
フィブリノゲンを産生するトランスジェニックカイコ

【課題】安全性の高いフィブリノゲンを低コストで大量に生産可能にする新規な手段を提供すること。
【解決手段】絹糸腺細胞内でフィブリノゲンのサブユニットAα鎖、Bβ鎖及びγ鎖を発現し、凝固活性を有するフィブリノゲンを繭糸中に産生するトランスジェニックカイコを提供した。好ましくは、該トランスジェニックカイコは、中部絹糸腺細胞内で前記サブユニットを発現し、繭糸のセリシン層中にフィブリノゲンを産生する。本発明のトランスジェニックカイコの繭からフィブリノゲンを回収することで、安全性の高いフィブリノゲンを低コストで大量に生産できる。
【解決手段】絹糸腺細胞内でフィブリノゲンのサブユニットAα鎖、Bβ鎖及びγ鎖を発現し、凝固活性を有するフィブリノゲンを繭糸中に産生するトランスジェニックカイコを提供した。好ましくは、該トランスジェニックカイコは、中部絹糸腺細胞内で前記サブユニットを発現し、繭糸のセリシン層中にフィブリノゲンを産生する。本発明のトランスジェニックカイコの繭からフィブリノゲンを回収することで、安全性の高いフィブリノゲンを低コストで大量に生産できる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、凝固活性を有するフィブリノゲンを繭糸中に産生するトランスジェニックカイコ、該カイコの製造方法、及びフィブリノゲンの製造方法に関する。
【背景技術】
【0002】
フィブリノゲンは血漿中に存在する血漿タンパク質の一種であり、分子量340kDaの糖タンパク質である。Aα鎖、Bβ鎖及びγ鎖の3本鎖がジスルフィド結合により相互に結合し、これが2量体を形成した(Aα−Bβ−γ)2という6量体の分子構造を有する。Aα鎖は67kDa、610アミノ酸残基からなり、糖鎖を有しない。Bβ鎖は56kDa、461アミノ酸残基よりなり、364位のAsnが糖鎖を有する。γ鎖は48kDa、411アミノ酸残基よりなり、52位のAsnが糖鎖を有する。
【0003】
フィブリノゲンは血液凝固に関わる。フィブリノゲンは生体内でトロンビンによりAα鎖、Bβ鎖が各々切断され、フィブリノペプチドA、フィブリノペプチドBが除去されて(α−β−γ)2へと変換される(フィブリノモノマー)。このフィブリノモノマーはCa2+の存在下で重合してフィブリンポリマーを形成する。さらにトロンビンにより血液凝固第XIII因子が活性化されると、そのトランスグルタミナーゼ活性によりフィブリンポリマー同士にペプチド結合が形成されて強固な交差架橋フィブリンになる。
【0004】
現在医療分野で用いられているフィブリノゲンは、ヒトの血漿より分離精製されて製造されている。ヒト血漿を原料にするため、ウイルス混入の危険性と隣り合わせであり、感染源の不活化処理等の工程が必須である。また、原料のヒト血漿の供給は献血に依存しており、常に安定供給することが必ずしも容易ではない。
【0005】
遺伝子組み換え技術を用いてフィブリノゲンを製造すれば、安全なフィブリノゲンを安定的に提供することができる。こうした試みは現在までにいくつか報告されているが、十分に効率的で満足できる手法は未だ提供されていない。例えば特許文献1及び非特許文献1には、ピキア酵母を用いて組換えフィブリノゲンを製造する方法が開示されているが、該方法では培養液中に分泌されたフィブリノゲンはプロテアーゼにより分解され、効率的な生産方法となっていない。特許文献2及び3には、動物培養細胞で組換えフィブリノゲンを製造する方法が開示されているが、商業的に製造するためには、細胞培養のための大規模な設備や培養密度のコントロールなど、コストや手間がかかる。また、動物細胞なので動物由来物質や感染性ウイルスが混入するおそれがあり、ヒト血液から製造する現在の方法と同様の問題点を抱えることになる。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2004−16055号公報
【特許文献2】特許第4573775号公報
【特許文献3】特表2009−528843号公報
【非特許文献】
【0007】
【非特許文献1】Protein Expression and Purification 59 (2008) 289-296
【発明の概要】
【発明が解決しようとする課題】
【0008】
従って、本願発明の目的は、安全性の高いフィブリノゲンを低コストで大量に生産可能にする新規な手段を提供することにある。
【課題を解決するための手段】
【0009】
本願発明者らは、カイコを用いた公知の組換えタンパク質生産技術を利用してフィブリノゲンを製造することに想到し、フィブリノゲンのAα鎖、Bβ鎖及びγ鎖の遺伝子をカイコ絹糸腺で発現させて繭からフィブリノゲンを回収することを試みた。Bβ鎖を発現するカイコとAα鎖及びγ鎖を発現するカイコとをそれぞれ作製し、交配により3鎖を発現するカイコを作出しようと試みたが、Bβ鎖発現カイコで繭へのBβ鎖分泌を確認することができなかった。しかしながら、このBβ鎖発現カイコをAα鎖/γ鎖発現カイコと交配させたところ、繭糸中に凝固活性を有するフィブリノゲンを分泌させることに成功し、さらに、該繭糸から効率よくフィブリノゲンを回収できる条件を見出し、本願発明を完成した。
【0010】
すなわち、本発明は、絹糸腺細胞内でフィブリノゲンのサブユニットAα鎖、Bβ鎖及びγ鎖を発現し、凝固活性を有するフィブリノゲンを繭糸中に産生するトランスジェニックカイコを提供する。また、本発明は、上記本発明のトランスジェニックカイコが生成する、凝固活性を有するフィブリノゲンを含有するカイコの繭を提供する。さらに、本発明は、上記本発明のトランスジェニックカイコの繭からフィブリノゲンを回収することを含む、フィブリノゲンの製造方法を提供する。さらに、本発明は、フィブリノゲンのサブユニットAα遺伝子、Bβ遺伝子及びγ遺伝子を、絹糸腺細胞内で機能するプロモーターと機能的に連結した形態でカイコに導入し、絹糸腺細胞内でAα鎖、Bβ鎖及びγ鎖を発現するカイコを選抜することを含む、凝固活性を有するフィブリノゲンを繭糸中に産生するトランスジェニックカイコの作出方法を提供する。
【発明の効果】
【0011】
本発明により、凝固活性を有するフィブリノゲンを繭糸に分泌するトランスジェニックカイコが初めて提供された。特に、中部絹糸腺でAα、Bβ及びγ遺伝子を発現させれば、繭糸のうち水に比較的易溶のセリシン層にフィブリノゲンが分泌されるので、活性を維持してフィブリノゲンを回収するのに有利である。カイコはタンパク質合成能力が極めて高い生物であり、飼育も容易である。献血により収集した血液材料からフィブリノゲンを製造するのとは異なり、ウイルス混入の危険性も排除できる。本発明によれば、安全性の高いフィブリノゲンを低コストで大量生産することが可能になる。
【図面の簡単な説明】
【0012】
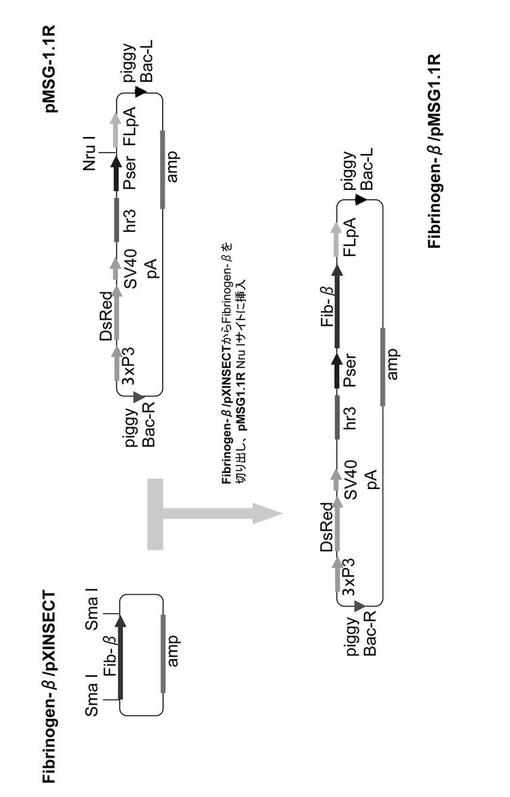
【図1】実施例でBβ導入カイコを作出するために用いたベクターの構築図である。
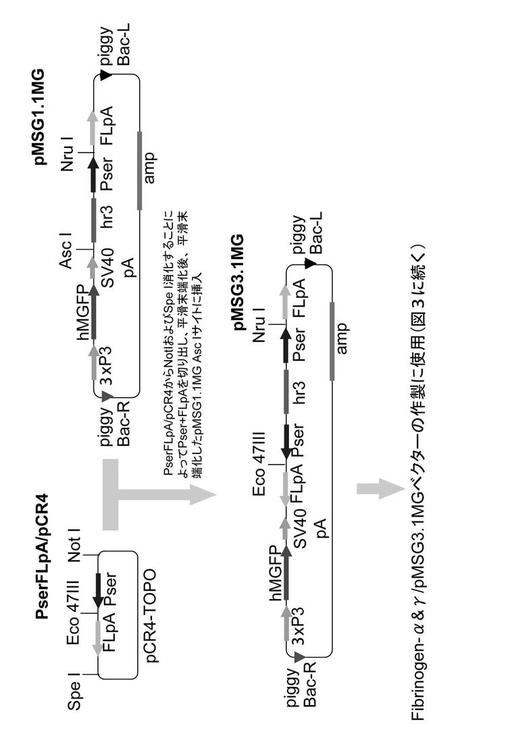
【図2】実施例でAα/γ導入カイコを作出するために用いたベクターの構築図である。
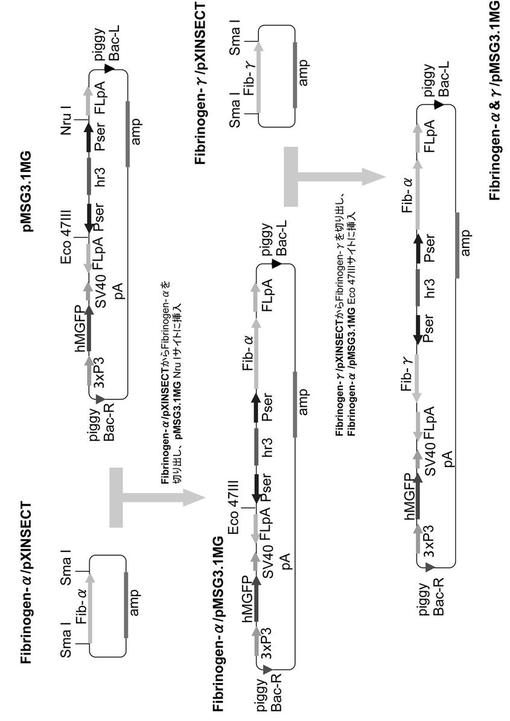
【図3】実施例でAα/γ導入カイコを作出するために用いたベクターの構築図である(図2の続き)。
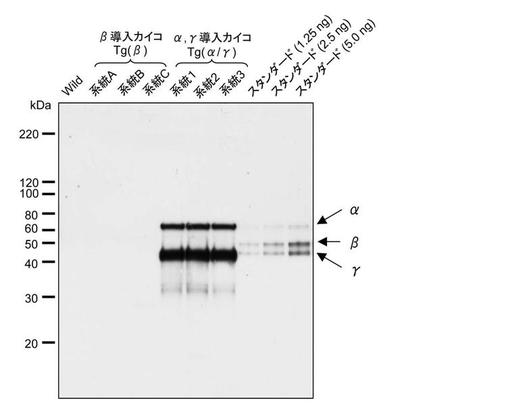
【図4】実施例で作出したBβ導入カイコとAα/γ導入カイコについて、繭糸中の各サブユニットタンパク質をウエスタンブロットにより検出した結果の一例を示す。
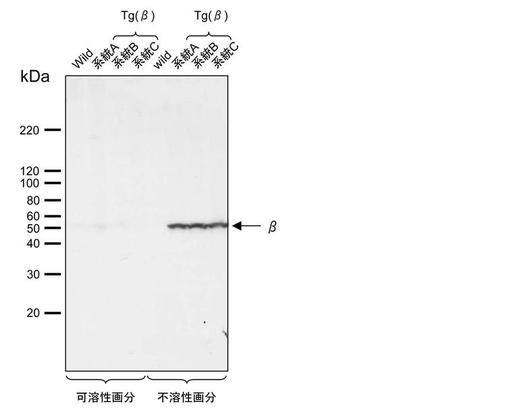
【図5】実施例で作出したBβ導入カイコについて、絹糸腺でのBβ鎖の発現をウエスタンブロットにより調べた結果の一例を示す。
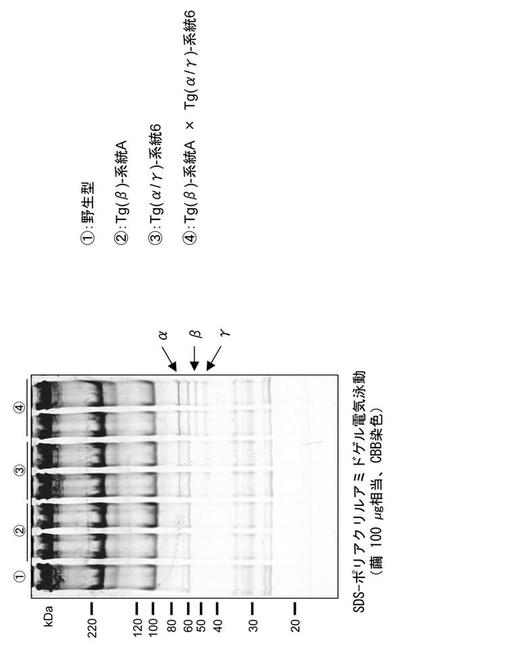
【図6】実施例で作出したトランスジェニックカイコの繭糸中に含まれるタンパク質を電気泳動しCBB(クマシーブリリアントブルー)染色した結果を示す図である。
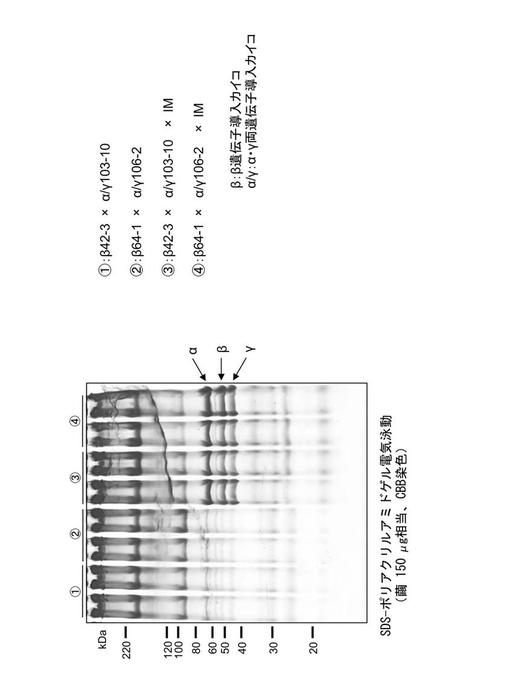
【図7】トランスアクチベーターIE1を導入したトランスジェニックカイコの繭ではフィブリノゲンの各サブユニットの蓄積量が増大していることを示す電気泳動像である。
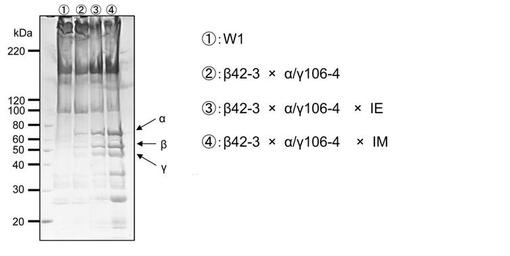
【図8】トランスアクチベーターIE1の発現量の違いが各サブユニットの発現量(繭糸中の蓄積量)に及ぼす影響を表す電気泳動像である。
【図9】トランスアクチベーターIE1を導入していないトランスジェニックカイコの繭を用いて、繭糸からのフィブリノゲンの抽出条件を検討した結果を示す図である。
【図10】トランスアクチベーターIE1を導入したトランスジェニックカイコの繭を用いて、繭糸からのフィブリノゲンの抽出条件を検討した結果を示す図である。
【発明を実施するための形態】
【0013】
本発明のトランスジェニックカイコは、絹糸腺細胞内でフィブリノゲンの3つのサブユニットAα鎖、Bβ鎖及びγ鎖を発現する。絹糸腺は絹糸を合成・分泌する器官であり、絹糸腺細胞内で組換えタンパク質を発現させれば絹糸と共に分泌され、繭に蓄積する。絹糸腺は前部、中部、後部の3部に分かれており、後部からフィブロインが、中部からセリシンが分泌される。フィブロインは絹糸の中心部を構成し、これの周囲をセリシンが覆う。後部絹糸腺でサブユニット遺伝子を発現させれば繭糸のフィブロイン層にフィブリノゲンが分泌され、中部絹糸腺でサブユニット遺伝子を発現させればセリシン層にフィブリノゲンが分泌される。セリシン層に分泌された組換えタンパク質は、繭糸を水や水性の緩衝液に浸漬するだけで容易に溶解するので、繭から活性を保持して回収することが容易である。本発明のトランスジェニックカイコも、中部絹糸腺で3つのサブユニット遺伝子を発現するものが好ましい。
【0014】
ヒトフィブリノゲンのアミノ酸配列及びこれをコードする塩基配列は公知であり、GenBankにも例えばNM_021871(Aα鎖)、NM_005141(Bβ鎖)、NM_000509(γ鎖)等のアクセッション番号で登録されている。これらの公知の配列を配列表の配列番号1、2(Aα鎖のcDNA配列及びアミノ酸配列)、配列番号5、6(Bβ鎖のcDNA配列及びアミノ酸配列)、配列番号9、10(γ鎖のcDNA配列及びアミノ酸配列)にそれぞれ示す。カイコ形質転換用ベクターに組み込むべきAα、Bβ、γ遺伝子は、本願配列表にも記載する公知の配列情報に基づいて適宜プライマーを設計し、ヒトのcDNAライブラリーから常法のPCRにより容易に増幅して得ることができる。
【0015】
フィブリノゲンサブユニット遺伝子をカイコに導入する際には、各サブユニットが有する本来のシグナルペプチド領域をヒトカルレティキュリンのシグナルペプチド領域と置き換えて用いることが好ましい。ヒトカルレティキュリンのシグナルペプチドは、カイコ中部絹糸腺細胞内で良好に機能するので、繭糸中に組換えフィブリノゲンを効率よく分泌させるのに有利である。シグナルペプチドをヒトカルレティキュリンのシグナルペプチドに置き換えた各サブユニットのcDNA配列及びアミノ酸配列を配列番号3、4(Aα鎖)、配列番号7、8(Bβ鎖)、配列番号11、12(γ鎖)にそれぞれ示す。配列番号3中の64nt〜111ntがヒトカルレティキュリンのシグナルペプチドをコードする領域である。なお、配列番号3、7、11に示す塩基配列は、ベクターに組み込むための配列(制限酵素認識部位など)や、翻訳を促進する目的で付加したBmNPVポリヘドリン5'-UTR(配列番号3では11nt〜60nt)を含んでいる。このような配列の修飾は、所望の付加配列を有するプライマーを用いたPCRにより容易に行うことができる。具体的な手順は下記実施例に詳述される通りである。
【0016】
3つのサブユニット遺伝子をカイコに導入する手順としては、3遺伝子を同時に導入する、3遺伝子のうちのいずれか1つを導入したカイコと残りの2遺伝子を同時に導入したカイコを交配する、1遺伝子を導入したカイコを順次交配する、などの手順が挙げられる。複数遺伝子を同時に導入する場合、単一のベクター上に複数の遺伝子を組み込んでもよいし、1遺伝子ずつ組み込んだ複数のベクターを同時にカイコに導入しても良い。下記実施例では、Bβ遺伝子のみを組み込んだベクターと、Aα遺伝子及びγ遺伝子の2つを組み込んだベクターを構築し、Bβ導入カイコとAα/γ導入カイコを得て両者を交配することにより、Aα/Bβ/γ導入カイコを取得しているが、3遺伝子の導入方法はこれに限定されるものではない。
