
ピラゾロスピロケトンアセチルCoAカルボキシラーゼ阻害剤
本発明は、式(1)の化合物または前記化合物の薬学的に許容できる塩(式中、R1、R2およびR3は、本明細書に記載された通りである)、その医薬組成物、および過体重の状態に罹った哺乳動物の治療におけるその使用を提供する。
【化1】

【化1】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アセチルCoAカルボキシラーゼの阻害剤として作用する置換ピラゾロスピロケトン化合物およびアセチルCoAカルボキシラーゼ酵素(複数可)の阻害によって調節される疾患、状態または障害の治療におけるそれらの使用に関する。
【背景技術】
【0002】
アセチルCoAカルボキシラーゼ(ACC)は、ほとんどの種に見られる酵素のファミリーであり、アセチルCoAからのマロニルCoAの生成を触媒することによって脂肪酸の合成および代謝に関連している。哺乳動物では、ACC酵素の2つのアイソフォームが確認されている。脂肪および肝臓などの脂質合成組織において高レベルで発現されるACC1は、長鎖脂肪酸の生合成の最初の始動反応を制御する。アセチルCoAがマロニルCoAを形成するためにカルボキシル化していなかったら、クレブス回路によって代謝される。肝臓ACCの微量成分であるが、心臓および骨格筋内で主要なアイソフォームであるACC2は、ミトコンドリアのシストリック(cystolic)表面でマロニルCoAの生成を触媒し、カルニチンパルミトイル転移酵素を阻害することによってβ酸化においてどれくらい脂肪酸が利用されているか調節する。したがって、脂肪酸利用を増大させ、かつ新規脂肪酸合成の増大を防止することによって、ACC阻害剤の慢性投与は、高脂肪食または低脂肪食を摂取している肥満対象における肝臓および脂肪組織TG貯蔵を激減させることもでき、体脂肪の選択的な減量をもたらす。
【0003】
Abu−Etheigaらによって実施された研究は、ACC2が脂肪酸酸化を制御する上で極めて重要な役割を果たすことを示唆している。したがって、ACC2の阻害が、肥満および肥満関連疾患、例えば2型糖尿病に対する治療の標的となると考えられる。Abu−Etheiga,L.ら、「Acetyl−CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high−fat/high−carbohydrate diets」PNAS、100(18)10207〜10212(2003)を参照のこと。Choi,C.S.ら、「Continuous fat oxidation in acetyl−CoA carboxylase 2 knockout mice increases total energy expenditure,reduces fat mass,and improves insulin sensitivity」PNAS、104(42)16480〜16485(2007)も参照のこと。肝臓脂質蓄積が肝臓インスリン抵抗性の原因であり、2型糖尿病の病因となることがますます明らかになっている。ACC1とACC2は両方とも肝細胞における脂肪酸化の調節に関与しているが、ラット肝臓において主要なアイソフォームであるACC1は脂肪酸合成の唯一の調節物質であることを、Salvageらは実証した。さらに、彼らのモデルでは、両アイソフォームの組み合わさった減少(combined reduction)には、肝臓マロニルCoAレベルの著しい低減、摂食状態における脂肪酸化の増大、脂質蓄積の低下、およびin vivoにおけるインスリン作用の改善が必要とされる。したがって、肝臓ACC1およびACC2阻害剤が非アルコール性脂肪肝疾患(NAFLD)および肝臓インスリン抵抗性の治療に有用である可能性があることを示している。Savage,D.B.ら、「Reversal of diet−induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl−CoA carboxylases 1 and 2」J Clin Invest doi:10.1172/JCI27300を参照のこと。Oh,Wら、「Glucose and fat metabolism in adipose tissue of acetyl−CoA carboxylase 2 knowckout mice」PNAS、102(5)1384〜1389(2005)も参照のこと。
【発明の概要】
【発明が解決しようとする課題】
【0004】
その結果、脂肪酸合成を阻害し、脂肪酸酸化を増大させることによって肥満および肥満関連疾患(NAFLDおよび2型糖尿病など)を治療するためにACC1およびACC2阻害剤を含有する医薬が必要とされている。
【課題を解決するための手段】
【0005】
本発明は、以下の式(1)の構造を有する化合物に関する。
【0006】
【化1】
【0007】
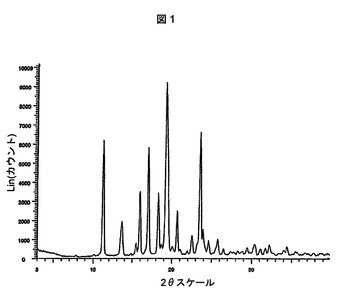
請求項1の化合物は、図1(11.2±0.2、15.4±0.2、17.0±0.2、18.3±0.2、19.3±0.2および20.6±0.2の回折角(2θ)にピークがある)によって表されるパターンと本質的に同じ粉末X線回折パターンを有する結晶形で存在することができる。本明細書において多形体A型と呼ぶ。
【0008】
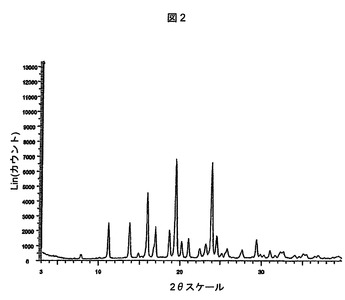
請求項1の化合物は、図2(7.8±0.2、11.2±0.2、13.7±0.2、15.9±0.2、18.7±0.2および20.2±0.2の回折角(2θ)にピークがある)によって表されるパターンと本質的に同じ粉末X線回折パターンを有する結晶形で存在することができる。本明細書において多形体B型と呼ぶ。
【0009】
本発明の別の態様は、(1)本発明の化合物(多形体A型およびB型を含む)、および(2)薬学的に許容できる賦形剤、希釈剤または担体を含む医薬組成物である。この組成物は、治療有効量の本発明の化合物を含むことが好ましい。この組成物は、少なくとも1種の別の薬剤(本明細書に記載されている)も含有することができる。好ましい薬剤には、抗肥満薬および/または抗糖尿病薬(以下に記載されている)が含まれる。
【0010】
本発明のさらに別の態様には、哺乳動物におけるアセチルCoAカルボキシラーゼ酵素(複数可)の阻害によって媒介される疾患、状態または障害を治療するための方法であって、かかる治療を必要とする哺乳動物、好ましくはヒトに、治療有効量の本発明の化合物、またはその医薬組成物を投与するステップを含む方法がある。
【0011】
アセチルCoAカルボキシラーゼの阻害剤によって媒介される疾患、障害または状態には、II型糖尿病および糖尿病関連疾患、例えば非アルコール性脂肪肝疾患(NAFLD)、肝臓インスリン抵抗性、高血糖、代謝症候群、耐糖能障害、糖尿病性神経障害、糖尿病性腎症、糖尿病性網膜症、肥満、異脂肪血症、高血圧、高インスリン血症およびインスリン抵抗性症候群が含まれる。好ましい疾患、障害または状態には、II型糖尿病、非アルコール性脂肪肝疾患(NAFLD)、肝臓インスリン抵抗性、高血糖、耐糖能障害、肥満、およびインスリン抵抗性症候群が含まれる。II型糖尿病、非アルコール性脂肪肝疾患(NAFLD)、肝臓インスリン抵抗性、高血糖および肥満がより好ましい。II型糖尿病が最も好ましい。
【0012】
好ましい実施形態は、動物における2型糖尿病および糖尿病関連障害の進行または発症を治療するまたは遅延させるための方法であって、かかる治療を必要とする動物に治療有効量の本発明の化合物またはその組成物を投与するステップを含む方法である。
【0013】
別の好ましい実施形態は、動物における肥満および肥満関連障害を治療するための方法であって、かかる治療を必要とする動物に治療有効量の本発明の化合物またはその組成物を投与するステップを含む方法である。
【0014】
さらに別の好ましい実施形態は、動物における非アルコール性脂肪肝疾患(NAFLD)または肝臓インスリン抵抗性を治療するための方法であって、かかる治療を必要とする動物に治療有効量の本発明の化合物またはその組成物を投与するステップを含む方法である。
【0015】
本発明の化合物は、他の薬剤(特に、本明細書の以下に記載の抗肥満および抗糖尿病薬)と併せて投与することができる。併用療法は、(a)本発明の化合物、本明細書に記載されている少なくとも1種の別の薬剤および薬学的に許容できる賦形剤、希釈剤もしくは担体を含む単一医薬組成物、または(b)(i)本発明の化合物および薬学的に許容できる賦形剤、希釈剤もしくは担体を含む第1の組成物と、(ii)本明細書に記載されている少なくとも1種の別の薬剤および薬学的に許容できる賦形剤、希釈剤もしくは担体を含む第2の組成物とを含む2種の別々の医薬組成物として投与することができる。医薬組成物は、同時にまたは順次および任意の順序で投与することができる。
【0016】
定義
X線回折ピーク位置に関して「本質的に同じ」という用語は、代表的なピーク位置および強度のばらつきを考慮していることを表す。例えば、ピーク位置(2θ)は、装置間の若干のばらつき、通常0.2°ほどを示すであろうことが当業者には理解されよう。さらに、相対ピーク強度は、装置間のばらつきと共に結晶化度、好ましい配向性、調製した試料の表面、および当業者に既知の他の要因によるばらつきを示すことになり、単に定性的尺度として見るべきであることが当業者には理解されよう。
【0017】
「治療有効量」という表現は、(i)特定の疾患、状態もしくは障害を治療もしくは予防する、(ii)特定の疾患、状態もしくは障害の1つもしくは複数の症候を減弱、回復、もしくは排除する、または(iii)本明細書に記載の特定の疾患、状態もしくは障害の1つもしくは複数の症候の発症を予防もしくは遅延する、本発明の化合物の量を表す。
【0018】
「動物」という用語は、ヒト(男性または女性)、コンパニオンアニマル(例えば、イヌ、ネコおよびウマ)、食物源の動物、動物園の動物、海生動物、鳥類および他の類似の動物種を意味する。「食用動物」は、ウシ、ブタ、ヒツジおよび家禽などの食物源の動物を意味する。
【0019】
「薬学的に許容できる」という表現は、物質または組成物が、製剤を構成する他の成分と、および/またはそれを用いて治療される哺乳動物と化学的および/または毒物学的に適合しなければならないことを示す。
【0020】
「治療すること(treating)」、「治療する(treat)」、または「治療(treatment)」という用語は、予防的な治療、すなわち、予防療法と待期療法の両方を包含する。
【0021】
「調節した(modulated)」または「調節すること(modulating)」、または「調節する(modulate(s))」という用語は、本明細書では、特に指示がない限り、本発明の化合物によるアセチルCoAカルボキシラーゼ(ACC)酵素(複数可)の阻害を意味する。
【0022】
「媒介した(mediated)」または「媒介すること(mediating)」または「媒介する(mediate(s))」という用語は、本明細書では、特に指示がない限り、特定の疾患、状態または障害の治療もしくは予防、(ii)特定の疾患、状態もしくは障害の1つもしくは複数の症候の減弱、回復もしくは排除、または(iii)アセチルCoAカルボキシラーゼ(ACC)酵素(複数可)を阻害することによる本明細書に記載の特定の疾患、状態もしくは障害の1つもしくは複数の症候の発症の予防もしくは遅延を意味する。
【0023】
「本発明の化合物」という用語は(特に具体的に特定しない限り)、式(I)の化合物ならびに、全ての互変異性体、配座異性体、および同位体的標識化合物を意味する。本発明の化合物の水和物および溶媒和物は、本発明の組成物と見なされ、その化合物は、それぞれ水または溶媒に関連している。
【図面の簡単な説明】
【0024】
【図1】式(I)の化合物のA型多形体の粉末X線回折パターン(pxrd)スペクトルを示す図である。
【図2】式(I)の化合物のB型多形体の粉末X線回折パターン(pxrd)スペクトルを示す図である。
【発明を実施するための形態】
【0025】
本発明の化合物は、特に本明細書に包含されている説明に照らして、化学分野でよく知られているものに類似したプロセスを含む合成経路によって合成することができる。出発物質は、一般にAldrich Chemicals(米国ウィスコンシン州Milwaukee)などの商業供給源から入手可能であり、または当業者によく知られている方法(例えば、Louis F.FieserおよびMary Fieser、Reagents for Organic Synthesis、1〜19巻、Wiley、New York(1967〜1999編)、もしくは補遺を含めたBeilsteins Handbuch der organischen Chemie、4、Aufl.編 Springer−Verlag、Berlin(Beilsteinオンラインデータベースによっても入手可能である)に一般に記載されている方法によって調製される)を用いて容易に調製される。
【0026】
例示のために、以下に示した反応スキームは、本発明の化合物ならびに重要な中間体を合成するための潜在的な経路を提供する。個々の反応ステップのより詳細な説明については、以下の実施例の項を参照のこと。他の合成経路を使用して本発明の化合物を合成するできることが当業者には理解されよう。特定の出発物質および試薬をスキームに示し、以下で論じているが、他の出発物質および試薬と容易に置き換えて、種々の誘導体および/または反応条件を得ることができる。さらに、後述の方法によって調製した多くの化合物は、当業者によく知られている従来の化学的手法を用いて本開示に照らしてさらに修飾することができる。
【0027】
本発明の化合物の調製では、中間体の離れた官能基(remote functionality)(例えば、第一級または第二級アミン)の保護が必要なこともある。このような保護の必要性は、離れた官能基の性質および調製方法の条件に応じて変わるものである。適当なアミノ保護基(NH−Pg)には、アセチル、トリフルオロアセチル、t−ブトキシカルボニル(BOC)、ベンジルオキシカルボニル(CBz)および9−フルオレニルメチレンオキシカルボニル(Fmoc)がある。同様に、「ヒドロキシ保護基」は、ヒドロキシ官能基を遮断または保護するヒドロキシ基の置換基を意味する。適当なヒドロキシル保護基(O−Pg)には、例えば、アリル、アセチル、シリル、ベンジル、パラメトキシベンジル、トリチル、などがある。このような保護の必要性は、当業者により容易に決定される。保護基およびそれらの使用の一般的な説明については、T.W.Greene、Protective Groups in Organic Synthesis、John Wiley & Sons、New York、1991を参照のこと。
【0028】
スキームIは、式(I)を有する本発明の化合物を得るために使用できる一般的な手順の概要である。
【0029】
【化2】
【0030】
中間体ヒドラゾン(1a)は、酢酸などの酸性環境において室温でメチルグリオキサル(SM−1)を1−t−ブチルヒドラジン(SM−2)で処理することによって形成することができる。還流させた水性酢酸中でヒドラゾン(1a)をオキサルアルデヒド(SM−3)で処理することによって、1−(4−ヒドロキシ−1H−ピラゾール−3−イル)エタノン中間体(1b)が得られる。あるいは、1H−ピラゾール中間体(1b)は、還流させた水性酢酸中でオキサルアルデヒド(SM−3)を1−t−ブチルヒドラジンオキサレートで処理することによって直接形成することもできる。アミノ保護ピラゾロスピロケトン中間体(1c)は、アミン(好ましくは、ピロリジン)の存在下において室温でアミノ保護4−ピペリドン(好ましくは、BOC保護基)を1−(4−ヒドロキシ−1H−ピラゾール−3−イル)エタノン中間体(1b)に加えることによって形成することができる。次いで保護基を除去して、ピラゾロスピロケトン中間体(1d)を得ることができる。アミノ保護基を除去するために使用した条件は、どの保護基を使用したかによって決まることになる。例えば、BOC保護基は、強酸(例えば、HCl)を用いた処理によって除去することができる。次いで最終化合物(I)は、1H−インダゾール−5−カルボン酸を用いた標準的なペプチドカップリング反応を使用することによって形成することができる。例えば、ピラゾロスピロケトン中間体(1d)および1H−インダゾール−5−カルボン酸は、例えばTHFおよび/またはDMFなどの適当な溶媒中に、ヒドロキシベンゾトリアゾール(HOBt)などの活性化剤の存在下または不在下、およびN,N−ジイソプロピルエチルアミン(DIEA)またはN−メチルモルフォリン(NMM)などの適当な塩基の存在下で、1H−インダゾール−5−カルボン酸とO−(7−アザベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロフォスフェート(HATU)などのペプチドカップリング試薬とを接触させることによって、活性カルボン酸エステルを形成し、次いで活性カルボン酸エステルとピラゾロスピロケトン中間体(1d)とを接触させることによって結合させて、式(1)の化合物を形成することができる。あるいは、式(1)の化合物は、まず例えば塩化チオニルと反応させることによって1H−インダゾール−5−カルボン酸を酸塩化物に転換し、次いで酸塩化物をピラゾロスピロケトン中間体(1d)と反応させて式(1)の化合物を形成することによって形成することができる。さらに別の代替法は、THFおよび/またはDMFなどの適当な溶媒中にN−メチルモルフォリンなどの適当な塩基の存在下で、1H−インダゾール−5−カルボン酸を2−クロロ−4,6−ジメトキシトリアジンで処理することを伴う。活性エステルには、THFおよび/またはDMFなどの適当な溶媒に溶かしたピラゾロスピロケトン中間体(1d)とN−メチルモルフォリンなどの塩基の溶液を加える。
【0031】
