
フェニルアラニン誘導体を調製するためのプロセス
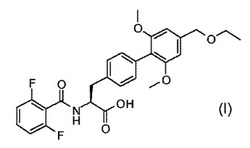
式(I)のフェニルアラニン誘導体を調製するための新規プロセス。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸を調製するための新規プロセス、及び該プロセスで用いられる中間生成物に関する。
【背景技術】
【0002】
2001年8月27日に出願された国際公開第02/18320号パンフレット(田辺製薬株式会社)には、α4(α4β7及びα4β1を含む)介在接着の阻害剤である新規フェニルアラニン誘導体が開示されている。特に、国際公開第02/18320号パンフレットは、本願に開示されている新規プロセスに関連する(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸(実施例12のN−(2,6−ジフルオロベンゾイル)−4−(2,6−ジメトキシ−4−エトキシメチルフェニル)−L−フェニルアラニン)を指す)について開示している。国際公開第02/18320号パンフレットは、更に、この対象化合物を調製するためのプロセスを開示している。
【0003】
2003年2月27日に出願された国際公開第03/072536号パンフレット(田辺製薬株式会社)は、前述の化合物(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸を含むフェニルアラニン誘導体を調製するための別のプロセスについて概説している。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】国際公開02/18320号
【特許文献2】国際公開03/072536号
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明の目的は、(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸(N−(2,6−ジフルオロベンゾイル)−4−(2,6−ジメトキシ−4−エトキシメチルフェニル)−L−フェニルアラニンとしても知られている)を調製するための別のプロセスを提供することである。
【課題を解決するための手段】
【0006】
本発明は、式(I)の化合物:
【化1】
【0007】
を調製するためのプロセスであって、
a)式(IIa)のエステル:
【化2】
【0008】
(式中、R1はC1−6アルキルである)
を加水分解する工程と、
(b)工程(a)から得られた生成物の溶媒和物を形成(溶媒和)する工程と、
(c)工程(b)から得られた前記溶媒和物を脱溶媒和して式(I)の化合物を得る工程と、
(d)任意で、工程(c)から得られた生成物を再結晶化させる工程とを含むプロセスを提供する。
【0009】
先行技術のプロセスに従って調製された式(I)の化合物と比較して、式(I)の化合物を調製するための本発明の別の単純化されたプロセスは、不純物プロファイルの改善された医薬製品を提供する。
【図面の簡単な説明】
【0010】
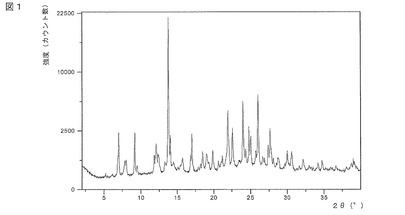
【図1】図1は、式(I)の化合物のアセトン溶媒和物の結晶質形態についてのXRPDデータを示す。
【図2a】図2aは、式(I)の化合物のアセトン溶媒和物の結晶質形態についてのFT−IRデータを示す(全スペクトル域4000〜675cm−1)。
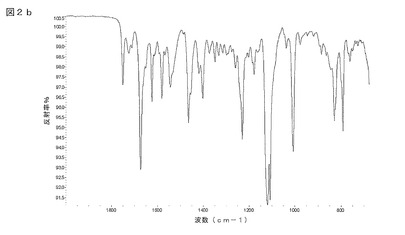
【図2b】図2bは、式(I)の化合物のアセトン溶媒和物の結晶質形態についてのFT−IRデータを示す(指紋領域2000〜675cm−1)。
【発明を実施するための形態】
【0011】
本発明は、式(I)の化合物:
【化3】
【0012】
を調製するためのプロセスであって、
a)式(IIa)のエステル:
【化4】
【0013】
(式中、R1はC1−6アルキルである)
を加水分解する工程と、
(b)工程(a)から得られた生成物の溶媒和物を形成(溶媒和)する工程と、
(c)工程(b)から得られた前記溶媒和物を脱溶媒和して式(I)の化合物を得る工程と、
(d)任意で、工程(c)から得られた生成物を再結晶化させる工程とを含むプロセスを提供する。
【0014】
先行技術のプロセスに従って調製された式(I)の化合物と比較して、式(I)の化合物を調製するための本発明の別の単純化されたプロセスは、不純物プロファイルの改善された医薬製品を提供する。
【0015】
本発明で使用する用語「アルキル」は、指定された数の炭素原子を含有している直鎖又は分岐鎖の炭化水素鎖を指す。例えば、C1−6アルキルは、少なくとも1個且つ多くても6個の炭素原子を含有している直鎖又は分岐鎖のアルキル鎖を意味する。本発明で使用する「C1−6アルキル」の例としては、メチル、エチル、n−プロピル及びn−ブチル、n−ペンチル及びn−ヘキシルが挙げられるが、これらに限定されない。
【0016】
本発明の1つの態様では、基R1はエチルである。
【0017】
工程(a)のエステルの加水分解は、酸性又は塩基性の条件下で行われ得る。
【0018】
本発明の1つの態様では、エステルを加水分解する工程は、塩基性条件下で行われる。好適な塩基としては、水酸化カリウム、水酸化ナトリウム及び水酸化リチウム等が挙げられるがこれらに限定されないアルカリ金属水酸化物が挙げられる。使用される塩基がアルカリ金属水酸化物である場合、エステルの加水分解は、カルボン酸塩中間体を介して進行する。このカルボン酸塩中間体は、溶媒から単離することができる。したがって、本発明の更なる態様では、アルカリ金属水酸化物を使用して塩基性条件下で工程(a)のエステルの加水分解を行って、溶媒から単離することができる適切なカルボン酸塩を得る。適切なカルボン酸塩は、一水和物又は二水和物等の水和物の形態で存在してもよい。本発明の更なる態様では、エステルの加水分解反応は、好適な塩基として水酸化カリウムを利用して行われる。
【0019】
本発明の更なる態様では、式(I)の化合物のカリウム塩:
【化5】
【0020】
が提供される。
【0021】
工程(a)のエステルの加水分解が塩基性条件下で行われる場合、反応混合物は、遊離酸を得るために酸性の後処理に供される。酸性の後処理で使用するための適切な酸としては、塩酸及び硫酸等であるがこれらに限定されない無機酸、並びにクエン酸等であるがこれらに限定されない、式(I)の化合物よりも低いpKa値を有する有機酸が挙げられる。
【0022】
工程(a)のエステルの加水分解を行うのに好適な酸としては、塩酸、硝酸、硫酸等であるがこれらに限定されない無機酸、及びトリフルオロ酢酸、p−トルエンスルホン酸等であるがこれらに限定されない有機酸を挙げることができる。
【0023】
工程(a)の酸性又は塩基性のエステルの加水分解は、好適な溶媒又は溶媒の混合物中で行うことができる。好適な溶媒としては、水及び有機溶媒が挙げられる。有機溶媒としては、エーテル(例えば、ジオキサン及びテトラヒドロフラン)、アセトニトリル及びケトン(例えば、アセトン及びメチルエチルケトン)が挙げられるが、これらに限定されない。
