
フルオロシチジン誘導体の製造プロセス
カペシタビンまたはその誘導体を製造するプロセスで、
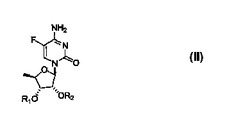
(a)式(II):
【化1】

(式中、各R1およびR2は、独立にヒドロキシル保護基を表す)の化合物を、式(III):X−C(=O)−R3(式中、Xはアシル活性化基)のアシル化試薬と有機溶媒中で反応させ、アシル化化合物を製造すること;(b)アシル化化合物を脱保護して式(I)の化合物を得ること;および(c)式(I)の化合物を溶媒で精製すること、を含む。
(a)式(II):
【化1】
(式中、各R1およびR2は、独立にヒドロキシル保護基を表す)の化合物を、式(III):X−C(=O)−R3(式中、Xはアシル活性化基)のアシル化試薬と有機溶媒中で反応させ、アシル化化合物を製造すること;(b)アシル化化合物を脱保護して式(I)の化合物を得ること;および(c)式(I)の化合物を溶媒で精製すること、を含む。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
関連出願への相互参照
本出願は、2009年7月23日出願の米国特許仮出願第61/227,971号の優先権を主張する。この仮出願の全体は参照により本明細書に組み込まれる。
【0002】
1.発明の分野
本出願は、5’−デオキシ−5−フルオロ−N4−n−ペンチルオキシカルボニルシチジン(カペシタビン)およびその誘導体の製造プロセスに関する。
2.関連技術の説明
カペシタビンは、抗悪性腫瘍活性を有するフロロピリミジンカルバマートで、ブランド名XELODA(登録商標)の商品として商業的に入手可能であり、次の化学構造を有する:
【0003】
【化1】
【0004】
カペシタビンの合成については、米国特許第5,472,949号;4,966,891号;5,453,497号;7,365,188号;および5,476,932号を含むいくつかの公表文献に記載されている。
しかし、カペシタビンおよびその誘導体の改良された製造プロセスの必要性が依然として存在する。
【発明の概要】
【0005】
本出願の1つの態様は、式(I):
【0006】
【化2】
【0007】
の精製された化合物の製造プロセスを提供することである。
(式中、R3はアルキル、シクロアルキル、アラルキル、アリール、またはアルコキシ、好ましくはC1〜C12アルキル、シクロアルキル、アラルキル、アリール、またはアルコキシ、さらに、より好ましくは、C1〜C6アルキルである。)
このプロセスは下記を含む。
(a)式(II):
【0008】
【化3】
【0009】
(式中、各R1およびR2は独立にヒドロキシル保護基を示す)の化合物を、式(III):X−C(=O)−R3(式中、Xはアシル活性化基、R3は上記で定義済み)のアシル化試薬と有機溶媒、例えば、CH2Cl2、THF、アセトニトリル、トルエン、または酢酸エチル、中で反応させ、式(IV):
【0010】
【化4】
【0011】
(式中、各R1、R2、およびR3は上記で定義済み)のアシル化化合物を作り;
(b)式(IV)のアシル化化合物を脱保護して式(I)の化合物を得て;さらに
(c)式(I)の化合物を溶媒を使って精製する。
好ましくは、ヒドロキシル保護基は、アセチルまたはベンゾイルである。
【0012】
上記式(III)のアシル化試薬中のXは、好ましくはハロゲン化物、さらに好ましくは、塩化物である。式(III)のアシル化試薬は、好ましくはn−ペンチルクロロホルマートである。
式(I)の化合物は、好ましくはカペシタビン、すなわち、上記式(I)のR3がペンチル基である化合物である。
【0013】
上記プロセスの反応ステップ(a)は塩基の存在下で行うことが好ましい。塩基は、好ましくは式(II)の化合物の3.5〜5.0モル当量、より好ましくは約4.0モル当量である。塩基は好ましくはピリジンである。
【0014】
上記プロセスの脱保護ステップ(b)は、塩基の存在下で行うことが好ましい。塩基は、好ましくは水酸化ナトリウムである。好ましい実施形態として、脱保護ステップ(b)は、約0〜10℃、さらに好ましくは0〜5℃の温度での加水分解反応により行われる。
【0015】
好ましい実施形態として、反応ステップ(a)および脱保護ステップ(b)が同じリアクター中で引き続いて行われる。言い換えると、本出願のプロセスは、1つのポットで行うことができる。
【0016】
上述のプロセスは、式(II)の化合物、または5−フルオロサイトシンもしくはその誘導体と5−デオキシフラノシドもしくはその誘導体の結合した如何なる化合物のシリル化ステップも含んでいない。
【0017】
上述のプロセスの精製ステップc)は、60℃未満の温度で行うことが好ましい。精製ステップで使われる溶媒は、水、ケトン、エステル(例えば、酢酸エチル)、アルコール、エーテル、およびこれらの組み合わせが可能である。例えば、溶媒は、水、n−ペンタノール、n−ペンタノールとn−ヘプタンの混合物、および酢酸エチルとn−ヘプタンの混合物でもよい。特に、精製ステップは、n−ペンタノール単独、またはn−ペンタノールと1つまたは複数の他の溶剤との混合物から式(I)の化合物を結晶化することを含む。
【0018】
本出願の別の態様は、下記の平均粒径分布を有するカペシタビンを提供する:
D90:250〜350ミクロン、D50:100〜120ミクロン、およびD10:25〜30ミクロン。
【0019】
さらに本出願の別の態様は、カペシタビンを製造するプロセスを提供する。このプロセスは、式(IV):
【0020】
【化5】
【0021】
の化合物を酵素で脱保護することを含み、式中、各R1およびR2は独立に、ヒドロキシル保護基、R3はアルキル、シクロアルキル、アラルキル、アリール、またはアルコキシ、好ましくは、C1〜C12アルキル、シクロアルキル、アラルキル、アリール、またはアルコキシ、より好ましくは、C1〜C6アルキルを表す。好ましくは、R1およびR2は両方同じヒドロキシル保護基、例えば、アセチルおよびベンゾイルを表す。
