
ビタミンD結合タンパク質に特異的に結合する化合物
【課題】DBPのアッセイや分離・精製などに用いることができる、新たなDBP結合化合物。
【解決手段】25−OH−ビタミンD3を除くビタミンDより選択されるDBPに結合可能なビタミンDを固相支持体に結合させてなるDBP結合化合物。
【解決手段】25−OH−ビタミンD3を除くビタミンDより選択されるDBPに結合可能なビタミンDを固相支持体に結合させてなるDBP結合化合物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ビタミンD結合タンパク質のアッセイや分離・精製などに用いることができる、ビタミンD結合タンパク質と特異的に結合可能な新規化合物に関する。
【背景技術】
【0002】
ビタミンD結合タンパク質(以下、「DBP」と記載)は動物血清中に存在し、25−OH−ビタミンD3の運び屋として、また、細胞骨格アクチンの調節タンパク質として知られている。
【0003】
DBPは、ヒトにおいてはGc−グロブリンまたは「グループ特異的成分」(非特許文献1)としても知られている分子量約52,000の糖タンパク質で3つのドメインを有することが知られており、1番目のドメインにビタミンD結合サイトが、2番目および3番目のドメインにわたってアクチン結合サイトが、また、3番目のドメインにはマクロファージ活性化サイトがある(非特許文献2)。
【0004】
DBPの研究のためには、DBPを分取・精製することが必要であるが、当該分取・精製の操作には従来から25−OH−ビタミンD3を固相支持体に結合させたDBP結合化合物が合成され使用されている。一つの態様において、当該DBP結合化合物は、エポキシ化した25−OH−ビタミンD3とSH基を導入したセファロースCL−6B樹脂を反応させることで合成される(非特許文献3)。また別の態様においては、当該DBP結合化合物は、アミノプロピル基を導入した25−OH−ビタミンD3とN−ヒドロキシスクシンイミドを導入したアガロース樹脂(Affi−Gel 10)を反応させることで合成される。(非特許文献4)。
【0005】
25−OH−ビタミンD3は、25位が水酸化されているビタミンD3であって、他のビタミンDと比べ、DBPに対する特異的結合力が極めて強いことが知られている。DBPと各種ビタミンDの結合力は、25−ヒドロキシビタミンD3の結合力を1とするとき、ビタミンD3で20分の1、ビタミンD3の代謝産物である1α,25−ジヒドロキシビタミンD3で668分の1であることが報告されている(非特許文献5,6)。この性質より、25−OH−ビタミンD3は上記DBP結合化合物に利用されている。
【0006】
しかしながら、25−OH−ビタミンD3は、生体内で生成されるプレビタミンD3が肝臓にて代謝され合成される希少な天然材料である。血清中に含まれる25−OH−ビタミンD3は、ビタミンD濃度の95%以上を占めるが、それでもその血中濃度は5ng/mL〜80ng/mLと非常に小さく、大量に入手することは困難である。そのため、25−OH−ビタミンD3を利用したDBP結合化合物を、安価かつ大量に生産することは困難であった。使用者は、使用済みのDBP結合化合物を再生して、長期の管理をしなければならず、使用者にとって大きな負担となっていた。
【0007】
そのため、当該分野においては、25−OH−ビタミンD3を結合させたDBP結合化合物に代わる、新たなDBP結合化合物が切望されていた。
【先行技術文献】
【非特許文献】
【0008】
【非特許文献1】J.G.Haddad, J. SteriodBiochem. Molec. Biol. (1995) 53, 579−582
【非特許文献2】堀 均ら,放射線生物研究 39(3),328−341(2004)
【非特許文献3】Rebecca P. et.al, ANALYTICAL BIOCHEMISTRY, 157, 262−269 (1986)
【非特許文献4】Swamy N.et.al, PROTEIN EXPRESSION AND PURIFICATION, 6, 185−188 (1995)
【非特許文献5】Bishop J.E.et.al, J. Bone Miner. Res., 9(8), 1277−88, 1994.
【非特許文献6】Bouillon R. et.al, Endocr. Rev., 16(2), 200−57, 1995.
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、DBPのアッセイや分離・精製などに用いることができる、ビタミンD結合タンパク質と特異的に結合可能な新規DBP結合化合物を提供する。
【課題を解決するための手段】
【0010】
本発明者らは、上記課題を解決すべく、鋭意検討した結果、25−OH−ビタミンD3と比べて、DBPとの結合力が弱いビタミンDであっても、当該ビタミンDと固相支持体とを結合させてなる化合物が、DBPに対して、25−OH−ビタミンD3を固相支持体に結合させてなる従来のDBP結合化合物と同程度またはそれ以上の結合特異性および結合力を有することを見出し、本発明を完成させるに至った。
【0011】
すなわち、本発明は以下のとおりである。
[1] 固相支持体と、ビタミンD結合タンパク質に結合可能なビタミンD3とが結合してなる、ビタミンD結合タンパク質結合化合物であって、該ビタミンD結合タンパク質に結合可能なビタミンD3が、25−OH−ビタミンD3を除くビタミンD3より選択される、上記ビタミンD結合タンパク質結合化合物。
[2] 固相支持体が、セファロース樹脂またはアガロース樹脂である、[1]のビタミンD結合タンパク質結合化合物。
[3] [1]または[2]のビタミンD結合タンパク質結合化合物を充填剤として含む、ビタミンD結合タンパク質精製用カラム。
[4] [1]または[2]のビタミンD結合タンパク質結合化合物または[3]のビタミンD結合タンパク質精製用カラムを用いた、ビタミンD結合タンパク質の分離または精製方法であって、
(i)ビタミンD結合タンパク質結合化合物とビタミンD結合タンパク質を含む試料とを接触させ、該ビタミンD結合タンパク質結合化合物に該ビタミンD結合タンパク質を結合させる工程;
(ii)該ビタミンD結合タンパク質結合化合物に結合しているビタミンD結合タンパク質を溶出する工程;ならびに
(iii)工程(ii)にて溶出されたビタミンD結合タンパク質を回収する工程。
[5] ビタミンD結合タンパク質結合化合物の製造方法であって、
固相支持体と、ビタミンD結合タンパク質に結合可能なビタミンD3とを結合させる工程を含み、該ビタミンD結合タンパク質に結合可能なビタミンD3が、25−OH−ビタミンD3を除くビタミンD3より選択される、上記方法。
[6] 固相支持体が、セファロース樹脂またはアガロース樹脂である、[5]の方法。
[7] 結合させる工程が、
(i)ビタミンD結合タンパク質に結合可能なビタミンD3をエポキシ化する工程;
(ii)固相支持体にチオール基を導入する工程;および
(iii)工程(i)で得られたビタミンD3と工程(ii)で得られた固相支持体を反応させ、該ビタミンD3と該固相支持体を結合させる工程、を含む、[5]または[6]の方法。
[8] 結合させる工程が、
(i)ビタミンD結合タンパク質に結合可能なビタミンD3をアミノプロピルエーテル化する工程;
(ii)固相支持体にN−ヒドロキシスクシンイミドを導入する工程;および
(iii)工程(i)で得られたビタミンD3と工程(ii)で得られた固相支持体を反応させ、該ビタミンD3と該固相支持体を結合させる工程、を含む、[5]または[6]の方法。
【発明の効果】
【0012】
本発明により、DBPのアッセイや分離・精製などに用いることができる、新たなDBP結合化合物を安価にかつ大量に提供することが可能となり、これによりDBPの研究分野において飛躍的な進展をもたらすことができる。
【図面の簡単な説明】
【0013】
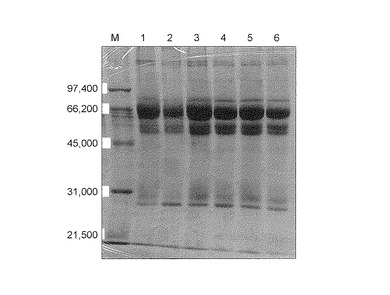
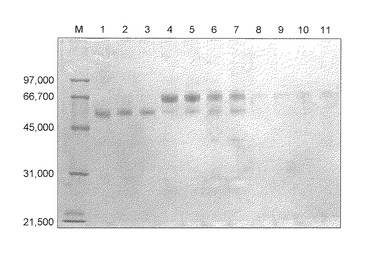
【図1】図1は、操作1の条件で回収したタンパク質濃縮液のSDS−PAGE(CBB染色)の結果を示す。各レーンはそれぞれ以下のサンプルを示す。M:マーカー;1:対照化合物(1)で処理したサンプル;2:従来化合物(1)で処理したサンプル;3−6:本発明化合物(1)で処理したサンプル。
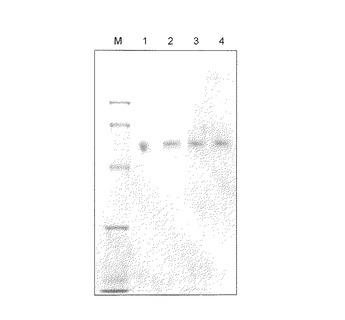
【図2】図2は、操作3の条件で回収したGcタイプ 1f1f、1s1sまたは22の血清に由来するタンパク質濃縮液のSDS−PAGE(CBB染色)の結果を示す。各レーンはそれぞれ以下のサンプルを示す。M:マーカー;1:シグマ社製のGcグロブリン;2:1f1fタイプ;3:1s1sタイプ;4:22タイプ。
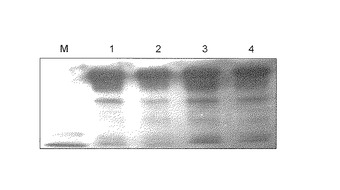
【図3】図3は、操作3の条件で回収したGcタイプ 1f1f、1s1sまたは22の血清に由来するタンパク質濃縮液のヒト抗IgG抗体を用いたウエスタンブロットの結果を示す。各レーンはそれぞれ以下のサンプルを示す。1:1f1fタイプ;2:1s1sタイプ;3:22タイプ;4:精製DBP(Gcタイプ 1f1f)。
【図4】図4は、操作3の条件で回収したGcタイプ 1f1f、1s1sまたは22の血清に由来するタンパク質濃縮液のPNAレクチンを用いたウエスタンブロットの結果を示す。各レーンはそれぞれ以下のサンプルを示す。1:1f1fタイプ;2:1s1sタイプ;3:22タイプ;4:精製DBP(Gcタイプ 1f1f)。
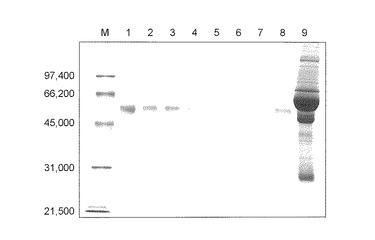
【図5】図5は、操作3の条件を用いて回収したGcタイプ 1s1sの血清に由来するタンパク質濃縮液のSDS−PAGE(CBB染色)の結果を示す。各レーンはそれぞれ以下のサンプルを示す。1:シグマ社製のGcグロブリン;2−3:本発明化合物(1)で処理したサンプル;4−5:本発明化合物(3)で処理したサンプル;6−7:対照化合物(2)で処理したサンプル;8:シグマ社製のGc精製品;9:血清。
【図6】図6は、操作3の条件を用いて回収したGcタイプ 1s1sの血清に由来するタンパク質濃縮液のSDS−PAGE(CBB染色)の結果を示す。各レーンはそれぞれ以下のサンプルを示す。1:シグマ社製のGcグロブリン;2−3:本発明化合物(1)で処理したサンプル;4−5:従来化合物(2)で処理したサンプル;6−7:本発明化合物(2)で処理したサンプル;8−9:本発明類似化合物で処理したサンプル;10−11:対照化合物(1)で処理したサンプル。
【発明を実施するための形態】
【0014】
本発明は、固相支持体と、DBPに結合可能なビタミンDとが結合してなる、DBPに特異的に結合する化合物に関する。
【0015】
本発明においてDBPは、ヒト由来のDBPおよび非ヒト動物由来のDBPを含む。
本発明において「DBPに結合可能なビタミンD」としては、25−OH−ビタミンD3を除く、DBPに対して親和性を有し得る任意のビタミンDが含まれる。
【0016】
本発明において「25−OH−ビタミンD3」とは、25位のみが水酸化されているビタミンD3および25位のみが水酸化されているその誘導体を意味する。25−OH−ビタミンD3は、DBPに対して高い親和性を有することから、DBPに特異的に結合する既存の化合物の製造に用いられている(Rebecca P.ら、(上掲)、Swamy N.ら、(上掲))。本発明における「DBPに結合可能なビタミンD」は、DBPに対して親和性を有し得る限り25−OH−ビタミンD3を除く全てのビタミンD類(ビタミンD3、ビタミンD4、ビタミンD5、ビタミンD6、ビタミンD7など)ならびにそれらのプロビタミン、プレビタミン、代謝産物、および誘導体より選択することができる。具体的には、ビタミンD3、24,25(OH)2ビタミンD3、25,26(OH)2ビタミンD3、1α,25−(OH)2ビタミンD3など、およびこれらの誘導体が挙げられるが、これらに限定されない。好ましくは、ビタミンD3を用いる。ビタミンD3は、高純度のものを大量に入手できるために、有利である。
【0017】
