
フマル酸エステルを含む制御放出医薬組成物
本発明は、活性成分としてフマル酸エステルを含む制御放出医薬組成物に関する。該組成物は、例えば、乾癬または他の過剰増殖性、炎症性、または自己免疫性疾患の治療における使用に適し、経口投与したときに消化管内で活性成分が局所的に高濃度となるのを避け、消化管関連副作用を減少させることができるように制御されるようにフマル酸エステルを放出するよう設計される。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は活性成分としてフマル酸エステルを含む制御放出医薬組成物に関する。該組成物は、例えば乾癬または他の過剰増殖性、炎症性、または自己免疫障害の治療に用いるのに適しており、経口投与したときに消化管内で活性物質が局所的に高濃度になるのを避けることができ、消化管関連副作用を低下させることができるように制御的にフマル酸エステルを放出するよう設計される。
【背景技術】
【0002】
フマル酸エステル、すなわち、ジメチルフマレートとエチル水素フマレートの合剤は、多年にわたり乾癬の治療に用いられてきた。該合剤は、商標Fumaderm(登録商標)で市販されている。該合剤は経口使用を意図した錠剤の形であり、2つの異なる用量強度(Fumaderm(登録商標)InitialおよびFumaderm(登録商標))が利用可能である。
【0003】
2つの強度は、増量するFumaderm(登録商標)Initialで出発し、次いで例えば3週間治療後にFumaderm(登録商標)に切り替える個別的な用法に適用することを意図している。Fumaderm(登録商標)initialおよびFumaderm(登録商標)は共に腸溶性錠剤である。
【0004】
別の市販組成物は、120mgのジメチルフマレートおよび95mgのカルシウムモノエチルフマレートを含むFumaraat 120(登録商標)(TioFarma、Oud-Beijerland、Netherlands)である。最近の刊行物(Litjens et al. Br. J. Clin. Pharmacol. 2004、vol. 58:4、pp. 429-432)において、Fumaraat 120(登録商標)の薬物動態プロフィールが健康対象について記載されている。その結果は、単経口用量のFumaraat 120(登録商標)は血清モノメチルフマレート濃度の上昇をもたらし、ジメチルフマレートとフマル酸の濃度はごくわずかであることを示す。結果は、ジメチルフマレートが著者によれば酸環境下ではなくアルカリ環境下で急速にモノメチルフマレートに加水分解されることを示す。該組成物は腸溶性であるため、フマレートの取り込みは、主に小腸で生じ、ジメチルフマレートは取り込み前にアルカリ環境下によりモノエステルに加水分解されるか、または循環中でエステラーゼにより急速に変換されるかもしれないと考えられる。さらに、この研究では、tmaxおよびCmaxは食物の影響を受け、すなわち、同時に食物摂取するとtmaxは延長し(絶食状態の平均は182分間であるが、摂食状態の平均は361分間である)(遅延時間は絶食時で90分間、摂食時で300分間である)、Cmaxは低下する(絶食時:0.84mg/l、摂食時:0.48mg/l)。2錠のFumaderm(登録商標)P forteを用いる健康対象における別の研究(Reddingius W. G. Bioanalysis and Pharmacokinetics of Fumarates in Humans. Dissertation ETH Zurich No. 12199(1997))は、Cmax値(モノエチル-またはモノメチルフマレートとして測定)が1.0〜2.4μg/mlの範囲、tmaxが4.8〜6.0時間の範囲であることを示した。
【0005】
US 6,277,882およびUS 6,355,676は、乾癬、乾癬性関節炎、神経皮膚炎、およびクローン限局性腸炎(enteritis regionalis Crohn)を治療するためのマイクロ錠剤を製造するためのアルカリ水素フマレートの使用とある種のフマル酸モノアルカリエステル塩の使用をそれぞれ開示する。US6,509,376は、マイクロ錠剤またはペレットの形の移植医学および自己免疫疾患の治療に用いる医薬製剤を製造するためのある種のジアルキルフマレートの使用を開示する。US 4,959,389は、フマル酸モノアルキルエステルの種々の塩を単独またはジアルキルフマレートと組み合わせて含む組成物を開示している。GB 1,153,927は、ジメチルマレイン酸無水物および/またはジメチルマレイン酸および/またはジメチルフマル酸化合物を含む医薬組成物に関する。BMC Dermatology、vol. 2、no.5、2002中のケースレポート「Treatment of disseminated granuloma with fumaric acid esters」は、フマル酸エステルを用いる治療に関する。
【0006】
しかしながら、例えばFumaderm(登録商標)のようなフマレートによる治療は、例えば、膨満感、下痢、急激な上腹部痛、鼓腸、および悪心のような消化管の副作用を生じることが多い。
【0007】
したがって、経口投与したときに消化管関連副作用の減少による治療の改善をもたらす1またはそれ以上の治療的または予防的に有効なフマル酸エステルを含む組成物を開発する必要がある。
【0008】
さらに、現在市販されている製品は、該エステルの一つ(すなわち、フマル酸のモノエチルエステルであるエチル水素フマレート)が3つの異なる塩の形(すなわち、カルシウム、マグネシウム、および亜鉛塩)で存在する2つの異なるエステルの組み合わせを含む。各個々の形がそれ自身治療的プロフィールを持つかもしれないが、可能であれば適切な治療効果を得るためにはるかに単純な生成物であることが好都合であろう。
【0009】
本発明者らは、制御された方法、すなわち市販の製品に比べて延長され、遅くなり、および/または遅延するように、活性物質が送達されるように設計された医薬組成物の投与により改良された治療方法が得られると予期する。さらに、異なるフマル酸エステルの組み合わせを用いる代わりにジメチルフマル酸のような1つのフマル酸エステルのみを用いることにより適切な治療反応を達成することができると予期される。
(図の簡単な説明)
【0010】
図1は、実施例5に記載のごとく製造したカプセル剤のin vitro溶出プロフィールの例を示す。
【0011】
図2は、実施例16に記載のごとく製造した錠剤試料(腸溶コーティングを施す前)のin vitro溶出プロフィールの例を示す。
【0012】
図3は、実施例17に記載のごとく製造した錠剤試料(腸溶コーティングを施す前)のin vitro溶出プロフィールの例を示す。
【発明の開示】
【発明が解決しようとする課題】
【0013】
したがって、本発明は、経口投与すると等価用量のFumaderm(登録商標)錠の経口投与後と比べてGI関連副作用の低下が生じる、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む医薬組成物に関する。
【0014】
上記のように、本発明者らは、消化管関連副作用を減少させる適切な方法は制御放出組成物の形で活性成分を投与することによると予期する。
【0015】
したがって、さらなる局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステルまたはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、試験の最初の2時間は0.1N塩酸を溶出溶媒に、次いで0.05Mリン酸緩衝液pH6.5を溶出溶媒に用いるin vitro溶出試験を行うと、該フマル酸エステルの該放出が以下のごとくである該医薬組成物に関する:
試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約70%w/wが放出される、および/または
試験開始後の最初の4時間以内に該フマル酸エステルの総量のほぼ約92%w/wが放出される、および/または
試験開始後の最初の5時間以内に該フマル酸エステルの総量のほぼ約94%w/wが放出される、および/または
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約95%w/wが放出される、および/または
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約98%w/wが放出される、および/または
試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約99%w/wが放出される、および/または
試験開始後の最初の12時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約99%w/wが放出される。
【0016】
本発明の文脈において、制御放出組成物は、同等の条件(例えば、in vivo試験では、用量等価、標準食有りまたは無しなど、またはin vitro試験では、溶出試験装置および用いる溶出溶媒の組成、容量、および温度、回転速度などを含む作業条件)下で試験したときに市販製品のFumaderm(登録商標)の放出に比べて延長され、遅くなり、および/または遅延するようにフマル酸エステルを放出するように設計された組成物である。
【0017】
in vivoの放出は、予め決定した時間で血漿濃度を測定し、目的とするフマル酸エステルまたは適切であればその代謝物の血漿濃度対時間プロフィールを得ることにより試験してよい(例えば、ジメチルフマレートの場合は、活性物質はメチル水素フマレート、すなわちフマル酸のモノメチルエステルであると予想される)。さらに、代謝はすでに消化管内で、または消化管粘膜の通過時に、または肝循環による初回通過時に生じると予期される。したがって、ジメチルフマレートを投与したときに血漿中で探す関連成分はフマル酸のジメチルエステルではなくモノメチルエステルであってよい。
【0018】
in vivoの活性物質の放出を測定しまたは指標を得るのに他の試験を用いてもよい。すなわち、動物(例えばマウス、ラット、イヌなど)をモデルとして用いてよい。動物に観察下で組成物を投与し、一定時間後動物を屠殺し、活性成分(または適切ならばその代謝物)の含有量を血漿や特定臓器について測定するか、または腸内容から抽出する。
【0019】
別の試験は、動物の腸の特定部分を用いることを含む。該部分を該部分により分離した2つのコンパートメント(ドナーとレシーバー)を含む適切な溶出装置中に置き、観察下で該組成物を1つのコンパートメント(ドナーコンパートメント)中の適切な媒質中に置く。該組成物は活性成分を放出し、次いで腸部分を横切って輸送される。したがって、適切な時間間隔で、活性物質(または適切であれば代謝物)の濃度をレシーバーコンパートメントで測定する。
【0020】
当業者は上記方法を特定の組成物に適用することができるであろう。
【0021】
in vitro方法に関してはよく確立された方法、具体的には例えばUnited States Pharmacopeia(USP)またはEuropean Pharmacopoeiaのような公式モノグラフに記載の方法が利用可能である。当業者は、in vitro試験を行うための具体的条件を選ぶ方法およびどの方法を選ぶかを理解しているであろう。例えば、USPは、in vitro試験を37±1.0、例えば37±0.5℃で行うことを規定している。適切な溶出試験は、例えばカプセルについて実施例29に記載されており、ここでは、溶出プロフィールはUnited States Pharmacopoeiaに記載のごとく試験の最初の2時間は0.1N塩酸を溶出溶媒に用い、次いで残りの試験時間は0.05Mリン酸緩衝液pH6.5を溶出溶媒に用い、100rpmの回転パスケットを用いて37℃で測定し、また例えば錠剤については実施例30に記載されており、ここでは、溶出プロフィールはUnited States Pharmacopoeiaに記載のごとく試験の最初の2時間は0.1N塩酸を溶出溶媒に用い、次いで残りの試験時間は0.05Mリン酸緩衝液pH6.5を溶出溶媒に用い、100rpmのパドル溶出装置を用いて37℃で測定する。
【0022】
上記のごとく、活性物質のin vivo放出は、市販のFumaderm(登録商標)組成物に比べて延長され、遅くなり、および/または遅延する。本発明の文脈に置いて、用語「延長された」は、活性物質がFumaderm(登録商標)より長い期間、例えばFumaderm(登録商標)より少なくとも1.2倍、例えば少なくとも1.5倍、少なくとも2倍、少なくとも3倍、少なくとも4倍、または少なくとも5倍長い期間放出されることを示すことを意図する。すなわち、例えば100%のジメチルフマレートがFumaderm(登録商標)錠から適切な試験の開始後3時間で放出されるならば、本発明組成物中の100%のジメチルフマレートが適切な試験の開始後少なくとも3.6時間で放出される。
【0023】
本発明の文脈において、用語「遅延する」は、活性成分の放出がFumaderm(登録商標)に比べて遅い時点で始まるか(例えば30分間またはそれ以上遅く、例えば45分間またはそれ以上遅く、1時間またはそれ以上遅く、1.5時間またはそれ以上遅く)、または最初の2時間の初期放出がFumaderm(登録商標)に比べて遙かに少ない(すなわち、Fumaderm(登録商標)の80%w/w以下、例えば70%w/w以下、60%w/w以下、または50%以下)ことを示すことを意図する。
【0024】
本発明において用いている消化管(GI)の副作用には、限定されるものではないが、下痢、腹痛(stomach ache、stomach pain、abdominal pain)、痙攣性腹痛、悪心、鼓腸(flatulence)、しぶり(tenesmus)、鼓腸(meteorism)、大便の頻度の増加、膨満感、および急激な上腹部痛が含まれよう。
【0025】
本発明の文脈において、GI関連副作用の減少は、本発明の組成物を投与した後に観察されるGI副作用の該治療患者ポピュレーションにおける重症度および/または発生率がFumaderm(登録商標)の場合と比べて低下することを意味することを意図する。すなわち、この定義によればGI関連副作用の減少は、上記のあらゆるGI副作用の発生率の実質的な減少、例えば少なくとも10%の減少、またはより好ましくは発生率の少なくとも20%の減少、またはさらにより好ましくは発生率の30%以上の減少と理解されよう。GI関連副作用の減少は、上記のあらゆるGI副作用の重症度の実質的な減少、例えば下痢、腹痛(stomach ache、stomach pain、abdominal pain)、痙攣性腹痛、悪心、鼓腸、しぶり、鼓腸、大便の頻度の増加、膨満感、または急激な上腹部痛の重症度および/または頻度の減少として表現することもできる。上記GI関連副作用の減少は、Fumaderm(登録商標)またはプラセボと本発明の組成物の投与を比較する臨床試験においてモニターすることができる。プラセボ対照臨床試験の場合、プラセボ群と比較した本発明組成物を投与された患者におけるGI関連副作用の発生率をFumaderm(登録商標)とプラセボを比較するヒストリカル臨床試験と比較することができる(例えば、Altmeyer et al、J. Am. Acad. Dermatol. 1994; 全参考文献:Altmeyer PJ et al、Antipsoriatic effect of fumaric acid derivatives. Results of a multicenter double-blind study in 100 patients. J. Am. Acad. Dermatol. 1994; 30:977-81参照)。典型的には、乾癬に罹患した患者がそのような試験に含まれ、典型的には体表面の10%以上が乾癬に罹患しているであろう(重症乾癬)。しかしながら、体表面積の2〜10%が罹患している患者も含むことができる(中等度の乾癬)。患者は乾癬面積重症度指数(PASI)に基づいて選ぶこともできる。典型的には、ある範囲内のPASI、例えば10〜40、または例えば12〜30、または例えば15〜25の患者が含まれる。あらゆるタイプの乾癬に罹患した患者が含まれてよい(慢性プラーク型、播種状滴状(exanthematic guttate)型、膿疱型、乾癬性紅皮症またはパルモプラナー(palmoplanar)型)が、ある場合には、慢性プラーク型の患者のみが含まれる。各治療群(本発明組成物およびFumaderm(登録商標)またはプラセボ)に約15〜20人の患者でほとんどの場合十分であったが、より好ましくは試験の各治療群に約30〜50人の患者が含まれる。全試験期間は1日間〜1週間であり得るが、より好ましくは該試験は8週間〜12週間または16週間まで行われよう。該副作用は、例えば各群で一定の副作用が報告される総回数(何人の患者が副作用を経験したかに関わらず)として評価することができるか、または副作用は一定の副作用を一定回数、例えば試験期間中に少なくとも1回または少なくとも2回、または少なくとも3回経験した患者数で評価することができる。さらに、副作用の重症度をモニターすることができるか、または該試験で副作用とみなすために副作用の一定の重症度を要求することができる。副作用の重症度を評価する好都合な方法は視覚アナログ(VAS)尺度による。
活性物質
【0026】
本発明組成物中の活性物質はあらゆるフマル酸エステルである。本発明のある態様において、該フマル酸エステルは、好ましくはジメチルフマレート、ジエチルフマレート、ジプロピルフマレート、ジブチルフマレート、ジペンチルフマレート、メチル-エチルフマレート、メチル-プロピルフマレート、メチル-ブチルフマレート、メチル-ペンチルフマレート、モノメチルフマレート、モノエチルフマレート、モノプロピルフマレート、モノブチルフマレート、およびモノペンチルフマレートからなる群から選ばれる(その医薬的に許容される塩を含む)。
【0027】
本発明の具体的態様において、該フマル酸エステルは、医薬的に許容される塩の形で存在するフマル酸のモノ-(C1-C5)アルキルエステルである。適切な塩は、例えば金属塩、例えばナトリウム、カリウム、カルシウム、マグネシウム、または亜鉛塩を含むアルキル金属塩およびアルキル土類金属塩から選ばれる塩である。
【0028】
用語(C1-C5)アルキルは、炭素数1〜5(含む)の分岐または非分岐アルキル基、例えばメチル、エチル、1-プロピル、2-プロピル、1-ブチル、2-ブチル、2-メチル-2-プロピル、2-メチル-1-プロピル、およびペンチルを表す。
【0029】
別の態様において、本発明組成物はジメチルフマレートを活性成分として含む。
【0030】
さらなる態様において、本発明組成物は、所望により医薬的に許容される塩、例えばナトリウム、カリウム、カルシウム、マグネシウムおよび/または亜鉛塩などの形のモノメチルフマレートを活性成分として含む。
【0031】
別の態様において、本発明組成物は、活性成分として本質的にジメチルフマレートからなる。
【0032】
別の態様において、本発明組成物は、活性成分としてジメチルフマレートからなる。
【0033】
さらなる態様において、本発明組成物は、活性成分として実質的に、所望により医薬的に許容される塩、例えばナトリウム、カリウム、カルシウム、マグネシウムおよび/または亜鉛塩などの形のモノメチルフマレートからなる。
【0034】
さらなる態様において、本発明組成物は、活性成分として、所望により医薬的に許容される塩、例えばナトリウム、カリウム、カルシウム、マグネシウムおよび/または亜鉛塩などの形のモノメチルフマレートからなる。
【0035】
さらなる態様において、本発明組成物は、重量比約1:10〜約10:1のジメチルフマレートおよびモノメチルフマレート(所望により医薬的に許容される塩、例えばそのナトリウム、カリウム、カルシウム、マグネシウムおよび/または亜鉛塩などの形の)を活性成分として含む。
【0036】
さらなる態様において、本発明組成物は、実質的に、重量比約1:10〜約10:1のジメチルフマレートおよびモノメチルフマレート(所望により医薬的に許容される塩、例えばそのナトリウム、カリウム、カルシウム、マグネシウムおよび/または亜鉛塩などの形の)を活性成分として含む。
【0037】
さらなる態様において、本発明組成物は、活性成分として重量比約1:10〜約10:1のジメチルフマレートおよびモノメチルフマレート(所望により医薬的に許容される塩、例えばそのナトリウム、カリウム、カルシウム、マグネシウムおよび/または亜鉛塩などの形の)からなる。
化粧品および/または医薬組成物
【0038】
本発明が解決する問題は、フマル酸エステルの経口投与による消化管の副作用の出現に関する。該組成物からの活性成分の放出を延長および/または遅延させることにより、消化管の特定部位における活性物質の局所濃度を減少させ(Fumaderm(登録商標)の比べて)、次いで消化管の副作用の減少をもたらすと予想される。したがって、上記のごとくフマル酸エステルの放出を延長および/または遅くすることができる組成物は本発明の範囲内である。
【0039】
そのような組成物は当業者によく知られており、例えば拡散制御ドラッグデリバリーシステム、浸透圧制御ドラッグデリバリーシステム、浸食(erodible)ドラッグデリバリーシステムなどが含まれる。さらに、特定の技術(例えば上記の)に基づいて活性成分の特定の放出特性を有する特定の組成物を提供することができる製薬会社がある。したがって、当業者は、特定の薬剤物質について特定の必要性を感じたら適切な生成物を得る方法が解るであろう。例として、Eurandは、特定の活性物質を含む、組成物からの活性物質の放出について特定の必要条件を有する制御放出医薬組成物を得るための技術的解答を提供するそのような会社の1つである(例えば、http://www.eurand.com参照)。別の会社には、いわゆるSQZgel(登録商標)(http://macromed.com)に関する技術を開発したMacroMed、Inc.がある。SQZgel(登録商標)の作用機序は、外部コーティングと組み合わせたpH感受性ポリマー混合物である。胃の酸性環境において、該ポリマーは水を吸収して膨潤し、薬剤を取り込む。pHがより高い腸に入ると、該ポリマーは徐々に縮むかまたは「絞られ」、「ダイアルドイン(dialed-in)」速度で(持続的活性組成物を放出する)、またはEgalet(登録商標)技術の重要な要素である特定の押出に基づく技術を有するEgalet a/s(http://www.eqalet.com)は、表面浸食性、疎水性のPEGステアレートからなる、活性成分を含む、生体分解性コートおよびマトリックスである。Egalet(登録商標)技術の1つに、コートとマトリックスからなる2コンポーネント生成モデルである2K Egalet(登録商標)持続放出系がある。該薬剤は、時間とともに持続放出のためにEgalet(登録商標)マトリックスを通して均一に分散する。本発明の文脈において例えばEurand技術のような技術が興味深い:Diffucaps(薬剤放出プロフィールは、中性コア、例えば糖スフェア、血漿、または顆粒上に活性薬剤、次いで速度調節機能膜を重層することによりもたらされる。Diffucaps/Surecapsビーズは、サイズが小さく直径が1mmまたはそれ以下である。種々の薬剤放出プロフィールのビーズを硬ゼラチンカプセルに組み込むことにより組み合わせ放出プロフィールを達成することができる。)、Diffutabs(Diffutab技術はマトリックス錠の拡散および浸食を通して薬剤放出を制御する親水性ポリマー混合物を組み込む)、Minitabs(Eurand Minitabsは薬剤放出速度を制御するゲル形成賦形剤を含む小さな(2mm x 2mm)錠剤である。さらなる膜をさらに放出速度を制御するために加えてよい。)、Orbexa(この技術は、規定ベースの(defined-based)造粒押出および球形化技術により制御されたサイズおよび密度のビーズを生成する。得られたビーズは、さらなる放出速度制御のために放出速度制御膜でコートすることができ、カプセルに充填するかサシェー型としてよい。)、SDS(EurandのSDS技術は、機能的ポリマーおよび機能的ポリマーと特定の添加剤の組み合わせ、例えば複合ポリマー物質を用い、腸管の最適吸収部位に薬剤を送達する。これを達成するために、Eurandは、最初に活性薬剤を組み込むDiffucapsまたはEurand Minitabsのような多粒子剤形を生成する。次に、この剤形を薬剤を所望の部位に送達するpH依存性/非依存性ポリマー膜でコートする。次にこれらを硬ゼラチンカプセルに充填する。)。
【0040】
本発明の組成物を製造するのに用いる別の興味深い技術に、WO 03/004001に記載のいわゆるMeltDose(登録商標)技術がある(例えば、http://www.lifecyclepharma.com参照)。MeltDose(登録商標)は、可溶化した個々の分子を錠剤に処方することを含む。個々の分子を処方することにより、低水溶性の薬剤の経口吸収の一次制限を除去し、優れたバイオアベイラビリティをもたらす。この技術を用いることにより、種々の医薬剤形、例えばペレットまたは錠剤の形に加工するのに適した粒子状物質を得ることができる。さらに、該技術は、活性物質の、例えば本明細書に記載の放出プロフィールのような適切な放出プロフィールを得ることができるので使用に適している。ある態様において、使用に適したペレットは、2000μm以上の平均粒子サイズを有してよい。別の態様において、使用に適したペレットは約0.01μm〜約250μmの平均粒子サイズを有してよい。
【0041】
本発明の文脈において使用する別の特定の適切な製剤方針は、例えば軟ゼラチンカプセルのような脂溶性環境中の製剤である。この製剤方針の適切な例には、Vegicaps Soft(Scherer)がある(100%植物由来であるにも関わらずカラギーナンおよびデンプンに基づく軟カプセル技術は、なお従来の軟ゼラチンカプセルの重要なすべての特性を提供する。これらには、飲み込み易さをもたらす軟質の柔軟な剤形が含まれる。)。(さらなる情報については、http://www.rpscherer.de/page.php?paqeID=94参照。)
【0042】
適切な製剤のさらなる具体例には、軟および硬ゼラチンカプセル中の活性物質とビタミンE濃縮物の製剤が含まれる。修飾形のこの製剤は、シクロスポリンに加えて他にコーン油モノ-ジ-トリグリセリド、ポリオキシル40水素化ひまし油NF、DL-α-トコフェロールUSP(ビタミンEファミリーの部分)、ゼラチンNF、グリセロール、四三酸化鉄、プロピレングリコールUSP、二酸化チタンUSP、カルミン、およびアルコールを含む市販のシクロスポリン製剤のNeoral(登録商標)の基礎原料である。
【0043】
適切な製剤(処方)の別の具体例には、軟または硬ゼラチンカプセル中に、エタノール、トコフェロールエチレングリコール1000 スクシネート(TPGS)、コーン油、およびワックスと活性物質の製剤が含まれる。この製品は半固体または固体剤形であり得る。この製剤の放出速度は腸中のリパーゼによる分解に依存する。
【0044】
適切な製剤のさらなる例には、軟または硬ゼラチンカプセル中に、エタノール、トコフェロールエチレングリコール1000 スクシネート(TPGS)、コーン油、およびポリグリコール化グリセリド(例えばGelucire)と活性物質の製剤が含まれる。この製品は半固体または固体剤形であり得る。この製剤の放出速度は腸中のリパーゼによる分解に依存する。
【0045】
適切な製剤のさらなる例には、経口パルス用量ドラッグデリバリーシステムがある。この剤形は、Schering Repetab錠の修飾形と考えることができる。本発明の組成物の部分は、錠剤のコア中に入れられる。
【0046】
該コアは、例えば常套的湿式造粒法または連続増粒法、例えば押出し、次いで顆粒を錠剤に圧縮することにより製造することができる。次に、該コアを適切な技術、好ましくはEudragitsのような腸溶コーティングポリマーを用いる空気サスペンジョンによりコートする。
【0047】
第1放出用量は、コア上に圧縮コートされるか、または腸溶コートでまたは腸溶コートの上部に空気懸濁コートされる。本発明の態様において、第1放出用量は、腸溶コートで空気懸濁コートされる。本発明のさらなる態様において、第1放出用量は、典型的には腸溶コートの分解前に本発明の組成物の放出を避けるために該コート上に圧縮コートされ、そのような分解は、典型的には胃(gastric ventricle)より高いpH値で生じ、すなわち、典型的には腸溶コートの分解は胃の通過後に生じる。
【0048】
適切な製剤のさらなる例には、経口持続ドラッグデリバリーシステムがある。本発明組成物の部分は錠剤のコア中に入れられる。
【0049】
該コアは、例えば常套的湿式造粒法または連続増粒法、例えば押出し、次いで顆粒を錠剤に圧縮することにより製造することができる。該コアを適切な技術、好ましくはエチルセルロースおよび親水性賦形剤、例えばヒドロキシルプロピルセルロース(HPC)を用いる空気サスペンジョンによりコートする。
【0050】
第1放出用量は、コア上に圧縮コートされるか、または腸溶コートでまたは腸溶コートの上部に空気サスペンジョンコートされる。本発明の好ましい態様において、第1放出用量は、腸溶コートで空気サスペンジョンコートされる。本発明のさらなる態様において、第1放出用量は、典型的には腸溶コートの分解前に本発明の組成物の放出を避けるために該コート上に圧縮コートされ、そのような分解は、典型的には胃(gastric ventricle)より高いpH値で生じ、すなわち、典型的には腸溶コートの分解は胃の通過後に生じる。
【0051】
適切な製剤のさらなる例は、例えばWO 03/080034に記載のような結晶工学により得られる(この内容は本明細書の一部を構成する。)。
【0052】
したがって、本発明組成物の別の態様において、親水性表面を有する微結晶の形の活性物質を含む。さらに、本発明の別の態様において、該微結晶は持続放出製剤を達成するために直接フィルムコートされる。
【0053】
適切な製剤の別の具体例には、本発明組成物と純粋なシクロデキストリンおよびシクロデキストリン誘導体(例えばアルキル-およびヒドロキシアルキル-誘導体、またはスルホブチル誘導体)の複合体(錯体)化が含まれる。該複合体化は、よく知られた方法に従って達成される。そのような複合体化は複合体化前の該組成物に比べて本発明組成物の高い溶解性と高い溶出速度をもたらすと予期される。さらに、そのような組成物は複合体化前の該組成物に比べて本発明組成物の高いバイオアベイラビリティをもたらすと予期される。
【0054】
具体的態様において、本発明は1日に1回、2回、またはそれ以上、例えば1日に1回または2回、または3回投与することができる制御放出医薬組成物に関する。さらに、該組成物はフマル酸エステルを比較的pH非依存性に、すなわち、消化管内のpHに依存しないで放出するよう設計してよい。そのような組成物の例には、例えば制御放出コーティングでコートされた固体剤形(例えば、錠剤、カプセル剤、ペレット、ビーズなど)の形の組成物がある。制御放出コーティングに適した物質には、例えばセルロース、およびメチルセルロース、エチルセルロース、セルロースアセテート、またはポリ(エチルレン-コ-ビニルアセテート)、ポリ(塩化ビニル)を含むセルロース誘導体がある。
【0055】
典型的には、該フマル酸エステルの放出は拡散制御膜でコートされた組成物から3段階で生じる:
i)第1に、水(GI管から)が周囲から剤形内に拡散し、
ii)第2に、該剤形内に存在する少なくともいくらかの該フマル酸エステルが水の作用により溶解し、
iii)該溶解したフマル酸エステルが剤形から周囲(すなわちGI管)中に拡散する。
【0056】
他の例には、例えばそれぞれマトリックス系の形の多数単位を含む剤形またはマトリックス錠が含まれる。活性物質を、例えば、セルロース、および微晶質セルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロースおよびメチルセルロースを含むセルロース誘導体、ポビドン、ポリ(エチレンオキシド)(PEO)、ポリエチレングリコール(PEG)、ポリ(ビニルアルコール)(PVA)、キサンタンガム、カラギーナン、および他の合成物質を含むマトリックス中に組み込む。医薬的に許容される賦形剤または添加剤として通常用いる物質をマトリックス組成物に加えてよい。
【0057】
適切な組成物の他の例には、活性物質が水膨潤性ネットワークポリマー中に組み込まれる、例えばヒドロゲル、すなわち単体システムがある。使用に適した物質には、例えば、親水性ビニルおよびアクリルポリマー、アルギネートのようなポリサッカライド、およびポリ(エチレンオキシド)がある。
【0058】
特定の態様において、本発明の組成物は該フマル酸エステルをpH制御放出(pH依存性放出としても知られる)する。通常、該放出は、もしあるいとしてもフマル酸エステルを少量しか胃(約pH3以下)内に放出せず、該フマル酸エステルを腸(pHが約6〜7に変化する)に放出するよう設計される。そのようなpH制御放出は、腸溶コーティングを有する本発明組成物(全組成物、または組成物が多粒子組成物である場合は個々の単位)を提供するか、または該フマル酸をpH依存性浸透圧メカニズムまたは適切な酵素の使用により放出する組成物を提供することにより得ることができる。
【0059】
