
ビンカアルカロイド、類似体および誘導体のリガンド結合体
本明細書では、患者において病原性細胞を治療するための化合物、製薬組成物、および方法が記載される。本明細書に記載する化合物は、細胞毒性薬物およびビタミン受容体結合リガンドの結合体を含む。該結合体は、1個以上のスペーサーリンカー、ヘテロ原子リンカー、および放出型リンカーから形成されるリンカーも含む。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、標的薬物送達で使用するための組成物および方法に関するものである。特に本発明は、ビンカアルカロイド、ならびにその類似体および誘導体のリガンド結合体、例えばビタミン受容体結合化合物およびビンカアルカロイドの結合体に関するものである。
【背景技術】
【0002】
哺乳類免疫系は、腫瘍細胞、他の病原性細胞、および侵入する外来病原体の認識および排除のための手段を提供する。免疫系は通常、強力な防御線を提供するが、癌細胞、他の病原性細胞、または感染性因子が宿主の免疫反応を逃れて、共存する宿主病原性と共に増殖または持続する多くの例がある。化学療法剤および放射線療法は、例えば分裂を繰り返す腫瘍を排除するために開発されてきた。しかしながら現在入手可能な化学療法剤および放射線療法投与計画の多くは、それらが病原性細胞を破壊するだけでなく、それらが正常な宿主細胞、例えば造血系の細胞にも影響を及ぼすために有害な副作用を有する。これらの抗癌剤の有害な副作用は、病原性細胞集団に対して選択的で、なおかつ宿主毒性の低い新たな治療法の開発の必要性を際立たせている。
【0003】
研究者らは、病原性細胞を、このような細胞に細胞毒性化合物を標的指向化することによって破壊する治療プロトコルを開発してきた。これらのプロトコルの多くは、毒素の正常細胞への送達を最小限に抑えることを試みて、病原性細胞に特有の、または病原性細胞によって過剰発現された抗原に結合する抗体に結合された毒素を利用する。この手法を使用して、病原性細胞上の特異性抗原に対する抗体より成る、ある免疫毒素が開発され、該抗体はリシン、緑膿菌外毒素、ジフテリア毒素、および腫瘍壊死因子などの毒素に結合されている。これらの免疫毒素は、抗体によって認識される特異性抗原を持つ病原性細胞、例えば腫瘍細胞を標的とする(非特許文献1;2、特許文献1参照)。
【0004】
宿主内の病原性細胞、例えば癌細胞または外来病原体の集団を標的とする別の手法は、独立した宿主毒性をも示しうる化合物投与の必要性を回避するために、病原性細胞に対する宿主免疫応答を向上させることである。免疫療法について報告された1つの方法は、抗体(例えば遺伝子組換え多重結合抗体)を腫瘍細胞の表面に結合させて、細胞表面で抗体の定常領域を提示して、それにより各種の免疫系媒介プロセスによって腫瘍細胞の死滅を誘発させることである(非特許文献3、特許文献2参照)。しかしながらこれらの手法は、腫瘍特異性抗原を定義することが困難なため、複雑になっている。
【特許文献1】Better,M.D.,国際公開WO91/07418号,1991年5月30日に公開。
【特許文献2】Soulillou,J.P.,米国特許第5,672,486号
【非特許文献1】Olsnes,S.,Immunol.Today,10,pp.291-295,1989
【非特許文献2】Melby,E.L.,Cancer Res.,53(8),pp.1755-1760,1993
【非特許文献3】De Vita,V.T.,Biologic Therapy of Cancer,2d ed.Philadelphia,Lippincott,1995
【発明の開示】
【課題を解決するための手段】
【0005】
ビンカアルカロイド、ならびにその類似体および誘導体の結合体を本明細書で説明する。結合体はリガンド、例えば、ビンカアルカロイドならびにその類似体および誘導体に、場合によりリンカーを介して共有結合した細胞表面受容体のリガンドを含む。本明細書に記載する結合体で有用なビンカアルカロイドとしては、アルカロイドのビンカインドール-ジヒドロインドールファミリーのすべてのメンバー、これに限定されるわけではないが例えば、ビンデシン、ビンブラスチン、ビンクリスチン、カタランチン、ビンドリン、ロイロシン、ビノレルビン、イミドカルブ、シブトラミン、トルトラズリル、ビンブラスチン酸など、ならびにその類似体および誘導体が挙げられる。
【0006】
一実施形態において、受容体結合型薬物送達結合体が記載される。薬物送達結合体は、リガンド(例えば細胞表面受容体のリガンド)、ビンカアルカロイド、および場合により2価リンカーを含み、一般に式
(B)−(L)−(D)
によって表される。式中、(B)は受容体結合部分を表し、これには例えば、限定されるわけではないが、ビタミン、およびそのビタミン受容体結合型類似体またはその誘導体、例えばビタミン受容体を結合できるビタミンならびにその類似体および誘導体が含まれ;(D)は、ビンカアルカロイド、あるいはその類似体または誘導体を表し;そして(L)は、2価リンカーを表す。2価リンカー(L)は複数のリンカーを含みうる。例えば2価リンカー(L)は、1個以上のスペーサーリンカー(ls)、および放出型リンカー(lr)を含むことができ、それぞれ他方およびリガンドならびにビンカアルカロイドに1個以上のヘテロ原子リンカー(lH)によって結合される。これらの各種リンカーは、2価リンカー(L)を構築するために選択され、いずれかの順序で配置されうる。実例として、2価リンカー(L)は次のうちの1つでありうる:

式中、a、b、c、d、およびeは整数、例えば0〜約4の範囲の整数であり、(ls)、(lH)、および(lr)はそれぞれ、スペーサーリンカー、放出型リンカー、ヘテロ原子リンカーである。2価リンカーのさらなる例証的な例は、米国特許出願公開第2005/0002942A1号および国際公開WO2006/012527号に記載されており、その開示の全体は参照により本明細書に組み入れられている。一変更形態において、1個以上の受容体結合リガンドが本明細書に記載する薬物送達結合体に含まれる。これらの受容体結合型リガンドのそれぞれが同じでありうる、または異なりうることが理解されるであろう。
【0007】
本明細書に記載する薬物送達結合体の例証的な一実施形態において、2価リンカーは、少なくとも1個の放出型なリンカー(lr)を含む。本明細書に記載する薬物送達結合体の別の例証的な実施形態において、2価リンカーは、少なくとも2個の放出型リンカー(lr)2を含む。別の例証的な態様において、2価リンカー(L)は、ジスルフィド放出型リンカーでない、少なくとも1個の放出型リンカー(lr)を含む。別の例証的な態様において、2価リンカー(L)は、1個の放出型リンカーがジスルフィド放出型リンカーでない、少なくとも2個の放出型リンカー(lr)2を含む。1個を超える放出型リンカーが2価リンカーに含まれるとき、これらの放出型リンカーは隣接しうることが認識される。2個の放出型リンカーが2価リンカーにおいて隣接しているとき、2個の放出型リンカーは連携してビンカアルカロイド、あるいはその類似体または誘導体の放出を引き起こしうることがさらに認識される。
【0008】
別の実施形態において、2価リンカーは、アミノ酸から形成されたペプチドである少なくとも1個のスペーサーリンカーを含む。一態様において、ペプチドは天然型アミノ酸、およびその立体異性体を含む。別の態様において、ペプチドは天然型アミノ酸、およびその立体異性体のみから形成される。
【0009】
本明細書に記載するリガンドは一般に、細胞表面受容体のリガンドを含む。本明細書に記載する結合体において有用な例証的なリガンドとしては、これに限定されるわけではないが、ビタミン、およびビタミン受容体に結合する他の部分、トランスポーター、もしくはビタミンあるいはその類似体または誘導体に特異的に結合する他の表面提示タンパク質、ライブラリースクリーニングから同定されたペプチドリガンド、腫瘍細胞特異性ペプチド、腫瘍細胞特異性アプタマー、腫瘍細胞特異性炭水化物、腫瘍細胞特異性モノクローナルまたはポリクローナル抗体、抗体のFabまたはscFv(すなわち単鎖可変領域)断片(例えば、EphA2、または転移性癌細胞で特異的に発現するタンパク質、または特異的に影響を受ける他のタンパク質、に対する抗体のFab断片)、コンビナトリアルライブラリーに由来する小型有機分子、成長因子(例えばEGF、FGF、インスリン、およびインスリン様成長因子)、および相同ポリペプチド、ソマトスタチンおよびその類似体、トランスフェリン、リポタンパク質複合体、胆汁塩、セレクチン、ステロイドホルモン、Arg-Gly-Asp含有ペプチド、レチノイド、各種のガレクチン、δ-オピオイド受容体リガンド、コレシストキニンA受容体リガンド、アンギオテンシンAT1またはAT2受容体に特異的なリガンド、ペルオキシソーム増殖因子活性化受容体λリガンド、β−ラクタム抗生物質(例えばペニシリン、抗菌薬を含む小型有機分子、ならびに腫瘍細胞の表面でまたは感染性生物で優先的に発現される受容体に特異的に結合する他の分子)、および受容体または他の細胞表面タンパク質の結晶構造に基づき特定の受容体の結合ポケット内に適合するように設計された抗菌薬および他の薬物、腫瘍抗原のリガンドまたは腫瘍細胞の表面で優先的に発現される他の分子のリガンド、あるいはこれらの分子のいずれかの断片が挙げられる。リガンド-ビンカ結合体の結合部位として機能しうる腫瘍特異性抗原は、タンパク質のエフリンファミリーのメンバーの細胞外エピトープ、例えばEphA2を含む。EphA2発現は、正常細胞の細胞-細胞接合部に限定されるが、転移性腫瘍細胞ではEphA2は細胞表面全体に分布している。それゆえ転移性細胞のEphA2は、例えばビンカアルカロイドに結合した抗体のFab断片への結合のために接近するが、これに対して該タンパク質は正常細胞のFab断片への結合のためには接近できず、そのため転移性癌細胞に対して特異的にリガンド-ビンカ結合体を生じる。
【0010】
他の実施形態において、製薬組成物が記載されている。製薬組成物は、本明細書に記載したリガンド-ビンカ結合体を、製薬的に許容される担体、賦形剤および/または希釈剤と組合せて含む。
【0011】
別の実施形態において、病原性細胞の集団を内部に持つ宿主動物において病原性細胞の集団を排除する方法が記載される。例証的な一態様において、病原性細胞集団のメンバーは、受容体結合部分あるいはその類似体または誘導体に対する接近可能な結合部位を有し、その結合部位は病原性細胞によって特異的に発現、過剰発現または優先的に発現される。該方法は、本明細書で記載するように、宿主に本明細書に記載する薬物送達結合体、あるいはその製薬組成物を投与する工程を含む。
【0012】
==関連出願==
本出願は、米国特許法119条(c)に基づいて、その開示全体が参照により本明細書に組み入れられている、2005年8月19日に出願された米国仮特許出願第60/709,936号の利益を主張する。
【発明を実施するための最良の形態】
【0013】
薬物、ならびにその類似体および誘導体のリガンド結合体を本明細書で説明する。結合体は、病原性細胞を含む細胞へ標的指向化されうる2つ以上の薬物に共有結合された、細胞表面受容体のリガンドを含めた、細胞受容体結合リガンドを含む。本明細書に記載する結合体は、リガンドを薬物に結合するための多価リンカーも含みうる。
【0014】
一実施形態において、受容体結合薬物送達結合体が記載される。薬物送達結合体は、細胞表面受容体のリガンド、ビンカアルカロイド、および場合により2価リンカーを含み、一般に式
(B)−(L)−(D)
によって表され、式中、(B)は受容体結合部分を表し、これには例えば、限定されるわけではないが、ビタミン、およびそのビタミン受容体結合類似体またはその誘導体、例えばビタミン受容体を結合できるビタミンならびにその類似体および誘導体が含まれ;(D)は、ビンカアルカロイド、あるいはその類似体または誘導体を表し;そして(L)は、2価リンカーを表す。2価リンカー(L)は複数のリンカーを含みうる。例えば2価リンカー(L)は、1個以上のスペーサーリンカー(ls)と、放出型リンカー(lr)とを含み、それぞれ相互に、そしてリガンドおよびビンカアルカロイドに1個以上のヘテロ原子リンカー(lH)によって結合されている。これらの各種リンカーは、2価リンカーを構築するために選択され、いずれの順序でも配置されうる。実例として、2価リンカー(L)は次のうちの1つでありうる:
式中、a、b、c、d、およびeは整数、例えば0〜約4の範囲の整数であり、(ls)、(lH)、および(lr)はそれぞれ、スペーサーリンカー、放出型リンカー、ヘテロ原子リンカーである。2価リンカーのさらなる例証的な例は、米国特許出願公開第2005/0002942A1号および国際公開WO2006/012527号に記載されており、その開示の全体は参照により本明細書に組み入れられている。
【0015】
受容体結合部分(B)、2価リンカー(L)、およびビンカアルカロイド薬物、あるいはその類似体または誘導体(D)を含む受容体結合薬物送達結合体が記載され、該受容体結合部分(B)および該ビンカアルカロイド薬物(D)はそれぞれヘテロ原子リンカー(lH)を介して2価リンカー(L)に結合される。2価リンカー(L)は、1個以上のスペーサーリンカー、ヘテロ原子リンカー、および放出型リンカー、ならびにその組合せをいずれの順序でも含む。
【0016】
例えば2価リンカー、または2価リンカーの一部を形成するためにリンカーが共有結合的に組み立てられる方法の例証的な一実施形態において、ヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーが結合されて式:
の2価基を形成し、式は-(ls)5-(ls)’-(lr)-(lH)-としても表されうる。その式では、(ls)5はペンタペプチドAla-Glu-Lys-Asp-Asp-OHであり、(ls)’はCH2CH2であり、(lr)はS-S-(CH2)2-O-C(O)であり、そして(lH)はOである。放出型リンカー(lr)は、ω-アミノ基にて(ls)5のLysに結合される。
【0017】
本明細書に記載する薬物送達結合体の説明的な一実施形態において、2価リンカーは、少なくとも1個の放出型なリンカー(lr)を含む。本明細書に記載する薬物送達結合体の別の例証的な実施形態において、2価リンカーは、少なくとも2個の放出型リンカー(lr)2を含む。別の説明的な態様において、2価リンカー(L)は、ジスルフィド放出型リンカーでない、少なくとも1個の放出型リンカー(lr)を含む。別の例証的な態様において、2価リンカー(L)は、1個の放出型リンカーがジスルフィド放出型リンカーでない、少なくとも2個の放出型リンカー(lr)2を含む。1個を超える放出型リンカーが2価リンカーに含まれるとき、これらの放出型リンカーは隣接しうることが認識される。2個の放出型リンカーが2価リンカーにおいて隣接しているとき、2個の放出型リンカーは連携してビンカアルカロイド、あるいはその類似体または誘導体の放出を引き起こしうることがさらに認識される。
【0018】
本明細書で使用され、また開裂性リンカーとしても公知である「放出型リンカー」という用語は、生理学的条件下で切断されうる少なくとも1個の結合(例えばpH不安定性、酸不安定性、酸化不安定性、または酵素不安定性結合)を含むリンカーを指す。結合切断を生じるこのような生理学的条件としては、例えば生理学的pHにて、またはサイトゾルpHよりも低いpHを有するエンドソームなどの細胞器官への細分化の結果として生じる標準化学加水分解反応が挙げられることが認識されるべきである。
【0019】
開裂性結合が放出型リンカーのどちらかまたは両方の端にて、本明細書に記載するように、放出型リンカー内の2個の隣接する原子を結合しうる、ならびに/あるいは他のリンカーまたは(B)および/または(D)を結合しうることが理解されるはずである。開裂性結合が放出型リンカー内の2個の隣接する原子を結合する場合、結合の切断後に、放出型リンカーは2個以上の断片に切断される。あるいは開裂性結合が放出型リンカーと別の部分、例えばヘテロ原子リンカー、スペーサーリンカー、別の放出型リンカー、薬物、あるいはその類似体または誘導体、あるいはビタミン、あるいはその類似体または誘導体との間にある場合、結合の切断後に、放出型リンカーは他の部分から分離される。
【0020】
開裂性結合の不安定性は例えば、開裂性結合におけるまたはその付近の置換的な変更、例えば開裂性ジスルフィド結合に隣接したアルファ分岐取り込みを含むこと、加水分解されうるケイ素-酸素結合を有する部分におけるケイ素上の置換基の疎水性の上昇、加水分解されうるケタールまたはアセタールの部分を形成するアルコキシ基のホモログ化などによって調整されうる。
【0021】
本明細書に記載する2価リンカーの開裂の例示的な機構は、次の1,4および1,6断片化機構を含み、
式中、Xは、外来性または内在性求核試薬、グルタチオン、または生物還元剤などであり、ZまたはZ’のどちらかは、ビタミン、あるいはその類似体または誘導体、あるいは薬物、あるいはその類似体または誘導体、あるいは2価リンカーの他の部分と結合したビタミンまたは薬物部分である。上の断片化機構が協奏機構として示されているが、多数の別個の工程が2価リンカーから、提示の最終生成物への最終的な断片化を実施するために起こりうることが理解されるものとする。例えば、結合開裂は酸触媒によるカルバメート部分の脱離反応によっても起こりうることが認識され、この脱離反応は上の例に示されたベータ硫黄または二硫化物のアリール基またはそのどちらかによってもたらされる安定化により隣接基に(anchimerically)補助されうる。この実施形態のこれらの変更態様において、放出型リンカーはカルバメート部分である。あるいは断片化は、ジスルフィド基に対する求核攻撃によって開始されて、開裂が引き起こされ、チオラートが形成される。チオラートによりカルボン酸またはカルバミン酸部分を分子間で置換して、対応するチアシクロプロパンを形成しうる。ベンジル含有2価リンカーの場合、例示のジスルフィド結合の切断の後、得られたフェニルチオラートはさらに断片化して、共鳴安定化中間体を形成することによってカルボン酸またはカルバミン酸を放出しうる。これらの場合にいずれにおいても、本明細書に記載する例証的な2価リンカーの放出性質は、存在する化学的、代謝的、生理学的、または生物学的条件に関連しうるどんな機構によっても実現されうる。
【0022】
放出型リンカーの結合開裂のための他の例証的な機構は、次のようなオキソニウム補助開裂:
を含み、式中、Zは、ビタミン、あるいはその類似体または誘導体、あるいは薬物、あるいはその類似体または誘導体であり、あるいはそれぞれが、2価リンカーの他の部分と併せたビタミンまたは薬物部分、例えば1個以上のスペーサーリンカー、ヘテロ原子リンカー、および/または他の放出型リンカーを含む薬物またはビタミン部分である。本実施形態において、酸触媒によるカルバメートの脱離反応は、CO2の放出およびZに結合した窒素含有部分の放出と、水、または他のいずれかのルイス塩基によって捕捉されうるベンジルカチオンの形成につながる。
【0023】
別の例証的な機構は、2価リンカーにおける結合の開裂に続いて、放出された官能基が、隣接基補助(anchimeric assisted)開裂または切断とも呼ばれる、さらなる結合の切断または開裂を化学的に補助するような方法で、放出型リンカー、スペーサーリンカー、およびヘテロ原子リンカーの配置をともなう。このような2価リンカーまたはその部分の例証的な実施形態は、式:
を有する化合物を含み、式中、Xはヘテロ原子、例えば窒素、酸素、または硫黄であり、nは、0、1、2、および3から選択される整数であり、Rは水素または置換基、例えばアリール環に正電荷を誘導的にまたは共鳴によって安定化できる置換基(例えばアルコキシなど)であり、ZまたはZ’のどちらかは、ビタミン、あるいはその類似体または誘導体、あるいは薬物、あるいはその類似体または誘導体、あるいは2価リンカーの他の部分と結合したビタミンまたは薬物部分である。これに限定されるわけではないが、ヒドロキシ、アルキル、アルコキシ、アルキルチオ、ハロなどを含む他の置換基が、アリール環、ベンジル炭素、カルバメート窒素、アルカン酸、またはメチレン架橋に存在しうることが認識される。補助開裂には、ベンジリウム中間体、ベンジン中間体、ラクトン環化、オキソニウム中間体、ベータ脱離などをともなう機構が含まれうる。放出型リンカーの開裂に続く断片化に加えて、放出型リンカーの初期開裂が隣接基補助機構によって促進されうることがさらに認識される。
【0024】
この本実施形態において、環化しうるヒドロキシアルカン酸は、例えばオキソニウムイオンによってメチレン架橋の開裂を促進し、結合開裂または放出型リンカーの結合開裂後の続いての断片化を促進する。あるいはメチレン架橋の酸触媒によるオキソニウムイオン補助開裂は、この例証的な2価リンカー、またはその断片の断片化のカスケードを開始しうる。あるいはカルバメートの酸触媒による加水分解は、環化しうるヒドロキシアルカン酸のベータ脱離を促進し、例えばオキソニウムイオンによってメチレン架橋の開裂を促進しうる。本明細書に記載する代謝的、生理学的、または細胞的条件下での結合切断または開裂の他の化学的機構がこのような断片化のカスケードを開始しうることが認識される。本明細書に記載する代謝的、生理学的、または細胞的条件下での結合切断または開裂の他の化学的機構がこのような断片化のカスケードを開始しうることが認識される。
【0025】
一実施形態において、本明細書に記載する2価リンカーは、次の式
の化合物であり、
式中、nは、1〜約4より選択される整数であり;RaおよびRbは、水素およびアルキル(例えば場合により分岐しているC1〜C4アルキルなどの低級アルキル)から成る群よりそれぞれ独立して選択され;あるいはRaおよびRbは、結合した炭素原子とひとまとめにされて、炭素環を形成し;Rは、場合により置換されたアルキル基、場合により置換されたアシル基、または適切に選択された窒素保護基であり;そして(*)は、薬物、ビタミン、造影剤、診断剤、他の2価リンカー、または結合体の他の部分への結合点を示す。
【0026】
別の実施形態において、本明細書に記載する2価リンカーは、次の式
の化合物を含み、式中、mは、1〜約4より選択される整数であり;Rは、場合により置換されたアルキル基、場合により置換されたアシル基、または適切に選択された窒素保護基であり;そして(*)は、薬物、ビタミン、造影剤、診断剤、他の2価リンカー、または結合体の他の部分への結合点を示す。
【0027】
別の実施形態において、本明細書に記載する2価リンカーは、次の式
の化合物を含み、式中、mは、1〜約4より選択される整数であり;Rは、場合により置換されたアルキル基、場合により置換されたアシル基、または適切に選択された窒素保護基であり;そして(*)は、薬物、ビタミン、造影剤、診断剤、他の2価リンカー、または結合体の他の部分への結合点を示す。
【0028】
別の実施形態において、2価リンカーにおける結合の開裂に続いて、放出された官能基が、隣接基補助開裂または切断とも呼ばれる、さらなる結合の切断または開裂を化学的に補助するような方法で、放出型リンカー、スペーサーリンカー、およびヘテロ原子リンカーが配置されうる。このような2価リンカーまたはその一部分の例証的な実施形態は、式:
を有する化合物を含み、式中、Xは、ヘテロ原子、例えば窒素、酸素、または硫黄であり、nは、0、1、2、および3から選択される整数であり、Rは水素または置換基、これは例えばアリール環に正電荷を誘導的にまたは共鳴によって安定化できる置換基(例えばアルコキシ)であり、記号(*)は、2価リンカーを形成している付加スペーサーリンカー、ヘテロ原子リンカー、または放出型リンカーへの結合点、あるいは薬物、あるいはその類似体または誘導体、あるいはビタミン、あるいはその類似体または誘導体への結合点を示す。これに限定されるわけではないが、ヒドロキシ、アルキル、アルコキシ、アルキルチオ、ハロなどを含む他の置換基が、アリール環、ベンジル炭素、アルカン酸、またはメチレン架橋に存在しうることが認識される。補助開裂には、ベンジリウム中間体、ベンジン中間体、ラクトン環化、オキソニウム中間体、ベータ脱離などをともなう機構が含まれうる。放出型リンカーの開裂に続く断片化に加えて、放出型リンカーの初期開裂が隣接基補助機構によって促進されうることがさらに認識される。
【0029】
別の実施形態において、2価リンカーは、2価3-チオスクシンイミド-1-イルアルキルオキシメチルオキシ基を形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、次の式
によって表される。式中、nは、1〜6の整数であり、アルキル基は場合により置換され、メチルは、付加アルキル、または場合により置換されたアリール基によって任意に置換され、そのそれぞれが独立して選択された基Rによって表される。記号(*)は、2価リンカー断片の、本明細書に記載する結合体の他の部分への結合点を示す。別の実施形態において、2価リンカーは、2価3-チオスクシンイミド-1-イルアルキルカルボニル基を形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、次の式
によって表される。式中、nは、1〜6の整数であり、アルキル基は場合により置換される。記号(*)は、2価リンカー断片の、本明細書に記載する結合体の他の部分への結合点を示す。別の実施形態において、2価リンカーは、2価3-チオアルキルスルホニルアルキル(2置換シリル)オキシ基を形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、2置換シリルは、アルキルおよび/または場合により置換されたアリール基によって置換される。
【0030】
別の実施形態において、2価リンカーは、2価ジチオアルキルカルボニルヒドラジド基、または2価3-チオもしくは3-ジチオスクシンイミド-1-イルアルキルカルボニルヒドラジドを形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、次の式
によって表される。式中、nは、1〜6の整数であり、アルキル基は場合により置換され、ヒドラジドは、(B)、(D)、または2価リンカー(L)の他の部分と共にヒドラゾンを形成する。記号(*)は、2価リンカー断片の、本明細書に記載する結合体の他の部分への結合点を示す。
【0031】
別の実施形態において、2価リンカーは、2価3-チオスクシンイミド-1-イルアルキルオキシアルキルオキシアルキリデン基を形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、次の式
によって表される。式中、各nは、1〜6より独立して選択された整数であり、各アルキル基は、独立して選択され、そして、例えばアルキルまたは場合により置換されたアリールによって場合により置換され、ここでアルキリデンは、(B)、(D)、または2価リンカー(L)の他の部分と共にヒドラゾンを形成する。記号(*)は、2価リンカー断片の、本明細書に記載する結合体の他の部分への結合点を示す。
【0032】
別の実施形態において、2価リンカーは、2価3-チオまたは3-ジチオアリールアルキルオキシカルボニル基、3-チオまたは3-ジチオアリールアルキルアミノカルボニル基、2価3-チオまたは3-ジチオアルキルオキシカルボニル、あるいは2価3-チオまたは3-ジチオアルキルアミノカルボニルを形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、ここでアルキルカルボニルは、(B)、(D)、または2価リンカー(L)の他の部分と共にカーボネート、カルバメート、または尿素を形成する。例示としては、アルキル基はエチルである。
【0033】
別の実施形態において、2価リンカーは、2価3-ジチオアルキルアミノ基を形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、ここでアミノは、(B)、(D)、または2価リンカー(L)の他の部分と共にビニル基含有アミドを形成する。例示としては、アルキル基はエチルである。
【0034】
別の実施形態において、2価リンカーは、2価1-アルコキシシクロアルキレンオキシ基、2価アルキレンアミノカルボニル(ジカルボキシルアリーレン)カルボキシラート基、2価3-ジチオアルコキシカルボニル基、2価3-ジチオアルキルオキシカルボニルヒドラジド基、2価を形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含む。
【0035】
別の実施形態において、2価リンカーは、アミノ酸で形成されたペプチドである少なくとも1個のスペーサーリンカーを含む。一態様において、ペプチドは天然型アミノ酸、およびその立体異性体を含む。別の態様において、ペプチドは天然型アミノ酸、およびその立体異性体のみから形成される。
【0036】
スペーサーリンカーおよび放出型リンカーのさらなる例証的な例を表1および2に示した。ここで(*)は、別のリンカーへの、ビンカアルカロイド、またはその類似体もしくは誘導体への結合点、あるいは受容体結合部分への結合点を示す。
表1 企図されるスペーサーリンカーおよびヘテロ原子リンカー、およびその組合せ
表2 企図される放出型リンカーおよびヘテロ原子リンカー、およびその組合せ
【0037】
本明細書で言及するように、本明細書に記載する結合体に使用できるビンカ薬物としては、アルカロイドのビンカインドール−ジヒドロインドールファミリーのすべてのメンバー、例えばビンデシン、ビンブラスチン、ビンクリスチン、カタランチン、ビンドリン、ロイロシン、ビノレルビン、イミドカルブ、シブトラミン、トルトラズリル、ビンブラスチン酸など、ならびにその類似体および誘導体が挙げられる。例証的には、そのような類似体および誘導体としては、米国特許第4,203,898号に記載されている3−カルボキスアジド;米国特許第4,166,810号に記載されている4-デスアセチルビンブラスチン-3-カルボキスヒドラジドのN2-アルキルおよび他の誘導体;Neuss et al.Tetrahedron Lett.783(1968)に記載されているロイロシンヒドラジド;Barnett et al.J.Med.Chem.21:88(1978)に記載されているヒドラジド誘導体;米国特許第3,392,173号および同第3,387,001号に記載されているC-4エステル誘導体;Langone et al.Anal.Biochem.95:214(1979)に記載されている酸化により生じるジカルボン酸誘導体;およびEP0247792A2に記載されているビンカヒドラジドが挙げられる。上記の特許および刊行物のそれぞれは、それがビンカ化合物を調製するための合成経路、および反応条件に関して開示する全てについて、参照により本明細書に組み入れられている。
【0038】
例証的な一実施形態において、ビンカ薬物は、式
の化合物であり、式中、
R1およびR2の一方がHであり、他方がエチルであり、かつR3はHであり、あるいはR1はエチルであり、R2およびR3はひとまとめにされて-O-を形成し;
R4、R7、およびR8は、H、アルキル、およびアシルからそれぞれ独立して選択され、
R5およびR6は、それぞれ独立して選択されたアルキルであり;
R9は、基-NHNHRであり、式中、Rは、H、アルキル、またはアシルであり;
R10は、Hまたはアシルであり;かつ
R11は、エチルである。
【0039】
一態様において、ビンカ薬物は、上式の化合物であり、式中、R4およびR8はそれぞれHであり;かつR5、R6、R9、およびR10はそれぞれメチルである。
【0040】
本明細書に記載する結合体において有用な細胞表面受容体のリガンドとしては、これに限定されるわけではないが、ビタミン、およびビタミン受容体に結合する他の部分、トランスポーター、あるいはビタミンまたはその類似体もしくは誘導体に特異的に結合する他の表面提示タンパク質、ライブラリースクリーニングから同定されたペプチドリガンド、腫瘍細胞特異性ペプチド、腫瘍細胞特異性アプタマー、腫瘍細胞特異性炭水化物、腫瘍細胞特異性モノクローナルまたはポリクローナル抗体、抗体のFabまたはscFv(すなわち単鎖可変領域)断片(例えば、EphA2に、または転移性癌細胞で特異的に発現または特異的に接近可能な他のタンパク質に対する抗体のFab断片)、コンビナトリアルライブラリーに由来する小型有機分子、成長因子(例えばEGF、FGF、インスリン、およびインスリン様成長因子)、および相同ポリペプチド、ソマトスタチンおよびその類似体、トランスフェリン、リポタンパク質複合体、胆汁塩、セレクチン、ステロイドホルモン、Arg-Gly-Asp含有ペプチド、レチノイド、各種のガレクチン、δ-オピオイド受容体リガンド、コレシストキニンA受容体リガンド、アンギオテンシンAT1またはAT2受容体に特異的なリガンド、ペルオキシソーム増殖因子活性化受容体λリガンド、β-ラクタム抗体(例えばペニシリン、抗菌薬を含む小型有機分子、ならびに腫瘍細胞の表面でまたは感染性生物で優先的に発現される受容体に特異的に結合する他の分子)、受容体または他の細胞表面タンパク質の結晶構造に基づき特定の受容体の結合ポケット内に適合するように設計された抗菌薬および他の薬物、腫瘍抗原のリガンドまたは腫瘍細胞の表面で優先的に発現される他の分子のリガンド、あるいはこれらのいずれかの分子の断片が挙げられる。リガンド-ビンカ結合体の結合部位として機能しうる腫瘍特異性抗原の例としては、タンパク質のエフリンファミリーのメンバーの細胞外エピトープ、例えばEphA2が挙げられる。EphA2発現は、正常細胞の細胞-細胞接合部に限定されるが、転移性腫瘍細胞ではEphA2は細胞表面全体に分布している。それゆえ転移性細胞のEphA2は、例えばビンカ化合物に結合した抗体のFab断片への結合のために接近可能であるが、これに対して該タンパク質は正常細胞のFab断片への結合のためには接近できず、そのため転移性癌細胞に対して特異的なリガンド-ビンカ結合体を生じる。
【0041】
本明細書に記載する方法および化合物にしたがって使用されうるビタミンとしては、カルニチン、イノシトール、リポ酸、ピリドキサール、アスコルビン酸、ナイアシン、パントテン酸、葉酸、リボフラビン、チアミン、ビオチン、ビタミンB12、ビタミンA、D、E、およびK,他の関連ビタミン分子、その類似体および誘導体、ならびにその組合せが挙げられる。これらのビタミン、ならびにその受容体結合類似体および誘導体は、薬物送達結合体を作製するために、本明細書に記載する2価リンカー(L)によってビンカ化合物に結合されうる例証的な標的実体を構成する。
【0042】
例証的な一態様において、ビタミンは葉酸、葉酸類似体、またはその他の葉酸塩受容体結合分子でありうる。使用されうる葉酸塩の類似体の例としては、フォリン酸、プテロイルポリグルタミン酸、プテロイン酸およびその他のアミノ酸誘導体、ならびに葉酸塩受容体結合プテリジン、例えばテトラヒドロプテリン、ジヒドロ葉酸塩、テトラヒドロ葉酸塩、ならびにそのデアザおよびジデアザ類似体が挙げられる。「デアザ」および「ジデアザ」類似体という用語は、天然型葉酸構造において1または2個の窒素原子に代わって置換された炭素原子を有する当分野で認識された類似体を指す。例えばデアザ類似体としては、1-デアザ、3-デアザ、5-デアザ、8-デアザ、および10デアザ類似体が挙げられる。ジデアザ類似体としては例えば、1,5-ジデアザ、5,10-ジデアザ、8,10-ジデアザ、および5,8-ジデアザ類似体が挙げられる。上記の葉酸類似体は、葉酸塩受容体に結合するその能力を反映して、慣例的に「葉酸塩」と呼ばれる。他の葉酸塩受容体結合類似体としては、アミノプテリン、アメトプテリン(メトトレキセート)、N10-メチル葉酸、2-デアミノ-ヒドロキシ葉酸、デアザ類似体、例えば1-デアザメトプテリンまたは3-デアザメトプテリン、および3’,5’-ジクロロ-4-アミノ-4-デオキシ-N10-メチルプテロイルグルタミン酸(ジクロロメトトレキセート)が挙げられる。薬物送達結合体の受容体媒介エンドサイトーシス輸送を開始させるために葉酸塩受容体に結合可能である他の適切なリガンドとしては、葉酸受容体に対する抗体が挙げられる。したがって例証的な一態様において、葉酸塩受容体に対する抗体と複合化したビンカ化合物は、複合体の膜透過輸送を引き起こすために使用されうる。
【0043】
ビタミン類似体および/または誘導体の例証的な実施形態も、ビオチンの類似体および誘導体、例えばビオシチン、ビオチンスルホキシド、オキシビオチンおよび他のビオチン受容体結合化合物などを含む。本明細書に記載する他のビタミンの類似体および誘導体も本明細書で企図されることが認識される。
【0044】
