
フェノールの製造方法
フェノール又は置換フェノールの製造方法において、一般式(I):

(式中、R1及びR2は、それぞれ独立に、1〜4個の炭素原子を有するアルキル基を表し、但し、R1とR2が結合して4〜10個の炭素原子を有する環式基を形成していてもよく、前記環式基は任意に置換されていてもよく、かつR3は、水素、1〜4個の炭素原子を有する1つ以上のアルキル基又はシクロヘキシル基を表す)を有するアルキル芳香族ヒドロペルオキシドを、元素周期表の3〜5族及び7〜14族の少なくとも1種の金属の酸化物と、元素周期表の6族の少なくとも1種の金属の酸化物とを含む触媒と接触させる。
(式中、R1及びR2は、それぞれ独立に、1〜4個の炭素原子を有するアルキル基を表し、但し、R1とR2が結合して4〜10個の炭素原子を有する環式基を形成していてもよく、前記環式基は任意に置換されていてもよく、かつR3は、水素、1〜4個の炭素原子を有する1つ以上のアルキル基又はシクロヘキシル基を表す)を有するアルキル芳香族ヒドロペルオキシドを、元素周期表の3〜5族及び7〜14族の少なくとも1種の金属の酸化物と、元素周期表の6族の少なくとも1種の金属の酸化物とを含む触媒と接触させる。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願の相互参照)
この出願は、2008年10月10日に出願された先行米国仮特許出願第61/104,292号、及び2008年12月17日に出願された欧州特許出願第08171948.6号の利益を主張する。これらの両出願内容は、参照によってその全体が本明細書に援用される。
(分野)
本発明は、フェノール又は置換フェノールの製造方法に関する。
【背景技術】
【0002】
(背景)
フェノールは化学工業で重要な製品である。例えば、フェノールは、フェノール樹脂、ビスフェノールA、ε-カプロラクタム、アジピン酸、アルキルフェノール、及び可塑剤の製造に有用である。
現在、フェノール製造の最も一般的な径路はホック(Hock)法である。これは3工程法であり、第1工程はベンゼンをプロピレンでアルキル化してクメンを生成することを含み、その後クメンを対応するヒドロペルオキシドに酸化してからクメンヒドロペルオキシドを開裂する。生成物は等モル量のフェノールとアセトンを含む。しかし、フェノールに対する世界需要はアセトンに対する需要より急速に増大している。さらに、供給不足が生じているため、プロピレンのコストが増加する可能性がある。
従って、供給原料としてのプロピレンの使用を回避又は低減し、かつアセトンではなく、メチルエチルケトン及び/又はシクロヘキサノン等の高級ケトンを共製造する方法は、フェノール製造の魅力的な代替経路であり得る。例えば、メチルエチルケトンは、ラッカー及び溶媒として使うため並びに潤滑油の脱ろうのための需要がある。さらに、工業溶媒として、及び酸化反応の活性化剤として、並びにアジピン酸、シクロヘキサノン樹脂、シクロヘキサノンオキシム、カプロラクタム及びナイロン6の製造で使用されるシクロヘキサノンの市場が増大している。
【0003】
sec-ブチルベンゼンを酸化してsec-ブチルベンゼンヒドロペルオキシドを得、このヒドロペルオキシドを所望のフェノールとメチルエチルケトンに分解する、ホック法の変形によってフェノールとメチルエチルケトンを共製造できることが知られている。ゼオライトβ又はMCM-22ファミリーの分子ふるい上で直鎖ブテンによるベンゼンのアルキル化によってsec-ブチルベンゼンを製造できる。該方法の詳細は、例えば、国際特許出願公開第WO2006/015826号で見つけられる。
同様に、米国特許第6,037,513号は、MCM-22ファミリーの分子ふるいと、パラジウム、ルテニウム、ニッケル、コバルト及びその混合物から選択される少なくとも1種の水素化金属とを含む二機能性触媒の存在下でベンゼンを水素と接触させることでシクロヘキシルベンゼンを製造できることを開示している。‘513特許は、結果として生じたシクロヘキシルベンゼンを対応するヒドロペルオキシドに酸化し、このペルオキシドを所望のフェノールとシクロヘキサノンに分解できることも開示している。
しかし、アルキルベンゼン前駆体としてsec-ブチルベンゼン及び/又はシクロヘキシルベンゼンを使用するフェノールの製造は、クメンを基礎とする方法では存在しないか又は深刻でない特定の問題を伴う。例えば、クメンに比し、sec-ブチルベンゼン及びシクロヘキシルベンゼンの対応するヒドロペルオキシドへの酸化は、触媒の非存在下では非常に遅く、かつ不純物の存在に非常に敏感である。結果として、米国特許第6,720,462号及び第6,852,893号は、触媒としてN-ヒドロキシフタルイミド等の環状イミドを用いてsec-ブチルベンゼン及びシクロヘキシルベンゼン等のアルキルベンゼンの酸化を促進することを提案している。
【0004】
ヒドロペルオキシド開裂工程について、現在の商業的なフェノール/アセトン法は、硫酸触媒が理論の92〜96%しかフェノール選択性をもたらさないにもかかわらず、ほとんど排他的に硫酸触媒を使用する。硫酸触媒されたクメンヒドロペルオキシド開裂における最も一般的な副反応には、1)α-メチルスチレン(フェノールをアルキル化して重生成物を形成し、フェノールの収率を減らし得る)を形成するカルビノール(酸化の副生物)の脱水素;2)ケトンの収率を減らすケトンのアルドール縮合;及び3)オリゴマーを形成するオレフィンのオリゴマー化があり、全て最終生成物の分離工程で高沸点残留物(「フェノールタール」)形成に寄与する。結果として、「フェノールタール」形成を減らすためクメンヒドロペルオキシド開裂は一般的に複数工程で行なわれる。さらに、開裂工程後に硫酸を厳密に中和して、開裂生成物のさらなる反応を回避しなければならない。
これらの全ての問題は、開裂プロセスに関与する複雑さ及び投資を増加するので、クメンヒドロペルオキシドからフェノールを製造するため硫酸の種々の代替品が提案されている。例えば、過塩素酸、リン酸、トルエンスルホン酸及びSO2などの他の均一酸触媒が有効であることも示されている。しかしながら、これらの全ての均一触媒は、硫酸と同じ下流の酸中和及び生成物精製の問題に悩まされる。これらの問題を最小限にするため、種々の固体酸触媒がクメンヒドロペルオキシドの不均一開裂のために提案されている。例えば、米国特許第4,490,565号は、クメンヒドロペルオキシドの開裂でのゼオライトβの使用を開示し、米国特許第4,490,566号は、拘束係数(Constraint Index)1〜12のゼオライト、例えばZSM-5の使用を開示し、EP-A-492807は、同方法でのフォージャサイトの使用を開示している。米国特許第4,870,217号にはクメンヒドロペルオキシドの酸触媒分解におけるスメクタイト粘土の使用が記載されている。
【0005】
米国特許第4,898,995号は、シリカ、アルミナ、チタニア及びジルコニア等の不活性担体上の、スルホン酸官能性を有するイオン交換樹脂又はヘテロポリ酸(例えば12-タングストリン酸)から成る不均一触媒上でクメンヒドロペルオキシドを反応させることによるフェノールとアセトンの共製造方法を開示している。該ヘテロポリ酸触媒は、通常、それらの水和物として使用され、そのままでは350℃を超える温度で本質的に不安定である。
米国特許第6,169,215号は、クメンヒドロペルオキシドからフェノール及びアセトンを製造する方法であって、IVB族金属酸化物源をVIB族金属オキシアニオン源と少なくとも400℃の温度でか焼して作られた固体酸触媒とクメンヒドロペルオキシドを接触させる工程を含む方法を開示している。IVB族金属酸化物はジルコニア及びチタニアから選択され、VIB族金属オキシアニオンはクロム、モリブデン及びタングステンのオキシアニオンから選択される。
sec-ブチルベンゼン及び/又はシクロヘキシルベンゼン等の他のアルキルベンゼンからフェノールを製造する場合、今日までヒドロペルオキシド開裂工程についてはあまり研究が行なわれていないが、ほとんどの提案が硫酸及び同様の均一触媒の使用に焦点を合わせている。しかし、いずれの実行可能な開裂方法も、ヒドロペルオキシドの製造がおそらく環状イミド等の触媒の使用を必要とするので、酸化工程の直接生成物が窒素化合物(該開裂工程で典型的に用いられる酸触媒にとって既知の毒物である)をかなり含み得るという事実に対処しなければならないことは明白である。
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明によれば、今や特定の混合金属酸化物が高級アルキルベンゼンのヒドロペルオキシドの開裂のための非常に活性な触媒であり、かつ98%以上の選択性でフェノールを製造できることが分かった。さらに、ヒドロペルオキシドを製造する際に使用する触媒に起因する窒素不純物を除去することが望ましいが、ヒドロペルオキシドを極性溶媒で希釈することによって該窒素不純物による触媒の被毒を軽減できること及び極性溶媒で洗浄することによって被毒触媒を効率的に復活させ得ることも分かった。さらに、触媒が固体なので、硫酸などの均一触媒に固有な下流の中和及び精製の問題が回避される。
【課題を解決するための手段】
【0007】
(概要)
一態様では、本発明は、フェノール又は置換フェノールの製造方法に属し、この方法は、下記一般式(I):
【0008】
【化1】
【0009】
(式中、R1及びR2は、それぞれ独立に、1〜4個の炭素原子を有するアルキル基を表し、但し、R1とR2が結合して4〜10個の炭素原子を有する環式基を形成していてもよく、前記環式基は任意に置換されていてもよく、かつR3は、水素、1〜4個の炭素原子を有する1つ以上のアルキル基又はシクロヘキシル基を表す)
を有するアルキル芳香族ヒドロペルオキシドを、元素周期表の3〜5族及び7〜14族の少なくとも1種の金属の酸化物と、元素周期表の6族の少なくとも1種の金属の酸化物とを含む触媒と接触させる工程を含む。
好都合には、触媒が、元素周期表の4族の少なくとも1種の金属の酸化物と、元素周期表の6族の少なくとも1種の金属の酸化物とを含む。一実施形態では、触媒が、酸化ジルコニウムと、モリブデン及び/又はタングステンの酸化物とを含む。
好都合には、触媒が、元素周期表の8〜11族の少なくとも1種の金属の酸化物をさらに含む。一実施形態では、触媒が、鉄及び/又は銅の酸化物をさらに含む。
好都合には、一般式(I)の前記アルキル芳香族ヒドロペルオキシドが、sec-ブチルベンゼンヒドロペルオキシド、p-メチル-sec-ブチルベンゼンヒドロペルオキシド、1,4-ジフェニルシクロヘキサンヒドロペルオキシド、sec-ペンチルベンゼンヒドロペルオキシド、sec-ヘキシルベンゼンヒドロペルオキシド、シクロペンチルベンゼンヒドロペルオキシド、シクロヘキシルベンゼンヒドロペルオキシド及びシクロオクチルベンゼンヒドロペルオキシドから選択される。一実施形態では、一般式(I)のアルキル芳香族ヒドロペルオキシドが、sec-ブチルベンゼンヒドロペルオキシド及びシクロヘキシルベンゼンヒドロペルオキシドから選択される。
好都合には、前記アルキル芳香族ヒドロペルオキシドを極性溶媒に溶解させる。
好都合には、前記接触工程が、約40℃〜約120℃の温度、約100〜約1000kPaの圧力、及びヒドロペルオキシドに基づいて約1〜約50時間-1の液空間速度(liquid hourly space velocity)(LHSV)で行なわれる。
さらなる態様では、本発明は、フェノール又は置換フェノールの製造方法に属し、この方法は、以下の工程:
(a)下記一般式(II):
【0010】
【化2】
【0011】
(式中、R1及びR2は、それぞれ独立に、1〜4個の炭素原子を有するアルキル基を表し、但し、R1とR2が結合して4〜10個の炭素原子を有する環式基を形成していてもよく、前記環式基は任意に置換されていてもよく、かつR3は、水素、1〜4個の炭素原子を有する1つ以上のアルキル基又はシクロヘキシル基を表す)
を有するアルキル芳香族化合物を、
下記一般式(III):
【0012】
【化3】
【0013】
(式中、R4及びR5は、それぞれ独立に、1〜20個の炭素原子を有するヒドロカルビル基及び置換ヒドロカルビル基、又は基SO3H、NH2、OH、及びNO2、又は原子H、F、Cl、Br、及びIから選択され、但し、R4とR5が相互に共有結合によって連結されていてもよく;
Q1及びQ2は、それぞれ独立に、C、CH、N、CR6から選択され;
X及びZは、それぞれ独立に、C、S、CH2、N、P及び周期表4族の元素から選択され;
Yは、O又はOHであり;
kは、0、1、又は2であり;
lは、0、1、又は2であり;
mは、1〜3であり;かつ
R6は、R4について列挙したいずれの構成要素であってもよい)
を有する環状イミドを含む触媒の存在下で酸素含有ガスと接触させる工程(ここで、前記接触工程は、前記アルキル芳香族化合物を下記一般式(I):
【0014】
【化4】
【0015】
(式中、R1、R2及びR3は、それぞれ前記定義どおりである)
を有するアルキル芳香族ヒドロペルオキシドに変換するための条件下で行なわれる);
及び
(b)前記アルキル芳香族ヒドロペルオキシドを、元素周期表の3〜5族及び7〜14族の少なくとも1種の金属酸化物と、元素周期表の6族の少なくとも1種の金属の酸化物とを含む酸化物触媒と接触させる工程
を含む。
好都合には、前記環状イミドが、下記一般式(IV):
【0016】
【化5】
【0017】
(式中、R7、R8、R9、及びR10は、それぞれ独立に、1〜20個の炭素原子を有するヒドロカルビル基及び置換ヒドロカルビル基、又は基SO3H、NH2、OH、及びNO2又は原子H、F、Cl、Br、及びIから選択され;
X及びZは、それぞれ独立に、C、S、CH2、N、P及び周期表4族の元素から選択され;
Yは、O又はOHであり;
kは、0、1、又は2であり;かつ
lは、0、1、又は2である)
に従う。
一実施形態では、前記環状イミドがN-ヒドロキシフタルイミドを含む。
好都合には、前記接触工程(a)が、前記アルキル芳香族ヒドロペルオキシド及び未反応環状イミド触媒を含む流出物を生成し、かつ本方法が、下記工程:
(c)前記接触工程(b)の前に前記流出物を処理して、前記流出物流中の未反応環状イミドの少なくとも一部を除去する工程
をさらに含む。
