
プライマーセット及び相同性組み換え方法
【課題】モーレラ属細菌において、オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子を欠失又は破壊して、ウラシル要求性を付与する形質転換に用いられるプライマーセットの提供。
【解決手段】プライマーセットは、モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子が欠失又は破壊されたウラシル要求性株を作出する際に用いられるプライマーセットであって、前記オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する上流領域を増幅する特定の配列で示されることを特徴とする。
【解決手段】プライマーセットは、モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子が欠失又は破壊されたウラシル要求性株を作出する際に用いられるプライマーセットであって、前記オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する上流領域を増幅する特定の配列で示されることを特徴とする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、相同性組み換えによるモーレラ属細菌における形質転換遺伝子発現のためのプロセスに用いられるプライマーセット及び該プライマーセットを用いた相同性組み換え方法に関し、詳しくは、モーレラ属細菌において、オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子を欠失又は破壊して、ウラシル要求性を付与する形質転換に用いられるプライマーセット及び該プライマーセットを用いた相同性組み換え方法に関する。
【背景技術】
【0002】
モーレラ属細菌は、ガスから酢酸やエタノール生産できる等、産業上有用な性質を有するが、例えばエタノール生産に着目した場合、生産効率に向上の余地がある。
【0003】
本発明者は、モーレラ属細菌を形質転換して、エタノールの生産効率を向上する等の有用な機能を付与することを検討したが、モーレラ属細菌の遺伝学的性質は、他の細菌と比べて特異的であり、また十分に解明されていないことなどから、形質転換は困難であった。例えば、モーレラ属細菌に対してニトロソグアニジン(NTG)など化学物質による変異処理を行った場合は、継代後もエタノール高生産を維持する株は得られず、あるいは、染色体外遺伝子としてプラスミドベクターの導入を試みた場合は、形質転換が確認された株は得られなかった。
【0004】
本発明者は、研究によって、これまでに、モーレラ属細菌であるMoorella sp. HUC22-1株を元株として用い、相同性組み換えによって、ウラシルの生合成系に関与する酵素であるオロチン酸ホスホリボシルトランスフェラーゼをコードする遺伝子(pyrE)を破壊し、ウラシル要求性モーレラ属細菌を得ることに成功している(特許文献1)。
【0005】
しかしながら、かかるウラシル要求性モーレラ属細菌は、pyrEを再度組み込んで相補を行うことが困難であったため、形質転換遺伝子を導入して、形質転換遺伝子を発現させる具体的手法を確立するまでには至っていなかった。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2010−17131号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明者は、更に研究を行い、相同性組み換えによるモーレラ属細菌における形質転換遺伝子発現のためのプロセスの確立を試み、オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子(pyrF)を破壊し、新規なウラシル要求性モーレラ属細菌を得ると共に、このウラシル要求性モーレラ属細菌の染色体に、pyrF及び形質転換遺伝子を導入して、形質転換遺伝子を発現させる具体的手法を見出して本発明を完成するに至った。
【0008】
そこで、本発明の課題は、モーレラ属細菌において、オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子を欠失又は破壊して、ウラシル要求性を付与する形質転換に用いられるプライマーセット及び該プライマーセットを用いた相同性組み換え方法を提供することにある。
【0009】
また本発明の他の課題は、以下の記載によって明らかとなる。
【課題を解決するための手段】
【0010】
上記課題は、以下の各発明によって解決される。
【0011】
(請求項1)
モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子が欠失又は破壊されたウラシル要求性株を作出する際に用いられるプライマーセットであって、
前記オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する上流領域を増幅する配列番号1及び配列番号2で示されることを特徴とするプライマーセット。
【0012】
(請求項2)
モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子が欠失又は破壊されたウラシル要求性株を作出する際に用いられるプライマーセットであって、
前記オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する下流領域を増幅する配列番号3及び配列番号4で示されることを特徴とするプライマーセット。
【0013】
(請求項3)
モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子が欠失又は破壊されたウラシル要求性株を作出する際に用いられるプライマーセットであって、
前記オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する上流領域を増幅する配列番号1及び配列番号2で示されプライマーセット、及び、該オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する下流領域を増幅する配列番号3及び配列番号4で示されるプライマーセットを備えることを特徴とするプライマーセット。
【0014】
(請求項4)
請求項1〜3の何れかに記載のプライマーセットを用いて、モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子を欠失又は破壊してウラシル要求性株を作出することを特徴とする相同性組み換え方法。
【発明の効果】
【0015】
本発明によれば、モーレラ属細菌において、オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子を欠失又は破壊して、ウラシル要求性を付与する形質転換に用いられるプライマーセット及び該プライマーセットを用いた相同性組み換え方法を提供することができる。
【図面の簡単な説明】
【0016】
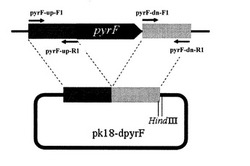
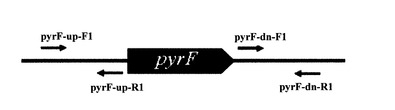
【図1】プライマーの位置を示す図
【図2】pyrF遺伝子破壊ベクターpk18-dpryFの構築方法を示す図
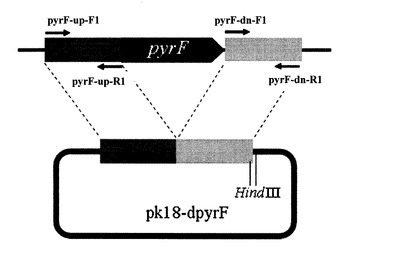
【図3】各プライマーの位置及び予想される断片の長さを示す図
【図4】ダイレクトPCRによるpyrF相補候補株の確認の結果を示す図
【図5】プラスミド構築のスキームを示す図
【図6】pyrF遺伝子上流領域の配列とプライマーの位置を示す図
【図7】KpnI処理による確認の結果を示す図
【図8】コロニーダイレクトPCR結果を示す図
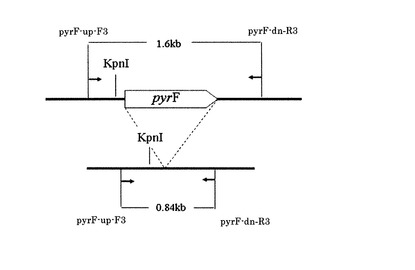
【図9】制限酵素KpnIサイトの位置を示す図
【図10】コロニーダイレクトPCR産物の制限酵素KpnI処理の結果を示す図
【発明を実施するための形態】
【0017】
以下に、本発明を実施するための形態を説明する。
【0018】
本発明に係るプライマーは、相同性組み換えによるモーレラ属細菌における形質転換遺伝子発現のためのプロセスに用いることができる。
【0019】
モーレラ(Moorella)属細菌としては、格別限定されるものではないが、Moorella thermoacetia、M.thermoautotrophica、M.glycerini等を好ましく例示でき、エタノール生産効率が高い菌株の作出の上では、水素と二酸化炭素、又は、一酸化炭素からエタノールを生産する能力を有するモーレラ属細菌であることが好ましく、更には、Moorella thermoacetica ATCC 39073株等を好ましく例示できる。
【0020】
ウラシル要求性とは、菌株がその生育にウラシルを栄養として要求する性質を指す。モーレラ属細菌は、通常ウラシルを生合成できるため、ウラシル要求性を有さないが、変異によりウラシルの生合成ができなくなった場合、ウラシルを栄養として要求するようになる。ウラシル要求性株とは、このようなウラシル要求性を有する菌株である。
【0021】
オロチジン−5−リン酸デカルボキシラーゼは、ウラシルの生合成に必要なピリミジン塩基の前駆体であるUMP(ウリジン一リン酸)の生合成に関与する酵素の一つであり、これをコードする遺伝子を欠いた株は、UMPを生合成できなくなるために、ウラシル要求性株となる。
【0022】
本発明の配列番号1〜4に示されるプライマーセットは、元株となるモーレラ属細菌において、オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子(以下pyrFという場合がある。)を欠失又は破壊して、ウラシル要求性を付与する相同性組換えに用いられる。
【0023】
具体的には、配列番号1及び配列番号2で示されるプライマーセットは、モーレラ属細菌の染色体上において、pyrFに隣接する上流領域を増幅する。
【0024】
一方、配列番号3及び配列番号4で示されるプライマーセットは、モーレラ属細菌の染色体上において、pyrFに隣接する下流領域を増幅する。
【0025】
配列番号1及び配列番号2で示されるプライマーセットを用いたPCRにより得られるPCR産物、及び、配列番号3及び配列番号4で示されるプライマーセットを用いたPCRにより得られる各PCR産物をライゲーションすることにより、pyrFが欠失または破壊されていると共に、pyrFに隣接した上流領域及び下流領域を含むDNA断片が得られる。
【0026】
得られたDNA断片をプラスミドに組み込んでpyrF破壊用プラスミドを構築し、元株となるモーレラ属細菌に導入する。これにより、元株において、相同性組み換えが誘発され、染色体上の遺伝子においてpyrFが欠失または破壊される。
【0027】
本発明において、pyrF破壊用プラスミドをモーレラ属細菌に導入する方法は、格別限定されず、例えば、エレクトロポレーション法等により、モーレラ属細菌に導入する方法を好ましく用いることができる。
【0028】