本明細書に記載する薬物送達結合体は、在来の合成方法によって調製されうる。合成方法は、ヘテロ原子リンカーの選択、ならびにスペーサーリンカーおよび放出型リンカーに存在する官能基に応じて選択されうる。一般に、関連する結合形成反応は、Richard C.Larock,“Comprehensive Organic Transformations,a guide to functional group preparations,”VCH Publishers,Inc.New York(1989)、およびTheodora E.Greene & Peter G.M.Wuts,“Protective Groups ion Organic Synthesis,”2d edition,John Wiley & Sons,Inc.New York(1991)に記載されており、その開示は全体が参照により本明細書に組み入れられている。さらなる合成経路および反応条件は、米国特許出願公開第2005/0002942A1号に記載されている。
【0045】
例示としては、本明細書に記載する薬物送達結合体は、線形および収束合成経路の両方を使用して調製されうる。このような経路で使用できる例証的な中間体としては、受容体結合部分、あるいはその類似体および誘導体、ならびにビンカアルカロイド、あるいはその類似体または誘導体への共有結合に適切なカップリング基をそれぞれの端に含む2価リンカーを含む中間体が挙げられる。このような経路で使用できる他の例証的な中間体としては、カップリング基を含む2価リンカーに結合された受容体結合部分、あるいはその類似体または誘導体を含む中間体が挙げられる。このような経路で使用できるその他の例証的な中間体としては、カップリング基を含む2価リンカーに結合された、ビンカアルカロイドあるいはその類似体または誘導体を含む中間体が挙げられる。どちらの場合でも、カップリング基は求核試薬、求電子試薬、またはそれらの前駆体でありうる。
【0046】
合成中間体の例証的な一実施形態において、カップリング基はMichael受容体であり、2価リンカーとしては、式-C(O)NHN=、-NHC(O)NHN=、または-CH2C(O)NHN=を有する放出型リンカーを含む。例証的な一態様において、カップリング基および2価リンカーはひとまとめにされて、式:
を有する化合物またはその保護誘導体を形成し、式中、(D)は、本明細書で例証するようなヒドラゾンを形成できるビンカアルカロイド、あるいはその類似体または誘導体であり;そしてnは、1、2、3、または4などの整数である。本明細書に記載する受容体結合薬物送達結合体中間体の別の例証的な態様において、第2のリンカーが、第2のリンカーに含まれるアルキルチオール求核試薬を介して上式に共有結合される。別の例証的な態様において、受容体結合部分、あるいはその類似体または誘導体は、その部分に含まれるアルキルチオール求核試薬を介して上式に共有結合される。
【0047】
別の例証的な実施形態において、カップリング基は例えば窒素、酸素、または硫黄等のヘテロ原子であり、2価リンカーは、受容体結合部分をカップリング基に共有結合する1個以上のヘテロ原子リンカーおよび1個以上のスペーサーリンカーを含む。例証的な一態様において、本明細書に記載する中間体は、式:
を有する化合物またはその保護誘導体を含み、式中、Xは、酸素、窒素、または硫黄であり、mは、1、2または3などの整数であり、かつ(B)、ls、およびlHは本明細書で定義する通りである。例証的な一態様において、lHは-NH-であり、mは1である。別の例証的な態様において、lHは-NH-であり、mは1であり、Xは-S-である。
【0048】
別の例証的な態様において、本明細書に記載する中間体は、式:
を有する化合物またはその保護誘導体を含み、式中、Yは、Hまたは置換基、例証的には、これに限定されるわけではないが、ニトロ、シアノ、ハロ、アルキルスルホニル、カルボン酸誘導体などを含む電子吸引性置換基であり、かつ(B)およびlsは本明細書で定義する通りである。
【0049】
本明細書に記載する中間体の別の例証的な実施形態において、カップリング基はMichael受容体であり、2価リンカーは、受容体結合部分をカップリング基に共有結合する1個以上のヘテロ原子リンカーおよび1個以上のスペーサーリンカーを含む。例証的な一態様において、カップリング基および2価リンカーはひとまとめにされて、式:
を有する化合物またはその保護誘導体を含み、式中、Xは、酸素、窒素、または硫黄であり、mおよびnは、独立して選択された整数、例えば1、2、または3であり、かつ(B)、lsおよび1Hは、本明細書で定義する通りである。別の例証的な態様において、ビンカアルカロイド、あるいはその類似体または誘導体は、ビンカアルカロイドに含まれるアルキルチオール求核試薬を介して上の式に共有結合される。
【0050】
別の例証的な態様において、中間体は、式:
を有する化合物またはその保護誘導体を含み、式中、AAは1個以上のアミノ酸であり、例示的には、天然型アミノ酸またはその立体異性体から例証的に選択され、Xは、窒素、酸素、または硫黄であり、Yは、水素または置換基、例証的には、これに限定されるわけではないが、ニトロ、シアノ、ハロ、アルキルスルホニル、カルボン酸誘導体などを含む電子吸引性置換基であり、nおよびmは、独立して選択される整数、例えば1、2、または3であり、pは、1、2、3、4、または5などの整数である。AAは、例えば一般式:-N(R)-(CR’R”)q-C(O)-を有する、他の任意のアミノ酸でもあり、式中、Rは、水素、アルキル、アシル、または適切な窒素保護基であり、R’およびR”は、水素または置換基であり、そのそれぞれが各存在に独立して選択され、qは、1、2、3、4、または5などの整数である。例証的には、R’およびR”は、これに限定されるわけではないが、天然型アミノ酸に存在する水素または側鎖、例えばメチル、ベンジル、ヒドロキシメチル、チオメチル、カルボキシル、カルボキシメチル、グアニジノプロピルなど、ならびにその誘導体および保護誘導体に独立して相当する。上記の式は、すべての立体異性変形を含む。例えばアミノ酸は、アスパラギン、アスパラギン酸、システイン、グルタミン酸、リジン、グルタミン、アルギニン、セリン、オルニチン、トレオニンなどから選択されうる。本明細書に記載するビタミン受容体結合薬物送達結合体中間体の別の例証的な態様において、薬物、あるいはその類似体または中間体は、アルキルチオール求核試薬を含む。
【0051】
上の中間体のそれぞれは、在来の合成経路を使用して調製されうる。さらなる合成経路および反応条件は、米国特許出願第10/765,336号および国際出願PCT/US/2005/026068号に記載されている。
【0052】
上の例証的な実施形態は、本明細書に記載する本発明の例示として意図され、本明細書に記載する本発明を制限するものとして、決して説明または解釈されるべきではない。例えば次の例証的なビタミン-薬物結合体によって一般に表される化合物は、本明細書によって記載されるように本明細書に含まれるものであり、
式中、R1およびR2はそれぞれ独立して、水素またはアルキル、例えばメチルであり;かつlHは、ヘテロ原子、例えば酸素、硫黄、任意に置換された窒素、または場合により保護された窒素などである。
【0053】
別の実施形態において、本明細書に記載する化合物は、ケタール基を含む放出型リンカーから形成された2価リンカーを含む。一態様において、ケタール基は、任意に置換された2-、または4-オキシベンズアルデヒド、例えば式:
の4-オキシベンズアルデヒドであり、式中、nは、1、2、3、および4から選択され;Raは、アルキルまたは場合により置換されたアリールアルキルであり、Raは、水素または任意選択の置換基であり;そして(*)原子は、受容体結合部分、2価リンカー、またはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される。
【0054】
別の実施形態において、本明細書に記載する化合物は、カーボネートを含む放出型リンカーから形成された2価リンカーを含む。一態様において、カーボネートは、ビスアルキルカーボネートである。別の態様において、カーボネートは、ジチオ基およびアミノ基またはヒドラジノ基を含むビスアルキルカーボネートである。別の態様において、カーボネートは、式:
の構造であり、式中、nおよびmは、1、2、3、および4よりそれぞれ独立して選択され;かつ(*)原子は、受容体結合部分、2価リンカー、またはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される。
【0055】
別の実施形態において、本明細書に記載する化合物は、2価ジチオアルキルアミノ基または2価ジチオベンジルオキシカルボニル基を含む放出型リンカーから形成された2価リンカーを含む。一態様において、2価ジチオアルキルアミノ基は、式:
の構造であり、式中、nは、1、2、3、および4より選択され;かつ(*)原子は、受容体結合部分、2価リンカー、またはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される。一態様において、2価ジチオベンジルオキシカルボニル基は、式:
の構造であり、式中、Rは、水素または任意選択の置換基であり;かつ(*)原子は、受容体結合部分、2価リンカー、またはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される。一態様において、2価ジチオベンジルオキシカルボニル基は、式:
の構造であり、式中、Rは、水素、アルキル、アルコキシ、シアノ、またはニトロであり;かつ(*)原子は、受容体結合部分、2価リンカー、またはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される。一態様において、2価ジチオベンジルオキシカルボニル基は、式:
の構造であり、式中、Rは、水素、アルキル、アルコキシ、シアノ、またはニトロであり;かつて(*)原子は、受容体結合部分、2価リンカー、またはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される。
【0056】
別の実施形態において、本明細書に記載する化合物としては、結合体を形成するために窒素を介して2価リンカーに結合されるカルボキサミドを含む、ビンカアルカロイド、あるいはその類似体または誘導体が挙げられる。別の実施形態において、本明細書に記載する化合物としては、結合体を形成するために窒素原子を介して2価リンカーに結合されるカルボキスヒドラジドを含む、ビンカアルカロイド、あるいはその類似体または誘導体が挙げられる。一態様において、その結合は末端窒素を介して作られる。別の実施形態において、本明細書に記載する化合物としては、結合体を形成するために酸素を介して2価リンカーに結合されるカルボキシラートを含む、ビンカアルカロイド、あるいはその類似体または誘導体が挙げられる。
【0057】
別の例証的な実施形態において、受容体結合部分(B)は、リンカー(L)-(D)が次の構造:
であるときに、葉酸ではない。
別の例証的な実施形態において、受容体結合部分(B)は、リンカー(L)-(D)が次の構造:
であるときに、葉酸ではない。
【0058】
他の実施形態において、製薬組成物が記載されている。製薬組成物は、本明細書に記載した薬物送達結合体を、そのための製薬的に許容される担体、賦形剤および/または希釈剤と組合せて含む。
【0059】
別の実施形態において、病原性細胞の集団を内部に持つ宿主動物において病原性細胞の集団を排除する方法が記載される。例証的な一態様において、病原性細胞集団のメンバーは、受容体結合部分、あるいはその類似体または誘導体に対するアクセス可能な結合部位を有し、その結合部位は病原性細胞によって独自に、発現、過剰発現または優先的に発現される。該方法は、本明細書で記載するように、宿主に本明細書に記載する薬物送達結合体、あるいはその製薬組成物を投与する工程を含む。
【0060】
本明細書に記載する薬物送達結合体は、ヒト臨床医学的および獣医学的用途の両方に使用されうる。それゆえ病原性細胞の集団を持ち、薬物送達結合体によって治療される宿主動物はヒトであり、あるいは獣医学的用途の場合には、実験用動物、農業用動物、家畜、または野生動物でありうる。本明細書に記載する薬物送達結合体は、これに限定されるわけではないが、ヒト、例えばげっ歯類(例えばマウス、ラット、ハムスターなど)、ウサギ、サル、チンパンジー等の実験用動物、例えばイヌ、ネコ、およびウサギ等の家畜、例えばウシ、ウマ、ブタ、ヒツジ、ヤギ等の農業用動物、および、例えばクマ、パンダ、ライオン、トラ、ヒョウ、ゾウ、シマウマ、キリン、ゴリラ、イルカ、およびクジラ等の飼育されている野生動物を含む宿主動物に投与されうる。
【0061】
本明細書に記載する薬物送達結合体は、宿主動物における各種の病状および病原性細胞を治療するために使用されうる。本明細書で使用するように「病原性細胞」は、癌細胞、感染性因子(例えば細菌およびウィルス)、細菌またはウィルス感染細胞、疾患状態を引き起こすことができる活性化マクロファージ、リガンド受容体(例えばビタミン受容体あるいはビタミンの類似体または誘導体を結合する受容体)を特異的に発現する、優先的に発現する、または過剰発現する他の任意の種類の病原性細胞を意味する。病原性細胞は、薬物送達結合体による治療が疾患症状の低減をもたらす疾患状態を引き起こすいかなる細胞も含みうる。病原性細胞は、移植片対宿主病の原因である免疫系の細胞などの、ある状況の下で病原性であるが、他の状況では病原性でない宿主細胞でもありうる。
【0062】
それゆえ病原性細胞の集団は、良性腫瘍および悪性腫瘍を含めて腫瘍原性である癌細胞集団でありうるか、またはそれは非腫瘍原性でありうる。癌細胞集団は、自然発生的に、または宿主動物の生殖細胞系に存在する変異または体細胞変異などのプロセスによって発生しうるか、あるいはそれは化学的に、ウィルスによって、または放射線によって誘発されうる。本発明は、癌腫、肉腫、リンパ腫、ホジキン病、黒色腫、中皮腫、バーキットリンパ腫、上咽頭癌、白血病、および骨髄腫などの癌を治療するために利用されうる。癌細胞集団としては、これに限定されるわけではないが、口腔癌、甲状腺癌、内分泌癌、皮膚癌、胃癌、食道癌、喉頭癌、膵臓癌、結腸癌、膀胱癌、骨癌、卵巣癌、子宮頸癌、子宮癌、乳癌、精巣癌、前立腺癌、直腸癌、腎臓癌、肝臓癌、および肺癌が挙げられる。
【0063】
病原性細胞集団が癌細胞集団である実施形態において、薬物送達結合体投与の効果は、腫瘤の縮小または消滅あるいは腫瘍細胞増殖の阻害によって測定される治療応答である。腫瘍の場合、消滅は、原発性腫瘍の細胞の、あるいは転移した腫瘍細胞の、または原発性腫瘍から解離する過程にある細胞の消滅でありうる。腫瘍の外科的除去、放射線療法、化学療法、または生物療法を含むいずれかの治療手法による腫瘍の除去後に腫瘍の再発を防止するための、薬物送達結合体による予防的治療も企図される。予防的治療は、薬物送達結合体を用いた初期治療、例えば連日多回容量処方での治療、および/または(複数の)初回治療後に数日または数ヶ月の間隔を置いた追加治療または一連の治療でありうる。したがって上記の任意の病原性細胞集団の排除は、病原性細胞の数の減少、病原性細胞の増殖の阻害、病原性細胞の再発を防止する予防的治療、または疾患の症状の軽減を引き起こす病原性細胞の治療を含む。
【0064】
癌細胞が排除される場合、本明細書に記載する方法は、腫瘍の外科的除去、放射線療法、化学療法、またはその他の免疫療法等の生物療法(これに限定されるわけではないが、モノクローナル抗体療法、免疫調節薬による治療、免疫エフェクタ細胞の養子移入、造血成長因子、サイトカインおよびワクチン接種による治療を含む)と組合せて使用されうる。
【0065】
本明細書に記載する方法は、各種の感染性疾患を引き起こす病原性細胞の集団にも利用できる。例えば本発明は、細菌、酵母を含む菌類、ウィルス、ウィルス感染細胞、マイコプラズマ、および寄生生物などの病原性細胞の集団に利用できる。本明細書に記載する薬物送達結合体によって治療されうる感染性生物は、グラム陰性またはグラム陽性球菌または桿菌である細菌などの生物を含む、動物において病原を引き起こす当分野で認識された任意の感染性生物である。例えばプロテウス種、クレブシエラ種、プロビデンシア種、エルシニア種、エルウィニア種、エンテロバクター種、サルモネラ種、セラチア種、アエロバクター種、エシェリキア種、シュードモナス種、赤痢菌種、ビブリオ種、アエロモナス種、カンピロバクター種、ストレプトコッカス種、スタフィロコッカス種、ラクトバシラス種、ミクロコッカス種、モラクセラ種、バシラス種、クロストリジウム種、コリネバクテリウム種、エベルセラ(Eberthella)種、ミクロコッカス種、マイコバクテリウム種、ナイセリア種、ヘモフィルス種、バクテロイデス種、リステリア種、エリジペロトリックス種、アシネトバクター種、ブルセラ種、パスツレラ種、ビブリオ種、フラボバクテリウム種、フゾバクテリウム種、ストレプトバシラス種、カリマトバクテリウム種、レジオネラ種、トレポネーマ種、ボレリア種、レプトスピラ種、アクチノミセス種、ノカルジア種、リケッチア種、および宿主動物において疾患を引き起こす他の任意の細菌種は、本明細書に記載する薬物送達結合体によって治療されうる。
【0066】
特に興味深いのは、抗生物質耐性ストレプトコッカス種およびスタフィロコッカス種などの抗生物質に対して耐性である細菌、または抗生物質に対して感受性であるが、抗生物質によって治療できる反復性感染を引き起こすので、耐性生物が最終的に発生する細菌である。抗生物質に対して感受性であるが、抗生物質によって治療できる反復性感染を引き起こすので、耐性生物が最終的に発生する細菌は、これらの抗生物質耐性細菌株の発生を回避するために、抗生物質の非存在下で、または宿主動物に通常投与されるよりも低い用量の抗生物質と組合せて、本明細書に記載する薬物送達結合体によって治療されうる。
【0067】
例えばDNAおよびRNAウィルス等のウィルスによって引き起こされた疾患も、本明細書に記載する薬物送達結合体によって治療されうる。このようなウィルスとしては、これに限定されるわけではないが、DNAウィルス、例えばパピローマウィルス、パルボウィルス、アデノウィルス、ヘルペスウィルス、およびワクシニアウィルス、ならびにRNAウィルス、例えばアレナウィルス、コロナウィルス、リノウィルス、呼吸器合胞体ウィルス、インフルエンザウィルス、ピコルナウィルス、パラミキソウィルス、レオウィルス、レトロウィルス、レンチウィルス、およびラブドウィルスが挙げられる。
【0068】
本明細書に記載する薬物送達結合体は、任意の、酵母を含む菌類、マイコプラズマ種、寄生生物、または動物で疾患を引き起こす他の感染性生物によって引き起こされた疾患を治療するためにも使用されうる。本明細書に記載する方法および薬物送達結合体によって治療されうる菌類の例としては、カビとして成長する、または酵母様菌類が挙げられ、これは、例えば白癬、ヒストプラスマ症、ブラストミセス症、アスペルギルス症、クリプトコッカス症、スポロトリクム症、コクシジオイデス症、パラコクシジオイデス症、ムコール症、色素酵母菌症、皮膚糸状菌症、プロトテカ症、フザリウム症、粃糠疹、菌腫、パラコクシジオイデス症、フェオフィホ真菌症、シュードアレシェリア症、スポロトリクム症、砂毛症、ニューモシスチス感染症、およびカンジダ症を引き起こす菌類を含む。
【0069】
本明細書に記載する薬物送達結合体は、これに限定されるわけではないが、サナダムシ(例えば条虫、矮小条虫、裂頭条虫属、およびエキノコッカス種)、吸虫(例えば肥大吸虫、異形吸虫、メタゴニムス、肝吸虫、肝蛭、肺吸虫、および住血吸虫種)、回虫(例えば蟯虫、鞭虫、豚回虫、鉤虫、アメリカ鉤虫、ストロンギロイデス、旋毛虫、ブケレリア、ブルギア、ロアオンコセルカ、およびドラクンクルス種)、アメーバ(例えばネグレリアおよびアカントアメーバ種)、ならびに原虫(例えばプラスモディウム、トリパノソーマ、リーシュマニア、トキソプラズマ、エントアメーバ、ジアルジア、イソスポラ、クリプトスポリジウム、およびエンテロシトゾーン種)によって引き起こされる感染を含む、寄生生物感染を治療するためにも使用されうる。
【0070】
薬物送達結合体が対象とする病原性細胞は、これらの細胞がリガンド受容体、例えばビタミン、あるいはその類似体または誘導体の受容体を優先的に発現する場合、内在性病原体を持つ細胞、例えばウィルス-、マイコプラズマ-、寄生生物-、または細菌-感染細胞でもありうる。
【0071】
一実施形態において、リガンドが、受容体、輸送体、またはリガンドを特異的に結合して、病原性細胞で優先的に発現される他の表面提示タンパク質へ結合する時に、薬物送達結合体は標的病原性細胞内へ内部移行されうる。そのような内部移行は例えば、受容体媒介エンドサイトーシスを通じて発生しうる。薬物送達結合体が放出型リンカーを含有する場合、リガンドおよびビンカ化合物は細胞内で解離して、ビンカはその細胞内標的に作用しうる。
【0072】
別の例証的な実施形態において、薬物送達結合体のリガンドは、ビンカ化合物を病原性細胞の表面に密接に配置する病原性細胞に結合しうる。ビンカ化合物は次に、放出型リンカーの開裂によって放出されうる。例えばビンカ化合物は、放出型リンカーがジスルフィド基である場合、タンパク質ジスルフィドイソメラーゼによって放出されうる。ビンカ化合物は次に、受容体結合薬物送達結合体が結合されている病原性細胞によって取り込まれうるか、またはビンカ化合物はそれに近接した別の病原性細胞によって取り込まれうる。あるいはビンカ化合物は、放出型リンカーがジスルフィド基である細胞内のタンパク質ジスルフィドイソメラーゼによって放出されうる。ビンカ化合物は、あるベータ離脱機構について上述したような加水分解機構、例えば酸触媒による加水分解によって放出され、あるいはオキソニウムイオンまたはラクトニウムイオン産生機構を通じた隣接基補助開裂によっても放出されうる。放出型リンカーの選択は、ビンカ化合物が結合体から放出される機構を決定するであろう。このような選択は、薬物送達結合体が使用される条件によって事前に定義されうることが認識される。
【0073】
リンカーが放出型リンカーを含まない別の例証的な実施形態において、薬物送達結合体のリガンド部分は、病原性細胞をビンカ化合物に結合できる他の分子による攻撃の標的とするために、病原性細胞の表面にビンカ化合物を有する病原性細胞に結合しうる。あるいは本実施形態において、薬物送達結合体は結合時に標的とした細胞内へ内部移行可能であり、リガンド部分およびビンカ化合物は細胞内でビンカ化合物と結合を維持することができ、ビンカ化合物はリガンド部分から解離せずにその効果を示す。
【0074】
なお別の実施形態において、または上記の実施形態と組合せて、薬物送達結合体がビタミン受容体または別のリガンド受容体を結合する場合、結合体は、血清中に存在する溶解性ビタミン受容体またはアルブミンの様な血清タンパク質に結合可能であり、そのため非結合ビンカ化合物と比較して結合体の長期の循環と、非結合ビンカ化合物と比較して病原性細胞集団に対して結合体の活性上昇を引き起こす。
【0075】
例えばビタミンの様なリガンドへの結合部位は、リガンドに特異的に結合できるリガンドの受容体を含み、ここで受容体または他のタンパク質は病原性細胞の集団で特異的に発現、過剰発現または優先的に発現される。病原性細胞によって特異的に発現、過剰発現または優先的に発現された表面提示タンパク質は通例、非病原性細胞には存在しないか、または低濃度で存在する受容体であり、病原性細胞の選択的排除の手段を与える受容体である。薬物送達結合体は、癌細胞または他の種類の病原性細胞上の受容体への高親和性結合が可能でありうる。高親和性結合はリガンドに固有でありうるか、または結合親和性は化学修飾リガンドの使用によって向上されうる。
【0076】
本明細書に記載する薬物送達結合体は、追加の薬物が標的指向化されるかどうかにかかわらず、その他の任意の既知の薬物と共に併用療法で投与されうる。例証的な追加の薬剤としては、これに限定されるわけではないが、ペプチド、オリゴペプチド、レトロ-インベルソオリゴペプチド、タンパク質、少なくとも1個の非ペプチド結合がペプチド結合を置換するタンパク質類似体、アポタンパク、糖タンパク質、酵素、補酵素、酵素阻害薬、アミノ酸およびその誘導体、受容体および他の膜タンパク質、抗原およびそれに対する抗体、ハプテンおよびそれに対する抗体、ホルモン、脂質、リン脂質、リポソーム、毒素、抗体、鎮痛剤、気管支拡張薬、ベータ遮断薬、抗菌剤、降圧剤、心血管作動薬(抗不整脈薬、強心配糖体、抗狭心症薬、血管拡張薬を含む)、中枢神経系剤(刺激薬、向精神薬、抗躁薬、および抑制薬を含む)、抗ウィルス剤、抗ヒスタミン薬、化学治療剤を含む制癌剤、精神安定剤、抗うつ薬、H-2拮抗薬、抗痙攣薬、制嘔吐剤、プロスタグランジンおよびプロスタグランジン類似体、筋弛緩剤、抗炎症性物質、刺激薬、うっ血除去薬、鎮吐薬、利尿剤、鎮痙薬、抗喘息薬、抗パーキンソン病薬、去痰薬、鎮咳剤、粘液溶解薬、ならびにミネラルおよび栄養添加物が挙げられる。
【0077】
別の例証的な態様において、追加の薬物は、内在性免疫応答を刺激できる化合物より選択されうる。適切な化合物としては、これに限定されるわけではないが、サイトカインまたは免疫細胞成長因子、例えばインターロイキン1〜18、幹細胞因子、塩基性FGF、EGF、G-CSF、GM-CSF、FLK-2リガンド、HILDA、MIP-1α、TGF-α、TGF-β、M-CSF、IFN-α、IFN-β、IFN-γ、溶解性CD23、LIF、およびその組合せが挙げられる。
【0078】
これらの免疫活性化因子の治療的に有効な組合せが使用されうる。一実施形態において、例えば約0.1MIU/m2/用量/日〜約15MIU/m2/用量/日の範囲の量の連日多回用量処方におけるIL-2の治療的有効量、および例えば約0.1MIU/m2/用量/日〜約7.5MIU/m2/用量/日の範囲の量の連日多回用量処方におけるIFN-αの治療的有効量は、病原性細胞を持つ宿主動物において病原性細胞を排除、減少または中和するために、薬物送達結合体と共に使用されうる(MIU=100万国際単位;m2=平均的なヒトのおおよその体表面積)。別の実施形態において、IL-12およびIFN-αは、インターロイキンおよびインターフェロンについての上記の治療的有効量で使用され、なお別の実施形態において、IL-15およびIFN-αは、インターロイキンおよびインターフェロンについての上記の治療的有効量で使用されうる。代わりの実施形態において、IL-2、INF-αまたはINF-γ、ならびにGM-CSFは組合せて、上記の治療的有効量で使用されうる。他のインターロイキンおよびインターフェロン、ならびにコロニー刺激因子の組み合わせを含むサイトカインの他の有効な任意の組合せも使用されうる。
【0079】
さらに追加の薬物は、当分野で既知の任意の薬物でありうる。それらの薬物は、細胞毒性または細胞増殖抑制性であり、腫瘍浸透性を向上させ、腫瘍細胞増殖を阻害し、アポトーシスを促進し、標的細胞の抗アポトーシス活性を低下させ、感染性因子によって引き起こされた疾患を治療するために使用され、病原性細胞に向けられた内在性免疫応答を向上させ、または任意の種類の病原性細胞によって引き起こされた疾患状態を治療するために有用である。例示的で適切な追加の薬物としては、アデノコルチコイドおよびコルチコステロイド、アルキル化剤、抗アンドロゲン、抗エストロゲン、アンドロゲン、アクラマイシンおよびアクラマイシン誘導体、エストロゲン、抗代謝産物(例えばシトシンアラビノシド、プリン類似体、ピリミジン類似体、およびメトトレキセート)、ブスルファン、カルボプラチン、クロランブシル、シスプラチンおよび他のプラチナ化合物、タモキシフェン、タキソール、パクリタキセル、パクリタキセル誘導体、タキソテール(登録商標)、シクロホスファミド、ダウノマイシン、リゾキシン、T2毒素、植物アルカロイド、プレドニゾン、ヒドロキシ尿素、テニポシド、マイトマイシン、ディスコデルモライド、非ビンカ微小管阻害薬、エポチロン、チューブリシン(tubulysin)、シクロプロピルベンズ[e]インドロン、seco-シクロプロピルベンズ[e]インドロン、O-Ac-seco-シクロブロピルベンズ[e]インドロン、ブレオマイシンおよび他の任意の抗生物質、窒素マスタード、ニトロソ尿素、コルヒチン、コルヒチン誘導体、アロコルヒチン、チオコルヒチン、トリチルシステイン、ハリコンドリンB、ドラスタチン(例えばドラスタチン10)、アマニチン(例えばα-アマニチン)、カンプトセシン、イリノテカン、および他のそのカンプトセシン誘導体、ゲルダナマイシンおよびゲルダナマイシン誘導体、エラムスチン、ノコダゾール、MAP4、コルセミド、ビンデシン、ビンブラスチン、ビンクリスチン、カタランチン、ビンドリン、ロイロシン、ビノレルビン、イミドカルブ、シブトラミン、トルトラズリル、ビンブラスチン酸、メイタンシンならびにその類似体および誘導体、ゲムシタビン、炎症性剤および炎症誘発剤、ペプチドおよびペプチドミメティックシグナル伝達阻害薬、および任意の他の当分野で認識される薬物または毒素が挙げられる。併用療法で使用されうる他の薬物としては、ペニシリン、セファロスポリン、バンコマイシン、エリスロマイシン、クリンダマイシン、リファンピン、クロラムフェニコール、アミノグリコシド抗生物質、ゲンタマイシン、アンホテリシンB、アシクロビル、トリフルリジン、ガンシクロビル、ジドブジン、アマンタジン、リバビリン、および他の当分野で認識される任意の抗菌化合物が挙げられる。上記の追加の薬物のいずれかの類似体または誘導体も併用療法で使用されうる。
【0080】
別の例証的な実施形態において、製薬組成物が提供される。製薬組成物は、1回以上の用量で投与されるときに宿主動物において病原性細胞の集団を排除するのに有効な薬物送達結合体の量を含む。薬物送達結合体は好ましくは、宿主動物に非経口的に、例えば皮内に、皮下に、筋肉内に、腹腔内に、静脈内に、または髄腔内に投与される。あるいは薬物送達結合体は、例えば経口投与の様な他の医療的に有用なプロセスによって宿主動物に投与することができ、そして任意の有効な用量および適切な治療剤形、例えば持続放出剤形が使用されうる。経口剤形に有用な賦形剤の例としては、これに限定されるわけではないが、コーンスターチ、ゼラチン、ラクトース、ステアリル酸マグネシウム、重炭酸ナトリウム、セルロース誘導体、およびナトリウムデンプングリコール酸が挙げられる。
【0081】
非経口剤形の例としては、等張性生理的食塩水、5%グルコースまたは製薬的に許容されるその他の周知の液体、例えば液体アルコール、グリコール、エステル、およびアミド中の活性物質の水性溶液が挙げられる。本発明による非経口剤形は、薬物送達結合体の用量を含む復元可能な凍結乾燥物の形でありうる。本実施形態の一態様において、例えばその開示が参照により本明細書に組み入れられている、米国特許第4,713,249号;同第5,266,333号;および同第5,417,982号に記載されている生分解性炭水化物マトリクスなどの、当分野で既知の多数の持続放出剤形のいずれも投与可能であり、あるいは低速ポンプ(例えば浸透圧ポンプ)が使用されうる。
【0082】
併用療法における追加の薬物は、薬物送達結合体の前、後、または薬物送達結合体と同時に宿主動物に投与可能であり、追加の薬物は、薬物送達結合体を含有する同じ組成物の一部として、または薬物送達結合体とは異なる組成物の一部として投与されうる。このような任意の併用療法が有効な用量での追加薬物投与に使用されうる。
【0083】
別の例証的な態様において、2つ以上の種類の薬物送達結合体が使用されうる。例えば宿主動物は、異なるリガンドを持つ結合体、例えば組合された葉酸塩-ビンカおよびビタミンB12-ビンカ結合体などを用いた同時投薬プロトコルで治療されうる。別の例証的な態様において、宿主動物は、2つ以上のリガンド、例えば複数の葉酸塩または複数のビタミンB12を1つの結合体中に含む結合体により、または葉酸塩およびビタミンB12リガンドの両方に結合されたビンカ化合物などの、同じ結合体中のリガンドの組合せを用いて治療されうる。さらに別の薬物送達結合体中に異なる種類のビンカ化合物を含む薬物送達結合体が使用されうる。
【0084】
薬物送達結合体の日単位の処方量は、宿主の状態、治療される疾患状態、結合体の分子量、その投与経路および組織分布、ならびに放射線療法または併用療法の追加の薬物などの他の治療処置の同時使用の可能性に応じて著しく変化しうる。宿主動物へ投与される有効量は、体表面積、体重、および患者の状態の医師による評価に基づく。有効用量は、例えば約1ng/kg〜約1mg/kg、約1μg/kg〜約500μg/kg、および約1μg/kg〜約100μg/kgの範囲で変化しうる。
【0085】
薬物送達結合体を投与するため、任意の有効な投薬計画が使用されうる。例えば薬物送達結合体は、1回用量として投与されうるか、または分割されて、連日多回用量処方として投与されうる。さらに時差投薬計画、例えば週に1〜3日を連日治療の代用として使用することができ、本発明を定義する目的で、このような間欠的または時差連日投薬計画は、毎日の治療と同等と見なされ、企図される。例証的な一実施形態において、宿主動物は、病原性細胞の集合を排除するために薬物送達結合体の複数回注射によって治療される。一実施形態において、宿主は薬物送達結合体を複数回(好ましくは約2〜50回まで)、例えば12〜72時間の間隔で、または48〜72時間の間隔で注射される。薬物送達結合体の追加の注射は宿主動物に、初回注射後の数日または数ヶ月の間隔で投与でき、追加の注射は病原性細胞によって引き起こされた疾患状態の再発を防止しうる。
【0086】
例証的な一態様において、薬物送達結合体で使用されうるビタミンあるいはその類似体または誘導体は、活性化マクロファージ上で特異的に発現される受容体、例えば葉酸塩あるいはその類似体または誘導体を結合する葉酸塩受容体に結合するものを含みうる。葉酸塩結合体は例えば、宿主の疾患状態を引き起こす活性化マクロファージの活性を死滅または抑制するために使用されうる。そのようなマクロファージを標的とする結合体は、活性化マクロファージ媒介疾患状態にある宿主動物に投与されるときに、活性化マクロファージを死滅させる、またはマクロファージ機能を抑制するために、結合体されたビンカ化合物を活性化マクロファージの集合に集中させ結合させるように作用する。活性化マクロファージ集合の排除、減少、または非活性化は、治療される疾患状態の活性化マクロファージ媒介病原特徴を停止または低減するように作用する。活性化マクロファージによって媒介されることが既知の疾患の例としては、関節リウマチ、潰瘍性大腸炎、クローン病、乾癬、骨髄炎、多発性硬化症、アテローム硬化症、肺線維症、サルコイドージス、全身性硬化症、移植臓器拒絶(GVHD)および慢性炎症が挙げられる。薬物送達結合体の投与は通例、疾患状態の症状が低減または排除されるまで継続される。
【0087】
活性化マクロファージを死滅させる、または活性化マクロファージの機能を抑制するために投与される薬物送達結合体は、宿主動物に非経口的に、例えば皮内に、皮下に、筋肉内に、腹腔内、または静脈内に製薬的に許容される担体と共に投与されうる。あるいは薬物送達結合体は宿主細胞に他の医療的に有用な手順で投与でき、有効用量は標準剤形または持続放出剤形で投与されうる。治療方法は単独で、または活性化マクロファージによって媒介される疾患状態の治療のために認められた他の治療方法と組合せて使用されうる。
【0088】
本明細書に記載する本発明は、次の実施例によってさらに例証される;しかしながらこれらの実施例は単に本発明の例証として意図され、決して本発明を制限すると解釈されるべきでないことが理解されなければならない。例えば本明細書に示す各化合物において、リンカーを形成するのに使用されるアミノ酸の立体化学は場合により、天然l配置または非天然d配置から選択されうる。加えて本明細書では、これに限定されるわけではないが、ビンブラスチンの各種の他の類似体および誘導体、各種の他のスペーサー、ヘテロ原子、およびリンカー組合せ、およびその他を含む多くの変更形態が企図されうる。本明細書に記載する各実施例の化合物は、示されたようにNMR、MS、および/またはUV分光法、および/またはHPLCによって特徴付けられ、特徴的な1H NMRシグナル、MSシグナルなどを含む選択された分析データは必要に応じて記載される。
【実施例】
【0089】
===方法実施例===
〔方法実施例1 マウスにおける腫瘍成長の阻害〕
本明細書に記載した化合物の抗腫瘍活性は、腫瘍担持動物へ静脈内(i.v.)投与したときに、皮下M109腫瘍を担持するBalb/cマウスにおいて評価した。右腋窩の皮下組織への1x106個のM109細胞の腫瘍播種の約11日後(t0の時点での腫瘍容積範囲=60〜80mm3)、マウス(5/グループ)に週3回(TIW)、規定の期間(例えば2〜3週間)にわたって、(a)体重1キログラムあたりの規定用量レベルの、本明細書に記載する薬物送達結合体、または(b)等用量体積のPBS(対照群)をi.v.注射した。腫瘍成長は、各処置グループで2日または3日間隔で測径器を使用して測定した。腫瘍体積は、式V=axb2/2を使用して計算し、式中、「a」は腫瘍の長さであり、「b」は幅である(ミリメートルで表す)。
