
ビフェニレン誘導体、その用途、及びその製造方法
【課題】優れた耐酸化性を有し、塗布法による半導体活性相形成が可能な、ビフェニレン誘導体、それを用いた耐酸化性有機半導体材料並びにそれからなる有機薄膜、発光材料、及び該ビフェニレン誘導体を簡便に経済的に製造する方法の提供。
【解決手段】式(1)で示されることを特徴とするビフェニレン誘導体。

(ここで、置換基R1〜R8は同一又は異なって、水素原子、フッ素原子、炭素数2〜20のアルキル基、炭素数2〜20のアルキニル基、炭素数2〜30のアルケニル基、又は炭素数4〜20のアリール基を示し、mは1又は2であり、nは0〜2の整数を示す。但し、R1及びR2は同時に水素原子であることはない。)
【解決手段】式(1)で示されることを特徴とするビフェニレン誘導体。
(ここで、置換基R1〜R8は同一又は異なって、水素原子、フッ素原子、炭素数2〜20のアルキル基、炭素数2〜20のアルキニル基、炭素数2〜30のアルケニル基、又は炭素数4〜20のアリール基を示し、mは1又は2であり、nは0〜2の整数を示す。但し、R1及びR2は同時に水素原子であることはない。)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、有機半導体等の電子材料への展開が可能なビフェニレン誘導体、その用途、及びそれらの製造方法に関する。
【背景技術】
【0002】
有機薄膜トランジスタに代表される有機半導体デバイスは、省エネルギー、低コスト、及びフレキシブルといった無機半導体デバイスにはない特徴を有することから近年注目されるようになった。有機薄膜トランジスタは有機半導体活性相、基板、絶縁相、電極等数種類の材料から構成されるが、中でも電荷のキャリアー移動を担う有機半導体活性相は該デバイスの中心的な役割を有している。この有機半導体活性相を構成する有機材料のキャリアー移動能により半導体デバイス性能が左右される。
【0003】
有機半導体活性相を作製する方法としては一般的に、高温真空下、有機材料を気化させて実施する真空蒸着法、及び有機材料を適当な溶媒に溶解させその溶液を塗布する塗布法が知られている。塗布法は高温高真空条件を用いることなく、印刷技術を用いても実施することができるため、デバイス作製の製造コストを大幅に削減することができることから、経済的に好ましいプロセスである。しかし、従来、有機半導体材料として高性能な材料ほど塗布での活性相形成が困難になるという問題があった。
【0004】
例えば、分子長軸を有するペンタセン等の結晶性材料はアモルファスシリコン並みの高いキャリアー移動度を有し、優れた半導体デバイス特性を発現することが報告されている(例えば、非特許文献1参照)。しかし、ペンタセンはその強い凝集性のため溶解性が低く、一般的には経済的な塗布法を適用することができない。また、ペンタセン等のポリアセンを溶解させ塗布法でデバイスを製造する試みも報告されているが(例えば、特許文献1参照)、元来難溶性のポリアセン類を溶解させるためには、高温加熱等の条件が必要とされ、さらにペンタセンの溶液は極めて容易に空気酸化されることから、塗布法の適用はプロセス的、経済的に困難を伴うものであった。また、ポリ−(3−ヘキシルチオフェン)等の自己組織化材料は溶媒に可溶であり、塗布によるデバイス作製が報告されているが、キャリアー移動度が結晶性低分子化合物より1桁低いことから(例えば、非特許文献2参照)、得られた有機半導体デバイスの特性が低いという問題があった。
【0005】
このような中で、ビフェニレンは剛直な共役縮環化合物であり、有機半導体材料として期待できる化合物である。しかし、これまでに報告されているビフェニレン誘導体は、有機半導体材料としては満足の行くものではなかった。
【0006】
例えば多置換多環芳香族化合物およびその製造方法には剛直なビフェニレン構造を有する化合物が記載されている(例えば、特許文献2〜4参照)が、分子長軸方向以外にも置換基を有することから結晶性が低下し、塗布法により均一な膜が得られず半導体材料としては適していない。また、カチオン重合性樹脂のための三元光開始剤システム(例えば、特許文献5参照)にはビフェニレン誘導体の構造が記載されているが、分子長軸が短いため半導体材料としては適していない。
【0007】
【非特許文献1】「ジャーナル オブ アプライドフィジックス」、(米国)、2002年、92巻、5259−5263頁
【非特許文献2】「サイエンス」、(米国)、1998年、280巻、1741−1744頁
【特許文献1】WO2003/016599パンフレット
【特許文献2】特開2004−256497号公報
【特許文献3】特開2006−156980号公報
【特許文献4】特開2006−328006号公報
【特許文献5】特表2005−523348号公報
【発明の開示】
【発明が解決しようとする課題】
【0008】
そこで、本発明は上記の従来技術が有する問題点に鑑み、優れた耐酸化性を有し、塗布法による半導体活性相形成が可能なビフェニレン誘導体、それを用いた耐酸化性有機半導体材料並びにそれからなる有機薄膜、発光材料及び該ビフェニレン誘導体を簡便に経済的に製造する方法を提供することを目的とする。さらに本発明は、該ビフェニレン誘導体の前駆化合物であるジハロビフェニル誘導体にも関するものである。
【課題を解決するための手段】
【0009】
本発明者らは上記課題を解決するため鋭意検討の結果、新規なビフェニレン誘導体及び前駆体であるジハロビフェニル誘導体を見出した。加えて、該ビフェニレン誘導体からなる耐酸化性有機半導体材料及びそれからなる有機薄膜、発光材料を見出した。さらに、該ビフェニレン誘導体を製造するに好適な製造方法を見出し、本発明を完成するに到った。
【0010】
以下に本発明を詳細に説明する。
(ビフェニレン誘導体)
本発明のビフェニレン誘導体は下記一般式(1)で示される。
【0011】
【化1】
(ここで、置換基R1〜R8は同一又は異なって、水素原子、フッ素原子、炭素数2〜20のアルキル基、炭素数2〜20のアルキニル基、炭素数2〜30のアルケニル基、又は炭素数4〜20のアリール基を示し、mは1又は2であり、nは0〜2の整数を示す。但し、R1及びR2は同時に水素原子であることはない。)
本発明の一般式(1)の置換基について、述べる。
【0012】
置換基R1〜R8における、炭素数2〜20のアルキル基は特に限定はなく、例えばプロピル基、ブチル基、tert−ブチル基、ペンチル基、ネオペンチル基、ヘキシル基、オクチル基、イソオクチル基、ノニル基、デシル基、ドデシル基、ペンタデシル基、オクタデシル基、2−エチルヘキシル基等のアルキル基;トリフルオロメチル基、ペンタフルオロエチル基、パーフルオロオクチル基、パーフルオロデシル基、パーフルオロドデシル基、パーフルオロオクタデシル基、パーフルオロシクロヘキシル基、パーフルオロシクロオクチル基等のパーフルオロアルキル基;トリフルオロエチル基、ペンタデカフルオロオクチル基、オクタデカフルオロデシル基、2−エチルパーフルオロヘキシル基等の一部の水素がフッ素に置換されたハロゲン化アルキル基を挙げることができ、好ましくは炭素数5〜20のアルキル基であり、さらに好ましくはオクチル基、ドデシル基、オクタデシル基、パーフルオロオクチル基、パーフルオロドデシル基、パーフルオロオクタデシル基、特に好ましくはドデシル基、パーフルオロドデシル基である。
【0013】
置換基R1〜R8における、炭素数2〜20のアルキニル基は特に限定はなく、例えばエチニル基、メチルエチニル基、イソプロピルエチニル基、tert−ブチルエチニル基、(オクチル)エチニル基、(デシル)エチニル基、(ドデシル)エチニル基、(オクタデシル)エチニル基、(トリフルオロメチル)エチニル基、(パーフルオロオクチル)エチニル基、(パーフルオロデシル)エチニル基、(パーフルオロドデシル)エチニル基、フェニルエチニル基、{p−(オクチル)フェニル}エチニル基、ナフチルエチニル基、アントラセニルエチニル基、ベンジルエチニル基、パーフルオロフェニルエチニル基、{p−(トリフルオロメチル)フェニル}エチニル基、{p−(パーフルオロオクチル)フェニル}エチニル基等を挙げることができ、好ましくは(オクチル)エチニル基、(デシル)エチニル基、(ドデシル)エチニル基、(パーフルオロオクチル)エチニル基、(パーフルオロデシル)エチニル基、(パーフルオロドデシル)エチニル基、フェニルエチニル基等である。
【0014】
置換基R1〜R8における、炭素数2〜30のアルケニル基は特に限定はなく、例えばエテニル基、メチルエテニル基、イソプロピルエテニル基、tert−ブチルエテニル基、(オクチル)エテニル基、(デシル)エテニル基、(ドデシル)エテニル基、(トリフルオロメチル)エテニル基、フェニルエテニル基、{p−(ヘキシル)フェニル}エテニル基、{p−(オクチル)フェニル}エテニル基、2−フェニル−1,2−ジフルオロエテニル基、2−フェニル−1,2−ジメチルエテニル基、ジフェニルエテニル基、トリフェニルエテニル基、ナフチルエテニル基、アントラセニルエテニル基、ベンジルエテニル基、フェニル(メチル)エテニル基、(パーフルオロフェニル)エテニル基、{p−(トリフルオロメチル)フェニル}エテニル基、(パーフルオロオクチル)エテニル基、(パーフルオロデシル)エテニル基、(パーフルオロドデシル)エテニル基、{5−(ヘキシル)チエニル−2−}エテニル基、{5−(パーフルオロヘキシル)チエニル−2−}エテニル基等を挙げることができ、好ましくは(オクチル)エテニル基、(デシル)エテニル基、(ドデシル)エテニル基、(パーフルオロオクチル)エテニル基、(パーフルオロデシル)エテニル基、(パーフルオロドデシル)エテニル基、{5−(ヘキシル)チエニル−2−}エテニル基、{5−(パーフルオロヘキシル)チエニル−2−}エテニル基、フェニルエテニル基等である。なお、該炭素数2〜30のアルケニル基はトランス体及びシス体の何れであってもよく、またそれらの任意の割合の混合物であってもよい。
【0015】
置換基R1〜R8における、炭素数4〜20のアリール基は特に限定はなく、例えばフェニル基、p−トリル基、p−(オクチル)フェニル基、m−(オクチル)フェニル基、p−(デシル)フェニル基、p−フルオロフェニル基、ペンタフルオロフェニル基、p−(トリフルオロメチル)フェニル基、p−(パーフルオロオクチル)フェニル基、m−(パーフルオロオクチル)フェニル基、p−(トリフルオロメチル)テトラフルオロフェニル基、p−メトキシフェニル基、p−フェノキシフェニル基、2−チエニル基、5−(ヘキシル)−2−チエニル基、5−(パーフルオロヘキシル)−2−チエニル基、ビフェニル基、パーフルオロビフェニル基、1−ナフチル基、2−ナフチル基、1−パーフルオロナフチル基、2−フルオレニル基、9,9−ジメチル−2−フルオレニル基、9−アントラセニル基等を挙げることができ、好ましくは、フェニル基、p−(オクチル)フェニル基、p−(トリフルオロメチル)フェニル基、p−(パーフルオロオクチル)フェニル基である。
【0016】
本発明の一般式(1)で示されるビフェニレン誘導体の置換基R1〜R8の置換様式として、R1〜R4が、同一又は異なって、水素原子、フッ素原子、炭素数2〜20のアルキル基、炭素数2〜20のアルキニル基、炭素数2〜30のアルケニル基、及び炭素数4〜20のアリール基からなる群から選ばれる少なくとも一種以上の基であり、且つR5〜R8が、同一又は異なって、水素原子、フッ素原子、及び炭素数4〜20のアリール基からなる群から選ばれる少なくとも一種以上の基であることが好ましい。
【0017】
さらに置換基R1〜R4が、同一又は異なって、水素原子、フッ素原子、炭素数2〜20のアルキル基、炭素数2〜20のアルキニル基、及び炭素数4〜20のアリール基からなる群から選ばれる少なくとも一種以上の基であり、且つR5〜R8が、同一又は異なって、水素原子及び炭素数4〜20のアリール基からなる群から選ばれる少なくとも一種以上の基であることがより好ましい。
【0018】
記号mは1又は2であり、好ましくは1である。また、記号nは0〜2の整数を示し、好ましくは1である。そして、特に好ましい記号m、nとしては、いずれも1である。
【0019】
これらの中でも本発明の一般式(1)で示されるビフェニレン誘導体は、該ビフェニレン誘導体及び該ビフェニレン誘導体を含む耐酸化性有機半導体材料及びその有機薄膜が、高い耐酸化性及びキャリアー移動度を発現することから、以下の化合物が好ましく、
【0020】
【化2】
【0021】
【化3】
【0022】
【化4】
【0023】
【化5】
【0024】
【化6】
【0025】
【化7】
【0026】
【化8】
【0027】
【化9】
特に好ましくは
【0028】
【化10】
【0029】
【化11】
である。
(ジハロビフェニル誘導体)
次に、本発明の一般式(1)で示されるビフェニレン誘導体の原料として用いられる下記一般式(2)で示されるジハロビフェニル誘導体について述べる。
【0030】
【化12】
(ここで、置換基X1及びX2は臭素原子、ヨウ素原子又は塩素原子を示し、置換基R1〜R8、並びに記号m及びnは、一般式(1)で示される置換基並びに記号と同意義を示す。)
一般式(2)におけるX1及びX2は臭素原子、ヨウ素原子又は塩素原子であり、その中でも好ましくは臭素原子又はヨウ素原子であり、より好ましくは臭素原子である。
【0031】
本発明の一般式(2)における置換基R1及びR2の好ましい例は、フッ素原子、炭素数2〜20のアルキル基、炭素数2〜20のアルキニル基であり、さらに好ましい例はフッ素原子、炭素数5〜20のアルキル基である。
【0032】
置換基R3及びR4の好ましい例は、水素原子、フッ素原子、炭素数2〜20のアルキル基、炭素数2〜20のアルキニル基であり、さらに好ましい例はフッ素原子、炭素数5〜20のアルキル基である。
【0033】
置換基R5〜R8の特に好ましい例は、水素原子、炭素数2〜20のアルキル基、炭素数4〜20のアリール基であり、さらに好ましい例は水素原子である。
【0034】
本発明の一般式(2)で示されるジハロビフェニル誘導体としては、以下の化合物が好ましく、
【0035】
【化13】
【0036】
【化14】
【0037】
【化15】
【0038】
【化16】
【0039】
【化17】
【0040】
【化18】
【0041】
【化19】
【0042】
【化20】
特に好ましくは
【0043】
【化21】
【0044】
【化22】
【0045】
【化23】
である。
(ビフェニレン誘導体の製造方法)
次に、本発明の一般式(1)で示されるビフェニレン誘導体の製造方法について述べる。
【0046】
本発明の一般式(1)で示されるビフェニレン誘導体は一般式(2)で示されるジハロビフェニル誘導体を、銅又は銅化合物と反応させることで製造することができる。
【0047】
該反応で用いられる銅又は銅化合物は特に限定はなく、ヨウ素又は臭素と反応するものであれば良く、例えば銅、塩化銅(I)、臭化銅(I)、ヨウ化銅(I)、塩化銅(II)、臭化銅(II)、ヨウ化銅(II)を挙げることができ、好ましくは銅、塩化銅(II)である。また、この銅は亜鉛及び/又はスズとの合金であっても何ら差し支えなく使用することができる。なお、係る銅及び/又は銅合金の形状としては粉体状が好ましい。
【0048】
この銅又は銅化合物との反応は溶媒を用いて、若しくは用いないで実施することができる。溶媒を用いる場合、その溶媒例としては、例えばN,N−ジメチルホルムアミド、N−メチルピロリドン、アセトニトリル、ジメチルスルホキサイド等の極性溶媒を挙げることができる。係る銅又は銅化合物との反応において、用いる銅又は銅化合物の量は一般式(2)で示されるジハロビフェニル誘導体1当量に対し、1〜50当量が好ましく、特に好ましくは5〜30当量であり、反応温度は30〜250℃が好ましく、特に好ましくは50〜220℃であり、反応時間は1〜120分が好ましく、特に好ましくは3〜80分である。
【0049】
反応は、好ましくは窒素又はアルゴン等の不活性雰囲気下で実施する。得られた、一般式(1)で示されるビフェニレン誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0050】
本発明の一般式(1)で示されるビフェニレン誘導体の、さらに好ましい製造方法について述べる。
【0051】
さらに好ましい製造方法としては、一般式(2)で示されるジハロビフェニル誘導体をジリチオ化及び/又はジグリニャール化した後、銅化合物と反応させて製造する方法を挙げることができる。なお、ここでジリチオ化とは、一般式(2)における2個のハロゲンX1、X2をそれぞれリチウムに置換することを意味し、ジグリニャール化とは、一般式(2)における2個のハロゲンX1及びX2をそれぞれハロゲン化マグネシウムに置換することを意味する。
【0052】
一般式(2)で示されるジハロビフェニル誘導体をジリチオ化する場合、用いるリチオ化剤は、一般式(2)におけるハロゲンX1及びX2をリチウムに置換することができるものである限り特に限定はなく、例えばn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、メチルリチウム、ヘキシルリチウム等のアルキルリチウム;フェニルリチウム、p−tert−ブチルフェニルリチウム、p−メトキシフェニルリチウム、p−フルオロフェニルリチウム等のアリールリチウム;リチウムジイソプロピルアミド、リチウムヘキサメチルジシラジド等のリチウムアミド;リチウムパウダー等のリチウム金属を挙げることができ、好ましくはアルキルリチウムであり、特に好ましくはn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウムである。
【0053】
該リチオ化剤の使用量は、一般式(2)で示されるジハロビフェニル誘導体1当量に対し、1.5〜4当量が好ましく、さらに好ましくは1.8〜3当量、特に好ましくは1.9〜2.6当量である。
【0054】
該ジリチオ化反応は、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテル、ジプロピルエーテル、ジイソプロピルエーテル、ジブチルエーテル、エチレングリコールジメチルエーテル等のジアルキルエーテル、テトラヒドロフラン(以下、THFと略す)、ジグライム、ジオキサン、トルエン、ペンタン、ヘキサン、シクロヘキサン等が挙げられ、好ましくはジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテル、ジプロピルエーテル、ジイソプロピルエーテル、ジブチルエーテル、エチレングリコールジメチルエーテル等のジアルキルエーテルであり、特に好ましくはジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテルである。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。
【0055】
該ジリチオ化反応温度は−80〜50℃が好ましく、特に好ましくは−30〜30℃である。反応時間は1〜240分が好ましく、特に好ましくは1〜90分である。なお、ジリチオ化反応の進行は、反応液の一部を取り出し、水で反応を停止させた後、薄層クロマトグラフィー、ガスクロマトグラフィーで分析することで監視することもできる。
【0056】
本発明の一般式(1)で示されるビフェニレン誘導体の製造方法では、ジアルキルエーテル中でジリチオ化を実施することで、ジリチオ化の反応温度を−30〜30℃に上げることができ、反応性の低い一般式(2)で示されるジハロビフェニル誘導体も効率良くジリチオ化できることを見出した。
【0057】
該ジリチオ化反応により生成したジリチウム塩は、次いで銅化合物と反応させる。係る銅化合物との反応は、前記リチオ化反応により生成したジリチウム塩を含む反応混合物に銅化合物を直接用いて反応させる方法、生成したジリチウム塩を一度単離した後、銅化合物と反応させる方法のいずれを用いてもよい。
【0058】
該ジリチウム塩と銅化合物との反応は、該ジリチウム塩に銅化合物を添加する方法、あるいは銅化合物に該ジリチウム塩を添加するいずれの方法を用いても実施することができる。
【0059】
ジリチウム塩と銅化合物との反応に用いられる銅化合物は特に限定はなく、例えば塩化銅(II)、臭化銅(II)、ヨウ化銅(II)、酢酸銅(II)、アセチルアセトナート銅(II)等の2価銅;塩化銅(I)、臭化銅(I)、ヨウ化銅(I)、酢酸銅(I)等の1価銅等を挙げることができ、好ましくは2価銅であり、特に好ましくは塩化銅(II)、臭化銅(II)である。生成ジリチウム塩と銅化合物との反応は好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばTHF、ジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテル、エチレングリコールジメチルエーテル、ジグライム、ジオキサン等が挙げられ、好ましくはTHF、ジエチルエーテルである。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。用いる銅化合物の量は、一般式(2)で示されるジハロビフェニル誘導体1当量に対し、0.9〜5当量が好ましく、特に好ましくは1.0〜3.5当量である。銅化合物との反応温度は−90〜50℃が好ましく、特に好ましくは−80〜30℃であり、反応時間は1〜30時間が好ましく、特に好ましくは1〜18時間である。
【0060】
これらの銅化合物は、そのまま用いることもできるし、上記のジリチオ化反応で挙げた溶媒と混合した状態で用いることもできる。
【0061】
本発明の一般式(1)のビフェニレン誘導体の製造は、好ましくは窒素又はアルゴン等の不活性雰囲気下で実施する。
【0062】
かくして得られた、一般式(1)で示されるビフェニレン誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0063】
一般式(2)で示されるジハロビフェニル誘導体をジグリニャール化する場合、用いるグリニャール化剤は、例えばマグネシウム金属、臭化エチルマグネシウム、臭化イソプロピルマグネシウム等のアルキルグリニャール試薬を挙げることができ、好ましくはマグネシウム金属である。マグネシウム金属の形態は特に限定はなく、例えば削り状、リボン状、粒状を挙げることができる。
【0064】
該グリニャール化剤は、例えばマグネシウム金属の場合、一般式(2)で示されるジハロビフェニル誘導体1当量に対し1.8〜10当量が好ましい。グリニャール化反応は、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばジリチオ化反応で用いた溶剤を挙げることができる。グリニャール化反応の温度は−20〜80℃が好ましく、反応時間は1〜120分が好ましい。
【0065】
該ジグリニャール化反応により生成したマグネシウム塩は、次いで銅化合物と反応させる。該銅化合物との反応方法及び用いる銅化合物は、ジリチオ化反応により生成したジリチウム塩と銅化合物とを反応させる場合と同様な方法で実施できる。又、反応雰囲気、反応生成物の精製も同様な方法で実施できる。
【0066】
本発明の一般式(1)で示されるビフェニレン誘導体の製造方法では、一般式(2)で示されるジハロビフェニル誘導体をジリチオ化及び/又はジグリニャール化した後、塩化亜鉛と反応させた後、銅化合物と処理することもできる。
【0067】
(ジハロビフェニル誘導体の製造方法)
一般式(2)で示されるジハロビフェニル誘導体は、下記一般式(3)で示される2,3−ジハロアントラセン誘導体と下記一般式(4)で示される2,3−ジハロアントラセン誘導体から誘導される3−ハロアントラセニル金属試薬をパラジウム及び/又はニッケル触媒存在下でクロスカップリングさせることで製造することができる。
【0068】
【化24】
(ここで、置換基R1、R2、R5、R6、及びX1、並びに記号mは、一般式(2)で示される置換基並びに記号と同意義を示し、置換基X3は臭素原子又はヨウ素原子を示す。)
【0069】
【化25】
(ここで、置換基R3、R4、R7、R8、及びX2、並びに記号nは、一般式(2)で示される置換基並びに記号と同意義を示し、置換基X4は臭素原子又はヨウ素原子を示す。)
置換基X3及びX4は臭素原子又はヨウ素原子であり、その中でも好ましくはヨウ素原子である。
【0070】
該3−ハロアントラセニル金属試薬は、その原料である一般式(4)で示される2,3−ジハロアントラセン誘導体を、例えばイソプロピルマグネシウムブロマイド等のグリニャール試薬あるいはn−ブチルリチウム等の有機リチウム試薬により一般式(4)のハロゲンであるX4とのハロゲン/金属交換反応を行った後、塩化亜鉛、トリメトキシボラン、トリ(イソプロポキシ)ボラン、2−イソプロポキシ−4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン等と反応させることで好適に調製することができる。なお、グリニャール試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ オルガニック ケミストリィー」、2000年、65巻、4618−4634頁に記載されている方法、有機リチウム試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ ケミカル リサーチ シノプシス」、1981年、185頁に記載されている−90℃以下でのハロゲンのリチオ化方法を用いることもできる。
【0071】
該3−ハロアントラセニル金属試薬と一般式(3)で示される2,3−ジハロアントラセン誘導体のクロスカップリング反応に用いる触媒はパラジウム及び/又はニッケル触媒であれば特に限定はなく、例えばテトラキス(トリフェニルホスフィン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム/トリフェニルホスフィン混合物、ビス(トリ−tert−ブチルホスフィン)パラジウム等の0価のパラジウム化合物;ジクロロビス(トリフェニルホスフィン)パラジウム、酢酸パラジウム/(トリ−tert−ブチルホスフィン)混合物、ジアセタトビス(トリフェニルホスフィン)パラジウム、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)パラジウム、ジクロロ(1,3−ビス(ジフェニルホスフィノ)プロパン)パラジウム、酢酸パラジウム/トリフェニルホスフィン混合物、酢酸パラジウム/2−(ジシクロヘキシルホスフィノ)−1,1’−ビフェニル混合物、ジクロロ(エチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルプロパンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム/トリフェニルホスフィン混合物等の2価のパラジウム化合物;ジクロロビス(トリフェニルホスフィン)ニッケル、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)ニッケル、ジクロロ(1,3−ビス(ジフェニルホスフィノ)プロパン)ニッケル、ジクロロ(エチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルプロパンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル/トリフェニルホスフィン混合物等の2価のニッケル化合物;ビス(1,5−シクロオクタジエン)ニッケル/トリフェニルホスフィン混合物等の0価のニッケル化合物を挙げることができ、中でも好ましくは0価あるいは2価のパラジウム化合物であり、特に好ましくはテトラキス(トリフェニルホスフィン)パラジウム、ジクロロビス(トリフェニルホスフィン)パラジウムである。
【0072】
該カップリング反応における、触媒の使用量は一般式(3)で示される2,3−ジハロアントラセン誘導体に対し、0.1〜20モル%の範囲が好ましい。また、一般式(4)から誘導される3−ハロアントラセニル金属試薬の使用量は一般式(3)で示される2,3−ジハロアントラセン誘導体1当量に対し、0.6〜1.5当量が好ましく、さらに好ましくは0.8〜1.4当量、特に好ましくは0.9〜1.2当量である。
【0073】
反応は好ましくは溶媒中で実施する。該溶媒として、例えばTHF、ジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテル、ジオキサン、エチレングリコールジメチルエーテル、エチレングリコール、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジイソプロピルアミン、ピペリジン、トルエン、キシレン、ヘキサン、シクロヘキサン、エタノール、水等を挙げることができる。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。例えばトルエン/水、トルエン/エタノール/水のような2乃至3成分系でも使用することができる。
【0074】
なお、一般式(3)と一般式(4)で示される2,3−ジハロアントラセン誘導体が同じ化合物であっても良い。
【0075】
なお、反応系中に塩基を存在させることもできる。この場合の塩基の種類としては特に限定はなく、例えば炭酸ナトリウム、炭酸水素ナトリウム、炭酸水素カリウム、炭酸カリウム、炭酸セシウム、りん酸カリウム、りん酸ナトリウム、水酸化ナトリウム、水酸化カリウム、フッ化カリウム等の無機塩基;ナトリウムメトキサイド、ナトリウムtert−ブトキサイド、カリウムtert−ブトキサイド等のアルコキサイド;トリエチルアミン、トリメチルアミン、トリプロピルアミン、トリブチルアミン、エチレンジアミン、N,N,N’,N’−テトラメチルエチレンジアミン、ジイソプロピルアミン、ピリジン、テトラブチルアンモニウムフルオライド等の有機塩基を好適なものとして挙げることができる。これらの塩基の使用量は一般式(3)で示される2,3−ジハロアントラセン誘導体1当量に対し、1.5〜10.0当量が好ましく、特に好ましくは2.0〜8.0当量である。さらにこれらの塩基と併用し、相間移動触媒を用いることもできる。相間移動触媒の種類は特に限定はなく、例えばトリオクチルメチルアンモニウムクロライド、テトラブチルアンモニウムクロライド、セチルピリジニウムクロライド、ベンジルトリメチルアンモニウムクロライド等を好適なものとして挙げることができる。これらの相間移動触媒の使用量は一般式(3)で示される2,3−ジハロアントラセン誘導体1当量に対し、0.1〜1.5当量が好ましく、特に好ましくは0.2〜0.8当量である。
【0076】