以上のようにして、pyrFを相同性組換えにより欠失または破壊してなるウラシル要求性モーレラ属細菌(pyrF破壊株)が得られる。
【0029】
pyrF遺伝子が破壊されたことを確認するためには、元株として用いたモーレラ属細菌のゲノムDNAと、pyrF破壊株のゲノムDNAをそれぞれ鋳型にPCR等を行って確認することができる。
【0030】
配列番号5〜配列番号10のプライマーは、作出した細胞からpyrFが欠失又は破壊されているかをPCRにより確認する際に好ましく使用することができる。
【0031】
得られたpyrF破壊株について、pyrF遺伝子を導入して、一度失われたピリミジン生合成経路を回復して、ウラシル要求性を消失させることが可能であること、即ち、相補が可能であることを確認することができる。
【0032】
相補の確認のためには、まず、配列番号1及び配列番号4で示されるプライマーセットを用いて、モーレラ属細菌における染色体上のオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子と、それに隣接する上流領域及び下流領域を増幅する。
【0033】
次いで、増幅されたDNA断片をプラスミドに組み込んで、上流領域及び下流領域からなる相同性部位間にpyrFを有する相補確認用プラスミドを構成する。
【0034】
相補確認用プラスミドをpyrF破壊株に導入し、ウラシル欠損培地での生育が確認されれば、pyrF破壊株が相補可能であることが確認される。
【0035】
本発明の配列番号1〜4に示されるプライマーセットを用いて、元株となるモーレラ属細菌Moorella thermoacetica ATCC 39073株のpyrF遺伝子を相同性組換えによって破壊しウラシル要求性を付与した菌株は、MTA−D−pF株(受託番号:NITE P−1057)として独立行政法人製品評価技術基盤機構特許微生物寄託センターに寄託されている。
【0036】
培養的な性質は、以下の通りである。
【0037】
改変型ATCC 1754 PETC 寒天培地(※1)において、嫌気的、55℃、
培養日数3〜5日で直径3〜5mmの円形コロニーを形成する。
【0038】
i)色: 茶色
ii)表面の形状:スムーズ
iii)透明度:不透明
【0039】
※1 改変型ATCC 1754 PETC 寒天培地
NH4Cl 1.0g
KCl 0.1g
MgSO4 0.2g
NaCl 0.8g
KH2PO4 0.1g
CaCl2 0.02g
Yeast Extract 1.0g
(Yeast Extract無添加の場合 Uracil 0.01g)
NaHCO3 2.0g
Cysteine−HCl 0.3g
Trace element solution(I) 10ml
Vitamin solution (II) 10ml
Distilled water 1000ml
(Agar(高温用) 20g)
(Fructose 5.0g)
pH 5.9(滅菌前)
滅菌温度・時間 121℃ 15分
【0040】
なお、上記の(I)Trace element solution及び(II) Vitamin solutionは以下の組成である。
【0041】
(I)Trace element solution
Nitrilotriacetic acid 2.0g
MnSO4・H2O 1.0g
Fe(SO4)2(NH4)2・6H2O 0.8g
CoCl2・6H2O 0.2g
ZnSO4・7H2O 0.0002g
CuCl2・2H2O 0.02g
NiCl2・6H2O 0.02g
Na2MoO4・2H2O 0.02g
Na2SeO4 0.02g
Na2WO4 0.02g
Distilled water 1000ml
(II)Vitamin solution
Biotin 2.0mg
Folic acid 2.0mg
Pyridoxine−HCl 10mg
Thiamine−HCl 5.0mg
Riboflavin 5.0mg
Nicotinic acid 5.0mg
D−Ca−pantothenate 5.0mg
Vitamin B12 0.1mg
p−Aminobenzoic acid 5.0mg
Thioctic acid 5.0mg
Distilled water 1000ml
【0042】
得られたpyrF破壊株は、相同性部位と、破壊遺伝子がわかっているので、例えばエタノール生産性の向上のような特定の機能を付与する遺伝子を導入する形質転換の元株として好適に用いることができる。
【0043】
例えば、2つの相同性部位の間にpyrF遺伝子及び導入したい形質転換遺伝子(特定の機能を付与する遺伝子)を有する形質転換遺伝子導入用ベクターを作製し、これをpyrF破壊株に導入することで、ピリミジン生合成経路が回復してウラシル要求性を消失させると共に、特定の形質転換遺伝子が発現可能なモーレラ属細菌が形成される。ウラシル欠損培地で生育可能な菌を分離する等、ウラシル要求性に基づいて菌の分離を行うことで、形質転換遺伝子が導入され、特定の機能が付与された形質転換株を容易に得ることができる。
【0044】
形質転換遺伝子導入用ベクターの作出に際しては、配列番号1及び配列番号4で示されるプライマーセットを好適に用いることができる。
【0045】
即ち、配列番号1及び配列番号4で示されるプライマーセットは、モーレラ属細菌における染色体上のオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子と、その上流及び下流に隣接する両隣接領域とを増幅する。
【0046】
配列番号1及び配列番号4で示されるプライマーセットを用いて、モーレラ属細菌における染色体を鋳型に増幅されたDNA断片において、pyrFの上流側又は下流側に形質転換遺伝子を組み込んで、両隣接領域間に、pyrF及び形質転換遺伝子が介在するDNA断片を作成する。
【0047】
作成されたDNA断片をプラスミドに組み込むことにより、形質転換遺伝子導入用ベクターを構成することができる。
【0048】
本発明において、形質転換遺伝子としては、元来モーレラ属細菌が保有していない遺伝子の他に、元来モーレラ属細菌が保有している遺伝子であってもよい。即ち、遺伝子保有数増加(発現増加)の目的で元来モーレラ属細菌が保有している遺伝子を追加的に組み込むことも好ましいことである。
【0049】
pyrF破壊株に形質転換遺伝子導入用ベクターを導入することにより、相同性組み換えが誘発され、pyrF破壊株の染色体上にpyrF及び形質転換遺伝子が組み込まれる。
【0050】
以上のようにして、pyrF破壊株の染色体上に、相同性組み換えによって形質転換遺伝子を導入してなる形質転換遺伝子導入モーレラ属細菌が得られる。
【0051】
かかる形質転換遺伝子導入モーレラ属細菌は、染色体上に、形質転換遺伝子を発現可能に保有することができる。
【実施例】
【0052】
(実施例1)
以下、モーレラ属細菌に対して使用する培地、試薬等は全て嫌気的、無菌的に調製したものを用い、また操作は嫌気雰囲気下で行った。
【0053】
1. Moorella thermoacetica ATCC 39073株のpyrF遺伝子破壊株(ΔpyrF株)の作製
【0054】
1.1. pyrF遺伝子破壊ベクターの構築
以下の手順で、Moorella thermoacetica ATCC 39073株のorotidine-5'-phosphate decarboxylase遺伝子pyrFを破壊するためのベクターを構築した。
【0055】
[pyrF遺伝子破壊ベクターpk18-dpryFの構築]
まず、表1に示したプライマーpyrF-up-F1(配列番号1)及びpyrF-up-R1(配列番号2)、並びに、pyrF-dn-F1(配列番号3)及びpyrF-dn-R1(配列番号4)の組み合わせを用いて、表2に示す条件でPCRを行い、pyrF遺伝子の上流及び下流それぞれ約1000 bpを増幅した。
【0056】
図1及び図2に示す通り、プライマーpyrF-up-F1(配列番号1)及びpyrF-up-R1(配列番号2)は、pyrF遺伝子の上流の隣接領域を増幅し、pyrF-dn-F1(配列番号3)及びpyrF-dn-R1(配列番号4)は、pyrF遺伝子の下流の隣接領域を増幅する。
【0057】
【表1】

【0058】
【表2】
【0059】
得られたPCR産物を、制限酵素SpeIで処理をした後に、MagExtractor Kit(東洋紡製)を用いて精製を行い、pyrF遺伝子上流領域のPCR産物を5μl、下流領域のPCR産物を5μl、Ligation high Ver.2(東洋紡製)を10μl混合し、16℃で3
0分間インキュベートし、ライゲーション産物を鋳型として、プライマーpyrF-up-F1(配列番号1)およびpyrF-dn-R1(配列番号4)を用いて表3に示す条件でPCRを行った。
【0060】
【表3】
【0061】
得られたPCR産物をMagExtractor Kit(東洋紡製)を用いてゲル抽出を行った後、2μlのSmaI処理をしたプラスミドpK18mobを、8μlのゲル抽出したPCR産物、1
0μlのLigation high Ver.2(東洋紡製)と混合し、16℃で1時間インキュベートし
、Escherichia coli HST08 Premiumコンピテントセル(タカラバイオ製)にライゲーション溶液10μlを添加して軽く攪拌した後、氷中に10分静置し、42℃で1分間ヒートショックを与えた後、すぐに氷中に静置した。
【0062】
SOC培地を1ml加え、37℃で1時間インキュベート後、LB寒天培地(カナマイシン、X−gal、IPTG含有)に塗沫し、37℃で一晩培養後、生えてきたコロニーを取得した。
【0063】
[pyrF遺伝子破壊ベクターの確認]
上記(pyrF遺伝子破壊ベクターpk18-dpryFの構築)で生育が見られたコロニーを、
カナマイシンを添加したLB培地に移植した後、コロニーダイレクトPCRを行い、インサートの確認を行った。プライマーは表1に示すpyrF-up-F1(配列番号1)、pyrF-dn-R1(配列番号4)を用いた。コロニーダイレクトPCRの条件を表4に示す。
【0064】
【表4】
【0065】
得られたPCR産物について、電気泳動によりバンドを確認した。
【0066】
バンドが確認できた株を、カナマイシンを添加したLB液体培地で一晩培養し、プラスミド抽出を行った。
【0067】
吸光度による濃度測定および電気泳動による確認後、シーケンスによる塩基配列の解読を行って目的のpyrF遺伝子破壊ベクターpk18-dpryFが構築できていることを確認した。
【0068】
1.2. M. thermoacetica ATCC 39073株のpyrF遺伝子破壊株(ΔpyrF株)の作製
1.1.で構築したpyrF遺伝子破壊ベクターpk18-dpryFを、以下の手順で、M. thermoacetica ATCC 39073株に導入し、ダブルクロスオーバーの相同性組換えによってpyrF遺伝子が破壊された株(ΔpyrF株)を選抜した。
【0069】
[M. thermoacetica ATCC 39073株へのpyrF遺伝子破壊ベクターpk18-dpryFの導入]
272mMスクロース、16mM HEPESの組成で、水酸化カリウムを用いてpH 7に合わせたHS bufferを調製し、20分間煮沸した後、さらに20分間N2ガ