【0090】
〔方法実施例2 マウスにおける腫瘍成長の阻害〕
本明細書に記載した化合物の抗腫瘍活性は、腫瘍担持動物へ静脈内(i.v.)投与したときに、皮下KB腫瘍を担持するnu/nuマウスにおいて評価した。右腋窩の皮下組織への1x106個のKB細胞の腫瘍播種の約8〜11日後(t0の時点の腫瘍容積範囲=60〜80mm3)、マウス(5/グループ)に週3回(TIW)、規定の期間(例えば2〜3週間)にわたって、(a)体重1キログラムあたりの規定用量レベルの本明細書に記載する薬物送達結合体、または(b)等用量体積のPBS(対照群)をi.v.注射した。腫瘍成長は、各処置グループで2日または3日間隔で測径器を使用して測定した。腫瘍体積は、式V=axb2/2を使用して計算し、式中、「a」は腫瘍の長さであり、「b」は幅である(ミリメートルで表す)。
【0091】
〔方法実施例3 細胞DNA合成の阻害〕
本明細書に記載する化合物は、薬物が葉酸塩受容体陽性KB細胞の成長を阻害する能力を予測するin vitro細胞毒性アッセイを使用して評価した。化合物は、本明細書に記載するプロトコルに従って調製した各化学療法薬に結合された葉酸塩を含んでいた。KB細胞を所定の期間にわたって37℃にて、少なくとも100倍過剰の葉酸の非存在下または存在下で、指示された濃度の葉酸-薬物結合体に暴露した。この細胞を次に新しい培地ですすぎ、新しい培地で72時間にわたって37℃でインキュベートした。細胞生存能力は、3H-チミジン取り込みアッセイを使用して評価した。
【0092】
本明細書の図に示すように、用量依存性細胞毒性は測定可能であり、大半の場合でIC50値(新たに合成されたDNAへの3H-チミジン取り込みを50%低下させるのに必要な薬物結合体の濃度)は低いナノモル範囲であった。さらにこれらの結合体の細胞毒性は、過剰な遊離葉酸の存在下で低減され、観察された細胞死滅は葉酸塩受容体への結合によって仲介されることを示す。
【0093】
〔方法実施例4 相対親和性アッセイ〕
葉酸塩と比較した葉酸塩受容体(FR)への本明細書に記載する化合物の親和性は、先に記載された方法(Westerhof,G.R.,J.H.Schornagel,et al.(1995)Mol.Pharm.48:459-471)に多少の変更を加えて決定した。簡潔にはFR陽性KB細胞を24ウェル細胞培養プレートに密集して播種して、プラスチックに18時間付着させた。使用済みのインキュベーション培地を指定されたウェルにて、試験品または葉酸の上昇濃度の非存在下または存在下で100nM 3H-葉酸を添加した葉酸塩を含まないRPMI(FFRPMI)と交換した。細胞を37℃にて60分間インキュベートして、次にPBS(pH7.4)によってすすぎ、次いでPBS(pH7.4)中に1% SDS 500μLを添加した。次に細胞溶解液を収集して、シンチレーションカクテル5mLを含有する個々のバイアルに添加し、次に放射能をカウントした。負の対照サンプルは、FFRPMI中に3H-葉酸のみを含有していた(競合物質なし)。正の対照サンプルは1mM葉酸の最終濃度を含有し、この正の対照サンプルで測定で得られたCMP(標識の非特異性結合を表す)はすべてのサンプルから引かれた。特に相対親和性は、KB細胞のFRに結合する3H-葉酸の50%を置換するのに必要な化合物のモル比の逆数として定義され、FRに対する葉酸の相対親和性は1に設定された。
【0094】
〔方法実施例5 4T−1腫瘍体積アッセイ〕
6〜7週齢マウス(メスBalb/c株)をインディアナ州インディアナポリスのHarlan,Inc.より入手した。マウスをHarlanの葉酸を含まない固形飼料で、本実験の開始前および本実験中の合計3週間にわたって維持した。葉酸塩受容体-陰性4T-1腫瘍細胞(動物あたり1x106細胞)を右腋窩の皮下組織に接種した。4T-1腫瘍平均容積が〜100mm3である、腫瘍接種の約5日後に、マウス(5/グループ)に、週3回(TIW)、3週間にわたって、3μmol/kgの薬物送達結合体、または等用量体積のPBS(対照群)をi.v.注射した。腫瘍成長は、各処置グループで2日または3日間隔で測径器を使用して測定した。腫瘍体積は、式V=axb2/2を使用して計算し、式中、「a」は腫瘍の長さであり、「b」は幅である(ミリメートルで表す)。
【0095】
〔方法実施例6 体重測定〕
マウスの重量変化率は、関連する腫瘍容積アッセイに記載したサンプルのグラフに示すように、腫瘍接種後(PTI)の指示された日にマウス(5マウス/グループ)において測定した。
【0096】
〔方法実施例7 葉酸塩-ペプチドの一般的な調製〕
ペプチドを含む本明細書に記載するリンカーは、酸感受性Fmoc-AA-Wang樹脂におけるFmoc方法などの標準方法を使用したポリマー支持連続手法によって調製した。例証的には、葉酸塩含有ペプチジル断片Pte-Glu-(AA)n-NH(CHR2)CO2H(3)は、Wang樹脂支持アミノ酸およびFmoc保護アミノ酸合成からスキーム1に示す方法によって調製する。
<スキーム1>
(a)20%ピペリジン/DMF;(b)Fmoc-AA-OH、PyBop、DIPEA、DMF;(c)Fmoc-Glu-O-t-BuまたはFmoc-Glu(γ-O-t-Bu)-OH、PyBop、DIPEA、DMF;(d)N10(TFA)-Pte-OH;PyBop、DIPEA、DMSO;(e)TFAA、(CH2SH)2、i-Pr3SiH;(f)NH4OH、pH9〜10。
【0097】
本明細書に記載するプロセスのこの例示的な実施形態において、R1は、Fmocであり、R2は、所望の適切に保護されたアミノ酸側鎖であり、Wangは、2-クロロトリチル樹脂であり、DIPEAは、ジイソプロピルエチルアミンである。標準カップリング手順、例えばPyBop、および本明細書に記載するまたは当分野で既知である他の手順が使用され、そこではカップリング剤が例証的に、効率的なカップリングを確実にするための活性化剤として利用される。Fmoc保護基は、標準条件下での各カップリング工程の後に、例えばピペリジン、テトラブチルアンモニウムフルオリド(TBAF)などによる処置時に除去される。適切に保護されたアミノ酸構築ブロック、例えばFmoc-Glu-OtBu、N10-TFA-P-te-OHなどが、スキーム1に記載され、Fmoc-AA-OHによる工程(b)で表されるように使用される。したがってAAは、適切に保護される任意のアミノ酸出発物質を指す。本明細書で使用するアミノ酸という用語は、1個以上の炭素によって隔離されたアミンおよびカルボン酸官能基の両方を有するいずれの試薬も指すものとし、天然型アルファおよびベータアミノ酸、ならびにこれらのアミノ酸のアミノ酸誘導体および類似体を含むことが理解される。特に保護される側鎖を有するアミノ酸、例えば保護セリン、トレオニン、システイン、アスパラギン酸なども、本明細書に記載する葉酸塩-ペプチド合成で使用されうる。さらなるガンマ、デルタ、またはより長い相同性アミノ酸も、本明細書に記載の葉酸塩-ペプチド合成で出発物質として含まれうる。さらに相同性側鎖、または交互の分岐構造を有するアミノ酸類似体、例えばノルロイシン、イソバリン、β-メチルトレオニン、β-メチルシステイン、β,β-ジメチルシステインなども、本明細書に記載する葉酸塩-ペプチド合成で出発物質として含まれうる。
【0098】
式Fmoc-AA-OHのFmoc-保護アミノ酸(AA)を含むカップリングシーケンス(工程(a)および(b))は、固体支持ペプチド(2)を調製するために「n」回実施され、ここでnは整数であり、0〜約100に等しくてもよい。最後のカップリング工程後の、残存するFmoc基が除去され(工程(a))、ペプチドが逐次的にグルタミン酸誘導体にカップリングされ(工程(c))、脱保護されて、TFA-保護プテロイン酸にカップリングされる(工程(d))。次にペプチドは、トリフルオロ酢酸、エタンジチオール、およびトリイソプロピルシランによる処理時にポリマー支持体から開裂される(工程(e))。これらの反応条件は、適切に保護されたアミノ酸側鎖の一部を形成しうるt-Bu、t-Boc、およびTrt保護基の同時除去を引き起こす。TFA保護基は、塩基による処理(工程(f)時に除去されて、葉酸塩含有ペプチジル断片(3)を与える。
【0099】
===化合物実施例===
〔実施例1]
方法実施例7の一般手順(スキーム1)により、Wang樹脂結合4-メトキシトリチル(MTT)保護Cys-NH2を次のシーケンスに従って反応させた。1)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;2)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;3)a.Fmoc-Arg(Pbf)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;4)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;5)a.Fmoc-GIu-OtBu、PyBOP、DIPEA;b.20%ピペリジン/DMF;6)N10-TFA-プテロイン酸、PyBOP、DIPEA、MTT、tBu、およびPbf保護基はTFA/H2O/TIPS/EDT(92.5:2.5:2.5:2.5)によって除去され、TFA保護基はpH=9.3のNH4OH水溶液によって除去した。選択された1H NMR(D2O)δ(ppm)8.68(s,1H,FA H-7),7.57(d,2H,J=8.4Hz,FA H-12&16),6.67(d,2H,J=9Hz,FA H-13&15),4.40-4.75(m,5H),4.35(m,2H),4.16(m,1H),3.02(m,2H),2.55-2.95(m,8H),2.42(m,2H),2.00-2.30(m,2H),1.55-1.90(m,2H),1.48(m,2H);MS(ESI,m+H+)1046。
【0100】
〔実施例2〕
方法実施例7の一般手順(スキーム1)により、Wang樹脂結合4-メトキシトリチル(MTT)保護Cys-NH2を次のシーケンスに従って反応させた。1)a.Fmoc-β-アミノアラニン(NH-MTT)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;2)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;3)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;4)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;5)a.Fmoc-Glu-OtBu、PyBOP、DIPEA;b.20%ピペリジン/DMF;6)N10-TFA-プテロイン酸、PyBOP、DIPEA。MTT、tBu、およびTFA保護基は、a.2%ヒドラジン/DMF;b.TFA/H2O/TIPS/EDT(92.5:2.5:2.5:2.5)によって除去した。次の表に示す試薬を調製で使用した:
【0101】
カップリング工程は次のように実施した:ペプチド合成容器に樹脂を添加し、アミノ酸溶液、DIPEA、およびPyBOPを添加する。アルゴンを1時間通気して、DMFおよびIPAで3回洗浄する。各アミノ酸カップリング前に、Fmoc脱保護のためにDMFに希釈した20%ピペリジンを3回(10分間)使用する。継続して6回のカップリング工程をすべて完了させる。最後に樹脂をDMFに希釈した2%ヒドラジンで3回(5分間)洗浄して、プテロイン酸のTFA保護基を開裂させる。
【0102】
次の試薬、92.5%(50ml)TFA、2.5%(1.34ml)H2O、2.5%(1.34ml)トリイソプロピルシラン、2.5%(1.34ml)エタンジチオール を使用して樹脂からペプチド類似体を開裂させ、開裂工程を次のように実施した:開裂試薬25mlを添加して、1.5時間通気し、試薬を排出し、残りの試薬で3回洗浄した。約5mLまで蒸発させて、エチルエーテル中に沈殿させる。遠心分離にかけた後、乾燥させる。精製は次のように実施した。カラム-Waters NovaPak C18 300xl9mm;緩衝液A=10mM酢酸アンモニウム、pH5;B=CAN;1%B〜20%B、15ml/分にて40分間、350mg(64%)まで;HPLC-RT 10.307分、純度100%、1H NMRスペクトルは割り当てられた構造と一致、そしてMS(ES-):1624.8、1463.2、1462.3、 977.1、976.2、975.1、974.1、486.8、477.8。
【0103】
〔実施例3〕
方法実施例7の一般手順(スキーム1)により、Wang樹脂結合MTT保護Cys-NH2を次のシーケンスに従って反応させた:1)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;2)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;3)a.Fmoc-Arg(Pbf)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;4)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;5)a.Fmoc-Glu(γ-OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;6)N10-TFA-プテロイン酸、PyBOP、DIPEA。MTT、tBu、およびPbf保護基はTFA/H2O/TIPS/EDT(92.5:2.5:2.5:2.5)によって除去され、TFA保護基はpH=9.3のNH4OH水溶液によって除去した。1H NMRスペクトルは、割り当てられた構造と一致していた。
【0104】
〔実施例4〕
方法実施例7の一般手順(スキーム1)により、Wang樹脂結合MTT保護D-Cys-NH2を次のシーケンスに従って反応させた:1)a.Fmoc-D-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;2)a.Fmoc-D-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;3)a.Fmoc-D-Arg(Pbf)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;4)a.Fmoc-D-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;5)a.Fmoc-D-Glu-OtBu、PyBOP、DIPEA;b.20%ピペリジン/DMF;6)N10-TFA-プテロイン酸、PyBOP、DIPEA。MTT、tBu、およびPbf保護基はTFA/H2O/TIPS/EDT(92.5:2.5:2.5:2.5)によって除去され、TFA保護基はpH=9.3のNH4OH水溶液によって除去した。1H NMRスペクトルは、割り当てられた構造と一致していた。
【0105】
〔実施例5〕
2-[(ベンゾトリアゾール-1-イル-(オキシカルボニルオキシ)-エチルジスルファニル]-ピリジンHCl(601mg)およびDIPEA 378μLを、0℃の、デスアセチルビンブラスチンヒドラジド(668mg)の5mLDMC溶液中に逐次的に添加した。反応物を室温まで加温して、3時間撹拌した。TLC(DCMに希釈した15% MeOH)は、完全な変換を示した。混合物をシリカゲルクロマトグラフィー(1:9 MeOH/DCM)によって精製した。合せた画分を蒸発させ、DCMに再溶解させて、10% Na2CO3で洗浄し、塩水につけ、乾燥(MgSO4)し、550mg(80%)まで蒸発させた;HPLC-RT 12.651分、純度91%、1H HMRスペクトルは割り当てられた構造と一致した。MS(ESI+):984.3、983.3、982.4、492.4、491.9、141.8。この手順のさらなる詳細は、参照によりその全体が本明細書に組み入れられている、米国特許出願公開第2005/0002942A1号に記載されている。
【0106】
〔実施例6〕
デスアセチルビンブラスチンモノヒドラジド(1当量)は、その開示が参照により本明細書に組み入れられているBarnett et al.,J Med.Chem.21:88-96(1978)に従って調製して、新しい蒸留THF中でトリフルオロ酢酸1当量によって処理した。10分間撹拌した後に、溶液をN-(4-アセチルフェニル)マレイミド1.05当量によって処理した。アシルヒドラゾン形成は45分後に完了して、溶媒を蒸発させた。
【0107】
ペプチジル断片Pte-Glu-Asp-Arg-Asp-Asp-Cys-OH(実施例1)(0.85当量)を水に溶解させて、pHは0.1N HClによって2.5に調整して、ペプチドを沈殿させた。ペプチジル断片を遠心分離によって収集し、乾燥し、DMSOに溶解した。得られた透明黄色の溶液に、ヒューニッヒ塩基(15当量)およびアシルヒドラゾンMicahel付加体を添加した。1時間後、最終結合体をHPLCによって精製した。図1Aおよび1Bは、葉酸塩対葉酸塩-デアセチルビンブラスチン結合体の相対結合親和性、結合体の3H-チミジン取り込みに対する効果をそれぞれ示す。図1Aおよび1Bは、結合体(14nM)のIC50と、葉酸塩が、葉酸塩受容体への結合について葉酸塩-デアセチルビンブラスチン結合体と競合することを示し、結合体の結合の特異性を証明している。アッセイは方法実施例3および4に従って実施した。
【0108】
図2は、Balb/cマウスにおけるM109腫瘍に対する実施例6(1.5μmol/kg)の活性を示す。アッセイは方法実施例1に従って実施した。実施例6は、固形腫瘍の成長を阻害する。図3は、FR陽性M109腫瘍に対し、3週間にわたりTIWで10μmol/kgが与えられた実施例6の活性を示し、実施例6化合物の投薬は点線によって示されるように第25日に終了した。アッセイは方法実施例1に従って実施した。実施例6は、固形腫瘍の成長を阻害する。
【0109】
図4Aおよび4Bは、FR陽性109腫瘍およびFR陰性4T-1腫瘍細胞それぞれに対する、3週間にわたる3μmol/kg TIWでの実施例6の活性を示す。アッセイは、方法実施例1および5にそれぞれ記載されたように実施した。実施例6は、固形M109腫瘍の成長を阻害するが、葉酸塩受容体(FR)陰性腫瘍は阻害しない。
【0110】
図5Aおよび図5Bは、FR陽性KB腫瘍に対する、およびnu/unマウスの体重(nu/unマウスはKB腫瘍体積アッセイに使用した)に対する、3週間にわたる10μmol/kg TIWでの実施例6の活性を示す。アッセイは方法実施例2および6にそれぞれ従って実施した。実施例6は、固形腫瘍の成長を阻害するが、マウスの体重に影響を及ぼさなかった。
【0111】
図6Aおよび6Bは、3週間にわたる1、5、および10μmol/kg TIWでの、FR陽性KB腫瘍(t0の時点での平均腫瘍体積=50〜100mm3)に、および実施例6の同じ濃度でのnu/nuマウスの体重(nu/nuマウスはKB腫瘍体積アッセイに使用した)にそれぞれ対する、実施例6の活性を示す。アッセイは方法実施例2および6にそれぞれ従って実施した。実施例6は、固形腫瘍の成長を阻害するが、マウスの体重にほとんど効果を持たない。
【0112】
図7Aおよび7Bは、FR陽性KB腫瘍(t0の時点での平均腫瘍体積=100〜150mm3)に対する、3週間にわたる10μmol/kg TIWでの実施例6の活性を示す。実施例6対非結合ビンブラスチンの、Balb/cマウスの体重に対する効果も示される。アッセイは方法実施例2および6にそれぞれ従って実施した。実施例6は、固形腫瘍の成長を阻害する。非結合ビンブラスチンは、最初にマウスの体重を減少させたが、マウスの体重は最終的に、おそらく腫瘍成長が理由で増加する。
【0113】
〔実施例7〕
THF中のペプチジル断片Pte-Glu-Asp-Arg-Asp-Asp-Cys-OH(実施例1)を、黄色溶液のチオスルホナートまたはピリジルジチオ活性化ビンブラスチン(実施例5)のどちらかによって処理して、アルゴン存在下でpH>6.5の0.1M NaHCO3への溶解を生じた。凍結乾燥およびHPLCにより、収率70%が得られた;選択された1H NMR(D2O)δ8.67(s,1H,FA H-7),7.50(br s,1H,VLB H-11’),7.30-7.40(br s,1H,VLB H-14’),7.35(d,2H,J=7.8Hz,FA H-12&16),7.25(m,1H,VLB H-13’),7.05(br s,1H,VLB H-12’),6.51(d,2H,J=8.7Hz,FA H-13&15),6.4(s,2H,VLB H-14&17),5.7(m,1H,VLB オレフィン),5.65(m,1H,VLB H-7),5.5(d,1H,VLB オレフィン),5.5(m,1H,VLB H-6),4.15(m,1H,VLB H-8’),3.82(s,3H,VLB C18-CO2CH3),3.69(s,3H,VLB C16-OCH3),2.8(s,3H,VLB N-CH3),1.35(br s,1H,VLB H-3’),1.15(m,1H,VLB H-2’),0.9(t,3H,J=7Hz,VLB H-21’),0.55(t,3H,J=6.9Hz,VLB H-21);LCMS(ESI,m+H+)1918。図8は、葉酸塩対実施例7結合体の相対親和性を示す。アッセイは方法実施例4に従って実施した。
【0114】
図9Aは、3H-チミジン取り込みに対する実施例7の効果、結合体(9nM)のIC50、および葉酸塩受容体への結合に関して葉酸塩が実施例7結合体と競合することを示し、結合体の結合特異性を証明している。図9Bは、実施例7の結合体(100nM 実施例7)による処理ためのパルス時間に対する、3H-チミジン取り込みに対する実施例7の効果を、および葉酸塩受容体への結合に関して葉酸塩が実施例7の結合体と競合することを示し、結合体の結合特異性を証明している(100nM 実施例7+100μM葉酸)。アッセイは方法実施例3に従って実施した。
【0115】
図10Aおよび10Bは、実施例7の結合体による処理のパルス時間に対する、3H-チミジン取り込みへの10および100nMの実施例7の効果を、そして葉酸塩受容体への結合に関して葉酸塩が実施例7の結合体と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例3に従って実施した。
【0116】
図11は、FR陽性KB腫瘍(t0の時点での平均腫瘍体積=50〜100mm3)に対する、3週間にわたる5μmol/kg TIWの実施例7の活性を示す。アッセイは方法実施例2に従って実施した。実施例7は、固形腫瘍の成長を阻害する。
【0117】
図12は、FR陽性KB腫瘍に対する3週間にわたる5μmol/kg TIWの実施例7の活性を示す(nu/nuマウスをKB腫瘍容積アッセイに使用した)。アッセイは方法実施例2に従って実施した。実施例7および8は、固形腫瘍の成長を阻害する。
【0118】
図13は、Balb/cマウスのM109腫瘍に対する実施例7(1.5μmol/kg)の活性を示す。 アッセイは方法実施例1に従って実施した。 実施例7は、固形腫瘍の成長を阻害する。
【0119】
図14Aおよび14Bは、Balb/cマウスのM109腫瘍に対する、およびBalb/cマウスの体重(Balb/cマウスはM109腫瘍体積アッセイに使用した)に対する、実施例7および8の活性を示す(それぞれ10μmol/kgにて)。アッセイは方法実施例1および6にそれぞれ従って実施した。実施例7および8は、固形腫瘍の成長を阻害し、Balb/cマウスの体重にほとんど効果を持たない。
【0120】
図15は、FR陽性KB腫瘍+/-40μmol/kg EC20(レニウム錯体)に対する、2週間にわたる2μmol/kg TIWの実施例7の活性を示す。実施例7は固形腫瘍の成長を阻害し、その阻害効果はEC20レニウム錯体によって防止(競合)される。EC20(レニウム錯体)は、レニウムにキレート化された次の式:
の化合物である。EC20の調製は、米国特許出願公開第2004/0033195A1号に記載されており、その合成手順の説明は参照により本明細書に組み入れられている。アッセイは方法実施例2に従って実施した。EC20は、葉酸塩受容体において実施例7の競合相手として作用し、結果は、実施例7の効果の特異性を示している。
【0121】
図16Aおよび図16Bは、FR陽性KB腫瘍に対する、3週間にわたる5μmol/kg TIWの実施例7および8の活性、ならびにnu/nuマウスの体重(nu/nuマウスはKB腫瘍体積アッセイに使用した)に対する、実施例7および8の効果を示す。アッセイは方法実施例2および6にそれぞれ従って実施した。結果は、nu/nuマウスにおける皮下FR陽性ヒト上咽頭KB腫瘍異種移植片に対して、実施例7が実施例6よりも高い成長阻害活性を有することを示す。実施例7および8は、nu/nuマウスの体重にはほとんど効果を持たない。
【0122】
実施例7は、次の表に示すように、s.c.KB腫瘍異種移植片を持つnu/nuマウスにおいて、非結合デスアセチルビンブラスチンヒドラジド(DAVLBH)よりも良好な治療指数を示した:
(a)CRは、対照群と比較して、試験化合物による処置に対し、完全寛解を示す動物の数(5回試験の合計)に相当する;(b)%T/Cは、完全寛解を示していない動物の対照群に対する腫瘍率である;(c)LCKは、完全寛解を示していない動物の細胞死滅の対数である;(d)死亡5匹。
【0123】
〔実施例9〕
方法実施例7の一般手順(スキーム1)により、Wang樹脂結合4-メトキシトリチル(MTT)保護CyS-NH2を次のシーケンスに従って反応させた。1)a.Fmoc-β-アミノアラニン(NH-IvDde)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;2)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;3)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;4)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;5)a.Fmoc-Glu-OtBu、PyBOP、DIPEA;b.20%ピペリジン/DMF;6)N10-TFA-プテロイン酸、PyBOP、DIPEA。MTT、tBu、およびTFA保護基は、a.2%ヒドラジン/DMF;b.TFA/H2O/TIPS/EDT(92.5:2.5:2.5:2.5)によって除去した。次の表に示す試薬を調製で使用した:
【0124】
カップリング工程は次のように実施した:ペプチド合成容器に樹脂を添加し、アミノ酸のDMF溶液、DIPEA、およびPyBOPを添加する。アルゴンを1時間通気して、DMFおよびIPA 10mLで3回洗浄する。各アミノ酸カップリング前に、Fmoc脱保護のためにDMF中の20%ピペリジンを3回x10mL(10分間)使用する。継続して6回のカップリング工程を完了させる。最後に樹脂を2%ヒドラジンのDMF希釈液によって3回x10mL(5分間)洗浄し、プテロイン酸のTFA保護基、およびβ-アミノアラニンのIvDde保護基を開裂させる。最後に、DIPEAおよびPyBopを使用し、β-アミノアラニンの遊離アミンをFmoc-チオプロピオン酸とDMF中でカップリングさせる。アルゴンを1時間通気して、DMFおよびIPA 10mLで3回洗浄する。樹脂をアルゴン存在下で30分間乾燥させる。
【0125】
次の試薬、92.5%(50ml)TFA、2.5%(1.34ml)H2O、2.5%(1.34ml)トリイソプロピルシラン、2.5%(1.34ml)エタンジチオールを使用して樹脂からペプチド類似体を開裂させ、開裂工程を次のように実施した:開裂試薬25mlを添加して、1.5時間通気し、排出させて、残りの試薬で3回洗浄した。約5mLまで蒸発させて、エチルエーテル中に沈殿させる。遠心分離にかけて、乾燥させる。精製は次のように実施した。カラム-Waters NovaPak C18 300xl9mm;緩衝液A=10mM酢酸アンモニウム、pH5;B=CAN;1%B〜20%B、15 ml/分にて40分間、450mg(65%)まで;1H HMRスペクトルは割り当てられた構造と一致。
【0126】
〔実施例10〕
ポリプロピレン遠心分離ボトル内で、実施例2(82mg、0.084mmol)を水5mLに溶解させて、アルゴンを10分間通気した。別のフラスコで0.1N NaHCO3溶液に10分間アルゴンを通気した。リンカー溶液のpHは、0.1N NaHCO3溶液を使用して約6.9に調整した。テトラヒドロフラン(THF)5mL中のビンブラスチンヒドラジド誘導体(実施例5、91mg、0.092mM)を上記溶液にゆっくり添加した。得られた透明溶液をアルゴン存在下で15分間〜1時間撹拌した。反応の進行は、分析HPLC(10mM酢酸アンモニウム、pH=7.0、およびアセトニトリル)によって監視した。THFを蒸発させて、水溶液を濾過して、分取精製HPLCカラム(XTerra Column,19X300mM)に注入した。1mMリン酸ナトリウムpH=7.0およびアセトニトリルによる溶出は、生成物を含有する純粋な画分を生じ、生成物は48時間の凍結乾燥後に単離した(78mg、50%);C83H103N19O26S2;正確な質量:1845.68;MW:1846.95;HPLC-RT 15.113分、純度100%、1H HMRスペクトルは割り当てられた構造と一致した。MS(ES-):1846.6、1845.5、933.3、924.2、923.3、922.5、615.6、 614.7、525.0。
【0127】
図21Aおよび図21Bは、葉酸塩対実施例10の相対結合親和性、および3H-チミジン取り込みに対する実施例10の効果、結合体のIC50(58nM)と、葉酸塩が葉酸塩受容体への結合で結合体と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例4および3にそれぞれ従って実施した。
【0128】
〔実施例11〕
実施例3を実施例5の代わりに使用することを除いて、実施例7に従って調製した。アルゴンを実施例3(302mg)の5ml水溶液に10分間通気した。この溶液のpHは、飽和NaHCO3溶液を使用して6.8〜7.0に調整した。5mLのTHF中の実施例5(258mg)を溶液に添加して、30分間撹拌した。溶媒を蒸発させて、得られた混合物を濾過した。濾液を分取精製HPLCによって(溶媒A-1mMリン酸塩緩衝液;溶媒B-アセトニトリル;Waters XTterra C18,19mmX300mm;勾配-30分間で5% B〜50% B)240mgまで精製した;1H HMRスペクトルは割り当てられた構造と一致;MS(ESI,m+H+)1917.9,960.9,960.2,959.3,813.1,812.3,803.0,295.0。
【0129】
〔実施例12〕
実施例4を実施例5の代わりに使用することを除いて、実施例7に従って調製した。アルゴンを実施例4(40mg)の水5ml中溶液に10分間通気した。この溶液のpHは、飽和NaHCO3溶液を使用して6.8〜7.0に調整した。THF 5mL中の実施例5(30mg)を溶液に添加して、30分間撹拌した。溶媒を蒸発させて、得られた混合物を濾過した。濾液を分取精製HPLCによって(溶媒A-1mMリン酸塩緩衝液;溶媒B-アセトニトリル;Waters XTterra C18,19mmX300mm;勾配-30分間で5% B〜50% B)43mgまで精製した;1H HMRスペクトルは割り当てられた構造と一致した。MS(ES-):1917.5,1916.5,1915.6,959.2,958.4。
【0130】
図26Aおよび図26Bは、FR陽性KB腫瘍に対する、ならびにnu/nuマウスの体重(nu/nuマウスはKB腫瘍体積アッセイに使用した)に対する、3週間にわたる2μmol/kg TIWの実施例11および12の活性を示す。アッセイは方法実施例2および6にそれぞれ従って実施した。実施例11および12は、固形腫瘍の成長を阻害するが、マウスの体重にほとんど効果を持たない。
【0131】
〔実施例13〕
ポリプロピレン遠心分離ボトル内で、実施例9(56mg)を水7.5mLに溶解させて、アルゴンを10分間通気した。別のフラスコで0.1N NaHCO3溶液に10分間アルゴンを通気した。実施例9溶液のpHは、0.1N NaHCO3溶液を使用して6.9に調整した。テトラヒドロフラン(THF)7.5mL中の実施例5(44mg)を実施例9溶液にゆっくり添加した。得られた透明溶液をアルゴン存在下で15分間〜1時間撹拌した。反応の進行は、分析HPLC(10mM酢酸アンモニウム、pH=7.0、およびアセトニトリル)によって監視した。THFを蒸発させて、水溶液を濾過して、分取精製HPLCによって精製した。pH=7.0の1mMリン酸ナトリウムおよびアセトニトリルによる溶離は純粋な画分を生じ、この画分をプールして、周囲温度にて蒸発させて、得られた水溶液は0.1N HClを使用してpH4.0に調整した。実施例13は、48時間の凍結乾燥後に単離した(61mg、64%)。1H HMRスペクトルおよびLCMSデータは、割り当てられた構造と一致した。
【0132】
〔実施例14〜32〕
本明細書に記載するプロセスおよび条件に従って調製した。要求されたチオスルホナートまたはピリジルジチオ活性化ブンブラスチン、およびマレイミド活性化ビンブラスチン誘導体の調製についてのさらなる詳細は、米国特許出願公開第2005/0002942A1号に記載されている。
【0133】
〔実施例14〕
【0134】
〔実施例15〕
【0135】
図17は、実施例14(a)および15(b)の3H-チミジン取り込みに対する効果(最初の2本の棒グラフ;それぞれ100nMにて1時間、72時間追跡、n=2)と、葉酸塩受容体への結合について葉酸塩が実施例14および15と競合することを示し、結合体の結合特異性を証明する(後の2本の棒グラフ)。アッセイは方法実施例3に従って実施した。
【0136】
〔実施例16]
図18は、実施例16の3H-チミジン取り込みに対する効果(2時間パルス、48時間追跡、n=2)と、葉酸塩受容体への結合について葉酸塩が実施例16と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例3に従って実施した。
【0137】
〔実施例17〕
図19Aは、非結合ビンカの3H-チミジン取り込みに対する効果を示す。図19Bは、実施例17の3H-チミジン取り込みに対する効果を示す。アッセイは方法実施例3に従って実施した。
【0138】
〔実施例18〕
【0139】
〔実施例19〕
【0140】
図20A、20B、および20Cは、実施例7と比較した、葉酸塩対実施例18および19の相対結合親和性(図20)と、3H-チミジン取り込みに対するその効果(図20Bおよび20C)と、葉酸塩受容体への結合について葉酸塩が結合体と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例3(図20Bおよび20C)および方法実施例4(図20)に従って実施した。
【0141】
〔実施例20〕
図22は、3H-チミジン取り込みへの実施例20の効果と、葉酸塩受容体への結合について葉酸塩が結合体と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例3に従って実施した。
【0142】
〔実施例21〕
図23Aおよび図23Bは、葉酸塩対実施例21の相対結合親和性と、3H-チミジン取り込みへの実施例21の効果と、葉酸塩受容体への結合について葉酸塩が結合体と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例4および3にそれぞれ従って実施した。
【0143】
〔実施例22〕
図24Aおよび24Bは、葉酸塩対実施例22の相対結合親和性と、3H-チミジン取り込みへの実施例22の効果を示す。アッセイは方法実施例4および3にそれぞれ従って実施した。
【0144】
実施例21および22の実施例7に対する活性の比較
図25Aおよび図25Bは、Balb/cマウスのM109腫瘍に対する、(それぞれ3μmol/kgにて)、およびBalb/cマウスの体重(Balb/cマウスはM109腫瘍体積アッセイに使用した)に対する、14Bと比較した実施例21および22の活性を示す。アッセイは方法実施例1および6にそれぞれ従って実施した。実施例21、22および7は、固形腫瘍の成長を阻害するが、マウスの体重にほとんど効果を持たない。