本発明において利用し得る「DBPに結合可能なビタミンD」それ自体はDBPに対して、必ずしも25−OH−ビタミンD3と同等またはそれを超越する結合力を持っていなくても良い。当該「DBPに結合可能なビタミンD」は、下記にて詳述する固相支持体と結合することによって、DBPに対して、25−OH−ビタミンD3と同等またはそれを超越する結合力を備えることができる。
【0018】
本発明において利用し得るビタミンDは、遺伝子組換え技術または化学合成技術を用いて人工的に製造されたものであっても良いし、植物または動物に由来する天然のものであっても良い。
【0019】
本発明において「固相支持体」には、固相支持体として利用し得ることが公知である様々な材料、例えば、セルロース、セルロース誘導体、セファロース、アガロース、金属、ガラス、セラミック、樹脂など(これらに限定されない)を用いることができる。好ましくは、セファロースおよびアガロースであり、さらに好ましくはセファロースである。固相支持体は市販のものを用いることが可能であり、例えば、セファロースCL−6B樹脂(シグマ社)を利用することができる。
【0020】
固相支持体は、本発明のDBP結合化合物の用途および使用方法に応じて、平板、ビーズ、粒子、球体など(これらに限定されない)任意の形状を有することが可能である。固相支持体の大きさは、特に限定されることなく本発明の化合物の用途および使用方法に応じて適宜決定することができる。好ましくは、固相支持体は、粒径75〜300μmのビーズの形状を有する。
【0021】
ビタミンDと固相支持体との結合は、共有結合または非共有結合を利用して行うことができる。ビタミンDと固相支持体との結合は、特に限定するものではないが、公知の手法を利用して行うことができる。例えば、ビタミンDをエポキシ化し、チオール基を導入した固相支持体と反応させ、両者を結合することができる(Rebecca P.et.al(上掲))。
【0022】
あるいは、ビタミンDをアミノプロピルエーテル化し、固相支持体のカルボキシル基をN−ヒドロキシスクシンイミド(NHS)を用いてエステル化して活性化エステル基とし、両者を反応させることによって結合することができる(Swamy N.et.al(上掲))。
【0023】
上記反応に際しては、3位のヒドロキシ基をエポキシ化またはアミノプロピルエーテル化したビタミンD3を利用することが好ましい。
【0024】
本発明のDBP結合化合物は、試料中に含まれるDBPと特異的に結合することが可能であり、DBPを分取および精製することに利用することができる。なお、本明細書において、本発明のDBP結合化合物とDBPとの「結合」を、「吸着」と記載する場合がある。この場合、「結合」と「吸着」なる記載は互換的に使用される。
【0025】
DBPを含む試料としては、ヒトおよび非ヒト動物に由来する、生体組織破砕液、体液(全血、血漿、血清、リンパ液など)を利用することができる(これらに限定されない)。DBPを含む試料はDBP結合化合物と接触させる前に、予め精製されていても良いし、精製されていなくても良い。好ましくは、DBPを含む試料は予め精製されている。ここでDBPを含む試料を予め精製するとは、DBP結合化合物と非特異的に結合する、試料中に含まれるタンパク質を除去することを意味する。「除去」とは、当該非特異的なタンパク質が全て試料中より除かれる場合だけでなく、その一部が試料中より除かれる場合も含む。当該精製方法は、DBP結合化合物と非特異的に結合する、試料中に含まれるタンパク質を除去し得る限り、特に限定されないが、例えば、使用するDBP結合化合物に含まれる固相支持体のみを、DBPを含む試料と接触させ、当該固相支持体に吸着するタンパク質を除去することによって行うことができる。あるいは、DBP含有溶液を陽イオン交換カラム入れて素通りさせることで行うことができる。当該操作は必要に応じて、1回以上行うことができる。あるいは、DBPを含む試料は、DBPを精製または粗精製したものであっても良い。
【0026】
本発明のDBP結合化合物とDBPの結合に際しては、上記DBPを含む試料を、適当な緩衝液を用いてpH6.0〜9.0、好ましくはpH6.5〜8.0に調整する。緩衝液としては、例えば、リン酸緩衝液(リン酸カリウム緩衝液、リン酸ナトリウム緩衝液等)、Tris緩衝液(STE緩衝液等)を用いることができる。緩衝液を添加してpHを調整したDBPを含む試料を、同じ緩衝液を用いて平衡化した本発明の化合物と接触させ、試料中のDBPを本発明の化合物に結合させる。両者の結合反応は、バッチ式で行っても良いし、連続式で行っても良い。両者の接触は、4℃〜40℃、好ましくは4℃または室温において、1分間〜4時間振とうしながら行う。バッチ式で行う場合には好ましくはおよそ5〜10分間振とうしながら行う。あるいは、両者の結合反応は、カラム法によって行っても良い。この場合、本発明のDBP結合化合物は、DBPを精製するための各種クロマトグラフィー用カラム、特にアフィニティーロマトグラフィー用カラム、の充填剤として利用することができる。
【0027】
次いで、本発明のDBP結合化合物に結合したDBPを溶出する。本発明のDBP結合化合物からDBPを溶出する際の溶出液としては、塩化ナトリウム溶液、塩化カリウム溶液等の塩溶液、リン酸、酢酸、クエン酸、トリス等の緩衝液、グアニジン等の変性剤およびこれらの混合溶液等を利用することができる。溶出は当業者に公知の一般的な手法を用いて行うことができ、例えば、溶出液の塩濃度を徐々に増やして溶出する方法、DBPが本発明のDBP結合化合物と結合するpHから反撥するpHへ徐々に溶出液のpHを変化させる方法またはそれらの組合せを用いて行うことができる。DBPの結合した本発明のDBP結合化合物を遠心分離して回収した後、溶出液を添加して、4℃〜40℃、好ましくは4℃または室温において、およそ1〜30分間振とうした後、遠心分離して本発明のDBP結合化合物を沈降させ、上清に含まれるDBPを回収する。
【0028】
回収されたDBPは、タンパク質精製に用いられる公知の方法、例えば、硫安塩析、有機溶媒(エタノール、メタノール、アセトン等)による沈殿分離、イオン交換クロマトグラフィー、等電点クロマトグラフィー、ゲルろ過クロマトグラフィー、疎水性クロマトグラフィー、吸着カラムクロマトグラフィー、基質または抗体などを利用したアフィニティークロマトグラフィー、逆相カラムクロマトグラフィーなどのクロマトグラフィー、精密ろ過、限外ろ過、逆浸透ろ過、ダイアフィルトレーション等の濾過処理など、を1つまたは複数組み合わせて用いて、さらに精製することが可能である。
【0029】
本発明のDBP結合化合物を用いることによって血清よりDBPを97%、98%、99%またはそれ以上の高純度で得ることができる。
【実施例】
【0030】
以下、本発明の実施例を示すが、本発明は特にこれにより限定されるものではない。
【0031】
(実施例1)樹脂の合成
(I)新規DBP結合化合物(1)(以下、本発明化合物(1)と記載)
(i)ビタミンD3のエポキシ化
50mgのビタミンD3(コレカルシフェロール:東京化成工業社製)をN,N−ジメチルホルムアミド(DMF)4.0mlに溶解させ、枝付きフラスコに入れ、N2で置換した。次いで0.06mlのエピブロモヒドリン(和光社製)と20mgのNaHとを順次加え室温で攪拌しながら、N2気流中でエポキシ化反応を行う。
【0032】
薄層クロマトグラフィーで反応完了を確認した後、反応液に氷水および酢酸エチルを加え生成物の溶媒抽出を行う。この抽出操作を3回行い、抽出液を集めて氷水で洗浄し、MgSO4を加えて乾燥後、減圧乾固して生成物を含む残渣を得た。
【0033】
得られた残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル系)に通し、原材料を除去し、エポキシ化ビタミンD3を回収し、減圧乾燥させ、(iii)のDBP結合化合物合成工程に用いた。
【0034】
(ii)セファロース樹脂へのチオール基の導入
セファロースCL−6B樹脂(シグマ社製)約10gに1,4−ブタンジオールジグリシジルエーテル10gおよびNaBH420mlを含む1M NaOH 10mlを加え室温で10〜12時間振とうして、セファロースCL−6B樹脂にエポキシ側鎖を導入する。この樹脂を蒸留水で洗浄した後、2.0MのNa2S2O3を15ml加え、室温で約8時間振とうしながら、当該樹脂にチオール基(S−S)を導入した。チオール基を導入したセファロースCL−6B樹脂を蒸留水、次いでSPBで洗浄した後、樹脂にDTT200mgと1mM EDTA−2Naを含む0.3M NaHCO320ml(PH8.5)を加え、室温で1時間振とうして、SH−activatedセファロースCL−6B樹脂を得た。0.5M NaClで洗浄、次いで0.3M NaHCO3(含1mM EDTA・2Na)で洗浄して、以下の本発明化合物(1)の合成工程に用いた。
【0035】
(iii)本発明化合物(1)の合成
上記(i)にて得られたエポキシ化ビタミンD3を1,4−ジオキサン20mlに溶解させ、1M NaOH 10mlおよび上記(ii)にて得られたSH−activatedセファロースCL−6B樹脂を加え、ウオータバス上、40℃、N2気流中で撹拌した。18時間後、60% アセトン/イオン交換水、アセトン、60% アセトン/イオン交換水、イオン交換水、0.05M NaCl、イオン交換水(各250 mL)で洗浄後、遠心して上清を取り除いた。本発明化合物(1)約10gを得た。
【0036】
(iv)樹脂保管
合成した本発明品は0.02% NaN3を入れたSTEバッファー(pH7.4, Tris・HCl 6.05g,EDTA・2Na 0.56g,NaCl 8.77gをSDWに溶かし、全量を1000mlとして濾過し、オートクレーブにかける。Triton X−100を1ml加えたもの)20mLを加えて、密栓し4℃で保管した。
【0037】
(II)新規DBP結合化合物(2)(以下、本発明化合物(2)と記載)
ビタミンD3(コレカルシフェロール:東京化成工業製)(100mg,0.26mmol)をt−BuOH/MeCN(9/1,v/v)混合液(3.0mL)に溶解し、氷上撹拌下、アクリロニトリル(300μL,0.56mmol,2.0eq.)を滴下した。NaHを加え、1時間後、冷Et2O、冷水を滴下し、減圧化で溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(hexane:EtOAc=10:1)に付し、98.4mg,(0.22mmol,86%)の固体1を得た。その固体124.9mg(0.06mmol)をEt2O(5.0mL)に溶解し、氷上撹拌下、LiAlH4/AlCl3 60.3mg,(1.07mmol, 3.9eq.)を加えた。1時間後、1N NaOHを滴下し、EtOAcで4回抽出し、有機層をまとめて、飽和食塩水で洗浄し、無水Na2SO4で乾燥させ、ろ過後、ろ液を減圧下で溶媒を留去し、固体2を得た。次の反応のためにAffiGel−10(4.0mL,45μmol)を1,4−ジオキサンで3回洗浄した。これに固体2(全量)を1,4−ジオキサン(5.0mL)で加え、プロペラで室温下、撹拌した。22時間後、エタノールアミン(2.5mL)を加え、さらに撹拌した。2時間後、1,4−ジオキサン/アセトン(1/1,v/v)で3回洗浄し、上清を取り除き、0.02% NaN3/STEバッファーを加え、ビタミンD3結合アガロース樹脂(本発明化合物(2))4.0mLを得た。
【0038】
(III)新規DBP結合化合物(3)(以下、本発明化合物(3)と記載)
Activated Thiol SepharoseCL−4B(GEヘルスケアバイオサイエンス社製)2.5gを蒸留水およびリン酸バッファーで洗浄し、遠心後、上清を取り除いた。DTT200mgと1mM EDTA−2Naを含む0.3M NaHCO320ml(PH8.4)を加え、室温で1時間振とうして、チオール基の還元反応を行った。生成したSH−activatedセファロースCL−4B樹脂を0.5M NaClで洗浄、次いで0.3M NaHCO3(含1mM EDTA・2Na)で洗浄して、これに上記本発明化合物(1)と同様にして得られた、エポキシ化ビタミンD3を1,4−ジオキサン(20ml)に溶解して加え、さらに1M NaOH 10mlを加え、ウオータバス上、40℃、N2気流中で撹拌して、ビタミンD3結合CL−4B樹脂(本発明化合物(3))を得た。
【0039】