【0022】
好ましくは、酵素はリパーゼである。反応温度は、好ましくは20〜60℃である。反応pH領域は、好ましくは4〜9である。R3は好ましくはペンチル基である。
【0023】
酵素は2’および3’位置の保護基を高い特異性で脱保護することができる。さらに、酵素加水分解を温和な条件で行うことができ、酵素は繰り返し使用可能である。
【0024】
本出願の別の態様は、下記を含むカペシタビンを提供する:
【0025】
【化6】
【0026】
【化7】
【0027】
【化8】
【0028】
【化9】
【0029】
【化10】
【0030】
したがって、本出願は、高純度(>99.5%)で、望ましくないアルファ型不純物のより少ない式(I)の化合物、特に、カペシタビンの工業規模で手軽な改良最終精製プロセスを提供する。
【0031】
本発明を特徴付ける種々の新規な特徴が特に本開示に添付の請求項において示され、本出願の一部を構成する。本発明、その操作の利点、およびその使用により得られる特異的物質のより深い理解のために、本発明の好ましい実施形態を図示し、説明した記述事項を提示する。
【発明を実施するための形態】
【0032】
好ましい実施形態の詳しい説明
以下の好ましい実施形態は、さらなる説明のために提供されており、本発明を制限するものではない。
【0033】
本出願の一実施形態では、カペシタビン製造プロセスは次のスキームにより図示できる:
【0034】
【化11】
【0035】
反応の完了後、粗製カペシタビンは水系で精製される。カペシタビンの純度は≧99.4%(HPLC面積百分率(A%)で)、不純物F≦0.3%、不純物G≦0.2%、不純物H≦0.3%、M2≦0.1%、不純物M≦0.10%および個別の最大不純物≦0.1%である。他に明示的に記載されていなければ、本出願で議論する純度は全てHPLC面積百分率(A%)に基づいている。
【0036】
【化12】
【0037】
【化13】
【0038】
【化14】
【0039】
【化15】
【0040】
【化16】
【0041】
反応完了後、粗製カペシタビンは、酢酸エチル系の下で精製可能である。カペシタビンの純度は≧99.5%、不純物F≦0.3%、不純物G≦0.2%、不純物≦0.3%、M2≦0.1%、不純物M≦0.10%および個別の最大不純物≦0.1%である。
【0042】
別の実施形態では、本発明の発明者らは、酵素を使ってカペシタビンの保護基を選択的に脱保護する新規プロセスを開発した。穏やかな条件下において酵素加水分解を行うことができ、酵素は、繰り返し使用可能である。さらに、酵素加水分解反応は脱保護ステップ中に作られる副産物および他の不純物の生成を防止することができる。
【0043】
酵素加水分解反応は、式(IV’)の化合物を酵素で処理し炭水化物成分の2’および3’位置を選択的に脱アシル化してカペシタビンを作ることを含む。
【0044】
【化17】
【0045】
式中、各R1およびR2は独立にヒドロキシル保護基である。好ましくは、R1=R2=アセチルまたはベンゾイルである。
【0046】
具体的実施形態として、本出願のプロセスは下記のスキームにより図示することができる:
【0047】
【化18】
【0048】
下記の実施例はさらに説明するために提供されるものであり、本発明の制限を意図するものではない。
【0049】
実施例
実施例1:2’,3’−ジ−O−アセチル−5’−デオキシ−5−フルオロシチジン(I)の製造および精製プロセス
5−フルオロサイトシン(1.2kg、9.30mol)、トリフル酸(5.0g)、ヘキサメチルジシラザン(1.06kg、6.57mol)およびアセトニトリル(4.3kg)を容器に加える。この混合物を加熱還流して、還流下約2時間保持する。溶液を室温に冷却し、β−アセチルフラノシド(2.528kg、9.71mol)およびトリフル酸(0.832kg、5.54mol)を添加する。得られた混合物を45〜55℃で約20時間加熱攪拌する。反応完了後、溶液を20〜30℃に冷却し、飽和重炭酸ナトリウム溶液で処理する。塩化メチレンを使って相分離後、有機層を集め、その後イソプロパノール(7.76kg)を使って適切な容量に移し替える。得られたイソプロパノール溶液を溶解する加熱還流する。50〜70℃で2’,3’−ジ−O−アセチル−5’−デオキシ−5−フルオロシチジンの種子添加後溶液は濁った状態になる。スラリーを室温に冷却し、n−ヘプタンを添加してさらに0.5時間攪拌する。溶液を10℃未満に冷却する。得られた固形物を濾過し、冷イソプロパノールで洗浄後、真空下乾燥して2’,3’−ジ−O−アセチル−5’−デオキシ−5−フルオロシチジンを得る。純度は≧99.5%で関連アルファ型不純物は≦0.2%である。収率は80%。1HNMR(CDCl3、400MHz)δ7.85(s、1H)、7.84(b、NH)、7.09(b、NH)、5.87(m、1H)、5.50(m、1H)、5.17(m、1H)、4.15(m、1H)、2.07(s、6H)、1.43(d、J=6.4Hz、3H)。
【0050】
実施例2:2’,3’−ジ−O−アセチル−5−デオキシ−5−フルオロ−N4−(ペンチル−オキシカルボニル)シチジン(II)の製造および精製プロセス