さらに反応系中にトリフェニルホスフィン等のホスフィンを存在させることもできる。これらのホスフィンの使用量は、該パラジウム及び/又はニッケル触媒1当量に対し、0.9〜8.0当量が好ましく、特に好ましくは1.0〜3.0当量である。
【0077】
反応の温度は10〜120℃が好ましく、特に好ましくは30〜100℃であり、反応時間は1〜48時間が好ましい。
【0078】
さらに、一般式(2)で示されるジハロビフェニル誘導体の別の製法について述べる。
【0079】
一般式(1)で示されるビフェニレン誘導体の前駆体である一般式(2)で示されるジハロビフェニル誘導体は、一般式(3)で示される2,3−ジハロアントラセン誘導体から誘導できる。
【0080】
即ち、一般式(2)で示されるジハロビフェニル誘導体は、一般式(3)で示される2,3−ジハロアントラセン誘導体をリチオ化剤又はグリニャール化剤を用いてホモカップリングすることで製造することができる。
【0081】
2,3−ジハロアントラセン誘導体のホモカップリング反応に用いるリチオ化剤は、一般式(3)におけるハロゲンX1又はX3をリチオ化することができるものである限り特に限定はなく、例えばn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、メチルリチウム、フェニルリチウム等の有機リチウム試薬;リチウムジイソプロピルアミド、リチウムヘキサメチルジシラジド等の有機リチウムアミド試薬;リチウム金属等を挙げることができ、好ましくは有機リチウム試薬であり、特に好ましくはn−ブチルリチウムである。
【0082】
該リチオ化反応は、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばTHF、ジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテル、ジオキサン、トルエン、ペンタン、ヘキサン、シクロヘキサン等であり、好ましくはTHFである。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。
【0083】
リチオ化剤は一般式(3)で示される2,3−ジハロアントラセン誘導体1当量に対し0.3〜1.2当量が好ましく、特に好ましくは0.4〜0.7当量である。リチオ化反応の温度は−100〜40℃が好ましく、特に好ましくは−90〜30℃であり、反応時間は1〜30時間が好ましく、特に好ましくは2〜20時間である。
【0084】
一方、一般式(3)で示される2,3−ジハロアントラセン誘導体のホモカップリング反応に用いるグリニャール化剤は、一般式(3)におけるハロゲンX1又はX3をグリニャール化することができるものである限り特に限定はなく、例えばマグネシウム金属、臭化エチルマグネシウム、臭化イソプロピルマグネシウム等のアルキルグリニャール試薬を挙げることができ、好ましくはマグネシウム金属である。マグネシウム金属の形態は特に限定はなく、例えば削り状、リボン状、粒状等を挙げることができる。
【0085】
該グリニャール化剤は、例えばマグネシウム金属の場合、一般式(3)で示される2,3−ジハロアントラセン誘導体1当量に対し0.4〜0.7当量が好ましい。グリニャール化反応は、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばリチオ化反応で用いた溶剤を挙げることができる。グリニャール化反応の温度は−20〜100℃が好ましく、反応時間は1〜30時間が好ましい。
【0086】
一般式(2)で示されるジハロビフェニル誘導体の製造は、好ましくは窒素又はアルゴン等の不活性雰囲気下で実施する。
【0087】
かくして得られた、一般式(2)で示されるジハロビフェニル誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0088】
(2,3−ジハロアントラセン誘導体の製造方法)
一般式(3)及び一般式(4)で示される2,3−ジハロアントラセン誘導体は、下記一般式(5)で示される2,3−ジハロアントラキノン誘導体から以下に述べる二通りの方法で製造することができる。
【0089】
【化26】
(ここで、置換基R1、R2、X1、及びX3、並びに記号mは、一般式(3)で示される置換基並びに記号と同意義を示す。)。
【0090】
(方法1)
一般式(3)で示される2,3−ジハロアントラセン誘導体における置換基R5及びR6が水素原子である場合、一般式(5)で示される2,3−ジハロアントラキノン誘導体をヒドリド還元剤を用いて還元し、さらに酸触媒で脱水することで製造することができる。
【0091】
用いるヒドリド還元剤は特に限定はなく、キノンをヒドロキシル基に還元できるものであれば良く、例えばジイソブチルアルミニウムヒドリド、ナトリウムボロヒドリド等を挙げることができる。該還元反応はTHF、ジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテル、トルエン、ヘキサン等の溶媒を用いて実施することが好ましい。反応温度は0〜60℃が好ましく、反応時間は1〜24時間が好ましい。ヒドリド還元剤の使用量は、一般式(5)で示される2,3−ジハロアントラキノン誘導体1当量に対し、1〜20当量が好ましい。
【0092】
次に該ヒドリド還元により得られたジヒドロキシル体を酸触媒を用いて脱水反応を行う。酸触媒としては、例えば塩酸、硫酸、トルエンスルホン酸、リン酸等が挙げられる。これらの酸触媒は水溶液としても用いることもできる。反応温度は10〜100℃が好ましく、反応時間は1〜24時間が好ましい。なお、先のヒドリド還元剤を用いる還元反応終了時に該酸触媒を添加し、そのまま脱水反応を実施することもできる。脱水反応に用いる酸触媒の使用量は、一般式(5)で示される2,3−ジハロアントラキノン誘導体1当量に対し、5〜80当量が好ましい。
【0093】
該ヒドリド還元反応及び脱水反応からなる工程は2〜4回繰り返すことで収率良く、一般式(3)で示される2,3−ジハロアントラセン誘導体を合成することができる。
【0094】
なお、一般式(5)で示される2,3−ジハロアントラキノン誘導体は既存の方法を用いて合成することができる。例えば、「ベリヒテ」(独国)、1933年、66B巻、1876−1891頁に記載されている方法を参考にして、4−ブロモ−5−ヨード無水フタル酸と1,2−ジアルキルベンゼンから合成することができる。
【0095】
(方法2)
一般式(3)で示される2,3−ジハロアントラセン誘導体における置換基R5及びR6が炭素数2〜20のアルキル基、フッ素原子、炭素数2〜20のアルキニル基、炭素数2〜30のアルケニル基、又は炭素数4〜20のアリール基である場合、一般式(5)で示される2,3−ジハロアントラキノン誘導体と下記一般式(6)及び/又は(7)で示される有機金属試薬と反応させた後、還元することで製造することができる。
【0096】
R5M1 (6)
R6M2 (7)
(ここで、置換基R5及びR6は、一般式(3)で示される置換基と同意義を示し、置換基M1及びM2は、リチウム又はマグネシウムハライドを示す。)
一般式(6)、(7)で示される有機金属試薬はキノンと反応できるものであれば特に限定はなく、例えば有機リチウム試薬、グリニャール試薬を挙げることができ、有機リチウム試薬の具体例としては、例えばオクチルリチウム、ドデシルリチウム、パーフルオロドデシルリチウム等のアルキルリチウム試薬;フェニルリチウム、トリルリチウム、p−オクチルフェニルリチウム、p−(パーフルオロオクチル)フェニルリチウム等のアリールリチウム試薬;エチニルリチウム、1−ドデシニルリチウム、1−(パーフルオロドデシニル)リチウム、フェニルエチニルリチウム等のアルキニルリチウム試薬;エテニルリチウム、1−ドデセニルリチウム、1−(パーフルオロドデセニル)リチウム、フェニルエテニルリチウム等のアルケニルリチウム試薬;等を挙げることができ、グリニャール試薬の具体例としては、例えばオクチルマグネシムブロミド、ドデシルマグネシウムブロミド、パーフルオロドデシルマグネシウムブロミド等のアルキルグリニャール試薬;フェニルマグネシウムブロミド、トリルマグネシウムブロミド、p−オクチルフェニルマグネシウムブロミド、p−(パーフルオロオクチル)フェニルマグネシウムブロミド等のアリールグリニャール試薬;エチニルマグネシウムブロミド、1−ドデシニルマグネシウムブロミド、1−(パーフルオロドデシニル)マグネシウムブロミド、フェニルエチニルマグネシウムブロミド等のアルキニルグルニャール試薬;エテニルマグネシウムブロミド、1−ドデセニルマグネシウムブロミド、1−(パーフルオロドデセニル)マグネシウムブロミド、フェニルエテニルマグネシウムブロミド等のアルケニルグルニャール試薬;等を挙げることができる。
【0097】
一般式(5)で示される2,3−ジハロアントラキノン誘導体と一般式(6)、(7)で示される有機金属試薬の反応はTHF、ジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテル、トルエン、ヘキサン等の溶媒を用いて実施することが好ましい。反応温度は−80〜60℃が好ましく、反応時間は5〜24時間が好ましい。該有機金属試薬の使用量は、一般式(5)で示される2,3−ジハロアントラキノン誘導体1当量に対し、2〜7当量が好ましい。次に該有機金属試薬との反応により得られたジヒドロキシル体を還元剤を用いて還元反応を行う。還元剤の具体例は、2塩化スズあるいは次亜リン酸ナトリウム/ヨウ化ナトリウムを用いることが好ましい。該還元反応は塩酸水溶液、酢酸等の溶媒を用いて実施することが好ましい。反応温度は20〜150℃が好ましく、反応時間は1〜5時間が好ましい。還元剤の使用量は、一般式(5)で示される2,3−ジハロアントラキノン誘導体1当量に対し、3〜10当量が好ましい。
【0098】
なお、9及び10位に置換基を有する一般式(3)で示される2,3−ジハロアントラセン誘導体は既存の方法を用いて合成することもできる。例えば、「シンレット」、2005年、217−222頁に記載されている方法を参考にして、2,3−ジハロアントラキノン誘導体から合成することができる。
(耐酸化性有機半導体材料)
次に、本発明の一般式(1)で示されるビフェニレン誘導体を含む耐酸化性有機半導体材料について述べる。該耐酸化性有機半導体材料は溶剤への溶解性、耐酸化性に優れ、好適な塗布性を有する。
【0099】
本発明の一般式(1)で示されるビフェニレン誘導体の溶解に用いる溶剤は、例えばo−ジクロロベンゼン、クロロベンゼン、1,2−ジクロロエタン、1,1,2,2−テトラクロロエタン、クロロホルム、ジクロロメタン等のハロゲン系溶剤;THF、ジオキサン等のエーテル系溶剤;トルエン、キシレン等の芳香族炭化水素系溶剤;酢酸エチル、γ−ブチロラクトン等のエステル系溶剤;N,N−ジメチルホルムアミド、N−メチルピロリドン等のアミド系溶剤;等であり、その中でも装置の腐食の観点から好ましくトルエン、キシレン等の芳香族炭化水素系溶剤である。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。
【0100】
上記に挙げた溶剤と一般式(1)で示されるビフェニレン誘導体を混合攪拌することにより、一般式(1)で示されるビフェニレン誘導体を含む耐酸化性有機半導体の溶液を調製することができる。この場合の温度は10〜130℃が好ましく、特に好ましくは20〜100℃である。得られる溶液の濃度は、溶剤及び温度により変えることができ、好ましくは0.01〜10.0重量%である。溶液の調製は空気中でも実施することができるが、好ましくは窒素、アルゴン等の不活性雰囲気下で調製する。
【0101】
一般式(1)で示されるビフェニレン誘導体を含む耐酸化性有機半導体材料の耐酸化性の評価は、該溶液を所定時間、空気と接触させる方法で実施することができる。まず用いる溶剤は予め脱気しておき、溶存酸素を除去する。空気との接触時間は、0.5分〜3時間が好適である。酸化の進行は、溶液の色の変化並びに薄層クロマトグラフィー、ガスクロマトグラフィー及び液体クロマトグラフィー分析による酸化物の検出により行うことができる。
【0102】
本発明の一般式(1)で示されるビフェニレン誘導体を含む耐酸化性有機半導体材料の溶液は、用いられる一般式(1)で示されるビフェニレン誘導体自体が適度の凝集性を有することから比較的に低温で溶剤へ溶解でき、且つ耐酸化性があることから、塗布法による有機薄膜の製造に好適に適用できる。即ち、雰囲気から厳密に空気を除く必要がないことから塗布工程を簡略化することができる。塗布は空気中でも実施できるが、好ましくは溶剤の乾燥を考慮して窒素気流下で行う。なお、好適な塗布性を得るために、本発明の一般式(1)で示されるビフェニレン誘導体を含む耐酸化性有機半導体材料の溶液の粘度は、0.005〜20ポアズの範囲にあることが好ましい。
(有機薄膜)
次に本発明の一般式(1)で示されるビフェニレン誘導体を含む耐酸化性有機半導体材料を用いた有機薄膜について述べる。係る有機薄膜は上記の耐酸化性有機半導体材料溶液の基板への塗布により製造することができる。
【0103】
基板への塗布による有機薄膜の製造は、該耐酸化性有機半導体材料溶液を基板上に塗布した後、加熱、気流、及び自然乾燥等の方法により溶剤を気化させることで実施することができる。該溶液中の一般式(1)で示されるビフェニレン誘導体の濃度は、特に限定はなく、例えば0.01〜10.0重量%であることが好ましい。塗布温度は特に限定はなく、例えば20〜130℃の間で好適に実施することができる。塗布の具体的方法は特に限定はなく、例えばスピンコート、キャストコート、ディップコート等を用いることができる。さらにスクリーン印刷、インクジェット印刷、グラビア印刷等の印刷技術を用いても作製することが可能である。使用する基板の材料は特に限定はなく、結晶性、非結晶性の種々の材料を用いることができる。また、基板は絶縁性あるいは誘電性を有する材料であっても良い。具体的な基板としては、例えばポリエチレンテレフタレート、ポリメチルメタクリレート、ポリエチレン、ポリプロピレン、ポリスチレン、環状ポリオレフィン、ポリイミド、ポリカーボネート、ポリビニルフェノール、ポリビニルアルコール、ポリエチレンナフタレート等のプラスチック基板;ガラス、石英、酸化アルミニウム、シリコン、酸化シリコン、二酸化タンタル、五酸化タンタル、インジウム錫酸化物等の無機材料基板;金、銅、クロム、チタン等の金属基板等を挙げることができる。またこれらの基板の表面は、例えばオクタデシルトリクロロシラン、オクチルトリクロロシラン、オクタデシルトリメトキシシラン等のシラン類で修飾処理したものであっても使用することができる。塗布した後の溶剤の乾燥は、常圧若しくは減圧で除去することができる、又、加熱により乾燥してもよい。さらに、溶剤の気化速度を調節することで本発明の一般式(1)で示されるビフェニレン誘導体の結晶成長を制御することができる。基板への塗布により得られる有機薄膜の膜厚は特に限定はなく、好ましくは1nm〜100μm、特に好ましくは10nm〜20μmである。なお、このようにして得られた薄膜は、60〜150℃に加熱することでより高度に配列させることもできる。
【0104】
(発光材料)
本発明で得られた一般式(1)で示されるビフェニレン誘導体は、ビフェニレン誘導体の溶液の基板への塗布により発光材料として用いることができ、とりわけ有機電界発光素子の材料として用いることができる。従って、一般式(1)で示されるビフェニレン誘導体は有機EL材料としての使用が期待される。
【0105】
一般式(1)で示されるビフェニレン誘導体の溶液にする際の溶剤は、特に限定はなく、例えばo−ジクロロベンゼン、クロロベンゼン、1,2−ジクロロエタン、1,1,2,2−テトラクロロエタン、クロロホルム等のハロゲン系溶剤;THF、ジオキサン等のエーテル系溶剤;トルエン、キシレン、メシチレン等の芳香族化合物の炭化水素系溶剤;酢酸エチル、γ−ブチロラクトン等のエステル系溶剤;N,N−ジメチルホルムアミド、N−メチルピロリドン等のアミド系溶剤;等が挙げられる。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。中でも、好ましくはクロロベンゼン、トルエン等である。
【0106】
上記に挙げた溶剤と一般式(1)で示されるビフェニレン誘導体を混合攪拌することにより、一般式(1)で示されるビフェニレン誘導体の溶液となるものである。混合攪拌する際の温度は10〜200℃が好ましく、特に好ましくは20〜150℃である。混合攪拌する際の一般式(1)で示されるビフェニレン誘導体の濃度は、溶剤及び温度により変えることができ、0.01〜10.0重量%であることが好ましい。溶液の調製は空気中でも実施することができるが、好ましくは窒素、アルゴン等の不活性雰囲気下で調製する。
【0107】
一般式(1)で示されるビフェニレン誘導体を含む溶液の塗布は空気中でも実施できるが、好ましくは溶剤の乾燥を考慮して窒素気流下で行う。なお、好適な塗布性を得るために、本発明の一般式(1)で示されるビフェニレン誘導体を含む溶液の粘度は、0.005〜20ポアズの範囲にあることが好ましい。
【0108】
次に本発明の一般式(1)で示されるビフェニレン誘導体を含む溶液の塗布による発光材料の作製について述べる。係る発光材料は上記の溶液の基板への塗布により製造することができる。
【0109】
基板への塗布による発光材料の製造は、前記溶液を基板上に塗布した後、加熱、気流及び自然乾燥等の方法により溶剤を気化させることで実施することができる。該溶液中の一般式(1)で示されるビフェニレン誘導体の濃度は、特に限定はなく、例えば0.01〜10.0重量%であることが好ましい。塗布温度は特に限定はなく、例えば20〜200℃の間、特に好ましくは30〜100℃の間で好適に実施することができる。塗布の具体的方法は特に限定はなく、例えばスピンコート、キャストコート及びディップコート等を用いることができる。さらにスクリーン印刷、インクジェット印刷、グラビア印刷等の印刷技術を用いても作製することが可能である。使用する基板の材料は特に限定はなく、結晶性、非結晶性の種々の材料を用いることができる。基板の具体例としては、例えばポリエチレンテレフタレート、ポリエチレンナフタレート、ポリメチルメタクリレート、ポリエチレン、ポリプロピレン、ポリスチレン、環状ポリオレフィン、ポリイミド、ポリカーボネート、ポリビニルフェノール、ポリビニルアルコール、ポリ(ジイソプロピルフマル酸)、ポリ(ジエチルフマル酸)等のプラスチック基板;ガラス、石英、酸化アルミニウム、シリコン、酸化シリコン、二酸化タンタル、五酸化タンタル、インジウム錫酸化物等の無機材料基板;金、銅、クロム、チタン、アルミニウム等の金属基板を好適に用いることができる。またこれらの基板の表面は、例えばオクタデシルトリクロロシラン、オクタデシルトリメトキシシラン等のシラン類;ヘキサメチルジシラザン等のシリルアミン類で修飾処理したものであっても使用することができる。さらに、基板は絶縁性あるいは誘電性を有する材料であっても良い。塗布した後の溶剤の乾燥は、常圧若しくは減圧で除去することができる、又、加熱、窒素気流により乾燥してもよい。さらに、溶剤の気化速度を調節することで一般式(1)で示されるビフェニレン誘導体の膜質を制御することができる。基板への塗布により得られる発光材料の膜厚は特に限定はなく、好ましくは1nm〜100μm、特に好ましくは10nm〜20μmである。
【0110】
また、本発明の一般式(1)で示されるビフェニレン誘導体を用いた発光材料は、真空蒸着法によっても薄膜を作製することができる。真空蒸着法による薄膜は、汎用の真空蒸着装置を用いることにより行うことができる。真空蒸着法で薄膜を作製する際の真空槽の真空度は、一般に用いられる拡散ポンプ、ターボ分子ポンプ、クライオポンプ等により到達し得る1.3×10−2〜1.3×10−5パスカルが好ましい。蒸着速度は、形成する膜圧によるが、0.005〜1.0nm/秒が好ましい。
【0111】
本発明の一般式(1)で示されるビフェニレン誘導体を用いた発光材料の発光量子収率は、好ましくは2〜80%、特に好ましくは3〜50%である。
【0112】
本発明の一般式(1)で示されるビフェニレン誘導体は平面剛直性の高い分子構造を有することから、優れた半導体特性を与えることが期待できる。該ビフェニレン誘導体はトルエン等の溶媒に溶解し、溶液状態にあっても容易に空気酸化されることはない。従って、塗布法により半導体薄膜を容易に作製できる。また、本発明の一般式(1)で示されるビフェニレン誘導体は、長い共役系を有することから、黄色から赤色の長波長の発光を与えるアントラセン環を含むことから発光材料としても期待することができる。したがって、本発明の一般式(1)で示されるビフェニレン誘導体は電子ペーパー、有機ELディスプレイ、液晶ディスプレイ、又はICタグ用等のトランジスタの有機半導体活性相用途、さらに有機ELディスプレイの発光材料、ホスト材料、有機半導体レーザー材料、有機薄膜太陽電池材料、発光有機トランジスタ、フォトニック結晶材料等に利用することができる。
【発明の効果】
【0113】
優れた耐酸化性を有し、塗布法による半導体活性相形成が可能な、ビフェニレン誘導体及びその用途を提供する。さらに本発明の製造法では置換基を導入したビフェニレン誘導体を製造することができ、新規な有機半導体材料を提供することができる。
【実施例】
【0114】
以下、実施例により本発明をさらに詳細に説明するが、本発明はこれら実施例にのみ限
定されるものではない。
【0115】
生成物の同定には1H NMRスペクトル及びマススペクトルを用いた。なお、1H NMRスペクトルは日本電子製JEOL GSX−270WB(270MHz)を用いて測定した。マススペクトル(MS)は日本電子製JEOL JMS−700を用いて、試料を直接導入し、電子衝突(EI)法(70エレクトロンボルト)又はFAB法(6キロエレクトロンボルト、キセノンガス、マトリックス(2−ニトロフェニルオクチルエーテル)で測定した。
【0116】
反応の進行の確認等は薄層クロマトグラフィー、ガスクロマトグラフィー及びガスクロマトグラフィー−マススペクトル(GCMS)分析を用いた。
【0117】
ガスクロマトグラフィー分析
装置 島津GC14B
カラム J&Wサイエンティフィック社製、DB−1,30m
ガスクロマトグラフィー−マススペクトル分析
装置 パーキンエルマーオートシステムXL(MS部;ターボマスゴールド)
カラム J&Wサイエンティフィック社製、DB−1,30m
反応用の溶媒は、断りのない限り市販品を用いた。なお、グリニャール試薬あるいはブチルリチウム等の有機金属試薬を用いた場合は、市販の脱水溶媒をそのまま用いた。
【0118】
発光材料としての評価は、薄膜状態での発光量子収率及び蛍光スペクトルを測定することで実施した。なお、発光量子収率は浜松ホトニクス製C9920−01を用い、蛍光スペクトルは日本分光製FP6500を用いて測定した。
【0119】
合成例1 (4−ブロモ−5−ヨード無水フタル酸の合成)
4−ブロモ−5−ヨード無水フタル酸は「ジャーナル オブ オーガニック ケミストリー」(米国)、1951年、16巻、1577−1581頁を参考に、以下の様に合成した。
【0120】
4−ブロモフタルイミド(東京化成工業製)9.95g(44.0mmol)を窒素ガスで置換した50mlの二口ナスフラスコに入れた。次いでヨウ素5.87g(23.1mmol)及び10%発煙硫酸(ヨツハタ化学工業製)12mlを加え、90℃で23時間反応を行った。反応混合物を室温に冷やして氷に注ぎ入れた後、ガラスフィルターでろ過し、黄色固体12.8gを得た。得られた固体を濃硫酸35mlに溶解させ、130℃で5時間反応を行った。反応混合物を氷冷後、氷水を加えて析出した固体をろ過し、フタル酸誘導体の固体13.8gを得た。次に得られた固体を、水酸化ナトリウム3.6gを水18mlに溶かした水溶液に室温で溶かした。この塩基性水溶液に酢酸を加えpHを3〜4に調整し、析出するフタル酸誘導体のモノナトリウム塩の白色沈殿をろ過した。得られた白色固体を水に懸濁させ、濃塩酸でpHを1以下にし、再びフタル酸誘導体として白色固体4.93gを得た。この固体をトルエン48mlに溶かし、無水酢酸8.7g(85.7mmol)を加え、105℃で4時間反応を行った。反応液を減圧濃縮して白色固体3.79gを得た。この固体から加熱トルエンに不溶な成分を除き、目的の4−ブロモ−5−ヨード無水フタル酸5.13g(14.5mmol)を得た(収率33%)。
1H NMR(CDCl3,22℃):δ=8.51(s,1H),8.23(s,1H)。
MS m/z: 353(M+,100%),309(M+−CO2,18%),282(M+−C2O3,10%),155(M+−C2O3−I,16%),74(M+−C2O3−I−Br,32%)。
【0121】
合成例2 (1,2―ジドデシルベンゼンの合成)
1,2−ジドデシルベンゼンは「日本化学会誌」1989年、983−987頁に従い以下の様に合成した。
【0122】
1,2−ジクロロベンゼン2.22g(15.1mmol)、ジクロロ〔1,3−ビス(ジフェニルホスフィノ)プロパン〕ニッケル(東京化成工業製)131mg(0.24mmol)、乾燥ジエチルエーテル11.5mlの混合液にドデシルマグネシウムブロミド(シグマ−アルドリッチ製、1.0mol/lジエチルエーテル溶液)45ml(45mmol)を窒素雰囲気中0℃で滴下した。35℃で20時間反応を行い、反応混合物を0℃に冷却し希塩酸を加え、ジエチルエーテルで抽出した。ジエチルエーテル溶液を水、飽和炭酸水素ナトリウム水溶液、水の順に洗浄し、塩化カルシウムで乾燥させた。得られた液体をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン)及び減圧蒸留で精製し、目的の1,2―ジドデシルベンゼン5.56g(13.4mmol)を得た(収率88%)。
1H NMR(CDCl3,22℃):δ=7.14−7.09(m,4H),2.59(t,J=7.8Hz,4H),1.55(m,4H),1.26(m,36H),0.88(t,J=6.8Hz,6H)。
MS m/z: 414(M+,100%),260(M+−C11H23,71%),106(M+−C22H46,98%)。
【0123】
合成例3 (2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノンの合成)(一般式(5)の化合物)
2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノンは「ベリヒテ」(独国)、1933年、66B巻、1876−1891頁を参考に以下の様に合成した。
【0124】
合成例1で得られた4−ブロモ−5−ヨード無水フタル酸2.82g(8.00mmol)、合成例2で得られた1,2−ジドデシルベンゼン3.32g(8.00mmol)、テトラクロロエタン5.0mlの混合液に塩化アルミニウム2.41g(18.1mmol)を加え、室温で3時間反応を行った。水を加えてクエンチし、さらに水洗浄を行い、真空加熱乾燥後、白色固体6.2gを得た。得られた固体を濃硫酸44mlに溶かし、80℃で1時間反応した。反応混合物を氷に注ぎ入れ、析出した固体をろ過して水で洗浄した。減圧乾燥後、シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:塩化メチレン=10:1)及びヘプタンからの再結晶で精製し、2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノンの黄色固体4.20g(5.60mmol)を得た(収率70%)。
1H NMR(CDCl3,22℃):δ=8.73(s,1H),8.45(s,1H),8.05(s,2H),2.75(m,4H),1.62(m,4H),1.26(m,36H),0.88(m,6H)。
MS m/z: 750(M+,100%),440(M+−C22H46,8%),313(M+−C22H46−I,2%),233(M+−C22H46−I−Br,1%)。
【0125】
合成例4 (2−ブロモ−3−ヨード−6,7−ジドデシルアントラセンの合成)(一般式(3)及び(4)の化合物)
窒素雰囲気下、100mlシュレンク反応容器に合成例3で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノン1.10g(1.47mmol)を入れた。次いでTHF17mlを加え、ヒドリド還元剤として水素化ジイソブチルアルミニウム(関東化学製、0.99mol/l、トルエン溶液)4.0ml(4.0mmol)を加え、室温で1.5時間還元反応を行った。次いで反応混合物に酸触媒として6M塩酸水溶液10mlを加え、65℃で3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。再びTHF17mlを加え、水素化ジイソブチルアルミニウム5.5ml(5.4mmol)を加え、室温で1.5時間還元反応を行った。次いで反応混合物に6M塩酸水溶液10mlを加え、3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(溶離液:ヘキサン)で精製し、2−ブロモ−3−ヨード−6,7−ジドデシルアントラセンの黄色固体629mg(0.87mmol)を得た(収率59%)。
1H NMR(CDCl3,22℃):δ=8.55(s,1H),8.27(s,1H),8.16(s,1H),8.15(s,1H),7.72(s,2H),2.78(m,4H),1.71(m,4H),1.27(m,36H)0.88(m,6H)。
MS m/z: 720(M+,100%),410(M+−C22H46,16%),283(M+−C22H46−I,4%),203(M+−C22H46−I−Br,5%)。
【0126】
