
フグ毒の凍結乾燥注射用粉末製剤及びその製造法
クリスタル・メス類、ヘロイン等のアヘン類、大麻類の薬物依存の断薬症状に使用し、迅速で顕著な効果を備えるフグ毒の凍結乾燥注射用粉末製剤及びその製造方法を提供する。安定性を備えるフグ毒の凍結乾燥注射用粉末製剤及びその製造方法において、本製剤は、フグ毒を主要有効成分とし、更に溶解補助剤、賦形剤、安定剤を含む。前記溶解補助剤はクエン酸、凍結乾燥賦形剤は、塩化ナトリウム(sodium)、マンニトール(mannitol)、二者の複合物の何れかとし、安定剤は、デキストラン(dextran)、トレハロース(trehalose)、二者の複合物の何れかとする。その内、フグ毒と賦形剤と安定剤の比率は、1:150〜3000:50〜500或50〜6000とする。好ましくは、塩酸リドカインの機能調節剤を含む。本発明の凍結乾燥注射用粉末製剤は、アヘン類及び大麻類の薬物依存症による断薬症状を乗り越えるために用いる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、フグ毒(TTX,テトロドトキシン)の凍結乾燥注射用粉末製剤及びその製造法に係る。具体的に述べると、本発明は、正確な一定量のフグ毒を主要有効成分とし、最適な骨格を形成する賦形剤と安定剤を含む凍結乾燥注射用粉末製剤に関する。本発明の凍結乾燥注射用粉末製剤は、クリスタル・メス(Crystal meth)類、ヘロイン(heroin)等のアヘン類、及び大麻類の薬物依存による断薬症状に使用するもので、迅速で顕著な効果を奏する。また、本発明の凍結乾燥注射用粉末製剤は、安定性、安全性が高く、更に人体に対する剌激性も少ないものである。
【背景技術】
【0002】
フグ毒は、自然の非タンパク性神経毒で、化学構造式は次に示されるとおりである。
【0003】
【化1】

化学名:オクタヒドロ-12-(ヒドロキシメチル)-2-イミノ-5,9:7,10a-ジメタノ-10aH-[1,3] ジオキソシノ[6,5-d]ピリミジン-4,7,10,11,12-ペンタオール
Octahydro-12-(hydroxymethyl)-2-imino-5,9:7,10a-dimethano-10aH-[1,3]dioxocino[6,5-d]pyrimidine-4,7,10,11,12-pentol
英文名:Tetrodotoxin(テトロドトキシン)、略してTTXと呼ぶ。
【0004】
研究によって、フグ毒が断薬剤として、クリスタル・メス類、ヘロイン等アヘン類及び大麻類の薬物依存による断薬症状に用いることができ、中毒発作を抑制し、断薬反応を取り除き、使用中止後も依存性を生じないことが発見されている。
【0005】
フグ毒の結晶は、熱に対して非常に安定しており、40℃の条件の下で6ヶ月間おいても、フグ毒の結晶の品質には基本的に変化はみられない(表1参照)。しかし、フグ毒の水溶液は、温度に非常に敏感で、温度の影響を極めて受け易く、分解を起こす。温度が高いほど、分解も早い。40℃の条件の下、通常のフグ毒の注射液は30日間放置後、その含量は、99.20%から65.57%に下がり、33.63%の低下が明らかである (表2参照)。
【0006】
【表1】
【0007】
【表2】
【0008】
水溶液中の不安定な薬物について言えば、安定した注射可能な製剤を得るために、それを用いて凍結乾燥させた製剤をつくることは、本分野が通常採用する一般的な方法である。例えば、特許文献1で開示された「安定性を備えるフグ毒の冷凍乾燥製剤」において、製剤は、乳糖(lactose)、蔗糖(sucrose)、麦芽糖(maltose)、セロビオース(cellobiose)等の二糖類(disacchrides)、縮合型葡萄糖(condensed glucose)やデキストラン(dextran)等の多糖類、或いは、ヒドロキシル(hydroxyl)澱粉、ヒドロキシプロピルシクロデキストリン(hydroxypropyl cyclodextrin)等の誘導体から選択した安定剤を含み、その用量は一製剤あたり5〜100mgとする。また、製剤中には更に、クエン酸(citric acid)、酒石酸(tartaric acid)、リンゴ酸(malic acid)、或いはラクトビオン酸(lactobionic acid)から選択した溶解補助剤を含み、その用量は一製剤あたり0.00005〜0.0005mgとする。特許文献1において指摘したとおり、クエン酸塩或いはマンニトール(mannitol)を賦形剤とした時、得られるフグ毒の凍結乾燥させた製剤は不安定であるため、フグ毒の含量は貯存過程において徐々に下降する。
【0009】
よって、安全性及び安定性を備え、品質をコントロールでき、長期的に貯存でき、フグ毒の臨床に応用する薬用製剤の研究開発が必要である。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】CN 1835754A
【発明の概要】
【発明が解決しようとする課題】
【0011】
安定したフグ毒製剤を得るために、出願人はフグ毒の凍結乾燥注射用粉末製剤及びその製作技術について深い研究を進めた結果、凍結乾燥注射用粉末製剤の骨格を形成する賦形剤、フグ毒の安定剤、溶解補助剤、処方溶液のpH範囲、真空冷凍昇華乾燥技術、注射用粉末製剤含水量の制御等の要因がいずれも注射用粉末製剤の安定性に影響を与えることが明らかにされた。特に、注射用粉末製剤の骨格を形成する担体の引湿性である。引湿性が強い補助剤は、貯蔵過程において時間が延びるほど製剤中の含水量はゆっくりと上昇する傾向がみられ、含水量の増加によって製剤中の主成分であるフグ毒の含量を下降させる。
【0012】
数多くの実験による研究から、本発明者は、等張化剤の塩化ナトリウム(sodium)或いはマンニトール(mannitol)を凍結乾燥の注射用粉末製剤の骨格を形成する補助剤とし、デキストラン(dextran)20またはトレハロース(trehalose)をフグ毒主薬の安定剤として選択し、しかも、冷凍乾燥前のpH値を3.5〜4.5に調節する時、白色のホットケーキ状の外観を備えたフグ毒凍結乾燥注射用粉末製剤が得られることを発見した。製剤の主な薬剤量が正確で、外観に優れ、品質安定性が良好、安全性が高く、人体に対して安全に注射する要望を満たすものである。
【課題を解決するための手段】
【0013】
そこで、本発明は、フグ毒、溶解補助剤、凍結乾燥賦形剤、及び安定剤を含み、人体に対して安全に注射できるフグ毒の凍結乾燥注射用粉末製剤を提供する。前記フグ毒の純度は>96%とし、より好ましくは、98%〜99.8%とする。前記凍結乾燥の賦形剤は塩化ナトリウム(sodium)、マンニトール(mannitol)、二者の複合物のいずれかとする。また、前記安定剤は、デキストラン(dextran)、トレハロース(trehalose)、二者の複合物のいずれかとし、前記溶解補助剤はクエン酸とする。
【0014】
本発明のフグ毒凍結乾燥注射用粉末製剤の、フグ毒:賦形剤:安定剤の比率は、好ましくは、1: 150〜3000: 50〜500或いは50〜6000とする。
【0015】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、一製剤あたりのフグ毒の含量は0.1から20.0μgとし、好ましくは、0.5から20.0μg、より好ましくは、0.5から12.0μgとする。
【0016】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、賦形剤としての塩化ナトリウム(sodium)の含量を一製剤あたり1.0〜30mgとし、好ましくは、5.0〜30mg、より好ましくは、5.0〜20mgとする。
【0017】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、賦形剤としてのマンニトール(mannitol)の含量を一製剤あたり1.0〜30mgとし、好ましくは、1.0〜20mg、より好ましくは、3.0〜10mgとする。
【0018】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、安定剤としてのデキストラン(dextran)の含量を一製剤あたり0.5〜5.0mgとし、好ましくは、2.0〜5.0mg、より好ましくは、3.0〜5.0mgとする。
【0019】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、安定剤としてのトレハロース(trehalose)の含量を一製剤あたり0.5〜60mgとし、好ましくは、2.0〜60mg、より好ましくは、10〜60mgとする。
【0020】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、クエン酸の含量を一製剤あたり0.001〜0.080mgとし、好ましくは、0.010〜0.080mgとし、より好ましくは、0.020〜0.060mgとする。
【0021】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、好ましくは、機能調節剤を含み、前記機能調節剤は、好ましくは、塩酸リドカインとする。
【0022】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、好ましくは、高純度窒素ガス或いは高純度炭酸ガスの惰性気体を充填する。
【0023】
本発明のフグ毒の凍結乾燥注射用粉末製剤は、好ましくは、筋肉或いは皮下に対して注射で薬剤を投与する形式を選択し、しかも、無菌注射を用いて水に溶解する場合、無菌注射の使用する水量は一製剤あたり0.5〜2.0mlとする。
【0024】
また、本発明が更に開示する「フグ毒の凍結乾燥注射用粉末製剤の製造法」での工程は、次のとおりである。
(1)予定量のフグ毒を直接、溶解補助剤を含む溶液中に入れ、pH値を3.0〜6.0に調節し、好ましくは、pH3.5〜4.5とする。ろ過して熱を取り除く。
(2)凍結乾燥賦形剤と安定剤、及び任意に選択した機能調節剤を直接、無菌注射用水中に入れ、活性炭素を加えて30分間撹拌した後、ろ過して熱を取り除く。
(3)(1)と(2)において得られた溶液を共に均等に混合し、ろ過して除菌し、定量をバイアル瓶に詰め、真空冷凍乾燥を行い、惰性気体を充填し、圧して緊密に蓋をし、凍結乾燥注射用粉末製剤を得る。
【0025】
本発明の製造法の(1)工程においては、好ましくは、超ろ過法によってろ過を行なう。
【0026】
本発明の製造法の(2)工程においては、好ましくは、活性炭素の用量を0.1〜6.0g/100mlとする。
【0027】
本発明の製造法の(3)工程においては、0.05μm〜0.20μmのサブミクロン級微細孔を備えるろ過膜或いは微細孔を備える荷電ろ過膜によるろ過を行なう。
【発明の効果】
【0028】
