
ブリンゾラミドの製造方法
本発明は、ブリンゾラミドの製造及び精製、並びにかかる方法において有用な新規化合物に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ブリンゾラミドの製造及び精製、並びにかかる方法において有用な新規化合物に関する。
【0002】
ブリンゾラミド[(4R)−4−(エチルアミノ)−3,4−ジヒドロ−2−(3−メトキシプロピル)−2H−チエノ[3,2−e]−1,2−チアジン−6−スルホンアミド−1,1−ジオキシド]は、緑内障の治療のために有用な炭酸アンヒドラーゼ阻害剤である。この生成物及びその製造は、例えば、US5240923、US5378703及びUS5461081に記載されている。
【0003】
本発明者らは、ブリンゾラミドを製造及び精製するための新規方法を確認した。更に、本発明者らは、かかる方法において有用な新規化合物を確認した。
【0004】
本発明の第1の態様は、
(a)化合物(I)
【化1】

[式中、Ac(P)は、アセチル基又はマスクされた、例えば保護若しくは還元されたアセチル基である]と、スルホン化試薬、特に亜硫酸塩とを反応させて、化合物(II)
【化2】
[式中、Ac(P)は、上記の通りであり、Mは、カチオン、特にNa+である]を得る工程、
(b)化合物(II)と3−メトキシアミノプロパンとを反応させて、化合物(III)
【化3】
[式中、Ac(P)は、上記の通りである]を得る工程、
(c)場合によって、化合物(III)のアセチル基をアセチル保護剤、特にエチレングリコール又は還元剤でマスクして、化合物(IV)
【化4】
[式中、AcPは、マスクされた、例えば保護又は還元されたアセチル基、特に、
【化5】
である]を得る工程、
(d)(i)化合物(IV)と有機金属(有機金属の)化合物とを反応させた後、SO2で処理し、続いてヒドロキシルアミン−O−スルホン酸で処理して、化合物(V)
【化6】
[式中、AcPは、上記の通りである]を得る工程、
(d)(ii)例えば化合物(V)を脱保護又は酸化させることによってアセチル基を再構成して、化合物(Va):
【化7】
[式中、Acは、アセチル基である]を得る工程、
(e)化合物(Va)と臭素化剤、特に過臭化臭化ピリジニウム(PBP)とを反応させて、化合物(VI)
【化8】
を得る工程、
(f)アルカリ性条件下で、化合物(VI)と、還元剤、特にキラル還元剤、より具体的には(+)−ジイソピノカンフェニルクロロボラン(DIPCl)とを反応させて、化合物(VII)
【化9】
を得る工程、
(g)(i)化合物(VII)を中間体(VIIa)、
【化10】
[式中、Xは、脱離基、特にスルホネート基である]に転化する工程、及び
(g)(ii)化合物(VIIa)とエチルアミンとを反応させて、化合物(VIII)
【化11】
を得る工程、
(h)場合によって、化合物(VIII)を、例えばキラル酒石酸、水及び/又はアルコール若しくは水性アルコール、特にイソプロパノールで精製して、望ましくない異性体を他の不純物と共に除去する工程
を含む、ブリンゾラミド(VIII)の製造方法に関する。
【0005】
工程(a)は、適切な反応条件下における、例えば還流下で10〜20時間の、3−アセチル−2,5−ジクロロチオフェン又はその保護アセチル誘導体、即ち化合物(I)と、スルホン化剤、特に亜硫酸塩、例えばアルカリ金属亜硫酸塩、例えば適切な溶媒中における亜硫酸ナトリウム、例えばエタノール/H2Oとの反応を含む。生成物が、好ましくは90%超の収率で得られる。
【0006】
工程(b)は、化合物(II)と3−メトキシアミノプロパンとを反応させることを含む。この反応工程のために、最初に、水分のない条件下で五塩化リン(Pcl5)、塩化チオニル、塩化スルフリル又はオキシ三塩化リンに化合物(II)を接触させる。次いで、例えば重炭酸ナトリウムによる中和の後、メトキシプロピルアミンを添加して、化合物(III)、即ち3−アセチル−5−クロロ−チオフェン−2−スルホニル−N−(3−メトキシプロピル)アミド又はそのアセチル保護誘導体を得る。反応は、例えば約1時間〜約5時間の冷却下で、水性溶媒中において実施され得る。好ましくは、生成物が、50%超の収率で得られる。
【0007】
ステップ(c)は、化合物(III)のアセチル基をマスクすることを含む。第1の方法の変形において、この工程は、非保護アセチル基が存在する場合に化合物(III)とアセチル保護剤とを反応させることを含む。好ましくは、アセチル保護剤は、例えば8〜15時間、加熱下で、スルホン酸、例えばp−トルエンスルホン酸若しくはカンファースルホン酸、ルイス酸、例えば三フッ化ホウ素エーテラート、又は酸性樹脂、例えばAmberlite IR120若しくはNafion Hの存在下で、化合物(III)と反応するエチレングリコールである。好ましくは、アセチル保護化合物(IV)が、80%以上の収率で得られる。
【0008】
第2の方法の変形において、この工程は、例えば0.5〜2時間、例えば5〜10℃で、冷却下で、化合物(III)と、還元剤、例えば、適切な溶媒、例えばアルコール中におけるNaBH4等のボラン還元剤とを反応させることを含む。それによって、アセチル基は、1−ヒドロキシ−エチル基に還元される。好ましくは、還元された化合物(IV)が、80%以上の収率で得られる。
【0009】
工程(d)(i)は、保護又は還元化合物(IV)と、有機金属(有機金属の)化合物、好ましくは有機リチウム化合物、より好ましくはアルキルリチウム化合物、例えばブチルリチウムとを反応させた後、SO2で処理し、続いてヒドロキシルアミン−O−スルホン酸で処理することを含む。好ましくは、保護雰囲気下で、無水溶媒、例えばテトラヒドロフラン中において、例えば−70℃未満における冷却下で有機リチウム及び二酸化硫黄との反応を実施する。ヒドロキシルアミン−O−スルホン酸との後続反応を、水性溶媒中において実施することができる。化合物(V)の収率は、好ましくは60%以上である。
【0010】
工程(d)(ii)は、アセチル基を再構成することを含む。第1の方法の変形において、この工程は、化合物(Va)を得るためにアセチル保護基を除去することを含む。好ましくは、アセチル基は、例えば、アセトニトリル/水等の適切な溶媒中におけるHClを添加することによって、酸性条件下で除去される。化合物(Va)の収率は、好ましくは80%以上である。
【0011】
第2の方法の変形において、この工程は、適切な時間、例えば0.5〜4時間、例えば10〜20℃における軽い冷却下で、適切な溶媒、例えばジクロロメタン中における適切な酸化剤、例えばデス−マーチンペルヨージナン(1,1,1−トリアセトキシ−1,1−ジヒドロ−1,2−ベンズヨードキソール−3[1H]オン)で1−ヒドロキシ−エチル基を酸化することを含む。化合物(Va)の収率は、好ましくは60%以上である。
【0012】
工程(e)は、好ましくは酸性条件下で、例えば酢酸エチル等の適切な溶媒中における硫酸の存在下で、化合物(Va)と、適切な臭素化剤、特に過臭化臭化ピリジニウム(PBP)とを反応させることを含む。化合物(VI)の収率は、好ましくは80%以上である。
【0013】
工程(f)は、アルカリ性条件下で化合物(VI)と還元剤とを反応させることを含む。好ましくは、還元剤は、立体選択的還元化合物(VII)においてR−鏡像異性体を与えるキラル還元剤である。より好ましくは、還元剤は、キラル有機ボラン化合物、例えば(+)−ジイソピノカンフェニルクロロボランである。別の場合、工程(f)は、化合物(VI)とNaBH4等の非キラル還元剤との反応を含むこともできる。しかし、非キラル還元剤の使用は、望ましくない異性体、例えばブリンゾラミドの(S)異性体を除去するために一層の努力を必要とするためあまり好ましくない。好ましくは、還元は、保護雰囲気下で、適切な溶媒、例えばt−ブチルメチルエーテル中において、無水条件下で実施される。好ましくは、化合物(VII)は、60%以上の収率で得られる。
【0014】
工程(g)(i)は、例えば、テトラヒドロフラン等の有機溶媒中におけるメタンスルホン酸無水物等の適切な酸無水物との、好ましくは冷却下における反応によって、化合物(VII)を中間体(VIIa)に転化することを含む。この反応混合物から、ブリンゾラミド、即ち化合物(VIII)が、好ましくは水性溶媒中におけるエチルアミンとの反応によっていかなる処理もなしで好ましくは得られる。粗ブリンゾラミド(VIII)の収率は、好ましくは60%以上である。
【0015】
任意の工程(h)は、不純物、特に望ましくない異性体を除去するために精製又はブリンゾラミドを含む。好ましくは、精製は、以下で詳述のように、キラル酒石酸による処理、高温の水による処理及び/又はアルコール若しくは水性アルコール、例えばイソプロピルアルコールからの再結晶を含んでよい。工程(b)からの純粋なブリンゾラミドの収率は、好ましくは80%以上である。
【図面の簡単な説明】
【0016】
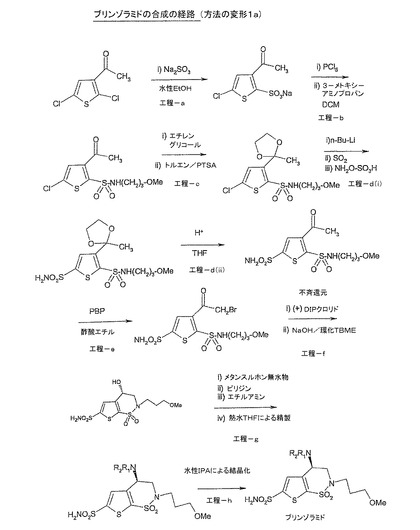
【図1】図1は、工程(f)において不斉還元を実施するためにキラル還元剤を用いる本発明の方法(変形1a)の好ましい一実施形態を示す。
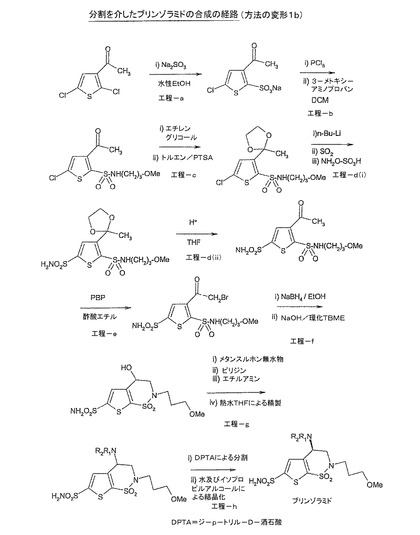
【図2】図2は、工程(f)における非キラル還元剤の使用、及び例えばジ−p−トリル−D−酒石酸(DTPA)を用いることによる後続の光学分割を含む本発明の方法(変形1b)の更なる一実施形態を示す。
【図3】図3は、アセチル基の還元/酸化工程を含む本発明の方法(変形2)の更なる好ましい一実施形態を示す。
【0017】
本発明は、段階II材料の製造の収率がより著しく高い費用効果的方法を用いるので、公知の方法に鑑みて明白な利点を示す。更に、上記の方法は、文献中において文書化されている方法と比較して、操作上、より単純で且つ安全である。
【0018】
更に、前記方法は、環化中間体によるリチウム化についての文献(参考文献:Org.Proc.Res.& Dev.,1999,3,114−120)中に報告されているように、リチウム化が、より短い反応時間で−40〜−20℃で進行し、更に、いかなるオルトリチウム化化合物の形成ももたらさないので、化合物Vの製造(方法の変形(2)の工程d(i))に関する著しい利点を示す。
