
フルオリナーゼの製造方法
【課題】 組換えフルオリナーゼの発現系を構築し、フルオリナーゼを簡易かつ大量に生産する手段を提供する。
【解決手段】 フルオリナーゼとシャペロンタンパク質を微生物内で共発現させる工程、及び前記微生物を破砕し、その破砕物からフルオリナーゼを採取する工程を含むことを特徴とするフルオリナーゼの製造方法。
【解決手段】 フルオリナーゼとシャペロンタンパク質を微生物内で共発現させる工程、及び前記微生物を破砕し、その破砕物からフルオリナーゼを採取する工程を含むことを特徴とするフルオリナーゼの製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、フルオリナーゼ(フッ素化酵素)の製造方法、並びに変異型フルオリナーゼ作製方法及びその方法によって作製された変異型フルオリナーゼに関する。
【背景技術】
【0002】
近年、放線菌Streptomyces cattleya (S.cattleya) が、フッ化物イオンを有機化合物に導入できるフルオリナーゼを有していることがわかり、その三次元構造と遺伝子の塩基配列が報告された(非特許文献1)。このようなフルオリナーゼの詳細な報告例は本発明者の知る限りこれまでに全くなく、これを応用することができれば、困難だと考えられていたフッ素化合物の酵素による合成が可能になる。
【0003】
このS.cattleya由来のフルオリナーゼを大量生産するためには、大腸菌などを用いた組換えフルオリナーゼの発現系の構築が必要であるが、未だこのような発現系の構築に成功したという報告はない。
【0004】
【非特許文献1】C. Dong, F. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O’ Hagan, and J. H. Naismith, Nature, 2004, 427, 561-565.
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、以上のような技術的背景の下、組換えフルオリナーゼの発現系を構築し、フルオリナーゼを簡易かつ大量に生産する手段を提供することを目的とする。また、天然型のフルオリナーゼの基質特異性を変化させ、より多様なフッ素化合物の合成手段を提供することも目的とする。
【課題を解決するための手段】
【0006】
本発明者は、上記課題を解決するため鋭意検討を重ねた結果、フルオリナーゼをシャペロンタンパク質と共に大腸菌内で発現させることにより、高い活性を持つフルオリナーゼを生産できることを見出した。また、フルオリナーゼの156位のフェニルアラニンをチロシンに置換しても酵素活性が維持されることを見出した。本発明は、以上の知見に基づき完成されたものである。
【0007】
即ち、本発明は、以下の(1)〜(7)を提供するものである。
(1)フルオリナーゼとシャペロンタンパク質を微生物内で共発現させる工程、及び前記微生物を破砕し、その破砕物からフルオリナーゼを採取する工程を含むことを特徴とするフルオリナーゼの製造方法。
(2)フルオリナーゼが、配列番号2に記載のアミノ酸配列で表されることを特徴とする(1)に記載のフルオリナーゼの製造方法。
【0008】
(3)シャペロンタンパク質が、GroEであることを特徴とする(1)又は(2)に記載のフルオリナーゼの製造方法。
(4)微生物が、大腸菌であることを特徴とする(1)乃至(3)のいずれかに記載のフルオリナーゼの製造方法。
(5)フルオリナーゼに対し、配列番号2に記載のアミノ酸配列における156番目のフェニルアラニンに相当するアミノ酸を他のアミノ酸に置換することを特徴とする変異型フルオリナーゼの作製方法。
【0009】
(6)他のアミノ酸が、チロシンであることを特徴とする(5)に記載の変異型フルオリナーゼの作製方法。
(7)(5)又は(6)に記載の方法によって作製された変異型フルオリナーゼ。
【発明の効果】
【0010】
本発明により活性の高いフルオリナーゼを簡易かつ大量に製造することが可能になる。また、本発明は、基質の取り込みに関与するアミノ酸を置換した変異型のフルオリナーゼも提供する。
【発明を実施するための最良の形態】
【0011】
以下、本発明を詳細に説明する。
(1)フルオリナーゼの製造方法
本発明のフルオリナーゼの製造方法は、フルオリナーゼとシャペロンタンパク質を微生物内で共発現させる工程、及び前記微生物を破砕し、その破砕物からフルオリナーゼを採取する工程を含むことを特徴とするものである。
【0012】
フルオリナーゼとしては、S.cattleya由来の5'-フルオロ-5'-デオキシアデノシン合成酵素を使用することができる。この酵素のアミノ酸配列及び酵素をコードする遺伝子の塩基配列をそれぞれ配列番号2及び配列番号1に示す。フルオリナーゼとしては、この5'-フルオロ-5'-デオキシアデノシン合成酵素のアミノ酸配列の一部を変更した酵素(例えば、活性を失わせない範囲で、1若しくは数個のアミノ酸を欠失、置換若しくは付加した酵素など)を使用してもよい。このようなアミノ酸配列の一部を変更したフルオリナーゼの一例として、配列番号2に記載のアミノ酸配列における156番目のフェニルアラニンをチロシンに置換した変異型フルオリナーゼを例示できる。
【0013】
シャペロンタンパク質としては、GroEを使用することができるが、これ以外のシャペロンタンパク質を使用してもよい。
微生物としては、大腸菌を使用することができるが、他の微生物を使用してもよい。
フルオリナーゼ遺伝子とシャペロンタンパク質遺伝子を微生物中で共発現させる方法は特に限定されず、常法に従って行うことができる。
微生物を破砕し、その破砕物からフルオリナーゼを採取する方法も特に限定されず、微生物の生産するタンパク質を採取する際に使用される一般的な方法を適用することができる。
【0014】
(2)変異型フルオリナーゼの作製方法
本発明の変異型フルオリナーゼの作製方法は、フルオリナーゼに対し、配列番号2に記載のアミノ酸配列における156番目のフェニルアラニンに相当するアミノ酸を他のアミノ酸に置換することを特徴とするものである。
フルオリナーゼとしては、S.cattleya由来の5'-フルオロ-5'-デオキシアデノシン合成酵素を使用することができる。また、この5'-フルオロ-5'-デオキシアデノシン合成酵素のアミノ酸配列の一部を変更した酵素を使用してもよい。
【0015】
「配列番号2に記載のアミノ酸配列における156番目のフェニルアラニンに相当するアミノ酸」とは、対象とするフルオリナーゼの立体構造を、配列番号2に記載のアミノ酸配列からなるフルオリナーゼ(S.cattleya由来の5'-フルオロ-5'-デオキシアデノシン合成酵素)の立体構造と比較した際に、配列番号2に記載のアミノ酸配列における156番目のフェニルアラニンと同様の位置に存在するフェニルアラニンを意味する。
フェニルアラニンと置き換える他のアミノ酸としては、チロシンを挙げることができるが、活性を維持できるのであればこれ以外のアミノ酸であってもよい。
【0016】
アミノ酸の置換は、常法に従って行うことができ、例えば、後述する実施例のように変異導入用のプラスミドを使用して行うことができる。
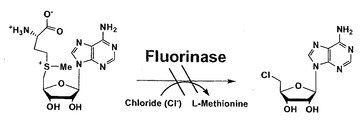
後述するように、配列番号2に記載のアミノ酸配列における156番目のフェニルアラニンは、基質の酵素への取り込みに重要な役割を果たしていると考えられる。従って、このフェニルアラニンを他のアミノ酸に置換した変異型フルオリナーゼは、本来の基質であるS-アデノシル-L-メチオニン以外の化合物に対してフッ化物イオンを導入できると推測される。
【実施例】
【0017】
以下、実施例により本発明を更に詳細に説明する。
1 実験材料および方法
1-1 化合物、使用菌株、培地および培養条件
【0018】
化合物
表1に本実施例で使用した基質、合成試薬を示した。S-アデノシル-L-メチオニン (SAM) は常温では不安定なため、20mM に希釈して-20℃に保存した。他の基質、試薬は各メーカーの指示に従い保存した。
【表1】

【0019】
菌体
表2に使用した菌株を示した。fla 遺伝子(フルオリナーゼをコードする遺伝子)供与体としてS.cattleya NBRC14057 を用いた。遺伝子組換えプラスミド調整用の宿主としてはEscherichia coli (E. coli ) JM109 株を使用し、タンパク質発現用宿主にはE. coli BL21(DE3) 株および E. coli BL21(DE3) pLysS 株を用いた。また、変異株作製にはE. coli ES1301 mutS 株を使用した。
【表2】
【0020】
プラスミド・プライマー
表3に使用したプラスミドを示した。PCR 産物のクローニングおよび組換え遺伝子の発現には、pET28b(+) (Novagen)を使用した。pET28b(+) はColE1 系の複製起点、カナマイシン耐性遺伝子、lacIq の他、ヒスチジンタグ融合型の遺伝子を含むマルチクローニングサイト上流にtac プロモーターが存在しており、IPTG による発現誘導調節可能な高コピーベクターである。また変異株作製にはpALTER1 を用いた。これは変異株作製キットAltered Sites II in vitro Mutagenesis System (Promega) に添付されている変異株作製用のプラスミドであり、ColE1 系の複製起点、アンピシリン耐性遺伝子、テタラサイクリン耐性遺伝子マルチクローニングサイト上流にtac プロモーターが存在しており、IPTG による発現誘導調節可能な高コピーベクターである。PGEL2 は東京工業大学大学院生命理工研究科和地研究室の村上智史氏が構築され、本実験のため、和地正明博士および荻野英賢博士の御好意により使用させていただいた。
【0021】
プライマーは各種必要に応じて設計し、グライナージャパン社より購入して用いた(表4)。
【表3】
【表4】
【0022】
培地
S.cattleya NBRC14057 の培養には227 培地 [0.4% Dried yeast extract (Wako)、1.0% Malt extract (Wako)、0.4% Glucose ] を用い、培養は28℃で行った。また大腸菌の培養には0.1% glucose を添加したLennox (L) 培地(DIFCO) を使用し、培養は30℃もしくは37℃で行った。227 培地は各成分を水道水に、L 培地は純水 (MILLIPORE) に溶解し、121℃で20 分間オートクレーブ滅菌したのち使用した。固形培地には終濃度1.5%の寒天 (Wako) を添加した。プラスミドの保持には適宜、抗生物質を添加した。
【0023】
1-2 クローニング技術
特に記述ないものはMolecular Cloning, 2ed.(J. Sambrook, E. F. Fritsch and T. Maniatis, Molecular cloning: A laboratory manual, 2nd edition. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press: 1989.)、およびExperiments in Molecular Genetics(J. H. Miller, Experiments in molecular genetics: Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press: 1972.)を参考にした。制限酵素およびDNA リガーゼ、T4DNAポリメラーゼはTAKARA、Promega より購入し、メーカーの指示に従って使用した。E.coli の形質転換はエレクトロポレーションで行った。エレクトロポレーションにはElectroporater (Invitrogen) を使用した。1500V、150Ωでパルスを付与することにより行った。コンピテントセルの調整は同社の説明書の指示に従った。大腸菌からのプラスミドDNA の調整は、アルカリSDS 法あるいはWizard Plus SV Minipreps DNA purification system (Promega) を用いて行った。DNA のアガロース電気泳動には、トリス-ホウ酸-EDTA 緩衝液(Promega)を用い、ゲル濃度は0.7%とした。泳動後のゲルはエチジウムブロミド染色を行い、DNA を検出した。DNA 分子量マーカーはλ/ Hind III(ニッポンジーン)を用いた。