【0024】
工程(a)の酸性又は塩基性のエステルの加水分解は、室温以下で実施することができる。
【0025】
工程(a)の生成物の溶媒和物を形成(溶媒和)する工程(b)は、工程(a)の生成物の溶液に溶媒和物が由来する溶媒を添加することを介して達成することができ、次いで、前記生成物を結晶化及び濾過により単離する。任意で、溶媒和物の結晶種を導入することにより結晶化を開始させてもよい。
【0026】
本発明の1つの態様では、工程(a)の生成物は、プロトン性であっても非プロトン性であってもよい極性溶媒で溶媒和することができる。本発明の更なる態様では、溶媒和は、溶媒和物として極性非プロトン性溶媒を使用して達成することができる。本発明の更なる態様では、工程(a)の生成物は、アセトン、酢酸、アセトニトリル、ニトロメタン、ジメチルスルホキシド及びジメチルホルムアミドからなる群から選択される溶媒で溶媒和される。本発明の別の態様では、工程(a)の生成物は、アセトンで溶媒和される。
【0027】
本発明の更なる態様では、式(I)の化合物のアセトン溶媒和物:
【化6】
【0028】
が提供される。
【0029】
式(I)の化合物のアセトン溶媒和物は、結晶質形態で存在してもよい。X線粉末回折(XRPD)によって及び/又はFT赤外分光法によって結晶質形態を特性評価することができる。式(I)の化合物の結晶質アセトン溶媒和物についての特性評価データを図1及び図2a/2bに示す。
【0030】
本発明は、図1に示されているのと実質的に同じX線粉末回折(XRPD)パターンであって、角度2θで表され且つCuKα線を使用して回折計で得られるXRPDパターン及び/又は図2a及び2bに示されるのと実質的に同じ赤外線スペクトルを特徴とする式(I)の化合物のアセトン溶媒和物の結晶質形態を提供する。
【0031】
XRPDデータは、X’Celerator検出器を装備したPANalytical X’Pert Pro粉末回折計で得られた。取得条件は、以下の通りである:放射線:Cu Kα、発生器電圧:40kV、発生器電流:45mA、開始角度:2.0°2θ、終了角度:40.0°2θ、刻み幅:0.0167°2θ。1工程当たりの時間は、31.750秒であった。数ミリグラムのサンプルをシリコンウェーファー(ゼロバックグラウンド)プレートに載せることによりサンプルを調製し、粉末の薄層を得た。
【0032】
特徴的なピーク位置及び計算されたd−スペーシングを表1に要約する。これらは、Highscoreソフトウェアを使用して生データから計算した。ピーク位置の実験誤差は、約±0.1°2θである。相対的なピーク強度は、定向性により変化する。
【表1】
【0033】
式(I)の化合物の結晶質アセトン溶媒和物の特徴的なXRPDピーク位置は、以下のとおりである:約7.0、9.2、13.7、14.0及び24.0°2θにおけるピーク。
【0034】
脱溶媒和工程(工程(c))は、工程(b)の溶媒和物を加熱することによって行い、式(I)の化合物を得ることができる。あるいは、工程(c)は、溶媒和物を除去することができる溶媒で溶媒和物を洗浄することにより達成することができる。したがって、本発明の1つの態様では、工程(c)において、脱溶媒和は、工程(b)の溶媒和物を乾燥又は洗浄することによって実施される。
【0035】
本発明の更なる態様では、脱溶媒和工程(c)は、室温と溶媒和物の沸点との間の温度で真空下にて工程(b)の溶媒和物を乾燥させることにより行われる。
【0036】
任意で、工程(c)から得られた式(I)の化合物を再結晶化(工程(d))によって更に精製してもよい。再結晶化は、冷却による再結晶化又は逆溶剤の添加による再結晶化等の様々な標準的な技術を使用して達成することができる。冷却による再結晶化では、高温で式(I)の結晶質化合物を好適な溶媒に溶解させ、次いで、溶液をゆっくり冷却し、任意で種を導入して、式(I)の化合物の結晶が得られ、これは、濾過により単離し、好適な溶媒で洗浄し、次いで乾燥させてもよい。逆溶剤の添加による再結晶では、式(I)の結晶質化合物を好適な溶媒に溶解させる。逆溶剤の添加は、溶液に対する化合物の溶解度を低下させて結晶の形成を促進する。任意で、溶剤系に種を導入してもよい。このように形成された式(I)の化合物の結晶は、濾過により単離し、好適な溶媒を用いて洗浄し、次いで乾燥させてもよい。
【0037】
本発明の更なる態様では、工程(c)から得られた式(I)の結晶質化合物を高温(例えば、約50℃)で酢酸エチルに溶解させてもよい。得られた溶液をヘプタンで処理し、冷却し、式(I)の化合物の結晶種を導入してもよい。次いで、得られた式(I)の化合物の結晶を濾過により単離し、好適な溶媒で洗浄し、乾燥させてもよい。
【0038】
当業者は、本明細書に記載される化学プロセスにおける次の段階に進む前に1つ以上の中間体生成物が単離されないように、前記プロセスにおける特定の工程をはめ込むことができることを理解するであろう。
【0039】
本発明の更なる態様では、式(I)の化合物:
【化7】
【0040】
を調製するためのプロセスであって、
a)水酸化カリウムを使用して式(II)のエステル:
【化8】
【0041】
を加水分解し、次いで、クエン酸を使用して酸性の後処理を行う工程と、
(b)工程(a)から得られた生成物のアセトン溶媒和物を形成する工程と、
(c)高温で真空下にて前記溶媒和物を乾燥させることを介して、工程(b)から得られたアセトン溶媒和物を脱溶媒和して、式(I)の化合物を得る工程とを含むプロセスが提供される。
【0042】
本発明の更なる態様では、式(I)の化合物を調製するためのプロセスは、酢酸エチル/ヘプタンから式(I)の化合物を再結晶化させる更なる工程を含む。
【0043】
式(II)の化合物は、国際公開第03/072537号パンフレット(田辺製薬株式会社)の工程1及び2に記載されている方法論に従って調製することができる。あるいは、式(II)の化合物は、国際公開第02/18320号パンフレット(田辺製薬株式会社)に記載されている通り調製することもできる。
【0044】
また、下記の反応スキーム(スキーム1):
【化9】
【0045】
(式中、R1は、C1−6アルキルである)
に従って式(IIa)の化合物を調製してもよい。
【0046】
好適な溶媒(MIBK等であるがこれに限定されない)又は溶媒の混合物(水及びMeTHF等であるがこれらに限定されない)中にて好適な塩基(炭酸カリウム等であるがこれに限定されない)の存在下で、式(IIIa)の化合物を式(IV)の化合物と反応させることにより、上記工程(i)の下で式(Va)の化合物を便利に調製することができる。
【0047】