本発明のフグ毒の凍結乾燥注射用粉末製剤は、クリスタル・メス(Crystal meth)類、ヘロイン(heroin)等のアヘン類、及び大麻類の薬物依存による断薬症状に使用するもので、迅速で顕著な効果を奏する。また、本発明の凍結乾燥注射用粉末製剤は、安定性、安全性が高く、更に人体に対する剌激性も少ないものである。
【図面の簡単な説明】
【0029】
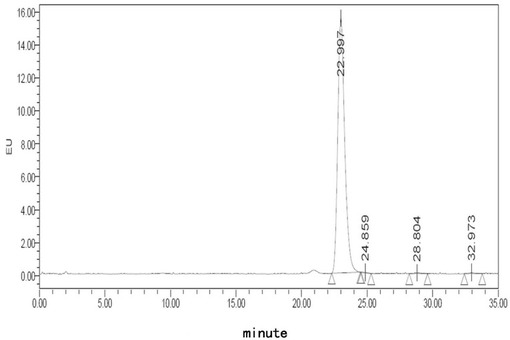
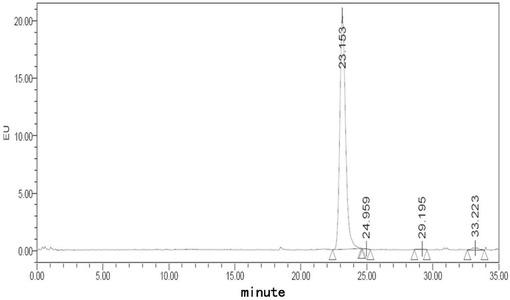
【図1】フグ毒注射液(40℃、0日)に対する蛍光検出HPLCチャートである。
【図2】フグ毒注射液(40℃、10日)に対する蛍光検出HPLCチャートである。
【図3】処方Alでのフグ毒の凍結乾燥注射用粉末製剤(40℃、0日)に対する蛍光検出HPLCチャートである。
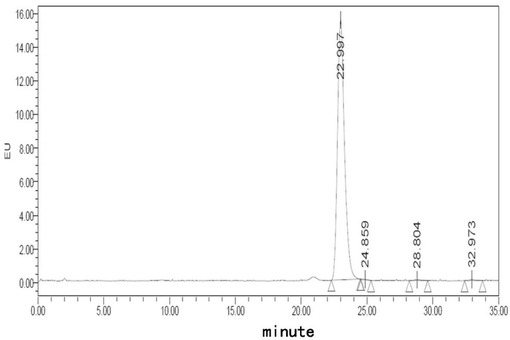
【図4】処方Alでのフグ毒の凍結乾燥注射用粉末製剤(40℃、10日)に対する蛍光検出HPLCチャートである。
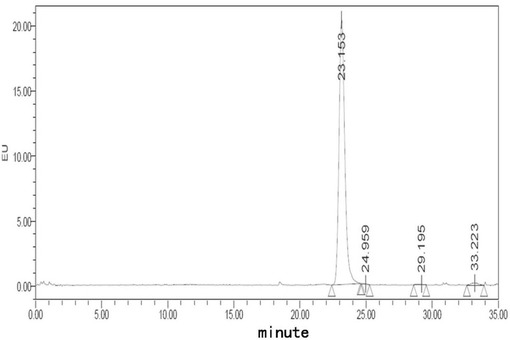
【図5】処方Blでのフグ毒の凍結乾燥注射用粉末製剤(40℃、0日)に対する蛍光検出HPLCチャートである。
【図6】処方Blでのフグ毒の凍結乾燥注射用粉末製剤(40℃、10日)に対する蛍光検出HPLCチャートである。
【図7】処方Clでのフグ毒の凍結乾燥注射用粉末製剤(40℃、0日)に対する蛍光検出HPLCチャートである。
【図8】処方Clでのフグ毒の凍結乾燥注射用粉末製剤(40℃、10日)に対する蛍光検出HPLCチャートである。
【図9】処方D5でのフグ毒の凍結乾燥注射用粉末製剤(40℃、0日)に対する蛍光検出HPLCチャートである。
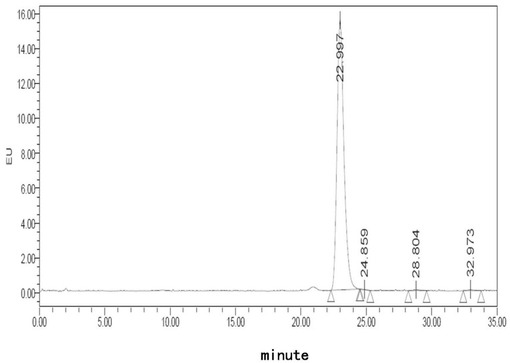
【図10】処方D5でのフグ毒の凍結乾燥注射用粉末製剤(40℃、10日)に対する蛍光検出HPLCチャートである。
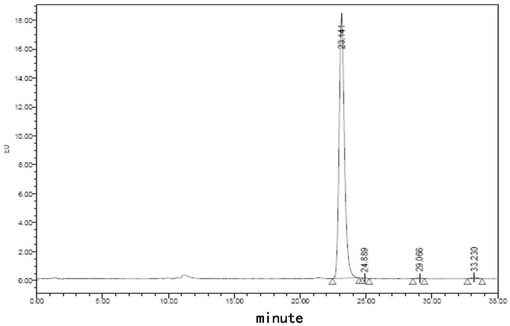
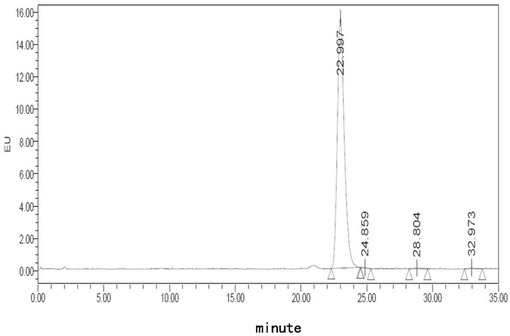
【図11】処方E5でのフグ毒の凍結乾燥注射用粉末製剤(40℃、0日)に対する蛍光検出HPLCチャートである。
【図12】処方E5でのフグ毒の凍結乾燥注射用粉末製剤(40℃、10日)に対する蛍光検出HPLCチャートである。
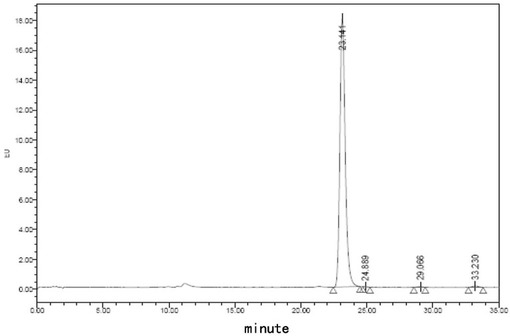
【図13】処方F5でのフグ毒の凍結乾燥注射用粉末製剤(40℃、0日)に対する蛍光検出HPLCチャートである。
【図14】処方F5でのフグ毒の凍結乾燥注射用粉末製剤(40℃、10日)に対する蛍光検出HPLCチャートである。
【発明を実施するための形態】
【0030】
本明細書において、本発明のフグ毒の凍結乾燥注射用粉末製剤に対するフグ毒含量の測定は、高速液体クロマトグラフィー(HPLC。中国薬典2005版二部付録VII)を用いて行なった。含量測定項目下に記録されているクロマトグラフにおいて、供試品溶液の保留時間の主ピークと、対照品溶液の保留時間の主ピークとは一致する。
【0031】
採用するクロマトグラフの条件は次のとおりである。オクチルシラン結合シリカゲル(octylsilane bonded silica gel)充填剤に用い、オクタンスルホン酸ナトリウムのリン酸塩緩衝液を移動相とする。流速を1分あたり0.3ml、励起波長を365nm、発射波長を510nm、ポストカラム誘導体化剤を4mol/Lの水酸化ナトリウム溶液とする。誘導体化剤の流速を1分あたり0.2〜0.5 ml、ポストカラム誘導化温度を100°C〜140°Cとする。理論段数は、フグ毒のピークに基づいて計算し、2000以下にはならない。
【0032】
具体的な測定方法は次のとおりである。
本発明のフグ毒の凍結乾燥注射用粉末製剤(フグ毒10μgを含む)、精密に水2.0mlを加えて溶解し、精密に20μlを取り出し、予測に基づくクロマトグラフの条件の下で投入し、蛍光検出クロマトグラフを記録する。別にフグ毒の対照品を適量取り出す。同法で測定する。外部標準法に基づくピーク面積により一瓶あたりの一製剤のフグ毒含量を算出する。フグ毒含量が表示量の90%〜110%である時、凍結乾燥注射用粉末製剤の医薬品要求に符合する。
【0033】
関連物質の測定:本発明のフグ毒の凍結乾燥注射用粉末製剤を取り、水を加えて1mlあたりフグ毒20μgを含む溶液を生成し、供試品溶液とする。精密にl.0mlを取り、25mlの瓶中に入れ、水を加えて目盛まで希釈し、振って均一にしたものを対照溶液とする。精密に供試品溶液と対照品溶液を各20μl取り出し、それぞれを液相クロマトグラフィー装置に注入し、主成分のピーク保留時間が2倍となるまで蛍光検出クロマトグラフを記録する。供試品溶液のクロマトグラフ中の各雑質ピーク面積の和が対照溶液ピーク面積を超えない時、医薬品要求に符合する。
【0034】
本発明では、蛍光検出による高速液体クロマトグラフィーを用いてフグ毒及び関連物質の含量に対する検出を行なう。通常の紫外可視吸光検出による高効液相クロマトグラフィーと比較すると、蛍光検出による高効液相クロマトグラフィーの方が敏感度が更に高く、測定の正確度も高いことから、フグ毒製剤の安定性を更に良好に測定できることが理解される。例えば、フグ毒製剤について言えば、紫外可視吸光検出による高速液体クロマトグラフィーの最低検出制限値は8.14ngであるが、蛍光検出による高速液体クロマトグラフィーでは0.40ngで、蛍光検出は紫外可視吸光検出の20倍も低いことがわかる。また、フグ毒製剤について言えば、紫外可視吸光検出による高速液体クロマトグラフィーの定量制限値は20.26ngであるが、蛍光検出による高速液体クロマトグラフィーでは0.81ngで、蛍光検出の定量制限値は紫外可視吸光検出の25倍となる。
【0035】
具体的に言えば、注射用フグ毒製剤の通常仕様は一瓶あたり10μgであり、サンプルを取り出し溶解した後、測定に用いる製剤を投入する。投入濃度を5μg /ml〜 20μg/ml、投入量を20μlとする時、サンプル中の雑質含量が1%であるなら、雑質量はわずか1〜4ngで、紫外可視吸光検出による液体クロマトグラフィーでの検出制限値を下回る。サンプル中の雑質が2%である時、雑質量はわずか2〜8ngで、紫外可視吸光検出による液体クロマトグラフィーの検出制限値を下回る。紫外可視吸光検出による液体クロマトグラフィーを用いて製剤中の関連雑質或いは含量を測定する場合、製品中の5%を下回る雑質は、検出制限値が要求を満足しないために検出されず、定量制限値を下回ることにより測定誤差が引き起こされる。それに対して、蛍光検出による液体クロマトグラフィーの検出制限値は0.40ngであるため、完全に検出要求を満たすものである。
【0036】
以上の測定条件を採用し、出願人は、フグ毒の凍結乾燥注射用粉末製剤の安定性に影響を及ぼす各種要素についての研究を行なった。
【0037】
次に、フグ毒凍結乾燥注射用粉末製剤の賦形剤と安定剤の選択についての説明を行なう。
【0038】
フグ毒の臨床使用量はマイクログラム級である。よって、凍結乾燥注射用粉末製剤の骨格を形成するための助剤となる賦形剤を添加する必要がある。それと同時に、フグ毒注射用粉末製剤の安定性の要望に基づき、安定剤の選択も極めて重要である。多数の実験を重ねた結果、出願人は、塩化ナトリウム(sodium)或いは/及びマンニトール(mannitol)をフグ毒の凍結乾燥注射用粉末製剤の賦形剤とし、デキストラン(dextran)20或いはトレハロース(trehalose)を安定剤として選択することにより良好な結果が得られることを発見した。次の表3及び表4には、塩化ナトリウム(sodium)及び/或いはマンニトール(mannitol)を賦形剤としデキストラン(dextran)20或いはトレハロース(trehalose)を安定剤とした本発明を代表するフグ毒の凍結乾燥注射用粉末製剤の処方及び40°C下での安定性試験結果を示す。
【0039】
【表3】
【0040】
【表4】
【0041】
その内、賦形剤とするマンニトール(mannitol)製品の外観は極めて良好で、しかも吸湿性が弱い。デキストラン(dextran)は、フグ毒の凍結乾燥製剤に対して顕著な保護作用を備えるが、吸湿性が強い。凍結乾燥の注射用粉末製剤の含水量をコントロールし、充満した凍結乾燥の骨格を形成するために、本発明製剤中のデキストラン(dextran)20含量を一製剤あたり4mgとし、マンニトール(mannitol)の含量を一製剤あたり6mgとする。また、等張化剤の塩化ナトリウム(sodium)を骨格形成の賦形剤とし、デキストラン(dextran)20或いはトレハロース(trehalose)を安定剤として組成する処方の場合、製剤の含水量は低く、外観は充満したホットケーキ状となる。実際の応用では水を加えた後は迅速に再溶解する。40°Cの高温下の10日間安定性試験は医薬品要求に符合するものである。
【0042】
次に、溶解補助剤と処方pH値の選択について説明を行なう。
【0043】
フグ毒は、相対分子質量を319.27とする海洋生物の非蛋白性神経毒であり、通常、両性分子の形で存在する。構造中のグアニジル(guanidyl)基は、その活性に必要な官能基である。水或いは有機溶剤には溶けず、強酸及び強アルカリに出会うと脱水分解し易く、弱酸性水溶液中では非常に安定する。よって、フグ毒から製造する凍結乾燥注射用粉末製剤は、適当な酸性溶媒を溶解補助剤として選択すべきである。出願人は『中国薬典2005版』『薬用補助剤帳』及び『中国薬用補助剤大全』等の資料を参考に、注射剤中に常用されるクエン酸、酢酸、アスコルビン酸(ascorbic acid)、及びリン酸二水素ナトリウム(sodium
dihydrogen phosphate)等注射剤の標準級弱酸性物質に対して分析と選択を行なった。『化学薬物製剤研究技術指導原則』に基づく原則を優先し、酢酸を揮発性酸として選択したが、冷凍乾燥過程中に揮発の損失があり、注射用粉末製剤に水を加えて再溶解した後のpH値制御が不利であった。アスコルビン酸は、光に当たると常温下で物理特性の安定が不充分となった。また、リン酸二水素ナトリウム(sodium dihydrogen phosphate)は酸性塩で、弱酸性、5%濃度下でのpH値がわずか4.56と、pH調節範囲が狭いため、フグ毒凍結乾燥注射用粉末製剤に用いる酸性溶媒には不適当である。クエン酸は非揮発性弱酸で、 pH 調節範囲には比較的大きな空間を有する。そこで、分析と比較を経て、クエン酸を本発明のフグ毒凍結乾燥注射用粉末製剤の溶解補助剤とした。本発明の凍結乾燥注射用粉末製剤において、溶解補助剤のクエン酸の用量は、好ましくは、一製剤あたり0.001mgから0.080mgとする。
【0044】