【0024】
1-3 プラスミドpETflA の構築
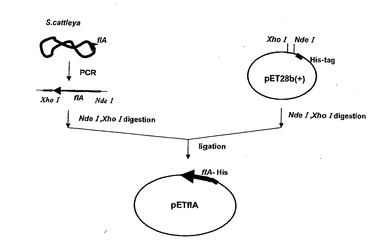
S.cattleya NBRC14057 株の菌体を鋳型として、先行論文(前述したC. Dongらの論文)に従いPCR プライマーHis-fla1、His-fla2(表4)を用いてS.cattleya ゲノムからfla 遺伝子の増幅をPCRにより行った(図1)。S.cattleya NBRC14057 株は28℃、120 rpm の条件で48 時間振とう培養したものを遠心により菌体を集菌した。これを滅菌水で希釈・懸濁したものをPCR の鋳型として用いた。PCR において、購入したオリゴヌクレオチド10μM に希釈したものをPCR プライマーとして用いた。使用したプライマーは表4に示した。PCR はiCycler (BIORAD)を用いて行った。耐熱性DNA ポリメラーゼ等はLA Taq polymerase (TaKaRa)を使用した。PCR の条件は94℃, 1min のち、94℃, 30sec., 59℃, 30sec., 72℃,2min を30 サイクル行い、最後に72℃, 5min の処理を施したものであり、最後は4℃に冷却した。得られたDNA 断片と高コピーのプラスミドベクターpET28b(+) をそれぞれ制限酵素NdeI、XhoIで消化し、リガーゼ (Takara) により結合することでフルオリナーゼタンパク質のN 末端側にヒスチジンタグが融合したタンパク質を発現させるプラスミドpETflA を構築した。この挿入の有無は制限消化により確認した。候補のクローンについて塩基配列を確認したが野生型の遺伝子は得られず、必ず変異が入っていた。そのため2つの変異遺伝子を制限消化により組み合わせることでサイレントな変異を持つクローンを得た。これをpETflA として用いることにした。
【0025】
1-4 発現タンパク質の確認
泳動用試料の調整
5ml のL 液体培地を用いて30℃で一晩培養した前培養液を、新たなL 液体培地5ml に1%植菌し30℃で24 時間培養した。対数増殖期前に終濃度0.1mM のIPTG を培地に添加した。遠心により集菌し、50mM pH7.5 Tris-HCl 緩衝液で2 度洗浄後、同緩衝液に懸濁し超音波処理により菌体を破砕した。適当量をSDS-PAGE 試料用緩衝液(Invitrogen)に懸濁し、これを100℃、3 分間加熱処理したものを用いた。
【0026】
SDS-ポリアクリルアミドゲル電気泳動(SDS-PAGE)
SDS-PAGE はLaemmli の方法(U. K. Laemmli, Nature, 1970, 227, 680.) を参考にした。泳動にはNuPAGE 10%Bis-Tris Gel(Invitrogen)を用いた。泳動後のタンパク質はクーマシーブリリアントブルーR-250(CBB)により染色した。なお分子量マーカーはタンパク質分子量マーカー「第一」・III(第一化学薬品)を使用した。
【0027】
Ni2+カラムによる酵素精製
前述の培養条件で菌体を調整・集菌した。各実験で使用する緩衝液で2 度洗浄後、同緩衝液に懸濁し超音波処理により菌体を破砕した。菌体破砕後、遠心により上清を取り出し、ヒスチジンタグと親和性のあるニッケルカラムNi-NTA spin kit(QIAGEN)を用いてアフィニテークロマトグラフィーにより酵素精製を行った。700mM のイミダゾールで溶出後、Microcon YM-10(MILLIPORE)により限外ろ過を行い、脱塩濃縮およびバッファー交換した。タンパク質の定量は、ウシ血清アルブミン (BSA) を標準としてBradford 法により行った。
【0028】
1-5 NMR による活性検出
1-4の培養条件で20μg/ml kanamycin を添加したE. coli BL21(DE3)pETflA 株の10ml 分の菌体を調整・集菌した。表5の各緩衝液で2 度洗浄後、同緩衝液に懸濁し超音波処理により菌体を破砕した。菌体破砕後遠心し、上清を反応溶液として用いて、終濃度10mM のNaF と800μM のSAM を投与し、200μl スケールで30℃にて一晩反応させた。NMR サンプルとして、この反応液と等量のd-DMSO(Merck 社)を加えてVXR-500(500MHz、Varian)により19F NMR を測定した。
【表5】
【0029】
1-6 変異導入用プラスミドpALTERflA の構築および各種変異株の作製
pETflA とpALTER ベクターをそれぞれ制限酵素SphI、DraIIIで消化し、これらをライゲーションさせることによりpALTERflA を構築した。このプラスミドに対して、Altered Sites II in vitro Mutagenesis System (Promega)を用いて各種変異導入プラスミド (S158A 変異株 (158 番目の残基をセリンからアラニンに置換したもの)、S158G、 S158C、F156A、F156G、F156C、F156T、F156Y、F156D )を作製した。変異導入は作製した各種プラスミドを鋳型として、DTCS Quick Start Kit (BECKMAN COULTER) によりdideoxy 反応を行い、CEQ8000 (BECKMAN COULTER) を用いて塩基配列を確認した。そして、変異導入したプラスミドはE. coli BL21(DE3)pLysS 株に形質転換し、1-4の方法でタンパク質発現の確認を行った。
【0030】
1-7 GroE タンパク質の導入
E. coli BL21(DE3) pLysS pALTERflA 由来の各菌体( wild type、S158A、S158G、 S158C、F156A、F156G、F156C、F156T、F156Y、F156D)にpSC101由来の複製起点を持つプラスミドpMW218 にgroE (groEL, groES) 遺伝子含むプラスミドpGEL2 を形質転換し、groE (groEL, groES) 遺伝子を大腸菌内でマルチコピーに導入した系を構築した。フルオリナーゼの発現は1-4の方法で確認した。
【0031】
1-8 HPLC による活性測定
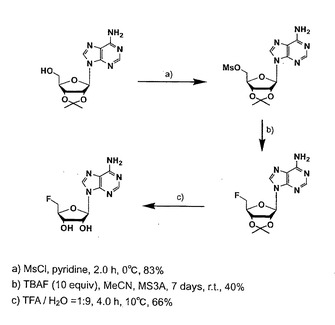
高速液体クロマトグラフィー(HPLC)を用いて、フルオリナーゼ反応の検出を行った。HPLC は島津製作所社製SPD-10Avp を用いた。カラムは逆相カラムShin-pack VP-ODS 150×4.6(島津製作所)を使用して、検出波長はアデニンの吸収波長である260nmとした。定量化するために先行論文(V. Reichelova, F. Albertioni, and J. Liliemark, J. Chromatography. 1994, 667, 37.)を参考に5’-FDAを化学合成し(図2)、分離条件を検討した。また、サンプルの標準物質として5’-クロロ-5’-デオキシアデノシン (5’-CDA) が適当であったのでこれを用いてピーク検出を行った。HPLC サンプルは、回収した反応液に等量のメタノールを加えて酵素を失活させ遠心により上清を取り出した。そして小型エバポレーターMicro Vac(TOMY)により溶媒を蒸発させ、この操作を二回繰り返したものを調整した。これを5’-CDA 添加したHPLC 溶出溶媒50μl で希釈し、10μl を測定に用いた。
【0032】
1-9 フルオリナーゼ反応
pH およびbuffer の検討
1-4の培養条件で10μg/ml tetracycline、20μg/ml chroramphenicol を添加したE. coli BL21(DE3) pLysS pALTERflA 株の20ml 分の菌体を調整・集菌した。50mM pH8.0 Tris-HCl 緩衝液で2 度洗浄後、同緩衝液に懸濁し超音波処理により菌体を破砕した。菌体破砕後、遠心により上清を取り出し、Ni2+カラムによるアフィニテークロマトグラフィーにより酵素精製を行った。この精製体を1-4の方法によりタンパク質定量し、表6の各種緩衝液を加えて1.0μg/ml の濃度に調整し、終濃度250μMのSAMと10mMのKF を添加して100μl スケールで37℃にて酵素反応を行った。反応温度はO’Hagan らの報告例(前述したC. Dongらの論文、及びS. L. Cobb, H. Deng, J. T. G. Hamilton, R. P. McGlinchey and D. O’ Hagan, 43 Chem. Commum., 2004, 592.)を参考にした。反応開始から5分後にそれぞれ反応液を回収し、HPLC による活性測定を行った。
【表6】
【0033】
野生株とGroE 導入株の活性比較
(1)酵素濃度の検討
1-4の培養条件で10μg/ml tetracycline、20μg/ml chroramphenicol を添加したE. coli BL21(DE3) pLysS pALTERflA 株(B株)および10μg/ml tetracycline、20μg/ml chroramphenicol、20μg/ml kanamycin を添加したGroE タンパク質を共発現させたE. coli BL21(DE3) pLysS pGEL2 pALTERflA株(G株)の20ml 分の菌体を調整・集菌した。50mM pH8.0 Tris-HCl 緩衝液で2 度洗浄後、同緩衝液に懸濁し超音波処理により菌体を破砕した。菌体破砕後、遠心により上清を取り出し、Ni2+カラムによるアフィニテークロマトグラフィーにより酵素精製を行った。この精製体を1-4 の方法でタンパク質定量し、50mM pH8.0 Tris-HCl 緩衝液に0.25μg/ml および1.0μg/ml の濃度に調整し、終濃度250μM のSAM と10mM のKF を添加して100μl スケールで37℃にて酵素反応を行った。反応開始から5分、15 分、60 分、180 分後にそれぞれ反応液を回収し、HPLC による活性測定を行った。
【0034】
(2)基質濃度の検討
上記と同様の方法でフルオリナーゼを精製した。50mM pH8.0 Tris-HCl 緩衝液で調整したB株およびG株由来の各1.0μg/ml のフルオリナーゼ溶液に、SAM 濃度の変化系には終濃度10mM のKF と、G株は10、12.5、15、20、40、80、100μM のSAM を、B株は15、25、40、50、80、100μM のSAM を添加して5分間37℃にて100μl スケールで酵素反応を行った。また、KF 濃度の変化系には終濃度250μM のSAM とG株は0.2、0.25、0.5、1.0、2.0、4.0、5.0、8.0 mM のKF を、B株は0.2、0.25、0.5、1.0、2.0、4.0 mM のKF を添加して5分間37℃にて100μl スケールで酵素反応を行った。反応後、HPLCによる活性測定を行った。
【0035】
超音波破砕液における活性検出
