
ビフェニル化合物の結晶性遊離塩基形
本発明は、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの2つの結晶性遊離塩基形を提供する。本発明は、前記結晶性遊離塩基を含むまたは前記結晶性遊離塩基を使用して調製される製薬組成物;前記結晶性遊離塩基を調製するためのプロセスおよび中間体;ならびに肺障害を処置するために前記結晶性遊離塩基を使用する方法も提供する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、肺障害を処置するために有用であることが期待される、ビフェニル化合物の新規結晶形に関するものである。本発明は、前記結晶性化合物を含むまたはこのような化合物から調製された製薬組成物、このような結晶性化合物を調製するためのプロセスおよび中間体ならびに肺障害を処置するためにこのような化合物を使用する方法にも関する。
【背景技術】
【0002】
Mammenらへの特許文献1は、慢性閉塞性肺疾患(COPD)および喘息などの肺障害を処置するのに有用であることが期待される新規ビフェニル化合物を開示している。特に、化合物ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルは、ムスカリン受容体アンタゴニストまたは抗コリン活性を所有するとして本出願に明確に記載されている。
【0003】
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの化学構造は、式Iによって表される:
【0004】
【化1】
【0005】
式Iの化合物は、市販のAutoNomソフトウェア(MDL,San Leandro,California)を使用して命名されている。
【0006】
肺または呼吸障害を処置するのに有用な治療剤は、吸入により気道中に直接、好都合に投与される。この点で、ドライパウダー吸入器(DPI)、定量吸入器(MDI)およびネブライザ吸入器を含む、吸入により治療剤を投与するための複数の種類の製薬吸入デバイスが開発されてきた。このようなデバイスでの使用のための製薬組成物および製剤を調製するとき、吸湿性でも潮解性でもなく、比較的高い融点を有し、これにより材料が著しく分解することなく微粉化される、結晶形の治療剤を有することが高度に所望である。結晶性遊離塩基形の式Iの化合物はAxtらへの特許文献2でI形およびII形として報告されているが、本発明の結晶性遊離塩基形は、より高い融点を含む、異なるおよび特に有用な特性を有する。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】米国特許出願公開第2005/0203133号明細書
【特許文献2】米国特許出願公開第2007/0112027号明細書
【発明の概要】
【課題を解決するための手段】
【0008】
本発明の1態様は、6.6±0.1、13.1±0.1、18.6±0.1、19.7±0.1、および20.2±0.1の2θ値に回折ピークを備える粉末X線回折パターンを特徴とする、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基形に関する。
【0009】
本発明の別の態様は、6.6±0.1、13.1±0.1、18.6±0.1、19.7±0.1、および20.2±0.1の2θ値に回折ピークを備える粉末X線回折パターンを特徴とする;ならびに8.8±0.1、10.1±0.1、11.4±0.1、11.6±0.1、14.8±0.1、15.2±0.1、16.1±0.1、16.4±0.1、16.9±0.1、17.5±0.1、18.2±0.1、19.3±0.1、19.9±0.1、20.8±0.1、21.1±0.1、21.7±0.1、および22.3±0.1から選択される2θ値に5つ以上の追加の回折ピークを有することをさらに特徴とする、III形と呼ばれる、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基に関する。
【0010】
本発明のなお別の態様は、6.6±0.1、13.1±0.1、18.6±0.1、19.7±0.1、および20.2±0.1の2θ値に回折ピークを備える粉末X線回折パターンを特徴とする;ならびに10.6±0.1、15.0±0.1、16.0±0.1、17.3±0.1、17.7±0.1、20.9±0.1、21.4±0.1、22.6±0.1、24.6±0.1、および27.8±0.1から選択される2θ値に5つ以上の追加の回折ピークを有することをさらに特徴とする、IV形と呼ばれる、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基に関する。
【0011】
本発明の別の態様は、本発明の結晶性遊離塩基および製薬的に許容され得る担体を含む製薬組成物に関する。本発明のまた別の態様は、1つ以上の他の治療剤と組合された本発明の結晶性遊離塩基を含む製薬組成物に関する。したがって、1実施形態において、本発明は、(a)製薬的に許容され得る担体および治療的有効量の本発明の結晶性遊離塩基;ならびに(b)治療的有効量の、コルチコステロイドなどのステロイド性抗炎症剤;β2アドレナリン受容体アゴニスト;ホスホジエステラーゼ−4阻害剤;またはその組合せ;から選択される薬剤を含み、結晶性遊離塩基および薬剤が共にまたは個別に製剤化される、製薬組成物に関する。薬剤が別々に製剤化されるとき、製薬的に許容され得る担体が含まれ得る。通例、本発明の結晶性遊離塩基および薬剤は治療的有効量で存在するであろう。
【0012】
本発明の別の態様は、約4から6の範囲のpHを有する、本発明の結晶性遊離塩基を含む等張食塩水溶液を含む製薬組成物に関するものである。特定の実施形態において、水性ネブライザ製剤は、クエン酸緩衝剤によって約5のpHまで緩衝される。
【0013】
1実施形態において、本発明は、製薬的に許容され得る担体および本発明の結晶性遊離塩基を含む製薬組成物を含有するドライパウダー吸入器を備える、薬物送達デバイスに関する。
【0014】
式Iの化合物は、ムスカリン受容体アンタゴニスト活性を有する。したがって、式Iの化合物の結晶性遊離塩基は、同じ活性を有し、それゆえ喘息および慢性閉塞性肺疾患などの肺障害を処置するのに有用性を見出すことが期待される。それゆえ本発明の別の態様は、患者に治療的有効量の本発明の結晶性遊離塩基を投与することを含む、肺障害を処置する方法に関する。本発明のなお別の態様は、患者に気管支拡張を生じる量の本発明の結晶性遊離塩基を投与することを含む、患者において気管支拡張を生じる方法に関する。1実施形態において、化合物は吸入によって投与される。本発明は、患者に治療的有効量の本発明の結晶性遊離塩基を投与することを含む、慢性閉塞性肺疾患または喘息を処置する方法も提供する。本発明の別の態様は、哺乳動物に治療的有効量の本発明の結晶性遊離塩基を投与することを含む、哺乳動物においてムスカリン受容体を拮抗する方法に関する。
【0015】
本発明は、式Iの化合物の結晶性遊離塩基形を調製するプロセスにも関する。本発明は、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基を形成することを含む、式Iの化合物を精製するプロセスも提供する。本発明はさらに、本明細書に記載するプロセスによって調製された生成物に関する。
【0016】
本発明は、微粉化形の式Iの化合物の結晶性遊離塩基に;ならびに製薬的に許容され得る担体および本発明の微粉化結晶性遊離塩基を含む製薬組成物にも関する。
【0017】
本発明は、療法でまたは医薬品として使用するための、式Iの化合物の結晶性遊離塩基形にも関する。加えて本発明は、医薬品の製造のための;とりわけ哺乳動物において肺障害を処置するまたはムスカリン受容体を拮抗する医薬品の製造のための、本発明の結晶性遊離塩基の使用に関する。
【図面の簡単な説明】
【0018】
本発明の各種の態様は、添付図面への参照により例証される。
【図1】図1は、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステル(式Iの化合物)のIII形の結晶性遊離塩基の粉末X線回折(PXRD)パターンを示す。III形の他の特徴は、示差走査熱量測定(DSC)サーモグラムを示す図4および熱重量分析(TGA)トレースを示す図6に提示されている。
【図2】図2は、式Iの化合物のIV形の結晶性遊離塩基形のPXRDパターンを示す。
【図3】図3は、III形およびIV形のPXRDパターンのオーバーレイを示す。IV形の他の特徴は、DSCサーモグラムを示す図5、およびTGAトレースを示す図7に提示されている。
【図4】図4は、III形の示差走査熱量測定(DSC)サーモグラムを示す。
【図5】図5は、IV形の示差走査熱量測定(DSC)サーモグラムを示す。
【図6】図6は、III形の熱重量分析(TGA)トレースを示す。
【図7】図7は、IV形の熱重量分析(TGA)トレースを示す。
【発明を実施するための形態】
【0019】
本発明は、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基形(式I)を提供する。驚くべきことに、本発明の結晶性遊離塩基形は大気中の水分に曝露されたときでも、潮解性でないことが見出されている。加えて、本発明の結晶性遊離塩基形は、許容され得るレベルの吸湿性および許容され得る融点を有する。たとえば結晶性遊離塩基III形は約125℃の融点を有し、結晶性遊離塩基IV形は約119℃の融点を有する。
【0020】
他の使用の中でも、本発明の結晶性遊離塩基形は、肺障害を処置する有用性を有することが期待される製薬組成物を調製するのに有用である。したがって、本発明の1態様は、製薬的に許容され得る担体および治療的有効量の本発明の結晶性遊離塩基を含む製薬組成物に関する。
【0021】
定義
本発明の化合物、組成物、方法およびプロセスについて記載するときに、以下の用語は、別途指摘されない限り、以下の意味を有する。加えて、本明細書で使用する場合、単数形「a」、「an」および「the」は、使用状況が別途明らかに指示しない限り、対応する複数形を含む。「備える、含む(comprising)」、「含む(including)」、および「有する(having)」という用語は、包括的であることが意図され、挙げられた要素以外に追加の要素があり得ることを意味する。本明細書で使用する成分の量、分子量などの特性、反応条件などを表現するすべての数は、別途指摘されない限り、すべての例において「約」という用語によって修飾されていることが理解されるはずである。したがって、本明細書に記述される数は、本発明によって得ようとされる望ましい特性に応じて変動し得る概数である。少なくとも、および請求項の範囲に対する均等論の適用を制限する試みとしてではなく、各数は、報告された有効数字に照らしておよび普通の丸め技法を適用することによって少なくとも解釈されるべきである。
【0022】
III形およびIV形はどちらも無水遊離塩基結晶多形である。「本発明の結晶性遊離塩基」が参照されるとき、用語はIII形およびIV形を含むことが理解される。
【0023】
本明細書で使用する場合、「式を有する」または「構造を有する」という句は、制限的であることが意図されず、「含む、備える」という用語が一般に使用されるのと同じ様式で使用される。
【0024】
「製薬的に許容され得る」という用語は、本明細書で使用されるときに生物学的にまたは別途許容され得ないわけではない材料を指す。たとえば「製薬的に許容され得る担体」という用語は、許容され得ない生物効果を引き起こすことなく、または組成物の他の構成要素と許容され得ない相互作用することなく、組成物中に組み入れられて患者に投与されることができる材料を指す。このような製薬的に許容され得る材料は通例、毒性および製造試験の要求される標準を満足しており、米国食品医薬品局によって好適な不活性成分として同定されたこのような材料を含む。
【0025】
「治療的有効量」という用語は、それを必要とする患者に投与されるときに処置をもたらすのに十分な量、すなわち望ましい治療効果を得るために必要とされる薬物の量を意味する。たとえば肺障害を処置するための治療的有効量は、たとえば喘息もしくは慢性閉塞性肺疾患(「COPD」)の症候を減少、抑制、消滅もしくは防止するために、または喘息もしくはCOPDの根底にある原因を処置するために必要とされる化合物の量である。1実施形態において、治療的有効量は、気管支拡張を生じるのに必要とされる量である。これに対して、「有効量」という用語は、必ずしも治療結果ではあり得ないが、望ましい結果を得るために十分な量を意味する。たとえばムスカリン受容体を含む系を試験するときに、「有効量」は受容体を拮抗するのに必要とされる量であり得る。
【0026】
「処置すること」または「処置」という用語は、本明細書で使用する場合:(a)疾患もしくは病状が出現するのを防止すること、すなわち患者の予防処置;(b)患者における疾患もしくは病状を消滅すること、または疾患もしくは病状の後退を引き起こすことなどによって、疾患もしくは病状を改善することと;(c)患者における疾患または病状の発症を減速もしくは停止することなどによって、疾患もしくは病状を抑制することと;または(d)患者における疾患もしくは病状の症候を緩和することと;を含む、哺乳動物(特にヒト)などの患者における疾患もしくは病状(COPDなど)を処置することまたは疾患もしくは病状の処置を意味する。たとえば「COPDを処置すること」という用語は、COPDが出現するのを防止すること、COPDを改善すること、COPDを抑制すること、およびCOPDの症候を改善することを含むであろう。「患者」という用語は、処置もしくは疾患防止を必要とする、または特異的疾患もしくは病状の疾患防止もしくは処置のために現在処置されている、ヒトなどのこのような哺乳動物を含むことが意図される。「患者」という用語は、本発明の化合物が評価されている試験対象またはアッセイで使用されている試験対象、たとえば動物モデルも含む。
【0027】
合成
本発明の結晶性遊離塩基形は、下および実施例に記載されているような、ただちに利用可能な開始材料から合成されることができる。各結晶性遊離塩基形を生じるために使用できる複数の方法があるが、しかし結晶含有量ならびに結晶の晶癖(サイズおよび形状)が、一部は調製方法、ならびに溶媒組成物に基づいて変動し得ることに注目される。
【0028】
特異的プロセス条件(すなわち結晶化温度、時間、反応物質のモル比、溶媒、圧力など)が与えられるが、別途明示されない限り、他のプロセス条件も使用できることが認識されるであろう。いくつかの例において、反応または結晶化は室温にて実行され、実際の温度測定は行われなかった。室温は実験室環境における周囲温度に一般に関連する範囲内の温度を意味するとして解釈することができ、通例、約25℃から約50℃の範囲内にあるであろうことが理解される。他の例において、反応または結晶化は室温にて実行され、温度は実際に測定および記録された。すべての重量、体積および等価物は、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステル(または塩形)開始材料に関係する。
【0029】
概して、結晶化は好適な不活性希釈剤または溶媒系において実行され、この例は、これに限定されるわけではないが、メタノール、エタノール、イソプロパノール、イソブタノール、酢酸エチル、アセトニトリル、ジクロロメタン、メチルt−ブチルエーテルなど、およびその混合物を含む。上述のいずれの結晶化の完了時にも、沈殿、濃縮、遠心分離などのいずれの従来手段によっても、結晶性化合物を反応混合物から単離することができる。
【0030】
本発明で用いられるビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステル、ならびに2リン酸塩などのその塩は、市販の開始材料および試薬から、実施例に記載された手順を使用して、またはMammenらへの米国特許公開第2005/0203133号およびAxtらへの米国特許公開第2007/0112027号に記載された手順を使用して、ただちに調製することができる。
【0031】
本発明の方法に記載されたモル比は、当業者に利用可能な各種の方法によってただちに決定できる。たとえばこのようなモル比は、1H NMRによってただちに決定することができる。代わりに元素分析およびHPLC法を使用して、モル比を決定することができる。
【0032】
III形
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルのIII形の結晶性遊離塩基は、エステルまたはエステルの2リン酸塩から調製することができる。
【0033】
1実施形態において、III形結晶性遊離塩基は、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルをアセトニトリルと接触させることによって調製される。通例、エステルのミリグラムの、アセトニトリルの総ミリリットルに対する比は約100:1であり、アセトニトリルは2ステップで添加される。概して、本反応は、0〜40℃の温度範囲を反復してサイクルしながら実行される。固体は次に、真空濾過によって単離され、乾燥される。
【0034】
別の実施形態において、III形結晶性遊離塩基は、III形結晶性遊離塩基の種晶およびエステルの2リン酸塩を使用して調製される。本方法は:a)結晶性遊離塩基III形の種晶を形成すること;b)ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの2リン酸塩を酢酸イソプロピルおよび水に溶解させて溶液を形成すること;c)ならびに種晶を溶液に添加することを包含する。さらに明確には、エステルの2リン酸塩(1重量)は、窒素下20±3℃にて酢酸イソプロピル(17.5体積)および水(10体積)中でスラリー化される。懸濁剤は53±3℃まで加温され、10M NaOH(0.5体積)が添加される。混合物はその温度にて短時間撹拌され、次に層が分離されて、塩基性水層が除去される。水(5体積)が有機層に添加され、撹拌される。層が分離され、水層が除去される。酢酸イソプロピル(17.5体積)が添加され、約10体積の蒸留液が常圧蒸留によって収集される。本ステップは追加の酢酸イソプロピル(10体積)を用いて反復される。2回目の蒸留の後、透明溶液の温度を53±3℃まで低下されて、次に酢酸イソプロピル(0.08体積)による結晶性遊離塩基III形(0.005重量;0.5重量%)の懸濁剤を用いて種晶が添加される。得られた懸濁剤は、53±3℃にて少なくとも2時間撹拌され、次に0.19℃/分のおよその冷却速度にて10±3℃まで冷却される。懸濁剤は10±3℃にて少なくとも2時間撹拌され、次に濾過により収集される。得られた濾過ケーキは酢酸イソプロピル(2×3体積)によって洗浄され、生成物は次に乾燥されてIII形結晶性遊離塩基を産する。
【0035】
IV形
1実施形態において、IV形結晶性遊離塩基はIII形結晶性遊離塩基の種晶を使用して調製される。本方法は:a)結晶性遊離塩基III形の種晶を形成すること;b)結晶性遊離塩基III形をアセトニトリルに溶解させて溶液を形成すること;c)および種晶を溶液に添加することを包含する。通例、種のエステルに対する重量比は、約2:250の範囲にある。通例、結晶性遊離塩基III形のグラムの、アセトニトリルの総ミリリットルに対する比は約2:10から3:30の範囲内にあり、2.5:16は1つの範囲である。アセトニトリルは通常、複数の一定分量で添加される。概して、本反応は、0〜40℃の温度範囲を反復してサイクルしながら実行される。固体は次に、真空濾過によって単離され、乾燥される。
【0036】
結晶特性
粉末X線回折の分野で周知であるように、粉末X線回折(PXRD)スペクトルの相対ピーク高さは、試料調製および機器形状に関するいくつかの因子に依存しているが、ピーク位置は実験の詳細事項に比較的非感受性である。結晶性遊離塩基III形およびIV形のPXRDパターンは、実施例5で記述されるように得られた。それゆえ1実施形態において、本発明の結晶性化合物は、あるピーク位置を有するPXRDパターンを特徴とする。
【0037】
本発明の各結晶性遊離塩基形は異なるPXRDパターンを呈するが、パターンはある共通のピークを有する。それゆえ1実施形態において、本発明は、6.6±0.1、13.1±0.1、18.6±0.1、19.7±0.1、および20.2±0.1から選択される2θ値に回折ピークを備える粉末X線回折パターンを特徴とする、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基形に関する。
【0038】
1実施形態において、結晶性遊離塩基III形は、6.6±0.1、13.1±0.1、18.6±0.1、19.7±0.1、および20.2±0.1の2θ値に回折ピークを備える粉末X線回折パターンを特徴とし;ならびに8.8±0.1、10.1±0.1、11.4±0.1、11.6±0.1、14.8±0.1、15.2±0.1、16.1±0.1、16.4±0.1、16.9±0.1、17.5±0.1、18.2±0.1、19.3±0.1、19.9±0.1、20.8±0.1、21.1±0.1、21.7±0.1、および22.3±0.1から選択される2θ値に5つ以上の追加の回折ピークを有することをさらに特徴とする。別の実施形態において、結晶性遊離塩基III形は、6.6±0.1、11.4±0.1、13.1±0.1、16.1±0.1、17.5±0.1、18.2±0.1、18.6±0.1、19.3±0.1、19.7±0.1、19.9±0.1、20.2±0.1、20.8±0.1、21.1±0.1、21.7±0.1、および22.3±0.1から選択される2θ値に回折ピークを備える粉末X線回折を特徴とする。また別の実施形態において、結晶性遊離塩基III形は、ピーク位置が図1に示すピーク位置に実質的に従うPXRDパターンを特徴とする。
【0039】
1実施形態において、結晶性遊離塩基IV形は、6.6±0.1、13.1±0.1、18.6±0.1、19.7±0.1、および20.2±0.1の2θ値に回折ピークを備える粉末X線回折パターンを特徴とし;ならびに10.6±0.1、15.0±0.1、16.0±0.1、17.3±0.1、17.7±0.1、20.9±0.1、21.4±0.1、22.6±0.1、24.6±0.1、および27.8±0.1から選択される2θ値に5つ以上の追加の回折ピークを有することをさらに特徴とする。別の実施形態において、結晶性遊離塩基IV形は、6.6±0.1、13.1±0.1、15.0±0.1、17.3±0.1、17.7±0.1、18.6±0.1、19.7±0.1、20.2±0.1、20.9±0.1、21.4±0.1、および22.6±0.1から選択される2θ値に回折ピークを備える粉末X線回折パターンを特徴とする。また別の実施形態において、結晶性遊離塩基IV形は、ピーク位置が図2に示すピーク位置に実質的に従うPXRDパターンを特徴とする。
【0040】
また別の実施形態において、本発明の結晶性遊離塩基は、示差走査熱量測定(DSC)サーモグラムを特徴とする。DSCサーモグラムは、実施例6で記述されるように得られた。本明細書で報告する融点は、DSC分析の間に登録された溶融開始に基づいて推定される。それゆえ1実施形態において、本発明の結晶性化合物は、そのDSCサーモグラフを特徴とする。1実施形態において、結晶性遊離塩基III形は、図4に見られるように、約123℃の吸熱熱流の開始および約125℃の融点を示すDSCサーモグラフを特徴とする。別の実施形態において、結晶性遊離塩基IV形は、図5に見られるように、約66℃の吸熱熱量の1つの開始、約119℃の吸熱熱量の第2の開始、および約119℃の融点を示すDSCサーモグラフを特徴とする。
【0041】
熱重量分析(TGA)は、実施例6に記載されているように、本発明の結晶性遊離塩基形に対して行われた。それゆえ1実施形態において、結晶性遊離塩基はTGAトレースを特徴とする。1実施形態において、結晶性遊離塩基III形は、図6に描かれたTGAトレースを特徴とする。別の実施形態において、結晶性遊離塩基IV形は、図7に描かれたTGAトレースを特徴とする。
【0042】
蒸気収着重量測定(GVS)アセスメントは、実施例7に記載されているように、本発明の結晶性遊離塩基形に対して行われた。本発明の結晶性遊離塩基形は、許容され得るレベルの吸湿性を有する可逆性収着/脱着プロフィールを有することが証明されている。たとえば結晶性遊離塩基III形は、25℃にて0%と90%の間の相対湿度で<2重量/重量%の可逆性水取込みを示した。加えて、結晶性遊離塩基III形は、高温および湿度への曝露時に安定であることが見出されている。
【0043】
本発明の結晶性遊離塩基形のこれらの特性は、下の実施例でさらに例証される。
【0044】
有用性
式Iの化合物はムスカリン受容体アンタゴニスト活性を所有し、およびそのため式Iの化合物の結晶性遊離塩基形は、ムスカリン受容体によって媒介される病状、すなわちムスカリン受容体アンタゴニストによる処置によって改善される病状を処置するように有用であることが期待される。このような病状は、1例として、慢性閉塞性肺疾患(たとえば慢性および喘鳴を伴う気管支炎ならびに気腫)、喘息、肺線維症、アレルギー性鼻炎、鼻漏などの可逆性気道閉塞に関連するものを含む、肺障害または疾患を含む。ムスカリン受容体アンタゴニストによって処置することができる他の病状は、過活動膀胱または排尿筋機能亢進ならびにその症候などの尿生殖路障害;過敏性腸症候群、憩室性疾患、アカラシア、胃腸過剰運動性障害および下痢などの胃腸管障害;洞性徐脈などの心不整脈;パーキンソン病;アルツハイマー病などの認知障害;月経困難(dismenorrhea);などである。
【0045】
したがって、1実施形態において、本発明は、肺障害を処置する方法であって、患者に治療的有効量のビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基を投与することを含む方法に関する。肺障害を処置するために使用されるとき、本発明の結晶性遊離塩基は通例、1日に付き複数回用量で、1日1回用量でまたは週1回用量で吸入により投与されるであろう。概して、肺障害を処置するための用量は、約10μg/日から200μg/日の範囲に及ぶであろう。
【0046】
吸入によって投与されるとき、本発明の結晶性遊離塩基は通例、気管支拡張を生じる効果を有するであろう。したがって、別の実施形態において、本発明は、患者に気管支拡張を生じる方法であって、患者に気管支拡張を生じる量のビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基を投与することを含む方法に関する。概して、気管支拡張を生じるための治療的有効用量は、約10μg/日から200μg/日の範囲に及ぶであろう。
