
ビフェニル−2−イルカルバミン酸エステルの調製方法
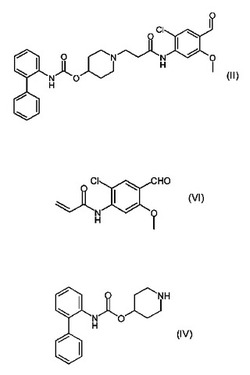
式(II)の化合物の調製方法であって、式(VI)の化合物を式(IV)と好適な溶媒中で反応させること含む上記方法。
【化1】

【化1】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ムスカリン性アゴニストおよびβ2アドレナリン受容体アゴニスト活性を有するビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの調製方法における新規で重要な工程に関する。
【背景技術】
【0002】
2004年2月13日に出願した国際特許出願WO2004/074246(Theravance Inc、 South San Francisco、 California、 US)には、慢性閉塞性肺疾患(COPD)および喘息などの肺疾患の治療に有用な新規のビフェニル化合物が開示されている。特に、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルはムスカリン性アゴニストおよびβ2アドレナリン受容体アゴニスト活性の両方を有する化合物として開示されている。ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの化学構造を以下の式(I)に示す。
【化1】
【0003】
WO2004/074246は式(I)の化合物の調製方法を開示する。
【0004】
式(I)の化合物の調製における重要な中間体はビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステル(1−3−{[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]アミノ}−3−オキソプロピル)−4−ピペリジニル2−ビフェニルイルカルバメート)としても知られている)であり、式(II)で表される。
【化2】
【0005】
WO2004/074246で開示する(調製95)式(II)の化合物の調製において重要な工程は、式(III)で表されるメチル4−(アクリロイルアミノ)−5−クロロ−2−(メチルオキシ)ベンゾエートと、
【化3】
【0006】
式(IV)で表されるビフェニル−2−イルカルバミン酸ピペリジン−4−イルエステルとの反応であり、
【化4】
【0007】
式(V)で表されるメチル4−{3−[4−(ビフェニル−2−イルカルバモイルオキシ)ピペリジン−1−イル]プロピオニルアミノ−5−クロロ−2−メトキシベンゾエートが生成する。
【化5】
【0008】
この反応において、式(III)の化合物はエステル酸化レベルにある。結果として、WO2004/074246は式(V)の化合物を式(II)の化合物(これら2つの化学式は上記に概説した)に変換するのに必要なさらなる2つの反応工程の概要を述べている。第1はアルコール酸化レベルへの還元であり、第2はアルデヒド酸化レベルへの還元である。したがって、式(III)の化合物と式(IV)の化合物間のカップリング反応から開始して、重要な中間体、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステルの調製には3工程が必要である。
【0009】
さらなる2つの最近の国際特許出願WO2006/023454およびWO2007/090859において、第1工程がメチル4−(アクリロイルアミノ)−5−クロロ−2−(メチルオキシ)ベンゾエートのビフェニル−2−イルカルバミン酸ピペリジン−4−イルエステルとの反応である、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステルの調製について、3工程の方法がまた開示されている。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】WO2004/074246
【特許文献2】WO2006/023454
【特許文献3】WO2007/090859
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明の目的は、式(II)の化合物、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの調製において重要な中間体を調製するための、新規で代替となり得る、より効率的で経済的な方法を提供することである。
【課題を解決するための手段】
【0012】
従って、本発明によると、式(II)の化合物の調製方法が提供され、
【化6】
【0013】
該方法は、式(VI)の化合物を、
【化7】
【0014】
式(IV)の化合物と、
【化8】
【0015】
好適な溶媒中で反応させることを含む。
【0016】
新規で代替となり得る本方法は、式(II)の化合物の調製に必要となる工程数を3工程から1工程へと減少させ多くの利点を有する。経済的に、本方法はサイクル時間の有意な減少、溶剤廃棄物の減少、および質量効率の上昇をもたらす。加えて、この改善された方法は水素化ホウ素リチウムおよび水素化アルミニウムリチウムなどの有害な還元剤の使用を無くすことで健康と安全について配慮し、かつ、工程中にでる金属廃棄物(マンガン、リチウム)を減少させる。水素化ホウ素リチウムは調製96(WO2004/074246)および調製5(WO2006/023454)に概説されている。水素化アルミニウムリチウムは調製15(WO2006/023454)に概説されている。二酸化マンガンは調製16(WO2006/023454)および実施例1(WO2007/090859)に概説されている。