本発明の化合物は、2種以上の結晶形で存在することができる。本発明の化合物の多形体(溶媒和物および水和物を含む)は、本発明を構成し、異なる条件下における本発明の化合物の結晶化によって調製することができる。例えば、再結晶のための異なる溶媒または異なる溶媒混合物の使用、異なる温度における結晶化、結晶化中に超高速から超低速までの範囲にわたる種々の冷却モードなどである。多形体は、本発明の化合物の加熱または融解とそれに続く段階的または高速冷却によって得ることもできる。多形体の存在は、固体プローブ核磁気共鳴(NMR)分光法、赤外(IR)分光法、示差走査熱分析、粉末X線回折またはこのような他の技術によって確認することができる。
【0032】
本発明には、1個または複数個の原子を通常自然界に見られる原子量または質量数と異なる原子量または質量数を有する原子と置き換えていることを除けば、式(1)によって記述されたものと等しい、同位体的標識化合物も含まれる。式(I)の化合物に取り込める同位体の例には、水素、炭素、窒素、酸素、硫黄およびフッ素の同位体、例えばそれぞれ2H、3H、13C、14C、15N、18O、17O、35S、36Cl、125I、129I、および18Fがある。いくつかの本発明の同位体的標識化合物、例えば3Hおよび14Cなどの放射性同位体を取り込んでいるものは、薬物および/または基質組織分布アッセイに有用である。トリチウム化(すなわち、3H)、および炭素−14(すなわち、14C)、同位体は、それらの調製の容易さおよび検出感度に関して特に好ましい。さらに、より重い同位体、例えば重水素(すなわち、2H)との置換は、より大きい代謝安定性の結果として生じるいくつかの治療的利点、例えばin vivo半減期の増大または必要投与量の低減をもたらす可能性があり、したがって、状況によっては好ましいこともある。本発明の同位体的標識化合物は、同位体的標識されていない試薬の代わりに容易に入手可能な同位体的標識された試薬を使用することにより、以下のスキームおよび/または実施例に開示されている手順を実施することによって、一般に調製することができる。
【0033】
本発明の化合物は、分離可能であり得る異なる安定な立体配座形態で存在することができる。例えば立体障害または環歪みのための、不斉単結合の周りの束縛回転が原因のねじれ非対称により、異なる配座の分離が可能となり得る。本発明の化合物は、式(1)の化合物の各配座異性体およびその混合物をさらに含む。
【0034】
本発明の化合物は、アセチルCoAカルボキシラーゼ酵素(複数可)(特に、ACC1およびACC2)の阻害によって調節される疾患、状態および/または障害を治療するのに有用である。したがって、本発明の別の実施形態は、治療有効量の本発明の化合物および薬学的に許容できる賦形剤、希釈剤または担体を含む医薬組成物である。本発明の化合物(そこに使用した組成物およびプロセスを含む)は、本明細書に記載の治療用途のための医薬の製造に使用することもできる。
【0035】
代表的な製剤は、本発明の化合物および担体、希釈剤または賦形剤を混合することによって調製する。適当な担体、希釈剤および賦形剤は、当業者によく知られており、炭水化物、蝋、水溶性および/または水膨潤性ポリマー、親水性または疎水性物質、ゼラチン、油、溶媒、水などの物質が含まれる。使用する特定の担体、希釈剤または賦形剤は、本発明の化合物を適用する手段および目的によって決まることになる。溶媒は、一般に哺乳動物に投与するのが安全と当業者によって認められた(GRAS)溶媒に基づいて選択される。一般に、安全な溶媒は、水などの非毒性水性溶媒および水に可溶性または混和性の他の非毒性溶媒である。適当な水性溶媒には、水、エタノール、プロピレングリコール、ポリエチレングリコール(例えば、PEG400、PEG300)など、およびその混合物がある。製剤は、1種または複数のバッファー、安定剤、界面活性剤、湿潤剤、平滑剤、乳化剤、懸濁化剤、保存剤、酸化防止剤、不透明化剤、流動促進剤、加工助剤、着色剤、甘味料、芳香剤、矯味矯臭剤および薬物(すなわち、本発明の化合物またはその医薬組成物)の洗練された体裁を与える、または医薬品(すなわち、医薬)の製造に役立つ他の既知の添加剤も含んでいてよい。
【0036】
製剤は、従来の溶解および混合手順を用いて調製することができる。例えば、原薬(すなわち、本発明の化合物または化合物の安定化形態(例えば、シクロデキストリン誘導体または他の既知の複合体形成剤との複合体))を、上記の1種または複数の賦形剤の存在下で適当な溶媒に溶解させる。低水溶性化合物の溶解速度は、参照により本明細書に組み込む、Takeuchi,H.らによって「Enhancement of the dissolution rate of a poorly water−soluble drug(tolbutamide) by a spray−drying solvent depostion method and disintegrants」J.Pharm.Pharmacol.、39、769〜773(1987)、および欧州特許第0901786B1号(米国特許出願公開第2002/009494号)に記載されているものなど、噴霧乾燥した分散体の使用によって高めることができる。本発明の化合物は、通常、容易に制御可能な薬物投与量を提供し、患者に洗練されかつ取り扱いやすい製品を与えるために、医薬剤形に製剤する。
【0037】
医薬組成物には、本発明の化合物の溶媒和物および水和物も含まれる。「溶媒和物」という用語は、本発明の化合物と1種または複数の溶媒分子との分子複合体を意味する。かかる溶媒分子は、レシピエントに対して無害であることが知られている、製薬技術分野で一般に使用されているもの、例えば、水、エタノール、エチレングリコールなどである。「水和物」という用語は、溶媒分子が水である複合体を意味する。溶媒和物および/または水和物は、結晶形で存在することが好ましい。メタノール、メチルt−ブチルエーテル、酢酸エチル、酢酸メチル、(S)−プロピレングリコール、(R)−プロピレングリコール、1,4−ブチン−ジオールなど、他の溶媒は、より望ましい溶媒和物の調製における中間溶媒和物として使用することができる。
【0038】
使用するための医薬組成物(または製剤)は、薬物を投与するために使用する方法に応じて種々の形に包装することができる。一般に、販売用の商品には、適切な形態の医薬製剤が入れられている容器が含まれる。適当な容器は、当業者によく知られており、ボトル(プラスチックおよびガラス)、サッシェ、アンプル、ビニール袋、金属シリンダーなどの材料が含まれる。容器は、包装の中身に不用心に近づくのを防ぐためにいたずら防止セットも含んでいてよい。その上、容器には、容器の中身を説明するラベルを付けている。ラベルは、適切な警告も含んでいてよい。
【0039】
本発明は、かかる治療を必要とする動物に、治療有効量の本発明の化合物または有効量の本発明の化合物と薬学的に許容できる賦形剤、希釈剤、もしくは担体とを含む医薬組成物を投与することを含む、動物におけるアセチルCoAカルボキシラーゼ酵素(複数可)の阻害によって調節される疾患、状態および/または障害を治療する方法をさらに提供する。この方法は、アセチルCoAカルボキシラーゼ酵素(複数可)の阻害から恩恵を受ける疾患、状態および/または障害を治療するのに特に有用である。
【0040】
本発明の一態様は、肥満、および肥満関連障害(例えば、過体重、体重増加、または体重維持)の治療である。
【0041】
肥満および過体重は一般に肥満度指数(BMI)によって定義され、これは全体脂肪と相関関係があり、疾患の相対危険度を推定する。BMIは、キログラム表示の体重を、メートル表示の身長の二乗で割って算出される(kg/m2)。過体重は通常、BMIが25〜29.9kg/m2と定義され、肥満は通常、BMIが30kg/m2と定義される。例えば、National Heart,Lung,and Blood Institute、Clinical Guidelines on the Identification,Evaluation,and Treatment of Overweight and Obesity in Adults、The Evidence Report、Washington,DC:U.S.Department of Health and Human Services、NIH publication no.98−4083(1998)を参照のこと。
【0042】
本発明の別の態様は、糖尿病または1型(インスリン依存真性糖尿病、「IDDM」とも呼ばれる)および2型(非インスリン依存真性糖尿病、「NIDDM」とも呼ばれる)糖尿病、耐糖能障害、インスリン抵抗性、高血糖、ならびに糖尿病性合併症(アテローム性動脈硬化症、冠状動脈性心臓病、発作、末梢血管疾患、腎症、高血圧、神経障害、および網膜症など)を含めた糖尿病関連障害の進行または発症を治療または遅延するためである。
【0043】
本発明のさらに別の態様では、代謝症候群などの肥満同時罹患状態(obesity comorbidities)の治療である。代謝症候群には、異脂肪血症、高血圧、インスリン抵抗性、糖尿病(例えば、2型糖尿病)、冠状動脈疾患および心不全などの疾患、状態または障害が含まれる。代謝症候群に関するより詳細な情報については、例えば、Zimmet,P.Z.ら、「The Metabolic Syndrome: Perhaps an Etiologic Mystery but Far From a Myth − Where Does the International Diabetes Federation Stand?」、Diabetes & Endocrinology、7(2)、(2005)、およびAlberti,K.G.ら、「The Metabolic Syndrome − A New Worldwide Definition」、Lancet、366、1059〜62(2005)を参照のこと。本発明の化合物の投与は、薬物を含有しないビヒクル対照と比較して、血漿レプチン、C反応性タンパク質(CRP)および/またはコレステロールの低下など少なくとも1種の心臓血管疾患危険因子で統計的に有意な(p<0.05)低減をもたらすことが好ましい。本発明の化合物の投与は、グルコース血清レベルでも統計的に有意な(p<0.05)低減をもたし得る。
【0044】
本発明のさらに別の態様では、非アルコール性脂肪肝疾患(NAFLD)および肝臓インスリン抵抗性の治療である。
【0045】
体重が約100kgの正常な成人ヒトの場合、体重1キログラム当たり約0.001mg〜約10mgの範囲の投与量が通常十分であり、約0.01mg/kg〜約5.0mg/kgが好ましく、約0.01mg/kg〜約1mg/kgがより好ましい。しかしながら、治療対象の年齢および体重、意図される投与経路、投与される特定の化合物などよって、一般的な投与量範囲にいくらかの変動性が求められてもよい。特定の患者についての投与量範囲および最適投与量の決定は、本開示の利点を有する当業者の能力の十分に範囲内である。本発明の化合物は、徐放、制御放出、および遅延放出製剤に使用でき、その形態も当業者によく知られているという点にも留意されたい。
【0046】
本発明の化合物は、本明細書に記載の疾患、状態および/または障害の治療のための他の薬剤と併用してもよい。したがって、本発明の化合物を他の薬剤と併せて投与することを含む治療の方法も提供している。本発明の化合物と併せて使用できる適当な薬剤には、抗肥満薬(食欲抑制薬を含む)、抗糖尿病薬、抗高血糖症薬、脂質低下剤、および抗高血圧剤がある。
【0047】
適当な抗肥満薬には、11β−ヒドロキシステロイドデヒドロゲナーゼ−1(11β−HSD1型)阻害剤、ステアロイルCoAデサチュラーゼ−1(SCD−1)阻害剤、MCR−4アゴニスト、コレシストキニン−A(CCK−A)アゴニスト、モノアミン再取り込み阻害剤(シブトラミンなど)、交感神経様作用薬、β3アドレナリンアゴニスト、ドーパミンアゴニスト(ブロモクリプチンなど)、メラノサイト刺激ホルモン類似体、5HT2cアゴニスト、メラニン凝集ホルモンアンタゴニスト、レプチン(OBタンパク質)、レプチン類似体、レプチンアゴニスト、ガラニンアンタゴニスト、リパーゼ阻害剤(テトラヒドロリプスタチン、すなわちオルリスタットなど)、食欲抑制薬(ボンベシンアゴニストなど)、ニューロペプチドYアンタゴニスト(例えば、NPY Y5アンタゴニスト)、PYY3−36(その類似体を含む)、甲状腺模倣剤、デヒドロエピアンドロステロンまたはその類似体、グルココルチコイドアゴニストまたはアンタゴニスト、オレキシンアンタゴニスト、グルカゴン様ペプチド−1アゴニスト、毛様体神経栄養因子(Regeneron Pharmaceuticals、Inc.、米国ニューヨーク州TarrytownおよびProcter&Gamble Company、米国オハイオ州Cincinnatiから入手可能なAxokine(商標)など)、ヒトアグーチ関連タンパク質(AGRP)阻害剤、グレリンアンタゴニスト、ヒスタミン3アンタゴニストまたはインバースアゴニスト、ニューロメジンUアゴニスト、MTP/ApoB阻害剤(例えば、ジルロタピドなどの消化管選択的MTP阻害剤)、オピオイドアンタゴニスト、オレキシンアンタゴニストなどがある。
【0048】
本発明の組み合わせた態様で使用するのに好ましい抗肥満薬には、消化管選択的MTP阻害剤(例えば、ジルロタピド、ミトラタピドおよびインプリタピド、R56918(CAS番号403987)およびCAS番号913541−47−6)、CCKaアゴニスト(例えば、PCT公開番号WO2005/116034または米国特許出願公開第2005−0267100A1号に記載されているN−ベンジル−2−[4−(1H−インドール−3−イルメチル)−5−オキソ−1−フェニル−4,5−ジヒドロ−2,3,6,10b−テトラアザ−ベンゾ[e]アズレン−6−イル]−N−イソプロピル−アセトアミド)、5HT2cアゴニスト(例えば、ロルカセリン)、MCR4アゴニスト(例えば、米国特許第6,818,658号に記載されている化合物)、リパーゼ阻害剤(例えば、セチリスタット)、PYY3−36(本明細書では「PYY3−36」にはペグ化PYY3−36、例えば、米国特許出願公開第2006/0178501号に記載されているものなどの類似体が含まれる)、オピオイドアンタゴニスト(例えば、ナルトレキソン)、オレオイル−エストロン(CAS番号180003−17−2)、オビネピチド(obinepitide)(TM30338)、プラムリンチド(Symlin(登録商標))、テソフェンシン(NS2330)、レプチン、リラグルチド、ブロモクリプチン、オルリスタット、エクセナチド(Byetta(登録商標))、AOD−9604(CAS番号221231−10−3)およびシブトラミンがある。本発明の化合物および併用療法は、運動および堅実な食事と併せて投与することが好ましい。
【0049】
適当な抗糖尿病薬には、ナトリウム−グルコース共輸送体(SGLT)阻害剤、ホスホジエステラーゼ(PDE)−10阻害剤、ジアシルグリセロールアシルトランスフェラーゼ(DGAT)1または2阻害剤、スルホニル尿素(例えば、アセトヘキサミド、クロルプロパミド、ジアビネース、グリベンクラミド、グリピジド、グリブリド、グリメピリド、グリクラジド、グリペンチド、グリキドン、グリソラミド、トラザミド、およびトルブタミド)、メグリチニド、α−アミラーゼ阻害剤(例えば、テンダミスタット、トレスタチンおよびAL−3688)、α−グルコシドヒドロラーゼ阻害剤(例えば、アカルボース)、α−グルコシダーゼ阻害剤(例えば、アジポシン、カミグリボース、エミグリテート、ミグリトール、ボグリボース、プラジミシン−Q、およびサルボスタチン)、PPARγアゴニスト(例えば、バラグリタゾン、シグリタゾン、ダルグリタゾン、エングリタゾン、イサグリタゾン、ピオグリタゾン、ロシグリタゾンおよびトログリタゾン)、PPARα/γアゴニスト(例えば、CLX−0940、GW−1536、GW−1929、GW−2433、KRP−297、L−796449、LR−90、MK−0767およびSB−219994)、ビグアナイド(例えば、メトホルミン)、グルカゴン様ペプチド1(GLP−1)アゴニスト(例えば、Byetta(商標)、エキセンディン−3およびエキセンディン−4)、タンパク質チロシンホスファターゼ−1B(PTP−1B)阻害剤(例えば、トロズスクエミン、ヒルチオサール抽出物、およびZhang,S.ら、Drug Discovery Today、12(9/10)、373〜381(2007)によって開示されている化合物)、SIRT−1阻害剤(例えば、レセルバトロール(reservatrol))、ジペプチジルペプチデアーゼIV(DPP−IV)阻害剤(例えば、シタグリプチン、ビルダグリプチン、アログリプチンおよびサクサグリプチン)、インスリン分泌促進物質、脂肪酸酸化阻害剤、A2アンタゴニスト、c−junアミノ末端キナーゼ(JNK)阻害剤、インスリン、インスリン様作用薬、グリコーゲンホスホリラーゼ阻害剤、VPAC2受容体アゴニストおよびグルコキナーゼ活性化剤がある。好ましい抗糖尿病薬は、メトホルミン、グルカゴン様ペプチド1(GLP−1)アゴニスト(例えば、Byetta(商標))およびDPP−IV阻害剤(例えば、シタグリプチン、ビルダグリプチン、アログリプチンおよびサクサグリプチン)である。
【0050】
上記の全ての米国特許および公開は、参照により本明細書に組み込まれている。
【0051】
本明細書の以下に記載されている実施例は、例示的な目的のために過ぎない。本明細書において反映されている組成物、方法、および種々のパラメーターは、本発明の種々の態様および実施形態を例示するものに過ぎず、決して特許請求した発明の範囲を限定するものではない。当業者は、反応の規模および使用した特定の装備に基づいて試薬、溶媒および条件をどのように最適化するのか知っているはずである。
【実施例】
【0052】
後述の化合物および中間体は、有機化学の命名法およびCAS索引規則に対するIUPAC(国際純正および応用化学連合)勧告に基づいて一般に命名した。特に指摘がない限り、全ての反応物は、商業的に得た。本明細書の以下に引用した全ての参考文献は、参照により組み込まれている。
【0053】