1-4の培養条件で10μg/ml tetracycline、20μg/ml chroramphenicol、20μg/ml kanamycin を添加したG 株の5ml 分の菌体を調整・集菌した。50mM pH8.0 Tris-HCl 緩衝液で2 度洗浄後、同緩衝液に10 倍濃縮で懸濁し超音波処理により菌体を破砕した。菌体破砕後、遠心により上清を取り出し、これを酵素反応液とした。そして、終濃度250μM のSAM と10mM のKF を添加して100μl スケールで37℃にて酵素反応を行った。反応開始から5分、15 分、60分、180 分後にそれぞれ反応液を回収した。回収後、HPLC による活性測定を
行った。
【0036】
超音波破砕液による野生株と変異株との活性比較
超音波破砕液における活性検出の項と同様の方法でG 株由来の各菌体( wild type、S158A、S158G、 S158C、F156A、F156G、F156C、F156T、F156Y、F156D)の超音波破砕液を作製した。培地に野生株には終濃度10μg/ml tetracycline、20μg/ml chroramphenicol、20μg/ml kanamycin を、変異株には125μg/ml ampicillin、20μg/ml chroramphenicol、20μg/ml kanamycin を添加した。菌体破砕後、遠心により上清を取り出し、これを酵素反応液とした。そして、終濃度250μM のSAM と10mM のKF を添加して100μl スケールで37℃にて酵素反応を行った。反応開始から60 分、180 分後にそれぞれ反応液を回収した。回収後、HPLC による活性測定を行った。
【0037】
酵素精製溶液による野生株と変異株との活性比較
G 株由来のF156Y 変異株から、野生株とGroE 導入株の活性比較と同様の方法でフルオリナーゼ変異株を精製した。50mM pH8.0 Tris-HCl 緩衝液で1.0μg/ml に調整した酵素溶液にSAM濃度変化系では、終濃度10mMのKFと15、25、15、40、50、80、100μM のSAM を添加して、5分間37℃にて100μlスケールで酵素反応を行った。KF 濃度変化系では、終濃度250μM のSAM と0.5、1.0、2.0、4.0、8.0、10、12、15、20 mM のKF を添加して、5分間37℃にて100μl スケールで酵素反応を行った。反応後、HPLC による活性測定を行った。
【0038】
塩化物イオンによるクロリナーゼ反応の試み
超音波破砕液により酵素反応を行った。超音波破砕液における活性検出の項と同様にG 株由来の各菌体( wild type、S158A、S158G、 S158C)の超音波破砕液を作製した。菌体破砕後、遠心により上清を取り出しこれを酵素反応液とした。そして、終濃度500μM のSAM と60mM のKCl を添加して酵素反応を行った。反応開始から180 分後、24 時間後にそれぞれ同量ずつ反応液を回収した。回収後、HPLC による活性測定を行った。
【0039】
2 結果と考察
2-1 pETflA の構築および大腸菌での組換えフルオリナーゼ発現
S.cattleya 由来のフルオリナーゼ反応系を構築するために、大腸菌内で組換えフルオリナーゼの発現を目指した。先行論文(前述したC. Dongらの論文)に従いプライマーを設計し、S.cattleya の菌体を鋳型としてflA 遺伝子をPCR で増幅した。増幅断片を制限酵素で消化後、pET28b(+) にクローン化し、pETflA を構築した。シークエンサーを用いて塩基配列を確認したが、変異がないflA 遺伝子を獲得することができなかった。これはGC リッチなflA 遺伝子配列がスムーズなポリメラーゼ反応に支障をきたし、変異が入りやくなったためであると考えられる。塩基配列解析結果から変異が一番少ない2つの変異遺伝子を制限消化により組み合わせることでサイレントな変異を持つクローンを得た。これをpETflA として用いることにした(data not shown)。
【0040】
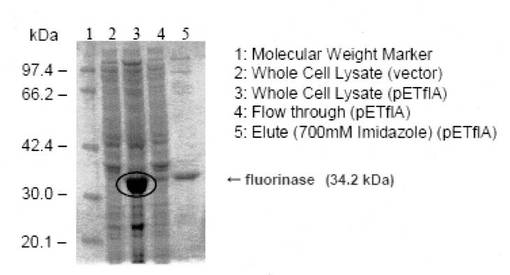
また、 pETflA をE.coli BL21(DE3)株に導入し、IPTG 誘導をかけた全菌体をSDS-PAGE に供したのちCBB 染色を施し発現タンパクを調べたところ大量発現しているタンパク質の存在が確認された(図3)。これは先行論文(前述したC. Dongらの論文)とほぼ一致する位置(34kDa)にバンドが出ていることから、フルオリナーゼが正しく発現されていることが示唆された。
【0041】
そして、この組換えフルオリナーゼはN 末端側にヒスチジンタグが融合しているので、Ni2+カラムによるアフィニテークロマトグラフィーで精製を行った。全菌体中に認められたフルオリナーゼのバンドに比べて少ない量ではあるが、ほぼ単一のバンドとして精製することができた。この回収量の低下は封入体形成などによるものと思われた。
【0042】
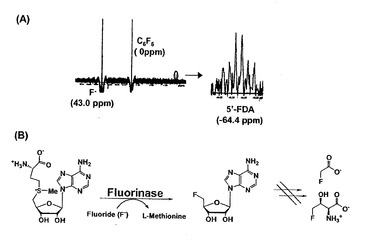
2-2 NMR による活性検出
E.coli BL21(DE3) pETflA 株の超音波破砕液にフルオリナーゼの基質であるSAM とNaF を加え一晩反応を行った。pH を6.0〜8.0 に調整し、19F NMR により5’-FDA の生成を確認したところ、pH6.0〜7.0 付近の反応溶液で弱いながらも5’-FDA と予想されるピーク(19F NMR: σ=-64.4 ppm (dt, J (F, 4’H)=24.2 z, J (F, 5’H)=47.3 Hz) )を確認した(図4)。これは化学合成した5’-FDAのピーク(19F NMR: σ=-64.4 ppm (dt, J (F, 4’H)=24.4 Hz, J (F, 5’H)=47.4 Hz) )とほぼ一致することから大腸菌においてもフルオリナーゼ反応が進行していると考えた。
【0043】
S.cattleya における生体内フッ素化合物の代謝においては、このSAM から5’-FDA へのフッ素化反応を経て、モノフルオロ酢酸(FAc)とフルオロスレオニン(4-FT)が生産されるが、この系は大腸菌内でフルオリナーゼのみを発現させているため下流の反応系はなく5’-FDA の生成で止まっていると考えられる(図4)。この結果から大腸菌による組換えフルオリナーゼを用いたin vitroフッ素化反応系は構築できることが確認できたが、NMR による5’-FDA の生成確認では弱いピークしか検出できなかった。この原因は大腸菌内でのフルオリナーゼ反応が弱いこともあるが、NMR が物質の検出系として向いてない点も挙げられる。NMR は検出にmg スケールの量を要するので、今回のような小さいスケールの酵素反応系の検出系には向いておらず、μg スケール以下で検出できる高感度の検出系の構築が必要であると考えられた。
【0044】
2-3 HPLC によるフルオリナーゼ活性検出系の構築
NMR に代わる高感度の5’-FDA の活性検出法としてHPLC を選択した。溶出溶媒等の検討を行ったところ、MeOH と10mM pH7.0 KH2PO4 を20:80に混合したものが適当であったのでこれを用いた。また定量化するために化学合成した5’-FDA を用いたところ、数ng スケールの5’-FDA を検出することが可能になり弱い酵素活性の反応でも定量化が可能になった。サンプルの標準物質としては5’-クロロ-5’-デオキシアデノシン (5’-CDA) を用いた。これは、5’-FDA と最大吸収波長が同じであること(260nm)とHPLC での溶出時間が十分離れて溶出時間比率も一定であるので(5’-FDA:10.0 分、5’-CDA: 20.5 分)、
標準物質として適当と考えた。
【0045】
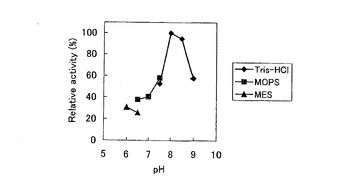
2-4 pH および緩衝液の検討
フルオリナーゼ反応を行うにあたり、反応溶液のpH および緩衝液を検討した。これは、先行論文(前述したC. Dongらの論文)ではHPLC を用いてpH8.0 付近でフルオリナーゼアッセイを行っているが、本実施例で行ったNMR における活性検出ではpH6.0〜7.0 付近でのみピークが認められたためである。したがって感度の高いHPLC による検出系を用いることで再度、至適pH や至適バッファー成分を検討することにした。表6の各緩衝溶液に置換した酵素溶液をそれぞれ5分間反応させ、生成した5’-FDA の量を比較した(図5) 。これによりpH8.0 で一番多く5’-FDAを生産していることが示され、NMR で検出できたpH の範囲とHPLC により最も生成物ピークが大きかったpH が異なる結果となった。これは、pH や緩衝液の成分により、NMR サンプル中の分子状態が変化してピークがブロード状態になるなど形状が変わり、ノイズ成分に隠れてしまったことが原因の一つとして考えられる。NMR の検出系が低感度であるため、HPLC による活性検出結果の方が信頼性が高いものと考えられた。したがって、フルオリナーゼ反応における至適pH は8.0 であると結論づけた。
【0046】
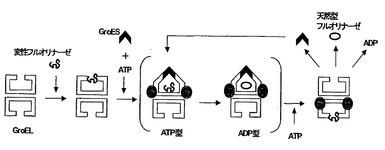
2-5 GroE タンパク質導入による活性の向上
E. coli BL21(DE3) pLysS pALTERflA 株(B株)由来のフルオリナーゼを用いて、HPLC による活性検出を行った。まず、酵素濃度を変えることにより5’-FDA の生産量の変化を観察した(図6(A))。酵素濃度を3段階に振ったところ、1.0μg/ml と2.5μg/ml の濃度では、その5’-FDA 生産量はほとんど変わらなかった。しかし、全体的に5’-FDA の生成量が少ないので失活したフルオリナーゼの割合が高いと考えられ、活性回復を図るためにin vitro タンパク質発現系にGroEタンパク質を共発現させることにした。GroEタンパク質はGroEL, GroES タンパク質からなる複合体タンパク質であり、大腸菌内でメジャーなシャペロンタンパク質である。GroE タンパク質を目的タンパク質と共発現させることで、そのリフォールディングが誘導され、酵素活性の回復が引き起こされることが知られている(図7)。この原理を利用してフルオリナーゼ活性向上のために、groE (groEL, groES ) 遺伝子を大腸菌内でマルチコピーに導入した系を構築した。GroE タンパク質を共発現した株(G 株)においてもB株と同様に酵素濃度を変えることにより5’-FDA の生産量の変化を観察した(図6(B))。B 株に比べて5’-FDA の生産量の増加が見られ、GroE タンパク質による活性回復が示唆された。
【0047】
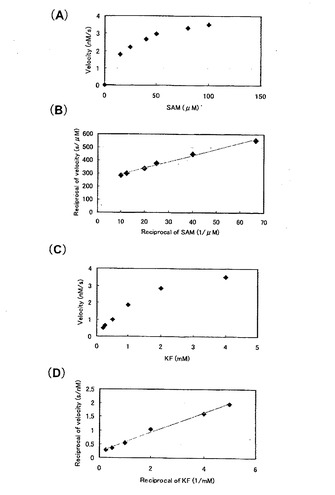
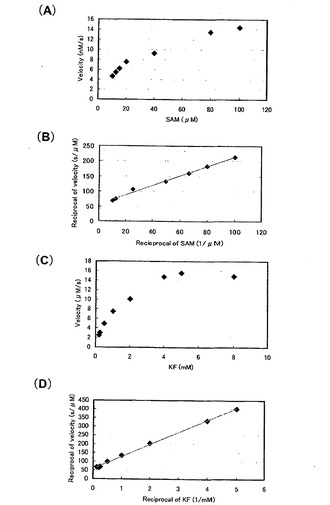
また、G 株でも酵素濃度を3段階に振ったところ、1.0μg/ml と2.5μg/ml の濃度ではその5’-FDA 生産量はほとんど変わらなかったので、これ以後反応溶液の酵素濃度は1.0μg/ml として測定を行った。
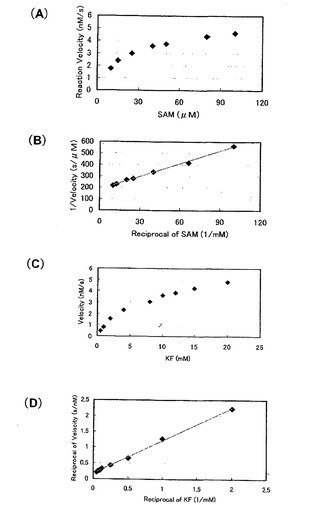
そして、定量的に酵素活性を評価するために、基質の濃度を変えて5’-FDA の生産量の変化を調べ (図8、図9)、Lineweaver-Burk プロットをとり各基質のミカエリス定数 (Km) と反応最大速度 (Vmax) を求めた(表7)。
【0048】
kcat / Km に注目すると、G 株はB 株と比べてSAM に関しては3.1 倍、KFでは5.1 倍の活性の向上が見られた。この要因としてVmax 値の向上が挙げられる。GroE マルチコピー系に用いたプラスミドはpSC101 由来の複製起点を持ち、コピー数は3〜5 と言われている。したがってG 株とB 株のVmax の比が約4倍となるのは、ちょうどgroE のコピー数の差と一致する(野生株1コピーに対してマルチコピー系は4〜6 コピーの試算になるため。)。このことよりgroE遺伝子を導入するプラスミドをより高コピーのベクターに変えることで反応系のVmax をさらに向上させることが期待できるので、より生産性の高い反応系を構築することができるのではないかと考えられる。
