
フルオロスルホニルイミドのアルカリ金属塩およびその製造方法
【課題】耐熱性に優れ、また、特定の不純物や水分含有量が低減されたフルオロスルホニルイミドのアルカリ金属塩、及び、反応溶液から溶媒を容易に除去することができるフルオロスルホニルイミドのアルカリ金属塩の製造方法を提供する。
【解決手段】本発明のフルオロスルホニルイミドのアルカリ金属塩は下記一般式(I)で表され、空気気流下、100℃で8時間保持したときの質量減少率が2%以下であり、本発明のフルオロスルホニルイミドのアルカリ金属塩の製造方法とは、フルオロスルホニルイミドのアルカリ金属塩を含む反応溶液中にガスをバブリングしながらフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程、及び/又は、薄膜蒸留によりフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程を含む。

【解決手段】本発明のフルオロスルホニルイミドのアルカリ金属塩は下記一般式(I)で表され、空気気流下、100℃で8時間保持したときの質量減少率が2%以下であり、本発明のフルオロスルホニルイミドのアルカリ金属塩の製造方法とは、フルオロスルホニルイミドのアルカリ金属塩を含む反応溶液中にガスをバブリングしながらフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程、及び/又は、薄膜蒸留によりフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程を含む。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、フルオロスルホニルイミドのアルカリ金属塩、詳しくは、N−(フルオロスルホニル)−N−(フルオロアルキルスルホニル)イミド、ビス(フルオロスルホニル)イミドのアルカリ金属塩とその製造方法に関する。
【背景技術】
【0002】
N−(フルオロスルホニル)−N−(フルオロアルキルスルホニル)イミドや、ビス(フルオロスルホニル)イミド等のフルオロスルホニルイミドの塩やその誘導体は、N(SO2F)基又はN(SO2F)2基を有する化合物の中間体として有用であり、また、電解質や、燃料電池の電解液への添加物や、選択的求電子フッ素化剤、光酸発生剤、熱酸発生剤、近赤外線吸収色素等として使用されるなど、様々な用途において有用な化合物である。
【0003】
従来、フルオロスルホニルイミド類は、フッ素化剤を使用して、クロロスルホニルイミドをハロゲン交換する方法や(非特許文献1,2)、尿素の存在下で、フルオロスルホン酸(HFSO3)を蒸留することによってビス(フルオロスルホニル)イミドとする方法(特許文献1)などにより調製されてきた。また、本発明者らは、所定の元素を含むフッ化物を使用してフルオロスルホニルイミドを製造する方法を提案している(特許文献2)。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特表平8−511274号公報
【特許文献2】国際公開第2009/123328号パンフレット
【非特許文献】
【0005】
【非特許文献1】John K. RuffおよびMax Lustig、Inorg.Synth. 11,138-140 (1968年)
【非特許文献2】Jean’ne M. Shreeveら、Inorg. Chem. 1998, 37 (24), 6295-6303
【発明の概要】
【発明が解決しようとする課題】
【0006】
上述のように、クロロスルホニルイミドのフッ素化については改良が重ねられ、その収率をある程度向上させることが可能になった。しかしながら、本発明者らは、フルオロスルホニルイミド塩の実操業レベルでの製造を検討する中で、フルオロスルホニルイミド塩の生成後、反応溶媒を除去する際、原料100質量%に対する溶媒の量が150質量%以下になると反応溶液から溶媒を除去し難いといった新たな問題があることを認識した。また、本発明者らは、フルオロスルホニルイミドが水との親和性が高く、生成物中の水分含有量を低減させ難いことも見出している。このような水分は、電気化学デバイスに用いた場合に、電解液の耐電圧性を低下させるのみならず、デバイスを構成する部材(電極等)を腐食させる原因にもなる。なお、溶媒や水分は加熱により生成物から除去することも考えられるが、フルオロスルホニルイミド塩の耐熱性はそれほど高いとはいえず、高温での加熱や長時間の加熱は、生成物の収率を低下させてしまい、結果として、製造コストが上昇してしまうといった問題もある。
【0007】
本発明は上記の様な事情に着目してなされたものであって、その目的は、スケールアップしても、反応溶液から溶媒を容易に除去することができるフルオロスルホニルイミドのアルカリ金属塩の製造方法と当該方法により得られるフルオロスルホニルイミドのアルカリ金属塩を提供することにある。また、本発明は、耐熱性の良好なフルオロスルホニルイミドのアルカリ金属塩、及び、特定の不純物や水分含有量が低減されたフルオロスルホニルイミドのアルカリ金属塩、及び、これを含む電解液を提供することも目的とする。
【課題を解決するための手段】
【0008】
本発明者らは、鋭意研究を重ねた結果、上記課題が下記方法により解決され、かつ、本発明の方法により得られたフルオロスルホニルイミドのアルカリ金属塩が耐熱性に優れること、また、特定の不純物や水分含有量が低減されたフルオロスルホニルイミドのアルカリ金属塩が耐熱性に優れることを見出し、本発明を完成した。
【0009】
すなわち、本発明のフルオロスルホニルイミドのアルカリ金属塩とは、下記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩であって、空気気流下、100℃で8時間保持したときの質量減少率が2%以下であるところに特徴を有する。
【0010】
【化1】
【0011】
(式(I)中、Maはアルカリ金属、Ra、Rbは、同一若しくは異なって、フッ素原子、または、1個以上の水素原子がフッ素原子で置換された炭素数1〜6の炭化水素基を表す。)
【0012】
このように、本発明のフルオロスルホニルイミドのアルカリ金属塩は、長時間加熱雰囲気にさらされても分解を生じ難く、耐熱性に優れるものである。
【0013】
上記フルオロスルホニルイミドのアルカリ金属塩は、空気気流下、25℃より10℃/分の昇温速度で加熱したときに、質量減少率が2%となる温度が210℃以上であるのが好ましい。
【0014】
また、本発明のフルオロスルホニルイミドのアルカリ金属塩は、硫酸イオン(SO42−)の含有量が3000ppm以下であるところに特徴を有する。本発明のフルオロスルホニルイミドのアルカリ金属塩に含まれるフッ化物イオン(F−)の含有量は1000ppm以下であるのが好ましく、また、水の含有量は500ppm以下であるのが好ましい。さらに、本発明のフルオロスルホニルイミドのアルカリ金属塩は、4000ppm以下の残留溶媒を含むものであるのが好ましい。
【0015】
本発明には、上記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩と媒体を含み、SO42−の含有量が1500ppm以下である電解液も含まれる。本発明の電解液は、水の含有量が250ppm以下であるのが好ましい。また、本発明の電解液は、2000ppm以下の残留溶媒を含むものであるのが好ましい。さらに、本発明の電解液は、F−の含有量が500ppm以下であるのが望ましい。
【0016】
上記目的を達成し得た本発明の製造方法とは、上記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩の製造方法であって、
(1)フルオロスルホニルイミドのアルカリ金属塩を含む反応溶液中にガスをバブリングしながらフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程、及び/又は、
(2)フルオロスルホニルイミドのアルカリ金属塩を含む反応溶液を薄膜蒸留により濃縮する工程
を含むところに特徴を有する。
【0017】
本発明者らは、上記問題点について検討を重ねたところ、反応溶媒の除去を困難にしている理由は、フルオロスルホニルイミドがアルカリ金属塩を形成したことによる沸点上昇や、フルオロスルホニルイミドのアルカリ金属塩が溶媒分子と溶媒和していることにあることが判明した。そこで、反応溶媒として、生成物と溶媒和を形成し難いものを使用することを検討した。しかしながら、クロロスルホニルイミドのフッ素化工程で使用できる溶媒は限られているため、反応溶媒を変更することは現実的ではない。また、フッ素化工程後、カチオン交換工程前のいずれかの工程で、反応溶媒を変更することも考えられるが、溶媒使用量の増加は、経済的にも環境的にも好ましくない。そこで、反応溶液から溶媒を効率よく除去する方法について更なる検討を重ねたところ、濃縮工程において、(1)バブリング法、及び/又は、(2)薄膜蒸留法を採用すれば、容易に反応溶媒を除去でき、効率よくフルオロスルホニルイミドのアルカリ金属塩を精製できることを見出した。すなわち、上記(1)の反応溶液にガスをバブリングする場合には、蒸発面積が増大させられるので、反応溶媒の蒸発を促進させることができる。したがって、反応溶液を高温に加熱しなくても、速やかに溶媒を除去できるのである。一方、(2)の薄膜蒸留法では、伝熱させるべき反応溶液を伝熱面に対して薄膜にすることにより、大きな伝熱係数を得ることができる。その結果、伝熱面を高温に加熱しなくても速やかに溶媒を除去できるのである。また、薄膜蒸留法では、伝熱面積の小さな装置でも効率よく溶媒を除去することができる。したがって、本発明法によれば、製造工程で反応溶液を長時間、高温に加熱する必要がないため、加熱によるフルオロスルホニルイミドのアルカリ金属塩の分解が抑制され、その結果、分解生成物に起因するフルオロスルホニルイミドのアルカリ金属塩の耐熱性の低下が抑制される。
【0018】
前記濃縮工程は150℃以下で行うのが好ましい。また、前記濃縮工程は40kPa以下で行うことが望ましい。前記反応溶液に含まれる反応溶媒は、エステル系溶媒及び/又はニトリル系溶媒であるのが好ましい。さらに、上述の各工程に加えて、濃縮工程で得られた濃縮液を100℃以下で加熱して乾燥する工程を含むことも本発明の推奨される実施態様である。
【0019】
上記製造方法は、さらに、反応溶媒の存在下で、クロロスルホニルイミドまたはその塩をフッ素化する工程を含むものであるのが望ましい。
【0020】
なお、本発明における「フルオロスルホニルイミド」との文言には、フルオロスルホニル基を2つ有するビス(フルオロスルホニル)イミドの他、フルオロスルホニル基と、フッ化アルキル基を有するN−(フルオロスルホニル)−N−(フルオロアルキルスルホニル)イミドが含まれる。出発原料である「クロロスルホニルイミド」も同様である。また、「フルオロアルキル」とは、炭素数1〜6のアルキル基において、1つ以上の水素原子がフッ素原子で置換されたものを意味し、例えば、フルオロメチル基、ジフルオロメチル基、トリフルオロメチル基、フルオロエチル基、ジフルオロエチル基、トリフルオロエチル基、ペンタフルオロエチル基等が含まれる。
【発明の効果】
【0021】
本発明によれば、反応溶液から溶媒を速やかに除去でき、効率よくフルオロスルホニルイミドのアルカリ金属塩を製造することができる。また、本発明のフルオロスルホニルイミドのアルカリ金属塩は耐熱性に優れるものである。
【図面の簡単な説明】
【0022】
【図1】図1は、実験例1−2〜1−5の結果を示すグラフである。
【発明を実施するための形態】
【0023】
<フルオロスルホニルイミドのアルカリ金属塩>
本発明のフルオロスルホニルイミドのアルカリ金属塩とは、下記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩であって、空気気流下、100℃で8時間保持したときの質量減少率が2%以下であるところに特徴を有する。
【0024】
【化2】
【0025】
(式(I)中、Maはアルカリ金属、Ra、Rbは、同一若しくは異なって、フッ素原子、または、1個以上の水素原子がフッ素原子で置換された炭素数1〜6の炭化水素基を表す。)
【0026】
このように、本発明のフルオロスルホニルイミドのアルカリ金属塩は、長時間、加熱雰囲気に曝されても分解し難いため、これをイオン伝導体材料として使用した各種電気化学デバイスは、長期に亘って安定した電気化学的特性を発揮することができる。また、各種電気化学デバイスは、広い温度領域で使用することができる。なお、優れた耐熱性は、後述するように、本発明のフルオロスルホニルイミドのアルカリ金属塩に含まれる不純物量が低減されていることに起因するものである。
【0027】
上記質量減少率が低いほど耐熱性は高く好ましいが、質量減少率は1%以下であるのが好ましい。
【0028】
また、本発明のフルオロスルホニルイミドのアルカリ金属塩は、空気気流下、25℃より10℃/分の昇温速度で加熱したときに、質量減少率が2%となる温度(以下、「2%質量減少温度」という場合がある)が210℃以上であるのが好ましい。ここで、2%質量減少温度とは、上記条件下で熱重量分析を行ったときに、初期の質量に対する質量減少率が2%となる温度を意味する。すなわち、2%質量減少温度が高いほど、高温に曝されてもフルオロスルホニルイミドのアルカリ金属塩が分解を生じ難く、安定であることを示す。2%質量減少温度は215℃以上であるのがより好ましい。
【0029】
さらに、本発明のフルオロスルホニルイミドのアルカリ金属塩は、上記2%質量減少温度と同様の条件で測定を行ったとき、質量減少率が1%となる温度(以下、「1%質量減少温度」という場合がある)が175℃以上であるのが好ましい。より好ましくは185℃以上である。
【0030】
上記質量減少率、2%、1%質量減少温度の測定に使用可能な装置としては、示差熱熱重量同時測定装置(例えばエスアイアイ・ナノテクノロジー株式会社製の「EXSTAR TG/DTA6200」)等が挙げられる。なお、測定試料が水分や溶媒を含んでいる場合には、正確な測定結果が得られ難い。したがって、本測定においては、含水率が1000ppm以下のフルオロスルホニルイミドのアルカリ金属塩を用いるのが好ましい。より好ましい含水率は500ppm以下であり、さらに好ましくは200ppm以下である。含水率が上記範囲であれば、測定結果への影響は無視できる程度となるからである。また、フルオロスルホニルイミドのアルカリ金属塩の製造直後に上記測定(質量減少率、2%、1%質量減少温度)を行わない場合は、測定試料を温度50℃、真空下で6時間保持し、試料に含まれる水分量や溶媒量を調整した後、上記測定に供することが推奨される。
【0031】
さらに、本発明のフルオロスルホニルイミドのアルカリ金属塩は、不純物の含有量が極低レベルに低減されている。上記不純物としては、例えば、後述する濃縮工程や乾燥工程での加熱により生じる分解生成物や精製段階で混入してくる不純物が挙げられ、例えば、フルオロスルホニルイミドのアルカリ金属塩の分解生成物に相当するフッ化物イオン(F-)や、硫酸イオン(SO42-)、また、フルオロスルホニルイミドの製造段階や精製段階で使用する溶媒(以下、残留溶媒と称する)及び水等が挙げられる。具体的に、本発明のフルオロスルホニルイミドのアルカリ金属塩(固体)に含まれる硫酸イオン(SO42-)の含有量は3000ppm以下であり、フッ化物イオン(F-)の含有量は1000ppm以下であるのが好ましい(いずれも質量基準。以下同様。)。
【0032】
上記フッ化物イオンおよび硫酸イオン等の不純物量の増加はフルオロスルホニルイミドの分解および純度の低下を意味する。したがって、不純物含有量が多い場合には、フルオロスルホニルイミドを各種用途に用いた場合に所期の特性が得られ難くなる。また、これらの不純物は、フルオロスルホニルイミドの分解を促進する逆効果を有する。したがって、不純物含有量が多い場合には初期の特性が維持し難いものとなる。さらに、フッ化物イオンは、フルオロスルホニルイミドのアルカリ金属塩が用いられる各種装置の周辺部材を腐食させる虞がある。したがって、これら不純物の含有量はできるだけ少ないのが好ましく、例えば、硫酸イオンの含有量はフルオロスルホニルイミドのアルカリ金属塩中1000ppm以下であるのが好ましく、より好ましくは500ppm以下であり、更に好ましくは300ppm以下である。一方、フッ化物イオンの含有量は、フルオロスルホニルイミドのアルカリ金属塩中800ppm以下であるのがより好ましい。より一層好ましくは500ppm以下であり、更に好ましくは300ppm以下である。なお、上記不純物は、本発明のフルオロスルホニルイミドのアルカリ金属塩中に含まれていないことが最も好ましいが、例えば、下限はいずれも1ppm程度であればよい。不純物の含有量が上記範囲であれば、本発明のフルオロスルホニルイミドのアルカリ金属塩を後述する各種電気化学デバイスに備えられるイオン伝導体材料として用いても、不純なイオン成分に由来する問題は生じ難い。
【0033】
また、本発明のフルオロスルホニルイミドのアルカリ金属塩(固体)の水の含有量は300ppm以下であるのが好ましい。上述のように、固体のフルオロスルホニルイミドのアルカリ金属塩に含まれる水分は、フルオロスルホニルイミドのみならず、これらが用いられるデバイスの構成部材を腐食させる原因となる。したがって、水の含有量は200ppm以下であるのがより好ましく、さらに好ましくは100ppm以下である。なお、水の含有量は少ないほど好ましく、実質的に0ppmであるのが好ましいが、0ppmにまで水分量を低減するのは技術的に難しく、また、経済的に好ましくない場合がある。よって水の含有量の下限は、1ppm程度であればよい。
【0034】
加えて、本発明のフルオロスルホニルイミドのアルカリ金属塩(固体)は、フルオロスルホニルイミドのアルカリ金属塩の製造段階で使用した残留溶媒の含有量が4000ppm以下であるのが好ましい。固体のフルオロスルホニルイミドのアルカリ金属塩に含まれる残留溶媒は耐熱性の低下、電気化学性能の低下の原因となる。したがって、斯かる残留溶媒の含有量は少ないほど好ましく、実質的に0ppmであるのが好ましいが、水の含有量の場合と同様の理由から、残留溶媒の量は3000ppm以下であるのがより好ましい。残留溶媒量の下限は1ppm程度であればよい。なお、上記残留溶媒には、後述するフルオロスルホニルイミドの製造段階で使用する溶媒が含まれる。
【0035】
さらに、本発明のフルオロスルホニルイミドのアルカリ金属塩は、上記フッ化物イオン、硫酸イオンに加えて、塩化物イオン(Cl-)、カリウムイオン(K+)などの他の不純物の含有量も少ないものであるのが好ましい。塩化物イオンの含有量は、200ppm以下であるのがより好ましく、より一層好ましくは100ppm以下である。一方、カリウムイオンの含有量は、10000ppm以下であるのが好ましく、より好ましくは8000ppm以下であり、より一層好ましくは4000ppm以下であり、更に好ましくは1000ppm以下、更に一層好ましくは500ppm以下であり、特に200ppm以下であるのがより好ましく、100ppm以下であるのが最も好ましい。なお、他の不純物の含有量は、合計で10000ppm以下であるのが好ましく、1000ppm以下であるのがより好ましく、500ppm以下であるのがさらに好ましい。なお、他の不純物の含有量の下限は合計で1ppm程度であればよい。
【0036】
上記不純物の種類や含有量は、後述するICP発光分光分析法、NMR測定あるいはイオンクロマトグラフィー等により分析することができる。水分含有量はカールフィッシャー水分計により測定でき、残留溶媒の含有量はガスクロマトグラフィーにより測定できる。
【0037】
<フルオロスルホニルイミドのアルカリ金属塩を含む組成物>
また、本発明には、上記フルオロスルホニルイミドのアルカリ金属塩を含む組成物も含まれる。本発明の組成物は、フルオロスルホニルイミドのアルカリ金属塩と、硫酸イオン、水、残留溶媒およびフッ素イオンの合計100質量%(以下、「本発明の組成物」という)に対する硫酸イオンの含有量が、3000ppm以下であるのが好ましい。より好ましくは1000ppm以下であり、より一層好ましくは500ppm以下であり、さらに好ましくは300ppm以下である。硫酸イオンは、本発明の組成物中に含まれていないことが最も好ましいが、例えば下限は1ppm程度であればよい。本発明の組成物を電気化学デバイス用の電解液に用いても、電解液の分解や電気化学デバイスの構成部材の腐食といった問題を生じ難いからである。
【0038】
また、本発明の組成物は、硫酸イオン以外の不純物含有量も低減されているのが好ましい。水、残留溶媒およびフッ素イオンも、硫酸イオン同様、本発明に係るフルオロスルホニルイミドのアルカリ金属塩を分解させたり、また、本発明の組成物を電気化学デバイス用の電解液として用いた場合には、当該デバイス等を劣化させる原因となる場合がある。よって、その含有量は以下の通りとするのが好ましい。すなわち、フッ素イオンの量は、本発明の組成物100質量%中1000ppm以下であるのが好ましく、より好ましくは800ppm以下であり、より一層好ましくは500ppm以下であり、さらに好ましくは300ppm以下である。水の含有量は、本発明の組成物100質量%中500ppm以下であるのが好ましく、より好ましくは300ppm以下であり、より一層好ましくは200ppm以下であり、さらに好ましくは100ppm以下である。残留溶媒の含有量は、本発明の組成物100質量%中4000ppm以下であるのが好ましく、より好ましくは3000ppm以下である。なお、本発明の組成物中における水、残留溶媒およびフッ素イオンの含有量は夫々0ppmであることが最も好ましいが、例えば、下限は夫々1ppm程度であればよい。水、残留溶媒およびフッ素イオンの含有量が上記範囲であれば、本発明の組成物を後述する各種電気化学デバイスに備えられる電解液に用いても、不純なイオン成分に由来する問題は生じ難い。
【0039】
本発明の組成物は、塩化物イオンやカリウムイオンを含んでいてもよい。ただし、塩化物イオンの含有量は、本発明の組成物と塩化物イオン及び/又はカリウムイオンの合計100質量%中200ppm以下であるのが好ましく、より好ましくは100ppm以下である。一方、カリウムイオンの含有量は、本発明の組成物と塩化物イオン及び/又はカリウムイオンの合計100質量%中10000ppm以下であるのが好ましく、より好ましくは8000ppm以下であり、より一層好ましくは4000ppm以下であり、更に好ましくは1000ppm以下、更に一層好ましくは500ppm以下であり、特に200ppm以下であるのがより好ましく、100ppm以下であるのが最も好ましい。また、これらのイオンの合計含有量は、本発明の組成物と塩化物イオン及び/又はカリウムイオンの合計100質量%中10000ppm以下であるのが好ましく、1000ppm以下であるのがより好ましく、500ppm以下であるのがさらに好ましい。なお、これらのイオンの含有量の下限は合計で1ppm程度であればよい。
【0040】
なお、本発明の組成物中における上記成分(硫酸イオン、フッ素イオン、および水および残留溶媒の含有量)の含有量は、ICP発光分光分析法、イオンクロマトグラフィー、カールフィッシャー水分計、ガスクロマトグラフィーにより測定できる。
なお、上記各種イオンの含有量は、上記測定方法により検出される値である。したがって、固体のフルオロスルホニルイミドのアルカリ金属塩中、または、本発明の組成物中では、上記イオンは対イオンとの塩として存在しているものと考えられる。
【0041】