【0017】
絹糸腺でフィブリノゲンサブユニットを発現させるためには、絹糸腺細胞内で機能するプロモーターと機能的に連結させた状態でAα、Bβ及びγ遺伝子をカイコに導入すればよい。ここで、「機能的に連結」とは、該プロモーターの支配を受けるようにその下流に遺伝子配列を連結することをいう。絹糸腺細胞内で機能するプロモーターは、絹糸腺細胞内で下流の遺伝子発現を開始させるものであればよく、他の組織・細胞内でも機能するものであっても良いが、目的とする絹糸腺細胞内で特異的に機能するプロモーターが好ましい。中部絹糸腺で機能するプロモーターの好ましい例としては、セリシン遺伝子(セリシン1遺伝子、セリシン2遺伝子など)を挙げることができ、後部絹糸腺で機能するプロモーターの好ましい例としては、フィブロイン重鎖遺伝子、フィブロイン軽鎖遺伝子、フィブロヘキサメリン遺伝子等のプロモーターを挙げることができるが、これらに限定されない。本発明では、フィブリノゲンサブユニット遺伝子を中部絹糸腺内で発現させることが好ましいので、セリシン遺伝子プロモーター等の中部絹糸腺で機能するプロモーターが好ましく用いられる。
【0018】
発現量を増大させる観点から、3つのサブユニット遺伝子はエンハンサーと組み合わせて導入することが好ましい。転写調節のシスエレメントであるエンハンサーは、プロモーター及びサブユニット遺伝子の近傍に存在すればよく、上流でも下流でもよいが、通常はプロモーターの上流に配置される。また、1つのエンハンサーで近傍の複数の遺伝子の転写を促進できるので、例えば2組のプロモーター+サブユニット遺伝子に対し1つのエンハンサーのみを組み合わせても良い。例えば、図3に示す遺伝子導入用ベクターの具体例では、2つのサブユニット遺伝子に対し1つのエンハンサーが挿入されている。エンハンサーとしては、採用するプロモーターの転写活性を増強させることができるものであれば特に限定されず、当業者であれば一過性発現系を用いてエンハンサーの転写活性増強効果を適宜評価し、好ましいエンハンサーを選択することができる。本発明で使用できるエンハンサーの好ましい具体例としては、バキュロウイルスの相同領域(homologous region)を挙げることができ、特にBmNPV由来のhr3を好ましく用いることができる(特許第4271122号公報参照)。hr3等のバキュロウイルス由来エンハンサーは、配列が公知であり(GenBank NC_001962, NC_001623など)、また市販の昆虫細胞用発現ベクター等でも用いられているため、バキュロウイルスゲノムや市販のベクター等から適宜PCRにより増幅して容易に入手することができる。
【0019】
また、同様に発現量を増大させる観点から、3つのサブユニット遺伝子はトランスアクチベーターと組み合わせて導入することが好ましい。トランスアクチベーターは、プロモーターに直接又は間接的に作用して遺伝子の転写を活性化する因子である。サブユニット遺伝子と同一のベクター上に挿入して同時に導入することもできるし、またサブユニット遺伝子とは別個にカイコに導入することもできる。例えば、サブユニット遺伝子を含むベクターとは別個のベクターにトランスアクチベーター遺伝子を挿入し、これをサブユニット遺伝子を含むベクターと共にカイコに導入しても良いし、あるいは、先に3つのサブユニット遺伝子を導入したAα/Bβ/γ導入カイコを作製し、これに改めて遺伝子工学的手法により又は別途作出されたトランスアクチベーター発現カイコとの交配によりトランスアクチベーター遺伝子を導入しても良い。これらの態様のいずれもが、「サブユニット遺伝子をトランスアクチベーターと組み合わせて導入する」ことに包含される。トランスアクチベーターとしては、採用するプロモーター(及び、エンハンサーに作用するトランスアクチベーターであれば採用するエンハンサー)の転写に及ぼす作用を増強できるものであれば特に限定されず、当業者であれば一過性発現系を用いて適宜活性を評価して好ましいトランスアクチベーターを選択することができる。本発明で使用できるトランスアクチベーターの好ましい具体例としては、バキュロウイルス由来の転写因子IE1を挙げることができる(特許第4271122号公報参照)。トランスアクチベーターを遺伝子工学的手法によりカイコに導入する際には、フィブリノゲンのサブユニット遺伝子に用いたプロモーターと同じプロモーターを使用することができる。IE1遺伝子の配列も公知であり(GenBank AY048770, M16820など)、また市販の昆虫細胞用発現ベクター等でも用いられているため、バキュロウイルスゲノムや市販のベクター等から適宜PCRにより増幅して容易に入手することができる。また、下記実施例でも用いているように、IE1発現カイコ系統が公知である(FEBS Journal 276, 5806-5820 (2009)、Biotechnol Bioeng 106, 860-870 (2010))。
【0020】
より好ましくは、エンハンサーとトランスアクチベーターの両者がフィブリノゲンサブユニット遺伝子と組み合わせて導入される。下記実施例に記載される通り、Aα/Bβ/γ導入カイコ(エンハンサーとしてhr3を利用)にIE1遺伝子を導入すると、IE1の発現量に応じて繭の重量は減少するが、3つのフィブリノゲンサブユニットの発現量は大きく増大し、繭糸中含量も大きく上昇するので、結果としてフィブリノゲンの生産効率をさらに向上することができる。
【0021】
本発明において、「組み合わせて導入される」とは、エンハンサー又はトランスアクチベーターが、絹糸腺細胞内におけるフィブリノゲンサブユニット遺伝子の発現増大に寄与するようにカイコに導入されることをいう。エンハンサーがサブユニット遺伝子と組み合わせて導入される場合、エンハンサーは、プロモーター及びサブユニット遺伝子の近傍に存在するようにして導入され、通常は、遺伝子導入用ベクター上のサブユニット遺伝子の近傍に組み込まれて該サブユニット遺伝子と同時にカイコに導入される。トランスアクチベーターは、上述した通り、エンハンサーとは異なりサブユニット遺伝子の近傍に位置する必要はないため、「トランスアクチベーターがサブユニット遺伝子と組み合わせて導入される」といった場合には、上述した態様、例えば3つのサブユニット遺伝子を発現するトランスジェニックカイコに改めてトランスアクチベーターを導入するという態様も包含される。
【0022】
カイコに外来遺伝子を導入する手法自体は公知であり、カイコ形質転換用のベクターも種々のものが公知である(例えば、Nature Biotechnology. 21, 52-56, 2003、J Biosci Bioeng 105, 595-603 (2008)、FEBS Journal 276, 5806-5820 (2009)、特開2002-306167号公報、特開2008-67612号公報等)。現在一般的に用いられるカイコ形質転換用ベクターは、昆虫由来のDNA型トランスポゾンを利用したベクターであり、最も代表的な例がpiggyBacを利用したプラスミドベクターである。該プラスミドベクターは、トランスポゾンpiggyBacの両末端に存在する2つの逆向き反復配列を含んでおり、カイコ染色体中に組み込みたい配列を該反復配列間に挿入する。これをトランスポゼース発現ヘルパープラスミドと共にカイコ卵に微量注入すると、トランスポゼースの働きで反復配列間の領域が転移するので、染色体中に該領域が組み込まれたカイコを得ることができる。本発明のトランスジェニックカイコを作製する際にも、このようなpiggyBacベクターを好ましく用いることができる。もっとも、使用する手法はこれに限定されるものではなく、公知のいかなる手法を用いても良い。
【0023】
カイコ形質転換用ベクターには、選抜の便宜のため、通常、サブユニット遺伝子と共にカイコ染色体に組み込まれるマーカー遺伝子が挿入されている。ベクターを導入したカイコ個体(卵、幼虫又は成虫)においてマーカー遺伝子の発現を確認することで、導入遺伝子の発現を間接的に確認できるので、マーカー遺伝子の発現に基づいて形質転換カイコを選抜すればよい。本発明のトランスジェニックカイコ作出方法における「絹糸腺細胞内でAα鎖、Bβ鎖及びγ鎖を発現するカイコを選抜する」工程は、このような、マーカー遺伝子の発現によりサブユニット遺伝子の発現を間接的に確認して選抜する工程であり得る。
【0024】
マーカーとしては、化学的処理や機械的処理によらずカイコを生かしたまま検出可能なマーカーが好ましく、例えば蛍光タンパク質を好ましく用いることができる。下記実施例に記載されるように、3つのサブユニット遺伝子を2グループに分けて2種類のトランスジェニックカイコを作出し、これらを交配させて3遺伝子を導入したカイコを用いる場合、蛍光波長の異なる2種類の蛍光タンパク質の遺伝子を用いて2種類のトランスジェニックカイコを作出すればよい。例えば、赤色蛍光タンパク質を一方のマーカーとし、緑色蛍光タンパク質を他方のマーカーとして用いると、交配により両者が導入されたカイコからは赤色と緑色が合わさった黄色蛍光が観察できるので、この黄色蛍光を指標として3遺伝子が導入されたカイコを選抜できる。
【0025】
図1〜3に示すベクター構築図は、下記実施例でトランスジェニックカイコを作出する際に用いたベクターである。使用したpMSG-1.1R及びpMSG1.1MGは、カイコのセリシン層に所望の組換えタンパク質を分泌させるためのベクターとして公知のものである(J Biosci Bioeng 105, 595-603 (2008)、FEBS Journal 276, 5806-5820 (2009))。これらのベクターには、エンハンサーhr3及びセリシン1プロモーターPserが含まれており、外来遺伝子cDNAを挿入するNruIサイトがPserとカイコフィブロインL鎖ポリA付加シグナルFLpAとの間に存在する(図1、図2参照)。図3中のpMSG3.1MGは、2つのサブユニット遺伝子を導入できるようにpMSG1.1MGを改変したベクターであり、第2のPser+FLpAの間に存在するEco47IIIサイトが2つ目のサブユニット遺伝子を挿入するサイトである。また、遺伝子導入のマーカーとして、蛍光タンパク質(DsRed, GFP)をコードする配列がプロモーター3xP3の下流に機能的に連結して組み込まれている。3xP3は眼や神経系で機能するプロモーターであり、これらの組織からの蛍光の有無に基づいて容易にトランスジェニックカイコを選抜することができる。
【0026】
このようにして構築したベクターは、下記実施例に具体的に記載する方法で、トランスポゼース発現ヘルパープラスミドと共にカイコ卵に微量注入することができる。使用するヘルパープラスミドは、カイコ卵中でプラスミド上からトランスポゼースを発現できるものであればよく、下記実施例で用いたpHA3PIG(Nat. Biotechnol. 18, 81-84 (2000))など公知のものを使用できる。ベクターとヘルパープラスミドは、通常1:1程度の割合で混合して注入に用いる。ベクターとヘルパープラスミドの濃度が各200μg/ml程度になるようにインジェクション用緩衝液(例えば、0.5 mMリン酸バッファー pH 7.0, 5 mM KClを含む緩衝液など)で調整し、これを産卵後2〜8時間の前胚盤葉期のカイコ卵に約15〜20 nl/卵の液量で注入すればよい。
【0027】
注入後の卵から孵化したF0世代をF0世代同士で又は野生株と交配してF1が得られる。F1孵化前の卵又は孵化後の幼虫に励起光を照射すれば、遺伝子が導入された卵又は幼虫では眼及び神経系でマーカーの蛍光タンパク質の蛍光が確認できるので、その個体を選抜すればよい。最終的には、PCRやサザンブロット等によりカイコゲノムへのサブユニット遺伝子の組み込みを確認してもよい。適宜、絹糸腺細胞内でのサブユニット遺伝子の発現量又は繭糸中へのサブユニットタンパク質の分泌量を常法により確認し、タンパク質発現量が十分に高い個体をさらに選抜してもよい。
【0028】
Aα、Bβ、γをそれぞれ発現する3つのトランスジェニックカイコ系統を作出した場合には、これら3系統を順次交配することにより、3遺伝子が導入された本発明のトランスジェニックカイコを得ることができる。1遺伝子を導入したカイコ系統と、他の2遺伝子を導入したカイコ系統を作出した場合には、両者を交配して3遺伝子が導入された本発明のトランスジェニックカイコを得ることができる。