一実施形態では、前記処理工程(c)が、好都合には前記環状イミドのpKa値以上のpKb値を有する塩基の水溶液と前記流出物を接触させて、前記未反応イミド触媒の少なくとも一部を含む水相と、前記アルキル芳香族ヒドロペルオキシドを含む有機相とを生成する工程を含む。
別の実施形態では、前記処理工程(c)が、前記流出物を固体吸着剤、例えば金属酸化物、金属炭酸塩及び/又は金属炭酸水素塩、粘土、及び/又はイオン交換樹脂などと接触させる工程を含む。
好都合には、本方法は、前記酸化物触媒をアセトン等の極性溶媒で洗浄することによって、前記酸化物触媒を定期的に復活させる工程をさらに含む。
【図面の簡単な説明】
【0018】
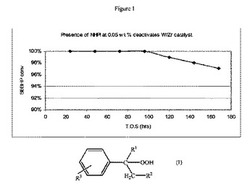
【図1】実施例9の方法に従うFe/W/Zr酸化物触媒上におけるsec-ブチルベンゼンヒドロペルオキシドの開裂について操業中の時間(time on stream)に対するsec-ブチルベンゼンヒドロペルオキシドの転化率のグラフである。
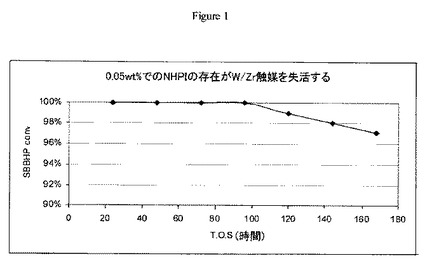
【図2】実施例10の方法に従う可変量のN-ヒドロキシフタルイミドの存在下でFe/W/Zr酸化物触媒上におけるsec-ブチルベンゼンヒドロペルオキシドの開裂について操業中の時間に対するsec-ブチルベンゼンヒドロペルオキシドの転化率のグラフである。
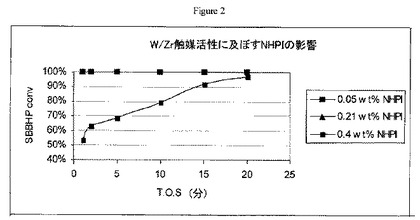
【図3】実施例11の方法に従うFe/W/Zr酸化物触媒上におけるアセトン中のsec-ブチルベンゼンヒドロペルオキシドの開裂について操業中の時間に対する生成物濃度のグラフである。
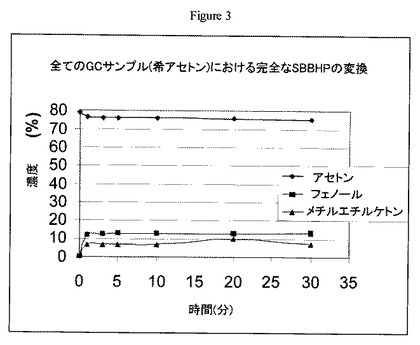
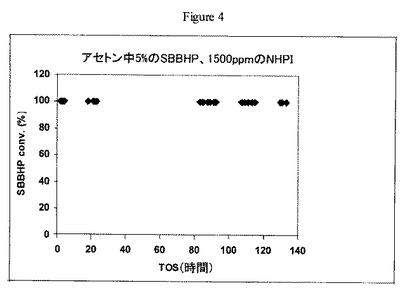
【図4】実施例12の方法に従うW/Zr酸化物触媒上における1500ppmのN-ヒドロキシフタルイミドを含むsec-ブチルベンゼンヒドロペルオキシドのアセトン溶液の開裂について操業中の時間に対するsec-ブチルベンゼンヒドロペルオキシドの転化率のグラフである。
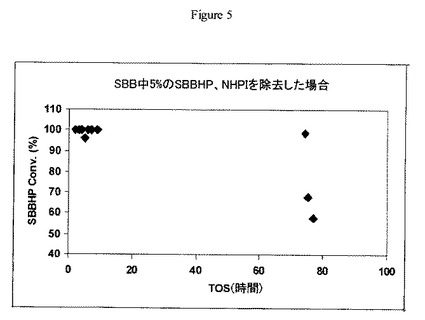
【図5】実施例13の方法に従うW/Zr酸化物触媒上における、sec-ブチルベンゼンで希釈され、かつ30ppm未満のN-ヒドロキシフタルイミドを含むsec-ブチルベンゼンヒドロペルオキシドの開裂について操業中の時間に対するsec-ブチルベンゼンヒドロペルオキシドの転化率のグラフである。
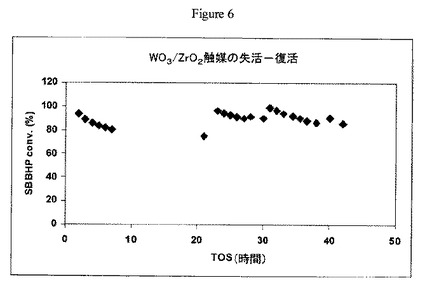
【図6】実施例14の方法に従うW/Zr酸化物触媒上における、sec-ブチルベンゼンで希釈され、かつ210ppmのN-ヒドロキシフタルイミドを含むsec-ブチルベンゼンヒドロペルオキシドの開裂について及び実施例15の方法に従う復活後の操業中の時間に対するsec-ブチルベンゼンヒドロペルオキシドの転化率のグラフである。
【発明を実施するための形態】
【0019】
(実施形態の詳細な説明)
本明細書では、フェノール又は置換フェノールの製造方法であって、下記一般式(I):
【0020】
【化6】
【0021】
(式中、R1及びR2は、それぞれ独立に、1〜4個の炭素原子を有するアルキル基を表し、但し、R1とR2が結合して4〜10個の炭素原子を有する環式基を形成していてもよく、前記環式基は任意に置換されていてもよく、かつR3は、水素、1〜4個の炭素原子を有する1つ以上のアルキル基又はシクロヘキシル基を表す)
を有するアルキル芳香族ヒドロペルオキシドを混合金属酸化物触媒と接触させる工程を含む方法について記述する。
適切なアルキル芳香族ヒドロペルオキシドの例として、sec-ブチルベンゼンヒドロペルオキシド、p-メチル-sec-ブチルベンゼンヒドロペルオキシド、1、4-ジフェニルシクロヘキサンヒドロペルオキシド、sec-ペンチルベンゼンヒドロペルオキシド、sec-ヘキシルベンゼンヒドロペルオキシド、シクロペンチルベンゼンヒドロペルオキシド、シクロヘキシルベンゼンヒドロペルオキシド及びシクロオクチルベンゼンヒドロペルオキシドが挙げられる。一般式(I)の好ましいアルキル芳香族ヒドロペルオキシドとして、sec-ブチルベンゼンヒドロペルオキシド及びシクロヘキシルベンゼンヒドロペルオキシドが挙げられる。
【0022】
(アルキル芳香族ヒドロペルオキシドの製造)
本方法で使用するアルキル芳香族ヒドロペルオキシドは、典型的に下記一般式(II):
【0023】
【化7】
【0024】
(式中、R1、R2及びR3は、式(I)の上記定義に帰する意味を有する)
を有するアルキル芳香族化合物の触媒酸化によって製造される。アルキル芳香族前駆体化合物は、今度は既知の芳香族アルキル化法によって製造される。例えば、sec-ブチルベンゼンヒドロペルオキシドは、国際特許公開第WO2006/015826号に記載されているようなMCM-22ファミリーの触媒の存在下で直鎖ブテンによるベンゼンのアルキル化の結果として生じるsec-ブチルベンゼン生成物の酸化によって好都合に製造される。同様に、シクロヘキシルベンゼンヒドロペルオキシドは、米国特許第6,037,513号に記載されているような、MCM-22ファミリーの分子ルふるいと水素化金属を含む二機能性触媒の存在下でのベンゼンのヒドロアルキル化の結果として生じるシクロヘキシルベンゼン生成物の酸化によって好都合に製造される。同様の方法を用いて、本方法で使用可能な他のヒドロペルオキシドを製造することができる。
所望のヒドロペルオキシドの製造で使用する酸化法は、通常、アルキル芳香族前駆体を、下記一般式(III):
【0025】
【化8】
【0026】
(式中、R4及びR5は、それぞれ独立に、1〜20個の炭素原子を有するヒドロカルビル基及び置換ヒドロカルビル基、又は基SO3H、NH2、OH、及びNO2又は原子H、F、Cl、Br、及びIから選択され、但し、R4とR5が相互に共有結合によって連結されていてもよく;
Q1及びQ2は、それぞれ独立に、C、CH、N、CR6から選択され;
X及びZは、それぞれ独立に、C、S、CH2、N、P及び周期表4族の元素から選択され;
Yは、O又はOHであり;
kは、0、1、又は2であり;
lは、0、1、又は2であり;
mは、1〜3であり;かつ
R6は、R4について列挙したいずれの構成要素であってもよい)
を有する環状イミドを含む触媒の存在下で酸素含有ガスと反応させる工程を含み、かつ前記接触工程は、アルキル芳香族化合物を所望のヒドロペルオキシドに変換するための条件下で行なわれる。
好都合には、環状イミドは下記一般式(IV):
【0027】
【化9】
【0028】
(式中、R7、R8、R9及びR10は、それぞれ独立に、1〜20個の炭素原子を有するヒドロカルビル基及び置換ヒドロカルビル基、又は基SO3H、NH2、OH、及びNO2又は原子H、F、Cl、Br、及びIから選択され;
X及びZは、それぞれ独立に、C、S、CH2、N、P及び周期表4族の元素から選択され;
Yは、O又はOHであり;
kは、0、1、又は2であり;かつ
lは、0、1、又は2である)
に従う。
【0029】
実際の一実施形態では、環状イミドがN-ヒドロキシフタルイミドを含む。
酸化工程の適切な条件は、約70℃〜約200℃、例えば約90℃〜約130℃の温度、及び約0.5〜約20気圧(50〜2000kPa)の圧力を含む。好都合には酸化反応を触媒蒸留装置内で行ない、通過毎の転化率を好ましくは50%未満に維持して、副生物の形成を最小限にする。
酸化工程は、アルキル芳香族前駆体化合物をその関連ヒドロペルオキシドに変換する。しかし、酸化プロセスは、副生物として水及び有機酸(例えば、酢酸又はギ酸)を発生する傾向もあり、該副生物が触媒を加水分解し、ヒドロペルオキシド種の分解をもたらすこともある。従って、一実施形態では、酸化工程で使用する条件、特に圧力と酸素濃度を制御して、反応媒体中の水及び有機酸の濃度を50ppm未満に維持するようにする。該条件は、典型的に酸化を相対的に低い圧力、例えば300kPa未満、例えば約100kPa〜約200kPaで行なうことを含む。さらに、0.1〜100%の広い酸素濃度範囲にわたって酸化を行なうことができるが、相対的に低い酸素濃度、例えば酸素含有ガス中21体積%以下、例えば約0.1〜約21体積%、一般的に約1〜約10体積%の酸素で操作することが好ましい。さらに、酸化工程中ストリッピングガスを反応媒体に通すことによって、所望の低レベルの水及び有機酸を維持するのを容易にする。一実施形態では、ストリッピングガスが酸素含有ガスと同一である。別の実施形態では、ストリッピングガスが酸素含有ガスと異なり、かつ反応媒体及び環状イミド触媒に不活性である。適切なストリッピングガスとして、ヘリウム及びアルゴン等の不活性ガスが挙げられる。
【0030】
低圧及び低酸素濃度で酸化プロセスを操作すること及び水と有機酸を反応媒体から放出することによるさらなる利点は、軽質ヒドロペルオキシド(例えば、エチル又はメチルヒドロペルオキシド)、軽質ケトン(例えば、メチルエチルケトン)、軽質アルデヒド(例えば、アセトアルデヒド)及び軽質アルコール(例えば、エタノール)が形成されるにつれて、それらが反応生成物から除去されることである。従って、軽質ヒドロペルオキシドは危険であり、それらの液体生成物中の濃度が高くなりすぎると安全に対する懸念をもたらす。また、軽質ヒドロペルオキシド、アルコール、アルデヒド及びケトンは有機酸及び水の形成の前駆体なので、これらの種を酸化媒体から除去すると、酸化反応速度及び環状イミド触媒の選択性と安定性を高める。実際に、データによると、100psig(790kPa)でNHPIによってsec-ブチルベンゼンの酸化を行なう場合、これらの軽質種及び水の99モル%より多くが反応器内に残るが、大気圧では、これらの種の95モル%より多くが酸化反応器から除去されることが分かる。
酸化反応の生成物には、所望のアルキルヒドロペルオキシドと共に未反応アルキル芳香族前駆体及び未反応環状イミド触媒が含まれる。未反応アルキル芳香族前駆体は蒸留で容易に除去され、酸化工程に再循環される。しかし、後述するように、未反応環状イミド触媒は、ヒドロペルオキシドをフェノールに開裂するために用いられる下流の混合金属酸化物触媒に対する有害物として作用し得る。さらに、環状イミドは費用がかかる傾向があるので、該未反応触媒を回収かつ再循環させるのが望ましい。従って、普通は酸化プロセスからの流出物を開裂工程に通す前に処理して未反応環状イミドのレベルを下げることが望ましいだろう。
【0031】
一実施形態では、酸化流出物の処理は、流出物を塩基、特に環状イミド触媒のpKa以上のpKb値を有する弱塩基の水溶液と接触させる工程を含み、これによって未反応イミド触媒が水相中に抽出され、前記酸化された炭化水素生成物を含み、かつレベルが減少した環状イミドを含む有機相を残す。一般的に、抽出は、有機相中のイミドのレベルを有機相の質量で100ppm未満、例えば50ppm未満、例えば10ppm未満に減らすように行なわれる。
弱塩基は水相中への抽出後にイミドの分解を触媒しにくいので、未反応イミド触媒の抽出では一般的に弱塩基の使用が望ましい。適切な弱塩基として、金属炭酸塩及び/又は金属炭酸水素塩、特にアルカリ金属炭酸塩及び/又はアルカリ金属炭酸水素塩、例えば炭酸ナトリウムが挙げられる。
環状イミド抽出工程で使用する条件を厳密に制御する必要はないが、一般的に約10℃〜約80℃、例えば約20℃〜約70℃の温度を含む。抽出時間は例えば約1分〜約30分、例えば約5分〜約10分であってよい。抽出工程で使用する塩基の量は、普通は未反応イミドに少なくとも等モル量の塩基を供給するのに十分な量、例えば未反応イミド1モル当たり1〜3モルの塩基である。一般に、抽出中は相を撹拌して相間の接触を最大にする。
塩基水溶液中への抽出後、水相を例えば酢酸で酸性にして未反応イミを沈殿させることによって、未反応環状イミドを容易に回収することができる。沈殿した未反応イミドを例えばろ過又は遠心分離によって水相から分離した後、所望により、酸化工程に再循環させてよい。
【0032】
別の実施形態では、酸化流出物の処理は、前記酸化炭化水素生成物に富み、かつ減少したか又はゼロレベルの環状イミドを含む処理流出物を生じさせるように、流出物を未反応イミド触媒の一部又は実質的に全ての除去に有効な固体吸着剤と接触させる工程を含む。この場合もやはり、吸着プロセスは、有機相中のイミドのレベルを有機相の100ppm未満、例えば50ppm未満、例えば10ppm未満に減らすように行なわれる。
適切な固体吸着剤は、金属炭酸塩及び/又は金属炭酸水素塩などの塩基特性を有するものであり、多孔性の担体、粘土、イオン交換樹脂及び金属酸化物、特に混合金属酸化物上に設けてよい。
環状イミド抽出工程で有効な吸着剤であるのに十分な塩基特性を有する金属酸化物は、これらの金属酸化物材料上のCO2及びNH3の化学吸着のモル比によって決定され得る。マイルドな酸であるCO2を用いて、被験金属酸化物上に存在する塩基サイトを滴定する。同様に、強塩基であるNH3を滴定して該材料上の酸性サイトを指し示す。該材料の表面積(多くの場合、金属酸化物の調製方法によってかなり影響を受ける)、化学吸着を試験する温度、及び化学吸着を試験する圧力などの多くの要因が化学吸着の実際量を決定する。本目的では、「塩基性」酸化物は、後述するように試験した場合、金属酸化物1g当たりのNH3の化学吸着に対する金属酸化物1g当たりのCO2の化学吸着のモル比が0.5より大きい、典型的に0.75より大きい、特に1.0より大きい酸化物と定義される。
【0033】