2’,3’−ジ−O−アセチル−5’−デオキシ−5−フルオロシチジン(0.2kg、0.6mol)、塩化メチレン(1.59Kg)およびピリジン(190.0g、2.4mol)を20〜30℃で容器に加える。混合物を5℃未満に冷却し、続いてn−ペンチルクロロホルマート(137.2g、0.9mol)を10℃未満の温度で添加する。得られた溶液を10℃未満の温度で少なくとも0.5時間攪拌する。反応完了後、水(2Kg)を添加し相分離させる。有機層を集め水(2kg)で3回洗浄する。次に有機層を集め真空下60℃未満の温度でトルエン(0.4Kg)で入れ替える。溶媒入れ替え後、n−ヘプタン(0.3kg)を40〜50℃で濁りが出るまで添加する。at40〜50℃で約1時間攪拌後、n−ヘプタン(0.4kg)を添加し、スラリーを10℃未満の温度に冷却する。溶液を少なくとも1時間攪拌し続ける。得られた固形物を濾過し、トルエン/n−ヘプタン(1:9)で洗浄後、真空下乾燥したて2’,3’−ジ−O−アセチル−5−デオキシ−5−フルオロ−N4−(ペンチル−オキシカルボニル)シチジンを得る。純度は≧99.5%で、最大不純物は≦0.2%である。収率:95%。1HNMR(CDCl3、400MHz)δ8.05(d、J=6.4Hz、1H)、5.93(m、1H)、5.52(m、1H)、5.15(m、1H)、4.24(m、1H)、4.15(m、2H)、2.06(s、6H)、1.68(m、2H)、1.47(d、J=6.4Hz、3H)、1.38(m、4H)、0.91(m、3H)。
【0051】
実施例3:水系によるカペシタビンの製造および精製プロセス
2’,3’−ジ−O−アセチル−5−デオキシ−5−フルオロ−N4−(ペンチル−オキシカルボニル)シチジン(20g、45.1mmol)、塩化メチレン(160g)およびメタノール(20mL)を5℃未満の温度で容器に加える。その後、25%NaOH(16g、100mmol)を5℃未満の温度で添加する。得られた溶液を5℃未満の温度に維持し少なくとも0.5時間攪拌する。反応の完了後、クエン酸(60g)を添加し反応を停止させ、相分離させる。有機層を集め、水性状態を継続して塩化メチレン(40mL)で洗浄する。相分離後、塩化メチレン層を集め、先の有機層と合わせる。得られた有機層を水(100g)で洗浄し、有機層を集める。有機層を濃縮し、次に真空下60℃で水(100g)と入れ替える。溶媒入れ替え後、得られた溶液を40〜55℃に加熱し、カペシタビンの種子添加を行う。混合物を20〜55℃で約1時間保持し、−5〜5℃に冷却する。スラリーを−5〜5℃で約2時間攪拌する。得られた固形物を濾過し、冷水で洗浄後、真空乾燥してカペシタビンを得る。純度は≧99.4%で、不純物F≦0.3%、不純物G≦0.2%、不純物H≦0.3%、M2≦0.1%、不純物M≦0.10%であり、最大個別不純物は≦0.1%である。収率:47%。
【0052】
【表1】
【0053】
実施例4:酢酸エチル系によるカペシタビンの製造と精製プロセス
2’,3’−ジ−O−アセチル−5−デオキシ−5−フルオロ−N4−(ペンチル−オキシカルボニル)シチジン(20g、45.1mmol)、塩化メチレン(160g)およびメタノール(20mL)を5℃未満の温度で容器に加える。続いて、25%NaOH(16g、100mmol)を5℃未満の温度で添加する。得られた溶液を5℃未満の温度に維持し、少なくとも0.5時間攪拌する。反応完了後、クエン酸(60g)を加えて反応を停止させ、相分離させる。有機層を集め、水性状態を継続し、塩化メチレン(40mL)で洗浄する、相分離後、塩化メチレン層を集め、先の有機層と合わせる。得られた有機層を水(100g)で洗浄し、有機層を集める。有機層を濃縮し、次に真空下60℃未満の温度で酢酸エチル(60mL)で入れ替える。溶媒入れ替え後、n−ヘプタン(20mL)を添加し、得られた溶液を40〜55℃で加熱し、カペシタビンの種子を添加する。混合物を40〜55℃で約1時間保持し、−5〜5℃に冷却する。スラリーを−5〜5℃で約2時間攪拌する。得られた固形物を濾過し、n−ヘプタンで洗浄後、真空下乾燥してカペシタビンを得る。純度は≧99.5%、不純物F≦0.3%、不純物G≦0.2%、不純物H≦0.3%、M2≦0.1%、不純物M≦0.10%であり、最大個別不純物は≦0.1%である。収率:85%。
【0054】
実施例5:2’,3’−ジ−O−アセチル−5’−デオキシ−5−フルオロシチジンからカペシタビンをワンポット反応で製造および精製するプロセス
2’,3’−ジ−O−アセチル−5’−デオキシ−5−フルオロシチジン(31.5kg、95.6mol)、塩化メチレン(230kg)およびピリジン(30kg、379.3mol)を20〜30℃で容器に添加する。混合物を5℃未満の温度に冷却し、続けて、n−ペンチルクロロホルマート(22kg、146.1mol)を10℃未満の温度で添加する。得られた溶液を10℃未満の温度で少なくとも0.5時間攪拌する。反応完了後、水(500g)を加え、相分離させる。有機層を集め、水(500g)でおよそ3回洗浄する。次に有機層を集め、容器に移す。その後、メタノール(38.7g)を5℃未満の温度で加える。続けて25%NaOH(36g、0.22mol)を5℃未満の温度で加える。得られた溶液を5℃未満の温度に維持し、少なくとも0.5時間攪拌する。A反応完了後、クエン酸(135g)を添加し反応を停止させ、相分離させる。有機層を集め、水性状態を継続し塩化メチレン(112g)で洗浄する。相分離後、塩化メチレン層を集め、先の有機層と合わせる。得られた有機層を水(225g)で洗浄し、有機層を集める。有機層を濃縮し、次に真空下60℃未満の温度でn−ペンタノール(225mL)で入れ替える。溶媒入れ替え後、得られた溶液を40〜55℃で加熱し、カペシタビンの種子添加を行う。混合物を40〜55℃で約1時間保持し、−5〜5℃に冷却する。スラリーを−5〜5℃で約2時間攪拌する。得られた固形物を濾過し、n−ヘプタンで洗浄後、真空乾燥してカペシタビンを得る。純度は≧99.5%、不純物≦0.3%、不純物G≦0.2%、不純物H≦0.3%、M2≦0.1%、不純物M≦0.10%であり、最大個別不純物は≦0.1%である。収率:77%。
【0055】
【表2】
【0056】
実施例6:n−ペンタノールおよび混合溶媒系によるカペシタビンの製造と精製プロセス