注射剤溶液中のpH値は、有効薬用成分の安定性と密切な関連を持つ重要な原因の一つである。本出願人は、設定したフグ毒凍結乾燥注射用粉末製剤の処方に対して、0.1%のクエン酸溶液を用いて異なるpH値に調節し、冷凍乾燥を経て注射用粉末製剤を作った。品質検査項目に基づき、外観、水を加えた後の再溶解性及びpH値、安定性等の指標に対して考察を行なった。主な安定性試験は、製剤を40°Cの高温下に置き、それぞれ0、5、10日目にサンプル検査を行い、蛍光検出による液体クロマトグラフィーを用いて測定する。面積正規化法を用いてフグ毒の含量と関連物質を算出する。実験結果は次の表5に示すとおりである。
【0045】
【表5】
【0046】
上述の実験結果より、フグ毒の注射用粉末製剤溶液中のpHがフグ毒の安定性に対して影響を及ぼすことが明らかに理解される。40°Cの条件下で、pH値が5以上である時、10日保持したフグ毒が分解されて生成した関連物質は対照溶液ピーク面積を上回り、新しい雑質ピークが出現した。
【0047】
繰り返し試験を行った結果、出願人は、本発明のフグ毒の凍結乾燥注射用粉末製剤のpH値を、好ましくは、3.5〜4.5とし、更に、4.0前後とすることが最も好適であることを理解した。
【0048】
次に、出願人が行った、フグ毒凍結乾燥注射用粉末製剤の異なる含水量が安定性に対して及ぼす影響についての研究結果を、表6に示す。
【0049】
【表6】
【0050】
実験結果:凍結乾燥注射用粉末製剤の含水量が5%を超える時、製剤の外観は40°Cの条件の下、萎縮を示し、安定性も劣ることが示された。凍結乾燥製剤の含水量が低いほど、製剤中のフグ毒の安定性が良好となる。本発明のフグ毒の凍結乾燥注射用粉末製剤の含水量は、好ましくは、3%以下とする。
【0051】
次に、凍結乾燥技術の選択について説明する。
【0052】
フグ毒の凍結乾燥注射用粉末製剤の冷凍乾燥技術は主に、事前凍結、主乾燥、及び後乾燥の三段階により構成される。処方製剤の共晶点及び賦形剤と安定剤の選択時に採用する凍結乾燥技術に基づき設計した五種類の凍結乾燥条件方案に対し試験を行なった(表7参照)。試験結果の比較分析を通して、本製剤での最良の冷凍乾燥技術の制御パラメータを確定する(表8参照)。
【0053】
【表7】
【0054】
【表8】
【0055】
方案1の試験結果より、製剤は事前凍結において、-10°Cでの低温真空乾燥を15時間行なった後、乾燥温度は直接、40°Cまで上昇し、製剤中のフグ毒含量が明らかに下降したことが理解される。これは、冷凍乾燥技術の設計がフグ毒凍結乾燥注射用粉末製剤の品質に顕著な影響を及ぼしたことを表している。方案4は、乾燥曲線の最適化を経て、主乾燥段階は段階別勾配を採用し、温度を後乾燥段階で採用した40°Cでの乾燥まで上昇させ、それによって得られた製剤は含水量の低いものとなる。凍結乾燥技術の過程は、製剤中のフグ毒含量に影響を及ぼすことはない。よって、方案4は、本製剤の冷凍乾燥技術に好適である。
【0056】
次に具体的な実施方案について説明を行なう。
【実施例1】
【0057】
実施例1は、賦形剤とする塩化ナトリウム(sodium)、安定剤とするデキストラン(dextran)20、溶解補助剤とするクエン酸を含むフグ毒の凍結乾燥注射用粉末製剤である。
【0058】
【表9】
【0059】
製造法は次のとおりである。
処方量のフグ毒を取り、10ml 0.1% のクエン酸溶液で溶解した後、処方量の塩化ナトリウム(sodium)を入れ、注射用水を加えて約250mlまで希釈する。0.1%クエン酸溶液を用いて指定pH値まで、超ろ過膜によってろ過を行い除熱しA グループの溶液を得る。また、別に処方量のデキストラン(dextran)20を取り、200ml注射用水を加えて溶解し、0.1%クエン酸溶液を用いて指定pH値に調節し、重量体積比0.1%〜1.0%の活性炭素を加えて60°Cの保温下で30分間撹拌し、ろ過によって除炭、除熱をし、室温まで冷却することによりBグループの溶液を得る。上述A、Bの二種溶液を均等に混合し、注射用水を用いて500mlに定容し、0.22μm 微細孔のろ過膜を用いてろ過を行い、サンプルを取り出してpH値、澄明度、含量等を測定し、合格後、無菌状態での個別包装を行なう。 -35°Cでの事前凍結を2〜6時間行なった後、 -10°C〜20°Cでの主乾燥を10〜20時間行い、20°C〜50°Cでの後乾燥を6〜10時間を行うと、製剤が得られる。製品外観は白色ホットケーキ状である。
【0060】
上述のフグ毒の凍結乾燥注射用粉末製剤において、機能調節剤とする塩酸リドカインを含むことを任意に選択可能とし、その用量を一製剤あたり5mgとする。
【実施例2】
【0061】
実施例2は、賦形剤とするマンニトール(mannitol)、安定剤とするデキストラン(dextran)20、溶解補助剤とするクエン酸を含むフグ毒の凍結乾燥注射用粉末製剤である。
【0062】
【表10】
【0063】
製造法は次のとおりである。
処方量のフグ毒を取り、10ml、0.1%のクエン酸溶液を用いて溶解した後、注射用水を加えて約250mlに希釈する。0.1%クエン酸溶液を指定のpH値に調節し、超ろ過膜でろ過し除熱することにより、A グループ溶液を得る。;また、処方量のデキストラン(dextran)20 とマンニトール(mannitol)を取り、200ml注射用水を加えて溶解し、0.1%クエン酸溶液を用いて指定のpH値に調節し、重量体積比0.2%の活性炭素を加えて60°Cの保温状態で30分間撹拌し、ろ過して除炭、除熱し、室温まで冷却することにより、Bグループ溶液を得る。前記A、B 二種の溶液を均等に混合し、注射用水を用いて500 mlに定容し、0.22μm 微細孔のろ過膜を用いてろ過し、サンプルを取り出しpH値、澄明度、含量等を測定する。合格後、無菌状態で個別包装する。-35°C での事前凍結を2〜6 時間行なった後、 -10°C〜20°C での主乾燥を10〜20時間行い、20°C〜50°Cでの後乾燥を6〜10時間行うことで、製剤が得られる。製品外観は白色ホットケーキ状である。
【0064】
前記フグ毒の凍結乾燥注射用粉末製剤において、機能調節剤の塩酸リドカインを含むことを任意で選択できる。また、その用量は一製剤あたり3.0mgとする。
【実施例3】
【0065】
実施例3は、賦形剤とする塩化ナトリウム(sodium)、安定剤とするトレハロース(trehalose)、溶解補助剤とするクエン酸を含むフグ毒の凍結乾燥注射用粉末製剤である。
【0066】
【表11】
【0067】
次に、製造法について説明する。処方量のフグ毒を取り、20ml、0.1%クエン酸溶液を用いて溶解した後、処方量の塩化ナトリウム(sodium)とトレハロース(trehalose)を入れ、注射用水を加えて約450mlに希釈する。0.1%クエン酸溶液を用いて指定のpH値に調節し、超ろ過膜でろ過をして除熱し、注射用水を500mlに定容する。0.22μm微細孔のろ過膜を用いてろ過し、サンプルを取りだしてpH値、澄明度、含量等を測定する。合格後、無菌状態で個別包装する。-35°C での事前凍結を2〜6時間行なった後、 -10°C〜20°C での主乾燥を10〜20時間行い、20°C〜50°Cでの後乾燥を6〜10時間行うことにより、製剤が得られる。製品の外観は白色ホットケーキ状である。
【0068】
前記フグ毒の凍結乾燥注射用粉末製剤において、機能調節剤の塩酸リドカインを含むことを任意で選択できる。また、その用量は一製剤あたり3.0mgとする。
【実施例4】
【0069】
実施例4は、マンニトール(mannitol)を賦形剤とし、トレハロース(trehalose)を安定剤としクエン酸を溶解補助剤とするフグ毒の凍結乾燥注射用粉末製剤について説明を行なう。
【0070】
【表12】
【0071】
次に、製造法について説明する。処方量のフグ毒を取り、20ml、0.1%クエン酸溶液を用いて溶解した後、処方量のトレハロース(trehalose)を入れ、注射用水を加えて約300mlに希釈する。0.1%クエン酸溶液を用いて指定のpH値に調節し、超ろ過膜でろ過をして除熱し、A グループの溶液を得る。また、別に処方量のマンニトール(mannitol)を取り、150ml注射用水を加えて溶解し、0.1%クエン酸溶液を用いて指定pH値に調節し、重量体積比0.1%〜1.0%の活性炭素を加えて60°Cの保温下で30分間撹拌し、ろ過によって除炭、除熱をし、室温まで冷却することによりBグループの溶液を得る。上述A、Bの二種溶液を均等に混合し、注射用水を用いて500mlに定容し、0.22μm 微細孔のろ過膜を用いてろ過を行い、サンプルを取り出してpH値、澄明度、含量等を測定し、合格後、無菌状態で個別包装する。 -35°Cでの事前凍結を2〜6時間行なった後、 -10°C〜20°Cでの主乾燥を10〜20時間行い、20°C〜50°Cでの後乾燥を6〜10時間を行うと、製剤が得られる。製品外観は白色ホットケーキ状である。
【0072】
前記フグ毒の凍結乾燥注射用粉末製剤において、機能調節剤の塩酸リドカインを含むことを任意で選択できる。また、その用量は一製剤あたり3.0mgとする。
【実施例5】
【0073】
実施例5は、マンニトール(mannitol)と塩化ナトリウム(sodium)を賦形剤とし、トレハロース(trehalose)を安定剤とし、クエン酸を溶解補助剤とするフグ毒の凍結乾燥注射用粉末製剤について説明を行なう。
【0074】
【表13】
【0075】
次に、製造法について説明する。処方量のフグ毒を取り、20ml、0.1%クエン酸溶液を用いて溶解した後、処方量の塩化ナトリウム(sodium)とトレハロース(trehalose)を入れ、注射用水を加えて約300mlに希釈する。0.1%クエン酸溶液を用いて指定のpH値に調節し、超ろ過膜でろ過をして除熱し、A グループの溶液を得る。また、別に処方量のマンニトール(mannitol)を取り、150ml注射用水を加えて溶解し、0.1%クエン酸溶液を用いて指定pH値に調節し、重量体積比0.1%〜1.0%の活性炭素を加えて、60°Cの保温下で30分間撹拌し、ろ過によって除炭、除熱をし、室温まで冷却することによりBグループの溶液を得る。上述A、Bの二種溶液を均等に混合し、注射用水を用いて500mlに定容し、0.22μm 微細孔のろ過膜を用いてろ過を行い、サンプルを取り出してpH値、澄明度、含量等を測定し、合格後、無菌状態で個別包装する。 -35°Cでの事前凍結を2〜6時間行なった後、 -10°C〜20°Cでの主乾燥を10〜20時間行い、20°C〜50°Cでの後乾燥を6〜10時間行うと、製剤が得られる。製品外観は白色ホットケーキ状である。
【0076】
前記フグ毒の凍結乾燥注射用粉末製剤において、機能調節剤の塩酸リドカインを含むことを任意で選択できる。また、その用量は一製剤あたり3.0mgとする。
【実施例6】
【0077】
実施例6は、マンニトール(mannitol)と塩化ナトリウム(sodium)を賦形剤とし、デキストラン(dextran)20を安定剤としクエン酸を溶解補助剤とするフグ毒の凍結乾燥注射用粉末製剤について説明を行なう。
【0078】
【表14】
【0079】
