
フッ素化法

【解決手段】 非フッ素化糖誘導体とフッ化物との反応を含んでなるフッ素化糖誘導体の製造に際して、上記反応を1000ppm超50000ppm未満の量の水を含有する溶媒中で実施する。
【発明の効果】 本発明の方法では、制御された量の水の存在下では、反応収率は減少するどころか実際に増加する。反応混合物は1000ppmを超える量の水を含んでいなければならないので、反応混合物に存在する水の量を一定に保つのが格段に容易であり、反応条件を一貫して再現できる。従来技術の方法で用いられている乾燥工程の幾つかを省くことができ、試薬のコスト及び合成装置の製造コストに関してプロセス全体のコストが低減する。
【発明の効果】 本発明の方法では、制御された量の水の存在下では、反応収率は減少するどころか実際に増加する。反応混合物は1000ppmを超える量の水を含んでいなければならないので、反応混合物に存在する水の量を一定に保つのが格段に容易であり、反応条件を一貫して再現できる。従来技術の方法で用いられている乾燥工程の幾つかを省くことができ、試薬のコスト及び合成装置の製造コストに関してプロセス全体のコストが低減する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は糖誘導体のフッ素化法に関するものであり、本発明は特にフッ素化グルコースの製造に関する。当該方法は、陽電子放射断層撮影法(PET)のような方法に使用される放射性フッ素化糖誘導体の製造に特に有用である。
【背景技術】
【0002】
PET用の[18F]標識トレーサー化合物の製造方法において最も重要な因子の一つは合成全体の非補正収率である。これはプロセス全体の化学収率だけでなく、合成時間によっても影響され、合成時間は[18F]の半減期が109.7分と比較的短いので重要である。
【0003】
[18F]フッ素イオンは通例ターゲットの[18O]水のサイクロトロン照射で生成する水溶液として得られる。[18F]フッ化物を反応性求核試薬に変換して求核放射性標識反応での使用に適するようにするための様々な工程を実施することが広く行われている。非放射性フッ素化に関して、これらの工程は[18F]フッ素イオンからの水の除去及び適当な対イオンの供給を含む(Handbook of Radiopharmaceuticals, 2003, Welch & Redvanly eds. ch.6, pp.195−227)。次いで、無水溶媒を用いて求核放射性フッ素化反応が行われる(Aigbirhio et al, J. Fluor. Chem. 70, 1995,pp.279−87)。フッ素イオンからの水の除去は「naked(裸の)」フッ素イオンの製造と呼ばれる。有意の量の水が存在するとフッ素イオンの溶媒和を生じ、保護糖前駆体での求核攻撃からフッ化物を遮蔽すると考えられている。そこで、当技術分野では、フッ化物の反応性を高めるとともに水の存在に起因する水酸化副生物を避けるために水の除去が必須な工程と考えられている(Moughamir et al, Tett. Letts. 39, 1998,pp.7305−6)。
【0004】
[18F]−フルオロデオキシグルコース([18F]−FDG)のような[18F]標識化合物の合成法に関する米国第6172207号には、水溶液にアセトニトリルを添加した後共沸して蒸発乾固することによってフッ素化剤を完全に無水にしなければならないと教示されている。
【0005】
[18F]−FDGの最も一般的な合成法はHamacher et al, J. Nucl. Med. 27: 235−238(1986)に記載された方法であり、1,3,4,6−テトラ−O−アセチル−2−O−トリフルオロメタンスルホニル−β−D−マンノピラノースと[18F]フッ化物との反応を無水溶媒中で実施する。
【特許文献1】米国第6172207号明細書
【非特許文献1】Handbook of Radiopharmaceuticals, 2003, Welch & Redvanly eds. ch.6, pp.195−227
【非特許文献2】Aigbirhio et al, J. Fluor. Chem. 70, 1995,pp.279−87
【非特許文献3】Moughamir et al, Tett. Letts. 39, 1998,pp.7305−6
【非特許文献4】Hamacher et al, J. Nucl. Med. 27: 235−238(1986)
【発明の開示】
【発明が解決しようとする課題】
【0006】
[18F]標識糖誘導体の製造に現在用いられている方法には幾つかの問題が存在する。その一つはフッ素イオン及び溶媒から残留水をすべて除去するのに時間がかかり、そのため合成全体の非補正収率に影響することである。また、残留水をすべて除去する必要があるとすると、自動合成装置での合成及び機械の複雑さが増す。例えば、合成に必要な乾燥サイクルの数が増し、合成装置で合成を行うために一段と強力なヒータが必要とされかねない。
【0007】
さらに、放射性フッ素化反応を一貫して再現できるようにするのは困難である。これは、溶媒中に少量(例えば約1000ppm以下)の残留水が存在することが多々あり、合成全体の非補正収率が標識反応の際に存在する残留水分量によって大きく変動するためである。水分量を1500ppm±200ppm、15%の偏差に維持できるという結果が得られている。750ppmでの水分量のバラツキの絶対値はパーセント単位で2倍の偏差をもつであろう。
【課題を解決するための手段】
【0008】
今回、本発明らは、糖誘導体のフッ素化を無水条件下で行う必要がないという驚くべき知見を得た。実際、反応混合物の水分量を慎重に制御すれば、プロセスの放射化学純度(ひいては全収率)が確かに向上する。
【0009】
これは、従来技術において上記反応を無水条件下で行う必要性が強調されていたことに鑑みればまさに予想外といえる。
【0010】
そこで、本発明の第一の態様では、非フッ素化糖誘導体とフッ化物との反応を含んでなるフッ素化糖誘導体の製造方法であって、上記反応を1000ppm超50000ppm未満の量の水を含有する溶媒中で実施することを特徴とする方法を提供する。
【0011】
本発明の方法は従来技術の方法に比して格段の利点を有する。まず、このような制御された量の水の存在下では、反応収率は減少するどころか実際に増加することが判明した。
【0012】
第二に、反応混合物は1000ppmを超える量の水を含んでいなければならないので、反応混合物に存在する水の量を一定に保つのが格段に容易である(例えば標識用溶媒に水を意図的に混入することによる)。これは、反応条件を一貫して再現できることを意味している。
【0013】
第三に、従来技術の方法で用いられている乾燥工程の幾つかを省くことができ、試薬のコスト及び合成装置の製造コストに関してプロセス全体のコストが低減する。プロセスが簡単なほどプロセス全体の信頼性に良い影響を与えると予想される。
【発明を実施するための最良の形態】
【0014】
本明細書において「非フッ素化糖誘導体」という用語は、OH基の1つが脱離基で置換された多糖類、オリゴ糖類、二糖類又は単糖類をいい、例えば国際公開第03/002157号に教示されているように固体担体に適宜結合していてもよい。本発明の方法は、グルコース、フルクトース、リボース、アラビノース、マンノース又はガラクトースのような単糖類のフッ素化に特に適している。
【0015】
「保護非フッ素化糖誘導体」では、糖の残りのOH基は適当な保護基で保護されている。
【0016】
「フッ素化糖誘導体」という用語は、OH基の1つがフッ素で置換された多糖類、オリゴ糖類、二糖類又は単糖類(例えばグルコース、フルクトース、リボース、アラビノース、マンノース又はガラクトースなど)をいう。
【0017】
「保護フッ素化糖誘導体」では、糖の残りのOH基は適当な保護基で保護されている。
【0018】
本発明で用いる保護糖誘導体に適した保護基は当技術分野で周知であり、例えば“Protecting Groups in Organic Synthesis”, Theodora W. Greene and Peter G. M. Wuts, published by John Wiley & Sons Incに記載されている。具体的にどのような保護基を選択するかはフッ素化生成物の所期の用途によって左右されるが、例えば、水酸基はアルキル又は芳香族エステルへの変換(例えば塩化アセチルのような塩化アルカノイルとの反応による)によって保護できる。別法として、水酸基はアルキル又はベンジルエーテルにのようなエーテルにも変換できる。
