
フォン・ヴィレブランド病の処置のためのFVIII変異タンパク質
本発明は、N−末端アミンではない所定の位置にて1つ以上の生体適合性ポリマー、例えば、ポリエチレングリコールに共有結合している第VIII因子変異タンパク質の投与によるフォン・ヴィレブランド病の処置に関する。変異タンパク質複合体はFVIII凝血促進活性を維持しており、フォン・ヴィレブランド因子を欠いている対象において改良された薬物動態学的特性を有する。

【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2008年6月4日に出願された米国仮出願第61/058,795号(この内容は、その全体において出典明示により本明細書に包含させる)の利益を主張する。
【0002】
技術分野
本発明は、フォン・ヴィレブランド病(vWD)の処置のために有用である第VIII因子(FVIII)変異タンパク質およびその誘導体に関する。FVIII変異タンパク質は、定義された位置にて、1つ以上の生体適合性ポリマー、例えば、ポリエチレングリコールに結合することができる。加えて、治療目的のためのそれらの関連製剤、用量および投与の方法を提供する。これらの修飾されたFVIII変異体ならびに関連組成物および方法は、フォン・ヴィレブランド病に罹患している個体に対する注射頻度の減少および免疫原性応答の減少をもたらす処置選択の提供に有用である。
【背景技術】
【0003】
本発明の背景
vWDは、先天性または後天性疾患の種々の病因群を表す用語である。多数の型のvWDの原因は、他の特性と共に、凝血促進FVIIIに結合し、血液循環中の天然FVIIIの半減期を延長させる一連の多量体血漿糖タンパク質であるフォン・ヴィレブランド因子(vWF)の機能に属する(例えば、Federici, Haemophilia 10 (suppl 4):169,2004; Denis, et al., Thromb. Haemost. 99:271, 2008、参照)。正常なヒトにおいて、FVIIIの半減期は、vWFの非存在下において約8分であり、vWFの存在下において8時間である。
【0004】
軽度の型(1型)において、vWDは、ヒト集団において100人に1人もの割合で罹患されており、そして男性および女性に等しく罹患されている非常に一般的なものである。
【0005】
2型vWDは、vWDの重度の型であり得、5つのサブタイプ:2A、2B、2C、2Mおよび2Nにおいて知られている。これらの中で、2N型はFVIIIのvWFへの結合の欠乏により特徴付けられる。したがって、2N型vWDを有する患者において、FVIIIは迅速に分解され、循環レベルが低下する。2N型vWFは、FVIIIへの結合を害するホモ接合体または化合物ヘテロ接合体vWF変異体による。vWFとの複合体ではない遊離FVIIIは循環から迅速に除去されるため、2N型vWDは常染色体劣性型の血友病Aを装う。しかしながら、患者は一般的に、FVIIIレベルは減少しているが、正常レベルのvWF−抗原およびvWF−血小板GP1b結合に対するリストセチン補因子活性(vWF:RCo活性)を有する。
【0006】
エリックフォン・ヴィレブランドの型でフィンランド人ファミリーにおいて初めて発見された3型vWDは、vWFのホモ接合体欠乏または二重ヘテロ接合体欠乏である。3型vWDは、ノンセンス変異体またはvWFをコードする核酸へのわずかな挿入または欠失によるフレームシフトにより、完全またはほぼ完全なvWFの欠乏を引き起こす。ほとんどの場合において、vWF:RCoおよびvWF:Agは検出することができず、FVIIIレベルは非常に少ない。3型vWDを有する患者は、血友病のような関節血症および関節または関節腔出血を有し得る。
【0007】
後天性vWDは、通常、抗−vWF抗体の発生、流体せん断応力−誘導タンパク質分解または血小板もしくは他の細胞への結合の増加による、自己免疫性クリアランスにより引き起こされる。後天性vWD症候群は、少ないレベルのvWF−Ag、vWF:RcoおよびFVIIIを有する3型vWDのものと同様である。3型vWDおよび後天性vWD患者は、vWDの特徴である粘膜出血だけでなく血友病Aの特徴である軟組織、筋肉および関節出血も有する。
【0008】
血友病Aは、5000人の男性あたり1人の推定発現率である最も一般的な遺伝的凝固障害である。血液凝固の内因経路の重要な成分であるFVIIIの欠乏または構造欠陥により引き起こされる。血友病Aに対する現在の処置はヒトFVIIIの静脈内注射を含む。ヒトFVIIIは、約300kDの一本鎖分子として組換え的に生産されている。それは構造ドメインA1−A2−B−A3−C1−C2からなる(Thompson, Semin. Hematol. 29:11-22, 2003)。金属イオンにより互いに保持されている2つの鎖を有する前駆体産物は、ゴルジ装置において200kD(重鎖)および80kD(軽鎖)の2つのポリペプチド鎖に処理される(Kaufman, et al., J. Biol. Chem. 263:6352, 1988; Andersson, et al., Proc. Natl. Acad. Sci. 83:2979, 1986)。
【0009】
B−ドメイン欠失FVIII(BDD、90kDのA1−A2重鎖プラス80kDの軽鎖)が血友病Aに対する補充療法のとき有効であることが示されているため、FVIIIのB−ドメインは重要ではないようである。B−ドメイン欠失FVIII配列は、14個のアミノ酸のB−ドメインのほとんどが欠失している。
【0010】
血友病A患者は現在、要求に応じてFVIIIの静脈内投与または1週間に数回の予防的治療投与により処置される。予防処置のために、15−25IU/kg体重にてFVIIIを1週間に3回投与する。患者において定期的に必要である。ヒトにおいて半減期が短いため、FVIIIは頻繁に投与されなければならない。全長タンパク質に関して300kD以上の大型サイズにもかかわらず、FVIIIはヒトにおいてわずか約11時間の半減期である(Ewenstein, et al., Semin. Hematol. 41:1-16, 2004)。頻繁な静脈内注射の必要性は患者のコンプライアンスに対する巨大な障壁を作る。長い半減期を有し、投与頻度が少なくてよいFVIII産物が開発されることが患者にとってより都合がよい。さらに、必要とされる投与がより少なくなるため、半減期が増加したとき処置費用を減少させることができる。
【0011】
現在の治療のさらなる不利益は、約25−30%の患者がFVIII活性を阻害する抗体を産生することである(Saenko, et al., Haemophilia 8:111, 2002)。阻害抗体の主なエピトープは、A2ドメインの残基484−508、A3ドメインの残基1811−1818およびC2ドメイン内に位置する。抗体産生は補充療法のようなFVIIIの使用を妨げ、このグループの患者に高用量の組換え第VIIa因子(FVIIa)および免疫寛容療法でのさらに費用のかかる処置を探させる。
【0012】
以下の試験によって、阻害抗体のFVIIIエピトープを同定した。25個の阻害血漿サンプルの試験において、11個はトロンビン生産73kDの軽鎖フラグメントA3C1C2へ、4個はA2ドメインへ、10個は両方へ結合することが見出された(Fulcher, et al., Proc. Natl. Acad. Sci. 2:7728-32, 1985)。他の試験において、患者由来のA2ドメインインヒビターの8個中6個が、組換えA2ポリペプチドにより中和された(Scandella, et al., Blood 82:1767-75, 1993)。患者由来のインヒビターの9個中6個に対するエピトープがA2残基379538にマップされていた(Scandella, et al., Proc. Natl. Acad. Sci. 85:6152-6, 1988)。18個の重鎖インヒビターに対するエピトープは、A2ドメインの同じN−末端18.3kD領域に局在していた(Scandella, et al., Blood 74:1618-26, 1989)。
【0013】
ヒトA2ドメイン残基387−604を相同ブタ配列で置き換えることにより生産された活性な組換えハイブリッドヒト/ブタFVIII分子は、患者A2インヒビターに抵抗性であり(Lubin, et al., J. Biol. Chem. 269:8639-41, 1994)、A2への結合に対する患者A2インヒビターと競合するマウスモノクローナル抗体mAB413IgGに抵抗性であった(Scandella, et al., Thromb Haemost. 67:665-71, 1992)。mAB413IgGおよび4個の患者インヒビターがA2ドメイン残基484−508がブタのもので置換されているハイブリッドヒト/ブタFVIIIを抑制しないことを実験において示されるとき、このA2ドメインエピトープはさらにA2ドメイン残基484−508に局在している(Healey, et al., J. Biol. Chem. 270:14505-14509, 1995)。このハイブリッドFVIIIは、また、スクリーニングされた23個の患者血漿の少なくとも半分に抵抗性であった(Barrow, et al., Blood 95:564-568, 2000)。アラニン走査変異誘発によって、残基487が試験された全5個の患者インヒビターへの結合に対して重要であることを同定されたが、残基484、487、489および492はmAB413IgGとの相互作用に極めて重要であった(Lubin, J. Biol. Chem. 272:30191-30195, 1997)。R484A/R489A変異体ではなくR484A/R489A/P492A変異体を受けているマウスにおける阻害抗体力価は、対照ヒトBDDFVIIIを受けているマウスよりも有意に低かった(Parker, et al., Blood 104:704-710, 2004)。要するに、A2ドメインの484−508領域は、FVIII活性のインヒビターに対する結合部位であるようである。
【0014】
FVIIIへの免疫応答の発達に加えて、慣用の治療での他の問題は、FVIIIの短いインビボ半減期のため、頻繁な投与が必要であることである。循環からのFVIIIのクリアランスに対するメカニズムが試験されている。
【0015】
循環からのFVIIIクリアランスは、広いリガンド特異性を有する肝臓クリアランス受容体である低密度のリポタンパク質受容体−関連タンパク質(LRP)への特異的結合に部分的に起因していた(Oldenburg, et al., Haemophilia 10 Suppl 4:133-139, 2004)。最近、低密度のリポタンパク質(LDL)受容体は、また、例えば、血漿制御レベルのFVIIIにおいてLRPと共同することにより、FVIIIクリアランスにおける役割を果たすことを示された(Bovenschen, et al., Blood 106:906-910, 2005)。両方の相互作用は、細胞表面ヘパリン硫酸プロテオグリカン(HSPG)へ結合することにより促進される。マウスにおける血漿半減期は、LRPがブロックされるとき3.3倍またはLRPおよび細胞表面HSPGの両方がブロックされるとき5.5倍延長され得る(Sarafanov, et al., J. Biol. Chem. 276:11970-11979, 2001)。HSPGは、細胞表面上でFVIIIを濃縮し、それをLRPに与えると仮説が立てられる。FVIII上のLRP結合部位は、A2残基484−509(Saenko, et al., J. Biol. Chem. 274:37685-37692, 1999)、A3残基1811−1818(Bovenschen, et al., J. Biol. Chem. 278:9370-9377, 2003)およびC2ドメインにおけるエピトープ(Lenting, et al., J. Biol. Chem. 274:23734-23739, 1999)に局在していた。
【0016】
FVIIIは、また、プロテアーゼの作用により循環から除去される。この作用を理解するためには、FVIIIが血液凝固に関与するメカニズムを理解しなければならい。FVIIIは、vWFへ結合している重鎖および軽鎖のヘテロダイマーとして循環する。vWF結合は、FVIII残基1649−1689(Foster, et al., J. Biol. Chem. 263:5230-5234, 1998)およびC1の部分(Jacquemin, et al., Blood 96:958-965, 2000)およびC2ドメイン(Spiegel, et al., J. Biol. Chem. 279:53691-53698, 2004)と関与する。FVIIIは、残基372、740および1689後のペプチド結合を開裂させ、A1、A2およびA3−C1−C2ドメインのヘテロトリマーを生産するトロンビンにより活性化される(Pittman, et al., Proc. Natl. Acad. Sci. 276:12434-12439, 2001)。活性化時、FVIIIはvWFから解離し、リン脂質に結合することにより血小板の細胞表面に濃縮される。リン脂質結合はFVIII残基2199、2200、2251および2252と関与する(Gilbert et al., J. Biol. Chem. 277:6374-6381, 2002)。それは、FVIII残基558−565(Fay, et al., J. Biol. Chem. 269:20522-20527, 1994)および1811−1818(Lenting, et al., J. Biol. Chem. 271:1935-1940, 1996)との相互作用を介してFIXへ、ならびにFVIII残基349−372との相互作用を介してFXへ(Nogami, et al., J. Biol. Chem. 279:15763-15771, 2004)結合し、内因性凝固経路の重要な成分であるFXのFIX活性化のための補因子として作用する。活性化FVIII(FVIIIa)は、FVIII残基336および562後の開裂を介してプロテアーゼ活性化タンパク質C(APC)により部分的に不活性化される(Regan, et al., J. Biol. Chem. 271:3982-3987, 1996)。しかしながら、不活性化の主な決定因子は、A1およびA3−C1−C2からのA2ドメインの解離である(Fay, et al., J. Biol. Chem. 266:8957-8962, 1991)。
【0017】
タンパク質のインビボ半減期を増加させるために証明されている1つの方法はペグ化である。ペグ化は長鎖ポリエチレングリコール(PEG)分子のタンパク質または他の分子への共有結合である。PEGは直線形態または分岐形態であり、異なる特徴を有する異なる分子を生産することができる。ペプチドまたはタンパク質の半減期を増加させること以外では、ペグ化は、抗体発生を減少させ、プロテアーゼ消化からタンパク質を保護し、腎臓濾液から物質を排出し続けるために使用されている(Harris, et al., Clinical Pharmacokinetics 40:539-551, 2001)。加えて、ペグ化はタンパク質の全ての安定性および溶解度も増加させ得る。最後に、ペグ化タンパク質の持続血漿濃度は、薬物のトラフ/ピークレベルを低減し、したがって早期の時点にて生理学的レベルを越えるタンパク質を導入する必要性を排除することにより、有害な副作用の程度を減少させ得る。
【0018】
大型ポリマー、例えば、PEGおよびデキストランにて一級アミン(N−末端およびリシン)を標的化することによるFVIIIのランダム修飾は、種々の程度の成功で試みられている(WO94/15625、米国特許4970300、米国特許6048720)。1994年の特許出願(WO94/15625)において記載されている非常に劇的な改善は、2倍の費用がかかるが、全長FVIIIを50倍モル過剰のPEGと反応後に4倍の半減期改善として示すことである。WO2004/075923は、ランダム修飾を介して生産されるFVIIIおよびポリエチレングリコールの複合体を記載している。ランダムペグ化タンパク質、例えば、インターフェロン−アルファ(Kozlowski, et al, BioDrugs 15:419-429, 2001)は治療剤としてこれまでに承認されている。
【0019】
しかしながら、このランダム手段は、ヘテロ二量体FVIIIに関してさらなる問題がある。FVIIIは158個のリシン、2つのN−末端ならびに多数のヒスチジン、セリン、スレオニンおよびチロシンを含む何百もの可能なペグ化の位置を有し、これら全てが主に一級アミンを標的にする試薬にてペグ化され得る。例えば、ペグ化インターフェロンアルファ−2bに関する主な位置異性体は、ヒスチジンであることが示された(Wang, et al., Biochemistry 39:10634-10640, 2000)。さらに、全長FVIIIの不均一過程は、ペグ化産物におけるさらなる複雑さとなる出発物質の混合物をもたらし得る。FVIII上のペグ化の位置を制御しないさらなる欠点は、PEGが重要な活性部位、または部位付近に結合するとき、とりわけ2つ以上のPEGまたは1つの大きなPEGがFVIIIと複合体化するとき、起こりうる活性の減少である。ランダムペグ化は常に多量の多様なペグ化産物を生産するため、モノ−ペグ化産物のみを得るための精製は劇的に低い全収率となる。最後に、製品プロフィールにおける大きな不均一性は一貫性のある合成および特性化をそれぞれ非常に不可能なことにさせる。良い製造物は一貫性のある、よく特徴付けられた産物である必要があるため、産物の不均一性は商業化への障壁である。すべてのこれらの理由のため、FVIIIをペグ化するためのさらなる特定の方法が望まれる。
【0020】
種々の部位特異的タンパク質ペグ化戦略は最近の文献に要約されている(Kochendoerfer, et al., Curr. Opin. Chem. Biol. 9:555-560, 2005)。1つの手段は、化学合成または組換え発現による非天然アミノ酸のタンパク質への取り込み、次に非天然アミノ酸と特異的に反応するPEG誘導体の付加を含む。例えば、非天然アミノ酸は、天然タンパク質に見られないケト基を含むものであり得る。しかしながら、タンパク質の化学合成は、FVIIIほどの大きさのタンパク質に対して実行不可能である。現在のペプチド合成の限界は約50残基である。いくつかのペプチドは大きなポリペプチドを形成するために結合することができるが、B−ドメイン欠失FVIIIでさえ生産するために20以上の結合が必要であるため、これでは理想的な反応条件下でさえ1%未満の回収率となる。非天然アミノ酸を有するタンパク質の組換え発現は、今までのところ主に非哺乳動物の発現系に限定されている。この手段は、哺乳動物系において発現する必要がある大型かつ複雑なタンパク質、例えば、FVIIIに対して問題のあることが予期される。
【0021】
タンパク質の部位特異的ペグ化のための他の手段は、PEG−アルデヒドにてN−末端骨格アミンを標的化することによる。しかしながら、他のアミン基を越える特異性を達成するためのこのプロセス下にて必要とされる低いpHは、FVIIIの安定性のために必要であるほぼ中性のpH範囲と適合性ではない(Wang, et al., Intl. J. Pharmaceutics 259, pp. 1-15, 2003)。さらに、FVIIIのN−末端ペグ化は、この領域が血漿クリアランスに関連しないとき、血漿半減期の改善を引き起こさないであろう。
【0022】
WO90/12874は、システインを挿入するか、またはシステインを他のアミノ酸と置換し、次にスルフヒドリル反応基を有するリガンドを付加することによる、ヒトIL−3、顆粒球コロニー刺激因子およびエリスロポエチンポリペプチドの部位特異的修飾を記載している。リガンドは選択的にシステイン残基に結合する。FVIIIまたはその変異体の修飾は記載されていない。
【0023】
EP0319315は、vWF結合の減少をもたらすvWF結合部位の欠失または変化を有するFVIII変異タンパク質を記載している。EP0319315は、さらにこのような変異タンパク質を投与することによるvWF阻害活性によるFVIII欠乏症の軽減を記載している。