本発明のフルオロスルホニルイミドのアルカリ金属塩は、上記一般式(I)で表されるものである。上記一般式(I)中、Maはアルカリ金属を表す。Maは、好ましくはLi、Na、Kであり、より好ましくはLiである。上記一般式(I)中、Ra、Rbは、同一若しくは異なって、フッ素原子、または、1個以上の水素原子がフッ素原子で置換された炭素数1〜6の炭化水素基を表す。上記炭化水素基としては、直鎖状の炭素数1〜6のフルオロアルキル基であるのが好ましく、例えば、フルオロメチル基、ジフルオロメチル基、トリフルオロメチル基、フルオロエチル基、ジフルオロエチル基、トリフルオロエチル基、ペンタフルオロエチル基等が挙げられる。これらの中でも、Ra、Rbとしては、フッ素原子、トリフルオロメチル基、ペンタフルオロエチル基が好ましい。
【0042】
本発明のビス(フルオロスルホニル)イミドや、N−(フルオロスルホニル)−N−(フルオロアルキルスルホニル)イミドのアルカリ金属塩は、各種電気化学デバイスのイオン伝導体材料として好適である。特に、カチオンがリチウムであるリチウムフルオロスルホニルイミドは、リチウム二次電池、キャパシタなどに用いられる電解質やイオン性液体、あるいは、フルオロスルホニル化合物の中間体などとして有用である。
【0043】
上記耐熱性を備えた本発明のフルオロスルホニルイミドの製造方法は特に限定されないが、不純物含有量を低減して耐熱性を向上させる観点からは、以下に説明する方法により製造することが推奨される。
【0044】
<フルオロスルホニルイミドのアルカリ金属塩の製造方法>
本発明のフルオロスルホニルイミドのアルカリ金属塩の製造方法とは、(1)フルオロスルホニルイミドのアルカリ金属塩を含む反応溶液中にガスをバブリングしながらフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程、及び/又は、(2)薄膜蒸留によりフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程、を含む点に特徴を有するものである。したがって、上記(1)及び/又は(2)の工程を含んでいればよく、その他の工程は特に限定されない。本発明においては、フルオロスルホニルイミドのアルカリ金属塩を合成する方法は特に限定されず、従来公知の方法は全て採用することが出来る。例えば、フルオロスルホニルイミドの合成方法としては、特許文献1に記載の、尿素の存在下で、フルオロスルホン酸(HFSO3)を蒸留することによって(フルオロスルホニル)イミドを得る方法、クロロスルホニルイミドからフッ素化剤を用いてフルオロスルホニルイミドを合成する方法等が挙げられる。また、フルオロスルホニルイミドのアルカリ金属塩を得る方法としては、上記方法により得られたフルオロスルホニルイミドのカチオンをアルカリ金属カチオンとカチオン交換する方法が挙げられる。以下では、上記方法の中でも、クロロスルホニルイミドからフッ素化剤を用いてフルオロスルホニルイミドを合成し(フッ素化工程)、次いで、カチオン交換反応(カチオン交換工程)により、フルオロスルホニルイミドのアルカリ金属塩を合成する方法について説明する。
【0045】
まず、フッ素化工程から説明する。
【0046】
[フッ素化工程]
フッ素化工程では、クロロスルホニルイミド又はその塩のフッ素化反応を行う。出発原料となるクロロスルホニルイミドは、市販のものを使用してもよく、また、公知の方法で合成したものを用いてもよい。
【0047】
クロロスルホニルイミドを合成する方法としては、例えば、塩化シアンに無水硫酸を反応させた後、生成物(クロロスルホニルイソシアネート)とクロロスルホン酸とを反応させる方法、アミド硫酸と塩化チオニルとを反応させた後、さらにクロロスルホン酸を反応させる方法(以上、ビス(クロロスルホニル)イミドの合成方法);クロロスルホニルイソシアネートとフッ化アルキルスルホン酸またはフルオロスルホン酸とを反応させる方法(N−(クロロスルホニル)−N−(フルオロアルキルスルホニル)イミド、または、N−(クロロスルホニル)−N−(フルオロスルホニル)イミドの合成方法);などが挙げられる。
【0048】
次いで、クロロスルホニルイミドのフッ素化反応を行う。なお、フッ素化反応のタイミングは特に限定されず、クロロスルホニルイミド(プロトン体)のフッ素化反応を行う態様;クロロスルホニルイミドのカチオン交換反応を行った後、クロロスルホニルイミド塩のフッ素化反応を行う態様;のいずれの態様であってもよい。
【0049】
上記クロロスルホニルイミド(プロトン体)またはその塩(以下、クロロスルホニルイミド類と言う)をフッ素化する方法としては、上記非特許文献1,2に記載のフッ素化剤(AsF3、SbF3)を使用して、クロロスルホニルイミドをハロゲン交換する方法、KFやCsF等の1価カチオンのイオン性フルオリドをフッ素化剤として用いて、ジ(クロロスルホニル)イミドをフッ素化する方法や、クロロスルホニルイミド類を、アルカリ金属のフッ化物や、第11族〜第15族、第4周期〜第6周期の元素よりなる群から選ばれる少なくとも1種の元素を含むフッ化物(好ましくはCuF2、ZnF2、SnF2、PbF2およびBiF3など)と反応させる方法が挙げられる。第11族〜第15族、第4周期〜第6周期の元素よりなる群から選ばれる少なくとも1種の元素を含むフッ化物(好ましくはCuF2、ZnF2、SnF2、PbF2およびBiF3等)を用いてジ(クロロスルホニルイミド)をフッ素化する方法が、反応収率の面では好ましい。また、KF、LiF、NaF等アルカリ金属のフッ化物をフッ素化剤としてジ(クロロスルホニルイミド)をフッ素化する方法では、フルオロスルホニルイミドのアルカリ金属塩を一段階で得ることができるため好ましい。
【0050】
フッ素化工程では、反応溶媒として、非プロトン性溶媒を用いるのが好ましい。具体的には、エチレンカーボネート、プロピレンカーボネート、ブチレンカーボネート、ジメチルカーボネート、エチルメチルカーボネート、ジエチルカーボネート等のカーボネート系溶媒;ジメトキシメタン、1,2−ジメトキシエタン、テトラヒドロフラン、2−メチルテトラヒドロフラン、1,3−ジオキサン、4−メチル−1,3−ジオキソラン等のエーテル系溶媒;ギ酸メチル、酢酸メチル、酢酸エチル、酢酸イソプロピル、酢酸ブチル、プロピオン酸メチル、γ−ブチロラクトン、γ−バレロラクトン等のエステル系溶媒;N,N−ジメチルホルムアミド等のアミド系溶媒;アセトニトリル、ブチロニトリル、イソブチロニトリル、バレロニトリル、ベンゾニトリル等のニトリル系溶媒;ニトロメタン、ニトロベンゼン等のニトロ系溶媒;スルホラン、3−メチルスルホラン、ジメチルスルホキシド等の硫黄系溶媒;N−メチルオキサゾリジノン;等が挙げられる。これらの溶媒は単独で用いてもよく、また2種以上を混合して用いてもよい。フッ素化反応を円滑に進行させる観点からは極性溶媒を使用することが推奨され、上記例示の溶媒の中でも、エステル系溶媒及び/又はニトリル系溶媒が好ましく、特に、ブチロニトリル、イソブチロニトリル、バレロニトリル、酢酸エチル、酢酸イソプロピル及び酢酸ブチルが好ましい。なお、精製時の作業性からは、沸点が低く、水と2層状態を形成し得る溶媒が好ましい。
【0051】
フッ素化反応の終了は、例えば、19F−NMRなどで確認することができる。すなわち、反応の進行によりフッ素に由来するケミカルシフトにピークが出現し、さらに、そのピークの相対強度(積分値)が増大する。したがって、19F−NMRにより反応の進行状態を追跡しながら、フッ素化反応の終了を確認すればよい。なお、反応時間が長すぎる場合には、副生物の生成が顕著となるので、目的物のピークの相対強度が最大となる時点(例えば、反応の開始から6時間〜12時間程度)でフッ素化反応を終了するのが好ましい。
【0052】
[カチオン交換工程]
次に、カチオン交換工程について説明する。クロロスルホニルイミド類またはフルオロスルホニルイミドまたはその塩(以下、フルオロスルホニルイミド類と言う場合がある)を、所望のカチオンを含む塩と反応させることで、カチオン交換することができる。カチオンとしては、Li,Na,K,Rb,Csなどのアルカリ金属、または、後述するオニウムカチオンが好ましい。アルカリ金属を含むフルオロスルホニルイミド塩は、高温で溶融させたり、あるいは、適当な有機溶媒に溶解させることで、各種電気化学デバイスのイオン伝導体材料として使用することができる。また、オニウムカチオンを含むフルオロスルホニルイミド塩は、常温で溶融した状態を安定に保つ常温溶融塩となり、長期間の使用に耐える電気化学デバイスのイオン伝導体材料や、有機合成における反応溶媒等として好適なものとなる。より好ましいフルオロスルホニルイミド塩としては、リチウムカチオンや、オニウムカチオンを含むフルオロスルホニルイミド塩が挙げられる。
【0053】
アルカリ金属を含む塩としては、LiOH、NaOH、KOH、RbOH、CsOH等の水酸化物;Li2CO3、Na2CO3、K2CO3、Rb2CO3、Cs2CO3等の炭酸塩、LiHCO3、NaHCO3、KHCO3、RbHCO3、CsHCO3等の炭酸水素塩;LiCl、NaCl、KCl、RbCl、CsCl等の塩化物;LiF、NaF、KF、RbF、CsF等のフッ化物;CH3OLi、EtOLi等のアルコキシド化合物;及び、EtLi、BuLiおよびt−BuLi(尚、Etはエチル基、Buはブチル基を示す)等のアルキルリチウム化合物;等のアルカリ金属塩が挙げられる。
【0054】
一方、オニウムカチオンとしては、一般式(II);L+−Rs(式中、Lは、C、Si、N、P、S又はOを表す。Rは、同一若しくは異なって、水素原子、フッ素原子、または、有機基であり、Rが有機基の場合、これらは互いに結合していてもよい。sは、2、3又は4であり、元素Lの価数によって決まる値である。尚、L−R間の結合は、単結合であっても良く、また二重結合であってもよい。)で表されるものが好適である。
【0055】
上記Rで示される「有機基」は、炭素原子を少なくとも1個有する基を意味する。上記「炭素原子を少なくとも1個有する基」は、炭素原子を少なくとも1個有していればよく、また、ハロゲン原子やヘテロ原子などの他の原子や、置換基などを有していてもよい。具体的な置換基としては、例えば、アミノ基、イミノ基、アミド基、エーテル結合を有する基、チオエーテル結合を有する基、エステル基、ヒドロキシル基、アルコキシ基、カルボキシル基、カルバモイル基、シアノ基、ジスルフィド基、ニトロ基、ニトロソ基、スルホニル基などが挙げられる。
【0056】
一般式(II)で表されるオニウムカチオンとしては、具体的には下記一般式;
【0057】
【化3】
【0058】
(式中、Rは、一般式(II)と同様)で表されるものが好適である。このようなオニウムカチオンは単独で用いてもよく、2種以上を併用してもよい。具体的なオニウムカチオンとしては、WO2009/123328号公報に記載される複素環オニウムカチオン、不飽和オニウムカチオン、飽和環オニウムカチオン、及び、鎖状オニウムカチオン等が挙げられる。
【0059】
なお、好ましいオニウムカチオンとしては、一般式(II);L+−RsにおいてLがN、Rが、水素、または、C1〜C8のアルキル基、sが4である鎖状オニウムカチオンや下記一般式で表される5種類のオニウムカチオンが挙げられる。
【0060】
【化4】
【0061】
上記一般式中、R1〜R12は、同一若しくは異なって、水素原子、フッ素原子、又は、有機基であり、有機基の場合、これらは互いに結合していてもよい。有機基としては、直鎖、分岐鎖又は環状の炭素数1〜18の飽和又は不飽和炭化水素基、炭化フッ素基等が好ましく、より好ましくは炭素数1〜8の飽和又は不飽和炭化水素基、炭化フッ素基である。これらの有機基は、水素原子、フッ素原子、窒素原子、酸素原子、硫黄原子や、アミノ基、イミノ基、アミド基、エーテル基、エステル基、ヒドロキシル基、カルボキシル基、カルバモイル基、シアノ基、スルホン基、スルフィド基等の官能基を含んでいてもよい。より好ましくは、R1〜R12は、水素原子、フッ素原子、シアノ基及びスルホン基等のいずれか1種以上を有するものである。なお、2以上の有機基が結合している場合は、当該結合は、有機基の主骨格間に形成されたものでも、また、有機基の主骨格と上述の官能基との間、あるいは、上記官能基間に形成されたものであっても良い。
【0062】
上記鎖状オニウムカチオンとしては、例えば、テトラメチルアンモニウム、テトラエチルアンモニウム、テトラプロピルアンモニウム、テトラブチルアンモニウム、テトラヘプチルアンモニウム、テトラヘキシルアンモニウム、テトラオクチルアンモニウム、トリエチルメチルアンモニウム、メトキシエチルジエチルメチルアンモニウム、トリメチルフェニルアンモニウム、ベンジルトリメチルアンモニウム、ベンジルトリエチルアンモニウム、ベンジルトリブチルアンモニウム、ジメチルジステアリルアンモニウム、ジアリルジメチルアンモニウム、(2−メトキシエトキシ)メチルトリメチルアンモニウム、ジエチルメチル(2−メトキシエチル)アンモニウム、テトラキス(ペンタフルオロエチル)アンモニウム等の第4級アンモニウム類、トリメチルアンモニウム、トリエチルアンモニウム、トリブチルアンモニウム、ジエチルメチルアンモニウム、ジメチルエチルアンモニウム、ジブチルメチルアンモニウム等の第3級アンモニウム類、ジメチルアンモニウム、ジエチルアンモニウム、ジブチルアンモニウム等の第2級アンモニウム類、メチルアンモニウム、エチルアンモニウム、ブチルアンモニウム、ヘキシルアンモニウム、オクチルアンモニウム等の第1級アンモニウム類、N−メトキシトリメチルアンモニウム、N−エトキシトリメチルアンモニウム、N−プロポキシトリメチルアンモニウム及びNH4等のアンモニウム化合物等が挙げられる。これら例示の鎖状オニウムカチオンの中でも、アンモニウム、トリメチルアンモニウム、トリエチルアンモニウム、トリブチルアンモニウム、トリエチルメチルアンモニウム、テトラエチルアンモニウムおよびジエチルメチル(2−メトキシエチル)アンモニウムが好ましい鎖状オニウムカチオンとして挙げられる。
【0063】
上記オニウムカチオンを含む塩としては、上記オニウムカチオンのハロゲン化物、水酸化物、炭酸化物および炭酸水素化物などが挙げられる。また、カチオン交換工程の反応系中でオニウムカチオンを含む塩が生成するような化合物を原料として用いてもよい。
【0064】
カチオン交換工程で使用可能な溶媒としては、上記フッ素化工程で例示したものが挙げられる。
【0065】
カチオン交換工程の実施時期は特に限定されるものではなく、状況に応じて任意の段階で実施することができる。例えば、フッ素化工程前に実施してもよく、また、フッ素化工程後に実施してもよいが、フッ素化工程の後に行うことが好ましい。
【0066】
またカチオン交換工程の実施回数も限定されず、1回、または2回以上繰り返し実施してもよい。例えば、1回のカチオン交換工程により、クロロスルホニルイミド類またはフルオロスルホニルイミド類のカチオンをアルカリ金属カチオンに交換してもよく、また、1回目のカチオン交換工程により、クロロスルホニルイミド類またはフルオロスルホニルイミド類のオニウム塩を得た後、2回目のカチオン交換工程でアルカリ金属塩を得てもよい。
【0067】
なお、フッ素化工程、カチオン交換工程のいずれにおいても、反応溶液に含まれるスルホニルイミド骨格を有する化合物(例えば、フルオロスルホニルイミド、フルオロスルホニルイミド塩など)の濃度は、1質量%〜70質量%とするのが好ましい。濃度が高すぎる場合には、反応が不均一になる虞があり、一方、低すぎる場合には、1バッチあたりの生産性が低く経済的でないからである。より好ましくは3質量%〜60質量%であり、さらに好ましくは5質量%〜50質量%である。
【0068】
また、上記フッ素化工程後には、アルカリ水溶液と接触させる工程を設けてもよい。このアルカリ接触工程を設けることにより、生成物中に含まれる不純物を除去することができる。なお「フッ素化工程後」とは、フッ素化工程直後のみに限られず、フッ素化工程に続けてカチオン交換工程を行った後も「フッ素化工程後」に含まれる。
【0069】
前記アルカリ水溶液としては、塩基性物質の水溶液を使用すればよく、塩基性物質としては、例えば、アンモニア;炭素原子数1〜8のアルキル基を有する第一級、第二級または第三級のアルキルアミン、炭素原子数1〜8のアルキレン基を有するアルキレンジアミンなどの脂肪族アミン;アルカノールアミン;脂環式アミン;芳香族アミン;これらのアミンのエチレンオキサイド付加物;ホルムアミジン;グアニジン;アミジン;複素環式アミン;アルカリ金属、または、アルカリ土類金属の水酸化物、炭酸塩、リン酸塩、ケイ酸塩、ホウ酸塩、ギ酸塩、酢酸塩、ステアリン酸塩、パルミチン酸塩、プロピオン酸塩、シュウ酸塩などが挙げられる。
【0070】
[濃縮工程]
濃縮工程は、カチオン交換工程後の反応溶液から溶媒を除去して、生成したフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程である。本発明では、(1)反応溶液中にガスをバブリングする方法(バブリング法)、及び/又は、(2)薄膜蒸留法により、濃縮工程を実施する。
【0071】
本発明において、濃縮工程とは、得られたフルオロスルホニルイミドのアルカリ金属塩溶液(反応溶液)から一部の溶媒を留去することに加えて、目的物であるフルオロスルホニルイミドのアルカリ金属塩が固体として得られるまで反応溶液から溶媒を留去することも含む。したがって、濃縮工程で得られる生成物は、フルオロスルホニルイミドのアルカリ金属塩の濃縮液、フルオロスルホニルイミドのアルカリ金属塩の固体(粉体)、又は、フルオロスルホニルイミドのアルカリ金属塩の一部が固体状態で存在する濃縮液(スラリー状の溶液)である。
【0072】
まず、(1)バブリング法を採用する濃縮工程について説明する。バブリング法では、反応溶液にガスを流通させることにより蒸発面積を増大させられるので、反応溶媒の蒸発が促進され、速やかに、反応溶液から反応溶媒を除去することができる。この場合、濃縮工程に使用できる反応装置は、反応溶液中にガスを導入する手段、反応溶媒を系外へ排出する手段を備えた装置であればよく、特に限定されない。例えば、槽型反応器、減圧可能な槽型反応器等が挙げられる。
【0073】
反応溶液に流通(バブリング)させるガスとしては、ヘリウム、窒素、アルゴン等の不活性ガス、乾燥空気、およびこれらの混合ガスなどが使用できる。製品品質及び安全性の観点からは、窒素ガスが好ましい。反応溶液へのガスの流通量は、反応溶液中のフルオロスルホニル化合物(フルオロスルホニルイミドのアルカリ金属塩)の濃度に応じて適宜決定すればよいが、例えば、フルオロスルホニルイミドのアルカリ金属塩溶液1gあたり0.001mL/分〜10000mL/分とするのが好ましい。より好ましくは0.005mL/分〜1000mL/分であり、更に好ましくは0.05mL/分〜100mL/分である。なお、反応溶媒の蒸発を促進する観点からは、反応溶液に供給したガスの気泡が、より小さな径となるようにするのが好ましい。なお、気泡を形成する手段は特に限定されず、例えば、供給ガスをガラスフィルター等のフィルターに通過させることで気泡を形成してもよいし、微細ガス発生装置等のガス発生装置を使用してもよい。
【0074】
また、フルオロスルホニルイミドのアルカリ金属塩の濃縮効率を一層高めるため、反応溶液を加熱しながら濃縮工程を行ってもよい。加熱温度は、使用する反応溶媒に応じて適宜設定すればよいが、フルオロスルホニルイミドのアルカリ金属塩の分解を抑制する観点からは、30℃以上、150℃以下とするのが好ましい。より好ましい温度は50℃以上であり、より好ましくは120℃以下である。温度が低すぎると反応溶媒の除去効率が得られず、一方、温度が高すぎるとフルオロスルホニルイミドのアルカリ金属塩が分解してしまう虞がある。
【0075】
バブリング法を採用する濃縮工程におけるその他の条件(例えば、反応溶液へのガスのバブリングに用いるノズル径等)は特に限定されない。上記以外の条件は、使用する装置のサイズや、反応溶液中のフルオロスルホニルイミドのアルカリ金属塩の濃度に応じて適宜決定すればよい。
【0076】
次に、(2)薄膜蒸留法を採用する濃縮工程について説明する。
【0077】
薄膜蒸留法とは、被処理液の薄膜を形成し、これを加熱して、被処理液に含まれる成分を蒸発分と非蒸発分とに分離する方法である。したがって、薄膜蒸留法を採用する濃縮工程では、薄膜蒸留により、カチオン交換工程後の反応溶液から溶媒を分離し、フルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する。
【0078】
この場合、濃縮工程は、薄膜蒸留器を使用して実施する。薄膜蒸留器としては、反応溶液の薄膜を形成する手段、形成された薄膜を加熱する手段、蒸発分(反応溶媒)を回収する手段、非蒸発分(フルオロスルホニルイミドのアルカリ金属塩)を回収する手段を備えた装置であればよい。また、薄膜蒸留器から抜き出された濃縮液を再び薄膜蒸留器に戻す循環手段を備えていてもよい。これにより、繰り返し濃縮を行うことができる。
【0079】
薄膜を形成する方法も特に限定されず、流下式、遠心式、攪拌式、回転式、ブレード式、上昇式等、従来公知の方法はいずれも採用することができる。具体的な薄膜蒸留器としては、例えば、「短行程蒸留装置」(UIC GmbH 社製)、「ワイプレン(登録商標)」、「エクセバ(登録商標)」(株式会社神鋼環境ソリューション製)、「コントロ」、「傾斜翼コントロ」、「セブコン(登録商標)」(以上、株式会社日立プラントテクノロジー製)、「ハイエバオレータ(登録商標)」(株式会社櫻製作所製)、「薄膜蒸留器」、「ビスコン」、「フィルムトルーダー」(以上、木村化工機株式会社製)、「Hi−Uブラッシャー」、「エバリアクター」、「リカバリー」(以上、関西化学機械製作株式会社製)、「NRH」(日南機械株式会社製)、「エバポール(登録商標)」(株式会社大河原製作所製)等が挙げられる。
【0080】
薄膜蒸留の際の温度は、使用する反応溶媒に応じて適宜設定すればよいが、フルオロスルホニルイミドのアルカリ金属塩の分解を抑制する観点からは、30℃以上、150℃以下とするのが好ましい。より好ましい温度は40℃以上であり、更に好ましくは50℃以上であり、より好ましくは120℃以下であり、更に好ましくは110℃以下である。薄膜温度が低すぎると反応溶媒の除去効率が得られず、一方、温度が高すぎるとフルオロスルホニルイミドのアルカリ金属塩が分解してしまう虞がある。また、薄膜蒸留法を採用する場合も、フルオロスルホニルイミドのアルカリ金属塩に与えられる熱量は上記範囲とするのが好ましい。
【0081】
また、フルオロスルホニルイミドのアルカリ金属塩の濃縮効率を一層高めるため、薄膜蒸留器内にガスを流通させながら濃縮工程を実施してもよい。ガスとしては、窒素、アルゴンなどの不活性ガスを用いるのが好ましく、より好ましくは窒素である。
【0082】
薄膜蒸留法による濃縮工程におけるその他の条件は特に限定されない。例えば、薄膜蒸留器内への反応溶液の供給速度は、使用する装置のサイズや、反応溶液中のフルオロスルホニルイミドのアルカリ金属塩の濃度に応じて適宜決定すればよい。
【0083】
バブリング法、薄膜蒸留法のいずれを採用する場合も、効率のよい濃縮工程を実施する観点からは、減圧下で濃縮工程を行ってもよい。減圧度をコントロールすることによって、低温であっても効率よく反応溶媒を除去でき、また、熱によるフルオロスルホニルイミドのアルカリ金属塩の分解も防ぐことができる。減圧度は反応溶媒の種類に応じて適宜調整すればよく特に限定はされないが、例えば、40kPa以下とするのが好ましい。より好ましくは15kPa以下であり、更に好ましくは5kPa以下である。
【0084】