【0047】
1実施形態において、本発明は、慢性閉塞性肺疾患または喘息を処置する方法であって、患者に治療的有効量のビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基を投与することを含む方法に関する。
【0048】
COPDまたは喘息を処置するために使用されるとき、本発明の結晶性遊離塩基は通例、1日に付き複数回用量でまたは1日1回用量で吸入により投与されるであろう。概して、COPDまたは喘息を処置するための用量は、約10μg/日から200μg/日の範囲に及ぶであろう。本明細書で使用する場合、COPDは慢性閉塞性気管支炎および気腫を含む(たとえばBarnes,Chronic Obstructive Pulmonary Disease,New England Journal of Medicine 343:269−78(2000)を参照)。
【0049】
肺障害を処置するために使用されるとき、本発明の結晶性遊離塩基は、場合により他の治療剤と組合せて投与される。したがって、特定の実施形態において、本発明の製薬組成物および方法は、治療的有効量のβ2−アドレナリン受容体アゴニスト、コルチコステロイド、非ステロイド性抗炎症剤、またはその組合せをさらに含む。
【0050】
別の実施形態において、本発明の結晶性遊離塩基は、生物系、および特にマウス、ラット、モルモット、ウサギ、イヌ、ブタ、ヒトなどの哺乳動物においてムスカリン受容体を拮抗するのに使用される。本実施形態において、治療的有効量の本発明の結晶性遊離塩基が哺乳動物に投与される。所望ならば次にムスカリン受容体を拮抗する効果を、従来の手順および機器を使用して決定することができる。
【0051】
ムスカリン受容体拮抗活性などの、本発明の結晶性遊離塩基の特性および有用性は、当業者に周知である各種のインビトロおよびインビボアッセイを使用して証明することができる。たとえば、代表的なアッセイは、以下の実施例でさらに詳細に記載されており、限定するのではなく例証として、(たとえばアッセイ1に記載されているような)hM1、hM2、hM3、hM4、およびhM5ムスカリン受容体結合を測定するアッセイを含む。本発明の結晶性遊離塩基のムスカリン受容体拮抗活性を決定するための有用な機能アッセイは、限定するのではなく例証として、細胞内環状アデノシン1リン酸(cAMP)におけるリガンド媒介変化、(cAMPを合成する)酵素アデニリルシクラーゼの活性におけるリガンド媒介変化、[35S]GTPγSのGDPとの受容体触媒交換を介した、グアノシン5’−O−(γ−チオ)3リン酸([35S]GTPγS)の単離膜中への組み入れにおけるリガンド媒介変化、(たとえば蛍光連結イメージング・プレート・リーダーまたはMolecular Devices,Inc.からのFLIPR(登録商標)によって測定された)遊離細胞内カルシウムイオンにおけるリガンド媒介変化などを測定するアッセイを含む。例示的なアッセイは、アッセイ2に記載されている。結晶性遊離塩基は、上に挙げたアッセイのいずれか、または類似の性質のアッセイにおいてムスカリン受容体の活性化を拮抗または低下することが期待され、通例、これらの試験では約0.1から100ナノモルの範囲に及ぶ濃度にて使用されるであろう。それゆえ上述のアッセイは、本発明の結晶性遊離塩基の治療有用性、たとえば気管支拡張活性を決定するのに有用である。
【0052】
本発明の結晶性遊離塩基の他の特性および有用性は、当業者に周知である各種のインビトロおよびインビボアッセイを使用して証明することができる。たとえば、結晶性遊離塩基のインビボ効力は、アイントホーフェンモデルなどの動物モデルで測定することができる。簡潔には、結晶性遊離塩基の気管支拡張活性は、気道抵抗の代替尺度として通気圧を使用する麻酔された動物(アイントホーフェンモデル)において評価される。たとえばEinthoven(1892)Pfugers Arch.51:367−445;およびMohammedら(2000)Pulm Pharmacol Ther.13(6):287−92、ならびにラットのアイトホーフェンモデルについて記載しているアッセイ3を参照。別の有用なインビボアッセイは、(たとえばアッセイ4に記載されているような)ラット唾液分泌抑制アッセイである。
【0053】
製薬組成物および製剤
本発明の結晶性遊離塩基は通例、患者に製薬組成物または製剤の形で投与される。このような製薬組成物は、これに限定されるわけではないが、吸入、経口、鼻内、局所(経皮を含む)および非経口の投与方式を含む、いずれかの許容され得る投与経路によって投与され得る。しかし、本発明の結晶性遊離塩基がいったん製剤化されると、これはもはや結晶形ではない場合がある、すなわち結晶性遊離塩基は好適な担体に溶解され得ることが当業者によって理解されるであろう。
【0054】
したがって、1実施形態において、本発明は、製薬的に許容され得る担体または賦形剤およびビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基を含む製薬組成物に関する。製薬組成物は所望ならば、他の治療剤および/または製剤化剤(formulating agent)を含有し得る。
【0055】
本発明の製薬組成物は通例、治療的有効量のビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基を活性剤として含有する。通例、このような製薬組成物は約0.01から約30重量%;たとえば約0.01から約10重量%の活性剤を含む、約0.01から約95重量%の活性剤を含有するであろう。
【0056】
いずれの従来の担体または賦形剤も本発明の製薬組成物で使用され得る。特定の担体もしくは賦形剤、または担体もしくは賦形剤の組合せの選出は、特定の患者または病状もしくは疾患状態の種類を処置するために使用されている投与方式に依存するであろう。この点で、特定の投与方式のための好適な製薬組成物の調製は、十分に製薬分野の当業者の範囲内である。加えて、このような組成物の成分は、たとえばSigma,P.O.Box 14508,St.Louis,MO 63178から市販されている。さらなる例証として、従来の製剤化技法はRemington:The Science and Practice of Pharmacy,20th Edition,Lippincott Williams & White,Baltimore,Maryland(2000);およびH.C.Anselら,Pharmaceutical Dosage Forms and Drug Delivery Systems,7th Edition,Lippincott Williams & White,Baltimore,Maryland(1999)に記載されている。
【0057】
製薬的に許容され得る担体として役目を果たすことができる材料の代表的な例は、これに限定されるわけではないが、以下の:ラクトース、グルコースおよびスクロースなどの糖;コーンスターチおよびジャガイモデンプンなどのデンプン;セルロースならびにナトリウムカルボキシメチルセルロース、エチルセルロースおよび酢酸セルロースなどのその誘導体;粉末トラガカント;麦芽;ゼラチン;タルク;ココアバターおよび坐剤ワックスなどの賦形剤;ピーナッツ油、綿実油、ベニバナ油、ゴマ油、オリーブ油、トウモロコシ油およびダイズ油などの油;プロピレングリコールなどのグリコール;グリセリン、ソルビトール、マンニトールおよびポリエチレングリコールなどのポリオール;オレイン酸エチルおよびラウリン酸エチルなどのエステル;寒天;水酸化マグネシウムおよび水酸化アルミニウムなどの緩衝剤;アルギン酸;発熱物質を含まない水;等張性生理食塩水;リンゲル液;エチルアルコール;リン酸緩衝生理食塩水;クロロフルオロカーボンおよびヒドロフルオロカーボンなどの圧縮噴射ガス;ならびに製薬組成物で用いられる他の非毒性適合性物質を含む。
【0058】
本発明の製薬組成物は通例、結晶性遊離塩基を製薬的に許容され得る担体および1つ以上の任意の成分と完全および密接に混合またはブレンドすることによって調製される。必要または所望ならば、得られた均一にブレンドされた混合物を次に、従来の手順および機器を使用して、錠剤、カプセル剤、丸剤に成形、またはキャニスター、カートリッジ、ディスペンサーなどに装入することができる。
【0059】
1実施形態において、本発明の製薬組成物は吸入投与に好適である。吸入投与の好適な製薬組成物は通例、エアゾール剤または粉剤の形であるだろう。このような組成物は概して、ネブライザ吸入器、定量吸入器(MDI)、ドライパウダー吸入器(DPI)などの周知の送達デバイスまたは類似の送達デバイスを使用して投与される。
【0060】
本発明の詳細な実施形態において、活性剤を含む製薬組成物は、ネブライザ吸入器を使用する吸入によって投与される。このようなネブライザデバイスは通例、活性剤を含む製薬組成物をミストとして噴霧して、患者の気道中に運ばれるようにする、高速気流を生じる。したがってネブライザ吸入器で使用するために製剤化されるとき、結晶性遊離塩基活性剤は通例、好適な担体に溶解されて溶液を形成する。好適なネブライザデバイスは、Respimat(登録商標)Soft Mist(商標)吸入器(Boehringer Ingelheim)、AERx(登録商標)肺送達システム(Aradigm Corp.)、およびPARI LC Plusリユーザブルネブライザ(Pari GmbH)を含む。
【0061】
ネブライザ吸入器での使用のための代表的な製薬組成物は、約0.05μg/mLから約10mg/mLの本発明の結晶性遊離塩基を含む等張性水溶液を含む。1実施形態において、水性ネブライザ製剤は等張性である。1実施形態において、このような溶液は約4〜6のpHを有する。特定の実施形態において、水性ネブライザ製剤は、クエン酸緩衝剤によって約5のpHまで緩衝される。別の特定の実施形態において、水性製剤は、約0.1mg/mLから約1.0mg/mLの、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの遊離塩基等価物を含有する。
【0062】
本発明の別の詳細な実施形態において、活性剤を含む製薬組成物は、DPIを使用する吸入によって投与される。このようなDPIは通例、活性剤を吸息の間に患者の気流に分散された自由流動パウダーとして投与する。自由流動パウダーを達成するために、結晶性遊離塩基活性剤は通例、ラクトース、デンプン、マンニトール、デキストロース、ポリ乳酸、ポリ乳酸−co−グリコリド、およびその組合せなどの好適な賦形剤を用いて製剤化される。微粉化は、結晶サイズを肺送達に好適なサイズまで減少させる一般的な方法である。通例、結晶性遊離塩基活性剤は微粉化され、好適な担体と組合されて呼吸可能なサイズの微粉化粒子の懸濁剤を形成し、「微粉化粒子」または「微粉化形」は、少なくとも約90%の粒子が約10μm未満の直径を有することを意味する。微細製粉、切刻み、圧壊、破砕、製粉、ふるい分け、粉砕、微粉砕などの粒径を減少させる他の方法も、所望の粒径を得ることができる限り使用され得る。
【0063】
DPIでの使用のための代表的な製薬組成物は、約1μmから約100μmの間の粒径を有する無水ラクトースおよび本発明の結晶性遊離塩基の微粉化粒子を含む。このようなドライパウダー製剤はたとえば、ラクトースを結晶性遊離塩基活性剤と組合せること、および次に構成要素を乾燥ブレンドすることによって作製することができる。代わりに、所望ならば、賦形剤を用いずに結晶性遊離塩基活性剤を製剤化することができる。製薬組成物は次に通例、ドライ・パウダー・ディスペンサー中に、またはドライパウダー送達デバイスでの使用のために吸入カートリッジもしくはカプセル剤の中に装入される。
【0064】
DPI送達デバイスの例は、Diskhaler(GlaxoSmithKline,Research Triangle Park,NC;たとえばNewellらへの米国特許第5,035,237号を参照);Diskus(GlaxoSmithKline;たとえばDaviesらへの米国特許第6,378,519号を参照);Turbuhaler(AstraZeneca,Wilmington,DE;たとえばWetterlinへの米国特許第4,524,769号を参照);Rotahaler(GlaxoSmithKline;たとえばHallworthらへの米国特許第4,353,365号を参照)およびHandihaler(Boehringer Ingelheim)を含む。好適なDPIデバイスのさらなる例は、Casperらへの米国特許第5,415,162号、Evansへの同第5,239,993号、およびArmstrongらへの同第5,715,810号、およびそこで引用された参考文献に記載されている。上述の特許の開示は、その全体が参照により本明細書に組み入れられている。
【0065】
本発明のまた別の詳細な実施形態において、結晶性遊離塩基活性剤を含む製薬組成物は、通例、測定された量の活性剤を、圧縮噴射ガスを使用して放出するMDIを使用する吸入によって投与される。したがって、MDIを使用して投与される製薬組成物は通例、液化噴射剤による結晶性遊離塩基活性剤の溶液または懸濁剤を含む。CCl3Fなどのクロロフルオロカーボン、ならびに1,1,1,2−テトラフルオロエタン(HFA 134a)および1,1,1,2,3,3,3−ヘプタフルオロ−n−プロパン(HFA 227)などのヒドロフルオロアルカン(HFA)を含む、いずれの好適な液化噴射剤も用いられ得る。オゾン層に影響を及ぼすクロロフルオロカーボンについての懸念のために、HFAを含有する製剤が概して好ましい。HFA製剤の追加の任意の構成要素は、エタノールまたはペンタンなどの共溶媒、ならびにトリオレイン酸ソルビタン、オレイン酸、レシチン、およびグリセリンなどの界面活性剤を含む。たとえばPurewalらへの米国特許第5,225,183号、EP 0717987 A2(Minnesota Mining and Manufacturing Company)、およびWO 92/22286(Minnesota Mining and Manufacturing Companyを参照、それらの開示はその全体が参照により本明細書に組み入れられている。
【0066】
定量吸入器での使用のための代表的な製薬組成物は、約0.01〜5重量%の本発明の遊離塩基結晶性化合物;約0〜20重量%のエタノール;および約0〜5重量%の界面活性剤を含む;残りはHFA噴射剤である。
【0067】
このような組成物は通例、冷却または加圧されたヒドロフルオロアルカンを結晶性遊離塩基活性剤、エタノール(存在する場合)および界面活性剤(存在する場合)を含有する好適な容器に添加することによって調製される。懸濁剤を調製するために、結晶性遊離塩基活性剤は微粉化され、次に噴射剤と組合される。製剤は次に、定量吸入器デバイスの部分を形成するエアゾールキャニスター中に装入される。HFA噴射剤を用いた使用のために明確に開発された定量吸入デバイスの例は、Mareckiへの米国特許第6,006,745号および同第Ashurstらへの6,143,277号に記載されている。代わりに懸濁製剤は、活性剤の微粉化粒子に界面活性剤のコーティングを噴霧乾燥することによって調製できる。たとえばWO 99/53901(Glaxo Group Ltd.)およびWO 00/61108(Glaxo Group Ltd.)を参照。上述の特許および刊行物の開示は、その全体が参照により本明細書に組み入れられている。
【0068】
吸入投薬に好適な呼吸可能な粒子、ならびに製剤およびデバイスを調製するプロセスの追加の例については、Gaoへの米国特許第6,268,533号、Trofastへの同第5,983,956号;Briggnerらへの同第5,874,063号;およびJakupovicらへの同第6,221,398号;ならびにWO 99/55319(Glaxo Group Ltd.)およびWO 00/30614(AstraZeneca AB)を参照;その開示は、その全体が参照により本明細書に組み入れられている。
【0069】
別の実施形態において、本発明の製薬組成物は経口投与に好適である。経口投与の好適な製薬組成物は、カプセル剤、錠剤、丸剤、ロゼンジ剤、カシェ剤、糖衣錠、粉剤、顆粒剤の形で;水性もしくは非水性液体による液剤もしくは懸濁剤として;または水中油型もしくは油中水型液体乳剤として;またはエリキシル剤もしくはシロップ剤としてなどであり得て;それぞれ所定量の本発明の結晶性遊離塩基を活性成分として含有する。製薬組成物は単位投薬形で包装され得る。
【0070】
固体投薬形(すなわちカプセル剤、錠剤、丸剤などとして)の経口投与が意図されるとき、本発明の製薬組成物は通例、活性成分としての本発明の結晶性遊離塩基ならびにクエン酸ナトリウムおよびリン酸2カルシウムなどの1つ以上の製薬的に許容され得る担体を含む。場合によりまたは代わりに、このような固体投薬形は:デンプン、ラクトース、スクロース、グルコース、マンニトール、および/またはケイ酸などの充填剤または増量剤;カルボキシメチルセルロース、アルギン酸塩、ゼラチン、ポリビニルピロリドン、スクロースおよび/またはアラビアゴムなどの結合剤;グリセロールなどの保湿剤;寒天、炭酸カルシウム、ジャガイモもしくはタピオカデンプン、アルギン酸、あるケイ酸塩、および/または炭酸ナトリウムなどの崩壊剤;パラフィンなどの溶解遅延剤;4級アンモニウム化合物などの吸収促進剤;セチルアルコールおよび/またはモノステアリン酸グリセロールなどの湿潤剤;カオリンおよび/またはベントナイト粘土などの吸収剤;タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、および/またはその混合物などの潤滑剤;着色剤;ならびに緩衝剤も含み得る。
【0071】
離型剤、湿潤剤、コーティング剤、甘味剤、香味剤および香料、保存料ならびに酸化防止剤も、本発明の製薬組成物中に存在することができる。製薬的に許容され得る酸化防止剤の例は:アスコルビン酸、塩酸システイン、重硫酸ナトリウム、メタ重硫酸ナトリウムなどの水溶性酸化防止剤;パルミチン酸アスコルビル、ブチル化ヒドロキシアニソール(BHA)、ブチル化ヒドロキシトルエン(BHT)、レシチン、没食子酸プロピル、アルファ−トコフェロールなどの油溶性酸化防止剤;およびクエン酸、エチレンジアミンテトラ酢酸(EDTA)、ソルビトール、酒石酸、リン酸などの金属キレート剤などを含む。錠剤、カプセル剤、丸剤などのコーティング剤は、酢酸フタル酸セルロース(CAP)、酢酸フタル酸ポリビニル(PVAP)、フタル酸ヒドロキシプロピルメチルセルロース、メタクリル酸−メタクリル酸エステルコポリマー、酢酸トリメリト酸セルロース(CAT)、カルボキシメチルエチルセルロース(CMEC)、酢酸コハク酸ヒドロキシプロピルメチルセルロース(HPMCAS)などの腸溶コーティングに使用されるコーティング剤を含む。
【0072】
所望ならば本発明の製薬組成物は、1例として、変動比のヒドロキシプロピルメチルセルロース;または他のポリマーマトリクス、リポソームおよび/もしくはミクロスフェアを使用して、活性成分の低速または制御放出を提供するようにも製剤化され得る。
【0073】
加えて、本発明の製薬組成物は、場合により乳白剤を含有し得て、製薬組成物が胃腸管のある部分において、場合により遅延方式で活性成分のみを、または活性成分を優先的に放出するように製剤化され得る。使用できる包埋組成物(embedding compositions)の例は、ポリマー物質およびワックスを含む。結晶性遊離塩基活性成分は、適切な場合、1つ以上の上記の賦形剤を用いたマイクロカプセル化形であることもできる。
【0074】
経口投与に好適な液体投薬形は、例証として、製薬的に許容され得る乳剤、マイクロエマルション、液剤、懸濁剤、シロップ剤およびエリキシル剤を含む。このような液体投薬形は通例、活性成分ならびにたとえば水または他の溶媒などの不活性希釈剤、エチルアルコール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3−ブチレングリコール、油(とりわけ綿実油、ラッカセイ油、トウモロコシ油、胚芽油、オリーブ油、ヒマシ油およびゴマ油)、グリセロール、テトラヒドロフリルアルコール、ポリエチレングリコールおよびソルビタンの脂肪酸エステル、ならびにその混合物などの可溶化剤および乳化剤を含む。懸濁剤は、活性成分に加えて、たとえばエトキシ化イソステアリルアルコール、ポリオキシエチレンソルビトールおよびソルビタンエステル、微結晶性セルロース、メタ水酸化アルミニウム、ベントナイト、寒天およびトラガカント、ならびにその混合物などの懸濁化剤を含有し得る。
【0075】
本発明の結晶性遊離塩基は、公知の経皮送達系および賦形剤を使用して経皮投与することもできる。たとえば結晶性遊離塩基は、プロピレングリコール、モノラウリン酸ポリエチレングリコール、アザシクロアルカン−2−オンなどの浸透促進剤と混和させて、パッチまたは類似の送達系に組み入れることができる。ゲル化剤、乳化剤および緩衝剤を含む追加の賦形剤は、所望ならばこのような経皮組成物で使用され得る。
【0076】
本発明の結晶性遊離塩基は、他の治療剤と共に同時投与することもできる。本併用療法は、共に製剤化された(たとえば単一の製剤に共に包装された)または別々に製剤化された(たとえば別々の単位投薬形として包装された)どちらかの、これらの第2の薬剤の1つ以上と組合された結晶性遊離塩基を使用することを包含する。同一製剤でまたは別々の単位投薬形で複数の薬剤を共に製剤化する方法は、当分野で周知である。「単位投薬形」という用語は、患者に投薬するのに好適な物理的に別個の単位を指し、すなわち各単位は、単独でまたは1つ以上の追加の単位と組合されてのどちらかで、所望の治療効果を生じるように計算された所定量の本発明の化合物を含有する。たとえばこのような単位投薬形は、カプセル剤、錠剤、丸剤などであり得る。
【0077】
追加の治療剤は、他の気管支拡張薬(たとえばPDE3阻害剤、アデノシン2b調節因子およびβ2アドレナリン受容体アゴニスト);抗炎症剤(たとえばコルチコステロイドなどのステロイド性抗炎症剤;非ステロイド性抗炎症剤(NSAID)、およびPDE4阻害剤);他のムスカリン受容体アンタゴニスト(すなわち抗コリン作用薬(antichlolinergic agents);抗感染剤(たとえばグラム陽性およびグラム陰性抗生剤または抗ウイルス薬);抗ヒスタミン薬;プロテアーゼ阻害剤;および求心性遮断薬(たとえばD2アゴニストおよびニューロキニン調節因子)から選択することができる。
【0078】
本発明の特定の1実施形態は、(a)製薬的に許容され得る担体および治療的有効量の本発明の結晶性遊離塩基;ならびに(b)製薬的に許容され得る担体および治療的有効量の、コルチコステロイドなどのステロイド性抗炎症剤;β2アドレナリン受容体アゴニスト;ホスホジエステラーゼ−4阻害剤;またはその組合せ;から選択される薬剤を含み、結晶性遊離塩基および薬剤が共にまたは個別に製剤化される、製薬組成物に関する。別の実施形態において、(b)は、製薬的に許容され得る担体ならびに治療的有効量のβ2アドレナリン受容体アゴニストおよびステロイド性抗炎症剤である。第2の薬剤は、製薬的に許容され得る塩または溶媒和物の形で、および適切な場合、光学的に純粋な立体異性体として使用することができる。
【0079】
本発明の結晶性遊離塩基と組合せて使用することができる代表的なβ2アドレナリン受容体アゴニストは、これに限定されるわけではないが、サルメテロール、サルブタモール、フォルモテロール、サルメファモール、フェノテロール、テルブタリン、アルブテロール、イソエタリン、メタプロテレノール、ビトルテロール、ピルブテロール、レバルブテロールなど、またはその製薬的に許容され得る塩を含む。使用することができる他のβ2アドレナリン受容体アゴニストは、これに限定されるわけではないが、WO 02/066422(Glaxo Group Ltd.)に記載された3−(4−{[6−({(2R)−2−ヒドロキシ−2−[4−ヒドロキシ−3−(ヒドロキシメチル)−フェニル]エチル}アミノ)−ヘキシル]オキシ}ブチル)ベンゼンスルホンアミドおよび3−(−3−{[7−({(2R)−2−ヒドロキシ−2−[4−ヒドロキシ−3−(ヒドロキシメチル)フェニル]エチル}−アミノ)ヘプチル]オキシ}−プロピル)ベンゼンスルホンアミドおよび関連化合物;WO 02/070490(Glaxo Group Ltd.)に記載された3−[3−(4−{[6−([(2R)−2−ヒドロキシ−2−[4−ヒドロキシ−3−(ヒドロキシメチル)フェニル]エチル}アミノ)ヘキシル]オキシ}ブチル)−フェニル]イミダゾリジン−2,4−ジオンおよび関連化合物;WO 02/076933(Glaxo Group Ltd.)に記載された3−(4−{[6−({(2R)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)−ベンゼンスルホンアミド、3−(4−{[6−({(2S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)−ベンゼンスルホンアミド、3−(4−{[6−({(2R/S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)−ベンゼンスルホンアミド、N−(t−ブチル)−3−(4−{[6−({(2R)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]−オキシ}ブチル)ベンゼンスルホンアミド、N−(tert−ブチル)−3−(4−{[6−({(2S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)−ヘキシル]オキシ}ブチル)−ベンゼンスルホンアミド、N−(tert−ブチル)−3−(4−{[6−({(2R/S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]−オキシ}ブチル)ベンゼンスルホンアミドおよび関連化合物;WO 03/024439(Glaxo Group Ltd.)に記載された4−{(1R)−2−[(6−{2−[(2,6−ジクロロベンジル)オキシ]エトキシ}ヘキシル)アミノ]−1−ヒドロキシエチル}−2−(ヒドロキシメチル)フェノールおよび関連化合物;Moranらに対する米国特許第6,576,793号に記載されたN−{2−[4−((R)−2−ヒドロキシ−2−フェニルエチルアミノ)フェニル]エチル}−(R)−2−ヒドロキシ−2−(3−ホルムアミド−4−ヒドロキシフェニル)エチルアミンおよび関連化合物;Moranらに対する米国特許第6,653,323号に記載されたN−{2−[4−(3−フェニル−4−メトキシフェニル)アミノフェニル]エチル}−(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2(1H)−キノリノン−5−イル)エチルアミンおよび関連化合物;ならびにその製薬的に許容され得る塩を含む。特定の実施形態において、β2−アドレナリン受容体アゴニストは、N−{2−[4−((R)−2−ヒドロキシ−2−フェニルエチルアミノ)フェニル]エチル}−(R)−2−ヒドロキシ−2−(3−ホルムアミド−4−ヒドロキシフェニル)エチルアミンの結晶性1塩酸塩である。β2−アドレナリン受容体アゴニストは用いられるときに、製薬組成物中に治療的有効量で存在するであろう。通例、β2アドレナリン受容体アゴニストは、用量当り約0.05μg〜500μgを提供するのに十分な量で存在するであろう。上述の特許および刊行物の開示は、その全体が参照により本明細書に組み入れられている。