【0019】
更に、本発明は、構造式(II):
【化12】
[式中、Ac(P)は、アセチル基又はマスクされた、例えば保護若しくは還元されたアセチル基であり、Mは、金属カチオン、特にNa+である]を有する新規化合物に関する。
【0020】
更に、本発明は、構造式(V):
【化13】
[式中、AcPは、マスクされた、例えば保護若しくは還元されたアセチル基、特に
【化14】
である]を有する化合物に関する。
【0021】
更に、本発明は、構造式(VI):
【化15】
を有する化合物に関する。
【0022】
更に、本発明は、式(VII):
【化16】
の構造式を有する化合物に関する。
【0023】
更に、本発明の更なる目的は、ブリンゾラミドの製造、特に上記の方法の化合物(II)、(V)、(VI)及び/又は(VII)の使用である。
【0024】
更に、本発明の更なる目的は、ブリンゾラミドの製造方法であり、化合物(II)、(V)、(VI)及び(VII)から選択される少なくとも1種の化合物が中間体として得られる製造方法である。
【0025】
また、本発明はブリンゾラミドを精製するための新規方法に関する。本実施形態における第1の態様は、粗ブリンゾラミドとキラル酒石酸とを含む溶液を形成すること、所望のブリンゾラミドタルトレートを溶液から沈殿させること、及び精製されたブリンゾラミド生成物を回収することを含む、不純物から、特にS−鏡像異性体からブリンゾラミドを精製するための方法である。
【0026】
キラル酒石酸は、好ましくはジ−p−トリル−D−酒石酸(DTPA)である。好ましくは、前記方法は、例えば、適切な溶媒、例えば水性メタノール中における加熱によって、(R及びS異性体を含む)粗ブリンゾラミド出発材料及びキラル酒石酸を接触させて溶液を形成することを含む。続いて、溶液温度を、例えば室温に低下させ、そこで十分な時間、例えば8〜24時間以上保持して、溶液からの所望のR−ブリンゾラミドタルトレートの沈殿を可能にする。沈殿した酒石酸塩は、溶液から単離される。続いて、単離された酒石酸塩を、例えば重炭酸塩で中和して、ブリンゾラミドの所望の遊離塩基を得ることができる。精製されたブリンゾラミド塩基は、好ましくは、望ましくないS−鏡像異性体の含有率が0.50質量%以下である。望ましくないS−鏡像異性体の含有率がより高い場合、以下に詳述のように更なる精製プロセスを実施しなければならない可能性がある。
【0027】
また、本発明の本実施形態は、キラル酒石酸によるブリンゾラミドの塩、特にDTPAによるブリンゾラミドの塩にも関する。好ましくは、望ましくないS−鏡像異性体の含有率が50質量%未満のブリンゾラミド塩である。
【0028】
更に、本発明の更なる一態様は、高温の水で粗ブリンゾラミドを処理すること、及び精製されたブリンゾラミド生成物を回収することを含む、不純物から、特にS−鏡像異性体からブリンゾラミドを精製するための方法に関する。
【0029】
驚くべきことに、適切な時間、例えば30分〜2時間、好ましくは45分〜1時間、高温の、例えば60〜80℃、好ましくは65〜70℃の水中で粗ブリンゾラミド生成物を懸濁、続いて約40〜50℃に冷却し、これらの条件で含有物を濾過することで、望ましくないS−鏡像異性体の量の実質的な減少をもたらすことが分かった。好ましくは、得られた精製ブリンゾラミド生成物中における望ましくないS−鏡像異性体の含有率は、0.50質量%以下である。必要な場合、水精製プロセスは、1回又は数回、例えば最高5回繰り返すことができる。
【0030】
本発明のなお更なる一態様は、イソプロピルアルコール中における粗ブリンゾラミドの再結晶、及び精製されたブリンゾラミド生成物の回収を含む、不純物から、特にS−鏡像異性体からブリンゾラミドを精製するための方法に関する。
【0031】
驚くべきことに、イソプロピルアルコールからの粗ブリンゾラミドの再結晶は、望ましくないS−鏡像異性体の含有率の実質的な減少をもたらすことが分かった。好ましくは、精製されたブリンゾラミド生成物中における望ましくないS−鏡像異性体の含有率は、0.50質量%以下である。必要な場合、イソプロピル再結晶手法を、例えば最高4回繰り返す。
【0032】
とりわけ好ましい実施形態において、粗ブリンゾラミドの精製は、以下の手法:
(i)キラル酒石酸による光学精製、
(ii)高温の水による処理、
(iii)イソプロピルアルコール中における再結晶
の内の少なくとも2つの組み合わせを含む。
【0033】
イソプロピルアルコール再結晶と組み合わせた高温の水中における処理が、とりわけ好ましい。
【0034】
更に、以下の実施例によって、本発明をより詳細に記載する。
【0035】
実施例1:ブリンゾラミド(VIII)の調製(方法の変形1)
工程(a):ナトリウム塩(II)の調製
3−アセチル−2,5−ジクロロチオフェン(I)(500g、2.56mol)をエタノール(2.50L)中に懸濁させた。亜硫酸ナトリウム溶液(5.0L、12.8mol)を25〜30℃でゆっくりと添加し、100〜110℃で16時間、還流まで加熱した。反応の完了を、HPLC[(3−アセチル−2,5−ジクロロチオフェンのHPLC限界:1.0%NMT(以下));実測:0.1%の3−アセチル−2,5−ジクロロチオフェン]によってモニタした。
【0036】
反応混合物を25〜30℃に冷却し、濾過し、無水メタノール(1.0L)で洗浄した。得られた残渣を別に保持し、濾液を減圧下で濃縮した。得られた粗生成物をメタノール(5vol)中で懸濁させ、1.0時間撹拌し、濾過し、無水メタノール(1vol)で洗浄した。濾液を減圧下で濃縮し、恒量が得られるまで60〜65℃で真空下において乾燥させた。
【0037】
化合物(II)の収量:640g(95.5%)
【0038】
工程(b):N−置換スルホンアミド(III)の調製
10〜20℃のジクロロメタン(6.50L;含水率0.5%NMT)中における化合物(II)(640g、2.44mol)の撹拌懸濁液に、五塩化リン溶液(PCl5)(7.20モル、12.80Lのジクロロメタン中において1.575kg)を1〜2時間の期間に亘ってゆっくりと滴下し、N2雰囲気下において10〜20℃で4.0〜5.0時間撹拌した。反応の完了を、HPLC分析[ナトリウム塩のHPLC限界:1.0%NMT]によってモニタした。
【0039】
反応混合物を冷却し、濾過し、濾液を飽和炭酸水素ナトリウム溶液(5.0L)で急冷した(水層のpHが7になるまで過剰なPCl5の痕跡を除去した)。濾液を洗浄し、分離し、硫酸ナトリウム上で乾燥して、濾過した。濾液を5〜10℃で撹拌し、その後メトキシプロピルアミン(435g、4.88mol)をゆっくりと添加して、5〜10℃で2時間撹拌した。反応の完了を、HPLC[(3−アセチル−5−クロロチオフェン−2−スルホニルクロリドのHPLC限界:1.0%NMT;実測:0.10%の3−アセチル−5−クロロチオフェン−2−スルホニルクロリド]によってモニタした。反応混合物を、連続的に撹拌しながら精製水(6×1.0vol)で洗浄した。2回の水洗(2×1.0vol)の後、溶液のpHをトリエチルアミンでpH8に調整した。残りの水洗(4×1.0vol)をpH8で行った。
【0040】
有機層を分離し、硫酸ナトリウム上で乾燥させ、完全に留去した。得られた生成物をt−ブチルメチルエーテル(3×1.0vol)で洗浄した後、ヘキサン(3×1.0vol)で洗浄し、恒量が得られるまで35〜40℃で乾燥させた。
【0041】
化合物(III)の収量:400g(52.6%)
【0042】
工程(c):保護されたN−置換スルホンアミド(IV)の調製
25〜30℃の乾燥トルエン(20vol)中における化合物(III)(400g、1.285mol)の懸濁液に、エチレングリコール(360ml、6.425mol)及びp−トルエン−スルホン酸(120.0g、0.642mol)を添加した。反応混合物を75〜80℃で10.0〜12.0時間撹拌した。反応の完了を、HPLC[(N−置換スルホンアミドのHPLC限界:1.0%NMT;移動相酢酸エチル:ヘキサン、1:9)]によってモニタした。
【0043】
反応混合物を25〜30℃に冷却し、有機層を分離して、飽和炭酸水素ナトリウム溶液(2×2.0L)及び冷水(2×2.0L)で洗浄した。有機層を分離し、硫酸ナトリウム上で乾燥させ、濾過し、減圧下で完全に留去した。得られた粗生成物は、淡黄色のシロップ状液体であった。
【0044】
化合物(IV)の収量:373g(81.7%)
【0045】
ステップ(d)(i):保護された5−スルホンアミド(V)の調製
無水テトラヒドロフラン(THF)(15vol)中における化合物(IV)(373g、1.046mol)の溶液を窒素下で−70℃まで冷却した。n−ブチルリチウム(1.28L、1.404モル、2.5Mの溶液)を、温度を−70℃未満に保持しながら2.5時間に亘って滴下した。反応混合物を−70℃未満で1.5〜2.0時間維持し、その後反応混合物中に二酸化硫黄(SO2)を、水中に急冷したアリコートがpH4.0を示すまで導入した。混合物を25〜30℃で15時間加温し、次いで濃縮した。
【0046】
残渣を水(2.5vol)中に溶解し、その溶液を、水(3.0vol)中における酢酸ナトリウム三水和物(1.175Kg、6.265mol)及びヒドロキシルアミン−O−スルホン酸(615g、4.202mol)の予冷却溶液に一部添加し、温度を25℃まで上昇させた。25〜30℃で15時間撹拌した後、溶液を重炭酸ナトリウムで中和し、次いで、pH9.0が達成されるまで50%の水酸化ナトリウム溶液で塩基性化した。溶液を酢酸エチルで抽出し、ブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過し、濃縮した。有機層を減圧下において50℃未満で完全に留去した。得られた粗生成物は、淡黄色の半固体であった。
【0047】
化合物(V)の収量:280g(66.6%)
【0048】
ステップ(d)(ii):脱保護された5−スルホンアミド(Va)の調製