2’,3’−ジ−O−アセチル−5’−デオキシ−5−フルオロシチジン(1.0kg、3.0mol)、塩化メチレン(7.0kg)およびピリジン(0.96kg、19.5mol)を20〜30℃で容器に添加する。混合物を5℃未満の温度に冷却し、続いてn−ペンチルクロロホルマート(0.69kg、4.6mol)を10℃未満の温度で加える。得られた溶液を10℃未満の温度で少なくとも0.5時間攪拌する。反応完了後、水を加え、相分離させる。有機層を集め、水でおよそ3回洗浄する。次に有機層を集め、容器に移す。次にメタノール(0.8kg)を5℃未満の温度で添加する。その後25%NaOH(0.8kg)を0〜10℃で加える。得られた溶液を0〜10℃で維持し、少なくとも0.5時間攪拌する。反応完了後、クエン酸(3kg)を加え手反応を停止させ、相分離させる。有機層を集め水性状態を継続して塩化メチレンで洗浄する。相分離後、塩化メチレン層を集め、先の有機層と合わせる。得られた有機層を水で洗浄し、有機層を集める。有機層を濃縮し、次いで真空下60℃未満の温度で、n−ペンタノール(3.3kg)で入れ替える。溶媒入れ替え後、n−ヘプタン(0.68kg)を添加し得られた溶液を40〜60℃で加熱し、カペシタビンの種子添加を行う。混合物を約1時間保持し、−5〜5℃に冷却する。スラリーを−5〜5℃で約2時間攪拌する。得られた固形物を濾過し、n−ヘプタンで洗浄後、真空乾燥してカペシタビン(0.9kg)を得る。収率:約80%。純度は≧99.5%で、不純物F≦0.3%、不純物G≦0.2%、不純物H≦0.3%、M2≦0.1%、不純物M≦0.10%であり、最大個別不純物は≦0.1%である。
【0057】
実施例7:結晶化母液からのカペシタビン単離
カペシタビンの結晶化母液(6L)を容器に加える。次に、残渣の最終容量が約1Lになるまで溶液を真空下60℃未満の温度で濃縮する。反応系を40〜50℃(目標は45℃)に冷却し、カペシタビンの種子添加を行う。混合物を40〜55℃で1時間保持し−5〜5℃冷却する。スラリーを−5〜5℃で約2時間攪拌する。得られた固形物を濾過し、n−ヘプタン(0.5kg)で洗浄後真空乾燥してカペシタビンを得る。純度は≧99.5%で、最大個別不純物は≦0.1%、水含量は≦0.05%である。収率:10%。
【0058】
実施例8:加水分解酵素触媒プロセスによるカペシタビンの合成
多質点系の適切なリアクターに化合物II(1.0g、1w/w)および19:1のn−BuOH−PPW(20.0mL、20v/w)含有共溶媒を室温で加える。この段階で溶液は0.5時間攪拌の間清澄である。別のリアクターでリパーゼ(2.0g、2w/w)およびセライト(2.0g、2w/w)またはシリカゲル(2.0g、2w/w)を含む混合試薬を調製する。その後、混合した固形物を前記溶液に数回に分けて加え、添加後45℃に加熱する。得られた溶液はスラリー混合物に見える。次に、50μL溶液を採取し1mLACNに入れて、IPCモニタリングを行い、固形物を濾過して濾液をHPLCにセットする。
【0059】
完了後、BuOH(10mL、10v/w)を溶液に添加し、スラリーをブフナー漏斗で濾過後、真空乾燥する。再使用するために固形物を集め、濾液を真空下濃縮して粗製APIを得る。
【0060】
本発明は上述の実施形態に限定されるのもではなく、これら実施形態は例としてのみ提示されている。しかしこれらは添付の特許請求項により定義される保護の範囲内で種々の改変が可能である。
【発明の詳細な説明】
【0001】
関連出願への相互参照
本出願は、2009年7月23日出願の米国特許仮出願第61/227,971号の優先権を主張する。この仮出願の全体は参照により本明細書に組み込まれる。
【0002】
1.発明の分野
本出願は、5’−デオキシ−5−フルオロ−N4−n−ペンチルオキシカルボニルシチジン(カペシタビン)およびその誘導体の製造プロセスに関する。
2.関連技術の説明
カペシタビンは、抗悪性腫瘍活性を有するフロロピリミジンカルバマートで、ブランド名XELODA(登録商標)の商品として商業的に入手可能であり、次の化学構造を有する:
【0003】
【化1】
【0004】
カペシタビンの合成については、米国特許第5,472,949号;4,966,891号;5,453,497号;7,365,188号;および5,476,932号を含むいくつかの公表文献に記載されている。
しかし、カペシタビンおよびその誘導体の改良された製造プロセスの必要性が依然として存在する。
【発明の概要】
【0005】
本出願の1つの態様は、式(I):
【0006】
【化2】
【0007】
の精製された化合物の製造プロセスを提供することである。
(式中、R3はアルキル、シクロアルキル、アラルキル、アリール、またはアルコキシ、好ましくはC1〜C12アルキル、シクロアルキル、アラルキル、アリール、またはアルコキシ、さらに、より好ましくは、C1〜C6アルキルである。)
このプロセスは下記を含む。
(a)式(II):
【0008】
【化3】
【0009】
(式中、各R1およびR2は独立にヒドロキシル保護基を示す)の化合物を、式(III):X−C(=O)−R3(式中、Xはアシル活性化基、R3は上記で定義済み)のアシル化試薬と有機溶媒、例えば、CH2Cl2、THF、アセトニトリル、トルエン、または酢酸エチル、中で反応させ、式(IV):
【0010】
【化4】
【0011】
(式中、各R1、R2、およびR3は上記で定義済み)のアシル化化合物を作り;
(b)式(IV)のアシル化化合物を脱保護して式(I)の化合物を得て;さらに
(c)式(I)の化合物を溶媒を使って精製する。
好ましくは、ヒドロキシル保護基は、アセチルまたはベンゾイルである。
【0012】
上記式(III)のアシル化試薬中のXは、好ましくはハロゲン化物、さらに好ましくは、塩化物である。式(III)のアシル化試薬は、好ましくはn−ペンチルクロロホルマートである。
