
フルクタン生合成の操作および植物バイオマスの増強
本発明は、植物の光合成細胞におけるフルクタン生合成を操作することができる遺伝的構築物であって、細菌FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む遺伝的構築物に関する。本発明はまた、植物におけるフルクタン生合成の改変に関し、特に、光合成細胞におけるフルクタン生合成を操作する方法に関する。本発明はまた、植物バイオマスの増加に関し、特に、植物におけるシュートおよび/または根の生長を含むバイオマス収量および/または収量安定性を増強する方法に関する。本発明はまた、生化学的経路の生産性を増強する方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、植物におけるフルクタン生合成の改変に関し、特に、光合成細胞におけるフルクタン生合成を操作する方法ならびに関連する核酸および構築物に関する。
【0002】
本発明はまた、植物バイオマスの増加に関し、特に、植物におけるシュートおよび/または根の生長を含むバイオマス収量および/または収量安定性を増強する方法ならびに関連する核酸および構築物にも関する。
【背景技術】
【0003】
フルクタンは水溶性炭水化物の一種であり、その主要機能は、植物生長のために容易に利用できるエネルギー貯蔵を提供することである。フルクタンは、寒冷および乾燥耐性、分げつ生存率の上昇、持続性の増強、切断または被食後の良好な再生長、ストレスからの回復の改善、早春における生長ならびに栄養価の上昇等、草本における様々な有利な特徴に関連する。
【0004】
草本および穀類におけるフルクタン合成および代謝は複雑である。フルクタンは、スクロースと結合した直鎖または分岐フルクトース鎖からなる。植物フルクタンの鎖長は、3から最大数百個のフルクトースユニットに及ぶ。異なる種類のフルクタンは、存在する結合の種類に基づいて識別することができる。ペレニアルライグラス(perennial ryegrass)において、3種類のフルクタンが同定された。すなわちイヌリン、イヌリンネオシリーズおよびレバンネオシリーズであり、このフルクタンプロファイルには4種類のフルクトシルトランスフェラーゼ(fructosyltransferse)(FT)酵素が関与する。酵素1−SST(スクロース:スクロース1−フルクトシルトランスフェラーゼ)はフルクタン生合成における最初のステップを触媒し、一方、残りの酵素は成長中のフルクトース鎖を伸長させる(1−FFT:フルクタン:フルクタン1−フルクトシルトランスフェラーゼ、6G−FFT:6−グルコースフルクトシルトランスフェラーゼおよび6−SFT:スクロース:フルクトース6−フルクトシルトランスフェラーゼ)。酵素1−FEHまたは6−FEH(フルクトエキソヒドロラーゼ(fructoexohydrolase))は、フルクトース分子を遊離させることによりフルクタン鎖長を短縮する。
【0005】
細菌は、高分子量フルクタンポリマーを合成するために1−SSTおよび1−FFT活性の両方を含む単一のFT酵素のみを利用する。大部分の細菌フルクトシルトランスフェラーゼは、フルクトース分子のβ−2,6結合によって特徴付けられるレバン型フルクタンを産生するが(レバンスクラーゼ(levansucrase))、一部の細菌からはイヌリン型フルクタン(β−2,1結合)を産生するイヌロスクラーゼ(inulosucrase)が単離されている。
【0006】
Bacillus subtilis由来のSacB遺伝子、Bacillus amyloliquefaciens由来のSacB遺伝子およびStreptococcus mutans由来のFTF遺伝子を含む、少なくとも3種類の細菌レバンスクラーゼがトランスジェニック植物において発現した。これら細菌レバンスクラーゼの植物における発現は、DP数千の非常に高分子量のフルクタンの合成をもたらす(総説として非特許文献1を参照)。
【0007】
フルクタンは、15%の植物種における主要な非構造炭水化物を表し、飼料の質に重要な役割を果たす。高フルクタン食を摂取する反芻家畜は、動物能力の改善を示す。
【0008】
草本において、FT遺伝子発現の遺伝子操作により、フルクタンのレベルおよび組成は茎および葉鞘で増加した。
【0009】
しかし、経路内酵素の活性を操作することによる生化学的経路の操作は、その道筋において様々な酵素およびそれらの基質が相互作用し得るため、困難なものとなるであろう。
【0010】
このため、特に植物における生化学的経路を操作するための改良法があることが望ましい。例えば、Lolium属、Festuca属およびBrachiaria属等の飼料草の種;サトウキビその他の草本;ならびにソルガムおよびコムギやトウモロコシ等、他の穀類を含む植物におけるフルクタン生合成を操作する方法があり、これにより例えば、
・牧草地の生産性の改善、動物の生産性の改善および/または環境汚染の抑制をもたらす、牧草品質および/または収量が改善された飼料草、
・バイオエタノール生産用等のバイオマス収量が増強されたスイッチグラス、Miscanthus属、ソルガム(穀物、飼料およびエネルギー用ソルガム)、サトウキビおよびエネルギー用サトウキビ等、バイオエネルギー用草本および作物、
・穀物および/またはバイオマス収量が増加したコムギ、イネ、トウモロコシ、オオムギ等、穀類
の生産を促進することが望ましい。
【0011】
フルクタン生合成経路に関与する数種の酵素をコードする核酸配列が特定の植物種で単離された。例えば、本出願人らの国際出願PCT/AU01/00705号明細書は、Lolium属およびFestuca属由来のフルクトシルトランスフェラーゼホモログについて記載している。しかし、依然として、植物におけるフルクタン生合成の改変に有用な、また植物体(plant)の様々な部分におけるフルクタン蓄積を遺伝子操作するための材料が必要である。
【0012】
国際特許出願PCT/AU2009/001211号明細書は、植物におけるフルクタン生合成の改変に有用な構築物について記載する。しかし、それら構築物は、好ましくは2以上のフルクタン生合成酵素の翻訳融合体をコードする融合遺伝子を含み、該遺伝子は、好ましくは植物フルクタン生合成遺伝子に相当する融合体を作り出す。
【先行技術文献】
【非特許文献】
【0013】
【非特許文献1】Cairns AJ. Fructan biosynthesis in transgenic plants. J Expt Biol(2003)54:549〜67
【発明の概要】
【発明が解決しようとする課題】
【0014】
本発明の目的は、先行技術に関連する問題または欠点の1または複数を克服、または少なくとも軽減することである。
【課題を解決するための手段】
【0015】
本出願人らは、細菌FT酵素をコードする遺伝子またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む構築物を用いて、光合成細胞におけるフルクタン生合成経路を空間的にリプログラミングすることにより、植物、例えば飼料草を栄養的に増強することおよび/または植物バイオマスを増加させることが可能であることを見出した。このプロセスを用いることによって、家畜によって先ず食べられる植物器官であり、通常フルクタンを蓄積できない葉身におけるフルクタン蓄積を駆動することが可能となる。このため、フルクタン、特に高い重合度を示すフルクタン(「高DPフルクタン」)の蓄積は、草食動物がより利用しやすい栄養を提供する。フルクタンは茎および葉鞘に蓄積するが、葉フルクタンはCO2同化が生長より優勢な期間においてのみ生成される。スクロースが合成される光合成細胞でフルクタン遺伝子を発現させることによって飼料草を栄養的に増強し、これによりフルクタン蓄積を葉身で優先的に駆動し、これを摂食する家畜にさらなるエネルギーを提供することができる。
【0016】
飼料草におけるフルクタンは、反芻動物に食べさせる食餌におけるすぐに利用できるエネルギーに大いに貢献する。反芻胃における発酵過程は、大量のすぐに利用できるエネルギーを必要とする。反芻胃におけるすぐに利用できるエネルギーの改善は、反芻胃の消化効率を上昇させることができる。反芻胃消化における効率の上昇は、反芻動物に与えた飼料タンパク質のミルクまたは肉への変換の改善および窒素性廃棄物の減少をもたらす。
【0017】
本出願人らは、寿命延長のために光合成細胞をリプログラミングすることにより、例えば葉の老化を遅延させることにより本発明の方法を促進することができ、植物バイオマスの増加に役立つことも見出した。
【0018】
本出願人らは、細菌FT遺伝子をコードする遺伝子またはその機能的に活性な断片もしくは変異体を含む構築物が、2以上のフルクタン生合成酵素の翻訳融合体をコードする融合遺伝子を含む構築物よりも優れた結果を示し得ることを見出した。例えば1−SSTおよび1−FFT活性の両方を含む細菌FT遺伝子の使用は、別々の遺伝子を融合するよりも技術的な困難が少なくなり、融合遺伝子を含む構築物と比べてより容易に植物に導入されるおよび/またはより良く機能する構築物をもたらすことができる。
【0019】
例えば、例えば1−SSTおよび1−FFT活性の両方を含む細菌FT遺伝子を発現することにより、植物ゲノムに独立的に組み込まれたこれら遺伝子の発現パターンの差に関連する課題を軽減し、その結果スクロース分子を、低い重合度(「低DPフルクタン」)および高い重合度(「高DPフルクタン」)を示すフルクタンに直接的に変換することができる。さらに、FTタンパク質は、効率的なフルクタン生合成の代謝チャネルを形成することができる。
【0020】
光合成細胞において低および高DPフルクタンの蓄積をもたらす光合成細胞におけるFT遺伝子の発現は、光合成抑制機構からの解放をもたらし、太陽エネルギーの獲得およびCO2固定の増加を促進することができる。
【0021】
このため、本出願人らは、光合成細胞の寿命延長のためのリプログラミングおよびフルクタン生合成の増強は太陽エネルギーの獲得を促進し、シュートおよび/または根の生長等、植物バイオマスの生産を増加させることを見出した。
【0022】
さらに、低および高DPフルクタンの蓄積は寒冷および乾燥等、非生物的ストレスに対する植物の耐性に関連づけられてきたため、また、根の生長増強および葉の老化遅延は植物の乾燥ストレス耐性に関係づけられてきたため、光合成細胞の寿命延長のためのリプログラミングおよびフルクタン生合成の増強は、収量安定性および植物の非生物的ストレス耐性を促進する。
【0023】
したがって、本発明はその一態様において、植物の光合成細胞におけるフルクタン生合成を操作する方法であって、細菌フルクトシルトランスフェラーゼ(FT)酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む有効量の遺伝的構築物を前記植物に導入するステップを含む方法を提供する。
【0024】
「フルクタン生合成の操作」とは、一般に、非形質転換対照植物と比べて形質転換植物におけるフルクタン生合成の増加を意味する。しかし、一部の適用では、非形質転換対照植物と比べて形質転換植物においてフルクタン生合成を低減させる、あるいは改変することが望ましいであろう。例えば、非形質転換対照植物と比べて形質転換植物のフルクタン生合成経路における特定の酵素の活性を増加または減少させることが望ましいであろう。
【0025】
「光合成細胞」とは、光合成が行われる植物細胞を意味する。このような細胞は一般にクロロフィル色素を含む、あるいは緑色細胞として公知のものである。大部分の光合成細胞は、植物の葉に含まれる。好ましくは、本発明の遺伝的構築物は、維管束鞘細胞で発現し、より好ましくは葉肉および/または柔組織維管束鞘細胞で発現する。
【0026】
「有効量」とは、前記植物またはそこから派生する植物体、植物種子もしくは他の植物部分において同定可能な表現型形質を生じるのに十分な量を意味する。このような量は、当業者であれば、植物の種類、投与経路その他の関連性のある要因を考慮に入れて容易に決定することができる。このような当業者は、適切な投与量および投与方法を容易に決定することができる。例えば、その開示全体を本明細書の一部を構成するものとしてここに援用するManiatisら、Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory、Cold Spring Harborを参照。
【0027】
「遺伝的構築物」とは、組換え核酸分子を意味する。
【0028】
「プロモーター」とは、作動的に連結した核酸配列の転写を導くのに十分な核酸配列を意味する。
【0029】
「作動的に連結した」とは、核酸(複数可)とプロモーター等の調節配列が、例えば転写活性化因子タンパク質等の適切な分子が前記調節配列に結合する場合、適切な条件下で前記核酸の発現を可能にする仕方で連結していることを意味する。好ましくは、作動的に連結したプロモーターは、関連する核酸の上流にある。
【0030】
「上流」とは、核酸に沿って3’→5’方向を意味する。
【0031】
「核酸」とは、遺伝情報を運ぶことができるヌクレオチド鎖を意味する。この用語は一般に、遺伝子またはその機能的に活性な断片もしくは変異体、および/またはその表現型に影響を及ぼす生物ゲノム内の他の配列を言う。用語「核酸」は、1本鎖または2本鎖であり、必要に応じて合成、非天然または変化したヌクレオチド塩基、合成核酸およびそれらの組合せを含むDNA(cDNAまたはゲノムDNA等)およびRNA(mRNAまたはマイクロRNA等)を含む。
【0032】
「細菌フルクトシルトランスフェラーゼ(FT)酵素をコードする核酸」または「細菌フルクトシルトランスフェラーゼ遺伝子」とは、フルクトシル部分をスクロース含有サッカライドからアクセプター分子に転移することによりオリゴフルクタンおよび/またはポリフルクタンの合成を触媒する、細菌に通常存在する酵素をコードする核酸を意味する。細菌FT酵素は、レバンスクラーゼ活性を含む、および/またはレバン型フルクタンを産生することができる。細菌FT酵素は、イヌロスクラーゼ活性を含む、および/またはイヌリン型フルクタンを産生することができる。好ましい細菌FTは、スクロース:スクロース1−フルクトシルトランスフェラーゼ(1−SST)およびフルクタン:フルクタン1−フルクトシルトランスフェラーゼ(1−FFT)酵素活性の両方を含む。SacB、LscまたはFTF遺伝子を用いてもよい。SacBまたはLsc遺伝子が特に好ましい。
【0033】
プロモーターに関する「機能的に活性な断片または変異体」とは、断片または変異体(アナログ、誘導体または突然変異体等)がそこに作動的に連結した核酸の転写を導くことができることを意味する。このような変異体は、自然発生的な対立遺伝子変異体および非自然発生的な変異体を含む。その改変が断片または変異体の機能欠失活性をもたらすことのない限りにおいて、1または複数のヌクレオチドの付加、欠失、置換および誘導体化が企図される。好ましくは、機能的に活性な断片または変異体は、該断片または変異体が対応する上述の配列の関連性のある部分に対して少なくとも約80%の同一性、より好ましくは少なくとも約90%の同一性、さらにより好ましくは少なくとも約95%の同一性、最も好ましくは少なくとも約98%の同一性を有する。好ましくは、断片は、少なくとも20ヌクレオチド、より好ましくは少なくとも50ヌクレオチド、より好ましくは少なくとも100ヌクレオチド、より好ましくは少なくとも200ヌクレオチド、より好ましくは少なくとも300ヌクレオチドのサイズを有する。
【0034】
細菌FT酵素をコードする核酸に関する「機能的に活性な」とは、本発明の方法により、例えばフルクタン生合成経路に関与することができる酵素に翻訳されることにより、断片または変異体(アナログ、誘導体または突然変異体等)が植物におけるフルクタン生合成を操作できることを意味する。このような変異体は、自然発生的な対立遺伝子変異体および非自然発生的な変異体を含む。その改変が断片または変異体の機能欠失活性をもたらさない限りにおいて、1または複数のヌクレオチドの付加、欠失、置換および誘導体化が企図される。好ましくは、機能的に活性な断片または変異体は、該断片または変異体が対応する上述の配列の関連性のある部分に対して少なくとも約80%の同一性、より好ましくは少なくとも約90%の同一性、さらにより好ましくは少なくとも約95%の同一性、最も好ましくは少なくとも約98%の同一性を有する。このような機能的に活性な変異体および断片は、例えば保存された核酸変化を有する機能的に活性な変異体および断片を含む。
【0035】
「保存された核酸変化」とは、遺伝暗号の縮重による、コードされているタンパク質におけるアミノ酸保存をもたらす核酸置換を意味する。このような機能的に活性な変異体および断片は、例えば対応するアミノ酸配列における1または複数の残基に保存されたアミノ酸置換をもたらす核酸変化を有する機能的に活性な変異体および断片も含む。
【0036】
「保存されたアミノ酸置換」とは、同一クラス内の別の1アミノ酸によるアミノ酸置換を意味し、クラスは次の通りである。
【0037】
非極性:Ala、Val、Leu、Ile、Pro、Met、Phe、Trp
電荷なし、極性:Gly、Ser、Thr、Cys、Tyr、Asn、Gln
酸性:Asp、Glu
塩基性:Lys、Arg、His
他の保存されたアミノ酸置換を次の通り行ってもよい。
【0038】
芳香族:Phe、Tyr、His
プロトンドナー:Asn、Gln、Lys、Arg、His、Trp
プロトンアクセプター:Glu、Asp、Thr、Ser、Tyr、Asn、Gln
特に好ましい断片および変異体は、1または複数の保存されたスクロース結合/加水分解ドメインを含む。このようなドメインの例を図1に示すが、これはLDVWDSWP、QEWSGSA、LRDPHおよびDEIERを含む。
【0039】
特に好ましい断片および変異体は触媒コアを含む。「触媒コア」とは、保存されたスクロース結合および/または加水分解ドメインを含む、シグナルペプチドならびにNおよびC末端可変領域を除いたポリペプチドの内部領域を意味する。触媒コアの例を図1に示すが、これはBacillus subtilis由来のSacBタンパク質のアミノ酸残基65〜468を含む。
【0040】
特に好ましい断片および変異体は、シグナルペプチドを欠く断片および変異体を含む。「シグナルペプチド」とは、N末端シグナル配列を意味する。シグナルペプチドの例を図1に示すが、これはBacillus subtilis由来のSacBタンパク質のアミノ酸1〜27を含む。
【0041】
特に好ましい断片および変異体は、単子葉植物および双子葉植物の翻訳開始等、植物に適応したコドン使用を有する。
【0042】
特に好ましい断片および変異体は、潜在性スプライス部位および/またはRNA不安定化配列エレメントが不活性化または除去されている。
【0043】
好ましくは、細菌FTをコードする核酸は、Bacillus subtilis、Bacillus amyloliquefaciens、Lactobacillus johnsoniiおよびStreptococcus mutans等、Bacillus属、Lactobacillus属およびStreptococcus属からなる群より選択された細菌種由来の遺伝子(単数または複数)から単離される、あるいはそれに相当する。さらにより好ましくは、細菌FT酵素をコードする核酸は、Bacillus subtilisもしくはBacillus amyloliquefaciens由来のSacB遺伝子、Lactobacillus johnsonii由来のLsc遺伝子またはStreptococcus mutans由来のFTF遺伝子から単離される、あるいはそれに相当する。
【0044】
特に好ましい一実施形態において、SacB遺伝子は、図2に示されている配列および図3に示されているポリペプチド配列をコードするヌクレオチド配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0045】
特に好ましい一実施形態において、Lsc遺伝子は、図4に示されている配列および図5に示されているポリペプチド配列をコードする核酸配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0046】
細菌レバンスクラーゼを発現するトランスジェニック植物が報告されており、一部の事例では発達異常の表現型を提示する。本出願人らは理論に制約されることを望まないが、この現象はレバンスクラーゼ酵素およびフルクタンポリマーの不適切な区画化に起因し得る。トランスジェニックタバコおよびジャガイモ植物における細菌SacB遺伝子のサイトゾルにおける発現は、植物の発達に特に破壊的な影響を与えることを示した(Caimiら、1997年)。しかし、液胞ターゲティングシグナルと融合した細菌フルクトシルトランスフェラーゼを発現する植物は、植物の発達に認識可能な効果を及ぼすことなくフルクタンを蓄積する(Ebskampら、1994年、Yeら、2001年)。
【0047】
発達異常の表現型を生じる可能性を低減させるため、細菌FT遺伝子を改変してそのターゲティングシグナル配列を変化させ、細菌FTタンパク質を高フルクタンレベルの存在する区画に方向付けることができる。
【0048】
より具体的には、細菌FT遺伝子のN−シグナルペプチドが除去され、細胞内ターゲティング配列、好ましくは液胞ターゲティング配列に置き換えられたキメラ配列を作製することができる。
【0049】
好ましい一実施形態において、液胞ターゲティング配列は、サツマイモのSPOR531等、プレプロスポラミンタンパク質をコードする遺伝子に由来する、あるいはそれに相当し得る。
【0050】
特に好ましい一実施形態において、液胞ターゲティング配列は、図6において太字・下線で示されている配列および図7において太字・下線で示されているポリペプチドをコードする核酸配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含むことができる。
【0051】
特に好ましい一実施形態において、細菌FT酵素をコードする核酸は、SPOR:SacBキメラ配列となることができる。好ましくは、SPOR:SacBキメラ配列は、図8に示されている配列および図9に示されているポリペプチド配列をコードする核酸配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0052】
特に好ましい一実施形態において、細菌FT酵素をコードする核酸は、SPOR:Lscキメラ配列となることができる。好ましくは、SPOR:Lscキメラ配列は、図9に示されている配列および図12に示されているポリペプチド配列をコードする核酸配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0053】
特に好ましい一実施形態において、遺伝的構築物は、図8と図11に示されている配列および図9と図12に示されているポリペプチドをコードする核酸配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0054】
別の好ましい一実施形態において、細菌FT遺伝子のN−シグナルペプチドが除去されて1−SST等、フルクトシルトランスフェラーゼ酵素の膜貫通ドメインに置き換えられたキメラ配列を作成することができる。
【0055】
特に好ましい一実施形態において、膜貫通ドメインは、図15において太字・斜字体で示されている配列および図16において太字・斜字体で示されているポリペプチドをコードする核酸配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0056】
特に好ましい一実施形態において、遺伝的構築物は、図17と図20に示されている配列および図18と図21に示されているポリペプチドをコードする核酸配列ならびにその機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0057】
「キメラ配列」とは、元々は別々のタンパク質をコードしていた2以上の連結された核酸またはその機能的に活性な断片もしくは変異体を含む融合遺伝子を発現することによる、組換えにより産生されたハイブリッドを意味する。
【0058】
「融合遺伝子」とは、融合タンパク質が好ましくは翻訳融合体として発現することが可能となる仕方で2以上の核酸が連結されたことを意味する。これは通常、第一のタンパク質をコードする核酸配列から終止コドンを除去し、次に第二のタンパク質の核酸配列をインフレームで付け加えることを含む。続いて、細胞により融合遺伝子を単一のタンパク質として発現させる。タンパク質は、両方のオリジナルタンパク質の完全配列、またはそのいずれかもしくは両者の機能的に活性な断片もしくは変異体を含むよう遺伝子操作されていてよい。
【0059】
遺伝的構築物は、連結された2核酸間のリンカーをコードする核酸配列を含んでいてもよい。「リンカー」は、融合タンパク質内の2個の隣接する活性断片の間に配置された、あるいはそれらに接続された任意の化学合成炭水化物・脂質・ポリペプチド分子(またはそれらの組合せ)である。本発明の好ましいリンカーは、アミノ酸の結合によって2個の活性断片と接続した1または複数のアミノ酸残基からなるポリペプチド鎖等、可動性リンカーである。例えば、(Gly4Ser)3リンカーが、融合タンパク質内の2個の活性断片間に配置されてよい。
【0060】
本発明の構築物および方法に用いられるプロモーターは、構成的、組織特異的または誘導性プロモーターとなることができる。好ましい一実施形態において、プロモーターは光調節プロモーター(light−regulated promoter)、より好ましくは光合成プロモーターとなることができる。「光調節プロモーター」とは、光刺激に応答して遺伝子発現を媒介することができるプロモーターを意味する。「光合成プロモーター」とは、植物における光合成経路に関与するタンパク質をコードする遺伝子の発現を媒介することができるプロモーターを意味する。
【0061】
成熟葉身は、葉鞘および茎よりも少量のフルクタンが蓄積する。葉身におけるフルクタンレベルを特異的に上昇させるため、光合成細胞においてフルクタン生合成遺伝子を協調的に発現させる戦略的アプローチが考案された。本出願人らは理論に制約されることを望まないが、光調節または光合成プロモーターの使用は次の利点を提供することができる。
【0062】
・光合成プロモーターは、葉身、茎の上部および外側を含む大きな細胞群(>55%の細胞)における活性を有する。
【0063】
・それは、スクロース産生細胞(葉肉および柔組織維管束鞘細胞)における活性を有する。
【0064】
・その発現パターンは、スクロース蓄積と時間的および空間的に重複する。
【0065】
・フルクトシルトランスフェラーゼ(Frutosyltransferase)活性はソースからスクロースを除去し、これにより光合成におけるフィードバック抑制を妨げ、CO2固定の増加を促進する。
【0066】
特に好ましい光調節プロモーターは、リブロース−1,5−二リン酸カルボキシラーゼ−オキシゲナーゼ(oxygtenase)−スモールサブユニット(RbcS)プロモーターおよびクロロフィルa/b結合タンパク質(CAB)プロモーターならびにそれらの機能的に活性な断片および変異体を含む。
【0067】
光調節プロモーターは、単子葉植物[トウモロコシ、イネ、コムギ、オオムギ、ソルガム、サトウキビ、エネルギー用サトウキビ、飼料草(例えば、Festuca属、Lolium属、Brachiaria属、Paspalum属、Pennisetum属)、バイオエネルギー用草本(例えば、スイッチグラス、Miscanthus属)等]、双子葉植物(シロイヌナズナ、ダイズ、キャノーラ、ワタ、アルファルファおよびタバコ等)および裸子植物を含む、任意の適切な植物種に由来することができる。
【0068】
単子葉植物の形質転換のため、好ましくは、光調節プロモーターは単子葉植物種、より具体的にはTriticum属またはLolium属の種、さらにより具体的にはTriticum aestivumまたはLolium perenneから単離される、あるいはそれらに由来するプロモーターに相当する。
【0069】
双子葉植物の形質転換のため、好ましくは、光調節プロモーターは双子葉植物種、より具体的にはArabidopsis属の種、さらにより具体的にはArabidopsis thalianaから単離される、あるいはそれらに由来するプロモーターに相当する。
【0070】
特に好ましい一実施形態において、RbcSプロモーターは、図23〜26のいずれか一図において小文字・斜字体で示されている配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0071】
特に好ましい別の一実施形態において、RbcSプロモーターは、図29〜36のいずれか一図において小文字・斜字体で示されている配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0072】
本発明の遺伝的構築物は、任意の適切な技法により植物に導入することができる。本発明の遺伝的構築物を植物細胞に組み入れるための技法(例えば形質導入、トランスフェクション、形質転換または遺伝子ターゲティング)は、当業者にとって周知のものである。このような技法は、アグロバクテリウムを介した導入、根粒菌を介した導入、組織、細胞およびプロトプラストへのエレクトロポレーション、プロトプラスト融合、生殖器官への注入、未熟胚への注入、細胞、組織、カルス、未熟胚および成熟胚への高速噴射による導入、遺伝子銃による形質転換、Whiskers形質転換ならびにそれらの組合せを含む。技法の選択は、主として形質転換される植物の種類に依存し、当業者であれば容易に決定することができる。
【0073】
本発明の遺伝的構築物を組み入れる細胞を後述の通り選択し、次に本技術分野で周知の技法を用いて適切な培地で培養して形質転換植物を再生することができる。温度やpHその他の培養条件は、当業者であれば明らかであろう。その結果生じた植物に、本技術分野で周知の方法を用いて有性生殖か無性生殖のいずれかを行わせて、形質転換植物の後代を作出することができる。
【0074】
本発明の方法は、単子葉植物[草本(例えば、ペレニアルライグラス、ヒロハノウシノケグサ(tall fescue)、イタリアンライグラス(Italian ryegrass)、オオウシノケグサ(red fescue)、クサヨシ(reed canarygrass)、ビッグブルーステム(big bluestem)、コードグラス(cordgrass)、ネピアグラス(napiergrass)、ワイルドライ(wildrye)、ワイルドシュガーケーン(wild sugarcane)、Miscanthus属、スイッチグラスを含む飼料草およびバイオエネルギー用草本)、コーンすなわちトウモロコシ、イネ、コムギ、オオムギ、ソルガム、サトウキビ、ライムギ、カラスムギ)およびエネルギー用作物(例えば、エネルギー用サトウキビ、エネルギー用ソルガム)等]、双子葉植物[シロイヌナズナ、タバコ、ダイズ、キャノーラ、アルファルファ、ジャガイモ、キャッサバ、クローバ(例えば、シロツメクサ、ムラサキツメクサ、ジモグリツメクサ(subterranean clover))、アブラナ科野菜(vegetable brassica)、レタス、ホウレンソウ等]および裸子植物を含む、種々の植物に適用することができる。
【0075】
本発明のさらに別の一態様において、細菌FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結した光調節プロモーターまたはその機能的に活性な断片もしくは変異体を含む、植物の光合成細胞におけるフルクタン生合成を操作することができる遺伝的構築物が提供される。
【0076】
好ましい一実施形態において、本発明に係る遺伝的構築物はベクターとなることができる。
【0077】
「ベクター」とは、遺伝子材料の標的細胞への導入に用いられる遺伝的構築物を意味する。
【0078】
ベクターは、任意の適切な種類のものとなることができ、ウイルスまたは非ウイルス性となることができる。ベクターは発現ベクターとなることができる。このようなベクターは、染色体、非染色体および合成核酸配列、例えば植物ウイルスの誘導体、細菌プラスミド、Agrobacterium tumefaciensから得られたTiプラスミドの誘導体、Agrobacterium rhizogenesから得られたRiプラスミドの誘導体、ファージDNA、酵母人工染色体、細菌人工染色体、バイナリー細菌人工染色体ならびにプラスミドとファージDNAの組合せに由来するベクターを含む。しかし、複製可能あるいは植物細胞に組み込み可能または機能する限りにおいて、その他のベクターを用いてもよい。
【0079】
本発明のこの態様の好ましい一実施形態において、遺伝的構築物はターミネーターをさらに含むことができ、前記プロモーター、遺伝子およびターミネーターは、作動可能に連結している。
【0080】
プロモーター、遺伝子およびターミネーターは、任意の適切な種類のものとなることができ、標的植物細胞で機能するのであれば、その標的植物細胞に対して内在性であっても外来性であってもよい。
【0081】
本発明の遺伝的構築物に用いることができる種々のターミネーターもまた、当業者にとって周知のものである。ターミネーターは、プロモーター配列と同じ遺伝子に由来してよいし、異なる遺伝子に由来してもよい。特に適切なターミネーターは、(CaMV)35SポリA等、ポリアデニル化シグナルならびにノパリン合成酵素(nos)およびオクトピン合成酵素(ocs)遺伝子由来の他のターミネーターである。
【0082】
プロモーター、遺伝子およびターミネーターに加えて、遺伝的構築物は、核酸の発現に必要なさらに別のエレメント、例えばベクター骨格、複製起点(ori)、多重クローニング部位、スペーサー配列、エンハンサー、イントロン(トウモロコシユビキチンUbiイントロン等)、抗生物質抵抗性遺伝子および他の選択マーカー遺伝子[ネオマイシンホスホトランスフェラーゼ(nptII)遺伝子、ハイグロマイシンホスホトランスフェラーゼ(hph)遺伝子、ホスフィノトリシンアセチルトランスフェラーゼ(barまたはpat)遺伝子]ならびにレポーター遺伝子(ベータ−グルクロニダーゼ(GUS)遺伝子(gusA)等)等]を様々な組合せで含むことができる。遺伝的構築物は、翻訳開始のためのリボソーム結合部位を含むこともできる。遺伝的構築物は、発現を増幅するための適切な配列を含むこともできる。
【0083】
特に、遺伝的構築物は、上文に記す通り、連結された2核酸の間にリンカーをコードする核酸配列をさらに含むことができる。
【0084】
当業者であれば、遺伝的構築物の様々な構成要素が、前記核酸の発現をもたらすよう作動可能に連結していることを理解できるであろう。本発明の遺伝的構築物の構成要素を作動可能に連結するための技法は、当業者にとって周知のものである。このような技法は、例えば1または複数の制限酵素部位を含む合成リンカー等、リンカーの使用を含む。
【0085】
好ましくは、遺伝的構築物は実質的に精製または単離されている。「実質的に精製された」とは、遺伝的構築物が、本発明の核酸またはプロモーターの由来する生物の自然発生的なゲノムにおいては、該核酸またはプロモーターに隣接する遺伝子を含まないことを意味する。したがって、この用語は、例えば、ベクター、自己複製プラスミドもしくはウイルス、または原核生物もしくは真核生物ゲノムDNAに組み入れられた、あるいは他の配列から独立した別々の分子(例えば、cDNAまたはPCRまたは制限エンドヌクレアーゼ消化によって生成されたゲノムもしくはcDNA断片)として存在する遺伝的構築物を含む。これは、追加的ポリペプチド配列をコードするハイブリッド遺伝子の一部である遺伝的構築物も含む。好ましくは、実質的に精製された遺伝的構築物は、少なくとも約90%の純度、より好ましくは少なくとも約95%の純度、さらにより好ましくは少なくとも約98%の純度である。
【0086】
用語「単離された」は、材料がその元々の環境(例えば、自然発生的であれば自然環境)から取り分けられたことを意味する。例えば、生きた植物に存在する自然発生的な核酸は単離されていないが、同じ核酸でも、自然システムにおいて共存している材料の一部または全部から分離された核酸は単離されている。このような核酸はベクターの一部であってよい、および/またはこのような核酸は組成物の一部であってよく、このようなベクターまたは組成物がその自然環境の一部ではないという点において、これは依然として単離されている。
【0087】
形質転換された宿主細胞を選抜するための表現型形質を提供するための選択マーカー遺伝子を使用する代わりとして、形質転換細胞における遺伝的構築物の存在は、PCR(ポリメラーゼ連鎖反応)、サザンブロットハイブリダイゼーション解析、組織化学的アッセイ(例えば、GUSアッセイ)、薄層クロマトグラフィー(TLC)、ノーザンブロットおよびウエスタンブロットハイブリダイゼーション解析等、本技術分野で周知の他の技法により決定することができる。
【0088】
本出願人は、本発明の方法が、非形質転換対照植物と比べて形質転換植物において増強されたバイオマスをもたらし得ることも見出した。これにより、この増強されたバイオマスは、形質転換植物を同定するための選抜ツールとして用いることができる。この構成は、後にこのようなマーカーを除去(そしてマーカーなし植物の作製のため)することが困難である場合、一部の状況において、形質転換体選抜のための抗生物質抵抗性または他のマーカーを含む必要がないという利点がある。
【0089】
「バイオマス増強」または「増強されたバイオマス」とは、非形質転換対照植物と比べて形質転換植物におけるシュートおよび/または根の生長を含むバイオマス収量の増強、増加または安定性増加を意味する。例えば、合計葉面積、累積葉面積、葉の生長動態(すなわち、経時的な葉の数)、シュートの数、分げつの数、根の数、根の質量もしくは重量、シュートの質量もしくは重量、根の長さ、シュートの長さ、走根の長さ、塊茎の数、塊茎の重量、花の数、果実の数、種子の数、種子の重量、果実の重量、開花している植物体のパーセンテージおよび花1個当たりまたは作付面積当たりの種子収量からなる群より選択された1または複数の生長特徴は、非形質転換対照植物と比べて形質転換植物において増強され、増加され、またはより安定的となることができる。
【0090】
この技法は、実質的に遺伝的に均一または遺伝的に同一である、あるいは形質転換前のバイオマスとの表現型の差が小さいことを示す植物に特に適用できる。
【0091】
したがって、本発明のさらに別の一態様において、植物におけるバイオマスを増強する方法であって、細菌FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結(liked)したプロモーターまたはその機能的に活性な断片もしくは変異体を含む有効量の遺伝的構築物を前記植物に導入するステップを含む方法が提供される。好ましくは、プロモーターは光調節プロモーターである。
【0092】
本発明の方法、核酸および遺伝的構築物は、他の遺伝的操作方法または他の導入核酸もしくは遺伝的構築物と組み合わせて用いて、これにより形質を積み重ねることができる。このため、積み重ねられた遺伝子(または積み重ねされた形質)を有するトランスジェニック植物体、植物細胞、植物種子または他の植物部分が作出される。例えば、本発明の技術は、除草剤抵抗性技術(例えば、グルホシネート、グリホサート)、害虫抵抗性技術(例えば、Bacillus thuringiensis)または老化遅延技術と組み合わせることができる。核酸または遺伝的構築物は、上文に記す通り、任意の適切な技法により植物に導入することができ、同時発生的に、連続的にまたは別々に導入することができる。
【0093】
本発明のさらになお別の一態様において、植物におけるバイオマスを増強する方法であって、有効量の
(a)植物の光合成細胞においてフルクタン生合成を操作することができる遺伝的構築物であって、細菌FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む遺伝的構築物と、
(b)植物の老化を操作することができる遺伝的構築物と、
を植物に導入するステップを含む方法が提供される。
【0094】
遺伝的構築物は、上文に記す通り任意の適切な技法により植物に導入することができ、同時発生的に、連続的にまたは別々に導入することができる。
【0095】
好ましくは、フルクタン生合成を操作することができる遺伝的構築物は、上文に記す通りである。
【0096】
好ましくは、植物の老化を操作することができる遺伝的構築物は、植物の光合成細胞の老化を操作することができる。
【0097】
好ましくは、老化を操作することができる遺伝的構築物は、サイトカイニンの生合成に関与する酵素をコードする遺伝子またはその機能的に活性な断片もしくは変異体に作動的に連結したmyb遺伝子プロモーターもしくは改変されたmyb遺伝子プロモーターまたはそれらの機能的に活性な断片もしくは変異体を含む。
【0098】
適切な遺伝的構築物またはベクターは、その開示全体を本明細書の一部を構成するものとしてここに援用する、国際特許出願PCT/AU01/01092号明細書および米国特許出願第11/789,526号明細書に記載されている。
【0099】
「老化を操作する」は一般に、非形質転換対照植物と比べた形質転換植物または形質転換植物の細胞もしくは器官、例えば光合成細胞における老化の遅延に関する。しかし、一部の適用では、植物における老化を促進、あるいは改変することが望ましいであろう。老化は、例えばアンチセンス遺伝子を利用することによって促進、あるいは改変することができる。
【0100】
myb遺伝子プロモーターは、任意の適切な種類のものとなることができる。好ましくは、myb遺伝子プロモーターは、myb32遺伝子プロモーターである。好ましくは、myb遺伝子プロモーターは、Arabidopsis属、より好ましくはArabidopsis thalianaに由来する。最も好ましくは、myb遺伝子プロモーターは、国際出願PCT/AU01/01092号明細書の配列番号1に示されている配列ならびにその機能的に活性な断片および変異体からなる群より選択されたヌクレオチド配列を含む。
【0101】
適切なプロモーターは、その開示全体を本明細書の一部を構成するものとしてここに援用するLiら、Cloning of three MYB−like genes from Arabidopsis(PGR99〜138)、Plant Physiology、121巻:313(1999年)に記載されている。
【0102】
「改変されたmyb遺伝子プロモーター」とは、前記プロモーターにおける1または複数の根特異的モチーフおよび/または花粉特異的モチーフを欠失するよう、あるいは不活性化するよう改変された、myb遺伝子に通常関連するプロモーターを意味する。
【0103】
好ましくは、改変されたmyb遺伝子プロモーターは、改変されたmyb32遺伝子プロモーターである。好ましくは、改変されたmyb遺伝子プロモーターは、Arabidopsis属、あるいはより好ましくはArabidopsis thalianaに由来するmyb遺伝子プロモーターから改変される。
【0104】
本発明に従って改変できる適切なプロモーターは、その開示全体を本明細書の一部を構成するものとしてここに援用するLiら、Cloning of three MYB−like genes from Arabidposis(PGR99〜138)Plant Physiology、121巻:313(1999年)に記載されている。
【0105】
「根特異的モチーフ」とは、植物の根において任意の関連遺伝子の発現を導く、3〜7ヌクレオチド、好ましくは4〜6ヌクレオチド、より好ましくは5ヌクレオチドの配列を意味する。
【0106】
好ましくは、根特異的モチーフは、コンセンサス配列ATATTまたはAATATを含む。
【0107】
「花粉特異的モチーフ」とは、植物の花粉において関連遺伝子の発現を導く、3〜7ヌクレオチド、好ましくは4〜6ヌクレオチド、より好ましくは4または5ヌクレオチドの配列を意味する。
【0108】
好ましくは、花粉特異的モチーフは、TTTCT、AGAAA、TTCTおよびAGAAからなる群より選択されたコンセンサス配列を含む。
【0109】
根または花粉特異的モチーフは、モチーフ内の1または複数のヌクレオチドを付加、欠失、置換または誘導体化することによって不活性化し、これにより好ましいコンセンサス配列を持たないようにすることができる。
【0110】
好ましくは、改変されたmyb遺伝子プロモーターは、米国特許出願第11/789,526号明細書の配列番号2、3および4に示されている配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択されたヌクレオチド配列を含む。
【0111】
「サイトカイニンの生合成に関与する酵素をコードする遺伝子」とは、カイネチン、ゼアチンおよびベンジルアデニン等、サイトカイニンの合成に関与する酵素をコードする遺伝子、例えば、sho遺伝子(例えば、ペチュニア由来の)等、イソペンチル(isopentyl)トランスフェラーゼ(ipt)またはipt様遺伝子をコードする遺伝子を意味する。好ましくは、遺伝子は、イソペンテニル(isopentenyl)トランスフェラーゼ(ipt)遺伝子またはsho遺伝子である。好ましい一実施形態において、遺伝子は、Agrobacterium属、より好ましくはAgrobacterium tumefaciens;Lotus属、より好ましくはLotus japonicus;およびPetunia属、より好ましくはPetunia hybridaからなる群より選択された種に由来する。
【0112】
最も好ましくは、遺伝子は、米国特許出願第11/789,526号明細書の配列番号5、7および9に示されている配列、米国特許出願第11/789,526号明細書の配列番号6、8および10に示されているポリペプチドをコードする配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択されたヌクレオチド配列を含む。
【0113】
本発明はまた、形質転換植物を選抜する方法であって、細菌FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む有効量の遺伝的構築物を前記植物に導入するステップと、バイオマスが増強された植物を選抜するステップとを含む方法も提供する。好ましくは、プロモーターは光調節プロモーターである。
【0114】
本発明のさらに別の一態様において、本発明に係る遺伝的構築物またはベクターを含み、非形質転換対照植物と比べてフルクタン生合成特徴が改変された、またはバイオマスが増強されたトランスジェニック植物細胞、植物体、植物種子または他の植物部分が提供される。
【0115】
「改変されたフルクタン生合成特徴」とは、形質転換植物が、非形質転換対照植物と比べてフルクタン生合成の増加を示すことおよび/またはより高レベルの可溶性炭水化物を含むことを意味する。
【0116】
好ましい一実施形態において、バイオマスが増強されたトランスジェニック植物細胞、植物体、植物種子または他の植物部分は、非形質転換対照植物と比べてバイオマスが少なくとも約15%、より好ましくは少なくとも約25%、より好ましくは少なくとも約35%、より好ましくは少なくとも約50%増加する。
【0117】
例えば、非形質転換対照植物と比べてバイオマスが約15%から500%の間、より好ましくは約25%から300%の間、より好ましくは約35%から200%の間、より好ましくは約50%から100%の間増加していてもよい。
【0118】
好ましい一実施形態において、フルクタン生合成特徴が改変されたトランスジェニック植物細胞、植物体、植物種子または他の植物部分は、非形質転換対照植物と比べて可溶性炭水化物が少なくとも約15%、より好ましくは少なくとも約25%、より好ましくは少なくとも約35%、より好ましくは少なくとも約50%増加する。
【0119】
例えば、非形質転換対照植物と比べて可溶性炭水化物が約15%から500%の間、より好ましくは約25%から300%の間、より好ましくは約35%から200%の間、より好ましくは約50%から100%の間増加する。
【0120】
好ましくは、トランスジェニック植物細胞、植物体、植物種子または他の植物部分は、本発明に係る方法によって作出される。