実施例1 (6,7,6’,7’−テトラドデシル−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)[一般式(2)で示されるジハロビフェニル誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に合成例4で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラセン205mg(0.285mmol)及びTHF8mlを添加した。この溶液を−55℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.81M)のTHF溶液0.70ml(0.57mmol)を滴下した。5分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)59.2mg(0.57mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮した。得られた固形物に、合成例4で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラセン210mg(0.292mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)16.5mg(0.014mmol)、トルエン5ml、及びエタノール1.2mlを添加した。さらに炭酸ナトリウム92.8mg(0.873mmol)と水1.6mlからなる水溶液を加え、60℃で24時間反応を実施した。室温まで冷却後、トルエン及び水を添加し分相した。有機相を濃縮し、得られた残渣をトルエン5mlに溶解後、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.02mlを添加し、室温で2時間撹拌した。このトルエン溶液を水で2回洗浄後、無水硫酸ナトリウムで乾燥した。有機相を濾過し、減圧濃縮し、得られた残渣をシリカゲルを充填したカラムで濾過した(溶媒、ヘキサン)。得られた粗固体をヘプタンから再結晶化し、目的物の黄色固体287mgを得た(収率85%)。
1H NMR(CDCl3,22℃):δ=8.33(s,2H),8.31(s,2H),8.29(s,2H),7.97(s,2H),7.78(s,2H),7.75(s,2H),2.79(m,8H),1.72(m,8H),1.28(m,72H)0.87(m,12H)。
1H NMRスペクトルを図1に示した。
MS m/z: 1185(M+,2%),592(M+/2,100)。
【0127】
1H NMR及びMS測定より、6,7,6’,7’−テトラドデシル−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお。その構造式を下記に示す。
【0128】
【化27】
実施例2 (テトラドデシルジナフトビフェニレンの合成)[一般式(1)で示されるビフェニレン誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に実施例1で合成した6,7,6’,7’−テトラドデシル−3,3’−ジブロモ−2,2’−ビアントラセニル240mg(0.202mmol)及びジエチルエーテル7mlを添加した。この混合物を0℃に冷却後、リチオ化剤であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.28ml(0.44mmol)を滴下した。1時間かけて5℃まで昇温しメタル化の熟成を行った。一方、別の100mlシュレンク反応容器に塩化銅(II)(和光純薬工業製)95.3mg(0.709mmol)及びTHF10mlを添加し、−78℃に冷却した。ここへ先のメタル化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。徐々に昇温し、一晩かけて室温まで反応温度を上げた。3M塩酸水溶液及びトルエンを添加した。分相し、有機相をさらに水で洗浄し、無水硫酸ナトリウムで乾燥した。有機相を濾過し、減圧濃縮し、得られた粗固体をトルエンから再結晶化し、目的物の黄色固体108mgを得た(収率52%)。
1H NMR(重トルエン,60℃):δ=7.95(s,4H),7.65(s,4H),7.40(s,4H),2.83(t,J=8.5Hz,8H),1.78(m,8H),1.31(m,72H)0.92(m,12H)。
1H NMRスペクトルを図2に示した。
FABMS m/z: 1026(M+)。
【0129】
1H NMR及びMS測定より、テトラドデシルジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0130】
【化28】
合成例5 (2−ブロモ−3−ヨード−6,7−(ドデシル)フルオロアントラセンの合成)(一般式(3)及び(4)の化合物)
合成例2で1,2−ジクロロベンゼンの代わりに、1−クロロ−2−フルオロベンゼン(東京化成工業製)を用いた以外は合成例2と同じ操作を繰り返して1−ドデシル−2−フルオロベンゼンを合成した(収率80%)。この1−ドデシル−2−フルオロベンゼンと合成例1で得られた4−ブロモ−5−ヨード無水フタル酸を用い、合成例3と同じ操作を繰り返し2−ブロモ−3−ヨード−6,7−(ドデシル)フルオロアントラキノンを得(収率54%)、さらに合成例4と同じ操作を繰り返して、2−ブロモ−3−ヨード−6,7−(ドデシル)フルオロアントラセンへ変換した(収率63%)。
【0131】
実施例3 (ジドデシルジフルオロ−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)[一般式(2)で示されるジハロビフェニル誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に合成例5で合成した2−ブロモ−3−ヨード−6,7−(ドデシル)フルオロアントラセン464mg(0.815mmol)及びTHF10mlを添加した。この溶液を−60℃に冷却し、イソプロピルマグネシウムブロマイド(関東化学製、0.65M)のTHF溶液1.3ml(0.84mmol)を滴下した。5分間熟成後、−78℃に冷却し、塩化亜鉛(シグマ−アルドリッチ製、1.0M)のジエチルエーテル溶液0.84ml(0.84mmol)を滴下した。徐々に室温まで昇温した後、生成したスラリー液を減圧濃縮した。得られた固形物に、合成例5で合成した2−ブロモ−3−ヨード−6−ドデシル−7−フルオロアントラセン471mg(0.827mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)47.1mg(0.041mmol)、及びTHF8mlを添加した。64℃で8時間反応を実施した。室温まで冷却後、トルエン及び水を添加し分相した。有機相を濃縮し、得られた残渣をトルエン5mlに溶解後、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.1mlを添加し、室温で2時間撹拌した。このトルエン溶液を水で2回洗浄後、有機相を減圧濃縮し、得られた残渣に飽和食塩水及びトルエンを添加した。分相し、有機相を水で洗浄し、無水硫酸ナトリウムで乾燥した。有機相を濾過し、減圧濃縮し、得られた残渣をシリカゲルを充填したカラムで濾過した(溶媒;ヘキサン)。濾液を減圧濃縮し、得られた粗固体をヘプタンから再結晶化し、ジドデシルジフルオロ−3,3’−ジブロモ−2,2’−ビアントラセニルの2種類の異性体の混合物からなる黄色固体440mgを得た(収率61%)。
MS m/z: 885(M+,4%),725(M+−2Br,100)。
【0132】
MS測定より、ジドデシルジフルオロ−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお、その構造式を下記に示す。
【0133】
【化29】
実施例4 (ジドデシルジフルオロジナフトビフェニレンの合成)[一般式(1)で示されるビフェニレン誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に、実施例3で合成したジドデシルジフルオロ−3,3’−ジブロモ−2,2’−ビアントラセニル388mg(0.438mmol)及びジエチルエーテル7mlを添加した。この混合物を0℃に冷却し、リチオ化剤であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.60ml(0.95mmol)を滴下した。0℃で80分間撹拌した(リチオ化反応)。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)177mg(1.31mmol)及びTHF15mlを添加し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温までゆっくり昇温し、3M塩酸水溶液を添加した。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、2種類のジドデシルジフルオロジナフトビフェニレンの2種類の異性体の混合物からなる黄色固体146mgを得た(収率46%)。
FABMS m/z: 725(M+)。
【0134】
MS測定より、ジドデシルジフルオロジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0135】
【化30】
合成例6 (4−ブロモ−1,2−ジヨードベンゼンの合成)
100mlシュレンク反応容器に、1,2−ジヨードベンゼン(東京化成工業製)5.56g(16.8mmol)及びジクロロメタン30mlを添加し、0℃に冷却した。鉄粉(シグマ−アルドリッチ製)67mg及びヨウ素10mg(0.04mmol)を添加後、臭素0.87ml(17mmol)を滴下した。0℃で8時間撹拌後、亜硫酸水素ナトリウム水溶液を添加し、反応を停止させた。有機相を水で洗浄後、無水硫酸ナトリウムで乾燥した。減圧濃縮し、得られた残渣をTHF:メタノール=1:1から2回再結晶化し、4−ブロモ−1,2−ジヨードベンゼン4.94gの白色結晶を得た(収率72%)。
【0136】
合成例7 (4−ブロモ−1,2−(パーフルオロドデシル)ベンゼンの合成)
4−ブロモ−1,2−(パーフルオロドデシル)ベンゼンは、「ジャーナル オブ フルオリン ケミストリィー」、1989年、43巻、207−228頁を参考に次のように合成した。
【0137】
窒素雰囲気下、100mlシュレンク反応容器に、銅粉(カッパーブロンズ)(シグマ−アルドリッチ製)2.85g(44.8mmol)、パーフルオロドデシルアイオダイド(シンクエスト製)18.4g(24.6mmol)、合成例6で合成した4−ブロモ−1,2−ジヨードベンゼン4.58g(11.2mmol)、及びジメチルスルホキシド(和光純薬工業製、脱水品)18mlを添加し、125℃に加熱し、8時間反応させた。室温に冷却後、水を添加し反応を停止させた。さらにジエチルエーテルを添加し、混合物をセライトを用いて濾過した。濾液をジエチルエーテル抽出し、合わせた有機相を水で洗浄し、無水硫酸マグネシウムで乾燥した。減圧濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィーで精製し(溶媒;ヘキサン)、4−ブロモ−1,2−(パーフルオロドデシル)ベンゼンの無色の液体6.24gを得た(収率40%)。
【0138】
合成例8 (2−ブロモ−3−ヨード−6,7−ジ(パーフルオロドデシル)アントラキノンの合成)(一般式(5)の化合物)
窒素雰囲気下、300mlシュレンク反応容器に、合成例7で得られた4−ブロモ−1,2−(パーフルオロドデシル)ベンゼン6.11g(4.39mmol)及びTHF80mlを添加した。この溶液を−50℃に冷却し、イソプロピルマグネシウムブロマイド(関東化学製、0.65M)のTHF溶液6.8ml(4.4mmol)を滴下した。−50℃で30分熟成後、ここに合成例1で合成した4−ブロモ−5−ヨード無水フタル酸1.48g(4.20mmol)とTHF20mlからなる溶液を滴下した。反応混合物を一晩かけて室温まで昇温した後、氷冷し3M塩酸水溶液を添加した。ジエチルエーテルで抽出し、合わせた有機相を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。減圧下濃縮し、白色固体7.00g得た。得られた固体に濃硫酸40mlを添加し、80℃で12時間反応した。反応混合物を氷に注ぎ入れ、析出した固体をろ過して水で洗浄した。乾燥後、シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:塩化メチレン=15:1)及びヘプタンからの再結晶で精製し、2−ブロモ−3−ヨード−6,7−ジ(パーフルオロドデシル)アントラキノンの固体1.86g(1.13mmol)を得た(収率27%)。
【0139】
合成例9 (2−ブロモ−3−ヨード−6,7−ジ(パーフルオロドデシル)アントラセンの合成)(一般式(3)及び(4)の化合物)
窒素雰囲気下、100mlシュレンク反応容器に合成例8で合成した2−ブロモ−3−ヨード−6,7−ジ(パーフルオロドデシル)アントラキノン1.81g(1.10mmol)を入れた。次いでTHF15mlを加え、ヒドリド還元剤である水素化ジイソブチルアルミニウム(関東化学製、0.99mol/l、トルエン溶液)3.5ml(3.5mmol)を加え、室温で1.5時間還元反応を行った。次いで反応混合物に酸触媒として6M塩酸水溶液10mlを加え、65℃で3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。再びTHF15mlを加え、水素化ジイソブチルアルミニウム3.5ml(3.5mmol)を加え、室温で1.5時間還元反応を行った。次いで反応混合物に6M塩酸水溶液10mlを加え、3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン)で精製し、2−ブロモ−3−ヨード−6,7−ジ(パーフルオロドデシル)アントラセンの黄色固体1.12gを得た(収率63%)。
【0140】
実施例5 (6,7,6’,7’−テトラ(パーフルオロドデシル)−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)[一般式(2)で示されるジハロビフェニル誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に合成例9で合成した2−ブロモ−3−ヨード−6,7−ジ(パーフルオロドデシル)アントラセン483mg(0.298mmol)及びTHF7mlを添加した。この溶液を−70℃に冷却し、イソプロピルマグネシウムブロマイド(関東化学製、0.65M)のTHF溶液0.46ml(0.30mmol)を滴下した。10分間熟成後、−78℃に冷却し、塩化亜鉛(シグマ−アルドリッチ製、1.0Mジエチルエーテル溶液、)0.30ml(0.30mmol)を滴下した。徐々に室温まで昇温した後、減圧濃縮した。得られた残渣に、合成例9で合成した2−ブロモ−3−ヨード−6,7−ジ(パーフルオロドデシル)アントラセン490mg(0.303mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)17.3mg(0.015mmol)、THF8mlを加え、64℃で10時間反応を実施した。容器を水冷し3M塩酸水溶液3mlを添加することで反応を停止させた。トルエンを添加後、分相し、有機相を食塩水で洗浄した。有機相を減圧濃縮し溶媒を留去し、さらに真空乾燥した。得られた残渣にトルエンを添加し、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)(0.06ml)を添加し、室温で2時間撹拌した。この溶液を水洗浄し、有機相を減圧濃縮析出した。残渣をシリカゲルカラムクロマトグラフィーで精製し(溶媒;ヘキサン及びヘキサン:クロロホルム=10:1)、6,7,6’,7’−テトラ(パーフルオロドデシル)−3,3’−ジブロモ−2,2’−ビアントラセニルの黄色固体507mgを得た(収率57%)。
FABMS m/z: 2985(M+)。
【0141】
MS測定より、6,7,6’,7’−テトラ(パーフルオロドデシル)−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお、その構造式を下記に示す。
【0142】
【化31】
実施例6 (テトラ(パーフルオロドデシル)ジナフトビフェニレンの合成)[一般式(1)で示されるビフェニレン誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に、実施例5で合成した6,7,6’,7’−テトラ(パーフルオロドデシル)−3,3’−ジブロモ−2,2’−ビアントラセニル500mg(0.167mmol)及びジエチルエーテル7mlを添加した。この混合物を0℃に冷却し、リチオ化剤であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.23ml(0.37mmol)を滴下し、0℃で90分間撹拌した(リチオ化反応)。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)67.4mg(0.501mmol)及びTHF16mlを添加し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温までゆっくり昇温し、3M塩酸水溶液を添加した。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、目的物の黄色固体184mgを得た(収率39%)。
FABMS m/z: 2825(M+)。
【0143】
MS測定より、テトラ(パーフルオロドデシル)ジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0144】
【化32】
合成例10 (4−ブロモ−1,2−ジフェニルベンゼンの合成)
窒素雰囲気下、200mlシュレンク反応容器に、合成例6で得られた4−ブロモ−1,2−ジヨードベンゼン2.15g(5.25mmol)にジヒドロキシフェニルボラン(和光純薬工業製)1.47g(12.1mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)458mg(0.40mmol)、炭酸ナトリウム3.34g(31.5mmol)、トルエン42ml、エタノール10.5ml、水13.3mlを加え、80℃で29時間反応させた。1M塩酸水溶液を加えて反応をクエンチし、トルエンで抽出した後、有機層を水洗浄して無水硫酸ナトリウムで乾燥した。溶媒を減圧留去し、シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン)で精製して目的の4−ブロモ−1,2−ジフェニルベンゼン1.46g(4.72mmol)を得た(収率90%)。
【0145】
合成例11 (2−ブロモ−3−ヨード−6,7−ジフェニルアントラキノンの合成)(一般式(5)の化合物)
窒素雰囲気下、300mlシュレンク反応容器に、合成例10で得られた4−ブロモ−1,2−ジフェニルベンゼン1.46g(4.72mmol)を入れた。次いでTHF28mlを加えて−78℃に冷却し、n−ブチルリチウム(関東化学製、1.59mol/l、ヘキサン溶液)3.0ml(4.77mmol)を加え、30分間反応させた。次いで合成例1で合成した4−ブロモ−5−ヨード無水フタル酸1.66g(4.72mmol)を加え、室温まで昇温した。水を加えてクエンチし得られた固体を濾過し、さらに水洗浄を行い、加熱真空乾燥後白色固体3.2gを得た。得られた固体に濃硫酸26mlを添加し、80℃で1時間反応した。反応混合物を氷に注ぎ入れ、析出した固体をろ過して水で洗浄した。乾燥後、シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:塩化メチレン=10:1)及びヘプタンからの再結晶で精製し、2−ブロモ−3−ヨード−6,7−ジフェニルアントラキノンの黄色固体298mg(0.53mmol)を得た(収率11%)。
【0146】
合成例12 (2−ブロモ−3−ヨード−6,7−ジフェニルアントラセンの合成)(一般式(3)及び(4)の化合物)
窒素雰囲気下、50mlシュレンク反応容器に、合成例11で合成した2−ブロモ−3−ヨード−6,7−ジフェニルアントラキノン243mg(0.43mmol)及びTHF5mlを加えた。ヒドリド還元剤である水素化ジイソブチルアルミニウム(関東化学製、0.99mol/l、トルエン溶液)1.20ml(1.20mmol)を滴下し、室温で1.5時間還元反応を行った。この反応混合物に酸触媒である6M塩酸水溶液3mlを加え、65℃で3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して、無水硫酸ナトリウムで乾燥、減圧濃縮した。得られた残渣に再びTHF5mlを加え、水素化ジイソブチルアルミニウム(関東化学製、0.99mol/l、トルエン溶液)1.20ml(1.20mmol)を加え、室温で1.5時間還元反応を行った。次いで反応混合物に6M塩酸水溶液3mlを加え、3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン)で精製し、2−ブロモ−3−ヨード−6,7−ジフェニルアントラセンの黄色固体128mg(0.239mmol)を得た(収率56%)。
【0147】
実施例7 (6,7,6’,7’−テトラフェニル−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)[一般式(2)で示されるジハロビフェニル誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に合成例12で合成した2−ブロモ−3−ヨード−6,7−ジフェニルアントラセン(一般式(3)の化合物)128mg(0.239mmol)及びTHF5mlを添加した。この溶液を−70℃に冷却し、イソプロピルマグネシウムブロマイド(関東化学製、0.65M)のTHF溶液0.74ml(0.48mmol)を滴下した。20分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)49.7mg(0.48mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮した。得られた残渣に、合成例12で合成した2−ブロモ−3−ヨード−6,7−ジフェニルアントラセン(一般式(4)の化合物)125mg(0.233mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)10.3mg(0.009mmol)、トルエン5ml、及びエタノール1.2mlを添加した。さらに炭酸ナトリウム78.5mg(0.741mmol)と水1.6mlからなる水溶液を加え、60℃で24時間反応を実施した。室温まで冷却後、トルエン及び水を添加し分相した。有機相を減圧濃縮し溶媒を留去し、さらに真空乾燥した。得られた残渣にトルエンを添加し、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)(0.03ml)を添加し、室温で2時間撹拌した。この溶液を水洗浄し、有機相を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製し(溶媒;ヘキサン(アントラセン誘導体を溶出)及びヘキサン:ジクロロメタン=10:1(目的物を溶出))、6,7,6’,7’−テトラフェニル−3,3’−ジブロモ−2,2’−ビアントラセニルの黄色固体87.2mgを得た(収率57%)。
MS m/z: 817(M+,2%),657(M+−2Br,100)。
【0148】
MS測定より、6,7,6’,7’−テトラフェニル−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお、その構造式を下記に示す。
【0149】
【化33】
実施例8 (テトラフェニルジナフトビフェニレンの合成)(一般式(1)で示されるビフェニレン誘導体)
窒素雰囲気下、50mlシュレンク反応容器に、実施例7で合成した6,7,6’,7’−テトラフェニル−3,3’−ジブロモ−2,2’−ビアントラセニル87.0mg(0.106mmol)及びジエチルエーテル4mlを添加した。この混合物を0℃に冷却し、リチオ化であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.16ml(0.25mmol)を滴下し、0℃で90分間撹拌した(リチオ化反応)。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)47.0mg(0.350mmol)及びTHF8mlを添加し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温までゆっくり昇温し、3M塩酸水溶液を添加した。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、目的物の黄色固体27mgを得た(収率39%)。
FABMS m/z: 657(M+)。
【0150】
MS測定より、テトラフェニルジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0151】
【化34】
合成例13 (4−ブロモ−1,2−ジ(ドデシン−1−イル)ベンゼンの合成)
窒素雰囲気下、300mlシュレンク反応容器に、合成例6で得られた4−ブロモ−1,2−ジヨードベンゼン1.84g(4.50mmol)、ヨウ化銅(I)(和光純薬工業製)90mg(0.47mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)272mg(0.24mmol)、THF59ml、トリエチルアミン2.29g(22.2mmol)、1−ドデシン1.57g(9.47mmol)を加えた。この反応混合物を室温で26時間反応させた。1M塩酸水溶液を加えて反応をクエンチし、トルエンで抽出した後、有機相を水洗浄して無水硫酸ナトリウムで乾燥した。溶媒を減圧留去し、シリカゲルカラムクロマトグラフィー(溶離液;ジエチルエーテル:ヘキサン=1:30の混合液)で精製して目的の4−ブロモ−1,2−ジ(ドデシン−1−イル)ベンゼン1.52g(3.14mmol)を得た(収率70%)。
【0152】
合成例14 (2−ブロモ−3−ヨード−6,7−ジ(ドデシン−1−イル)アントラキノンの合成)(一般式(5)の化合物)
窒素雰囲気下、200mlシュレンク反応容器に、合成例13で得られた4−ブロモ−1,2−ジ(ドデシン−1−イル)ベンゼン1.52g(3.14mmol)を入れた。次いでTHF19mlを加えて−78℃に冷却し、n−ブチルリチウム(関東化学製、1.59mol/l、ヘキサン溶液)2.0ml(3.18mmol)を加え、30分間反応させた。次いで合成例1で得られた4−ブロモ−5−ヨードフタル酸無水物1.10g(3.14mmol)を加え、室温まで昇温した。水を加えてクエンチし得られた固体を濾過し、さらに水洗浄を行い、加熱真空乾燥後白色固体2.3g(3.1mmol)を得た。この固体に濃硫酸17mlを添加し、80℃で1時間反応した。反応混合物を氷に注ぎ入れ、析出した固体をろ過して水で洗浄した。乾燥後、シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:塩化メチレン=10:1)及びヘプタンからの再結晶で精製し、2−ブロモ−3−ヨード−6,7−ジ(ドデシン−1−イル)アントラキノンの黄色固体245mg(0.33mmol)を得た(収率11%)。
【0153】
合成例15 (2−ブロモ−3−ヨード−6,7−ジ(ドデシン−1−イル)アントラセンの合成)(一般式(3)及び(4)の化合物)
窒素雰囲気下、50mlシュレンク反応容器に、合成例14で合成した2−ブロモ−3−ヨード−6,7−ジ(ドデシン−1−イル)アントラキノン245mg(0.33mmol)及びTHF4mlを加えた。ヒドリド還元剤である水素化ジイソブチルアルミニウム(関東化学製、0.99mol/l、トルエン溶液)0.70ml(0.70mmol)を滴下し、室温で1.5時間還元反応を行った。この反応混合物に酸触媒である6M塩酸水溶液2mlを加え、65℃で3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。得られた残渣に再びTHF4mlを加え、水素化ジイソブチルアルミニウム0.70ml(0.70mmol)を加え、室温で1.5時間還元反応を行った。次いで反応混合物に6M塩酸水溶液2mlを加え、3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン)で精製し、2−ブロモ−3−ヨード−6,7−ジ(ドデシン−1−イル)アントラセンの黄色固体102mg(0.143mmol)を得た(収率42%)。
【0154】
実施例9 (6,7,6’,7’−テトラ(ドデシン−1−イル)−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)(一般式(2)で示されるジハロビフェニル誘導体)
窒素雰囲気下、100mlシュレンク反応容器に合成例15で合成した2−ブロモ−3−ヨード−6,7−ジ(ドデシン−1−イル)アントラセン102mg(0.143mmol)(一般式(3)の化合物)及びTHF5mlを添加した。この溶液を−70℃に冷却し、イソプロピルマグネシウムブロマイド(関東化学製、0.65M)のTHF溶液0.45ml(0.29mmol)を滴下した。10分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)29.7mg(0.29mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮した。得られた残渣に、合成例15で合成した2−ブロモ−3−ヨード−6,7−ジ(ドデシン−1−イル)アントラセン(一般式(4)の化合物)99.3mg(0.140mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)8.1mg(0.007mmol)、トルエン5ml、及びエタノール1.2mlを添加した。さらに炭酸ナトリウム44.5mg(0.420mmol)と水1.6mlからなる水溶液を加え、60℃で24時間反応を実施した。室温に冷却し、トルエンを添加後、分相し、有機相を食塩水で洗浄した。有機相を減圧濃縮し溶媒を留去し、さらに真空乾燥した。得られた残渣にトルエンを添加し、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)(0.03ml)を添加し、室温で1時間撹拌した。この溶液を水洗浄し、有機相を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製し(溶媒;ヘキサン(アントラセン誘導体を溶出)及びヘキサン:クロロホルム=10:1(目的物を溶出))、6,7,6’,7’−テトラ(ドデシン−1−イル)−3,3’−ジブロモ−2,2’−ビアントラセニルの黄色固体93.3mgを得た(収率57%)。
FABMS m/z: 1169(M+,100%),1089(M+−Br,7)。