フラッシュクロマトグラフィーは、Stillら、J.Org.Chem.、1978、43、2923によって記載されている方法に基づいて行った。
【0054】
本明細書で論じている全てのBiotage(登録商標)精製は、KP−SILシリカ(40〜63μM、60オングストローム)を含有する40Mまたは40S Biotage(登録商標)カラム(Bioatge AB、Uppsala、スウェーデン)を用いて行った。
【0055】
本明細書で論じている全てのCombiflash(登録商標)精製は、充填RediSep(登録商標)シリカカラムを利用したCombiFlash(登録商標)Companion system(Teledyne Isco、米国ネブラスカ州Lincoln)を用いて行った。
【0056】
質量スペクトルは、Waters(Waters Corp.、米国マサチューセッツ州Milford)Micromass Platform II分光計で記録した。特に指定のない限り、質量スペクトルは、Waters(米国マサチューセッツ州Milford)Micromass Platform II分光計で記録した。
【0057】
プロトンNMR化学シフトは、テトラメチルシランから低磁場に百万分率で示され、Varian Unity 400または500MHz(メガヘルツ)分光計(Varian Inc.、米国カリフォルニア州Palo Alto)で記録した。NMR化学シフトは、テトラメチルシラン(プロトンの場合)またはフルオロトリクロロメタン(フッ素の場合)から低磁場に百万分率で示される。
【0058】
重要な中間体および出発物質
1H−インダゾール−5−カルボン酸は、Tyger Scientific、Inc.、米国ニュージャージー州Ewingから入手可能である。
【0059】
中間体2’−tert−ブチル−2’H−スピロ[ピペリジン−4,5’−ピラノ[3,2−c]ピラゾール]−7’(6’H)−オン塩酸塩(I−1a)の調製:
【0060】
【化3】
【0061】
ピルブアルデヒド(26.2mL、160mmol)のH2O(120mL)溶液を、tert−ブチルヒドラジン・HCl(20g、124mmol)のH2O(500mL)溶液に20分にわたって加えた。この溶液を室温で5時間撹拌した。次いで反応混合物を酢酸エチル(5回)を用いて抽出した。合わせた有機層を乾燥(Na2SO4)させ、減圧下で濃縮した。次いで残留物を、酢酸エチル/ヘプタン(10:90〜40:60)の勾配を用いて溶離するフラッシュクロマトグラフィー(シリカゲル)によって精製して、2−オキソプロパナールtert−ブチルヒドラゾン13.1g(74%)を琥珀色油としてもたらした。
【0062】
グリオキサール(2.9mL、25.3mmol)の40%水溶液を、水(14mL)に溶かした2−オキソプロパナールtert−ブチルヒドラゾン(1.20g、8.44mmol)に加えた。次いで混合物を5時間還流させながら加熱した。反応混合物を室温まで冷却し、EtOAcを用いて4回抽出した。合わせた有機層を乾燥(Na2SO4)させ、ろ過し、減圧下で濃縮した。残留物をフラッシュクロマトグラフィー(シリカゲル)によって精製し、ヘプタン/酢酸エチル(100:0〜90:10)の勾配を用いて溶離して、1−(4−ヒドロキシ−1−tert−ブチル−1H−ピラゾール−3−イル)エタノン1.06g(69%)を無色油として得た。
【0063】
1−(4−ヒドロキシ−1−tert−ブチル−1H−ピラゾール−3−イル)エタノン(6.63g、36.4mmol)のMeOH(73mL)溶液に、ピロリジン(3.6mL、43.7mmol)および1−(N−Boc)−4−ピペリドン(8.7g、43.7mmol)を加えた。暗赤色溶液を室温で終夜撹拌した。溶液を濃縮し、残留物をEtOAcに溶解し、1N NaOHおよびブラインで洗浄した。層を分離し、次いで有機層を取っておいた。合わせた水層を酢酸エチルで抽出した。層を分離し、有機層を1N NaOHおよびブラインで洗浄した。全ての有機層を合わせ、次いで1N HCl、およびブラインで洗浄し、乾燥(Na2SO4)させ、ろ過し、減圧下で濃縮して赤色ガム(11g)を生じさせた。赤色ガムを粉砕し、25%EtOAc/ヘキサン(125mL)中で加熱し、ガムは黄色固体に変化したが、還流させながら完全に溶解しなかった。混合物を室温まで冷却し、ろ過して生成物であるオフホワイト固体(1.69g)を得た。ろ液を約5〜10mL(ヘキサン、EtOAcおよびアセトンを含む)まで濃縮し、次いで追加量の2%EtOAc/ヘキサン(100mL)を加え、その後固体が沈殿し始めた。混合物を終夜撹拌した。固体をろ過して、別の所望の生成物3.89gをオフホワイト固体として得た。全部で、5.58g(42%)のtert−ブチル2’−tert−ブチル−7’−オキソ−6’,7’−ジヒドロ−1H,2’H−スピロ[ピペリジン−4,5’−ピラノ[3,2−c]ピラゾール]−1−カルボキシレートをオフホワイト固体として単離した。
【0064】
tert−ブチル2’−tert−ブチル−7’−オキソ−6,7’−ジヒドロ−1H,2’H−スピロ[ピペリジン−4,5’−ピラノ[3,2−c]ピラゾール]−1−カルボキシレート(2.73g、7.5mmol)の1,4−ジオキサン(15mL)溶液に室温でHCl溶液(1,4−ジオキサン中4M、15mL、60mmol)を加えた。混合物を室温で3時間撹拌した。次いで反応混合物を濃縮乾固した。得られたピンク色固体(2.6g)を2−メチルテトラヒドロフラン(20mL)および少量のEtOH(1mL)と一緒に粉砕した。固体をろ過し、2−メチルテトラヒドロフラン(20mL)で洗浄し、50℃で真空乾燥させて、表題化合物(I−1a)2.15g(95%)を白色固体としてを得た。
【0065】
(実施例1)
2’−tert−ブチル−1−(1H−インダゾール−5−イルカルボニル)−2’H−スピロ[ピペリジン−4,5’−ピラノ[3,2−c]ピラゾール]−7’(6’H)−オン(I)の調製:
【0066】
【化4】
【0067】
N−ジメチルホルムアミド(1mL)に溶かした1H−インダゾール−5−カルボン酸(27mg、0.17mmol)、2−クロロ−4,6−ジメトキシ−1,3,5−トリアジン(36mg、0.20mmol)およびN−メチルモルフォリン(NMM)(19uL、0.17mmol)の混合物を室温で35分間撹拌してからNMM(3当量)、続いて2’−tert−ブチル−2’H−スピロ[ピペリジン−4,5’−ピラノ[3,2−c]ピラゾール]−7’(6’H)−オン・HCl(I−1a:50mg、0.17mmol)を加えた。混合物を室温で終夜撹拌した。溶媒を減圧下で除去し、残留物をCH2Cl2に溶解させ、飽和水性NH4Clで洗浄した。水相をCH2Cl2(2回)を用いて逆抽出した。合わせた有機抽出物を水および飽和水性NaClで洗浄してからMgSO4上で乾燥させた。物質をろ過し、濃縮し、分取薄層クロマトグラフィー(95:5 CHCl3/MeOH)によって精製した。所望の物質を続いてEt2Oと一緒に粉砕し、ろ過し、固体を真空下に50℃で乾燥させて、所望の生成物(21mg、31%)を生じさせた。
1H NMR (500 MHz, DMSO-d6) δ ppm 13.26 (1 H, br. s.), 8.14 (1 H, s), 7.86 (1 H, s), 7.81 (1 H,
s), 7.58 (1 H, d, J=8.54 Hz), 7.40 (1 H, br. s.), 3.18 (2 H, br. s.), 2.75 (2
H, s), 1.99 (2 H, s), 1.88 (2 H, br. s.), 1.74 (2 H, t), 1.51 (9 H, s).
【0068】
(実施例2)
あるいは、化合物(I)は、結晶性生成物(本明細書において「A型」と呼ぶ)を生成する以下の手順を用いて調製することができる。
【0069】
400L反応器に、2’−tert−ブチル−2’H−スピロ[ピペリジン−4,5’−ピラノ[3,2−c]ピラゾール]−7’(6’H)−オン・HCl(I−1a:6.6kg、22.0モル)、1H−インダゾール−5−カルボン酸(3.26kg、20.1モル)、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩(4.85kg、25.3モル)、アセトニトリル(124L)、およびピリジン(13.9L、172モル)を入れた。溶液を周囲温度で16時間撹拌し、次いで酢酸エチル(250L)で希釈し、10wt%の水性クエン酸(100L)で2回洗浄した。51Lの溶液体積まで蒸留するために有機層を加熱し、次いで酢酸エチル(約85L)を加え、76℃の内部温度が得られるまで蒸留した(溶液体積約55L)。次いで溶液を3時間にわたって周囲温度まで冷却し、固体をヌッチェフィルターに通してろ過し、酢酸エチル(17L)で洗浄し、50℃で12時間真空下で乾燥させた。化合物(I)を白色結晶性固体(3.89kg、9.55モル、48%)として単離した。融点:265℃。
1H NMR (400 MHz, DMSO-d6) δ ppm 13.2 (1H, s), 8.11 (1H, s), 7.83 (1H, s), 7.77 (1H, s), 7.55 (1
H, d, J=8.0 Hz), 7.37 (1 H, dd, J=8.0, 1.2 Hz), 4.35-3.45 (2 H, m), 3.30-3.15
(2 H, m), 2.72 (2H, s), 1.98-1.86 (2 H, m),1.73 (2 H, td, J=12.0, 4.0 Hz), 1.48
(9 H, s).
13C NMR (100 MHz, DMSO-d6) δ ppm: 186.5, 170.2, 147.6, 140.6, 134.9, 134.2, 128.6, 125.9, 122.8,
120.5, 114.3, 110.7, 81.6, 61.0, 49.0, 34.0, 29.7.
【0070】
式(I)の化合物のA型多形体に関するX線粉末回折パターンを、銅放射線によるSiemens D5000回折計を用いて作成した。この機器は、線状焦点X線管を備えていた。管電圧およびアンペア数を、それぞれ38kVおよび38mAに設定した。発散および散乱スリットを1mmに設定し、受信スリットを0.6mmに設定した。Sol−Xエネルギー分散X線検出器を用いて回折CuKα1放射線(λ=1.54056Å)を検出した。3.0〜40°2θから2.4°2θ/分(1秒/0.04°2θステップ)でθ2θ連続スキャンを使用した。アルミナ標準物質(NIST標準参照物質1976)を分析して機器アライメントを確認した。データを捕集し、BRUKER AXS DIFFRAC PLUSソフトウェアバージョン2.0を用いて分析した。分析用に試料を石英ホルダーに取り付けることによって準備した。Bruker Instruments社がSiemens社を買収したことを留意されたい。したがって、Bruker D5000機器は、本質的にSiemens D5000と同じである。以下の表は、A型結晶について観察されたバックグラウンドに対して5倍閾値を有するピークをまとめて示している。A型の特徴ピーク(2θ)は、11.2±0.2、15.4±0.2、17.0±0.2、18.3±0.2、19.3±0.2および20.6±0.2である。
【0071】
【表1】
【0072】
アセトンに溶かしたヒドロキシプロピルメチルセルロースアセテートサクシネート(HPMCAS)を用いて化合物(I)を噴霧乾燥分散(SDD)させた(25重量%化合物(I))場合に、化合物(I)の溶解が11倍増大したことが観察された。0.5重量%Methocel(商標)に懸濁させたモデル絶食十二指腸溶液(0.5重量%タウロコール酸ナトリウム/1−パルミトイル−2−オレオイル−sn−グリセロ−3−ホスホコリンを溶かしたリン酸緩衝溶液、pH6.5)中600μg(有効成分))/mLで、化合物およびSDDを比較し試験した。
【0073】
(実施例3)
実施例3は、式(I)の化合物の異なる多形形態(本明細書において「B型」と呼ぶ)を提供する。
【0074】
実施例2からのA型(20mg)を、電磁撹拌棒およびアセトン2mL(2mL)を含有する4mLバイアルに加えた。固体を25℃で3週間撹拌した。固体をPTFEフィルター上でろ過し、MTBE1mLで洗浄した。B型約10mgを白色結晶性固体として単離した。
【0075】
式(I)の化合物のB型に関するX線粉末回折パターンを、銅放射線によるSiemens D5000回折計および実施例2に上述されている条件を用いて作成した。以下の表は、B型多形体について観察されたバックグラウンドに対して5倍閾値を有するピークをまとめて示している。B型の特徴ピーク(2θ)は、7.8±0.2、11.2±0.2、13.7±0.2、15.9±0.2、18.7±0.2および20.2±0.2である。
【0076】
【表2】
【0077】
薬理学的データ
生物学的プロトコル
動物、特に哺乳動物(例えば、ヒト)における疾患(例えば本明細書において詳述しているもの)の治療における本発明の化合物の有用性は、後述のin vitroおよびin vivoアッセイを含めた当業者に既知の従来のアッセイにおけるその活性によって実証することができる。かかるアッセイは、それによって本発明の化合物の活性が他の既知の化合物の活性と比較できる手段も提供する。
【0078】
ACC1およびACC2の活性の直接阻害
本発明の化合物のACC阻害活性を標準手順に基づく方法によって実証した。例えば式(1)の化合物に関するACC活性の直接阻害を、ラット肝臓ACCおよび組換えヒトACC2の調製物を用いて決定した。
【0079】
[1]ラット肝臓ACCの調製。標準手順、例えばThampyおよびWakil(J.Biol.Chem.260:6318〜6323;1985)によって記載されているものに基づいて以下の方法を用いてラット肝臓からラット肝臓ACCを得た。
【0080】
重さが150〜200gの雄CDラットを18〜24時間絶食させ、その後高スクロース食餌(AIN−76Aげっ歯類食餌、Cat#D10001、Research Diets Inc.、米国ニュージャージー州New Brunswick)を3日間与え、その時点でそれらをCO2窒息によって殺した。肝臓を取り出し、氷冷リン酸緩衝溶液(PBS)中ですすぎ、Waring(登録商標)ブレンダー中の5倍の体積量の均質化バッファー(50mMリン酸カリウム、pH7.5、10mM EDTA、10mM 2−メルカプトエタノール、2mMベンズアミジン、0.2mMフェニルメチルスルホニルフルオライド(PMSF)、各5mg/Lのロイペプチン、アプロチニン、および抗トリプシン)中で、1分間4℃でホモジナイズした。後続の全ての操作を4℃で実施した。50%PEG溶液を加えることによってポリエチレングリコール(PEG)に対してホモジネートを3%とし、20,000xgで15分間遠心分離した。50%PEG溶液を加えて得られた上清を5%PEGに調節し、5分間撹拌した。20,000×gで20分間の遠心分離によってペレット(ACC活性を含有する)を捕集し、氷冷二重蒸留水ですすいで過剰PEGを除去し、均質化バッファーを用いて元のホモジネート体積の4分の1に再懸濁させた。硫酸アンモニウム(200g/リットル)を撹拌しながらゆっくり加えた。45分後、20,000×gで30分間の遠心分離によって酵素を捕集し、10mLの50mM HEPES、pH7.5、0.1mM DTT、1.0mM EDTA、および10%グリセリン中に再懸濁させ、同じバッファーで平衡化したSephadex(商標)G−25カラム(2.5cm×50cm)(Pharmacia、米国ニュージャージー州Piscataway現GE Healthcare)上で脱塩する。脱塩した酵素調製物をアリコート中に−70℃で保管した。即時使用の前に冷凍ラット肝臓ACCアリコートを解凍し、50mM HEPES、pH7.5、10mM MgCl2、10mMクエン酸三カリウム、2.0mMジチオスレイトール(DTT)、および0.75mg/mLの脂肪酸を含まないウシ血清アルブミン(BSA)を含有するバッファー中に500μg/mLまで希釈し、37℃で30分間プレインキュベートした。
【0081】
[2]ラット肝臓ACC阻害の測定。ACC活性の測定およびACC阻害の評価のために、試験化合物をジメチルスルホキシド(DMSO)中に溶解し、アリコート1μLをクリアボトム、96穴プレート(Perkin−Elmer PN#1450−514)に加えた。対照穴は、DMSO単独1μLまたは高阻害化合物1μLを含有する。上記のようにラット肝臓から得た酵素を、酵素バッファー中37℃で30分間活性化させてから化合物プレートに加えた。全穴に、50mM HEPES、pH7.5、7.5mM MgCl2、7.5mMクエン酸三カリウム、2mM DTT、50mg/mL BSAを含有するバッファーに溶かした活性酵素(1.33X)75μLを与える。活性酵素を化合物と一緒に10分間プレインキュベートしてから、50mM HEPES、pH7.5、7.5mM MgCl2、7.5mMクエン酸三カリウム、2mM DTT、50mg/mL BSA、120μMアセチルCoA、8.0mM ATP、38.4mM KHCO3、および1.6mM NaH[14C]O3(100μCi/μL)を含有する基質溶液25μLを加えることによって反応を開始した。反応中の最終基質濃度は、30μMアセチルCoA、9.6mM KHCO3、0.4mM NaH[14C]O3、および2mM ATPであった。3N HCl 25μLを加えることによって10分後に反応を停止させ、プレートを50℃で最低20時間乾燥させた。水30μLを乾燥させたプレートに加え、5分間混合した。Optiphase Supermix液体シンチレーション流体(Perkin Elmer、米国マサチューセッツ州Waltham)95μLを加え、プレートを20分間混合する。Wallac Trilux 1450 Microbeta LSCルミネセンスカウンターを用いてMCoA中への14Cの取り込みを測定した。
【0082】