金属酸化物1g当たりのNH3の化学吸着に対する金属酸化物1g当たりのCO2の化学吸着のモル比を決定する試験は、周囲圧力でMettler TGA/SDTA 851熱重量分析システムを用いて行なわれる。金属酸化物サンプルを空気流中で約500℃(表1で指摘されている場合を除き)に約3時間(少なくとも一定のサンプル重量が得られるまで)か焼する。そして、空気流(ヘリウムを使用することもできる)中でサンプルの温度を化学吸着の所望温度に下げる。次に、サンプルをヘリウム流中で所望温度にて平衡させて秤量する。二酸化炭素の化学吸着を100℃で測定し、アンモニアの化学吸着を250℃で測定する。秤量後、一定重量が得られるまでサンプルをヘリウムと二酸化炭素又はアンモニアを含むガス混合物のいくつかのパルス(約12秒/パルス)にさらす。ガス混合物は、約10重量%の二酸化炭素又はアンモニアを含み、残りはヘリウムである。被験ガス混合物の各パルス後、金属酸化物サンプルに約3分間ヘリウム流を流す。各試験ではガス混合物の約20の個別パルスを使用する。か焼後の金属酸化物サンプル重量に基づいて金属酸化物1g当たりのmgを単位としたサンプルの重量の増加を用いて金属酸化物1g当たりに吸着したCO2又はNH3のモル数を決定する。
いくつかの代表的金属酸化物種について吸着質1g当たりのNH3の化学吸着に対するCO2の化学吸着のモル比下を表1に示す。
【0034】
表1
【0035】
環状イミド抽出工程で固体吸着剤として使うのに適した金属酸化物には、周期表の2族、3族、4族、ランタニド系列、又はアクチニド系列の金属の酸化物及び混合酸化物が含まれる。一実施形態では、吸着剤は、2種以上の金属酸化物、好ましくは1種の4族の金属酸化物と、2族、3族、ランタニド系列、及びアクチニド系列の金属酸化物から選択される1種以上の金属酸化物とを含む。酸化物は、種々の方法を用いて調製され得るが、一般的に溶液からの沈殿及び/又はか焼によって適切な前駆体の変換によって調製される。適切な前駆体としては、金属塩、例えばハロゲン化物、硫酸塩、リン酸塩、ハロゲン化物、硝酸塩、オキシ塩化物、アルコキシド及び酢酸塩が挙げられる。
一実施形態では、金属酸化物は、該金属の塩を水などの溶媒中に含む溶液を調製することによって製造される。結果として生じた溶液を次に、例えば沈殿試薬、典型的には水酸化ナトリウム又は水酸化アンモニウム等の塩基の添加によってのような、固体酸化物材料の沈殿を引き起こすのに十分な条件にさらす。沈殿中、溶液は一般的に200℃以下、例えば約0℃〜約200℃、例えば約20℃〜約100℃の範囲の温度で維持される。結果として生じるゲルを好ましくは次に少なくとも80℃、好ましくは少なくとも100℃の温度で、例えば10日間まで、例えば5日間まで、例えば3日間まで熱水処理する。結果として生じる材料を次に例えばろ過又は遠心分離によって回収し、洗浄し、乾燥させる。結果として生じる粒子材料を典型的には次に普通は酸化雰囲気内で、少なくとも400℃、例えば約400℃〜約800℃の温度にて、48時間まで、例えば約0.5時間〜約24時間、例えば約1時間〜約10時間か焼する。
【0036】
環状イミド抽出工程で2種以上の金属酸化物を使用する場合、それらを共沈殿させるか又は別々に沈殿させ、か焼固体粒子として含む加工のいずれの後の段階でも相互に混ぜ合わせてよい。
固体吸着剤として使うのに適したイオン交換樹脂には、酸性又は塩基性種を除去するために常用される当該樹脂、例えばAmberlystイオン交換樹脂がある。
固体吸着剤を用いた環状イミドの吸着に適した条件は、約10℃〜約130℃、例えば約20℃〜約80℃の温度、約1分〜約30分、例えば約5分〜約10分の時間を含む。
固体吸着剤による除去後、極性溶媒、例えばエタノール又はアセトンで吸着剤を洗浄することによって、未反応環状イミドを容易に回収することができる。エタノールとイミドの存在は、再循環触媒の酸化活性又は選択性に悪影響を与えないことが分かっているので、回収したイミドを次に、エタノールの事前除去をするか又はしないで酸化工程に再循環させることができる。
【0037】
(ヒドロペルオキシド開裂)
本方法のヒドロペルオキシド開裂工程は、普通は未反応アルキル芳香族前駆体の除去後及び必要に応じて流出物を前処理して環状イミドのレベルを100ppm未満に減らした後に、酸化工程からの流出物を混合金属酸化物触媒と接触させることによって行なわれる。特に、混合金属酸化物触媒は、元素周期表の3〜5族及び7〜14族の少なくとも1種の金属の酸化物、好都合には、元素周期表の4族の少なくとも1種の金属の酸化物と、元素周期表の6族の少なくとも1種の金属の酸化物とを含む。一実施形態では、触媒が酸化ジルコニウムと、モリブデン及び/又はタングステンの酸化物とを含む。
好都合には、触媒が元素周期表の8〜11族の少なくとも1種の金属の酸化物、例えば鉄及び/又は銅の酸化物をさらに含む。
混合金属酸化物触媒は、3〜5族及び7〜14族の少なくとも1種の金属のイオン源を含む第1溶液、例えば水溶液を、少なくとも1種の4族金属のイオン源を含む第2溶液、この場合もやはり例えば水溶液及び必要に応じて少なくとも1種の8〜11族金属のイオン源を含む第3溶液と混ぜ合わせることによって都合良く調製される。この混合は、液体媒体から固体として混合金属材料の共沈殿を引き起こすのに十分な条件下で起こり得る。或いは、3〜5族及び7〜14族の金属のイオン源、4族金属のイオン源及び必要に応じて8〜11族金属のイオン源を単一溶液中に混ぜ合わせてよい。この溶液を次に例えば該溶液に沈殿試薬の添加によって、固体混合酸化物材料の共沈殿を引き起こすのに十分な条件に供してよい。便宜上、沈殿は7より高いpHで行なわれる。例えば、沈殿試薬は水酸化ナトリウム又は水酸化アンモニウム等の塩基であってよい。
【0038】
沈殿中に液体媒体を維持する温度は一般的に約200℃未満、例えば約0℃〜約200℃の範囲である。沈殿に特有の温度範囲は約20℃〜約100℃である。結果として生じるゲルを好ましくは次に少なくとも80℃、好ましくは少なくとも100℃の温度で熱水処理する。熱水処理は、典型的に容器内で周囲圧力にて行なわれる。一実施形態では、ゲルを10日間まで、例えば5日間まで、例えば3日間まで熱水処理する。
混合金属酸化物に水和した前駆体を次に例えばろ過又は遠心分離によって回収し、洗浄し、乾燥させる。結果として生じる材料を次に、例えば酸化雰囲気内で少なくとも400℃、例えば少なくとも500℃、例えば約600℃〜約900℃、特に約650℃〜約800℃の温度でか焼して混合金属酸化物触媒を形成することができる。か焼時間は典型的に48時間まで、例えば約0.5〜24時間、例えば約1.0〜10時間である。一実施形態では、約700℃で約1〜約3時間か焼を行なう。
開裂反応は、ヒドロペルオキシドを液相内で約20℃〜約150℃、例えば約40℃〜約120℃の温度、及び/又は約50〜約2500kPa、例えば約100〜約1000kPaの圧力で及び/又はヒドロペルオキシドに基づいて約0.1〜約1000時間-1、好ましくは約1〜約50時間-1の液空間速度(LHSV)で混合金属酸化物触媒と接触させることによって都合良く作用する。開裂反応は、触媒蒸留装置内で好都合に行なわれる。
典型的に、開裂反応に不活性な有機溶媒、例えばメチルエチルケトン、フェノール、シクロヘキシルベンゼン、シクロヘキサノン及びsec-ブチルベンゼンでヒドロペルオキシドを希釈して熱除去を助ける。さらに好ましくは、極性溶媒の存在が酸化反応により残存している環状イミドによる混合金属酸化物触媒開裂触媒の被毒を軽減し得ることが分かっているので、開裂反応のため、アルキル芳香族ヒドロペルオキシドを極性溶媒、例えばアセトンに溶解させる。
【0039】
開裂反応への供給原料中の未反応環状イミドの存否に関係なく、混合金属酸化物は経時的にその活性を失い、ヒドロペルオキシドのフェノールへの変換度の減少をもたらす傾向があることが分かっている。しかし、触媒をアセトン等の極性溶媒で洗浄することによって触媒を定期的に復活させることで触媒の開裂活性を回復させ得ることが分かる。
本発明により酸化されるアルキル芳香族化合物がシクロヘキシルベンゼンである場合、酸化生成物はシクロヘキシルベンゼンヒドロペルオキシドを含み、開裂生成物はフェノール及びシクロヘキサノンを含む。開裂工程からの粗製シクロヘキサノン及び粗製フェノールをさらなる精製に供して、精製シクロヘキサノン及び精製フェノールを生成し得る。適切な精製プロセスには、限定するものではないが、シクロヘキサノン及びフェノールを他の種から分離するための一連の蒸留塔が含まれる。粗製又は精製シクロヘキサノン自体を脱水素に供して、それをフェノールに変換することができる。例えば、白金、ニッケル又はパラジウム等の触媒上で該脱水素を行なうことができる。
ここで、以下の非限定例を参照して本発明をさらに詳細に説明する。
【実施例】
【0040】
実施例1:Mo/Zr酸化物触媒の合成
500グラムのZrOCl2・8H2Oを3リットルの蒸留水に撹拌しながら溶かした。260グラムの濃水酸化アンモニウム、66グラムの(NH4)6Mo7O24・4H2O及び3リットルの蒸留水を含む別の溶液を調製した。両溶液を60℃に加熱し、ノズルミキシングを利用して加熱溶液を50ml/分の速度で混ぜ合わせた。濃水酸化アンモニウムを添加して最終複合物のpHを約9に調整した。結果として生じたスラリーを次にポリプロピレンボトルに取り、蒸し器(100℃)内に72時間置いた。生じた生成物をろ過で回収し、過剰の水で洗浄し、85℃で一晩乾燥させた。この触媒のサンプルを空気流内で3時間800℃にか焼した。このか焼触媒は、触媒中の総重量Mo及びZrの重量で16%のMoを含んでいた。
【0041】
実施例2:Cu/W/Zr酸化物触媒の合成
500グラムのZrOCl2・8H2Oを3.0リットルの蒸留水に撹拌しながら溶かし、この溶液に6.8グラムのCuSO4・5H2Oを加えた。260グラムの濃水酸化アンモニウム、54グラムの(NH4)6H2W12O40・xH2O及び3リットルの蒸留水を含む別の溶液を調製した。両溶液を40℃に加熱し、ノズルミキシングを利用して加熱溶液を50ml/分の速度で混ぜ合わせた。濃水酸化アンモニウムを添加して最終複合物のpHを約9に調整した。結果として生じたスラリーを次にポリプロピレンボトルに取り、蒸し器(100℃)内に72時間置いた。生じた生成物をろ過で回収し、過剰の水で洗浄し、85℃で一晩乾燥させた。この触媒のサンプルを空気流内で3時間700℃にか焼した。このか焼触媒は、触媒中の総重量W、Zr及びCuの重量で1%のCuと16%のWを含んでいた。
【0042】
実施例3:Fe/W/Zr酸化物触媒の合成
1000グラムのZrOCl2・8H2Oを3.0リットルの蒸留水に撹拌しながら溶かしてからこの溶液に15.2グラムのFeSO4・7H2Oを加えた。400グラムの濃水酸化アンモニウム、108グラムの(NH4)6H2W12O40・xH2O及び2940mlの蒸留水を含む別の溶液を調製した。両溶液を60℃に加熱し、ノズルミキシングを利用して加熱溶液を50ml/分の速度で混ぜ合わせた。濃水酸化アンモニウムを添加して最終複合物のpHを約9に調整した。結果として生じたスラリーを次にポリプロピレンボトルに取り、蒸し器(100℃)内に72時間置いた。生じた生成物をろ過で回収し、過剰の水で洗浄し、85℃で一晩乾燥させた。この触媒のサンプルを空気流内で3時間800℃にか焼した。このか焼触媒は73m2/gの表面積を有し、触媒中の総重量W、Zr及びFeの重量で1%のFeと16%のWを含んでいた。
【0043】
実施例4:W/Zr酸化物触媒
1000グラムのZrOCl2・8H2Oを3.0リットルの蒸留水に撹拌しながら溶かした。400グラムの濃水酸化アンモニウム、108グラムの(NH4)6H2W12O40・xH2O及び3.0リットルの蒸留水を含む別の溶液を調製した。両溶液を60℃に加熱し、ノズルミキシングを利用して加熱溶液を50ml/分の速度で混ぜ合わせた。濃水酸化アンモニウムを添加して最終複合物のpHを約9に調整した。結果として生じたスラリーを次にポリプロピレンボトルに取り、蒸し器(100℃)内に72時間置いた。生じた生成物をろ過で回収し、過剰の水で洗浄し、85℃で一晩乾燥させた。この触媒のサンプルを空気流内で3時間800℃にか焼した。このか焼触媒は73m2/gの表面積を有し、触媒中の総重量W及びZrの重量で16%のWを含んでいた。
【0044】
実施例5:Fe/W/Zr酸化物触媒
500グラムのZrOCl2・8H2Oを3.0リットルの蒸留水に撹拌しながら溶かしてからこの溶液に7.6グラムのFeSO4・7H2Oを加えた。260グラムの濃水酸化アンモニウム、54グラムの(NH4)6H2W12O40・xH2O及び2940mlの蒸留水を含む別の溶液を調製した。両溶液を60℃に加熱し、ノズルミキシングを利用して加熱溶液を50ml/分の速度で混ぜ合わせた。濃水酸化アンモニウムを添加して最終複合物のpHを約9に調整した。結果として生じたスラリーを次にポリプロピレンボトルに取り、蒸し器(100℃)内に72時間置いた。生じた生成物をろ過で回収し、過剰の水で洗浄し、85℃で一晩乾燥させた。この触媒のサンプルを空気流内で3時間800℃にか焼した。このか焼触媒は71m2/gの表面積を有し、触媒中の総重量W、Zr及びFeの重量で1%のFeと16%のWを含んでいた。
【0045】
1
実施例6:実施例1のMo/Zr酸化物触媒を用いたシクロヘキシルベンゼンヒドロペルオキシドの開裂
実施例1で調製した触媒を40〜80メッシュサイズにペレット化し、60〜80メッシュの砂と混合し(v/v 1:1)、1/4インチ(0.6cm)径のステンレススチール管形反応器に装填した。反応器を70℃で維持し、ISCOポンプを用いて30分間3cc/分で液体アセトンを反応器管に通した。アセトンの供給を止め、アセトン中シクロヘキシルベンゼンヒドロペルオキシドの5wt%溶液を別のISCOポンプを用いて0.45cc/分の速度で反応器に供給した。生成物をチルドノックアウトポット(chilled knock-out pot)に収集し、定期的にGC分析用サンプルを抽出した。実験の完了時、異なるシクロヘキシルベンゼンヒドロペルオキシドの流速を利用して滞留時間を変えた。
【0046】
実施例7:実施例2のCu/W/Zr酸化物触媒を用いたシクロヘキシルベンゼンヒドロペルオキシドの開裂
実施例2で調製した触媒を40〜80メッシュサイズにペレット化し、60〜80メッシュの砂と混合し(v/v 1:1)、1/4インチ(0.6cm)径のステンレススチール管形反応器に装填した。反応器を70℃で維持し、ISCOポンプを用いて30分間3cc/分で液体アセトンを反応器管に通した。アセトンの供給を止め、アセトン中シクロヘキシルベンゼンヒドロペルオキシドの5wt%溶液を別のISCOポンプを用いて0.45cc/分の速度で反応器に供給した。生成物をチルドノックアウトポットに収集し、定期的にGC分析用サンプルを抽出した。実験の完了時、異なるシクロヘキシルベンゼンヒドロペルオキシドの流速を利用して滞留時間を変えた。