スズキカップリング反応条件下で式(Va)の化合物を式(VI)の化合物とカップリングさせることにより、上記工程(ii)の下で式(IIa)の化合物を便利に調製することができる。スズキカップリング反応で使用するための好適な触媒の例としては、酢酸パラジウム、塩化パラジウム及びジクロロビス(トリフェニルホスフィン)パラジウム等であるがこれらに限定されないパラジウム触媒が挙げられる。酢酸パラジウム又は塩化パラジウム等であるがこれらに限定されない、リガンドを有しないパラジウム(II)触媒の存在下で反応を実施する場合、反応を促進するために、ホスフィン(トリフェニルホスフィン、トリ−オルト−トリルホスフィン、トリ−tert−ブチルホスフィン又はジ−フェニルシクロ−ヘキシルホスフィン等であるがこれらに限定されない)又は亜リン酸エステル(亜リン酸トリエチル等であるがこれらに限定されない)を添加することが必要である。スズキカップリング反応において使用することができる好適な塩基の例としては、アルカリ金属炭酸塩等であるがこれに限定されない無機塩基、及びアルキルアミン(ジイソプロピルアミン、トリエチルアミン及びジイソプロピルエチルアミン)等であるがこれらに限定されない有機塩基が挙げられる。工程(ii)下におけるスズキカップリング反応は、好適な溶媒又は溶媒の混合物(水及びMeTHF等であるがこれらに限定されない)中で実施すべきである。
【0048】
下記の反応スキーム(スキーム2):
【化10】
【0049】
(式中、R1はエチルである)
に従って式(II)の化合物を調製してもよい。
【0050】
本発明の更なる態様では、式(V)の化合物:
【化11】
【0051】
を式(VI)の化合物:
【化12】
【0052】
とカップリングさせることを含む式(II)の化合物を調製するためのプロセスを提供する。
【0053】
式(V)の化合物及び式(VI)の化合物に好適なカップリング条件としては、スキーム2に示す条件が挙げられる。
【0054】
本発明の更なる態様では、式(V)の化合物:
【化13】
【0055】
が提供される。
【0056】
単離及び精製されたバッチにおいて、式(V)の化合物についての1H NMR特性評価データを作成した。内部標準としてTMSを使用して、400MHzでBruker Avance400に1H−NMRスペクトルを記録した。
【0057】
1H NMR(400MHz,DMSO−D6)δ ppm 1.17(t,J=7.09Hz,3H)2.96(dd,J=13.82,9.90Hz,1H)3.11(dd,J=13.82,5.26Hz,1H)4.12(q,J=7.09Hz,2H)4.63(ddd,J=9.78,7.82,5.38Hz,1H)7.15(t,J=7.95Hz,2H)7.25(d,J=8.31Hz,2H)7.47−7.55(m,3H)9.23(d,J=7.83Hz,1H)。
【実施例】
【0058】
実験
分析機器
内部標準としてTMSを使用して、400MHzでBruker Avance400に1H−NMRスペクトルを記録した。
【0059】
Perkin Elmer Universal ATR(減衰全反射)サンプリングアクセサリを備えたPerkin Elmer Spectrum One FT−IR分光計を用いて、波数範囲4000〜650cm−1にわたって赤外吸収スペクトルを記録した。
【0060】
具体例
以下の非限定的な例において本発明を例証する。
【0061】
エチル(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパノエートの調製
炭酸カリウム(18.8Kg、136.04mol)の水(70L)溶液に、エチル−4−ブロモ−L−フェニルアラニネート塩酸塩(14Kg、45.37mol、DowPharmによって供給)及びMe−THF(70L)を添加する。二相混合物を10±3℃に冷却し、温度を15℃未満に維持しながら2,6−ジフルオロベンゾイル塩化物(8.4Kg、47.58mol、Shanghai Chemspecによって供給)を添加する。次いで、25±3℃まで加温しながら30分間反応物を撹拌する。次いで、相を分離する。エチル4−ブロモ−N−[(2,6−ジフルオロフェニル)カルボニル]−L−フェニルアラニネートを含有している有機相に、4−[(エチルオキシ)メチル]−2,6−ビス(メチルオキシ)フェニル]ボロン酸(11.4Kg、47.5mol、Juzenによって供給)を添加する。次いで、Me−THF(28L)及び水(18.2L)で有機相を希釈する。塩化パラジウム(23.8g、0.13mol)及びトリフェニルホスフィン(71.4g、0.27mol)を添加し、窒素で容器を3回パージして、微量の空気を全て除去する。ジイソプロピルアミン(9.5L、67.93mol)を添加し、パージを繰り返す。次いで、反応混合物を約3時間75±3℃(還流)に加熱する。HPLCが完了したら、溶液を60±3℃に冷却し、L−システイン(2.8Kg)を添加する。反応混合物を2時間60±3℃で加熱する。この時間後、反応混合物を25±3℃に冷却する。2Mの塩酸(28L)を添加する。10分間撹拌した後、層を分離させる。次いで、飽和重炭酸ナトリウム水溶液(28L)で有機相を洗浄する。再度層を分離させ、有機層をDomnic Hunterフィルターカートリッジに通し、Me−THF(7L)で洗浄する。次いで、常圧蒸留を介して有機相を28Lまで濃縮する。イソプロピルアルコール(84L)を添加し、溶液を28Lまで濃縮する。再度イソプロピルアルコール(84L)を添加し、溶液を84Lまで濃縮する。Me−THFレベルが確実に<0.2当量になるようにサンプルを採取する。内容物を55℃超に維持しながらヘプタン(95%)(84L)を添加し、溶液を45±3℃に冷却した後、エチル(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパノエートの種(70g)を添加し、スラリーを約30分間エージングする。薄いスラリーを38℃に冷却し、1時間保持する。次いで、前記溶液を45℃に再加熱し、45分間保持する。得られたスラリーを2時間にわたって10℃に冷却し、1時間保持する。次いで、濾過によって固体を回収し、イソプロピルアルコール:ヘプタン(95%)(1:4、2×28L)で洗浄する。次いで、生成物を50℃にて真空内で乾燥させて、生成物(20.35Kg、85%)を得る。
【0062】
1H NMR(400MHz,DMSO−D6)δ ppm 1.17(dt,J=16.08,7.00Hz,6H)3.08(ddd,J=19.81,14.06,5.50Hz,2H)3.53(q,J=7.01Hz,2H)3.65(s,6H)4.04−4.16(m,2H)4.47(s,2H)4.60−4.68(m,1H)6.69(s,2H)7.09−7.18(m,4H)7.24(d,J=8.07Hz,2H)7.51(ddd,J=14.92,8.31,6.60Hz,1H)9.31(d,J=7.58Hz,1H)。
【0063】
(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸の調製