腸溶コーティング物質として用いるのに適した物質の例には、ポリアクリルアミド、フタレート誘導体、例えば、炭水化物の酸フタレート、アミロースアセテートフタレート、セルロースアセテートフタレート、他のセルロースエステルフタレート、セルロースエーテルフタレート、ヒドロキシプロピルセルロースフタレート、ヒドロキシプロピルエチルセルロースフタレート、ヒドロキシプロピルメチルセルロースフタレート、メチルセルロースフタレート、ポリビニルアセテートフタレート、ポリアクリルメタクリル酸コポリマー、セラックおよびビニルアセテート、およびクロトン酸コポリマーなどが含まれる。
【0060】
pH非依存性放出を有する上記組成物は、例えば腸溶コーティングの外層を有する該組成物を与えることにより該フマル酸エステルを放出するよう製剤化してもよい。
【0061】
さらに、該組成物は、フマル酸エステルの放出に初期遅延が得られるような方法で製剤化してよい。そのような遅延は、例えば時間制御的に分解する(例えば浸食する)最外層のコーティングを選ぶことにより得ることができ、この最外層のコーティングが浸食されて除かれた時にのみフマル酸エステルの放出が始まる。
【0062】
フマル酸エステルの適切な放出が得られるよう設計された種々の本発明組成物について以下に説明する。上記説明および医薬の制御放出の分野のハンドブックに基づいて当業者は必要な放出プロフィールを達成するために種々の製剤方針を選ぶ方法がわかるであろう。
1日に2回またはそれ以上投与するよう設計された組成物
pH非依存性放出
【0063】
フマル酸エステルをpH非依存性に放出し、放出パターンが1日に2回またはそれ以上投与する組成物に適している具体的態様を以下に説明する。適切な製剤方針の例には、例えば上記のあらゆる製剤方針を含む、拡散コーティング、例えば制御放出拡散コーティング、マトリックス粒子またはマトリックス錠、ハイドロゲル、パルス用量ドラッグデリバリーシステム、ビタミンE濃縮物またはエタノール、TPGS、コーン油、およびワックスなどとの合剤により提供される組成物がある。
【0064】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、水を溶出溶媒に用いてin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物に関する:
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約60%w/w、例えば約30%〜約60%w/w、約40%〜約55%w/w、または約50%が放出される、および/または
試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、約60%〜約80%w/w、または約75%が放出される、および/または
試験開始後の最初の12時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約80%w/w、例えば約80%w/wまたはそれ以上、約85%w/wまたはそれ以上、約90%w/wまたはそれ以上、または約95%w/wまたはそれ以上が放出される、および/または
該組成物中に含まれるフマル酸エステルの総量が試験開始後の最初の12時間以内に放出される。
pH制御放出
【0065】
フマル酸エステルがpH依存性に放出され、放出パターンが1日に2回またはそれ以上投与する組成物に適している特定の態様について以下に説明する。適切な製剤方針の例には、例えばZentner et al(US 6,537,584)およびBae(US 5,484,610)に記載のタイプの腸溶コーティングまたはハイドロゲルを用いて提供される組成物がある(この内容は本明細書の一部を構成する。)。適切な製剤方針の例には、例えば上記のあらゆる製剤方針を含む、所望により腸溶コーティングを有する、拡散コーティング、例えば制御放出拡散コーティング、マトリックス粒子またはマトリックス錠、ハイドロゲル、パルス用量ドラッグデリバリーシステム、ビタミンE濃縮物またはエタノール、TPGS、コーン油、およびワックスなどとの合剤により提供される組成物がある。
【0066】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、試験の最初の2時間は0.1N塩酸を溶出溶媒に、次いで0.05Mリン酸緩衝液pH6.5または6.8を溶出溶媒に用いてin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物を提供する:
試験開始後の最初の2時間以内に該フマル酸エステルの総量の少なくとも約1%w/w、例えば少なくとも約2%w/w、少なくとも約3%w/w、または約5%w/wが放出される、および/または
試験開始後の最初の3時間以内に該フマル酸エステルの総量のほぼ約35%w/w、例えば約15%〜約35%w/w、約20%〜約30%w/w、または約25%w/wが放出される、および/または
試験開始後の最初の3時間以内に該フマル酸エステルの総量の約10%〜約70%w/w、約10%〜約65%w/w、約10%〜約60%w/w、約15%〜約50%w/w、約15%〜約35%w/w、約20%〜約30%w/w、または約20%w/w、または約25%w/が放出される、および/または
試験開始後の最初の4時間以内に該フマル酸エステルの総量のほぼ約92%w/w、例えば約10%〜約92%w/w、約20%〜約85%w/w、約20%〜約80%w/w、約20%〜約70%w/w、約25%〜約60%w/w、約25%〜約55%w/w、約30%〜約50%w/w、または約35%w/w、または約40%w/w、または約45%w/wが放出される、および/または
試験開始後の最初の5時間以内に該フマル酸エステルの総量のほぼ約94%w/w、例えば約15%〜約94%w/w、約25%〜約90%w/w、約30%〜約85%w/w、約35%〜約80%w/w、約35%〜約75%w/w、約40%〜約70%w/w、約45%〜約70%w/w、約55%〜約70%w/w、約60%〜約70%w/w、または約45%w/w、または約50%w/w、または約55%w/w、または約60%w/w、または約65%w/wが放出される、および/または
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約60%w/w、例えば約30%〜約60%w/w、約40%〜約55%w/w、または約50%w/wが放出される、および/または
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約95%w/w、例えば約35%〜約95%w/w、約40%〜約90%w/w、約45%〜約85%w/w、約50%〜約85%w/w、約55%〜約85%w/w、約60%〜約85%w/w、約65%〜約85%w/w、約70%〜約85%w/w、約75%〜約85%w/w、または約65%w/w、または約70%w/w、または約75%w/w、または約80%w/wが放出される、および/または
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約98%w/w、例えば約45%〜約98%w/w、約50%〜約98%w/w、約55%〜約98%w/w、約60%〜約98%w/w、約65%〜約98%w/w、約70%〜約98%w/w、約75%〜約95%w/w、約80%〜約95%w/w、約85%〜約95%w/w、または約75%w/w、または約80%w/w、または約85%w/w、または約90%w/wが放出される、および/または
試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、約60%〜約80%w/w、または約75%w/wが放出される、および/または
試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約99%w/w、例えば約60%〜約99%w/w、約70%〜約99%w/w、約80%〜約99%w/w、約90%〜約99%w/w、または約95%w/wが放出される。
【0067】
本発明の別の局面において、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、最初の2時間は0.1N塩酸を、次いで0.05Mリン酸緩衝液pH6.5または6.8を用いてUSPに従って測定したときにin vitro溶出プロフィールに従って予め決定された期間にわたりフマル酸のジ-(C1-C5)アルキルエステルおよびモノ-(C1-C5)アルキルエステル、またはその医薬的に許容される塩を放出するのに適合した制御放出剤形からなることを特徴とする該組成物を提供する:
ここで、試験開始後の最初の2時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ5%w/wが放出される、および/または
試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量の約20%〜約75%w/wが放出される、および/または
試験開始後の最初の4時間以内に該組成物中に含まれる該フマル酸エステルの総量の約50%〜約90%w/wが放出される、および/または
試験開始後の最初の5時間以内に該組成物中に含まれる該フマル酸エステルの総量の約60%〜約90%w/wが放出される、および/または
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量の約70%〜約95%w/wが放出される、および/または
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量の約75%〜約97%w/wが放出される。
【0068】
本発明のさらなる局面において、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、最初の2時間は0.1N塩酸を、次いで0.05Mリン酸緩衝液pH6.5または6.8を用いてUSPに従って測定したときにin vitro溶出プロフィールに従って予め決定された期間にわたりフマル酸のジ-(C1-C5)アルキルエステルおよびモノ-(C1-C5)アルキルエステル、またはその医薬的に許容される塩を放出するのに適合した制御放出剤形からなることを特徴とする該組成物を提供する:
ここで、試験開始後の最初の2時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ5%w/wが放出される、
試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量の約20%〜約75%w/wが放出される、
試験開始後の最初の4時間以内に該組成物中に含まれる該フマル酸エステルの総量の約50%〜約90%w/wが放出される、
試験開始後の最初の5時間以内に該組成物中に含まれる該フマル酸エステルの総量の約60%〜約90%w/wが放出される、
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量の約70%〜約95%w/wが放出される、
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量の約75%〜約97%w/wが放出される、
試験開始後の最初の8時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも85%w/wが放出される。
【0069】
本発明の別のさらなる局面において、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む制御放出医薬組成物であって、最初の2時間は0.1N塩酸を、次いで0.05Mリン酸緩衝液pH6.5または6.8を用いてUSPに従って測定したときにin vitro溶出プロフィールに従って予め決定された期間にわたりフマル酸のジ-(C1-C5)アルキルエステルおよびモノ-(C1-C5)アルキルエステル、またはその医薬的に許容される塩を放出するのに適合した制御放出剤形からなることを特徴とする該組成物を提供する:
ここで、試験開始後の最初の2時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ5%w/wが放出される、および/または
試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量の約20%〜約50%w/wが放出される、および/または
試験開始後の最初の4時間以内に該組成物中に含まれる該フマル酸エステルの総量の約45%〜約70%w/wが放出される、および/または
試験開始後の最初の5時間以内に該組成物中に含まれる該フマル酸エステルの総量の約65%〜約85%w/wが放出される、および/または
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量の約75%〜約90%w/wが放出される。
【0070】
本発明のさらに別の局面において、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む制御放出医薬組成物であって、最初の2時間は0.1N塩酸を、次いで0.05Mリン酸緩衝液pH6.5または6.8を用いてUSPに従って測定したときにin vitro溶出プロフィールに従って予め決定された期間にわたりフマル酸のジ-(C1-C5)アルキルエステルおよびモノ-(C1-C5)アルキルエステル、またはその医薬的に許容される塩を放出するのに適合した制御放出剤形からなることを特徴とする該組成物を提供する:
ここで、試験開始後の最初の2時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ5%w/wが放出される、
試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量の約20%〜約50%w/wが放出される、
試験開始後の最初の4時間以内に該組成物中に含まれる該フマル酸エステルの総量の約45%〜約70%w/wが放出される、
試験開始後の最初の5時間以内に該組成物中に含まれる該フマル酸エステルの総量の約65%〜約85%w/wが放出される、
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量の約75%〜約90%w/wが放出される、
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも80%が放出される。
【0071】
本発明のさらに別の局面において、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む制御放出医薬組成物であって、最初の2時間は0.1N塩酸を、次いで0.05Mリン酸緩衝液pH6.5または6.8を用いてUSPに従って測定したときにin vitro溶出プロフィールに従って予め決定された期間にわたりフマル酸のジ-(C1-C5)アルキルエステルおよびモノ-(C1-C5)アルキルエステル、またはその医薬的に許容される塩を放出するのに適合した制御放出剤形からなることを特徴とする該組成物を提供する:
ここで、試験開始後の最初の2時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ5%w/wが放出される、および/または
試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量の約50%〜約75%w/wが放出される、および/または
試験開始後の最初の4時間以内に該組成物中に含まれる該フマル酸エステルの総量の約70%〜約90%w/wが放出される、および/または
試験開始後の最初の5時間以内に該組成物中に含まれる該フマル酸エステルの総量の約80%〜約90%w/wが放出される。
【0072】
本発明のさらに別の局面において、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む制御放出医薬組成物であって、最初の2時間は0.1N塩酸を、次いで0.05Mリン酸緩衝液pH6.5または6.8を用いてUSPに従って測定したときにin vitro溶出プロフィールに従って予め決定された期間にわたりフマル酸のジ-(C1-C5)アルキルエステルおよびモノ-(C1-C5)アルキルエステル、またはその医薬的に許容される塩を放出するのに適合した制御放出剤形からなることを特徴とする該組成物を提供する:
ここで、試験開始後の最初の2時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ5%w/wが放出される、
試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量の約50%〜約75%w/wが放出される、
試験開始後の最初の4時間以内に該組成物中に含まれる該フマル酸エステルの総量の約70%〜約90%w/wが放出される、
試験開始後の最初の5時間以内に該組成物中に含まれる該フマル酸エステルの総量の約80%〜約90%w/wが放出される、
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約90%w/wが放出される。
徐々のpH変化に及ぶ放出(「ハーフチェンジ」法)
【0073】
フマル酸エステルがpH依存性に放出され、放出パターンが1日に2回またはそれ以上投与する組成物に適している特定の態様について以下に説明する。適切な製剤方針の例には、例えばZentner et al(US 6,537,584)およびBae(US 5,484,610)に記載のタイプの腸溶コーティングまたはハイドロゲルを用いて提供される組成物がある(この内容は本明細書の一部を構成する。)。適切な製剤方針の例には、例えば上記のあらゆる製剤方針を含む、所望により腸溶コーティングを有する、拡散コーティング、例えば制御放出拡散コーティング、マトリックス粒子またはマトリックス錠、ハイドロゲル、パルス用量ドラッグデリバリーシステム、ビタミンE濃縮物またはエタノール、TPGS、コーン油、およびワックスなどとの合剤により提供される組成物がある。
【0074】
「ハーフチェンジ」法は、腸溶コートまたは持続放出製剤について特に開発された。この方法は、1時間毎に溶出溶媒の半分を中性溶出溶媒の一定分量で置換することを含む(十二指腸から回腸へのpH値のわずかな変化についてGI通過をシミュレートするため)。該アプローチを下記表に説明する。
【0075】
【表1】
【0076】
模擬(シミュレート)胃液の組成は、例えばUnited States Pharmacopeia(USP)2005にみることができる:
タンパク質1mgあたり800〜2500単位の活性を含むブタ胃粘膜由来の精製ペプシン3.2gおよびNaCl 2.0gを7.0mlの塩化水素酸に溶解し、十分な水を加えて1000mLとする。得られた試験溶液はpH約1.2である。
【0077】
模擬胃液の別の組成はGerman E DIN 19738(Deutsche Industrie Norm)にみることができる:
100mLの合成/模擬胃液は、290mgのNaCl、70mgのKCl、27mgのKH2PO4を含み、十分なHClでpH2.0に調整する。さらに、該胃液は、100mgのペプシンと300mgのムチンを含む。
【0078】
模擬腸液の組成は、例えばUnited States Pharmacopeia(USP)2005にみることができる:
6.8gのリン酸2水素カリウムを250mLの水に溶解する。混合し、77mLの0.2N水酸化ナトリウムおよび500mLの水を加える。10.0gのパンクレアチンを加え、該溶液を混合し、該溶液を混合し、次いで0.2 N水酸化ナトリウムまたは0.2N塩酸を加えてpH6.8±0.1に調整する。得られた溶液を水で1000mLに希釈する。
【0079】
模擬腸液の別の組成はGerman E DIN 19738(Deutsche Industrie Norm)に記載されている:
100mLの合成/模擬腸液は、30mgのKCl、50mgのCaCl2、20mgのMgCl2を含み、十分なNaHCO3でpH7.5に調整する。さらに、該腸液は30mgのトリプシン、900mgのパンクレアチン、900mgの凍結乾燥胆汁、および30mgの尿素を含む。
【0080】
本発明の好ましい態様において、該「ハーフチェンジ」法は、USP 2005に記載の模擬胃液と模擬腸液を用いて実施される。
【0081】
本発明の別の態様において、該「ハーフチェンジ」法は、USP 2005に記載の模擬胃液と模擬腸液を用いるが該タンパク質不含(すなわち、模擬胃液中にペプシン不含、模擬腸液中にパンクレアチン不含)で実施される。
【0082】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、「ハーフチェンジ」法に従ってin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物に関する:
試験開始後の最初の3時間以内に該フマル酸エステルの総量の約20%〜約40%w/w、約20%〜約35%w/w、または約30%w/wが放出される、および/または
試験開始後の最初の3時間以内に該フマル酸エステルの総量の少なくとも約12%w/w、例えば約12%〜約50%w/w、約15%〜約45%w/w、約20%〜約40%w/w、約20%〜約35%w/w、約22%〜約35%w/w、または約25%w/w、または約30%w/wが放出される、および/または
試験開始後の最初の4時間以内に該フマル酸エステルの総量の約25%〜約40%w/w、約30%〜約40%w/w、または約40%w/wが放出される、および/または
試験開始後の最初の4時間以内に該フマル酸エステルの総量の少なくとも約76%w/w、例えば約76%〜約95%w/w、約80%〜約90%w/w、または約80%w/w、または約85%w/wが放出される、および/または
試験開始後の最初の4時間以内に該フマル酸エステルの総量のほぼ約40%w/w、例えば約10%〜約40%w/w、約15%〜約35%w/w、約20%〜約30%w/w、または約25%w/w、または約30%w/wが放出される、および/または
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約81%w/w、例えば約81%〜約96%w/w、約85%〜約95%w/w、約85%〜約90%w/w、または約80%w/w、または約85%w/w、または約90%w/wが放出される、および/または
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約50%w/w、例えば約20%〜約50%w/w、約25%〜約45%w/w、約30%〜約45%w/w、または約40%w/w、または約45%w/wが放出される、および/または
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約82%w/w、例えば、約82%〜約99%w/w、約85%〜約99%w/w、約85%〜約95%w/w、または約90%w/wが放出される、および/または
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約65%w/w、例えば約25%〜約65%w/w、約30%〜約65%w/w、約35%〜約60%w/w、約40%〜約60%w/w、約50%〜約6O%w/w、または約55%w/w、または約60%w/wが放出される、および/または
試験開始後の最初の8時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、約60%〜約80%w/w、または約75%w/wが放出される、および/または
試験開始後の最初の8時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約92%w/w、例えば約30%〜約92%w/w、約35%〜約90%w/w、約40%〜約85%w/w、約45%〜約80%w/w、約50%〜約75%w/w、約55%〜約75%w/w、約60%〜約75%w/w、または約65%w/w、または約70%w/wが放出される、および/または
試験開始後の最初の12時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約80%w/w、例えば約80%w/wまたはそれ以上、約85%w/wまたはそれ以上、約90%w/wまたはそれ以上、または約95%w/wまたはそれ以上が放出される。
徐 放
【0083】
フマル酸エステルが徐々にまたは遅延して放出され、放出パターンが1日に2回またはそれ以上投与する組成物に適している特定の態様について以下に説明する。適切な製剤方針の例には上記のあらゆるものがある。
【0084】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、水を溶出溶媒に用いてin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物に関する:
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約35%w/w、例えば約15%〜約35%w/w、例えば約20%〜約30%w/w、または約25%w/wが放出される、および/または
試験開始後の以最初の8時間内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約60%w/w、例えば約30%〜約60%w/w、例えば約40%〜約55%w/w、または約50%w/wが放出される、および/または
試験開始後の最初の10時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、例えば約60%〜約80%w/w、または約75%w/wが放出される,および/または
試験開始後の最初の12時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約80%w/w、例えば約80%w/wまたはそれ以上、例えば約85%w/wまたはそれ以上、約90%w/wまたはそれ以上、または約95%w/wまたはそれ以上が放出される。
1日1回投与するよう設計された組成物
pH非依存性放出
【0085】
フマル酸エステルをpH非依存性に放出し、放出パターンが1日に1回投与する組成物に適している具体的態様を以下に説明する。適切な製剤方針の例には、例えば上記のあらゆる製剤方針を含む、拡散コーティング、例えば制御放出拡散コーティング、マトリックス粒子またはマトリックス錠、ハイドロゲル、パルス用量ドラッグデリバリーシステム、ビタミンE濃縮物またはエタノール、TPGS、コーン油、およびワックスなどとの合剤により提供される組成物がある。
【0086】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、水を溶出溶媒に用いてin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物に関する:
試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約60%w/w、例えば約30%〜約60%w/w、約40%〜約55%w/w、または約50%w/wが放出される、および/または
試験開始後の最初の13.5時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、約60%〜約80%w/w、または約75%w/wが放出される、および/または
試験開始後の最初の18時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約80%w/w、例えば約80%w/wまたはそれ以上、約85%w/wまたはそれ以上、約90%w/wまたはそれ以上、または約95%w/wまたはそれ以上が放出される、および/または
該組成物に含まれるフマル酸エステルは試験開始後の最初の18時間以内に放出される。
pH制御放出
【0087】
フマル酸エステルがpH依存性に放出され、放出パターンが1日に1回投与する組成物に適している特定の態様について以下に説明する。適切な製剤方針の例には、例えばZentner et al(US 6,537,584)およびBae(US 5,484,610)に記載のタイプの腸溶コーティングまたはハイドロゲルを用いて提供される組成物がある。適切な製剤方針のさらなる例には、例えば上記のあらゆる製剤方針を含む、所望により腸溶コーティングを有する、拡散コーティング、例えば制御放出拡散コーティング、マトリックス粒子またはマトリックス錠、ハイドロゲル、パルス用量ドラッグデリバリーシステム、ビタミンE濃縮物またはエタノール、TPGS、コーン油、およびワックスなどとの合剤により提供される組成物がある。
【0088】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、試験の最初の2時間は0.1N塩酸を溶出溶媒に、次いで0.05Mリン酸緩衝液pH6.5または6.8を溶出溶媒に用いてin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物を提供する:
試験開始後の最初の2時間以内に該フマル酸エステルの総量の少なくとも約1%w/w、例えば少なくとも約2%w/w、少なくとも約3%w/w、または約5%w/wが放出される、および/または
試験開始後の最初の4時間以内に該フマル酸エステルの総量のほぼ約90%w/w、例えば約5%〜約90%w/w、約5%〜約85%w/w、約10%〜約80%w/w、約10%〜約70%w/w、約10%〜約65%w/w、約10%〜約60%w/w、約15%〜約50%w/w、約15%〜約35%w/w、約20%〜約30%w/w、または約20%w/w、または約25%w/wが放出される、および/または
試験開始後の最初の4.5時間以内に該フマル酸エステルの総量のほぼ約35%w/w、例えば約15%〜約35%w/w、約20%〜約30%w/w、または約25%w/wが放出される、および/または
試験開始後の最初の5時間以内に該フマル酸エステルの総量のほぼ約92%w/w、例えば約10%〜約92%w/w、約20%〜約85%w/w、約20%〜約80%w/w、約20%〜約70%w/w、約25%〜約60%w/w、約25%〜約55%w/w、約30%〜約50%w/w、または約35%w/w、または約40%w/w、または約45%w/wが放出される、および/または
試験開始後の最初の6時間以内に該フマル酸エステルの総量のほぼ約94%w/w、例えば約15%〜約94%w/w、約25%〜約90%w/w、約30%〜約85%w/w、約35%〜約80%w/w、約35%〜約75%w/w、約40%〜約70%w/w、約45%〜約70%w/w、約55%〜約70%w/w、約60%〜約70%w/w、または約45%w/w、または約50%w/w、または約55%w/w、または約60%w/w、または約65%w/wが放出される、および/または
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約95%w/w、例えば約35%〜約95%w/w、約40%〜約90%w/w、約45%〜約85%w/w、約50%〜約85%w/w、約55%〜約85%w/w、約60%〜約85%w/w、約65%〜約85%w/w、約70%〜約85%w/w、約75%〜約85%w/w、または約65%w/w、または約70%w/w、または約75%w/w、または約80%w/wが放出される、および/または
試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約98%w/w、例えば約45%〜約98%w/w、約50%〜約98%w/w、約55%〜約98%w/w、約60%〜約98%w/w、約65%〜約98%w/w、約70%〜約98%w/w、約75%〜約95%w/w、約80%〜約95%w/w、約85%〜約95%w/w、または約75%w/w、または約80%w/w、または約85%w/w、または約90%w/wが放出される、および/または
試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約60%w/w、例えば約30%〜約60%w/w、約40%〜約55%w/w、または約50%w/wが放出される、および/または
試験開始後の最初の12時間以内に該組成物中に含まれる該フマル酸エステルの総量のがほぼ約99%w/w、例えば約60%〜約99%w/w、約70%〜約99%w/w、約80%〜約99%w/w、約90%〜約99%w/w、または約95%w/w放出される、および/または
試験開始後の最初の13.5時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、約60%〜約80%w/w、または約75%w/wが放出される。
徐々のpH変化における放出(「ハーフチェンジ」法)
【0089】
フマル酸エステルがpH依存性に放出され、放出パターンが1日に1回投与する組成物に適している特定の態様について以下に説明する。適切な製剤方針の例には、例えばZentner et al(US 6,537,584)およびBae(US 5,484,610)に記載のタイプの腸溶コーティングまたはハイドロゲルを用いて提供される組成物がある(この内容は本明細書の一部を構成する。)。適切な製剤方針の例には、例えば上記のあらゆる製剤方針を含む、所望により腸溶コーティングを有する、拡散コーティング、例えば制御放出拡散コーティング、マトリックス粒子またはマトリックス錠、ハイドロゲル、パルス用量ドラッグデリバリーシステム、ビタミンE濃縮物またはエタノール、TPGS、コーン油、およびワックスなどとの合剤により提供される組成物がある。
【0090】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、ハーフチェンジ法に従ってin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物を提供する:
試験開始後の最初の3時間以内に該フマル酸エステルの総量の少なくとも約12%w/w、例えば約12%〜約60%w/w、約15%〜約50%w/w、約20%〜約40%w/w、約20%〜約35%w/w、または約25%w/w、または約30%w/wが放出される、および/または
試験開始後の最初の4時間以内に該フマル酸エステルの総量のほぼ約35%w/w、例えば約15%〜約35%w/w、約20%〜約30%w/w、または約25%w/wが放出される、および/または
試験開始後の最初の5時間以内に該フマル酸エステルの総量のほぼ約45%w/w、例えば約10%〜約45%w/w、約15%〜約40%w/w、約15%〜約35%w/w、約20%〜約30%w/w、または約25%w/w、または約30%w/wが放出される、および/または
試験開始後の最初の7時間以内に該フマル酸エステルの総量のほぼ約65%w/w、例えば約20%〜約65%w/w、約20%〜約60%w/w、約20%〜約50%w/w、約25%〜約45%w/w、約30%〜約45%w/w、または約40%w/w、または約45%w/wが放出される、および/または
試験開始後の最初の8時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約92%w/w、例えば約25%〜約92%w/w、約25%〜約90%w/w、約30%〜約80%w/w、約35%〜約70%w/w、約40%〜約65%w/w、約45%〜約60%w/w、約50%〜約60%w/w、または約55%w/w、または約60%w/wが放出される、および/または
試験開始後の最初の8時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約60%w/w、例えば約30%〜約60%w/w、約40%〜約55%w/w、または約50%w/wが放出される、および/または
試験開始後の最初の12時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約99%w/w、例えば約30%〜約99%w/w、約30%〜約95%w/w、約35%〜約90%w/w、約40%〜約85%w/w、約45%〜約80%w/w、約50%〜約75%w/w、約55%〜約75%w/w、約60%〜約75%w/w、または約65%w/w、または約70%w/wが放出される、および/または
試験開始後の最初の12.5時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、約60%〜約80%w/w、または約75%w/wが放出される、および/または
試験開始後の最初の18時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約80%w/w、例えば約80%w/wまたはそれ以上、約85%w/wまたはそれ以上、約90%w/wまたはそれ以上、または約95%w/wまたはそれ以上が放出される。
徐 放
【0091】
フマル酸エステルが徐々にまたは遅延して放出され、放出パターンが1日に1回投与する組成物に適している特定の態様について以下に説明する。適切な製剤方針の例には、上記のあらゆる組成物がある。
【0092】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、水を溶出溶媒として用いるin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物を提供する:
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約35%w/w、例えば約15%〜約35%w/w、約20%〜約30%w/w、または約25%w/wが放出される、および/または
試験開始後の最初の11時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約60%w/w、例えば約30%〜約60%w/w、約40%〜約55%w/w、または約50%w/wが放出される、および/または
試験開始後の最初の14時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、約60%〜約80%w/w、または約75%w/wが放出される、および/または
試験開始後の以最初の18時間内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約80%w/w、例えば約80%w/wまたはそれ以上、約85%w/wまたはそれ以上、約90%w/wまたはそれ以上、または約95%w/wまたはそれ以上が放出される。
【0093】
典型的には、上記のごとく本発明組成物は、持続的に活性物質(すなわち、フマル酸に代謝され、次いで急速な排除プロセスを受けるフマル酸のモノアルキルエステル)を送達するよう設計される。本明細書に記載の特徴的in vitro放出パターンに加えて、そのような持続放出は臨床試験後に得られる薬物動態パラメーターにも反映される。したがって、フマル酸のモノアルキルエステル(投与されたジアルキルエステルが加水分解または代謝されると血漿中に出現する)のCmaxは、同様または等価用量を投与した場合に以前に論文に記載されたものと同じ桁数であると予期される(すなわち、約0.4〜約2.0mg/Lの範囲のモノメチルエステルのCmaxは120〜240mgジメチルフマレートの経口用量に対応する)。しかしながら、1日に多くの頻度での投与(1日に2〜4錠を1〜3回)を避けるために、濃度が治療領域内である時間を延長することが目的である。したがって、W50(すなわち、血漿濃度がCmaxの50%またはそれ以上である期間)が市販の処置に比べて少なくとも10%、例えば少なくとも20%、少なくとも30%、少なくとも40%、または少なくとも50%延長されると予期される。適切なW50は、少なくとも2時間、例えば約2〜約15時間、または約2.5〜約10時間、または約3〜約8時間の範囲内であると考えられる。
【0094】
さらに、本発明の制御放出組成物は、血漿プロフィールの個体間および/または個体内変動の減少、および該組成物が食物と共に摂取するか食物なしに摂取するかに対する依存性の減少(医薬組成物を食物摂取と同時にかまたは同時でなく投与したときのモノメチルフマレートの血漿濃度プロフィールの変動の減少)をもたらす。したがって、本発明の制御放出組成物は、Fumaderm(登録商標)に比べ、投与頻度の減少および/または平均総1日用量の減少、および/または活性物質の同じ総1日用量での効果の増加をもたらし得る。
【0095】
種々の動力学的モデル、例えばゼロ次(1)、1次(2)、平方根(Higuchiの方程式)(3)は、薬物放出動力学の解釈に適用することができる。
1:Mt = M0 + k0*t
2:InMt = InM + k1*t
3:Mt = M0 + kH*t1/2
【0096】
これら方程式において、Mtは、あらゆる特定の時点で放出された薬剤の累積量であり、M0は、医薬組成物に組み込まれる活性物質の用量である。k0、k1、およびkHは、それぞれゼロ次、1次、およびHiguchiの方程式の速度定数である。
【0097】
本発明のある局面は、ゼロ次溶出放出プロフィールに関する。別の局面は1次溶出放出プロフィールに関する。さらなる局面は、平方根(Higuchiの方程式)溶出放出プロフィールに関する。
【0098】
本発明のある局面において、活性物質として10%〜90%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩、2%〜40%(重量)の医薬的に許容されるポリマー、および1%〜40%(重量)の親水性賦形剤、および所望により医薬的に許容される賦形剤または添加剤を含む制御放出医薬組成物を提供する。
【0099】
本発明の別の局面において、活性物質として40%〜60%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩、15%〜25%(重量)医薬的に許容されるポリマー、および2%〜15%(重量)親水性賦形剤、および所望により医薬的に許容される賦形剤または添加剤を含む制御放出医薬組成物を提供する。
【0100】
本発明のさらなる局面において、活性物質として65%〜80%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩、10%〜25%(重量)医薬的に許容されるポリマー、および2%〜15%(重量)親水性賦形剤、および所望により医薬的に許容される賦形剤または添加剤を含む制御放出医薬組成物を提供する。
【0101】
「医薬的に許容されるポリマー」の例には、限定されるものではないがエチルセルロース、またはメタクリル酸/アクリル酸コポリマー、例えばアンモニオメタクリレートコポリマータイプAおよびB、またはメタクリル酸コポリマーAおよびBが含まれる。
【0102】
「親水性賦形剤」の例には、限定されるものではないがポリエチレングリコール(PEG)、ポビドン、ヒドロキシルプロピルセルロース(HPC)、ヒドロキシエチルデンプン(HES)もしくはヒドロキシプロピルメチルセルロース(HPMC)、または同様の特性を有する物質、またはその混合物が含まれる。
【0103】
本発明のさらなる局面において、医薬的に許容されるポリマーがエチルセルロースである制御放出医薬組成物を提供する。
【0104】
本発明の別の局面において、親水性賦形剤がヒドロキシルプロピルセルロースである制御放出医薬組成物を提供する。
【0105】
本発明の別の局面において、親水性賦形剤がポリエチレングリコールである制御放出医薬組成物を提供する。
【0106】
本発明のさらに別の局面において、活性物質として10%〜90%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩、および重量比1:9〜9:1の2%〜40%(重量)メタクリル酸コポリマーAおよびB、および所望により医薬的に許容される賦形剤または添加剤を含む制御放出医薬組成物を提供する。
【0107】
本発明さらなる局面において、50%〜90%の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む制御放出医薬組成物を提供する。
【0108】
本発明の範囲を限定しない本発明を例示する種々の制御放出製剤を以下に記載する(全ての濃度は最終錠剤に基づく):
1)顆粒
顆粒は、濃度約10〜約90%、特に約50〜約70%の活性物質を、造粒用賦形剤、例えば医薬的に許容されるポリマー、例えばエチルセルロース、例えばEthocel(登録商標)NFプレミアム、またはメタクリル酸/アクリル酸コポリマー、例えばアンモニオメタクリレートコポリマータイプAおよびB(重量比1:9〜9:1)またはメタクリル酸コポリマーAおよびB(重量比1:9〜9:1)と濃度約2〜約40%で混合し造粒することにより製造してよい。濃度約1〜約40%の、親水性賦形剤、例えばポリエチレングリコール(PEG)、ポビドン、ヒドロキシルプロピルセルロース(HPC)、ヒドロキシエチルデンプン(HES)もしくはヒドロキシプロピルメチルセルロース(HPMC)、および/または濃度約0.01〜約3%のHLB値8以上の医薬的に許容される界面活性剤を混合してよい。
2)微晶質製剤
【0109】
結晶化は、例えば+70℃〜-20℃のような適切な温度のイソプロパノールのような再結晶のためのあらゆる適切な有機溶媒中で実施する。ヒドロコロイド(例えばHPMC)または界面活性剤(例えばポリソルベート)は、再結晶化中に結晶の成長を操作するのに適切な濃度で用いることができる。あらゆる造粒/コーティング用賦形剤、例えば医薬的に許容されるポリマー、例えば、濃度約10〜約50%、特に約20〜約35%のエチルセルロース、ポリメタクリレート、例えばアンモニオメタクリレートコポリマータイプAおよびB、またはメタクリル酸コポリマーAおよびBを用いてよい。親水性賦形剤は例えばPEG 400であり得る。
3)カプセル剤およびサシェー剤
【0110】
カプセル(例えば、ゼラチン、HPMC、またはデンプン誘導体のカプセル)またはサシェーは、コートした(コート)微結晶またはコート顆粒、および必要ならば適切な量の充填用賦形剤、例えば糖アルコール、例えばマンニトールおよび/または流動促進剤で充填してよい。
4)錠剤
【0111】
錠剤は微結晶または顆粒で作られてよい。大規模に、特に回転装置(rotary machine)を用いて錠剤を製造する時は、さらに流動活性を増加しまたは錠剤化挙動を改善するための賦形剤が必要かもしれない。必要であれば充填用および結合用(binding)賦形剤として、例えば、濃度約1〜約60%の微晶質セルロース、例えばAvicel(登録商標)102、およびセルロース、濃度約5〜約60%の、結晶、スプレー乾燥または造粒ラクトース1水和物、例えばTablettose(登録商標)、および無水ラクトース1水和物、濃度約0〜約40%の糖アルコール、例えばソルビトールおよびマンニトール、および濃度約0〜約40%の修飾デンプンを挙げることができる。さらに、崩壊剤、例えばデンプンおよびデンプン誘導体、例えばナトリウムデンプングリコレート(濃度約0.2〜約10%)、クロスポビドン(濃度約0.2〜約10%)、ナトリウムカルボキシメチルセルロース(濃度約0.1〜約10%)、流動促進剤、例えばコロイド無水および含水シリカ(濃度約0.2〜約4%)、および潤滑剤、例えばステアリン酸マグネシウム、ベヘン酸カルシウム、およびカルシウムアラキネート(濃度約0.2〜約3%)またはナトリウムステアリルフマレート(濃度約1〜約8%)を加えることができる。
用 量
【0112】
存在するフマル酸の含有量が異なる組成物を提供するだけでなく、本発明は、例えば、種々の量のフマル酸を有する組成物を含む2またはそれ以上の容器を含むキットも提供する。そのようなキットは、時間とともに増加する用量が必要な状況に用いるのに適している。用量の通常のアップスケールを以下に示す。
【0113】
Aは、低強度、例えば約30mgジメチルフマレート(または別のフマル酸エステルの対応する有効用量)に対応する。
【0114】
Bは、高強度、例えば約120mgジメチルフマレート(または別のフマル酸エステルの対応する有効用量)に対応する。
【0115】
本発明のある局面において、剤形中のフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩の量が活性物質90mg〜360mg、例えば活性物質90、120、180、240、または360mgである制御放出医薬組成物を提供する。本発明のさらなる局面において、活性物質の量は活性物質120、180、または240mgである。本発明のさらなる局面において、活性物質の量は180または360mgである。
【0116】
患者を治療するのに投与される本発明の制御放出医薬組成物の1日投与量は、限定されるものではないが体重および年齢、および治療する病状または疾患の根本原因を含む多くの因子に依存し、決定する医師の技術内にある。本発明のある局面において、1日投与量は、例えば1〜3用量で投与する活性物質240〜360mg、別の局面では1〜3用量で投与する活性物質360〜480mg、別の局面では1〜3用量で投与する活性物質480〜600mg、別の局面では1〜3用量で投与する活性物質600〜720mg、別の局面では1〜3用量で投与する活性物質720〜840mg、別の局面では1〜3用量で投与する活性物質840〜960mgで、さらに別の局面では1〜3用量で投与する活性物質960〜1080mgであり得る。
本発明のある局面において、該制御放出医薬組成物はカプセル形である。
【0117】
本発明の別の局面において、錠剤形、例えば患者が飲み込むのを簡単および好都合にする形を有する錠剤、例えばいかなる尖った角もない丸または棒状の形を有する錠剤の形の該制御放出医薬組成物が提供される。
【0118】
本発明の別の局面において、2またはそれ以上の部分に分割されるよう設計された錠剤の形の医薬組成物を提供する。
【0119】
本発明の組成物は、食事と一緒に、または例えば食事前少なくとも約30分〜食事後約2時間の範囲に対応する時間内に食事に関連して投与するか、または該組成物をその日の時間内のあらゆる特定の時点で投与してよい。
【0120】
ある態様において、総1日投与量は、就寝時刻、例えば就寝時刻前約30分またはそれ以内、就寝時刻前約60分またはそれ以内、就寝時刻前約90分またはそれ以内、就寝時刻前約120分またはそれ以内、または就寝時刻前約180分またはそれ以内に投与される。
【0121】
本発明の組成物およびキットは1またはそれ以上の以下の病状の治療に用いるのに適していると予期される:
a. 乾癬
b. 乾癬性関節炎
c. 神経皮膚炎
d. 炎症性腸疾患、例えば
i. クローン病
ii. 潰瘍性大腸炎
e. 自己免疫疾患:
i. 多発性関節炎
ii. 多発性硬化症(MS)
iii. 若年発症糖尿病
iv. 橋本甲状腺炎
v. バセドウ病
vi. SLE(全身性エリテマトーデス)
vii. シェーグレン症候群
viii. 悪性貧血
ix. 慢性活動性(狼瘡)肝炎
x. 関節リウマチ(RA)
xi. 視神経炎。
【0122】
さらに、本発明の新規組成物またはキットは以下の治療に用いることができよう:
1. 痛み、例えば神経根痛、神経根障害関連痛、ニューロパシー痛、または坐骨神経痛/坐骨神経痛の痛み
2. 臓器移植(拒絶の予防)
3. サルコイドーシス
4. リポイド類壊死症
5. 環状肉芽腫。
【0123】
乾癬は以下の病状に関連する可能性があると提唱されてきた:クローン病(Najarian DJ、Gottlieb AB、Connections between psoriasis and Crohn's disease. J Am Acad Dermatol. 2003 Jun;48(6):805-21)、セリアック病(Ojetti V et al、High prevalence of celiac disease in psoriasis. Am J Gastroenterol. 2003 Nov;98(ll):2574-5.)、精神的または心理的疾患、例えば鬱病または人生の危機(Gupta MA、Gupta AK、Psychiatric and psychological co-morbidity in patients with dermatologic disorders:epidemiology and management. Am J Clin Dermatol. 2003;4(12):833-42、およびMallbris L et al、psoriasis phenotype at disease onset:clinical characterization of 400 adult cases. J Invest Dermatol. 2005 Mar; 124(3):499-504.)、肥満、糖尿病、アルコールの過剰摂取/アルコール中毒、および乾癬性関節炎。
【0124】
すなわち、本発明は、ある局面において、乾癬、乾癬性関節炎、神経皮膚炎、炎症性腸疾患、例えばクローン病および潰瘍性大腸炎、自己免疫疾患、例えば多発性関節炎、多発性硬化症(MS)、若年発症糖尿病、橋本甲状腺炎、バセドウ病、SLE(全身性エリテマトーデス)、シェーグレン症候群、悪性貧血、慢性活動性(狼瘡)肝炎、関節リウマチ(RA)および視神経炎、痛み、例えば神経根痛、神経根障害関連痛、ニューロパシー痛、または坐骨神経痛/坐骨神経痛の痛み、臓器移植(拒絶の予防)、サルコイドーシス、リポイド類壊死症、または環状肉芽腫の治療方法であって、有効量の本発明制御放出医薬組成物をそれを必要とする患者に経口投与することを含む方法に関する。
【0125】
本発明は、別の局面において、乾癬、乾癬性関節炎、神経皮膚炎、炎症性腸疾患、例えばクローン病および潰瘍性大腸炎、自己免疫疾患、例えば多発性関節炎、多発性硬化症(MS)、若年発症糖尿病、橋本甲状腺炎、バセドウ病、SLE(全身性エリテマトーデス)、シェーグレン症候群、悪性貧血、慢性活動性(狼瘡)肝炎、関節リウマチ(RA)および視神経炎、痛み、例えば神経根痛、神経根障害関連痛、ニューロパシー痛または坐骨神経痛/坐骨神経痛の痛み、臓器移植(拒絶の予防)、サルコイドーシス、リポイド類壊死症または環状肉芽腫を治療するための医薬を製造するための本発明制御放出医薬組成物の使用に関する。
【0126】
さらに、本発明は、本発明の組成物またはキットを用いる、上記病状の1つ、より具体的には乾癬または乾癬性関節炎に罹患した個体の治療にも関するものであり、該個体はさらに以下で治療される。
a)局所抗乾癬薬、例えば1)ビタミンDまたはその誘導体(カルシポトリオール、カルシポトリエン)、2)コルチコステロイド(例えば、ベタメタゾン、デソキシメタゾン、フルオシノロン、モメタゾン、ヒドロコーチゾンアセポネート、フルチカゾン、クロベタゾール、クロベタゾン、ヒドロコーチゾンブチレート、デソニド、トリアムシノロン、またはヒドロコーチゾン)、3)タザロテン、4)ジトラノール、5)タクロリムス(FK-506)、および他のカルシネウリン阻害剤、例えばピメクロリムス、または6)1〜5のあらゆる組み合わせ、および/または
b)経口抗乾癬薬、例えば1)経口レチノイド(例えばアシトレチンまたはエトレチネート)(PUVAと組み合わせてまたは組み合わせずに)、2)シクロスポリンおよび他のカルシネウリン阻害剤、例えばISA247、タクロリムスおよびピメクロリムス、3)メトトレキセート、4)ヒドロキシウレア、5)アザチオプリン、6)スルファサラジン、7)フマレート誘導体(例えば、Fumaderm(登録商標)またはBG-12)、8)ロシグリタゾン(Avandia)および他のペルオキシソーム増殖因子活性化γ(PPARγ)アゴニストまたはモジュレーター、例えばピオグリタゾン、ファルグリタザル、GW1929、GW7845、MC-555、MBX-102/MBX-10、MBX-1828、MBX-2044、CLX-0921、R-483、レグリタザル、ナベグリタザル(LY-519818/LY-818)、ネトグルタゾン(MCC-555)、CS-7017、トログリタゾン、シグリタゾン、テサグルタザル、イサグリタゾン、バラグリタゾン、ムラグリタザル、TAK-654、LBM642、DRF 4158、EML 4156、T-174、TY-51501、TY-12780、VDO-52、またはAMG-131(T131)、または1〜8のあらゆる組み合わせ、および/または
c)非経口投与抗乾癬薬、例えば1)アレファセプト(Amevive)、2)エタネルセプト(Enbrel)、3)エファリズマブ(Raptiva)、4)オネルセプト、5)アダリムマブ(Humira)、または1〜5のあらゆる組み合わせ、および/または
d)腸内または非経口経路で投与される、上記c)項に記載していないTNFα阻害剤(例えば、CDP 870またはインフリキシマブ(Remicade))、および/または
e)チソカリシトレート、および/またはNCX 1022および/またはIDEC-131および/またはMEDI-507、および/または
f)NSAID、またはCOXまたはLOX阻害剤、例えば、COX-2阻害剤またはCOX/5-LOX阻害剤、および/または
g)抗糖尿病薬または抗肥満薬、例えばビグアニド、例えばメトホルミン;メトホルミンXR;スルホニルウレア、例えばクロルプロパミド、グリピジド、グリクラジド、グリブリド/グリベンクラミドまたはグリメピリド;グルコバンス(メトホルミン+フリブリド);メタグリプ(グリピジド+メトホルミン);ペルオキシソーム増殖因子活性化γ(PPARγ)アゴニストまたはモジュレーター、例えばロシグリタゾン(Avandia)、ピオグリタゾン、ファルグリタザル、GW1929、GW7845、MC-555、MBX-102/MBX-10、MBX-1828、MBX-2044、CLX-0921、R-483、レグリタザル、ナベグリタザル(LY-519818/LY-818)、ネトグリタゾン(MCC-555)、CS-7017、トログリタゾン、シグリタゾン、テサグリタザル、イサグリタゾン、ベラグリタゾン、ムラグリタザル、TAK-654、LBM642、DRF 4158、EML 4156、T-174、TY-51501、TY-12780、VDO-52、またはAMG-131(T131);アバンダメト(ロシグリタゾン+メトホルミン);アクトス(ピオグリタゾン+メトホルミン);アバンダリル(ロシグリタゾンマレエート+グリメピリド);アクトス(ピグリタゾン+メトホルミン);アバンダリル(ロシグリタゾンマレエート+グリメピリド);ベンゾイミダゾール、例えばFK-614;CS-917;TA-1095;ONO-5129;TAK-559;TAK-677/AJ-9667;d-フェニルアラニン誘導因子、例えばセナグリニド;c-3347;NBI-6024;イングリホリブ;BVT 3498;LY 929;SGLT2阻害剤;CS 011;BIM 51077;R1438;R1439;R1440;R1498;R1499;AVE0847;AVE 2268;AVE 5688;AVE 8134;TA-6666;AZD 6370;SSR 162369;TLK-17411;NN 2501;MK 431;KGA-2727;MK-767;CS-872;β-3レセプターアンタゴニスト、例えばN-5984;α-グルコシダーゼ阻害剤、例えばアカルボース、ボグリボーズ、またはミグリトール;グリニチド/メグリチニド類似体またはカルバモイルメチル安息香酸(bensoeic acid)誘導体、例えばミチグリニド、レパグリニド、またはナテグリニド;DPP-IV阻害剤、例えばLAF 237(ビルダグリプチン)、DPP728、P93/01、P32/98、PT-630、またはサキサグリプチン;GLP-1またはGLP-1類似体、例えばエキセナチド、エキセナチド-LAR、リラグルチド(NN 2211)、ZP10/AVE0010、LY307161、ベタトロピン、CJC-1131、GTP-010、SUN E7001、またはAZM 134;プラムリニチドアセテート;インスリンまたはインスリン類似体、例えばヒューマログ(インスリンリスプロ)、ヒューマリン、ノボリン、ノボログ/ノボラピッド(インスリンアスパート)、アピドラ(インスリングルイシン)、ランタス(インスリングラルギン)、エクスベラ、レベミル/NN 304(インスリンデテミル)、AERx/NN 1998、インスマン、肺インスリン、またはNN 344;シブトラミン、または他のセロトニンおよびノルアドレナリンのシナプス前再取り込み遮断薬;オルリスタット、および他のGIリパーゼ阻害剤;β3-アドレナリンレセプターアゴニスト;脱共役タンパク質;PPARγの(特異的)アンタゴニスト(ペルオキシソーム増殖因子活性化レセプターγ);インスリン分泌促進剤;リモナバントおよび他のCB1エンドカンナビノイドアンタゴニスト;ブプロピオン;トピラメート;レプチンアゴニスト;毛様体神経栄養因子;ヒトペプチドホルモン断片のペプチド類似体177-191;コレシストキニン-Aレセプターアゴニスト;メラノコルチン-3アゴニスト;ノルアドレナリン作動薬、例えばフェンテルミン、ジエチルプロピオン、フェンジメトラジン、またはベンズフェタミン;または上記抗糖尿病薬または抗肥満薬のあらゆる組み合わせ、および/または
h)物質乱用、例えばアルコール乱用の治療に潜在的に有用な薬剤、例えばナルトレゾン、アカンプロセート、ジスルフィラム、またはビビトレックス(ナルトレゾン長時間作用注射)、および/または
i)クローン病の治療に潜在的に有用な薬剤、例えば
1. 5-ASA化合物、例えばスルファサラジン、経口5-ASA製剤、または直腸5-ASA製剤、
2. グルココルチコステロイド、例えば全身性ステロイド(例えば、ブデソニドまたはプレドニゾロン)または局所作用ステロイド(例えば、ブデソニド)、
3. 抗生物質、例えばメトロニダゾールまたはキノロン(例えば、シプロフロキサシン、オフロキサシン、ノルフルキサシン、レボフロキサシン、またはモキシフロキサシン)、
4. 免疫抑制剤、例えばアザチオプリン、6-メルカプトプリン、またはメトトレキセート、
5. 栄養療法、例えば元素または高分子製剤、またはプレ-およびプロバイオティクス、
6. 生物療法、例えば、TNF-α阻害剤、例えばインフリキシマブ、アダリムマブ、CDP870、CDP571、エタネルセプト、またはオネルセプト、
7. 対症療法剤、例えば抗下痢剤または抗痙攣剤。
【0127】
適切なNSAIDの例には、ピロキシカム、ジクロフェナク、ナブメトン、ナプロキセン、フルルビプロフェン、フェノプオフェン、ケトプロフェン、およびイブプロフェンを含むプロピオン酸、メフェナム酸を含むフェナメート、パラセタモール、インドメタシン、スリンダク、メロキシカム、アパゾン、フェニルブタゾンを含むピラゾロン、アスピリンを含むサリチル酸塩がある。
【0128】
適切なCOX-2阻害剤の例には、ロフェコキシブ(Vioxx)、バルデコキシブ(Bextra)、セレコキシブ(Celebrex)、エトリコキシブ(Arcoxia)、ルミラコキシブ(Prexige)、パレコキシブ(Dynastat)、デラコキシブ(Deram)、チラコキシブ、メロキシカム、ニメソリド、(1,1-ジメチルヘプチル)-6a,7,10,10a-テトラヒドロ-1-ヒドロキシ-6,6ジメチル-6H-ジベンゾ[b,d]ピランカルボン酸(CT-3)、2(5H)-フラノン、5,5-ジメチル(l-メチルエトキシ)[4(メチルスルホニル)フェニル]-(DFP);カプロフェン(RIMADYL)、(アセチルオキシ)-安息香酸、3-[(ニトロオキシ)メチル]フェニルエステル(NCX4016)、P54(CAS Reg. No.130996 0) 2,6-ビス(1,1-ジメチルエチル)[(E)-(2-エチル-1,1-ジオキソイソチアゾリジニリデン)メチル]フェノール(S-2474)、5(R)-チオスルホンアミド-3(2H)-ベンゾフラノン(SVT-2016)、およびN-[3-(ホニル-アミノ)オキソフェノキシ-4H ベンゾピラン イル]メタンスルホンアミド(「T-614」);またはその医薬的に許容される塩がある。
【0129】
適切なCOX/5-LOX阻害剤の例には、リコフェロン(ML-3000または[2,2-ジメチル-6-(4-クロロフェニル)-7-フェニル-2,3,ジヒドロ-1H-ピロリジン-5-イル]-酢酸)、ジ-tert-ブチルフェノール、例えば(E)-(5)-(3,5-ジ-tert-ブチル-4-ヒドロキシベンジリデンス)-2-エチル-l,2-イソチアゾリジン-1,1-ジオキシド(S-2474)、ダルブフェロンまたはテブフェロン、および薬理学的に活性な代謝物ならびに誘導体、例えばジヒドロ-ジメチル-ベンゾフランおよびPGV-20229、ジヒドロ-ジメチル-ベンゾフラン、チオフェン誘導化合物、例えばRWJ-63556、N-ヒドロキシ-N-メチル-4-(2,3-ビス-(4-メトキシフェニル)-チオフェン-5-イル)-ブタンアミド(S19812)、メトキシテトラヒドロピラン誘導体、酸化キサントン、例えば1,3,6,7-テトラヒドロキシキサントン(ノルアチリオール(norathyriol))-ピラゾールチオカルバメート、ピラゾール、例えば修飾形のフェニドン含有化合物、またはトリ-フルオロ-ベンゾール置換ピラゾリン誘導体BW-755C、テポキサリンおよび誘導体、およびジ-tert-ブチルピリミジンがある。
【0130】
そのような併用療法は、本発明の組成物またはキットを用いずに治療される個体に比べて個体に対する治療反応の改善および/または利便性の増大をもたらすと予期される。
【0131】
さらなる局面において、本発明はあらゆる上記病状a〜eおよび1〜5の経口治療に伴う副作用を減少させる方法であって、該病状を治療するための医薬成分が1またはそれ以上の以下の薬剤と組み合わせて用いられる方法に関する:
a)制酸剤、例えば1)水酸化マグネシウム、2)三ケイ酸マグネシウム、3)水酸化アルミニウムゲル、3)炭酸水素ナトリウム、4)マガルドラト、または1〜5のあらゆる組み合わせ、および/または
b)ヒスタミンH-2アンタゴニスト、例えば1)シメチジン、2)ラニチジン、3)ニザチジン、4)ファモチジン、5)ロキサチジン、6)ラフタジン、または1〜6のあらゆる組み合わせ、および/または
c)細胞保護剤、例えば1)スクラルファート、2)トリカリウムジクチトラトビスムテート、3)カルベノキソロン、4)プロスタグランジンE-2類似体、例えばミソプロストール、5)エカベト、6)セトラキセートHCl、7)テプレノン、8)トロキシピド、9)ジシクロミン塩酸、10)ソファルコンまたは1〜10のあらゆる組み合わせ、および/または
d)プロトンポンプ阻害剤(PPI)、例えば1)オメプラゾール、2)エソメプラゾール、3)ランソプラゾール、4)パントプラゾール、5)ラベプラゾール、6)CS-526/R-105266、7)AZD 0865、8)ソラプラザン、または1〜8のあらゆる組み合わせ、および/または
e)NSAIDまたはCOXまたはLOX阻害剤、例えば、COX-2阻害剤またはCOX/5-LOX阻害剤、および/または
f)ペントキシフィリン(例えば用量範囲400〜800mg/日で)。
【0132】
具体的態様において、活性物質はフマル酸エステル含有化合物である。具体的には、フマル酸エステル含有化合物は、Fumaderm(登録商標)またはFumaraat(登録商標)またはPanaclar(登録商標)(BG-12)に含まれるか、US 6,277,882、US 6,355,676もしくはUS 6,509,376に記載の、または本発明の製剤に含まれる塩のいずれかおよびすべてである。活性医薬成分は、本発明の製剤、またはあらゆるFumaderm(登録商標)またはFumaraat(登録商標)またはPanaclar(登録商標)製剤において、または例えば、US 6,277,882、US 6,355,676またはUS 6,509,376に記載のごとく得ることができよう。