【0047】
実施例8:実施例3のFe/W/Zr酸化物触媒を用いたシクロヘキシルベンゼンヒドロペルオキシドの開裂
実施例3で調製した触媒を40〜80メッシュサイズにペレット化し、60〜80メッシュの砂と混合し(v/v 1:1)、1/4インチ(0.6cm)径のステンレススチール管形反応器に装填した。反応器を70℃で維持し、ISCOポンプを用いて30分間3cc/分で液体アセトンを反応器管に通した。アセトンの供給を止め、アセトン中シクロヘキシルベンゼンヒドロペルオキシドの5wt%溶液を別のISCOポンプを用いて0.45cc/分の速度で反応器に供給した。生成物をチルドノックアウトポットに収集し、定期的にGC分析用サンプルを抽出した。実験の完了時、異なるシクロヘキシルベンゼンヒドロペルオキシドの流速を利用して滞留時間を変えた。
実施例6〜8の結果を下表2に示す。この表から、実施例1〜3の混合金属酸化物が、シクロヘキシルベンゼンヒドロペルオキシド(CHBHP)のフェノール及びシクロヘキサノンへの開裂にとって非常に活性かつ選択的な触媒であることが分かるだろう。
【0048】
表2
【0049】
実施例9:実施例5のFe/W/Zr酸化物触媒上のSec-ブチルベンゼンヒドロペルオキシドの開裂−供給原料中のNHPIを除去しない場合
実施例5で調製した触媒(1.5g)を20〜40メッシュサイズにペレット化し、60〜80メッシュの砂(1.5g)と混合し(v/v 1:1)、3/8インチ(1cm)径のステンレススチール管形反応器に装填した。反応器を90℃に加熱し、ISCOポンプを用いて135分間1cc/分で液体メチルエチルケトン(MEK)を反応器管を介して導入しながら90℃で維持した。MEK供給を止め、MEK中sec-ブチルベンゼンヒドロペルオキシド(SBBHP)の5wt%溶液を別のISCOポンプを用いて0.25cc/分の速度で反応器に導入した。sec-ブチルベンゼンヒドロペルオキシドは、ヒドロペルオキシドをMEKに溶かして開裂反応器に溶液を供給する前に触媒を除去せずに0.11wt%のN-ヒドロキシフタルイミド触媒の存在下でsec-ブチルベンゼンを酸化することで製造された。従って開裂工程に供給されたSBBHP/MEK溶液は、500ppmの未反応N-ヒドロキシフタルイミド触媒を含んでいた。開裂反応の生成物をチルドノックアウトポットに収集し、GC分析用サンプルを抽出した。結果を図1に示す。図1は、SBBHP転化率の減少によって示唆されるように、約95時間の操業中の時間後に触媒が失活し始めたことを示している。
【0050】
実施例10:実施例5のFe/W/Zr酸化物触媒上のSec-ブチルヒドロペルオキシド開裂−供給原料中のNHPI濃度の影響
空気撹拌機、温度計、冷却水冷凝縮器、及び窒素誘導器を備えた100mlの丸底三口フラスコにアセトン(39.5g,50ml)及び0.5gの実施例5の粉末触媒を添加した。アセトン及び触媒をアセトン還流下で加熱し(56℃)、かつ還流しながら、0.05wt%のNHPIを含む10cc(10.42g)の70wt.%のsec-ブチルベンゼンヒドロペルオキシド濃縮物をシリンジポンプを用いて20分で30cc/時間の速度にて添加した。1、3、6、10、20及び30分撹拌後にGCサンプルを収集した。GCサンプルは、ろ過して固体触媒を除去した後に明黄色液体だった。このプロセスを0.21wt%及び0.4wt%のNHPIを含有するsec-ブチルベンゼンヒドロペルオキシドサンプルで繰り返し、結果を図2に示す。
【0051】
実施例11:実施例5のFe/W/Zr酸化物触媒上のSec-ブチルヒドロペルオキシド開裂−供給原料からNHPIを除去した場合
空気撹拌機、温度計、冷却水冷凝縮器、及び窒素誘導器を備えた100mlの丸底三口フラスコにアセトン(39.5g,50ml)及び0.5gの実施例5の粉末触媒を添加した。アセトン及び触媒をアセトン還流下で加熱し(56℃)、かつ還流しながら、10cc(10.42g)の70wt.%のsec-ブチルベンゼンヒドロペルオキシド濃縮物(希Na2CO3溶液による苛性洗浄でNHPIを除去した)をシリンジポンプを用いて20分で30cc/時間の速度にて添加した。この添加時間中、反応器の温度が59℃に上昇した。1、3、6、10、20及び30分撹拌後にGCサンプルを収集した。GCサンプルは、ろ過して固体触媒を除去した後に明黄色液体だった。結果を図3に示す。図3から、sec-ブチルベンゼンヒドロペルオキシドからフェノール及びメチルエチルケトンに実質的に完全に変換したことが分かるだろう。
【0052】
実施例12:実施例4のW/Zr酸化物触媒上のSec-ブチルヒドロペルオキシド開裂−1500ppmのNHPIを含有するアセトン中5%のSBBHPの場合
実施例4で調製した触媒(1.5g)を40〜80メッシュサイズにペレット化し、60〜80メッシュの砂と混合し(v/v 1:1)、1/4インチ(0.6cm)径のステンレススチール管形反応器に装填した。反応器を70℃で維持し、ISCOポンプを用いて30分間3cc/分で液体アセトンを反応器管に通した。アセトン供給を止め、1500ppmのNHPIを含有するアセトン中5wt%のsec-ブチルベンゼンヒドロペルオキシド(SBBHP)溶液を別のISCOポンプを用いて0.5cc/分の速度で反応器に通した。生成物をチルドノックアウトポットに収集し、定期的にGC分析用サンプルを抽出した。結果を図4に示す。操業中の時間130時間後でさえ触媒失活の徴候が見られなかった。
【0053】
実施例13:実施例4のW/Zr酸化物触媒上のSec-ブチルヒドロペルオキシド開裂−NHPIを除去したSBB中5%のSBBHPの場合
実施例4で調製した触媒(1.5g)を40〜80メッシュサイズにペレット化し、60〜80メッシュの砂と混合し(v/v 1:1)、1/4インチ(0.6cm)径のステンレススチール管形反応器に装填した。反応器を70℃で維持し、ISCOポンプを用いて30分間3cc/分で液体アセトンを反応器管に通した。アセトン供給を止め、30ppm未満のNHPIを含有するsec-ブチルベンゼン(SBB)中5wt%のsec-ブチルベンゼンヒドロペルオキシド(SBBHP)溶液を別のISCOポンプを用いて0.25cc/分の速度で反応器に通した。生成物をチルドノックアウトポットに収集し、定期的にGC分析用サンプルを抽出した。結果を図3に示す。図3から、SBBHP転化率の減少によって示唆されるように、操業中の時間約70時間後に触媒が失活し始めたことが分かるだろう。
【0054】
実施例14:実施例4のW/Zr酸化物触媒上のSec-ブチルヒドロペルオキシド開裂−310ppmのNHPIを含有するSBB/アセトン中5%のSBBHPの場合
実施例4で調製した触媒(1.5g)を40〜80メッシュサイズにペレット化し、60〜80メッシュの砂と混合し(v/v 1:1)、1/4インチ(0.6cm)径のステンレススチール管形反応器に装填した。反応器を70℃で維持し、ISCOポンプを用いて30分間3cc/分で液体アセトンを反応器管に通した。アセトン供給を止め、310ppmのNHPIを含有する98/2(wt/wt)のSBB/アセトン中5wt%のSBBHP溶液を別のISCOポンプを用いて0.25cc/分の速度で反応器に通した。生成物をチルドノックアウトポットに収集し、定期的にGC分析用サンプルを抽出した。結果を図4に示す。図4から、操業中の時間約2時間後に触媒が失活し始めたことが分かるだろう。
【0055】
実施例15:失活した実施例14のW/Zr酸化物触媒のアセトン溶媒を用いた復活
実施例14の実験の一部として、触媒の復活を現場で行なった。SBBHP転化率が80%未満に低下したらすぐに、NHPIを含有するヒドロペルオキシド供給を止めた。反応器を70℃で維持しながら、アセトンを3cc/分で30分間触媒に通した。次にアセトン流を止めてヒドロペルオキシドの供給を再開した。SBBHP転化率が戻ってほとんど100%に増えたことによって示唆されるように、触媒活性が回復した。このような失活−復活のサイクルを3回行なうと(図4)、最初の触媒活性が各復活サイクル後に実質的に回復した。
本発明を特定の実施形態に関連して記載及び説明したが、当業者は、必ずしも本明細書で説明していない変形に本発明が役立つことを認めるだろう。従って、この理由のため、本発明の真の範囲を決定する目的のためには添付の特許請求の範囲をもっぱら参照すべきである。
【技術分野】
【0001】
(関連出願の相互参照)
この出願は、2008年10月10日に出願された先行米国仮特許出願第61/104,292号、及び2008年12月17日に出願された欧州特許出願第08171948.6号の利益を主張する。これらの両出願内容は、参照によってその全体が本明細書に援用される。
(分野)
本発明は、フェノール又は置換フェノールの製造方法に関する。
【背景技術】
【0002】
(背景)
フェノールは化学工業で重要な製品である。例えば、フェノールは、フェノール樹脂、ビスフェノールA、ε-カプロラクタム、アジピン酸、アルキルフェノール、及び可塑剤の製造に有用である。
現在、フェノール製造の最も一般的な径路はホック(Hock)法である。これは3工程法であり、第1工程はベンゼンをプロピレンでアルキル化してクメンを生成することを含み、その後クメンを対応するヒドロペルオキシドに酸化してからクメンヒドロペルオキシドを開裂する。生成物は等モル量のフェノールとアセトンを含む。しかし、フェノールに対する世界需要はアセトンに対する需要より急速に増大している。さらに、供給不足が生じているため、プロピレンのコストが増加する可能性がある。
従って、供給原料としてのプロピレンの使用を回避又は低減し、かつアセトンではなく、メチルエチルケトン及び/又はシクロヘキサノン等の高級ケトンを共製造する方法は、フェノール製造の魅力的な代替経路であり得る。例えば、メチルエチルケトンは、ラッカー及び溶媒として使うため並びに潤滑油の脱ろうのための需要がある。さらに、工業溶媒として、及び酸化反応の活性化剤として、並びにアジピン酸、シクロヘキサノン樹脂、シクロヘキサノンオキシム、カプロラクタム及びナイロン6の製造で使用されるシクロヘキサノンの市場が増大している。
【0003】
sec-ブチルベンゼンを酸化してsec-ブチルベンゼンヒドロペルオキシドを得、このヒドロペルオキシドを所望のフェノールとメチルエチルケトンに分解する、ホック法の変形によってフェノールとメチルエチルケトンを共製造できることが知られている。ゼオライトβ又はMCM-22ファミリーの分子ふるい上で直鎖ブテンによるベンゼンのアルキル化によってsec-ブチルベンゼンを製造できる。該方法の詳細は、例えば、国際特許出願公開第WO2006/015826号で見つけられる。
同様に、米国特許第6,037,513号は、MCM-22ファミリーの分子ふるいと、パラジウム、ルテニウム、ニッケル、コバルト及びその混合物から選択される少なくとも1種の水素化金属とを含む二機能性触媒の存在下でベンゼンを水素と接触させることでシクロヘキシルベンゼンを製造できることを開示している。‘513特許は、結果として生じたシクロヘキシルベンゼンを対応するヒドロペルオキシドに酸化し、このペルオキシドを所望のフェノールとシクロヘキサノンに分解できることも開示している。
しかし、アルキルベンゼン前駆体としてsec-ブチルベンゼン及び/又はシクロヘキシルベンゼンを使用するフェノールの製造は、クメンを基礎とする方法では存在しないか又は深刻でない特定の問題を伴う。例えば、クメンに比し、sec-ブチルベンゼン及びシクロヘキシルベンゼンの対応するヒドロペルオキシドへの酸化は、触媒の非存在下では非常に遅く、かつ不純物の存在に非常に敏感である。結果として、米国特許第6,720,462号及び第6,852,893号は、触媒としてN-ヒドロキシフタルイミド等の環状イミドを用いてsec-ブチルベンゼン及びシクロヘキシルベンゼン等のアルキルベンゼンの酸化を促進することを提案している。
【0004】
ヒドロペルオキシド開裂工程について、現在の商業的なフェノール/アセトン法は、硫酸触媒が理論の92〜96%しかフェノール選択性をもたらさないにもかかわらず、ほとんど排他的に硫酸触媒を使用する。硫酸触媒されたクメンヒドロペルオキシド開裂における最も一般的な副反応には、1)α-メチルスチレン(フェノールをアルキル化して重生成物を形成し、フェノールの収率を減らし得る)を形成するカルビノール(酸化の副生物)の脱水素;2)ケトンの収率を減らすケトンのアルドール縮合;及び3)オリゴマーを形成するオレフィンのオリゴマー化があり、全て最終生成物の分離工程で高沸点残留物(「フェノールタール」)形成に寄与する。結果として、「フェノールタール」形成を減らすためクメンヒドロペルオキシド開裂は一般的に複数工程で行なわれる。さらに、開裂工程後に硫酸を厳密に中和して、開裂生成物のさらなる反応を回避しなければならない。
これらの全ての問題は、開裂プロセスに関与する複雑さ及び投資を増加するので、クメンヒドロペルオキシドからフェノールを製造するため硫酸の種々の代替品が提案されている。例えば、過塩素酸、リン酸、トルエンスルホン酸及びSO2などの他の均一酸触媒が有効であることも示されている。しかしながら、これらの全ての均一触媒は、硫酸と同じ下流の酸中和及び生成物精製の問題に悩まされる。これらの問題を最小限にするため、種々の固体酸触媒がクメンヒドロペルオキシドの不均一開裂のために提案されている。例えば、米国特許第4,490,565号は、クメンヒドロペルオキシドの開裂でのゼオライトβの使用を開示し、米国特許第4,490,566号は、拘束係数(Constraint Index)1〜12のゼオライト、例えばZSM-5の使用を開示し、EP-A-492807は、同方法でのフォージャサイトの使用を開示している。米国特許第4,870,217号にはクメンヒドロペルオキシドの酸触媒分解におけるスメクタイト粘土の使用が記載されている。
【0005】
米国特許第4,898,995号は、シリカ、アルミナ、チタニア及びジルコニア等の不活性担体上の、スルホン酸官能性を有するイオン交換樹脂又はヘテロポリ酸(例えば12-タングストリン酸)から成る不均一触媒上でクメンヒドロペルオキシドを反応させることによるフェノールとアセトンの共製造方法を開示している。該ヘテロポリ酸触媒は、通常、それらの水和物として使用され、そのままでは350℃を超える温度で本質的に不安定である。
