
ビスホスホネート錯体
【課題】骨癌や骨粗鬆症のような骨疾患及び骨関連疾患の周囲にある柔組織の治療に有用なPdまたはPt錯体の提供。
【解決手段】式II:

[式中、Mは、Pt(II)又はPd(II)である]で示される錯体等、およびビスホスホネート化合物、特に、ビスホスホネート錯体、又はその医薬的に許容される塩。
【解決手段】式II:
[式中、Mは、Pt(II)又はPd(II)である]で示される錯体等、およびビスホスホネート化合物、特に、ビスホスホネート錯体、又はその医薬的に許容される塩。
【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明は、特別なビスホスホネート化合物、そして特に、骨癌や骨粗鬆症のような骨疾患及び骨関連疾患の周囲にある柔組織の治療に有用であるビスホスホネート抱合体へ向けられる。
【0002】
背景技術
ビスホスホネートは、骨粗鬆症、ページェット病、及び悪性の高カルシウム血症が含まれる、異常に促進された骨吸収に関連したいくつかの疾患の治療においてきわめて有望な治療効果を示した医薬品のクラスを代表する。Fleish H., Ann Med, 29, 55-62 (1997) 及び FleishH., Drugs, 42, 919-944 (1991)。より最近、ビスホスホネートは、骨へ拡がった前立腺癌のある患者において骨格の合併症(例、病的な骨折、脊髄圧迫、骨手術又は放射線療法の必要性)が発症するリスクを低下させるのに有効であること(Saad F et al., J National Cancer Institute 94: 1458-1468, 2002);そしてRAS依存型悪性疾患(例、小細胞肺癌)の増殖を阻害すること(Matsumoto, et al., Am. Soc. of Clin. Oncology, 2003, Abst. No. 2750)が示された。ビスホスホネートは、抗血管形成活性を有することも示されている。Wood et al, J Pharmacol Exp Ther 2002 Sep; 302(3): 1055-61。ビスホスホネートは、通常、骨髄腫骨疾患の治療と乳癌の溶骨性転移に抗して使用され、臨床試験は、転移性前立腺癌における疼痛を緩和するためのその使用を示唆した。
【0003】
白金ベースの薬剤は、化学療法の適用に広く利用されている。例えば、シスプラチンは、共有結合性、交差性又は鎖内DNA付加物の形成を介して腫瘍細胞を殺す(Sherman et al. Chem. Rev., 87, 1153-81 (1987); Chu, J. Biol. Chem., 269, 787-90 (1994))。そのような白金ベースの薬剤での治療は、それによりDNA合成の阻害をもたらす(Howle et al., Biochem. Pharmacol., 19, 2757-62 (1970); Salles et al., Biochem. Biophys. Res. Commun., 112, 555-63 (1983))。従って、DNAを活発に合成する細胞は、シスプラチンに対して高感受性である(Roberts et al., Prog. Nucl. Acid Res. Mol. Biol., 22, 71-133 (1979); Pinto et al., Proc. Nat. Acad Sci. (Wash.) 82, 4616-19 (1985))。一般に、そのような細胞は、G2において増殖阻止を経験して、最終的にはアポトーシスを蒙る。このアポトーシス効果は、DNA合成を阻害するのに不十分な薬物濃度で観察され(Sorenson et al., J. Natl. Cancer Inst., 82, 749-55 (1990))、白金剤が多数の機序を介して新生物細胞に作用することを示唆する。細胞周期のG1期にあるときに高められた白金感受性を明示する細胞もある(Krishnaswamy et al., Mutation Res., 293, 161-72 (1993); Donaldson et al., Int. J. Cancer, 57, 847-55 (1994))。そのような細胞は、G0/G1−Sブロックより解放されると、細胞周期の残りを通して最大に感作されたままである。
【0004】
米国特許第6,087,349号は、ビスホスホネートがタンパク質プレニルトランスフェラーゼ阻害剤として作用し得ることを開示する。
米国特許第4,746,654号は、抗炎症剤として有用なビスホスホネートを開示する。
【0005】
オーストラリア特許A−5 1534/85は、異常なカルシウム及びリンの代謝を治療するのに有用で、関節炎を治療するのに有用なビスホスホネートを開示する。
米国特許第3,683,080号は、リン酸カルシウムの動物組織における異常な沈着及び動員を阻害するのに有用なポリホスホネート、特にジホスホネートを開示する。
【0006】
DE3,719,513−A(Derwent 89−000580/01)は、カルシウム代謝の障害の治療に有用なジホスホン酸誘導体を開示する。
WO88/06158は、ビニリデンジホスホネートと活性化メチレンの反応を開示する。
【0007】
国際特許出願番号PCT/US90/01106に代わる国際特許公開公報番号WO90/12017は、抗関節炎剤としてのジェミナルビスホスホン酸とその誘導体を開示する。
【0008】
米国特許出願番号20020022603は、両性イオンリン脂質及びビスホスホネートの組成物と、GI毒性の低下したビスリン酸送達系としてのそのような組成物の使用を開示する。
【0009】
米国特許出願番号20030032628は、湿式造粒の錠剤製剤化によって調製する、ビスホスホン酸とその塩の医薬組成物を開示する。これらの医薬組成物は、結合剤の添加なしに調製される;むしろ、薬物それ自身が結合剤として作用すると言われる。
【0010】
米国特許出願番号20020002140は、顕著に高められた腸吸収性と高められたバイオアベイラビリティを有すると言われる、ビスホスホネート化合物のグリコシド及びオルトエステルグリコシド誘導体を開示する。
【0011】
米国特許第5,133,972号は、経皮送達のリン酸化合物、特にビスホスホネートを開示する。関連して、米国特許第6,018,679号は、皮膚刺激や他の有害な効果を引き起こすことが可能な化合物をイオン泳動的に除去する方法を開示する。
【0012】
米国特許第6,114,316号は、過剰なプロテイナーゼ活性に関連した組織破壊状態を生物系において治療するか又は予防する相乗的なプロテイナーゼ阻害量においてテトラサイクリンとビスホスホネートを組み合わせる組成物を開示する。
【0013】
米国特許第6,214,812号は、骨組織との結合時に抗菌及び/又は細胞傷害成分を放出することが可能であると言われるビスホスホネート抱合体を開示する。
米国特許第6,436,386号は、ヒドロキシアパタイト標的指向性ポリマー構造とその生物学的に活性な抱合体を開示し、ここでヒドロキシアパタイト標的指向性部分は、ビスホスホネートであってよい。この抱合体は、生物学的に活性な物質を骨表面へつなげるための手段を提供すると言われる。
【0014】
当該技術分野には、様々な種類のビスホスホネート化合物、抱合体、製剤、組合せ、及びそれらの使用について記載する多数の他の参考文献を見出すことができる。しかしながら、そこには、白金、パラジウム、又は同様の部分を含んでなる、治療上有用であるビスホスホネート錯体を合成する方法が開示されていない。当該技術分野で知られている他のビスホスホネート錯体が治療薬剤としてのその使用に関して、特に癌の治療へのその使用に関して、より特別には、骨組織に影響を及ぼす癌の治療へのその使用に関してより優れた特性を保有することは知られていなかった。
【0015】
発明の要約
本発明は、ビスホスホネート錯体と、標的指向される細胞増殖抑制剤及び/又は細胞傷害剤としてのその使用に関する。望ましくは、本発明のビスホスホネート錯体は、細胞、例えば骨に関連した癌性細胞に標的指向するために使用することができる。
【0016】
第一の側面において、本発明は、式I:
【0017】
【化1】
【0018】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第一の側面の好ましい態様では、MがPt(II)である。
【0019】
第二の側面において、本発明は、式II:
【0020】
【化2】
【0021】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第二の側面の好ましい態様では、MがPt(II)である。
【0022】
第三の側面において、本発明は、式III:
【0023】
【化3】
【0024】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第三の側面の好ましい態様では、MがPt(II)である。
【0025】
第四の側面において、本発明は、式IV:
【0026】
【化4】
【0027】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第四の側面の好ましい態様では、MがPt(II)である。
【0028】
第五の側面において、本発明は、式V:
【0029】
【化5】
【0030】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第五の側面の好ましい態様では、MがPt(II)である。
【0031】
第六の側面において、本発明は、式VI:
【0032】
【化6】
【0033】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第六の側面の好ましい態様では、MがPt(II)である。
【0034】
第七の側面において、本発明は、式VII:
【0035】
【化7】
【0036】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第七の側面の好ましい態様では、MがPt(II)である。
【0037】
第八の側面において、本発明は、式VIII:
【0038】
【化8】
【0039】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第八の側面の好ましい態様では、MがPt(II)である。
【0040】
第九の側面において、本発明は、式Iの実施例について記載する合成手順を実質的に本明細書に記載のように実施することを含んでなる、式Iによる化合物を合成する方法に関する。前記第九の側面の好ましい態様では、MがPt(II)である。
【0041】
第十の側面において、本発明は、式IIの実施例について記載する合成手順を実質的に本明細書に記載のように実施することを含んでなる、式IIによる化合物を合成する方法に関する。前記第十の側面の好ましい態様では、MがPt(II)である。
【0042】
第十一の側面において、本発明は、式IIIの実施例について記載する合成手順を実質的に本明細書に記載のように実施することを含んでなる、式IIIによる化合物を合成する方法に関する。前記側面の好ましい態様では、MがPt(II)である。
【0043】