エチル(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパノエート(15Kg)をテトラヒドロフラン(37.5L)に取り、炭を含有するCUNOフィルタ(R55SP)に通した。テトラヒドロフラン(37.5L)及び水(45L)を添加し、得られた混合物を10±3℃に冷却した。KOH水溶液(4.65Kg、45%w/w)を添加し、反応が完了するまで混合物を10±3℃で攪拌した。クエン酸水溶液(18.15Kg、50%w/v)を充填し、次いで、トルエン(75L)を充填した。反応混合物を50±3℃に加熱し、水相を排出して廃棄した。有機相を50±3℃にて水(2×30L)で洗浄した。次いで、常圧蒸留によって有機相を75Lまで濃縮する。トルエン(45L)及びアセトン(75L)を充填し、溶液を120Lまで濃縮した。再度アセトン(75L)を充填し、再度溶液を105Lまで濃縮した。温度を55±3℃に維持しながらトルエン(75L)を充填した。溶液を35℃に冷却し、(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸(アセトン溶媒和物)(75g)の種を導入し、4時間にわたって0±3℃に冷却し、1時間この温度で保持した。固体生成物を濾過により単離し、冷(<5℃)トルエン/アセトン(45L、10:1)、冷(<5℃)トルエン(45L)で洗浄し、70℃で真空内乾燥させて、生成物(10.1Kg、71%)を得た。
【0064】
(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸の再結晶化
(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸(9.38Kg)を清浄な反応器に充填し、次いで、酢酸エチル(46.9L)を充填した。溶液を50℃に加熱し、濾過して、予め加温しておいた(35℃)結晶化容器に入れた。酢酸エチル(9.4L)によるライン洗浄を行った。合わせた酢酸エチル溶液を50℃に加熱し、攪拌して、確実に完全に溶解させた。温度を50℃に維持しながら濾過したヘプタン(9.4L)を添加し、次いで、溶液を30℃に冷却し、1:9の酢酸エチル:ヘプタン(0.47L)でスラリー化された(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸(47g)の種を導入した。スラリーを30℃で2時間エージングした。濾過したヘプタン(75L)を3時間にわたって添加した。次いで、スラリーを1時間にわたって0℃に冷却した。混合物を1時間0℃でエージングし、次いで、固体を濾取し、イソプロピルエーテル(29.6L)で洗浄し、50±3℃で真空下乾燥させて、生成物(8.55Kg、91%)を得た。
【0065】
約754、768、800、820、849、866、1006、1100、1122、1157、1188、1225、1242、1268、1292、1317、1352、1417、1466、1530、1580、1624、1650、1662、1711、1728、2938、3302cm−1に顕著な吸収帯を備えた赤外吸収スペクトルを有することを特徴としていた。
【技術分野】
【0001】
本発明は、(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸を調製するための新規プロセス、及び該プロセスで用いられる中間生成物に関する。
【背景技術】
【0002】
2001年8月27日に出願された国際公開第02/18320号パンフレット(田辺製薬株式会社)には、α4(α4β7及びα4β1を含む)介在接着の阻害剤である新規フェニルアラニン誘導体が開示されている。特に、国際公開第02/18320号パンフレットは、本願に開示されている新規プロセスに関連する(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸(実施例12のN−(2,6−ジフルオロベンゾイル)−4−(2,6−ジメトキシ−4−エトキシメチルフェニル)−L−フェニルアラニン)を指す)について開示している。国際公開第02/18320号パンフレットは、更に、この対象化合物を調製するためのプロセスを開示している。
【0003】
2003年2月27日に出願された国際公開第03/072536号パンフレット(田辺製薬株式会社)は、前述の化合物(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸を含むフェニルアラニン誘導体を調製するための別のプロセスについて概説している。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】国際公開02/18320号
【特許文献2】国際公開03/072536号
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明の目的は、(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸(N−(2,6−ジフルオロベンゾイル)−4−(2,6−ジメトキシ−4−エトキシメチルフェニル)−L−フェニルアラニンとしても知られている)を調製するための別のプロセスを提供することである。
【課題を解決するための手段】
【0006】
本発明は、式(I)の化合物:
【化1】
【0007】
を調製するためのプロセスであって、
a)式(IIa)のエステル:
【化2】
【0008】
(式中、R1はC1−6アルキルである)
を加水分解する工程と、
(b)工程(a)から得られた生成物の溶媒和物を形成(溶媒和)する工程と、
(c)工程(b)から得られた前記溶媒和物を脱溶媒和して式(I)の化合物を得る工程と、
(d)任意で、工程(c)から得られた生成物を再結晶化させる工程とを含むプロセスを提供する。
【0009】
先行技術のプロセスに従って調製された式(I)の化合物と比較して、式(I)の化合物を調製するための本発明の別の単純化されたプロセスは、不純物プロファイルの改善された医薬製品を提供する。
【図面の簡単な説明】
【0010】
【図1】図1は、式(I)の化合物のアセトン溶媒和物の結晶質形態についてのXRPDデータを示す。
【図2a】図2aは、式(I)の化合物のアセトン溶媒和物の結晶質形態についてのFT−IRデータを示す(全スペクトル域4000〜675cm−1)。
【図2b】図2bは、式(I)の化合物のアセトン溶媒和物の結晶質形態についてのFT−IRデータを示す(指紋領域2000〜675cm−1)。
【発明を実施するための形態】
【0011】
本発明は、式(I)の化合物:
【化3】
【0012】
を調製するためのプロセスであって、
a)式(IIa)のエステル:
【化4】
【0013】
(式中、R1はC1−6アルキルである)
を加水分解する工程と、
(b)工程(a)から得られた生成物の溶媒和物を形成(溶媒和)する工程と、
(c)工程(b)から得られた前記溶媒和物を脱溶媒和して式(I)の化合物を得る工程と、
(d)任意で、工程(c)から得られた生成物を再結晶化させる工程とを含むプロセスを提供する。
【0014】
先行技術のプロセスに従って調製された式(I)の化合物と比較して、式(I)の化合物を調製するための本発明の別の単純化されたプロセスは、不純物プロファイルの改善された医薬製品を提供する。