【0155】
MS測定より、6,7,6’,7’−テトラ(ドデシン−1−イル)−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお、その構造式を下記に示す。
【0156】
【化35】
実施例10 (テトラ(ドデシン−1−イル)ジナフトビフェニレンの合成)(一般式(1)で示されるビフェニレン誘導体)
窒素雰囲気下、50mlシュレンク反応容器に、実施例9で合成した6,7,6’,7’−テトラ(ドデシン−1−イル)−3,3’−ジブロモ−2,2’−ビアントラセニル93.0mg(0.080mmol)及びジエチルエーテル5mlを添加した。この混合物を0℃に冷却し、リチオ化剤であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.23ml(0.13mmol)を滴下し、0℃で30分間及び10℃で60分間撹拌した(リチオ化反応)。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)35.5mg(0.264mmol)及びTHF10mlを添加し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温までゆっくり昇温し、3M塩酸水溶液を添加した。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、目的物の黄色固体28.1mgを得た(収率35%)。
FABMS m/z: 1010(M+)。
【0157】
MS測定より、テトラ(ドデシン−1−イル)ジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0158】
【化36】
合成例16 (2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ジフェニルアントラセンの合成)(一般式(3)及び(4)の化合物)
窒素雰囲気下、100mlシュレンク反応容器に合成例3で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノン541mg(0.722mmol)及びTHF20mlを添加した。−78℃に冷却後、フェニルリチウム(関東化学製、1.0mol/l、シクロヘキサン/ジエチルエーテル溶液)1.5ml(1.5mmol)(一般式(6)及び(7)の化合物)を加えた後、一晩かけて室温まで昇温した。次いで反応混合物に3M塩酸水溶液及びジエチルエーテルを加えた後、分相し、さらにジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。得られた残渣に酢酸30ml、ヨウ化ナトリウム749mg(5.0mmol)、及び次亜りん酸ナトリウム・1水和物727mg(6.86mmol)を加え、1時間加熱還流下で反応を行った(還元反応)。反応混合物を室温まで冷やし、トルエンで抽出した。トルエン溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:トルエン=30:1)で精製し、2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ジフェニルアントラセンの黄色固体453mgを得た(収率72%)。
【0159】
実施例11 (6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラフェニル−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)[一般式(2)で示されるジハロビフェニル誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に合成例16で合成した2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ジフェニルアントラセン205mg(0.235mmol)及びTHF6mlを添加した。この混合物を−60℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.81M)のTHF溶液0.58ml(0.47mmol)を滴下した。10分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)48.8mg(0.47mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮した。得られた固形物に、合成例16で合成した2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ジフェニルアントラセン210mg(0.241mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)13.9mg(0.012mmol)、トルエン5ml、及びエタノール1.2mlを添加した。さらに炭酸ナトリウム76.6mg(0.723mmol)と水1.6mlからなる水溶液を加え、60℃で24時間反応を実施した。室温まで冷却後、トルエン及び水を添加し分相した。有機相を濃縮し、得られた残渣をトルエン5mlに溶解後、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.02mlを添加し、室温で2時間撹拌した。このトルエン溶液を水で2回洗浄後、有機相を減圧濃縮し、得られた残渣に飽和食塩水及びトルエンを添加した。分相し、有機相を水で洗浄し、無水硫酸ナトリウムで乾燥した。有機相を濾過し、減圧濃縮し、得られた残渣をシリカゲルを充填したカラムで濾過した(溶媒:ヘキサン)。得られた粗固体をヘプタンから再結晶化し、目的物の黄色固体217mgを得た(収率62%)。
FABMS m/z: 1490(M+,100%),1419(M+−Br,5)。
【0160】
MS測定より、6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラフェニル−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお。その構造式を下記に示す。
【0161】
【化37】
実施例12 (テトラドデシルテトラフェニルジナフトビフェニレンの合成)[一般式(1)で示されるビフェニレン誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に実施例11で合成した6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラフェニル−3,3’−ジブロモ−2,2’−ビアントラセニル210mg(0.141mmol)及びジエチルエーテル7mlを添加した。この混合物を0℃に冷却後、リチオ化剤であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.20ml(0.31mmol)を滴下した。1時間かけて5℃まで昇温しメタル化の熟成を行った。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)64.5mg(0.479mmol)及びTHF15mlを添加し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温までゆっくり昇温し、3M塩酸水溶液を添加した。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、目的物の黄色固体97.5mgを得た(収率52%)。
FABMS m/z: 1330(M+)。
【0162】
MS測定より、テトラドデシルテトラフェニルジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0163】
【化38】
合成例17 (2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエチニル)アントラセンの合成)(一般式(3)及び(4)の化合物の合成)
窒素雰囲気下、100mlシュレンク反応容器にフェニルアセチレン(東京化成工業製)178mg(1.74mmol)及びTHF20mlを添加した。n−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液1.05ml(1.67mmol)を滴下し、20分間撹拌した。合成例3で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノン523mg(0.697mmol)を加えた後、混合物を室温で一晩反応させた。次いで反応混合物に3M塩酸水溶液及びジエチルエーテルを加えた後、分相し、さらにジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。得られた残渣に酢酸30ml、ヨウ化ナトリウム749mg(5.0mmol)、及び次亜りん酸ナトリウム・1水和物727mg(6.86mmol)を加え、1時間加熱還流下で反応を行った。反応混合物を室温まで冷やし、トルエンで抽出した。トルエン溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:トルエン=30:1の混合液)で精製し、2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエチニル)アントラセンの黄色固体393mgを得た(収率61%)。
【0164】
実施例13 (6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラキス(フェニルエチニル)−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)(一般式(2)で示されるジハロビフェニル誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例17で合成した2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエチニル)アントラセン190mg(0.206mmol)及びTHF6mlを添加した。この混合物を−60℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.81M)のTHF溶液0.51ml(0.41mmol)を滴下した。5分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)42.6mg(0.41mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて室温で30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮し、得られた固形物に、合成例17で合成した2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエチニル)アントラセン195mg(0.212mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)13.9mg(0.012mmol)、トルエン5ml、及びエタノール1.2mlを添加した。さらに炭酸ナトリウム67.4mg(0.636mmol)と水1.6mlからなる水溶液を加え、60℃で24時間反応を実施した。室温まで冷却後、トルエン及び水を添加し分相した。有機相を濃縮し、得られた残渣をトルエン5mlに溶解後、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.02mlを添加し、室温で2時間撹拌した。このトルエン溶液を水で2回洗浄後、有機相を減圧濃縮し、得られた残渣に飽和食塩水及びトルエンを添加した。分相し、有機相を水で洗浄し、無水硫酸ナトリウムで乾燥した。有機相を濾過し、減圧濃縮し、得られた残渣をシリカゲルを充填したカラムで濾過した(溶媒:ヘキサン)。得られた粗固体をヘプタンから再結晶化し、目的物の黄色固体212mgを得た(収率65%)。
MS m/z: 1586(M+,2%),793(M+/2,100)。
【0165】
MS測定より、6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラキス(フェニルエチニル)−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお。その構造式を下記に示す。
【0166】
【化39】
実施例14 (テトラドデシルテトラキス(フェニルエチニル)ジナフトビフェニレンの合成)(一般式(1)で示されるビフェニレン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に実施例13で合成した6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラキス(フェニルエチニル)−3,3’−ジブロモ−2,2’−ビアントラセニル208mg(0.131mmol)及びジエチルエーテル7mlを添加した。この混合物を0℃に冷却後、リチオ化剤であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.20ml(0.31mmol)を滴下した。1時間かけて5℃まで昇温しメタル化の熟成を行った。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)64.5mg(0.479mmol)及びTHF15mlを添加し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温までゆっくり昇温し、3M塩酸水溶液を添加した。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、目的物の橙色固体80.3mgを得た(収率43%)。
FABMS m/z: 1426(M+)。
【0167】
MS測定より、テトラドデシルテトラキス(フェニルエチニル)ジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0168】
【化40】
合成例18 (2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエテニル)アントラセンの合成)(一般式(3)及び(4)の化合物の合成)
窒素雰囲気下、100mlシュレンク反応容器にマグネシウム54.7mg(2.25mmol)及びTHF20mlを添加した。ヨウ素5mgを加えた後、β−ブロモスチレン(東京化成工業製)40mgを添加し、撹拌下マグネシウムを活性化させた。β−ブロモスチレン359mg(1.96mmol、計2.17mmol)を緩く還流が起こる程度に滴下した。滴下終了後、30分間撹拌を継続した。合成例3で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノン678mg(0.904mmol)を加えた後、混合物を室温で一晩反応させた。次いで反応混合物に3M塩酸水溶液及びジエチルエーテルを加えた後、分相し、さらにジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。得られた残渣に酢酸30ml、ヨウ化ナトリウム749mg(5.0mmol)、及び次亜りん酸ナトリウム・1水和物727mg(6.86mmol)を加え、1時間加熱還流下で反応を行った。反応混合物を室温まで冷やし、トルエンで抽出した。トルエン溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:トルエン=30:1の混合液)で精製し、2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエテニル)アントラセンの黄色固体468mgを得た(収率56%)。
【0169】
実施例15 (6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラキス(フェニルエテニル)−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)(一般式(2)で示されるジハロビフェニル誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例18で合成した2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエテニル)アントラセン225mg(0.243mmol)及びTHF7mlを添加した。この混合物を−60℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.81M)のTHF溶液0.60ml(0.49mmol)を滴下した。5分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)50.9mg(0.490mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて室温で30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮し、得られた固形物に、合成例18で合成した2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエテニル)アントラセン235mg(0.254mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)13.9mg(0.012mmol)、トルエン5ml、及びエタノール1.2mlを添加した。さらに炭酸ナトリウム80.8mg(0.762mmol)と水1.6mlからなる水溶液を加え、60℃で24時間反応を実施した。室温まで冷却後、トルエン及び水を添加し分相した。有機相を濃縮し、得られた残渣をトルエン5mlに溶解後、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.02mlを添加し、室温で2時間撹拌した。このトルエン溶液を水で2回洗浄後、有機相を減圧濃縮し、得られた残渣に飽和食塩水及びトルエンを添加した。分相し、有機相を水で洗浄し、無水硫酸ナトリウムで乾燥した。有機相を濾過し、減圧濃縮し、得られた残渣をシリカゲルを充填したカラムで濾過した(溶媒:ヘキサン)。得られた粗固体をヘプタンから再結晶化し、目的物の黄色固体259mgを得た(収率67%)。
MS m/z: 1594(M+,2%),797(M+/2,100)。
【0170】
MS測定より、6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラキス(フェニルエテニル)−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお。その構造式を下記に示す。
【0171】
【化41】
実施例16 (テトラドデシルテトラキス(フェニルエテニル)ジナフトビフェニレンの合成)(一般式(1)で示されるビフェニレン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に実施例15で合成した6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラキス(フェニルエテニル)−3,3’−ジブロモ−2,2’−ビアントラセニル254mg(0.159mmol)及びジエチルエーテル7mlを添加した。この混合物を0℃に冷却後、リチオ化剤であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.22ml(0.35mmol)を滴下した。1時間かけて5℃まで昇温しメタル化の熟成を行った。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)64.1mg(0.477mmol)及びTHF15mlを添加し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温までゆっくり昇温し、3M塩酸水溶液を添加した。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、目的物の橙色固体93.5mgを得た(収率41%)。
【0172】
FABMS m/z: 1434(M+)。
【0173】
MS測定より、テトラドデシルテトラキス(フェニルエテニル)ジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0174】
【化42】
実施例17 (耐酸化性評価)
窒素雰囲気下、100mlシュレンク容器にトルエン10.4gを添加し、凍結(液体窒素)−減圧−窒素置換−融解から成るサイクルを3回繰り返すことで溶存酸素を除去した。そこへ実施例2で得られたテトラドデシルジナフトビフェニレンの黄色固体10.5mgを添加し、110℃に加熱溶解させると黄色透明溶液となった。次にこのシュレンク容器の上部の栓を開け、1時間、外気に接触させることで空気を導入し、さらに110℃で撹拌した。で酸化に由来する新たなピークの出現はなかった。
【0175】
さらにこの溶液を110℃、1時間、撹拌下で空気を導入しても溶液の色の変化はみられず、耐酸化性に優れるものであった。
【0176】
比較例1
窒素雰囲気下、100mlシュレンク容器にトルエン28.9gを添加し、凍結(液体窒素)−減圧−窒素置換−融解から成るサイクルを3回繰り返すことで溶存酸素を除去した。そこへペンタセン(東京化成工業製)3.0mgを添加し、110℃に加熱し溶解させると赤紫色溶液となった。次にこのシュレンク容器の上部の栓を開け、1時間、空気を導入すると溶液の色が赤ピンク色に変化していた。さらに110℃で撹拌した。ガスクロマトグラフィー及びガスクロマトグラフィー−マススペクトル(GCMS)分析から、6,13−ペンタセンキノンが生成していることがわかった。
【0177】
さらにこの溶液を110℃、1時間、撹拌下で空気を導入すると溶液の色が黄色に変化していた。
【0178】
実施例18 (有機薄膜の作製)
窒素雰囲気下、実施例2で得られたテトラドデシルジナフトビフェニレン7.2mgをトルエン(10.2g)と混合し、80℃で1時間撹拌し、テトラドデシルジナフトビフェニレンの黄色透明溶液を調製した。
【0179】
空気雰囲気下、凹面のある石英基板を80℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、膜厚420nmの有機薄膜を作製した。この有機薄膜の成分をガスクロマトグラフィーで分析した結果、テトラドデシルジナフトビフェニレン以外にピークはなく、酸化されていなかった。従って、空気中でも酸化されることなくテトラドデシルジナフトビフェニレンの有機薄膜を作製できることがわかった。
【0180】
実施例19 (有機薄膜の作製、発光材料としての評価)
窒素雰囲気下、実施例2で得られたテトラドデシルジナフトビフェニレン26mgをジクロロエタン5gと混合し、80℃で1時間撹拌し、テトラドデシルジナフトビフェニレンの黄色溶液を調製した。
【0181】
空気雰囲気下、石英基板を70℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、膜厚287nmの有機薄膜を作製した。この有機薄膜を用いて、発光量子収率及び蛍光スペクトルを測定した。結果を表1に示す。これらの結果からテトラドデシルジナフトビフェニレンは発光材料として利用できるものであった。
【0182】
実施例20 (有機薄膜の作製、発光材料としての評価)
窒素雰囲気下、実施例12で得られたテトラドデシルテトラフェニルジナフトビフェニレン36mgをジクロロエタン5gと混合し、80℃で1時間撹拌し、テトラドデシルテトラフェニルジナフトビフェニレンの黄色溶液を調製した。
【0183】
空気雰囲気下、石英基板を70℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、膜厚398nmの有機薄膜を作製した。この有機薄膜を用いて、発光量子収率及び蛍光スペクトルを測定した。結果を表1に示す。これらの結果からテトラドデシルテトラフェニルジナフトビフェニレンは発光材料として利用できるものであった。
【0184】
実施例21 (有機薄膜の作製、発光材料としての評価)
窒素雰囲気下、実施例14で得られたテトラドデシルテトラキス(フェニルエチニル)ジナフトビフェニレン36mgをジクロロエタン5gと混合し、80℃で1時間撹拌し、テトラドデシルテトラキス(フェニルエチニル)ジナフトビフェニレンの橙色溶液を調製した。
【0185】
空気雰囲気下、石英基板を70℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、膜厚378nmの有機薄膜を作製した。この有機薄膜を用いて、発光量子収率及び蛍光スペクトルを測定した。結果を表1に示す。これらの結果からテトラドデシルテトラキス(フェニルエチニル)ジナフトビフェニレンは発光材料として利用できるものであった。
【0186】
実施例22 (有機薄膜の作製、発光材料としての評価)
窒素雰囲気下、実施例16で得られたテトラドデシルテトラキス(フェニルエテニル)ジナフトビフェニレン32mgをジクロロエタン5gと混合し、80℃で1時間撹拌し、テトラドデシルテトラキス(フェニルエテニル)ジナフトビフェニレンの橙色溶液を調製した。
【0187】
空気雰囲気下、石英基板を70℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、膜厚356nmの有機薄膜を作製した。この有機薄膜を用いて、発光量子収率及び蛍光スペクトルを測定した。結果を表1に示す。これらの結果からテトラドデシルテトラキス(フェニルエテニル)ジナフトビフェニレンは発光材料として利用できるものであった。
【0188】
【表1】
比較例2(テトラドデシルジベンゾビフェニレン(本願:n=m=0)の合成及び評価)
1) (3,3’−ジブロモ−6,7,6’,7’−テトラドデシル−2,2’−ビナフチルの合成)
窒素雰囲気下、100mlシュレンク反応容器に2,3−ジブロモ−6,7−ジドデシルナフタレン389mg(0.625mmol)及びTHF9mlを加えた。−78℃に冷却し、n−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.20ml(0.31mmol)を滴下した。20分間反応後、冷却用バスを外し、室温で1時間撹拌した。3M塩酸水溶液及びトルエン添加し分相した。有機相を無水硫酸ナトリウムで乾燥し、減圧濃縮した。残渣をヘプタンから再結晶精製し、白色固体204mgを得た(収率61%)。
【0189】
2) (テトラドデシルジベンゾビフェニレンの合成)
窒素雰囲気下、100mlシュレンク反応容器に上記1)で得た3,3’−ジブロモ−6,7,6’,7’−テトラドデシル−2,2’−ビナフチル201mg(0.185mmol)及びジエチルエーテル5mlを加えた。0℃に冷却し、n−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.25ml(0.40mmol)を滴下し、0℃で30分間及び10℃で60分間撹拌した。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)74.6mg(0.555mmol)及びTHF10mlを投入し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温まで昇温し、3M塩酸水溶液を加えて反応を停止させた。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、テトラドデシルジベンゾビフェニレンの淡黄色固体83.9mgを得た(収率49%)。
【0190】
なお、その構造式を下記に示す。
【0191】
【化43】
3)テトラドデシルジベンゾビフェニレンの評価
窒素雰囲気下、100mlシュレンク容器にトルエン10.2gを添加し、凍結(液体窒素)−減圧−窒素置換−融解から成るサイクルを3回繰り返すことで溶存酸素を除去した。そこへ上記2)で得たテトラドデシルジベンゾビフェニレン7.2mgを添加し、80℃で1時間撹拌し、テトラドデシルジベンゾビフェニレンの薄黄色透明溶液を調製した。
【0192】
次に窒素雰囲気下、凹面のある石英基板を80℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥した。しかし、材料がある部分とない部分が存在し、均一な膜は得られず、塗布による均質な薄膜作製は困難であった。
【図面の簡単な説明】
【0193】
【図1】実施例1で合成した6,7,6’,7’−テトラドデシル−3,3’−ジブロモ−2,2’−ビアントラセニルの1H NMRスペクトル
【図2】実施例2で合成したテトラドデシルジナフトビフェニレンの1H NMRスペクトル
【技術分野】
【0001】
本発明は、有機半導体等の電子材料への展開が可能なビフェニレン誘導体、その用途、及びそれらの製造方法に関する。
【背景技術】
【0002】
有機薄膜トランジスタに代表される有機半導体デバイスは、省エネルギー、低コスト、及びフレキシブルといった無機半導体デバイスにはない特徴を有することから近年注目されるようになった。有機薄膜トランジスタは有機半導体活性相、基板、絶縁相、電極等数種類の材料から構成されるが、中でも電荷のキャリアー移動を担う有機半導体活性相は該デバイスの中心的な役割を有している。この有機半導体活性相を構成する有機材料のキャリアー移動能により半導体デバイス性能が左右される。
【0003】
有機半導体活性相を作製する方法としては一般的に、高温真空下、有機材料を気化させて実施する真空蒸着法、及び有機材料を適当な溶媒に溶解させその溶液を塗布する塗布法が知られている。塗布法は高温高真空条件を用いることなく、印刷技術を用いても実施することができるため、デバイス作製の製造コストを大幅に削減することができることから、経済的に好ましいプロセスである。しかし、従来、有機半導体材料として高性能な材料ほど塗布での活性相形成が困難になるという問題があった。
【0004】