【0019】
出発原料及び反応生成物が共に保護糖誘導体であるのが好ましい。
【0020】
好適な脱離基は当技術分野で周知であり、トルエンスルホネート及びメタンスルホネートが挙げられる。ただし、脱離基がトリフルオロメタンスルホネート(トリフレート)基であるのが特に好ましい。
【0021】
フッ素化反応は概して求核置換反応であり、脱離基のフルオロ置換はSN2機構による糖の立体化学の反転を起こすことがある。そこで、最初の非フッ素化糖誘導体が生成物とは異なる糖の誘導体となることが多々ある。
【0022】
好ましい生成物は保護フッ素化グルコース誘導体であり、対応マンノース誘導体(例えばテトラアセチルマンノース誘導体)から調製できる。
【0023】
この反応は1,3,4,6−テトラ−O−アセチル−2−トリフルオロメタンスルホニル−β−D−マンノピラノース(すなわちテトラアセチルマンノーストリフレート)からの2−フルオロ−1,3,4,6−テトラ−O−アセチル−D−グルコース(すなわちテトラアセチルフルオログルコース又はpFDG)の調製に特に適している。
【0024】
好適な溶媒としては、アセトニトリル、ジメチルホルムアミド、ジメチルスルホキシド、テトラヒドロフラン、ジオキサン、1,2−ジメトキシエタン、スルホラン及び/又はN−メチルピロリジノンのような非プロトン性有機溶媒又はそのいずれかの混合物が挙げられる。ただし、アセトニトリルが反応に特に適した溶媒であることが判明した。
【0025】
溶媒に1000ppm以上50000ppm未満の水を含有させることによって反応収率が向上するが、水分量が約1000〜15000ppmのときに、さらに大きな向上が達成される。最良の結果は、水分量約2000〜7000ppm、好ましくは2500〜5000ppmの溶媒を用いて得られた。一実施形態では、好ましい水分量は3000ppm〜6000ppmである。
【0026】
本明細書で用いる「ppm」という用語は、溶媒の水分量を表す場合にはμg水/gを意味する。
【0027】
溶媒中の水の正確な量は、含水溶媒を所望の水分量に達するまで乾燥させるか或いは乾燥溶媒に適量の水を添加することによって達成できる。フッ化物は水溶液中で製造してもよく、この場合、溶媒の添加と溶媒/水混液の蒸発を繰返し行うか或いはフッ化物水溶液を所望の有機溶媒で希釈することによって、所望の水分量のフッ化物溶液を得ることができる。官能化ポリスチレン樹脂(例えば、エポキシド、メチルイソシアネート又は酸無水物官能化樹脂など)のようなスカベンジャー樹脂を用いてフッ化物溶液から水を除去することによって、溶媒の水分量を低減することもできる。好適な樹脂は、例えばNovabiochem社から市販されている。適当な触媒、例えば4−ジメチルアミノピリジン(4−DMAP)を用いることによってスカベンジャー樹脂の性能を向上させることができる。
【0028】
この実施形態では、容器中でスカベンジャー樹脂をフッ化物溶液と混合し、スカベンジャー樹脂を濾過により分離することによって乾燥工程を実施し得る。スカベンジャー樹脂を自動化合成装置で用いる場合に特に好適な別法では、スカベンジャー樹脂を容器に収容し、これにフッ化物溶液を流してもよい。フッ化物溶液は、乾燥に十分な時間スカベンジャー樹脂に滞留できるように、例えば0.1mL/分〜100mL/分の流速で連続流として又は回分式にスカベンジャー樹脂に流せばよい。
【0029】
スカベンジャー樹脂のこの用途は新規であり、本発明の別の態様では、放射性フッ化物、特に[18F]フッ化物の溶液の水分量を低減する方法であって、当該方法が上記溶液をスカベンジャー樹脂に接触させることを含んでなる方法を提供する。好適には、上記溶液は、アセトニトリル、ジメチルホルムアミド、ジメチルスルホキシド、テトラヒドロフラン、ジオキサン、1,2−ジメトキシエタン、スルホラン及びN−メチルピロリジノンのような非プロトン性有機溶媒中のフッ化物を含む。
【0030】
反応は液相中で実施してもよいし、或いは非フッ素化糖誘導体を固体担体に結合して以下の式(I)の樹脂−リンカー−ベクター(RLV)を形成してもよい。
【0031】
固体担体−リンカー−X−保護非フッ素化糖誘導体 (I)
式中、固体担体は適当な担体であり、
保護非フッ素化糖誘導体は上記で定義した通りであり、
Xは保護非フッ素化糖誘導体の特定の部位での求核置換を促進する基、例えば−SO2O−であり、
リンカーは、反応性を最大に高めるため反応性部位を固体担体構造から十分に離隔させるのに役立つ適当な有機基であり、例えば0〜4個のアリール基(例えばフェニル)及び/又はC1〜C6アルキル又はハロアルキル(特にフルオロアルキル)鎖並びに適宜追加される1〜4個のアミド又はスルホンアミド基のような官能基である。
【0032】
RLV系は国際公開第03/002157号に詳細に記載されており、好適なリンカーの詳細についても記載されている。
【0033】
式(I)のRLVをフッ化物溶液と接触させると、固体担体から糖が置換され、保護フッ素化糖誘導体を与える。
【0034】
好適な固体担体は国際公開第03/002157号にも記載されており、ポリスチレン(ポリエチレングリコールなどでブロックグラフト化したものでもよい)、ポリアクリルアミド又はポリプロピレンのようなポリマー或いはガラス又はケイ素をかかるポリマーで被覆したものが挙げられる。或いは、例えば国際公開第03/002157号に記載されるように樹脂を用いてもよい。固体担体は、ビーズやピンのような小さな離散粒子の形態でも、或いはカートリッジの内面や微細加工容器に設けられたコーティングの形態であってもよい。本発明の方法を固体担体で実施することによって、追加の分離段階を必要とせずに純粋な形の生成物を得ることができる。これはフッ素化が放射性フッ素化のときに特に有利である。プロセスにおける時間の節約は非補正放射化学収率の向上をもたらすからである。
【0035】
反応は通常5℃〜180℃、特に75℃〜125℃の温度で実施される。
【0036】
本発明の方法は自動化合成の一部として実施できる。これは、反応を溶液中で行おうと非フッ素化糖を固相に結合させようと当てはまる。
【0037】
非フッ素化糖誘導体と反応させるフッ化物はイオン性化合物であってもよいし、適当な対イオンと共に供給してもよい。ただし、対イオンが反応溶媒中でフッ化物の溶解性を維持するのに十分な溶解性をもつことが重要である。従って、好適な対イオンとしては、ルビジウム又はセシウムのような大きなソフト金属イオン或いはテトラアルキルアンモニウム及びテトラアルキルホスホニウムのような非金属イオンが挙げられる。カリウムイオンも対イオンとして用いることができ、この場合、フッ化物の反応性を高めるため、4,7,13,16,21,24−ヘキサオキサ−1,10−ジアザビシクロ−[8,8,8]−ヘキサコサン(Kryptofix(商標)2.2.2という商標で市販)のような相間移動触媒を添加してカリウム塩を有機溶媒中に溶解させてもよい。
【0038】
本発明の方法は放射性フッ素化誘導体、特に[18F]標識誘導体の製造に特に適しており、フッ化物は[18F]フッ素イオンを含んでいてもよい。
【0039】
上記で簡単に説明した通り、[18F]フッ素イオンは[18O]水ターゲットの照射によって調製することができ、これを本発明の方法における最初の工程としてもよい。
【0040】
本発明の方法は[18F]−pFDGのような放射性フッ素化糖誘導体の製造に特に有用であり、[18F]−pFDGを脱保護すれば周知のPETトレーサーである[18F]−FDGのような化合物が得られる。脱保護はこの方法の追加工程とし得る。生成物のフッ素化糖の保護基がアセチル誘導体のようなエステルである場合、脱保護は酸又は塩基加水分解によって実施し得る。
【0041】
その他の追加工程としては、溶液からの過剰の[18F]フッ化物の除去及び有機溶媒の除去が挙げられる。過剰の[18F]フッ化物は、例えばイオン交換クロマトグラフィー又は固相吸着剤のような常法で除去できる。好適なイオン交換樹脂としてはBIO−RAD AG 1−X8(商標)及びWaters QMA(商標)が挙げられ、好適な固相吸着剤としてはアルミナが挙げられる。