(IV)ビタミンD2を含む化合物(以下、本発明類似化合物と記載)
上記本発明化合物(1)と同様に、ビタミンD2(カルシフェロール:東京化成工業社製)100mgをエポキシ化して、エポキシ化ビタミンD2とした。さらに、上記本発明化合物(1)と同様に合成したSH−activatedセファロースCL−6B樹脂とを同様な操作で反応させ、ビタミンD2結合CL−6B樹脂(本発明類似化合物)を得た。
【0040】
(V)従来公知のDBP結合化合物(1)(以下、従来化合物(1)と記載)
25−OH−ビタミンD3とセファロースCL−6B樹脂からなる従来公知のDBP結合化合物として、徳島大学大学院ソシオテクノサイエンス研究部 堀均博士より恵与された25−OH−ビタミンD2結合CL−6B樹脂(従来化合物(1))を使用した。
【0041】
(VI)連結部の構造が異なる従来公知のDBP結合化合物(2)(以下、従来化合物(2)と記載)
洗浄したAffiGel−10 4.0mlを準備し、これに25−OH−ビタミンD3(カルシフェディジール:東京化成工業製)100.0mg,(0.25mmol)をt−BuOH/MeCN(9/1,v/v)混合液(3.0mL)に溶解し、上記(II)新規DBP結合化合物(2)と同様に操作して得た固体(全量)を1,4−ジオキサン(5.0mL)で加え、さらに同様に処理して25−OH−ビタミンD3結合アガロース樹脂4.0mL(従来化合物(2))を得た。従来化合物(2)は、ビタミンD3と固相支持体の連結部に、エーテル結合と3つのヒドロキシ側鎖を有する。
【0042】
(VII)対照化合物(1)
対照化合物(1)として、セファロースCL−6B樹脂をそのまま使用した。
【0043】
(VIII)対照化合物(2)
対照化合物(2)としてはActivated Thiol SepharoseCL−4BセファロースCL−4B樹脂をそのまま使用した。
【0044】
(実施例2)本発明化合物と従来化合物の比較
血清(Gcタイプ 1f1f)から硫安分画した粗Gcグロブリンを、25−OH−ビタミンD3とセファロース樹脂からなる従来化合物(1)(徳島大学大学院ソシオテクノサイエンス研究部 堀均博士より恵与のもの)をアフィニティカラムとして使って一次精製し、次にヒドロキシアパタイトカラムで二次精製し、精製DBPを得た。この精製DBPを用いて、実施例1で製造した本発明化合物(1)および従来化合物(1)のタンパク質吸着能を比較した。
【0045】
(i)使用前の樹脂の調整
1.5mlのエッペンチューブに加えた樹脂は、使用する前にPH7.4のSTEバッファーを500μL加え、30秒間振とう後、3分間遠心分離(3,000rpm)した。この洗浄を5回繰り返し、樹脂の保存のために加えたアジ化ナトリウムを除いた。
【0046】
(ii)吸着および洗浄、溶出操作
上記(i)の操作でアジ化ナトリウムを除いた本発明化合物(1)および従来化合物(1)を各0.5g含むエッペンチューブに100μg相当量の精製DBP溶液18.31μlとSTEバッファー981.7μlを加えて、タンパク質を吸着させるため常温にて1時間振とうした。以下、4℃、2分間遠心分離(13,000rpm)し、上澄み液を除いた後、化合物を洗浄するためSTEバッファー500μlを加え、キュートミキサー(1,800rpm)で60秒間振とうした。その後、2分間遠心分離(13,000rpm)し上澄み液を除いた。この洗浄操作を5回繰り返した後に、溶出液としてpH5.0の0.5M酢酸バッファー500μLを加えキュートミキサー(1,800rpm)で60秒間振とうし、4℃、2分間遠心分離(13,000rpm)した。この溶出操作を3回繰り返し、溶出液を得た。
【0047】
(iii)タンパク質の定量
各溶出液600μlを正確にとり、タンパク質定量用BCA(ビシンコニン酸)試薬2.4mlを加えて37℃で30分間加温した後、562nmの吸光度を測定した。溶出液中のタンパク質量は、BSA(シグマ社製)を標準品とした検量線から求めた。
【0048】
その結果、溶出液中のタンパク質量は、本発明化合物(1)では46.5μg/ml、また、従来化合物(1)では36.5μg/mlとなった。
この結果は、従来化合物(1)と同様、本発明化合物(1)がDBPと結合することを示す。
【0049】
(実施例3)DBPおよびBSAの吸着比較
(i)吸着および洗浄、溶出操作
アジ化ナトリウムを除いた本発明化合物(1)を各0.5g含むエッペンチューブに100μg相当量の精製DBP溶液あるいはBSA溶液を加え、さらに、全量が1000μlとなるようにSTEバッファーを加えて、以下、1時間振とうし、実施例2(ii)と同様に、洗浄、溶出操作を行い、溶出液を得た。
【0050】
(ii)タンパク質定量
各溶出液600μLを正確にとり、タンパク質定量用BCA液(Pierce BCA Protein Assay Kit、Thermo SCIENTIFIC社製)2.4mlを加えて37℃で30分間加温した後、562nmの吸光度を測定した。回収液中のタンパク質濃度を、BSAを標準品とした検量線から求めた。
【0051】
結果を以下の表1に示す。なお表1中、用いた化合物は、それぞれ別個の操作により合成された化合物を意味する。
【0052】
【表1】

【0053】
DBPサンプルでの回収タンパク質量は本発明化合物(1)では42.42μg、従来化合物(1)では35.76μgであった。一方、BSAサンプルでの回収タンパク質量はブランク値の範囲内でタンパク質量は算出できなかった。
【0054】
この結果は、本発明化合物がDBPを特異的に結合することが可能であり、また従来化合物(1)よりも優れた結合能を有することを示す。
【0055】
実施例4:血清からのタンパク質の回収
(i)吸着および洗浄、溶出操作
(操作1)
Gcタイプ 1s1sのボランティアからヒト血液を20ml採取し、室温で30分静置し、その後遠心により血清を回収し、4℃で保管する。室温に戻して30分間、静置し、10分間遠心分離(3,000rpm)して血清(約10ml)を回収し、4℃に保管した。
【0056】
各エッペンチューブに本発明化合物(1)、従来化合物(1)および対照品化合物(1)をそれぞれ0.5g分注し、アジ化ナトリウムを除いた後、250μlの血清と250μlのSTEバッファー(10mM Tris,pH 8.0;1mM EDTA,100mM NaCl)を加え、実施例2(ii)と同様な操作を行い、溶出液を得た。
【0057】
得られた溶出液の500μLをアミコン(millipore社製アミコンウルトラ0.5 100000M.W.CO)上で10分間、4℃、13,000Gで、限外濾過した。さらに残りの溶出液を加えて同様に処理した。このアミコンの中に、10mM リン酸ナトリウムバッファー(pH7.0)500μLを加えて、4℃で、10分間、13,000Gで、バッファー置換を行う。これを5回行い、その後、4℃で、3分間、遠心分離(1000G)して、タンパク質濃縮液を得た。
【0058】
(操作2)
対照化合物(1)0.5gをとり、アジ化ナトリウムを除いた後、1s1sタイプのヒト血清250μLとリン酸ナトリウムバッファー250μLを加えて、常温にて1時間振とうし、4℃で、2分間、遠心分離(13,000rpm)し、上澄み液を回収した(前処理操作)。
【0059】
この上澄み液500μLずつを、アジ化ナトリウムを除いた本発明化合物(1)、従来化合物(1)および対照化合物(1)をそれぞれ0.5g含むエッペンチューブに加え、1時間振とうし、その後、4℃で、2分間、遠心分離(13,000rpm)し、上澄み液を除いた。化合物を洗浄するため、500μLのSTEバッファー(PH7.4)を加え、キュートミキサー(1,800rpm)で30秒間振とうし、さらに、4℃で、2分間、遠心分離(13,000rpm)を行い、上澄み液を除いた。この洗浄操作を10回繰り返した後に、0.5M酢酸バッファー(pH5.0)500μLを加え、60秒間振とう、2分間、遠心分離(13,000rpm)を行い、上澄み液を回収した。この回収操作を3回繰り返し、上澄み液を合わせて溶出液とした。以下、上記(操作1)と同様な限外濾過およびバッファー置換を行い、タンパク質濃縮液を得た。
【0060】
(ii)タンパク質定量
操作1または操作2の条件で回収したタンパク質濃縮液を用いて、本発明化合物(1)、対照化合物(1)および従来化合物(1)のタンパク質吸着能について、BCA法を用いたタンパク質定量によって解析した。すなわち、BSA溶液を2.0,1.0,0.5,0.25,0,1mg/mlの濃度で用意した。タンパク質濃縮液はそれぞれ30倍に希釈した。調製したBSAおよびタンパク質濃縮液60μLを96穴プレートに正確に加え、さらにBCA液(Pierce BCA Protein Assay Kit、Thermo SCIENTIFIC社製)を240μLずつ加える。37℃、30分間インキュベーションした後、吸光度を波長570nmで測定し、タンパク濃度を算出した。
【0061】
結果を以下の表2および表3に示す。なお表2および3中、用いた化合物は、それぞれ別個の操作により合成された化合物を意味する。
【0062】
【表2】
【0063】
【表3】
【0064】
上記(操作1)において、対照化合物も含め、全ての樹脂で数百μgレベルのタンパク質を得た(表2参照)。非特異的なタンパク質が大量に混入しており、各樹脂のDBP結合能については判断できなかった。
【0065】
上記(操作2)において、本発明化合物および従来化合物では14μg〜28μgのタンパク質の吸着が得られた。対照化合物ではタンパク質の吸着は得られなかった(表3参照)。これは(操作2)において、本発明化合物(1)および従来化合物(1)がDBPを回収できたことを示唆する。
【0066】
(iii)SDS−PAGE(CBB染色)
上記(操作1)と(操作2)の結果を確認するために、(操作1)の濃縮液のSDS−PAGEを行い、ゲルをCBB染色した。すなわち、CBB溶液(CBB 0.25gをメタノール:酢酸:mQ(5:1:4)に溶かしたもの)を500〜1000μLをゲルにかけ、2〜3分程度染色した後、脱色液に10分間ほど浸して振盪し脱色した後、一晩、風乾した。
【0067】
その結果、対照化合物(1)、従来化合物(1)および4ロットの本発明化合物(1)のいずれのサンプルでも多くのタンパク質バンドが見られた(図1参照)。上記(操作1)では、洗浄不足のため非特異的タンパク質が大量に混入するため、従来化合物(1)でもDBPバンドを確認することができなかった。
【0068】
(iv)HPLC解析
さらに、上記(操作1)と(操作2)の違いを確認するため、各濃縮液をリン酸ナトリウムバッファーでタンパク濃度250ng/μLに希釈して、下記の条件でHPLCを行った。DBP標準品にシグマ社製Gcグロブリンを用いて、標準品の保持時間と重なるピークの保持時間およびそのピークの面積を全タンパク質の面積で割り、見かけ上のGc%を調べた。
・移動相:50mM SPB+0.3M NaCl
・流量:0.7ml/min
・注入量:タンパク質2.5μg相当(10μL inject)
・カラム:東ソーTSKgel G3000SWXL
・カラム温度:25℃
・検出波長:225nm
結果を以下の表4に示す。なお表4中、用いた化合物は、それぞれ別個の操作により合成された化合物を意味する。
【0069】
【表4】
【0070】
【表5】
【0071】
上記(操作1)によるタンパク質濃縮液で当該ピークの保持時間は、いずれの樹脂の場合も標準品には一致しなかった(表4参照)。一方、(操作2)によるタンパク質濃縮液において、本発明化合物(1)および従来化合物(1)で処理したサンプルでは当該ピークの保持時間が、完全に標準品と一致した(表5参照)。
【0072】
タンパク定量、電気泳動およびHPLC分析から、(操作2)の条件で、不純タンパク質が除去でき、本発明化合物(1)および従来化合物(1)を用いてDBPを精製できることが示された。
【0073】
(実施例5)Gcタイプ 1f1f、1s1sおよび22の血清からの直接DBP濃縮
本発明化合物(1)によるDBPの精製能を高めるため、下記操作3の条件を見出した。Gcタイプの異なる3種類1f1f、1s1sおよび22のヒト血清を用いて、操作3の条件を用いてDBP精製を行い、サンプルのタンパク質定量、SDS−PAGE,ウエスタンブロッティングを行った。
【0074】