米国特許第6,169,215号は、クメンヒドロペルオキシドからフェノール及びアセトンを製造する方法であって、IVB族金属酸化物源をVIB族金属オキシアニオン源と少なくとも400℃の温度でか焼して作られた固体酸触媒とクメンヒドロペルオキシドを接触させる工程を含む方法を開示している。IVB族金属酸化物はジルコニア及びチタニアから選択され、VIB族金属オキシアニオンはクロム、モリブデン及びタングステンのオキシアニオンから選択される。
sec-ブチルベンゼン及び/又はシクロヘキシルベンゼン等の他のアルキルベンゼンからフェノールを製造する場合、今日までヒドロペルオキシド開裂工程についてはあまり研究が行なわれていないが、ほとんどの提案が硫酸及び同様の均一触媒の使用に焦点を合わせている。しかし、いずれの実行可能な開裂方法も、ヒドロペルオキシドの製造がおそらく環状イミド等の触媒の使用を必要とするので、酸化工程の直接生成物が窒素化合物(該開裂工程で典型的に用いられる酸触媒にとって既知の毒物である)をかなり含み得るという事実に対処しなければならないことは明白である。
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明によれば、今や特定の混合金属酸化物が高級アルキルベンゼンのヒドロペルオキシドの開裂のための非常に活性な触媒であり、かつ98%以上の選択性でフェノールを製造できることが分かった。さらに、ヒドロペルオキシドを製造する際に使用する触媒に起因する窒素不純物を除去することが望ましいが、ヒドロペルオキシドを極性溶媒で希釈することによって該窒素不純物による触媒の被毒を軽減できること及び極性溶媒で洗浄することによって被毒触媒を効率的に復活させ得ることも分かった。さらに、触媒が固体なので、硫酸などの均一触媒に固有な下流の中和及び精製の問題が回避される。
【課題を解決するための手段】
【0007】
(概要)
一態様では、本発明は、フェノール又は置換フェノールの製造方法に属し、この方法は、下記一般式(I):
【0008】
【化1】
【0009】
(式中、R1及びR2は、それぞれ独立に、1〜4個の炭素原子を有するアルキル基を表し、但し、R1とR2が結合して4〜10個の炭素原子を有する環式基を形成していてもよく、前記環式基は任意に置換されていてもよく、かつR3は、水素、1〜4個の炭素原子を有する1つ以上のアルキル基又はシクロヘキシル基を表す)
を有するアルキル芳香族ヒドロペルオキシドを、元素周期表の3〜5族及び7〜14族の少なくとも1種の金属の酸化物と、元素周期表の6族の少なくとも1種の金属の酸化物とを含む触媒と接触させる工程を含む。
好都合には、触媒が、元素周期表の4族の少なくとも1種の金属の酸化物と、元素周期表の6族の少なくとも1種の金属の酸化物とを含む。一実施形態では、触媒が、酸化ジルコニウムと、モリブデン及び/又はタングステンの酸化物とを含む。
好都合には、触媒が、元素周期表の8〜11族の少なくとも1種の金属の酸化物をさらに含む。一実施形態では、触媒が、鉄及び/又は銅の酸化物をさらに含む。
好都合には、一般式(I)の前記アルキル芳香族ヒドロペルオキシドが、sec-ブチルベンゼンヒドロペルオキシド、p-メチル-sec-ブチルベンゼンヒドロペルオキシド、1,4-ジフェニルシクロヘキサンヒドロペルオキシド、sec-ペンチルベンゼンヒドロペルオキシド、sec-ヘキシルベンゼンヒドロペルオキシド、シクロペンチルベンゼンヒドロペルオキシド、シクロヘキシルベンゼンヒドロペルオキシド及びシクロオクチルベンゼンヒドロペルオキシドから選択される。一実施形態では、一般式(I)のアルキル芳香族ヒドロペルオキシドが、sec-ブチルベンゼンヒドロペルオキシド及びシクロヘキシルベンゼンヒドロペルオキシドから選択される。
好都合には、前記アルキル芳香族ヒドロペルオキシドを極性溶媒に溶解させる。
好都合には、前記接触工程が、約40℃〜約120℃の温度、約100〜約1000kPaの圧力、及びヒドロペルオキシドに基づいて約1〜約50時間-1の液空間速度(liquid hourly space velocity)(LHSV)で行なわれる。
さらなる態様では、本発明は、フェノール又は置換フェノールの製造方法に属し、この方法は、以下の工程:
(a)下記一般式(II):
【0010】
【化2】
【0011】
(式中、R1及びR2は、それぞれ独立に、1〜4個の炭素原子を有するアルキル基を表し、但し、R1とR2が結合して4〜10個の炭素原子を有する環式基を形成していてもよく、前記環式基は任意に置換されていてもよく、かつR3は、水素、1〜4個の炭素原子を有する1つ以上のアルキル基又はシクロヘキシル基を表す)
を有するアルキル芳香族化合物を、
下記一般式(III):
【0012】
【化3】
【0013】
(式中、R4及びR5は、それぞれ独立に、1〜20個の炭素原子を有するヒドロカルビル基及び置換ヒドロカルビル基、又は基SO3H、NH2、OH、及びNO2、又は原子H、F、Cl、Br、及びIから選択され、但し、R4とR5が相互に共有結合によって連結されていてもよく;
Q1及びQ2は、それぞれ独立に、C、CH、N、CR6から選択され;
X及びZは、それぞれ独立に、C、S、CH2、N、P及び周期表4族の元素から選択され;
Yは、O又はOHであり;
kは、0、1、又は2であり;
lは、0、1、又は2であり;
mは、1〜3であり;かつ
R6は、R4について列挙したいずれの構成要素であってもよい)
を有する環状イミドを含む触媒の存在下で酸素含有ガスと接触させる工程(ここで、前記接触工程は、前記アルキル芳香族化合物を下記一般式(I):
【0014】
【化4】
【0015】
(式中、R1、R2及びR3は、それぞれ前記定義どおりである)
を有するアルキル芳香族ヒドロペルオキシドに変換するための条件下で行なわれる);
及び
(b)前記アルキル芳香族ヒドロペルオキシドを、元素周期表の3〜5族及び7〜14族の少なくとも1種の金属酸化物と、元素周期表の6族の少なくとも1種の金属の酸化物とを含む酸化物触媒と接触させる工程
を含む。
好都合には、前記環状イミドが、下記一般式(IV):
【0016】
【化5】
【0017】
(式中、R7、R8、R9、及びR10は、それぞれ独立に、1〜20個の炭素原子を有するヒドロカルビル基及び置換ヒドロカルビル基、又は基SO3H、NH2、OH、及びNO2又は原子H、F、Cl、Br、及びIから選択され;
X及びZは、それぞれ独立に、C、S、CH2、N、P及び周期表4族の元素から選択され;
Yは、O又はOHであり;
kは、0、1、又は2であり;かつ
lは、0、1、又は2である)
に従う。
一実施形態では、前記環状イミドがN-ヒドロキシフタルイミドを含む。
好都合には、前記接触工程(a)が、前記アルキル芳香族ヒドロペルオキシド及び未反応環状イミド触媒を含む流出物を生成し、かつ本方法が、下記工程:
(c)前記接触工程(b)の前に前記流出物を処理して、前記流出物流中の未反応環状イミドの少なくとも一部を除去する工程
をさらに含む。
一実施形態では、前記処理工程(c)が、好都合には前記環状イミドのpKa値以上のpKb値を有する塩基の水溶液と前記流出物を接触させて、前記未反応イミド触媒の少なくとも一部を含む水相と、前記アルキル芳香族ヒドロペルオキシドを含む有機相とを生成する工程を含む。
別の実施形態では、前記処理工程(c)が、前記流出物を固体吸着剤、例えば金属酸化物、金属炭酸塩及び/又は金属炭酸水素塩、粘土、及び/又はイオン交換樹脂などと接触させる工程を含む。
好都合には、本方法は、前記酸化物触媒をアセトン等の極性溶媒で洗浄することによって、前記酸化物触媒を定期的に復活させる工程をさらに含む。
【図面の簡単な説明】
【0018】
【図1】実施例9の方法に従うFe/W/Zr酸化物触媒上におけるsec-ブチルベンゼンヒドロペルオキシドの開裂について操業中の時間(time on stream)に対するsec-ブチルベンゼンヒドロペルオキシドの転化率のグラフである。
【図2】実施例10の方法に従う可変量のN-ヒドロキシフタルイミドの存在下でFe/W/Zr酸化物触媒上におけるsec-ブチルベンゼンヒドロペルオキシドの開裂について操業中の時間に対するsec-ブチルベンゼンヒドロペルオキシドの転化率のグラフである。
【図3】実施例11の方法に従うFe/W/Zr酸化物触媒上におけるアセトン中のsec-ブチルベンゼンヒドロペルオキシドの開裂について操業中の時間に対する生成物濃度のグラフである。
【図4】実施例12の方法に従うW/Zr酸化物触媒上における1500ppmのN-ヒドロキシフタルイミドを含むsec-ブチルベンゼンヒドロペルオキシドのアセトン溶液の開裂について操業中の時間に対するsec-ブチルベンゼンヒドロペルオキシドの転化率のグラフである。
【図5】実施例13の方法に従うW/Zr酸化物触媒上における、sec-ブチルベンゼンで希釈され、かつ30ppm未満のN-ヒドロキシフタルイミドを含むsec-ブチルベンゼンヒドロペルオキシドの開裂について操業中の時間に対するsec-ブチルベンゼンヒドロペルオキシドの転化率のグラフである。
【図6】実施例14の方法に従うW/Zr酸化物触媒上における、sec-ブチルベンゼンで希釈され、かつ210ppmのN-ヒドロキシフタルイミドを含むsec-ブチルベンゼンヒドロペルオキシドの開裂について及び実施例15の方法に従う復活後の操業中の時間に対するsec-ブチルベンゼンヒドロペルオキシドの転化率のグラフである。
【発明を実施するための形態】
【0019】
(実施形態の詳細な説明)
本明細書では、フェノール又は置換フェノールの製造方法であって、下記一般式(I):
【0020】
【化6】
【0021】
(式中、R1及びR2は、それぞれ独立に、1〜4個の炭素原子を有するアルキル基を表し、但し、R1とR2が結合して4〜10個の炭素原子を有する環式基を形成していてもよく、前記環式基は任意に置換されていてもよく、かつR3は、水素、1〜4個の炭素原子を有する1つ以上のアルキル基又はシクロヘキシル基を表す)
を有するアルキル芳香族ヒドロペルオキシドを混合金属酸化物触媒と接触させる工程を含む方法について記述する。
適切なアルキル芳香族ヒドロペルオキシドの例として、sec-ブチルベンゼンヒドロペルオキシド、p-メチル-sec-ブチルベンゼンヒドロペルオキシド、1、4-ジフェニルシクロヘキサンヒドロペルオキシド、sec-ペンチルベンゼンヒドロペルオキシド、sec-ヘキシルベンゼンヒドロペルオキシド、シクロペンチルベンゼンヒドロペルオキシド、シクロヘキシルベンゼンヒドロペルオキシド及びシクロオクチルベンゼンヒドロペルオキシドが挙げられる。一般式(I)の好ましいアルキル芳香族ヒドロペルオキシドとして、sec-ブチルベンゼンヒドロペルオキシド及びシクロヘキシルベンゼンヒドロペルオキシドが挙げられる。
【0022】
(アルキル芳香族ヒドロペルオキシドの製造)
本方法で使用するアルキル芳香族ヒドロペルオキシドは、典型的に下記一般式(II):
【0023】
【化7】
【0024】
(式中、R1、R2及びR3は、式(I)の上記定義に帰する意味を有する)
を有するアルキル芳香族化合物の触媒酸化によって製造される。アルキル芳香族前駆体化合物は、今度は既知の芳香族アルキル化法によって製造される。例えば、sec-ブチルベンゼンヒドロペルオキシドは、国際特許公開第WO2006/015826号に記載されているようなMCM-22ファミリーの触媒の存在下で直鎖ブテンによるベンゼンのアルキル化の結果として生じるsec-ブチルベンゼン生成物の酸化によって好都合に製造される。同様に、シクロヘキシルベンゼンヒドロペルオキシドは、米国特許第6,037,513号に記載されているような、MCM-22ファミリーの分子ルふるいと水素化金属を含む二機能性触媒の存在下でのベンゼンのヒドロアルキル化の結果として生じるシクロヘキシルベンゼン生成物の酸化によって好都合に製造される。同様の方法を用いて、本方法で使用可能な他のヒドロペルオキシドを製造することができる。
所望のヒドロペルオキシドの製造で使用する酸化法は、通常、アルキル芳香族前駆体を、下記一般式(III):
【0025】
【化8】
【0026】
(式中、R4及びR5は、それぞれ独立に、1〜20個の炭素原子を有するヒドロカルビル基及び置換ヒドロカルビル基、又は基SO3H、NH2、OH、及びNO2又は原子H、F、Cl、Br、及びIから選択され、但し、R4とR5が相互に共有結合によって連結されていてもよく;
Q1及びQ2は、それぞれ独立に、C、CH、N、CR6から選択され;
X及びZは、それぞれ独立に、C、S、CH2、N、P及び周期表4族の元素から選択され;
Yは、O又はOHであり;
kは、0、1、又は2であり;
lは、0、1、又は2であり;
mは、1〜3であり;かつ
R6は、R4について列挙したいずれの構成要素であってもよい)
を有する環状イミドを含む触媒の存在下で酸素含有ガスと反応させる工程を含み、かつ前記接触工程は、アルキル芳香族化合物を所望のヒドロペルオキシドに変換するための条件下で行なわれる。
好都合には、環状イミドは下記一般式(IV):
【0027】
【化9】
【0028】
(式中、R7、R8、R9及びR10は、それぞれ独立に、1〜20個の炭素原子を有するヒドロカルビル基及び置換ヒドロカルビル基、又は基SO3H、NH2、OH、及びNO2又は原子H、F、Cl、Br、及びIから選択され;
X及びZは、それぞれ独立に、C、S、CH2、N、P及び周期表4族の元素から選択され;
Yは、O又はOHであり;
kは、0、1、又は2であり;かつ
lは、0、1、又は2である)
に従う。
【0029】
実際の一実施形態では、環状イミドがN-ヒドロキシフタルイミドを含む。
酸化工程の適切な条件は、約70℃〜約200℃、例えば約90℃〜約130℃の温度、及び約0.5〜約20気圧(50〜2000kPa)の圧力を含む。好都合には酸化反応を触媒蒸留装置内で行ない、通過毎の転化率を好ましくは50%未満に維持して、副生物の形成を最小限にする。