式(I)の化合物は、好ましくはカペシタビン、すなわち、上記式(I)のR3がペンチル基である化合物である。
【0013】
上記プロセスの反応ステップ(a)は塩基の存在下で行うことが好ましい。塩基は、好ましくは式(II)の化合物の3.5〜5.0モル当量、より好ましくは約4.0モル当量である。塩基は好ましくはピリジンである。
【0014】
上記プロセスの脱保護ステップ(b)は、塩基の存在下で行うことが好ましい。塩基は、好ましくは水酸化ナトリウムである。好ましい実施形態として、脱保護ステップ(b)は、約0〜10℃、さらに好ましくは0〜5℃の温度での加水分解反応により行われる。
【0015】
好ましい実施形態として、反応ステップ(a)および脱保護ステップ(b)が同じリアクター中で引き続いて行われる。言い換えると、本出願のプロセスは、1つのポットで行うことができる。
【0016】
上述のプロセスは、式(II)の化合物、または5−フルオロサイトシンもしくはその誘導体と5−デオキシフラノシドもしくはその誘導体の結合した如何なる化合物のシリル化ステップも含んでいない。
【0017】
上述のプロセスの精製ステップc)は、60℃未満の温度で行うことが好ましい。精製ステップで使われる溶媒は、水、ケトン、エステル(例えば、酢酸エチル)、アルコール、エーテル、およびこれらの組み合わせが可能である。例えば、溶媒は、水、n−ペンタノール、n−ペンタノールとn−ヘプタンの混合物、および酢酸エチルとn−ヘプタンの混合物でもよい。特に、精製ステップは、n−ペンタノール単独、またはn−ペンタノールと1つまたは複数の他の溶剤との混合物から式(I)の化合物を結晶化することを含む。
【0018】
本出願の別の態様は、下記の平均粒径分布を有するカペシタビンを提供する:
D90:250〜350ミクロン、D50:100〜120ミクロン、およびD10:25〜30ミクロン。
【0019】
さらに本出願の別の態様は、カペシタビンを製造するプロセスを提供する。このプロセスは、式(IV):
【0020】
【化5】
【0021】
の化合物を酵素で脱保護することを含み、式中、各R1およびR2は独立に、ヒドロキシル保護基、R3はアルキル、シクロアルキル、アラルキル、アリール、またはアルコキシ、好ましくは、C1〜C12アルキル、シクロアルキル、アラルキル、アリール、またはアルコキシ、より好ましくは、C1〜C6アルキルを表す。好ましくは、R1およびR2は両方同じヒドロキシル保護基、例えば、アセチルおよびベンゾイルを表す。
【0022】
好ましくは、酵素はリパーゼである。反応温度は、好ましくは20〜60℃である。反応pH領域は、好ましくは4〜9である。R3は好ましくはペンチル基である。
【0023】
酵素は2’および3’位置の保護基を高い特異性で脱保護することができる。さらに、酵素加水分解を温和な条件で行うことができ、酵素は繰り返し使用可能である。
【0024】
本出願の別の態様は、下記を含むカペシタビンを提供する:
【0025】
【化6】
【0026】
【化7】
【0027】
【化8】
【0028】
【化9】
【0029】
【化10】
【0030】
したがって、本出願は、高純度(>99.5%)で、望ましくないアルファ型不純物のより少ない式(I)の化合物、特に、カペシタビンの工業規模で手軽な改良最終精製プロセスを提供する。
【0031】
本発明を特徴付ける種々の新規な特徴が特に本開示に添付の請求項において示され、本出願の一部を構成する。本発明、その操作の利点、およびその使用により得られる特異的物質のより深い理解のために、本発明の好ましい実施形態を図示し、説明した記述事項を提示する。
【発明を実施するための形態】
【0032】
好ましい実施形態の詳しい説明
以下の好ましい実施形態は、さらなる説明のために提供されており、本発明を制限するものではない。
【0033】
本出願の一実施形態では、カペシタビン製造プロセスは次のスキームにより図示できる:
【0034】
【化11】
【0035】
反応の完了後、粗製カペシタビンは水系で精製される。カペシタビンの純度は≧99.4%(HPLC面積百分率(A%)で)、不純物F≦0.3%、不純物G≦0.2%、不純物H≦0.3%、M2≦0.1%、不純物M≦0.10%および個別の最大不純物≦0.1%である。他に明示的に記載されていなければ、本出願で議論する純度は全てHPLC面積百分率(A%)に基づいている。
【0036】
【化12】
【0037】
【化13】
【0038】
【化14】
【0039】
【化15】
【0040】
【化16】
【0041】
反応完了後、粗製カペシタビンは、酢酸エチル系の下で精製可能である。カペシタビンの純度は≧99.5%、不純物F≦0.3%、不純物G≦0.2%、不純物≦0.3%、M2≦0.1%、不純物M≦0.10%および個別の最大不純物≦0.1%である。
【0042】
別の実施形態では、本発明の発明者らは、酵素を使ってカペシタビンの保護基を選択的に脱保護する新規プロセスを開発した。穏やかな条件下において酵素加水分解を行うことができ、酵素は、繰り返し使用可能である。さらに、酵素加水分解反応は脱保護ステップ中に作られる副産物および他の不純物の生成を防止することができる。
【0043】
酵素加水分解反応は、式(IV’)の化合物を酵素で処理し炭水化物成分の2’および3’位置を選択的に脱アシル化してカペシタビンを作ることを含む。
【0044】
【化17】
【0045】
式中、各R1およびR2は独立にヒドロキシル保護基である。好ましくは、R1=R2=アセチルまたはベンゾイルである。
【0046】
具体的実施形態として、本出願のプロセスは下記のスキームにより図示することができる:
【0047】
【化18】
【0048】
下記の実施例はさらに説明するために提供されるものであり、本発明の制限を意図するものではない。
【0049】
実施例