アセトニトリル(5vol)中における化合物(V)(280g、0.700モル)の溶液に、2NのHCl(5vol)を添加した。混合物を75〜80℃で4.0時間還流させた。反応をTLC[TLC移動相酢酸エチル:ヘキサン、1:1]によってモニタした。
【0049】
反応後、真空下でアセトニトリルを完全に除去し、反応混合物を飽和炭酸水素ナトリウム溶液でpH8.0に塩基性化した。水層を酢酸エチル(3×3.0vol)で抽出し、硫酸ナトリウム上で乾燥させ、濾過した。有機層を減圧下において50℃未満で完全に留去した。得られた粗生成物は、淡黄色のシロップ状液体であった。
【0050】
化合物(Va)の収量:210g(84.2%)
【0051】
工程(e):ブロモ化合物(VI)の調製
化合物(Va)(210g、0.588モル)を、酢酸エチル(10vol)中に懸濁させ、撹拌した。淡黄色の懸濁液を30分間で0〜5℃に冷却し、過臭化臭化ピリジニウム(188g、0.588mol)を一部添加した。上記の懸濁液に、硫酸(0.5vol)を15〜25分に亘って滴下漏斗を介して加え、温度を5℃まで上昇させた。硫酸の添加後、反応混合物を徐々に25〜30℃にし、この温度で1時間撹拌した。反応をTLC[移動相酢酸エチル:ヘキサン、3:7]によってモニタした。
【0052】
反応の完了後、反応混合物に氷冷した水を添加した。有機層を分離し、水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、濾過し、溶媒を真空(50℃未満)中において除去して、粗生成物を黄色のシロップとして生じた。
【0053】
化合物(VI)の収量:210g(82.00%)
【0054】
ステップ(f):環化生成物(VII)の調製
立体選択的還元によって化合物(VII)の所望の異性体を得るために(+)−ジイソピノカンフェニルボラン(DIPCl)等のキラル還元剤を用いて化合物(VI)の環化を実施することができる。
【0055】
化合物(VI)(210g、0.484モル)をt−ブチルメチルエーテル(15vol)中で懸濁させ、窒素雰囲気下で撹拌した。撹拌懸濁液を−40℃まで冷却し、(+)−ジイソピノカンフェニルボラン(DIPCl)(762g(ヘキサン溶液中において60〜65%)、1.063モル)を30分の期間に亘ってカニューレを介して添加した。温度の上昇(−30℃まで)を観察した。反応混合物を−25〜−20℃で3.0〜4.0時間維持し、TLC[移動相酢酸エチル:ヘキサン、3:7]によってモニタした。
【0056】
還元の完了後、1Mの水酸化ナトリウム水溶液(20vol)を15〜20分に亘って滴下漏斗から加えた。反応混合物を、25〜30℃で終夜撹拌し、完了についてTLC[移動相酢酸エチル:ヘキサン、1:1]によってモニタした。完了後、水相及び有機相を分離し、水相をt−ブチルメチルエーテル(4.0vol)で抽出し、5N塩酸溶液を用いてpH1に酸性化し、酢酸エチル(3×6.0vol)で抽出した。組み合わせた酢酸エチル抽出物を飽和ブライン溶液(2×2.5vol)で洗浄し、硫酸ナトリウム上で乾燥させ、濾過した。有機層を減圧下において(50℃未満で)完全に留去した。得られた粗生成物は、淡黄色のシロップであった。
【0057】
化合物(VII)の収量:112g(65.2%)
【0058】
別の場合、ナトリウムテトラヒドロボラン等の非キラル還元剤を用いて工程(f)を実施することもできる。この場合、後続の光学分割工程、即ち、望ましくない異性体の除去を実施しなければならない。
【0059】
工程(g):粗ブリンゾラミド(VIII)の調製
化合物(VII)(112g、0.313mol)をテトラヒドロフラン(10vol)中に懸濁させた。メタンスルホン酸無水物(65.7g、0.374モル)を1ロット添加し、撹拌した。反応混合物を0〜5℃に冷却し、ピリジン(2.5vol)を45分に亘ってゆっくりと添加した。完了についてTLC[移動相ジクロロメタン:メタノール、9.5:0.5]によって反応をモニタした。
【0060】
反応生成物として、中間体化合物(VIIa)を得た。
【0061】
上記の反応混合物に、70%のエチルアミン水溶液(400ml、10vol)を0〜5℃でゆっくりと添加した。完全に添加した後、反応混合物を25〜30℃で12.0時間撹拌した。完了についてTLC[移動相ジクロロメタン:メタノール、9.5:0.5]によって反応をモニタした。有機層(エチルアミン及びテトラヒドロフラン混合物)を減圧下において50℃未満で完全に留去した。水相を濃塩酸で酸性化し、pHを15〜25℃で3.0に調整した。水相をt−ブチルメチルエーテルで2回(2×2.0vol)抽出して、存在する場合あらゆる有機不純物を除去した。t−ブチルメチルエーテル層を1NのHCl溶液で抽出し、層を分離した。
【0062】
水層を組み合わせ、活性炭で処理し、65〜70℃で加熱した。含有物を65〜70℃で30分間撹拌し、65〜70℃でhyflo supercelを通して濾過し、予熱した水(65〜70℃)で洗浄した。濾液を25〜30℃に冷却し、pHを固体の重炭酸ナトリウムで8に調整した。反応混合物を結晶化のために12時間撹拌した。結晶化が起きなかった場合、反応塊をブフナー漏斗を通して濾過し、酢酸エチル(3×5.0vol)で抽出し、硫酸ナトリウム上で乾燥させ、濾過した。有機層を完全に減圧下において(50℃未満で)留去した。粗生成物を淡黄色のシロップとして得て、それをヘキサン(4×1.0vol)で洗浄し、静置して固体に結晶化した残渣を生じた。
【0063】
粗化合物(VIII)の収量:80g(66.3%)
【0064】
工程(h):純粋なブリンゾラミド(VIII)の調製
粗化合物(VIII)(80.0g、0.208モル)を水(5.0vol)中に懸濁させ、65〜70℃に1時間加熱し、(熱間)濾過し、30分間吸引して乾燥させた。そのプロセスを、他の異性体含有率が0.50%NMTになるまでキラルによって繰り返した。
【0065】
このようにして得られた生成物をイソプロピルアルコール(8.0vol)中に懸濁させ、65〜70℃に加熱し、30分間維持し、熱条件下で濾過した。暗色の溶液に活性炭素(10.0質量%)を添加し、65〜70℃で30分間加熱した。溶液を熱条件下で濾過し、(65〜70℃で予熱した)イソプロピルアルコールで洗浄した。溶液を別のフラスコに移し、25〜30℃で1時間撹拌した。結晶質固体沈殿物を濾過し、30分間吸引して乾燥させ、恒量が得られるまで65〜70℃で真空下において乾燥させた。
【0066】
純粋化合物(VIII)の収量:60g(86%)
【0067】
実施例2:
ブリンゾラミドの調製(方法の変形2)
工程(a):ナトリウム塩(II)の調製
3−アセチル−2,5−ジクロロチオフェン(I)(500g、2.56mol)をエタノール(2.50L)中に懸濁させた。(水中に亜硫酸ナトリウム(即ち、5.0Lの水中に1.61Kg、12.8モル)を溶解させることによって得られた)亜硫酸ナトリウム溶液を25〜30℃でゆっくりと添加し、100〜110℃で還流まで加熱した。反応混合物を100〜110℃で16時間還流させた。反応の完了を、HPLC[(3−アセチル−2,5−ジクロロチオフェンのHPLC限界:1.0%NMT);実測:0.1%の3−アセチル−2,5−ジクロロチオフェン]によってモニタした。
【0068】
反応混合物を25〜30℃に冷却し、濾過し、無水メタノール(1.0L)で洗浄した。濾液を減圧下において(60〜65℃で)濃縮した。得られた粗生成物を無水メタノール(6.0L)中で懸濁させ、25〜30℃で30分間撹拌した。未溶解の固体を濾過し、濾液を完全に減圧下において60〜65℃で濃縮した。材料を、恒量が得られるまで60〜65℃で真空下において乾燥させた。
【0069】
収量:640gの化合物(II)(95.5%)
【0070】
工程(b):N−置換スルホンアミド(III)の調製
ジクロロメタン(6.50L;含水率0.5%NMT)中における化合物(II)(640g、2.44mol)の撹拌懸濁液に、五塩化リン(PCl5)溶液(12.80Lのジクロロメタン中において1.575Kg)を、10〜20℃で1〜2時間の間隔でゆっくりと添加した。反応を、温度を保持しながらN2下で4.0時間撹拌した。反応の完了を、HPLC分析[ナトリウム塩のHPLC限界:1.0%NMT]によってモニタした。
【0071】
冷却した反応混合物を濾過して(未溶解の塩を除去し)、濾液を飽和炭酸水素ナトリウム溶液(5.0L)で急冷して(水層のpHが7になるまで過剰なPCl5痕跡を除去し)、水で洗浄した。有機層を分離し、硫酸ナトリウム上で乾燥させ、濾過した。撹拌された有機層にメトキシプロピルアミン(435g、4.88mol)をゆっくりと加え、反応混合物を5〜10℃で2.0時間撹拌した。反応の完了をHPLC[(3−アセチル−5−クロロチオフェン−2−スルホニルクロリドについての限界:1.0%NMT);実測:0.1%の3−アセチル−5−クロロチオフェン−2−スルホニルクロリド]によってモニタした。反応を1NのHCl溶液(2vol)及び水で洗浄した。有機層を分離し、硫酸ナトリウム上で乾燥させ、濾過し、濃縮して、生成物を生じた。得られた生成物をヘキサン(2.0L)で洗浄し、恒量が得られるまで45〜50℃で乾燥させた。
【0072】
化合物(III)の収量:350g(46.2%)
【0073】
ステップ(c):還元スルホンアミド(IV)の調製
無水エタノール(15vol)中における化合物(III)(25g、0.080mol)の溶液に、水素化ホウ素ナトリウム(1.2g、0.032モル)を5〜10℃で添加した。反応混合物を5〜10℃で45分〜1時間撹拌した。反応の完了を、TLC[(N−置換スルホンアミドのTLC限界:1.0%NMT;移動相酢酸エチル:ヘキサン、3:7における)]によってモニタした。
【0074】
反応の完了後、エタノールを除去し、残渣をジクロロメタン(5vol)中に取った。ジクロロメタン溶液を水及びブライン(3vol)で洗浄した。有機層を分離し、硫酸ナトリウム上で乾燥させ、濾過し、減圧下において50℃未満で留去した。粗生成物を淡黄色のシロップとして得た。
【0075】
化合物(IV)の収量:21.0g(83.0%)
【0076】