スで置換した。
【0070】
80%の水素、20%の二酸化炭素の混合ガスを基質とし、改変型ATCC 1754
PETC培地、あるいは、グリシンを終濃度5g/Lになるように添加した改変型AT
CC 1754 PETC培地でM. thermoacetica ATCC39073株を培養した。
【0071】
菌体濃度がOD600で約0.3になるまで培養し、培養液約100ml分を集菌した後、HS bufferで菌体を2度洗浄した。
【0072】
洗浄した菌体を適当な量のHS buffer(約3ml)に懸濁し、懸濁液380μ
lとプラスミド20μlを混合した。
【0073】
Bio-Rad Gene Pulser(登録商標)、及び0.2cm gapのキュベット(バイオラ
ッド製)を用いて、1.5kV、500Ω、50μFまたは2.0kV、500Ω、50μFでエレクトロポレーションを行った。
【0074】
エレクトロポレーション後の懸濁液を、ピルビン酸40mMの終濃度で添加した5mlの培地に植菌し、55℃で2日間培養後に、ウラシル10μg/ml、5−フルオロオロ
チン酸(5−FOA)0.2%の終濃度で添加した寒天培地に植菌し、ロールチューブを作製した。
【0075】
[ダイレクトPCRによるpyrF遺伝子破壊株(ΔpyrF株)の確認]
上記寒天培地に形成された20個のコロニーを、5 mlのウラシル10μg/ml、5−FOA 0.2%の終濃度で添加した液体培地に植菌し、培養3日目に培地が濁っていることが確認できた6株を選択し、培養液1mlを集菌した。
【0076】
アクロモペプチダーゼ(20mg/ml)+リゾチーム(20mg/ml)の入ったTE
buffer 20μlで懸濁し、37℃で5分間インキュベートし、DMSOを20μl添加して懸濁し、PCRの鋳型とした。
【0077】
表5に示すプライマーのうち、pyrF-up-F2(配列番号5)及びpyrF-dn-R2(配列番号6)、pyrF-up-F3(配列番号7)及びpyrF-dn-R3(配列番号8)、又は、pyrF-F(配列番号9)及びpyrF-R(配列番号10)の組み合わせを用い、各々の組合せについて、表6に示す条件でコロニーダイレクトPCRを行い、電気泳動によりバンドを確認した。図3は各プライマーの位置及び予想される断片の長さを示している。図3は各プライマーの位置及び予想される断片の長さを示している。
【0078】
【表5】
【0079】
【表6】
【0080】
<評価>
ロールチューブ法によるコロニー形成を試みた結果、多数のコロニーを得た。そこで、コロニーを20株選択し、液体培地(10μg/mlウラシル、0.2% 5−FOA)にて培養した。菌体の生育が確認できた培養液から菌体を集菌し、ダイレクトPCRによる確認を行った。
【0081】
培養3日目に増殖が確認できた6株について、プライマーpyrF-up-F2及びpyrF-dn-R2、又は、pyrF-up-F3及びpyrF-dn-R3の組み合わせを用い、pyrFの外側からPCRを行ったところ、6株中1株において野生株よりも短いバンドが確認できた。
【0082】
さらに、プライマーpyrF-F及びpyrF−Rの組み合わせを用い、pyrFの内
側をPCRしたところ前述の株においてはバンドが確認できなかった。
【0083】
これらの結果から、野生株よりも短いバンドを確認できた株がpyrF遺伝子破壊株(ΔpyrF株)である可能性があると判断し、染色体抽出を行った後にもう一度PCRを行った。その結果、ダイレクトPCRの際と同様のバンドパターンが得られた。
【0084】
さらに、pyrF遺伝子が破壊された部分には制限酵素SpeIサイトが1か所付与されることから(プライマーpyrF-up-R1、pyrF-dn-F1参照)、上記のPCR産物についてSpeIで処理したところ、SpeIサイトで切断が起こり、バンドが2本になることを確認した。
【0085】
次に、pyrF遺伝子破壊候補株についてウラシル要求性試験を行った。酵母エキスを除いた改変型ATCC 1754 PETC培地にpyrF遺伝子破壊株を植菌し、10μg/mlの終濃度でウラシルを添加した場合と添加しなかった場合で増殖の確認を行っ
た。
【0086】
その結果、ウラシルを添加したサンプルでは培養2日目には菌体の増殖が確認できたが、ウラシルを添加しなかったサンプルにおいては増殖が確認できなかった。これらの結果から、pyrF遺伝子の破壊が確認できた。
【0087】
このpyrF遺伝子破壊株は、MTA−D−pF株(受託番号:NITE P−1057)として独立行政法人製品評価技術基盤機構特許微生物寄託センターに寄託されている。
【0088】
2. M. thermoacetica ATCC 39073 pyrF遺伝子破壊株(ΔpyrF株)の相補試験1
【0089】
2.1. pyrF遺伝子相補ベクター(遺伝子発現ベクター)の構築
以下の手順で、M. thermoacetica ATCC 39073 pyrF遺伝子破壊株(ΔpyrF株)の相補試験を行うために、pyrF遺伝子相補ベクターを構築した。
【0090】
[pyrF遺伝子相補ベクターの構築]
表1に示したプライマーpyrF-up-F1(配列番号1)とpyrF-dn-R1(配列番号4)の組み合わせを用いて、表7に示す条件でPCRを行い、pyrF遺伝子翻訳領域とその5’側の約1000bp、3’側の約1000bpを含む約2.7kbpの遺伝子断片を増幅した。
【0091】
【表7】
【0092】
得られたPCR産物についてMagExtractor Kit(東洋紡製)を用いてゲル抽出を行った。
【0093】
2μlのEcoRV処理をしたプラスミドpBluescript II KS+、または、2μlのSmaI処理をしたプラスミドpK18mobを、8μlのゲル抽出したPCR産物、10μlのLigation high Ver.2(東洋紡製)と混合し、16℃で1時間インキュベートした。
【0094】
Escherichia coli HST08 Premiumコンピテントセル(タカラバイオ製)にライゲーション溶液10μlを添加して軽く攪拌し、氷中に10分静置した後、42℃で1分間ヒートショックを与えた後、すぐに氷中に静置した。
【0095】
SOC培地を1ml加え、37℃で1時間インキュベート後、pBluescript II KS+を用いた場合はLB寒天培地(アンピシリン、X−gal、IPTG含有)に、pK18mobを用
いた場合はLB寒天培地(カナマイシン、X−gal、IPTG含有)に塗沫し、37℃で一晩培養後、生えてきたコロニーを取得した。
【0096】
[pyrF遺伝子相補ベクターの確認]
上記で生育が見られたコロニーをアンピシリン、あるいはカナマイシンを添加したLB寒天培地に移植した後、コロニーダイレクトPCRを行い、インサートの確認を行った。プライマーは表1に示すpyrF-up-F1(配列番号1)、pyrF-dn-R1(配列番号4)を用いた。コロニーダイレクトPCRの条件を表8に示す。
【0097】
【表8】
【0098】
さらに、電気泳動によりバンドが確認できた株について、pBluescript II KS+を用いた場合はアンピシリンを、pK18mobを用いた場合はカナマイシン(pK18-epyrF)を添加した
LB液体培地で一晩培養し、プラスミド抽出を行った。
【0099】
吸光度による濃度測定、及び、EcoRI又はPstI処理サンプルの電気泳動を行い、さらに、シーケンスによる塩基配列の解読を行って目的のpyrF遺伝子相補ベクターpK18-epyrFおよびpBS-epyrFが構築できていることを確認した。
【0100】
2.2. M. thermoacetica ATCC 39073 pyrF遺伝子破壊株(ΔpyrF株)へのpyr
F遺伝子相補ベクターpBS-epyrFの導入
以下の手順で、1.2.で構築したM. thermoacetica ATCC 39073 pyrF遺伝子破壊株(
ΔpyrF株)に、2.1.で構築したpyrF遺伝子相補ベクターpBS-epyrFを導入し、相同組換えによる相補試験を行った。
【0101】
[エレクトロポレーションによるpyrF遺伝子相補ベクターpBS-epyrFの導入]
272mMスクロース、16mM HEPESの組成で、水酸化カリウムを用いてpH 6.7に合わせたHS bufferを調製し、20分間煮沸した後、さらに20分間N2ガスで置換した。
【0102】
80%水素、20%二酸化炭素の混合ガスを基質とし、ウラシルを終濃度が10μg/m
lになるように添加した完全合成培地でΔpyrF株を培養した。
【0103】
菌体濃度がOD600で約0.1になるまで培養し、培養液約100ml分を集菌した後、水酸化カリウムでpH7に調整した272mMスクロースバッファーで2度洗浄した。
【0104】
洗浄した菌体を適当な量のHS buffer(約3ml)に懸濁し、懸濁液380μlとプラスミド20μlを混合した。
【0105】
Bio-Rad Gene Pulser(登録商標)、 及び0.2cm gapのキュベット (バイオ
ラッド製)を用いて、1.5kV、500Ω、50μF、または2.0kV、500Ω、50μFでエレクトロポレーションを行った。
【0106】
エレクトロポレーション後の懸濁液をウラシル10μg/ml、ピルビン酸40mMの
終濃度で添加した5mlの完全合成培地に植菌し、55℃で2日間培養後、完全合成培地で洗浄し、完全合成培地と寒天を含む培地に植菌し、ロールチューブを作製した。
【0107】
[ダイレクトPCRによるpyrF遺伝子相補株の確認]
上記で得られたコロニーを5mlの完全合成培地に植菌し、培養4日目に培地が濁っているサンプルを選択し、培養液1mlを集菌した。
【0108】
アクロモペプチダーゼ(20mg/ml)+リゾチーム(20mg/ml)の入ったTE
buffer10μlで懸濁し、37℃で5分間インキュベートし、DMSOを10μl添加して懸濁し、PCRの鋳型とした。
【0109】
表5に示すpyrF-up-F3(配列番号7)、pyrF-dn-R3(配列番号8)の組み合わせのプライマーを用い、表9に示す条件でコロニーダイレクトPCRを行った。
【0110】
電気泳動によりバンドを確認し、pyrF遺伝子相補株が得られたと判断した。
【0111】
【表9】
【0112】
<評価>
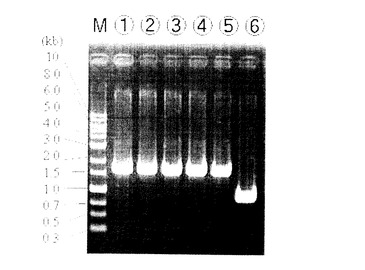
培養4日目で培養液が白く濁っていたサンプル4株についてダイレクトPCRによる確認を実施した。結果を図4に示す。
【0113】
図4の電気泳動結果において、レーン1〜4は相補株、レーン5は野生株、レーン6はpyrF破壊株であり、今回確認した4株全てが野生株と同じ約1.6kbpのサイズの位置にバンドを示した。これは、相補プラスミドがエレクトロポレーションによりpyrF遺伝子破壊株の細胞内へ取り込まれ、相同組換えによりpyrFが元の場所に挿入されたことを意味している。尚、pyrF破壊株ではバンドの長さは約0.9 bpであり、
予想された大きさであった。
【0114】
3. M. thermoacetica ATCC 39073 pyrF遺伝子破壊株(ΔpyrF株)の相補試験2
【0115】
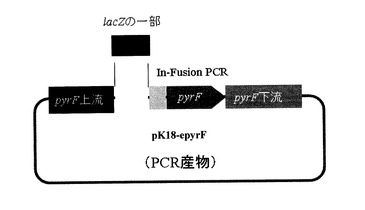