次に、製造法について説明する。処方量のフグ毒を取り、20ml、0.1%クエン酸溶液を用いて溶解した後、処方量のマンニトール(mannitol)と塩化ナトリウム(sodium)を入れ、注射用水を加えて約300mlに希釈する。0.1%クエン酸溶液を用いて指定のpH値に調節し、超ろ過膜でろ過をして除熱し、A グループの溶液を得る。また、別に処方量のデキストラン(dextran)20を取り、150ml注射用水を加えて溶解し、0.1%クエン酸溶液を用いて指定pH値に調節し、重量体積比0.1%〜1.0%の活性炭素を加えて、60°Cの保温下で30分間撹拌し、ろ過によって除炭、除熱をし、室温まで冷却することによりBグループの溶液を得る。上述A、Bの二種溶液を均等に混合し、注射用水を用いて500mlに定容し、0.22μm 微細孔のろ過膜を用いてろ過を行い、サンプルを取り出してpH値、澄明度、含量等を測定し、合格後、無菌状態で個別包装する。 -35°Cでの事前凍結を2〜6時間行なった後、 -10°C〜20°Cでの主乾燥を10〜20時間行い、20°C〜50°Cでの後乾燥を6〜10時間を行うと、製剤が得られる。製品外観は白色ホットケーキ状である。
【0080】
前記フグ毒の凍結乾燥注射用粉末製剤において、機能調節剤の塩酸リドカインを含むことを任意で選択できる。また、その用量は一製剤あたり3.0mgとする。
【実施例7】
【0081】
実施例7の安定性試験について説明を行なう。
【0082】
前記実施例によって作られたフグ毒の凍結乾燥注射用粉末製剤に対し40°Cの高温、10日の安定性試験を行なう。試験の具体的な操作内容は次のとおりである。
【0083】
各処方の凍結乾燥製品を温度40 ± 2°C、相対湿度75% ± 5%の薬品安定性実験箱内に置き、10日後に取り出し、蛍光検出による高速液体クロマトグラフィーの測定を行なう。テトロドトキシンの純度変化は製剤の安定レベルを示す。
【0084】
前記安定性試験において、本発明のフグ毒の凍結乾燥注射用粉末製剤は、良好な安定性を示した。最適処方の具体的な試験結果は次のとおりである。
【0085】
【表15】
【0086】
<応用実施例>
本発明を応用した製剤を実施し、治癒した薬物中毒患者例は合計80 である。その内、男性は49例、女性は31例、最年長者は41歳、最年少者は18歳、薬物吸引歴の最長は13年、最短は2年、一日の薬物吸引量最高は2〜3g、最低は0.1g、薬物使用種類は、ヘロイン、ヘロインと鎮静睡眠薬の併用、薬物吸引方式はヘロインを火であぶって吸引、筋肉注射或いは静脈注射であった。80例の患者は何れも、DSM−III−R精神活性物質による精神障害に当てはまる。アヘン類依存症検査、尿液のモルヒネ類物質検査が何れもプラスで、その他の深刻な身体の疾病を伴わず、治療前に重度の薬物依存、断薬症状と診断された中毒患者を選んだ。本製剤の用量は一日あたり10ug〜20ug、使用日数は3〜7日とした。
【0087】
アヘン類薬物の依存患者が薬物をやめた後に現れる断薬症状は、人により異なる。患者の中毒レベル、断薬症状の発作レベルに基づき、それぞれ適時に本発明の製剤を注射した。統計結果に示すとおり、製剤を一回使用後の0.5〜3時間以内において、95%の患者の断薬症状が消滅し、患者は始終、意識のはっきりした状態を保ち苦痛はなかった。初めて断薬症状が消滅した後はその状態が6〜12時間続き、次に断薬症状が現れた時、以前と比較し明らかに軽減した。一部の薬物吸引時間が長く、薬物を静脈注射し重度の中毒患者に対しては、24時間以内に本発明の製剤を二回注射した。全患者は、意識のはっきりした状態で苦痛なく楽に、最も激しい断薬症状期間を乗り切ることができる。断薬症状が消滅した後の患者は、精神状態も良好で、これに伴う食欲、体力も迅速に回復する。一般には、3〜6回製剤を使用した治療の後、再度の治療は不要である。全ての病例において製剤使用過程での副症状及び不適感は何れもみられなかった。
【0088】
次に、典型的な実施例について説明を行う。
【0089】
患者E.S。男、32歳、薬物吸引年数10年、一日あたり平均使用量1.0グラム、薬物種類はヘロイン、使用薬物方式は筋肉注射または静脈注射。患者は薬物の使用をやめた後、自ら希望して断薬治療を受けている。同日19: 00 、明らかな断薬症状が現れ、21:00、症状が更にひどくなった。患者の身体に、鼻水、涙、あくび、骨と筋肉の痛み、身震い、腹痛、下痢、むかつき、嘔吐、皮膚に鳥肌が出る、毛が逆立つ、身体が曲がった姿勢となる等の特徴が現れ、断薬症状が“+ +”〜“+ + +”に達した時、即刻、本製剤を筋肉注射すると、0.5時間後に、断薬症状が明らかに抑制され、2.5時間後に静かに眠りに入り、いずれの苦痛もなかった。10 時間後、患者に再び断薬症状が現れたが、断薬症状のレベルは以前と比較し明らかに軽減されていた。翌日9:30、再び本製剤を注射。注射後の患者には自己満足がみられ、明らかな不適応はなかった。約12時間間隔をあけた後、患者に再び断薬症状が現れ、下痢を伴った。21:00、再び本製剤を注射すると、患者は気分がよくなり、一晩、落ち着いていた。以後、毎日、患者が明らかに好転したという自覚症状が出るまで本製剤を注射した。下痢は完全になくなり、食欲も回復、排便も正常になり、5日目には断薬症状は現れず、患者にはその他の不適応症状もなかった。治療は計5日、本製剤を6回使用した。
【技術分野】
【0001】
本発明は、フグ毒(TTX,テトロドトキシン)の凍結乾燥注射用粉末製剤及びその製造法に係る。具体的に述べると、本発明は、正確な一定量のフグ毒を主要有効成分とし、最適な骨格を形成する賦形剤と安定剤を含む凍結乾燥注射用粉末製剤に関する。本発明の凍結乾燥注射用粉末製剤は、クリスタル・メス(Crystal meth)類、ヘロイン(heroin)等のアヘン類、及び大麻類の薬物依存による断薬症状に使用するもので、迅速で顕著な効果を奏する。また、本発明の凍結乾燥注射用粉末製剤は、安定性、安全性が高く、更に人体に対する剌激性も少ないものである。
【背景技術】
【0002】
フグ毒は、自然の非タンパク性神経毒で、化学構造式は次に示されるとおりである。
【0003】
【化1】
化学名:オクタヒドロ-12-(ヒドロキシメチル)-2-イミノ-5,9:7,10a-ジメタノ-10aH-[1,3] ジオキソシノ[6,5-d]ピリミジン-4,7,10,11,12-ペンタオール
Octahydro-12-(hydroxymethyl)-2-imino-5,9:7,10a-dimethano-10aH-[1,3]dioxocino[6,5-d]pyrimidine-4,7,10,11,12-pentol
英文名:Tetrodotoxin(テトロドトキシン)、略してTTXと呼ぶ。
【0004】
研究によって、フグ毒が断薬剤として、クリスタル・メス類、ヘロイン等アヘン類及び大麻類の薬物依存による断薬症状に用いることができ、中毒発作を抑制し、断薬反応を取り除き、使用中止後も依存性を生じないことが発見されている。
【0005】
フグ毒の結晶は、熱に対して非常に安定しており、40℃の条件の下で6ヶ月間おいても、フグ毒の結晶の品質には基本的に変化はみられない(表1参照)。しかし、フグ毒の水溶液は、温度に非常に敏感で、温度の影響を極めて受け易く、分解を起こす。温度が高いほど、分解も早い。40℃の条件の下、通常のフグ毒の注射液は30日間放置後、その含量は、99.20%から65.57%に下がり、33.63%の低下が明らかである (表2参照)。
【0006】
【表1】
【0007】
【表2】
【0008】
水溶液中の不安定な薬物について言えば、安定した注射可能な製剤を得るために、それを用いて凍結乾燥させた製剤をつくることは、本分野が通常採用する一般的な方法である。例えば、特許文献1で開示された「安定性を備えるフグ毒の冷凍乾燥製剤」において、製剤は、乳糖(lactose)、蔗糖(sucrose)、麦芽糖(maltose)、セロビオース(cellobiose)等の二糖類(disacchrides)、縮合型葡萄糖(condensed glucose)やデキストラン(dextran)等の多糖類、或いは、ヒドロキシル(hydroxyl)澱粉、ヒドロキシプロピルシクロデキストリン(hydroxypropyl cyclodextrin)等の誘導体から選択した安定剤を含み、その用量は一製剤あたり5〜100mgとする。また、製剤中には更に、クエン酸(citric acid)、酒石酸(tartaric acid)、リンゴ酸(malic acid)、或いはラクトビオン酸(lactobionic acid)から選択した溶解補助剤を含み、その用量は一製剤あたり0.00005〜0.0005mgとする。特許文献1において指摘したとおり、クエン酸塩或いはマンニトール(mannitol)を賦形剤とした時、得られるフグ毒の凍結乾燥させた製剤は不安定であるため、フグ毒の含量は貯存過程において徐々に下降する。
【0009】
よって、安全性及び安定性を備え、品質をコントロールでき、長期的に貯存でき、フグ毒の臨床に応用する薬用製剤の研究開発が必要である。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】CN 1835754A
【発明の概要】
【発明が解決しようとする課題】
【0011】
安定したフグ毒製剤を得るために、出願人はフグ毒の凍結乾燥注射用粉末製剤及びその製作技術について深い研究を進めた結果、凍結乾燥注射用粉末製剤の骨格を形成する賦形剤、フグ毒の安定剤、溶解補助剤、処方溶液のpH範囲、真空冷凍昇華乾燥技術、注射用粉末製剤含水量の制御等の要因がいずれも注射用粉末製剤の安定性に影響を与えることが明らかにされた。特に、注射用粉末製剤の骨格を形成する担体の引湿性である。引湿性が強い補助剤は、貯蔵過程において時間が延びるほど製剤中の含水量はゆっくりと上昇する傾向がみられ、含水量の増加によって製剤中の主成分であるフグ毒の含量を下降させる。
【0012】
数多くの実験による研究から、本発明者は、等張化剤の塩化ナトリウム(sodium)或いはマンニトール(mannitol)を凍結乾燥の注射用粉末製剤の骨格を形成する補助剤とし、デキストラン(dextran)20またはトレハロース(trehalose)をフグ毒主薬の安定剤として選択し、しかも、冷凍乾燥前のpH値を3.5〜4.5に調節する時、白色のホットケーキ状の外観を備えたフグ毒凍結乾燥注射用粉末製剤が得られることを発見した。製剤の主な薬剤量が正確で、外観に優れ、品質安定性が良好、安全性が高く、人体に対して安全に注射する要望を満たすものである。
【課題を解決するための手段】
【0013】
そこで、本発明は、フグ毒、溶解補助剤、凍結乾燥賦形剤、及び安定剤を含み、人体に対して安全に注射できるフグ毒の凍結乾燥注射用粉末製剤を提供する。前記フグ毒の純度は>96%とし、より好ましくは、98%〜99.8%とする。前記凍結乾燥の賦形剤は塩化ナトリウム(sodium)、マンニトール(mannitol)、二者の複合物のいずれかとする。また、前記安定剤は、デキストラン(dextran)、トレハロース(trehalose)、二者の複合物のいずれかとし、前記溶解補助剤はクエン酸とする。