ステップ(d)(i):還元5−スルホンアミド(V)の調製
無水テトラヒドロフラン(15vol)中における化合物(IV)(20g、0.063mol)の溶液を、N2下でドライアイス/2−プロパノールを用いて−40℃まで冷却した。n−ブチルリチウム(79ml、0.190mol)を、添加中に温度を−20℃未満で保持しながら30分〜1時間に亘って滴下した。反応混合物を、−40℃未満で30分〜1時間維持した。反応混合物中に二酸化硫黄(SO2)を、水中に急冷したアリコートがpH3.0〜4.0を示すまで導入した。混合物を25〜30℃で15時間加温し、次いで濃縮した。
【0077】
残渣を水(100ml)中に溶解し、この溶液に、(0℃で)水(0.3L)中における酢酸ナトリウム三水和物(31.5g、0.38mol)及びヒドロキシルアミン−O−スルホン酸(29g、0.252mol)の溶液を一部添加し、温度を25℃まで上昇させた。25〜30℃で15時間撹拌した後、溶液を重炭酸ナトリウムで中和し、次いで、pH8〜9が達成されるまで50%の水酸化ナトリウム溶液で塩基性化した。溶液を酢酸エチルで抽出し、ブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過し、次いで濃縮した。有機層を減圧下において50℃未満で完全に留去した。粗生成物を淡黄色のシロップとして得て、それをDCM/ヘキサンで結晶化して、淡黄色の固体として化合物を生じた。
【0078】
化合物(V)の収量:16.06g(70%)
【0079】
ステップ(d)(ii):5−スルホンアミド(Va)の調製
ジクロロメタン(15vol)中における化合物(V)(10g、0.028mol)の懸濁液に、デス−マーチンペルヨージナン(5volのジクロロメタン中における11.80g、0.028mol)の懸濁液を、10〜20℃で1.0〜2.0時間ゆっくりと添加した。反応をTLC[還元5−スルホンアミドのTLC限界:1.0%NMT;移動相ジクロロメタン:メタノール、9.5:0.5における)]によってモニタした。
【0080】
反応の完了後、反応混合物を精製水(20vol)で急冷し、45分〜1.0時間撹拌した。有機層を分離し、飽和炭酸水素ナトリウム溶液(2×5.0vol)及びチオ硫酸ナトリウム溶液(10%)で洗浄し、硫酸ナトリウム上で乾燥させ、濾過し、減圧下において50℃未満で完全に留去した。得られた粗生成物は、淡黄色のシロップであった。粗生成物は、ジクロロメタン及びヘキサンから結晶化して、淡黄色/薄茶色の結晶質固体として純粋化合物を生じた。
【0081】
化合物(Va)の収量:6g(65%)
【0082】
更なる工程を、実施例1に記載したように実施した。
【0083】
実施例3:
ブリンゾラミドの光学分割のための方法
粗ブリンゾラミド(0.065mol)を5.0%の水性メタノール(4.0vol)中に溶解し、50℃で1時間加熱した。同時に、メタノール(1.0vol)中に溶解した溶液ジ−p−トリル−D−酒石酸(DPTA)を調製して、25〜30℃で20〜30分間撹拌した。DPTA溶液を、撹拌しながら50℃で粗ブリンゾラミドを含有する溶液にゆっくりと添加した。混合物を50℃で1.0時間維持した。1時間後、溶液をゆっくりと25〜30℃にし、25〜30℃で12時間連続的に撹拌した。沈殿する固体を濾過し、冷たいメタノール(1.0vol)で洗浄し、乾燥させて酒石酸塩を得た。
【0084】
酒石酸塩を飽和炭酸水素ナトリウム溶液(60vol)と混合し、得られた懸濁液を1.0時間撹拌し、次いで酢酸エチル(3×60vol)で抽出した。抽出物を硫酸ナトリウム上で乾燥させ、濾過し、乾燥状態に蒸発させて、ブリンゾラミドの遊離塩基を得た。遊離塩基を、キラルHPLC(限界NMT:0.50%(S−異性体))によってS−異性体含有率について確認した。
【0085】
望ましくないS−異性体の含有率が0.50%より高い場合、以下に記載される通りのイソプロパノールからの水精製プロセス及び/又は再結晶を実施して、望ましくない異性体を減少させることができる。
【0086】
実施例4:
a)水精製プロセス
ブリンゾラミド塩基を精製水(10vol)中に懸濁させ、65〜70℃に45分〜1.0時間加熱した。混合物を50℃まで冷却し、含有物を熱条件下で濾過し、精製水(2.0vol)で洗浄し、1.0時間吸引して乾燥させた。そのプロセスを、0.50%NMTのS−異性体含有率の限界が得られるまで(例えば、4〜5回)繰り返すことができる。繰り返された水精製によって、最高10.00〜20.00%のS−異性体含有率を0.50%NMTの限界に減少させることができる。
【0087】
b)イソプロパノールからの再結晶
粗ブリンゾラミド(80.0g、0.208mol)をイソプロピルアルコール(8.0vol)中に懸濁させ、65〜70℃に加熱し、30分間維持し、熱条件下で濾過した。暗色の溶液に、活性炭素(10.0質量%)を添加し、65〜70℃で30分間加熱した。溶液を熱条件下で濾過し、(65〜70℃で予熱した)イソプロピルアルコールで洗浄した。溶液を別のフラスコに移し、25〜30℃で1時間撹拌した。結晶質固体が沈殿した。沈澱物を濾過し、30分間吸引して乾燥させ、恒量が得られるまで65〜70℃で真空下において乾燥させた。そのプロセスを、生成物が0.50%以下の望ましくない異性体を含むまで繰り返すことができる。
【0088】
収量:60gの純粋なブリンゾラミド(VIII)(86%)
【技術分野】
【0001】
本発明は、ブリンゾラミドの製造及び精製、並びにかかる方法において有用な新規化合物に関する。
【0002】
ブリンゾラミド[(4R)−4−(エチルアミノ)−3,4−ジヒドロ−2−(3−メトキシプロピル)−2H−チエノ[3,2−e]−1,2−チアジン−6−スルホンアミド−1,1−ジオキシド]は、緑内障の治療のために有用な炭酸アンヒドラーゼ阻害剤である。この生成物及びその製造は、例えば、US5240923、US5378703及びUS5461081に記載されている。
【0003】
本発明者らは、ブリンゾラミドを製造及び精製するための新規方法を確認した。更に、本発明者らは、かかる方法において有用な新規化合物を確認した。
【0004】
本発明の第1の態様は、
(a)化合物(I)
【化1】
[式中、Ac(P)は、アセチル基又はマスクされた、例えば保護若しくは還元されたアセチル基である]と、スルホン化試薬、特に亜硫酸塩とを反応させて、化合物(II)
【化2】
[式中、Ac(P)は、上記の通りであり、Mは、カチオン、特にNa+である]を得る工程、
(b)化合物(II)と3−メトキシアミノプロパンとを反応させて、化合物(III)
【化3】
[式中、Ac(P)は、上記の通りである]を得る工程、
(c)場合によって、化合物(III)のアセチル基をアセチル保護剤、特にエチレングリコール又は還元剤でマスクして、化合物(IV)
【化4】
[式中、AcPは、マスクされた、例えば保護又は還元されたアセチル基、特に、
【化5】
である]を得る工程、
(d)(i)化合物(IV)と有機金属(有機金属の)化合物とを反応させた後、SO2で処理し、続いてヒドロキシルアミン−O−スルホン酸で処理して、化合物(V)
【化6】
[式中、AcPは、上記の通りである]を得る工程、
(d)(ii)例えば化合物(V)を脱保護又は酸化させることによってアセチル基を再構成して、化合物(Va):
【化7】
[式中、Acは、アセチル基である]を得る工程、
(e)化合物(Va)と臭素化剤、特に過臭化臭化ピリジニウム(PBP)とを反応させて、化合物(VI)
【化8】
を得る工程、
(f)アルカリ性条件下で、化合物(VI)と、還元剤、特にキラル還元剤、より具体的には(+)−ジイソピノカンフェニルクロロボラン(DIPCl)とを反応させて、化合物(VII)
【化9】
を得る工程、
(g)(i)化合物(VII)を中間体(VIIa)、
【化10】
[式中、Xは、脱離基、特にスルホネート基である]に転化する工程、及び
(g)(ii)化合物(VIIa)とエチルアミンとを反応させて、化合物(VIII)
【化11】
を得る工程、
(h)場合によって、化合物(VIII)を、例えばキラル酒石酸、水及び/又はアルコール若しくは水性アルコール、特にイソプロパノールで精製して、望ましくない異性体を他の不純物と共に除去する工程
を含む、ブリンゾラミド(VIII)の製造方法に関する。
【0005】
工程(a)は、適切な反応条件下における、例えば還流下で10〜20時間の、3−アセチル−2,5−ジクロロチオフェン又はその保護アセチル誘導体、即ち化合物(I)と、スルホン化剤、特に亜硫酸塩、例えばアルカリ金属亜硫酸塩、例えば適切な溶媒中における亜硫酸ナトリウム、例えばエタノール/H2Oとの反応を含む。生成物が、好ましくは90%超の収率で得られる。
【0006】
工程(b)は、化合物(II)と3−メトキシアミノプロパンとを反応させることを含む。この反応工程のために、最初に、水分のない条件下で五塩化リン(Pcl5)、塩化チオニル、塩化スルフリル又はオキシ三塩化リンに化合物(II)を接触させる。次いで、例えば重炭酸ナトリウムによる中和の後、メトキシプロピルアミンを添加して、化合物(III)、即ち3−アセチル−5−クロロ−チオフェン−2−スルホニル−N−(3−メトキシプロピル)アミド又はそのアセチル保護誘導体を得る。反応は、例えば約1時間〜約5時間の冷却下で、水性溶媒中において実施され得る。好ましくは、生成物が、50%超の収率で得られる。
【0007】
ステップ(c)は、化合物(III)のアセチル基をマスクすることを含む。第1の方法の変形において、この工程は、非保護アセチル基が存在する場合に化合物(III)とアセチル保護剤とを反応させることを含む。好ましくは、アセチル保護剤は、例えば8〜15時間、加熱下で、スルホン酸、例えばp−トルエンスルホン酸若しくはカンファースルホン酸、ルイス酸、例えば三フッ化ホウ素エーテラート、又は酸性樹脂、例えばAmberlite IR120若しくはNafion Hの存在下で、化合物(III)と反応するエチレングリコールである。好ましくは、アセチル保護化合物(IV)が、80%以上の収率で得られる。
【0008】