(i)吸着および洗浄、溶出操作
(操作3)
3本のエッペンチューブに本発明化合物(1)0.5gずつを分注し、アジ化ナトリウムを除いた後、各エッペンチューブに各々3タイプの血清250μlを加え、さらにSTEバッファー250μlを加えて、キュートミキサー(1,800rpm)で5分間振とうした。次に、4℃で、2分間、遠心分離(13,000rpm)し、上澄み液を除いた。さらに、STEバッファー(PH7.4)を500μl加え、キュートミキサー(1,800rpm)で10秒間振とうし、化合物の洗浄を行い、4℃で、2分間、遠心分離(13,000rpm)し、上澄み液を除いた。この洗浄操作を15回行った。次に、0.5M酢酸バッファー(pH5.0)500μlを加え、キュートミキサー(1,800rpm)で10秒間振とうしタンパク質を溶出させ、4℃で、2分間、遠心分離(13,000rpm)した。これを2回行い、上澄み液を回収液とした。以下、限外濾過、バッファー置換を行い、タンパク質濃縮液を得た。
【0075】
(ii)タンパク質定量とSDS−PAGE(CBB染色)
操作3の条件で回収したタンパク質濃縮液を用いて、本発明化合物(1)のタンパク質吸着能について、BCA法を用いたタンパク質定量によって解析した。
結果を以下の表6に示す。
【0076】
【表6】
【0077】
結果、血清250μL当り24〜27μgのタンパク質が得られた(表6参照)。
【0078】
次に、操作3の条件で回収したタンパク質濃縮液のSDS−PAGE(CBB染色)を行った。
結果を図2に示す。結果、Gcタイプに関係なく同一の単一バンドを得た(図2)。
【0079】
シグマ社製のGcグロブリンはバンドの位置がサンプルとは異なるが、これは他のサンプルと異なり、操作3の条件を用いて精製されていないためであり、シグマ社製のGcグロブリンも(操作3)の条件を用いて精製することによって、血清由来のGcグロブリンとバンド位置は一致する(図5レーン1および2参照)。
【0080】
(iii)ウエスタンブロット
操作3の条件で回収したタンパク質濃縮液を用いて、ヒト抗IgG抗体およびPNAレクチンを用いたウエスタンブロットを行った。標準品には精製DBP(実施例2参照)を使用した。転写後、膜を0.1% TBSTで10分間、3回洗浄を行い、1.0% BSA in TBSTでブロッキングを行った後、0.1% TBSTで10分間、3回洗浄を行った。以下、室温で1時間、振盪させて一次抗体と反応させた後、0.1% TBSTで10分間、3回洗浄を行い、室温で二次抗体と1時間、振盪反応させた。反応後、同様に0.1% TBSTで10分間、3回洗浄を行った。この膜を暗室に運び、プラスティック容器内で膜に1分間程ECL液をまんべんなくかけ、それをプラスティックシートにはさみ、フィルムをシートの上に30秒〜3分間置いた。
【0081】
以下、現像液に1分間漬け、水洗いし、停止液に1分間漬けて現像を行った。
結果を、図3および図4に示す。結果、バンドはヒト抗IgG抗体を用いたウエスタンブロットで染色され、それは精製DBPと一致した(図3参照)。また、PNAレクチンを用いたウエスタンブロットでもバンドは染色され、精製DBPと一致した(図4参照)。 これより、本発明化合物(1)を用いて、血清中のDBPを回収できることが明らかとなった。
【0082】
(iv)HPLC解析
1f1f、1s1s、2.2の3タイプのGcサンプルについて、実施例4(iv)と同じ条件でHPLC法を行い、HPLC法で精製DBPの純度分析が可能かどうかを調べた。標準品としてシグマ社製Gcグロブリンを使用し、サンプルの保持時間(RT)が標準品のそれと±0.02min以内であることを事前に調査し、また、シンメトリー係数を調べ、その値が1.00〜1.30を示した場合、分析の信頼性があるものとした。この2つの条件をクリアにしたサンプルは正しい評価ができるとして、次式からピーク面積法によりタンパク質当りのDBP純度を求めた。
DBP純度=主ピーク面積÷9〜16分間に見られる全体のピーク面積×100%
結果を以下の表7に示す。
【0083】
【表7】
【0084】
結果、3タイプのGcサンプル全てにおいて、保持時間が標準品の保持時間と、0.002〜0.014分以内で一致した(表7参照)。また、シンメトリー係数は1.167〜1.186で、分析の信頼性があることを確認した。
【0085】
DBP純度を求めた結果、いずれのサンプルも全タンパク質ピーク面積の98.7%以上であった。これはシグマ社製Gcグロブリンの純度98.16%と同等あるいはそれ以上である。
【0086】
以上の結果は、HPLC法によって、本発明化合物(1)の処理で得られたサンプル中のDBPの純度評価が可能であることを示す。また、本発明化合物(1)を用いることにより、Gcタイプを問わず、血清より高純度のDBPを回収できることを示す。
【0087】
(実施例6)本発明化合物と本発明類似化合物の比較。
実施例1で合成したビタミンD2結合CL−6B樹脂である本発明類似化合物、さらにビタミンD3結合アガロース樹脂(本発明化合物(2))および連結部分の構造が異なるビタミンD3結合CL−4B樹脂(本発明化合物(3))についてDBPを吸着するかどうかを、タンパク定量法およびSDS−PAGEで検討した。
【0088】
(i)吸着および洗浄、溶出操作
1s1sのヒト血清を使い、(操作3)の条件に従って回収したタンパク質濃縮液、ならびに(操作3)において、本発明化合物(1)に代えて、本発明類似化合物、本発明化合物(2)、本発明化合物(3)、対照化合物(1)もしくは(2)、または従来化合物(2)を用いる以外は同様の条件を用いて調製された各タンパク質濃縮液を、以下(ii)−(iv)の実験に用いた。
【0089】
(ii)本発明類似品化合物
本発明類似化合物を用いて回収したタンパク質濃縮液を用いて、本発明類似化合物のタンパク質吸着能について、BCA法を用いたタンパク質定量によって解析した。結果を以下の表8に示す。なお表8中、用いた化合物は、それぞれ別個の操作により合成された化合物を意味する。
【0090】
【表8】
【0091】
BCA法によるタンパク質定量の結果、本発明類似化合物によって27〜38μg程度のタンパク質が得られた(表8参照)。
【0092】
しかし、SDS−PAGE(CBB染色)においては、DBPのバンドは認められなかった。本発明類似化合物にはDBPが吸着しないことが明らかとなった(図6レーン8および9参照)。
【0093】
(iii)本発明化合物(2)
従来化合物(2)である25(OH)ビタミンD3結合アガロース樹脂は、DBPのアフィニティカラムとして使用される(非特許文献4)。そこで、従来化合物(2)または本発明化合物(2)を用いて回収したタンパク質濃縮液を用いて、従来化合物(2)および本発明化合物(2)のそれぞれのタンパク質吸着能について、BCA法を用いたタンパク質定量によって解析した。
【0094】
結果を以下の表9に示す。なお表9中、用いた化合物は、それぞれ別個の操作により合成された化合物を意味する。
【0095】
【表9】
【0096】
BCA法によるタンパク質定量の結果、本発明化合物(2)および従来化合物(2)には本発明化合物(1)よりも多くのタンパク質が吸着した(表8および9参照)。タンパク質濃縮液には不純タンパク質の混入があるが、このサンプルのSDS−PAGE(BCC染色)をおこなった結果、本発明化合物(2)と従来化合物(2)には、同様の2つのバンドが見られ、下側のバンドが本発明化合物(1)のDBPバンドに一致した。したがって、本発明化合物(2)を用いて、DBPを吸着できることが示された(図6レーン4−7参照)。
【0097】
(iv)本発明化合物(3)
本発明化合物(3)を用いて回収したタンパク質濃縮液を用いて、本発明化合物(3)のタンパク質吸着能について、BCA法を用いたタンパク質定量によって解析した。結果を、以下の表10に示す。なお表10中、用いた化合物は、それぞれ別個の操作により合成された化合物を意味する。
【0098】
【表10】
【0099】
結果、本発明化合物(3)を用いることによって、本発明化合物(1)よりも少ない、5.1〜6.0μg程度のタンパク量が回収された(表10参照)。そこで、SDS−PAGE(CBB染色)を行ったこころ、本発明化合物(1)に一致した1つバンドが見られ、DBPが吸着することが示された(図5レーン4および5参照)。
【産業上の利用可能性】
【0100】
本発明により、DBPのアッセイや分離・精製などに用いることができる、新たなDBP結合化合物を安価にかつ大量に供給することが可能となるために、これによりDBPの研究利用や臨床利用など様々な分野において重要な貢献をすることが期待できる。
【技術分野】
【0001】
本発明は、ビタミンD結合タンパク質のアッセイや分離・精製などに用いることができる、ビタミンD結合タンパク質と特異的に結合可能な新規化合物に関する。
【背景技術】
【0002】
ビタミンD結合タンパク質(以下、「DBP」と記載)は動物血清中に存在し、25−OH−ビタミンD3の運び屋として、また、細胞骨格アクチンの調節タンパク質として知られている。
【0003】
DBPは、ヒトにおいてはGc−グロブリンまたは「グループ特異的成分」(非特許文献1)としても知られている分子量約52,000の糖タンパク質で3つのドメインを有することが知られており、1番目のドメインにビタミンD結合サイトが、2番目および3番目のドメインにわたってアクチン結合サイトが、また、3番目のドメインにはマクロファージ活性化サイトがある(非特許文献2)。
【0004】
DBPの研究のためには、DBPを分取・精製することが必要であるが、当該分取・精製の操作には従来から25−OH−ビタミンD3を固相支持体に結合させたDBP結合化合物が合成され使用されている。一つの態様において、当該DBP結合化合物は、エポキシ化した25−OH−ビタミンD3とSH基を導入したセファロースCL−6B樹脂を反応させることで合成される(非特許文献3)。また別の態様においては、当該DBP結合化合物は、アミノプロピル基を導入した25−OH−ビタミンD3とN−ヒドロキシスクシンイミドを導入したアガロース樹脂(Affi−Gel 10)を反応させることで合成される。(非特許文献4)。
【0005】
25−OH−ビタミンD3は、25位が水酸化されているビタミンD3であって、他のビタミンDと比べ、DBPに対する特異的結合力が極めて強いことが知られている。DBPと各種ビタミンDの結合力は、25−ヒドロキシビタミンD3の結合力を1とするとき、ビタミンD3で20分の1、ビタミンD3の代謝産物である1α,25−ジヒドロキシビタミンD3で668分の1であることが報告されている(非特許文献5,6)。この性質より、25−OH−ビタミンD3は上記DBP結合化合物に利用されている。
【0006】
しかしながら、25−OH−ビタミンD3は、生体内で生成されるプレビタミンD3が肝臓にて代謝され合成される希少な天然材料である。血清中に含まれる25−OH−ビタミンD3は、ビタミンD濃度の95%以上を占めるが、それでもその血中濃度は5ng/mL〜80ng/mLと非常に小さく、大量に入手することは困難である。そのため、25−OH−ビタミンD3を利用したDBP結合化合物を、安価かつ大量に生産することは困難であった。使用者は、使用済みのDBP結合化合物を再生して、長期の管理をしなければならず、使用者にとって大きな負担となっていた。
【0007】
そのため、当該分野においては、25−OH−ビタミンD3を結合させたDBP結合化合物に代わる、新たなDBP結合化合物が切望されていた。
【先行技術文献】
【非特許文献】
【0008】
【非特許文献1】J.G.Haddad, J. SteriodBiochem. Molec. Biol. (1995) 53, 579−582
【非特許文献2】堀 均ら,放射線生物研究 39(3),328−341(2004)
【非特許文献3】Rebecca P. et.al, ANALYTICAL BIOCHEMISTRY, 157, 262−269 (1986)
【非特許文献4】Swamy N.et.al, PROTEIN EXPRESSION AND PURIFICATION, 6, 185−188 (1995)
【非特許文献5】Bishop J.E.et.al, J. Bone Miner. Res., 9(8), 1277−88, 1994.