【0145】
〔実施例23〕
図27Aおよび図27Bは、葉酸塩対実施例23の相対結合親和性と、3H-チミジン取り込みへの実施例23の効果と、結合体のIC50(15nM)と、葉酸塩受容体への結合について葉酸塩が結合体と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例4および3にそれぞれ従って実施した。
【0146】
〔実施例24〕
図28Aおよび図28Bは、葉酸塩対実施例24の相対結合親和性と、3H-チミジン取り込みへの実施例24の効果、結合体のIC50(9nM)と、葉酸塩受容体への結合について葉酸塩が結合体と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例4および3にそれぞれ従って実施した。
【0147】
〔実施例25〕
C116H140N30O32S2;mol重量:2530.67;正確な質量:2528.97;C,55.05;H,5.58;N,16.60;O,20.23;S,2.53。
【0148】
〔実施例26〕
【0149】
〔実施例27〕
【0150】
〔実施例28〕
【0151】
〔実施例29〕
【0152】
〔実施例30〕
【0153】
〔実施例31〕
【0154】
〔実施例32〕
【0155】
次の表は、KB細胞への活性、葉酸塩受容体競合、および本明細書に記載する選択された薬物送達結合体への相対葉酸塩受容体親和性の要約である:
【図面の簡単な説明】
【0156】
【図1A】37℃、1時間における、葉酸受容体の実施例6(■、0.35)対葉酸(●、1.0)の相対結合親和性を示す。
【図1B】過剰な葉酸を含む(■)および含まない(●)場合の3H-チミジン取り込みに対する実施例6の活性を示す;実施例6のIC50=14nM。
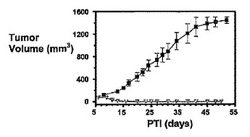
【図2】未処置対照群(●)と対比した、Balb/cマウスのM109腫瘍に対して1.5μmol/kgで与えられたTIW(7回用量)の実施例6(■)の活性を示す。
【図3】未処置対照群(a)と対比した、FR陽性M109腫瘍に3週間にわたって10μmol/kgで与えられたTIWの実施例6(b)の活性を示す(垂直点線は最終の処置日を示す)。
【図4A】未処置対照群(■)と対比した、FR陽性M109腫瘍に対する3週間にわたる3μmol/kg、TIWでの実施例6(▲)の活性を示す。
【図4B】未処置対照群(a)と対比し、FR陰性4T-1腫瘍細胞に対する3週間にわたる3μmol/kg、TIWの実施例6(b)の活性がないことを示す。
【図5A】未処置対照群(■)と対比した、nu/nuマウスにおけるFR陽性KB腫瘍に対する3週間にわたる10μmol/kg、TIWの実施例6(●)の活性を示す。
【図5B】未処置対照群(■)と対比し、nu/nuマウスの体重に対する3週間にわたる10μmol/kgTIWの実施例6(●)による効果がないことを示す。
【図6A】未処置対照群(a)と対比した、nu/nuマウスにおけるFR陽性KB腫瘍に対する3週間にわたる(垂直点線は最終処置日を示した)1μmol/kg(b)、5μmol/kg(c)、および10μmol/kg(d)TIWの実施例6の活性を示す;t0の時点で平均腫瘍体積=50〜100mm3。
【図6B】未処置対照群(a)と対比し、nu/nuマウスの体重に対する3週間にわたる1μmol/kg(b)、5μmol/kg(c)、および10μmol/kg(d)TIWの実施例6による効果がないことを示す。
【図7A】未処置対照群(a)と対比した、nu/nuマウスにおける大型FR陽性KB腫瘍に対する3週間にわたる(垂直点線は最終処置日を示した)10μmol/kg TIWでの実施例6(b)の活性を示す;t0の時点で平均腫瘍体積=100〜150mm3。
【図7B】非結合デスアセチルビンブラスチンモノヒドラジド(b)と対比し、nu/nuマウスの体重に対する3週間にわたる(垂直点線は最終処置日を示した)10μmol/kg TIWの実施例6(a)による効果がないこと示す。
【図8】葉酸受容体における葉酸(●)と対比した、実施例7(b、0.2)の相対結合親和性を示す。
【図9A】過剰な葉酸を含む(a)および含まない(b)場合の、FR陽性KB細胞中への3H-チミジン取り込みに対する実施例7の活性を示す;実施例7のIC50=9nM。
【図9B】治療のパルス時間に対しての、過剰な葉酸を含む(a)および含まない(b)場合の、FR陽性KB細胞への3H-チミジン取り込みでの100nMにおける実施例7の活性に対するインキュベーション時間の効果を示す。
【図10A】治療のパルス時間に対しての、過剰な葉酸を含む(a)および含まない(b)場合の、48時間で収集された2002KB細胞への3H-チミジン取り込みでの10nMにおける実施例7の活性に対するインキュベーション時間の効果を示す。
【図10B】治療のパルス時間に対しての、過剰な葉酸を含む(a)および含まない(b)、48時間で収集された2002KB細胞への3H-チミジン取り込みでの100nMにおける実施例7の活性に対するインキュベーション時間の効果を示す。
【図11】未処置対照群(■)に対しての、FR陽性KB腫瘍に対する3週間にわたる5μmol/kg TIWの実施例7(▼)の活性を示す;t0の時点の平均腫瘍体積=50〜100mm3。
【図12】未処置対照群(a)と対比した、nu/nuマウスにおけるFR陽性KB腫瘍に対する3週間にわたる(垂直点線は最終処置日を示した)5μmol/kg TIWの実施例6および14B(それぞれ(b)および(c))の活性を示す;t0の時点で平均腫瘍体積=50〜80mm3;実施例7は5/5の完全寛解を示す。
【図13】未処置対照群(■)と対比した、Balb/cマウスのM109腫瘍に対する1.5μmol/kg TIWの実施例7(■)の活性を示す。
【図14A】未処置対照群(a)と対比した、Balb/cマウスのM109腫瘍に対する3週間にわたる(垂直点線は最終処置日を示した)それぞれ10μmol/kgでの実施例6および7(それぞれ(b)および(c))の活性を示す;t0の時点で平均腫瘍体積=50〜80mm3。
【図14B】Balb/cマウスの体重に対し、3週間にわたる(垂直点線は最終処置日を示した)(それぞれ10μmol/kgでの)実施例6および7(それぞれ(b)および(c))による効果がないことを示す。
【図15】未処置対照群(a)と対比した、40μmol/kg EC20(レニウム錯体)を含む(b)および(c)含まない場合のFR陽性KB腫瘍に対する2週間にわたる2μmol/kg TIWでの実施例7の活性を示す;実施例7のみが4/5の完全寛解を示した;実施例7+EC20は0/5の完全寛解を示した。
【図16A】未処置対照群(a)と対比した、nu/nuマウスにおけるFR陽性KB腫瘍に対する3週間にわたる5μmol/kg TIWの実施例6および7(それぞれ(b)および(c))の活性を示す。
【図16B】nu/nuマウスの体重に対する3週間にわたる(それぞれ5μmol/kgでの)実施例6および7(それぞれ(b)および(c))の効果がないことを示す。
【図17】FR陽性KB細胞への3H-チミジン取り込みに対する、実施例14(a)および15(b)単独(左側棒グラフ:それぞれ100nMにて1時間、72時間追跡、n=2)の、過剰な葉酸を含む同じ条件下での実施例14(a)および15(b)(右側棒グラフ)と対比した活性を示す。
【図18】KB細胞での3H-チミジン取り込みに対する実施例16の活性を示す;IC50は約250nMである。
【図19A】KB細胞での3H-チミジン取り込みに対する実施例5の活性を示す。
【図19B】KB細胞での3H-チミジン取り込みに対する実施例17の活性を示す。
【図20A】葉酸受容体における、葉酸(a、1.0)に対しての、実施例19(b、0.046)、実施例18(c、0.13)、および実施例7(d)の相対結合親和性を示す。
【図20B】過剰な葉酸を含む(b)および含まない(a)場合のKB細胞での3H-チミジン取り込みに対する実施例7の活性を示す;実施例7のIC50は約16nMである;そして過剰な葉酸を含む(d)および含まない(c)2002KB細胞での3H-チミジン取り込みに対する実施例19の活性を示す;実施例19のIC50は約100nMである。
【図20C】過剰な葉酸を含む(b)および含まない(a)2002KB細胞での3H-チミジン取り込みに対する実施例18の活性を示す;実施例18のIC50は約6nMである。
【図21A】葉酸受容体における実施例10(■、0.24)対葉酸(●、1.0)の相対結合親和性を示す。
【図21B】過剰な葉酸を含む(〇)および含まない(●)場合のKB細胞への3H-チミジン取り込みに対する実施例10の活性を示す;実施例10のIC50は約58nMである。
【図22】過剰な葉酸を含む(〇)および含まない(●)場合のKB細胞への3H-チミジン取り込みに対する実施例20の活性を示す;実施例20のIC50は約58nMである。
【図23A】葉酸受容体における実施例21(■、0.16)対葉酸(●、1.0)の相対結合親和性を示す。
【図23B】過剰な葉酸を含む(〇)および含まない(●)KB細胞への3H-チミジン取り込みに対する実施例21の活性を示す。
【図24A】葉酸受容体における実施例22(○、0.26)対葉酸(●、1.0)に対しての、の相対結合親和性を示す。
【図24B】過剰な葉酸を含む(〇)および含まない(●)KB細胞への3H-チミジン取り込みに対する実施例22の活性を示す。
【図25】Balb/cマウスのM109腫瘍に対して、およびBalb/cマウスの体重(Balb/cマウスはM109腫瘍体積アッセイに使用した)に対する、7と比較した実施例21および22の活性を示す(それぞれ3μmol/kgにて)。図25Aは、未処置対照群(■)と対比した、Balb/cマウスにおけるFR陽性M109腫瘍に対する3週間にわたるそれぞれ3μmol/kg TIWの実施例7(●)、実施例21(▲)、および実施例22(▼)の活性を示す。図25Bは、未処置対照群(■)と対比した、Balb/cマウスの体重に対する3週間にわたるそれぞれ3μmol/kg TIWでの実施例7(●)、実施例21(▲)、および実施例22(▼)による効果の非存在を示す。
【図26】FR陽性KB腫瘍に対する、およびnu/unマウスの体重(nu/unマウスはKB腫瘍体積アッセイに使用した)に対する、3週間にわたる2μmol/kg TIWにおける実施例11および12の活性を示す。図26Aは、未処置対照群(■)と対比した、nu/nuマウスにおけるFR陽性KB腫瘍に対する3週間にわたる2μmol/kg TIWの実施例11(●)および実施例12(▼)の活性を示す。図26Bは、未処置対照群(■)と対比し、nu/nuマウスの体重に対する3週間にわたる2μmol/kg TIWの実施例11(●)および実施例12(▼)による効果がないことを示す。
【図27】葉酸塩対実施例23の相対結合親和性、および3H-チミジン取り込みに対する実施例23の効果、結合体のIC50(15nM)と、葉酸塩が葉酸塩受容体への結合に際し結合体と競合することを示し、結合体の結合特異性を示している。アッセイは方法実施例4および3にそれぞれ従って実施した。図27Aは、葉酸受容体における実施例23(■、0.51)対葉酸(●、1.0)の相対結合親和性を示す。図27Bは、過剰な葉酸を含む(〇)および含まない(●)場合のKB細胞への3H-チミジン取り込みに対する実施例23の活性を示す;実施例23のIC50は約15nMである。
【図28】実施例24に対する葉酸塩の相対親和性効果、および3H-チミジン取り込みに対する実施例24の効果、結合体のIC50(9nM)と、葉酸塩が葉酸塩受容体への結合に際し結合体と競合することを示し、結合体の結合特異性を証明している。図28Aは、葉酸受容体における実施例24(■、0.45)対葉酸(●、1.0)の相対結合親和性を示す。図28Bは、過剰な葉酸を含む(〇)および含まない(●)KB細胞での3H-チミジン取り込みに対する実施例24の活性を示す;実施例24のIC50は約9nMである。
【技術分野】
【0001】
本発明は、標的薬物送達で使用するための組成物および方法に関するものである。特に本発明は、ビンカアルカロイド、ならびにその類似体および誘導体のリガンド結合体、例えばビタミン受容体結合化合物およびビンカアルカロイドの結合体に関するものである。
【背景技術】
【0002】
哺乳類免疫系は、腫瘍細胞、他の病原性細胞、および侵入する外来病原体の認識および排除のための手段を提供する。免疫系は通常、強力な防御線を提供するが、癌細胞、他の病原性細胞、または感染性因子が宿主の免疫反応を逃れて、共存する宿主病原性と共に増殖または持続する多くの例がある。化学療法剤および放射線療法は、例えば分裂を繰り返す腫瘍を排除するために開発されてきた。しかしながら現在入手可能な化学療法剤および放射線療法投与計画の多くは、それらが病原性細胞を破壊するだけでなく、それらが正常な宿主細胞、例えば造血系の細胞にも影響を及ぼすために有害な副作用を有する。これらの抗癌剤の有害な副作用は、病原性細胞集団に対して選択的で、なおかつ宿主毒性の低い新たな治療法の開発の必要性を際立たせている。
【0003】
研究者らは、病原性細胞を、このような細胞に細胞毒性化合物を標的指向化することによって破壊する治療プロトコルを開発してきた。これらのプロトコルの多くは、毒素の正常細胞への送達を最小限に抑えることを試みて、病原性細胞に特有の、または病原性細胞によって過剰発現された抗原に結合する抗体に結合された毒素を利用する。この手法を使用して、病原性細胞上の特異性抗原に対する抗体より成る、ある免疫毒素が開発され、該抗体はリシン、緑膿菌外毒素、ジフテリア毒素、および腫瘍壊死因子などの毒素に結合されている。これらの免疫毒素は、抗体によって認識される特異性抗原を持つ病原性細胞、例えば腫瘍細胞を標的とする(非特許文献1;2、特許文献1参照)。
【0004】
宿主内の病原性細胞、例えば癌細胞または外来病原体の集団を標的とする別の手法は、独立した宿主毒性をも示しうる化合物投与の必要性を回避するために、病原性細胞に対する宿主免疫応答を向上させることである。免疫療法について報告された1つの方法は、抗体(例えば遺伝子組換え多重結合抗体)を腫瘍細胞の表面に結合させて、細胞表面で抗体の定常領域を提示して、それにより各種の免疫系媒介プロセスによって腫瘍細胞の死滅を誘発させることである(非特許文献3、特許文献2参照)。しかしながらこれらの手法は、腫瘍特異性抗原を定義することが困難なため、複雑になっている。
【特許文献1】Better,M.D.,国際公開WO91/07418号,1991年5月30日に公開。
【特許文献2】Soulillou,J.P.,米国特許第5,672,486号
【非特許文献1】Olsnes,S.,Immunol.Today,10,pp.291-295,1989
【非特許文献2】Melby,E.L.,Cancer Res.,53(8),pp.1755-1760,1993
【非特許文献3】De Vita,V.T.,Biologic Therapy of Cancer,2d ed.Philadelphia,Lippincott,1995
【発明の開示】
【課題を解決するための手段】
【0005】
ビンカアルカロイド、ならびにその類似体および誘導体の結合体を本明細書で説明する。結合体はリガンド、例えば、ビンカアルカロイドならびにその類似体および誘導体に、場合によりリンカーを介して共有結合した細胞表面受容体のリガンドを含む。本明細書に記載する結合体で有用なビンカアルカロイドとしては、アルカロイドのビンカインドール-ジヒドロインドールファミリーのすべてのメンバー、これに限定されるわけではないが例えば、ビンデシン、ビンブラスチン、ビンクリスチン、カタランチン、ビンドリン、ロイロシン、ビノレルビン、イミドカルブ、シブトラミン、トルトラズリル、ビンブラスチン酸など、ならびにその類似体および誘導体が挙げられる。
【0006】
一実施形態において、受容体結合型薬物送達結合体が記載される。薬物送達結合体は、リガンド(例えば細胞表面受容体のリガンド)、ビンカアルカロイド、および場合により2価リンカーを含み、一般に式
(B)−(L)−(D)
によって表される。式中、(B)は受容体結合部分を表し、これには例えば、限定されるわけではないが、ビタミン、およびそのビタミン受容体結合型類似体またはその誘導体、例えばビタミン受容体を結合できるビタミンならびにその類似体および誘導体が含まれ;(D)は、ビンカアルカロイド、あるいはその類似体または誘導体を表し;そして(L)は、2価リンカーを表す。2価リンカー(L)は複数のリンカーを含みうる。例えば2価リンカー(L)は、1個以上のスペーサーリンカー(ls)、および放出型リンカー(lr)を含むことができ、それぞれ他方およびリガンドならびにビンカアルカロイドに1個以上のヘテロ原子リンカー(lH)によって結合される。これらの各種リンカーは、2価リンカー(L)を構築するために選択され、いずれかの順序で配置されうる。実例として、2価リンカー(L)は次のうちの1つでありうる:
式中、a、b、c、d、およびeは整数、例えば0〜約4の範囲の整数であり、(ls)、(lH)、および(lr)はそれぞれ、スペーサーリンカー、放出型リンカー、ヘテロ原子リンカーである。2価リンカーのさらなる例証的な例は、米国特許出願公開第2005/0002942A1号および国際公開WO2006/012527号に記載されており、その開示の全体は参照により本明細書に組み入れられている。一変更形態において、1個以上の受容体結合リガンドが本明細書に記載する薬物送達結合体に含まれる。これらの受容体結合型リガンドのそれぞれが同じでありうる、または異なりうることが理解されるであろう。
【0007】
本明細書に記載する薬物送達結合体の例証的な一実施形態において、2価リンカーは、少なくとも1個の放出型なリンカー(lr)を含む。本明細書に記載する薬物送達結合体の別の例証的な実施形態において、2価リンカーは、少なくとも2個の放出型リンカー(lr)2を含む。別の例証的な態様において、2価リンカー(L)は、ジスルフィド放出型リンカーでない、少なくとも1個の放出型リンカー(lr)を含む。別の例証的な態様において、2価リンカー(L)は、1個の放出型リンカーがジスルフィド放出型リンカーでない、少なくとも2個の放出型リンカー(lr)2を含む。1個を超える放出型リンカーが2価リンカーに含まれるとき、これらの放出型リンカーは隣接しうることが認識される。2個の放出型リンカーが2価リンカーにおいて隣接しているとき、2個の放出型リンカーは連携してビンカアルカロイド、あるいはその類似体または誘導体の放出を引き起こしうることがさらに認識される。
【0008】
別の実施形態において、2価リンカーは、アミノ酸から形成されたペプチドである少なくとも1個のスペーサーリンカーを含む。一態様において、ペプチドは天然型アミノ酸、およびその立体異性体を含む。別の態様において、ペプチドは天然型アミノ酸、およびその立体異性体のみから形成される。
【0009】
本明細書に記載するリガンドは一般に、細胞表面受容体のリガンドを含む。本明細書に記載する結合体において有用な例証的なリガンドとしては、これに限定されるわけではないが、ビタミン、およびビタミン受容体に結合する他の部分、トランスポーター、もしくはビタミンあるいはその類似体または誘導体に特異的に結合する他の表面提示タンパク質、ライブラリースクリーニングから同定されたペプチドリガンド、腫瘍細胞特異性ペプチド、腫瘍細胞特異性アプタマー、腫瘍細胞特異性炭水化物、腫瘍細胞特異性モノクローナルまたはポリクローナル抗体、抗体のFabまたはscFv(すなわち単鎖可変領域)断片(例えば、EphA2、または転移性癌細胞で特異的に発現するタンパク質、または特異的に影響を受ける他のタンパク質、に対する抗体のFab断片)、コンビナトリアルライブラリーに由来する小型有機分子、成長因子(例えばEGF、FGF、インスリン、およびインスリン様成長因子)、および相同ポリペプチド、ソマトスタチンおよびその類似体、トランスフェリン、リポタンパク質複合体、胆汁塩、セレクチン、ステロイドホルモン、Arg-Gly-Asp含有ペプチド、レチノイド、各種のガレクチン、δ-オピオイド受容体リガンド、コレシストキニンA受容体リガンド、アンギオテンシンAT1またはAT2受容体に特異的なリガンド、ペルオキシソーム増殖因子活性化受容体λリガンド、β−ラクタム抗生物質(例えばペニシリン、抗菌薬を含む小型有機分子、ならびに腫瘍細胞の表面でまたは感染性生物で優先的に発現される受容体に特異的に結合する他の分子)、および受容体または他の細胞表面タンパク質の結晶構造に基づき特定の受容体の結合ポケット内に適合するように設計された抗菌薬および他の薬物、腫瘍抗原のリガンドまたは腫瘍細胞の表面で優先的に発現される他の分子のリガンド、あるいはこれらの分子のいずれかの断片が挙げられる。リガンド-ビンカ結合体の結合部位として機能しうる腫瘍特異性抗原は、タンパク質のエフリンファミリーのメンバーの細胞外エピトープ、例えばEphA2を含む。EphA2発現は、正常細胞の細胞-細胞接合部に限定されるが、転移性腫瘍細胞ではEphA2は細胞表面全体に分布している。それゆえ転移性細胞のEphA2は、例えばビンカアルカロイドに結合した抗体のFab断片への結合のために接近するが、これに対して該タンパク質は正常細胞のFab断片への結合のためには接近できず、そのため転移性癌細胞に対して特異的にリガンド-ビンカ結合体を生じる。
【0010】
他の実施形態において、製薬組成物が記載されている。製薬組成物は、本明細書に記載したリガンド-ビンカ結合体を、製薬的に許容される担体、賦形剤および/または希釈剤と組合せて含む。
【0011】
別の実施形態において、病原性細胞の集団を内部に持つ宿主動物において病原性細胞の集団を排除する方法が記載される。例証的な一態様において、病原性細胞集団のメンバーは、受容体結合部分あるいはその類似体または誘導体に対する接近可能な結合部位を有し、その結合部位は病原性細胞によって特異的に発現、過剰発現または優先的に発現される。該方法は、本明細書で記載するように、宿主に本明細書に記載する薬物送達結合体、あるいはその製薬組成物を投与する工程を含む。
【0012】
==関連出願==
本出願は、米国特許法119条(c)に基づいて、その開示全体が参照により本明細書に組み入れられている、2005年8月19日に出願された米国仮特許出願第60/709,936号の利益を主張する。
【発明を実施するための最良の形態】
【0013】
薬物、ならびにその類似体および誘導体のリガンド結合体を本明細書で説明する。結合体は、病原性細胞を含む細胞へ標的指向化されうる2つ以上の薬物に共有結合された、細胞表面受容体のリガンドを含めた、細胞受容体結合リガンドを含む。本明細書に記載する結合体は、リガンドを薬物に結合するための多価リンカーも含みうる。
【0014】
一実施形態において、受容体結合薬物送達結合体が記載される。薬物送達結合体は、細胞表面受容体のリガンド、ビンカアルカロイド、および場合により2価リンカーを含み、一般に式
(B)−(L)−(D)
によって表され、式中、(B)は受容体結合部分を表し、これには例えば、限定されるわけではないが、ビタミン、およびそのビタミン受容体結合類似体またはその誘導体、例えばビタミン受容体を結合できるビタミンならびにその類似体および誘導体が含まれ;(D)は、ビンカアルカロイド、あるいはその類似体または誘導体を表し;そして(L)は、2価リンカーを表す。2価リンカー(L)は複数のリンカーを含みうる。例えば2価リンカー(L)は、1個以上のスペーサーリンカー(ls)と、放出型リンカー(lr)とを含み、それぞれ相互に、そしてリガンドおよびビンカアルカロイドに1個以上のヘテロ原子リンカー(lH)によって結合されている。これらの各種リンカーは、2価リンカーを構築するために選択され、いずれの順序でも配置されうる。実例として、2価リンカー(L)は次のうちの1つでありうる:
式中、a、b、c、d、およびeは整数、例えば0〜約4の範囲の整数であり、(ls)、(lH)、および(lr)はそれぞれ、スペーサーリンカー、放出型リンカー、ヘテロ原子リンカーである。2価リンカーのさらなる例証的な例は、米国特許出願公開第2005/0002942A1号および国際公開WO2006/012527号に記載されており、その開示の全体は参照により本明細書に組み入れられている。
【0015】
受容体結合部分(B)、2価リンカー(L)、およびビンカアルカロイド薬物、あるいはその類似体または誘導体(D)を含む受容体結合薬物送達結合体が記載され、該受容体結合部分(B)および該ビンカアルカロイド薬物(D)はそれぞれヘテロ原子リンカー(lH)を介して2価リンカー(L)に結合される。2価リンカー(L)は、1個以上のスペーサーリンカー、ヘテロ原子リンカー、および放出型リンカー、ならびにその組合せをいずれの順序でも含む。
【0016】
例えば2価リンカー、または2価リンカーの一部を形成するためにリンカーが共有結合的に組み立てられる方法の例証的な一実施形態において、ヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーが結合されて式:
の2価基を形成し、式は-(ls)5-(ls)’-(lr)-(lH)-としても表されうる。その式では、(ls)5はペンタペプチドAla-Glu-Lys-Asp-Asp-OHであり、(ls)’はCH2CH2であり、(lr)はS-S-(CH2)2-O-C(O)であり、そして(lH)はOである。放出型リンカー(lr)は、ω-アミノ基にて(ls)5のLysに結合される。
【0017】
本明細書に記載する薬物送達結合体の説明的な一実施形態において、2価リンカーは、少なくとも1個の放出型なリンカー(lr)を含む。本明細書に記載する薬物送達結合体の別の例証的な実施形態において、2価リンカーは、少なくとも2個の放出型リンカー(lr)2を含む。別の説明的な態様において、2価リンカー(L)は、ジスルフィド放出型リンカーでない、少なくとも1個の放出型リンカー(lr)を含む。別の例証的な態様において、2価リンカー(L)は、1個の放出型リンカーがジスルフィド放出型リンカーでない、少なくとも2個の放出型リンカー(lr)2を含む。1個を超える放出型リンカーが2価リンカーに含まれるとき、これらの放出型リンカーは隣接しうることが認識される。2個の放出型リンカーが2価リンカーにおいて隣接しているとき、2個の放出型リンカーは連携してビンカアルカロイド、あるいはその類似体または誘導体の放出を引き起こしうることがさらに認識される。
【0018】
本明細書で使用され、また開裂性リンカーとしても公知である「放出型リンカー」という用語は、生理学的条件下で切断されうる少なくとも1個の結合(例えばpH不安定性、酸不安定性、酸化不安定性、または酵素不安定性結合)を含むリンカーを指す。結合切断を生じるこのような生理学的条件としては、例えば生理学的pHにて、またはサイトゾルpHよりも低いpHを有するエンドソームなどの細胞器官への細分化の結果として生じる標準化学加水分解反応が挙げられることが認識されるべきである。
【0019】
開裂性結合が放出型リンカーのどちらかまたは両方の端にて、本明細書に記載するように、放出型リンカー内の2個の隣接する原子を結合しうる、ならびに/あるいは他のリンカーまたは(B)および/または(D)を結合しうることが理解されるはずである。開裂性結合が放出型リンカー内の2個の隣接する原子を結合する場合、結合の切断後に、放出型リンカーは2個以上の断片に切断される。あるいは開裂性結合が放出型リンカーと別の部分、例えばヘテロ原子リンカー、スペーサーリンカー、別の放出型リンカー、薬物、あるいはその類似体または誘導体、あるいはビタミン、あるいはその類似体または誘導体との間にある場合、結合の切断後に、放出型リンカーは他の部分から分離される。
【0020】
開裂性結合の不安定性は例えば、開裂性結合におけるまたはその付近の置換的な変更、例えば開裂性ジスルフィド結合に隣接したアルファ分岐取り込みを含むこと、加水分解されうるケイ素-酸素結合を有する部分におけるケイ素上の置換基の疎水性の上昇、加水分解されうるケタールまたはアセタールの部分を形成するアルコキシ基のホモログ化などによって調整されうる。
【0021】
本明細書に記載する2価リンカーの開裂の例示的な機構は、次の1,4および1,6断片化機構を含み、
式中、Xは、外来性または内在性求核試薬、グルタチオン、または生物還元剤などであり、ZまたはZ’のどちらかは、ビタミン、あるいはその類似体または誘導体、あるいは薬物、あるいはその類似体または誘導体、あるいは2価リンカーの他の部分と結合したビタミンまたは薬物部分である。上の断片化機構が協奏機構として示されているが、多数の別個の工程が2価リンカーから、提示の最終生成物への最終的な断片化を実施するために起こりうることが理解されるものとする。例えば、結合開裂は酸触媒によるカルバメート部分の脱離反応によっても起こりうることが認識され、この脱離反応は上の例に示されたベータ硫黄または二硫化物のアリール基またはそのどちらかによってもたらされる安定化により隣接基に(anchimerically)補助されうる。この実施形態のこれらの変更態様において、放出型リンカーはカルバメート部分である。あるいは断片化は、ジスルフィド基に対する求核攻撃によって開始されて、開裂が引き起こされ、チオラートが形成される。チオラートによりカルボン酸またはカルバミン酸部分を分子間で置換して、対応するチアシクロプロパンを形成しうる。ベンジル含有2価リンカーの場合、例示のジスルフィド結合の切断の後、得られたフェニルチオラートはさらに断片化して、共鳴安定化中間体を形成することによってカルボン酸またはカルバミン酸を放出しうる。これらの場合にいずれにおいても、本明細書に記載する例証的な2価リンカーの放出性質は、存在する化学的、代謝的、生理学的、または生物学的条件に関連しうるどんな機構によっても実現されうる。
【0022】
放出型リンカーの結合開裂のための他の例証的な機構は、次のようなオキソニウム補助開裂:
を含み、式中、Zは、ビタミン、あるいはその類似体または誘導体、あるいは薬物、あるいはその類似体または誘導体であり、あるいはそれぞれが、2価リンカーの他の部分と併せたビタミンまたは薬物部分、例えば1個以上のスペーサーリンカー、ヘテロ原子リンカー、および/または他の放出型リンカーを含む薬物またはビタミン部分である。本実施形態において、酸触媒によるカルバメートの脱離反応は、CO2の放出およびZに結合した窒素含有部分の放出と、水、または他のいずれかのルイス塩基によって捕捉されうるベンジルカチオンの形成につながる。
【0023】
別の例証的な機構は、2価リンカーにおける結合の開裂に続いて、放出された官能基が、隣接基補助(anchimeric assisted)開裂または切断とも呼ばれる、さらなる結合の切断または開裂を化学的に補助するような方法で、放出型リンカー、スペーサーリンカー、およびヘテロ原子リンカーの配置をともなう。このような2価リンカーまたはその部分の例証的な実施形態は、式:
を有する化合物を含み、式中、Xはヘテロ原子、例えば窒素、酸素、または硫黄であり、nは、0、1、2、および3から選択される整数であり、Rは水素または置換基、例えばアリール環に正電荷を誘導的にまたは共鳴によって安定化できる置換基(例えばアルコキシなど)であり、ZまたはZ’のどちらかは、ビタミン、あるいはその類似体または誘導体、あるいは薬物、あるいはその類似体または誘導体、あるいは2価リンカーの他の部分と結合したビタミンまたは薬物部分である。これに限定されるわけではないが、ヒドロキシ、アルキル、アルコキシ、アルキルチオ、ハロなどを含む他の置換基が、アリール環、ベンジル炭素、カルバメート窒素、アルカン酸、またはメチレン架橋に存在しうることが認識される。補助開裂には、ベンジリウム中間体、ベンジン中間体、ラクトン環化、オキソニウム中間体、ベータ脱離などをともなう機構が含まれうる。放出型リンカーの開裂に続く断片化に加えて、放出型リンカーの初期開裂が隣接基補助機構によって促進されうることがさらに認識される。
【0024】
この本実施形態において、環化しうるヒドロキシアルカン酸は、例えばオキソニウムイオンによってメチレン架橋の開裂を促進し、結合開裂または放出型リンカーの結合開裂後の続いての断片化を促進する。あるいはメチレン架橋の酸触媒によるオキソニウムイオン補助開裂は、この例証的な2価リンカー、またはその断片の断片化のカスケードを開始しうる。あるいはカルバメートの酸触媒による加水分解は、環化しうるヒドロキシアルカン酸のベータ脱離を促進し、例えばオキソニウムイオンによってメチレン架橋の開裂を促進しうる。本明細書に記載する代謝的、生理学的、または細胞的条件下での結合切断または開裂の他の化学的機構がこのような断片化のカスケードを開始しうることが認識される。本明細書に記載する代謝的、生理学的、または細胞的条件下での結合切断または開裂の他の化学的機構がこのような断片化のカスケードを開始しうることが認識される。
【0025】
一実施形態において、本明細書に記載する2価リンカーは、次の式
の化合物であり、
式中、nは、1〜約4より選択される整数であり;RaおよびRbは、水素およびアルキル(例えば場合により分岐しているC1〜C4アルキルなどの低級アルキル)から成る群よりそれぞれ独立して選択され;あるいはRaおよびRbは、結合した炭素原子とひとまとめにされて、炭素環を形成し;Rは、場合により置換されたアルキル基、場合により置換されたアシル基、または適切に選択された窒素保護基であり;そして(*)は、薬物、ビタミン、造影剤、診断剤、他の2価リンカー、または結合体の他の部分への結合点を示す。
【0026】
別の実施形態において、本明細書に記載する2価リンカーは、次の式
の化合物を含み、式中、mは、1〜約4より選択される整数であり;Rは、場合により置換されたアルキル基、場合により置換されたアシル基、または適切に選択された窒素保護基であり;そして(*)は、薬物、ビタミン、造影剤、診断剤、他の2価リンカー、または結合体の他の部分への結合点を示す。
【0027】
別の実施形態において、本明細書に記載する2価リンカーは、次の式
の化合物を含み、式中、mは、1〜約4より選択される整数であり;Rは、場合により置換されたアルキル基、場合により置換されたアシル基、または適切に選択された窒素保護基であり;そして(*)は、薬物、ビタミン、造影剤、診断剤、他の2価リンカー、または結合体の他の部分への結合点を示す。
【0028】
別の実施形態において、2価リンカーにおける結合の開裂に続いて、放出された官能基が、隣接基補助開裂または切断とも呼ばれる、さらなる結合の切断または開裂を化学的に補助するような方法で、放出型リンカー、スペーサーリンカー、およびヘテロ原子リンカーが配置されうる。このような2価リンカーまたはその一部分の例証的な実施形態は、式:
を有する化合物を含み、式中、Xは、ヘテロ原子、例えば窒素、酸素、または硫黄であり、nは、0、1、2、および3から選択される整数であり、Rは水素または置換基、これは例えばアリール環に正電荷を誘導的にまたは共鳴によって安定化できる置換基(例えばアルコキシ)であり、記号(*)は、2価リンカーを形成している付加スペーサーリンカー、ヘテロ原子リンカー、または放出型リンカーへの結合点、あるいは薬物、あるいはその類似体または誘導体、あるいはビタミン、あるいはその類似体または誘導体への結合点を示す。これに限定されるわけではないが、ヒドロキシ、アルキル、アルコキシ、アルキルチオ、ハロなどを含む他の置換基が、アリール環、ベンジル炭素、アルカン酸、またはメチレン架橋に存在しうることが認識される。補助開裂には、ベンジリウム中間体、ベンジン中間体、ラクトン環化、オキソニウム中間体、ベータ脱離などをともなう機構が含まれうる。放出型リンカーの開裂に続く断片化に加えて、放出型リンカーの初期開裂が隣接基補助機構によって促進されうることがさらに認識される。
【0029】
別の実施形態において、2価リンカーは、2価3-チオスクシンイミド-1-イルアルキルオキシメチルオキシ基を形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、次の式
によって表される。式中、nは、1〜6の整数であり、アルキル基は場合により置換され、メチルは、付加アルキル、または場合により置換されたアリール基によって任意に置換され、そのそれぞれが独立して選択された基Rによって表される。記号(*)は、2価リンカー断片の、本明細書に記載する結合体の他の部分への結合点を示す。別の実施形態において、2価リンカーは、2価3-チオスクシンイミド-1-イルアルキルカルボニル基を形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、次の式