【0080】
本発明の結晶性遊離塩基と組合せて使用することができる代表的なステロイド性抗炎症剤は、これに限定されるわけではないが、メチルプレドニゾロン、プレドニゾロン、デキサメタゾン、プロピオン酸フルチカゾン、6α,9α−ジフルオロ−17α−[(2−フラニルカルボニル)オキシ]−11β−ヒドロキシ−16α−メチル−3−オキソアンドロスタ−1,4−ジエン−17β−カルボチオ酸S−フルオロメチルエステル、6α,9α−ジフルオロ−11β−ヒドロキシ−16α−メチル−3−オキソ−17α−プロピオニルオキシ−アンドロスタ−1,4−ジエン−17β−カルボチオ酸S−(2−オキソ−テトラヒドロフラン−3S−イル)エステル、ベクロメタゾンエステル(たとえば17−プロピオン酸エステルまたは17,21−ジプロピオン酸エステル)、ブデソニド、フルニソリド、モメタゾンエステル(たとえばフロ酸エステル)、トリアムシノロンアセトニド、ロフレポニド、シクレソニド、プロピオン酸ブチキソコルト、RPR−106541,ST−126など、またはその製薬的に許容され得る塩を含む。ステロイド性抗炎症剤は用いられるときに、組成物中に治療的有効量で存在するであろう。通例、ステロイド性抗炎症剤は、用量当り約0.05μg〜500μgを提供するのに十分な量で存在するであろう。
【0081】
例示的な組合せは、β2アドレナリン受容体アゴニストとしてのサルメテロール、およびステロイド性抗炎症剤としてのプロピオン酸フルチカゾンと同時投与される本発明の結晶性遊離塩基である。別の例示的な組合せは、β2アドレナリン受容体アゴニストとしてのN−{2−[4−((R)−2−ヒドロキシ−2−フェニルエチルアミノ)フェニル]エチル}−(R)−2−ヒドロキシ−2−(3−ホルムアミド−4−ヒドロキシフェニル)エチルアミンの結晶性1塩酸塩、およびステロイド性抗炎症剤としての6α,9α−ジフルオロ−17α−[(2−フラニルカルボニル)オキシ]−11β−ヒドロキシ−16α−メチル−3−オキソアンドロスタ−1,4−ジエン−17β−カルボチオ酸S−フルオロメチルエステルと同時投与される本発明の結晶性遊離塩基である。上記のように、これらの薬剤は共にまたは別々に製剤化することができる。
【0082】
他の好適な組合せはたとえば、他の抗炎症剤、たとえばNSAID(たとえばクロモグリク酸ナトリウム、ネドクロミルナトリウム、ならびにテオフィリン、PDE4阻害剤および混合PDE3/PDE4阻害剤などのホスホジエステラーゼ(PDE)阻害剤);ロイコトリエンアンタゴニスト(たとえばモンテルカスト(monteleukast));ロイコトリエン合成の阻害剤;iNOS阻害剤;トリプターゼおよびエラスターゼ阻害剤などのプロテアーゼ阻害剤;ベータ−2インテグリンアンタゴニストおよびアデノシン受容体アゴニストもしくはアンタゴニスト(たとえばアデノシン2aアゴニスト);サイトカインアンタゴニスト(たとえばインターロイキン抗体(αIL抗体)、明確にはαIL−4療法、αIL−13療法、またはその組合せなどのケモカインアンタゴニスト);またはサイトカイン合成の阻害剤を含む。
【0083】
本発明の結晶性遊離塩基と組合せて使用することができる代表的なホスホジエステラーゼ−4(PDE4)阻害剤または混合PDE3/PDE4阻害剤は、これに限定されるわけではないが、シス4−シアノ−4−(3−シクロペンチルオキシ−4−メトキシフェニル)シクロヘキサン−1−カルボン酸、2−カルボメトキシ−4−シアノ−4−(3−シクロプロピルメトキシ−4−ジフルオロメトキシフェニル)シクロヘキサン−1−オン;シス−[4−シアノ−4−(3−シクロプロピルメトキシ−4−ジフルオロメトキシフェニル)シクロヘキサン−1−オール];シス−4−シアノ−4−[3−(シクロペンチルオキシ)−4−メトキシフェニル]シクロヘキサン−1−カルボン酸など、またはその製薬的に許容され得る塩を含む。他の代表的なPDE4または混合PDE4/PDE3阻害剤は、AWD−12−281(elbion);NCS−613(INSERM);D−4418(Chiroscience and Schering−Plough);CI−1018またはPD−168787(Pfizer);WO99/16766(Kyowa Hakko)に記載されているベンゾジオキソール化合物;K−34(Kyowa Hakko);V−11294A(Napp);ロフルミラスト(Byk−Gulden);WO99/47505(Byk−Gulden)に記載されているフタラジノン化合物;プマフェントリン(Byk−Gulden、現在Altana);アロフィリン(Almirall−Prodesfarma);VM554/UM565(Vernalis);T−440(Tanabe Seiyaku);およびT2585(Tanabe Seiyaku)を含む。
【0084】
本発明の結晶性遊離塩基と組合せて使用することができる代表的なムスカリンアンタゴニスト(すなわち抗コリン剤)は、これに限定されるわけではないが、アトロピン、硫酸アトロピン、酸化アトロピン、硝酸メチルアトロピン、臭化水素酸ホマトロピン、臭化水素酸ヒヨスチアミン(d、l)、臭化水素酸スコポラミン、臭化イプラトロピウム、臭化オキシトロピウム、臭化チオトロピウム、メタンテリン、臭化プロパンテリン、臭化メチルアニソトロピン、臭化クリジニウム、コピロレート(ロビヌル)、ヨウ化イソプロパミド、臭化メペンゾラート、塩化トリジヘキセチル(パシロン)、メチル硫酸ヘキソシクリウム、塩酸シクロペントレート、トロピカミド、塩酸トリヘキシフェニジル、ピレンゼピン、テレンゼピン、AF−DX116およびメトクトラミンなど、もしくはその製薬的に許容され得る塩;または塩として挙げられた化合物では、その代わりの製薬的に許容され得る塩を含む。
【0085】
本発明の結晶性遊離塩基と組合せて使用することができる代表的な抗ヒスタミン薬(すなわちH1受容体アンタゴニスト)は、これに限定されるわけではないが、マレイン酸カルビノキサミン、フマル酸クレマスチン、塩酸ジフェニルヒドラミンおよびジメンヒドリナートなどのエタノールアミン;マレイン酸ピリラミン(pyrilamine amleate)、塩酸トリペレナミンおよびクエン酸トリペレナミンなどのエチレンジアミン;クロルフェニラミンおよびアクリバスチンなどのアルキルアミン;塩酸ヒドロキシジン、パモ酸ヒドロキシジン、塩酸シクリジン、乳酸シクリジン、塩酸メクリジンおよび塩酸セチリジンなどのピペラジン;アステミゾール、塩酸レボカバスチン、ロラタジンまたはそのデスカルボエトキシ類似体、テルフェナジンおよび塩酸フェキソフェナジンなどのピペリジン;塩酸アゼラスチン;など、もしくはその製薬的に許容され得る塩;または塩として挙げられた化合物では、その代わりの製薬的に許容され得る塩を含む。
【0086】
別途指摘されない限り、本発明の結晶性遊離塩基と組合せて投与される他の治療剤の例示的な好適な用量は、約0.05μg/日〜100mg/日の範囲にある。
【0087】
以下の製剤は、本発明の代表的な製薬組成物、ならびに例示的な調製方法を例証する。1つ以上の第2の薬剤は場合により、本発明の結晶性遊離塩基(第1の活性剤)と共に製剤化することができる。代わって、第2の薬剤は別々に製剤化されて、第1の活性剤と同時にまたは連続して、共に投与することができる。たとえば、1実施形態において、単一のドライパウダー製剤は、本発明の結晶性遊離塩基および1つ以上の第2の薬剤をどちらも含むように製造することができる。別の実施形態において、1つの製剤が本発明の結晶性遊離塩基を含有するように製造され、別々の製剤が第2の薬剤を含有するように製造される。このようなドライパウダー製剤は次に、別々のブリスターパックに包装されて、単一のDPIデバイスによって投与することができる。
【0088】
吸入投与のための例示的なドライパウダー製剤
0.2mgの本発明の結晶性遊離塩基は微粉化され、次に25mgのラクトースとブレンドされる。ブレンドされた混合物は次に、ゼラチン吸入カートリッジに装入される。カートリッジの内容物は、粉剤吸入器を使用して投与される。
【0089】
ドライパウダー吸入器による投与のための例示的なドライパウダー製剤
本発明の微粉化結晶性遊離塩基(活性剤)のラクトースに対する、1:200のバルク製剤比を有するドライパウダーが調製される。パウダーは、用量当り約10μg〜100μgの活性剤を送達することができるドライパウダー吸入デバイスに充填される。
【0090】
定量吸入器による投与のための例示的な製剤
5重量%の本発明の結晶性遊離塩基(活性剤)および0.1重量%のレシチンを含有する懸濁剤は、10gの結晶性遊離塩基を10μm未満の平均径を有する微粉化粒子として、200mLの脱塩水に溶解された0.2gのレシチンから形成された溶液に分散させることによって調製される。懸濁剤は噴霧乾燥され、得られた材料は1.5μm未満の平均直径を有する粒子に微粉化される。粒子は、加圧された1,1,1,2−テトラフルオロエタンと共にカートリッジに装入される。
【0091】
代わって、5重量%の本発明の結晶性遊離塩基、0.5重量%のレシチン、および0.5重量%のトレハロースを含有する懸濁剤は、5gの結晶性遊離塩基を10μm未満の平均径を有する微粉化粒子として、100mLの脱塩水に溶解された0.5gのトレハロースおよび0.5gのレシチンから形成されたコロイド状溶液に分散させることによって調製される。懸濁剤は噴霧乾燥され、得られた材料は1.5μm未満の平均直径を有する粒子に微粉化される。粒子は、加圧された1,1,1,2−テトラフルオロエタンと共にキャニスターに装入される。
【0092】
ネブライザによる投与のための例示的な水性エアゾール製剤
製薬組成物は、0.5mgの本発明の結晶性遊離塩基(活性剤)を、クエン酸によって酸性化された1mLの0.9%塩化ナトリウム溶液に溶解させることによって調製される。混合物は、活性剤が溶解するまで撹拌および超音波処理される。溶液のpHは、NaOHの低速での添加によって、3から8の範囲の値(通例、約5)に調整される。
【0093】
経口投与のための例示的な硬ゼラチンカプセル製剤
以下の成分は完全にブレンドされ、次に硬ゼラチンカプセル中に装入される:カプセル当り合計460mgの組成物のために、250mgの本発明の結晶性遊離塩基、200mgのラクトース(噴霧乾燥)、および10mgのステアリン酸マグネシウム。
【0094】
経口投与のための例示的な懸濁製剤
以下の成分は混合されて、10mLの懸濁剤当り100mgの活性成分を含有する懸濁剤を形成する。
【0095】
【表1】
【0096】
例示的な注射用製剤
以下の成分がブレンドされ、pHは0.5N HClまたは0.5N NaOHを使用して4±0.5に調整される。
【0097】
【表2】
【実施例】
【0098】
以下の調製および実施例が提供されて、本発明の詳細な実施形態を例証する。しかし、これらの詳細な実施形態は、明確に指摘されない限り、本発明の範囲を決して制限することを意図されない。以下の省略形は、別途指摘されない限り以下の意味を有し、本明細書で使用され、定義されていないその他の省略形は、その標準の意味を有する:
AC アデニリルシクラーゼ
BSA ウシ血清アルブミン
cAMP 3’−5’環状アデノシン1リン酸
CHO チャイニーズハムスター卵巣
cM5 クローン化チンパンジーM5受容体
DCM ジクロロメタン
dPBS ダルベッコリン酸緩衝生理食塩水
EDTA エチレンジアミンテトラ酢酸
EtOAc 酢酸エチル
FBS ウシ胎仔血清
FLIPR 蛍光測定イメージング・プレート・リーダー
HBSS ハンクス緩衝塩溶液
HEPES 4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸
hM1 クローン化ヒトM1受容体
hM2 クローン化ヒトM2受容体
hM3 クローン化ヒトM3受容体
hM4 クローン化ヒトM4受容体
hM5 クローン化ヒトM5受容体
HOBT N−ヒドロキシベンゾトリアゾール
HPLC 高速液体クロマトグラフィー
MCh メチルコリン
MeCN アセトニトリル
本明細書で使用されるが、定義されていないその他の省略形は、その標準の、概して許容される意味を有する。別途記されない限り、試薬、開始材料および溶媒は、市販品供給者(Sigma−Aldrich、Flukaなど)から購入されて、さらに精製せずに使用された。
【0099】
調製1
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステル
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの2リン酸塩(16g)を、水(100mL)およびEtOAc(200mL)の2相性混合物に溶解させた。NaOH(2N、75mL)が5分間の期間にわたって添加された。次に混合物は30分間撹拌された。相は分離され、水相はEtOAc(200mL)によって抽出された。合せた有機相は濃縮された。DCM(100mL)が添加され、混合物は蒸発乾固された。固体は乾燥器内で約48時間乾燥されて、表題化合物(9.6g)が得られた。
【0100】
(実施例1)
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基(III形)
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステル(102.4mg)をMeCN(500μL)に溶解させた。溶液は室温にて80分間撹拌され、白色固体沈殿が形成された。混合物は振とう器ブロックに配置され、48時間にわたって熱サイクル処理された(1時間ブロックで0〜40℃)。白色で濃密な静止した固体が観察された。MeCN(500μL)が添加され、スラリーは移動性となった。次に混合物は再度、振とう器ブロックに2時間配置された。固体は、焼結漏斗を使用して真空濾過により単離され、次に完全真空下で40℃のピストン乾燥器に15.5時間配置されて、76.85mgの表題結晶性化合物を産した。
【0101】
(実施例2)
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基(III形)
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの2リン酸塩(C35H43N5O4・2H3PO4;MW 793.75;632.9g)は、窒素下の室温にて酢酸イソプロピル(11.08L)および水(6.33L)中でスラリー化された。懸濁剤は53±3℃まで加温され、混合物の温度を50℃超に維持しながら、10M NaOH(317mL)が攪拌混合物に添加された。混合物が53±3℃にて約5分間撹拌されてから、層が沈降した。次に層が分離されて、水層が除去された。混合物の温度が50℃超に維持されながら、水(3.16L)が有機層に添加された。混合物が53±3℃にて5分間撹拌されると、層が静置された。層が分離され、水層が除去された。酢酸イソプロピル(6.33L)が添加され、次に約10体積の蒸留液が常圧蒸留によって収集された。本ステップは追加の酢酸イソプロピル(3.2L)を用いて反復された。2回目の蒸留の後、透明溶液の温度が53±3℃まで低下されて、次にビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステル結晶性遊離塩基(III形;3.2g)の酢酸イソプロピル(51mL)による懸濁剤によって種晶が添加された。得られた懸濁剤は53±3℃にて2時間撹拌され、次に4時間にわたって10±3℃まで冷却された。懸濁剤は10±3℃にて少なくとも2時間撹拌され、次に固体が濾過によって収集された。得られた濾過ケーキは酢酸イソプロピル(2×1.9L)で洗浄され、生成物は真空中で50℃にて乾燥されて、表題結晶性化合物を産した(C35H43N5O4;MW 597.76;382.5g、収率80.3%)。
【0102】
(実施例3)
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基(III形)の再結晶化
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基(III形;C35H43N5O4;MW 597.76;372.5g)は、窒素下20±3℃にてトルエン(5.6L)中でスラリー化された。懸濁剤は、82±3℃まで加温され、完全な溶解が観察されるまで本温度で維持された。溶液は次に、晶析装置容器で清澄にされて、トルエン(373μL)によるすすぎが続けられた。晶析装置容器内で固体が観察され、容器は82±3℃まで再加熱されて溶解がもたらされ、次に58±3℃まで冷却されて、トルエン(8μL)中で事前に超音波処理された(約1分間)結晶性遊離塩基(III形;1.9g)によって種晶が添加された。得られた懸濁剤は、58±3℃にて少なくとも4時間静置され、次に2時間にわたって20±3℃まで冷却された(およそ0.33℃/分の冷却速度)。懸濁剤は20±3℃にて少なくとも1時間撹拌され、次に固体が濾過によって収集された。得られた濾過ケーキはトルエン(2×1.2L)によって洗浄され、生成物は真空中で52±3℃にて乾燥されて、表題結晶性化合物を産した(345.3g、収率92.7%)。
【0103】
(実施例4)
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基(IV形)
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステル(調製1に記載されたように調製;2.5g)をMeCN(10mL)に溶解させて、粘性油性薄黄色材料を産した。追加のMeCN(5mL)が添加されて、材料を希釈した。溶液に結晶性遊離塩基(20mg;実施例1に記載されたように調製されたIII形)によって種晶が添加され、室温にて90分間撹拌された。大量の白色沈殿(小型結晶)が観察された。スラリーは偏光顕微鏡下で分析され、複屈折であることが見出された。
【0104】
追加のMeCN(3mL)が添加され、スラリーがMetz Syn10ブロックに配置されて、800rpmにて一晩、熱サイクル処理された(1時間ブロックで0〜40℃)。Metz Syn10は、静止した10位置の並列反応ステーションである。溶液/スラリーのかき混ぜは、クロス磁気撹拌棒によった。振とう器ブロックは、外部ユラボ浴によって加熱および冷却される別個の機器であった。材料は0℃にて除去された。スラリーが沈降して、白色沈殿の上に薄黄色溶液が残っているのが観察された。スラリーは撹拌され、再度、振とう器ブロックに配置されて熱サイクル処理された。材料は40℃にて除去され、室温にて高いかき混ぜ速度にて80分間撹拌された。スラリーは再度分析され、複屈折であることが見出された。濾過ケーキは、焼結漏斗を使用して真空濾過によって単離された。MeCN(3mL)が使用されて濾紙が濡らされて、濾過の前に濾過ケーキがMeCNによって洗浄された。ケーキは真空下にて40分間脱液されて、2.3gの流動性白色パウダーを産した。材料は40℃にて65時間、ピストン乾燥器に配置され、2.2gの表題結晶性化合物を白色パウダー(99.6%純度)として産した。
【0105】
生成物のラマンスペクトルの大部分は、III形開始材料のラマンスペクトルと一致していた。しかし、3個のシフトが注目された。
【0106】
III形 生成物
878 cm−1 881 cm−1
775 cm−1 772 cm−1
485 cm−1 488 cm−1
生成物は次に、粉末X線回折、示差走査熱量測定、および熱重量分析によって分析された。生成物がIII形開始材料とは異なる遊離塩基結晶形であることが決定され、IV形と呼ばれた。
【0107】
(実施例5)
粉末X線回折
結晶性遊離塩基III形(実施例1より)およびIV形(実施例4より)の粉末X線回折(PXRD)パターンは、XCelerator検出器を装備したPANalytical X’Pert Pro粉末回折計(diffractomer)で取得された。取得条件は、放射線:Cu Kα;発電機電圧:40kV;発電機電流:45mA;開始角度2.0°2θ;終了角度40.0°2θ、ステップサイズ:0.0167°2θであった。ステップ当りの時間は、31.750秒であった。試料は、2、3ミリグラムの試料をシリコンウェハ(ゼロ背景)プレート上に載せて、粉末薄膜を生じることによって調製された。
【0108】
特徴的なピーク位置および計算されたd間隔が下にまとめられており、これらのピークが14%を超える相対強度を有することのみが報告される。これらはHighscoreソフトウェアを使用して生データから計算された。ピーク位置の実験誤差は、約±0.1°2θである。相対ピーク強度は、好ましい配向のために変動するであろう。
【0109】
【表3】
【0110】
結晶性遊離塩基III形の代表的なPXRDパターンは図1に示されている。結晶性遊離塩基IV形の代表的なPXRDパターンは図2に示されている。
【0111】
(実施例6)
熱分析
結晶性遊離塩基III形(実施例1より)およびIV形(実施例4より)の示差走査熱量測定(DSC)サーモグラムは、TA Instruments熱量計を使用して得られた。試料はアルミニウム皿に秤量され、皿の蓋が上に配置されて、皿を密封することなく軽く圧着された。実験は、10℃/分の加熱速度を使用して実行された。
【0112】
結晶性遊離塩基III形の代表的なDSCサーモグラフは図4に示されている。DSCサーモグラフは、III形が123.1℃における吸熱熱流の開始(エンタルピー67.7J/g)を示すDSCサーモグラフを特徴とすることを証明している。
【0113】
結晶性遊離塩基IV形の代表的なDSCサーモグラフは図5に示されている。DSCサーモグラフは、IV形が小規模な吸熱および主吸熱、すなわち65.6℃にて出現する小規模な第1の吸熱熱流の開始(エンタルピー0.8J/g)および118.8℃にて出現する主な第2の吸熱熱流の開始(エンタルピー66.8J/g)を示すDSCサーモグラフを特徴とすることを証明する。
【0114】
熱重量分析(TGA)データは、TA Instruments Q500計器を使用して得られた。試料は、開放されたアルミニウム皿内で10℃/分の加熱速度にて200℃まで加熱された。
【0115】
結晶性遊離塩基III形の代表的なTGAトレースは図6に示されており、試料分解前にごくわずかな重量損失が観察されたことを指摘している。結晶性遊離塩基IV形の代表的なTGAトレースは図7に示されており、試料溶融前に約0.3%の重量損失が観察され、これが残留溶媒の損失と一致することを指摘している。
【0116】
(実施例7)
蒸気収着重量測定アセスメント
蒸気収着重量測定(GVS)試験は、25℃にて水蒸気散布を使用した全収着等温線の発生のためにSurface Measurements System DVS−1計器を使用して行われた。約7mgの試料サイズは、風袋を差し引いた透明で乾燥した試料メッシュ皿に配置され、内蔵天秤で秤量された。目標相対湿度(RH)は10%刻みで、30%〜90%、次に90%〜0%および0%〜30%であった。平衡点は、0.02dm/dt漸近線設定を使用して自動的に決定された。
【0117】
25℃にて行われた結晶性遊離塩基III形の試料に対するGVS試験は、材料が0%RH〜90%RHの範囲にわたって水分を吸収する傾向が低いことを証明した。試料は、25℃にて0〜90%のRHの間で<2%重量/重量の可逆的吸水を示した。本GVSトレースは、III形が広範な湿度範囲に曝露されたときに、許容され得る重量増加を有することを証明している。
【0118】
(実施例8)
微粉化
結晶性遊離塩基III形の試料は、APTM 4”微粉装置およびレーザー光回折によって(buy)決定された粒径のどちらかを使用して微粉化された。
【0119】
【表4】
【0120】
参考のために、投入した結晶性遊離塩基III形の粒径はX10=5.58μm X50=18.2μm、およびX90=49.7μmであった。微粉化は、呼吸可能なサイズ範囲の粒子を産した。微粉化は結晶性の低下を生じたが、微粉化前材料の本質的な特徴を維持した。40℃/20%相対湿度、40℃/75%相対湿度(キャップなし)、および50℃/周囲湿度での3カ月にわたる保管後に、PXRDでは変化が観察されなかった。
【0121】
結晶性遊離塩基III形のDSCサーモグラフは、微粉化の前後に、125℃にて鋭い溶融を示した。おそらく結晶化のために、87℃にて微粉化材料に追加の小規模な熱的イベントがあった。40℃/20%RH、40℃/75%RH(露出)、および50℃/周囲湿度での3カ月間の保管の後に、微粉化材料は125℃にて鋭い溶融を示し、アモルファス内容物の証拠はなかった。
【0122】
(実施例9)
ラクトース適合性
結晶性遊離塩基III形の2つの製剤が、40℃/20%相対湿度(RH)、40℃/75%RH(キャップなし)、および50℃/周囲湿度での3カ月にわたる安定性について評価された。0.08重量/重量%(10μg DPI用量)および2重量/重量%(250μg DPI用量)製剤は、ラクトースのみとの、またはラクトースおよび1重量/重量%のステアリン酸マグネシウムとのブレンドとして調製された。すべての製剤の安定性は、許容され得ることが見出された。
【0123】
(実施例10)
pH溶解度および安定性
結晶性遊離塩基III形は、pH7までの培地で良好な溶解度(およそ2mg/mLを超える)を示した。水溶解度は、8.9の天然pHで0.66mg/mLであった。人工肺液への溶解度は0.46mg/mLであり、4時間から24時間の間の溶解度測定では変化は見られなかった。
【0124】
結晶性遊離塩基III形溶液は、50℃において、または露光下でpH4からpH6の緩衝液中にて7日間まで安定である。溶液は、水および生理食塩水中にて室温で遮光されて7日間安定である。
【0125】
アッセイ1
放射性リガンド結合アッセイ
hM1、hM2、hM3およびhM4ムスカリン受容体サブタイプを発現する細胞からの膜調製