【非特許文献6】Bouillon R. et.al, Endocr. Rev., 16(2), 200−57, 1995.
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、DBPのアッセイや分離・精製などに用いることができる、ビタミンD結合タンパク質と特異的に結合可能な新規DBP結合化合物を提供する。
【課題を解決するための手段】
【0010】
本発明者らは、上記課題を解決すべく、鋭意検討した結果、25−OH−ビタミンD3と比べて、DBPとの結合力が弱いビタミンDであっても、当該ビタミンDと固相支持体とを結合させてなる化合物が、DBPに対して、25−OH−ビタミンD3を固相支持体に結合させてなる従来のDBP結合化合物と同程度またはそれ以上の結合特異性および結合力を有することを見出し、本発明を完成させるに至った。
【0011】
すなわち、本発明は以下のとおりである。
[1] 固相支持体と、ビタミンD結合タンパク質に結合可能なビタミンD3とが結合してなる、ビタミンD結合タンパク質結合化合物であって、該ビタミンD結合タンパク質に結合可能なビタミンD3が、25−OH−ビタミンD3を除くビタミンD3より選択される、上記ビタミンD結合タンパク質結合化合物。
[2] 固相支持体が、セファロース樹脂またはアガロース樹脂である、[1]のビタミンD結合タンパク質結合化合物。
[3] [1]または[2]のビタミンD結合タンパク質結合化合物を充填剤として含む、ビタミンD結合タンパク質精製用カラム。
[4] [1]または[2]のビタミンD結合タンパク質結合化合物または[3]のビタミンD結合タンパク質精製用カラムを用いた、ビタミンD結合タンパク質の分離または精製方法であって、
(i)ビタミンD結合タンパク質結合化合物とビタミンD結合タンパク質を含む試料とを接触させ、該ビタミンD結合タンパク質結合化合物に該ビタミンD結合タンパク質を結合させる工程;
(ii)該ビタミンD結合タンパク質結合化合物に結合しているビタミンD結合タンパク質を溶出する工程;ならびに
(iii)工程(ii)にて溶出されたビタミンD結合タンパク質を回収する工程。
[5] ビタミンD結合タンパク質結合化合物の製造方法であって、
固相支持体と、ビタミンD結合タンパク質に結合可能なビタミンD3とを結合させる工程を含み、該ビタミンD結合タンパク質に結合可能なビタミンD3が、25−OH−ビタミンD3を除くビタミンD3より選択される、上記方法。
[6] 固相支持体が、セファロース樹脂またはアガロース樹脂である、[5]の方法。
[7] 結合させる工程が、
(i)ビタミンD結合タンパク質に結合可能なビタミンD3をエポキシ化する工程;
(ii)固相支持体にチオール基を導入する工程;および
(iii)工程(i)で得られたビタミンD3と工程(ii)で得られた固相支持体を反応させ、該ビタミンD3と該固相支持体を結合させる工程、を含む、[5]または[6]の方法。
[8] 結合させる工程が、
(i)ビタミンD結合タンパク質に結合可能なビタミンD3をアミノプロピルエーテル化する工程;
(ii)固相支持体にN−ヒドロキシスクシンイミドを導入する工程;および
(iii)工程(i)で得られたビタミンD3と工程(ii)で得られた固相支持体を反応させ、該ビタミンD3と該固相支持体を結合させる工程、を含む、[5]または[6]の方法。
【発明の効果】
【0012】
本発明により、DBPのアッセイや分離・精製などに用いることができる、新たなDBP結合化合物を安価にかつ大量に提供することが可能となり、これによりDBPの研究分野において飛躍的な進展をもたらすことができる。
【図面の簡単な説明】
【0013】
【図1】図1は、操作1の条件で回収したタンパク質濃縮液のSDS−PAGE(CBB染色)の結果を示す。各レーンはそれぞれ以下のサンプルを示す。M:マーカー;1:対照化合物(1)で処理したサンプル;2:従来化合物(1)で処理したサンプル;3−6:本発明化合物(1)で処理したサンプル。
【図2】図2は、操作3の条件で回収したGcタイプ 1f1f、1s1sまたは22の血清に由来するタンパク質濃縮液のSDS−PAGE(CBB染色)の結果を示す。各レーンはそれぞれ以下のサンプルを示す。M:マーカー;1:シグマ社製のGcグロブリン;2:1f1fタイプ;3:1s1sタイプ;4:22タイプ。
【図3】図3は、操作3の条件で回収したGcタイプ 1f1f、1s1sまたは22の血清に由来するタンパク質濃縮液のヒト抗IgG抗体を用いたウエスタンブロットの結果を示す。各レーンはそれぞれ以下のサンプルを示す。1:1f1fタイプ;2:1s1sタイプ;3:22タイプ;4:精製DBP(Gcタイプ 1f1f)。
【図4】図4は、操作3の条件で回収したGcタイプ 1f1f、1s1sまたは22の血清に由来するタンパク質濃縮液のPNAレクチンを用いたウエスタンブロットの結果を示す。各レーンはそれぞれ以下のサンプルを示す。1:1f1fタイプ;2:1s1sタイプ;3:22タイプ;4:精製DBP(Gcタイプ 1f1f)。
【図5】図5は、操作3の条件を用いて回収したGcタイプ 1s1sの血清に由来するタンパク質濃縮液のSDS−PAGE(CBB染色)の結果を示す。各レーンはそれぞれ以下のサンプルを示す。1:シグマ社製のGcグロブリン;2−3:本発明化合物(1)で処理したサンプル;4−5:本発明化合物(3)で処理したサンプル;6−7:対照化合物(2)で処理したサンプル;8:シグマ社製のGc精製品;9:血清。
【図6】図6は、操作3の条件を用いて回収したGcタイプ 1s1sの血清に由来するタンパク質濃縮液のSDS−PAGE(CBB染色)の結果を示す。各レーンはそれぞれ以下のサンプルを示す。1:シグマ社製のGcグロブリン;2−3:本発明化合物(1)で処理したサンプル;4−5:従来化合物(2)で処理したサンプル;6−7:本発明化合物(2)で処理したサンプル;8−9:本発明類似化合物で処理したサンプル;10−11:対照化合物(1)で処理したサンプル。
【発明を実施するための形態】
【0014】
本発明は、固相支持体と、DBPに結合可能なビタミンDとが結合してなる、DBPに特異的に結合する化合物に関する。
【0015】
本発明においてDBPは、ヒト由来のDBPおよび非ヒト動物由来のDBPを含む。
本発明において「DBPに結合可能なビタミンD」としては、25−OH−ビタミンD3を除く、DBPに対して親和性を有し得る任意のビタミンDが含まれる。
【0016】
本発明において「25−OH−ビタミンD3」とは、25位のみが水酸化されているビタミンD3および25位のみが水酸化されているその誘導体を意味する。25−OH−ビタミンD3は、DBPに対して高い親和性を有することから、DBPに特異的に結合する既存の化合物の製造に用いられている(Rebecca P.ら、(上掲)、Swamy N.ら、(上掲))。本発明における「DBPに結合可能なビタミンD」は、DBPに対して親和性を有し得る限り25−OH−ビタミンD3を除く全てのビタミンD類(ビタミンD3、ビタミンD4、ビタミンD5、ビタミンD6、ビタミンD7など)ならびにそれらのプロビタミン、プレビタミン、代謝産物、および誘導体より選択することができる。具体的には、ビタミンD3、24,25(OH)2ビタミンD3、25,26(OH)2ビタミンD3、1α,25−(OH)2ビタミンD3など、およびこれらの誘導体が挙げられるが、これらに限定されない。好ましくは、ビタミンD3を用いる。ビタミンD3は、高純度のものを大量に入手できるために、有利である。
【0017】
本発明において利用し得る「DBPに結合可能なビタミンD」それ自体はDBPに対して、必ずしも25−OH−ビタミンD3と同等またはそれを超越する結合力を持っていなくても良い。当該「DBPに結合可能なビタミンD」は、下記にて詳述する固相支持体と結合することによって、DBPに対して、25−OH−ビタミンD3と同等またはそれを超越する結合力を備えることができる。
【0018】
本発明において利用し得るビタミンDは、遺伝子組換え技術または化学合成技術を用いて人工的に製造されたものであっても良いし、植物または動物に由来する天然のものであっても良い。
【0019】
本発明において「固相支持体」には、固相支持体として利用し得ることが公知である様々な材料、例えば、セルロース、セルロース誘導体、セファロース、アガロース、金属、ガラス、セラミック、樹脂など(これらに限定されない)を用いることができる。好ましくは、セファロースおよびアガロースであり、さらに好ましくはセファロースである。固相支持体は市販のものを用いることが可能であり、例えば、セファロースCL−6B樹脂(シグマ社)を利用することができる。
【0020】
固相支持体は、本発明のDBP結合化合物の用途および使用方法に応じて、平板、ビーズ、粒子、球体など(これらに限定されない)任意の形状を有することが可能である。固相支持体の大きさは、特に限定されることなく本発明の化合物の用途および使用方法に応じて適宜決定することができる。好ましくは、固相支持体は、粒径75〜300μmのビーズの形状を有する。
【0021】
ビタミンDと固相支持体との結合は、共有結合または非共有結合を利用して行うことができる。ビタミンDと固相支持体との結合は、特に限定するものではないが、公知の手法を利用して行うことができる。例えば、ビタミンDをエポキシ化し、チオール基を導入した固相支持体と反応させ、両者を結合することができる(Rebecca P.et.al(上掲))。
【0022】
あるいは、ビタミンDをアミノプロピルエーテル化し、固相支持体のカルボキシル基をN−ヒドロキシスクシンイミド(NHS)を用いてエステル化して活性化エステル基とし、両者を反応させることによって結合することができる(Swamy N.et.al(上掲))。
【0023】
上記反応に際しては、3位のヒドロキシ基をエポキシ化またはアミノプロピルエーテル化したビタミンD3を利用することが好ましい。
【0024】
本発明のDBP結合化合物は、試料中に含まれるDBPと特異的に結合することが可能であり、DBPを分取および精製することに利用することができる。なお、本明細書において、本発明のDBP結合化合物とDBPとの「結合」を、「吸着」と記載する場合がある。この場合、「結合」と「吸着」なる記載は互換的に使用される。
【0025】
DBPを含む試料としては、ヒトおよび非ヒト動物に由来する、生体組織破砕液、体液(全血、血漿、血清、リンパ液など)を利用することができる(これらに限定されない)。DBPを含む試料はDBP結合化合物と接触させる前に、予め精製されていても良いし、精製されていなくても良い。好ましくは、DBPを含む試料は予め精製されている。ここでDBPを含む試料を予め精製するとは、DBP結合化合物と非特異的に結合する、試料中に含まれるタンパク質を除去することを意味する。「除去」とは、当該非特異的なタンパク質が全て試料中より除かれる場合だけでなく、その一部が試料中より除かれる場合も含む。当該精製方法は、DBP結合化合物と非特異的に結合する、試料中に含まれるタンパク質を除去し得る限り、特に限定されないが、例えば、使用するDBP結合化合物に含まれる固相支持体のみを、DBPを含む試料と接触させ、当該固相支持体に吸着するタンパク質を除去することによって行うことができる。あるいは、DBP含有溶液を陽イオン交換カラム入れて素通りさせることで行うことができる。当該操作は必要に応じて、1回以上行うことができる。あるいは、DBPを含む試料は、DBPを精製または粗精製したものであっても良い。
【0026】