【0029】
本発明のトランスジェニックカイコの繭には、3つのサブユニットが6量体となった活性のあるフィブリノゲンが含まれている。6量体を形成していることは、非還元状態で電気泳動してサイズで確認することができる。この繭からフィブリノゲンを回収することで、ヒトに感染力のあるウイルス等の混入の恐れがない安全なフィブリノゲンを大量に得ることができる。後部絹糸腺でサブユニット遺伝子を発現させ、繭糸のフィブロイン層にフィブリノゲンを分泌させた場合、フィブロインを溶解させればフィブリノゲンを回収できる。フィブロインの溶解には、通常、リチウムチオシアネート、グアニジンチオシアネート、臭化リチウム等のカオトロピック塩、又は塩化カルシウムとエタノールの混合液等が用いられる。中部絹糸腺でサブユニット遺伝子を発現させ、繭糸のセリシン層にフィブリノゲンを分泌させた場合、セリシン層は水に比較的易溶なので、水系の緩衝液に繭糸を浸漬すればフィブリノゲンを容易に抽出できる。繭糸からの抽出の際は、繭をそのまま抽出液に浸漬しても良いし、適宜裁断、粉砕等してから浸漬しても良い。
【0030】
セリシン層に分泌されたフィブリノゲンは、基本的には単に水系の緩衝液中に浸漬するのみで抽出、回収可能であるが、とりわけ好ましい抽出条件としては、1〜4 M 尿素、25〜100 mM Tris-HCl (pH 6.5〜8.5)、0.01〜2.0% 界面活性剤、0〜0.25M NaClを含有する緩衝液中で、4℃〜10℃程度の低温にて10時間〜24時間程度、好ましくは12時間〜18時間程度抽出するという条件が挙げられる。この条件によれば、セリシンの溶出を抑えつつ後の精製工程にも有利な状態でフィブリノゲンを効率よく抽出することができる。尿素濃度としては1〜3 Mがより好ましい。界面活性剤としては、ポリオキシエチレンオクチルフェニルエーテル(商品名 Triton X-100)、ポリエチレングリコール−p−オクチルフェニルエーテル(商品名 NP-40)、3‐[(3‐コラミドプロピル)ジメチルアンモニオ]‐1‐プロパンスルホナート(商品名 CHAPS)等を使用することができ、2種以上を混合して用いても良い。緩衝液のNaCl濃度は低くすることが好ましく、例えば0.1M以下、あるいは0.01M以下としてよい。
【0031】
緩衝液中に溶出したフィブリノゲンは、適宜限外濾過とバッファー交換を行ない、必要に応じ沈殿物を除去して濃縮フィブリノゲン溶液を得ることができる。このようにして本発明のトランスジェニックカイコの繭から回収されたフィブリノゲンは、下記実施例に記載されるように、トロンビンと混合して37℃で1時間反応させると、反応液の粘度の上昇が認められ、凝固活性を有することが確認されている。
【実施例】
【0032】
以下、本発明を実施例に基づきより具体的に説明する。もっとも、本発明は下記実施例に限定されるものではない。なお、下記実施例及び図面中では、「Aα鎖」を単に「α鎖」と、「Bβ鎖」を単に「β鎖」と呼ぶことがある。
【0033】
1.ヒトフィブリノゲンcDNA断片の調製
ヒト肝臓のcDNAライブラリーから、フィブリノゲンの3つのサブユニットAα、Bβ、γのcDNA断片をPCRにより増幅し、適宜サブクローニングしながらシグナル配列の入れ替え、開始コドンATGの付加、UTR配列の付加を順次行ない、カイコ中部絹糸腺用の発現ベクターに挿入するための各サブユニットcDNA断片を調製した。以下、各サブユニットごとにcDNA断片の調製を詳述する。なお、PCR反応は50μlで行い、Ex Taqは5ユニット、KODは1ユニットを用いた。
【0034】
(1) Aα鎖cDNAの取得
cDNAライブラリーから次の反応条件でAα鎖cDNA断片(以下Alpha)を増幅した。1回目のPCR産物をアガロースゲルで電気泳動し、Alphaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0035】
【表1】

【0036】
2回目PCR産物のアガロースゲル電気泳動を行い、Alphaと推定されるバンドをゲルから回収、精製し、pBluescriptII SK+のBamHI、HindIIIサイトに挿入した。塩基配列を確認し、pSK-alphaとした。
【0037】
次いで、Alphaのシグナル配列改変を行なった。成熟型と同様にC末端の15アミノ酸が除去された配列となるように、成熟型の位置にリバースプライマーFibrinogen Alpha C1を設定した。1回目のPCR産物のアガロースゲル電気泳動を行い、Alphaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0038】
【表2】
【0039】
2回目PCR産物のアガロースゲル電気泳動を行い、Alphaと推定されるバンドをゲルから回収、精製した。Ex Taqを使用して2回目PCR精製産物の3'末端にAを付加し、pCR2.1-TOPOにライゲーションを行なった。塩基配列を確認し、pCR-ATGalphaとした。
【0040】
次いで、以下の通りにUTR配列の付加を行なった。1回目PCR産物のアガロースゲル電気泳動を行い、Alphaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0041】
【表3】
【0042】
2回目PCR産物のアガロースゲル電気泳動を行い、Alphaと推定されるバンドをゲルから回収、精製した。この精製断片をカイコ発現ベクターへの組み込み用ベクターであるpENTR/D-TOPOにライゲーションした。塩基配列を確認し、pENTR-UTRalphaとした。
【0043】
(2) Bβ鎖cDNAの取得
cDNAライブラリーから次の反応条件でBβ鎖cDNA断片(以下Beta)を増幅した。1回目PCR産物のアガロースゲル電気泳動を行い、Betaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0044】
【表4】
【0045】
2回目PCR産物のアガロースゲル電気泳動を行い、Betaと推定されるバンドをゲルから回収、精製した。pBluescriptII SK+のEcoRI、XhoIサイトに挿入した。塩基配列を確認し、pSK-betaとした。
【0046】
次いで、Betaのシグナル配列改変を行なった。1回目のPCR産物をアガロースゲルで電気泳動し、Betaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0047】
【表5】
【0048】
2回目PCR産物のアガロースゲル電気泳動を行い、Betaと推定されるバンドをゲルから回収した。Ex Taqを使用して2回目PCR精製産物の3'末端にAを付加し、pCR2.1-TOPOにライゲーションを行った。塩基配列を確認し、pCR-ATGbetatとした。
【0049】
次いで、以下の通りにUTR配列の付加を行なった。1回目のPCR産物のアガロースゲル電気泳動を行い、Betaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0050】
【表6】
【0051】
2回目PCR産物のアガロースゲル電気泳動を行い、Betaと推定されるバンドをゲルから回収、精製し、カイコ発現ベクターへの組み込み用ベクターであるpENTR/D-TOPOにライゲーションを行った。塩基配列を確認し、pENTR-UTRbetaとした。
【0052】
(3) γ鎖cDNAの取得
cDNAライブラリーから次の反応条件でγ鎖cDNA断片(以下Gamma)を増幅した。1回目PCR産物のアガロースゲル電気泳動を行い、Gammaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0053】
【表7】
【0054】
2回目PCR産物のアガロースゲル電気泳動を行い、Gammaと推定されるバンドをゲルから回収、精製し、pBluescriptII SK+のEcoRI、HindIIIサイトに挿入した。塩基配列を確認し、pSK-gammaとした。
【0055】
次いで、Gammaのシグナル配列改変を行なった。1回目PCR産物のアガロースゲル電気泳動を行い、Gammaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0056】
【表8】
【0057】
2回目PCR産物のアガロースゲル電気泳動を行い、Gammaと推定されるバンドをゲルから回収、精製した。Ex Taqを使用して2回目PCR精製産物の3'末端にAを付加し、pCR2.1-TOPOにライゲーションを行った。塩基配列を確認し、pCR-ATGgammaとした。
【0058】
次いで、以下の通りにUTR配列の付加を行なった。1回目PCR産物のアガロースゲル電気泳動を行い、Gammaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0059】
【表9】
【0060】
2回目PCR産物のアガロースゲル電気泳動を行い、Gammaと推定されるバンドをゲルから回収、精製し、カイコ発現ベクターへの組み込み用ベクターであるpENTR/D-TOPOにライゲーションした。塩基配列を確認し、pENTR-UTRgammaとした。
【0061】
2.ヒトフィブリノゲン発現ベクターの構築
インビトロジェン社GATEWAYシステムを用いて、エントリーベクターpENTR/D-TOPOにクローニングした断片をpXINSECT-DEST38にサブクローニングした。ここから挿入断片を切り出して、公知のカイコ中部絹糸腺用発現ベクターに挿入した。Bβ鎖を発現するベクターと、Aα鎖及びγ鎖を発現するベクターの2つを作製した。詳細を以下に記載する。
【0062】
(1) Bβ鎖発現ベクターFibrinogen-β/pMSG1.1R(図1参照)
上記で調製したpENTR-UTRbetaより、GATEWAYシステムを用いてpXINSECT-DEST38に挿入遺伝子をサブクローニングした。得られたFibrinogen-β/pXINSECTからSmaI消化によってFibrinogen-β cDNAを切り出し、公知のカイコ形質転換用ベクターpMSG1.1R(J Biosci Bioeng 105, 595-603 (2008))のNruIサイトに挿入し、カイコ形質転換用のFibrinogen-β/pMSG1.1Rを完成した(図1)。なお、pMSG1.1Rベクターは、中部絹糸腺細胞内で機能するセリシン1遺伝子プロモーターPser1により挿入遺伝子を中部絹糸腺内で発現させるベクターであり、エンハンサーとしてバキュロウイルス由来のhr3を、また導入遺伝子の発現マーカーとして、カイコの眼及び神経系で機能するプロモーター3xP3に制御された赤色蛍光タンパク質遺伝子DsRedを含む。
【0063】
(2) Aα鎖/γ鎖発現ベクターFibrinogen-α&γ/pMSG-MG(図2、3参照)
上記で調製したpENTR-UTRalpha及びpENTR-UTRgammaより、GATEWAYシステムを用いてpXINSECT-DEST38に挿入遺伝子をそれぞれサブクローニングした(Fibrinogen-α/pXINSECT及びFibrinogen-γ/pXINSECT)。
【0064】
一方、公知のカイコ形質転換用ベクターpMSG1.1MG(FEBS Journal 276, 5806-5820 (2009))をもとに、2種類の遺伝子を発現させるための遺伝子導入用ベクターpMSG3.1MGを以下の通り構築した(図2)。なお、pMSG1.1MGは、上記で用いたpMSG1.1Rの導入遺伝子発現マーカーDsRedを緑色蛍光タンパク質遺伝子hMGFPに入れ替えたものである。
【0065】
セリシン1プロモーター、Eco47III制限酵素切断サイト、フィブロインL鎖ポリA付加シグナルからなる配列が挿入されたpCR4-TOPOベクター(PserFLpA/pCR4)から、ベクター由来の制限酵素サイトであるNotI及びSpeIによってインサートDNAを切り出し、切り出したインサートDNAを平滑末端化した。続いて、pMSG1.1MGをAscI消化(AscIサイトはSV40ポリA付加シグナル及びhr3間にある)した後、平滑末端化処理をし、そのサイトに平滑末端化したインサートDNAを挿入した。以上の操作により、pMSG3.1MGを完成した(図2)。
【0066】