例えば、分子長軸を有するペンタセン等の結晶性材料はアモルファスシリコン並みの高いキャリアー移動度を有し、優れた半導体デバイス特性を発現することが報告されている(例えば、非特許文献1参照)。しかし、ペンタセンはその強い凝集性のため溶解性が低く、一般的には経済的な塗布法を適用することができない。また、ペンタセン等のポリアセンを溶解させ塗布法でデバイスを製造する試みも報告されているが(例えば、特許文献1参照)、元来難溶性のポリアセン類を溶解させるためには、高温加熱等の条件が必要とされ、さらにペンタセンの溶液は極めて容易に空気酸化されることから、塗布法の適用はプロセス的、経済的に困難を伴うものであった。また、ポリ−(3−ヘキシルチオフェン)等の自己組織化材料は溶媒に可溶であり、塗布によるデバイス作製が報告されているが、キャリアー移動度が結晶性低分子化合物より1桁低いことから(例えば、非特許文献2参照)、得られた有機半導体デバイスの特性が低いという問題があった。
【0005】
このような中で、ビフェニレンは剛直な共役縮環化合物であり、有機半導体材料として期待できる化合物である。しかし、これまでに報告されているビフェニレン誘導体は、有機半導体材料としては満足の行くものではなかった。
【0006】
例えば多置換多環芳香族化合物およびその製造方法には剛直なビフェニレン構造を有する化合物が記載されている(例えば、特許文献2〜4参照)が、分子長軸方向以外にも置換基を有することから結晶性が低下し、塗布法により均一な膜が得られず半導体材料としては適していない。また、カチオン重合性樹脂のための三元光開始剤システム(例えば、特許文献5参照)にはビフェニレン誘導体の構造が記載されているが、分子長軸が短いため半導体材料としては適していない。
【0007】
【非特許文献1】「ジャーナル オブ アプライドフィジックス」、(米国)、2002年、92巻、5259−5263頁
【非特許文献2】「サイエンス」、(米国)、1998年、280巻、1741−1744頁
【特許文献1】WO2003/016599パンフレット
【特許文献2】特開2004−256497号公報
【特許文献3】特開2006−156980号公報
【特許文献4】特開2006−328006号公報
【特許文献5】特表2005−523348号公報
【発明の開示】
【発明が解決しようとする課題】
【0008】
そこで、本発明は上記の従来技術が有する問題点に鑑み、優れた耐酸化性を有し、塗布法による半導体活性相形成が可能なビフェニレン誘導体、それを用いた耐酸化性有機半導体材料並びにそれからなる有機薄膜、発光材料及び該ビフェニレン誘導体を簡便に経済的に製造する方法を提供することを目的とする。さらに本発明は、該ビフェニレン誘導体の前駆化合物であるジハロビフェニル誘導体にも関するものである。
【課題を解決するための手段】
【0009】
本発明者らは上記課題を解決するため鋭意検討の結果、新規なビフェニレン誘導体及び前駆体であるジハロビフェニル誘導体を見出した。加えて、該ビフェニレン誘導体からなる耐酸化性有機半導体材料及びそれからなる有機薄膜、発光材料を見出した。さらに、該ビフェニレン誘導体を製造するに好適な製造方法を見出し、本発明を完成するに到った。
【0010】
以下に本発明を詳細に説明する。
(ビフェニレン誘導体)
本発明のビフェニレン誘導体は下記一般式(1)で示される。
【0011】
【化1】
(ここで、置換基R1〜R8は同一又は異なって、水素原子、フッ素原子、炭素数2〜20のアルキル基、炭素数2〜20のアルキニル基、炭素数2〜30のアルケニル基、又は炭素数4〜20のアリール基を示し、mは1又は2であり、nは0〜2の整数を示す。但し、R1及びR2は同時に水素原子であることはない。)
本発明の一般式(1)の置換基について、述べる。
【0012】
置換基R1〜R8における、炭素数2〜20のアルキル基は特に限定はなく、例えばプロピル基、ブチル基、tert−ブチル基、ペンチル基、ネオペンチル基、ヘキシル基、オクチル基、イソオクチル基、ノニル基、デシル基、ドデシル基、ペンタデシル基、オクタデシル基、2−エチルヘキシル基等のアルキル基;トリフルオロメチル基、ペンタフルオロエチル基、パーフルオロオクチル基、パーフルオロデシル基、パーフルオロドデシル基、パーフルオロオクタデシル基、パーフルオロシクロヘキシル基、パーフルオロシクロオクチル基等のパーフルオロアルキル基;トリフルオロエチル基、ペンタデカフルオロオクチル基、オクタデカフルオロデシル基、2−エチルパーフルオロヘキシル基等の一部の水素がフッ素に置換されたハロゲン化アルキル基を挙げることができ、好ましくは炭素数5〜20のアルキル基であり、さらに好ましくはオクチル基、ドデシル基、オクタデシル基、パーフルオロオクチル基、パーフルオロドデシル基、パーフルオロオクタデシル基、特に好ましくはドデシル基、パーフルオロドデシル基である。
【0013】
置換基R1〜R8における、炭素数2〜20のアルキニル基は特に限定はなく、例えばエチニル基、メチルエチニル基、イソプロピルエチニル基、tert−ブチルエチニル基、(オクチル)エチニル基、(デシル)エチニル基、(ドデシル)エチニル基、(オクタデシル)エチニル基、(トリフルオロメチル)エチニル基、(パーフルオロオクチル)エチニル基、(パーフルオロデシル)エチニル基、(パーフルオロドデシル)エチニル基、フェニルエチニル基、{p−(オクチル)フェニル}エチニル基、ナフチルエチニル基、アントラセニルエチニル基、ベンジルエチニル基、パーフルオロフェニルエチニル基、{p−(トリフルオロメチル)フェニル}エチニル基、{p−(パーフルオロオクチル)フェニル}エチニル基等を挙げることができ、好ましくは(オクチル)エチニル基、(デシル)エチニル基、(ドデシル)エチニル基、(パーフルオロオクチル)エチニル基、(パーフルオロデシル)エチニル基、(パーフルオロドデシル)エチニル基、フェニルエチニル基等である。
【0014】
置換基R1〜R8における、炭素数2〜30のアルケニル基は特に限定はなく、例えばエテニル基、メチルエテニル基、イソプロピルエテニル基、tert−ブチルエテニル基、(オクチル)エテニル基、(デシル)エテニル基、(ドデシル)エテニル基、(トリフルオロメチル)エテニル基、フェニルエテニル基、{p−(ヘキシル)フェニル}エテニル基、{p−(オクチル)フェニル}エテニル基、2−フェニル−1,2−ジフルオロエテニル基、2−フェニル−1,2−ジメチルエテニル基、ジフェニルエテニル基、トリフェニルエテニル基、ナフチルエテニル基、アントラセニルエテニル基、ベンジルエテニル基、フェニル(メチル)エテニル基、(パーフルオロフェニル)エテニル基、{p−(トリフルオロメチル)フェニル}エテニル基、(パーフルオロオクチル)エテニル基、(パーフルオロデシル)エテニル基、(パーフルオロドデシル)エテニル基、{5−(ヘキシル)チエニル−2−}エテニル基、{5−(パーフルオロヘキシル)チエニル−2−}エテニル基等を挙げることができ、好ましくは(オクチル)エテニル基、(デシル)エテニル基、(ドデシル)エテニル基、(パーフルオロオクチル)エテニル基、(パーフルオロデシル)エテニル基、(パーフルオロドデシル)エテニル基、{5−(ヘキシル)チエニル−2−}エテニル基、{5−(パーフルオロヘキシル)チエニル−2−}エテニル基、フェニルエテニル基等である。なお、該炭素数2〜30のアルケニル基はトランス体及びシス体の何れであってもよく、またそれらの任意の割合の混合物であってもよい。
【0015】
置換基R1〜R8における、炭素数4〜20のアリール基は特に限定はなく、例えばフェニル基、p−トリル基、p−(オクチル)フェニル基、m−(オクチル)フェニル基、p−(デシル)フェニル基、p−フルオロフェニル基、ペンタフルオロフェニル基、p−(トリフルオロメチル)フェニル基、p−(パーフルオロオクチル)フェニル基、m−(パーフルオロオクチル)フェニル基、p−(トリフルオロメチル)テトラフルオロフェニル基、p−メトキシフェニル基、p−フェノキシフェニル基、2−チエニル基、5−(ヘキシル)−2−チエニル基、5−(パーフルオロヘキシル)−2−チエニル基、ビフェニル基、パーフルオロビフェニル基、1−ナフチル基、2−ナフチル基、1−パーフルオロナフチル基、2−フルオレニル基、9,9−ジメチル−2−フルオレニル基、9−アントラセニル基等を挙げることができ、好ましくは、フェニル基、p−(オクチル)フェニル基、p−(トリフルオロメチル)フェニル基、p−(パーフルオロオクチル)フェニル基である。
【0016】
本発明の一般式(1)で示されるビフェニレン誘導体の置換基R1〜R8の置換様式として、R1〜R4が、同一又は異なって、水素原子、フッ素原子、炭素数2〜20のアルキル基、炭素数2〜20のアルキニル基、炭素数2〜30のアルケニル基、及び炭素数4〜20のアリール基からなる群から選ばれる少なくとも一種以上の基であり、且つR5〜R8が、同一又は異なって、水素原子、フッ素原子、及び炭素数4〜20のアリール基からなる群から選ばれる少なくとも一種以上の基であることが好ましい。
【0017】
さらに置換基R1〜R4が、同一又は異なって、水素原子、フッ素原子、炭素数2〜20のアルキル基、炭素数2〜20のアルキニル基、及び炭素数4〜20のアリール基からなる群から選ばれる少なくとも一種以上の基であり、且つR5〜R8が、同一又は異なって、水素原子及び炭素数4〜20のアリール基からなる群から選ばれる少なくとも一種以上の基であることがより好ましい。
【0018】
記号mは1又は2であり、好ましくは1である。また、記号nは0〜2の整数を示し、好ましくは1である。そして、特に好ましい記号m、nとしては、いずれも1である。
【0019】
これらの中でも本発明の一般式(1)で示されるビフェニレン誘導体は、該ビフェニレン誘導体及び該ビフェニレン誘導体を含む耐酸化性有機半導体材料及びその有機薄膜が、高い耐酸化性及びキャリアー移動度を発現することから、以下の化合物が好ましく、
【0020】
【化2】
【0021】
【化3】
【0022】
【化4】
【0023】
【化5】
【0024】
【化6】
【0025】
【化7】
【0026】
【化8】
【0027】
【化9】
特に好ましくは
【0028】
【化10】
【0029】
【化11】
である。
(ジハロビフェニル誘導体)
次に、本発明の一般式(1)で示されるビフェニレン誘導体の原料として用いられる下記一般式(2)で示されるジハロビフェニル誘導体について述べる。
【0030】
【化12】
(ここで、置換基X1及びX2は臭素原子、ヨウ素原子又は塩素原子を示し、置換基R1〜R8、並びに記号m及びnは、一般式(1)で示される置換基並びに記号と同意義を示す。)
一般式(2)におけるX1及びX2は臭素原子、ヨウ素原子又は塩素原子であり、その中でも好ましくは臭素原子又はヨウ素原子であり、より好ましくは臭素原子である。
【0031】
本発明の一般式(2)における置換基R1及びR2の好ましい例は、フッ素原子、炭素数2〜20のアルキル基、炭素数2〜20のアルキニル基であり、さらに好ましい例はフッ素原子、炭素数5〜20のアルキル基である。
【0032】
置換基R3及びR4の好ましい例は、水素原子、フッ素原子、炭素数2〜20のアルキル基、炭素数2〜20のアルキニル基であり、さらに好ましい例はフッ素原子、炭素数5〜20のアルキル基である。
【0033】
置換基R5〜R8の特に好ましい例は、水素原子、炭素数2〜20のアルキル基、炭素数4〜20のアリール基であり、さらに好ましい例は水素原子である。
【0034】
本発明の一般式(2)で示されるジハロビフェニル誘導体としては、以下の化合物が好ましく、
【0035】
【化13】
【0036】
【化14】
【0037】
【化15】
【0038】
【化16】
【0039】
【化17】
【0040】
【化18】
【0041】
【化19】
【0042】
【化20】
特に好ましくは
【0043】
【化21】
【0044】
【化22】
【0045】
【化23】
である。
(ビフェニレン誘導体の製造方法)
次に、本発明の一般式(1)で示されるビフェニレン誘導体の製造方法について述べる。
【0046】
本発明の一般式(1)で示されるビフェニレン誘導体は一般式(2)で示されるジハロビフェニル誘導体を、銅又は銅化合物と反応させることで製造することができる。
【0047】
該反応で用いられる銅又は銅化合物は特に限定はなく、ヨウ素又は臭素と反応するものであれば良く、例えば銅、塩化銅(I)、臭化銅(I)、ヨウ化銅(I)、塩化銅(II)、臭化銅(II)、ヨウ化銅(II)を挙げることができ、好ましくは銅、塩化銅(II)である。また、この銅は亜鉛及び/又はスズとの合金であっても何ら差し支えなく使用することができる。なお、係る銅及び/又は銅合金の形状としては粉体状が好ましい。
【0048】
この銅又は銅化合物との反応は溶媒を用いて、若しくは用いないで実施することができる。溶媒を用いる場合、その溶媒例としては、例えばN,N−ジメチルホルムアミド、N−メチルピロリドン、アセトニトリル、ジメチルスルホキサイド等の極性溶媒を挙げることができる。係る銅又は銅化合物との反応において、用いる銅又は銅化合物の量は一般式(2)で示されるジハロビフェニル誘導体1当量に対し、1〜50当量が好ましく、特に好ましくは5〜30当量であり、反応温度は30〜250℃が好ましく、特に好ましくは50〜220℃であり、反応時間は1〜120分が好ましく、特に好ましくは3〜80分である。
【0049】
反応は、好ましくは窒素又はアルゴン等の不活性雰囲気下で実施する。得られた、一般式(1)で示されるビフェニレン誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0050】
本発明の一般式(1)で示されるビフェニレン誘導体の、さらに好ましい製造方法について述べる。
【0051】
さらに好ましい製造方法としては、一般式(2)で示されるジハロビフェニル誘導体をジリチオ化及び/又はジグリニャール化した後、銅化合物と反応させて製造する方法を挙げることができる。なお、ここでジリチオ化とは、一般式(2)における2個のハロゲンX1、X2をそれぞれリチウムに置換することを意味し、ジグリニャール化とは、一般式(2)における2個のハロゲンX1及びX2をそれぞれハロゲン化マグネシウムに置換することを意味する。
【0052】
一般式(2)で示されるジハロビフェニル誘導体をジリチオ化する場合、用いるリチオ化剤は、一般式(2)におけるハロゲンX1及びX2をリチウムに置換することができるものである限り特に限定はなく、例えばn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、メチルリチウム、ヘキシルリチウム等のアルキルリチウム;フェニルリチウム、p−tert−ブチルフェニルリチウム、p−メトキシフェニルリチウム、p−フルオロフェニルリチウム等のアリールリチウム;リチウムジイソプロピルアミド、リチウムヘキサメチルジシラジド等のリチウムアミド;リチウムパウダー等のリチウム金属を挙げることができ、好ましくはアルキルリチウムであり、特に好ましくはn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウムである。
【0053】
該リチオ化剤の使用量は、一般式(2)で示されるジハロビフェニル誘導体1当量に対し、1.5〜4当量が好ましく、さらに好ましくは1.8〜3当量、特に好ましくは1.9〜2.6当量である。
【0054】
該ジリチオ化反応は、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテル、ジプロピルエーテル、ジイソプロピルエーテル、ジブチルエーテル、エチレングリコールジメチルエーテル等のジアルキルエーテル、テトラヒドロフラン(以下、THFと略す)、ジグライム、ジオキサン、トルエン、ペンタン、ヘキサン、シクロヘキサン等が挙げられ、好ましくはジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテル、ジプロピルエーテル、ジイソプロピルエーテル、ジブチルエーテル、エチレングリコールジメチルエーテル等のジアルキルエーテルであり、特に好ましくはジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテルである。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。
【0055】
該ジリチオ化反応温度は−80〜50℃が好ましく、特に好ましくは−30〜30℃である。反応時間は1〜240分が好ましく、特に好ましくは1〜90分である。なお、ジリチオ化反応の進行は、反応液の一部を取り出し、水で反応を停止させた後、薄層クロマトグラフィー、ガスクロマトグラフィーで分析することで監視することもできる。
【0056】
本発明の一般式(1)で示されるビフェニレン誘導体の製造方法では、ジアルキルエーテル中でジリチオ化を実施することで、ジリチオ化の反応温度を−30〜30℃に上げることができ、反応性の低い一般式(2)で示されるジハロビフェニル誘導体も効率良くジリチオ化できることを見出した。
【0057】
該ジリチオ化反応により生成したジリチウム塩は、次いで銅化合物と反応させる。係る銅化合物との反応は、前記リチオ化反応により生成したジリチウム塩を含む反応混合物に銅化合物を直接用いて反応させる方法、生成したジリチウム塩を一度単離した後、銅化合物と反応させる方法のいずれを用いてもよい。
【0058】
該ジリチウム塩と銅化合物との反応は、該ジリチウム塩に銅化合物を添加する方法、あるいは銅化合物に該ジリチウム塩を添加するいずれの方法を用いても実施することができる。
【0059】
ジリチウム塩と銅化合物との反応に用いられる銅化合物は特に限定はなく、例えば塩化銅(II)、臭化銅(II)、ヨウ化銅(II)、酢酸銅(II)、アセチルアセトナート銅(II)等の2価銅;塩化銅(I)、臭化銅(I)、ヨウ化銅(I)、酢酸銅(I)等の1価銅等を挙げることができ、好ましくは2価銅であり、特に好ましくは塩化銅(II)、臭化銅(II)である。生成ジリチウム塩と銅化合物との反応は好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばTHF、ジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテル、エチレングリコールジメチルエーテル、ジグライム、ジオキサン等が挙げられ、好ましくはTHF、ジエチルエーテルである。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。用いる銅化合物の量は、一般式(2)で示されるジハロビフェニル誘導体1当量に対し、0.9〜5当量が好ましく、特に好ましくは1.0〜3.5当量である。銅化合物との反応温度は−90〜50℃が好ましく、特に好ましくは−80〜30℃であり、反応時間は1〜30時間が好ましく、特に好ましくは1〜18時間である。
【0060】
これらの銅化合物は、そのまま用いることもできるし、上記のジリチオ化反応で挙げた溶媒と混合した状態で用いることもできる。
【0061】
本発明の一般式(1)のビフェニレン誘導体の製造は、好ましくは窒素又はアルゴン等の不活性雰囲気下で実施する。
【0062】
かくして得られた、一般式(1)で示されるビフェニレン誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0063】
一般式(2)で示されるジハロビフェニル誘導体をジグリニャール化する場合、用いるグリニャール化剤は、例えばマグネシウム金属、臭化エチルマグネシウム、臭化イソプロピルマグネシウム等のアルキルグリニャール試薬を挙げることができ、好ましくはマグネシウム金属である。マグネシウム金属の形態は特に限定はなく、例えば削り状、リボン状、粒状を挙げることができる。
【0064】
該グリニャール化剤は、例えばマグネシウム金属の場合、一般式(2)で示されるジハロビフェニル誘導体1当量に対し1.8〜10当量が好ましい。グリニャール化反応は、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばジリチオ化反応で用いた溶剤を挙げることができる。グリニャール化反応の温度は−20〜80℃が好ましく、反応時間は1〜120分が好ましい。
【0065】
該ジグリニャール化反応により生成したマグネシウム塩は、次いで銅化合物と反応させる。該銅化合物との反応方法及び用いる銅化合物は、ジリチオ化反応により生成したジリチウム塩と銅化合物とを反応させる場合と同様な方法で実施できる。又、反応雰囲気、反応生成物の精製も同様な方法で実施できる。
【0066】
本発明の一般式(1)で示されるビフェニレン誘導体の製造方法では、一般式(2)で示されるジハロビフェニル誘導体をジリチオ化及び/又はジグリニャール化した後、塩化亜鉛と反応させた後、銅化合物と処理することもできる。
【0067】
(ジハロビフェニル誘導体の製造方法)
一般式(2)で示されるジハロビフェニル誘導体は、下記一般式(3)で示される2,3−ジハロアントラセン誘導体と下記一般式(4)で示される2,3−ジハロアントラセン誘導体から誘導される3−ハロアントラセニル金属試薬をパラジウム及び/又はニッケル触媒存在下でクロスカップリングさせることで製造することができる。
【0068】
【化24】
(ここで、置換基R1、R2、R5、R6、及びX1、並びに記号mは、一般式(2)で示される置換基並びに記号と同意義を示し、置換基X3は臭素原子又はヨウ素原子を示す。)
【0069】
【化25】
(ここで、置換基R3、R4、R7、R8、及びX2、並びに記号nは、一般式(2)で示される置換基並びに記号と同意義を示し、置換基X4は臭素原子又はヨウ素原子を示す。)
置換基X3及びX4は臭素原子又はヨウ素原子であり、その中でも好ましくはヨウ素原子である。
【0070】
該3−ハロアントラセニル金属試薬は、その原料である一般式(4)で示される2,3−ジハロアントラセン誘導体を、例えばイソプロピルマグネシウムブロマイド等のグリニャール試薬あるいはn−ブチルリチウム等の有機リチウム試薬により一般式(4)のハロゲンであるX4とのハロゲン/金属交換反応を行った後、塩化亜鉛、トリメトキシボラン、トリ(イソプロポキシ)ボラン、2−イソプロポキシ−4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン等と反応させることで好適に調製することができる。なお、グリニャール試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ オルガニック ケミストリィー」、2000年、65巻、4618−4634頁に記載されている方法、有機リチウム試薬によるハロゲン/金属交換反応は、例えば「ジャーナル オブ ケミカル リサーチ シノプシス」、1981年、185頁に記載されている−90℃以下でのハロゲンのリチオ化方法を用いることもできる。
【0071】
該3−ハロアントラセニル金属試薬と一般式(3)で示される2,3−ジハロアントラセン誘導体のクロスカップリング反応に用いる触媒はパラジウム及び/又はニッケル触媒であれば特に限定はなく、例えばテトラキス(トリフェニルホスフィン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム/トリフェニルホスフィン混合物、ビス(トリ−tert−ブチルホスフィン)パラジウム等の0価のパラジウム化合物;ジクロロビス(トリフェニルホスフィン)パラジウム、酢酸パラジウム/(トリ−tert−ブチルホスフィン)混合物、ジアセタトビス(トリフェニルホスフィン)パラジウム、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)パラジウム、ジクロロ(1,3−ビス(ジフェニルホスフィノ)プロパン)パラジウム、酢酸パラジウム/トリフェニルホスフィン混合物、酢酸パラジウム/2−(ジシクロヘキシルホスフィノ)−1,1’−ビフェニル混合物、ジクロロ(エチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルプロパンジアミン)パラジウム、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)パラジウム/トリフェニルホスフィン混合物等の2価のパラジウム化合物;ジクロロビス(トリフェニルホスフィン)ニッケル、ジクロロ(1,2−ビス(ジフェニルホスフィノ)エタン)ニッケル、ジクロロ(1,3−ビス(ジフェニルホスフィノ)プロパン)ニッケル、ジクロロ(エチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルプロパンジアミン)ニッケル、ジクロロ(N,N,N’,N’−テトラメチルエチレンジアミン)ニッケル/トリフェニルホスフィン混合物等の2価のニッケル化合物;ビス(1,5−シクロオクタジエン)ニッケル/トリフェニルホスフィン混合物等の0価のニッケル化合物を挙げることができ、中でも好ましくは0価あるいは2価のパラジウム化合物であり、特に好ましくはテトラキス(トリフェニルホスフィン)パラジウム、ジクロロビス(トリフェニルホスフィン)パラジウムである。
【0072】
該カップリング反応における、触媒の使用量は一般式(3)で示される2,3−ジハロアントラセン誘導体に対し、0.1〜20モル%の範囲が好ましい。また、一般式(4)から誘導される3−ハロアントラセニル金属試薬の使用量は一般式(3)で示される2,3−ジハロアントラセン誘導体1当量に対し、0.6〜1.5当量が好ましく、さらに好ましくは0.8〜1.4当量、特に好ましくは0.9〜1.2当量である。
【0073】
反応は好ましくは溶媒中で実施する。該溶媒として、例えばTHF、ジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテル、ジオキサン、エチレングリコールジメチルエーテル、エチレングリコール、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジイソプロピルアミン、ピペリジン、トルエン、キシレン、ヘキサン、シクロヘキサン、エタノール、水等を挙げることができる。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。例えばトルエン/水、トルエン/エタノール/水のような2乃至3成分系でも使用することができる。
【0074】
なお、一般式(3)と一般式(4)で示される2,3−ジハロアントラセン誘導体が同じ化合物であっても良い。
【0075】
なお、反応系中に塩基を存在させることもできる。この場合の塩基の種類としては特に限定はなく、例えば炭酸ナトリウム、炭酸水素ナトリウム、炭酸水素カリウム、炭酸カリウム、炭酸セシウム、りん酸カリウム、りん酸ナトリウム、水酸化ナトリウム、水酸化カリウム、フッ化カリウム等の無機塩基;ナトリウムメトキサイド、ナトリウムtert−ブトキサイド、カリウムtert−ブトキサイド等のアルコキサイド;トリエチルアミン、トリメチルアミン、トリプロピルアミン、トリブチルアミン、エチレンジアミン、N,N,N’,N’−テトラメチルエチレンジアミン、ジイソプロピルアミン、ピリジン、テトラブチルアンモニウムフルオライド等の有機塩基を好適なものとして挙げることができる。これらの塩基の使用量は一般式(3)で示される2,3−ジハロアントラセン誘導体1当量に対し、1.5〜10.0当量が好ましく、特に好ましくは2.0〜8.0当量である。さらにこれらの塩基と併用し、相間移動触媒を用いることもできる。相間移動触媒の種類は特に限定はなく、例えばトリオクチルメチルアンモニウムクロライド、テトラブチルアンモニウムクロライド、セチルピリジニウムクロライド、ベンジルトリメチルアンモニウムクロライド等を好適なものとして挙げることができる。これらの相間移動触媒の使用量は一般式(3)で示される2,3−ジハロアントラセン誘導体1当量に対し、0.1〜1.5当量が好ましく、特に好ましくは0.2〜0.8当量である。
【0076】
さらに反応系中にトリフェニルホスフィン等のホスフィンを存在させることもできる。これらのホスフィンの使用量は、該パラジウム及び/又はニッケル触媒1当量に対し、0.9〜8.0当量が好ましく、特に好ましくは1.0〜3.0当量である。
【0077】
反応の温度は10〜120℃が好ましく、特に好ましくは30〜100℃であり、反応時間は1〜48時間が好ましい。
【0078】
さらに、一般式(2)で示されるジハロビフェニル誘導体の別の製法について述べる。
【0079】
一般式(1)で示されるビフェニレン誘導体の前駆体である一般式(2)で示されるジハロビフェニル誘導体は、一般式(3)で示される2,3−ジハロアントラセン誘導体から誘導できる。
【0080】
即ち、一般式(2)で示されるジハロビフェニル誘導体は、一般式(3)で示される2,3−ジハロアントラセン誘導体をリチオ化剤又はグリニャール化剤を用いてホモカップリングすることで製造することができる。
【0081】
2,3−ジハロアントラセン誘導体のホモカップリング反応に用いるリチオ化剤は、一般式(3)におけるハロゲンX1又はX3をリチオ化することができるものである限り特に限定はなく、例えばn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、メチルリチウム、フェニルリチウム等の有機リチウム試薬;リチウムジイソプロピルアミド、リチウムヘキサメチルジシラジド等の有機リチウムアミド試薬;リチウム金属等を挙げることができ、好ましくは有機リチウム試薬であり、特に好ましくはn−ブチルリチウムである。
【0082】
該リチオ化反応は、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばTHF、ジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテル、ジオキサン、トルエン、ペンタン、ヘキサン、シクロヘキサン等であり、好ましくはTHFである。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。
【0083】
リチオ化剤は一般式(3)で示される2,3−ジハロアントラセン誘導体1当量に対し0.3〜1.2当量が好ましく、特に好ましくは0.4〜0.7当量である。リチオ化反応の温度は−100〜40℃が好ましく、特に好ましくは−90〜30℃であり、反応時間は1〜30時間が好ましく、特に好ましくは2〜20時間である。
【0084】
一方、一般式(3)で示される2,3−ジハロアントラセン誘導体のホモカップリング反応に用いるグリニャール化剤は、一般式(3)におけるハロゲンX1又はX3をグリニャール化することができるものである限り特に限定はなく、例えばマグネシウム金属、臭化エチルマグネシウム、臭化イソプロピルマグネシウム等のアルキルグリニャール試薬を挙げることができ、好ましくはマグネシウム金属である。マグネシウム金属の形態は特に限定はなく、例えば削り状、リボン状、粒状等を挙げることができる。