3.1. 形質転換DNA挿入用相補ベクターの構築
ΔpyrF株に対するpyrF遺伝子相補試験を行う際に、pyrF上流に形質転換遺伝子の一部を挿入し、相同組換えによる染色体加工が可能であることを示すためのプラスミドを構築する。具体的には、Thermoanaerobacter ethanolicus 39E株のlacZ遺伝子の一部(約500bp)を2.1.で構築したpyrF遺伝子相補ベクターpK18-epyrFの
pyrF遺伝子上流へ挿入した。手順を以下に示す。
【0116】
[In−Fusion PCRによる形質転換DNA挿入用相補ベクターの構築]
表10に示すプライマーpyrF-1-R(配列番号11)及びpyrF-1-F(配列番号12)、pyrF-2-R(配列番号13)及びpyrF-2-F(配列番号14)、又は、pyrF-3-R(配列番号15)及びpyrF-3-F(配列番号16)の各組み合わせを用いて、pyrF遺伝子領域を含むベクター領域をPCR増幅した。何れの組合せにおいても、PCRの条件は表11に示した通りである。
【0117】
【表10】
【0118】
【表11】
【0119】
表10に示したプライマーlacZ-500-F(配列番号17)とlacZ-500-R(配列暗号18)を用いて、lacZ遺伝子領域をPCR増幅した。PCRの条件は表12に示した通りである。
【0120】
【表12】
【0121】
各PCR産物をゲル抽出し、表13に示した条件で、In−Fusion PCR(In-Fusion・Advantage PCR Cloning Kit、タカラバイオ製)を行った。
【0122】
【表13】
【0123】
In−Fusionサンプルに滅菌水を50μl加えて希釈し、希釈したサンプル10μlを形質転換に用いた。
【0124】
ダイレクトPCRによりコロニーを選択した。
【0125】
<評価>
pyrFの上流領域へlacZ遺伝子の一部を挿入するために、2.1.で構築したpK18-epyrFを鋳型とし、プライマーpyrF-1-R及びpyrF-1-F、pyrF-2-R及びpyrF-2-F、又は、pyrF-3-R及びpyrF-3-Fの組み合わせを用いてインバースPCRを行った。その際に、プロモーター領域が300bp、203bp、147bpとなるようにプライマーの位置をずらして3パターンのPCR産物を得た(図5及び図6)。さらに、In−FusionPCRを用いてプラスミドへlacZ遺伝子を挿入するため、プライマーにはlacZ遺伝子と相同配列を付加しておいた。表10において配列番号11〜16のSequence下線部はプロモーター領域である。下線以外の部分がプライマーの位置を決める配列であり、配列番号11は図6における丸囲み数字1、配列番号12は図6における丸囲み数字2、配列番号13は図6における丸囲み数字3、配列番号14は図6における丸囲み数字4、配列番号15は図6における丸囲み数字5、配列番号16は図6における丸囲み数字6に、矢印の向きで対応している。
【0126】
In−Fusion PCRおよび大腸菌における形質転換の結果、複数のコロニーを
得た。lacZ-500-FおよびlacZ-500-RをプライマーとしたコロニーダイレクトPCRの結果、全ての株において目的のバンドが確認できた。
【0127】
それらの株の中から3株ずつを培養し、プラスミドを抽出した。プライマーpyrF-1-R及びpyrF-1-Fの組み合わせによるプラスミドをpK18-pyz-1とし、プライマーpyrF-2-R及びpyrF-2-Fの組み合わせによるプラスミドをpK18-pyz-2とし、さらに、プライマーpyrF-3-R及びpyrF-3-Fの組み合わせによるプラスミドをpK18-pyz-3とする。
【0128】
制限酵素KpnI処理による確認の結果、図7に示す通り、全ての株においてlacZ遺伝子の挿入が確認できた。
【0129】
図7の電気泳動結果において、レーン1、2、3は、プロモーター領域300bp(pK18-pyz-1)、レーン4、5、6は、プロモーター領域203bp(pK18-pyz-2)、レーン7、8、9は、プロモーター領域147bp(pK18-pyz-3)に各々対応している。レーン10は、pK18-epyrFである。
【0130】
3.2. M. thermoacetica ATCC 39073 pyrF遺伝子破壊株(ΔpyrF株)への形質転
換DNA挿入用相補ベクターpK18-pyz-1、pK18-pyz-2、pK18-pyz-3の導入
以下の手順で、1.2.で構築したM. thermoacetica ATCC 39073 pyrF遺伝子破壊株(
ΔpyrF株)に、3.1.で構築した形質転換DNA挿入用相補ベクターpK18-pyz-1、pK18-pyz-2、pK18-pyz-3を導入し、相同組換えによる相補試験を行った。
【0131】
[エレクトロポレーションによる形質転換DNA挿入用相補ベクターpK18-pyz-1、pK18-pyz-2、pK18-pyz-3の導入]
272mMスクロース、16mM HEPESの組成で、水酸化カリウムを用いてpH 6.7に合わせたHS bufferを調製し、20分間煮沸した後、さらに20分間N
2ガスで置換した。
【0132】
80%水素、20%二酸化炭素の混合ガスを基質とし、ウラシルを終濃度が10μg/m
lになるように添加した完全合成培地でΔpyrF株を培養した。
【0133】
菌体濃度がOD600で約0.1になるまで培養し、培養液約50ml分を集菌した後、水酸化カリウムでpH7に調整した272mMスクロースバッファーで2度洗浄し、洗浄した菌体を適当な量のHS buffer(約3ml)に懸濁した。
【0134】
懸濁液380μlとプラスミド5〜10μlを混合し、Bio-Rad Gene Pulser(登録商
標)、及び0.2cm gapのキュベット (バイオラッド製)を用いて、1.5 kV、500Ω、50μF、または2.0kV、500Ω、50μFでエレクトロポレーションを行った。
【0135】
エレクトロポレーション後の懸濁液を、ウラシル10μg/mlの終濃度で添加した5
mlの完全合成培地に植菌し、80%水素、20%二酸化炭素の混合ガスを基質として培養し、55℃で2日間培養後、完全合成培地で洗浄し、完全合成培地と寒天を含む培地に植菌し、ロールチューブを作製した。
【0136】
[ダイレクトPCRによるpyrF遺伝子相補株の確認]
上記で得られたコロニーを5mlの完全合成培地に植菌し、培養後に培地が濁っているサンプルを選択し、培養液1mlを集菌した。
【0137】
アクロモペプチダーゼ(20mg/ml)+リゾチーム(20mg/ml)の入ったTE
buffer 10μlで懸濁し、37℃で5分間インキュベートした後、DMSOを
10μl添加して懸濁し、PCRの鋳型とした。
【0138】
表5に示したpyrF-up-F3(配列番号7)、pyrF-dn-R3(配列番号8)の組み合わせのプライマーを用い、表14に示す条件でコロニーダイレクトPCRを行った。
【0139】
電気泳動によりバンドを確認し、pyrF遺伝子相補株が得られたと判断した。
【0140】
【表14】
【0141】
<評価>
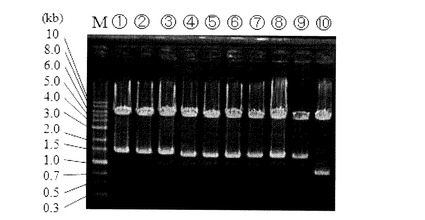
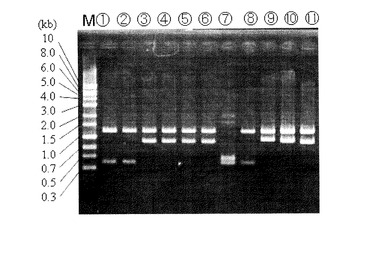
計20コロニー取得し、そのうち8株についてダイレクトPCRによる確認を実施した(図8)。
【0142】
図8の電気泳動結果において、レーン1〜8は、コロニー単離株、レーン9は、ΔpyrF株ダイレクト、レーン10は、ATCC39073野生株ダイレクト、レーン11は、ATCC39073野生株の抽出DNA、レーン12は、形質転換DNA挿入用相補ベクターpK18-pyz-1、レーン13は、pyrF遺伝子破壊ベクターpk18-dpryFに各々対応している。
【0143】
図8に示される通り、4株(レーン3、4、5、6)で野生株(レーン10、11)より大きいサイズの約2.1kbpの位置にバンドが見られ、約500bpのLacZ遺伝子(形質転換遺伝子)の導入が確認された。残り4株については野生株と同じ約1.6kbpのサイズの位置にバンドを示した。
【0144】
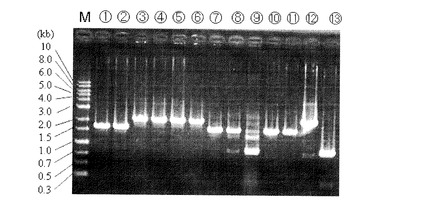
次に、6株分のPCR産物を精製し、制限酵素KpnIで処理して電気泳動にて確認した。KpnIの制限酵素サイトは図9の位置にある。PCR産物をKpnIで処理すると2本のバンドが見えるが、形質転換遺伝子が挿入されていた場合には下側のバンドが野生株の時よりも大きい位置に現れる。結果を図10に示す。
【0145】
図10において、レーン1〜6は、コロニー単離株、レーン7は、ΔpyrF株ダイレクト、レーン8は、ATCC39073野生株の抽出DNA、レーン9は、形質転換DNA挿入用
相補ベクターpK18-pyz-1、レーン10は、形質転換DNA挿入用相補ベクターpK18-pyz-2、レーン11は、形質転換DNA挿入用相補ベクターpK18-pyz-3に各々対応している。
【0146】
図10に示される通り、レーン1、2は野生株と同じバンドが確認され、レーン3、4、5、6では野生株より大きい位置にバンドが見られ、形質転換遺伝子LacZの挿入が確認された。
【0147】
さらに、ダイレクトPCRではなく、増殖によって得られた菌体からTotal DNAを抽出し、同様の条件でPCRを行って形質転換遺伝子LacZの挿入を確認した結果、ダイレクトPCRの時と同じ結果が得られた。
【技術分野】
【0001】
本発明は、相同性組み換えによるモーレラ属細菌における形質転換遺伝子発現のためのプロセスに用いられるプライマーセット及び該プライマーセットを用いた相同性組み換え方法に関し、詳しくは、モーレラ属細菌において、オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子を欠失又は破壊して、ウラシル要求性を付与する形質転換に用いられるプライマーセット及び該プライマーセットを用いた相同性組み換え方法に関する。
【背景技術】
【0002】
モーレラ属細菌は、ガスから酢酸やエタノール生産できる等、産業上有用な性質を有するが、例えばエタノール生産に着目した場合、生産効率に向上の余地がある。
【0003】
本発明者は、モーレラ属細菌を形質転換して、エタノールの生産効率を向上する等の有用な機能を付与することを検討したが、モーレラ属細菌の遺伝学的性質は、他の細菌と比べて特異的であり、また十分に解明されていないことなどから、形質転換は困難であった。例えば、モーレラ属細菌に対してニトロソグアニジン(NTG)など化学物質による変異処理を行った場合は、継代後もエタノール高生産を維持する株は得られず、あるいは、染色体外遺伝子としてプラスミドベクターの導入を試みた場合は、形質転換が確認された株は得られなかった。
【0004】
本発明者は、研究によって、これまでに、モーレラ属細菌であるMoorella sp. HUC22-1株を元株として用い、相同性組み換えによって、ウラシルの生合成系に関与する酵素であるオロチン酸ホスホリボシルトランスフェラーゼをコードする遺伝子(pyrE)を破壊し、ウラシル要求性モーレラ属細菌を得ることに成功している(特許文献1)。
【0005】