なお、反応溶媒の量が多い場合は、濃縮工程の前に、一部の反応溶媒を除去しておいてもよい。フルオロスルホニルイミドのアルカリ金属塩と溶媒との相互作用が顕著になって反応溶液から反応溶媒の除去が困難になるのは、フルオロスルホニルイミドのアルカリ金属塩に対する反応溶媒の量が150質量%以下となる時点からであるので、可能な限り反応溶液量を低減しておくことで、効率よく濃縮工程が実施できるからである。また、濃縮工程は、反応溶液を攪拌しながら実施してもよい。濃縮工程では、上記バブリング法、薄膜蒸留法の両方を順に実施してもよく(実施の順序は特に限定されない)、また、いずれか一方を単独で実施してもよい。なお、フルオロスルホニルイミドのアルカリ金属塩の熱分解を防ぐ観点からは、より短時間で濃縮工程を実施できる薄膜蒸留法が好ましい。
【0085】
濃縮工程における加熱によるフルオロスルホニルイミドのアルカリ金属塩の分解を防ぐ観点からは、上記バブリング法、薄膜蒸留法のいずれを採用する場合も、濃縮工程で加えられる熱量は、フルオロスルホニルイミドのアルカリ金属塩1gあたり1,000,000J以下とするのが好ましく、より好ましくは500,000J以下であり、さらに好ましくは100,000J以下である。なお、上記熱量には、濃縮工程に供する前に一部の反応溶媒を除去するために反応溶液に与えられる熱量は含まれない。
【0086】
本発明では、濃縮工程で反応溶液に与えられた熱量を、フルオロスルホニルイミドのアルカリ金属塩に与ええられた熱量とする。なお、上記熱量は、濃縮工程で使用する装置の消費電力(装置メーカーの公証値を参照すればよい)、反応溶液に含まれるフルオロスルホニルイミドのアルカリ金属塩の量および加熱時間に基づいて求めればよく、具体的には、濃縮工程で反応溶液に与えられた熱量を算出し、これを、フルオロスルホニルイミドのアルカリ金属塩1gあたりに与えられた熱量に換算すればよい。
【0087】
[乾燥、粉体化工程]
濃縮工程で得られたフルオロスルホニルイミドのアルカリ金属塩濃縮液は、そのまま製品とすることもできるが、保存時の安定性を高め、また、製品の流通を容易にするため、フルオロスルホニルイミドのアルカリ金属塩を粉体化してもよい(粉体化、乾燥工程)。なお、濃縮工程で、固体状態のフルオロスルホニルイミドのアルカリ金属塩を得た場合には、得られた固体をそのまま乾燥装置で乾燥してもよく、また、フルオロスルホニルイミドのアルカリ金属塩が可溶な溶媒に溶解させた後、乾燥、粉体化工程に供してもよい。
【0088】
フルオロスルホニルイミドのアルカリ金属塩を乾燥し、粉体化する方法は特に限定されず、(1)フルオロスルホニルイミドのアルカリ金属塩が析出するまで上記濃縮工程を継続し、これを分離し、乾燥して粉体化する方法、(2)濃縮工程で得られた濃縮液をそのまま、あるいは、必要により30℃以下に冷却しながら静置して、フルオロスルホニルイミドのアルカリ金属塩を析出させ、これを分離し、乾燥して粉体化する方法、(3)濃縮液に溶媒を添加してフルオロスルホニルイミドのアルカリ金属塩を析出させ、これをろ別して分離し、乾燥して粉体化する方法、等が挙げられる。
【0089】
上記(3)で使用可能な溶媒は、上述した反応溶媒以外の溶媒であって、フルオロスルホニルイミドのアルカリ金属塩と溶媒和を形成し難いものであればよい。具体的に(3)の方法で使用可能な溶媒としては、トルエン、o−キシレン、m−キシレン、p−キシレン、エチルベンゼン、イソプロピルベンゼン、1,2,4−トリメチルベンゼン、1,3,5−トリメチルベンゼン、1,2,3−トリメチルベンゼン、クロロベンゼン、ジクロロベンゼン等の芳香族系炭化水素溶媒、ヘキサン、ヘプタン、オクタン、ノナン、デカン、ウンデカン、ドデカン、デカリン、ジクロロメタン等の脂肪族炭化水素溶媒が挙げられる。また、これらの溶媒は、濃縮液1質量部に対して、20質量倍以下、より好ましくは10質量倍以下の量で加えるのが好ましい。
【0090】
次いで、傾斜法、遠心分離法、濾過法等により、析出したフルオロスルホニルイミドのアルカリ金属塩を反応溶媒などから分離し、乾燥させる。フルオロスルホニルイミドのアルカリ金属塩の乾燥方法は特に限定されず、従来公知の乾燥装置が使用できる。乾燥時の温度は0℃〜100℃とするのが好ましい。より好ましくは10℃以上、さらに好ましくは20℃以上であり、より好ましくは80℃以下、更に好ましくは60℃以下である。
【0091】
また、フルオロスルホニルイミドのアルカリ金属塩の乾燥は、乾燥装置にガスを供給しながら行ってもよい。使用可能なガスとしては、濃縮工程で使用したものが挙げられるが、例えば、窒素、アルゴンなどの不活性ガスや乾燥空気が挙げられる。
【0092】
[回収工程]
本発明の製造方法では、上記各工程で生成物から分離されたフルオロスルホニルイミドのアルカリ金属塩やスルホニルイミド骨格を有する化合物を回収する工程を設けてもよい。特に、薄膜蒸留法を採用する濃縮工程で排出される廃液や、上記粉体化、乾燥工程で析出したフルオロスルホニルイミドのアルカリ金属塩を除去した溶液(母液)には、溶解したフルオロスルホニルイミドのアルカリ金属塩が含まれているので、これを回収することでフルオロスルホニルイミドのアルカリ金属塩の収率を向上させることができる。
【0093】
また、上記乾燥、粉体化工程で得られたフルオロスルホニルイミドのアルカリ金属塩の純度が低い場合には、これを単独で精製してもよいが、固体状態(粉体)のフルオロスルホニルイミドのアルカリ金属塩を回収溶液(上記廃液や母液)と混合してもよい。上記乾燥、粉体化工程における操作は、晶析、再沈殿法などの精製操作にも相当するので、廃液や母液からのフルオロスルホニルイミドのアルカリ金属塩の回収と共に、フルオロスルホニルイミドのアルカリ金属塩の純度を向上させられるからである。
【0094】
なお、回収したフルオロスルホニルイミドのアルカリ金属塩の精製方法は特に限定されず、各工程から回収した溶液を単独で、あるいは、混合して精製してフルオロスルホニルイミドのアルカリ金属塩を回収してもよく、また、回収溶液は、カチオン交換工程、濃縮工程、粉体化、乾燥工程のいずれかの工程に供給してもよい。なお、生産性の観点からは、回収溶液は濃縮工程に供給するのが好ましい。
【0095】
上記方法により得られたフルオロスルホニルイミドのアルカリ金属塩は、必要に応じて純度をさらに向上させるための精製工程に供してもよい。精製工程としては、従来公知の精製方法はいずれも採用可能である。
【0096】
<電解液>
本発明には、上記フルオロスルホニルイミドのアルカリ金属塩と媒体とを含む電解液も含まれる。本発明のフルオロスルホニルイミドのアルカリ金属塩は、上述のように、不純なイオンや水分の含有量が低減されているので、電解液の分解が生じ難いものである。したがって、本発明の電解液をイオン伝導体として備えた電気化学デバイスは長期に亘って安定した特性を有する電気化学デバイスとすることができる。
【0097】
上記媒体としては、非プロトン性溶媒、ポリマー等が挙げられる。非プロトン性有機溶媒としては、誘電率が大きく、電解質塩(フルオロスルホニルイミドのアルカリ金属塩、後述する他の電解質)の溶解性が高く、沸点が60℃以上であり、且つ、電気化学的安定範囲が広い溶媒が好適である。より好ましくは、含有水分量が低い有機溶媒(非水系溶媒)である。具体的に、含水量は250ppm以下であるのが好ましく、100ppm以下であるのがより好ましく、50ppm以下であるのがさらに好ましい。含有水分量が低い有機溶媒としては、市販の脱水溶媒等を使用することができる。このような有機溶媒としては、エチレングリコールジメチルエーテル(1,2−ジメトキシエタン)、エチレングリコールジエチルエーテル、テトラヒドロフラン、2−メチルテトラヒドロフラン、2,6−ジメチルテトラヒドロフラン、テトラヒドロピラン、クラウンエーテル、トリエチレングリコールジメチルエーテル、テトラエチレングリコールジメチルエ−テル、1,4−ジオキサン、1,3−ジオキソラン等のエーテル類;炭酸ジメチル、炭酸エチルメチル(メチルエチルカーボネート)、炭酸ジエチル(ジエチルカーボネート)、炭酸ジフェニル、炭酸メチルフェニル等の鎖状炭酸エステル類;炭酸エチレン(エチレンカーボネート)、炭酸プロピレン(プロピレンカーボネート)、2,3−ジメチル炭酸エチレン、炭酸ブチレン、炭酸ビニレン、2−ビニル炭酸エチレン等の環状炭酸エステル類;蟻酸メチル、酢酸メチル、プロピオン酸、プロピオン酸メチル、酢酸エチル、酢酸プロピル、酢酸ブチル、酢酸アミル等の脂肪族カルボン酸エステル類;安息香酸メチル、安息香酸エチル等の芳香族カルボン酸エステル類;γ−ブチロラクトン、γ−バレロラクトン、δ−バレロラクトン等のカルボン酸エステル類;リン酸トリメチル、リン酸エチルジメチル、リン酸ジエチルメチル、リン酸トリエチル等のリン酸エステル類;アセトニトリル、プロピオニトリル、メトキシプロピオニトリル、グルタロニトリル、アジポニトリル、2−メチルグルタロニトリル、バレロニトリル、ブチロニトリル、イソブチロニトリル等のニトリル類;N−メチルホルムアミド、N−エチルホルムアミド、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジノン、N−メチルピロリドン、N−ビニルピロリドン等のアミド類;ジメチルスルホン、エチルメチルスルホン、ジエチルスルホン、スルホラン、3−メチルスルホラン、2,4−ジメチルスルホラン等の硫黄化合物類:エチレングリコール、プロピレングリコール、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル等のアルコール類;ジメチルスルホキシド、メチルエチルスルホキシド、ジエチルスルホキシド等のスルホキシド類;ベンゾニトリル、トルニトリル等の芳香族ニトリル類;ニトロメタン、1,3−ジメチル−2−イミダゾリジノン、1,3−ジメチル−3,4,5,6−テトラヒドロ−2(1H)−ピリミジノン、3−メチル−2−オキサゾリジノン等を挙げることができ、これらの1種又は2種以上が好適である。これらの中でも、炭酸エステル類、脂肪族カルボン酸エステル類、カルボン酸エステル類、エーテル類がより好ましく、炭酸エステル類がさらに好ましい。
【0098】
媒体として用いられるポリマーには、ポリエチレンオキシド(PEO)、ポリプロピレンオキシドなどのポリエーテル系ポリマー、ポリメチルメタクリレート(PMMA)などのメタクリル系ポリマー、ポリアクリロニトリル(PAN)等のニトリル系ポリマー、ポリフッ化ビニリデン(PVDF)、ポリフッ化ビニリデン−ヘキサフルオロプロピレンなどのフッ素系ポリマー、および、これらの共重合体等が含まれる。また、これらのポリマーと他の有機溶媒とを混合したポリマーゲルも本発明に係る媒体として用いることができる。他の有機溶媒としては上述の非プロトン性溶媒が挙げられる。
【0099】
上記ポリマーゲルを媒体とする場合の電解液の製造方法としては、従来公知の方法で成膜したポリマーに、上述の非プロトン性溶媒にフルオロスルホニルイミドのアルカリ金属塩等の電解質を溶解させた溶液を滴下して、電解質並びに非プロトン性溶媒を含浸、担持させる方法;ポリマーの融点以上の温度でポリマーと電解質とを溶融、混合した後、成膜し、ここに非プロトン性溶媒を含浸させる方法;予め非プロトン性溶媒に溶解させた電解質溶液とポリマーとを混合した後、これをキャスト法やコーティング法により成膜し、非プロトン性溶媒を揮発させる方法(以上、ゲル電解質);ポリマーの融点以上の温度でポリマーと電解質とを溶融し、混合して成形する方法(真性ポリマー電解質);等が挙げられる。
【0100】
媒体の使用量は、電解質(フルオロスルホニルイミドのアルカリ金属塩と他の電解質)と媒体の合計量100質量部に対して、50質量部〜99.9質量部であるのが好ましい。より好ましくは60質量部〜99.5質量部であり、さらに好ましくは70質量部〜99質量部である。媒体量が少なすぎると、充分なイオン伝導度が得られ難い場合があり、一方、多すぎると、溶媒の揮発によるイオン伝導性材料中のイオン濃度が変化し易くなり、安定したイオン伝導度が得られ難い場合がある。
【0101】
本発明に係る電解液には、上記フルオロスルホニルイミドのアルカリ金属塩のみが電解質として含まれていてもよいが、フルオロスルホニルイミドのアルカリ金属塩に加え、これ以外の他の電解質が含まれていてもよい。他の電解質を用いることで、電解液中のイオンの絶対量を増加させることができ、電気伝導度の向上を図ることができる。
【0102】
他の電解質としては、電解液中での解離定数が大きく、また、上記非プロトン性溶媒と溶媒和し難いアニオンを有するものが好ましい。他の電解質を構成するカチオン種としては、例えば、Li+、Na+、K+等のアルカリ金属イオン、Ca2+、Mg2+等のアルカリ土類金属イオンおよびオニウムカチオンが挙げられ、特に、リチウムイオンが好ましい。一方、アニオン種としては、PF6-、BF4-、ClO4-、AlCl4-、C[(CN)3]-、N[(CN)2]-、B[(CN)4]-、N[(SO2CF3)2]-、CF3(SO3)-、C[(CF3SO2)3]-、AsF6-、SbF6-およびジシアノトリアゾレートイオン(DCTA)などが挙げられる。これらの中でも、PF6-、BF4-がより好ましく、PF6-が特に好ましい。
【0103】
上記他の電解質の存在量としては、フルオロスルホニルイミドのアルカリ金属塩と他の電解質との合計100質量%中、0.1質量%、99質量%以下であることが好適である。他の電解質量が少なすぎる場合には、他の電解質を用いた効果(たとえばイオンの絶対量が充分なものとならず、電気伝導度が小さくなる)が得られ難い場合があり、他の電解質量が多すぎる場合には、イオンの移動が大きく阻害される虞がある。より好ましくは1質量%以上、さらに好ましくは5質量%以上であり、より好ましくは95質量%以下、さらに好ましくは90質量%以下である。
【0104】
なお、本発明に係る電解液中における電解質濃度(フルオロスルホニルイミドのアルカリ金属塩と他の電解質の総量)は、0.1質量%以上が好ましく、また、飽和濃度以下が好ましい。0.1質量%未満であると、イオン伝導度が低くなるため好ましくない。より好ましくは、0.5質量%以上であり、さらに好ましくは1質量%以上である。また、電解液中における電解質濃度は、50質量%未満であるのがより好ましく、さらに好ましくは40質量%以下、更に一層好ましくは30質量%以下である。
【0105】
本発明の電解液は、硫酸イオンの含有量が1500ppm以下であるのが好ましい。上述のように硫酸イオンは本発明の電解液にとって不純物に相当し、斯かる不純物の含有量が多い場合には、本発明の電解液を蓄電デバイスに用いた場合に所期の特性が得られ難い場合がある。本発明の電解液中の硫酸イオン量は500ppm以下であるのがより好ましく、さらに好ましくは250ppm以下である。なお、硫酸イオン含有量は低いほどよく、0ppmであるのが最も好ましいが、例えば、硫酸イオン含有量の下限は0.5ppm程度であればよく、また1ppm以下であってもよい。この範囲であれば上述のような問題が生じ難いからである。
【0106】
本発明の電解液は、硫酸イオンに加えて、水、溶媒、フッ素イオン、塩化物イオン、カリウムイオンの含有量も低減されたものであるのが好ましい。具体的に、水の含有量は、本発明の電解液中250ppm以下であるのが好ましく、より好ましくは150ppm以下であり、さらに好ましくは50ppm以下である。残留溶媒の含有量は、本発明の組成物中2000ppm以下であるのが好ましく、より好ましくは1500ppm以下である。フッ素イオンの含有量は本発明の電解液中500ppm以下であるのが好ましく、より好ましくは400ppm以下であり、さらに好ましくは150ppm以下である。塩化物イオンの含有量は本発明の電解液中100ppm以下であるのが好ましく、より好ましくは50ppm以下である。カリウムイオンの含有量は本発明の電解液中5000ppm以下であるのが好ましく、より好ましくは4000ppm以下であり、より一層好ましくは2000ppm以下であり、更に好ましくは500ppm以下、更に一層好ましくは250ppm以下、特に100ppm以下であるのが好ましく、50ppm以下であるのが最も好ましい。
【0107】
なお、本発明の電解液中における水、溶媒、フッ素イオン、塩化物イオンおよびカリウムイオンの含有量はいずれも0ppmであることが最も好ましいが、例えば、下限は夫々0.5ppm程度であればよく、また、1ppm程度であってもよい。水、溶媒およびフッ素イオンの含有量の含有量が上記範囲であれば、本発明の電解液を各種電気化学デバイスに用いても、これらの不純物に由来する問題が生じ難いからである。
【0108】
尚、本発明の電解液に媒体として含まれるものは上記残留溶媒には含まれない。すなわち、上記残留溶媒に含まれる溶媒とは、フルオロスルホニルイミドのアルカリ金属塩の製造工程で使用された反応溶媒や水である。ちなみに、製造工程で使用された反応溶媒と電解液の媒体として含まれる溶媒とが同一である場合、当該溶媒は、残留溶媒には含まれないものとする。
【0109】
本発明の電解液中に含まれる上記イオン、水、残留溶媒の含有量は、例えばICP発光分光分析法、イオンクロマトグラフィー、カールフィッシャー水分計、ガスクロマトグラフィーにより測定することができる。
【実施例】
【0110】
以下、実験例を挙げて本発明をより具体的に説明するが、本発明はもとより下記実験例によって制限を受けるものではなく、前・後記の趣旨に適合し得る範囲で適当に変更を加えて実施することも勿論可能であり、それらはいずれも本発明の技術的範囲に包含される。
【0111】
[NMR測定]
1H−NMR、19F−NMRの測定は、Varian社製の「Unity Plus−400」を使用して行った(内部標準物質:トリフルオロメチルベンゼン、溶媒:重アセトニトリル、積算回数:16回)。
【0112】
[ICP発光分光分析法]
下記例で得られたフルオロスルホニルイミド塩0.1gを超純水9.9gと混合した濃度1質量%の水溶液を測定試料とし、マルチタイプICP発光分光分析装置(島津製作所製「ICPE−9000」)を使用した。
【0113】
実験例1
実験例1−1
〔フッ素化工程〕
攪拌装置を備えたパイレックス(登録商標)製反応容器A(内容量10L)に、窒素気流下で酢酸ブチル1800gを加え、ここに200g(934mmol)のビス(クロロスルホニル)イミドを室温(25℃)で滴下した。
【0114】
得られたビス(クロロスルホニル)イミドの酢酸ブチル溶液に、室温で、フッ化亜鉛101g(982mmol、ビス(クロロスルホニル)イミドに対して1.05当量)を一度に加え、これが完全に溶解するまで室温で6時間攪拌した。
【0115】
〔カチオン交換工程1−アンモニウム塩の合成〕
攪拌装置を備えたパイレックス(登録商標)製反応容器B(内容量10L)に、25質量%アンモニア水540g(7928mmol、ビス(クロロスルホニル)イミドに対して8.49当量)を加えた。アンモニア水の攪拌下、室温で、反応容器Bに、反応容器Aの反応溶液を滴下して加えた。反応溶液の滴下終了後、攪拌を停止し、水層と酢酸ブチル層の2層に分かれた反応溶液から、塩化亜鉛などの副生物を含む水層を除去し、有機層として、アンモニウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液を得た。得られた有機層を試料として、19F-NMR(溶媒:重アセトニトリル)測定を行った。得られたチャートにおいて、内部標準物質として加えたトリフルオロメチルベンゼンの量、及び、これに由来するピークの積分値と、目的生成物に由来するピークの積分値との比較から、有機層に含まれるアンモニウムビス(フルオロスルホニル)イミドの粗収量を求めた(756mmol)。
19F-NMR(溶媒:重アセトニトリル):δ56.0
【0116】
〔カチオン交換工程2−リチウム塩の合成〕
得られた有機層に含まれるアンモニウムビス(フルオロスルホニル)イミドに対して、リチウムの量が2当量となるように、15質量%の水酸化リチウム水溶液242g(Liとして1516mmol)を加え、室温で10分間攪拌した。その後、反応溶液から水層を除去して、リチウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液を得た。
【0117】
得られた有機層を試料とし、ICP発光分光分析法により、フルオロスルホニルイミドのアンモニウムカチオンがリチウムイオンに交換されていることを確認した。また、有機層中のリチウムビス(フルオロスルホニル)イミド濃度は7質量%であった(収量:127g、収率:73%)。
【0118】
なお、フルオロスルホニルイミドの濃度は、得られた有機層を試料として、19F-NMR(溶媒:重アセトニトリル)測定を行い、測定結果のチャートにおいて、内部標準物質として加えたトリフルオロメチルベンゼンの量、及び、これに由来するピークの積分値と、目的生成物に由来するピークの積分値との比較から求めた。
【0119】
[濃縮工程]
まず、カチオン交換工程で得られたリチウムビス(フルオロスルホニル)イミド溶液をロータリーエバポレーター(「REN−1000」、IWAKI社製)に加えて、減圧下で溶媒を留去し、リチウムビス(フルオロスルホニル)イミド溶液282gを得た(濃度:45質量%)。
【0120】
次いで、ガス導入管と減圧装置を備えたロータリーエバポレーター(「REN−1000」、IWAKI製)に、45質量%の濃度のリチウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液200gを入れたフラスコ(容量:500mL)を装着した。フラスコ内の液中に500mL/分で窒素ガスを吹き込みながら、60℃に設定した恒温水槽で加熱しながら回転を開始させた(100rpm)。次いで、装置内を933Paまで徐々に減圧し、12時間濃縮工程を行った。得られた溶液の濃度は72質量%であった。濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、72,000Jであった。
【0121】
[粉体化、乾燥工程]
得られた濃縮液125gに、トルエン125gを加えて、25℃で1時間静置し、リチウムビス(フルオロスルホニル)イミドの固体を析出させた。得られた固体をろ取し、これを50℃で真空乾燥することで、リチウムビス(フルオロスルホニル)イミドを得た(収量:68g、収率:76%(濃縮工程より))。
【0122】
実験例1−2
外径15mmの試験管にガス導入管及び減圧装置を装着した。試験管の中に、実験例1−1のカチオン交換工程2で得られた45質量%の濃度のリチウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液5gを入れ、試験管内を2666Paまで徐々に減圧した。減圧度を保ったまま更に標準状態で1mL/分の流量で窒素ガスを液中に吹き込みながら、65℃に設定した恒温水槽で加熱しながら濃縮を開始した。2.5時間濃縮工程を行ったところ、得られた溶液のリチウムビス(フルオロスルホニル)イミドの濃度は85質量%であった。濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、17,000Jであった。
【0123】
実験例1−3
濃縮工程において、窒素ガスに代えて空気を使用したこと(供給速度:標準状態で約1mL/分)、恒温水槽の温度を75℃に変更したこと、減圧度を667Paとしたこと以外は、実験例1−2と同様にして、リチウムビス(フルオロスルホニル)イミドの製造を行った。このときの濃縮工程の実施時間は2時間であり、濃縮工程で得られた溶液のリチウムビス(フルオロスルホニル)イミド濃度は88質量%であった。濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、15,000Jであった。
【0124】
実験例1−4