によって表される。式中、nは、1〜6の整数であり、アルキル基は場合により置換される。記号(*)は、2価リンカー断片の、本明細書に記載する結合体の他の部分への結合点を示す。別の実施形態において、2価リンカーは、2価3-チオアルキルスルホニルアルキル(2置換シリル)オキシ基を形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、2置換シリルは、アルキルおよび/または場合により置換されたアリール基によって置換される。
【0030】
別の実施形態において、2価リンカーは、2価ジチオアルキルカルボニルヒドラジド基、または2価3-チオもしくは3-ジチオスクシンイミド-1-イルアルキルカルボニルヒドラジドを形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、次の式
によって表される。式中、nは、1〜6の整数であり、アルキル基は場合により置換され、ヒドラジドは、(B)、(D)、または2価リンカー(L)の他の部分と共にヒドラゾンを形成する。記号(*)は、2価リンカー断片の、本明細書に記載する結合体の他の部分への結合点を示す。
【0031】
別の実施形態において、2価リンカーは、2価3-チオスクシンイミド-1-イルアルキルオキシアルキルオキシアルキリデン基を形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、次の式
によって表される。式中、各nは、1〜6より独立して選択された整数であり、各アルキル基は、独立して選択され、そして、例えばアルキルまたは場合により置換されたアリールによって場合により置換され、ここでアルキリデンは、(B)、(D)、または2価リンカー(L)の他の部分と共にヒドラゾンを形成する。記号(*)は、2価リンカー断片の、本明細書に記載する結合体の他の部分への結合点を示す。
【0032】
別の実施形態において、2価リンカーは、2価3-チオまたは3-ジチオアリールアルキルオキシカルボニル基、3-チオまたは3-ジチオアリールアルキルアミノカルボニル基、2価3-チオまたは3-ジチオアルキルオキシカルボニル、あるいは2価3-チオまたは3-ジチオアルキルアミノカルボニルを形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、ここでアルキルカルボニルは、(B)、(D)、または2価リンカー(L)の他の部分と共にカーボネート、カルバメート、または尿素を形成する。例示としては、アルキル基はエチルである。
【0033】
別の実施形態において、2価リンカーは、2価3-ジチオアルキルアミノ基を形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、ここでアミノは、(B)、(D)、または2価リンカー(L)の他の部分と共にビニル基含有アミドを形成する。例示としては、アルキル基はエチルである。
【0034】
別の実施形態において、2価リンカーは、2価1-アルコキシシクロアルキレンオキシ基、2価アルキレンアミノカルボニル(ジカルボキシルアリーレン)カルボキシラート基、2価3-ジチオアルコキシカルボニル基、2価3-ジチオアルキルオキシカルボニルヒドラジド基、2価を形成するために結合されたヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含む。
【0035】
別の実施形態において、2価リンカーは、アミノ酸で形成されたペプチドである少なくとも1個のスペーサーリンカーを含む。一態様において、ペプチドは天然型アミノ酸、およびその立体異性体を含む。別の態様において、ペプチドは天然型アミノ酸、およびその立体異性体のみから形成される。
【0036】
スペーサーリンカーおよび放出型リンカーのさらなる例証的な例を表1および2に示した。ここで(*)は、別のリンカーへの、ビンカアルカロイド、またはその類似体もしくは誘導体への結合点、あるいは受容体結合部分への結合点を示す。
表1 企図されるスペーサーリンカーおよびヘテロ原子リンカー、およびその組合せ
表2 企図される放出型リンカーおよびヘテロ原子リンカー、およびその組合せ
【0037】
本明細書で言及するように、本明細書に記載する結合体に使用できるビンカ薬物としては、アルカロイドのビンカインドール−ジヒドロインドールファミリーのすべてのメンバー、例えばビンデシン、ビンブラスチン、ビンクリスチン、カタランチン、ビンドリン、ロイロシン、ビノレルビン、イミドカルブ、シブトラミン、トルトラズリル、ビンブラスチン酸など、ならびにその類似体および誘導体が挙げられる。例証的には、そのような類似体および誘導体としては、米国特許第4,203,898号に記載されている3−カルボキスアジド;米国特許第4,166,810号に記載されている4-デスアセチルビンブラスチン-3-カルボキスヒドラジドのN2-アルキルおよび他の誘導体;Neuss et al.Tetrahedron Lett.783(1968)に記載されているロイロシンヒドラジド;Barnett et al.J.Med.Chem.21:88(1978)に記載されているヒドラジド誘導体;米国特許第3,392,173号および同第3,387,001号に記載されているC-4エステル誘導体;Langone et al.Anal.Biochem.95:214(1979)に記載されている酸化により生じるジカルボン酸誘導体;およびEP0247792A2に記載されているビンカヒドラジドが挙げられる。上記の特許および刊行物のそれぞれは、それがビンカ化合物を調製するための合成経路、および反応条件に関して開示する全てについて、参照により本明細書に組み入れられている。
【0038】
例証的な一実施形態において、ビンカ薬物は、式
の化合物であり、式中、
R1およびR2の一方がHであり、他方がエチルであり、かつR3はHであり、あるいはR1はエチルであり、R2およびR3はひとまとめにされて-O-を形成し;
R4、R7、およびR8は、H、アルキル、およびアシルからそれぞれ独立して選択され、
R5およびR6は、それぞれ独立して選択されたアルキルであり;
R9は、基-NHNHRであり、式中、Rは、H、アルキル、またはアシルであり;
R10は、Hまたはアシルであり;かつ
R11は、エチルである。
【0039】
一態様において、ビンカ薬物は、上式の化合物であり、式中、R4およびR8はそれぞれHであり;かつR5、R6、R9、およびR10はそれぞれメチルである。
【0040】
本明細書に記載する結合体において有用な細胞表面受容体のリガンドとしては、これに限定されるわけではないが、ビタミン、およびビタミン受容体に結合する他の部分、トランスポーター、あるいはビタミンまたはその類似体もしくは誘導体に特異的に結合する他の表面提示タンパク質、ライブラリースクリーニングから同定されたペプチドリガンド、腫瘍細胞特異性ペプチド、腫瘍細胞特異性アプタマー、腫瘍細胞特異性炭水化物、腫瘍細胞特異性モノクローナルまたはポリクローナル抗体、抗体のFabまたはscFv(すなわち単鎖可変領域)断片(例えば、EphA2に、または転移性癌細胞で特異的に発現または特異的に接近可能な他のタンパク質に対する抗体のFab断片)、コンビナトリアルライブラリーに由来する小型有機分子、成長因子(例えばEGF、FGF、インスリン、およびインスリン様成長因子)、および相同ポリペプチド、ソマトスタチンおよびその類似体、トランスフェリン、リポタンパク質複合体、胆汁塩、セレクチン、ステロイドホルモン、Arg-Gly-Asp含有ペプチド、レチノイド、各種のガレクチン、δ-オピオイド受容体リガンド、コレシストキニンA受容体リガンド、アンギオテンシンAT1またはAT2受容体に特異的なリガンド、ペルオキシソーム増殖因子活性化受容体λリガンド、β-ラクタム抗体(例えばペニシリン、抗菌薬を含む小型有機分子、ならびに腫瘍細胞の表面でまたは感染性生物で優先的に発現される受容体に特異的に結合する他の分子)、受容体または他の細胞表面タンパク質の結晶構造に基づき特定の受容体の結合ポケット内に適合するように設計された抗菌薬および他の薬物、腫瘍抗原のリガンドまたは腫瘍細胞の表面で優先的に発現される他の分子のリガンド、あるいはこれらのいずれかの分子の断片が挙げられる。リガンド-ビンカ結合体の結合部位として機能しうる腫瘍特異性抗原の例としては、タンパク質のエフリンファミリーのメンバーの細胞外エピトープ、例えばEphA2が挙げられる。EphA2発現は、正常細胞の細胞-細胞接合部に限定されるが、転移性腫瘍細胞ではEphA2は細胞表面全体に分布している。それゆえ転移性細胞のEphA2は、例えばビンカ化合物に結合した抗体のFab断片への結合のために接近可能であるが、これに対して該タンパク質は正常細胞のFab断片への結合のためには接近できず、そのため転移性癌細胞に対して特異的なリガンド-ビンカ結合体を生じる。
【0041】
本明細書に記載する方法および化合物にしたがって使用されうるビタミンとしては、カルニチン、イノシトール、リポ酸、ピリドキサール、アスコルビン酸、ナイアシン、パントテン酸、葉酸、リボフラビン、チアミン、ビオチン、ビタミンB12、ビタミンA、D、E、およびK,他の関連ビタミン分子、その類似体および誘導体、ならびにその組合せが挙げられる。これらのビタミン、ならびにその受容体結合類似体および誘導体は、薬物送達結合体を作製するために、本明細書に記載する2価リンカー(L)によってビンカ化合物に結合されうる例証的な標的実体を構成する。
【0042】
例証的な一態様において、ビタミンは葉酸、葉酸類似体、またはその他の葉酸塩受容体結合分子でありうる。使用されうる葉酸塩の類似体の例としては、フォリン酸、プテロイルポリグルタミン酸、プテロイン酸およびその他のアミノ酸誘導体、ならびに葉酸塩受容体結合プテリジン、例えばテトラヒドロプテリン、ジヒドロ葉酸塩、テトラヒドロ葉酸塩、ならびにそのデアザおよびジデアザ類似体が挙げられる。「デアザ」および「ジデアザ」類似体という用語は、天然型葉酸構造において1または2個の窒素原子に代わって置換された炭素原子を有する当分野で認識された類似体を指す。例えばデアザ類似体としては、1-デアザ、3-デアザ、5-デアザ、8-デアザ、および10デアザ類似体が挙げられる。ジデアザ類似体としては例えば、1,5-ジデアザ、5,10-ジデアザ、8,10-ジデアザ、および5,8-ジデアザ類似体が挙げられる。上記の葉酸類似体は、葉酸塩受容体に結合するその能力を反映して、慣例的に「葉酸塩」と呼ばれる。他の葉酸塩受容体結合類似体としては、アミノプテリン、アメトプテリン(メトトレキセート)、N10-メチル葉酸、2-デアミノ-ヒドロキシ葉酸、デアザ類似体、例えば1-デアザメトプテリンまたは3-デアザメトプテリン、および3’,5’-ジクロロ-4-アミノ-4-デオキシ-N10-メチルプテロイルグルタミン酸(ジクロロメトトレキセート)が挙げられる。薬物送達結合体の受容体媒介エンドサイトーシス輸送を開始させるために葉酸塩受容体に結合可能である他の適切なリガンドとしては、葉酸受容体に対する抗体が挙げられる。したがって例証的な一態様において、葉酸塩受容体に対する抗体と複合化したビンカ化合物は、複合体の膜透過輸送を引き起こすために使用されうる。
【0043】
ビタミン類似体および/または誘導体の例証的な実施形態も、ビオチンの類似体および誘導体、例えばビオシチン、ビオチンスルホキシド、オキシビオチンおよび他のビオチン受容体結合化合物などを含む。本明細書に記載する他のビタミンの類似体および誘導体も本明細書で企図されることが認識される。
【0044】
本明細書に記載する薬物送達結合体は、在来の合成方法によって調製されうる。合成方法は、ヘテロ原子リンカーの選択、ならびにスペーサーリンカーおよび放出型リンカーに存在する官能基に応じて選択されうる。一般に、関連する結合形成反応は、Richard C.Larock,“Comprehensive Organic Transformations,a guide to functional group preparations,”VCH Publishers,Inc.New York(1989)、およびTheodora E.Greene & Peter G.M.Wuts,“Protective Groups ion Organic Synthesis,”2d edition,John Wiley & Sons,Inc.New York(1991)に記載されており、その開示は全体が参照により本明細書に組み入れられている。さらなる合成経路および反応条件は、米国特許出願公開第2005/0002942A1号に記載されている。
【0045】
例示としては、本明細書に記載する薬物送達結合体は、線形および収束合成経路の両方を使用して調製されうる。このような経路で使用できる例証的な中間体としては、受容体結合部分、あるいはその類似体および誘導体、ならびにビンカアルカロイド、あるいはその類似体または誘導体への共有結合に適切なカップリング基をそれぞれの端に含む2価リンカーを含む中間体が挙げられる。このような経路で使用できる他の例証的な中間体としては、カップリング基を含む2価リンカーに結合された受容体結合部分、あるいはその類似体または誘導体を含む中間体が挙げられる。このような経路で使用できるその他の例証的な中間体としては、カップリング基を含む2価リンカーに結合された、ビンカアルカロイドあるいはその類似体または誘導体を含む中間体が挙げられる。どちらの場合でも、カップリング基は求核試薬、求電子試薬、またはそれらの前駆体でありうる。
【0046】
合成中間体の例証的な一実施形態において、カップリング基はMichael受容体であり、2価リンカーとしては、式-C(O)NHN=、-NHC(O)NHN=、または-CH2C(O)NHN=を有する放出型リンカーを含む。例証的な一態様において、カップリング基および2価リンカーはひとまとめにされて、式:
を有する化合物またはその保護誘導体を形成し、式中、(D)は、本明細書で例証するようなヒドラゾンを形成できるビンカアルカロイド、あるいはその類似体または誘導体であり;そしてnは、1、2、3、または4などの整数である。本明細書に記載する受容体結合薬物送達結合体中間体の別の例証的な態様において、第2のリンカーが、第2のリンカーに含まれるアルキルチオール求核試薬を介して上式に共有結合される。別の例証的な態様において、受容体結合部分、あるいはその類似体または誘導体は、その部分に含まれるアルキルチオール求核試薬を介して上式に共有結合される。
【0047】
別の例証的な実施形態において、カップリング基は例えば窒素、酸素、または硫黄等のヘテロ原子であり、2価リンカーは、受容体結合部分をカップリング基に共有結合する1個以上のヘテロ原子リンカーおよび1個以上のスペーサーリンカーを含む。例証的な一態様において、本明細書に記載する中間体は、式:
を有する化合物またはその保護誘導体を含み、式中、Xは、酸素、窒素、または硫黄であり、mは、1、2または3などの整数であり、かつ(B)、ls、およびlHは本明細書で定義する通りである。例証的な一態様において、lHは-NH-であり、mは1である。別の例証的な態様において、lHは-NH-であり、mは1であり、Xは-S-である。
【0048】
別の例証的な態様において、本明細書に記載する中間体は、式:
を有する化合物またはその保護誘導体を含み、式中、Yは、Hまたは置換基、例証的には、これに限定されるわけではないが、ニトロ、シアノ、ハロ、アルキルスルホニル、カルボン酸誘導体などを含む電子吸引性置換基であり、かつ(B)およびlsは本明細書で定義する通りである。
【0049】
本明細書に記載する中間体の別の例証的な実施形態において、カップリング基はMichael受容体であり、2価リンカーは、受容体結合部分をカップリング基に共有結合する1個以上のヘテロ原子リンカーおよび1個以上のスペーサーリンカーを含む。例証的な一態様において、カップリング基および2価リンカーはひとまとめにされて、式:
を有する化合物またはその保護誘導体を含み、式中、Xは、酸素、窒素、または硫黄であり、mおよびnは、独立して選択された整数、例えば1、2、または3であり、かつ(B)、lsおよび1Hは、本明細書で定義する通りである。別の例証的な態様において、ビンカアルカロイド、あるいはその類似体または誘導体は、ビンカアルカロイドに含まれるアルキルチオール求核試薬を介して上の式に共有結合される。
【0050】
別の例証的な態様において、中間体は、式:
を有する化合物またはその保護誘導体を含み、式中、AAは1個以上のアミノ酸であり、例示的には、天然型アミノ酸またはその立体異性体から例証的に選択され、Xは、窒素、酸素、または硫黄であり、Yは、水素または置換基、例証的には、これに限定されるわけではないが、ニトロ、シアノ、ハロ、アルキルスルホニル、カルボン酸誘導体などを含む電子吸引性置換基であり、nおよびmは、独立して選択される整数、例えば1、2、または3であり、pは、1、2、3、4、または5などの整数である。AAは、例えば一般式:-N(R)-(CR’R”)q-C(O)-を有する、他の任意のアミノ酸でもあり、式中、Rは、水素、アルキル、アシル、または適切な窒素保護基であり、R’およびR”は、水素または置換基であり、そのそれぞれが各存在に独立して選択され、qは、1、2、3、4、または5などの整数である。例証的には、R’およびR”は、これに限定されるわけではないが、天然型アミノ酸に存在する水素または側鎖、例えばメチル、ベンジル、ヒドロキシメチル、チオメチル、カルボキシル、カルボキシメチル、グアニジノプロピルなど、ならびにその誘導体および保護誘導体に独立して相当する。上記の式は、すべての立体異性変形を含む。例えばアミノ酸は、アスパラギン、アスパラギン酸、システイン、グルタミン酸、リジン、グルタミン、アルギニン、セリン、オルニチン、トレオニンなどから選択されうる。本明細書に記載するビタミン受容体結合薬物送達結合体中間体の別の例証的な態様において、薬物、あるいはその類似体または中間体は、アルキルチオール求核試薬を含む。
【0051】
上の中間体のそれぞれは、在来の合成経路を使用して調製されうる。さらなる合成経路および反応条件は、米国特許出願第10/765,336号および国際出願PCT/US/2005/026068号に記載されている。
【0052】
上の例証的な実施形態は、本明細書に記載する本発明の例示として意図され、本明細書に記載する本発明を制限するものとして、決して説明または解釈されるべきではない。例えば次の例証的なビタミン-薬物結合体によって一般に表される化合物は、本明細書によって記載されるように本明細書に含まれるものであり、
式中、R1およびR2はそれぞれ独立して、水素またはアルキル、例えばメチルであり;かつlHは、ヘテロ原子、例えば酸素、硫黄、任意に置換された窒素、または場合により保護された窒素などである。
【0053】
別の実施形態において、本明細書に記載する化合物は、ケタール基を含む放出型リンカーから形成された2価リンカーを含む。一態様において、ケタール基は、任意に置換された2-、または4-オキシベンズアルデヒド、例えば式:
の4-オキシベンズアルデヒドであり、式中、nは、1、2、3、および4から選択され;Raは、アルキルまたは場合により置換されたアリールアルキルであり、Raは、水素または任意選択の置換基であり;そして(*)原子は、受容体結合部分、2価リンカー、またはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される。
【0054】
別の実施形態において、本明細書に記載する化合物は、カーボネートを含む放出型リンカーから形成された2価リンカーを含む。一態様において、カーボネートは、ビスアルキルカーボネートである。別の態様において、カーボネートは、ジチオ基およびアミノ基またはヒドラジノ基を含むビスアルキルカーボネートである。別の態様において、カーボネートは、式:
の構造であり、式中、nおよびmは、1、2、3、および4よりそれぞれ独立して選択され;かつ(*)原子は、受容体結合部分、2価リンカー、またはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される。
【0055】
別の実施形態において、本明細書に記載する化合物は、2価ジチオアルキルアミノ基または2価ジチオベンジルオキシカルボニル基を含む放出型リンカーから形成された2価リンカーを含む。一態様において、2価ジチオアルキルアミノ基は、式:
の構造であり、式中、nは、1、2、3、および4より選択され;かつ(*)原子は、受容体結合部分、2価リンカー、またはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される。一態様において、2価ジチオベンジルオキシカルボニル基は、式:
の構造であり、式中、Rは、水素または任意選択の置換基であり;かつ(*)原子は、受容体結合部分、2価リンカー、またはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される。一態様において、2価ジチオベンジルオキシカルボニル基は、式:
の構造であり、式中、Rは、水素、アルキル、アルコキシ、シアノ、またはニトロであり;かつ(*)原子は、受容体結合部分、2価リンカー、またはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される。一態様において、2価ジチオベンジルオキシカルボニル基は、式:
の構造であり、式中、Rは、水素、アルキル、アルコキシ、シアノ、またはニトロであり;かつて(*)原子は、受容体結合部分、2価リンカー、またはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される。
【0056】
別の実施形態において、本明細書に記載する化合物としては、結合体を形成するために窒素を介して2価リンカーに結合されるカルボキサミドを含む、ビンカアルカロイド、あるいはその類似体または誘導体が挙げられる。別の実施形態において、本明細書に記載する化合物としては、結合体を形成するために窒素原子を介して2価リンカーに結合されるカルボキスヒドラジドを含む、ビンカアルカロイド、あるいはその類似体または誘導体が挙げられる。一態様において、その結合は末端窒素を介して作られる。別の実施形態において、本明細書に記載する化合物としては、結合体を形成するために酸素を介して2価リンカーに結合されるカルボキシラートを含む、ビンカアルカロイド、あるいはその類似体または誘導体が挙げられる。
【0057】
別の例証的な実施形態において、受容体結合部分(B)は、リンカー(L)-(D)が次の構造:
であるときに、葉酸ではない。
別の例証的な実施形態において、受容体結合部分(B)は、リンカー(L)-(D)が次の構造:
であるときに、葉酸ではない。
【0058】
他の実施形態において、製薬組成物が記載されている。製薬組成物は、本明細書に記載した薬物送達結合体を、そのための製薬的に許容される担体、賦形剤および/または希釈剤と組合せて含む。
【0059】
別の実施形態において、病原性細胞の集団を内部に持つ宿主動物において病原性細胞の集団を排除する方法が記載される。例証的な一態様において、病原性細胞集団のメンバーは、受容体結合部分、あるいはその類似体または誘導体に対するアクセス可能な結合部位を有し、その結合部位は病原性細胞によって独自に、発現、過剰発現または優先的に発現される。該方法は、本明細書で記載するように、宿主に本明細書に記載する薬物送達結合体、あるいはその製薬組成物を投与する工程を含む。
【0060】
本明細書に記載する薬物送達結合体は、ヒト臨床医学的および獣医学的用途の両方に使用されうる。それゆえ病原性細胞の集団を持ち、薬物送達結合体によって治療される宿主動物はヒトであり、あるいは獣医学的用途の場合には、実験用動物、農業用動物、家畜、または野生動物でありうる。本明細書に記載する薬物送達結合体は、これに限定されるわけではないが、ヒト、例えばげっ歯類(例えばマウス、ラット、ハムスターなど)、ウサギ、サル、チンパンジー等の実験用動物、例えばイヌ、ネコ、およびウサギ等の家畜、例えばウシ、ウマ、ブタ、ヒツジ、ヤギ等の農業用動物、および、例えばクマ、パンダ、ライオン、トラ、ヒョウ、ゾウ、シマウマ、キリン、ゴリラ、イルカ、およびクジラ等の飼育されている野生動物を含む宿主動物に投与されうる。
【0061】
本明細書に記載する薬物送達結合体は、宿主動物における各種の病状および病原性細胞を治療するために使用されうる。本明細書で使用するように「病原性細胞」は、癌細胞、感染性因子(例えば細菌およびウィルス)、細菌またはウィルス感染細胞、疾患状態を引き起こすことができる活性化マクロファージ、リガンド受容体(例えばビタミン受容体あるいはビタミンの類似体または誘導体を結合する受容体)を特異的に発現する、優先的に発現する、または過剰発現する他の任意の種類の病原性細胞を意味する。病原性細胞は、薬物送達結合体による治療が疾患症状の低減をもたらす疾患状態を引き起こすいかなる細胞も含みうる。病原性細胞は、移植片対宿主病の原因である免疫系の細胞などの、ある状況の下で病原性であるが、他の状況では病原性でない宿主細胞でもありうる。
【0062】
それゆえ病原性細胞の集団は、良性腫瘍および悪性腫瘍を含めて腫瘍原性である癌細胞集団でありうるか、またはそれは非腫瘍原性でありうる。癌細胞集団は、自然発生的に、または宿主動物の生殖細胞系に存在する変異または体細胞変異などのプロセスによって発生しうるか、あるいはそれは化学的に、ウィルスによって、または放射線によって誘発されうる。本発明は、癌腫、肉腫、リンパ腫、ホジキン病、黒色腫、中皮腫、バーキットリンパ腫、上咽頭癌、白血病、および骨髄腫などの癌を治療するために利用されうる。癌細胞集団としては、これに限定されるわけではないが、口腔癌、甲状腺癌、内分泌癌、皮膚癌、胃癌、食道癌、喉頭癌、膵臓癌、結腸癌、膀胱癌、骨癌、卵巣癌、子宮頸癌、子宮癌、乳癌、精巣癌、前立腺癌、直腸癌、腎臓癌、肝臓癌、および肺癌が挙げられる。
【0063】
病原性細胞集団が癌細胞集団である実施形態において、薬物送達結合体投与の効果は、腫瘤の縮小または消滅あるいは腫瘍細胞増殖の阻害によって測定される治療応答である。腫瘍の場合、消滅は、原発性腫瘍の細胞の、あるいは転移した腫瘍細胞の、または原発性腫瘍から解離する過程にある細胞の消滅でありうる。腫瘍の外科的除去、放射線療法、化学療法、または生物療法を含むいずれかの治療手法による腫瘍の除去後に腫瘍の再発を防止するための、薬物送達結合体による予防的治療も企図される。予防的治療は、薬物送達結合体を用いた初期治療、例えば連日多回容量処方での治療、および/または(複数の)初回治療後に数日または数ヶ月の間隔を置いた追加治療または一連の治療でありうる。したがって上記の任意の病原性細胞集団の排除は、病原性細胞の数の減少、病原性細胞の増殖の阻害、病原性細胞の再発を防止する予防的治療、または疾患の症状の軽減を引き起こす病原性細胞の治療を含む。
【0064】
癌細胞が排除される場合、本明細書に記載する方法は、腫瘍の外科的除去、放射線療法、化学療法、またはその他の免疫療法等の生物療法(これに限定されるわけではないが、モノクローナル抗体療法、免疫調節薬による治療、免疫エフェクタ細胞の養子移入、造血成長因子、サイトカインおよびワクチン接種による治療を含む)と組合せて使用されうる。
【0065】
本明細書に記載する方法は、各種の感染性疾患を引き起こす病原性細胞の集団にも利用できる。例えば本発明は、細菌、酵母を含む菌類、ウィルス、ウィルス感染細胞、マイコプラズマ、および寄生生物などの病原性細胞の集団に利用できる。本明細書に記載する薬物送達結合体によって治療されうる感染性生物は、グラム陰性またはグラム陽性球菌または桿菌である細菌などの生物を含む、動物において病原を引き起こす当分野で認識された任意の感染性生物である。例えばプロテウス種、クレブシエラ種、プロビデンシア種、エルシニア種、エルウィニア種、エンテロバクター種、サルモネラ種、セラチア種、アエロバクター種、エシェリキア種、シュードモナス種、赤痢菌種、ビブリオ種、アエロモナス種、カンピロバクター種、ストレプトコッカス種、スタフィロコッカス種、ラクトバシラス種、ミクロコッカス種、モラクセラ種、バシラス種、クロストリジウム種、コリネバクテリウム種、エベルセラ(Eberthella)種、ミクロコッカス種、マイコバクテリウム種、ナイセリア種、ヘモフィルス種、バクテロイデス種、リステリア種、エリジペロトリックス種、アシネトバクター種、ブルセラ種、パスツレラ種、ビブリオ種、フラボバクテリウム種、フゾバクテリウム種、ストレプトバシラス種、カリマトバクテリウム種、レジオネラ種、トレポネーマ種、ボレリア種、レプトスピラ種、アクチノミセス種、ノカルジア種、リケッチア種、および宿主動物において疾患を引き起こす他の任意の細菌種は、本明細書に記載する薬物送達結合体によって治療されうる。
【0066】
特に興味深いのは、抗生物質耐性ストレプトコッカス種およびスタフィロコッカス種などの抗生物質に対して耐性である細菌、または抗生物質に対して感受性であるが、抗生物質によって治療できる反復性感染を引き起こすので、耐性生物が最終的に発生する細菌である。抗生物質に対して感受性であるが、抗生物質によって治療できる反復性感染を引き起こすので、耐性生物が最終的に発生する細菌は、これらの抗生物質耐性細菌株の発生を回避するために、抗生物質の非存在下で、または宿主動物に通常投与されるよりも低い用量の抗生物質と組合せて、本明細書に記載する薬物送達結合体によって治療されうる。
【0067】
例えばDNAおよびRNAウィルス等のウィルスによって引き起こされた疾患も、本明細書に記載する薬物送達結合体によって治療されうる。このようなウィルスとしては、これに限定されるわけではないが、DNAウィルス、例えばパピローマウィルス、パルボウィルス、アデノウィルス、ヘルペスウィルス、およびワクシニアウィルス、ならびにRNAウィルス、例えばアレナウィルス、コロナウィルス、リノウィルス、呼吸器合胞体ウィルス、インフルエンザウィルス、ピコルナウィルス、パラミキソウィルス、レオウィルス、レトロウィルス、レンチウィルス、およびラブドウィルスが挙げられる。
【0068】
本明細書に記載する薬物送達結合体は、任意の、酵母を含む菌類、マイコプラズマ種、寄生生物、または動物で疾患を引き起こす他の感染性生物によって引き起こされた疾患を治療するためにも使用されうる。本明細書に記載する方法および薬物送達結合体によって治療されうる菌類の例としては、カビとして成長する、または酵母様菌類が挙げられ、これは、例えば白癬、ヒストプラスマ症、ブラストミセス症、アスペルギルス症、クリプトコッカス症、スポロトリクム症、コクシジオイデス症、パラコクシジオイデス症、ムコール症、色素酵母菌症、皮膚糸状菌症、プロトテカ症、フザリウム症、粃糠疹、菌腫、パラコクシジオイデス症、フェオフィホ真菌症、シュードアレシェリア症、スポロトリクム症、砂毛症、ニューモシスチス感染症、およびカンジダ症を引き起こす菌類を含む。
【0069】
本明細書に記載する薬物送達結合体は、これに限定されるわけではないが、サナダムシ(例えば条虫、矮小条虫、裂頭条虫属、およびエキノコッカス種)、吸虫(例えば肥大吸虫、異形吸虫、メタゴニムス、肝吸虫、肝蛭、肺吸虫、および住血吸虫種)、回虫(例えば蟯虫、鞭虫、豚回虫、鉤虫、アメリカ鉤虫、ストロンギロイデス、旋毛虫、ブケレリア、ブルギア、ロアオンコセルカ、およびドラクンクルス種)、アメーバ(例えばネグレリアおよびアカントアメーバ種)、ならびに原虫(例えばプラスモディウム、トリパノソーマ、リーシュマニア、トキソプラズマ、エントアメーバ、ジアルジア、イソスポラ、クリプトスポリジウム、およびエンテロシトゾーン種)によって引き起こされる感染を含む、寄生生物感染を治療するためにも使用されうる。
【0070】
薬物送達結合体が対象とする病原性細胞は、これらの細胞がリガンド受容体、例えばビタミン、あるいはその類似体または誘導体の受容体を優先的に発現する場合、内在性病原体を持つ細胞、例えばウィルス-、マイコプラズマ-、寄生生物-、または細菌-感染細胞でもありうる。
【0071】
一実施形態において、リガンドが、受容体、輸送体、またはリガンドを特異的に結合して、病原性細胞で優先的に発現される他の表面提示タンパク質へ結合する時に、薬物送達結合体は標的病原性細胞内へ内部移行されうる。そのような内部移行は例えば、受容体媒介エンドサイトーシスを通じて発生しうる。薬物送達結合体が放出型リンカーを含有する場合、リガンドおよびビンカ化合物は細胞内で解離して、ビンカはその細胞内標的に作用しうる。
【0072】
別の例証的な実施形態において、薬物送達結合体のリガンドは、ビンカ化合物を病原性細胞の表面に密接に配置する病原性細胞に結合しうる。ビンカ化合物は次に、放出型リンカーの開裂によって放出されうる。例えばビンカ化合物は、放出型リンカーがジスルフィド基である場合、タンパク質ジスルフィドイソメラーゼによって放出されうる。ビンカ化合物は次に、受容体結合薬物送達結合体が結合されている病原性細胞によって取り込まれうるか、またはビンカ化合物はそれに近接した別の病原性細胞によって取り込まれうる。あるいはビンカ化合物は、放出型リンカーがジスルフィド基である細胞内のタンパク質ジスルフィドイソメラーゼによって放出されうる。ビンカ化合物は、あるベータ離脱機構について上述したような加水分解機構、例えば酸触媒による加水分解によって放出され、あるいはオキソニウムイオンまたはラクトニウムイオン産生機構を通じた隣接基補助開裂によっても放出されうる。放出型リンカーの選択は、ビンカ化合物が結合体から放出される機構を決定するであろう。このような選択は、薬物送達結合体が使用される条件によって事前に定義されうることが認識される。
【0073】
リンカーが放出型リンカーを含まない別の例証的な実施形態において、薬物送達結合体のリガンド部分は、病原性細胞をビンカ化合物に結合できる他の分子による攻撃の標的とするために、病原性細胞の表面にビンカ化合物を有する病原性細胞に結合しうる。あるいは本実施形態において、薬物送達結合体は結合時に標的とした細胞内へ内部移行可能であり、リガンド部分およびビンカ化合物は細胞内でビンカ化合物と結合を維持することができ、ビンカ化合物はリガンド部分から解離せずにその効果を示す。
【0074】
なお別の実施形態において、または上記の実施形態と組合せて、薬物送達結合体がビタミン受容体または別のリガンド受容体を結合する場合、結合体は、血清中に存在する溶解性ビタミン受容体またはアルブミンの様な血清タンパク質に結合可能であり、そのため非結合ビンカ化合物と比較して結合体の長期の循環と、非結合ビンカ化合物と比較して病原性細胞集団に対して結合体の活性上昇を引き起こす。
【0075】
例えばビタミンの様なリガンドへの結合部位は、リガンドに特異的に結合できるリガンドの受容体を含み、ここで受容体または他のタンパク質は病原性細胞の集団で特異的に発現、過剰発現または優先的に発現される。病原性細胞によって特異的に発現、過剰発現または優先的に発現された表面提示タンパク質は通例、非病原性細胞には存在しないか、または低濃度で存在する受容体であり、病原性細胞の選択的排除の手段を与える受容体である。薬物送達結合体は、癌細胞または他の種類の病原性細胞上の受容体への高親和性結合が可能でありうる。高親和性結合はリガンドに固有でありうるか、または結合親和性は化学修飾リガンドの使用によって向上されうる。
【0076】
本明細書に記載する薬物送達結合体は、追加の薬物が標的指向化されるかどうかにかかわらず、その他の任意の既知の薬物と共に併用療法で投与されうる。例証的な追加の薬剤としては、これに限定されるわけではないが、ペプチド、オリゴペプチド、レトロ-インベルソオリゴペプチド、タンパク質、少なくとも1個の非ペプチド結合がペプチド結合を置換するタンパク質類似体、アポタンパク、糖タンパク質、酵素、補酵素、酵素阻害薬、アミノ酸およびその誘導体、受容体および他の膜タンパク質、抗原およびそれに対する抗体、ハプテンおよびそれに対する抗体、ホルモン、脂質、リン脂質、リポソーム、毒素、抗体、鎮痛剤、気管支拡張薬、ベータ遮断薬、抗菌剤、降圧剤、心血管作動薬(抗不整脈薬、強心配糖体、抗狭心症薬、血管拡張薬を含む)、中枢神経系剤(刺激薬、向精神薬、抗躁薬、および抑制薬を含む)、抗ウィルス剤、抗ヒスタミン薬、化学治療剤を含む制癌剤、精神安定剤、抗うつ薬、H-2拮抗薬、抗痙攣薬、制嘔吐剤、プロスタグランジンおよびプロスタグランジン類似体、筋弛緩剤、抗炎症性物質、刺激薬、うっ血除去薬、鎮吐薬、利尿剤、鎮痙薬、抗喘息薬、抗パーキンソン病薬、去痰薬、鎮咳剤、粘液溶解薬、ならびにミネラルおよび栄養添加物が挙げられる。
【0077】
別の例証的な態様において、追加の薬物は、内在性免疫応答を刺激できる化合物より選択されうる。適切な化合物としては、これに限定されるわけではないが、サイトカインまたは免疫細胞成長因子、例えばインターロイキン1〜18、幹細胞因子、塩基性FGF、EGF、G-CSF、GM-CSF、FLK-2リガンド、HILDA、MIP-1α、TGF-α、TGF-β、M-CSF、IFN-α、IFN-β、IFN-γ、溶解性CD23、LIF、およびその組合せが挙げられる。
