
フェニルアラニンまたはアラニンの電気化学的測定方法
【課題】血液中の濃度が格段に低いPheのセンサによる測定における妨害物質の有効な除去方法を提供し、血液中のPhe濃度を高い精度で測定する方法を提供する。
【解決手段】フェニルアラニン脱水素酵素またはアラニン脱水素酵素とジアホラーゼとを固定化した作用極及び対極を有する電極系を含むPheまたはAlaセンサを用いて血液中のフェニルアラニンまたはアラニン濃度を測定する方法。以下の(1)〜(6)の工程を含む。
(1)被検体である血液を補酵素及び電子メディエーターを含有する試薬液と混合して測定液を調製し、
(2)前記測定液を前記電極系に供給し、
(3)前記電極系の作用極にプラス電位を印加して、前記測定液に含有されるアスコルビン酸及び電子メディエーターを酸化し、
(4)プラス電位の印加を終了し、酵素反応を所定時間実施し、
(5)所定時間経過後、前記電極系の作用極にプラス電位を印加して、電流値を測定し、
(6)測定した電流値またはその積分値に基づいて前記被検体である血液中のフェニルアラニンまたはアラニン濃度を算出する
【解決手段】フェニルアラニン脱水素酵素またはアラニン脱水素酵素とジアホラーゼとを固定化した作用極及び対極を有する電極系を含むPheまたはAlaセンサを用いて血液中のフェニルアラニンまたはアラニン濃度を測定する方法。以下の(1)〜(6)の工程を含む。
(1)被検体である血液を補酵素及び電子メディエーターを含有する試薬液と混合して測定液を調製し、
(2)前記測定液を前記電極系に供給し、
(3)前記電極系の作用極にプラス電位を印加して、前記測定液に含有されるアスコルビン酸及び電子メディエーターを酸化し、
(4)プラス電位の印加を終了し、酵素反応を所定時間実施し、
(5)所定時間経過後、前記電極系の作用極にプラス電位を印加して、電流値を測定し、
(6)測定した電流値またはその積分値に基づいて前記被検体である血液中のフェニルアラニンまたはアラニン濃度を算出する
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、フェニルアラニンまたはアラニンの電気化学的測定方法に関する。
【背景技術】
【0002】
アミノ酸は生体内における重要代謝物質であり、血中の遊離アミノ酸濃度を計測することは、医療分野における疾患の診断や病状の判定に非常に有用である。特にフェニルアラニン(Phe)は必須アミノ酸の1つであり、重要な代謝物質であるだけではなく、先天性代謝異常症の代表であるフェニルケトン尿症のバイオマーカーとしても注目される。通常、人の血中Phe濃度は60μM程度であるが、重篤なフェニルケトン尿症患者の場合、血中Phe濃度は1 mM以上にまで上昇する。現在、フェニルケトン尿症は高速液体クロマトグラフィー分析、あるいはPhe選択性の高い酵素を用いる分光法を用いる新生児マススクリーンによって検査されている。浅野らが開発したBacillus badius由来のフェニルアラニン脱水素酵素(PheDH)がこの酵素法で活躍している(非特許文献1、2)。
【0003】
【化1】

【0004】
ところで、フェニルケトン尿症患者が在宅での食餌療法などのために血中Pheの簡易測定を行うには、上記高速液体クロマトグラフィー分析や分光法の利用は難しく、それに代わる方法として、電気化学センサ(以下、単にセンサと呼ぶことがある)が適している。フェニルアラニンセンサ(以下、Pheセンサと呼ぶことがある)としては、例えば、特許文献1に記載のものがある。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2009-69085号公報
【特許文献2】特開2005-121551号公報
【特許文献3】特開平10-90214号公報
【特許文献4】特開2000‐97899号公報
【特許文献5】特開2000-230916号公報
【非特許文献】
【0006】
【非特許文献1】K. Nakamura, T. Fujii, Y. Kato, Y. Asano and A.J.L. Cooper, Quantitation of l-amino acids by substrate recycling between an aminotransferase and a dehydrogenase: application to the determination of l-phenylalanine in human blood, Anal. Biochem., 234, 19-22 (1996).
【非特許文献2】S. Tachibana, M. Suzuki, Y. Asano, Application of an enzyme chip to the microquantification of l-phenylalanine, Anal. Biochem., 359, 72-78 (2006).
【非特許文献3】M.N.Szentirmay and C.R.Martin, Anal. Chem., 1984, 56, 1898-1902
【非特許文献4】A.Koshy, E.Zilkha, T.P.Obrenovitch, H.P.Bennetto, D.A.Richards, L,Symon: Anal. Lett., 26, 831 (1993)
【発明の概要】
【発明が解決しようとする課題】
【0007】
特許文献1に記載のPheセンサは、フェニルアラニン脱水素酵素とジアホラーゼとを固定化した作用極及び対極からなる電極系と、補酵素及び電子メディエーターを含有する試薬液とを備えたものである。特許文献1によれば、このPheセンサは、フェニルアラニンの検出限界濃度が低く、短時間にて分析測定できるとともに繰り返し使用も可能なフェニルアラニンセンサである。
【0008】
Pheセンサが測定対象とする血液中には、Phe以外に種々の物質が含まれている。センサによる測定においては、血液中に含まれるアスコルビン酸がPhe測定の妨害物質となる可能性がある。事実、本発明者らの予備的な検討によれば、センサによるPhe測定においては、アスコルビン酸が妨害物質または夾雑物質となり、Phe量の正確な測定を妨げた。
【0009】
従来、血液中の成分をセンサによって測定することは既に行われており、代表的な成分が血糖値である。血糖、即ち、血液中のグルコースの濃度をセンサにより測定する場合にアスコルビン酸などの物質が妨害物質となること及び妨害物質の影響を回避する方法が種々提案されている。例えば、特許文献2には、多孔質材料を用いて、妨害物質を除去した後に試料をセンサに供給することが記載されている。さらに、特許文献2の先行技術として高分子膜を用いて妨害物質を除去する方法(非特許文献3)、前電解により妨害物質を除去する方法(非特許文献4)も記載されている。
【0010】
また、特許文献3には、γ−アミノ酪酸(GABA)のセンサによる測定において、アスコルビン酸を前電解により酸化することで妨害を除くことが記載されている。特許文献4には、グルタミン酸のセンサによる測定において、アスコルビン酸を前電解により酸化することで妨害を除くことが記載されている。
【0011】
一方で、特許文献5には、以下のように、妨害物質除去のための前電解についての問題も指摘されていた。『オンライン型センサでは、検出したい物質以外の妨害物質を選択的に取り除くために、電気化学的に活性な妨害物質の場合は前電解用電極を、酵素反応を起こす妨害物質については、前電解用電極に酵素を固定化する方法や、酵素を固定化した反応器を上流に設け、妨害物質を100%分解する方法(例えば、O.Niwa, T.Horiuchi, R.Kurita and K.Torimitsu, Anal.Chem., 70巻, 1126−1132頁, 1998年)を用いていた。しかしながら、試料と検出器の間に、前電解用電極等を設置したり、酵素反応容器を設けると、試料と検出器の間の内容積が増大し、応答速度が遅くなる。その結果、センサは速い時間分解能が得られないという問題があった。一方、マイクロマシン技術を用いて作製されたオンライン型センサは、内容積を極めて小さくできるため、従来のオンライン型センサと比較すると速い応答を示す。しかし、妨害物質を除去する場合、電解用電極の面積が小さいことや、反応器の容積が小さいことにより妨害物質を100%除去することは困難であるという問題がある。』(段落0003)