【表7】
【0049】
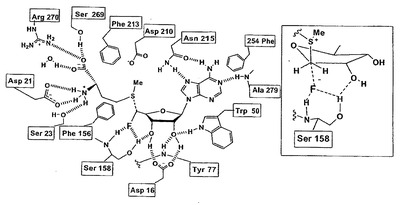
2-6 変異株の作製
今回フルオリナーゼ変異株を作製するにあたり、報告された基質結合部位(図10) に着目した。X 線結晶構造解析より、基質結合部位においてフッ化物イオンは水分子から隔離されるようなポケット構造に収まり、SAM で蓋をされ基質結合部位に収まると考えられている。そして、コンピューターシミュレーションからSAM が結合してしまうとフッ化物イオンは入り込めなくなるため、反応は進行しないと察せられている。このことより、このフッ化物イオンが取り込まれるポケット構造を変化させれば基質特異性など性質が大きく変化すると予測し、フッ化物イオンに接近しているアミノ酸残基に注目した。その1つとしてSer158 残基に着目し、セリンを3つのアミノ酸に置換したもの(S158A, S158G, S158C)を作製した。S158A およびS158G においては、フッ化物イオンが入り込むポケット構造を広げることを狙った。S158C は、セリン残基のO原子とシステイン残基のS 原子のように同族元素で大きさが異なる原子がどのような影響を及ぼすのか検討するために調整した。さらにもう1つのアミノ酸残基に着目した。それはSer158 残基と近傍に存在するPhe156 残基である。図10よりPhe156 残基は基質と分子間力がないので、基質と酵素との分子間力の影響を比較・検討することに適当であると考え、6つの各種変異株を作製した ( F156A, F156G, F156C, F156D, F156T, F156Y) 。
【0050】
2-7 超音波破砕液での活性測定
フルオリナーゼの活性測定は2-5項で示したように精製酵素を用いた条件の方が正確で信頼性の高いデータが期待できるが、変異株の活性測定の場合、多検体の処理を要する点やスクリーニング的要素を含む点から考えると効率が悪い。そこで超音波破砕液を用いて酵素活性の比較を行うことにした。
【0051】
まず始めに5ml の菌体培養液を10 倍濃縮した超音波破砕液で酵素反応を行い、精製酵素を用いた場合と5’-FDA の生産量の変化を比較した(図11)。幸いにも超音波破砕液を用いた場合でも5’-FDA のピークはHPLC で独立したピークとして検出可能で十分な活性測定が行えた(data not shown)。また、精製酵素を用いた場合とほぼ同等の活性で検出することができ、超音波破砕液を用いた場合でも活性比較の議論が可能であることが分かった。
【0052】
2-8 超音波破砕液での野生株と変異株との活性検討・比較
G 株由来の各菌体(Wild type、S158A、S158G、 S158C、F156A、F156G、F156C、F156T、F156Y、F156D)の超音波破砕液を調整して酵素反応を行い、5’-FDA の生産量の変化を観察した(図12)。その結果、ほとんどすべての変異株でフルオリナーゼ活性を検出することはできなかった。しかし、一つの変異株F156Y は野生株の1/2 程度の5’-FDA を生産しておりフルオリナーゼ活性を有していることが示された。この株は156 番目の残基であるフェニルアラニンをチロシンに変えたものであり、構造の変化が比較的少ないためフルオリナーゼ活性を示すことができたと考えられる。
【0053】
他の変異株において活性が見られなかった原因としては、S158 変異株はフッ化物イオンと分子間力が働いているアミノ酸残基を変えてしまったため、フッ化物イオンが本来取り込まれるべきポケット構造に収まらなかったことが考えられる。F156 変異株においては、フェニルアラニンの構造的な大きさが影響していると察せられる。このF156 残基は基質と分子間力が働いていないが、その構造の大きさが基質の酵素への取り込みに大きな役割を果たしていると示唆される。唯一、構造変化が少ないF156Y 変異株で活性が見られたことからこれが言えると考える。
【0054】
この超音波破砕液でのアッセイ系で一つの変異株の活性が見られたので、これを精製してさらに詳細な活性比較を行うことにした。
【0055】
2-9 精製フルオリナーゼによる野生株と変異株との活性比較
G 株由来のF156Y 株より精製したフルオリナーゼ変異体を用いて、定量的に酵素活性を評価・比較するために基質濃度を変化させて酵素反応を行い(図13)、Lineweaver-BurkプロットをとりKm値およびVmax値を求めた(表8)。kcat / Km 値より、F156Y 変異株は野生株と比べてSAM に関しては2.5 倍、KFでは14.8 倍の活性の低下が見られた。特にフッ化物イオンに対するKm 値に着目すると、F156Y 変異株は野生株に比べて5.0 倍大きいことから、フッ素イオンと酵素との親和性がそれだけ低くなったと考えられ、F156 残基は基質との分子間力はないがフッ化物イオンが収まるポケット部位に構造的な影響を与えていることが示唆された。
【表8】
【0056】
2-10 塩化物イオンによるクロリナーゼ反応の試み
フルオリナーゼ反応において、基質の一つであるKF の代わりにKCl を投与することにより、5’-CDA の生産を行うことを試みた。使用した株はG 株由来の野生株とS158A、S158G、 S158C のSer158 番目を他のアミノ酸に変えた変異株である。X 線結晶構造解析より、基質結合部位においてフッ化物イオンは水分子から隔離されるようなポケット構造に収まり、SAM で蓋をされ基質結合部位に収まると考えられている。このポケット構造は狭いため、塩化物イオンや臭化物イオンは入り込まず、フッ化物イオンのみが収まると考えられている。Ser158 残基はこのこのポケット構造近傍に存在しているため、この側鎖を他のアミノ酸に置換させて、ポケット構造を広くした。これにより、このポケットに塩化物イオンも取り込まれ、塩素化反応が進むのではないかと期待された。しかし、超音波破砕液において過剰に基質を投与して、フッ素化反応に比べて長時間酵素反応を試みたが、HPLC で5’-CDA のピークを検出することはできなかった(図14)。このことより、フッ化物イオンが入り込む分子ポケット構造だけではなく、Ser158 残基とフッ化物イオンとの分子間力が大きく酵素反応に寄与していると示唆された。フルオリナーゼ反応の反応機構はSN2 反応であると考えられている。Ser158 残基とフッ化物イオンとの分子間力を考慮しなければ、塩化物イオンは求核剤としてフッ化物イオンより強いため、塩素化反応はフッ素化反応より起こりやすいと考えられる。しかし、この反応が進まなかったことより、この酵素反応には、Ser158 残基とフッ化物イオンとの分子間力が大きく影響を及ぼしていると察せられた。
【0057】
また、最近Deng らは野生型のフルオリナーゼを用いて、クロリナーゼ反応が起こることを報告しているが(H. Deng, S. L. Cobb, A. R. McEwan, R. P. McGlinchey, J. H. Naismith, D. O’ Hagan, D. A. Robinson, and J. B. Spencer, Angew. Chem., Int. Ed., 2006, 45, 759.)、反応の平衡は分解に強く傾いており、5’-CDAをさらに酵素反応で他の化合物を強く引き込まないと生成が確認できないと述べている。変異型酵素においてもこのような工夫が必要なのかもしれない。
【図面の簡単な説明】
【0058】
【図1】フルオリナーゼを発現させるプラスミドpETflAの構築方法を示す図。フルオリナーゼタンパク質をプラスミドベクターpET28b(+)のマルチクローニングサイトに挿入した。。
【図2】5'-FDAの化学合成法を示す図。
【図3】E.coli BL21(DE3)pETflA株における発現タンパク質の解析結果を示す図。E.coli BL21(DE3)pETflA株の全菌体、Ni2+カラムにより精製を行ったものを10%SDS-PAGEに供した後、CBB染色を行った。
【図4】NMRによるフルオリナーゼ活性の検出結果を示す図。(A)pH6.0にて一晩フルオリナーゼ反応を行ったものを19F NMRで活性を測定した。(B)大腸菌内でのフッ素化反応。フルオリナーゼのみを過剰に発現させているため、FAcや4-FTの生産は行われていない。
【図5】緩衝液のpHとフルオリナーゼ活性との関係を示す図。酵素反応のpHおよび緩衝液を変化させ、HPLCを用いて活性測定を行った。pH 8.0 Tris-HCl緩衝液での条件が最適であることが示された。
【図6】(A)B株由来のフルオリナーゼによる5'-FDA生産量を示す図及び(B)GroEタンパク質を導入したG株由来のフルオリナーゼによる5'-FDA生産量を示す図。B株:E.coli BL21(DE3)pLysS pALTERflA株、G株:B株にGroEタンパク質を共発現させた株。
【図7】GroEタンパク質の作用機構を示す図。ATPの補充により繰り返しフルオリナーゼのリフォーディングが誘導され、酵素活性が回復する。
【図8】B株における反応初速度と基質量との関係を示す図。(A)SAM濃度を変化させた。(B)グラフ(A)の両軸を逆数にした。(C)KF濃度を変化させた。(D)グラフ(C)の両軸を逆数にした。
【図9】G株における反応初速度と基質量との関係を示す図。(A)SAM濃度を変化させた。(B)グラフ(A)の両軸を逆数にした。(C)KF濃度を変化させた。(D)グラフ(C)の両軸を逆数にした。
【図10】フルオリナーゼの基質結合部位およびSer158残基近傍を示す図。フッ化物イオンの近傍にはSer158残基とPhe156残基が存在する。
【図11】菌体破砕液と精製酵素の5'-FDA生産量を示す図。
【図12】野生株と各種変異株の5'-FDA生産量を比較した図。超音波破砕液にて酵素反応を3時間行った結果。各種変異株のうちF156Yだけがフルオリナーゼ活性を示した。
【図13】F156Y変異株における反応初速度と基質量との関係を示す図。(A)SAM濃度を変化させた。(B)グラフ(A)の両軸を逆数にした。(C)KF濃度を変化させた。(D)グラフ(C)の両軸を逆数にした。
【図14】フルオリナーゼによる塩素化反応を表わす図。野生株及び各種変異株由来のフルオリナーゼ溶液にSAMとKClを添加して酵素反応を試みたが、反応は進行しなかった。
【技術分野】
【0001】
本発明は、フルオリナーゼ(フッ素化酵素)の製造方法、並びに変異型フルオリナーゼ作製方法及びその方法によって作製された変異型フルオリナーゼに関する。
【背景技術】
【0002】
近年、放線菌Streptomyces cattleya (S.cattleya) が、フッ化物イオンを有機化合物に導入できるフルオリナーゼを有していることがわかり、その三次元構造と遺伝子の塩基配列が報告された(非特許文献1)。このようなフルオリナーゼの詳細な報告例は本発明者の知る限りこれまでに全くなく、これを応用することができれば、困難だと考えられていたフッ素化合物の酵素による合成が可能になる。
【0003】
このS.cattleya由来のフルオリナーゼを大量生産するためには、大腸菌などを用いた組換えフルオリナーゼの発現系の構築が必要であるが、未だこのような発現系の構築に成功したという報告はない。
【0004】
【非特許文献1】C. Dong, F. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O’ Hagan, and J. H. Naismith, Nature, 2004, 427, 561-565.