第十二の側面において、本発明は、細胞増殖抑制的に、及び/又は細胞傷害的に有効な量の白金又はパラジウム、又は白金含有又はパラジウム含有部分をその必要な被検者へ送達する方法に関し、前記方法は、式Iによる化合物又はその医薬的に許容される塩の治療有効量をその必要な前記被検者へ投与することを含んでなる。好ましくは、式IのMは、白金である。また好ましくは、前記方法は、前記化合物を前記被検者内の骨組織にか又は骨組織へ送達することを含む。より好ましくは、前記方法は、癌細胞がその上か又はその中に存在する骨組織にか又は骨組織へ送達することを含む。なおより好ましくは、前記化合物の前記治療有効量は、前記癌細胞を治療する、即ちそれを阻害する、及び/又は殺すのに有効な量である。
【0044】
第十三の側面において、本発明は、細胞増殖抑制的に、及び/又は細胞傷害的に有効な量の白金又はパラジウム、又はパラジウム含有又は白金含有部分をその必要な被検者へ送達する方法に関し、前記方法は、式IIによる化合物又はその医薬的に許容される塩の治療有効量をその必要な前記被検者へ投与することを含んでなる。好ましくは、式IIのMは、白金である。また好ましくは、前記方法は、前記化合物を前記被検者内の骨組織にか又は骨組織へ送達することを含む。より好ましくは、前記方法は、癌細胞がその上か又はその中に存在する骨組織にか又は骨組織へ送達することを含む。なおより好ましくは、前記化合物の前記治療有効量は、前記癌細胞を治療する、即ちそれを阻害する、及び/又は殺すのに有効な量である。
【0045】
第十四の側面において、本発明は、細胞増殖抑制的に、及び/又は細胞傷害的に有効な量の白金又はパラジウム、又はパラジウム含有又は白金含有部分をその必要な被検者へ送達する方法に関し、前記方法は、式IIIによる化合物又はその医薬的に許容される塩の治療有効量をその必要な前記被検者へ投与することを含んでなる。好ましくは、式IIIのMは、白金である。また好ましくは、前記方法は、前記化合物を前記被検者内の骨組織にか又は骨組織へ送達することを含む。より好ましくは、前記方法は、癌細胞がその上か又はその中に存在する骨組織にか又は骨組織へ送達することを含む。なおより好ましくは、前記化合物の前記治療有効量は、前記癌細胞を治療する、即ちそれを阻害する、及び/又は殺すのに有効な量である。
【図面の簡単な説明】
【0046】
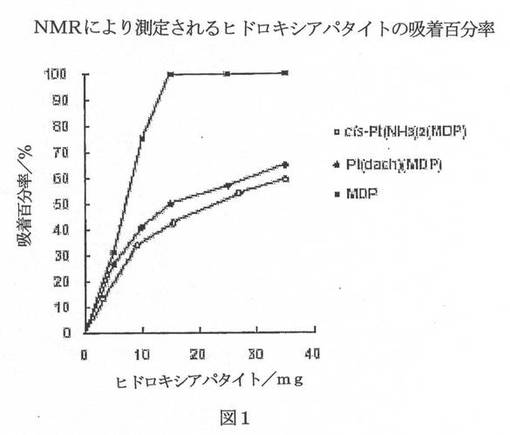
【図1】図1は、NMRにより測定される、試験化合物のヒドロキシアパタイトへの吸着百分率を示すグラフである。
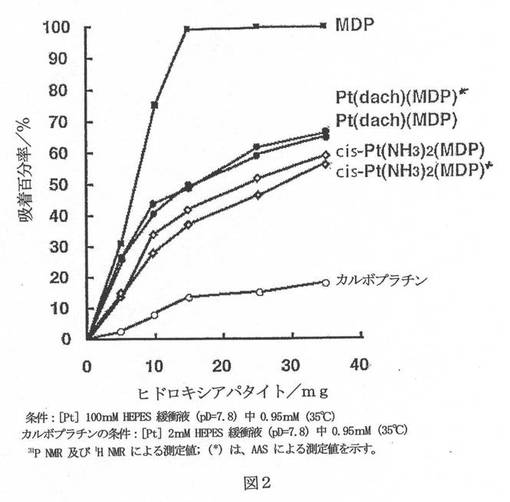
【図2】図2は、31P NMR、1H NMR、及びAASにより測定される、試験化合物のヒドロキシアパタイトへの吸着百分率を示すグラフである。
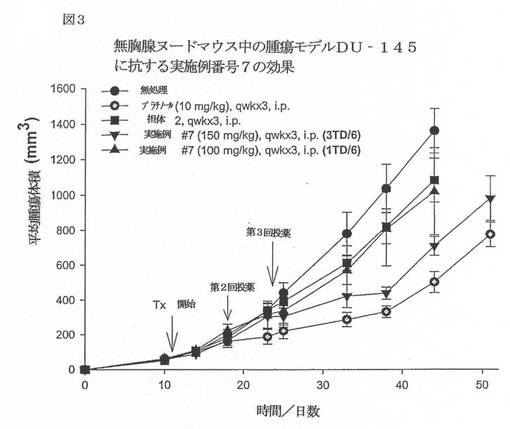
【図3】図3は、無胸腺ヌードマウス中の腫瘍モデルDU−145に抗する実施例番号7の効果を例示するグラフである。
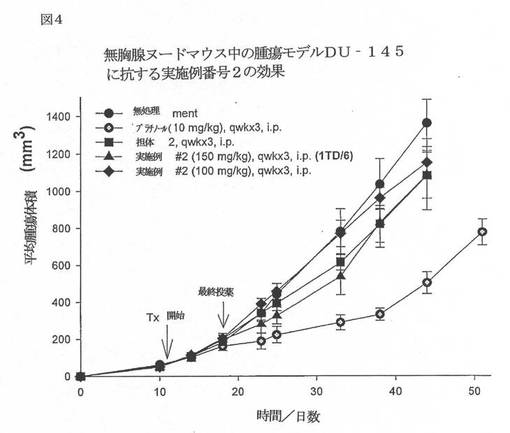
【図4】図4は、無胸腺ヌードマウス中の腫瘍モデルDU−145に抗する実施例番号2の効果を例示するグラフである。
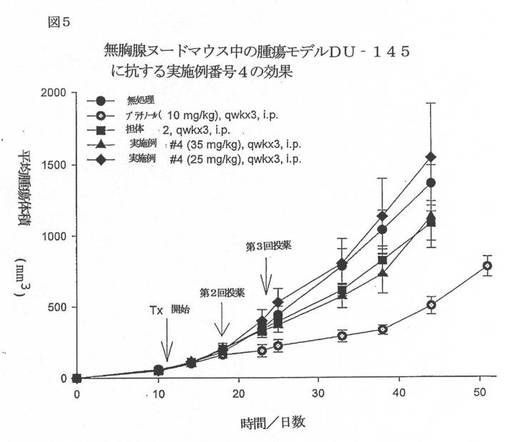
【図5】図5は、無胸腺ヌードマウス中の腫瘍モデルDU−145に抗する実施例番号4の効果を例示するグラフである。
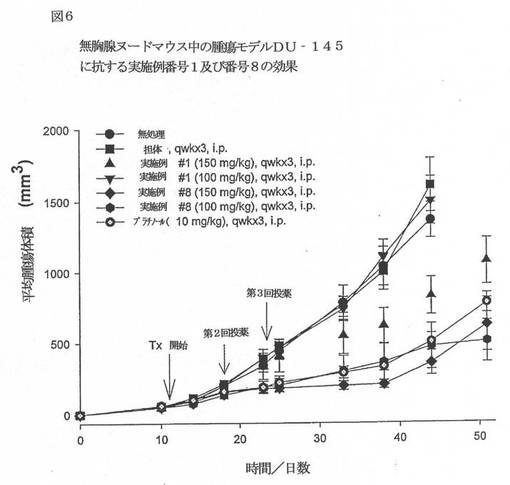
【図6】図6は、無胸腺ヌードマウス中の腫瘍モデルDU−145に抗する実施例番号1及び番号8の効果を例示するグラフである。
【0047】
好ましい態様の詳細な説明
以下の実験結果は、例示の目的のために提供されるのであって、本発明の範囲を限定することを企図しない。
【0048】
実施例1:cis−Pt(NH3)2(MDP)
a.材料
ビスホスホネート(メチレンジホスフィン酸(MDP))は、Tokyo-Kasei(東京化成)より、K2PtCl4は、Tanaka(田中化学研究所)より、ジメチルアセトアミド(DMA)と他の試薬は、Nakarai Tesque(ナカライテスク)より購入した。すべての化学品は、入手可能な最高級品であり、さらに精製せずに使用した。水は脱イオン化し、二回蒸留して、最後はMilli−Qにより精製した。
【0049】
b.手順
【0050】
【化9】
【0051】
b.1.cis−Pt(NH3)2I2
文献(S. C. Dhara, Indian J. Chem, 1970, 8)に従って、20ml H2O中のK2PtCl4 2g(4.8ミリモル)へKI 3.3g(19.8ミリモル)を加えた。この溶液を水浴中で5分間撹拌してから、47mLの0.21M NH3水溶液へ加えた。室温で約3時間静置後、沈積した黄色い粉末を濾過して取り、温水、EtOH、及びエーテルで洗浄した。収率:91%。元素分析:計算値:H:1.25%,N:5.80%;実測値:H:1.08%,N:5.58%。IR:3260cm−1,3200cm−1,1282cm−1,1270cm−1。
【0052】
b.2.cis−Pt(NH3)2(MDP)
ジメチルアセトアミド 5ml中の工程b.1.からの0.20g(0.5ミリモル)のcis−Pt(NH3)2I2を0.149g(0.48ミリモル)のAg2SO4の20mL H2O懸濁液へ加え、暗所で約4時間撹拌した。膜フィルターによる濾過後、20mL H2O中のBa(OH)2・8H2O 0.15g(0.48ミリモル)及びMDP 0.12g(0.70ミリモル)を加えた。一晩撹拌後、この溶液を蒸発により濃縮して、生じる粉末を、H2O−EtOHを使用して再沈殿させた。収率:35%。
【0053】
元素分析:計算値:C:2.98%,H:2.50%,N:6.95%;実測値:C:3.14%,H:2.50%,N:6.83%。
【0054】
【化10】
【0055】
実施例2:Pt(dach)(MDP)
a.材料
1R,2R−1,2−シクロヘキサンジアミン(dach)は、Tokyo-Kasei より;K2PtCl4は、Tanaka より;ジメチルアセトアミド(DMA)と他の試薬は、Nakarai Tesque より購入した。すべての化学品は、入手可能な最高級品であり、さらに精製せずに使用した。水は脱イオン化し、蒸留して、最後はMilli−Qにより精製した。
【0056】
b.手順
【0057】
【化11】
【0058】
b.1.Pt(dach)2I2
20ml H2O中のK2PtCl4 1.25g(3ミリモル)へKI 2.0g(12ミリモル)を加えた。この溶液を水浴中(50℃)で約5分間撹拌してから、1R,2R−1,2−シクロヘキサンジアミン 0.34g(3ミリモル)へ加えた。この反応混合物を室温で一晩撹拌し、沈積した黄色い粉末を濾過して取り、温水、次いでEtOH、最後はジエチルエーテルで洗浄した。収率:91%。
【0059】
元素分析:計算値:C:12.80%,H:2.51%,N:4.98%;実測値:C:12.66%,H:2.27%,N:5.01%。
b.2.Pt(dach)(MDP)
ジメチルアセトアミド 5ml中の工程b.1.からの0.28g(0.5ミリモル)のPt(dach)2I2を0.149g(0.48ミリモル)のAg2SO4の20mL H2O懸濁液へ加え、暗所で約4時間撹拌した。膜フィルターによる濾過後、20mL H2O中のBa(OH)2・8H2O 0.156g(0.48ミリモル)及びMDP 0.129g(0.75ミリモル)を加えた。約2時間撹拌後、この反応溶液混合物へ約5分にわたり撹拌しながら1mlの0.5M H2SO4水溶液を加えた。この混合物を濾過し、蒸発により約5mlへ濃縮して、MeOHを使用して白い粉末を再沈殿させた。収率:35%。
【0060】
元素分析:計算値:C:17.40%,H:3.75%,N:5.80%;実測値:C:17.47%,H:3.44%,N:5.89%。
【0061】
【化12】
【0062】
実施例3:N−(9−アントラニル)メチル−1,2−エタンジアミン二塩酸塩(Aten・2HCl)
a.材料
N−(9−アントラニル)アルデヒドは、Tokyo-Kasei より購入し;K2PtCl4は、Tanaka より購入し;PdCl2は、Kishida Chemical(キシダ化学)より購入し;2,2’−ビピリジン(bpy)は、Wako(和光純薬工業)より;DMSOと他の試薬は、Nakarai Tesque より購入した。すべての化学品は、入手可能な最高級品であり、さらに精製せずに使用した。水は脱イオン化し、二回蒸留して、最後はMilli−Qにより精製した。
【0063】
b.手順
【0064】
【化13】
【0065】
200ml 1:1 ジオキサン−CHCl3中のN−(9−アントラニル)アルデヒド 2.06g(10ミリモル)へエチレンジアミン 6.00g(10ミリモル)を加えて、この溶液を約3時間還流した。室温へ冷却後、この溶液を蒸発により濃縮して、MeOH中0.45g(12ミリモル)のNaBH4を加えた。一晩撹拌後、6N HClを加えてpHを1へ調整して、液体を蒸発させた。NaOH水溶液を加え、この塩基性溶液をCHCl3で抽出して、Na2SO4を使用して乾燥させた。CHCl3相を蒸発させて、油性残渣をMeOH/6N HClで処理した。生じる黄色がかった粉末を濾過により回収して、EtOH−H2Oより再結晶させた。収量:2.2g(65%)。
【0066】
元素分析 C17H20N2Cl2・0.75H2O:計算値:C:60.63%,H:6.43%,N:8.32%;実測値:C:60.40%,H:6.45%,N:7.81%。
【0067】
【化14】
【0068】
実施例4:Pt(bpy)(Aten)錯体
a.Pt(bpy)Cl2
10mL H2O、30mL DMSOの溶液中1.411g(3.4ミリモル)のK2PtCl4を0.531g(3.4ミリモル)のbpy(50mL DMSO)へ加え、この混合物を撹拌しながら80℃まで約3時間加熱した。この反応物を一晩撹拌し、生じる黄色い針状物を濾過して取り、H2Oとエーテルで洗浄した。収量:1.2g(81%)。
【0069】
元素分析 C10H8N2Cl2Pt:計算値:C:28.45%,H:1.91%,N:6.58%;実測値:C:28.65%,H:1.95%,N:6.64%。
b.[Pt(bpy)(Aten)]Cl2
【0070】
【化15】
【0071】
文献(Goto, M., et al., Bull. Chem. Soc. Jpn. 2000, 73, 97-105)に従って、Pt(bpy)Cl2(0.42g,1.0ミリモル)の20ml H2O懸濁液へ0.44g(1.3ミリモル)のAten・2HCl・0.75H2Oと0.16g(1.5ミリモル)のNa2CO3を加え、この混合物を80℃で約2.5時間撹拌した。次いで、この混合物をまだ熱い間に濾過して、少量の不溶性材料を除去した。室温へ冷却後、薄黄色の沈殿が生じ、これをフィルターで採取して、デシケーター中で乾燥させた。収量:0.56g(79%)。
【0072】
元素分析 C27H26N4Cl2Pt・2H2O:計算値:C:45.77%,H:4.27%,N:7.91%;実測値:C:45.51%,H:4.05%,N:7.61%。
【0073】
実施例5:Pd(bpy)(Aten)錯体
a.Pd(bpy)Cl2