【0078】
これらの免疫活性化因子の治療的に有効な組合せが使用されうる。一実施形態において、例えば約0.1MIU/m2/用量/日〜約15MIU/m2/用量/日の範囲の量の連日多回用量処方におけるIL-2の治療的有効量、および例えば約0.1MIU/m2/用量/日〜約7.5MIU/m2/用量/日の範囲の量の連日多回用量処方におけるIFN-αの治療的有効量は、病原性細胞を持つ宿主動物において病原性細胞を排除、減少または中和するために、薬物送達結合体と共に使用されうる(MIU=100万国際単位;m2=平均的なヒトのおおよその体表面積)。別の実施形態において、IL-12およびIFN-αは、インターロイキンおよびインターフェロンについての上記の治療的有効量で使用され、なお別の実施形態において、IL-15およびIFN-αは、インターロイキンおよびインターフェロンについての上記の治療的有効量で使用されうる。代わりの実施形態において、IL-2、INF-αまたはINF-γ、ならびにGM-CSFは組合せて、上記の治療的有効量で使用されうる。他のインターロイキンおよびインターフェロン、ならびにコロニー刺激因子の組み合わせを含むサイトカインの他の有効な任意の組合せも使用されうる。
【0079】
さらに追加の薬物は、当分野で既知の任意の薬物でありうる。それらの薬物は、細胞毒性または細胞増殖抑制性であり、腫瘍浸透性を向上させ、腫瘍細胞増殖を阻害し、アポトーシスを促進し、標的細胞の抗アポトーシス活性を低下させ、感染性因子によって引き起こされた疾患を治療するために使用され、病原性細胞に向けられた内在性免疫応答を向上させ、または任意の種類の病原性細胞によって引き起こされた疾患状態を治療するために有用である。例示的で適切な追加の薬物としては、アデノコルチコイドおよびコルチコステロイド、アルキル化剤、抗アンドロゲン、抗エストロゲン、アンドロゲン、アクラマイシンおよびアクラマイシン誘導体、エストロゲン、抗代謝産物(例えばシトシンアラビノシド、プリン類似体、ピリミジン類似体、およびメトトレキセート)、ブスルファン、カルボプラチン、クロランブシル、シスプラチンおよび他のプラチナ化合物、タモキシフェン、タキソール、パクリタキセル、パクリタキセル誘導体、タキソテール(登録商標)、シクロホスファミド、ダウノマイシン、リゾキシン、T2毒素、植物アルカロイド、プレドニゾン、ヒドロキシ尿素、テニポシド、マイトマイシン、ディスコデルモライド、非ビンカ微小管阻害薬、エポチロン、チューブリシン(tubulysin)、シクロプロピルベンズ[e]インドロン、seco-シクロプロピルベンズ[e]インドロン、O-Ac-seco-シクロブロピルベンズ[e]インドロン、ブレオマイシンおよび他の任意の抗生物質、窒素マスタード、ニトロソ尿素、コルヒチン、コルヒチン誘導体、アロコルヒチン、チオコルヒチン、トリチルシステイン、ハリコンドリンB、ドラスタチン(例えばドラスタチン10)、アマニチン(例えばα-アマニチン)、カンプトセシン、イリノテカン、および他のそのカンプトセシン誘導体、ゲルダナマイシンおよびゲルダナマイシン誘導体、エラムスチン、ノコダゾール、MAP4、コルセミド、ビンデシン、ビンブラスチン、ビンクリスチン、カタランチン、ビンドリン、ロイロシン、ビノレルビン、イミドカルブ、シブトラミン、トルトラズリル、ビンブラスチン酸、メイタンシンならびにその類似体および誘導体、ゲムシタビン、炎症性剤および炎症誘発剤、ペプチドおよびペプチドミメティックシグナル伝達阻害薬、および任意の他の当分野で認識される薬物または毒素が挙げられる。併用療法で使用されうる他の薬物としては、ペニシリン、セファロスポリン、バンコマイシン、エリスロマイシン、クリンダマイシン、リファンピン、クロラムフェニコール、アミノグリコシド抗生物質、ゲンタマイシン、アンホテリシンB、アシクロビル、トリフルリジン、ガンシクロビル、ジドブジン、アマンタジン、リバビリン、および他の当分野で認識される任意の抗菌化合物が挙げられる。上記の追加の薬物のいずれかの類似体または誘導体も併用療法で使用されうる。
【0080】
別の例証的な実施形態において、製薬組成物が提供される。製薬組成物は、1回以上の用量で投与されるときに宿主動物において病原性細胞の集団を排除するのに有効な薬物送達結合体の量を含む。薬物送達結合体は好ましくは、宿主動物に非経口的に、例えば皮内に、皮下に、筋肉内に、腹腔内に、静脈内に、または髄腔内に投与される。あるいは薬物送達結合体は、例えば経口投与の様な他の医療的に有用なプロセスによって宿主動物に投与することができ、そして任意の有効な用量および適切な治療剤形、例えば持続放出剤形が使用されうる。経口剤形に有用な賦形剤の例としては、これに限定されるわけではないが、コーンスターチ、ゼラチン、ラクトース、ステアリル酸マグネシウム、重炭酸ナトリウム、セルロース誘導体、およびナトリウムデンプングリコール酸が挙げられる。
【0081】
非経口剤形の例としては、等張性生理的食塩水、5%グルコースまたは製薬的に許容されるその他の周知の液体、例えば液体アルコール、グリコール、エステル、およびアミド中の活性物質の水性溶液が挙げられる。本発明による非経口剤形は、薬物送達結合体の用量を含む復元可能な凍結乾燥物の形でありうる。本実施形態の一態様において、例えばその開示が参照により本明細書に組み入れられている、米国特許第4,713,249号;同第5,266,333号;および同第5,417,982号に記載されている生分解性炭水化物マトリクスなどの、当分野で既知の多数の持続放出剤形のいずれも投与可能であり、あるいは低速ポンプ(例えば浸透圧ポンプ)が使用されうる。
【0082】
併用療法における追加の薬物は、薬物送達結合体の前、後、または薬物送達結合体と同時に宿主動物に投与可能であり、追加の薬物は、薬物送達結合体を含有する同じ組成物の一部として、または薬物送達結合体とは異なる組成物の一部として投与されうる。このような任意の併用療法が有効な用量での追加薬物投与に使用されうる。
【0083】
別の例証的な態様において、2つ以上の種類の薬物送達結合体が使用されうる。例えば宿主動物は、異なるリガンドを持つ結合体、例えば組合された葉酸塩-ビンカおよびビタミンB12-ビンカ結合体などを用いた同時投薬プロトコルで治療されうる。別の例証的な態様において、宿主動物は、2つ以上のリガンド、例えば複数の葉酸塩または複数のビタミンB12を1つの結合体中に含む結合体により、または葉酸塩およびビタミンB12リガンドの両方に結合されたビンカ化合物などの、同じ結合体中のリガンドの組合せを用いて治療されうる。さらに別の薬物送達結合体中に異なる種類のビンカ化合物を含む薬物送達結合体が使用されうる。
【0084】
薬物送達結合体の日単位の処方量は、宿主の状態、治療される疾患状態、結合体の分子量、その投与経路および組織分布、ならびに放射線療法または併用療法の追加の薬物などの他の治療処置の同時使用の可能性に応じて著しく変化しうる。宿主動物へ投与される有効量は、体表面積、体重、および患者の状態の医師による評価に基づく。有効用量は、例えば約1ng/kg〜約1mg/kg、約1μg/kg〜約500μg/kg、および約1μg/kg〜約100μg/kgの範囲で変化しうる。
【0085】
薬物送達結合体を投与するため、任意の有効な投薬計画が使用されうる。例えば薬物送達結合体は、1回用量として投与されうるか、または分割されて、連日多回用量処方として投与されうる。さらに時差投薬計画、例えば週に1〜3日を連日治療の代用として使用することができ、本発明を定義する目的で、このような間欠的または時差連日投薬計画は、毎日の治療と同等と見なされ、企図される。例証的な一実施形態において、宿主動物は、病原性細胞の集合を排除するために薬物送達結合体の複数回注射によって治療される。一実施形態において、宿主は薬物送達結合体を複数回(好ましくは約2〜50回まで)、例えば12〜72時間の間隔で、または48〜72時間の間隔で注射される。薬物送達結合体の追加の注射は宿主動物に、初回注射後の数日または数ヶ月の間隔で投与でき、追加の注射は病原性細胞によって引き起こされた疾患状態の再発を防止しうる。
【0086】
例証的な一態様において、薬物送達結合体で使用されうるビタミンあるいはその類似体または誘導体は、活性化マクロファージ上で特異的に発現される受容体、例えば葉酸塩あるいはその類似体または誘導体を結合する葉酸塩受容体に結合するものを含みうる。葉酸塩結合体は例えば、宿主の疾患状態を引き起こす活性化マクロファージの活性を死滅または抑制するために使用されうる。そのようなマクロファージを標的とする結合体は、活性化マクロファージ媒介疾患状態にある宿主動物に投与されるときに、活性化マクロファージを死滅させる、またはマクロファージ機能を抑制するために、結合体されたビンカ化合物を活性化マクロファージの集合に集中させ結合させるように作用する。活性化マクロファージ集合の排除、減少、または非活性化は、治療される疾患状態の活性化マクロファージ媒介病原特徴を停止または低減するように作用する。活性化マクロファージによって媒介されることが既知の疾患の例としては、関節リウマチ、潰瘍性大腸炎、クローン病、乾癬、骨髄炎、多発性硬化症、アテローム硬化症、肺線維症、サルコイドージス、全身性硬化症、移植臓器拒絶(GVHD)および慢性炎症が挙げられる。薬物送達結合体の投与は通例、疾患状態の症状が低減または排除されるまで継続される。
【0087】
活性化マクロファージを死滅させる、または活性化マクロファージの機能を抑制するために投与される薬物送達結合体は、宿主動物に非経口的に、例えば皮内に、皮下に、筋肉内に、腹腔内、または静脈内に製薬的に許容される担体と共に投与されうる。あるいは薬物送達結合体は宿主細胞に他の医療的に有用な手順で投与でき、有効用量は標準剤形または持続放出剤形で投与されうる。治療方法は単独で、または活性化マクロファージによって媒介される疾患状態の治療のために認められた他の治療方法と組合せて使用されうる。
【0088】
本明細書に記載する本発明は、次の実施例によってさらに例証される;しかしながらこれらの実施例は単に本発明の例証として意図され、決して本発明を制限すると解釈されるべきでないことが理解されなければならない。例えば本明細書に示す各化合物において、リンカーを形成するのに使用されるアミノ酸の立体化学は場合により、天然l配置または非天然d配置から選択されうる。加えて本明細書では、これに限定されるわけではないが、ビンブラスチンの各種の他の類似体および誘導体、各種の他のスペーサー、ヘテロ原子、およびリンカー組合せ、およびその他を含む多くの変更形態が企図されうる。本明細書に記載する各実施例の化合物は、示されたようにNMR、MS、および/またはUV分光法、および/またはHPLCによって特徴付けられ、特徴的な1H NMRシグナル、MSシグナルなどを含む選択された分析データは必要に応じて記載される。
【実施例】
【0089】
===方法実施例===
〔方法実施例1 マウスにおける腫瘍成長の阻害〕
本明細書に記載した化合物の抗腫瘍活性は、腫瘍担持動物へ静脈内(i.v.)投与したときに、皮下M109腫瘍を担持するBalb/cマウスにおいて評価した。右腋窩の皮下組織への1x106個のM109細胞の腫瘍播種の約11日後(t0の時点での腫瘍容積範囲=60〜80mm3)、マウス(5/グループ)に週3回(TIW)、規定の期間(例えば2〜3週間)にわたって、(a)体重1キログラムあたりの規定用量レベルの、本明細書に記載する薬物送達結合体、または(b)等用量体積のPBS(対照群)をi.v.注射した。腫瘍成長は、各処置グループで2日または3日間隔で測径器を使用して測定した。腫瘍体積は、式V=axb2/2を使用して計算し、式中、「a」は腫瘍の長さであり、「b」は幅である(ミリメートルで表す)。
【0090】
〔方法実施例2 マウスにおける腫瘍成長の阻害〕
本明細書に記載した化合物の抗腫瘍活性は、腫瘍担持動物へ静脈内(i.v.)投与したときに、皮下KB腫瘍を担持するnu/nuマウスにおいて評価した。右腋窩の皮下組織への1x106個のKB細胞の腫瘍播種の約8〜11日後(t0の時点の腫瘍容積範囲=60〜80mm3)、マウス(5/グループ)に週3回(TIW)、規定の期間(例えば2〜3週間)にわたって、(a)体重1キログラムあたりの規定用量レベルの本明細書に記載する薬物送達結合体、または(b)等用量体積のPBS(対照群)をi.v.注射した。腫瘍成長は、各処置グループで2日または3日間隔で測径器を使用して測定した。腫瘍体積は、式V=axb2/2を使用して計算し、式中、「a」は腫瘍の長さであり、「b」は幅である(ミリメートルで表す)。
【0091】
〔方法実施例3 細胞DNA合成の阻害〕
本明細書に記載する化合物は、薬物が葉酸塩受容体陽性KB細胞の成長を阻害する能力を予測するin vitro細胞毒性アッセイを使用して評価した。化合物は、本明細書に記載するプロトコルに従って調製した各化学療法薬に結合された葉酸塩を含んでいた。KB細胞を所定の期間にわたって37℃にて、少なくとも100倍過剰の葉酸の非存在下または存在下で、指示された濃度の葉酸-薬物結合体に暴露した。この細胞を次に新しい培地ですすぎ、新しい培地で72時間にわたって37℃でインキュベートした。細胞生存能力は、3H-チミジン取り込みアッセイを使用して評価した。
【0092】
本明細書の図に示すように、用量依存性細胞毒性は測定可能であり、大半の場合でIC50値(新たに合成されたDNAへの3H-チミジン取り込みを50%低下させるのに必要な薬物結合体の濃度)は低いナノモル範囲であった。さらにこれらの結合体の細胞毒性は、過剰な遊離葉酸の存在下で低減され、観察された細胞死滅は葉酸塩受容体への結合によって仲介されることを示す。
【0093】
〔方法実施例4 相対親和性アッセイ〕
葉酸塩と比較した葉酸塩受容体(FR)への本明細書に記載する化合物の親和性は、先に記載された方法(Westerhof,G.R.,J.H.Schornagel,et al.(1995)Mol.Pharm.48:459-471)に多少の変更を加えて決定した。簡潔にはFR陽性KB細胞を24ウェル細胞培養プレートに密集して播種して、プラスチックに18時間付着させた。使用済みのインキュベーション培地を指定されたウェルにて、試験品または葉酸の上昇濃度の非存在下または存在下で100nM 3H-葉酸を添加した葉酸塩を含まないRPMI(FFRPMI)と交換した。細胞を37℃にて60分間インキュベートして、次にPBS(pH7.4)によってすすぎ、次いでPBS(pH7.4)中に1% SDS 500μLを添加した。次に細胞溶解液を収集して、シンチレーションカクテル5mLを含有する個々のバイアルに添加し、次に放射能をカウントした。負の対照サンプルは、FFRPMI中に3H-葉酸のみを含有していた(競合物質なし)。正の対照サンプルは1mM葉酸の最終濃度を含有し、この正の対照サンプルで測定で得られたCMP(標識の非特異性結合を表す)はすべてのサンプルから引かれた。特に相対親和性は、KB細胞のFRに結合する3H-葉酸の50%を置換するのに必要な化合物のモル比の逆数として定義され、FRに対する葉酸の相対親和性は1に設定された。
【0094】
〔方法実施例5 4T−1腫瘍体積アッセイ〕
6〜7週齢マウス(メスBalb/c株)をインディアナ州インディアナポリスのHarlan,Inc.より入手した。マウスをHarlanの葉酸を含まない固形飼料で、本実験の開始前および本実験中の合計3週間にわたって維持した。葉酸塩受容体-陰性4T-1腫瘍細胞(動物あたり1x106細胞)を右腋窩の皮下組織に接種した。4T-1腫瘍平均容積が〜100mm3である、腫瘍接種の約5日後に、マウス(5/グループ)に、週3回(TIW)、3週間にわたって、3μmol/kgの薬物送達結合体、または等用量体積のPBS(対照群)をi.v.注射した。腫瘍成長は、各処置グループで2日または3日間隔で測径器を使用して測定した。腫瘍体積は、式V=axb2/2を使用して計算し、式中、「a」は腫瘍の長さであり、「b」は幅である(ミリメートルで表す)。
【0095】
〔方法実施例6 体重測定〕
マウスの重量変化率は、関連する腫瘍容積アッセイに記載したサンプルのグラフに示すように、腫瘍接種後(PTI)の指示された日にマウス(5マウス/グループ)において測定した。
【0096】
〔方法実施例7 葉酸塩-ペプチドの一般的な調製〕
ペプチドを含む本明細書に記載するリンカーは、酸感受性Fmoc-AA-Wang樹脂におけるFmoc方法などの標準方法を使用したポリマー支持連続手法によって調製した。例証的には、葉酸塩含有ペプチジル断片Pte-Glu-(AA)n-NH(CHR2)CO2H(3)は、Wang樹脂支持アミノ酸およびFmoc保護アミノ酸合成からスキーム1に示す方法によって調製する。
<スキーム1>
(a)20%ピペリジン/DMF;(b)Fmoc-AA-OH、PyBop、DIPEA、DMF;(c)Fmoc-Glu-O-t-BuまたはFmoc-Glu(γ-O-t-Bu)-OH、PyBop、DIPEA、DMF;(d)N10(TFA)-Pte-OH;PyBop、DIPEA、DMSO;(e)TFAA、(CH2SH)2、i-Pr3SiH;(f)NH4OH、pH9〜10。
【0097】
本明細書に記載するプロセスのこの例示的な実施形態において、R1は、Fmocであり、R2は、所望の適切に保護されたアミノ酸側鎖であり、Wangは、2-クロロトリチル樹脂であり、DIPEAは、ジイソプロピルエチルアミンである。標準カップリング手順、例えばPyBop、および本明細書に記載するまたは当分野で既知である他の手順が使用され、そこではカップリング剤が例証的に、効率的なカップリングを確実にするための活性化剤として利用される。Fmoc保護基は、標準条件下での各カップリング工程の後に、例えばピペリジン、テトラブチルアンモニウムフルオリド(TBAF)などによる処置時に除去される。適切に保護されたアミノ酸構築ブロック、例えばFmoc-Glu-OtBu、N10-TFA-P-te-OHなどが、スキーム1に記載され、Fmoc-AA-OHによる工程(b)で表されるように使用される。したがってAAは、適切に保護される任意のアミノ酸出発物質を指す。本明細書で使用するアミノ酸という用語は、1個以上の炭素によって隔離されたアミンおよびカルボン酸官能基の両方を有するいずれの試薬も指すものとし、天然型アルファおよびベータアミノ酸、ならびにこれらのアミノ酸のアミノ酸誘導体および類似体を含むことが理解される。特に保護される側鎖を有するアミノ酸、例えば保護セリン、トレオニン、システイン、アスパラギン酸なども、本明細書に記載する葉酸塩-ペプチド合成で使用されうる。さらなるガンマ、デルタ、またはより長い相同性アミノ酸も、本明細書に記載の葉酸塩-ペプチド合成で出発物質として含まれうる。さらに相同性側鎖、または交互の分岐構造を有するアミノ酸類似体、例えばノルロイシン、イソバリン、β-メチルトレオニン、β-メチルシステイン、β,β-ジメチルシステインなども、本明細書に記載する葉酸塩-ペプチド合成で出発物質として含まれうる。
【0098】
式Fmoc-AA-OHのFmoc-保護アミノ酸(AA)を含むカップリングシーケンス(工程(a)および(b))は、固体支持ペプチド(2)を調製するために「n」回実施され、ここでnは整数であり、0〜約100に等しくてもよい。最後のカップリング工程後の、残存するFmoc基が除去され(工程(a))、ペプチドが逐次的にグルタミン酸誘導体にカップリングされ(工程(c))、脱保護されて、TFA-保護プテロイン酸にカップリングされる(工程(d))。次にペプチドは、トリフルオロ酢酸、エタンジチオール、およびトリイソプロピルシランによる処理時にポリマー支持体から開裂される(工程(e))。これらの反応条件は、適切に保護されたアミノ酸側鎖の一部を形成しうるt-Bu、t-Boc、およびTrt保護基の同時除去を引き起こす。TFA保護基は、塩基による処理(工程(f)時に除去されて、葉酸塩含有ペプチジル断片(3)を与える。
【0099】
===化合物実施例===
〔実施例1]
方法実施例7の一般手順(スキーム1)により、Wang樹脂結合4-メトキシトリチル(MTT)保護Cys-NH2を次のシーケンスに従って反応させた。1)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;2)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;3)a.Fmoc-Arg(Pbf)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;4)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;5)a.Fmoc-GIu-OtBu、PyBOP、DIPEA;b.20%ピペリジン/DMF;6)N10-TFA-プテロイン酸、PyBOP、DIPEA、MTT、tBu、およびPbf保護基はTFA/H2O/TIPS/EDT(92.5:2.5:2.5:2.5)によって除去され、TFA保護基はpH=9.3のNH4OH水溶液によって除去した。選択された1H NMR(D2O)δ(ppm)8.68(s,1H,FA H-7),7.57(d,2H,J=8.4Hz,FA H-12&16),6.67(d,2H,J=9Hz,FA H-13&15),4.40-4.75(m,5H),4.35(m,2H),4.16(m,1H),3.02(m,2H),2.55-2.95(m,8H),2.42(m,2H),2.00-2.30(m,2H),1.55-1.90(m,2H),1.48(m,2H);MS(ESI,m+H+)1046。
【0100】
〔実施例2〕
方法実施例7の一般手順(スキーム1)により、Wang樹脂結合4-メトキシトリチル(MTT)保護Cys-NH2を次のシーケンスに従って反応させた。1)a.Fmoc-β-アミノアラニン(NH-MTT)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;2)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;3)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;4)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;5)a.Fmoc-Glu-OtBu、PyBOP、DIPEA;b.20%ピペリジン/DMF;6)N10-TFA-プテロイン酸、PyBOP、DIPEA。MTT、tBu、およびTFA保護基は、a.2%ヒドラジン/DMF;b.TFA/H2O/TIPS/EDT(92.5:2.5:2.5:2.5)によって除去した。次の表に示す試薬を調製で使用した:
【0101】
カップリング工程は次のように実施した:ペプチド合成容器に樹脂を添加し、アミノ酸溶液、DIPEA、およびPyBOPを添加する。アルゴンを1時間通気して、DMFおよびIPAで3回洗浄する。各アミノ酸カップリング前に、Fmoc脱保護のためにDMFに希釈した20%ピペリジンを3回(10分間)使用する。継続して6回のカップリング工程をすべて完了させる。最後に樹脂をDMFに希釈した2%ヒドラジンで3回(5分間)洗浄して、プテロイン酸のTFA保護基を開裂させる。
【0102】
次の試薬、92.5%(50ml)TFA、2.5%(1.34ml)H2O、2.5%(1.34ml)トリイソプロピルシラン、2.5%(1.34ml)エタンジチオール を使用して樹脂からペプチド類似体を開裂させ、開裂工程を次のように実施した:開裂試薬25mlを添加して、1.5時間通気し、試薬を排出し、残りの試薬で3回洗浄した。約5mLまで蒸発させて、エチルエーテル中に沈殿させる。遠心分離にかけた後、乾燥させる。精製は次のように実施した。カラム-Waters NovaPak C18 300xl9mm;緩衝液A=10mM酢酸アンモニウム、pH5;B=CAN;1%B〜20%B、15ml/分にて40分間、350mg(64%)まで;HPLC-RT 10.307分、純度100%、1H NMRスペクトルは割り当てられた構造と一致、そしてMS(ES-):1624.8、1463.2、1462.3、 977.1、976.2、975.1、974.1、486.8、477.8。
【0103】
〔実施例3〕
方法実施例7の一般手順(スキーム1)により、Wang樹脂結合MTT保護Cys-NH2を次のシーケンスに従って反応させた:1)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;2)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;3)a.Fmoc-Arg(Pbf)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;4)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;5)a.Fmoc-Glu(γ-OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;6)N10-TFA-プテロイン酸、PyBOP、DIPEA。MTT、tBu、およびPbf保護基はTFA/H2O/TIPS/EDT(92.5:2.5:2.5:2.5)によって除去され、TFA保護基はpH=9.3のNH4OH水溶液によって除去した。1H NMRスペクトルは、割り当てられた構造と一致していた。
【0104】
〔実施例4〕
方法実施例7の一般手順(スキーム1)により、Wang樹脂結合MTT保護D-Cys-NH2を次のシーケンスに従って反応させた:1)a.Fmoc-D-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;2)a.Fmoc-D-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;3)a.Fmoc-D-Arg(Pbf)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;4)a.Fmoc-D-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;5)a.Fmoc-D-Glu-OtBu、PyBOP、DIPEA;b.20%ピペリジン/DMF;6)N10-TFA-プテロイン酸、PyBOP、DIPEA。MTT、tBu、およびPbf保護基はTFA/H2O/TIPS/EDT(92.5:2.5:2.5:2.5)によって除去され、TFA保護基はpH=9.3のNH4OH水溶液によって除去した。1H NMRスペクトルは、割り当てられた構造と一致していた。
【0105】
〔実施例5〕
2-[(ベンゾトリアゾール-1-イル-(オキシカルボニルオキシ)-エチルジスルファニル]-ピリジンHCl(601mg)およびDIPEA 378μLを、0℃の、デスアセチルビンブラスチンヒドラジド(668mg)の5mLDMC溶液中に逐次的に添加した。反応物を室温まで加温して、3時間撹拌した。TLC(DCMに希釈した15% MeOH)は、完全な変換を示した。混合物をシリカゲルクロマトグラフィー(1:9 MeOH/DCM)によって精製した。合せた画分を蒸発させ、DCMに再溶解させて、10% Na2CO3で洗浄し、塩水につけ、乾燥(MgSO4)し、550mg(80%)まで蒸発させた;HPLC-RT 12.651分、純度91%、1H HMRスペクトルは割り当てられた構造と一致した。MS(ESI+):984.3、983.3、982.4、492.4、491.9、141.8。この手順のさらなる詳細は、参照によりその全体が本明細書に組み入れられている、米国特許出願公開第2005/0002942A1号に記載されている。
【0106】
〔実施例6〕
デスアセチルビンブラスチンモノヒドラジド(1当量)は、その開示が参照により本明細書に組み入れられているBarnett et al.,J Med.Chem.21:88-96(1978)に従って調製して、新しい蒸留THF中でトリフルオロ酢酸1当量によって処理した。10分間撹拌した後に、溶液をN-(4-アセチルフェニル)マレイミド1.05当量によって処理した。アシルヒドラゾン形成は45分後に完了して、溶媒を蒸発させた。
【0107】
ペプチジル断片Pte-Glu-Asp-Arg-Asp-Asp-Cys-OH(実施例1)(0.85当量)を水に溶解させて、pHは0.1N HClによって2.5に調整して、ペプチドを沈殿させた。ペプチジル断片を遠心分離によって収集し、乾燥し、DMSOに溶解した。得られた透明黄色の溶液に、ヒューニッヒ塩基(15当量)およびアシルヒドラゾンMicahel付加体を添加した。1時間後、最終結合体をHPLCによって精製した。図1Aおよび1Bは、葉酸塩対葉酸塩-デアセチルビンブラスチン結合体の相対結合親和性、結合体の3H-チミジン取り込みに対する効果をそれぞれ示す。図1Aおよび1Bは、結合体(14nM)のIC50と、葉酸塩が、葉酸塩受容体への結合について葉酸塩-デアセチルビンブラスチン結合体と競合することを示し、結合体の結合の特異性を証明している。アッセイは方法実施例3および4に従って実施した。
【0108】
図2は、Balb/cマウスにおけるM109腫瘍に対する実施例6(1.5μmol/kg)の活性を示す。アッセイは方法実施例1に従って実施した。実施例6は、固形腫瘍の成長を阻害する。図3は、FR陽性M109腫瘍に対し、3週間にわたりTIWで10μmol/kgが与えられた実施例6の活性を示し、実施例6化合物の投薬は点線によって示されるように第25日に終了した。アッセイは方法実施例1に従って実施した。実施例6は、固形腫瘍の成長を阻害する。
【0109】
図4Aおよび4Bは、FR陽性109腫瘍およびFR陰性4T-1腫瘍細胞それぞれに対する、3週間にわたる3μmol/kg TIWでの実施例6の活性を示す。アッセイは、方法実施例1および5にそれぞれ記載されたように実施した。実施例6は、固形M109腫瘍の成長を阻害するが、葉酸塩受容体(FR)陰性腫瘍は阻害しない。
【0110】
図5Aおよび図5Bは、FR陽性KB腫瘍に対する、およびnu/unマウスの体重(nu/unマウスはKB腫瘍体積アッセイに使用した)に対する、3週間にわたる10μmol/kg TIWでの実施例6の活性を示す。アッセイは方法実施例2および6にそれぞれ従って実施した。実施例6は、固形腫瘍の成長を阻害するが、マウスの体重に影響を及ぼさなかった。
【0111】
図6Aおよび6Bは、3週間にわたる1、5、および10μmol/kg TIWでの、FR陽性KB腫瘍(t0の時点での平均腫瘍体積=50〜100mm3)に、および実施例6の同じ濃度でのnu/nuマウスの体重(nu/nuマウスはKB腫瘍体積アッセイに使用した)にそれぞれ対する、実施例6の活性を示す。アッセイは方法実施例2および6にそれぞれ従って実施した。実施例6は、固形腫瘍の成長を阻害するが、マウスの体重にほとんど効果を持たない。
【0112】
図7Aおよび7Bは、FR陽性KB腫瘍(t0の時点での平均腫瘍体積=100〜150mm3)に対する、3週間にわたる10μmol/kg TIWでの実施例6の活性を示す。実施例6対非結合ビンブラスチンの、Balb/cマウスの体重に対する効果も示される。アッセイは方法実施例2および6にそれぞれ従って実施した。実施例6は、固形腫瘍の成長を阻害する。非結合ビンブラスチンは、最初にマウスの体重を減少させたが、マウスの体重は最終的に、おそらく腫瘍成長が理由で増加する。
【0113】
〔実施例7〕
THF中のペプチジル断片Pte-Glu-Asp-Arg-Asp-Asp-Cys-OH(実施例1)を、黄色溶液のチオスルホナートまたはピリジルジチオ活性化ビンブラスチン(実施例5)のどちらかによって処理して、アルゴン存在下でpH>6.5の0.1M NaHCO3への溶解を生じた。凍結乾燥およびHPLCにより、収率70%が得られた;選択された1H NMR(D2O)δ8.67(s,1H,FA H-7),7.50(br s,1H,VLB H-11’),7.30-7.40(br s,1H,VLB H-14’),7.35(d,2H,J=7.8Hz,FA H-12&16),7.25(m,1H,VLB H-13’),7.05(br s,1H,VLB H-12’),6.51(d,2H,J=8.7Hz,FA H-13&15),6.4(s,2H,VLB H-14&17),5.7(m,1H,VLB オレフィン),5.65(m,1H,VLB H-7),5.5(d,1H,VLB オレフィン),5.5(m,1H,VLB H-6),4.15(m,1H,VLB H-8’),3.82(s,3H,VLB C18-CO2CH3),3.69(s,3H,VLB C16-OCH3),2.8(s,3H,VLB N-CH3),1.35(br s,1H,VLB H-3’),1.15(m,1H,VLB H-2’),0.9(t,3H,J=7Hz,VLB H-21’),0.55(t,3H,J=6.9Hz,VLB H-21);LCMS(ESI,m+H+)1918。図8は、葉酸塩対実施例7結合体の相対親和性を示す。アッセイは方法実施例4に従って実施した。
【0114】
図9Aは、3H-チミジン取り込みに対する実施例7の効果、結合体(9nM)のIC50、および葉酸塩受容体への結合に関して葉酸塩が実施例7結合体と競合することを示し、結合体の結合特異性を証明している。図9Bは、実施例7の結合体(100nM 実施例7)による処理ためのパルス時間に対する、3H-チミジン取り込みに対する実施例7の効果を、および葉酸塩受容体への結合に関して葉酸塩が実施例7の結合体と競合することを示し、結合体の結合特異性を証明している(100nM 実施例7+100μM葉酸)。アッセイは方法実施例3に従って実施した。
【0115】
図10Aおよび10Bは、実施例7の結合体による処理のパルス時間に対する、3H-チミジン取り込みへの10および100nMの実施例7の効果を、そして葉酸塩受容体への結合に関して葉酸塩が実施例7の結合体と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例3に従って実施した。
【0116】
図11は、FR陽性KB腫瘍(t0の時点での平均腫瘍体積=50〜100mm3)に対する、3週間にわたる5μmol/kg TIWの実施例7の活性を示す。アッセイは方法実施例2に従って実施した。実施例7は、固形腫瘍の成長を阻害する。
【0117】
図12は、FR陽性KB腫瘍に対する3週間にわたる5μmol/kg TIWの実施例7の活性を示す(nu/nuマウスをKB腫瘍容積アッセイに使用した)。アッセイは方法実施例2に従って実施した。実施例7および8は、固形腫瘍の成長を阻害する。
【0118】
図13は、Balb/cマウスのM109腫瘍に対する実施例7(1.5μmol/kg)の活性を示す。 アッセイは方法実施例1に従って実施した。 実施例7は、固形腫瘍の成長を阻害する。
【0119】
図14Aおよび14Bは、Balb/cマウスのM109腫瘍に対する、およびBalb/cマウスの体重(Balb/cマウスはM109腫瘍体積アッセイに使用した)に対する、実施例7および8の活性を示す(それぞれ10μmol/kgにて)。アッセイは方法実施例1および6にそれぞれ従って実施した。実施例7および8は、固形腫瘍の成長を阻害し、Balb/cマウスの体重にほとんど効果を持たない。
【0120】
図15は、FR陽性KB腫瘍+/-40μmol/kg EC20(レニウム錯体)に対する、2週間にわたる2μmol/kg TIWの実施例7の活性を示す。実施例7は固形腫瘍の成長を阻害し、その阻害効果はEC20レニウム錯体によって防止(競合)される。EC20(レニウム錯体)は、レニウムにキレート化された次の式:
の化合物である。EC20の調製は、米国特許出願公開第2004/0033195A1号に記載されており、その合成手順の説明は参照により本明細書に組み入れられている。アッセイは方法実施例2に従って実施した。EC20は、葉酸塩受容体において実施例7の競合相手として作用し、結果は、実施例7の効果の特異性を示している。
【0121】
図16Aおよび図16Bは、FR陽性KB腫瘍に対する、3週間にわたる5μmol/kg TIWの実施例7および8の活性、ならびにnu/nuマウスの体重(nu/nuマウスはKB腫瘍体積アッセイに使用した)に対する、実施例7および8の効果を示す。アッセイは方法実施例2および6にそれぞれ従って実施した。結果は、nu/nuマウスにおける皮下FR陽性ヒト上咽頭KB腫瘍異種移植片に対して、実施例7が実施例6よりも高い成長阻害活性を有することを示す。実施例7および8は、nu/nuマウスの体重にはほとんど効果を持たない。
【0122】