しかしながら、かかるウラシル要求性モーレラ属細菌は、pyrEを再度組み込んで相補を行うことが困難であったため、形質転換遺伝子を導入して、形質転換遺伝子を発現させる具体的手法を確立するまでには至っていなかった。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2010−17131号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明者は、更に研究を行い、相同性組み換えによるモーレラ属細菌における形質転換遺伝子発現のためのプロセスの確立を試み、オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子(pyrF)を破壊し、新規なウラシル要求性モーレラ属細菌を得ると共に、このウラシル要求性モーレラ属細菌の染色体に、pyrF及び形質転換遺伝子を導入して、形質転換遺伝子を発現させる具体的手法を見出して本発明を完成するに至った。
【0008】
そこで、本発明の課題は、モーレラ属細菌において、オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子を欠失又は破壊して、ウラシル要求性を付与する形質転換に用いられるプライマーセット及び該プライマーセットを用いた相同性組み換え方法を提供することにある。
【0009】
また本発明の他の課題は、以下の記載によって明らかとなる。
【課題を解決するための手段】
【0010】
上記課題は、以下の各発明によって解決される。
【0011】
(請求項1)
モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子が欠失又は破壊されたウラシル要求性株を作出する際に用いられるプライマーセットであって、
前記オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する上流領域を増幅する配列番号1及び配列番号2で示されることを特徴とするプライマーセット。
【0012】
(請求項2)
モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子が欠失又は破壊されたウラシル要求性株を作出する際に用いられるプライマーセットであって、
前記オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する下流領域を増幅する配列番号3及び配列番号4で示されることを特徴とするプライマーセット。
【0013】
(請求項3)
モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子が欠失又は破壊されたウラシル要求性株を作出する際に用いられるプライマーセットであって、
前記オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する上流領域を増幅する配列番号1及び配列番号2で示されプライマーセット、及び、該オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する下流領域を増幅する配列番号3及び配列番号4で示されるプライマーセットを備えることを特徴とするプライマーセット。
【0014】
(請求項4)
請求項1〜3の何れかに記載のプライマーセットを用いて、モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子を欠失又は破壊してウラシル要求性株を作出することを特徴とする相同性組み換え方法。
【発明の効果】
【0015】
本発明によれば、モーレラ属細菌において、オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子を欠失又は破壊して、ウラシル要求性を付与する形質転換に用いられるプライマーセット及び該プライマーセットを用いた相同性組み換え方法を提供することができる。
【図面の簡単な説明】
【0016】
【図1】プライマーの位置を示す図
【図2】pyrF遺伝子破壊ベクターpk18-dpryFの構築方法を示す図
【図3】各プライマーの位置及び予想される断片の長さを示す図
【図4】ダイレクトPCRによるpyrF相補候補株の確認の結果を示す図
【図5】プラスミド構築のスキームを示す図
【図6】pyrF遺伝子上流領域の配列とプライマーの位置を示す図
【図7】KpnI処理による確認の結果を示す図
【図8】コロニーダイレクトPCR結果を示す図
【図9】制限酵素KpnIサイトの位置を示す図
【図10】コロニーダイレクトPCR産物の制限酵素KpnI処理の結果を示す図
【発明を実施するための形態】
【0017】
以下に、本発明を実施するための形態を説明する。
【0018】
本発明に係るプライマーは、相同性組み換えによるモーレラ属細菌における形質転換遺伝子発現のためのプロセスに用いることができる。
【0019】
モーレラ(Moorella)属細菌としては、格別限定されるものではないが、Moorella thermoacetia、M.thermoautotrophica、M.glycerini等を好ましく例示でき、エタノール生産効率が高い菌株の作出の上では、水素と二酸化炭素、又は、一酸化炭素からエタノールを生産する能力を有するモーレラ属細菌であることが好ましく、更には、Moorella thermoacetica ATCC 39073株等を好ましく例示できる。
【0020】
ウラシル要求性とは、菌株がその生育にウラシルを栄養として要求する性質を指す。モーレラ属細菌は、通常ウラシルを生合成できるため、ウラシル要求性を有さないが、変異によりウラシルの生合成ができなくなった場合、ウラシルを栄養として要求するようになる。ウラシル要求性株とは、このようなウラシル要求性を有する菌株である。
【0021】
オロチジン−5−リン酸デカルボキシラーゼは、ウラシルの生合成に必要なピリミジン塩基の前駆体であるUMP(ウリジン一リン酸)の生合成に関与する酵素の一つであり、これをコードする遺伝子を欠いた株は、UMPを生合成できなくなるために、ウラシル要求性株となる。
【0022】
本発明の配列番号1〜4に示されるプライマーセットは、元株となるモーレラ属細菌において、オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子(以下pyrFという場合がある。)を欠失又は破壊して、ウラシル要求性を付与する相同性組換えに用いられる。
【0023】
具体的には、配列番号1及び配列番号2で示されるプライマーセットは、モーレラ属細菌の染色体上において、pyrFに隣接する上流領域を増幅する。
【0024】
一方、配列番号3及び配列番号4で示されるプライマーセットは、モーレラ属細菌の染色体上において、pyrFに隣接する下流領域を増幅する。
【0025】
配列番号1及び配列番号2で示されるプライマーセットを用いたPCRにより得られるPCR産物、及び、配列番号3及び配列番号4で示されるプライマーセットを用いたPCRにより得られる各PCR産物をライゲーションすることにより、pyrFが欠失または破壊されていると共に、pyrFに隣接した上流領域及び下流領域を含むDNA断片が得られる。
【0026】
得られたDNA断片をプラスミドに組み込んでpyrF破壊用プラスミドを構築し、元株となるモーレラ属細菌に導入する。これにより、元株において、相同性組み換えが誘発され、染色体上の遺伝子においてpyrFが欠失または破壊される。
【0027】
本発明において、pyrF破壊用プラスミドをモーレラ属細菌に導入する方法は、格別限定されず、例えば、エレクトロポレーション法等により、モーレラ属細菌に導入する方法を好ましく用いることができる。
【0028】
以上のようにして、pyrFを相同性組換えにより欠失または破壊してなるウラシル要求性モーレラ属細菌(pyrF破壊株)が得られる。
【0029】
pyrF遺伝子が破壊されたことを確認するためには、元株として用いたモーレラ属細菌のゲノムDNAと、pyrF破壊株のゲノムDNAをそれぞれ鋳型にPCR等を行って確認することができる。
【0030】
配列番号5〜配列番号10のプライマーは、作出した細胞からpyrFが欠失又は破壊されているかをPCRにより確認する際に好ましく使用することができる。
【0031】
得られたpyrF破壊株について、pyrF遺伝子を導入して、一度失われたピリミジン生合成経路を回復して、ウラシル要求性を消失させることが可能であること、即ち、相補が可能であることを確認することができる。
【0032】
相補の確認のためには、まず、配列番号1及び配列番号4で示されるプライマーセットを用いて、モーレラ属細菌における染色体上のオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子と、それに隣接する上流領域及び下流領域を増幅する。
【0033】
次いで、増幅されたDNA断片をプラスミドに組み込んで、上流領域及び下流領域からなる相同性部位間にpyrFを有する相補確認用プラスミドを構成する。
【0034】
相補確認用プラスミドをpyrF破壊株に導入し、ウラシル欠損培地での生育が確認されれば、pyrF破壊株が相補可能であることが確認される。
【0035】
本発明の配列番号1〜4に示されるプライマーセットを用いて、元株となるモーレラ属細菌Moorella thermoacetica ATCC 39073株のpyrF遺伝子を相同性組換えによって破壊しウラシル要求性を付与した菌株は、MTA−D−pF株(受託番号:NITE P−1057)として独立行政法人製品評価技術基盤機構特許微生物寄託センターに寄託されている。
【0036】
培養的な性質は、以下の通りである。
【0037】
改変型ATCC 1754 PETC 寒天培地(※1)において、嫌気的、55℃、
培養日数3〜5日で直径3〜5mmの円形コロニーを形成する。
【0038】
i)色: 茶色
ii)表面の形状:スムーズ
iii)透明度:不透明
【0039】
※1 改変型ATCC 1754 PETC 寒天培地
NH4Cl 1.0g
KCl 0.1g
MgSO4 0.2g
NaCl 0.8g
KH2PO4 0.1g
CaCl2 0.02g
Yeast Extract 1.0g
(Yeast Extract無添加の場合 Uracil 0.01g)
NaHCO3 2.0g
Cysteine−HCl 0.3g
Trace element solution(I) 10ml
Vitamin solution (II) 10ml
Distilled water 1000ml
(Agar(高温用) 20g)
(Fructose 5.0g)
pH 5.9(滅菌前)
滅菌温度・時間 121℃ 15分
【0040】
なお、上記の(I)Trace element solution及び(II) Vitamin solutionは以下の組成である。
【0041】
(I)Trace element solution
Nitrilotriacetic acid 2.0g
MnSO4・H2O 1.0g
Fe(SO4)2(NH4)2・6H2O 0.8g
CoCl2・6H2O 0.2g
ZnSO4・7H2O 0.0002g
