
ブレビバチルス属細菌を用いたプロテインA様蛋白質の生産方法
【課題】本発明は、プロテインA様蛋白質の生産のための効率的で経済的な方法に関する。
【解決手段】プロテインA様蛋白質の遺伝子組換え技術を用いた生産は、大腸菌、枯草菌などの宿主が使用されているが生産性の低さにより高価格の大きな原因となっている。従って大腸菌や枯草菌以外の組換えDNA技術を用いたプロテインA様蛋白質の安価で大量な生産を可能にする技術の早急な確立が強く望まれている。本発明は、プロテインA様蛋白質の大量生産方法であり、組換えブレビバチルス属細菌により該蛋白質を培養液中へ大量に分泌発現させ、培養液中からその蓄積された該プロテインA様蛋白質を分離回収することからなる方法などを提供する。
【解決手段】プロテインA様蛋白質の遺伝子組換え技術を用いた生産は、大腸菌、枯草菌などの宿主が使用されているが生産性の低さにより高価格の大きな原因となっている。従って大腸菌や枯草菌以外の組換えDNA技術を用いたプロテインA様蛋白質の安価で大量な生産を可能にする技術の早急な確立が強く望まれている。本発明は、プロテインA様蛋白質の大量生産方法であり、組換えブレビバチルス属細菌により該蛋白質を培養液中へ大量に分泌発現させ、培養液中からその蓄積された該プロテインA様蛋白質を分離回収することからなる方法などを提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ブレビバチルス属細菌を使用した、イムノグロブリン結合能を有するプロテインA様蛋白質の製造方法に関する。詳細には、本発明は、遺伝子組換え技術により、ブレビバチルス属細菌にプロテインA様蛋白質を多量に分泌発現させること、その発現されたプロテインA様蛋白質を、プロテアーゼ等による分解を受けることなく高純度で分離回収すること、及び、その分離回収されたプロテインA様蛋白質を、抗体精製用のカラム樹脂等の用途へ有効に使用することに関する。
【0002】
日本国特許出願2004−198831号(2004年7月6日出願)の明細書、請求の範囲、図面および要約を含む全開示内容は、これら全開示内容を参照することによって本出願に合体される。
【背景技術】
【0003】
抗体(イムノグロブリン、またはIgとも呼ばれる)蛋白質は生体における有害な抗原の補足及び排除機能を有するため古くから医薬品として利用されてきた。近年では遺伝子工学技術や細胞融合技術の進歩により特異的な抗原に反応する抗体を分子設計し、動物細胞内で発現させることにより均一で高い抗原性を有するモノクローナル抗体の作製が可能となった。これらの抗体蛋白質は細胞培養液中に分泌されることから、比較的容易に分離精製回収することができる。
【0004】
一般的に、イムノアッセイやイムノブロット解析に利用される抗体蛋白質の場合、通常の蛋白質精製において利用される方法、すなわち硫酸アンモニウムによる沈殿法やイオン交換クロマトグラフィー等を用いることによって、血清、腹水または細胞培養液のような天然生体サンプルから、十分な収量や純度で得ることができる。
【0005】
一方、高純度を必要とする医薬品または診断薬等に利用される抗体蛋白質の場合、上記のような方法を用いて分離精製するには、様々な分離抽出条件の検討や数多くの他のクロマトグラフィーとの併用を必要とし、また、それぞれの抗体蛋白質に関して精製条件の最適化が必要となり、非常な労力を要する。従って、高純度を必要とする抗体蛋白質の精製の場合、他の夾雑物質から簡便に分離精製するために、抗体蛋白質を特異的に吸着することが可能なアフィニティークロマトグラフィーを用いるのが一般的である。
【0006】
抗体結合能を有するアフィニティークロマトグラフィーとして最も良く利用されているのが、プロテインA、プロテインG及びプロテインLなどの蛋白質を適当な樹脂に固定化した担体によるクロマトグラフィーである。これらの蛋白質の中で特に精製用担体のリガンドとしてよく利用されているのがプロテインAである。プロテインAはグラム陽性細菌スタフィロコッカス・アウレウス(Staphylococcusaureus)によって生産される細胞壁蛋白質の1種であり、約42,000の分子量であることが明らかとなっている。その構造は7つの機能ドメイン(アミノ末端からシグナル配列S、イムノグロブリン結合ドメインE、イムノグロブリン結合ドメインD,イムノグロブリン結合ドメインA、イムノグロブリン結合ドメインB、イムノグロブリン結合ドメインC、スタフィロコッカス・アウレウス細菌細胞壁結合ドメインX)(非特許文献1、2、3参照)から構成されている。プロテインAの5つのイムノグロブリン結合ドメイン(ドメインE、D、A、B、C)は、それぞれイムノグロブリン分子内のFc部分で結合することができる(非特許文献3参照)。
【0007】
このプロテインAのイムノグロブリン結合ドメインに対する相対親和性は、pH、スタフィロコッカス・アウレウス菌株種(Cheung, A.ら Infec. Immun. 1987. 55:843-847)、またイムノグロブリンのクラス(IgG,IgM,IgA,IgD,IgE)及びサブクラス(IgG1,IgG2,IgG3,IgG4,IgA1,IgA2)などの多くの因子に依存することが知られ、特にイムノグロブリンのクラスではヒトIgG1、IgG2、IgG4及びマウスIgG2a、IgG2b,IgG3のFc部分と強い結合を示す。以上のような性質を持つプロテインAは、イムノグロブリンとしての抗原結合能力、親和力や性質を損なうことなくイムノグロブリンに結合することができるため、様々な診断、医薬品及び基礎研究に用いられるイムノグロブリン、特にIgGに対し精製用担体のリガンドとして広く使用されている。
【0008】
また最近では、感作末梢血リンパ球の腫瘍細胞毒性作用を阻害する血清阻止因子(特異抗原、抗体、抗グロブリン及び免疫複合体で構成される)を、プロテインAに吸着させ腫瘍患者の血清から除去するといった、癌治療への応用にも興味が持たれている(特許文献1−3参照)。さらにIgG結合活性とは別にプロテインAは多クローン抗体合成を活性化する働きも有していることから、当初の精製樹脂リガンド用途だけでなく様々なバイオテクノロジー分野における使用用途が期待されている。
【0009】
初期のプロテインAの生産方法においてはスタフィロコッカス・アウレウス株の培養液から直接、分離精製を行ってきたが、この菌自身の病原性の問題や不純物の混入の問題により、現在では大腸菌(Escherichia coli)(特許文献1−3)やグラム陽性細菌である枯草菌(Bacillus subtilis)(特許文献4−5)を用いた組換えDNA技術を用いた生産方法へ移行している。ところが、大腸菌における組換え型プロテインAの生産性は極めて低く、また、発現蛋白質の多くがインクルージョンボディーを形成したり、細胞内で分解されるため、分離回収が容易ではない(非特許文献4)。一方、スタフィロコッカス・アウレウスと同様にグラム陽性細菌である枯草菌を用いたプロテインAの生産では、プロテインAのN末端へ枯草菌分泌蛋白質のシグナル配列を付加することで培地中へ分泌発現させる方法が取られており、大腸菌での生産系と比較した場合、分離精製等が容易であり、生産性も高いことが報告されている(約47−100mg/L)(Fahnestock, S, R.ら J. Bacteriol. 1986. 165:796-804)。ところが、枯草菌において生産されたプロテインAは、枯草菌が本来持つ菌体外プロテアーゼによる分解を受ける。そのため、枯草菌の数種の菌体外プロテアーゼ欠損株(非特許文献5)を宿主として用いることも試みられてきたが、プロテインAの分解を抑えることは未だ達成できていない。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】日本国特願平07−187019号公報
【特許文献2】米国特許番号US5151350号公報
【特許文献3】欧州特許番号EP0107509号公報
【特許文献4】米国特許番号US4617266号公報
【特許文献5】欧州特許番号EP0124374号公報
【非特許文献】
【0011】
【非特許文献1】Lofdahl, Sら.Proc. Natl. Acad. Sci. USA. 1983. 80:697-701.
【非特許文献2】Shuttleworth, H. Lら.Gene. 1987. 58:283-295.
【非特許文献3】Uhlen, M.ら.J. Bio. Chem. 1984. 259:1695-1702.
【非特許文献4】Nilsson, Bら. Protein Eng. 1987. 1:107-113.
【非特許文献5】Fahnestock, S. Rら. Appl.Envron. Microbiol. 1987. 53:379-384.
【非特許文献6】Brigido, Mら, J. Basic Microbiology. 1991. 31: 337-345.
【非特許文献7】Sjostrom, J, -Eら. J. bacteriol. 1975. 123:905-915.
【非特許文献8】Bjorck, L.ら. 1984. J. Immunol. 133, 969-974.
【非特許文献9】Kastern, W.ら. J Biol Chem. 1992. 267:12820-12825
【非特許文献10】Udaka, S.ら. Method Enzymol. 1993. 217:23-33.
【発明の概要】
【発明が解決しようとする課題】
【0012】
このような背景から、大腸菌や枯草菌を用いた生産方法を上回る、効率の良いプロテインAの生産技術の確立が、強く望まれていた。
【0013】
本発明は、大腸菌や枯草菌を用いた生産方法を上回る、効率の良いプロテインAの生産技術の提供を一つの課題とする。
【課題を解決するための手段】
【0014】
本発明者は、プロテインAのような機能蛋白質を安定、大量に生産する技術を確立するため、ブレビバチルス属細菌を宿主に使い鋭意研究を行った結果、プロテインAを効率良く培養液中へ大量に分泌発現、そして安定に蓄積させ、容易に高純度で分離回収できることを見いだした。
【発明の効果】
【0015】
本発明に従えば、ブレビバチルス属細菌を宿主に用いることにより、大腸菌や枯草菌などの微生物を宿主とした場合に報告されている生産量を大幅に上回るプロテインAを培養液中へ分泌生産させ、イムノグロブリン結合機能を損なわずに容易に高純度にまで精製することができる。従って、本発明により、現在まで高価格の一因となっていたプロテインAの低生産性や、精製工程の複雑さが解消される。
【0016】
本発明は、以下の1または複数の対象を含む。
(1)本発明は、プロテインA様蛋白質またはその部分的配列をコードするDNA配列、およびその配列に作動可能に連結されたブレビバチルス属細菌で機能しうるプロモーターを含む、DNA配列を提供する。
(3)本発明は、前記DNA配列を含む、発現ベクターを提供する。
(4)本発明は、前記発現ベクターを含む、ブレビバチルス属細菌形質転換体を提供する。
(6)本発明は、前記形質転換体を培養すること、および該形質転換体によって分泌生産されるプロテインA様蛋白質、またはその部分的配列を有する蛋白質を回収することを含む、プロテインA様蛋白質またはその部分的配列を有する蛋白質の製造方法を提供する。
(7)本発明は、前記製造方法によってプロテインA様蛋白質またはその部分的配列を有する蛋白質を製造すること、および該プロテイン様蛋白質またはその部分的配列を有する蛋白質を適当な基材に固定化すること、を含む、イムノグロブリン吸着担体の製造方法を提供する。
【図面の簡単な説明】
【0017】
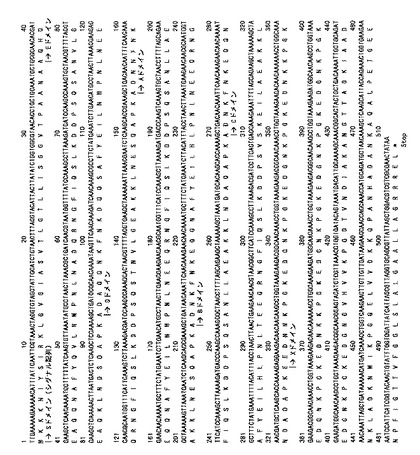
【図1】図1は、スタフィロコッカス・アウレウス・コワン(Cowan)I株のプロテインAの塩基配列とアミノ酸配列とを示す図である(番号はアミノ酸残基数を示す)。
【図2】図2は、スタフィロコッカス・アウレウス株のプロテインAの遺伝子配列とアミノ酸配列とを示す図である(番号はアミノ酸残基数を示す)。
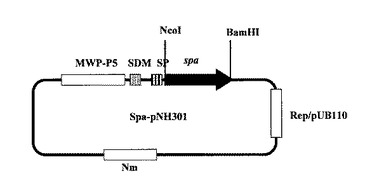
【図3】図3は、プロテインA(SPA)発現ベクター(Spa−pNH301)を示す図である。
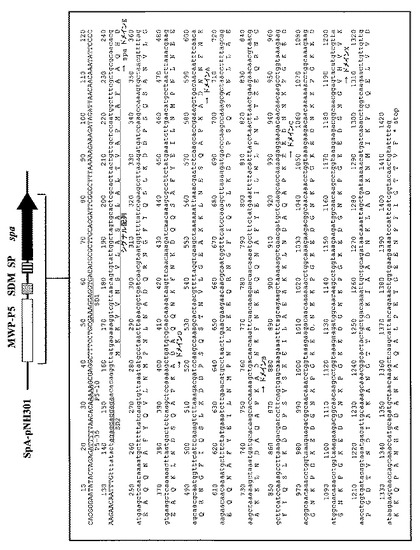
【図4】図4は、プロテインA(SPA)発現ベクター(Spa−pNH301)のプロモーター配列からプロテインA(SPA)をコードする領域までの塩基配列およびアミノ酸配列を示す図である。
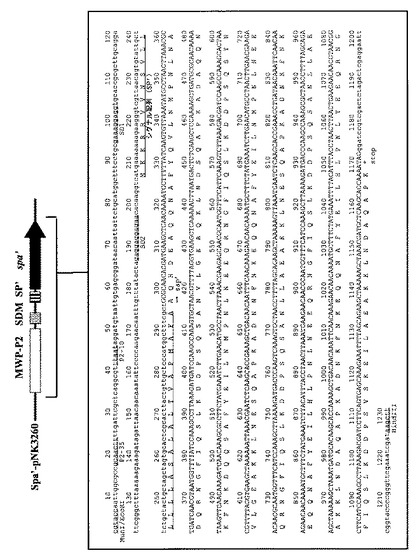
【図5】図5は、ブレビバチルス・チョウシネンシスHPD31−OK株により生産されたプロテインA(SPA)のSDS−PAGEによる解析結果を示す図である。
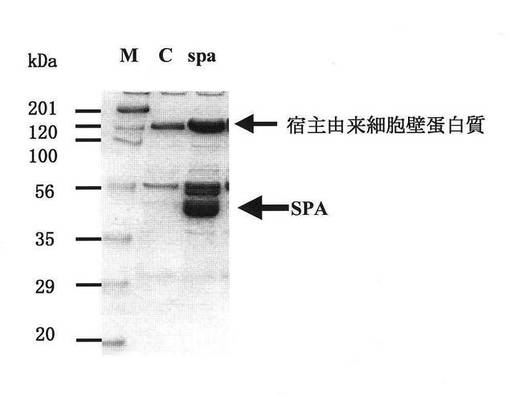
【図6】図6は、ブレビバチルス・チョウシネンシスHPD31−OK株により生産されたプロテインA(SPA)の、抗体結合試験の結果を示す図である。
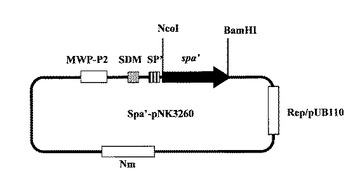
【図7】図7は、プロテインA(SPA’)発現ベクター(Spa’−pNK3260)を示す図である。
【図8】図8は、プロテインA(SPA’)発現ベクター(Spa’−pNK3260)のプロモーター配列、シャインダルガノ配列、シグナルペプチドおよびプロテインA(SPA’)をコードするDNA配列を示す図である。
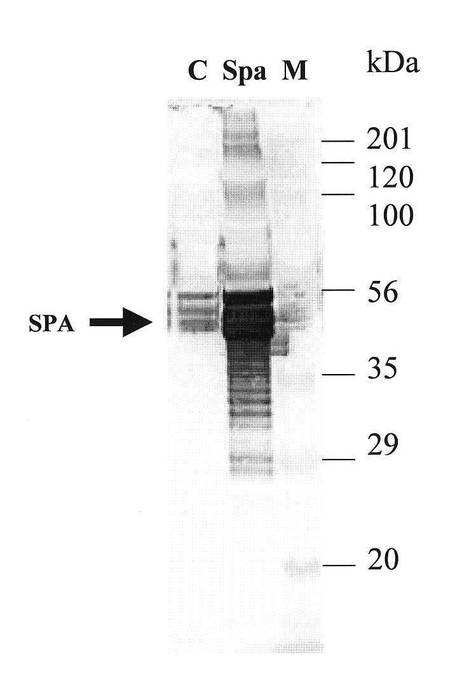
【図9】図9は、ブレビバチルス・チョウシネンシスHPD31−OK株により生産されたプロテインA(SPA’)の培養液中での挙動およびその蓄積量を、SDS−PAGEにより解析した結果を示す図である。
【発明を実施するための形態】
【0018】
本発明者らは、組換えDNA技術が利用できる細菌の中でも、グラム陽性細菌であるブレビバチルス属細菌を用いることで、活性型のプロテインAを大量に培養液中へ分泌発現させることができ、大腸菌及び枯草菌における低生産性の問題、並びに枯草菌における発現したプロテインAの分解に対する問題が効果的に改善されることを発見した。すなわち、ブレビバチルス属細菌を用いることで、枯草菌で得られる発現量を容易に確保し、さらにこれを培地中へ蓄積させることができる。以下、実施形態に基づいて本発明について詳しく説明する。
【0019】
1.プロテインA
プロテインAは、前述のように、グラム陽性細菌スタフィロコッカス・アウレウスによって生産される細胞壁蛋白質の1種であり、例えば、図1(配列番号2)にて示されるアミノ酸配列からなるスタフィロコッカス・アウレウス・コワン(Cowan)I株(JCM2179)由来のもの(非特許文献2)、図2(配列番号4)にて示されるアミノ酸配列からなるもの(非特許文献6、Finck-Barbancon,V.ら FEMS Microbiol. Lett. 1992. 91:1-8)、Woods46株由来のもの(非特許文献3)、8325−4株由来のもの(非特許文献3)、および既にクローニングされているプラスミドDNAすなわちpSP1やpSP3等(非特許文献7)にコードされているspa遺伝子産物等を指す。
【0020】