【0014】
本発明のフグ毒凍結乾燥注射用粉末製剤の、フグ毒:賦形剤:安定剤の比率は、好ましくは、1: 150〜3000: 50〜500或いは50〜6000とする。
【0015】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、一製剤あたりのフグ毒の含量は0.1から20.0μgとし、好ましくは、0.5から20.0μg、より好ましくは、0.5から12.0μgとする。
【0016】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、賦形剤としての塩化ナトリウム(sodium)の含量を一製剤あたり1.0〜30mgとし、好ましくは、5.0〜30mg、より好ましくは、5.0〜20mgとする。
【0017】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、賦形剤としてのマンニトール(mannitol)の含量を一製剤あたり1.0〜30mgとし、好ましくは、1.0〜20mg、より好ましくは、3.0〜10mgとする。
【0018】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、安定剤としてのデキストラン(dextran)の含量を一製剤あたり0.5〜5.0mgとし、好ましくは、2.0〜5.0mg、より好ましくは、3.0〜5.0mgとする。
【0019】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、安定剤としてのトレハロース(trehalose)の含量を一製剤あたり0.5〜60mgとし、好ましくは、2.0〜60mg、より好ましくは、10〜60mgとする。
【0020】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、クエン酸の含量を一製剤あたり0.001〜0.080mgとし、好ましくは、0.010〜0.080mgとし、より好ましくは、0.020〜0.060mgとする。
【0021】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、好ましくは、機能調節剤を含み、前記機能調節剤は、好ましくは、塩酸リドカインとする。
【0022】
本発明のフグ毒の凍結乾燥注射用粉末製剤において、好ましくは、高純度窒素ガス或いは高純度炭酸ガスの惰性気体を充填する。
【0023】
本発明のフグ毒の凍結乾燥注射用粉末製剤は、好ましくは、筋肉或いは皮下に対して注射で薬剤を投与する形式を選択し、しかも、無菌注射を用いて水に溶解する場合、無菌注射の使用する水量は一製剤あたり0.5〜2.0mlとする。
【0024】
また、本発明が更に開示する「フグ毒の凍結乾燥注射用粉末製剤の製造法」での工程は、次のとおりである。
(1)予定量のフグ毒を直接、溶解補助剤を含む溶液中に入れ、pH値を3.0〜6.0に調節し、好ましくは、pH3.5〜4.5とする。ろ過して熱を取り除く。
(2)凍結乾燥賦形剤と安定剤、及び任意に選択した機能調節剤を直接、無菌注射用水中に入れ、活性炭素を加えて30分間撹拌した後、ろ過して熱を取り除く。
(3)(1)と(2)において得られた溶液を共に均等に混合し、ろ過して除菌し、定量をバイアル瓶に詰め、真空冷凍乾燥を行い、惰性気体を充填し、圧して緊密に蓋をし、凍結乾燥注射用粉末製剤を得る。
【0025】
本発明の製造法の(1)工程においては、好ましくは、超ろ過法によってろ過を行なう。
【0026】
本発明の製造法の(2)工程においては、好ましくは、活性炭素の用量を0.1〜6.0g/100mlとする。
【0027】
本発明の製造法の(3)工程においては、0.05μm〜0.20μmのサブミクロン級微細孔を備えるろ過膜或いは微細孔を備える荷電ろ過膜によるろ過を行なう。
【発明の効果】
【0028】
本発明のフグ毒の凍結乾燥注射用粉末製剤は、クリスタル・メス(Crystal meth)類、ヘロイン(heroin)等のアヘン類、及び大麻類の薬物依存による断薬症状に使用するもので、迅速で顕著な効果を奏する。また、本発明の凍結乾燥注射用粉末製剤は、安定性、安全性が高く、更に人体に対する剌激性も少ないものである。
【図面の簡単な説明】
【0029】
【図1】フグ毒注射液(40℃、0日)に対する蛍光検出HPLCチャートである。
【図2】フグ毒注射液(40℃、10日)に対する蛍光検出HPLCチャートである。
【図3】処方Alでのフグ毒の凍結乾燥注射用粉末製剤(40℃、0日)に対する蛍光検出HPLCチャートである。
【図4】処方Alでのフグ毒の凍結乾燥注射用粉末製剤(40℃、10日)に対する蛍光検出HPLCチャートである。
【図5】処方Blでのフグ毒の凍結乾燥注射用粉末製剤(40℃、0日)に対する蛍光検出HPLCチャートである。
【図6】処方Blでのフグ毒の凍結乾燥注射用粉末製剤(40℃、10日)に対する蛍光検出HPLCチャートである。
【図7】処方Clでのフグ毒の凍結乾燥注射用粉末製剤(40℃、0日)に対する蛍光検出HPLCチャートである。
【図8】処方Clでのフグ毒の凍結乾燥注射用粉末製剤(40℃、10日)に対する蛍光検出HPLCチャートである。
【図9】処方D5でのフグ毒の凍結乾燥注射用粉末製剤(40℃、0日)に対する蛍光検出HPLCチャートである。
【図10】処方D5でのフグ毒の凍結乾燥注射用粉末製剤(40℃、10日)に対する蛍光検出HPLCチャートである。
【図11】処方E5でのフグ毒の凍結乾燥注射用粉末製剤(40℃、0日)に対する蛍光検出HPLCチャートである。
【図12】処方E5でのフグ毒の凍結乾燥注射用粉末製剤(40℃、10日)に対する蛍光検出HPLCチャートである。
【図13】処方F5でのフグ毒の凍結乾燥注射用粉末製剤(40℃、0日)に対する蛍光検出HPLCチャートである。
【図14】処方F5でのフグ毒の凍結乾燥注射用粉末製剤(40℃、10日)に対する蛍光検出HPLCチャートである。
【発明を実施するための形態】
【0030】
本明細書において、本発明のフグ毒の凍結乾燥注射用粉末製剤に対するフグ毒含量の測定は、高速液体クロマトグラフィー(HPLC。中国薬典2005版二部付録VII)を用いて行なった。含量測定項目下に記録されているクロマトグラフにおいて、供試品溶液の保留時間の主ピークと、対照品溶液の保留時間の主ピークとは一致する。
【0031】
採用するクロマトグラフの条件は次のとおりである。オクチルシラン結合シリカゲル(octylsilane bonded silica gel)充填剤に用い、オクタンスルホン酸ナトリウムのリン酸塩緩衝液を移動相とする。流速を1分あたり0.3ml、励起波長を365nm、発射波長を510nm、ポストカラム誘導体化剤を4mol/Lの水酸化ナトリウム溶液とする。誘導体化剤の流速を1分あたり0.2〜0.5 ml、ポストカラム誘導化温度を100°C〜140°Cとする。理論段数は、フグ毒のピークに基づいて計算し、2000以下にはならない。
【0032】
具体的な測定方法は次のとおりである。
本発明のフグ毒の凍結乾燥注射用粉末製剤(フグ毒10μgを含む)、精密に水2.0mlを加えて溶解し、精密に20μlを取り出し、予測に基づくクロマトグラフの条件の下で投入し、蛍光検出クロマトグラフを記録する。別にフグ毒の対照品を適量取り出す。同法で測定する。外部標準法に基づくピーク面積により一瓶あたりの一製剤のフグ毒含量を算出する。フグ毒含量が表示量の90%〜110%である時、凍結乾燥注射用粉末製剤の医薬品要求に符合する。
【0033】
関連物質の測定:本発明のフグ毒の凍結乾燥注射用粉末製剤を取り、水を加えて1mlあたりフグ毒20μgを含む溶液を生成し、供試品溶液とする。精密にl.0mlを取り、25mlの瓶中に入れ、水を加えて目盛まで希釈し、振って均一にしたものを対照溶液とする。精密に供試品溶液と対照品溶液を各20μl取り出し、それぞれを液相クロマトグラフィー装置に注入し、主成分のピーク保留時間が2倍となるまで蛍光検出クロマトグラフを記録する。供試品溶液のクロマトグラフ中の各雑質ピーク面積の和が対照溶液ピーク面積を超えない時、医薬品要求に符合する。
【0034】
本発明では、蛍光検出による高速液体クロマトグラフィーを用いてフグ毒及び関連物質の含量に対する検出を行なう。通常の紫外可視吸光検出による高効液相クロマトグラフィーと比較すると、蛍光検出による高効液相クロマトグラフィーの方が敏感度が更に高く、測定の正確度も高いことから、フグ毒製剤の安定性を更に良好に測定できることが理解される。例えば、フグ毒製剤について言えば、紫外可視吸光検出による高速液体クロマトグラフィーの最低検出制限値は8.14ngであるが、蛍光検出による高速液体クロマトグラフィーでは0.40ngで、蛍光検出は紫外可視吸光検出の20倍も低いことがわかる。また、フグ毒製剤について言えば、紫外可視吸光検出による高速液体クロマトグラフィーの定量制限値は20.26ngであるが、蛍光検出による高速液体クロマトグラフィーでは0.81ngで、蛍光検出の定量制限値は紫外可視吸光検出の25倍となる。
【0035】
具体的に言えば、注射用フグ毒製剤の通常仕様は一瓶あたり10μgであり、サンプルを取り出し溶解した後、測定に用いる製剤を投入する。投入濃度を5μg /ml〜 20μg/ml、投入量を20μlとする時、サンプル中の雑質含量が1%であるなら、雑質量はわずか1〜4ngで、紫外可視吸光検出による液体クロマトグラフィーでの検出制限値を下回る。サンプル中の雑質が2%である時、雑質量はわずか2〜8ngで、紫外可視吸光検出による液体クロマトグラフィーの検出制限値を下回る。紫外可視吸光検出による液体クロマトグラフィーを用いて製剤中の関連雑質或いは含量を測定する場合、製品中の5%を下回る雑質は、検出制限値が要求を満足しないために検出されず、定量制限値を下回ることにより測定誤差が引き起こされる。それに対して、蛍光検出による液体クロマトグラフィーの検出制限値は0.40ngであるため、完全に検出要求を満たすものである。
【0036】
以上の測定条件を採用し、出願人は、フグ毒の凍結乾燥注射用粉末製剤の安定性に影響を及ぼす各種要素についての研究を行なった。
【0037】
次に、フグ毒凍結乾燥注射用粉末製剤の賦形剤と安定剤の選択についての説明を行なう。
【0038】