【0085】
該グリニャール化剤は、例えばマグネシウム金属の場合、一般式(3)で示される2,3−ジハロアントラセン誘導体1当量に対し0.4〜0.7当量が好ましい。グリニャール化反応は、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなく、例えばリチオ化反応で用いた溶剤を挙げることができる。グリニャール化反応の温度は−20〜100℃が好ましく、反応時間は1〜30時間が好ましい。
【0086】
一般式(2)で示されるジハロビフェニル誘導体の製造は、好ましくは窒素又はアルゴン等の不活性雰囲気下で実施する。
【0087】
かくして得られた、一般式(2)で示されるジハロビフェニル誘導体は、さらに精製することができる。精製する方法は特に限定はなく、例えばカラムクロマトグラフィー、再結晶化、あるいは昇華による方法を挙げることができる。
【0088】
(2,3−ジハロアントラセン誘導体の製造方法)
一般式(3)及び一般式(4)で示される2,3−ジハロアントラセン誘導体は、下記一般式(5)で示される2,3−ジハロアントラキノン誘導体から以下に述べる二通りの方法で製造することができる。
【0089】
【化26】
(ここで、置換基R1、R2、X1、及びX3、並びに記号mは、一般式(3)で示される置換基並びに記号と同意義を示す。)。
【0090】
(方法1)
一般式(3)で示される2,3−ジハロアントラセン誘導体における置換基R5及びR6が水素原子である場合、一般式(5)で示される2,3−ジハロアントラキノン誘導体をヒドリド還元剤を用いて還元し、さらに酸触媒で脱水することで製造することができる。
【0091】
用いるヒドリド還元剤は特に限定はなく、キノンをヒドロキシル基に還元できるものであれば良く、例えばジイソブチルアルミニウムヒドリド、ナトリウムボロヒドリド等を挙げることができる。該還元反応はTHF、ジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテル、トルエン、ヘキサン等の溶媒を用いて実施することが好ましい。反応温度は0〜60℃が好ましく、反応時間は1〜24時間が好ましい。ヒドリド還元剤の使用量は、一般式(5)で示される2,3−ジハロアントラキノン誘導体1当量に対し、1〜20当量が好ましい。
【0092】
次に該ヒドリド還元により得られたジヒドロキシル体を酸触媒を用いて脱水反応を行う。酸触媒としては、例えば塩酸、硫酸、トルエンスルホン酸、リン酸等が挙げられる。これらの酸触媒は水溶液としても用いることもできる。反応温度は10〜100℃が好ましく、反応時間は1〜24時間が好ましい。なお、先のヒドリド還元剤を用いる還元反応終了時に該酸触媒を添加し、そのまま脱水反応を実施することもできる。脱水反応に用いる酸触媒の使用量は、一般式(5)で示される2,3−ジハロアントラキノン誘導体1当量に対し、5〜80当量が好ましい。
【0093】
該ヒドリド還元反応及び脱水反応からなる工程は2〜4回繰り返すことで収率良く、一般式(3)で示される2,3−ジハロアントラセン誘導体を合成することができる。
【0094】
なお、一般式(5)で示される2,3−ジハロアントラキノン誘導体は既存の方法を用いて合成することができる。例えば、「ベリヒテ」(独国)、1933年、66B巻、1876−1891頁に記載されている方法を参考にして、4−ブロモ−5−ヨード無水フタル酸と1,2−ジアルキルベンゼンから合成することができる。
【0095】
(方法2)
一般式(3)で示される2,3−ジハロアントラセン誘導体における置換基R5及びR6が炭素数2〜20のアルキル基、フッ素原子、炭素数2〜20のアルキニル基、炭素数2〜30のアルケニル基、又は炭素数4〜20のアリール基である場合、一般式(5)で示される2,3−ジハロアントラキノン誘導体と下記一般式(6)及び/又は(7)で示される有機金属試薬と反応させた後、還元することで製造することができる。
【0096】
R5M1 (6)
R6M2 (7)
(ここで、置換基R5及びR6は、一般式(3)で示される置換基と同意義を示し、置換基M1及びM2は、リチウム又はマグネシウムハライドを示す。)
一般式(6)、(7)で示される有機金属試薬はキノンと反応できるものであれば特に限定はなく、例えば有機リチウム試薬、グリニャール試薬を挙げることができ、有機リチウム試薬の具体例としては、例えばオクチルリチウム、ドデシルリチウム、パーフルオロドデシルリチウム等のアルキルリチウム試薬;フェニルリチウム、トリルリチウム、p−オクチルフェニルリチウム、p−(パーフルオロオクチル)フェニルリチウム等のアリールリチウム試薬;エチニルリチウム、1−ドデシニルリチウム、1−(パーフルオロドデシニル)リチウム、フェニルエチニルリチウム等のアルキニルリチウム試薬;エテニルリチウム、1−ドデセニルリチウム、1−(パーフルオロドデセニル)リチウム、フェニルエテニルリチウム等のアルケニルリチウム試薬;等を挙げることができ、グリニャール試薬の具体例としては、例えばオクチルマグネシムブロミド、ドデシルマグネシウムブロミド、パーフルオロドデシルマグネシウムブロミド等のアルキルグリニャール試薬;フェニルマグネシウムブロミド、トリルマグネシウムブロミド、p−オクチルフェニルマグネシウムブロミド、p−(パーフルオロオクチル)フェニルマグネシウムブロミド等のアリールグリニャール試薬;エチニルマグネシウムブロミド、1−ドデシニルマグネシウムブロミド、1−(パーフルオロドデシニル)マグネシウムブロミド、フェニルエチニルマグネシウムブロミド等のアルキニルグルニャール試薬;エテニルマグネシウムブロミド、1−ドデセニルマグネシウムブロミド、1−(パーフルオロドデセニル)マグネシウムブロミド、フェニルエテニルマグネシウムブロミド等のアルケニルグルニャール試薬;等を挙げることができる。
【0097】
一般式(5)で示される2,3−ジハロアントラキノン誘導体と一般式(6)、(7)で示される有機金属試薬の反応はTHF、ジエチルエーテル、メチルtert−ブチルエーテル、エチルtert−ブチルエーテル、トルエン、ヘキサン等の溶媒を用いて実施することが好ましい。反応温度は−80〜60℃が好ましく、反応時間は5〜24時間が好ましい。該有機金属試薬の使用量は、一般式(5)で示される2,3−ジハロアントラキノン誘導体1当量に対し、2〜7当量が好ましい。次に該有機金属試薬との反応により得られたジヒドロキシル体を還元剤を用いて還元反応を行う。還元剤の具体例は、2塩化スズあるいは次亜リン酸ナトリウム/ヨウ化ナトリウムを用いることが好ましい。該還元反応は塩酸水溶液、酢酸等の溶媒を用いて実施することが好ましい。反応温度は20〜150℃が好ましく、反応時間は1〜5時間が好ましい。還元剤の使用量は、一般式(5)で示される2,3−ジハロアントラキノン誘導体1当量に対し、3〜10当量が好ましい。
【0098】
なお、9及び10位に置換基を有する一般式(3)で示される2,3−ジハロアントラセン誘導体は既存の方法を用いて合成することもできる。例えば、「シンレット」、2005年、217−222頁に記載されている方法を参考にして、2,3−ジハロアントラキノン誘導体から合成することができる。
(耐酸化性有機半導体材料)
次に、本発明の一般式(1)で示されるビフェニレン誘導体を含む耐酸化性有機半導体材料について述べる。該耐酸化性有機半導体材料は溶剤への溶解性、耐酸化性に優れ、好適な塗布性を有する。
【0099】
本発明の一般式(1)で示されるビフェニレン誘導体の溶解に用いる溶剤は、例えばo−ジクロロベンゼン、クロロベンゼン、1,2−ジクロロエタン、1,1,2,2−テトラクロロエタン、クロロホルム、ジクロロメタン等のハロゲン系溶剤;THF、ジオキサン等のエーテル系溶剤;トルエン、キシレン等の芳香族炭化水素系溶剤;酢酸エチル、γ−ブチロラクトン等のエステル系溶剤;N,N−ジメチルホルムアミド、N−メチルピロリドン等のアミド系溶剤;等であり、その中でも装置の腐食の観点から好ましくトルエン、キシレン等の芳香族炭化水素系溶剤である。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。
【0100】
上記に挙げた溶剤と一般式(1)で示されるビフェニレン誘導体を混合攪拌することにより、一般式(1)で示されるビフェニレン誘導体を含む耐酸化性有機半導体の溶液を調製することができる。この場合の温度は10〜130℃が好ましく、特に好ましくは20〜100℃である。得られる溶液の濃度は、溶剤及び温度により変えることができ、好ましくは0.01〜10.0重量%である。溶液の調製は空気中でも実施することができるが、好ましくは窒素、アルゴン等の不活性雰囲気下で調製する。
【0101】
一般式(1)で示されるビフェニレン誘導体を含む耐酸化性有機半導体材料の耐酸化性の評価は、該溶液を所定時間、空気と接触させる方法で実施することができる。まず用いる溶剤は予め脱気しておき、溶存酸素を除去する。空気との接触時間は、0.5分〜3時間が好適である。酸化の進行は、溶液の色の変化並びに薄層クロマトグラフィー、ガスクロマトグラフィー及び液体クロマトグラフィー分析による酸化物の検出により行うことができる。
【0102】
本発明の一般式(1)で示されるビフェニレン誘導体を含む耐酸化性有機半導体材料の溶液は、用いられる一般式(1)で示されるビフェニレン誘導体自体が適度の凝集性を有することから比較的に低温で溶剤へ溶解でき、且つ耐酸化性があることから、塗布法による有機薄膜の製造に好適に適用できる。即ち、雰囲気から厳密に空気を除く必要がないことから塗布工程を簡略化することができる。塗布は空気中でも実施できるが、好ましくは溶剤の乾燥を考慮して窒素気流下で行う。なお、好適な塗布性を得るために、本発明の一般式(1)で示されるビフェニレン誘導体を含む耐酸化性有機半導体材料の溶液の粘度は、0.005〜20ポアズの範囲にあることが好ましい。
(有機薄膜)
次に本発明の一般式(1)で示されるビフェニレン誘導体を含む耐酸化性有機半導体材料を用いた有機薄膜について述べる。係る有機薄膜は上記の耐酸化性有機半導体材料溶液の基板への塗布により製造することができる。
【0103】
基板への塗布による有機薄膜の製造は、該耐酸化性有機半導体材料溶液を基板上に塗布した後、加熱、気流、及び自然乾燥等の方法により溶剤を気化させることで実施することができる。該溶液中の一般式(1)で示されるビフェニレン誘導体の濃度は、特に限定はなく、例えば0.01〜10.0重量%であることが好ましい。塗布温度は特に限定はなく、例えば20〜130℃の間で好適に実施することができる。塗布の具体的方法は特に限定はなく、例えばスピンコート、キャストコート、ディップコート等を用いることができる。さらにスクリーン印刷、インクジェット印刷、グラビア印刷等の印刷技術を用いても作製することが可能である。使用する基板の材料は特に限定はなく、結晶性、非結晶性の種々の材料を用いることができる。また、基板は絶縁性あるいは誘電性を有する材料であっても良い。具体的な基板としては、例えばポリエチレンテレフタレート、ポリメチルメタクリレート、ポリエチレン、ポリプロピレン、ポリスチレン、環状ポリオレフィン、ポリイミド、ポリカーボネート、ポリビニルフェノール、ポリビニルアルコール、ポリエチレンナフタレート等のプラスチック基板;ガラス、石英、酸化アルミニウム、シリコン、酸化シリコン、二酸化タンタル、五酸化タンタル、インジウム錫酸化物等の無機材料基板;金、銅、クロム、チタン等の金属基板等を挙げることができる。またこれらの基板の表面は、例えばオクタデシルトリクロロシラン、オクチルトリクロロシラン、オクタデシルトリメトキシシラン等のシラン類で修飾処理したものであっても使用することができる。塗布した後の溶剤の乾燥は、常圧若しくは減圧で除去することができる、又、加熱により乾燥してもよい。さらに、溶剤の気化速度を調節することで本発明の一般式(1)で示されるビフェニレン誘導体の結晶成長を制御することができる。基板への塗布により得られる有機薄膜の膜厚は特に限定はなく、好ましくは1nm〜100μm、特に好ましくは10nm〜20μmである。なお、このようにして得られた薄膜は、60〜150℃に加熱することでより高度に配列させることもできる。
【0104】
(発光材料)
本発明で得られた一般式(1)で示されるビフェニレン誘導体は、ビフェニレン誘導体の溶液の基板への塗布により発光材料として用いることができ、とりわけ有機電界発光素子の材料として用いることができる。従って、一般式(1)で示されるビフェニレン誘導体は有機EL材料としての使用が期待される。
【0105】
一般式(1)で示されるビフェニレン誘導体の溶液にする際の溶剤は、特に限定はなく、例えばo−ジクロロベンゼン、クロロベンゼン、1,2−ジクロロエタン、1,1,2,2−テトラクロロエタン、クロロホルム等のハロゲン系溶剤;THF、ジオキサン等のエーテル系溶剤;トルエン、キシレン、メシチレン等の芳香族化合物の炭化水素系溶剤;酢酸エチル、γ−ブチロラクトン等のエステル系溶剤;N,N−ジメチルホルムアミド、N−メチルピロリドン等のアミド系溶剤;等が挙げられる。又、これら溶剤は1種若しくは2種以上の混合物を用いても良い。中でも、好ましくはクロロベンゼン、トルエン等である。
【0106】
上記に挙げた溶剤と一般式(1)で示されるビフェニレン誘導体を混合攪拌することにより、一般式(1)で示されるビフェニレン誘導体の溶液となるものである。混合攪拌する際の温度は10〜200℃が好ましく、特に好ましくは20〜150℃である。混合攪拌する際の一般式(1)で示されるビフェニレン誘導体の濃度は、溶剤及び温度により変えることができ、0.01〜10.0重量%であることが好ましい。溶液の調製は空気中でも実施することができるが、好ましくは窒素、アルゴン等の不活性雰囲気下で調製する。
【0107】
一般式(1)で示されるビフェニレン誘導体を含む溶液の塗布は空気中でも実施できるが、好ましくは溶剤の乾燥を考慮して窒素気流下で行う。なお、好適な塗布性を得るために、本発明の一般式(1)で示されるビフェニレン誘導体を含む溶液の粘度は、0.005〜20ポアズの範囲にあることが好ましい。
【0108】
次に本発明の一般式(1)で示されるビフェニレン誘導体を含む溶液の塗布による発光材料の作製について述べる。係る発光材料は上記の溶液の基板への塗布により製造することができる。
【0109】
基板への塗布による発光材料の製造は、前記溶液を基板上に塗布した後、加熱、気流及び自然乾燥等の方法により溶剤を気化させることで実施することができる。該溶液中の一般式(1)で示されるビフェニレン誘導体の濃度は、特に限定はなく、例えば0.01〜10.0重量%であることが好ましい。塗布温度は特に限定はなく、例えば20〜200℃の間、特に好ましくは30〜100℃の間で好適に実施することができる。塗布の具体的方法は特に限定はなく、例えばスピンコート、キャストコート及びディップコート等を用いることができる。さらにスクリーン印刷、インクジェット印刷、グラビア印刷等の印刷技術を用いても作製することが可能である。使用する基板の材料は特に限定はなく、結晶性、非結晶性の種々の材料を用いることができる。基板の具体例としては、例えばポリエチレンテレフタレート、ポリエチレンナフタレート、ポリメチルメタクリレート、ポリエチレン、ポリプロピレン、ポリスチレン、環状ポリオレフィン、ポリイミド、ポリカーボネート、ポリビニルフェノール、ポリビニルアルコール、ポリ(ジイソプロピルフマル酸)、ポリ(ジエチルフマル酸)等のプラスチック基板;ガラス、石英、酸化アルミニウム、シリコン、酸化シリコン、二酸化タンタル、五酸化タンタル、インジウム錫酸化物等の無機材料基板;金、銅、クロム、チタン、アルミニウム等の金属基板を好適に用いることができる。またこれらの基板の表面は、例えばオクタデシルトリクロロシラン、オクタデシルトリメトキシシラン等のシラン類;ヘキサメチルジシラザン等のシリルアミン類で修飾処理したものであっても使用することができる。さらに、基板は絶縁性あるいは誘電性を有する材料であっても良い。塗布した後の溶剤の乾燥は、常圧若しくは減圧で除去することができる、又、加熱、窒素気流により乾燥してもよい。さらに、溶剤の気化速度を調節することで一般式(1)で示されるビフェニレン誘導体の膜質を制御することができる。基板への塗布により得られる発光材料の膜厚は特に限定はなく、好ましくは1nm〜100μm、特に好ましくは10nm〜20μmである。
【0110】
また、本発明の一般式(1)で示されるビフェニレン誘導体を用いた発光材料は、真空蒸着法によっても薄膜を作製することができる。真空蒸着法による薄膜は、汎用の真空蒸着装置を用いることにより行うことができる。真空蒸着法で薄膜を作製する際の真空槽の真空度は、一般に用いられる拡散ポンプ、ターボ分子ポンプ、クライオポンプ等により到達し得る1.3×10−2〜1.3×10−5パスカルが好ましい。蒸着速度は、形成する膜圧によるが、0.005〜1.0nm/秒が好ましい。
【0111】
本発明の一般式(1)で示されるビフェニレン誘導体を用いた発光材料の発光量子収率は、好ましくは2〜80%、特に好ましくは3〜50%である。
【0112】
本発明の一般式(1)で示されるビフェニレン誘導体は平面剛直性の高い分子構造を有することから、優れた半導体特性を与えることが期待できる。該ビフェニレン誘導体はトルエン等の溶媒に溶解し、溶液状態にあっても容易に空気酸化されることはない。従って、塗布法により半導体薄膜を容易に作製できる。また、本発明の一般式(1)で示されるビフェニレン誘導体は、長い共役系を有することから、黄色から赤色の長波長の発光を与えるアントラセン環を含むことから発光材料としても期待することができる。したがって、本発明の一般式(1)で示されるビフェニレン誘導体は電子ペーパー、有機ELディスプレイ、液晶ディスプレイ、又はICタグ用等のトランジスタの有機半導体活性相用途、さらに有機ELディスプレイの発光材料、ホスト材料、有機半導体レーザー材料、有機薄膜太陽電池材料、発光有機トランジスタ、フォトニック結晶材料等に利用することができる。
【発明の効果】
【0113】
優れた耐酸化性を有し、塗布法による半導体活性相形成が可能な、ビフェニレン誘導体及びその用途を提供する。さらに本発明の製造法では置換基を導入したビフェニレン誘導体を製造することができ、新規な有機半導体材料を提供することができる。
【実施例】
【0114】
以下、実施例により本発明をさらに詳細に説明するが、本発明はこれら実施例にのみ限
定されるものではない。
【0115】
生成物の同定には1H NMRスペクトル及びマススペクトルを用いた。なお、1H NMRスペクトルは日本電子製JEOL GSX−270WB(270MHz)を用いて測定した。マススペクトル(MS)は日本電子製JEOL JMS−700を用いて、試料を直接導入し、電子衝突(EI)法(70エレクトロンボルト)又はFAB法(6キロエレクトロンボルト、キセノンガス、マトリックス(2−ニトロフェニルオクチルエーテル)で測定した。
【0116】
反応の進行の確認等は薄層クロマトグラフィー、ガスクロマトグラフィー及びガスクロマトグラフィー−マススペクトル(GCMS)分析を用いた。
【0117】
ガスクロマトグラフィー分析
装置 島津GC14B
カラム J&Wサイエンティフィック社製、DB−1,30m
ガスクロマトグラフィー−マススペクトル分析
装置 パーキンエルマーオートシステムXL(MS部;ターボマスゴールド)
カラム J&Wサイエンティフィック社製、DB−1,30m
反応用の溶媒は、断りのない限り市販品を用いた。なお、グリニャール試薬あるいはブチルリチウム等の有機金属試薬を用いた場合は、市販の脱水溶媒をそのまま用いた。
【0118】
発光材料としての評価は、薄膜状態での発光量子収率及び蛍光スペクトルを測定することで実施した。なお、発光量子収率は浜松ホトニクス製C9920−01を用い、蛍光スペクトルは日本分光製FP6500を用いて測定した。
【0119】
合成例1 (4−ブロモ−5−ヨード無水フタル酸の合成)
4−ブロモ−5−ヨード無水フタル酸は「ジャーナル オブ オーガニック ケミストリー」(米国)、1951年、16巻、1577−1581頁を参考に、以下の様に合成した。
【0120】
4−ブロモフタルイミド(東京化成工業製)9.95g(44.0mmol)を窒素ガスで置換した50mlの二口ナスフラスコに入れた。次いでヨウ素5.87g(23.1mmol)及び10%発煙硫酸(ヨツハタ化学工業製)12mlを加え、90℃で23時間反応を行った。反応混合物を室温に冷やして氷に注ぎ入れた後、ガラスフィルターでろ過し、黄色固体12.8gを得た。得られた固体を濃硫酸35mlに溶解させ、130℃で5時間反応を行った。反応混合物を氷冷後、氷水を加えて析出した固体をろ過し、フタル酸誘導体の固体13.8gを得た。次に得られた固体を、水酸化ナトリウム3.6gを水18mlに溶かした水溶液に室温で溶かした。この塩基性水溶液に酢酸を加えpHを3〜4に調整し、析出するフタル酸誘導体のモノナトリウム塩の白色沈殿をろ過した。得られた白色固体を水に懸濁させ、濃塩酸でpHを1以下にし、再びフタル酸誘導体として白色固体4.93gを得た。この固体をトルエン48mlに溶かし、無水酢酸8.7g(85.7mmol)を加え、105℃で4時間反応を行った。反応液を減圧濃縮して白色固体3.79gを得た。この固体から加熱トルエンに不溶な成分を除き、目的の4−ブロモ−5−ヨード無水フタル酸5.13g(14.5mmol)を得た(収率33%)。
1H NMR(CDCl3,22℃):δ=8.51(s,1H),8.23(s,1H)。
MS m/z: 353(M+,100%),309(M+−CO2,18%),282(M+−C2O3,10%),155(M+−C2O3−I,16%),74(M+−C2O3−I−Br,32%)。
【0121】
合成例2 (1,2―ジドデシルベンゼンの合成)
1,2−ジドデシルベンゼンは「日本化学会誌」1989年、983−987頁に従い以下の様に合成した。
【0122】
1,2−ジクロロベンゼン2.22g(15.1mmol)、ジクロロ〔1,3−ビス(ジフェニルホスフィノ)プロパン〕ニッケル(東京化成工業製)131mg(0.24mmol)、乾燥ジエチルエーテル11.5mlの混合液にドデシルマグネシウムブロミド(シグマ−アルドリッチ製、1.0mol/lジエチルエーテル溶液)45ml(45mmol)を窒素雰囲気中0℃で滴下した。35℃で20時間反応を行い、反応混合物を0℃に冷却し希塩酸を加え、ジエチルエーテルで抽出した。ジエチルエーテル溶液を水、飽和炭酸水素ナトリウム水溶液、水の順に洗浄し、塩化カルシウムで乾燥させた。得られた液体をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン)及び減圧蒸留で精製し、目的の1,2―ジドデシルベンゼン5.56g(13.4mmol)を得た(収率88%)。
1H NMR(CDCl3,22℃):δ=7.14−7.09(m,4H),2.59(t,J=7.8Hz,4H),1.55(m,4H),1.26(m,36H),0.88(t,J=6.8Hz,6H)。
MS m/z: 414(M+,100%),260(M+−C11H23,71%),106(M+−C22H46,98%)。
【0123】
合成例3 (2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノンの合成)(一般式(5)の化合物)
2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノンは「ベリヒテ」(独国)、1933年、66B巻、1876−1891頁を参考に以下の様に合成した。
【0124】
合成例1で得られた4−ブロモ−5−ヨード無水フタル酸2.82g(8.00mmol)、合成例2で得られた1,2−ジドデシルベンゼン3.32g(8.00mmol)、テトラクロロエタン5.0mlの混合液に塩化アルミニウム2.41g(18.1mmol)を加え、室温で3時間反応を行った。水を加えてクエンチし、さらに水洗浄を行い、真空加熱乾燥後、白色固体6.2gを得た。得られた固体を濃硫酸44mlに溶かし、80℃で1時間反応した。反応混合物を氷に注ぎ入れ、析出した固体をろ過して水で洗浄した。減圧乾燥後、シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:塩化メチレン=10:1)及びヘプタンからの再結晶で精製し、2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノンの黄色固体4.20g(5.60mmol)を得た(収率70%)。
1H NMR(CDCl3,22℃):δ=8.73(s,1H),8.45(s,1H),8.05(s,2H),2.75(m,4H),1.62(m,4H),1.26(m,36H),0.88(m,6H)。
MS m/z: 750(M+,100%),440(M+−C22H46,8%),313(M+−C22H46−I,2%),233(M+−C22H46−I−Br,1%)。
【0125】
合成例4 (2−ブロモ−3−ヨード−6,7−ジドデシルアントラセンの合成)(一般式(3)及び(4)の化合物)
窒素雰囲気下、100mlシュレンク反応容器に合成例3で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノン1.10g(1.47mmol)を入れた。次いでTHF17mlを加え、ヒドリド還元剤として水素化ジイソブチルアルミニウム(関東化学製、0.99mol/l、トルエン溶液)4.0ml(4.0mmol)を加え、室温で1.5時間還元反応を行った。次いで反応混合物に酸触媒として6M塩酸水溶液10mlを加え、65℃で3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。再びTHF17mlを加え、水素化ジイソブチルアルミニウム5.5ml(5.4mmol)を加え、室温で1.5時間還元反応を行った。次いで反応混合物に6M塩酸水溶液10mlを加え、3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(溶離液:ヘキサン)で精製し、2−ブロモ−3−ヨード−6,7−ジドデシルアントラセンの黄色固体629mg(0.87mmol)を得た(収率59%)。
1H NMR(CDCl3,22℃):δ=8.55(s,1H),8.27(s,1H),8.16(s,1H),8.15(s,1H),7.72(s,2H),2.78(m,4H),1.71(m,4H),1.27(m,36H)0.88(m,6H)。
MS m/z: 720(M+,100%),410(M+−C22H46,16%),283(M+−C22H46−I,4%),203(M+−C22H46−I−Br,5%)。
【0126】
実施例1 (6,7,6’,7’−テトラドデシル−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)[一般式(2)で示されるジハロビフェニル誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に合成例4で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラセン205mg(0.285mmol)及びTHF8mlを添加した。この溶液を−55℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.81M)のTHF溶液0.70ml(0.57mmol)を滴下した。5分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)59.2mg(0.57mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮した。得られた固形物に、合成例4で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラセン210mg(0.292mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)16.5mg(0.014mmol)、トルエン5ml、及びエタノール1.2mlを添加した。さらに炭酸ナトリウム92.8mg(0.873mmol)と水1.6mlからなる水溶液を加え、60℃で24時間反応を実施した。室温まで冷却後、トルエン及び水を添加し分相した。有機相を濃縮し、得られた残渣をトルエン5mlに溶解後、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.02mlを添加し、室温で2時間撹拌した。このトルエン溶液を水で2回洗浄後、無水硫酸ナトリウムで乾燥した。有機相を濾過し、減圧濃縮し、得られた残渣をシリカゲルを充填したカラムで濾過した(溶媒、ヘキサン)。得られた粗固体をヘプタンから再結晶化し、目的物の黄色固体287mgを得た(収率85%)。
1H NMR(CDCl3,22℃):δ=8.33(s,2H),8.31(s,2H),8.29(s,2H),7.97(s,2H),7.78(s,2H),7.75(s,2H),2.79(m,8H),1.72(m,8H),1.28(m,72H)0.87(m,12H)。
1H NMRスペクトルを図1に示した。
MS m/z: 1185(M+,2%),592(M+/2,100)。
【0127】
1H NMR及びMS測定より、6,7,6’,7’−テトラドデシル−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお。その構造式を下記に示す。
【0128】
【化27】
実施例2 (テトラドデシルジナフトビフェニレンの合成)[一般式(1)で示されるビフェニレン誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に実施例1で合成した6,7,6’,7’−テトラドデシル−3,3’−ジブロモ−2,2’−ビアントラセニル240mg(0.202mmol)及びジエチルエーテル7mlを添加した。この混合物を0℃に冷却後、リチオ化剤であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.28ml(0.44mmol)を滴下した。1時間かけて5℃まで昇温しメタル化の熟成を行った。一方、別の100mlシュレンク反応容器に塩化銅(II)(和光純薬工業製)95.3mg(0.709mmol)及びTHF10mlを添加し、−78℃に冷却した。ここへ先のメタル化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。徐々に昇温し、一晩かけて室温まで反応温度を上げた。3M塩酸水溶液及びトルエンを添加した。分相し、有機相をさらに水で洗浄し、無水硫酸ナトリウムで乾燥した。有機相を濾過し、減圧濃縮し、得られた粗固体をトルエンから再結晶化し、目的物の黄色固体108mgを得た(収率52%)。