【0017】
式(IV)の化合物で処理した場合の、式(VI)の化合物の予想外の安定性と、その予想外の反応選択性が組み合わさることにより、この単一工程の方法の開発に成功した。
【発明を実施するための形態】
【0018】
式(VI)の化合物と式(IV)との反応は好適な溶媒中で行う。好適な溶媒とは、非プロトン性およびプロトン性溶媒を含んでもよい。好適な非プロトン性溶媒の例は、これらに限定されないが、アセトニトリル、2−メチルテトラヒドロフラン、テトラヒドロフラン、酢酸エチル、ジメチルホルムアミドおよびトルエンを含む。好適なプロトン性溶媒の例は、これらに限定されないが、エタノール、メタノール、イソプロピルアルコール、およびフェノールを含む。本発明のさらなる態様では、反応は溶媒として2−メチルテトラヒドロフラン中で行う。
【0019】
場合により、本方法は好適な有機酸源の添加をさらに含んでもよい。好適な有機酸源の例は、酢酸、ギ酸および安息香酸などの有機カルボン酸を含む。好適な有機カルボン酸を本方法に添加することで反応の不純物プロファイルが向上する。従って、本発明のさらなる態様では、本方法は、有機酸源のさらなる添加を含む。本発明のさらに別の態様では、有機酸源が有機カルボン酸である。本発明のさらに別の態様では、有機カルボン酸が酢酸である。
【0020】
反応は周囲温度と選択された溶媒の還流温度間の温度で実施することができ、この温度を反応が完了するまで維持する。
【0021】
冷却結晶化または貧溶媒添加結晶化などの種々の標準的な結晶化技術を用いて反応の生成物を溶液から結晶させてもよい。冷却結晶化においては、溶解した不純化合物を含有する反応混合物をゆっくり冷却し、場合により種結晶を添加して、溶液から分離をすることになる必要な化合物が形成される。結晶化した後、濾過して結晶を単離し、好適な溶媒を用いて洗浄し、乾燥することができる。
【0022】
本発明のさらなる態様では、溶媒として2−メチルテトラヒドロフランを用いてその中で反応させる場合、有機酸源として酢酸を添加し、反応混合物を60℃まで冷却し、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステルを種結晶として添加し、60℃で30分熟成させてから4時間かけて20℃まで冷却する。
【0023】
目的の化合物を分離または精製する場合、貧溶媒添加結晶化を冷却結晶化の代替法として使用することができる。貧溶媒添加結晶化においては、不純化合物を好適な溶媒中で溶解する。貧溶媒を添加することで溶液中の目的の化合物の溶解度が下がり、求める化合物の結晶形成を促進する。結晶化した後、濾過して結晶を単離し、好適な溶媒を用いて洗浄し、乾燥することができる。
【0024】
本発明のさらなる態様では、溶媒としてトルエンを用いてその中で反応させる場合、混合反応物を50℃で濃縮し、結晶化させるために変性エタノール貧溶媒を添加する。混合物を60℃で4時間熟成し、次に4時間かけて20℃まで冷却する。
【0025】
本発明のさらなる態様では、式(VI)の化合物を提供する。
【化9】
【0026】
本発明のさらに別の態様では、N−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]−2−プロペンアミドを提供する。
【0027】
本発明は、以下の実施例によって例示される。
【実施例】
【0028】
工程A:4−ブロモ−2−クロロ−5−メトキシアニリンの調製
【化10】
【0029】
1,3−ジブロモ−5,5−ジメチルヒダントイン(例えば、Aldrichから市販されている)(9.1g、32mmol)を酢酸エチル(150ml)中の6−クロロ−3−メトキシアニリン(例えば、Apollo Scientificから市販されている)(10.0g、63mmol)に−5℃で20分にわたって添加した。得られた溶液を−5℃で1時間撹拌し、水(40ml)中の炭酸カリウム(6g、43mmol)の溶液で洗浄し、次に水(20ml)で洗浄した。得られた溶液を減圧下で濃縮し、薄い茶色の固体として4−ブロモ−2−クロロ−5−メトキシアニリンを得た(14.3g、95%)。
【0030】
1H NMR(400MHz、CDCl3)δH(ppm)7.38(1H、s)、6.34(1H、s),4.02‐4.14(2H、br s)、3.83(3H、s)
m/z(ES+)236(M+H)
工程B:4−アミノ−5−クロロ−2−メトキシベンズアルデヒドの調製
【化11】
【0031】
イソプロピルマグネシウムクロリド(テトラヒドロフラン溶液中2M、23ml、46mmol)(例えば、Aldrichから市販されている)を−10℃でテトラヒドロフラン溶液中の4−ブロモ−2−クロロ−5−メトキシアニリン(工程A)(10g、42mmol)の撹拌溶液に5分かけて添加した。得られた溶液を50分かけて0℃として濃厚スラリーを得、次に−25℃まで冷却し、n−ブチルリチウム(ヘキサン溶液中1.6M、90ml、144mmol)を20分かけて添加し、そしてテトラヒドロフラン(20ml)を添加した。溶液を30分かけて−10℃まで加熱し、次にN,N−ジメチルホルムアミド(16ml、207mmol)を5分かけて添加し、得られた濃厚スラリーを20分かけて0℃まで加熱した。<10℃で反応を維持して水(50ml)中のクエン酸(22g、105mmol)の溶液を15分かけて慎重に添加した。スラリーを30分間20℃で熟成し、真空下で濾過した。濾過ケーキを水(100ml)で洗浄し、真空下で16時間、40℃で乾燥し、薄黄色固体として4−アミノ−5−クロロ−2−メトキシベンズアルデヒド(6.1g、80%th)を得た。