本発明のDBP結合化合物とDBPの結合に際しては、上記DBPを含む試料を、適当な緩衝液を用いてpH6.0〜9.0、好ましくはpH6.5〜8.0に調整する。緩衝液としては、例えば、リン酸緩衝液(リン酸カリウム緩衝液、リン酸ナトリウム緩衝液等)、Tris緩衝液(STE緩衝液等)を用いることができる。緩衝液を添加してpHを調整したDBPを含む試料を、同じ緩衝液を用いて平衡化した本発明の化合物と接触させ、試料中のDBPを本発明の化合物に結合させる。両者の結合反応は、バッチ式で行っても良いし、連続式で行っても良い。両者の接触は、4℃〜40℃、好ましくは4℃または室温において、1分間〜4時間振とうしながら行う。バッチ式で行う場合には好ましくはおよそ5〜10分間振とうしながら行う。あるいは、両者の結合反応は、カラム法によって行っても良い。この場合、本発明のDBP結合化合物は、DBPを精製するための各種クロマトグラフィー用カラム、特にアフィニティーロマトグラフィー用カラム、の充填剤として利用することができる。
【0027】
次いで、本発明のDBP結合化合物に結合したDBPを溶出する。本発明のDBP結合化合物からDBPを溶出する際の溶出液としては、塩化ナトリウム溶液、塩化カリウム溶液等の塩溶液、リン酸、酢酸、クエン酸、トリス等の緩衝液、グアニジン等の変性剤およびこれらの混合溶液等を利用することができる。溶出は当業者に公知の一般的な手法を用いて行うことができ、例えば、溶出液の塩濃度を徐々に増やして溶出する方法、DBPが本発明のDBP結合化合物と結合するpHから反撥するpHへ徐々に溶出液のpHを変化させる方法またはそれらの組合せを用いて行うことができる。DBPの結合した本発明のDBP結合化合物を遠心分離して回収した後、溶出液を添加して、4℃〜40℃、好ましくは4℃または室温において、およそ1〜30分間振とうした後、遠心分離して本発明のDBP結合化合物を沈降させ、上清に含まれるDBPを回収する。
【0028】
回収されたDBPは、タンパク質精製に用いられる公知の方法、例えば、硫安塩析、有機溶媒(エタノール、メタノール、アセトン等)による沈殿分離、イオン交換クロマトグラフィー、等電点クロマトグラフィー、ゲルろ過クロマトグラフィー、疎水性クロマトグラフィー、吸着カラムクロマトグラフィー、基質または抗体などを利用したアフィニティークロマトグラフィー、逆相カラムクロマトグラフィーなどのクロマトグラフィー、精密ろ過、限外ろ過、逆浸透ろ過、ダイアフィルトレーション等の濾過処理など、を1つまたは複数組み合わせて用いて、さらに精製することが可能である。
【0029】
本発明のDBP結合化合物を用いることによって血清よりDBPを97%、98%、99%またはそれ以上の高純度で得ることができる。
【実施例】
【0030】
以下、本発明の実施例を示すが、本発明は特にこれにより限定されるものではない。
【0031】
(実施例1)樹脂の合成
(I)新規DBP結合化合物(1)(以下、本発明化合物(1)と記載)
(i)ビタミンD3のエポキシ化
50mgのビタミンD3(コレカルシフェロール:東京化成工業社製)をN,N−ジメチルホルムアミド(DMF)4.0mlに溶解させ、枝付きフラスコに入れ、N2で置換した。次いで0.06mlのエピブロモヒドリン(和光社製)と20mgのNaHとを順次加え室温で攪拌しながら、N2気流中でエポキシ化反応を行う。
【0032】
薄層クロマトグラフィーで反応完了を確認した後、反応液に氷水および酢酸エチルを加え生成物の溶媒抽出を行う。この抽出操作を3回行い、抽出液を集めて氷水で洗浄し、MgSO4を加えて乾燥後、減圧乾固して生成物を含む残渣を得た。
【0033】
得られた残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル系)に通し、原材料を除去し、エポキシ化ビタミンD3を回収し、減圧乾燥させ、(iii)のDBP結合化合物合成工程に用いた。
【0034】
(ii)セファロース樹脂へのチオール基の導入
セファロースCL−6B樹脂(シグマ社製)約10gに1,4−ブタンジオールジグリシジルエーテル10gおよびNaBH420mlを含む1M NaOH 10mlを加え室温で10〜12時間振とうして、セファロースCL−6B樹脂にエポキシ側鎖を導入する。この樹脂を蒸留水で洗浄した後、2.0MのNa2S2O3を15ml加え、室温で約8時間振とうしながら、当該樹脂にチオール基(S−S)を導入した。チオール基を導入したセファロースCL−6B樹脂を蒸留水、次いでSPBで洗浄した後、樹脂にDTT200mgと1mM EDTA−2Naを含む0.3M NaHCO320ml(PH8.5)を加え、室温で1時間振とうして、SH−activatedセファロースCL−6B樹脂を得た。0.5M NaClで洗浄、次いで0.3M NaHCO3(含1mM EDTA・2Na)で洗浄して、以下の本発明化合物(1)の合成工程に用いた。
【0035】
(iii)本発明化合物(1)の合成
上記(i)にて得られたエポキシ化ビタミンD3を1,4−ジオキサン20mlに溶解させ、1M NaOH 10mlおよび上記(ii)にて得られたSH−activatedセファロースCL−6B樹脂を加え、ウオータバス上、40℃、N2気流中で撹拌した。18時間後、60% アセトン/イオン交換水、アセトン、60% アセトン/イオン交換水、イオン交換水、0.05M NaCl、イオン交換水(各250 mL)で洗浄後、遠心して上清を取り除いた。本発明化合物(1)約10gを得た。
【0036】
(iv)樹脂保管
合成した本発明品は0.02% NaN3を入れたSTEバッファー(pH7.4, Tris・HCl 6.05g,EDTA・2Na 0.56g,NaCl 8.77gをSDWに溶かし、全量を1000mlとして濾過し、オートクレーブにかける。Triton X−100を1ml加えたもの)20mLを加えて、密栓し4℃で保管した。
【0037】
(II)新規DBP結合化合物(2)(以下、本発明化合物(2)と記載)
ビタミンD3(コレカルシフェロール:東京化成工業製)(100mg,0.26mmol)をt−BuOH/MeCN(9/1,v/v)混合液(3.0mL)に溶解し、氷上撹拌下、アクリロニトリル(300μL,0.56mmol,2.0eq.)を滴下した。NaHを加え、1時間後、冷Et2O、冷水を滴下し、減圧化で溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(hexane:EtOAc=10:1)に付し、98.4mg,(0.22mmol,86%)の固体1を得た。その固体124.9mg(0.06mmol)をEt2O(5.0mL)に溶解し、氷上撹拌下、LiAlH4/AlCl3 60.3mg,(1.07mmol, 3.9eq.)を加えた。1時間後、1N NaOHを滴下し、EtOAcで4回抽出し、有機層をまとめて、飽和食塩水で洗浄し、無水Na2SO4で乾燥させ、ろ過後、ろ液を減圧下で溶媒を留去し、固体2を得た。次の反応のためにAffiGel−10(4.0mL,45μmol)を1,4−ジオキサンで3回洗浄した。これに固体2(全量)を1,4−ジオキサン(5.0mL)で加え、プロペラで室温下、撹拌した。22時間後、エタノールアミン(2.5mL)を加え、さらに撹拌した。2時間後、1,4−ジオキサン/アセトン(1/1,v/v)で3回洗浄し、上清を取り除き、0.02% NaN3/STEバッファーを加え、ビタミンD3結合アガロース樹脂(本発明化合物(2))4.0mLを得た。
【0038】
(III)新規DBP結合化合物(3)(以下、本発明化合物(3)と記載)
Activated Thiol SepharoseCL−4B(GEヘルスケアバイオサイエンス社製)2.5gを蒸留水およびリン酸バッファーで洗浄し、遠心後、上清を取り除いた。DTT200mgと1mM EDTA−2Naを含む0.3M NaHCO320ml(PH8.4)を加え、室温で1時間振とうして、チオール基の還元反応を行った。生成したSH−activatedセファロースCL−4B樹脂を0.5M NaClで洗浄、次いで0.3M NaHCO3(含1mM EDTA・2Na)で洗浄して、これに上記本発明化合物(1)と同様にして得られた、エポキシ化ビタミンD3を1,4−ジオキサン(20ml)に溶解して加え、さらに1M NaOH 10mlを加え、ウオータバス上、40℃、N2気流中で撹拌して、ビタミンD3結合CL−4B樹脂(本発明化合物(3))を得た。
【0039】
(IV)ビタミンD2を含む化合物(以下、本発明類似化合物と記載)
上記本発明化合物(1)と同様に、ビタミンD2(カルシフェロール:東京化成工業社製)100mgをエポキシ化して、エポキシ化ビタミンD2とした。さらに、上記本発明化合物(1)と同様に合成したSH−activatedセファロースCL−6B樹脂とを同様な操作で反応させ、ビタミンD2結合CL−6B樹脂(本発明類似化合物)を得た。
【0040】
(V)従来公知のDBP結合化合物(1)(以下、従来化合物(1)と記載)
25−OH−ビタミンD3とセファロースCL−6B樹脂からなる従来公知のDBP結合化合物として、徳島大学大学院ソシオテクノサイエンス研究部 堀均博士より恵与された25−OH−ビタミンD2結合CL−6B樹脂(従来化合物(1))を使用した。
【0041】
(VI)連結部の構造が異なる従来公知のDBP結合化合物(2)(以下、従来化合物(2)と記載)
洗浄したAffiGel−10 4.0mlを準備し、これに25−OH−ビタミンD3(カルシフェディジール:東京化成工業製)100.0mg,(0.25mmol)をt−BuOH/MeCN(9/1,v/v)混合液(3.0mL)に溶解し、上記(II)新規DBP結合化合物(2)と同様に操作して得た固体(全量)を1,4−ジオキサン(5.0mL)で加え、さらに同様に処理して25−OH−ビタミンD3結合アガロース樹脂4.0mL(従来化合物(2))を得た。従来化合物(2)は、ビタミンD3と固相支持体の連結部に、エーテル結合と3つのヒドロキシ側鎖を有する。
【0042】
(VII)対照化合物(1)
対照化合物(1)として、セファロースCL−6B樹脂をそのまま使用した。
【0043】
(VIII)対照化合物(2)
対照化合物(2)としてはActivated Thiol SepharoseCL−4BセファロースCL−4B樹脂をそのまま使用した。
【0044】
(実施例2)本発明化合物と従来化合物の比較
血清(Gcタイプ 1f1f)から硫安分画した粗Gcグロブリンを、25−OH−ビタミンD3とセファロース樹脂からなる従来化合物(1)(徳島大学大学院ソシオテクノサイエンス研究部 堀均博士より恵与のもの)をアフィニティカラムとして使って一次精製し、次にヒドロキシアパタイトカラムで二次精製し、精製DBPを得た。この精製DBPを用いて、実施例1で製造した本発明化合物(1)および従来化合物(1)のタンパク質吸着能を比較した。
【0045】
(i)使用前の樹脂の調整
1.5mlのエッペンチューブに加えた樹脂は、使用する前にPH7.4のSTEバッファーを500μL加え、30秒間振とう後、3分間遠心分離(3,000rpm)した。この洗浄を5回繰り返し、樹脂の保存のために加えたアジ化ナトリウムを除いた。
【0046】
(ii)吸着および洗浄、溶出操作
上記(i)の操作でアジ化ナトリウムを除いた本発明化合物(1)および従来化合物(1)を各0.5g含むエッペンチューブに100μg相当量の精製DBP溶液18.31μlとSTEバッファー981.7μlを加えて、タンパク質を吸着させるため常温にて1時間振とうした。以下、4℃、2分間遠心分離(13,000rpm)し、上澄み液を除いた後、化合物を洗浄するためSTEバッファー500μlを加え、キュートミキサー(1,800rpm)で60秒間振とうした。その後、2分間遠心分離(13,000rpm)し上澄み液を除いた。この洗浄操作を5回繰り返した後に、溶出液としてpH5.0の0.5M酢酸バッファー500μLを加えキュートミキサー(1,800rpm)で60秒間振とうし、4℃、2分間遠心分離(13,000rpm)した。この溶出操作を3回繰り返し、溶出液を得た。