【0042】
有機溶媒は、真空昇温下での蒸発或いは溶液に窒素又はアルゴンのような不活性ガス気流を通すことによっても除去できる。
【0043】
これら工程の最終生成物である[18F]トレーサー化合物は、患者への投与のために製剤化してもよく、例えば水溶液として、[18F]標識トレーサーを無菌等張食塩水(エタノールのような適当な有機溶媒を10%以下含んでもよい。)或いはリン酸緩衝液のような適当な緩衝溶液中に溶解することによって調製してもよい。その他アスコルビン酸のような添加剤を用いてもよく、アスコルビン酸は放射線分解を低減する。
【0044】
上述の通り本発明の方法で調製できる特に好ましい化合物は[18F]−pFDGであり、そこで、本発明の第二の態様では、テトラアセチルマンノーストリフレートと[18F]フッ化物との反応を含む[18F]−pFDGの製造方法であって、上記フッ化物を1000ppm超50000ppm未満の量の水を含有する溶媒中に溶解されていることを特徴とする方法を提供する。本発明のこの態様の一実施形態では、テトラアセチルマンノーストリフレート(1当量)を、Kryptofix(商標)2.2.2(0.9〜1.1モル当量、好適には0.98〜0.99モル当量)、炭酸カリウム(0.4〜0.6モル当量、好適には0.50〜0.60モル当量)の存在下、1000ppm超50000ppm未満の量の水を含有するアセトニトリル中で[18F]フッ化物と反応させる。
【0045】
本発明の好ましい特徴は第一の態様で説明した通りである。特に、この方法は[18O]−水ターゲットの照射によって[18F]フッ化物を生成させる最初の工程と、酸又はアルカリ加水分解によって[18F]−pFDGを[18F]−FDGへと変換する追加工程を含んでもよい。
【0046】
以下の実施例及び図面を参照して本発明をさらに詳しく説明する。
【実施例】
【0047】
実施例1−糖の18F標識に対する水分量変化の影響
本例では、18F−での3通りの糖標識法を用いて、反応混合物の水分量変化の影響を評価した。
【0048】
a)樹脂−リンカーベクター(RLV)標識
18F−をTracerlab MX(商標)に導入し、2−[18F]フルオロ−2−デオキシグルコースの製造で用いられる標準的乾燥工程を用いて乾燥した。Kryptofix(商標)2.2.2/炭酸カリウムを用いてフッ化物をアセトニトリル中に溶解させた。乾燥工程を終え、フッ化物をアセトニトリル中に溶解した後、アセトニトリル中の乾燥18F−試料を水分量分析用に採取し、カールフィッシャー滴定計で測定した。乾燥プロセスで通常得られるよりも水分量を高くするため、必要に応じて、追加の水を導入した。
【0049】
1mLのHi Trap(登録商標)カートリッジに、0.003mmol/gの置換度の固相結合保護マンノース前駆体約370mgを充填した。カートリッジの一端はループを介してシリンジドライバーに接続した。カートリッジのもう一方の端はモレキュラーシーブベントを備えたN2充填バイアルに接続した。ホットエアガンを用いて、カートリッジを80℃のカートリッジ外部温度に加熱した。
【0050】
この系に次いで乾燥アセトニトリル6×0.5mLを注入して不純物及び水(例えば不完全な乾燥のため自然に樹脂に存在する)を洗い流し、アセトニトリルは廃棄した。この装置に、乾燥18F−溶液(450μL)を収容したオートサンプラーバイアルを装着した。次いで、シリンジドライバーで乾燥フッ化物を流速180μL/分で5サイクル前後に移動させた。乾燥フッ化物が固相マンノース前駆体と反応して保護[18F]−2−デオキシグルコース誘導体が遊離した(これを脱保護すると2−[18F]−2−デオキシグルコースが得られる)。
【0051】
次にオートサンプラーバイアルから5μL試料を薄層クロマトグラフィー(TLC)分析用に採取し、シリカゲル60 F254プレートにスポットし、90/10比に調製したアセトニトリル/水溶媒で展開した。放射化学純度はPerkin Elmer Instant Imagerで求めた。
【0052】
b)テトラアセチルマンノーストリフレート標識
HPLCグレードのH2O600μLに溶解した32mgのK2CO3に、2.5mLアセトニトリルに溶解したKryptofix(商標)2.2.2を加えた溶液を調製した。その0.6mLを、18O濃縮水中の約40MBqの18F−と共にガラス状炭素反応器に加えた。ヒータコントローラを95℃に設定し、反応容器を合計35分間加熱してフッ化物を乾燥した。水とアセトニトリルを窒素気流中で蒸発させた。
【0053】
フッ化物からの水の共沸除去を促すため、1mLのアセトニトリルを2分間隔で計3回注入したが、最初の添加は乾燥プロセスで20分であった。35分後にヒータを切り、反応容器外からの圧縮気流で反応容器を約45℃に冷却した。
【0054】
次いでCH3CN2.0mL中のマンノーストリフレート25mgを乾燥フッ化物に添加し、混合した。ヒータコントローラを85℃に設定した。設定温度に達して2分後にTLC分析用試料を採取した。ヒータを切り、反応容器外部からの圧縮気流で反応容器を約45℃に冷却した。
【0055】
TLC分析用試料をシリカゲルにスポットし、90/10比に調製したアセトニトリル/水溶媒で展開した。放射化学純度はPerkin Elmer Instant Imagerで求めた。次に反応容器の蓋を取り、50μL試料を採取してカールフィッシャー滴定計で水分量を分析した。
【0056】
c)自動化標識
内蔵加熱反応容器及び使い捨てポリプロピレンカセットと共に25個の三方弁を備えたプロトタイプ自動化合成プラットフォームで標識を行った。カセット流路は中間体又は最終生成物のSPE精製も可能にする。
【0057】
フッ化物はまずQMAカートリッジでトラップし、20mgのKryptofix(商標)2.2.2と4.1mgのK2CO3と320μLのCH3CNと80μLのH2Oの溶液で溶出した。これを次いで窒素気流中105℃/120℃で約6分間乾燥し、アセトニトリル中の約20mg/mLテトラアセチルマンノーストリフレート1.5mLに再溶解した。
【0058】
標識反応は標識温度105又は125℃、反応時間90又は270秒で行った。標識後、2[18F]−2−デオキシグルコースをTLCで分析した。TLCプレートはシリカゲル60 F254であり、95%アセトニトリル、5%水を展開溶媒として用いた。放射化学純度はPerkin Elmer Instant Imagerで求めた。
【0059】
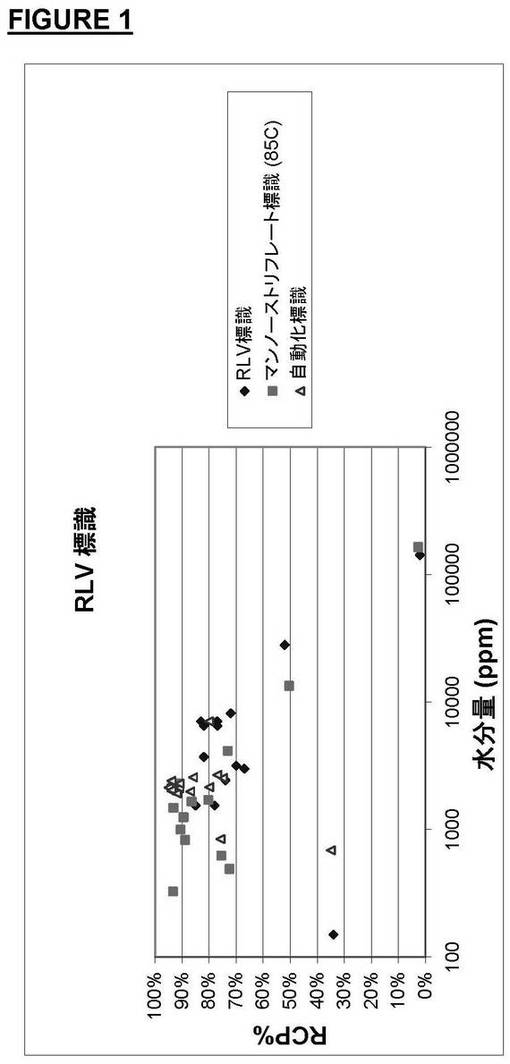
実施例1の3通りの実験の結果を図1に示すが、この図から、反応混合物の水分量が1000ppm未満のときは生成物の放射化学純度が比較的低かったのに対して、水分量が1000〜5000ppmのときは生成物の放射化学純度が大幅に向上したことが分かる。このプロットは溶媒中の最適水分量が約2000〜7000ppmであったことを示している。
【0060】
実施例2−標識プロセスでの[18F]−pFDG及びpGlucoseの生成の相関
RLVの18−フッ化物標識を、Kryptofix(商標)2.2.2、炭酸カリウム及び様々な量の水の存在下、アセトニトリル中で実施した。標識後、得られた混合物を逆相HPLCに付して、Phenomenex Luna 5μm C18カラム(4.6mm×150mm)を用いて10分間流速1mL/分で90%溶媒A/10%溶媒Bから5%溶媒A/95%溶媒Bへの勾配(溶媒A=0.1%トリフルオロ酢酸水溶液、溶媒B=0.1%トリフルオロ酢酸アセトニトリル溶液)を流した。保持時間3分の保護グルコースのピークと6.6分の保護FDGのピーク(主に[19F]−FDGの存在に起因し、その量は[18F]−FDGに比例する)の積分値を求め、それらの相関関係を求めた。
【0061】
反応混合物中に大量の水が存在すると、トリフレート基での求核置換の結果、([18F]−pFDGではなく)保護グルコースが大量に生成すると一般に考えられている。したがって、生成混合物中の保護グルコース誘導体濃度に対する[18F]−pFDG濃度のグラフをプロットしたグラフの傾きは負となり、水分量が高いほど保護グルコースレベルは高く[18F]−pFDGレベルは低くなると予想されていた。
【0062】