本発明にいうプロテインA様蛋白質には、プロテインA、またはプロテインAと実質的に同一の蛋白質が含まれる。また、プロテインA様蛋白質には、当業者にとって公知の配列比較アルゴリズムを使用して、プロテインAのアミノ酸配列と比較され、そして最大の一致のためにアラインメントされるときに、少なくとも60%、好ましくは80%、より好ましくは90−95%、そして最も好ましくは少なくとも99%のアミノ酸残基の同一性を有するアミノ酸配列を有し、かつ、イムノグロブリン結合活性を有する蛋白質が含まれる。ここで、同一性を有するアミノ酸配列は、好ましくは50残基長以上であり、より好ましくは100残基長以上であり、さらに好ましくは150残基長以上であり、最も好ましい実施態様では、完全長に渡り同一性を有する。
【0021】
パーセント配列同一性を決定するために好適であるアルゴリズムの例は、BLASTアルゴリズムであり、これはAltschul et al., J. Mol. Biol. 215: 403-410(1990)に記載されている。BLAST分析を実施するためのソフトウェアは、National Center for Biotechnology Information(http://www.ncbi.nlm.nih.gov/)を通して公衆が利用可能である。
【0022】
プロテインA様蛋白質は、例えば、配列番号1または配列番号3に示したDNA配列に対して相補的な配列を有するDNAに、ストリンジェント条件下でハイブリダイズするDNAによってコードされるアミノ酸配列からなる蛋白質であり得る。該ストリンジェント条件下のハイブリダイゼーション条件の例は:好ましくは約7%のドデシル硫酸ナトリウム(SDS)、約0.5MのNaPO4、1mMのEDTA中で約50℃でハイブリダイゼーション、および約2XSSC、約0.1%のSDS中で50℃の洗浄;より望ましくは約7%のドデシル硫酸ナトリウム(SDS)、約0.5MのNaPO4、約1mMのEDTAで50℃でハイブリダイゼーション、および約1XSSC、約0.1%のSDSで約50℃の洗浄;より望ましくは約7%のドデシル硫酸ナトリウム(SDS)、約0.5MのNaPO4、約1mMのEDTAで約50℃でハイブリダイゼーション、および約0.5XSSC、約0.1%のSDSで約50℃の洗浄;より好ましくは約7%のドデシル硫酸ナトリウム(SDS)、約0.5MのNaPO4、約1mMのEDTAで約50℃でハイブリダイゼーション、および約0.1XSSC、約0.1%のSDSで約50℃の洗浄;並びになおより好ましくは約7%のドデシル硫酸ナトリウム(SDS)、約0.5MのNaPO4、約1mMのEDTAで約50℃で、約0.1XSSC、約0.1%のSDSで約65℃の洗浄である。もっとも、該条件は、ヌクレオチド鎖の長さ、該配列、および異なる環境パラメーターに依存して異なり得る。より長い配列は、より高い温度で特異的にハイブリダイズする。核酸のハイブリダイゼーションの詳細なガイドは、例えばTijssen(1993) Laboratory Techniques in Biochemistry and Molecular Biology-Hybridization with Nucleic Acid Probes part I chapter 2''Overview of principles of hybridization and the starategy of nucleic acid probe assay''Elsvier, New Yorkに見出される。
【0023】
このように、図1(配列番号1および2)および図2(配列番号3および4)にて示されるプロテインAをコードするDNA配列、およびプロテインAのアミノ酸配列を知ることにより、「プロテインA様蛋白質」、およびそれをコードするDNA配列の存在を認識することは、遺伝子工学分野の技術者にとって容易である。
【0024】
「プロテインA様蛋白質」には、例えば、プロテインAを構成するイムノグロブリン結合ドメイン(E、D、A、B、C)を任意の順列に並べ替えたものも含まれる。
【0025】
さらに、「プロテインA様蛋白質」には、例えば、グループC及びGのストレプトコッカス属細菌(Streptococcal)の有するプロテインG(非特許文献8)、またはペプトストレプトコッカス・マグニウス(Peptostreptococcus magnus)のプロテインL(非特許文献9)といったプロテインAと同様なイムノグロブリン結合機能を有する蛋白質も含まれる。
【0026】
2.プロテインA様蛋白質の部分的配列
プロテインA様蛋白質の「部分的配列」とは、上記プロテインA様蛋白質を構成するアミノ酸配列の任意の一部分から構成されるものであって、かつ、イムノグロブリン結合活性を有する蛋白質を指し、具体的には、例えば、プロテインAから先述のシグナル配列Sおよび細胞壁結合ドメインXの一部を除いて得られる、配列番号8の24番目のAla以降で示されるアミノ酸配列(実施例1の「SPA」に対応。図3および4、配列番号7および8参照。)、および、プロテインAから先述のシグナル配列Sおよび細胞壁結合ドメインXの全部を除いて得られる、配列番号19の31番目のAla以降で示されるアミノ酸配列(実施例5の「SPA’」に対応。図7および8、配列番号18および19参照。)のそれぞれが、プロテインA様蛋白質の「部分的配列」に対応する。
【0027】
さらに上記プロテインGやLが有するイムノグロブリン結合ドメインを構成するアミノ酸配列も「部分的配列」として挙げることができる。
【0028】
本明細書にいうプロテインAのイムノグロブリン結合ドメインとは、例えば、図1における37アミノ酸残基から327アミノ酸残基位(ドメインEからC)、図2における37アミノ酸残基から355アミノ酸残基(ドメインEからC)のことを指す。
【0029】
3.プロテインA様蛋白質をコードするDNA配列
本発明に用いられる、プロテインA様蛋白質をコードするDNA配列は、そのDNA配列を翻訳したアミノ酸配列が、プロテインA様蛋白質を構成するものであればいずれでも良い。そのようなDNA配列は、通常用いられる公知の方法、例えば、ポリメラーゼ・チェーン・リアクション(以下、PCRと略す)法を利用して取得できる。また、公知の化学合成法で合成することも可能であり(Nucleic acids Res.1984. 12:4359)、さらに、DNAライブラリーから得ることもできる。当該DNA配列は、コドンが縮重コドンで置換されていても良く、ブレビバチルス属内で翻訳されたときに同一のアミノ酸をコードしている限り、本来のDNA配列と同一である必要性は無い。
【0030】
4.発現ベクター
本発明の「発現ベクター」は、プロテインA様蛋白質またはその部分的配列をコードするDNA配列、およびその配列に作動可能に連結されたブレビバチルス属細菌で機能しうるプロモーターを含む。当該プロモーターは、ブレビバチルス属細菌で機能しうるものであればいかなるものでも良いが、大腸菌、枯草菌、ブレビバチルス属、スタフィロコッカス属、ストレプトコッカス属、ストレプトミセス属(Streptomyces)、コリネバクテリウム属(Corynebacterium)等細菌由来でブレビバチルス属細菌内にて作動可能なプロモーターが好ましく、ブレビバチルス属細菌細胞壁蛋白質middle wall protein(MWP)、同蛋白質であるouter wallprotein(OWP)(非特許文献10)または、ブレビバチルス・チョウシネンシスHPD31細胞壁蛋白質HWP(Ebisu. Sら. J. Bacteriol. 1990. 172:1312-1320)をコードする遺伝子のプロモーターがより好ましい。実施例では、実施例1に示すブレビバチルス・ブレビス細胞壁蛋白質MWPのP5プロモーター領域「MWP−P5」(図3、4、配列番号7、8参照)、および、実施例5に示すブレビバチルス・ブレビス細胞壁蛋白質MWPのP2プロモーター領域「MWP−P2」(図7、8、配列番号18、19参照)のそれぞれが、「ブレビバチルス属細菌で機能しうるプロモーター」に対応する。
【0031】
また、「発現ベクター」は、該プロモーターの下流に、ブレビバチルス属細菌で機能しうるシャインダルガノ配列及びシグナル配列をさらに含むのが好ましい。発現ベクターは、所望によりマーカー配列を含んでもよい。
【0032】
上記のプロモーター配列に続く「シャインダルガノ配列」は、大腸菌、枯草菌、ブレビバチルス属、スタフィロコッカス属、ストレプトコッカス属、ストレプトミセス属(Streptomyces)、コリネバクテリウム属(Corynebacterium)等細菌由来でブレビバチルス属細菌内にて作動可能なシャインダルガノ配列が好ましく、ブレビバチルス属細菌細胞壁蛋白質middle wall protein(MWP)、同蛋白質であるouterwallprotein(OWP)、または、ブレビバチルス・チョウシネンシスHPD31細胞壁蛋白質HWPをコードする遺伝子の上流に存在するシャインダルガノ配列がより好ましい。
【0033】
上記のシャインダルガノ配列に続く、分泌シグナルペプチドをコードするDNA配列は、以下の分泌シグナルペプチドをコードするDNA配列であれば特に制限は無く、ブレビバチルス・ブレビス内で翻訳されたときに同一のアミノ酸をコードしている限り、本来のDNA配列と同一である必要性は無い。分泌シグナルペプチドとしては、例えば、大腸菌、枯草菌、ブレビバチルス属、スタフィロコッカス属、ストレプトコッカス属、ストレプトミセス属(Streptomyces)、コリネバクテリウム属(Corynebacterium)等細菌由来で、ブレビバチルス属細菌内にて作動可能な分泌シグナルペプチドが好ましく、ブレビバチルス属細菌細胞壁蛋白質middle wall protein(MWP)、同蛋白質であるouter wallprotein(OWP)または、ブレビバチルス・チョウシネンシスHPD31細胞壁蛋白質HWPの分泌シグナルペプチドがより好ましい。また従来の分泌シグナルペプチドのアミノ酸配列を改良したものでも構わない。具体的に言えばmiddle wallprotein(MWP)のシグナルペプチド、Met−Lys−Lys−Val−Val−Asn−Ser−Val−Leu−Ala−Ser−Ala−Leu−Ala−Leu−Thr−Val−Ala−Pro−Met−Ala−Phe−Ala(配列番号11)をMet−Lys−Lys−Arg−Arg−Val−Val−Asn−Asn−Ser−Val−Leu−Leu−Leu−Leu−Leu−Leu−Ala−Ser−Ala−Leu−Ala−Leu−Thr−Val−Ala−Pro−Met−Ala−Phe−Ala(配列番号12)の下線部ように塩基性や疎水性アミノ酸残基など付加または消失させた分泌シグナルペプチドでも構わない。また従来からブレビバチルス属の分泌蛋白質において使われている分泌シグナルペプチドでも構わない。さらに該プロテインAが本来有するシグナルペプチド(図1、2)、すなわち、Met−Lys−Lys−Lys−Asn−Ile−Tyr−Ser−Ile−Arg−Lys−Leu−Gly−Val−Gly−Ile−Ala−Ser−Val−Thr−Leu−Gly−Thr−Leu−Leu−Ile−Ser−Gly−Gly−Val−Thr−Pro−Ala−Ala−Asn−Ala でも構わない。
【0034】
上記のプロモーター配列、シャインダルガノ配列、および分泌シグナルペプチドをコードするDNA配列は、例えば、ブレビバチルス属細菌から得ることができる。好ましくは、ブレビバチルス・ブレビス47(JCM6285)(特開昭60−58074号公報参照)、ブレビバチルス・ブレビス47K(FERM BP−2308)(非特許文献10参照)、ブレビバチルス・ブレビス47−5(FERM BP−1664)、ブレビバチルス・チョウシネンシスHPD31(FERM BP−1087)(特開平4−278091号公報参照)、ブレビバチルス・チョウシネンシスHPD31−S(FERM BP−6623)、またはブレビバチルス・チョウシネンシスHPD31−OK(FERM BP−4573)(特開平6−296485号公報参照)の染色体DNAを鋳型として、公知のPCR法で特異的に増やすことにより取得できる。
【0035】
本発明の「発現ベクター」においては、上記の任意のプロモーターと、上記の任意のシャインダルガノ配列、上記の任意のシグナルペプチド配列と、該プロテインA様蛋白質またはプロテインA様蛋白質の部分的配列をコードするDNA配列とが、ブレビバチルス属細菌内において作動可能に連結されていることが好ましい。
【0036】
ベクターとしては、プラスミドベクターが好ましい。ブレビバチルス属細菌の遺伝子の発現に有用なプラスミドベクターとして具体的には、例えば、枯草菌ベクターとして公知であるpUB110、またはpHY500(特開平2−31682号公報)、pNY700(特開平4−278091号公報)、pHY4831(J. Bacteriol. 1987. 1239-1245)、pNU200(鵜高重三、日本農芸化学会誌1987. 61: 669-676)、pNU100(Appl. Microbiol. Biotechnol., 1989, 30:75-80)、pNU211(J. Biochem., 1992, 112:488-491)、pNU211R2L5(特開平7−170984号公報)、pNH301(Shiga. Y.ら. Appl. Environ. Microbiol. 1992. 58:525-531.)、pNH326、pNH400(Ishihara. Tら、1995. J. Bacteriol, 177:745-749)、pHT210(特開平6−133782号公報)、pHT110R2L5(Appl. Microbiol. Biotechnol., 1994, 42:358-363)、または大腸菌とブレビバチルス属細菌とのシャトルベクターであるpNCO2(特開2002−238569号公報)が使用可能であるが、これらに限定されることはない。また、ブレビバチルス属細菌で機能するプロモーターとシャインダルガノ配列と目的蛋白質をコードするDNA配列とを含んだ発現ベクター、または、それらの各配列を含む遺伝子断片を染色体中へ直接組み込み、発現させる方法(特開平9−135693号公報)を用いても良い。そのような方法は、枯草菌や酵母で既に用いられている公知な方法である。
【0037】
本発明において、プロテインA様蛋白質またはその部分的配列からなる蛋白質は、分泌形態または非分泌形態のいずれで生産されても良いが、分離精製の容易さから培養液中へ分泌させる形態が好ましい。
【0038】
プロテインA様蛋白質またはその部分的配列からなる蛋白質が分泌形態で生産される場合、該ポリペプチドをコードするDNAの上流にブレビバチルス属で機能するシグナルペプチドをコードするDNAを付加または連結するのが好ましい。
【0039】
5.形質転換体
本発明はまた、上記発現ベクターで形質転換されている、ブレビバチルス属細菌形質転換体を提供する。
【0040】