【0121】
本発明はまた、本発明の植物細胞に由来し、本発明の遺伝的構築物またはベクターを含むトランスジェニック植物体、植物種子または他の植物部分も提供する。
【0122】
本発明はまた、本発明の植物に由来し、本発明の遺伝的構築物またはベクターを含むトランスジェニック植物体、植物種子または他の植物部分も提供する。
【0123】
好ましくは、トランスジェニック植物細胞、植物体、植物種子または他の植物部分は、Lolium属の種、より好ましくはLolium perenneまたはLolium arundinaceumである。
【0124】
好ましくは、トランスジェニック植物細胞、植物体、植物種子または他の植物部分は、穀物類、より好ましくはTriticum属の種、より好ましくはコムギ(Triticum aestivum)である。
【0125】
例えば、本発明は、草食動物の栄養改善のための、葉身におけるフルクタンが増加し、活発に生長し、非生物的ストレスに対しより高い耐性を有するトランスジェニック−ペレニアルライグラス植物の作出を可能にする。
【0126】
本発明はまた、例えばバイオ燃料生産または動物の食餌に利用可能な炭水化物の質量増加のための、フルクタンが増加し、活発に生長し、非生物的ストレスに対する耐性を有するトランスジェニックコムギ植物の作出も可能にする。
【0127】
「植物細胞」とは、半透膜によって仕切られた、色素体を含む任意の自己増殖細胞を意味する。このような細胞は、さらなる増殖が望ましい場合、細胞壁も必要とする。本明細書における植物細胞は、藻類、ラン藻、種子懸濁培養物、胚、成長点領域、カルス組織、葉、根、シュート、配偶体、胞子体、花粉および小胞子を含むが、これらに限定されるものではない。
【0128】
「トランスジェニック」とは、技巧的手法によって細胞に挿入され、その細胞から発生した生物のゲノムの一部となったDNA配列を含む、任意の細胞を意味する。本明細書において、トランスジェニック生物は一般にトランスジェニック植物であり、DNA(導入遺伝子)は、技巧的手法によって核または色素体ゲノムのいずれかに挿入される。
【0129】
次に、添付の実施例および図面を参照することにより本発明をさらに十分に説明する。なお、次の記述は具体例としてのものであるに過ぎず、決して上述の本発明の一般性を限定するものと考えるべきではないことを理解するべきである。
【図面の簡単な説明】
【0130】
【図1】図1は、Bacillus subtilisから得られたSacBタンパク質、GH68ファミリーメンバーの略図である。示されている4個の異なる領域は、N末端シグナル配列、N末端可変領域、触媒コアおよびC末端可変領域である。触媒三連構造(D86、D247およびE342)およびスクロース結合(W85、W163およびR246)を含むアミノ酸残基。
【図2】図2は、Bacillus subtilisから得られたSacB遺伝子(レバンスクラーゼ)のヌクレオチド配列を示す図である。N末端シグナルペプチドをコードするヌクレオチド配列を太字で示す。
【図3】図3は、Bacillus subtilisから得られたSacBタンパク質(レバンスクラーゼ)のアミノ酸配列を示す図である。N末端シグナルペプチドを太字で示す。
【図4】図4は、Lactobacillus johnsonii NCC 533から得られたLsc遺伝子(イヌロスクラーゼ)のヌクレオチド配列を示す図である。N末端シグナルペプチドをコードするヌクレオチド配列を太字で示す。
【図5】図5は、Lactobacillus johnsonii NCC 533から得られたLscタンパク質(イヌロスクラーゼ)のアミノ酸配列を示す図である。N末端シグナルペプチドを太字で示す。
【図6】図6は、I.batatasから得られたプレプロスポラミンタンパク質であるSPOR531のヌクレオチド配列を示す図である。液胞ターゲティングシグナル配列を太字・下線で示す。
【図7】図7は、I.batatasから得られたプレプロスポラミンタンパク質であるSPOR531のアミノ酸配列を示す図である。液胞ターゲティングシグナル配列を太字・下線で示す。
【図8】図8は、SPOR:SacBキメラヌクレオチド配列を示す図である。SacBのN末端シグナル配列をSPORの液胞ターゲティングシグナル(太字・下線で示す)に置き換えてある。
【図9】図9は、SPOR:SacBキメラタンパク質配列を示す図である。SacBのN末端シグナル配列をSPORの液胞ターゲティングシグナル(太字・下線で示す)に置き換えてある。
【図10】図10は、膜タンパク質の二次構造予測ソフトウェアSOSUI、http://bp.nuap.nagoya−u.ac.jp/sosui/を用いたSPOR−SacB融合タンパク質の二次構造予測を示す図である。
【図11】図11は、SPOR:Lscキメラヌクレオチド配列を示す図である。LscのN末端シグナル配列をSPORの液胞ターゲティングシグナル(太字・下線で示す)に置き換えてある。
【図12】図12は、SPOR:Lscキメラタンパク質配列を示す図である。LscのN末端シグナル配列をSPORの液胞ターゲティングシグナル(太字・下線で示す)に置き換えてある。
【図13】図13は、膜タンパク質の二次構造予測ソフトウェアSOSUI、http://bp.nuap.nagoya−u.ac.jp/sosui/を用いたSPOR−Lsc融合タンパク質の二次構造予測を示す図である。
【図14】図14は、膜タンパク質の二次構造予測ソフトウェアSOSUI、http://bp.nuap.nagoya−u.ac.jp/sosui/を用いたLp1−SSTの二次構造予測を示す図である。
【図15】図15は、L.perenneから得られたLp1−SSTヌクレオチド配列を示す図である。Lp1−SST膜貫通ドメインのコード配列を太字・斜字体で示す。
【図16】図16は、L.perenneから得られたLp1−SSTタンパク質配列を示す図である。Lp1−SST膜貫通ドメインを太字・斜字体で示す。
【図17】図17は、Lp1−SST−SacBキメラヌクレオチド配列を示す図である。SacBのN末端シグナルのコード配列をLp1−SST膜貫通ドメインのコード配列(太字・斜字体で示す)に置き換えてある。
【図18】図18は、Lp1−SST−SacBキメラタンパク質配列を示す図である。SacBのN末端シグナル配列をLp1−SST膜貫通ドメイン(太字・斜字体で示す)に置き換えてある。
【図19】図19は、膜タンパク質の二次構造予測ソフトウェアSOSUI、http://bp.nuap.nagoya−u.ac.jp/sosui/を用いたLp1−SST−SacB融合タンパク質の二次構造予測を示す図である。
【図20】図20は、Lp1−SST−Lscキメラヌクレオチド配列を示す図である。LscのN末端シグナルのコード配列をLp1−SST膜貫通ドメインのコード配列(太字・斜字体で示す)に置き換えてある。
【図21】図21は、Lp1−SST−Lscキメラタンパク質配列を示す図である。LscのN末端シグナル配列をLp1−SST膜貫通ドメイン(太字・斜字体で示す)に置き換えてある。
【図22】図22は、膜タンパク質の二次構造予測ソフトウェアSOSUI、http://bp.nuap.nagoya−u.ac.jp/sosui/を用いたLp1−SST−Lsc融合タンパク質の二次構造予測を示す図である。
【図23−1】図23は、AtRbcS::SPOR−SacB::NOS発現カセットのヌクレオチド配列を示す図である。
【図23−2】図23は、AtRbcS::SPOR−SacB::NOS発現カセットのヌクレオチド配列を示す図である。
【図24−1】図24は、AtRbcS::SPOR−Lsc::NOS発現カセットのヌクレオチド配列を示す図である。
【図24−2】図24は、AtRbcS::SPOR−Lsc::NOS発現カセットのヌクレオチド配列を示す図である。
【図25−1】図25は、AtRbcS::Lp1−SST−SacB::NOS発現カセットのヌクレオチド配列を示す図である。
【図25−2】図25は、AtRbcS::Lp1−SST−SacB::NOS発現カセットのヌクレオチド配列を示す図である。
【図26−1】図26は、AtRbcS::Lp1−SST−Lsc::NOS発現カセットのヌクレオチド配列を示す図である。
【図26−2】図26は、AtRbcS::Lp1−SST−Lsc::NOS発現カセットのヌクレオチド配列を示す図である。
【図27】図27は、Gateway pDestinationベクターを示す図である。
【図28A】図28は、双子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.AtRbcS::SPOR−SacB::NOS;B.AtRbcS::SPOR−Lsc::NOS;C.AtRbcS::Lp1−SST−SacB::NOS、およびD.AtRbcS::Lp1−SST−Lsc::NOS。
【図28B】図28は、双子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.AtRbcS::SPOR−SacB::NOS;B.AtRbcS::SPOR−Lsc::NOS;C.AtRbcS::Lp1−SST−SacB::NOS、およびD.AtRbcS::Lp1−SST−Lsc::NOS。
【図28C】図28は、双子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.AtRbcS::SPOR−SacB::NOS;B.AtRbcS::SPOR−Lsc::NOS;C.AtRbcS::Lp1−SST−SacB::NOS、およびD.AtRbcS::Lp1−SST−Lsc::NOS。
【図28D】図28は、双子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.AtRbcS::SPOR−SacB::NOS;B.AtRbcS::SPOR−Lsc::NOS;C.AtRbcS::Lp1−SST−SacB::NOS、およびD.AtRbcS::Lp1−SST−Lsc::NOS。
【図29】図29は、TaRbcSp::SPOR−SacB::TaRbcst発現カセットのヌクレオチド配列を示す図である。
【図30−1】図30は、TaRbcSp::SPOR−SacB::TaRbcst TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図30−2】図30は、TaRbcSp::SPOR−SacB::TaRbcst TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図31−1】図31は、TaRbcSp::SPOR−Lsc::TaRbcst発現カセットのヌクレオチド配列を示す図である。
【図31−2】図31は、TaRbcSp::SPOR−Lsc::TaRbcst発現カセットのヌクレオチド配列を示す図である。
【図32−1】図32は、TaRbcSp::SPOR−Lsc::TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図32−2】図32は、TaRbcSp::SPOR−Lsc::TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図33−1】図33は、TaRbcSp::Lp1−SST−SacB::TaRbcst発現カセットのヌクレオチド配列を示す図である。
【図33−2】図33は、TaRbcSp::Lp1−SST−SacB::TaRbcst発現カセットのヌクレオチド配列を示す図である。
【図34−1】図34は、TaRbcSp::Lp1−SST−SacB::TaRbcst TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図34−2】図34は、TaRbcSp::Lp1−SST−SacB::TaRbcst TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図35−1】図35は、TaRbcSp::Lp1−SST−Lsc::TaRbcst発現カセットのヌクレオチド配列を示す図である。
【図35−2】図35は、TaRbcSp::Lp1−SST−Lsc::TaRbcst発現カセットのヌクレオチド配列を示す図である。
【図36−1】図36は、TaRbcSp::SPOR−Lsc::TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図36−2】図36は、TaRbcSp::SPOR−Lsc::TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図36−3】図36は、TaRbcSp::SPOR−Lsc::TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図37】図37は、Gateway pDestinationベクター、pBS:ubi::bar::NOSを示す図である。
【図38】図38は、単子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.TaRbcS::SPOR−SacB::TaRbcS、およびB.TaRbcS::SPOR−SacB::TaRbcS+AtMYB32::IPT::35S
【図39】図39は、単子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.TaRbcS::SPOR−Lsc::TaRbcS、およびB.TaRbcS::SPOR−Lsc::TaRbcS+AtMYB32::IPT::35S
【図40】図40は、単子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.TaRbcS::Lp1−SST−SacB::TaRbcS、およびB.TaRbcS::Lp1−SST−SacB::TaRbcS+AtMYB32::IPT::35S
【図41】図41は、単子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.TaRbcS::Lp1−SST−Lsc::TaRbcS、およびB.TaRbcS::Lp1−SST−Lsc::TaRbcS+AtMYB32::IPT::35S
【図42】図42は、Nicotiana tabacumの葉肉由来プロトプラストの単離を示す図である。A)〜B)4〜6週目のin vitro葉材料の解剖、前酵素消化;C)4〜6週目のin vitro葉材料の消化、16時間のインキュベーション;D)プロトプラスト懸濁液の回収;E)プロトプラスト豊富界面の分離;F)〜G)インタクトな葉緑体豊富プロトプラスト
【図43】図43は、一過性発現解析のためのNicotiana tabacumの葉肉由来プロトプラストの単離を示す図である。A)〜B)インタクトな葉緑体豊富プロトプラスト;C)液体濃縮培地におけるプロトプラストの培養;D)生存プロトプラスト、単離48時間後
【図44】図44は、安定的な形質転換のためのNicotiana tabacumの葉肉由来プロトプラストの単離を示す図である。A)〜B)インタクトな葉緑体豊富プロトプラスト;C)プロトプラストを包埋したsea plaqueアガロースプラグ、0日目;D)生存プロトプラスト、単離および包埋6日後;E)〜F)包埋され、解放されたプロトプラスト由来の微小カルス、単離および包埋4週間後
【図45】図45は、Nicotiana tabacumの葉肉プロトプラスト由来の微小カルスからシュートの再生を示す図である。A)液体成長培地Aに解放された微小カルス、単離および包埋6〜7週間後;B〜C)固形成長培地におけるカルスの増殖;D)〜E)葉肉プロトプラスト由来のカルスからシュートの誘導および再生;F)再生シュートから根の発達;G)〜H)ガラス温室内に封じ込めた植物体の生長および発達
【図46】図46は、非形質転換プロトプラスト生存率の評価を示す図である。単離48時間後。
【図47】図47は、PEG形質転換プロトプラストの生存率の評価を示す図である。単離およびトランスフェクション48時間後。
【図48】図48は、タバコリーフディスクのアグロバクテリウムを介した形質転換を示す図である。A)形質転換リーフディスクの共存培養、0日目;B)ステージ1シュートの開始、共存培養3日後
【図49−1】図49は、タバコ形質転換リーフディスクにおけるgfp発現の検出を示す図である。A)非形質転換リーフディスク、白色光;B)非形質転換リーフディスク、gfp2フィルター;C)非形質転換リーフディスク、gfp3フィルター;D)&G)Turbo gfp形質転換リーフディスク、白色光;E)&H)Turbo gfp形質転換リーフディスク、gfp2フィルター;F)&I)Turbo gfp形質転換リーフディスク、gfp3フィルター。
【図49−2】図49は、タバコ形質転換リーフディスクにおけるgfp発現の検出を示す図である。A)非形質転換リーフディスク、白色光;B)非形質転換リーフディスク、gfp2フィルター;C)非形質転換リーフディスク、gfp3フィルター;D)&G)Turbo gfp形質転換リーフディスク、白色光;E)&H)Turbo gfp形質転換リーフディスク、gfp2フィルター;F)&I)Turbo gfp形質転換リーフディスク、gfp3フィルター。
【図50】図50は、RT−PCR試料および対照の電気泳動を示す図である。レーン1および13:1kb+DNAラダーレーン2:トランスフェクトしたプロトプラスト1(試料9A)から得られたmRNAの逆転写によって生成したcDNA(2μL)から、遺伝子特異的プライマーによるsacB転写産物の増幅レーン3:cDNA(1μL)からsacB転写産物の増幅(試料9A)レーン4:逆転写酵素なしで行った対照反応(試料9A)レーン5:鋳型なしで行った対照反応(sacBプライマー)レーン6:トランスフェクトしたプロトプラスト1(試料12A)から得られたmRNAの逆転写によって生成したcDNA(2μL)から、遺伝子特異的プライマーによるsacB転写産物の増幅レーン7:cDNA(1μL)からsacB転写産物の増幅(試料12A)レーン8:逆転写酵素なしで行った対照反応(試料12A)レーン9:非トランスフェクトプロトプラストから得られたmRNAの逆転写によって生成したcDNA(2μL)から、遺伝子特異的プライマーによる18S転写産物の増幅レーン10:非トランスフェクトプロトプラストから得られたmRNAの逆転写によって生成したcDNA(1μL)から、遺伝子特異的プライマーによる18S転写産物の増幅レーン11:逆転写酵素なしで行った対照反応(非トランスフェクトプロトプラスト)レーン12:鋳型なしで行った対照反応(18Sプライマー)。
【発明を実施するための形態】
【0131】
(実施例1)
細菌フルクタン生合成遺伝子の単離
図1は、Bacillus subtilisから得られたSacBタンパク質の略図を提示する。示されている4個の異なる領域は、N末端シグナル配列、N末端可変領域、触媒コアおよびC末端可変領域である。構造的には、細菌イヌロスクラーゼおよびレバンスクラーゼの多くは、N末端シグナルペプチド、触媒三連構造を共有する。この配列は配列改変の際に除去される。スクロース結合に関与する残基は触媒コア配列内部に位置するが、これは改変の際に手をつけずにおく。
【0132】
細菌レバンスクラーゼ(SacB)およびイヌロスクラーゼ(Lsc)のヌクレオチドおよびタンパク質配列をそれぞれ図2〜5に提供する。しかし、植物形質転換のため、細菌レバンスクラーゼおよびイヌロスクラーゼ配列は次の仕方によっても改変される。
【0133】
・細菌N−シグナルペプチドの除去、
・翻訳開始等、コドン使用の単子葉植物および双子葉植物への適応、
・潜在性スプライス部位およびRNA不安定化配列エレメントの除去、
・単子葉植物および双子葉植物の液胞ターゲティング配列ならびに植物1−SST特異的膜貫通ドメイン等、推定細胞内ターゲティング配列を用いてコード配列をさらに改変する。
【0134】
これらの変化の概略を次の実施例に記す。
【0135】
(実施例2)
細菌フルクタン生合成遺伝子の改変
細菌FT遺伝子の特異的細胞区画へのターゲティング
細菌FT遺伝子をサイトゾルから離れてスクロースおよびフルクタンの両方が蓄積する区画に方向付けるため、サツマイモ(Ipomoea batatas)のプレプロスポラミンタンパク質(SPOR531)由来の推定液胞ターゲティング配列を用いてSacBおよびLscのコード配列を改変する。スポラミン前駆体のプロペプチドは、スポラミンの液胞へのターゲティングに必要とされる(Hattoriら、1985年)。スポラミンの液胞ターゲティング情報はアミノ末端プロペプチドにコードされ、これは図5および図6に示されている。
【0136】
配列改変は、SacBおよびLsc細菌フルクタン(fuctan)生合成遺伝子の両方からN−シグナルペプチドを除去すること、およびSPOR531液胞ターゲティングシグナルを付加することに関与する(それぞれ図7〜8および図10〜11)。
【0137】
膜タンパク質の二次構造予測ソフトウェアSOSUI、http://bp.nuap.nagoya−u.ac.jp/sosui/を用いた細胞内局在の予測および改変されたタンパク質のトポロジーは、液胞ターゲティングシグナルによって誘発された膜貫通局在を示す(図9および図12)。
【0138】
細菌FT遺伝子へのLp1−SSTタンパク質由来膜貫通ドメインの付加
SOSUIソフトウェアは、Lolium perenne 1−SST遺伝子の二次構造を予測するためにも用いた。N末端に膜貫通ドメインを示唆するこの構造を図13に示す。膜貫通ドメインコード配列およびタンパク質配列をそれぞれ図14および図15に示す。
【0139】
配列改変は、SacBおよびLsc細菌フルクタン生合成遺伝子の両方からN−シグナルペプチドを除去すること、およびLp1−SST膜貫通ドメインを付加することに関与する(それぞれ図16〜17および図19〜20)。膜タンパク質の二次構造予測ソフトウェアSOSUIを用いて、SacBおよびLscの改変された配列の細胞内(subsellular)局在およびタンパク質トポロジーを評価し、それらの予測二次構造をそれぞれ図18および図21に提示する。
【0140】
(実施例3)
双子葉植物における安定的な形質転換のためのベクターの作製
発現構築物の合成
双子葉植物に形質転換するための、光合成(photosyntheic)プロモーター、実施例2に示されている改変された細菌フルクタン生合成遺伝子およびNOSターミネーター配列を利用した発現構築物を人工合成する。
【0141】
光合成プロモーターの使用はフルクタンを蓄積する組織において遺伝子を発現させ、一方改変された配列はタンパク質を特異的植物細胞区画に標的化させる。
【0142】
リブロース−1,5−二リン酸カルボキシラーゼ−オキシゲナーゼ−スモールサブユニット(RbcS)は、高等植物における十分に特徴付けられた光調節遺伝子である。Arabidopsis thalianaリブロース−1,5−二リン酸カルボキシラーゼ−オキシゲナーゼ−スモールサブユニット(AtRbcS)プロモーター配列の1700bp断片は、以前にクローニングされている。発現ベクターで用いるため、AtRbcSプロモーター由来のTATAシグナルを含む、より短い断片を増幅するためのプライマーを設計する。
【0143】
改変された細菌フルクタン生合成遺伝子のための新たな予測配列は、植物における発現のためにコドン使用を変更させ、潜在性スプライス部位およびRNA不安定化配列エレメントを除去しつつ人工合成し、植物細胞におけるその性能を最適化する。
【0144】
図23〜26は、それぞれ発現カセットAtRbcS::SPOR−SacB::NOS、AtRbcS::SPOR−Lsc::NOS、AtRbcS::Lp1−SST−SacB::NOSおよびAtRbcS::Lp1−SST−Lsc::NOSを表し、これらはまだコドン最適化または不安定化エレメントの除去を行っていない。
【0145】
シロイヌナズナ光合成プロモーターによって駆動される改変された細菌FT遺伝子を含む双子葉植物形質転換のための構築物の作製
各合成発現カセットを、Gateway法に使用可能なpDONORベクター内に配置して、植物の形質転換用の最終的な目的ベクターに組み換える。
【0146】
35Sp:hph:35St選択マーカーカセットを含む、Gateway法に使用可能な目的ベクターを作製した。pPZP200_35Sp_hph_35St_R4/R3(図27)。
【0147】
Gateway LR組換え反応は、次の双子葉植物の形質転換用のdestinationベクターを作製する。
AtRbcS::SPOR−SacB::NOS(図28A)
AtRbcS::SPOR−Lsc::NOS(図28B)
AtRbcS::Lp1−SST−SacB::NOS(図28c)
AtRbcS::Lp1−SST−Lsc::NOS(図28d)
(実施例4)
単子葉植物における安定的な形質転換のためのベクターの作製
発現構築物の合成
単子葉植物の形質転換用の、パンコムギ光合成プロモーター(TaRbcsp)、実施例2に示されている改変された細菌フルクタン生合成遺伝子およびTaRbcSターミネーター配列を利用した発現構築物を人工合成する。光合成プロモーターの使用はフルクタンを蓄積する組織において遺伝子を発現させ、一方改変された配列はタンパク質を特異的植物細胞区画に標的化させる。
【0148】
パンコムギ(Triticum aestivum)のTaRbcS調節配列(プロモーターおよびターミネーター)は、以前にクローニングされている(Zengら、1995年;Sasanuma、2001年)。発現ベクターで用いるため、TaRbcS遺伝子(NCBI受託番号AB042069)由来のTATAシグナルを含む、以前に発表された配列から得られた695bpプロモーター断片を増幅する。
【0149】
改変された細菌フルクタン生合成遺伝子のための新たな予測配列を、植物における発現のためにコドン使用を変更させ、潜在性スプライス部位およびRNA不安定化配列エレメントを除去しつつ人工合成して、植物細胞におけるその性能を最適化する。
【0150】
上に概略されている方法を用いて発現カセットを合成し、フルクタン生合成遺伝子およびLXR(商標)技術の両方を含むトランスジェニック植物を作出する。LXR(商標)技術は、AtMYB32遺伝子プロモーター制御下における葉老化遅延のための1種類の候補遺伝子(IPT)を含む発現カセットに基づく。発現カセットAtMYB3p::IPT::35Stは、国際特許出願PCT/AU01/01092号明細書に記載されている。トランスジェニックLXR(商標)植物の表現型は、植物の加齢に関連する葉の黄化およびクロロフィル喪失の減少を含み、これは光合成能の上昇を導き、分げつ化および植物バイオマスの改善をもたらす。
【0151】
2種の技術の統合は、光合成プロモーター活性化の拡大によるフルクタンの発現上昇をもたらし、植物における生産性の範囲を増加させることにより種々の適用の効力に有意な影響を与えることができる。
【0152】
図29〜36は、それぞれ発現カセットTaRbcS::SPOR−SacB::TaRbcS、TaRbcS::SPOR−SacB::TaRbcS+AtMYB32::IPT::35S、TaRbcS::SPOR−Lsc::TaRbcS、TaRbcS::SPOR−Lsc::TaRbcS+AtMYB32::IPT::35S、TaRbcS::Lp1−SST−SacB::TaRbcS、TaRbcS::Lp1−SST−SacB::TaRbcS+AtMYB32::IPT::35S、TaRbcS::Lp1−SST−Lsc::TaRbcSおよびTaRbcS::Lp1−SST−Lsc::TaRbcS+AtMYB32::IPT::35Sを表し、これらはまだコドン最適化または不安定化エレメントを除去されていない。
Triticum属の光合成プロモーターによって駆動される改変された細菌FT遺伝子を含む単子葉植物形質転換のための構築物の作製
各合成発現カセットを、Gateway法に使用可能なpDONORベクター内に配置して、植物の形質転換用の最終的な目的ベクターに組み換える。
【0153】
Ubi::bar::NOS選択マーカーカセットを含む、Gateway法に使用可能な目的ベクターを作製した。pBS::Ubi::bar::NOS_R4/R3(図37)。Gateway LR組換え反応は、次の単子葉植物の形質転換用のdestinationベクターを作製する。
TaRbcS::SPOR−SacB::TaRbcS(図38A)
TaRbcS::SPOR−SacB::TaRbcS+AtMYB32::IPT::35S(図B);
TaRbcS::SPOR−Lsc::TaRbcS(図39A)
TaRbcS::SPOR−Lsc::TaRbcS+AtMYB32::IPT::35S(図39B)
TaRbcS::Lp1−SST−SacB::TaRbcS(図40A)
TaRbcS::Lp1−SST−SacB::TaRbcS+AtMYB32::IPT::35S(図B)
TaRbcS::Lp1−SST−Lsc::TaRbcS(図41A)
TaRbcS::Lp1−SST−Lsc::TaRbcS+AtMYB32::IPT::35S(図41B)
(実施例5)
双子葉植物用構築物 − N.tabacumプロトプラストおよびA.thaliana
直接送達バージョン(プロトプラストにおける一過性発現)ならびにシロイヌナズナ(A.thaliana)およびタバコ(N.tabacum)への安定的な送達のためのバイナリー形質転換ベクターバージョンの、次の構築物を作製した。
1.AtRbcS::1−SST−SacB*
2.AtRbcS::SPOR−SacB*
3.AtRbcS::1−SST−Lsc*
4.AtRbcS::SPOR−Lsc*
*細菌レバンスクラーゼおよびイヌロスクラーゼ配列は、次の仕方で改変する:
・細菌N−シグナルペプチドの除去、
・翻訳開始等、コドン使用の単子葉植物および双子葉植物への適応、
・潜在性スプライス部位およびRNA不安定化配列エレメントの除去、
・単子葉植物および双子葉植物の液胞ターゲティング配列ならびに植物1−SSTおよびFFT特異的膜貫通ドメイン等、推定細胞内ターゲティング配列を用いてコード配列をさらに改変する。
【0154】
(実施例6)
ポリエチレングリコールを介したタバコ葉肉由来プロトプラストの形質転換
本実施例は、上文に記載されている発現カセットのタバコプロトプラストへの送達を説明する(図42〜45を参照)。
【0155】
I.直接遺伝子導入のための葉肉由来プロトプラストの単離
A.葉肉由来プロトプラストを得るためのin vitroシュート培養物の消化
酵素溶液
K4培地[0.3Mではなく0.4Mスクロースを含むK3培地]に溶解した1.0%(w/v)セルラーゼ「Onozuka」R10および1.0%(w/v)Macerozyme(登録商標)R10。酵素標品の混入デンプンをペレット化させるため、スピンダウンする(Sorvall遠心分離機、SS34ローター;7,000rpmで10分間)。KOHでpH5.6に調整し、濾過滅菌(0.2μm孔径)する。4℃で長くて3〜4週間保存する。
【0156】
材料
・固形MS培地とNicotiana tabacumシュート培養物の入った400ml培養容器
・90×20mm滅菌ペトリ皿
・ピンセット
・メス
・滅菌メス刃;#11または#22 。
【0157】
溶液
・酵素溶液;K4培地に溶解した1%セルラーゼおよび1%マセロザイム
・滅菌水 。
【0158】
手順
1.プレート底部をたっぷりと覆うのに十分な量の酵素溶液(15mlで十分と思われる)を滅菌90×20mmペトリ皿にデカントする。
2.4〜6週目のシュート培養物から健常で十分に開いた葉を2〜4枚、空の90×20mmペトリ皿に移す。
3.1枚の葉の背軸面を上にして、周囲の葉組織の断裂を最小限に抑えつつ鋭利な滅菌刃を確実に用いて慎重に主脈を除去する。これを残り3枚の葉に繰り返す。1回当たり少量の葉材料(最大4枚)を取り扱い、層流による乾燥効果を最小限に抑える。
4.半分にした葉を穏やかに積み重ね、鋭利な滅菌刃を用いて1〜2mm片にスライスする。
5.葉切片を酵素溶液の入ったペトリ皿に慎重に移す(背軸面を下に)。皿をParafilm(登録商標)で封じ、16〜18時間、25℃、暗所で振盪せずに一晩インキュベートする。
【0159】
B.葉肉由来プロトプラストの単離
材料
・滅菌5mlピペット
・滅菌10mlピペット
・ピペットボーイ
・滅菌したプロトプラスト濾過ユニット:100mlガラスビーカー上に載せた100μmステンレス鋼メッシュ篩
・滅菌14mlプラスチック製丸底遠心チューブ
・Clements Orbital 500卓上遠心分離機
・ウォーターバス 。
【0160】
培地
・消化中のNicotiana tabacum葉の入った90×20mm滅菌ペトリ皿(複数可)
・0.6%Sea Plaque(商標)アガロースを含む、1:1混合のK3:H固形培地 。
【0161】
溶液
・オートクレーブしたW5溶液
・オートクレーブしたK3溶液 。
【0162】
手順
6.一晩消化した標本を穏やかに撹拌して酵素溶液中にプロトプラストを遊離させる。
【0163】
撹拌は穏やかに、だが徹底的に行い、左右に(水平に)動かして行うこと。
7.プレートをやや傾け、消化中の懸濁液(酵素溶液および植物デブリ)を移し易くする。10ml滅菌ピペットを用いて消化中の懸濁液を滅菌したプロトプラスト濾過ユニットに移し、プロトプラスト懸濁液を植物デブリから分離する。
8.濾過ユニットを穏やかにタップして篩内にたまった余分な液体を出す。
9.プロトプラスト懸濁液を穏やかに混合し、14ml滅菌プラスチック製丸底遠心チューブに分配し、約8mlまで(最大9ml)充填する。
10.懸濁液を再分配して均一な容量分配を得る(最大9ml)。
11.各懸濁液に1.5mlのW5溶液を慎重に重層する。
W5溶液を分注し易くするため、懸濁液の入ったチューブを傾けて置き、ピペットチップをチューブ開口部近くの壁面に触れさせ、その後懸濁液面のすぐ上までゆっくりと下げる。一滴ずつ加えながら、ピペットチップが可能な限り懸濁液面に近づいた状態を確実に維持しつつW5溶液をゆっくりと分注する。これを正確に行えば、プロトプラスト懸濁液の撹拌、ひいてはW5溶液との混合は最小限となるであろう。
12.慎重に蓋をし、チューブを5分間、70gで遠心分離する(Clements Orbital 500卓上遠心分離機、スイングアウトローター、400rpm)。プロトプラストは、界面に浮遊している筈である。
13.プロトプラストの入ったチューブを直立に維持し、滅菌5mlピペットをプロトプラスト層のすぐ上のポイントまで慎重に下げて、下相を可能な限り取らないよう界面におけるプロトプラストを採取する。
W5溶液も同時に採取される。
14.プロトプラストを採取し、「1本」の新しい14ml遠心チューブに移す。プロトプラスト採取が完了したら、ピペッティングで穏やかに吸って吐くことによりプロトプラスト懸濁液を穏やかに混合する。
15.プロトプラスト懸濁液の100μlアリコートを取り分け、900μlのW5溶液の入ったチューブに移すことにより、プロトプラスト収量を決定する。血球計算板でプロトプラストを計数し、1ml当たりのプロトプラスト数を決定する。
16.1ml当たり約1×106(最大1.5×105)個のプロトプラストを得るのに必要な全容量を算出する。新しい14ml丸底遠心チューブにプロトプラスト懸濁液を分配し、等容量が得られたことを確実にする。
17.10mlピペットを用いて、プロトプラストの入った各チューブを全容量最大10mlまでW5溶液で充填する。プロトプラストが乱れるのを最小限に抑えるため、充填の際チューブ壁に沿わせてW5溶液を散布する。
18.蓋をし、蓋閉めしたチューブを穏やかに1回反転することによってプロトプラストを再懸濁する。
19.プロトプラストをペレット化し[70g(Clements Orbital 500卓上遠心分離機、400rpm)5分間でスピンする]、その後W5溶液を全て除去して純粋なプロトプラスト懸濁液を残す。
20.穏やかに振盪することによりプロトプラスト懸濁液を再懸濁する。
21.プロトプラストの入った各チューブを全容量5mlまでW5溶液で充填し、室温で最短で1時間、最長で4時間インキュベートする。
22.1〜4時間のインキュベーション時間の間に、単離プロトプラストに直接遺伝子導入するための調製物に次の構成要素を構成する。
【0164】
・40%PEG溶液を−20℃保存から取り出し、室温で保存する。直接遺伝子導入に進む30分前に、PEG溶液を温水の入ったビーカー内でインキュベートする。
【0165】
・K3:H固形培地を電子レンジで融解させる。完全に融解したら、使用準備が整うまで40℃のウォーターバス内に置く。
【0166】
II.ポリエチレングリコールを用いたプロトプラストへの直接遺伝子導入
DNA形質転換
100%(v/v)エタノールで沈殿および洗浄することによりプラスミドDNAを滅菌し、層流フード内で乾かす[70%エタノールにおけるプラスミドDNAの沈殿も可能であるが、DNAペレットは乾かすのにさらに時間がかかる]。一過性形質転換のため、終濃度0.7μg/μlになるようDNAペレットを30μlの滅菌再蒸留水に再懸濁する。DNAの物理的構造は、一過性のためにはスーパーコイルとなるべきであり、安定的な形質転換のためには直鎖化するべきである(対象遺伝子の外側で)。形質転換プラスミドDNAにキャリアーDNA(例えば、魚精子DNA)を添加すると、通常、より良好で安定的な形質転換頻度が得られる。安定的な形質転換のため、上に示す通り10μgの直鎖化プラスミドDNAおよび40μgの剪断した魚精子DNAを共沈殿し、乾かし、30μlの滅菌再蒸留水に溶解する。
【0167】
形質転換バッファー
15mM MgCl2、0.1%(w/v)モルホリノエタンスルホン酸(MES)および0.5Mマンニトール。蒸留水に溶解した後、KOHでpH5.8に調整し、オートクレーブする。4℃で保存する。
【0168】
PEG溶液
0.4Mマンニトールおよび0.1M Ca(NO3)2中の40%(w/v)PEG4000。PEGを0.4Mマンニトールおよび0.1M Ca(NO3)2に溶解する(これら2種の構成要素の終濃度は、PEGの容量のため低くなる)。pH8〜9に調整し、オートクレーブする(pHはこの溶液中で安定化するのに数時間、例えば一晩かかり、オートクレーブ後にpH6〜7に下がる)。
【0169】
材料
・滅菌1mlピペット
・滅菌5mlピペット
・滅菌10mlピペット
・ピペットボーイ
・滅菌14mlプラスチック製丸底遠心チューブ
・Clements Orbital 500卓上遠心分離機
・ウォーターバス
・50×10mmペトリ皿 。
【0170】
培地
・単離プロトプラスト懸濁液、ペレット化の入った14ml丸底遠心チューブ
・0.6%Sea Plaque(商標)アガロースを含む1:1混合のK3:H固形培地 。
【0171】
溶液
・オートクレーブしたW5溶液
・オートクレーブしたK3溶液
・オートクレーブしたH溶液
・40%PEG溶液
・形質転換バッファー
・30μlの滅菌再蒸留水に溶解した10μgの形質転換DNA 。
【0172】
手順
1.プロトプラストをペレット化し[70g(Clements Orbital 500卓上遠心分離機、400rpm)で5分間スピンする]、その後W5溶液を全て除去し、純粋なプロトプラスト懸濁液を残す。
2.1mlピペットを用いて、300μl(約7滴)の形質転換バッファーを単離プロトプラストの入った各14ml丸底遠心チューブに加える(滴下する)。
3.チューブ底を穏やかにタップすることによってペレットを慎重に再懸濁する。
4.各プロトプラスト懸濁液に10μg(30μl)の形質転換DNAを加え、その後300μl(1mlピペットを分注に用いる場合、約7滴)の予熱したPEG溶液を加える。チューブ底を穏やかにタップすることによってプロトプラスト懸濁液を混合する。
形質転換バッファーへのプロトプラスト再懸濁と形質転換DNAおよびPEGの添加との間の時間間隔は、最短を維持しなければならない。
5.形質転換混合物を15分間、室温で撹拌せずにインキュベートする。
6.10mlピペットを用いて、次の間隔で10mlのW5溶液を各チューブに徐々に加える。
【0173】
・1ml(約12滴)を各チューブに滴下する。全チューブを穏やかに反転して混合する。
【0174】
・1ml(約12滴)を各チューブに滴下する。全チューブを穏やかに反転して混合する。
【0175】
・1ml(約12滴)を各チューブに滴下する。全チューブを穏やかに反転して混合する。
【0176】
・2mlを穏やかな流れとして各チューブに加える。全チューブを穏やかに反転して混合する。
【0177】
・2mlを穏やかな流れとして各チューブに加える。全チューブを穏やかに反転して混合する。
【0178】
・3mlを穏やかな流れとして各チューブに加える。全チューブを穏やかに反転して混合する。
【0179】
分注し易くするため、分注前に、各チューブを必要容量のW5溶液で充填するのに各間隔で必要とされる合計容量を10mlピペットで採取する。各間隔でこれを繰り返す。
7.プロトプラストをペレット化し[70g(Clements Orbital 500卓上遠心分離機、400rpm)で10分間スピンする]、その後全W5溶液を除去し、純粋なプロトプラスト懸濁液を残す。次に進む前に、全チューブ底を1回タップする。
【0180】
一過性形質転換用手順
8.合計容量が最大5mlとなるよう、等容量のK3培地およびH培地(各溶液2.5ml)にプロトプラストペレットを再懸濁する。
9.液体K3:H+プロトプラスト懸濁液の混合液を50×10mmペトリ皿の中央にゆっくりと移す。
10.全皿をParafilm(登録商標)で封じ、一過性発現解析に進む前にプロトプラストを24〜72時間、薄明かりの下で24℃にて培養する。
【0181】
安定的な形質転換用手順
11.次の「パートIII.葉肉由来プロトプラストの培養および植物体の再生」に続ける。
【0182】
III.葉肉由来プロトプラストの培養および植物体の再生
ステップ1&2のため、プロトプラストの入った各チューブは、一度に1チューブで取り扱わなければならない。
【0183】
材料
・滅菌1mlピペット
・滅菌5mlピペット
・滅菌10mlピペット
・ピペットボーイ
・Clements Orbital 500卓上遠心分離機
・ウォーターバス
・50×10mmペトリ皿
・オートクレーブしたステンレス鋼製スパチュラ
培地
・単離プロトプラスト懸濁液、ペレット化の入った14ml丸底遠心チューブ
・0.6%Sea Plaque(商標)アガロースを含む1:1混合のK3:H固形培地、40℃
・オートクレーブしたK3溶液
・20mlのA培地の入った250ml培養容器
・固形MS Morpho培地の入った12ウェルCostar(登録商標)プレート
・固形MS Morpho培地の入った250ml培養容器
・固形MS培地の入った250ml培養容器 。
【0184】
手順
1.プロトプラストペレットの近くに0.5mlのK3培地を加えて、プロトプラストを再懸濁する。
2.K3+プロトプラスト懸濁液の混合液を50×10mmペトリ皿の中央にゆっくりと移す。
3.次に進む前に、プロトプラストの入った全チューブに対しステップ1および2を繰り返す。
4.一度に1プレートで、予熱した0.6%Sea Plaque(商標)アガロースを含む1:1混合のK3:H培地を5ml加える。穏やかな旋回運動でプレートを1回だけ振盪して培地中のプロトプラスト懸濁液を均等に分布させる。全プレートに対しこれを繰り返す。
5.培地が固形化するまで(外気温に依存して10〜30分間)、プレートを放置し手をつけない。
6.全皿をParafilm(登録商標)で封じ、プロトプラストを24時間、完全な暗所で24℃にて培養し、続いて6日間連続的な薄明かり(5μmol・m−2s1、Osram L36 W/21 Lumilux白チューブ)の下で培養すると、最初の、そして複数の細胞分裂が行われる。
7.滅菌スパチュラを用いて、プロトプラストを含むsea plaqueアガロースプラグを四分円に分割し、適切な抗生物質を添加した20mlのA培地の入った250mlプラスチック製培養容器内に置く(250ml容器につき1個の四分円)。ロータリーシェーカー上で80rpm、1.25cm throw、24℃、連続的な薄明かりでインキュベートする。
8.2週間毎に液体A培地+適切な抗生物質を交換し、プロトプラスト由来コロニーの生長をモニターする。
9.プロトプラスト由来コロニーが直径約2〜3mmになったとき(液体A培地中で5〜6週間インキュベーション)、コロニーをMS Morpho固形培地の入った24ウェルCostar(登録商標)プレートの個々のウェルに移す。
10.プレート(複数可)を1〜2週間、24℃で連続的な薄明かり(5μmol・m−2s−1、Osram L36 W/21 Lumilux白チューブ)の下でインキュベートすると、カルスは増殖して直径8〜10mmのサイズに達する。
11.プロトプラスト由来カルスが直径約1〜2cmになったとき、カルスを固形MS Morpho培地の入った個々の250ml培養容器に移す。容器を24℃で16時間・明/8時間・暗条件下(20μmol・m−2s−1、Osram L36 W/21 Lumilux白チューブ)でインキュベートする。1〜2週間以内に、複数のシュートを目視できるようになる。
12.長さ3〜4cmのシュートを固形MS培地の入った250ml培養容器に移し、根形成を助長する。容器を24℃、16時間・明/8時間・暗条件下(20μmol・m−2s−1、Osram L36 W/21 Lumilux白チューブ)でインキュベートする。3週間以内に、根形成の徴候を目視できるようになる。
13.確立された根システムを備える小植物を、タバコ葉肉プロトプラストの供給源としてのin vitro植物培養物として維持する。
【0185】
(実施例7)
エバンスブルー染色を用いたタバコプロトプラスト生存率の評価
図46および図47を参照。
【0186】
背景情報
・エバンスブルー染色(EVB;6,6’−[(3,3’−ジメチル1[1,1’−ビフェニル]−4,4’−ジイル)ビス(アゾ)ビス[4−アミノ−5−ヒドロキシ−1,3−ナフタレンジスルホン酸]四ナトリウム塩)
・非蛍光色素
・作用方法:生細胞は、エバンスブルーを排除する能力を細胞膜に保持し、その本来の色を保つ。塩または浸透圧ストレスにより損傷を受けた細胞は、エバンスブルーを排除することができず紺青に染色され、検鏡により容易に識別できる。
【0187】
・調製方法:400mg/l原液(溶媒:0.65Mマンニトール)
・染色方法:エバンスブルー原液を等容量のプロトプラスト懸濁液に加えて穏やかに混合し、室温で10分間インキュベートし、その後顕微鏡で可視化する。
【0188】
・検出方法:Leica MZFL III光学解剖顕微鏡
(実施例8)
Nicotiana tabacumプロトプラストにおける遺伝子発現解析
実験目標:トランスフェクトしたタバコプロトプラストにおけるクローニングした遺伝子AtRbcS::1−SST−SacBおよびAtRbcS::SPOR−SacBの発現をRT−PCRを用いて試験した。
【0189】
材料と方法:3プレートのプロトプラストの命名
i)トランスフェクトしたプロトプラスト(試料9A):AtRbcS::1−SST−SacB
ii)トランスフェクトしたプロトプラスト(試料12A):AtRbcS::SPOR−SacB
iii)非トランスフェクトプロトプラスト 。
【0190】
発現解析に関与するステップ:
1.プライマー設計およびPCRの最適化
2.全RNAの単離
3.RT反応
4.qRT−PCRアッセイ
プライマー設計およびPCR産物の同一性:ビーコン設計ソフトウェア(Premier Biosoft International)および遺伝子バンクにおいて利用できる遺伝子配列を用いてプライマー対を設計して対象遺伝子を増幅した。その結果生じるPCR産物のサイズが200〜250bpの範囲になるよう遺伝子特異的プライマーを選択した。融解曲線解析およびゲル電気泳動に基づくサイズによりPCR産物を同定した。
全RNA単離:1処理当たり1×106個のプロトプラストからPROMEGA製のSV全RNA単離システムにより全RNAを単離する。http://www.promega.com/tbs/tm048/tm048.pdf
【0191】
【表1】