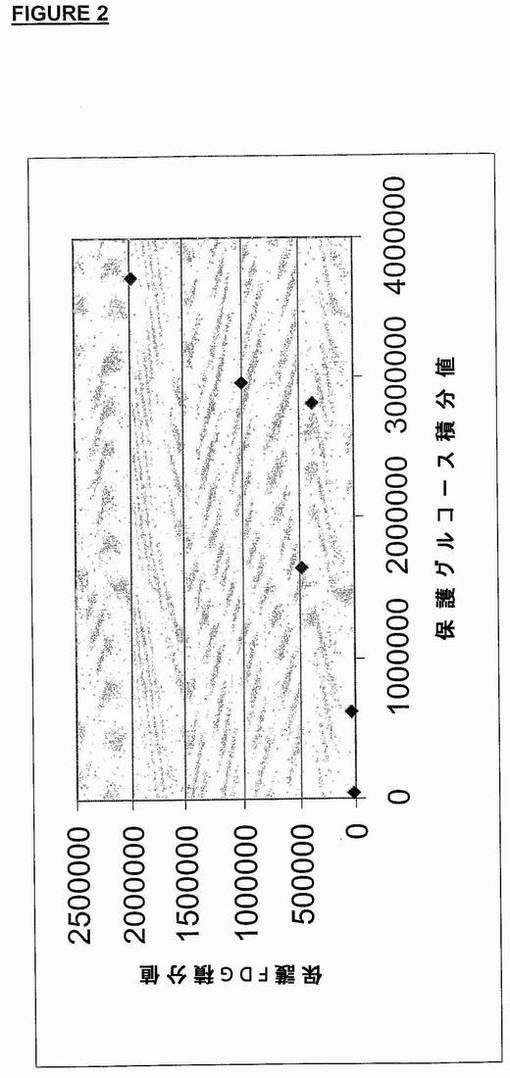
しかし、樹脂に結合させたテトラアセチルマンノーストリフレートの標識実験から、HPLCの2つのピークの間に良好な正の相関関係があることが判明した(図2参照)。これは高濃度の水が存在するとこれら2種類の生成物の形成が抑制されることを示している。
【0063】
実施例3:1,3,4,6−テトラ−O−アセチル−2−フルオロ−β−D−マンノピラノースの自動化合成
標識化の最初に放射性反応混合物をサンプリングするのには問題があった。そのため、水分量を標識反応の最後にRCP(ITLCで測定)と共に測定した。これから、以下に述べる通り水封鎖を計算に入れることよって標識開始時の水分量を算出した。
【0064】
放射性標識実験
1,3,4,6−テトラ−O−アセチル−2−フルオロ−β−D−マンノピラノースの合成は、使い捨てカセットが取り付けられるように設計された自動合成装置で実施した。このカセットは、固相抽出カートリッジ用のスペースとシリンジと共に各種試薬の収容用バイアルを備えた25弁の使い捨てカセットからなる。
【0065】
合成シーケンスを行って2mL水中の約50MBqの18−フッ化物をWaters Access PlusQMAカートリッジ(その炭酸塩の形態)でトラップし、次にアセトニトリル/水中のkryptofix及び炭酸塩の溶液(20.3mgのkryptofix 222、4.3mgの炭酸カリウム、320mLのアセトニトリル、80mLの水)でカートリッジを溶出して加熱反応器に回収した。これを乾燥窒素気流中で加熱して乾燥し、次に所定の水分量のアセトニトリル中マンノーストリフレート溶液を反応器に添加した。
【0066】
外部ヒータ温度125℃でさらに80秒間反応せしめた後、0.6mLを回収して廃棄し(ラインから残留水を除去するため)、残余を生成物バイアルに移した。50μL溶液を用いたカールフィッシャー滴定で生成物バイアルの水分量を決定し、インスタント薄層クロマトグラフィー(ITLC)でRCPを測定した。TLCはシリカTLCプレートで実施し、スポットを95%アセトニトリル、5%水で溶出し、TLCを用いて18−フッ化物及び1,3,4,6−テトラ−O−アセチル−2−フルオロβ−D−マンノピラノース(いずれの場合も2成分のみ)の相対比を計測した。
【0067】
水封鎖測定
3回のコールド(非放射性)実験を行って、マンノーストリフレートとの反応でどの程度の水がなくなったかを調べるために所定体積の反応液を標識反応の前後にサンプリングした。これによって、測定した水分量に水封鎖を計算に入れることができた。
【0068】
上記放射性標識実験と同様の合成シーケンスを行ってWaters Access PlusQMAカートリッジ(その炭酸塩の形態)に2mLの水を流し、次にアセトニトリル/水中のkryptofixと炭酸塩の溶液(20.3mgのkryptofix 222、4.3mgの炭酸カリウム、320mLのアセトニトリル、80mLの水)でカートリッジを溶出して加熱反応器に回収した。これを乾燥窒素気流中で加熱して乾燥し、次に所定の水分量のアセトニトリル中マンノーストリフレート溶液を反応器に添加した。
【0069】
マンノーストリフレート溶液を反応器に添加してすぐに0.6mLを回収して生成物バイアル中に注入した。標識反応を外部ヒータ温度125℃でさらに80秒間進行させ、残りの溶液を別の生成物バイアルに移した。50μLの溶液を用いたカールフィッシャー滴定器で各バイアルの水分量を測定した。
【0070】
水封鎖実験の結果を表1に示す。表1はアセトニトリル溶媒中の水の存在量を示す。
【0071】
【表1】

低レベル及び中間レベルの水分量では、マンノーストリフレートとの反応による水の有意な封鎖は認められなかった。しかし、高レベルの水分量では水分量が約7%低下した。
【0072】
放射標識結果
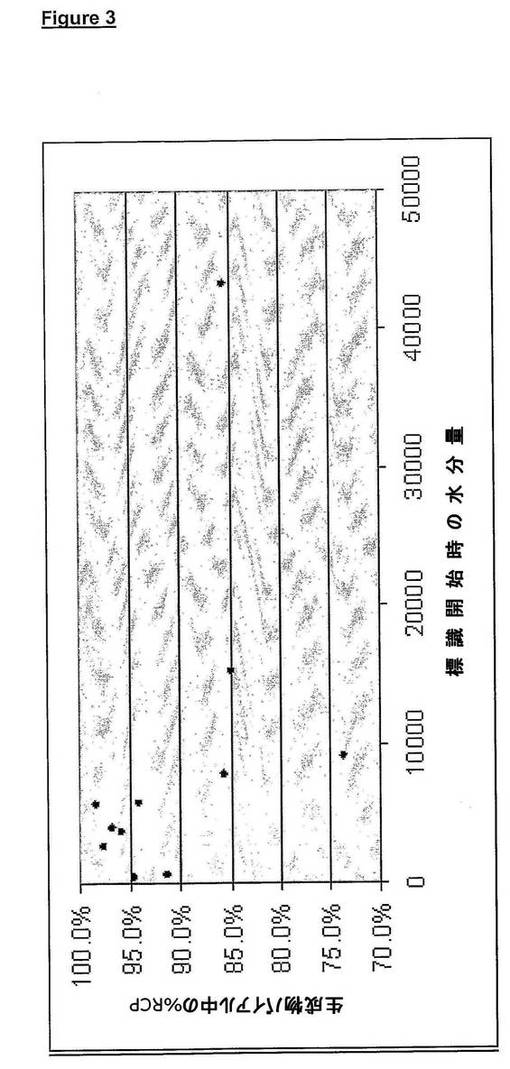
各放射標識反応の水分量を測定し、マンノーストリフレートによる水の封鎖を計算に入れて標識前の水分量を補正した。各水分量で得られたRCPを表2に示すとともに、図3に図示する。
【0073】
【表2】
これらの結果は3000〜6000ppmが好ましい水分量であることを裏付ける。RCP73.6%での疑わしい結果を無視すれば、非常に湿分の多い反応条件であったとしても、反応は約85%RCPに近づく。
【図面の簡単な説明】
【0074】
【図1】[18F]−pFDG生成物の放射化学純度と溶媒の水分量との関係を示すプロット。
【図2】樹脂−リンカー−ベクターでの標識プロセスにおける[18F]−pFDGとpGlucoseの生成の関係を示すグラフ。
【図3】自動化合成における[18F]−pFDG生成物の放射化学純度と溶媒の水分量との関係を示すプロット。
【技術分野】
【0001】
本発明は糖誘導体のフッ素化法に関するものであり、本発明は特にフッ素化グルコースの製造に関する。当該方法は、陽電子放射断層撮影法(PET)のような方法に使用される放射性フッ素化糖誘導体の製造に特に有用である。
【背景技術】
【0002】
PET用の[18F]標識トレーサー化合物の製造方法において最も重要な因子の一つは合成全体の非補正収率である。これはプロセス全体の化学収率だけでなく、合成時間によっても影響され、合成時間は[18F]の半減期が109.7分と比較的短いので重要である。
【0003】
[18F]フッ素イオンは通例ターゲットの[18O]水のサイクロトロン照射で生成する水溶液として得られる。[18F]フッ化物を反応性求核試薬に変換して求核放射性標識反応での使用に適するようにするための様々な工程を実施することが広く行われている。非放射性フッ素化に関して、これらの工程は[18F]フッ素イオンからの水の除去及び適当な対イオンの供給を含む(Handbook of Radiopharmaceuticals, 2003, Welch & Redvanly eds. ch.6, pp.195−227)。次いで、無水溶媒を用いて求核放射性フッ素化反応が行われる(Aigbirhio et al, J. Fluor. Chem. 70, 1995,pp.279−87)。フッ素イオンからの水の除去は「naked(裸の)」フッ素イオンの製造と呼ばれる。有意の量の水が存在するとフッ素イオンの溶媒和を生じ、保護糖前駆体での求核攻撃からフッ化物を遮蔽すると考えられている。そこで、当技術分野では、フッ化物の反応性を高めるとともに水の存在に起因する水酸化副生物を避けるために水の除去が必須な工程と考えられている(Moughamir et al, Tett. Letts. 39, 1998,pp.7305−6)。
【0004】
[18F]−フルオロデオキシグルコース([18F]−FDG)のような[18F]標識化合物の合成法に関する米国第6172207号には、水溶液にアセトニトリルを添加した後共沸して蒸発乾固することによってフッ素化剤を完全に無水にしなければならないと教示されている。
【0005】
[18F]−FDGの最も一般的な合成法はHamacher et al, J. Nucl. Med. 27: 235−238(1986)に記載された方法であり、1,3,4,6−テトラ−O−アセチル−2−O−トリフルオロメタンスルホニル−β−D−マンノピラノースと[18F]フッ化物との反応を無水溶媒中で実施する。
【特許文献1】米国第6172207号明細書
【非特許文献1】Handbook of Radiopharmaceuticals, 2003, Welch & Redvanly eds. ch.6, pp.195−227
【非特許文献2】Aigbirhio et al, J. Fluor. Chem. 70, 1995,pp.279−87
【非特許文献3】Moughamir et al, Tett. Letts. 39, 1998,pp.7305−6
【非特許文献4】Hamacher et al, J. Nucl. Med. 27: 235−238(1986)
【発明の開示】
【発明が解決しようとする課題】
【0006】
[18F]標識糖誘導体の製造に現在用いられている方法には幾つかの問題が存在する。その一つはフッ素イオン及び溶媒から残留水をすべて除去するのに時間がかかり、そのため合成全体の非補正収率に影響することである。また、残留水をすべて除去する必要があるとすると、自動合成装置での合成及び機械の複雑さが増す。例えば、合成に必要な乾燥サイクルの数が増し、合成装置で合成を行うために一段と強力なヒータが必要とされかねない。