【0024】
Rottensteiner et al.は、ポリエチレングリコールまたはポリシアル酸との複合体を形成するためのFVIIIにおけるリシン残基のランダム化学修飾を記載している。Blood 110(11)、3150A (2007)。Rottensteiner et al.は、さらにランダム修飾されたFVIIIが2N型vWDに有用であり得ることを示唆している。
【0025】
上記理由のため、より良いインビボ作用持続期間および減少した免疫原性を有するが、機能活性を維持している改良されたFVIII変異体の必要性がある。さらに、このようなタンパク質は一貫した方法で均質製品として生産されることが望ましい。
【発明の概要】
【0026】
発明の概要
本発明の目的は、改良された薬物動態学的特性および治療特性を有する生体適合性ポリマー−複合体化機能性FVIIIポリペプチドの投与を含むvWDを処置する方法を提供することである。
【0027】
また、本発明の目的は、FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量の複合体を必要とする対象に投与することを含むvWDを処置するための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号(numerical position)により識別される特定のアミノ酸残基であり、N−末端アミンではない方法を提供することである。フォン・ヴィレブランド病はフォン・ヴィレブランド因子の欠乏および/または異常により特徴付けることができる。
【0028】
また、本発明のさらなる目的は、FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる複合体を製造することを含むvWDを処置するための薬物を製造するための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号により識別される特定のアミノ酸残基であり、N−末端アミンではない方法を提供することである。
【0029】
本発明のさらなる目的は、FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量のFVIIIのシステイン置換変異体を必要とする対象に投与することを含むvWDを処置するための方法であって、該変異体はFVIII配列におけるアミノ酸に対して置換されたシステイン残基を有することにより特徴付けられ、該置換は配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にして、システイン残基がFVIIIに存在しないアミノ酸位置にてシステイン残基をもたらし、該システイン付加変異体は該置換システイン残基へ共有結合している生体適合性ポリマーを有することによりさらに特徴付けられる方法を提供することである。
【0030】
本発明のさらなる目的は、改良された薬物動態学的特性を有する生体適合性ポリマー−複合体化Bドメイン欠失FVIIIタンパク質を必要とする対象に投与することを含むvWDを処置するための方法を提供することである。
【0031】
本発明のさらなる目的は、低密度のリポタンパク質受容体−関連タンパク質(LRP)、低密度のリポタンパク質(LDL)受容体、へパラン硫酸プロテオグリカン(HSPG)および/またはFVIIIに対する阻害抗体への減少した結合性を有する生体適合性ポリマー−複合体化機能性FVIIIポリペプチドを必要とする対象に投与することを含むvWDを処置するための方法を提供する。
【0032】
本発明のさらなる目的は、一貫した方法で均質製品として生産することができる、より良いインビボ作用持続期間および減少した免疫原性を有する治療有効量の改良されたFVIII変異体の必要とする対象への投与を含むvWDを処置するための方法を提供する。
【0033】
本発明の1つの局面において、FVIII凝血促進活性を有し、ポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含む治療有効量の複合体を必要とする対象に投与することを含むvWDを処置するための方法であって、該所定の位置はN−末端アミンではない方法を提供する。
【0034】
本発明のさらなる局面において、手術前にFVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量の複合体を対象に手術前に投与することを含み、それによって一過性の出血を弱める予防処置のための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号により識別される特定のアミノ酸残基であり、N−末端アミンではない方法を提供する。対象はvWD、例えば、3型vWDを有することができる。有利には、複合体を手術前24時間以内に、好ましくは8時間以内、より好ましくは手術0.5から2時間前に投与する。
【0035】
本発明のさらなる局面において、FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量の複合体を必要とする対象に投与することを含み、それによって一過性の出血を弱める外傷の処置のための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号により識別される特定のアミノ酸残基であり、N−末端アミンではない方法を提供する。対象は3型vWDを含むvWDを含むことができる。
【図面の簡単な説明】
【0036】
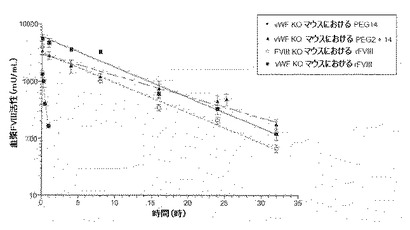
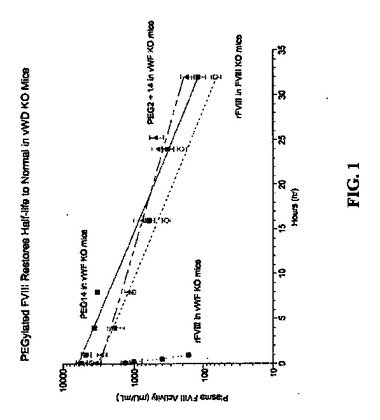
【図1】vWDノックアウト(KO)マウスにおけるFVIII半減期が正常に戻るペグ化FVIIIの効果。図は、i)vWF KOマウスへのrFVIIIの投与(黒丸)、ii)FVIII KOマウスへのrFVIIIの投与(白丸)、iii)vWF KOマウスへのペグ化rFVIIIの投与(64kD PEG14、黒四角)、およびiv)vWF KOマウスへの異なったペグ化rFVIIIの投与(64kD PEG2+14、黒三角)による血漿FVIII活性の経時変化を説明する。
【発明を実施するための形態】
【0037】
本発明の詳細な説明
本発明は、FVIII活性を有するポリペプチドがN−末端アミンではない所定の位置にて生体適合性ポリマーに共有結合することができ、実質的に凝固活性を維持している発見に基づく。さらに、これらのポリペプチド複合体は、改善された循環時間および減少した抗原性を有する。
【0038】
本発明は、さらに、所定の位置にて生体適合性ポリマーに共有結合したFVIII変異タンパク質が、修飾されていないFVIIIよりも、vWFを欠いている対象の循環においてより長い半減期の凝血促進活性を有する発見に基づく。本発明の複合体を使用するvWFを実質的に欠いている対象の処置は、FVIIIへのランダムポリマー結合またはN−末端での結合を有する先行技術の複合体の使用を越えて有利であり得る。部位特異的結合は生物学的活性のために必要である領域を避ける修飾を設計することで可能であり、それにより実質的なFVIII活性を維持する。また、FVIIIクリアランスに関連する部位における結合をブロックするようにポリマーを結合するように設計することが可能である。部位特異的結合は、また、ランダムポリマー結合により当分野で生産される不均一な複合体よりも、均一な生産物ができる。軽鎖のN−末端アミンにおける結合を避けることにより、本発明の複合体は、FVIIIポリペプチドの活性部位へのリガンドの結合による活性の喪失の可能性を避ける。
【0039】
定義
生体適合性ポリマー。生体適合性ポリマーは、ポリアルキレンオキシド、例えば、限定はしないが、ポリエチレングリコール(PEG)、デキストラン、コロミン酸または他の炭水化物ベースのポリマー、アミノ酸のポリマー、ビオチン誘導体、ポリビニルアルコール(PVA)、ポリカルボキシレート、ポリビニルピロリドン、ポリエチレン−コ−マレイン酸無水物、ポリスチレン−コ−リンゴ酸無水物、ポリオキサゾリン、ポリアクリロイルモルホリン、ヘパリン、アルブミン、セルロース、キトサンの加水分解物、デンプン、例えば、ヒドロキシエチル−デンプンおよびヒドロキシプロピル−デンプン、グリコーゲン、アガロースおよびそれらの誘導体、グアーガム、プルラン、イヌリン、キサンタンゴム、カラギーナン、ペクチン、アルギン酸加水分解物、他の生物ポリマーおよびそれらのあらゆる同等物を含む。ポリマーの例は、ポリエチレングリコール、例えば、メトキシポリエチレングリコール(mPEG)である。他の有用なポリアルキレングリコール化合物は、ポリプロピレングリコール(PPG)、ポリブチレングリコール(PBG)、PEG−グリシジルエーテル(Epox−PEG)、PEG−オキシカルボニルイミダゾール(CDI−PEG)、分岐ポリエチレングリコール、直鎖ポリエチレングリコール、フォーク状ポリエチレングリコールおよび多分岐または“超分岐”ポリエチレングリコール(星形−PEG)である。
【0040】
ポリエチレングリコール(PEG)。本明細書において使用される“PEG”および“ポリエチレングリコール”は相互互換であり、あらゆる水溶性ポリ(エチレンオキシド)を含む。一般的に、本発明において使用するためのPEGは、構造“−−(OCH2CH2)n−−”((n)は2から4000である)を含む。本明細書において使用されるPEGは、また、末端の酸素が置換されているか否かに依存して“−−CH2CH2−−O(CH2CH2O)n−−CH2CH2−−”および“−(OCH2CH2)nO−−”を含む。本明細書および本特許請求の範囲を通して、“PEG”なる用語が、例えば、ヒドロキシルまたはC1−20アルコキシ基に限定することなく、種々の末端または“末端キャップ”基を有する構造を含むことを認識すべきである。“PEG”なる用語は、また、−OCH 2CH2−−繰り返しサブユニットの大部分、すなわち、50%以上を含むポリマーを意味する。特定の形態に対して、PEGは非常に種々の分子量、ならびに構造または形状、例えば、分岐状、直鎖状、フォーク状および多分岐状であり得る。
【0041】
ペグ化。ペグ化はポリエチレングリコール(PEG)が分子、例えば、タンパク質に共有結合するプロセスである。
【0042】
活性化または活性な官能基。官能基、例えば、生体適合性ポリマーが活性化として記載されているとき、官能基は他の分子上の求電子または求核原子と容易に反応する。
【0043】
Bドメイン欠失FVIII(BDD)。本明細書において使用されるBDDは、FVIIIのB−ドメインの全14アミノ酸の欠失を含むアミノ酸配列を有することにより特徴付けられる。B−ドメインの最初の4アミノ酸(SFSQ、配列番号:2)はB−ドメインの最後の10残基(NPPVLKRHQR、配列番号:3)に結合する(Lind, et al, Eur. J. Biochem. 232:19-27, 1995)。本明細書において使用されるBDDは配列番号:4のアミノ酸配列を有する。BDDポリペプチドの例はWO2006/053299(これは出典明示により本明細書に包含させる)に記載されている。
【0044】
FVIII。血液凝固第VIII因子(FVIII)は肝臓により合成された糖タンパク質であり、血流に放出される。循環血液において、それはフォン・ヴィレブランド因子(vWF、FVIII−関連抗原としても知られている)に結合し、安定な複合体を形成する。トロンビンにより活性化されると、それは複合体から解離し、凝固カスケードにおける他の凝固因子と相互作用し、最後に血栓の形成を誘導する。ヒト全長FVIIIは配列番号:1のアミノ酸配列を有するが、対立遺伝子多型が起こりうる。
【0045】
機能性FVIIIポリペプチド。本明細書において使用される機能性FVIIIポリペプチドは、インビボまたはインビトロにて、例えば、血友病Aにより特徴付けられるヒトFVIII欠乏を修正することが可能である機能性ポリペプチドまたはポリペプチドの組合せを示す。FVIIIは、天然状態における多数の分解物または加工された形態を有する。これらは、本明細書において証明されるとおり一本鎖タンパク質である前駆体からタンパク質分解により得られる。機能性FVIIIポリペプチドは、このような一本鎖タンパク質を含み、ヒトFVIII欠乏を修正する生物学的活性を有するこれらの種々の分解産物も提供する。アレル変異体が恐らく存在する。機能性FVIIIポリペプチドは、ヒトFVIIIの機能性セグメントを含み、必須の特徴的なヒトFVIII機能活性が同じやり方で影響を受けずに維持されている限り、FVIIIの誘導体となるすべてのこのようなアレル変異体、グリコシル化バージョン、修飾物およびフラグメントを含む。このような必要な機能活性を有するFVIIIの誘導体は、本明細書に記載されている直接的インビトロ試験により容易に同定することができる。さらに、機能性FVIIIポリペプチドは、FIXa、カルシウムおよびリン脂質の存在下でのFXaへの因子X(FX)の変換を触媒すること、ならびに血友病A罹患個体由来の血漿における凝固障害を修正することが可能である。ヒトFVIIIアミノ酸配列およびその機能性領域の配列の公開から、DNAの制限酵素切断またはヒトFVIIIタンパク質のタンパク質分解もしくは他の分解を介して得ることができるフラグメントは、当業者に明らかである。機能性FVIIIポリペプチドの例は、WO2006/053299(これを出典明示により本明細書に包含させる)に記載されている。
【0046】
FIX。本明細書において使用されるFIXは、ヒト凝固因子IXとしても知られている凝固因子IX、または血漿トロンボプラスチン成分を意味する。
【0047】
FX。本明細書において使用されるFXは、名称、ヒト凝固因子Xおよび名称、スチュアートプラウナー因子としても知られている凝固因子Xを意味する。
【0048】
薬物動態学。“薬物動態学”(“PK”)は、体内の薬物の吸収、分配、代謝および除去の特性を述べるために使用される用語である。薬物の薬物動態学の改善は、薬物を治療剤としてインビボにてより有効にさせる特性、とりわけ体内の有効な持続時間の改善を意味する。
【0049】
変異タンパク質。変異タンパク質は、タンパク質またはポリペプチドにおける実験誘導変異の結果として生じる遺伝的に加工されたタンパク質である。
【0050】
タンパク質。本明細書において使用されるタンパク質およびポリペプチドは同義である。
【0051】
FVIIIクリアランス受容体。本明細書において使用されるFVIIIクリアランス受容体は、1つ以上の他の分子と結合または関連して、循環からFVIIIクリアランスをもたらす機能性FVIIIポリペプチド上の受容体領域を意味する。FVIIIクリアランス受容体は、LRP、LDL受容体および/またはHSPGに結合するFVIII分子の領域を含むが、これらに限定されない。
【0052】
すべての機能性FVIIIポリペプチドが所定の位置にて変異されていてよく、本発明の方法にしたがって生体適合性ポリマーへ所定の位置にて共有結合していてよいと想定する。有用なポリペプチドは、配列番号:1に示されるアミノ酸配列を有する全長FVIIIおよび配列番号:4に示されるアミノ酸配列を有するBDD FVIIIを含むが、これらに限定されない。
【0053】
本発明の複合体において使用される生体適合性ポリマーは、上記のあらゆるポリマーであり得る。生体適合性ポリマーは、薬物動態学において所望の改善を提供するように選択される。例えば、ポリマーの独自性、サイズおよび構造は、FVIII活性を有するポリペプチドの循環半減期を改善させるか、または活性において許容されない減少なくポリペプチドの抗原性を減少させるように選択される。ポリマーはPEGを含んでいてよく、一例として、PEGとして分子量の少なくとも50%を有し得る。1つの態様において、ポリマーは、末端キャップ分子、例えば、ヒドロキシル、アルコキシ、置換アルコキシ、アルケノキシ、置換アルケノキシ、アルキノキシ、置換アルキノキシ、アリールオキシおよび置換アリールオキシで末端を覆われているポリエチレングリコールである。他の態様において、ポリマーはメトキシポリエチレングリコールを含み得る。さらなる態様において、ポリマーは、3kDから100kDまたは5kDから64kDまたは5kDから43kDの範囲のサイズを有するメトキシポリエチレングリコールを含み得る。
【0054】
ポリマーは反応性部分を有し得る。例えば、1つの態様において、ポリマーは、機能性FVIIIポリペプチド上の遊離システインと反応し、共有結合を形成することができるスルフヒドリル反応性部分を有する。このようなスルフヒドリル反応性部分は、チオール、トリフラート、トレシラート、アジリジン、オキシラン、S−ピリジルまたはマレイミド部分を含む。1つの態様において、ポリマーは直鎖状であり、スルフヒドリル(例えば、メトキシ)に対して強く反応しない一方の末端に“キャップ”および他方の末端にスルフヒドリル反応性部分を有する。1つの態様において、複合体はPEG−マレイミドを含み、5kDから64kDの範囲のサイズを有する。
【0055】
有用な生体適合性ポリマーを選択するためのさらなるアドバイスは、以下の実施例において提供される。
【0056】
FVIII活性を有するポリペプチドをコードする核酸配列の部位特異的変異体は、当分野で既知のいずれかの方法により行われ得る。方法は、ポリマーの共有結合に対して選択される部位にシステインコドンを導入するための変異誘発を含む。これは、市販されている部位特異的変異誘発キット、例えば、Stratagene cQuickChangeTM II 部位特異的変異誘発キット、Clontech Transformer 部位特異的変異誘発キットナンバーK1600−1、Invitrogen GenTaylor 部位特異的変異誘発システムナンバー12397014、Promega Altered Sites II インビトロ変異誘発システムキットナンバーQ6210またはTakara Mirus Bio LA PCR 変異誘発キットナンバーTAK RR016を使用して達成され得る。
【0057】
本発明の複合体は、最初に機能性FVIIIポリペプチドの表面上の1つ以上のアミノ酸に対するコドンをシステインに対するコドンで置換し、組換え発現系においてシステイン変異タンパク質を生産し、変異タンパク質をシステイン−特異的ポリマー試薬と反応させ、そして、変異タンパク質を精製することにより製造され得る。
【0058】
このシステムにおいて、システイン部位へのポリマーの付加はポリマー上のマレイミド活性官能基を介して成し遂げることができる。この技術の例は以下に提供されている。使用されるスルフヒドリル反応性ポリマーの量は、誘導体化されるシステインのモル量と少なくとも等モルであり、好ましくは過剰に存在する。例えば、少なくとも5倍モル過剰のスルフヒドリル反応性ポリマーが使用されるか、または少なくとも10倍過剰のこのようなポリマーが使用される。共有結合のために有用な他の条件は当業者に知られている。
【0059】
以下の実施例において、変異タンパク質は当分野の慣用の様式において名付けられる。変異体を名付けるための慣例は、配列番号:1において提供されている全長FVIIIの成熟体に対するアミノ酸配列に基づく。分泌タンパク質として、FVIIIは翻訳プロセス中にタンパク質分解的に開裂されるシグナル配列を含む。19のアミノ酸シグナル配列の除去後、分泌されたFVIII産物の第1のアミノ酸はアラニンである。
【0060】
一般的に、かつ本明細書に使用されているとおり、BDD FVIIIにおける変異アミノ酸に言及しているとき、変異アミノ酸は全長FVIII配列における位置により示される。例えば、以下に記載されているPEG6変異タンパク質は、全長配列における1808と類似である位置にてリシン(K)をシステイン(C)に変化しているとき、K1808Cと示される。
【0061】
ポリマーの共有結合に関する所定の位置は、FVIII活性に関連していないポリペプチドの表面に暴露されている部位から選択されるのが最も良い。このような部位は、また、FVIIIが循環から不活性化されるか、除外されるメカニズムに関連することが知られている部位から選択されるのが最も良い。これらの部位の選択は以下に詳細に記載されている。好ましい部位は、(a)低密度のリポタンパク質受容体関連タンパク質、(b)ヘパリン硫酸プロテオグリカン、(c)低密度のリポタンパク質受容体、および/または(d)FVIII阻害抗体に対する結合部位中またはその付近のアミノ酸残基を含む。“結合部位中またはその付近”とは、生体適合性ポリマーの共有結合が結合部位の立体障害をもたらすであろう、結合部位に非常に近接している残基を意味する。このような部位は、例えば、結合部位の20Å以内であると予期される。