PdCl2 0.89g(5.0ミリモル)とNaCl 0.58g(10.0ミリモル)を50mL H2Oに懸濁させて、約1時間撹拌した。濾過後、この溶液を20mL MeOH中0.78g(5.0ミリモル)bpyの溶液へ加えて、生じる溶液を一晩撹拌した。黄色がかった粉末が沈殿し、これを回収して、H2OとEtOHで洗浄した。収量:1.56g(93%)。
【0074】
元素分析 C10H8N2Cl2Pd:計算値:C:36.01%,H:2.42%,N:8.40%;実測値:C:35.94%,H:2.14%,N:8.31%。
b.[Pd(bpy)(Aten)]Cl2
【0075】
【化16】
【0076】
直前の工程からの0.33g(1.0ミリモル)のPd(bpy)Cl2を10ml H2Oに懸濁させた。0.44g(1.3ミリモル)のAten・2HCl・0.75H2Oと0.16g(1.5ミリモル)のNa2CO3を加えてから、この混合物を80℃まで約2時間加熱した。次いで、この溶液をまだ熱い間に濾過して、蒸発により濃縮した。室温で静置させると、黄色い粉末が沈積した。収量:0.43g(68%)。
【0077】
元素分析 C27H26N4Cl2Pd・3H2O:計算値:C:50.84%,H:5.06%,N:8.78%;実測値:C:50.89%,H:4.69%,N:8.80%。
【0078】
実施例6:Pd(bpy)(AtC3)
【0079】
【化17】
【0080】
a.AtC3・2HCl
200ml 1:1 ジオキサン−CHCl3中のN−(9−アントラニル)アルデヒド 2.06g(10ミリモル)へ1,3−ジアミンプロパン 7.41g(100ミリモル)を加えて、この溶液を約3時間還流した。室温へ冷却後、この溶液を蒸発により濃縮して、MeOH中0.45g(12ミリモル)のNaBH4を加えた。一晩撹拌後、6N HClを加えてpHを1へ調整して、溶液を蒸発させた。NaOH水溶液を加え、この塩基性溶液をCHCl3で抽出して、Na2SO4を使用して乾燥させた。CHCl3相を蒸発させて、油性残渣をMeOH/6N HClで処理した。生じる黄色がかった粉末を濾過により回収して、EtOH−H2Oより再結晶させた。収率:70%。
【0081】
元素分析 C18H22N2Cl2:計算値:C:64.10%,H:6.57%,N:8.31%;実測値:C:64.40%,H:6.38%,N:7.98%。
【0082】
【化18】
【0083】
b.[Pd(bpy)(AtC3)]Cl2
直前の工程からの0.33g(1.0ミリモル)のPd(bpy)Cl2を10ml H2Oに懸濁させた。0.44g(1.3ミリモル)のAtC3・2HClと0.16g(1.5ミリモル)のNa2CO3を加えてから、この混合物を80℃まで約2時間加熱した。次いで、この溶液をまだ熱い間に濾過して、蒸発により濃縮した。室温で静置させると、黄色い粉末が沈積した。収量:0.40g(58%)。
【0084】
元素分析 C28H28N4Pd1Cl2・H2O:計算値:C:54.60%,H:4.91%,N:9.10%;実測値:C:54.40%,H:4.48%,N:8.98%。
【0085】
実施例7:Pt(bpy)(AtC3)
【0086】
【化19】
【0087】
[Pt(bpy)(AtC3)]Cl2の合成
文献(Goto, M., et al., Bull. Chem. Soc. Jpn. 2000, 73, 97-105)に従って、Pt(bpy)Cl2(0.42g,1.0ミリモル)の20mlのH2O懸濁液へ0.44g(1.3ミリモル)のAtC3・2HClと0.16g(1.5ミリモル)のNa2CO3を加え、この混合物を80℃で約3時間撹拌した。次いで、この混合物をまだ熱い間に濾過して、少量の不溶性材料を除去した。室温へ冷却後、薄黄色の沈殿が生じ、これをフィルターで採取して、デシケーター中で乾燥させた。収量:0.60g(73%)。
【0088】
元素分析 C28H28N4Pt1Cl2:計算値:C:48.99%,H:4.11%,N:8.16%;実測値:C:49.13,48.78%,H:4.37,4.29%,N:8.08,8.01%。
【0089】
【化20】
【0090】
実施例8:cis−Pt(NH3)2(ピロリン酸)
【0091】
【化21】
【0092】
cis−Pt(NH3)2(ピロリン酸)の合成
ジメチルアセトアミド 5ml中0.482g(1ミリモル)のcis−Pt(NH3)2I2を0.306g(0.98ミリモル)のAg2SO4の40mL H2O懸濁液へ加え、暗所で約4時間撹拌した。膜フィルターによる濾過後、80mL H2O中のBa(OH)2・8H2O 0.305g(1.00ミリモル)を加えて、30分間撹拌した。濾過後、この溶液のpHをNaOH水溶液により3〜4へ調整し、1:1 ピロリン酸(2.0ミリモル):H2O 0.5gを加えて、1時間撹拌した。濾過後、この溶液を蒸発により濃縮して、生じる粉末を、MeOHを加えることによって沈殿させた。この緑がかった粉末を温水に溶かした。濾過した溶液を真空で濃縮して、MeOHを加えた。沈積した白い粉末をエーテルで洗浄した。収率:30%。
【0093】
元素分析 H8N2O6P2Pt・1H2O:計算値:H:1.91%,N:6.65%;実測値:H:2.28,2.27%,N:6.90,6.62%。
31P NMR(D2O,85% H3PO4):δ(ppm)0ppm。
【0094】
実施例9:Pt(NH3)−IP6 Pt(NH3)2・IP6・10Na・7H2O
【0095】
【化22】
【0096】
cis−Pt(NH3)2・IP6の合成
N,N−ジメチルアセトアミド 5mlに溶かしたcis−Pt(NH3)2・I2 0.483g(1.0ミリモル)をAgNO3 0.340g(2.0ミリモル)のH2O 40ml懸濁液へ加えて、暗所で一晩撹拌した。膜フィルターによる濾過後、H2O 30ml中のIP6・12Na 1.013g(1.0ミリモル)を加えて、3時間撹拌した。この溶液を蒸発により濃縮して、生じる粉末を、MeOHを加えることによって再沈殿させた。収率:54%。
【0097】
元素分析 C6H26N2O31P6Pt1Na10:計算値:C:5.84%,H:2.11%,N:2.27%;実測値:C:5.95%,H:2.15%,N:2.12%。
【0098】
【化23】
【0099】
XRF 計算値:P 6.00,Na 10.0,Pt 1.00;実測値:P 6.00,Na 10.9,Pt 0.94。
実施例10:Pt(dach)−IP6
【0100】
【化24】
【0101】
cis−Pt(dach)・IP6の合成
N,N−ジメチルアセトアミド 5mlに溶かしたcis−Pt(dach)I2 0.563g(1.0ミリモル)をAgNO3 0.340g(2.0ミリモル)のH2O 20ml懸濁液へ加えて、暗所で一晩撹拌した。膜フィルターによる濾過後、H2O 30ml中のIP6・12Na 1.013g(1.0ミリモル)を加えて、3時間撹拌した。この溶液を蒸発により濃縮して、生じる粉末を、MeOH 100mlを加えることによって再沈殿させた。収率:73%。
【0102】
元素分析 C12H44N2O36P6Pt1Na10:計算値:C:10.26%,H:3.14%,N:2.00%;実測値:C:10.13%,H:2.88%,N:1.71%。
【0103】
XRF 計算値:P 6.00,Na 10.0,Pt 1.00;実測値:P 6.00,Na 10.3,Pt 1.42。
【0104】
【化25】
【0105】
実施例11:Pt(NH3)2(NDP)
【0106】
【化26】
【0107】
cis−Pt(NH3)2・NDPの合成
ジメチルアセトアミド 5ml中の工程b.1.からの0.20g(0.5ミリモル)のcis−Pt(NH3)2I2を0.149g(0.48ミリモル)のAg2SO4の20mL H2O懸濁液へ加え、暗所で約4時間撹拌した。膜フィルターによる濾過後、20mL H2O中のBa(OH)2・8H2O 0.15g(0.48ミリモル)及びイミノ二リン酸ナトリウム(NDP)0.19g(0.70ミリモル)を加えた。一晩撹拌後、1M HClO4水溶液1.4mlを加え、この溶液を蒸発により濃縮して、生じる粉末を、H2O−EtOHを使用して再沈殿させた。収率:35%。
【0108】
元素分析 H9N3O6P2Pt1・1H2O:計算値:H:2.24%,N:10.40%;実測値:H:1.99%,N:10.01%。
31P NMR(D2O,85% H3PO4):δ(ppm)+10。
【0109】
実施例12:Pt(NH3)2(MDPOH)
【0110】
【化27】
【0111】
cis−Pt(NH3)2・MDPOHの合成
ジメチルアセトアミド 5ml中の0.20g(0.5ミリモル)のcis−Pt(NH3)2I2を0.149g(0.48ミリモル)のAg2SO4の20mL H2O懸濁液へ加え、暗所で約4時間撹拌した。膜フィルターによる濾過後、20mL H2O中のBa(OH)2・8H2O 0.15g(0.48ミリモル)及び60% 1−ヒドロキシエタン−1,1−ビス(ホスホン酸)(エチドロン酸、MDPOH)0.24g(0.70ミリモル)を加えた。一晩撹拌後、この溶液を蒸発により濃縮して、生じる粉末を、H2O−EtOHを使用して再沈殿させた。収率:65%。
【0112】
元素分析 C2H12N2O7P2Pt1:計算値:C:5.54%,H:2.50%,N:6.95%;実測値:C:5.46%,H:2.30%,N:6.76%。
実施例13:in vitro アッセイ−細胞増殖阻害
a.材料と方法
KB細胞は、Human Science Research Resource Bank(ヒューマンサイエンス研究資源バンク;大阪、日本)より購入した。この細胞を、10% FBS(Bio Whittaker)を含有するEarle’s MEM(GIBCO BRL)において5% CO2下に37℃で培養した。24時間後、この細胞へ試験化合物を指定濃度で加えた。72時間後、この細胞をトリパンブルーで染色して、手動で計数した。細胞の増殖を50%阻害するのに必要とされる錯体の濃度(水溶液)としてIC50を計算する。この結果を表1、2、及び3に示す。
【0113】
b.結果
表1
【0114】
【表1】
【0115】
表2
【0116】
【表2】
【0117】
表3
【0118】
【表3】
【0119】
同様のやり方で、本発明の化合物の実施例の代表的な番号について、いくつかの異なる癌を代表する細胞種の成長/増殖を阻害するその能力を試験した。この結果を、様々な細胞系に対する各化合物の阻害定数(IC50,μM)を提供する以下の表4〜13に提供する。
【0120】
表4 乳癌細胞系
【0121】
【表4】
【0122】
表5 脳腫瘍細胞系
【0123】
【表5】
【0124】
表6 結腸癌細胞系
【0125】
【表6】
【0126】
表7 肺癌細胞系
【0127】
【表7】
【0128】
表8 メラノーマ細胞系
【0129】
【表8】
【0130】
表9 卵巣癌細胞系
【0131】
【表9】
【0132】
表10 腎癌
【0133】
【表10】
【0134】
表11 ヒト胃癌
【0135】
【表11】
【0136】
表12 前立腺癌
【0137】
【表12】
【0138】
表13 平均IC50(全細胞種)
【0139】
【表13】
【0140】
上記のデータは、本発明の化合物が癌性細胞の有意な阻害を明示することを示す。