【0007】
さらに、放射性フッ素化反応を一貫して再現できるようにするのは困難である。これは、溶媒中に少量(例えば約1000ppm以下)の残留水が存在することが多々あり、合成全体の非補正収率が標識反応の際に存在する残留水分量によって大きく変動するためである。水分量を1500ppm±200ppm、15%の偏差に維持できるという結果が得られている。750ppmでの水分量のバラツキの絶対値はパーセント単位で2倍の偏差をもつであろう。
【課題を解決するための手段】
【0008】
今回、本発明らは、糖誘導体のフッ素化を無水条件下で行う必要がないという驚くべき知見を得た。実際、反応混合物の水分量を慎重に制御すれば、プロセスの放射化学純度(ひいては全収率)が確かに向上する。
【0009】
これは、従来技術において上記反応を無水条件下で行う必要性が強調されていたことに鑑みればまさに予想外といえる。
【0010】
そこで、本発明の第一の態様では、非フッ素化糖誘導体とフッ化物との反応を含んでなるフッ素化糖誘導体の製造方法であって、上記反応を1000ppm超50000ppm未満の量の水を含有する溶媒中で実施することを特徴とする方法を提供する。
【0011】
本発明の方法は従来技術の方法に比して格段の利点を有する。まず、このような制御された量の水の存在下では、反応収率は減少するどころか実際に増加することが判明した。
【0012】
第二に、反応混合物は1000ppmを超える量の水を含んでいなければならないので、反応混合物に存在する水の量を一定に保つのが格段に容易である(例えば標識用溶媒に水を意図的に混入することによる)。これは、反応条件を一貫して再現できることを意味している。
【0013】
第三に、従来技術の方法で用いられている乾燥工程の幾つかを省くことができ、試薬のコスト及び合成装置の製造コストに関してプロセス全体のコストが低減する。プロセスが簡単なほどプロセス全体の信頼性に良い影響を与えると予想される。
【発明を実施するための最良の形態】
【0014】
本明細書において「非フッ素化糖誘導体」という用語は、OH基の1つが脱離基で置換された多糖類、オリゴ糖類、二糖類又は単糖類をいい、例えば国際公開第03/002157号に教示されているように固体担体に適宜結合していてもよい。本発明の方法は、グルコース、フルクトース、リボース、アラビノース、マンノース又はガラクトースのような単糖類のフッ素化に特に適している。
【0015】
「保護非フッ素化糖誘導体」では、糖の残りのOH基は適当な保護基で保護されている。
【0016】
「フッ素化糖誘導体」という用語は、OH基の1つがフッ素で置換された多糖類、オリゴ糖類、二糖類又は単糖類(例えばグルコース、フルクトース、リボース、アラビノース、マンノース又はガラクトースなど)をいう。
【0017】
「保護フッ素化糖誘導体」では、糖の残りのOH基は適当な保護基で保護されている。
【0018】
本発明で用いる保護糖誘導体に適した保護基は当技術分野で周知であり、例えば“Protecting Groups in Organic Synthesis”, Theodora W. Greene and Peter G. M. Wuts, published by John Wiley & Sons Incに記載されている。具体的にどのような保護基を選択するかはフッ素化生成物の所期の用途によって左右されるが、例えば、水酸基はアルキル又は芳香族エステルへの変換(例えば塩化アセチルのような塩化アルカノイルとの反応による)によって保護できる。別法として、水酸基はアルキル又はベンジルエーテルにのようなエーテルにも変換できる。
【0019】
出発原料及び反応生成物が共に保護糖誘導体であるのが好ましい。
【0020】
好適な脱離基は当技術分野で周知であり、トルエンスルホネート及びメタンスルホネートが挙げられる。ただし、脱離基がトリフルオロメタンスルホネート(トリフレート)基であるのが特に好ましい。
【0021】
フッ素化反応は概して求核置換反応であり、脱離基のフルオロ置換はSN2機構による糖の立体化学の反転を起こすことがある。そこで、最初の非フッ素化糖誘導体が生成物とは異なる糖の誘導体となることが多々ある。
【0022】
好ましい生成物は保護フッ素化グルコース誘導体であり、対応マンノース誘導体(例えばテトラアセチルマンノース誘導体)から調製できる。
【0023】
この反応は1,3,4,6−テトラ−O−アセチル−2−トリフルオロメタンスルホニル−β−D−マンノピラノース(すなわちテトラアセチルマンノーストリフレート)からの2−フルオロ−1,3,4,6−テトラ−O−アセチル−D−グルコース(すなわちテトラアセチルフルオログルコース又はpFDG)の調製に特に適している。
【0024】
好適な溶媒としては、アセトニトリル、ジメチルホルムアミド、ジメチルスルホキシド、テトラヒドロフラン、ジオキサン、1,2−ジメトキシエタン、スルホラン及び/又はN−メチルピロリジノンのような非プロトン性有機溶媒又はそのいずれかの混合物が挙げられる。ただし、アセトニトリルが反応に特に適した溶媒であることが判明した。
【0025】
溶媒に1000ppm以上50000ppm未満の水を含有させることによって反応収率が向上するが、水分量が約1000〜15000ppmのときに、さらに大きな向上が達成される。最良の結果は、水分量約2000〜7000ppm、好ましくは2500〜5000ppmの溶媒を用いて得られた。一実施形態では、好ましい水分量は3000ppm〜6000ppmである。
【0026】
本明細書で用いる「ppm」という用語は、溶媒の水分量を表す場合にはμg水/gを意味する。
【0027】
溶媒中の水の正確な量は、含水溶媒を所望の水分量に達するまで乾燥させるか或いは乾燥溶媒に適量の水を添加することによって達成できる。フッ化物は水溶液中で製造してもよく、この場合、溶媒の添加と溶媒/水混液の蒸発を繰返し行うか或いはフッ化物水溶液を所望の有機溶媒で希釈することによって、所望の水分量のフッ化物溶液を得ることができる。官能化ポリスチレン樹脂(例えば、エポキシド、メチルイソシアネート又は酸無水物官能化樹脂など)のようなスカベンジャー樹脂を用いてフッ化物溶液から水を除去することによって、溶媒の水分量を低減することもできる。好適な樹脂は、例えばNovabiochem社から市販されている。適当な触媒、例えば4−ジメチルアミノピリジン(4−DMAP)を用いることによってスカベンジャー樹脂の性能を向上させることができる。
【0028】
この実施形態では、容器中でスカベンジャー樹脂をフッ化物溶液と混合し、スカベンジャー樹脂を濾過により分離することによって乾燥工程を実施し得る。スカベンジャー樹脂を自動化合成装置で用いる場合に特に好適な別法では、スカベンジャー樹脂を容器に収容し、これにフッ化物溶液を流してもよい。フッ化物溶液は、乾燥に十分な時間スカベンジャー樹脂に滞留できるように、例えば0.1mL/分〜100mL/分の流速で連続流として又は回分式にスカベンジャー樹脂に流せばよい。
【0029】
スカベンジャー樹脂のこの用途は新規であり、本発明の別の態様では、放射性フッ化物、特に[18F]フッ化物の溶液の水分量を低減する方法であって、当該方法が上記溶液をスカベンジャー樹脂に接触させることを含んでなる方法を提供する。好適には、上記溶液は、アセトニトリル、ジメチルホルムアミド、ジメチルスルホキシド、テトラヒドロフラン、ジオキサン、1,2−ジメトキシエタン、スルホラン及びN−メチルピロリジノンのような非プロトン性有機溶媒中のフッ化物を含む。
【0030】
反応は液相中で実施してもよいし、或いは非フッ素化糖誘導体を固体担体に結合して以下の式(I)の樹脂−リンカー−ベクター(RLV)を形成してもよい。
【0031】
固体担体−リンカー−X−保護非フッ素化糖誘導体 (I)
式中、固体担体は適当な担体であり、
保護非フッ素化糖誘導体は上記で定義した通りであり、
Xは保護非フッ素化糖誘導体の特定の部位での求核置換を促進する基、例えば−SO2O−であり、