濃縮工程において、窒素ガスに代えて空気を使用したこと(供給速度:標準状態で約1mL/分)、恒温水槽の温度を50℃に変更したこと、減圧度を667Paとしたこと以外は、実験例1−2と同様にして、リチウムビス(フルオロスルホニル)イミドの製造を行った。このときの濃縮工程の実施時間は10時間であり、濃縮工程で得られた溶液のリチウムビス(フルオロスルホニル)イミド濃度は84質量%であった。濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、50,000Jであった。
【0125】
実験例1−5
濃縮工程において、窒素ガスを吹き込まなかったこと、恒温水槽の温度を50℃としたこと、減圧度を667Paとしたこと以外は、実験例1−2と同様にして、20時間濃縮工程を行った。得られた溶液の濃度は60質量%であった。濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、100,000Jであった。
【0126】
表1に実験例1−1〜1−5の濃縮条件と結果、図1に、実験例1−2〜1−5の結果を示す。図1では、濃縮時間に対して反応溶媒量をプロットした。
【0127】
【表1】
【0128】
実験例1−2〜1−4と実験例1−5の対比より、バブリングしながら実施する濃縮工程を含む本発明法によれば、生成物であるフルオロスルホニルイミドのアルカリ金属塩を短時間で濃縮できることが分かる。また、濃縮工程を減圧下で行うことで、より一層効率よくフルオロスルホニルイミドのアルカリ金属塩を濃縮できることが分かる。さらに、実験例1−1と1−5の対比より、バブリングしながら濃縮工程を実施すれば、スケールアップしても効率よくフルオロスルホニルイミドのアルカリ金属塩を濃縮できることが分かる。
【0129】
実験例2
実験例2−1
〔フッ素化工程〕
攪拌装置を備えたパイレックス(登録商標)製反応容器A(内容量5L)に、窒素気流下で酢酸ブチル900gを加え、ここに100g(467mmol)のビス(クロロスルホニル)イミドを室温(25℃)で滴下した。
【0130】
得られたビス(クロロスルホニル)イミドの酢酸ブチル溶液に、室温で、フッ化亜鉛50.5g(491mmol、ビス(クロロスルホニル)イミドに対して1.05当量)を一度に加え、これが完全に溶解するまで室温で6時間攪拌した。
【0131】
〔カチオン交換工程1−アンモニウム塩の合成〕
攪拌装置を備えたパイレックス(登録商標)製反応容器B(内容量1L)に、25質量%アンモニア水270g(3964mmol、ビス(クロロスルホニル)イミドに対して8.49当量)を加えた。アンモニア水の攪拌下、室温で、反応容器Bに、反応容器Aの反応溶液を滴下して加えた。反応溶液の滴下終了後、攪拌を停止し、水層と酢酸ブチル層の2層に分かれた反応溶液から、塩化亜鉛などの副生物を含む水層を除去し、有機層として、アンモニウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液を得た。得られた有機層を試料として、19F-NMR(溶媒:重アセトニトリル)測定を行った。得られたチャートにおいて、内部標準物質として加えたトリフルオロメチルベンゼンの量、及び、これに由来するピークの積分値と、目的生成物に由来するピークの積分値との比較から、有機層に含まれるアンモニウムビス(フルオロスルホニル)イミドの粗収量を求めた(378mmol)。
19F-NMR(溶媒:重アセトニトリル):δ56.0
【0132】
〔カチオン交換工程2−リチウム塩の合成〕
得られた有機層に含まれるアンモニウムビス(フルオロスルホニル)イミドに対して、リチウムの量が2当量となるように、15質量%の水酸化リチウム水溶液121g(Liとして758mmol)を加え、室温で10分間攪拌した。その後、反応溶液から水層を除去して、リチウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液を得た。
【0133】
得られた有機層を試料とし、ICP発光分光分析法により、フルオロスルホニルイミドのプロトンがリチウムイオンに交換されていることを確認した。また、有機層中のリチウムビス(フルオロスルホニル)イミド濃度は7質量%であった(収量:63.5g、収率:73%)。
【0134】
なお、フルオロスルホニルイミドの濃度は、得られた有機層を試料として、19F-NMR(溶媒:重アセトニトリル)測定を行い、測定結果のチャートにおいて、内部標準物質として加えたトリフルオロメチルベンゼンの量、及び、これに由来するピークの積分値と、目的生成物に由来するピークの積分値との比較から求めた。
【0135】
[濃縮工程]
カチオン交換工程で得られたリチウムビス(フルオロスルホニル)イミド溶液をロータリーエバポレーター(「REN−1000」、IWAKI社製)に加えて、減圧下で溶媒を留去し、リチウムビス(フルオロスルホニル)イミド溶液141gを得た(濃度:45質量%)。
【0136】
次いで、短行程蒸留装置(型式「KDL1」、攪拌式、蒸発面積:0.01m2、UIC GmbH 社製)を使用して、加熱面の温度(薄膜温度)100℃、圧力1.333kPa、ロータ回転数300rpmで、45質量%のリチウムビス(フルオロスルホニル)イミド溶液120gを供給速度2g/分で、装置内に供給し、薄膜蒸留を実施した後、装置を停止し、濃縮液を抜き出した。得られた溶液のリチウムビス(フルオロスルホニル)イミド濃度は80質量%であった。濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、20,000Jであった。
【0137】
[乾燥、粉体化工程]
次いで、得られた濃縮液67.5gに、トルエン600gを加えて、25℃で1時間静置し、リチウムビス(フルオロスルホニル)イミドの固体を析出させた。得られた固体をろ取し、これを50℃で真空乾燥することで、リチウムビス(フルオロスルホニル)イミドを得た(収量:38g、濃縮工程からの収率:56%)。このとき、ろ液には、濃縮工程からの収率で44質量%に相当するリチウムビス(フルオロスルホニル)イミドが含まれていた。なお、ろ液に含まれるリチウムビス(フルオロスルホニル)イミドは、ろ液を再び濃縮工程、粉体化、乾燥工程に供することで回収することができる。
【0138】
実験例2−2
濃縮工程において、薄膜温度を75℃に変更したこと以外は、実験例2−1と同様にして、リチウムビス(フルオロスルホニル)イミドの製造を行った。このときのリチウムビス(フルオロスルホニル)イミド溶液の供給速度は2g/分(薄膜蒸留時間:1時間)であり、濃縮工程で得られた溶液の濃度は63質量%であった。また、濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、15,000Jであった。
【0139】
実験例2−3
濃縮工程において、薄膜温度を50℃に変更したこと以外は、実験例2−1と同様にして、リチウムビス(フルオロスルホニル)イミドの製造を行った。このときのリチウムビス(フルオロスルホニル)イミド溶液の供給速度は2g/分(薄膜蒸留時間:1時間)であり、濃縮工程で得られた溶液の濃度は58質量%であった。また、濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、10,000Jであった。
【0140】
実験例2−4
短行程蒸留装置に代えて、ロータリーエバポレーター(「REN−1000」、IWAKI社製)を使用した。45質量%の濃度のリチウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液10gを入れたフラスコをロータリーエバポレーターに装着し、これを50℃に設定した恒温水槽で加熱しながら回転を開始し(100rpm)、系内を667Paにまで減圧して、20時間濃縮工程を行った。得られた溶液の濃度は60質量%であった。また、濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、100,000Jであった。
【0141】
実験例2−1〜2−3と実験例2−4の対比より、薄膜蒸留による濃縮工程を含む実験例2−1〜2−3では、薄膜蒸留法を採用しない実験例2−4に比べて、生成物であるリチウムビス(フルオロスルホニル)イミドを短時間で濃縮できることが分かる。
【0142】
実験例3
実験例2−1と同様にしてフッ素化工程およびカチオン交換工程1,2を実施して得られたリチウムビス(フルオロスルホニル)イミド溶液をロータリーエバポレーター(「REN−1000」、IWAKI社製)に加えて、減圧下で溶媒を留去し、リチウムビス(フルオロスルホニル)イミド溶液140gを得た(濃度45質量%)。得られた溶液を、テフロン(登録商標)コーティングされたステンレス鋼製のバット(長さ:29cm、幅22cm、高さ5cm)に流し込み、これを棚段減圧乾燥機に入れ、温度50℃、減圧度150Paで7日間乾燥を行って、リチウムビス(フルオロスルホニル)イミドの固体を得た(収量:63g)。濃縮時に加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、1,200,000Jであった。
【0143】
実験例4
下記方法に従って、実験例2−1および実験例3で得られたフルオロスルホニルイミドのアルカリ金属塩の耐熱性(質量減少率、2%質量減少温度)、および、不純物含有量を測定した。
【0144】
上記手順によりNMR測定を行い、実験例2−1および実験例3で得られたフルオロスルホニルイミドのアルカリ金属塩には、未反応の原料や反応途中の化合物は含まれておらず、いずれもフッ素化、カチオン交換されたものであることを確認した。
【0145】
[耐熱性1 質量減少率]
アルミパンに約20mgの試料を量り取り、示差熱熱重量同時測定装置(「EXSTAR TG/DTA6200」、エスアイアイ・ナノテクノロジー株式会社製)を使用して、乾燥空気気流下(流量200mL/分)、100℃で8時間試料を保持した後の質量減少量を測定した。
【0146】
[耐熱性2 2%及び1%質量減少温度]
アルミパンに量り取った約20mgの試料を、示差熱熱重量同時測定装置(「EXSTAR TG/DTA6200」、エスアイアイ・ナノテクノロジー株式会社製)を使用して、乾燥空気の流量200mL/分、測定温度範囲25℃〜450℃、昇温速度10℃/分で加熱し、質量減少の様子を観察した。
【0147】
なお、耐熱性1,2で使用した試料について、平沼産業株式会社製の「AQ−2000」により水分量を測定したところ、それぞれ64ppm、68ppmであった。
【0148】
[不純物含有量]
上記実験例で得られたフルオロスルホニルイミドのアルカリ金属塩0.01gを超純水(18.2Ω・cm超)で1000倍に希釈して測定溶液とし、イオンクロマトグラフィーシステム ICS−3000(日本ダイオネクス株式会社製)を用いて、フルオロスルホニルイミドのアルカリ金属塩中に含まれるハロゲン化物イオン(F-,Cl-)、および硫酸イオン(SO42-)の量を測定した。
分離モード:イオン交換
溶離液:7〜20mM KOH水溶液
検出器:電気伝導度検出器
カラム:アニオン分析用カラム Ion PAC AS−17C(日本ダイオネクス株式会社製)
なお、カリウムイオン、ナトリウムイオンおよび亜鉛イオン量は、上記ICP発光分光分析法により測定した。
【0149】
【表2】
【0150】
表2より、棚段式減圧乾燥機による濃縮、乾燥を行った実験例3のリチウムビス(フルオロスルホニル)イミドは、実験例2−1に比べて不純物含有量が多く、耐熱性も低いものであった。これは、棚段式減圧乾燥機ではリチウムビス(フルオロスルホニル)イミドから溶媒を除去し、乾燥させるために長時間加熱しなければならず(7日間)、この際にリチウムビス(フルオロスルホニル)イミドがわずかに分解してしまい、生じた分解生成物がリチウムビス(フルオロスルホニル)イミドの耐熱性を低下させているものと考えられる。
【0151】
これに対して、本発明の方法を採用した実験例2−1では、実験例3に比べてリチウムビス(フルオロスルホニル)イミドの耐熱性が30℃以上向上していることが分かる。また、薄膜蒸留法による濃縮工程を採用した実験例2−1のリチウムビス(フルオロスルホニル)イミドは、100℃で8時間保持した後も質量減少は、0%であった。
【0152】
これは、実験例2−1では、実験例3に比べて短い加熱時間でリチウムビス(フルオロスルホニル)イミドを乾燥することができたため、生成物の分解が抑制され、その結果、分解生成物(不純物)に起因する生成物の分解が生じ難くなり、良好な耐熱性を示しているものと考えられる。
【0153】
実験例5
実験例2−1と同様にしてフッ素化工程およびカチオン交換工程1,2を実施して、リチウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液を得た。
実験例5−1〜5−16の各実験例では、実験例2−1と同様にして、フルオロスルホニルイミドのアルカリ金属塩の濃縮工程、乾燥、粉体化工程を行った。
【0154】
得られたフルオロスルホニルイミドのアルカリ金属塩の不純物含有量を、実験例4と同様にして測定した。尚、水の含有量は、下記方法に従って測定した。結果を表3に示す。実験例2−1、実験例3の結果も併せて表3に示す。
【0155】
また、上記NMR測定により、実験例5−1〜5−16で得られたフルオロスルホニルイミドのアルカリ金属塩にも、未反応の原料や反応途中の化合物は含まれておらず、いずれもフッ素化、カチオン交換されたものであることを確認した。
【0156】
[水分含有量]
平沼産業(株)製カールフィッシャー水分測定装置「AQ−2100」を用いて行った。測定試料は、各実験例で得られたフルオロスルホニルイミドのアルカリ金属塩0.3gをメタノールで10倍に希釈して調製した。なお、試料の調整及び測定などの一連の操作は、ドライルーム(温度:25℃、露点:−70℃〜−50℃)で行った。試料注入量は試料の水分含有量に応じて0.1ml〜3mlとし、発生液には「ハイドラナール(登録商標) クローマットAK」(Sigma Aldrich社製)を使用し、対極液には「ハイドラナール(登録商標) クローマットCG−K」(Sigma Aldrich社製)を使用した。試料は、外気に触れないよう注射器を用いて試料注入口より注入した。同様にして、希釈に使用したメタノールの水分含有量を測定し、試料溶液の水分含有量(測定値)から、メタノールの水分含有量を差し引くことで、フルオロスルホニルイミドのアルカリ金属塩の水分含有量を求めた。
【0157】
【表3】
【0158】
表3中、収率とは、濃縮工程からの収率を意味する。尚、実験例5−3〜5−6、5−15〜5−16および実験例3では収率を算出しなかった。また、「純度100%」とは、19F-NMR(溶媒:重アセトニトリル)測定により得られたチャートに、フルオロスルホニルイミド及び内部標準物質に由来するピーク以外にピークが確認されなかったことを意味する。
【0159】
表3より、棚段式減圧乾燥機による濃縮、乾燥を行った実験例3のリチウムビス(フルオロスルホニル)イミドに比べて、薄膜蒸留による濃縮工程を採用した実験例5−1〜5−16では、各種不純物量が低減されていた。この結果より、本発明法によれば、不純物の生成を抑制でき、各種不純物の含有量の少ないフルオロスルホニルイミドのアルカリ金属塩が得られることが分かる。
なお、実験例5では、いずれの実験例も不純物イオン量は少ないものの、実験例間においてその含有量にややバラツキが見られている。これは、フルオロスルホニルイミドが熱による影響を受け易いため、同じ操作を行っていても不純物の生成量に多少のバラツキが見られたものと考えられる。
【0160】
実験例6
実験例2−1、3及び5で得られたフルオロスルホニルイミドのアルカリ金属塩に含まれる残留溶媒量を、下記方法により測定した。
【0161】
[残留溶媒量]
上記実験例で得られたフルオロスルホニルイミドのアルカリ金属塩0.05gにジメチルスルホキシド水溶液(ジメチルスルホキシド/超純水=20/80、体積比)200μl、20質量%塩化ナトリウム水溶液2mlを加えて測定溶液とし、これをバイアル瓶に入れ密閉し、ヘッドスペース−ガスクロマトグラフィーシステム(「Agilent6890」、Agilent社製)により、フルオロスルホニルイミドのアルカリ金属塩中に含まれる残留溶媒量を測定した。
装置:Agilent6890
カラム:HP−5(長さ:30m、カラム内径:0.32mm、膜厚:0.25μm)(Agilent社製)
カラム温度条件:60℃(2分保持)、30℃/分で300℃まで昇温、300℃(2分保持)
ヘッドスペース条件:80℃(30分保持)
インジェクター温度:250℃
検出器:FID(300℃)
結果を表4に示す。また、実験例2−1及び実験例3における残留溶媒量も併せて表4に示す。
【0162】
【表4】
【0163】
表3,4より、本願発明法によれば、不純物の生成を抑制しつつ、棚段式減圧乾燥機を使用した実験例3と同程度にまで残留溶媒量を低減できることが分かる。
【0164】
実験例7
実験例4と同様の方法に従って、実験例5で得られたフルオロスルホニルイミドのアルカリ金属塩の耐熱性(質量減少率、2%質量減少温度)を測定した。結果を表5に示す。比較のため、各実験例における不純物量、及び、実験例2−1、実験例3の結果を表5に併せて示す。
【0165】
【表5】
【0166】
薄膜蒸留法による濃縮工程を採用した実験例5−3,5−5,5−13でも、不純物量の生成が抑制されており、これらの実験例におけるリチウムビス(フルオロスルホニル)イミドは良好な耐熱性を示していた。
【0167】
実験例8
実験例2−1で得られたリチウムビス(フルオロスルホニル)イミド1gを、エチレンカーボネート/エチルメチルカーボネートの混合溶液9g(50/50、体積比)に溶解し、電解液1を作製した。同様にして実験例3で得られたリチウムビス(フルオロスルホニル)イミドを用いて電解液2を作製した。
【0168】
電解液1、2を夫々20mlの褐色のスクリュー管に入れて密閉し、外部からの水分の侵入を防止した。各電解液を入れたスクリュー管を、温度25℃の環境下で2ヶ月間保存し、電解液1、2の経時変化を評価した。
【0169】
実験例2−1の生成物を原料として作製した電解液1には外観上の変化は認められず、無色透明の液体であったが、実験例3の生成物を原料として作製した電解液2には黄橙色の着色が見られた。また、上記不純物含有量の測定方法に従い、保存前後の電解液1,2を超純水で希釈した測定溶液についてイオンクロマトグラフィーにより、不純物含有量の測定を行った。結果を表6に示す。
【0170】
【表6】
【0171】
表6に示されるように、実験例2−1から得られた電解液1では、フッ化物イオン量が2ppm(保存前:1ppm)、硫酸イオン量が14ppm(保存前:14ppm)であり、保存前とほぼ同等の値であった。一方、実験例3から得られた電解液2では、フッ化物イオン量が153ppm(保存前:122ppm)、硫酸イオン量が413ppm(339ppm)と、保管前に比べて不純物が増加していることが確認された。この結果より、本発明の電解液は、保存安定性に優れることがわかった。
【産業上の利用可能性】
【0172】
本発明によれば、スケールアップしても、反応溶液から溶媒を速やかに除去することができ、効率よくフルオロスルホニルイミドのアルカリ金属塩を製造できる。また、本発明法では高温に加熱する必要がないので、フルオロスルホニルイミドのアルカリ金属塩の熱分解による収率の低下も抑制することができ、その結果、フルオロスルホニルイミドのアルカリ金属塩の耐熱性の低下を抑制することができる。さらに、本発明のフルオロスルホニルイミドのアルカリ金属塩は、各種不純物量が低減されているので、各種電気化学デバイスのイオン伝導体などとして有用である。加えて、本発明の製造方法は、工業的なフルオロスルホニルイミドのアルカリ金属塩の製造方法として好適である。
【技術分野】
【0001】
本発明は、フルオロスルホニルイミドのアルカリ金属塩、詳しくは、N−(フルオロスルホニル)−N−(フルオロアルキルスルホニル)イミド、ビス(フルオロスルホニル)イミドのアルカリ金属塩とその製造方法に関する。
【背景技術】
【0002】
N−(フルオロスルホニル)−N−(フルオロアルキルスルホニル)イミドや、ビス(フルオロスルホニル)イミド等のフルオロスルホニルイミドの塩やその誘導体は、N(SO2F)基又はN(SO2F)2基を有する化合物の中間体として有用であり、また、電解質や、燃料電池の電解液への添加物や、選択的求電子フッ素化剤、光酸発生剤、熱酸発生剤、近赤外線吸収色素等として使用されるなど、様々な用途において有用な化合物である。
【0003】
従来、フルオロスルホニルイミド類は、フッ素化剤を使用して、クロロスルホニルイミドをハロゲン交換する方法や(非特許文献1,2)、尿素の存在下で、フルオロスルホン酸(HFSO3)を蒸留することによってビス(フルオロスルホニル)イミドとする方法(特許文献1)などにより調製されてきた。また、本発明者らは、所定の元素を含むフッ化物を使用してフルオロスルホニルイミドを製造する方法を提案している(特許文献2)。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特表平8−511274号公報
【特許文献2】国際公開第2009/123328号パンフレット
【非特許文献】
【0005】
【非特許文献1】John K. RuffおよびMax Lustig、Inorg.Synth. 11,138-140 (1968年)
【非特許文献2】Jean’ne M. Shreeveら、Inorg. Chem. 1998, 37 (24), 6295-6303
【発明の概要】
【発明が解決しようとする課題】
【0006】
上述のように、クロロスルホニルイミドのフッ素化については改良が重ねられ、その収率をある程度向上させることが可能になった。しかしながら、本発明者らは、フルオロスルホニルイミド塩の実操業レベルでの製造を検討する中で、フルオロスルホニルイミド塩の生成後、反応溶媒を除去する際、原料100質量%に対する溶媒の量が150質量%以下になると反応溶液から溶媒を除去し難いといった新たな問題があることを認識した。また、本発明者らは、フルオロスルホニルイミドが水との親和性が高く、生成物中の水分含有量を低減させ難いことも見出している。このような水分は、電気化学デバイスに用いた場合に、電解液の耐電圧性を低下させるのみならず、デバイスを構成する部材(電極等)を腐食させる原因にもなる。なお、溶媒や水分は加熱により生成物から除去することも考えられるが、フルオロスルホニルイミド塩の耐熱性はそれほど高いとはいえず、高温での加熱や長時間の加熱は、生成物の収率を低下させてしまい、結果として、製造コストが上昇してしまうといった問題もある。
【0007】
本発明は上記の様な事情に着目してなされたものであって、その目的は、スケールアップしても、反応溶液から溶媒を容易に除去することができるフルオロスルホニルイミドのアルカリ金属塩の製造方法と当該方法により得られるフルオロスルホニルイミドのアルカリ金属塩を提供することにある。また、本発明は、耐熱性の良好なフルオロスルホニルイミドのアルカリ金属塩、及び、特定の不純物や水分含有量が低減されたフルオロスルホニルイミドのアルカリ金属塩、及び、これを含む電解液を提供することも目的とする。
【課題を解決するための手段】
【0008】
本発明者らは、鋭意研究を重ねた結果、上記課題が下記方法により解決され、かつ、本発明の方法により得られたフルオロスルホニルイミドのアルカリ金属塩が耐熱性に優れること、また、特定の不純物や水分含有量が低減されたフルオロスルホニルイミドのアルカリ金属塩が耐熱性に優れることを見出し、本発明を完成した。
【0009】
すなわち、本発明のフルオロスルホニルイミドのアルカリ金属塩とは、下記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩であって、空気気流下、100℃で8時間保持したときの質量減少率が2%以下であるところに特徴を有する。