Fibrinogen-α/pXINSECTよりSmaI消化によってFibrinogen-α cDNAを切り出し、このcDNAを上記で作製したpMSG3.1MGベクターのNruIサイトに挿入し、Fibrinogen-α/pMSG3.1MGを得た。Fibrinogen-γ/pXINSECTよりSmaI消化によってFibrinogen-γ cDNAを切り出し、このcDNAをFibrinogen-α/pMSG3.1MGのEco47IIIサイトに挿入し、Fibrinogen-α&γ/pMSG3.1MGを完成した(図3)。
【0067】
3.トランスジェニックカイコの作出
(1) Bβ導入カイコの作出
上記で構築した遺伝子導入用ベクターFibrinogen-β/pMSG1.1Rを塩化セシウム超遠心法で精製した後、ヘルパープラスミドであるpHA3PIG(Nat. Biotechnol. 18, 81-84 (2000))とプラスミド量が1:1になるように混合した。エタノール沈殿により濃縮し、遺伝子導入用ベクターとpHA3PIGの濃度がそれぞれ200μg/mlになるようにインジェクションバッファー(0.5 mMリン酸バッファー pH 7.0, 5 mM KCl)に溶解し、卵注入用のDNA溶液を得た。このDNA溶液を、産卵後2〜8時間の前胚盤葉期のカイコ卵(カイコ胚)に約15〜20 nl/卵の液量で微量注入し、卵を25℃でインキュベートした。合計3,032個の注入卵から600個が孵化した。ここから得られた生殖可能な成虫を交配し、112グループのF1卵塊を得た。産卵日から5〜6日目のF1卵塊を蛍光実体顕微鏡で観察し、マーカー遺伝子の発現すなわち眼や神経系からの赤色蛍光を確認できた卵を選抜した。Bβ発現カイコの卵を含む卵塊が7グループ得られた。これらを孵化させ飼育したところ、複数の卵塊に由来するトランスジェニックカイコが正常に発育し、生殖能力を有する成虫になった。各成虫を野生型のカイコと交配し、Bβ導入カイコを6系統得た。
【0068】
(2) Aα/γ導入カイコの作出
上記と同様にして、遺伝子導入用ベクターFibrinogen-α&γ/pMSG3.1MGを精製し、ヘルパープラスミドと共にカイコ卵に微量注入した。合計3,336個の注入卵から1,050個が孵化した。ここから得られた生殖可能な成虫を交配し、233グループのF1卵塊を得た。眼や神経系からの緑色蛍光に基づき卵を選抜し、Aα/γ発現カイコの卵を含む卵塊を17グループ得た。正常に発育した成虫を野生型カイコと交配し、Aα/γ導入カイコを14系統得た。
【0069】
(3) フィブリノゲンサブユニットの発現の確認
上記で得られたBβ導入カイコ系統及びAα/γ導入カイコ系統について、繭中への目的タンパク質の分泌をウエスタンブロットにより調べた。繭を裁断して8M尿素を含む緩衝液(8 M尿素、50 mMトリス塩緩衝液、pH 8.0)に浸漬し、80℃、5分間加温した後、遠心して得られた上清をウエスタンブロットに付した。結果の一部を図4に示す。Aα/γ導入カイコについては、Aα鎖及びγ鎖の両者が繭中に分泌されていることが確認できた。しかしながら、Bβ導入カイコでは繭中にBβ鎖を検出することができなかった。
【0070】
Bβ鎖が絹糸腺細胞内でタンパク質として発現しているかどうかを調べるため、5齢幼虫の絹糸腺の発現解析を行なった。野生型カイコ及びBβ導入カイコの5齢幼虫より絹糸腺を回収して1×TBSで抽出を行ない、遠心後の上清を可溶性画分とした。沈殿をさらに1×SDS-PAGEサンプルバッファーで抽出し、不溶性画分とした。それぞれを泳動してウエスタンブロットを行なった結果、不溶性画分においてBβ鎖のシグナルが検出された(図5)。以上より、Bβ導入カイコではBβ鎖は正常に合成されているが、絹糸腺細胞内にとどまっていることが確認された。
【0071】
(4) Aα/Bβ/γ導入カイコの作出
繭へのBβ鎖分泌が確認できないBβ導入カイコと、繭へのAα鎖及びγ鎖分泌が確認できたAα/γ導入カイコを交配し、Aα/Bβ/γ導入カイコを複数系統得た。これら複数系統のカイコについては、卵又は幼虫の段階で励起光を照射し、赤色蛍光と緑色蛍光が合わさって黄色の蛍光が観察されることを確認した。このカイコの繭に含まれるタンパク質を上記と同様に抽出してSDS-ポリアクリルアミドゲルで泳動しCBB染色して観察したところ、Aα鎖、Bβ鎖、γ鎖いずれも明瞭なバンドが検出された(図6)。
【0072】
4.フィブリノゲンの発現量を増加させるための転写因子の利用
セリシンプロモーターに、バキュロウイルス(BmNPV)由来のエンハンサーであるhr3と、同じくBmNPV由来のトランスアクチベーターであるIE1遺伝子を組み合わせると、セリシンプロモーターの活性が大幅に増大することが知られている(特許第4271122号公報参照)。カイコでの組換えタンパク質発現にも応用されており、IE1タンパク質を中部絹糸腺で発現するトランスジェニックカイコ系統が知られている(FEBS Journal 276, 5806-5820 (2009)、Biotechnol Bioeng 106, 860-870 (2010))。公知のIE1発現カイコを用いて、Aα/Bβ/γ導入カイコでの導入遺伝子の発現量の増加を試みた。
【0073】
IM1カイコはセリシン1プロモーターの制御下でIE1が導入されたカイコであり、転写因子IE1を高発現する(Biotechnol Bioeng 106, 860-870 (2010))。上記で作製したAα/Bβ/γ導入カイコとIM1カイコを交配してAα/Bβ/γ×IMカイコを取得し、このカイコの繭を得て各サブユニットの発現量(繭糸中の各サブユニットの含量)を調べた。その結果、フィブリノゲンの発現量は大幅に上昇したが、繭重量は3〜4割程度まで減少した(表10及び図7)。
【0074】
【表10】
【0075】
IE1カイコは上記のIM1カイコよりもIE1の発現量が低い系統である(FEBS Journal 276, 5806-5820 (2009))。IE1カイコと交配させてAα/Bβ/γ×IEカイコを取得し、このカイコの繭を得て各サブユニットの発現量(繭糸中の各サブユニットの含量)及び繭重量を調べた。繭重量の減少は回避でき、またフィブリノゲン発現量もAα/Bβ/γカイコと比較して増加していたが、Aα/Bβ/γ×IMカイコよりもフィブリノゲン発現量は低かった(表11及び図8)。
【0076】
【表11】
【0077】
Aα/Bβ/γ×IMカイコとAα/Bβ/γ×IEカイコとを比較すると、繭1個当たりのトータルでは、前者のAα/Bβ/γ×IMカイコの方がフィブリノゲンの生産量が高いという結果であった。フィブリノゲンの生産には、トランスアクチベーターを高発現するIM1カイコと交配を行なった方が有利である。
【0078】
5.繭からの組換えフィブリノゲン抽出条件の検討
Aα/Bβ/γ導入カイコの繭からフィブリノゲンを効率よく抽出するため、抽出条件を検討した。
【0079】
トランスアクチベーターを導入していないAα/Bβ/γ導入カイコの繭を用いて、フィブリノゲンの抽出条件を検討した。繭を裁断し、緩衝液に浸漬して抽出処理を行なった。PBS抽出が困難だったため、NaClや界面活性剤を含む種々の組成のバッファーを用いて、図9に示す通りに検討した。後の精製のことを考慮してなるべくセリシンが抽出されない条件にしたいこと、変性を抑えるためUrea濃度を下げたいことなどを考えると、図9中の8の条件(2M Urea, 50mM Tris-HCl(pH7.5), 1% Triton X-100, 抽出温度4℃、16時間)が最も良いと考えられた。
【0080】
トランスアクチベーターを導入したAα/Bβ/γ×IMカイコの繭を用いて、図10の通りフィブリノゲン抽出条件を検討した(全て4℃、16時間抽出)。先に検討したAα/Bβ/γカイコと比較してフィブリノゲンの発現量が高いため、より穏やかな条件でも効率よく抽出可能であり、Triton X-100濃度を0.1%に下げてもよく抽出された。
【0081】
Aα/Bβ/γ×IMカイコの繭を6M Urea, 50mM Tris-HCl(pH7.5)で抽出して非還元条件(2-メルカプトエタノールなし)で泳動した。分子量が大きいため明瞭なバンドにはならなかったが、そのサイズから6量体を形成していると考えられた(図は省略)。
【0082】
6.フィブリノゲンの凝固活性の確認
カイコで製造したヒトフィブリノゲンがトロンビンと反応して凝固(fibrin clot)を作ることができるかを確認した。
【0083】
β42-3×α/γ106-2×IMの繭を2M Urea, 50mM Tris-HCl(pH7.5), 0.1% Triton X-100で4℃にて一晩抽出した。限外濾過(amicon ultra-15 10,000NMWL, millipore社)にて抽出液を15mlから400μlまで濃縮した。次いで、15mlの100mM Tris-HCl(pH8.0), 200mM NaCl, 500nM CaCl2を加え、再度400μlまで濃縮(バッファー交換)した。この段階で一部のタンパク質が不溶化し沈殿が見られた。沈殿を除去し、終濃度10 U/mlになるようにトロンビン(Calbiochem社)を加えて37℃で1時間インキュベートしたところ、吸い上げたピペットの先端から滴下し難くなる程度まで液の粘度が上昇した。これにより、カイコで製造したフィブリノゲンの凝固活性が確認された。
【技術分野】
【0001】
本発明は、凝固活性を有するフィブリノゲンを繭糸中に産生するトランスジェニックカイコ、該カイコの製造方法、及びフィブリノゲンの製造方法に関する。
【背景技術】
【0002】
フィブリノゲンは血漿中に存在する血漿タンパク質の一種であり、分子量340kDaの糖タンパク質である。Aα鎖、Bβ鎖及びγ鎖の3本鎖がジスルフィド結合により相互に結合し、これが2量体を形成した(Aα−Bβ−γ)2という6量体の分子構造を有する。Aα鎖は67kDa、610アミノ酸残基からなり、糖鎖を有しない。Bβ鎖は56kDa、461アミノ酸残基よりなり、364位のAsnが糖鎖を有する。γ鎖は48kDa、411アミノ酸残基よりなり、52位のAsnが糖鎖を有する。
【0003】
フィブリノゲンは血液凝固に関わる。フィブリノゲンは生体内でトロンビンによりAα鎖、Bβ鎖が各々切断され、フィブリノペプチドA、フィブリノペプチドBが除去されて(α−β−γ)2へと変換される(フィブリノモノマー)。このフィブリノモノマーはCa2+の存在下で重合してフィブリンポリマーを形成する。さらにトロンビンにより血液凝固第XIII因子が活性化されると、そのトランスグルタミナーゼ活性によりフィブリンポリマー同士にペプチド結合が形成されて強固な交差架橋フィブリンになる。
【0004】
現在医療分野で用いられているフィブリノゲンは、ヒトの血漿より分離精製されて製造されている。ヒト血漿を原料にするため、ウイルス混入の危険性と隣り合わせであり、感染源の不活化処理等の工程が必須である。また、原料のヒト血漿の供給は献血に依存しており、常に安定供給することが必ずしも容易ではない。
【0005】
遺伝子組み換え技術を用いてフィブリノゲンを製造すれば、安全なフィブリノゲンを安定的に提供することができる。こうした試みは現在までにいくつか報告されているが、十分に効率的で満足できる手法は未だ提供されていない。例えば特許文献1及び非特許文献1には、ピキア酵母を用いて組換えフィブリノゲンを製造する方法が開示されているが、該方法では培養液中に分泌されたフィブリノゲンはプロテアーゼにより分解され、効率的な生産方法となっていない。特許文献2及び3には、動物培養細胞で組換えフィブリノゲンを製造する方法が開示されているが、商業的に製造するためには、細胞培養のための大規模な設備や培養密度のコントロールなど、コストや手間がかかる。また、動物細胞なので動物由来物質や感染性ウイルスが混入するおそれがあり、ヒト血液から製造する現在の方法と同様の問題点を抱えることになる。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2004−16055号公報
【特許文献2】特許第4573775号公報
【特許文献3】特表2009−528843号公報
【非特許文献】
【0007】
【非特許文献1】Protein Expression and Purification 59 (2008) 289-296
【発明の概要】
【発明が解決しようとする課題】
【0008】
従って、本願発明の目的は、安全性の高いフィブリノゲンを低コストで大量に生産可能にする新規な手段を提供することにある。
【課題を解決するための手段】