【0062】
本発明の1つの態様において、生体適合性ポリマーは、(a)FVIIIを分解することができるプロテアーゼに対する結合部位、および/または(b)FVIII阻害抗体に対する結合部位中またはその付近のアミノ酸残基にて機能性FVIIIポリペプチドに共有結合している。プロテアーゼは活性化タンパク質C(APC)であり得る。他の態様において、低密度のリポタンパク質受容体関連タンパク質のポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように(例えば、2分の1未満)、生体適合性ポリマーが機能性FVIIIポリペプチド上の所定の位置にて共有結合している。1つの態様において、生体適合性ポリマーは、ヘパリン硫酸プロテオグリカンのポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように(例えば、2分の1未満)、機能性FVIIIポリペプチド上の所定の位置にて共有結合している。さらなる態様において、生体適合性ポリマーは、FVIII阻害抗体のポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように(例えば、複合体化していないときのポリペプチドへの結合の2分の1未満)、機能性FVIIIポリペプチド上の所定の位置にて共有結合している。他の態様において、生体適合性ポリマーは、低密度のリポタンパク質受容体のポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように(例えば、2分の1未満)、機能性FVIIIポリペプチド上の所定の位置にて共有結合している。他の態様において、生体適合性ポリマーは、ポリペプチドが複合体化していないときよりも少ないポリペプチドを血漿プロテアーゼが分解するように、機能性FVIIIポリペプチド上の所定の位置にて共有結合している。さらなる態様において、血漿プロテアーゼによるポリペプチドの分解は、同じ条件下で同じ期間にわたって測定したとき、複合体化していないときのポリペプチドの分解の2分の1未満である。
【0063】
FVIIIに対するLRP、LDL受容体またはHSPG結合親和性は、表面プラズモン共鳴技術(Biacore)を使用して決定することができる。例えば、FVIIIをBiacoreTMチップに直接的に、またはFVIII抗体を介して間接的に被覆し、種々の濃度のLRPをチップ上を通過させ、相互作用のオン速度(on-rate)およびオフ速度(off-rate)両方を測定することができる(Bovenschen, et al., J. Biol. Chem. 278:9370-9377, 2003)。2つの速度の比率は親和性の基準を与える。ペグ化時に親和性の2倍、5倍、10倍または30倍の減少が望ましい。
【0064】
プロテアーゼAPCによるFVIIIの分解は当業者に知られている方法により測定することができる。
【0065】
1つの態様において、方法はFVIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118および2284のうち1つ以上にて、ポリペプチドへ共有結合している生体適合性ポリマーを投与することを含む。他の態様において、生体適合性ポリマーが、FVIIIアミノ酸位置377、378、468、491、504、556、1795、1796、1803、1804、1808、1810、1864、1903、1911および2284のうち1つ以上にて、ポリペプチドへ共有結合しており、さらに(1)複合体の低密度のリポタンパク質受容体関連タンパク質への結合が非複合体化ポリペプチドの低密度のリポタンパク質受容体関連タンパク質への結合より少ないか;(2)複合体の低密度のリポタンパク質受容体への結合が非複合体化ポリペプチドの低密度のリポタンパク質受容体への結合より少ないか;または(3)複合体の低密度のリポタンパク質受容体関連タンパク質および低密度のリポタンパク質受容体両方への結合が非複合体化ポリペプチドの低密度のリポタンパク質受容体関連タンパク質および低密度のリポタンパク質受容体への結合への結合より少ない。
【0066】
さらなる態様において、方法は、FVIIIアミノ酸位置377、378、468、491、504、556および711のうち1つ以上にてポリペプチドへ共有結合している生体適合性ポリマーを投与することを含み、複合体のヘパリン硫酸プロテオグリカンへの結合が非複合体化ポリペプチドのヘパリン硫酸プロテオグリカンへの結合より少ない。さらなる態様において、生体適合性ポリマーは、FVIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118および2284のうち1つ以上にてポリペプチドへ共有結合しており、複合体は非複合体化ポリペプチドよりFVIII阻害抗体への結合が少ない。さらなる態様において、生体適合性ポリマーは、FVIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118および2284のうち1つ以上にて、例えば、位置377、378、468、491、504、556および711のうち1つ以上にてポリペプチドへ共有結合しており、複合体は非複合体化ポリペプチドよりもFVIIIを分解できる血漿プロテアーゼから分解されない。血漿プロテアーゼは活性化タンパク質Cであり得る。
【0067】
さらなる態様において、方法は、アミノ酸位置129、491、1804および/または1808にてB−ドメイン欠失FVIIIに共有結合している生体適合性ポリマーを投与することを含む。さらなる態様において、生体適合性ポリマーはFVIIIアミノ酸位置1804にてポリペプチドに結合しており、ポリエチレングリコールを含む。生体適合性ポリマー結合のための1つ以上の所定の位置は部位特異的システイン変異により制御され得る。
【0068】
機能性FVIIIポリペプチド上の1つ以上、例えば、1つまたは2つの位置は、ポリマー結合に関する所定の位置であり得る。特定の態様において、ポリペプチドはモノ−ペグ化またはジ−ペグ化される。
【0069】
本発明は、また、所定の位置にてコード配列をシステイン残基と置き換えるように機能性FVIIIポリペプチドをコードする核酸配列を変異させ;変異核酸配列を発現させ、システイン増大変異タンパク質を生産し;変異タンパク質を精製し;複合体が形成されるように、実質的に還元されたシステイン残基のみにてポリペプチドと反応するように活性化されている生体適合性ポリマーと変異タンパク質を反応させ;そして、複合体を精製することを含む、複合体の製造のための方法に関する。他の態様において、本発明は、(a)部位特異的FVIII変異タンパク質を発現させ、ここで、変異タンパク質はFVIII変異タンパク質の暴露されている表面上のアミノ酸残基の代わりにシステインを有し、システインはキャップされている;(b)システイン変異タンパク質を穏やかに還元させ、キャップを放出する条件下で、システイン変異タンパク質を還元剤と接触させ;(c)システイン変異タンパク質からキャップおよび還元剤を除去し;そして(d)還元剤の除去から少なくとも約5分、少なくとも15分、少なくとも30分後、ペグ化FVIII変異タンパク質が生産される条件下で、システイン変異タンパク質をスルフヒドリル結合部分を含むPEGで処理することを含む、FVIII変異タンパク質の部位特異的ペグ化のための方法を提供する。PEGのスルフヒドリル結合部分は、チオール、トリフラート、トレシラート、アジリジン、オキシラン、S−ピリジルおよびマレイミド部分からなる群から選択される。
【0070】
本発明は、また、治療有効量本発明の複合体および薬学的に許容されるアジュバントを含む非経口投与用医薬組成物を含む。薬学的に許容されるアジュバントは、活性成分に加えられ得る物質であり、製造物を製剤化または安定化するための助けとなり、患者に有意な有害な毒性効果を引き起こさない。このようなアジュバントの例は当業者に知られており、水、糖、例えば、マルトースまたはスクロース、アルブミン、塩などを含む。他のアジュバントは、例えば、Remington’s Pharmaceutical Sciences by E. W. Martinに記載されている。このような組成物は、宿主への有効な投与のために適当な薬学的に許容される組成物を製造するために、有効量の本複合体を適当な量のビヒクルと共に含む。例えば、複合体は、出血の発現の重症度で変化し得る用量で血友病Aに罹患している対象に非経腸的に投与され得る。血友病Aに対して静脈内に投与される平均用量は、術前適応に対してキログラムあたり40単位、軽度の出血に対してキログラムあたり15から20単位および維持投与のために8時間にわたっての投与に対してキログラムあたり20から40単位の範囲である。vWDの処置に対して、用量はキログラムあたり25−400IUであり得る。vWDに対する他の有効な用量は、25−50、25−100、50−75、50−100、100−200、150−200、200−300、250−300、300−350、300−400、25−250、100−400および200−400IU/kgである。低用量が予防のために有用であり、高用量がFVIIIインヒビターを有する患者における免疫寛容誘導のために有用である。
【0071】
1つの態様において、本発明の方法は、1つ以上の表面BDDアミノ酸をシステインで置換し、哺乳動物発現系においてシステイン変異タンパク質を生産し、培養培地由来のシステインにより発現中にキャップされているシステインを還元し、BDDジスルフィドが再形成できるように還元剤を除去し、そしてシステイン−特異的生体適合性ポリマー試薬、例えば、PEG−マレイミドと反応させることを含む。このような試薬の例は、例えば、Nektar カタログナンバー 2D2M0H01 mPEG-MAL MW 5,000 Da、2D2M0P01 mPEG-MAL MW 20 kD、2D3X0P01 mPEG2-MAL MW 40 kDの下にNektar Therapeutics of San Carlos, CAから入手できる5、22または43kD、または、NOF カタログナンバー Sunbright ME-120MAおよびSunbright ME-300MAの下にNOF Corporation, Tokyo, Japanから入手できる12または33kDのPEGサイズを有するPEG−マレイミドである。ペグ化生産物は、未反応PEGを除去するためにイオン交換クロマトグラフィーを使用して、未反応BDDを除去するためにサイズ排除クロマトグラフィーを使用して精製される。この方法は、同定するために、および、FVIIIとの好ましくない相互作用、例えば、受容体−介在クリアランス、阻害抗体結合およびタンパク質分解酵素による分解から選択的に保護するために使用することができる。PEG試薬が、我々の研究室において6kDとして分析されたものが5kDとしてNektarまたはNOFにより提供され、ならびに、同様にPEG試薬が、我々の研究室において22kDとして分析されたものが直鎖20kDとして提供され、43kDとして分析されたものが40kDとして提供され、および64kDとして分析されたものが60kDとして提供されたことを、我々は指摘する。混乱を避けるため、我々は、5kD PEG(我々は製造業者がそれを確認したとおりの5kDとして報告する)を除いて、本明細書の記載において我々の研究室において分析されているとおりの分子量を使用する。
【0072】
BDDの位置491および1808(上記)でのシステイン変異体に加えて、位置487、496、504、468、1810、1812、1813、1815、1795、1796、1803および1804を、可能性があるペグ化時にLRP結合の妨害を可能にするために、システインに変異させた。また、位置377、378および556を、ペグ化時にLRPおよびHSPG結合両方の妨害を可能にするために、システインに変異させた。これらの位置における大きなPEG(>40kD)での部位特異的ペグ化が、天然のグリコシル化部位(41、239および2118)およびLRP結合部位におけるペグ化と共に、BDDの表面を完全に覆い、BDDに関する新規クリアランスメカニズムを同定することができるように、位置81、129、422、523、570、1864、1911、2091および2284をBDDにおいて等間隔に選択した。
【0073】
1つの態様において、細胞培養培地は、ジスルフィド結合を形成することにより変異タンパク質のシステイン残基を“キャップ”するシステインを含む。複合体の製造において、組換え系において生産されるシステイン変異タンパク質は培地由来のシステインでキャップされ、このキャップはシステイン−特異的ポリマー試薬を加える前にキャップを放出する穏やかな還元により除去される。FVIIIの部位特異的変異体の当分野で既知の他の方法も、当業者に明らかであるため使用され得る。
【0074】
FVIIIの構造活性相関分析
FVIIIおよびBDD FVIIIは、生物学的反応に関連する多数の異なる部位を有する非常に大きな複合体分子である。薬物動態学的特性を改善するためにそれらを共有結合により修飾する以前の試みは、混同した結果であった。分子を特異的に変異させ、次に部位特異的方法においてポリマーを付加させることができることは、驚くべきことであった。さらに、非特異的付加および活性の減少を引き起こすこれまでのポリマー複合体における問題を考慮すると、改良された薬物動態学的特性および維持されている活性の結果も驚く
【0075】
1つの態様において、本発明は、システイン−特異的リガンド、例えば、PEG−マレイミドを使用する部位特異的変異誘発に関する。非変異BDDは、PEG−マレイミドと反応するために利用できるシステインを有さないため、変異システイン位置のみがペグ化の位置である。より具体的には、BDD FVIIIは19個のシステインを有し、このうち16個はジスルフィドを形成し、他の3個は遊離システインである(McMullen, et al., Protein Sci. 4:740-746, 1995)。BDDの構造モデルは、全3個の遊離システインが覆い隠されていることを示唆する(Stoliova-McPhie, et al., Blood 99:1215-1223, 2002)。酸化されているシステインはPEGマレイミドによりペグ化されないため、BDDにおいてジスルフィドを形成する16個のシステインは、最初に還元されることなくペグ化されない。BDDの構造モデルに基づいて、BDDにおける3個の遊離システインは、最初にタンパク質を変性させ、これらのシステインがPEG試薬に暴露されることなくペグ化され得ない。したがって、機能を破壊するようなBDD構造が劇的に変化することなく、天然のシステイン残基にてペグ化によりBDDの特異的なペグ化が達成されることは実行可能であると思えない。
【0076】
全長FVIIIのBドメインにおける4個のシステインのレドックス状態は未知である。Bドメインにおける4個のシステインのペグ化は、それらがジスルフィドを形成しておらず、表面に暴露されているとき可能であり得る。しかしながら、全長FVIIIおよびBDDは同様の薬物動態学(PK)プロフィールおよび同様のインビボ半減期(Gruppo, et al., Haemophilia 9:251-260, 2003)を有するため、PEGが偶然に非Bドメイン領域も保護しないかぎり、Bドメインペグ化が血漿半減期の改善をもたらすことは起こりそうにない。
【0077】
FVIII活性を維持し、薬物動態学を改善するポリマー結合に関するFVIII活性を有するポリペプチドにおける所定の位置を決定するために、以下の指針をBDD FVIIIに基づいて示す。修飾はクリアランス、不活性化および免疫原性メカニズム、例えば、LRP、HSPG、APCおよび阻害抗体結合部位を標的とすべきである。Stoilova-McPhie, et al., (Blood 99:1215-23, 2002)はBDDの構造を示す。例えば、半減期を延長するために、1個のPEGをA2残基484−509およびA3残基1811−1818におけるLRP結合部位、または部位付近における特定の部位に導入することができる。これらの部位への巨大なPEGの導入は、FVIIIがLRPに結合する能力を破壊し、循環からのFVIIIのクリアランスを減少させるはずである。活性に有意に影響せずに半減期を延長するために、PEGを残基1648に導入することができるとも思われ、それは全長分子中のBドメインとA3ドメインの連結部分にあり、BDDではA2及びA3ドメインの間の14−アミノ酸リンカー(linker)中にある。
【0078】
ペグ化の特異性は、組換えDNA変異誘発技術を使用してA2またはA3ドメインへ単一システイン残基を設計し、次にシステイン−特異的PEG試薬、例えば、PEG−マレイミドにて導入されたシステインの部位特異的ペグ化により達成することができる。484−509および1811−1818におけるペグ化のさらなる利点は、これらの2つのエピトープが患者における阻害抗原部位の3つの主なクラスの2つを示すことである。循環半減期の改善および免疫原性応答の減少における最大の効果を達成するために、A2およびA3 LRP結合部位の両方をペグ化し、ジペグ化生成物を得ることができる。1811−1818領域はFIX結合にも関連しているため、この領域内のペグ化は活性の有意な喪失を引き起こし得ることに注意すべきである。また、558−565内の部位特異的ペグ化はHSPG結合を破壊し、この領域がFIXに結合するような活性を減少させ得る。
【0079】
FVIIIの新規クリアランスメカニズムを確認するために、さらなる表面部位をペグ化することができる。A2ドメインは活性化時にFVIIIから解離され、小さなサイズであるため、FVIII分子の残りよりも恐らく速く循環から除去されるという点において、A2ドメインのペグ化はさらなる利点を提供し得る。他方では、ペグ化A2は、腎臓クリアランスから逃れるために十分な大きさであり、FVIIIの残りに匹敵する血漿半減期を有し得、したがってインビボにて活性化FVIIIを再構成することができる。
【0080】
A2およびA3領域におけるペグ化部位の同定。推定A2 LRP結合領域、または領域付近における5つの位置(PEG1−5位置に対応するY487、L491、K496、L504およびQ468)を、高い表面暴露およびそれらのCαからCβへの軌道の外側への向きに基づき、部位特異的ペグ化のための例として選択した。さらに、これらの残基は分子の三次元構造において互いからほぼ等距離であり、それらは一緒になってこの領域全体を表す。推定A3 LRP結合領域、または領域付近における8つの位置(PEG6−14に対応する1808、1810、1812、1813、1815、1795、1796、1803、1804)を部位特異的ペグ化のための例として選択した。PEG6(K1808)は1811−1818および1810における天然のN結合グリコシル化部位に隣接している。位置1810(PEG7)におけるペグ化は糖をPEGで置換されるであろう。PEG8位置T1812における変異体も、グリコシル化部位を破壊するであろう。PEG9位置(K1813)は内側に向いていると予測されたが、構造モデルが正しくない場合にそれを選択した。PEG10(Y1815)はLRP結合ループ内の巨大な疎水性アミノ酸であり、疎水性アミノ酸は一般的にタンパク質−タンパク質相互作用の中心に見られるため、重要な相互作用残基であり得る。1811−1818領域はLRPおよびFIX結合の両方に含まれると報告されているため、このループ内におけるペグ化はおそらく活性の減少をもたらすと考えられた。したがって、PEG11−PEG14(1795、1796、1803、1804)は、異なるPEGサイズによってLRPおよびFIX結合を解離することができるように、1811−1818ループ付近であるがループ内にないように設計した。
【0081】