フグ毒の臨床使用量はマイクログラム級である。よって、凍結乾燥注射用粉末製剤の骨格を形成するための助剤となる賦形剤を添加する必要がある。それと同時に、フグ毒注射用粉末製剤の安定性の要望に基づき、安定剤の選択も極めて重要である。多数の実験を重ねた結果、出願人は、塩化ナトリウム(sodium)或いは/及びマンニトール(mannitol)をフグ毒の凍結乾燥注射用粉末製剤の賦形剤とし、デキストラン(dextran)20或いはトレハロース(trehalose)を安定剤として選択することにより良好な結果が得られることを発見した。次の表3及び表4には、塩化ナトリウム(sodium)及び/或いはマンニトール(mannitol)を賦形剤としデキストラン(dextran)20或いはトレハロース(trehalose)を安定剤とした本発明を代表するフグ毒の凍結乾燥注射用粉末製剤の処方及び40°C下での安定性試験結果を示す。
【0039】
【表3】
【0040】
【表4】
【0041】
その内、賦形剤とするマンニトール(mannitol)製品の外観は極めて良好で、しかも吸湿性が弱い。デキストラン(dextran)は、フグ毒の凍結乾燥製剤に対して顕著な保護作用を備えるが、吸湿性が強い。凍結乾燥の注射用粉末製剤の含水量をコントロールし、充満した凍結乾燥の骨格を形成するために、本発明製剤中のデキストラン(dextran)20含量を一製剤あたり4mgとし、マンニトール(mannitol)の含量を一製剤あたり6mgとする。また、等張化剤の塩化ナトリウム(sodium)を骨格形成の賦形剤とし、デキストラン(dextran)20或いはトレハロース(trehalose)を安定剤として組成する処方の場合、製剤の含水量は低く、外観は充満したホットケーキ状となる。実際の応用では水を加えた後は迅速に再溶解する。40°Cの高温下の10日間安定性試験は医薬品要求に符合するものである。
【0042】
次に、溶解補助剤と処方pH値の選択について説明を行なう。
【0043】
フグ毒は、相対分子質量を319.27とする海洋生物の非蛋白性神経毒であり、通常、両性分子の形で存在する。構造中のグアニジル(guanidyl)基は、その活性に必要な官能基である。水或いは有機溶剤には溶けず、強酸及び強アルカリに出会うと脱水分解し易く、弱酸性水溶液中では非常に安定する。よって、フグ毒から製造する凍結乾燥注射用粉末製剤は、適当な酸性溶媒を溶解補助剤として選択すべきである。出願人は『中国薬典2005版』『薬用補助剤帳』及び『中国薬用補助剤大全』等の資料を参考に、注射剤中に常用されるクエン酸、酢酸、アスコルビン酸(ascorbic acid)、及びリン酸二水素ナトリウム(sodium
dihydrogen phosphate)等注射剤の標準級弱酸性物質に対して分析と選択を行なった。『化学薬物製剤研究技術指導原則』に基づく原則を優先し、酢酸を揮発性酸として選択したが、冷凍乾燥過程中に揮発の損失があり、注射用粉末製剤に水を加えて再溶解した後のpH値制御が不利であった。アスコルビン酸は、光に当たると常温下で物理特性の安定が不充分となった。また、リン酸二水素ナトリウム(sodium dihydrogen phosphate)は酸性塩で、弱酸性、5%濃度下でのpH値がわずか4.56と、pH調節範囲が狭いため、フグ毒凍結乾燥注射用粉末製剤に用いる酸性溶媒には不適当である。クエン酸は非揮発性弱酸で、 pH 調節範囲には比較的大きな空間を有する。そこで、分析と比較を経て、クエン酸を本発明のフグ毒凍結乾燥注射用粉末製剤の溶解補助剤とした。本発明の凍結乾燥注射用粉末製剤において、溶解補助剤のクエン酸の用量は、好ましくは、一製剤あたり0.001mgから0.080mgとする。
【0044】
注射剤溶液中のpH値は、有効薬用成分の安定性と密切な関連を持つ重要な原因の一つである。本出願人は、設定したフグ毒凍結乾燥注射用粉末製剤の処方に対して、0.1%のクエン酸溶液を用いて異なるpH値に調節し、冷凍乾燥を経て注射用粉末製剤を作った。品質検査項目に基づき、外観、水を加えた後の再溶解性及びpH値、安定性等の指標に対して考察を行なった。主な安定性試験は、製剤を40°Cの高温下に置き、それぞれ0、5、10日目にサンプル検査を行い、蛍光検出による液体クロマトグラフィーを用いて測定する。面積正規化法を用いてフグ毒の含量と関連物質を算出する。実験結果は次の表5に示すとおりである。
【0045】
【表5】
【0046】
上述の実験結果より、フグ毒の注射用粉末製剤溶液中のpHがフグ毒の安定性に対して影響を及ぼすことが明らかに理解される。40°Cの条件下で、pH値が5以上である時、10日保持したフグ毒が分解されて生成した関連物質は対照溶液ピーク面積を上回り、新しい雑質ピークが出現した。
【0047】
繰り返し試験を行った結果、出願人は、本発明のフグ毒の凍結乾燥注射用粉末製剤のpH値を、好ましくは、3.5〜4.5とし、更に、4.0前後とすることが最も好適であることを理解した。
【0048】
次に、出願人が行った、フグ毒凍結乾燥注射用粉末製剤の異なる含水量が安定性に対して及ぼす影響についての研究結果を、表6に示す。
【0049】
【表6】
【0050】
実験結果:凍結乾燥注射用粉末製剤の含水量が5%を超える時、製剤の外観は40°Cの条件の下、萎縮を示し、安定性も劣ることが示された。凍結乾燥製剤の含水量が低いほど、製剤中のフグ毒の安定性が良好となる。本発明のフグ毒の凍結乾燥注射用粉末製剤の含水量は、好ましくは、3%以下とする。
【0051】
次に、凍結乾燥技術の選択について説明する。
【0052】
フグ毒の凍結乾燥注射用粉末製剤の冷凍乾燥技術は主に、事前凍結、主乾燥、及び後乾燥の三段階により構成される。処方製剤の共晶点及び賦形剤と安定剤の選択時に採用する凍結乾燥技術に基づき設計した五種類の凍結乾燥条件方案に対し試験を行なった(表7参照)。試験結果の比較分析を通して、本製剤での最良の冷凍乾燥技術の制御パラメータを確定する(表8参照)。
【0053】
【表7】
【0054】
【表8】
【0055】
方案1の試験結果より、製剤は事前凍結において、-10°Cでの低温真空乾燥を15時間行なった後、乾燥温度は直接、40°Cまで上昇し、製剤中のフグ毒含量が明らかに下降したことが理解される。これは、冷凍乾燥技術の設計がフグ毒凍結乾燥注射用粉末製剤の品質に顕著な影響を及ぼしたことを表している。方案4は、乾燥曲線の最適化を経て、主乾燥段階は段階別勾配を採用し、温度を後乾燥段階で採用した40°Cでの乾燥まで上昇させ、それによって得られた製剤は含水量の低いものとなる。凍結乾燥技術の過程は、製剤中のフグ毒含量に影響を及ぼすことはない。よって、方案4は、本製剤の冷凍乾燥技術に好適である。
【0056】
次に具体的な実施方案について説明を行なう。
【実施例1】
【0057】
実施例1は、賦形剤とする塩化ナトリウム(sodium)、安定剤とするデキストラン(dextran)20、溶解補助剤とするクエン酸を含むフグ毒の凍結乾燥注射用粉末製剤である。
【0058】
【表9】
【0059】
製造法は次のとおりである。
処方量のフグ毒を取り、10ml 0.1% のクエン酸溶液で溶解した後、処方量の塩化ナトリウム(sodium)を入れ、注射用水を加えて約250mlまで希釈する。0.1%クエン酸溶液を用いて指定pH値まで、超ろ過膜によってろ過を行い除熱しA グループの溶液を得る。また、別に処方量のデキストラン(dextran)20を取り、200ml注射用水を加えて溶解し、0.1%クエン酸溶液を用いて指定pH値に調節し、重量体積比0.1%〜1.0%の活性炭素を加えて60°Cの保温下で30分間撹拌し、ろ過によって除炭、除熱をし、室温まで冷却することによりBグループの溶液を得る。上述A、Bの二種溶液を均等に混合し、注射用水を用いて500mlに定容し、0.22μm 微細孔のろ過膜を用いてろ過を行い、サンプルを取り出してpH値、澄明度、含量等を測定し、合格後、無菌状態での個別包装を行なう。 -35°Cでの事前凍結を2〜6時間行なった後、 -10°C〜20°Cでの主乾燥を10〜20時間行い、20°C〜50°Cでの後乾燥を6〜10時間を行うと、製剤が得られる。製品外観は白色ホットケーキ状である。
【0060】
上述のフグ毒の凍結乾燥注射用粉末製剤において、機能調節剤とする塩酸リドカインを含むことを任意に選択可能とし、その用量を一製剤あたり5mgとする。
【実施例2】
【0061】
実施例2は、賦形剤とするマンニトール(mannitol)、安定剤とするデキストラン(dextran)20、溶解補助剤とするクエン酸を含むフグ毒の凍結乾燥注射用粉末製剤である。
【0062】
【表10】
【0063】
製造法は次のとおりである。
処方量のフグ毒を取り、10ml、0.1%のクエン酸溶液を用いて溶解した後、注射用水を加えて約250mlに希釈する。0.1%クエン酸溶液を指定のpH値に調節し、超ろ過膜でろ過し除熱することにより、A グループ溶液を得る。;また、処方量のデキストラン(dextran)20 とマンニトール(mannitol)を取り、200ml注射用水を加えて溶解し、0.1%クエン酸溶液を用いて指定のpH値に調節し、重量体積比0.2%の活性炭素を加えて60°Cの保温状態で30分間撹拌し、ろ過して除炭、除熱し、室温まで冷却することにより、Bグループ溶液を得る。前記A、B 二種の溶液を均等に混合し、注射用水を用いて500 mlに定容し、0.22μm 微細孔のろ過膜を用いてろ過し、サンプルを取り出しpH値、澄明度、含量等を測定する。合格後、無菌状態で個別包装する。-35°C での事前凍結を2〜6 時間行なった後、 -10°C〜20°C での主乾燥を10〜20時間行い、20°C〜50°Cでの後乾燥を6〜10時間行うことで、製剤が得られる。製品外観は白色ホットケーキ状である。
【0064】
前記フグ毒の凍結乾燥注射用粉末製剤において、機能調節剤の塩酸リドカインを含むことを任意で選択できる。また、その用量は一製剤あたり3.0mgとする。
【実施例3】
【0065】
実施例3は、賦形剤とする塩化ナトリウム(sodium)、安定剤とするトレハロース(trehalose)、溶解補助剤とするクエン酸を含むフグ毒の凍結乾燥注射用粉末製剤である。
【0066】
【表11】
【0067】
次に、製造法について説明する。処方量のフグ毒を取り、20ml、0.1%クエン酸溶液を用いて溶解した後、処方量の塩化ナトリウム(sodium)とトレハロース(trehalose)を入れ、注射用水を加えて約450mlに希釈する。0.1%クエン酸溶液を用いて指定のpH値に調節し、超ろ過膜でろ過をして除熱し、注射用水を500mlに定容する。0.22μm微細孔のろ過膜を用いてろ過し、サンプルを取りだしてpH値、澄明度、含量等を測定する。合格後、無菌状態で個別包装する。-35°C での事前凍結を2〜6時間行なった後、 -10°C〜20°C での主乾燥を10〜20時間行い、20°C〜50°Cでの後乾燥を6〜10時間行うことにより、製剤が得られる。製品の外観は白色ホットケーキ状である。
【0068】
前記フグ毒の凍結乾燥注射用粉末製剤において、機能調節剤の塩酸リドカインを含むことを任意で選択できる。また、その用量は一製剤あたり3.0mgとする。
【実施例4】
【0069】
実施例4は、マンニトール(mannitol)を賦形剤とし、トレハロース(trehalose)を安定剤としクエン酸を溶解補助剤とするフグ毒の凍結乾燥注射用粉末製剤について説明を行なう。