CuCl2・2H2O 0.02g
NiCl2・6H2O 0.02g
Na2MoO4・2H2O 0.02g
Na2SeO4 0.02g
Na2WO4 0.02g
Distilled water 1000ml
(II)Vitamin solution
Biotin 2.0mg
Folic acid 2.0mg
Pyridoxine−HCl 10mg
Thiamine−HCl 5.0mg
Riboflavin 5.0mg
Nicotinic acid 5.0mg
D−Ca−pantothenate 5.0mg
Vitamin B12 0.1mg
p−Aminobenzoic acid 5.0mg
Thioctic acid 5.0mg
Distilled water 1000ml
【0042】
得られたpyrF破壊株は、相同性部位と、破壊遺伝子がわかっているので、例えばエタノール生産性の向上のような特定の機能を付与する遺伝子を導入する形質転換の元株として好適に用いることができる。
【0043】
例えば、2つの相同性部位の間にpyrF遺伝子及び導入したい形質転換遺伝子(特定の機能を付与する遺伝子)を有する形質転換遺伝子導入用ベクターを作製し、これをpyrF破壊株に導入することで、ピリミジン生合成経路が回復してウラシル要求性を消失させると共に、特定の形質転換遺伝子が発現可能なモーレラ属細菌が形成される。ウラシル欠損培地で生育可能な菌を分離する等、ウラシル要求性に基づいて菌の分離を行うことで、形質転換遺伝子が導入され、特定の機能が付与された形質転換株を容易に得ることができる。
【0044】
形質転換遺伝子導入用ベクターの作出に際しては、配列番号1及び配列番号4で示されるプライマーセットを好適に用いることができる。
【0045】
即ち、配列番号1及び配列番号4で示されるプライマーセットは、モーレラ属細菌における染色体上のオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子と、その上流及び下流に隣接する両隣接領域とを増幅する。
【0046】
配列番号1及び配列番号4で示されるプライマーセットを用いて、モーレラ属細菌における染色体を鋳型に増幅されたDNA断片において、pyrFの上流側又は下流側に形質転換遺伝子を組み込んで、両隣接領域間に、pyrF及び形質転換遺伝子が介在するDNA断片を作成する。
【0047】
作成されたDNA断片をプラスミドに組み込むことにより、形質転換遺伝子導入用ベクターを構成することができる。
【0048】
本発明において、形質転換遺伝子としては、元来モーレラ属細菌が保有していない遺伝子の他に、元来モーレラ属細菌が保有している遺伝子であってもよい。即ち、遺伝子保有数増加(発現増加)の目的で元来モーレラ属細菌が保有している遺伝子を追加的に組み込むことも好ましいことである。
【0049】
pyrF破壊株に形質転換遺伝子導入用ベクターを導入することにより、相同性組み換えが誘発され、pyrF破壊株の染色体上にpyrF及び形質転換遺伝子が組み込まれる。
【0050】
以上のようにして、pyrF破壊株の染色体上に、相同性組み換えによって形質転換遺伝子を導入してなる形質転換遺伝子導入モーレラ属細菌が得られる。
【0051】
かかる形質転換遺伝子導入モーレラ属細菌は、染色体上に、形質転換遺伝子を発現可能に保有することができる。
【実施例】
【0052】
(実施例1)
以下、モーレラ属細菌に対して使用する培地、試薬等は全て嫌気的、無菌的に調製したものを用い、また操作は嫌気雰囲気下で行った。
【0053】
1. Moorella thermoacetica ATCC 39073株のpyrF遺伝子破壊株(ΔpyrF株)の作製
【0054】
1.1. pyrF遺伝子破壊ベクターの構築
以下の手順で、Moorella thermoacetica ATCC 39073株のorotidine-5'-phosphate decarboxylase遺伝子pyrFを破壊するためのベクターを構築した。
【0055】
[pyrF遺伝子破壊ベクターpk18-dpryFの構築]
まず、表1に示したプライマーpyrF-up-F1(配列番号1)及びpyrF-up-R1(配列番号2)、並びに、pyrF-dn-F1(配列番号3)及びpyrF-dn-R1(配列番号4)の組み合わせを用いて、表2に示す条件でPCRを行い、pyrF遺伝子の上流及び下流それぞれ約1000 bpを増幅した。
【0056】
図1及び図2に示す通り、プライマーpyrF-up-F1(配列番号1)及びpyrF-up-R1(配列番号2)は、pyrF遺伝子の上流の隣接領域を増幅し、pyrF-dn-F1(配列番号3)及びpyrF-dn-R1(配列番号4)は、pyrF遺伝子の下流の隣接領域を増幅する。
【0057】
【表1】
【0058】
【表2】
【0059】
得られたPCR産物を、制限酵素SpeIで処理をした後に、MagExtractor Kit(東洋紡製)を用いて精製を行い、pyrF遺伝子上流領域のPCR産物を5μl、下流領域のPCR産物を5μl、Ligation high Ver.2(東洋紡製)を10μl混合し、16℃で3
0分間インキュベートし、ライゲーション産物を鋳型として、プライマーpyrF-up-F1(配列番号1)およびpyrF-dn-R1(配列番号4)を用いて表3に示す条件でPCRを行った。
【0060】
【表3】
【0061】
得られたPCR産物をMagExtractor Kit(東洋紡製)を用いてゲル抽出を行った後、2μlのSmaI処理をしたプラスミドpK18mobを、8μlのゲル抽出したPCR産物、1
0μlのLigation high Ver.2(東洋紡製)と混合し、16℃で1時間インキュベートし
、Escherichia coli HST08 Premiumコンピテントセル(タカラバイオ製)にライゲーション溶液10μlを添加して軽く攪拌した後、氷中に10分静置し、42℃で1分間ヒートショックを与えた後、すぐに氷中に静置した。
【0062】
SOC培地を1ml加え、37℃で1時間インキュベート後、LB寒天培地(カナマイシン、X−gal、IPTG含有)に塗沫し、37℃で一晩培養後、生えてきたコロニーを取得した。
【0063】
[pyrF遺伝子破壊ベクターの確認]
上記(pyrF遺伝子破壊ベクターpk18-dpryFの構築)で生育が見られたコロニーを、
カナマイシンを添加したLB培地に移植した後、コロニーダイレクトPCRを行い、インサートの確認を行った。プライマーは表1に示すpyrF-up-F1(配列番号1)、pyrF-dn-R1(配列番号4)を用いた。コロニーダイレクトPCRの条件を表4に示す。
【0064】
【表4】
【0065】
得られたPCR産物について、電気泳動によりバンドを確認した。
【0066】
バンドが確認できた株を、カナマイシンを添加したLB液体培地で一晩培養し、プラスミド抽出を行った。
【0067】
吸光度による濃度測定および電気泳動による確認後、シーケンスによる塩基配列の解読を行って目的のpyrF遺伝子破壊ベクターpk18-dpryFが構築できていることを確認した。
【0068】
1.2. M. thermoacetica ATCC 39073株のpyrF遺伝子破壊株(ΔpyrF株)の作製
1.1.で構築したpyrF遺伝子破壊ベクターpk18-dpryFを、以下の手順で、M. thermoacetica ATCC 39073株に導入し、ダブルクロスオーバーの相同性組換えによってpyrF遺伝子が破壊された株(ΔpyrF株)を選抜した。
【0069】
[M. thermoacetica ATCC 39073株へのpyrF遺伝子破壊ベクターpk18-dpryFの導入]
272mMスクロース、16mM HEPESの組成で、水酸化カリウムを用いてpH 7に合わせたHS bufferを調製し、20分間煮沸した後、さらに20分間N2ガ
スで置換した。
【0070】
80%の水素、20%の二酸化炭素の混合ガスを基質とし、改変型ATCC 1754
PETC培地、あるいは、グリシンを終濃度5g/Lになるように添加した改変型AT
CC 1754 PETC培地でM. thermoacetica ATCC39073株を培養した。
【0071】
菌体濃度がOD600で約0.3になるまで培養し、培養液約100ml分を集菌した後、HS bufferで菌体を2度洗浄した。
【0072】
洗浄した菌体を適当な量のHS buffer(約3ml)に懸濁し、懸濁液380μ
lとプラスミド20μlを混合した。
【0073】
Bio-Rad Gene Pulser(登録商標)、及び0.2cm gapのキュベット(バイオラ
ッド製)を用いて、1.5kV、500Ω、50μFまたは2.0kV、500Ω、50μFでエレクトロポレーションを行った。
【0074】
エレクトロポレーション後の懸濁液を、ピルビン酸40mMの終濃度で添加した5mlの培地に植菌し、55℃で2日間培養後に、ウラシル10μg/ml、5−フルオロオロ
チン酸(5−FOA)0.2%の終濃度で添加した寒天培地に植菌し、ロールチューブを作製した。
【0075】
[ダイレクトPCRによるpyrF遺伝子破壊株(ΔpyrF株)の確認]
上記寒天培地に形成された20個のコロニーを、5 mlのウラシル10μg/ml、5−FOA 0.2%の終濃度で添加した液体培地に植菌し、培養3日目に培地が濁っていることが確認できた6株を選択し、培養液1mlを集菌した。
【0076】
アクロモペプチダーゼ(20mg/ml)+リゾチーム(20mg/ml)の入ったTE
buffer 20μlで懸濁し、37℃で5分間インキュベートし、DMSOを20μl添加して懸濁し、PCRの鋳型とした。
【0077】
表5に示すプライマーのうち、pyrF-up-F2(配列番号5)及びpyrF-dn-R2(配列番号6)、pyrF-up-F3(配列番号7)及びpyrF-dn-R3(配列番号8)、又は、pyrF-F(配列番号9)及びpyrF-R(配列番号10)の組み合わせを用い、各々の組合せについて、表6に示す条件でコロニーダイレクトPCRを行い、電気泳動によりバンドを確認した。図3は各プライマーの位置及び予想される断片の長さを示している。図3は各プライマーの位置及び予想される断片の長さを示している。
【0078】
【表5】
【0079】
【表6】
【0080】
<評価>
ロールチューブ法によるコロニー形成を試みた結果、多数のコロニーを得た。そこで、コロニーを20株選択し、液体培地(10μg/mlウラシル、0.2% 5−FOA)にて培養した。菌体の生育が確認できた培養液から菌体を集菌し、ダイレクトPCRによる確認を行った。
【0081】
培養3日目に増殖が確認できた6株について、プライマーpyrF-up-F2及びpyrF-dn-R2、又は、pyrF-up-F3及びpyrF-dn-R3の組み合わせを用い、pyrFの外側からPCRを行ったところ、6株中1株において野生株よりも短いバンドが確認できた。
【0082】
さらに、プライマーpyrF-F及びpyrF−Rの組み合わせを用い、pyrFの内
側をPCRしたところ前述の株においてはバンドが確認できなかった。
【0083】
これらの結果から、野生株よりも短いバンドを確認できた株がpyrF遺伝子破壊株(ΔpyrF株)である可能性があると判断し、染色体抽出を行った後にもう一度PCRを行った。その結果、ダイレクトPCRの際と同様のバンドパターンが得られた。