第2の方法の変形において、この工程は、例えば0.5〜2時間、例えば5〜10℃で、冷却下で、化合物(III)と、還元剤、例えば、適切な溶媒、例えばアルコール中におけるNaBH4等のボラン還元剤とを反応させることを含む。それによって、アセチル基は、1−ヒドロキシ−エチル基に還元される。好ましくは、還元された化合物(IV)が、80%以上の収率で得られる。
【0009】
工程(d)(i)は、保護又は還元化合物(IV)と、有機金属(有機金属の)化合物、好ましくは有機リチウム化合物、より好ましくはアルキルリチウム化合物、例えばブチルリチウムとを反応させた後、SO2で処理し、続いてヒドロキシルアミン−O−スルホン酸で処理することを含む。好ましくは、保護雰囲気下で、無水溶媒、例えばテトラヒドロフラン中において、例えば−70℃未満における冷却下で有機リチウム及び二酸化硫黄との反応を実施する。ヒドロキシルアミン−O−スルホン酸との後続反応を、水性溶媒中において実施することができる。化合物(V)の収率は、好ましくは60%以上である。
【0010】
工程(d)(ii)は、アセチル基を再構成することを含む。第1の方法の変形において、この工程は、化合物(Va)を得るためにアセチル保護基を除去することを含む。好ましくは、アセチル基は、例えば、アセトニトリル/水等の適切な溶媒中におけるHClを添加することによって、酸性条件下で除去される。化合物(Va)の収率は、好ましくは80%以上である。
【0011】
第2の方法の変形において、この工程は、適切な時間、例えば0.5〜4時間、例えば10〜20℃における軽い冷却下で、適切な溶媒、例えばジクロロメタン中における適切な酸化剤、例えばデス−マーチンペルヨージナン(1,1,1−トリアセトキシ−1,1−ジヒドロ−1,2−ベンズヨードキソール−3[1H]オン)で1−ヒドロキシ−エチル基を酸化することを含む。化合物(Va)の収率は、好ましくは60%以上である。
【0012】
工程(e)は、好ましくは酸性条件下で、例えば酢酸エチル等の適切な溶媒中における硫酸の存在下で、化合物(Va)と、適切な臭素化剤、特に過臭化臭化ピリジニウム(PBP)とを反応させることを含む。化合物(VI)の収率は、好ましくは80%以上である。
【0013】
工程(f)は、アルカリ性条件下で化合物(VI)と還元剤とを反応させることを含む。好ましくは、還元剤は、立体選択的還元化合物(VII)においてR−鏡像異性体を与えるキラル還元剤である。より好ましくは、還元剤は、キラル有機ボラン化合物、例えば(+)−ジイソピノカンフェニルクロロボランである。別の場合、工程(f)は、化合物(VI)とNaBH4等の非キラル還元剤との反応を含むこともできる。しかし、非キラル還元剤の使用は、望ましくない異性体、例えばブリンゾラミドの(S)異性体を除去するために一層の努力を必要とするためあまり好ましくない。好ましくは、還元は、保護雰囲気下で、適切な溶媒、例えばt−ブチルメチルエーテル中において、無水条件下で実施される。好ましくは、化合物(VII)は、60%以上の収率で得られる。
【0014】
工程(g)(i)は、例えば、テトラヒドロフラン等の有機溶媒中におけるメタンスルホン酸無水物等の適切な酸無水物との、好ましくは冷却下における反応によって、化合物(VII)を中間体(VIIa)に転化することを含む。この反応混合物から、ブリンゾラミド、即ち化合物(VIII)が、好ましくは水性溶媒中におけるエチルアミンとの反応によっていかなる処理もなしで好ましくは得られる。粗ブリンゾラミド(VIII)の収率は、好ましくは60%以上である。
【0015】
任意の工程(h)は、不純物、特に望ましくない異性体を除去するために精製又はブリンゾラミドを含む。好ましくは、精製は、以下で詳述のように、キラル酒石酸による処理、高温の水による処理及び/又はアルコール若しくは水性アルコール、例えばイソプロピルアルコールからの再結晶を含んでよい。工程(b)からの純粋なブリンゾラミドの収率は、好ましくは80%以上である。
【図面の簡単な説明】
【0016】
【図1】図1は、工程(f)において不斉還元を実施するためにキラル還元剤を用いる本発明の方法(変形1a)の好ましい一実施形態を示す。
【図2】図2は、工程(f)における非キラル還元剤の使用、及び例えばジ−p−トリル−D−酒石酸(DTPA)を用いることによる後続の光学分割を含む本発明の方法(変形1b)の更なる一実施形態を示す。
【図3】図3は、アセチル基の還元/酸化工程を含む本発明の方法(変形2)の更なる好ましい一実施形態を示す。
【0017】
本発明は、段階II材料の製造の収率がより著しく高い費用効果的方法を用いるので、公知の方法に鑑みて明白な利点を示す。更に、上記の方法は、文献中において文書化されている方法と比較して、操作上、より単純で且つ安全である。
【0018】
更に、前記方法は、環化中間体によるリチウム化についての文献(参考文献:Org.Proc.Res.& Dev.,1999,3,114−120)中に報告されているように、リチウム化が、より短い反応時間で−40〜−20℃で進行し、更に、いかなるオルトリチウム化化合物の形成ももたらさないので、化合物Vの製造(方法の変形(2)の工程d(i))に関する著しい利点を示す。
【0019】
更に、本発明は、構造式(II):
【化12】
[式中、Ac(P)は、アセチル基又はマスクされた、例えば保護若しくは還元されたアセチル基であり、Mは、金属カチオン、特にNa+である]を有する新規化合物に関する。
【0020】
更に、本発明は、構造式(V):
【化13】
[式中、AcPは、マスクされた、例えば保護若しくは還元されたアセチル基、特に
【化14】
である]を有する化合物に関する。
【0021】
更に、本発明は、構造式(VI):
【化15】
を有する化合物に関する。
【0022】
更に、本発明は、式(VII):
【化16】
の構造式を有する化合物に関する。
【0023】
更に、本発明の更なる目的は、ブリンゾラミドの製造、特に上記の方法の化合物(II)、(V)、(VI)及び/又は(VII)の使用である。
【0024】
更に、本発明の更なる目的は、ブリンゾラミドの製造方法であり、化合物(II)、(V)、(VI)及び(VII)から選択される少なくとも1種の化合物が中間体として得られる製造方法である。
【0025】
また、本発明はブリンゾラミドを精製するための新規方法に関する。本実施形態における第1の態様は、粗ブリンゾラミドとキラル酒石酸とを含む溶液を形成すること、所望のブリンゾラミドタルトレートを溶液から沈殿させること、及び精製されたブリンゾラミド生成物を回収することを含む、不純物から、特にS−鏡像異性体からブリンゾラミドを精製するための方法である。
【0026】
キラル酒石酸は、好ましくはジ−p−トリル−D−酒石酸(DTPA)である。好ましくは、前記方法は、例えば、適切な溶媒、例えば水性メタノール中における加熱によって、(R及びS異性体を含む)粗ブリンゾラミド出発材料及びキラル酒石酸を接触させて溶液を形成することを含む。続いて、溶液温度を、例えば室温に低下させ、そこで十分な時間、例えば8〜24時間以上保持して、溶液からの所望のR−ブリンゾラミドタルトレートの沈殿を可能にする。沈殿した酒石酸塩は、溶液から単離される。続いて、単離された酒石酸塩を、例えば重炭酸塩で中和して、ブリンゾラミドの所望の遊離塩基を得ることができる。精製されたブリンゾラミド塩基は、好ましくは、望ましくないS−鏡像異性体の含有率が0.50質量%以下である。望ましくないS−鏡像異性体の含有率がより高い場合、以下に詳述のように更なる精製プロセスを実施しなければならない可能性がある。
【0027】
また、本発明の本実施形態は、キラル酒石酸によるブリンゾラミドの塩、特にDTPAによるブリンゾラミドの塩にも関する。好ましくは、望ましくないS−鏡像異性体の含有率が50質量%未満のブリンゾラミド塩である。
【0028】
更に、本発明の更なる一態様は、高温の水で粗ブリンゾラミドを処理すること、及び精製されたブリンゾラミド生成物を回収することを含む、不純物から、特にS−鏡像異性体からブリンゾラミドを精製するための方法に関する。
【0029】
驚くべきことに、適切な時間、例えば30分〜2時間、好ましくは45分〜1時間、高温の、例えば60〜80℃、好ましくは65〜70℃の水中で粗ブリンゾラミド生成物を懸濁、続いて約40〜50℃に冷却し、これらの条件で含有物を濾過することで、望ましくないS−鏡像異性体の量の実質的な減少をもたらすことが分かった。好ましくは、得られた精製ブリンゾラミド生成物中における望ましくないS−鏡像異性体の含有率は、0.50質量%以下である。必要な場合、水精製プロセスは、1回又は数回、例えば最高5回繰り返すことができる。
【0030】
本発明のなお更なる一態様は、イソプロピルアルコール中における粗ブリンゾラミドの再結晶、及び精製されたブリンゾラミド生成物の回収を含む、不純物から、特にS−鏡像異性体からブリンゾラミドを精製するための方法に関する。
【0031】
驚くべきことに、イソプロピルアルコールからの粗ブリンゾラミドの再結晶は、望ましくないS−鏡像異性体の含有率の実質的な減少をもたらすことが分かった。好ましくは、精製されたブリンゾラミド生成物中における望ましくないS−鏡像異性体の含有率は、0.50質量%以下である。必要な場合、イソプロピル再結晶手法を、例えば最高4回繰り返す。
【0032】
とりわけ好ましい実施形態において、粗ブリンゾラミドの精製は、以下の手法:
(i)キラル酒石酸による光学精製、
(ii)高温の水による処理、
(iii)イソプロピルアルコール中における再結晶
の内の少なくとも2つの組み合わせを含む。
【0033】
イソプロピルアルコール再結晶と組み合わせた高温の水中における処理が、とりわけ好ましい。
【0034】
更に、以下の実施例によって、本発明をより詳細に記載する。
【0035】
実施例1:ブリンゾラミド(VIII)の調製(方法の変形1)
工程(a):ナトリウム塩(II)の調製