【0015】
本発明で使用する用語「アルキル」は、指定された数の炭素原子を含有している直鎖又は分岐鎖の炭化水素鎖を指す。例えば、C1−6アルキルは、少なくとも1個且つ多くても6個の炭素原子を含有している直鎖又は分岐鎖のアルキル鎖を意味する。本発明で使用する「C1−6アルキル」の例としては、メチル、エチル、n−プロピル及びn−ブチル、n−ペンチル及びn−ヘキシルが挙げられるが、これらに限定されない。
【0016】
本発明の1つの態様では、基R1はエチルである。
【0017】
工程(a)のエステルの加水分解は、酸性又は塩基性の条件下で行われ得る。
【0018】
本発明の1つの態様では、エステルを加水分解する工程は、塩基性条件下で行われる。好適な塩基としては、水酸化カリウム、水酸化ナトリウム及び水酸化リチウム等が挙げられるがこれらに限定されないアルカリ金属水酸化物が挙げられる。使用される塩基がアルカリ金属水酸化物である場合、エステルの加水分解は、カルボン酸塩中間体を介して進行する。このカルボン酸塩中間体は、溶媒から単離することができる。したがって、本発明の更なる態様では、アルカリ金属水酸化物を使用して塩基性条件下で工程(a)のエステルの加水分解を行って、溶媒から単離することができる適切なカルボン酸塩を得る。適切なカルボン酸塩は、一水和物又は二水和物等の水和物の形態で存在してもよい。本発明の更なる態様では、エステルの加水分解反応は、好適な塩基として水酸化カリウムを利用して行われる。
【0019】
本発明の更なる態様では、式(I)の化合物のカリウム塩:
【化5】
【0020】
が提供される。
【0021】
工程(a)のエステルの加水分解が塩基性条件下で行われる場合、反応混合物は、遊離酸を得るために酸性の後処理に供される。酸性の後処理で使用するための適切な酸としては、塩酸及び硫酸等であるがこれらに限定されない無機酸、並びにクエン酸等であるがこれらに限定されない、式(I)の化合物よりも低いpKa値を有する有機酸が挙げられる。
【0022】
工程(a)のエステルの加水分解を行うのに好適な酸としては、塩酸、硝酸、硫酸等であるがこれらに限定されない無機酸、及びトリフルオロ酢酸、p−トルエンスルホン酸等であるがこれらに限定されない有機酸を挙げることができる。
【0023】
工程(a)の酸性又は塩基性のエステルの加水分解は、好適な溶媒又は溶媒の混合物中で行うことができる。好適な溶媒としては、水及び有機溶媒が挙げられる。有機溶媒としては、エーテル(例えば、ジオキサン及びテトラヒドロフラン)、アセトニトリル及びケトン(例えば、アセトン及びメチルエチルケトン)が挙げられるが、これらに限定されない。
【0024】
工程(a)の酸性又は塩基性のエステルの加水分解は、室温以下で実施することができる。
【0025】
工程(a)の生成物の溶媒和物を形成(溶媒和)する工程(b)は、工程(a)の生成物の溶液に溶媒和物が由来する溶媒を添加することを介して達成することができ、次いで、前記生成物を結晶化及び濾過により単離する。任意で、溶媒和物の結晶種を導入することにより結晶化を開始させてもよい。
【0026】
本発明の1つの態様では、工程(a)の生成物は、プロトン性であっても非プロトン性であってもよい極性溶媒で溶媒和することができる。本発明の更なる態様では、溶媒和は、溶媒和物として極性非プロトン性溶媒を使用して達成することができる。本発明の更なる態様では、工程(a)の生成物は、アセトン、酢酸、アセトニトリル、ニトロメタン、ジメチルスルホキシド及びジメチルホルムアミドからなる群から選択される溶媒で溶媒和される。本発明の別の態様では、工程(a)の生成物は、アセトンで溶媒和される。
【0027】
本発明の更なる態様では、式(I)の化合物のアセトン溶媒和物:
【化6】
【0028】
が提供される。
【0029】
式(I)の化合物のアセトン溶媒和物は、結晶質形態で存在してもよい。X線粉末回折(XRPD)によって及び/又はFT赤外分光法によって結晶質形態を特性評価することができる。式(I)の化合物の結晶質アセトン溶媒和物についての特性評価データを図1及び図2a/2bに示す。
【0030】
本発明は、図1に示されているのと実質的に同じX線粉末回折(XRPD)パターンであって、角度2θで表され且つCuKα線を使用して回折計で得られるXRPDパターン及び/又は図2a及び2bに示されるのと実質的に同じ赤外線スペクトルを特徴とする式(I)の化合物のアセトン溶媒和物の結晶質形態を提供する。
【0031】
XRPDデータは、X’Celerator検出器を装備したPANalytical X’Pert Pro粉末回折計で得られた。取得条件は、以下の通りである:放射線:Cu Kα、発生器電圧:40kV、発生器電流:45mA、開始角度:2.0°2θ、終了角度:40.0°2θ、刻み幅:0.0167°2θ。1工程当たりの時間は、31.750秒であった。数ミリグラムのサンプルをシリコンウェーファー(ゼロバックグラウンド)プレートに載せることによりサンプルを調製し、粉末の薄層を得た。
【0032】
特徴的なピーク位置及び計算されたd−スペーシングを表1に要約する。これらは、Highscoreソフトウェアを使用して生データから計算した。ピーク位置の実験誤差は、約±0.1°2θである。相対的なピーク強度は、定向性により変化する。
【表1】
【0033】
式(I)の化合物の結晶質アセトン溶媒和物の特徴的なXRPDピーク位置は、以下のとおりである:約7.0、9.2、13.7、14.0及び24.0°2θにおけるピーク。
【0034】
脱溶媒和工程(工程(c))は、工程(b)の溶媒和物を加熱することによって行い、式(I)の化合物を得ることができる。あるいは、工程(c)は、溶媒和物を除去することができる溶媒で溶媒和物を洗浄することにより達成することができる。したがって、本発明の1つの態様では、工程(c)において、脱溶媒和は、工程(b)の溶媒和物を乾燥又は洗浄することによって実施される。
【0035】
本発明の更なる態様では、脱溶媒和工程(c)は、室温と溶媒和物の沸点との間の温度で真空下にて工程(b)の溶媒和物を乾燥させることにより行われる。
【0036】
任意で、工程(c)から得られた式(I)の化合物を再結晶化(工程(d))によって更に精製してもよい。再結晶化は、冷却による再結晶化又は逆溶剤の添加による再結晶化等の様々な標準的な技術を使用して達成することができる。冷却による再結晶化では、高温で式(I)の結晶質化合物を好適な溶媒に溶解させ、次いで、溶液をゆっくり冷却し、任意で種を導入して、式(I)の化合物の結晶が得られ、これは、濾過により単離し、好適な溶媒で洗浄し、次いで乾燥させてもよい。逆溶剤の添加による再結晶では、式(I)の結晶質化合物を好適な溶媒に溶解させる。逆溶剤の添加は、溶液に対する化合物の溶解度を低下させて結晶の形成を促進する。任意で、溶剤系に種を導入してもよい。このように形成された式(I)の化合物の結晶は、濾過により単離し、好適な溶媒を用いて洗浄し、次いで乾燥させてもよい。
【0037】