【0084】
さらに、pyrF遺伝子が破壊された部分には制限酵素SpeIサイトが1か所付与されることから(プライマーpyrF-up-R1、pyrF-dn-F1参照)、上記のPCR産物についてSpeIで処理したところ、SpeIサイトで切断が起こり、バンドが2本になることを確認した。
【0085】
次に、pyrF遺伝子破壊候補株についてウラシル要求性試験を行った。酵母エキスを除いた改変型ATCC 1754 PETC培地にpyrF遺伝子破壊株を植菌し、10μg/mlの終濃度でウラシルを添加した場合と添加しなかった場合で増殖の確認を行っ
た。
【0086】
その結果、ウラシルを添加したサンプルでは培養2日目には菌体の増殖が確認できたが、ウラシルを添加しなかったサンプルにおいては増殖が確認できなかった。これらの結果から、pyrF遺伝子の破壊が確認できた。
【0087】
このpyrF遺伝子破壊株は、MTA−D−pF株(受託番号:NITE P−1057)として独立行政法人製品評価技術基盤機構特許微生物寄託センターに寄託されている。
【0088】
2. M. thermoacetica ATCC 39073 pyrF遺伝子破壊株(ΔpyrF株)の相補試験1
【0089】
2.1. pyrF遺伝子相補ベクター(遺伝子発現ベクター)の構築
以下の手順で、M. thermoacetica ATCC 39073 pyrF遺伝子破壊株(ΔpyrF株)の相補試験を行うために、pyrF遺伝子相補ベクターを構築した。
【0090】
[pyrF遺伝子相補ベクターの構築]
表1に示したプライマーpyrF-up-F1(配列番号1)とpyrF-dn-R1(配列番号4)の組み合わせを用いて、表7に示す条件でPCRを行い、pyrF遺伝子翻訳領域とその5’側の約1000bp、3’側の約1000bpを含む約2.7kbpの遺伝子断片を増幅した。
【0091】
【表7】
【0092】
得られたPCR産物についてMagExtractor Kit(東洋紡製)を用いてゲル抽出を行った。
【0093】
2μlのEcoRV処理をしたプラスミドpBluescript II KS+、または、2μlのSmaI処理をしたプラスミドpK18mobを、8μlのゲル抽出したPCR産物、10μlのLigation high Ver.2(東洋紡製)と混合し、16℃で1時間インキュベートした。
【0094】
Escherichia coli HST08 Premiumコンピテントセル(タカラバイオ製)にライゲーション溶液10μlを添加して軽く攪拌し、氷中に10分静置した後、42℃で1分間ヒートショックを与えた後、すぐに氷中に静置した。
【0095】
SOC培地を1ml加え、37℃で1時間インキュベート後、pBluescript II KS+を用いた場合はLB寒天培地(アンピシリン、X−gal、IPTG含有)に、pK18mobを用
いた場合はLB寒天培地(カナマイシン、X−gal、IPTG含有)に塗沫し、37℃で一晩培養後、生えてきたコロニーを取得した。
【0096】
[pyrF遺伝子相補ベクターの確認]
上記で生育が見られたコロニーをアンピシリン、あるいはカナマイシンを添加したLB寒天培地に移植した後、コロニーダイレクトPCRを行い、インサートの確認を行った。プライマーは表1に示すpyrF-up-F1(配列番号1)、pyrF-dn-R1(配列番号4)を用いた。コロニーダイレクトPCRの条件を表8に示す。
【0097】
【表8】
【0098】
さらに、電気泳動によりバンドが確認できた株について、pBluescript II KS+を用いた場合はアンピシリンを、pK18mobを用いた場合はカナマイシン(pK18-epyrF)を添加した
LB液体培地で一晩培養し、プラスミド抽出を行った。
【0099】
吸光度による濃度測定、及び、EcoRI又はPstI処理サンプルの電気泳動を行い、さらに、シーケンスによる塩基配列の解読を行って目的のpyrF遺伝子相補ベクターpK18-epyrFおよびpBS-epyrFが構築できていることを確認した。
【0100】
2.2. M. thermoacetica ATCC 39073 pyrF遺伝子破壊株(ΔpyrF株)へのpyr
F遺伝子相補ベクターpBS-epyrFの導入
以下の手順で、1.2.で構築したM. thermoacetica ATCC 39073 pyrF遺伝子破壊株(
ΔpyrF株)に、2.1.で構築したpyrF遺伝子相補ベクターpBS-epyrFを導入し、相同組換えによる相補試験を行った。
【0101】
[エレクトロポレーションによるpyrF遺伝子相補ベクターpBS-epyrFの導入]
272mMスクロース、16mM HEPESの組成で、水酸化カリウムを用いてpH 6.7に合わせたHS bufferを調製し、20分間煮沸した後、さらに20分間N2ガスで置換した。
【0102】
80%水素、20%二酸化炭素の混合ガスを基質とし、ウラシルを終濃度が10μg/m
lになるように添加した完全合成培地でΔpyrF株を培養した。
【0103】
菌体濃度がOD600で約0.1になるまで培養し、培養液約100ml分を集菌した後、水酸化カリウムでpH7に調整した272mMスクロースバッファーで2度洗浄した。
【0104】
洗浄した菌体を適当な量のHS buffer(約3ml)に懸濁し、懸濁液380μlとプラスミド20μlを混合した。
【0105】
Bio-Rad Gene Pulser(登録商標)、 及び0.2cm gapのキュベット (バイオ
ラッド製)を用いて、1.5kV、500Ω、50μF、または2.0kV、500Ω、50μFでエレクトロポレーションを行った。
【0106】
エレクトロポレーション後の懸濁液をウラシル10μg/ml、ピルビン酸40mMの
終濃度で添加した5mlの完全合成培地に植菌し、55℃で2日間培養後、完全合成培地で洗浄し、完全合成培地と寒天を含む培地に植菌し、ロールチューブを作製した。
【0107】
[ダイレクトPCRによるpyrF遺伝子相補株の確認]
上記で得られたコロニーを5mlの完全合成培地に植菌し、培養4日目に培地が濁っているサンプルを選択し、培養液1mlを集菌した。
【0108】
アクロモペプチダーゼ(20mg/ml)+リゾチーム(20mg/ml)の入ったTE
buffer10μlで懸濁し、37℃で5分間インキュベートし、DMSOを10μl添加して懸濁し、PCRの鋳型とした。
【0109】
表5に示すpyrF-up-F3(配列番号7)、pyrF-dn-R3(配列番号8)の組み合わせのプライマーを用い、表9に示す条件でコロニーダイレクトPCRを行った。
【0110】
電気泳動によりバンドを確認し、pyrF遺伝子相補株が得られたと判断した。
【0111】
【表9】
【0112】
<評価>
培養4日目で培養液が白く濁っていたサンプル4株についてダイレクトPCRによる確認を実施した。結果を図4に示す。
【0113】
図4の電気泳動結果において、レーン1〜4は相補株、レーン5は野生株、レーン6はpyrF破壊株であり、今回確認した4株全てが野生株と同じ約1.6kbpのサイズの位置にバンドを示した。これは、相補プラスミドがエレクトロポレーションによりpyrF遺伝子破壊株の細胞内へ取り込まれ、相同組換えによりpyrFが元の場所に挿入されたことを意味している。尚、pyrF破壊株ではバンドの長さは約0.9 bpであり、
予想された大きさであった。
【0114】
3. M. thermoacetica ATCC 39073 pyrF遺伝子破壊株(ΔpyrF株)の相補試験2
【0115】
3.1. 形質転換DNA挿入用相補ベクターの構築
ΔpyrF株に対するpyrF遺伝子相補試験を行う際に、pyrF上流に形質転換遺伝子の一部を挿入し、相同組換えによる染色体加工が可能であることを示すためのプラスミドを構築する。具体的には、Thermoanaerobacter ethanolicus 39E株のlacZ遺伝子の一部(約500bp)を2.1.で構築したpyrF遺伝子相補ベクターpK18-epyrFの
pyrF遺伝子上流へ挿入した。手順を以下に示す。
【0116】
[In−Fusion PCRによる形質転換DNA挿入用相補ベクターの構築]
表10に示すプライマーpyrF-1-R(配列番号11)及びpyrF-1-F(配列番号12)、pyrF-2-R(配列番号13)及びpyrF-2-F(配列番号14)、又は、pyrF-3-R(配列番号15)及びpyrF-3-F(配列番号16)の各組み合わせを用いて、pyrF遺伝子領域を含むベクター領域をPCR増幅した。何れの組合せにおいても、PCRの条件は表11に示した通りである。
【0117】
【表10】
【0118】
【表11】
【0119】
表10に示したプライマーlacZ-500-F(配列番号17)とlacZ-500-R(配列暗号18)を用いて、lacZ遺伝子領域をPCR増幅した。PCRの条件は表12に示した通りである。
【0120】
【表12】
【0121】
各PCR産物をゲル抽出し、表13に示した条件で、In−Fusion PCR(In-Fusion・Advantage PCR Cloning Kit、タカラバイオ製)を行った。
【0122】
【表13】
【0123】
In−Fusionサンプルに滅菌水を50μl加えて希釈し、希釈したサンプル10μlを形質転換に用いた。
【0124】
ダイレクトPCRによりコロニーを選択した。
【0125】
<評価>
pyrFの上流領域へlacZ遺伝子の一部を挿入するために、2.1.で構築したpK18-epyrFを鋳型とし、プライマーpyrF-1-R及びpyrF-1-F、pyrF-2-R及びpyrF-2-F、又は、pyrF-3-R及びpyrF-3-Fの組み合わせを用いてインバースPCRを行った。その際に、プロモーター領域が300bp、203bp、147bpとなるようにプライマーの位置をずらして3パターンのPCR産物を得た(図5及び図6)。さらに、In−FusionPCRを用いてプラスミドへlacZ遺伝子を挿入するため、プライマーにはlacZ遺伝子と相同配列を付加しておいた。表10において配列番号11〜16のSequence下線部はプロモーター領域である。下線以外の部分がプライマーの位置を決める配列であり、配列番号11は図6における丸囲み数字1、配列番号12は図6における丸囲み数字2、配列番号13は図6における丸囲み数字3、配列番号14は図6における丸囲み数字4、配列番号15は図6における丸囲み数字5、配列番号16は図6における丸囲み数字6に、矢印の向きで対応している。
【0126】
In−Fusion PCRおよび大腸菌における形質転換の結果、複数のコロニーを
得た。lacZ-500-FおよびlacZ-500-RをプライマーとしたコロニーダイレクトPCRの結果、全ての株において目的のバンドが確認できた。
【0127】
それらの株の中から3株ずつを培養し、プラスミドを抽出した。プライマーpyrF-1-R及びpyrF-1-Fの組み合わせによるプラスミドをpK18-pyz-1とし、プライマーpyrF-2-R及びpyrF-2-Fの組み合わせによるプラスミドをpK18-pyz-2とし、さらに、プライマーpyrF-3-R及びpyrF-3-Fの組み合わせによるプラスミドをpK18-pyz-3とする。