3−アセチル−2,5−ジクロロチオフェン(I)(500g、2.56mol)をエタノール(2.50L)中に懸濁させた。亜硫酸ナトリウム溶液(5.0L、12.8mol)を25〜30℃でゆっくりと添加し、100〜110℃で16時間、還流まで加熱した。反応の完了を、HPLC[(3−アセチル−2,5−ジクロロチオフェンのHPLC限界:1.0%NMT(以下));実測:0.1%の3−アセチル−2,5−ジクロロチオフェン]によってモニタした。
【0036】
反応混合物を25〜30℃に冷却し、濾過し、無水メタノール(1.0L)で洗浄した。得られた残渣を別に保持し、濾液を減圧下で濃縮した。得られた粗生成物をメタノール(5vol)中で懸濁させ、1.0時間撹拌し、濾過し、無水メタノール(1vol)で洗浄した。濾液を減圧下で濃縮し、恒量が得られるまで60〜65℃で真空下において乾燥させた。
【0037】
化合物(II)の収量:640g(95.5%)
【0038】
工程(b):N−置換スルホンアミド(III)の調製
10〜20℃のジクロロメタン(6.50L;含水率0.5%NMT)中における化合物(II)(640g、2.44mol)の撹拌懸濁液に、五塩化リン溶液(PCl5)(7.20モル、12.80Lのジクロロメタン中において1.575kg)を1〜2時間の期間に亘ってゆっくりと滴下し、N2雰囲気下において10〜20℃で4.0〜5.0時間撹拌した。反応の完了を、HPLC分析[ナトリウム塩のHPLC限界:1.0%NMT]によってモニタした。
【0039】
反応混合物を冷却し、濾過し、濾液を飽和炭酸水素ナトリウム溶液(5.0L)で急冷した(水層のpHが7になるまで過剰なPCl5の痕跡を除去した)。濾液を洗浄し、分離し、硫酸ナトリウム上で乾燥して、濾過した。濾液を5〜10℃で撹拌し、その後メトキシプロピルアミン(435g、4.88mol)をゆっくりと添加して、5〜10℃で2時間撹拌した。反応の完了を、HPLC[(3−アセチル−5−クロロチオフェン−2−スルホニルクロリドのHPLC限界:1.0%NMT;実測:0.10%の3−アセチル−5−クロロチオフェン−2−スルホニルクロリド]によってモニタした。反応混合物を、連続的に撹拌しながら精製水(6×1.0vol)で洗浄した。2回の水洗(2×1.0vol)の後、溶液のpHをトリエチルアミンでpH8に調整した。残りの水洗(4×1.0vol)をpH8で行った。
【0040】
有機層を分離し、硫酸ナトリウム上で乾燥させ、完全に留去した。得られた生成物をt−ブチルメチルエーテル(3×1.0vol)で洗浄した後、ヘキサン(3×1.0vol)で洗浄し、恒量が得られるまで35〜40℃で乾燥させた。
【0041】
化合物(III)の収量:400g(52.6%)
【0042】
工程(c):保護されたN−置換スルホンアミド(IV)の調製
25〜30℃の乾燥トルエン(20vol)中における化合物(III)(400g、1.285mol)の懸濁液に、エチレングリコール(360ml、6.425mol)及びp−トルエン−スルホン酸(120.0g、0.642mol)を添加した。反応混合物を75〜80℃で10.0〜12.0時間撹拌した。反応の完了を、HPLC[(N−置換スルホンアミドのHPLC限界:1.0%NMT;移動相酢酸エチル:ヘキサン、1:9)]によってモニタした。
【0043】
反応混合物を25〜30℃に冷却し、有機層を分離して、飽和炭酸水素ナトリウム溶液(2×2.0L)及び冷水(2×2.0L)で洗浄した。有機層を分離し、硫酸ナトリウム上で乾燥させ、濾過し、減圧下で完全に留去した。得られた粗生成物は、淡黄色のシロップ状液体であった。
【0044】
化合物(IV)の収量:373g(81.7%)
【0045】
ステップ(d)(i):保護された5−スルホンアミド(V)の調製
無水テトラヒドロフラン(THF)(15vol)中における化合物(IV)(373g、1.046mol)の溶液を窒素下で−70℃まで冷却した。n−ブチルリチウム(1.28L、1.404モル、2.5Mの溶液)を、温度を−70℃未満に保持しながら2.5時間に亘って滴下した。反応混合物を−70℃未満で1.5〜2.0時間維持し、その後反応混合物中に二酸化硫黄(SO2)を、水中に急冷したアリコートがpH4.0を示すまで導入した。混合物を25〜30℃で15時間加温し、次いで濃縮した。
【0046】
残渣を水(2.5vol)中に溶解し、その溶液を、水(3.0vol)中における酢酸ナトリウム三水和物(1.175Kg、6.265mol)及びヒドロキシルアミン−O−スルホン酸(615g、4.202mol)の予冷却溶液に一部添加し、温度を25℃まで上昇させた。25〜30℃で15時間撹拌した後、溶液を重炭酸ナトリウムで中和し、次いで、pH9.0が達成されるまで50%の水酸化ナトリウム溶液で塩基性化した。溶液を酢酸エチルで抽出し、ブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過し、濃縮した。有機層を減圧下において50℃未満で完全に留去した。得られた粗生成物は、淡黄色の半固体であった。
【0047】
化合物(V)の収量:280g(66.6%)
【0048】
ステップ(d)(ii):脱保護された5−スルホンアミド(Va)の調製
アセトニトリル(5vol)中における化合物(V)(280g、0.700モル)の溶液に、2NのHCl(5vol)を添加した。混合物を75〜80℃で4.0時間還流させた。反応をTLC[TLC移動相酢酸エチル:ヘキサン、1:1]によってモニタした。
【0049】
反応後、真空下でアセトニトリルを完全に除去し、反応混合物を飽和炭酸水素ナトリウム溶液でpH8.0に塩基性化した。水層を酢酸エチル(3×3.0vol)で抽出し、硫酸ナトリウム上で乾燥させ、濾過した。有機層を減圧下において50℃未満で完全に留去した。得られた粗生成物は、淡黄色のシロップ状液体であった。
【0050】
化合物(Va)の収量:210g(84.2%)
【0051】
工程(e):ブロモ化合物(VI)の調製
化合物(Va)(210g、0.588モル)を、酢酸エチル(10vol)中に懸濁させ、撹拌した。淡黄色の懸濁液を30分間で0〜5℃に冷却し、過臭化臭化ピリジニウム(188g、0.588mol)を一部添加した。上記の懸濁液に、硫酸(0.5vol)を15〜25分に亘って滴下漏斗を介して加え、温度を5℃まで上昇させた。硫酸の添加後、反応混合物を徐々に25〜30℃にし、この温度で1時間撹拌した。反応をTLC[移動相酢酸エチル:ヘキサン、3:7]によってモニタした。
【0052】
反応の完了後、反応混合物に氷冷した水を添加した。有機層を分離し、水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、濾過し、溶媒を真空(50℃未満)中において除去して、粗生成物を黄色のシロップとして生じた。
【0053】
化合物(VI)の収量:210g(82.00%)
【0054】
ステップ(f):環化生成物(VII)の調製
立体選択的還元によって化合物(VII)の所望の異性体を得るために(+)−ジイソピノカンフェニルボラン(DIPCl)等のキラル還元剤を用いて化合物(VI)の環化を実施することができる。
【0055】
化合物(VI)(210g、0.484モル)をt−ブチルメチルエーテル(15vol)中で懸濁させ、窒素雰囲気下で撹拌した。撹拌懸濁液を−40℃まで冷却し、(+)−ジイソピノカンフェニルボラン(DIPCl)(762g(ヘキサン溶液中において60〜65%)、1.063モル)を30分の期間に亘ってカニューレを介して添加した。温度の上昇(−30℃まで)を観察した。反応混合物を−25〜−20℃で3.0〜4.0時間維持し、TLC[移動相酢酸エチル:ヘキサン、3:7]によってモニタした。
【0056】
還元の完了後、1Mの水酸化ナトリウム水溶液(20vol)を15〜20分に亘って滴下漏斗から加えた。反応混合物を、25〜30℃で終夜撹拌し、完了についてTLC[移動相酢酸エチル:ヘキサン、1:1]によってモニタした。完了後、水相及び有機相を分離し、水相をt−ブチルメチルエーテル(4.0vol)で抽出し、5N塩酸溶液を用いてpH1に酸性化し、酢酸エチル(3×6.0vol)で抽出した。組み合わせた酢酸エチル抽出物を飽和ブライン溶液(2×2.5vol)で洗浄し、硫酸ナトリウム上で乾燥させ、濾過した。有機層を減圧下において(50℃未満で)完全に留去した。得られた粗生成物は、淡黄色のシロップであった。
【0057】
化合物(VII)の収量:112g(65.2%)
【0058】
別の場合、ナトリウムテトラヒドロボラン等の非キラル還元剤を用いて工程(f)を実施することもできる。この場合、後続の光学分割工程、即ち、望ましくない異性体の除去を実施しなければならない。
【0059】
工程(g):粗ブリンゾラミド(VIII)の調製
化合物(VII)(112g、0.313mol)をテトラヒドロフラン(10vol)中に懸濁させた。メタンスルホン酸無水物(65.7g、0.374モル)を1ロット添加し、撹拌した。反応混合物を0〜5℃に冷却し、ピリジン(2.5vol)を45分に亘ってゆっくりと添加した。完了についてTLC[移動相ジクロロメタン:メタノール、9.5:0.5]によって反応をモニタした。
【0060】
反応生成物として、中間体化合物(VIIa)を得た。
【0061】
上記の反応混合物に、70%のエチルアミン水溶液(400ml、10vol)を0〜5℃でゆっくりと添加した。完全に添加した後、反応混合物を25〜30℃で12.0時間撹拌した。完了についてTLC[移動相ジクロロメタン:メタノール、9.5:0.5]によって反応をモニタした。有機層(エチルアミン及びテトラヒドロフラン混合物)を減圧下において50℃未満で完全に留去した。水相を濃塩酸で酸性化し、pHを15〜25℃で3.0に調整した。水相をt−ブチルメチルエーテルで2回(2×2.0vol)抽出して、存在する場合あらゆる有機不純物を除去した。t−ブチルメチルエーテル層を1NのHCl溶液で抽出し、層を分離した。
【0062】
水層を組み合わせ、活性炭で処理し、65〜70℃で加熱した。含有物を65〜70℃で30分間撹拌し、65〜70℃でhyflo supercelを通して濾過し、予熱した水(65〜70℃)で洗浄した。濾液を25〜30℃に冷却し、pHを固体の重炭酸ナトリウムで8に調整した。反応混合物を結晶化のために12時間撹拌した。結晶化が起きなかった場合、反応塊をブフナー漏斗を通して濾過し、酢酸エチル(3×5.0vol)で抽出し、硫酸ナトリウム上で乾燥させ、濾過した。有機層を完全に減圧下において(50℃未満で)留去した。粗生成物を淡黄色のシロップとして得て、それをヘキサン(4×1.0vol)で洗浄し、静置して固体に結晶化した残渣を生じた。