【0010】
【化1】
【0011】
(式(I)中、Maはアルカリ金属、Ra、Rbは、同一若しくは異なって、フッ素原子、または、1個以上の水素原子がフッ素原子で置換された炭素数1〜6の炭化水素基を表す。)
【0012】
このように、本発明のフルオロスルホニルイミドのアルカリ金属塩は、長時間加熱雰囲気にさらされても分解を生じ難く、耐熱性に優れるものである。
【0013】
上記フルオロスルホニルイミドのアルカリ金属塩は、空気気流下、25℃より10℃/分の昇温速度で加熱したときに、質量減少率が2%となる温度が210℃以上であるのが好ましい。
【0014】
また、本発明のフルオロスルホニルイミドのアルカリ金属塩は、硫酸イオン(SO42−)の含有量が3000ppm以下であるところに特徴を有する。本発明のフルオロスルホニルイミドのアルカリ金属塩に含まれるフッ化物イオン(F−)の含有量は1000ppm以下であるのが好ましく、また、水の含有量は500ppm以下であるのが好ましい。さらに、本発明のフルオロスルホニルイミドのアルカリ金属塩は、4000ppm以下の残留溶媒を含むものであるのが好ましい。
【0015】
本発明には、上記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩と媒体を含み、SO42−の含有量が1500ppm以下である電解液も含まれる。本発明の電解液は、水の含有量が250ppm以下であるのが好ましい。また、本発明の電解液は、2000ppm以下の残留溶媒を含むものであるのが好ましい。さらに、本発明の電解液は、F−の含有量が500ppm以下であるのが望ましい。
【0016】
上記目的を達成し得た本発明の製造方法とは、上記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩の製造方法であって、
(1)フルオロスルホニルイミドのアルカリ金属塩を含む反応溶液中にガスをバブリングしながらフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程、及び/又は、
(2)フルオロスルホニルイミドのアルカリ金属塩を含む反応溶液を薄膜蒸留により濃縮する工程
を含むところに特徴を有する。
【0017】
本発明者らは、上記問題点について検討を重ねたところ、反応溶媒の除去を困難にしている理由は、フルオロスルホニルイミドがアルカリ金属塩を形成したことによる沸点上昇や、フルオロスルホニルイミドのアルカリ金属塩が溶媒分子と溶媒和していることにあることが判明した。そこで、反応溶媒として、生成物と溶媒和を形成し難いものを使用することを検討した。しかしながら、クロロスルホニルイミドのフッ素化工程で使用できる溶媒は限られているため、反応溶媒を変更することは現実的ではない。また、フッ素化工程後、カチオン交換工程前のいずれかの工程で、反応溶媒を変更することも考えられるが、溶媒使用量の増加は、経済的にも環境的にも好ましくない。そこで、反応溶液から溶媒を効率よく除去する方法について更なる検討を重ねたところ、濃縮工程において、(1)バブリング法、及び/又は、(2)薄膜蒸留法を採用すれば、容易に反応溶媒を除去でき、効率よくフルオロスルホニルイミドのアルカリ金属塩を精製できることを見出した。すなわち、上記(1)の反応溶液にガスをバブリングする場合には、蒸発面積が増大させられるので、反応溶媒の蒸発を促進させることができる。したがって、反応溶液を高温に加熱しなくても、速やかに溶媒を除去できるのである。一方、(2)の薄膜蒸留法では、伝熱させるべき反応溶液を伝熱面に対して薄膜にすることにより、大きな伝熱係数を得ることができる。その結果、伝熱面を高温に加熱しなくても速やかに溶媒を除去できるのである。また、薄膜蒸留法では、伝熱面積の小さな装置でも効率よく溶媒を除去することができる。したがって、本発明法によれば、製造工程で反応溶液を長時間、高温に加熱する必要がないため、加熱によるフルオロスルホニルイミドのアルカリ金属塩の分解が抑制され、その結果、分解生成物に起因するフルオロスルホニルイミドのアルカリ金属塩の耐熱性の低下が抑制される。
【0018】
前記濃縮工程は150℃以下で行うのが好ましい。また、前記濃縮工程は40kPa以下で行うことが望ましい。前記反応溶液に含まれる反応溶媒は、エステル系溶媒及び/又はニトリル系溶媒であるのが好ましい。さらに、上述の各工程に加えて、濃縮工程で得られた濃縮液を100℃以下で加熱して乾燥する工程を含むことも本発明の推奨される実施態様である。
【0019】
上記製造方法は、さらに、反応溶媒の存在下で、クロロスルホニルイミドまたはその塩をフッ素化する工程を含むものであるのが望ましい。
【0020】
なお、本発明における「フルオロスルホニルイミド」との文言には、フルオロスルホニル基を2つ有するビス(フルオロスルホニル)イミドの他、フルオロスルホニル基と、フッ化アルキル基を有するN−(フルオロスルホニル)−N−(フルオロアルキルスルホニル)イミドが含まれる。出発原料である「クロロスルホニルイミド」も同様である。また、「フルオロアルキル」とは、炭素数1〜6のアルキル基において、1つ以上の水素原子がフッ素原子で置換されたものを意味し、例えば、フルオロメチル基、ジフルオロメチル基、トリフルオロメチル基、フルオロエチル基、ジフルオロエチル基、トリフルオロエチル基、ペンタフルオロエチル基等が含まれる。
【発明の効果】
【0021】
本発明によれば、反応溶液から溶媒を速やかに除去でき、効率よくフルオロスルホニルイミドのアルカリ金属塩を製造することができる。また、本発明のフルオロスルホニルイミドのアルカリ金属塩は耐熱性に優れるものである。
【図面の簡単な説明】
【0022】
【図1】図1は、実験例1−2〜1−5の結果を示すグラフである。
【発明を実施するための形態】
【0023】
<フルオロスルホニルイミドのアルカリ金属塩>
本発明のフルオロスルホニルイミドのアルカリ金属塩とは、下記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩であって、空気気流下、100℃で8時間保持したときの質量減少率が2%以下であるところに特徴を有する。
【0024】
【化2】
【0025】
(式(I)中、Maはアルカリ金属、Ra、Rbは、同一若しくは異なって、フッ素原子、または、1個以上の水素原子がフッ素原子で置換された炭素数1〜6の炭化水素基を表す。)
【0026】
このように、本発明のフルオロスルホニルイミドのアルカリ金属塩は、長時間、加熱雰囲気に曝されても分解し難いため、これをイオン伝導体材料として使用した各種電気化学デバイスは、長期に亘って安定した電気化学的特性を発揮することができる。また、各種電気化学デバイスは、広い温度領域で使用することができる。なお、優れた耐熱性は、後述するように、本発明のフルオロスルホニルイミドのアルカリ金属塩に含まれる不純物量が低減されていることに起因するものである。
【0027】
上記質量減少率が低いほど耐熱性は高く好ましいが、質量減少率は1%以下であるのが好ましい。
【0028】
また、本発明のフルオロスルホニルイミドのアルカリ金属塩は、空気気流下、25℃より10℃/分の昇温速度で加熱したときに、質量減少率が2%となる温度(以下、「2%質量減少温度」という場合がある)が210℃以上であるのが好ましい。ここで、2%質量減少温度とは、上記条件下で熱重量分析を行ったときに、初期の質量に対する質量減少率が2%となる温度を意味する。すなわち、2%質量減少温度が高いほど、高温に曝されてもフルオロスルホニルイミドのアルカリ金属塩が分解を生じ難く、安定であることを示す。2%質量減少温度は215℃以上であるのがより好ましい。
【0029】
さらに、本発明のフルオロスルホニルイミドのアルカリ金属塩は、上記2%質量減少温度と同様の条件で測定を行ったとき、質量減少率が1%となる温度(以下、「1%質量減少温度」という場合がある)が175℃以上であるのが好ましい。より好ましくは185℃以上である。
【0030】
上記質量減少率、2%、1%質量減少温度の測定に使用可能な装置としては、示差熱熱重量同時測定装置(例えばエスアイアイ・ナノテクノロジー株式会社製の「EXSTAR TG/DTA6200」)等が挙げられる。なお、測定試料が水分や溶媒を含んでいる場合には、正確な測定結果が得られ難い。したがって、本測定においては、含水率が1000ppm以下のフルオロスルホニルイミドのアルカリ金属塩を用いるのが好ましい。より好ましい含水率は500ppm以下であり、さらに好ましくは200ppm以下である。含水率が上記範囲であれば、測定結果への影響は無視できる程度となるからである。また、フルオロスルホニルイミドのアルカリ金属塩の製造直後に上記測定(質量減少率、2%、1%質量減少温度)を行わない場合は、測定試料を温度50℃、真空下で6時間保持し、試料に含まれる水分量や溶媒量を調整した後、上記測定に供することが推奨される。
【0031】
さらに、本発明のフルオロスルホニルイミドのアルカリ金属塩は、不純物の含有量が極低レベルに低減されている。上記不純物としては、例えば、後述する濃縮工程や乾燥工程での加熱により生じる分解生成物や精製段階で混入してくる不純物が挙げられ、例えば、フルオロスルホニルイミドのアルカリ金属塩の分解生成物に相当するフッ化物イオン(F-)や、硫酸イオン(SO42-)、また、フルオロスルホニルイミドの製造段階や精製段階で使用する溶媒(以下、残留溶媒と称する)及び水等が挙げられる。具体的に、本発明のフルオロスルホニルイミドのアルカリ金属塩(固体)に含まれる硫酸イオン(SO42-)の含有量は3000ppm以下であり、フッ化物イオン(F-)の含有量は1000ppm以下であるのが好ましい(いずれも質量基準。以下同様。)。
【0032】
上記フッ化物イオンおよび硫酸イオン等の不純物量の増加はフルオロスルホニルイミドの分解および純度の低下を意味する。したがって、不純物含有量が多い場合には、フルオロスルホニルイミドを各種用途に用いた場合に所期の特性が得られ難くなる。また、これらの不純物は、フルオロスルホニルイミドの分解を促進する逆効果を有する。したがって、不純物含有量が多い場合には初期の特性が維持し難いものとなる。さらに、フッ化物イオンは、フルオロスルホニルイミドのアルカリ金属塩が用いられる各種装置の周辺部材を腐食させる虞がある。したがって、これら不純物の含有量はできるだけ少ないのが好ましく、例えば、硫酸イオンの含有量はフルオロスルホニルイミドのアルカリ金属塩中1000ppm以下であるのが好ましく、より好ましくは500ppm以下であり、更に好ましくは300ppm以下である。一方、フッ化物イオンの含有量は、フルオロスルホニルイミドのアルカリ金属塩中800ppm以下であるのがより好ましい。より一層好ましくは500ppm以下であり、更に好ましくは300ppm以下である。なお、上記不純物は、本発明のフルオロスルホニルイミドのアルカリ金属塩中に含まれていないことが最も好ましいが、例えば、下限はいずれも1ppm程度であればよい。不純物の含有量が上記範囲であれば、本発明のフルオロスルホニルイミドのアルカリ金属塩を後述する各種電気化学デバイスに備えられるイオン伝導体材料として用いても、不純なイオン成分に由来する問題は生じ難い。
【0033】
また、本発明のフルオロスルホニルイミドのアルカリ金属塩(固体)の水の含有量は300ppm以下であるのが好ましい。上述のように、固体のフルオロスルホニルイミドのアルカリ金属塩に含まれる水分は、フルオロスルホニルイミドのみならず、これらが用いられるデバイスの構成部材を腐食させる原因となる。したがって、水の含有量は200ppm以下であるのがより好ましく、さらに好ましくは100ppm以下である。なお、水の含有量は少ないほど好ましく、実質的に0ppmであるのが好ましいが、0ppmにまで水分量を低減するのは技術的に難しく、また、経済的に好ましくない場合がある。よって水の含有量の下限は、1ppm程度であればよい。
【0034】
加えて、本発明のフルオロスルホニルイミドのアルカリ金属塩(固体)は、フルオロスルホニルイミドのアルカリ金属塩の製造段階で使用した残留溶媒の含有量が4000ppm以下であるのが好ましい。固体のフルオロスルホニルイミドのアルカリ金属塩に含まれる残留溶媒は耐熱性の低下、電気化学性能の低下の原因となる。したがって、斯かる残留溶媒の含有量は少ないほど好ましく、実質的に0ppmであるのが好ましいが、水の含有量の場合と同様の理由から、残留溶媒の量は3000ppm以下であるのがより好ましい。残留溶媒量の下限は1ppm程度であればよい。なお、上記残留溶媒には、後述するフルオロスルホニルイミドの製造段階で使用する溶媒が含まれる。
【0035】
さらに、本発明のフルオロスルホニルイミドのアルカリ金属塩は、上記フッ化物イオン、硫酸イオンに加えて、塩化物イオン(Cl-)、カリウムイオン(K+)などの他の不純物の含有量も少ないものであるのが好ましい。塩化物イオンの含有量は、200ppm以下であるのがより好ましく、より一層好ましくは100ppm以下である。一方、カリウムイオンの含有量は、10000ppm以下であるのが好ましく、より好ましくは8000ppm以下であり、より一層好ましくは4000ppm以下であり、更に好ましくは1000ppm以下、更に一層好ましくは500ppm以下であり、特に200ppm以下であるのがより好ましく、100ppm以下であるのが最も好ましい。なお、他の不純物の含有量は、合計で10000ppm以下であるのが好ましく、1000ppm以下であるのがより好ましく、500ppm以下であるのがさらに好ましい。なお、他の不純物の含有量の下限は合計で1ppm程度であればよい。
【0036】
上記不純物の種類や含有量は、後述するICP発光分光分析法、NMR測定あるいはイオンクロマトグラフィー等により分析することができる。水分含有量はカールフィッシャー水分計により測定でき、残留溶媒の含有量はガスクロマトグラフィーにより測定できる。
【0037】
<フルオロスルホニルイミドのアルカリ金属塩を含む組成物>
また、本発明には、上記フルオロスルホニルイミドのアルカリ金属塩を含む組成物も含まれる。本発明の組成物は、フルオロスルホニルイミドのアルカリ金属塩と、硫酸イオン、水、残留溶媒およびフッ素イオンの合計100質量%(以下、「本発明の組成物」という)に対する硫酸イオンの含有量が、3000ppm以下であるのが好ましい。より好ましくは1000ppm以下であり、より一層好ましくは500ppm以下であり、さらに好ましくは300ppm以下である。硫酸イオンは、本発明の組成物中に含まれていないことが最も好ましいが、例えば下限は1ppm程度であればよい。本発明の組成物を電気化学デバイス用の電解液に用いても、電解液の分解や電気化学デバイスの構成部材の腐食といった問題を生じ難いからである。
【0038】
また、本発明の組成物は、硫酸イオン以外の不純物含有量も低減されているのが好ましい。水、残留溶媒およびフッ素イオンも、硫酸イオン同様、本発明に係るフルオロスルホニルイミドのアルカリ金属塩を分解させたり、また、本発明の組成物を電気化学デバイス用の電解液として用いた場合には、当該デバイス等を劣化させる原因となる場合がある。よって、その含有量は以下の通りとするのが好ましい。すなわち、フッ素イオンの量は、本発明の組成物100質量%中1000ppm以下であるのが好ましく、より好ましくは800ppm以下であり、より一層好ましくは500ppm以下であり、さらに好ましくは300ppm以下である。水の含有量は、本発明の組成物100質量%中500ppm以下であるのが好ましく、より好ましくは300ppm以下であり、より一層好ましくは200ppm以下であり、さらに好ましくは100ppm以下である。残留溶媒の含有量は、本発明の組成物100質量%中4000ppm以下であるのが好ましく、より好ましくは3000ppm以下である。なお、本発明の組成物中における水、残留溶媒およびフッ素イオンの含有量は夫々0ppmであることが最も好ましいが、例えば、下限は夫々1ppm程度であればよい。水、残留溶媒およびフッ素イオンの含有量が上記範囲であれば、本発明の組成物を後述する各種電気化学デバイスに備えられる電解液に用いても、不純なイオン成分に由来する問題は生じ難い。
【0039】
本発明の組成物は、塩化物イオンやカリウムイオンを含んでいてもよい。ただし、塩化物イオンの含有量は、本発明の組成物と塩化物イオン及び/又はカリウムイオンの合計100質量%中200ppm以下であるのが好ましく、より好ましくは100ppm以下である。一方、カリウムイオンの含有量は、本発明の組成物と塩化物イオン及び/又はカリウムイオンの合計100質量%中10000ppm以下であるのが好ましく、より好ましくは8000ppm以下であり、より一層好ましくは4000ppm以下であり、更に好ましくは1000ppm以下、更に一層好ましくは500ppm以下であり、特に200ppm以下であるのがより好ましく、100ppm以下であるのが最も好ましい。また、これらのイオンの合計含有量は、本発明の組成物と塩化物イオン及び/又はカリウムイオンの合計100質量%中10000ppm以下であるのが好ましく、1000ppm以下であるのがより好ましく、500ppm以下であるのがさらに好ましい。なお、これらのイオンの含有量の下限は合計で1ppm程度であればよい。
【0040】
なお、本発明の組成物中における上記成分(硫酸イオン、フッ素イオン、および水および残留溶媒の含有量)の含有量は、ICP発光分光分析法、イオンクロマトグラフィー、カールフィッシャー水分計、ガスクロマトグラフィーにより測定できる。
なお、上記各種イオンの含有量は、上記測定方法により検出される値である。したがって、固体のフルオロスルホニルイミドのアルカリ金属塩中、または、本発明の組成物中では、上記イオンは対イオンとの塩として存在しているものと考えられる。
【0041】
本発明のフルオロスルホニルイミドのアルカリ金属塩は、上記一般式(I)で表されるものである。上記一般式(I)中、Maはアルカリ金属を表す。Maは、好ましくはLi、Na、Kであり、より好ましくはLiである。上記一般式(I)中、Ra、Rbは、同一若しくは異なって、フッ素原子、または、1個以上の水素原子がフッ素原子で置換された炭素数1〜6の炭化水素基を表す。上記炭化水素基としては、直鎖状の炭素数1〜6のフルオロアルキル基であるのが好ましく、例えば、フルオロメチル基、ジフルオロメチル基、トリフルオロメチル基、フルオロエチル基、ジフルオロエチル基、トリフルオロエチル基、ペンタフルオロエチル基等が挙げられる。これらの中でも、Ra、Rbとしては、フッ素原子、トリフルオロメチル基、ペンタフルオロエチル基が好ましい。
【0042】
本発明のビス(フルオロスルホニル)イミドや、N−(フルオロスルホニル)−N−(フルオロアルキルスルホニル)イミドのアルカリ金属塩は、各種電気化学デバイスのイオン伝導体材料として好適である。特に、カチオンがリチウムであるリチウムフルオロスルホニルイミドは、リチウム二次電池、キャパシタなどに用いられる電解質やイオン性液体、あるいは、フルオロスルホニル化合物の中間体などとして有用である。
【0043】
上記耐熱性を備えた本発明のフルオロスルホニルイミドの製造方法は特に限定されないが、不純物含有量を低減して耐熱性を向上させる観点からは、以下に説明する方法により製造することが推奨される。
【0044】
<フルオロスルホニルイミドのアルカリ金属塩の製造方法>
本発明のフルオロスルホニルイミドのアルカリ金属塩の製造方法とは、(1)フルオロスルホニルイミドのアルカリ金属塩を含む反応溶液中にガスをバブリングしながらフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程、及び/又は、(2)薄膜蒸留によりフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程、を含む点に特徴を有するものである。したがって、上記(1)及び/又は(2)の工程を含んでいればよく、その他の工程は特に限定されない。本発明においては、フルオロスルホニルイミドのアルカリ金属塩を合成する方法は特に限定されず、従来公知の方法は全て採用することが出来る。例えば、フルオロスルホニルイミドの合成方法としては、特許文献1に記載の、尿素の存在下で、フルオロスルホン酸(HFSO3)を蒸留することによって(フルオロスルホニル)イミドを得る方法、クロロスルホニルイミドからフッ素化剤を用いてフルオロスルホニルイミドを合成する方法等が挙げられる。また、フルオロスルホニルイミドのアルカリ金属塩を得る方法としては、上記方法により得られたフルオロスルホニルイミドのカチオンをアルカリ金属カチオンとカチオン交換する方法が挙げられる。以下では、上記方法の中でも、クロロスルホニルイミドからフッ素化剤を用いてフルオロスルホニルイミドを合成し(フッ素化工程)、次いで、カチオン交換反応(カチオン交換工程)により、フルオロスルホニルイミドのアルカリ金属塩を合成する方法について説明する。
【0045】
まず、フッ素化工程から説明する。
【0046】
[フッ素化工程]
フッ素化工程では、クロロスルホニルイミド又はその塩のフッ素化反応を行う。出発原料となるクロロスルホニルイミドは、市販のものを使用してもよく、また、公知の方法で合成したものを用いてもよい。
【0047】
クロロスルホニルイミドを合成する方法としては、例えば、塩化シアンに無水硫酸を反応させた後、生成物(クロロスルホニルイソシアネート)とクロロスルホン酸とを反応させる方法、アミド硫酸と塩化チオニルとを反応させた後、さらにクロロスルホン酸を反応させる方法(以上、ビス(クロロスルホニル)イミドの合成方法);クロロスルホニルイソシアネートとフッ化アルキルスルホン酸またはフルオロスルホン酸とを反応させる方法(N−(クロロスルホニル)−N−(フルオロアルキルスルホニル)イミド、または、N−(クロロスルホニル)−N−(フルオロスルホニル)イミドの合成方法);などが挙げられる。
【0048】
次いで、クロロスルホニルイミドのフッ素化反応を行う。なお、フッ素化反応のタイミングは特に限定されず、クロロスルホニルイミド(プロトン体)のフッ素化反応を行う態様;クロロスルホニルイミドのカチオン交換反応を行った後、クロロスルホニルイミド塩のフッ素化反応を行う態様;のいずれの態様であってもよい。
【0049】
上記クロロスルホニルイミド(プロトン体)またはその塩(以下、クロロスルホニルイミド類と言う)をフッ素化する方法としては、上記非特許文献1,2に記載のフッ素化剤(AsF3、SbF3)を使用して、クロロスルホニルイミドをハロゲン交換する方法、KFやCsF等の1価カチオンのイオン性フルオリドをフッ素化剤として用いて、ジ(クロロスルホニル)イミドをフッ素化する方法や、クロロスルホニルイミド類を、アルカリ金属のフッ化物や、第11族〜第15族、第4周期〜第6周期の元素よりなる群から選ばれる少なくとも1種の元素を含むフッ化物(好ましくはCuF2、ZnF2、SnF2、PbF2およびBiF3など)と反応させる方法が挙げられる。第11族〜第15族、第4周期〜第6周期の元素よりなる群から選ばれる少なくとも1種の元素を含むフッ化物(好ましくはCuF2、ZnF2、SnF2、PbF2およびBiF3等)を用いてジ(クロロスルホニルイミド)をフッ素化する方法が、反応収率の面では好ましい。また、KF、LiF、NaF等アルカリ金属のフッ化物をフッ素化剤としてジ(クロロスルホニルイミド)をフッ素化する方法では、フルオロスルホニルイミドのアルカリ金属塩を一段階で得ることができるため好ましい。
【0050】