逆転写酵素反応
QIAGEN RTキットを用いてRT反応を行った。プライマーミックス(QIAGEN)を用いることにより4通りのRT反応を行った(9、12、対照およびWTタバコRNA)。
http://www1.qiagen.com/products/pcr/QuantiTectPcrSystems/QuantiTectRevTranscriptionKit.aspx#Tabs=t2
上述の試料それぞれの複製物を、遺伝子特異的プライマーを用いたRT反応に付した。具体的には、試料9Aおよび12AはsacBリバースプライマーを用いて、対照(非トランスフェクト)試料は18Sリバースプライマーで転写した。
【0192】
PCR
反応を次の通りセットアップした。
2×GoTaq(登録商標)Green MasterMix(Promega) 10μL
cDNA鋳型 2.0μL
フォワードプライマー(10μM) 0.5μL
リバースプライマー(10μM) 0.5μL
ヌクレアーゼを含まない水 7.0μL 。
【0193】
反応サイクルは次の通りである。
ステップ1:95℃ 2分間
ステップ2:95℃ 30秒間
ステップ3:55℃ 30秒間
ステップ4:72℃ 60秒間
ステップ2から25サイクル(18S)、あるいはステップ2から35サイクル(sacB)
ステップ5:4℃ ホールド 。
【0194】
1×TBEバッファー中で1%(w/v)アガロースにより電気泳動し、SYBR(50μL/L)で染色した後、UV光の下で反応産物を可視化する。
【0195】
トランスフェクトしたプロトプラストcDNA試料(9および12)の両方でsacB転写産物の検出が示され、一方、非トランスフェクトプロトプラストcDNA試料から18Sを増幅することによって、用いた方法を検証した(図50)。
【0196】
光調節プロモーターの制御下におけるキメラsacB遺伝子の発現をトランスフェクトしたプロトプラストにおいて観察した。産物は、RTなし対照および鋳型なし対照では観察されなかった。sacB遺伝子発現は、試料9Aおよび12Aに用いたベクターでトランスフェクトしたプロトプラストにおいて検出することができた。
【0197】
(実施例9)
Agrobacterium tumefaciensを介したタバコリーフディスクの形質転換
本実施例は、上文に記載の発現カセットを用いて遺伝子操作したバイナリーベクターを運ぶアグロバクテリウムを用いたタバコリーフディスクの安定的な形質転換を説明する(図48および図49を参照)。
【0198】
序文
Agrobacterium tumefaciensを介したリーフディスク形質転換の利用は、トランスジェニック植物を作出する効率的な方法である。A.tumefaciensは、植物に感染して細菌DNAを植物ゲノムに組み入れるための遺伝的機構を含む天然の双子葉植物病原体である。この能力の結果、A.tumefaciensを、例えば特定の対象DNAをタバコに組み入れるためのクローニング媒体として採用することができる。
【0199】
この方法は、タバコにおけるモデルシステムの作製に用いて、対象遺伝子のcDNA異種クローンの機能を評価することができる。
【0200】
材料および化学薬品
装置および器具
水平流の層流フード(シリーズHWS180、CLYDE−APAC、Evans Deakin Pty.Ltd.の一部門、ウッドビルノース、南オーストラリア州5012、オーストラリア)、ロータリーシェーカー(Infors型RC−406、Infors AG、CH−4103ボットミンゲン、スイス)、スイングアウトローターを備える卓上遠心分離機(Clements Orbital 500)、ピンセット(先曲がり(bend)、cat no.2108/160、Crown Scientific、ロウビル、ビクトリア州3178、オーストラリア)、滅菌外科手術用メス刃(サイズ11、cat no.1838、Laboratory Supply Pty.Ltd.、ミルペラ(Milperra)DC、ニューサウスウェールズ州1891、オーストラリア)を備えるメス柄(No3、cat no.SHN3、Crown Scientific、ロウビル、ビクトリア州3178、オーストラリア)を用いた。
【0201】
原液
全培地に必要なマクロ成分、ミクロ成分およびビタミンは、それらを添加する際に有用となるよう、濃縮ストック(マクロ成分ストック:10倍濃縮;ミクロ成分およびビタミンストック:100倍濃縮)として調製する必要がある。ミクロ成分以外の全ストックは、室温で調製する。ミクロ成分ストックの調製は、混合前に構成要素の加熱を必要とする。Na2−EDTAおよびFeSO4×7H2Oは、混合前にそれぞれ400mLの蒸留水(全容量1000mLに対し)に溶解する必要がある。溶解した溶液を混合し、溶液が黄色に変色するまでca.60℃で加熱する。残りの構成要素を添加する前に溶液を冷ます。溶液を蒸留水で1000mLとする。全ストックを4℃で保存する。ホルモン[2,4−D(2,4−ジクロロフェノキシ酢酸)およびカイネチン]を1M KOHに溶解し、蒸留水で希釈して100mg/リットル濃縮ストックを調製する。
【0202】
培地
それらの個々の原料を終濃度で用いた培地の組成を別表1に示す:MSミクロ、MSマクロ、B5ビタミン、Lauria Bertani培地、洗浄培地、PC培地、SEL培地、RM培地およびSEM培地。
【0203】
化学薬品
2,4−D:2,4−ジクロロフェノキシ酢酸、活性炭、チメンチン(同じ濃度のセフォタキシムに代替可能)、BAP(6−ベンジルアミノプリン)、ゼアチン、AgNO3、リファンピシン、寒天(Difco、Bacto−Agar、cat.no:0140−01)はゲル化剤とし用いる。他の化学薬品(PEG4000、Tween80、KOH、NH4OH、NaCl、KCl、Ca(NO3)2、MgCl2およびCaCl2)は、BDHから購入し、MES(2−[N−モルホリノ]エタンスルホン酸(ethanesulfuric acid))はSigma(Cat.No.N−8250)から、カナマイシン硫酸塩はSigmaから、ハイグロマイシンBはCalbiochemから、Ca(OCl)2(ほぼ65%)およびホスフィノトリシン(phosphinotricin)は、Roth and Riedel de Haenからそれぞれ購入される。スクロースはFluka(cat.no:84100)から購入した。Parafilm(登録商標)「M」(American National Can(商標)、グリニッジ、コネチカット州06836、米国)は、封密用テープとして用いた。滅菌使い捨て瓶上型フィルター(0.2μm vacucap90;cat.no.4622、Gelman Sciences(登録商標)Pty.Ltd.、チェルトナム、ビクトリア州3192、オーストラリア)および使い捨てフィルターユニット(0.2μm;cat.no.16534、Sartorius AG、37070ゲッティンゲン、ドイツ)をフィルター滅菌に用いた。滅菌使い捨てピペット:1mL_(TRP(登録商標);cat.no.94001)、5mL_(TRP(登録商標);cat.no.94005)および10ml_(TRP(登録商標);cat.no.94010)は全てLife Technologies Pty. Ltd.、モルグレイブ、3170オーストラリア製であり、ねじ蓋付き滅菌プラスチック製遠心チューブ(14mL、TRP(登録商標);cat.no.91016、Life Technologies Pty.Ltd.、モルグレイブ、3170オーストラリア)、滅菌プラスチック製ペトリ皿(90×14mm;cat.no.82.9923.484および60×14mm、cat.no.83.1801.011、Sarstedt(登録商標)Australia Pty.Ltd.、テクノロジーパーク、南オーストラリア州5095、オーストラリアならびに90×20mm;cat.no.664160、Greiner Labortechnik GmbH、72636フリッケンハウゼン、ドイツ)およびオートクレーブ可能培養容器(250mL、cat.no.75.9922.519、Sarstedt Australia Pty.Ltd、テクノロジーパーク、南オーストラリア州5095、オーストラリア)を用いた。
【0204】
植物材料
A)N.tabacum cv.Petit Havana SR1の滅菌シュート培養物を利用することができる。これは、次亜塩素酸塩溶液[1.4%(w/v)Ca(OCl)2、0.05%(v/v)Tween80]中で15分間表面滅菌し、滅菌蒸留水中で3〜4回リンスした後、0.8%(w/v)寒天で固形化した半分に薄めたMS培地(別表1)上に発芽のため播種した、対応する種子から確立する。2〜3枚の葉をつけたシュートを切り出し、250mL培養容器内の0.8%(w/v)寒天で固形化したMS培地上で25℃、16h/d明期(20μmol・m−2s−1、Osram L36 W/21 Lumilux白チューブ)で培養する。根の生えたシュートを、6週間の間隔で使用前に茎切断として数回継代培養する。
B)ガラス温室で育成したN.tobacumを利用してもよいが、後述[I.B)]の通り葉の表面滅菌が必要である。植え付けが深過ぎないこと、また湿り気が十分であることを確実にしつつ、種子を滅菌土壌に植える。16h/d明期(20umolm“2s”1、Osram L36 W/21 Lumilux白チューブ)条件下、25℃で培養し、Osmocoteスローリリース肥料を用いて施肥する。
C)リーフディスク形質転換に利用したAgrobacterium tumefaciens株はAGL1である。
【0205】
I.タバコリーフディスクをベクターpBinhph200で形質転換するためのAgrobacterium tumefaciensの調製
形質転換したAgrobacterium tumefaciens株AGL1の前培養をタバコ形質転換日の2日前に開始し、滅菌条件が維持されていることを確実にする(pBinhph200のIDカードについては別表2を参照)。
1.−70℃で凍結したグリセロール細菌ストックの表面を接種(innoculation)用ループで掻き取り、滅菌チューブ内の2mLのLB(20mg/mLリファンピシン+10mg/Lスペクチノマイシンを含む)に接種する。24時間、28℃、150rpmでインキュベートする。
2.4mLのLB+抗生物質(12mL滅菌チューブ内)に0.25mLの前記24時間前培養物を接種する。6〜7時間、28℃でインキュベートする。
3.25mLの抗生物質なしLB(150mL滅菌フラスコ内)に0.025mlの前記6〜7時間前培養物を接種し、一晩28℃、150rpmでインキュベートする。
4.25mlの前記一晩前培養物に25mlのLBを加え、28℃でさらに90分間、150rpmで培養を継続する。
5.50mlの前培養物を遠心チューブに移し、Clements卓上遠心分離機において12分間、2,000rpm、室温でスピンする。
6.上清を除去し、ペレットを20mLのWMに穏やかに再懸濁する。OD600を測定する。細菌懸濁液にさらにWMを加え、最終OD600を0.45とする。この調製物をステップIII)の1に用いる。
【0206】
II.リーフディスクの調製
A)タバコシュート培養物を用いたリーフディスクの調製
1.層流において、MS培地上で培養したタバコ植物体から4〜6枚の葉を収集する。葉をWMの入った1.5×9cmペトリ皿に置く。メスを用いて主脈を除去し、葉組織をほぼ1cm2四方に切る。この時点で、組織またはリーフディスクは、Agrobacterium tumefaciens(AGL1)による形質転換の準備が整った。ディスクは1時間以内に形質転換しなければならない。
B)非滅菌タバコ組織を用いたリーフディスクの調製
1.ガラス温室で育成したタバコ植物体から4枚の若い葉(ほぼ8cmの長さ)を収集する。
2.葉を70%エタノールの入った滅菌ビーカー内に置き、アルミホイルで覆い、円軌道シェーカー(Bio−Line円軌道シェーカー、Edwards Instrument Company)上で1分間、穏やかに旋回させる。
3.70%エタノールを除去し、1%Ca(OCl)2に交換する。組織を8分間旋回させる。葉を滅菌水中で少なくとも3回洗浄する。
4.層流において主脈を除去し、残りの葉組織をほぼ1cm2四方に切る。調製したディスクをWMの入った1.5×9cmペトリ皿内に置く。この時点でディスクは、Agrobacterium tumefaciens(AGL1)による形質転換の準備が整った。
【0207】
III.リーフディスクとAgrobacterium tumefaciensのインキュベーションおよび共存培養
1.WM(リーフディスク調製における最後のステップのもの)をアグロバクテリウム培養物に交換し、1〜2分間インキュベートする。
2.細菌懸濁液を除去し、外植片をWMで短時間リンスする。PC培地上に播く前に、滅菌ナプキンで外植片を拭き取る。3日間共存培養するため、育成室(16h/d明期(20μmol・m−2s−1、Osram L36 W/21 Lumilux白チューブ)条件、25℃)内に置く。
【0208】
IV.アグロバクテリウムを介した形質転換後のリーフディスクの前再生
1.外植片(5/プレート)をSEL培地に移し、育成室にさらに7日間戻しておく。
【0209】
V.再生
1.外植片(5/プレート)をRMに移し、育成室に戻す。シュート形成は3〜6週間以内に起こるであろう。4週間後に外植片を新しいRMに移す。
2.カルスが大きくなり過ぎたら、特に培地に接触していないシュートがある場合、メスを用いてカルスを分割する。できるだけ多くのシュートを選抜に曝露させる。
【0210】
VI.シュートの伸長および根の発達
1.選抜8週間後、あるいは選抜中の非形質転換対照外植片が死滅したとき、緑色のシュート(5/プレート)を9×2cmペトリ皿内のSEM培地(IBA(1mg/L)を含む)に移す。4〜5週間で根が出てくる筈である。
【0211】
VII.in vitro小植物発達
1.根が出たら、根の生えた小植物を組織培養容器内のSEM培地に移す。
【0212】
参考文献
Stewart、C.N.Jr.、Adang、M.J.、All、J.N.、Rayner、P.L.、Ramachandran、S.およびParrott、W.A.(1996年)Insect control and dosage effects in transgenic canola containing a synthetic Bacillus thuringiensis crylAc gene. Plant Physiol、112巻:115〜120頁 。
【0213】
別表
【0214】
【表2】
【0215】
【表3】
【0216】
【表4】
【0217】
【表5】
(実施例10)
同じ発現カセットで改変したバイナリーベクターを有するAgrobacterium tumefaciensを用いたフローラルディップ法によるシロイヌナズナ(ARABIDPOSIS)の安定的な形質転換
電気的形質転換受容性のAgrobacterium tumefaciens細胞の調製
実験手順
1.−80℃凍結グリセロールストックから、Agrobacterium tumefaciens(AGL1株)を20mg/Lリファンピシンおよび100mg/Lアンピシリンを含むMGL寒天上に画線培養し、27℃で2日間インキュベートする。
2.5mlのMGLを50ml Falconチューブ内で測定し、リファンピシン20mg/Lおよびアンピシリン100mg/Lの終濃度となるよう加える。Agrobacterium tumefaciens AGL1のシングルコロニーを接種する。
3.150rpmの円軌道シェーカー上の傾いた試験管立てにおいて(ca.30度)27℃で24時間インキュベートする。
4.夕方、20mg/Lリファンピシンおよび100mg/Lアンピシリン(amplicillin)(500mlフラスコ内)を含む100mlのMGLに、500μlの一晩培養物を接種する。
5.27℃で一晩、150rpmの円軌道シェーカー上で0.4〜0.6(最大0.6)の間のOD600が得られるまでインキュベートする。過増殖した場合は下のコメントを参照。
6.オートクレーブしたJA10遠心チューブに細胞を移し、氷上で10分間冷やす。
7.JA10ローターを用いて10分間、9000rpm、4℃で遠心分離する。
8.培養に用いた500mlフラスコに注ぐことによって、上清を慎重に廃棄する(ペレットは非常に安定的というわけではない)。
9.20mlの氷冷10%グリセロールをJA10遠心チューブ内のペレットに加え、ボルテックス撹拌することによりペレットを再懸濁する。
10.懸濁液をJA20遠心チューブに注ぎ、JA20ローターを用いて10分間、10000rpm、4℃でスピンする。
11.500mlフラスコに注ぐことにより上清を廃棄する。
12.15mlの氷冷10%グリセロールをペレットに加え、ピペッティングにより再懸濁する。JA20ローターにおいて10分間、10000rpm、4℃で遠心分離する。
13.ステップ11および12を2回繰り返す。
14.ペレットを1mlの氷冷10%グリセロールに再懸濁し(ピペットまたはボルテックスによる)、滅菌マイクロチューブに移す。
15.微量遠心管において3分間、13000rpm、4℃でスピンする
16.最後に、ペレットを1mlの10%氷冷グリセロールに再懸濁する。
17.標識された1.7mlマイクロチューブに50μlバッチのアリコートを作成し、液体窒素中でスナップ凍結する。
18.液体窒素からチューブを取り出し、コンピテント細胞を−80℃で保存する。
【0218】
コメント
A.tumefaciens細菌培養物を取り扱う間、ゴム手袋を着用する。全細菌廃棄物を500mlフラスコ内に集め、オートクレーブする。A.tumefaciensの100ml培養物が過増殖したら、20mg/Lリファンピシンを含む新しいMGL培地で1/3〜1/4希釈し、さらに1〜2時間インキュベートする。
【0219】
Agrobacterium tumefaciensのエレクトロポレーションによる形質転換
実験手順
1.ジーンパルサーキュベットホルダーを氷上で予冷する。
2.コンピテントアグロバクテリウム(AGL1)細胞のアリコートを−80℃から取り出し、氷上で解凍する。
3.ジーンパルサー背面のメインスイッチを入れる。電圧を2.5kVに調整し(ディスプレイ上に「2.5」と記録されるまで「raise」ボタンを使う)、静電容量25μFDおよび抵抗600Ωにする。
4.50μl以上の容量の解凍した細胞に0.1μgのDNAを加える。
5.ピペッティングにより混合し、細胞/DNA混合液を予冷した0.2cm gapジーンパルサーキュベットに移す。
6.両方の電極に細胞が触れるよう、細胞をキュベット底面に慎重にタップまたは振盪する。
7.キュベットの外側をティッシュペーパーで乾かし、キュベットをキュベットホルダー内に置く。キュベット上の切れ込みは正確な方向付けを確実にする。キュベットがチャンバー底の接触点の間に収容されるまでキュベットホルダーをチャンバー内にスライドさせる。
8.ビープ音がするまで両方の赤いボタンを押すことによって細胞にパルスを与える。この機械は、充電の間はCHGを表示し、放電の際にビープ音を出す。細胞を戻して1分間氷上に置いて回復を助ける。
9.ガラス製注入器を用いて1mlのLB培地をキュベット内の細胞に加える。
10.懸濁液を吸って吐いて混合し、滅菌15mlチューブに移す。
11.傾いた試験管立て(ca.30度)を用いて150rpmの円軌道シェーカー上で1〜2時間、27℃でインキュベートする。
12.9mlのLBを細胞懸濁液に加えて徹底的に混合し、20mg/Lリファンピシンおよび適切な抗生物質(例えば、pPZPシリーズのベクターに対しては100mg/Lスペクチノマイシン)を含むLBプレート上にこの培養物100μlを播く。この懸濁液100μlを900μlのLBブロスの入った1.7mlマイクロチューブに移し、徹底的に混合し、適切な抗生物質を含む別のLBプレート上にその100μlを播く。上述の懸濁液から100μlを取り分け、新しい1.7mlマイクロチューブ内に置き、900μlのLBブロスを加える。徹底的に混合し、その100μlを適切な抗生物質を含む別のLBプレート上に播く。
13.プレートをパラフィルムで封じ、大きなシングルコロニーが目視できるようになるまで27℃で2〜3日間インキュベートする。
【0220】
形質転換アグロバクテリウムの保存
実験手順
1.50ml滅菌チューブ内で20mg/Lリファンピシンおよび適切な選抜抗生物質(すなわち、pZPシリーズに対しては100mg/Lスペクチノマイシン、pBinシリーズに対しては50mg/Lカナマイシン)を含む5mlのLBブロスにシングルコロニーを接種する。翌日に増殖の程度を密接にモニターできるよう、この操作は早朝に行うとよい。
2.チューブを暗所において27℃で24〜36時間、250rpmで振盪しつつインキュベートする。インキュベーションの最初の24時間の後、培養物の増殖を定期的に観察する。細胞が活発に増殖したら(非常に目に見える)インキュベーションをやめる。濁りの最初の徴候が見えた直後に、急速な増殖が行われるであろう。増殖時間は、個々の株および形質転換体に依存する。
3.各培養物をチェックして、AGL1が望ましいバイナリーベクターを含むことを立証するべきである。この操作は、セクション5.2に記されているプロトコールを用いて行う。
4.キュベット内に培養物の500μlアリコートを作成する。適切な抗生物質を含む500μlのLBブロスでブランク作成して、各読み込み値の間からOD600読み込み値を測定する。
5.OD600読み込み値が0.8から1.0の間の範囲となるまで培養物を増殖させる。
6.滅菌15mlチューブ内に、5.0mlの培養物および5.0mlの保存ストックを加える。ステップ7に進む前に徹底的に混合する。
7.しっかりと標識した滅菌クリオチューブ内に500μlのアリコートを作成する。液体窒素中でスナップ凍結する前に全チューブを反転する。必要とされるまで−80℃で保存する。PCR陰性であることが示された場合、全ストックを廃棄する。
【0221】
アグロバクテリウムのPCR解析
1.1μlのアグロバクテリウム培養物を滅菌PCRチューブ内の9μlの滅菌MQ H2Oに加える。
2.細胞を98℃で5分間インキュベートする。チューブを氷に移す。
3.10μlの調製済2×PCRマスターミックスを加える。10μlの2×PCRマスターミックスは、次のものを含む。
10×Dynazymeバッファー 2μl
10mM dNTP 1.0μl
10μMフォワードプライマー(10μM) 1.0μl
10μMリバースプライマー(10μM) 1.0μl
Dynazyme IIポリメラーゼ(polymerise) 1μl
MQ水 3μl
4.陽性対照(50ngのオリジナルプラスミドDNA)および陰性対照(鋳型DNAなし)を含む。標準PCR条件用いて合計35サイクル行う。
【0222】
1.95℃、3分間
2.94℃、30秒間
3.55℃、30秒間
4.72℃、1分間
5.72℃、10分間
ステップ2〜4を合計35回繰り返す。
5.PCR産物を1%アガロースゲルで解析する。
【0223】
コメント
アグロバクテリウムを取り扱う間、手袋を着用する。あらゆる細菌およびDNA廃棄物を集めてオートクレーブする。ジーンパルサーキュベットは再使用可能である。蓋は70%EtOHに浸し、キュベットは水を張って閉鎖した入れ物の中でオートクレーブしてアグロバクテリウムを除去する。その後、キュベットは70%EtOH中に保存し、乾かした後に再使用することができる。
【0224】
Arabidopsis thalianaのin planta形質転換
実験手順
植物材料の調製
1.実生用方形ケース(punnet)に種子栽培ミックスを充填し、盛り土を形成する。2層の防鳥ネットで覆い、ゴムバンドで各端を固定する。トレイに張った水の中に方形ケースを入れることによって、土壌を水で満たす。方形ケース当たりほぼ40体の植物を得るのに十分な種子を播く。
2.方形ケースを4℃に2〜3日間置くことにより、種子を春化処理する。方形ケースを22℃、蛍光灯(連続照明、55μmol・m−2s−1)下の育成チャンバーに移し、1週間に1回、Miracle−GroまたはAquasolを与える。
3.一次花序(bolt)が現れたら摘心し、二次花序を概ね2〜10cmの丈になるまで生長させる(概ね4〜6日間かかり、植物体は多数の未開花の花蕾をつけ、長角果(silique)は殆どない筈である)。ピンセットを用いて長角果または開花した花を全て慎重に取り除く。気孔が開くよう浸潤前に植物に十分に水をやる。浸潤前に土壌を水で飽和させて細菌溶液が土壌に吸収されるのを最小限に抑える。
4.LWSに詳細を入力し、バーコードを作成する。LWSバーコード詳細によって方形ケースを標識する。
【0225】
Agrobacterium tumefaciensの調製
1.朝に、適切な選抜抗生物質(すなわち、pPZPベクターに対し100mg/mlのスペクチノマイシン)を含む200mlのLB培地に、アグロバクテリウム保存ストック(セクション5.1)の単一のスターター培養物500μlを接種する。250rpmの円軌道シェーカー上で24時間、27℃でインキュベートする。およそ2個の方形ケースの植物を浸潤させるには200mlの培養物で十分である。
2.一晩培養した培養物を500ml遠心分離ボトルにおいて5500g、室温、15分間遠心分離して細胞をペレット化させる。可能な限り液体を除去しつつ上清を廃棄する。OD600読み込み値が約0.7〜0.9となるようペレットを浸潤培地(別表1を参照)に再懸濁する。
【0226】
アグロ浸潤
1.アグロバクテリウム溶液の半量を250ml容器内に置く。
2.方形ケースを逆さにし、ロゼット葉を含む植物体全体を細菌溶液に浸漬し、穏やかに振盪して気泡を追い出す。植物を2分間共培養する。
3.方形ケースを取り出して短時間水を抜くが、植物の周りの薄い水膜はそのままにしておく。植物をプラスチックフィルムで覆って湿度を維持し、育成室に戻して直射光を避ける。廃液をオートクレーブし、正しい処分のための化学廃棄物ドラム缶に捨てる。
4.A.thalianaの全方形ケースに対してステップ1〜3を繰り返して形質転換する。
5.詳細をLWSに入力し、バーコードを作成する。個々の方形ケースをLWSバーコード詳細で標識する。
6.翌日、鉢の覆いを取って直射光に戻し置く。植物が十分に発達した長角果をつけるまで植物に水をやる。
【0227】
種子採取
1.植物が完全に乾ききったら、長角果をつけた茎を取ってこれを紙袋の中に入れ、1週間37℃で乾かしておく。袋をLWSバーコードで標識する。乾いた長角果を紙袋の中で押しつぶす。この操作は長角果を粉々にして種子を遊離させる。
2.200ミクロンの篩を新しいA4用紙1枚の上に置き、そこに種子および押しつぶされた長角果を移し入れる。篩を穏やかにタップし、その下にある用紙の上に種子を落とす。篩の中に残った植物材料を廃棄する。植物材料の大部分が除去されるまでこのプロセスを繰り返す(注記:植物材料は次のステップにおける汚染源となり得る)。種子を1.7mlマイクロチューブ内に入れ、LWSバーコード詳細で標識する。チューブを小型のマニラ封筒の中に置き、LWSバーコードで標識する。このバーコードは、元々の形質転換事象と関連することに留意する。
3.種子を4℃保存に移す前に、種子を−20℃で24時間保存する。
【0228】
陽性T1トランスジェニックArabidopsis thaliana植物の選抜
種子の表面滅菌
1.層流フードで作業しつつ、滅菌する種子(150×15mmプレートにつき、40mg=ほぼ2000個の種子)を2.0mlマイクロチューブ内に置く。
2.1000μlの70%エタノールを加え、2分間放置する。
3.エタノールを除去し、1000μlの種子滅菌用溶液(4%塩素:水:5%SDSがそれぞれ8:15:1の比率)を加えてボルテックス撹拌によって徹底的に混合する。
4.Ratek「観覧車(ferris wheel)」上にチューブを置き、種子と溶液を確実に混合し、10分間放置する。
5.層流において滅菌用溶液を除去して滅菌水に交換する。チューブ(複数可)をボルテックス撹拌し、ベンチトップ遠心分離機で30秒間スピンして種子を沈降させる。水を除去し、さらに1mlの滅菌水に交換する。種子洗浄ステップは、目視できる泡が出なくなるまで繰り返すべきである(少なくとも4回)。最後の洗浄の後、約200μlの水を残して種子を浸す。
【0229】
T1形質転換体の選抜
1.適切な選抜抗生物質(例えば、PZP200シリーズに対し8mg/Lのハイグロマイシン)を含む選抜発芽培地(SGM)を用いて150×15mmプレートを調製する。チメンチン(250mg/L)を入れて、アグロバクテリウムの増殖を阻害する。各プレート当たり約125mlのSGMが必要とされる。
2.層流フード内で作業しつつ、SGM寒天プレートの表面にわたって平行に滅菌メスをすべらせる(図4を参照)。この操作は種子を広げるのに役立つ。
3.先端を切り取った滅菌1mlチップを用いて、滅菌種子をピペットで取ってプレート上に置く。滅菌使い捨て延展器(spreader)を用いて種子を分布させる。
4.種子を4℃で2日間低温処理し、次に連続的な蛍光灯(55μmol・m−2s−1)下、22℃で育成する。
5.推定形質転換体は、6〜8葉ステージになったら土壌に移すことができる。ピンセット1本を用いて、根を確実にインタクトに保ちつつ植物を組織培養培地から慎重に取り出す。ARASYSTEMを用いて湿ったin−vitro混合土壌に移植する(別表2の図5を参照)。プラスチックチューブで覆う。新しいLWSバーコードを作成し、チューブを標識する。数日間チューブの上端をプラスチックラップで覆い、回復を助ける。
【0230】
導入遺伝子組み込みの立証:ゲノムDNAソースとしてのアルカリ処理した葉組織
推定形質転換体を土壌に移して3日後、次のプロトコールを用いて個々の植物体を導入遺伝子の存在に関して分子的に特徴付けることができる。
1.試験するアルカリ処理した葉組織それぞれに対し、1×PCRバッファーミックスを調製する(詳細は後述を参照)。
2.1.7mlマイクロチューブ内で、200μlの0.25M NaOHを小型の若い葉(T1植物から採取)に加える。
3.チューブを沸騰水に2分間浸漬する。注記:煮沸の間に蓋がはじけ開くのを防ぐため、蓋をマイクロチューブ蓋ロックで固定する、あるいは蓋を細針で刺し通すこと。
4.煮沸後、チューブを水から取り出して、200μlの0.25M HClおよび100μlの0.25%(v/v)igepal[0.5M Tris HCl、pH8.0]を加える。チューブを沸騰水にさらに4分間浸漬する。
5.アルカリ処理した葉のごく一部(ほぼ2mm2)を取り出し、予め調製したPCRミックス中に置く。
【0231】
10×PCR反応バッファー(Mg(SO4)・7H2O含有) 5.0ml
10mM dNTP 1.0μl
10μMフォワードプライマー 1.0μl
10μMリバースプライマー 1.0μl
PWO DNAポリメラーゼ 1.0μl
MQ H2O 41.5μl
6.標準PCR条件を用いて35サイクルを行う。
【0232】
1.95℃、3分間
2.94℃、30秒間
3.55℃、30秒間
4.72℃、1分間
5.72℃、10分間
ステップ2〜4を合計35回繰り返す。
7.PCR産物を1%アガロースゲルで解析する。
【0233】
注記:初回にアルカリ処理した葉組織を用いてインサートが増幅されない場合、組織をさらに2分間再度煮沸して導入遺伝子のPCR増幅を繰り返す。この第二のPCRが失敗したら、Qiagen植物ゲノムDNA抽出キットを用いて葉組織から少量のゲノムDNAを抽出する。LWSをアップデートする。
【0234】
種子採取
1.植物が完全に乾ききったら、長角果をつけた茎を取ってこれを紙袋の中に入れ、1週間室温で乾かしておく。袋をLWSバーコードで標識する。乾いた長角果を紙袋の中で押しつぶす。この操作は長角果を粉々にして種子を遊離させる。
2.200ミクロンの篩を新しいA4用紙1枚の上に置き、そこに種子および押しつぶされた長角果を移し入れる。篩を穏やかにタップし、その下にある用紙の上に種子を落とす。篩の中に残った植物材料を廃棄する。植物材料の大部分が取り除かれるまでこのプロセスを繰り返す(注記:植物材料は次のステップにおける汚染源となり得る)。種子を1.7mlマイクロチューブ内に入れ、LWSバーコード詳細で標識する。チューブを小型のマニラ封筒の中に置き、LWSバーコードで標識する。このバーコードは、元々の形質転換事象に関連することに留意する。
3.種子を4℃で保存する前に、T2種子を−20℃で24時間保存する(陽性トランスジェニックT2植物の選抜における真菌汚染の見込みを低減させるのに有用である)。
【0235】
コメント
ARASYSTEMトレイ、ホルダー等を含む、アグロバクテリウムと接触したあらゆる入れ物は、市販の漂白剤および70%エタノールを用いて徹底的に消毒しなければならない。
【0236】
この実験に依存し、T1植物を表現型特性評価、すなわちレポーター遺伝子解析に用いることができ、これらの系統はT1ステージを越えて継続する必要はないと思われる。
【0237】
ホモ接合型T3種子の作出
序文
詳細に後述するプロトコールは、シングルコピーの導入遺伝子を有するホモ接合型植物の選抜に用いた方法を説明する。浸潤方法を用いた導入遺伝子のArabidopsis thalianaへの組み込みは、子房の受精前に雌しべで起こる。したがって、浸潤によって産生されたあらゆる種子で導入遺伝子を有するものはT1であると考えられる。T1種子が発芽してT1植物を産生し、これは次にT2種子を産生する。目標は、単一のインサートを有し導入遺伝子を発現する独立したトランスジェニック植物を、構築物につき少なくとも5個体得ることである。T1種子用に作成した各LWSバーコードは、別個の形質転換事象を表す。各T1植物のT2種子を収集するとき、新たなLWSバーコード番号を付与する。このLWSバーコードは、元々の形質転換事象に関する。
【0238】
T3種子作出のためのT2形質転換体の選抜
1.層流フード内で作業しつつ、約100個のT2種子を表面滅菌する(セクション7.1を参照)。
2.250mg/Lチメンチン(timetin)および8mg/Lハイグロマイシン(pPZP200−35s−hph−35stに対する選抜剤)を含む選抜SGM培地上に、1プレート当たり約25個の種子を播く(合計4枚)。
3.種子を4℃で2日間低温処理し、次に連続照明(55μmol.m−2.s−1)、22℃の育成室に移す。
4.2週間後、ハイグロマイシンに対して抵抗性または感受性を有する植物の分離解析を行う。
5.推定形質転換体が6〜8葉ステージになったら、Arasystemを用いて少なくとも10個体の植物を土壌に移す。新たなLWSバーコードを作成し(元々の形質転換と関連する)、各植物体をバーコードで個々に標識する。チューブをプラスチックフィルムで数日間覆って回復を助ける。
6.個々の植物それぞれが組み込まれた導入遺伝子を有することを確認するため、アルカリ処理した葉組織の方法を用いる(セクション7.3)。LWSをアップデートする。
7.植物が完全に乾ききったら、長角果をつけた茎を取ってそれを紙袋に入れ、1週間37℃で乾かしておく。袋をLWSバーコードで標識する。乾いた長角果を紙袋の中で押しつぶす。この操作は長角果を粉々にして種子を遊離させる。
8.200ミクロンの篩を新しいA4用紙1枚の上に置き、そこに種子および押しつぶされた長角果を移し入れる。篩を穏やかにタップし、その下にある用紙の上に種子を落とす。篩の中に残った植物材料を廃棄する。植物材料の大部分が取り除かれるまでこのプロセスを繰り返す(注記:植物材料は次のステップにおける汚染源となり得る)。種子を1.7mlマイクロチューブ内に入れ、LWSバーコード詳細で標識する。チューブを小型のマニラ封筒の中に置き、LWSバーコードで標識する。このバーコードは、元々の形質転換事象に関連することに留意すること。
9.種子を4℃保存に移す前に、種子を−20℃で24時間保存する(陽性トランスジェニックT3植物の選抜における真菌汚染の見込みの減少に役立つ)。
【0239】
分離解析
1.ハイグロマイシンに対して抵抗性または感受性のいずれかを有する各系統から得られたT2植物の総数を採点する。