宿主細胞としては、任意のブレビバチルス属細菌を使用し得る。ブレビバチルス属細菌は、限定されないがBrevibacillus agri、B.borstelensis、B. brevis、B. centrosporus、B. choshinensis、B. formosus、B. invocatus、B. laterosporus、B. limnophilus、B. parabrevis、B. reuszeri、B. thermoruberを含む。好ましくは、ブレビバチルス属細菌が、ブレビバチルス・ブレビス47株(JCM6285)、ブレビバチルス・ブレビス47K株(FERM BP−2308)、ブレビバチルス・ブレビス47−5Q株(JCM8970)、ブレビバチルス・チョウシネンシスHPD31株(FERM BP−1087)およびブレビバチルス・チョウシネンシスHPD31−OK株(FERM BP−4573)からなる群より選択される。特に上記のブレビバチルス・ブレビス47株、ブレビバチルス・ブレビス47−5Q株やブレビバチルス・チョウシネンシスHPD31株、ブレビバチルス・チョウシネンシスHPD31−S株が適している。
【0041】
生産量の向上などの目的に応じて、上記ブレビバチルス属細菌のプロテアーゼ欠損株や高発現株のような変異株を使用しても良い。具体的に挙げれば、ブレビバチルス・チョウシネンシスHPD31由来のプロテアーゼ変異株であるブレビバチルス・チョウシネンシスHPD31−OK(特開平6−296485号公報)や、ヒト唾液アミラーゼ高生産株として取得されたブレビバチルス・ブレビス47K(Konishi, H.ら. Appl Microbiol. Biotechnol. 1990. 34:297-302)が使用できる。また前記ブレビバチルス属細菌群に含まれるいずれかの株の変異体を使用してもよい。
【0042】
上記微生物のうち、ブレビバチルス・ブレビス47K(FERM BP−2308)、ブレビバチルス・ブレビス47−5(FERM BP−1664)、ブレビバチルス・チョウシネンシスHPD31(FERM BP−1087)、ブレビバチルス・チョウシネンシスHPD31−S(FERM BP−6623)、及びブレビバチルス・チョウシネンシスHPD31−OK(FERM BP−4573)は、独立行政法人産業技術総合研究所特許生物寄託センター(IPOD;〒305-8566 茨城県つくば市東1丁目1−1中央第6)に、上記それぞれの受託番号にて寄託されている。また、ブレビバチルス・ブレビス47(JCM6285)及びブレビバチルス・ブレビス47−5Q株(JCM8970)は、独立行政法人理化学研究所バイオリソースセンター微生物材料開発室(JCM;〒351-0198 埼玉県和光市広沢2−1)から入手することができる。
【0043】
6.蛋白質の発現調節
ブレビバチルス属細菌を含めた微生物において異種蛋白質を高発現させた場合、正しくフォールディングされずに不活性型の蛋白質を形成することが多く、特にジスルフィド結合の多い蛋白質を高発現させた場合、細胞内外にて不溶性化することも多い。一方で、目的蛋白質を発現させる際、シャペロン蛋白質やジスルフィド結合異性化酵素および/またはプロリン異性化酵素などを作用させることによって、目的蛋白質の不溶性化や分泌効率の低下を抑えられることが知られている。広く試みられている方法は、PDI(プロテインジスルフィドイソメラーゼ)および/またはDsbAなどのジスルフィド酸化還元活性を有する蛋白質を作用させる方法(特開昭63−294796号公報、特開平5−336986号公報)である。
【0044】
さらに、ジスルフィド酸化還元活性を有する蛋白質をコードする遺伝子を宿主生物に導入し、目的蛋白質とジスルフィド酸化還元活性を有する蛋白質とを同時に発現させて正しいジスルフィド結合を有する蛋白質を生産する方法も知られている(特開2000−83670号公報、特表2001−514490号公報等)。
【0045】
本発明によるプロテインA様蛋白質またはその部分的配列からなる蛋白質の発現の場合も、過度な蛋白質合成が行われることによる宿主細胞への負担を軽減し、蛋白質分泌をスムーズに行わせるために、当該蛋白質発現の際、数種類のシャペロン蛋白質、ジスルフィド結合酸化還元酵素、および/またはジスルフィド異性化酵素のようなフォールディングを促進する酵素を同時発現させることも可能である。具体的に挙げれば、ブレビバチルス属細菌において当該蛋白質発現時に、蛋白質のジスルフィド結合に関与し、プロテインジスルフィドイソメラーゼの類縁と考えられている大腸菌のDsbA(Bardwell, J.C.A.ら. Cell. 1991. 67:582-589、Kamitani. Sら. EMBO. J. 1992. 11:57-62.)および/または、DnaK、DnaJ、GrpE(特開平9−180558号公報)などのシャペロン蛋白質を同時に発現させることもできる。その他、ポリペプチドの正確なジスルフィド結合に関与している酵素PDI(特願2001−567367号公報)、ジスルフィド酸化還元酵素(特開2003−169675号公報)(Kontinen, V, P. ら Molecular Microbiology. 1993. 8:727-737)、および/またはジスルフィド異性化酵素のようなフォールディングを促進する酵素を当該蛋白質と同時に発現させ、更に分泌効率を向上させることもできる。
【0046】
7.形質転換体
本発明において用いられるブレビバチルス属細菌の宿主細胞の形質転換は、公知のTakahahiらの方法(Takahashi. Wら. J. Bacteriol. 1983. 156:1130-1134)や、Takagiらの方法(Takagi. H.ら. 1989. Agric. Biol. Chem, 53:3099-3100)、またはOkamotoらの方法(Okamoto. A.ら 1997. Biosci. Biotechnol. Biochem. 61:202-203) により実施することができる。
【0047】
得られた形質転換体の培養に用いる培地は、プロテインA様蛋白質またはその部分的配列からなる蛋白質を高効率、高収量で生産できるものであれば特に制限は無い。具体的にはグルコース、蔗糖、グリセロール、ポリペプトン、肉エキス、酵母エキス、カザミノ酸などの炭素源や窒素源を使用することが出来る。その他、カリウム塩、ナトリウム塩、リン酸塩、マグネシウム塩、マンガン塩、亜鉛塩、鉄塩等の無機塩類が必要に応じて添加される。栄養要求性の宿主細胞を用いる場合は、生育に要求される栄養物質を添加すればよい。また、必要であればペニシリン、エリスロマイシン、クロラムフェニコール、ネオマイシンなどの抗生物質が添加されても良い。さらに、菌体内外に存在する宿主由来のプロテアーゼによる当該目的蛋白質の分解、低分子化を抑えるために、公知の各種プロテアーゼ阻害剤、すなわちPhenylmethane sulfonyl fluoride (PMSF)、Benzamidine、4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF)、Antipain、Chymostatin、Leupeptin、Pepstatin A、Phosphoramidon、Aprotinin、Ethylenediaminetetra acetic acid (EDTA)、および/または、その他市販されているプロテアーゼ阻害剤を適当な濃度で添加しても良い。
【0048】
培養温度は約15−42℃、好ましくは約28−37℃であり、通気攪拌条件で好気的に培養を行うことが望ましいが、必要であれば通気を遮断し嫌気的に培養してもよい。
【0049】
8.プロテインA様蛋白質の取得
本発明の実施形態によれば、形質転換されたブレビバチルス属細菌を培養することにより、プロテインA様蛋白質またはその部分的配列からなる蛋白質は、菌体外、すなわち培養上清中に大量に蓄積されるので、培養上清中から活性型の当該蛋白質を採取、精製することが出来る。また菌体内、及び菌表層に残った当該蛋白質も、公知の方法、例えば超音波やフレンチプレス、アルカリやSDS処理などを利用した方法により菌を破砕し抽出出来る。得られた当該蛋白質は、公知の蛋白質精製方法、例えば硫酸アンモニウムや硫酸ナトリウムなどを用いた塩析、エタノールやアセトン等による濃縮、及び、ゲル濾過、イオン交換、ハイドロキシアパタイト、当該蛋白質のもつ抗体結合活性を利用した各種クロマトグラフィーおよび/または、当該蛋白質が有する親和性を利用したクロマトグラフィーなどを用いて、効果的に精製することができる。
【0050】
本発明の「発現ベクター」により形質転換されたブレビバチルス属細菌形質転換体は、当該蛋白質を安定に発現し、大量に培養上清中へ分泌蓄積することができる。具体的には、適当な培地で培養することによって、SDS−PAGEにおいて分子量4万から5万付近に現れる活性型のプロテインAを培養上清中へ大量に分泌、蓄積させることができる。実施形態による当該蛋白質の生産方法によれば、少なくとも約150mg/L培養液、好ましくは約200mg/L培養液、より好ましくは約500mg/L培養液、もっとも好ましくは約1000mg/L以上の生産量を達成できる。もっとも、生産量は、培養条件等によって変化しうる。
【0051】
9.プロテイン様蛋白質の基材への固定
本発明におけるプロテイン様蛋白質またはその部分的配列からなる蛋白質を固定する「基材」としては、活性炭、ガラスビーズ、シリカゲルなどの無機基材、架橋ポリビニルアルコール、架橋ポリアクリレート、架橋ポリアクリルアミド、架橋ポリスチレンなどの合成高分子、または樹脂、結晶性セルロース、架橋セルロース、架橋アガロース、架橋デキストリンなどの多糖類からなる有機基材、セルロース、ポリビニルアルコール、エチレン−酢酸ビニル共重合体けん化物、ポリアクリルアミド、ポリアクリル酸、ポリメタクリル酸、ポリメタクリル酸メチル、ポリアクリル酸グラフト化ポリエチレン、ポリアクリルアミドグラフト化ポリエチレン、ガラス、さらには、これらの組み合わせによって得られうる有機−有機、有機−無機などの複合基材が代表例として挙げられるが、本発明はこれらのみに限定されるものではない。好ましくは、該基材は水、ナイロン6、ナイロン6,6、ナイロン11、ポリエチレン、ポリ(塩化ビニリデン)、ポリ(塩化ビニル)、ポリ(酢酸ビニル)、ポリスチレン、スチレン−ジビニルベンゼン共重合体、スチレン−ジビニルベンゼン、ポリ(トリフルオロエチレン)、ポリ(クロロトリフィルオロエチレン)、ポリ(テレフタル酸エチレン)、ポリプロピレン、ポリ(アクリル酸メチル)、ポリアクリル酸エステル、ポリ(メタクリル酸メチル)、ポリメタクリル酸エステル、架橋ポリアクリレート、架橋ポリアミドなどの合成高分子化合物からなる群より選択される。基材の形状としては、球状、粒状、平膜状、繊維状、中空糸状等いずれも有効に用いられるが、吸着性能面から球状または粒状がより好ましく用いられる。水不溶性多孔質体が球状または粒状である場合、平均粒径は約5μmから1000μmであることが好ましく、より好ましくは約20〜800μm、最も好ましくは約30〜600μmである。
【0052】
本発明において、プロテインA様蛋白質またはその部分的配列からなる蛋白質の該基材への固定化は、共有または非共有結合、例えばアフィニティ、会合、抗原抗体反応、水素結合、コンジュゲーションなどにより実施しうる。また、当業者に周知の手段により、当該蛋白質に対してアミノ酸残基の付加、置換、欠失などの分子改変を施し、該基材への固定化を容易にすることも可能である。例えば、プロテインA様蛋白質の分子内にシステイン残基を導入することによって、プロテインA様蛋白質を前記基材に容易に固定化することができる。
【0053】
本発明の製造方法によって得られるイムノグロブリン吸着担体は、イムノグロブリン、特にIgGの精製用担体として好適に用いられ得る。また、血漿からのIgGの除去等、疾病治療への応用も可能である。
【実施例】
【0054】
以下、参考例及び実施例に基づいて本発明を具体的に説明するが、これらが本発明の範囲を制限するものでない。本発明の実施にあたり、組換えDNAの作製や操作などは特に断わらない限り下記の実験書に従って実施した。 (1) T. Maniatis, E. F. Fritsch, J. Sambrook著、「モレキュラー・クローニング/ア・ラボラトリー・マニュアル(Molecular Cloning/A Laboratory Manual)」、第2版(1989)、Cold Spring Harbor Laboratory 刊(米国)。 (2) 村松正實編著「ラボマニュアル遺伝子工学」、第3版(1996)、丸善株式会社刊。
【0055】
(実施例1) スタフィロコッカス・アウレウス ATCC6538P株由来のプロテインAをコードするDNA配列のクローニング
スタフィロコッカス・アウレウスATCC6538P株を、T2液体培地(ポリペプトン 1%、イーストエキストラクト 0.2%、グルコース1%、魚肉エキス0.5%、pH7.0)で37℃一晩振とう培養した。得られた培養液から菌体を遠心分離により回収後、10mMのトリス−塩酸緩衝液(pH8.0)で2度洗浄した。菌体を同緩衝液に懸濁後、1%SDSで溶菌し、60℃にて30分間加熱後、フェノール抽出及びエタノール沈殿等の定法により全ゲノムDNAを抽出した。
【0056】
次に、プロテインA遺伝子のDNA配列情報(非特許文献6)を基に、2つのオリゴヌクレオチドプライマー5'-TTGCTCCCATGGCTTTCGCTGCGCAACACGATGAAGCT-3' (配列番号5)と5'-CGGGATCCCTAAAATACAGTTGTACCGATGAATGGATT-3'(配列番号6)を調製した。上記のゲノムDNAを鋳型とし、これら2つのオリゴヌクレオチドプライマーを用いてPCRを行い、プロテインAからシグナルシーケンス(Sドメイン)及び細胞壁結合ドメイン(Xドメイン)の一部を除いた部分(これ以降SPAと称する)をコードするDNA断片(約1.2kbp(キロ塩基対))を増幅した。
【0057】
得られたDNA断片は、制限酵素NcoI及びBamHIにより消化した後、アガロースゲルより分離回収した。
【0058】
一方、ブレビバチルス発現ベクターであるpNH301(Shiga. Y.ら. Appl. Environ. Microbiol. 1992. 58:525-531.)もまた同様に制限酵素NcoI及びBamHIにより消化後、精製回収してアルカリフォスファターゼ処理により脱リン酸化処理を行った。
【0059】
制限酵素処理したSPAをコードする上記DNA断片と上記発現ベクターpNH301とをT4DNAライゲースを用いて連結し、SPA発現プラスミドSpa−pNH301を構築した(図3、4;配列番号7、8)。図3および図4において、「MWP−P5」はブレビバチルス・ブレビス細胞壁蛋白質MWPのP5プロモーター領域、「SDM」はブレビバチルス・ブレビス細胞壁蛋白質MWPのSD配列、「SP」はブレビバチルス・ブレビス細胞壁蛋白質MWPのシグナルペプチド配列、「spa」は「SPA」をコードするDNA配列、「Nm」はネオマイシン耐性遺伝子コード領域、「Rep/pUB110」はベクターpNH301の複製開始点を意味する。また図4において、「P5−35」および「P5−10」は、それぞれ、ブレビバチルス・ブレビス細胞壁蛋白質MWPのP5プロモーターの−35領域および−10領域意味する。公知の方法により、このSpa−pNH301を用いてブレビバチルス・ブレビス47K、またはブレビバチルス・チョウシネンシスHPD31−OK株の形質転換を行った。