【0032】
1H NMR(400MHz、DMSO-d6)δH(ppm)9.95(1H,s)、7.49(1H、s)、6.50-6.57(2H、br s)、6.44(1H,s)、3.81(3H、s)
m/z(ES+)186(M+H)
工程C:N−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]−2−プロペンアミドの調製
【化12】
【0033】
調製1
アクリル酸(例えば、Aldrichから市販されている)(46ml、0.67mol)を酢酸エチル(0.85L)中の4−アミノ−5−クロロ−2−メトキシベンズアルデヒド(工程B)(50.0g、0.27mol)およびトリエチルアミン(204g、2.02mol)の撹拌懸濁液に25℃でゆっくりと添加した。反応温度を30℃から40℃の範囲で維持してプロパンホスホン酸無水物(酢酸エチル中50%、429g、0.67mol)を30分かけて添加した。混合物を30℃から40℃の範囲でさらに1時間撹拌し、次に25℃まで冷却し、水(0.26L)で希釈し、32%塩酸(108g)でpH2〜3まで酸性化した。有機相を分離し、水(0.23L)と32%水酸化ナトリウム(14g)の混合液で洗浄した。水相は約pH7であった。有機相を水(0.23L)で洗浄し、次に減圧下(約300ミリバール)で濃縮して0.56kgの蒸留物を除去した。メチルシクロヘキサン(335g)を添加し、さらに286gの蒸留物を減圧下で除去した。メチルシクロヘキサン(111g)を添加し、さらに得られた懸濁液を20℃まで冷却し、濾過してメチルシクロヘキサンで洗浄した。濾過ケーキを減圧下において40℃で12時間乾燥し、N−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]−2−プロペンアミド(46g、71%)を得た。
【0034】
調製2
反応温度が20℃を超えないようにしながら、3−クロロプロピオニルクロリド(98.4ml、1.0mol)を30分かけて4−アミノ−5−クロロ−2−メトキシベンズアルデヒドの撹拌懸濁液に添加した。添加完了後、混合物を20℃でさらに2時間撹拌し、次に濾過した。濾液を減圧下で150mlまで濃縮し、次に酢酸エチル(100ml)と水(400ml)で希釈した。混合物を20℃で1時間撹拌し、次に濾過してオフホワイト色の固体として3−クロロ−N−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]プロピオンアミドを得、これを単離せずにテトラヒドロフラン(730ml)中に懸濁してジイソプロピルエチルアミン(154ml、0.88mol)で処理した。得られた混合物を45℃で46時間撹拌し、次に減圧下で濃縮し残渣を得、これを酢酸エチル(300ml)で希釈し、2M塩酸(4x100ml)で洗浄し、さらに減圧下で濃縮してN−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]−2−プロペンアミド(37.8g、62%)を得た。
【0035】
1H NMR(400MHz、CDCl3)δH(ppm)10.31(1H、s)、8.43(1H、s)、8.02(1H、br s)、7.82(1H、 s)、6.47−6.53(1H、dd)、6.28−6.38(1H、dd)、5.88−5.93(1H、dd)、3.96(3H、s)
m/z(ES+)240(M+H)
工程D:ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステルの調製
【化13】
【0036】
調製1
ビフェニル−2−イルカルバミン酸ピペリジン−4−イルエステル(WO2004/074246Aの調製8に従い調製することができる)(1.03kg、3.48mol)を、2−メチルテトラヒドロフラン(8.1L)中のN−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]−2−プロペンアミド(工程C(調製1)またはC(調製2)に従い調製することができる)(0.81kg、3.38mol)および酢酸(0.39L、6.62mol)の撹拌溶液に60℃で5分間かけて少しずつ加えた。得られた溶液を75℃に加熱し、2時間この温度を保持した。溶液を60℃まで冷却し、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステル(4.0g)を種結晶として添加して、60℃で30分熟成させてから4時間かけて20℃まで冷却した。得られた懸濁液を真空下で濾過して濾過ケーキをIMS(3x1.6L)で洗浄した。固体を真空オーブン中で50℃で10時間乾燥させ白色の固体としてビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステルを得た(1.50kg、82%th)。
【0037】