2.T−DNAが1遺伝子座に挿入されている場合、抵抗性植物の感受性植物に対する比率は3:1となる筈である。T−DNA遺伝子座が2遺伝子座に挿入されている場合、抵抗性植物の感受性植物に対する比率は15:1となる筈である。T−DNAが3以上の遺伝子座に挿入されている場合、抵抗性植物の感受性植物に対する比率は>15:1となる筈である。
3.カイ2乗(χ2)統計検定を用いて、分離データが特定の仮説にどの程度適合するか決定する。
4.導入遺伝子のシングルコピーを含むことをカイ2乗解析が示すトランスジェニック系統の育成を継続する。
5.LWSをアップデートする。
導入遺伝子組み込みの立証:ゲノムDNAソースとしてのアルカリ処理した葉組織
1.各T2植物につき小型の葉を1枚収集する。
2.アルカリ処理した葉のプロトコール(セクション7.3)に従い、導入遺伝子の存在を決定する。
3.LWSをアップデートする。
【0240】
ホモ接合型T3系統の選抜
1.単一の導入遺伝子挿入を含むことを示すT2トランスジェニック系統の育成を継続する。
2.T2種子を採取し(セクション8.2)、LWSをアップデートして新たなバーコードを作成する。
3.ほぼ40個のT3種子をSGM上で発芽させる。
4.2週間後、ハイグロマイシンに対し抵抗性または感受性のいずれかを有する各系統由来のT3植物の総数を採点する。
5.ホモ接合型T3系統は、感受性植物の不在によって示されるであろう。
6.推定形質転換体が6〜8葉ステージになったら、Arasystemを用いて少なくとも20個体の植物を土壌に移す。新たなLWSバーコードを作成し、各植物を個々にバーコードで標識する。チューブをプラスチックフィルムで数日間覆い回復を助ける。
7.系統の単一挿入がホモ接合型であることをさらに検証するため、アルカリ処理した葉組織の方法を用いて(セクション7.3)、全植物が導入遺伝子を含むことを確認する。LWSをアップデートする。
8.推定ホモ接合型系統から十分な材料を収集してサザンハイブリダイゼーションを行い、導入遺伝子の組み込み数を確認する。LWSをアップデートする。
9.セクション8.2に記されているプロトコールに従って、種子をT3ホモ接合型系統から収集する。
【0241】
【化1】
【技術分野】
【0001】
本発明は、植物におけるフルクタン生合成の改変に関し、特に、光合成細胞におけるフルクタン生合成を操作する方法ならびに関連する核酸および構築物に関する。
【0002】
本発明はまた、植物バイオマスの増加に関し、特に、植物におけるシュートおよび/または根の生長を含むバイオマス収量および/または収量安定性を増強する方法ならびに関連する核酸および構築物にも関する。
【背景技術】
【0003】
フルクタンは水溶性炭水化物の一種であり、その主要機能は、植物生長のために容易に利用できるエネルギー貯蔵を提供することである。フルクタンは、寒冷および乾燥耐性、分げつ生存率の上昇、持続性の増強、切断または被食後の良好な再生長、ストレスからの回復の改善、早春における生長ならびに栄養価の上昇等、草本における様々な有利な特徴に関連する。
【0004】
草本および穀類におけるフルクタン合成および代謝は複雑である。フルクタンは、スクロースと結合した直鎖または分岐フルクトース鎖からなる。植物フルクタンの鎖長は、3から最大数百個のフルクトースユニットに及ぶ。異なる種類のフルクタンは、存在する結合の種類に基づいて識別することができる。ペレニアルライグラス(perennial ryegrass)において、3種類のフルクタンが同定された。すなわちイヌリン、イヌリンネオシリーズおよびレバンネオシリーズであり、このフルクタンプロファイルには4種類のフルクトシルトランスフェラーゼ(fructosyltransferse)(FT)酵素が関与する。酵素1−SST(スクロース:スクロース1−フルクトシルトランスフェラーゼ)はフルクタン生合成における最初のステップを触媒し、一方、残りの酵素は成長中のフルクトース鎖を伸長させる(1−FFT:フルクタン:フルクタン1−フルクトシルトランスフェラーゼ、6G−FFT:6−グルコースフルクトシルトランスフェラーゼおよび6−SFT:スクロース:フルクトース6−フルクトシルトランスフェラーゼ)。酵素1−FEHまたは6−FEH(フルクトエキソヒドロラーゼ(fructoexohydrolase))は、フルクトース分子を遊離させることによりフルクタン鎖長を短縮する。
【0005】
細菌は、高分子量フルクタンポリマーを合成するために1−SSTおよび1−FFT活性の両方を含む単一のFT酵素のみを利用する。大部分の細菌フルクトシルトランスフェラーゼは、フルクトース分子のβ−2,6結合によって特徴付けられるレバン型フルクタンを産生するが(レバンスクラーゼ(levansucrase))、一部の細菌からはイヌリン型フルクタン(β−2,1結合)を産生するイヌロスクラーゼ(inulosucrase)が単離されている。
【0006】
Bacillus subtilis由来のSacB遺伝子、Bacillus amyloliquefaciens由来のSacB遺伝子およびStreptococcus mutans由来のFTF遺伝子を含む、少なくとも3種類の細菌レバンスクラーゼがトランスジェニック植物において発現した。これら細菌レバンスクラーゼの植物における発現は、DP数千の非常に高分子量のフルクタンの合成をもたらす(総説として非特許文献1を参照)。
【0007】
フルクタンは、15%の植物種における主要な非構造炭水化物を表し、飼料の質に重要な役割を果たす。高フルクタン食を摂取する反芻家畜は、動物能力の改善を示す。
【0008】
草本において、FT遺伝子発現の遺伝子操作により、フルクタンのレベルおよび組成は茎および葉鞘で増加した。
【0009】
しかし、経路内酵素の活性を操作することによる生化学的経路の操作は、その道筋において様々な酵素およびそれらの基質が相互作用し得るため、困難なものとなるであろう。
【0010】
このため、特に植物における生化学的経路を操作するための改良法があることが望ましい。例えば、Lolium属、Festuca属およびBrachiaria属等の飼料草の種;サトウキビその他の草本;ならびにソルガムおよびコムギやトウモロコシ等、他の穀類を含む植物におけるフルクタン生合成を操作する方法があり、これにより例えば、
・牧草地の生産性の改善、動物の生産性の改善および/または環境汚染の抑制をもたらす、牧草品質および/または収量が改善された飼料草、
・バイオエタノール生産用等のバイオマス収量が増強されたスイッチグラス、Miscanthus属、ソルガム(穀物、飼料およびエネルギー用ソルガム)、サトウキビおよびエネルギー用サトウキビ等、バイオエネルギー用草本および作物、
・穀物および/またはバイオマス収量が増加したコムギ、イネ、トウモロコシ、オオムギ等、穀類
の生産を促進することが望ましい。
【0011】
フルクタン生合成経路に関与する数種の酵素をコードする核酸配列が特定の植物種で単離された。例えば、本出願人らの国際出願PCT/AU01/00705号明細書は、Lolium属およびFestuca属由来のフルクトシルトランスフェラーゼホモログについて記載している。しかし、依然として、植物におけるフルクタン生合成の改変に有用な、また植物体(plant)の様々な部分におけるフルクタン蓄積を遺伝子操作するための材料が必要である。
【0012】
国際特許出願PCT/AU2009/001211号明細書は、植物におけるフルクタン生合成の改変に有用な構築物について記載する。しかし、それら構築物は、好ましくは2以上のフルクタン生合成酵素の翻訳融合体をコードする融合遺伝子を含み、該遺伝子は、好ましくは植物フルクタン生合成遺伝子に相当する融合体を作り出す。
【先行技術文献】
【非特許文献】
【0013】
【非特許文献1】Cairns AJ. Fructan biosynthesis in transgenic plants. J Expt Biol(2003)54:549〜67
【発明の概要】
【発明が解決しようとする課題】
【0014】
本発明の目的は、先行技術に関連する問題または欠点の1または複数を克服、または少なくとも軽減することである。
【課題を解決するための手段】
【0015】
本出願人らは、細菌FT酵素をコードする遺伝子またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む構築物を用いて、光合成細胞におけるフルクタン生合成経路を空間的にリプログラミングすることにより、植物、例えば飼料草を栄養的に増強することおよび/または植物バイオマスを増加させることが可能であることを見出した。このプロセスを用いることによって、家畜によって先ず食べられる植物器官であり、通常フルクタンを蓄積できない葉身におけるフルクタン蓄積を駆動することが可能となる。このため、フルクタン、特に高い重合度を示すフルクタン(「高DPフルクタン」)の蓄積は、草食動物がより利用しやすい栄養を提供する。フルクタンは茎および葉鞘に蓄積するが、葉フルクタンはCO2同化が生長より優勢な期間においてのみ生成される。スクロースが合成される光合成細胞でフルクタン遺伝子を発現させることによって飼料草を栄養的に増強し、これによりフルクタン蓄積を葉身で優先的に駆動し、これを摂食する家畜にさらなるエネルギーを提供することができる。
【0016】
飼料草におけるフルクタンは、反芻動物に食べさせる食餌におけるすぐに利用できるエネルギーに大いに貢献する。反芻胃における発酵過程は、大量のすぐに利用できるエネルギーを必要とする。反芻胃におけるすぐに利用できるエネルギーの改善は、反芻胃の消化効率を上昇させることができる。反芻胃消化における効率の上昇は、反芻動物に与えた飼料タンパク質のミルクまたは肉への変換の改善および窒素性廃棄物の減少をもたらす。
【0017】
本出願人らは、寿命延長のために光合成細胞をリプログラミングすることにより、例えば葉の老化を遅延させることにより本発明の方法を促進することができ、植物バイオマスの増加に役立つことも見出した。
【0018】
本出願人らは、細菌FT遺伝子をコードする遺伝子またはその機能的に活性な断片もしくは変異体を含む構築物が、2以上のフルクタン生合成酵素の翻訳融合体をコードする融合遺伝子を含む構築物よりも優れた結果を示し得ることを見出した。例えば1−SSTおよび1−FFT活性の両方を含む細菌FT遺伝子の使用は、別々の遺伝子を融合するよりも技術的な困難が少なくなり、融合遺伝子を含む構築物と比べてより容易に植物に導入されるおよび/またはより良く機能する構築物をもたらすことができる。
【0019】
例えば、例えば1−SSTおよび1−FFT活性の両方を含む細菌FT遺伝子を発現することにより、植物ゲノムに独立的に組み込まれたこれら遺伝子の発現パターンの差に関連する課題を軽減し、その結果スクロース分子を、低い重合度(「低DPフルクタン」)および高い重合度(「高DPフルクタン」)を示すフルクタンに直接的に変換することができる。さらに、FTタンパク質は、効率的なフルクタン生合成の代謝チャネルを形成することができる。
【0020】
光合成細胞において低および高DPフルクタンの蓄積をもたらす光合成細胞におけるFT遺伝子の発現は、光合成抑制機構からの解放をもたらし、太陽エネルギーの獲得およびCO2固定の増加を促進することができる。
【0021】
このため、本出願人らは、光合成細胞の寿命延長のためのリプログラミングおよびフルクタン生合成の増強は太陽エネルギーの獲得を促進し、シュートおよび/または根の生長等、植物バイオマスの生産を増加させることを見出した。
【0022】
さらに、低および高DPフルクタンの蓄積は寒冷および乾燥等、非生物的ストレスに対する植物の耐性に関連づけられてきたため、また、根の生長増強および葉の老化遅延は植物の乾燥ストレス耐性に関係づけられてきたため、光合成細胞の寿命延長のためのリプログラミングおよびフルクタン生合成の増強は、収量安定性および植物の非生物的ストレス耐性を促進する。
【0023】
したがって、本発明はその一態様において、植物の光合成細胞におけるフルクタン生合成を操作する方法であって、細菌フルクトシルトランスフェラーゼ(FT)酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む有効量の遺伝的構築物を前記植物に導入するステップを含む方法を提供する。
【0024】
「フルクタン生合成の操作」とは、一般に、非形質転換対照植物と比べて形質転換植物におけるフルクタン生合成の増加を意味する。しかし、一部の適用では、非形質転換対照植物と比べて形質転換植物においてフルクタン生合成を低減させる、あるいは改変することが望ましいであろう。例えば、非形質転換対照植物と比べて形質転換植物のフルクタン生合成経路における特定の酵素の活性を増加または減少させることが望ましいであろう。
【0025】
「光合成細胞」とは、光合成が行われる植物細胞を意味する。このような細胞は一般にクロロフィル色素を含む、あるいは緑色細胞として公知のものである。大部分の光合成細胞は、植物の葉に含まれる。好ましくは、本発明の遺伝的構築物は、維管束鞘細胞で発現し、より好ましくは葉肉および/または柔組織維管束鞘細胞で発現する。
【0026】
「有効量」とは、前記植物またはそこから派生する植物体、植物種子もしくは他の植物部分において同定可能な表現型形質を生じるのに十分な量を意味する。このような量は、当業者であれば、植物の種類、投与経路その他の関連性のある要因を考慮に入れて容易に決定することができる。このような当業者は、適切な投与量および投与方法を容易に決定することができる。例えば、その開示全体を本明細書の一部を構成するものとしてここに援用するManiatisら、Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory、Cold Spring Harborを参照。
【0027】
「遺伝的構築物」とは、組換え核酸分子を意味する。
【0028】
「プロモーター」とは、作動的に連結した核酸配列の転写を導くのに十分な核酸配列を意味する。
【0029】
「作動的に連結した」とは、核酸(複数可)とプロモーター等の調節配列が、例えば転写活性化因子タンパク質等の適切な分子が前記調節配列に結合する場合、適切な条件下で前記核酸の発現を可能にする仕方で連結していることを意味する。好ましくは、作動的に連結したプロモーターは、関連する核酸の上流にある。
【0030】
「上流」とは、核酸に沿って3’→5’方向を意味する。
【0031】
「核酸」とは、遺伝情報を運ぶことができるヌクレオチド鎖を意味する。この用語は一般に、遺伝子またはその機能的に活性な断片もしくは変異体、および/またはその表現型に影響を及ぼす生物ゲノム内の他の配列を言う。用語「核酸」は、1本鎖または2本鎖であり、必要に応じて合成、非天然または変化したヌクレオチド塩基、合成核酸およびそれらの組合せを含むDNA(cDNAまたはゲノムDNA等)およびRNA(mRNAまたはマイクロRNA等)を含む。
【0032】
「細菌フルクトシルトランスフェラーゼ(FT)酵素をコードする核酸」または「細菌フルクトシルトランスフェラーゼ遺伝子」とは、フルクトシル部分をスクロース含有サッカライドからアクセプター分子に転移することによりオリゴフルクタンおよび/またはポリフルクタンの合成を触媒する、細菌に通常存在する酵素をコードする核酸を意味する。細菌FT酵素は、レバンスクラーゼ活性を含む、および/またはレバン型フルクタンを産生することができる。細菌FT酵素は、イヌロスクラーゼ活性を含む、および/またはイヌリン型フルクタンを産生することができる。好ましい細菌FTは、スクロース:スクロース1−フルクトシルトランスフェラーゼ(1−SST)およびフルクタン:フルクタン1−フルクトシルトランスフェラーゼ(1−FFT)酵素活性の両方を含む。SacB、LscまたはFTF遺伝子を用いてもよい。SacBまたはLsc遺伝子が特に好ましい。
【0033】
プロモーターに関する「機能的に活性な断片または変異体」とは、断片または変異体(アナログ、誘導体または突然変異体等)がそこに作動的に連結した核酸の転写を導くことができることを意味する。このような変異体は、自然発生的な対立遺伝子変異体および非自然発生的な変異体を含む。その改変が断片または変異体の機能欠失活性をもたらすことのない限りにおいて、1または複数のヌクレオチドの付加、欠失、置換および誘導体化が企図される。好ましくは、機能的に活性な断片または変異体は、該断片または変異体が対応する上述の配列の関連性のある部分に対して少なくとも約80%の同一性、より好ましくは少なくとも約90%の同一性、さらにより好ましくは少なくとも約95%の同一性、最も好ましくは少なくとも約98%の同一性を有する。好ましくは、断片は、少なくとも20ヌクレオチド、より好ましくは少なくとも50ヌクレオチド、より好ましくは少なくとも100ヌクレオチド、より好ましくは少なくとも200ヌクレオチド、より好ましくは少なくとも300ヌクレオチドのサイズを有する。
【0034】
細菌FT酵素をコードする核酸に関する「機能的に活性な」とは、本発明の方法により、例えばフルクタン生合成経路に関与することができる酵素に翻訳されることにより、断片または変異体(アナログ、誘導体または突然変異体等)が植物におけるフルクタン生合成を操作できることを意味する。このような変異体は、自然発生的な対立遺伝子変異体および非自然発生的な変異体を含む。その改変が断片または変異体の機能欠失活性をもたらさない限りにおいて、1または複数のヌクレオチドの付加、欠失、置換および誘導体化が企図される。好ましくは、機能的に活性な断片または変異体は、該断片または変異体が対応する上述の配列の関連性のある部分に対して少なくとも約80%の同一性、より好ましくは少なくとも約90%の同一性、さらにより好ましくは少なくとも約95%の同一性、最も好ましくは少なくとも約98%の同一性を有する。このような機能的に活性な変異体および断片は、例えば保存された核酸変化を有する機能的に活性な変異体および断片を含む。
【0035】
「保存された核酸変化」とは、遺伝暗号の縮重による、コードされているタンパク質におけるアミノ酸保存をもたらす核酸置換を意味する。このような機能的に活性な変異体および断片は、例えば対応するアミノ酸配列における1または複数の残基に保存されたアミノ酸置換をもたらす核酸変化を有する機能的に活性な変異体および断片も含む。
【0036】
「保存されたアミノ酸置換」とは、同一クラス内の別の1アミノ酸によるアミノ酸置換を意味し、クラスは次の通りである。
【0037】
非極性:Ala、Val、Leu、Ile、Pro、Met、Phe、Trp
電荷なし、極性:Gly、Ser、Thr、Cys、Tyr、Asn、Gln
酸性:Asp、Glu
塩基性:Lys、Arg、His
他の保存されたアミノ酸置換を次の通り行ってもよい。
【0038】
芳香族:Phe、Tyr、His
プロトンドナー:Asn、Gln、Lys、Arg、His、Trp
プロトンアクセプター:Glu、Asp、Thr、Ser、Tyr、Asn、Gln
特に好ましい断片および変異体は、1または複数の保存されたスクロース結合/加水分解ドメインを含む。このようなドメインの例を図1に示すが、これはLDVWDSWP、QEWSGSA、LRDPHおよびDEIERを含む。
【0039】
特に好ましい断片および変異体は触媒コアを含む。「触媒コア」とは、保存されたスクロース結合および/または加水分解ドメインを含む、シグナルペプチドならびにNおよびC末端可変領域を除いたポリペプチドの内部領域を意味する。触媒コアの例を図1に示すが、これはBacillus subtilis由来のSacBタンパク質のアミノ酸残基65〜468を含む。
【0040】
特に好ましい断片および変異体は、シグナルペプチドを欠く断片および変異体を含む。「シグナルペプチド」とは、N末端シグナル配列を意味する。シグナルペプチドの例を図1に示すが、これはBacillus subtilis由来のSacBタンパク質のアミノ酸1〜27を含む。
【0041】
特に好ましい断片および変異体は、単子葉植物および双子葉植物の翻訳開始等、植物に適応したコドン使用を有する。
【0042】
特に好ましい断片および変異体は、潜在性スプライス部位および/またはRNA不安定化配列エレメントが不活性化または除去されている。
【0043】
好ましくは、細菌FTをコードする核酸は、Bacillus subtilis、Bacillus amyloliquefaciens、Lactobacillus johnsoniiおよびStreptococcus mutans等、Bacillus属、Lactobacillus属およびStreptococcus属からなる群より選択された細菌種由来の遺伝子(単数または複数)から単離される、あるいはそれに相当する。さらにより好ましくは、細菌FT酵素をコードする核酸は、Bacillus subtilisもしくはBacillus amyloliquefaciens由来のSacB遺伝子、Lactobacillus johnsonii由来のLsc遺伝子またはStreptococcus mutans由来のFTF遺伝子から単離される、あるいはそれに相当する。
【0044】
特に好ましい一実施形態において、SacB遺伝子は、図2に示されている配列および図3に示されているポリペプチド配列をコードするヌクレオチド配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0045】
特に好ましい一実施形態において、Lsc遺伝子は、図4に示されている配列および図5に示されているポリペプチド配列をコードする核酸配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0046】
細菌レバンスクラーゼを発現するトランスジェニック植物が報告されており、一部の事例では発達異常の表現型を提示する。本出願人らは理論に制約されることを望まないが、この現象はレバンスクラーゼ酵素およびフルクタンポリマーの不適切な区画化に起因し得る。トランスジェニックタバコおよびジャガイモ植物における細菌SacB遺伝子のサイトゾルにおける発現は、植物の発達に特に破壊的な影響を与えることを示した(Caimiら、1997年)。しかし、液胞ターゲティングシグナルと融合した細菌フルクトシルトランスフェラーゼを発現する植物は、植物の発達に認識可能な効果を及ぼすことなくフルクタンを蓄積する(Ebskampら、1994年、Yeら、2001年)。
【0047】
発達異常の表現型を生じる可能性を低減させるため、細菌FT遺伝子を改変してそのターゲティングシグナル配列を変化させ、細菌FTタンパク質を高フルクタンレベルの存在する区画に方向付けることができる。
【0048】
より具体的には、細菌FT遺伝子のN−シグナルペプチドが除去され、細胞内ターゲティング配列、好ましくは液胞ターゲティング配列に置き換えられたキメラ配列を作製することができる。
【0049】
好ましい一実施形態において、液胞ターゲティング配列は、サツマイモのSPOR531等、プレプロスポラミンタンパク質をコードする遺伝子に由来する、あるいはそれに相当し得る。
【0050】
特に好ましい一実施形態において、液胞ターゲティング配列は、図6において太字・下線で示されている配列および図7において太字・下線で示されているポリペプチドをコードする核酸配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含むことができる。
【0051】
特に好ましい一実施形態において、細菌FT酵素をコードする核酸は、SPOR:SacBキメラ配列となることができる。好ましくは、SPOR:SacBキメラ配列は、図8に示されている配列および図9に示されているポリペプチド配列をコードする核酸配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0052】
特に好ましい一実施形態において、細菌FT酵素をコードする核酸は、SPOR:Lscキメラ配列となることができる。好ましくは、SPOR:Lscキメラ配列は、図9に示されている配列および図12に示されているポリペプチド配列をコードする核酸配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0053】
特に好ましい一実施形態において、遺伝的構築物は、図8と図11に示されている配列および図9と図12に示されているポリペプチドをコードする核酸配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0054】
別の好ましい一実施形態において、細菌FT遺伝子のN−シグナルペプチドが除去されて1−SST等、フルクトシルトランスフェラーゼ酵素の膜貫通ドメインに置き換えられたキメラ配列を作成することができる。
【0055】
特に好ましい一実施形態において、膜貫通ドメインは、図15において太字・斜字体で示されている配列および図16において太字・斜字体で示されているポリペプチドをコードする核酸配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0056】
特に好ましい一実施形態において、遺伝的構築物は、図17と図20に示されている配列および図18と図21に示されているポリペプチドをコードする核酸配列ならびにその機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0057】
「キメラ配列」とは、元々は別々のタンパク質をコードしていた2以上の連結された核酸またはその機能的に活性な断片もしくは変異体を含む融合遺伝子を発現することによる、組換えにより産生されたハイブリッドを意味する。
【0058】
「融合遺伝子」とは、融合タンパク質が好ましくは翻訳融合体として発現することが可能となる仕方で2以上の核酸が連結されたことを意味する。これは通常、第一のタンパク質をコードする核酸配列から終止コドンを除去し、次に第二のタンパク質の核酸配列をインフレームで付け加えることを含む。続いて、細胞により融合遺伝子を単一のタンパク質として発現させる。タンパク質は、両方のオリジナルタンパク質の完全配列、またはそのいずれかもしくは両者の機能的に活性な断片もしくは変異体を含むよう遺伝子操作されていてよい。
【0059】
遺伝的構築物は、連結された2核酸間のリンカーをコードする核酸配列を含んでいてもよい。「リンカー」は、融合タンパク質内の2個の隣接する活性断片の間に配置された、あるいはそれらに接続された任意の化学合成炭水化物・脂質・ポリペプチド分子(またはそれらの組合せ)である。本発明の好ましいリンカーは、アミノ酸の結合によって2個の活性断片と接続した1または複数のアミノ酸残基からなるポリペプチド鎖等、可動性リンカーである。例えば、(Gly4Ser)3リンカーが、融合タンパク質内の2個の活性断片間に配置されてよい。
【0060】
本発明の構築物および方法に用いられるプロモーターは、構成的、組織特異的または誘導性プロモーターとなることができる。好ましい一実施形態において、プロモーターは光調節プロモーター(light−regulated promoter)、より好ましくは光合成プロモーターとなることができる。「光調節プロモーター」とは、光刺激に応答して遺伝子発現を媒介することができるプロモーターを意味する。「光合成プロモーター」とは、植物における光合成経路に関与するタンパク質をコードする遺伝子の発現を媒介することができるプロモーターを意味する。
【0061】
成熟葉身は、葉鞘および茎よりも少量のフルクタンが蓄積する。葉身におけるフルクタンレベルを特異的に上昇させるため、光合成細胞においてフルクタン生合成遺伝子を協調的に発現させる戦略的アプローチが考案された。本出願人らは理論に制約されることを望まないが、光調節または光合成プロモーターの使用は次の利点を提供することができる。
【0062】
・光合成プロモーターは、葉身、茎の上部および外側を含む大きな細胞群(>55%の細胞)における活性を有する。
【0063】
・それは、スクロース産生細胞(葉肉および柔組織維管束鞘細胞)における活性を有する。
【0064】
・その発現パターンは、スクロース蓄積と時間的および空間的に重複する。
【0065】
・フルクトシルトランスフェラーゼ(Frutosyltransferase)活性はソースからスクロースを除去し、これにより光合成におけるフィードバック抑制を妨げ、CO2固定の増加を促進する。
【0066】
特に好ましい光調節プロモーターは、リブロース−1,5−二リン酸カルボキシラーゼ−オキシゲナーゼ(oxygtenase)−スモールサブユニット(RbcS)プロモーターおよびクロロフィルa/b結合タンパク質(CAB)プロモーターならびにそれらの機能的に活性な断片および変異体を含む。
【0067】
光調節プロモーターは、単子葉植物[トウモロコシ、イネ、コムギ、オオムギ、ソルガム、サトウキビ、エネルギー用サトウキビ、飼料草(例えば、Festuca属、Lolium属、Brachiaria属、Paspalum属、Pennisetum属)、バイオエネルギー用草本(例えば、スイッチグラス、Miscanthus属)等]、双子葉植物(シロイヌナズナ、ダイズ、キャノーラ、ワタ、アルファルファおよびタバコ等)および裸子植物を含む、任意の適切な植物種に由来することができる。
【0068】
単子葉植物の形質転換のため、好ましくは、光調節プロモーターは単子葉植物種、より具体的にはTriticum属またはLolium属の種、さらにより具体的にはTriticum aestivumまたはLolium perenneから単離される、あるいはそれらに由来するプロモーターに相当する。
【0069】
双子葉植物の形質転換のため、好ましくは、光調節プロモーターは双子葉植物種、より具体的にはArabidopsis属の種、さらにより具体的にはArabidopsis thalianaから単離される、あるいはそれらに由来するプロモーターに相当する。
【0070】
特に好ましい一実施形態において、RbcSプロモーターは、図23〜26のいずれか一図において小文字・斜字体で示されている配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0071】
特に好ましい別の一実施形態において、RbcSプロモーターは、図29〜36のいずれか一図において小文字・斜字体で示されている配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択された配列を含む。
【0072】
本発明の遺伝的構築物は、任意の適切な技法により植物に導入することができる。本発明の遺伝的構築物を植物細胞に組み入れるための技法(例えば形質導入、トランスフェクション、形質転換または遺伝子ターゲティング)は、当業者にとって周知のものである。このような技法は、アグロバクテリウムを介した導入、根粒菌を介した導入、組織、細胞およびプロトプラストへのエレクトロポレーション、プロトプラスト融合、生殖器官への注入、未熟胚への注入、細胞、組織、カルス、未熟胚および成熟胚への高速噴射による導入、遺伝子銃による形質転換、Whiskers形質転換ならびにそれらの組合せを含む。技法の選択は、主として形質転換される植物の種類に依存し、当業者であれば容易に決定することができる。
【0073】
本発明の遺伝的構築物を組み入れる細胞を後述の通り選択し、次に本技術分野で周知の技法を用いて適切な培地で培養して形質転換植物を再生することができる。温度やpHその他の培養条件は、当業者であれば明らかであろう。その結果生じた植物に、本技術分野で周知の方法を用いて有性生殖か無性生殖のいずれかを行わせて、形質転換植物の後代を作出することができる。
【0074】
本発明の方法は、単子葉植物[草本(例えば、ペレニアルライグラス、ヒロハノウシノケグサ(tall fescue)、イタリアンライグラス(Italian ryegrass)、オオウシノケグサ(red fescue)、クサヨシ(reed canarygrass)、ビッグブルーステム(big bluestem)、コードグラス(cordgrass)、ネピアグラス(napiergrass)、ワイルドライ(wildrye)、ワイルドシュガーケーン(wild sugarcane)、Miscanthus属、スイッチグラスを含む飼料草およびバイオエネルギー用草本)、コーンすなわちトウモロコシ、イネ、コムギ、オオムギ、ソルガム、サトウキビ、ライムギ、カラスムギ)およびエネルギー用作物(例えば、エネルギー用サトウキビ、エネルギー用ソルガム)等]、双子葉植物[シロイヌナズナ、タバコ、ダイズ、キャノーラ、アルファルファ、ジャガイモ、キャッサバ、クローバ(例えば、シロツメクサ、ムラサキツメクサ、ジモグリツメクサ(subterranean clover))、アブラナ科野菜(vegetable brassica)、レタス、ホウレンソウ等]および裸子植物を含む、種々の植物に適用することができる。
【0075】
本発明のさらに別の一態様において、細菌FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結した光調節プロモーターまたはその機能的に活性な断片もしくは変異体を含む、植物の光合成細胞におけるフルクタン生合成を操作することができる遺伝的構築物が提供される。
【0076】
好ましい一実施形態において、本発明に係る遺伝的構築物はベクターとなることができる。
【0077】
「ベクター」とは、遺伝子材料の標的細胞への導入に用いられる遺伝的構築物を意味する。
【0078】
ベクターは、任意の適切な種類のものとなることができ、ウイルスまたは非ウイルス性となることができる。ベクターは発現ベクターとなることができる。このようなベクターは、染色体、非染色体および合成核酸配列、例えば植物ウイルスの誘導体、細菌プラスミド、Agrobacterium tumefaciensから得られたTiプラスミドの誘導体、Agrobacterium rhizogenesから得られたRiプラスミドの誘導体、ファージDNA、酵母人工染色体、細菌人工染色体、バイナリー細菌人工染色体ならびにプラスミドとファージDNAの組合せに由来するベクターを含む。しかし、複製可能あるいは植物細胞に組み込み可能または機能する限りにおいて、その他のベクターを用いてもよい。
【0079】
本発明のこの態様の好ましい一実施形態において、遺伝的構築物はターミネーターをさらに含むことができ、前記プロモーター、遺伝子およびターミネーターは、作動可能に連結している。
【0080】
プロモーター、遺伝子およびターミネーターは、任意の適切な種類のものとなることができ、標的植物細胞で機能するのであれば、その標的植物細胞に対して内在性であっても外来性であってもよい。
【0081】
本発明の遺伝的構築物に用いることができる種々のターミネーターもまた、当業者にとって周知のものである。ターミネーターは、プロモーター配列と同じ遺伝子に由来してよいし、異なる遺伝子に由来してもよい。特に適切なターミネーターは、(CaMV)35SポリA等、ポリアデニル化シグナルならびにノパリン合成酵素(nos)およびオクトピン合成酵素(ocs)遺伝子由来の他のターミネーターである。
【0082】
プロモーター、遺伝子およびターミネーターに加えて、遺伝的構築物は、核酸の発現に必要なさらに別のエレメント、例えばベクター骨格、複製起点(ori)、多重クローニング部位、スペーサー配列、エンハンサー、イントロン(トウモロコシユビキチンUbiイントロン等)、抗生物質抵抗性遺伝子および他の選択マーカー遺伝子[ネオマイシンホスホトランスフェラーゼ(nptII)遺伝子、ハイグロマイシンホスホトランスフェラーゼ(hph)遺伝子、ホスフィノトリシンアセチルトランスフェラーゼ(barまたはpat)遺伝子]ならびにレポーター遺伝子(ベータ−グルクロニダーゼ(GUS)遺伝子(gusA)等)等]を様々な組合せで含むことができる。遺伝的構築物は、翻訳開始のためのリボソーム結合部位を含むこともできる。遺伝的構築物は、発現を増幅するための適切な配列を含むこともできる。
【0083】
特に、遺伝的構築物は、上文に記す通り、連結された2核酸の間にリンカーをコードする核酸配列をさらに含むことができる。
【0084】
当業者であれば、遺伝的構築物の様々な構成要素が、前記核酸の発現をもたらすよう作動可能に連結していることを理解できるであろう。本発明の遺伝的構築物の構成要素を作動可能に連結するための技法は、当業者にとって周知のものである。このような技法は、例えば1または複数の制限酵素部位を含む合成リンカー等、リンカーの使用を含む。
【0085】