【0060】
(実施例2) ブレビバチルス属細菌によるプロテインAの発現試験
実施例1にて得られた形質転換体、及びその対照となるベクターpNH301のみを有するブレビバチルス・チョウシネンシスHPD31−OK株のそれぞれを、生産培地3YC(ポリペプトンS 3%、酵母エキス0.5%、グルコース3%、MgSO4・7H2O 0.01%、CaCl2・7H2O 0.01%、MnSO4・4H2O 0.001%、FeSO4・7H2O 0.001%、ZnSO4・7H2O 0.0001% pH7.0)にネオマイシン60mg/Lを添加した培地にて、30℃で、好気的条件下、3日間培養を行った。培養液は遠心分離(10,000rpm、4℃、5分間)により菌体を除去した後、定法により、10−20%グラジエントゲルを用いた還元条件下でのSDS−PAGEに供した。電気泳動後、ゲルをCBB染色することにより、SPAのバンドを検出した(図5)。SDS−PAGEによる解析の結果、その培養上清液中に大量のSPAを確認することが出来た。
【0061】
なお、プロテインAの細胞壁結合ドメイン(Xドメイン)を含めた完全長型のプロテインAを発現させる場合には、以下の方法を採用することができる。実施例1で記載したスタフィロコッカス・アウレウスから調製したゲノムDNAを鋳型とし、2つのオリゴヌクレオチドプライマー5'- TTGCTCCCATGGCTTTCGCTGCGCAACACGATGAAGCT-3' (配列番号5)と、5'-CGCGGATCCTTATAGTTCGCGACGACG-3'(配列番号9)または5'- CGCGGATCCTCAACGTATATAAGTTAAAAT-3'(配列番号10)とを用い、PCRによりDNA断片を増幅する。得られたプロテインAをコードするDNA断片は、実施例1にて記載された方法によりpNH301のNcoIとBamHIとの間へ連結する。得られたプラスミドをブレビバチルス・ブレビス47Kまたはブレビバチルス・チョウシネンシスHPD31−OK株へ形質転換することで形質転換体を取得する。この形質転換体を実施例2に記載された培養方法にて培養した後、培養液中へ分泌されたプロテインAを確認する。
【0062】
(実施例3) ブレビバチルス属細菌により生産されたプロテインAの抗体結合能の測定実施例1で得られた形質転換体により生産されたSPAが抗体結合能を有するか確認するため、マウス抗ヒトIgG抗体及びアルカリフォスファターゼ標識ウサギ抗マウスIgG抗体を用いて結合試験を行った。
【0063】
実施例1にて得られた、Spa−pNH301、及びその対照となるpNH301をそれぞれ有するブレビバチルス・チョウシネンシスHPD31−OK株を、実施例2と同様に培養し、それぞれの培養上清液をSDS−PAGEに供した後、定法によりPVDFメンブランへ転写し、3%スキムミルクにてブロッキングを行い、Fahnestockら(Fahnestock. S. Rら. J. Bacteriol. 1986. 165:796-804.)らの方法に準じて抗体結合試験を行った。検出は、AP発色キット(バイオラッド社製)を用い、その取扱い説明書に従って実施した。その結果、比較対照であるベクターpNH301のみを有する形質転換体ではバンドは認められなかった。一方、SPA発現ベクターSpa−pNH301を有する形質転換体では、SPAと同じ移動度、すなわちSDS−PAGE上、CBB染色で濃いバンドの認められた42kDa付近に強い発色が認められた(図6)。図6において、「M」は分子量マーカー、「C」はベクターpNH301を有するブレビバチルス・チョウシネンシスHPD31−OK株、SpaはSPA発現ベクターSpa−pNH301を有するブレビバチルス・チョウシネンシスHPD31−OK株のレーンを示す。これらの結果から、SPA発現ベクターSpa−pNH301を有するブレビバチルス・チョウシネンシスHPD31−OK株により生産された分子量約42kDaの蛋白質は、抗体結合活性を有することが確認された。
【0064】
(実施例4) ブレビバチルス発現ベクターpNK3260の構築
pNH326(Ishihara. Tら、1995. J. Bacteriol, 177:745-749)に含まれるMWPのP5プロモーターをMWPのP2プロモーターに変換して、ブレビバチルス発現ベクターpNK3260を以下のように構築した。
【0065】
まず、pNH326を鋳型として、2つのオリゴヌクレオチドプライマー5'-GGAATTCTGATTTCACTTTTTGCATTCTACA-3'(配列番号13)および5'-AGTGCACTCGCACTTACTGT-3'(配列番号14)を用いてPCRを行い、pNH326のうちMWPのP5プロモーターを除く部分を増幅し、その末端を制限酵素EcoRIとHindIIIとで消化した。次に、MWPのP2プロモーターを含む2本鎖DNA断片5'- GGTACCAATTGGCGCCGCAACTTTTGATTCGCTCAGGCGTTTAATAGGATGTAATTGTGAGCGGATAACAATTATTCTGCATGGCTTTCCTGCGAAAGGAGGTGCACCGCGCTTGCAGGATTCGGGCTTTAAAAAGAAAGATAGATTAACAACAAATATTCCCCAAGAACAATTTGTTTATACTGGAGGAGGAGAACACAAGGTCATGAAAAAAAGAAGGGTCGTTAACAGTGTATTGCTTCTGCTACTGCTAGCTAGTGCACTCGCACTTACTGTTGCTCCCATGGCTTTCGCTGCAGGATCCGTCGACTCTAGACTCGAGGAATTCGGTACCCCGGGTTCGAAATCGATAAGCTTCTGT- 3'(配列番号15)を定法に従い調製し、その末端を制限酵素MunIおよびHindIIIで消化した。これら2つのDNA断片をT4DNAライゲースを用いて連結し、pNK3260を構築した。
【0066】
(実施例5) スタフィロコッカス・アウレウス・コワンI株(JCM2179)由来のプロテインAをコードするDNA配列のクローニング
実施例1と同様に、スタフィロコッカス・アウレウス・コワンI株(JCM2179)から全ゲノムDNAを抽出した。次に、プロテインA遺伝子のDNA配列情報(非特許文献2)を基に、2つのオリゴヌクレオチドプライマー5'- TTGCTCCCATGGCTTTCGCTGCGCAACACGATGAAGCTCAACAA-3' (配列番号16)と 5'-CGGGATCCCTATTTTGGTGCTTGAGCATCGTTTAGCTTTTTAGCTTCTGCTAAAATTTTC-3'(配列番号17)を調製した。上記のゲノムDNAを鋳型とし、これら2つのオリゴヌクレオチドプライマーを用いてPCRを行い、プロテインAからシグナルシーケンス(Sドメイン)及び細胞壁結合ドメイン(Xドメイン)を除いた部分(これ以降SPA’と称する)をコードするDNA断片(約0.9kbp)を増幅した。得られたDNA断片は、制限酵素NcoI及びBamHIにより消化した後、アガロースゲルより分離回収した。
【0067】
一方、実施例4で構築したブレビバチルス発現ベクターpNK3260もまた同様に制限酵素NcoI及びBamHIにより消化後、精製回収してアルカリフォスファターゼ処理により脱リン酸化処理を行った。
【0068】
制限酵素処理後の、SPA’をコードする上記DNA断片と上記発現ベクターpNK3260とをT4DNAライゲースを用いて連結し、SPA’発現プラスミドSpa’−pNK3260を構築した(図7、8;配列番号18、19)。図7および図8において、「MWP−P2」はブレビバチルス・ブレビス細胞壁蛋白質MWPのP2プロモーター領域、「SDM」はブレビバチルス・ブレビス細胞壁蛋白質MWPのSD配列、「SP’」はブレビバチルス・ブレビス細胞壁蛋白質MWPのシグナルペプチド配列を一部改変した改変型シグナルペプチド配列、「spa’」はSPA’をコードするDNA配列、「Nm」はネオマイシン耐性遺伝子コード領域、「Rep/pUB110」はベクターpNK3260の複製開始点を意味する。また図8において、「P2−35」および「P2−10」は、それぞれ、ブレビバチルス・ブレビス細胞壁蛋白質MWPのP2プロモーターの−35領域および−10領域意味する。
【0069】
公知の方法により、このSpa’−pNK3260を用いてブレビバチルス・チョウシネンシスHPD31−OK株の形質転換を行った。
【0070】
(実施例6) ブレビバチルス属細菌により分泌発現されたプロテインAの培養液中の挙動
実施例5にて得られた形質転換体を、生産培地3YC(ポリペプトンS 3%、酵母エキス0.5%、グルコース3%、MgSO4・7H2O 0.01%、CaCl2・7H2O 0.01%、MnSO4・4H2O 0.001%、FeSO4・7H2O 0.001%、ZnSO4・7H2O 0.0001% pH7.0)にネオマイシン60mg/Lを添加した培地にて、30℃の、好気的条件下で培養した。培養開始から、24、48、72、78時間後に培養液を分取し、遠心分離(10,000rpm、4℃、5分間)により菌体を除去した後、定法により、10−20%グラジエントゲルを用いた還元条件下でのSDS−PAGEに供した。電気泳動後、ゲルをCBB染色することにより、SPA’のバンドを検出した(図9)。図9において、「レーン番号1」は分子量マーカーを、「レーン番号2」は対照として用いたRepligen社製プロテインA(rPA−50)0.52μgを、「レーン番号3」は培養開始から24時間経過後のSPA’発現ベクターSpa’−pNK3260を有するブレビバチルス・チョウシネンシスHPD31−OK株の培養上清1μlを、「レーン番号4」は培養開始から48時間経過後の同培養上清1μlを、「レーン番号5」は培養開始から72時間経過後の同培養上清1μlを、「レーン番号6」は培養開始から78時間経過後の同培養上清1μlを、それぞれ泳動したレーンを示す。
【0071】
SDS−PAGEによる解析の結果、目的とするSPA’は、培養開始後48時間目(レーン番号4)には大量に発現し、それ以降もSPA’の濃度は上昇しつづけ、最終的に培養上清中に約2g/Lの濃度で蓄積していた。培養上清中のSPA’の濃度は、レーン番号2で泳動したRepligen社製プロテインA(rPA−50)0.52μgのバンドを対照として、ChemiDoc XRSシステム(Bio−Rad社)により測定した。
【0072】
(実施例7) 形質転換体により生産されたプロテインAのN末端アミノ酸配列の確認
図9に示したSDS−PAGEゲル中のレーン番号6において、分子量33kDa付近に見られるSPA’のバンドについて、そのN末端から10残基のアミノ酸配列を、定法に従い解析した。その結果、配列番号2に示したプロテインAのアミノ酸配列の37番目のAla以降の配列と一致し、分泌シグナル配列が正確に除去されていることが確認された。
【0073】
(実施例8) 形質転換体により生産されたプロテインAの抗体結合活性
実施例6と同様に78時間培養して得られた培養液の上清1Lを、陽イオン交換クロマトグラフィー(CM−Sepharose:アマシャムバイオサイエンス)に供し、pH7.0において0−1M塩化ナトリウムの濃度勾配により分離した。次にSPA’画分を回収し、疎水クロマトグラフィー(Phenyl−Sepharose:アマシャムバイオサイエンス)に供し、pH7.0において1−0M硫酸アンモニウムの濃度勾配により分離した。さらに、SPA’画分を回収し、ゲルろ過クロマトグラフィー(HiLoad 16/60 Superdex 75 pg:アマシャムバイオサイエンス)に供し、SPA’画分を回収した。以上の精製操作により、SDS−PAGEにおいて単一バンドを示す、約100mgのSPA’を調製した。
【0074】
上記で調製したSPA’のヒトIgG結合活性を以下の様に評価した。まず、5μg/mLとなるようにSPA’をPBS緩衝液(タカラバイオ)で希釈し、96穴イムノプレート(NUNC)に100μLずつ分注した。37℃で1時間反応させた後に、PBS緩衝液(250μL)にて二回洗浄し、3%ウシ血清アルブミン/PBS溶液を250μL加え、4℃で一晩ブロッキングした。次に、0.1%BSAを含有するPBS緩衝液で調製した25μg/mLヒトIgG(シグマ)溶液100μlを加えて、37℃で、1.5時間反応後、0.01%Tween20を含むPBS緩衝液で洗浄した。これに、PBS緩衝液にて2000倍に希釈したHRP標識プロテインL(0.3mg/ml;シグマ社)溶液を100μl加え、37℃で、1.5時間反応後、0.01%Tween20を含むPBS緩衝液で洗浄した。さらに、発色基質[2,2’−アジノジ(3−エチルベンゾチアゾリンー6−スルフォン酸)アンモニウム塩]溶液(シグマ社)を100μl加え、暗所にて20分間反応後、405nmにおける吸光度を測定した。この時、対照としてRepligen社製プロテインA(rPA−50)についても同様の操作を行い、測定値を比較したところ、上記で調製したSPA’の単位質量当たりのヒトIgG結合活性は、上記Repligen社製プロテインAの約97%であり、両者はほぼ同等の活性を有することが確認された。
【0075】
以上の結果から、実施例によるプロテインAの生産方法によれば、従来報告されていた大腸菌、枯草菌における組換え型プロテインAの発現量を上回る生産性を達成でき、従来問題となっていた低生産性の問題を解決できることを示している。
【技術分野】
【0001】
本発明は、ブレビバチルス属細菌を使用した、イムノグロブリン結合能を有するプロテインA様蛋白質の製造方法に関する。詳細には、本発明は、遺伝子組換え技術により、ブレビバチルス属細菌にプロテインA様蛋白質を多量に分泌発現させること、その発現されたプロテインA様蛋白質を、プロテアーゼ等による分解を受けることなく高純度で分離回収すること、及び、その分離回収されたプロテインA様蛋白質を、抗体精製用のカラム樹脂等の用途へ有効に使用することに関する。
【0002】
日本国特許出願2004−198831号(2004年7月6日出願)の明細書、請求の範囲、図面および要約を含む全開示内容は、これら全開示内容を参照することによって本出願に合体される。
【背景技術】
【0003】
抗体(イムノグロブリン、またはIgとも呼ばれる)蛋白質は生体における有害な抗原の補足及び排除機能を有するため古くから医薬品として利用されてきた。近年では遺伝子工学技術や細胞融合技術の進歩により特異的な抗原に反応する抗体を分子設計し、動物細胞内で発現させることにより均一で高い抗原性を有するモノクローナル抗体の作製が可能となった。これらの抗体蛋白質は細胞培養液中に分泌されることから、比較的容易に分離精製回収することができる。
【0004】
一般的に、イムノアッセイやイムノブロット解析に利用される抗体蛋白質の場合、通常の蛋白質精製において利用される方法、すなわち硫酸アンモニウムによる沈殿法やイオン交換クロマトグラフィー等を用いることによって、血清、腹水または細胞培養液のような天然生体サンプルから、十分な収量や純度で得ることができる。
【0005】