実施例14:in vitro アッセイ−Pt錯体のヒドロキシアパタイト、Ca10(PO4)6(OH)2への吸着
5、10、15、25、35、及び100mg試料のヒドロキシアパタイト(Bio-Rad Macro-Prep Ceramic Hydroxyapatite, I型、40μm)のそれぞれを2mlのHEPES緩衝液へpD=7.8で加えて、生じる混合物を37℃で約24時間振り混ぜた。次いで、HEPES緩衝液にpD=7.8で溶かした各錯体の100μlを加えて、この混合物を37℃で1.5時間振り混ぜた。すべての懸濁液を濾過して、これらの溶液を白金の31P NMR(Varian VXR−300S)及び Atomic Absorption Spectrometry(日立、Z−5710 AAS)によって測定するか、又はカルボプラチンでは、1H NMRによって測定した。ヒドロキシアパタイトに対する吸着百分率を以下のように計算した:
NMRより:結合百分率/%=[(A−B)/A]x100
AASより:結合百分率/%=[(C−D)/C]x100
A=Pt(II)錯体のインテグレーション(integration)強度
B=ヒドロキシアパタイトと反応後のPt(II)錯体のインテグレーション強度
C=Pt(II)錯体の濃度
D=ヒドロキシアパタイトと反応後のPt(II)錯体の濃度
図1及び2は、31P NMR、1H NMR、及びAASより計算したヒドロキシアパタイトへの吸着百分率を図示する。これらの結果は、cis−Pt(NH3)2(MDP)とPt(dach)(MDP)がPt(II)錯体としてヒドロキシアパタイトへ吸着することを示した。Pt(dach)(MDP)は、cis−Pt(NH3)2(MDP)よりややよく吸着して、Pt(dach)(MDP)とcis−Pt(NH3)2(MDP)は、ともにカルボプラチンよりきわめて有意によく吸着した。Pt(II)錯体を100mgのヒドロキシアパタイトと反応させるとき、cis−Pt(NH3)2(MDP)の吸着百分率は69.3%であり、Pt(dach)(MDP)のそれは79.6%であった。
【0141】
実施例15−in vivo 腫瘍アッセイ
6〜8週齢の雄性NCr−ヌードマウスに水(逆浸透、0.17% Cl)と18%タンパク質;5%脂肪;5%繊維;8%灰分;及び3%ミネラルからなる滅菌済み標準齧歯動物(NIH31)食を自由摂取させた。マウスは、22℃(72°F)、湿度40%〜60%、12時間の明周期でミクロアイソレーターに収容した。マウスの脇腹に5x106個のDU145ヒト前立腺癌細胞を皮下移植した。はじめは週2回、次いで毎日腫瘍をモニターすると、この新生物が所望のサイズ、ほぼ100mm3(100mg)に達した。DU145前立腺癌がこのサイズに達したとき、この動物を様々な処理群へ対マッチさせた。式:腫瘍重量(mg)=(w2XL)/2(ここで、w=腫瘍の幅で、L=腫瘍の長さ(mm)である)を使用して、推定腫瘍重量を計算した。
【0142】
本発明の代表的な化合物をこの動物へ腹腔内注射して、有意な抗腫瘍活性を保有することを見出した。以下の図3〜6を参照のこと。
一般に、本発明の化合物は、無機酸及び有機酸を使用することより誘導される塩のように、その医薬的に許容される酸付加塩の形態で単離することができる。そのような酸の例は、塩酸、硝酸、硫酸、リン酸、ギ酸、酢酸、トリフルオロ酢酸、プロピオン酸、マレイン酸、コハク酸、D−酒石酸、L−酒石酸、マロン酸、メタンスルホン酸、等である。さらに、カルボキシのような酸性官能基を含有する一定の化合物をその無機塩の形態で単離してよく、ここで対イオンは、ナトリウム、カリウム、リチウム、カルシウム、マグネシウム、等より、並びに有機塩基より選択してよい。
【0143】
医薬的に許容される塩は、本発明の化合物(例、以下の化合物C)の約1当量を取り、所望される塩の適切な対応する酸の約1当量以上とそれを接触させることによって生成することができる。生じる塩の後処理及び単離は、当業者によく知られている。
【0144】
本発明の化合物は、経口、非経口(例、筋肉内、腹腔内、静脈内、又は皮下の注射又はインプラント)、経鼻、膣、直腸、舌下、又は局所の投与経路によって投与してよく、医薬的に許容される担体とともに製剤化してそれぞれの投与経路に適した剤形を提供することができる。従って、本発明には、その範囲内に、本発明の少なくとも1つの化合物を有効成分として医薬的に許容される担体と一緒に含んでなる医薬組成物が含まれる。
【0145】
経口投与用の固体剤形には、カプセル剤、錠剤、丸剤、散剤、及び顆粒剤が含まれる。そのような固体剤形では、ショ糖、乳糖、又はデンプンのような少なくとも1つの不活性な医薬的に許容される担体と活性化合物を混合する。そのような剤形は、通常の実践であるように、そのような不活性希釈剤とは別の追加物質、例えば、ステアリン酸マグネシウムのような滑沢剤も含んでよい。カプセル剤、錠剤、及び丸剤の場合、剤形は、緩衝剤も含んでよい。錠剤と丸剤は、追加的に腸溶コーティング剤とともに調製してよい。
【0146】
経口投与用の液体剤形には、当該技術分野で一般的に使用される、水のような不活性希釈剤を含有する医薬的に許容される乳剤、溶液剤、懸濁液剤、シロップ剤、エリキシル剤が含まれる。そのような不活性希釈剤以外に、組成物には、湿潤剤、乳化剤、及び懸濁剤、並びに甘味剤、芳香剤、及び発香剤のようなアジュバントも含めてよい。
【0147】
本発明による非経口投与用の調製物には、無菌の水系又は非水系溶液剤、懸濁液剤、又は乳剤が含まれる。非水系溶媒又は担体の例は、プロピレングリコール、ポリエチレングリコール、オリーブ油又はとうもろこし油のような植物油、ゼラチン、及びオレイン酸エチルのような注射可能な有機エステルである。そのような剤形は、保存剤、湿潤剤、乳化剤、及び分散剤のようなアジュバントも含有してよい。それらは、例えば、細菌保持フィルターを通した濾過によって、滅菌剤を組成物へ取り込ませることによって、組成物に照射することによって、又は組成物を加熱することによって滅菌してよい。それらは、使用直前に滅菌水や他の無菌の注射可能な媒体に溶かすことができる、無菌の固体組成物の形態で製造してもよい。
【0148】
直腸又は膣からの投与用の組成物は、好ましくは、活性物質に加えて、ココア脂や坐剤ワックスのような賦形剤を含有し得る坐剤である。
経鼻又は舌下投与用の組成物も、当該技術分野でよく知られた標準の賦形剤とともに調製する。
【0149】
一般に、本発明の組成物中の有効成分の有効投与量は変動してよいが、有効成分の量は、好適な剤形が得られるようにすることが必要である。選択される投与量は、所望される治療効果、投与経路、及び治療の期間に依存し、このいずれも当業者の知識の領域内にある。一般に、1日あたり体重1kgにつき0.0001〜100mgの間の投与量レベルをヒトや他の動物、例えば哺乳動物へ投与する。
【0150】
好ましい投与量範囲は、1日あたり体重1kgにつき0.01〜10.0mgであり、これは単回用量として投与しても、頻回用量へ分割してもよい。
本発明の様々な態様を詳しく記載してきたが、本発明のさらなる修飾及び適応が当業者に思いつくことは明らかである。しかしながら、そのような修飾及び適応が本発明の精神及び範囲の内にあることは、はっきりと理解されるべきである。本明細書に引用する参考文献は、いずれもそのまま参照により本明細書に組み込まれる。
【技術分野】
【0001】
発明の分野
本発明は、特別なビスホスホネート化合物、そして特に、骨癌や骨粗鬆症のような骨疾患及び骨関連疾患の周囲にある柔組織の治療に有用であるビスホスホネート抱合体へ向けられる。
【0002】
背景技術
ビスホスホネートは、骨粗鬆症、ページェット病、及び悪性の高カルシウム血症が含まれる、異常に促進された骨吸収に関連したいくつかの疾患の治療においてきわめて有望な治療効果を示した医薬品のクラスを代表する。Fleish H., Ann Med, 29, 55-62 (1997) 及び FleishH., Drugs, 42, 919-944 (1991)。より最近、ビスホスホネートは、骨へ拡がった前立腺癌のある患者において骨格の合併症(例、病的な骨折、脊髄圧迫、骨手術又は放射線療法の必要性)が発症するリスクを低下させるのに有効であること(Saad F et al., J National Cancer Institute 94: 1458-1468, 2002);そしてRAS依存型悪性疾患(例、小細胞肺癌)の増殖を阻害すること(Matsumoto, et al., Am. Soc. of Clin. Oncology, 2003, Abst. No. 2750)が示された。ビスホスホネートは、抗血管形成活性を有することも示されている。Wood et al, J Pharmacol Exp Ther 2002 Sep; 302(3): 1055-61。ビスホスホネートは、通常、骨髄腫骨疾患の治療と乳癌の溶骨性転移に抗して使用され、臨床試験は、転移性前立腺癌における疼痛を緩和するためのその使用を示唆した。
【0003】
白金ベースの薬剤は、化学療法の適用に広く利用されている。例えば、シスプラチンは、共有結合性、交差性又は鎖内DNA付加物の形成を介して腫瘍細胞を殺す(Sherman et al. Chem. Rev., 87, 1153-81 (1987); Chu, J. Biol. Chem., 269, 787-90 (1994))。そのような白金ベースの薬剤での治療は、それによりDNA合成の阻害をもたらす(Howle et al., Biochem. Pharmacol., 19, 2757-62 (1970); Salles et al., Biochem. Biophys. Res. Commun., 112, 555-63 (1983))。従って、DNAを活発に合成する細胞は、シスプラチンに対して高感受性である(Roberts et al., Prog. Nucl. Acid Res. Mol. Biol., 22, 71-133 (1979); Pinto et al., Proc. Nat. Acad Sci. (Wash.) 82, 4616-19 (1985))。一般に、そのような細胞は、G2において増殖阻止を経験して、最終的にはアポトーシスを蒙る。このアポトーシス効果は、DNA合成を阻害するのに不十分な薬物濃度で観察され(Sorenson et al., J. Natl. Cancer Inst., 82, 749-55 (1990))、白金剤が多数の機序を介して新生物細胞に作用することを示唆する。細胞周期のG1期にあるときに高められた白金感受性を明示する細胞もある(Krishnaswamy et al., Mutation Res., 293, 161-72 (1993); Donaldson et al., Int. J. Cancer, 57, 847-55 (1994))。そのような細胞は、G0/G1−Sブロックより解放されると、細胞周期の残りを通して最大に感作されたままである。
【0004】
米国特許第6,087,349号は、ビスホスホネートがタンパク質プレニルトランスフェラーゼ阻害剤として作用し得ることを開示する。
米国特許第4,746,654号は、抗炎症剤として有用なビスホスホネートを開示する。
【0005】