本発明の更なる態様では、工程(c)から得られた式(I)の結晶質化合物を高温(例えば、約50℃)で酢酸エチルに溶解させてもよい。得られた溶液をヘプタンで処理し、冷却し、式(I)の化合物の結晶種を導入してもよい。次いで、得られた式(I)の化合物の結晶を濾過により単離し、好適な溶媒で洗浄し、乾燥させてもよい。
【0038】
当業者は、本明細書に記載される化学プロセスにおける次の段階に進む前に1つ以上の中間体生成物が単離されないように、前記プロセスにおける特定の工程をはめ込むことができることを理解するであろう。
【0039】
本発明の更なる態様では、式(I)の化合物:
【化7】
【0040】
を調製するためのプロセスであって、
a)水酸化カリウムを使用して式(II)のエステル:
【化8】
【0041】
を加水分解し、次いで、クエン酸を使用して酸性の後処理を行う工程と、
(b)工程(a)から得られた生成物のアセトン溶媒和物を形成する工程と、
(c)高温で真空下にて前記溶媒和物を乾燥させることを介して、工程(b)から得られたアセトン溶媒和物を脱溶媒和して、式(I)の化合物を得る工程とを含むプロセスが提供される。
【0042】
本発明の更なる態様では、式(I)の化合物を調製するためのプロセスは、酢酸エチル/ヘプタンから式(I)の化合物を再結晶化させる更なる工程を含む。
【0043】
式(II)の化合物は、国際公開第03/072537号パンフレット(田辺製薬株式会社)の工程1及び2に記載されている方法論に従って調製することができる。あるいは、式(II)の化合物は、国際公開第02/18320号パンフレット(田辺製薬株式会社)に記載されている通り調製することもできる。
【0044】
また、下記の反応スキーム(スキーム1):
【化9】
【0045】
(式中、R1は、C1−6アルキルである)
に従って式(IIa)の化合物を調製してもよい。
【0046】
好適な溶媒(MIBK等であるがこれに限定されない)又は溶媒の混合物(水及びMeTHF等であるがこれらに限定されない)中にて好適な塩基(炭酸カリウム等であるがこれに限定されない)の存在下で、式(IIIa)の化合物を式(IV)の化合物と反応させることにより、上記工程(i)の下で式(Va)の化合物を便利に調製することができる。
【0047】
スズキカップリング反応条件下で式(Va)の化合物を式(VI)の化合物とカップリングさせることにより、上記工程(ii)の下で式(IIa)の化合物を便利に調製することができる。スズキカップリング反応で使用するための好適な触媒の例としては、酢酸パラジウム、塩化パラジウム及びジクロロビス(トリフェニルホスフィン)パラジウム等であるがこれらに限定されないパラジウム触媒が挙げられる。酢酸パラジウム又は塩化パラジウム等であるがこれらに限定されない、リガンドを有しないパラジウム(II)触媒の存在下で反応を実施する場合、反応を促進するために、ホスフィン(トリフェニルホスフィン、トリ−オルト−トリルホスフィン、トリ−tert−ブチルホスフィン又はジ−フェニルシクロ−ヘキシルホスフィン等であるがこれらに限定されない)又は亜リン酸エステル(亜リン酸トリエチル等であるがこれらに限定されない)を添加することが必要である。スズキカップリング反応において使用することができる好適な塩基の例としては、アルカリ金属炭酸塩等であるがこれに限定されない無機塩基、及びアルキルアミン(ジイソプロピルアミン、トリエチルアミン及びジイソプロピルエチルアミン)等であるがこれらに限定されない有機塩基が挙げられる。工程(ii)下におけるスズキカップリング反応は、好適な溶媒又は溶媒の混合物(水及びMeTHF等であるがこれらに限定されない)中で実施すべきである。
【0048】
下記の反応スキーム(スキーム2):
【化10】
【0049】
(式中、R1はエチルである)
に従って式(II)の化合物を調製してもよい。
【0050】
本発明の更なる態様では、式(V)の化合物:
【化11】
【0051】
を式(VI)の化合物:
【化12】
【0052】
とカップリングさせることを含む式(II)の化合物を調製するためのプロセスを提供する。
【0053】
式(V)の化合物及び式(VI)の化合物に好適なカップリング条件としては、スキーム2に示す条件が挙げられる。
【0054】
本発明の更なる態様では、式(V)の化合物:
【化13】
【0055】
が提供される。
【0056】
単離及び精製されたバッチにおいて、式(V)の化合物についての1H NMR特性評価データを作成した。内部標準としてTMSを使用して、400MHzでBruker Avance400に1H−NMRスペクトルを記録した。
【0057】
1H NMR(400MHz,DMSO−D6)δ ppm 1.17(t,J=7.09Hz,3H)2.96(dd,J=13.82,9.90Hz,1H)3.11(dd,J=13.82,5.26Hz,1H)4.12(q,J=7.09Hz,2H)4.63(ddd,J=9.78,7.82,5.38Hz,1H)7.15(t,J=7.95Hz,2H)7.25(d,J=8.31Hz,2H)7.47−7.55(m,3H)9.23(d,J=7.83Hz,1H)。
【実施例】
【0058】
実験
分析機器
内部標準としてTMSを使用して、400MHzでBruker Avance400に1H−NMRスペクトルを記録した。
【0059】
Perkin Elmer Universal ATR(減衰全反射)サンプリングアクセサリを備えたPerkin Elmer Spectrum One FT−IR分光計を用いて、波数範囲4000〜650cm−1にわたって赤外吸収スペクトルを記録した。
【0060】
具体例
以下の非限定的な例において本発明を例証する。
【0061】
エチル(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパノエートの調製
炭酸カリウム(18.8Kg、136.04mol)の水(70L)溶液に、エチル−4−ブロモ−L−フェニルアラニネート塩酸塩(14Kg、45.37mol、DowPharmによって供給)及びMe−THF(70L)を添加する。二相混合物を10±3℃に冷却し、温度を15℃未満に維持しながら2,6−ジフルオロベンゾイル塩化物(8.4Kg、47.58mol、Shanghai Chemspecによって供給)を添加する。次いで、25±3℃まで加温しながら30分間反応物を撹拌する。次いで、相を分離する。エチル4−ブロモ−N−[(2,6−ジフルオロフェニル)カルボニル]−L−フェニルアラニネートを含有している有機相に、4−[(エチルオキシ)メチル]−2,6−ビス(メチルオキシ)フェニル]ボロン酸(11.4Kg、47.5mol、Juzenによって供給)を添加する。次いで、Me−THF(28L)及び水(18.2L)で有機相を希釈する。塩化パラジウム(23.8g、0.13mol)及びトリフェニルホスフィン(71.4g、0.27mol)を添加し、窒素で容器を3回パージして、微量の空気を全て除去する。ジイソプロピルアミン(9.5L、67.93mol)を添加し、パージを繰り返す。次いで、反応混合物を約3時間75±3℃(還流)に加熱する。