実施例1:2’,3’−ジ−O−アセチル−5’−デオキシ−5−フルオロシチジン(I)の製造および精製プロセス
5−フルオロサイトシン(1.2kg、9.30mol)、トリフル酸(5.0g)、ヘキサメチルジシラザン(1.06kg、6.57mol)およびアセトニトリル(4.3kg)を容器に加える。この混合物を加熱還流して、還流下約2時間保持する。溶液を室温に冷却し、β−アセチルフラノシド(2.528kg、9.71mol)およびトリフル酸(0.832kg、5.54mol)を添加する。得られた混合物を45〜55℃で約20時間加熱攪拌する。反応完了後、溶液を20〜30℃に冷却し、飽和重炭酸ナトリウム溶液で処理する。塩化メチレンを使って相分離後、有機層を集め、その後イソプロパノール(7.76kg)を使って適切な容量に移し替える。得られたイソプロパノール溶液を溶解する加熱還流する。50〜70℃で2’,3’−ジ−O−アセチル−5’−デオキシ−5−フルオロシチジンの種子添加後溶液は濁った状態になる。スラリーを室温に冷却し、n−ヘプタンを添加してさらに0.5時間攪拌する。溶液を10℃未満に冷却する。得られた固形物を濾過し、冷イソプロパノールで洗浄後、真空下乾燥して2’,3’−ジ−O−アセチル−5’−デオキシ−5−フルオロシチジンを得る。純度は≧99.5%で関連アルファ型不純物は≦0.2%である。収率は80%。1HNMR(CDCl3、400MHz)δ7.85(s、1H)、7.84(b、NH)、7.09(b、NH)、5.87(m、1H)、5.50(m、1H)、5.17(m、1H)、4.15(m、1H)、2.07(s、6H)、1.43(d、J=6.4Hz、3H)。
【0050】
実施例2:2’,3’−ジ−O−アセチル−5−デオキシ−5−フルオロ−N4−(ペンチル−オキシカルボニル)シチジン(II)の製造および精製プロセス
2’,3’−ジ−O−アセチル−5’−デオキシ−5−フルオロシチジン(0.2kg、0.6mol)、塩化メチレン(1.59Kg)およびピリジン(190.0g、2.4mol)を20〜30℃で容器に加える。混合物を5℃未満に冷却し、続いてn−ペンチルクロロホルマート(137.2g、0.9mol)を10℃未満の温度で添加する。得られた溶液を10℃未満の温度で少なくとも0.5時間攪拌する。反応完了後、水(2Kg)を添加し相分離させる。有機層を集め水(2kg)で3回洗浄する。次に有機層を集め真空下60℃未満の温度でトルエン(0.4Kg)で入れ替える。溶媒入れ替え後、n−ヘプタン(0.3kg)を40〜50℃で濁りが出るまで添加する。at40〜50℃で約1時間攪拌後、n−ヘプタン(0.4kg)を添加し、スラリーを10℃未満の温度に冷却する。溶液を少なくとも1時間攪拌し続ける。得られた固形物を濾過し、トルエン/n−ヘプタン(1:9)で洗浄後、真空下乾燥したて2’,3’−ジ−O−アセチル−5−デオキシ−5−フルオロ−N4−(ペンチル−オキシカルボニル)シチジンを得る。純度は≧99.5%で、最大不純物は≦0.2%である。収率:95%。1HNMR(CDCl3、400MHz)δ8.05(d、J=6.4Hz、1H)、5.93(m、1H)、5.52(m、1H)、5.15(m、1H)、4.24(m、1H)、4.15(m、2H)、2.06(s、6H)、1.68(m、2H)、1.47(d、J=6.4Hz、3H)、1.38(m、4H)、0.91(m、3H)。
【0051】
実施例3:水系によるカペシタビンの製造および精製プロセス
2’,3’−ジ−O−アセチル−5−デオキシ−5−フルオロ−N4−(ペンチル−オキシカルボニル)シチジン(20g、45.1mmol)、塩化メチレン(160g)およびメタノール(20mL)を5℃未満の温度で容器に加える。その後、25%NaOH(16g、100mmol)を5℃未満の温度で添加する。得られた溶液を5℃未満の温度に維持し少なくとも0.5時間攪拌する。反応の完了後、クエン酸(60g)を添加し反応を停止させ、相分離させる。有機層を集め、水性状態を継続して塩化メチレン(40mL)で洗浄する。相分離後、塩化メチレン層を集め、先の有機層と合わせる。得られた有機層を水(100g)で洗浄し、有機層を集める。有機層を濃縮し、次に真空下60℃で水(100g)と入れ替える。溶媒入れ替え後、得られた溶液を40〜55℃に加熱し、カペシタビンの種子添加を行う。混合物を20〜55℃で約1時間保持し、−5〜5℃に冷却する。スラリーを−5〜5℃で約2時間攪拌する。得られた固形物を濾過し、冷水で洗浄後、真空乾燥してカペシタビンを得る。純度は≧99.4%で、不純物F≦0.3%、不純物G≦0.2%、不純物H≦0.3%、M2≦0.1%、不純物M≦0.10%であり、最大個別不純物は≦0.1%である。収率:47%。
【0052】
【表1】
【0053】
実施例4:酢酸エチル系によるカペシタビンの製造と精製プロセス
2’,3’−ジ−O−アセチル−5−デオキシ−5−フルオロ−N4−(ペンチル−オキシカルボニル)シチジン(20g、45.1mmol)、塩化メチレン(160g)およびメタノール(20mL)を5℃未満の温度で容器に加える。続いて、25%NaOH(16g、100mmol)を5℃未満の温度で添加する。得られた溶液を5℃未満の温度に維持し、少なくとも0.5時間攪拌する。反応完了後、クエン酸(60g)を加えて反応を停止させ、相分離させる。有機層を集め、水性状態を継続し、塩化メチレン(40mL)で洗浄する、相分離後、塩化メチレン層を集め、先の有機層と合わせる。得られた有機層を水(100g)で洗浄し、有機層を集める。有機層を濃縮し、次に真空下60℃未満の温度で酢酸エチル(60mL)で入れ替える。溶媒入れ替え後、n−ヘプタン(20mL)を添加し、得られた溶液を40〜55℃で加熱し、カペシタビンの種子を添加する。混合物を40〜55℃で約1時間保持し、−5〜5℃に冷却する。スラリーを−5〜5℃で約2時間攪拌する。得られた固形物を濾過し、n−ヘプタンで洗浄後、真空下乾燥してカペシタビンを得る。純度は≧99.5%、不純物F≦0.3%、不純物G≦0.2%、不純物H≦0.3%、M2≦0.1%、不純物M≦0.10%であり、最大個別不純物は≦0.1%である。収率:85%。