クローン化ヒトhM1、hM2、hM3およびhM4ムスカリン受容体サブタイプをそれぞれ安定発現するCHO株化細胞は、10%FBSおよび250μg/mLジェネティシンを補ったHAMのF−12から成る培地でコンフルエント近くまで培養された。細胞は5%CO2、37℃のインキュベータで培養され、dPBS中2mM EDTAによって浮上された。細胞は、650xgにおける5分間の遠心分離によって収集され、細胞ペレットは−80℃にて凍結保管されるか、または膜がただちに調製されるかのどちらかであった。膜調製では、細胞ペレットは溶解緩衝液に再懸濁され、Polytron PT−2100組織破砕装置(Kinematica AG;20秒×2バースト)によってホモジナイズされた。粗膜は、40,000xgで4℃にて15分間遠心分離された。膜ペレットは次に再懸濁緩衝液によって再懸濁され、Polytron組織破砕装置によって再度ホモジナイズされた。膜懸濁剤のタンパク質濃度は、Lowry,O.ら、Journal of Biochemistry 193:265(1951)に記載された方法によって決定された。すべての膜は、一定分量で−80℃にて凍結保管されるか、またはただちに使用された。一定分量の調製hM5受容体膜は、Perkin Elmerから直接購入されて、使用するまで−80℃にて保管された。
【0126】
ムスカリン受容体サブタイプhM1、hM2、hM3、hM4およびhM5に対する放射性リガンド結合アッセイ
放射性リガンド結合アッセイは、96ウェル・マイクロタイター・プレートにおいて、1000μLのアッセイ全量で行われた。hM1、hM2、hM3、hM4またはhM5ムスカリンサブタイプのいずれかを安定発現するCHO細胞膜は、アッセイ緩衝液で以下の特異的目標タンパク質濃度(μg/ウェル):hM1では10μg、hM2では10〜15μg、hM3では10〜20μg、hM4では10〜20μg、およびhM5では10〜12μgに希釈された。膜は、アッセイプレート添加の前に、Polytron組織破砕装置を使用して短時間(10秒)ホモジナイズされた。放射性リガンドのKD値を決定するための飽和結合試験は、L−[N−メチル−3H]スコポラミンメチルクロリド([3H]−NMS)(TRK666、84.0Ci/mmol,Amersham Pharmacia Biotech,Buckinghamshire,England)を0.001nM〜20nMの範囲に及ぶ濃度で使用して行われた。試験化合物のKi値を決定するための置換アッセイは、[3H]−NMSを1nMおよび11の異なる試験化合物濃度にて用いて行われた。試験化合物は、最初に希釈緩衝液に40μMの濃度で溶解され、次に希釈緩衝液によって、400fM〜4μMの範囲に及ぶ最終濃度に5倍ずつ連続希釈された。アッセイプレートへの添加順序および体積は、以下の通りであった:0.1%BSAを有する825μLアッセイ緩衝液、25μL放射性リガンド、100μL希釈試験化合物、および50μL膜。アッセイプレートを37℃にて6時間インキュベートした。結合反応は、0.3%ポリエチレンイミン(PEI)中で予備処理されたGF/Bガラス繊維フィルタプレート(Perkin Elmer Inc.,Wellesley,MA)での高速濾過によって終結された。フィルタプレートは洗浄緩衝液(10mM HEPES)で3回すすがれて、未結合放射能が除去された。プレートが次に空気乾燥されて、50μL Microscint−20液体シンチレーション液(PerkinElmer Inc.,Wellesley,MA)が各ウェルに添加された。プレートは次に、PerkinElmer Topcount液体シンチレーションカウンタ(PerkinElmer Inc.,Wellesley,MA)でカウントされた。結合データは、GraphPad Prismソフトウェアパッケージ(GraphPad Software,Inc.,San Diego,CA)による非線形回帰分析によって、1部位競合モデルを使用して分析された。試験化合物のKi値は、観察されたIC50値から、放射性リガンドのKD値はCheng−Prusoffの等式(Cheng Y;Prusoff W.H.Biochemical Pharmacology 22(23):3099−108(1973))を使用して計算された。Ki値はpKi値に変換されて、幾何平均および95%信頼区間が決定された。これらの要約統計量は次に、データ報告のために再度Ki値に変換された。
【0127】
本アッセイにおいて、より低いKi値は、試験化合物が試験された受容体に対してより高い結合親和性を有することを指摘している。式Iの化合物は、本アッセイまたは類似のアッセイで試験されるときに、M3ムスカリン受容体サブタイプに対して約5nM未満のKi値を有することが見出された。
【0128】
アッセイ2
ムスカリン受容体機能的効力アッセイ
cAMP蓄積のアゴニスト媒介阻害の遮断
本アッセイにおいて、試験化合物の機能的効力は、試験化合物がhM2受容体を発現するCHO−K1細胞におけるフォルスコリン媒介cAMP蓄積のオキソトレモリン阻害を遮断する能力を測定することによって決定される。
【0129】
cAMPアッセイは、ラジオイムノアッセイ形式で、125I−cAMPを有するFlashplateアデニリルシクラーゼ活性化アッセイシステム(NEN SMP004B,PerkinElmer Life Sciences Inc.,Boston,MA)を製造者の説明書に従って使用して行われる。
【0130】
細胞は、dPBSによって1回洗浄され、アッセイ1に記載されているようにトリプシン−EDTA溶液(0.05%トリプシン/0.53mM EDTA)によって浮上させる。分離された細胞は、50mL dPBS中の650×gでの5分間の遠心分離によって2回洗浄される。細胞ペレットは次に10mL dPBSに再懸濁され、細胞はCoulter Z1 Dual Particleカウンター(Beckman Coulter,Fullerton,CA)によってカウントされる。細胞は650×gにて5分間にわたって再度遠心分離され、刺激緩衝液に1.6×106〜2.8×106細胞/mLのアッセイ濃度まで再懸濁される。
【0131】
試験化合物は、最初に400μMの濃度まで希釈緩衝液(1mg/mL BSA(0.1%)を補ったdPBS)で希釈され、次に希釈緩衝液によって100μM〜0.1nMの範囲に及ぶ最終モル濃度まで連続希釈される。オキソトレモリンは同様の方式で希釈される。
【0132】
AC活性のオキソトレモリン阻害を測定するために、25μLのフォルスコリン(dPBSで希釈された25μM最終濃度)、25μLの希釈オキソトレモリン、および50μLの細胞がアゴニスト・アッセイ・ウェルに添加される。試験化合物がオキソトレモリン阻害AC活性を遮断する能力を測定するために、25μLのフォルスコリンおよびオキソトレモリン(それぞれ25μMおよび5μMの最終濃度、dPBSで希釈)、25μLの希釈試験化合物、および50μLの細胞が残りのアッセイウェルに添加される。
【0133】
反応物は37℃にて10分間インキュベートされ、100μLの氷冷検出緩衝液の添加により停止される。プレートは密封され、室温にて一晩インキュベートされて、翌朝、PerkinElmer TopCount液体シンチレーションカウンタ(PerkinElmer Inc.,Wellesley,MA)でカウントされる。産生されたcAMPの量(pmol/ウェル)は、製造者のユーザマニュアルに記載されているように、試料およびcAMP標準で観察されたカウントに基づいて計算される。データは、非線形回帰分析によって、1部位競合等式を使用してGraphPad Prism Software package(GraphPad Software,Inc.,San Diego,CA)を用いて分析される。Cheng−Prusoffの等式を使用して、オキソトレモリン濃度応答曲線のEC50およびオキソトレモリンアッセイ濃度をそれぞれKDおよび[L]として用いてKiを計算する。KiはpKi値に変換されて、幾何平均および95%信頼区間が決定される。これらの要約統計量は次に、データ報告のために再度Ki値に変換される。
【0134】
本アッセイにおいて、より低いKi値は、試験化合物が試験された受容体に対してより高い機能的活性を有することを指摘している。式Iの化合物は、本アッセイまたは類似のアッセイで試験されたときに、hM2受容体を発現するCHO−K1細胞におけるフォルスコリン媒介cAMP蓄積のオキソトレモリン阻害の遮断のために、約5nM未満のKi値を有することが見出された。
【0135】
アゴニスト媒介[35S]GTPγS結合の遮断
第2の機能的アッセイにおいて、試験化合物の機能的効力は、化合物がhM2受容体を発現するCHO−K1細胞におけるオキソトレモリン刺激[35S]GTPγS結合を遮断する能力を測定することによって決定することができる。
【0136】
使用時に凍結膜は解凍され、次にアッセイ緩衝液中でウェル当り5〜10μgのタンパク質の最終目標組織濃度で希釈される。膜は、Polytron PT−2100組織破砕装置を使用して短時間ホモジナイズされ、次にアッセイプレートに添加される。
【0137】
アゴニストオキソトレモリンによる[35S]GTPγS結合の刺激のためのEC90値(90%最大応答に有効な濃度)は、各実験で決定される。
【0138】
試験化合物を阻害するオキソトレモリン刺激[35S]GTPγS結合の能力を決定するために、以下が96ウェルプレートの各ウェルに添加される:25μLの、[35S]GTPγS(0.4nM)を有するアッセイ緩衝液、25μLのオキソトレモリン(EC90)およびGDP(3μM)、25μLの希釈試験化合物ならびに25μLの、hM2受容体を発現するCHO細胞膜。次にアッセイプレートを37℃にて60分間インキュベートする。アッセイプレートは、PerkinElmer 96ウェル収集装置を使用して、1%BSA−前処理GF/Bフィルタで濾過される。プレートは氷冷洗浄緩衝液で3×3秒すすがれ、次に空気または真空乾燥される。Microscint−20シンチレーション液(50μL)が各ウェルに添加され、各プレートは密封されて、トップカウンター(PerkinElmer)で放射能カウントされる。データは、非線形回帰分析によって、1部位競合等式を使用してGraphPad Prism Software package(GraphPad Software,Inc.,San Diego,CA)を用いて分析される。Cheng−Prusoffの等式を使用して、試験化合物の濃度応答曲線のIC50値およびアッセイにおけるオキソトレモリン濃度をそれぞれKDおよび[L]リガンド濃度として用いて、Kiを計算する。
【0139】
本アッセイにおいて、より低いKi値は、試験化合物が試験された受容体に対してより高い機能的活性を有することを指摘している。式Iの化合物は、本アッセイまたは類似のアッセイで試験されたときに、hM2受容体を発現するCHO−K1細胞におけるオキソトレモリン刺激[35S]GTPγS−結合の遮断のために、約5nM未満のKi値を有することが見出された。
【0140】
FLIPRアッセイを介したアゴニスト媒介カルシウム放出の遮断
Gqタンパク質に結合するムスカリン受容体サブタイプ(M1、M3およびM5受容体)は、受容体へのアゴニスト結合時にホスホリパーゼC(PLC)経路を活性化する。結果として、活性化されたPLCはホスファチルイノシトール2リン酸(PIP2)をジアシルグリセロール(DAG)およびホスファチジル−1,4,5−3リン酸(IP3)に加水分解して、これが次に細胞内貯蔵、すなわち小胞体および筋小胞体からカルシウム放出を発生する。FLIPR(Molecular Devices,Sunnyvale,CA)アッセイは、遊離カルシウムを結合するときに蛍光を発するカルシウム感受性染料(Fluo−4AM,Molecular Probes,Eugene,OR)を使用することにより、細胞内カルシウムのこのような増加を利用する。本蛍光イベントは、ヒトM1およびM3、ならびにチンパンジーM5受容体でクローン化された細胞の単層からの蛍光の変化を検出するFLIPRによってリアルタイムで測定される。アンタゴニスト効力は、アンタゴニストが細胞内カルシウムのアゴニスト媒介上昇を阻害する能力によって決定することができる。
【0141】
FLIPRカルシウム刺激アッセイでは、hM1、hM3およびcM5受容体を安定発現するCHO細胞が96ウェルFLIPRプレートに、アッセイが行われる前の晩に播種される。播種された細胞は、Cellwash(MTX Labsystems,Inc.)によりFLIPR緩衝液(カルシウムおよびマグネシウムを含まないHBSS中の10mM HEPES、pH7.4、2mM塩化カルシウム、2.5mMプロベネシド)を用いて2回洗浄されて、成長培地が除去され、50μL/ウェルのFLIPR緩衝液が残る。細胞は次に、50μL/ウェルの4μM FLUO−4AM(2×溶液が作製された)によって37℃、5%二酸化炭素で40分間インキュベートされる。染料インキュベーション期間の後、細胞はFLIPR緩衝液で2回洗浄され、50μL/ウェルの最終体積が残る。
【0142】
アンタゴニスト効力を決定するために、オキソトレモリンの細胞内Ca2+放出の第1の用量依存性刺激が最初に決定されるので、EC90濃度でのオキソトレモリン刺激に対するアンタゴニスト効力を後に測定することができる。細胞は最初に化合物希釈緩衝液によって20分間インキュベートされ、FLIPRによって行われるアゴニスト添加が続く。オキソトレモリンのEC90値は、式ECF=((F/100−F)^1/H)*EC50と併せて、下のFLIPR測定およびデータ整理の項で詳説される方法に従って生成される。オキソトレモリンのEC90濃度がアンタゴニスト阻害アッセイプレートの各ウェルに添加されるように、3×ECFのオキソトレモリン濃度が刺激プレート中で調製される。
【0143】
FLIPRに使用されるパラメータは:0.4秒の照射期間、0.5ワットのレーザー強度、488nmの励起波長、および550nmの発光波長である。ベースラインは、アゴニスト添加前の10秒間にわたって蛍光変化を測定することによって決定される。アゴニスト刺激の後、FLIPRは蛍光変化を0.5〜1秒ごとに1.5分間にわたって連続測定して、最大蛍光変化を捕捉する。
【0144】
蛍光変化は、各ウェルについて最大蛍光からベースライン蛍光を引いたものとして表現される。生データは、S字状用量応答の組込みモデルを使用するGraphPad Prism(GraphPad Software,Inc.,San Diego,CA)を用いた非線形回帰により、薬物濃度の対数に対して分析される。アンタゴニストKi値は、KDとしてのオキソトレモリンEC50値およびCheng−Prusoffの等式(Cheng&Prusoff,1973)によるリガンド濃度のオキソトレモリンEC90を使用してPrismによって決定される。
【0145】
本アッセイにおいて、より低いKi値は、試験化合物が試験された受容体に対してより高い機能的活性を有することを指摘している。式Iの化合物は、本アッセイまたは類似のアッセイで試験されたときに、hM3受容体を安定に発現するCHO細胞におけるアゴニスト媒介カルシウム放出の遮断のために、約5nM未満のKi値を有することが見出された。
【0146】
アッセイ3
ラット・アイントホーフェン・アッセイ
本インビボアッセイが使用されて、ムスカリン受容体アンタゴニスト活性を呈する試験化合物の気管支保護効果が影響評価される。すべての試験化合物は滅菌水で希釈されて、吸入経路(IH)を介して投薬される。ラット(スプラーグ−ドーリー、オス、250〜350g)、LC Starネブライザセットから発生され、気体の混合物(5%CO2/95%周囲空気)によって操縦されたエアゾールに曝露される。各試験化合物溶液は、マウス6匹を保持することができるパイ形状投薬チャンバにおいて10分間の期間にわたって噴霧される。化合物の吸入後、所定の時点でアイントホーフェンアッセイが行われる。
【0147】
肺評価開始の30分前に、動物をイナクチン(チオブタバルビタール、120mg/kgIP)を介して麻酔する。頸静脈に生理食塩水を充填したポリエチレンカテーテル(PE−50)が挿入され、MChを注入するために使用される。次に気管が切開され、14G針によってカニューレ処置されて、肺評価の間のラットの人工呼吸に使用される。手術がいったん完了したら、2.5mlの量を超えない1ml/100g体重の心拍出量、および1分当り90拍動の速度に設定されたピストン呼吸装置を使用して、ラットに人工呼吸させる。
【0148】
各呼吸によって発生する圧力の変化が測定される。ベースライン値が少なくとも2.5分間にわたって収集され、次にラットは2倍ずつ漸進的に増加される気管支収縮剤MCh(5、10、20、40および80μg/ml)を用いて、非累積的に攻撃される。MChは注射器ポンプから2mL/kg/分の速度で2.5分間注入される。動物は試験完了時に安楽死させる。
【0149】
処置動物の人工呼吸圧の変化(cm H2O)は、対照動物に関係するMCh応答の阻害%として表される。本アッセイにおいて、より高い阻害%は、試験化合物が気管支保護効果を有することを指摘している。式Iの化合物は、本アッセイにおいて100μg/mlの用量で試験されるとき、35%を超える阻害、おそらく70%を超える阻害、およびなおさらにおそらく90%を超える阻害を呈することが期待される。
【0150】
1.5時間ID50の決定
標準ムスカリンアンタゴニストは、ラット・アイントホーフェン・アッセイで投薬1.5時間後に評価された。試験された5つの標準の効力(ID50)の順序は:イプラトロピウム(4.4μg/ml)>チオトロピウム(6μg/ml)>デス−メチル−チオトロピウム(12μg/ml)>グリコピロレート(15μg/ml)>LAS−34237(24μg/ml)として決定された。試験化合物の効力は、投薬1.5時間後に同様に決定される。
【0151】
6および24時間ID50の決定
標準のチオトロピウムおよびイプラトロピウムも、ラット・アイントホーフェン・アッセイで投薬24時間および/または6時間後に評価された。イプラトロピウム(10および30μg/ml)は投薬6時間後には、その1.5時間の効力と比較して3倍弱かった。本時点(6時間)で観察された活性の消失は、診療所での比較的短い作用期間と一致している。チオトロピウムは、ピーク気管支保護による緩慢な効果の開始が投薬6時間後に達成されることを示した。その6時間および24時間の効力値は、相互から有意に異なっておらず、その1.5時間の効力と比較して約2倍強力であった。試験化合物の作用の開始、ならびに6および24時間の効力値は同様に決定される。
【0152】
アッセイ4
ラット唾液分泌抑制アッセイ
ラット(スプラーグ−ドーリー、オス、250〜350g)は、アッセイ3で記載されたように、投薬、麻酔およびカニューレ処置される。所定の時点および手術後に、動物は下りスロープに仰向けに頭を20度傾斜させて配置される。事前に秤量されたガーゼパッドが動物の口内に挿入され、ムスカリンアゴニストのピロカルピン(PILO)(3mg/kg、iv.)が投与される。PILO後の10分間に産生された唾液は、PILO前後のガーゼパッドの重量を決定することによって、重量測定法で測定される。唾液分泌抑制効果は、対照動物に関係する唾液分泌の阻害%として表される。
1、6および24時間ID50の決定
ラット唾液分泌抑制アッセイは、試験化合物の全身曝露を影響評価して、肺選択指数(LSI)を計算するために開発された。標準のチオトロピウムは、本モデルにおいて投薬1時間、6時間、および24時間後に評価された。チオトロピウムは、投薬6時間後にピロカルピン誘発唾液分泌を最も強力に阻害することが見出された。本発見は、アイントホーフェンアッセイで観察されたピーク効果と一致している。
【0153】
本モデルは、Rechter,“Estimation of anticholinergic drug effects in mice by antagonism against pilocarpine−induced salivation”Ata Pharmacol Toxicol 24:243−254(1996)に記載された手順の修正版である。ビヒクルで処置された動物の唾液の平均重量は、各処置前時間に計算され、各用量の対応する処置前時間における唾液分布の阻害%が算出される。
【0154】
本アッセイで試験される本発明の例示的な化合物は、100μg/ml未満のID50値(24時間にて測定)を呈することが期待され、いくつかの化合物は30μg/ml未満の、いくつかは20μg/ml未満の、およびいくつかのは15μg/ml未満のID50値を呈することが期待される。
【0155】
唾液分泌抑制ID50の気管支保護ID50に対する比を使用して、試験化合物の見かけの肺選択指数が算出される。概して、約5を超える見かけの肺選択指数を有する化合物が好ましい。
【0156】
本発明はその詳細な態様または実施形態への参照により記載されてきたが、本発明の真の精神および範囲から逸脱することなく、各種の変更が行われることができるか、または均等物が置換されることができることが当業者によって理解されるであろう。加えて、本明細書で引用されたすべての刊行物、特許および特許出願は、適用可能な特許法および規則によって許容される範囲で、各文書が参照により本明細書に個別に組み入れられているのと同じ範囲まで、その全体が参照により本明細書に組み入れられている。
【技術分野】
【0001】
本発明は、肺障害を処置するために有用であることが期待される、ビフェニル化合物の新規結晶形に関するものである。本発明は、前記結晶性化合物を含むまたはこのような化合物から調製された製薬組成物、このような結晶性化合物を調製するためのプロセスおよび中間体ならびに肺障害を処置するためにこのような化合物を使用する方法にも関する。
【背景技術】
【0002】
Mammenらへの特許文献1は、慢性閉塞性肺疾患(COPD)および喘息などの肺障害を処置するのに有用であることが期待される新規ビフェニル化合物を開示している。特に、化合物ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルは、ムスカリン受容体アンタゴニストまたは抗コリン活性を所有するとして本出願に明確に記載されている。
【0003】
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの化学構造は、式Iによって表される:
【0004】
【化1】
【0005】
式Iの化合物は、市販のAutoNomソフトウェア(MDL,San Leandro,California)を使用して命名されている。
【0006】
肺または呼吸障害を処置するのに有用な治療剤は、吸入により気道中に直接、好都合に投与される。この点で、ドライパウダー吸入器(DPI)、定量吸入器(MDI)およびネブライザ吸入器を含む、吸入により治療剤を投与するための複数の種類の製薬吸入デバイスが開発されてきた。このようなデバイスでの使用のための製薬組成物および製剤を調製するとき、吸湿性でも潮解性でもなく、比較的高い融点を有し、これにより材料が著しく分解することなく微粉化される、結晶形の治療剤を有することが高度に所望である。結晶性遊離塩基形の式Iの化合物はAxtらへの特許文献2でI形およびII形として報告されているが、本発明の結晶性遊離塩基形は、より高い融点を含む、異なるおよび特に有用な特性を有する。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】米国特許出願公開第2005/0203133号明細書
【特許文献2】米国特許出願公開第2007/0112027号明細書
【発明の概要】
【課題を解決するための手段】
【0008】
本発明の1態様は、6.6±0.1、13.1±0.1、18.6±0.1、19.7±0.1、および20.2±0.1の2θ値に回折ピークを備える粉末X線回折パターンを特徴とする、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基形に関する。
【0009】
本発明の別の態様は、6.6±0.1、13.1±0.1、18.6±0.1、19.7±0.1、および20.2±0.1の2θ値に回折ピークを備える粉末X線回折パターンを特徴とする;ならびに8.8±0.1、10.1±0.1、11.4±0.1、11.6±0.1、14.8±0.1、15.2±0.1、16.1±0.1、16.4±0.1、16.9±0.1、17.5±0.1、18.2±0.1、19.3±0.1、19.9±0.1、20.8±0.1、21.1±0.1、21.7±0.1、および22.3±0.1から選択される2θ値に5つ以上の追加の回折ピークを有することをさらに特徴とする、III形と呼ばれる、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基に関する。
【0010】
本発明のなお別の態様は、6.6±0.1、13.1±0.1、18.6±0.1、19.7±0.1、および20.2±0.1の2θ値に回折ピークを備える粉末X線回折パターンを特徴とする;ならびに10.6±0.1、15.0±0.1、16.0±0.1、17.3±0.1、17.7±0.1、20.9±0.1、21.4±0.1、22.6±0.1、24.6±0.1、および27.8±0.1から選択される2θ値に5つ以上の追加の回折ピークを有することをさらに特徴とする、IV形と呼ばれる、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基に関する。
【0011】
本発明の別の態様は、本発明の結晶性遊離塩基および製薬的に許容され得る担体を含む製薬組成物に関する。本発明のまた別の態様は、1つ以上の他の治療剤と組合された本発明の結晶性遊離塩基を含む製薬組成物に関する。したがって、1実施形態において、本発明は、(a)製薬的に許容され得る担体および治療的有効量の本発明の結晶性遊離塩基;ならびに(b)治療的有効量の、コルチコステロイドなどのステロイド性抗炎症剤;β2アドレナリン受容体アゴニスト;ホスホジエステラーゼ−4阻害剤;またはその組合せ;から選択される薬剤を含み、結晶性遊離塩基および薬剤が共にまたは個別に製剤化される、製薬組成物に関する。薬剤が別々に製剤化されるとき、製薬的に許容され得る担体が含まれ得る。通例、本発明の結晶性遊離塩基および薬剤は治療的有効量で存在するであろう。
【0012】
本発明の別の態様は、約4から6の範囲のpHを有する、本発明の結晶性遊離塩基を含む等張食塩水溶液を含む製薬組成物に関するものである。特定の実施形態において、水性ネブライザ製剤は、クエン酸緩衝剤によって約5のpHまで緩衝される。
【0013】
1実施形態において、本発明は、製薬的に許容され得る担体および本発明の結晶性遊離塩基を含む製薬組成物を含有するドライパウダー吸入器を備える、薬物送達デバイスに関する。