【0012】
また、Pheセンサが測定対象とする血液中のPhe濃度は、前述のように、通常は、60μM程度(990μg/dL)であり、重篤なフェニルケトン尿症患者の場合でも1mM程度である。血糖値測定の場合、血液中のグルコース濃度は、健常人で3.3〜5.6mM(60〜100mg/dl)であるのに対し、血液中のPhe濃度は桁違いに小さい。その一方で、血液中に含まれるアスコルビン酸は、電気化学酸化され易く、しかもその濃度が、11〜80μM(200〜1400μg/dL)であるため、血糖値測定の場合に比べて、Phe濃度測定に対するアスコルビン酸の夾雑による妨害影響は、格段に大きい。また、アラニン(Ala)センサに付いても同様の問題がある。
【0013】
そこで本発明の目的は、血液中の濃度が格段に低いPheセンサまたはAlaセンサによる測定における妨害物質の有効な除去方法を提供し、血液中のPhe濃度またはAla濃度を高い精度で測定する方法を提供することにある。
【0014】
特に本発明は、特許文献1に記載のフェニルアラニン脱水素酵素とジアホラーゼとを固定化した作用極及び対極からなる電極系と、補酵素及び電子メディエーターを含有する試薬液とを備えたPheセンサまたはAlaセンサを用いて、血液中のPhe濃度またはAla濃度を高い精度で測定する方法を提供することにある。
【課題を解決するための手段】
【0015】
本発明は、以下のとおりである。
[1]
フェニルアラニン脱水素酵素またはアラニン脱水素酵素とジアホラーゼとを固定化した作用極及び対極を有する電極系を含むフェニルアラニンまたはアラニンセンサを用いて血液中のフェニルアラニンまたはアラニン濃度を測定する方法であって、
(1)被検体である血液を補酵素及び電子メディエーターを含有する試薬液と混合して測定液を調製し、
(2)前記測定液を前記電極系に供給し、
(3)前記電極系の作用極にプラス電位を印加して、前記測定液に含有されるアスコルビン酸及び電子メディエーターを酸化し、
(4)プラス電位の印加を終了し、酵素反応を所定時間実施し、
(5)所定時間経過後、前記電極系の作用極にプラス電位を印加して、電流値を測定し、
(6)測定した電流値またはその積分値に基づいて前記被検体である血液中のフェニルアラニンまたはアラニン濃度を算出する
ことを含む前記測定方法。
[2]
工程(3)におけるプラス電位の印加は、前記測定液に含有されるアスコルビン酸の全量が酸化される条件で実施する、[1]に記載の方法。
[3]
工程(3)におけるプラス電位は、0.3〜0.5Vである、[1]に記載の方法。
[4]
酵素反応のための所定時間は、10〜60秒間である、[1]〜[3]のいずれかに記載の方法。
[5]
工程(5)におけるプラス電位は、電子メディエーターの還元体を酸化するに適した電位から選択する、[1]に記載の方法。
[6]
工程(6)におけるフェニルアラニンまたはアラニン濃度の算出は、プラス電位印加開始後0.1〜60秒の範囲のいずれかの時間に計測される電流値に基づき、かつ予め作成した検量線を基準にして行う、[1]に記載の方法。
[7]
工程(6)におけるフェニルアラニンまたはアラニン濃度の算出は、プラス電位印加開始後、60秒間に計測された全てまたは一部の電流値から求めた電気量に基づき、かつ予め作成した検量線を基準にして行う、[1]に記載の方法。
[8]
被検体である血液の量は0.2〜4μLの範囲であり、試薬液の量は0.3〜12μLの範囲である[1]〜[8]のいずれかに記載の製造方法。
【発明の効果】
【0016】
本発明によれば、血液中の濃度が格段に低いPheセンサまたはAlaセンサによる測定における妨害物質の有効な除去方法を提供することができ、かつこの妨害物質の除去方法を利用して、血液中のPhe濃度またはAla濃度を高い精度で測定する方法を提供することができる。
【図面の簡単な説明】
【0017】
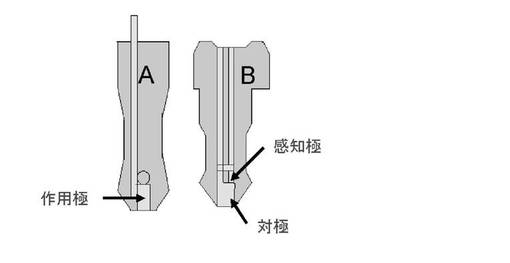
【図1】本発明で用いたセンサチップの説明図である。
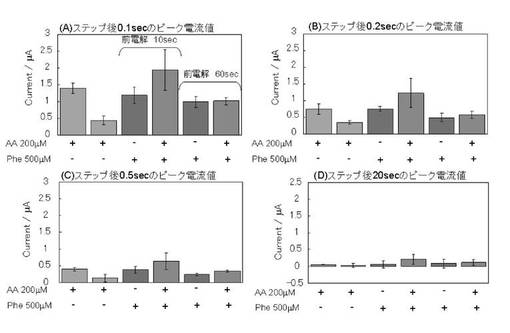
【図2−1】実施例1のPheDH(フェニルアラニン脱水素酵素)を用いた結果(前電解の効果)を示す。
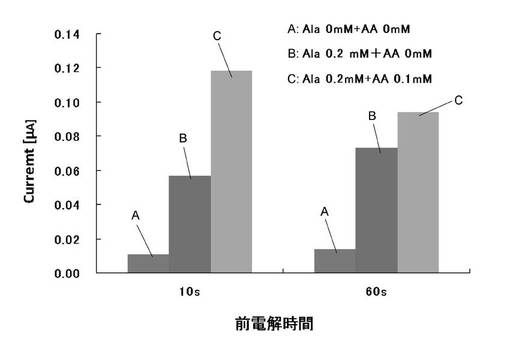
【図2−2】実施例1のAlaDH(アラニン脱水素酵素)を用いた結果(前電解の効果)を示す。
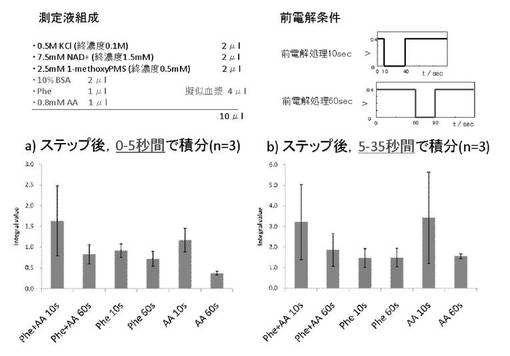
【図3】実施例1の結果(電流値の積分)を示す。
【図4】実施例1の結果(電流値でのPhe検量線)を示す。
【図5】実施例1の結果(電流積分値での検量線)を示す。
【図6】実施例3の結果を示す。ヒト血漿(HS1)及び500μMのPheを付加したヒト血漿(HS1+Phe500μM)を測定した際の時間-電流曲線である。
【図7】実施例3の結果を示す。Phe測定用検量線とHS1及びHS1+Phe500μMの測定点(n=3)を示す。HS1+Phe500μMに対する測定値の変動係数は13%であった。
【発明を実施するための形態】
【0018】
本発明は、フェニルアラニン脱水素酵素またはアラニン脱水素酵素とジアホラーゼとを固定化した作用極及び対極を有する電極系を含むPheまたはAlaセンサを用いて血液中のフェニルアラニンまたはアラニン濃度を測定する方法である。
【0019】
フェニルアラニン脱水素酵素は、例えば、浅野らが開発したBacillus badius 由来のフェニルアラニン脱水素酵素(PheDH)(非特許文献1)を用いることができる。アラニン脱水素酵素も、ユニチカなどで製造販売されている公知の酵素を用いることができる。また、アラニン脱水素酵素については、Sakamoto, Y., Nagata, S., et al.; J. Fermet. Bioeng., 69,154 (1990)を参照できる。
【0020】
ジアホラーゼは、NAD(P)H + Acceptor(ox.) + H+ →NAD(P)+ + Acceptor(red.)の反応を触媒する酵素で、例えば市販のClostridium kluyveri由来のジアホラーゼ(DI)を用いることができる。
【0021】
作用極となる電極は、例えば、金電極であることができる。但し、金電極に限定されず、金以外の電極反応に対して不活性な材料も電極として用いることができる。対極は特に限定されないが、作用極と同様に例えば、金電極であることができ、金以外の電極反応に対して不活性な材料も電極として用いることができる。
【0022】
作用極及び対極を有する電極系は、微量の血液サンプルでの測定を可能とするという観点から、センサチップであることが好ましい。センサチップは、例えば、樹脂製基材にスクリーン印刷等で上記金電極を設けたものであることができる。但し、これに限定される意図ではない。実施例においては、インサート精密射出成形された特殊プラスチック樹脂(マイクロレシコ(登録商標))と金メッキ電極から成る、作用極、対極、感知極をもつPhe計測用酵素チップを用いた。
【0023】
作用極となる電極上に、フェニルアラニン脱水素酵素またはアラニン脱水素酵素とジアホラーゼとを固定化する方法は、特に制限はないが、例えば、メルカプトカルボン酸の金電極表面上の自己集積化膜等を用いた共有結合法が簡便で同時固定化できるので好ましい。また、2つの酵素を混合して滴下吸着させる方法は、最も簡単で安価な固定化法であり、使い捨ての酵素チップの作製に好ましい。
【0024】
上記作用極は、2種類の異なる酵素を同一電極上に固定化したものであり、フェニルアラニン脱水素酵素またはアラニン脱水素酵素とジアホラーゼとが近接して存在することから、補酵素の酸化還元と電子メディエーターの酸化還元とが互いに近接した位置で、かつ電極表面近傍で生じることから、補酵素の酸化還元と電子メディエーターの酸化還元に基づく電流値の感度を高めることができる。さらに、後述する前電解によるアスコルビン酸の影響の排除も短時間の電解で効率よく実施できるという利点もある。
【0025】
さらに本発明の方法は、被検体である血液の量及び試薬液の量を、例えば、それぞれ0.2〜4μL及び0.3〜12μLの範囲の微量に限定でき、その結果、電流値の感度及び前電解の効率を高めることもできる。
【0026】