【0009】
本願発明者らは、カイコを用いた公知の組換えタンパク質生産技術を利用してフィブリノゲンを製造することに想到し、フィブリノゲンのAα鎖、Bβ鎖及びγ鎖の遺伝子をカイコ絹糸腺で発現させて繭からフィブリノゲンを回収することを試みた。Bβ鎖を発現するカイコとAα鎖及びγ鎖を発現するカイコとをそれぞれ作製し、交配により3鎖を発現するカイコを作出しようと試みたが、Bβ鎖発現カイコで繭へのBβ鎖分泌を確認することができなかった。しかしながら、このBβ鎖発現カイコをAα鎖/γ鎖発現カイコと交配させたところ、繭糸中に凝固活性を有するフィブリノゲンを分泌させることに成功し、さらに、該繭糸から効率よくフィブリノゲンを回収できる条件を見出し、本願発明を完成した。
【0010】
すなわち、本発明は、絹糸腺細胞内でフィブリノゲンのサブユニットAα鎖、Bβ鎖及びγ鎖を発現し、凝固活性を有するフィブリノゲンを繭糸中に産生するトランスジェニックカイコを提供する。また、本発明は、上記本発明のトランスジェニックカイコが生成する、凝固活性を有するフィブリノゲンを含有するカイコの繭を提供する。さらに、本発明は、上記本発明のトランスジェニックカイコの繭からフィブリノゲンを回収することを含む、フィブリノゲンの製造方法を提供する。さらに、本発明は、フィブリノゲンのサブユニットAα遺伝子、Bβ遺伝子及びγ遺伝子を、絹糸腺細胞内で機能するプロモーターと機能的に連結した形態でカイコに導入し、絹糸腺細胞内でAα鎖、Bβ鎖及びγ鎖を発現するカイコを選抜することを含む、凝固活性を有するフィブリノゲンを繭糸中に産生するトランスジェニックカイコの作出方法を提供する。
【発明の効果】
【0011】
本発明により、凝固活性を有するフィブリノゲンを繭糸に分泌するトランスジェニックカイコが初めて提供された。特に、中部絹糸腺でAα、Bβ及びγ遺伝子を発現させれば、繭糸のうち水に比較的易溶のセリシン層にフィブリノゲンが分泌されるので、活性を維持してフィブリノゲンを回収するのに有利である。カイコはタンパク質合成能力が極めて高い生物であり、飼育も容易である。献血により収集した血液材料からフィブリノゲンを製造するのとは異なり、ウイルス混入の危険性も排除できる。本発明によれば、安全性の高いフィブリノゲンを低コストで大量生産することが可能になる。
【図面の簡単な説明】
【0012】
【図1】実施例でBβ導入カイコを作出するために用いたベクターの構築図である。
【図2】実施例でAα/γ導入カイコを作出するために用いたベクターの構築図である。
【図3】実施例でAα/γ導入カイコを作出するために用いたベクターの構築図である(図2の続き)。
【図4】実施例で作出したBβ導入カイコとAα/γ導入カイコについて、繭糸中の各サブユニットタンパク質をウエスタンブロットにより検出した結果の一例を示す。
【図5】実施例で作出したBβ導入カイコについて、絹糸腺でのBβ鎖の発現をウエスタンブロットにより調べた結果の一例を示す。
【図6】実施例で作出したトランスジェニックカイコの繭糸中に含まれるタンパク質を電気泳動しCBB(クマシーブリリアントブルー)染色した結果を示す図である。
【図7】トランスアクチベーターIE1を導入したトランスジェニックカイコの繭ではフィブリノゲンの各サブユニットの蓄積量が増大していることを示す電気泳動像である。
【図8】トランスアクチベーターIE1の発現量の違いが各サブユニットの発現量(繭糸中の蓄積量)に及ぼす影響を表す電気泳動像である。
【図9】トランスアクチベーターIE1を導入していないトランスジェニックカイコの繭を用いて、繭糸からのフィブリノゲンの抽出条件を検討した結果を示す図である。
【図10】トランスアクチベーターIE1を導入したトランスジェニックカイコの繭を用いて、繭糸からのフィブリノゲンの抽出条件を検討した結果を示す図である。
【発明を実施するための形態】
【0013】
本発明のトランスジェニックカイコは、絹糸腺細胞内でフィブリノゲンの3つのサブユニットAα鎖、Bβ鎖及びγ鎖を発現する。絹糸腺は絹糸を合成・分泌する器官であり、絹糸腺細胞内で組換えタンパク質を発現させれば絹糸と共に分泌され、繭に蓄積する。絹糸腺は前部、中部、後部の3部に分かれており、後部からフィブロインが、中部からセリシンが分泌される。フィブロインは絹糸の中心部を構成し、これの周囲をセリシンが覆う。後部絹糸腺でサブユニット遺伝子を発現させれば繭糸のフィブロイン層にフィブリノゲンが分泌され、中部絹糸腺でサブユニット遺伝子を発現させればセリシン層にフィブリノゲンが分泌される。セリシン層に分泌された組換えタンパク質は、繭糸を水や水性の緩衝液に浸漬するだけで容易に溶解するので、繭から活性を保持して回収することが容易である。本発明のトランスジェニックカイコも、中部絹糸腺で3つのサブユニット遺伝子を発現するものが好ましい。
【0014】
ヒトフィブリノゲンのアミノ酸配列及びこれをコードする塩基配列は公知であり、GenBankにも例えばNM_021871(Aα鎖)、NM_005141(Bβ鎖)、NM_000509(γ鎖)等のアクセッション番号で登録されている。これらの公知の配列を配列表の配列番号1、2(Aα鎖のcDNA配列及びアミノ酸配列)、配列番号5、6(Bβ鎖のcDNA配列及びアミノ酸配列)、配列番号9、10(γ鎖のcDNA配列及びアミノ酸配列)にそれぞれ示す。カイコ形質転換用ベクターに組み込むべきAα、Bβ、γ遺伝子は、本願配列表にも記載する公知の配列情報に基づいて適宜プライマーを設計し、ヒトのcDNAライブラリーから常法のPCRにより容易に増幅して得ることができる。
【0015】
フィブリノゲンサブユニット遺伝子をカイコに導入する際には、各サブユニットが有する本来のシグナルペプチド領域をヒトカルレティキュリンのシグナルペプチド領域と置き換えて用いることが好ましい。ヒトカルレティキュリンのシグナルペプチドは、カイコ中部絹糸腺細胞内で良好に機能するので、繭糸中に組換えフィブリノゲンを効率よく分泌させるのに有利である。シグナルペプチドをヒトカルレティキュリンのシグナルペプチドに置き換えた各サブユニットのcDNA配列及びアミノ酸配列を配列番号3、4(Aα鎖)、配列番号7、8(Bβ鎖)、配列番号11、12(γ鎖)にそれぞれ示す。配列番号3中の64nt〜111ntがヒトカルレティキュリンのシグナルペプチドをコードする領域である。なお、配列番号3、7、11に示す塩基配列は、ベクターに組み込むための配列(制限酵素認識部位など)や、翻訳を促進する目的で付加したBmNPVポリヘドリン5'-UTR(配列番号3では11nt〜60nt)を含んでいる。このような配列の修飾は、所望の付加配列を有するプライマーを用いたPCRにより容易に行うことができる。具体的な手順は下記実施例に詳述される通りである。
【0016】
3つのサブユニット遺伝子をカイコに導入する手順としては、3遺伝子を同時に導入する、3遺伝子のうちのいずれか1つを導入したカイコと残りの2遺伝子を同時に導入したカイコを交配する、1遺伝子を導入したカイコを順次交配する、などの手順が挙げられる。複数遺伝子を同時に導入する場合、単一のベクター上に複数の遺伝子を組み込んでもよいし、1遺伝子ずつ組み込んだ複数のベクターを同時にカイコに導入しても良い。下記実施例では、Bβ遺伝子のみを組み込んだベクターと、Aα遺伝子及びγ遺伝子の2つを組み込んだベクターを構築し、Bβ導入カイコとAα/γ導入カイコを得て両者を交配することにより、Aα/Bβ/γ導入カイコを取得しているが、3遺伝子の導入方法はこれに限定されるものではない。
【0017】
絹糸腺でフィブリノゲンサブユニットを発現させるためには、絹糸腺細胞内で機能するプロモーターと機能的に連結させた状態でAα、Bβ及びγ遺伝子をカイコに導入すればよい。ここで、「機能的に連結」とは、該プロモーターの支配を受けるようにその下流に遺伝子配列を連結することをいう。絹糸腺細胞内で機能するプロモーターは、絹糸腺細胞内で下流の遺伝子発現を開始させるものであればよく、他の組織・細胞内でも機能するものであっても良いが、目的とする絹糸腺細胞内で特異的に機能するプロモーターが好ましい。中部絹糸腺で機能するプロモーターの好ましい例としては、セリシン遺伝子(セリシン1遺伝子、セリシン2遺伝子など)を挙げることができ、後部絹糸腺で機能するプロモーターの好ましい例としては、フィブロイン重鎖遺伝子、フィブロイン軽鎖遺伝子、フィブロヘキサメリン遺伝子等のプロモーターを挙げることができるが、これらに限定されない。本発明では、フィブリノゲンサブユニット遺伝子を中部絹糸腺内で発現させることが好ましいので、セリシン遺伝子プロモーター等の中部絹糸腺で機能するプロモーターが好ましく用いられる。
【0018】
発現量を増大させる観点から、3つのサブユニット遺伝子はエンハンサーと組み合わせて導入することが好ましい。転写調節のシスエレメントであるエンハンサーは、プロモーター及びサブユニット遺伝子の近傍に存在すればよく、上流でも下流でもよいが、通常はプロモーターの上流に配置される。また、1つのエンハンサーで近傍の複数の遺伝子の転写を促進できるので、例えば2組のプロモーター+サブユニット遺伝子に対し1つのエンハンサーのみを組み合わせても良い。例えば、図3に示す遺伝子導入用ベクターの具体例では、2つのサブユニット遺伝子に対し1つのエンハンサーが挿入されている。エンハンサーとしては、採用するプロモーターの転写活性を増強させることができるものであれば特に限定されず、当業者であれば一過性発現系を用いてエンハンサーの転写活性増強効果を適宜評価し、好ましいエンハンサーを選択することができる。本発明で使用できるエンハンサーの好ましい具体例としては、バキュロウイルスの相同領域(homologous region)を挙げることができ、特にBmNPV由来のhr3を好ましく用いることができる(特許第4271122号公報参照)。hr3等のバキュロウイルス由来エンハンサーは、配列が公知であり(GenBank NC_001962, NC_001623など)、また市販の昆虫細胞用発現ベクター等でも用いられているため、バキュロウイルスゲノムや市販のベクター等から適宜PCRにより増幅して容易に入手することができる。
【0019】
また、同様に発現量を増大させる観点から、3つのサブユニット遺伝子はトランスアクチベーターと組み合わせて導入することが好ましい。トランスアクチベーターは、プロモーターに直接又は間接的に作用して遺伝子の転写を活性化する因子である。サブユニット遺伝子と同一のベクター上に挿入して同時に導入することもできるし、またサブユニット遺伝子とは別個にカイコに導入することもできる。例えば、サブユニット遺伝子を含むベクターとは別個のベクターにトランスアクチベーター遺伝子を挿入し、これをサブユニット遺伝子を含むベクターと共にカイコに導入しても良いし、あるいは、先に3つのサブユニット遺伝子を導入したAα/Bβ/γ導入カイコを作製し、これに改めて遺伝子工学的手法により又は別途作出されたトランスアクチベーター発現カイコとの交配によりトランスアクチベーター遺伝子を導入しても良い。これらの態様のいずれもが、「サブユニット遺伝子をトランスアクチベーターと組み合わせて導入する」ことに包含される。トランスアクチベーターとしては、採用するプロモーター(及び、エンハンサーに作用するトランスアクチベーターであれば採用するエンハンサー)の転写に及ぼす作用を増強できるものであれば特に限定されず、当業者であれば一過性発現系を用いて適宜活性を評価して好ましいトランスアクチベーターを選択することができる。本発明で使用できるトランスアクチベーターの好ましい具体例としては、バキュロウイルス由来の転写因子IE1を挙げることができる(特許第4271122号公報参照)。トランスアクチベーターを遺伝子工学的手法によりカイコに導入する際には、フィブリノゲンのサブユニット遺伝子に用いたプロモーターと同じプロモーターを使用することができる。IE1遺伝子の配列も公知であり(GenBank AY048770, M16820など)、また市販の昆虫細胞用発現ベクター等でも用いられているため、バキュロウイルスゲノムや市販のベクター等から適宜PCRにより増幅して容易に入手することができる。また、下記実施例でも用いているように、IE1発現カイコ系統が公知である(FEBS Journal 276, 5806-5820 (2009)、Biotechnol Bioeng 106, 860-870 (2010))。
【0020】