HPLCが完了したら、溶液を60±3℃に冷却し、L−システイン(2.8Kg)を添加する。反応混合物を2時間60±3℃で加熱する。この時間後、反応混合物を25±3℃に冷却する。2Mの塩酸(28L)を添加する。10分間撹拌した後、層を分離させる。次いで、飽和重炭酸ナトリウム水溶液(28L)で有機相を洗浄する。再度層を分離させ、有機層をDomnic Hunterフィルターカートリッジに通し、Me−THF(7L)で洗浄する。次いで、常圧蒸留を介して有機相を28Lまで濃縮する。イソプロピルアルコール(84L)を添加し、溶液を28Lまで濃縮する。再度イソプロピルアルコール(84L)を添加し、溶液を84Lまで濃縮する。Me−THFレベルが確実に<0.2当量になるようにサンプルを採取する。内容物を55℃超に維持しながらヘプタン(95%)(84L)を添加し、溶液を45±3℃に冷却した後、エチル(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパノエートの種(70g)を添加し、スラリーを約30分間エージングする。薄いスラリーを38℃に冷却し、1時間保持する。次いで、前記溶液を45℃に再加熱し、45分間保持する。得られたスラリーを2時間にわたって10℃に冷却し、1時間保持する。次いで、濾過によって固体を回収し、イソプロピルアルコール:ヘプタン(95%)(1:4、2×28L)で洗浄する。次いで、生成物を50℃にて真空内で乾燥させて、生成物(20.35Kg、85%)を得る。
【0062】
1H NMR(400MHz,DMSO−D6)δ ppm 1.17(dt,J=16.08,7.00Hz,6H)3.08(ddd,J=19.81,14.06,5.50Hz,2H)3.53(q,J=7.01Hz,2H)3.65(s,6H)4.04−4.16(m,2H)4.47(s,2H)4.60−4.68(m,1H)6.69(s,2H)7.09−7.18(m,4H)7.24(d,J=8.07Hz,2H)7.51(ddd,J=14.92,8.31,6.60Hz,1H)9.31(d,J=7.58Hz,1H)。
【0063】
(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸の調製
エチル(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパノエート(15Kg)をテトラヒドロフラン(37.5L)に取り、炭を含有するCUNOフィルタ(R55SP)に通した。テトラヒドロフラン(37.5L)及び水(45L)を添加し、得られた混合物を10±3℃に冷却した。KOH水溶液(4.65Kg、45%w/w)を添加し、反応が完了するまで混合物を10±3℃で攪拌した。クエン酸水溶液(18.15Kg、50%w/v)を充填し、次いで、トルエン(75L)を充填した。反応混合物を50±3℃に加熱し、水相を排出して廃棄した。有機相を50±3℃にて水(2×30L)で洗浄した。次いで、常圧蒸留によって有機相を75Lまで濃縮する。トルエン(45L)及びアセトン(75L)を充填し、溶液を120Lまで濃縮した。再度アセトン(75L)を充填し、再度溶液を105Lまで濃縮した。温度を55±3℃に維持しながらトルエン(75L)を充填した。溶液を35℃に冷却し、(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸(アセトン溶媒和物)(75g)の種を導入し、4時間にわたって0±3℃に冷却し、1時間この温度で保持した。固体生成物を濾過により単離し、冷(<5℃)トルエン/アセトン(45L、10:1)、冷(<5℃)トルエン(45L)で洗浄し、70℃で真空内乾燥させて、生成物(10.1Kg、71%)を得た。
【0064】
(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸の再結晶化
(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸(9.38Kg)を清浄な反応器に充填し、次いで、酢酸エチル(46.9L)を充填した。溶液を50℃に加熱し、濾過して、予め加温しておいた(35℃)結晶化容器に入れた。酢酸エチル(9.4L)によるライン洗浄を行った。合わせた酢酸エチル溶液を50℃に加熱し、攪拌して、確実に完全に溶解させた。温度を50℃に維持しながら濾過したヘプタン(9.4L)を添加し、次いで、溶液を30℃に冷却し、1:9の酢酸エチル:ヘプタン(0.47L)でスラリー化された(2S)−2−{[(2,6−ジフルオロフェニル)カルボニル]アミノ}−3−[4’−[(エチルオキシ)メチル]−2’,6’−ビス(メチルオキシ)−4−ビフェニリル]プロパン酸(47g)の種を導入した。スラリーを30℃で2時間エージングした。濾過したヘプタン(75L)を3時間にわたって添加した。次いで、スラリーを1時間にわたって0℃に冷却した。混合物を1時間0℃でエージングし、次いで、固体を濾取し、イソプロピルエーテル(29.6L)で洗浄し、50±3℃で真空下乾燥させて、生成物(8.55Kg、91%)を得た。
【0065】
約754、768、800、820、849、866、1006、1100、1122、1157、1188、1225、1242、1268、1292、1317、1352、1417、1466、1530、1580、1624、1650、1662、1711、1728、2938、3302cm−1に顕著な吸収帯を備えた赤外吸収スペクトルを有することを特徴としていた。
【特許請求の範囲】
【請求項1】
式(I)の化合物:
【化1】
を調製するためのプロセスであって、
a)式(IIa)のエステル:
【化2】
(式中、R1はC1−6アルキルである)
を加水分解する工程と、
(b)工程(a)から得られた生成物の溶媒和物を形成(溶媒和)する工程と、
(c)工程(b)から得られた前記溶媒和物を脱溶媒和して式(I)の化合物を得る工程と、
(d)任意で、工程(c)から得られた生成物を再結晶化させる工程とを含む、プロセス。
【請求項2】
R1がエチルである、請求項1に記載のプロセス。
【請求項3】
塩基性条件下でエステルの加水分解工程(工程(a))が行われる、請求項1又は2に記載のプロセス。
【請求項4】
アルカリ金属水酸化物を使用して塩基性条件下で工程(a)のエステルの加水分解を行って、溶媒から単離することができる適切なカルボン酸塩を得る、請求項1から3のいずれか一項に記載のプロセス。
【請求項5】
水酸化カリウムを利用して工程(a)のエステルの加水分解を行う、請求項4に記載のプロセス。
【請求項6】
工程(b)において、プロトン性であっても非プロトン性であってもよい極性溶媒で工程(a)の生成物を溶媒和することができる、請求項1〜5のいずれかに記載のプロセス。
【請求項7】
工程(b)において、アセトン、酢酸、アセトニトリル、ニトロメタン、ジメチルスルホキシド及びジメチルホルムアミドからなる群より選択される溶媒で工程(a)の生成物を溶媒和する、請求項6に記載のプロセス。
【請求項8】
アセトンで工程(a)の生成物を溶媒和する、請求項6又は7に記載のプロセス。