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、以上のような技術的背景の下、組換えフルオリナーゼの発現系を構築し、フルオリナーゼを簡易かつ大量に生産する手段を提供することを目的とする。また、天然型のフルオリナーゼの基質特異性を変化させ、より多様なフッ素化合物の合成手段を提供することも目的とする。
【課題を解決するための手段】
【0006】
本発明者は、上記課題を解決するため鋭意検討を重ねた結果、フルオリナーゼをシャペロンタンパク質と共に大腸菌内で発現させることにより、高い活性を持つフルオリナーゼを生産できることを見出した。また、フルオリナーゼの156位のフェニルアラニンをチロシンに置換しても酵素活性が維持されることを見出した。本発明は、以上の知見に基づき完成されたものである。
【0007】
即ち、本発明は、以下の(1)〜(7)を提供するものである。
(1)フルオリナーゼとシャペロンタンパク質を微生物内で共発現させる工程、及び前記微生物を破砕し、その破砕物からフルオリナーゼを採取する工程を含むことを特徴とするフルオリナーゼの製造方法。
(2)フルオリナーゼが、配列番号2に記載のアミノ酸配列で表されることを特徴とする(1)に記載のフルオリナーゼの製造方法。
【0008】
(3)シャペロンタンパク質が、GroEであることを特徴とする(1)又は(2)に記載のフルオリナーゼの製造方法。
(4)微生物が、大腸菌であることを特徴とする(1)乃至(3)のいずれかに記載のフルオリナーゼの製造方法。
(5)フルオリナーゼに対し、配列番号2に記載のアミノ酸配列における156番目のフェニルアラニンに相当するアミノ酸を他のアミノ酸に置換することを特徴とする変異型フルオリナーゼの作製方法。
【0009】
(6)他のアミノ酸が、チロシンであることを特徴とする(5)に記載の変異型フルオリナーゼの作製方法。
(7)(5)又は(6)に記載の方法によって作製された変異型フルオリナーゼ。
【発明の効果】
【0010】
本発明により活性の高いフルオリナーゼを簡易かつ大量に製造することが可能になる。また、本発明は、基質の取り込みに関与するアミノ酸を置換した変異型のフルオリナーゼも提供する。
【発明を実施するための最良の形態】
【0011】
以下、本発明を詳細に説明する。
(1)フルオリナーゼの製造方法
本発明のフルオリナーゼの製造方法は、フルオリナーゼとシャペロンタンパク質を微生物内で共発現させる工程、及び前記微生物を破砕し、その破砕物からフルオリナーゼを採取する工程を含むことを特徴とするものである。
【0012】
フルオリナーゼとしては、S.cattleya由来の5'-フルオロ-5'-デオキシアデノシン合成酵素を使用することができる。この酵素のアミノ酸配列及び酵素をコードする遺伝子の塩基配列をそれぞれ配列番号2及び配列番号1に示す。フルオリナーゼとしては、この5'-フルオロ-5'-デオキシアデノシン合成酵素のアミノ酸配列の一部を変更した酵素(例えば、活性を失わせない範囲で、1若しくは数個のアミノ酸を欠失、置換若しくは付加した酵素など)を使用してもよい。このようなアミノ酸配列の一部を変更したフルオリナーゼの一例として、配列番号2に記載のアミノ酸配列における156番目のフェニルアラニンをチロシンに置換した変異型フルオリナーゼを例示できる。
【0013】
シャペロンタンパク質としては、GroEを使用することができるが、これ以外のシャペロンタンパク質を使用してもよい。
微生物としては、大腸菌を使用することができるが、他の微生物を使用してもよい。
フルオリナーゼ遺伝子とシャペロンタンパク質遺伝子を微生物中で共発現させる方法は特に限定されず、常法に従って行うことができる。
微生物を破砕し、その破砕物からフルオリナーゼを採取する方法も特に限定されず、微生物の生産するタンパク質を採取する際に使用される一般的な方法を適用することができる。
【0014】
(2)変異型フルオリナーゼの作製方法
本発明の変異型フルオリナーゼの作製方法は、フルオリナーゼに対し、配列番号2に記載のアミノ酸配列における156番目のフェニルアラニンに相当するアミノ酸を他のアミノ酸に置換することを特徴とするものである。
フルオリナーゼとしては、S.cattleya由来の5'-フルオロ-5'-デオキシアデノシン合成酵素を使用することができる。また、この5'-フルオロ-5'-デオキシアデノシン合成酵素のアミノ酸配列の一部を変更した酵素を使用してもよい。
【0015】
「配列番号2に記載のアミノ酸配列における156番目のフェニルアラニンに相当するアミノ酸」とは、対象とするフルオリナーゼの立体構造を、配列番号2に記載のアミノ酸配列からなるフルオリナーゼ(S.cattleya由来の5'-フルオロ-5'-デオキシアデノシン合成酵素)の立体構造と比較した際に、配列番号2に記載のアミノ酸配列における156番目のフェニルアラニンと同様の位置に存在するフェニルアラニンを意味する。
フェニルアラニンと置き換える他のアミノ酸としては、チロシンを挙げることができるが、活性を維持できるのであればこれ以外のアミノ酸であってもよい。
【0016】
アミノ酸の置換は、常法に従って行うことができ、例えば、後述する実施例のように変異導入用のプラスミドを使用して行うことができる。
後述するように、配列番号2に記載のアミノ酸配列における156番目のフェニルアラニンは、基質の酵素への取り込みに重要な役割を果たしていると考えられる。従って、このフェニルアラニンを他のアミノ酸に置換した変異型フルオリナーゼは、本来の基質であるS-アデノシル-L-メチオニン以外の化合物に対してフッ化物イオンを導入できると推測される。
【実施例】
【0017】
以下、実施例により本発明を更に詳細に説明する。
1 実験材料および方法
1-1 化合物、使用菌株、培地および培養条件
【0018】
化合物
表1に本実施例で使用した基質、合成試薬を示した。S-アデノシル-L-メチオニン (SAM) は常温では不安定なため、20mM に希釈して-20℃に保存した。他の基質、試薬は各メーカーの指示に従い保存した。
【表1】
【0019】
菌体
表2に使用した菌株を示した。fla 遺伝子(フルオリナーゼをコードする遺伝子)供与体としてS.cattleya NBRC14057 を用いた。遺伝子組換えプラスミド調整用の宿主としてはEscherichia coli (E. coli ) JM109 株を使用し、タンパク質発現用宿主にはE. coli BL21(DE3) 株および E. coli BL21(DE3) pLysS 株を用いた。また、変異株作製にはE. coli ES1301 mutS 株を使用した。
【表2】
【0020】
プラスミド・プライマー
表3に使用したプラスミドを示した。PCR 産物のクローニングおよび組換え遺伝子の発現には、pET28b(+) (Novagen)を使用した。pET28b(+) はColE1 系の複製起点、カナマイシン耐性遺伝子、lacIq の他、ヒスチジンタグ融合型の遺伝子を含むマルチクローニングサイト上流にtac プロモーターが存在しており、IPTG による発現誘導調節可能な高コピーベクターである。また変異株作製にはpALTER1 を用いた。これは変異株作製キットAltered Sites II in vitro Mutagenesis System (Promega) に添付されている変異株作製用のプラスミドであり、ColE1 系の複製起点、アンピシリン耐性遺伝子、テタラサイクリン耐性遺伝子マルチクローニングサイト上流にtac プロモーターが存在しており、IPTG による発現誘導調節可能な高コピーベクターである。PGEL2 は東京工業大学大学院生命理工研究科和地研究室の村上智史氏が構築され、本実験のため、和地正明博士および荻野英賢博士の御好意により使用させていただいた。
【0021】
プライマーは各種必要に応じて設計し、グライナージャパン社より購入して用いた(表4)。
【表3】
【表4】
【0022】
培地
S.cattleya NBRC14057 の培養には227 培地 [0.4% Dried yeast extract (Wako)、1.0% Malt extract (Wako)、0.4% Glucose ] を用い、培養は28℃で行った。また大腸菌の培養には0.1% glucose を添加したLennox (L) 培地(DIFCO) を使用し、培養は30℃もしくは37℃で行った。227 培地は各成分を水道水に、L 培地は純水 (MILLIPORE) に溶解し、121℃で20 分間オートクレーブ滅菌したのち使用した。固形培地には終濃度1.5%の寒天 (Wako) を添加した。プラスミドの保持には適宜、抗生物質を添加した。
【0023】
1-2 クローニング技術
特に記述ないものはMolecular Cloning, 2ed.(J. Sambrook, E. F. Fritsch and T. Maniatis, Molecular cloning: A laboratory manual, 2nd edition. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press: 1989.)、およびExperiments in Molecular Genetics(J. H. Miller, Experiments in molecular genetics: Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press: 1972.)を参考にした。制限酵素およびDNA リガーゼ、T4DNAポリメラーゼはTAKARA、Promega より購入し、メーカーの指示に従って使用した。E.coli の形質転換はエレクトロポレーションで行った。エレクトロポレーションにはElectroporater (Invitrogen) を使用した。1500V、150Ωでパルスを付与することにより行った。コンピテントセルの調整は同社の説明書の指示に従った。大腸菌からのプラスミドDNA の調整は、アルカリSDS 法あるいはWizard Plus SV Minipreps DNA purification system (Promega) を用いて行った。DNA のアガロース電気泳動には、トリス-ホウ酸-EDTA 緩衝液(Promega)を用い、ゲル濃度は0.7%とした。泳動後のゲルはエチジウムブロミド染色を行い、DNA を検出した。DNA 分子量マーカーはλ/ Hind III(ニッポンジーン)を用いた。
【0024】
1-3 プラスミドpETflA の構築
S.cattleya NBRC14057 株の菌体を鋳型として、先行論文(前述したC. Dongらの論文)に従いPCR プライマーHis-fla1、His-fla2(表4)を用いてS.cattleya ゲノムからfla 遺伝子の増幅をPCRにより行った(図1)。S.cattleya NBRC14057 株は28℃、120 rpm の条件で48 時間振とう培養したものを遠心により菌体を集菌した。これを滅菌水で希釈・懸濁したものをPCR の鋳型として用いた。PCR において、購入したオリゴヌクレオチド10μM に希釈したものをPCR プライマーとして用いた。使用したプライマーは表4に示した。PCR はiCycler (BIORAD)を用いて行った。耐熱性DNA ポリメラーゼ等はLA Taq polymerase (TaKaRa)を使用した。PCR の条件は94℃, 1min のち、94℃, 30sec., 59℃, 30sec., 72℃,2min を30 サイクル行い、最後に72℃, 5min の処理を施したものであり、最後は4℃に冷却した。得られたDNA 断片と高コピーのプラスミドベクターpET28b(+) をそれぞれ制限酵素NdeI、XhoIで消化し、リガーゼ (Takara) により結合することでフルオリナーゼタンパク質のN 末端側にヒスチジンタグが融合したタンパク質を発現させるプラスミドpETflA を構築した。この挿入の有無は制限消化により確認した。候補のクローンについて塩基配列を確認したが野生型の遺伝子は得られず、必ず変異が入っていた。そのため2つの変異遺伝子を制限消化により組み合わせることでサイレントな変異を持つクローンを得た。これをpETflA として用いることにした。