[3]ヒトACC2阻害の測定。精製した組換えヒトACC2(hrACC2)を用いてヒトACC2阻害を測定した。簡単に言えば、ACC2の完全長CytomaxクローンをCambridge Bioscience Limitedから購入し、配列決定し、PCDNA5 FRT TO−TOPO(Invitrogen、米国カリフォルニア州Carlsbad)中にサブクローニングした。テトラサイクリン誘導によってACC2をCHO細胞中で発現させ、1μg/mLテトラサイクリン(Invitrogen、米国カリフォルニア州Carlsbad)を含む、グルタミン、ビオチン、ハイグロマイシンおよびブラストサイジンを含んだDMEM/F12 5リットル中に回収した。次いでACC2を含有するならし培地をSoftlink Soft Release Avidinカラム(Promega、米国ウィスコンシン州Madison)に加え、5mMビオチンで溶離した。ACC2 4mgを濃度0.05mg/mL(A280によって決定した)、推定純度95%(A280によって決定した)で溶離した。精製したACC2を50mMトリス、200mM NaCl、4mM DTT、2mM EDTA、および5%グリセリン中で透析した。プールしたタンパク質を凍結し、融解時に活性の損失なしに−80℃で保管した。ACC2活性の測定およびACC2阻害の評価のために、試験化合物をDMSO中に溶解し、最終DMSO濃度1%の5倍ストックとしてrhACC2酵素に加えた。50μM ATP反応のための製造条件を用いたTranscreener ADP検出FPアッセイキット(Bellbrook Labs、米国ウィスコンシン州Madison)を使用して、rhACC2をCostar#3767(Costar、米国マサチューセッツ州Canbridge)384穴プレート中で検定した。アッセイの最終条件は、50mM HEPES、pH7.5、5mM MgCl2、5mMクエン酸三カリウム、2mM DTT、0.5mg/mL BSA、30μMアセチルCoA、50μM ATP、および8mM KHCO3であった。通常、10μL反応を室温で1時間実施し、10μlのTranscreener停止および検出バッファーを加え、さらに1時間インキュベートした。620励起Cy5 FPジェネラルデュアルミラー、620励起Cy5 FPフィルター、688発光(S)および688(P)発光フィルターを用いたEnvision Fluorescenceリーダー(Perkinelmer)上でデータを取得した。
【0083】
上記ラット肝臓ACC放射酵素アッセイおよび組換えhACC2 transcreenerアッセイを用いた結果を、式(I)の化合物に関して以下の表にまとめて示す。
【0084】
【表3】
【0085】
実験動物におけるACC阻害の急性in vivo評価
本発明の化合物のACC阻害活性は、治療した動物からの肝臓および筋肉組織のマロニルCoAレベルを低減させるそれらの能力の評価によってin vivoで確認することができる。
【0086】
実験動物におけるマロニルCoA生成阻害の測定。この方法では、適宜の標準固形飼料および水で維持した雄スプレーグ・ドーリーネズミ(225〜275g)を、研究する前に無作為化した。動物は、実験を開始する18時間前に餌を与えるか、または絶食させた。明期に入って2時間後に、動物に5mL/kgの体積(0.5%メチルセルロース、ビヒクル)または適切な化合物(ビヒクル中で調製)を経口投与した。ビヒクルを与えた対照は、ベースライン組織マロニルCoAレベルを決定するために含め、一方絶食した動物は、マロニルCoAレベルに対して絶食が与えた影響を決定するために含めた。化合物投与の1時間後、動物をCO2で窒息させ、組織を取り出した。特に、血液を心臓穿刺によって捕集し、EDTAを含有するBD Microtainerチューブ中に入れ(BD Biosciences、米国ニュージャージー州)、混合し、氷上に置いた。血漿を用いて薬物暴露を決定した。肝臓および四頭筋を取り出し、直ちに凍結クランプし、金属箔で包み、液体窒素中で保管した。
【0087】
組織を液体N2下で微粉砕して、サンプリングにおける均一性を確保した。FastPrep FP120(Thermo Scientific、速度=5.5、45秒間)中のLysing Matrix A(MP Biomedicals、PN 6910)に溶かした5倍体積量の10%トリカルボン酸を用いて組織(150〜200mg)からマロニルCoAを抽出した。15000×gで30分間遠心分離(Eppendorf Centrifuge 5402)した後にマロニルCoAを含有する上清を細胞片から取り出した。分析が完了するまで試料を−80℃で安定して凍結させた。
【0088】
肝臓および筋肉組織中のマロニルCoAレベルの分析は、以下の方法論を用いて評価することができる。
【0089】
この方法は、以下の物質を利用する:Isotec(米国オハイオ州Miamisburg)から購入したマロニルCoAテトラリチウム塩およびマロニル−13C3−CoAトリリチウム塩、過塩素酸ナトリウム(Sigma、cat no.410241)、トリクロロ酢酸(ACROS、cat no.42145)、リン酸(J.T.Baker、cat no.0260−01)、ギ酸アンモニウム(Fluka、cat no.17843)、メタノール(HPLCグレード、J.T.Baker、cat no.9093−33)、および水(HPLCグレード、J.T.Baker、4218−03)を使用して必要とする移動相を作成した。Strata−Xオンライン固相抽出カラム、25μm、20mm×2.0mm I.D(cat no.00M−S033−B0−CB)をPhenomenex(米国カリフォルニア州Torrance)から得た。SunFire C18逆相カラム、3.5μm、100mm×3.0mm I.D.(cat no.186002543)をWaters Corporation(米国マサチューセッツ州Milford)から購入した。
【0090】
この方法は、以下の装備を利用して行うことができる。Agilent 1100バイナリーポンプ、Agilent 1100クォータナリーポンプおよび2個のValco Cheminert 6ポート2位置弁を用いた二次元クロマトグラフィー。10℃で保持したPeltier冷却スタックおよび20μLサンプリングループを備えたLEAP HTC PALオートサンプラーによって試料を導入した。オートサンプラーのためのニードル洗浄溶液は、Wash 1が10%トリクロロ酢酸水溶液(w/v)であり、Wash 2が90:10 メタノール:水である。MicroTech Scientific Micro−LC Column Ovenを用いて分析用カラム(Sunfire)を35℃で保持した。Turbo Ion Sprayを備えたABI Sciex API3000三連四重極質量分析計で溶離液を分析した。
【0091】
オンライン固相抽出および逆相クロマトグラフィーに関して異なる勾配溶離条件を用いて同時に二次元クロマトグラフィーを行った。この方法の一般的な計画は、第1次元をサンプル洗浄および目的の分析物の捕捉のために利用し、続いて第1次元から第2次元上への溶離のために両方の次元を短時間カップリングするようなものであった。両次元を続いて脱共役させて、定量化のために第2次元から分析物を勾配溶離させ、同時に配列中の次の試料のために第1次元を調製した。両次元を簡単に連結した場合、第1次元中の移動相の流れを第2次元上への分析物溶離のために逆流させて、最適ピーク幅、ピーク形状、および溶離時間を可能にする。
【0092】
HPLC系の第1次元は、Phenomenex strata−Xオンライン固相抽出カラムと溶媒Aが100mM過塩素酸ナトリウム/0.1%(v/v)リン酸および溶媒Bがメタノールからなる移動相とを利用した。
【0093】
HPLC系の第2次元は、Waters SunFire C18逆相カラムと溶媒Aが100mMギ酸アンモニウムおよび溶媒Bがメタノールからなる移動相とを利用した。勾配の初期条件を2分間保持し、この間に分析物を分析用カラムに移した。分析用で維持している間、オンラインSPEカラムから分析物を溶離するのに初期条件は十分な強度であったことが重要であった。その後、洗浄および再平衡ステップの前に4.5分で74.5%Aまで勾配が直線的に上がった。
【0094】
質量分析は、HPLCと連結すると、複雑な基質中の分析物を定量的に測定する場合に高選択的および高感度な方法になり得るが、依然として干渉および抑制を受ける。二次元HPLCを質量分析計と連結することによって、これらの干渉は著しく減少する。さらに、三連四重極質量分析計の多重反応モニタリング(MRM)特色を利用することによって、信号対雑音比が著しく改善した。
【0095】
このアッセイでは、質量分析計を、TurbolonSpray電圧2250Vで陽イオンモードにおいて操作した。噴霧ガスを450℃まで加熱した。デクラスタリング電位(DP)、集束電位(FP)、および衝突エネルギー(CE)を、それぞれ60、340、および42Vに設定した。四重極1(Q1)分解能をユニット分解能に設定し、四重極3(Q3)を低に設定した。CADガスを8に設定した。モニターしたMRM転移は、停止時間200msでのマロニルCoA:854.1→347.0m/z(L.Gaoら(2007)J.Chromatogr.B 853、303〜313)、およびマロニル−13C3−CoA:857.1→350.0m/zに関してであった。溶離液を、予想した分析物の溶離時間付近で質量分析計に分流し、他の場合には、供給源の保持および器械使用の頑健性の向上を助けるために廃棄した。得られたクロマトグラムを、Analystソフトウェア(Applied Biosystems)を用いて組み込んだ。マロニルCoAの組織濃度を、10%トリクロロ酢酸水溶液で調製した標準曲線から計算した。
【0096】
組織抽出物中のマロニルCoAの定量化のための標準曲線を含む試料を10%(w/v)トリクロロ酢酸(TCA)で調製し、0.01〜1pmol/μLの範囲であった。マロニル−13C3−CoA(最終濃度0.4pmol/μL)を各標準曲線成分および試料に内部標準として加えた。
【0097】
アッセイ内品質対照を6個調製した。絶食させた動物から調製したプールした抽出物から3個、および餌を与えた動物から作製したプールから3個。これらは、0、0.1または0.3pmol/μLの12C−マロニルCoAならびにマロニル−13C3−CoA(0.4pmol/μL)を加えた別々の試料として実行した。各アッセイ内品質対照は、水性組織抽出物を85%含有し、残りの部分が内部標準(0.4pmol/μL)および12C−マロニルCoAによって占められていた。アッセイ間対照を各実験に含めた。それらは1個の絶食させたおよび1個の餌を与えたプールした四頭筋の試料および/または1個の絶食させたおよび1個の餌を与えたプールした肝臓の試料で構成される。かかる全ての対照に、マロニル−13C3−CoA(0.4pmol/μL)を加えている。
【0098】
式(I)の化合物を上記in vivo試験で使用して、肝臓および筋肉組織中のマロニルCoAレベルに対するそれらの効果を調べた。結果を以下の表に示す。
【0099】
【表4】
【技術分野】
【0001】
本発明は、アセチルCoAカルボキシラーゼの阻害剤として作用する置換ピラゾロスピロケトン化合物およびアセチルCoAカルボキシラーゼ酵素(複数可)の阻害によって調節される疾患、状態または障害の治療におけるそれらの使用に関する。
【背景技術】
【0002】
アセチルCoAカルボキシラーゼ(ACC)は、ほとんどの種に見られる酵素のファミリーであり、アセチルCoAからのマロニルCoAの生成を触媒することによって脂肪酸の合成および代謝に関連している。哺乳動物では、ACC酵素の2つのアイソフォームが確認されている。脂肪および肝臓などの脂質合成組織において高レベルで発現されるACC1は、長鎖脂肪酸の生合成の最初の始動反応を制御する。アセチルCoAがマロニルCoAを形成するためにカルボキシル化していなかったら、クレブス回路によって代謝される。肝臓ACCの微量成分であるが、心臓および骨格筋内で主要なアイソフォームであるACC2は、ミトコンドリアのシストリック(cystolic)表面でマロニルCoAの生成を触媒し、カルニチンパルミトイル転移酵素を阻害することによってβ酸化においてどれくらい脂肪酸が利用されているか調節する。したがって、脂肪酸利用を増大させ、かつ新規脂肪酸合成の増大を防止することによって、ACC阻害剤の慢性投与は、高脂肪食または低脂肪食を摂取している肥満対象における肝臓および脂肪組織TG貯蔵を激減させることもでき、体脂肪の選択的な減量をもたらす。
【0003】
Abu−Etheigaらによって実施された研究は、ACC2が脂肪酸酸化を制御する上で極めて重要な役割を果たすことを示唆している。したがって、ACC2の阻害が、肥満および肥満関連疾患、例えば2型糖尿病に対する治療の標的となると考えられる。Abu−Etheiga,L.ら、「Acetyl−CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high−fat/high−carbohydrate diets」PNAS、100(18)10207〜10212(2003)を参照のこと。Choi,C.S.ら、「Continuous fat oxidation in acetyl−CoA carboxylase 2 knockout mice increases total energy expenditure,reduces fat mass,and improves insulin sensitivity」PNAS、104(42)16480〜16485(2007)も参照のこと。肝臓脂質蓄積が肝臓インスリン抵抗性の原因であり、2型糖尿病の病因となることがますます明らかになっている。ACC1とACC2は両方とも肝細胞における脂肪酸化の調節に関与しているが、ラット肝臓において主要なアイソフォームであるACC1は脂肪酸合成の唯一の調節物質であることを、Salvageらは実証した。さらに、彼らのモデルでは、両アイソフォームの組み合わさった減少(combined reduction)には、肝臓マロニルCoAレベルの著しい低減、摂食状態における脂肪酸化の増大、脂質蓄積の低下、およびin vivoにおけるインスリン作用の改善が必要とされる。したがって、肝臓ACC1およびACC2阻害剤が非アルコール性脂肪肝疾患(NAFLD)および肝臓インスリン抵抗性の治療に有用である可能性があることを示している。Savage,D.B.ら、「Reversal of diet−induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl−CoA carboxylases 1 and 2」J Clin Invest doi:10.1172/JCI27300を参照のこと。Oh,Wら、「Glucose and fat metabolism in adipose tissue of acetyl−CoA carboxylase 2 knowckout mice」PNAS、102(5)1384〜1389(2005)も参照のこと。
【発明の概要】
【発明が解決しようとする課題】
【0004】
その結果、脂肪酸合成を阻害し、脂肪酸酸化を増大させることによって肥満および肥満関連疾患(NAFLDおよび2型糖尿病など)を治療するためにACC1およびACC2阻害剤を含有する医薬が必要とされている。
【課題を解決するための手段】
【0005】
本発明は、以下の式(1)の構造を有する化合物に関する。
【0006】
【化1】
【0007】
請求項1の化合物は、図1(11.2±0.2、15.4±0.2、17.0±0.2、18.3±0.2、19.3±0.2および20.6±0.2の回折角(2θ)にピークがある)によって表されるパターンと本質的に同じ粉末X線回折パターンを有する結晶形で存在することができる。本明細書において多形体A型と呼ぶ。
【0008】
請求項1の化合物は、図2(7.8±0.2、11.2±0.2、13.7±0.2、15.9±0.2、18.7±0.2および20.2±0.2の回折角(2θ)にピークがある)によって表されるパターンと本質的に同じ粉末X線回折パターンを有する結晶形で存在することができる。本明細書において多形体B型と呼ぶ。
【0009】
本発明の別の態様は、(1)本発明の化合物(多形体A型およびB型を含む)、および(2)薬学的に許容できる賦形剤、希釈剤または担体を含む医薬組成物である。この組成物は、治療有効量の本発明の化合物を含むことが好ましい。この組成物は、少なくとも1種の別の薬剤(本明細書に記載されている)も含有することができる。好ましい薬剤には、抗肥満薬および/または抗糖尿病薬(以下に記載されている)が含まれる。
【0010】
本発明のさらに別の態様には、哺乳動物におけるアセチルCoAカルボキシラーゼ酵素(複数可)の阻害によって媒介される疾患、状態または障害を治療するための方法であって、かかる治療を必要とする哺乳動物、好ましくはヒトに、治療有効量の本発明の化合物、またはその医薬組成物を投与するステップを含む方法がある。
【0011】
アセチルCoAカルボキシラーゼの阻害剤によって媒介される疾患、障害または状態には、II型糖尿病および糖尿病関連疾患、例えば非アルコール性脂肪肝疾患(NAFLD)、肝臓インスリン抵抗性、高血糖、代謝症候群、耐糖能障害、糖尿病性神経障害、糖尿病性腎症、糖尿病性網膜症、肥満、異脂肪血症、高血圧、高インスリン血症およびインスリン抵抗性症候群が含まれる。好ましい疾患、障害または状態には、II型糖尿病、非アルコール性脂肪肝疾患(NAFLD)、肝臓インスリン抵抗性、高血糖、耐糖能障害、肥満、およびインスリン抵抗性症候群が含まれる。II型糖尿病、非アルコール性脂肪肝疾患(NAFLD)、肝臓インスリン抵抗性、高血糖および肥満がより好ましい。II型糖尿病が最も好ましい。
【0012】
好ましい実施形態は、動物における2型糖尿病および糖尿病関連障害の進行または発症を治療するまたは遅延させるための方法であって、かかる治療を必要とする動物に治療有効量の本発明の化合物またはその組成物を投与するステップを含む方法である。