本発明の方法は以下の(1)〜(6)の工程を含む。
【0027】
工程(1)
被検体である血液を補酵素及び電子メディエーターを含有する試薬液と混合して測定液を調製する。
【0028】
補酵素としては、例えば、酸化型ニコチンアミドアデニンジヌクレオチド(NAD+)又は酸化型ニコチンアミドアデニンヌクレオチドリン酸(NADP+)を用いることができる。
【0029】
電子メディエーターは、ジアホラーゼ(DI)に触媒されて、NADH又はNADPHなどにより電気化学的に還元され、かつ電極において酸化される可逆的反応物質であれば特に限定されない。そのような電子メディエーターの例としては、フェロセン誘導体、キノン類、フェリシアン化カリウム、オスミウム錯体、ルテニウム錯体、フェノチアジン誘導体、フェナジンメトサルフェート誘導体、p−アミノフェノール、メルドラブルー、2,6−ジクロロフェノールインドフェノールを挙げることができる。フェロセン誘導体の代表例にはフェロセンメタノールがあり、キノン類の代表例にはベンゾキノンがある。例えば、フェロセンメタノールは下記化学式のように、可逆的に酸化及び還元される。
【0030】
【化2】
【0031】
本発明の測定原理をフェニルアラニン脱水素酵素を用いる場合を例に、以下に示す。
【0032】
【化3】
【0033】
工程(2)
前記測定液を前記電極系に供給する。測定液の電極系への供給方法は特に制限されていない。微量の血液サンプルでの測定が可能なセンサチップを用いる場合には、センサチップの先端に設けられた電極系に通じる導管に測定液を接触させることにより毛細管現象で、測定液がセンサチップ内部の電極系に導かれる。被検体である血液の量は、例えば、0.2〜4μLの範囲であり、試薬液の量は、例えば、0.3〜12μLの範囲とすることができる。
【0034】
工程(3)
前記電極系の作用極にプラス電位を印加して、前記測定液に含有されるアスコルビン酸及び電子メディエーターを酸化する。この工程でのプラス電位の印加は、測定液に含有される血液に由来するアスコルビン酸の全量が酸化される条件で実施することが、フェニルアラニン濃度またはアラニン濃度を精度良く測定するという観点から適当である。プラス電位は、アスコルビン酸を酸化するという観点から0.3〜0.5Vの範囲であることが適当である。ここでのプラス電位は、対極に対する電位である。また、0.3〜0.5Vであれば、フェロセンメタノールなどの電子メディエーターも酸化されて酸化体とすることができる。酸化時間は、作用極の電極面積、測定液量、使用した血液量(アスコルビン酸量)等に応じて適宜決定できるが、例えば、20〜100秒間の範囲とすることができる。但し、この範囲に限定される意図ではなく、測定液に含有されるアスコルビン酸及び電子メディエーターのほぼ全量が酸化される条件であればよい。尚、アスコルビン酸及び電子メディエーターのほぼ全量の酸化とは、後の測定に大きな誤差を生じない範囲で、アスコルビン酸及び還元型の電子メディエーターが残存する場合を含むものである。
【0035】
工程(4)
プラス電位の印加を終了し、酵素反応を所定時間実施する。酵素反応のための所定時間は、電極に固定化されている2つの酵素の量や比活性、さらには反応温度などを考慮して適宜決定できるが、例えば、10〜60秒間の範囲とすることができる。但し、この範囲に限定される意図ではない。
【0036】
この酵素反応では、フェニルアラニン(基質)とフェニルアラニン脱水素酵素(PheDH)との酵素反応により還元型ニコチンアミドアデニンジヌクレオチド(NADH)が生じる。基質 がアラニンの場合は、アラニン(基質)とアラニン脱水素酵素(AlaDH)との酵素反応により還元型ニコチンアミドアデニンジヌクレオチド(NADH)が生じる。さらにこの還元型ニコチンアミドアデニンジヌクレオチド(NADH)からジアホラーゼ(DI)の作用により電子メディエーターに電子移動して、還元型の電子メディエーターになる。酵素反応の間に、還元型の電子メディエーターが測定液中に蓄積する。また、メディエーターの還元に使用されなかった還元型ニコチンアミドアデニンヌクレオチドリン酸も測定液中に蓄積(残存)する場合がある。
【0037】
工程(5)
酵素反応のための所定時間経過後、電極系の作用極にプラス電位を印加して、電流値を測定する。工程(5)におけるプラス電位は、電子メディエーターの還元体を酸化するに適した電位から選択する。上記のように酵素反応において、還元型の電子メディエーターが測定液中に蓄積する。また、メディエーターの還元に使用されなかった還元型ニコチンアミドアデニンヌクレオチドリン酸も測定液中に蓄積(残存)する場合がある。
【0038】
少なくとも還元型の電子メディエーターが存在する状態において電極系に所定のプラス電圧を印加すると、還元型の電子メディエーターが酸化されて、還元型の電子メディエーターの濃度に応じた酸化電流が応答電流として出力される。応答電流として出力される酸化電流は、酵素反応の条件や作用極に固定化される酵素量等を選択することで、フェニルアラニン脱水素酵素(PheDH)の基質であるフェニルアラニンの濃度に比例する。基質 がアラニンの場合は、アラニン脱水素酵素(PheDH)の基質であるアラニンの濃度に比例する。
【0039】
工程(6)
測定した電流値に基づいて前記被検体である血液中のフェニルアラニン濃度またはアラニン濃度を算出する。フェニルアラニン濃度またはアラニン濃度の算出は、電流値から直接求めることもできるが、電流値を積分して求めた電気量から求めることもできる。電流値から直接求める場合には、フェニルアラニン濃度またはアラニン濃度の算出は、プラス電位印加開始後0.1〜60秒の範囲のいずれかの時間に計測される電流値に基づき、かつ予め作成した検量線を基準にして行うことが好ましい。前電解が例えば0.4V、60秒間のように十分であれば夾雑するアスコルビン酸の影響を受けることなく、プラス電位印加開始後0.1〜0.5秒の間に計測された電流値に基づくことで、速やかに精度良くフェニルアラニン濃度またはアラニン濃度を算出できる。また、例えばプラス電位印加開始後5秒後に計測された電流値に基づくことで、より広い濃度範囲で直線性の良い検量線を作成でき、さらに高精度にフェニルアラニン濃度またはアラニン濃度を算出できることからより好ましい。
【0040】
また、電流値を積分して求めた電気量からフェニルアラニン濃度またはアラニン濃度を求める場合には、所謂、電位ステップクロノアンペロメトリー(CA)を用いることが好ましく、具体的には、プラス電位印加開始後、60秒経過後の間に計測された全てまたは一部の電流値から求めた電気量に基づき、かつ予め作成した検量線を基準にして行うことが好ましい。より好ましくは、プラス電位印加開始後、5秒間に計測された全ての電流値から求めた電気量に基づくか、またはプラス電位印加開始後、5〜10秒の間に計測された全ての電流値から求めた電気量に基づくことが、速やかに、高精度にフェニルアラニン濃度またはアラニン濃度を算出できることからより好ましい。
【実施例】
【0041】
以下、本発明を実施例によりさらに詳細に説明する。
【0042】
実施例1
(1)実験方法
酵素センサの試作のため、図1に示す作用極、対極、感知極をもつ新型金電極チップを用いた。先ず3.6mg/mLのPheDH/Tricine-KOH(pH8.9)溶液(PheDH)と3.6m/mLのDI/PB(pH 7.0)溶液(DI)の混合酵素溶液2μLを作用極上に滴下し、乾燥させることにより2つの酵素を物理吸着させた。次いで、この作用極が貼られた樹脂部(A)を、対極および感知極が貼られた樹脂部(B)に重ねて、35℃で加温し接合させることによって一体型の金電極酵素センサチップを作製した。
【0043】
種々の濃度のPhe、200μMのアスコルビン酸(AA)、5%のウシ血清アルブミン(BSA)を含むPBS溶液を擬似血漿サンプルとして4μL用意し、それを2.5mMのNAD+、0.17MのKCl、およびメディエーターである1-methoxy-PMSを0.83mM溶かしたGlycine-KOH緩衝液(pH9.5)6μLと混合して測定溶液とした(合計10μL)。この測定溶液をセンサチップの先端から毛管現象により吸い上げ、感知極が通電により測定溶液の充填を感知したところで自動的に対極に対して作用極に0.4Vの電圧を10秒間あるいは60秒間印加し、メディエーターをすべて酸化状態にすると同時に、妨害物質であるAAを前電解することによって測定溶液中から除去した(前電解)。