実施例7は、次の表に示すように、s.c.KB腫瘍異種移植片を持つnu/nuマウスにおいて、非結合デスアセチルビンブラスチンヒドラジド(DAVLBH)よりも良好な治療指数を示した:
(a)CRは、対照群と比較して、試験化合物による処置に対し、完全寛解を示す動物の数(5回試験の合計)に相当する;(b)%T/Cは、完全寛解を示していない動物の対照群に対する腫瘍率である;(c)LCKは、完全寛解を示していない動物の細胞死滅の対数である;(d)死亡5匹。
【0123】
〔実施例9〕
方法実施例7の一般手順(スキーム1)により、Wang樹脂結合4-メトキシトリチル(MTT)保護CyS-NH2を次のシーケンスに従って反応させた。1)a.Fmoc-β-アミノアラニン(NH-IvDde)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;2)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;3)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;4)a.Fmoc-Asp(OtBu)-OH、PyBOP、DIPEA;b.20%ピペリジン/DMF;5)a.Fmoc-Glu-OtBu、PyBOP、DIPEA;b.20%ピペリジン/DMF;6)N10-TFA-プテロイン酸、PyBOP、DIPEA。MTT、tBu、およびTFA保護基は、a.2%ヒドラジン/DMF;b.TFA/H2O/TIPS/EDT(92.5:2.5:2.5:2.5)によって除去した。次の表に示す試薬を調製で使用した:
【0124】
カップリング工程は次のように実施した:ペプチド合成容器に樹脂を添加し、アミノ酸のDMF溶液、DIPEA、およびPyBOPを添加する。アルゴンを1時間通気して、DMFおよびIPA 10mLで3回洗浄する。各アミノ酸カップリング前に、Fmoc脱保護のためにDMF中の20%ピペリジンを3回x10mL(10分間)使用する。継続して6回のカップリング工程を完了させる。最後に樹脂を2%ヒドラジンのDMF希釈液によって3回x10mL(5分間)洗浄し、プテロイン酸のTFA保護基、およびβ-アミノアラニンのIvDde保護基を開裂させる。最後に、DIPEAおよびPyBopを使用し、β-アミノアラニンの遊離アミンをFmoc-チオプロピオン酸とDMF中でカップリングさせる。アルゴンを1時間通気して、DMFおよびIPA 10mLで3回洗浄する。樹脂をアルゴン存在下で30分間乾燥させる。
【0125】
次の試薬、92.5%(50ml)TFA、2.5%(1.34ml)H2O、2.5%(1.34ml)トリイソプロピルシラン、2.5%(1.34ml)エタンジチオールを使用して樹脂からペプチド類似体を開裂させ、開裂工程を次のように実施した:開裂試薬25mlを添加して、1.5時間通気し、排出させて、残りの試薬で3回洗浄した。約5mLまで蒸発させて、エチルエーテル中に沈殿させる。遠心分離にかけて、乾燥させる。精製は次のように実施した。カラム-Waters NovaPak C18 300xl9mm;緩衝液A=10mM酢酸アンモニウム、pH5;B=CAN;1%B〜20%B、15 ml/分にて40分間、450mg(65%)まで;1H HMRスペクトルは割り当てられた構造と一致。
【0126】
〔実施例10〕
ポリプロピレン遠心分離ボトル内で、実施例2(82mg、0.084mmol)を水5mLに溶解させて、アルゴンを10分間通気した。別のフラスコで0.1N NaHCO3溶液に10分間アルゴンを通気した。リンカー溶液のpHは、0.1N NaHCO3溶液を使用して約6.9に調整した。テトラヒドロフラン(THF)5mL中のビンブラスチンヒドラジド誘導体(実施例5、91mg、0.092mM)を上記溶液にゆっくり添加した。得られた透明溶液をアルゴン存在下で15分間〜1時間撹拌した。反応の進行は、分析HPLC(10mM酢酸アンモニウム、pH=7.0、およびアセトニトリル)によって監視した。THFを蒸発させて、水溶液を濾過して、分取精製HPLCカラム(XTerra Column,19X300mM)に注入した。1mMリン酸ナトリウムpH=7.0およびアセトニトリルによる溶出は、生成物を含有する純粋な画分を生じ、生成物は48時間の凍結乾燥後に単離した(78mg、50%);C83H103N19O26S2;正確な質量:1845.68;MW:1846.95;HPLC-RT 15.113分、純度100%、1H HMRスペクトルは割り当てられた構造と一致した。MS(ES-):1846.6、1845.5、933.3、924.2、923.3、922.5、615.6、 614.7、525.0。
【0127】
図21Aおよび図21Bは、葉酸塩対実施例10の相対結合親和性、および3H-チミジン取り込みに対する実施例10の効果、結合体のIC50(58nM)と、葉酸塩が葉酸塩受容体への結合で結合体と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例4および3にそれぞれ従って実施した。
【0128】
〔実施例11〕
実施例3を実施例5の代わりに使用することを除いて、実施例7に従って調製した。アルゴンを実施例3(302mg)の5ml水溶液に10分間通気した。この溶液のpHは、飽和NaHCO3溶液を使用して6.8〜7.0に調整した。5mLのTHF中の実施例5(258mg)を溶液に添加して、30分間撹拌した。溶媒を蒸発させて、得られた混合物を濾過した。濾液を分取精製HPLCによって(溶媒A-1mMリン酸塩緩衝液;溶媒B-アセトニトリル;Waters XTterra C18,19mmX300mm;勾配-30分間で5% B〜50% B)240mgまで精製した;1H HMRスペクトルは割り当てられた構造と一致;MS(ESI,m+H+)1917.9,960.9,960.2,959.3,813.1,812.3,803.0,295.0。
【0129】
〔実施例12〕
実施例4を実施例5の代わりに使用することを除いて、実施例7に従って調製した。アルゴンを実施例4(40mg)の水5ml中溶液に10分間通気した。この溶液のpHは、飽和NaHCO3溶液を使用して6.8〜7.0に調整した。THF 5mL中の実施例5(30mg)を溶液に添加して、30分間撹拌した。溶媒を蒸発させて、得られた混合物を濾過した。濾液を分取精製HPLCによって(溶媒A-1mMリン酸塩緩衝液;溶媒B-アセトニトリル;Waters XTterra C18,19mmX300mm;勾配-30分間で5% B〜50% B)43mgまで精製した;1H HMRスペクトルは割り当てられた構造と一致した。MS(ES-):1917.5,1916.5,1915.6,959.2,958.4。
【0130】
図26Aおよび図26Bは、FR陽性KB腫瘍に対する、ならびにnu/nuマウスの体重(nu/nuマウスはKB腫瘍体積アッセイに使用した)に対する、3週間にわたる2μmol/kg TIWの実施例11および12の活性を示す。アッセイは方法実施例2および6にそれぞれ従って実施した。実施例11および12は、固形腫瘍の成長を阻害するが、マウスの体重にほとんど効果を持たない。
【0131】
〔実施例13〕
ポリプロピレン遠心分離ボトル内で、実施例9(56mg)を水7.5mLに溶解させて、アルゴンを10分間通気した。別のフラスコで0.1N NaHCO3溶液に10分間アルゴンを通気した。実施例9溶液のpHは、0.1N NaHCO3溶液を使用して6.9に調整した。テトラヒドロフラン(THF)7.5mL中の実施例5(44mg)を実施例9溶液にゆっくり添加した。得られた透明溶液をアルゴン存在下で15分間〜1時間撹拌した。反応の進行は、分析HPLC(10mM酢酸アンモニウム、pH=7.0、およびアセトニトリル)によって監視した。THFを蒸発させて、水溶液を濾過して、分取精製HPLCによって精製した。pH=7.0の1mMリン酸ナトリウムおよびアセトニトリルによる溶離は純粋な画分を生じ、この画分をプールして、周囲温度にて蒸発させて、得られた水溶液は0.1N HClを使用してpH4.0に調整した。実施例13は、48時間の凍結乾燥後に単離した(61mg、64%)。1H HMRスペクトルおよびLCMSデータは、割り当てられた構造と一致した。
【0132】
〔実施例14〜32〕
本明細書に記載するプロセスおよび条件に従って調製した。要求されたチオスルホナートまたはピリジルジチオ活性化ブンブラスチン、およびマレイミド活性化ビンブラスチン誘導体の調製についてのさらなる詳細は、米国特許出願公開第2005/0002942A1号に記載されている。
【0133】
〔実施例14〕
【0134】
〔実施例15〕
【0135】
図17は、実施例14(a)および15(b)の3H-チミジン取り込みに対する効果(最初の2本の棒グラフ;それぞれ100nMにて1時間、72時間追跡、n=2)と、葉酸塩受容体への結合について葉酸塩が実施例14および15と競合することを示し、結合体の結合特異性を証明する(後の2本の棒グラフ)。アッセイは方法実施例3に従って実施した。
【0136】
〔実施例16]
図18は、実施例16の3H-チミジン取り込みに対する効果(2時間パルス、48時間追跡、n=2)と、葉酸塩受容体への結合について葉酸塩が実施例16と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例3に従って実施した。
【0137】
〔実施例17〕
図19Aは、非結合ビンカの3H-チミジン取り込みに対する効果を示す。図19Bは、実施例17の3H-チミジン取り込みに対する効果を示す。アッセイは方法実施例3に従って実施した。
【0138】
〔実施例18〕
【0139】
〔実施例19〕
【0140】
図20A、20B、および20Cは、実施例7と比較した、葉酸塩対実施例18および19の相対結合親和性(図20)と、3H-チミジン取り込みに対するその効果(図20Bおよび20C)と、葉酸塩受容体への結合について葉酸塩が結合体と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例3(図20Bおよび20C)および方法実施例4(図20)に従って実施した。
【0141】
〔実施例20〕
図22は、3H-チミジン取り込みへの実施例20の効果と、葉酸塩受容体への結合について葉酸塩が結合体と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例3に従って実施した。
【0142】
〔実施例21〕
図23Aおよび図23Bは、葉酸塩対実施例21の相対結合親和性と、3H-チミジン取り込みへの実施例21の効果と、葉酸塩受容体への結合について葉酸塩が結合体と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例4および3にそれぞれ従って実施した。
【0143】
〔実施例22〕
図24Aおよび24Bは、葉酸塩対実施例22の相対結合親和性と、3H-チミジン取り込みへの実施例22の効果を示す。アッセイは方法実施例4および3にそれぞれ従って実施した。
【0144】
実施例21および22の実施例7に対する活性の比較
図25Aおよび図25Bは、Balb/cマウスのM109腫瘍に対する、(それぞれ3μmol/kgにて)、およびBalb/cマウスの体重(Balb/cマウスはM109腫瘍体積アッセイに使用した)に対する、14Bと比較した実施例21および22の活性を示す。アッセイは方法実施例1および6にそれぞれ従って実施した。実施例21、22および7は、固形腫瘍の成長を阻害するが、マウスの体重にほとんど効果を持たない。
【0145】
〔実施例23〕
図27Aおよび図27Bは、葉酸塩対実施例23の相対結合親和性と、3H-チミジン取り込みへの実施例23の効果と、結合体のIC50(15nM)と、葉酸塩受容体への結合について葉酸塩が結合体と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例4および3にそれぞれ従って実施した。
【0146】
〔実施例24〕
図28Aおよび図28Bは、葉酸塩対実施例24の相対結合親和性と、3H-チミジン取り込みへの実施例24の効果、結合体のIC50(9nM)と、葉酸塩受容体への結合について葉酸塩が結合体と競合することを示し、結合体の結合特異性を証明している。アッセイは方法実施例4および3にそれぞれ従って実施した。
【0147】
〔実施例25〕
C116H140N30O32S2;mol重量:2530.67;正確な質量:2528.97;C,55.05;H,5.58;N,16.60;O,20.23;S,2.53。
【0148】
〔実施例26〕
【0149】
〔実施例27〕
【0150】
〔実施例28〕
【0151】
〔実施例29〕
【0152】
〔実施例30〕
【0153】
〔実施例31〕
【0154】
〔実施例32〕
【0155】
次の表は、KB細胞への活性、葉酸塩受容体競合、および本明細書に記載する選択された薬物送達結合体への相対葉酸塩受容体親和性の要約である:
【図面の簡単な説明】
【0156】
【図1A】37℃、1時間における、葉酸受容体の実施例6(■、0.35)対葉酸(●、1.0)の相対結合親和性を示す。
【図1B】過剰な葉酸を含む(■)および含まない(●)場合の3H-チミジン取り込みに対する実施例6の活性を示す;実施例6のIC50=14nM。
【図2】未処置対照群(●)と対比した、Balb/cマウスのM109腫瘍に対して1.5μmol/kgで与えられたTIW(7回用量)の実施例6(■)の活性を示す。
【図3】未処置対照群(a)と対比した、FR陽性M109腫瘍に3週間にわたって10μmol/kgで与えられたTIWの実施例6(b)の活性を示す(垂直点線は最終の処置日を示す)。
【図4A】未処置対照群(■)と対比した、FR陽性M109腫瘍に対する3週間にわたる3μmol/kg、TIWでの実施例6(▲)の活性を示す。
【図4B】未処置対照群(a)と対比し、FR陰性4T-1腫瘍細胞に対する3週間にわたる3μmol/kg、TIWの実施例6(b)の活性がないことを示す。
【図5A】未処置対照群(■)と対比した、nu/nuマウスにおけるFR陽性KB腫瘍に対する3週間にわたる10μmol/kg、TIWの実施例6(●)の活性を示す。
【図5B】未処置対照群(■)と対比し、nu/nuマウスの体重に対する3週間にわたる10μmol/kgTIWの実施例6(●)による効果がないことを示す。
【図6A】未処置対照群(a)と対比した、nu/nuマウスにおけるFR陽性KB腫瘍に対する3週間にわたる(垂直点線は最終処置日を示した)1μmol/kg(b)、5μmol/kg(c)、および10μmol/kg(d)TIWの実施例6の活性を示す;t0の時点で平均腫瘍体積=50〜100mm3。
【図6B】未処置対照群(a)と対比し、nu/nuマウスの体重に対する3週間にわたる1μmol/kg(b)、5μmol/kg(c)、および10μmol/kg(d)TIWの実施例6による効果がないことを示す。
【図7A】未処置対照群(a)と対比した、nu/nuマウスにおける大型FR陽性KB腫瘍に対する3週間にわたる(垂直点線は最終処置日を示した)10μmol/kg TIWでの実施例6(b)の活性を示す;t0の時点で平均腫瘍体積=100〜150mm3。
【図7B】非結合デスアセチルビンブラスチンモノヒドラジド(b)と対比し、nu/nuマウスの体重に対する3週間にわたる(垂直点線は最終処置日を示した)10μmol/kg TIWの実施例6(a)による効果がないこと示す。
【図8】葉酸受容体における葉酸(●)と対比した、実施例7(b、0.2)の相対結合親和性を示す。
【図9A】過剰な葉酸を含む(a)および含まない(b)場合の、FR陽性KB細胞中への3H-チミジン取り込みに対する実施例7の活性を示す;実施例7のIC50=9nM。
【図9B】治療のパルス時間に対しての、過剰な葉酸を含む(a)および含まない(b)場合の、FR陽性KB細胞への3H-チミジン取り込みでの100nMにおける実施例7の活性に対するインキュベーション時間の効果を示す。
【図10A】治療のパルス時間に対しての、過剰な葉酸を含む(a)および含まない(b)場合の、48時間で収集された2002KB細胞への3H-チミジン取り込みでの10nMにおける実施例7の活性に対するインキュベーション時間の効果を示す。
【図10B】治療のパルス時間に対しての、過剰な葉酸を含む(a)および含まない(b)、48時間で収集された2002KB細胞への3H-チミジン取り込みでの100nMにおける実施例7の活性に対するインキュベーション時間の効果を示す。
【図11】未処置対照群(■)に対しての、FR陽性KB腫瘍に対する3週間にわたる5μmol/kg TIWの実施例7(▼)の活性を示す;t0の時点の平均腫瘍体積=50〜100mm3。
【図12】未処置対照群(a)と対比した、nu/nuマウスにおけるFR陽性KB腫瘍に対する3週間にわたる(垂直点線は最終処置日を示した)5μmol/kg TIWの実施例6および14B(それぞれ(b)および(c))の活性を示す;t0の時点で平均腫瘍体積=50〜80mm3;実施例7は5/5の完全寛解を示す。
【図13】未処置対照群(■)と対比した、Balb/cマウスのM109腫瘍に対する1.5μmol/kg TIWの実施例7(■)の活性を示す。
【図14A】未処置対照群(a)と対比した、Balb/cマウスのM109腫瘍に対する3週間にわたる(垂直点線は最終処置日を示した)それぞれ10μmol/kgでの実施例6および7(それぞれ(b)および(c))の活性を示す;t0の時点で平均腫瘍体積=50〜80mm3。
【図14B】Balb/cマウスの体重に対し、3週間にわたる(垂直点線は最終処置日を示した)(それぞれ10μmol/kgでの)実施例6および7(それぞれ(b)および(c))による効果がないことを示す。
【図15】未処置対照群(a)と対比した、40μmol/kg EC20(レニウム錯体)を含む(b)および(c)含まない場合のFR陽性KB腫瘍に対する2週間にわたる2μmol/kg TIWでの実施例7の活性を示す;実施例7のみが4/5の完全寛解を示した;実施例7+EC20は0/5の完全寛解を示した。
【図16A】未処置対照群(a)と対比した、nu/nuマウスにおけるFR陽性KB腫瘍に対する3週間にわたる5μmol/kg TIWの実施例6および7(それぞれ(b)および(c))の活性を示す。
【図16B】nu/nuマウスの体重に対する3週間にわたる(それぞれ5μmol/kgでの)実施例6および7(それぞれ(b)および(c))の効果がないことを示す。
【図17】FR陽性KB細胞への3H-チミジン取り込みに対する、実施例14(a)および15(b)単独(左側棒グラフ:それぞれ100nMにて1時間、72時間追跡、n=2)の、過剰な葉酸を含む同じ条件下での実施例14(a)および15(b)(右側棒グラフ)と対比した活性を示す。
【図18】KB細胞での3H-チミジン取り込みに対する実施例16の活性を示す;IC50は約250nMである。
【図19A】KB細胞での3H-チミジン取り込みに対する実施例5の活性を示す。
【図19B】KB細胞での3H-チミジン取り込みに対する実施例17の活性を示す。
【図20A】葉酸受容体における、葉酸(a、1.0)に対しての、実施例19(b、0.046)、実施例18(c、0.13)、および実施例7(d)の相対結合親和性を示す。
【図20B】過剰な葉酸を含む(b)および含まない(a)場合のKB細胞での3H-チミジン取り込みに対する実施例7の活性を示す;実施例7のIC50は約16nMである;そして過剰な葉酸を含む(d)および含まない(c)2002KB細胞での3H-チミジン取り込みに対する実施例19の活性を示す;実施例19のIC50は約100nMである。
【図20C】過剰な葉酸を含む(b)および含まない(a)2002KB細胞での3H-チミジン取り込みに対する実施例18の活性を示す;実施例18のIC50は約6nMである。
【図21A】葉酸受容体における実施例10(■、0.24)対葉酸(●、1.0)の相対結合親和性を示す。
【図21B】過剰な葉酸を含む(〇)および含まない(●)場合のKB細胞への3H-チミジン取り込みに対する実施例10の活性を示す;実施例10のIC50は約58nMである。
【図22】過剰な葉酸を含む(〇)および含まない(●)場合のKB細胞への3H-チミジン取り込みに対する実施例20の活性を示す;実施例20のIC50は約58nMである。
【図23A】葉酸受容体における実施例21(■、0.16)対葉酸(●、1.0)の相対結合親和性を示す。
【図23B】過剰な葉酸を含む(〇)および含まない(●)KB細胞への3H-チミジン取り込みに対する実施例21の活性を示す。
【図24A】葉酸受容体における実施例22(○、0.26)対葉酸(●、1.0)に対しての、の相対結合親和性を示す。
【図24B】過剰な葉酸を含む(〇)および含まない(●)KB細胞への3H-チミジン取り込みに対する実施例22の活性を示す。
【図25】Balb/cマウスのM109腫瘍に対して、およびBalb/cマウスの体重(Balb/cマウスはM109腫瘍体積アッセイに使用した)に対する、7と比較した実施例21および22の活性を示す(それぞれ3μmol/kgにて)。図25Aは、未処置対照群(■)と対比した、Balb/cマウスにおけるFR陽性M109腫瘍に対する3週間にわたるそれぞれ3μmol/kg TIWの実施例7(●)、実施例21(▲)、および実施例22(▼)の活性を示す。図25Bは、未処置対照群(■)と対比した、Balb/cマウスの体重に対する3週間にわたるそれぞれ3μmol/kg TIWでの実施例7(●)、実施例21(▲)、および実施例22(▼)による効果の非存在を示す。
【図26】FR陽性KB腫瘍に対する、およびnu/unマウスの体重(nu/unマウスはKB腫瘍体積アッセイに使用した)に対する、3週間にわたる2μmol/kg TIWにおける実施例11および12の活性を示す。図26Aは、未処置対照群(■)と対比した、nu/nuマウスにおけるFR陽性KB腫瘍に対する3週間にわたる2μmol/kg TIWの実施例11(●)および実施例12(▼)の活性を示す。図26Bは、未処置対照群(■)と対比し、nu/nuマウスの体重に対する3週間にわたる2μmol/kg TIWの実施例11(●)および実施例12(▼)による効果がないことを示す。
【図27】葉酸塩対実施例23の相対結合親和性、および3H-チミジン取り込みに対する実施例23の効果、結合体のIC50(15nM)と、葉酸塩が葉酸塩受容体への結合に際し結合体と競合することを示し、結合体の結合特異性を示している。アッセイは方法実施例4および3にそれぞれ従って実施した。図27Aは、葉酸受容体における実施例23(■、0.51)対葉酸(●、1.0)の相対結合親和性を示す。図27Bは、過剰な葉酸を含む(〇)および含まない(●)場合のKB細胞への3H-チミジン取り込みに対する実施例23の活性を示す;実施例23のIC50は約15nMである。
【図28】実施例24に対する葉酸塩の相対親和性効果、および3H-チミジン取り込みに対する実施例24の効果、結合体のIC50(9nM)と、葉酸塩が葉酸塩受容体への結合に際し結合体と競合することを示し、結合体の結合特異性を証明している。図28Aは、葉酸受容体における実施例24(■、0.45)対葉酸(●、1.0)の相対結合親和性を示す。図28Bは、過剰な葉酸を含む(〇)および含まない(●)KB細胞での3H-チミジン取り込みに対する実施例24の活性を示す;実施例24のIC50は約9nMである。
【特許請求の範囲】
【請求項1】
受容体結合薬物送達結合体であって:
(a)受容体結合部分と;
(b)2価リンカーと;
(c)ビンカアルカロイド、あるいはその類似体または誘導体と、を含み;
受容体結合部分は2価リンカーに共有結合され;
ビンカアルカロイド、あるいはその類似体または誘導体は、2価リンカーに共有結合され;かつ
2価リンカーが、スペーサーリンカー、放出型リンカー、およびヘテロ原子リンカー、ならびにその組合せから成る群より選択される1つ以上の構成要素を含む、
受容体結合薬物送達結合体。
【請求項2】
式:(B)-(L)-(D)を有するビタミン受容体結合薬物送達結合体であって、
式中、(L)は、(ls)a、(lH)b、および(lr)c、ならびにその組合せから成る群より選択され;
ここで(lr)は放出型リンカーであり、(ls)はスペーサーリンカーであり、かつ(lH)はヘテロ原子リンカーであり;
(B)はビタミン受容体結合部分であり、かつ(D)は薬物、あるいはその類似体または誘導体であり;かつ
a、b、およびcはそれぞれ独立して、0、1、2、3、または4である、
ビタミン受容体結合薬物送達結合体。
【請求項3】
式:(B)-(L)-(D)を有するビタミン受容体結合薬物送達結合体であって、
式中、(L)は、(ls)aおよび(lH)b、ならびにその組合せから成る群より選択され;
ここで(ls)はスペーサーリンカーであり、(lH)はヘテロ原子リンカーであり;
かつaおよびbはそれぞれ独立して、0、1、2、3、または4であり;かつ
(B)はビタミン受容体結合部分であり、かつ(D)はビンカアルカロイド、あるいはその類似体または誘導体である、
ビタミン受容体結合薬物送達結合体。
【請求項4】
2価リンカーが、ケタールを含む放出型リンカーを含む、請求項1、2、または3に記載の薬物送達結合体。
【請求項5】
少なくとも1個のスペーサーリンカーがペプチドである、請求項4に記載の薬物送達結合体。
【請求項6】
2価リンカーが、式:
の放出型リンカーを含み;
nが、1、2、3、および4から選択され;
Raが、アルキルまたは場合により置換されたアリールアルキルであり、Raが、水素または任意選択の置換基であり;かつ
(*)原子が、受容体結合部分、2価リンカー、あるいはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項7】
2価リンカーが、カーボネートを含む放出型リンカーを含む、請求項1、2、または3に記載の薬物送達結合体。
【請求項8】
少なくとも1個のスペーサーリンカーがペプチドである、請求項4に記載の薬物送達結合体。
【請求項9】
2価リンカーが、式:
の放出型リンカーを含み、
式中、nおよびmが、1、2、3、および4よりそれぞれ独立して選択される整数であり;かつ
(*)原子が、受容体結合部分、2価リンカー、あるいはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項10】
2価リンカーが、2価ジチオアルキルアミノ基または2価ジチオベンジルオキシカルボニル基を含む放出型リンカーを含む、請求項1、2、または3に記載の薬物送達結合体。
【請求項11】
少なくとも1個のスペーサーリンカーがペプチドである、請求項4に記載の薬物送達結合体。
【請求項12】
2価リンカーが、式:
の放出型リンカーを含み、
式中、nが、1、2、3、および4から選択され;かつ
(*)原子が、受容体結合部分、2価リンカー、あるいはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項13】
2価リンカーが、式:
の放出型リンカーを含み、
式中、Rが、水素または任意の置換基であり;かつ
(*)原子が、受容体結合部分、2価リンカー、あるいはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項14】
2価リンカーが、式:
の放出型リンカーを含み、
式中、Rが、水素、アルキル、アルコキシ、シアノ、またはニトロであり;かつ
(*)原子が、受容体結合部分、2価リンカー、あるいはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項15】
2価リンカーが、式:
の放出型リンカーを含み、
式中、Rが、水素、アルキル、アルコキシ、シアノ、またはニトロであり;かつ
(*)原子が、受容体結合部分、2価リンカー、あるいはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項16】
ビンカアルカロイド、あるいはその類似体または誘導体が、窒素を介して2価リンカーに結合されたカルボキサミドを含む、請求項1、2、または3に記載の薬物送達結合体。
【請求項17】
少なくとも1個のスペーサーリンカーがペプチドである、請求項4に記載の薬物送達結合体。
【請求項18】
ビンカアルカロイド、あるいはその類似体または誘導体が、酸素を介して2価リンカーに結合されたカルボキシラートを含む、請求項1、2、または3に記載の薬物送達結合体。
【請求項19】
少なくとも1個のスペーサーリンカーがペプチドである、請求項4に記載の薬物送達結合体。
【請求項20】
2価リンカーが、ジスルフィドでない少なくとも1個の放出型リンカーを含む、請求項1、2、または3に記載の薬物送達結合体。
【請求項21】
ヘテロ原子リンカーが窒素、酸素、または硫黄原子、あるいは-NHR1NHR2-、-SO-、-S(O)2-、および-NR3O-から成る式の群より選択され、ここでR1、R2、およびR3が、水素、アルキル、アリール、アリールアルキル、置換アリール、置換アリールアルキル、ヘテロアリール、置換ヘテロアリール、およびアルコキシアルキルから成る群よりそれぞれ独立して選択される、請求項1、2、または3に記載の薬物送達結合体。
【請求項22】
スペーサーリンカーが、カルボニル、チオノカルボニル、アルキレン、シクロアルキレン、アルキレンシクロアルキル、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、1-アルキレンスクシンイミド-3-イル、1-(カルボニルアルキル)スクシンイミド-3-イル、アルキレンスルホキシル、スルホニルアルキル、アルキレンスルホキシアルキル、アルキレンスルホニルアルキル、カルボニルテトラヒドロ-2H-ピラニル、カルボニルテトラヒドロフラニル、1-(カルボニルテトラヒドロ-2H-ピラニル)スクシンイミド-3-イル、および1-(カルボニルテトラヒドロフラニル)スクシンイミド-3-イルから成る群より選択され、ここで前記スペーサーリンカーのそれぞれが1個以上の置換基X1によって場合により置換され;
ここで各置換基X1は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、置換アリール、アリールアルキル、置換アリールアルキル、ヘテロアリール、置換ヘテロアリール、カルボキシ、カルボキシアルキル、アルキルカルボキシラート、アルキルアルカノアート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、およびR7-アシルアミノアルキルから成る群より独立して選択され、ここでR4およびR5は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択され、ここでR6およびR7は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項23】
ヘテロ原子リンカーが窒素であり、ここで置換基X1およびヘテロ原子リンカーが、それらが結合するスペーサーリンカーとひとまとめにされて複素環を形成する、請求項22に記載の薬物送達結合体。
【請求項24】
複素環がピロリジン、ピペリジン、オキサゾリジン、イソオキサゾリジン、チアゾリジン、イソチアゾリジン、ピロリジノン、ピペリジノン、オキサゾリジノン、イソオキサゾリジノン、チアゾリジノン、イソチアゾリジノン、およびスクシンイミドから成る群より選択される、請求項23に記載の薬物送達結合体。
【請求項25】
放出型リンカーが、メチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、1-アルコキシシクロアルキレンカルボニル、カルボニルアリールカルボニル、カルボニル(カルボキシアリール)カルボニル、カルボニル(ビスカルボキシアリール)カルボニル、ハロアルキレンカルボニル、アルキレン(ジアルキルシリル)、アルキレン(アルキルアリールシリル)、アルキレン(ジアリールシリル)、(ジアルキルシリル)アリール、(アルキルアリールシリル)アリール、(ジアリールシリル)アリール、オキシカルボニルオキシ、オキシカルボニルオキシアルキル、スルホニルアルキル、イミノアルキリデニル、カルボニルアルキリデンイミニル、イミノシクロアルキリデニル、カルボニルシクロアルキリデンイミニル、アルキレンスルホニル、アルキレンチオ、アルキレンアリールチオ、およびカルボニルアルキルチオから成る群より選択され、ここで前記放出型リンカーのそれぞれが1個以上の置換基X2によって場合により置換され;
ここで各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、置換アリール、アリールアルキル、置換アリールアルキル、ヘテロアリール、置換ヘテロアリール、カルボキシ、カルボキシアルキル、アルキルカルボキシラート、アルキルアルカノアート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、およびR7-アシルアミノアルキルから成る群より独立して選択され、ここでR4およびR5は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択され、かつここでR6およびR7は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項26】
ヘテロ原子リンカーが窒素であり、ここで置換基X2およびヘテロ原子リンカーが、それらが結合する放出型リンカーとひとまとめにされて複素環を形成する、請求項25に記載の薬物送達結合体。
【請求項27】
複素環がピロリジン、ピペリジン、オキサゾリジン、イソオキサゾリジン、チアゾリジン、イソチアゾリジン、ピロリジノン、ピペリジノン、オキサゾリジノン、イソオキサゾリジノン、チアゾリジノン、イソチアゾリジノン、およびスクシンイミドから成る群より選択される、請求項26に記載の薬物送達結合体。
【請求項28】
ヘテロ原子リンカーが窒素であり、放出型リンカーおよびヘテロ原子リンカーがひとまとめにされてアルキレンアジリジン-1-イル、アルキレンカルボニルアジリジン-1-イル、カルボニルアルキルアジリジン-1-イル、アルキレンスルホキシルアジリジン-1-イル、スルホキシアルキルアジリジン-1-イル、スルホニルアルキルアジリジン-1-イル、またはアルキレンスルホニルアジリジン-1-イルを含む2価基を形成し、前記放出型リンカーのそれぞれが1個以上の置換基X2によって場合により置換され;
ここで各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、置換アリール、アリールアルキル、置換アリールアルキル、ヘテロアリール、置換ヘテロアリール、カルボキシ、カルボキシアルキル、アルキルカルボキシラート、アルキルアルカノアート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、およびR7-アシルアミノアルキルから成る群より独立して選択され、ここでR4およびR5は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択され、かつここでR6およびR7は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項29】
ヘテロ原子リンカーが窒素であり、かつ放出型リンカーおよびヘテロ原子リンカーがひとまとめにされてアルキレンアジリジン-1-イル、カルボニルアルキルアジリジン-1-イル、スルホキシルアルキルアジリジン-1-イル、またはスルホニルアルキルアジリジン-1-イルを含む2価基を形成する、請求項28に記載の薬物送達結合体。
【請求項30】
スペーサーリンカーが、カルボニル、チオノカルボニル、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、および1-(カルボニルアルキル)スクシンイミド-3-イルから成る群より選択され、ここで前記スペーサーリンカーそれぞれが1個以上の置換基X1によって場合により置換され;
ここで各置換基X1は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、置換アリール、アリールアルキル、置換アリールアルキル、ヘテロアリール、置換ヘテロアリール、カルボキシ、カルボキシアルキル、アルキルカルボキシラート、アルキルアルカノアート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、およびR7-アシルアミノアルキルから成る群より独立して選択され、ここでR4およびR5は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択され、かつここでR6およびR7は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択され;