1H NMR(重トルエン,60℃):δ=7.95(s,4H),7.65(s,4H),7.40(s,4H),2.83(t,J=8.5Hz,8H),1.78(m,8H),1.31(m,72H)0.92(m,12H)。
1H NMRスペクトルを図2に示した。
FABMS m/z: 1026(M+)。
【0129】
1H NMR及びMS測定より、テトラドデシルジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0130】
【化28】
合成例5 (2−ブロモ−3−ヨード−6,7−(ドデシル)フルオロアントラセンの合成)(一般式(3)及び(4)の化合物)
合成例2で1,2−ジクロロベンゼンの代わりに、1−クロロ−2−フルオロベンゼン(東京化成工業製)を用いた以外は合成例2と同じ操作を繰り返して1−ドデシル−2−フルオロベンゼンを合成した(収率80%)。この1−ドデシル−2−フルオロベンゼンと合成例1で得られた4−ブロモ−5−ヨード無水フタル酸を用い、合成例3と同じ操作を繰り返し2−ブロモ−3−ヨード−6,7−(ドデシル)フルオロアントラキノンを得(収率54%)、さらに合成例4と同じ操作を繰り返して、2−ブロモ−3−ヨード−6,7−(ドデシル)フルオロアントラセンへ変換した(収率63%)。
【0131】
実施例3 (ジドデシルジフルオロ−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)[一般式(2)で示されるジハロビフェニル誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に合成例5で合成した2−ブロモ−3−ヨード−6,7−(ドデシル)フルオロアントラセン464mg(0.815mmol)及びTHF10mlを添加した。この溶液を−60℃に冷却し、イソプロピルマグネシウムブロマイド(関東化学製、0.65M)のTHF溶液1.3ml(0.84mmol)を滴下した。5分間熟成後、−78℃に冷却し、塩化亜鉛(シグマ−アルドリッチ製、1.0M)のジエチルエーテル溶液0.84ml(0.84mmol)を滴下した。徐々に室温まで昇温した後、生成したスラリー液を減圧濃縮した。得られた固形物に、合成例5で合成した2−ブロモ−3−ヨード−6−ドデシル−7−フルオロアントラセン471mg(0.827mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)47.1mg(0.041mmol)、及びTHF8mlを添加した。64℃で8時間反応を実施した。室温まで冷却後、トルエン及び水を添加し分相した。有機相を濃縮し、得られた残渣をトルエン5mlに溶解後、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.1mlを添加し、室温で2時間撹拌した。このトルエン溶液を水で2回洗浄後、有機相を減圧濃縮し、得られた残渣に飽和食塩水及びトルエンを添加した。分相し、有機相を水で洗浄し、無水硫酸ナトリウムで乾燥した。有機相を濾過し、減圧濃縮し、得られた残渣をシリカゲルを充填したカラムで濾過した(溶媒;ヘキサン)。濾液を減圧濃縮し、得られた粗固体をヘプタンから再結晶化し、ジドデシルジフルオロ−3,3’−ジブロモ−2,2’−ビアントラセニルの2種類の異性体の混合物からなる黄色固体440mgを得た(収率61%)。
MS m/z: 885(M+,4%),725(M+−2Br,100)。
【0132】
MS測定より、ジドデシルジフルオロ−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお、その構造式を下記に示す。
【0133】
【化29】
実施例4 (ジドデシルジフルオロジナフトビフェニレンの合成)[一般式(1)で示されるビフェニレン誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に、実施例3で合成したジドデシルジフルオロ−3,3’−ジブロモ−2,2’−ビアントラセニル388mg(0.438mmol)及びジエチルエーテル7mlを添加した。この混合物を0℃に冷却し、リチオ化剤であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.60ml(0.95mmol)を滴下した。0℃で80分間撹拌した(リチオ化反応)。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)177mg(1.31mmol)及びTHF15mlを添加し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温までゆっくり昇温し、3M塩酸水溶液を添加した。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、2種類のジドデシルジフルオロジナフトビフェニレンの2種類の異性体の混合物からなる黄色固体146mgを得た(収率46%)。
FABMS m/z: 725(M+)。
【0134】
MS測定より、ジドデシルジフルオロジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0135】
【化30】
合成例6 (4−ブロモ−1,2−ジヨードベンゼンの合成)
100mlシュレンク反応容器に、1,2−ジヨードベンゼン(東京化成工業製)5.56g(16.8mmol)及びジクロロメタン30mlを添加し、0℃に冷却した。鉄粉(シグマ−アルドリッチ製)67mg及びヨウ素10mg(0.04mmol)を添加後、臭素0.87ml(17mmol)を滴下した。0℃で8時間撹拌後、亜硫酸水素ナトリウム水溶液を添加し、反応を停止させた。有機相を水で洗浄後、無水硫酸ナトリウムで乾燥した。減圧濃縮し、得られた残渣をTHF:メタノール=1:1から2回再結晶化し、4−ブロモ−1,2−ジヨードベンゼン4.94gの白色結晶を得た(収率72%)。
【0136】
合成例7 (4−ブロモ−1,2−(パーフルオロドデシル)ベンゼンの合成)
4−ブロモ−1,2−(パーフルオロドデシル)ベンゼンは、「ジャーナル オブ フルオリン ケミストリィー」、1989年、43巻、207−228頁を参考に次のように合成した。
【0137】
窒素雰囲気下、100mlシュレンク反応容器に、銅粉(カッパーブロンズ)(シグマ−アルドリッチ製)2.85g(44.8mmol)、パーフルオロドデシルアイオダイド(シンクエスト製)18.4g(24.6mmol)、合成例6で合成した4−ブロモ−1,2−ジヨードベンゼン4.58g(11.2mmol)、及びジメチルスルホキシド(和光純薬工業製、脱水品)18mlを添加し、125℃に加熱し、8時間反応させた。室温に冷却後、水を添加し反応を停止させた。さらにジエチルエーテルを添加し、混合物をセライトを用いて濾過した。濾液をジエチルエーテル抽出し、合わせた有機相を水で洗浄し、無水硫酸マグネシウムで乾燥した。減圧濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィーで精製し(溶媒;ヘキサン)、4−ブロモ−1,2−(パーフルオロドデシル)ベンゼンの無色の液体6.24gを得た(収率40%)。
【0138】
合成例8 (2−ブロモ−3−ヨード−6,7−ジ(パーフルオロドデシル)アントラキノンの合成)(一般式(5)の化合物)
窒素雰囲気下、300mlシュレンク反応容器に、合成例7で得られた4−ブロモ−1,2−(パーフルオロドデシル)ベンゼン6.11g(4.39mmol)及びTHF80mlを添加した。この溶液を−50℃に冷却し、イソプロピルマグネシウムブロマイド(関東化学製、0.65M)のTHF溶液6.8ml(4.4mmol)を滴下した。−50℃で30分熟成後、ここに合成例1で合成した4−ブロモ−5−ヨード無水フタル酸1.48g(4.20mmol)とTHF20mlからなる溶液を滴下した。反応混合物を一晩かけて室温まで昇温した後、氷冷し3M塩酸水溶液を添加した。ジエチルエーテルで抽出し、合わせた有機相を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。減圧下濃縮し、白色固体7.00g得た。得られた固体に濃硫酸40mlを添加し、80℃で12時間反応した。反応混合物を氷に注ぎ入れ、析出した固体をろ過して水で洗浄した。乾燥後、シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:塩化メチレン=15:1)及びヘプタンからの再結晶で精製し、2−ブロモ−3−ヨード−6,7−ジ(パーフルオロドデシル)アントラキノンの固体1.86g(1.13mmol)を得た(収率27%)。
【0139】
合成例9 (2−ブロモ−3−ヨード−6,7−ジ(パーフルオロドデシル)アントラセンの合成)(一般式(3)及び(4)の化合物)
窒素雰囲気下、100mlシュレンク反応容器に合成例8で合成した2−ブロモ−3−ヨード−6,7−ジ(パーフルオロドデシル)アントラキノン1.81g(1.10mmol)を入れた。次いでTHF15mlを加え、ヒドリド還元剤である水素化ジイソブチルアルミニウム(関東化学製、0.99mol/l、トルエン溶液)3.5ml(3.5mmol)を加え、室温で1.5時間還元反応を行った。次いで反応混合物に酸触媒として6M塩酸水溶液10mlを加え、65℃で3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。再びTHF15mlを加え、水素化ジイソブチルアルミニウム3.5ml(3.5mmol)を加え、室温で1.5時間還元反応を行った。次いで反応混合物に6M塩酸水溶液10mlを加え、3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン)で精製し、2−ブロモ−3−ヨード−6,7−ジ(パーフルオロドデシル)アントラセンの黄色固体1.12gを得た(収率63%)。
【0140】
実施例5 (6,7,6’,7’−テトラ(パーフルオロドデシル)−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)[一般式(2)で示されるジハロビフェニル誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に合成例9で合成した2−ブロモ−3−ヨード−6,7−ジ(パーフルオロドデシル)アントラセン483mg(0.298mmol)及びTHF7mlを添加した。この溶液を−70℃に冷却し、イソプロピルマグネシウムブロマイド(関東化学製、0.65M)のTHF溶液0.46ml(0.30mmol)を滴下した。10分間熟成後、−78℃に冷却し、塩化亜鉛(シグマ−アルドリッチ製、1.0Mジエチルエーテル溶液、)0.30ml(0.30mmol)を滴下した。徐々に室温まで昇温した後、減圧濃縮した。得られた残渣に、合成例9で合成した2−ブロモ−3−ヨード−6,7−ジ(パーフルオロドデシル)アントラセン490mg(0.303mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)17.3mg(0.015mmol)、THF8mlを加え、64℃で10時間反応を実施した。容器を水冷し3M塩酸水溶液3mlを添加することで反応を停止させた。トルエンを添加後、分相し、有機相を食塩水で洗浄した。有機相を減圧濃縮し溶媒を留去し、さらに真空乾燥した。得られた残渣にトルエンを添加し、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)(0.06ml)を添加し、室温で2時間撹拌した。この溶液を水洗浄し、有機相を減圧濃縮析出した。残渣をシリカゲルカラムクロマトグラフィーで精製し(溶媒;ヘキサン及びヘキサン:クロロホルム=10:1)、6,7,6’,7’−テトラ(パーフルオロドデシル)−3,3’−ジブロモ−2,2’−ビアントラセニルの黄色固体507mgを得た(収率57%)。
FABMS m/z: 2985(M+)。
【0141】
MS測定より、6,7,6’,7’−テトラ(パーフルオロドデシル)−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお、その構造式を下記に示す。
【0142】
【化31】
実施例6 (テトラ(パーフルオロドデシル)ジナフトビフェニレンの合成)[一般式(1)で示されるビフェニレン誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に、実施例5で合成した6,7,6’,7’−テトラ(パーフルオロドデシル)−3,3’−ジブロモ−2,2’−ビアントラセニル500mg(0.167mmol)及びジエチルエーテル7mlを添加した。この混合物を0℃に冷却し、リチオ化剤であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.23ml(0.37mmol)を滴下し、0℃で90分間撹拌した(リチオ化反応)。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)67.4mg(0.501mmol)及びTHF16mlを添加し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温までゆっくり昇温し、3M塩酸水溶液を添加した。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、目的物の黄色固体184mgを得た(収率39%)。
FABMS m/z: 2825(M+)。
【0143】
MS測定より、テトラ(パーフルオロドデシル)ジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0144】
【化32】
合成例10 (4−ブロモ−1,2−ジフェニルベンゼンの合成)
窒素雰囲気下、200mlシュレンク反応容器に、合成例6で得られた4−ブロモ−1,2−ジヨードベンゼン2.15g(5.25mmol)にジヒドロキシフェニルボラン(和光純薬工業製)1.47g(12.1mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)458mg(0.40mmol)、炭酸ナトリウム3.34g(31.5mmol)、トルエン42ml、エタノール10.5ml、水13.3mlを加え、80℃で29時間反応させた。1M塩酸水溶液を加えて反応をクエンチし、トルエンで抽出した後、有機層を水洗浄して無水硫酸ナトリウムで乾燥した。溶媒を減圧留去し、シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン)で精製して目的の4−ブロモ−1,2−ジフェニルベンゼン1.46g(4.72mmol)を得た(収率90%)。
【0145】
合成例11 (2−ブロモ−3−ヨード−6,7−ジフェニルアントラキノンの合成)(一般式(5)の化合物)
窒素雰囲気下、300mlシュレンク反応容器に、合成例10で得られた4−ブロモ−1,2−ジフェニルベンゼン1.46g(4.72mmol)を入れた。次いでTHF28mlを加えて−78℃に冷却し、n−ブチルリチウム(関東化学製、1.59mol/l、ヘキサン溶液)3.0ml(4.77mmol)を加え、30分間反応させた。次いで合成例1で合成した4−ブロモ−5−ヨード無水フタル酸1.66g(4.72mmol)を加え、室温まで昇温した。水を加えてクエンチし得られた固体を濾過し、さらに水洗浄を行い、加熱真空乾燥後白色固体3.2gを得た。得られた固体に濃硫酸26mlを添加し、80℃で1時間反応した。反応混合物を氷に注ぎ入れ、析出した固体をろ過して水で洗浄した。乾燥後、シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:塩化メチレン=10:1)及びヘプタンからの再結晶で精製し、2−ブロモ−3−ヨード−6,7−ジフェニルアントラキノンの黄色固体298mg(0.53mmol)を得た(収率11%)。
【0146】
合成例12 (2−ブロモ−3−ヨード−6,7−ジフェニルアントラセンの合成)(一般式(3)及び(4)の化合物)
窒素雰囲気下、50mlシュレンク反応容器に、合成例11で合成した2−ブロモ−3−ヨード−6,7−ジフェニルアントラキノン243mg(0.43mmol)及びTHF5mlを加えた。ヒドリド還元剤である水素化ジイソブチルアルミニウム(関東化学製、0.99mol/l、トルエン溶液)1.20ml(1.20mmol)を滴下し、室温で1.5時間還元反応を行った。この反応混合物に酸触媒である6M塩酸水溶液3mlを加え、65℃で3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して、無水硫酸ナトリウムで乾燥、減圧濃縮した。得られた残渣に再びTHF5mlを加え、水素化ジイソブチルアルミニウム(関東化学製、0.99mol/l、トルエン溶液)1.20ml(1.20mmol)を加え、室温で1.5時間還元反応を行った。次いで反応混合物に6M塩酸水溶液3mlを加え、3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン)で精製し、2−ブロモ−3−ヨード−6,7−ジフェニルアントラセンの黄色固体128mg(0.239mmol)を得た(収率56%)。
【0147】
実施例7 (6,7,6’,7’−テトラフェニル−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)[一般式(2)で示されるジハロビフェニル誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に合成例12で合成した2−ブロモ−3−ヨード−6,7−ジフェニルアントラセン(一般式(3)の化合物)128mg(0.239mmol)及びTHF5mlを添加した。この溶液を−70℃に冷却し、イソプロピルマグネシウムブロマイド(関東化学製、0.65M)のTHF溶液0.74ml(0.48mmol)を滴下した。20分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)49.7mg(0.48mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮した。得られた残渣に、合成例12で合成した2−ブロモ−3−ヨード−6,7−ジフェニルアントラセン(一般式(4)の化合物)125mg(0.233mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)10.3mg(0.009mmol)、トルエン5ml、及びエタノール1.2mlを添加した。さらに炭酸ナトリウム78.5mg(0.741mmol)と水1.6mlからなる水溶液を加え、60℃で24時間反応を実施した。室温まで冷却後、トルエン及び水を添加し分相した。有機相を減圧濃縮し溶媒を留去し、さらに真空乾燥した。得られた残渣にトルエンを添加し、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)(0.03ml)を添加し、室温で2時間撹拌した。この溶液を水洗浄し、有機相を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製し(溶媒;ヘキサン(アントラセン誘導体を溶出)及びヘキサン:ジクロロメタン=10:1(目的物を溶出))、6,7,6’,7’−テトラフェニル−3,3’−ジブロモ−2,2’−ビアントラセニルの黄色固体87.2mgを得た(収率57%)。
MS m/z: 817(M+,2%),657(M+−2Br,100)。
【0148】
MS測定より、6,7,6’,7’−テトラフェニル−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお、その構造式を下記に示す。
【0149】
【化33】
実施例8 (テトラフェニルジナフトビフェニレンの合成)(一般式(1)で示されるビフェニレン誘導体)
窒素雰囲気下、50mlシュレンク反応容器に、実施例7で合成した6,7,6’,7’−テトラフェニル−3,3’−ジブロモ−2,2’−ビアントラセニル87.0mg(0.106mmol)及びジエチルエーテル4mlを添加した。この混合物を0℃に冷却し、リチオ化であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.16ml(0.25mmol)を滴下し、0℃で90分間撹拌した(リチオ化反応)。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)47.0mg(0.350mmol)及びTHF8mlを添加し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温までゆっくり昇温し、3M塩酸水溶液を添加した。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、目的物の黄色固体27mgを得た(収率39%)。
FABMS m/z: 657(M+)。
【0150】
MS測定より、テトラフェニルジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0151】
【化34】
合成例13 (4−ブロモ−1,2−ジ(ドデシン−1−イル)ベンゼンの合成)
窒素雰囲気下、300mlシュレンク反応容器に、合成例6で得られた4−ブロモ−1,2−ジヨードベンゼン1.84g(4.50mmol)、ヨウ化銅(I)(和光純薬工業製)90mg(0.47mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)272mg(0.24mmol)、THF59ml、トリエチルアミン2.29g(22.2mmol)、1−ドデシン1.57g(9.47mmol)を加えた。この反応混合物を室温で26時間反応させた。1M塩酸水溶液を加えて反応をクエンチし、トルエンで抽出した後、有機相を水洗浄して無水硫酸ナトリウムで乾燥した。溶媒を減圧留去し、シリカゲルカラムクロマトグラフィー(溶離液;ジエチルエーテル:ヘキサン=1:30の混合液)で精製して目的の4−ブロモ−1,2−ジ(ドデシン−1−イル)ベンゼン1.52g(3.14mmol)を得た(収率70%)。
【0152】
合成例14 (2−ブロモ−3−ヨード−6,7−ジ(ドデシン−1−イル)アントラキノンの合成)(一般式(5)の化合物)
窒素雰囲気下、200mlシュレンク反応容器に、合成例13で得られた4−ブロモ−1,2−ジ(ドデシン−1−イル)ベンゼン1.52g(3.14mmol)を入れた。次いでTHF19mlを加えて−78℃に冷却し、n−ブチルリチウム(関東化学製、1.59mol/l、ヘキサン溶液)2.0ml(3.18mmol)を加え、30分間反応させた。次いで合成例1で得られた4−ブロモ−5−ヨードフタル酸無水物1.10g(3.14mmol)を加え、室温まで昇温した。水を加えてクエンチし得られた固体を濾過し、さらに水洗浄を行い、加熱真空乾燥後白色固体2.3g(3.1mmol)を得た。この固体に濃硫酸17mlを添加し、80℃で1時間反応した。反応混合物を氷に注ぎ入れ、析出した固体をろ過して水で洗浄した。乾燥後、シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:塩化メチレン=10:1)及びヘプタンからの再結晶で精製し、2−ブロモ−3−ヨード−6,7−ジ(ドデシン−1−イル)アントラキノンの黄色固体245mg(0.33mmol)を得た(収率11%)。
【0153】
合成例15 (2−ブロモ−3−ヨード−6,7−ジ(ドデシン−1−イル)アントラセンの合成)(一般式(3)及び(4)の化合物)
窒素雰囲気下、50mlシュレンク反応容器に、合成例14で合成した2−ブロモ−3−ヨード−6,7−ジ(ドデシン−1−イル)アントラキノン245mg(0.33mmol)及びTHF4mlを加えた。ヒドリド還元剤である水素化ジイソブチルアルミニウム(関東化学製、0.99mol/l、トルエン溶液)0.70ml(0.70mmol)を滴下し、室温で1.5時間還元反応を行った。この反応混合物に酸触媒である6M塩酸水溶液2mlを加え、65℃で3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。得られた残渣に再びTHF4mlを加え、水素化ジイソブチルアルミニウム0.70ml(0.70mmol)を加え、室温で1.5時間還元反応を行った。次いで反応混合物に6M塩酸水溶液2mlを加え、3時間脱水反応を行った。反応混合物を室温まで冷やし、ジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン)で精製し、2−ブロモ−3−ヨード−6,7−ジ(ドデシン−1−イル)アントラセンの黄色固体102mg(0.143mmol)を得た(収率42%)。
【0154】
実施例9 (6,7,6’,7’−テトラ(ドデシン−1−イル)−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)(一般式(2)で示されるジハロビフェニル誘導体)
窒素雰囲気下、100mlシュレンク反応容器に合成例15で合成した2−ブロモ−3−ヨード−6,7−ジ(ドデシン−1−イル)アントラセン102mg(0.143mmol)(一般式(3)の化合物)及びTHF5mlを添加した。この溶液を−70℃に冷却し、イソプロピルマグネシウムブロマイド(関東化学製、0.65M)のTHF溶液0.45ml(0.29mmol)を滴下した。10分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)29.7mg(0.29mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮した。得られた残渣に、合成例15で合成した2−ブロモ−3−ヨード−6,7−ジ(ドデシン−1−イル)アントラセン(一般式(4)の化合物)99.3mg(0.140mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)8.1mg(0.007mmol)、トルエン5ml、及びエタノール1.2mlを添加した。さらに炭酸ナトリウム44.5mg(0.420mmol)と水1.6mlからなる水溶液を加え、60℃で24時間反応を実施した。室温に冷却し、トルエンを添加後、分相し、有機相を食塩水で洗浄した。有機相を減圧濃縮し溶媒を留去し、さらに真空乾燥した。得られた残渣にトルエンを添加し、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)(0.03ml)を添加し、室温で1時間撹拌した。この溶液を水洗浄し、有機相を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製し(溶媒;ヘキサン(アントラセン誘導体を溶出)及びヘキサン:クロロホルム=10:1(目的物を溶出))、6,7,6’,7’−テトラ(ドデシン−1−イル)−3,3’−ジブロモ−2,2’−ビアントラセニルの黄色固体93.3mgを得た(収率57%)。
FABMS m/z: 1169(M+,100%),1089(M+−Br,7)。
【0155】
MS測定より、6,7,6’,7’−テトラ(ドデシン−1−イル)−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお、その構造式を下記に示す。
【0156】
【化35】
実施例10 (テトラ(ドデシン−1−イル)ジナフトビフェニレンの合成)(一般式(1)で示されるビフェニレン誘導体)
窒素雰囲気下、50mlシュレンク反応容器に、実施例9で合成した6,7,6’,7’−テトラ(ドデシン−1−イル)−3,3’−ジブロモ−2,2’−ビアントラセニル93.0mg(0.080mmol)及びジエチルエーテル5mlを添加した。この混合物を0℃に冷却し、リチオ化剤であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.23ml(0.13mmol)を滴下し、0℃で30分間及び10℃で60分間撹拌した(リチオ化反応)。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)35.5mg(0.264mmol)及びTHF10mlを添加し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温までゆっくり昇温し、3M塩酸水溶液を添加した。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、目的物の黄色固体28.1mgを得た(収率35%)。
FABMS m/z: 1010(M+)。
【0157】
MS測定より、テトラ(ドデシン−1−イル)ジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0158】
【化36】
合成例16 (2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ジフェニルアントラセンの合成)(一般式(3)及び(4)の化合物)
窒素雰囲気下、100mlシュレンク反応容器に合成例3で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノン541mg(0.722mmol)及びTHF20mlを添加した。−78℃に冷却後、フェニルリチウム(関東化学製、1.0mol/l、シクロヘキサン/ジエチルエーテル溶液)1.5ml(1.5mmol)(一般式(6)及び(7)の化合物)を加えた後、一晩かけて室温まで昇温した。次いで反応混合物に3M塩酸水溶液及びジエチルエーテルを加えた後、分相し、さらにジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。得られた残渣に酢酸30ml、ヨウ化ナトリウム749mg(5.0mmol)、及び次亜りん酸ナトリウム・1水和物727mg(6.86mmol)を加え、1時間加熱還流下で反応を行った(還元反応)。反応混合物を室温まで冷やし、トルエンで抽出した。トルエン溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:トルエン=30:1)で精製し、2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ジフェニルアントラセンの黄色固体453mgを得た(収率72%)。