【0013】
別の好ましい実施形態は、動物における肥満および肥満関連障害を治療するための方法であって、かかる治療を必要とする動物に治療有効量の本発明の化合物またはその組成物を投与するステップを含む方法である。
【0014】
さらに別の好ましい実施形態は、動物における非アルコール性脂肪肝疾患(NAFLD)または肝臓インスリン抵抗性を治療するための方法であって、かかる治療を必要とする動物に治療有効量の本発明の化合物またはその組成物を投与するステップを含む方法である。
【0015】
本発明の化合物は、他の薬剤(特に、本明細書の以下に記載の抗肥満および抗糖尿病薬)と併せて投与することができる。併用療法は、(a)本発明の化合物、本明細書に記載されている少なくとも1種の別の薬剤および薬学的に許容できる賦形剤、希釈剤もしくは担体を含む単一医薬組成物、または(b)(i)本発明の化合物および薬学的に許容できる賦形剤、希釈剤もしくは担体を含む第1の組成物と、(ii)本明細書に記載されている少なくとも1種の別の薬剤および薬学的に許容できる賦形剤、希釈剤もしくは担体を含む第2の組成物とを含む2種の別々の医薬組成物として投与することができる。医薬組成物は、同時にまたは順次および任意の順序で投与することができる。
【0016】
定義
X線回折ピーク位置に関して「本質的に同じ」という用語は、代表的なピーク位置および強度のばらつきを考慮していることを表す。例えば、ピーク位置(2θ)は、装置間の若干のばらつき、通常0.2°ほどを示すであろうことが当業者には理解されよう。さらに、相対ピーク強度は、装置間のばらつきと共に結晶化度、好ましい配向性、調製した試料の表面、および当業者に既知の他の要因によるばらつきを示すことになり、単に定性的尺度として見るべきであることが当業者には理解されよう。
【0017】
「治療有効量」という表現は、(i)特定の疾患、状態もしくは障害を治療もしくは予防する、(ii)特定の疾患、状態もしくは障害の1つもしくは複数の症候を減弱、回復、もしくは排除する、または(iii)本明細書に記載の特定の疾患、状態もしくは障害の1つもしくは複数の症候の発症を予防もしくは遅延する、本発明の化合物の量を表す。
【0018】
「動物」という用語は、ヒト(男性または女性)、コンパニオンアニマル(例えば、イヌ、ネコおよびウマ)、食物源の動物、動物園の動物、海生動物、鳥類および他の類似の動物種を意味する。「食用動物」は、ウシ、ブタ、ヒツジおよび家禽などの食物源の動物を意味する。
【0019】
「薬学的に許容できる」という表現は、物質または組成物が、製剤を構成する他の成分と、および/またはそれを用いて治療される哺乳動物と化学的および/または毒物学的に適合しなければならないことを示す。
【0020】
「治療すること(treating)」、「治療する(treat)」、または「治療(treatment)」という用語は、予防的な治療、すなわち、予防療法と待期療法の両方を包含する。
【0021】
「調節した(modulated)」または「調節すること(modulating)」、または「調節する(modulate(s))」という用語は、本明細書では、特に指示がない限り、本発明の化合物によるアセチルCoAカルボキシラーゼ(ACC)酵素(複数可)の阻害を意味する。
【0022】
「媒介した(mediated)」または「媒介すること(mediating)」または「媒介する(mediate(s))」という用語は、本明細書では、特に指示がない限り、特定の疾患、状態または障害の治療もしくは予防、(ii)特定の疾患、状態もしくは障害の1つもしくは複数の症候の減弱、回復もしくは排除、または(iii)アセチルCoAカルボキシラーゼ(ACC)酵素(複数可)を阻害することによる本明細書に記載の特定の疾患、状態もしくは障害の1つもしくは複数の症候の発症の予防もしくは遅延を意味する。
【0023】
「本発明の化合物」という用語は(特に具体的に特定しない限り)、式(I)の化合物ならびに、全ての互変異性体、配座異性体、および同位体的標識化合物を意味する。本発明の化合物の水和物および溶媒和物は、本発明の組成物と見なされ、その化合物は、それぞれ水または溶媒に関連している。
【図面の簡単な説明】
【0024】
【図1】式(I)の化合物のA型多形体の粉末X線回折パターン(pxrd)スペクトルを示す図である。
【図2】式(I)の化合物のB型多形体の粉末X線回折パターン(pxrd)スペクトルを示す図である。
【発明を実施するための形態】
【0025】
本発明の化合物は、特に本明細書に包含されている説明に照らして、化学分野でよく知られているものに類似したプロセスを含む合成経路によって合成することができる。出発物質は、一般にAldrich Chemicals(米国ウィスコンシン州Milwaukee)などの商業供給源から入手可能であり、または当業者によく知られている方法(例えば、Louis F.FieserおよびMary Fieser、Reagents for Organic Synthesis、1〜19巻、Wiley、New York(1967〜1999編)、もしくは補遺を含めたBeilsteins Handbuch der organischen Chemie、4、Aufl.編 Springer−Verlag、Berlin(Beilsteinオンラインデータベースによっても入手可能である)に一般に記載されている方法によって調製される)を用いて容易に調製される。
【0026】
例示のために、以下に示した反応スキームは、本発明の化合物ならびに重要な中間体を合成するための潜在的な経路を提供する。個々の反応ステップのより詳細な説明については、以下の実施例の項を参照のこと。他の合成経路を使用して本発明の化合物を合成するできることが当業者には理解されよう。特定の出発物質および試薬をスキームに示し、以下で論じているが、他の出発物質および試薬と容易に置き換えて、種々の誘導体および/または反応条件を得ることができる。さらに、後述の方法によって調製した多くの化合物は、当業者によく知られている従来の化学的手法を用いて本開示に照らしてさらに修飾することができる。
【0027】
本発明の化合物の調製では、中間体の離れた官能基(remote functionality)(例えば、第一級または第二級アミン)の保護が必要なこともある。このような保護の必要性は、離れた官能基の性質および調製方法の条件に応じて変わるものである。適当なアミノ保護基(NH−Pg)には、アセチル、トリフルオロアセチル、t−ブトキシカルボニル(BOC)、ベンジルオキシカルボニル(CBz)および9−フルオレニルメチレンオキシカルボニル(Fmoc)がある。同様に、「ヒドロキシ保護基」は、ヒドロキシ官能基を遮断または保護するヒドロキシ基の置換基を意味する。適当なヒドロキシル保護基(O−Pg)には、例えば、アリル、アセチル、シリル、ベンジル、パラメトキシベンジル、トリチル、などがある。このような保護の必要性は、当業者により容易に決定される。保護基およびそれらの使用の一般的な説明については、T.W.Greene、Protective Groups in Organic Synthesis、John Wiley & Sons、New York、1991を参照のこと。
【0028】
スキームIは、式(I)を有する本発明の化合物を得るために使用できる一般的な手順の概要である。
【0029】
【化2】
【0030】
中間体ヒドラゾン(1a)は、酢酸などの酸性環境において室温でメチルグリオキサル(SM−1)を1−t−ブチルヒドラジン(SM−2)で処理することによって形成することができる。還流させた水性酢酸中でヒドラゾン(1a)をオキサルアルデヒド(SM−3)で処理することによって、1−(4−ヒドロキシ−1H−ピラゾール−3−イル)エタノン中間体(1b)が得られる。あるいは、1H−ピラゾール中間体(1b)は、還流させた水性酢酸中でオキサルアルデヒド(SM−3)を1−t−ブチルヒドラジンオキサレートで処理することによって直接形成することもできる。アミノ保護ピラゾロスピロケトン中間体(1c)は、アミン(好ましくは、ピロリジン)の存在下において室温でアミノ保護4−ピペリドン(好ましくは、BOC保護基)を1−(4−ヒドロキシ−1H−ピラゾール−3−イル)エタノン中間体(1b)に加えることによって形成することができる。次いで保護基を除去して、ピラゾロスピロケトン中間体(1d)を得ることができる。アミノ保護基を除去するために使用した条件は、どの保護基を使用したかによって決まることになる。例えば、BOC保護基は、強酸(例えば、HCl)を用いた処理によって除去することができる。次いで最終化合物(I)は、1H−インダゾール−5−カルボン酸を用いた標準的なペプチドカップリング反応を使用することによって形成することができる。例えば、ピラゾロスピロケトン中間体(1d)および1H−インダゾール−5−カルボン酸は、例えばTHFおよび/またはDMFなどの適当な溶媒中に、ヒドロキシベンゾトリアゾール(HOBt)などの活性化剤の存在下または不在下、およびN,N−ジイソプロピルエチルアミン(DIEA)またはN−メチルモルフォリン(NMM)などの適当な塩基の存在下で、1H−インダゾール−5−カルボン酸とO−(7−アザベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロフォスフェート(HATU)などのペプチドカップリング試薬とを接触させることによって、活性カルボン酸エステルを形成し、次いで活性カルボン酸エステルとピラゾロスピロケトン中間体(1d)とを接触させることによって結合させて、式(1)の化合物を形成することができる。あるいは、式(1)の化合物は、まず例えば塩化チオニルと反応させることによって1H−インダゾール−5−カルボン酸を酸塩化物に転換し、次いで酸塩化物をピラゾロスピロケトン中間体(1d)と反応させて式(1)の化合物を形成することによって形成することができる。さらに別の代替法は、THFおよび/またはDMFなどの適当な溶媒中にN−メチルモルフォリンなどの適当な塩基の存在下で、1H−インダゾール−5−カルボン酸を2−クロロ−4,6−ジメトキシトリアジンで処理することを伴う。活性エステルには、THFおよび/またはDMFなどの適当な溶媒に溶かしたピラゾロスピロケトン中間体(1d)とN−メチルモルフォリンなどの塩基の溶液を加える。
【0031】
本発明の化合物は、2種以上の結晶形で存在することができる。本発明の化合物の多形体(溶媒和物および水和物を含む)は、本発明を構成し、異なる条件下における本発明の化合物の結晶化によって調製することができる。例えば、再結晶のための異なる溶媒または異なる溶媒混合物の使用、異なる温度における結晶化、結晶化中に超高速から超低速までの範囲にわたる種々の冷却モードなどである。多形体は、本発明の化合物の加熱または融解とそれに続く段階的または高速冷却によって得ることもできる。多形体の存在は、固体プローブ核磁気共鳴(NMR)分光法、赤外(IR)分光法、示差走査熱分析、粉末X線回折またはこのような他の技術によって確認することができる。
【0032】
本発明には、1個または複数個の原子を通常自然界に見られる原子量または質量数と異なる原子量または質量数を有する原子と置き換えていることを除けば、式(1)によって記述されたものと等しい、同位体的標識化合物も含まれる。式(I)の化合物に取り込める同位体の例には、水素、炭素、窒素、酸素、硫黄およびフッ素の同位体、例えばそれぞれ2H、3H、13C、14C、15N、18O、17O、35S、36Cl、125I、129I、および18Fがある。いくつかの本発明の同位体的標識化合物、例えば3Hおよび14Cなどの放射性同位体を取り込んでいるものは、薬物および/または基質組織分布アッセイに有用である。トリチウム化(すなわち、3H)、および炭素−14(すなわち、14C)、同位体は、それらの調製の容易さおよび検出感度に関して特に好ましい。さらに、より重い同位体、例えば重水素(すなわち、2H)との置換は、より大きい代謝安定性の結果として生じるいくつかの治療的利点、例えばin vivo半減期の増大または必要投与量の低減をもたらす可能性があり、したがって、状況によっては好ましいこともある。本発明の同位体的標識化合物は、同位体的標識されていない試薬の代わりに容易に入手可能な同位体的標識された試薬を使用することにより、以下のスキームおよび/または実施例に開示されている手順を実施することによって、一般に調製することができる。
【0033】
本発明の化合物は、分離可能であり得る異なる安定な立体配座形態で存在することができる。例えば立体障害または環歪みのための、不斉単結合の周りの束縛回転が原因のねじれ非対称により、異なる配座の分離が可能となり得る。本発明の化合物は、式(1)の化合物の各配座異性体およびその混合物をさらに含む。
【0034】
本発明の化合物は、アセチルCoAカルボキシラーゼ酵素(複数可)(特に、ACC1およびACC2)の阻害によって調節される疾患、状態および/または障害を治療するのに有用である。したがって、本発明の別の実施形態は、治療有効量の本発明の化合物および薬学的に許容できる賦形剤、希釈剤または担体を含む医薬組成物である。本発明の化合物(そこに使用した組成物およびプロセスを含む)は、本明細書に記載の治療用途のための医薬の製造に使用することもできる。
【0035】
代表的な製剤は、本発明の化合物および担体、希釈剤または賦形剤を混合することによって調製する。適当な担体、希釈剤および賦形剤は、当業者によく知られており、炭水化物、蝋、水溶性および/または水膨潤性ポリマー、親水性または疎水性物質、ゼラチン、油、溶媒、水などの物質が含まれる。使用する特定の担体、希釈剤または賦形剤は、本発明の化合物を適用する手段および目的によって決まることになる。溶媒は、一般に哺乳動物に投与するのが安全と当業者によって認められた(GRAS)溶媒に基づいて選択される。一般に、安全な溶媒は、水などの非毒性水性溶媒および水に可溶性または混和性の他の非毒性溶媒である。適当な水性溶媒には、水、エタノール、プロピレングリコール、ポリエチレングリコール(例えば、PEG400、PEG300)など、およびその混合物がある。製剤は、1種または複数のバッファー、安定剤、界面活性剤、湿潤剤、平滑剤、乳化剤、懸濁化剤、保存剤、酸化防止剤、不透明化剤、流動促進剤、加工助剤、着色剤、甘味料、芳香剤、矯味矯臭剤および薬物(すなわち、本発明の化合物またはその医薬組成物)の洗練された体裁を与える、または医薬品(すなわち、医薬)の製造に役立つ他の既知の添加剤も含んでいてよい。
【0036】
製剤は、従来の溶解および混合手順を用いて調製することができる。例えば、原薬(すなわち、本発明の化合物または化合物の安定化形態(例えば、シクロデキストリン誘導体または他の既知の複合体形成剤との複合体))を、上記の1種または複数の賦形剤の存在下で適当な溶媒に溶解させる。低水溶性化合物の溶解速度は、参照により本明細書に組み込む、Takeuchi,H.らによって「Enhancement of the dissolution rate of a poorly water−soluble drug(tolbutamide) by a spray−drying solvent depostion method and disintegrants」J.Pharm.Pharmacol.、39、769〜773(1987)、および欧州特許第0901786B1号(米国特許出願公開第2002/009494号)に記載されているものなど、噴霧乾燥した分散体の使用によって高めることができる。本発明の化合物は、通常、容易に制御可能な薬物投与量を提供し、患者に洗練されかつ取り扱いやすい製品を与えるために、医薬剤形に製剤する。
【0037】
医薬組成物には、本発明の化合物の溶媒和物および水和物も含まれる。「溶媒和物」という用語は、本発明の化合物と1種または複数の溶媒分子との分子複合体を意味する。かかる溶媒分子は、レシピエントに対して無害であることが知られている、製薬技術分野で一般に使用されているもの、例えば、水、エタノール、エチレングリコールなどである。「水和物」という用語は、溶媒分子が水である複合体を意味する。溶媒和物および/または水和物は、結晶形で存在することが好ましい。メタノール、メチルt−ブチルエーテル、酢酸エチル、酢酸メチル、(S)−プロピレングリコール、(R)−プロピレングリコール、1,4−ブチン−ジオールなど、他の溶媒は、より望ましい溶媒和物の調製における中間溶媒和物として使用することができる。
【0038】
使用するための医薬組成物(または製剤)は、薬物を投与するために使用する方法に応じて種々の形に包装することができる。一般に、販売用の商品には、適切な形態の医薬製剤が入れられている容器が含まれる。適当な容器は、当業者によく知られており、ボトル(プラスチックおよびガラス)、サッシェ、アンプル、ビニール袋、金属シリンダーなどの材料が含まれる。容器は、包装の中身に不用心に近づくのを防ぐためにいたずら防止セットも含んでいてよい。その上、容器には、容器の中身を説明するラベルを付けている。ラベルは、適切な警告も含んでいてよい。
【0039】
本発明は、かかる治療を必要とする動物に、治療有効量の本発明の化合物または有効量の本発明の化合物と薬学的に許容できる賦形剤、希釈剤、もしくは担体とを含む医薬組成物を投与することを含む、動物におけるアセチルCoAカルボキシラーゼ酵素(複数可)の阻害によって調節される疾患、状態および/または障害を治療する方法をさらに提供する。この方法は、アセチルCoAカルボキシラーゼ酵素(複数可)の阻害から恩恵を受ける疾患、状態および/または障害を治療するのに特に有用である。
【0040】
本発明の一態様は、肥満、および肥満関連障害(例えば、過体重、体重増加、または体重維持)の治療である。
【0041】