【0047】
(iii)タンパク質の定量
各溶出液600μlを正確にとり、タンパク質定量用BCA(ビシンコニン酸)試薬2.4mlを加えて37℃で30分間加温した後、562nmの吸光度を測定した。溶出液中のタンパク質量は、BSA(シグマ社製)を標準品とした検量線から求めた。
【0048】
その結果、溶出液中のタンパク質量は、本発明化合物(1)では46.5μg/ml、また、従来化合物(1)では36.5μg/mlとなった。
この結果は、従来化合物(1)と同様、本発明化合物(1)がDBPと結合することを示す。
【0049】
(実施例3)DBPおよびBSAの吸着比較
(i)吸着および洗浄、溶出操作
アジ化ナトリウムを除いた本発明化合物(1)を各0.5g含むエッペンチューブに100μg相当量の精製DBP溶液あるいはBSA溶液を加え、さらに、全量が1000μlとなるようにSTEバッファーを加えて、以下、1時間振とうし、実施例2(ii)と同様に、洗浄、溶出操作を行い、溶出液を得た。
【0050】
(ii)タンパク質定量
各溶出液600μLを正確にとり、タンパク質定量用BCA液(Pierce BCA Protein Assay Kit、Thermo SCIENTIFIC社製)2.4mlを加えて37℃で30分間加温した後、562nmの吸光度を測定した。回収液中のタンパク質濃度を、BSAを標準品とした検量線から求めた。
【0051】
結果を以下の表1に示す。なお表1中、用いた化合物は、それぞれ別個の操作により合成された化合物を意味する。
【0052】
【表1】
【0053】
DBPサンプルでの回収タンパク質量は本発明化合物(1)では42.42μg、従来化合物(1)では35.76μgであった。一方、BSAサンプルでの回収タンパク質量はブランク値の範囲内でタンパク質量は算出できなかった。
【0054】
この結果は、本発明化合物がDBPを特異的に結合することが可能であり、また従来化合物(1)よりも優れた結合能を有することを示す。
【0055】
実施例4:血清からのタンパク質の回収
(i)吸着および洗浄、溶出操作
(操作1)
Gcタイプ 1s1sのボランティアからヒト血液を20ml採取し、室温で30分静置し、その後遠心により血清を回収し、4℃で保管する。室温に戻して30分間、静置し、10分間遠心分離(3,000rpm)して血清(約10ml)を回収し、4℃に保管した。
【0056】
各エッペンチューブに本発明化合物(1)、従来化合物(1)および対照品化合物(1)をそれぞれ0.5g分注し、アジ化ナトリウムを除いた後、250μlの血清と250μlのSTEバッファー(10mM Tris,pH 8.0;1mM EDTA,100mM NaCl)を加え、実施例2(ii)と同様な操作を行い、溶出液を得た。
【0057】
得られた溶出液の500μLをアミコン(millipore社製アミコンウルトラ0.5 100000M.W.CO)上で10分間、4℃、13,000Gで、限外濾過した。さらに残りの溶出液を加えて同様に処理した。このアミコンの中に、10mM リン酸ナトリウムバッファー(pH7.0)500μLを加えて、4℃で、10分間、13,000Gで、バッファー置換を行う。これを5回行い、その後、4℃で、3分間、遠心分離(1000G)して、タンパク質濃縮液を得た。
【0058】
(操作2)
対照化合物(1)0.5gをとり、アジ化ナトリウムを除いた後、1s1sタイプのヒト血清250μLとリン酸ナトリウムバッファー250μLを加えて、常温にて1時間振とうし、4℃で、2分間、遠心分離(13,000rpm)し、上澄み液を回収した(前処理操作)。
【0059】
この上澄み液500μLずつを、アジ化ナトリウムを除いた本発明化合物(1)、従来化合物(1)および対照化合物(1)をそれぞれ0.5g含むエッペンチューブに加え、1時間振とうし、その後、4℃で、2分間、遠心分離(13,000rpm)し、上澄み液を除いた。化合物を洗浄するため、500μLのSTEバッファー(PH7.4)を加え、キュートミキサー(1,800rpm)で30秒間振とうし、さらに、4℃で、2分間、遠心分離(13,000rpm)を行い、上澄み液を除いた。この洗浄操作を10回繰り返した後に、0.5M酢酸バッファー(pH5.0)500μLを加え、60秒間振とう、2分間、遠心分離(13,000rpm)を行い、上澄み液を回収した。この回収操作を3回繰り返し、上澄み液を合わせて溶出液とした。以下、上記(操作1)と同様な限外濾過およびバッファー置換を行い、タンパク質濃縮液を得た。
【0060】
(ii)タンパク質定量
操作1または操作2の条件で回収したタンパク質濃縮液を用いて、本発明化合物(1)、対照化合物(1)および従来化合物(1)のタンパク質吸着能について、BCA法を用いたタンパク質定量によって解析した。すなわち、BSA溶液を2.0,1.0,0.5,0.25,0,1mg/mlの濃度で用意した。タンパク質濃縮液はそれぞれ30倍に希釈した。調製したBSAおよびタンパク質濃縮液60μLを96穴プレートに正確に加え、さらにBCA液(Pierce BCA Protein Assay Kit、Thermo SCIENTIFIC社製)を240μLずつ加える。37℃、30分間インキュベーションした後、吸光度を波長570nmで測定し、タンパク濃度を算出した。
【0061】
結果を以下の表2および表3に示す。なお表2および3中、用いた化合物は、それぞれ別個の操作により合成された化合物を意味する。
【0062】
【表2】
【0063】
【表3】
【0064】
上記(操作1)において、対照化合物も含め、全ての樹脂で数百μgレベルのタンパク質を得た(表2参照)。非特異的なタンパク質が大量に混入しており、各樹脂のDBP結合能については判断できなかった。
【0065】
上記(操作2)において、本発明化合物および従来化合物では14μg〜28μgのタンパク質の吸着が得られた。対照化合物ではタンパク質の吸着は得られなかった(表3参照)。これは(操作2)において、本発明化合物(1)および従来化合物(1)がDBPを回収できたことを示唆する。
【0066】
(iii)SDS−PAGE(CBB染色)
上記(操作1)と(操作2)の結果を確認するために、(操作1)の濃縮液のSDS−PAGEを行い、ゲルをCBB染色した。すなわち、CBB溶液(CBB 0.25gをメタノール:酢酸:mQ(5:1:4)に溶かしたもの)を500〜1000μLをゲルにかけ、2〜3分程度染色した後、脱色液に10分間ほど浸して振盪し脱色した後、一晩、風乾した。
【0067】
その結果、対照化合物(1)、従来化合物(1)および4ロットの本発明化合物(1)のいずれのサンプルでも多くのタンパク質バンドが見られた(図1参照)。上記(操作1)では、洗浄不足のため非特異的タンパク質が大量に混入するため、従来化合物(1)でもDBPバンドを確認することができなかった。
【0068】
(iv)HPLC解析
さらに、上記(操作1)と(操作2)の違いを確認するため、各濃縮液をリン酸ナトリウムバッファーでタンパク濃度250ng/μLに希釈して、下記の条件でHPLCを行った。DBP標準品にシグマ社製Gcグロブリンを用いて、標準品の保持時間と重なるピークの保持時間およびそのピークの面積を全タンパク質の面積で割り、見かけ上のGc%を調べた。
・移動相:50mM SPB+0.3M NaCl
・流量:0.7ml/min
・注入量:タンパク質2.5μg相当(10μL inject)
・カラム:東ソーTSKgel G3000SWXL
・カラム温度:25℃
・検出波長:225nm
結果を以下の表4に示す。なお表4中、用いた化合物は、それぞれ別個の操作により合成された化合物を意味する。
【0069】
【表4】
【0070】
【表5】
【0071】
上記(操作1)によるタンパク質濃縮液で当該ピークの保持時間は、いずれの樹脂の場合も標準品には一致しなかった(表4参照)。一方、(操作2)によるタンパク質濃縮液において、本発明化合物(1)および従来化合物(1)で処理したサンプルでは当該ピークの保持時間が、完全に標準品と一致した(表5参照)。
【0072】
タンパク定量、電気泳動およびHPLC分析から、(操作2)の条件で、不純タンパク質が除去でき、本発明化合物(1)および従来化合物(1)を用いてDBPを精製できることが示された。
【0073】
(実施例5)Gcタイプ 1f1f、1s1sおよび22の血清からの直接DBP濃縮
本発明化合物(1)によるDBPの精製能を高めるため、下記操作3の条件を見出した。Gcタイプの異なる3種類1f1f、1s1sおよび22のヒト血清を用いて、操作3の条件を用いてDBP精製を行い、サンプルのタンパク質定量、SDS−PAGE,ウエスタンブロッティングを行った。
【0074】
(i)吸着および洗浄、溶出操作
(操作3)
3本のエッペンチューブに本発明化合物(1)0.5gずつを分注し、アジ化ナトリウムを除いた後、各エッペンチューブに各々3タイプの血清250μlを加え、さらにSTEバッファー250μlを加えて、キュートミキサー(1,800rpm)で5分間振とうした。次に、4℃で、2分間、遠心分離(13,000rpm)し、上澄み液を除いた。さらに、STEバッファー(PH7.4)を500μl加え、キュートミキサー(1,800rpm)で10秒間振とうし、化合物の洗浄を行い、4℃で、2分間、遠心分離(13,000rpm)し、上澄み液を除いた。この洗浄操作を15回行った。次に、0.5M酢酸バッファー(pH5.0)500μlを加え、キュートミキサー(1,800rpm)で10秒間振とうしタンパク質を溶出させ、4℃で、2分間、遠心分離(13,000rpm)した。これを2回行い、上澄み液を回収液とした。以下、限外濾過、バッファー置換を行い、タンパク質濃縮液を得た。
【0075】
(ii)タンパク質定量とSDS−PAGE(CBB染色)
操作3の条件で回収したタンパク質濃縮液を用いて、本発明化合物(1)のタンパク質吸着能について、BCA法を用いたタンパク質定量によって解析した。
結果を以下の表6に示す。
【0076】
【表6】
【0077】
結果、血清250μL当り24〜27μgのタンパク質が得られた(表6参照)。
【0078】
次に、操作3の条件で回収したタンパク質濃縮液のSDS−PAGE(CBB染色)を行った。
結果を図2に示す。結果、Gcタイプに関係なく同一の単一バンドを得た(図2)。
【0079】
シグマ社製のGcグロブリンはバンドの位置がサンプルとは異なるが、これは他のサンプルと異なり、操作3の条件を用いて精製されていないためであり、シグマ社製のGcグロブリンも(操作3)の条件を用いて精製することによって、血清由来のGcグロブリンとバンド位置は一致する(図5レーン1および2参照)。
【0080】
(iii)ウエスタンブロット
操作3の条件で回収したタンパク質濃縮液を用いて、ヒト抗IgG抗体およびPNAレクチンを用いたウエスタンブロットを行った。標準品には精製DBP(実施例2参照)を使用した。転写後、膜を0.1% TBSTで10分間、3回洗浄を行い、1.0% BSA in TBSTでブロッキングを行った後、0.1% TBSTで10分間、3回洗浄を行った。以下、室温で1時間、振盪させて一次抗体と反応させた後、0.1% TBSTで10分間、3回洗浄を行い、室温で二次抗体と1時間、振盪反応させた。反応後、同様に0.1% TBSTで10分間、3回洗浄を行った。この膜を暗室に運び、プラスティック容器内で膜に1分間程ECL液をまんべんなくかけ、それをプラスティックシートにはさみ、フィルムをシートの上に30秒〜3分間置いた。