【0044】
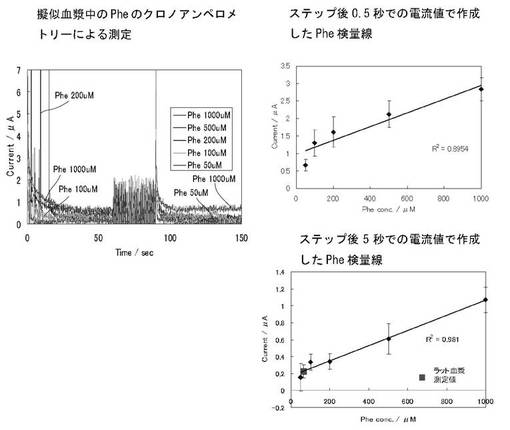
次いで0Vで30秒間保ち(室温)、この間に酵素反応を進行させた。その後、再度0.4Vに電位ステップして60秒間電流を計測した。電流値は0.1秒ごとに計測した。この第2の0.4Vへの電位ステップ時の計測された酸化電流について、ステップ後0.5秒後及び5秒後の電流を読み取って、あるいは0〜5秒間の電流値を積分して、あるいは5〜10秒の間の電流値を積分して、あるいは5〜35秒の間の電流値を積分した。さらに、擬似血漿中のPhe濃度に対する検量線を作成した。
【0045】
(2)結果
1)前電解時間の検討
前電解時間を10秒間とした場合及び前電解時間を60秒間とした場合について、AAを含有する場合としない場合について、電流値を図2-1に示す。ステップ後、0.1 秒後の電流値、0.2秒後の電流値、0.5秒後の電流値、20秒後の電流値をそれぞれ示す。また、各条件において、Pheを含まず、AAを含む測定溶液を用い、前電解時間を10秒間とした場合または前電解時間を60秒間とした場合の結果も併せて図2-1に示す。
【0046】
図2-1に示す結果から、上記条件では、前電解時間が10秒間では、AAの除去には不十分であり、前電解時間が60秒間であれば、AAの除去に十分であることが分かる。さらに、前電解時間が60秒間とし、少なくともステップ後0.1秒後〜0.5秒後の電流値を用いることで、AAの妨害を回避しつつPhe濃度を測定することができる。
【0047】
2)電流値の積分についての検討
ステップ後0〜5秒間の電流値を積分して、あるいはステップ後5〜35秒の間の電流値を積分した結果を図3に示す。前電解時間を60秒間とし、ステップ後0〜5秒間の電流値を積分した場合、AAの妨害を回避しつつPhe濃度を測定することができることが分かる。前電解時間を60秒間とし、ステップ後5〜35秒の間の電流値を積分した場合でも、AAの妨害を回避しつつPhe濃度を測定することができることが分かる。
【0048】
60秒間の前電解が、Phe以外のアミノ酸を測る電気化学酵素センサのセンシング時のAAの除去にも有効であるかを検討するため、PheDHの代わりに、AlaDH(アラニン脱水素酵素)を用いて、ジアホラーゼと組み合わせて、Alaセンサを作製し、同様に前電解時間を10秒とした場合と60秒とした場合で、AAを含有する場合と含有しない場合について、Alaセンシングを行った際の、ステップ後10秒後における酸化電流値を図2-2に示す。
【0049】
図2-2の結果から、Alaセンシングにおいても、前電解時間が10秒間では、AA自体の酸化電流がAlaの測定電流の上に大きく上乗せされるのが観測され、AAの除去には不十分であることが示された。一方、前電解時間が60秒間の場合には、このAAの上乗せ電流がかなり減少し、AAの除去が進むことが示された。
【0050】
この結果は、PheおよびAlaだけでなく、同様な原理で種々のアミノ酸を測る電気化学酵素センサについて、AAの妨害除去に60秒間以上の前電解が有効であることを示した。
【0051】
3)検量線
種々の濃度のPhe(0〜1000μM)を含む擬似血漿サンプル(原液は200μMのアスコルビン酸と5%のウシ血清アルブミン(BSA)を含むPBS溶液)に対する酵素センサチップのCA応答を図4に示す。図からわかるように、擬似血漿中のPhe濃度の増加に伴い酸化電流の増加が観測された。0.4 Vへの第2ステップ後0.5秒後における酸化電流値を読み取って作成したPheに対する検量線と第2ステップ後5秒後における酸化電流値を読み取って作成した検量線もあわせて示す。その結果、AA存在下においても50〜1000μMでフェニルアラニンの計測が可能であることが示された。
【0052】
また、図5に、0.4 Vへの電位ステップ時の計測された酸化電流について、0〜5秒間の電流値を積分して、あるいは5〜10秒の間の電流値を積分して、あるいは5〜35秒の間の電流値を積分して擬似血漿中のPhe濃度に対する検量線を作成した結果を示す。
【0053】
実施例2
血液中のPheを実測した。実施例1における擬似血漿の代わりに血漿を用いて測定溶液を調製し、計測された電流値(ステップ後5秒で読み取り)あるいは電流積分値(ステップ後5〜10秒の間の電気量)の3回の測定結果の平均値から検量線によりPhe濃度を求めた。その結果、それぞれの検量線を用いた場合のサンプル血漿中のPhe濃度は、65μMおよび89μMと求められた。なお、同一血漿のPheをHPLC分析した結果は、69μMであった。
【0054】
実施例3
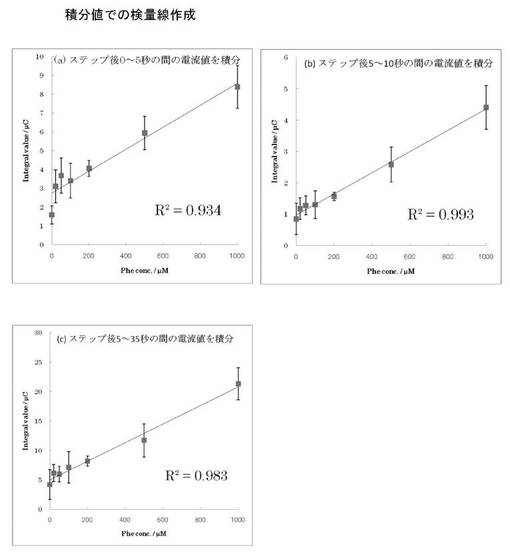
ヒト血漿サンプル(HS1と呼ぶ。コージンバイオより購入)を用いたPhe付加試験を行った。実験は、HPLC分析によりPhe濃度が既知のHS1溶液1mLに対してPhe 8.3 mgを加え混合し、HS1溶液を用いて100倍に希釈することでPheが500μM付加された血漿溶液を調製した(HS1+Pheと呼ぶ)。その後、NAD+、KCl、およびメディエーターである1-methoxy-PMSを溶かしたGlycine-KOH緩衝液 (pH 9.5)6μLに対して、限外ろ過フィルターを用いて遠心により除タンパク質処理をしたHS1+PheまたはHS1溶液を4μL取り、混合して測定溶液とした。この測定溶液を実施例1で用いたのと同様のセンサチップの先端から毛管現象により吸い上げ、感知極が通電により測定溶液の充填を感知したところで自動的に対極に対して作用極に0.4 Vの電圧を60秒間印加し、アスコルビン酸を完全に電解させると同時にメディエーターをすべて酸化状態にした。次いで0 Vで30秒間酵素反応を進行させた後、再度0.4 Vに電位ステップし5秒後の酸化電流を測定した。酸化電流の測定結果を図6に示す。
【0055】
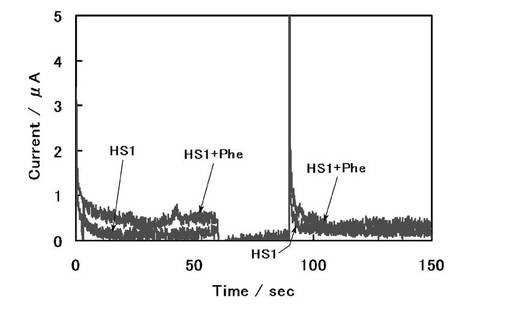
図6からわかるように、Pheを500μM付加したHS1+Phe溶液において酸化触媒電流の増加が観測された。0.4 Vへの第2ステップ後5秒における酸化電流値を読み取ってPheに対する検量線を作成し、図7に示す。図7のPheに対する検量線を用いて、それぞれの血漿サンプルのPhe濃度を定量した。その結果、Pheを500μM付加したHS1+Phe溶液のPhe濃度は585μM(CV値 : 10%)、通常のHS1溶液のPhe濃度は101.4μM(CV値 : 22%)と求められた。HPLCにより求められているHS1溶液のPhe濃度は65.8μMであり、本センサの測定値はHPLCでの分析結果に比べるとやや高い値を示したが、血漿中のPhe濃度についても測定が可能であることが示された。
【産業上の利用可能性】
【0056】
本発明は、フェニルアラニンまたはアラニンの測定が必要とされる健康及び医療の分野に有用である。また食品及び飲料中のフェニルアラニンまたはアラニン測定にも有用と考えられる。
【技術分野】
【0001】
本発明は、フェニルアラニンまたはアラニンの電気化学的測定方法に関する。
【背景技術】
【0002】
アミノ酸は生体内における重要代謝物質であり、血中の遊離アミノ酸濃度を計測することは、医療分野における疾患の診断や病状の判定に非常に有用である。特にフェニルアラニン(Phe)は必須アミノ酸の1つであり、重要な代謝物質であるだけではなく、先天性代謝異常症の代表であるフェニルケトン尿症のバイオマーカーとしても注目される。通常、人の血中Phe濃度は60μM程度であるが、重篤なフェニルケトン尿症患者の場合、血中Phe濃度は1 mM以上にまで上昇する。現在、フェニルケトン尿症は高速液体クロマトグラフィー分析、あるいはPhe選択性の高い酵素を用いる分光法を用いる新生児マススクリーンによって検査されている。浅野らが開発したBacillus badius由来のフェニルアラニン脱水素酵素(PheDH)がこの酵素法で活躍している(非特許文献1、2)。
【0003】
【化1】
【0004】