一方、高純度を必要とする医薬品または診断薬等に利用される抗体蛋白質の場合、上記のような方法を用いて分離精製するには、様々な分離抽出条件の検討や数多くの他のクロマトグラフィーとの併用を必要とし、また、それぞれの抗体蛋白質に関して精製条件の最適化が必要となり、非常な労力を要する。従って、高純度を必要とする抗体蛋白質の精製の場合、他の夾雑物質から簡便に分離精製するために、抗体蛋白質を特異的に吸着することが可能なアフィニティークロマトグラフィーを用いるのが一般的である。
【0006】
抗体結合能を有するアフィニティークロマトグラフィーとして最も良く利用されているのが、プロテインA、プロテインG及びプロテインLなどの蛋白質を適当な樹脂に固定化した担体によるクロマトグラフィーである。これらの蛋白質の中で特に精製用担体のリガンドとしてよく利用されているのがプロテインAである。プロテインAはグラム陽性細菌スタフィロコッカス・アウレウス(Staphylococcusaureus)によって生産される細胞壁蛋白質の1種であり、約42,000の分子量であることが明らかとなっている。その構造は7つの機能ドメイン(アミノ末端からシグナル配列S、イムノグロブリン結合ドメインE、イムノグロブリン結合ドメインD,イムノグロブリン結合ドメインA、イムノグロブリン結合ドメインB、イムノグロブリン結合ドメインC、スタフィロコッカス・アウレウス細菌細胞壁結合ドメインX)(非特許文献1、2、3参照)から構成されている。プロテインAの5つのイムノグロブリン結合ドメイン(ドメインE、D、A、B、C)は、それぞれイムノグロブリン分子内のFc部分で結合することができる(非特許文献3参照)。
【0007】
このプロテインAのイムノグロブリン結合ドメインに対する相対親和性は、pH、スタフィロコッカス・アウレウス菌株種(Cheung, A.ら Infec. Immun. 1987. 55:843-847)、またイムノグロブリンのクラス(IgG,IgM,IgA,IgD,IgE)及びサブクラス(IgG1,IgG2,IgG3,IgG4,IgA1,IgA2)などの多くの因子に依存することが知られ、特にイムノグロブリンのクラスではヒトIgG1、IgG2、IgG4及びマウスIgG2a、IgG2b,IgG3のFc部分と強い結合を示す。以上のような性質を持つプロテインAは、イムノグロブリンとしての抗原結合能力、親和力や性質を損なうことなくイムノグロブリンに結合することができるため、様々な診断、医薬品及び基礎研究に用いられるイムノグロブリン、特にIgGに対し精製用担体のリガンドとして広く使用されている。
【0008】
また最近では、感作末梢血リンパ球の腫瘍細胞毒性作用を阻害する血清阻止因子(特異抗原、抗体、抗グロブリン及び免疫複合体で構成される)を、プロテインAに吸着させ腫瘍患者の血清から除去するといった、癌治療への応用にも興味が持たれている(特許文献1−3参照)。さらにIgG結合活性とは別にプロテインAは多クローン抗体合成を活性化する働きも有していることから、当初の精製樹脂リガンド用途だけでなく様々なバイオテクノロジー分野における使用用途が期待されている。
【0009】
初期のプロテインAの生産方法においてはスタフィロコッカス・アウレウス株の培養液から直接、分離精製を行ってきたが、この菌自身の病原性の問題や不純物の混入の問題により、現在では大腸菌(Escherichia coli)(特許文献1−3)やグラム陽性細菌である枯草菌(Bacillus subtilis)(特許文献4−5)を用いた組換えDNA技術を用いた生産方法へ移行している。ところが、大腸菌における組換え型プロテインAの生産性は極めて低く、また、発現蛋白質の多くがインクルージョンボディーを形成したり、細胞内で分解されるため、分離回収が容易ではない(非特許文献4)。一方、スタフィロコッカス・アウレウスと同様にグラム陽性細菌である枯草菌を用いたプロテインAの生産では、プロテインAのN末端へ枯草菌分泌蛋白質のシグナル配列を付加することで培地中へ分泌発現させる方法が取られており、大腸菌での生産系と比較した場合、分離精製等が容易であり、生産性も高いことが報告されている(約47−100mg/L)(Fahnestock, S, R.ら J. Bacteriol. 1986. 165:796-804)。ところが、枯草菌において生産されたプロテインAは、枯草菌が本来持つ菌体外プロテアーゼによる分解を受ける。そのため、枯草菌の数種の菌体外プロテアーゼ欠損株(非特許文献5)を宿主として用いることも試みられてきたが、プロテインAの分解を抑えることは未だ達成できていない。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】日本国特願平07−187019号公報
【特許文献2】米国特許番号US5151350号公報
【特許文献3】欧州特許番号EP0107509号公報
【特許文献4】米国特許番号US4617266号公報
【特許文献5】欧州特許番号EP0124374号公報
【非特許文献】
【0011】
【非特許文献1】Lofdahl, Sら.Proc. Natl. Acad. Sci. USA. 1983. 80:697-701.
【非特許文献2】Shuttleworth, H. Lら.Gene. 1987. 58:283-295.
【非特許文献3】Uhlen, M.ら.J. Bio. Chem. 1984. 259:1695-1702.
【非特許文献4】Nilsson, Bら. Protein Eng. 1987. 1:107-113.
【非特許文献5】Fahnestock, S. Rら. Appl.Envron. Microbiol. 1987. 53:379-384.
【非特許文献6】Brigido, Mら, J. Basic Microbiology. 1991. 31: 337-345.
【非特許文献7】Sjostrom, J, -Eら. J. bacteriol. 1975. 123:905-915.
【非特許文献8】Bjorck, L.ら. 1984. J. Immunol. 133, 969-974.
【非特許文献9】Kastern, W.ら. J Biol Chem. 1992. 267:12820-12825
【非特許文献10】Udaka, S.ら. Method Enzymol. 1993. 217:23-33.
【発明の概要】
【発明が解決しようとする課題】
【0012】
このような背景から、大腸菌や枯草菌を用いた生産方法を上回る、効率の良いプロテインAの生産技術の確立が、強く望まれていた。
【0013】
本発明は、大腸菌や枯草菌を用いた生産方法を上回る、効率の良いプロテインAの生産技術の提供を一つの課題とする。
【課題を解決するための手段】
【0014】
本発明者は、プロテインAのような機能蛋白質を安定、大量に生産する技術を確立するため、ブレビバチルス属細菌を宿主に使い鋭意研究を行った結果、プロテインAを効率良く培養液中へ大量に分泌発現、そして安定に蓄積させ、容易に高純度で分離回収できることを見いだした。
【発明の効果】
【0015】
本発明に従えば、ブレビバチルス属細菌を宿主に用いることにより、大腸菌や枯草菌などの微生物を宿主とした場合に報告されている生産量を大幅に上回るプロテインAを培養液中へ分泌生産させ、イムノグロブリン結合機能を損なわずに容易に高純度にまで精製することができる。従って、本発明により、現在まで高価格の一因となっていたプロテインAの低生産性や、精製工程の複雑さが解消される。
【0016】
本発明は、以下の1または複数の対象を含む。
(1)本発明は、プロテインA様蛋白質またはその部分的配列をコードするDNA配列、およびその配列に作動可能に連結されたブレビバチルス属細菌で機能しうるプロモーターを含む、DNA配列を提供する。
(3)本発明は、前記DNA配列を含む、発現ベクターを提供する。
(4)本発明は、前記発現ベクターを含む、ブレビバチルス属細菌形質転換体を提供する。
(6)本発明は、前記形質転換体を培養すること、および該形質転換体によって分泌生産されるプロテインA様蛋白質、またはその部分的配列を有する蛋白質を回収することを含む、プロテインA様蛋白質またはその部分的配列を有する蛋白質の製造方法を提供する。
(7)本発明は、前記製造方法によってプロテインA様蛋白質またはその部分的配列を有する蛋白質を製造すること、および該プロテイン様蛋白質またはその部分的配列を有する蛋白質を適当な基材に固定化すること、を含む、イムノグロブリン吸着担体の製造方法を提供する。
【図面の簡単な説明】
【0017】
【図1】図1は、スタフィロコッカス・アウレウス・コワン(Cowan)I株のプロテインAの塩基配列とアミノ酸配列とを示す図である(番号はアミノ酸残基数を示す)。
【図2】図2は、スタフィロコッカス・アウレウス株のプロテインAの遺伝子配列とアミノ酸配列とを示す図である(番号はアミノ酸残基数を示す)。
【図3】図3は、プロテインA(SPA)発現ベクター(Spa−pNH301)を示す図である。
【図4】図4は、プロテインA(SPA)発現ベクター(Spa−pNH301)のプロモーター配列からプロテインA(SPA)をコードする領域までの塩基配列およびアミノ酸配列を示す図である。
【図5】図5は、ブレビバチルス・チョウシネンシスHPD31−OK株により生産されたプロテインA(SPA)のSDS−PAGEによる解析結果を示す図である。
【図6】図6は、ブレビバチルス・チョウシネンシスHPD31−OK株により生産されたプロテインA(SPA)の、抗体結合試験の結果を示す図である。
【図7】図7は、プロテインA(SPA’)発現ベクター(Spa’−pNK3260)を示す図である。
【図8】図8は、プロテインA(SPA’)発現ベクター(Spa’−pNK3260)のプロモーター配列、シャインダルガノ配列、シグナルペプチドおよびプロテインA(SPA’)をコードするDNA配列を示す図である。
【図9】図9は、ブレビバチルス・チョウシネンシスHPD31−OK株により生産されたプロテインA(SPA’)の培養液中での挙動およびその蓄積量を、SDS−PAGEにより解析した結果を示す図である。
【発明を実施するための形態】
【0018】
本発明者らは、組換えDNA技術が利用できる細菌の中でも、グラム陽性細菌であるブレビバチルス属細菌を用いることで、活性型のプロテインAを大量に培養液中へ分泌発現させることができ、大腸菌及び枯草菌における低生産性の問題、並びに枯草菌における発現したプロテインAの分解に対する問題が効果的に改善されることを発見した。すなわち、ブレビバチルス属細菌を用いることで、枯草菌で得られる発現量を容易に確保し、さらにこれを培地中へ蓄積させることができる。以下、実施形態に基づいて本発明について詳しく説明する。
【0019】
1.プロテインA
プロテインAは、前述のように、グラム陽性細菌スタフィロコッカス・アウレウスによって生産される細胞壁蛋白質の1種であり、例えば、図1(配列番号2)にて示されるアミノ酸配列からなるスタフィロコッカス・アウレウス・コワン(Cowan)I株(JCM2179)由来のもの(非特許文献2)、図2(配列番号4)にて示されるアミノ酸配列からなるもの(非特許文献6、Finck-Barbancon,V.ら FEMS Microbiol. Lett. 1992. 91:1-8)、Woods46株由来のもの(非特許文献3)、8325−4株由来のもの(非特許文献3)、および既にクローニングされているプラスミドDNAすなわちpSP1やpSP3等(非特許文献7)にコードされているspa遺伝子産物等を指す。
【0020】
本発明にいうプロテインA様蛋白質には、プロテインA、またはプロテインAと実質的に同一の蛋白質が含まれる。また、プロテインA様蛋白質には、当業者にとって公知の配列比較アルゴリズムを使用して、プロテインAのアミノ酸配列と比較され、そして最大の一致のためにアラインメントされるときに、少なくとも60%、好ましくは80%、より好ましくは90−95%、そして最も好ましくは少なくとも99%のアミノ酸残基の同一性を有するアミノ酸配列を有し、かつ、イムノグロブリン結合活性を有する蛋白質が含まれる。ここで、同一性を有するアミノ酸配列は、好ましくは50残基長以上であり、より好ましくは100残基長以上であり、さらに好ましくは150残基長以上であり、最も好ましい実施態様では、完全長に渡り同一性を有する。
【0021】
パーセント配列同一性を決定するために好適であるアルゴリズムの例は、BLASTアルゴリズムであり、これはAltschul et al., J. Mol. Biol. 215: 403-410(1990)に記載されている。BLAST分析を実施するためのソフトウェアは、National Center for Biotechnology Information(http://www.ncbi.nlm.nih.gov/)を通して公衆が利用可能である。
【0022】
プロテインA様蛋白質は、例えば、配列番号1または配列番号3に示したDNA配列に対して相補的な配列を有するDNAに、ストリンジェント条件下でハイブリダイズするDNAによってコードされるアミノ酸配列からなる蛋白質であり得る。該ストリンジェント条件下のハイブリダイゼーション条件の例は:好ましくは約7%のドデシル硫酸ナトリウム(SDS)、約0.5MのNaPO4、1mMのEDTA中で約50℃でハイブリダイゼーション、および約2XSSC、約0.1%のSDS中で50℃の洗浄;より望ましくは約7%のドデシル硫酸ナトリウム(SDS)、約0.5MのNaPO4、約1mMのEDTAで50℃でハイブリダイゼーション、および約1XSSC、約0.1%のSDSで約50℃の洗浄;より望ましくは約7%のドデシル硫酸ナトリウム(SDS)、約0.5MのNaPO4、約1mMのEDTAで約50℃でハイブリダイゼーション、および約0.5XSSC、約0.1%のSDSで約50℃の洗浄;より好ましくは約7%のドデシル硫酸ナトリウム(SDS)、約0.5MのNaPO4、約1mMのEDTAで約50℃でハイブリダイゼーション、および約0.1XSSC、約0.1%のSDSで約50℃の洗浄;並びになおより好ましくは約7%のドデシル硫酸ナトリウム(SDS)、約0.5MのNaPO4、約1mMのEDTAで約50℃で、約0.1XSSC、約0.1%のSDSで約65℃の洗浄である。もっとも、該条件は、ヌクレオチド鎖の長さ、該配列、および異なる環境パラメーターに依存して異なり得る。より長い配列は、より高い温度で特異的にハイブリダイズする。核酸のハイブリダイゼーションの詳細なガイドは、例えばTijssen(1993) Laboratory Techniques in Biochemistry and Molecular Biology-Hybridization with Nucleic Acid Probes part I chapter 2''Overview of principles of hybridization and the starategy of nucleic acid probe assay''Elsvier, New Yorkに見出される。
【0023】
このように、図1(配列番号1および2)および図2(配列番号3および4)にて示されるプロテインAをコードするDNA配列、およびプロテインAのアミノ酸配列を知ることにより、「プロテインA様蛋白質」、およびそれをコードするDNA配列の存在を認識することは、遺伝子工学分野の技術者にとって容易である。