【0054】
実施例5:2’,3’−ジ−O−アセチル−5’−デオキシ−5−フルオロシチジンからカペシタビンをワンポット反応で製造および精製するプロセス
2’,3’−ジ−O−アセチル−5’−デオキシ−5−フルオロシチジン(31.5kg、95.6mol)、塩化メチレン(230kg)およびピリジン(30kg、379.3mol)を20〜30℃で容器に添加する。混合物を5℃未満の温度に冷却し、続けて、n−ペンチルクロロホルマート(22kg、146.1mol)を10℃未満の温度で添加する。得られた溶液を10℃未満の温度で少なくとも0.5時間攪拌する。反応完了後、水(500g)を加え、相分離させる。有機層を集め、水(500g)でおよそ3回洗浄する。次に有機層を集め、容器に移す。その後、メタノール(38.7g)を5℃未満の温度で加える。続けて25%NaOH(36g、0.22mol)を5℃未満の温度で加える。得られた溶液を5℃未満の温度に維持し、少なくとも0.5時間攪拌する。A反応完了後、クエン酸(135g)を添加し反応を停止させ、相分離させる。有機層を集め、水性状態を継続し塩化メチレン(112g)で洗浄する。相分離後、塩化メチレン層を集め、先の有機層と合わせる。得られた有機層を水(225g)で洗浄し、有機層を集める。有機層を濃縮し、次に真空下60℃未満の温度でn−ペンタノール(225mL)で入れ替える。溶媒入れ替え後、得られた溶液を40〜55℃で加熱し、カペシタビンの種子添加を行う。混合物を40〜55℃で約1時間保持し、−5〜5℃に冷却する。スラリーを−5〜5℃で約2時間攪拌する。得られた固形物を濾過し、n−ヘプタンで洗浄後、真空乾燥してカペシタビンを得る。純度は≧99.5%、不純物≦0.3%、不純物G≦0.2%、不純物H≦0.3%、M2≦0.1%、不純物M≦0.10%であり、最大個別不純物は≦0.1%である。収率:77%。
【0055】
【表2】
【0056】
実施例6:n−ペンタノールおよび混合溶媒系によるカペシタビンの製造と精製プロセス
2’,3’−ジ−O−アセチル−5’−デオキシ−5−フルオロシチジン(1.0kg、3.0mol)、塩化メチレン(7.0kg)およびピリジン(0.96kg、19.5mol)を20〜30℃で容器に添加する。混合物を5℃未満の温度に冷却し、続いてn−ペンチルクロロホルマート(0.69kg、4.6mol)を10℃未満の温度で加える。得られた溶液を10℃未満の温度で少なくとも0.5時間攪拌する。反応完了後、水を加え、相分離させる。有機層を集め、水でおよそ3回洗浄する。次に有機層を集め、容器に移す。次にメタノール(0.8kg)を5℃未満の温度で添加する。その後25%NaOH(0.8kg)を0〜10℃で加える。得られた溶液を0〜10℃で維持し、少なくとも0.5時間攪拌する。反応完了後、クエン酸(3kg)を加え手反応を停止させ、相分離させる。有機層を集め水性状態を継続して塩化メチレンで洗浄する。相分離後、塩化メチレン層を集め、先の有機層と合わせる。得られた有機層を水で洗浄し、有機層を集める。有機層を濃縮し、次いで真空下60℃未満の温度で、n−ペンタノール(3.3kg)で入れ替える。溶媒入れ替え後、n−ヘプタン(0.68kg)を添加し得られた溶液を40〜60℃で加熱し、カペシタビンの種子添加を行う。混合物を約1時間保持し、−5〜5℃に冷却する。スラリーを−5〜5℃で約2時間攪拌する。得られた固形物を濾過し、n−ヘプタンで洗浄後、真空乾燥してカペシタビン(0.9kg)を得る。収率:約80%。純度は≧99.5%で、不純物F≦0.3%、不純物G≦0.2%、不純物H≦0.3%、M2≦0.1%、不純物M≦0.10%であり、最大個別不純物は≦0.1%である。
【0057】
実施例7:結晶化母液からのカペシタビン単離
カペシタビンの結晶化母液(6L)を容器に加える。次に、残渣の最終容量が約1Lになるまで溶液を真空下60℃未満の温度で濃縮する。反応系を40〜50℃(目標は45℃)に冷却し、カペシタビンの種子添加を行う。混合物を40〜55℃で1時間保持し−5〜5℃冷却する。スラリーを−5〜5℃で約2時間攪拌する。得られた固形物を濾過し、n−ヘプタン(0.5kg)で洗浄後真空乾燥してカペシタビンを得る。純度は≧99.5%で、最大個別不純物は≦0.1%、水含量は≦0.05%である。収率:10%。
【0058】
実施例8:加水分解酵素触媒プロセスによるカペシタビンの合成
多質点系の適切なリアクターに化合物II(1.0g、1w/w)および19:1のn−BuOH−PPW(20.0mL、20v/w)含有共溶媒を室温で加える。この段階で溶液は0.5時間攪拌の間清澄である。別のリアクターでリパーゼ(2.0g、2w/w)およびセライト(2.0g、2w/w)またはシリカゲル(2.0g、2w/w)を含む混合試薬を調製する。その後、混合した固形物を前記溶液に数回に分けて加え、添加後45℃に加熱する。得られた溶液はスラリー混合物に見える。次に、50μL溶液を採取し1mLACNに入れて、IPCモニタリングを行い、固形物を濾過して濾液をHPLCにセットする。
【0059】
完了後、BuOH(10mL、10v/w)を溶液に添加し、スラリーをブフナー漏斗で濾過後、真空乾燥する。再使用するために固形物を集め、濾液を真空下濃縮して粗製APIを得る。
【0060】
本発明は上述の実施形態に限定されるのもではなく、これら実施形態は例としてのみ提示されている。しかしこれらは添付の特許請求項により定義される保護の範囲内で種々の改変が可能である。
【特許請求の範囲】
【請求項1】
式(I):
【化1】