酸化工程は、アルキル芳香族前駆体化合物をその関連ヒドロペルオキシドに変換する。しかし、酸化プロセスは、副生物として水及び有機酸(例えば、酢酸又はギ酸)を発生する傾向もあり、該副生物が触媒を加水分解し、ヒドロペルオキシド種の分解をもたらすこともある。従って、一実施形態では、酸化工程で使用する条件、特に圧力と酸素濃度を制御して、反応媒体中の水及び有機酸の濃度を50ppm未満に維持するようにする。該条件は、典型的に酸化を相対的に低い圧力、例えば300kPa未満、例えば約100kPa〜約200kPaで行なうことを含む。さらに、0.1〜100%の広い酸素濃度範囲にわたって酸化を行なうことができるが、相対的に低い酸素濃度、例えば酸素含有ガス中21体積%以下、例えば約0.1〜約21体積%、一般的に約1〜約10体積%の酸素で操作することが好ましい。さらに、酸化工程中ストリッピングガスを反応媒体に通すことによって、所望の低レベルの水及び有機酸を維持するのを容易にする。一実施形態では、ストリッピングガスが酸素含有ガスと同一である。別の実施形態では、ストリッピングガスが酸素含有ガスと異なり、かつ反応媒体及び環状イミド触媒に不活性である。適切なストリッピングガスとして、ヘリウム及びアルゴン等の不活性ガスが挙げられる。
【0030】
低圧及び低酸素濃度で酸化プロセスを操作すること及び水と有機酸を反応媒体から放出することによるさらなる利点は、軽質ヒドロペルオキシド(例えば、エチル又はメチルヒドロペルオキシド)、軽質ケトン(例えば、メチルエチルケトン)、軽質アルデヒド(例えば、アセトアルデヒド)及び軽質アルコール(例えば、エタノール)が形成されるにつれて、それらが反応生成物から除去されることである。従って、軽質ヒドロペルオキシドは危険であり、それらの液体生成物中の濃度が高くなりすぎると安全に対する懸念をもたらす。また、軽質ヒドロペルオキシド、アルコール、アルデヒド及びケトンは有機酸及び水の形成の前駆体なので、これらの種を酸化媒体から除去すると、酸化反応速度及び環状イミド触媒の選択性と安定性を高める。実際に、データによると、100psig(790kPa)でNHPIによってsec-ブチルベンゼンの酸化を行なう場合、これらの軽質種及び水の99モル%より多くが反応器内に残るが、大気圧では、これらの種の95モル%より多くが酸化反応器から除去されることが分かる。
酸化反応の生成物には、所望のアルキルヒドロペルオキシドと共に未反応アルキル芳香族前駆体及び未反応環状イミド触媒が含まれる。未反応アルキル芳香族前駆体は蒸留で容易に除去され、酸化工程に再循環される。しかし、後述するように、未反応環状イミド触媒は、ヒドロペルオキシドをフェノールに開裂するために用いられる下流の混合金属酸化物触媒に対する有害物として作用し得る。さらに、環状イミドは費用がかかる傾向があるので、該未反応触媒を回収かつ再循環させるのが望ましい。従って、普通は酸化プロセスからの流出物を開裂工程に通す前に処理して未反応環状イミドのレベルを下げることが望ましいだろう。
【0031】
一実施形態では、酸化流出物の処理は、流出物を塩基、特に環状イミド触媒のpKa以上のpKb値を有する弱塩基の水溶液と接触させる工程を含み、これによって未反応イミド触媒が水相中に抽出され、前記酸化された炭化水素生成物を含み、かつレベルが減少した環状イミドを含む有機相を残す。一般的に、抽出は、有機相中のイミドのレベルを有機相の質量で100ppm未満、例えば50ppm未満、例えば10ppm未満に減らすように行なわれる。
弱塩基は水相中への抽出後にイミドの分解を触媒しにくいので、未反応イミド触媒の抽出では一般的に弱塩基の使用が望ましい。適切な弱塩基として、金属炭酸塩及び/又は金属炭酸水素塩、特にアルカリ金属炭酸塩及び/又はアルカリ金属炭酸水素塩、例えば炭酸ナトリウムが挙げられる。
環状イミド抽出工程で使用する条件を厳密に制御する必要はないが、一般的に約10℃〜約80℃、例えば約20℃〜約70℃の温度を含む。抽出時間は例えば約1分〜約30分、例えば約5分〜約10分であってよい。抽出工程で使用する塩基の量は、普通は未反応イミドに少なくとも等モル量の塩基を供給するのに十分な量、例えば未反応イミド1モル当たり1〜3モルの塩基である。一般に、抽出中は相を撹拌して相間の接触を最大にする。
塩基水溶液中への抽出後、水相を例えば酢酸で酸性にして未反応イミを沈殿させることによって、未反応環状イミドを容易に回収することができる。沈殿した未反応イミドを例えばろ過又は遠心分離によって水相から分離した後、所望により、酸化工程に再循環させてよい。
【0032】
別の実施形態では、酸化流出物の処理は、前記酸化炭化水素生成物に富み、かつ減少したか又はゼロレベルの環状イミドを含む処理流出物を生じさせるように、流出物を未反応イミド触媒の一部又は実質的に全ての除去に有効な固体吸着剤と接触させる工程を含む。この場合もやはり、吸着プロセスは、有機相中のイミドのレベルを有機相の100ppm未満、例えば50ppm未満、例えば10ppm未満に減らすように行なわれる。
適切な固体吸着剤は、金属炭酸塩及び/又は金属炭酸水素塩などの塩基特性を有するものであり、多孔性の担体、粘土、イオン交換樹脂及び金属酸化物、特に混合金属酸化物上に設けてよい。
環状イミド抽出工程で有効な吸着剤であるのに十分な塩基特性を有する金属酸化物は、これらの金属酸化物材料上のCO2及びNH3の化学吸着のモル比によって決定され得る。マイルドな酸であるCO2を用いて、被験金属酸化物上に存在する塩基サイトを滴定する。同様に、強塩基であるNH3を滴定して該材料上の酸性サイトを指し示す。該材料の表面積(多くの場合、金属酸化物の調製方法によってかなり影響を受ける)、化学吸着を試験する温度、及び化学吸着を試験する圧力などの多くの要因が化学吸着の実際量を決定する。本目的では、「塩基性」酸化物は、後述するように試験した場合、金属酸化物1g当たりのNH3の化学吸着に対する金属酸化物1g当たりのCO2の化学吸着のモル比が0.5より大きい、典型的に0.75より大きい、特に1.0より大きい酸化物と定義される。
【0033】
金属酸化物1g当たりのNH3の化学吸着に対する金属酸化物1g当たりのCO2の化学吸着のモル比を決定する試験は、周囲圧力でMettler TGA/SDTA 851熱重量分析システムを用いて行なわれる。金属酸化物サンプルを空気流中で約500℃(表1で指摘されている場合を除き)に約3時間(少なくとも一定のサンプル重量が得られるまで)か焼する。そして、空気流(ヘリウムを使用することもできる)中でサンプルの温度を化学吸着の所望温度に下げる。次に、サンプルをヘリウム流中で所望温度にて平衡させて秤量する。二酸化炭素の化学吸着を100℃で測定し、アンモニアの化学吸着を250℃で測定する。秤量後、一定重量が得られるまでサンプルをヘリウムと二酸化炭素又はアンモニアを含むガス混合物のいくつかのパルス(約12秒/パルス)にさらす。ガス混合物は、約10重量%の二酸化炭素又はアンモニアを含み、残りはヘリウムである。被験ガス混合物の各パルス後、金属酸化物サンプルに約3分間ヘリウム流を流す。各試験ではガス混合物の約20の個別パルスを使用する。か焼後の金属酸化物サンプル重量に基づいて金属酸化物1g当たりのmgを単位としたサンプルの重量の増加を用いて金属酸化物1g当たりに吸着したCO2又はNH3のモル数を決定する。
いくつかの代表的金属酸化物種について吸着質1g当たりのNH3の化学吸着に対するCO2の化学吸着のモル比下を表1に示す。
【0034】
表1
【0035】
環状イミド抽出工程で固体吸着剤として使うのに適した金属酸化物には、周期表の2族、3族、4族、ランタニド系列、又はアクチニド系列の金属の酸化物及び混合酸化物が含まれる。一実施形態では、吸着剤は、2種以上の金属酸化物、好ましくは1種の4族の金属酸化物と、2族、3族、ランタニド系列、及びアクチニド系列の金属酸化物から選択される1種以上の金属酸化物とを含む。酸化物は、種々の方法を用いて調製され得るが、一般的に溶液からの沈殿及び/又はか焼によって適切な前駆体の変換によって調製される。適切な前駆体としては、金属塩、例えばハロゲン化物、硫酸塩、リン酸塩、ハロゲン化物、硝酸塩、オキシ塩化物、アルコキシド及び酢酸塩が挙げられる。
一実施形態では、金属酸化物は、該金属の塩を水などの溶媒中に含む溶液を調製することによって製造される。結果として生じた溶液を次に、例えば沈殿試薬、典型的には水酸化ナトリウム又は水酸化アンモニウム等の塩基の添加によってのような、固体酸化物材料の沈殿を引き起こすのに十分な条件にさらす。沈殿中、溶液は一般的に200℃以下、例えば約0℃〜約200℃、例えば約20℃〜約100℃の範囲の温度で維持される。結果として生じるゲルを好ましくは次に少なくとも80℃、好ましくは少なくとも100℃の温度で、例えば10日間まで、例えば5日間まで、例えば3日間まで熱水処理する。結果として生じる材料を次に例えばろ過又は遠心分離によって回収し、洗浄し、乾燥させる。結果として生じる粒子材料を典型的には次に普通は酸化雰囲気内で、少なくとも400℃、例えば約400℃〜約800℃の温度にて、48時間まで、例えば約0.5時間〜約24時間、例えば約1時間〜約10時間か焼する。
【0036】
環状イミド抽出工程で2種以上の金属酸化物を使用する場合、それらを共沈殿させるか又は別々に沈殿させ、か焼固体粒子として含む加工のいずれの後の段階でも相互に混ぜ合わせてよい。
固体吸着剤として使うのに適したイオン交換樹脂には、酸性又は塩基性種を除去するために常用される当該樹脂、例えばAmberlystイオン交換樹脂がある。
固体吸着剤を用いた環状イミドの吸着に適した条件は、約10℃〜約130℃、例えば約20℃〜約80℃の温度、約1分〜約30分、例えば約5分〜約10分の時間を含む。
固体吸着剤による除去後、極性溶媒、例えばエタノール又はアセトンで吸着剤を洗浄することによって、未反応環状イミドを容易に回収することができる。エタノールとイミドの存在は、再循環触媒の酸化活性又は選択性に悪影響を与えないことが分かっているので、回収したイミドを次に、エタノールの事前除去をするか又はしないで酸化工程に再循環させることができる。
【0037】
(ヒドロペルオキシド開裂)
本方法のヒドロペルオキシド開裂工程は、普通は未反応アルキル芳香族前駆体の除去後及び必要に応じて流出物を前処理して環状イミドのレベルを100ppm未満に減らした後に、酸化工程からの流出物を混合金属酸化物触媒と接触させることによって行なわれる。特に、混合金属酸化物触媒は、元素周期表の3〜5族及び7〜14族の少なくとも1種の金属の酸化物、好都合には、元素周期表の4族の少なくとも1種の金属の酸化物と、元素周期表の6族の少なくとも1種の金属の酸化物とを含む。一実施形態では、触媒が酸化ジルコニウムと、モリブデン及び/又はタングステンの酸化物とを含む。
好都合には、触媒が元素周期表の8〜11族の少なくとも1種の金属の酸化物、例えば鉄及び/又は銅の酸化物をさらに含む。
混合金属酸化物触媒は、3〜5族及び7〜14族の少なくとも1種の金属のイオン源を含む第1溶液、例えば水溶液を、少なくとも1種の4族金属のイオン源を含む第2溶液、この場合もやはり例えば水溶液及び必要に応じて少なくとも1種の8〜11族金属のイオン源を含む第3溶液と混ぜ合わせることによって都合良く調製される。この混合は、液体媒体から固体として混合金属材料の共沈殿を引き起こすのに十分な条件下で起こり得る。或いは、3〜5族及び7〜14族の金属のイオン源、4族金属のイオン源及び必要に応じて8〜11族金属のイオン源を単一溶液中に混ぜ合わせてよい。この溶液を次に例えば該溶液に沈殿試薬の添加によって、固体混合酸化物材料の共沈殿を引き起こすのに十分な条件に供してよい。便宜上、沈殿は7より高いpHで行なわれる。例えば、沈殿試薬は水酸化ナトリウム又は水酸化アンモニウム等の塩基であってよい。
【0038】
沈殿中に液体媒体を維持する温度は一般的に約200℃未満、例えば約0℃〜約200℃の範囲である。沈殿に特有の温度範囲は約20℃〜約100℃である。結果として生じるゲルを好ましくは次に少なくとも80℃、好ましくは少なくとも100℃の温度で熱水処理する。熱水処理は、典型的に容器内で周囲圧力にて行なわれる。一実施形態では、ゲルを10日間まで、例えば5日間まで、例えば3日間まで熱水処理する。
混合金属酸化物に水和した前駆体を次に例えばろ過又は遠心分離によって回収し、洗浄し、乾燥させる。結果として生じる材料を次に、例えば酸化雰囲気内で少なくとも400℃、例えば少なくとも500℃、例えば約600℃〜約900℃、特に約650℃〜約800℃の温度でか焼して混合金属酸化物触媒を形成することができる。か焼時間は典型的に48時間まで、例えば約0.5〜24時間、例えば約1.0〜10時間である。一実施形態では、約700℃で約1〜約3時間か焼を行なう。
開裂反応は、ヒドロペルオキシドを液相内で約20℃〜約150℃、例えば約40℃〜約120℃の温度、及び/又は約50〜約2500kPa、例えば約100〜約1000kPaの圧力で及び/又はヒドロペルオキシドに基づいて約0.1〜約1000時間-1、好ましくは約1〜約50時間-1の液空間速度(LHSV)で混合金属酸化物触媒と接触させることによって都合良く作用する。開裂反応は、触媒蒸留装置内で好都合に行なわれる。
典型的に、開裂反応に不活性な有機溶媒、例えばメチルエチルケトン、フェノール、シクロヘキシルベンゼン、シクロヘキサノン及びsec-ブチルベンゼンでヒドロペルオキシドを希釈して熱除去を助ける。さらに好ましくは、極性溶媒の存在が酸化反応により残存している環状イミドによる混合金属酸化物触媒開裂触媒の被毒を軽減し得ることが分かっているので、開裂反応のため、アルキル芳香族ヒドロペルオキシドを極性溶媒、例えばアセトンに溶解させる。
【0039】
開裂反応への供給原料中の未反応環状イミドの存否に関係なく、混合金属酸化物は経時的にその活性を失い、ヒドロペルオキシドのフェノールへの変換度の減少をもたらす傾向があることが分かっている。しかし、触媒をアセトン等の極性溶媒で洗浄することによって触媒を定期的に復活させることで触媒の開裂活性を回復させ得ることが分かる。