好ましくは、遺伝的構築物は実質的に精製または単離されている。「実質的に精製された」とは、遺伝的構築物が、本発明の核酸またはプロモーターの由来する生物の自然発生的なゲノムにおいては、該核酸またはプロモーターに隣接する遺伝子を含まないことを意味する。したがって、この用語は、例えば、ベクター、自己複製プラスミドもしくはウイルス、または原核生物もしくは真核生物ゲノムDNAに組み入れられた、あるいは他の配列から独立した別々の分子(例えば、cDNAまたはPCRまたは制限エンドヌクレアーゼ消化によって生成されたゲノムもしくはcDNA断片)として存在する遺伝的構築物を含む。これは、追加的ポリペプチド配列をコードするハイブリッド遺伝子の一部である遺伝的構築物も含む。好ましくは、実質的に精製された遺伝的構築物は、少なくとも約90%の純度、より好ましくは少なくとも約95%の純度、さらにより好ましくは少なくとも約98%の純度である。
【0086】
用語「単離された」は、材料がその元々の環境(例えば、自然発生的であれば自然環境)から取り分けられたことを意味する。例えば、生きた植物に存在する自然発生的な核酸は単離されていないが、同じ核酸でも、自然システムにおいて共存している材料の一部または全部から分離された核酸は単離されている。このような核酸はベクターの一部であってよい、および/またはこのような核酸は組成物の一部であってよく、このようなベクターまたは組成物がその自然環境の一部ではないという点において、これは依然として単離されている。
【0087】
形質転換された宿主細胞を選抜するための表現型形質を提供するための選択マーカー遺伝子を使用する代わりとして、形質転換細胞における遺伝的構築物の存在は、PCR(ポリメラーゼ連鎖反応)、サザンブロットハイブリダイゼーション解析、組織化学的アッセイ(例えば、GUSアッセイ)、薄層クロマトグラフィー(TLC)、ノーザンブロットおよびウエスタンブロットハイブリダイゼーション解析等、本技術分野で周知の他の技法により決定することができる。
【0088】
本出願人は、本発明の方法が、非形質転換対照植物と比べて形質転換植物において増強されたバイオマスをもたらし得ることも見出した。これにより、この増強されたバイオマスは、形質転換植物を同定するための選抜ツールとして用いることができる。この構成は、後にこのようなマーカーを除去(そしてマーカーなし植物の作製のため)することが困難である場合、一部の状況において、形質転換体選抜のための抗生物質抵抗性または他のマーカーを含む必要がないという利点がある。
【0089】
「バイオマス増強」または「増強されたバイオマス」とは、非形質転換対照植物と比べて形質転換植物におけるシュートおよび/または根の生長を含むバイオマス収量の増強、増加または安定性増加を意味する。例えば、合計葉面積、累積葉面積、葉の生長動態(すなわち、経時的な葉の数)、シュートの数、分げつの数、根の数、根の質量もしくは重量、シュートの質量もしくは重量、根の長さ、シュートの長さ、走根の長さ、塊茎の数、塊茎の重量、花の数、果実の数、種子の数、種子の重量、果実の重量、開花している植物体のパーセンテージおよび花1個当たりまたは作付面積当たりの種子収量からなる群より選択された1または複数の生長特徴は、非形質転換対照植物と比べて形質転換植物において増強され、増加され、またはより安定的となることができる。
【0090】
この技法は、実質的に遺伝的に均一または遺伝的に同一である、あるいは形質転換前のバイオマスとの表現型の差が小さいことを示す植物に特に適用できる。
【0091】
したがって、本発明のさらに別の一態様において、植物におけるバイオマスを増強する方法であって、細菌FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結(liked)したプロモーターまたはその機能的に活性な断片もしくは変異体を含む有効量の遺伝的構築物を前記植物に導入するステップを含む方法が提供される。好ましくは、プロモーターは光調節プロモーターである。
【0092】
本発明の方法、核酸および遺伝的構築物は、他の遺伝的操作方法または他の導入核酸もしくは遺伝的構築物と組み合わせて用いて、これにより形質を積み重ねることができる。このため、積み重ねられた遺伝子(または積み重ねされた形質)を有するトランスジェニック植物体、植物細胞、植物種子または他の植物部分が作出される。例えば、本発明の技術は、除草剤抵抗性技術(例えば、グルホシネート、グリホサート)、害虫抵抗性技術(例えば、Bacillus thuringiensis)または老化遅延技術と組み合わせることができる。核酸または遺伝的構築物は、上文に記す通り、任意の適切な技法により植物に導入することができ、同時発生的に、連続的にまたは別々に導入することができる。
【0093】
本発明のさらになお別の一態様において、植物におけるバイオマスを増強する方法であって、有効量の
(a)植物の光合成細胞においてフルクタン生合成を操作することができる遺伝的構築物であって、細菌FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む遺伝的構築物と、
(b)植物の老化を操作することができる遺伝的構築物と、
を植物に導入するステップを含む方法が提供される。
【0094】
遺伝的構築物は、上文に記す通り任意の適切な技法により植物に導入することができ、同時発生的に、連続的にまたは別々に導入することができる。
【0095】
好ましくは、フルクタン生合成を操作することができる遺伝的構築物は、上文に記す通りである。
【0096】
好ましくは、植物の老化を操作することができる遺伝的構築物は、植物の光合成細胞の老化を操作することができる。
【0097】
好ましくは、老化を操作することができる遺伝的構築物は、サイトカイニンの生合成に関与する酵素をコードする遺伝子またはその機能的に活性な断片もしくは変異体に作動的に連結したmyb遺伝子プロモーターもしくは改変されたmyb遺伝子プロモーターまたはそれらの機能的に活性な断片もしくは変異体を含む。
【0098】
適切な遺伝的構築物またはベクターは、その開示全体を本明細書の一部を構成するものとしてここに援用する、国際特許出願PCT/AU01/01092号明細書および米国特許出願第11/789,526号明細書に記載されている。
【0099】
「老化を操作する」は一般に、非形質転換対照植物と比べた形質転換植物または形質転換植物の細胞もしくは器官、例えば光合成細胞における老化の遅延に関する。しかし、一部の適用では、植物における老化を促進、あるいは改変することが望ましいであろう。老化は、例えばアンチセンス遺伝子を利用することによって促進、あるいは改変することができる。
【0100】
myb遺伝子プロモーターは、任意の適切な種類のものとなることができる。好ましくは、myb遺伝子プロモーターは、myb32遺伝子プロモーターである。好ましくは、myb遺伝子プロモーターは、Arabidopsis属、より好ましくはArabidopsis thalianaに由来する。最も好ましくは、myb遺伝子プロモーターは、国際出願PCT/AU01/01092号明細書の配列番号1に示されている配列ならびにその機能的に活性な断片および変異体からなる群より選択されたヌクレオチド配列を含む。
【0101】
適切なプロモーターは、その開示全体を本明細書の一部を構成するものとしてここに援用するLiら、Cloning of three MYB−like genes from Arabidopsis(PGR99〜138)、Plant Physiology、121巻:313(1999年)に記載されている。
【0102】
「改変されたmyb遺伝子プロモーター」とは、前記プロモーターにおける1または複数の根特異的モチーフおよび/または花粉特異的モチーフを欠失するよう、あるいは不活性化するよう改変された、myb遺伝子に通常関連するプロモーターを意味する。
【0103】
好ましくは、改変されたmyb遺伝子プロモーターは、改変されたmyb32遺伝子プロモーターである。好ましくは、改変されたmyb遺伝子プロモーターは、Arabidopsis属、あるいはより好ましくはArabidopsis thalianaに由来するmyb遺伝子プロモーターから改変される。
【0104】
本発明に従って改変できる適切なプロモーターは、その開示全体を本明細書の一部を構成するものとしてここに援用するLiら、Cloning of three MYB−like genes from Arabidposis(PGR99〜138)Plant Physiology、121巻:313(1999年)に記載されている。
【0105】
「根特異的モチーフ」とは、植物の根において任意の関連遺伝子の発現を導く、3〜7ヌクレオチド、好ましくは4〜6ヌクレオチド、より好ましくは5ヌクレオチドの配列を意味する。
【0106】
好ましくは、根特異的モチーフは、コンセンサス配列ATATTまたはAATATを含む。
【0107】
「花粉特異的モチーフ」とは、植物の花粉において関連遺伝子の発現を導く、3〜7ヌクレオチド、好ましくは4〜6ヌクレオチド、より好ましくは4または5ヌクレオチドの配列を意味する。
【0108】
好ましくは、花粉特異的モチーフは、TTTCT、AGAAA、TTCTおよびAGAAからなる群より選択されたコンセンサス配列を含む。
【0109】
根または花粉特異的モチーフは、モチーフ内の1または複数のヌクレオチドを付加、欠失、置換または誘導体化することによって不活性化し、これにより好ましいコンセンサス配列を持たないようにすることができる。
【0110】
好ましくは、改変されたmyb遺伝子プロモーターは、米国特許出願第11/789,526号明細書の配列番号2、3および4に示されている配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択されたヌクレオチド配列を含む。
【0111】
「サイトカイニンの生合成に関与する酵素をコードする遺伝子」とは、カイネチン、ゼアチンおよびベンジルアデニン等、サイトカイニンの合成に関与する酵素をコードする遺伝子、例えば、sho遺伝子(例えば、ペチュニア由来の)等、イソペンチル(isopentyl)トランスフェラーゼ(ipt)またはipt様遺伝子をコードする遺伝子を意味する。好ましくは、遺伝子は、イソペンテニル(isopentenyl)トランスフェラーゼ(ipt)遺伝子またはsho遺伝子である。好ましい一実施形態において、遺伝子は、Agrobacterium属、より好ましくはAgrobacterium tumefaciens;Lotus属、より好ましくはLotus japonicus;およびPetunia属、より好ましくはPetunia hybridaからなる群より選択された種に由来する。
【0112】
最も好ましくは、遺伝子は、米国特許出願第11/789,526号明細書の配列番号5、7および9に示されている配列、米国特許出願第11/789,526号明細書の配列番号6、8および10に示されているポリペプチドをコードする配列ならびにそれらの機能的に活性な断片および変異体からなる群より選択されたヌクレオチド配列を含む。
【0113】
本発明はまた、形質転換植物を選抜する方法であって、細菌FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む有効量の遺伝的構築物を前記植物に導入するステップと、バイオマスが増強された植物を選抜するステップとを含む方法も提供する。好ましくは、プロモーターは光調節プロモーターである。
【0114】
本発明のさらに別の一態様において、本発明に係る遺伝的構築物またはベクターを含み、非形質転換対照植物と比べてフルクタン生合成特徴が改変された、またはバイオマスが増強されたトランスジェニック植物細胞、植物体、植物種子または他の植物部分が提供される。
【0115】
「改変されたフルクタン生合成特徴」とは、形質転換植物が、非形質転換対照植物と比べてフルクタン生合成の増加を示すことおよび/またはより高レベルの可溶性炭水化物を含むことを意味する。
【0116】
好ましい一実施形態において、バイオマスが増強されたトランスジェニック植物細胞、植物体、植物種子または他の植物部分は、非形質転換対照植物と比べてバイオマスが少なくとも約15%、より好ましくは少なくとも約25%、より好ましくは少なくとも約35%、より好ましくは少なくとも約50%増加する。
【0117】
例えば、非形質転換対照植物と比べてバイオマスが約15%から500%の間、より好ましくは約25%から300%の間、より好ましくは約35%から200%の間、より好ましくは約50%から100%の間増加していてもよい。
【0118】
好ましい一実施形態において、フルクタン生合成特徴が改変されたトランスジェニック植物細胞、植物体、植物種子または他の植物部分は、非形質転換対照植物と比べて可溶性炭水化物が少なくとも約15%、より好ましくは少なくとも約25%、より好ましくは少なくとも約35%、より好ましくは少なくとも約50%増加する。
【0119】
例えば、非形質転換対照植物と比べて可溶性炭水化物が約15%から500%の間、より好ましくは約25%から300%の間、より好ましくは約35%から200%の間、より好ましくは約50%から100%の間増加する。
【0120】
好ましくは、トランスジェニック植物細胞、植物体、植物種子または他の植物部分は、本発明に係る方法によって作出される。
【0121】
本発明はまた、本発明の植物細胞に由来し、本発明の遺伝的構築物またはベクターを含むトランスジェニック植物体、植物種子または他の植物部分も提供する。
【0122】
本発明はまた、本発明の植物に由来し、本発明の遺伝的構築物またはベクターを含むトランスジェニック植物体、植物種子または他の植物部分も提供する。
【0123】
好ましくは、トランスジェニック植物細胞、植物体、植物種子または他の植物部分は、Lolium属の種、より好ましくはLolium perenneまたはLolium arundinaceumである。
【0124】
好ましくは、トランスジェニック植物細胞、植物体、植物種子または他の植物部分は、穀物類、より好ましくはTriticum属の種、より好ましくはコムギ(Triticum aestivum)である。
【0125】
例えば、本発明は、草食動物の栄養改善のための、葉身におけるフルクタンが増加し、活発に生長し、非生物的ストレスに対しより高い耐性を有するトランスジェニック−ペレニアルライグラス植物の作出を可能にする。
【0126】
本発明はまた、例えばバイオ燃料生産または動物の食餌に利用可能な炭水化物の質量増加のための、フルクタンが増加し、活発に生長し、非生物的ストレスに対する耐性を有するトランスジェニックコムギ植物の作出も可能にする。
【0127】
「植物細胞」とは、半透膜によって仕切られた、色素体を含む任意の自己増殖細胞を意味する。このような細胞は、さらなる増殖が望ましい場合、細胞壁も必要とする。本明細書における植物細胞は、藻類、ラン藻、種子懸濁培養物、胚、成長点領域、カルス組織、葉、根、シュート、配偶体、胞子体、花粉および小胞子を含むが、これらに限定されるものではない。
【0128】
「トランスジェニック」とは、技巧的手法によって細胞に挿入され、その細胞から発生した生物のゲノムの一部となったDNA配列を含む、任意の細胞を意味する。本明細書において、トランスジェニック生物は一般にトランスジェニック植物であり、DNA(導入遺伝子)は、技巧的手法によって核または色素体ゲノムのいずれかに挿入される。
【0129】
次に、添付の実施例および図面を参照することにより本発明をさらに十分に説明する。なお、次の記述は具体例としてのものであるに過ぎず、決して上述の本発明の一般性を限定するものと考えるべきではないことを理解するべきである。
【図面の簡単な説明】
【0130】
【図1】図1は、Bacillus subtilisから得られたSacBタンパク質、GH68ファミリーメンバーの略図である。示されている4個の異なる領域は、N末端シグナル配列、N末端可変領域、触媒コアおよびC末端可変領域である。触媒三連構造(D86、D247およびE342)およびスクロース結合(W85、W163およびR246)を含むアミノ酸残基。
【図2】図2は、Bacillus subtilisから得られたSacB遺伝子(レバンスクラーゼ)のヌクレオチド配列を示す図である。N末端シグナルペプチドをコードするヌクレオチド配列を太字で示す。
【図3】図3は、Bacillus subtilisから得られたSacBタンパク質(レバンスクラーゼ)のアミノ酸配列を示す図である。N末端シグナルペプチドを太字で示す。
【図4】図4は、Lactobacillus johnsonii NCC 533から得られたLsc遺伝子(イヌロスクラーゼ)のヌクレオチド配列を示す図である。N末端シグナルペプチドをコードするヌクレオチド配列を太字で示す。
【図5】図5は、Lactobacillus johnsonii NCC 533から得られたLscタンパク質(イヌロスクラーゼ)のアミノ酸配列を示す図である。N末端シグナルペプチドを太字で示す。
【図6】図6は、I.batatasから得られたプレプロスポラミンタンパク質であるSPOR531のヌクレオチド配列を示す図である。液胞ターゲティングシグナル配列を太字・下線で示す。
【図7】図7は、I.batatasから得られたプレプロスポラミンタンパク質であるSPOR531のアミノ酸配列を示す図である。液胞ターゲティングシグナル配列を太字・下線で示す。
【図8】図8は、SPOR:SacBキメラヌクレオチド配列を示す図である。SacBのN末端シグナル配列をSPORの液胞ターゲティングシグナル(太字・下線で示す)に置き換えてある。
【図9】図9は、SPOR:SacBキメラタンパク質配列を示す図である。SacBのN末端シグナル配列をSPORの液胞ターゲティングシグナル(太字・下線で示す)に置き換えてある。
【図10】図10は、膜タンパク質の二次構造予測ソフトウェアSOSUI、http://bp.nuap.nagoya−u.ac.jp/sosui/を用いたSPOR−SacB融合タンパク質の二次構造予測を示す図である。
【図11】図11は、SPOR:Lscキメラヌクレオチド配列を示す図である。LscのN末端シグナル配列をSPORの液胞ターゲティングシグナル(太字・下線で示す)に置き換えてある。
【図12】図12は、SPOR:Lscキメラタンパク質配列を示す図である。LscのN末端シグナル配列をSPORの液胞ターゲティングシグナル(太字・下線で示す)に置き換えてある。
【図13】図13は、膜タンパク質の二次構造予測ソフトウェアSOSUI、http://bp.nuap.nagoya−u.ac.jp/sosui/を用いたSPOR−Lsc融合タンパク質の二次構造予測を示す図である。
【図14】図14は、膜タンパク質の二次構造予測ソフトウェアSOSUI、http://bp.nuap.nagoya−u.ac.jp/sosui/を用いたLp1−SSTの二次構造予測を示す図である。
【図15】図15は、L.perenneから得られたLp1−SSTヌクレオチド配列を示す図である。Lp1−SST膜貫通ドメインのコード配列を太字・斜字体で示す。
【図16】図16は、L.perenneから得られたLp1−SSTタンパク質配列を示す図である。Lp1−SST膜貫通ドメインを太字・斜字体で示す。
【図17】図17は、Lp1−SST−SacBキメラヌクレオチド配列を示す図である。SacBのN末端シグナルのコード配列をLp1−SST膜貫通ドメインのコード配列(太字・斜字体で示す)に置き換えてある。
【図18】図18は、Lp1−SST−SacBキメラタンパク質配列を示す図である。SacBのN末端シグナル配列をLp1−SST膜貫通ドメイン(太字・斜字体で示す)に置き換えてある。
【図19】図19は、膜タンパク質の二次構造予測ソフトウェアSOSUI、http://bp.nuap.nagoya−u.ac.jp/sosui/を用いたLp1−SST−SacB融合タンパク質の二次構造予測を示す図である。
【図20】図20は、Lp1−SST−Lscキメラヌクレオチド配列を示す図である。LscのN末端シグナルのコード配列をLp1−SST膜貫通ドメインのコード配列(太字・斜字体で示す)に置き換えてある。
【図21】図21は、Lp1−SST−Lscキメラタンパク質配列を示す図である。LscのN末端シグナル配列をLp1−SST膜貫通ドメイン(太字・斜字体で示す)に置き換えてある。
【図22】図22は、膜タンパク質の二次構造予測ソフトウェアSOSUI、http://bp.nuap.nagoya−u.ac.jp/sosui/を用いたLp1−SST−Lsc融合タンパク質の二次構造予測を示す図である。
【図23−1】図23は、AtRbcS::SPOR−SacB::NOS発現カセットのヌクレオチド配列を示す図である。
【図23−2】図23は、AtRbcS::SPOR−SacB::NOS発現カセットのヌクレオチド配列を示す図である。
【図24−1】図24は、AtRbcS::SPOR−Lsc::NOS発現カセットのヌクレオチド配列を示す図である。
【図24−2】図24は、AtRbcS::SPOR−Lsc::NOS発現カセットのヌクレオチド配列を示す図である。
【図25−1】図25は、AtRbcS::Lp1−SST−SacB::NOS発現カセットのヌクレオチド配列を示す図である。
【図25−2】図25は、AtRbcS::Lp1−SST−SacB::NOS発現カセットのヌクレオチド配列を示す図である。
【図26−1】図26は、AtRbcS::Lp1−SST−Lsc::NOS発現カセットのヌクレオチド配列を示す図である。
【図26−2】図26は、AtRbcS::Lp1−SST−Lsc::NOS発現カセットのヌクレオチド配列を示す図である。
【図27】図27は、Gateway pDestinationベクターを示す図である。
【図28A】図28は、双子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.AtRbcS::SPOR−SacB::NOS;B.AtRbcS::SPOR−Lsc::NOS;C.AtRbcS::Lp1−SST−SacB::NOS、およびD.AtRbcS::Lp1−SST−Lsc::NOS。
【図28B】図28は、双子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.AtRbcS::SPOR−SacB::NOS;B.AtRbcS::SPOR−Lsc::NOS;C.AtRbcS::Lp1−SST−SacB::NOS、およびD.AtRbcS::Lp1−SST−Lsc::NOS。
【図28C】図28は、双子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.AtRbcS::SPOR−SacB::NOS;B.AtRbcS::SPOR−Lsc::NOS;C.AtRbcS::Lp1−SST−SacB::NOS、およびD.AtRbcS::Lp1−SST−Lsc::NOS。
【図28D】図28は、双子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.AtRbcS::SPOR−SacB::NOS;B.AtRbcS::SPOR−Lsc::NOS;C.AtRbcS::Lp1−SST−SacB::NOS、およびD.AtRbcS::Lp1−SST−Lsc::NOS。
【図29】図29は、TaRbcSp::SPOR−SacB::TaRbcst発現カセットのヌクレオチド配列を示す図である。
【図30−1】図30は、TaRbcSp::SPOR−SacB::TaRbcst TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図30−2】図30は、TaRbcSp::SPOR−SacB::TaRbcst TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図31−1】図31は、TaRbcSp::SPOR−Lsc::TaRbcst発現カセットのヌクレオチド配列を示す図である。
【図31−2】図31は、TaRbcSp::SPOR−Lsc::TaRbcst発現カセットのヌクレオチド配列を示す図である。
【図32−1】図32は、TaRbcSp::SPOR−Lsc::TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図32−2】図32は、TaRbcSp::SPOR−Lsc::TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図33−1】図33は、TaRbcSp::Lp1−SST−SacB::TaRbcst発現カセットのヌクレオチド配列を示す図である。
【図33−2】図33は、TaRbcSp::Lp1−SST−SacB::TaRbcst発現カセットのヌクレオチド配列を示す図である。
【図34−1】図34は、TaRbcSp::Lp1−SST−SacB::TaRbcst TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図34−2】図34は、TaRbcSp::Lp1−SST−SacB::TaRbcst TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図35−1】図35は、TaRbcSp::Lp1−SST−Lsc::TaRbcst発現カセットのヌクレオチド配列を示す図である。
【図35−2】図35は、TaRbcSp::Lp1−SST−Lsc::TaRbcst発現カセットのヌクレオチド配列を示す図である。
【図36−1】図36は、TaRbcSp::SPOR−Lsc::TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図36−2】図36は、TaRbcSp::SPOR−Lsc::TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図36−3】図36は、TaRbcSp::SPOR−Lsc::TaRbcst+AtMYB32::IPT::35S発現カセットのヌクレオチド配列を示す図である。
【図37】図37は、Gateway pDestinationベクター、pBS:ubi::bar::NOSを示す図である。
【図38】図38は、単子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.TaRbcS::SPOR−SacB::TaRbcS、およびB.TaRbcS::SPOR−SacB::TaRbcS+AtMYB32::IPT::35S
【図39】図39は、単子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.TaRbcS::SPOR−Lsc::TaRbcS、およびB.TaRbcS::SPOR−Lsc::TaRbcS+AtMYB32::IPT::35S
【図40】図40は、単子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.TaRbcS::Lp1−SST−SacB::TaRbcS、およびB.TaRbcS::Lp1−SST−SacB::TaRbcS+AtMYB32::IPT::35S
【図41】図41は、単子葉植物形質転換用のGateway pDestinationベクターを示す図である。A.TaRbcS::Lp1−SST−Lsc::TaRbcS、およびB.TaRbcS::Lp1−SST−Lsc::TaRbcS+AtMYB32::IPT::35S
【図42】図42は、Nicotiana tabacumの葉肉由来プロトプラストの単離を示す図である。A)〜B)4〜6週目のin vitro葉材料の解剖、前酵素消化;C)4〜6週目のin vitro葉材料の消化、16時間のインキュベーション;D)プロトプラスト懸濁液の回収;E)プロトプラスト豊富界面の分離;F)〜G)インタクトな葉緑体豊富プロトプラスト
【図43】図43は、一過性発現解析のためのNicotiana tabacumの葉肉由来プロトプラストの単離を示す図である。A)〜B)インタクトな葉緑体豊富プロトプラスト;C)液体濃縮培地におけるプロトプラストの培養;D)生存プロトプラスト、単離48時間後
【図44】図44は、安定的な形質転換のためのNicotiana tabacumの葉肉由来プロトプラストの単離を示す図である。A)〜B)インタクトな葉緑体豊富プロトプラスト;C)プロトプラストを包埋したsea plaqueアガロースプラグ、0日目;D)生存プロトプラスト、単離および包埋6日後;E)〜F)包埋され、解放されたプロトプラスト由来の微小カルス、単離および包埋4週間後
【図45】図45は、Nicotiana tabacumの葉肉プロトプラスト由来の微小カルスからシュートの再生を示す図である。A)液体成長培地Aに解放された微小カルス、単離および包埋6〜7週間後;B〜C)固形成長培地におけるカルスの増殖;D)〜E)葉肉プロトプラスト由来のカルスからシュートの誘導および再生;F)再生シュートから根の発達;G)〜H)ガラス温室内に封じ込めた植物体の生長および発達
【図46】図46は、非形質転換プロトプラスト生存率の評価を示す図である。単離48時間後。
【図47】図47は、PEG形質転換プロトプラストの生存率の評価を示す図である。単離およびトランスフェクション48時間後。
【図48】図48は、タバコリーフディスクのアグロバクテリウムを介した形質転換を示す図である。A)形質転換リーフディスクの共存培養、0日目;B)ステージ1シュートの開始、共存培養3日後
【図49−1】図49は、タバコ形質転換リーフディスクにおけるgfp発現の検出を示す図である。A)非形質転換リーフディスク、白色光;B)非形質転換リーフディスク、gfp2フィルター;C)非形質転換リーフディスク、gfp3フィルター;D)&G)Turbo gfp形質転換リーフディスク、白色光;E)&H)Turbo gfp形質転換リーフディスク、gfp2フィルター;F)&I)Turbo gfp形質転換リーフディスク、gfp3フィルター。
【図49−2】図49は、タバコ形質転換リーフディスクにおけるgfp発現の検出を示す図である。A)非形質転換リーフディスク、白色光;B)非形質転換リーフディスク、gfp2フィルター;C)非形質転換リーフディスク、gfp3フィルター;D)&G)Turbo gfp形質転換リーフディスク、白色光;E)&H)Turbo gfp形質転換リーフディスク、gfp2フィルター;F)&I)Turbo gfp形質転換リーフディスク、gfp3フィルター。
【図50】図50は、RT−PCR試料および対照の電気泳動を示す図である。レーン1および13:1kb+DNAラダーレーン2:トランスフェクトしたプロトプラスト1(試料9A)から得られたmRNAの逆転写によって生成したcDNA(2μL)から、遺伝子特異的プライマーによるsacB転写産物の増幅レーン3:cDNA(1μL)からsacB転写産物の増幅(試料9A)レーン4:逆転写酵素なしで行った対照反応(試料9A)レーン5:鋳型なしで行った対照反応(sacBプライマー)レーン6:トランスフェクトしたプロトプラスト1(試料12A)から得られたmRNAの逆転写によって生成したcDNA(2μL)から、遺伝子特異的プライマーによるsacB転写産物の増幅レーン7:cDNA(1μL)からsacB転写産物の増幅(試料12A)レーン8:逆転写酵素なしで行った対照反応(試料12A)レーン9:非トランスフェクトプロトプラストから得られたmRNAの逆転写によって生成したcDNA(2μL)から、遺伝子特異的プライマーによる18S転写産物の増幅レーン10:非トランスフェクトプロトプラストから得られたmRNAの逆転写によって生成したcDNA(1μL)から、遺伝子特異的プライマーによる18S転写産物の増幅レーン11:逆転写酵素なしで行った対照反応(非トランスフェクトプロトプラスト)レーン12:鋳型なしで行った対照反応(18Sプライマー)。
【発明を実施するための形態】
【0131】
(実施例1)
細菌フルクタン生合成遺伝子の単離
図1は、Bacillus subtilisから得られたSacBタンパク質の略図を提示する。示されている4個の異なる領域は、N末端シグナル配列、N末端可変領域、触媒コアおよびC末端可変領域である。構造的には、細菌イヌロスクラーゼおよびレバンスクラーゼの多くは、N末端シグナルペプチド、触媒三連構造を共有する。この配列は配列改変の際に除去される。スクロース結合に関与する残基は触媒コア配列内部に位置するが、これは改変の際に手をつけずにおく。
【0132】
細菌レバンスクラーゼ(SacB)およびイヌロスクラーゼ(Lsc)のヌクレオチドおよびタンパク質配列をそれぞれ図2〜5に提供する。しかし、植物形質転換のため、細菌レバンスクラーゼおよびイヌロスクラーゼ配列は次の仕方によっても改変される。
【0133】
・細菌N−シグナルペプチドの除去、
・翻訳開始等、コドン使用の単子葉植物および双子葉植物への適応、
・潜在性スプライス部位およびRNA不安定化配列エレメントの除去、
・単子葉植物および双子葉植物の液胞ターゲティング配列ならびに植物1−SST特異的膜貫通ドメイン等、推定細胞内ターゲティング配列を用いてコード配列をさらに改変する。
【0134】
これらの変化の概略を次の実施例に記す。
【0135】
(実施例2)
細菌フルクタン生合成遺伝子の改変
細菌FT遺伝子の特異的細胞区画へのターゲティング
細菌FT遺伝子をサイトゾルから離れてスクロースおよびフルクタンの両方が蓄積する区画に方向付けるため、サツマイモ(Ipomoea batatas)のプレプロスポラミンタンパク質(SPOR531)由来の推定液胞ターゲティング配列を用いてSacBおよびLscのコード配列を改変する。スポラミン前駆体のプロペプチドは、スポラミンの液胞へのターゲティングに必要とされる(Hattoriら、1985年)。スポラミンの液胞ターゲティング情報はアミノ末端プロペプチドにコードされ、これは図5および図6に示されている。
【0136】
配列改変は、SacBおよびLsc細菌フルクタン(fuctan)生合成遺伝子の両方からN−シグナルペプチドを除去すること、およびSPOR531液胞ターゲティングシグナルを付加することに関与する(それぞれ図7〜8および図10〜11)。
【0137】
膜タンパク質の二次構造予測ソフトウェアSOSUI、http://bp.nuap.nagoya−u.ac.jp/sosui/を用いた細胞内局在の予測および改変されたタンパク質のトポロジーは、液胞ターゲティングシグナルによって誘発された膜貫通局在を示す(図9および図12)。
【0138】
細菌FT遺伝子へのLp1−SSTタンパク質由来膜貫通ドメインの付加
SOSUIソフトウェアは、Lolium perenne 1−SST遺伝子の二次構造を予測するためにも用いた。N末端に膜貫通ドメインを示唆するこの構造を図13に示す。膜貫通ドメインコード配列およびタンパク質配列をそれぞれ図14および図15に示す。
【0139】
配列改変は、SacBおよびLsc細菌フルクタン生合成遺伝子の両方からN−シグナルペプチドを除去すること、およびLp1−SST膜貫通ドメインを付加することに関与する(それぞれ図16〜17および図19〜20)。膜タンパク質の二次構造予測ソフトウェアSOSUIを用いて、SacBおよびLscの改変された配列の細胞内(subsellular)局在およびタンパク質トポロジーを評価し、それらの予測二次構造をそれぞれ図18および図21に提示する。
【0140】
(実施例3)
双子葉植物における安定的な形質転換のためのベクターの作製
発現構築物の合成
双子葉植物に形質転換するための、光合成(photosyntheic)プロモーター、実施例2に示されている改変された細菌フルクタン生合成遺伝子およびNOSターミネーター配列を利用した発現構築物を人工合成する。
【0141】
光合成プロモーターの使用はフルクタンを蓄積する組織において遺伝子を発現させ、一方改変された配列はタンパク質を特異的植物細胞区画に標的化させる。
【0142】
リブロース−1,5−二リン酸カルボキシラーゼ−オキシゲナーゼ−スモールサブユニット(RbcS)は、高等植物における十分に特徴付けられた光調節遺伝子である。Arabidopsis thalianaリブロース−1,5−二リン酸カルボキシラーゼ−オキシゲナーゼ−スモールサブユニット(AtRbcS)プロモーター配列の1700bp断片は、以前にクローニングされている。発現ベクターで用いるため、AtRbcSプロモーター由来のTATAシグナルを含む、より短い断片を増幅するためのプライマーを設計する。