肥満および過体重は一般に肥満度指数(BMI)によって定義され、これは全体脂肪と相関関係があり、疾患の相対危険度を推定する。BMIは、キログラム表示の体重を、メートル表示の身長の二乗で割って算出される(kg/m2)。過体重は通常、BMIが25〜29.9kg/m2と定義され、肥満は通常、BMIが30kg/m2と定義される。例えば、National Heart,Lung,and Blood Institute、Clinical Guidelines on the Identification,Evaluation,and Treatment of Overweight and Obesity in Adults、The Evidence Report、Washington,DC:U.S.Department of Health and Human Services、NIH publication no.98−4083(1998)を参照のこと。
【0042】
本発明の別の態様は、糖尿病または1型(インスリン依存真性糖尿病、「IDDM」とも呼ばれる)および2型(非インスリン依存真性糖尿病、「NIDDM」とも呼ばれる)糖尿病、耐糖能障害、インスリン抵抗性、高血糖、ならびに糖尿病性合併症(アテローム性動脈硬化症、冠状動脈性心臓病、発作、末梢血管疾患、腎症、高血圧、神経障害、および網膜症など)を含めた糖尿病関連障害の進行または発症を治療または遅延するためである。
【0043】
本発明のさらに別の態様では、代謝症候群などの肥満同時罹患状態(obesity comorbidities)の治療である。代謝症候群には、異脂肪血症、高血圧、インスリン抵抗性、糖尿病(例えば、2型糖尿病)、冠状動脈疾患および心不全などの疾患、状態または障害が含まれる。代謝症候群に関するより詳細な情報については、例えば、Zimmet,P.Z.ら、「The Metabolic Syndrome: Perhaps an Etiologic Mystery but Far From a Myth − Where Does the International Diabetes Federation Stand?」、Diabetes & Endocrinology、7(2)、(2005)、およびAlberti,K.G.ら、「The Metabolic Syndrome − A New Worldwide Definition」、Lancet、366、1059〜62(2005)を参照のこと。本発明の化合物の投与は、薬物を含有しないビヒクル対照と比較して、血漿レプチン、C反応性タンパク質(CRP)および/またはコレステロールの低下など少なくとも1種の心臓血管疾患危険因子で統計的に有意な(p<0.05)低減をもたらすことが好ましい。本発明の化合物の投与は、グルコース血清レベルでも統計的に有意な(p<0.05)低減をもたし得る。
【0044】
本発明のさらに別の態様では、非アルコール性脂肪肝疾患(NAFLD)および肝臓インスリン抵抗性の治療である。
【0045】
体重が約100kgの正常な成人ヒトの場合、体重1キログラム当たり約0.001mg〜約10mgの範囲の投与量が通常十分であり、約0.01mg/kg〜約5.0mg/kgが好ましく、約0.01mg/kg〜約1mg/kgがより好ましい。しかしながら、治療対象の年齢および体重、意図される投与経路、投与される特定の化合物などよって、一般的な投与量範囲にいくらかの変動性が求められてもよい。特定の患者についての投与量範囲および最適投与量の決定は、本開示の利点を有する当業者の能力の十分に範囲内である。本発明の化合物は、徐放、制御放出、および遅延放出製剤に使用でき、その形態も当業者によく知られているという点にも留意されたい。
【0046】
本発明の化合物は、本明細書に記載の疾患、状態および/または障害の治療のための他の薬剤と併用してもよい。したがって、本発明の化合物を他の薬剤と併せて投与することを含む治療の方法も提供している。本発明の化合物と併せて使用できる適当な薬剤には、抗肥満薬(食欲抑制薬を含む)、抗糖尿病薬、抗高血糖症薬、脂質低下剤、および抗高血圧剤がある。
【0047】
適当な抗肥満薬には、11β−ヒドロキシステロイドデヒドロゲナーゼ−1(11β−HSD1型)阻害剤、ステアロイルCoAデサチュラーゼ−1(SCD−1)阻害剤、MCR−4アゴニスト、コレシストキニン−A(CCK−A)アゴニスト、モノアミン再取り込み阻害剤(シブトラミンなど)、交感神経様作用薬、β3アドレナリンアゴニスト、ドーパミンアゴニスト(ブロモクリプチンなど)、メラノサイト刺激ホルモン類似体、5HT2cアゴニスト、メラニン凝集ホルモンアンタゴニスト、レプチン(OBタンパク質)、レプチン類似体、レプチンアゴニスト、ガラニンアンタゴニスト、リパーゼ阻害剤(テトラヒドロリプスタチン、すなわちオルリスタットなど)、食欲抑制薬(ボンベシンアゴニストなど)、ニューロペプチドYアンタゴニスト(例えば、NPY Y5アンタゴニスト)、PYY3−36(その類似体を含む)、甲状腺模倣剤、デヒドロエピアンドロステロンまたはその類似体、グルココルチコイドアゴニストまたはアンタゴニスト、オレキシンアンタゴニスト、グルカゴン様ペプチド−1アゴニスト、毛様体神経栄養因子(Regeneron Pharmaceuticals、Inc.、米国ニューヨーク州TarrytownおよびProcter&Gamble Company、米国オハイオ州Cincinnatiから入手可能なAxokine(商標)など)、ヒトアグーチ関連タンパク質(AGRP)阻害剤、グレリンアンタゴニスト、ヒスタミン3アンタゴニストまたはインバースアゴニスト、ニューロメジンUアゴニスト、MTP/ApoB阻害剤(例えば、ジルロタピドなどの消化管選択的MTP阻害剤)、オピオイドアンタゴニスト、オレキシンアンタゴニストなどがある。
【0048】
本発明の組み合わせた態様で使用するのに好ましい抗肥満薬には、消化管選択的MTP阻害剤(例えば、ジルロタピド、ミトラタピドおよびインプリタピド、R56918(CAS番号403987)およびCAS番号913541−47−6)、CCKaアゴニスト(例えば、PCT公開番号WO2005/116034または米国特許出願公開第2005−0267100A1号に記載されているN−ベンジル−2−[4−(1H−インドール−3−イルメチル)−5−オキソ−1−フェニル−4,5−ジヒドロ−2,3,6,10b−テトラアザ−ベンゾ[e]アズレン−6−イル]−N−イソプロピル−アセトアミド)、5HT2cアゴニスト(例えば、ロルカセリン)、MCR4アゴニスト(例えば、米国特許第6,818,658号に記載されている化合物)、リパーゼ阻害剤(例えば、セチリスタット)、PYY3−36(本明細書では「PYY3−36」にはペグ化PYY3−36、例えば、米国特許出願公開第2006/0178501号に記載されているものなどの類似体が含まれる)、オピオイドアンタゴニスト(例えば、ナルトレキソン)、オレオイル−エストロン(CAS番号180003−17−2)、オビネピチド(obinepitide)(TM30338)、プラムリンチド(Symlin(登録商標))、テソフェンシン(NS2330)、レプチン、リラグルチド、ブロモクリプチン、オルリスタット、エクセナチド(Byetta(登録商標))、AOD−9604(CAS番号221231−10−3)およびシブトラミンがある。本発明の化合物および併用療法は、運動および堅実な食事と併せて投与することが好ましい。
【0049】
適当な抗糖尿病薬には、ナトリウム−グルコース共輸送体(SGLT)阻害剤、ホスホジエステラーゼ(PDE)−10阻害剤、ジアシルグリセロールアシルトランスフェラーゼ(DGAT)1または2阻害剤、スルホニル尿素(例えば、アセトヘキサミド、クロルプロパミド、ジアビネース、グリベンクラミド、グリピジド、グリブリド、グリメピリド、グリクラジド、グリペンチド、グリキドン、グリソラミド、トラザミド、およびトルブタミド)、メグリチニド、α−アミラーゼ阻害剤(例えば、テンダミスタット、トレスタチンおよびAL−3688)、α−グルコシドヒドロラーゼ阻害剤(例えば、アカルボース)、α−グルコシダーゼ阻害剤(例えば、アジポシン、カミグリボース、エミグリテート、ミグリトール、ボグリボース、プラジミシン−Q、およびサルボスタチン)、PPARγアゴニスト(例えば、バラグリタゾン、シグリタゾン、ダルグリタゾン、エングリタゾン、イサグリタゾン、ピオグリタゾン、ロシグリタゾンおよびトログリタゾン)、PPARα/γアゴニスト(例えば、CLX−0940、GW−1536、GW−1929、GW−2433、KRP−297、L−796449、LR−90、MK−0767およびSB−219994)、ビグアナイド(例えば、メトホルミン)、グルカゴン様ペプチド1(GLP−1)アゴニスト(例えば、Byetta(商標)、エキセンディン−3およびエキセンディン−4)、タンパク質チロシンホスファターゼ−1B(PTP−1B)阻害剤(例えば、トロズスクエミン、ヒルチオサール抽出物、およびZhang,S.ら、Drug Discovery Today、12(9/10)、373〜381(2007)によって開示されている化合物)、SIRT−1阻害剤(例えば、レセルバトロール(reservatrol))、ジペプチジルペプチデアーゼIV(DPP−IV)阻害剤(例えば、シタグリプチン、ビルダグリプチン、アログリプチンおよびサクサグリプチン)、インスリン分泌促進物質、脂肪酸酸化阻害剤、A2アンタゴニスト、c−junアミノ末端キナーゼ(JNK)阻害剤、インスリン、インスリン様作用薬、グリコーゲンホスホリラーゼ阻害剤、VPAC2受容体アゴニストおよびグルコキナーゼ活性化剤がある。好ましい抗糖尿病薬は、メトホルミン、グルカゴン様ペプチド1(GLP−1)アゴニスト(例えば、Byetta(商標))およびDPP−IV阻害剤(例えば、シタグリプチン、ビルダグリプチン、アログリプチンおよびサクサグリプチン)である。
【0050】
上記の全ての米国特許および公開は、参照により本明細書に組み込まれている。
【0051】
本明細書の以下に記載されている実施例は、例示的な目的のために過ぎない。本明細書において反映されている組成物、方法、および種々のパラメーターは、本発明の種々の態様および実施形態を例示するものに過ぎず、決して特許請求した発明の範囲を限定するものではない。当業者は、反応の規模および使用した特定の装備に基づいて試薬、溶媒および条件をどのように最適化するのか知っているはずである。
【実施例】
【0052】
後述の化合物および中間体は、有機化学の命名法およびCAS索引規則に対するIUPAC(国際純正および応用化学連合)勧告に基づいて一般に命名した。特に指摘がない限り、全ての反応物は、商業的に得た。本明細書の以下に引用した全ての参考文献は、参照により組み込まれている。
【0053】
フラッシュクロマトグラフィーは、Stillら、J.Org.Chem.、1978、43、2923によって記載されている方法に基づいて行った。
【0054】
本明細書で論じている全てのBiotage(登録商標)精製は、KP−SILシリカ(40〜63μM、60オングストローム)を含有する40Mまたは40S Biotage(登録商標)カラム(Bioatge AB、Uppsala、スウェーデン)を用いて行った。
【0055】
本明細書で論じている全てのCombiflash(登録商標)精製は、充填RediSep(登録商標)シリカカラムを利用したCombiFlash(登録商標)Companion system(Teledyne Isco、米国ネブラスカ州Lincoln)を用いて行った。
【0056】
質量スペクトルは、Waters(Waters Corp.、米国マサチューセッツ州Milford)Micromass Platform II分光計で記録した。特に指定のない限り、質量スペクトルは、Waters(米国マサチューセッツ州Milford)Micromass Platform II分光計で記録した。
【0057】
プロトンNMR化学シフトは、テトラメチルシランから低磁場に百万分率で示され、Varian Unity 400または500MHz(メガヘルツ)分光計(Varian Inc.、米国カリフォルニア州Palo Alto)で記録した。NMR化学シフトは、テトラメチルシラン(プロトンの場合)またはフルオロトリクロロメタン(フッ素の場合)から低磁場に百万分率で示される。
【0058】
重要な中間体および出発物質
1H−インダゾール−5−カルボン酸は、Tyger Scientific、Inc.、米国ニュージャージー州Ewingから入手可能である。
【0059】
中間体2’−tert−ブチル−2’H−スピロ[ピペリジン−4,5’−ピラノ[3,2−c]ピラゾール]−7’(6’H)−オン塩酸塩(I−1a)の調製:
【0060】
【化3】
【0061】
ピルブアルデヒド(26.2mL、160mmol)のH2O(120mL)溶液を、tert−ブチルヒドラジン・HCl(20g、124mmol)のH2O(500mL)溶液に20分にわたって加えた。この溶液を室温で5時間撹拌した。次いで反応混合物を酢酸エチル(5回)を用いて抽出した。合わせた有機層を乾燥(Na2SO4)させ、減圧下で濃縮した。次いで残留物を、酢酸エチル/ヘプタン(10:90〜40:60)の勾配を用いて溶離するフラッシュクロマトグラフィー(シリカゲル)によって精製して、2−オキソプロパナールtert−ブチルヒドラゾン13.1g(74%)を琥珀色油としてもたらした。
【0062】
グリオキサール(2.9mL、25.3mmol)の40%水溶液を、水(14mL)に溶かした2−オキソプロパナールtert−ブチルヒドラゾン(1.20g、8.44mmol)に加えた。次いで混合物を5時間還流させながら加熱した。反応混合物を室温まで冷却し、EtOAcを用いて4回抽出した。合わせた有機層を乾燥(Na2SO4)させ、ろ過し、減圧下で濃縮した。残留物をフラッシュクロマトグラフィー(シリカゲル)によって精製し、ヘプタン/酢酸エチル(100:0〜90:10)の勾配を用いて溶離して、1−(4−ヒドロキシ−1−tert−ブチル−1H−ピラゾール−3−イル)エタノン1.06g(69%)を無色油として得た。
【0063】
1−(4−ヒドロキシ−1−tert−ブチル−1H−ピラゾール−3−イル)エタノン(6.63g、36.4mmol)のMeOH(73mL)溶液に、ピロリジン(3.6mL、43.7mmol)および1−(N−Boc)−4−ピペリドン(8.7g、43.7mmol)を加えた。暗赤色溶液を室温で終夜撹拌した。溶液を濃縮し、残留物をEtOAcに溶解し、1N NaOHおよびブラインで洗浄した。層を分離し、次いで有機層を取っておいた。合わせた水層を酢酸エチルで抽出した。層を分離し、有機層を1N NaOHおよびブラインで洗浄した。全ての有機層を合わせ、次いで1N HCl、およびブラインで洗浄し、乾燥(Na2SO4)させ、ろ過し、減圧下で濃縮して赤色ガム(11g)を生じさせた。赤色ガムを粉砕し、25%EtOAc/ヘキサン(125mL)中で加熱し、ガムは黄色固体に変化したが、還流させながら完全に溶解しなかった。混合物を室温まで冷却し、ろ過して生成物であるオフホワイト固体(1.69g)を得た。ろ液を約5〜10mL(ヘキサン、EtOAcおよびアセトンを含む)まで濃縮し、次いで追加量の2%EtOAc/ヘキサン(100mL)を加え、その後固体が沈殿し始めた。混合物を終夜撹拌した。固体をろ過して、別の所望の生成物3.89gをオフホワイト固体として得た。全部で、5.58g(42%)のtert−ブチル2’−tert−ブチル−7’−オキソ−6’,7’−ジヒドロ−1H,2’H−スピロ[ピペリジン−4,5’−ピラノ[3,2−c]ピラゾール]−1−カルボキシレートをオフホワイト固体として単離した。
【0064】
tert−ブチル2’−tert−ブチル−7’−オキソ−6,7’−ジヒドロ−1H,2’H−スピロ[ピペリジン−4,5’−ピラノ[3,2−c]ピラゾール]−1−カルボキシレート(2.73g、7.5mmol)の1,4−ジオキサン(15mL)溶液に室温でHCl溶液(1,4−ジオキサン中4M、15mL、60mmol)を加えた。混合物を室温で3時間撹拌した。次いで反応混合物を濃縮乾固した。得られたピンク色固体(2.6g)を2−メチルテトラヒドロフラン(20mL)および少量のEtOH(1mL)と一緒に粉砕した。固体をろ過し、2−メチルテトラヒドロフラン(20mL)で洗浄し、50℃で真空乾燥させて、表題化合物(I−1a)2.15g(95%)を白色固体としてを得た。
【0065】
(実施例1)
2’−tert−ブチル−1−(1H−インダゾール−5−イルカルボニル)−2’H−スピロ[ピペリジン−4,5’−ピラノ[3,2−c]ピラゾール]−7’(6’H)−オン(I)の調製:
【0066】
【化4】
【0067】
N−ジメチルホルムアミド(1mL)に溶かした1H−インダゾール−5−カルボン酸(27mg、0.17mmol)、2−クロロ−4,6−ジメトキシ−1,3,5−トリアジン(36mg、0.20mmol)およびN−メチルモルフォリン(NMM)(19uL、0.17mmol)の混合物を室温で35分間撹拌してからNMM(3当量)、続いて2’−tert−ブチル−2’H−スピロ[ピペリジン−4,5’−ピラノ[3,2−c]ピラゾール]−7’(6’H)−オン・HCl(I−1a:50mg、0.17mmol)を加えた。混合物を室温で終夜撹拌した。溶媒を減圧下で除去し、残留物をCH2Cl2に溶解させ、飽和水性NH4Clで洗浄した。水相をCH2Cl2(2回)を用いて逆抽出した。合わせた有機抽出物を水および飽和水性NaClで洗浄してからMgSO4上で乾燥させた。物質をろ過し、濃縮し、分取薄層クロマトグラフィー(95:5 CHCl3/MeOH)によって精製した。所望の物質を続いてEt2Oと一緒に粉砕し、ろ過し、固体を真空下に50℃で乾燥させて、所望の生成物(21mg、31%)を生じさせた。
1H NMR (500 MHz, DMSO-d6) δ ppm 13.26 (1 H, br. s.), 8.14 (1 H, s), 7.86 (1 H, s), 7.81 (1 H,
s), 7.58 (1 H, d, J=8.54 Hz), 7.40 (1 H, br. s.), 3.18 (2 H, br. s.), 2.75 (2
H, s), 1.99 (2 H, s), 1.88 (2 H, br. s.), 1.74 (2 H, t), 1.51 (9 H, s).