【0133】
本発明は、記載した具体的態様に限定されず、もちろん変化し得ると理解すべきである。本発明の範囲は添付の特許請求の範囲にのみ限定されるので、本明細書で用いる専門用語は、特定の態様を説明するためのみのものであり、限定を意図するものではないと理解すべきである。値の範囲が示されている場合は、該範囲の上限と下限の間のそれぞれの間の値(文脈が明確にそうでないと記載していない限り、下限の単位の1/10まで)、およびあらゆる他の記載されるかまたは範囲が記載された間の値も本発明内に含まれると理解すべきである。このより小さい範囲の上限および下限は、独立して該より小さい範囲に含まれ、本発明内に含まれる(記載した範囲内のあらゆる具体的に除外した制限を受ける)。記載した範囲が該限界の1または両方を含む場合は、その含まれる限界のいずれかまたは両方を排除する範囲も本発明に含まれる。特記しない限り、本明細書で用いるすべての技術的および科学的用語は、本発明が属する技術分野の当業者が通常理解するのと同じ意味を有する。本明細書に記載のものと同様または等価なあらゆる方法および物質を本発明の実施または試験に用いることもできるが、好ましい方法および物質を記載する。本明細書に記載のすべての刊行物は、本明細書の一部を構成し、該刊行物が引用している方法および/または物質を開示し説明する。本明細書および添付の特許請求の範囲で用いている単数形の「a」、「an」および「the」は、文脈が明確にそうでないと記載していない限り複数形の言及を含むと理解すべきである。本明細書に記載の特許および刊行物は、単に本出願の出願日以前の開示内容を示すものである。本明細書のいかなる内容も、本発明が以前の発明に基づいてそのような特許または刊行物に先行することはできないと認めるものと解釈すべきでない。さらに、示した刊行物の日付は、実際の刊行日と異なることがあり、別々に確認する必要がありうる。この開示を読んだ当業者に明らかであろうように、本明細書に記載し、例示した個々の各態様は、別個の要素と特徴を有し、本発明の範囲や精神から離れることなく他のあらゆる種々の態様の特徴から容易に分離または結合してよい。本明細書に示す図面は必ずしも一定の縮尺を示すものではなく、ある要素および特徴は明確さのために誇張されている。
【0134】
前記発明について理解を明確にするために例示および実施例により詳細に説明したが、本発明の開示に照らして添付の特許請求の範囲の精神や範囲から離れることなくある種の変更および修飾を行ってよいことは当業者は容易に明白であろう。
【実施例】
【0135】
実施例1
錠剤の製造
200g顆粒を、150g微晶質セルロース(例えば、Avicel(登録商標) 102)、97.5gラクトース(例えば、Tablettose(登録商標))、10gナトリウムカルボキシメチルセルロース(例えば、Ac-Di-Sol(登録商標))、および25gデンプンと30分間混合する。次に、10gステアリン酸マグネシウムおよび7.5g非晶質二酸化ケイ素(例えば、Aerosil(登録商標) 200)を加え、粉末混合物を5分間混合する。
【0136】
この粉末混合物を打錠器で圧縮して錠剤にする(錠剤直径10mm、表面約280-300mm2)。該錠剤を実施例4に記載のごとくパンコーティングまたは流動コーティング(fluid-bed coating)法を用いて腸溶コートする。
実施例2
錠剤の製造
【0137】
200g微結晶を150g微晶質セルロース(例えば、/Avicel(登録商標) 102)、130gラクトース(例えば、Tablettose(登録商標))、10gナトリウムカルボキシメチルセルロース(例えば、Ac-Di-Sol(登録商標))、および25mgデンプンと30分間混合する。次に、10gステアリン酸マグネシウムおよび7.5g非晶質二酸化ケイ素を加え、次いで粉末混合物を5分間混合物する。この粉末混合物を打錠器で圧縮して錠剤にする(錠剤直径10mm、表面約280-300mm2)。該錠剤を実施例4に記載のごとくパンコーティングまたは流動コーティング(fluid-bed coating)法を用いて腸溶コートする。
実施例3
カプセルの製造
【0138】
顆粒または微結晶をHPMCカプセルに充填し、該カプセルを以下に記載のごとく腸溶コートする。パンコーティング器を用い、Eudragit(登録商標) L30D-55を60℃〜80℃の乾燥温度でポリマー物質20mg/mm2の量でカプセル上にスプレーする。色素およびタルクを適当量加える。
実施例4
錠剤の腸溶コーティング
【0139】
パンコーティング器を用い、Eudragit(登録商標) L30D-55を60℃〜80℃の乾燥温度でポリマー物質6mg/mm2の量で錠剤上にスプレーする。色素およびタルクを適当量加える。
実施例5
カプセルの製造
【0140】
実施例15に記載のごとく製造した156mgの微結晶を硬ゼラチンカプセルサイズ0に充填する。カプセル剤を、各カプセル面を4回、アセトン中の5% HPMCP(Pharmacoat HP 50(登録商標))の溶液に浸漬して腸溶コートする。
実施例6
顆粒の製造
【0141】
造粒法を用い、50gジメチルフマレート(以下のDMF中)を1gエチルセルロース(例えば、Ethocel(登録商標)NFプレミアム)と混合し、これを10mLエタノール96%に溶解し、シーブ1.0mmを通し、次いで50℃〜60℃で30分間乾燥する。この顆粒を実施例1および3に記載したのと同じ方法を用いて錠剤およびカプセルに製造する。
実施例7
顆粒の製造
【0142】
造粒法を用い、50g DMFを1gポリビニルアセテート(PVA)(例えば、Kollicoat(登録商標) SR30)と混合し、これを10mLエタノール96%に溶解し、シーブ1.00mmを通し、次いで50℃〜60℃で30分間乾燥する。
実施例8
顆粒の製造
【0143】
造粒法を用い、50g DMFを15gの粉末Eudragit(登録商標) RL 100と混合する。適切な量の2-プロパノールを加え、シーブ1.00mmを通し、次いで60℃で乾燥する。この顆粒を実施例1および3に記載したのと同じ方法を用いて錠剤およびカプセルに製造する。
実施例9
コート顆粒の製造
【0144】
造粒法を用い、50g DMFを直接5g Eudragit(登録商標) RL30Dと混合し、シーブ(1.00mm)を通し、次いで80℃で乾燥する。シーブを通した後、顆粒を流動コーティング器(Mini-Glatt)を用い、15gのEudragit(登録商標) RL30D/RS30D、1:1混合物でコートする。コート顆粒を実施例1および3に記載したのと同じ方法を用いて錠剤およびカプセルに製造することができる。
実施例10
コート顆粒の製造
【0145】
造粒法を用い、50g DMFを20%エチルセルロース(例えば、Ethocel(登録商標)NFプレミアム)と混合し、これを適切な量のエタノール96%に溶解する。15%ポリエチレングリコール6000を造粒液に加える。混合物をシーブ1.00mmに通し、50℃〜60℃で30分間乾燥する。シーブを通した後、顆粒を流動コーティング(Mini-Glatt)を用い、20mg/mm2顆粒表面積の量のエチルセルロースとポリエチレングリコール6000の2:1混合物でコートする。この顆粒を実施例1および3に記載したのと同じ方法を用いて錠剤またはカプセルに製造することができる。
実施例11
コート顆粒の製造
【0146】
造粒法を用い、50g DMFを10%エチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)と混合し、次いで適切な量のエタノール96%に溶解する。6%ポビドン(例えば、Kollidon(登録商標) 25)を造粒液に加える。混合物をシーブ1.00mmに通し、50℃〜60℃で30分間乾燥する。シーブを通した後、顆粒を流動コーティング(Mini-Glatt)を用い、20mg/mm2顆粒表面積の量のエチルセルロースとポビドンの3:2混合物でコートする。
この顆粒を実施例1および3に記載した方法を用いて錠剤またはカプセルに製造することができる。
実施例12
コート顆粒の製造
【0147】
造粒法を用い、50g DMFを10%エチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)と混合し、次いで適切な量のエタノール96%に溶解する。5%ヒドロキシプロピルセルロース(HPC)(例えば、Klucel(登録商標))を造粒液に加える。混合物をシーブ1.00mmに通し、50℃〜60℃で30分間乾燥する。シーブを通した後、顆粒を流動コーティング(Mini-Glatt)を用い、20mg/mm2顆粒表面積の量のエチルセルロースとHPCの2:1混合物でコートする。
この顆粒を実施例1および3に記載した方法を用いて錠剤またはカプセルに製造することができる。
実施例13
コート顆粒の製造
【0148】
造粒法を用い、50g DMFを直接適切な量のEudragit(登録商標) NE30Dの水性分散液と混合し、シーブ(1.00mm)を通し、次いで80℃で乾燥する。シーブを通した後、顆粒を流動コーティング器(Mini-Glatt)を用い、15gのEudragit(登録商標) RL30D/RS30D、1:1混合物でコートする。コート顆粒を実施例1および3に記載した方法を用いて錠剤およびカプセルに製造することができる。
実施例14
コート顆粒の製造
【0149】
造粒法を用い、50g DMFを直接適切な量のEudragit(登録商標) RL30Dの水性分散液と混合し、シーブ(1.00mm)を通し、次いで80℃で乾燥する。シーブを通した後、顆粒を流動コーティング器(Mini-Glatt)を用い、Eudragit(登録商標) NE30Dでコートする。コート顆粒を実施例1および3に記載した方法を用いて錠剤およびカプセルに製造することができる。
実施例15
コート微結晶の製造
【0150】
300mL 2-プロパノール中の50g DMFの飽和溶液を60℃で調製し、永続的に撹拌しながら徐々に冷却する。沈殿した結晶を濾過して除去し、50℃で乾燥する。結晶をシーブに通し、315〜710μm分画をパンコーティング器または流動コーティング器(Mini-Glatt)を用いるコーティング工程に用いる。500gエタノール中の、12gエチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)および3gポリエチレングリコール400のコーティング溶液を粉末表面上に60℃でスプレーする。乾燥後、コートした結晶を1.00mmシーブに通す。このコートDMF結晶を実施例2および3に記載の方法を用いて錠剤およびカプセルに製造することができる。
実施例16
錠剤の製造
【0151】
造粒法を用い、50g DMFを2gエチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)および3gポリエチレングリコール400と混合し、これを150mLエタノール96%に溶解し、1.0mmシーブを通し、50〜60℃で30分間乾燥し、再度1.0mmシーブに通す。プラセボ顆粒を以下のごとく製造する:Tablettose(登録商標)およびAvicel(登録商標)102を等量混合し、水(q.s.)に溶解した2%ポビドン(例えば、Kollidon(登録商標)25)で顆粒とし、1.0mmシーブを通し、50℃〜60℃で30分間乾燥し、再度1.0mmシーブに通す。DMF-顆粒60部およびプラセボ顆粒38部をTurbulaシェーカーミキサー中で30分間混合する。Aerosil(登録商標)200を1部およびステアリン酸マグネシウム1部を加え、混合物を再度5分間混合する。混合物を直径10mm、重量約260mg、および硬さ約50Nの錠剤に圧縮する。錠剤を実施例4に記載の方法を用いて腸溶コートする。
実施例17
錠剤の製造
【0152】
造粒法を用い、50g DMFを12gエチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)および3gポリエチレングリコール400と混合し、これを150mLエタノール96%に溶解し、1.0mmシーブに通し、50〜60℃で30分間乾燥し、再度1.0mmシーブに通す。プラセボ顆粒を以下のごとく製造する:Tablettose(登録商標)およびAvicel(登録商標)102を等量混合し、水(q.s.)に溶解した2%ポビドン(例えば、Kollidon(登録商標)25)で顆粒とし、1.0mmシーブに通し、50℃〜60℃で30分間乾燥し、再度1.0mmシーブに通す。DMF-顆粒60部およびプラセボ顆粒37部をTurbulaシェーカーミキサー中で30分間混合する。カルボキシメチルセルロース(例えば、Ac-Di-Sol(登録商標))1部、Aerosil(登録商標)200を1部およびステアリン酸マグネシウム1部を加え、混合物を再度5分間混合する。
混合物を直径10mm、重量約260mg、および硬さ約50Nの錠剤に圧縮する。錠剤を実施例4に記載の方法を用いて腸溶コートする。
実施例18
コート微結晶の製造
【0153】
300ml 2-プロパノール中の50g DMFの飽和溶液を60℃で調製し、永続的に撹拌しながら徐々に冷却する。沈殿した結晶を濾過して除去し、50℃で乾燥する。結晶をシーブに通し、315〜710μm分画をパンコーティング器または流動コーティング器(Mini-Glatt)を用いるコーティング工程に用いる。500gエタノール中の、12gエチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)および3gポビドン(PVP)のコーティング溶液を結晶表面上に60℃でスプレーする。乾燥後、コートした結晶を1.00mmシーブに通す。コートDMF結晶を実施例2および3に記載の方法を用いて錠剤およびカプセルに製造することができる。
実施例19
コート微結晶の製造
【0154】
300ml 2-プロパノール中の50g DMFの飽和溶液を60℃で調製し、永続的に撹拌しながら徐々に冷却する。沈殿した結晶を濾過して除去し、50℃で乾燥する。結晶をシーブに通し、315〜710μm分画をパンコーティング器または流動コーティング器(Mini-Glatt)を用いるコーティング工程に用いる。500gエタノール中の、12gエチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)および3gヒドロキシルプロピルセルロース(HPC)のコーティング溶液を粉末表面上に60℃でスプレーする。乾燥後、コートした結晶を1.00mmシーブに通す。コートDMF結晶を実施例2および3に記載の方法を用いて錠剤およびカプセルに製造することができる。
実施例20
微結晶の製造
【0155】
DMF結晶を実施例15に記載のごとく製造する。ただし、結晶質量に対し2%のエチルセルロースを結晶が沈殿する前に2-プロパノールに直接加える。
実施例21
コート微結晶の製造
【0156】
実施例15に記載のごとく調製した50g DMF結晶を流動コーティング器(Mini-Glatt)を用い、80℃の温度で20gのEudragit(登録商標) RL30D/RS30D、1:1混合物の水性分散液でコートする。コートDMF結晶を実施例2および3に記載した方法を用いて錠剤およびカプセルに製造する。
実施例22
錠剤の製造
【0157】
実施例15に記載のごとく調製したDMF結晶を、25%固体Eudragit(登録商標)RS PO/RL PO(比1:2)と直接混合し、実施例2に記載のごとく錠剤を製造する。
実施例23
コート微結晶の製造
【0158】
実施例15に記載のごとく調製したDMF結晶を、流動コーティング器(Mini-Glatt)を用い、5%(結晶の質量に対して)の量のポリビニルアセテートの水性分散液(例えば、Kollicoat(登録商標) SR30D)でコートする。コートDMF結晶を実施例2および3に記載の方法を用いて錠剤およびカプセルに製造することができる。
実施例24
顆粒の製造
【0159】
造粒法を用い、50g DMFを15%エチルセルロース(例えば、Ethocel(登録商標)NFプレミアム)と混合し、これを適切な量のエタノール96%に溶解する。10%ポリエチレングリコール6000を造粒液に加える。混合物をシーブ1.00mmに通し、50℃〜60℃で30分間乾燥する。この顆粒を実施例1および3に記載した方法を用いて錠剤またはカプセルに製造することができる。
実施例25
顆粒の製造
【0160】
造粒法を用い、50gジエチルフマレート(DEF)を15%エチルセルロース(例えば、Ethocel(登録商標)NFプレミアム)と混合し、これを適切な量のエタノール96%に溶解する。10%ポリエチレングリコール6000を造粒液に加える。混合物をシーブ1.00mmに通し、50℃〜60℃で30分間乾燥する。この顆粒を実施例1および3に記載した方法を用いて錠剤またはカプセルに製造することができる。
実施例26
錠剤の製造
【0161】
PEG 6000の代わりに10%ポビドン(例えば、Kollidon(登録商標) 25)を加える以外は実施例24に記載のごとく顆粒を製造する。この混合物を実施例1および3に記載した方法を用いて錠剤またはカプセルに製造することができる。
実施例27
錠剤の製造
【0162】
PEG 6000の代わりに10%ヒドロキシルプロピルメチルセルロースを加える以外は実施例24に記載のごとく顆粒を製造する。この混合物を実施例1および3に記載した方法を用いて錠剤またはカプセルに製造することができる。
実施例28
【0163】
実施例15に記載のごとく製造した50g DMF結晶を流動コーティング器(Mini-Glatt)を用い、80℃の温度で20gのEudragit(登録商標) RL30D/RS30D、1:1混合物の水性分散液でコートする。コート結晶を以下に記載のごとくパンコーティング器で腸溶コートする。Eudragit(登録商標) L30D-55を6mgポリマー物質/mm2の量で乾燥温度60℃〜80℃でコート結晶上にスプレーする。
【0164】
この二重コートDMF結晶を硬ゼラチンカプセルまたは軟ゼラチンカプセルに充填するか、または実施例2に記載の方法を用いて錠剤とする。
実施例29
錠剤の製造
【0165】
造粒法を用い、50g DMFを12gエチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)および3gヒドロキシプロピルセルロース(例えば、Klucel(登録商標))と混合し、これを150mLエタノール96%に溶解し、1.0mmシーブに通し、50〜60℃で30分間乾燥し、再度1.0mmシーブに通す。
【0166】
Tablettose(登録商標)およびAvicel(登録商標)102を等量混合し、水(q.s.)に溶解した2%ポビドン(例えば、Kollidon(登録商標)25)で顆粒とする。DMF-顆粒60部およびプラセボ顆粒38部をTurbulaシェーカーミキサー中で30分間混合する。Aerosil(登録商標)200を1部およびステアリン酸マグネシウム1部を加え、混合物を再度5分間混合する。混合物を直径10mm、重量約260mg、および硬さ約50Nの錠剤に圧縮する。錠剤を実施例4に記載の方法を用いて腸溶コートする。
実施例30
カプセル剤のpH制御放出溶出プロフィールの測定
【0167】
該溶出プロフィールを、電気モーター(100rpm)で駆動する6個のバスケット撹拌エレメントと容量1Lの6個のいわゆるLevyグラスを取り付けた回転バスケットを用い、United States Pharmacopoeiaに記載のごとく測定する。Levyグラスに0,1N HCl(水浴の温度は37℃±0.5℃)を満たし、カプセル剤を該バスケットに適用する。2時間後、該酸を容器から除去し、溶出溶媒(USPリン酸緩衝液、pH6.5)で置換し、さらに6時間試験する。試料(5ml)を、該酸媒質から0、60、および120分間後、ならびに溶出溶媒をUSP緩衝液で置換後溶出溶媒から30、60、90、120、180、240、300、および360分間後に取り出す。各試料採取後に取り出した緩衝溶液の量を置換する代わりに、放出されたDMFの量を計算する時に緩衝液の減少を考慮する。DMFの量を、25℃に調節したMerck LiChroCART RP8 5μM、20cmカラムを用いるHPLC(Kontron XXX)により測定する。移動相は、アセトニトリルとリン酸でpH3.2に調節した0.0725mol/l NaH2PO4*H2O緩衝液の混合物(35:65)からなる。UV検出器を波長230nmおよび流速1.0mL/分に設定する。DMFピークは約5分間の保持時間後に検出することができる。
実施例31
非腸溶性錠剤のpH制御放出溶出プロフィールの測定
【0168】
溶出プロフィールを、電気モーターで駆動する撹拌エレメントとして6個のパドルと容量1Lの6個のいわゆるLevyグラスを用いて測定する。パドルの回転速度は100rpmである。LevyグラスにUSPリン酸緩衝液pH6.5(水浴の温度は37℃±0.5℃)を満たし、錠剤を該バスケットに適用する。試料(5ml)を、溶出溶媒をUSP緩衝液で置換後に溶出溶媒から0、30、60、90、120、180、240、300、および360分間後に取り出す。各試料採取後に取り出した緩衝溶液の量を置換する代わりに、放出されたDMFの量を計算する時に緩衝液の減少を考慮する。DMFの量を、25℃に調節したMerck LiChroCART RP8 5μM、20cmカラムを用いるHPLC(Kontron XXX)により測定する。移動相は、アセトニトリルとリン酸でpH3.2に調節した0.0725mol/l NaH2PO4*H2O緩衝液の混合物(35:65)からなる。UV検出器を波長230nmおよび流速1.0mL/分に設定する。DMFピークは約5分間の保持時間後に検出することができる。
実施例32
【0169】
実施例5に記載のごとく製造したカプセル剤の溶出プロフィールを実施例30に記載のごとく測定する。溶出プロフィールを図1に示す。
実施例33
【0170】
実施例16に記載のごとく製造した錠剤(腸溶コーティングを適用する前)の溶出プロフィールを実施例31に記載のごとく測定する。溶出プロフィールを図2に示す。
実施例34
【0171】
実施例17に記載のごとく製造した錠剤(腸溶コーティングを適用する前)の溶出プロフィールを実施例31に記載のごとく測定する。溶出プロフィールを図3に示す。
【図面の簡単な説明】
【0172】
【図1】実施例5に記載のごとく製造したカプセル剤のin vitro溶出プロフィールの例を示す。
【図2】実施例16に記載のごとく製造した錠剤試料(腸溶コーティングを施す前)のin vitro溶出プロフィールの例を示す。
【図3】実施例17に記載のごとく製造した錠剤試料(腸溶コーティングを施す前)のin vitro溶出プロフィールの例を示す。
【技術分野】
【0001】
本発明は活性成分としてフマル酸エステルを含む制御放出医薬組成物に関する。該組成物は、例えば乾癬または他の過剰増殖性、炎症性、または自己免疫障害の治療に用いるのに適しており、経口投与したときに消化管内で活性物質が局所的に高濃度になるのを避けることができ、消化管関連副作用を低下させることができるように制御的にフマル酸エステルを放出するよう設計される。
【背景技術】
【0002】
フマル酸エステル、すなわち、ジメチルフマレートとエチル水素フマレートの合剤は、多年にわたり乾癬の治療に用いられてきた。該合剤は、商標Fumaderm(登録商標)で市販されている。該合剤は経口使用を意図した錠剤の形であり、2つの異なる用量強度(Fumaderm(登録商標)InitialおよびFumaderm(登録商標))が利用可能である。
【0003】
2つの強度は、増量するFumaderm(登録商標)Initialで出発し、次いで例えば3週間治療後にFumaderm(登録商標)に切り替える個別的な用法に適用することを意図している。Fumaderm(登録商標)initialおよびFumaderm(登録商標)は共に腸溶性錠剤である。
【0004】
別の市販組成物は、120mgのジメチルフマレートおよび95mgのカルシウムモノエチルフマレートを含むFumaraat 120(登録商標)(TioFarma、Oud-Beijerland、Netherlands)である。最近の刊行物(Litjens et al. Br. J. Clin. Pharmacol. 2004、vol. 58:4、pp. 429-432)において、Fumaraat 120(登録商標)の薬物動態プロフィールが健康対象について記載されている。その結果は、単経口用量のFumaraat 120(登録商標)は血清モノメチルフマレート濃度の上昇をもたらし、ジメチルフマレートとフマル酸の濃度はごくわずかであることを示す。結果は、ジメチルフマレートが著者によれば酸環境下ではなくアルカリ環境下で急速にモノメチルフマレートに加水分解されることを示す。該組成物は腸溶性であるため、フマレートの取り込みは、主に小腸で生じ、ジメチルフマレートは取り込み前にアルカリ環境下によりモノエステルに加水分解されるか、または循環中でエステラーゼにより急速に変換されるかもしれないと考えられる。さらに、この研究では、tmaxおよびCmaxは食物の影響を受け、すなわち、同時に食物摂取するとtmaxは延長し(絶食状態の平均は182分間であるが、摂食状態の平均は361分間である)(遅延時間は絶食時で90分間、摂食時で300分間である)、Cmaxは低下する(絶食時:0.84mg/l、摂食時:0.48mg/l)。2錠のFumaderm(登録商標)P forteを用いる健康対象における別の研究(Reddingius W. G. Bioanalysis and Pharmacokinetics of Fumarates in Humans. Dissertation ETH Zurich No. 12199(1997))は、Cmax値(モノエチル-またはモノメチルフマレートとして測定)が1.0〜2.4μg/mlの範囲、tmaxが4.8〜6.0時間の範囲であることを示した。
【0005】
US 6,277,882およびUS 6,355,676は、乾癬、乾癬性関節炎、神経皮膚炎、およびクローン限局性腸炎(enteritis regionalis Crohn)を治療するためのマイクロ錠剤を製造するためのアルカリ水素フマレートの使用とある種のフマル酸モノアルカリエステル塩の使用をそれぞれ開示する。US6,509,376は、マイクロ錠剤またはペレットの形の移植医学および自己免疫疾患の治療に用いる医薬製剤を製造するためのある種のジアルキルフマレートの使用を開示する。US 4,959,389は、フマル酸モノアルキルエステルの種々の塩を単独またはジアルキルフマレートと組み合わせて含む組成物を開示している。GB 1,153,927は、ジメチルマレイン酸無水物および/またはジメチルマレイン酸および/またはジメチルフマル酸化合物を含む医薬組成物に関する。BMC Dermatology、vol. 2、no.5、2002中のケースレポート「Treatment of disseminated granuloma with fumaric acid esters」は、フマル酸エステルを用いる治療に関する。
【0006】
しかしながら、例えばFumaderm(登録商標)のようなフマレートによる治療は、例えば、膨満感、下痢、急激な上腹部痛、鼓腸、および悪心のような消化管の副作用を生じることが多い。
【0007】
したがって、経口投与したときに消化管関連副作用の減少による治療の改善をもたらす1またはそれ以上の治療的または予防的に有効なフマル酸エステルを含む組成物を開発する必要がある。
【0008】
さらに、現在市販されている製品は、該エステルの一つ(すなわち、フマル酸のモノエチルエステルであるエチル水素フマレート)が3つの異なる塩の形(すなわち、カルシウム、マグネシウム、および亜鉛塩)で存在する2つの異なるエステルの組み合わせを含む。各個々の形がそれ自身治療的プロフィールを持つかもしれないが、可能であれば適切な治療効果を得るためにはるかに単純な生成物であることが好都合であろう。
【0009】
本発明者らは、制御された方法、すなわち市販の製品に比べて延長され、遅くなり、および/または遅延するように、活性物質が送達されるように設計された医薬組成物の投与により改良された治療方法が得られると予期する。さらに、異なるフマル酸エステルの組み合わせを用いる代わりにジメチルフマル酸のような1つのフマル酸エステルのみを用いることにより適切な治療反応を達成することができると予期される。
(図の簡単な説明)
【0010】
図1は、実施例5に記載のごとく製造したカプセル剤のin vitro溶出プロフィールの例を示す。
【0011】
図2は、実施例16に記載のごとく製造した錠剤試料(腸溶コーティングを施す前)のin vitro溶出プロフィールの例を示す。
【0012】
図3は、実施例17に記載のごとく製造した錠剤試料(腸溶コーティングを施す前)のin vitro溶出プロフィールの例を示す。
【発明の開示】
【発明が解決しようとする課題】
【0013】
したがって、本発明は、経口投与すると等価用量のFumaderm(登録商標)錠の経口投与後と比べてGI関連副作用の低下が生じる、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む医薬組成物に関する。
【0014】
上記のように、本発明者らは、消化管関連副作用を減少させる適切な方法は制御放出組成物の形で活性成分を投与することによると予期する。
【0015】
したがって、さらなる局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステルまたはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、試験の最初の2時間は0.1N塩酸を溶出溶媒に、次いで0.05Mリン酸緩衝液pH6.5を溶出溶媒に用いるin vitro溶出試験を行うと、該フマル酸エステルの該放出が以下のごとくである該医薬組成物に関する:
試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約70%w/wが放出される、および/または
試験開始後の最初の4時間以内に該フマル酸エステルの総量のほぼ約92%w/wが放出される、および/または
試験開始後の最初の5時間以内に該フマル酸エステルの総量のほぼ約94%w/wが放出される、および/または
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約95%w/wが放出される、および/または
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約98%w/wが放出される、および/または
試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約99%w/wが放出される、および/または
試験開始後の最初の12時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約99%w/wが放出される。
【0016】
本発明の文脈において、制御放出組成物は、同等の条件(例えば、in vivo試験では、用量等価、標準食有りまたは無しなど、またはin vitro試験では、溶出試験装置および用いる溶出溶媒の組成、容量、および温度、回転速度などを含む作業条件)下で試験したときに市販製品のFumaderm(登録商標)の放出に比べて延長され、遅くなり、および/または遅延するようにフマル酸エステルを放出するように設計された組成物である。
【0017】
in vivoの放出は、予め決定した時間で血漿濃度を測定し、目的とするフマル酸エステルまたは適切であればその代謝物の血漿濃度対時間プロフィールを得ることにより試験してよい(例えば、ジメチルフマレートの場合は、活性物質はメチル水素フマレート、すなわちフマル酸のモノメチルエステルであると予想される)。さらに、代謝はすでに消化管内で、または消化管粘膜の通過時に、または肝循環による初回通過時に生じると予期される。したがって、ジメチルフマレートを投与したときに血漿中で探す関連成分はフマル酸のジメチルエステルではなくモノメチルエステルであってよい。
【0018】
in vivoの活性物質の放出を測定しまたは指標を得るのに他の試験を用いてもよい。すなわち、動物(例えばマウス、ラット、イヌなど)をモデルとして用いてよい。動物に観察下で組成物を投与し、一定時間後動物を屠殺し、活性成分(または適切ならばその代謝物)の含有量を血漿や特定臓器について測定するか、または腸内容から抽出する。
【0019】
別の試験は、動物の腸の特定部分を用いることを含む。該部分を該部分により分離した2つのコンパートメント(ドナーとレシーバー)を含む適切な溶出装置中に置き、観察下で該組成物を1つのコンパートメント(ドナーコンパートメント)中の適切な媒質中に置く。該組成物は活性成分を放出し、次いで腸部分を横切って輸送される。したがって、適切な時間間隔で、活性物質(または適切であれば代謝物)の濃度をレシーバーコンパートメントで測定する。
【0020】
当業者は上記方法を特定の組成物に適用することができるであろう。
【0021】