【0025】
1-4 発現タンパク質の確認
泳動用試料の調整
5ml のL 液体培地を用いて30℃で一晩培養した前培養液を、新たなL 液体培地5ml に1%植菌し30℃で24 時間培養した。対数増殖期前に終濃度0.1mM のIPTG を培地に添加した。遠心により集菌し、50mM pH7.5 Tris-HCl 緩衝液で2 度洗浄後、同緩衝液に懸濁し超音波処理により菌体を破砕した。適当量をSDS-PAGE 試料用緩衝液(Invitrogen)に懸濁し、これを100℃、3 分間加熱処理したものを用いた。
【0026】
SDS-ポリアクリルアミドゲル電気泳動(SDS-PAGE)
SDS-PAGE はLaemmli の方法(U. K. Laemmli, Nature, 1970, 227, 680.) を参考にした。泳動にはNuPAGE 10%Bis-Tris Gel(Invitrogen)を用いた。泳動後のタンパク質はクーマシーブリリアントブルーR-250(CBB)により染色した。なお分子量マーカーはタンパク質分子量マーカー「第一」・III(第一化学薬品)を使用した。
【0027】
Ni2+カラムによる酵素精製
前述の培養条件で菌体を調整・集菌した。各実験で使用する緩衝液で2 度洗浄後、同緩衝液に懸濁し超音波処理により菌体を破砕した。菌体破砕後、遠心により上清を取り出し、ヒスチジンタグと親和性のあるニッケルカラムNi-NTA spin kit(QIAGEN)を用いてアフィニテークロマトグラフィーにより酵素精製を行った。700mM のイミダゾールで溶出後、Microcon YM-10(MILLIPORE)により限外ろ過を行い、脱塩濃縮およびバッファー交換した。タンパク質の定量は、ウシ血清アルブミン (BSA) を標準としてBradford 法により行った。
【0028】
1-5 NMR による活性検出
1-4の培養条件で20μg/ml kanamycin を添加したE. coli BL21(DE3)pETflA 株の10ml 分の菌体を調整・集菌した。表5の各緩衝液で2 度洗浄後、同緩衝液に懸濁し超音波処理により菌体を破砕した。菌体破砕後遠心し、上清を反応溶液として用いて、終濃度10mM のNaF と800μM のSAM を投与し、200μl スケールで30℃にて一晩反応させた。NMR サンプルとして、この反応液と等量のd-DMSO(Merck 社)を加えてVXR-500(500MHz、Varian)により19F NMR を測定した。
【表5】
【0029】
1-6 変異導入用プラスミドpALTERflA の構築および各種変異株の作製
pETflA とpALTER ベクターをそれぞれ制限酵素SphI、DraIIIで消化し、これらをライゲーションさせることによりpALTERflA を構築した。このプラスミドに対して、Altered Sites II in vitro Mutagenesis System (Promega)を用いて各種変異導入プラスミド (S158A 変異株 (158 番目の残基をセリンからアラニンに置換したもの)、S158G、 S158C、F156A、F156G、F156C、F156T、F156Y、F156D )を作製した。変異導入は作製した各種プラスミドを鋳型として、DTCS Quick Start Kit (BECKMAN COULTER) によりdideoxy 反応を行い、CEQ8000 (BECKMAN COULTER) を用いて塩基配列を確認した。そして、変異導入したプラスミドはE. coli BL21(DE3)pLysS 株に形質転換し、1-4の方法でタンパク質発現の確認を行った。
【0030】
1-7 GroE タンパク質の導入
E. coli BL21(DE3) pLysS pALTERflA 由来の各菌体( wild type、S158A、S158G、 S158C、F156A、F156G、F156C、F156T、F156Y、F156D)にpSC101由来の複製起点を持つプラスミドpMW218 にgroE (groEL, groES) 遺伝子含むプラスミドpGEL2 を形質転換し、groE (groEL, groES) 遺伝子を大腸菌内でマルチコピーに導入した系を構築した。フルオリナーゼの発現は1-4の方法で確認した。
【0031】
1-8 HPLC による活性測定
高速液体クロマトグラフィー(HPLC)を用いて、フルオリナーゼ反応の検出を行った。HPLC は島津製作所社製SPD-10Avp を用いた。カラムは逆相カラムShin-pack VP-ODS 150×4.6(島津製作所)を使用して、検出波長はアデニンの吸収波長である260nmとした。定量化するために先行論文(V. Reichelova, F. Albertioni, and J. Liliemark, J. Chromatography. 1994, 667, 37.)を参考に5’-FDAを化学合成し(図2)、分離条件を検討した。また、サンプルの標準物質として5’-クロロ-5’-デオキシアデノシン (5’-CDA) が適当であったのでこれを用いてピーク検出を行った。HPLC サンプルは、回収した反応液に等量のメタノールを加えて酵素を失活させ遠心により上清を取り出した。そして小型エバポレーターMicro Vac(TOMY)により溶媒を蒸発させ、この操作を二回繰り返したものを調整した。これを5’-CDA 添加したHPLC 溶出溶媒50μl で希釈し、10μl を測定に用いた。
【0032】
1-9 フルオリナーゼ反応
pH およびbuffer の検討
1-4の培養条件で10μg/ml tetracycline、20μg/ml chroramphenicol を添加したE. coli BL21(DE3) pLysS pALTERflA 株の20ml 分の菌体を調整・集菌した。50mM pH8.0 Tris-HCl 緩衝液で2 度洗浄後、同緩衝液に懸濁し超音波処理により菌体を破砕した。菌体破砕後、遠心により上清を取り出し、Ni2+カラムによるアフィニテークロマトグラフィーにより酵素精製を行った。この精製体を1-4の方法によりタンパク質定量し、表6の各種緩衝液を加えて1.0μg/ml の濃度に調整し、終濃度250μMのSAMと10mMのKF を添加して100μl スケールで37℃にて酵素反応を行った。反応温度はO’Hagan らの報告例(前述したC. Dongらの論文、及びS. L. Cobb, H. Deng, J. T. G. Hamilton, R. P. McGlinchey and D. O’ Hagan, 43 Chem. Commum., 2004, 592.)を参考にした。反応開始から5分後にそれぞれ反応液を回収し、HPLC による活性測定を行った。
【表6】
【0033】
野生株とGroE 導入株の活性比較
(1)酵素濃度の検討
1-4の培養条件で10μg/ml tetracycline、20μg/ml chroramphenicol を添加したE. coli BL21(DE3) pLysS pALTERflA 株(B株)および10μg/ml tetracycline、20μg/ml chroramphenicol、20μg/ml kanamycin を添加したGroE タンパク質を共発現させたE. coli BL21(DE3) pLysS pGEL2 pALTERflA株(G株)の20ml 分の菌体を調整・集菌した。50mM pH8.0 Tris-HCl 緩衝液で2 度洗浄後、同緩衝液に懸濁し超音波処理により菌体を破砕した。菌体破砕後、遠心により上清を取り出し、Ni2+カラムによるアフィニテークロマトグラフィーにより酵素精製を行った。この精製体を1-4 の方法でタンパク質定量し、50mM pH8.0 Tris-HCl 緩衝液に0.25μg/ml および1.0μg/ml の濃度に調整し、終濃度250μM のSAM と10mM のKF を添加して100μl スケールで37℃にて酵素反応を行った。反応開始から5分、15 分、60 分、180 分後にそれぞれ反応液を回収し、HPLC による活性測定を行った。
【0034】
(2)基質濃度の検討
上記と同様の方法でフルオリナーゼを精製した。50mM pH8.0 Tris-HCl 緩衝液で調整したB株およびG株由来の各1.0μg/ml のフルオリナーゼ溶液に、SAM 濃度の変化系には終濃度10mM のKF と、G株は10、12.5、15、20、40、80、100μM のSAM を、B株は15、25、40、50、80、100μM のSAM を添加して5分間37℃にて100μl スケールで酵素反応を行った。また、KF 濃度の変化系には終濃度250μM のSAM とG株は0.2、0.25、0.5、1.0、2.0、4.0、5.0、8.0 mM のKF を、B株は0.2、0.25、0.5、1.0、2.0、4.0 mM のKF を添加して5分間37℃にて100μl スケールで酵素反応を行った。反応後、HPLCによる活性測定を行った。
【0035】
超音波破砕液における活性検出
1-4の培養条件で10μg/ml tetracycline、20μg/ml chroramphenicol、20μg/ml kanamycin を添加したG 株の5ml 分の菌体を調整・集菌した。50mM pH8.0 Tris-HCl 緩衝液で2 度洗浄後、同緩衝液に10 倍濃縮で懸濁し超音波処理により菌体を破砕した。菌体破砕後、遠心により上清を取り出し、これを酵素反応液とした。そして、終濃度250μM のSAM と10mM のKF を添加して100μl スケールで37℃にて酵素反応を行った。反応開始から5分、15 分、60分、180 分後にそれぞれ反応液を回収した。回収後、HPLC による活性測定を
行った。
【0036】
超音波破砕液による野生株と変異株との活性比較
超音波破砕液における活性検出の項と同様の方法でG 株由来の各菌体( wild type、S158A、S158G、 S158C、F156A、F156G、F156C、F156T、F156Y、F156D)の超音波破砕液を作製した。培地に野生株には終濃度10μg/ml tetracycline、20μg/ml chroramphenicol、20μg/ml kanamycin を、変異株には125μg/ml ampicillin、20μg/ml chroramphenicol、20μg/ml kanamycin を添加した。菌体破砕後、遠心により上清を取り出し、これを酵素反応液とした。そして、終濃度250μM のSAM と10mM のKF を添加して100μl スケールで37℃にて酵素反応を行った。反応開始から60 分、180 分後にそれぞれ反応液を回収した。回収後、HPLC による活性測定を行った。
【0037】
酵素精製溶液による野生株と変異株との活性比較