【0143】
改変された細菌フルクタン生合成遺伝子のための新たな予測配列は、植物における発現のためにコドン使用を変更させ、潜在性スプライス部位およびRNA不安定化配列エレメントを除去しつつ人工合成し、植物細胞におけるその性能を最適化する。
【0144】
図23〜26は、それぞれ発現カセットAtRbcS::SPOR−SacB::NOS、AtRbcS::SPOR−Lsc::NOS、AtRbcS::Lp1−SST−SacB::NOSおよびAtRbcS::Lp1−SST−Lsc::NOSを表し、これらはまだコドン最適化または不安定化エレメントの除去を行っていない。
【0145】
シロイヌナズナ光合成プロモーターによって駆動される改変された細菌FT遺伝子を含む双子葉植物形質転換のための構築物の作製
各合成発現カセットを、Gateway法に使用可能なpDONORベクター内に配置して、植物の形質転換用の最終的な目的ベクターに組み換える。
【0146】
35Sp:hph:35St選択マーカーカセットを含む、Gateway法に使用可能な目的ベクターを作製した。pPZP200_35Sp_hph_35St_R4/R3(図27)。
【0147】
Gateway LR組換え反応は、次の双子葉植物の形質転換用のdestinationベクターを作製する。
AtRbcS::SPOR−SacB::NOS(図28A)
AtRbcS::SPOR−Lsc::NOS(図28B)
AtRbcS::Lp1−SST−SacB::NOS(図28c)
AtRbcS::Lp1−SST−Lsc::NOS(図28d)
(実施例4)
単子葉植物における安定的な形質転換のためのベクターの作製
発現構築物の合成
単子葉植物の形質転換用の、パンコムギ光合成プロモーター(TaRbcsp)、実施例2に示されている改変された細菌フルクタン生合成遺伝子およびTaRbcSターミネーター配列を利用した発現構築物を人工合成する。光合成プロモーターの使用はフルクタンを蓄積する組織において遺伝子を発現させ、一方改変された配列はタンパク質を特異的植物細胞区画に標的化させる。
【0148】
パンコムギ(Triticum aestivum)のTaRbcS調節配列(プロモーターおよびターミネーター)は、以前にクローニングされている(Zengら、1995年;Sasanuma、2001年)。発現ベクターで用いるため、TaRbcS遺伝子(NCBI受託番号AB042069)由来のTATAシグナルを含む、以前に発表された配列から得られた695bpプロモーター断片を増幅する。
【0149】
改変された細菌フルクタン生合成遺伝子のための新たな予測配列を、植物における発現のためにコドン使用を変更させ、潜在性スプライス部位およびRNA不安定化配列エレメントを除去しつつ人工合成して、植物細胞におけるその性能を最適化する。
【0150】
上に概略されている方法を用いて発現カセットを合成し、フルクタン生合成遺伝子およびLXR(商標)技術の両方を含むトランスジェニック植物を作出する。LXR(商標)技術は、AtMYB32遺伝子プロモーター制御下における葉老化遅延のための1種類の候補遺伝子(IPT)を含む発現カセットに基づく。発現カセットAtMYB3p::IPT::35Stは、国際特許出願PCT/AU01/01092号明細書に記載されている。トランスジェニックLXR(商標)植物の表現型は、植物の加齢に関連する葉の黄化およびクロロフィル喪失の減少を含み、これは光合成能の上昇を導き、分げつ化および植物バイオマスの改善をもたらす。
【0151】
2種の技術の統合は、光合成プロモーター活性化の拡大によるフルクタンの発現上昇をもたらし、植物における生産性の範囲を増加させることにより種々の適用の効力に有意な影響を与えることができる。
【0152】
図29〜36は、それぞれ発現カセットTaRbcS::SPOR−SacB::TaRbcS、TaRbcS::SPOR−SacB::TaRbcS+AtMYB32::IPT::35S、TaRbcS::SPOR−Lsc::TaRbcS、TaRbcS::SPOR−Lsc::TaRbcS+AtMYB32::IPT::35S、TaRbcS::Lp1−SST−SacB::TaRbcS、TaRbcS::Lp1−SST−SacB::TaRbcS+AtMYB32::IPT::35S、TaRbcS::Lp1−SST−Lsc::TaRbcSおよびTaRbcS::Lp1−SST−Lsc::TaRbcS+AtMYB32::IPT::35Sを表し、これらはまだコドン最適化または不安定化エレメントを除去されていない。
Triticum属の光合成プロモーターによって駆動される改変された細菌FT遺伝子を含む単子葉植物形質転換のための構築物の作製
各合成発現カセットを、Gateway法に使用可能なpDONORベクター内に配置して、植物の形質転換用の最終的な目的ベクターに組み換える。
【0153】
Ubi::bar::NOS選択マーカーカセットを含む、Gateway法に使用可能な目的ベクターを作製した。pBS::Ubi::bar::NOS_R4/R3(図37)。Gateway LR組換え反応は、次の単子葉植物の形質転換用のdestinationベクターを作製する。
TaRbcS::SPOR−SacB::TaRbcS(図38A)
TaRbcS::SPOR−SacB::TaRbcS+AtMYB32::IPT::35S(図B);
TaRbcS::SPOR−Lsc::TaRbcS(図39A)
TaRbcS::SPOR−Lsc::TaRbcS+AtMYB32::IPT::35S(図39B)
TaRbcS::Lp1−SST−SacB::TaRbcS(図40A)
TaRbcS::Lp1−SST−SacB::TaRbcS+AtMYB32::IPT::35S(図B)
TaRbcS::Lp1−SST−Lsc::TaRbcS(図41A)
TaRbcS::Lp1−SST−Lsc::TaRbcS+AtMYB32::IPT::35S(図41B)
(実施例5)
双子葉植物用構築物 − N.tabacumプロトプラストおよびA.thaliana
直接送達バージョン(プロトプラストにおける一過性発現)ならびにシロイヌナズナ(A.thaliana)およびタバコ(N.tabacum)への安定的な送達のためのバイナリー形質転換ベクターバージョンの、次の構築物を作製した。
1.AtRbcS::1−SST−SacB*
2.AtRbcS::SPOR−SacB*
3.AtRbcS::1−SST−Lsc*
4.AtRbcS::SPOR−Lsc*
*細菌レバンスクラーゼおよびイヌロスクラーゼ配列は、次の仕方で改変する:
・細菌N−シグナルペプチドの除去、
・翻訳開始等、コドン使用の単子葉植物および双子葉植物への適応、
・潜在性スプライス部位およびRNA不安定化配列エレメントの除去、
・単子葉植物および双子葉植物の液胞ターゲティング配列ならびに植物1−SSTおよびFFT特異的膜貫通ドメイン等、推定細胞内ターゲティング配列を用いてコード配列をさらに改変する。
【0154】
(実施例6)
ポリエチレングリコールを介したタバコ葉肉由来プロトプラストの形質転換
本実施例は、上文に記載されている発現カセットのタバコプロトプラストへの送達を説明する(図42〜45を参照)。
【0155】
I.直接遺伝子導入のための葉肉由来プロトプラストの単離
A.葉肉由来プロトプラストを得るためのin vitroシュート培養物の消化
酵素溶液
K4培地[0.3Mではなく0.4Mスクロースを含むK3培地]に溶解した1.0%(w/v)セルラーゼ「Onozuka」R10および1.0%(w/v)Macerozyme(登録商標)R10。酵素標品の混入デンプンをペレット化させるため、スピンダウンする(Sorvall遠心分離機、SS34ローター;7,000rpmで10分間)。KOHでpH5.6に調整し、濾過滅菌(0.2μm孔径)する。4℃で長くて3〜4週間保存する。
【0156】
材料
・固形MS培地とNicotiana tabacumシュート培養物の入った400ml培養容器
・90×20mm滅菌ペトリ皿
・ピンセット
・メス
・滅菌メス刃;#11または#22 。
【0157】
溶液
・酵素溶液;K4培地に溶解した1%セルラーゼおよび1%マセロザイム
・滅菌水 。
【0158】
手順
1.プレート底部をたっぷりと覆うのに十分な量の酵素溶液(15mlで十分と思われる)を滅菌90×20mmペトリ皿にデカントする。
2.4〜6週目のシュート培養物から健常で十分に開いた葉を2〜4枚、空の90×20mmペトリ皿に移す。
3.1枚の葉の背軸面を上にして、周囲の葉組織の断裂を最小限に抑えつつ鋭利な滅菌刃を確実に用いて慎重に主脈を除去する。これを残り3枚の葉に繰り返す。1回当たり少量の葉材料(最大4枚)を取り扱い、層流による乾燥効果を最小限に抑える。
4.半分にした葉を穏やかに積み重ね、鋭利な滅菌刃を用いて1〜2mm片にスライスする。
5.葉切片を酵素溶液の入ったペトリ皿に慎重に移す(背軸面を下に)。皿をParafilm(登録商標)で封じ、16〜18時間、25℃、暗所で振盪せずに一晩インキュベートする。
【0159】
B.葉肉由来プロトプラストの単離
材料
・滅菌5mlピペット
・滅菌10mlピペット
・ピペットボーイ
・滅菌したプロトプラスト濾過ユニット:100mlガラスビーカー上に載せた100μmステンレス鋼メッシュ篩
・滅菌14mlプラスチック製丸底遠心チューブ
・Clements Orbital 500卓上遠心分離機
・ウォーターバス 。
【0160】
培地
・消化中のNicotiana tabacum葉の入った90×20mm滅菌ペトリ皿(複数可)
・0.6%Sea Plaque(商標)アガロースを含む、1:1混合のK3:H固形培地 。
【0161】
溶液
・オートクレーブしたW5溶液
・オートクレーブしたK3溶液 。
【0162】
手順
6.一晩消化した標本を穏やかに撹拌して酵素溶液中にプロトプラストを遊離させる。
【0163】
撹拌は穏やかに、だが徹底的に行い、左右に(水平に)動かして行うこと。
7.プレートをやや傾け、消化中の懸濁液(酵素溶液および植物デブリ)を移し易くする。10ml滅菌ピペットを用いて消化中の懸濁液を滅菌したプロトプラスト濾過ユニットに移し、プロトプラスト懸濁液を植物デブリから分離する。
8.濾過ユニットを穏やかにタップして篩内にたまった余分な液体を出す。
9.プロトプラスト懸濁液を穏やかに混合し、14ml滅菌プラスチック製丸底遠心チューブに分配し、約8mlまで(最大9ml)充填する。
10.懸濁液を再分配して均一な容量分配を得る(最大9ml)。
11.各懸濁液に1.5mlのW5溶液を慎重に重層する。
W5溶液を分注し易くするため、懸濁液の入ったチューブを傾けて置き、ピペットチップをチューブ開口部近くの壁面に触れさせ、その後懸濁液面のすぐ上までゆっくりと下げる。一滴ずつ加えながら、ピペットチップが可能な限り懸濁液面に近づいた状態を確実に維持しつつW5溶液をゆっくりと分注する。これを正確に行えば、プロトプラスト懸濁液の撹拌、ひいてはW5溶液との混合は最小限となるであろう。
12.慎重に蓋をし、チューブを5分間、70gで遠心分離する(Clements Orbital 500卓上遠心分離機、スイングアウトローター、400rpm)。プロトプラストは、界面に浮遊している筈である。
13.プロトプラストの入ったチューブを直立に維持し、滅菌5mlピペットをプロトプラスト層のすぐ上のポイントまで慎重に下げて、下相を可能な限り取らないよう界面におけるプロトプラストを採取する。
W5溶液も同時に採取される。
14.プロトプラストを採取し、「1本」の新しい14ml遠心チューブに移す。プロトプラスト採取が完了したら、ピペッティングで穏やかに吸って吐くことによりプロトプラスト懸濁液を穏やかに混合する。
15.プロトプラスト懸濁液の100μlアリコートを取り分け、900μlのW5溶液の入ったチューブに移すことにより、プロトプラスト収量を決定する。血球計算板でプロトプラストを計数し、1ml当たりのプロトプラスト数を決定する。
16.1ml当たり約1×106(最大1.5×105)個のプロトプラストを得るのに必要な全容量を算出する。新しい14ml丸底遠心チューブにプロトプラスト懸濁液を分配し、等容量が得られたことを確実にする。
17.10mlピペットを用いて、プロトプラストの入った各チューブを全容量最大10mlまでW5溶液で充填する。プロトプラストが乱れるのを最小限に抑えるため、充填の際チューブ壁に沿わせてW5溶液を散布する。
18.蓋をし、蓋閉めしたチューブを穏やかに1回反転することによってプロトプラストを再懸濁する。
19.プロトプラストをペレット化し[70g(Clements Orbital 500卓上遠心分離機、400rpm)5分間でスピンする]、その後W5溶液を全て除去して純粋なプロトプラスト懸濁液を残す。
20.穏やかに振盪することによりプロトプラスト懸濁液を再懸濁する。
21.プロトプラストの入った各チューブを全容量5mlまでW5溶液で充填し、室温で最短で1時間、最長で4時間インキュベートする。
22.1〜4時間のインキュベーション時間の間に、単離プロトプラストに直接遺伝子導入するための調製物に次の構成要素を構成する。
【0164】
・40%PEG溶液を−20℃保存から取り出し、室温で保存する。直接遺伝子導入に進む30分前に、PEG溶液を温水の入ったビーカー内でインキュベートする。
【0165】
・K3:H固形培地を電子レンジで融解させる。完全に融解したら、使用準備が整うまで40℃のウォーターバス内に置く。
【0166】
II.ポリエチレングリコールを用いたプロトプラストへの直接遺伝子導入
DNA形質転換
100%(v/v)エタノールで沈殿および洗浄することによりプラスミドDNAを滅菌し、層流フード内で乾かす[70%エタノールにおけるプラスミドDNAの沈殿も可能であるが、DNAペレットは乾かすのにさらに時間がかかる]。一過性形質転換のため、終濃度0.7μg/μlになるようDNAペレットを30μlの滅菌再蒸留水に再懸濁する。DNAの物理的構造は、一過性のためにはスーパーコイルとなるべきであり、安定的な形質転換のためには直鎖化するべきである(対象遺伝子の外側で)。形質転換プラスミドDNAにキャリアーDNA(例えば、魚精子DNA)を添加すると、通常、より良好で安定的な形質転換頻度が得られる。安定的な形質転換のため、上に示す通り10μgの直鎖化プラスミドDNAおよび40μgの剪断した魚精子DNAを共沈殿し、乾かし、30μlの滅菌再蒸留水に溶解する。
【0167】
形質転換バッファー
15mM MgCl2、0.1%(w/v)モルホリノエタンスルホン酸(MES)および0.5Mマンニトール。蒸留水に溶解した後、KOHでpH5.8に調整し、オートクレーブする。4℃で保存する。
【0168】
PEG溶液
0.4Mマンニトールおよび0.1M Ca(NO3)2中の40%(w/v)PEG4000。PEGを0.4Mマンニトールおよび0.1M Ca(NO3)2に溶解する(これら2種の構成要素の終濃度は、PEGの容量のため低くなる)。pH8〜9に調整し、オートクレーブする(pHはこの溶液中で安定化するのに数時間、例えば一晩かかり、オートクレーブ後にpH6〜7に下がる)。
【0169】
材料
・滅菌1mlピペット
・滅菌5mlピペット
・滅菌10mlピペット
・ピペットボーイ
・滅菌14mlプラスチック製丸底遠心チューブ
・Clements Orbital 500卓上遠心分離機
・ウォーターバス
・50×10mmペトリ皿 。
【0170】
培地
・単離プロトプラスト懸濁液、ペレット化の入った14ml丸底遠心チューブ
・0.6%Sea Plaque(商標)アガロースを含む1:1混合のK3:H固形培地 。
【0171】
溶液
・オートクレーブしたW5溶液
・オートクレーブしたK3溶液
・オートクレーブしたH溶液
・40%PEG溶液
・形質転換バッファー
・30μlの滅菌再蒸留水に溶解した10μgの形質転換DNA 。
【0172】
手順
1.プロトプラストをペレット化し[70g(Clements Orbital 500卓上遠心分離機、400rpm)で5分間スピンする]、その後W5溶液を全て除去し、純粋なプロトプラスト懸濁液を残す。
2.1mlピペットを用いて、300μl(約7滴)の形質転換バッファーを単離プロトプラストの入った各14ml丸底遠心チューブに加える(滴下する)。
3.チューブ底を穏やかにタップすることによってペレットを慎重に再懸濁する。
4.各プロトプラスト懸濁液に10μg(30μl)の形質転換DNAを加え、その後300μl(1mlピペットを分注に用いる場合、約7滴)の予熱したPEG溶液を加える。チューブ底を穏やかにタップすることによってプロトプラスト懸濁液を混合する。
形質転換バッファーへのプロトプラスト再懸濁と形質転換DNAおよびPEGの添加との間の時間間隔は、最短を維持しなければならない。
5.形質転換混合物を15分間、室温で撹拌せずにインキュベートする。
6.10mlピペットを用いて、次の間隔で10mlのW5溶液を各チューブに徐々に加える。
【0173】
・1ml(約12滴)を各チューブに滴下する。全チューブを穏やかに反転して混合する。
【0174】
・1ml(約12滴)を各チューブに滴下する。全チューブを穏やかに反転して混合する。
【0175】
・1ml(約12滴)を各チューブに滴下する。全チューブを穏やかに反転して混合する。
【0176】
・2mlを穏やかな流れとして各チューブに加える。全チューブを穏やかに反転して混合する。
【0177】
・2mlを穏やかな流れとして各チューブに加える。全チューブを穏やかに反転して混合する。
【0178】
・3mlを穏やかな流れとして各チューブに加える。全チューブを穏やかに反転して混合する。
【0179】
分注し易くするため、分注前に、各チューブを必要容量のW5溶液で充填するのに各間隔で必要とされる合計容量を10mlピペットで採取する。各間隔でこれを繰り返す。
7.プロトプラストをペレット化し[70g(Clements Orbital 500卓上遠心分離機、400rpm)で10分間スピンする]、その後全W5溶液を除去し、純粋なプロトプラスト懸濁液を残す。次に進む前に、全チューブ底を1回タップする。
【0180】
一過性形質転換用手順
8.合計容量が最大5mlとなるよう、等容量のK3培地およびH培地(各溶液2.5ml)にプロトプラストペレットを再懸濁する。
9.液体K3:H+プロトプラスト懸濁液の混合液を50×10mmペトリ皿の中央にゆっくりと移す。
10.全皿をParafilm(登録商標)で封じ、一過性発現解析に進む前にプロトプラストを24〜72時間、薄明かりの下で24℃にて培養する。
【0181】
安定的な形質転換用手順
11.次の「パートIII.葉肉由来プロトプラストの培養および植物体の再生」に続ける。
【0182】
III.葉肉由来プロトプラストの培養および植物体の再生
ステップ1&2のため、プロトプラストの入った各チューブは、一度に1チューブで取り扱わなければならない。
【0183】
材料
・滅菌1mlピペット
・滅菌5mlピペット
・滅菌10mlピペット
・ピペットボーイ
・Clements Orbital 500卓上遠心分離機
・ウォーターバス
・50×10mmペトリ皿
・オートクレーブしたステンレス鋼製スパチュラ
培地
・単離プロトプラスト懸濁液、ペレット化の入った14ml丸底遠心チューブ
・0.6%Sea Plaque(商標)アガロースを含む1:1混合のK3:H固形培地、40℃
・オートクレーブしたK3溶液
・20mlのA培地の入った250ml培養容器
・固形MS Morpho培地の入った12ウェルCostar(登録商標)プレート
・固形MS Morpho培地の入った250ml培養容器
・固形MS培地の入った250ml培養容器 。
【0184】
手順
1.プロトプラストペレットの近くに0.5mlのK3培地を加えて、プロトプラストを再懸濁する。
2.K3+プロトプラスト懸濁液の混合液を50×10mmペトリ皿の中央にゆっくりと移す。
3.次に進む前に、プロトプラストの入った全チューブに対しステップ1および2を繰り返す。
4.一度に1プレートで、予熱した0.6%Sea Plaque(商標)アガロースを含む1:1混合のK3:H培地を5ml加える。穏やかな旋回運動でプレートを1回だけ振盪して培地中のプロトプラスト懸濁液を均等に分布させる。全プレートに対しこれを繰り返す。
5.培地が固形化するまで(外気温に依存して10〜30分間)、プレートを放置し手をつけない。
6.全皿をParafilm(登録商標)で封じ、プロトプラストを24時間、完全な暗所で24℃にて培養し、続いて6日間連続的な薄明かり(5μmol・m−2s1、Osram L36 W/21 Lumilux白チューブ)の下で培養すると、最初の、そして複数の細胞分裂が行われる。
7.滅菌スパチュラを用いて、プロトプラストを含むsea plaqueアガロースプラグを四分円に分割し、適切な抗生物質を添加した20mlのA培地の入った250mlプラスチック製培養容器内に置く(250ml容器につき1個の四分円)。ロータリーシェーカー上で80rpm、1.25cm throw、24℃、連続的な薄明かりでインキュベートする。
8.2週間毎に液体A培地+適切な抗生物質を交換し、プロトプラスト由来コロニーの生長をモニターする。
9.プロトプラスト由来コロニーが直径約2〜3mmになったとき(液体A培地中で5〜6週間インキュベーション)、コロニーをMS Morpho固形培地の入った24ウェルCostar(登録商標)プレートの個々のウェルに移す。
10.プレート(複数可)を1〜2週間、24℃で連続的な薄明かり(5μmol・m−2s−1、Osram L36 W/21 Lumilux白チューブ)の下でインキュベートすると、カルスは増殖して直径8〜10mmのサイズに達する。
11.プロトプラスト由来カルスが直径約1〜2cmになったとき、カルスを固形MS Morpho培地の入った個々の250ml培養容器に移す。容器を24℃で16時間・明/8時間・暗条件下(20μmol・m−2s−1、Osram L36 W/21 Lumilux白チューブ)でインキュベートする。1〜2週間以内に、複数のシュートを目視できるようになる。
12.長さ3〜4cmのシュートを固形MS培地の入った250ml培養容器に移し、根形成を助長する。容器を24℃、16時間・明/8時間・暗条件下(20μmol・m−2s−1、Osram L36 W/21 Lumilux白チューブ)でインキュベートする。3週間以内に、根形成の徴候を目視できるようになる。
13.確立された根システムを備える小植物を、タバコ葉肉プロトプラストの供給源としてのin vitro植物培養物として維持する。
【0185】
(実施例7)
エバンスブルー染色を用いたタバコプロトプラスト生存率の評価
図46および図47を参照。
【0186】
背景情報
・エバンスブルー染色(EVB;6,6’−[(3,3’−ジメチル1[1,1’−ビフェニル]−4,4’−ジイル)ビス(アゾ)ビス[4−アミノ−5−ヒドロキシ−1,3−ナフタレンジスルホン酸]四ナトリウム塩)
・非蛍光色素
・作用方法:生細胞は、エバンスブルーを排除する能力を細胞膜に保持し、その本来の色を保つ。塩または浸透圧ストレスにより損傷を受けた細胞は、エバンスブルーを排除することができず紺青に染色され、検鏡により容易に識別できる。
【0187】
・調製方法:400mg/l原液(溶媒:0.65Mマンニトール)
・染色方法:エバンスブルー原液を等容量のプロトプラスト懸濁液に加えて穏やかに混合し、室温で10分間インキュベートし、その後顕微鏡で可視化する。
【0188】
・検出方法:Leica MZFL III光学解剖顕微鏡
(実施例8)
Nicotiana tabacumプロトプラストにおける遺伝子発現解析
実験目標:トランスフェクトしたタバコプロトプラストにおけるクローニングした遺伝子AtRbcS::1−SST−SacBおよびAtRbcS::SPOR−SacBの発現をRT−PCRを用いて試験した。
【0189】
材料と方法:3プレートのプロトプラストの命名
i)トランスフェクトしたプロトプラスト(試料9A):AtRbcS::1−SST−SacB
ii)トランスフェクトしたプロトプラスト(試料12A):AtRbcS::SPOR−SacB
iii)非トランスフェクトプロトプラスト 。
【0190】
発現解析に関与するステップ:
1.プライマー設計およびPCRの最適化
2.全RNAの単離
3.RT反応
4.qRT−PCRアッセイ
プライマー設計およびPCR産物の同一性:ビーコン設計ソフトウェア(Premier Biosoft International)および遺伝子バンクにおいて利用できる遺伝子配列を用いてプライマー対を設計して対象遺伝子を増幅した。その結果生じるPCR産物のサイズが200〜250bpの範囲になるよう遺伝子特異的プライマーを選択した。融解曲線解析およびゲル電気泳動に基づくサイズによりPCR産物を同定した。
全RNA単離:1処理当たり1×106個のプロトプラストからPROMEGA製のSV全RNA単離システムにより全RNAを単離する。http://www.promega.com/tbs/tm048/tm048.pdf
【0191】
【表1】
逆転写酵素反応
QIAGEN RTキットを用いてRT反応を行った。プライマーミックス(QIAGEN)を用いることにより4通りのRT反応を行った(9、12、対照およびWTタバコRNA)。
http://www1.qiagen.com/products/pcr/QuantiTectPcrSystems/QuantiTectRevTranscriptionKit.aspx#Tabs=t2
上述の試料それぞれの複製物を、遺伝子特異的プライマーを用いたRT反応に付した。具体的には、試料9Aおよび12AはsacBリバースプライマーを用いて、対照(非トランスフェクト)試料は18Sリバースプライマーで転写した。
【0192】
PCR
反応を次の通りセットアップした。
2×GoTaq(登録商標)Green MasterMix(Promega) 10μL
cDNA鋳型 2.0μL
フォワードプライマー(10μM) 0.5μL
リバースプライマー(10μM) 0.5μL
ヌクレアーゼを含まない水 7.0μL 。
【0193】
反応サイクルは次の通りである。
ステップ1:95℃ 2分間
ステップ2:95℃ 30秒間
ステップ3:55℃ 30秒間
ステップ4:72℃ 60秒間
ステップ2から25サイクル(18S)、あるいはステップ2から35サイクル(sacB)
ステップ5:4℃ ホールド 。
【0194】
1×TBEバッファー中で1%(w/v)アガロースにより電気泳動し、SYBR(50μL/L)で染色した後、UV光の下で反応産物を可視化する。
【0195】
トランスフェクトしたプロトプラストcDNA試料(9および12)の両方でsacB転写産物の検出が示され、一方、非トランスフェクトプロトプラストcDNA試料から18Sを増幅することによって、用いた方法を検証した(図50)。
【0196】
光調節プロモーターの制御下におけるキメラsacB遺伝子の発現をトランスフェクトしたプロトプラストにおいて観察した。産物は、RTなし対照および鋳型なし対照では観察されなかった。sacB遺伝子発現は、試料9Aおよび12Aに用いたベクターでトランスフェクトしたプロトプラストにおいて検出することができた。
【0197】
(実施例9)
Agrobacterium tumefaciensを介したタバコリーフディスクの形質転換
本実施例は、上文に記載の発現カセットを用いて遺伝子操作したバイナリーベクターを運ぶアグロバクテリウムを用いたタバコリーフディスクの安定的な形質転換を説明する(図48および図49を参照)。
【0198】
序文
Agrobacterium tumefaciensを介したリーフディスク形質転換の利用は、トランスジェニック植物を作出する効率的な方法である。A.tumefaciensは、植物に感染して細菌DNAを植物ゲノムに組み入れるための遺伝的機構を含む天然の双子葉植物病原体である。この能力の結果、A.tumefaciensを、例えば特定の対象DNAをタバコに組み入れるためのクローニング媒体として採用することができる。
【0199】
この方法は、タバコにおけるモデルシステムの作製に用いて、対象遺伝子のcDNA異種クローンの機能を評価することができる。
【0200】
材料および化学薬品
装置および器具
水平流の層流フード(シリーズHWS180、CLYDE−APAC、Evans Deakin Pty.Ltd.の一部門、ウッドビルノース、南オーストラリア州5012、オーストラリア)、ロータリーシェーカー(Infors型RC−406、Infors AG、CH−4103ボットミンゲン、スイス)、スイングアウトローターを備える卓上遠心分離機(Clements Orbital 500)、ピンセット(先曲がり(bend)、cat no.2108/160、Crown Scientific、ロウビル、ビクトリア州3178、オーストラリア)、滅菌外科手術用メス刃(サイズ11、cat no.1838、Laboratory Supply Pty.Ltd.、ミルペラ(Milperra)DC、ニューサウスウェールズ州1891、オーストラリア)を備えるメス柄(No3、cat no.SHN3、Crown Scientific、ロウビル、ビクトリア州3178、オーストラリア)を用いた。
【0201】
原液
全培地に必要なマクロ成分、ミクロ成分およびビタミンは、それらを添加する際に有用となるよう、濃縮ストック(マクロ成分ストック:10倍濃縮;ミクロ成分およびビタミンストック:100倍濃縮)として調製する必要がある。ミクロ成分以外の全ストックは、室温で調製する。ミクロ成分ストックの調製は、混合前に構成要素の加熱を必要とする。Na2−EDTAおよびFeSO4×7H2Oは、混合前にそれぞれ400mLの蒸留水(全容量1000mLに対し)に溶解する必要がある。溶解した溶液を混合し、溶液が黄色に変色するまでca.60℃で加熱する。残りの構成要素を添加する前に溶液を冷ます。溶液を蒸留水で1000mLとする。全ストックを4℃で保存する。ホルモン[2,4−D(2,4−ジクロロフェノキシ酢酸)およびカイネチン]を1M KOHに溶解し、蒸留水で希釈して100mg/リットル濃縮ストックを調製する。
【0202】
培地
それらの個々の原料を終濃度で用いた培地の組成を別表1に示す:MSミクロ、MSマクロ、B5ビタミン、Lauria Bertani培地、洗浄培地、PC培地、SEL培地、RM培地およびSEM培地。
【0203】
化学薬品
2,4−D:2,4−ジクロロフェノキシ酢酸、活性炭、チメンチン(同じ濃度のセフォタキシムに代替可能)、BAP(6−ベンジルアミノプリン)、ゼアチン、AgNO3、リファンピシン、寒天(Difco、Bacto−Agar、cat.no:0140−01)はゲル化剤とし用いる。他の化学薬品(PEG4000、Tween80、KOH、NH4OH、NaCl、KCl、Ca(NO3)2、MgCl2およびCaCl2)は、BDHから購入し、MES(2−[N−モルホリノ]エタンスルホン酸(ethanesulfuric acid))はSigma(Cat.No.N−8250)から、カナマイシン硫酸塩はSigmaから、ハイグロマイシンBはCalbiochemから、Ca(OCl)2(ほぼ65%)およびホスフィノトリシン(phosphinotricin)は、Roth and Riedel de Haenからそれぞれ購入される。スクロースはFluka(cat.no:84100)から購入した。Parafilm(登録商標)「M」(American National Can(商標)、グリニッジ、コネチカット州06836、米国)は、封密用テープとして用いた。滅菌使い捨て瓶上型フィルター(0.2μm vacucap90;cat.no.4622、Gelman Sciences(登録商標)Pty.Ltd.、チェルトナム、ビクトリア州3192、オーストラリア)および使い捨てフィルターユニット(0.2μm;cat.no.16534、Sartorius AG、37070ゲッティンゲン、ドイツ)をフィルター滅菌に用いた。滅菌使い捨てピペット:1mL_(TRP(登録商標);cat.no.94001)、5mL_(TRP(登録商標);cat.no.94005)および10ml_(TRP(登録商標);cat.no.94010)は全てLife Technologies Pty. Ltd.、モルグレイブ、3170オーストラリア製であり、ねじ蓋付き滅菌プラスチック製遠心チューブ(14mL、TRP(登録商標);cat.no.91016、Life Technologies Pty.Ltd.、モルグレイブ、3170オーストラリア)、滅菌プラスチック製ペトリ皿(90×14mm;cat.no.82.9923.484および60×14mm、cat.no.83.1801.011、Sarstedt(登録商標)Australia Pty.Ltd.、テクノロジーパーク、南オーストラリア州5095、オーストラリアならびに90×20mm;cat.no.664160、Greiner Labortechnik GmbH、72636フリッケンハウゼン、ドイツ)およびオートクレーブ可能培養容器(250mL、cat.no.75.9922.519、Sarstedt Australia Pty.Ltd、テクノロジーパーク、南オーストラリア州5095、オーストラリア)を用いた。
【0204】
植物材料
A)N.tabacum cv.Petit Havana SR1の滅菌シュート培養物を利用することができる。これは、次亜塩素酸塩溶液[1.4%(w/v)Ca(OCl)2、0.05%(v/v)Tween80]中で15分間表面滅菌し、滅菌蒸留水中で3〜4回リンスした後、0.8%(w/v)寒天で固形化した半分に薄めたMS培地(別表1)上に発芽のため播種した、対応する種子から確立する。2〜3枚の葉をつけたシュートを切り出し、250mL培養容器内の0.8%(w/v)寒天で固形化したMS培地上で25℃、16h/d明期(20μmol・m−2s−1、Osram L36 W/21 Lumilux白チューブ)で培養する。根の生えたシュートを、6週間の間隔で使用前に茎切断として数回継代培養する。
B)ガラス温室で育成したN.tobacumを利用してもよいが、後述[I.B)]の通り葉の表面滅菌が必要である。植え付けが深過ぎないこと、また湿り気が十分であることを確実にしつつ、種子を滅菌土壌に植える。16h/d明期(20umolm“2s”1、Osram L36 W/21 Lumilux白チューブ)条件下、25℃で培養し、Osmocoteスローリリース肥料を用いて施肥する。
C)リーフディスク形質転換に利用したAgrobacterium tumefaciens株はAGL1である。
【0205】
I.タバコリーフディスクをベクターpBinhph200で形質転換するためのAgrobacterium tumefaciensの調製
形質転換したAgrobacterium tumefaciens株AGL1の前培養をタバコ形質転換日の2日前に開始し、滅菌条件が維持されていることを確実にする(pBinhph200のIDカードについては別表2を参照)。
1.−70℃で凍結したグリセロール細菌ストックの表面を接種(innoculation)用ループで掻き取り、滅菌チューブ内の2mLのLB(20mg/mLリファンピシン+10mg/Lスペクチノマイシンを含む)に接種する。24時間、28℃、150rpmでインキュベートする。
2.4mLのLB+抗生物質(12mL滅菌チューブ内)に0.25mLの前記24時間前培養物を接種する。6〜7時間、28℃でインキュベートする。
3.25mLの抗生物質なしLB(150mL滅菌フラスコ内)に0.025mlの前記6〜7時間前培養物を接種し、一晩28℃、150rpmでインキュベートする。
4.25mlの前記一晩前培養物に25mlのLBを加え、28℃でさらに90分間、150rpmで培養を継続する。
5.50mlの前培養物を遠心チューブに移し、Clements卓上遠心分離機において12分間、2,000rpm、室温でスピンする。
6.上清を除去し、ペレットを20mLのWMに穏やかに再懸濁する。OD600を測定する。細菌懸濁液にさらにWMを加え、最終OD600を0.45とする。この調製物をステップIII)の1に用いる。