in vitro方法に関してはよく確立された方法、具体的には例えばUnited States Pharmacopeia(USP)またはEuropean Pharmacopoeiaのような公式モノグラフに記載の方法が利用可能である。当業者は、in vitro試験を行うための具体的条件を選ぶ方法およびどの方法を選ぶかを理解しているであろう。例えば、USPは、in vitro試験を37±1.0、例えば37±0.5℃で行うことを規定している。適切な溶出試験は、例えばカプセルについて実施例29に記載されており、ここでは、溶出プロフィールはUnited States Pharmacopoeiaに記載のごとく試験の最初の2時間は0.1N塩酸を溶出溶媒に用い、次いで残りの試験時間は0.05Mリン酸緩衝液pH6.5を溶出溶媒に用い、100rpmの回転パスケットを用いて37℃で測定し、また例えば錠剤については実施例30に記載されており、ここでは、溶出プロフィールはUnited States Pharmacopoeiaに記載のごとく試験の最初の2時間は0.1N塩酸を溶出溶媒に用い、次いで残りの試験時間は0.05Mリン酸緩衝液pH6.5を溶出溶媒に用い、100rpmのパドル溶出装置を用いて37℃で測定する。
【0022】
上記のごとく、活性物質のin vivo放出は、市販のFumaderm(登録商標)組成物に比べて延長され、遅くなり、および/または遅延する。本発明の文脈に置いて、用語「延長された」は、活性物質がFumaderm(登録商標)より長い期間、例えばFumaderm(登録商標)より少なくとも1.2倍、例えば少なくとも1.5倍、少なくとも2倍、少なくとも3倍、少なくとも4倍、または少なくとも5倍長い期間放出されることを示すことを意図する。すなわち、例えば100%のジメチルフマレートがFumaderm(登録商標)錠から適切な試験の開始後3時間で放出されるならば、本発明組成物中の100%のジメチルフマレートが適切な試験の開始後少なくとも3.6時間で放出される。
【0023】
本発明の文脈において、用語「遅延する」は、活性成分の放出がFumaderm(登録商標)に比べて遅い時点で始まるか(例えば30分間またはそれ以上遅く、例えば45分間またはそれ以上遅く、1時間またはそれ以上遅く、1.5時間またはそれ以上遅く)、または最初の2時間の初期放出がFumaderm(登録商標)に比べて遙かに少ない(すなわち、Fumaderm(登録商標)の80%w/w以下、例えば70%w/w以下、60%w/w以下、または50%以下)ことを示すことを意図する。
【0024】
本発明において用いている消化管(GI)の副作用には、限定されるものではないが、下痢、腹痛(stomach ache、stomach pain、abdominal pain)、痙攣性腹痛、悪心、鼓腸(flatulence)、しぶり(tenesmus)、鼓腸(meteorism)、大便の頻度の増加、膨満感、および急激な上腹部痛が含まれよう。
【0025】
本発明の文脈において、GI関連副作用の減少は、本発明の組成物を投与した後に観察されるGI副作用の該治療患者ポピュレーションにおける重症度および/または発生率がFumaderm(登録商標)の場合と比べて低下することを意味することを意図する。すなわち、この定義によればGI関連副作用の減少は、上記のあらゆるGI副作用の発生率の実質的な減少、例えば少なくとも10%の減少、またはより好ましくは発生率の少なくとも20%の減少、またはさらにより好ましくは発生率の30%以上の減少と理解されよう。GI関連副作用の減少は、上記のあらゆるGI副作用の重症度の実質的な減少、例えば下痢、腹痛(stomach ache、stomach pain、abdominal pain)、痙攣性腹痛、悪心、鼓腸、しぶり、鼓腸、大便の頻度の増加、膨満感、または急激な上腹部痛の重症度および/または頻度の減少として表現することもできる。上記GI関連副作用の減少は、Fumaderm(登録商標)またはプラセボと本発明の組成物の投与を比較する臨床試験においてモニターすることができる。プラセボ対照臨床試験の場合、プラセボ群と比較した本発明組成物を投与された患者におけるGI関連副作用の発生率をFumaderm(登録商標)とプラセボを比較するヒストリカル臨床試験と比較することができる(例えば、Altmeyer et al、J. Am. Acad. Dermatol. 1994; 全参考文献:Altmeyer PJ et al、Antipsoriatic effect of fumaric acid derivatives. Results of a multicenter double-blind study in 100 patients. J. Am. Acad. Dermatol. 1994; 30:977-81参照)。典型的には、乾癬に罹患した患者がそのような試験に含まれ、典型的には体表面の10%以上が乾癬に罹患しているであろう(重症乾癬)。しかしながら、体表面積の2〜10%が罹患している患者も含むことができる(中等度の乾癬)。患者は乾癬面積重症度指数(PASI)に基づいて選ぶこともできる。典型的には、ある範囲内のPASI、例えば10〜40、または例えば12〜30、または例えば15〜25の患者が含まれる。あらゆるタイプの乾癬に罹患した患者が含まれてよい(慢性プラーク型、播種状滴状(exanthematic guttate)型、膿疱型、乾癬性紅皮症またはパルモプラナー(palmoplanar)型)が、ある場合には、慢性プラーク型の患者のみが含まれる。各治療群(本発明組成物およびFumaderm(登録商標)またはプラセボ)に約15〜20人の患者でほとんどの場合十分であったが、より好ましくは試験の各治療群に約30〜50人の患者が含まれる。全試験期間は1日間〜1週間であり得るが、より好ましくは該試験は8週間〜12週間または16週間まで行われよう。該副作用は、例えば各群で一定の副作用が報告される総回数(何人の患者が副作用を経験したかに関わらず)として評価することができるか、または副作用は一定の副作用を一定回数、例えば試験期間中に少なくとも1回または少なくとも2回、または少なくとも3回経験した患者数で評価することができる。さらに、副作用の重症度をモニターすることができるか、または該試験で副作用とみなすために副作用の一定の重症度を要求することができる。副作用の重症度を評価する好都合な方法は視覚アナログ(VAS)尺度による。
活性物質
【0026】
本発明組成物中の活性物質はあらゆるフマル酸エステルである。本発明のある態様において、該フマル酸エステルは、好ましくはジメチルフマレート、ジエチルフマレート、ジプロピルフマレート、ジブチルフマレート、ジペンチルフマレート、メチル-エチルフマレート、メチル-プロピルフマレート、メチル-ブチルフマレート、メチル-ペンチルフマレート、モノメチルフマレート、モノエチルフマレート、モノプロピルフマレート、モノブチルフマレート、およびモノペンチルフマレートからなる群から選ばれる(その医薬的に許容される塩を含む)。
【0027】
本発明の具体的態様において、該フマル酸エステルは、医薬的に許容される塩の形で存在するフマル酸のモノ-(C1-C5)アルキルエステルである。適切な塩は、例えば金属塩、例えばナトリウム、カリウム、カルシウム、マグネシウム、または亜鉛塩を含むアルキル金属塩およびアルキル土類金属塩から選ばれる塩である。
【0028】
用語(C1-C5)アルキルは、炭素数1〜5(含む)の分岐または非分岐アルキル基、例えばメチル、エチル、1-プロピル、2-プロピル、1-ブチル、2-ブチル、2-メチル-2-プロピル、2-メチル-1-プロピル、およびペンチルを表す。
【0029】
別の態様において、本発明組成物はジメチルフマレートを活性成分として含む。
【0030】
さらなる態様において、本発明組成物は、所望により医薬的に許容される塩、例えばナトリウム、カリウム、カルシウム、マグネシウムおよび/または亜鉛塩などの形のモノメチルフマレートを活性成分として含む。
【0031】
別の態様において、本発明組成物は、活性成分として本質的にジメチルフマレートからなる。
【0032】
別の態様において、本発明組成物は、活性成分としてジメチルフマレートからなる。
【0033】
さらなる態様において、本発明組成物は、活性成分として実質的に、所望により医薬的に許容される塩、例えばナトリウム、カリウム、カルシウム、マグネシウムおよび/または亜鉛塩などの形のモノメチルフマレートからなる。
【0034】
さらなる態様において、本発明組成物は、活性成分として、所望により医薬的に許容される塩、例えばナトリウム、カリウム、カルシウム、マグネシウムおよび/または亜鉛塩などの形のモノメチルフマレートからなる。
【0035】
さらなる態様において、本発明組成物は、重量比約1:10〜約10:1のジメチルフマレートおよびモノメチルフマレート(所望により医薬的に許容される塩、例えばそのナトリウム、カリウム、カルシウム、マグネシウムおよび/または亜鉛塩などの形の)を活性成分として含む。
【0036】
さらなる態様において、本発明組成物は、実質的に、重量比約1:10〜約10:1のジメチルフマレートおよびモノメチルフマレート(所望により医薬的に許容される塩、例えばそのナトリウム、カリウム、カルシウム、マグネシウムおよび/または亜鉛塩などの形の)を活性成分として含む。
【0037】
さらなる態様において、本発明組成物は、活性成分として重量比約1:10〜約10:1のジメチルフマレートおよびモノメチルフマレート(所望により医薬的に許容される塩、例えばそのナトリウム、カリウム、カルシウム、マグネシウムおよび/または亜鉛塩などの形の)からなる。
化粧品および/または医薬組成物
【0038】
本発明が解決する問題は、フマル酸エステルの経口投与による消化管の副作用の出現に関する。該組成物からの活性成分の放出を延長および/または遅延させることにより、消化管の特定部位における活性物質の局所濃度を減少させ(Fumaderm(登録商標)の比べて)、次いで消化管の副作用の減少をもたらすと予想される。したがって、上記のごとくフマル酸エステルの放出を延長および/または遅くすることができる組成物は本発明の範囲内である。
【0039】
そのような組成物は当業者によく知られており、例えば拡散制御ドラッグデリバリーシステム、浸透圧制御ドラッグデリバリーシステム、浸食(erodible)ドラッグデリバリーシステムなどが含まれる。さらに、特定の技術(例えば上記の)に基づいて活性成分の特定の放出特性を有する特定の組成物を提供することができる製薬会社がある。したがって、当業者は、特定の薬剤物質について特定の必要性を感じたら適切な生成物を得る方法が解るであろう。例として、Eurandは、特定の活性物質を含む、組成物からの活性物質の放出について特定の必要条件を有する制御放出医薬組成物を得るための技術的解答を提供するそのような会社の1つである(例えば、http://www.eurand.com参照)。別の会社には、いわゆるSQZgel(登録商標)(http://macromed.com)に関する技術を開発したMacroMed、Inc.がある。SQZgel(登録商標)の作用機序は、外部コーティングと組み合わせたpH感受性ポリマー混合物である。胃の酸性環境において、該ポリマーは水を吸収して膨潤し、薬剤を取り込む。pHがより高い腸に入ると、該ポリマーは徐々に縮むかまたは「絞られ」、「ダイアルドイン(dialed-in)」速度で(持続的活性組成物を放出する)、またはEgalet(登録商標)技術の重要な要素である特定の押出に基づく技術を有するEgalet a/s(http://www.eqalet.com)は、表面浸食性、疎水性のPEGステアレートからなる、活性成分を含む、生体分解性コートおよびマトリックスである。Egalet(登録商標)技術の1つに、コートとマトリックスからなる2コンポーネント生成モデルである2K Egalet(登録商標)持続放出系がある。該薬剤は、時間とともに持続放出のためにEgalet(登録商標)マトリックスを通して均一に分散する。本発明の文脈において例えばEurand技術のような技術が興味深い:Diffucaps(薬剤放出プロフィールは、中性コア、例えば糖スフェア、血漿、または顆粒上に活性薬剤、次いで速度調節機能膜を重層することによりもたらされる。Diffucaps/Surecapsビーズは、サイズが小さく直径が1mmまたはそれ以下である。種々の薬剤放出プロフィールのビーズを硬ゼラチンカプセルに組み込むことにより組み合わせ放出プロフィールを達成することができる。)、Diffutabs(Diffutab技術はマトリックス錠の拡散および浸食を通して薬剤放出を制御する親水性ポリマー混合物を組み込む)、Minitabs(Eurand Minitabsは薬剤放出速度を制御するゲル形成賦形剤を含む小さな(2mm x 2mm)錠剤である。さらなる膜をさらに放出速度を制御するために加えてよい。)、Orbexa(この技術は、規定ベースの(defined-based)造粒押出および球形化技術により制御されたサイズおよび密度のビーズを生成する。得られたビーズは、さらなる放出速度制御のために放出速度制御膜でコートすることができ、カプセルに充填するかサシェー型としてよい。)、SDS(EurandのSDS技術は、機能的ポリマーおよび機能的ポリマーと特定の添加剤の組み合わせ、例えば複合ポリマー物質を用い、腸管の最適吸収部位に薬剤を送達する。これを達成するために、Eurandは、最初に活性薬剤を組み込むDiffucapsまたはEurand Minitabsのような多粒子剤形を生成する。次に、この剤形を薬剤を所望の部位に送達するpH依存性/非依存性ポリマー膜でコートする。次にこれらを硬ゼラチンカプセルに充填する。)。
【0040】
本発明の組成物を製造するのに用いる別の興味深い技術に、WO 03/004001に記載のいわゆるMeltDose(登録商標)技術がある(例えば、http://www.lifecyclepharma.com参照)。MeltDose(登録商標)は、可溶化した個々の分子を錠剤に処方することを含む。個々の分子を処方することにより、低水溶性の薬剤の経口吸収の一次制限を除去し、優れたバイオアベイラビリティをもたらす。この技術を用いることにより、種々の医薬剤形、例えばペレットまたは錠剤の形に加工するのに適した粒子状物質を得ることができる。さらに、該技術は、活性物質の、例えば本明細書に記載の放出プロフィールのような適切な放出プロフィールを得ることができるので使用に適している。ある態様において、使用に適したペレットは、2000μm以上の平均粒子サイズを有してよい。別の態様において、使用に適したペレットは約0.01μm〜約250μmの平均粒子サイズを有してよい。
【0041】
本発明の文脈において使用する別の特定の適切な製剤方針は、例えば軟ゼラチンカプセルのような脂溶性環境中の製剤である。この製剤方針の適切な例には、Vegicaps Soft(Scherer)がある(100%植物由来であるにも関わらずカラギーナンおよびデンプンに基づく軟カプセル技術は、なお従来の軟ゼラチンカプセルの重要なすべての特性を提供する。これらには、飲み込み易さをもたらす軟質の柔軟な剤形が含まれる。)。(さらなる情報については、http://www.rpscherer.de/page.php?paqeID=94参照。)
【0042】
適切な製剤のさらなる具体例には、軟および硬ゼラチンカプセル中の活性物質とビタミンE濃縮物の製剤が含まれる。修飾形のこの製剤は、シクロスポリンに加えて他にコーン油モノ-ジ-トリグリセリド、ポリオキシル40水素化ひまし油NF、DL-α-トコフェロールUSP(ビタミンEファミリーの部分)、ゼラチンNF、グリセロール、四三酸化鉄、プロピレングリコールUSP、二酸化チタンUSP、カルミン、およびアルコールを含む市販のシクロスポリン製剤のNeoral(登録商標)の基礎原料である。
【0043】
適切な製剤(処方)の別の具体例には、軟または硬ゼラチンカプセル中に、エタノール、トコフェロールエチレングリコール1000 スクシネート(TPGS)、コーン油、およびワックスと活性物質の製剤が含まれる。この製品は半固体または固体剤形であり得る。この製剤の放出速度は腸中のリパーゼによる分解に依存する。
【0044】
適切な製剤のさらなる例には、軟または硬ゼラチンカプセル中に、エタノール、トコフェロールエチレングリコール1000 スクシネート(TPGS)、コーン油、およびポリグリコール化グリセリド(例えばGelucire)と活性物質の製剤が含まれる。この製品は半固体または固体剤形であり得る。この製剤の放出速度は腸中のリパーゼによる分解に依存する。
【0045】
適切な製剤のさらなる例には、経口パルス用量ドラッグデリバリーシステムがある。この剤形は、Schering Repetab錠の修飾形と考えることができる。本発明の組成物の部分は、錠剤のコア中に入れられる。
【0046】
該コアは、例えば常套的湿式造粒法または連続増粒法、例えば押出し、次いで顆粒を錠剤に圧縮することにより製造することができる。次に、該コアを適切な技術、好ましくはEudragitsのような腸溶コーティングポリマーを用いる空気サスペンジョンによりコートする。
【0047】
第1放出用量は、コア上に圧縮コートされるか、または腸溶コートでまたは腸溶コートの上部に空気懸濁コートされる。本発明の態様において、第1放出用量は、腸溶コートで空気懸濁コートされる。本発明のさらなる態様において、第1放出用量は、典型的には腸溶コートの分解前に本発明の組成物の放出を避けるために該コート上に圧縮コートされ、そのような分解は、典型的には胃(gastric ventricle)より高いpH値で生じ、すなわち、典型的には腸溶コートの分解は胃の通過後に生じる。
【0048】
適切な製剤のさらなる例には、経口持続ドラッグデリバリーシステムがある。本発明組成物の部分は錠剤のコア中に入れられる。
【0049】
該コアは、例えば常套的湿式造粒法または連続増粒法、例えば押出し、次いで顆粒を錠剤に圧縮することにより製造することができる。該コアを適切な技術、好ましくはエチルセルロースおよび親水性賦形剤、例えばヒドロキシルプロピルセルロース(HPC)を用いる空気サスペンジョンによりコートする。
【0050】
第1放出用量は、コア上に圧縮コートされるか、または腸溶コートでまたは腸溶コートの上部に空気サスペンジョンコートされる。本発明の好ましい態様において、第1放出用量は、腸溶コートで空気サスペンジョンコートされる。本発明のさらなる態様において、第1放出用量は、典型的には腸溶コートの分解前に本発明の組成物の放出を避けるために該コート上に圧縮コートされ、そのような分解は、典型的には胃(gastric ventricle)より高いpH値で生じ、すなわち、典型的には腸溶コートの分解は胃の通過後に生じる。
【0051】
適切な製剤のさらなる例は、例えばWO 03/080034に記載のような結晶工学により得られる(この内容は本明細書の一部を構成する。)。
【0052】
したがって、本発明組成物の別の態様において、親水性表面を有する微結晶の形の活性物質を含む。さらに、本発明の別の態様において、該微結晶は持続放出製剤を達成するために直接フィルムコートされる。
【0053】
適切な製剤の別の具体例には、本発明組成物と純粋なシクロデキストリンおよびシクロデキストリン誘導体(例えばアルキル-およびヒドロキシアルキル-誘導体、またはスルホブチル誘導体)の複合体(錯体)化が含まれる。該複合体化は、よく知られた方法に従って達成される。そのような複合体化は複合体化前の該組成物に比べて本発明組成物の高い溶解性と高い溶出速度をもたらすと予期される。さらに、そのような組成物は複合体化前の該組成物に比べて本発明組成物の高いバイオアベイラビリティをもたらすと予期される。
【0054】
具体的態様において、本発明は1日に1回、2回、またはそれ以上、例えば1日に1回または2回、または3回投与することができる制御放出医薬組成物に関する。さらに、該組成物はフマル酸エステルを比較的pH非依存性に、すなわち、消化管内のpHに依存しないで放出するよう設計してよい。そのような組成物の例には、例えば制御放出コーティングでコートされた固体剤形(例えば、錠剤、カプセル剤、ペレット、ビーズなど)の形の組成物がある。制御放出コーティングに適した物質には、例えばセルロース、およびメチルセルロース、エチルセルロース、セルロースアセテート、またはポリ(エチルレン-コ-ビニルアセテート)、ポリ(塩化ビニル)を含むセルロース誘導体がある。
【0055】
典型的には、該フマル酸エステルの放出は拡散制御膜でコートされた組成物から3段階で生じる:
i)第1に、水(GI管から)が周囲から剤形内に拡散し、
ii)第2に、該剤形内に存在する少なくともいくらかの該フマル酸エステルが水の作用により溶解し、
iii)該溶解したフマル酸エステルが剤形から周囲(すなわちGI管)中に拡散する。
【0056】
他の例には、例えばそれぞれマトリックス系の形の多数単位を含む剤形またはマトリックス錠が含まれる。活性物質を、例えば、セルロース、および微晶質セルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロースおよびメチルセルロースを含むセルロース誘導体、ポビドン、ポリ(エチレンオキシド)(PEO)、ポリエチレングリコール(PEG)、ポリ(ビニルアルコール)(PVA)、キサンタンガム、カラギーナン、および他の合成物質を含むマトリックス中に組み込む。医薬的に許容される賦形剤または添加剤として通常用いる物質をマトリックス組成物に加えてよい。
【0057】
適切な組成物の他の例には、活性物質が水膨潤性ネットワークポリマー中に組み込まれる、例えばヒドロゲル、すなわち単体システムがある。使用に適した物質には、例えば、親水性ビニルおよびアクリルポリマー、アルギネートのようなポリサッカライド、およびポリ(エチレンオキシド)がある。
【0058】
特定の態様において、本発明の組成物は該フマル酸エステルをpH制御放出(pH依存性放出としても知られる)する。通常、該放出は、もしあるいとしてもフマル酸エステルを少量しか胃(約pH3以下)内に放出せず、該フマル酸エステルを腸(pHが約6〜7に変化する)に放出するよう設計される。そのようなpH制御放出は、腸溶コーティングを有する本発明組成物(全組成物、または組成物が多粒子組成物である場合は個々の単位)を提供するか、または該フマル酸をpH依存性浸透圧メカニズムまたは適切な酵素の使用により放出する組成物を提供することにより得ることができる。
【0059】
腸溶コーティング物質として用いるのに適した物質の例には、ポリアクリルアミド、フタレート誘導体、例えば、炭水化物の酸フタレート、アミロースアセテートフタレート、セルロースアセテートフタレート、他のセルロースエステルフタレート、セルロースエーテルフタレート、ヒドロキシプロピルセルロースフタレート、ヒドロキシプロピルエチルセルロースフタレート、ヒドロキシプロピルメチルセルロースフタレート、メチルセルロースフタレート、ポリビニルアセテートフタレート、ポリアクリルメタクリル酸コポリマー、セラックおよびビニルアセテート、およびクロトン酸コポリマーなどが含まれる。
【0060】
pH非依存性放出を有する上記組成物は、例えば腸溶コーティングの外層を有する該組成物を与えることにより該フマル酸エステルを放出するよう製剤化してもよい。
【0061】
さらに、該組成物は、フマル酸エステルの放出に初期遅延が得られるような方法で製剤化してよい。そのような遅延は、例えば時間制御的に分解する(例えば浸食する)最外層のコーティングを選ぶことにより得ることができ、この最外層のコーティングが浸食されて除かれた時にのみフマル酸エステルの放出が始まる。
【0062】
フマル酸エステルの適切な放出が得られるよう設計された種々の本発明組成物について以下に説明する。上記説明および医薬の制御放出の分野のハンドブックに基づいて当業者は必要な放出プロフィールを達成するために種々の製剤方針を選ぶ方法がわかるであろう。
1日に2回またはそれ以上投与するよう設計された組成物
pH非依存性放出
【0063】
フマル酸エステルをpH非依存性に放出し、放出パターンが1日に2回またはそれ以上投与する組成物に適している具体的態様を以下に説明する。適切な製剤方針の例には、例えば上記のあらゆる製剤方針を含む、拡散コーティング、例えば制御放出拡散コーティング、マトリックス粒子またはマトリックス錠、ハイドロゲル、パルス用量ドラッグデリバリーシステム、ビタミンE濃縮物またはエタノール、TPGS、コーン油、およびワックスなどとの合剤により提供される組成物がある。
【0064】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、水を溶出溶媒に用いてin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物に関する:
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約60%w/w、例えば約30%〜約60%w/w、約40%〜約55%w/w、または約50%が放出される、および/または
試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、約60%〜約80%w/w、または約75%が放出される、および/または
試験開始後の最初の12時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約80%w/w、例えば約80%w/wまたはそれ以上、約85%w/wまたはそれ以上、約90%w/wまたはそれ以上、または約95%w/wまたはそれ以上が放出される、および/または
該組成物中に含まれるフマル酸エステルの総量が試験開始後の最初の12時間以内に放出される。
pH制御放出
【0065】
フマル酸エステルがpH依存性に放出され、放出パターンが1日に2回またはそれ以上投与する組成物に適している特定の態様について以下に説明する。適切な製剤方針の例には、例えばZentner et al(US 6,537,584)およびBae(US 5,484,610)に記載のタイプの腸溶コーティングまたはハイドロゲルを用いて提供される組成物がある(この内容は本明細書の一部を構成する。)。適切な製剤方針の例には、例えば上記のあらゆる製剤方針を含む、所望により腸溶コーティングを有する、拡散コーティング、例えば制御放出拡散コーティング、マトリックス粒子またはマトリックス錠、ハイドロゲル、パルス用量ドラッグデリバリーシステム、ビタミンE濃縮物またはエタノール、TPGS、コーン油、およびワックスなどとの合剤により提供される組成物がある。
【0066】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、試験の最初の2時間は0.1N塩酸を溶出溶媒に、次いで0.05Mリン酸緩衝液pH6.5または6.8を溶出溶媒に用いてin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物を提供する:
試験開始後の最初の2時間以内に該フマル酸エステルの総量の少なくとも約1%w/w、例えば少なくとも約2%w/w、少なくとも約3%w/w、または約5%w/wが放出される、および/または
試験開始後の最初の3時間以内に該フマル酸エステルの総量のほぼ約35%w/w、例えば約15%〜約35%w/w、約20%〜約30%w/w、または約25%w/wが放出される、および/または
試験開始後の最初の3時間以内に該フマル酸エステルの総量の約10%〜約70%w/w、約10%〜約65%w/w、約10%〜約60%w/w、約15%〜約50%w/w、約15%〜約35%w/w、約20%〜約30%w/w、または約20%w/w、または約25%w/が放出される、および/または
試験開始後の最初の4時間以内に該フマル酸エステルの総量のほぼ約92%w/w、例えば約10%〜約92%w/w、約20%〜約85%w/w、約20%〜約80%w/w、約20%〜約70%w/w、約25%〜約60%w/w、約25%〜約55%w/w、約30%〜約50%w/w、または約35%w/w、または約40%w/w、または約45%w/wが放出される、および/または
試験開始後の最初の5時間以内に該フマル酸エステルの総量のほぼ約94%w/w、例えば約15%〜約94%w/w、約25%〜約90%w/w、約30%〜約85%w/w、約35%〜約80%w/w、約35%〜約75%w/w、約40%〜約70%w/w、約45%〜約70%w/w、約55%〜約70%w/w、約60%〜約70%w/w、または約45%w/w、または約50%w/w、または約55%w/w、または約60%w/w、または約65%w/wが放出される、および/または
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約60%w/w、例えば約30%〜約60%w/w、約40%〜約55%w/w、または約50%w/wが放出される、および/または
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約95%w/w、例えば約35%〜約95%w/w、約40%〜約90%w/w、約45%〜約85%w/w、約50%〜約85%w/w、約55%〜約85%w/w、約60%〜約85%w/w、約65%〜約85%w/w、約70%〜約85%w/w、約75%〜約85%w/w、または約65%w/w、または約70%w/w、または約75%w/w、または約80%w/wが放出される、および/または
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約98%w/w、例えば約45%〜約98%w/w、約50%〜約98%w/w、約55%〜約98%w/w、約60%〜約98%w/w、約65%〜約98%w/w、約70%〜約98%w/w、約75%〜約95%w/w、約80%〜約95%w/w、約85%〜約95%w/w、または約75%w/w、または約80%w/w、または約85%w/w、または約90%w/wが放出される、および/または
試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、約60%〜約80%w/w、または約75%w/wが放出される、および/または
試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約99%w/w、例えば約60%〜約99%w/w、約70%〜約99%w/w、約80%〜約99%w/w、約90%〜約99%w/w、または約95%w/wが放出される。
【0067】
本発明の別の局面において、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、最初の2時間は0.1N塩酸を、次いで0.05Mリン酸緩衝液pH6.5または6.8を用いてUSPに従って測定したときにin vitro溶出プロフィールに従って予め決定された期間にわたりフマル酸のジ-(C1-C5)アルキルエステルおよびモノ-(C1-C5)アルキルエステル、またはその医薬的に許容される塩を放出するのに適合した制御放出剤形からなることを特徴とする該組成物を提供する:
ここで、試験開始後の最初の2時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ5%w/wが放出される、および/または
試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量の約20%〜約75%w/wが放出される、および/または
試験開始後の最初の4時間以内に該組成物中に含まれる該フマル酸エステルの総量の約50%〜約90%w/wが放出される、および/または
試験開始後の最初の5時間以内に該組成物中に含まれる該フマル酸エステルの総量の約60%〜約90%w/wが放出される、および/または
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量の約70%〜約95%w/wが放出される、および/または
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量の約75%〜約97%w/wが放出される。
【0068】
本発明のさらなる局面において、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、最初の2時間は0.1N塩酸を、次いで0.05Mリン酸緩衝液pH6.5または6.8を用いてUSPに従って測定したときにin vitro溶出プロフィールに従って予め決定された期間にわたりフマル酸のジ-(C1-C5)アルキルエステルおよびモノ-(C1-C5)アルキルエステル、またはその医薬的に許容される塩を放出するのに適合した制御放出剤形からなることを特徴とする該組成物を提供する:
ここで、試験開始後の最初の2時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ5%w/wが放出される、
試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量の約20%〜約75%w/wが放出される、
試験開始後の最初の4時間以内に該組成物中に含まれる該フマル酸エステルの総量の約50%〜約90%w/wが放出される、
試験開始後の最初の5時間以内に該組成物中に含まれる該フマル酸エステルの総量の約60%〜約90%w/wが放出される、
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量の約70%〜約95%w/wが放出される、
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量の約75%〜約97%w/wが放出される、
試験開始後の最初の8時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも85%w/wが放出される。
【0069】