1H NMR(400MHz、DMSO−d6)δH(ppm)10.70(1H、s)、10.19(1H、s)、8.68(1H、s)、8.25(1H、s)、7.72(1H、s)、7.43−7.28(9H、m)、4.48−4.54(1H、br m)、3.88(3H、s)、2.70−2.82(2H、br s)、2.63(4H、s)、2.20−2.30(2H、br m)、1.72−1.82(2H、br m)、1.49−1.56(2H、br m)
m/z(ES+)536(M+H)
調製2
ビフェニル−2−イルカルバミン酸ピペリジン−4−イルエステル(WO2004/074246Aの調製8に従い調製することができる)(63.0kg、212.57mol)を、2−メチルテトラヒドロフラン(430kg)中のN−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]−2−プロペンアミド(工程C(調製1)またはC(調製2)に従い調製することができる)(50.0kg、208.62mol)および酢酸(12.6kg、209.83mol)の撹拌懸濁液に25℃で添加した。次に混合物を60分かけて50℃まで加熱し、この温度を2時間保持した。得られた懸濁液を90分かけて20℃まで冷却し、この温度で4時間保持した。懸濁液を真空下で濾過して濾過ケーキをIMS(3x78.9kg)で洗浄した。固体を真空オーブン中で50℃で10時間乾燥し、白色の固体としてビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステルを得た(90.7kg、80.6%th)。
【0038】
IRピーク波数(cm−1) 3413、2775、1731、1698、1677、1575、1515、1443、1447、1401、1376、1206、1043、1001、758、702
LC Rt=4.58分
装置
1H NMRスペクトルはCDCl3またはDMSO−d6のいずれかにおいて、Bruker DPX400、400MHz機器に記録した。
【0039】
質量スペクトルは、正イオンエレクロトロスプレー、質量範囲100−1000(ZQ)または150−1500(LCT)amuで動作するWaters LCT質量分析計に記録した。
【0040】
IRスペクトルは、16回積算、2.0cm−1分解能を用いて、ATR固体試料としてPerkin Elmer Spectrum FTIR計測器に記録した。
【0041】
ATR=全反射測定法
HPLCクロマトグラムは、以下の条件で、Hewlett Packard Agilent 1100シリーズHPLCに記録した。
【表1】
【技術分野】
【0001】
本発明は、ムスカリン性アゴニストおよびβ2アドレナリン受容体アゴニスト活性を有するビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの調製方法における新規で重要な工程に関する。
【背景技術】
【0002】
2004年2月13日に出願した国際特許出願WO2004/074246(Theravance Inc、 South San Francisco、 California、 US)には、慢性閉塞性肺疾患(COPD)および喘息などの肺疾患の治療に有用な新規のビフェニル化合物が開示されている。特に、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルはムスカリン性アゴニストおよびβ2アドレナリン受容体アゴニスト活性の両方を有する化合物として開示されている。ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの化学構造を以下の式(I)に示す。
【化1】
【0003】
WO2004/074246は式(I)の化合物の調製方法を開示する。
【0004】
式(I)の化合物の調製における重要な中間体はビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステル(1−3−{[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]アミノ}−3−オキソプロピル)−4−ピペリジニル2−ビフェニルイルカルバメート)としても知られている)であり、式(II)で表される。
【化2】
【0005】
WO2004/074246で開示する(調製95)式(II)の化合物の調製において重要な工程は、式(III)で表されるメチル4−(アクリロイルアミノ)−5−クロロ−2−(メチルオキシ)ベンゾエートと、
【化3】
【0006】
式(IV)で表されるビフェニル−2−イルカルバミン酸ピペリジン−4−イルエステルとの反応であり、
【化4】
【0007】
式(V)で表されるメチル4−{3−[4−(ビフェニル−2−イルカルバモイルオキシ)ピペリジン−1−イル]プロピオニルアミノ−5−クロロ−2−メトキシベンゾエートが生成する。
【化5】
【0008】
この反応において、式(III)の化合物はエステル酸化レベルにある。結果として、WO2004/074246は式(V)の化合物を式(II)の化合物(これら2つの化学式は上記に概説した)に変換するのに必要なさらなる2つの反応工程の概要を述べている。第1はアルコール酸化レベルへの還元であり、第2はアルデヒド酸化レベルへの還元である。したがって、式(III)の化合物と式(IV)の化合物間のカップリング反応から開始して、重要な中間体、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステルの調製には3工程が必要である。
【0009】