リンカーは、反応性を最大に高めるため反応性部位を固体担体構造から十分に離隔させるのに役立つ適当な有機基であり、例えば0〜4個のアリール基(例えばフェニル)及び/又はC1〜C6アルキル又はハロアルキル(特にフルオロアルキル)鎖並びに適宜追加される1〜4個のアミド又はスルホンアミド基のような官能基である。
【0032】
RLV系は国際公開第03/002157号に詳細に記載されており、好適なリンカーの詳細についても記載されている。
【0033】
式(I)のRLVをフッ化物溶液と接触させると、固体担体から糖が置換され、保護フッ素化糖誘導体を与える。
【0034】
好適な固体担体は国際公開第03/002157号にも記載されており、ポリスチレン(ポリエチレングリコールなどでブロックグラフト化したものでもよい)、ポリアクリルアミド又はポリプロピレンのようなポリマー或いはガラス又はケイ素をかかるポリマーで被覆したものが挙げられる。或いは、例えば国際公開第03/002157号に記載されるように樹脂を用いてもよい。固体担体は、ビーズやピンのような小さな離散粒子の形態でも、或いはカートリッジの内面や微細加工容器に設けられたコーティングの形態であってもよい。本発明の方法を固体担体で実施することによって、追加の分離段階を必要とせずに純粋な形の生成物を得ることができる。これはフッ素化が放射性フッ素化のときに特に有利である。プロセスにおける時間の節約は非補正放射化学収率の向上をもたらすからである。
【0035】
反応は通常5℃〜180℃、特に75℃〜125℃の温度で実施される。
【0036】
本発明の方法は自動化合成の一部として実施できる。これは、反応を溶液中で行おうと非フッ素化糖を固相に結合させようと当てはまる。
【0037】
非フッ素化糖誘導体と反応させるフッ化物はイオン性化合物であってもよいし、適当な対イオンと共に供給してもよい。ただし、対イオンが反応溶媒中でフッ化物の溶解性を維持するのに十分な溶解性をもつことが重要である。従って、好適な対イオンとしては、ルビジウム又はセシウムのような大きなソフト金属イオン或いはテトラアルキルアンモニウム及びテトラアルキルホスホニウムのような非金属イオンが挙げられる。カリウムイオンも対イオンとして用いることができ、この場合、フッ化物の反応性を高めるため、4,7,13,16,21,24−ヘキサオキサ−1,10−ジアザビシクロ−[8,8,8]−ヘキサコサン(Kryptofix(商標)2.2.2という商標で市販)のような相間移動触媒を添加してカリウム塩を有機溶媒中に溶解させてもよい。
【0038】
本発明の方法は放射性フッ素化誘導体、特に[18F]標識誘導体の製造に特に適しており、フッ化物は[18F]フッ素イオンを含んでいてもよい。
【0039】
上記で簡単に説明した通り、[18F]フッ素イオンは[18O]水ターゲットの照射によって調製することができ、これを本発明の方法における最初の工程としてもよい。
【0040】
本発明の方法は[18F]−pFDGのような放射性フッ素化糖誘導体の製造に特に有用であり、[18F]−pFDGを脱保護すれば周知のPETトレーサーである[18F]−FDGのような化合物が得られる。脱保護はこの方法の追加工程とし得る。生成物のフッ素化糖の保護基がアセチル誘導体のようなエステルである場合、脱保護は酸又は塩基加水分解によって実施し得る。
【0041】
その他の追加工程としては、溶液からの過剰の[18F]フッ化物の除去及び有機溶媒の除去が挙げられる。過剰の[18F]フッ化物は、例えばイオン交換クロマトグラフィー又は固相吸着剤のような常法で除去できる。好適なイオン交換樹脂としてはBIO−RAD AG 1−X8(商標)及びWaters QMA(商標)が挙げられ、好適な固相吸着剤としてはアルミナが挙げられる。
【0042】
有機溶媒は、真空昇温下での蒸発或いは溶液に窒素又はアルゴンのような不活性ガス気流を通すことによっても除去できる。
【0043】
これら工程の最終生成物である[18F]トレーサー化合物は、患者への投与のために製剤化してもよく、例えば水溶液として、[18F]標識トレーサーを無菌等張食塩水(エタノールのような適当な有機溶媒を10%以下含んでもよい。)或いはリン酸緩衝液のような適当な緩衝溶液中に溶解することによって調製してもよい。その他アスコルビン酸のような添加剤を用いてもよく、アスコルビン酸は放射線分解を低減する。
【0044】
上述の通り本発明の方法で調製できる特に好ましい化合物は[18F]−pFDGであり、そこで、本発明の第二の態様では、テトラアセチルマンノーストリフレートと[18F]フッ化物との反応を含む[18F]−pFDGの製造方法であって、上記フッ化物を1000ppm超50000ppm未満の量の水を含有する溶媒中に溶解されていることを特徴とする方法を提供する。本発明のこの態様の一実施形態では、テトラアセチルマンノーストリフレート(1当量)を、Kryptofix(商標)2.2.2(0.9〜1.1モル当量、好適には0.98〜0.99モル当量)、炭酸カリウム(0.4〜0.6モル当量、好適には0.50〜0.60モル当量)の存在下、1000ppm超50000ppm未満の量の水を含有するアセトニトリル中で[18F]フッ化物と反応させる。
【0045】
本発明の好ましい特徴は第一の態様で説明した通りである。特に、この方法は[18O]−水ターゲットの照射によって[18F]フッ化物を生成させる最初の工程と、酸又はアルカリ加水分解によって[18F]−pFDGを[18F]−FDGへと変換する追加工程を含んでもよい。
【0046】
以下の実施例及び図面を参照して本発明をさらに詳しく説明する。
【実施例】
【0047】
実施例1−糖の18F標識に対する水分量変化の影響
本例では、18F−での3通りの糖標識法を用いて、反応混合物の水分量変化の影響を評価した。
【0048】
a)樹脂−リンカーベクター(RLV)標識
18F−をTracerlab MX(商標)に導入し、2−[18F]フルオロ−2−デオキシグルコースの製造で用いられる標準的乾燥工程を用いて乾燥した。Kryptofix(商標)2.2.2/炭酸カリウムを用いてフッ化物をアセトニトリル中に溶解させた。乾燥工程を終え、フッ化物をアセトニトリル中に溶解した後、アセトニトリル中の乾燥18F−試料を水分量分析用に採取し、カールフィッシャー滴定計で測定した。乾燥プロセスで通常得られるよりも水分量を高くするため、必要に応じて、追加の水を導入した。
【0049】
1mLのHi Trap(登録商標)カートリッジに、0.003mmol/gの置換度の固相結合保護マンノース前駆体約370mgを充填した。カートリッジの一端はループを介してシリンジドライバーに接続した。カートリッジのもう一方の端はモレキュラーシーブベントを備えたN2充填バイアルに接続した。ホットエアガンを用いて、カートリッジを80℃のカートリッジ外部温度に加熱した。
【0050】
この系に次いで乾燥アセトニトリル6×0.5mLを注入して不純物及び水(例えば不完全な乾燥のため自然に樹脂に存在する)を洗い流し、アセトニトリルは廃棄した。この装置に、乾燥18F−溶液(450μL)を収容したオートサンプラーバイアルを装着した。次いで、シリンジドライバーで乾燥フッ化物を流速180μL/分で5サイクル前後に移動させた。乾燥フッ化物が固相マンノース前駆体と反応して保護[18F]−2−デオキシグルコース誘導体が遊離した(これを脱保護すると2−[18F]−2−デオキシグルコースが得られる)。
【0051】
次にオートサンプラーバイアルから5μL試料を薄層クロマトグラフィー(TLC)分析用に採取し、シリカゲル60 F254プレートにスポットし、90/10比に調製したアセトニトリル/水溶媒で展開した。放射化学純度はPerkin Elmer Instant Imagerで求めた。
【0052】
b)テトラアセチルマンノーストリフレート標識
HPLCグレードのH2O600μLに溶解した32mgのK2CO3に、2.5mLアセトニトリルに溶解したKryptofix(商標)2.2.2を加えた溶液を調製した。その0.6mLを、18O濃縮水中の約40MBqの18F−と共にガラス状炭素反応器に加えた。ヒータコントローラを95℃に設定し、反応容器を合計35分間加熱してフッ化物を乾燥した。水とアセトニトリルを窒素気流中で蒸発させた。
【0053】
フッ化物からの水の共沸除去を促すため、1mLのアセトニトリルを2分間隔で計3回注入したが、最初の添加は乾燥プロセスで20分であった。35分後にヒータを切り、反応容器外からの圧縮気流で反応容器を約45℃に冷却した。