本発明の別のさらなる局面において、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む制御放出医薬組成物であって、最初の2時間は0.1N塩酸を、次いで0.05Mリン酸緩衝液pH6.5または6.8を用いてUSPに従って測定したときにin vitro溶出プロフィールに従って予め決定された期間にわたりフマル酸のジ-(C1-C5)アルキルエステルおよびモノ-(C1-C5)アルキルエステル、またはその医薬的に許容される塩を放出するのに適合した制御放出剤形からなることを特徴とする該組成物を提供する:
ここで、試験開始後の最初の2時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ5%w/wが放出される、および/または
試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量の約20%〜約50%w/wが放出される、および/または
試験開始後の最初の4時間以内に該組成物中に含まれる該フマル酸エステルの総量の約45%〜約70%w/wが放出される、および/または
試験開始後の最初の5時間以内に該組成物中に含まれる該フマル酸エステルの総量の約65%〜約85%w/wが放出される、および/または
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量の約75%〜約90%w/wが放出される。
【0070】
本発明のさらに別の局面において、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む制御放出医薬組成物であって、最初の2時間は0.1N塩酸を、次いで0.05Mリン酸緩衝液pH6.5または6.8を用いてUSPに従って測定したときにin vitro溶出プロフィールに従って予め決定された期間にわたりフマル酸のジ-(C1-C5)アルキルエステルおよびモノ-(C1-C5)アルキルエステル、またはその医薬的に許容される塩を放出するのに適合した制御放出剤形からなることを特徴とする該組成物を提供する:
ここで、試験開始後の最初の2時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ5%w/wが放出される、
試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量の約20%〜約50%w/wが放出される、
試験開始後の最初の4時間以内に該組成物中に含まれる該フマル酸エステルの総量の約45%〜約70%w/wが放出される、
試験開始後の最初の5時間以内に該組成物中に含まれる該フマル酸エステルの総量の約65%〜約85%w/wが放出される、
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量の約75%〜約90%w/wが放出される、
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも80%が放出される。
【0071】
本発明のさらに別の局面において、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む制御放出医薬組成物であって、最初の2時間は0.1N塩酸を、次いで0.05Mリン酸緩衝液pH6.5または6.8を用いてUSPに従って測定したときにin vitro溶出プロフィールに従って予め決定された期間にわたりフマル酸のジ-(C1-C5)アルキルエステルおよびモノ-(C1-C5)アルキルエステル、またはその医薬的に許容される塩を放出するのに適合した制御放出剤形からなることを特徴とする該組成物を提供する:
ここで、試験開始後の最初の2時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ5%w/wが放出される、および/または
試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量の約50%〜約75%w/wが放出される、および/または
試験開始後の最初の4時間以内に該組成物中に含まれる該フマル酸エステルの総量の約70%〜約90%w/wが放出される、および/または
試験開始後の最初の5時間以内に該組成物中に含まれる該フマル酸エステルの総量の約80%〜約90%w/wが放出される。
【0072】
本発明のさらに別の局面において、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む制御放出医薬組成物であって、最初の2時間は0.1N塩酸を、次いで0.05Mリン酸緩衝液pH6.5または6.8を用いてUSPに従って測定したときにin vitro溶出プロフィールに従って予め決定された期間にわたりフマル酸のジ-(C1-C5)アルキルエステルおよびモノ-(C1-C5)アルキルエステル、またはその医薬的に許容される塩を放出するのに適合した制御放出剤形からなることを特徴とする該組成物を提供する:
ここで、試験開始後の最初の2時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ5%w/wが放出される、
試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量の約50%〜約75%w/wが放出される、
試験開始後の最初の4時間以内に該組成物中に含まれる該フマル酸エステルの総量の約70%〜約90%w/wが放出される、
試験開始後の最初の5時間以内に該組成物中に含まれる該フマル酸エステルの総量の約80%〜約90%w/wが放出される、
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約90%w/wが放出される。
徐々のpH変化に及ぶ放出(「ハーフチェンジ」法)
【0073】
フマル酸エステルがpH依存性に放出され、放出パターンが1日に2回またはそれ以上投与する組成物に適している特定の態様について以下に説明する。適切な製剤方針の例には、例えばZentner et al(US 6,537,584)およびBae(US 5,484,610)に記載のタイプの腸溶コーティングまたはハイドロゲルを用いて提供される組成物がある(この内容は本明細書の一部を構成する。)。適切な製剤方針の例には、例えば上記のあらゆる製剤方針を含む、所望により腸溶コーティングを有する、拡散コーティング、例えば制御放出拡散コーティング、マトリックス粒子またはマトリックス錠、ハイドロゲル、パルス用量ドラッグデリバリーシステム、ビタミンE濃縮物またはエタノール、TPGS、コーン油、およびワックスなどとの合剤により提供される組成物がある。
【0074】
「ハーフチェンジ」法は、腸溶コートまたは持続放出製剤について特に開発された。この方法は、1時間毎に溶出溶媒の半分を中性溶出溶媒の一定分量で置換することを含む(十二指腸から回腸へのpH値のわずかな変化についてGI通過をシミュレートするため)。該アプローチを下記表に説明する。
【0075】
【表1】
【0076】
模擬(シミュレート)胃液の組成は、例えばUnited States Pharmacopeia(USP)2005にみることができる:
タンパク質1mgあたり800〜2500単位の活性を含むブタ胃粘膜由来の精製ペプシン3.2gおよびNaCl 2.0gを7.0mlの塩化水素酸に溶解し、十分な水を加えて1000mLとする。得られた試験溶液はpH約1.2である。
【0077】
模擬胃液の別の組成はGerman E DIN 19738(Deutsche Industrie Norm)にみることができる:
100mLの合成/模擬胃液は、290mgのNaCl、70mgのKCl、27mgのKH2PO4を含み、十分なHClでpH2.0に調整する。さらに、該胃液は、100mgのペプシンと300mgのムチンを含む。
【0078】
模擬腸液の組成は、例えばUnited States Pharmacopeia(USP)2005にみることができる:
6.8gのリン酸2水素カリウムを250mLの水に溶解する。混合し、77mLの0.2N水酸化ナトリウムおよび500mLの水を加える。10.0gのパンクレアチンを加え、該溶液を混合し、該溶液を混合し、次いで0.2 N水酸化ナトリウムまたは0.2N塩酸を加えてpH6.8±0.1に調整する。得られた溶液を水で1000mLに希釈する。
【0079】
模擬腸液の別の組成はGerman E DIN 19738(Deutsche Industrie Norm)に記載されている:
100mLの合成/模擬腸液は、30mgのKCl、50mgのCaCl2、20mgのMgCl2を含み、十分なNaHCO3でpH7.5に調整する。さらに、該腸液は30mgのトリプシン、900mgのパンクレアチン、900mgの凍結乾燥胆汁、および30mgの尿素を含む。
【0080】
本発明の好ましい態様において、該「ハーフチェンジ」法は、USP 2005に記載の模擬胃液と模擬腸液を用いて実施される。
【0081】
本発明の別の態様において、該「ハーフチェンジ」法は、USP 2005に記載の模擬胃液と模擬腸液を用いるが該タンパク質不含(すなわち、模擬胃液中にペプシン不含、模擬腸液中にパンクレアチン不含)で実施される。
【0082】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、「ハーフチェンジ」法に従ってin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物に関する:
試験開始後の最初の3時間以内に該フマル酸エステルの総量の約20%〜約40%w/w、約20%〜約35%w/w、または約30%w/wが放出される、および/または
試験開始後の最初の3時間以内に該フマル酸エステルの総量の少なくとも約12%w/w、例えば約12%〜約50%w/w、約15%〜約45%w/w、約20%〜約40%w/w、約20%〜約35%w/w、約22%〜約35%w/w、または約25%w/w、または約30%w/wが放出される、および/または
試験開始後の最初の4時間以内に該フマル酸エステルの総量の約25%〜約40%w/w、約30%〜約40%w/w、または約40%w/wが放出される、および/または
試験開始後の最初の4時間以内に該フマル酸エステルの総量の少なくとも約76%w/w、例えば約76%〜約95%w/w、約80%〜約90%w/w、または約80%w/w、または約85%w/wが放出される、および/または
試験開始後の最初の4時間以内に該フマル酸エステルの総量のほぼ約40%w/w、例えば約10%〜約40%w/w、約15%〜約35%w/w、約20%〜約30%w/w、または約25%w/w、または約30%w/wが放出される、および/または
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約81%w/w、例えば約81%〜約96%w/w、約85%〜約95%w/w、約85%〜約90%w/w、または約80%w/w、または約85%w/w、または約90%w/wが放出される、および/または
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約50%w/w、例えば約20%〜約50%w/w、約25%〜約45%w/w、約30%〜約45%w/w、または約40%w/w、または約45%w/wが放出される、および/または
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約82%w/w、例えば、約82%〜約99%w/w、約85%〜約99%w/w、約85%〜約95%w/w、または約90%w/wが放出される、および/または
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約65%w/w、例えば約25%〜約65%w/w、約30%〜約65%w/w、約35%〜約60%w/w、約40%〜約60%w/w、約50%〜約6O%w/w、または約55%w/w、または約60%w/wが放出される、および/または
試験開始後の最初の8時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、約60%〜約80%w/w、または約75%w/wが放出される、および/または
試験開始後の最初の8時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約92%w/w、例えば約30%〜約92%w/w、約35%〜約90%w/w、約40%〜約85%w/w、約45%〜約80%w/w、約50%〜約75%w/w、約55%〜約75%w/w、約60%〜約75%w/w、または約65%w/w、または約70%w/wが放出される、および/または
試験開始後の最初の12時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約80%w/w、例えば約80%w/wまたはそれ以上、約85%w/wまたはそれ以上、約90%w/wまたはそれ以上、または約95%w/wまたはそれ以上が放出される。
徐 放
【0083】
フマル酸エステルが徐々にまたは遅延して放出され、放出パターンが1日に2回またはそれ以上投与する組成物に適している特定の態様について以下に説明する。適切な製剤方針の例には上記のあらゆるものがある。
【0084】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、水を溶出溶媒に用いてin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物に関する:
試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約35%w/w、例えば約15%〜約35%w/w、例えば約20%〜約30%w/w、または約25%w/wが放出される、および/または
試験開始後の以最初の8時間内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約60%w/w、例えば約30%〜約60%w/w、例えば約40%〜約55%w/w、または約50%w/wが放出される、および/または
試験開始後の最初の10時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、例えば約60%〜約80%w/w、または約75%w/wが放出される,および/または
試験開始後の最初の12時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約80%w/w、例えば約80%w/wまたはそれ以上、例えば約85%w/wまたはそれ以上、約90%w/wまたはそれ以上、または約95%w/wまたはそれ以上が放出される。
1日1回投与するよう設計された組成物
pH非依存性放出
【0085】
フマル酸エステルをpH非依存性に放出し、放出パターンが1日に1回投与する組成物に適している具体的態様を以下に説明する。適切な製剤方針の例には、例えば上記のあらゆる製剤方針を含む、拡散コーティング、例えば制御放出拡散コーティング、マトリックス粒子またはマトリックス錠、ハイドロゲル、パルス用量ドラッグデリバリーシステム、ビタミンE濃縮物またはエタノール、TPGS、コーン油、およびワックスなどとの合剤により提供される組成物がある。
【0086】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、水を溶出溶媒に用いてin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物に関する:
試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約60%w/w、例えば約30%〜約60%w/w、約40%〜約55%w/w、または約50%w/wが放出される、および/または
試験開始後の最初の13.5時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、約60%〜約80%w/w、または約75%w/wが放出される、および/または
試験開始後の最初の18時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約80%w/w、例えば約80%w/wまたはそれ以上、約85%w/wまたはそれ以上、約90%w/wまたはそれ以上、または約95%w/wまたはそれ以上が放出される、および/または
該組成物に含まれるフマル酸エステルは試験開始後の最初の18時間以内に放出される。
pH制御放出
【0087】
フマル酸エステルがpH依存性に放出され、放出パターンが1日に1回投与する組成物に適している特定の態様について以下に説明する。適切な製剤方針の例には、例えばZentner et al(US 6,537,584)およびBae(US 5,484,610)に記載のタイプの腸溶コーティングまたはハイドロゲルを用いて提供される組成物がある。適切な製剤方針のさらなる例には、例えば上記のあらゆる製剤方針を含む、所望により腸溶コーティングを有する、拡散コーティング、例えば制御放出拡散コーティング、マトリックス粒子またはマトリックス錠、ハイドロゲル、パルス用量ドラッグデリバリーシステム、ビタミンE濃縮物またはエタノール、TPGS、コーン油、およびワックスなどとの合剤により提供される組成物がある。
【0088】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、試験の最初の2時間は0.1N塩酸を溶出溶媒に、次いで0.05Mリン酸緩衝液pH6.5または6.8を溶出溶媒に用いてin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物を提供する:
試験開始後の最初の2時間以内に該フマル酸エステルの総量の少なくとも約1%w/w、例えば少なくとも約2%w/w、少なくとも約3%w/w、または約5%w/wが放出される、および/または
試験開始後の最初の4時間以内に該フマル酸エステルの総量のほぼ約90%w/w、例えば約5%〜約90%w/w、約5%〜約85%w/w、約10%〜約80%w/w、約10%〜約70%w/w、約10%〜約65%w/w、約10%〜約60%w/w、約15%〜約50%w/w、約15%〜約35%w/w、約20%〜約30%w/w、または約20%w/w、または約25%w/wが放出される、および/または
試験開始後の最初の4.5時間以内に該フマル酸エステルの総量のほぼ約35%w/w、例えば約15%〜約35%w/w、約20%〜約30%w/w、または約25%w/wが放出される、および/または
試験開始後の最初の5時間以内に該フマル酸エステルの総量のほぼ約92%w/w、例えば約10%〜約92%w/w、約20%〜約85%w/w、約20%〜約80%w/w、約20%〜約70%w/w、約25%〜約60%w/w、約25%〜約55%w/w、約30%〜約50%w/w、または約35%w/w、または約40%w/w、または約45%w/wが放出される、および/または
試験開始後の最初の6時間以内に該フマル酸エステルの総量のほぼ約94%w/w、例えば約15%〜約94%w/w、約25%〜約90%w/w、約30%〜約85%w/w、約35%〜約80%w/w、約35%〜約75%w/w、約40%〜約70%w/w、約45%〜約70%w/w、約55%〜約70%w/w、約60%〜約70%w/w、または約45%w/w、または約50%w/w、または約55%w/w、または約60%w/w、または約65%w/wが放出される、および/または
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約95%w/w、例えば約35%〜約95%w/w、約40%〜約90%w/w、約45%〜約85%w/w、約50%〜約85%w/w、約55%〜約85%w/w、約60%〜約85%w/w、約65%〜約85%w/w、約70%〜約85%w/w、約75%〜約85%w/w、または約65%w/w、または約70%w/w、または約75%w/w、または約80%w/wが放出される、および/または
試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約98%w/w、例えば約45%〜約98%w/w、約50%〜約98%w/w、約55%〜約98%w/w、約60%〜約98%w/w、約65%〜約98%w/w、約70%〜約98%w/w、約75%〜約95%w/w、約80%〜約95%w/w、約85%〜約95%w/w、または約75%w/w、または約80%w/w、または約85%w/w、または約90%w/wが放出される、および/または
試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約60%w/w、例えば約30%〜約60%w/w、約40%〜約55%w/w、または約50%w/wが放出される、および/または
試験開始後の最初の12時間以内に該組成物中に含まれる該フマル酸エステルの総量のがほぼ約99%w/w、例えば約60%〜約99%w/w、約70%〜約99%w/w、約80%〜約99%w/w、約90%〜約99%w/w、または約95%w/w放出される、および/または
試験開始後の最初の13.5時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、約60%〜約80%w/w、または約75%w/wが放出される。
徐々のpH変化における放出(「ハーフチェンジ」法)
【0089】
フマル酸エステルがpH依存性に放出され、放出パターンが1日に1回投与する組成物に適している特定の態様について以下に説明する。適切な製剤方針の例には、例えばZentner et al(US 6,537,584)およびBae(US 5,484,610)に記載のタイプの腸溶コーティングまたはハイドロゲルを用いて提供される組成物がある(この内容は本明細書の一部を構成する。)。適切な製剤方針の例には、例えば上記のあらゆる製剤方針を含む、所望により腸溶コーティングを有する、拡散コーティング、例えば制御放出拡散コーティング、マトリックス粒子またはマトリックス錠、ハイドロゲル、パルス用量ドラッグデリバリーシステム、ビタミンE濃縮物またはエタノール、TPGS、コーン油、およびワックスなどとの合剤により提供される組成物がある。
【0090】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、ハーフチェンジ法に従ってin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物を提供する:
試験開始後の最初の3時間以内に該フマル酸エステルの総量の少なくとも約12%w/w、例えば約12%〜約60%w/w、約15%〜約50%w/w、約20%〜約40%w/w、約20%〜約35%w/w、または約25%w/w、または約30%w/wが放出される、および/または
試験開始後の最初の4時間以内に該フマル酸エステルの総量のほぼ約35%w/w、例えば約15%〜約35%w/w、約20%〜約30%w/w、または約25%w/wが放出される、および/または
試験開始後の最初の5時間以内に該フマル酸エステルの総量のほぼ約45%w/w、例えば約10%〜約45%w/w、約15%〜約40%w/w、約15%〜約35%w/w、約20%〜約30%w/w、または約25%w/w、または約30%w/wが放出される、および/または
試験開始後の最初の7時間以内に該フマル酸エステルの総量のほぼ約65%w/w、例えば約20%〜約65%w/w、約20%〜約60%w/w、約20%〜約50%w/w、約25%〜約45%w/w、約30%〜約45%w/w、または約40%w/w、または約45%w/wが放出される、および/または
試験開始後の最初の8時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約92%w/w、例えば約25%〜約92%w/w、約25%〜約90%w/w、約30%〜約80%w/w、約35%〜約70%w/w、約40%〜約65%w/w、約45%〜約60%w/w、約50%〜約60%w/w、または約55%w/w、または約60%w/wが放出される、および/または
試験開始後の最初の8時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約60%w/w、例えば約30%〜約60%w/w、約40%〜約55%w/w、または約50%w/wが放出される、および/または
試験開始後の最初の12時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約99%w/w、例えば約30%〜約99%w/w、約30%〜約95%w/w、約35%〜約90%w/w、約40%〜約85%w/w、約45%〜約80%w/w、約50%〜約75%w/w、約55%〜約75%w/w、約60%〜約75%w/w、または約65%w/w、または約70%w/wが放出される、および/または
試験開始後の最初の12.5時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、約60%〜約80%w/w、または約75%w/wが放出される、および/または
試験開始後の最初の18時間以内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約80%w/w、例えば約80%w/wまたはそれ以上、約85%w/wまたはそれ以上、約90%w/wまたはそれ以上、または約95%w/wまたはそれ以上が放出される。
徐 放
【0091】
フマル酸エステルが徐々にまたは遅延して放出され、放出パターンが1日に1回投与する組成物に適している特定の態様について以下に説明する。適切な製剤方針の例には、上記のあらゆる組成物がある。
【0092】
したがって、ある局面において、本発明は、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、水を溶出溶媒として用いるin vitro溶出試験を行ったときに該フマル酸エステルの該放出が以下の通りである該医薬組成物を提供する:
試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約35%w/w、例えば約15%〜約35%w/w、約20%〜約30%w/w、または約25%w/wが放出される、および/または
試験開始後の最初の11時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約60%w/w、例えば約30%〜約60%w/w、約40%〜約55%w/w、または約50%w/wが放出される、および/または
試験開始後の最初の14時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約85%w/w、例えば約50%〜約85%w/w、約60%〜約80%w/w、または約75%w/wが放出される、および/または
試験開始後の以最初の18時間内に該組成物中に含まれる該フマル酸エステルの総量の少なくとも約80%w/w、例えば約80%w/wまたはそれ以上、約85%w/wまたはそれ以上、約90%w/wまたはそれ以上、または約95%w/wまたはそれ以上が放出される。
【0093】
典型的には、上記のごとく本発明組成物は、持続的に活性物質(すなわち、フマル酸に代謝され、次いで急速な排除プロセスを受けるフマル酸のモノアルキルエステル)を送達するよう設計される。本明細書に記載の特徴的in vitro放出パターンに加えて、そのような持続放出は臨床試験後に得られる薬物動態パラメーターにも反映される。したがって、フマル酸のモノアルキルエステル(投与されたジアルキルエステルが加水分解または代謝されると血漿中に出現する)のCmaxは、同様または等価用量を投与した場合に以前に論文に記載されたものと同じ桁数であると予期される(すなわち、約0.4〜約2.0mg/Lの範囲のモノメチルエステルのCmaxは120〜240mgジメチルフマレートの経口用量に対応する)。しかしながら、1日に多くの頻度での投与(1日に2〜4錠を1〜3回)を避けるために、濃度が治療領域内である時間を延長することが目的である。したがって、W50(すなわち、血漿濃度がCmaxの50%またはそれ以上である期間)が市販の処置に比べて少なくとも10%、例えば少なくとも20%、少なくとも30%、少なくとも40%、または少なくとも50%延長されると予期される。適切なW50は、少なくとも2時間、例えば約2〜約15時間、または約2.5〜約10時間、または約3〜約8時間の範囲内であると考えられる。
【0094】
さらに、本発明の制御放出組成物は、血漿プロフィールの個体間および/または個体内変動の減少、および該組成物が食物と共に摂取するか食物なしに摂取するかに対する依存性の減少(医薬組成物を食物摂取と同時にかまたは同時でなく投与したときのモノメチルフマレートの血漿濃度プロフィールの変動の減少)をもたらす。したがって、本発明の制御放出組成物は、Fumaderm(登録商標)に比べ、投与頻度の減少および/または平均総1日用量の減少、および/または活性物質の同じ総1日用量での効果の増加をもたらし得る。
【0095】
種々の動力学的モデル、例えばゼロ次(1)、1次(2)、平方根(Higuchiの方程式)(3)は、薬物放出動力学の解釈に適用することができる。
1:Mt = M0 + k0*t
2:InMt = InM + k1*t
3:Mt = M0 + kH*t1/2
【0096】
これら方程式において、Mtは、あらゆる特定の時点で放出された薬剤の累積量であり、M0は、医薬組成物に組み込まれる活性物質の用量である。k0、k1、およびkHは、それぞれゼロ次、1次、およびHiguchiの方程式の速度定数である。
【0097】
本発明のある局面は、ゼロ次溶出放出プロフィールに関する。別の局面は1次溶出放出プロフィールに関する。さらなる局面は、平方根(Higuchiの方程式)溶出放出プロフィールに関する。
【0098】
本発明のある局面において、活性物質として10%〜90%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩、2%〜40%(重量)の医薬的に許容されるポリマー、および1%〜40%(重量)の親水性賦形剤、および所望により医薬的に許容される賦形剤または添加剤を含む制御放出医薬組成物を提供する。
【0099】
本発明の別の局面において、活性物質として40%〜60%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩、15%〜25%(重量)医薬的に許容されるポリマー、および2%〜15%(重量)親水性賦形剤、および所望により医薬的に許容される賦形剤または添加剤を含む制御放出医薬組成物を提供する。
【0100】
本発明のさらなる局面において、活性物質として65%〜80%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩、10%〜25%(重量)医薬的に許容されるポリマー、および2%〜15%(重量)親水性賦形剤、および所望により医薬的に許容される賦形剤または添加剤を含む制御放出医薬組成物を提供する。
【0101】
「医薬的に許容されるポリマー」の例には、限定されるものではないがエチルセルロース、またはメタクリル酸/アクリル酸コポリマー、例えばアンモニオメタクリレートコポリマータイプAおよびB、またはメタクリル酸コポリマーAおよびBが含まれる。
【0102】
「親水性賦形剤」の例には、限定されるものではないがポリエチレングリコール(PEG)、ポビドン、ヒドロキシルプロピルセルロース(HPC)、ヒドロキシエチルデンプン(HES)もしくはヒドロキシプロピルメチルセルロース(HPMC)、または同様の特性を有する物質、またはその混合物が含まれる。
【0103】
本発明のさらなる局面において、医薬的に許容されるポリマーがエチルセルロースである制御放出医薬組成物を提供する。
【0104】
本発明の別の局面において、親水性賦形剤がヒドロキシルプロピルセルロースである制御放出医薬組成物を提供する。
【0105】
本発明の別の局面において、親水性賦形剤がポリエチレングリコールである制御放出医薬組成物を提供する。
【0106】
本発明のさらに別の局面において、活性物質として10%〜90%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩、および重量比1:9〜9:1の2%〜40%(重量)メタクリル酸コポリマーAおよびB、および所望により医薬的に許容される賦形剤または添加剤を含む制御放出医薬組成物を提供する。
【0107】
本発明さらなる局面において、50%〜90%の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む制御放出医薬組成物を提供する。
【0108】
本発明の範囲を限定しない本発明を例示する種々の制御放出製剤を以下に記載する(全ての濃度は最終錠剤に基づく):
1)顆粒
顆粒は、濃度約10〜約90%、特に約50〜約70%の活性物質を、造粒用賦形剤、例えば医薬的に許容されるポリマー、例えばエチルセルロース、例えばEthocel(登録商標)NFプレミアム、またはメタクリル酸/アクリル酸コポリマー、例えばアンモニオメタクリレートコポリマータイプAおよびB(重量比1:9〜9:1)またはメタクリル酸コポリマーAおよびB(重量比1:9〜9:1)と濃度約2〜約40%で混合し造粒することにより製造してよい。濃度約1〜約40%の、親水性賦形剤、例えばポリエチレングリコール(PEG)、ポビドン、ヒドロキシルプロピルセルロース(HPC)、ヒドロキシエチルデンプン(HES)もしくはヒドロキシプロピルメチルセルロース(HPMC)、および/または濃度約0.01〜約3%のHLB値8以上の医薬的に許容される界面活性剤を混合してよい。