さらなる2つの最近の国際特許出願WO2006/023454およびWO2007/090859において、第1工程がメチル4−(アクリロイルアミノ)−5−クロロ−2−(メチルオキシ)ベンゾエートのビフェニル−2−イルカルバミン酸ピペリジン−4−イルエステルとの反応である、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステルの調製について、3工程の方法がまた開示されている。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】WO2004/074246
【特許文献2】WO2006/023454
【特許文献3】WO2007/090859
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明の目的は、式(II)の化合物、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの調製において重要な中間体を調製するための、新規で代替となり得る、より効率的で経済的な方法を提供することである。
【課題を解決するための手段】
【0012】
従って、本発明によると、式(II)の化合物の調製方法が提供され、
【化6】
【0013】
該方法は、式(VI)の化合物を、
【化7】
【0014】
式(IV)の化合物と、
【化8】
【0015】
好適な溶媒中で反応させることを含む。
【0016】
新規で代替となり得る本方法は、式(II)の化合物の調製に必要となる工程数を3工程から1工程へと減少させ多くの利点を有する。経済的に、本方法はサイクル時間の有意な減少、溶剤廃棄物の減少、および質量効率の上昇をもたらす。加えて、この改善された方法は水素化ホウ素リチウムおよび水素化アルミニウムリチウムなどの有害な還元剤の使用を無くすことで健康と安全について配慮し、かつ、工程中にでる金属廃棄物(マンガン、リチウム)を減少させる。水素化ホウ素リチウムは調製96(WO2004/074246)および調製5(WO2006/023454)に概説されている。水素化アルミニウムリチウムは調製15(WO2006/023454)に概説されている。二酸化マンガンは調製16(WO2006/023454)および実施例1(WO2007/090859)に概説されている。
【0017】
式(IV)の化合物で処理した場合の、式(VI)の化合物の予想外の安定性と、その予想外の反応選択性が組み合わさることにより、この単一工程の方法の開発に成功した。
【発明を実施するための形態】
【0018】
式(VI)の化合物と式(IV)との反応は好適な溶媒中で行う。好適な溶媒とは、非プロトン性およびプロトン性溶媒を含んでもよい。好適な非プロトン性溶媒の例は、これらに限定されないが、アセトニトリル、2−メチルテトラヒドロフラン、テトラヒドロフラン、酢酸エチル、ジメチルホルムアミドおよびトルエンを含む。好適なプロトン性溶媒の例は、これらに限定されないが、エタノール、メタノール、イソプロピルアルコール、およびフェノールを含む。本発明のさらなる態様では、反応は溶媒として2−メチルテトラヒドロフラン中で行う。
【0019】
場合により、本方法は好適な有機酸源の添加をさらに含んでもよい。好適な有機酸源の例は、酢酸、ギ酸および安息香酸などの有機カルボン酸を含む。好適な有機カルボン酸を本方法に添加することで反応の不純物プロファイルが向上する。従って、本発明のさらなる態様では、本方法は、有機酸源のさらなる添加を含む。本発明のさらに別の態様では、有機酸源が有機カルボン酸である。本発明のさらに別の態様では、有機カルボン酸が酢酸である。
【0020】
反応は周囲温度と選択された溶媒の還流温度間の温度で実施することができ、この温度を反応が完了するまで維持する。
【0021】
冷却結晶化または貧溶媒添加結晶化などの種々の標準的な結晶化技術を用いて反応の生成物を溶液から結晶させてもよい。冷却結晶化においては、溶解した不純化合物を含有する反応混合物をゆっくり冷却し、場合により種結晶を添加して、溶液から分離をすることになる必要な化合物が形成される。結晶化した後、濾過して結晶を単離し、好適な溶媒を用いて洗浄し、乾燥することができる。
【0022】
本発明のさらなる態様では、溶媒として2−メチルテトラヒドロフランを用いてその中で反応させる場合、有機酸源として酢酸を添加し、反応混合物を60℃まで冷却し、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステルを種結晶として添加し、60℃で30分熟成させてから4時間かけて20℃まで冷却する。
【0023】
目的の化合物を分離または精製する場合、貧溶媒添加結晶化を冷却結晶化の代替法として使用することができる。貧溶媒添加結晶化においては、不純化合物を好適な溶媒中で溶解する。貧溶媒を添加することで溶液中の目的の化合物の溶解度が下がり、求める化合物の結晶形成を促進する。結晶化した後、濾過して結晶を単離し、好適な溶媒を用いて洗浄し、乾燥することができる。
【0024】
本発明のさらなる態様では、溶媒としてトルエンを用いてその中で反応させる場合、混合反応物を50℃で濃縮し、結晶化させるために変性エタノール貧溶媒を添加する。混合物を60℃で4時間熟成し、次に4時間かけて20℃まで冷却する。
【0025】
本発明のさらなる態様では、式(VI)の化合物を提供する。
【化9】
【0026】