G 株由来のF156Y 変異株から、野生株とGroE 導入株の活性比較と同様の方法でフルオリナーゼ変異株を精製した。50mM pH8.0 Tris-HCl 緩衝液で1.0μg/ml に調整した酵素溶液にSAM濃度変化系では、終濃度10mMのKFと15、25、15、40、50、80、100μM のSAM を添加して、5分間37℃にて100μlスケールで酵素反応を行った。KF 濃度変化系では、終濃度250μM のSAM と0.5、1.0、2.0、4.0、8.0、10、12、15、20 mM のKF を添加して、5分間37℃にて100μl スケールで酵素反応を行った。反応後、HPLC による活性測定を行った。
【0038】
塩化物イオンによるクロリナーゼ反応の試み
超音波破砕液により酵素反応を行った。超音波破砕液における活性検出の項と同様にG 株由来の各菌体( wild type、S158A、S158G、 S158C)の超音波破砕液を作製した。菌体破砕後、遠心により上清を取り出しこれを酵素反応液とした。そして、終濃度500μM のSAM と60mM のKCl を添加して酵素反応を行った。反応開始から180 分後、24 時間後にそれぞれ同量ずつ反応液を回収した。回収後、HPLC による活性測定を行った。
【0039】
2 結果と考察
2-1 pETflA の構築および大腸菌での組換えフルオリナーゼ発現
S.cattleya 由来のフルオリナーゼ反応系を構築するために、大腸菌内で組換えフルオリナーゼの発現を目指した。先行論文(前述したC. Dongらの論文)に従いプライマーを設計し、S.cattleya の菌体を鋳型としてflA 遺伝子をPCR で増幅した。増幅断片を制限酵素で消化後、pET28b(+) にクローン化し、pETflA を構築した。シークエンサーを用いて塩基配列を確認したが、変異がないflA 遺伝子を獲得することができなかった。これはGC リッチなflA 遺伝子配列がスムーズなポリメラーゼ反応に支障をきたし、変異が入りやくなったためであると考えられる。塩基配列解析結果から変異が一番少ない2つの変異遺伝子を制限消化により組み合わせることでサイレントな変異を持つクローンを得た。これをpETflA として用いることにした(data not shown)。
【0040】
また、 pETflA をE.coli BL21(DE3)株に導入し、IPTG 誘導をかけた全菌体をSDS-PAGE に供したのちCBB 染色を施し発現タンパクを調べたところ大量発現しているタンパク質の存在が確認された(図3)。これは先行論文(前述したC. Dongらの論文)とほぼ一致する位置(34kDa)にバンドが出ていることから、フルオリナーゼが正しく発現されていることが示唆された。
【0041】
そして、この組換えフルオリナーゼはN 末端側にヒスチジンタグが融合しているので、Ni2+カラムによるアフィニテークロマトグラフィーで精製を行った。全菌体中に認められたフルオリナーゼのバンドに比べて少ない量ではあるが、ほぼ単一のバンドとして精製することができた。この回収量の低下は封入体形成などによるものと思われた。
【0042】
2-2 NMR による活性検出
E.coli BL21(DE3) pETflA 株の超音波破砕液にフルオリナーゼの基質であるSAM とNaF を加え一晩反応を行った。pH を6.0〜8.0 に調整し、19F NMR により5’-FDA の生成を確認したところ、pH6.0〜7.0 付近の反応溶液で弱いながらも5’-FDA と予想されるピーク(19F NMR: σ=-64.4 ppm (dt, J (F, 4’H)=24.2 z, J (F, 5’H)=47.3 Hz) )を確認した(図4)。これは化学合成した5’-FDAのピーク(19F NMR: σ=-64.4 ppm (dt, J (F, 4’H)=24.4 Hz, J (F, 5’H)=47.4 Hz) )とほぼ一致することから大腸菌においてもフルオリナーゼ反応が進行していると考えた。
【0043】
S.cattleya における生体内フッ素化合物の代謝においては、このSAM から5’-FDA へのフッ素化反応を経て、モノフルオロ酢酸(FAc)とフルオロスレオニン(4-FT)が生産されるが、この系は大腸菌内でフルオリナーゼのみを発現させているため下流の反応系はなく5’-FDA の生成で止まっていると考えられる(図4)。この結果から大腸菌による組換えフルオリナーゼを用いたin vitroフッ素化反応系は構築できることが確認できたが、NMR による5’-FDA の生成確認では弱いピークしか検出できなかった。この原因は大腸菌内でのフルオリナーゼ反応が弱いこともあるが、NMR が物質の検出系として向いてない点も挙げられる。NMR は検出にmg スケールの量を要するので、今回のような小さいスケールの酵素反応系の検出系には向いておらず、μg スケール以下で検出できる高感度の検出系の構築が必要であると考えられた。
【0044】
2-3 HPLC によるフルオリナーゼ活性検出系の構築
NMR に代わる高感度の5’-FDA の活性検出法としてHPLC を選択した。溶出溶媒等の検討を行ったところ、MeOH と10mM pH7.0 KH2PO4 を20:80に混合したものが適当であったのでこれを用いた。また定量化するために化学合成した5’-FDA を用いたところ、数ng スケールの5’-FDA を検出することが可能になり弱い酵素活性の反応でも定量化が可能になった。サンプルの標準物質としては5’-クロロ-5’-デオキシアデノシン (5’-CDA) を用いた。これは、5’-FDA と最大吸収波長が同じであること(260nm)とHPLC での溶出時間が十分離れて溶出時間比率も一定であるので(5’-FDA:10.0 分、5’-CDA: 20.5 分)、
標準物質として適当と考えた。
【0045】
2-4 pH および緩衝液の検討
フルオリナーゼ反応を行うにあたり、反応溶液のpH および緩衝液を検討した。これは、先行論文(前述したC. Dongらの論文)ではHPLC を用いてpH8.0 付近でフルオリナーゼアッセイを行っているが、本実施例で行ったNMR における活性検出ではpH6.0〜7.0 付近でのみピークが認められたためである。したがって感度の高いHPLC による検出系を用いることで再度、至適pH や至適バッファー成分を検討することにした。表6の各緩衝溶液に置換した酵素溶液をそれぞれ5分間反応させ、生成した5’-FDA の量を比較した(図5) 。これによりpH8.0 で一番多く5’-FDAを生産していることが示され、NMR で検出できたpH の範囲とHPLC により最も生成物ピークが大きかったpH が異なる結果となった。これは、pH や緩衝液の成分により、NMR サンプル中の分子状態が変化してピークがブロード状態になるなど形状が変わり、ノイズ成分に隠れてしまったことが原因の一つとして考えられる。NMR の検出系が低感度であるため、HPLC による活性検出結果の方が信頼性が高いものと考えられた。したがって、フルオリナーゼ反応における至適pH は8.0 であると結論づけた。
【0046】
2-5 GroE タンパク質導入による活性の向上
E. coli BL21(DE3) pLysS pALTERflA 株(B株)由来のフルオリナーゼを用いて、HPLC による活性検出を行った。まず、酵素濃度を変えることにより5’-FDA の生産量の変化を観察した(図6(A))。酵素濃度を3段階に振ったところ、1.0μg/ml と2.5μg/ml の濃度では、その5’-FDA 生産量はほとんど変わらなかった。しかし、全体的に5’-FDA の生成量が少ないので失活したフルオリナーゼの割合が高いと考えられ、活性回復を図るためにin vitro タンパク質発現系にGroEタンパク質を共発現させることにした。GroEタンパク質はGroEL, GroES タンパク質からなる複合体タンパク質であり、大腸菌内でメジャーなシャペロンタンパク質である。GroE タンパク質を目的タンパク質と共発現させることで、そのリフォールディングが誘導され、酵素活性の回復が引き起こされることが知られている(図7)。この原理を利用してフルオリナーゼ活性向上のために、groE (groEL, groES ) 遺伝子を大腸菌内でマルチコピーに導入した系を構築した。GroE タンパク質を共発現した株(G 株)においてもB株と同様に酵素濃度を変えることにより5’-FDA の生産量の変化を観察した(図6(B))。B 株に比べて5’-FDA の生産量の増加が見られ、GroE タンパク質による活性回復が示唆された。
【0047】
また、G 株でも酵素濃度を3段階に振ったところ、1.0μg/ml と2.5μg/ml の濃度ではその5’-FDA 生産量はほとんど変わらなかったので、これ以後反応溶液の酵素濃度は1.0μg/ml として測定を行った。
そして、定量的に酵素活性を評価するために、基質の濃度を変えて5’-FDA の生産量の変化を調べ (図8、図9)、Lineweaver-Burk プロットをとり各基質のミカエリス定数 (Km) と反応最大速度 (Vmax) を求めた(表7)。
【0048】
kcat / Km に注目すると、G 株はB 株と比べてSAM に関しては3.1 倍、KFでは5.1 倍の活性の向上が見られた。この要因としてVmax 値の向上が挙げられる。GroE マルチコピー系に用いたプラスミドはpSC101 由来の複製起点を持ち、コピー数は3〜5 と言われている。したがってG 株とB 株のVmax の比が約4倍となるのは、ちょうどgroE のコピー数の差と一致する(野生株1コピーに対してマルチコピー系は4〜6 コピーの試算になるため。)。このことよりgroE遺伝子を導入するプラスミドをより高コピーのベクターに変えることで反応系のVmax をさらに向上させることが期待できるので、より生産性の高い反応系を構築することができるのではないかと考えられる。
【表7】
【0049】
2-6 変異株の作製
今回フルオリナーゼ変異株を作製するにあたり、報告された基質結合部位(図10) に着目した。X 線結晶構造解析より、基質結合部位においてフッ化物イオンは水分子から隔離されるようなポケット構造に収まり、SAM で蓋をされ基質結合部位に収まると考えられている。そして、コンピューターシミュレーションからSAM が結合してしまうとフッ化物イオンは入り込めなくなるため、反応は進行しないと察せられている。このことより、このフッ化物イオンが取り込まれるポケット構造を変化させれば基質特異性など性質が大きく変化すると予測し、フッ化物イオンに接近しているアミノ酸残基に注目した。その1つとしてSer158 残基に着目し、セリンを3つのアミノ酸に置換したもの(S158A, S158G, S158C)を作製した。S158A およびS158G においては、フッ化物イオンが入り込むポケット構造を広げることを狙った。S158C は、セリン残基のO原子とシステイン残基のS 原子のように同族元素で大きさが異なる原子がどのような影響を及ぼすのか検討するために調整した。さらにもう1つのアミノ酸残基に着目した。それはSer158 残基と近傍に存在するPhe156 残基である。図10よりPhe156 残基は基質と分子間力がないので、基質と酵素との分子間力の影響を比較・検討することに適当であると考え、6つの各種変異株を作製した ( F156A, F156G, F156C, F156D, F156T, F156Y) 。
【0050】
2-7 超音波破砕液での活性測定
フルオリナーゼの活性測定は2-5項で示したように精製酵素を用いた条件の方が正確で信頼性の高いデータが期待できるが、変異株の活性測定の場合、多検体の処理を要する点やスクリーニング的要素を含む点から考えると効率が悪い。そこで超音波破砕液を用いて酵素活性の比較を行うことにした。
【0051】