【0063】
粗化合物(VIII)の収量:80g(66.3%)
【0064】
工程(h):純粋なブリンゾラミド(VIII)の調製
粗化合物(VIII)(80.0g、0.208モル)を水(5.0vol)中に懸濁させ、65〜70℃に1時間加熱し、(熱間)濾過し、30分間吸引して乾燥させた。そのプロセスを、他の異性体含有率が0.50%NMTになるまでキラルによって繰り返した。
【0065】
このようにして得られた生成物をイソプロピルアルコール(8.0vol)中に懸濁させ、65〜70℃に加熱し、30分間維持し、熱条件下で濾過した。暗色の溶液に活性炭素(10.0質量%)を添加し、65〜70℃で30分間加熱した。溶液を熱条件下で濾過し、(65〜70℃で予熱した)イソプロピルアルコールで洗浄した。溶液を別のフラスコに移し、25〜30℃で1時間撹拌した。結晶質固体沈殿物を濾過し、30分間吸引して乾燥させ、恒量が得られるまで65〜70℃で真空下において乾燥させた。
【0066】
純粋化合物(VIII)の収量:60g(86%)
【0067】
実施例2:
ブリンゾラミドの調製(方法の変形2)
工程(a):ナトリウム塩(II)の調製
3−アセチル−2,5−ジクロロチオフェン(I)(500g、2.56mol)をエタノール(2.50L)中に懸濁させた。(水中に亜硫酸ナトリウム(即ち、5.0Lの水中に1.61Kg、12.8モル)を溶解させることによって得られた)亜硫酸ナトリウム溶液を25〜30℃でゆっくりと添加し、100〜110℃で還流まで加熱した。反応混合物を100〜110℃で16時間還流させた。反応の完了を、HPLC[(3−アセチル−2,5−ジクロロチオフェンのHPLC限界:1.0%NMT);実測:0.1%の3−アセチル−2,5−ジクロロチオフェン]によってモニタした。
【0068】
反応混合物を25〜30℃に冷却し、濾過し、無水メタノール(1.0L)で洗浄した。濾液を減圧下において(60〜65℃で)濃縮した。得られた粗生成物を無水メタノール(6.0L)中で懸濁させ、25〜30℃で30分間撹拌した。未溶解の固体を濾過し、濾液を完全に減圧下において60〜65℃で濃縮した。材料を、恒量が得られるまで60〜65℃で真空下において乾燥させた。
【0069】
収量:640gの化合物(II)(95.5%)
【0070】
工程(b):N−置換スルホンアミド(III)の調製
ジクロロメタン(6.50L;含水率0.5%NMT)中における化合物(II)(640g、2.44mol)の撹拌懸濁液に、五塩化リン(PCl5)溶液(12.80Lのジクロロメタン中において1.575Kg)を、10〜20℃で1〜2時間の間隔でゆっくりと添加した。反応を、温度を保持しながらN2下で4.0時間撹拌した。反応の完了を、HPLC分析[ナトリウム塩のHPLC限界:1.0%NMT]によってモニタした。
【0071】
冷却した反応混合物を濾過して(未溶解の塩を除去し)、濾液を飽和炭酸水素ナトリウム溶液(5.0L)で急冷して(水層のpHが7になるまで過剰なPCl5痕跡を除去し)、水で洗浄した。有機層を分離し、硫酸ナトリウム上で乾燥させ、濾過した。撹拌された有機層にメトキシプロピルアミン(435g、4.88mol)をゆっくりと加え、反応混合物を5〜10℃で2.0時間撹拌した。反応の完了をHPLC[(3−アセチル−5−クロロチオフェン−2−スルホニルクロリドについての限界:1.0%NMT);実測:0.1%の3−アセチル−5−クロロチオフェン−2−スルホニルクロリド]によってモニタした。反応を1NのHCl溶液(2vol)及び水で洗浄した。有機層を分離し、硫酸ナトリウム上で乾燥させ、濾過し、濃縮して、生成物を生じた。得られた生成物をヘキサン(2.0L)で洗浄し、恒量が得られるまで45〜50℃で乾燥させた。
【0072】
化合物(III)の収量:350g(46.2%)
【0073】
ステップ(c):還元スルホンアミド(IV)の調製
無水エタノール(15vol)中における化合物(III)(25g、0.080mol)の溶液に、水素化ホウ素ナトリウム(1.2g、0.032モル)を5〜10℃で添加した。反応混合物を5〜10℃で45分〜1時間撹拌した。反応の完了を、TLC[(N−置換スルホンアミドのTLC限界:1.0%NMT;移動相酢酸エチル:ヘキサン、3:7における)]によってモニタした。
【0074】
反応の完了後、エタノールを除去し、残渣をジクロロメタン(5vol)中に取った。ジクロロメタン溶液を水及びブライン(3vol)で洗浄した。有機層を分離し、硫酸ナトリウム上で乾燥させ、濾過し、減圧下において50℃未満で留去した。粗生成物を淡黄色のシロップとして得た。
【0075】
化合物(IV)の収量:21.0g(83.0%)
【0076】
ステップ(d)(i):還元5−スルホンアミド(V)の調製
無水テトラヒドロフラン(15vol)中における化合物(IV)(20g、0.063mol)の溶液を、N2下でドライアイス/2−プロパノールを用いて−40℃まで冷却した。n−ブチルリチウム(79ml、0.190mol)を、添加中に温度を−20℃未満で保持しながら30分〜1時間に亘って滴下した。反応混合物を、−40℃未満で30分〜1時間維持した。反応混合物中に二酸化硫黄(SO2)を、水中に急冷したアリコートがpH3.0〜4.0を示すまで導入した。混合物を25〜30℃で15時間加温し、次いで濃縮した。
【0077】
残渣を水(100ml)中に溶解し、この溶液に、(0℃で)水(0.3L)中における酢酸ナトリウム三水和物(31.5g、0.38mol)及びヒドロキシルアミン−O−スルホン酸(29g、0.252mol)の溶液を一部添加し、温度を25℃まで上昇させた。25〜30℃で15時間撹拌した後、溶液を重炭酸ナトリウムで中和し、次いで、pH8〜9が達成されるまで50%の水酸化ナトリウム溶液で塩基性化した。溶液を酢酸エチルで抽出し、ブラインで洗浄し、硫酸ナトリウムで乾燥させ、濾過し、次いで濃縮した。有機層を減圧下において50℃未満で完全に留去した。粗生成物を淡黄色のシロップとして得て、それをDCM/ヘキサンで結晶化して、淡黄色の固体として化合物を生じた。
【0078】
化合物(V)の収量:16.06g(70%)
【0079】
ステップ(d)(ii):5−スルホンアミド(Va)の調製
ジクロロメタン(15vol)中における化合物(V)(10g、0.028mol)の懸濁液に、デス−マーチンペルヨージナン(5volのジクロロメタン中における11.80g、0.028mol)の懸濁液を、10〜20℃で1.0〜2.0時間ゆっくりと添加した。反応をTLC[還元5−スルホンアミドのTLC限界:1.0%NMT;移動相ジクロロメタン:メタノール、9.5:0.5における)]によってモニタした。
【0080】
反応の完了後、反応混合物を精製水(20vol)で急冷し、45分〜1.0時間撹拌した。有機層を分離し、飽和炭酸水素ナトリウム溶液(2×5.0vol)及びチオ硫酸ナトリウム溶液(10%)で洗浄し、硫酸ナトリウム上で乾燥させ、濾過し、減圧下において50℃未満で完全に留去した。得られた粗生成物は、淡黄色のシロップであった。粗生成物は、ジクロロメタン及びヘキサンから結晶化して、淡黄色/薄茶色の結晶質固体として純粋化合物を生じた。
【0081】
化合物(Va)の収量:6g(65%)
【0082】
更なる工程を、実施例1に記載したように実施した。
【0083】
実施例3:
ブリンゾラミドの光学分割のための方法
粗ブリンゾラミド(0.065mol)を5.0%の水性メタノール(4.0vol)中に溶解し、50℃で1時間加熱した。同時に、メタノール(1.0vol)中に溶解した溶液ジ−p−トリル−D−酒石酸(DPTA)を調製して、25〜30℃で20〜30分間撹拌した。DPTA溶液を、撹拌しながら50℃で粗ブリンゾラミドを含有する溶液にゆっくりと添加した。混合物を50℃で1.0時間維持した。1時間後、溶液をゆっくりと25〜30℃にし、25〜30℃で12時間連続的に撹拌した。沈殿する固体を濾過し、冷たいメタノール(1.0vol)で洗浄し、乾燥させて酒石酸塩を得た。
【0084】
酒石酸塩を飽和炭酸水素ナトリウム溶液(60vol)と混合し、得られた懸濁液を1.0時間撹拌し、次いで酢酸エチル(3×60vol)で抽出した。抽出物を硫酸ナトリウム上で乾燥させ、濾過し、乾燥状態に蒸発させて、ブリンゾラミドの遊離塩基を得た。遊離塩基を、キラルHPLC(限界NMT:0.50%(S−異性体))によってS−異性体含有率について確認した。
【0085】
望ましくないS−異性体の含有率が0.50%より高い場合、以下に記載される通りのイソプロパノールからの水精製プロセス及び/又は再結晶を実施して、望ましくない異性体を減少させることができる。
【0086】
実施例4:
a)水精製プロセス
ブリンゾラミド塩基を精製水(10vol)中に懸濁させ、65〜70℃に45分〜1.0時間加熱した。混合物を50℃まで冷却し、含有物を熱条件下で濾過し、精製水(2.0vol)で洗浄し、1.0時間吸引して乾燥させた。そのプロセスを、0.50%NMTのS−異性体含有率の限界が得られるまで(例えば、4〜5回)繰り返すことができる。繰り返された水精製によって、最高10.00〜20.00%のS−異性体含有率を0.50%NMTの限界に減少させることができる。
【0087】
b)イソプロパノールからの再結晶
粗ブリンゾラミド(80.0g、0.208mol)をイソプロピルアルコール(8.0vol)中に懸濁させ、65〜70℃に加熱し、30分間維持し、熱条件下で濾過した。暗色の溶液に、活性炭素(10.0質量%)を添加し、65〜70℃で30分間加熱した。溶液を熱条件下で濾過し、(65〜70℃で予熱した)イソプロピルアルコールで洗浄した。溶液を別のフラスコに移し、25〜30℃で1時間撹拌した。結晶質固体が沈殿した。沈澱物を濾過し、30分間吸引して乾燥させ、恒量が得られるまで65〜70℃で真空下において乾燥させた。そのプロセスを、生成物が0.50%以下の望ましくない異性体を含むまで繰り返すことができる。