より好ましくは、エンハンサーとトランスアクチベーターの両者がフィブリノゲンサブユニット遺伝子と組み合わせて導入される。下記実施例に記載される通り、Aα/Bβ/γ導入カイコ(エンハンサーとしてhr3を利用)にIE1遺伝子を導入すると、IE1の発現量に応じて繭の重量は減少するが、3つのフィブリノゲンサブユニットの発現量は大きく増大し、繭糸中含量も大きく上昇するので、結果としてフィブリノゲンの生産効率をさらに向上することができる。
【0021】
本発明において、「組み合わせて導入される」とは、エンハンサー又はトランスアクチベーターが、絹糸腺細胞内におけるフィブリノゲンサブユニット遺伝子の発現増大に寄与するようにカイコに導入されることをいう。エンハンサーがサブユニット遺伝子と組み合わせて導入される場合、エンハンサーは、プロモーター及びサブユニット遺伝子の近傍に存在するようにして導入され、通常は、遺伝子導入用ベクター上のサブユニット遺伝子の近傍に組み込まれて該サブユニット遺伝子と同時にカイコに導入される。トランスアクチベーターは、上述した通り、エンハンサーとは異なりサブユニット遺伝子の近傍に位置する必要はないため、「トランスアクチベーターがサブユニット遺伝子と組み合わせて導入される」といった場合には、上述した態様、例えば3つのサブユニット遺伝子を発現するトランスジェニックカイコに改めてトランスアクチベーターを導入するという態様も包含される。
【0022】
カイコに外来遺伝子を導入する手法自体は公知であり、カイコ形質転換用のベクターも種々のものが公知である(例えば、Nature Biotechnology. 21, 52-56, 2003、J Biosci Bioeng 105, 595-603 (2008)、FEBS Journal 276, 5806-5820 (2009)、特開2002-306167号公報、特開2008-67612号公報等)。現在一般的に用いられるカイコ形質転換用ベクターは、昆虫由来のDNA型トランスポゾンを利用したベクターであり、最も代表的な例がpiggyBacを利用したプラスミドベクターである。該プラスミドベクターは、トランスポゾンpiggyBacの両末端に存在する2つの逆向き反復配列を含んでおり、カイコ染色体中に組み込みたい配列を該反復配列間に挿入する。これをトランスポゼース発現ヘルパープラスミドと共にカイコ卵に微量注入すると、トランスポゼースの働きで反復配列間の領域が転移するので、染色体中に該領域が組み込まれたカイコを得ることができる。本発明のトランスジェニックカイコを作製する際にも、このようなpiggyBacベクターを好ましく用いることができる。もっとも、使用する手法はこれに限定されるものではなく、公知のいかなる手法を用いても良い。
【0023】
カイコ形質転換用ベクターには、選抜の便宜のため、通常、サブユニット遺伝子と共にカイコ染色体に組み込まれるマーカー遺伝子が挿入されている。ベクターを導入したカイコ個体(卵、幼虫又は成虫)においてマーカー遺伝子の発現を確認することで、導入遺伝子の発現を間接的に確認できるので、マーカー遺伝子の発現に基づいて形質転換カイコを選抜すればよい。本発明のトランスジェニックカイコ作出方法における「絹糸腺細胞内でAα鎖、Bβ鎖及びγ鎖を発現するカイコを選抜する」工程は、このような、マーカー遺伝子の発現によりサブユニット遺伝子の発現を間接的に確認して選抜する工程であり得る。
【0024】
マーカーとしては、化学的処理や機械的処理によらずカイコを生かしたまま検出可能なマーカーが好ましく、例えば蛍光タンパク質を好ましく用いることができる。下記実施例に記載されるように、3つのサブユニット遺伝子を2グループに分けて2種類のトランスジェニックカイコを作出し、これらを交配させて3遺伝子を導入したカイコを用いる場合、蛍光波長の異なる2種類の蛍光タンパク質の遺伝子を用いて2種類のトランスジェニックカイコを作出すればよい。例えば、赤色蛍光タンパク質を一方のマーカーとし、緑色蛍光タンパク質を他方のマーカーとして用いると、交配により両者が導入されたカイコからは赤色と緑色が合わさった黄色蛍光が観察できるので、この黄色蛍光を指標として3遺伝子が導入されたカイコを選抜できる。
【0025】
図1〜3に示すベクター構築図は、下記実施例でトランスジェニックカイコを作出する際に用いたベクターである。使用したpMSG-1.1R及びpMSG1.1MGは、カイコのセリシン層に所望の組換えタンパク質を分泌させるためのベクターとして公知のものである(J Biosci Bioeng 105, 595-603 (2008)、FEBS Journal 276, 5806-5820 (2009))。これらのベクターには、エンハンサーhr3及びセリシン1プロモーターPserが含まれており、外来遺伝子cDNAを挿入するNruIサイトがPserとカイコフィブロインL鎖ポリA付加シグナルFLpAとの間に存在する(図1、図2参照)。図3中のpMSG3.1MGは、2つのサブユニット遺伝子を導入できるようにpMSG1.1MGを改変したベクターであり、第2のPser+FLpAの間に存在するEco47IIIサイトが2つ目のサブユニット遺伝子を挿入するサイトである。また、遺伝子導入のマーカーとして、蛍光タンパク質(DsRed, GFP)をコードする配列がプロモーター3xP3の下流に機能的に連結して組み込まれている。3xP3は眼や神経系で機能するプロモーターであり、これらの組織からの蛍光の有無に基づいて容易にトランスジェニックカイコを選抜することができる。
【0026】
このようにして構築したベクターは、下記実施例に具体的に記載する方法で、トランスポゼース発現ヘルパープラスミドと共にカイコ卵に微量注入することができる。使用するヘルパープラスミドは、カイコ卵中でプラスミド上からトランスポゼースを発現できるものであればよく、下記実施例で用いたpHA3PIG(Nat. Biotechnol. 18, 81-84 (2000))など公知のものを使用できる。ベクターとヘルパープラスミドは、通常1:1程度の割合で混合して注入に用いる。ベクターとヘルパープラスミドの濃度が各200μg/ml程度になるようにインジェクション用緩衝液(例えば、0.5 mMリン酸バッファー pH 7.0, 5 mM KClを含む緩衝液など)で調整し、これを産卵後2〜8時間の前胚盤葉期のカイコ卵に約15〜20 nl/卵の液量で注入すればよい。
【0027】
注入後の卵から孵化したF0世代をF0世代同士で又は野生株と交配してF1が得られる。F1孵化前の卵又は孵化後の幼虫に励起光を照射すれば、遺伝子が導入された卵又は幼虫では眼及び神経系でマーカーの蛍光タンパク質の蛍光が確認できるので、その個体を選抜すればよい。最終的には、PCRやサザンブロット等によりカイコゲノムへのサブユニット遺伝子の組み込みを確認してもよい。適宜、絹糸腺細胞内でのサブユニット遺伝子の発現量又は繭糸中へのサブユニットタンパク質の分泌量を常法により確認し、タンパク質発現量が十分に高い個体をさらに選抜してもよい。
【0028】
Aα、Bβ、γをそれぞれ発現する3つのトランスジェニックカイコ系統を作出した場合には、これら3系統を順次交配することにより、3遺伝子が導入された本発明のトランスジェニックカイコを得ることができる。1遺伝子を導入したカイコ系統と、他の2遺伝子を導入したカイコ系統を作出した場合には、両者を交配して3遺伝子が導入された本発明のトランスジェニックカイコを得ることができる。
【0029】
本発明のトランスジェニックカイコの繭には、3つのサブユニットが6量体となった活性のあるフィブリノゲンが含まれている。6量体を形成していることは、非還元状態で電気泳動してサイズで確認することができる。この繭からフィブリノゲンを回収することで、ヒトに感染力のあるウイルス等の混入の恐れがない安全なフィブリノゲンを大量に得ることができる。後部絹糸腺でサブユニット遺伝子を発現させ、繭糸のフィブロイン層にフィブリノゲンを分泌させた場合、フィブロインを溶解させればフィブリノゲンを回収できる。フィブロインの溶解には、通常、リチウムチオシアネート、グアニジンチオシアネート、臭化リチウム等のカオトロピック塩、又は塩化カルシウムとエタノールの混合液等が用いられる。中部絹糸腺でサブユニット遺伝子を発現させ、繭糸のセリシン層にフィブリノゲンを分泌させた場合、セリシン層は水に比較的易溶なので、水系の緩衝液に繭糸を浸漬すればフィブリノゲンを容易に抽出できる。繭糸からの抽出の際は、繭をそのまま抽出液に浸漬しても良いし、適宜裁断、粉砕等してから浸漬しても良い。
【0030】
セリシン層に分泌されたフィブリノゲンは、基本的には単に水系の緩衝液中に浸漬するのみで抽出、回収可能であるが、とりわけ好ましい抽出条件としては、1〜4 M 尿素、25〜100 mM Tris-HCl (pH 6.5〜8.5)、0.01〜2.0% 界面活性剤、0〜0.25M NaClを含有する緩衝液中で、4℃〜10℃程度の低温にて10時間〜24時間程度、好ましくは12時間〜18時間程度抽出するという条件が挙げられる。この条件によれば、セリシンの溶出を抑えつつ後の精製工程にも有利な状態でフィブリノゲンを効率よく抽出することができる。尿素濃度としては1〜3 Mがより好ましい。界面活性剤としては、ポリオキシエチレンオクチルフェニルエーテル(商品名 Triton X-100)、ポリエチレングリコール−p−オクチルフェニルエーテル(商品名 NP-40)、3‐[(3‐コラミドプロピル)ジメチルアンモニオ]‐1‐プロパンスルホナート(商品名 CHAPS)等を使用することができ、2種以上を混合して用いても良い。緩衝液のNaCl濃度は低くすることが好ましく、例えば0.1M以下、あるいは0.01M以下としてよい。
【0031】
緩衝液中に溶出したフィブリノゲンは、適宜限外濾過とバッファー交換を行ない、必要に応じ沈殿物を除去して濃縮フィブリノゲン溶液を得ることができる。このようにして本発明のトランスジェニックカイコの繭から回収されたフィブリノゲンは、下記実施例に記載されるように、トロンビンと混合して37℃で1時間反応させると、反応液の粘度の上昇が認められ、凝固活性を有することが確認されている。
【実施例】
【0032】
以下、本発明を実施例に基づきより具体的に説明する。もっとも、本発明は下記実施例に限定されるものではない。なお、下記実施例及び図面中では、「Aα鎖」を単に「α鎖」と、「Bβ鎖」を単に「β鎖」と呼ぶことがある。
【0033】
1.ヒトフィブリノゲンcDNA断片の調製
ヒト肝臓のcDNAライブラリーから、フィブリノゲンの3つのサブユニットAα、Bβ、γのcDNA断片をPCRにより増幅し、適宜サブクローニングしながらシグナル配列の入れ替え、開始コドンATGの付加、UTR配列の付加を順次行ない、カイコ中部絹糸腺用の発現ベクターに挿入するための各サブユニットcDNA断片を調製した。以下、各サブユニットごとにcDNA断片の調製を詳述する。なお、PCR反応は50μlで行い、Ex Taqは5ユニット、KODは1ユニットを用いた。
【0034】
(1) Aα鎖cDNAの取得
cDNAライブラリーから次の反応条件でAα鎖cDNA断片(以下Alpha)を増幅した。1回目のPCR産物をアガロースゲルで電気泳動し、Alphaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0035】
【表1】
【0036】
2回目PCR産物のアガロースゲル電気泳動を行い、Alphaと推定されるバンドをゲルから回収、精製し、pBluescriptII SK+のBamHI、HindIIIサイトに挿入した。塩基配列を確認し、pSK-alphaとした。
【0037】
次いで、Alphaのシグナル配列改変を行なった。成熟型と同様にC末端の15アミノ酸が除去された配列となるように、成熟型の位置にリバースプライマーFibrinogen Alpha C1を設定した。1回目のPCR産物のアガロースゲル電気泳動を行い、Alphaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0038】
【表2】
【0039】
2回目PCR産物のアガロースゲル電気泳動を行い、Alphaと推定されるバンドをゲルから回収、精製した。Ex Taqを使用して2回目PCR精製産物の3'末端にAを付加し、pCR2.1-TOPOにライゲーションを行なった。塩基配列を確認し、pCR-ATGalphaとした。
【0040】
次いで、以下の通りにUTR配列の付加を行なった。1回目PCR産物のアガロースゲル電気泳動を行い、Alphaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0041】
