請求項8】
前記冷却された溶液は、アジテーションされる一方で最大で35℃の温度に保たれる、請求項6に記載の方法。
【請求項9】
前記加熱された溶液を、約−200℃ないし0℃の温度に冷やす、請求項6に記載の方法。
【請求項10】
ビシファジンを多形相B固晶として生産する方法であって、
ビシファジン塩酸塩の多形相A固晶を提供するステップと、
約−200℃ないし50℃の温度で前記結晶をアジテーションすることで、前記多形相A固晶を多形相B結晶に変換するステップと、を含む方法。
【請求項11】
前記アジテーションをグラインディングによって実行する、請求項10の方法。
【請求項12】
前記アジテーションは、約−200℃ないし約35℃の温度で実行される、請求項10に記載の方法。
【請求項13】
前記アジテーションは、約−200℃ないし0℃の温度で実行される、請求項12に記載の方法。
【請求項14】
経口単位投薬形態である医薬組成物であって、ビシファジン塩酸塩の多形相B固晶と、医薬的に許容される不活性の担体または希釈剤とを含む、医薬組成物。
【請求項15】
多形相B結晶状態の前記ビシファジン塩酸塩が25mgないし約600mgの量で前記経口単位投薬形態で存在する、請求項14の経口単位投薬形態。
【請求項16】
前記経口単位投薬形態は、約25mgないし600mgの前記多形相B結晶を含有する、請求項15に記載の医薬組成物。
【請求項17】
前記経口単位投薬形態は、錠剤またはカプセルである、請求項15に記載の組成物。
【請求項18】
前記処置を必要としている患者の痛みを抑える方法であって、
前記患者に対して、多形相Bの結晶構造を持つビシファジン塩酸塩と不活性担体または希釈剤とを含む組成物を投与するステップを含み、前記組成物は前記痛みを緩和するのに有効な量で投与される、方法。
【請求項19】
多形相Bの結晶形態を持つ前記ビシファジン塩酸塩は、1日あたり0.5mg/kgないし約20mg/kgの量で投与される、請求項18に記載の組成物。
【請求項20】
前記ビシファジンは、約25ないし600mg含む全ての単位投薬形態で投与される、請求項19に記載の方法。
【請求項21】
多形相Aまたは多形相AからBまでの混合物のいずれかに含まれるビシファジン塩酸塩から純粋な多形相B結晶を生産する方法であって、
多形相Aまたは多形相AからBまでの混合物のいずれかに含まれるビシファジン塩酸塩を、有機溶媒に添加してスラリーを形成するステップと、
最大で約35℃の温度で、またはビシファジン塩酸塩の多形相B結晶を形成するのに十分な時間を少なくとも時間として、前記スラリーに運動エネルギーを与える、方法。
【請求項22】
前記運動エネルギーは、アジテーションによって与えられる請求項21に記載の方法。
【請求項23】
前記アジテーションは、撹拌によって実行される、請求項22に記載の方法。
【図1】

【公表番号】特表2006−519162(P2006−519162A)
【公表日】平成18年8月24日(2006.8.24)
【国際特許分類】
【出願番号】特願2004−551708(P2004−551708)
【出願日】平成15年11月5日(2003.11.5)
【国際出願番号】PCT/US2003/035099
【国際公開番号】WO2004/043920
【国際公開日】平成16年5月27日(2004.5.27)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
パイレックス
【出願人】(505166502)ディオーブイ ファーマシューティカル,インク. (5)
【Fターム(参考)】
【公表日】平成18年8月24日(2006.8.24)
【国際特許分類】
【出願日】平成15年11月5日(2003.11.5)
【国際出願番号】PCT/US2003/035099
【国際公開番号】WO2004/043920
【国際公開日】平成16年5月27日(2004.5.27)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
パイレックス
【出願人】(505166502)ディオーブイ ファーマシューティカル,インク. (5)
【Fターム(参考)】
[ Back to top ]