【0014】
式Iの化合物は、ムスカリン受容体アンタゴニスト活性を有する。したがって、式Iの化合物の結晶性遊離塩基は、同じ活性を有し、それゆえ喘息および慢性閉塞性肺疾患などの肺障害を処置するのに有用性を見出すことが期待される。それゆえ本発明の別の態様は、患者に治療的有効量の本発明の結晶性遊離塩基を投与することを含む、肺障害を処置する方法に関する。本発明のなお別の態様は、患者に気管支拡張を生じる量の本発明の結晶性遊離塩基を投与することを含む、患者において気管支拡張を生じる方法に関する。1実施形態において、化合物は吸入によって投与される。本発明は、患者に治療的有効量の本発明の結晶性遊離塩基を投与することを含む、慢性閉塞性肺疾患または喘息を処置する方法も提供する。本発明の別の態様は、哺乳動物に治療的有効量の本発明の結晶性遊離塩基を投与することを含む、哺乳動物においてムスカリン受容体を拮抗する方法に関する。
【0015】
本発明は、式Iの化合物の結晶性遊離塩基形を調製するプロセスにも関する。本発明は、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基を形成することを含む、式Iの化合物を精製するプロセスも提供する。本発明はさらに、本明細書に記載するプロセスによって調製された生成物に関する。
【0016】
本発明は、微粉化形の式Iの化合物の結晶性遊離塩基に;ならびに製薬的に許容され得る担体および本発明の微粉化結晶性遊離塩基を含む製薬組成物にも関する。
【0017】
本発明は、療法でまたは医薬品として使用するための、式Iの化合物の結晶性遊離塩基形にも関する。加えて本発明は、医薬品の製造のための;とりわけ哺乳動物において肺障害を処置するまたはムスカリン受容体を拮抗する医薬品の製造のための、本発明の結晶性遊離塩基の使用に関する。
【図面の簡単な説明】
【0018】
本発明の各種の態様は、添付図面への参照により例証される。
【図1】図1は、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステル(式Iの化合物)のIII形の結晶性遊離塩基の粉末X線回折(PXRD)パターンを示す。III形の他の特徴は、示差走査熱量測定(DSC)サーモグラムを示す図4および熱重量分析(TGA)トレースを示す図6に提示されている。
【図2】図2は、式Iの化合物のIV形の結晶性遊離塩基形のPXRDパターンを示す。
【図3】図3は、III形およびIV形のPXRDパターンのオーバーレイを示す。IV形の他の特徴は、DSCサーモグラムを示す図5、およびTGAトレースを示す図7に提示されている。
【図4】図4は、III形の示差走査熱量測定(DSC)サーモグラムを示す。
【図5】図5は、IV形の示差走査熱量測定(DSC)サーモグラムを示す。
【図6】図6は、III形の熱重量分析(TGA)トレースを示す。
【図7】図7は、IV形の熱重量分析(TGA)トレースを示す。
【発明を実施するための形態】
【0019】
本発明は、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基形(式I)を提供する。驚くべきことに、本発明の結晶性遊離塩基形は大気中の水分に曝露されたときでも、潮解性でないことが見出されている。加えて、本発明の結晶性遊離塩基形は、許容され得るレベルの吸湿性および許容され得る融点を有する。たとえば結晶性遊離塩基III形は約125℃の融点を有し、結晶性遊離塩基IV形は約119℃の融点を有する。
【0020】
他の使用の中でも、本発明の結晶性遊離塩基形は、肺障害を処置する有用性を有することが期待される製薬組成物を調製するのに有用である。したがって、本発明の1態様は、製薬的に許容され得る担体および治療的有効量の本発明の結晶性遊離塩基を含む製薬組成物に関する。
【0021】
定義
本発明の化合物、組成物、方法およびプロセスについて記載するときに、以下の用語は、別途指摘されない限り、以下の意味を有する。加えて、本明細書で使用する場合、単数形「a」、「an」および「the」は、使用状況が別途明らかに指示しない限り、対応する複数形を含む。「備える、含む(comprising)」、「含む(including)」、および「有する(having)」という用語は、包括的であることが意図され、挙げられた要素以外に追加の要素があり得ることを意味する。本明細書で使用する成分の量、分子量などの特性、反応条件などを表現するすべての数は、別途指摘されない限り、すべての例において「約」という用語によって修飾されていることが理解されるはずである。したがって、本明細書に記述される数は、本発明によって得ようとされる望ましい特性に応じて変動し得る概数である。少なくとも、および請求項の範囲に対する均等論の適用を制限する試みとしてではなく、各数は、報告された有効数字に照らしておよび普通の丸め技法を適用することによって少なくとも解釈されるべきである。
【0022】
III形およびIV形はどちらも無水遊離塩基結晶多形である。「本発明の結晶性遊離塩基」が参照されるとき、用語はIII形およびIV形を含むことが理解される。
【0023】
本明細書で使用する場合、「式を有する」または「構造を有する」という句は、制限的であることが意図されず、「含む、備える」という用語が一般に使用されるのと同じ様式で使用される。
【0024】
「製薬的に許容され得る」という用語は、本明細書で使用されるときに生物学的にまたは別途許容され得ないわけではない材料を指す。たとえば「製薬的に許容され得る担体」という用語は、許容され得ない生物効果を引き起こすことなく、または組成物の他の構成要素と許容され得ない相互作用することなく、組成物中に組み入れられて患者に投与されることができる材料を指す。このような製薬的に許容され得る材料は通例、毒性および製造試験の要求される標準を満足しており、米国食品医薬品局によって好適な不活性成分として同定されたこのような材料を含む。
【0025】
「治療的有効量」という用語は、それを必要とする患者に投与されるときに処置をもたらすのに十分な量、すなわち望ましい治療効果を得るために必要とされる薬物の量を意味する。たとえば肺障害を処置するための治療的有効量は、たとえば喘息もしくは慢性閉塞性肺疾患(「COPD」)の症候を減少、抑制、消滅もしくは防止するために、または喘息もしくはCOPDの根底にある原因を処置するために必要とされる化合物の量である。1実施形態において、治療的有効量は、気管支拡張を生じるのに必要とされる量である。これに対して、「有効量」という用語は、必ずしも治療結果ではあり得ないが、望ましい結果を得るために十分な量を意味する。たとえばムスカリン受容体を含む系を試験するときに、「有効量」は受容体を拮抗するのに必要とされる量であり得る。
【0026】
「処置すること」または「処置」という用語は、本明細書で使用する場合:(a)疾患もしくは病状が出現するのを防止すること、すなわち患者の予防処置;(b)患者における疾患もしくは病状を消滅すること、または疾患もしくは病状の後退を引き起こすことなどによって、疾患もしくは病状を改善することと;(c)患者における疾患または病状の発症を減速もしくは停止することなどによって、疾患もしくは病状を抑制することと;または(d)患者における疾患もしくは病状の症候を緩和することと;を含む、哺乳動物(特にヒト)などの患者における疾患もしくは病状(COPDなど)を処置することまたは疾患もしくは病状の処置を意味する。たとえば「COPDを処置すること」という用語は、COPDが出現するのを防止すること、COPDを改善すること、COPDを抑制すること、およびCOPDの症候を改善することを含むであろう。「患者」という用語は、処置もしくは疾患防止を必要とする、または特異的疾患もしくは病状の疾患防止もしくは処置のために現在処置されている、ヒトなどのこのような哺乳動物を含むことが意図される。「患者」という用語は、本発明の化合物が評価されている試験対象またはアッセイで使用されている試験対象、たとえば動物モデルも含む。
【0027】
合成
本発明の結晶性遊離塩基形は、下および実施例に記載されているような、ただちに利用可能な開始材料から合成されることができる。各結晶性遊離塩基形を生じるために使用できる複数の方法があるが、しかし結晶含有量ならびに結晶の晶癖(サイズおよび形状)が、一部は調製方法、ならびに溶媒組成物に基づいて変動し得ることに注目される。
【0028】
特異的プロセス条件(すなわち結晶化温度、時間、反応物質のモル比、溶媒、圧力など)が与えられるが、別途明示されない限り、他のプロセス条件も使用できることが認識されるであろう。いくつかの例において、反応または結晶化は室温にて実行され、実際の温度測定は行われなかった。室温は実験室環境における周囲温度に一般に関連する範囲内の温度を意味するとして解釈することができ、通例、約25℃から約50℃の範囲内にあるであろうことが理解される。他の例において、反応または結晶化は室温にて実行され、温度は実際に測定および記録された。すべての重量、体積および等価物は、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステル(または塩形)開始材料に関係する。
【0029】
概して、結晶化は好適な不活性希釈剤または溶媒系において実行され、この例は、これに限定されるわけではないが、メタノール、エタノール、イソプロパノール、イソブタノール、酢酸エチル、アセトニトリル、ジクロロメタン、メチルt−ブチルエーテルなど、およびその混合物を含む。上述のいずれの結晶化の完了時にも、沈殿、濃縮、遠心分離などのいずれの従来手段によっても、結晶性化合物を反応混合物から単離することができる。
【0030】
本発明で用いられるビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステル、ならびに2リン酸塩などのその塩は、市販の開始材料および試薬から、実施例に記載された手順を使用して、またはMammenらへの米国特許公開第2005/0203133号およびAxtらへの米国特許公開第2007/0112027号に記載された手順を使用して、ただちに調製することができる。
【0031】
本発明の方法に記載されたモル比は、当業者に利用可能な各種の方法によってただちに決定できる。たとえばこのようなモル比は、1H NMRによってただちに決定することができる。代わりに元素分析およびHPLC法を使用して、モル比を決定することができる。
【0032】
III形
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルのIII形の結晶性遊離塩基は、エステルまたはエステルの2リン酸塩から調製することができる。
【0033】
1実施形態において、III形結晶性遊離塩基は、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルをアセトニトリルと接触させることによって調製される。通例、エステルのミリグラムの、アセトニトリルの総ミリリットルに対する比は約100:1であり、アセトニトリルは2ステップで添加される。概して、本反応は、0〜40℃の温度範囲を反復してサイクルしながら実行される。固体は次に、真空濾過によって単離され、乾燥される。
【0034】
別の実施形態において、III形結晶性遊離塩基は、III形結晶性遊離塩基の種晶およびエステルの2リン酸塩を使用して調製される。本方法は:a)結晶性遊離塩基III形の種晶を形成すること;b)ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの2リン酸塩を酢酸イソプロピルおよび水に溶解させて溶液を形成すること;c)ならびに種晶を溶液に添加することを包含する。さらに明確には、エステルの2リン酸塩(1重量)は、窒素下20±3℃にて酢酸イソプロピル(17.5体積)および水(10体積)中でスラリー化される。懸濁剤は53±3℃まで加温され、10M NaOH(0.5体積)が添加される。混合物はその温度にて短時間撹拌され、次に層が分離されて、塩基性水層が除去される。水(5体積)が有機層に添加され、撹拌される。層が分離され、水層が除去される。酢酸イソプロピル(17.5体積)が添加され、約10体積の蒸留液が常圧蒸留によって収集される。本ステップは追加の酢酸イソプロピル(10体積)を用いて反復される。2回目の蒸留の後、透明溶液の温度を53±3℃まで低下されて、次に酢酸イソプロピル(0.08体積)による結晶性遊離塩基III形(0.005重量;0.5重量%)の懸濁剤を用いて種晶が添加される。得られた懸濁剤は、53±3℃にて少なくとも2時間撹拌され、次に0.19℃/分のおよその冷却速度にて10±3℃まで冷却される。懸濁剤は10±3℃にて少なくとも2時間撹拌され、次に濾過により収集される。得られた濾過ケーキは酢酸イソプロピル(2×3体積)によって洗浄され、生成物は次に乾燥されてIII形結晶性遊離塩基を産する。
【0035】
IV形
1実施形態において、IV形結晶性遊離塩基はIII形結晶性遊離塩基の種晶を使用して調製される。本方法は:a)結晶性遊離塩基III形の種晶を形成すること;b)結晶性遊離塩基III形をアセトニトリルに溶解させて溶液を形成すること;c)および種晶を溶液に添加することを包含する。通例、種のエステルに対する重量比は、約2:250の範囲にある。通例、結晶性遊離塩基III形のグラムの、アセトニトリルの総ミリリットルに対する比は約2:10から3:30の範囲内にあり、2.5:16は1つの範囲である。アセトニトリルは通常、複数の一定分量で添加される。概して、本反応は、0〜40℃の温度範囲を反復してサイクルしながら実行される。固体は次に、真空濾過によって単離され、乾燥される。
【0036】
結晶特性
粉末X線回折の分野で周知であるように、粉末X線回折(PXRD)スペクトルの相対ピーク高さは、試料調製および機器形状に関するいくつかの因子に依存しているが、ピーク位置は実験の詳細事項に比較的非感受性である。結晶性遊離塩基III形およびIV形のPXRDパターンは、実施例5で記述されるように得られた。それゆえ1実施形態において、本発明の結晶性化合物は、あるピーク位置を有するPXRDパターンを特徴とする。
【0037】
本発明の各結晶性遊離塩基形は異なるPXRDパターンを呈するが、パターンはある共通のピークを有する。それゆえ1実施形態において、本発明は、6.6±0.1、13.1±0.1、18.6±0.1、19.7±0.1、および20.2±0.1から選択される2θ値に回折ピークを備える粉末X線回折パターンを特徴とする、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基形に関する。
【0038】
1実施形態において、結晶性遊離塩基III形は、6.6±0.1、13.1±0.1、18.6±0.1、19.7±0.1、および20.2±0.1の2θ値に回折ピークを備える粉末X線回折パターンを特徴とし;ならびに8.8±0.1、10.1±0.1、11.4±0.1、11.6±0.1、14.8±0.1、15.2±0.1、16.1±0.1、16.4±0.1、16.9±0.1、17.5±0.1、18.2±0.1、19.3±0.1、19.9±0.1、20.8±0.1、21.1±0.1、21.7±0.1、および22.3±0.1から選択される2θ値に5つ以上の追加の回折ピークを有することをさらに特徴とする。別の実施形態において、結晶性遊離塩基III形は、6.6±0.1、11.4±0.1、13.1±0.1、16.1±0.1、17.5±0.1、18.2±0.1、18.6±0.1、19.3±0.1、19.7±0.1、19.9±0.1、20.2±0.1、20.8±0.1、21.1±0.1、21.7±0.1、および22.3±0.1から選択される2θ値に回折ピークを備える粉末X線回折を特徴とする。また別の実施形態において、結晶性遊離塩基III形は、ピーク位置が図1に示すピーク位置に実質的に従うPXRDパターンを特徴とする。
【0039】
1実施形態において、結晶性遊離塩基IV形は、6.6±0.1、13.1±0.1、18.6±0.1、19.7±0.1、および20.2±0.1の2θ値に回折ピークを備える粉末X線回折パターンを特徴とし;ならびに10.6±0.1、15.0±0.1、16.0±0.1、17.3±0.1、17.7±0.1、20.9±0.1、21.4±0.1、22.6±0.1、24.6±0.1、および27.8±0.1から選択される2θ値に5つ以上の追加の回折ピークを有することをさらに特徴とする。別の実施形態において、結晶性遊離塩基IV形は、6.6±0.1、13.1±0.1、15.0±0.1、17.3±0.1、17.7±0.1、18.6±0.1、19.7±0.1、20.2±0.1、20.9±0.1、21.4±0.1、および22.6±0.1から選択される2θ値に回折ピークを備える粉末X線回折パターンを特徴とする。また別の実施形態において、結晶性遊離塩基IV形は、ピーク位置が図2に示すピーク位置に実質的に従うPXRDパターンを特徴とする。
【0040】
また別の実施形態において、本発明の結晶性遊離塩基は、示差走査熱量測定(DSC)サーモグラムを特徴とする。DSCサーモグラムは、実施例6で記述されるように得られた。本明細書で報告する融点は、DSC分析の間に登録された溶融開始に基づいて推定される。それゆえ1実施形態において、本発明の結晶性化合物は、そのDSCサーモグラフを特徴とする。1実施形態において、結晶性遊離塩基III形は、図4に見られるように、約123℃の吸熱熱流の開始および約125℃の融点を示すDSCサーモグラフを特徴とする。別の実施形態において、結晶性遊離塩基IV形は、図5に見られるように、約66℃の吸熱熱量の1つの開始、約119℃の吸熱熱量の第2の開始、および約119℃の融点を示すDSCサーモグラフを特徴とする。
【0041】
熱重量分析(TGA)は、実施例6に記載されているように、本発明の結晶性遊離塩基形に対して行われた。それゆえ1実施形態において、結晶性遊離塩基はTGAトレースを特徴とする。1実施形態において、結晶性遊離塩基III形は、図6に描かれたTGAトレースを特徴とする。別の実施形態において、結晶性遊離塩基IV形は、図7に描かれたTGAトレースを特徴とする。
【0042】
蒸気収着重量測定(GVS)アセスメントは、実施例7に記載されているように、本発明の結晶性遊離塩基形に対して行われた。本発明の結晶性遊離塩基形は、許容され得るレベルの吸湿性を有する可逆性収着/脱着プロフィールを有することが証明されている。たとえば結晶性遊離塩基III形は、25℃にて0%と90%の間の相対湿度で<2重量/重量%の可逆性水取込みを示した。加えて、結晶性遊離塩基III形は、高温および湿度への曝露時に安定であることが見出されている。
【0043】
本発明の結晶性遊離塩基形のこれらの特性は、下の実施例でさらに例証される。
【0044】
有用性
式Iの化合物はムスカリン受容体アンタゴニスト活性を所有し、およびそのため式Iの化合物の結晶性遊離塩基形は、ムスカリン受容体によって媒介される病状、すなわちムスカリン受容体アンタゴニストによる処置によって改善される病状を処置するように有用であることが期待される。このような病状は、1例として、慢性閉塞性肺疾患(たとえば慢性および喘鳴を伴う気管支炎ならびに気腫)、喘息、肺線維症、アレルギー性鼻炎、鼻漏などの可逆性気道閉塞に関連するものを含む、肺障害または疾患を含む。ムスカリン受容体アンタゴニストによって処置することができる他の病状は、過活動膀胱または排尿筋機能亢進ならびにその症候などの尿生殖路障害;過敏性腸症候群、憩室性疾患、アカラシア、胃腸過剰運動性障害および下痢などの胃腸管障害;洞性徐脈などの心不整脈;パーキンソン病;アルツハイマー病などの認知障害;月経困難(dismenorrhea);などである。
【0045】
したがって、1実施形態において、本発明は、肺障害を処置する方法であって、患者に治療的有効量のビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基を投与することを含む方法に関する。肺障害を処置するために使用されるとき、本発明の結晶性遊離塩基は通例、1日に付き複数回用量で、1日1回用量でまたは週1回用量で吸入により投与されるであろう。概して、肺障害を処置するための用量は、約10μg/日から200μg/日の範囲に及ぶであろう。
【0046】
吸入によって投与されるとき、本発明の結晶性遊離塩基は通例、気管支拡張を生じる効果を有するであろう。したがって、別の実施形態において、本発明は、患者に気管支拡張を生じる方法であって、患者に気管支拡張を生じる量のビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基を投与することを含む方法に関する。概して、気管支拡張を生じるための治療的有効用量は、約10μg/日から200μg/日の範囲に及ぶであろう。
【0047】
1実施形態において、本発明は、慢性閉塞性肺疾患または喘息を処置する方法であって、患者に治療的有効量のビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基を投与することを含む方法に関する。
【0048】
COPDまたは喘息を処置するために使用されるとき、本発明の結晶性遊離塩基は通例、1日に付き複数回用量でまたは1日1回用量で吸入により投与されるであろう。概して、COPDまたは喘息を処置するための用量は、約10μg/日から200μg/日の範囲に及ぶであろう。本明細書で使用する場合、COPDは慢性閉塞性気管支炎および気腫を含む(たとえばBarnes,Chronic Obstructive Pulmonary Disease,New England Journal of Medicine 343:269−78(2000)を参照)。
【0049】
肺障害を処置するために使用されるとき、本発明の結晶性遊離塩基は、場合により他の治療剤と組合せて投与される。したがって、特定の実施形態において、本発明の製薬組成物および方法は、治療的有効量のβ2−アドレナリン受容体アゴニスト、コルチコステロイド、非ステロイド性抗炎症剤、またはその組合せをさらに含む。
【0050】
別の実施形態において、本発明の結晶性遊離塩基は、生物系、および特にマウス、ラット、モルモット、ウサギ、イヌ、ブタ、ヒトなどの哺乳動物においてムスカリン受容体を拮抗するのに使用される。本実施形態において、治療的有効量の本発明の結晶性遊離塩基が哺乳動物に投与される。所望ならば次にムスカリン受容体を拮抗する効果を、従来の手順および機器を使用して決定することができる。
【0051】
ムスカリン受容体拮抗活性などの、本発明の結晶性遊離塩基の特性および有用性は、当業者に周知である各種のインビトロおよびインビボアッセイを使用して証明することができる。たとえば、代表的なアッセイは、以下の実施例でさらに詳細に記載されており、限定するのではなく例証として、(たとえばアッセイ1に記載されているような)hM1、hM2、hM3、hM4、およびhM5ムスカリン受容体結合を測定するアッセイを含む。本発明の結晶性遊離塩基のムスカリン受容体拮抗活性を決定するための有用な機能アッセイは、限定するのではなく例証として、細胞内環状アデノシン1リン酸(cAMP)におけるリガンド媒介変化、(cAMPを合成する)酵素アデニリルシクラーゼの活性におけるリガンド媒介変化、[35S]GTPγSのGDPとの受容体触媒交換を介した、グアノシン5’−O−(γ−チオ)3リン酸([35S]GTPγS)の単離膜中への組み入れにおけるリガンド媒介変化、(たとえば蛍光連結イメージング・プレート・リーダーまたはMolecular Devices,Inc.からのFLIPR(登録商標)によって測定された)遊離細胞内カルシウムイオンにおけるリガンド媒介変化などを測定するアッセイを含む。例示的なアッセイは、アッセイ2に記載されている。結晶性遊離塩基は、上に挙げたアッセイのいずれか、または類似の性質のアッセイにおいてムスカリン受容体の活性化を拮抗または低下することが期待され、通例、これらの試験では約0.1から100ナノモルの範囲に及ぶ濃度にて使用されるであろう。それゆえ上述のアッセイは、本発明の結晶性遊離塩基の治療有用性、たとえば気管支拡張活性を決定するのに有用である。
【0052】
本発明の結晶性遊離塩基の他の特性および有用性は、当業者に周知である各種のインビトロおよびインビボアッセイを使用して証明することができる。たとえば、結晶性遊離塩基のインビボ効力は、アイントホーフェンモデルなどの動物モデルで測定することができる。簡潔には、結晶性遊離塩基の気管支拡張活性は、気道抵抗の代替尺度として通気圧を使用する麻酔された動物(アイントホーフェンモデル)において評価される。たとえばEinthoven(1892)Pfugers Arch.51:367−445;およびMohammedら(2000)Pulm Pharmacol Ther.13(6):287−92、ならびにラットのアイトホーフェンモデルについて記載しているアッセイ3を参照。別の有用なインビボアッセイは、(たとえばアッセイ4に記載されているような)ラット唾液分泌抑制アッセイである。
【0053】
製薬組成物および製剤
本発明の結晶性遊離塩基は通例、患者に製薬組成物または製剤の形で投与される。このような製薬組成物は、これに限定されるわけではないが、吸入、経口、鼻内、局所(経皮を含む)および非経口の投与方式を含む、いずれかの許容され得る投与経路によって投与され得る。しかし、本発明の結晶性遊離塩基がいったん製剤化されると、これはもはや結晶形ではない場合がある、すなわち結晶性遊離塩基は好適な担体に溶解され得ることが当業者によって理解されるであろう。
【0054】
したがって、1実施形態において、本発明は、製薬的に許容され得る担体または賦形剤およびビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基を含む製薬組成物に関する。製薬組成物は所望ならば、他の治療剤および/または製剤化剤(formulating agent)を含有し得る。
【0055】
本発明の製薬組成物は通例、治療的有効量のビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基を活性剤として含有する。通例、このような製薬組成物は約0.01から約30重量%;たとえば約0.01から約10重量%の活性剤を含む、約0.01から約95重量%の活性剤を含有するであろう。
【0056】