【表3】
【0042】
2回目PCR産物のアガロースゲル電気泳動を行い、Alphaと推定されるバンドをゲルから回収、精製した。この精製断片をカイコ発現ベクターへの組み込み用ベクターであるpENTR/D-TOPOにライゲーションした。塩基配列を確認し、pENTR-UTRalphaとした。
【0043】
(2) Bβ鎖cDNAの取得
cDNAライブラリーから次の反応条件でBβ鎖cDNA断片(以下Beta)を増幅した。1回目PCR産物のアガロースゲル電気泳動を行い、Betaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0044】
【表4】
【0045】
2回目PCR産物のアガロースゲル電気泳動を行い、Betaと推定されるバンドをゲルから回収、精製した。pBluescriptII SK+のEcoRI、XhoIサイトに挿入した。塩基配列を確認し、pSK-betaとした。
【0046】
次いで、Betaのシグナル配列改変を行なった。1回目のPCR産物をアガロースゲルで電気泳動し、Betaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0047】
【表5】
【0048】
2回目PCR産物のアガロースゲル電気泳動を行い、Betaと推定されるバンドをゲルから回収した。Ex Taqを使用して2回目PCR精製産物の3'末端にAを付加し、pCR2.1-TOPOにライゲーションを行った。塩基配列を確認し、pCR-ATGbetatとした。
【0049】
次いで、以下の通りにUTR配列の付加を行なった。1回目のPCR産物のアガロースゲル電気泳動を行い、Betaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0050】
【表6】
【0051】
2回目PCR産物のアガロースゲル電気泳動を行い、Betaと推定されるバンドをゲルから回収、精製し、カイコ発現ベクターへの組み込み用ベクターであるpENTR/D-TOPOにライゲーションを行った。塩基配列を確認し、pENTR-UTRbetaとした。
【0052】
(3) γ鎖cDNAの取得
cDNAライブラリーから次の反応条件でγ鎖cDNA断片(以下Gamma)を増幅した。1回目PCR産物のアガロースゲル電気泳動を行い、Gammaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0053】
【表7】
【0054】
2回目PCR産物のアガロースゲル電気泳動を行い、Gammaと推定されるバンドをゲルから回収、精製し、pBluescriptII SK+のEcoRI、HindIIIサイトに挿入した。塩基配列を確認し、pSK-gammaとした。
【0055】
次いで、Gammaのシグナル配列改変を行なった。1回目PCR産物のアガロースゲル電気泳動を行い、Gammaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0056】
【表8】
【0057】
2回目PCR産物のアガロースゲル電気泳動を行い、Gammaと推定されるバンドをゲルから回収、精製した。Ex Taqを使用して2回目PCR精製産物の3'末端にAを付加し、pCR2.1-TOPOにライゲーションを行った。塩基配列を確認し、pCR-ATGgammaとした。
【0058】
次いで、以下の通りにUTR配列の付加を行なった。1回目PCR産物のアガロースゲル電気泳動を行い、Gammaと推定されるバンドをゲルから回収、精製し、2回目PCRの鋳型とした。
【0059】
【表9】
【0060】
2回目PCR産物のアガロースゲル電気泳動を行い、Gammaと推定されるバンドをゲルから回収、精製し、カイコ発現ベクターへの組み込み用ベクターであるpENTR/D-TOPOにライゲーションした。塩基配列を確認し、pENTR-UTRgammaとした。
【0061】
2.ヒトフィブリノゲン発現ベクターの構築
インビトロジェン社GATEWAYシステムを用いて、エントリーベクターpENTR/D-TOPOにクローニングした断片をpXINSECT-DEST38にサブクローニングした。ここから挿入断片を切り出して、公知のカイコ中部絹糸腺用発現ベクターに挿入した。Bβ鎖を発現するベクターと、Aα鎖及びγ鎖を発現するベクターの2つを作製した。詳細を以下に記載する。
【0062】
(1) Bβ鎖発現ベクターFibrinogen-β/pMSG1.1R(図1参照)
上記で調製したpENTR-UTRbetaより、GATEWAYシステムを用いてpXINSECT-DEST38に挿入遺伝子をサブクローニングした。得られたFibrinogen-β/pXINSECTからSmaI消化によってFibrinogen-β cDNAを切り出し、公知のカイコ形質転換用ベクターpMSG1.1R(J Biosci Bioeng 105, 595-603 (2008))のNruIサイトに挿入し、カイコ形質転換用のFibrinogen-β/pMSG1.1Rを完成した(図1)。なお、pMSG1.1Rベクターは、中部絹糸腺細胞内で機能するセリシン1遺伝子プロモーターPser1により挿入遺伝子を中部絹糸腺内で発現させるベクターであり、エンハンサーとしてバキュロウイルス由来のhr3を、また導入遺伝子の発現マーカーとして、カイコの眼及び神経系で機能するプロモーター3xP3に制御された赤色蛍光タンパク質遺伝子DsRedを含む。
【0063】
(2) Aα鎖/γ鎖発現ベクターFibrinogen-α&γ/pMSG-MG(図2、3参照)
上記で調製したpENTR-UTRalpha及びpENTR-UTRgammaより、GATEWAYシステムを用いてpXINSECT-DEST38に挿入遺伝子をそれぞれサブクローニングした(Fibrinogen-α/pXINSECT及びFibrinogen-γ/pXINSECT)。
【0064】
一方、公知のカイコ形質転換用ベクターpMSG1.1MG(FEBS Journal 276, 5806-5820 (2009))をもとに、2種類の遺伝子を発現させるための遺伝子導入用ベクターpMSG3.1MGを以下の通り構築した(図2)。なお、pMSG1.1MGは、上記で用いたpMSG1.1Rの導入遺伝子発現マーカーDsRedを緑色蛍光タンパク質遺伝子hMGFPに入れ替えたものである。
【0065】
セリシン1プロモーター、Eco47III制限酵素切断サイト、フィブロインL鎖ポリA付加シグナルからなる配列が挿入されたpCR4-TOPOベクター(PserFLpA/pCR4)から、ベクター由来の制限酵素サイトであるNotI及びSpeIによってインサートDNAを切り出し、切り出したインサートDNAを平滑末端化した。続いて、pMSG1.1MGをAscI消化(AscIサイトはSV40ポリA付加シグナル及びhr3間にある)した後、平滑末端化処理をし、そのサイトに平滑末端化したインサートDNAを挿入した。以上の操作により、pMSG3.1MGを完成した(図2)。
【0066】
Fibrinogen-α/pXINSECTよりSmaI消化によってFibrinogen-α cDNAを切り出し、このcDNAを上記で作製したpMSG3.1MGベクターのNruIサイトに挿入し、Fibrinogen-α/pMSG3.1MGを得た。Fibrinogen-γ/pXINSECTよりSmaI消化によってFibrinogen-γ cDNAを切り出し、このcDNAをFibrinogen-α/pMSG3.1MGのEco47IIIサイトに挿入し、Fibrinogen-α&γ/pMSG3.1MGを完成した(図3)。
【0067】
3.トランスジェニックカイコの作出
(1) Bβ導入カイコの作出
上記で構築した遺伝子導入用ベクターFibrinogen-β/pMSG1.1Rを塩化セシウム超遠心法で精製した後、ヘルパープラスミドであるpHA3PIG(Nat. Biotechnol. 18, 81-84 (2000))とプラスミド量が1:1になるように混合した。エタノール沈殿により濃縮し、遺伝子導入用ベクターとpHA3PIGの濃度がそれぞれ200μg/mlになるようにインジェクションバッファー(0.5 mMリン酸バッファー pH 7.0, 5 mM KCl)に溶解し、卵注入用のDNA溶液を得た。このDNA溶液を、産卵後2〜8時間の前胚盤葉期のカイコ卵(カイコ胚)に約15〜20 nl/卵の液量で微量注入し、卵を25℃でインキュベートした。合計3,032個の注入卵から600個が孵化した。ここから得られた生殖可能な成虫を交配し、112グループのF1卵塊を得た。産卵日から5〜6日目のF1卵塊を蛍光実体顕微鏡で観察し、マーカー遺伝子の発現すなわち眼や神経系からの赤色蛍光を確認できた卵を選抜した。Bβ発現カイコの卵を含む卵塊が7グループ得られた。これらを孵化させ飼育したところ、複数の卵塊に由来するトランスジェニックカイコが正常に発育し、生殖能力を有する成虫になった。各成虫を野生型のカイコと交配し、Bβ導入カイコを6系統得た。
【0068】
(2) Aα/γ導入カイコの作出
上記と同様にして、遺伝子導入用ベクターFibrinogen-α&γ/pMSG3.1MGを精製し、ヘルパープラスミドと共にカイコ卵に微量注入した。合計3,336個の注入卵から1,050個が孵化した。ここから得られた生殖可能な成虫を交配し、233グループのF1卵塊を得た。眼や神経系からの緑色蛍光に基づき卵を選抜し、Aα/γ発現カイコの卵を含む卵塊を17グループ得た。正常に発育した成虫を野生型カイコと交配し、Aα/γ導入カイコを14系統得た。
【0069】
(3) フィブリノゲンサブユニットの発現の確認
上記で得られたBβ導入カイコ系統及びAα/γ導入カイコ系統について、繭中への目的タンパク質の分泌をウエスタンブロットにより調べた。繭を裁断して8M尿素を含む緩衝液(8 M尿素、50 mMトリス塩緩衝液、pH 8.0)に浸漬し、80℃、5分間加温した後、遠心して得られた上清をウエスタンブロットに付した。結果の一部を図4に示す。Aα/γ導入カイコについては、Aα鎖及びγ鎖の両者が繭中に分泌されていることが確認できた。しかしながら、Bβ導入カイコでは繭中にBβ鎖を検出することができなかった。
【0070】
Bβ鎖が絹糸腺細胞内でタンパク質として発現しているかどうかを調べるため、5齢幼虫の絹糸腺の発現解析を行なった。野生型カイコ及びBβ導入カイコの5齢幼虫より絹糸腺を回収して1×TBSで抽出を行ない、遠心後の上清を可溶性画分とした。沈殿をさらに1×SDS-PAGEサンプルバッファーで抽出し、不溶性画分とした。それぞれを泳動してウエスタンブロットを行なった結果、不溶性画分においてBβ鎖のシグナルが検出された(図5)。以上より、Bβ導入カイコではBβ鎖は正常に合成されているが、絹糸腺細胞内にとどまっていることが確認された。
【0071】
(4) Aα/Bβ/γ導入カイコの作出
繭へのBβ鎖分泌が確認できないBβ導入カイコと、繭へのAα鎖及びγ鎖分泌が確認できたAα/γ導入カイコを交配し、Aα/Bβ/γ導入カイコを複数系統得た。これら複数系統のカイコについては、卵又は幼虫の段階で励起光を照射し、赤色蛍光と緑色蛍光が合わさって黄色の蛍光が観察されることを確認した。このカイコの繭に含まれるタンパク質を上記と同様に抽出してSDS-ポリアクリルアミドゲルで泳動しCBB染色して観察したところ、Aα鎖、Bβ鎖、γ鎖いずれも明瞭なバンドが検出された(図6)。
【0072】
4.フィブリノゲンの発現量を増加させるための転写因子の利用
セリシンプロモーターに、バキュロウイルス(BmNPV)由来のエンハンサーであるhr3と、同じくBmNPV由来のトランスアクチベーターであるIE1遺伝子を組み合わせると、セリシンプロモーターの活性が大幅に増大することが知られている(特許第4271122号公報参照)。カイコでの組換えタンパク質発現にも応用されており、IE1タンパク質を中部絹糸腺で発現するトランスジェニックカイコ系統が知られている(FEBS Journal 276, 5806-5820 (2009)、Biotechnol Bioeng 106, 860-870 (2010))。公知のIE1発現カイコを用いて、Aα/Bβ/γ導入カイコでの導入遺伝子の発現量の増加を試みた。
【0073】