【0054】
次いでCH3CN2.0mL中のマンノーストリフレート25mgを乾燥フッ化物に添加し、混合した。ヒータコントローラを85℃に設定した。設定温度に達して2分後にTLC分析用試料を採取した。ヒータを切り、反応容器外部からの圧縮気流で反応容器を約45℃に冷却した。
【0055】
TLC分析用試料をシリカゲルにスポットし、90/10比に調製したアセトニトリル/水溶媒で展開した。放射化学純度はPerkin Elmer Instant Imagerで求めた。次に反応容器の蓋を取り、50μL試料を採取してカールフィッシャー滴定計で水分量を分析した。
【0056】
c)自動化標識
内蔵加熱反応容器及び使い捨てポリプロピレンカセットと共に25個の三方弁を備えたプロトタイプ自動化合成プラットフォームで標識を行った。カセット流路は中間体又は最終生成物のSPE精製も可能にする。
【0057】
フッ化物はまずQMAカートリッジでトラップし、20mgのKryptofix(商標)2.2.2と4.1mgのK2CO3と320μLのCH3CNと80μLのH2Oの溶液で溶出した。これを次いで窒素気流中105℃/120℃で約6分間乾燥し、アセトニトリル中の約20mg/mLテトラアセチルマンノーストリフレート1.5mLに再溶解した。
【0058】
標識反応は標識温度105又は125℃、反応時間90又は270秒で行った。標識後、2[18F]−2−デオキシグルコースをTLCで分析した。TLCプレートはシリカゲル60 F254であり、95%アセトニトリル、5%水を展開溶媒として用いた。放射化学純度はPerkin Elmer Instant Imagerで求めた。
【0059】
実施例1の3通りの実験の結果を図1に示すが、この図から、反応混合物の水分量が1000ppm未満のときは生成物の放射化学純度が比較的低かったのに対して、水分量が1000〜5000ppmのときは生成物の放射化学純度が大幅に向上したことが分かる。このプロットは溶媒中の最適水分量が約2000〜7000ppmであったことを示している。
【0060】
実施例2−標識プロセスでの[18F]−pFDG及びpGlucoseの生成の相関
RLVの18−フッ化物標識を、Kryptofix(商標)2.2.2、炭酸カリウム及び様々な量の水の存在下、アセトニトリル中で実施した。標識後、得られた混合物を逆相HPLCに付して、Phenomenex Luna 5μm C18カラム(4.6mm×150mm)を用いて10分間流速1mL/分で90%溶媒A/10%溶媒Bから5%溶媒A/95%溶媒Bへの勾配(溶媒A=0.1%トリフルオロ酢酸水溶液、溶媒B=0.1%トリフルオロ酢酸アセトニトリル溶液)を流した。保持時間3分の保護グルコースのピークと6.6分の保護FDGのピーク(主に[19F]−FDGの存在に起因し、その量は[18F]−FDGに比例する)の積分値を求め、それらの相関関係を求めた。
【0061】
反応混合物中に大量の水が存在すると、トリフレート基での求核置換の結果、([18F]−pFDGではなく)保護グルコースが大量に生成すると一般に考えられている。したがって、生成混合物中の保護グルコース誘導体濃度に対する[18F]−pFDG濃度のグラフをプロットしたグラフの傾きは負となり、水分量が高いほど保護グルコースレベルは高く[18F]−pFDGレベルは低くなると予想されていた。
【0062】
しかし、樹脂に結合させたテトラアセチルマンノーストリフレートの標識実験から、HPLCの2つのピークの間に良好な正の相関関係があることが判明した(図2参照)。これは高濃度の水が存在するとこれら2種類の生成物の形成が抑制されることを示している。
【0063】
実施例3:1,3,4,6−テトラ−O−アセチル−2−フルオロ−β−D−マンノピラノースの自動化合成
標識化の最初に放射性反応混合物をサンプリングするのには問題があった。そのため、水分量を標識反応の最後にRCP(ITLCで測定)と共に測定した。これから、以下に述べる通り水封鎖を計算に入れることよって標識開始時の水分量を算出した。
【0064】
放射性標識実験
1,3,4,6−テトラ−O−アセチル−2−フルオロ−β−D−マンノピラノースの合成は、使い捨てカセットが取り付けられるように設計された自動合成装置で実施した。このカセットは、固相抽出カートリッジ用のスペースとシリンジと共に各種試薬の収容用バイアルを備えた25弁の使い捨てカセットからなる。
【0065】
合成シーケンスを行って2mL水中の約50MBqの18−フッ化物をWaters Access PlusQMAカートリッジ(その炭酸塩の形態)でトラップし、次にアセトニトリル/水中のkryptofix及び炭酸塩の溶液(20.3mgのkryptofix 222、4.3mgの炭酸カリウム、320mLのアセトニトリル、80mLの水)でカートリッジを溶出して加熱反応器に回収した。これを乾燥窒素気流中で加熱して乾燥し、次に所定の水分量のアセトニトリル中マンノーストリフレート溶液を反応器に添加した。
【0066】
外部ヒータ温度125℃でさらに80秒間反応せしめた後、0.6mLを回収して廃棄し(ラインから残留水を除去するため)、残余を生成物バイアルに移した。50μL溶液を用いたカールフィッシャー滴定で生成物バイアルの水分量を決定し、インスタント薄層クロマトグラフィー(ITLC)でRCPを測定した。TLCはシリカTLCプレートで実施し、スポットを95%アセトニトリル、5%水で溶出し、TLCを用いて18−フッ化物及び1,3,4,6−テトラ−O−アセチル−2−フルオロβ−D−マンノピラノース(いずれの場合も2成分のみ)の相対比を計測した。
【0067】
水封鎖測定
3回のコールド(非放射性)実験を行って、マンノーストリフレートとの反応でどの程度の水がなくなったかを調べるために所定体積の反応液を標識反応の前後にサンプリングした。これによって、測定した水分量に水封鎖を計算に入れることができた。
【0068】
上記放射性標識実験と同様の合成シーケンスを行ってWaters Access PlusQMAカートリッジ(その炭酸塩の形態)に2mLの水を流し、次にアセトニトリル/水中のkryptofixと炭酸塩の溶液(20.3mgのkryptofix 222、4.3mgの炭酸カリウム、320mLのアセトニトリル、80mLの水)でカートリッジを溶出して加熱反応器に回収した。これを乾燥窒素気流中で加熱して乾燥し、次に所定の水分量のアセトニトリル中マンノーストリフレート溶液を反応器に添加した。
【0069】
マンノーストリフレート溶液を反応器に添加してすぐに0.6mLを回収して生成物バイアル中に注入した。標識反応を外部ヒータ温度125℃でさらに80秒間進行させ、残りの溶液を別の生成物バイアルに移した。50μLの溶液を用いたカールフィッシャー滴定器で各バイアルの水分量を測定した。
【0070】
水封鎖実験の結果を表1に示す。表1はアセトニトリル溶媒中の水の存在量を示す。
【0071】
【表1】
低レベル及び中間レベルの水分量では、マンノーストリフレートとの反応による水の有意な封鎖は認められなかった。しかし、高レベルの水分量では水分量が約7%低下した。
【0072】
放射標識結果
各放射標識反応の水分量を測定し、マンノーストリフレートによる水の封鎖を計算に入れて標識前の水分量を補正した。各水分量で得られたRCPを表2に示すとともに、図3に図示する。
【0073】
【表2】
これらの結果は3000〜6000ppmが好ましい水分量であることを裏付ける。RCP73.6%での疑わしい結果を無視すれば、非常に湿分の多い反応条件であったとしても、反応は約85%RCPに近づく。
【図面の簡単な説明】
【0074】
【図1】[18F]−pFDG生成物の放射化学純度と溶媒の水分量との関係を示すプロット。
【図2】樹脂−リンカー−ベクターでの標識プロセスにおける[18F]−pFDGとpGlucoseの生成の関係を示すグラフ。
【図3】自動化合成における[18F]−pFDG生成物の放射化学純度と溶媒の水分量との関係を示すプロット。
【特許請求の範囲】
【請求項1】
非フッ素化糖誘導体とフッ化物との反応を含んでなるフッ素化糖誘導体の製造方法であって、上記反応を1000ppm超50000ppm未満の量の水を含有する溶媒中で実施することを特徴とする方法。
【請求項2】