ところで、フェニルケトン尿症患者が在宅での食餌療法などのために血中Pheの簡易測定を行うには、上記高速液体クロマトグラフィー分析や分光法の利用は難しく、それに代わる方法として、電気化学センサ(以下、単にセンサと呼ぶことがある)が適している。フェニルアラニンセンサ(以下、Pheセンサと呼ぶことがある)としては、例えば、特許文献1に記載のものがある。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2009-69085号公報
【特許文献2】特開2005-121551号公報
【特許文献3】特開平10-90214号公報
【特許文献4】特開2000‐97899号公報
【特許文献5】特開2000-230916号公報
【非特許文献】
【0006】
【非特許文献1】K. Nakamura, T. Fujii, Y. Kato, Y. Asano and A.J.L. Cooper, Quantitation of l-amino acids by substrate recycling between an aminotransferase and a dehydrogenase: application to the determination of l-phenylalanine in human blood, Anal. Biochem., 234, 19-22 (1996).
【非特許文献2】S. Tachibana, M. Suzuki, Y. Asano, Application of an enzyme chip to the microquantification of l-phenylalanine, Anal. Biochem., 359, 72-78 (2006).
【非特許文献3】M.N.Szentirmay and C.R.Martin, Anal. Chem., 1984, 56, 1898-1902
【非特許文献4】A.Koshy, E.Zilkha, T.P.Obrenovitch, H.P.Bennetto, D.A.Richards, L,Symon: Anal. Lett., 26, 831 (1993)
【発明の概要】
【発明が解決しようとする課題】
【0007】
特許文献1に記載のPheセンサは、フェニルアラニン脱水素酵素とジアホラーゼとを固定化した作用極及び対極からなる電極系と、補酵素及び電子メディエーターを含有する試薬液とを備えたものである。特許文献1によれば、このPheセンサは、フェニルアラニンの検出限界濃度が低く、短時間にて分析測定できるとともに繰り返し使用も可能なフェニルアラニンセンサである。
【0008】
Pheセンサが測定対象とする血液中には、Phe以外に種々の物質が含まれている。センサによる測定においては、血液中に含まれるアスコルビン酸がPhe測定の妨害物質となる可能性がある。事実、本発明者らの予備的な検討によれば、センサによるPhe測定においては、アスコルビン酸が妨害物質または夾雑物質となり、Phe量の正確な測定を妨げた。
【0009】
従来、血液中の成分をセンサによって測定することは既に行われており、代表的な成分が血糖値である。血糖、即ち、血液中のグルコースの濃度をセンサにより測定する場合にアスコルビン酸などの物質が妨害物質となること及び妨害物質の影響を回避する方法が種々提案されている。例えば、特許文献2には、多孔質材料を用いて、妨害物質を除去した後に試料をセンサに供給することが記載されている。さらに、特許文献2の先行技術として高分子膜を用いて妨害物質を除去する方法(非特許文献3)、前電解により妨害物質を除去する方法(非特許文献4)も記載されている。
【0010】
また、特許文献3には、γ−アミノ酪酸(GABA)のセンサによる測定において、アスコルビン酸を前電解により酸化することで妨害を除くことが記載されている。特許文献4には、グルタミン酸のセンサによる測定において、アスコルビン酸を前電解により酸化することで妨害を除くことが記載されている。
【0011】
一方で、特許文献5には、以下のように、妨害物質除去のための前電解についての問題も指摘されていた。『オンライン型センサでは、検出したい物質以外の妨害物質を選択的に取り除くために、電気化学的に活性な妨害物質の場合は前電解用電極を、酵素反応を起こす妨害物質については、前電解用電極に酵素を固定化する方法や、酵素を固定化した反応器を上流に設け、妨害物質を100%分解する方法(例えば、O.Niwa, T.Horiuchi, R.Kurita and K.Torimitsu, Anal.Chem., 70巻, 1126−1132頁, 1998年)を用いていた。しかしながら、試料と検出器の間に、前電解用電極等を設置したり、酵素反応容器を設けると、試料と検出器の間の内容積が増大し、応答速度が遅くなる。その結果、センサは速い時間分解能が得られないという問題があった。一方、マイクロマシン技術を用いて作製されたオンライン型センサは、内容積を極めて小さくできるため、従来のオンライン型センサと比較すると速い応答を示す。しかし、妨害物質を除去する場合、電解用電極の面積が小さいことや、反応器の容積が小さいことにより妨害物質を100%除去することは困難であるという問題がある。』(段落0003)
【0012】
また、Pheセンサが測定対象とする血液中のPhe濃度は、前述のように、通常は、60μM程度(990μg/dL)であり、重篤なフェニルケトン尿症患者の場合でも1mM程度である。血糖値測定の場合、血液中のグルコース濃度は、健常人で3.3〜5.6mM(60〜100mg/dl)であるのに対し、血液中のPhe濃度は桁違いに小さい。その一方で、血液中に含まれるアスコルビン酸は、電気化学酸化され易く、しかもその濃度が、11〜80μM(200〜1400μg/dL)であるため、血糖値測定の場合に比べて、Phe濃度測定に対するアスコルビン酸の夾雑による妨害影響は、格段に大きい。また、アラニン(Ala)センサに付いても同様の問題がある。
【0013】
そこで本発明の目的は、血液中の濃度が格段に低いPheセンサまたはAlaセンサによる測定における妨害物質の有効な除去方法を提供し、血液中のPhe濃度またはAla濃度を高い精度で測定する方法を提供することにある。
【0014】
特に本発明は、特許文献1に記載のフェニルアラニン脱水素酵素とジアホラーゼとを固定化した作用極及び対極からなる電極系と、補酵素及び電子メディエーターを含有する試薬液とを備えたPheセンサまたはAlaセンサを用いて、血液中のPhe濃度またはAla濃度を高い精度で測定する方法を提供することにある。
【課題を解決するための手段】
【0015】
本発明は、以下のとおりである。
[1]
フェニルアラニン脱水素酵素またはアラニン脱水素酵素とジアホラーゼとを固定化した作用極及び対極を有する電極系を含むフェニルアラニンまたはアラニンセンサを用いて血液中のフェニルアラニンまたはアラニン濃度を測定する方法であって、
(1)被検体である血液を補酵素及び電子メディエーターを含有する試薬液と混合して測定液を調製し、
(2)前記測定液を前記電極系に供給し、
(3)前記電極系の作用極にプラス電位を印加して、前記測定液に含有されるアスコルビン酸及び電子メディエーターを酸化し、
(4)プラス電位の印加を終了し、酵素反応を所定時間実施し、
(5)所定時間経過後、前記電極系の作用極にプラス電位を印加して、電流値を測定し、
(6)測定した電流値またはその積分値に基づいて前記被検体である血液中のフェニルアラニンまたはアラニン濃度を算出する
ことを含む前記測定方法。
[2]
工程(3)におけるプラス電位の印加は、前記測定液に含有されるアスコルビン酸の全量が酸化される条件で実施する、[1]に記載の方法。
[3]
工程(3)におけるプラス電位は、0.3〜0.5Vである、[1]に記載の方法。
[4]
酵素反応のための所定時間は、10〜60秒間である、[1]〜[3]のいずれかに記載の方法。
[5]
工程(5)におけるプラス電位は、電子メディエーターの還元体を酸化するに適した電位から選択する、[1]に記載の方法。
[6]
工程(6)におけるフェニルアラニンまたはアラニン濃度の算出は、プラス電位印加開始後0.1〜60秒の範囲のいずれかの時間に計測される電流値に基づき、かつ予め作成した検量線を基準にして行う、[1]に記載の方法。
[7]
工程(6)におけるフェニルアラニンまたはアラニン濃度の算出は、プラス電位印加開始後、60秒間に計測された全てまたは一部の電流値から求めた電気量に基づき、かつ予め作成した検量線を基準にして行う、[1]に記載の方法。