オーストラリア特許A−5 1534/85は、異常なカルシウム及びリンの代謝を治療するのに有用で、関節炎を治療するのに有用なビスホスホネートを開示する。
米国特許第3,683,080号は、リン酸カルシウムの動物組織における異常な沈着及び動員を阻害するのに有用なポリホスホネート、特にジホスホネートを開示する。
【0006】
DE3,719,513−A(Derwent 89−000580/01)は、カルシウム代謝の障害の治療に有用なジホスホン酸誘導体を開示する。
WO88/06158は、ビニリデンジホスホネートと活性化メチレンの反応を開示する。
【0007】
国際特許出願番号PCT/US90/01106に代わる国際特許公開公報番号WO90/12017は、抗関節炎剤としてのジェミナルビスホスホン酸とその誘導体を開示する。
【0008】
米国特許出願番号20020022603は、両性イオンリン脂質及びビスホスホネートの組成物と、GI毒性の低下したビスリン酸送達系としてのそのような組成物の使用を開示する。
【0009】
米国特許出願番号20030032628は、湿式造粒の錠剤製剤化によって調製する、ビスホスホン酸とその塩の医薬組成物を開示する。これらの医薬組成物は、結合剤の添加なしに調製される;むしろ、薬物それ自身が結合剤として作用すると言われる。
【0010】
米国特許出願番号20020002140は、顕著に高められた腸吸収性と高められたバイオアベイラビリティを有すると言われる、ビスホスホネート化合物のグリコシド及びオルトエステルグリコシド誘導体を開示する。
【0011】
米国特許第5,133,972号は、経皮送達のリン酸化合物、特にビスホスホネートを開示する。関連して、米国特許第6,018,679号は、皮膚刺激や他の有害な効果を引き起こすことが可能な化合物をイオン泳動的に除去する方法を開示する。
【0012】
米国特許第6,114,316号は、過剰なプロテイナーゼ活性に関連した組織破壊状態を生物系において治療するか又は予防する相乗的なプロテイナーゼ阻害量においてテトラサイクリンとビスホスホネートを組み合わせる組成物を開示する。
【0013】
米国特許第6,214,812号は、骨組織との結合時に抗菌及び/又は細胞傷害成分を放出することが可能であると言われるビスホスホネート抱合体を開示する。
米国特許第6,436,386号は、ヒドロキシアパタイト標的指向性ポリマー構造とその生物学的に活性な抱合体を開示し、ここでヒドロキシアパタイト標的指向性部分は、ビスホスホネートであってよい。この抱合体は、生物学的に活性な物質を骨表面へつなげるための手段を提供すると言われる。
【0014】
当該技術分野には、様々な種類のビスホスホネート化合物、抱合体、製剤、組合せ、及びそれらの使用について記載する多数の他の参考文献を見出すことができる。しかしながら、そこには、白金、パラジウム、又は同様の部分を含んでなる、治療上有用であるビスホスホネート錯体を合成する方法が開示されていない。当該技術分野で知られている他のビスホスホネート錯体が治療薬剤としてのその使用に関して、特に癌の治療へのその使用に関して、より特別には、骨組織に影響を及ぼす癌の治療へのその使用に関してより優れた特性を保有することは知られていなかった。
【0015】
発明の要約
本発明は、ビスホスホネート錯体と、標的指向される細胞増殖抑制剤及び/又は細胞傷害剤としてのその使用に関する。望ましくは、本発明のビスホスホネート錯体は、細胞、例えば骨に関連した癌性細胞に標的指向するために使用することができる。
【0016】
第一の側面において、本発明は、式I:
【0017】
【化1】
【0018】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第一の側面の好ましい態様では、MがPt(II)である。
【0019】
第二の側面において、本発明は、式II:
【0020】
【化2】
【0021】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第二の側面の好ましい態様では、MがPt(II)である。
【0022】
第三の側面において、本発明は、式III:
【0023】
【化3】
【0024】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第三の側面の好ましい態様では、MがPt(II)である。
【0025】
第四の側面において、本発明は、式IV:
【0026】
【化4】
【0027】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第四の側面の好ましい態様では、MがPt(II)である。
【0028】
第五の側面において、本発明は、式V:
【0029】
【化5】
【0030】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第五の側面の好ましい態様では、MがPt(II)である。
【0031】
第六の側面において、本発明は、式VI:
【0032】
【化6】
【0033】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第六の側面の好ましい態様では、MがPt(II)である。
【0034】
第七の側面において、本発明は、式VII:
【0035】
【化7】
【0036】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第七の側面の好ましい態様では、MがPt(II)である。
【0037】
第八の側面において、本発明は、式VIII:
【0038】
【化8】
【0039】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩に関する。
前記第八の側面の好ましい態様では、MがPt(II)である。
【0040】
第九の側面において、本発明は、式Iの実施例について記載する合成手順を実質的に本明細書に記載のように実施することを含んでなる、式Iによる化合物を合成する方法に関する。前記第九の側面の好ましい態様では、MがPt(II)である。
【0041】
第十の側面において、本発明は、式IIの実施例について記載する合成手順を実質的に本明細書に記載のように実施することを含んでなる、式IIによる化合物を合成する方法に関する。前記第十の側面の好ましい態様では、MがPt(II)である。
【0042】
第十一の側面において、本発明は、式IIIの実施例について記載する合成手順を実質的に本明細書に記載のように実施することを含んでなる、式IIIによる化合物を合成する方法に関する。前記側面の好ましい態様では、MがPt(II)である。
【0043】
第十二の側面において、本発明は、細胞増殖抑制的に、及び/又は細胞傷害的に有効な量の白金又はパラジウム、又は白金含有又はパラジウム含有部分をその必要な被検者へ送達する方法に関し、前記方法は、式Iによる化合物又はその医薬的に許容される塩の治療有効量をその必要な前記被検者へ投与することを含んでなる。好ましくは、式IのMは、白金である。また好ましくは、前記方法は、前記化合物を前記被検者内の骨組織にか又は骨組織へ送達することを含む。より好ましくは、前記方法は、癌細胞がその上か又はその中に存在する骨組織にか又は骨組織へ送達することを含む。なおより好ましくは、前記化合物の前記治療有効量は、前記癌細胞を治療する、即ちそれを阻害する、及び/又は殺すのに有効な量である。
【0044】
第十三の側面において、本発明は、細胞増殖抑制的に、及び/又は細胞傷害的に有効な量の白金又はパラジウム、又はパラジウム含有又は白金含有部分をその必要な被検者へ送達する方法に関し、前記方法は、式IIによる化合物又はその医薬的に許容される塩の治療有効量をその必要な前記被検者へ投与することを含んでなる。好ましくは、式IIのMは、白金である。また好ましくは、前記方法は、前記化合物を前記被検者内の骨組織にか又は骨組織へ送達することを含む。より好ましくは、前記方法は、癌細胞がその上か又はその中に存在する骨組織にか又は骨組織へ送達することを含む。なおより好ましくは、前記化合物の前記治療有効量は、前記癌細胞を治療する、即ちそれを阻害する、及び/又は殺すのに有効な量である。
【0045】
第十四の側面において、本発明は、細胞増殖抑制的に、及び/又は細胞傷害的に有効な量の白金又はパラジウム、又はパラジウム含有又は白金含有部分をその必要な被検者へ送達する方法に関し、前記方法は、式IIIによる化合物又はその医薬的に許容される塩の治療有効量をその必要な前記被検者へ投与することを含んでなる。好ましくは、式IIIのMは、白金である。また好ましくは、前記方法は、前記化合物を前記被検者内の骨組織にか又は骨組織へ送達することを含む。より好ましくは、前記方法は、癌細胞がその上か又はその中に存在する骨組織にか又は骨組織へ送達することを含む。なおより好ましくは、前記化合物の前記治療有効量は、前記癌細胞を治療する、即ちそれを阻害する、及び/又は殺すのに有効な量である。
【図面の簡単な説明】
【0046】
【図1】図1は、NMRにより測定される、試験化合物のヒドロキシアパタイトへの吸着百分率を示すグラフである。
【図2】図2は、31P NMR、1H NMR、及びAASにより測定される、試験化合物のヒドロキシアパタイトへの吸着百分率を示すグラフである。
【図3】図3は、無胸腺ヌードマウス中の腫瘍モデルDU−145に抗する実施例番号7の効果を例示するグラフである。
【図4】図4は、無胸腺ヌードマウス中の腫瘍モデルDU−145に抗する実施例番号2の効果を例示するグラフである。
【図5】図5は、無胸腺ヌードマウス中の腫瘍モデルDU−145に抗する実施例番号4の効果を例示するグラフである。
【図6】図6は、無胸腺ヌードマウス中の腫瘍モデルDU−145に抗する実施例番号1及び番号8の効果を例示するグラフである。
【0047】
好ましい態様の詳細な説明
以下の実験結果は、例示の目的のために提供されるのであって、本発明の範囲を限定することを企図しない。
【0048】
実施例1:cis−Pt(NH3)2(MDP)
a.材料
ビスホスホネート(メチレンジホスフィン酸(MDP))は、Tokyo-Kasei(東京化成)より、K2PtCl4は、Tanaka(田中化学研究所)より、ジメチルアセトアミド(DMA)と他の試薬は、Nakarai Tesque(ナカライテスク)より購入した。すべての化学品は、入手可能な最高級品であり、さらに精製せずに使用した。水は脱イオン化し、二回蒸留して、最後はMilli−Qにより精製した。
【0049】
b.手順
【0050】
【化9】
【0051】
b.1.cis−Pt(NH3)2I2
文献(S. C. Dhara, Indian J. Chem, 1970, 8)に従って、20ml H2O中のK2PtCl4 2g(4.8ミリモル)へKI 3.3g(19.8ミリモル)を加えた。この溶液を水浴中で5分間撹拌してから、47mLの0.21M NH3水溶液へ加えた。室温で約3時間静置後、沈積した黄色い粉末を濾過して取り、温水、EtOH、及びエーテルで洗浄した。収率:91%。元素分析:計算値:H:1.25%,N:5.80%;実測値:H:1.08%,N:5.58%。IR:3260cm−1,3200cm−1,1282cm−1,1270cm−1。
【0052】
b.2.cis−Pt(NH3)2(MDP)