2)微晶質製剤
【0109】
結晶化は、例えば+70℃〜-20℃のような適切な温度のイソプロパノールのような再結晶のためのあらゆる適切な有機溶媒中で実施する。ヒドロコロイド(例えばHPMC)または界面活性剤(例えばポリソルベート)は、再結晶化中に結晶の成長を操作するのに適切な濃度で用いることができる。あらゆる造粒/コーティング用賦形剤、例えば医薬的に許容されるポリマー、例えば、濃度約10〜約50%、特に約20〜約35%のエチルセルロース、ポリメタクリレート、例えばアンモニオメタクリレートコポリマータイプAおよびB、またはメタクリル酸コポリマーAおよびBを用いてよい。親水性賦形剤は例えばPEG 400であり得る。
3)カプセル剤およびサシェー剤
【0110】
カプセル(例えば、ゼラチン、HPMC、またはデンプン誘導体のカプセル)またはサシェーは、コートした(コート)微結晶またはコート顆粒、および必要ならば適切な量の充填用賦形剤、例えば糖アルコール、例えばマンニトールおよび/または流動促進剤で充填してよい。
4)錠剤
【0111】
錠剤は微結晶または顆粒で作られてよい。大規模に、特に回転装置(rotary machine)を用いて錠剤を製造する時は、さらに流動活性を増加しまたは錠剤化挙動を改善するための賦形剤が必要かもしれない。必要であれば充填用および結合用(binding)賦形剤として、例えば、濃度約1〜約60%の微晶質セルロース、例えばAvicel(登録商標)102、およびセルロース、濃度約5〜約60%の、結晶、スプレー乾燥または造粒ラクトース1水和物、例えばTablettose(登録商標)、および無水ラクトース1水和物、濃度約0〜約40%の糖アルコール、例えばソルビトールおよびマンニトール、および濃度約0〜約40%の修飾デンプンを挙げることができる。さらに、崩壊剤、例えばデンプンおよびデンプン誘導体、例えばナトリウムデンプングリコレート(濃度約0.2〜約10%)、クロスポビドン(濃度約0.2〜約10%)、ナトリウムカルボキシメチルセルロース(濃度約0.1〜約10%)、流動促進剤、例えばコロイド無水および含水シリカ(濃度約0.2〜約4%)、および潤滑剤、例えばステアリン酸マグネシウム、ベヘン酸カルシウム、およびカルシウムアラキネート(濃度約0.2〜約3%)またはナトリウムステアリルフマレート(濃度約1〜約8%)を加えることができる。
用 量
【0112】
存在するフマル酸の含有量が異なる組成物を提供するだけでなく、本発明は、例えば、種々の量のフマル酸を有する組成物を含む2またはそれ以上の容器を含むキットも提供する。そのようなキットは、時間とともに増加する用量が必要な状況に用いるのに適している。用量の通常のアップスケールを以下に示す。
【0113】
Aは、低強度、例えば約30mgジメチルフマレート(または別のフマル酸エステルの対応する有効用量)に対応する。
【0114】
Bは、高強度、例えば約120mgジメチルフマレート(または別のフマル酸エステルの対応する有効用量)に対応する。
【0115】
本発明のある局面において、剤形中のフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩の量が活性物質90mg〜360mg、例えば活性物質90、120、180、240、または360mgである制御放出医薬組成物を提供する。本発明のさらなる局面において、活性物質の量は活性物質120、180、または240mgである。本発明のさらなる局面において、活性物質の量は180または360mgである。
【0116】
患者を治療するのに投与される本発明の制御放出医薬組成物の1日投与量は、限定されるものではないが体重および年齢、および治療する病状または疾患の根本原因を含む多くの因子に依存し、決定する医師の技術内にある。本発明のある局面において、1日投与量は、例えば1〜3用量で投与する活性物質240〜360mg、別の局面では1〜3用量で投与する活性物質360〜480mg、別の局面では1〜3用量で投与する活性物質480〜600mg、別の局面では1〜3用量で投与する活性物質600〜720mg、別の局面では1〜3用量で投与する活性物質720〜840mg、別の局面では1〜3用量で投与する活性物質840〜960mgで、さらに別の局面では1〜3用量で投与する活性物質960〜1080mgであり得る。
本発明のある局面において、該制御放出医薬組成物はカプセル形である。
【0117】
本発明の別の局面において、錠剤形、例えば患者が飲み込むのを簡単および好都合にする形を有する錠剤、例えばいかなる尖った角もない丸または棒状の形を有する錠剤の形の該制御放出医薬組成物が提供される。
【0118】
本発明の別の局面において、2またはそれ以上の部分に分割されるよう設計された錠剤の形の医薬組成物を提供する。
【0119】
本発明の組成物は、食事と一緒に、または例えば食事前少なくとも約30分〜食事後約2時間の範囲に対応する時間内に食事に関連して投与するか、または該組成物をその日の時間内のあらゆる特定の時点で投与してよい。
【0120】
ある態様において、総1日投与量は、就寝時刻、例えば就寝時刻前約30分またはそれ以内、就寝時刻前約60分またはそれ以内、就寝時刻前約90分またはそれ以内、就寝時刻前約120分またはそれ以内、または就寝時刻前約180分またはそれ以内に投与される。
【0121】
本発明の組成物およびキットは1またはそれ以上の以下の病状の治療に用いるのに適していると予期される:
a. 乾癬
b. 乾癬性関節炎
c. 神経皮膚炎
d. 炎症性腸疾患、例えば
i. クローン病
ii. 潰瘍性大腸炎
e. 自己免疫疾患:
i. 多発性関節炎
ii. 多発性硬化症(MS)
iii. 若年発症糖尿病
iv. 橋本甲状腺炎
v. バセドウ病
vi. SLE(全身性エリテマトーデス)
vii. シェーグレン症候群
viii. 悪性貧血
ix. 慢性活動性(狼瘡)肝炎
x. 関節リウマチ(RA)
xi. 視神経炎。
【0122】
さらに、本発明の新規組成物またはキットは以下の治療に用いることができよう:
1. 痛み、例えば神経根痛、神経根障害関連痛、ニューロパシー痛、または坐骨神経痛/坐骨神経痛の痛み
2. 臓器移植(拒絶の予防)
3. サルコイドーシス
4. リポイド類壊死症
5. 環状肉芽腫。
【0123】
乾癬は以下の病状に関連する可能性があると提唱されてきた:クローン病(Najarian DJ、Gottlieb AB、Connections between psoriasis and Crohn's disease. J Am Acad Dermatol. 2003 Jun;48(6):805-21)、セリアック病(Ojetti V et al、High prevalence of celiac disease in psoriasis. Am J Gastroenterol. 2003 Nov;98(ll):2574-5.)、精神的または心理的疾患、例えば鬱病または人生の危機(Gupta MA、Gupta AK、Psychiatric and psychological co-morbidity in patients with dermatologic disorders:epidemiology and management. Am J Clin Dermatol. 2003;4(12):833-42、およびMallbris L et al、psoriasis phenotype at disease onset:clinical characterization of 400 adult cases. J Invest Dermatol. 2005 Mar; 124(3):499-504.)、肥満、糖尿病、アルコールの過剰摂取/アルコール中毒、および乾癬性関節炎。
【0124】
すなわち、本発明は、ある局面において、乾癬、乾癬性関節炎、神経皮膚炎、炎症性腸疾患、例えばクローン病および潰瘍性大腸炎、自己免疫疾患、例えば多発性関節炎、多発性硬化症(MS)、若年発症糖尿病、橋本甲状腺炎、バセドウ病、SLE(全身性エリテマトーデス)、シェーグレン症候群、悪性貧血、慢性活動性(狼瘡)肝炎、関節リウマチ(RA)および視神経炎、痛み、例えば神経根痛、神経根障害関連痛、ニューロパシー痛、または坐骨神経痛/坐骨神経痛の痛み、臓器移植(拒絶の予防)、サルコイドーシス、リポイド類壊死症、または環状肉芽腫の治療方法であって、有効量の本発明制御放出医薬組成物をそれを必要とする患者に経口投与することを含む方法に関する。
【0125】
本発明は、別の局面において、乾癬、乾癬性関節炎、神経皮膚炎、炎症性腸疾患、例えばクローン病および潰瘍性大腸炎、自己免疫疾患、例えば多発性関節炎、多発性硬化症(MS)、若年発症糖尿病、橋本甲状腺炎、バセドウ病、SLE(全身性エリテマトーデス)、シェーグレン症候群、悪性貧血、慢性活動性(狼瘡)肝炎、関節リウマチ(RA)および視神経炎、痛み、例えば神経根痛、神経根障害関連痛、ニューロパシー痛または坐骨神経痛/坐骨神経痛の痛み、臓器移植(拒絶の予防)、サルコイドーシス、リポイド類壊死症または環状肉芽腫を治療するための医薬を製造するための本発明制御放出医薬組成物の使用に関する。
【0126】
さらに、本発明は、本発明の組成物またはキットを用いる、上記病状の1つ、より具体的には乾癬または乾癬性関節炎に罹患した個体の治療にも関するものであり、該個体はさらに以下で治療される。
a)局所抗乾癬薬、例えば1)ビタミンDまたはその誘導体(カルシポトリオール、カルシポトリエン)、2)コルチコステロイド(例えば、ベタメタゾン、デソキシメタゾン、フルオシノロン、モメタゾン、ヒドロコーチゾンアセポネート、フルチカゾン、クロベタゾール、クロベタゾン、ヒドロコーチゾンブチレート、デソニド、トリアムシノロン、またはヒドロコーチゾン)、3)タザロテン、4)ジトラノール、5)タクロリムス(FK-506)、および他のカルシネウリン阻害剤、例えばピメクロリムス、または6)1〜5のあらゆる組み合わせ、および/または
b)経口抗乾癬薬、例えば1)経口レチノイド(例えばアシトレチンまたはエトレチネート)(PUVAと組み合わせてまたは組み合わせずに)、2)シクロスポリンおよび他のカルシネウリン阻害剤、例えばISA247、タクロリムスおよびピメクロリムス、3)メトトレキセート、4)ヒドロキシウレア、5)アザチオプリン、6)スルファサラジン、7)フマレート誘導体(例えば、Fumaderm(登録商標)またはBG-12)、8)ロシグリタゾン(Avandia)および他のペルオキシソーム増殖因子活性化γ(PPARγ)アゴニストまたはモジュレーター、例えばピオグリタゾン、ファルグリタザル、GW1929、GW7845、MC-555、MBX-102/MBX-10、MBX-1828、MBX-2044、CLX-0921、R-483、レグリタザル、ナベグリタザル(LY-519818/LY-818)、ネトグルタゾン(MCC-555)、CS-7017、トログリタゾン、シグリタゾン、テサグルタザル、イサグリタゾン、バラグリタゾン、ムラグリタザル、TAK-654、LBM642、DRF 4158、EML 4156、T-174、TY-51501、TY-12780、VDO-52、またはAMG-131(T131)、または1〜8のあらゆる組み合わせ、および/または
c)非経口投与抗乾癬薬、例えば1)アレファセプト(Amevive)、2)エタネルセプト(Enbrel)、3)エファリズマブ(Raptiva)、4)オネルセプト、5)アダリムマブ(Humira)、または1〜5のあらゆる組み合わせ、および/または
d)腸内または非経口経路で投与される、上記c)項に記載していないTNFα阻害剤(例えば、CDP 870またはインフリキシマブ(Remicade))、および/または
e)チソカリシトレート、および/またはNCX 1022および/またはIDEC-131および/またはMEDI-507、および/または
f)NSAID、またはCOXまたはLOX阻害剤、例えば、COX-2阻害剤またはCOX/5-LOX阻害剤、および/または
g)抗糖尿病薬または抗肥満薬、例えばビグアニド、例えばメトホルミン;メトホルミンXR;スルホニルウレア、例えばクロルプロパミド、グリピジド、グリクラジド、グリブリド/グリベンクラミドまたはグリメピリド;グルコバンス(メトホルミン+フリブリド);メタグリプ(グリピジド+メトホルミン);ペルオキシソーム増殖因子活性化γ(PPARγ)アゴニストまたはモジュレーター、例えばロシグリタゾン(Avandia)、ピオグリタゾン、ファルグリタザル、GW1929、GW7845、MC-555、MBX-102/MBX-10、MBX-1828、MBX-2044、CLX-0921、R-483、レグリタザル、ナベグリタザル(LY-519818/LY-818)、ネトグリタゾン(MCC-555)、CS-7017、トログリタゾン、シグリタゾン、テサグリタザル、イサグリタゾン、ベラグリタゾン、ムラグリタザル、TAK-654、LBM642、DRF 4158、EML 4156、T-174、TY-51501、TY-12780、VDO-52、またはAMG-131(T131);アバンダメト(ロシグリタゾン+メトホルミン);アクトス(ピオグリタゾン+メトホルミン);アバンダリル(ロシグリタゾンマレエート+グリメピリド);アクトス(ピグリタゾン+メトホルミン);アバンダリル(ロシグリタゾンマレエート+グリメピリド);ベンゾイミダゾール、例えばFK-614;CS-917;TA-1095;ONO-5129;TAK-559;TAK-677/AJ-9667;d-フェニルアラニン誘導因子、例えばセナグリニド;c-3347;NBI-6024;イングリホリブ;BVT 3498;LY 929;SGLT2阻害剤;CS 011;BIM 51077;R1438;R1439;R1440;R1498;R1499;AVE0847;AVE 2268;AVE 5688;AVE 8134;TA-6666;AZD 6370;SSR 162369;TLK-17411;NN 2501;MK 431;KGA-2727;MK-767;CS-872;β-3レセプターアンタゴニスト、例えばN-5984;α-グルコシダーゼ阻害剤、例えばアカルボース、ボグリボーズ、またはミグリトール;グリニチド/メグリチニド類似体またはカルバモイルメチル安息香酸(bensoeic acid)誘導体、例えばミチグリニド、レパグリニド、またはナテグリニド;DPP-IV阻害剤、例えばLAF 237(ビルダグリプチン)、DPP728、P93/01、P32/98、PT-630、またはサキサグリプチン;GLP-1またはGLP-1類似体、例えばエキセナチド、エキセナチド-LAR、リラグルチド(NN 2211)、ZP10/AVE0010、LY307161、ベタトロピン、CJC-1131、GTP-010、SUN E7001、またはAZM 134;プラムリニチドアセテート;インスリンまたはインスリン類似体、例えばヒューマログ(インスリンリスプロ)、ヒューマリン、ノボリン、ノボログ/ノボラピッド(インスリンアスパート)、アピドラ(インスリングルイシン)、ランタス(インスリングラルギン)、エクスベラ、レベミル/NN 304(インスリンデテミル)、AERx/NN 1998、インスマン、肺インスリン、またはNN 344;シブトラミン、または他のセロトニンおよびノルアドレナリンのシナプス前再取り込み遮断薬;オルリスタット、および他のGIリパーゼ阻害剤;β3-アドレナリンレセプターアゴニスト;脱共役タンパク質;PPARγの(特異的)アンタゴニスト(ペルオキシソーム増殖因子活性化レセプターγ);インスリン分泌促進剤;リモナバントおよび他のCB1エンドカンナビノイドアンタゴニスト;ブプロピオン;トピラメート;レプチンアゴニスト;毛様体神経栄養因子;ヒトペプチドホルモン断片のペプチド類似体177-191;コレシストキニン-Aレセプターアゴニスト;メラノコルチン-3アゴニスト;ノルアドレナリン作動薬、例えばフェンテルミン、ジエチルプロピオン、フェンジメトラジン、またはベンズフェタミン;または上記抗糖尿病薬または抗肥満薬のあらゆる組み合わせ、および/または
h)物質乱用、例えばアルコール乱用の治療に潜在的に有用な薬剤、例えばナルトレゾン、アカンプロセート、ジスルフィラム、またはビビトレックス(ナルトレゾン長時間作用注射)、および/または
i)クローン病の治療に潜在的に有用な薬剤、例えば
1. 5-ASA化合物、例えばスルファサラジン、経口5-ASA製剤、または直腸5-ASA製剤、
2. グルココルチコステロイド、例えば全身性ステロイド(例えば、ブデソニドまたはプレドニゾロン)または局所作用ステロイド(例えば、ブデソニド)、
3. 抗生物質、例えばメトロニダゾールまたはキノロン(例えば、シプロフロキサシン、オフロキサシン、ノルフルキサシン、レボフロキサシン、またはモキシフロキサシン)、
4. 免疫抑制剤、例えばアザチオプリン、6-メルカプトプリン、またはメトトレキセート、
5. 栄養療法、例えば元素または高分子製剤、またはプレ-およびプロバイオティクス、
6. 生物療法、例えば、TNF-α阻害剤、例えばインフリキシマブ、アダリムマブ、CDP870、CDP571、エタネルセプト、またはオネルセプト、
7. 対症療法剤、例えば抗下痢剤または抗痙攣剤。
【0127】
適切なNSAIDの例には、ピロキシカム、ジクロフェナク、ナブメトン、ナプロキセン、フルルビプロフェン、フェノプオフェン、ケトプロフェン、およびイブプロフェンを含むプロピオン酸、メフェナム酸を含むフェナメート、パラセタモール、インドメタシン、スリンダク、メロキシカム、アパゾン、フェニルブタゾンを含むピラゾロン、アスピリンを含むサリチル酸塩がある。
【0128】
適切なCOX-2阻害剤の例には、ロフェコキシブ(Vioxx)、バルデコキシブ(Bextra)、セレコキシブ(Celebrex)、エトリコキシブ(Arcoxia)、ルミラコキシブ(Prexige)、パレコキシブ(Dynastat)、デラコキシブ(Deram)、チラコキシブ、メロキシカム、ニメソリド、(1,1-ジメチルヘプチル)-6a,7,10,10a-テトラヒドロ-1-ヒドロキシ-6,6ジメチル-6H-ジベンゾ[b,d]ピランカルボン酸(CT-3)、2(5H)-フラノン、5,5-ジメチル(l-メチルエトキシ)[4(メチルスルホニル)フェニル]-(DFP);カプロフェン(RIMADYL)、(アセチルオキシ)-安息香酸、3-[(ニトロオキシ)メチル]フェニルエステル(NCX4016)、P54(CAS Reg. No.130996 0) 2,6-ビス(1,1-ジメチルエチル)[(E)-(2-エチル-1,1-ジオキソイソチアゾリジニリデン)メチル]フェノール(S-2474)、5(R)-チオスルホンアミド-3(2H)-ベンゾフラノン(SVT-2016)、およびN-[3-(ホニル-アミノ)オキソフェノキシ-4H ベンゾピラン イル]メタンスルホンアミド(「T-614」);またはその医薬的に許容される塩がある。
【0129】
適切なCOX/5-LOX阻害剤の例には、リコフェロン(ML-3000または[2,2-ジメチル-6-(4-クロロフェニル)-7-フェニル-2,3,ジヒドロ-1H-ピロリジン-5-イル]-酢酸)、ジ-tert-ブチルフェノール、例えば(E)-(5)-(3,5-ジ-tert-ブチル-4-ヒドロキシベンジリデンス)-2-エチル-l,2-イソチアゾリジン-1,1-ジオキシド(S-2474)、ダルブフェロンまたはテブフェロン、および薬理学的に活性な代謝物ならびに誘導体、例えばジヒドロ-ジメチル-ベンゾフランおよびPGV-20229、ジヒドロ-ジメチル-ベンゾフラン、チオフェン誘導化合物、例えばRWJ-63556、N-ヒドロキシ-N-メチル-4-(2,3-ビス-(4-メトキシフェニル)-チオフェン-5-イル)-ブタンアミド(S19812)、メトキシテトラヒドロピラン誘導体、酸化キサントン、例えば1,3,6,7-テトラヒドロキシキサントン(ノルアチリオール(norathyriol))-ピラゾールチオカルバメート、ピラゾール、例えば修飾形のフェニドン含有化合物、またはトリ-フルオロ-ベンゾール置換ピラゾリン誘導体BW-755C、テポキサリンおよび誘導体、およびジ-tert-ブチルピリミジンがある。
【0130】
そのような併用療法は、本発明の組成物またはキットを用いずに治療される個体に比べて個体に対する治療反応の改善および/または利便性の増大をもたらすと予期される。
【0131】
さらなる局面において、本発明はあらゆる上記病状a〜eおよび1〜5の経口治療に伴う副作用を減少させる方法であって、該病状を治療するための医薬成分が1またはそれ以上の以下の薬剤と組み合わせて用いられる方法に関する:
a)制酸剤、例えば1)水酸化マグネシウム、2)三ケイ酸マグネシウム、3)水酸化アルミニウムゲル、3)炭酸水素ナトリウム、4)マガルドラト、または1〜5のあらゆる組み合わせ、および/または
b)ヒスタミンH-2アンタゴニスト、例えば1)シメチジン、2)ラニチジン、3)ニザチジン、4)ファモチジン、5)ロキサチジン、6)ラフタジン、または1〜6のあらゆる組み合わせ、および/または
c)細胞保護剤、例えば1)スクラルファート、2)トリカリウムジクチトラトビスムテート、3)カルベノキソロン、4)プロスタグランジンE-2類似体、例えばミソプロストール、5)エカベト、6)セトラキセートHCl、7)テプレノン、8)トロキシピド、9)ジシクロミン塩酸、10)ソファルコンまたは1〜10のあらゆる組み合わせ、および/または
d)プロトンポンプ阻害剤(PPI)、例えば1)オメプラゾール、2)エソメプラゾール、3)ランソプラゾール、4)パントプラゾール、5)ラベプラゾール、6)CS-526/R-105266、7)AZD 0865、8)ソラプラザン、または1〜8のあらゆる組み合わせ、および/または
e)NSAIDまたはCOXまたはLOX阻害剤、例えば、COX-2阻害剤またはCOX/5-LOX阻害剤、および/または
f)ペントキシフィリン(例えば用量範囲400〜800mg/日で)。
【0132】
具体的態様において、活性物質はフマル酸エステル含有化合物である。具体的には、フマル酸エステル含有化合物は、Fumaderm(登録商標)またはFumaraat(登録商標)またはPanaclar(登録商標)(BG-12)に含まれるか、US 6,277,882、US 6,355,676もしくはUS 6,509,376に記載の、または本発明の製剤に含まれる塩のいずれかおよびすべてである。活性医薬成分は、本発明の製剤、またはあらゆるFumaderm(登録商標)またはFumaraat(登録商標)またはPanaclar(登録商標)製剤において、または例えば、US 6,277,882、US 6,355,676またはUS 6,509,376に記載のごとく得ることができよう。
【0133】
本発明は、記載した具体的態様に限定されず、もちろん変化し得ると理解すべきである。本発明の範囲は添付の特許請求の範囲にのみ限定されるので、本明細書で用いる専門用語は、特定の態様を説明するためのみのものであり、限定を意図するものではないと理解すべきである。値の範囲が示されている場合は、該範囲の上限と下限の間のそれぞれの間の値(文脈が明確にそうでないと記載していない限り、下限の単位の1/10まで)、およびあらゆる他の記載されるかまたは範囲が記載された間の値も本発明内に含まれると理解すべきである。このより小さい範囲の上限および下限は、独立して該より小さい範囲に含まれ、本発明内に含まれる(記載した範囲内のあらゆる具体的に除外した制限を受ける)。記載した範囲が該限界の1または両方を含む場合は、その含まれる限界のいずれかまたは両方を排除する範囲も本発明に含まれる。特記しない限り、本明細書で用いるすべての技術的および科学的用語は、本発明が属する技術分野の当業者が通常理解するのと同じ意味を有する。本明細書に記載のものと同様または等価なあらゆる方法および物質を本発明の実施または試験に用いることもできるが、好ましい方法および物質を記載する。本明細書に記載のすべての刊行物は、本明細書の一部を構成し、該刊行物が引用している方法および/または物質を開示し説明する。本明細書および添付の特許請求の範囲で用いている単数形の「a」、「an」および「the」は、文脈が明確にそうでないと記載していない限り複数形の言及を含むと理解すべきである。本明細書に記載の特許および刊行物は、単に本出願の出願日以前の開示内容を示すものである。本明細書のいかなる内容も、本発明が以前の発明に基づいてそのような特許または刊行物に先行することはできないと認めるものと解釈すべきでない。さらに、示した刊行物の日付は、実際の刊行日と異なることがあり、別々に確認する必要がありうる。この開示を読んだ当業者に明らかであろうように、本明細書に記載し、例示した個々の各態様は、別個の要素と特徴を有し、本発明の範囲や精神から離れることなく他のあらゆる種々の態様の特徴から容易に分離または結合してよい。本明細書に示す図面は必ずしも一定の縮尺を示すものではなく、ある要素および特徴は明確さのために誇張されている。
【0134】
前記発明について理解を明確にするために例示および実施例により詳細に説明したが、本発明の開示に照らして添付の特許請求の範囲の精神や範囲から離れることなくある種の変更および修飾を行ってよいことは当業者は容易に明白であろう。
【実施例】
【0135】
実施例1
錠剤の製造
200g顆粒を、150g微晶質セルロース(例えば、Avicel(登録商標) 102)、97.5gラクトース(例えば、Tablettose(登録商標))、10gナトリウムカルボキシメチルセルロース(例えば、Ac-Di-Sol(登録商標))、および25gデンプンと30分間混合する。次に、10gステアリン酸マグネシウムおよび7.5g非晶質二酸化ケイ素(例えば、Aerosil(登録商標) 200)を加え、粉末混合物を5分間混合する。
【0136】
この粉末混合物を打錠器で圧縮して錠剤にする(錠剤直径10mm、表面約280-300mm2)。該錠剤を実施例4に記載のごとくパンコーティングまたは流動コーティング(fluid-bed coating)法を用いて腸溶コートする。
実施例2
錠剤の製造
【0137】
200g微結晶を150g微晶質セルロース(例えば、/Avicel(登録商標) 102)、130gラクトース(例えば、Tablettose(登録商標))、10gナトリウムカルボキシメチルセルロース(例えば、Ac-Di-Sol(登録商標))、および25mgデンプンと30分間混合する。次に、10gステアリン酸マグネシウムおよび7.5g非晶質二酸化ケイ素を加え、次いで粉末混合物を5分間混合物する。この粉末混合物を打錠器で圧縮して錠剤にする(錠剤直径10mm、表面約280-300mm2)。該錠剤を実施例4に記載のごとくパンコーティングまたは流動コーティング(fluid-bed coating)法を用いて腸溶コートする。
実施例3
カプセルの製造
【0138】
顆粒または微結晶をHPMCカプセルに充填し、該カプセルを以下に記載のごとく腸溶コートする。パンコーティング器を用い、Eudragit(登録商標) L30D-55を60℃〜80℃の乾燥温度でポリマー物質20mg/mm2の量でカプセル上にスプレーする。色素およびタルクを適当量加える。
実施例4
錠剤の腸溶コーティング
【0139】
パンコーティング器を用い、Eudragit(登録商標) L30D-55を60℃〜80℃の乾燥温度でポリマー物質6mg/mm2の量で錠剤上にスプレーする。色素およびタルクを適当量加える。
実施例5
カプセルの製造
【0140】
実施例15に記載のごとく製造した156mgの微結晶を硬ゼラチンカプセルサイズ0に充填する。カプセル剤を、各カプセル面を4回、アセトン中の5% HPMCP(Pharmacoat HP 50(登録商標))の溶液に浸漬して腸溶コートする。
実施例6
顆粒の製造
【0141】
造粒法を用い、50gジメチルフマレート(以下のDMF中)を1gエチルセルロース(例えば、Ethocel(登録商標)NFプレミアム)と混合し、これを10mLエタノール96%に溶解し、シーブ1.0mmを通し、次いで50℃〜60℃で30分間乾燥する。この顆粒を実施例1および3に記載したのと同じ方法を用いて錠剤およびカプセルに製造する。
実施例7
顆粒の製造
【0142】
造粒法を用い、50g DMFを1gポリビニルアセテート(PVA)(例えば、Kollicoat(登録商標) SR30)と混合し、これを10mLエタノール96%に溶解し、シーブ1.00mmを通し、次いで50℃〜60℃で30分間乾燥する。
実施例8
顆粒の製造
【0143】
造粒法を用い、50g DMFを15gの粉末Eudragit(登録商標) RL 100と混合する。適切な量の2-プロパノールを加え、シーブ1.00mmを通し、次いで60℃で乾燥する。この顆粒を実施例1および3に記載したのと同じ方法を用いて錠剤およびカプセルに製造する。
実施例9
コート顆粒の製造
【0144】
造粒法を用い、50g DMFを直接5g Eudragit(登録商標) RL30Dと混合し、シーブ(1.00mm)を通し、次いで80℃で乾燥する。シーブを通した後、顆粒を流動コーティング器(Mini-Glatt)を用い、15gのEudragit(登録商標) RL30D/RS30D、1:1混合物でコートする。コート顆粒を実施例1および3に記載したのと同じ方法を用いて錠剤およびカプセルに製造することができる。
実施例10
コート顆粒の製造
【0145】
造粒法を用い、50g DMFを20%エチルセルロース(例えば、Ethocel(登録商標)NFプレミアム)と混合し、これを適切な量のエタノール96%に溶解する。15%ポリエチレングリコール6000を造粒液に加える。混合物をシーブ1.00mmに通し、50℃〜60℃で30分間乾燥する。シーブを通した後、顆粒を流動コーティング(Mini-Glatt)を用い、20mg/mm2顆粒表面積の量のエチルセルロースとポリエチレングリコール6000の2:1混合物でコートする。この顆粒を実施例1および3に記載したのと同じ方法を用いて錠剤またはカプセルに製造することができる。
実施例11
コート顆粒の製造
【0146】
造粒法を用い、50g DMFを10%エチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)と混合し、次いで適切な量のエタノール96%に溶解する。6%ポビドン(例えば、Kollidon(登録商標) 25)を造粒液に加える。混合物をシーブ1.00mmに通し、50℃〜60℃で30分間乾燥する。シーブを通した後、顆粒を流動コーティング(Mini-Glatt)を用い、20mg/mm2顆粒表面積の量のエチルセルロースとポビドンの3:2混合物でコートする。
この顆粒を実施例1および3に記載した方法を用いて錠剤またはカプセルに製造することができる。
実施例12
コート顆粒の製造
【0147】
造粒法を用い、50g DMFを10%エチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)と混合し、次いで適切な量のエタノール96%に溶解する。5%ヒドロキシプロピルセルロース(HPC)(例えば、Klucel(登録商標))を造粒液に加える。混合物をシーブ1.00mmに通し、50℃〜60℃で30分間乾燥する。シーブを通した後、顆粒を流動コーティング(Mini-Glatt)を用い、20mg/mm2顆粒表面積の量のエチルセルロースとHPCの2:1混合物でコートする。
この顆粒を実施例1および3に記載した方法を用いて錠剤またはカプセルに製造することができる。
実施例13
コート顆粒の製造
【0148】
造粒法を用い、50g DMFを直接適切な量のEudragit(登録商標) NE30Dの水性分散液と混合し、シーブ(1.00mm)を通し、次いで80℃で乾燥する。シーブを通した後、顆粒を流動コーティング器(Mini-Glatt)を用い、15gのEudragit(登録商標) RL30D/RS30D、1:1混合物でコートする。コート顆粒を実施例1および3に記載した方法を用いて錠剤およびカプセルに製造することができる。
実施例14
コート顆粒の製造
【0149】
造粒法を用い、50g DMFを直接適切な量のEudragit(登録商標) RL30Dの水性分散液と混合し、シーブ(1.00mm)を通し、次いで80℃で乾燥する。シーブを通した後、顆粒を流動コーティング器(Mini-Glatt)を用い、Eudragit(登録商標) NE30Dでコートする。コート顆粒を実施例1および3に記載した方法を用いて錠剤およびカプセルに製造することができる。
実施例15
コート微結晶の製造
【0150】
300mL 2-プロパノール中の50g DMFの飽和溶液を60℃で調製し、永続的に撹拌しながら徐々に冷却する。沈殿した結晶を濾過して除去し、50℃で乾燥する。結晶をシーブに通し、315〜710μm分画をパンコーティング器または流動コーティング器(Mini-Glatt)を用いるコーティング工程に用いる。500gエタノール中の、12gエチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)および3gポリエチレングリコール400のコーティング溶液を粉末表面上に60℃でスプレーする。乾燥後、コートした結晶を1.00mmシーブに通す。このコートDMF結晶を実施例2および3に記載の方法を用いて錠剤およびカプセルに製造することができる。
実施例16
錠剤の製造
【0151】