いずれの従来の担体または賦形剤も本発明の製薬組成物で使用され得る。特定の担体もしくは賦形剤、または担体もしくは賦形剤の組合せの選出は、特定の患者または病状もしくは疾患状態の種類を処置するために使用されている投与方式に依存するであろう。この点で、特定の投与方式のための好適な製薬組成物の調製は、十分に製薬分野の当業者の範囲内である。加えて、このような組成物の成分は、たとえばSigma,P.O.Box 14508,St.Louis,MO 63178から市販されている。さらなる例証として、従来の製剤化技法はRemington:The Science and Practice of Pharmacy,20th Edition,Lippincott Williams & White,Baltimore,Maryland(2000);およびH.C.Anselら,Pharmaceutical Dosage Forms and Drug Delivery Systems,7th Edition,Lippincott Williams & White,Baltimore,Maryland(1999)に記載されている。
【0057】
製薬的に許容され得る担体として役目を果たすことができる材料の代表的な例は、これに限定されるわけではないが、以下の:ラクトース、グルコースおよびスクロースなどの糖;コーンスターチおよびジャガイモデンプンなどのデンプン;セルロースならびにナトリウムカルボキシメチルセルロース、エチルセルロースおよび酢酸セルロースなどのその誘導体;粉末トラガカント;麦芽;ゼラチン;タルク;ココアバターおよび坐剤ワックスなどの賦形剤;ピーナッツ油、綿実油、ベニバナ油、ゴマ油、オリーブ油、トウモロコシ油およびダイズ油などの油;プロピレングリコールなどのグリコール;グリセリン、ソルビトール、マンニトールおよびポリエチレングリコールなどのポリオール;オレイン酸エチルおよびラウリン酸エチルなどのエステル;寒天;水酸化マグネシウムおよび水酸化アルミニウムなどの緩衝剤;アルギン酸;発熱物質を含まない水;等張性生理食塩水;リンゲル液;エチルアルコール;リン酸緩衝生理食塩水;クロロフルオロカーボンおよびヒドロフルオロカーボンなどの圧縮噴射ガス;ならびに製薬組成物で用いられる他の非毒性適合性物質を含む。
【0058】
本発明の製薬組成物は通例、結晶性遊離塩基を製薬的に許容され得る担体および1つ以上の任意の成分と完全および密接に混合またはブレンドすることによって調製される。必要または所望ならば、得られた均一にブレンドされた混合物を次に、従来の手順および機器を使用して、錠剤、カプセル剤、丸剤に成形、またはキャニスター、カートリッジ、ディスペンサーなどに装入することができる。
【0059】
1実施形態において、本発明の製薬組成物は吸入投与に好適である。吸入投与の好適な製薬組成物は通例、エアゾール剤または粉剤の形であるだろう。このような組成物は概して、ネブライザ吸入器、定量吸入器(MDI)、ドライパウダー吸入器(DPI)などの周知の送達デバイスまたは類似の送達デバイスを使用して投与される。
【0060】
本発明の詳細な実施形態において、活性剤を含む製薬組成物は、ネブライザ吸入器を使用する吸入によって投与される。このようなネブライザデバイスは通例、活性剤を含む製薬組成物をミストとして噴霧して、患者の気道中に運ばれるようにする、高速気流を生じる。したがってネブライザ吸入器で使用するために製剤化されるとき、結晶性遊離塩基活性剤は通例、好適な担体に溶解されて溶液を形成する。好適なネブライザデバイスは、Respimat(登録商標)Soft Mist(商標)吸入器(Boehringer Ingelheim)、AERx(登録商標)肺送達システム(Aradigm Corp.)、およびPARI LC Plusリユーザブルネブライザ(Pari GmbH)を含む。
【0061】
ネブライザ吸入器での使用のための代表的な製薬組成物は、約0.05μg/mLから約10mg/mLの本発明の結晶性遊離塩基を含む等張性水溶液を含む。1実施形態において、水性ネブライザ製剤は等張性である。1実施形態において、このような溶液は約4〜6のpHを有する。特定の実施形態において、水性ネブライザ製剤は、クエン酸緩衝剤によって約5のpHまで緩衝される。別の特定の実施形態において、水性製剤は、約0.1mg/mLから約1.0mg/mLの、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの遊離塩基等価物を含有する。
【0062】
本発明の別の詳細な実施形態において、活性剤を含む製薬組成物は、DPIを使用する吸入によって投与される。このようなDPIは通例、活性剤を吸息の間に患者の気流に分散された自由流動パウダーとして投与する。自由流動パウダーを達成するために、結晶性遊離塩基活性剤は通例、ラクトース、デンプン、マンニトール、デキストロース、ポリ乳酸、ポリ乳酸−co−グリコリド、およびその組合せなどの好適な賦形剤を用いて製剤化される。微粉化は、結晶サイズを肺送達に好適なサイズまで減少させる一般的な方法である。通例、結晶性遊離塩基活性剤は微粉化され、好適な担体と組合されて呼吸可能なサイズの微粉化粒子の懸濁剤を形成し、「微粉化粒子」または「微粉化形」は、少なくとも約90%の粒子が約10μm未満の直径を有することを意味する。微細製粉、切刻み、圧壊、破砕、製粉、ふるい分け、粉砕、微粉砕などの粒径を減少させる他の方法も、所望の粒径を得ることができる限り使用され得る。
【0063】
DPIでの使用のための代表的な製薬組成物は、約1μmから約100μmの間の粒径を有する無水ラクトースおよび本発明の結晶性遊離塩基の微粉化粒子を含む。このようなドライパウダー製剤はたとえば、ラクトースを結晶性遊離塩基活性剤と組合せること、および次に構成要素を乾燥ブレンドすることによって作製することができる。代わりに、所望ならば、賦形剤を用いずに結晶性遊離塩基活性剤を製剤化することができる。製薬組成物は次に通例、ドライ・パウダー・ディスペンサー中に、またはドライパウダー送達デバイスでの使用のために吸入カートリッジもしくはカプセル剤の中に装入される。
【0064】
DPI送達デバイスの例は、Diskhaler(GlaxoSmithKline,Research Triangle Park,NC;たとえばNewellらへの米国特許第5,035,237号を参照);Diskus(GlaxoSmithKline;たとえばDaviesらへの米国特許第6,378,519号を参照);Turbuhaler(AstraZeneca,Wilmington,DE;たとえばWetterlinへの米国特許第4,524,769号を参照);Rotahaler(GlaxoSmithKline;たとえばHallworthらへの米国特許第4,353,365号を参照)およびHandihaler(Boehringer Ingelheim)を含む。好適なDPIデバイスのさらなる例は、Casperらへの米国特許第5,415,162号、Evansへの同第5,239,993号、およびArmstrongらへの同第5,715,810号、およびそこで引用された参考文献に記載されている。上述の特許の開示は、その全体が参照により本明細書に組み入れられている。
【0065】
本発明のまた別の詳細な実施形態において、結晶性遊離塩基活性剤を含む製薬組成物は、通例、測定された量の活性剤を、圧縮噴射ガスを使用して放出するMDIを使用する吸入によって投与される。したがって、MDIを使用して投与される製薬組成物は通例、液化噴射剤による結晶性遊離塩基活性剤の溶液または懸濁剤を含む。CCl3Fなどのクロロフルオロカーボン、ならびに1,1,1,2−テトラフルオロエタン(HFA 134a)および1,1,1,2,3,3,3−ヘプタフルオロ−n−プロパン(HFA 227)などのヒドロフルオロアルカン(HFA)を含む、いずれの好適な液化噴射剤も用いられ得る。オゾン層に影響を及ぼすクロロフルオロカーボンについての懸念のために、HFAを含有する製剤が概して好ましい。HFA製剤の追加の任意の構成要素は、エタノールまたはペンタンなどの共溶媒、ならびにトリオレイン酸ソルビタン、オレイン酸、レシチン、およびグリセリンなどの界面活性剤を含む。たとえばPurewalらへの米国特許第5,225,183号、EP 0717987 A2(Minnesota Mining and Manufacturing Company)、およびWO 92/22286(Minnesota Mining and Manufacturing Companyを参照、それらの開示はその全体が参照により本明細書に組み入れられている。
【0066】
定量吸入器での使用のための代表的な製薬組成物は、約0.01〜5重量%の本発明の遊離塩基結晶性化合物;約0〜20重量%のエタノール;および約0〜5重量%の界面活性剤を含む;残りはHFA噴射剤である。
【0067】
このような組成物は通例、冷却または加圧されたヒドロフルオロアルカンを結晶性遊離塩基活性剤、エタノール(存在する場合)および界面活性剤(存在する場合)を含有する好適な容器に添加することによって調製される。懸濁剤を調製するために、結晶性遊離塩基活性剤は微粉化され、次に噴射剤と組合される。製剤は次に、定量吸入器デバイスの部分を形成するエアゾールキャニスター中に装入される。HFA噴射剤を用いた使用のために明確に開発された定量吸入デバイスの例は、Mareckiへの米国特許第6,006,745号および同第Ashurstらへの6,143,277号に記載されている。代わりに懸濁製剤は、活性剤の微粉化粒子に界面活性剤のコーティングを噴霧乾燥することによって調製できる。たとえばWO 99/53901(Glaxo Group Ltd.)およびWO 00/61108(Glaxo Group Ltd.)を参照。上述の特許および刊行物の開示は、その全体が参照により本明細書に組み入れられている。
【0068】
吸入投薬に好適な呼吸可能な粒子、ならびに製剤およびデバイスを調製するプロセスの追加の例については、Gaoへの米国特許第6,268,533号、Trofastへの同第5,983,956号;Briggnerらへの同第5,874,063号;およびJakupovicらへの同第6,221,398号;ならびにWO 99/55319(Glaxo Group Ltd.)およびWO 00/30614(AstraZeneca AB)を参照;その開示は、その全体が参照により本明細書に組み入れられている。
【0069】
別の実施形態において、本発明の製薬組成物は経口投与に好適である。経口投与の好適な製薬組成物は、カプセル剤、錠剤、丸剤、ロゼンジ剤、カシェ剤、糖衣錠、粉剤、顆粒剤の形で;水性もしくは非水性液体による液剤もしくは懸濁剤として;または水中油型もしくは油中水型液体乳剤として;またはエリキシル剤もしくはシロップ剤としてなどであり得て;それぞれ所定量の本発明の結晶性遊離塩基を活性成分として含有する。製薬組成物は単位投薬形で包装され得る。
【0070】
固体投薬形(すなわちカプセル剤、錠剤、丸剤などとして)の経口投与が意図されるとき、本発明の製薬組成物は通例、活性成分としての本発明の結晶性遊離塩基ならびにクエン酸ナトリウムおよびリン酸2カルシウムなどの1つ以上の製薬的に許容され得る担体を含む。場合によりまたは代わりに、このような固体投薬形は:デンプン、ラクトース、スクロース、グルコース、マンニトール、および/またはケイ酸などの充填剤または増量剤;カルボキシメチルセルロース、アルギン酸塩、ゼラチン、ポリビニルピロリドン、スクロースおよび/またはアラビアゴムなどの結合剤;グリセロールなどの保湿剤;寒天、炭酸カルシウム、ジャガイモもしくはタピオカデンプン、アルギン酸、あるケイ酸塩、および/または炭酸ナトリウムなどの崩壊剤;パラフィンなどの溶解遅延剤;4級アンモニウム化合物などの吸収促進剤;セチルアルコールおよび/またはモノステアリン酸グリセロールなどの湿潤剤;カオリンおよび/またはベントナイト粘土などの吸収剤;タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、および/またはその混合物などの潤滑剤;着色剤;ならびに緩衝剤も含み得る。
【0071】
離型剤、湿潤剤、コーティング剤、甘味剤、香味剤および香料、保存料ならびに酸化防止剤も、本発明の製薬組成物中に存在することができる。製薬的に許容され得る酸化防止剤の例は:アスコルビン酸、塩酸システイン、重硫酸ナトリウム、メタ重硫酸ナトリウムなどの水溶性酸化防止剤;パルミチン酸アスコルビル、ブチル化ヒドロキシアニソール(BHA)、ブチル化ヒドロキシトルエン(BHT)、レシチン、没食子酸プロピル、アルファ−トコフェロールなどの油溶性酸化防止剤;およびクエン酸、エチレンジアミンテトラ酢酸(EDTA)、ソルビトール、酒石酸、リン酸などの金属キレート剤などを含む。錠剤、カプセル剤、丸剤などのコーティング剤は、酢酸フタル酸セルロース(CAP)、酢酸フタル酸ポリビニル(PVAP)、フタル酸ヒドロキシプロピルメチルセルロース、メタクリル酸−メタクリル酸エステルコポリマー、酢酸トリメリト酸セルロース(CAT)、カルボキシメチルエチルセルロース(CMEC)、酢酸コハク酸ヒドロキシプロピルメチルセルロース(HPMCAS)などの腸溶コーティングに使用されるコーティング剤を含む。
【0072】
所望ならば本発明の製薬組成物は、1例として、変動比のヒドロキシプロピルメチルセルロース;または他のポリマーマトリクス、リポソームおよび/もしくはミクロスフェアを使用して、活性成分の低速または制御放出を提供するようにも製剤化され得る。
【0073】
加えて、本発明の製薬組成物は、場合により乳白剤を含有し得て、製薬組成物が胃腸管のある部分において、場合により遅延方式で活性成分のみを、または活性成分を優先的に放出するように製剤化され得る。使用できる包埋組成物(embedding compositions)の例は、ポリマー物質およびワックスを含む。結晶性遊離塩基活性成分は、適切な場合、1つ以上の上記の賦形剤を用いたマイクロカプセル化形であることもできる。
【0074】
経口投与に好適な液体投薬形は、例証として、製薬的に許容され得る乳剤、マイクロエマルション、液剤、懸濁剤、シロップ剤およびエリキシル剤を含む。このような液体投薬形は通例、活性成分ならびにたとえば水または他の溶媒などの不活性希釈剤、エチルアルコール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3−ブチレングリコール、油(とりわけ綿実油、ラッカセイ油、トウモロコシ油、胚芽油、オリーブ油、ヒマシ油およびゴマ油)、グリセロール、テトラヒドロフリルアルコール、ポリエチレングリコールおよびソルビタンの脂肪酸エステル、ならびにその混合物などの可溶化剤および乳化剤を含む。懸濁剤は、活性成分に加えて、たとえばエトキシ化イソステアリルアルコール、ポリオキシエチレンソルビトールおよびソルビタンエステル、微結晶性セルロース、メタ水酸化アルミニウム、ベントナイト、寒天およびトラガカント、ならびにその混合物などの懸濁化剤を含有し得る。
【0075】
本発明の結晶性遊離塩基は、公知の経皮送達系および賦形剤を使用して経皮投与することもできる。たとえば結晶性遊離塩基は、プロピレングリコール、モノラウリン酸ポリエチレングリコール、アザシクロアルカン−2−オンなどの浸透促進剤と混和させて、パッチまたは類似の送達系に組み入れることができる。ゲル化剤、乳化剤および緩衝剤を含む追加の賦形剤は、所望ならばこのような経皮組成物で使用され得る。
【0076】
本発明の結晶性遊離塩基は、他の治療剤と共に同時投与することもできる。本併用療法は、共に製剤化された(たとえば単一の製剤に共に包装された)または別々に製剤化された(たとえば別々の単位投薬形として包装された)どちらかの、これらの第2の薬剤の1つ以上と組合された結晶性遊離塩基を使用することを包含する。同一製剤でまたは別々の単位投薬形で複数の薬剤を共に製剤化する方法は、当分野で周知である。「単位投薬形」という用語は、患者に投薬するのに好適な物理的に別個の単位を指し、すなわち各単位は、単独でまたは1つ以上の追加の単位と組合されてのどちらかで、所望の治療効果を生じるように計算された所定量の本発明の化合物を含有する。たとえばこのような単位投薬形は、カプセル剤、錠剤、丸剤などであり得る。
【0077】
追加の治療剤は、他の気管支拡張薬(たとえばPDE3阻害剤、アデノシン2b調節因子およびβ2アドレナリン受容体アゴニスト);抗炎症剤(たとえばコルチコステロイドなどのステロイド性抗炎症剤;非ステロイド性抗炎症剤(NSAID)、およびPDE4阻害剤);他のムスカリン受容体アンタゴニスト(すなわち抗コリン作用薬(antichlolinergic agents);抗感染剤(たとえばグラム陽性およびグラム陰性抗生剤または抗ウイルス薬);抗ヒスタミン薬;プロテアーゼ阻害剤;および求心性遮断薬(たとえばD2アゴニストおよびニューロキニン調節因子)から選択することができる。
【0078】
本発明の特定の1実施形態は、(a)製薬的に許容され得る担体および治療的有効量の本発明の結晶性遊離塩基;ならびに(b)製薬的に許容され得る担体および治療的有効量の、コルチコステロイドなどのステロイド性抗炎症剤;β2アドレナリン受容体アゴニスト;ホスホジエステラーゼ−4阻害剤;またはその組合せ;から選択される薬剤を含み、結晶性遊離塩基および薬剤が共にまたは個別に製剤化される、製薬組成物に関する。別の実施形態において、(b)は、製薬的に許容され得る担体ならびに治療的有効量のβ2アドレナリン受容体アゴニストおよびステロイド性抗炎症剤である。第2の薬剤は、製薬的に許容され得る塩または溶媒和物の形で、および適切な場合、光学的に純粋な立体異性体として使用することができる。
【0079】
本発明の結晶性遊離塩基と組合せて使用することができる代表的なβ2アドレナリン受容体アゴニストは、これに限定されるわけではないが、サルメテロール、サルブタモール、フォルモテロール、サルメファモール、フェノテロール、テルブタリン、アルブテロール、イソエタリン、メタプロテレノール、ビトルテロール、ピルブテロール、レバルブテロールなど、またはその製薬的に許容され得る塩を含む。使用することができる他のβ2アドレナリン受容体アゴニストは、これに限定されるわけではないが、WO 02/066422(Glaxo Group Ltd.)に記載された3−(4−{[6−({(2R)−2−ヒドロキシ−2−[4−ヒドロキシ−3−(ヒドロキシメチル)−フェニル]エチル}アミノ)−ヘキシル]オキシ}ブチル)ベンゼンスルホンアミドおよび3−(−3−{[7−({(2R)−2−ヒドロキシ−2−[4−ヒドロキシ−3−(ヒドロキシメチル)フェニル]エチル}−アミノ)ヘプチル]オキシ}−プロピル)ベンゼンスルホンアミドおよび関連化合物;WO 02/070490(Glaxo Group Ltd.)に記載された3−[3−(4−{[6−([(2R)−2−ヒドロキシ−2−[4−ヒドロキシ−3−(ヒドロキシメチル)フェニル]エチル}アミノ)ヘキシル]オキシ}ブチル)−フェニル]イミダゾリジン−2,4−ジオンおよび関連化合物;WO 02/076933(Glaxo Group Ltd.)に記載された3−(4−{[6−({(2R)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)−ベンゼンスルホンアミド、3−(4−{[6−({(2S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)−ベンゼンスルホンアミド、3−(4−{[6−({(2R/S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)−ベンゼンスルホンアミド、N−(t−ブチル)−3−(4−{[6−({(2R)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]−オキシ}ブチル)ベンゼンスルホンアミド、N−(tert−ブチル)−3−(4−{[6−({(2S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)−ヘキシル]オキシ}ブチル)−ベンゼンスルホンアミド、N−(tert−ブチル)−3−(4−{[6−({(2R/S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]−オキシ}ブチル)ベンゼンスルホンアミドおよび関連化合物;WO 03/024439(Glaxo Group Ltd.)に記載された4−{(1R)−2−[(6−{2−[(2,6−ジクロロベンジル)オキシ]エトキシ}ヘキシル)アミノ]−1−ヒドロキシエチル}−2−(ヒドロキシメチル)フェノールおよび関連化合物;Moranらに対する米国特許第6,576,793号に記載されたN−{2−[4−((R)−2−ヒドロキシ−2−フェニルエチルアミノ)フェニル]エチル}−(R)−2−ヒドロキシ−2−(3−ホルムアミド−4−ヒドロキシフェニル)エチルアミンおよび関連化合物;Moranらに対する米国特許第6,653,323号に記載されたN−{2−[4−(3−フェニル−4−メトキシフェニル)アミノフェニル]エチル}−(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2(1H)−キノリノン−5−イル)エチルアミンおよび関連化合物;ならびにその製薬的に許容され得る塩を含む。特定の実施形態において、β2−アドレナリン受容体アゴニストは、N−{2−[4−((R)−2−ヒドロキシ−2−フェニルエチルアミノ)フェニル]エチル}−(R)−2−ヒドロキシ−2−(3−ホルムアミド−4−ヒドロキシフェニル)エチルアミンの結晶性1塩酸塩である。β2−アドレナリン受容体アゴニストは用いられるときに、製薬組成物中に治療的有効量で存在するであろう。通例、β2アドレナリン受容体アゴニストは、用量当り約0.05μg〜500μgを提供するのに十分な量で存在するであろう。上述の特許および刊行物の開示は、その全体が参照により本明細書に組み入れられている。
【0080】
本発明の結晶性遊離塩基と組合せて使用することができる代表的なステロイド性抗炎症剤は、これに限定されるわけではないが、メチルプレドニゾロン、プレドニゾロン、デキサメタゾン、プロピオン酸フルチカゾン、6α,9α−ジフルオロ−17α−[(2−フラニルカルボニル)オキシ]−11β−ヒドロキシ−16α−メチル−3−オキソアンドロスタ−1,4−ジエン−17β−カルボチオ酸S−フルオロメチルエステル、6α,9α−ジフルオロ−11β−ヒドロキシ−16α−メチル−3−オキソ−17α−プロピオニルオキシ−アンドロスタ−1,4−ジエン−17β−カルボチオ酸S−(2−オキソ−テトラヒドロフラン−3S−イル)エステル、ベクロメタゾンエステル(たとえば17−プロピオン酸エステルまたは17,21−ジプロピオン酸エステル)、ブデソニド、フルニソリド、モメタゾンエステル(たとえばフロ酸エステル)、トリアムシノロンアセトニド、ロフレポニド、シクレソニド、プロピオン酸ブチキソコルト、RPR−106541,ST−126など、またはその製薬的に許容され得る塩を含む。ステロイド性抗炎症剤は用いられるときに、組成物中に治療的有効量で存在するであろう。通例、ステロイド性抗炎症剤は、用量当り約0.05μg〜500μgを提供するのに十分な量で存在するであろう。
【0081】
例示的な組合せは、β2アドレナリン受容体アゴニストとしてのサルメテロール、およびステロイド性抗炎症剤としてのプロピオン酸フルチカゾンと同時投与される本発明の結晶性遊離塩基である。別の例示的な組合せは、β2アドレナリン受容体アゴニストとしてのN−{2−[4−((R)−2−ヒドロキシ−2−フェニルエチルアミノ)フェニル]エチル}−(R)−2−ヒドロキシ−2−(3−ホルムアミド−4−ヒドロキシフェニル)エチルアミンの結晶性1塩酸塩、およびステロイド性抗炎症剤としての6α,9α−ジフルオロ−17α−[(2−フラニルカルボニル)オキシ]−11β−ヒドロキシ−16α−メチル−3−オキソアンドロスタ−1,4−ジエン−17β−カルボチオ酸S−フルオロメチルエステルと同時投与される本発明の結晶性遊離塩基である。上記のように、これらの薬剤は共にまたは別々に製剤化することができる。
【0082】
他の好適な組合せはたとえば、他の抗炎症剤、たとえばNSAID(たとえばクロモグリク酸ナトリウム、ネドクロミルナトリウム、ならびにテオフィリン、PDE4阻害剤および混合PDE3/PDE4阻害剤などのホスホジエステラーゼ(PDE)阻害剤);ロイコトリエンアンタゴニスト(たとえばモンテルカスト(monteleukast));ロイコトリエン合成の阻害剤;iNOS阻害剤;トリプターゼおよびエラスターゼ阻害剤などのプロテアーゼ阻害剤;ベータ−2インテグリンアンタゴニストおよびアデノシン受容体アゴニストもしくはアンタゴニスト(たとえばアデノシン2aアゴニスト);サイトカインアンタゴニスト(たとえばインターロイキン抗体(αIL抗体)、明確にはαIL−4療法、αIL−13療法、またはその組合せなどのケモカインアンタゴニスト);またはサイトカイン合成の阻害剤を含む。
【0083】
本発明の結晶性遊離塩基と組合せて使用することができる代表的なホスホジエステラーゼ−4(PDE4)阻害剤または混合PDE3/PDE4阻害剤は、これに限定されるわけではないが、シス4−シアノ−4−(3−シクロペンチルオキシ−4−メトキシフェニル)シクロヘキサン−1−カルボン酸、2−カルボメトキシ−4−シアノ−4−(3−シクロプロピルメトキシ−4−ジフルオロメトキシフェニル)シクロヘキサン−1−オン;シス−[4−シアノ−4−(3−シクロプロピルメトキシ−4−ジフルオロメトキシフェニル)シクロヘキサン−1−オール];シス−4−シアノ−4−[3−(シクロペンチルオキシ)−4−メトキシフェニル]シクロヘキサン−1−カルボン酸など、またはその製薬的に許容され得る塩を含む。他の代表的なPDE4または混合PDE4/PDE3阻害剤は、AWD−12−281(elbion);NCS−613(INSERM);D−4418(Chiroscience and Schering−Plough);CI−1018またはPD−168787(Pfizer);WO99/16766(Kyowa Hakko)に記載されているベンゾジオキソール化合物;K−34(Kyowa Hakko);V−11294A(Napp);ロフルミラスト(Byk−Gulden);WO99/47505(Byk−Gulden)に記載されているフタラジノン化合物;プマフェントリン(Byk−Gulden、現在Altana);アロフィリン(Almirall−Prodesfarma);VM554/UM565(Vernalis);T−440(Tanabe Seiyaku);およびT2585(Tanabe Seiyaku)を含む。
【0084】