[8]
被検体である血液の量は0.2〜4μLの範囲であり、試薬液の量は0.3〜12μLの範囲である[1]〜[8]のいずれかに記載の製造方法。
【発明の効果】
【0016】
本発明によれば、血液中の濃度が格段に低いPheセンサまたはAlaセンサによる測定における妨害物質の有効な除去方法を提供することができ、かつこの妨害物質の除去方法を利用して、血液中のPhe濃度またはAla濃度を高い精度で測定する方法を提供することができる。
【図面の簡単な説明】
【0017】
【図1】本発明で用いたセンサチップの説明図である。
【図2−1】実施例1のPheDH(フェニルアラニン脱水素酵素)を用いた結果(前電解の効果)を示す。
【図2−2】実施例1のAlaDH(アラニン脱水素酵素)を用いた結果(前電解の効果)を示す。
【図3】実施例1の結果(電流値の積分)を示す。
【図4】実施例1の結果(電流値でのPhe検量線)を示す。
【図5】実施例1の結果(電流積分値での検量線)を示す。
【図6】実施例3の結果を示す。ヒト血漿(HS1)及び500μMのPheを付加したヒト血漿(HS1+Phe500μM)を測定した際の時間-電流曲線である。
【図7】実施例3の結果を示す。Phe測定用検量線とHS1及びHS1+Phe500μMの測定点(n=3)を示す。HS1+Phe500μMに対する測定値の変動係数は13%であった。
【発明を実施するための形態】
【0018】
本発明は、フェニルアラニン脱水素酵素またはアラニン脱水素酵素とジアホラーゼとを固定化した作用極及び対極を有する電極系を含むPheまたはAlaセンサを用いて血液中のフェニルアラニンまたはアラニン濃度を測定する方法である。
【0019】
フェニルアラニン脱水素酵素は、例えば、浅野らが開発したBacillus badius 由来のフェニルアラニン脱水素酵素(PheDH)(非特許文献1)を用いることができる。アラニン脱水素酵素も、ユニチカなどで製造販売されている公知の酵素を用いることができる。また、アラニン脱水素酵素については、Sakamoto, Y., Nagata, S., et al.; J. Fermet. Bioeng., 69,154 (1990)を参照できる。
【0020】
ジアホラーゼは、NAD(P)H + Acceptor(ox.) + H+ →NAD(P)+ + Acceptor(red.)の反応を触媒する酵素で、例えば市販のClostridium kluyveri由来のジアホラーゼ(DI)を用いることができる。
【0021】
作用極となる電極は、例えば、金電極であることができる。但し、金電極に限定されず、金以外の電極反応に対して不活性な材料も電極として用いることができる。対極は特に限定されないが、作用極と同様に例えば、金電極であることができ、金以外の電極反応に対して不活性な材料も電極として用いることができる。
【0022】
作用極及び対極を有する電極系は、微量の血液サンプルでの測定を可能とするという観点から、センサチップであることが好ましい。センサチップは、例えば、樹脂製基材にスクリーン印刷等で上記金電極を設けたものであることができる。但し、これに限定される意図ではない。実施例においては、インサート精密射出成形された特殊プラスチック樹脂(マイクロレシコ(登録商標))と金メッキ電極から成る、作用極、対極、感知極をもつPhe計測用酵素チップを用いた。
【0023】
作用極となる電極上に、フェニルアラニン脱水素酵素またはアラニン脱水素酵素とジアホラーゼとを固定化する方法は、特に制限はないが、例えば、メルカプトカルボン酸の金電極表面上の自己集積化膜等を用いた共有結合法が簡便で同時固定化できるので好ましい。また、2つの酵素を混合して滴下吸着させる方法は、最も簡単で安価な固定化法であり、使い捨ての酵素チップの作製に好ましい。
【0024】
上記作用極は、2種類の異なる酵素を同一電極上に固定化したものであり、フェニルアラニン脱水素酵素またはアラニン脱水素酵素とジアホラーゼとが近接して存在することから、補酵素の酸化還元と電子メディエーターの酸化還元とが互いに近接した位置で、かつ電極表面近傍で生じることから、補酵素の酸化還元と電子メディエーターの酸化還元に基づく電流値の感度を高めることができる。さらに、後述する前電解によるアスコルビン酸の影響の排除も短時間の電解で効率よく実施できるという利点もある。
【0025】
さらに本発明の方法は、被検体である血液の量及び試薬液の量を、例えば、それぞれ0.2〜4μL及び0.3〜12μLの範囲の微量に限定でき、その結果、電流値の感度及び前電解の効率を高めることもできる。
【0026】
本発明の方法は以下の(1)〜(6)の工程を含む。
【0027】
工程(1)
被検体である血液を補酵素及び電子メディエーターを含有する試薬液と混合して測定液を調製する。
【0028】
補酵素としては、例えば、酸化型ニコチンアミドアデニンジヌクレオチド(NAD+)又は酸化型ニコチンアミドアデニンヌクレオチドリン酸(NADP+)を用いることができる。
【0029】
電子メディエーターは、ジアホラーゼ(DI)に触媒されて、NADH又はNADPHなどにより電気化学的に還元され、かつ電極において酸化される可逆的反応物質であれば特に限定されない。そのような電子メディエーターの例としては、フェロセン誘導体、キノン類、フェリシアン化カリウム、オスミウム錯体、ルテニウム錯体、フェノチアジン誘導体、フェナジンメトサルフェート誘導体、p−アミノフェノール、メルドラブルー、2,6−ジクロロフェノールインドフェノールを挙げることができる。フェロセン誘導体の代表例にはフェロセンメタノールがあり、キノン類の代表例にはベンゾキノンがある。例えば、フェロセンメタノールは下記化学式のように、可逆的に酸化及び還元される。
【0030】
【化2】
【0031】
本発明の測定原理をフェニルアラニン脱水素酵素を用いる場合を例に、以下に示す。
【0032】
【化3】
【0033】
工程(2)
前記測定液を前記電極系に供給する。測定液の電極系への供給方法は特に制限されていない。微量の血液サンプルでの測定が可能なセンサチップを用いる場合には、センサチップの先端に設けられた電極系に通じる導管に測定液を接触させることにより毛細管現象で、測定液がセンサチップ内部の電極系に導かれる。被検体である血液の量は、例えば、0.2〜4μLの範囲であり、試薬液の量は、例えば、0.3〜12μLの範囲とすることができる。
【0034】
工程(3)
前記電極系の作用極にプラス電位を印加して、前記測定液に含有されるアスコルビン酸及び電子メディエーターを酸化する。この工程でのプラス電位の印加は、測定液に含有される血液に由来するアスコルビン酸の全量が酸化される条件で実施することが、フェニルアラニン濃度またはアラニン濃度を精度良く測定するという観点から適当である。プラス電位は、アスコルビン酸を酸化するという観点から0.3〜0.5Vの範囲であることが適当である。ここでのプラス電位は、対極に対する電位である。また、0.3〜0.5Vであれば、フェロセンメタノールなどの電子メディエーターも酸化されて酸化体とすることができる。酸化時間は、作用極の電極面積、測定液量、使用した血液量(アスコルビン酸量)等に応じて適宜決定できるが、例えば、20〜100秒間の範囲とすることができる。但し、この範囲に限定される意図ではなく、測定液に含有されるアスコルビン酸及び電子メディエーターのほぼ全量が酸化される条件であればよい。尚、アスコルビン酸及び電子メディエーターのほぼ全量の酸化とは、後の測定に大きな誤差を生じない範囲で、アスコルビン酸及び還元型の電子メディエーターが残存する場合を含むものである。
【0035】
工程(4)
プラス電位の印加を終了し、酵素反応を所定時間実施する。酵素反応のための所定時間は、電極に固定化されている2つの酵素の量や比活性、さらには反応温度などを考慮して適宜決定できるが、例えば、10〜60秒間の範囲とすることができる。但し、この範囲に限定される意図ではない。
【0036】