LRP結合部位の両方を同時にブロックするために、例えば、PEG2およびPEG6位置におけるダブルペグ化(double PEGylation)を生じさせることができる。
【0082】
558−565領域がHSPGおよびFIX両方へ結合することが示されているため、部位をこの領域内に設計しなかった。代わりに、PEG15−PEG17(377、378および556)を、結合したPEGが両方の相互作用を妨げ、それらの間の起こりうる相互作用を崩壊させることができるように、A2 LRPおよびHSPG結合領域間に設計した。表面暴露されており外側に向いているさらなる部位も、LRPおよびHPSG結合領域内または領域付近で選択することができる。新規クリアランスメカニズムを同定するために、FVIIIを系統的にペグ化することができる。PEG1−17に加えて、PEG18−20に対応する3つの他の天然グリコシル化部位、すなわち、N41、N239およびN2118は、表面暴露されているはずであるから、ペグ化のための連結点として使用することができる。PEG2、PEG6および4つのグリコシル化部位のCβ原子から20オングストローム半径内の表面領域は、vWF、FIX、FX、リン脂質およびトロンビンに関する機能性相互作用部位に加えてBDDモデル上にマッピングされた。
【0083】
次にY81、F129、K422、K523、K570、N1864、T1911、Q2091およびQ2284に対応するPEG21−29を、それらのCβ原子のそれぞれから20オングストローム半径で、残っているBDD表面のほぼ全体を覆う能力に基づいて選択した。これらの位置も、完全に暴露されており、外側を向いており、天然のシステインから遠く離れており、正しくないジスルフィド形成の可能性が最小になるため、選択した。大型PEG、例えば、64kD分岐状PEGは約20オングストローム半径を有する球を覆う可能性を有すると予期されるため、20オングストローム半径を選択する。PEG2およびPEG6ならびにグリコシル化部位PEG18、19および20と一緒のPEG21−29のペグ化はFVIIIの非機能性表面のほぼ全体を保護する可能性がある。
【0084】
増強された特性、例えば、改良されたPKプロファイル、より良い安定性または減少した免疫原性を誘導するペグ化の位置は、最大に増強された特性を有する複数−ペグ化された生成物を生産するために組み合わせることができる。PEG30およびPEG31は、A2およびA3ドメイン各々における暴露されているジスルフィドを除去することにより設計した。PEG30またはC630Aは、ペグ化のため、そのジスルフィドパートナーC711を自由にさせる。同様に、PEG31、C1899AはC1903がペグ化されることを可能にする。
【実施例】
【0085】
実施例
本発明をさらに理解するために、以下の実施例を説明する。これらの実施例は説明の目的のためのみであり、いかなる方法によっても本発明の範囲を限定すると解釈してはならない。本明細書に記載されているすべての刊行物を、出典明示よりそれら全体を包含する。
【0086】
実施例1.変異誘発
ペグ化のために選択された部位にシステインコドンを導入することにより、FVIIIの部位特異的ペグ化のための基質を生成することができる。Stratagene cQuickChangeTM II 部位特異的変異誘発キットを使用して、すべてのPEG変異体を生産した(Stratagene Corporation, La Jolla, CA)。cQuikChangeTM部位特異的な変異誘発方法は、PfuTurbo(登録商標)DNAポリメラーゼおよび温度サイクラー(temperature cycler)を使用して行う。所望の変異体を含む2個の相補的オリゴヌクレオチドプライマーを、プライマーを置き換えないであろうPfuTurbo(登録商標)を使用して延長させる。野生型FVIII遺伝子を含むdsDNAを鋳型として使用する。複数回伸長サイクル後、生成物をメチル化DNAに対して特異的であるDpnI エンドヌクレアーゼで消化する。変異体を含む新しく合成されたDNAはメチル化されていないが、親の野生型DNAをメチル化されている。次に消化されたDNAを使用して、XL−1 Blue 超−コンピテント細胞を形質転換させる。
【0087】
変異誘発反応をpSK207+BDD C2.6またはpSK207+BDDのいずれかで行った。FVIIIの部位特異的な変異誘発、変異タンパク質の精製、ペグ化および活性測定の記載は、WO2006/053299(これを出典明示により本明細書に包含させる)において見出すことができる。変異タンパク質の要約を表1に提供する。
表1
【表1】
【0088】
実施例2.vWF結合ELISA
FVIIIを、溶液中の重度の血友病血漿中のvWfに結合させる。次にFVIII−vWf複合体を、vWf−特異的モノクローナル抗体で被覆されているマイクロタイタープレート上に捕獲する。vWfに結合したFVIIIを、FVIIIポリクローナル抗体およびセイヨウワサビペルオキシダーゼ−抗−ウサギ複合体で検出する。ペルオキシダーゼ−複合体化抗体複合体は、基質を添加すると呈色反応を生じる。4パラメーターフィットモデルを使用して、サンプル濃度を標準曲線から補間する。FVIII結合の結果をμg/mLで報告する。ペグ化により活性のいずれにおいても有意な影響はなく、これはBドメインにおけるペグ化と一致する。結果は表2に見出すことができる。
表2
【表2】
【0089】
実施例3.薬物動態活性
ペグ化FVIIIおよびBドメイン欠失FVIII(BDD−FVIII)のPKを、FVIIIノックアウト(KO)マウスにおいて測定した。マウスに、BDD−FVIII(200IU/kg)、アミノ酸位置1804に導入されたシステイン変異体において64kD PEG(64kD PEG14)と複合体化したBDD−FVIII(108IU/kg)、または位置491および1804にてそれぞれシステイン変異体において64kD PEG(64kD PEG2+14)と複合体化したBDD−FVIII(194IU/kg)を静脈内(i.v.)注射によって与えた。血液試料を、5分、4時間、8時間、16時間、24時間、32時間および48時間に処理されたマウス(5匹マウス/処置/時点)から回収した。血漿FVIII活性をCoatest アッセイにより測定した。終末相半減期を、WinNonLinにおける活性vs時間曲線のnon-compartment modelingにより測定した。FVIII KOマウスにおけるBDD−FVIIIに対するt1/2は6時間であるが、64kD PEG(64kD PEG14)または128kD PEG(64kD PEG2+14)と複合体化したFVIIIに対するt1/2はそれぞれ12.43時間および12.75時間である。したがって、ペグ化FVIIIの半減期は、FVIII KOマウスにおけるBDD−FVIIIと比較して、約2倍に増加した。
【0090】
vWF KOマウスにおいて証明されるとおり、循環におけるvWFの欠如はペグ化FVIIIの半減期延長への限界を排除した。マウスにBDD−FVIII(200IU/kg)、64kD PEG14(520IU/kg)、または64kD PEG2+14(400IU/kg)をi.v.投与により投与した。血液試料を、BDDFVIII処置マウスから5分、15分、30分、1時間、2時間、4時間、6時間および8時間に、およびペグ化FVIII処置マウスから5分、4時間、8時間、16時間、24時間、32時間および48時間に回収した(5匹マウス/処置/時点)。vWF KOマウスにおいて約2%の正常レベルである、内因性マウスFVIIIからバックグラウンド活性を除去するために、注入されたヒトFVIIIの血漿活性をCapture Coatestにより測定した。最初に、血漿におけるBDD−FVIIIおよびペグ化FVIIIをヒトFVIIIのA3ドメインに特異的であるmAb R8B12(2ug/mL)により捕獲し、次にCoatestにより測定した。vWFの保護なしに迅速にクリアされ、18分と短いt1/2であるBDD−FVIIIと対照的に、64kD PEG14および64kD PEG2+14のt1/2はそれぞれ5.7時間および8.2時間である(図1)。したがって、FVIII KOマウスにおけるvWFの存在下において観察されるBDD−FVIIIと比較してPEG−FVIIIのt1/2の最大2倍増加と対照的に、64kD PEG14および64kD PEG2+14のt1/2は、vWF KOマウスにおけるvWFの非存在下において、19から27倍延長される。さらに、PEG−FVIIIのt1/2の延長はPEGのサイズに比例する。
【0091】
本明細書に記載されている全ての刊行物および特許は、出典明示により本明細書に包含させる。本発明の精神および範囲から逸脱することなく、記載されている本発明の方法の種々の修飾および変化が当業者に明らかである。
【0092】
本発明は特定の態様と関連して記載されているが、本発明はこのような特定の態様に不都合に限定されるべきでないと理解すべきである。実際に、生化学の分野または関連分野の当業者に明らかである本発明を実施するための上記様式の種々の修飾は、特許請求の範囲内であると意図される。当業者は、わずかに日常の実験を使用して、本明細書に記載されている本発明の特定の態様における多数の均等を認識するか、または確かめることができる。このような均等は特許請求の範囲に包含されることを意図する。
【技術分野】
【0001】
本出願は、2008年6月4日に出願された米国仮出願第61/058,795号(この内容は、その全体において出典明示により本明細書に包含させる)の利益を主張する。
【0002】
技術分野
本発明は、フォン・ヴィレブランド病(vWD)の処置のために有用である第VIII因子(FVIII)変異タンパク質およびその誘導体に関する。FVIII変異タンパク質は、定義された位置にて、1つ以上の生体適合性ポリマー、例えば、ポリエチレングリコールに結合することができる。加えて、治療目的のためのそれらの関連製剤、用量および投与の方法を提供する。これらの修飾されたFVIII変異体ならびに関連組成物および方法は、フォン・ヴィレブランド病に罹患している個体に対する注射頻度の減少および免疫原性応答の減少をもたらす処置選択の提供に有用である。
【背景技術】
【0003】
本発明の背景
vWDは、先天性または後天性疾患の種々の病因群を表す用語である。多数の型のvWDの原因は、他の特性と共に、凝血促進FVIIIに結合し、血液循環中の天然FVIIIの半減期を延長させる一連の多量体血漿糖タンパク質であるフォン・ヴィレブランド因子(vWF)の機能に属する(例えば、Federici, Haemophilia 10 (suppl 4):169,2004; Denis, et al., Thromb. Haemost. 99:271, 2008、参照)。正常なヒトにおいて、FVIIIの半減期は、vWFの非存在下において約8分であり、vWFの存在下において8時間である。
【0004】
軽度の型(1型)において、vWDは、ヒト集団において100人に1人もの割合で罹患されており、そして男性および女性に等しく罹患されている非常に一般的なものである。
【0005】
2型vWDは、vWDの重度の型であり得、5つのサブタイプ:2A、2B、2C、2Mおよび2Nにおいて知られている。これらの中で、2N型はFVIIIのvWFへの結合の欠乏により特徴付けられる。したがって、2N型vWDを有する患者において、FVIIIは迅速に分解され、循環レベルが低下する。2N型vWFは、FVIIIへの結合を害するホモ接合体または化合物ヘテロ接合体vWF変異体による。vWFとの複合体ではない遊離FVIIIは循環から迅速に除去されるため、2N型vWDは常染色体劣性型の血友病Aを装う。しかしながら、患者は一般的に、FVIIIレベルは減少しているが、正常レベルのvWF−抗原およびvWF−血小板GP1b結合に対するリストセチン補因子活性(vWF:RCo活性)を有する。
【0006】
エリックフォン・ヴィレブランドの型でフィンランド人ファミリーにおいて初めて発見された3型vWDは、vWFのホモ接合体欠乏または二重ヘテロ接合体欠乏である。3型vWDは、ノンセンス変異体またはvWFをコードする核酸へのわずかな挿入または欠失によるフレームシフトにより、完全またはほぼ完全なvWFの欠乏を引き起こす。ほとんどの場合において、vWF:RCoおよびvWF:Agは検出することができず、FVIIIレベルは非常に少ない。3型vWDを有する患者は、血友病のような関節血症および関節または関節腔出血を有し得る。
【0007】
後天性vWDは、通常、抗−vWF抗体の発生、流体せん断応力−誘導タンパク質分解または血小板もしくは他の細胞への結合の増加による、自己免疫性クリアランスにより引き起こされる。後天性vWD症候群は、少ないレベルのvWF−Ag、vWF:RcoおよびFVIIIを有する3型vWDのものと同様である。3型vWDおよび後天性vWD患者は、vWDの特徴である粘膜出血だけでなく血友病Aの特徴である軟組織、筋肉および関節出血も有する。
【0008】
血友病Aは、5000人の男性あたり1人の推定発現率である最も一般的な遺伝的凝固障害である。血液凝固の内因経路の重要な成分であるFVIIIの欠乏または構造欠陥により引き起こされる。血友病Aに対する現在の処置はヒトFVIIIの静脈内注射を含む。ヒトFVIIIは、約300kDの一本鎖分子として組換え的に生産されている。それは構造ドメインA1−A2−B−A3−C1−C2からなる(Thompson, Semin. Hematol. 29:11-22, 2003)。金属イオンにより互いに保持されている2つの鎖を有する前駆体産物は、ゴルジ装置において200kD(重鎖)および80kD(軽鎖)の2つのポリペプチド鎖に処理される(Kaufman, et al., J. Biol. Chem. 263:6352, 1988; Andersson, et al., Proc. Natl. Acad. Sci. 83:2979, 1986)。
【0009】
B−ドメイン欠失FVIII(BDD、90kDのA1−A2重鎖プラス80kDの軽鎖)が血友病Aに対する補充療法のとき有効であることが示されているため、FVIIIのB−ドメインは重要ではないようである。B−ドメイン欠失FVIII配列は、14個のアミノ酸のB−ドメインのほとんどが欠失している。
【0010】
血友病A患者は現在、要求に応じてFVIIIの静脈内投与または1週間に数回の予防的治療投与により処置される。予防処置のために、15−25IU/kg体重にてFVIIIを1週間に3回投与する。患者において定期的に必要である。ヒトにおいて半減期が短いため、FVIIIは頻繁に投与されなければならない。全長タンパク質に関して300kD以上の大型サイズにもかかわらず、FVIIIはヒトにおいてわずか約11時間の半減期である(Ewenstein, et al., Semin. Hematol. 41:1-16, 2004)。頻繁な静脈内注射の必要性は患者のコンプライアンスに対する巨大な障壁を作る。長い半減期を有し、投与頻度が少なくてよいFVIII産物が開発されることが患者にとってより都合がよい。さらに、必要とされる投与がより少なくなるため、半減期が増加したとき処置費用を減少させることができる。
【0011】
現在の治療のさらなる不利益は、約25−30%の患者がFVIII活性を阻害する抗体を産生することである(Saenko, et al., Haemophilia 8:111, 2002)。阻害抗体の主なエピトープは、A2ドメインの残基484−508、A3ドメインの残基1811−1818およびC2ドメイン内に位置する。抗体産生は補充療法のようなFVIIIの使用を妨げ、このグループの患者に高用量の組換え第VIIa因子(FVIIa)および免疫寛容療法でのさらに費用のかかる処置を探させる。
【0012】
以下の試験によって、阻害抗体のFVIIIエピトープを同定した。25個の阻害血漿サンプルの試験において、11個はトロンビン生産73kDの軽鎖フラグメントA3C1C2へ、4個はA2ドメインへ、10個は両方へ結合することが見出された(Fulcher, et al., Proc. Natl. Acad. Sci. 2:7728-32, 1985)。他の試験において、患者由来のA2ドメインインヒビターの8個中6個が、組換えA2ポリペプチドにより中和された(Scandella, et al., Blood 82:1767-75, 1993)。患者由来のインヒビターの9個中6個に対するエピトープがA2残基379538にマップされていた(Scandella, et al., Proc. Natl. Acad. Sci. 85:6152-6, 1988)。18個の重鎖インヒビターに対するエピトープは、A2ドメインの同じN−末端18.3kD領域に局在していた(Scandella, et al., Blood 74:1618-26, 1989)。
【0013】
ヒトA2ドメイン残基387−604を相同ブタ配列で置き換えることにより生産された活性な組換えハイブリッドヒト/ブタFVIII分子は、患者A2インヒビターに抵抗性であり(Lubin, et al., J. Biol. Chem. 269:8639-41, 1994)、A2への結合に対する患者A2インヒビターと競合するマウスモノクローナル抗体mAB413IgGに抵抗性であった(Scandella, et al., Thromb Haemost. 67:665-71, 1992)。mAB413IgGおよび4個の患者インヒビターがA2ドメイン残基484−508がブタのもので置換されているハイブリッドヒト/ブタFVIIIを抑制しないことを実験において示されるとき、このA2ドメインエピトープはさらにA2ドメイン残基484−508に局在している(Healey, et al., J. Biol. Chem. 270:14505-14509, 1995)。このハイブリッドFVIIIは、また、スクリーニングされた23個の患者血漿の少なくとも半分に抵抗性であった(Barrow, et al., Blood 95:564-568, 2000)。アラニン走査変異誘発によって、残基487が試験された全5個の患者インヒビターへの結合に対して重要であることを同定されたが、残基484、487、489および492はmAB413IgGとの相互作用に極めて重要であった(Lubin, J. Biol. Chem. 272:30191-30195, 1997)。R484A/R489A変異体ではなくR484A/R489A/P492A変異体を受けているマウスにおける阻害抗体力価は、対照ヒトBDDFVIIIを受けているマウスよりも有意に低かった(Parker, et al., Blood 104:704-710, 2004)。要するに、A2ドメインの484−508領域は、FVIII活性のインヒビターに対する結合部位であるようである。
【0014】
FVIIIへの免疫応答の発達に加えて、慣用の治療での他の問題は、FVIIIの短いインビボ半減期のため、頻繁な投与が必要であることである。循環からのFVIIIのクリアランスに対するメカニズムが試験されている。
【0015】
循環からのFVIIIクリアランスは、広いリガンド特異性を有する肝臓クリアランス受容体である低密度のリポタンパク質受容体−関連タンパク質(LRP)への特異的結合に部分的に起因していた(Oldenburg, et al., Haemophilia 10 Suppl 4:133-139, 2004)。最近、低密度のリポタンパク質(LDL)受容体は、また、例えば、血漿制御レベルのFVIIIにおいてLRPと共同することにより、FVIIIクリアランスにおける役割を果たすことを示された(Bovenschen, et al., Blood 106:906-910, 2005)。両方の相互作用は、細胞表面ヘパリン硫酸プロテオグリカン(HSPG)へ結合することにより促進される。マウスにおける血漿半減期は、LRPがブロックされるとき3.3倍またはLRPおよび細胞表面HSPGの両方がブロックされるとき5.5倍延長され得る(Sarafanov, et al., J. Biol. Chem. 276:11970-11979, 2001)。HSPGは、細胞表面上でFVIIIを濃縮し、それをLRPに与えると仮説が立てられる。FVIII上のLRP結合部位は、A2残基484−509(Saenko, et al., J. Biol. Chem. 274:37685-37692, 1999)、A3残基1811−1818(Bovenschen, et al., J. Biol. Chem. 278:9370-9377, 2003)およびC2ドメインにおけるエピトープ(Lenting, et al., J. Biol. Chem. 274:23734-23739, 1999)に局在していた。