【0068】
(実施例2)
あるいは、化合物(I)は、結晶性生成物(本明細書において「A型」と呼ぶ)を生成する以下の手順を用いて調製することができる。
【0069】
400L反応器に、2’−tert−ブチル−2’H−スピロ[ピペリジン−4,5’−ピラノ[3,2−c]ピラゾール]−7’(6’H)−オン・HCl(I−1a:6.6kg、22.0モル)、1H−インダゾール−5−カルボン酸(3.26kg、20.1モル)、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩(4.85kg、25.3モル)、アセトニトリル(124L)、およびピリジン(13.9L、172モル)を入れた。溶液を周囲温度で16時間撹拌し、次いで酢酸エチル(250L)で希釈し、10wt%の水性クエン酸(100L)で2回洗浄した。51Lの溶液体積まで蒸留するために有機層を加熱し、次いで酢酸エチル(約85L)を加え、76℃の内部温度が得られるまで蒸留した(溶液体積約55L)。次いで溶液を3時間にわたって周囲温度まで冷却し、固体をヌッチェフィルターに通してろ過し、酢酸エチル(17L)で洗浄し、50℃で12時間真空下で乾燥させた。化合物(I)を白色結晶性固体(3.89kg、9.55モル、48%)として単離した。融点:265℃。
1H NMR (400 MHz, DMSO-d6) δ ppm 13.2 (1H, s), 8.11 (1H, s), 7.83 (1H, s), 7.77 (1H, s), 7.55 (1
H, d, J=8.0 Hz), 7.37 (1 H, dd, J=8.0, 1.2 Hz), 4.35-3.45 (2 H, m), 3.30-3.15
(2 H, m), 2.72 (2H, s), 1.98-1.86 (2 H, m),1.73 (2 H, td, J=12.0, 4.0 Hz), 1.48
(9 H, s).
13C NMR (100 MHz, DMSO-d6) δ ppm: 186.5, 170.2, 147.6, 140.6, 134.9, 134.2, 128.6, 125.9, 122.8,
120.5, 114.3, 110.7, 81.6, 61.0, 49.0, 34.0, 29.7.
【0070】
式(I)の化合物のA型多形体に関するX線粉末回折パターンを、銅放射線によるSiemens D5000回折計を用いて作成した。この機器は、線状焦点X線管を備えていた。管電圧およびアンペア数を、それぞれ38kVおよび38mAに設定した。発散および散乱スリットを1mmに設定し、受信スリットを0.6mmに設定した。Sol−Xエネルギー分散X線検出器を用いて回折CuKα1放射線(λ=1.54056Å)を検出した。3.0〜40°2θから2.4°2θ/分(1秒/0.04°2θステップ)でθ2θ連続スキャンを使用した。アルミナ標準物質(NIST標準参照物質1976)を分析して機器アライメントを確認した。データを捕集し、BRUKER AXS DIFFRAC PLUSソフトウェアバージョン2.0を用いて分析した。分析用に試料を石英ホルダーに取り付けることによって準備した。Bruker Instruments社がSiemens社を買収したことを留意されたい。したがって、Bruker D5000機器は、本質的にSiemens D5000と同じである。以下の表は、A型結晶について観察されたバックグラウンドに対して5倍閾値を有するピークをまとめて示している。A型の特徴ピーク(2θ)は、11.2±0.2、15.4±0.2、17.0±0.2、18.3±0.2、19.3±0.2および20.6±0.2である。
【0071】
【表1】
【0072】
アセトンに溶かしたヒドロキシプロピルメチルセルロースアセテートサクシネート(HPMCAS)を用いて化合物(I)を噴霧乾燥分散(SDD)させた(25重量%化合物(I))場合に、化合物(I)の溶解が11倍増大したことが観察された。0.5重量%Methocel(商標)に懸濁させたモデル絶食十二指腸溶液(0.5重量%タウロコール酸ナトリウム/1−パルミトイル−2−オレオイル−sn−グリセロ−3−ホスホコリンを溶かしたリン酸緩衝溶液、pH6.5)中600μg(有効成分))/mLで、化合物およびSDDを比較し試験した。
【0073】
(実施例3)
実施例3は、式(I)の化合物の異なる多形形態(本明細書において「B型」と呼ぶ)を提供する。
【0074】
実施例2からのA型(20mg)を、電磁撹拌棒およびアセトン2mL(2mL)を含有する4mLバイアルに加えた。固体を25℃で3週間撹拌した。固体をPTFEフィルター上でろ過し、MTBE1mLで洗浄した。B型約10mgを白色結晶性固体として単離した。
【0075】
式(I)の化合物のB型に関するX線粉末回折パターンを、銅放射線によるSiemens D5000回折計および実施例2に上述されている条件を用いて作成した。以下の表は、B型多形体について観察されたバックグラウンドに対して5倍閾値を有するピークをまとめて示している。B型の特徴ピーク(2θ)は、7.8±0.2、11.2±0.2、13.7±0.2、15.9±0.2、18.7±0.2および20.2±0.2である。
【0076】
【表2】
【0077】
薬理学的データ
生物学的プロトコル
動物、特に哺乳動物(例えば、ヒト)における疾患(例えば本明細書において詳述しているもの)の治療における本発明の化合物の有用性は、後述のin vitroおよびin vivoアッセイを含めた当業者に既知の従来のアッセイにおけるその活性によって実証することができる。かかるアッセイは、それによって本発明の化合物の活性が他の既知の化合物の活性と比較できる手段も提供する。
【0078】
ACC1およびACC2の活性の直接阻害
本発明の化合物のACC阻害活性を標準手順に基づく方法によって実証した。例えば式(1)の化合物に関するACC活性の直接阻害を、ラット肝臓ACCおよび組換えヒトACC2の調製物を用いて決定した。
【0079】
[1]ラット肝臓ACCの調製。標準手順、例えばThampyおよびWakil(J.Biol.Chem.260:6318〜6323;1985)によって記載されているものに基づいて以下の方法を用いてラット肝臓からラット肝臓ACCを得た。
【0080】
重さが150〜200gの雄CDラットを18〜24時間絶食させ、その後高スクロース食餌(AIN−76Aげっ歯類食餌、Cat#D10001、Research Diets Inc.、米国ニュージャージー州New Brunswick)を3日間与え、その時点でそれらをCO2窒息によって殺した。肝臓を取り出し、氷冷リン酸緩衝溶液(PBS)中ですすぎ、Waring(登録商標)ブレンダー中の5倍の体積量の均質化バッファー(50mMリン酸カリウム、pH7.5、10mM EDTA、10mM 2−メルカプトエタノール、2mMベンズアミジン、0.2mMフェニルメチルスルホニルフルオライド(PMSF)、各5mg/Lのロイペプチン、アプロチニン、および抗トリプシン)中で、1分間4℃でホモジナイズした。後続の全ての操作を4℃で実施した。50%PEG溶液を加えることによってポリエチレングリコール(PEG)に対してホモジネートを3%とし、20,000xgで15分間遠心分離した。50%PEG溶液を加えて得られた上清を5%PEGに調節し、5分間撹拌した。20,000×gで20分間の遠心分離によってペレット(ACC活性を含有する)を捕集し、氷冷二重蒸留水ですすいで過剰PEGを除去し、均質化バッファーを用いて元のホモジネート体積の4分の1に再懸濁させた。硫酸アンモニウム(200g/リットル)を撹拌しながらゆっくり加えた。45分後、20,000×gで30分間の遠心分離によって酵素を捕集し、10mLの50mM HEPES、pH7.5、0.1mM DTT、1.0mM EDTA、および10%グリセリン中に再懸濁させ、同じバッファーで平衡化したSephadex(商標)G−25カラム(2.5cm×50cm)(Pharmacia、米国ニュージャージー州Piscataway現GE Healthcare)上で脱塩する。脱塩した酵素調製物をアリコート中に−70℃で保管した。即時使用の前に冷凍ラット肝臓ACCアリコートを解凍し、50mM HEPES、pH7.5、10mM MgCl2、10mMクエン酸三カリウム、2.0mMジチオスレイトール(DTT)、および0.75mg/mLの脂肪酸を含まないウシ血清アルブミン(BSA)を含有するバッファー中に500μg/mLまで希釈し、37℃で30分間プレインキュベートした。
【0081】
[2]ラット肝臓ACC阻害の測定。ACC活性の測定およびACC阻害の評価のために、試験化合物をジメチルスルホキシド(DMSO)中に溶解し、アリコート1μLをクリアボトム、96穴プレート(Perkin−Elmer PN#1450−514)に加えた。対照穴は、DMSO単独1μLまたは高阻害化合物1μLを含有する。上記のようにラット肝臓から得た酵素を、酵素バッファー中37℃で30分間活性化させてから化合物プレートに加えた。全穴に、50mM HEPES、pH7.5、7.5mM MgCl2、7.5mMクエン酸三カリウム、2mM DTT、50mg/mL BSAを含有するバッファーに溶かした活性酵素(1.33X)75μLを与える。活性酵素を化合物と一緒に10分間プレインキュベートしてから、50mM HEPES、pH7.5、7.5mM MgCl2、7.5mMクエン酸三カリウム、2mM DTT、50mg/mL BSA、120μMアセチルCoA、8.0mM ATP、38.4mM KHCO3、および1.6mM NaH[14C]O3(100μCi/μL)を含有する基質溶液25μLを加えることによって反応を開始した。反応中の最終基質濃度は、30μMアセチルCoA、9.6mM KHCO3、0.4mM NaH[14C]O3、および2mM ATPであった。3N HCl 25μLを加えることによって10分後に反応を停止させ、プレートを50℃で最低20時間乾燥させた。水30μLを乾燥させたプレートに加え、5分間混合した。Optiphase Supermix液体シンチレーション流体(Perkin Elmer、米国マサチューセッツ州Waltham)95μLを加え、プレートを20分間混合する。Wallac Trilux 1450 Microbeta LSCルミネセンスカウンターを用いてMCoA中への14Cの取り込みを測定した。
【0082】
[3]ヒトACC2阻害の測定。精製した組換えヒトACC2(hrACC2)を用いてヒトACC2阻害を測定した。簡単に言えば、ACC2の完全長CytomaxクローンをCambridge Bioscience Limitedから購入し、配列決定し、PCDNA5 FRT TO−TOPO(Invitrogen、米国カリフォルニア州Carlsbad)中にサブクローニングした。テトラサイクリン誘導によってACC2をCHO細胞中で発現させ、1μg/mLテトラサイクリン(Invitrogen、米国カリフォルニア州Carlsbad)を含む、グルタミン、ビオチン、ハイグロマイシンおよびブラストサイジンを含んだDMEM/F12 5リットル中に回収した。次いでACC2を含有するならし培地をSoftlink Soft Release Avidinカラム(Promega、米国ウィスコンシン州Madison)に加え、5mMビオチンで溶離した。ACC2 4mgを濃度0.05mg/mL(A280によって決定した)、推定純度95%(A280によって決定した)で溶離した。精製したACC2を50mMトリス、200mM NaCl、4mM DTT、2mM EDTA、および5%グリセリン中で透析した。プールしたタンパク質を凍結し、融解時に活性の損失なしに−80℃で保管した。ACC2活性の測定およびACC2阻害の評価のために、試験化合物をDMSO中に溶解し、最終DMSO濃度1%の5倍ストックとしてrhACC2酵素に加えた。50μM ATP反応のための製造条件を用いたTranscreener ADP検出FPアッセイキット(Bellbrook Labs、米国ウィスコンシン州Madison)を使用して、rhACC2をCostar#3767(Costar、米国マサチューセッツ州Canbridge)384穴プレート中で検定した。アッセイの最終条件は、50mM HEPES、pH7.5、5mM MgCl2、5mMクエン酸三カリウム、2mM DTT、0.5mg/mL BSA、30μMアセチルCoA、50μM ATP、および8mM KHCO3であった。通常、10μL反応を室温で1時間実施し、10μlのTranscreener停止および検出バッファーを加え、さらに1時間インキュベートした。620励起Cy5 FPジェネラルデュアルミラー、620励起Cy5 FPフィルター、688発光(S)および688(P)発光フィルターを用いたEnvision Fluorescenceリーダー(Perkinelmer)上でデータを取得した。
【0083】
上記ラット肝臓ACC放射酵素アッセイおよび組換えhACC2 transcreenerアッセイを用いた結果を、式(I)の化合物に関して以下の表にまとめて示す。
【0084】
【表3】
【0085】
実験動物におけるACC阻害の急性in vivo評価
本発明の化合物のACC阻害活性は、治療した動物からの肝臓および筋肉組織のマロニルCoAレベルを低減させるそれらの能力の評価によってin vivoで確認することができる。
【0086】
実験動物におけるマロニルCoA生成阻害の測定。この方法では、適宜の標準固形飼料および水で維持した雄スプレーグ・ドーリーネズミ(225〜275g)を、研究する前に無作為化した。動物は、実験を開始する18時間前に餌を与えるか、または絶食させた。明期に入って2時間後に、動物に5mL/kgの体積(0.5%メチルセルロース、ビヒクル)または適切な化合物(ビヒクル中で調製)を経口投与した。ビヒクルを与えた対照は、ベースライン組織マロニルCoAレベルを決定するために含め、一方絶食した動物は、マロニルCoAレベルに対して絶食が与えた影響を決定するために含めた。化合物投与の1時間後、動物をCO2で窒息させ、組織を取り出した。特に、血液を心臓穿刺によって捕集し、EDTAを含有するBD Microtainerチューブ中に入れ(BD Biosciences、米国ニュージャージー州)、混合し、氷上に置いた。血漿を用いて薬物暴露を決定した。肝臓および四頭筋を取り出し、直ちに凍結クランプし、金属箔で包み、液体窒素中で保管した。
【0087】
組織を液体N2下で微粉砕して、サンプリングにおける均一性を確保した。FastPrep FP120(Thermo Scientific、速度=5.5、45秒間)中のLysing Matrix A(MP Biomedicals、PN 6910)に溶かした5倍体積量の10%トリカルボン酸を用いて組織(150〜200mg)からマロニルCoAを抽出した。15000×gで30分間遠心分離(Eppendorf Centrifuge 5402)した後にマロニルCoAを含有する上清を細胞片から取り出した。分析が完了するまで試料を−80℃で安定して凍結させた。
【0088】
肝臓および筋肉組織中のマロニルCoAレベルの分析は、以下の方法論を用いて評価することができる。
【0089】
この方法は、以下の物質を利用する:Isotec(米国オハイオ州Miamisburg)から購入したマロニルCoAテトラリチウム塩およびマロニル−13C3−CoAトリリチウム塩、過塩素酸ナトリウム(Sigma、cat no.410241)、トリクロロ酢酸(ACROS、cat no.42145)、リン酸(J.T.Baker、cat no.0260−01)、ギ酸アンモニウム(Fluka、cat no.17843)、メタノール(HPLCグレード、J.T.Baker、cat no.9093−33)、および水(HPLCグレード、J.T.Baker、4218−03)を使用して必要とする移動相を作成した。Strata−Xオンライン固相抽出カラム、25μm、20mm×2.0mm I.D(cat no.00M−S033−B0−CB)をPhenomenex(米国カリフォルニア州Torrance)から得た。SunFire C18逆相カラム、3.5μm、100mm×3.0mm I.D.(cat no.186002543)をWaters Corporation(米国マサチューセッツ州Milford)から購入した。
【0090】
この方法は、以下の装備を利用して行うことができる。Agilent 1100バイナリーポンプ、Agilent 1100クォータナリーポンプおよび2個のValco Cheminert 6ポート2位置弁を用いた二次元クロマトグラフィー。10℃で保持したPeltier冷却スタックおよび20μLサンプリングループを備えたLEAP HTC PALオートサンプラーによって試料を導入した。オートサンプラーのためのニードル洗浄溶液は、Wash 1が10%トリクロロ酢酸水溶液(w/v)であり、Wash 2が90:10 メタノール:水である。MicroTech Scientific Micro−LC Column Ovenを用いて分析用カラム(Sunfire)を35℃で保持した。Turbo Ion Sprayを備えたABI Sciex API3000三連四重極質量分析計で溶離液を分析した。
【0091】
オンライン固相抽出および逆相クロマトグラフィーに関して異なる勾配溶離条件を用いて同時に二次元クロマトグラフィーを行った。この方法の一般的な計画は、第1次元をサンプル洗浄および目的の分析物の捕捉のために利用し、続いて第1次元から第2次元上への溶離のために両方の次元を短時間カップリングするようなものであった。両次元を続いて脱共役させて、定量化のために第2次元から分析物を勾配溶離させ、同時に配列中の次の試料のために第1次元を調製した。両次元を簡単に連結した場合、第1次元中の移動相の流れを第2次元上への分析物溶離のために逆流させて、最適ピーク幅、ピーク形状、および溶離時間を可能にする。