フッ素化工程では、反応溶媒として、非プロトン性溶媒を用いるのが好ましい。具体的には、エチレンカーボネート、プロピレンカーボネート、ブチレンカーボネート、ジメチルカーボネート、エチルメチルカーボネート、ジエチルカーボネート等のカーボネート系溶媒;ジメトキシメタン、1,2−ジメトキシエタン、テトラヒドロフラン、2−メチルテトラヒドロフラン、1,3−ジオキサン、4−メチル−1,3−ジオキソラン等のエーテル系溶媒;ギ酸メチル、酢酸メチル、酢酸エチル、酢酸イソプロピル、酢酸ブチル、プロピオン酸メチル、γ−ブチロラクトン、γ−バレロラクトン等のエステル系溶媒;N,N−ジメチルホルムアミド等のアミド系溶媒;アセトニトリル、ブチロニトリル、イソブチロニトリル、バレロニトリル、ベンゾニトリル等のニトリル系溶媒;ニトロメタン、ニトロベンゼン等のニトロ系溶媒;スルホラン、3−メチルスルホラン、ジメチルスルホキシド等の硫黄系溶媒;N−メチルオキサゾリジノン;等が挙げられる。これらの溶媒は単独で用いてもよく、また2種以上を混合して用いてもよい。フッ素化反応を円滑に進行させる観点からは極性溶媒を使用することが推奨され、上記例示の溶媒の中でも、エステル系溶媒及び/又はニトリル系溶媒が好ましく、特に、ブチロニトリル、イソブチロニトリル、バレロニトリル、酢酸エチル、酢酸イソプロピル及び酢酸ブチルが好ましい。なお、精製時の作業性からは、沸点が低く、水と2層状態を形成し得る溶媒が好ましい。
【0051】
フッ素化反応の終了は、例えば、19F−NMRなどで確認することができる。すなわち、反応の進行によりフッ素に由来するケミカルシフトにピークが出現し、さらに、そのピークの相対強度(積分値)が増大する。したがって、19F−NMRにより反応の進行状態を追跡しながら、フッ素化反応の終了を確認すればよい。なお、反応時間が長すぎる場合には、副生物の生成が顕著となるので、目的物のピークの相対強度が最大となる時点(例えば、反応の開始から6時間〜12時間程度)でフッ素化反応を終了するのが好ましい。
【0052】
[カチオン交換工程]
次に、カチオン交換工程について説明する。クロロスルホニルイミド類またはフルオロスルホニルイミドまたはその塩(以下、フルオロスルホニルイミド類と言う場合がある)を、所望のカチオンを含む塩と反応させることで、カチオン交換することができる。カチオンとしては、Li,Na,K,Rb,Csなどのアルカリ金属、または、後述するオニウムカチオンが好ましい。アルカリ金属を含むフルオロスルホニルイミド塩は、高温で溶融させたり、あるいは、適当な有機溶媒に溶解させることで、各種電気化学デバイスのイオン伝導体材料として使用することができる。また、オニウムカチオンを含むフルオロスルホニルイミド塩は、常温で溶融した状態を安定に保つ常温溶融塩となり、長期間の使用に耐える電気化学デバイスのイオン伝導体材料や、有機合成における反応溶媒等として好適なものとなる。より好ましいフルオロスルホニルイミド塩としては、リチウムカチオンや、オニウムカチオンを含むフルオロスルホニルイミド塩が挙げられる。
【0053】
アルカリ金属を含む塩としては、LiOH、NaOH、KOH、RbOH、CsOH等の水酸化物;Li2CO3、Na2CO3、K2CO3、Rb2CO3、Cs2CO3等の炭酸塩、LiHCO3、NaHCO3、KHCO3、RbHCO3、CsHCO3等の炭酸水素塩;LiCl、NaCl、KCl、RbCl、CsCl等の塩化物;LiF、NaF、KF、RbF、CsF等のフッ化物;CH3OLi、EtOLi等のアルコキシド化合物;及び、EtLi、BuLiおよびt−BuLi(尚、Etはエチル基、Buはブチル基を示す)等のアルキルリチウム化合物;等のアルカリ金属塩が挙げられる。
【0054】
一方、オニウムカチオンとしては、一般式(II);L+−Rs(式中、Lは、C、Si、N、P、S又はOを表す。Rは、同一若しくは異なって、水素原子、フッ素原子、または、有機基であり、Rが有機基の場合、これらは互いに結合していてもよい。sは、2、3又は4であり、元素Lの価数によって決まる値である。尚、L−R間の結合は、単結合であっても良く、また二重結合であってもよい。)で表されるものが好適である。
【0055】
上記Rで示される「有機基」は、炭素原子を少なくとも1個有する基を意味する。上記「炭素原子を少なくとも1個有する基」は、炭素原子を少なくとも1個有していればよく、また、ハロゲン原子やヘテロ原子などの他の原子や、置換基などを有していてもよい。具体的な置換基としては、例えば、アミノ基、イミノ基、アミド基、エーテル結合を有する基、チオエーテル結合を有する基、エステル基、ヒドロキシル基、アルコキシ基、カルボキシル基、カルバモイル基、シアノ基、ジスルフィド基、ニトロ基、ニトロソ基、スルホニル基などが挙げられる。
【0056】
一般式(II)で表されるオニウムカチオンとしては、具体的には下記一般式;
【0057】
【化3】
【0058】
(式中、Rは、一般式(II)と同様)で表されるものが好適である。このようなオニウムカチオンは単独で用いてもよく、2種以上を併用してもよい。具体的なオニウムカチオンとしては、WO2009/123328号公報に記載される複素環オニウムカチオン、不飽和オニウムカチオン、飽和環オニウムカチオン、及び、鎖状オニウムカチオン等が挙げられる。
【0059】
なお、好ましいオニウムカチオンとしては、一般式(II);L+−RsにおいてLがN、Rが、水素、または、C1〜C8のアルキル基、sが4である鎖状オニウムカチオンや下記一般式で表される5種類のオニウムカチオンが挙げられる。
【0060】
【化4】
【0061】
上記一般式中、R1〜R12は、同一若しくは異なって、水素原子、フッ素原子、又は、有機基であり、有機基の場合、これらは互いに結合していてもよい。有機基としては、直鎖、分岐鎖又は環状の炭素数1〜18の飽和又は不飽和炭化水素基、炭化フッ素基等が好ましく、より好ましくは炭素数1〜8の飽和又は不飽和炭化水素基、炭化フッ素基である。これらの有機基は、水素原子、フッ素原子、窒素原子、酸素原子、硫黄原子や、アミノ基、イミノ基、アミド基、エーテル基、エステル基、ヒドロキシル基、カルボキシル基、カルバモイル基、シアノ基、スルホン基、スルフィド基等の官能基を含んでいてもよい。より好ましくは、R1〜R12は、水素原子、フッ素原子、シアノ基及びスルホン基等のいずれか1種以上を有するものである。なお、2以上の有機基が結合している場合は、当該結合は、有機基の主骨格間に形成されたものでも、また、有機基の主骨格と上述の官能基との間、あるいは、上記官能基間に形成されたものであっても良い。
【0062】
上記鎖状オニウムカチオンとしては、例えば、テトラメチルアンモニウム、テトラエチルアンモニウム、テトラプロピルアンモニウム、テトラブチルアンモニウム、テトラヘプチルアンモニウム、テトラヘキシルアンモニウム、テトラオクチルアンモニウム、トリエチルメチルアンモニウム、メトキシエチルジエチルメチルアンモニウム、トリメチルフェニルアンモニウム、ベンジルトリメチルアンモニウム、ベンジルトリエチルアンモニウム、ベンジルトリブチルアンモニウム、ジメチルジステアリルアンモニウム、ジアリルジメチルアンモニウム、(2−メトキシエトキシ)メチルトリメチルアンモニウム、ジエチルメチル(2−メトキシエチル)アンモニウム、テトラキス(ペンタフルオロエチル)アンモニウム等の第4級アンモニウム類、トリメチルアンモニウム、トリエチルアンモニウム、トリブチルアンモニウム、ジエチルメチルアンモニウム、ジメチルエチルアンモニウム、ジブチルメチルアンモニウム等の第3級アンモニウム類、ジメチルアンモニウム、ジエチルアンモニウム、ジブチルアンモニウム等の第2級アンモニウム類、メチルアンモニウム、エチルアンモニウム、ブチルアンモニウム、ヘキシルアンモニウム、オクチルアンモニウム等の第1級アンモニウム類、N−メトキシトリメチルアンモニウム、N−エトキシトリメチルアンモニウム、N−プロポキシトリメチルアンモニウム及びNH4等のアンモニウム化合物等が挙げられる。これら例示の鎖状オニウムカチオンの中でも、アンモニウム、トリメチルアンモニウム、トリエチルアンモニウム、トリブチルアンモニウム、トリエチルメチルアンモニウム、テトラエチルアンモニウムおよびジエチルメチル(2−メトキシエチル)アンモニウムが好ましい鎖状オニウムカチオンとして挙げられる。
【0063】
上記オニウムカチオンを含む塩としては、上記オニウムカチオンのハロゲン化物、水酸化物、炭酸化物および炭酸水素化物などが挙げられる。また、カチオン交換工程の反応系中でオニウムカチオンを含む塩が生成するような化合物を原料として用いてもよい。
【0064】
カチオン交換工程で使用可能な溶媒としては、上記フッ素化工程で例示したものが挙げられる。
【0065】
カチオン交換工程の実施時期は特に限定されるものではなく、状況に応じて任意の段階で実施することができる。例えば、フッ素化工程前に実施してもよく、また、フッ素化工程後に実施してもよいが、フッ素化工程の後に行うことが好ましい。
【0066】
またカチオン交換工程の実施回数も限定されず、1回、または2回以上繰り返し実施してもよい。例えば、1回のカチオン交換工程により、クロロスルホニルイミド類またはフルオロスルホニルイミド類のカチオンをアルカリ金属カチオンに交換してもよく、また、1回目のカチオン交換工程により、クロロスルホニルイミド類またはフルオロスルホニルイミド類のオニウム塩を得た後、2回目のカチオン交換工程でアルカリ金属塩を得てもよい。
【0067】
なお、フッ素化工程、カチオン交換工程のいずれにおいても、反応溶液に含まれるスルホニルイミド骨格を有する化合物(例えば、フルオロスルホニルイミド、フルオロスルホニルイミド塩など)の濃度は、1質量%〜70質量%とするのが好ましい。濃度が高すぎる場合には、反応が不均一になる虞があり、一方、低すぎる場合には、1バッチあたりの生産性が低く経済的でないからである。より好ましくは3質量%〜60質量%であり、さらに好ましくは5質量%〜50質量%である。
【0068】
また、上記フッ素化工程後には、アルカリ水溶液と接触させる工程を設けてもよい。このアルカリ接触工程を設けることにより、生成物中に含まれる不純物を除去することができる。なお「フッ素化工程後」とは、フッ素化工程直後のみに限られず、フッ素化工程に続けてカチオン交換工程を行った後も「フッ素化工程後」に含まれる。
【0069】
前記アルカリ水溶液としては、塩基性物質の水溶液を使用すればよく、塩基性物質としては、例えば、アンモニア;炭素原子数1〜8のアルキル基を有する第一級、第二級または第三級のアルキルアミン、炭素原子数1〜8のアルキレン基を有するアルキレンジアミンなどの脂肪族アミン;アルカノールアミン;脂環式アミン;芳香族アミン;これらのアミンのエチレンオキサイド付加物;ホルムアミジン;グアニジン;アミジン;複素環式アミン;アルカリ金属、または、アルカリ土類金属の水酸化物、炭酸塩、リン酸塩、ケイ酸塩、ホウ酸塩、ギ酸塩、酢酸塩、ステアリン酸塩、パルミチン酸塩、プロピオン酸塩、シュウ酸塩などが挙げられる。
【0070】
[濃縮工程]
濃縮工程は、カチオン交換工程後の反応溶液から溶媒を除去して、生成したフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程である。本発明では、(1)反応溶液中にガスをバブリングする方法(バブリング法)、及び/又は、(2)薄膜蒸留法により、濃縮工程を実施する。
【0071】
本発明において、濃縮工程とは、得られたフルオロスルホニルイミドのアルカリ金属塩溶液(反応溶液)から一部の溶媒を留去することに加えて、目的物であるフルオロスルホニルイミドのアルカリ金属塩が固体として得られるまで反応溶液から溶媒を留去することも含む。したがって、濃縮工程で得られる生成物は、フルオロスルホニルイミドのアルカリ金属塩の濃縮液、フルオロスルホニルイミドのアルカリ金属塩の固体(粉体)、又は、フルオロスルホニルイミドのアルカリ金属塩の一部が固体状態で存在する濃縮液(スラリー状の溶液)である。
【0072】
まず、(1)バブリング法を採用する濃縮工程について説明する。バブリング法では、反応溶液にガスを流通させることにより蒸発面積を増大させられるので、反応溶媒の蒸発が促進され、速やかに、反応溶液から反応溶媒を除去することができる。この場合、濃縮工程に使用できる反応装置は、反応溶液中にガスを導入する手段、反応溶媒を系外へ排出する手段を備えた装置であればよく、特に限定されない。例えば、槽型反応器、減圧可能な槽型反応器等が挙げられる。
【0073】
反応溶液に流通(バブリング)させるガスとしては、ヘリウム、窒素、アルゴン等の不活性ガス、乾燥空気、およびこれらの混合ガスなどが使用できる。製品品質及び安全性の観点からは、窒素ガスが好ましい。反応溶液へのガスの流通量は、反応溶液中のフルオロスルホニル化合物(フルオロスルホニルイミドのアルカリ金属塩)の濃度に応じて適宜決定すればよいが、例えば、フルオロスルホニルイミドのアルカリ金属塩溶液1gあたり0.001mL/分〜10000mL/分とするのが好ましい。より好ましくは0.005mL/分〜1000mL/分であり、更に好ましくは0.05mL/分〜100mL/分である。なお、反応溶媒の蒸発を促進する観点からは、反応溶液に供給したガスの気泡が、より小さな径となるようにするのが好ましい。なお、気泡を形成する手段は特に限定されず、例えば、供給ガスをガラスフィルター等のフィルターに通過させることで気泡を形成してもよいし、微細ガス発生装置等のガス発生装置を使用してもよい。
【0074】
また、フルオロスルホニルイミドのアルカリ金属塩の濃縮効率を一層高めるため、反応溶液を加熱しながら濃縮工程を行ってもよい。加熱温度は、使用する反応溶媒に応じて適宜設定すればよいが、フルオロスルホニルイミドのアルカリ金属塩の分解を抑制する観点からは、30℃以上、150℃以下とするのが好ましい。より好ましい温度は50℃以上であり、より好ましくは120℃以下である。温度が低すぎると反応溶媒の除去効率が得られず、一方、温度が高すぎるとフルオロスルホニルイミドのアルカリ金属塩が分解してしまう虞がある。
【0075】
バブリング法を採用する濃縮工程におけるその他の条件(例えば、反応溶液へのガスのバブリングに用いるノズル径等)は特に限定されない。上記以外の条件は、使用する装置のサイズや、反応溶液中のフルオロスルホニルイミドのアルカリ金属塩の濃度に応じて適宜決定すればよい。
【0076】
次に、(2)薄膜蒸留法を採用する濃縮工程について説明する。
【0077】
薄膜蒸留法とは、被処理液の薄膜を形成し、これを加熱して、被処理液に含まれる成分を蒸発分と非蒸発分とに分離する方法である。したがって、薄膜蒸留法を採用する濃縮工程では、薄膜蒸留により、カチオン交換工程後の反応溶液から溶媒を分離し、フルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する。
【0078】
この場合、濃縮工程は、薄膜蒸留器を使用して実施する。薄膜蒸留器としては、反応溶液の薄膜を形成する手段、形成された薄膜を加熱する手段、蒸発分(反応溶媒)を回収する手段、非蒸発分(フルオロスルホニルイミドのアルカリ金属塩)を回収する手段を備えた装置であればよい。また、薄膜蒸留器から抜き出された濃縮液を再び薄膜蒸留器に戻す循環手段を備えていてもよい。これにより、繰り返し濃縮を行うことができる。
【0079】
薄膜を形成する方法も特に限定されず、流下式、遠心式、攪拌式、回転式、ブレード式、上昇式等、従来公知の方法はいずれも採用することができる。具体的な薄膜蒸留器としては、例えば、「短行程蒸留装置」(UIC GmbH 社製)、「ワイプレン(登録商標)」、「エクセバ(登録商標)」(株式会社神鋼環境ソリューション製)、「コントロ」、「傾斜翼コントロ」、「セブコン(登録商標)」(以上、株式会社日立プラントテクノロジー製)、「ハイエバオレータ(登録商標)」(株式会社櫻製作所製)、「薄膜蒸留器」、「ビスコン」、「フィルムトルーダー」(以上、木村化工機株式会社製)、「Hi−Uブラッシャー」、「エバリアクター」、「リカバリー」(以上、関西化学機械製作株式会社製)、「NRH」(日南機械株式会社製)、「エバポール(登録商標)」(株式会社大河原製作所製)等が挙げられる。
【0080】
薄膜蒸留の際の温度は、使用する反応溶媒に応じて適宜設定すればよいが、フルオロスルホニルイミドのアルカリ金属塩の分解を抑制する観点からは、30℃以上、150℃以下とするのが好ましい。より好ましい温度は40℃以上であり、更に好ましくは50℃以上であり、より好ましくは120℃以下であり、更に好ましくは110℃以下である。薄膜温度が低すぎると反応溶媒の除去効率が得られず、一方、温度が高すぎるとフルオロスルホニルイミドのアルカリ金属塩が分解してしまう虞がある。また、薄膜蒸留法を採用する場合も、フルオロスルホニルイミドのアルカリ金属塩に与えられる熱量は上記範囲とするのが好ましい。
【0081】
また、フルオロスルホニルイミドのアルカリ金属塩の濃縮効率を一層高めるため、薄膜蒸留器内にガスを流通させながら濃縮工程を実施してもよい。ガスとしては、窒素、アルゴンなどの不活性ガスを用いるのが好ましく、より好ましくは窒素である。
【0082】
薄膜蒸留法による濃縮工程におけるその他の条件は特に限定されない。例えば、薄膜蒸留器内への反応溶液の供給速度は、使用する装置のサイズや、反応溶液中のフルオロスルホニルイミドのアルカリ金属塩の濃度に応じて適宜決定すればよい。
【0083】
バブリング法、薄膜蒸留法のいずれを採用する場合も、効率のよい濃縮工程を実施する観点からは、減圧下で濃縮工程を行ってもよい。減圧度をコントロールすることによって、低温であっても効率よく反応溶媒を除去でき、また、熱によるフルオロスルホニルイミドのアルカリ金属塩の分解も防ぐことができる。減圧度は反応溶媒の種類に応じて適宜調整すればよく特に限定はされないが、例えば、40kPa以下とするのが好ましい。より好ましくは15kPa以下であり、更に好ましくは5kPa以下である。
【0084】
なお、反応溶媒の量が多い場合は、濃縮工程の前に、一部の反応溶媒を除去しておいてもよい。フルオロスルホニルイミドのアルカリ金属塩と溶媒との相互作用が顕著になって反応溶液から反応溶媒の除去が困難になるのは、フルオロスルホニルイミドのアルカリ金属塩に対する反応溶媒の量が150質量%以下となる時点からであるので、可能な限り反応溶液量を低減しておくことで、効率よく濃縮工程が実施できるからである。また、濃縮工程は、反応溶液を攪拌しながら実施してもよい。濃縮工程では、上記バブリング法、薄膜蒸留法の両方を順に実施してもよく(実施の順序は特に限定されない)、また、いずれか一方を単独で実施してもよい。なお、フルオロスルホニルイミドのアルカリ金属塩の熱分解を防ぐ観点からは、より短時間で濃縮工程を実施できる薄膜蒸留法が好ましい。
【0085】
濃縮工程における加熱によるフルオロスルホニルイミドのアルカリ金属塩の分解を防ぐ観点からは、上記バブリング法、薄膜蒸留法のいずれを採用する場合も、濃縮工程で加えられる熱量は、フルオロスルホニルイミドのアルカリ金属塩1gあたり1,000,000J以下とするのが好ましく、より好ましくは500,000J以下であり、さらに好ましくは100,000J以下である。なお、上記熱量には、濃縮工程に供する前に一部の反応溶媒を除去するために反応溶液に与えられる熱量は含まれない。
【0086】
本発明では、濃縮工程で反応溶液に与えられた熱量を、フルオロスルホニルイミドのアルカリ金属塩に与ええられた熱量とする。なお、上記熱量は、濃縮工程で使用する装置の消費電力(装置メーカーの公証値を参照すればよい)、反応溶液に含まれるフルオロスルホニルイミドのアルカリ金属塩の量および加熱時間に基づいて求めればよく、具体的には、濃縮工程で反応溶液に与えられた熱量を算出し、これを、フルオロスルホニルイミドのアルカリ金属塩1gあたりに与えられた熱量に換算すればよい。
【0087】
[乾燥、粉体化工程]
濃縮工程で得られたフルオロスルホニルイミドのアルカリ金属塩濃縮液は、そのまま製品とすることもできるが、保存時の安定性を高め、また、製品の流通を容易にするため、フルオロスルホニルイミドのアルカリ金属塩を粉体化してもよい(粉体化、乾燥工程)。なお、濃縮工程で、固体状態のフルオロスルホニルイミドのアルカリ金属塩を得た場合には、得られた固体をそのまま乾燥装置で乾燥してもよく、また、フルオロスルホニルイミドのアルカリ金属塩が可溶な溶媒に溶解させた後、乾燥、粉体化工程に供してもよい。
【0088】
フルオロスルホニルイミドのアルカリ金属塩を乾燥し、粉体化する方法は特に限定されず、(1)フルオロスルホニルイミドのアルカリ金属塩が析出するまで上記濃縮工程を継続し、これを分離し、乾燥して粉体化する方法、(2)濃縮工程で得られた濃縮液をそのまま、あるいは、必要により30℃以下に冷却しながら静置して、フルオロスルホニルイミドのアルカリ金属塩を析出させ、これを分離し、乾燥して粉体化する方法、(3)濃縮液に溶媒を添加してフルオロスルホニルイミドのアルカリ金属塩を析出させ、これをろ別して分離し、乾燥して粉体化する方法、等が挙げられる。
【0089】
上記(3)で使用可能な溶媒は、上述した反応溶媒以外の溶媒であって、フルオロスルホニルイミドのアルカリ金属塩と溶媒和を形成し難いものであればよい。具体的に(3)の方法で使用可能な溶媒としては、トルエン、o−キシレン、m−キシレン、p−キシレン、エチルベンゼン、イソプロピルベンゼン、1,2,4−トリメチルベンゼン、1,3,5−トリメチルベンゼン、1,2,3−トリメチルベンゼン、クロロベンゼン、ジクロロベンゼン等の芳香族系炭化水素溶媒、ヘキサン、ヘプタン、オクタン、ノナン、デカン、ウンデカン、ドデカン、デカリン、ジクロロメタン等の脂肪族炭化水素溶媒が挙げられる。また、これらの溶媒は、濃縮液1質量部に対して、20質量倍以下、より好ましくは10質量倍以下の量で加えるのが好ましい。
【0090】
次いで、傾斜法、遠心分離法、濾過法等により、析出したフルオロスルホニルイミドのアルカリ金属塩を反応溶媒などから分離し、乾燥させる。フルオロスルホニルイミドのアルカリ金属塩の乾燥方法は特に限定されず、従来公知の乾燥装置が使用できる。乾燥時の温度は0℃〜100℃とするのが好ましい。より好ましくは10℃以上、さらに好ましくは20℃以上であり、より好ましくは80℃以下、更に好ましくは60℃以下である。
【0091】
また、フルオロスルホニルイミドのアルカリ金属塩の乾燥は、乾燥装置にガスを供給しながら行ってもよい。使用可能なガスとしては、濃縮工程で使用したものが挙げられるが、例えば、窒素、アルゴンなどの不活性ガスや乾燥空気が挙げられる。
【0092】
[回収工程]
本発明の製造方法では、上記各工程で生成物から分離されたフルオロスルホニルイミドのアルカリ金属塩やスルホニルイミド骨格を有する化合物を回収する工程を設けてもよい。特に、薄膜蒸留法を採用する濃縮工程で排出される廃液や、上記粉体化、乾燥工程で析出したフルオロスルホニルイミドのアルカリ金属塩を除去した溶液(母液)には、溶解したフルオロスルホニルイミドのアルカリ金属塩が含まれているので、これを回収することでフルオロスルホニルイミドのアルカリ金属塩の収率を向上させることができる。
【0093】
また、上記乾燥、粉体化工程で得られたフルオロスルホニルイミドのアルカリ金属塩の純度が低い場合には、これを単独で精製してもよいが、固体状態(粉体)のフルオロスルホニルイミドのアルカリ金属塩を回収溶液(上記廃液や母液)と混合してもよい。上記乾燥、粉体化工程における操作は、晶析、再沈殿法などの精製操作にも相当するので、廃液や母液からのフルオロスルホニルイミドのアルカリ金属塩の回収と共に、フルオロスルホニルイミドのアルカリ金属塩の純度を向上させられるからである。
【0094】
なお、回収したフルオロスルホニルイミドのアルカリ金属塩の精製方法は特に限定されず、各工程から回収した溶液を単独で、あるいは、混合して精製してフルオロスルホニルイミドのアルカリ金属塩を回収してもよく、また、回収溶液は、カチオン交換工程、濃縮工程、粉体化、乾燥工程のいずれかの工程に供給してもよい。なお、生産性の観点からは、回収溶液は濃縮工程に供給するのが好ましい。