【0206】
II.リーフディスクの調製
A)タバコシュート培養物を用いたリーフディスクの調製
1.層流において、MS培地上で培養したタバコ植物体から4〜6枚の葉を収集する。葉をWMの入った1.5×9cmペトリ皿に置く。メスを用いて主脈を除去し、葉組織をほぼ1cm2四方に切る。この時点で、組織またはリーフディスクは、Agrobacterium tumefaciens(AGL1)による形質転換の準備が整った。ディスクは1時間以内に形質転換しなければならない。
B)非滅菌タバコ組織を用いたリーフディスクの調製
1.ガラス温室で育成したタバコ植物体から4枚の若い葉(ほぼ8cmの長さ)を収集する。
2.葉を70%エタノールの入った滅菌ビーカー内に置き、アルミホイルで覆い、円軌道シェーカー(Bio−Line円軌道シェーカー、Edwards Instrument Company)上で1分間、穏やかに旋回させる。
3.70%エタノールを除去し、1%Ca(OCl)2に交換する。組織を8分間旋回させる。葉を滅菌水中で少なくとも3回洗浄する。
4.層流において主脈を除去し、残りの葉組織をほぼ1cm2四方に切る。調製したディスクをWMの入った1.5×9cmペトリ皿内に置く。この時点でディスクは、Agrobacterium tumefaciens(AGL1)による形質転換の準備が整った。
【0207】
III.リーフディスクとAgrobacterium tumefaciensのインキュベーションおよび共存培養
1.WM(リーフディスク調製における最後のステップのもの)をアグロバクテリウム培養物に交換し、1〜2分間インキュベートする。
2.細菌懸濁液を除去し、外植片をWMで短時間リンスする。PC培地上に播く前に、滅菌ナプキンで外植片を拭き取る。3日間共存培養するため、育成室(16h/d明期(20μmol・m−2s−1、Osram L36 W/21 Lumilux白チューブ)条件、25℃)内に置く。
【0208】
IV.アグロバクテリウムを介した形質転換後のリーフディスクの前再生
1.外植片(5/プレート)をSEL培地に移し、育成室にさらに7日間戻しておく。
【0209】
V.再生
1.外植片(5/プレート)をRMに移し、育成室に戻す。シュート形成は3〜6週間以内に起こるであろう。4週間後に外植片を新しいRMに移す。
2.カルスが大きくなり過ぎたら、特に培地に接触していないシュートがある場合、メスを用いてカルスを分割する。できるだけ多くのシュートを選抜に曝露させる。
【0210】
VI.シュートの伸長および根の発達
1.選抜8週間後、あるいは選抜中の非形質転換対照外植片が死滅したとき、緑色のシュート(5/プレート)を9×2cmペトリ皿内のSEM培地(IBA(1mg/L)を含む)に移す。4〜5週間で根が出てくる筈である。
【0211】
VII.in vitro小植物発達
1.根が出たら、根の生えた小植物を組織培養容器内のSEM培地に移す。
【0212】
参考文献
Stewart、C.N.Jr.、Adang、M.J.、All、J.N.、Rayner、P.L.、Ramachandran、S.およびParrott、W.A.(1996年)Insect control and dosage effects in transgenic canola containing a synthetic Bacillus thuringiensis crylAc gene. Plant Physiol、112巻:115〜120頁 。
【0213】
別表
【0214】
【表2】
【0215】
【表3】
【0216】
【表4】
【0217】
【表5】
(実施例10)
同じ発現カセットで改変したバイナリーベクターを有するAgrobacterium tumefaciensを用いたフローラルディップ法によるシロイヌナズナ(ARABIDPOSIS)の安定的な形質転換
電気的形質転換受容性のAgrobacterium tumefaciens細胞の調製
実験手順
1.−80℃凍結グリセロールストックから、Agrobacterium tumefaciens(AGL1株)を20mg/Lリファンピシンおよび100mg/Lアンピシリンを含むMGL寒天上に画線培養し、27℃で2日間インキュベートする。
2.5mlのMGLを50ml Falconチューブ内で測定し、リファンピシン20mg/Lおよびアンピシリン100mg/Lの終濃度となるよう加える。Agrobacterium tumefaciens AGL1のシングルコロニーを接種する。
3.150rpmの円軌道シェーカー上の傾いた試験管立てにおいて(ca.30度)27℃で24時間インキュベートする。
4.夕方、20mg/Lリファンピシンおよび100mg/Lアンピシリン(amplicillin)(500mlフラスコ内)を含む100mlのMGLに、500μlの一晩培養物を接種する。
5.27℃で一晩、150rpmの円軌道シェーカー上で0.4〜0.6(最大0.6)の間のOD600が得られるまでインキュベートする。過増殖した場合は下のコメントを参照。
6.オートクレーブしたJA10遠心チューブに細胞を移し、氷上で10分間冷やす。
7.JA10ローターを用いて10分間、9000rpm、4℃で遠心分離する。
8.培養に用いた500mlフラスコに注ぐことによって、上清を慎重に廃棄する(ペレットは非常に安定的というわけではない)。
9.20mlの氷冷10%グリセロールをJA10遠心チューブ内のペレットに加え、ボルテックス撹拌することによりペレットを再懸濁する。
10.懸濁液をJA20遠心チューブに注ぎ、JA20ローターを用いて10分間、10000rpm、4℃でスピンする。
11.500mlフラスコに注ぐことにより上清を廃棄する。
12.15mlの氷冷10%グリセロールをペレットに加え、ピペッティングにより再懸濁する。JA20ローターにおいて10分間、10000rpm、4℃で遠心分離する。
13.ステップ11および12を2回繰り返す。
14.ペレットを1mlの氷冷10%グリセロールに再懸濁し(ピペットまたはボルテックスによる)、滅菌マイクロチューブに移す。
15.微量遠心管において3分間、13000rpm、4℃でスピンする
16.最後に、ペレットを1mlの10%氷冷グリセロールに再懸濁する。
17.標識された1.7mlマイクロチューブに50μlバッチのアリコートを作成し、液体窒素中でスナップ凍結する。
18.液体窒素からチューブを取り出し、コンピテント細胞を−80℃で保存する。
【0218】
コメント
A.tumefaciens細菌培養物を取り扱う間、ゴム手袋を着用する。全細菌廃棄物を500mlフラスコ内に集め、オートクレーブする。A.tumefaciensの100ml培養物が過増殖したら、20mg/Lリファンピシンを含む新しいMGL培地で1/3〜1/4希釈し、さらに1〜2時間インキュベートする。
【0219】
Agrobacterium tumefaciensのエレクトロポレーションによる形質転換
実験手順
1.ジーンパルサーキュベットホルダーを氷上で予冷する。
2.コンピテントアグロバクテリウム(AGL1)細胞のアリコートを−80℃から取り出し、氷上で解凍する。
3.ジーンパルサー背面のメインスイッチを入れる。電圧を2.5kVに調整し(ディスプレイ上に「2.5」と記録されるまで「raise」ボタンを使う)、静電容量25μFDおよび抵抗600Ωにする。
4.50μl以上の容量の解凍した細胞に0.1μgのDNAを加える。
5.ピペッティングにより混合し、細胞/DNA混合液を予冷した0.2cm gapジーンパルサーキュベットに移す。
6.両方の電極に細胞が触れるよう、細胞をキュベット底面に慎重にタップまたは振盪する。
7.キュベットの外側をティッシュペーパーで乾かし、キュベットをキュベットホルダー内に置く。キュベット上の切れ込みは正確な方向付けを確実にする。キュベットがチャンバー底の接触点の間に収容されるまでキュベットホルダーをチャンバー内にスライドさせる。
8.ビープ音がするまで両方の赤いボタンを押すことによって細胞にパルスを与える。この機械は、充電の間はCHGを表示し、放電の際にビープ音を出す。細胞を戻して1分間氷上に置いて回復を助ける。
9.ガラス製注入器を用いて1mlのLB培地をキュベット内の細胞に加える。
10.懸濁液を吸って吐いて混合し、滅菌15mlチューブに移す。
11.傾いた試験管立て(ca.30度)を用いて150rpmの円軌道シェーカー上で1〜2時間、27℃でインキュベートする。
12.9mlのLBを細胞懸濁液に加えて徹底的に混合し、20mg/Lリファンピシンおよび適切な抗生物質(例えば、pPZPシリーズのベクターに対しては100mg/Lスペクチノマイシン)を含むLBプレート上にこの培養物100μlを播く。この懸濁液100μlを900μlのLBブロスの入った1.7mlマイクロチューブに移し、徹底的に混合し、適切な抗生物質を含む別のLBプレート上にその100μlを播く。上述の懸濁液から100μlを取り分け、新しい1.7mlマイクロチューブ内に置き、900μlのLBブロスを加える。徹底的に混合し、その100μlを適切な抗生物質を含む別のLBプレート上に播く。
13.プレートをパラフィルムで封じ、大きなシングルコロニーが目視できるようになるまで27℃で2〜3日間インキュベートする。
【0220】
形質転換アグロバクテリウムの保存
実験手順
1.50ml滅菌チューブ内で20mg/Lリファンピシンおよび適切な選抜抗生物質(すなわち、pZPシリーズに対しては100mg/Lスペクチノマイシン、pBinシリーズに対しては50mg/Lカナマイシン)を含む5mlのLBブロスにシングルコロニーを接種する。翌日に増殖の程度を密接にモニターできるよう、この操作は早朝に行うとよい。
2.チューブを暗所において27℃で24〜36時間、250rpmで振盪しつつインキュベートする。インキュベーションの最初の24時間の後、培養物の増殖を定期的に観察する。細胞が活発に増殖したら(非常に目に見える)インキュベーションをやめる。濁りの最初の徴候が見えた直後に、急速な増殖が行われるであろう。増殖時間は、個々の株および形質転換体に依存する。
3.各培養物をチェックして、AGL1が望ましいバイナリーベクターを含むことを立証するべきである。この操作は、セクション5.2に記されているプロトコールを用いて行う。
4.キュベット内に培養物の500μlアリコートを作成する。適切な抗生物質を含む500μlのLBブロスでブランク作成して、各読み込み値の間からOD600読み込み値を測定する。
5.OD600読み込み値が0.8から1.0の間の範囲となるまで培養物を増殖させる。
6.滅菌15mlチューブ内に、5.0mlの培養物および5.0mlの保存ストックを加える。ステップ7に進む前に徹底的に混合する。
7.しっかりと標識した滅菌クリオチューブ内に500μlのアリコートを作成する。液体窒素中でスナップ凍結する前に全チューブを反転する。必要とされるまで−80℃で保存する。PCR陰性であることが示された場合、全ストックを廃棄する。
【0221】
アグロバクテリウムのPCR解析
1.1μlのアグロバクテリウム培養物を滅菌PCRチューブ内の9μlの滅菌MQ H2Oに加える。
2.細胞を98℃で5分間インキュベートする。チューブを氷に移す。
3.10μlの調製済2×PCRマスターミックスを加える。10μlの2×PCRマスターミックスは、次のものを含む。
10×Dynazymeバッファー 2μl
10mM dNTP 1.0μl
10μMフォワードプライマー(10μM) 1.0μl
10μMリバースプライマー(10μM) 1.0μl
Dynazyme IIポリメラーゼ(polymerise) 1μl
MQ水 3μl
4.陽性対照(50ngのオリジナルプラスミドDNA)および陰性対照(鋳型DNAなし)を含む。標準PCR条件用いて合計35サイクル行う。
【0222】
1.95℃、3分間
2.94℃、30秒間
3.55℃、30秒間
4.72℃、1分間
5.72℃、10分間
ステップ2〜4を合計35回繰り返す。
5.PCR産物を1%アガロースゲルで解析する。
【0223】
コメント
アグロバクテリウムを取り扱う間、手袋を着用する。あらゆる細菌およびDNA廃棄物を集めてオートクレーブする。ジーンパルサーキュベットは再使用可能である。蓋は70%EtOHに浸し、キュベットは水を張って閉鎖した入れ物の中でオートクレーブしてアグロバクテリウムを除去する。その後、キュベットは70%EtOH中に保存し、乾かした後に再使用することができる。
【0224】
Arabidopsis thalianaのin planta形質転換
実験手順
植物材料の調製
1.実生用方形ケース(punnet)に種子栽培ミックスを充填し、盛り土を形成する。2層の防鳥ネットで覆い、ゴムバンドで各端を固定する。トレイに張った水の中に方形ケースを入れることによって、土壌を水で満たす。方形ケース当たりほぼ40体の植物を得るのに十分な種子を播く。
2.方形ケースを4℃に2〜3日間置くことにより、種子を春化処理する。方形ケースを22℃、蛍光灯(連続照明、55μmol・m−2s−1)下の育成チャンバーに移し、1週間に1回、Miracle−GroまたはAquasolを与える。
3.一次花序(bolt)が現れたら摘心し、二次花序を概ね2〜10cmの丈になるまで生長させる(概ね4〜6日間かかり、植物体は多数の未開花の花蕾をつけ、長角果(silique)は殆どない筈である)。ピンセットを用いて長角果または開花した花を全て慎重に取り除く。気孔が開くよう浸潤前に植物に十分に水をやる。浸潤前に土壌を水で飽和させて細菌溶液が土壌に吸収されるのを最小限に抑える。
4.LWSに詳細を入力し、バーコードを作成する。LWSバーコード詳細によって方形ケースを標識する。
【0225】
Agrobacterium tumefaciensの調製
1.朝に、適切な選抜抗生物質(すなわち、pPZPベクターに対し100mg/mlのスペクチノマイシン)を含む200mlのLB培地に、アグロバクテリウム保存ストック(セクション5.1)の単一のスターター培養物500μlを接種する。250rpmの円軌道シェーカー上で24時間、27℃でインキュベートする。およそ2個の方形ケースの植物を浸潤させるには200mlの培養物で十分である。
2.一晩培養した培養物を500ml遠心分離ボトルにおいて5500g、室温、15分間遠心分離して細胞をペレット化させる。可能な限り液体を除去しつつ上清を廃棄する。OD600読み込み値が約0.7〜0.9となるようペレットを浸潤培地(別表1を参照)に再懸濁する。
【0226】
アグロ浸潤
1.アグロバクテリウム溶液の半量を250ml容器内に置く。
2.方形ケースを逆さにし、ロゼット葉を含む植物体全体を細菌溶液に浸漬し、穏やかに振盪して気泡を追い出す。植物を2分間共培養する。
3.方形ケースを取り出して短時間水を抜くが、植物の周りの薄い水膜はそのままにしておく。植物をプラスチックフィルムで覆って湿度を維持し、育成室に戻して直射光を避ける。廃液をオートクレーブし、正しい処分のための化学廃棄物ドラム缶に捨てる。
4.A.thalianaの全方形ケースに対してステップ1〜3を繰り返して形質転換する。
5.詳細をLWSに入力し、バーコードを作成する。個々の方形ケースをLWSバーコード詳細で標識する。
6.翌日、鉢の覆いを取って直射光に戻し置く。植物が十分に発達した長角果をつけるまで植物に水をやる。
【0227】
種子採取
1.植物が完全に乾ききったら、長角果をつけた茎を取ってこれを紙袋の中に入れ、1週間37℃で乾かしておく。袋をLWSバーコードで標識する。乾いた長角果を紙袋の中で押しつぶす。この操作は長角果を粉々にして種子を遊離させる。
2.200ミクロンの篩を新しいA4用紙1枚の上に置き、そこに種子および押しつぶされた長角果を移し入れる。篩を穏やかにタップし、その下にある用紙の上に種子を落とす。篩の中に残った植物材料を廃棄する。植物材料の大部分が除去されるまでこのプロセスを繰り返す(注記:植物材料は次のステップにおける汚染源となり得る)。種子を1.7mlマイクロチューブ内に入れ、LWSバーコード詳細で標識する。チューブを小型のマニラ封筒の中に置き、LWSバーコードで標識する。このバーコードは、元々の形質転換事象と関連することに留意する。
3.種子を4℃保存に移す前に、種子を−20℃で24時間保存する。
【0228】
陽性T1トランスジェニックArabidopsis thaliana植物の選抜
種子の表面滅菌
1.層流フードで作業しつつ、滅菌する種子(150×15mmプレートにつき、40mg=ほぼ2000個の種子)を2.0mlマイクロチューブ内に置く。
2.1000μlの70%エタノールを加え、2分間放置する。
3.エタノールを除去し、1000μlの種子滅菌用溶液(4%塩素:水:5%SDSがそれぞれ8:15:1の比率)を加えてボルテックス撹拌によって徹底的に混合する。
4.Ratek「観覧車(ferris wheel)」上にチューブを置き、種子と溶液を確実に混合し、10分間放置する。
5.層流において滅菌用溶液を除去して滅菌水に交換する。チューブ(複数可)をボルテックス撹拌し、ベンチトップ遠心分離機で30秒間スピンして種子を沈降させる。水を除去し、さらに1mlの滅菌水に交換する。種子洗浄ステップは、目視できる泡が出なくなるまで繰り返すべきである(少なくとも4回)。最後の洗浄の後、約200μlの水を残して種子を浸す。
【0229】
T1形質転換体の選抜
1.適切な選抜抗生物質(例えば、PZP200シリーズに対し8mg/Lのハイグロマイシン)を含む選抜発芽培地(SGM)を用いて150×15mmプレートを調製する。チメンチン(250mg/L)を入れて、アグロバクテリウムの増殖を阻害する。各プレート当たり約125mlのSGMが必要とされる。
2.層流フード内で作業しつつ、SGM寒天プレートの表面にわたって平行に滅菌メスをすべらせる(図4を参照)。この操作は種子を広げるのに役立つ。
3.先端を切り取った滅菌1mlチップを用いて、滅菌種子をピペットで取ってプレート上に置く。滅菌使い捨て延展器(spreader)を用いて種子を分布させる。
4.種子を4℃で2日間低温処理し、次に連続的な蛍光灯(55μmol・m−2s−1)下、22℃で育成する。
5.推定形質転換体は、6〜8葉ステージになったら土壌に移すことができる。ピンセット1本を用いて、根を確実にインタクトに保ちつつ植物を組織培養培地から慎重に取り出す。ARASYSTEMを用いて湿ったin−vitro混合土壌に移植する(別表2の図5を参照)。プラスチックチューブで覆う。新しいLWSバーコードを作成し、チューブを標識する。数日間チューブの上端をプラスチックラップで覆い、回復を助ける。
【0230】
導入遺伝子組み込みの立証:ゲノムDNAソースとしてのアルカリ処理した葉組織
推定形質転換体を土壌に移して3日後、次のプロトコールを用いて個々の植物体を導入遺伝子の存在に関して分子的に特徴付けることができる。
1.試験するアルカリ処理した葉組織それぞれに対し、1×PCRバッファーミックスを調製する(詳細は後述を参照)。
2.1.7mlマイクロチューブ内で、200μlの0.25M NaOHを小型の若い葉(T1植物から採取)に加える。
3.チューブを沸騰水に2分間浸漬する。注記:煮沸の間に蓋がはじけ開くのを防ぐため、蓋をマイクロチューブ蓋ロックで固定する、あるいは蓋を細針で刺し通すこと。
4.煮沸後、チューブを水から取り出して、200μlの0.25M HClおよび100μlの0.25%(v/v)igepal[0.5M Tris HCl、pH8.0]を加える。チューブを沸騰水にさらに4分間浸漬する。
5.アルカリ処理した葉のごく一部(ほぼ2mm2)を取り出し、予め調製したPCRミックス中に置く。
【0231】
10×PCR反応バッファー(Mg(SO4)・7H2O含有) 5.0ml
10mM dNTP 1.0μl
10μMフォワードプライマー 1.0μl
10μMリバースプライマー 1.0μl
PWO DNAポリメラーゼ 1.0μl
MQ H2O 41.5μl
6.標準PCR条件を用いて35サイクルを行う。
【0232】
1.95℃、3分間
2.94℃、30秒間
3.55℃、30秒間
4.72℃、1分間
5.72℃、10分間
ステップ2〜4を合計35回繰り返す。
7.PCR産物を1%アガロースゲルで解析する。
【0233】
注記:初回にアルカリ処理した葉組織を用いてインサートが増幅されない場合、組織をさらに2分間再度煮沸して導入遺伝子のPCR増幅を繰り返す。この第二のPCRが失敗したら、Qiagen植物ゲノムDNA抽出キットを用いて葉組織から少量のゲノムDNAを抽出する。LWSをアップデートする。
【0234】
種子採取
1.植物が完全に乾ききったら、長角果をつけた茎を取ってこれを紙袋の中に入れ、1週間室温で乾かしておく。袋をLWSバーコードで標識する。乾いた長角果を紙袋の中で押しつぶす。この操作は長角果を粉々にして種子を遊離させる。
2.200ミクロンの篩を新しいA4用紙1枚の上に置き、そこに種子および押しつぶされた長角果を移し入れる。篩を穏やかにタップし、その下にある用紙の上に種子を落とす。篩の中に残った植物材料を廃棄する。植物材料の大部分が取り除かれるまでこのプロセスを繰り返す(注記:植物材料は次のステップにおける汚染源となり得る)。種子を1.7mlマイクロチューブ内に入れ、LWSバーコード詳細で標識する。チューブを小型のマニラ封筒の中に置き、LWSバーコードで標識する。このバーコードは、元々の形質転換事象に関連することに留意する。
3.種子を4℃で保存する前に、T2種子を−20℃で24時間保存する(陽性トランスジェニックT2植物の選抜における真菌汚染の見込みを低減させるのに有用である)。
【0235】
コメント
ARASYSTEMトレイ、ホルダー等を含む、アグロバクテリウムと接触したあらゆる入れ物は、市販の漂白剤および70%エタノールを用いて徹底的に消毒しなければならない。
【0236】
この実験に依存し、T1植物を表現型特性評価、すなわちレポーター遺伝子解析に用いることができ、これらの系統はT1ステージを越えて継続する必要はないと思われる。
【0237】
ホモ接合型T3種子の作出
序文
詳細に後述するプロトコールは、シングルコピーの導入遺伝子を有するホモ接合型植物の選抜に用いた方法を説明する。浸潤方法を用いた導入遺伝子のArabidopsis thalianaへの組み込みは、子房の受精前に雌しべで起こる。したがって、浸潤によって産生されたあらゆる種子で導入遺伝子を有するものはT1であると考えられる。T1種子が発芽してT1植物を産生し、これは次にT2種子を産生する。目標は、単一のインサートを有し導入遺伝子を発現する独立したトランスジェニック植物を、構築物につき少なくとも5個体得ることである。T1種子用に作成した各LWSバーコードは、別個の形質転換事象を表す。各T1植物のT2種子を収集するとき、新たなLWSバーコード番号を付与する。このLWSバーコードは、元々の形質転換事象に関する。
【0238】
T3種子作出のためのT2形質転換体の選抜
1.層流フード内で作業しつつ、約100個のT2種子を表面滅菌する(セクション7.1を参照)。
2.250mg/Lチメンチン(timetin)および8mg/Lハイグロマイシン(pPZP200−35s−hph−35stに対する選抜剤)を含む選抜SGM培地上に、1プレート当たり約25個の種子を播く(合計4枚)。
3.種子を4℃で2日間低温処理し、次に連続照明(55μmol.m−2.s−1)、22℃の育成室に移す。
4.2週間後、ハイグロマイシンに対して抵抗性または感受性を有する植物の分離解析を行う。
5.推定形質転換体が6〜8葉ステージになったら、Arasystemを用いて少なくとも10個体の植物を土壌に移す。新たなLWSバーコードを作成し(元々の形質転換と関連する)、各植物体をバーコードで個々に標識する。チューブをプラスチックフィルムで数日間覆って回復を助ける。
6.個々の植物それぞれが組み込まれた導入遺伝子を有することを確認するため、アルカリ処理した葉組織の方法を用いる(セクション7.3)。LWSをアップデートする。
7.植物が完全に乾ききったら、長角果をつけた茎を取ってそれを紙袋に入れ、1週間37℃で乾かしておく。袋をLWSバーコードで標識する。乾いた長角果を紙袋の中で押しつぶす。この操作は長角果を粉々にして種子を遊離させる。
8.200ミクロンの篩を新しいA4用紙1枚の上に置き、そこに種子および押しつぶされた長角果を移し入れる。篩を穏やかにタップし、その下にある用紙の上に種子を落とす。篩の中に残った植物材料を廃棄する。植物材料の大部分が取り除かれるまでこのプロセスを繰り返す(注記:植物材料は次のステップにおける汚染源となり得る)。種子を1.7mlマイクロチューブ内に入れ、LWSバーコード詳細で標識する。チューブを小型のマニラ封筒の中に置き、LWSバーコードで標識する。このバーコードは、元々の形質転換事象に関連することに留意すること。
9.種子を4℃保存に移す前に、種子を−20℃で24時間保存する(陽性トランスジェニックT3植物の選抜における真菌汚染の見込みの減少に役立つ)。
【0239】
分離解析
1.ハイグロマイシンに対して抵抗性または感受性のいずれかを有する各系統から得られたT2植物の総数を採点する。
2.T−DNAが1遺伝子座に挿入されている場合、抵抗性植物の感受性植物に対する比率は3:1となる筈である。T−DNA遺伝子座が2遺伝子座に挿入されている場合、抵抗性植物の感受性植物に対する比率は15:1となる筈である。T−DNAが3以上の遺伝子座に挿入されている場合、抵抗性植物の感受性植物に対する比率は>15:1となる筈である。
3.カイ2乗(χ2)統計検定を用いて、分離データが特定の仮説にどの程度適合するか決定する。
4.導入遺伝子のシングルコピーを含むことをカイ2乗解析が示すトランスジェニック系統の育成を継続する。
5.LWSをアップデートする。
導入遺伝子組み込みの立証:ゲノムDNAソースとしてのアルカリ処理した葉組織
1.各T2植物につき小型の葉を1枚収集する。
2.アルカリ処理した葉のプロトコール(セクション7.3)に従い、導入遺伝子の存在を決定する。
3.LWSをアップデートする。
【0240】
ホモ接合型T3系統の選抜
1.単一の導入遺伝子挿入を含むことを示すT2トランスジェニック系統の育成を継続する。
2.T2種子を採取し(セクション8.2)、LWSをアップデートして新たなバーコードを作成する。
3.ほぼ40個のT3種子をSGM上で発芽させる。
4.2週間後、ハイグロマイシンに対し抵抗性または感受性のいずれかを有する各系統由来のT3植物の総数を採点する。
5.ホモ接合型T3系統は、感受性植物の不在によって示されるであろう。
6.推定形質転換体が6〜8葉ステージになったら、Arasystemを用いて少なくとも20個体の植物を土壌に移す。新たなLWSバーコードを作成し、各植物を個々にバーコードで標識する。チューブをプラスチックフィルムで数日間覆い回復を助ける。
7.系統の単一挿入がホモ接合型であることをさらに検証するため、アルカリ処理した葉組織の方法を用いて(セクション7.3)、全植物が導入遺伝子を含むことを確認する。LWSをアップデートする。
8.推定ホモ接合型系統から十分な材料を収集してサザンハイブリダイゼーションを行い、導入遺伝子の組み込み数を確認する。LWSをアップデートする。
9.セクション8.2に記されているプロトコールに従って、種子をT3ホモ接合型系統から収集する。
【0241】
【化1】
【特許請求の範囲】
【請求項1】
植物の光合成細胞におけるフルクタン生合成を操作する方法であって、前記方法は、細菌フルクトシルトランスフェラーゼ(FT)酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む有効量の遺伝的構築物を前記植物に導入するステップを含む、方法。
【請求項2】
前記プロモーターが構成的、組織特異的および誘導性プロモーターからなる群より選択される、請求項1に記載の方法。
【請求項3】
前記プロモーターが光調節プロモーターである、請求項2に記載の方法。
【請求項4】
前記細菌FT酵素がスクロース:スクロース1−フルクトシルトランスフェラーゼ(1−SST)酵素活性およびフルクタン:フルクタン1−フルクトシルトランスフェラーゼ(1−FFT)酵素活性の両方を含む、請求項1に記載の方法。
【請求項5】
細菌FT酵素をコードする前記核酸がSacB、LscおよびFTF遺伝子からなる群より選択される、請求項4に記載の方法。
【請求項6】
植物における生化学的経路の生産性を増強する方法であって、前記方法は、FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む有効量の遺伝的構築物を前記植物に導入するステップを含む、方法。
【請求項7】
植物におけるバイオマスを増強する方法であって、前記方法は、1または複数のフルクタン生合成酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む有効量の遺伝的構築物を前記植物に導入するステップを含む、方法。
【請求項8】
形質転換植物を選抜する方法であって、前記方法は、1または複数のフルクタン生合成酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む有効量の遺伝的構築物を前記植物に導入するステップと、バイオマスが増強された植物を選抜するステップとを含む、方法。
【請求項9】
植物の光合成細胞におけるフルクタン生合成を操作することができる遺伝的構築物であって、細菌FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む、遺伝的構築物。
【請求項10】
前記プロモーターが構成的、組織特異的および誘導性プロモーターからなる群より選択される、請求項9に記載の遺伝的構築物。
【請求項11】
前記プロモーターが光調節プロモーターである、請求項10に記載の遺伝的構築物。
【請求項12】
前記細菌FT酵素がスクロース:スクロース1−フルクトシルトランスフェラーゼ(1−SST)酵素活性およびフルクタン:フルクタン1−フルクトシルトランスフェラーゼ(1−FFT)酵素活性の両方を含む、請求項9に記載の遺伝的構築物。
【請求項13】
細菌FT酵素をコードする前記核酸がSacB、LscおよびFTF遺伝子からなる群より選択される、請求項12に記載の遺伝的構築物。
【請求項14】
植物におけるバイオマスを増強する方法であって、有効量の
(a)前記植物の光合成細胞におけるフルクタン生合成を操作することができる遺伝的構築物であって、細菌FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む、遺伝的構築物と、
(b)前記植物の老化を操作することができる遺伝的構築物と
を前記植物に導入するステップを含む、方法。
【請求項15】
前記プロモーターが光調節プロモーターである、請求項14に記載の方法。
【請求項16】
老化を操作することができる前記遺伝的構築物が、サイトカイニンの生合成に関与する酵素をコードする遺伝子またはその機能的に活性な断片もしくは変異体に作動的に連結したmyb遺伝子プロモーターもしくは改変されたmyb遺伝子プロモーターまたはそれらの機能的に活性な断片もしくは変異体を含む、請求項14に記載の方法。
【請求項17】
請求項9に記載の遺伝的構築物を含み、非形質転換対照植物と比べてフルクタン生合成特徴が改変された、またはバイオマスが増強されたトランスジェニック植物細胞、植物体、植物種子または他の植物部分。
【請求項18】
非形質転換対照植物と比べてバイオマスが少なくとも約50%増加した、請求項17に記載のトランスジェニック植物細胞、植物体、植物種子または他の植物部分。
【請求項19】
非形質転換対照植物と比べて可溶性炭水化物が少なくとも約50%増加した、請求項17に記載のトランスジェニック植物細胞、植物体、植物種子または他の植物部分。
【請求項1】
植物の光合成細胞におけるフルクタン生合成を操作する方法であって、前記方法は、細菌フルクトシルトランスフェラーゼ(FT)酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む有効量の遺伝的構築物を前記植物に導入するステップを含む、方法。
【請求項2】
前記プロモーターが構成的、組織特異的および誘導性プロモーターからなる群より選択される、請求項1に記載の方法。
【請求項3】
前記プロモーターが光調節プロモーターである、請求項2に記載の方法。
【請求項4】
前記細菌FT酵素がスクロース:スクロース1−フルクトシルトランスフェラーゼ(1−SST)酵素活性およびフルクタン:フルクタン1−フルクトシルトランスフェラーゼ(1−FFT)酵素活性の両方を含む、請求項1に記載の方法。
【請求項5】
細菌FT酵素をコードする前記核酸がSacB、LscおよびFTF遺伝子からなる群より選択される、請求項4に記載の方法。
【請求項6】
植物における生化学的経路の生産性を増強する方法であって、前記方法は、FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む有効量の遺伝的構築物を前記植物に導入するステップを含む、方法。
【請求項7】
植物におけるバイオマスを増強する方法であって、前記方法は、1または複数のフルクタン生合成酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む有効量の遺伝的構築物を前記植物に導入するステップを含む、方法。
【請求項8】
形質転換植物を選抜する方法であって、前記方法は、1または複数のフルクタン生合成酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む有効量の遺伝的構築物を前記植物に導入するステップと、バイオマスが増強された植物を選抜するステップとを含む、方法。
【請求項9】
植物の光合成細胞におけるフルクタン生合成を操作することができる遺伝的構築物であって、細菌FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む、遺伝的構築物。
【請求項10】
前記プロモーターが構成的、組織特異的および誘導性プロモーターからなる群より選択される、請求項9に記載の遺伝的構築物。
【請求項11】
前記プロモーターが光調節プロモーターである、請求項10に記載の遺伝的構築物。
【請求項12】
前記細菌FT酵素がスクロース:スクロース1−フルクトシルトランスフェラーゼ(1−SST)酵素活性およびフルクタン:フルクタン1−フルクトシルトランスフェラーゼ(1−FFT)酵素活性の両方を含む、請求項9に記載の遺伝的構築物。
【請求項13】
細菌FT酵素をコードする前記核酸がSacB、LscおよびFTF遺伝子からなる群より選択される、請求項12に記載の遺伝的構築物。
【請求項14】
植物におけるバイオマスを増強する方法であって、有効量の
(a)前記植物の光合成細胞におけるフルクタン生合成を操作することができる遺伝的構築物であって、細菌FT酵素をコードする核酸またはその機能的に活性な断片もしくは変異体に作動的に連結したプロモーターまたはその機能的に活性な断片もしくは変異体を含む、遺伝的構築物と、
(b)前記植物の老化を操作することができる遺伝的構築物と
を前記植物に導入するステップを含む、方法。
【請求項15】
前記プロモーターが光調節プロモーターである、請求項14に記載の方法。
【請求項16】
老化を操作することができる前記遺伝的構築物が、サイトカイニンの生合成に関与する酵素をコードする遺伝子またはその機能的に活性な断片もしくは変異体に作動的に連結したmyb遺伝子プロモーターもしくは改変されたmyb遺伝子プロモーターまたはそれらの機能的に活性な断片もしくは変異体を含む、請求項14に記載の方法。
【請求項17】
請求項9に記載の遺伝的構築物を含み、非形質転換対照植物と比べてフルクタン生合成特徴が改変された、またはバイオマスが増強されたトランスジェニック植物細胞、植物体、植物種子または他の植物部分。
【請求項18】
非形質転換対照植物と比べてバイオマスが少なくとも約50%増加した、請求項17に記載のトランスジェニック植物細胞、植物体、植物種子または他の植物部分。
【請求項19】
非形質転換対照植物と比べて可溶性炭水化物が少なくとも約50%増加した、請求項17に記載のトランスジェニック植物細胞、植物体、植物種子または他の植物部分。
【図1】