【0024】
「プロテインA様蛋白質」には、例えば、プロテインAを構成するイムノグロブリン結合ドメイン(E、D、A、B、C)を任意の順列に並べ替えたものも含まれる。
【0025】
さらに、「プロテインA様蛋白質」には、例えば、グループC及びGのストレプトコッカス属細菌(Streptococcal)の有するプロテインG(非特許文献8)、またはペプトストレプトコッカス・マグニウス(Peptostreptococcus magnus)のプロテインL(非特許文献9)といったプロテインAと同様なイムノグロブリン結合機能を有する蛋白質も含まれる。
【0026】
2.プロテインA様蛋白質の部分的配列
プロテインA様蛋白質の「部分的配列」とは、上記プロテインA様蛋白質を構成するアミノ酸配列の任意の一部分から構成されるものであって、かつ、イムノグロブリン結合活性を有する蛋白質を指し、具体的には、例えば、プロテインAから先述のシグナル配列Sおよび細胞壁結合ドメインXの一部を除いて得られる、配列番号8の24番目のAla以降で示されるアミノ酸配列(実施例1の「SPA」に対応。図3および4、配列番号7および8参照。)、および、プロテインAから先述のシグナル配列Sおよび細胞壁結合ドメインXの全部を除いて得られる、配列番号19の31番目のAla以降で示されるアミノ酸配列(実施例5の「SPA’」に対応。図7および8、配列番号18および19参照。)のそれぞれが、プロテインA様蛋白質の「部分的配列」に対応する。
【0027】
さらに上記プロテインGやLが有するイムノグロブリン結合ドメインを構成するアミノ酸配列も「部分的配列」として挙げることができる。
【0028】
本明細書にいうプロテインAのイムノグロブリン結合ドメインとは、例えば、図1における37アミノ酸残基から327アミノ酸残基位(ドメインEからC)、図2における37アミノ酸残基から355アミノ酸残基(ドメインEからC)のことを指す。
【0029】
3.プロテインA様蛋白質をコードするDNA配列
本発明に用いられる、プロテインA様蛋白質をコードするDNA配列は、そのDNA配列を翻訳したアミノ酸配列が、プロテインA様蛋白質を構成するものであればいずれでも良い。そのようなDNA配列は、通常用いられる公知の方法、例えば、ポリメラーゼ・チェーン・リアクション(以下、PCRと略す)法を利用して取得できる。また、公知の化学合成法で合成することも可能であり(Nucleic acids Res.1984. 12:4359)、さらに、DNAライブラリーから得ることもできる。当該DNA配列は、コドンが縮重コドンで置換されていても良く、ブレビバチルス属内で翻訳されたときに同一のアミノ酸をコードしている限り、本来のDNA配列と同一である必要性は無い。
【0030】
4.発現ベクター
本発明の「発現ベクター」は、プロテインA様蛋白質またはその部分的配列をコードするDNA配列、およびその配列に作動可能に連結されたブレビバチルス属細菌で機能しうるプロモーターを含む。当該プロモーターは、ブレビバチルス属細菌で機能しうるものであればいかなるものでも良いが、大腸菌、枯草菌、ブレビバチルス属、スタフィロコッカス属、ストレプトコッカス属、ストレプトミセス属(Streptomyces)、コリネバクテリウム属(Corynebacterium)等細菌由来でブレビバチルス属細菌内にて作動可能なプロモーターが好ましく、ブレビバチルス属細菌細胞壁蛋白質middle wall protein(MWP)、同蛋白質であるouter wallprotein(OWP)(非特許文献10)または、ブレビバチルス・チョウシネンシスHPD31細胞壁蛋白質HWP(Ebisu. Sら. J. Bacteriol. 1990. 172:1312-1320)をコードする遺伝子のプロモーターがより好ましい。実施例では、実施例1に示すブレビバチルス・ブレビス細胞壁蛋白質MWPのP5プロモーター領域「MWP−P5」(図3、4、配列番号7、8参照)、および、実施例5に示すブレビバチルス・ブレビス細胞壁蛋白質MWPのP2プロモーター領域「MWP−P2」(図7、8、配列番号18、19参照)のそれぞれが、「ブレビバチルス属細菌で機能しうるプロモーター」に対応する。
【0031】
また、「発現ベクター」は、該プロモーターの下流に、ブレビバチルス属細菌で機能しうるシャインダルガノ配列及びシグナル配列をさらに含むのが好ましい。発現ベクターは、所望によりマーカー配列を含んでもよい。
【0032】
上記のプロモーター配列に続く「シャインダルガノ配列」は、大腸菌、枯草菌、ブレビバチルス属、スタフィロコッカス属、ストレプトコッカス属、ストレプトミセス属(Streptomyces)、コリネバクテリウム属(Corynebacterium)等細菌由来でブレビバチルス属細菌内にて作動可能なシャインダルガノ配列が好ましく、ブレビバチルス属細菌細胞壁蛋白質middle wall protein(MWP)、同蛋白質であるouterwallprotein(OWP)、または、ブレビバチルス・チョウシネンシスHPD31細胞壁蛋白質HWPをコードする遺伝子の上流に存在するシャインダルガノ配列がより好ましい。
【0033】
上記のシャインダルガノ配列に続く、分泌シグナルペプチドをコードするDNA配列は、以下の分泌シグナルペプチドをコードするDNA配列であれば特に制限は無く、ブレビバチルス・ブレビス内で翻訳されたときに同一のアミノ酸をコードしている限り、本来のDNA配列と同一である必要性は無い。分泌シグナルペプチドとしては、例えば、大腸菌、枯草菌、ブレビバチルス属、スタフィロコッカス属、ストレプトコッカス属、ストレプトミセス属(Streptomyces)、コリネバクテリウム属(Corynebacterium)等細菌由来で、ブレビバチルス属細菌内にて作動可能な分泌シグナルペプチドが好ましく、ブレビバチルス属細菌細胞壁蛋白質middle wall protein(MWP)、同蛋白質であるouter wallprotein(OWP)または、ブレビバチルス・チョウシネンシスHPD31細胞壁蛋白質HWPの分泌シグナルペプチドがより好ましい。また従来の分泌シグナルペプチドのアミノ酸配列を改良したものでも構わない。具体的に言えばmiddle wallprotein(MWP)のシグナルペプチド、Met−Lys−Lys−Val−Val−Asn−Ser−Val−Leu−Ala−Ser−Ala−Leu−Ala−Leu−Thr−Val−Ala−Pro−Met−Ala−Phe−Ala(配列番号11)をMet−Lys−Lys−Arg−Arg−Val−Val−Asn−Asn−Ser−Val−Leu−Leu−Leu−Leu−Leu−Leu−Ala−Ser−Ala−Leu−Ala−Leu−Thr−Val−Ala−Pro−Met−Ala−Phe−Ala(配列番号12)の下線部ように塩基性や疎水性アミノ酸残基など付加または消失させた分泌シグナルペプチドでも構わない。また従来からブレビバチルス属の分泌蛋白質において使われている分泌シグナルペプチドでも構わない。さらに該プロテインAが本来有するシグナルペプチド(図1、2)、すなわち、Met−Lys−Lys−Lys−Asn−Ile−Tyr−Ser−Ile−Arg−Lys−Leu−Gly−Val−Gly−Ile−Ala−Ser−Val−Thr−Leu−Gly−Thr−Leu−Leu−Ile−Ser−Gly−Gly−Val−Thr−Pro−Ala−Ala−Asn−Ala でも構わない。
【0034】
上記のプロモーター配列、シャインダルガノ配列、および分泌シグナルペプチドをコードするDNA配列は、例えば、ブレビバチルス属細菌から得ることができる。好ましくは、ブレビバチルス・ブレビス47(JCM6285)(特開昭60−58074号公報参照)、ブレビバチルス・ブレビス47K(FERM BP−2308)(非特許文献10参照)、ブレビバチルス・ブレビス47−5(FERM BP−1664)、ブレビバチルス・チョウシネンシスHPD31(FERM BP−1087)(特開平4−278091号公報参照)、ブレビバチルス・チョウシネンシスHPD31−S(FERM BP−6623)、またはブレビバチルス・チョウシネンシスHPD31−OK(FERM BP−4573)(特開平6−296485号公報参照)の染色体DNAを鋳型として、公知のPCR法で特異的に増やすことにより取得できる。
【0035】
本発明の「発現ベクター」においては、上記の任意のプロモーターと、上記の任意のシャインダルガノ配列、上記の任意のシグナルペプチド配列と、該プロテインA様蛋白質またはプロテインA様蛋白質の部分的配列をコードするDNA配列とが、ブレビバチルス属細菌内において作動可能に連結されていることが好ましい。
【0036】
ベクターとしては、プラスミドベクターが好ましい。ブレビバチルス属細菌の遺伝子の発現に有用なプラスミドベクターとして具体的には、例えば、枯草菌ベクターとして公知であるpUB110、またはpHY500(特開平2−31682号公報)、pNY700(特開平4−278091号公報)、pHY4831(J. Bacteriol. 1987. 1239-1245)、pNU200(鵜高重三、日本農芸化学会誌1987. 61: 669-676)、pNU100(Appl. Microbiol. Biotechnol., 1989, 30:75-80)、pNU211(J. Biochem., 1992, 112:488-491)、pNU211R2L5(特開平7−170984号公報)、pNH301(Shiga. Y.ら. Appl. Environ. Microbiol. 1992. 58:525-531.)、pNH326、pNH400(Ishihara. Tら、1995. J. Bacteriol, 177:745-749)、pHT210(特開平6−133782号公報)、pHT110R2L5(Appl. Microbiol. Biotechnol., 1994, 42:358-363)、または大腸菌とブレビバチルス属細菌とのシャトルベクターであるpNCO2(特開2002−238569号公報)が使用可能であるが、これらに限定されることはない。また、ブレビバチルス属細菌で機能するプロモーターとシャインダルガノ配列と目的蛋白質をコードするDNA配列とを含んだ発現ベクター、または、それらの各配列を含む遺伝子断片を染色体中へ直接組み込み、発現させる方法(特開平9−135693号公報)を用いても良い。そのような方法は、枯草菌や酵母で既に用いられている公知な方法である。
【0037】
本発明において、プロテインA様蛋白質またはその部分的配列からなる蛋白質は、分泌形態または非分泌形態のいずれで生産されても良いが、分離精製の容易さから培養液中へ分泌させる形態が好ましい。
【0038】
プロテインA様蛋白質またはその部分的配列からなる蛋白質が分泌形態で生産される場合、該ポリペプチドをコードするDNAの上流にブレビバチルス属で機能するシグナルペプチドをコードするDNAを付加または連結するのが好ましい。
【0039】
5.形質転換体
本発明はまた、上記発現ベクターで形質転換されている、ブレビバチルス属細菌形質転換体を提供する。
【0040】
宿主細胞としては、任意のブレビバチルス属細菌を使用し得る。ブレビバチルス属細菌は、限定されないがBrevibacillus agri、B.borstelensis、B. brevis、B. centrosporus、B. choshinensis、B. formosus、B. invocatus、B. laterosporus、B. limnophilus、B. parabrevis、B. reuszeri、B. thermoruberを含む。好ましくは、ブレビバチルス属細菌が、ブレビバチルス・ブレビス47株(JCM6285)、ブレビバチルス・ブレビス47K株(FERM BP−2308)、ブレビバチルス・ブレビス47−5Q株(JCM8970)、ブレビバチルス・チョウシネンシスHPD31株(FERM BP−1087)およびブレビバチルス・チョウシネンシスHPD31−OK株(FERM BP−4573)からなる群より選択される。特に上記のブレビバチルス・ブレビス47株、ブレビバチルス・ブレビス47−5Q株やブレビバチルス・チョウシネンシスHPD31株、ブレビバチルス・チョウシネンシスHPD31−S株が適している。
【0041】
生産量の向上などの目的に応じて、上記ブレビバチルス属細菌のプロテアーゼ欠損株や高発現株のような変異株を使用しても良い。具体的に挙げれば、ブレビバチルス・チョウシネンシスHPD31由来のプロテアーゼ変異株であるブレビバチルス・チョウシネンシスHPD31−OK(特開平6−296485号公報)や、ヒト唾液アミラーゼ高生産株として取得されたブレビバチルス・ブレビス47K(Konishi, H.ら. Appl Microbiol. Biotechnol. 1990. 34:297-302)が使用できる。また前記ブレビバチルス属細菌群に含まれるいずれかの株の変異体を使用してもよい。
【0042】
上記微生物のうち、ブレビバチルス・ブレビス47K(FERM BP−2308)、ブレビバチルス・ブレビス47−5(FERM BP−1664)、ブレビバチルス・チョウシネンシスHPD31(FERM BP−1087)、ブレビバチルス・チョウシネンシスHPD31−S(FERM BP−6623)、及びブレビバチルス・チョウシネンシスHPD31−OK(FERM BP−4573)は、独立行政法人産業技術総合研究所特許生物寄託センター(IPOD;〒305-8566 茨城県つくば市東1丁目1−1中央第6)に、上記それぞれの受託番号にて寄託されている。また、ブレビバチルス・ブレビス47(JCM6285)及びブレビバチルス・ブレビス47−5Q株(JCM8970)は、独立行政法人理化学研究所バイオリソースセンター微生物材料開発室(JCM;〒351-0198 埼玉県和光市広沢2−1)から入手することができる。
【0043】
6.蛋白質の発現調節
ブレビバチルス属細菌を含めた微生物において異種蛋白質を高発現させた場合、正しくフォールディングされずに不活性型の蛋白質を形成することが多く、特にジスルフィド結合の多い蛋白質を高発現させた場合、細胞内外にて不溶性化することも多い。一方で、目的蛋白質を発現させる際、シャペロン蛋白質やジスルフィド結合異性化酵素および/またはプロリン異性化酵素などを作用させることによって、目的蛋白質の不溶性化や分泌効率の低下を抑えられることが知られている。広く試みられている方法は、PDI(プロテインジスルフィドイソメラーゼ)および/またはDsbAなどのジスルフィド酸化還元活性を有する蛋白質を作用させる方法(特開昭63−294796号公報、特開平5−336986号公報)である。
【0044】
さらに、ジスルフィド酸化還元活性を有する蛋白質をコードする遺伝子を宿主生物に導入し、目的蛋白質とジスルフィド酸化還元活性を有する蛋白質とを同時に発現させて正しいジスルフィド結合を有する蛋白質を生産する方法も知られている(特開2000−83670号公報、特表2001−514490号公報等)。
【0045】