【0095】
上記方法により得られたフルオロスルホニルイミドのアルカリ金属塩は、必要に応じて純度をさらに向上させるための精製工程に供してもよい。精製工程としては、従来公知の精製方法はいずれも採用可能である。
【0096】
<電解液>
本発明には、上記フルオロスルホニルイミドのアルカリ金属塩と媒体とを含む電解液も含まれる。本発明のフルオロスルホニルイミドのアルカリ金属塩は、上述のように、不純なイオンや水分の含有量が低減されているので、電解液の分解が生じ難いものである。したがって、本発明の電解液をイオン伝導体として備えた電気化学デバイスは長期に亘って安定した特性を有する電気化学デバイスとすることができる。
【0097】
上記媒体としては、非プロトン性溶媒、ポリマー等が挙げられる。非プロトン性有機溶媒としては、誘電率が大きく、電解質塩(フルオロスルホニルイミドのアルカリ金属塩、後述する他の電解質)の溶解性が高く、沸点が60℃以上であり、且つ、電気化学的安定範囲が広い溶媒が好適である。より好ましくは、含有水分量が低い有機溶媒(非水系溶媒)である。具体的に、含水量は250ppm以下であるのが好ましく、100ppm以下であるのがより好ましく、50ppm以下であるのがさらに好ましい。含有水分量が低い有機溶媒としては、市販の脱水溶媒等を使用することができる。このような有機溶媒としては、エチレングリコールジメチルエーテル(1,2−ジメトキシエタン)、エチレングリコールジエチルエーテル、テトラヒドロフラン、2−メチルテトラヒドロフラン、2,6−ジメチルテトラヒドロフラン、テトラヒドロピラン、クラウンエーテル、トリエチレングリコールジメチルエーテル、テトラエチレングリコールジメチルエ−テル、1,4−ジオキサン、1,3−ジオキソラン等のエーテル類;炭酸ジメチル、炭酸エチルメチル(メチルエチルカーボネート)、炭酸ジエチル(ジエチルカーボネート)、炭酸ジフェニル、炭酸メチルフェニル等の鎖状炭酸エステル類;炭酸エチレン(エチレンカーボネート)、炭酸プロピレン(プロピレンカーボネート)、2,3−ジメチル炭酸エチレン、炭酸ブチレン、炭酸ビニレン、2−ビニル炭酸エチレン等の環状炭酸エステル類;蟻酸メチル、酢酸メチル、プロピオン酸、プロピオン酸メチル、酢酸エチル、酢酸プロピル、酢酸ブチル、酢酸アミル等の脂肪族カルボン酸エステル類;安息香酸メチル、安息香酸エチル等の芳香族カルボン酸エステル類;γ−ブチロラクトン、γ−バレロラクトン、δ−バレロラクトン等のカルボン酸エステル類;リン酸トリメチル、リン酸エチルジメチル、リン酸ジエチルメチル、リン酸トリエチル等のリン酸エステル類;アセトニトリル、プロピオニトリル、メトキシプロピオニトリル、グルタロニトリル、アジポニトリル、2−メチルグルタロニトリル、バレロニトリル、ブチロニトリル、イソブチロニトリル等のニトリル類;N−メチルホルムアミド、N−エチルホルムアミド、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリジノン、N−メチルピロリドン、N−ビニルピロリドン等のアミド類;ジメチルスルホン、エチルメチルスルホン、ジエチルスルホン、スルホラン、3−メチルスルホラン、2,4−ジメチルスルホラン等の硫黄化合物類:エチレングリコール、プロピレングリコール、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル等のアルコール類;ジメチルスルホキシド、メチルエチルスルホキシド、ジエチルスルホキシド等のスルホキシド類;ベンゾニトリル、トルニトリル等の芳香族ニトリル類;ニトロメタン、1,3−ジメチル−2−イミダゾリジノン、1,3−ジメチル−3,4,5,6−テトラヒドロ−2(1H)−ピリミジノン、3−メチル−2−オキサゾリジノン等を挙げることができ、これらの1種又は2種以上が好適である。これらの中でも、炭酸エステル類、脂肪族カルボン酸エステル類、カルボン酸エステル類、エーテル類がより好ましく、炭酸エステル類がさらに好ましい。
【0098】
媒体として用いられるポリマーには、ポリエチレンオキシド(PEO)、ポリプロピレンオキシドなどのポリエーテル系ポリマー、ポリメチルメタクリレート(PMMA)などのメタクリル系ポリマー、ポリアクリロニトリル(PAN)等のニトリル系ポリマー、ポリフッ化ビニリデン(PVDF)、ポリフッ化ビニリデン−ヘキサフルオロプロピレンなどのフッ素系ポリマー、および、これらの共重合体等が含まれる。また、これらのポリマーと他の有機溶媒とを混合したポリマーゲルも本発明に係る媒体として用いることができる。他の有機溶媒としては上述の非プロトン性溶媒が挙げられる。
【0099】
上記ポリマーゲルを媒体とする場合の電解液の製造方法としては、従来公知の方法で成膜したポリマーに、上述の非プロトン性溶媒にフルオロスルホニルイミドのアルカリ金属塩等の電解質を溶解させた溶液を滴下して、電解質並びに非プロトン性溶媒を含浸、担持させる方法;ポリマーの融点以上の温度でポリマーと電解質とを溶融、混合した後、成膜し、ここに非プロトン性溶媒を含浸させる方法;予め非プロトン性溶媒に溶解させた電解質溶液とポリマーとを混合した後、これをキャスト法やコーティング法により成膜し、非プロトン性溶媒を揮発させる方法(以上、ゲル電解質);ポリマーの融点以上の温度でポリマーと電解質とを溶融し、混合して成形する方法(真性ポリマー電解質);等が挙げられる。
【0100】
媒体の使用量は、電解質(フルオロスルホニルイミドのアルカリ金属塩と他の電解質)と媒体の合計量100質量部に対して、50質量部〜99.9質量部であるのが好ましい。より好ましくは60質量部〜99.5質量部であり、さらに好ましくは70質量部〜99質量部である。媒体量が少なすぎると、充分なイオン伝導度が得られ難い場合があり、一方、多すぎると、溶媒の揮発によるイオン伝導性材料中のイオン濃度が変化し易くなり、安定したイオン伝導度が得られ難い場合がある。
【0101】
本発明に係る電解液には、上記フルオロスルホニルイミドのアルカリ金属塩のみが電解質として含まれていてもよいが、フルオロスルホニルイミドのアルカリ金属塩に加え、これ以外の他の電解質が含まれていてもよい。他の電解質を用いることで、電解液中のイオンの絶対量を増加させることができ、電気伝導度の向上を図ることができる。
【0102】
他の電解質としては、電解液中での解離定数が大きく、また、上記非プロトン性溶媒と溶媒和し難いアニオンを有するものが好ましい。他の電解質を構成するカチオン種としては、例えば、Li+、Na+、K+等のアルカリ金属イオン、Ca2+、Mg2+等のアルカリ土類金属イオンおよびオニウムカチオンが挙げられ、特に、リチウムイオンが好ましい。一方、アニオン種としては、PF6-、BF4-、ClO4-、AlCl4-、C[(CN)3]-、N[(CN)2]-、B[(CN)4]-、N[(SO2CF3)2]-、CF3(SO3)-、C[(CF3SO2)3]-、AsF6-、SbF6-およびジシアノトリアゾレートイオン(DCTA)などが挙げられる。これらの中でも、PF6-、BF4-がより好ましく、PF6-が特に好ましい。
【0103】
上記他の電解質の存在量としては、フルオロスルホニルイミドのアルカリ金属塩と他の電解質との合計100質量%中、0.1質量%、99質量%以下であることが好適である。他の電解質量が少なすぎる場合には、他の電解質を用いた効果(たとえばイオンの絶対量が充分なものとならず、電気伝導度が小さくなる)が得られ難い場合があり、他の電解質量が多すぎる場合には、イオンの移動が大きく阻害される虞がある。より好ましくは1質量%以上、さらに好ましくは5質量%以上であり、より好ましくは95質量%以下、さらに好ましくは90質量%以下である。
【0104】
なお、本発明に係る電解液中における電解質濃度(フルオロスルホニルイミドのアルカリ金属塩と他の電解質の総量)は、0.1質量%以上が好ましく、また、飽和濃度以下が好ましい。0.1質量%未満であると、イオン伝導度が低くなるため好ましくない。より好ましくは、0.5質量%以上であり、さらに好ましくは1質量%以上である。また、電解液中における電解質濃度は、50質量%未満であるのがより好ましく、さらに好ましくは40質量%以下、更に一層好ましくは30質量%以下である。
【0105】
本発明の電解液は、硫酸イオンの含有量が1500ppm以下であるのが好ましい。上述のように硫酸イオンは本発明の電解液にとって不純物に相当し、斯かる不純物の含有量が多い場合には、本発明の電解液を蓄電デバイスに用いた場合に所期の特性が得られ難い場合がある。本発明の電解液中の硫酸イオン量は500ppm以下であるのがより好ましく、さらに好ましくは250ppm以下である。なお、硫酸イオン含有量は低いほどよく、0ppmであるのが最も好ましいが、例えば、硫酸イオン含有量の下限は0.5ppm程度であればよく、また1ppm以下であってもよい。この範囲であれば上述のような問題が生じ難いからである。
【0106】
本発明の電解液は、硫酸イオンに加えて、水、溶媒、フッ素イオン、塩化物イオン、カリウムイオンの含有量も低減されたものであるのが好ましい。具体的に、水の含有量は、本発明の電解液中250ppm以下であるのが好ましく、より好ましくは150ppm以下であり、さらに好ましくは50ppm以下である。残留溶媒の含有量は、本発明の組成物中2000ppm以下であるのが好ましく、より好ましくは1500ppm以下である。フッ素イオンの含有量は本発明の電解液中500ppm以下であるのが好ましく、より好ましくは400ppm以下であり、さらに好ましくは150ppm以下である。塩化物イオンの含有量は本発明の電解液中100ppm以下であるのが好ましく、より好ましくは50ppm以下である。カリウムイオンの含有量は本発明の電解液中5000ppm以下であるのが好ましく、より好ましくは4000ppm以下であり、より一層好ましくは2000ppm以下であり、更に好ましくは500ppm以下、更に一層好ましくは250ppm以下、特に100ppm以下であるのが好ましく、50ppm以下であるのが最も好ましい。
【0107】
なお、本発明の電解液中における水、溶媒、フッ素イオン、塩化物イオンおよびカリウムイオンの含有量はいずれも0ppmであることが最も好ましいが、例えば、下限は夫々0.5ppm程度であればよく、また、1ppm程度であってもよい。水、溶媒およびフッ素イオンの含有量の含有量が上記範囲であれば、本発明の電解液を各種電気化学デバイスに用いても、これらの不純物に由来する問題が生じ難いからである。
【0108】
尚、本発明の電解液に媒体として含まれるものは上記残留溶媒には含まれない。すなわち、上記残留溶媒に含まれる溶媒とは、フルオロスルホニルイミドのアルカリ金属塩の製造工程で使用された反応溶媒や水である。ちなみに、製造工程で使用された反応溶媒と電解液の媒体として含まれる溶媒とが同一である場合、当該溶媒は、残留溶媒には含まれないものとする。
【0109】
本発明の電解液中に含まれる上記イオン、水、残留溶媒の含有量は、例えばICP発光分光分析法、イオンクロマトグラフィー、カールフィッシャー水分計、ガスクロマトグラフィーにより測定することができる。
【実施例】
【0110】
以下、実験例を挙げて本発明をより具体的に説明するが、本発明はもとより下記実験例によって制限を受けるものではなく、前・後記の趣旨に適合し得る範囲で適当に変更を加えて実施することも勿論可能であり、それらはいずれも本発明の技術的範囲に包含される。
【0111】
[NMR測定]
1H−NMR、19F−NMRの測定は、Varian社製の「Unity Plus−400」を使用して行った(内部標準物質:トリフルオロメチルベンゼン、溶媒:重アセトニトリル、積算回数:16回)。
【0112】
[ICP発光分光分析法]
下記例で得られたフルオロスルホニルイミド塩0.1gを超純水9.9gと混合した濃度1質量%の水溶液を測定試料とし、マルチタイプICP発光分光分析装置(島津製作所製「ICPE−9000」)を使用した。
【0113】
実験例1
実験例1−1
〔フッ素化工程〕
攪拌装置を備えたパイレックス(登録商標)製反応容器A(内容量10L)に、窒素気流下で酢酸ブチル1800gを加え、ここに200g(934mmol)のビス(クロロスルホニル)イミドを室温(25℃)で滴下した。
【0114】
得られたビス(クロロスルホニル)イミドの酢酸ブチル溶液に、室温で、フッ化亜鉛101g(982mmol、ビス(クロロスルホニル)イミドに対して1.05当量)を一度に加え、これが完全に溶解するまで室温で6時間攪拌した。
【0115】
〔カチオン交換工程1−アンモニウム塩の合成〕
攪拌装置を備えたパイレックス(登録商標)製反応容器B(内容量10L)に、25質量%アンモニア水540g(7928mmol、ビス(クロロスルホニル)イミドに対して8.49当量)を加えた。アンモニア水の攪拌下、室温で、反応容器Bに、反応容器Aの反応溶液を滴下して加えた。反応溶液の滴下終了後、攪拌を停止し、水層と酢酸ブチル層の2層に分かれた反応溶液から、塩化亜鉛などの副生物を含む水層を除去し、有機層として、アンモニウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液を得た。得られた有機層を試料として、19F-NMR(溶媒:重アセトニトリル)測定を行った。得られたチャートにおいて、内部標準物質として加えたトリフルオロメチルベンゼンの量、及び、これに由来するピークの積分値と、目的生成物に由来するピークの積分値との比較から、有機層に含まれるアンモニウムビス(フルオロスルホニル)イミドの粗収量を求めた(756mmol)。
19F-NMR(溶媒:重アセトニトリル):δ56.0
【0116】
〔カチオン交換工程2−リチウム塩の合成〕
得られた有機層に含まれるアンモニウムビス(フルオロスルホニル)イミドに対して、リチウムの量が2当量となるように、15質量%の水酸化リチウム水溶液242g(Liとして1516mmol)を加え、室温で10分間攪拌した。その後、反応溶液から水層を除去して、リチウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液を得た。
【0117】
得られた有機層を試料とし、ICP発光分光分析法により、フルオロスルホニルイミドのアンモニウムカチオンがリチウムイオンに交換されていることを確認した。また、有機層中のリチウムビス(フルオロスルホニル)イミド濃度は7質量%であった(収量:127g、収率:73%)。
【0118】
なお、フルオロスルホニルイミドの濃度は、得られた有機層を試料として、19F-NMR(溶媒:重アセトニトリル)測定を行い、測定結果のチャートにおいて、内部標準物質として加えたトリフルオロメチルベンゼンの量、及び、これに由来するピークの積分値と、目的生成物に由来するピークの積分値との比較から求めた。
【0119】
[濃縮工程]
まず、カチオン交換工程で得られたリチウムビス(フルオロスルホニル)イミド溶液をロータリーエバポレーター(「REN−1000」、IWAKI社製)に加えて、減圧下で溶媒を留去し、リチウムビス(フルオロスルホニル)イミド溶液282gを得た(濃度:45質量%)。
【0120】
次いで、ガス導入管と減圧装置を備えたロータリーエバポレーター(「REN−1000」、IWAKI製)に、45質量%の濃度のリチウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液200gを入れたフラスコ(容量:500mL)を装着した。フラスコ内の液中に500mL/分で窒素ガスを吹き込みながら、60℃に設定した恒温水槽で加熱しながら回転を開始させた(100rpm)。次いで、装置内を933Paまで徐々に減圧し、12時間濃縮工程を行った。得られた溶液の濃度は72質量%であった。濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、72,000Jであった。
【0121】
[粉体化、乾燥工程]
得られた濃縮液125gに、トルエン125gを加えて、25℃で1時間静置し、リチウムビス(フルオロスルホニル)イミドの固体を析出させた。得られた固体をろ取し、これを50℃で真空乾燥することで、リチウムビス(フルオロスルホニル)イミドを得た(収量:68g、収率:76%(濃縮工程より))。
【0122】
実験例1−2
外径15mmの試験管にガス導入管及び減圧装置を装着した。試験管の中に、実験例1−1のカチオン交換工程2で得られた45質量%の濃度のリチウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液5gを入れ、試験管内を2666Paまで徐々に減圧した。減圧度を保ったまま更に標準状態で1mL/分の流量で窒素ガスを液中に吹き込みながら、65℃に設定した恒温水槽で加熱しながら濃縮を開始した。2.5時間濃縮工程を行ったところ、得られた溶液のリチウムビス(フルオロスルホニル)イミドの濃度は85質量%であった。濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、17,000Jであった。
【0123】
実験例1−3
濃縮工程において、窒素ガスに代えて空気を使用したこと(供給速度:標準状態で約1mL/分)、恒温水槽の温度を75℃に変更したこと、減圧度を667Paとしたこと以外は、実験例1−2と同様にして、リチウムビス(フルオロスルホニル)イミドの製造を行った。このときの濃縮工程の実施時間は2時間であり、濃縮工程で得られた溶液のリチウムビス(フルオロスルホニル)イミド濃度は88質量%であった。濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、15,000Jであった。
【0124】
実験例1−4
濃縮工程において、窒素ガスに代えて空気を使用したこと(供給速度:標準状態で約1mL/分)、恒温水槽の温度を50℃に変更したこと、減圧度を667Paとしたこと以外は、実験例1−2と同様にして、リチウムビス(フルオロスルホニル)イミドの製造を行った。このときの濃縮工程の実施時間は10時間であり、濃縮工程で得られた溶液のリチウムビス(フルオロスルホニル)イミド濃度は84質量%であった。濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、50,000Jであった。
【0125】
実験例1−5
濃縮工程において、窒素ガスを吹き込まなかったこと、恒温水槽の温度を50℃としたこと、減圧度を667Paとしたこと以外は、実験例1−2と同様にして、20時間濃縮工程を行った。得られた溶液の濃度は60質量%であった。濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、100,000Jであった。
【0126】
表1に実験例1−1〜1−5の濃縮条件と結果、図1に、実験例1−2〜1−5の結果を示す。図1では、濃縮時間に対して反応溶媒量をプロットした。
【0127】
【表1】
【0128】
実験例1−2〜1−4と実験例1−5の対比より、バブリングしながら実施する濃縮工程を含む本発明法によれば、生成物であるフルオロスルホニルイミドのアルカリ金属塩を短時間で濃縮できることが分かる。また、濃縮工程を減圧下で行うことで、より一層効率よくフルオロスルホニルイミドのアルカリ金属塩を濃縮できることが分かる。さらに、実験例1−1と1−5の対比より、バブリングしながら濃縮工程を実施すれば、スケールアップしても効率よくフルオロスルホニルイミドのアルカリ金属塩を濃縮できることが分かる。
【0129】
実験例2
実験例2−1
〔フッ素化工程〕
攪拌装置を備えたパイレックス(登録商標)製反応容器A(内容量5L)に、窒素気流下で酢酸ブチル900gを加え、ここに100g(467mmol)のビス(クロロスルホニル)イミドを室温(25℃)で滴下した。
【0130】
得られたビス(クロロスルホニル)イミドの酢酸ブチル溶液に、室温で、フッ化亜鉛50.5g(491mmol、ビス(クロロスルホニル)イミドに対して1.05当量)を一度に加え、これが完全に溶解するまで室温で6時間攪拌した。
【0131】
〔カチオン交換工程1−アンモニウム塩の合成〕
攪拌装置を備えたパイレックス(登録商標)製反応容器B(内容量1L)に、25質量%アンモニア水270g(3964mmol、ビス(クロロスルホニル)イミドに対して8.49当量)を加えた。アンモニア水の攪拌下、室温で、反応容器Bに、反応容器Aの反応溶液を滴下して加えた。反応溶液の滴下終了後、攪拌を停止し、水層と酢酸ブチル層の2層に分かれた反応溶液から、塩化亜鉛などの副生物を含む水層を除去し、有機層として、アンモニウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液を得た。得られた有機層を試料として、19F-NMR(溶媒:重アセトニトリル)測定を行った。得られたチャートにおいて、内部標準物質として加えたトリフルオロメチルベンゼンの量、及び、これに由来するピークの積分値と、目的生成物に由来するピークの積分値との比較から、有機層に含まれるアンモニウムビス(フルオロスルホニル)イミドの粗収量を求めた(378mmol)。
19F-NMR(溶媒:重アセトニトリル):δ56.0
【0132】
〔カチオン交換工程2−リチウム塩の合成〕
得られた有機層に含まれるアンモニウムビス(フルオロスルホニル)イミドに対して、リチウムの量が2当量となるように、15質量%の水酸化リチウム水溶液121g(Liとして758mmol)を加え、室温で10分間攪拌した。その後、反応溶液から水層を除去して、リチウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液を得た。
【0133】
得られた有機層を試料とし、ICP発光分光分析法により、フルオロスルホニルイミドのプロトンがリチウムイオンに交換されていることを確認した。また、有機層中のリチウムビス(フルオロスルホニル)イミド濃度は7質量%であった(収量:63.5g、収率:73%)。
【0134】
なお、フルオロスルホニルイミドの濃度は、得られた有機層を試料として、19F-NMR(溶媒:重アセトニトリル)測定を行い、測定結果のチャートにおいて、内部標準物質として加えたトリフルオロメチルベンゼンの量、及び、これに由来するピークの積分値と、目的生成物に由来するピークの積分値との比較から求めた。
【0135】
[濃縮工程]
カチオン交換工程で得られたリチウムビス(フルオロスルホニル)イミド溶液をロータリーエバポレーター(「REN−1000」、IWAKI社製)に加えて、減圧下で溶媒を留去し、リチウムビス(フルオロスルホニル)イミド溶液141gを得た(濃度:45質量%)。
【0136】
次いで、短行程蒸留装置(型式「KDL1」、攪拌式、蒸発面積:0.01m2、UIC GmbH 社製)を使用して、加熱面の温度(薄膜温度)100℃、圧力1.333kPa、ロータ回転数300rpmで、45質量%のリチウムビス(フルオロスルホニル)イミド溶液120gを供給速度2g/分で、装置内に供給し、薄膜蒸留を実施した後、装置を停止し、濃縮液を抜き出した。得られた溶液のリチウムビス(フルオロスルホニル)イミド濃度は80質量%であった。濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、20,000Jであった。
【0137】
[乾燥、粉体化工程]
次いで、得られた濃縮液67.5gに、トルエン600gを加えて、25℃で1時間静置し、リチウムビス(フルオロスルホニル)イミドの固体を析出させた。得られた固体をろ取し、これを50℃で真空乾燥することで、リチウムビス(フルオロスルホニル)イミドを得た(収量:38g、濃縮工程からの収率:56%)。このとき、ろ液には、濃縮工程からの収率で44質量%に相当するリチウムビス(フルオロスルホニル)イミドが含まれていた。なお、ろ液に含まれるリチウムビス(フルオロスルホニル)イミドは、ろ液を再び濃縮工程、粉体化、乾燥工程に供することで回収することができる。