本発明により酸化されるアルキル芳香族化合物がシクロヘキシルベンゼンである場合、酸化生成物はシクロヘキシルベンゼンヒドロペルオキシドを含み、開裂生成物はフェノール及びシクロヘキサノンを含む。開裂工程からの粗製シクロヘキサノン及び粗製フェノールをさらなる精製に供して、精製シクロヘキサノン及び精製フェノールを生成し得る。適切な精製プロセスには、限定するものではないが、シクロヘキサノン及びフェノールを他の種から分離するための一連の蒸留塔が含まれる。粗製又は精製シクロヘキサノン自体を脱水素に供して、それをフェノールに変換することができる。例えば、白金、ニッケル又はパラジウム等の触媒上で該脱水素を行なうことができる。
ここで、以下の非限定例を参照して本発明をさらに詳細に説明する。
【実施例】
【0040】
実施例1:Mo/Zr酸化物触媒の合成
500グラムのZrOCl2・8H2Oを3リットルの蒸留水に撹拌しながら溶かした。260グラムの濃水酸化アンモニウム、66グラムの(NH4)6Mo7O24・4H2O及び3リットルの蒸留水を含む別の溶液を調製した。両溶液を60℃に加熱し、ノズルミキシングを利用して加熱溶液を50ml/分の速度で混ぜ合わせた。濃水酸化アンモニウムを添加して最終複合物のpHを約9に調整した。結果として生じたスラリーを次にポリプロピレンボトルに取り、蒸し器(100℃)内に72時間置いた。生じた生成物をろ過で回収し、過剰の水で洗浄し、85℃で一晩乾燥させた。この触媒のサンプルを空気流内で3時間800℃にか焼した。このか焼触媒は、触媒中の総重量Mo及びZrの重量で16%のMoを含んでいた。
【0041】
実施例2:Cu/W/Zr酸化物触媒の合成
500グラムのZrOCl2・8H2Oを3.0リットルの蒸留水に撹拌しながら溶かし、この溶液に6.8グラムのCuSO4・5H2Oを加えた。260グラムの濃水酸化アンモニウム、54グラムの(NH4)6H2W12O40・xH2O及び3リットルの蒸留水を含む別の溶液を調製した。両溶液を40℃に加熱し、ノズルミキシングを利用して加熱溶液を50ml/分の速度で混ぜ合わせた。濃水酸化アンモニウムを添加して最終複合物のpHを約9に調整した。結果として生じたスラリーを次にポリプロピレンボトルに取り、蒸し器(100℃)内に72時間置いた。生じた生成物をろ過で回収し、過剰の水で洗浄し、85℃で一晩乾燥させた。この触媒のサンプルを空気流内で3時間700℃にか焼した。このか焼触媒は、触媒中の総重量W、Zr及びCuの重量で1%のCuと16%のWを含んでいた。
【0042】
実施例3:Fe/W/Zr酸化物触媒の合成
1000グラムのZrOCl2・8H2Oを3.0リットルの蒸留水に撹拌しながら溶かしてからこの溶液に15.2グラムのFeSO4・7H2Oを加えた。400グラムの濃水酸化アンモニウム、108グラムの(NH4)6H2W12O40・xH2O及び2940mlの蒸留水を含む別の溶液を調製した。両溶液を60℃に加熱し、ノズルミキシングを利用して加熱溶液を50ml/分の速度で混ぜ合わせた。濃水酸化アンモニウムを添加して最終複合物のpHを約9に調整した。結果として生じたスラリーを次にポリプロピレンボトルに取り、蒸し器(100℃)内に72時間置いた。生じた生成物をろ過で回収し、過剰の水で洗浄し、85℃で一晩乾燥させた。この触媒のサンプルを空気流内で3時間800℃にか焼した。このか焼触媒は73m2/gの表面積を有し、触媒中の総重量W、Zr及びFeの重量で1%のFeと16%のWを含んでいた。
【0043】
実施例4:W/Zr酸化物触媒
1000グラムのZrOCl2・8H2Oを3.0リットルの蒸留水に撹拌しながら溶かした。400グラムの濃水酸化アンモニウム、108グラムの(NH4)6H2W12O40・xH2O及び3.0リットルの蒸留水を含む別の溶液を調製した。両溶液を60℃に加熱し、ノズルミキシングを利用して加熱溶液を50ml/分の速度で混ぜ合わせた。濃水酸化アンモニウムを添加して最終複合物のpHを約9に調整した。結果として生じたスラリーを次にポリプロピレンボトルに取り、蒸し器(100℃)内に72時間置いた。生じた生成物をろ過で回収し、過剰の水で洗浄し、85℃で一晩乾燥させた。この触媒のサンプルを空気流内で3時間800℃にか焼した。このか焼触媒は73m2/gの表面積を有し、触媒中の総重量W及びZrの重量で16%のWを含んでいた。
【0044】
実施例5:Fe/W/Zr酸化物触媒
500グラムのZrOCl2・8H2Oを3.0リットルの蒸留水に撹拌しながら溶かしてからこの溶液に7.6グラムのFeSO4・7H2Oを加えた。260グラムの濃水酸化アンモニウム、54グラムの(NH4)6H2W12O40・xH2O及び2940mlの蒸留水を含む別の溶液を調製した。両溶液を60℃に加熱し、ノズルミキシングを利用して加熱溶液を50ml/分の速度で混ぜ合わせた。濃水酸化アンモニウムを添加して最終複合物のpHを約9に調整した。結果として生じたスラリーを次にポリプロピレンボトルに取り、蒸し器(100℃)内に72時間置いた。生じた生成物をろ過で回収し、過剰の水で洗浄し、85℃で一晩乾燥させた。この触媒のサンプルを空気流内で3時間800℃にか焼した。このか焼触媒は71m2/gの表面積を有し、触媒中の総重量W、Zr及びFeの重量で1%のFeと16%のWを含んでいた。
【0045】
1
実施例6:実施例1のMo/Zr酸化物触媒を用いたシクロヘキシルベンゼンヒドロペルオキシドの開裂
実施例1で調製した触媒を40〜80メッシュサイズにペレット化し、60〜80メッシュの砂と混合し(v/v 1:1)、1/4インチ(0.6cm)径のステンレススチール管形反応器に装填した。反応器を70℃で維持し、ISCOポンプを用いて30分間3cc/分で液体アセトンを反応器管に通した。アセトンの供給を止め、アセトン中シクロヘキシルベンゼンヒドロペルオキシドの5wt%溶液を別のISCOポンプを用いて0.45cc/分の速度で反応器に供給した。生成物をチルドノックアウトポット(chilled knock-out pot)に収集し、定期的にGC分析用サンプルを抽出した。実験の完了時、異なるシクロヘキシルベンゼンヒドロペルオキシドの流速を利用して滞留時間を変えた。
【0046】
実施例7:実施例2のCu/W/Zr酸化物触媒を用いたシクロヘキシルベンゼンヒドロペルオキシドの開裂
実施例2で調製した触媒を40〜80メッシュサイズにペレット化し、60〜80メッシュの砂と混合し(v/v 1:1)、1/4インチ(0.6cm)径のステンレススチール管形反応器に装填した。反応器を70℃で維持し、ISCOポンプを用いて30分間3cc/分で液体アセトンを反応器管に通した。アセトンの供給を止め、アセトン中シクロヘキシルベンゼンヒドロペルオキシドの5wt%溶液を別のISCOポンプを用いて0.45cc/分の速度で反応器に供給した。生成物をチルドノックアウトポットに収集し、定期的にGC分析用サンプルを抽出した。実験の完了時、異なるシクロヘキシルベンゼンヒドロペルオキシドの流速を利用して滞留時間を変えた。
【0047】
実施例8:実施例3のFe/W/Zr酸化物触媒を用いたシクロヘキシルベンゼンヒドロペルオキシドの開裂
実施例3で調製した触媒を40〜80メッシュサイズにペレット化し、60〜80メッシュの砂と混合し(v/v 1:1)、1/4インチ(0.6cm)径のステンレススチール管形反応器に装填した。反応器を70℃で維持し、ISCOポンプを用いて30分間3cc/分で液体アセトンを反応器管に通した。アセトンの供給を止め、アセトン中シクロヘキシルベンゼンヒドロペルオキシドの5wt%溶液を別のISCOポンプを用いて0.45cc/分の速度で反応器に供給した。生成物をチルドノックアウトポットに収集し、定期的にGC分析用サンプルを抽出した。実験の完了時、異なるシクロヘキシルベンゼンヒドロペルオキシドの流速を利用して滞留時間を変えた。
実施例6〜8の結果を下表2に示す。この表から、実施例1〜3の混合金属酸化物が、シクロヘキシルベンゼンヒドロペルオキシド(CHBHP)のフェノール及びシクロヘキサノンへの開裂にとって非常に活性かつ選択的な触媒であることが分かるだろう。
【0048】
表2
【0049】
実施例9:実施例5のFe/W/Zr酸化物触媒上のSec-ブチルベンゼンヒドロペルオキシドの開裂−供給原料中のNHPIを除去しない場合
実施例5で調製した触媒(1.5g)を20〜40メッシュサイズにペレット化し、60〜80メッシュの砂(1.5g)と混合し(v/v 1:1)、3/8インチ(1cm)径のステンレススチール管形反応器に装填した。反応器を90℃に加熱し、ISCOポンプを用いて135分間1cc/分で液体メチルエチルケトン(MEK)を反応器管を介して導入しながら90℃で維持した。MEK供給を止め、MEK中sec-ブチルベンゼンヒドロペルオキシド(SBBHP)の5wt%溶液を別のISCOポンプを用いて0.25cc/分の速度で反応器に導入した。sec-ブチルベンゼンヒドロペルオキシドは、ヒドロペルオキシドをMEKに溶かして開裂反応器に溶液を供給する前に触媒を除去せずに0.11wt%のN-ヒドロキシフタルイミド触媒の存在下でsec-ブチルベンゼンを酸化することで製造された。従って開裂工程に供給されたSBBHP/MEK溶液は、500ppmの未反応N-ヒドロキシフタルイミド触媒を含んでいた。開裂反応の生成物をチルドノックアウトポットに収集し、GC分析用サンプルを抽出した。結果を図1に示す。図1は、SBBHP転化率の減少によって示唆されるように、約95時間の操業中の時間後に触媒が失活し始めたことを示している。
【0050】
実施例10:実施例5のFe/W/Zr酸化物触媒上のSec-ブチルヒドロペルオキシド開裂−供給原料中のNHPI濃度の影響
空気撹拌機、温度計、冷却水冷凝縮器、及び窒素誘導器を備えた100mlの丸底三口フラスコにアセトン(39.5g,50ml)及び0.5gの実施例5の粉末触媒を添加した。アセトン及び触媒をアセトン還流下で加熱し(56℃)、かつ還流しながら、0.05wt%のNHPIを含む10cc(10.42g)の70wt.%のsec-ブチルベンゼンヒドロペルオキシド濃縮物をシリンジポンプを用いて20分で30cc/時間の速度にて添加した。1、3、6、10、20及び30分撹拌後にGCサンプルを収集した。GCサンプルは、ろ過して固体触媒を除去した後に明黄色液体だった。このプロセスを0.21wt%及び0.4wt%のNHPIを含有するsec-ブチルベンゼンヒドロペルオキシドサンプルで繰り返し、結果を図2に示す。
【0051】
実施例11:実施例5のFe/W/Zr酸化物触媒上のSec-ブチルヒドロペルオキシド開裂−供給原料からNHPIを除去した場合
空気撹拌機、温度計、冷却水冷凝縮器、及び窒素誘導器を備えた100mlの丸底三口フラスコにアセトン(39.5g,50ml)及び0.5gの実施例5の粉末触媒を添加した。アセトン及び触媒をアセトン還流下で加熱し(56℃)、かつ還流しながら、10cc(10.42g)の70wt.%のsec-ブチルベンゼンヒドロペルオキシド濃縮物(希Na2CO3溶液による苛性洗浄でNHPIを除去した)をシリンジポンプを用いて20分で30cc/時間の速度にて添加した。この添加時間中、反応器の温度が59℃に上昇した。1、3、6、10、20及び30分撹拌後にGCサンプルを収集した。GCサンプルは、ろ過して固体触媒を除去した後に明黄色液体だった。結果を図3に示す。図3から、sec-ブチルベンゼンヒドロペルオキシドからフェノール及びメチルエチルケトンに実質的に完全に変換したことが分かるだろう。
【0052】
実施例12:実施例4のW/Zr酸化物触媒上のSec-ブチルヒドロペルオキシド開裂−1500ppmのNHPIを含有するアセトン中5%のSBBHPの場合
実施例4で調製した触媒(1.5g)を40〜80メッシュサイズにペレット化し、60〜80メッシュの砂と混合し(v/v 1:1)、1/4インチ(0.6cm)径のステンレススチール管形反応器に装填した。反応器を70℃で維持し、ISCOポンプを用いて30分間3cc/分で液体アセトンを反応器管に通した。アセトン供給を止め、1500ppmのNHPIを含有するアセトン中5wt%のsec-ブチルベンゼンヒドロペルオキシド(SBBHP)溶液を別のISCOポンプを用いて0.5cc/分の速度で反応器に通した。生成物をチルドノックアウトポットに収集し、定期的にGC分析用サンプルを抽出した。結果を図4に示す。操業中の時間130時間後でさえ触媒失活の徴候が見られなかった。
【0053】
実施例13:実施例4のW/Zr酸化物触媒上のSec-ブチルヒドロペルオキシド開裂−NHPIを除去したSBB中5%のSBBHPの場合
実施例4で調製した触媒(1.5g)を40〜80メッシュサイズにペレット化し、60〜80メッシュの砂と混合し(v/v 1:1)、1/4インチ(0.6cm)径のステンレススチール管形反応器に装填した。反応器を70℃で維持し、ISCOポンプを用いて30分間3cc/分で液体アセトンを反応器管に通した。アセトン供給を止め、30ppm未満のNHPIを含有するsec-ブチルベンゼン(SBB)中5wt%のsec-ブチルベンゼンヒドロペルオキシド(SBBHP)溶液を別のISCOポンプを用いて0.25cc/分の速度で反応器に通した。生成物をチルドノックアウトポットに収集し、定期的にGC分析用サンプルを抽出した。結果を図3に示す。図3から、SBBHP転化率の減少によって示唆されるように、操業中の時間約70時間後に触媒が失活し始めたことが分かるだろう。
【0054】
実施例14:実施例4のW/Zr酸化物触媒上のSec-ブチルヒドロペルオキシド開裂−310ppmのNHPIを含有するSBB/アセトン中5%のSBBHPの場合
実施例4で調製した触媒(1.5g)を40〜80メッシュサイズにペレット化し、60〜80メッシュの砂と混合し(v/v 1:1)、1/4インチ(0.6cm)径のステンレススチール管形反応器に装填した。反応器を70℃で維持し、ISCOポンプを用いて30分間3cc/分で液体アセトンを反応器管に通した。アセトン供給を止め、310ppmのNHPIを含有する98/2(wt/wt)のSBB/アセトン中5wt%のSBBHP溶液を別のISCOポンプを用いて0.25cc/分の速度で反応器に通した。生成物をチルドノックアウトポットに収集し、定期的にGC分析用サンプルを抽出した。結果を図4に示す。図4から、操業中の時間約2時間後に触媒が失活し始めたことが分かるだろう。
【0055】
実施例15:失活した実施例14のW/Zr酸化物触媒のアセトン溶媒を用いた復活