本発明のさらに別の態様では、N−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]−2−プロペンアミドを提供する。
【0027】
本発明は、以下の実施例によって例示される。
【実施例】
【0028】
工程A:4−ブロモ−2−クロロ−5−メトキシアニリンの調製
【化10】
【0029】
1,3−ジブロモ−5,5−ジメチルヒダントイン(例えば、Aldrichから市販されている)(9.1g、32mmol)を酢酸エチル(150ml)中の6−クロロ−3−メトキシアニリン(例えば、Apollo Scientificから市販されている)(10.0g、63mmol)に−5℃で20分にわたって添加した。得られた溶液を−5℃で1時間撹拌し、水(40ml)中の炭酸カリウム(6g、43mmol)の溶液で洗浄し、次に水(20ml)で洗浄した。得られた溶液を減圧下で濃縮し、薄い茶色の固体として4−ブロモ−2−クロロ−5−メトキシアニリンを得た(14.3g、95%)。
【0030】
1H NMR(400MHz、CDCl3)δH(ppm)7.38(1H、s)、6.34(1H、s),4.02‐4.14(2H、br s)、3.83(3H、s)
m/z(ES+)236(M+H)
工程B:4−アミノ−5−クロロ−2−メトキシベンズアルデヒドの調製
【化11】
【0031】
イソプロピルマグネシウムクロリド(テトラヒドロフラン溶液中2M、23ml、46mmol)(例えば、Aldrichから市販されている)を−10℃でテトラヒドロフラン溶液中の4−ブロモ−2−クロロ−5−メトキシアニリン(工程A)(10g、42mmol)の撹拌溶液に5分かけて添加した。得られた溶液を50分かけて0℃として濃厚スラリーを得、次に−25℃まで冷却し、n−ブチルリチウム(ヘキサン溶液中1.6M、90ml、144mmol)を20分かけて添加し、そしてテトラヒドロフラン(20ml)を添加した。溶液を30分かけて−10℃まで加熱し、次にN,N−ジメチルホルムアミド(16ml、207mmol)を5分かけて添加し、得られた濃厚スラリーを20分かけて0℃まで加熱した。<10℃で反応を維持して水(50ml)中のクエン酸(22g、105mmol)の溶液を15分かけて慎重に添加した。スラリーを30分間20℃で熟成し、真空下で濾過した。濾過ケーキを水(100ml)で洗浄し、真空下で16時間、40℃で乾燥し、薄黄色固体として4−アミノ−5−クロロ−2−メトキシベンズアルデヒド(6.1g、80%th)を得た。
【0032】
1H NMR(400MHz、DMSO-d6)δH(ppm)9.95(1H,s)、7.49(1H、s)、6.50-6.57(2H、br s)、6.44(1H,s)、3.81(3H、s)
m/z(ES+)186(M+H)
工程C:N−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]−2−プロペンアミドの調製
【化12】
【0033】
調製1
アクリル酸(例えば、Aldrichから市販されている)(46ml、0.67mol)を酢酸エチル(0.85L)中の4−アミノ−5−クロロ−2−メトキシベンズアルデヒド(工程B)(50.0g、0.27mol)およびトリエチルアミン(204g、2.02mol)の撹拌懸濁液に25℃でゆっくりと添加した。反応温度を30℃から40℃の範囲で維持してプロパンホスホン酸無水物(酢酸エチル中50%、429g、0.67mol)を30分かけて添加した。混合物を30℃から40℃の範囲でさらに1時間撹拌し、次に25℃まで冷却し、水(0.26L)で希釈し、32%塩酸(108g)でpH2〜3まで酸性化した。有機相を分離し、水(0.23L)と32%水酸化ナトリウム(14g)の混合液で洗浄した。水相は約pH7であった。有機相を水(0.23L)で洗浄し、次に減圧下(約300ミリバール)で濃縮して0.56kgの蒸留物を除去した。メチルシクロヘキサン(335g)を添加し、さらに286gの蒸留物を減圧下で除去した。メチルシクロヘキサン(111g)を添加し、さらに得られた懸濁液を20℃まで冷却し、濾過してメチルシクロヘキサンで洗浄した。濾過ケーキを減圧下において40℃で12時間乾燥し、N−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]−2−プロペンアミド(46g、71%)を得た。
【0034】
調製2
反応温度が20℃を超えないようにしながら、3−クロロプロピオニルクロリド(98.4ml、1.0mol)を30分かけて4−アミノ−5−クロロ−2−メトキシベンズアルデヒドの撹拌懸濁液に添加した。添加完了後、混合物を20℃でさらに2時間撹拌し、次に濾過した。濾液を減圧下で150mlまで濃縮し、次に酢酸エチル(100ml)と水(400ml)で希釈した。混合物を20℃で1時間撹拌し、次に濾過してオフホワイト色の固体として3−クロロ−N−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]プロピオンアミドを得、これを単離せずにテトラヒドロフラン(730ml)中に懸濁してジイソプロピルエチルアミン(154ml、0.88mol)で処理した。得られた混合物を45℃で46時間撹拌し、次に減圧下で濃縮し残渣を得、これを酢酸エチル(300ml)で希釈し、2M塩酸(4x100ml)で洗浄し、さらに減圧下で濃縮してN−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]−2−プロペンアミド(37.8g、62%)を得た。