まず始めに5ml の菌体培養液を10 倍濃縮した超音波破砕液で酵素反応を行い、精製酵素を用いた場合と5’-FDA の生産量の変化を比較した(図11)。幸いにも超音波破砕液を用いた場合でも5’-FDA のピークはHPLC で独立したピークとして検出可能で十分な活性測定が行えた(data not shown)。また、精製酵素を用いた場合とほぼ同等の活性で検出することができ、超音波破砕液を用いた場合でも活性比較の議論が可能であることが分かった。
【0052】
2-8 超音波破砕液での野生株と変異株との活性検討・比較
G 株由来の各菌体(Wild type、S158A、S158G、 S158C、F156A、F156G、F156C、F156T、F156Y、F156D)の超音波破砕液を調整して酵素反応を行い、5’-FDA の生産量の変化を観察した(図12)。その結果、ほとんどすべての変異株でフルオリナーゼ活性を検出することはできなかった。しかし、一つの変異株F156Y は野生株の1/2 程度の5’-FDA を生産しておりフルオリナーゼ活性を有していることが示された。この株は156 番目の残基であるフェニルアラニンをチロシンに変えたものであり、構造の変化が比較的少ないためフルオリナーゼ活性を示すことができたと考えられる。
【0053】
他の変異株において活性が見られなかった原因としては、S158 変異株はフッ化物イオンと分子間力が働いているアミノ酸残基を変えてしまったため、フッ化物イオンが本来取り込まれるべきポケット構造に収まらなかったことが考えられる。F156 変異株においては、フェニルアラニンの構造的な大きさが影響していると察せられる。このF156 残基は基質と分子間力が働いていないが、その構造の大きさが基質の酵素への取り込みに大きな役割を果たしていると示唆される。唯一、構造変化が少ないF156Y 変異株で活性が見られたことからこれが言えると考える。
【0054】
この超音波破砕液でのアッセイ系で一つの変異株の活性が見られたので、これを精製してさらに詳細な活性比較を行うことにした。
【0055】
2-9 精製フルオリナーゼによる野生株と変異株との活性比較
G 株由来のF156Y 株より精製したフルオリナーゼ変異体を用いて、定量的に酵素活性を評価・比較するために基質濃度を変化させて酵素反応を行い(図13)、Lineweaver-BurkプロットをとりKm値およびVmax値を求めた(表8)。kcat / Km 値より、F156Y 変異株は野生株と比べてSAM に関しては2.5 倍、KFでは14.8 倍の活性の低下が見られた。特にフッ化物イオンに対するKm 値に着目すると、F156Y 変異株は野生株に比べて5.0 倍大きいことから、フッ素イオンと酵素との親和性がそれだけ低くなったと考えられ、F156 残基は基質との分子間力はないがフッ化物イオンが収まるポケット部位に構造的な影響を与えていることが示唆された。
【表8】
【0056】
2-10 塩化物イオンによるクロリナーゼ反応の試み
フルオリナーゼ反応において、基質の一つであるKF の代わりにKCl を投与することにより、5’-CDA の生産を行うことを試みた。使用した株はG 株由来の野生株とS158A、S158G、 S158C のSer158 番目を他のアミノ酸に変えた変異株である。X 線結晶構造解析より、基質結合部位においてフッ化物イオンは水分子から隔離されるようなポケット構造に収まり、SAM で蓋をされ基質結合部位に収まると考えられている。このポケット構造は狭いため、塩化物イオンや臭化物イオンは入り込まず、フッ化物イオンのみが収まると考えられている。Ser158 残基はこのこのポケット構造近傍に存在しているため、この側鎖を他のアミノ酸に置換させて、ポケット構造を広くした。これにより、このポケットに塩化物イオンも取り込まれ、塩素化反応が進むのではないかと期待された。しかし、超音波破砕液において過剰に基質を投与して、フッ素化反応に比べて長時間酵素反応を試みたが、HPLC で5’-CDA のピークを検出することはできなかった(図14)。このことより、フッ化物イオンが入り込む分子ポケット構造だけではなく、Ser158 残基とフッ化物イオンとの分子間力が大きく酵素反応に寄与していると示唆された。フルオリナーゼ反応の反応機構はSN2 反応であると考えられている。Ser158 残基とフッ化物イオンとの分子間力を考慮しなければ、塩化物イオンは求核剤としてフッ化物イオンより強いため、塩素化反応はフッ素化反応より起こりやすいと考えられる。しかし、この反応が進まなかったことより、この酵素反応には、Ser158 残基とフッ化物イオンとの分子間力が大きく影響を及ぼしていると察せられた。
【0057】
また、最近Deng らは野生型のフルオリナーゼを用いて、クロリナーゼ反応が起こることを報告しているが(H. Deng, S. L. Cobb, A. R. McEwan, R. P. McGlinchey, J. H. Naismith, D. O’ Hagan, D. A. Robinson, and J. B. Spencer, Angew. Chem., Int. Ed., 2006, 45, 759.)、反応の平衡は分解に強く傾いており、5’-CDAをさらに酵素反応で他の化合物を強く引き込まないと生成が確認できないと述べている。変異型酵素においてもこのような工夫が必要なのかもしれない。
【図面の簡単な説明】
【0058】
【図1】フルオリナーゼを発現させるプラスミドpETflAの構築方法を示す図。フルオリナーゼタンパク質をプラスミドベクターpET28b(+)のマルチクローニングサイトに挿入した。。
【図2】5'-FDAの化学合成法を示す図。
【図3】E.coli BL21(DE3)pETflA株における発現タンパク質の解析結果を示す図。E.coli BL21(DE3)pETflA株の全菌体、Ni2+カラムにより精製を行ったものを10%SDS-PAGEに供した後、CBB染色を行った。
【図4】NMRによるフルオリナーゼ活性の検出結果を示す図。(A)pH6.0にて一晩フルオリナーゼ反応を行ったものを19F NMRで活性を測定した。(B)大腸菌内でのフッ素化反応。フルオリナーゼのみを過剰に発現させているため、FAcや4-FTの生産は行われていない。
【図5】緩衝液のpHとフルオリナーゼ活性との関係を示す図。酵素反応のpHおよび緩衝液を変化させ、HPLCを用いて活性測定を行った。pH 8.0 Tris-HCl緩衝液での条件が最適であることが示された。
【図6】(A)B株由来のフルオリナーゼによる5'-FDA生産量を示す図及び(B)GroEタンパク質を導入したG株由来のフルオリナーゼによる5'-FDA生産量を示す図。B株:E.coli BL21(DE3)pLysS pALTERflA株、G株:B株にGroEタンパク質を共発現させた株。
【図7】GroEタンパク質の作用機構を示す図。ATPの補充により繰り返しフルオリナーゼのリフォーディングが誘導され、酵素活性が回復する。
【図8】B株における反応初速度と基質量との関係を示す図。(A)SAM濃度を変化させた。(B)グラフ(A)の両軸を逆数にした。(C)KF濃度を変化させた。(D)グラフ(C)の両軸を逆数にした。
【図9】G株における反応初速度と基質量との関係を示す図。(A)SAM濃度を変化させた。(B)グラフ(A)の両軸を逆数にした。(C)KF濃度を変化させた。(D)グラフ(C)の両軸を逆数にした。
【図10】フルオリナーゼの基質結合部位およびSer158残基近傍を示す図。フッ化物イオンの近傍にはSer158残基とPhe156残基が存在する。
【図11】菌体破砕液と精製酵素の5'-FDA生産量を示す図。
【図12】野生株と各種変異株の5'-FDA生産量を比較した図。超音波破砕液にて酵素反応を3時間行った結果。各種変異株のうちF156Yだけがフルオリナーゼ活性を示した。
【図13】F156Y変異株における反応初速度と基質量との関係を示す図。(A)SAM濃度を変化させた。(B)グラフ(A)の両軸を逆数にした。(C)KF濃度を変化させた。(D)グラフ(C)の両軸を逆数にした。
【図14】フルオリナーゼによる塩素化反応を表わす図。野生株及び各種変異株由来のフルオリナーゼ溶液にSAMとKClを添加して酵素反応を試みたが、反応は進行しなかった。
【特許請求の範囲】
【請求項1】
フルオリナーゼとシャペロンタンパク質を微生物内で共発現させる工程、及び前記微生物を破砕し、その破砕物からフルオリナーゼを採取する工程を含むことを特徴とするフルオリナーゼの製造方法。
【請求項2】
フルオリナーゼが、配列番号2に記載のアミノ酸配列で表されることを特徴とする請求項1に記載のフルオリナーゼの製造方法。
【請求項3】
シャペロンタンパク質が、GroEであることを特徴とする請求項1又は2に記載のフルオリナーゼの製造方法。
【請求項4】
微生物が、大腸菌であることを特徴とする請求項1乃至3のいずれか一項に記載のフルオリナーゼの製造方法。
【請求項5】
フルオリナーゼに対し、配列番号2に記載のアミノ酸配列における156番目のフェニルアラニンに相当するアミノ酸を他のアミノ酸に置換することを特徴とする変異型フルオリナーゼの作製方法。
【請求項6】
他のアミノ酸が、チロシンであることを特徴とする請求項5に記載の変異型フルオリナーゼの作製方法。
【請求項7】
請求項5又は6に記載の方法によって作製された変異型フルオリナーゼ。
【請求項1】
フルオリナーゼとシャペロンタンパク質を微生物内で共発現させる工程、及び前記微生物を破砕し、その破砕物からフルオリナーゼを採取する工程を含むことを特徴とするフルオリナーゼの製造方法。
【請求項2】
フルオリナーゼが、配列番号2に記載のアミノ酸配列で表されることを特徴とする請求項1に記載のフルオリナーゼの製造方法。
【請求項3】
シャペロンタンパク質が、GroEであることを特徴とする請求項1又は2に記載のフルオリナーゼの製造方法。
【請求項4】
微生物が、大腸菌であることを特徴とする請求項1乃至3のいずれか一項に記載のフルオリナーゼの製造方法。
【請求項5】
フルオリナーゼに対し、配列番号2に記載のアミノ酸配列における156番目のフェニルアラニンに相当するアミノ酸を他のアミノ酸に置換することを特徴とする変異型フルオリナーゼの作製方法。
【請求項6】
他のアミノ酸が、チロシンであることを特徴とする請求項5に記載の変異型フルオリナーゼの作製方法。
【請求項7】
請求項5又は6に記載の方法によって作製された変異型フルオリナーゼ。
【図1】

【図2】
【図4】
【図5】
【図6】

【図7】
【図8】
【図9】
【図10】
【図11】
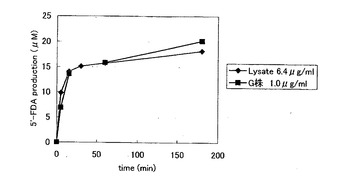
【図12】

【図13】
【図14】
【図3】
【図2】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図3】
【公開番号】特開2008−113626(P2008−113626A)
【公開日】平成20年5月22日(2008.5.22)
【国際特許分類】
【出願番号】特願2006−301793(P2006−301793)
【出願日】平成18年11月7日(2006.11.7)
【出願人】(304021417)国立大学法人東京工業大学 (1,821)
【出願人】(000002853)ダイキン工業株式会社 (7,604)
【Fターム(参考)】
【公開日】平成20年5月22日(2008.5.22)
【国際特許分類】
【出願日】平成18年11月7日(2006.11.7)
【出願人】(304021417)国立大学法人東京工業大学 (1,821)
【出願人】(000002853)ダイキン工業株式会社 (7,604)
【Fターム(参考)】
[ Back to top ]