実施例14の実験の一部として、触媒の復活を現場で行なった。SBBHP転化率が80%未満に低下したらすぐに、NHPIを含有するヒドロペルオキシド供給を止めた。反応器を70℃で維持しながら、アセトンを3cc/分で30分間触媒に通した。次にアセトン流を止めてヒドロペルオキシドの供給を再開した。SBBHP転化率が戻ってほとんど100%に増えたことによって示唆されるように、触媒活性が回復した。このような失活−復活のサイクルを3回行なうと(図4)、最初の触媒活性が各復活サイクル後に実質的に回復した。
本発明を特定の実施形態に関連して記載及び説明したが、当業者は、必ずしも本明細書で説明していない変形に本発明が役立つことを認めるだろう。従って、この理由のため、本発明の真の範囲を決定する目的のためには添付の特許請求の範囲をもっぱら参照すべきである。
【特許請求の範囲】
【請求項1】
フェノール又は置換フェノールの製造方法であって、下記一般式(I):
【化1】
(式中、R1及びR2は、それぞれ独立に、1〜4個の炭素原子を有するアルキル基を表し、但し、R1とR2が結合して4〜10個の炭素原子を有する環式基を形成していてもよく、前記環式基は任意に置換されていてもよく、かつR3は、水素、1〜4個の炭素原子を有する1つ以上のアルキル基又はシクロヘキシル基を表す)
を有するアルキル芳香族ヒドロペルオキシドを、元素周期表の3〜5族及び7〜14族の少なくとも1種の金属の酸化物と、元素周期表の6族の少なくとも1種の金属の酸化物とを含む第1触媒と接触させる工程を含む方法。
【請求項2】
前記第1触媒が、元素周期表の4族の少なくとも1種の金属の酸化物と、元素周期表の6族の少なくとも1種の金属の酸化物とを含む、請求項1に記載の方法。
【請求項3】
前記第1触媒が、酸化ジルコニウムと、モリブデン及び/又はタングステンの酸化物とを含む、請求項1又は2に記載の方法。
【請求項4】
前記第1触媒が、元素周期表の8〜11族の少なくとも1種の金属の酸化物をさらに含む、請求項2又は3に記載の方法。
【請求項5】
前記第1触媒が、鉄及び/又は銅の酸化物をさらに含む、請求項2〜4のいずれか1項に記載の方法。
【請求項6】
一般式(I)の前記アルキル芳香族ヒドロペルオキシドが、sec-ブチルベンゼンヒドロペルオキシド、p-メチル-sec-ブチルベンゼンヒドロペルオキシド、1,4-ジフェニルシクロヘキサンヒドロペルオキシド、sec-ペンチルベンゼンヒドロペルオキシド、sec-ヘキシルベンゼンヒドロペルオキシド、シクロペンチルベンゼンヒドロペルオキシド、シクロヘキシルベンゼンヒドロペルオキシド及びシクロオクチルベンゼンヒドロペルオキシドから選択され、好ましくはsec-ブチルベンゼンヒドロペルオキシド及びシクロヘキシルベンゼンヒドロペルオキシドから選択される、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記接触工程が、40℃〜120℃の温度、100〜1000kPaの圧力、及びヒドロペルオキシドに基づいて1〜50時間-1の液空間速度(LHSV)で行なわれる、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
一般式(I)を有する前記アルキル芳香族ヒドロペルオキシドが、下記一般式(II):
【化2】
(式中、R1、R2及びR3は、式(I)の定義どおりである)
を有するアルキル芳香族化合物を、下記一般式(III):
【化3】
(式中、R4及びR5は、それぞれ独立に、1〜20個の炭素原子を有するヒドロカルビル基及び置換ヒドロカルビル基、又は基SO3H、NH2、OH、及びNO2又は原子H、F、Cl、Br、及びIから選択され、但し、R4とR5が相互に共有結合によって連結されていてもよく;
Q1及びQ2は、それぞれ独立に、C、CH、N、CR6から選択され;
X及びZは、それぞれ独立に、C、S、CH2、N、P及び周期表4族の元素から選択され;
Yは、O又はOHであり;
kは、0、1、又は2であり;
lは、0、1、又は2であり;
mは、1〜3であり;かつ
R6は、R4について列挙したいずれの構成要素であってもよい)
を有する環状イミドを含む第2触媒の存在下で酸素含有ガスで酸化することによって製造される、請求項1〜7のいずれか1項に記載の方法。
【請求項9】
前記環状イミドが、下記一般式(IV):
【化4】
(式中、R7、R8、R9、及びR10は、それぞれ独立に、1〜20個の炭素原子を有するヒドロカルビル基及び置換ヒドロカルビル基、又は基SO3H、NH2、OH、及びNO2、又は原子H、F、Cl、Br、及びIから選択され;
X及びZは、それぞれ独立に、C、S、CH2、N、P及び周期表4族の元素から選択され;
Yは、O又はOHであり;
kは、0、1、又は2であり;かつ
lは、0、1、又は2である)
に従う、請求項8に記載の方法。
【請求項10】
前記環状イミドがN-ヒドロキシフタルイミドを含む、請求項8又は9に記載の方法。
【請求項11】
前記酸化工程が、前記アルキル芳香族ヒドロペルオキシド及び未反応環状イミド触媒を含む流出物を生成し、かつ該方法が、前記接触工程前に前記流出物を処理して、前記流出物中の前記未反応環状イミド触媒の少なくとも一部を除去する工程をさらに含む、請求項8〜10のいずれか1項に記載の方法。
【請求項12】
前記処理工程が、前記流出物を塩基の水溶液と接触させて、前記未反応イミド触媒の少なくとも一部を含む水相と、前記アルキル芳香族ヒドロペルオキシドを含む有機相とを生成する工程とを含む、請求項11に記載の方法。
【請求項13】
前記流出物を、前記環状イミドのpKa値以上のpKb値を有する弱塩基の水溶液と接触させる、請求項12に記載の方法。
【請求項14】
前記流出物を金属炭酸塩及び/又は金属炭酸水素塩の水溶液と接触させる、請求項12又は13に記載の方法。
【請求項15】
前記処理工程が、前記流出物を固体吸着剤と接触させる工程を含む、請求項11に記載の方法。
【請求項16】
前記固体吸着剤が、金属酸化物、金属炭酸塩及び/又は金属炭酸水素塩、粘土、及び/又はイオン交換樹脂を含む、請求項15に記載の方法。
【請求項17】
前記固体吸着剤が、金属酸化物1g当たりのNH3吸着に対する金属酸化物1g当たりのCO2吸着のモル比が0.5より大きい金属酸化物を含む、請求項15又は16に記載の方法。
【請求項18】
前記接触工程前に、前記アルキル芳香族ヒドロペルオキシドを極性溶媒、好ましくはアセトンに溶解させる、請求項1〜17のいずれか1項に記載の方法。
【請求項19】
前記第1触媒を極性溶媒、好ましくはアセトンで洗浄することによって、前記第1触媒を定期的に復活させる工程をさらに含む、請求項1〜18のいずれか1項に記載の方法。
【請求項20】
前記アルキル芳香族ヒドロペルオキシドがシクロヘキシルベンゼンヒドロペルオキシドであり、かつその変換生成物がフェノール及びシクロヘキサノンを含み、該方法が前記シクロヘキサノンを脱水素してさらなるフェノールを生成する工程をさらに含む、請求項1〜19のいずれか1項に記載の方法。
【請求項1】
フェノール又は置換フェノールの製造方法であって、下記一般式(I):
【化1】
(式中、R1及びR2は、それぞれ独立に、1〜4個の炭素原子を有するアルキル基を表し、但し、R1とR2が結合して4〜10個の炭素原子を有する環式基を形成していてもよく、前記環式基は任意に置換されていてもよく、かつR3は、水素、1〜4個の炭素原子を有する1つ以上のアルキル基又はシクロヘキシル基を表す)
を有するアルキル芳香族ヒドロペルオキシドを、元素周期表の3〜5族及び7〜14族の少なくとも1種の金属の酸化物と、元素周期表の6族の少なくとも1種の金属の酸化物とを含む第1触媒と接触させる工程を含む方法。
【請求項2】
前記第1触媒が、元素周期表の4族の少なくとも1種の金属の酸化物と、元素周期表の6族の少なくとも1種の金属の酸化物とを含む、請求項1に記載の方法。
【請求項3】
前記第1触媒が、酸化ジルコニウムと、モリブデン及び/又はタングステンの酸化物とを含む、請求項1又は2に記載の方法。
【請求項4】
前記第1触媒が、元素周期表の8〜11族の少なくとも1種の金属の酸化物をさらに含む、請求項2又は3に記載の方法。
【請求項5】
前記第1触媒が、鉄及び/又は銅の酸化物をさらに含む、請求項2〜4のいずれか1項に記載の方法。
【請求項6】
一般式(I)の前記アルキル芳香族ヒドロペルオキシドが、sec-ブチルベンゼンヒドロペルオキシド、p-メチル-sec-ブチルベンゼンヒドロペルオキシド、1,4-ジフェニルシクロヘキサンヒドロペルオキシド、sec-ペンチルベンゼンヒドロペルオキシド、sec-ヘキシルベンゼンヒドロペルオキシド、シクロペンチルベンゼンヒドロペルオキシド、シクロヘキシルベンゼンヒドロペルオキシド及びシクロオクチルベンゼンヒドロペルオキシドから選択され、好ましくはsec-ブチルベンゼンヒドロペルオキシド及びシクロヘキシルベンゼンヒドロペルオキシドから選択される、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記接触工程が、40℃〜120℃の温度、100〜1000kPaの圧力、及びヒドロペルオキシドに基づいて1〜50時間-1の液空間速度(LHSV)で行なわれる、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
一般式(I)を有する前記アルキル芳香族ヒドロペルオキシドが、下記一般式(II):
【化2】
(式中、R1、R2及びR3は、式(I)の定義どおりである)
を有するアルキル芳香族化合物を、下記一般式(III):
【化3】
(式中、R4及びR5は、それぞれ独立に、1〜20個の炭素原子を有するヒドロカルビル基及び置換ヒドロカルビル基、又は基SO3H、NH2、OH、及びNO2又は原子H、F、Cl、Br、及びIから選択され、但し、R4とR5が相互に共有結合によって連結されていてもよく;
Q1及びQ2は、それぞれ独立に、C、CH、N、CR6から選択され;
X及びZは、それぞれ独立に、C、S、CH2、N、P及び周期表4族の元素から選択され;
Yは、O又はOHであり;
kは、0、1、又は2であり;
lは、0、1、又は2であり;
mは、1〜3であり;かつ
R6は、R4について列挙したいずれの構成要素であってもよい)
を有する環状イミドを含む第2触媒の存在下で酸素含有ガスで酸化することによって製造される、請求項1〜7のいずれか1項に記載の方法。
【請求項9】
前記環状イミドが、下記一般式(IV):
【化4】
(式中、R7、R8、R9、及びR10は、それぞれ独立に、1〜20個の炭素原子を有するヒドロカルビル基及び置換ヒドロカルビル基、又は基SO3H、NH2、OH、及びNO2、又は原子H、F、Cl、Br、及びIから選択され;
X及びZは、それぞれ独立に、C、S、CH2、N、P及び周期表4族の元素から選択され;
Yは、O又はOHであり;
kは、0、1、又は2であり;かつ
lは、0、1、又は2である)
に従う、請求項8に記載の方法。
【請求項10】
前記環状イミドがN-ヒドロキシフタルイミドを含む、請求項8又は9に記載の方法。
【請求項11】
前記酸化工程が、前記アルキル芳香族ヒドロペルオキシド及び未反応環状イミド触媒を含む流出物を生成し、かつ該方法が、前記接触工程前に前記流出物を処理して、前記流出物中の前記未反応環状イミド触媒の少なくとも一部を除去する工程をさらに含む、請求項8〜10のいずれか1項に記載の方法。
【請求項12】
前記処理工程が、前記流出物を塩基の水溶液と接触させて、前記未反応イミド触媒の少なくとも一部を含む水相と、前記アルキル芳香族ヒドロペルオキシドを含む有機相とを生成する工程とを含む、請求項11に記載の方法。
【請求項13】
前記流出物を、前記環状イミドのpKa値以上のpKb値を有する弱塩基の水溶液と接触させる、請求項12に記載の方法。
【請求項14】
前記流出物を金属炭酸塩及び/又は金属炭酸水素塩の水溶液と接触させる、請求項12又は13に記載の方法。
【請求項15】
前記処理工程が、前記流出物を固体吸着剤と接触させる工程を含む、請求項11に記載の方法。
【請求項16】
前記固体吸着剤が、金属酸化物、金属炭酸塩及び/又は金属炭酸水素塩、粘土、及び/又はイオン交換樹脂を含む、請求項15に記載の方法。
【請求項17】
前記固体吸着剤が、金属酸化物1g当たりのNH3吸着に対する金属酸化物1g当たりのCO2吸着のモル比が0.5より大きい金属酸化物を含む、請求項15又は16に記載の方法。
【請求項18】
前記接触工程前に、前記アルキル芳香族ヒドロペルオキシドを極性溶媒、好ましくはアセトンに溶解させる、請求項1〜17のいずれか1項に記載の方法。
【請求項19】
前記第1触媒を極性溶媒、好ましくはアセトンで洗浄することによって、前記第1触媒を定期的に復活させる工程をさらに含む、請求項1〜18のいずれか1項に記載の方法。
【請求項20】
前記アルキル芳香族ヒドロペルオキシドがシクロヘキシルベンゼンヒドロペルオキシドであり、かつその変換生成物がフェノール及びシクロヘキサノンを含み、該方法が前記シクロヘキサノンを脱水素してさらなるフェノールを生成する工程をさらに含む、請求項1〜19のいずれか1項に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2012−505214(P2012−505214A)
【公表日】平成24年3月1日(2012.3.1)
【国際特許分類】
【出願番号】特願2011−531040(P2011−531040)
【出願日】平成21年7月14日(2009.7.14)
【国際出願番号】PCT/US2009/050489
【国際公開番号】WO2010/042261
【国際公開日】平成22年4月15日(2010.4.15)
【出願人】(599134676)エクソンモービル・ケミカル・パテンツ・インク (301)
【Fターム(参考)】
【公表日】平成24年3月1日(2012.3.1)
【国際特許分類】
【出願日】平成21年7月14日(2009.7.14)
【国際出願番号】PCT/US2009/050489
【国際公開番号】WO2010/042261
【国際公開日】平成22年4月15日(2010.4.15)
【出願人】(599134676)エクソンモービル・ケミカル・パテンツ・インク (301)
【Fターム(参考)】
[ Back to top ]