【0035】
1H NMR(400MHz、CDCl3)δH(ppm)10.31(1H、s)、8.43(1H、s)、8.02(1H、br s)、7.82(1H、 s)、6.47−6.53(1H、dd)、6.28−6.38(1H、dd)、5.88−5.93(1H、dd)、3.96(3H、s)
m/z(ES+)240(M+H)
工程D:ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステルの調製
【化13】
【0036】
調製1
ビフェニル−2−イルカルバミン酸ピペリジン−4−イルエステル(WO2004/074246Aの調製8に従い調製することができる)(1.03kg、3.48mol)を、2−メチルテトラヒドロフラン(8.1L)中のN−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]−2−プロペンアミド(工程C(調製1)またはC(調製2)に従い調製することができる)(0.81kg、3.38mol)および酢酸(0.39L、6.62mol)の撹拌溶液に60℃で5分間かけて少しずつ加えた。得られた溶液を75℃に加熱し、2時間この温度を保持した。溶液を60℃まで冷却し、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステル(4.0g)を種結晶として添加して、60℃で30分熟成させてから4時間かけて20℃まで冷却した。得られた懸濁液を真空下で濾過して濾過ケーキをIMS(3x1.6L)で洗浄した。固体を真空オーブン中で50℃で10時間乾燥させ白色の固体としてビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステルを得た(1.50kg、82%th)。
【0037】
1H NMR(400MHz、DMSO−d6)δH(ppm)10.70(1H、s)、10.19(1H、s)、8.68(1H、s)、8.25(1H、s)、7.72(1H、s)、7.43−7.28(9H、m)、4.48−4.54(1H、br m)、3.88(3H、s)、2.70−2.82(2H、br s)、2.63(4H、s)、2.20−2.30(2H、br m)、1.72−1.82(2H、br m)、1.49−1.56(2H、br m)
m/z(ES+)536(M+H)
調製2
ビフェニル−2−イルカルバミン酸ピペリジン−4−イルエステル(WO2004/074246Aの調製8に従い調製することができる)(63.0kg、212.57mol)を、2−メチルテトラヒドロフラン(430kg)中のN−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]−2−プロペンアミド(工程C(調製1)またはC(調製2)に従い調製することができる)(50.0kg、208.62mol)および酢酸(12.6kg、209.83mol)の撹拌懸濁液に25℃で添加した。次に混合物を60分かけて50℃まで加熱し、この温度を2時間保持した。得られた懸濁液を90分かけて20℃まで冷却し、この温度で4時間保持した。懸濁液を真空下で濾過して濾過ケーキをIMS(3x78.9kg)で洗浄した。固体を真空オーブン中で50℃で10時間乾燥し、白色の固体としてビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステルを得た(90.7kg、80.6%th)。
【0038】
IRピーク波数(cm−1) 3413、2775、1731、1698、1677、1575、1515、1443、1447、1401、1376、1206、1043、1001、758、702
LC Rt=4.58分
装置
1H NMRスペクトルはCDCl3またはDMSO−d6のいずれかにおいて、Bruker DPX400、400MHz機器に記録した。
【0039】
質量スペクトルは、正イオンエレクロトロスプレー、質量範囲100−1000(ZQ)または150−1500(LCT)amuで動作するWaters LCT質量分析計に記録した。
【0040】
IRスペクトルは、16回積算、2.0cm−1分解能を用いて、ATR固体試料としてPerkin Elmer Spectrum FTIR計測器に記録した。
【0041】
ATR=全反射測定法
HPLCクロマトグラムは、以下の条件で、Hewlett Packard Agilent 1100シリーズHPLCに記録した。
【表1】
【特許請求の範囲】
【請求項1】
式(II)の化合物を調製するための方法であって、
【化1】
式(VI)の化合物を、
【化2】
式(IV)の化合物と、
【化3】
好適な溶媒中で反応させることを含む、上記方法。
【請求項2】
前記反応が非プロトン性溶媒中で行われる、請求項1に記載の方法。
【請求項3】
前記非プロトン性溶媒が2−メチルテトラヒドロフランである、請求項2に記載の方法。
【請求項4】
前記方法が有機酸源の添加をさらに含む、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
前記有機酸源が有機カルボン酸である、請求項4に記載の方法。
【請求項6】
前記有機カルボン酸が酢酸である、請求項5に記載の方法。
【請求項7】
前記反応が、周囲温度と前記選択された溶媒の還流温度の間の温度で実施される、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