【0088】
収量:60gの純粋なブリンゾラミド(VIII)(86%)
【特許請求の範囲】
【請求項1】
(a)化合物(I)
【化1】
[式中、Ac(P)は、アセチル基又はマスクされた、例えば保護若しくは還元されたアセチル基である]と、スルホン化試薬、特に亜硫酸塩とを反応させて、化合物(II)
【化2】
[式中、Ac(P)は、上記の通りであり、Mは、カチオン、特にNa+である]を得る工程、
(b)化合物(II)と3−メトキシアミノプロパンとを反応させて、化合物(III)
【化3】
[式中、Ac(P)は、上記の通りである]を得る工程、
(c)場合によって、化合物(III)と、アセチル保護剤、特にエチレングリコールとを反応させて、化合物(IV)
【化4】
[式中、AcPは、マスクされた、例えば保護又は還元されたアセチル基、特に、
【化5】
である]を得る工程、
(d)(i)化合物(IV)と有機金属化合物とを反応させた後、SO2で処理し、続いてヒドロキシルアミン−O−スルホン酸で処理して、化合物(V)
【化6】
[式中、AcPは、上記の通りである]を得る工程、
(d)(ii)例えば化合物(V)を脱保護又は酸化させることによってアセチル基を再構成して、化合物(Va):
【化7】
[式中、Acは、アセチル基である]を得る工程、
(e)化合物(Va)と臭素化剤、特に過臭化臭化ピリジニウム(PBP)とを反応させて、化合物(VI)
【化8】
を得る工程、
(f)アルカリ性条件下で、化合物(VI)と、還元剤、特にキラル還元剤、より具体的には(+)−ジイソピノカンフェニルクロロボラン(DIPCl)とを反応させて、化合物(VII)
【化9】
を得る工程、
(g)(i)化合物(VII)を中間体(VIIa)、
【化10】
[式中、Xは、脱離基、特にスルホネート基である]に転化する工程、及び
(g)(ii)化合物(VIIa)とエチルアミンとを反応させて、化合物(VIII)
【化11】
を得る工程、
(h)場合によって、化合物(VIII)を精製して、望ましくない異性体を他の不純物と共に除去する工程
を含む、ブリンゾラミドの製造方法。
【請求項2】
構造式(II)
【化12】
[式中、Ac(P)は、アセチル基又はマスクされた、例えば保護若しくは還元されたアセチル基であり、Mは、金属カチオン、特にNa+である]を有する化合物。
【請求項3】
構造式(V)
【化13】
[式中、AcPは、マスクされた、例えば保護若しくは還元されたアセチル基、特に
【化14】
である]を有する化合物。
【請求項4】
構造式(VI)
【化15】
を有する化合物。
【請求項5】
構造式(VII)
【化16】
を有する化合物。
【請求項6】
請求項2から5までのいずれか一項に記載の化合物を、ブリンゾラミドの製造のために用いる使用。
【請求項7】
請求項2から5までのいずれか一項に記載の少なくとも1種の化合物が、中間体として得られる、ブリンゾラミドの製造方法。
【請求項8】
粗ブリンゾラミドとキラル酒石酸とを含む溶液を形成すること、所望のブリンゾラミドタルトレートを溶液から沈殿させること、及び精製されたブリンゾラミド生成物を回収することを含む、不純物から、特にS−鏡像異性体からブリンゾラミドを精製するための方法。
【請求項9】
キラル酒石酸が、ジ−p−トリル−D−酒石酸(DTPA)である、請求項8に記載の方法。
【請求項10】
精製されたブリンゾラミド生成物中における望ましくないS−鏡像異性体の含有率が、0.50質量%以下である、請求項8又は9に記載の方法。
【請求項11】
キラル酒石酸によるブリンゾラミドの塩、特にDTPAによるブリンゾラミドの塩。
【請求項12】
高温の水で粗ブリンゾラミドを処理すること、及び精製されたブリンゾラミド生成物を回収することを含む、不純物から、特にS−鏡像異性体からブリンゾラミドを精製するための方法。
【請求項13】
イソプロピルアルコール中における粗ブリンゾラミドの再結晶、及び精製されたブリンゾラミド生成物の回収を含む、不純物から、特にS−鏡像異性体からブリンゾラミドを精製するための方法。
【請求項14】
精製されたブリンゾラミド生成物中における望ましくないS−鏡像異性体の含有率が、0.50質量%以下である、請求項12又は13に記載の方法。
【請求項1】
(a)化合物(I)
【化1】
[式中、Ac(P)は、アセチル基又はマスクされた、例えば保護若しくは還元されたアセチル基である]と、スルホン化試薬、特に亜硫酸塩とを反応させて、化合物(II)
【化2】
[式中、Ac(P)は、上記の通りであり、Mは、カチオン、特にNa+である]を得る工程、
(b)化合物(II)と3−メトキシアミノプロパンとを反応させて、化合物(III)
【化3】
[式中、Ac(P)は、上記の通りである]を得る工程、
(c)場合によって、化合物(III)と、アセチル保護剤、特にエチレングリコールとを反応させて、化合物(IV)
【化4】
[式中、AcPは、マスクされた、例えば保護又は還元されたアセチル基、特に、
【化5】
である]を得る工程、
(d)(i)化合物(IV)と有機金属化合物とを反応させた後、SO2で処理し、続いてヒドロキシルアミン−O−スルホン酸で処理して、化合物(V)
【化6】
[式中、AcPは、上記の通りである]を得る工程、
(d)(ii)例えば化合物(V)を脱保護又は酸化させることによってアセチル基を再構成して、化合物(Va):
【化7】
[式中、Acは、アセチル基である]を得る工程、
(e)化合物(Va)と臭素化剤、特に過臭化臭化ピリジニウム(PBP)とを反応させて、化合物(VI)
【化8】
を得る工程、
(f)アルカリ性条件下で、化合物(VI)と、還元剤、特にキラル還元剤、より具体的には(+)−ジイソピノカンフェニルクロロボラン(DIPCl)とを反応させて、化合物(VII)
【化9】
を得る工程、
(g)(i)化合物(VII)を中間体(VIIa)、
【化10】
[式中、Xは、脱離基、特にスルホネート基である]に転化する工程、及び
(g)(ii)化合物(VIIa)とエチルアミンとを反応させて、化合物(VIII)
【化11】
を得る工程、
(h)場合によって、化合物(VIII)を精製して、望ましくない異性体を他の不純物と共に除去する工程
を含む、ブリンゾラミドの製造方法。
【請求項2】
構造式(II)
【化12】
[式中、Ac(P)は、アセチル基又はマスクされた、例えば保護若しくは還元されたアセチル基であり、Mは、金属カチオン、特にNa+である]を有する化合物。
【請求項3】
構造式(V)
【化13】
[式中、AcPは、マスクされた、例えば保護若しくは還元されたアセチル基、特に
【化14】
である]を有する化合物。
【請求項4】
構造式(VI)
【化15】
を有する化合物。
【請求項5】
構造式(VII)
【化16】
を有する化合物。
【請求項6】
請求項2から5までのいずれか一項に記載の化合物を、ブリンゾラミドの製造のために用いる使用。
【請求項7】
請求項2から5までのいずれか一項に記載の少なくとも1種の化合物が、中間体として得られる、ブリンゾラミドの製造方法。
【請求項8】
粗ブリンゾラミドとキラル酒石酸とを含む溶液を形成すること、所望のブリンゾラミドタルトレートを溶液から沈殿させること、及び精製されたブリンゾラミド生成物を回収することを含む、不純物から、特にS−鏡像異性体からブリンゾラミドを精製するための方法。
【請求項9】
キラル酒石酸が、ジ−p−トリル−D−酒石酸(DTPA)である、請求項8に記載の方法。
【請求項10】
精製されたブリンゾラミド生成物中における望ましくないS−鏡像異性体の含有率が、0.50質量%以下である、請求項8又は9に記載の方法。
【請求項11】
キラル酒石酸によるブリンゾラミドの塩、特にDTPAによるブリンゾラミドの塩。
【請求項12】
高温の水で粗ブリンゾラミドを処理すること、及び精製されたブリンゾラミド生成物を回収することを含む、不純物から、特にS−鏡像異性体からブリンゾラミドを精製するための方法。
【請求項13】
イソプロピルアルコール中における粗ブリンゾラミドの再結晶、及び精製されたブリンゾラミド生成物の回収を含む、不純物から、特にS−鏡像異性体からブリンゾラミドを精製するための方法。
【請求項14】
精製されたブリンゾラミド生成物中における望ましくないS−鏡像異性体の含有率が、0.50質量%以下である、請求項12又は13に記載の方法。
【図1】

【図2】
【図3】

【図2】
【図3】
【公表番号】特表2012−520266(P2012−520266A)
【公表日】平成24年9月6日(2012.9.6)
【国際特許分類】
【出願番号】特願2011−553471(P2011−553471)
【出願日】平成22年3月12日(2010.3.12)
【国際出願番号】PCT/EP2010/053216
【国際公開番号】WO2010/103115
【国際公開日】平成22年9月16日(2010.9.16)
【出願人】(511223202)アザド ファーマスーティカル イングリーディエンツ アクチエンゲゼルシャフト (1)
【氏名又は名称原語表記】AZAD Pharmaceutical Ingredients AG
【住所又は居所原語表記】Friedbergstrasse 68, CH−8200 Schaffhausen, Switzerland
【Fターム(参考)】
【公表日】平成24年9月6日(2012.9.6)
【国際特許分類】
【出願日】平成22年3月12日(2010.3.12)
【国際出願番号】PCT/EP2010/053216
【国際公開番号】WO2010/103115
【国際公開日】平成22年9月16日(2010.9.16)
【出願人】(511223202)アザド ファーマスーティカル イングリーディエンツ アクチエンゲゼルシャフト (1)
【氏名又は名称原語表記】AZAD Pharmaceutical Ingredients AG
【住所又は居所原語表記】Friedbergstrasse 68, CH−8200 Schaffhausen, Switzerland
【Fターム(参考)】
[ Back to top ]