【0016】
FVIIIは、また、プロテアーゼの作用により循環から除去される。この作用を理解するためには、FVIIIが血液凝固に関与するメカニズムを理解しなければならい。FVIIIは、vWFへ結合している重鎖および軽鎖のヘテロダイマーとして循環する。vWF結合は、FVIII残基1649−1689(Foster, et al., J. Biol. Chem. 263:5230-5234, 1998)およびC1の部分(Jacquemin, et al., Blood 96:958-965, 2000)およびC2ドメイン(Spiegel, et al., J. Biol. Chem. 279:53691-53698, 2004)と関与する。FVIIIは、残基372、740および1689後のペプチド結合を開裂させ、A1、A2およびA3−C1−C2ドメインのヘテロトリマーを生産するトロンビンにより活性化される(Pittman, et al., Proc. Natl. Acad. Sci. 276:12434-12439, 2001)。活性化時、FVIIIはvWFから解離し、リン脂質に結合することにより血小板の細胞表面に濃縮される。リン脂質結合はFVIII残基2199、2200、2251および2252と関与する(Gilbert et al., J. Biol. Chem. 277:6374-6381, 2002)。それは、FVIII残基558−565(Fay, et al., J. Biol. Chem. 269:20522-20527, 1994)および1811−1818(Lenting, et al., J. Biol. Chem. 271:1935-1940, 1996)との相互作用を介してFIXへ、ならびにFVIII残基349−372との相互作用を介してFXへ(Nogami, et al., J. Biol. Chem. 279:15763-15771, 2004)結合し、内因性凝固経路の重要な成分であるFXのFIX活性化のための補因子として作用する。活性化FVIII(FVIIIa)は、FVIII残基336および562後の開裂を介してプロテアーゼ活性化タンパク質C(APC)により部分的に不活性化される(Regan, et al., J. Biol. Chem. 271:3982-3987, 1996)。しかしながら、不活性化の主な決定因子は、A1およびA3−C1−C2からのA2ドメインの解離である(Fay, et al., J. Biol. Chem. 266:8957-8962, 1991)。
【0017】
タンパク質のインビボ半減期を増加させるために証明されている1つの方法はペグ化である。ペグ化は長鎖ポリエチレングリコール(PEG)分子のタンパク質または他の分子への共有結合である。PEGは直線形態または分岐形態であり、異なる特徴を有する異なる分子を生産することができる。ペプチドまたはタンパク質の半減期を増加させること以外では、ペグ化は、抗体発生を減少させ、プロテアーゼ消化からタンパク質を保護し、腎臓濾液から物質を排出し続けるために使用されている(Harris, et al., Clinical Pharmacokinetics 40:539-551, 2001)。加えて、ペグ化はタンパク質の全ての安定性および溶解度も増加させ得る。最後に、ペグ化タンパク質の持続血漿濃度は、薬物のトラフ/ピークレベルを低減し、したがって早期の時点にて生理学的レベルを越えるタンパク質を導入する必要性を排除することにより、有害な副作用の程度を減少させ得る。
【0018】
大型ポリマー、例えば、PEGおよびデキストランにて一級アミン(N−末端およびリシン)を標的化することによるFVIIIのランダム修飾は、種々の程度の成功で試みられている(WO94/15625、米国特許4970300、米国特許6048720)。1994年の特許出願(WO94/15625)において記載されている非常に劇的な改善は、2倍の費用がかかるが、全長FVIIIを50倍モル過剰のPEGと反応後に4倍の半減期改善として示すことである。WO2004/075923は、ランダム修飾を介して生産されるFVIIIおよびポリエチレングリコールの複合体を記載している。ランダムペグ化タンパク質、例えば、インターフェロン−アルファ(Kozlowski, et al, BioDrugs 15:419-429, 2001)は治療剤としてこれまでに承認されている。
【0019】
しかしながら、このランダム手段は、ヘテロ二量体FVIIIに関してさらなる問題がある。FVIIIは158個のリシン、2つのN−末端ならびに多数のヒスチジン、セリン、スレオニンおよびチロシンを含む何百もの可能なペグ化の位置を有し、これら全てが主に一級アミンを標的にする試薬にてペグ化され得る。例えば、ペグ化インターフェロンアルファ−2bに関する主な位置異性体は、ヒスチジンであることが示された(Wang, et al., Biochemistry 39:10634-10640, 2000)。さらに、全長FVIIIの不均一過程は、ペグ化産物におけるさらなる複雑さとなる出発物質の混合物をもたらし得る。FVIII上のペグ化の位置を制御しないさらなる欠点は、PEGが重要な活性部位、または部位付近に結合するとき、とりわけ2つ以上のPEGまたは1つの大きなPEGがFVIIIと複合体化するとき、起こりうる活性の減少である。ランダムペグ化は常に多量の多様なペグ化産物を生産するため、モノ−ペグ化産物のみを得るための精製は劇的に低い全収率となる。最後に、製品プロフィールにおける大きな不均一性は一貫性のある合成および特性化をそれぞれ非常に不可能なことにさせる。良い製造物は一貫性のある、よく特徴付けられた産物である必要があるため、産物の不均一性は商業化への障壁である。すべてのこれらの理由のため、FVIIIをペグ化するためのさらなる特定の方法が望まれる。
【0020】
種々の部位特異的タンパク質ペグ化戦略は最近の文献に要約されている(Kochendoerfer, et al., Curr. Opin. Chem. Biol. 9:555-560, 2005)。1つの手段は、化学合成または組換え発現による非天然アミノ酸のタンパク質への取り込み、次に非天然アミノ酸と特異的に反応するPEG誘導体の付加を含む。例えば、非天然アミノ酸は、天然タンパク質に見られないケト基を含むものであり得る。しかしながら、タンパク質の化学合成は、FVIIIほどの大きさのタンパク質に対して実行不可能である。現在のペプチド合成の限界は約50残基である。いくつかのペプチドは大きなポリペプチドを形成するために結合することができるが、B−ドメイン欠失FVIIIでさえ生産するために20以上の結合が必要であるため、これでは理想的な反応条件下でさえ1%未満の回収率となる。非天然アミノ酸を有するタンパク質の組換え発現は、今までのところ主に非哺乳動物の発現系に限定されている。この手段は、哺乳動物系において発現する必要がある大型かつ複雑なタンパク質、例えば、FVIIIに対して問題のあることが予期される。
【0021】
タンパク質の部位特異的ペグ化のための他の手段は、PEG−アルデヒドにてN−末端骨格アミンを標的化することによる。しかしながら、他のアミン基を越える特異性を達成するためのこのプロセス下にて必要とされる低いpHは、FVIIIの安定性のために必要であるほぼ中性のpH範囲と適合性ではない(Wang, et al., Intl. J. Pharmaceutics 259, pp. 1-15, 2003)。さらに、FVIIIのN−末端ペグ化は、この領域が血漿クリアランスに関連しないとき、血漿半減期の改善を引き起こさないであろう。
【0022】
WO90/12874は、システインを挿入するか、またはシステインを他のアミノ酸と置換し、次にスルフヒドリル反応基を有するリガンドを付加することによる、ヒトIL−3、顆粒球コロニー刺激因子およびエリスロポエチンポリペプチドの部位特異的修飾を記載している。リガンドは選択的にシステイン残基に結合する。FVIIIまたはその変異体の修飾は記載されていない。
【0023】
EP0319315は、vWF結合の減少をもたらすvWF結合部位の欠失または変化を有するFVIII変異タンパク質を記載している。EP0319315は、さらにこのような変異タンパク質を投与することによるvWF阻害活性によるFVIII欠乏症の軽減を記載している。
【0024】
Rottensteiner et al.は、ポリエチレングリコールまたはポリシアル酸との複合体を形成するためのFVIIIにおけるリシン残基のランダム化学修飾を記載している。Blood 110(11)、3150A (2007)。Rottensteiner et al.は、さらにランダム修飾されたFVIIIが2N型vWDに有用であり得ることを示唆している。
【0025】
上記理由のため、より良いインビボ作用持続期間および減少した免疫原性を有するが、機能活性を維持している改良されたFVIII変異体の必要性がある。さらに、このようなタンパク質は一貫した方法で均質製品として生産されることが望ましい。
【発明の概要】
【0026】
発明の概要
本発明の目的は、改良された薬物動態学的特性および治療特性を有する生体適合性ポリマー−複合体化機能性FVIIIポリペプチドの投与を含むvWDを処置する方法を提供することである。
【0027】
また、本発明の目的は、FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量の複合体を必要とする対象に投与することを含むvWDを処置するための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号(numerical position)により識別される特定のアミノ酸残基であり、N−末端アミンではない方法を提供することである。フォン・ヴィレブランド病はフォン・ヴィレブランド因子の欠乏および/または異常により特徴付けることができる。
【0028】
また、本発明のさらなる目的は、FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる複合体を製造することを含むvWDを処置するための薬物を製造するための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号により識別される特定のアミノ酸残基であり、N−末端アミンではない方法を提供することである。
【0029】
本発明のさらなる目的は、FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量のFVIIIのシステイン置換変異体を必要とする対象に投与することを含むvWDを処置するための方法であって、該変異体はFVIII配列におけるアミノ酸に対して置換されたシステイン残基を有することにより特徴付けられ、該置換は配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にして、システイン残基がFVIIIに存在しないアミノ酸位置にてシステイン残基をもたらし、該システイン付加変異体は該置換システイン残基へ共有結合している生体適合性ポリマーを有することによりさらに特徴付けられる方法を提供することである。
【0030】
本発明のさらなる目的は、改良された薬物動態学的特性を有する生体適合性ポリマー−複合体化Bドメイン欠失FVIIIタンパク質を必要とする対象に投与することを含むvWDを処置するための方法を提供することである。
【0031】
本発明のさらなる目的は、低密度のリポタンパク質受容体−関連タンパク質(LRP)、低密度のリポタンパク質(LDL)受容体、へパラン硫酸プロテオグリカン(HSPG)および/またはFVIIIに対する阻害抗体への減少した結合性を有する生体適合性ポリマー−複合体化機能性FVIIIポリペプチドを必要とする対象に投与することを含むvWDを処置するための方法を提供する。
【0032】
本発明のさらなる目的は、一貫した方法で均質製品として生産することができる、より良いインビボ作用持続期間および減少した免疫原性を有する治療有効量の改良されたFVIII変異体の必要とする対象への投与を含むvWDを処置するための方法を提供する。
【0033】
本発明の1つの局面において、FVIII凝血促進活性を有し、ポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含む治療有効量の複合体を必要とする対象に投与することを含むvWDを処置するための方法であって、該所定の位置はN−末端アミンではない方法を提供する。
【0034】
本発明のさらなる局面において、手術前にFVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量の複合体を対象に手術前に投与することを含み、それによって一過性の出血を弱める予防処置のための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号により識別される特定のアミノ酸残基であり、N−末端アミンではない方法を提供する。対象はvWD、例えば、3型vWDを有することができる。有利には、複合体を手術前24時間以内に、好ましくは8時間以内、より好ましくは手術0.5から2時間前に投与する。
【0035】
本発明のさらなる局面において、FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量の複合体を必要とする対象に投与することを含み、それによって一過性の出血を弱める外傷の処置のための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号により識別される特定のアミノ酸残基であり、N−末端アミンではない方法を提供する。対象は3型vWDを含むvWDを含むことができる。
【図面の簡単な説明】
【0036】
【図1】vWDノックアウト(KO)マウスにおけるFVIII半減期が正常に戻るペグ化FVIIIの効果。図は、i)vWF KOマウスへのrFVIIIの投与(黒丸)、ii)FVIII KOマウスへのrFVIIIの投与(白丸)、iii)vWF KOマウスへのペグ化rFVIIIの投与(64kD PEG14、黒四角)、およびiv)vWF KOマウスへの異なったペグ化rFVIIIの投与(64kD PEG2+14、黒三角)による血漿FVIII活性の経時変化を説明する。
【発明を実施するための形態】
【0037】
本発明の詳細な説明
本発明は、FVIII活性を有するポリペプチドがN−末端アミンではない所定の位置にて生体適合性ポリマーに共有結合することができ、実質的に凝固活性を維持している発見に基づく。さらに、これらのポリペプチド複合体は、改善された循環時間および減少した抗原性を有する。
【0038】
本発明は、さらに、所定の位置にて生体適合性ポリマーに共有結合したFVIII変異タンパク質が、修飾されていないFVIIIよりも、vWFを欠いている対象の循環においてより長い半減期の凝血促進活性を有する発見に基づく。本発明の複合体を使用するvWFを実質的に欠いている対象の処置は、FVIIIへのランダムポリマー結合またはN−末端での結合を有する先行技術の複合体の使用を越えて有利であり得る。部位特異的結合は生物学的活性のために必要である領域を避ける修飾を設計することで可能であり、それにより実質的なFVIII活性を維持する。また、FVIIIクリアランスに関連する部位における結合をブロックするようにポリマーを結合するように設計することが可能である。部位特異的結合は、また、ランダムポリマー結合により当分野で生産される不均一な複合体よりも、均一な生産物ができる。軽鎖のN−末端アミンにおける結合を避けることにより、本発明の複合体は、FVIIIポリペプチドの活性部位へのリガンドの結合による活性の喪失の可能性を避ける。
【0039】
定義
生体適合性ポリマー。生体適合性ポリマーは、ポリアルキレンオキシド、例えば、限定はしないが、ポリエチレングリコール(PEG)、デキストラン、コロミン酸または他の炭水化物ベースのポリマー、アミノ酸のポリマー、ビオチン誘導体、ポリビニルアルコール(PVA)、ポリカルボキシレート、ポリビニルピロリドン、ポリエチレン−コ−マレイン酸無水物、ポリスチレン−コ−リンゴ酸無水物、ポリオキサゾリン、ポリアクリロイルモルホリン、ヘパリン、アルブミン、セルロース、キトサンの加水分解物、デンプン、例えば、ヒドロキシエチル−デンプンおよびヒドロキシプロピル−デンプン、グリコーゲン、アガロースおよびそれらの誘導体、グアーガム、プルラン、イヌリン、キサンタンゴム、カラギーナン、ペクチン、アルギン酸加水分解物、他の生物ポリマーおよびそれらのあらゆる同等物を含む。ポリマーの例は、ポリエチレングリコール、例えば、メトキシポリエチレングリコール(mPEG)である。他の有用なポリアルキレングリコール化合物は、ポリプロピレングリコール(PPG)、ポリブチレングリコール(PBG)、PEG−グリシジルエーテル(Epox−PEG)、PEG−オキシカルボニルイミダゾール(CDI−PEG)、分岐ポリエチレングリコール、直鎖ポリエチレングリコール、フォーク状ポリエチレングリコールおよび多分岐または“超分岐”ポリエチレングリコール(星形−PEG)である。
【0040】
ポリエチレングリコール(PEG)。本明細書において使用される“PEG”および“ポリエチレングリコール”は相互互換であり、あらゆる水溶性ポリ(エチレンオキシド)を含む。一般的に、本発明において使用するためのPEGは、構造“−−(OCH2CH2)n−−”((n)は2から4000である)を含む。本明細書において使用されるPEGは、また、末端の酸素が置換されているか否かに依存して“−−CH2CH2−−O(CH2CH2O)n−−CH2CH2−−”および“−(OCH2CH2)nO−−”を含む。本明細書および本特許請求の範囲を通して、“PEG”なる用語が、例えば、ヒドロキシルまたはC1−20アルコキシ基に限定することなく、種々の末端または“末端キャップ”基を有する構造を含むことを認識すべきである。“PEG”なる用語は、また、−OCH 2CH2−−繰り返しサブユニットの大部分、すなわち、50%以上を含むポリマーを意味する。特定の形態に対して、PEGは非常に種々の分子量、ならびに構造または形状、例えば、分岐状、直鎖状、フォーク状および多分岐状であり得る。
【0041】
ペグ化。ペグ化はポリエチレングリコール(PEG)が分子、例えば、タンパク質に共有結合するプロセスである。
【0042】
活性化または活性な官能基。官能基、例えば、生体適合性ポリマーが活性化として記載されているとき、官能基は他の分子上の求電子または求核原子と容易に反応する。
【0043】
Bドメイン欠失FVIII(BDD)。本明細書において使用されるBDDは、FVIIIのB−ドメインの全14アミノ酸の欠失を含むアミノ酸配列を有することにより特徴付けられる。B−ドメインの最初の4アミノ酸(SFSQ、配列番号:2)はB−ドメインの最後の10残基(NPPVLKRHQR、配列番号:3)に結合する(Lind, et al, Eur. J. Biochem. 232:19-27, 1995)。本明細書において使用されるBDDは配列番号:4のアミノ酸配列を有する。BDDポリペプチドの例はWO2006/053299(これは出典明示により本明細書に包含させる)に記載されている。
【0044】
FVIII。血液凝固第VIII因子(FVIII)は肝臓により合成された糖タンパク質であり、血流に放出される。循環血液において、それはフォン・ヴィレブランド因子(vWF、FVIII−関連抗原としても知られている)に結合し、安定な複合体を形成する。トロンビンにより活性化されると、それは複合体から解離し、凝固カスケードにおける他の凝固因子と相互作用し、最後に血栓の形成を誘導する。ヒト全長FVIIIは配列番号:1のアミノ酸配列を有するが、対立遺伝子多型が起こりうる。