本発明の結晶性遊離塩基と組合せて使用することができる代表的なムスカリンアンタゴニスト(すなわち抗コリン剤)は、これに限定されるわけではないが、アトロピン、硫酸アトロピン、酸化アトロピン、硝酸メチルアトロピン、臭化水素酸ホマトロピン、臭化水素酸ヒヨスチアミン(d、l)、臭化水素酸スコポラミン、臭化イプラトロピウム、臭化オキシトロピウム、臭化チオトロピウム、メタンテリン、臭化プロパンテリン、臭化メチルアニソトロピン、臭化クリジニウム、コピロレート(ロビヌル)、ヨウ化イソプロパミド、臭化メペンゾラート、塩化トリジヘキセチル(パシロン)、メチル硫酸ヘキソシクリウム、塩酸シクロペントレート、トロピカミド、塩酸トリヘキシフェニジル、ピレンゼピン、テレンゼピン、AF−DX116およびメトクトラミンなど、もしくはその製薬的に許容され得る塩;または塩として挙げられた化合物では、その代わりの製薬的に許容され得る塩を含む。
【0085】
本発明の結晶性遊離塩基と組合せて使用することができる代表的な抗ヒスタミン薬(すなわちH1受容体アンタゴニスト)は、これに限定されるわけではないが、マレイン酸カルビノキサミン、フマル酸クレマスチン、塩酸ジフェニルヒドラミンおよびジメンヒドリナートなどのエタノールアミン;マレイン酸ピリラミン(pyrilamine amleate)、塩酸トリペレナミンおよびクエン酸トリペレナミンなどのエチレンジアミン;クロルフェニラミンおよびアクリバスチンなどのアルキルアミン;塩酸ヒドロキシジン、パモ酸ヒドロキシジン、塩酸シクリジン、乳酸シクリジン、塩酸メクリジンおよび塩酸セチリジンなどのピペラジン;アステミゾール、塩酸レボカバスチン、ロラタジンまたはそのデスカルボエトキシ類似体、テルフェナジンおよび塩酸フェキソフェナジンなどのピペリジン;塩酸アゼラスチン;など、もしくはその製薬的に許容され得る塩;または塩として挙げられた化合物では、その代わりの製薬的に許容され得る塩を含む。
【0086】
別途指摘されない限り、本発明の結晶性遊離塩基と組合せて投与される他の治療剤の例示的な好適な用量は、約0.05μg/日〜100mg/日の範囲にある。
【0087】
以下の製剤は、本発明の代表的な製薬組成物、ならびに例示的な調製方法を例証する。1つ以上の第2の薬剤は場合により、本発明の結晶性遊離塩基(第1の活性剤)と共に製剤化することができる。代わって、第2の薬剤は別々に製剤化されて、第1の活性剤と同時にまたは連続して、共に投与することができる。たとえば、1実施形態において、単一のドライパウダー製剤は、本発明の結晶性遊離塩基および1つ以上の第2の薬剤をどちらも含むように製造することができる。別の実施形態において、1つの製剤が本発明の結晶性遊離塩基を含有するように製造され、別々の製剤が第2の薬剤を含有するように製造される。このようなドライパウダー製剤は次に、別々のブリスターパックに包装されて、単一のDPIデバイスによって投与することができる。
【0088】
吸入投与のための例示的なドライパウダー製剤
0.2mgの本発明の結晶性遊離塩基は微粉化され、次に25mgのラクトースとブレンドされる。ブレンドされた混合物は次に、ゼラチン吸入カートリッジに装入される。カートリッジの内容物は、粉剤吸入器を使用して投与される。
【0089】
ドライパウダー吸入器による投与のための例示的なドライパウダー製剤
本発明の微粉化結晶性遊離塩基(活性剤)のラクトースに対する、1:200のバルク製剤比を有するドライパウダーが調製される。パウダーは、用量当り約10μg〜100μgの活性剤を送達することができるドライパウダー吸入デバイスに充填される。
【0090】
定量吸入器による投与のための例示的な製剤
5重量%の本発明の結晶性遊離塩基(活性剤)および0.1重量%のレシチンを含有する懸濁剤は、10gの結晶性遊離塩基を10μm未満の平均径を有する微粉化粒子として、200mLの脱塩水に溶解された0.2gのレシチンから形成された溶液に分散させることによって調製される。懸濁剤は噴霧乾燥され、得られた材料は1.5μm未満の平均直径を有する粒子に微粉化される。粒子は、加圧された1,1,1,2−テトラフルオロエタンと共にカートリッジに装入される。
【0091】
代わって、5重量%の本発明の結晶性遊離塩基、0.5重量%のレシチン、および0.5重量%のトレハロースを含有する懸濁剤は、5gの結晶性遊離塩基を10μm未満の平均径を有する微粉化粒子として、100mLの脱塩水に溶解された0.5gのトレハロースおよび0.5gのレシチンから形成されたコロイド状溶液に分散させることによって調製される。懸濁剤は噴霧乾燥され、得られた材料は1.5μm未満の平均直径を有する粒子に微粉化される。粒子は、加圧された1,1,1,2−テトラフルオロエタンと共にキャニスターに装入される。
【0092】
ネブライザによる投与のための例示的な水性エアゾール製剤
製薬組成物は、0.5mgの本発明の結晶性遊離塩基(活性剤)を、クエン酸によって酸性化された1mLの0.9%塩化ナトリウム溶液に溶解させることによって調製される。混合物は、活性剤が溶解するまで撹拌および超音波処理される。溶液のpHは、NaOHの低速での添加によって、3から8の範囲の値(通例、約5)に調整される。
【0093】
経口投与のための例示的な硬ゼラチンカプセル製剤
以下の成分は完全にブレンドされ、次に硬ゼラチンカプセル中に装入される:カプセル当り合計460mgの組成物のために、250mgの本発明の結晶性遊離塩基、200mgのラクトース(噴霧乾燥)、および10mgのステアリン酸マグネシウム。
【0094】
経口投与のための例示的な懸濁製剤
以下の成分は混合されて、10mLの懸濁剤当り100mgの活性成分を含有する懸濁剤を形成する。
【0095】
【表1】
【0096】
例示的な注射用製剤
以下の成分がブレンドされ、pHは0.5N HClまたは0.5N NaOHを使用して4±0.5に調整される。
【0097】
【表2】
【実施例】
【0098】
以下の調製および実施例が提供されて、本発明の詳細な実施形態を例証する。しかし、これらの詳細な実施形態は、明確に指摘されない限り、本発明の範囲を決して制限することを意図されない。以下の省略形は、別途指摘されない限り以下の意味を有し、本明細書で使用され、定義されていないその他の省略形は、その標準の意味を有する:
AC アデニリルシクラーゼ
BSA ウシ血清アルブミン
cAMP 3’−5’環状アデノシン1リン酸
CHO チャイニーズハムスター卵巣
cM5 クローン化チンパンジーM5受容体
DCM ジクロロメタン
dPBS ダルベッコリン酸緩衝生理食塩水
EDTA エチレンジアミンテトラ酢酸
EtOAc 酢酸エチル
FBS ウシ胎仔血清
FLIPR 蛍光測定イメージング・プレート・リーダー
HBSS ハンクス緩衝塩溶液
HEPES 4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸
hM1 クローン化ヒトM1受容体
hM2 クローン化ヒトM2受容体
hM3 クローン化ヒトM3受容体
hM4 クローン化ヒトM4受容体
hM5 クローン化ヒトM5受容体
HOBT N−ヒドロキシベンゾトリアゾール
HPLC 高速液体クロマトグラフィー
MCh メチルコリン
MeCN アセトニトリル
本明細書で使用されるが、定義されていないその他の省略形は、その標準の、概して許容される意味を有する。別途記されない限り、試薬、開始材料および溶媒は、市販品供給者(Sigma−Aldrich、Flukaなど)から購入されて、さらに精製せずに使用された。
【0099】
調製1
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステル
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの2リン酸塩(16g)を、水(100mL)およびEtOAc(200mL)の2相性混合物に溶解させた。NaOH(2N、75mL)が5分間の期間にわたって添加された。次に混合物は30分間撹拌された。相は分離され、水相はEtOAc(200mL)によって抽出された。合せた有機相は濃縮された。DCM(100mL)が添加され、混合物は蒸発乾固された。固体は乾燥器内で約48時間乾燥されて、表題化合物(9.6g)が得られた。
【0100】
(実施例1)
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基(III形)
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステル(102.4mg)をMeCN(500μL)に溶解させた。溶液は室温にて80分間撹拌され、白色固体沈殿が形成された。混合物は振とう器ブロックに配置され、48時間にわたって熱サイクル処理された(1時間ブロックで0〜40℃)。白色で濃密な静止した固体が観察された。MeCN(500μL)が添加され、スラリーは移動性となった。次に混合物は再度、振とう器ブロックに2時間配置された。固体は、焼結漏斗を使用して真空濾過により単離され、次に完全真空下で40℃のピストン乾燥器に15.5時間配置されて、76.85mgの表題結晶性化合物を産した。
【0101】
(実施例2)
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基(III形)
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの2リン酸塩(C35H43N5O4・2H3PO4;MW 793.75;632.9g)は、窒素下の室温にて酢酸イソプロピル(11.08L)および水(6.33L)中でスラリー化された。懸濁剤は53±3℃まで加温され、混合物の温度を50℃超に維持しながら、10M NaOH(317mL)が攪拌混合物に添加された。混合物が53±3℃にて約5分間撹拌されてから、層が沈降した。次に層が分離されて、水層が除去された。混合物の温度が50℃超に維持されながら、水(3.16L)が有機層に添加された。混合物が53±3℃にて5分間撹拌されると、層が静置された。層が分離され、水層が除去された。酢酸イソプロピル(6.33L)が添加され、次に約10体積の蒸留液が常圧蒸留によって収集された。本ステップは追加の酢酸イソプロピル(3.2L)を用いて反復された。2回目の蒸留の後、透明溶液の温度が53±3℃まで低下されて、次にビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステル結晶性遊離塩基(III形;3.2g)の酢酸イソプロピル(51mL)による懸濁剤によって種晶が添加された。得られた懸濁剤は53±3℃にて2時間撹拌され、次に4時間にわたって10±3℃まで冷却された。懸濁剤は10±3℃にて少なくとも2時間撹拌され、次に固体が濾過によって収集された。得られた濾過ケーキは酢酸イソプロピル(2×1.9L)で洗浄され、生成物は真空中で50℃にて乾燥されて、表題結晶性化合物を産した(C35H43N5O4;MW 597.76;382.5g、収率80.3%)。
【0102】
(実施例3)
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基(III形)の再結晶化
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基(III形;C35H43N5O4;MW 597.76;372.5g)は、窒素下20±3℃にてトルエン(5.6L)中でスラリー化された。懸濁剤は、82±3℃まで加温され、完全な溶解が観察されるまで本温度で維持された。溶液は次に、晶析装置容器で清澄にされて、トルエン(373μL)によるすすぎが続けられた。晶析装置容器内で固体が観察され、容器は82±3℃まで再加熱されて溶解がもたらされ、次に58±3℃まで冷却されて、トルエン(8μL)中で事前に超音波処理された(約1分間)結晶性遊離塩基(III形;1.9g)によって種晶が添加された。得られた懸濁剤は、58±3℃にて少なくとも4時間静置され、次に2時間にわたって20±3℃まで冷却された(およそ0.33℃/分の冷却速度)。懸濁剤は20±3℃にて少なくとも1時間撹拌され、次に固体が濾過によって収集された。得られた濾過ケーキはトルエン(2×1.2L)によって洗浄され、生成物は真空中で52±3℃にて乾燥されて、表題結晶性化合物を産した(345.3g、収率92.7%)。
【0103】
(実施例4)
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基(IV形)
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステル(調製1に記載されたように調製;2.5g)をMeCN(10mL)に溶解させて、粘性油性薄黄色材料を産した。追加のMeCN(5mL)が添加されて、材料を希釈した。溶液に結晶性遊離塩基(20mg;実施例1に記載されたように調製されたIII形)によって種晶が添加され、室温にて90分間撹拌された。大量の白色沈殿(小型結晶)が観察された。スラリーは偏光顕微鏡下で分析され、複屈折であることが見出された。
【0104】
追加のMeCN(3mL)が添加され、スラリーがMetz Syn10ブロックに配置されて、800rpmにて一晩、熱サイクル処理された(1時間ブロックで0〜40℃)。Metz Syn10は、静止した10位置の並列反応ステーションである。溶液/スラリーのかき混ぜは、クロス磁気撹拌棒によった。振とう器ブロックは、外部ユラボ浴によって加熱および冷却される別個の機器であった。材料は0℃にて除去された。スラリーが沈降して、白色沈殿の上に薄黄色溶液が残っているのが観察された。スラリーは撹拌され、再度、振とう器ブロックに配置されて熱サイクル処理された。材料は40℃にて除去され、室温にて高いかき混ぜ速度にて80分間撹拌された。スラリーは再度分析され、複屈折であることが見出された。濾過ケーキは、焼結漏斗を使用して真空濾過によって単離された。MeCN(3mL)が使用されて濾紙が濡らされて、濾過の前に濾過ケーキがMeCNによって洗浄された。ケーキは真空下にて40分間脱液されて、2.3gの流動性白色パウダーを産した。材料は40℃にて65時間、ピストン乾燥器に配置され、2.2gの表題結晶性化合物を白色パウダー(99.6%純度)として産した。
【0105】
生成物のラマンスペクトルの大部分は、III形開始材料のラマンスペクトルと一致していた。しかし、3個のシフトが注目された。
【0106】
III形 生成物
878 cm−1 881 cm−1
775 cm−1 772 cm−1
485 cm−1 488 cm−1
生成物は次に、粉末X線回折、示差走査熱量測定、および熱重量分析によって分析された。生成物がIII形開始材料とは異なる遊離塩基結晶形であることが決定され、IV形と呼ばれた。
【0107】
(実施例5)
粉末X線回折
結晶性遊離塩基III形(実施例1より)およびIV形(実施例4より)の粉末X線回折(PXRD)パターンは、XCelerator検出器を装備したPANalytical X’Pert Pro粉末回折計(diffractomer)で取得された。取得条件は、放射線:Cu Kα;発電機電圧:40kV;発電機電流:45mA;開始角度2.0°2θ;終了角度40.0°2θ、ステップサイズ:0.0167°2θであった。ステップ当りの時間は、31.750秒であった。試料は、2、3ミリグラムの試料をシリコンウェハ(ゼロ背景)プレート上に載せて、粉末薄膜を生じることによって調製された。
【0108】
特徴的なピーク位置および計算されたd間隔が下にまとめられており、これらのピークが14%を超える相対強度を有することのみが報告される。これらはHighscoreソフトウェアを使用して生データから計算された。ピーク位置の実験誤差は、約±0.1°2θである。相対ピーク強度は、好ましい配向のために変動するであろう。
【0109】
【表3】
【0110】
結晶性遊離塩基III形の代表的なPXRDパターンは図1に示されている。結晶性遊離塩基IV形の代表的なPXRDパターンは図2に示されている。
【0111】
(実施例6)
熱分析
結晶性遊離塩基III形(実施例1より)およびIV形(実施例4より)の示差走査熱量測定(DSC)サーモグラムは、TA Instruments熱量計を使用して得られた。試料はアルミニウム皿に秤量され、皿の蓋が上に配置されて、皿を密封することなく軽く圧着された。実験は、10℃/分の加熱速度を使用して実行された。
【0112】
結晶性遊離塩基III形の代表的なDSCサーモグラフは図4に示されている。DSCサーモグラフは、III形が123.1℃における吸熱熱流の開始(エンタルピー67.7J/g)を示すDSCサーモグラフを特徴とすることを証明している。
【0113】
結晶性遊離塩基IV形の代表的なDSCサーモグラフは図5に示されている。DSCサーモグラフは、IV形が小規模な吸熱および主吸熱、すなわち65.6℃にて出現する小規模な第1の吸熱熱流の開始(エンタルピー0.8J/g)および118.8℃にて出現する主な第2の吸熱熱流の開始(エンタルピー66.8J/g)を示すDSCサーモグラフを特徴とすることを証明する。
【0114】
熱重量分析(TGA)データは、TA Instruments Q500計器を使用して得られた。試料は、開放されたアルミニウム皿内で10℃/分の加熱速度にて200℃まで加熱された。
【0115】
結晶性遊離塩基III形の代表的なTGAトレースは図6に示されており、試料分解前にごくわずかな重量損失が観察されたことを指摘している。結晶性遊離塩基IV形の代表的なTGAトレースは図7に示されており、試料溶融前に約0.3%の重量損失が観察され、これが残留溶媒の損失と一致することを指摘している。
【0116】
(実施例7)
蒸気収着重量測定アセスメント
蒸気収着重量測定(GVS)試験は、25℃にて水蒸気散布を使用した全収着等温線の発生のためにSurface Measurements System DVS−1計器を使用して行われた。約7mgの試料サイズは、風袋を差し引いた透明で乾燥した試料メッシュ皿に配置され、内蔵天秤で秤量された。目標相対湿度(RH)は10%刻みで、30%〜90%、次に90%〜0%および0%〜30%であった。平衡点は、0.02dm/dt漸近線設定を使用して自動的に決定された。
【0117】
25℃にて行われた結晶性遊離塩基III形の試料に対するGVS試験は、材料が0%RH〜90%RHの範囲にわたって水分を吸収する傾向が低いことを証明した。試料は、25℃にて0〜90%のRHの間で<2%重量/重量の可逆的吸水を示した。本GVSトレースは、III形が広範な湿度範囲に曝露されたときに、許容され得る重量増加を有することを証明している。
【0118】
(実施例8)
微粉化
結晶性遊離塩基III形の試料は、APTM 4”微粉装置およびレーザー光回折によって(buy)決定された粒径のどちらかを使用して微粉化された。
【0119】
【表4】
【0120】
参考のために、投入した結晶性遊離塩基III形の粒径はX10=5.58μm X50=18.2μm、およびX90=49.7μmであった。微粉化は、呼吸可能なサイズ範囲の粒子を産した。微粉化は結晶性の低下を生じたが、微粉化前材料の本質的な特徴を維持した。40℃/20%相対湿度、40℃/75%相対湿度(キャップなし)、および50℃/周囲湿度での3カ月にわたる保管後に、PXRDでは変化が観察されなかった。
【0121】
結晶性遊離塩基III形のDSCサーモグラフは、微粉化の前後に、125℃にて鋭い溶融を示した。おそらく結晶化のために、87℃にて微粉化材料に追加の小規模な熱的イベントがあった。40℃/20%RH、40℃/75%RH(露出)、および50℃/周囲湿度での3カ月間の保管の後に、微粉化材料は125℃にて鋭い溶融を示し、アモルファス内容物の証拠はなかった。
【0122】
(実施例9)
ラクトース適合性
結晶性遊離塩基III形の2つの製剤が、40℃/20%相対湿度(RH)、40℃/75%RH(キャップなし)、および50℃/周囲湿度での3カ月にわたる安定性について評価された。0.08重量/重量%(10μg DPI用量)および2重量/重量%(250μg DPI用量)製剤は、ラクトースのみとの、またはラクトースおよび1重量/重量%のステアリン酸マグネシウムとのブレンドとして調製された。すべての製剤の安定性は、許容され得ることが見出された。
【0123】
(実施例10)
pH溶解度および安定性
結晶性遊離塩基III形は、pH7までの培地で良好な溶解度(およそ2mg/mLを超える)を示した。水溶解度は、8.9の天然pHで0.66mg/mLであった。人工肺液への溶解度は0.46mg/mLであり、4時間から24時間の間の溶解度測定では変化は見られなかった。
【0124】
結晶性遊離塩基III形溶液は、50℃において、または露光下でpH4からpH6の緩衝液中にて7日間まで安定である。溶液は、水および生理食塩水中にて室温で遮光されて7日間安定である。
【0125】
アッセイ1
放射性リガンド結合アッセイ
hM1、hM2、hM3およびhM4ムスカリン受容体サブタイプを発現する細胞からの膜調製
クローン化ヒトhM1、hM2、hM3およびhM4ムスカリン受容体サブタイプをそれぞれ安定発現するCHO株化細胞は、10%FBSおよび250μg/mLジェネティシンを補ったHAMのF−12から成る培地でコンフルエント近くまで培養された。細胞は5%CO2、37℃のインキュベータで培養され、dPBS中2mM EDTAによって浮上された。細胞は、650xgにおける5分間の遠心分離によって収集され、細胞ペレットは−80℃にて凍結保管されるか、または膜がただちに調製されるかのどちらかであった。膜調製では、細胞ペレットは溶解緩衝液に再懸濁され、Polytron PT−2100組織破砕装置(Kinematica AG;20秒×2バースト)によってホモジナイズされた。粗膜は、40,000xgで4℃にて15分間遠心分離された。膜ペレットは次に再懸濁緩衝液によって再懸濁され、Polytron組織破砕装置によって再度ホモジナイズされた。膜懸濁剤のタンパク質濃度は、Lowry,O.ら、Journal of Biochemistry 193:265(1951)に記載された方法によって決定された。すべての膜は、一定分量で−80℃にて凍結保管されるか、またはただちに使用された。一定分量の調製hM5受容体膜は、Perkin Elmerから直接購入されて、使用するまで−80℃にて保管された。
【0126】
ムスカリン受容体サブタイプhM1、hM2、hM3、hM4およびhM5に対する放射性リガンド結合アッセイ
放射性リガンド結合アッセイは、96ウェル・マイクロタイター・プレートにおいて、1000μLのアッセイ全量で行われた。hM1、hM2、hM3、hM4またはhM5ムスカリンサブタイプのいずれかを安定発現するCHO細胞膜は、アッセイ緩衝液で以下の特異的目標タンパク質濃度(μg/ウェル):hM1では10μg、hM2では10〜15μg、hM3では10〜20μg、hM4では10〜20μg、およびhM5では10〜12μgに希釈された。膜は、アッセイプレート添加の前に、Polytron組織破砕装置を使用して短時間(10秒)ホモジナイズされた。放射性リガンドのKD値を決定するための飽和結合試験は、L−[N−メチル−3H]スコポラミンメチルクロリド([3H]−NMS)(TRK666、84.0Ci/mmol,Amersham Pharmacia Biotech,Buckinghamshire,England)を0.001nM〜20nMの範囲に及ぶ濃度で使用して行われた。試験化合物のKi値を決定するための置換アッセイは、[3H]−NMSを1nMおよび11の異なる試験化合物濃度にて用いて行われた。試験化合物は、最初に希釈緩衝液に40μMの濃度で溶解され、次に希釈緩衝液によって、400fM〜4μMの範囲に及ぶ最終濃度に5倍ずつ連続希釈された。アッセイプレートへの添加順序および体積は、以下の通りであった:0.1%BSAを有する825μLアッセイ緩衝液、25μL放射性リガンド、100μL希釈試験化合物、および50μL膜。アッセイプレートを37℃にて6時間インキュベートした。結合反応は、0.3%ポリエチレンイミン(PEI)中で予備処理されたGF/Bガラス繊維フィルタプレート(Perkin Elmer Inc.,Wellesley,MA)での高速濾過によって終結された。フィルタプレートは洗浄緩衝液(10mM HEPES)で3回すすがれて、未結合放射能が除去された。プレートが次に空気乾燥されて、50μL Microscint−20液体シンチレーション液(PerkinElmer Inc.,Wellesley,MA)が各ウェルに添加された。プレートは次に、PerkinElmer Topcount液体シンチレーションカウンタ(PerkinElmer Inc.,Wellesley,MA)でカウントされた。結合データは、GraphPad Prismソフトウェアパッケージ(GraphPad Software,Inc.,San Diego,CA)による非線形回帰分析によって、1部位競合モデルを使用して分析された。試験化合物のKi値は、観察されたIC50値から、放射性リガンドのKD値はCheng−Prusoffの等式(Cheng Y;Prusoff W.H.Biochemical Pharmacology 22(23):3099−108(1973))を使用して計算された。Ki値はpKi値に変換されて、幾何平均および95%信頼区間が決定された。これらの要約統計量は次に、データ報告のために再度Ki値に変換された。
【0127】
本アッセイにおいて、より低いKi値は、試験化合物が試験された受容体に対してより高い結合親和性を有することを指摘している。式Iの化合物は、本アッセイまたは類似のアッセイで試験されるときに、M3ムスカリン受容体サブタイプに対して約5nM未満のKi値を有することが見出された。
【0128】
アッセイ2
ムスカリン受容体機能的効力アッセイ
cAMP蓄積のアゴニスト媒介阻害の遮断
本アッセイにおいて、試験化合物の機能的効力は、試験化合物がhM2受容体を発現するCHO−K1細胞におけるフォルスコリン媒介cAMP蓄積のオキソトレモリン阻害を遮断する能力を測定することによって決定される。
【0129】
cAMPアッセイは、ラジオイムノアッセイ形式で、125I−cAMPを有するFlashplateアデニリルシクラーゼ活性化アッセイシステム(NEN SMP004B,PerkinElmer Life Sciences Inc.,Boston,MA)を製造者の説明書に従って使用して行われる。
【0130】
細胞は、dPBSによって1回洗浄され、アッセイ1に記載されているようにトリプシン−EDTA溶液(0.05%トリプシン/0.53mM EDTA)によって浮上させる。分離された細胞は、50mL dPBS中の650×gでの5分間の遠心分離によって2回洗浄される。細胞ペレットは次に10mL dPBSに再懸濁され、細胞はCoulter Z1 Dual Particleカウンター(Beckman Coulter,Fullerton,CA)によってカウントされる。細胞は650×gにて5分間にわたって再度遠心分離され、刺激緩衝液に1.6×106〜2.8×106細胞/mLのアッセイ濃度まで再懸濁される。