本発明によるプロテインA様蛋白質またはその部分的配列からなる蛋白質の発現の場合も、過度な蛋白質合成が行われることによる宿主細胞への負担を軽減し、蛋白質分泌をスムーズに行わせるために、当該蛋白質発現の際、数種類のシャペロン蛋白質、ジスルフィド結合酸化還元酵素、および/またはジスルフィド異性化酵素のようなフォールディングを促進する酵素を同時発現させることも可能である。具体的に挙げれば、ブレビバチルス属細菌において当該蛋白質発現時に、蛋白質のジスルフィド結合に関与し、プロテインジスルフィドイソメラーゼの類縁と考えられている大腸菌のDsbA(Bardwell, J.C.A.ら. Cell. 1991. 67:582-589、Kamitani. Sら. EMBO. J. 1992. 11:57-62.)および/または、DnaK、DnaJ、GrpE(特開平9−180558号公報)などのシャペロン蛋白質を同時に発現させることもできる。その他、ポリペプチドの正確なジスルフィド結合に関与している酵素PDI(特願2001−567367号公報)、ジスルフィド酸化還元酵素(特開2003−169675号公報)(Kontinen, V, P. ら Molecular Microbiology. 1993. 8:727-737)、および/またはジスルフィド異性化酵素のようなフォールディングを促進する酵素を当該蛋白質と同時に発現させ、更に分泌効率を向上させることもできる。
【0046】
7.形質転換体
本発明において用いられるブレビバチルス属細菌の宿主細胞の形質転換は、公知のTakahahiらの方法(Takahashi. Wら. J. Bacteriol. 1983. 156:1130-1134)や、Takagiらの方法(Takagi. H.ら. 1989. Agric. Biol. Chem, 53:3099-3100)、またはOkamotoらの方法(Okamoto. A.ら 1997. Biosci. Biotechnol. Biochem. 61:202-203) により実施することができる。
【0047】
得られた形質転換体の培養に用いる培地は、プロテインA様蛋白質またはその部分的配列からなる蛋白質を高効率、高収量で生産できるものであれば特に制限は無い。具体的にはグルコース、蔗糖、グリセロール、ポリペプトン、肉エキス、酵母エキス、カザミノ酸などの炭素源や窒素源を使用することが出来る。その他、カリウム塩、ナトリウム塩、リン酸塩、マグネシウム塩、マンガン塩、亜鉛塩、鉄塩等の無機塩類が必要に応じて添加される。栄養要求性の宿主細胞を用いる場合は、生育に要求される栄養物質を添加すればよい。また、必要であればペニシリン、エリスロマイシン、クロラムフェニコール、ネオマイシンなどの抗生物質が添加されても良い。さらに、菌体内外に存在する宿主由来のプロテアーゼによる当該目的蛋白質の分解、低分子化を抑えるために、公知の各種プロテアーゼ阻害剤、すなわちPhenylmethane sulfonyl fluoride (PMSF)、Benzamidine、4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF)、Antipain、Chymostatin、Leupeptin、Pepstatin A、Phosphoramidon、Aprotinin、Ethylenediaminetetra acetic acid (EDTA)、および/または、その他市販されているプロテアーゼ阻害剤を適当な濃度で添加しても良い。
【0048】
培養温度は約15−42℃、好ましくは約28−37℃であり、通気攪拌条件で好気的に培養を行うことが望ましいが、必要であれば通気を遮断し嫌気的に培養してもよい。
【0049】
8.プロテインA様蛋白質の取得
本発明の実施形態によれば、形質転換されたブレビバチルス属細菌を培養することにより、プロテインA様蛋白質またはその部分的配列からなる蛋白質は、菌体外、すなわち培養上清中に大量に蓄積されるので、培養上清中から活性型の当該蛋白質を採取、精製することが出来る。また菌体内、及び菌表層に残った当該蛋白質も、公知の方法、例えば超音波やフレンチプレス、アルカリやSDS処理などを利用した方法により菌を破砕し抽出出来る。得られた当該蛋白質は、公知の蛋白質精製方法、例えば硫酸アンモニウムや硫酸ナトリウムなどを用いた塩析、エタノールやアセトン等による濃縮、及び、ゲル濾過、イオン交換、ハイドロキシアパタイト、当該蛋白質のもつ抗体結合活性を利用した各種クロマトグラフィーおよび/または、当該蛋白質が有する親和性を利用したクロマトグラフィーなどを用いて、効果的に精製することができる。
【0050】
本発明の「発現ベクター」により形質転換されたブレビバチルス属細菌形質転換体は、当該蛋白質を安定に発現し、大量に培養上清中へ分泌蓄積することができる。具体的には、適当な培地で培養することによって、SDS−PAGEにおいて分子量4万から5万付近に現れる活性型のプロテインAを培養上清中へ大量に分泌、蓄積させることができる。実施形態による当該蛋白質の生産方法によれば、少なくとも約150mg/L培養液、好ましくは約200mg/L培養液、より好ましくは約500mg/L培養液、もっとも好ましくは約1000mg/L以上の生産量を達成できる。もっとも、生産量は、培養条件等によって変化しうる。
【0051】
9.プロテイン様蛋白質の基材への固定
本発明におけるプロテイン様蛋白質またはその部分的配列からなる蛋白質を固定する「基材」としては、活性炭、ガラスビーズ、シリカゲルなどの無機基材、架橋ポリビニルアルコール、架橋ポリアクリレート、架橋ポリアクリルアミド、架橋ポリスチレンなどの合成高分子、または樹脂、結晶性セルロース、架橋セルロース、架橋アガロース、架橋デキストリンなどの多糖類からなる有機基材、セルロース、ポリビニルアルコール、エチレン−酢酸ビニル共重合体けん化物、ポリアクリルアミド、ポリアクリル酸、ポリメタクリル酸、ポリメタクリル酸メチル、ポリアクリル酸グラフト化ポリエチレン、ポリアクリルアミドグラフト化ポリエチレン、ガラス、さらには、これらの組み合わせによって得られうる有機−有機、有機−無機などの複合基材が代表例として挙げられるが、本発明はこれらのみに限定されるものではない。好ましくは、該基材は水、ナイロン6、ナイロン6,6、ナイロン11、ポリエチレン、ポリ(塩化ビニリデン)、ポリ(塩化ビニル)、ポリ(酢酸ビニル)、ポリスチレン、スチレン−ジビニルベンゼン共重合体、スチレン−ジビニルベンゼン、ポリ(トリフルオロエチレン)、ポリ(クロロトリフィルオロエチレン)、ポリ(テレフタル酸エチレン)、ポリプロピレン、ポリ(アクリル酸メチル)、ポリアクリル酸エステル、ポリ(メタクリル酸メチル)、ポリメタクリル酸エステル、架橋ポリアクリレート、架橋ポリアミドなどの合成高分子化合物からなる群より選択される。基材の形状としては、球状、粒状、平膜状、繊維状、中空糸状等いずれも有効に用いられるが、吸着性能面から球状または粒状がより好ましく用いられる。水不溶性多孔質体が球状または粒状である場合、平均粒径は約5μmから1000μmであることが好ましく、より好ましくは約20〜800μm、最も好ましくは約30〜600μmである。
【0052】
本発明において、プロテインA様蛋白質またはその部分的配列からなる蛋白質の該基材への固定化は、共有または非共有結合、例えばアフィニティ、会合、抗原抗体反応、水素結合、コンジュゲーションなどにより実施しうる。また、当業者に周知の手段により、当該蛋白質に対してアミノ酸残基の付加、置換、欠失などの分子改変を施し、該基材への固定化を容易にすることも可能である。例えば、プロテインA様蛋白質の分子内にシステイン残基を導入することによって、プロテインA様蛋白質を前記基材に容易に固定化することができる。
【0053】
本発明の製造方法によって得られるイムノグロブリン吸着担体は、イムノグロブリン、特にIgGの精製用担体として好適に用いられ得る。また、血漿からのIgGの除去等、疾病治療への応用も可能である。
【実施例】
【0054】
以下、参考例及び実施例に基づいて本発明を具体的に説明するが、これらが本発明の範囲を制限するものでない。本発明の実施にあたり、組換えDNAの作製や操作などは特に断わらない限り下記の実験書に従って実施した。 (1) T. Maniatis, E. F. Fritsch, J. Sambrook著、「モレキュラー・クローニング/ア・ラボラトリー・マニュアル(Molecular Cloning/A Laboratory Manual)」、第2版(1989)、Cold Spring Harbor Laboratory 刊(米国)。 (2) 村松正實編著「ラボマニュアル遺伝子工学」、第3版(1996)、丸善株式会社刊。
【0055】
(実施例1) スタフィロコッカス・アウレウス ATCC6538P株由来のプロテインAをコードするDNA配列のクローニング
スタフィロコッカス・アウレウスATCC6538P株を、T2液体培地(ポリペプトン 1%、イーストエキストラクト 0.2%、グルコース1%、魚肉エキス0.5%、pH7.0)で37℃一晩振とう培養した。得られた培養液から菌体を遠心分離により回収後、10mMのトリス−塩酸緩衝液(pH8.0)で2度洗浄した。菌体を同緩衝液に懸濁後、1%SDSで溶菌し、60℃にて30分間加熱後、フェノール抽出及びエタノール沈殿等の定法により全ゲノムDNAを抽出した。
【0056】
次に、プロテインA遺伝子のDNA配列情報(非特許文献6)を基に、2つのオリゴヌクレオチドプライマー5'-TTGCTCCCATGGCTTTCGCTGCGCAACACGATGAAGCT-3' (配列番号5)と5'-CGGGATCCCTAAAATACAGTTGTACCGATGAATGGATT-3'(配列番号6)を調製した。上記のゲノムDNAを鋳型とし、これら2つのオリゴヌクレオチドプライマーを用いてPCRを行い、プロテインAからシグナルシーケンス(Sドメイン)及び細胞壁結合ドメイン(Xドメイン)の一部を除いた部分(これ以降SPAと称する)をコードするDNA断片(約1.2kbp(キロ塩基対))を増幅した。
【0057】
得られたDNA断片は、制限酵素NcoI及びBamHIにより消化した後、アガロースゲルより分離回収した。
【0058】
一方、ブレビバチルス発現ベクターであるpNH301(Shiga. Y.ら. Appl. Environ. Microbiol. 1992. 58:525-531.)もまた同様に制限酵素NcoI及びBamHIにより消化後、精製回収してアルカリフォスファターゼ処理により脱リン酸化処理を行った。
【0059】
制限酵素処理したSPAをコードする上記DNA断片と上記発現ベクターpNH301とをT4DNAライゲースを用いて連結し、SPA発現プラスミドSpa−pNH301を構築した(図3、4;配列番号7、8)。図3および図4において、「MWP−P5」はブレビバチルス・ブレビス細胞壁蛋白質MWPのP5プロモーター領域、「SDM」はブレビバチルス・ブレビス細胞壁蛋白質MWPのSD配列、「SP」はブレビバチルス・ブレビス細胞壁蛋白質MWPのシグナルペプチド配列、「spa」は「SPA」をコードするDNA配列、「Nm」はネオマイシン耐性遺伝子コード領域、「Rep/pUB110」はベクターpNH301の複製開始点を意味する。また図4において、「P5−35」および「P5−10」は、それぞれ、ブレビバチルス・ブレビス細胞壁蛋白質MWPのP5プロモーターの−35領域および−10領域意味する。公知の方法により、このSpa−pNH301を用いてブレビバチルス・ブレビス47K、またはブレビバチルス・チョウシネンシスHPD31−OK株の形質転換を行った。
【0060】
(実施例2) ブレビバチルス属細菌によるプロテインAの発現試験
実施例1にて得られた形質転換体、及びその対照となるベクターpNH301のみを有するブレビバチルス・チョウシネンシスHPD31−OK株のそれぞれを、生産培地3YC(ポリペプトンS 3%、酵母エキス0.5%、グルコース3%、MgSO4・7H2O 0.01%、CaCl2・7H2O 0.01%、MnSO4・4H2O 0.001%、FeSO4・7H2O 0.001%、ZnSO4・7H2O 0.0001% pH7.0)にネオマイシン60mg/Lを添加した培地にて、30℃で、好気的条件下、3日間培養を行った。培養液は遠心分離(10,000rpm、4℃、5分間)により菌体を除去した後、定法により、10−20%グラジエントゲルを用いた還元条件下でのSDS−PAGEに供した。電気泳動後、ゲルをCBB染色することにより、SPAのバンドを検出した(図5)。SDS−PAGEによる解析の結果、その培養上清液中に大量のSPAを確認することが出来た。
【0061】
なお、プロテインAの細胞壁結合ドメイン(Xドメイン)を含めた完全長型のプロテインAを発現させる場合には、以下の方法を採用することができる。実施例1で記載したスタフィロコッカス・アウレウスから調製したゲノムDNAを鋳型とし、2つのオリゴヌクレオチドプライマー5'- TTGCTCCCATGGCTTTCGCTGCGCAACACGATGAAGCT-3' (配列番号5)と、5'-CGCGGATCCTTATAGTTCGCGACGACG-3'(配列番号9)または5'- CGCGGATCCTCAACGTATATAAGTTAAAAT-3'(配列番号10)とを用い、PCRによりDNA断片を増幅する。得られたプロテインAをコードするDNA断片は、実施例1にて記載された方法によりpNH301のNcoIとBamHIとの間へ連結する。得られたプラスミドをブレビバチルス・ブレビス47Kまたはブレビバチルス・チョウシネンシスHPD31−OK株へ形質転換することで形質転換体を取得する。この形質転換体を実施例2に記載された培養方法にて培養した後、培養液中へ分泌されたプロテインAを確認する。
【0062】
(実施例3) ブレビバチルス属細菌により生産されたプロテインAの抗体結合能の測定実施例1で得られた形質転換体により生産されたSPAが抗体結合能を有するか確認するため、マウス抗ヒトIgG抗体及びアルカリフォスファターゼ標識ウサギ抗マウスIgG抗体を用いて結合試験を行った。
【0063】
実施例1にて得られた、Spa−pNH301、及びその対照となるpNH301をそれぞれ有するブレビバチルス・チョウシネンシスHPD31−OK株を、実施例2と同様に培養し、それぞれの培養上清液をSDS−PAGEに供した後、定法によりPVDFメンブランへ転写し、3%スキムミルクにてブロッキングを行い、Fahnestockら(Fahnestock. S. Rら. J. Bacteriol. 1986. 165:796-804.)らの方法に準じて抗体結合試験を行った。検出は、AP発色キット(バイオラッド社製)を用い、その取扱い説明書に従って実施した。その結果、比較対照であるベクターpNH301のみを有する形質転換体ではバンドは認められなかった。一方、SPA発現ベクターSpa−pNH301を有する形質転換体では、SPAと同じ移動度、すなわちSDS−PAGE上、CBB染色で濃いバンドの認められた42kDa付近に強い発色が認められた(図6)。図6において、「M」は分子量マーカー、「C」はベクターpNH301を有するブレビバチルス・チョウシネンシスHPD31−OK株、SpaはSPA発現ベクターSpa−pNH301を有するブレビバチルス・チョウシネンシスHPD31−OK株のレーンを示す。これらの結果から、SPA発現ベクターSpa−pNH301を有するブレビバチルス・チョウシネンシスHPD31−OK株により生産された分子量約42kDaの蛋白質は、抗体結合活性を有することが確認された。