この酵素反応では、フェニルアラニン(基質)とフェニルアラニン脱水素酵素(PheDH)との酵素反応により還元型ニコチンアミドアデニンジヌクレオチド(NADH)が生じる。基質 がアラニンの場合は、アラニン(基質)とアラニン脱水素酵素(AlaDH)との酵素反応により還元型ニコチンアミドアデニンジヌクレオチド(NADH)が生じる。さらにこの還元型ニコチンアミドアデニンジヌクレオチド(NADH)からジアホラーゼ(DI)の作用により電子メディエーターに電子移動して、還元型の電子メディエーターになる。酵素反応の間に、還元型の電子メディエーターが測定液中に蓄積する。また、メディエーターの還元に使用されなかった還元型ニコチンアミドアデニンヌクレオチドリン酸も測定液中に蓄積(残存)する場合がある。
【0037】
工程(5)
酵素反応のための所定時間経過後、電極系の作用極にプラス電位を印加して、電流値を測定する。工程(5)におけるプラス電位は、電子メディエーターの還元体を酸化するに適した電位から選択する。上記のように酵素反応において、還元型の電子メディエーターが測定液中に蓄積する。また、メディエーターの還元に使用されなかった還元型ニコチンアミドアデニンヌクレオチドリン酸も測定液中に蓄積(残存)する場合がある。
【0038】
少なくとも還元型の電子メディエーターが存在する状態において電極系に所定のプラス電圧を印加すると、還元型の電子メディエーターが酸化されて、還元型の電子メディエーターの濃度に応じた酸化電流が応答電流として出力される。応答電流として出力される酸化電流は、酵素反応の条件や作用極に固定化される酵素量等を選択することで、フェニルアラニン脱水素酵素(PheDH)の基質であるフェニルアラニンの濃度に比例する。基質 がアラニンの場合は、アラニン脱水素酵素(PheDH)の基質であるアラニンの濃度に比例する。
【0039】
工程(6)
測定した電流値に基づいて前記被検体である血液中のフェニルアラニン濃度またはアラニン濃度を算出する。フェニルアラニン濃度またはアラニン濃度の算出は、電流値から直接求めることもできるが、電流値を積分して求めた電気量から求めることもできる。電流値から直接求める場合には、フェニルアラニン濃度またはアラニン濃度の算出は、プラス電位印加開始後0.1〜60秒の範囲のいずれかの時間に計測される電流値に基づき、かつ予め作成した検量線を基準にして行うことが好ましい。前電解が例えば0.4V、60秒間のように十分であれば夾雑するアスコルビン酸の影響を受けることなく、プラス電位印加開始後0.1〜0.5秒の間に計測された電流値に基づくことで、速やかに精度良くフェニルアラニン濃度またはアラニン濃度を算出できる。また、例えばプラス電位印加開始後5秒後に計測された電流値に基づくことで、より広い濃度範囲で直線性の良い検量線を作成でき、さらに高精度にフェニルアラニン濃度またはアラニン濃度を算出できることからより好ましい。
【0040】
また、電流値を積分して求めた電気量からフェニルアラニン濃度またはアラニン濃度を求める場合には、所謂、電位ステップクロノアンペロメトリー(CA)を用いることが好ましく、具体的には、プラス電位印加開始後、60秒経過後の間に計測された全てまたは一部の電流値から求めた電気量に基づき、かつ予め作成した検量線を基準にして行うことが好ましい。より好ましくは、プラス電位印加開始後、5秒間に計測された全ての電流値から求めた電気量に基づくか、またはプラス電位印加開始後、5〜10秒の間に計測された全ての電流値から求めた電気量に基づくことが、速やかに、高精度にフェニルアラニン濃度またはアラニン濃度を算出できることからより好ましい。
【実施例】
【0041】
以下、本発明を実施例によりさらに詳細に説明する。
【0042】
実施例1
(1)実験方法
酵素センサの試作のため、図1に示す作用極、対極、感知極をもつ新型金電極チップを用いた。先ず3.6mg/mLのPheDH/Tricine-KOH(pH8.9)溶液(PheDH)と3.6m/mLのDI/PB(pH 7.0)溶液(DI)の混合酵素溶液2μLを作用極上に滴下し、乾燥させることにより2つの酵素を物理吸着させた。次いで、この作用極が貼られた樹脂部(A)を、対極および感知極が貼られた樹脂部(B)に重ねて、35℃で加温し接合させることによって一体型の金電極酵素センサチップを作製した。
【0043】
種々の濃度のPhe、200μMのアスコルビン酸(AA)、5%のウシ血清アルブミン(BSA)を含むPBS溶液を擬似血漿サンプルとして4μL用意し、それを2.5mMのNAD+、0.17MのKCl、およびメディエーターである1-methoxy-PMSを0.83mM溶かしたGlycine-KOH緩衝液(pH9.5)6μLと混合して測定溶液とした(合計10μL)。この測定溶液をセンサチップの先端から毛管現象により吸い上げ、感知極が通電により測定溶液の充填を感知したところで自動的に対極に対して作用極に0.4Vの電圧を10秒間あるいは60秒間印加し、メディエーターをすべて酸化状態にすると同時に、妨害物質であるAAを前電解することによって測定溶液中から除去した(前電解)。
【0044】
次いで0Vで30秒間保ち(室温)、この間に酵素反応を進行させた。その後、再度0.4Vに電位ステップして60秒間電流を計測した。電流値は0.1秒ごとに計測した。この第2の0.4Vへの電位ステップ時の計測された酸化電流について、ステップ後0.5秒後及び5秒後の電流を読み取って、あるいは0〜5秒間の電流値を積分して、あるいは5〜10秒の間の電流値を積分して、あるいは5〜35秒の間の電流値を積分した。さらに、擬似血漿中のPhe濃度に対する検量線を作成した。
【0045】
(2)結果
1)前電解時間の検討
前電解時間を10秒間とした場合及び前電解時間を60秒間とした場合について、AAを含有する場合としない場合について、電流値を図2-1に示す。ステップ後、0.1 秒後の電流値、0.2秒後の電流値、0.5秒後の電流値、20秒後の電流値をそれぞれ示す。また、各条件において、Pheを含まず、AAを含む測定溶液を用い、前電解時間を10秒間とした場合または前電解時間を60秒間とした場合の結果も併せて図2-1に示す。
【0046】
図2-1に示す結果から、上記条件では、前電解時間が10秒間では、AAの除去には不十分であり、前電解時間が60秒間であれば、AAの除去に十分であることが分かる。さらに、前電解時間が60秒間とし、少なくともステップ後0.1秒後〜0.5秒後の電流値を用いることで、AAの妨害を回避しつつPhe濃度を測定することができる。
【0047】
2)電流値の積分についての検討
ステップ後0〜5秒間の電流値を積分して、あるいはステップ後5〜35秒の間の電流値を積分した結果を図3に示す。前電解時間を60秒間とし、ステップ後0〜5秒間の電流値を積分した場合、AAの妨害を回避しつつPhe濃度を測定することができることが分かる。前電解時間を60秒間とし、ステップ後5〜35秒の間の電流値を積分した場合でも、AAの妨害を回避しつつPhe濃度を測定することができることが分かる。
【0048】
60秒間の前電解が、Phe以外のアミノ酸を測る電気化学酵素センサのセンシング時のAAの除去にも有効であるかを検討するため、PheDHの代わりに、AlaDH(アラニン脱水素酵素)を用いて、ジアホラーゼと組み合わせて、Alaセンサを作製し、同様に前電解時間を10秒とした場合と60秒とした場合で、AAを含有する場合と含有しない場合について、Alaセンシングを行った際の、ステップ後10秒後における酸化電流値を図2-2に示す。
【0049】
図2-2の結果から、Alaセンシングにおいても、前電解時間が10秒間では、AA自体の酸化電流がAlaの測定電流の上に大きく上乗せされるのが観測され、AAの除去には不十分であることが示された。一方、前電解時間が60秒間の場合には、このAAの上乗せ電流がかなり減少し、AAの除去が進むことが示された。
【0050】
この結果は、PheおよびAlaだけでなく、同様な原理で種々のアミノ酸を測る電気化学酵素センサについて、AAの妨害除去に60秒間以上の前電解が有効であることを示した。
【0051】
3)検量線
種々の濃度のPhe(0〜1000μM)を含む擬似血漿サンプル(原液は200μMのアスコルビン酸と5%のウシ血清アルブミン(BSA)を含むPBS溶液)に対する酵素センサチップのCA応答を図4に示す。図からわかるように、擬似血漿中のPhe濃度の増加に伴い酸化電流の増加が観測された。0.4 Vへの第2ステップ後0.5秒後における酸化電流値を読み取って作成したPheに対する検量線と第2ステップ後5秒後における酸化電流値を読み取って作成した検量線もあわせて示す。その結果、AA存在下においても50〜1000μMでフェニルアラニンの計測が可能であることが示された。