【0131】
試験化合物は、最初に400μMの濃度まで希釈緩衝液(1mg/mL BSA(0.1%)を補ったdPBS)で希釈され、次に希釈緩衝液によって100μM〜0.1nMの範囲に及ぶ最終モル濃度まで連続希釈される。オキソトレモリンは同様の方式で希釈される。
【0132】
AC活性のオキソトレモリン阻害を測定するために、25μLのフォルスコリン(dPBSで希釈された25μM最終濃度)、25μLの希釈オキソトレモリン、および50μLの細胞がアゴニスト・アッセイ・ウェルに添加される。試験化合物がオキソトレモリン阻害AC活性を遮断する能力を測定するために、25μLのフォルスコリンおよびオキソトレモリン(それぞれ25μMおよび5μMの最終濃度、dPBSで希釈)、25μLの希釈試験化合物、および50μLの細胞が残りのアッセイウェルに添加される。
【0133】
反応物は37℃にて10分間インキュベートされ、100μLの氷冷検出緩衝液の添加により停止される。プレートは密封され、室温にて一晩インキュベートされて、翌朝、PerkinElmer TopCount液体シンチレーションカウンタ(PerkinElmer Inc.,Wellesley,MA)でカウントされる。産生されたcAMPの量(pmol/ウェル)は、製造者のユーザマニュアルに記載されているように、試料およびcAMP標準で観察されたカウントに基づいて計算される。データは、非線形回帰分析によって、1部位競合等式を使用してGraphPad Prism Software package(GraphPad Software,Inc.,San Diego,CA)を用いて分析される。Cheng−Prusoffの等式を使用して、オキソトレモリン濃度応答曲線のEC50およびオキソトレモリンアッセイ濃度をそれぞれKDおよび[L]として用いてKiを計算する。KiはpKi値に変換されて、幾何平均および95%信頼区間が決定される。これらの要約統計量は次に、データ報告のために再度Ki値に変換される。
【0134】
本アッセイにおいて、より低いKi値は、試験化合物が試験された受容体に対してより高い機能的活性を有することを指摘している。式Iの化合物は、本アッセイまたは類似のアッセイで試験されたときに、hM2受容体を発現するCHO−K1細胞におけるフォルスコリン媒介cAMP蓄積のオキソトレモリン阻害の遮断のために、約5nM未満のKi値を有することが見出された。
【0135】
アゴニスト媒介[35S]GTPγS結合の遮断
第2の機能的アッセイにおいて、試験化合物の機能的効力は、化合物がhM2受容体を発現するCHO−K1細胞におけるオキソトレモリン刺激[35S]GTPγS結合を遮断する能力を測定することによって決定することができる。
【0136】
使用時に凍結膜は解凍され、次にアッセイ緩衝液中でウェル当り5〜10μgのタンパク質の最終目標組織濃度で希釈される。膜は、Polytron PT−2100組織破砕装置を使用して短時間ホモジナイズされ、次にアッセイプレートに添加される。
【0137】
アゴニストオキソトレモリンによる[35S]GTPγS結合の刺激のためのEC90値(90%最大応答に有効な濃度)は、各実験で決定される。
【0138】
試験化合物を阻害するオキソトレモリン刺激[35S]GTPγS結合の能力を決定するために、以下が96ウェルプレートの各ウェルに添加される:25μLの、[35S]GTPγS(0.4nM)を有するアッセイ緩衝液、25μLのオキソトレモリン(EC90)およびGDP(3μM)、25μLの希釈試験化合物ならびに25μLの、hM2受容体を発現するCHO細胞膜。次にアッセイプレートを37℃にて60分間インキュベートする。アッセイプレートは、PerkinElmer 96ウェル収集装置を使用して、1%BSA−前処理GF/Bフィルタで濾過される。プレートは氷冷洗浄緩衝液で3×3秒すすがれ、次に空気または真空乾燥される。Microscint−20シンチレーション液(50μL)が各ウェルに添加され、各プレートは密封されて、トップカウンター(PerkinElmer)で放射能カウントされる。データは、非線形回帰分析によって、1部位競合等式を使用してGraphPad Prism Software package(GraphPad Software,Inc.,San Diego,CA)を用いて分析される。Cheng−Prusoffの等式を使用して、試験化合物の濃度応答曲線のIC50値およびアッセイにおけるオキソトレモリン濃度をそれぞれKDおよび[L]リガンド濃度として用いて、Kiを計算する。
【0139】
本アッセイにおいて、より低いKi値は、試験化合物が試験された受容体に対してより高い機能的活性を有することを指摘している。式Iの化合物は、本アッセイまたは類似のアッセイで試験されたときに、hM2受容体を発現するCHO−K1細胞におけるオキソトレモリン刺激[35S]GTPγS−結合の遮断のために、約5nM未満のKi値を有することが見出された。
【0140】
FLIPRアッセイを介したアゴニスト媒介カルシウム放出の遮断
Gqタンパク質に結合するムスカリン受容体サブタイプ(M1、M3およびM5受容体)は、受容体へのアゴニスト結合時にホスホリパーゼC(PLC)経路を活性化する。結果として、活性化されたPLCはホスファチルイノシトール2リン酸(PIP2)をジアシルグリセロール(DAG)およびホスファチジル−1,4,5−3リン酸(IP3)に加水分解して、これが次に細胞内貯蔵、すなわち小胞体および筋小胞体からカルシウム放出を発生する。FLIPR(Molecular Devices,Sunnyvale,CA)アッセイは、遊離カルシウムを結合するときに蛍光を発するカルシウム感受性染料(Fluo−4AM,Molecular Probes,Eugene,OR)を使用することにより、細胞内カルシウムのこのような増加を利用する。本蛍光イベントは、ヒトM1およびM3、ならびにチンパンジーM5受容体でクローン化された細胞の単層からの蛍光の変化を検出するFLIPRによってリアルタイムで測定される。アンタゴニスト効力は、アンタゴニストが細胞内カルシウムのアゴニスト媒介上昇を阻害する能力によって決定することができる。
【0141】
FLIPRカルシウム刺激アッセイでは、hM1、hM3およびcM5受容体を安定発現するCHO細胞が96ウェルFLIPRプレートに、アッセイが行われる前の晩に播種される。播種された細胞は、Cellwash(MTX Labsystems,Inc.)によりFLIPR緩衝液(カルシウムおよびマグネシウムを含まないHBSS中の10mM HEPES、pH7.4、2mM塩化カルシウム、2.5mMプロベネシド)を用いて2回洗浄されて、成長培地が除去され、50μL/ウェルのFLIPR緩衝液が残る。細胞は次に、50μL/ウェルの4μM FLUO−4AM(2×溶液が作製された)によって37℃、5%二酸化炭素で40分間インキュベートされる。染料インキュベーション期間の後、細胞はFLIPR緩衝液で2回洗浄され、50μL/ウェルの最終体積が残る。
【0142】
アンタゴニスト効力を決定するために、オキソトレモリンの細胞内Ca2+放出の第1の用量依存性刺激が最初に決定されるので、EC90濃度でのオキソトレモリン刺激に対するアンタゴニスト効力を後に測定することができる。細胞は最初に化合物希釈緩衝液によって20分間インキュベートされ、FLIPRによって行われるアゴニスト添加が続く。オキソトレモリンのEC90値は、式ECF=((F/100−F)^1/H)*EC50と併せて、下のFLIPR測定およびデータ整理の項で詳説される方法に従って生成される。オキソトレモリンのEC90濃度がアンタゴニスト阻害アッセイプレートの各ウェルに添加されるように、3×ECFのオキソトレモリン濃度が刺激プレート中で調製される。
【0143】
FLIPRに使用されるパラメータは:0.4秒の照射期間、0.5ワットのレーザー強度、488nmの励起波長、および550nmの発光波長である。ベースラインは、アゴニスト添加前の10秒間にわたって蛍光変化を測定することによって決定される。アゴニスト刺激の後、FLIPRは蛍光変化を0.5〜1秒ごとに1.5分間にわたって連続測定して、最大蛍光変化を捕捉する。
【0144】
蛍光変化は、各ウェルについて最大蛍光からベースライン蛍光を引いたものとして表現される。生データは、S字状用量応答の組込みモデルを使用するGraphPad Prism(GraphPad Software,Inc.,San Diego,CA)を用いた非線形回帰により、薬物濃度の対数に対して分析される。アンタゴニストKi値は、KDとしてのオキソトレモリンEC50値およびCheng−Prusoffの等式(Cheng&Prusoff,1973)によるリガンド濃度のオキソトレモリンEC90を使用してPrismによって決定される。
【0145】
本アッセイにおいて、より低いKi値は、試験化合物が試験された受容体に対してより高い機能的活性を有することを指摘している。式Iの化合物は、本アッセイまたは類似のアッセイで試験されたときに、hM3受容体を安定に発現するCHO細胞におけるアゴニスト媒介カルシウム放出の遮断のために、約5nM未満のKi値を有することが見出された。
【0146】
アッセイ3
ラット・アイントホーフェン・アッセイ
本インビボアッセイが使用されて、ムスカリン受容体アンタゴニスト活性を呈する試験化合物の気管支保護効果が影響評価される。すべての試験化合物は滅菌水で希釈されて、吸入経路(IH)を介して投薬される。ラット(スプラーグ−ドーリー、オス、250〜350g)、LC Starネブライザセットから発生され、気体の混合物(5%CO2/95%周囲空気)によって操縦されたエアゾールに曝露される。各試験化合物溶液は、マウス6匹を保持することができるパイ形状投薬チャンバにおいて10分間の期間にわたって噴霧される。化合物の吸入後、所定の時点でアイントホーフェンアッセイが行われる。
【0147】
肺評価開始の30分前に、動物をイナクチン(チオブタバルビタール、120mg/kgIP)を介して麻酔する。頸静脈に生理食塩水を充填したポリエチレンカテーテル(PE−50)が挿入され、MChを注入するために使用される。次に気管が切開され、14G針によってカニューレ処置されて、肺評価の間のラットの人工呼吸に使用される。手術がいったん完了したら、2.5mlの量を超えない1ml/100g体重の心拍出量、および1分当り90拍動の速度に設定されたピストン呼吸装置を使用して、ラットに人工呼吸させる。
【0148】
各呼吸によって発生する圧力の変化が測定される。ベースライン値が少なくとも2.5分間にわたって収集され、次にラットは2倍ずつ漸進的に増加される気管支収縮剤MCh(5、10、20、40および80μg/ml)を用いて、非累積的に攻撃される。MChは注射器ポンプから2mL/kg/分の速度で2.5分間注入される。動物は試験完了時に安楽死させる。
【0149】
処置動物の人工呼吸圧の変化(cm H2O)は、対照動物に関係するMCh応答の阻害%として表される。本アッセイにおいて、より高い阻害%は、試験化合物が気管支保護効果を有することを指摘している。式Iの化合物は、本アッセイにおいて100μg/mlの用量で試験されるとき、35%を超える阻害、おそらく70%を超える阻害、およびなおさらにおそらく90%を超える阻害を呈することが期待される。
【0150】
1.5時間ID50の決定
標準ムスカリンアンタゴニストは、ラット・アイントホーフェン・アッセイで投薬1.5時間後に評価された。試験された5つの標準の効力(ID50)の順序は:イプラトロピウム(4.4μg/ml)>チオトロピウム(6μg/ml)>デス−メチル−チオトロピウム(12μg/ml)>グリコピロレート(15μg/ml)>LAS−34237(24μg/ml)として決定された。試験化合物の効力は、投薬1.5時間後に同様に決定される。
【0151】
6および24時間ID50の決定
標準のチオトロピウムおよびイプラトロピウムも、ラット・アイントホーフェン・アッセイで投薬24時間および/または6時間後に評価された。イプラトロピウム(10および30μg/ml)は投薬6時間後には、その1.5時間の効力と比較して3倍弱かった。本時点(6時間)で観察された活性の消失は、診療所での比較的短い作用期間と一致している。チオトロピウムは、ピーク気管支保護による緩慢な効果の開始が投薬6時間後に達成されることを示した。その6時間および24時間の効力値は、相互から有意に異なっておらず、その1.5時間の効力と比較して約2倍強力であった。試験化合物の作用の開始、ならびに6および24時間の効力値は同様に決定される。
【0152】
アッセイ4
ラット唾液分泌抑制アッセイ
ラット(スプラーグ−ドーリー、オス、250〜350g)は、アッセイ3で記載されたように、投薬、麻酔およびカニューレ処置される。所定の時点および手術後に、動物は下りスロープに仰向けに頭を20度傾斜させて配置される。事前に秤量されたガーゼパッドが動物の口内に挿入され、ムスカリンアゴニストのピロカルピン(PILO)(3mg/kg、iv.)が投与される。PILO後の10分間に産生された唾液は、PILO前後のガーゼパッドの重量を決定することによって、重量測定法で測定される。唾液分泌抑制効果は、対照動物に関係する唾液分泌の阻害%として表される。
1、6および24時間ID50の決定
ラット唾液分泌抑制アッセイは、試験化合物の全身曝露を影響評価して、肺選択指数(LSI)を計算するために開発された。標準のチオトロピウムは、本モデルにおいて投薬1時間、6時間、および24時間後に評価された。チオトロピウムは、投薬6時間後にピロカルピン誘発唾液分泌を最も強力に阻害することが見出された。本発見は、アイントホーフェンアッセイで観察されたピーク効果と一致している。
【0153】
本モデルは、Rechter,“Estimation of anticholinergic drug effects in mice by antagonism against pilocarpine−induced salivation”Ata Pharmacol Toxicol 24:243−254(1996)に記載された手順の修正版である。ビヒクルで処置された動物の唾液の平均重量は、各処置前時間に計算され、各用量の対応する処置前時間における唾液分布の阻害%が算出される。
【0154】
本アッセイで試験される本発明の例示的な化合物は、100μg/ml未満のID50値(24時間にて測定)を呈することが期待され、いくつかの化合物は30μg/ml未満の、いくつかは20μg/ml未満の、およびいくつかのは15μg/ml未満のID50値を呈することが期待される。
【0155】
唾液分泌抑制ID50の気管支保護ID50に対する比を使用して、試験化合物の見かけの肺選択指数が算出される。概して、約5を超える見かけの肺選択指数を有する化合物が好ましい。
【0156】
本発明はその詳細な態様または実施形態への参照により記載されてきたが、本発明の真の精神および範囲から逸脱することなく、各種の変更が行われることができるか、または均等物が置換されることができることが当業者によって理解されるであろう。加えて、本明細書で引用されたすべての刊行物、特許および特許出願は、適用可能な特許法および規則によって許容される範囲で、各文書が参照により本明細書に個別に組み入れられているのと同じ範囲まで、その全体が参照により本明細書に組み入れられている。
【特許請求の範囲】
【請求項1】
6.6±0.1、13.1±0.1、18.6±0.1、19.7±0.1、および20.2±0.1の2θ値に回折ピークを備える粉末X線回折を特徴とする、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基。
【請求項2】
8.8±0.1、10.1±0.1、11.4±0.1、11.6±0.1、14.8±0.1、15.2±0.1、16.1±0.1、16.4±0.1、16.9±0.1、17.5±0.1、18.2±0.1、19.3±0.1、19.9±0.1、20.8±0.1、21.1±0.1、21.7±0.1、および22.3±0.1から選択される2θ値に5つ以上の追加の回折ピークを有することをさらに特徴とする、請求項1に記載の結晶性遊離塩基。
【請求項3】
6.6±0.1、11.4±0.1、13.1±0.1、16.1±0.1、17.5±0.1、18.2±0.1、18.6±0.1、19.3±0.1、19.7±0.1、19.9±0.1、20.2±0.1、20.8±0.1、21.1±0.1、21.7±0.1、および22.3±0.1から選択される2θ値に回折ピークを備える粉末X線回折パターンを特徴とする、請求項2に記載の結晶性化合物。
【請求項4】
ピーク位置が図1に示されたパターンのピーク位置と実質的に一致する粉末X線回折パターンをさらに特徴とする、請求項2に記載の結晶性化合物。
【請求項5】
約125℃の融点を示す示差走査熱量測定サーモグラムをさらに特徴とする、請求項2に記載の化合物。
【請求項6】
図4に示されたものと実質的に一致する示差走査熱量測定サーモグラムをさらに特徴とする、請求項2に記載の化合物。
【請求項7】
10.6±0.1、15.0±0.1、16.0±0.1、17.3±0.1、17.7±0.1、20.9±0.1、21.4±0.1、22.6±0.1、24.6±0.1、および27.8±0.1から選択される2θ値に5つ以上の追加の回折ピークを有することをさらに特徴とする、請求項1に記載の結晶性遊離塩基。
【請求項8】
6.6±0.1、13.1±0.1、15.0±0.1、17.3±0.1、17.7±0.1、18.6±0.1、19.7±0.1、20.2±0.1、20.9±0.1、21.4±0.1、および22.6±0.1から選択される2θ値に回折ピークを備える粉末X線回折パターンを特徴とする、請求項7に記載の結晶性化合物。
【請求項9】
ピーク位置が図2に示されたパターンのピーク位置と実質的に一致する粉末X線回折パターンをさらに特徴とする、請求項7に記載の結晶性化合物。
【請求項10】
約119℃の融点を示す示差走査熱量測定サーモグラムをさらに特徴とする、請求項7に記載の化合物。
【請求項11】
図5に示されたものと実質的に一致する示差走査熱量測定サーモグラムをさらに特徴とする、請求項7に記載の化合物。
【請求項12】
製薬的に許容され得る担体および請求項1〜11のいずれか1項に記載の化合物を含む製薬組成物。
【請求項13】
β2アドレナリン受容体アゴニスト、ステロイド性抗炎症剤、ホスホジエステラーゼ−4阻害剤、およびその組合せから選択される薬剤をさらに含み;前記結晶形および前記薬剤が共にまたは別々に製剤化される、請求項12に記載の組成物。
【請求項14】
β2アドレナリン受容体アゴニストおよびステロイド性抗炎症剤を含む、請求項13に記載の組成物。
【請求項15】
微粉化形の、請求項1〜11のいずれか1項に記載の化合物。
【請求項16】
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルをアセトニトリルと接触させることを含む、請求項2に記載の結晶性遊離塩基III形を調製するプロセス。
【請求項17】
a)結晶性遊離塩基III形の種晶を形成すること;b)前記結晶性遊離塩基III形をアセトニトリルに溶解させて溶液を形成すること;c)および前記種晶を前記溶液に添加すること;を含む、請求項7に記載の結晶性遊離塩基IV形を調製するプロセス。
【請求項18】
請求項1〜11のいずれか1項に記載の化合物を形成することを含む、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルを精製するプロセス。
【請求項19】
療法での使用のための、請求項1〜11のいずれか1項に記載の化合物。
【請求項20】
慢性閉塞性肺疾患もしくは喘息の処置での使用のための、または気管支拡張を生じるための、請求項19に記載の化合物。
【請求項21】
慢性閉塞性肺疾患もしくは喘息の処置のための、または気管支拡張を生じるための、医薬の製造のための、請求項1〜11のいずれか1項に記載の化合物の使用。
【請求項1】
6.6±0.1、13.1±0.1、18.6±0.1、19.7±0.1、および20.2±0.1の2θ値に回折ピークを備える粉末X線回折を特徴とする、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルの結晶性遊離塩基。
【請求項2】
8.8±0.1、10.1±0.1、11.4±0.1、11.6±0.1、14.8±0.1、15.2±0.1、16.1±0.1、16.4±0.1、16.9±0.1、17.5±0.1、18.2±0.1、19.3±0.1、19.9±0.1、20.8±0.1、21.1±0.1、21.7±0.1、および22.3±0.1から選択される2θ値に5つ以上の追加の回折ピークを有することをさらに特徴とする、請求項1に記載の結晶性遊離塩基。
【請求項3】
6.6±0.1、11.4±0.1、13.1±0.1、16.1±0.1、17.5±0.1、18.2±0.1、18.6±0.1、19.3±0.1、19.7±0.1、19.9±0.1、20.2±0.1、20.8±0.1、21.1±0.1、21.7±0.1、および22.3±0.1から選択される2θ値に回折ピークを備える粉末X線回折パターンを特徴とする、請求項2に記載の結晶性化合物。
【請求項4】
ピーク位置が図1に示されたパターンのピーク位置と実質的に一致する粉末X線回折パターンをさらに特徴とする、請求項2に記載の結晶性化合物。
【請求項5】
約125℃の融点を示す示差走査熱量測定サーモグラムをさらに特徴とする、請求項2に記載の化合物。
【請求項6】
図4に示されたものと実質的に一致する示差走査熱量測定サーモグラムをさらに特徴とする、請求項2に記載の化合物。
【請求項7】
10.6±0.1、15.0±0.1、16.0±0.1、17.3±0.1、17.7±0.1、20.9±0.1、21.4±0.1、22.6±0.1、24.6±0.1、および27.8±0.1から選択される2θ値に5つ以上の追加の回折ピークを有することをさらに特徴とする、請求項1に記載の結晶性遊離塩基。
【請求項8】
6.6±0.1、13.1±0.1、15.0±0.1、17.3±0.1、17.7±0.1、18.6±0.1、19.7±0.1、20.2±0.1、20.9±0.1、21.4±0.1、および22.6±0.1から選択される2θ値に回折ピークを備える粉末X線回折パターンを特徴とする、請求項7に記載の結晶性化合物。
【請求項9】
ピーク位置が図2に示されたパターンのピーク位置と実質的に一致する粉末X線回折パターンをさらに特徴とする、請求項7に記載の結晶性化合物。
【請求項10】
約119℃の融点を示す示差走査熱量測定サーモグラムをさらに特徴とする、請求項7に記載の化合物。
【請求項11】
図5に示されたものと実質的に一致する示差走査熱量測定サーモグラムをさらに特徴とする、請求項7に記載の化合物。
【請求項12】
製薬的に許容され得る担体および請求項1〜11のいずれか1項に記載の化合物を含む製薬組成物。
【請求項13】
β2アドレナリン受容体アゴニスト、ステロイド性抗炎症剤、ホスホジエステラーゼ−4阻害剤、およびその組合せから選択される薬剤をさらに含み;前記結晶形および前記薬剤が共にまたは別々に製剤化される、請求項12に記載の組成物。
【請求項14】
β2アドレナリン受容体アゴニストおよびステロイド性抗炎症剤を含む、請求項13に記載の組成物。
【請求項15】
微粉化形の、請求項1〜11のいずれか1項に記載の化合物。
【請求項16】
ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルをアセトニトリルと接触させることを含む、請求項2に記載の結晶性遊離塩基III形を調製するプロセス。
【請求項17】
a)結晶性遊離塩基III形の種晶を形成すること;b)前記結晶性遊離塩基III形をアセトニトリルに溶解させて溶液を形成すること;c)および前記種晶を前記溶液に添加すること;を含む、請求項7に記載の結晶性遊離塩基IV形を調製するプロセス。
【請求項18】
請求項1〜11のいずれか1項に記載の化合物を形成することを含む、ビフェニル−2−イルカルバミン酸1−(2−{[4−(4−カルバモイルピペリジン−1−イルメチル)ベンゾイル]メチルアミノ}エチル)ピペリジン−4−イルエステルを精製するプロセス。
【請求項19】
療法での使用のための、請求項1〜11のいずれか1項に記載の化合物。
【請求項20】
慢性閉塞性肺疾患もしくは喘息の処置での使用のための、または気管支拡張を生じるための、請求項19に記載の化合物。
【請求項21】
慢性閉塞性肺疾患もしくは喘息の処置のための、または気管支拡張を生じるための、医薬の製造のための、請求項1〜11のいずれか1項に記載の化合物の使用。
【図1】

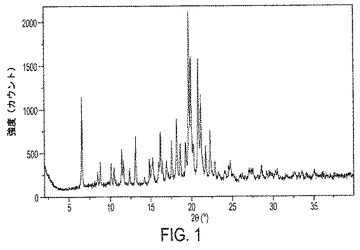
【図2】

【図3】
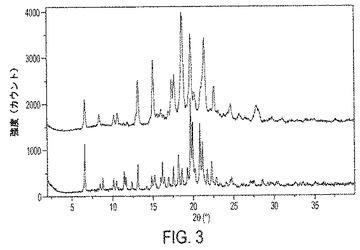
【図4】
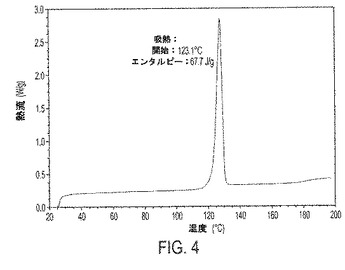
【図5】
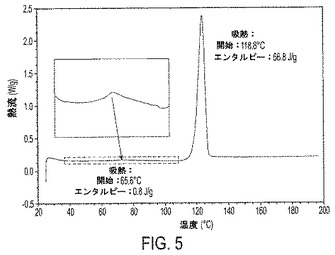
【図6】
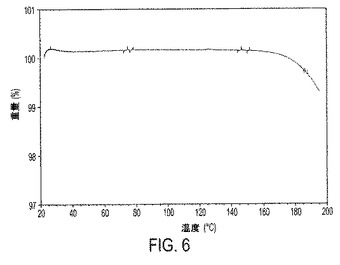
【図7】
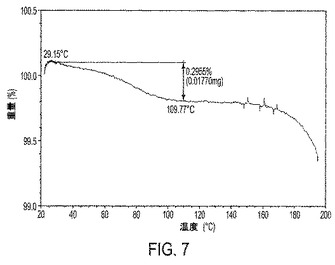
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2012−533550(P2012−533550A)
【公表日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願番号】特願2012−520734(P2012−520734)
【出願日】平成22年7月14日(2010.7.14)
【国際出願番号】PCT/US2010/041903
【国際公開番号】WO2011/008809
【国際公開日】平成23年1月20日(2011.1.20)
【出願人】(500154711)セラヴァンス, インコーポレーテッド (129)
【Fターム(参考)】
【公表日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願日】平成22年7月14日(2010.7.14)
【国際出願番号】PCT/US2010/041903
【国際公開番号】WO2011/008809
【国際公開日】平成23年1月20日(2011.1.20)
【出願人】(500154711)セラヴァンス, インコーポレーテッド (129)
【Fターム(参考)】
[ Back to top ]