【0045】
機能性FVIIIポリペプチド。本明細書において使用される機能性FVIIIポリペプチドは、インビボまたはインビトロにて、例えば、血友病Aにより特徴付けられるヒトFVIII欠乏を修正することが可能である機能性ポリペプチドまたはポリペプチドの組合せを示す。FVIIIは、天然状態における多数の分解物または加工された形態を有する。これらは、本明細書において証明されるとおり一本鎖タンパク質である前駆体からタンパク質分解により得られる。機能性FVIIIポリペプチドは、このような一本鎖タンパク質を含み、ヒトFVIII欠乏を修正する生物学的活性を有するこれらの種々の分解産物も提供する。アレル変異体が恐らく存在する。機能性FVIIIポリペプチドは、ヒトFVIIIの機能性セグメントを含み、必須の特徴的なヒトFVIII機能活性が同じやり方で影響を受けずに維持されている限り、FVIIIの誘導体となるすべてのこのようなアレル変異体、グリコシル化バージョン、修飾物およびフラグメントを含む。このような必要な機能活性を有するFVIIIの誘導体は、本明細書に記載されている直接的インビトロ試験により容易に同定することができる。さらに、機能性FVIIIポリペプチドは、FIXa、カルシウムおよびリン脂質の存在下でのFXaへの因子X(FX)の変換を触媒すること、ならびに血友病A罹患個体由来の血漿における凝固障害を修正することが可能である。ヒトFVIIIアミノ酸配列およびその機能性領域の配列の公開から、DNAの制限酵素切断またはヒトFVIIIタンパク質のタンパク質分解もしくは他の分解を介して得ることができるフラグメントは、当業者に明らかである。機能性FVIIIポリペプチドの例は、WO2006/053299(これを出典明示により本明細書に包含させる)に記載されている。
【0046】
FIX。本明細書において使用されるFIXは、ヒト凝固因子IXとしても知られている凝固因子IX、または血漿トロンボプラスチン成分を意味する。
【0047】
FX。本明細書において使用されるFXは、名称、ヒト凝固因子Xおよび名称、スチュアートプラウナー因子としても知られている凝固因子Xを意味する。
【0048】
薬物動態学。“薬物動態学”(“PK”)は、体内の薬物の吸収、分配、代謝および除去の特性を述べるために使用される用語である。薬物の薬物動態学の改善は、薬物を治療剤としてインビボにてより有効にさせる特性、とりわけ体内の有効な持続時間の改善を意味する。
【0049】
変異タンパク質。変異タンパク質は、タンパク質またはポリペプチドにおける実験誘導変異の結果として生じる遺伝的に加工されたタンパク質である。
【0050】
タンパク質。本明細書において使用されるタンパク質およびポリペプチドは同義である。
【0051】
FVIIIクリアランス受容体。本明細書において使用されるFVIIIクリアランス受容体は、1つ以上の他の分子と結合または関連して、循環からFVIIIクリアランスをもたらす機能性FVIIIポリペプチド上の受容体領域を意味する。FVIIIクリアランス受容体は、LRP、LDL受容体および/またはHSPGに結合するFVIII分子の領域を含むが、これらに限定されない。
【0052】
すべての機能性FVIIIポリペプチドが所定の位置にて変異されていてよく、本発明の方法にしたがって生体適合性ポリマーへ所定の位置にて共有結合していてよいと想定する。有用なポリペプチドは、配列番号:1に示されるアミノ酸配列を有する全長FVIIIおよび配列番号:4に示されるアミノ酸配列を有するBDD FVIIIを含むが、これらに限定されない。
【0053】
本発明の複合体において使用される生体適合性ポリマーは、上記のあらゆるポリマーであり得る。生体適合性ポリマーは、薬物動態学において所望の改善を提供するように選択される。例えば、ポリマーの独自性、サイズおよび構造は、FVIII活性を有するポリペプチドの循環半減期を改善させるか、または活性において許容されない減少なくポリペプチドの抗原性を減少させるように選択される。ポリマーはPEGを含んでいてよく、一例として、PEGとして分子量の少なくとも50%を有し得る。1つの態様において、ポリマーは、末端キャップ分子、例えば、ヒドロキシル、アルコキシ、置換アルコキシ、アルケノキシ、置換アルケノキシ、アルキノキシ、置換アルキノキシ、アリールオキシおよび置換アリールオキシで末端を覆われているポリエチレングリコールである。他の態様において、ポリマーはメトキシポリエチレングリコールを含み得る。さらなる態様において、ポリマーは、3kDから100kDまたは5kDから64kDまたは5kDから43kDの範囲のサイズを有するメトキシポリエチレングリコールを含み得る。
【0054】
ポリマーは反応性部分を有し得る。例えば、1つの態様において、ポリマーは、機能性FVIIIポリペプチド上の遊離システインと反応し、共有結合を形成することができるスルフヒドリル反応性部分を有する。このようなスルフヒドリル反応性部分は、チオール、トリフラート、トレシラート、アジリジン、オキシラン、S−ピリジルまたはマレイミド部分を含む。1つの態様において、ポリマーは直鎖状であり、スルフヒドリル(例えば、メトキシ)に対して強く反応しない一方の末端に“キャップ”および他方の末端にスルフヒドリル反応性部分を有する。1つの態様において、複合体はPEG−マレイミドを含み、5kDから64kDの範囲のサイズを有する。
【0055】
有用な生体適合性ポリマーを選択するためのさらなるアドバイスは、以下の実施例において提供される。
【0056】
FVIII活性を有するポリペプチドをコードする核酸配列の部位特異的変異体は、当分野で既知のいずれかの方法により行われ得る。方法は、ポリマーの共有結合に対して選択される部位にシステインコドンを導入するための変異誘発を含む。これは、市販されている部位特異的変異誘発キット、例えば、Stratagene cQuickChangeTM II 部位特異的変異誘発キット、Clontech Transformer 部位特異的変異誘発キットナンバーK1600−1、Invitrogen GenTaylor 部位特異的変異誘発システムナンバー12397014、Promega Altered Sites II インビトロ変異誘発システムキットナンバーQ6210またはTakara Mirus Bio LA PCR 変異誘発キットナンバーTAK RR016を使用して達成され得る。
【0057】
本発明の複合体は、最初に機能性FVIIIポリペプチドの表面上の1つ以上のアミノ酸に対するコドンをシステインに対するコドンで置換し、組換え発現系においてシステイン変異タンパク質を生産し、変異タンパク質をシステイン−特異的ポリマー試薬と反応させ、そして、変異タンパク質を精製することにより製造され得る。
【0058】
このシステムにおいて、システイン部位へのポリマーの付加はポリマー上のマレイミド活性官能基を介して成し遂げることができる。この技術の例は以下に提供されている。使用されるスルフヒドリル反応性ポリマーの量は、誘導体化されるシステインのモル量と少なくとも等モルであり、好ましくは過剰に存在する。例えば、少なくとも5倍モル過剰のスルフヒドリル反応性ポリマーが使用されるか、または少なくとも10倍過剰のこのようなポリマーが使用される。共有結合のために有用な他の条件は当業者に知られている。
【0059】
以下の実施例において、変異タンパク質は当分野の慣用の様式において名付けられる。変異体を名付けるための慣例は、配列番号:1において提供されている全長FVIIIの成熟体に対するアミノ酸配列に基づく。分泌タンパク質として、FVIIIは翻訳プロセス中にタンパク質分解的に開裂されるシグナル配列を含む。19のアミノ酸シグナル配列の除去後、分泌されたFVIII産物の第1のアミノ酸はアラニンである。
【0060】
一般的に、かつ本明細書に使用されているとおり、BDD FVIIIにおける変異アミノ酸に言及しているとき、変異アミノ酸は全長FVIII配列における位置により示される。例えば、以下に記載されているPEG6変異タンパク質は、全長配列における1808と類似である位置にてリシン(K)をシステイン(C)に変化しているとき、K1808Cと示される。
【0061】
ポリマーの共有結合に関する所定の位置は、FVIII活性に関連していないポリペプチドの表面に暴露されている部位から選択されるのが最も良い。このような部位は、また、FVIIIが循環から不活性化されるか、除外されるメカニズムに関連することが知られている部位から選択されるのが最も良い。これらの部位の選択は以下に詳細に記載されている。好ましい部位は、(a)低密度のリポタンパク質受容体関連タンパク質、(b)ヘパリン硫酸プロテオグリカン、(c)低密度のリポタンパク質受容体、および/または(d)FVIII阻害抗体に対する結合部位中またはその付近のアミノ酸残基を含む。“結合部位中またはその付近”とは、生体適合性ポリマーの共有結合が結合部位の立体障害をもたらすであろう、結合部位に非常に近接している残基を意味する。このような部位は、例えば、結合部位の20Å以内であると予期される。
【0062】
本発明の1つの態様において、生体適合性ポリマーは、(a)FVIIIを分解することができるプロテアーゼに対する結合部位、および/または(b)FVIII阻害抗体に対する結合部位中またはその付近のアミノ酸残基にて機能性FVIIIポリペプチドに共有結合している。プロテアーゼは活性化タンパク質C(APC)であり得る。他の態様において、低密度のリポタンパク質受容体関連タンパク質のポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように(例えば、2分の1未満)、生体適合性ポリマーが機能性FVIIIポリペプチド上の所定の位置にて共有結合している。1つの態様において、生体適合性ポリマーは、ヘパリン硫酸プロテオグリカンのポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように(例えば、2分の1未満)、機能性FVIIIポリペプチド上の所定の位置にて共有結合している。さらなる態様において、生体適合性ポリマーは、FVIII阻害抗体のポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように(例えば、複合体化していないときのポリペプチドへの結合の2分の1未満)、機能性FVIIIポリペプチド上の所定の位置にて共有結合している。他の態様において、生体適合性ポリマーは、低密度のリポタンパク質受容体のポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように(例えば、2分の1未満)、機能性FVIIIポリペプチド上の所定の位置にて共有結合している。他の態様において、生体適合性ポリマーは、ポリペプチドが複合体化していないときよりも少ないポリペプチドを血漿プロテアーゼが分解するように、機能性FVIIIポリペプチド上の所定の位置にて共有結合している。さらなる態様において、血漿プロテアーゼによるポリペプチドの分解は、同じ条件下で同じ期間にわたって測定したとき、複合体化していないときのポリペプチドの分解の2分の1未満である。
【0063】
FVIIIに対するLRP、LDL受容体またはHSPG結合親和性は、表面プラズモン共鳴技術(Biacore)を使用して決定することができる。例えば、FVIIIをBiacoreTMチップに直接的に、またはFVIII抗体を介して間接的に被覆し、種々の濃度のLRPをチップ上を通過させ、相互作用のオン速度(on-rate)およびオフ速度(off-rate)両方を測定することができる(Bovenschen, et al., J. Biol. Chem. 278:9370-9377, 2003)。2つの速度の比率は親和性の基準を与える。ペグ化時に親和性の2倍、5倍、10倍または30倍の減少が望ましい。
【0064】
プロテアーゼAPCによるFVIIIの分解は当業者に知られている方法により測定することができる。
【0065】
1つの態様において、方法はFVIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118および2284のうち1つ以上にて、ポリペプチドへ共有結合している生体適合性ポリマーを投与することを含む。他の態様において、生体適合性ポリマーが、FVIIIアミノ酸位置377、378、468、491、504、556、1795、1796、1803、1804、1808、1810、1864、1903、1911および2284のうち1つ以上にて、ポリペプチドへ共有結合しており、さらに(1)複合体の低密度のリポタンパク質受容体関連タンパク質への結合が非複合体化ポリペプチドの低密度のリポタンパク質受容体関連タンパク質への結合より少ないか;(2)複合体の低密度のリポタンパク質受容体への結合が非複合体化ポリペプチドの低密度のリポタンパク質受容体への結合より少ないか;または(3)複合体の低密度のリポタンパク質受容体関連タンパク質および低密度のリポタンパク質受容体両方への結合が非複合体化ポリペプチドの低密度のリポタンパク質受容体関連タンパク質および低密度のリポタンパク質受容体への結合への結合より少ない。
【0066】
さらなる態様において、方法は、FVIIIアミノ酸位置377、378、468、491、504、556および711のうち1つ以上にてポリペプチドへ共有結合している生体適合性ポリマーを投与することを含み、複合体のヘパリン硫酸プロテオグリカンへの結合が非複合体化ポリペプチドのヘパリン硫酸プロテオグリカンへの結合より少ない。さらなる態様において、生体適合性ポリマーは、FVIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118および2284のうち1つ以上にてポリペプチドへ共有結合しており、複合体は非複合体化ポリペプチドよりFVIII阻害抗体への結合が少ない。さらなる態様において、生体適合性ポリマーは、FVIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118および2284のうち1つ以上にて、例えば、位置377、378、468、491、504、556および711のうち1つ以上にてポリペプチドへ共有結合しており、複合体は非複合体化ポリペプチドよりもFVIIIを分解できる血漿プロテアーゼから分解されない。血漿プロテアーゼは活性化タンパク質Cであり得る。
【0067】
さらなる態様において、方法は、アミノ酸位置129、491、1804および/または1808にてB−ドメイン欠失FVIIIに共有結合している生体適合性ポリマーを投与することを含む。さらなる態様において、生体適合性ポリマーはFVIIIアミノ酸位置1804にてポリペプチドに結合しており、ポリエチレングリコールを含む。生体適合性ポリマー結合のための1つ以上の所定の位置は部位特異的システイン変異により制御され得る。
【0068】
機能性FVIIIポリペプチド上の1つ以上、例えば、1つまたは2つの位置は、ポリマー結合に関する所定の位置であり得る。特定の態様において、ポリペプチドはモノ−ペグ化またはジ−ペグ化される。
【0069】
本発明は、また、所定の位置にてコード配列をシステイン残基と置き換えるように機能性FVIIIポリペプチドをコードする核酸配列を変異させ;変異核酸配列を発現させ、システイン増大変異タンパク質を生産し;変異タンパク質を精製し;複合体が形成されるように、実質的に還元されたシステイン残基のみにてポリペプチドと反応するように活性化されている生体適合性ポリマーと変異タンパク質を反応させ;そして、複合体を精製することを含む、複合体の製造のための方法に関する。他の態様において、本発明は、(a)部位特異的FVIII変異タンパク質を発現させ、ここで、変異タンパク質はFVIII変異タンパク質の暴露されている表面上のアミノ酸残基の代わりにシステインを有し、システインはキャップされている;(b)システイン変異タンパク質を穏やかに還元させ、キャップを放出する条件下で、システイン変異タンパク質を還元剤と接触させ;(c)システイン変異タンパク質からキャップおよび還元剤を除去し;そして(d)還元剤の除去から少なくとも約5分、少なくとも15分、少なくとも30分後、ペグ化FVIII変異タンパク質が生産される条件下で、システイン変異タンパク質をスルフヒドリル結合部分を含むPEGで処理することを含む、FVIII変異タンパク質の部位特異的ペグ化のための方法を提供する。PEGのスルフヒドリル結合部分は、チオール、トリフラート、トレシラート、アジリジン、オキシラン、S−ピリジルおよびマレイミド部分からなる群から選択される。
【0070】
本発明は、また、治療有効量本発明の複合体および薬学的に許容されるアジュバントを含む非経口投与用医薬組成物を含む。薬学的に許容されるアジュバントは、活性成分に加えられ得る物質であり、製造物を製剤化または安定化するための助けとなり、患者に有意な有害な毒性効果を引き起こさない。このようなアジュバントの例は当業者に知られており、水、糖、例えば、マルトースまたはスクロース、アルブミン、塩などを含む。他のアジュバントは、例えば、Remington’s Pharmaceutical Sciences by E. W. Martinに記載されている。このような組成物は、宿主への有効な投与のために適当な薬学的に許容される組成物を製造するために、有効量の本複合体を適当な量のビヒクルと共に含む。例えば、複合体は、出血の発現の重症度で変化し得る用量で血友病Aに罹患している対象に非経腸的に投与され得る。血友病Aに対して静脈内に投与される平均用量は、術前適応に対してキログラムあたり40単位、軽度の出血に対してキログラムあたり15から20単位および維持投与のために8時間にわたっての投与に対してキログラムあたり20から40単位の範囲である。vWDの処置に対して、用量はキログラムあたり25−400IUであり得る。vWDに対する他の有効な用量は、25−50、25−100、50−75、50−100、100−200、150−200、200−300、250−300、300−350、300−400、25−250、100−400および200−400IU/kgである。低用量が予防のために有用であり、高用量がFVIIIインヒビターを有する患者における免疫寛容誘導のために有用である。
【0071】
1つの態様において、本発明の方法は、1つ以上の表面BDDアミノ酸をシステインで置換し、哺乳動物発現系においてシステイン変異タンパク質を生産し、培養培地由来のシステインにより発現中にキャップされているシステインを還元し、BDDジスルフィドが再形成できるように還元剤を除去し、そしてシステイン−特異的生体適合性ポリマー試薬、例えば、PEG−マレイミドと反応させることを含む。このような試薬の例は、例えば、Nektar カタログナンバー 2D2M0H01 mPEG-MAL MW 5,000 Da、2D2M0P01 mPEG-MAL MW 20 kD、2D3X0P01 mPEG2-MAL MW 40 kDの下にNektar Therapeutics of San Carlos, CAから入手できる5、22または43kD、または、NOF カタログナンバー Sunbright ME-120MAおよびSunbright ME-300MAの下にNOF Corporation, Tokyo, Japanから入手できる12または33kDのPEGサイズを有するPEG−マレイミドである。ペグ化生産物は、未反応PEGを除去するためにイオン交換クロマトグラフィーを使用して、未反応BDDを除去するためにサイズ排除クロマトグラフィーを使用して精製される。この方法は、同定するために、および、FVIIIとの好ましくない相互作用、例えば、受容体−介在クリアランス、阻害抗体結合およびタンパク質分解酵素による分解から選択的に保護するために使用することができる。PEG試薬が、我々の研究室において6kDとして分析されたものが5kDとしてNektarまたはNOFにより提供され、ならびに、同様にPEG試薬が、我々の研究室において22kDとして分析されたものが直鎖20kDとして提供され、43kDとして分析されたものが40kDとして提供され、および64kDとして分析されたものが60kDとして提供されたことを、我々は指摘する。混乱を避けるため、我々は、5kD PEG(我々は製造業者がそれを確認したとおりの5kDとして報告する)を除いて、本明細書の記載において我々の研究室において分析されているとおりの分子量を使用する。