【0064】
(実施例4) ブレビバチルス発現ベクターpNK3260の構築
pNH326(Ishihara. Tら、1995. J. Bacteriol, 177:745-749)に含まれるMWPのP5プロモーターをMWPのP2プロモーターに変換して、ブレビバチルス発現ベクターpNK3260を以下のように構築した。
【0065】
まず、pNH326を鋳型として、2つのオリゴヌクレオチドプライマー5'-GGAATTCTGATTTCACTTTTTGCATTCTACA-3'(配列番号13)および5'-AGTGCACTCGCACTTACTGT-3'(配列番号14)を用いてPCRを行い、pNH326のうちMWPのP5プロモーターを除く部分を増幅し、その末端を制限酵素EcoRIとHindIIIとで消化した。次に、MWPのP2プロモーターを含む2本鎖DNA断片5'- GGTACCAATTGGCGCCGCAACTTTTGATTCGCTCAGGCGTTTAATAGGATGTAATTGTGAGCGGATAACAATTATTCTGCATGGCTTTCCTGCGAAAGGAGGTGCACCGCGCTTGCAGGATTCGGGCTTTAAAAAGAAAGATAGATTAACAACAAATATTCCCCAAGAACAATTTGTTTATACTGGAGGAGGAGAACACAAGGTCATGAAAAAAAGAAGGGTCGTTAACAGTGTATTGCTTCTGCTACTGCTAGCTAGTGCACTCGCACTTACTGTTGCTCCCATGGCTTTCGCTGCAGGATCCGTCGACTCTAGACTCGAGGAATTCGGTACCCCGGGTTCGAAATCGATAAGCTTCTGT- 3'(配列番号15)を定法に従い調製し、その末端を制限酵素MunIおよびHindIIIで消化した。これら2つのDNA断片をT4DNAライゲースを用いて連結し、pNK3260を構築した。
【0066】
(実施例5) スタフィロコッカス・アウレウス・コワンI株(JCM2179)由来のプロテインAをコードするDNA配列のクローニング
実施例1と同様に、スタフィロコッカス・アウレウス・コワンI株(JCM2179)から全ゲノムDNAを抽出した。次に、プロテインA遺伝子のDNA配列情報(非特許文献2)を基に、2つのオリゴヌクレオチドプライマー5'- TTGCTCCCATGGCTTTCGCTGCGCAACACGATGAAGCTCAACAA-3' (配列番号16)と 5'-CGGGATCCCTATTTTGGTGCTTGAGCATCGTTTAGCTTTTTAGCTTCTGCTAAAATTTTC-3'(配列番号17)を調製した。上記のゲノムDNAを鋳型とし、これら2つのオリゴヌクレオチドプライマーを用いてPCRを行い、プロテインAからシグナルシーケンス(Sドメイン)及び細胞壁結合ドメイン(Xドメイン)を除いた部分(これ以降SPA’と称する)をコードするDNA断片(約0.9kbp)を増幅した。得られたDNA断片は、制限酵素NcoI及びBamHIにより消化した後、アガロースゲルより分離回収した。
【0067】
一方、実施例4で構築したブレビバチルス発現ベクターpNK3260もまた同様に制限酵素NcoI及びBamHIにより消化後、精製回収してアルカリフォスファターゼ処理により脱リン酸化処理を行った。
【0068】
制限酵素処理後の、SPA’をコードする上記DNA断片と上記発現ベクターpNK3260とをT4DNAライゲースを用いて連結し、SPA’発現プラスミドSpa’−pNK3260を構築した(図7、8;配列番号18、19)。図7および図8において、「MWP−P2」はブレビバチルス・ブレビス細胞壁蛋白質MWPのP2プロモーター領域、「SDM」はブレビバチルス・ブレビス細胞壁蛋白質MWPのSD配列、「SP’」はブレビバチルス・ブレビス細胞壁蛋白質MWPのシグナルペプチド配列を一部改変した改変型シグナルペプチド配列、「spa’」はSPA’をコードするDNA配列、「Nm」はネオマイシン耐性遺伝子コード領域、「Rep/pUB110」はベクターpNK3260の複製開始点を意味する。また図8において、「P2−35」および「P2−10」は、それぞれ、ブレビバチルス・ブレビス細胞壁蛋白質MWPのP2プロモーターの−35領域および−10領域意味する。
【0069】
公知の方法により、このSpa’−pNK3260を用いてブレビバチルス・チョウシネンシスHPD31−OK株の形質転換を行った。
【0070】
(実施例6) ブレビバチルス属細菌により分泌発現されたプロテインAの培養液中の挙動
実施例5にて得られた形質転換体を、生産培地3YC(ポリペプトンS 3%、酵母エキス0.5%、グルコース3%、MgSO4・7H2O 0.01%、CaCl2・7H2O 0.01%、MnSO4・4H2O 0.001%、FeSO4・7H2O 0.001%、ZnSO4・7H2O 0.0001% pH7.0)にネオマイシン60mg/Lを添加した培地にて、30℃の、好気的条件下で培養した。培養開始から、24、48、72、78時間後に培養液を分取し、遠心分離(10,000rpm、4℃、5分間)により菌体を除去した後、定法により、10−20%グラジエントゲルを用いた還元条件下でのSDS−PAGEに供した。電気泳動後、ゲルをCBB染色することにより、SPA’のバンドを検出した(図9)。図9において、「レーン番号1」は分子量マーカーを、「レーン番号2」は対照として用いたRepligen社製プロテインA(rPA−50)0.52μgを、「レーン番号3」は培養開始から24時間経過後のSPA’発現ベクターSpa’−pNK3260を有するブレビバチルス・チョウシネンシスHPD31−OK株の培養上清1μlを、「レーン番号4」は培養開始から48時間経過後の同培養上清1μlを、「レーン番号5」は培養開始から72時間経過後の同培養上清1μlを、「レーン番号6」は培養開始から78時間経過後の同培養上清1μlを、それぞれ泳動したレーンを示す。
【0071】
SDS−PAGEによる解析の結果、目的とするSPA’は、培養開始後48時間目(レーン番号4)には大量に発現し、それ以降もSPA’の濃度は上昇しつづけ、最終的に培養上清中に約2g/Lの濃度で蓄積していた。培養上清中のSPA’の濃度は、レーン番号2で泳動したRepligen社製プロテインA(rPA−50)0.52μgのバンドを対照として、ChemiDoc XRSシステム(Bio−Rad社)により測定した。
【0072】
(実施例7) 形質転換体により生産されたプロテインAのN末端アミノ酸配列の確認
図9に示したSDS−PAGEゲル中のレーン番号6において、分子量33kDa付近に見られるSPA’のバンドについて、そのN末端から10残基のアミノ酸配列を、定法に従い解析した。その結果、配列番号2に示したプロテインAのアミノ酸配列の37番目のAla以降の配列と一致し、分泌シグナル配列が正確に除去されていることが確認された。
【0073】
(実施例8) 形質転換体により生産されたプロテインAの抗体結合活性
実施例6と同様に78時間培養して得られた培養液の上清1Lを、陽イオン交換クロマトグラフィー(CM−Sepharose:アマシャムバイオサイエンス)に供し、pH7.0において0−1M塩化ナトリウムの濃度勾配により分離した。次にSPA’画分を回収し、疎水クロマトグラフィー(Phenyl−Sepharose:アマシャムバイオサイエンス)に供し、pH7.0において1−0M硫酸アンモニウムの濃度勾配により分離した。さらに、SPA’画分を回収し、ゲルろ過クロマトグラフィー(HiLoad 16/60 Superdex 75 pg:アマシャムバイオサイエンス)に供し、SPA’画分を回収した。以上の精製操作により、SDS−PAGEにおいて単一バンドを示す、約100mgのSPA’を調製した。
【0074】
上記で調製したSPA’のヒトIgG結合活性を以下の様に評価した。まず、5μg/mLとなるようにSPA’をPBS緩衝液(タカラバイオ)で希釈し、96穴イムノプレート(NUNC)に100μLずつ分注した。37℃で1時間反応させた後に、PBS緩衝液(250μL)にて二回洗浄し、3%ウシ血清アルブミン/PBS溶液を250μL加え、4℃で一晩ブロッキングした。次に、0.1%BSAを含有するPBS緩衝液で調製した25μg/mLヒトIgG(シグマ)溶液100μlを加えて、37℃で、1.5時間反応後、0.01%Tween20を含むPBS緩衝液で洗浄した。これに、PBS緩衝液にて2000倍に希釈したHRP標識プロテインL(0.3mg/ml;シグマ社)溶液を100μl加え、37℃で、1.5時間反応後、0.01%Tween20を含むPBS緩衝液で洗浄した。さらに、発色基質[2,2’−アジノジ(3−エチルベンゾチアゾリンー6−スルフォン酸)アンモニウム塩]溶液(シグマ社)を100μl加え、暗所にて20分間反応後、405nmにおける吸光度を測定した。この時、対照としてRepligen社製プロテインA(rPA−50)についても同様の操作を行い、測定値を比較したところ、上記で調製したSPA’の単位質量当たりのヒトIgG結合活性は、上記Repligen社製プロテインAの約97%であり、両者はほぼ同等の活性を有することが確認された。
【0075】
以上の結果から、実施例によるプロテインAの生産方法によれば、従来報告されていた大腸菌、枯草菌における組換え型プロテインAの発現量を上回る生産性を達成でき、従来問題となっていた低生産性の問題を解決できることを示している。
【特許請求の範囲】
【請求項1】
プロテインA様蛋白質、または、その配列の任意の一部分から構成されイムノグロブリン結合活性を有する蛋白質をコードするDNA、およびその配列に作動可能に連結されたブレビバチルス(Brevibacillus)属細菌で機能しうるプロモーターを含む、DNA。
【請求項2】
該プロモーターが、ブレビバチルス属細菌の細胞壁蛋白質のプロモーターであり、該プロモーターの下流に、ブレビバチルス属細菌で機能しうるシャインダルガノ配列、及びブレビバチルス属細菌で機能しうる分泌シグナルペプチドをコードするDNAをさらに含む、請求項1に記載のDNA。
【請求項3】
発現した該蛋白質がSDS−PAGEにおいて単一バンドを示す、請求項1または2に記載のDNA。
【請求項4】
請求項1〜3のいずれか1項に記載のDNAを含む、発現ベクター。
【請求項5】
請求項4に記載の発現ベクターを含む、ブレビバチルス属細菌形質転換体。
【請求項6】
該ブレビバチルス属細菌が、ブレビバチルス・ブレビス(Brevibacillus brevis)47株(JCM6285)、ブレビバチルス・ブレビス47K株(FERM BP−2308)、ブレビバチルス・ブレビス47−5Q株(JCM8970)、ブレビバチルス・チョウシネンシス(Brevibacillus choshinensis)HPD31(FERM BP−1087)株およびブレビバチルス・チョウシネンシスHPD31−OK株(FERM BP−4573)、並びにこれらの株由来の変異体からなる群より選択される、請求項5に記載の形質転換体。
【請求項7】
培養上清中に約2g/Lの濃度で該蛋白質を蓄積する、請求項5または6に記載の形質転換体。
【請求項8】
請求項5〜7のいずれか1項に記載の形質転換体を培養する工程、および、該形質転換体によって分泌生産されるプロテインA様蛋白質またはその配列の任意の一部分から構成されイムノグロブリン結合活性を有する蛋白質を回収する工程を含む、蛋白質の製造方法。
【請求項9】
請求項8に記載の製造方法によってプロテインA様蛋白質またはその配列の任意の一部分から構成されイムノグロブリン結合活性を有する蛋白質を製造する工程、および該蛋白質を適当な基材に固定化する工程を含む、イムノグロブリン吸着担体の製造方法。
【請求項1】
プロテインA様蛋白質、または、その配列の任意の一部分から構成されイムノグロブリン結合活性を有する蛋白質をコードするDNA、およびその配列に作動可能に連結されたブレビバチルス(Brevibacillus)属細菌で機能しうるプロモーターを含む、DNA。
【請求項2】
該プロモーターが、ブレビバチルス属細菌の細胞壁蛋白質のプロモーターであり、該プロモーターの下流に、ブレビバチルス属細菌で機能しうるシャインダルガノ配列、及びブレビバチルス属細菌で機能しうる分泌シグナルペプチドをコードするDNAをさらに含む、請求項1に記載のDNA。
【請求項3】
発現した該蛋白質がSDS−PAGEにおいて単一バンドを示す、請求項1または2に記載のDNA。
【請求項4】
請求項1〜3のいずれか1項に記載のDNAを含む、発現ベクター。
【請求項5】
請求項4に記載の発現ベクターを含む、ブレビバチルス属細菌形質転換体。
【請求項6】
該ブレビバチルス属細菌が、ブレビバチルス・ブレビス(Brevibacillus brevis)47株(JCM6285)、ブレビバチルス・ブレビス47K株(FERM BP−2308)、ブレビバチルス・ブレビス47−5Q株(JCM8970)、ブレビバチルス・チョウシネンシス(Brevibacillus choshinensis)HPD31(FERM BP−1087)株およびブレビバチルス・チョウシネンシスHPD31−OK株(FERM BP−4573)、並びにこれらの株由来の変異体からなる群より選択される、請求項5に記載の形質転換体。
【請求項7】
培養上清中に約2g/Lの濃度で該蛋白質を蓄積する、請求項5または6に記載の形質転換体。
【請求項8】
請求項5〜7のいずれか1項に記載の形質転換体を培養する工程、および、該形質転換体によって分泌生産されるプロテインA様蛋白質またはその配列の任意の一部分から構成されイムノグロブリン結合活性を有する蛋白質を回収する工程を含む、蛋白質の製造方法。
【請求項9】
請求項8に記載の製造方法によってプロテインA様蛋白質またはその配列の任意の一部分から構成されイムノグロブリン結合活性を有する蛋白質を製造する工程、および該蛋白質を適当な基材に固定化する工程を含む、イムノグロブリン吸着担体の製造方法。
【図1】


【図2】

【図3】
【図4】
【図7】
【図8】
【図5】
【図6】
【図9】
【図2】
【図3】
【図4】
【図7】
【図8】
【図5】
【図6】
【図9】
【公開番号】特開2010−246569(P2010−246569A)
【公開日】平成22年11月4日(2010.11.4)
【国際特許分類】
【出願番号】特願2010−179378(P2010−179378)
【出願日】平成22年8月10日(2010.8.10)
【分割の表示】特願2006−528867(P2006−528867)の分割
【原出願日】平成17年7月1日(2005.7.1)
【出願人】(000000941)株式会社カネカ (3,932)
【Fターム(参考)】
【公開日】平成22年11月4日(2010.11.4)
【国際特許分類】
【出願日】平成22年8月10日(2010.8.10)
【分割の表示】特願2006−528867(P2006−528867)の分割
【原出願日】平成17年7月1日(2005.7.1)
【出願人】(000000941)株式会社カネカ (3,932)
【Fターム(参考)】
[ Back to top ]