かつここでスペーサーリンカーが放出型リンカーに結合されて、アジリジンアミドを形成する、
請求項1、2、または3に記載の薬物送達結合体。
【請求項31】
ビンカアルカロイドがビンブラスチン、デスアセチルビンブラスチン、ビンデシン、またはチオビンデシンである、請求項1、2、または3に記載の薬物送達結合体。
【請求項32】
ビンカアルカロイドが二重結合窒素原子を含むその誘導体であり、放出型リンカーがアルキレンカルボニルアミノおよび1-(アルキレンカルボニルアミノ)スクシンイミド-3-イルから成る群より選択され、かつ放出型リンカーが薬物窒素に結合されて、ヒドラゾンを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項33】
ビンカアルカロイドが硫黄原子を含むその誘導体であり、放出型リンカーがアルキレンチオおよびカルボニルアルキルチオから成る群より選択され、かつ放出型リンカーが薬物硫黄に結合されて、ジスルフィドを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項34】
ヘテロ原子リンカーが酸素であり、放出型リンカーがアルキレン(ジアルキルシリル)、アルキレン(アルキルアリールシリル)、アルキレン(ジアリールシリル)、(ジアルキルシリル)アリール、(アルキルアリールシリル)アリールおよび(ジアリールシリル)アリールから成る群より選択され、前記放出型リンカーのそれぞれが1個以上の置換基X2によって場合により置換され、かつ放出型リンカーが酸素に結合されて、シラノールを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項35】
ヘテロ原子リンカーが酸素であり、スペーサーリンカーが1個以上の置換基X1によって場合により置換された1-アルキレンスクシンイミド-3-イルであり、かつ放出型リンカーがメチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、1-アルコキシシクロアルキレンカルボニルから成る群より選択され、前記放出型リンカーのそれぞれが1個以上の置換基X2によって場合により置換され;
ここで各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、置換アリール、アリールアルキル、置換アリールアルキル、ヘテロアリール、置換ヘテロアリール、カルボキシ、カルボキシアルキル、アルキルカルボキシラート、アルキルアルカノアート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、およびR7-アシルアミノアルキルから成る群より独立して選択され、ここでR4およびR5は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択され、かつここでR6およびR7は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択され;
かつここでスペーサーリンカーおよび放出型リンカーがそれぞれヘテロ原子リンカーに結合されて、スクシンイミド-1-イルアルキルアセタールまたはケタールを形成する、
請求項1、2、または3に記載の薬物送達結合体。
【請求項36】
2価リンカーがヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、メチルがアルキルまたは置換アリールによって場合により置換される3-チオスクシンイミド-1-イルアルコキシメチルオキシを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項37】
2価リンカーがヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、カルボニルが薬物、あるいはその類似体または誘導体と共にアシルアジリジンを形成する3-チオスクシンイミド-1-イルアルキルカルボニルを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項38】
2価リンカーがヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて1-アルコキシシクロアルキルレンオキシを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項39】
2価リンカーがスペーサーリンカー、ヘテロ原子リンカー、および放出型リンカーを含み、これらがひとまとめにされてアルキレンアミノカルボニル(ジカルボキシルアリーレン)カルボキシラートを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項40】
2価リンカーがヘテロ原子リンカー、スペーサーリンカー、ヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、2置換シリルがアルキルまたは場合により置換アリールによって置換される3-チオアルキルスルホニルアルキル(2置換シリル)オキシを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項41】
2価リンカーが、天然型アミノ酸およびその立体異性体から成る群より選択される複数のスペーサーリンカーを含む、請求項1に記載の薬物送達結合体。
【請求項42】
2価リンカーが放出型リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、カルボニルが薬物、あるいはその類似体または誘導体と共にカーボネートを形成する3-ジチオアルキルオキシカルボニルを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項43】
2価リンカーが放出型リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、カルボニルが薬物、あるいはその類似体または誘導体と共にカーボネートを形成しかつアリールが場合により置換される3-ジチオアリールアルキルオキシカルボニルを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項44】
2価リンカーが、放出型リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、アミノがビンカアルカロイド、あるいはその類似体または誘導体と共にビニル性アミドを形成する3-ジチオアルキルアミノを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項45】
アルキルがエチルである、請求項44に記載の薬物送達結合体。
【請求項46】
2価リンカーが放出型リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、カルボニルがビンカアルカロイド、あるいはその類似体または誘導体と共にカルバメートを形成する3-ジチオアルキルアミノカルボニルを形成する、請求項1に記載の薬物送達結合体。
【請求項47】
アルキルがエチルである、請求項46に記載の薬物送達結合体。
【請求項48】
2価リンカーが放出型リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、カルボニルがビンカアルカロイド、あるいはその類似体または誘導体と共にカルバメートまたはカルバモイルアジリジンを形成する、3-ジチオアリールアルキルオキシカルボニルを形成する、請求項1に記載の薬物送達結合体。
【請求項49】
請求項1、2、または3に記載の薬物送達結合体、およびそのための製薬的に許容される担体、希釈剤、または賦形剤を含む製薬組成物。
【請求項50】
病原性細胞群を内部に持つ宿主動物において病原性細胞群を排除する方法であって、病原性細胞集団のメンバーがビタミン、あるいはその類似体または誘導体に対して接近可能な結合部位を有し、結合部位が病原性細胞によって特異的に発現、過剰発現または優先的に発現され、前記宿主に請求項1、2、または3に記載の薬物送達結合体、あるいはその製薬組成物を投与する工程を含む、方法。
【請求項1】
受容体結合薬物送達結合体であって:
(a)受容体結合部分と;
(b)2価リンカーと;
(c)ビンカアルカロイド、あるいはその類似体または誘導体と、を含み;
受容体結合部分は2価リンカーに共有結合され;
ビンカアルカロイド、あるいはその類似体または誘導体は、2価リンカーに共有結合され;かつ
2価リンカーが、スペーサーリンカー、放出型リンカー、およびヘテロ原子リンカー、ならびにその組合せから成る群より選択される1つ以上の構成要素を含む、
受容体結合薬物送達結合体。
【請求項2】
式:(B)-(L)-(D)を有するビタミン受容体結合薬物送達結合体であって、
式中、(L)は、(ls)a、(lH)b、および(lr)c、ならびにその組合せから成る群より選択され;
ここで(lr)は放出型リンカーであり、(ls)はスペーサーリンカーであり、かつ(lH)はヘテロ原子リンカーであり;
(B)はビタミン受容体結合部分であり、かつ(D)は薬物、あるいはその類似体または誘導体であり;かつ
a、b、およびcはそれぞれ独立して、0、1、2、3、または4である、
ビタミン受容体結合薬物送達結合体。
【請求項3】
式:(B)-(L)-(D)を有するビタミン受容体結合薬物送達結合体であって、
式中、(L)は、(ls)aおよび(lH)b、ならびにその組合せから成る群より選択され;
ここで(ls)はスペーサーリンカーであり、(lH)はヘテロ原子リンカーであり;
かつaおよびbはそれぞれ独立して、0、1、2、3、または4であり;かつ
(B)はビタミン受容体結合部分であり、かつ(D)はビンカアルカロイド、あるいはその類似体または誘導体である、
ビタミン受容体結合薬物送達結合体。
【請求項4】
2価リンカーが、ケタールを含む放出型リンカーを含む、請求項1、2、または3に記載の薬物送達結合体。
【請求項5】
少なくとも1個のスペーサーリンカーがペプチドである、請求項4に記載の薬物送達結合体。
【請求項6】
2価リンカーが、式:
の放出型リンカーを含み;
nが、1、2、3、および4から選択され;
Raが、アルキルまたは場合により置換されたアリールアルキルであり、Raが、水素または任意選択の置換基であり;かつ
(*)原子が、受容体結合部分、2価リンカー、あるいはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項7】
2価リンカーが、カーボネートを含む放出型リンカーを含む、請求項1、2、または3に記載の薬物送達結合体。
【請求項8】
少なくとも1個のスペーサーリンカーがペプチドである、請求項4に記載の薬物送達結合体。
【請求項9】
2価リンカーが、式:
の放出型リンカーを含み、
式中、nおよびmが、1、2、3、および4よりそれぞれ独立して選択される整数であり;かつ
(*)原子が、受容体結合部分、2価リンカー、あるいはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項10】
2価リンカーが、2価ジチオアルキルアミノ基または2価ジチオベンジルオキシカルボニル基を含む放出型リンカーを含む、請求項1、2、または3に記載の薬物送達結合体。
【請求項11】
少なくとも1個のスペーサーリンカーがペプチドである、請求項4に記載の薬物送達結合体。
【請求項12】
2価リンカーが、式:
の放出型リンカーを含み、
式中、nが、1、2、3、および4から選択され;かつ
(*)原子が、受容体結合部分、2価リンカー、あるいはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項13】
2価リンカーが、式:
の放出型リンカーを含み、
式中、Rが、水素または任意の置換基であり;かつ
(*)原子が、受容体結合部分、2価リンカー、あるいはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項14】
2価リンカーが、式:
の放出型リンカーを含み、
式中、Rが、水素、アルキル、アルコキシ、シアノ、またはニトロであり;かつ
(*)原子が、受容体結合部分、2価リンカー、あるいはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項15】
2価リンカーが、式:
の放出型リンカーを含み、
式中、Rが、水素、アルキル、アルコキシ、シアノ、またはニトロであり;かつ
(*)原子が、受容体結合部分、2価リンカー、あるいはビンカアルカロイド、あるいはその類似体または誘導体にそれぞれ結合される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項16】
ビンカアルカロイド、あるいはその類似体または誘導体が、窒素を介して2価リンカーに結合されたカルボキサミドを含む、請求項1、2、または3に記載の薬物送達結合体。
【請求項17】
少なくとも1個のスペーサーリンカーがペプチドである、請求項4に記載の薬物送達結合体。
【請求項18】
ビンカアルカロイド、あるいはその類似体または誘導体が、酸素を介して2価リンカーに結合されたカルボキシラートを含む、請求項1、2、または3に記載の薬物送達結合体。
【請求項19】
少なくとも1個のスペーサーリンカーがペプチドである、請求項4に記載の薬物送達結合体。
【請求項20】
2価リンカーが、ジスルフィドでない少なくとも1個の放出型リンカーを含む、請求項1、2、または3に記載の薬物送達結合体。
【請求項21】
ヘテロ原子リンカーが窒素、酸素、または硫黄原子、あるいは-NHR1NHR2-、-SO-、-S(O)2-、および-NR3O-から成る式の群より選択され、ここでR1、R2、およびR3が、水素、アルキル、アリール、アリールアルキル、置換アリール、置換アリールアルキル、ヘテロアリール、置換ヘテロアリール、およびアルコキシアルキルから成る群よりそれぞれ独立して選択される、請求項1、2、または3に記載の薬物送達結合体。
【請求項22】
スペーサーリンカーが、カルボニル、チオノカルボニル、アルキレン、シクロアルキレン、アルキレンシクロアルキル、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、1-アルキレンスクシンイミド-3-イル、1-(カルボニルアルキル)スクシンイミド-3-イル、アルキレンスルホキシル、スルホニルアルキル、アルキレンスルホキシアルキル、アルキレンスルホニルアルキル、カルボニルテトラヒドロ-2H-ピラニル、カルボニルテトラヒドロフラニル、1-(カルボニルテトラヒドロ-2H-ピラニル)スクシンイミド-3-イル、および1-(カルボニルテトラヒドロフラニル)スクシンイミド-3-イルから成る群より選択され、ここで前記スペーサーリンカーのそれぞれが1個以上の置換基X1によって場合により置換され;
ここで各置換基X1は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、置換アリール、アリールアルキル、置換アリールアルキル、ヘテロアリール、置換ヘテロアリール、カルボキシ、カルボキシアルキル、アルキルカルボキシラート、アルキルアルカノアート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、およびR7-アシルアミノアルキルから成る群より独立して選択され、ここでR4およびR5は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択され、ここでR6およびR7は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項23】
ヘテロ原子リンカーが窒素であり、ここで置換基X1およびヘテロ原子リンカーが、それらが結合するスペーサーリンカーとひとまとめにされて複素環を形成する、請求項22に記載の薬物送達結合体。
【請求項24】
複素環がピロリジン、ピペリジン、オキサゾリジン、イソオキサゾリジン、チアゾリジン、イソチアゾリジン、ピロリジノン、ピペリジノン、オキサゾリジノン、イソオキサゾリジノン、チアゾリジノン、イソチアゾリジノン、およびスクシンイミドから成る群より選択される、請求項23に記載の薬物送達結合体。
【請求項25】
放出型リンカーが、メチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、1-アルコキシシクロアルキレンカルボニル、カルボニルアリールカルボニル、カルボニル(カルボキシアリール)カルボニル、カルボニル(ビスカルボキシアリール)カルボニル、ハロアルキレンカルボニル、アルキレン(ジアルキルシリル)、アルキレン(アルキルアリールシリル)、アルキレン(ジアリールシリル)、(ジアルキルシリル)アリール、(アルキルアリールシリル)アリール、(ジアリールシリル)アリール、オキシカルボニルオキシ、オキシカルボニルオキシアルキル、スルホニルアルキル、イミノアルキリデニル、カルボニルアルキリデンイミニル、イミノシクロアルキリデニル、カルボニルシクロアルキリデンイミニル、アルキレンスルホニル、アルキレンチオ、アルキレンアリールチオ、およびカルボニルアルキルチオから成る群より選択され、ここで前記放出型リンカーのそれぞれが1個以上の置換基X2によって場合により置換され;
ここで各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、置換アリール、アリールアルキル、置換アリールアルキル、ヘテロアリール、置換ヘテロアリール、カルボキシ、カルボキシアルキル、アルキルカルボキシラート、アルキルアルカノアート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、およびR7-アシルアミノアルキルから成る群より独立して選択され、ここでR4およびR5は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択され、かつここでR6およびR7は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項26】
ヘテロ原子リンカーが窒素であり、ここで置換基X2およびヘテロ原子リンカーが、それらが結合する放出型リンカーとひとまとめにされて複素環を形成する、請求項25に記載の薬物送達結合体。
【請求項27】
複素環がピロリジン、ピペリジン、オキサゾリジン、イソオキサゾリジン、チアゾリジン、イソチアゾリジン、ピロリジノン、ピペリジノン、オキサゾリジノン、イソオキサゾリジノン、チアゾリジノン、イソチアゾリジノン、およびスクシンイミドから成る群より選択される、請求項26に記載の薬物送達結合体。
【請求項28】
ヘテロ原子リンカーが窒素であり、放出型リンカーおよびヘテロ原子リンカーがひとまとめにされてアルキレンアジリジン-1-イル、アルキレンカルボニルアジリジン-1-イル、カルボニルアルキルアジリジン-1-イル、アルキレンスルホキシルアジリジン-1-イル、スルホキシアルキルアジリジン-1-イル、スルホニルアルキルアジリジン-1-イル、またはアルキレンスルホニルアジリジン-1-イルを含む2価基を形成し、前記放出型リンカーのそれぞれが1個以上の置換基X2によって場合により置換され;
ここで各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、置換アリール、アリールアルキル、置換アリールアルキル、ヘテロアリール、置換ヘテロアリール、カルボキシ、カルボキシアルキル、アルキルカルボキシラート、アルキルアルカノアート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、およびR7-アシルアミノアルキルから成る群より独立して選択され、ここでR4およびR5は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択され、かつここでR6およびR7は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択される、
請求項1、2、または3に記載の薬物送達結合体。
【請求項29】
ヘテロ原子リンカーが窒素であり、かつ放出型リンカーおよびヘテロ原子リンカーがひとまとめにされてアルキレンアジリジン-1-イル、カルボニルアルキルアジリジン-1-イル、スルホキシルアルキルアジリジン-1-イル、またはスルホニルアルキルアジリジン-1-イルを含む2価基を形成する、請求項28に記載の薬物送達結合体。
【請求項30】
スペーサーリンカーが、カルボニル、チオノカルボニル、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、および1-(カルボニルアルキル)スクシンイミド-3-イルから成る群より選択され、ここで前記スペーサーリンカーそれぞれが1個以上の置換基X1によって場合により置換され;
ここで各置換基X1は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、置換アリール、アリールアルキル、置換アリールアルキル、ヘテロアリール、置換ヘテロアリール、カルボキシ、カルボキシアルキル、アルキルカルボキシラート、アルキルアルカノアート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、およびR7-アシルアミノアルキルから成る群より独立して選択され、ここでR4およびR5は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択され、かつここでR6およびR7は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択され;
かつここでスペーサーリンカーが放出型リンカーに結合されて、アジリジンアミドを形成する、
請求項1、2、または3に記載の薬物送達結合体。
【請求項31】
ビンカアルカロイドがビンブラスチン、デスアセチルビンブラスチン、ビンデシン、またはチオビンデシンである、請求項1、2、または3に記載の薬物送達結合体。
【請求項32】
ビンカアルカロイドが二重結合窒素原子を含むその誘導体であり、放出型リンカーがアルキレンカルボニルアミノおよび1-(アルキレンカルボニルアミノ)スクシンイミド-3-イルから成る群より選択され、かつ放出型リンカーが薬物窒素に結合されて、ヒドラゾンを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項33】
ビンカアルカロイドが硫黄原子を含むその誘導体であり、放出型リンカーがアルキレンチオおよびカルボニルアルキルチオから成る群より選択され、かつ放出型リンカーが薬物硫黄に結合されて、ジスルフィドを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項34】
ヘテロ原子リンカーが酸素であり、放出型リンカーがアルキレン(ジアルキルシリル)、アルキレン(アルキルアリールシリル)、アルキレン(ジアリールシリル)、(ジアルキルシリル)アリール、(アルキルアリールシリル)アリールおよび(ジアリールシリル)アリールから成る群より選択され、前記放出型リンカーのそれぞれが1個以上の置換基X2によって場合により置換され、かつ放出型リンカーが酸素に結合されて、シラノールを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項35】
ヘテロ原子リンカーが酸素であり、スペーサーリンカーが1個以上の置換基X1によって場合により置換された1-アルキレンスクシンイミド-3-イルであり、かつ放出型リンカーがメチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、1-アルコキシシクロアルキレンカルボニルから成る群より選択され、前記放出型リンカーのそれぞれが1個以上の置換基X2によって場合により置換され;
ここで各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、置換アリール、アリールアルキル、置換アリールアルキル、ヘテロアリール、置換ヘテロアリール、カルボキシ、カルボキシアルキル、アルキルカルボキシラート、アルキルアルカノアート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、およびR7-アシルアミノアルキルから成る群より独立して選択され、ここでR4およびR5は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択され、かつここでR6およびR7は、アミノ酸、アミノ酸誘導体、およびペプチドから成る群よりそれぞれ独立して選択され;
かつここでスペーサーリンカーおよび放出型リンカーがそれぞれヘテロ原子リンカーに結合されて、スクシンイミド-1-イルアルキルアセタールまたはケタールを形成する、
請求項1、2、または3に記載の薬物送達結合体。
【請求項36】
2価リンカーがヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、メチルがアルキルまたは置換アリールによって場合により置換される3-チオスクシンイミド-1-イルアルコキシメチルオキシを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項37】
2価リンカーがヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、カルボニルが薬物、あるいはその類似体または誘導体と共にアシルアジリジンを形成する3-チオスクシンイミド-1-イルアルキルカルボニルを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項38】
2価リンカーがヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて1-アルコキシシクロアルキルレンオキシを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項39】
2価リンカーがスペーサーリンカー、ヘテロ原子リンカー、および放出型リンカーを含み、これらがひとまとめにされてアルキレンアミノカルボニル(ジカルボキシルアリーレン)カルボキシラートを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項40】
2価リンカーがヘテロ原子リンカー、スペーサーリンカー、ヘテロ原子リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、2置換シリルがアルキルまたは場合により置換アリールによって置換される3-チオアルキルスルホニルアルキル(2置換シリル)オキシを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項41】
2価リンカーが、天然型アミノ酸およびその立体異性体から成る群より選択される複数のスペーサーリンカーを含む、請求項1に記載の薬物送達結合体。
【請求項42】
2価リンカーが放出型リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、カルボニルが薬物、あるいはその類似体または誘導体と共にカーボネートを形成する3-ジチオアルキルオキシカルボニルを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項43】
2価リンカーが放出型リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、カルボニルが薬物、あるいはその類似体または誘導体と共にカーボネートを形成しかつアリールが場合により置換される3-ジチオアリールアルキルオキシカルボニルを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項44】
2価リンカーが、放出型リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、アミノがビンカアルカロイド、あるいはその類似体または誘導体と共にビニル性アミドを形成する3-ジチオアルキルアミノを形成する、請求項1、2、または3に記載の薬物送達結合体。
【請求項45】
アルキルがエチルである、請求項44に記載の薬物送達結合体。
【請求項46】
2価リンカーが放出型リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、カルボニルがビンカアルカロイド、あるいはその類似体または誘導体と共にカルバメートを形成する3-ジチオアルキルアミノカルボニルを形成する、請求項1に記載の薬物送達結合体。
【請求項47】
アルキルがエチルである、請求項46に記載の薬物送達結合体。
【請求項48】
2価リンカーが放出型リンカー、スペーサーリンカー、および放出型リンカーを含み、これらがひとまとめにされて、カルボニルがビンカアルカロイド、あるいはその類似体または誘導体と共にカルバメートまたはカルバモイルアジリジンを形成する、3-ジチオアリールアルキルオキシカルボニルを形成する、請求項1に記載の薬物送達結合体。
【請求項49】
請求項1、2、または3に記載の薬物送達結合体、およびそのための製薬的に許容される担体、希釈剤、または賦形剤を含む製薬組成物。
【請求項50】
病原性細胞群を内部に持つ宿主動物において病原性細胞群を排除する方法であって、病原性細胞集団のメンバーがビタミン、あるいはその類似体または誘導体に対して接近可能な結合部位を有し、結合部位が病原性細胞によって特異的に発現、過剰発現または優先的に発現され、前記宿主に請求項1、2、または3に記載の薬物送達結合体、あるいはその製薬組成物を投与する工程を含む、方法。
【図1A】

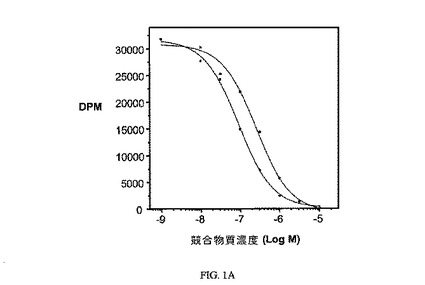
【図1B】
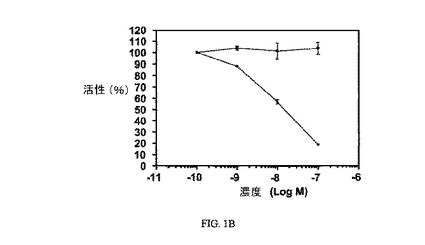
【図2】
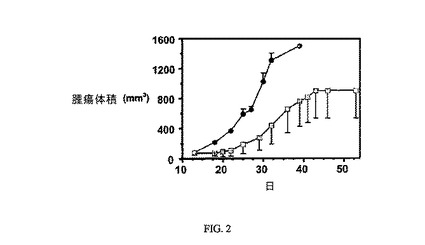
【図3】
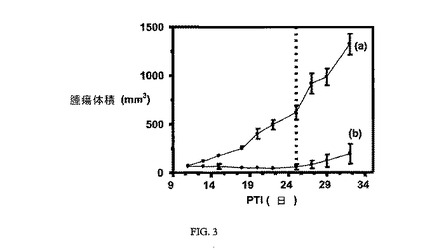
【図4A】
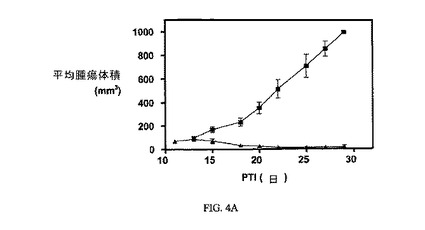
【図4B】
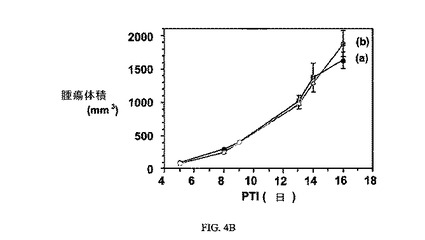
【図5A】
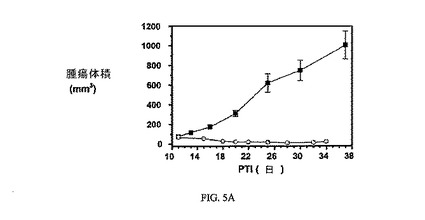
【図5B】
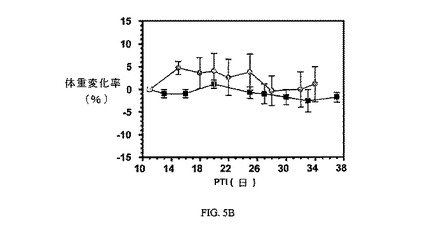
【図6A】
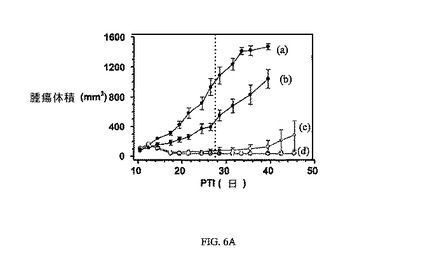
【図6B】
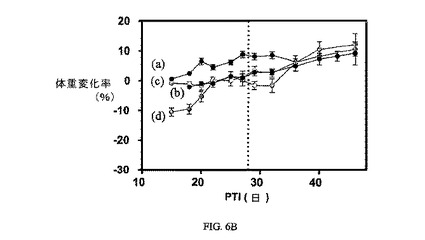
【図7A】
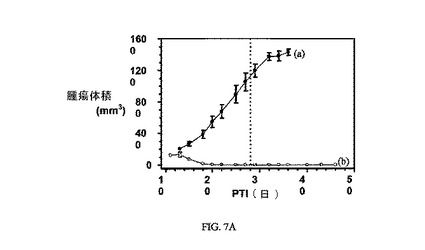
【図7B】
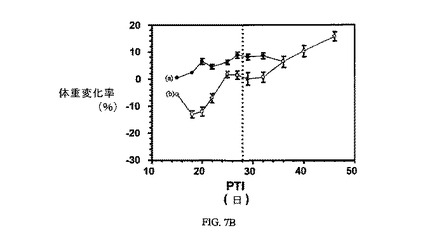
【図8】
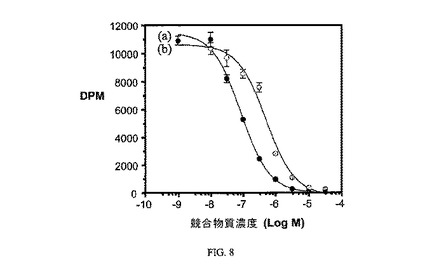
【図9A】
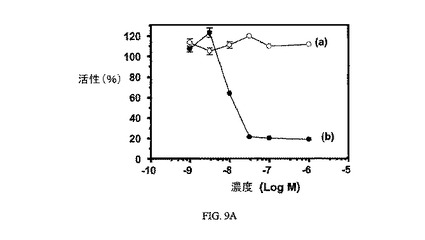
【図9B】
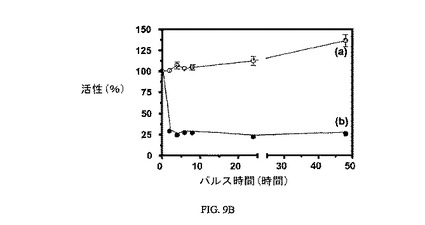
【図10A】
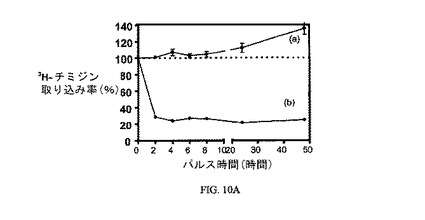
【図10B】
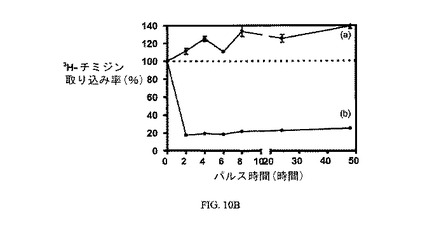
【図11】
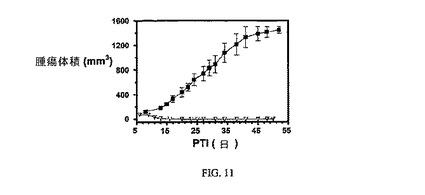
【図12】
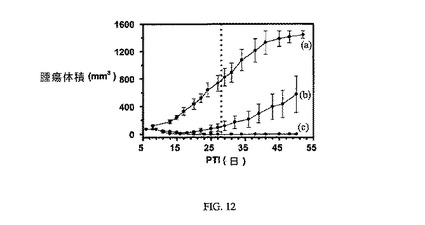
【図13】
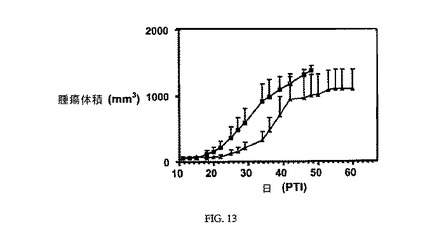
【図14A】
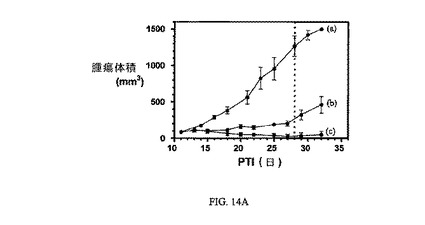
【図14B】
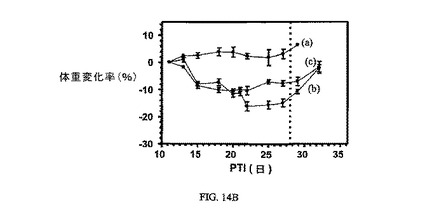
【図15】
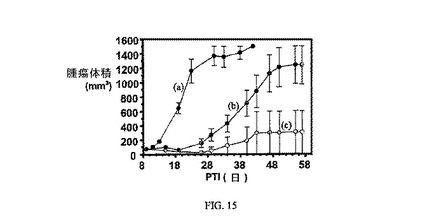
【図16A】
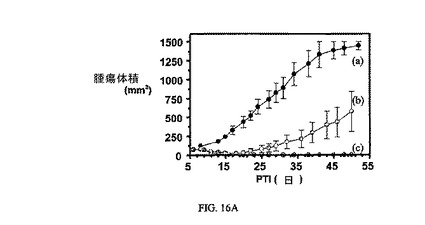
【図16B】
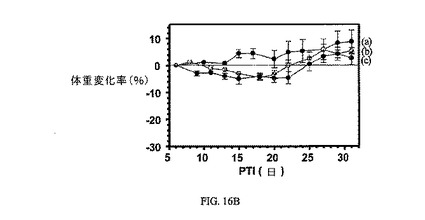
【図17】
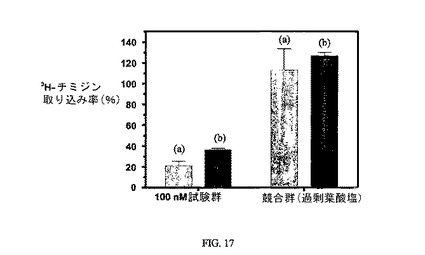
【図18】
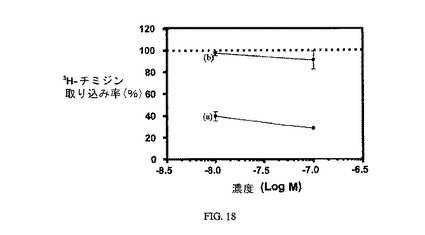
【図19A】
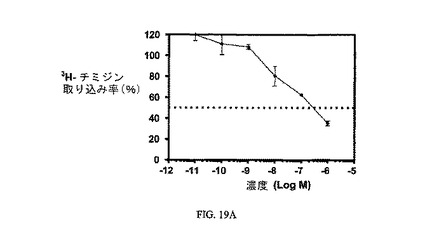
【図19B】
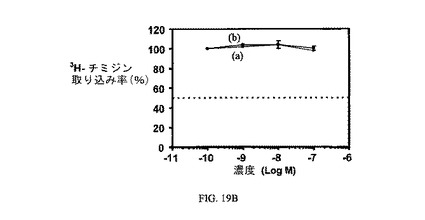
【図20A】
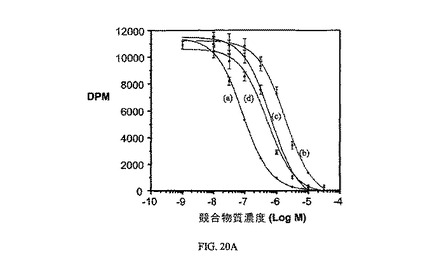
【図20B】
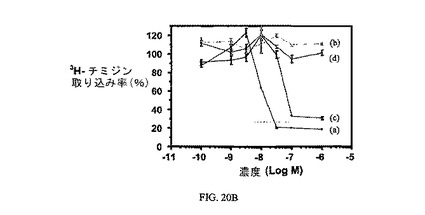
【図20C】
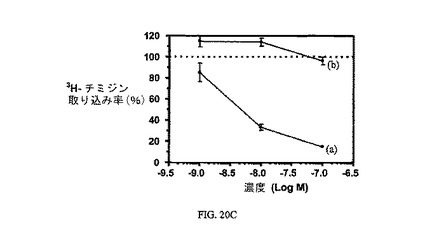
【図21A】
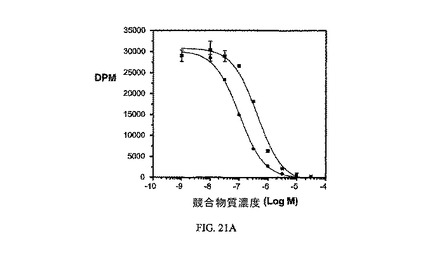
【図21B】
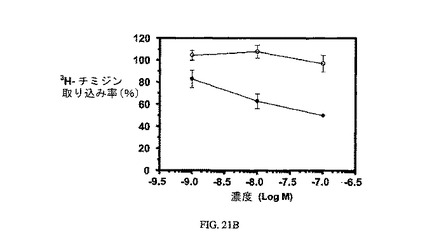
【図22】
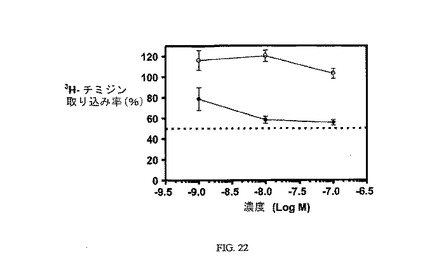
【図23A】
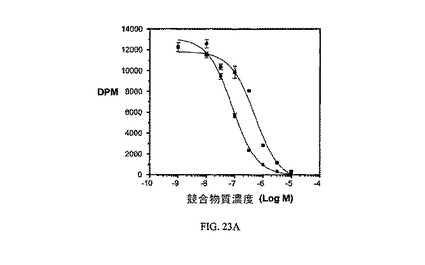
【図23B】
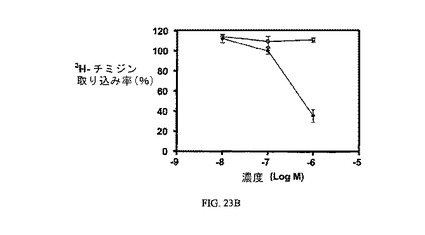
【図24A】
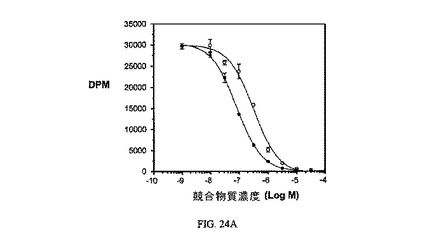
【図24B】
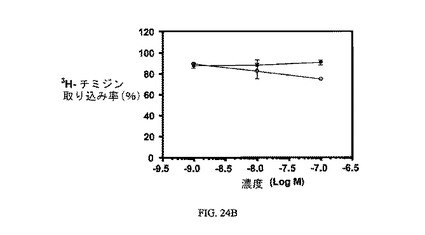
【図25A】
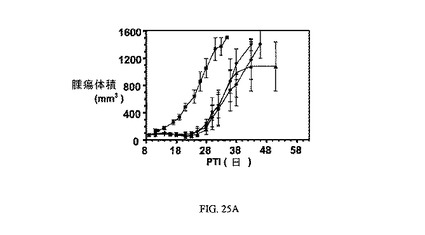
【図25B】
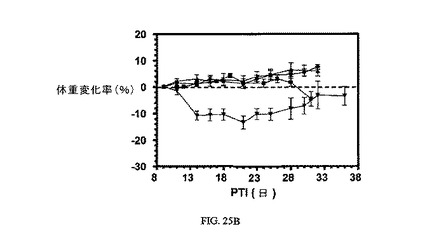
【図26A】
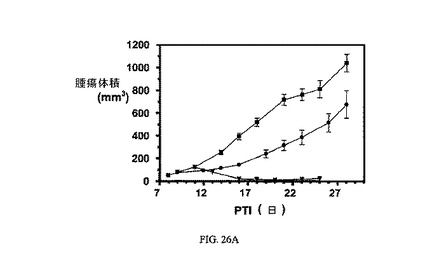
【図26B】
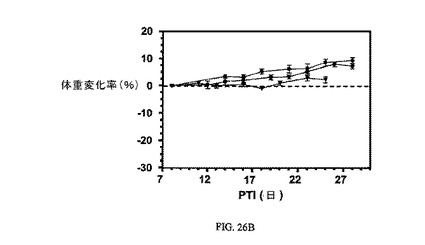
【図27A】
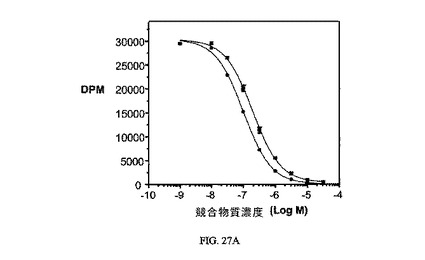
【図27B】
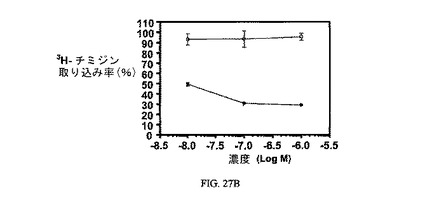
【図28A】
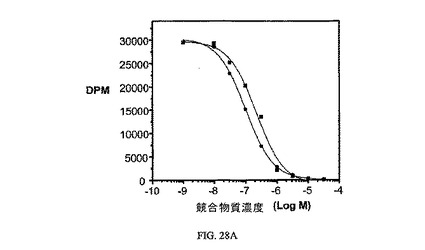
【図28B】
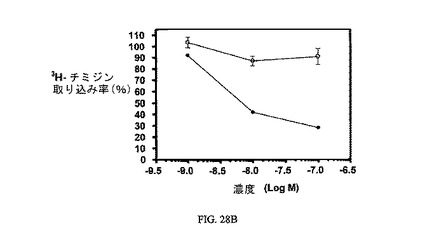
【図1B】
【図2】
【図3】
【図4A】
【図4B】
【図5A】
【図5B】
【図6A】
【図6B】
【図7A】
【図7B】
【図8】
【図9A】
【図9B】
【図10A】
【図10B】
【図11】
【図12】
【図13】
【図14A】
【図14B】
【図15】
【図16A】
【図16B】
【図17】
【図18】
【図19A】
【図19B】
【図20A】
【図20B】
【図20C】
【図21A】
【図21B】
【図22】
【図23A】
【図23B】
【図24A】
【図24B】
【図25A】
【図25B】
【図26A】
【図26B】
【図27A】
【図27B】
【図28A】
【図28B】
【公表番号】特表2009−504783(P2009−504783A)
【公表日】平成21年2月5日(2009.2.5)
【国際特許分類】
【出願番号】特願2008−527204(P2008−527204)
【出願日】平成18年8月18日(2006.8.18)
【国際出願番号】PCT/US2006/032560
【国際公開番号】WO2007/022493
【国際公開日】平成19年2月22日(2007.2.22)
【出願人】(504389588)エンドサイト,インコーポレイテッド (16)
【Fターム(参考)】
【公表日】平成21年2月5日(2009.2.5)
【国際特許分類】
【出願日】平成18年8月18日(2006.8.18)
【国際出願番号】PCT/US2006/032560
【国際公開番号】WO2007/022493
【国際公開日】平成19年2月22日(2007.2.22)
【出願人】(504389588)エンドサイト,インコーポレイテッド (16)
【Fターム(参考)】
[ Back to top ]