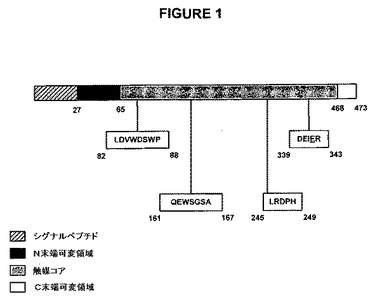
【図2】

【図3】

【図4】

【図5】

【図6】
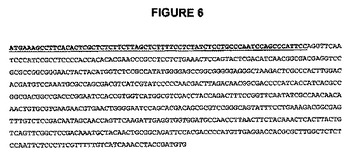
【図7】

【図8】

【図9】

【図11】

【図12】

【図15】

【図16】

【図17】

【図18】

【図20】

【図21】

【図23−1】

【図23−2】

【図24−1】

【図24−2】

【図25−1】

【図25−2】
【図26−1】

【図26−2】

【図29】

【図30−1】

【図30−2】

【図31−1】

【図31−2】
【図32−1】

【図32−2】

【図33−1】

【図33−2】

【図34−1】

【図34−2】

【図35−1】

【図35−2】

【図36−1】

【図36−2】

【図36−3】

【図42】
【図43】
【図44】
【図45】
【図46】

【図47】

【図48】
【図50】
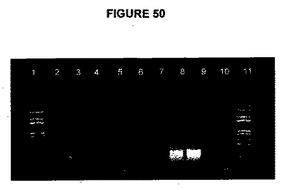
【図10】

【図13】

【図14】
【図19】
【図22】
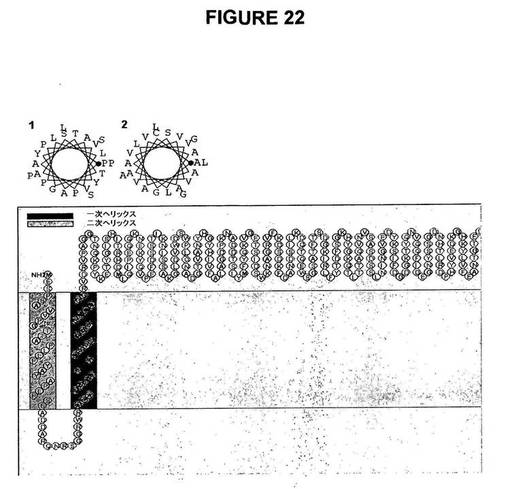
【図27】
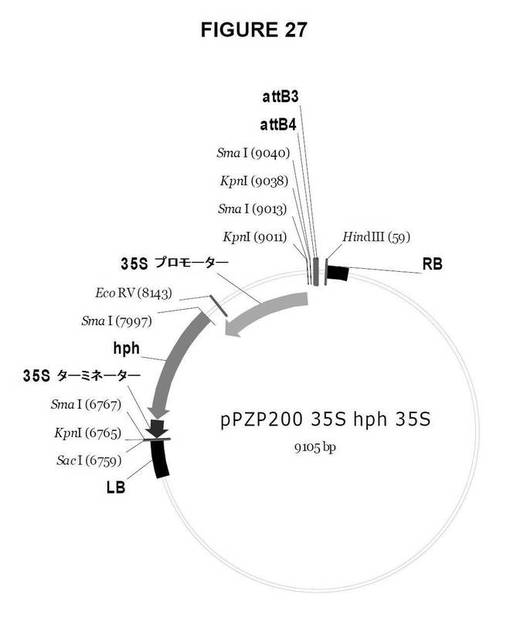
【図28A】
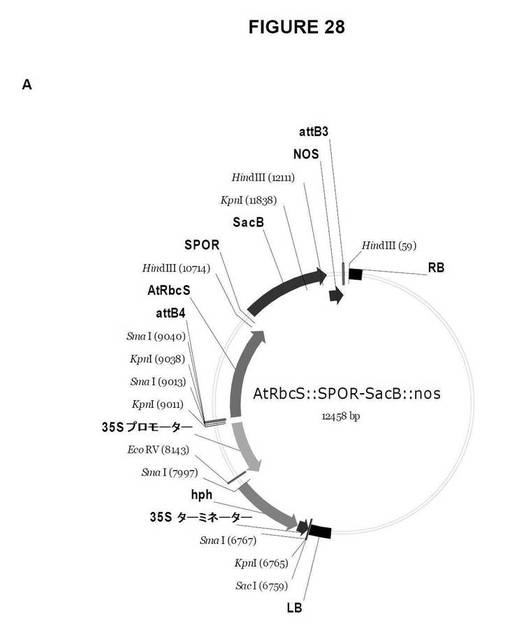
【図28B】
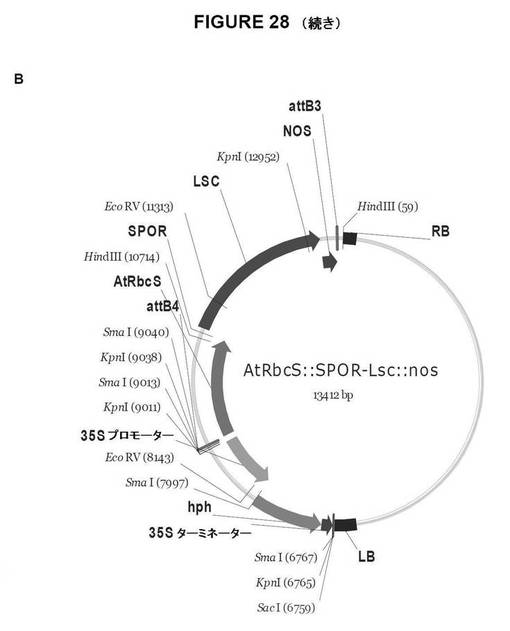
【図28C】
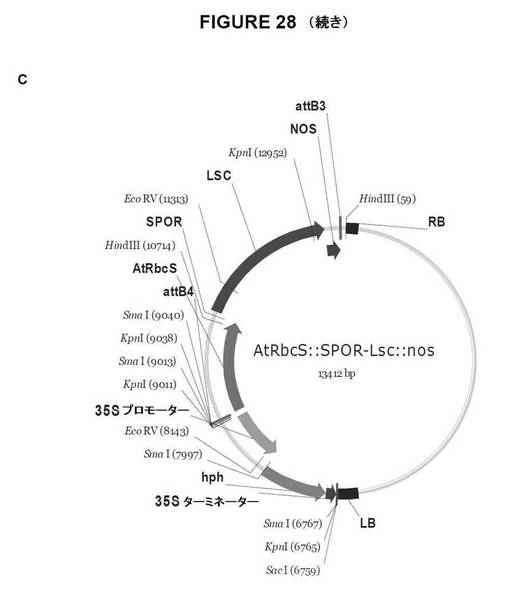
【図28D】
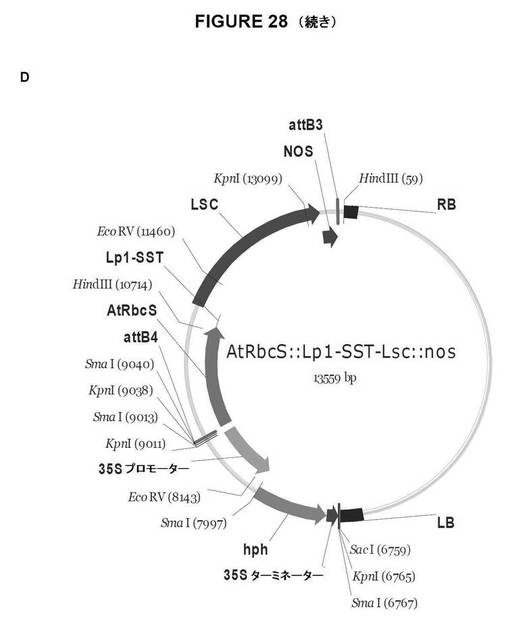
【図37】
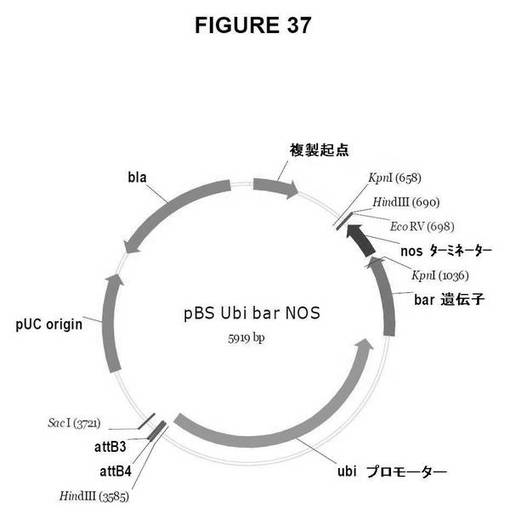
【図38】
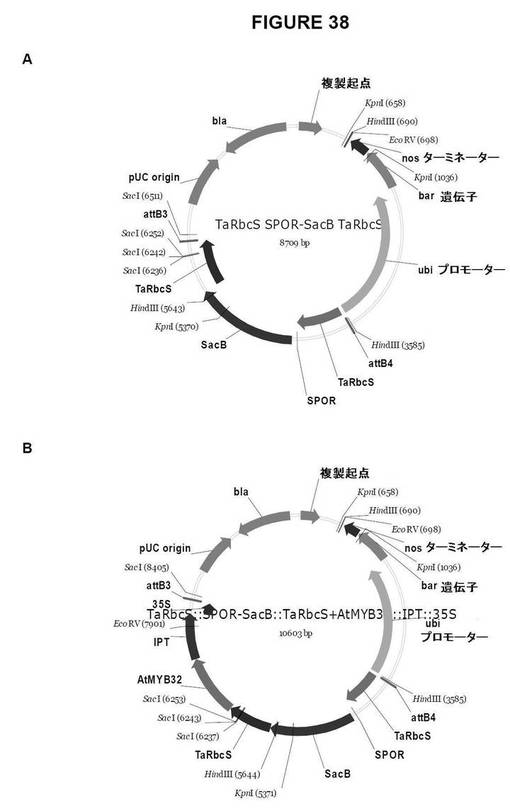
【図39】
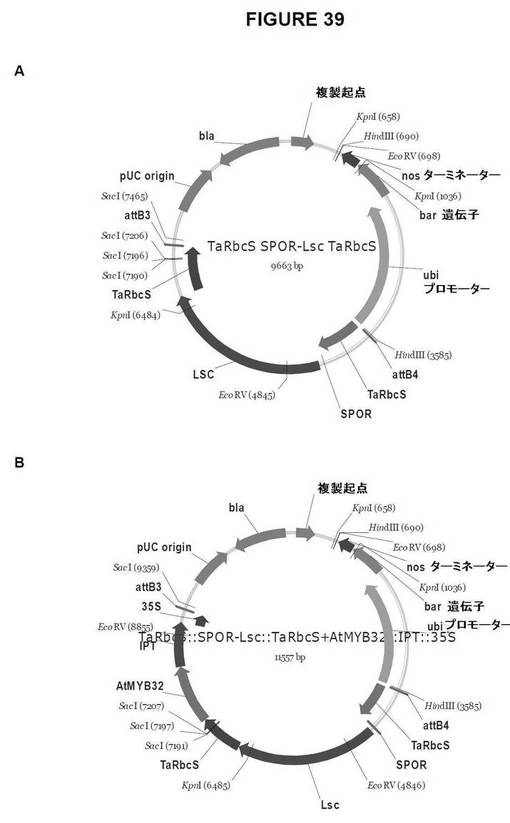
【図40】
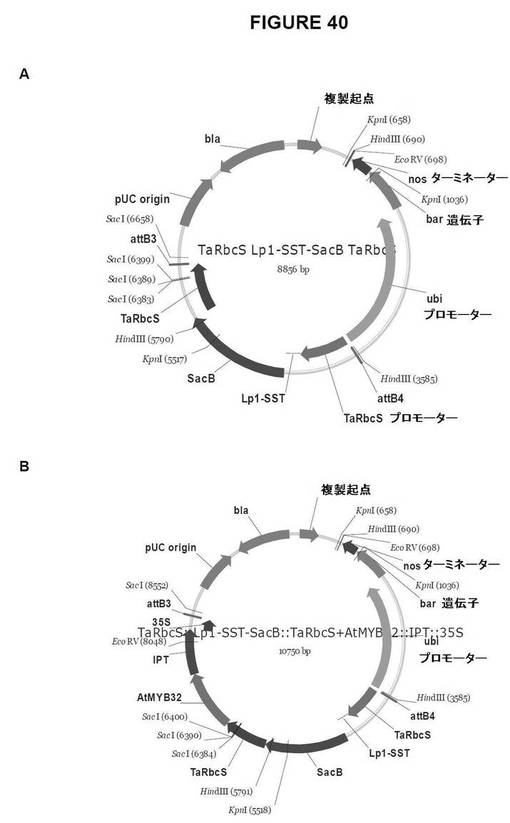
【図41】
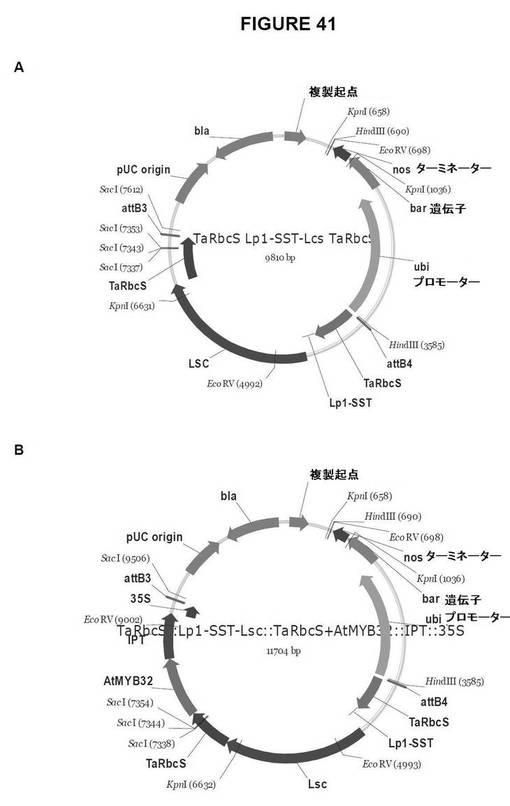
【図49−1】
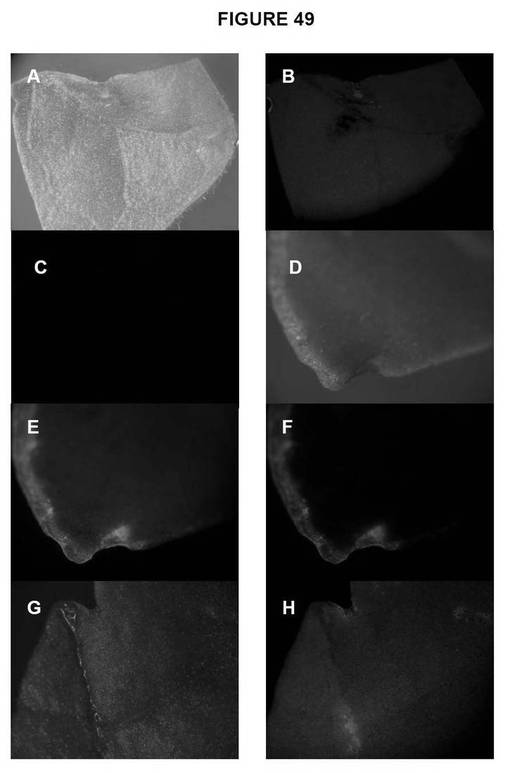
【図49−2】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図11】
【図12】
【図15】
【図16】
【図17】
【図18】
【図20】
【図21】
【図23−1】
【図23−2】
【図24−1】
【図24−2】
【図25−1】
【図25−2】
【図26−1】
【図26−2】
【図29】
【図30−1】
【図30−2】
【図31−1】
【図31−2】
【図32−1】
【図32−2】
【図33−1】
【図33−2】
【図34−1】
【図34−2】
【図35−1】
【図35−2】
【図36−1】
【図36−2】
【図36−3】
【図42】
【図43】
【図44】
【図45】
【図46】
【図47】
【図48】
【図50】
【図10】
【図13】
【図14】
【図19】
【図22】
【図27】
【図28A】
【図28B】
【図28C】
【図28D】
【図37】
【図38】
【図39】
【図40】
【図41】
【図49−1】
【図49−2】
【公表番号】特表2012−525119(P2012−525119A)
【公表日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願番号】特願2012−507544(P2012−507544)
【出願日】平成22年4月27日(2010.4.27)
【国際出願番号】PCT/AU2010/000481
【国際公開番号】WO2010/124324
【国際公開日】平成22年11月4日(2010.11.4)
【出願人】(503087223)アグリカルチャー ビクトリア サービシーズ プロプライエタリー リミテッド (3)
【Fターム(参考)】
【公表日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願日】平成22年4月27日(2010.4.27)
【国際出願番号】PCT/AU2010/000481
【国際公開番号】WO2010/124324
【国際公開日】平成22年11月4日(2010.11.4)
【出願人】(503087223)アグリカルチャー ビクトリア サービシーズ プロプライエタリー リミテッド (3)
【Fターム(参考)】
[ Back to top ]