【0092】
HPLC系の第1次元は、Phenomenex strata−Xオンライン固相抽出カラムと溶媒Aが100mM過塩素酸ナトリウム/0.1%(v/v)リン酸および溶媒Bがメタノールからなる移動相とを利用した。
【0093】
HPLC系の第2次元は、Waters SunFire C18逆相カラムと溶媒Aが100mMギ酸アンモニウムおよび溶媒Bがメタノールからなる移動相とを利用した。勾配の初期条件を2分間保持し、この間に分析物を分析用カラムに移した。分析用で維持している間、オンラインSPEカラムから分析物を溶離するのに初期条件は十分な強度であったことが重要であった。その後、洗浄および再平衡ステップの前に4.5分で74.5%Aまで勾配が直線的に上がった。
【0094】
質量分析は、HPLCと連結すると、複雑な基質中の分析物を定量的に測定する場合に高選択的および高感度な方法になり得るが、依然として干渉および抑制を受ける。二次元HPLCを質量分析計と連結することによって、これらの干渉は著しく減少する。さらに、三連四重極質量分析計の多重反応モニタリング(MRM)特色を利用することによって、信号対雑音比が著しく改善した。
【0095】
このアッセイでは、質量分析計を、TurbolonSpray電圧2250Vで陽イオンモードにおいて操作した。噴霧ガスを450℃まで加熱した。デクラスタリング電位(DP)、集束電位(FP)、および衝突エネルギー(CE)を、それぞれ60、340、および42Vに設定した。四重極1(Q1)分解能をユニット分解能に設定し、四重極3(Q3)を低に設定した。CADガスを8に設定した。モニターしたMRM転移は、停止時間200msでのマロニルCoA:854.1→347.0m/z(L.Gaoら(2007)J.Chromatogr.B 853、303〜313)、およびマロニル−13C3−CoA:857.1→350.0m/zに関してであった。溶離液を、予想した分析物の溶離時間付近で質量分析計に分流し、他の場合には、供給源の保持および器械使用の頑健性の向上を助けるために廃棄した。得られたクロマトグラムを、Analystソフトウェア(Applied Biosystems)を用いて組み込んだ。マロニルCoAの組織濃度を、10%トリクロロ酢酸水溶液で調製した標準曲線から計算した。
【0096】
組織抽出物中のマロニルCoAの定量化のための標準曲線を含む試料を10%(w/v)トリクロロ酢酸(TCA)で調製し、0.01〜1pmol/μLの範囲であった。マロニル−13C3−CoA(最終濃度0.4pmol/μL)を各標準曲線成分および試料に内部標準として加えた。
【0097】
アッセイ内品質対照を6個調製した。絶食させた動物から調製したプールした抽出物から3個、および餌を与えた動物から作製したプールから3個。これらは、0、0.1または0.3pmol/μLの12C−マロニルCoAならびにマロニル−13C3−CoA(0.4pmol/μL)を加えた別々の試料として実行した。各アッセイ内品質対照は、水性組織抽出物を85%含有し、残りの部分が内部標準(0.4pmol/μL)および12C−マロニルCoAによって占められていた。アッセイ間対照を各実験に含めた。それらは1個の絶食させたおよび1個の餌を与えたプールした四頭筋の試料および/または1個の絶食させたおよび1個の餌を与えたプールした肝臓の試料で構成される。かかる全ての対照に、マロニル−13C3−CoA(0.4pmol/μL)を加えている。
【0098】
式(I)の化合物を上記in vivo試験で使用して、肝臓および筋肉組織中のマロニルCoAレベルに対するそれらの効果を調べた。結果を以下の表に示す。
【0099】
【表4】
【特許請求の範囲】
【請求項1】
式(I)の化合物。
【化1】
【請求項2】
11.2±0.2、15.4±0.2、17.0±0.2、18.3±0.2、19.3±0.2および20.6±0.2の回折角(2θ)でピークを含む粉末X線回折パターンを有する結晶形である、請求項1に記載の化合物。
【請求項3】
7.8±0.2、11.2±0.2、13.7±0.2、15.9±0.2、18.7±0.2および20.2±0.2の回折角(2θ)でピークを含む粉末X線回折パターンを有する結晶形である、請求項1に記載の化合物。
【請求項4】
(i)前記請求項のいずれか一項に記載の化合物、および(ii)薬学的に許容できる賦形剤、希釈剤または担体を含む医薬組成物。
【請求項5】
前記化合物が治療有効量で存在する、請求項4に記載の組成物。
【請求項6】
抗肥満薬および抗糖尿病薬からなる群から選択される少なくとも1種の別の薬剤をさらに含む、請求項5に記載の組成物。
【請求項7】
前記抗肥満薬が、ジルロタピド、ミトラタピド、インプリタピド、R56918(CAS番号403987)、CAS番号913541−47−6、ロルカセリン、セチリスタット、PYY3−36、ナルトレキソン、オレオイル−エストロン、オビネピチド、プラムリンチド、テソフェンシン、レプチン、リラグルチド、ブロモクリプチン、オルリスタット、エクセナチド、AOD−9604(CAS番号221231−10−3)およびシブトラミンからなる群から選択される、請求項6に記載の組成物。
【請求項8】
前記抗糖尿病薬が、メトホルミン、アセトヘキサミド、クロルプロパミド、ジアビネース、グリベンクラミド、グリピジド、グリブリド、グリメピリド、グリクラジド、グリペンチド、グリキドン、グリソラミド、トラザミド、トルブタミド、テンダミスタット、トレスタチン、アカルボース、アジポシン、カミグリボース、エミグリテート、ミグリトール、ボグリボース、プラジミシン−Q、サルボスタチン、バラグリタゾン、シグリタゾン、ダルグリタゾン、エングリタゾン、イサグリタゾン、ピオグリタゾン、ロシグリタゾン、トログリタゾン、エキセンディン−3、エキセンディン−4、トロズスクエミン、レセルバトロール、ヒルチオサール抽出物、シタグリプチン、ビルダグリプチン、アログリプチンおよびサクサグリプチンからなる群から選択される、請求項6に記載の組成物。
【請求項9】
動物における肥満および肥満関連障害を治療するための方法であって、かかる治療を必要とする動物に、治療有効量の請求項1、2または3に記載の化合物を投与するステップを含む方法。
【請求項10】
動物における2型糖尿病および糖尿病関連障害の進行または発症を治療するまたは遅延させるための方法であって、かかる治療を必要とする動物に、治療有効量の請求項1、2または3に記載の化合物を投与するステップを含む方法。
【請求項11】
動物における非アルコール性脂肪肝疾患(NAFLD)または肝臓インスリン抵抗性を治療するための方法であって、かかる治療を必要とする動物に、治療有効量の請求項1、2または3に記載の化合物を投与するステップを含む方法。
【請求項12】
動物における肥満および肥満関連障害を治療するための方法であって、かかる治療を必要とする動物に、請求項5から8のいずれか一項に記載の医薬組成物を投与するステップを含む方法。
【請求項13】
動物における2型糖尿病および糖尿病関連障害の進行または発症を治療するまたは遅延させるための方法であって、かかる治療を必要とする動物に、請求項5から8のいずれか一項に記載の医薬組成物を投与するステップを含む方法。
【請求項14】
動物における非アルコール性脂肪肝疾患(NAFLD)または肝臓インスリン抵抗性を治療するための方法であって、かかる治療を必要とする動物に、請求項5から8のいずれか一項に記載の医薬組成物を投与するステップを含む方法。
【請求項15】
動物におけるアセチルCoAカルボキシラーゼ酵素(複数可)の阻害によって調節される疾患、状態または障害を治療するための方法であって、かかる治療を必要とする動物に、
(i)治療量の請求項1、2または3に記載の化合物、および薬学的に許容できる賦形剤、希釈剤または担体を含む第1の組成物と、
(ii)抗肥満薬および抗糖尿病薬からなる群から選択される少なくとも1種の別の薬剤、および薬学的に許容できる賦形剤、希釈剤または担体を含む第2の組成物と
を含む2種の別々の医薬組成物を投与するステップを含み、
アセチルCoAカルボキシラーゼ酵素(複数可)の阻害によって調節される前記疾患、状態または障害が、肥満、肥満関連障害、2型糖尿病、糖尿病関連障害、非アルコール性脂肪肝疾患(NAFLD)および肝臓インスリン抵抗性からなる群から選択される、方法。
【請求項16】
前記抗肥満薬が、ジルロタピド、ミトラタピド、インプリタピド、R56918(CAS番号403987)、CAS番号913541−47−6、ロルカセリン、セチリスタット、PYY3−36、ナルトレキソン、オレオイル−エストロン、オビネピチド、プラムリンチド、テソフェンシン、レプチン、リラグルチド、ブロモクリプチン、オルリスタット、エクセナチド、AOD−9604(CAS番号221231−10−3)およびシブトラミンからなる群から選択され、前記抗糖尿病薬が、メトホルミン、アセトヘキサミド、クロルプロパミド、ジアビネース、グリベンクラミド、グリピジド、グリブリド、グリメピリド、グリクラジド、グリペンチド、グリキドン、グリソラミド、トラザミド、トルブタミド、テンダミスタット、トレスタチン、アカルボース、アジポシン、カミグリボース、エミグリテート、ミグリトール、ボグリボース、プラジミシン−Q、サルボスタチン、バラグリタゾン、シグリタゾン、ダルグリタゾン、エングリタゾン、イサグリタゾン、ピオグリタゾン、ロシグリタゾン、トログリタゾン、エキセンディン−3、エキセンディン−4、トロズスクエミン、レセルバトロール、ヒルチオサール抽出物、シタグリプチン、ビルダグリプチン、アログリプチンおよびサクサグリプチンからなる群から選択される、請求項15に記載の方法。
【請求項17】
前記第1の組成物および前記第2の組成物を同時に投与する、請求項15または16に記載の方法。
【請求項18】
前記第1の組成物および前記第2の組成物を順次および任意の順序で投与する、請求項15または16に記載の方法。
【請求項19】
アセチルCoAカルボキシラーゼ酵素(複数可)の阻害によって調節される疾患、状態または障害を治療するための医薬の製造における請求項1、2または3に記載の化合物の使用。
【請求項1】
式(I)の化合物。
【化1】
【請求項2】
11.2±0.2、15.4±0.2、17.0±0.2、18.3±0.2、19.3±0.2および20.6±0.2の回折角(2θ)でピークを含む粉末X線回折パターンを有する結晶形である、請求項1に記載の化合物。
【請求項3】
7.8±0.2、11.2±0.2、13.7±0.2、15.9±0.2、18.7±0.2および20.2±0.2の回折角(2θ)でピークを含む粉末X線回折パターンを有する結晶形である、請求項1に記載の化合物。
【請求項4】
(i)前記請求項のいずれか一項に記載の化合物、および(ii)薬学的に許容できる賦形剤、希釈剤または担体を含む医薬組成物。
【請求項5】
前記化合物が治療有効量で存在する、請求項4に記載の組成物。
【請求項6】
抗肥満薬および抗糖尿病薬からなる群から選択される少なくとも1種の別の薬剤をさらに含む、請求項5に記載の組成物。
【請求項7】
前記抗肥満薬が、ジルロタピド、ミトラタピド、インプリタピド、R56918(CAS番号403987)、CAS番号913541−47−6、ロルカセリン、セチリスタット、PYY3−36、ナルトレキソン、オレオイル−エストロン、オビネピチド、プラムリンチド、テソフェンシン、レプチン、リラグルチド、ブロモクリプチン、オルリスタット、エクセナチド、AOD−9604(CAS番号221231−10−3)およびシブトラミンからなる群から選択される、請求項6に記載の組成物。
【請求項8】
前記抗糖尿病薬が、メトホルミン、アセトヘキサミド、クロルプロパミド、ジアビネース、グリベンクラミド、グリピジド、グリブリド、グリメピリド、グリクラジド、グリペンチド、グリキドン、グリソラミド、トラザミド、トルブタミド、テンダミスタット、トレスタチン、アカルボース、アジポシン、カミグリボース、エミグリテート、ミグリトール、ボグリボース、プラジミシン−Q、サルボスタチン、バラグリタゾン、シグリタゾン、ダルグリタゾン、エングリタゾン、イサグリタゾン、ピオグリタゾン、ロシグリタゾン、トログリタゾン、エキセンディン−3、エキセンディン−4、トロズスクエミン、レセルバトロール、ヒルチオサール抽出物、シタグリプチン、ビルダグリプチン、アログリプチンおよびサクサグリプチンからなる群から選択される、請求項6に記載の組成物。
【請求項9】
動物における肥満および肥満関連障害を治療するための方法であって、かかる治療を必要とする動物に、治療有効量の請求項1、2または3に記載の化合物を投与するステップを含む方法。
【請求項10】
動物における2型糖尿病および糖尿病関連障害の進行または発症を治療するまたは遅延させるための方法であって、かかる治療を必要とする動物に、治療有効量の請求項1、2または3に記載の化合物を投与するステップを含む方法。
【請求項11】
動物における非アルコール性脂肪肝疾患(NAFLD)または肝臓インスリン抵抗性を治療するための方法であって、かかる治療を必要とする動物に、治療有効量の請求項1、2または3に記載の化合物を投与するステップを含む方法。
【請求項12】
動物における肥満および肥満関連障害を治療するための方法であって、かかる治療を必要とする動物に、請求項5から8のいずれか一項に記載の医薬組成物を投与するステップを含む方法。
【請求項13】
動物における2型糖尿病および糖尿病関連障害の進行または発症を治療するまたは遅延させるための方法であって、かかる治療を必要とする動物に、請求項5から8のいずれか一項に記載の医薬組成物を投与するステップを含む方法。
【請求項14】
動物における非アルコール性脂肪肝疾患(NAFLD)または肝臓インスリン抵抗性を治療するための方法であって、かかる治療を必要とする動物に、請求項5から8のいずれか一項に記載の医薬組成物を投与するステップを含む方法。
【請求項15】
動物におけるアセチルCoAカルボキシラーゼ酵素(複数可)の阻害によって調節される疾患、状態または障害を治療するための方法であって、かかる治療を必要とする動物に、
(i)治療量の請求項1、2または3に記載の化合物、および薬学的に許容できる賦形剤、希釈剤または担体を含む第1の組成物と、
(ii)抗肥満薬および抗糖尿病薬からなる群から選択される少なくとも1種の別の薬剤、および薬学的に許容できる賦形剤、希釈剤または担体を含む第2の組成物と
を含む2種の別々の医薬組成物を投与するステップを含み、
アセチルCoAカルボキシラーゼ酵素(複数可)の阻害によって調節される前記疾患、状態または障害が、肥満、肥満関連障害、2型糖尿病、糖尿病関連障害、非アルコール性脂肪肝疾患(NAFLD)および肝臓インスリン抵抗性からなる群から選択される、方法。
【請求項16】
前記抗肥満薬が、ジルロタピド、ミトラタピド、インプリタピド、R56918(CAS番号403987)、CAS番号913541−47−6、ロルカセリン、セチリスタット、PYY3−36、ナルトレキソン、オレオイル−エストロン、オビネピチド、プラムリンチド、テソフェンシン、レプチン、リラグルチド、ブロモクリプチン、オルリスタット、エクセナチド、AOD−9604(CAS番号221231−10−3)およびシブトラミンからなる群から選択され、前記抗糖尿病薬が、メトホルミン、アセトヘキサミド、クロルプロパミド、ジアビネース、グリベンクラミド、グリピジド、グリブリド、グリメピリド、グリクラジド、グリペンチド、グリキドン、グリソラミド、トラザミド、トルブタミド、テンダミスタット、トレスタチン、アカルボース、アジポシン、カミグリボース、エミグリテート、ミグリトール、ボグリボース、プラジミシン−Q、サルボスタチン、バラグリタゾン、シグリタゾン、ダルグリタゾン、エングリタゾン、イサグリタゾン、ピオグリタゾン、ロシグリタゾン、トログリタゾン、エキセンディン−3、エキセンディン−4、トロズスクエミン、レセルバトロール、ヒルチオサール抽出物、シタグリプチン、ビルダグリプチン、アログリプチンおよびサクサグリプチンからなる群から選択される、請求項15に記載の方法。
【請求項17】
前記第1の組成物および前記第2の組成物を同時に投与する、請求項15または16に記載の方法。
【請求項18】
前記第1の組成物および前記第2の組成物を順次および任意の順序で投与する、請求項15または16に記載の方法。
【請求項19】
アセチルCoAカルボキシラーゼ酵素(複数可)の阻害によって調節される疾患、状態または障害を治療するための医薬の製造における請求項1、2または3に記載の化合物の使用。
【図1】

【図2】
【図2】
【公表番号】特表2011−521940(P2011−521940A)
【公表日】平成23年7月28日(2011.7.28)
【国際特許分類】
【出願番号】特願2011−511102(P2011−511102)
【出願日】平成21年5月18日(2009.5.18)
【国際出願番号】PCT/IB2009/005659
【国際公開番号】WO2009/144555
【国際公開日】平成21年12月3日(2009.12.3)
【出願人】(593141953)ファイザー・インク (302)
【Fターム(参考)】
【公表日】平成23年7月28日(2011.7.28)
【国際特許分類】
【出願日】平成21年5月18日(2009.5.18)
【国際出願番号】PCT/IB2009/005659
【国際公開番号】WO2009/144555
【国際公開日】平成21年12月3日(2009.12.3)
【出願人】(593141953)ファイザー・インク (302)
【Fターム(参考)】
[ Back to top ]