ジメチルアセトアミド 5ml中の工程b.1.からの0.20g(0.5ミリモル)のcis−Pt(NH3)2I2を0.149g(0.48ミリモル)のAg2SO4の20mL H2O懸濁液へ加え、暗所で約4時間撹拌した。膜フィルターによる濾過後、20mL H2O中のBa(OH)2・8H2O 0.15g(0.48ミリモル)及びMDP 0.12g(0.70ミリモル)を加えた。一晩撹拌後、この溶液を蒸発により濃縮して、生じる粉末を、H2O−EtOHを使用して再沈殿させた。収率:35%。
【0053】
元素分析:計算値:C:2.98%,H:2.50%,N:6.95%;実測値:C:3.14%,H:2.50%,N:6.83%。
【0054】
【化10】
【0055】
実施例2:Pt(dach)(MDP)
a.材料
1R,2R−1,2−シクロヘキサンジアミン(dach)は、Tokyo-Kasei より;K2PtCl4は、Tanaka より;ジメチルアセトアミド(DMA)と他の試薬は、Nakarai Tesque より購入した。すべての化学品は、入手可能な最高級品であり、さらに精製せずに使用した。水は脱イオン化し、蒸留して、最後はMilli−Qにより精製した。
【0056】
b.手順
【0057】
【化11】
【0058】
b.1.Pt(dach)2I2
20ml H2O中のK2PtCl4 1.25g(3ミリモル)へKI 2.0g(12ミリモル)を加えた。この溶液を水浴中(50℃)で約5分間撹拌してから、1R,2R−1,2−シクロヘキサンジアミン 0.34g(3ミリモル)へ加えた。この反応混合物を室温で一晩撹拌し、沈積した黄色い粉末を濾過して取り、温水、次いでEtOH、最後はジエチルエーテルで洗浄した。収率:91%。
【0059】
元素分析:計算値:C:12.80%,H:2.51%,N:4.98%;実測値:C:12.66%,H:2.27%,N:5.01%。
b.2.Pt(dach)(MDP)
ジメチルアセトアミド 5ml中の工程b.1.からの0.28g(0.5ミリモル)のPt(dach)2I2を0.149g(0.48ミリモル)のAg2SO4の20mL H2O懸濁液へ加え、暗所で約4時間撹拌した。膜フィルターによる濾過後、20mL H2O中のBa(OH)2・8H2O 0.156g(0.48ミリモル)及びMDP 0.129g(0.75ミリモル)を加えた。約2時間撹拌後、この反応溶液混合物へ約5分にわたり撹拌しながら1mlの0.5M H2SO4水溶液を加えた。この混合物を濾過し、蒸発により約5mlへ濃縮して、MeOHを使用して白い粉末を再沈殿させた。収率:35%。
【0060】
元素分析:計算値:C:17.40%,H:3.75%,N:5.80%;実測値:C:17.47%,H:3.44%,N:5.89%。
【0061】
【化12】
【0062】
実施例3:N−(9−アントラニル)メチル−1,2−エタンジアミン二塩酸塩(Aten・2HCl)
a.材料
N−(9−アントラニル)アルデヒドは、Tokyo-Kasei より購入し;K2PtCl4は、Tanaka より購入し;PdCl2は、Kishida Chemical(キシダ化学)より購入し;2,2’−ビピリジン(bpy)は、Wako(和光純薬工業)より;DMSOと他の試薬は、Nakarai Tesque より購入した。すべての化学品は、入手可能な最高級品であり、さらに精製せずに使用した。水は脱イオン化し、二回蒸留して、最後はMilli−Qにより精製した。
【0063】
b.手順
【0064】
【化13】
【0065】
200ml 1:1 ジオキサン−CHCl3中のN−(9−アントラニル)アルデヒド 2.06g(10ミリモル)へエチレンジアミン 6.00g(10ミリモル)を加えて、この溶液を約3時間還流した。室温へ冷却後、この溶液を蒸発により濃縮して、MeOH中0.45g(12ミリモル)のNaBH4を加えた。一晩撹拌後、6N HClを加えてpHを1へ調整して、液体を蒸発させた。NaOH水溶液を加え、この塩基性溶液をCHCl3で抽出して、Na2SO4を使用して乾燥させた。CHCl3相を蒸発させて、油性残渣をMeOH/6N HClで処理した。生じる黄色がかった粉末を濾過により回収して、EtOH−H2Oより再結晶させた。収量:2.2g(65%)。
【0066】
元素分析 C17H20N2Cl2・0.75H2O:計算値:C:60.63%,H:6.43%,N:8.32%;実測値:C:60.40%,H:6.45%,N:7.81%。
【0067】
【化14】
【0068】
実施例4:Pt(bpy)(Aten)錯体
a.Pt(bpy)Cl2
10mL H2O、30mL DMSOの溶液中1.411g(3.4ミリモル)のK2PtCl4を0.531g(3.4ミリモル)のbpy(50mL DMSO)へ加え、この混合物を撹拌しながら80℃まで約3時間加熱した。この反応物を一晩撹拌し、生じる黄色い針状物を濾過して取り、H2Oとエーテルで洗浄した。収量:1.2g(81%)。
【0069】
元素分析 C10H8N2Cl2Pt:計算値:C:28.45%,H:1.91%,N:6.58%;実測値:C:28.65%,H:1.95%,N:6.64%。
b.[Pt(bpy)(Aten)]Cl2
【0070】
【化15】
【0071】
文献(Goto, M., et al., Bull. Chem. Soc. Jpn. 2000, 73, 97-105)に従って、Pt(bpy)Cl2(0.42g,1.0ミリモル)の20ml H2O懸濁液へ0.44g(1.3ミリモル)のAten・2HCl・0.75H2Oと0.16g(1.5ミリモル)のNa2CO3を加え、この混合物を80℃で約2.5時間撹拌した。次いで、この混合物をまだ熱い間に濾過して、少量の不溶性材料を除去した。室温へ冷却後、薄黄色の沈殿が生じ、これをフィルターで採取して、デシケーター中で乾燥させた。収量:0.56g(79%)。
【0072】
元素分析 C27H26N4Cl2Pt・2H2O:計算値:C:45.77%,H:4.27%,N:7.91%;実測値:C:45.51%,H:4.05%,N:7.61%。
【0073】
実施例5:Pd(bpy)(Aten)錯体
a.Pd(bpy)Cl2
PdCl2 0.89g(5.0ミリモル)とNaCl 0.58g(10.0ミリモル)を50mL H2Oに懸濁させて、約1時間撹拌した。濾過後、この溶液を20mL MeOH中0.78g(5.0ミリモル)bpyの溶液へ加えて、生じる溶液を一晩撹拌した。黄色がかった粉末が沈殿し、これを回収して、H2OとEtOHで洗浄した。収量:1.56g(93%)。
【0074】
元素分析 C10H8N2Cl2Pd:計算値:C:36.01%,H:2.42%,N:8.40%;実測値:C:35.94%,H:2.14%,N:8.31%。
b.[Pd(bpy)(Aten)]Cl2
【0075】
【化16】
【0076】
直前の工程からの0.33g(1.0ミリモル)のPd(bpy)Cl2を10ml H2Oに懸濁させた。0.44g(1.3ミリモル)のAten・2HCl・0.75H2Oと0.16g(1.5ミリモル)のNa2CO3を加えてから、この混合物を80℃まで約2時間加熱した。次いで、この溶液をまだ熱い間に濾過して、蒸発により濃縮した。室温で静置させると、黄色い粉末が沈積した。収量:0.43g(68%)。
【0077】
元素分析 C27H26N4Cl2Pd・3H2O:計算値:C:50.84%,H:5.06%,N:8.78%;実測値:C:50.89%,H:4.69%,N:8.80%。
【0078】
実施例6:Pd(bpy)(AtC3)
【0079】
【化17】
【0080】
a.AtC3・2HCl
200ml 1:1 ジオキサン−CHCl3中のN−(9−アントラニル)アルデヒド 2.06g(10ミリモル)へ1,3−ジアミンプロパン 7.41g(100ミリモル)を加えて、この溶液を約3時間還流した。室温へ冷却後、この溶液を蒸発により濃縮して、MeOH中0.45g(12ミリモル)のNaBH4を加えた。一晩撹拌後、6N HClを加えてpHを1へ調整して、溶液を蒸発させた。NaOH水溶液を加え、この塩基性溶液をCHCl3で抽出して、Na2SO4を使用して乾燥させた。CHCl3相を蒸発させて、油性残渣をMeOH/6N HClで処理した。生じる黄色がかった粉末を濾過により回収して、EtOH−H2Oより再結晶させた。収率:70%。
【0081】
元素分析 C18H22N2Cl2:計算値:C:64.10%,H:6.57%,N:8.31%;実測値:C:64.40%,H:6.38%,N:7.98%。
【0082】
【化18】
【0083】
b.[Pd(bpy)(AtC3)]Cl2
直前の工程からの0.33g(1.0ミリモル)のPd(bpy)Cl2を10ml H2Oに懸濁させた。0.44g(1.3ミリモル)のAtC3・2HClと0.16g(1.5ミリモル)のNa2CO3を加えてから、この混合物を80℃まで約2時間加熱した。次いで、この溶液をまだ熱い間に濾過して、蒸発により濃縮した。室温で静置させると、黄色い粉末が沈積した。収量:0.40g(58%)。
【0084】
元素分析 C28H28N4Pd1Cl2・H2O:計算値:C:54.60%,H:4.91%,N:9.10%;実測値:C:54.40%,H:4.48%,N:8.98%。
【0085】
実施例7:Pt(bpy)(AtC3)
【0086】
【化19】
【0087】
[Pt(bpy)(AtC3)]Cl2の合成
文献(Goto, M., et al., Bull. Chem. Soc. Jpn. 2000, 73, 97-105)に従って、Pt(bpy)Cl2(0.42g,1.0ミリモル)の20mlのH2O懸濁液へ0.44g(1.3ミリモル)のAtC3・2HClと0.16g(1.5ミリモル)のNa2CO3を加え、この混合物を80℃で約3時間撹拌した。次いで、この混合物をまだ熱い間に濾過して、少量の不溶性材料を除去した。室温へ冷却後、薄黄色の沈殿が生じ、これをフィルターで採取して、デシケーター中で乾燥させた。収量:0.60g(73%)。
【0088】
元素分析 C28H28N4Pt1Cl2:計算値:C:48.99%,H:4.11%,N:8.16%;実測値:C:49.13,48.78%,H:4.37,4.29%,N:8.08,8.01%。
【0089】
【化20】
【0090】
実施例8:cis−Pt(NH3)2(ピロリン酸)
【0091】
【化21】
【0092】
cis−Pt(NH3)2(ピロリン酸)の合成
ジメチルアセトアミド 5ml中0.482g(1ミリモル)のcis−Pt(NH3)2I2を0.306g(0.98ミリモル)のAg2SO4の40mL H2O懸濁液へ加え、暗所で約4時間撹拌した。膜フィルターによる濾過後、80mL H2O中のBa(OH)2・8H2O 0.305g(1.00ミリモル)を加えて、30分間撹拌した。濾過後、この溶液のpHをNaOH水溶液により3〜4へ調整し、1:1 ピロリン酸(2.0ミリモル):H2O 0.5gを加えて、1時間撹拌した。濾過後、この溶液を蒸発により濃縮して、生じる粉末を、MeOHを加えることによって沈殿させた。この緑がかった粉末を温水に溶かした。濾過した溶液を真空で濃縮して、MeOHを加えた。沈積した白い粉末をエーテルで洗浄した。収率:30%。
【0093】
元素分析 H8N2O6P2Pt・1H2O:計算値:H:1.91%,N:6.65%;実測値:H:2.28,2.27%,N:6.90,6.62%。