(式中、R3は、アルキル、シクロアルキル、アラルキル、アリール、またはアルコキシである)
の精製された化合物を製造するプロセスであって、
(a)式(II):
【化2】
(式中、各R1およびR2は独立にヒドロキシル保護基を表す)の化合物を、
式(III):X−C(=O)−R3(式中、Xはアシル活性化基およびR3は上記で定義済み)のアシル化試薬と有機溶媒中で反応させ、式(IV):
【化3】
(式中、各R1、R2、およびR3は上記で定義済み)のアシル化化合物を製造すること;
(b)式(IV)のアシル化化合物を脱保護して式(I)の化合物を得ること;さらに
(c)式(I)の化合物を溶媒で精製すること、
を含むプロセス。
【請求項2】
Xがハロゲン化物である請求項1記載のプロセス。
【請求項3】
R3がC1〜C6アルキルである請求項1記載のプロセス。
【請求項4】
R3がペンチル基である請求項1記載のプロセス。
【請求項5】
反応ステップ(a)が、式(II)の化合物の3.5〜5.0モル当量の塩基の存在下で行われる請求項1記載のプロセス。
【請求項6】
塩基が、式(II)の化合物の3.5〜4.5モル当量のピリジンである請求項5記載のプロセス。
【請求項7】
脱保護ステップ(b)が、約0〜10℃の温度での加水分解反応により行われる請求項1記載のプロセス。
【請求項8】
溶媒がn−ペンタノールである請求項1記載のプロセス。
【請求項9】
精製ステップ(c)が60℃未満の温度で行われる請求項1記載のプロセス。
【請求項10】
反応ステップ(a)および脱保護ステップ(b)が、同じリアクターで連続的に行われる請求項1記載のプロセス。
【請求項11】
D90が250〜350ミクロン、D50が100〜120ミクロンおよびD10が25〜30ミクロンの平均粒形を有するカペシタビン。
【請求項12】
カペシタビンの製造プロセスであって、式(IV):
【化4】
の化合物の脱保護を酵素で行うことを含むプロセス。
(式中、各R1およびR2は独立にヒドロキシル保護基、R3はアルキル、シクロアルキル、アラルキル、アリール、またはアルコキシを表す)
【請求項13】
酵素がリパーゼである請求項15記載のプロセス。
【請求項14】
R3がペンチル基である請求項15記載のプロセス。
【請求項15】
【化5】
【化6】
【化7】
【化8】
【化9】
を含むカペシタビン。
【請求項1】
式(I):
【化1】
(式中、R3は、アルキル、シクロアルキル、アラルキル、アリール、またはアルコキシである)
の精製された化合物を製造するプロセスであって、
(a)式(II):
【化2】
(式中、各R1およびR2は独立にヒドロキシル保護基を表す)の化合物を、
式(III):X−C(=O)−R3(式中、Xはアシル活性化基およびR3は上記で定義済み)のアシル化試薬と有機溶媒中で反応させ、式(IV):
【化3】
(式中、各R1、R2、およびR3は上記で定義済み)のアシル化化合物を製造すること;
(b)式(IV)のアシル化化合物を脱保護して式(I)の化合物を得ること;さらに
(c)式(I)の化合物を溶媒で精製すること、
を含むプロセス。
【請求項2】
Xがハロゲン化物である請求項1記載のプロセス。
【請求項3】
R3がC1〜C6アルキルである請求項1記載のプロセス。
【請求項4】
R3がペンチル基である請求項1記載のプロセス。
【請求項5】
反応ステップ(a)が、式(II)の化合物の3.5〜5.0モル当量の塩基の存在下で行われる請求項1記載のプロセス。
【請求項6】
塩基が、式(II)の化合物の3.5〜4.5モル当量のピリジンである請求項5記載のプロセス。
【請求項7】
脱保護ステップ(b)が、約0〜10℃の温度での加水分解反応により行われる請求項1記載のプロセス。
【請求項8】
溶媒がn−ペンタノールである請求項1記載のプロセス。
【請求項9】
精製ステップ(c)が60℃未満の温度で行われる請求項1記載のプロセス。
【請求項10】
反応ステップ(a)および脱保護ステップ(b)が、同じリアクターで連続的に行われる請求項1記載のプロセス。
【請求項11】
D90が250〜350ミクロン、D50が100〜120ミクロンおよびD10が25〜30ミクロンの平均粒形を有するカペシタビン。
【請求項12】
カペシタビンの製造プロセスであって、式(IV):
【化4】
の化合物の脱保護を酵素で行うことを含むプロセス。
(式中、各R1およびR2は独立にヒドロキシル保護基、R3はアルキル、シクロアルキル、アラルキル、アリール、またはアルコキシを表す)
【請求項13】
酵素がリパーゼである請求項15記載のプロセス。
【請求項14】
R3がペンチル基である請求項15記載のプロセス。
【請求項15】
【化5】
【化6】
【化7】
【化8】
【化9】
を含むカペシタビン。
【公表番号】特表2012−533618(P2012−533618A)
【公表日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願番号】特願2012−521599(P2012−521599)
【出願日】平成22年7月21日(2010.7.21)
【国際出願番号】PCT/SG2010/000276
【国際公開番号】WO2011/010967
【国際公開日】平成23年1月27日(2011.1.27)
【出願人】(503345569)サイノファーム タイワン リミテッド (18)
【Fターム(参考)】
【公表日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願日】平成22年7月21日(2010.7.21)
【国際出願番号】PCT/SG2010/000276
【国際公開番号】WO2011/010967
【国際公開日】平成23年1月27日(2011.1.27)
【出願人】(503345569)サイノファーム タイワン リミテッド (18)
【Fターム(参考)】
[ Back to top ]