【0070】
【表12】
【0071】
次に、製造法について説明する。処方量のフグ毒を取り、20ml、0.1%クエン酸溶液を用いて溶解した後、処方量のトレハロース(trehalose)を入れ、注射用水を加えて約300mlに希釈する。0.1%クエン酸溶液を用いて指定のpH値に調節し、超ろ過膜でろ過をして除熱し、A グループの溶液を得る。また、別に処方量のマンニトール(mannitol)を取り、150ml注射用水を加えて溶解し、0.1%クエン酸溶液を用いて指定pH値に調節し、重量体積比0.1%〜1.0%の活性炭素を加えて60°Cの保温下で30分間撹拌し、ろ過によって除炭、除熱をし、室温まで冷却することによりBグループの溶液を得る。上述A、Bの二種溶液を均等に混合し、注射用水を用いて500mlに定容し、0.22μm 微細孔のろ過膜を用いてろ過を行い、サンプルを取り出してpH値、澄明度、含量等を測定し、合格後、無菌状態で個別包装する。 -35°Cでの事前凍結を2〜6時間行なった後、 -10°C〜20°Cでの主乾燥を10〜20時間行い、20°C〜50°Cでの後乾燥を6〜10時間を行うと、製剤が得られる。製品外観は白色ホットケーキ状である。
【0072】
前記フグ毒の凍結乾燥注射用粉末製剤において、機能調節剤の塩酸リドカインを含むことを任意で選択できる。また、その用量は一製剤あたり3.0mgとする。
【実施例5】
【0073】
実施例5は、マンニトール(mannitol)と塩化ナトリウム(sodium)を賦形剤とし、トレハロース(trehalose)を安定剤とし、クエン酸を溶解補助剤とするフグ毒の凍結乾燥注射用粉末製剤について説明を行なう。
【0074】
【表13】
【0075】
次に、製造法について説明する。処方量のフグ毒を取り、20ml、0.1%クエン酸溶液を用いて溶解した後、処方量の塩化ナトリウム(sodium)とトレハロース(trehalose)を入れ、注射用水を加えて約300mlに希釈する。0.1%クエン酸溶液を用いて指定のpH値に調節し、超ろ過膜でろ過をして除熱し、A グループの溶液を得る。また、別に処方量のマンニトール(mannitol)を取り、150ml注射用水を加えて溶解し、0.1%クエン酸溶液を用いて指定pH値に調節し、重量体積比0.1%〜1.0%の活性炭素を加えて、60°Cの保温下で30分間撹拌し、ろ過によって除炭、除熱をし、室温まで冷却することによりBグループの溶液を得る。上述A、Bの二種溶液を均等に混合し、注射用水を用いて500mlに定容し、0.22μm 微細孔のろ過膜を用いてろ過を行い、サンプルを取り出してpH値、澄明度、含量等を測定し、合格後、無菌状態で個別包装する。 -35°Cでの事前凍結を2〜6時間行なった後、 -10°C〜20°Cでの主乾燥を10〜20時間行い、20°C〜50°Cでの後乾燥を6〜10時間行うと、製剤が得られる。製品外観は白色ホットケーキ状である。
【0076】
前記フグ毒の凍結乾燥注射用粉末製剤において、機能調節剤の塩酸リドカインを含むことを任意で選択できる。また、その用量は一製剤あたり3.0mgとする。
【実施例6】
【0077】
実施例6は、マンニトール(mannitol)と塩化ナトリウム(sodium)を賦形剤とし、デキストラン(dextran)20を安定剤としクエン酸を溶解補助剤とするフグ毒の凍結乾燥注射用粉末製剤について説明を行なう。
【0078】
【表14】
【0079】
次に、製造法について説明する。処方量のフグ毒を取り、20ml、0.1%クエン酸溶液を用いて溶解した後、処方量のマンニトール(mannitol)と塩化ナトリウム(sodium)を入れ、注射用水を加えて約300mlに希釈する。0.1%クエン酸溶液を用いて指定のpH値に調節し、超ろ過膜でろ過をして除熱し、A グループの溶液を得る。また、別に処方量のデキストラン(dextran)20を取り、150ml注射用水を加えて溶解し、0.1%クエン酸溶液を用いて指定pH値に調節し、重量体積比0.1%〜1.0%の活性炭素を加えて、60°Cの保温下で30分間撹拌し、ろ過によって除炭、除熱をし、室温まで冷却することによりBグループの溶液を得る。上述A、Bの二種溶液を均等に混合し、注射用水を用いて500mlに定容し、0.22μm 微細孔のろ過膜を用いてろ過を行い、サンプルを取り出してpH値、澄明度、含量等を測定し、合格後、無菌状態で個別包装する。 -35°Cでの事前凍結を2〜6時間行なった後、 -10°C〜20°Cでの主乾燥を10〜20時間行い、20°C〜50°Cでの後乾燥を6〜10時間を行うと、製剤が得られる。製品外観は白色ホットケーキ状である。
【0080】
前記フグ毒の凍結乾燥注射用粉末製剤において、機能調節剤の塩酸リドカインを含むことを任意で選択できる。また、その用量は一製剤あたり3.0mgとする。
【実施例7】
【0081】
実施例7の安定性試験について説明を行なう。
【0082】
前記実施例によって作られたフグ毒の凍結乾燥注射用粉末製剤に対し40°Cの高温、10日の安定性試験を行なう。試験の具体的な操作内容は次のとおりである。
【0083】
各処方の凍結乾燥製品を温度40 ± 2°C、相対湿度75% ± 5%の薬品安定性実験箱内に置き、10日後に取り出し、蛍光検出による高速液体クロマトグラフィーの測定を行なう。テトロドトキシンの純度変化は製剤の安定レベルを示す。
【0084】
前記安定性試験において、本発明のフグ毒の凍結乾燥注射用粉末製剤は、良好な安定性を示した。最適処方の具体的な試験結果は次のとおりである。
【0085】
【表15】
【0086】
<応用実施例>
本発明を応用した製剤を実施し、治癒した薬物中毒患者例は合計80 である。その内、男性は49例、女性は31例、最年長者は41歳、最年少者は18歳、薬物吸引歴の最長は13年、最短は2年、一日の薬物吸引量最高は2〜3g、最低は0.1g、薬物使用種類は、ヘロイン、ヘロインと鎮静睡眠薬の併用、薬物吸引方式はヘロインを火であぶって吸引、筋肉注射或いは静脈注射であった。80例の患者は何れも、DSM−III−R精神活性物質による精神障害に当てはまる。アヘン類依存症検査、尿液のモルヒネ類物質検査が何れもプラスで、その他の深刻な身体の疾病を伴わず、治療前に重度の薬物依存、断薬症状と診断された中毒患者を選んだ。本製剤の用量は一日あたり10ug〜20ug、使用日数は3〜7日とした。
【0087】
アヘン類薬物の依存患者が薬物をやめた後に現れる断薬症状は、人により異なる。患者の中毒レベル、断薬症状の発作レベルに基づき、それぞれ適時に本発明の製剤を注射した。統計結果に示すとおり、製剤を一回使用後の0.5〜3時間以内において、95%の患者の断薬症状が消滅し、患者は始終、意識のはっきりした状態を保ち苦痛はなかった。初めて断薬症状が消滅した後はその状態が6〜12時間続き、次に断薬症状が現れた時、以前と比較し明らかに軽減した。一部の薬物吸引時間が長く、薬物を静脈注射し重度の中毒患者に対しては、24時間以内に本発明の製剤を二回注射した。全患者は、意識のはっきりした状態で苦痛なく楽に、最も激しい断薬症状期間を乗り切ることができる。断薬症状が消滅した後の患者は、精神状態も良好で、これに伴う食欲、体力も迅速に回復する。一般には、3〜6回製剤を使用した治療の後、再度の治療は不要である。全ての病例において製剤使用過程での副症状及び不適感は何れもみられなかった。
【0088】
次に、典型的な実施例について説明を行う。
【0089】
患者E.S。男、32歳、薬物吸引年数10年、一日あたり平均使用量1.0グラム、薬物種類はヘロイン、使用薬物方式は筋肉注射または静脈注射。患者は薬物の使用をやめた後、自ら希望して断薬治療を受けている。同日19: 00 、明らかな断薬症状が現れ、21:00、症状が更にひどくなった。患者の身体に、鼻水、涙、あくび、骨と筋肉の痛み、身震い、腹痛、下痢、むかつき、嘔吐、皮膚に鳥肌が出る、毛が逆立つ、身体が曲がった姿勢となる等の特徴が現れ、断薬症状が“+ +”〜“+ + +”に達した時、即刻、本製剤を筋肉注射すると、0.5時間後に、断薬症状が明らかに抑制され、2.5時間後に静かに眠りに入り、いずれの苦痛もなかった。10 時間後、患者に再び断薬症状が現れたが、断薬症状のレベルは以前と比較し明らかに軽減されていた。翌日9:30、再び本製剤を注射。注射後の患者には自己満足がみられ、明らかな不適応はなかった。約12時間間隔をあけた後、患者に再び断薬症状が現れ、下痢を伴った。21:00、再び本製剤を注射すると、患者は気分がよくなり、一晩、落ち着いていた。以後、毎日、患者が明らかに好転したという自覚症状が出るまで本製剤を注射した。下痢は完全になくなり、食欲も回復、排便も正常になり、5日目には断薬症状は現れず、患者にはその他の不適応症状もなかった。治療は計5日、本製剤を6回使用した。
【特許請求の範囲】
【請求項1】
フグ毒、溶解補助剤、凍結乾燥賦形剤、安定剤を含み、
フグ毒の純度は、96%以上とし、好ましくは、98%〜99.8%とし、
前記凍結乾燥賦形剤は、塩化ナトリウム(sodium)、マンニトール(mannitol)、二者の複合物のいずれかとし、前記安定剤は、デキストラン(dextran)、トレハロース(trehalose)、二者の複合物のいずれかとし、前記溶解補助剤は、クエン酸とし、前記製剤乾燥法は、真空冷凍昇華乾燥とすることを特徴とするフグ毒の凍結乾燥注射用粉末製剤。
【請求項2】
前記フグ毒と賦形剤と安定剤の比率は、1: 150〜3000: 50〜500或いは50〜6000とすることを特徴とする請求項1に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項3】
前記フグ毒の含量は、一製剤あたり0.1〜20.0μgとし、好ましくは、一製剤あたり0.5〜20.0μgとし、更に好ましくは、一製剤あたり0.5〜12.0μgとすることを特徴とする請求項1に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項4】
前記賦形剤とする塩化ナトリウム(sodium)の含量は、一製剤あたり1.0〜30mgとし、好ましくは、一製剤あたり5.0〜30mgとし、更に好ましくは、一製剤あたり5.0〜20mgとすることを特徴とする請求項1に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項5】
前記賦形剤とするマンニトール(mannitol)の含量は、一製剤あたり1.0〜30mgとし、好ましくは、一製剤あたり1.0〜20mgとし、更に好ましくは、一製剤あたり3.0〜10mgとすることを特徴とする請求項1に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項6】