31P NMR(D2O,85% H3PO4):δ(ppm)0ppm。
【0094】
実施例9:Pt(NH3)−IP6 Pt(NH3)2・IP6・10Na・7H2O
【0095】
【化22】
【0096】
cis−Pt(NH3)2・IP6の合成
N,N−ジメチルアセトアミド 5mlに溶かしたcis−Pt(NH3)2・I2 0.483g(1.0ミリモル)をAgNO3 0.340g(2.0ミリモル)のH2O 40ml懸濁液へ加えて、暗所で一晩撹拌した。膜フィルターによる濾過後、H2O 30ml中のIP6・12Na 1.013g(1.0ミリモル)を加えて、3時間撹拌した。この溶液を蒸発により濃縮して、生じる粉末を、MeOHを加えることによって再沈殿させた。収率:54%。
【0097】
元素分析 C6H26N2O31P6Pt1Na10:計算値:C:5.84%,H:2.11%,N:2.27%;実測値:C:5.95%,H:2.15%,N:2.12%。
【0098】
【化23】
【0099】
XRF 計算値:P 6.00,Na 10.0,Pt 1.00;実測値:P 6.00,Na 10.9,Pt 0.94。
実施例10:Pt(dach)−IP6
【0100】
【化24】
【0101】
cis−Pt(dach)・IP6の合成
N,N−ジメチルアセトアミド 5mlに溶かしたcis−Pt(dach)I2 0.563g(1.0ミリモル)をAgNO3 0.340g(2.0ミリモル)のH2O 20ml懸濁液へ加えて、暗所で一晩撹拌した。膜フィルターによる濾過後、H2O 30ml中のIP6・12Na 1.013g(1.0ミリモル)を加えて、3時間撹拌した。この溶液を蒸発により濃縮して、生じる粉末を、MeOH 100mlを加えることによって再沈殿させた。収率:73%。
【0102】
元素分析 C12H44N2O36P6Pt1Na10:計算値:C:10.26%,H:3.14%,N:2.00%;実測値:C:10.13%,H:2.88%,N:1.71%。
【0103】
XRF 計算値:P 6.00,Na 10.0,Pt 1.00;実測値:P 6.00,Na 10.3,Pt 1.42。
【0104】
【化25】
【0105】
実施例11:Pt(NH3)2(NDP)
【0106】
【化26】
【0107】
cis−Pt(NH3)2・NDPの合成
ジメチルアセトアミド 5ml中の工程b.1.からの0.20g(0.5ミリモル)のcis−Pt(NH3)2I2を0.149g(0.48ミリモル)のAg2SO4の20mL H2O懸濁液へ加え、暗所で約4時間撹拌した。膜フィルターによる濾過後、20mL H2O中のBa(OH)2・8H2O 0.15g(0.48ミリモル)及びイミノ二リン酸ナトリウム(NDP)0.19g(0.70ミリモル)を加えた。一晩撹拌後、1M HClO4水溶液1.4mlを加え、この溶液を蒸発により濃縮して、生じる粉末を、H2O−EtOHを使用して再沈殿させた。収率:35%。
【0108】
元素分析 H9N3O6P2Pt1・1H2O:計算値:H:2.24%,N:10.40%;実測値:H:1.99%,N:10.01%。
31P NMR(D2O,85% H3PO4):δ(ppm)+10。
【0109】
実施例12:Pt(NH3)2(MDPOH)
【0110】
【化27】
【0111】
cis−Pt(NH3)2・MDPOHの合成
ジメチルアセトアミド 5ml中の0.20g(0.5ミリモル)のcis−Pt(NH3)2I2を0.149g(0.48ミリモル)のAg2SO4の20mL H2O懸濁液へ加え、暗所で約4時間撹拌した。膜フィルターによる濾過後、20mL H2O中のBa(OH)2・8H2O 0.15g(0.48ミリモル)及び60% 1−ヒドロキシエタン−1,1−ビス(ホスホン酸)(エチドロン酸、MDPOH)0.24g(0.70ミリモル)を加えた。一晩撹拌後、この溶液を蒸発により濃縮して、生じる粉末を、H2O−EtOHを使用して再沈殿させた。収率:65%。
【0112】
元素分析 C2H12N2O7P2Pt1:計算値:C:5.54%,H:2.50%,N:6.95%;実測値:C:5.46%,H:2.30%,N:6.76%。
実施例13:in vitro アッセイ−細胞増殖阻害
a.材料と方法
KB細胞は、Human Science Research Resource Bank(ヒューマンサイエンス研究資源バンク;大阪、日本)より購入した。この細胞を、10% FBS(Bio Whittaker)を含有するEarle’s MEM(GIBCO BRL)において5% CO2下に37℃で培養した。24時間後、この細胞へ試験化合物を指定濃度で加えた。72時間後、この細胞をトリパンブルーで染色して、手動で計数した。細胞の増殖を50%阻害するのに必要とされる錯体の濃度(水溶液)としてIC50を計算する。この結果を表1、2、及び3に示す。
【0113】
b.結果
表1
【0114】
【表1】
【0115】
表2
【0116】
【表2】
【0117】
表3
【0118】
【表3】
【0119】
同様のやり方で、本発明の化合物の実施例の代表的な番号について、いくつかの異なる癌を代表する細胞種の成長/増殖を阻害するその能力を試験した。この結果を、様々な細胞系に対する各化合物の阻害定数(IC50,μM)を提供する以下の表4〜13に提供する。
【0120】
表4 乳癌細胞系
【0121】
【表4】
【0122】
表5 脳腫瘍細胞系
【0123】
【表5】
【0124】
表6 結腸癌細胞系
【0125】
【表6】
【0126】
表7 肺癌細胞系
【0127】
【表7】
【0128】
表8 メラノーマ細胞系
【0129】
【表8】
【0130】
表9 卵巣癌細胞系
【0131】
【表9】
【0132】
表10 腎癌
【0133】
【表10】
【0134】
表11 ヒト胃癌
【0135】
【表11】
【0136】
表12 前立腺癌
【0137】
【表12】
【0138】
表13 平均IC50(全細胞種)
【0139】
【表13】
【0140】
上記のデータは、本発明の化合物が癌性細胞の有意な阻害を明示することを示す。
実施例14:in vitro アッセイ−Pt錯体のヒドロキシアパタイト、Ca10(PO4)6(OH)2への吸着
5、10、15、25、35、及び100mg試料のヒドロキシアパタイト(Bio-Rad Macro-Prep Ceramic Hydroxyapatite, I型、40μm)のそれぞれを2mlのHEPES緩衝液へpD=7.8で加えて、生じる混合物を37℃で約24時間振り混ぜた。次いで、HEPES緩衝液にpD=7.8で溶かした各錯体の100μlを加えて、この混合物を37℃で1.5時間振り混ぜた。すべての懸濁液を濾過して、これらの溶液を白金の31P NMR(Varian VXR−300S)及び Atomic Absorption Spectrometry(日立、Z−5710 AAS)によって測定するか、又はカルボプラチンでは、1H NMRによって測定した。ヒドロキシアパタイトに対する吸着百分率を以下のように計算した:
NMRより:結合百分率/%=[(A−B)/A]x100
AASより:結合百分率/%=[(C−D)/C]x100
A=Pt(II)錯体のインテグレーション(integration)強度
B=ヒドロキシアパタイトと反応後のPt(II)錯体のインテグレーション強度
C=Pt(II)錯体の濃度
D=ヒドロキシアパタイトと反応後のPt(II)錯体の濃度
図1及び2は、31P NMR、1H NMR、及びAASより計算したヒドロキシアパタイトへの吸着百分率を図示する。これらの結果は、cis−Pt(NH3)2(MDP)とPt(dach)(MDP)がPt(II)錯体としてヒドロキシアパタイトへ吸着することを示した。Pt(dach)(MDP)は、cis−Pt(NH3)2(MDP)よりややよく吸着して、Pt(dach)(MDP)とcis−Pt(NH3)2(MDP)は、ともにカルボプラチンよりきわめて有意によく吸着した。Pt(II)錯体を100mgのヒドロキシアパタイトと反応させるとき、cis−Pt(NH3)2(MDP)の吸着百分率は69.3%であり、Pt(dach)(MDP)のそれは79.6%であった。
【0141】
実施例15−in vivo 腫瘍アッセイ
6〜8週齢の雄性NCr−ヌードマウスに水(逆浸透、0.17% Cl)と18%タンパク質;5%脂肪;5%繊維;8%灰分;及び3%ミネラルからなる滅菌済み標準齧歯動物(NIH31)食を自由摂取させた。マウスは、22℃(72°F)、湿度40%〜60%、12時間の明周期でミクロアイソレーターに収容した。マウスの脇腹に5x106個のDU145ヒト前立腺癌細胞を皮下移植した。はじめは週2回、次いで毎日腫瘍をモニターすると、この新生物が所望のサイズ、ほぼ100mm3(100mg)に達した。DU145前立腺癌がこのサイズに達したとき、この動物を様々な処理群へ対マッチさせた。式:腫瘍重量(mg)=(w2XL)/2(ここで、w=腫瘍の幅で、L=腫瘍の長さ(mm)である)を使用して、推定腫瘍重量を計算した。
【0142】
本発明の代表的な化合物をこの動物へ腹腔内注射して、有意な抗腫瘍活性を保有することを見出した。以下の図3〜6を参照のこと。
一般に、本発明の化合物は、無機酸及び有機酸を使用することより誘導される塩のように、その医薬的に許容される酸付加塩の形態で単離することができる。そのような酸の例は、塩酸、硝酸、硫酸、リン酸、ギ酸、酢酸、トリフルオロ酢酸、プロピオン酸、マレイン酸、コハク酸、D−酒石酸、L−酒石酸、マロン酸、メタンスルホン酸、等である。さらに、カルボキシのような酸性官能基を含有する一定の化合物をその無機塩の形態で単離してよく、ここで対イオンは、ナトリウム、カリウム、リチウム、カルシウム、マグネシウム、等より、並びに有機塩基より選択してよい。
【0143】
医薬的に許容される塩は、本発明の化合物(例、以下の化合物C)の約1当量を取り、所望される塩の適切な対応する酸の約1当量以上とそれを接触させることによって生成することができる。生じる塩の後処理及び単離は、当業者によく知られている。
【0144】
本発明の化合物は、経口、非経口(例、筋肉内、腹腔内、静脈内、又は皮下の注射又はインプラント)、経鼻、膣、直腸、舌下、又は局所の投与経路によって投与してよく、医薬的に許容される担体とともに製剤化してそれぞれの投与経路に適した剤形を提供することができる。従って、本発明には、その範囲内に、本発明の少なくとも1つの化合物を有効成分として医薬的に許容される担体と一緒に含んでなる医薬組成物が含まれる。
【0145】
経口投与用の固体剤形には、カプセル剤、錠剤、丸剤、散剤、及び顆粒剤が含まれる。そのような固体剤形では、ショ糖、乳糖、又はデンプンのような少なくとも1つの不活性な医薬的に許容される担体と活性化合物を混合する。そのような剤形は、通常の実践であるように、そのような不活性希釈剤とは別の追加物質、例えば、ステアリン酸マグネシウムのような滑沢剤も含んでよい。カプセル剤、錠剤、及び丸剤の場合、剤形は、緩衝剤も含んでよい。錠剤と丸剤は、追加的に腸溶コーティング剤とともに調製してよい。
【0146】