【0128】
制限酵素KpnI処理による確認の結果、図7に示す通り、全ての株においてlacZ遺伝子の挿入が確認できた。
【0129】
図7の電気泳動結果において、レーン1、2、3は、プロモーター領域300bp(pK18-pyz-1)、レーン4、5、6は、プロモーター領域203bp(pK18-pyz-2)、レーン7、8、9は、プロモーター領域147bp(pK18-pyz-3)に各々対応している。レーン10は、pK18-epyrFである。
【0130】
3.2. M. thermoacetica ATCC 39073 pyrF遺伝子破壊株(ΔpyrF株)への形質転
換DNA挿入用相補ベクターpK18-pyz-1、pK18-pyz-2、pK18-pyz-3の導入
以下の手順で、1.2.で構築したM. thermoacetica ATCC 39073 pyrF遺伝子破壊株(
ΔpyrF株)に、3.1.で構築した形質転換DNA挿入用相補ベクターpK18-pyz-1、pK18-pyz-2、pK18-pyz-3を導入し、相同組換えによる相補試験を行った。
【0131】
[エレクトロポレーションによる形質転換DNA挿入用相補ベクターpK18-pyz-1、pK18-pyz-2、pK18-pyz-3の導入]
272mMスクロース、16mM HEPESの組成で、水酸化カリウムを用いてpH 6.7に合わせたHS bufferを調製し、20分間煮沸した後、さらに20分間N
2ガスで置換した。
【0132】
80%水素、20%二酸化炭素の混合ガスを基質とし、ウラシルを終濃度が10μg/m
lになるように添加した完全合成培地でΔpyrF株を培養した。
【0133】
菌体濃度がOD600で約0.1になるまで培養し、培養液約50ml分を集菌した後、水酸化カリウムでpH7に調整した272mMスクロースバッファーで2度洗浄し、洗浄した菌体を適当な量のHS buffer(約3ml)に懸濁した。
【0134】
懸濁液380μlとプラスミド5〜10μlを混合し、Bio-Rad Gene Pulser(登録商
標)、及び0.2cm gapのキュベット (バイオラッド製)を用いて、1.5 kV、500Ω、50μF、または2.0kV、500Ω、50μFでエレクトロポレーションを行った。
【0135】
エレクトロポレーション後の懸濁液を、ウラシル10μg/mlの終濃度で添加した5
mlの完全合成培地に植菌し、80%水素、20%二酸化炭素の混合ガスを基質として培養し、55℃で2日間培養後、完全合成培地で洗浄し、完全合成培地と寒天を含む培地に植菌し、ロールチューブを作製した。
【0136】
[ダイレクトPCRによるpyrF遺伝子相補株の確認]
上記で得られたコロニーを5mlの完全合成培地に植菌し、培養後に培地が濁っているサンプルを選択し、培養液1mlを集菌した。
【0137】
アクロモペプチダーゼ(20mg/ml)+リゾチーム(20mg/ml)の入ったTE
buffer 10μlで懸濁し、37℃で5分間インキュベートした後、DMSOを
10μl添加して懸濁し、PCRの鋳型とした。
【0138】
表5に示したpyrF-up-F3(配列番号7)、pyrF-dn-R3(配列番号8)の組み合わせのプライマーを用い、表14に示す条件でコロニーダイレクトPCRを行った。
【0139】
電気泳動によりバンドを確認し、pyrF遺伝子相補株が得られたと判断した。
【0140】
【表14】
【0141】
<評価>
計20コロニー取得し、そのうち8株についてダイレクトPCRによる確認を実施した(図8)。
【0142】
図8の電気泳動結果において、レーン1〜8は、コロニー単離株、レーン9は、ΔpyrF株ダイレクト、レーン10は、ATCC39073野生株ダイレクト、レーン11は、ATCC39073野生株の抽出DNA、レーン12は、形質転換DNA挿入用相補ベクターpK18-pyz-1、レーン13は、pyrF遺伝子破壊ベクターpk18-dpryFに各々対応している。
【0143】
図8に示される通り、4株(レーン3、4、5、6)で野生株(レーン10、11)より大きいサイズの約2.1kbpの位置にバンドが見られ、約500bpのLacZ遺伝子(形質転換遺伝子)の導入が確認された。残り4株については野生株と同じ約1.6kbpのサイズの位置にバンドを示した。
【0144】
次に、6株分のPCR産物を精製し、制限酵素KpnIで処理して電気泳動にて確認した。KpnIの制限酵素サイトは図9の位置にある。PCR産物をKpnIで処理すると2本のバンドが見えるが、形質転換遺伝子が挿入されていた場合には下側のバンドが野生株の時よりも大きい位置に現れる。結果を図10に示す。
【0145】
図10において、レーン1〜6は、コロニー単離株、レーン7は、ΔpyrF株ダイレクト、レーン8は、ATCC39073野生株の抽出DNA、レーン9は、形質転換DNA挿入用
相補ベクターpK18-pyz-1、レーン10は、形質転換DNA挿入用相補ベクターpK18-pyz-2、レーン11は、形質転換DNA挿入用相補ベクターpK18-pyz-3に各々対応している。
【0146】
図10に示される通り、レーン1、2は野生株と同じバンドが確認され、レーン3、4、5、6では野生株より大きい位置にバンドが見られ、形質転換遺伝子LacZの挿入が確認された。
【0147】
さらに、ダイレクトPCRではなく、増殖によって得られた菌体からTotal DNAを抽出し、同様の条件でPCRを行って形質転換遺伝子LacZの挿入を確認した結果、ダイレクトPCRの時と同じ結果が得られた。
【特許請求の範囲】
【請求項1】
モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子が欠失又は破壊されたウラシル要求性株を作出する際に用いられるプライマーセットであって、
前記オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する上流領域を増幅する配列番号1及び配列番号2で示されることを特徴とするプライマーセット。
【請求項2】
モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子が欠失又は破壊されたウラシル要求性株を作出する際に用いられるプライマーセットであって、
前記オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する下流領域を増幅する配列番号3及び配列番号4で示されることを特徴とするプライマーセット。
【請求項3】
モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子が欠失又は破壊されたウラシル要求性株を作出する際に用いられるプライマーセットであって、
前記オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する上流領域を増幅する配列番号1及び配列番号2で示されプライマーセット、及び、該オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する下流領域を増幅する配列番号3及び配列番号4で示されるプライマーセットを備えることを特徴とするプライマーセット。
【請求項4】
請求項1〜3の何れかに記載のプライマーセットを用いて、モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子を欠失又は破壊してウラシル要求性株を作出することを特徴とする相同性組み換え方法。
【請求項1】
モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子が欠失又は破壊されたウラシル要求性株を作出する際に用いられるプライマーセットであって、
前記オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する上流領域を増幅する配列番号1及び配列番号2で示されることを特徴とするプライマーセット。
【請求項2】
モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子が欠失又は破壊されたウラシル要求性株を作出する際に用いられるプライマーセットであって、
前記オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する下流領域を増幅する配列番号3及び配列番号4で示されることを特徴とするプライマーセット。
【請求項3】
モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子が欠失又は破壊されたウラシル要求性株を作出する際に用いられるプライマーセットであって、
前記オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する上流領域を増幅する配列番号1及び配列番号2で示されプライマーセット、及び、該オロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子に隣接する下流領域を増幅する配列番号3及び配列番号4で示されるプライマーセットを備えることを特徴とするプライマーセット。
【請求項4】
請求項1〜3の何れかに記載のプライマーセットを用いて、モーレラ属細菌において、相同性組換えによってオロチジン−5−リン酸デカルボキシラーゼをコードする遺伝子を欠失又は破壊してウラシル要求性株を作出することを特徴とする相同性組み換え方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】

【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2012−249573(P2012−249573A)
【公開日】平成24年12月20日(2012.12.20)
【国際特許分類】
【出願番号】特願2011−124459(P2011−124459)
【出願日】平成23年6月2日(2011.6.2)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度独立行政法人科学技術振興機構 研究成果最適展開支援事業 産業技術力強化法第19条の適用を受ける特許出願
【出願人】(000005902)三井造船株式会社 (1,723)
【出願人】(504136568)国立大学法人広島大学 (924)
【出願人】(304019399)国立大学法人岐阜大学 (289)
【Fターム(参考)】
【公開日】平成24年12月20日(2012.12.20)
【国際特許分類】
【出願日】平成23年6月2日(2011.6.2)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度独立行政法人科学技術振興機構 研究成果最適展開支援事業 産業技術力強化法第19条の適用を受ける特許出願
【出願人】(000005902)三井造船株式会社 (1,723)
【出願人】(504136568)国立大学法人広島大学 (924)
【出願人】(304019399)国立大学法人岐阜大学 (289)
【Fターム(参考)】
[ Back to top ]