前記安定剤とするデキストラン(dextran)の含量は、一製剤あたり0.5〜5.0mgとし、好ましくは、一製剤あたり2.0〜5.0mgとし、更に好ましくは、一製剤あたり3.0〜5.0mgとすることを特徴とする請求項1に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項7】
前記安定剤とするトレハロース(trehalose)の含量は、一製剤あたり0.5〜60mgとし、好ましくは、一製剤あたり2〜60mgとし、更に好ましくは、一製剤あたり10〜60mgとすることを特徴とする請求項1に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項8】
前記クエン酸の含量は、一製剤あたり0.001〜0.080mgとし、好ましくは、一製剤あたり0.010〜0.080mgとし、更に好ましくは、一製剤あたり0.020〜0.060mgとすることを特徴とする請求項1に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項9】
前記フグ毒の凍結乾燥注射用粉末製剤には、更に機能調節剤の塩酸リドカインを含むことを特徴とする請求項1乃至8のいずれか一項に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項10】
前記フグ毒の凍結乾燥注射用粉末製剤には、高純度窒素ガス或いは高純度炭酸ガス等の惰性気体を充填することを特徴とする請求項1乃至9のいずれか一項に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項11】
前記フグ毒の凍結乾燥注射用粉末製剤は、筋肉注射或いは皮下注射によって投薬することを特徴とする請求項1乃至10のいずれか一項に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項12】
前記フグ毒の凍結乾燥注射用粉末製剤は、
(1)予定量のフグ毒及び任意に選択した機能調節剤を直接、溶解補助剤を含む溶液中に溶かし、pH値を3.0〜6.0に調節し、好ましくは、3.5〜4.5に調節し、ろ過によって除熱し、
(2)凍結乾燥賦形剤と安定剤を直接、無菌注射用水中に入れて溶かし、活性炭素を加えて30分間撹拌した後、ろ過で除熱し、
(3)(1)と(2)から得られた溶液を合併し、均等に混合し、ろ過によって除菌し、定量をバイアル瓶中に詰め、真空冷凍乾燥し、惰性気体を充填し、圧して緊密に蓋をすることにより、凍結乾燥注射用粉末製剤を得る
工程を含むことを特徴とする請求項1乃至11のいずれか一項に記載のフグ毒の凍結乾燥注射用粉末製剤の製造法。
【請求項13】
前記(1)工程は、超ろ過法によってろ過を行なうことを特徴とする請求項12に記載のフグ毒の凍結乾燥注射用粉末製剤の製造法。
【請求項14】
前記(2)工程における活性炭素の用量は、100mlあたり0.1〜6.0gとすることを特徴とする請求項12または13に記載のフグ毒の凍結乾燥注射用粉末製剤の製造法。
【請求項15】
前記(3)工程は、0.05μm〜0.20μmサブミクロン級の微細孔を備えるろ過膜、或いは荷電微細孔を備えるろ過膜によってろ過を行なうことを特徴とする請求項12乃至14のいずれか一項に記載のフグ毒の凍結乾燥注射用粉末製剤の製造法。
【請求項16】
前記(3)工程における製品の事前凍結温度は、-20°C〜-40°C、時間は、2〜3時間とし、主乾燥温度は、-10°Cで
6〜10時間、10°Cで 2〜5時間、20°Cで 2〜5時間とし、後乾燥温度は、30〜50°Cで 4〜10時間とすることを特徴とする請求項12に記載の真空冷凍乾燥法。
【請求項1】
フグ毒、溶解補助剤、凍結乾燥賦形剤、安定剤を含み、
フグ毒の純度は、96%以上とし、好ましくは、98%〜99.8%とし、
前記凍結乾燥賦形剤は、塩化ナトリウム(sodium)、マンニトール(mannitol)、二者の複合物のいずれかとし、前記安定剤は、デキストラン(dextran)、トレハロース(trehalose)、二者の複合物のいずれかとし、前記溶解補助剤は、クエン酸とし、前記製剤乾燥法は、真空冷凍昇華乾燥とすることを特徴とするフグ毒の凍結乾燥注射用粉末製剤。
【請求項2】
前記フグ毒と賦形剤と安定剤の比率は、1: 150〜3000: 50〜500或いは50〜6000とすることを特徴とする請求項1に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項3】
前記フグ毒の含量は、一製剤あたり0.1〜20.0μgとし、好ましくは、一製剤あたり0.5〜20.0μgとし、更に好ましくは、一製剤あたり0.5〜12.0μgとすることを特徴とする請求項1に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項4】
前記賦形剤とする塩化ナトリウム(sodium)の含量は、一製剤あたり1.0〜30mgとし、好ましくは、一製剤あたり5.0〜30mgとし、更に好ましくは、一製剤あたり5.0〜20mgとすることを特徴とする請求項1に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項5】
前記賦形剤とするマンニトール(mannitol)の含量は、一製剤あたり1.0〜30mgとし、好ましくは、一製剤あたり1.0〜20mgとし、更に好ましくは、一製剤あたり3.0〜10mgとすることを特徴とする請求項1に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項6】
前記安定剤とするデキストラン(dextran)の含量は、一製剤あたり0.5〜5.0mgとし、好ましくは、一製剤あたり2.0〜5.0mgとし、更に好ましくは、一製剤あたり3.0〜5.0mgとすることを特徴とする請求項1に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項7】
前記安定剤とするトレハロース(trehalose)の含量は、一製剤あたり0.5〜60mgとし、好ましくは、一製剤あたり2〜60mgとし、更に好ましくは、一製剤あたり10〜60mgとすることを特徴とする請求項1に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項8】
前記クエン酸の含量は、一製剤あたり0.001〜0.080mgとし、好ましくは、一製剤あたり0.010〜0.080mgとし、更に好ましくは、一製剤あたり0.020〜0.060mgとすることを特徴とする請求項1に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項9】
前記フグ毒の凍結乾燥注射用粉末製剤には、更に機能調節剤の塩酸リドカインを含むことを特徴とする請求項1乃至8のいずれか一項に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項10】
前記フグ毒の凍結乾燥注射用粉末製剤には、高純度窒素ガス或いは高純度炭酸ガス等の惰性気体を充填することを特徴とする請求項1乃至9のいずれか一項に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項11】
前記フグ毒の凍結乾燥注射用粉末製剤は、筋肉注射或いは皮下注射によって投薬することを特徴とする請求項1乃至10のいずれか一項に記載のフグ毒の凍結乾燥注射用粉末製剤。
【請求項12】
前記フグ毒の凍結乾燥注射用粉末製剤は、
(1)予定量のフグ毒及び任意に選択した機能調節剤を直接、溶解補助剤を含む溶液中に溶かし、pH値を3.0〜6.0に調節し、好ましくは、3.5〜4.5に調節し、ろ過によって除熱し、
(2)凍結乾燥賦形剤と安定剤を直接、無菌注射用水中に入れて溶かし、活性炭素を加えて30分間撹拌した後、ろ過で除熱し、
(3)(1)と(2)から得られた溶液を合併し、均等に混合し、ろ過によって除菌し、定量をバイアル瓶中に詰め、真空冷凍乾燥し、惰性気体を充填し、圧して緊密に蓋をすることにより、凍結乾燥注射用粉末製剤を得る
工程を含むことを特徴とする請求項1乃至11のいずれか一項に記載のフグ毒の凍結乾燥注射用粉末製剤の製造法。
【請求項13】
前記(1)工程は、超ろ過法によってろ過を行なうことを特徴とする請求項12に記載のフグ毒の凍結乾燥注射用粉末製剤の製造法。
【請求項14】
前記(2)工程における活性炭素の用量は、100mlあたり0.1〜6.0gとすることを特徴とする請求項12または13に記載のフグ毒の凍結乾燥注射用粉末製剤の製造法。
【請求項15】
前記(3)工程は、0.05μm〜0.20μmサブミクロン級の微細孔を備えるろ過膜、或いは荷電微細孔を備えるろ過膜によってろ過を行なうことを特徴とする請求項12乃至14のいずれか一項に記載のフグ毒の凍結乾燥注射用粉末製剤の製造法。
【請求項16】
前記(3)工程における製品の事前凍結温度は、-20°C〜-40°C、時間は、2〜3時間とし、主乾燥温度は、-10°Cで
6〜10時間、10°Cで 2〜5時間、20°Cで 2〜5時間とし、後乾燥温度は、30〜50°Cで 4〜10時間とすることを特徴とする請求項12に記載の真空冷凍乾燥法。
【図1】

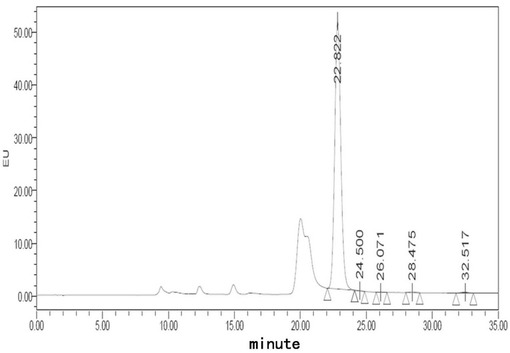
【図2】
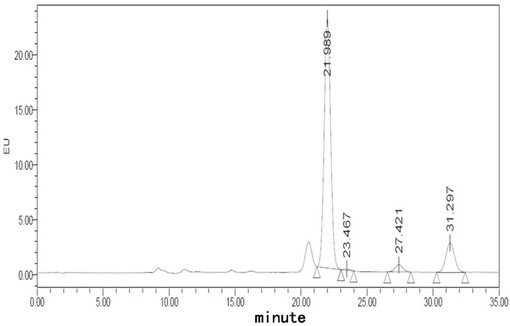
【図3】
【図4】
【図5】
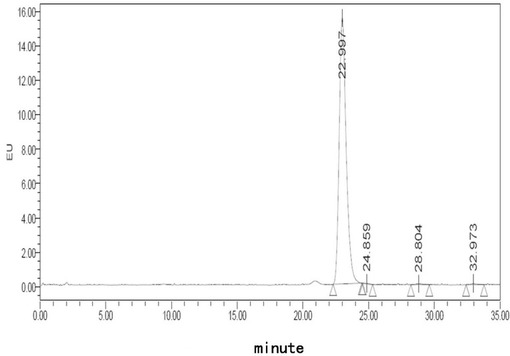
【図6】
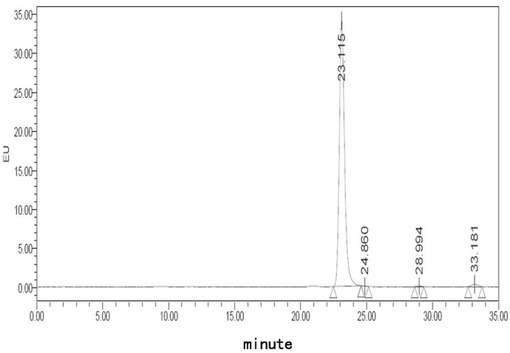
【図7】
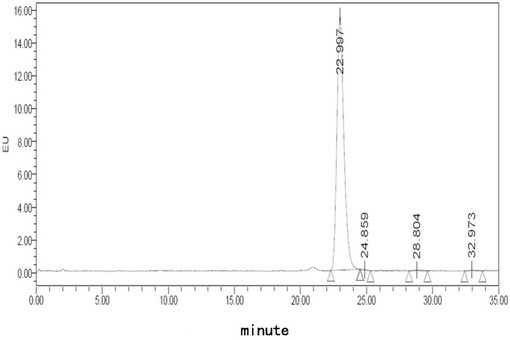
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公表番号】特表2012−502932(P2012−502932A)
【公表日】平成24年2月2日(2012.2.2)
【国際特許分類】
【出願番号】特願2011−527192(P2011−527192)
【出願日】平成21年9月17日(2009.9.17)
【国際出願番号】PCT/CN2009/074006
【国際公開番号】WO2010/031346
【国際公開日】平成22年3月25日(2010.3.25)
【出願人】(511068670)厦門朝陽生物工程有限公司 (1)
【出願人】(511068681)国家海洋局第三海洋研究所 (1)
【Fターム(参考)】
【公表日】平成24年2月2日(2012.2.2)
【国際特許分類】
【出願日】平成21年9月17日(2009.9.17)
【国際出願番号】PCT/CN2009/074006
【国際公開番号】WO2010/031346
【国際公開日】平成22年3月25日(2010.3.25)
【出願人】(511068670)厦門朝陽生物工程有限公司 (1)
【出願人】(511068681)国家海洋局第三海洋研究所 (1)
【Fターム(参考)】
[ Back to top ]