経口投与用の液体剤形には、当該技術分野で一般的に使用される、水のような不活性希釈剤を含有する医薬的に許容される乳剤、溶液剤、懸濁液剤、シロップ剤、エリキシル剤が含まれる。そのような不活性希釈剤以外に、組成物には、湿潤剤、乳化剤、及び懸濁剤、並びに甘味剤、芳香剤、及び発香剤のようなアジュバントも含めてよい。
【0147】
本発明による非経口投与用の調製物には、無菌の水系又は非水系溶液剤、懸濁液剤、又は乳剤が含まれる。非水系溶媒又は担体の例は、プロピレングリコール、ポリエチレングリコール、オリーブ油又はとうもろこし油のような植物油、ゼラチン、及びオレイン酸エチルのような注射可能な有機エステルである。そのような剤形は、保存剤、湿潤剤、乳化剤、及び分散剤のようなアジュバントも含有してよい。それらは、例えば、細菌保持フィルターを通した濾過によって、滅菌剤を組成物へ取り込ませることによって、組成物に照射することによって、又は組成物を加熱することによって滅菌してよい。それらは、使用直前に滅菌水や他の無菌の注射可能な媒体に溶かすことができる、無菌の固体組成物の形態で製造してもよい。
【0148】
直腸又は膣からの投与用の組成物は、好ましくは、活性物質に加えて、ココア脂や坐剤ワックスのような賦形剤を含有し得る坐剤である。
経鼻又は舌下投与用の組成物も、当該技術分野でよく知られた標準の賦形剤とともに調製する。
【0149】
一般に、本発明の組成物中の有効成分の有効投与量は変動してよいが、有効成分の量は、好適な剤形が得られるようにすることが必要である。選択される投与量は、所望される治療効果、投与経路、及び治療の期間に依存し、このいずれも当業者の知識の領域内にある。一般に、1日あたり体重1kgにつき0.0001〜100mgの間の投与量レベルをヒトや他の動物、例えば哺乳動物へ投与する。
【0150】
好ましい投与量範囲は、1日あたり体重1kgにつき0.01〜10.0mgであり、これは単回用量として投与しても、頻回用量へ分割してもよい。
本発明の様々な態様を詳しく記載してきたが、本発明のさらなる修飾及び適応が当業者に思いつくことは明らかである。しかしながら、そのような修飾及び適応が本発明の精神及び範囲の内にあることは、はっきりと理解されるべきである。本明細書に引用する参考文献は、いずれもそのまま参照により本明細書に組み込まれる。
【特許請求の範囲】
【請求項1】
式II:
【化1】
[式中、Mは、Pd(II)である]による錯体、又はその医薬的に許容される塩。
【請求項2】
式IV:
【化2】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩。
【請求項3】
式V:
【化3】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩。
【請求項4】
MがPt(II)である、請求項2又は3に記載の錯体、又はその医薬的に許容される塩。
【請求項5】
請求項1〜3のいずれかに記載の化合物又はその医薬的に許容される塩の治療有効量を含む、細胞増殖抑制剤又は細胞傷害剤。
【請求項6】
被検者内の骨組織にか又は骨組織へ送達する、請求項5記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項7】
癌細胞がその上か又はその中に存在する、被検者内の骨組織にか又は骨組織へ送達する、請求項6記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項8】
前記治療有効量が前記癌細胞を治療するのに有効な量である、請求項5に記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項9】
白金含有錯体を含む、請求項5〜8のいずれかに記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項10】
パラジウム含有錯体を含む、請求項5〜8のいずれかに記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項11】
請求項3に記載の化合物又はその医薬的に許容される塩の治療有効量を含む、細胞増殖抑制剤又は細胞傷害剤。
【請求項12】
被検者内の骨組織にか又は骨組織へ送達する、請求項11記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項13】
癌細胞がその上か又はその中に存在する、被検者内の骨組織にか又は骨組織へ送達する、請求項12記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項14】
前記治療有効量が前記癌細胞を治療するのに有効な量である、請求項11に記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項15】
白金含有錯体を含む、請求項11〜14のいずれかに記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項16】
パラジウム含有錯体を含む、請求項11〜14のいずれかに記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項17】
前記治療有効量が骨格の合併症を抑制するのに有効な量である、請求項5に記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項18】
前記治療有効量が被検者において望まれないか又は不適正な血管形成活性を阻害するのに有効な量である、請求項5記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項19】
前記治療有効量が骨髄腫骨疾患、乳癌の転移、前立腺癌の転移を治療するのに有効な量である、請求項5記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項20】
前記治療有効量が骨疼痛を治療するのに有効な量である、請求項5記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項21】
前記治療有効量がタンパク質プレニルトランスフェラーゼのプレニル化を阻害するのに有効な量である、請求項5記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項22】
Mがパラジウムである、請求項1に記載の錯体。
【請求項1】
式II:
【化1】
[式中、Mは、Pd(II)である]による錯体、又はその医薬的に許容される塩。
【請求項2】
式IV:
【化2】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩。
【請求項3】
式V:
【化3】
[式中、Mは、Pt(II)又はPd(II)である]による錯体、又はその医薬的に許容される塩。
【請求項4】
MがPt(II)である、請求項2又は3に記載の錯体、又はその医薬的に許容される塩。
【請求項5】
請求項1〜3のいずれかに記載の化合物又はその医薬的に許容される塩の治療有効量を含む、細胞増殖抑制剤又は細胞傷害剤。
【請求項6】
被検者内の骨組織にか又は骨組織へ送達する、請求項5記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項7】
癌細胞がその上か又はその中に存在する、被検者内の骨組織にか又は骨組織へ送達する、請求項6記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項8】
前記治療有効量が前記癌細胞を治療するのに有効な量である、請求項5に記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項9】
白金含有錯体を含む、請求項5〜8のいずれかに記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項10】
パラジウム含有錯体を含む、請求項5〜8のいずれかに記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項11】
請求項3に記載の化合物又はその医薬的に許容される塩の治療有効量を含む、細胞増殖抑制剤又は細胞傷害剤。
【請求項12】
被検者内の骨組織にか又は骨組織へ送達する、請求項11記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項13】
癌細胞がその上か又はその中に存在する、被検者内の骨組織にか又は骨組織へ送達する、請求項12記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項14】
前記治療有効量が前記癌細胞を治療するのに有効な量である、請求項11に記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項15】
白金含有錯体を含む、請求項11〜14のいずれかに記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項16】
パラジウム含有錯体を含む、請求項11〜14のいずれかに記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項17】
前記治療有効量が骨格の合併症を抑制するのに有効な量である、請求項5に記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項18】
前記治療有効量が被検者において望まれないか又は不適正な血管形成活性を阻害するのに有効な量である、請求項5記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項19】
前記治療有効量が骨髄腫骨疾患、乳癌の転移、前立腺癌の転移を治療するのに有効な量である、請求項5記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項20】
前記治療有効量が骨疼痛を治療するのに有効な量である、請求項5記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項21】
前記治療有効量がタンパク質プレニルトランスフェラーゼのプレニル化を阻害するのに有効な量である、請求項5記載の細胞増殖抑制剤又は細胞傷害剤。
【請求項22】
Mがパラジウムである、請求項1に記載の錯体。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2010−18618(P2010−18618A)
【公開日】平成22年1月28日(2010.1.28)
【国際特許分類】
【出願番号】特願2009−199381(P2009−199381)
【出願日】平成21年8月31日(2009.8.31)
【分割の表示】特願2006−516615(P2006−516615)の分割
【原出願日】平成16年6月28日(2004.6.28)
【出願人】(506001631)
【Fターム(参考)】
【公開日】平成22年1月28日(2010.1.28)
【国際特許分類】
【出願日】平成21年8月31日(2009.8.31)
【分割の表示】特願2006−516615(P2006−516615)の分割
【原出願日】平成16年6月28日(2004.6.28)
【出願人】(506001631)
【Fターム(参考)】
[ Back to top ]