前記フッ素化糖誘導体及び非フッ素化糖誘導体が単糖類である、請求項1記載の方法。
【請求項3】
前記フッ素化糖誘導体及び非フッ素化糖誘導体が保護されている、請求項1又は請求項2記載の方法。
【請求項4】
前記フッ素化糖誘導体及び非フッ素化糖誘導体がアセチル基で保護されている、請求項3記載の方法。
【請求項5】
保護フッ素化グルコース誘導体の製造のための請求項1乃至請求項4のいずれか1項記載の方法であって、前記非フッ素化糖誘導体が保護マンノース誘導体である方法。
【請求項6】
1,3,4,6−テトラ−O−アセチル−2−トリフルオロメタンスルホニル−β−D−マンノピラノース(すなわちテトラアセチルマンノーストリフレート)から2−フルオロ−1,3,4,6−テトラ−O−アセチル−D−グルコース(すなわちテトラアセチルフルオログルコース又はpFDG)を製造するための請求項5記載の方法。
【請求項7】
前記溶媒がアセトニトリル、ジメチルホルムアミド、ジメチルスルホキシド、テトラヒドロフラン、ジオキサン、1,2−ジメトキシエタン、スルホラン及びN−メチルピロリジノンから選択される、請求項1乃至請求項6のいずれか1項記載の方法。
【請求項8】
前記溶媒がアセトニトリルである、請求項7記載の方法。
【請求項9】
前記溶媒の水分量が約1000〜15000ppmである、請求項1乃至請求項8のいずれか1項記載の方法。
【請求項10】
前記溶媒の水分量が約2000〜7000ppmである、請求項9記載の方法。
【請求項11】
溶媒の水分量が3000ppm〜6000ppmである、請求項9記載の方法。
【請求項12】
液相中で実施される、請求項1乃至請求項11のいずれか1項記載の方法。
【請求項13】
自動化された請求項1乃至請求項12のいずれか1項記載の方法。
【請求項14】
前記フッ化物がカリウム対イオンとのイオン性フッ化物であって、4,7,13,16,21,24−ヘキサオキサ−1,10−ジアザビシクロ−[8,8,8]−ヘキサコサンのような相間移動触媒が前記フッ化物に添加される、請求項1乃至請求項13のいずれか1項記載の方法。
【請求項15】
放射性標識糖誘導体の製造のための1乃至請求項14のいずれか1項記載の方法。
【請求項16】
放射性標識糖誘導体が[18F]標識糖誘導体である、請求項15記載の方法。
【請求項17】
[18F]−pFDGの製造のための請求項1乃至請求項16のいずれか1項記載の方法であって、当該方法がテトラアセチルマンノーストリフレートと[18F]フッ化物との反応を含む方法。
【請求項18】
以下の(i)〜(iv)の1以上の追加工程を任意の順序でさらに含む、請求項1乃至請求項17のいずれか1項記載の方法。
(i)溶液からの過剰フッ化物の除去、
(ii)脱保護フッ化物を得るための保護フッ素化糖誘導体の脱保護、
(iii)有機溶媒の除去、及び
(iv)水溶液中の脱保護フッ化物糖誘導体の製剤化。
【請求項19】
放射性フッ化物、特に[18F]フッ化物の溶液の水分量を低減する方法であって、当該方法が上記溶液をスカベンジャー樹脂に接触させることを含んでなる方法。
【請求項20】
前記溶液がアセトニトリル、ジメチルホルムアミド、ジメチルスルホキシド、テトラヒドロフラン、ジオキサン、1,2−ジメトキシエタン、スルホラン及びN−メチルピロリジノンから選択される非プロトン性有機溶媒中のフッ化物を含む、請求項19記載の方法。
【請求項21】
前記溶液がアセトニトリル中のフッ化物を含む、請求項19又は請求項20記載の方法。
【請求項1】
非フッ素化糖誘導体とフッ化物との反応を含んでなるフッ素化糖誘導体の製造方法であって、上記反応を1000ppm超50000ppm未満の量の水を含有する溶媒中で実施することを特徴とする方法。
【請求項2】
前記フッ素化糖誘導体及び非フッ素化糖誘導体が単糖類である、請求項1記載の方法。
【請求項3】
前記フッ素化糖誘導体及び非フッ素化糖誘導体が保護されている、請求項1又は請求項2記載の方法。
【請求項4】
前記フッ素化糖誘導体及び非フッ素化糖誘導体がアセチル基で保護されている、請求項3記載の方法。
【請求項5】
保護フッ素化グルコース誘導体の製造のための請求項1乃至請求項4のいずれか1項記載の方法であって、前記非フッ素化糖誘導体が保護マンノース誘導体である方法。
【請求項6】
1,3,4,6−テトラ−O−アセチル−2−トリフルオロメタンスルホニル−β−D−マンノピラノース(すなわちテトラアセチルマンノーストリフレート)から2−フルオロ−1,3,4,6−テトラ−O−アセチル−D−グルコース(すなわちテトラアセチルフルオログルコース又はpFDG)を製造するための請求項5記載の方法。
【請求項7】
前記溶媒がアセトニトリル、ジメチルホルムアミド、ジメチルスルホキシド、テトラヒドロフラン、ジオキサン、1,2−ジメトキシエタン、スルホラン及びN−メチルピロリジノンから選択される、請求項1乃至請求項6のいずれか1項記載の方法。
【請求項8】
前記溶媒がアセトニトリルである、請求項7記載の方法。
【請求項9】
前記溶媒の水分量が約1000〜15000ppmである、請求項1乃至請求項8のいずれか1項記載の方法。
【請求項10】
前記溶媒の水分量が約2000〜7000ppmである、請求項9記載の方法。
【請求項11】
溶媒の水分量が3000ppm〜6000ppmである、請求項9記載の方法。
【請求項12】
液相中で実施される、請求項1乃至請求項11のいずれか1項記載の方法。
【請求項13】
自動化された請求項1乃至請求項12のいずれか1項記載の方法。
【請求項14】
前記フッ化物がカリウム対イオンとのイオン性フッ化物であって、4,7,13,16,21,24−ヘキサオキサ−1,10−ジアザビシクロ−[8,8,8]−ヘキサコサンのような相間移動触媒が前記フッ化物に添加される、請求項1乃至請求項13のいずれか1項記載の方法。
【請求項15】
放射性標識糖誘導体の製造のための1乃至請求項14のいずれか1項記載の方法。
【請求項16】
放射性標識糖誘導体が[18F]標識糖誘導体である、請求項15記載の方法。
【請求項17】
[18F]−pFDGの製造のための請求項1乃至請求項16のいずれか1項記載の方法であって、当該方法がテトラアセチルマンノーストリフレートと[18F]フッ化物との反応を含む方法。
【請求項18】
以下の(i)〜(iv)の1以上の追加工程を任意の順序でさらに含む、請求項1乃至請求項17のいずれか1項記載の方法。
(i)溶液からの過剰フッ化物の除去、
(ii)脱保護フッ化物を得るための保護フッ素化糖誘導体の脱保護、
(iii)有機溶媒の除去、及び
(iv)水溶液中の脱保護フッ化物糖誘導体の製剤化。
【請求項19】
放射性フッ化物、特に[18F]フッ化物の溶液の水分量を低減する方法であって、当該方法が上記溶液をスカベンジャー樹脂に接触させることを含んでなる方法。
【請求項20】
前記溶液がアセトニトリル、ジメチルホルムアミド、ジメチルスルホキシド、テトラヒドロフラン、ジオキサン、1,2−ジメトキシエタン、スルホラン及びN−メチルピロリジノンから選択される非プロトン性有機溶媒中のフッ化物を含む、請求項19記載の方法。
【請求項21】
前記溶液がアセトニトリル中のフッ化物を含む、請求項19又は請求項20記載の方法。
【図1】

【図2】
【図3】
【図2】
【図3】
【公表番号】特表2008−520636(P2008−520636A)
【公表日】平成20年6月19日(2008.6.19)
【国際特許分類】
【出願番号】特願2007−542100(P2007−542100)
【出願日】平成17年11月18日(2005.11.18)
【国際出願番号】PCT/GB2005/004451
【国際公開番号】WO2006/054098
【国際公開日】平成18年5月26日(2006.5.26)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
【公表日】平成20年6月19日(2008.6.19)
【国際特許分類】
【出願日】平成17年11月18日(2005.11.18)
【国際出願番号】PCT/GB2005/004451
【国際公開番号】WO2006/054098
【国際公開日】平成18年5月26日(2006.5.26)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
[ Back to top ]