式(II)の化合物を調製するための請求項1に記載の方法であって、
【化4】
式(VI)の化合物を、
【化5】
式(IV)の化合物と、
【化6】
溶媒としての2−メチルテトラヒドロフラン、および有機カルボン酸源としての酢酸の存在下で反応させることを含み、ここで、前記反応が75℃の温度で実施される、上記方法。
【請求項9】
反応後、前記反応混合物を60℃まで冷却し、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステルを種結晶として添加し、60℃で30分熟成させ、次に4時間かけて20℃まで冷却する、請求項8に記載の方法。
【請求項10】
式(II)の化合物を調製するための請求項1に記載の方法であって、
【化7】
式(VI)の化合物を、
【化8】
式(IV)の化合物と、
【化9】
溶媒としての2−メチルテトラヒドロフラン、および有機カルボン酸源としての酢酸の存在下で反応させることを含み、ここで、前記反応が50℃の温度で実施される、上記方法。
【請求項11】
反応後、前記反応混合物を90分かけて20℃まで冷却し、次に20℃で4時間維持する、請求項10記載の方法。
【請求項12】
式(VI)の化合物。
【化10】
【請求項13】
N−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]−2−プロペンアミド。
【請求項1】
式(II)の化合物を調製するための方法であって、
【化1】
式(VI)の化合物を、
【化2】
式(IV)の化合物と、
【化3】
好適な溶媒中で反応させることを含む、上記方法。
【請求項2】
前記反応が非プロトン性溶媒中で行われる、請求項1に記載の方法。
【請求項3】
前記非プロトン性溶媒が2−メチルテトラヒドロフランである、請求項2に記載の方法。
【請求項4】
前記方法が有機酸源の添加をさらに含む、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
前記有機酸源が有機カルボン酸である、請求項4に記載の方法。
【請求項6】
前記有機カルボン酸が酢酸である、請求項5に記載の方法。
【請求項7】
前記反応が、周囲温度と前記選択された溶媒の還流温度の間の温度で実施される、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
式(II)の化合物を調製するための請求項1に記載の方法であって、
【化4】
式(VI)の化合物を、
【化5】
式(IV)の化合物と、
【化6】
溶媒としての2−メチルテトラヒドロフラン、および有機カルボン酸源としての酢酸の存在下で反応させることを含み、ここで、前記反応が75℃の温度で実施される、上記方法。
【請求項9】
反応後、前記反応混合物を60℃まで冷却し、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイルエチル]ピペリジン−4−イルエステルを種結晶として添加し、60℃で30分熟成させ、次に4時間かけて20℃まで冷却する、請求項8に記載の方法。
【請求項10】
式(II)の化合物を調製するための請求項1に記載の方法であって、
【化7】
式(VI)の化合物を、
【化8】
式(IV)の化合物と、
【化9】
溶媒としての2−メチルテトラヒドロフラン、および有機カルボン酸源としての酢酸の存在下で反応させることを含み、ここで、前記反応が50℃の温度で実施される、上記方法。
【請求項11】
反応後、前記反応混合物を90分かけて20℃まで冷却し、次に20℃で4時間維持する、請求項10記載の方法。
【請求項12】
式(VI)の化合物。
【化10】
【請求項13】
N−[2−クロロ−4−ホルミル−5−(メチルオキシ)フェニル]−2−プロペンアミド。
【公表番号】特表2012−523447(P2012−523447A)
【公表日】平成24年10月4日(2012.10.4)
【国際特許分類】
【出願番号】特願2012−505154(P2012−505154)
【出願日】平成22年4月14日(2010.4.14)
【国際出願番号】PCT/EP2010/054893
【国際公開番号】WO2010/119064
【国際公開日】平成22年10月21日(2010.10.21)
【出願人】(397009934)グラクソ グループ リミテッド (832)
【氏名又は名称原語表記】GLAXO GROUP LIMITED
【住所又は居所原語表記】Glaxo Wellcome House,Berkeley Avenue Greenford,Middlesex UB6 0NN,Great Britain
【Fターム(参考)】
【公表日】平成24年10月4日(2012.10.4)
【国際特許分類】
【出願日】平成22年4月14日(2010.4.14)
【国際出願番号】PCT/EP2010/054893
【国際公開番号】WO2010/119064
【国際公開日】平成22年10月21日(2010.10.21)
【出願人】(397009934)グラクソ グループ リミテッド (832)
【氏名又は名称原語表記】GLAXO GROUP LIMITED
【住所又は居所原語表記】Glaxo Wellcome House,Berkeley Avenue Greenford,Middlesex UB6 0NN,Great Britain
【Fターム(参考)】
[ Back to top ]