【0072】
BDDの位置491および1808(上記)でのシステイン変異体に加えて、位置487、496、504、468、1810、1812、1813、1815、1795、1796、1803および1804を、可能性があるペグ化時にLRP結合の妨害を可能にするために、システインに変異させた。また、位置377、378および556を、ペグ化時にLRPおよびHSPG結合両方の妨害を可能にするために、システインに変異させた。これらの位置における大きなPEG(>40kD)での部位特異的ペグ化が、天然のグリコシル化部位(41、239および2118)およびLRP結合部位におけるペグ化と共に、BDDの表面を完全に覆い、BDDに関する新規クリアランスメカニズムを同定することができるように、位置81、129、422、523、570、1864、1911、2091および2284をBDDにおいて等間隔に選択した。
【0073】
1つの態様において、細胞培養培地は、ジスルフィド結合を形成することにより変異タンパク質のシステイン残基を“キャップ”するシステインを含む。複合体の製造において、組換え系において生産されるシステイン変異タンパク質は培地由来のシステインでキャップされ、このキャップはシステイン−特異的ポリマー試薬を加える前にキャップを放出する穏やかな還元により除去される。FVIIIの部位特異的変異体の当分野で既知の他の方法も、当業者に明らかであるため使用され得る。
【0074】
FVIIIの構造活性相関分析
FVIIIおよびBDD FVIIIは、生物学的反応に関連する多数の異なる部位を有する非常に大きな複合体分子である。薬物動態学的特性を改善するためにそれらを共有結合により修飾する以前の試みは、混同した結果であった。分子を特異的に変異させ、次に部位特異的方法においてポリマーを付加させることができることは、驚くべきことであった。さらに、非特異的付加および活性の減少を引き起こすこれまでのポリマー複合体における問題を考慮すると、改良された薬物動態学的特性および維持されている活性の結果も驚く
【0075】
1つの態様において、本発明は、システイン−特異的リガンド、例えば、PEG−マレイミドを使用する部位特異的変異誘発に関する。非変異BDDは、PEG−マレイミドと反応するために利用できるシステインを有さないため、変異システイン位置のみがペグ化の位置である。より具体的には、BDD FVIIIは19個のシステインを有し、このうち16個はジスルフィドを形成し、他の3個は遊離システインである(McMullen, et al., Protein Sci. 4:740-746, 1995)。BDDの構造モデルは、全3個の遊離システインが覆い隠されていることを示唆する(Stoliova-McPhie, et al., Blood 99:1215-1223, 2002)。酸化されているシステインはPEGマレイミドによりペグ化されないため、BDDにおいてジスルフィドを形成する16個のシステインは、最初に還元されることなくペグ化されない。BDDの構造モデルに基づいて、BDDにおける3個の遊離システインは、最初にタンパク質を変性させ、これらのシステインがPEG試薬に暴露されることなくペグ化され得ない。したがって、機能を破壊するようなBDD構造が劇的に変化することなく、天然のシステイン残基にてペグ化によりBDDの特異的なペグ化が達成されることは実行可能であると思えない。
【0076】
全長FVIIIのBドメインにおける4個のシステインのレドックス状態は未知である。Bドメインにおける4個のシステインのペグ化は、それらがジスルフィドを形成しておらず、表面に暴露されているとき可能であり得る。しかしながら、全長FVIIIおよびBDDは同様の薬物動態学(PK)プロフィールおよび同様のインビボ半減期(Gruppo, et al., Haemophilia 9:251-260, 2003)を有するため、PEGが偶然に非Bドメイン領域も保護しないかぎり、Bドメインペグ化が血漿半減期の改善をもたらすことは起こりそうにない。
【0077】
FVIII活性を維持し、薬物動態学を改善するポリマー結合に関するFVIII活性を有するポリペプチドにおける所定の位置を決定するために、以下の指針をBDD FVIIIに基づいて示す。修飾はクリアランス、不活性化および免疫原性メカニズム、例えば、LRP、HSPG、APCおよび阻害抗体結合部位を標的とすべきである。Stoilova-McPhie, et al., (Blood 99:1215-23, 2002)はBDDの構造を示す。例えば、半減期を延長するために、1個のPEGをA2残基484−509およびA3残基1811−1818におけるLRP結合部位、または部位付近における特定の部位に導入することができる。これらの部位への巨大なPEGの導入は、FVIIIがLRPに結合する能力を破壊し、循環からのFVIIIのクリアランスを減少させるはずである。活性に有意に影響せずに半減期を延長するために、PEGを残基1648に導入することができるとも思われ、それは全長分子中のBドメインとA3ドメインの連結部分にあり、BDDではA2及びA3ドメインの間の14−アミノ酸リンカー(linker)中にある。
【0078】
ペグ化の特異性は、組換えDNA変異誘発技術を使用してA2またはA3ドメインへ単一システイン残基を設計し、次にシステイン−特異的PEG試薬、例えば、PEG−マレイミドにて導入されたシステインの部位特異的ペグ化により達成することができる。484−509および1811−1818におけるペグ化のさらなる利点は、これらの2つのエピトープが患者における阻害抗原部位の3つの主なクラスの2つを示すことである。循環半減期の改善および免疫原性応答の減少における最大の効果を達成するために、A2およびA3 LRP結合部位の両方をペグ化し、ジペグ化生成物を得ることができる。1811−1818領域はFIX結合にも関連しているため、この領域内のペグ化は活性の有意な喪失を引き起こし得ることに注意すべきである。また、558−565内の部位特異的ペグ化はHSPG結合を破壊し、この領域がFIXに結合するような活性を減少させ得る。
【0079】
FVIIIの新規クリアランスメカニズムを確認するために、さらなる表面部位をペグ化することができる。A2ドメインは活性化時にFVIIIから解離され、小さなサイズであるため、FVIII分子の残りよりも恐らく速く循環から除去されるという点において、A2ドメインのペグ化はさらなる利点を提供し得る。他方では、ペグ化A2は、腎臓クリアランスから逃れるために十分な大きさであり、FVIIIの残りに匹敵する血漿半減期を有し得、したがってインビボにて活性化FVIIIを再構成することができる。
【0080】
A2およびA3領域におけるペグ化部位の同定。推定A2 LRP結合領域、または領域付近における5つの位置(PEG1−5位置に対応するY487、L491、K496、L504およびQ468)を、高い表面暴露およびそれらのCαからCβへの軌道の外側への向きに基づき、部位特異的ペグ化のための例として選択した。さらに、これらの残基は分子の三次元構造において互いからほぼ等距離であり、それらは一緒になってこの領域全体を表す。推定A3 LRP結合領域、または領域付近における8つの位置(PEG6−14に対応する1808、1810、1812、1813、1815、1795、1796、1803、1804)を部位特異的ペグ化のための例として選択した。PEG6(K1808)は1811−1818および1810における天然のN結合グリコシル化部位に隣接している。位置1810(PEG7)におけるペグ化は糖をPEGで置換されるであろう。PEG8位置T1812における変異体も、グリコシル化部位を破壊するであろう。PEG9位置(K1813)は内側に向いていると予測されたが、構造モデルが正しくない場合にそれを選択した。PEG10(Y1815)はLRP結合ループ内の巨大な疎水性アミノ酸であり、疎水性アミノ酸は一般的にタンパク質−タンパク質相互作用の中心に見られるため、重要な相互作用残基であり得る。1811−1818領域はLRPおよびFIX結合の両方に含まれると報告されているため、このループ内におけるペグ化はおそらく活性の減少をもたらすと考えられた。したがって、PEG11−PEG14(1795、1796、1803、1804)は、異なるPEGサイズによってLRPおよびFIX結合を解離することができるように、1811−1818ループ付近であるがループ内にないように設計した。
【0081】
LRP結合部位の両方を同時にブロックするために、例えば、PEG2およびPEG6位置におけるダブルペグ化(double PEGylation)を生じさせることができる。
【0082】
558−565領域がHSPGおよびFIX両方へ結合することが示されているため、部位をこの領域内に設計しなかった。代わりに、PEG15−PEG17(377、378および556)を、結合したPEGが両方の相互作用を妨げ、それらの間の起こりうる相互作用を崩壊させることができるように、A2 LRPおよびHSPG結合領域間に設計した。表面暴露されており外側に向いているさらなる部位も、LRPおよびHPSG結合領域内または領域付近で選択することができる。新規クリアランスメカニズムを同定するために、FVIIIを系統的にペグ化することができる。PEG1−17に加えて、PEG18−20に対応する3つの他の天然グリコシル化部位、すなわち、N41、N239およびN2118は、表面暴露されているはずであるから、ペグ化のための連結点として使用することができる。PEG2、PEG6および4つのグリコシル化部位のCβ原子から20オングストローム半径内の表面領域は、vWF、FIX、FX、リン脂質およびトロンビンに関する機能性相互作用部位に加えてBDDモデル上にマッピングされた。
【0083】
次にY81、F129、K422、K523、K570、N1864、T1911、Q2091およびQ2284に対応するPEG21−29を、それらのCβ原子のそれぞれから20オングストローム半径で、残っているBDD表面のほぼ全体を覆う能力に基づいて選択した。これらの位置も、完全に暴露されており、外側を向いており、天然のシステインから遠く離れており、正しくないジスルフィド形成の可能性が最小になるため、選択した。大型PEG、例えば、64kD分岐状PEGは約20オングストローム半径を有する球を覆う可能性を有すると予期されるため、20オングストローム半径を選択する。PEG2およびPEG6ならびにグリコシル化部位PEG18、19および20と一緒のPEG21−29のペグ化はFVIIIの非機能性表面のほぼ全体を保護する可能性がある。
【0084】
増強された特性、例えば、改良されたPKプロファイル、より良い安定性または減少した免疫原性を誘導するペグ化の位置は、最大に増強された特性を有する複数−ペグ化された生成物を生産するために組み合わせることができる。PEG30およびPEG31は、A2およびA3ドメイン各々における暴露されているジスルフィドを除去することにより設計した。PEG30またはC630Aは、ペグ化のため、そのジスルフィドパートナーC711を自由にさせる。同様に、PEG31、C1899AはC1903がペグ化されることを可能にする。
【実施例】
【0085】
実施例
本発明をさらに理解するために、以下の実施例を説明する。これらの実施例は説明の目的のためのみであり、いかなる方法によっても本発明の範囲を限定すると解釈してはならない。本明細書に記載されているすべての刊行物を、出典明示よりそれら全体を包含する。
【0086】
実施例1.変異誘発
ペグ化のために選択された部位にシステインコドンを導入することにより、FVIIIの部位特異的ペグ化のための基質を生成することができる。Stratagene cQuickChangeTM II 部位特異的変異誘発キットを使用して、すべてのPEG変異体を生産した(Stratagene Corporation, La Jolla, CA)。cQuikChangeTM部位特異的な変異誘発方法は、PfuTurbo(登録商標)DNAポリメラーゼおよび温度サイクラー(temperature cycler)を使用して行う。所望の変異体を含む2個の相補的オリゴヌクレオチドプライマーを、プライマーを置き換えないであろうPfuTurbo(登録商標)を使用して延長させる。野生型FVIII遺伝子を含むdsDNAを鋳型として使用する。複数回伸長サイクル後、生成物をメチル化DNAに対して特異的であるDpnI エンドヌクレアーゼで消化する。変異体を含む新しく合成されたDNAはメチル化されていないが、親の野生型DNAをメチル化されている。次に消化されたDNAを使用して、XL−1 Blue 超−コンピテント細胞を形質転換させる。
【0087】
変異誘発反応をpSK207+BDD C2.6またはpSK207+BDDのいずれかで行った。FVIIIの部位特異的な変異誘発、変異タンパク質の精製、ペグ化および活性測定の記載は、WO2006/053299(これを出典明示により本明細書に包含させる)において見出すことができる。変異タンパク質の要約を表1に提供する。
表1
【表1】
【0088】
実施例2.vWF結合ELISA
FVIIIを、溶液中の重度の血友病血漿中のvWfに結合させる。次にFVIII−vWf複合体を、vWf−特異的モノクローナル抗体で被覆されているマイクロタイタープレート上に捕獲する。vWfに結合したFVIIIを、FVIIIポリクローナル抗体およびセイヨウワサビペルオキシダーゼ−抗−ウサギ複合体で検出する。ペルオキシダーゼ−複合体化抗体複合体は、基質を添加すると呈色反応を生じる。4パラメーターフィットモデルを使用して、サンプル濃度を標準曲線から補間する。FVIII結合の結果をμg/mLで報告する。ペグ化により活性のいずれにおいても有意な影響はなく、これはBドメインにおけるペグ化と一致する。結果は表2に見出すことができる。
表2
【表2】
【0089】
実施例3.薬物動態活性
ペグ化FVIIIおよびBドメイン欠失FVIII(BDD−FVIII)のPKを、FVIIIノックアウト(KO)マウスにおいて測定した。マウスに、BDD−FVIII(200IU/kg)、アミノ酸位置1804に導入されたシステイン変異体において64kD PEG(64kD PEG14)と複合体化したBDD−FVIII(108IU/kg)、または位置491および1804にてそれぞれシステイン変異体において64kD PEG(64kD PEG2+14)と複合体化したBDD−FVIII(194IU/kg)を静脈内(i.v.)注射によって与えた。血液試料を、5分、4時間、8時間、16時間、24時間、32時間および48時間に処理されたマウス(5匹マウス/処置/時点)から回収した。血漿FVIII活性をCoatest アッセイにより測定した。終末相半減期を、WinNonLinにおける活性vs時間曲線のnon-compartment modelingにより測定した。FVIII KOマウスにおけるBDD−FVIIIに対するt1/2は6時間であるが、64kD PEG(64kD PEG14)または128kD PEG(64kD PEG2+14)と複合体化したFVIIIに対するt1/2はそれぞれ12.43時間および12.75時間である。したがって、ペグ化FVIIIの半減期は、FVIII KOマウスにおけるBDD−FVIIIと比較して、約2倍に増加した。
【0090】
vWF KOマウスにおいて証明されるとおり、循環におけるvWFの欠如はペグ化FVIIIの半減期延長への限界を排除した。マウスにBDD−FVIII(200IU/kg)、64kD PEG14(520IU/kg)、または64kD PEG2+14(400IU/kg)をi.v.投与により投与した。血液試料を、BDDFVIII処置マウスから5分、15分、30分、1時間、2時間、4時間、6時間および8時間に、およびペグ化FVIII処置マウスから5分、4時間、8時間、16時間、24時間、32時間および48時間に回収した(5匹マウス/処置/時点)。vWF KOマウスにおいて約2%の正常レベルである、内因性マウスFVIIIからバックグラウンド活性を除去するために、注入されたヒトFVIIIの血漿活性をCapture Coatestにより測定した。最初に、血漿におけるBDD−FVIIIおよびペグ化FVIIIをヒトFVIIIのA3ドメインに特異的であるmAb R8B12(2ug/mL)により捕獲し、次にCoatestにより測定した。vWFの保護なしに迅速にクリアされ、18分と短いt1/2であるBDD−FVIIIと対照的に、64kD PEG14および64kD PEG2+14のt1/2はそれぞれ5.7時間および8.2時間である(図1)。したがって、FVIII KOマウスにおけるvWFの存在下において観察されるBDD−FVIIIと比較してPEG−FVIIIのt1/2の最大2倍増加と対照的に、64kD PEG14および64kD PEG2+14のt1/2は、vWF KOマウスにおけるvWFの非存在下において、19から27倍延長される。さらに、PEG−FVIIIのt1/2の延長はPEGのサイズに比例する。
【0091】
本明細書に記載されている全ての刊行物および特許は、出典明示により本明細書に包含させる。本発明の精神および範囲から逸脱することなく、記載されている本発明の方法の種々の修飾および変化が当業者に明らかである。
【0092】
本発明は特定の態様と関連して記載されているが、本発明はこのような特定の態様に不都合に限定されるべきでないと理解すべきである。実際に、生化学の分野または関連分野の当業者に明らかである本発明を実施するための上記様式の種々の修飾は、特許請求の範囲内であると意図される。当業者は、わずかに日常の実験を使用して、本明細書に記載されている本発明の特定の態様における多数の均等を認識するか、または確かめることができる。このような均等は特許請求の範囲に包含されることを意図する。
【特許請求の範囲】
【請求項1】
FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量の複合体を必要とする対象に投与することを含む、フォン・ヴィレブランド病を処置するための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号(numerical position)により識別される特定のアミノ酸残基であり、N−末端アミンではない方法。
【請求項2】
生体適合性ポリマーがポリエチレングリコールを含む、請求項1に記載の方法。
【請求項3】
ポリエチレングリコールがメトキシポリエチレングリコールを含む、請求項2に記載の方法。
【請求項4】
メトキシポリエチレングリコールが5kDから64kDの範囲のサイズを有する、請求項3に記載の方法。
【請求項5】
生体適合性ポリマーが(a)FVIIIクリアランス受容体に対する結合部位、(b)FVIIIを分解することができるプロテアーゼに対する結合部位および/または(c)FVIII阻害抗体に対する結合部位中またはその付近のアミノ酸残基にて機能性FVIIIポリペプチドに共有結合している、請求項1に記載の方法。
【請求項6】
低密度のリポタンパク質受容体関連タンパク質のポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように、生体適合性ポリマーが機能性FVIIIポリペプチド上の所定の位置にて共有結合している、請求項1に記載の方法。
【請求項7】
低密度のリポタンパク質受容体関連タンパク質の複合体への結合が、複合体化していないときのポリペプチドへの結合の2分の1未満である、請求項6に記載の方法。
【請求項8】
へパラン硫酸プロテオグリカンのポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように、生体適合性ポリマーが機能性FVIIIポリペプチド上の所定の位置にて共有結合している、請求項1に記載の方法。
【請求項9】
ヘパリン硫酸プロテオグリカンの複合体への結合が、複合体化していないときのポリペプチドへの結合の2分の1未満である、請求項8に記載の方法。
【請求項10】