【0081】
以下、現像液に1分間漬け、水洗いし、停止液に1分間漬けて現像を行った。
結果を、図3および図4に示す。結果、バンドはヒト抗IgG抗体を用いたウエスタンブロットで染色され、それは精製DBPと一致した(図3参照)。また、PNAレクチンを用いたウエスタンブロットでもバンドは染色され、精製DBPと一致した(図4参照)。 これより、本発明化合物(1)を用いて、血清中のDBPを回収できることが明らかとなった。
【0082】
(iv)HPLC解析
1f1f、1s1s、2.2の3タイプのGcサンプルについて、実施例4(iv)と同じ条件でHPLC法を行い、HPLC法で精製DBPの純度分析が可能かどうかを調べた。標準品としてシグマ社製Gcグロブリンを使用し、サンプルの保持時間(RT)が標準品のそれと±0.02min以内であることを事前に調査し、また、シンメトリー係数を調べ、その値が1.00〜1.30を示した場合、分析の信頼性があるものとした。この2つの条件をクリアにしたサンプルは正しい評価ができるとして、次式からピーク面積法によりタンパク質当りのDBP純度を求めた。
DBP純度=主ピーク面積÷9〜16分間に見られる全体のピーク面積×100%
結果を以下の表7に示す。
【0083】
【表7】
【0084】
結果、3タイプのGcサンプル全てにおいて、保持時間が標準品の保持時間と、0.002〜0.014分以内で一致した(表7参照)。また、シンメトリー係数は1.167〜1.186で、分析の信頼性があることを確認した。
【0085】
DBP純度を求めた結果、いずれのサンプルも全タンパク質ピーク面積の98.7%以上であった。これはシグマ社製Gcグロブリンの純度98.16%と同等あるいはそれ以上である。
【0086】
以上の結果は、HPLC法によって、本発明化合物(1)の処理で得られたサンプル中のDBPの純度評価が可能であることを示す。また、本発明化合物(1)を用いることにより、Gcタイプを問わず、血清より高純度のDBPを回収できることを示す。
【0087】
(実施例6)本発明化合物と本発明類似化合物の比較。
実施例1で合成したビタミンD2結合CL−6B樹脂である本発明類似化合物、さらにビタミンD3結合アガロース樹脂(本発明化合物(2))および連結部分の構造が異なるビタミンD3結合CL−4B樹脂(本発明化合物(3))についてDBPを吸着するかどうかを、タンパク定量法およびSDS−PAGEで検討した。
【0088】
(i)吸着および洗浄、溶出操作
1s1sのヒト血清を使い、(操作3)の条件に従って回収したタンパク質濃縮液、ならびに(操作3)において、本発明化合物(1)に代えて、本発明類似化合物、本発明化合物(2)、本発明化合物(3)、対照化合物(1)もしくは(2)、または従来化合物(2)を用いる以外は同様の条件を用いて調製された各タンパク質濃縮液を、以下(ii)−(iv)の実験に用いた。
【0089】
(ii)本発明類似品化合物
本発明類似化合物を用いて回収したタンパク質濃縮液を用いて、本発明類似化合物のタンパク質吸着能について、BCA法を用いたタンパク質定量によって解析した。結果を以下の表8に示す。なお表8中、用いた化合物は、それぞれ別個の操作により合成された化合物を意味する。
【0090】
【表8】
【0091】
BCA法によるタンパク質定量の結果、本発明類似化合物によって27〜38μg程度のタンパク質が得られた(表8参照)。
【0092】
しかし、SDS−PAGE(CBB染色)においては、DBPのバンドは認められなかった。本発明類似化合物にはDBPが吸着しないことが明らかとなった(図6レーン8および9参照)。
【0093】
(iii)本発明化合物(2)
従来化合物(2)である25(OH)ビタミンD3結合アガロース樹脂は、DBPのアフィニティカラムとして使用される(非特許文献4)。そこで、従来化合物(2)または本発明化合物(2)を用いて回収したタンパク質濃縮液を用いて、従来化合物(2)および本発明化合物(2)のそれぞれのタンパク質吸着能について、BCA法を用いたタンパク質定量によって解析した。
【0094】
結果を以下の表9に示す。なお表9中、用いた化合物は、それぞれ別個の操作により合成された化合物を意味する。
【0095】
【表9】
【0096】
BCA法によるタンパク質定量の結果、本発明化合物(2)および従来化合物(2)には本発明化合物(1)よりも多くのタンパク質が吸着した(表8および9参照)。タンパク質濃縮液には不純タンパク質の混入があるが、このサンプルのSDS−PAGE(BCC染色)をおこなった結果、本発明化合物(2)と従来化合物(2)には、同様の2つのバンドが見られ、下側のバンドが本発明化合物(1)のDBPバンドに一致した。したがって、本発明化合物(2)を用いて、DBPを吸着できることが示された(図6レーン4−7参照)。
【0097】
(iv)本発明化合物(3)
本発明化合物(3)を用いて回収したタンパク質濃縮液を用いて、本発明化合物(3)のタンパク質吸着能について、BCA法を用いたタンパク質定量によって解析した。結果を、以下の表10に示す。なお表10中、用いた化合物は、それぞれ別個の操作により合成された化合物を意味する。
【0098】
【表10】
【0099】
結果、本発明化合物(3)を用いることによって、本発明化合物(1)よりも少ない、5.1〜6.0μg程度のタンパク量が回収された(表10参照)。そこで、SDS−PAGE(CBB染色)を行ったこころ、本発明化合物(1)に一致した1つバンドが見られ、DBPが吸着することが示された(図5レーン4および5参照)。
【産業上の利用可能性】
【0100】
本発明により、DBPのアッセイや分離・精製などに用いることができる、新たなDBP結合化合物を安価にかつ大量に供給することが可能となるために、これによりDBPの研究利用や臨床利用など様々な分野において重要な貢献をすることが期待できる。
【特許請求の範囲】
【請求項1】
固相支持体と、ビタミンD結合タンパク質に結合可能なビタミンD3とが結合してなる、ビタミンD結合タンパク質結合化合物であって、該ビタミンD結合タンパク質に結合可能なビタミンD3が、25−OH−ビタミンD3を除くビタミンD3より選択される、上記ビタミンD結合タンパク質結合化合物。
【請求項2】
固相支持体が、セファロース樹脂またはアガロース樹脂である、請求項1に記載のビタミンD結合タンパク質結合化合物。
【請求項3】
請求項1または2に記載のビタミンD結合タンパク質結合化合物を充填剤として含む、ビタミンD結合タンパク質精製用カラム。
【請求項4】
請求項1または2に記載のビタミンD結合タンパク質結合化合物または請求項3に記載のビタミンD結合タンパク質精製用カラムを用いた、ビタミンD結合タンパク質の分離または精製方法であって、
(i)ビタミンD結合タンパク質結合化合物とビタミンD結合タンパク質を含む試料とを接触させ、該ビタミンD結合タンパク質結合化合物に該ビタミンD結合タンパク質を結合させる工程;
(ii)該ビタミンD結合タンパク質結合化合物に結合しているビタミンD結合タンパク質を溶出する工程;ならびに
(iii)工程(ii)にて溶出されたビタミンD結合タンパク質を回収する工程。
【請求項5】
ビタミンD結合タンパク質結合化合物の製造方法であって、
固相支持体と、ビタミンD結合タンパク質に結合可能なビタミンD3とを結合させる工程を含み、該ビタミンD結合タンパク質に結合可能なビタミンD3が、25−OH−ビタミンD3を除くビタミンD3より選択される、上記方法。
【請求項6】
固相支持体が、セファロース樹脂またはアガロース樹脂である、請求項5に記載の方法。
【請求項7】
結合させる工程が、
(i)ビタミンD結合タンパク質に結合可能なビタミンD3をエポキシ化する工程;
(ii)固相支持体にチオール基を導入する工程;および
(iii)工程(i)で得られたビタミンD3と工程(ii)で得られた固相支持体を反応させ、該ビタミンD3と該固相支持体を結合させる工程、を含む、請求項5または6に記載の方法。
【請求項8】
結合させる工程が、
(i)ビタミンD結合タンパク質に結合可能なビタミンD3をアミノプロピルエーテル化する工程;
(ii)固相支持体にN−ヒドロキシスクシンイミドを導入する工程;および
(iii)工程(i)で得られたビタミンD3と工程(ii)で得られた固相支持体を反応させ、該ビタミンD3と該固相支持体を結合させる工程、を含む、請求項5または6に記載の方法。
【請求項1】
固相支持体と、ビタミンD結合タンパク質に結合可能なビタミンD3とが結合してなる、ビタミンD結合タンパク質結合化合物であって、該ビタミンD結合タンパク質に結合可能なビタミンD3が、25−OH−ビタミンD3を除くビタミンD3より選択される、上記ビタミンD結合タンパク質結合化合物。
【請求項2】
固相支持体が、セファロース樹脂またはアガロース樹脂である、請求項1に記載のビタミンD結合タンパク質結合化合物。
【請求項3】
請求項1または2に記載のビタミンD結合タンパク質結合化合物を充填剤として含む、ビタミンD結合タンパク質精製用カラム。
【請求項4】
請求項1または2に記載のビタミンD結合タンパク質結合化合物または請求項3に記載のビタミンD結合タンパク質精製用カラムを用いた、ビタミンD結合タンパク質の分離または精製方法であって、
(i)ビタミンD結合タンパク質結合化合物とビタミンD結合タンパク質を含む試料とを接触させ、該ビタミンD結合タンパク質結合化合物に該ビタミンD結合タンパク質を結合させる工程;
(ii)該ビタミンD結合タンパク質結合化合物に結合しているビタミンD結合タンパク質を溶出する工程;ならびに
(iii)工程(ii)にて溶出されたビタミンD結合タンパク質を回収する工程。
【請求項5】
ビタミンD結合タンパク質結合化合物の製造方法であって、
固相支持体と、ビタミンD結合タンパク質に結合可能なビタミンD3とを結合させる工程を含み、該ビタミンD結合タンパク質に結合可能なビタミンD3が、25−OH−ビタミンD3を除くビタミンD3より選択される、上記方法。
【請求項6】
固相支持体が、セファロース樹脂またはアガロース樹脂である、請求項5に記載の方法。
【請求項7】
結合させる工程が、
(i)ビタミンD結合タンパク質に結合可能なビタミンD3をエポキシ化する工程;
(ii)固相支持体にチオール基を導入する工程;および
(iii)工程(i)で得られたビタミンD3と工程(ii)で得られた固相支持体を反応させ、該ビタミンD3と該固相支持体を結合させる工程、を含む、請求項5または6に記載の方法。
【請求項8】
結合させる工程が、
(i)ビタミンD結合タンパク質に結合可能なビタミンD3をアミノプロピルエーテル化する工程;
(ii)固相支持体にN−ヒドロキシスクシンイミドを導入する工程;および
(iii)工程(i)で得られたビタミンD3と工程(ii)で得られた固相支持体を反応させ、該ビタミンD3と該固相支持体を結合させる工程、を含む、請求項5または6に記載の方法。
【図1】

【図2】
【図3】
【図4】

【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2012−122038(P2012−122038A)
【公開日】平成24年6月28日(2012.6.28)
【国際特許分類】
【出願番号】特願2010−276128(P2010−276128)
【出願日】平成22年12月10日(2010.12.10)
【出願人】(510327161)
【Fターム(参考)】
【公開日】平成24年6月28日(2012.6.28)
【国際特許分類】
【出願日】平成22年12月10日(2010.12.10)
【出願人】(510327161)
【Fターム(参考)】
[ Back to top ]