IM1カイコはセリシン1プロモーターの制御下でIE1が導入されたカイコであり、転写因子IE1を高発現する(Biotechnol Bioeng 106, 860-870 (2010))。上記で作製したAα/Bβ/γ導入カイコとIM1カイコを交配してAα/Bβ/γ×IMカイコを取得し、このカイコの繭を得て各サブユニットの発現量(繭糸中の各サブユニットの含量)を調べた。その結果、フィブリノゲンの発現量は大幅に上昇したが、繭重量は3〜4割程度まで減少した(表10及び図7)。
【0074】
【表10】
【0075】
IE1カイコは上記のIM1カイコよりもIE1の発現量が低い系統である(FEBS Journal 276, 5806-5820 (2009))。IE1カイコと交配させてAα/Bβ/γ×IEカイコを取得し、このカイコの繭を得て各サブユニットの発現量(繭糸中の各サブユニットの含量)及び繭重量を調べた。繭重量の減少は回避でき、またフィブリノゲン発現量もAα/Bβ/γカイコと比較して増加していたが、Aα/Bβ/γ×IMカイコよりもフィブリノゲン発現量は低かった(表11及び図8)。
【0076】
【表11】
【0077】
Aα/Bβ/γ×IMカイコとAα/Bβ/γ×IEカイコとを比較すると、繭1個当たりのトータルでは、前者のAα/Bβ/γ×IMカイコの方がフィブリノゲンの生産量が高いという結果であった。フィブリノゲンの生産には、トランスアクチベーターを高発現するIM1カイコと交配を行なった方が有利である。
【0078】
5.繭からの組換えフィブリノゲン抽出条件の検討
Aα/Bβ/γ導入カイコの繭からフィブリノゲンを効率よく抽出するため、抽出条件を検討した。
【0079】
トランスアクチベーターを導入していないAα/Bβ/γ導入カイコの繭を用いて、フィブリノゲンの抽出条件を検討した。繭を裁断し、緩衝液に浸漬して抽出処理を行なった。PBS抽出が困難だったため、NaClや界面活性剤を含む種々の組成のバッファーを用いて、図9に示す通りに検討した。後の精製のことを考慮してなるべくセリシンが抽出されない条件にしたいこと、変性を抑えるためUrea濃度を下げたいことなどを考えると、図9中の8の条件(2M Urea, 50mM Tris-HCl(pH7.5), 1% Triton X-100, 抽出温度4℃、16時間)が最も良いと考えられた。
【0080】
トランスアクチベーターを導入したAα/Bβ/γ×IMカイコの繭を用いて、図10の通りフィブリノゲン抽出条件を検討した(全て4℃、16時間抽出)。先に検討したAα/Bβ/γカイコと比較してフィブリノゲンの発現量が高いため、より穏やかな条件でも効率よく抽出可能であり、Triton X-100濃度を0.1%に下げてもよく抽出された。
【0081】
Aα/Bβ/γ×IMカイコの繭を6M Urea, 50mM Tris-HCl(pH7.5)で抽出して非還元条件(2-メルカプトエタノールなし)で泳動した。分子量が大きいため明瞭なバンドにはならなかったが、そのサイズから6量体を形成していると考えられた(図は省略)。
【0082】
6.フィブリノゲンの凝固活性の確認
カイコで製造したヒトフィブリノゲンがトロンビンと反応して凝固(fibrin clot)を作ることができるかを確認した。
【0083】
β42-3×α/γ106-2×IMの繭を2M Urea, 50mM Tris-HCl(pH7.5), 0.1% Triton X-100で4℃にて一晩抽出した。限外濾過(amicon ultra-15 10,000NMWL, millipore社)にて抽出液を15mlから400μlまで濃縮した。次いで、15mlの100mM Tris-HCl(pH8.0), 200mM NaCl, 500nM CaCl2を加え、再度400μlまで濃縮(バッファー交換)した。この段階で一部のタンパク質が不溶化し沈殿が見られた。沈殿を除去し、終濃度10 U/mlになるようにトロンビン(Calbiochem社)を加えて37℃で1時間インキュベートしたところ、吸い上げたピペットの先端から滴下し難くなる程度まで液の粘度が上昇した。これにより、カイコで製造したフィブリノゲンの凝固活性が確認された。
【特許請求の範囲】
【請求項1】
絹糸腺細胞内でフィブリノゲンのサブユニットAα鎖、Bβ鎖及びγ鎖を発現し、凝固活性を有するフィブリノゲンを繭糸中に産生するトランスジェニックカイコ。
【請求項2】
中部絹糸腺細胞内で前記サブユニットを発現し、繭糸のセリシン層中にフィブリノゲンを産生する請求項1記載のトランスジェニックカイコ。
【請求項3】
Aα鎖遺伝子、Bβ鎖遺伝子及びγ鎖遺伝子がセリシン遺伝子のプロモーターと機能的に連結されて導入されている請求項2記載のトランスジェニックカイコ。
【請求項4】
Aα鎖遺伝子、Bβ鎖遺伝子及びγ鎖遺伝子がエンハンサーと組み合わせて導入されている請求項3記載のトランスジェニックカイコ。
【請求項5】
前記エンハンサーがバキュロウイルス由来のhr3である請求項4記載のトランスジェニックカイコ。
【請求項6】
Aα鎖遺伝子、Bβ鎖遺伝子及びγ鎖遺伝子がトランスアクチベーターと組み合わせて導入されている請求項3ないし5のいずれか1項に記載のトランスジェニックカイコ。
【請求項7】
前記トランスアクチベーターがバキュロウイルス由来の転写因子IE1である請求項6記載のトランスジェニックカイコ。
【請求項8】
請求項1ないし7のいずれか1項に記載のトランスジェニックカイコが生成する、凝固活性を有するフィブリノゲンを含有するカイコの繭。
【請求項9】
請求項8記載のトランスジェニックカイコの繭からフィブリノゲンを回収することを含む、フィブリノゲンの製造方法。
【請求項10】
1〜4 M 尿素、25〜100 mM Tris-HCl (pH 6.5〜8.5)、0.01〜2.0% 界面活性剤、0〜0.25 M NaClを含有する緩衝液中に前記カイコの繭糸を浸漬し、緩衝液中に溶出したフィブリノゲンを回収する請求項9記載の方法。
【請求項11】
前記界面活性剤がポリオキシエチレンオクチルフェニルエーテル、ポリエチレングリコール‐p‐オクチルフェニルエーテル及び3‐[(3‐コラミドプロピル)ジメチルアンモニオ]‐1‐プロパンスルホナートから成る群より選択される少なくとも1種である請求項10記載の方法。
【請求項12】
フィブリノゲンのサブユニットAα遺伝子、Bβ遺伝子及びγ遺伝子を、絹糸腺細胞内で機能するプロモーターと機能的に連結した形態でカイコに導入し、絹糸腺細胞内でAα鎖、Bβ鎖及びγ鎖を発現するカイコを選抜することを含む、凝固活性を有するフィブリノゲンを繭糸中に産生するトランスジェニックカイコの作出方法。
【請求項13】
前記絹糸腺が中部絹糸腺であり、前記プロモーターがセリシン遺伝子のプロモーターである請求項12記載の方法。
【請求項14】
Aα遺伝子、Bβ遺伝子及びγ遺伝子がエンハンサーと共に導入される請求項12又は13記載の方法。
【請求項15】
前記エンハンサーがバキュロウイルス由来のhr3である請求項14記載の方法。
【請求項16】
トランスアクチベーターをさらにカイコに導入することを含む請求項12ないし15のいずれか1項に記載の方法。
【請求項17】
前記トランスアクチベーターがバキュロウイルス由来の転写因子IE1である請求項16記載の方法。
【請求項18】
Aα遺伝子、Bβ遺伝子及びγ遺伝子のいずれか1つを絹糸腺細胞内で機能するプロモーターの下流に含むベクターを導入した第1のカイコと、前記遺伝子のうちの残りの2つを絹糸腺細胞内で機能するプロモーターの下流に含むベクターを導入した第2のカイコとを交配させることにより、前記3つのサブユニット遺伝子をカイコに導入する請求項12ないし17のいずれか1項に記載の方法。
【請求項19】
Bβ遺伝子を含むベクターを導入した第1のカイコと、Aα遺伝子及びγ遺伝子を含むベクターを導入した第2のカイコとを交配させる請求項18記載の方法。
【請求項1】
絹糸腺細胞内でフィブリノゲンのサブユニットAα鎖、Bβ鎖及びγ鎖を発現し、凝固活性を有するフィブリノゲンを繭糸中に産生するトランスジェニックカイコ。
【請求項2】
中部絹糸腺細胞内で前記サブユニットを発現し、繭糸のセリシン層中にフィブリノゲンを産生する請求項1記載のトランスジェニックカイコ。
【請求項3】
Aα鎖遺伝子、Bβ鎖遺伝子及びγ鎖遺伝子がセリシン遺伝子のプロモーターと機能的に連結されて導入されている請求項2記載のトランスジェニックカイコ。
【請求項4】
Aα鎖遺伝子、Bβ鎖遺伝子及びγ鎖遺伝子がエンハンサーと組み合わせて導入されている請求項3記載のトランスジェニックカイコ。
【請求項5】
前記エンハンサーがバキュロウイルス由来のhr3である請求項4記載のトランスジェニックカイコ。
【請求項6】
Aα鎖遺伝子、Bβ鎖遺伝子及びγ鎖遺伝子がトランスアクチベーターと組み合わせて導入されている請求項3ないし5のいずれか1項に記載のトランスジェニックカイコ。
【請求項7】
前記トランスアクチベーターがバキュロウイルス由来の転写因子IE1である請求項6記載のトランスジェニックカイコ。
【請求項8】
請求項1ないし7のいずれか1項に記載のトランスジェニックカイコが生成する、凝固活性を有するフィブリノゲンを含有するカイコの繭。
【請求項9】
請求項8記載のトランスジェニックカイコの繭からフィブリノゲンを回収することを含む、フィブリノゲンの製造方法。
【請求項10】
1〜4 M 尿素、25〜100 mM Tris-HCl (pH 6.5〜8.5)、0.01〜2.0% 界面活性剤、0〜0.25 M NaClを含有する緩衝液中に前記カイコの繭糸を浸漬し、緩衝液中に溶出したフィブリノゲンを回収する請求項9記載の方法。
【請求項11】
前記界面活性剤がポリオキシエチレンオクチルフェニルエーテル、ポリエチレングリコール‐p‐オクチルフェニルエーテル及び3‐[(3‐コラミドプロピル)ジメチルアンモニオ]‐1‐プロパンスルホナートから成る群より選択される少なくとも1種である請求項10記載の方法。
【請求項12】
フィブリノゲンのサブユニットAα遺伝子、Bβ遺伝子及びγ遺伝子を、絹糸腺細胞内で機能するプロモーターと機能的に連結した形態でカイコに導入し、絹糸腺細胞内でAα鎖、Bβ鎖及びγ鎖を発現するカイコを選抜することを含む、凝固活性を有するフィブリノゲンを繭糸中に産生するトランスジェニックカイコの作出方法。
【請求項13】
前記絹糸腺が中部絹糸腺であり、前記プロモーターがセリシン遺伝子のプロモーターである請求項12記載の方法。
【請求項14】
Aα遺伝子、Bβ遺伝子及びγ遺伝子がエンハンサーと共に導入される請求項12又は13記載の方法。
【請求項15】
前記エンハンサーがバキュロウイルス由来のhr3である請求項14記載の方法。
【請求項16】
トランスアクチベーターをさらにカイコに導入することを含む請求項12ないし15のいずれか1項に記載の方法。
【請求項17】
前記トランスアクチベーターがバキュロウイルス由来の転写因子IE1である請求項16記載の方法。
【請求項18】
Aα遺伝子、Bβ遺伝子及びγ遺伝子のいずれか1つを絹糸腺細胞内で機能するプロモーターの下流に含むベクターを導入した第1のカイコと、前記遺伝子のうちの残りの2つを絹糸腺細胞内で機能するプロモーターの下流に含むベクターを導入した第2のカイコとを交配させることにより、前記3つのサブユニット遺伝子をカイコに導入する請求項12ないし17のいずれか1項に記載の方法。
【請求項19】
Bβ遺伝子を含むベクターを導入した第1のカイコと、Aα遺伝子及びγ遺伝子を含むベクターを導入した第2のカイコとを交配させる請求項18記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】

【図10】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2012−182995(P2012−182995A)
【公開日】平成24年9月27日(2012.9.27)
【国際特許分類】
【出願番号】特願2011−46386(P2011−46386)
【出願日】平成23年3月3日(2011.3.3)
【出願人】(000231637)日本製粉株式会社 (144)
【出願人】(399032282)株式会社 免疫生物研究所 (14)
【Fターム(参考)】
【公開日】平成24年9月27日(2012.9.27)
【国際特許分類】
【出願日】平成23年3月3日(2011.3.3)
【出願人】(000231637)日本製粉株式会社 (144)
【出願人】(399032282)株式会社 免疫生物研究所 (14)
【Fターム(参考)】
[ Back to top ]