【0159】
実施例11 (6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラフェニル−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)[一般式(2)で示されるジハロビフェニル誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に合成例16で合成した2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ジフェニルアントラセン205mg(0.235mmol)及びTHF6mlを添加した。この混合物を−60℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.81M)のTHF溶液0.58ml(0.47mmol)を滴下した。10分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)48.8mg(0.47mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮した。得られた固形物に、合成例16で合成した2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ジフェニルアントラセン210mg(0.241mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)13.9mg(0.012mmol)、トルエン5ml、及びエタノール1.2mlを添加した。さらに炭酸ナトリウム76.6mg(0.723mmol)と水1.6mlからなる水溶液を加え、60℃で24時間反応を実施した。室温まで冷却後、トルエン及び水を添加し分相した。有機相を濃縮し、得られた残渣をトルエン5mlに溶解後、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.02mlを添加し、室温で2時間撹拌した。このトルエン溶液を水で2回洗浄後、有機相を減圧濃縮し、得られた残渣に飽和食塩水及びトルエンを添加した。分相し、有機相を水で洗浄し、無水硫酸ナトリウムで乾燥した。有機相を濾過し、減圧濃縮し、得られた残渣をシリカゲルを充填したカラムで濾過した(溶媒:ヘキサン)。得られた粗固体をヘプタンから再結晶化し、目的物の黄色固体217mgを得た(収率62%)。
FABMS m/z: 1490(M+,100%),1419(M+−Br,5)。
【0160】
MS測定より、6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラフェニル−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお。その構造式を下記に示す。
【0161】
【化37】
実施例12 (テトラドデシルテトラフェニルジナフトビフェニレンの合成)[一般式(1)で示されるビフェニレン誘導体の合成]
窒素雰囲気下、100mlシュレンク反応容器に実施例11で合成した6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラフェニル−3,3’−ジブロモ−2,2’−ビアントラセニル210mg(0.141mmol)及びジエチルエーテル7mlを添加した。この混合物を0℃に冷却後、リチオ化剤であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.20ml(0.31mmol)を滴下した。1時間かけて5℃まで昇温しメタル化の熟成を行った。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)64.5mg(0.479mmol)及びTHF15mlを添加し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温までゆっくり昇温し、3M塩酸水溶液を添加した。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、目的物の黄色固体97.5mgを得た(収率52%)。
FABMS m/z: 1330(M+)。
【0162】
MS測定より、テトラドデシルテトラフェニルジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0163】
【化38】
合成例17 (2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエチニル)アントラセンの合成)(一般式(3)及び(4)の化合物の合成)
窒素雰囲気下、100mlシュレンク反応容器にフェニルアセチレン(東京化成工業製)178mg(1.74mmol)及びTHF20mlを添加した。n−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液1.05ml(1.67mmol)を滴下し、20分間撹拌した。合成例3で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノン523mg(0.697mmol)を加えた後、混合物を室温で一晩反応させた。次いで反応混合物に3M塩酸水溶液及びジエチルエーテルを加えた後、分相し、さらにジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。得られた残渣に酢酸30ml、ヨウ化ナトリウム749mg(5.0mmol)、及び次亜りん酸ナトリウム・1水和物727mg(6.86mmol)を加え、1時間加熱還流下で反応を行った。反応混合物を室温まで冷やし、トルエンで抽出した。トルエン溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:トルエン=30:1の混合液)で精製し、2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエチニル)アントラセンの黄色固体393mgを得た(収率61%)。
【0164】
実施例13 (6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラキス(フェニルエチニル)−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)(一般式(2)で示されるジハロビフェニル誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例17で合成した2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエチニル)アントラセン190mg(0.206mmol)及びTHF6mlを添加した。この混合物を−60℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.81M)のTHF溶液0.51ml(0.41mmol)を滴下した。5分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)42.6mg(0.41mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて室温で30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮し、得られた固形物に、合成例17で合成した2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエチニル)アントラセン195mg(0.212mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)13.9mg(0.012mmol)、トルエン5ml、及びエタノール1.2mlを添加した。さらに炭酸ナトリウム67.4mg(0.636mmol)と水1.6mlからなる水溶液を加え、60℃で24時間反応を実施した。室温まで冷却後、トルエン及び水を添加し分相した。有機相を濃縮し、得られた残渣をトルエン5mlに溶解後、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.02mlを添加し、室温で2時間撹拌した。このトルエン溶液を水で2回洗浄後、有機相を減圧濃縮し、得られた残渣に飽和食塩水及びトルエンを添加した。分相し、有機相を水で洗浄し、無水硫酸ナトリウムで乾燥した。有機相を濾過し、減圧濃縮し、得られた残渣をシリカゲルを充填したカラムで濾過した(溶媒:ヘキサン)。得られた粗固体をヘプタンから再結晶化し、目的物の黄色固体212mgを得た(収率65%)。
MS m/z: 1586(M+,2%),793(M+/2,100)。
【0165】
MS測定より、6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラキス(フェニルエチニル)−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお。その構造式を下記に示す。
【0166】
【化39】
実施例14 (テトラドデシルテトラキス(フェニルエチニル)ジナフトビフェニレンの合成)(一般式(1)で示されるビフェニレン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に実施例13で合成した6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラキス(フェニルエチニル)−3,3’−ジブロモ−2,2’−ビアントラセニル208mg(0.131mmol)及びジエチルエーテル7mlを添加した。この混合物を0℃に冷却後、リチオ化剤であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.20ml(0.31mmol)を滴下した。1時間かけて5℃まで昇温しメタル化の熟成を行った。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)64.5mg(0.479mmol)及びTHF15mlを添加し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温までゆっくり昇温し、3M塩酸水溶液を添加した。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、目的物の橙色固体80.3mgを得た(収率43%)。
FABMS m/z: 1426(M+)。
【0167】
MS測定より、テトラドデシルテトラキス(フェニルエチニル)ジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0168】
【化40】
合成例18 (2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエテニル)アントラセンの合成)(一般式(3)及び(4)の化合物の合成)
窒素雰囲気下、100mlシュレンク反応容器にマグネシウム54.7mg(2.25mmol)及びTHF20mlを添加した。ヨウ素5mgを加えた後、β−ブロモスチレン(東京化成工業製)40mgを添加し、撹拌下マグネシウムを活性化させた。β−ブロモスチレン359mg(1.96mmol、計2.17mmol)を緩く還流が起こる程度に滴下した。滴下終了後、30分間撹拌を継続した。合成例3で合成した2−ブロモ−3−ヨード−6,7−ジドデシルアントラキノン678mg(0.904mmol)を加えた後、混合物を室温で一晩反応させた。次いで反応混合物に3M塩酸水溶液及びジエチルエーテルを加えた後、分相し、さらにジエチルエーテルで抽出した。ジエチルエーテル溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。得られた残渣に酢酸30ml、ヨウ化ナトリウム749mg(5.0mmol)、及び次亜りん酸ナトリウム・1水和物727mg(6.86mmol)を加え、1時間加熱還流下で反応を行った。反応混合物を室温まで冷やし、トルエンで抽出した。トルエン溶液を飽和食塩水で洗浄して無水硫酸ナトリウムで乾燥し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:トルエン=30:1の混合液)で精製し、2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエテニル)アントラセンの黄色固体468mgを得た(収率56%)。
【0169】
実施例15 (6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラキス(フェニルエテニル)−3,3’−ジブロモ−2,2’−ビアントラセニルの合成)(一般式(2)で示されるジハロビフェニル誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に合成例18で合成した2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエテニル)アントラセン225mg(0.243mmol)及びTHF7mlを添加した。この混合物を−60℃に冷却し、イソプロピルマグネシウムブロマイド(東京化成工業製、0.81M)のTHF溶液0.60ml(0.49mmol)を滴下した。5分間熟成後、−78℃に冷却し、トリメトキシボラン(和光純薬工業製)50.9mg(0.490mmol)を滴下した。徐々に室温まで昇温した後、3M塩酸水溶液を加えて室温で30分間攪拌後、トルエンを添加し分相した。有機相を減圧濃縮し、得られた固形物に、合成例18で合成した2−ブロモ−3−ヨード−6,7−ジドデシル−9,10−ビス(フェニルエテニル)アントラセン235mg(0.254mmol)、テトラキス(トリフェニルホスフィン)パラジウム(東京化成工業製)13.9mg(0.012mmol)、トルエン5ml、及びエタノール1.2mlを添加した。さらに炭酸ナトリウム80.8mg(0.762mmol)と水1.6mlからなる水溶液を加え、60℃で24時間反応を実施した。室温まで冷却後、トルエン及び水を添加し分相した。有機相を濃縮し、得られた残渣をトルエン5mlに溶解後、70%tert−ブチルハイドロパーオキサイド溶液(和光純薬工業製)0.02mlを添加し、室温で2時間撹拌した。このトルエン溶液を水で2回洗浄後、有機相を減圧濃縮し、得られた残渣に飽和食塩水及びトルエンを添加した。分相し、有機相を水で洗浄し、無水硫酸ナトリウムで乾燥した。有機相を濾過し、減圧濃縮し、得られた残渣をシリカゲルを充填したカラムで濾過した(溶媒:ヘキサン)。得られた粗固体をヘプタンから再結晶化し、目的物の黄色固体259mgを得た(収率67%)。
MS m/z: 1594(M+,2%),797(M+/2,100)。
【0170】
MS測定より、6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラキス(フェニルエテニル)−3,3’−ジブロモ−2,2’−ビアントラセニルが得られたことを確認した。なお。その構造式を下記に示す。
【0171】
【化41】
実施例16 (テトラドデシルテトラキス(フェニルエテニル)ジナフトビフェニレンの合成)(一般式(1)で示されるビフェニレン誘導体の合成)
窒素雰囲気下、100mlシュレンク反応容器に実施例15で合成した6,7,6’,7’−テトラドデシル−9,10,9’,10’−テトラキス(フェニルエテニル)−3,3’−ジブロモ−2,2’−ビアントラセニル254mg(0.159mmol)及びジエチルエーテル7mlを添加した。この混合物を0℃に冷却後、リチオ化剤であるn−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.22ml(0.35mmol)を滴下した。1時間かけて5℃まで昇温しメタル化の熟成を行った。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)64.1mg(0.477mmol)及びTHF15mlを添加し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温までゆっくり昇温し、3M塩酸水溶液を添加した。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、目的物の橙色固体93.5mgを得た(収率41%)。
【0172】
FABMS m/z: 1434(M+)。
【0173】
MS測定より、テトラドデシルテトラキス(フェニルエテニル)ジナフトビフェニレンが得られたことを確認した。なお、その構造式を下記に示す。
【0174】
【化42】
実施例17 (耐酸化性評価)
窒素雰囲気下、100mlシュレンク容器にトルエン10.4gを添加し、凍結(液体窒素)−減圧−窒素置換−融解から成るサイクルを3回繰り返すことで溶存酸素を除去した。そこへ実施例2で得られたテトラドデシルジナフトビフェニレンの黄色固体10.5mgを添加し、110℃に加熱溶解させると黄色透明溶液となった。次にこのシュレンク容器の上部の栓を開け、1時間、外気に接触させることで空気を導入し、さらに110℃で撹拌した。で酸化に由来する新たなピークの出現はなかった。
【0175】
さらにこの溶液を110℃、1時間、撹拌下で空気を導入しても溶液の色の変化はみられず、耐酸化性に優れるものであった。
【0176】
比較例1
窒素雰囲気下、100mlシュレンク容器にトルエン28.9gを添加し、凍結(液体窒素)−減圧−窒素置換−融解から成るサイクルを3回繰り返すことで溶存酸素を除去した。そこへペンタセン(東京化成工業製)3.0mgを添加し、110℃に加熱し溶解させると赤紫色溶液となった。次にこのシュレンク容器の上部の栓を開け、1時間、空気を導入すると溶液の色が赤ピンク色に変化していた。さらに110℃で撹拌した。ガスクロマトグラフィー及びガスクロマトグラフィー−マススペクトル(GCMS)分析から、6,13−ペンタセンキノンが生成していることがわかった。
【0177】
さらにこの溶液を110℃、1時間、撹拌下で空気を導入すると溶液の色が黄色に変化していた。
【0178】
実施例18 (有機薄膜の作製)
窒素雰囲気下、実施例2で得られたテトラドデシルジナフトビフェニレン7.2mgをトルエン(10.2g)と混合し、80℃で1時間撹拌し、テトラドデシルジナフトビフェニレンの黄色透明溶液を調製した。
【0179】
空気雰囲気下、凹面のある石英基板を80℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、膜厚420nmの有機薄膜を作製した。この有機薄膜の成分をガスクロマトグラフィーで分析した結果、テトラドデシルジナフトビフェニレン以外にピークはなく、酸化されていなかった。従って、空気中でも酸化されることなくテトラドデシルジナフトビフェニレンの有機薄膜を作製できることがわかった。
【0180】
実施例19 (有機薄膜の作製、発光材料としての評価)
窒素雰囲気下、実施例2で得られたテトラドデシルジナフトビフェニレン26mgをジクロロエタン5gと混合し、80℃で1時間撹拌し、テトラドデシルジナフトビフェニレンの黄色溶液を調製した。
【0181】
空気雰囲気下、石英基板を70℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、膜厚287nmの有機薄膜を作製した。この有機薄膜を用いて、発光量子収率及び蛍光スペクトルを測定した。結果を表1に示す。これらの結果からテトラドデシルジナフトビフェニレンは発光材料として利用できるものであった。
【0182】
実施例20 (有機薄膜の作製、発光材料としての評価)
窒素雰囲気下、実施例12で得られたテトラドデシルテトラフェニルジナフトビフェニレン36mgをジクロロエタン5gと混合し、80℃で1時間撹拌し、テトラドデシルテトラフェニルジナフトビフェニレンの黄色溶液を調製した。
【0183】
空気雰囲気下、石英基板を70℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、膜厚398nmの有機薄膜を作製した。この有機薄膜を用いて、発光量子収率及び蛍光スペクトルを測定した。結果を表1に示す。これらの結果からテトラドデシルテトラフェニルジナフトビフェニレンは発光材料として利用できるものであった。
【0184】
実施例21 (有機薄膜の作製、発光材料としての評価)
窒素雰囲気下、実施例14で得られたテトラドデシルテトラキス(フェニルエチニル)ジナフトビフェニレン36mgをジクロロエタン5gと混合し、80℃で1時間撹拌し、テトラドデシルテトラキス(フェニルエチニル)ジナフトビフェニレンの橙色溶液を調製した。
【0185】
空気雰囲気下、石英基板を70℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、膜厚378nmの有機薄膜を作製した。この有機薄膜を用いて、発光量子収率及び蛍光スペクトルを測定した。結果を表1に示す。これらの結果からテトラドデシルテトラキス(フェニルエチニル)ジナフトビフェニレンは発光材料として利用できるものであった。
【0186】
実施例22 (有機薄膜の作製、発光材料としての評価)
窒素雰囲気下、実施例16で得られたテトラドデシルテトラキス(フェニルエテニル)ジナフトビフェニレン32mgをジクロロエタン5gと混合し、80℃で1時間撹拌し、テトラドデシルテトラキス(フェニルエテニル)ジナフトビフェニレンの橙色溶液を調製した。
【0187】
空気雰囲気下、石英基板を70℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥し、膜厚356nmの有機薄膜を作製した。この有機薄膜を用いて、発光量子収率及び蛍光スペクトルを測定した。結果を表1に示す。これらの結果からテトラドデシルテトラキス(フェニルエテニル)ジナフトビフェニレンは発光材料として利用できるものであった。
【0188】
【表1】
比較例2(テトラドデシルジベンゾビフェニレン(本願:n=m=0)の合成及び評価)
1) (3,3’−ジブロモ−6,7,6’,7’−テトラドデシル−2,2’−ビナフチルの合成)
窒素雰囲気下、100mlシュレンク反応容器に2,3−ジブロモ−6,7−ジドデシルナフタレン389mg(0.625mmol)及びTHF9mlを加えた。−78℃に冷却し、n−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.20ml(0.31mmol)を滴下した。20分間反応後、冷却用バスを外し、室温で1時間撹拌した。3M塩酸水溶液及びトルエン添加し分相した。有機相を無水硫酸ナトリウムで乾燥し、減圧濃縮した。残渣をヘプタンから再結晶精製し、白色固体204mgを得た(収率61%)。
【0189】
2) (テトラドデシルジベンゾビフェニレンの合成)
窒素雰囲気下、100mlシュレンク反応容器に上記1)で得た3,3’−ジブロモ−6,7,6’,7’−テトラドデシル−2,2’−ビナフチル201mg(0.185mmol)及びジエチルエーテル5mlを加えた。0℃に冷却し、n−ブチルリチウム(関東化学製、1.59M)のヘキサン溶液0.25ml(0.40mmol)を滴下し、0℃で30分間及び10℃で60分間撹拌した。別の100mlシュレンク反応容器に、塩化銅(II)(和光純薬工業製)74.6mg(0.555mmol)及びTHF10mlを投入し、−78℃に冷却した。ここへ先のジリチオ化物のジエチルエーテル溶液をテフロン(登録商標)キャヌラーを用いて移液した。15時間かけて室温まで昇温し、3M塩酸水溶液を加えて反応を停止させた。分相し、有機相をさらに飽和食塩水で洗浄した。懸濁している有機相を濾過し、固体を濾別した。この固体を水及びヘキサンで洗浄し、得られた固体をトルエンから再結晶化し、テトラドデシルジベンゾビフェニレンの淡黄色固体83.9mgを得た(収率49%)。
【0190】
なお、その構造式を下記に示す。
【0191】
【化43】
3)テトラドデシルジベンゾビフェニレンの評価
窒素雰囲気下、100mlシュレンク容器にトルエン10.2gを添加し、凍結(液体窒素)−減圧−窒素置換−融解から成るサイクルを3回繰り返すことで溶存酸素を除去した。そこへ上記2)で得たテトラドデシルジベンゾビフェニレン7.2mgを添加し、80℃で1時間撹拌し、テトラドデシルジベンゾビフェニレンの薄黄色透明溶液を調製した。
【0192】
次に窒素雰囲気下、凹面のある石英基板を80℃に加熱し、この基板上に上記の溶液をスポイトを用いて塗布し常圧下で乾燥した。しかし、材料がある部分とない部分が存在し、均一な膜は得られず、塗布による均質な薄膜作製は困難であった。
【図面の簡単な説明】
【0193】
【図1】実施例1で合成した6,7,6’,7’−テトラドデシル−3,3’−ジブロモ−2,2’−ビアントラセニルの1H NMRスペクトル
【図2】実施例2で合成したテトラドデシルジナフトビフェニレンの1H NMRスペクトル
【特許請求の範囲】
【請求項1】
下記一般式(1)で示されることを特徴とするビフェニレン誘導体。
【化1】
(ここで、置換基R1〜R8は同一又は異なって、水素原子、フッ素原子、炭素数2〜20のアルキル基、炭素数2〜20のアルキニル基、炭素数2〜30のアルケニル基、又は炭素数4〜20のアリール基を示し、mは1又は2であり、nは0〜2の整数を示す。但し、R1及びR2は同時に水素原子であることはない。)
【請求項2】
m及びnが1であることを特徴とする請求項1に記載のビフェニレン誘導体。
【請求項3】
請求項1又は2に記載のビフェニレン誘導体を含む耐酸化性有機半導体材料。
【請求項4】
請求項3に記載の耐酸化性有機半導体材料からなる有機薄膜。
【請求項5】
請求項3に記載の耐酸化性有機半導体材料からなる発光材料。
【請求項6】
下記一般式(2)で示されることを特徴とするジハロビフェニル誘導体。
【化2】
(ここで、置換基X1及びX2は臭素原子、ヨウ素原子又は塩素原子を示し、置換基R1〜R8、並びに記号m及びnは請求項1に記載の一般式(1)で示される置換基並びに記号と同意義を示す。)
【請求項7】
m及びnが1であることを特徴とする請求項6に記載のジハロビフェニル誘導体。
【請求項8】
請求項6に記載のジハロビフェニル誘導体を銅化合物又は銅と反応させることを特徴とする請求項1又は2に記載のビフェニレン誘導体の製造方法。
【請求項9】
請求項6に記載のジハロビフェニル誘導体をジリチオ化及び/又はジグリニャール化した後、銅化合物と反応させることを特徴とする請求項8に記載のビフェニレン誘導体の製造方法。
【請求項10】
銅化合物が、2価の銅化合物であることを特徴とする請求項8又は9に記載のビフェニレン誘導体の製造方法。
【請求項11】
銅化合物が、塩化銅(II)又は臭化銅(II)であることを特徴とする請求項8〜10のいずれかに記載のビフェニレン誘導体の製造方法。
【請求項12】
ジアルキルエーテル中でジリチオ化することを特徴とする請求項8〜11のいずれかに記載のビフェニレン誘導体の製造方法。
【請求項1】
下記一般式(1)で示されることを特徴とするビフェニレン誘導体。
【化1】
(ここで、置換基R1〜R8は同一又は異なって、水素原子、フッ素原子、炭素数2〜20のアルキル基、炭素数2〜20のアルキニル基、炭素数2〜30のアルケニル基、又は炭素数4〜20のアリール基を示し、mは1又は2であり、nは0〜2の整数を示す。但し、R1及びR2は同時に水素原子であることはない。)
【請求項2】
m及びnが1であることを特徴とする請求項1に記載のビフェニレン誘導体。
【請求項3】
請求項1又は2に記載のビフェニレン誘導体を含む耐酸化性有機半導体材料。
【請求項4】
請求項3に記載の耐酸化性有機半導体材料からなる有機薄膜。
【請求項5】
請求項3に記載の耐酸化性有機半導体材料からなる発光材料。
【請求項6】
下記一般式(2)で示されることを特徴とするジハロビフェニル誘導体。
【化2】
(ここで、置換基X1及びX2は臭素原子、ヨウ素原子又は塩素原子を示し、置換基R1〜R8、並びに記号m及びnは請求項1に記載の一般式(1)で示される置換基並びに記号と同意義を示す。)
【請求項7】
m及びnが1であることを特徴とする請求項6に記載のジハロビフェニル誘導体。
【請求項8】
請求項6に記載のジハロビフェニル誘導体を銅化合物又は銅と反応させることを特徴とする請求項1又は2に記載のビフェニレン誘導体の製造方法。
【請求項9】
請求項6に記載のジハロビフェニル誘導体をジリチオ化及び/又はジグリニャール化した後、銅化合物と反応させることを特徴とする請求項8に記載のビフェニレン誘導体の製造方法。
【請求項10】
銅化合物が、2価の銅化合物であることを特徴とする請求項8又は9に記載のビフェニレン誘導体の製造方法。
【請求項11】
銅化合物が、塩化銅(II)又は臭化銅(II)であることを特徴とする請求項8〜10のいずれかに記載のビフェニレン誘導体の製造方法。
【請求項12】
ジアルキルエーテル中でジリチオ化することを特徴とする請求項8〜11のいずれかに記載のビフェニレン誘導体の製造方法。
【図1】

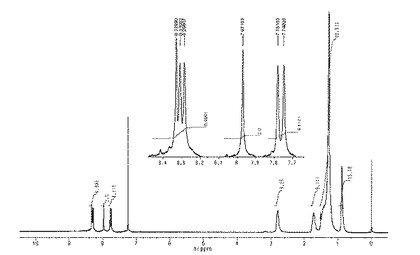
【図2】
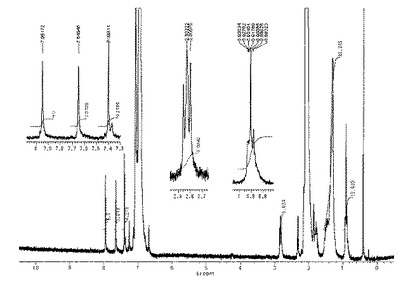
【図2】
【公開番号】特開2009−227652(P2009−227652A)
【公開日】平成21年10月8日(2009.10.8)
【国際特許分類】
【出願番号】特願2008−236973(P2008−236973)
【出願日】平成20年9月16日(2008.9.16)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
【公開日】平成21年10月8日(2009.10.8)
【国際特許分類】
【出願日】平成20年9月16日(2008.9.16)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
[ Back to top ]