【0138】
実験例2−2
濃縮工程において、薄膜温度を75℃に変更したこと以外は、実験例2−1と同様にして、リチウムビス(フルオロスルホニル)イミドの製造を行った。このときのリチウムビス(フルオロスルホニル)イミド溶液の供給速度は2g/分(薄膜蒸留時間:1時間)であり、濃縮工程で得られた溶液の濃度は63質量%であった。また、濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、15,000Jであった。
【0139】
実験例2−3
濃縮工程において、薄膜温度を50℃に変更したこと以外は、実験例2−1と同様にして、リチウムビス(フルオロスルホニル)イミドの製造を行った。このときのリチウムビス(フルオロスルホニル)イミド溶液の供給速度は2g/分(薄膜蒸留時間:1時間)であり、濃縮工程で得られた溶液の濃度は58質量%であった。また、濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、10,000Jであった。
【0140】
実験例2−4
短行程蒸留装置に代えて、ロータリーエバポレーター(「REN−1000」、IWAKI社製)を使用した。45質量%の濃度のリチウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液10gを入れたフラスコをロータリーエバポレーターに装着し、これを50℃に設定した恒温水槽で加熱しながら回転を開始し(100rpm)、系内を667Paにまで減圧して、20時間濃縮工程を行った。得られた溶液の濃度は60質量%であった。また、濃縮工程で加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、100,000Jであった。
【0141】
実験例2−1〜2−3と実験例2−4の対比より、薄膜蒸留による濃縮工程を含む実験例2−1〜2−3では、薄膜蒸留法を採用しない実験例2−4に比べて、生成物であるリチウムビス(フルオロスルホニル)イミドを短時間で濃縮できることが分かる。
【0142】
実験例3
実験例2−1と同様にしてフッ素化工程およびカチオン交換工程1,2を実施して得られたリチウムビス(フルオロスルホニル)イミド溶液をロータリーエバポレーター(「REN−1000」、IWAKI社製)に加えて、減圧下で溶媒を留去し、リチウムビス(フルオロスルホニル)イミド溶液140gを得た(濃度45質量%)。得られた溶液を、テフロン(登録商標)コーティングされたステンレス鋼製のバット(長さ:29cm、幅22cm、高さ5cm)に流し込み、これを棚段減圧乾燥機に入れ、温度50℃、減圧度150Paで7日間乾燥を行って、リチウムビス(フルオロスルホニル)イミドの固体を得た(収量:63g)。濃縮時に加えられた熱量は、リチウムビス(フルオロスルホニル)イミド1gに対して、1,200,000Jであった。
【0143】
実験例4
下記方法に従って、実験例2−1および実験例3で得られたフルオロスルホニルイミドのアルカリ金属塩の耐熱性(質量減少率、2%質量減少温度)、および、不純物含有量を測定した。
【0144】
上記手順によりNMR測定を行い、実験例2−1および実験例3で得られたフルオロスルホニルイミドのアルカリ金属塩には、未反応の原料や反応途中の化合物は含まれておらず、いずれもフッ素化、カチオン交換されたものであることを確認した。
【0145】
[耐熱性1 質量減少率]
アルミパンに約20mgの試料を量り取り、示差熱熱重量同時測定装置(「EXSTAR TG/DTA6200」、エスアイアイ・ナノテクノロジー株式会社製)を使用して、乾燥空気気流下(流量200mL/分)、100℃で8時間試料を保持した後の質量減少量を測定した。
【0146】
[耐熱性2 2%及び1%質量減少温度]
アルミパンに量り取った約20mgの試料を、示差熱熱重量同時測定装置(「EXSTAR TG/DTA6200」、エスアイアイ・ナノテクノロジー株式会社製)を使用して、乾燥空気の流量200mL/分、測定温度範囲25℃〜450℃、昇温速度10℃/分で加熱し、質量減少の様子を観察した。
【0147】
なお、耐熱性1,2で使用した試料について、平沼産業株式会社製の「AQ−2000」により水分量を測定したところ、それぞれ64ppm、68ppmであった。
【0148】
[不純物含有量]
上記実験例で得られたフルオロスルホニルイミドのアルカリ金属塩0.01gを超純水(18.2Ω・cm超)で1000倍に希釈して測定溶液とし、イオンクロマトグラフィーシステム ICS−3000(日本ダイオネクス株式会社製)を用いて、フルオロスルホニルイミドのアルカリ金属塩中に含まれるハロゲン化物イオン(F-,Cl-)、および硫酸イオン(SO42-)の量を測定した。
分離モード:イオン交換
溶離液:7〜20mM KOH水溶液
検出器:電気伝導度検出器
カラム:アニオン分析用カラム Ion PAC AS−17C(日本ダイオネクス株式会社製)
なお、カリウムイオン、ナトリウムイオンおよび亜鉛イオン量は、上記ICP発光分光分析法により測定した。
【0149】
【表2】
【0150】
表2より、棚段式減圧乾燥機による濃縮、乾燥を行った実験例3のリチウムビス(フルオロスルホニル)イミドは、実験例2−1に比べて不純物含有量が多く、耐熱性も低いものであった。これは、棚段式減圧乾燥機ではリチウムビス(フルオロスルホニル)イミドから溶媒を除去し、乾燥させるために長時間加熱しなければならず(7日間)、この際にリチウムビス(フルオロスルホニル)イミドがわずかに分解してしまい、生じた分解生成物がリチウムビス(フルオロスルホニル)イミドの耐熱性を低下させているものと考えられる。
【0151】
これに対して、本発明の方法を採用した実験例2−1では、実験例3に比べてリチウムビス(フルオロスルホニル)イミドの耐熱性が30℃以上向上していることが分かる。また、薄膜蒸留法による濃縮工程を採用した実験例2−1のリチウムビス(フルオロスルホニル)イミドは、100℃で8時間保持した後も質量減少は、0%であった。
【0152】
これは、実験例2−1では、実験例3に比べて短い加熱時間でリチウムビス(フルオロスルホニル)イミドを乾燥することができたため、生成物の分解が抑制され、その結果、分解生成物(不純物)に起因する生成物の分解が生じ難くなり、良好な耐熱性を示しているものと考えられる。
【0153】
実験例5
実験例2−1と同様にしてフッ素化工程およびカチオン交換工程1,2を実施して、リチウムビス(フルオロスルホニル)イミドの酢酸ブチル溶液を得た。
実験例5−1〜5−16の各実験例では、実験例2−1と同様にして、フルオロスルホニルイミドのアルカリ金属塩の濃縮工程、乾燥、粉体化工程を行った。
【0154】
得られたフルオロスルホニルイミドのアルカリ金属塩の不純物含有量を、実験例4と同様にして測定した。尚、水の含有量は、下記方法に従って測定した。結果を表3に示す。実験例2−1、実験例3の結果も併せて表3に示す。
【0155】
また、上記NMR測定により、実験例5−1〜5−16で得られたフルオロスルホニルイミドのアルカリ金属塩にも、未反応の原料や反応途中の化合物は含まれておらず、いずれもフッ素化、カチオン交換されたものであることを確認した。
【0156】
[水分含有量]
平沼産業(株)製カールフィッシャー水分測定装置「AQ−2100」を用いて行った。測定試料は、各実験例で得られたフルオロスルホニルイミドのアルカリ金属塩0.3gをメタノールで10倍に希釈して調製した。なお、試料の調整及び測定などの一連の操作は、ドライルーム(温度:25℃、露点:−70℃〜−50℃)で行った。試料注入量は試料の水分含有量に応じて0.1ml〜3mlとし、発生液には「ハイドラナール(登録商標) クローマットAK」(Sigma Aldrich社製)を使用し、対極液には「ハイドラナール(登録商標) クローマットCG−K」(Sigma Aldrich社製)を使用した。試料は、外気に触れないよう注射器を用いて試料注入口より注入した。同様にして、希釈に使用したメタノールの水分含有量を測定し、試料溶液の水分含有量(測定値)から、メタノールの水分含有量を差し引くことで、フルオロスルホニルイミドのアルカリ金属塩の水分含有量を求めた。
【0157】
【表3】
【0158】
表3中、収率とは、濃縮工程からの収率を意味する。尚、実験例5−3〜5−6、5−15〜5−16および実験例3では収率を算出しなかった。また、「純度100%」とは、19F-NMR(溶媒:重アセトニトリル)測定により得られたチャートに、フルオロスルホニルイミド及び内部標準物質に由来するピーク以外にピークが確認されなかったことを意味する。
【0159】
表3より、棚段式減圧乾燥機による濃縮、乾燥を行った実験例3のリチウムビス(フルオロスルホニル)イミドに比べて、薄膜蒸留による濃縮工程を採用した実験例5−1〜5−16では、各種不純物量が低減されていた。この結果より、本発明法によれば、不純物の生成を抑制でき、各種不純物の含有量の少ないフルオロスルホニルイミドのアルカリ金属塩が得られることが分かる。
なお、実験例5では、いずれの実験例も不純物イオン量は少ないものの、実験例間においてその含有量にややバラツキが見られている。これは、フルオロスルホニルイミドが熱による影響を受け易いため、同じ操作を行っていても不純物の生成量に多少のバラツキが見られたものと考えられる。
【0160】
実験例6
実験例2−1、3及び5で得られたフルオロスルホニルイミドのアルカリ金属塩に含まれる残留溶媒量を、下記方法により測定した。
【0161】
[残留溶媒量]
上記実験例で得られたフルオロスルホニルイミドのアルカリ金属塩0.05gにジメチルスルホキシド水溶液(ジメチルスルホキシド/超純水=20/80、体積比)200μl、20質量%塩化ナトリウム水溶液2mlを加えて測定溶液とし、これをバイアル瓶に入れ密閉し、ヘッドスペース−ガスクロマトグラフィーシステム(「Agilent6890」、Agilent社製)により、フルオロスルホニルイミドのアルカリ金属塩中に含まれる残留溶媒量を測定した。
装置:Agilent6890
カラム:HP−5(長さ:30m、カラム内径:0.32mm、膜厚:0.25μm)(Agilent社製)
カラム温度条件:60℃(2分保持)、30℃/分で300℃まで昇温、300℃(2分保持)
ヘッドスペース条件:80℃(30分保持)
インジェクター温度:250℃
検出器:FID(300℃)
結果を表4に示す。また、実験例2−1及び実験例3における残留溶媒量も併せて表4に示す。
【0162】
【表4】
【0163】
表3,4より、本願発明法によれば、不純物の生成を抑制しつつ、棚段式減圧乾燥機を使用した実験例3と同程度にまで残留溶媒量を低減できることが分かる。
【0164】
実験例7
実験例4と同様の方法に従って、実験例5で得られたフルオロスルホニルイミドのアルカリ金属塩の耐熱性(質量減少率、2%質量減少温度)を測定した。結果を表5に示す。比較のため、各実験例における不純物量、及び、実験例2−1、実験例3の結果を表5に併せて示す。
【0165】
【表5】
【0166】
薄膜蒸留法による濃縮工程を採用した実験例5−3,5−5,5−13でも、不純物量の生成が抑制されており、これらの実験例におけるリチウムビス(フルオロスルホニル)イミドは良好な耐熱性を示していた。
【0167】
実験例8
実験例2−1で得られたリチウムビス(フルオロスルホニル)イミド1gを、エチレンカーボネート/エチルメチルカーボネートの混合溶液9g(50/50、体積比)に溶解し、電解液1を作製した。同様にして実験例3で得られたリチウムビス(フルオロスルホニル)イミドを用いて電解液2を作製した。
【0168】
電解液1、2を夫々20mlの褐色のスクリュー管に入れて密閉し、外部からの水分の侵入を防止した。各電解液を入れたスクリュー管を、温度25℃の環境下で2ヶ月間保存し、電解液1、2の経時変化を評価した。
【0169】
実験例2−1の生成物を原料として作製した電解液1には外観上の変化は認められず、無色透明の液体であったが、実験例3の生成物を原料として作製した電解液2には黄橙色の着色が見られた。また、上記不純物含有量の測定方法に従い、保存前後の電解液1,2を超純水で希釈した測定溶液についてイオンクロマトグラフィーにより、不純物含有量の測定を行った。結果を表6に示す。
【0170】
【表6】
【0171】
表6に示されるように、実験例2−1から得られた電解液1では、フッ化物イオン量が2ppm(保存前:1ppm)、硫酸イオン量が14ppm(保存前:14ppm)であり、保存前とほぼ同等の値であった。一方、実験例3から得られた電解液2では、フッ化物イオン量が153ppm(保存前:122ppm)、硫酸イオン量が413ppm(339ppm)と、保管前に比べて不純物が増加していることが確認された。この結果より、本発明の電解液は、保存安定性に優れることがわかった。
【産業上の利用可能性】
【0172】
本発明によれば、スケールアップしても、反応溶液から溶媒を速やかに除去することができ、効率よくフルオロスルホニルイミドのアルカリ金属塩を製造できる。また、本発明法では高温に加熱する必要がないので、フルオロスルホニルイミドのアルカリ金属塩の熱分解による収率の低下も抑制することができ、その結果、フルオロスルホニルイミドのアルカリ金属塩の耐熱性の低下を抑制することができる。さらに、本発明のフルオロスルホニルイミドのアルカリ金属塩は、各種不純物量が低減されているので、各種電気化学デバイスのイオン伝導体などとして有用である。加えて、本発明の製造方法は、工業的なフルオロスルホニルイミドのアルカリ金属塩の製造方法として好適である。
【特許請求の範囲】
【請求項1】
下記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩であって、空気気流下、100℃で8時間保持したときの質量減少率が2%以下であることを特徴とするフルオロスルホニルイミドのアルカリ金属塩。
【化1】
(式(I)中、Maはアルカリ金属、Ra、Rbは、同一若しくは異なって、フッ素原子、または、1個以上の水素原子がフッ素原子で置換された炭素数1〜6の炭化水素基を表す。)
【請求項2】
下記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩であって、空気気流下、25℃より10℃/分の昇温速度で加熱したときに、質量減少率が2%となる温度が210℃以上であることを特徴とするフルオロスルホニルイミドのアルカリ金属塩。
【化2】
(式(I)中、Maはアルカリ金属、Ra、Rbは、同一若しくは異なって、フッ素原子、または、1個以上の水素原子がフッ素原子で置換された炭素数1〜6の炭化水素基を表す。)
【請求項3】
下記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩であって、SO42-の含有量が3000ppm以下であることを特徴とするフルオロスルホニルイミドのアルカリ金属塩。
【化3】
(式(I)中、Maはアルカリ金属、Ra、Rbは、同一若しくは異なって、フッ素原子、または、1個以上の水素原子がフッ素原子で置換された炭素数1〜6の炭化水素基を表す。)
【請求項4】
さらに、F-の含有量が1000ppm以下である請求項3に記載のフルオロスルホニルイミドのアルカリ金属塩。
【請求項5】
水の含有量が500ppm以下である請求項3又は4に記載のフルオロスルホニルイミドのアルカリ金属塩。
【請求項6】
4000ppm以下の残留溶媒を含む請求項3〜5のいずれかに記載のフルオロスルホニルイミドのアルカリ金属塩。
【請求項7】
下記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩と媒体を含み、SO42-の含有量が1500ppm以下であることを特徴とする電解液。
【化4】
(式(I)中、Maはアルカリ金属、Ra、Rbは、同一若しくは異なって、フッ素原子、または、1個以上の水素原子がフッ素原子で置換された炭素数1〜6の炭化水素基を表す。)
【請求項8】
水の含有量が250ppm以下である請求項7に記載の電解液。
【請求項9】
2000ppm以下の残留溶媒を含む請求項7または8に記載の電解液。
【請求項10】
さらに、F-の含有量が500ppm以下である請求項7〜9のいずれかに記載の電解液。
【請求項11】
請求項1〜6のいずれかに記載のフルオロスルホニルイミドのアルカリ金属塩の製造方法であって、
フルオロスルホニルイミドのアルカリ金属塩を含む反応溶液中にガスをバブリングしながらフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程、及び/又は、
薄膜蒸留によりフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程
を含むことを特徴とするフルオロスルホニルイミドのアルカリ金属塩の製造方法。
【請求項12】
前記濃縮工程を150℃以下で行う請求項11に記載のフルオロスルホニルイミドのアルカリ金属塩の製造方法。
【請求項13】
前記濃縮工程を40kPa以下で行う請求項11または12に記載のフルオロスルホニルイミドのアルカリ金属塩の製造方法。
【請求項14】
前記反応溶液に含まれる反応溶媒が、エステル系溶媒及び/又はニトリル系溶媒である請求項11〜13のいずれかに記載のフルオロスルホニルイミドのアルカリ金属塩の製造方法。
【請求項15】
請求項1〜6のいずれかに記載のフルオロスルホニルイミドのアルカリ金属塩の製造方法であって、
薄膜蒸留によりフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程を含むことを特徴とするフルオロスルホニルイミドのアルカリ金属塩の製造方法。
【請求項16】
濃縮工程で得られた濃縮液を100℃以下で加熱して乾燥する工程を含む請求項11〜15のいずれかに記載のフルオロスルホニルイミドのアルカリ金属塩の製造方法。
【請求項17】
反応溶媒の存在下で、クロロスルホニルイミドまたはその塩をフッ素化する工程を含む請求項11〜16のいずれかに記載のフルオロスルホニルイミドのアルカリ金属塩の製造方法。
【請求項1】
下記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩であって、空気気流下、100℃で8時間保持したときの質量減少率が2%以下であることを特徴とするフルオロスルホニルイミドのアルカリ金属塩。
【化1】
(式(I)中、Maはアルカリ金属、Ra、Rbは、同一若しくは異なって、フッ素原子、または、1個以上の水素原子がフッ素原子で置換された炭素数1〜6の炭化水素基を表す。)
【請求項2】
下記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩であって、空気気流下、25℃より10℃/分の昇温速度で加熱したときに、質量減少率が2%となる温度が210℃以上であることを特徴とするフルオロスルホニルイミドのアルカリ金属塩。
【化2】
(式(I)中、Maはアルカリ金属、Ra、Rbは、同一若しくは異なって、フッ素原子、または、1個以上の水素原子がフッ素原子で置換された炭素数1〜6の炭化水素基を表す。)
【請求項3】
下記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩であって、SO42-の含有量が3000ppm以下であることを特徴とするフルオロスルホニルイミドのアルカリ金属塩。
【化3】
(式(I)中、Maはアルカリ金属、Ra、Rbは、同一若しくは異なって、フッ素原子、または、1個以上の水素原子がフッ素原子で置換された炭素数1〜6の炭化水素基を表す。)
【請求項4】
さらに、F-の含有量が1000ppm以下である請求項3に記載のフルオロスルホニルイミドのアルカリ金属塩。
【請求項5】
水の含有量が500ppm以下である請求項3又は4に記載のフルオロスルホニルイミドのアルカリ金属塩。
【請求項6】
4000ppm以下の残留溶媒を含む請求項3〜5のいずれかに記載のフルオロスルホニルイミドのアルカリ金属塩。
【請求項7】
下記一般式(I)で表されるフルオロスルホニルイミドのアルカリ金属塩と媒体を含み、SO42-の含有量が1500ppm以下であることを特徴とする電解液。
【化4】
(式(I)中、Maはアルカリ金属、Ra、Rbは、同一若しくは異なって、フッ素原子、または、1個以上の水素原子がフッ素原子で置換された炭素数1〜6の炭化水素基を表す。)
【請求項8】
水の含有量が250ppm以下である請求項7に記載の電解液。
【請求項9】
2000ppm以下の残留溶媒を含む請求項7または8に記載の電解液。
【請求項10】
さらに、F-の含有量が500ppm以下である請求項7〜9のいずれかに記載の電解液。
【請求項11】
請求項1〜6のいずれかに記載のフルオロスルホニルイミドのアルカリ金属塩の製造方法であって、
フルオロスルホニルイミドのアルカリ金属塩を含む反応溶液中にガスをバブリングしながらフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程、及び/又は、
薄膜蒸留によりフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程
を含むことを特徴とするフルオロスルホニルイミドのアルカリ金属塩の製造方法。
【請求項12】
前記濃縮工程を150℃以下で行う請求項11に記載のフルオロスルホニルイミドのアルカリ金属塩の製造方法。
【請求項13】
前記濃縮工程を40kPa以下で行う請求項11または12に記載のフルオロスルホニルイミドのアルカリ金属塩の製造方法。
【請求項14】
前記反応溶液に含まれる反応溶媒が、エステル系溶媒及び/又はニトリル系溶媒である請求項11〜13のいずれかに記載のフルオロスルホニルイミドのアルカリ金属塩の製造方法。
【請求項15】
請求項1〜6のいずれかに記載のフルオロスルホニルイミドのアルカリ金属塩の製造方法であって、
薄膜蒸留によりフルオロスルホニルイミドのアルカリ金属塩溶液を濃縮する工程を含むことを特徴とするフルオロスルホニルイミドのアルカリ金属塩の製造方法。
【請求項16】
濃縮工程で得られた濃縮液を100℃以下で加熱して乾燥する工程を含む請求項11〜15のいずれかに記載のフルオロスルホニルイミドのアルカリ金属塩の製造方法。
【請求項17】
反応溶媒の存在下で、クロロスルホニルイミドまたはその塩をフッ素化する工程を含む請求項11〜16のいずれかに記載のフルオロスルホニルイミドのアルカリ金属塩の製造方法。
【図1】

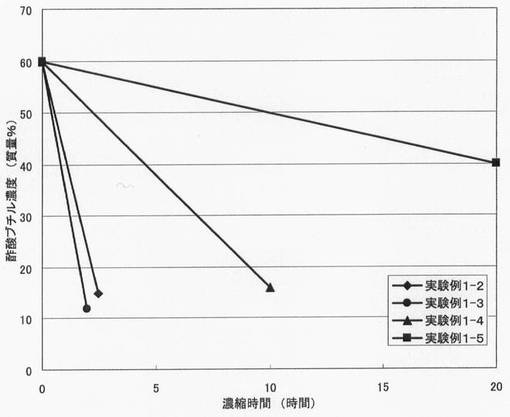
【公開番号】特開2013−18702(P2013−18702A)
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願番号】特願2012−183450(P2012−183450)
【出願日】平成24年8月22日(2012.8.22)
【分割の表示】特願2012−517349(P2012−517349)の分割
【原出願日】平成23年5月27日(2011.5.27)
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願日】平成24年8月22日(2012.8.22)
【分割の表示】特願2012−517349(P2012−517349)の分割
【原出願日】平成23年5月27日(2011.5.27)
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
[ Back to top ]