【0052】
また、図5に、0.4 Vへの電位ステップ時の計測された酸化電流について、0〜5秒間の電流値を積分して、あるいは5〜10秒の間の電流値を積分して、あるいは5〜35秒の間の電流値を積分して擬似血漿中のPhe濃度に対する検量線を作成した結果を示す。
【0053】
実施例2
血液中のPheを実測した。実施例1における擬似血漿の代わりに血漿を用いて測定溶液を調製し、計測された電流値(ステップ後5秒で読み取り)あるいは電流積分値(ステップ後5〜10秒の間の電気量)の3回の測定結果の平均値から検量線によりPhe濃度を求めた。その結果、それぞれの検量線を用いた場合のサンプル血漿中のPhe濃度は、65μMおよび89μMと求められた。なお、同一血漿のPheをHPLC分析した結果は、69μMであった。
【0054】
実施例3
ヒト血漿サンプル(HS1と呼ぶ。コージンバイオより購入)を用いたPhe付加試験を行った。実験は、HPLC分析によりPhe濃度が既知のHS1溶液1mLに対してPhe 8.3 mgを加え混合し、HS1溶液を用いて100倍に希釈することでPheが500μM付加された血漿溶液を調製した(HS1+Pheと呼ぶ)。その後、NAD+、KCl、およびメディエーターである1-methoxy-PMSを溶かしたGlycine-KOH緩衝液 (pH 9.5)6μLに対して、限外ろ過フィルターを用いて遠心により除タンパク質処理をしたHS1+PheまたはHS1溶液を4μL取り、混合して測定溶液とした。この測定溶液を実施例1で用いたのと同様のセンサチップの先端から毛管現象により吸い上げ、感知極が通電により測定溶液の充填を感知したところで自動的に対極に対して作用極に0.4 Vの電圧を60秒間印加し、アスコルビン酸を完全に電解させると同時にメディエーターをすべて酸化状態にした。次いで0 Vで30秒間酵素反応を進行させた後、再度0.4 Vに電位ステップし5秒後の酸化電流を測定した。酸化電流の測定結果を図6に示す。
【0055】
図6からわかるように、Pheを500μM付加したHS1+Phe溶液において酸化触媒電流の増加が観測された。0.4 Vへの第2ステップ後5秒における酸化電流値を読み取ってPheに対する検量線を作成し、図7に示す。図7のPheに対する検量線を用いて、それぞれの血漿サンプルのPhe濃度を定量した。その結果、Pheを500μM付加したHS1+Phe溶液のPhe濃度は585μM(CV値 : 10%)、通常のHS1溶液のPhe濃度は101.4μM(CV値 : 22%)と求められた。HPLCにより求められているHS1溶液のPhe濃度は65.8μMであり、本センサの測定値はHPLCでの分析結果に比べるとやや高い値を示したが、血漿中のPhe濃度についても測定が可能であることが示された。
【産業上の利用可能性】
【0056】
本発明は、フェニルアラニンまたはアラニンの測定が必要とされる健康及び医療の分野に有用である。また食品及び飲料中のフェニルアラニンまたはアラニン測定にも有用と考えられる。
【特許請求の範囲】
【請求項1】
フェニルアラニン脱水素酵素またはアラニン脱水素酵素とジアホラーゼとを固定化した作用極及び対極を有する電極系を含むフェニルアラニンまたはアラニンセンサを用いて血液中のフェニルアラニンまたはアラニン濃度を測定する方法であって、
(1)被検体である血液を補酵素及び電子メディエーターを含有する試薬液と混合して測定液を調製し、
(2)前記測定液を前記電極系に供給し、
(3)前記電極系の作用極にプラス電位を印加して、前記測定液に含有されるアスコルビン酸及び電子メディエーターを酸化し、
(4)プラス電位の印加を終了し、酵素反応を所定時間実施し、
(5)所定時間経過後、前記電極系の作用極にプラス電位を印加して、電流値を測定し、
(6)測定した電流値またはその積分値に基づいて前記被検体である血液中のフェニルアラニンまたはアラニン濃度を算出する
ことを含む前記測定方法。
【請求項2】
工程(3)におけるプラス電位の印加は、前記測定液に含有されるアスコルビン酸の全量が酸化される条件で実施する、請求項1に記載の方法。
【請求項3】
工程(3)におけるプラス電位は、0.3〜0.5Vである、請求項1に記載の方法。
【請求項4】
酵素反応のための所定時間は、10〜60秒間である、請求項1〜3のいずれかに記載の方法。
【請求項5】
工程(5)におけるプラス電位は、電子メディエーターの還元体を酸化するに適した電位から選択する、請求項1に記載の方法。
【請求項6】
工程(6)におけるフェニルアラニンまたはアラニン濃度の算出は、プラス電位印加開始後0.1〜60秒の範囲のいずれかの時間に計測される電流値に基づき、かつ予め作成した検量線を基準にして行う、請求項1に記載の方法。
【請求項7】
工程(6)におけるフェニルアラニンまたはアラニン濃度の算出は、プラス電位印加開始後、60秒間に計測された全てまたは一部の電流値から求めた電気量に基づき、かつ予め作成した検量線を基準にして行う、請求項1に記載の方法。
【請求項8】
被検体である血液の量は0.2〜4μLの範囲であり、試薬液の量は0.3〜12μLの範囲である請求項1〜8のいずれかに記載の製造方法。
【請求項1】
フェニルアラニン脱水素酵素またはアラニン脱水素酵素とジアホラーゼとを固定化した作用極及び対極を有する電極系を含むフェニルアラニンまたはアラニンセンサを用いて血液中のフェニルアラニンまたはアラニン濃度を測定する方法であって、
(1)被検体である血液を補酵素及び電子メディエーターを含有する試薬液と混合して測定液を調製し、
(2)前記測定液を前記電極系に供給し、
(3)前記電極系の作用極にプラス電位を印加して、前記測定液に含有されるアスコルビン酸及び電子メディエーターを酸化し、
(4)プラス電位の印加を終了し、酵素反応を所定時間実施し、
(5)所定時間経過後、前記電極系の作用極にプラス電位を印加して、電流値を測定し、
(6)測定した電流値またはその積分値に基づいて前記被検体である血液中のフェニルアラニンまたはアラニン濃度を算出する
ことを含む前記測定方法。
【請求項2】
工程(3)におけるプラス電位の印加は、前記測定液に含有されるアスコルビン酸の全量が酸化される条件で実施する、請求項1に記載の方法。
【請求項3】
工程(3)におけるプラス電位は、0.3〜0.5Vである、請求項1に記載の方法。
【請求項4】
酵素反応のための所定時間は、10〜60秒間である、請求項1〜3のいずれかに記載の方法。
【請求項5】
工程(5)におけるプラス電位は、電子メディエーターの還元体を酸化するに適した電位から選択する、請求項1に記載の方法。
【請求項6】
工程(6)におけるフェニルアラニンまたはアラニン濃度の算出は、プラス電位印加開始後0.1〜60秒の範囲のいずれかの時間に計測される電流値に基づき、かつ予め作成した検量線を基準にして行う、請求項1に記載の方法。
【請求項7】
工程(6)におけるフェニルアラニンまたはアラニン濃度の算出は、プラス電位印加開始後、60秒間に計測された全てまたは一部の電流値から求めた電気量に基づき、かつ予め作成した検量線を基準にして行う、請求項1に記載の方法。
【請求項8】
被検体である血液の量は0.2〜4μLの範囲であり、試薬液の量は0.3〜12μLの範囲である請求項1〜8のいずれかに記載の製造方法。
【図1】

【図2−1】
【図2−2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2−1】
【図2−2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2012−78338(P2012−78338A)
【公開日】平成24年4月19日(2012.4.19)
【国際特許分類】
【出願番号】特願2011−66547(P2011−66547)
【出願日】平成23年3月24日(2011.3.24)
【出願人】(305060567)国立大学法人富山大学 (194)
【公開日】平成24年4月19日(2012.4.19)
【国際特許分類】
【出願日】平成23年3月24日(2011.3.24)
【出願人】(305060567)国立大学法人富山大学 (194)
[ Back to top ]