造粒法を用い、50g DMFを2gエチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)および3gポリエチレングリコール400と混合し、これを150mLエタノール96%に溶解し、1.0mmシーブを通し、50〜60℃で30分間乾燥し、再度1.0mmシーブに通す。プラセボ顆粒を以下のごとく製造する:Tablettose(登録商標)およびAvicel(登録商標)102を等量混合し、水(q.s.)に溶解した2%ポビドン(例えば、Kollidon(登録商標)25)で顆粒とし、1.0mmシーブを通し、50℃〜60℃で30分間乾燥し、再度1.0mmシーブに通す。DMF-顆粒60部およびプラセボ顆粒38部をTurbulaシェーカーミキサー中で30分間混合する。Aerosil(登録商標)200を1部およびステアリン酸マグネシウム1部を加え、混合物を再度5分間混合する。混合物を直径10mm、重量約260mg、および硬さ約50Nの錠剤に圧縮する。錠剤を実施例4に記載の方法を用いて腸溶コートする。
実施例17
錠剤の製造
【0152】
造粒法を用い、50g DMFを12gエチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)および3gポリエチレングリコール400と混合し、これを150mLエタノール96%に溶解し、1.0mmシーブに通し、50〜60℃で30分間乾燥し、再度1.0mmシーブに通す。プラセボ顆粒を以下のごとく製造する:Tablettose(登録商標)およびAvicel(登録商標)102を等量混合し、水(q.s.)に溶解した2%ポビドン(例えば、Kollidon(登録商標)25)で顆粒とし、1.0mmシーブに通し、50℃〜60℃で30分間乾燥し、再度1.0mmシーブに通す。DMF-顆粒60部およびプラセボ顆粒37部をTurbulaシェーカーミキサー中で30分間混合する。カルボキシメチルセルロース(例えば、Ac-Di-Sol(登録商標))1部、Aerosil(登録商標)200を1部およびステアリン酸マグネシウム1部を加え、混合物を再度5分間混合する。
混合物を直径10mm、重量約260mg、および硬さ約50Nの錠剤に圧縮する。錠剤を実施例4に記載の方法を用いて腸溶コートする。
実施例18
コート微結晶の製造
【0153】
300ml 2-プロパノール中の50g DMFの飽和溶液を60℃で調製し、永続的に撹拌しながら徐々に冷却する。沈殿した結晶を濾過して除去し、50℃で乾燥する。結晶をシーブに通し、315〜710μm分画をパンコーティング器または流動コーティング器(Mini-Glatt)を用いるコーティング工程に用いる。500gエタノール中の、12gエチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)および3gポビドン(PVP)のコーティング溶液を結晶表面上に60℃でスプレーする。乾燥後、コートした結晶を1.00mmシーブに通す。コートDMF結晶を実施例2および3に記載の方法を用いて錠剤およびカプセルに製造することができる。
実施例19
コート微結晶の製造
【0154】
300ml 2-プロパノール中の50g DMFの飽和溶液を60℃で調製し、永続的に撹拌しながら徐々に冷却する。沈殿した結晶を濾過して除去し、50℃で乾燥する。結晶をシーブに通し、315〜710μm分画をパンコーティング器または流動コーティング器(Mini-Glatt)を用いるコーティング工程に用いる。500gエタノール中の、12gエチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)および3gヒドロキシルプロピルセルロース(HPC)のコーティング溶液を粉末表面上に60℃でスプレーする。乾燥後、コートした結晶を1.00mmシーブに通す。コートDMF結晶を実施例2および3に記載の方法を用いて錠剤およびカプセルに製造することができる。
実施例20
微結晶の製造
【0155】
DMF結晶を実施例15に記載のごとく製造する。ただし、結晶質量に対し2%のエチルセルロースを結晶が沈殿する前に2-プロパノールに直接加える。
実施例21
コート微結晶の製造
【0156】
実施例15に記載のごとく調製した50g DMF結晶を流動コーティング器(Mini-Glatt)を用い、80℃の温度で20gのEudragit(登録商標) RL30D/RS30D、1:1混合物の水性分散液でコートする。コートDMF結晶を実施例2および3に記載した方法を用いて錠剤およびカプセルに製造する。
実施例22
錠剤の製造
【0157】
実施例15に記載のごとく調製したDMF結晶を、25%固体Eudragit(登録商標)RS PO/RL PO(比1:2)と直接混合し、実施例2に記載のごとく錠剤を製造する。
実施例23
コート微結晶の製造
【0158】
実施例15に記載のごとく調製したDMF結晶を、流動コーティング器(Mini-Glatt)を用い、5%(結晶の質量に対して)の量のポリビニルアセテートの水性分散液(例えば、Kollicoat(登録商標) SR30D)でコートする。コートDMF結晶を実施例2および3に記載の方法を用いて錠剤およびカプセルに製造することができる。
実施例24
顆粒の製造
【0159】
造粒法を用い、50g DMFを15%エチルセルロース(例えば、Ethocel(登録商標)NFプレミアム)と混合し、これを適切な量のエタノール96%に溶解する。10%ポリエチレングリコール6000を造粒液に加える。混合物をシーブ1.00mmに通し、50℃〜60℃で30分間乾燥する。この顆粒を実施例1および3に記載した方法を用いて錠剤またはカプセルに製造することができる。
実施例25
顆粒の製造
【0160】
造粒法を用い、50gジエチルフマレート(DEF)を15%エチルセルロース(例えば、Ethocel(登録商標)NFプレミアム)と混合し、これを適切な量のエタノール96%に溶解する。10%ポリエチレングリコール6000を造粒液に加える。混合物をシーブ1.00mmに通し、50℃〜60℃で30分間乾燥する。この顆粒を実施例1および3に記載した方法を用いて錠剤またはカプセルに製造することができる。
実施例26
錠剤の製造
【0161】
PEG 6000の代わりに10%ポビドン(例えば、Kollidon(登録商標) 25)を加える以外は実施例24に記載のごとく顆粒を製造する。この混合物を実施例1および3に記載した方法を用いて錠剤またはカプセルに製造することができる。
実施例27
錠剤の製造
【0162】
PEG 6000の代わりに10%ヒドロキシルプロピルメチルセルロースを加える以外は実施例24に記載のごとく顆粒を製造する。この混合物を実施例1および3に記載した方法を用いて錠剤またはカプセルに製造することができる。
実施例28
【0163】
実施例15に記載のごとく製造した50g DMF結晶を流動コーティング器(Mini-Glatt)を用い、80℃の温度で20gのEudragit(登録商標) RL30D/RS30D、1:1混合物の水性分散液でコートする。コート結晶を以下に記載のごとくパンコーティング器で腸溶コートする。Eudragit(登録商標) L30D-55を6mgポリマー物質/mm2の量で乾燥温度60℃〜80℃でコート結晶上にスプレーする。
【0164】
この二重コートDMF結晶を硬ゼラチンカプセルまたは軟ゼラチンカプセルに充填するか、または実施例2に記載の方法を用いて錠剤とする。
実施例29
錠剤の製造
【0165】
造粒法を用い、50g DMFを12gエチルセルロース(例えば、Ethocel(登録商標) NFプレミアム)および3gヒドロキシプロピルセルロース(例えば、Klucel(登録商標))と混合し、これを150mLエタノール96%に溶解し、1.0mmシーブに通し、50〜60℃で30分間乾燥し、再度1.0mmシーブに通す。
【0166】
Tablettose(登録商標)およびAvicel(登録商標)102を等量混合し、水(q.s.)に溶解した2%ポビドン(例えば、Kollidon(登録商標)25)で顆粒とする。DMF-顆粒60部およびプラセボ顆粒38部をTurbulaシェーカーミキサー中で30分間混合する。Aerosil(登録商標)200を1部およびステアリン酸マグネシウム1部を加え、混合物を再度5分間混合する。混合物を直径10mm、重量約260mg、および硬さ約50Nの錠剤に圧縮する。錠剤を実施例4に記載の方法を用いて腸溶コートする。
実施例30
カプセル剤のpH制御放出溶出プロフィールの測定
【0167】
該溶出プロフィールを、電気モーター(100rpm)で駆動する6個のバスケット撹拌エレメントと容量1Lの6個のいわゆるLevyグラスを取り付けた回転バスケットを用い、United States Pharmacopoeiaに記載のごとく測定する。Levyグラスに0,1N HCl(水浴の温度は37℃±0.5℃)を満たし、カプセル剤を該バスケットに適用する。2時間後、該酸を容器から除去し、溶出溶媒(USPリン酸緩衝液、pH6.5)で置換し、さらに6時間試験する。試料(5ml)を、該酸媒質から0、60、および120分間後、ならびに溶出溶媒をUSP緩衝液で置換後溶出溶媒から30、60、90、120、180、240、300、および360分間後に取り出す。各試料採取後に取り出した緩衝溶液の量を置換する代わりに、放出されたDMFの量を計算する時に緩衝液の減少を考慮する。DMFの量を、25℃に調節したMerck LiChroCART RP8 5μM、20cmカラムを用いるHPLC(Kontron XXX)により測定する。移動相は、アセトニトリルとリン酸でpH3.2に調節した0.0725mol/l NaH2PO4*H2O緩衝液の混合物(35:65)からなる。UV検出器を波長230nmおよび流速1.0mL/分に設定する。DMFピークは約5分間の保持時間後に検出することができる。
実施例31
非腸溶性錠剤のpH制御放出溶出プロフィールの測定
【0168】
溶出プロフィールを、電気モーターで駆動する撹拌エレメントとして6個のパドルと容量1Lの6個のいわゆるLevyグラスを用いて測定する。パドルの回転速度は100rpmである。LevyグラスにUSPリン酸緩衝液pH6.5(水浴の温度は37℃±0.5℃)を満たし、錠剤を該バスケットに適用する。試料(5ml)を、溶出溶媒をUSP緩衝液で置換後に溶出溶媒から0、30、60、90、120、180、240、300、および360分間後に取り出す。各試料採取後に取り出した緩衝溶液の量を置換する代わりに、放出されたDMFの量を計算する時に緩衝液の減少を考慮する。DMFの量を、25℃に調節したMerck LiChroCART RP8 5μM、20cmカラムを用いるHPLC(Kontron XXX)により測定する。移動相は、アセトニトリルとリン酸でpH3.2に調節した0.0725mol/l NaH2PO4*H2O緩衝液の混合物(35:65)からなる。UV検出器を波長230nmおよび流速1.0mL/分に設定する。DMFピークは約5分間の保持時間後に検出することができる。
実施例32
【0169】
実施例5に記載のごとく製造したカプセル剤の溶出プロフィールを実施例30に記載のごとく測定する。溶出プロフィールを図1に示す。
実施例33
【0170】
実施例16に記載のごとく製造した錠剤(腸溶コーティングを適用する前)の溶出プロフィールを実施例31に記載のごとく測定する。溶出プロフィールを図2に示す。
実施例34
【0171】
実施例17に記載のごとく製造した錠剤(腸溶コーティングを適用する前)の溶出プロフィールを実施例31に記載のごとく測定する。溶出プロフィールを図3に示す。
【図面の簡単な説明】
【0172】
【図1】実施例5に記載のごとく製造したカプセル剤のin vitro溶出プロフィールの例を示す。
【図2】実施例16に記載のごとく製造した錠剤試料(腸溶コーティングを施す前)のin vitro溶出プロフィールの例を示す。
【図3】実施例17に記載のごとく製造した錠剤試料(腸溶コーティングを施す前)のin vitro溶出プロフィールの例を示す。
【特許請求の範囲】
【請求項1】
経口投与すると等価用量のFumaderm(登録商標)錠の経口投与後と比べてGI関連副作用の低下が生じる、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む医薬組成物。
【請求項2】
制御放出組成物の形の請求項1記載の医薬組成物。
【請求項3】
活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、
該フマル酸エステルの該放出が、試験の最初の2時間は0.1N塩酸を溶出溶媒に、次いで0.05Mリン酸緩衝液pH6.5を溶出溶媒に用いるin vitro溶出試験を行うと、試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約70%w/wが放出されるものである該医薬組成物。
【請求項4】
活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、該フマル酸エステルの該放出が、試験の最初の2時間は0.1N塩酸を溶出溶媒に、次いで0.05Mリン酸緩衝液pH6.5を溶出溶媒に用いるin vitro溶出試験を行うと、試験開始後の最初の4時間以内に該フマル酸エステルの総量のほぼ約92%w/wが放出されるものである該医薬組成物。
【請求項5】
該フマル酸エステルの該放出が、試験開始後の最初の4時間以内に該フマル酸エステルの総量のほぼ約92%w/wが放出されるものである請求項3記載の制御放出組成物。
【請求項6】
該フマル酸エステルの該放出が、試験開始後の最初の5時間以内に該フマル酸エステルの総量のほぼ約94%w/wが放出されるものである請求項3〜5のいずれかに記載の制御放出組成物。
【請求項7】
該フマル酸エステルの該放出が、試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約95%w/wが放出されるものである請求項3〜6のいずれかに記載の制御放出組成物。
【請求項8】
該フマル酸エステルの該放出が、試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約98%w/wが放出されるものである請求項3〜7のいずれかに記載の制御放出医薬組成物。
【請求項9】
該フマル酸エステルの該放出が、試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約99%w/wが放出されるものである請求項3〜8のいずれかに記載の制御放出組成物。
【請求項10】
該フマル酸エステルの該放出が、試験開始後の最初の12時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約99%w/wが放出されるものである請求項3〜9のいずれかに記載の制御放出組成物。
【請求項11】
該放出が、ゼロ次、1次、または平方根(Higuchiの方程式)動力学放出プロフィールを有する先の請求項のいずれかに記載の制御放出医薬組成物。
【請求項12】
該放出が平方根(Higuchiの方程式)動力学放出プロフィールを有する請求項11記載の制御放出医薬組成物。
【請求項13】
経口投与すると等価用量のFumaderm(登録商標)錠の経口投与後と比べてGI関連副作用の低下が生じる請求項3〜12のいずれかに記載の制御放出医薬組成物。
【請求項14】
活性成分として10%〜90%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩、2%〜40%(重量)医薬的に許容されるポリマー、および1%〜40%(重量)親水性賦形剤、および所望により医薬的に許容される賦形剤または添加剤を含む制御放出医薬組成物。
【請求項15】
活性物質として40%〜60%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩、15%〜25%(重量)医薬的に許容されるポリマー、および2%〜15%(重量)親水性賦形剤、および所望により医薬的に許容される賦形剤または添加剤を含む請求項14記載の制御放出医薬組成物。
【請求項16】
活性物質として65%〜80%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩、10%〜25%(重量)医薬的に許容されるポリマー、および2%〜15%(重量)親水性賦形剤、および所望により医薬的に許容される賦形剤または添加剤を含む請求項14記載の制御放出医薬組成物。
【請求項17】
該医薬的に許容されるポリマーがエチルセルロースである請求項14〜16のいずれかに記載の制御放出医薬組成物。
【請求項18】
該親水性賦形剤がポリエチレングリコールである請求項14〜17のいずれかに記載の制御放出医薬組成物。
【請求項19】
該親水性賦形剤がヒドロキシルプロピルセルロースである請求項14〜17のいずれかに記載の制御放出医薬組成物。
【請求項20】
活性物質として10%〜90%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステルまたはその医薬的に許容される塩、および2%〜40%(重量)メタクリル酸コポリマーAおよびBを重量比1:9〜9:1で、および所望により医薬的に許容される賦形剤または添加剤を含む制御放出医薬組成物。
【請求項21】
50%〜90%の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む請求項14および16〜20のいずれかに記載の制御放出医薬組成物。
【請求項22】
請求項3〜13のいずれかに記載の溶出プロフィールを有する請求項14〜21のいずれかに記載の制御放出医薬組成物。
【請求項23】
該フマル酸エステルが、ジメチルフマレート、ジエチルフマレート、ジプロピルフマレート、ジブチルフマレート、ジペンチルフマレート、メチル-エチル-フマレート、メチル-プロピルフマレート、メチル-ブチルフマレート、メチル-ペンチルフマレート、モノメチルフマレート、モノエチルフマレート、モノプロピルフマレート、モノブチルフマレート、およびモノペンチルフマレートからなる群から選ばれる(その医薬的に許容される塩を含む)先の請求項のいずれかに記載の制御放出医薬組成物。
【請求項24】
該フマル酸エステルが医薬的に許容される塩の形で存在するフマル酸のモノ-(C1-C5)アルキルエステルである先の請求項のいずれかに記載の制御放出医薬組成物。
【請求項25】
該塩がアルキル金属塩およびアルキル土類金属塩から選ばれる塩のような金属塩である請求項24記載の制御放出医薬組成物。
【請求項26】
該塩がナトリウム、カリウム、カルシウム、マグネシウム、または亜鉛塩である請求項24または25に記載の制御放出医薬組成物。
【請求項27】
ジメチルフマレートを活性物質として含む先の請求項のいずれかに記載の制御放出医薬組成物。
【請求項28】
モノメチルフマレートを活性物質として含む先の請求項のいずれかに記載の制御放出医薬組成物。
【請求項29】
モノメチルフマレートがそのナトリウム、カリウム、カルシウム、マグネシウムおよび/または亜鉛塩の形で存在する請求項28記載の制御放出医薬組成物。
【請求項30】
1日1回、2回、または3回投与するための先の請求項のいずれかに記載の制御放出組成物。
【請求項31】
1日1回投与するための請求項30記載の制御放出組成物。
【請求項32】
1日2回投与するための請求項30記載の制御放出組成物。
【請求項33】
剤形中のフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩の量が活性物質90mg〜360mgである先の請求項のいずれかに記載の制御放出医薬組成物。
【請求項34】
剤形中の量が活性物質90、120、180、240、または360mgである請求項33記載の制御放出医薬組成物。
【請求項35】
剤形中の量が活性物質120mgである請求項33記載の制御放出医薬組成物。
【請求項36】
剤形中の量が活性物質180mgである請求項33記載の制御放出医薬組成物。
【請求項37】
剤形中の量が活性物質240mgである請求項33記載の制御放出医薬組成物。
【請求項38】
剤形中の量が活性物質360mgである請求項33記載の制御放出医薬組成物。
【請求項39】
錠剤形の先の請求項のいずれかに記載の制御放出医薬組成物。
【請求項40】
該錠剤が患者が飲み込みやすい好都合な形を有する請求項39記載の制御放出医薬組成物。
【請求項41】
該錠剤がいかなる鋭角もない棒状形または円形である請求項39〜40のいずれかに記載の制御放出医薬組成物。
【請求項42】
該錠剤が2またはそれ以上の部分に分割されるよう設計される請求項39〜41のいずれかに記載の制御放出医薬組成物。
【請求項43】
カプセルの形の請求項1〜38のいずれかに記載の制御放出医薬組成物。
【請求項44】
有効量の請求項1〜43のいずれかに記載の医薬組成物を治療を必要とする患者に投与することを含む以下の疾患の治療方法:
乾癬、乾癬性関節炎、神経皮膚炎、炎症性腸疾患、例えばクローン病および潰瘍性大腸炎、自己免疫疾患、例えば多発性関節炎、多発性硬化症(MS)、若年発症糖尿病、橋本甲状腺炎、バセドウ病、SLE(全身性エリテマトーデス)、シェーグレン症候群、悪性貧血、慢性活動性(狼瘡)肝炎、関節リウマチ(RA)および視神経炎、痛み、例えば神経根痛、神経根障害関連痛、ニューロパシー痛もしくは坐骨神経痛/坐骨神経痛の痛み、臓器移植(拒絶の予防)、サルコイドーシス、リポイド類壊死症、または環状肉芽腫。
【請求項45】
以下の疾患を治療するための医薬を製造するための請求項1〜43のいずれかに記載の医薬組成物の使用:乾癬、乾癬性関節炎、神経皮膚炎、炎症性腸疾患、例えばクローン病および潰瘍性大腸炎、自己免疫疾患、例えば多発性関節炎、多発性硬化症(MS)、若年発症糖尿病、橋本甲状腺炎、バセドウ病、SLE(全身性エリテマトーデス)、シェーグレン症候群、悪性貧血、慢性活動性(狼瘡)肝炎、関節リウマチ(RA)および視神経炎、痛み、例えば神経根痛、神経根障害関連痛、ニューロパシー痛もしくは坐骨神経痛/坐骨神経痛の痛み、臓器移植(拒絶の予防)、サルコイドーシス、リポイド類壊死症、または環状肉芽腫。
【請求項1】
経口投与すると等価用量のFumaderm(登録商標)錠の経口投与後と比べてGI関連副作用の低下が生じる、活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む医薬組成物。
【請求項2】
制御放出組成物の形の請求項1記載の医薬組成物。
【請求項3】
活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、
該フマル酸エステルの該放出が、試験の最初の2時間は0.1N塩酸を溶出溶媒に、次いで0.05Mリン酸緩衝液pH6.5を溶出溶媒に用いるin vitro溶出試験を行うと、試験開始後の最初の3時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約70%w/wが放出されるものである該医薬組成物。
【請求項4】
活性物質としてフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む経口使用用の制御放出医薬組成物であって、該フマル酸エステルの該放出が、試験の最初の2時間は0.1N塩酸を溶出溶媒に、次いで0.05Mリン酸緩衝液pH6.5を溶出溶媒に用いるin vitro溶出試験を行うと、試験開始後の最初の4時間以内に該フマル酸エステルの総量のほぼ約92%w/wが放出されるものである該医薬組成物。
【請求項5】
該フマル酸エステルの該放出が、試験開始後の最初の4時間以内に該フマル酸エステルの総量のほぼ約92%w/wが放出されるものである請求項3記載の制御放出組成物。
【請求項6】
該フマル酸エステルの該放出が、試験開始後の最初の5時間以内に該フマル酸エステルの総量のほぼ約94%w/wが放出されるものである請求項3〜5のいずれかに記載の制御放出組成物。
【請求項7】
該フマル酸エステルの該放出が、試験開始後の最初の6時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約95%w/wが放出されるものである請求項3〜6のいずれかに記載の制御放出組成物。
【請求項8】
該フマル酸エステルの該放出が、試験開始後の最初の7時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約98%w/wが放出されるものである請求項3〜7のいずれかに記載の制御放出医薬組成物。
【請求項9】
該フマル酸エステルの該放出が、試験開始後の最初の9時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約99%w/wが放出されるものである請求項3〜8のいずれかに記載の制御放出組成物。
【請求項10】
該フマル酸エステルの該放出が、試験開始後の最初の12時間以内に該組成物中に含まれる該フマル酸エステルの総量のほぼ約99%w/wが放出されるものである請求項3〜9のいずれかに記載の制御放出組成物。
【請求項11】
該放出が、ゼロ次、1次、または平方根(Higuchiの方程式)動力学放出プロフィールを有する先の請求項のいずれかに記載の制御放出医薬組成物。
【請求項12】
該放出が平方根(Higuchiの方程式)動力学放出プロフィールを有する請求項11記載の制御放出医薬組成物。
【請求項13】
経口投与すると等価用量のFumaderm(登録商標)錠の経口投与後と比べてGI関連副作用の低下が生じる請求項3〜12のいずれかに記載の制御放出医薬組成物。
【請求項14】
活性成分として10%〜90%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩、2%〜40%(重量)医薬的に許容されるポリマー、および1%〜40%(重量)親水性賦形剤、および所望により医薬的に許容される賦形剤または添加剤を含む制御放出医薬組成物。
【請求項15】
活性物質として40%〜60%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩、15%〜25%(重量)医薬的に許容されるポリマー、および2%〜15%(重量)親水性賦形剤、および所望により医薬的に許容される賦形剤または添加剤を含む請求項14記載の制御放出医薬組成物。
【請求項16】
活性物質として65%〜80%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩、10%〜25%(重量)医薬的に許容されるポリマー、および2%〜15%(重量)親水性賦形剤、および所望により医薬的に許容される賦形剤または添加剤を含む請求項14記載の制御放出医薬組成物。
【請求項17】
該医薬的に許容されるポリマーがエチルセルロースである請求項14〜16のいずれかに記載の制御放出医薬組成物。
【請求項18】
該親水性賦形剤がポリエチレングリコールである請求項14〜17のいずれかに記載の制御放出医薬組成物。
【請求項19】
該親水性賦形剤がヒドロキシルプロピルセルロースである請求項14〜17のいずれかに記載の制御放出医薬組成物。
【請求項20】
活性物質として10%〜90%(重量)の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステルまたはその医薬的に許容される塩、および2%〜40%(重量)メタクリル酸コポリマーAおよびBを重量比1:9〜9:1で、および所望により医薬的に許容される賦形剤または添加剤を含む制御放出医薬組成物。
【請求項21】
50%〜90%の、フマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩を含む請求項14および16〜20のいずれかに記載の制御放出医薬組成物。
【請求項22】
請求項3〜13のいずれかに記載の溶出プロフィールを有する請求項14〜21のいずれかに記載の制御放出医薬組成物。
【請求項23】
該フマル酸エステルが、ジメチルフマレート、ジエチルフマレート、ジプロピルフマレート、ジブチルフマレート、ジペンチルフマレート、メチル-エチル-フマレート、メチル-プロピルフマレート、メチル-ブチルフマレート、メチル-ペンチルフマレート、モノメチルフマレート、モノエチルフマレート、モノプロピルフマレート、モノブチルフマレート、およびモノペンチルフマレートからなる群から選ばれる(その医薬的に許容される塩を含む)先の請求項のいずれかに記載の制御放出医薬組成物。
【請求項24】
該フマル酸エステルが医薬的に許容される塩の形で存在するフマル酸のモノ-(C1-C5)アルキルエステルである先の請求項のいずれかに記載の制御放出医薬組成物。
【請求項25】
該塩がアルキル金属塩およびアルキル土類金属塩から選ばれる塩のような金属塩である請求項24記載の制御放出医薬組成物。
【請求項26】
該塩がナトリウム、カリウム、カルシウム、マグネシウム、または亜鉛塩である請求項24または25に記載の制御放出医薬組成物。
【請求項27】
ジメチルフマレートを活性物質として含む先の請求項のいずれかに記載の制御放出医薬組成物。
【請求項28】
モノメチルフマレートを活性物質として含む先の請求項のいずれかに記載の制御放出医薬組成物。
【請求項29】
モノメチルフマレートがそのナトリウム、カリウム、カルシウム、マグネシウムおよび/または亜鉛塩の形で存在する請求項28記載の制御放出医薬組成物。
【請求項30】
1日1回、2回、または3回投与するための先の請求項のいずれかに記載の制御放出組成物。
【請求項31】
1日1回投与するための請求項30記載の制御放出組成物。
【請求項32】
1日2回投与するための請求項30記載の制御放出組成物。
【請求項33】
剤形中のフマル酸のジ-(C1-C5)アルキルエステルおよびフマル酸のモノ-(C1-C5)アルキルエステルから選ばれる1またはそれ以上のフマル酸エステル、またはその医薬的に許容される塩の量が活性物質90mg〜360mgである先の請求項のいずれかに記載の制御放出医薬組成物。
【請求項34】
剤形中の量が活性物質90、120、180、240、または360mgである請求項33記載の制御放出医薬組成物。
【請求項35】
剤形中の量が活性物質120mgである請求項33記載の制御放出医薬組成物。
【請求項36】
剤形中の量が活性物質180mgである請求項33記載の制御放出医薬組成物。
【請求項37】
剤形中の量が活性物質240mgである請求項33記載の制御放出医薬組成物。
【請求項38】
剤形中の量が活性物質360mgである請求項33記載の制御放出医薬組成物。
【請求項39】
錠剤形の先の請求項のいずれかに記載の制御放出医薬組成物。
【請求項40】
該錠剤が患者が飲み込みやすい好都合な形を有する請求項39記載の制御放出医薬組成物。
【請求項41】
該錠剤がいかなる鋭角もない棒状形または円形である請求項39〜40のいずれかに記載の制御放出医薬組成物。
【請求項42】
該錠剤が2またはそれ以上の部分に分割されるよう設計される請求項39〜41のいずれかに記載の制御放出医薬組成物。
【請求項43】
カプセルの形の請求項1〜38のいずれかに記載の制御放出医薬組成物。
【請求項44】
有効量の請求項1〜43のいずれかに記載の医薬組成物を治療を必要とする患者に投与することを含む以下の疾患の治療方法:
乾癬、乾癬性関節炎、神経皮膚炎、炎症性腸疾患、例えばクローン病および潰瘍性大腸炎、自己免疫疾患、例えば多発性関節炎、多発性硬化症(MS)、若年発症糖尿病、橋本甲状腺炎、バセドウ病、SLE(全身性エリテマトーデス)、シェーグレン症候群、悪性貧血、慢性活動性(狼瘡)肝炎、関節リウマチ(RA)および視神経炎、痛み、例えば神経根痛、神経根障害関連痛、ニューロパシー痛もしくは坐骨神経痛/坐骨神経痛の痛み、臓器移植(拒絶の予防)、サルコイドーシス、リポイド類壊死症、または環状肉芽腫。
【請求項45】
以下の疾患を治療するための医薬を製造するための請求項1〜43のいずれかに記載の医薬組成物の使用:乾癬、乾癬性関節炎、神経皮膚炎、炎症性腸疾患、例えばクローン病および潰瘍性大腸炎、自己免疫疾患、例えば多発性関節炎、多発性硬化症(MS)、若年発症糖尿病、橋本甲状腺炎、バセドウ病、SLE(全身性エリテマトーデス)、シェーグレン症候群、悪性貧血、慢性活動性(狼瘡)肝炎、関節リウマチ(RA)および視神経炎、痛み、例えば神経根痛、神経根障害関連痛、ニューロパシー痛もしくは坐骨神経痛/坐骨神経痛の痛み、臓器移植(拒絶の予防)、サルコイドーシス、リポイド類壊死症、または環状肉芽腫。
【図1】

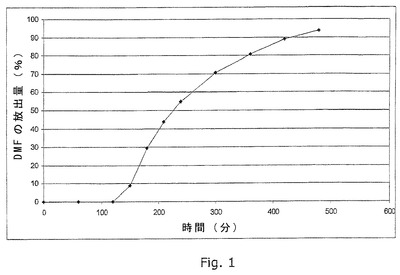
【図2】
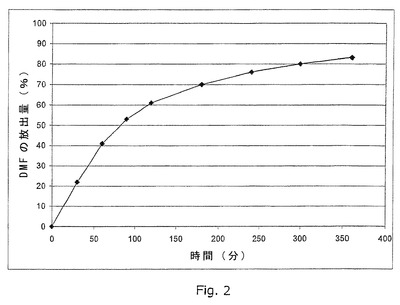
【図3】
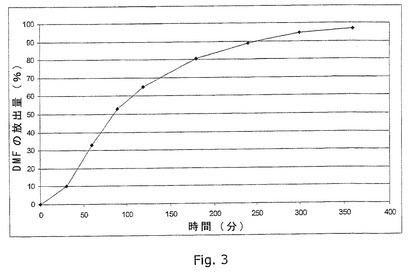
【図2】
【図3】
【公表番号】特表2008−515822(P2008−515822A)
【公表日】平成20年5月15日(2008.5.15)
【国際特許分類】
【出願番号】特願2007−535023(P2007−535023)
【出願日】平成17年10月7日(2005.10.7)
【国際出願番号】PCT/DK2005/000648
【国際公開番号】WO2006/037342
【国際公開日】平成18年4月13日(2006.4.13)
【出願人】(506387214)アディテック・ファルマ・アクチボラゲット (3)
【氏名又は名称原語表記】AdiTech Pharma AB
【Fターム(参考)】
【公表日】平成20年5月15日(2008.5.15)
【国際特許分類】
【出願日】平成17年10月7日(2005.10.7)
【国際出願番号】PCT/DK2005/000648
【国際公開番号】WO2006/037342
【国際公開日】平成18年4月13日(2006.4.13)
【出願人】(506387214)アディテック・ファルマ・アクチボラゲット (3)
【氏名又は名称原語表記】AdiTech Pharma AB
【Fターム(参考)】
[ Back to top ]