【請求項9】
工程(c)において、工程(b)の溶媒和物を乾燥させるか又は洗浄することによって脱溶媒和を行う、請求項1〜8のいずれか一項に記載のプロセス。
【請求項10】
脱溶媒和工程(c)が、室温と溶媒和物の沸点との間の温度で真空下にて工程(b)の溶媒和物を乾燥させることにより行われる、請求項9に記載のプロセス。
【請求項11】
式(I)の化合物:
【化3】
を調製するためのプロセスであって、
a)水酸化カリウムを使用して式(II)のエステル:
【化4】
を加水分解し、次いで、クエン酸を使用して酸性の後処理を行う工程と、
(b)工程(a)から得られた生成物のアセトン溶媒和物を形成する工程と、
(c)高温で真空下にて前記溶媒和物を乾燥させることを介して、工程(b)から得られたアセトン溶媒和物を脱溶媒和して、式(I)の化合物を得る工程とを含む、プロセス。
【請求項12】
式(I)の化合物のカリウム塩:
【化5】
。
【請求項13】
式(I)の化合物のアセトン溶媒和物:
【化6】
。
【請求項14】
式(I)の化合物のアセトン溶媒和物の結晶質形態であって、
【化7】
図1に示されているのと実質的に同じX線粉末回折(XRPD)パターンであって、角度2θで表され且つCuKα線を使用して回折計で得られるXRPDパターン及び/又は図2a及び2bに示されるのと実質的に同じ赤外線スペクトルを特徴とする、結晶質形態。
【請求項15】
約7.0、9.2、13.7、14.0及び24.0°2θにおいて特徴的なXPRDピークを有する、請求項14に記載の結晶質形態。
【請求項16】
式(V)の化合物:
【化8】
を式(VI)の化合物:
【化9】
とカップリングさせることによって式(II)の化合物を調製する、請求項11に記載のプロセス。
【請求項17】
式(V)の化合物:
【化10】
【請求項1】
式(I)の化合物:
【化1】
を調製するためのプロセスであって、
a)式(IIa)のエステル:
【化2】
(式中、R1はC1−6アルキルである)
を加水分解する工程と、
(b)工程(a)から得られた生成物の溶媒和物を形成(溶媒和)する工程と、
(c)工程(b)から得られた前記溶媒和物を脱溶媒和して式(I)の化合物を得る工程と、
(d)任意で、工程(c)から得られた生成物を再結晶化させる工程とを含む、プロセス。
【請求項2】
R1がエチルである、請求項1に記載のプロセス。
【請求項3】
塩基性条件下でエステルの加水分解工程(工程(a))が行われる、請求項1又は2に記載のプロセス。
【請求項4】
アルカリ金属水酸化物を使用して塩基性条件下で工程(a)のエステルの加水分解を行って、溶媒から単離することができる適切なカルボン酸塩を得る、請求項1から3のいずれか一項に記載のプロセス。
【請求項5】
水酸化カリウムを利用して工程(a)のエステルの加水分解を行う、請求項4に記載のプロセス。
【請求項6】
工程(b)において、プロトン性であっても非プロトン性であってもよい極性溶媒で工程(a)の生成物を溶媒和することができる、請求項1〜5のいずれかに記載のプロセス。
【請求項7】
工程(b)において、アセトン、酢酸、アセトニトリル、ニトロメタン、ジメチルスルホキシド及びジメチルホルムアミドからなる群より選択される溶媒で工程(a)の生成物を溶媒和する、請求項6に記載のプロセス。
【請求項8】
アセトンで工程(a)の生成物を溶媒和する、請求項6又は7に記載のプロセス。
【請求項9】
工程(c)において、工程(b)の溶媒和物を乾燥させるか又は洗浄することによって脱溶媒和を行う、請求項1〜8のいずれか一項に記載のプロセス。
【請求項10】
脱溶媒和工程(c)が、室温と溶媒和物の沸点との間の温度で真空下にて工程(b)の溶媒和物を乾燥させることにより行われる、請求項9に記載のプロセス。
【請求項11】
式(I)の化合物:
【化3】
を調製するためのプロセスであって、
a)水酸化カリウムを使用して式(II)のエステル:
【化4】
を加水分解し、次いで、クエン酸を使用して酸性の後処理を行う工程と、
(b)工程(a)から得られた生成物のアセトン溶媒和物を形成する工程と、
(c)高温で真空下にて前記溶媒和物を乾燥させることを介して、工程(b)から得られたアセトン溶媒和物を脱溶媒和して、式(I)の化合物を得る工程とを含む、プロセス。
【請求項12】
式(I)の化合物のカリウム塩:
【化5】
。
【請求項13】
式(I)の化合物のアセトン溶媒和物:
【化6】
。
【請求項14】
式(I)の化合物のアセトン溶媒和物の結晶質形態であって、
【化7】
図1に示されているのと実質的に同じX線粉末回折(XRPD)パターンであって、角度2θで表され且つCuKα線を使用して回折計で得られるXRPDパターン及び/又は図2a及び2bに示されるのと実質的に同じ赤外線スペクトルを特徴とする、結晶質形態。
【請求項15】
約7.0、9.2、13.7、14.0及び24.0°2θにおいて特徴的なXPRDピークを有する、請求項14に記載の結晶質形態。
【請求項16】
式(V)の化合物:
【化8】
を式(VI)の化合物:
【化9】
とカップリングさせることによって式(II)の化合物を調製する、請求項11に記載のプロセス。
【請求項17】
式(V)の化合物:
【化10】
【図1】

【図2a】

【図2b】
【図2a】
【図2b】
【公表番号】特表2013−508333(P2013−508333A)
【公表日】平成25年3月7日(2013.3.7)
【国際特許分類】
【出願番号】特願2012−534664(P2012−534664)
【出願日】平成22年10月19日(2010.10.19)
【国際出願番号】PCT/EP2010/065710
【国際公開番号】WO2011/048091
【国際公開日】平成23年4月28日(2011.4.28)
【出願人】(397009934)グラクソ グループ リミテッド (832)
【氏名又は名称原語表記】GLAXO GROUP LIMITED
【住所又は居所原語表記】Glaxo Wellcome House,Berkeley Avenue Greenford,Middlesex UB6 0NN,Great Britain
【出願人】(000002956)田辺三菱製薬株式会社 (225)
【Fターム(参考)】
【公表日】平成25年3月7日(2013.3.7)
【国際特許分類】
【出願日】平成22年10月19日(2010.10.19)
【国際出願番号】PCT/EP2010/065710
【国際公開番号】WO2011/048091
【国際公開日】平成23年4月28日(2011.4.28)
【出願人】(397009934)グラクソ グループ リミテッド (832)
【氏名又は名称原語表記】GLAXO GROUP LIMITED
【住所又は居所原語表記】Glaxo Wellcome House,Berkeley Avenue Greenford,Middlesex UB6 0NN,Great Britain
【出願人】(000002956)田辺三菱製薬株式会社 (225)
【Fターム(参考)】
[ Back to top ]