FVIII阻害抗体のポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように、生体適合性ポリマーが機能性FVIIIポリペプチド上の所定の位置にて共有結合している、請求項1に記載の方法。
【請求項11】
FVIII阻害抗体の複合体への結合が、複合体化していないときのポリペプチドへの結合の2分の1未満である、請求項10に記載の方法。
【請求項12】
低密度のリポタンパク質受容体のポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように、生体適合性ポリマーが機能性FVIIIポリペプチド上の所定の位置にて共有結合している、請求項1に記載の方法。
【請求項13】
低密度のリポタンパク質受容体の複合体への結合が、複合体化していないときのポリペプチドへの結合の2分の1未満である、請求項12に記載の方法。
【請求項14】
血漿プロテアーゼがポリペプチドが、複合体化していないときよりポリペプチドを分解しないように生体適合性ポリマーが機能性FVIIIポリペプチド上の所定の位置にて共有結合している、請求項1に記載の方法。
【請求項15】
血漿プロテアーゼによるポリペプチドの分解が同じ条件下で同じ期間にわたって測定したとき複合体化していないときのポリペプチドの分解の2分の1未満である、請求項14に記載の方法。
【請求項16】
配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にしてFVIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118および2284のうち1つにて、生体適合性ポリマーがポリペプチドへ共有結合している、請求項1に記載の方法。
【請求項17】
配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にしてFVIIIアミノ酸位置377、378、468、491、504、556、1795、1796、1803、1804、1808、1810、1864、1903、1911および2284のうち1つ以上にて、生体適合性ポリマーがポリペプチドへ共有結合しており、さらに(1)複合体の低密度のリポタンパク質受容体関連タンパク質への結合が非複合体化ポリペプチドの低密度のリポタンパク質受容体関連タンパク質への結合より少ないか;(2)複合体の低密度のリポタンパク質受容体への結合が非複合体化ポリペプチドの低密度のリポタンパク質受容体への結合より少ないか;または(3)複合体の低密度のリポタンパク質受容体関連タンパク質および低密度のリポタンパク質受容体両方への結合が非複合体化ポリペプチドの低密度のリポタンパク質受容体関連タンパク質および低密度のリポタンパク質受容体への結合より少ない、請求項1に記載の方法。
【請求項18】
配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にして、FVIIIアミノ酸位置377、378、468、491、504、556および711のうち1つ以上にて、生体適合性ポリマーがポリペプチドへ共有結合しており、さらに、複合体のヘパリン硫酸プロテオグリカンへの結合が非複合体化ポリペプチドのヘパリン硫酸プロテオグリカンへの結合より少ない、請求項1に記載の方法。
【請求項19】
配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にして、FVIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118および2284のうち1つ以上にて、生体適合性ポリマーがポリペプチドへ共有結合しており、複合体が非複合体化ポリペプチドよりもFVIII阻害抗体への結合が少ない、請求項1に記載の方法。
【請求項20】
配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にして、FVIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118および2284のうち1つ以上にて、生体適合性ポリマーがポリペプチドへ共有結合しており、複合体が非複合体化ポリペプチドよりもFVIIIを分解できる血漿プロテアーゼから分解されない、請求項1に記載の方法。
【請求項21】
血漿プロテアーゼが活性化タンパク質Cである、請求項20に記載の方法。
【請求項22】
機能性FVIIIポリペプチドがBドメイン欠失FVIIIである、請求項1に記載の方法。
【請求項23】
生体適合性ポリマーが配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にして、アミノ酸位置129、491、1804および/または1808にてBドメイン欠失FVIIIに共有結合している、請求項22に記載の方法。
【請求項24】
生体適合性ポリマーが配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にして、FVIIIアミノ酸位置1804にてポリペプチドに結合しており、ポリエチレングリコールを含む、請求項1に記載の方法。
【請求項25】
生体適合性ポリマー結合のための1つ以上の所定の位置がシステイン残基である、請求項1に記載の方法。
【請求項26】
フォン・ヴィレブランド病がフォン・ヴィレブランド因子の欠乏および/または異常により特徴付けられる、請求項1に記載の方法。
【請求項27】
フォン・ヴィレブランド病がN2型である、請求項1に記載の方法。
【請求項28】
フォン・ヴィレブランド病が3型である、請求項1に記載の方法。
【請求項29】
FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる複合体を製造することを含む、フォン・ヴィレブランド病を処置するための薬物を製造するための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号により識別される特定のアミノ酸残基であり、N−末端アミンではない方法。
【請求項30】
FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量のFVIIIのシステイン置換変異体を必要とする対象に投与することを含む、フォン・ヴィレブランド病を処置するための方法であって、該変異体はFVIII配列におけるアミノ酸に対して置換されたシステイン残基を有することにより特徴付けられ、該置換は配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にして、システイン残基がFVIIIに存在しないアミノ酸位置にてシステイン残基をもたらし、該システイン付加変異体は該置換システイン残基へ共有結合している生体適合性ポリマーを有することによりさらに特徴付けられる方法。
【請求項31】
生体適合性ポリマーがポリエチレングリコールを含む、請求項30に記載の方法。
【請求項32】
FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量の複合体を必要とする対象に手術前に投与することを含み、それによって一過性の出血を弱める予防処置のための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号により識別される特定のアミノ酸残基であり、N−末端アミンではない方法。
【請求項33】
対象が3型vWDを有する、請求項32に記載の方法。
【請求項34】
生体適合性ポリマーがポリエチレングリコールを含む、請求項32に記載の方法。
【請求項35】
生体適合性ポリマー結合のための1つ以上の所定の位置がシステイン残基である、請求項32に記載の方法。
【請求項36】
FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量の複合体を外傷対象に投与することを含み、それによって一過性の出血を弱める外傷の処置のための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号により識別される特定のアミノ酸残基であり、N−末端アミンではない方法。
【請求項37】
対象が3型vWDを有する、請求項36に記載の方法。
【請求項38】
生体適合性ポリマーがポリエチレングリコールを含む、請求項36に記載の方法。
【請求項39】
生体適合性ポリマー結合のための1つ以上の所定の位置がシステイン残基である、請求項36に記載の方法。
【請求項1】
FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量の複合体を必要とする対象に投与することを含む、フォン・ヴィレブランド病を処置するための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号(numerical position)により識別される特定のアミノ酸残基であり、N−末端アミンではない方法。
【請求項2】
生体適合性ポリマーがポリエチレングリコールを含む、請求項1に記載の方法。
【請求項3】
ポリエチレングリコールがメトキシポリエチレングリコールを含む、請求項2に記載の方法。
【請求項4】
メトキシポリエチレングリコールが5kDから64kDの範囲のサイズを有する、請求項3に記載の方法。
【請求項5】
生体適合性ポリマーが(a)FVIIIクリアランス受容体に対する結合部位、(b)FVIIIを分解することができるプロテアーゼに対する結合部位および/または(c)FVIII阻害抗体に対する結合部位中またはその付近のアミノ酸残基にて機能性FVIIIポリペプチドに共有結合している、請求項1に記載の方法。
【請求項6】
低密度のリポタンパク質受容体関連タンパク質のポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように、生体適合性ポリマーが機能性FVIIIポリペプチド上の所定の位置にて共有結合している、請求項1に記載の方法。
【請求項7】
低密度のリポタンパク質受容体関連タンパク質の複合体への結合が、複合体化していないときのポリペプチドへの結合の2分の1未満である、請求項6に記載の方法。
【請求項8】
へパラン硫酸プロテオグリカンのポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように、生体適合性ポリマーが機能性FVIIIポリペプチド上の所定の位置にて共有結合している、請求項1に記載の方法。
【請求項9】
ヘパリン硫酸プロテオグリカンの複合体への結合が、複合体化していないときのポリペプチドへの結合の2分の1未満である、請求項8に記載の方法。
【請求項10】
FVIII阻害抗体のポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように、生体適合性ポリマーが機能性FVIIIポリペプチド上の所定の位置にて共有結合している、請求項1に記載の方法。
【請求項11】
FVIII阻害抗体の複合体への結合が、複合体化していないときのポリペプチドへの結合の2分の1未満である、請求項10に記載の方法。
【請求項12】
低密度のリポタンパク質受容体のポリペプチドへの結合が、複合体化していないときのポリペプチドへの結合よりも少ないように、生体適合性ポリマーが機能性FVIIIポリペプチド上の所定の位置にて共有結合している、請求項1に記載の方法。
【請求項13】
低密度のリポタンパク質受容体の複合体への結合が、複合体化していないときのポリペプチドへの結合の2分の1未満である、請求項12に記載の方法。
【請求項14】
血漿プロテアーゼがポリペプチドが、複合体化していないときよりポリペプチドを分解しないように生体適合性ポリマーが機能性FVIIIポリペプチド上の所定の位置にて共有結合している、請求項1に記載の方法。
【請求項15】
血漿プロテアーゼによるポリペプチドの分解が同じ条件下で同じ期間にわたって測定したとき複合体化していないときのポリペプチドの分解の2分の1未満である、請求項14に記載の方法。
【請求項16】
配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にしてFVIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118および2284のうち1つにて、生体適合性ポリマーがポリペプチドへ共有結合している、請求項1に記載の方法。
【請求項17】
配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にしてFVIIIアミノ酸位置377、378、468、491、504、556、1795、1796、1803、1804、1808、1810、1864、1903、1911および2284のうち1つ以上にて、生体適合性ポリマーがポリペプチドへ共有結合しており、さらに(1)複合体の低密度のリポタンパク質受容体関連タンパク質への結合が非複合体化ポリペプチドの低密度のリポタンパク質受容体関連タンパク質への結合より少ないか;(2)複合体の低密度のリポタンパク質受容体への結合が非複合体化ポリペプチドの低密度のリポタンパク質受容体への結合より少ないか;または(3)複合体の低密度のリポタンパク質受容体関連タンパク質および低密度のリポタンパク質受容体両方への結合が非複合体化ポリペプチドの低密度のリポタンパク質受容体関連タンパク質および低密度のリポタンパク質受容体への結合より少ない、請求項1に記載の方法。
【請求項18】
配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にして、FVIIIアミノ酸位置377、378、468、491、504、556および711のうち1つ以上にて、生体適合性ポリマーがポリペプチドへ共有結合しており、さらに、複合体のヘパリン硫酸プロテオグリカンへの結合が非複合体化ポリペプチドのヘパリン硫酸プロテオグリカンへの結合より少ない、請求項1に記載の方法。
【請求項19】
配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にして、FVIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118および2284のうち1つ以上にて、生体適合性ポリマーがポリペプチドへ共有結合しており、複合体が非複合体化ポリペプチドよりもFVIII阻害抗体への結合が少ない、請求項1に記載の方法。
【請求項20】
配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にして、FVIIIアミノ酸位置81、129、377、378、468、487、491、504、556、570、711、1648、1795、1796、1803、1804、1808、1810、1864、1903、1911、2091、2118および2284のうち1つ以上にて、生体適合性ポリマーがポリペプチドへ共有結合しており、複合体が非複合体化ポリペプチドよりもFVIIIを分解できる血漿プロテアーゼから分解されない、請求項1に記載の方法。
【請求項21】
血漿プロテアーゼが活性化タンパク質Cである、請求項20に記載の方法。
【請求項22】
機能性FVIIIポリペプチドがBドメイン欠失FVIIIである、請求項1に記載の方法。
【請求項23】
生体適合性ポリマーが配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にして、アミノ酸位置129、491、1804および/または1808にてBドメイン欠失FVIIIに共有結合している、請求項22に記載の方法。
【請求項24】
生体適合性ポリマーが配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にして、FVIIIアミノ酸位置1804にてポリペプチドに結合しており、ポリエチレングリコールを含む、請求項1に記載の方法。
【請求項25】
生体適合性ポリマー結合のための1つ以上の所定の位置がシステイン残基である、請求項1に記載の方法。
【請求項26】
フォン・ヴィレブランド病がフォン・ヴィレブランド因子の欠乏および/または異常により特徴付けられる、請求項1に記載の方法。
【請求項27】
フォン・ヴィレブランド病がN2型である、請求項1に記載の方法。
【請求項28】
フォン・ヴィレブランド病が3型である、請求項1に記載の方法。
【請求項29】
FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる複合体を製造することを含む、フォン・ヴィレブランド病を処置するための薬物を製造するための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号により識別される特定のアミノ酸残基であり、N−末端アミンではない方法。
【請求項30】
FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量のFVIIIのシステイン置換変異体を必要とする対象に投与することを含む、フォン・ヴィレブランド病を処置するための方法であって、該変異体はFVIII配列におけるアミノ酸に対して置換されたシステイン残基を有することにより特徴付けられ、該置換は配列番号:1の全長ヒトFVIIIアミノ酸配列である成熟体を基準にして、システイン残基がFVIIIに存在しないアミノ酸位置にてシステイン残基をもたらし、該システイン付加変異体は該置換システイン残基へ共有結合している生体適合性ポリマーを有することによりさらに特徴付けられる方法。
【請求項31】
生体適合性ポリマーがポリエチレングリコールを含む、請求項30に記載の方法。
【請求項32】
FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量の複合体を必要とする対象に手術前に投与することを含み、それによって一過性の出血を弱める予防処置のための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号により識別される特定のアミノ酸残基であり、N−末端アミンではない方法。
【請求項33】
対象が3型vWDを有する、請求項32に記載の方法。
【請求項34】
生体適合性ポリマーがポリエチレングリコールを含む、請求項32に記載の方法。
【請求項35】
生体適合性ポリマー結合のための1つ以上の所定の位置がシステイン残基である、請求項32に記載の方法。
【請求項36】
FVIII凝血促進活性を有し、ヒトFVIII欠乏症を修正することができる治療有効量の複合体を外傷対象に投与することを含み、それによって一過性の出血を弱める外傷の処置のための方法であって、該複合体はポリペプチド上の1つ以上の所定の位置にて1つ以上の生体適合性ポリマーへ共有結合している機能性FVIIIポリペプチドを含み、該所定の位置はポリペプチドのアミノ酸配列における位置番号により識別される特定のアミノ酸残基であり、N−末端アミンではない方法。
【請求項37】
対象が3型vWDを有する、請求項36に記載の方法。
【請求項38】
生体適合性ポリマーがポリエチレングリコールを含む、請求項36に記載の方法。
【請求項39】
生体適合性ポリマー結合のための1つ以上の所定の位置がシステイン残基である、請求項36に記載の方法。
【図1】

【公表番号】特表2011−523663(P2011−523663A)
【公表日】平成23年8月18日(2011.8.18)
【国際特許分類】
【出願番号】特願2011−512670(P2011−512670)
【出願日】平成21年6月4日(2009.6.4)
【国際出願番号】PCT/US2009/046327
【国際公開番号】WO2009/149303
【国際公開日】平成21年12月10日(2009.12.10)
【出願人】(503106111)バイエル・ヘルスケア・エルエルシー (154)
【Fターム(参考)】
【公表日】平成23年8月18日(2011.8.18)
【国際特許分類】
【出願日】平成21年6月4日(2009.6.4)
【国際出願番号】PCT/US2009/046327
【国際公開番号】WO2009/149303
【国際公開日】平成21年12月10日(2009.12.10)
【出願人】(503106111)バイエル・ヘルスケア・エルエルシー (154)
【Fターム(参考)】
[ Back to top ]